User login
The fog may be lifting
One of the common symptoms described by postconcussion patients is that their heads feel a bit foggy. It may not be simply by chance that “foggy” is the best word to describe the atmosphere surrounding the entire field of concussion diagnosis and management.
Back in the Dark Ages, when the diagnosis of concussion was a simpler binary call, the issue of management seldom created much discussion. If the patient lost consciousness or was amnesic, he (it was less frequently she) could return to activity when his headache was gone and he could remember what he was supposed to do when the quarterback called for a “Red 34, Drive Right Smash” play. That may have even been during the second half of the game in which he was injured.
As it became more widely understood that the diagnosis of concussion didn’t require loss of consciousness and that repeated concussions could have serious sequelae, management became a bit fuzzier. No one had thought much about the recuperative process. Into this vacuum came a wide variety of researchers and providers. Not surprisingly, much of their advice was based on unproven assumptions, including the concept of “brain rest.”
It has taken time, but fortunately, folks with patience and wisdom have questioned these assumptions and begun collecting data. The result of these investigations and others has prompted the American Academy of Pediatrics to publish an updated set of guidelines on concussion management that includes the observation that extended school absence may slow the rehabilitation process (Pediatrics. 2018 Dec. doi: 10.1542/peds.2018-3074).
It is becoming clear that management of concussion can be rather complex and must be individualized to each patient. In my experience, the postconcussion period can unmask behavioral, cognitive, and emotional problems that were preexisting but had received little or no attention. For example, the trauma of the event may trigger anxiety about further injury or exacerbate depression that had been building for years. The student who “couldn’t do algebra” following a head injury may have had a lifelong learning disability that had gone unnoticed. The student athlete with prolonged postconcussion symptoms may indeed have another more serious problem. Hopefully, the new guidelines from the AAP will be a first step toward a more thoughtful and scientifically driven approach to concussion management.

It would be nice if that approach could filter down to the management of the more common but less dramatic pediatric injuries. There is hope. Choosing Wisely – a patient/parent–targeted initiative by the American Board of Internal Medicine Foundation in cooperation with the AAP – points out that, although half of the pediatric head injury patients seen in emergency departments received CT scan, only a third of those studies were indicated. Parents are encouraged to learn more about the risks of CT scans and question the physician when one is recommended.
But, doctors’ habits and old wives’ tales die slowly. I hope that you no longer recommend that parents keep their children awake after a head injury, or wake them every hour to check their pupils. Those counterproductive recommendations make about as much sense as staying out of the swimming pool for an hour after eating a chocolate chip cookie.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
One of the common symptoms described by postconcussion patients is that their heads feel a bit foggy. It may not be simply by chance that “foggy” is the best word to describe the atmosphere surrounding the entire field of concussion diagnosis and management.
Back in the Dark Ages, when the diagnosis of concussion was a simpler binary call, the issue of management seldom created much discussion. If the patient lost consciousness or was amnesic, he (it was less frequently she) could return to activity when his headache was gone and he could remember what he was supposed to do when the quarterback called for a “Red 34, Drive Right Smash” play. That may have even been during the second half of the game in which he was injured.
As it became more widely understood that the diagnosis of concussion didn’t require loss of consciousness and that repeated concussions could have serious sequelae, management became a bit fuzzier. No one had thought much about the recuperative process. Into this vacuum came a wide variety of researchers and providers. Not surprisingly, much of their advice was based on unproven assumptions, including the concept of “brain rest.”
It has taken time, but fortunately, folks with patience and wisdom have questioned these assumptions and begun collecting data. The result of these investigations and others has prompted the American Academy of Pediatrics to publish an updated set of guidelines on concussion management that includes the observation that extended school absence may slow the rehabilitation process (Pediatrics. 2018 Dec. doi: 10.1542/peds.2018-3074).
It is becoming clear that management of concussion can be rather complex and must be individualized to each patient. In my experience, the postconcussion period can unmask behavioral, cognitive, and emotional problems that were preexisting but had received little or no attention. For example, the trauma of the event may trigger anxiety about further injury or exacerbate depression that had been building for years. The student who “couldn’t do algebra” following a head injury may have had a lifelong learning disability that had gone unnoticed. The student athlete with prolonged postconcussion symptoms may indeed have another more serious problem. Hopefully, the new guidelines from the AAP will be a first step toward a more thoughtful and scientifically driven approach to concussion management.
It would be nice if that approach could filter down to the management of the more common but less dramatic pediatric injuries. There is hope. Choosing Wisely – a patient/parent–targeted initiative by the American Board of Internal Medicine Foundation in cooperation with the AAP – points out that, although half of the pediatric head injury patients seen in emergency departments received CT scan, only a third of those studies were indicated. Parents are encouraged to learn more about the risks of CT scans and question the physician when one is recommended.
But, doctors’ habits and old wives’ tales die slowly. I hope that you no longer recommend that parents keep their children awake after a head injury, or wake them every hour to check their pupils. Those counterproductive recommendations make about as much sense as staying out of the swimming pool for an hour after eating a chocolate chip cookie.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
One of the common symptoms described by postconcussion patients is that their heads feel a bit foggy. It may not be simply by chance that “foggy” is the best word to describe the atmosphere surrounding the entire field of concussion diagnosis and management.
Back in the Dark Ages, when the diagnosis of concussion was a simpler binary call, the issue of management seldom created much discussion. If the patient lost consciousness or was amnesic, he (it was less frequently she) could return to activity when his headache was gone and he could remember what he was supposed to do when the quarterback called for a “Red 34, Drive Right Smash” play. That may have even been during the second half of the game in which he was injured.
As it became more widely understood that the diagnosis of concussion didn’t require loss of consciousness and that repeated concussions could have serious sequelae, management became a bit fuzzier. No one had thought much about the recuperative process. Into this vacuum came a wide variety of researchers and providers. Not surprisingly, much of their advice was based on unproven assumptions, including the concept of “brain rest.”
It has taken time, but fortunately, folks with patience and wisdom have questioned these assumptions and begun collecting data. The result of these investigations and others has prompted the American Academy of Pediatrics to publish an updated set of guidelines on concussion management that includes the observation that extended school absence may slow the rehabilitation process (Pediatrics. 2018 Dec. doi: 10.1542/peds.2018-3074).
It is becoming clear that management of concussion can be rather complex and must be individualized to each patient. In my experience, the postconcussion period can unmask behavioral, cognitive, and emotional problems that were preexisting but had received little or no attention. For example, the trauma of the event may trigger anxiety about further injury or exacerbate depression that had been building for years. The student who “couldn’t do algebra” following a head injury may have had a lifelong learning disability that had gone unnoticed. The student athlete with prolonged postconcussion symptoms may indeed have another more serious problem. Hopefully, the new guidelines from the AAP will be a first step toward a more thoughtful and scientifically driven approach to concussion management.
It would be nice if that approach could filter down to the management of the more common but less dramatic pediatric injuries. There is hope. Choosing Wisely – a patient/parent–targeted initiative by the American Board of Internal Medicine Foundation in cooperation with the AAP – points out that, although half of the pediatric head injury patients seen in emergency departments received CT scan, only a third of those studies were indicated. Parents are encouraged to learn more about the risks of CT scans and question the physician when one is recommended.
But, doctors’ habits and old wives’ tales die slowly. I hope that you no longer recommend that parents keep their children awake after a head injury, or wake them every hour to check their pupils. Those counterproductive recommendations make about as much sense as staying out of the swimming pool for an hour after eating a chocolate chip cookie.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
2019 ID update for dermatologists: Ticks are the “ride of choice” for arthropods
ORLANDO – New tricks from ticks, near-zero Zika, and the perils of personal grooming: Dermatologists have a lot to think about along the infectious disease spectrum in 2019, according to Justin Finch, MD, speaking at the Orlando Dermatology Aesthetic and Clinical Conference.

Anaphylaxis from alpha-gal syndrome is on the rise, caused in part by the geographic spread of the Lone Star tick. Beginning in 2006, isolated cases of an anaphylactic reaction to cetuximab, the epidermal growth factor receptor antagonist used to treat certain cancers, began to be seen in a curious geographic distribution. “The anaphylaxis cases were restricted to the southeastern United States, the home of the Lone Star tick,” said Dr. Finch, of the department of dermatology at the University of Connecticut, Farmington.
With some detective work, physicians and epidemiologists eventually determined that patients were reacting to an oligosaccharide called galactose-alpha–1,3-galactose (alpha-gal) found in cetuximab. This protein is also found in the meat of nonprimate mammals; individuals in the southeastern United States, where the Lone Star tick is endemic, had been sensitized via exposure to alpha-gal from Lone Star tick bites.
“Alpha-gal syndrome is on the rise,” said Dr. Finch, driven by the increased spread of this tick. Individuals who are sensitized develop delayed anaphylaxis 2-7 hours after ingesting red meat such as beef, pork, or lamb. “Ask about it,” said Dr. Finch, in patients who develop urticaria, dyspnea, angioedema, or hypotension without a clear offender. Because of the delay between allergen ingestion and anaphylaxis, it can be hard to connect the dots.
A number of drugs other than cetuximab contain alpha-gal, so patients must also be told to avoid these agents, said Dr. Finch, who noted that alpha-gal syndrome isn’t the only emerging culprit for tick-borne diseases. “The tick is the ride of choice for arthropod-borne diseases in the U.S.,” he added. “Year after year, tick-borne diseases top mosquito-borne diseases in the U.S.” Zika’s explosion in 2016 made that year the exception to the rule.
Now, Zika virus may be on the wane – the number of case reports have plummeted both in the United States and in Central and South America this past year – but it hasn’t completely gone away. “It looks like it fell off all the maps,” but the virus is still present at low levels, he said.
When Zika virus is symptomatic, there’s often a nonspecific maculopapular rash. Critically, Dr. Finch said, “women with a rash are four times as likely to have adverse congenital outcomes. This is the important point for us to take home as dermatologists. ... It’s really important to have a high index of suspicion and to screen these women as they are coming into our clinic.”
Turning back to ticks, Lyme disease continues to be a problem in endemic areas in the Northeast, the mid-Atlantic region, and the Midwest, said Dr. Finch, so it’s a perennial on the differential diagnosis for dermatologists.
An Asian tick new to North America was seen for the first time in New Jersey in the summer of 2017. The Asian longhorned tick carries a phlebovirus that causes severe fever with thrombocytopenia syndrome, a disease with a 15% fatality rate. The reservoir host of this virus in Asia isn’t known, said Dr. Finch, adding that no cases of the virus have yet been seen in the United States. As of November 2018, according to the Centers for Disease Control and Prevention, the tick had been found in nine states (Arkansas, Connecticut, Maryland, North Carolina, New Jersey, New York, Pennsylvania, Virginia, and West Virginia).
“What’s not on the rise? Pubic lice. We are destroying their natural habitat!” said Dr. Finch, citing surveys about personal grooming that show that more than 90% of women remove at least some of their pubic hair. Most college campuses are currently reporting essentially no cases of pubic lice, he noted.
However, the same personal grooming practices may be contributing to increases in molluscum contagiosum, herpes simplex virus, some strains of human papillomavirus, and cutaneous Streptococcus pyogenes infections, he said.
Another STI has had a resurgence in geographic pockets around the nation and among specific populations, said Dr. Finch. Syphilis is on the rise among gay and bisexual men and African Americans. Known as the “great imitator,” syphilis should be on the differential for dermatologists when the clinical picture isn’t quite adding up. “Think of this, and screen with an RPR [rapid plasma reagin],” he said.
Finally, an old enemy is back: A total of 11 measles outbreaks were reported in 2018. “We need to know about measles because of the complications,” said Dr. Finch. Even years later, such dire sequelae as subacute sclerosing panencephalitis can crop up, he added.
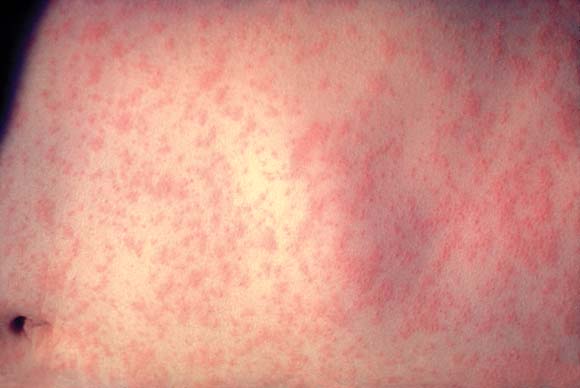
After a 2-week incubation period, measles begins with a fever and cough, congestion, and conjunctivitis. The rash begins on the head and spreads inferiorly by day 3. As the rash blooms, the classic morbilliform eruption becomes apparent. A biopsy of affected skin will be nonspecific; measles is diagnosed with a nasopharyngeal culture and serologic assay. Dr. Finch pointed out that dermatologists are unlikely to see measles in its earliest stages because their expertise will be called on only after it becomes clear that the patient is not experiencing just a mild illness with a viral exanthem.
When there’s suspicion for measles, a full-body skin exam is needed. “Koplik’s spots – the gray white papules on the buccal mucosa – are not pathognomonic in themselves, but in the clinical scenario of a person with measles” they can help the dermatologist make a definitive call, he said.
Vitamin A can be given to a patient with active measles, but prevention via immunization at age 12 months and 5 years is the only way to stop the disease, Dr. Finch noted.
Dr. Finch reported that he has no relevant conflicts of interest.
ORLANDO – New tricks from ticks, near-zero Zika, and the perils of personal grooming: Dermatologists have a lot to think about along the infectious disease spectrum in 2019, according to Justin Finch, MD, speaking at the Orlando Dermatology Aesthetic and Clinical Conference.
Anaphylaxis from alpha-gal syndrome is on the rise, caused in part by the geographic spread of the Lone Star tick. Beginning in 2006, isolated cases of an anaphylactic reaction to cetuximab, the epidermal growth factor receptor antagonist used to treat certain cancers, began to be seen in a curious geographic distribution. “The anaphylaxis cases were restricted to the southeastern United States, the home of the Lone Star tick,” said Dr. Finch, of the department of dermatology at the University of Connecticut, Farmington.
With some detective work, physicians and epidemiologists eventually determined that patients were reacting to an oligosaccharide called galactose-alpha–1,3-galactose (alpha-gal) found in cetuximab. This protein is also found in the meat of nonprimate mammals; individuals in the southeastern United States, where the Lone Star tick is endemic, had been sensitized via exposure to alpha-gal from Lone Star tick bites.
“Alpha-gal syndrome is on the rise,” said Dr. Finch, driven by the increased spread of this tick. Individuals who are sensitized develop delayed anaphylaxis 2-7 hours after ingesting red meat such as beef, pork, or lamb. “Ask about it,” said Dr. Finch, in patients who develop urticaria, dyspnea, angioedema, or hypotension without a clear offender. Because of the delay between allergen ingestion and anaphylaxis, it can be hard to connect the dots.
A number of drugs other than cetuximab contain alpha-gal, so patients must also be told to avoid these agents, said Dr. Finch, who noted that alpha-gal syndrome isn’t the only emerging culprit for tick-borne diseases. “The tick is the ride of choice for arthropod-borne diseases in the U.S.,” he added. “Year after year, tick-borne diseases top mosquito-borne diseases in the U.S.” Zika’s explosion in 2016 made that year the exception to the rule.
Now, Zika virus may be on the wane – the number of case reports have plummeted both in the United States and in Central and South America this past year – but it hasn’t completely gone away. “It looks like it fell off all the maps,” but the virus is still present at low levels, he said.
When Zika virus is symptomatic, there’s often a nonspecific maculopapular rash. Critically, Dr. Finch said, “women with a rash are four times as likely to have adverse congenital outcomes. This is the important point for us to take home as dermatologists. ... It’s really important to have a high index of suspicion and to screen these women as they are coming into our clinic.”
Turning back to ticks, Lyme disease continues to be a problem in endemic areas in the Northeast, the mid-Atlantic region, and the Midwest, said Dr. Finch, so it’s a perennial on the differential diagnosis for dermatologists.
An Asian tick new to North America was seen for the first time in New Jersey in the summer of 2017. The Asian longhorned tick carries a phlebovirus that causes severe fever with thrombocytopenia syndrome, a disease with a 15% fatality rate. The reservoir host of this virus in Asia isn’t known, said Dr. Finch, adding that no cases of the virus have yet been seen in the United States. As of November 2018, according to the Centers for Disease Control and Prevention, the tick had been found in nine states (Arkansas, Connecticut, Maryland, North Carolina, New Jersey, New York, Pennsylvania, Virginia, and West Virginia).
“What’s not on the rise? Pubic lice. We are destroying their natural habitat!” said Dr. Finch, citing surveys about personal grooming that show that more than 90% of women remove at least some of their pubic hair. Most college campuses are currently reporting essentially no cases of pubic lice, he noted.
However, the same personal grooming practices may be contributing to increases in molluscum contagiosum, herpes simplex virus, some strains of human papillomavirus, and cutaneous Streptococcus pyogenes infections, he said.
Another STI has had a resurgence in geographic pockets around the nation and among specific populations, said Dr. Finch. Syphilis is on the rise among gay and bisexual men and African Americans. Known as the “great imitator,” syphilis should be on the differential for dermatologists when the clinical picture isn’t quite adding up. “Think of this, and screen with an RPR [rapid plasma reagin],” he said.
Finally, an old enemy is back: A total of 11 measles outbreaks were reported in 2018. “We need to know about measles because of the complications,” said Dr. Finch. Even years later, such dire sequelae as subacute sclerosing panencephalitis can crop up, he added.
After a 2-week incubation period, measles begins with a fever and cough, congestion, and conjunctivitis. The rash begins on the head and spreads inferiorly by day 3. As the rash blooms, the classic morbilliform eruption becomes apparent. A biopsy of affected skin will be nonspecific; measles is diagnosed with a nasopharyngeal culture and serologic assay. Dr. Finch pointed out that dermatologists are unlikely to see measles in its earliest stages because their expertise will be called on only after it becomes clear that the patient is not experiencing just a mild illness with a viral exanthem.
When there’s suspicion for measles, a full-body skin exam is needed. “Koplik’s spots – the gray white papules on the buccal mucosa – are not pathognomonic in themselves, but in the clinical scenario of a person with measles” they can help the dermatologist make a definitive call, he said.
Vitamin A can be given to a patient with active measles, but prevention via immunization at age 12 months and 5 years is the only way to stop the disease, Dr. Finch noted.
Dr. Finch reported that he has no relevant conflicts of interest.
ORLANDO – New tricks from ticks, near-zero Zika, and the perils of personal grooming: Dermatologists have a lot to think about along the infectious disease spectrum in 2019, according to Justin Finch, MD, speaking at the Orlando Dermatology Aesthetic and Clinical Conference.
Anaphylaxis from alpha-gal syndrome is on the rise, caused in part by the geographic spread of the Lone Star tick. Beginning in 2006, isolated cases of an anaphylactic reaction to cetuximab, the epidermal growth factor receptor antagonist used to treat certain cancers, began to be seen in a curious geographic distribution. “The anaphylaxis cases were restricted to the southeastern United States, the home of the Lone Star tick,” said Dr. Finch, of the department of dermatology at the University of Connecticut, Farmington.
With some detective work, physicians and epidemiologists eventually determined that patients were reacting to an oligosaccharide called galactose-alpha–1,3-galactose (alpha-gal) found in cetuximab. This protein is also found in the meat of nonprimate mammals; individuals in the southeastern United States, where the Lone Star tick is endemic, had been sensitized via exposure to alpha-gal from Lone Star tick bites.
“Alpha-gal syndrome is on the rise,” said Dr. Finch, driven by the increased spread of this tick. Individuals who are sensitized develop delayed anaphylaxis 2-7 hours after ingesting red meat such as beef, pork, or lamb. “Ask about it,” said Dr. Finch, in patients who develop urticaria, dyspnea, angioedema, or hypotension without a clear offender. Because of the delay between allergen ingestion and anaphylaxis, it can be hard to connect the dots.
A number of drugs other than cetuximab contain alpha-gal, so patients must also be told to avoid these agents, said Dr. Finch, who noted that alpha-gal syndrome isn’t the only emerging culprit for tick-borne diseases. “The tick is the ride of choice for arthropod-borne diseases in the U.S.,” he added. “Year after year, tick-borne diseases top mosquito-borne diseases in the U.S.” Zika’s explosion in 2016 made that year the exception to the rule.
Now, Zika virus may be on the wane – the number of case reports have plummeted both in the United States and in Central and South America this past year – but it hasn’t completely gone away. “It looks like it fell off all the maps,” but the virus is still present at low levels, he said.
When Zika virus is symptomatic, there’s often a nonspecific maculopapular rash. Critically, Dr. Finch said, “women with a rash are four times as likely to have adverse congenital outcomes. This is the important point for us to take home as dermatologists. ... It’s really important to have a high index of suspicion and to screen these women as they are coming into our clinic.”
Turning back to ticks, Lyme disease continues to be a problem in endemic areas in the Northeast, the mid-Atlantic region, and the Midwest, said Dr. Finch, so it’s a perennial on the differential diagnosis for dermatologists.
An Asian tick new to North America was seen for the first time in New Jersey in the summer of 2017. The Asian longhorned tick carries a phlebovirus that causes severe fever with thrombocytopenia syndrome, a disease with a 15% fatality rate. The reservoir host of this virus in Asia isn’t known, said Dr. Finch, adding that no cases of the virus have yet been seen in the United States. As of November 2018, according to the Centers for Disease Control and Prevention, the tick had been found in nine states (Arkansas, Connecticut, Maryland, North Carolina, New Jersey, New York, Pennsylvania, Virginia, and West Virginia).
“What’s not on the rise? Pubic lice. We are destroying their natural habitat!” said Dr. Finch, citing surveys about personal grooming that show that more than 90% of women remove at least some of their pubic hair. Most college campuses are currently reporting essentially no cases of pubic lice, he noted.
However, the same personal grooming practices may be contributing to increases in molluscum contagiosum, herpes simplex virus, some strains of human papillomavirus, and cutaneous Streptococcus pyogenes infections, he said.
Another STI has had a resurgence in geographic pockets around the nation and among specific populations, said Dr. Finch. Syphilis is on the rise among gay and bisexual men and African Americans. Known as the “great imitator,” syphilis should be on the differential for dermatologists when the clinical picture isn’t quite adding up. “Think of this, and screen with an RPR [rapid plasma reagin],” he said.
Finally, an old enemy is back: A total of 11 measles outbreaks were reported in 2018. “We need to know about measles because of the complications,” said Dr. Finch. Even years later, such dire sequelae as subacute sclerosing panencephalitis can crop up, he added.
After a 2-week incubation period, measles begins with a fever and cough, congestion, and conjunctivitis. The rash begins on the head and spreads inferiorly by day 3. As the rash blooms, the classic morbilliform eruption becomes apparent. A biopsy of affected skin will be nonspecific; measles is diagnosed with a nasopharyngeal culture and serologic assay. Dr. Finch pointed out that dermatologists are unlikely to see measles in its earliest stages because their expertise will be called on only after it becomes clear that the patient is not experiencing just a mild illness with a viral exanthem.
When there’s suspicion for measles, a full-body skin exam is needed. “Koplik’s spots – the gray white papules on the buccal mucosa – are not pathognomonic in themselves, but in the clinical scenario of a person with measles” they can help the dermatologist make a definitive call, he said.
Vitamin A can be given to a patient with active measles, but prevention via immunization at age 12 months and 5 years is the only way to stop the disease, Dr. Finch noted.
Dr. Finch reported that he has no relevant conflicts of interest.
EXPERT ANALYSIS FROM ODAC 2019
The other side of activity
While the increasing prevalence of obesity has been obvious for nearly half a century, it is only in the last decade or two that the focus has broadened to include the associated decline in physical activity.

A recent paper attempts to sharpen that focus by examining the timeline of that decline (Pediatrics 2019 Jan. doi: 10.1542/peds.2018-0994.). Using a device incorporating five sensors, one of which was an accelerometer, the investigators collected data from 600 children from five European countries accumulating more than 1,200 observations. What they discovered was that their subjects’ physical activity declined by 75 minutes per day from ages 6 to 11 years of age while sedentary behavior increased more than 100 minutes over that same interval. This observation is concerning because previous attention has focused intervention on adolescents assuming that the erosion of physical activity was occurring primarily during the teen years.
Not surprisingly the authors suggest that more studies should be performed to aid in the design of more sharply targeted interventions. While more information may be helpful, their current findings and an abundance of anecdotal observations suggest that to be effective that intervention must begin well before children reach school age.
What should this intervention look like? Currently, the emphasis seems to have been on programs that encourage activity. The National Football League is promoting its NFL Play 60 initiative. The Afterschool Alliance has its Kids on the Move programs. Former First Lady Michelle Obama has been the spokesperson and driving force behind Let’s Move. And, the American Academy of Pediatrics has recently been encouraging both parents and pediatricians to appreciate The Power of Play to encourage children to get into more physical activity. All of these initiatives are well meaning, but I suspect their effectiveness is usually limited to the public awareness they generate.
We seem to have forgotten that there are two sides to the equation. The accelerometer study from Europe should remind us that our initiatives should also be addressing the problem of epidemic inactivity with equal vigor. Creating programs that focus on increasing activity can be expensive. There may be costs for equipment, spaces to be maintained, and staff to be paid. On the other hand, curbing sedentary behavior requires only an adult with the courage to say, “No.” “No, we will have the television for only an hour today.” “No, you can’t play your video game until after dinner.”
While addressing the disciplinary side of the activity-inactivity dichotomy may be relatively inexpensive, it does seem to have a cost on parents. It requires them to buy into the idea that, given even the most-limited supply of objects and infrastructure, most children can keep themselves entertained and active. There does seem to be a small subset of children who enter the world with a sedentary mindset, possibly inherited from their parents. This unfortunate minority will require some creative intervention to achieve a healthy level of activity.
However, most young children who have become accustomed to being amused by sedentary “activities” such as television and video games still retain their innate creativity and natural inclination to be physically active. Unfortunately, unmasking these health-sustaining attributes may require a long and unpleasant weaning period that many parents don’t seem to have the patience to endure. The longer the child has been allowed to engage in sedentary behaviors, the longer this adjustment period will be, yet another argument for early intervention.
Encouraging physical activity is something we should be doing every day in our offices, but it must go hand in hand with an equivalent emphasis on helping parents create a discipline framework that discourages sedentary behavior.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
While the increasing prevalence of obesity has been obvious for nearly half a century, it is only in the last decade or two that the focus has broadened to include the associated decline in physical activity.
A recent paper attempts to sharpen that focus by examining the timeline of that decline (Pediatrics 2019 Jan. doi: 10.1542/peds.2018-0994.). Using a device incorporating five sensors, one of which was an accelerometer, the investigators collected data from 600 children from five European countries accumulating more than 1,200 observations. What they discovered was that their subjects’ physical activity declined by 75 minutes per day from ages 6 to 11 years of age while sedentary behavior increased more than 100 minutes over that same interval. This observation is concerning because previous attention has focused intervention on adolescents assuming that the erosion of physical activity was occurring primarily during the teen years.
Not surprisingly the authors suggest that more studies should be performed to aid in the design of more sharply targeted interventions. While more information may be helpful, their current findings and an abundance of anecdotal observations suggest that to be effective that intervention must begin well before children reach school age.
What should this intervention look like? Currently, the emphasis seems to have been on programs that encourage activity. The National Football League is promoting its NFL Play 60 initiative. The Afterschool Alliance has its Kids on the Move programs. Former First Lady Michelle Obama has been the spokesperson and driving force behind Let’s Move. And, the American Academy of Pediatrics has recently been encouraging both parents and pediatricians to appreciate The Power of Play to encourage children to get into more physical activity. All of these initiatives are well meaning, but I suspect their effectiveness is usually limited to the public awareness they generate.
We seem to have forgotten that there are two sides to the equation. The accelerometer study from Europe should remind us that our initiatives should also be addressing the problem of epidemic inactivity with equal vigor. Creating programs that focus on increasing activity can be expensive. There may be costs for equipment, spaces to be maintained, and staff to be paid. On the other hand, curbing sedentary behavior requires only an adult with the courage to say, “No.” “No, we will have the television for only an hour today.” “No, you can’t play your video game until after dinner.”
While addressing the disciplinary side of the activity-inactivity dichotomy may be relatively inexpensive, it does seem to have a cost on parents. It requires them to buy into the idea that, given even the most-limited supply of objects and infrastructure, most children can keep themselves entertained and active. There does seem to be a small subset of children who enter the world with a sedentary mindset, possibly inherited from their parents. This unfortunate minority will require some creative intervention to achieve a healthy level of activity.
However, most young children who have become accustomed to being amused by sedentary “activities” such as television and video games still retain their innate creativity and natural inclination to be physically active. Unfortunately, unmasking these health-sustaining attributes may require a long and unpleasant weaning period that many parents don’t seem to have the patience to endure. The longer the child has been allowed to engage in sedentary behaviors, the longer this adjustment period will be, yet another argument for early intervention.
Encouraging physical activity is something we should be doing every day in our offices, but it must go hand in hand with an equivalent emphasis on helping parents create a discipline framework that discourages sedentary behavior.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
While the increasing prevalence of obesity has been obvious for nearly half a century, it is only in the last decade or two that the focus has broadened to include the associated decline in physical activity.
A recent paper attempts to sharpen that focus by examining the timeline of that decline (Pediatrics 2019 Jan. doi: 10.1542/peds.2018-0994.). Using a device incorporating five sensors, one of which was an accelerometer, the investigators collected data from 600 children from five European countries accumulating more than 1,200 observations. What they discovered was that their subjects’ physical activity declined by 75 minutes per day from ages 6 to 11 years of age while sedentary behavior increased more than 100 minutes over that same interval. This observation is concerning because previous attention has focused intervention on adolescents assuming that the erosion of physical activity was occurring primarily during the teen years.
Not surprisingly the authors suggest that more studies should be performed to aid in the design of more sharply targeted interventions. While more information may be helpful, their current findings and an abundance of anecdotal observations suggest that to be effective that intervention must begin well before children reach school age.
What should this intervention look like? Currently, the emphasis seems to have been on programs that encourage activity. The National Football League is promoting its NFL Play 60 initiative. The Afterschool Alliance has its Kids on the Move programs. Former First Lady Michelle Obama has been the spokesperson and driving force behind Let’s Move. And, the American Academy of Pediatrics has recently been encouraging both parents and pediatricians to appreciate The Power of Play to encourage children to get into more physical activity. All of these initiatives are well meaning, but I suspect their effectiveness is usually limited to the public awareness they generate.
We seem to have forgotten that there are two sides to the equation. The accelerometer study from Europe should remind us that our initiatives should also be addressing the problem of epidemic inactivity with equal vigor. Creating programs that focus on increasing activity can be expensive. There may be costs for equipment, spaces to be maintained, and staff to be paid. On the other hand, curbing sedentary behavior requires only an adult with the courage to say, “No.” “No, we will have the television for only an hour today.” “No, you can’t play your video game until after dinner.”
While addressing the disciplinary side of the activity-inactivity dichotomy may be relatively inexpensive, it does seem to have a cost on parents. It requires them to buy into the idea that, given even the most-limited supply of objects and infrastructure, most children can keep themselves entertained and active. There does seem to be a small subset of children who enter the world with a sedentary mindset, possibly inherited from their parents. This unfortunate minority will require some creative intervention to achieve a healthy level of activity.
However, most young children who have become accustomed to being amused by sedentary “activities” such as television and video games still retain their innate creativity and natural inclination to be physically active. Unfortunately, unmasking these health-sustaining attributes may require a long and unpleasant weaning period that many parents don’t seem to have the patience to endure. The longer the child has been allowed to engage in sedentary behaviors, the longer this adjustment period will be, yet another argument for early intervention.
Encouraging physical activity is something we should be doing every day in our offices, but it must go hand in hand with an equivalent emphasis on helping parents create a discipline framework that discourages sedentary behavior.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
Precision medicine in rheumatology: Enormous opportunity exists
Advances in precision medicine present enormous opportunity for rheumatology, but optimizing its benefits requires more input from the specialty and a sharper focus on related training for rheumatologists, according to Judith A. James, MD, PhD.

Precision medicine is getting a great deal of attention and is an exciting area, but it is already widely used in the field; think treat-to-target in rheumatoid arthritis, autoantibody testing for patient stratification across various conditions, and individual monitoring and dose escalation to achieve optimal uric acid levels in gout patients, Dr. James, professor of medicine and associate vice provost of clinical and translational science at the University of Oklahoma, Oklahoma City, said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
“We have historically ... actually had the highest number of FDA approved biomarker tests in rheumatology compared to all other specialties until this last couple of years where we’re starting to see this explosion of genetic testing in oncology – and we’ve been doing genetic testing,” she said.
However, there is a great deal more work to be done.
“We still have a long way to go to go to get the right drug at the right dose at the right time in the right patient in order to optimize outcomes in all of these diseases that we are responsible for as rheumatologists,” she said.
The fields of oncology and hematology have been intensely focused on precision medicine – the development of unique therapies based on specific genetic abnormalities in an individual’s tumor – and this focus is apparent in practice patterns: A recent survey of 132 medical oncologists and hematologists/oncologists showed that nearly 90% had ordered DNA sequencing, about 65% do so monthly, and 25% do so weekly.
“Those numbers are just going to continue to climb, and I think will see this in other disciplines as well,” she said.
The possibilities for improved outcomes in rheumatologic conditions using tailored treatments based on individual characteristics are practically limitless, she said, noting the heterogeneity of many rheumatologic conditions.
This is particularly true for systemic lupus erythematosus (SLE) patients, she said.
Identifying patient subsets based on organ involvement, demographics, and biomarkers, for example, could lead to personalized treatments with different doses, routes of administration, and concurrent medications, she explained.
Genetics in SLE
Dr. James highlighted the role of genetics and the value of precision medicine in the SLE setting in a large transancestral association study published in 2017. The investigators analyzed Immunochip genotype data from 27,574 SLE cases and controls and identified 58 distinct non–human leukocyte antigen (HLA) regions in Americans with European ancestry, 9 in those with African ancestry, and 16 in those with Hispanic ancestry. The investigators found that these non-HLA regions included 24 novel to SLE, and in their analysis the researchers were able to refine association signals in previously established regions, extend associations to additional ancestries, and reveal a complex multigenic effect just outside of the HLA region (Nature Commun. 2017;8:16021).
The findings led to a “cumulative hit hypothesis” for autoimmune disease, and help to clarify genetic architecture and ethnic disparities in SLE, they concluded.
“So we now have over a hundred genetic regions that have been associated with lupus, compared to healthy controls,” Dr. James said.
A frustration with genetic data such as these, however, is the challenge of “getting it into the clinic,” she noted.
“I think that looking at individual [single nuclear polymorphisms] is probably not what we’re going to be doing, but we’re seeing a lot of interest in the idea of genetic load,” she said, explaining that it may soon be possible to use genetic load information to evaluate patient risk.
A recent study at her institution looked at lupus risk from another angle: She and her colleagues recontacted family members from Oklahoma Lupus Genetics studies to look more closely at which blood relatives of SLE patients transitioned to SLE, and what factors were associated with that transition when compared with relatives who remained unaffected (Arthritis Rheumatol. 2017;69[3]:630-42).
Among the findings was a higher risk of transitioning among family members with both a positive antinuclear antibody test and a baseline Connective Tissue Disease Screening Questionnaire score indicative of connective tissue disease.
“We also found, of course, biomarkers, or blood markers, that helped us identify the individuals who were at the highest risk of transitioning, so we think a blood test might really be helpful,” she said.
That study also suggested that there may be ways to intervene in SLE patients’ relatives at increased risk for also developing lupus. For example, those who transitioned had increased levels of soluble tumor necrosis factor receptors and the interferon-driven chemokine MCP-3; a prevention trial is now underway, she noted.
Beyond genetics
Genetics are just one piece of the precision medicine puzzle, and other areas of investigation that may help to divide patients into subgroups for more precise treatment include genomics, soluble mediators, and immunophenotyping, Dr. James said.
“It may be that we need different pieces of all of these things to help guide our treatment in lupus patients,” she said.
Longitudinal clinical and blood transcriptional profiling of patients in the Dallas Pediatric SLE cohort, for example, identified a molecular classification system for SLE patients. The analysis of 972 samples from 158 SLE patients and 48 healthy controls, which were collected for up to 4 years, showed that an interferon response signature was present in 784 of the samples.
The investigators found that a plasmablast signature, which is found more in African-American patients than in other populations, best correlates with disease activity and that a neutrophil-related signature is associated with progression to active lupus nephritis (Cell. 2016;165[3]:551-65).
“This is something that will potentially be helpful [in the clinic], and we need to test this in the adult population,” Dr. James said.
The investigators also were able to stratify patients, based on individual immunoprofiling, into seven major groups based on molecular correlates. They concluded that such stratification could help improve the outcomes of clinical trials in SLE.
In another study, researchers looked at longitudinal gene expression in SLE patients by stratifying each of two independent sets of patients (a pediatric cohort and an adult cohort) into three clinically differentiated disease clusters defined by mechanisms of disease progression (Arthritis Rheumatol. Dec 2018;70[12]:2025-35).
The clusters included one showing a correlation between the percentage of neutrophils and disease activity progression, one showing a correlation between the percentage of lymphocytes and disease activity progression, and a third for which the percentage of neutrophils correlated to a lesser degree with disease activity but was functionally more heterogeneous. Patients in the two neutrophil‐driven clusters had an increased risk of developing proliferative nephritis.
The results have implications for treatment, trial design, and understanding of disease etiology, the investigators concluded.
“This may help us in the future as we think about which medicine to start patients on, and which medicines to start patients on first,” Dr. James said.
It is clear that precision medicine will play an increasingly important role in rheumatology, Dr. James said, when considering the context of other findings in recent years, such as those from studies looking at soluble mediators of inflammation associated with disease flare, as well as those that involved extensive immunophenotyping and showed widely divergent transcriptional patterns based on ancestral backgrounds. Other research, such as the BOLD (Biomarkers of Lupus Disease) study, looked at various mechanisms of disease flare.
Numerous types of personalized therapies are being considered in rheumatology, ranging from expanded regulatory T cells to chimeric antigen receptor T cell therapy to risk profiling for disease prevention, just to name a few. Going forward it will be important to perform more systems biology analyses to assemble precision medicine–related data that can inform clinical diagnosis, prognosis, and therapy selection and optimization, she said.
The future of personalized therapies in rheumatology will require more input from rheumatologists on large-scale precision medicine projects such as the National Institutes of Health’s All of Us Research Project and the Million Veteran Program, as well as other similar programs of major health systems, she noted, adding that different types of training and interaction with molecular pathologists, genetic counselors, health coaches, and other key players also are needed.
Dr. James reported having no relevant disclosures.
Advances in precision medicine present enormous opportunity for rheumatology, but optimizing its benefits requires more input from the specialty and a sharper focus on related training for rheumatologists, according to Judith A. James, MD, PhD.
Precision medicine is getting a great deal of attention and is an exciting area, but it is already widely used in the field; think treat-to-target in rheumatoid arthritis, autoantibody testing for patient stratification across various conditions, and individual monitoring and dose escalation to achieve optimal uric acid levels in gout patients, Dr. James, professor of medicine and associate vice provost of clinical and translational science at the University of Oklahoma, Oklahoma City, said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
“We have historically ... actually had the highest number of FDA approved biomarker tests in rheumatology compared to all other specialties until this last couple of years where we’re starting to see this explosion of genetic testing in oncology – and we’ve been doing genetic testing,” she said.
However, there is a great deal more work to be done.
“We still have a long way to go to go to get the right drug at the right dose at the right time in the right patient in order to optimize outcomes in all of these diseases that we are responsible for as rheumatologists,” she said.
The fields of oncology and hematology have been intensely focused on precision medicine – the development of unique therapies based on specific genetic abnormalities in an individual’s tumor – and this focus is apparent in practice patterns: A recent survey of 132 medical oncologists and hematologists/oncologists showed that nearly 90% had ordered DNA sequencing, about 65% do so monthly, and 25% do so weekly.
“Those numbers are just going to continue to climb, and I think will see this in other disciplines as well,” she said.
The possibilities for improved outcomes in rheumatologic conditions using tailored treatments based on individual characteristics are practically limitless, she said, noting the heterogeneity of many rheumatologic conditions.
This is particularly true for systemic lupus erythematosus (SLE) patients, she said.
Identifying patient subsets based on organ involvement, demographics, and biomarkers, for example, could lead to personalized treatments with different doses, routes of administration, and concurrent medications, she explained.
Genetics in SLE
Dr. James highlighted the role of genetics and the value of precision medicine in the SLE setting in a large transancestral association study published in 2017. The investigators analyzed Immunochip genotype data from 27,574 SLE cases and controls and identified 58 distinct non–human leukocyte antigen (HLA) regions in Americans with European ancestry, 9 in those with African ancestry, and 16 in those with Hispanic ancestry. The investigators found that these non-HLA regions included 24 novel to SLE, and in their analysis the researchers were able to refine association signals in previously established regions, extend associations to additional ancestries, and reveal a complex multigenic effect just outside of the HLA region (Nature Commun. 2017;8:16021).
The findings led to a “cumulative hit hypothesis” for autoimmune disease, and help to clarify genetic architecture and ethnic disparities in SLE, they concluded.
“So we now have over a hundred genetic regions that have been associated with lupus, compared to healthy controls,” Dr. James said.
A frustration with genetic data such as these, however, is the challenge of “getting it into the clinic,” she noted.
“I think that looking at individual [single nuclear polymorphisms] is probably not what we’re going to be doing, but we’re seeing a lot of interest in the idea of genetic load,” she said, explaining that it may soon be possible to use genetic load information to evaluate patient risk.
A recent study at her institution looked at lupus risk from another angle: She and her colleagues recontacted family members from Oklahoma Lupus Genetics studies to look more closely at which blood relatives of SLE patients transitioned to SLE, and what factors were associated with that transition when compared with relatives who remained unaffected (Arthritis Rheumatol. 2017;69[3]:630-42).
Among the findings was a higher risk of transitioning among family members with both a positive antinuclear antibody test and a baseline Connective Tissue Disease Screening Questionnaire score indicative of connective tissue disease.
“We also found, of course, biomarkers, or blood markers, that helped us identify the individuals who were at the highest risk of transitioning, so we think a blood test might really be helpful,” she said.
That study also suggested that there may be ways to intervene in SLE patients’ relatives at increased risk for also developing lupus. For example, those who transitioned had increased levels of soluble tumor necrosis factor receptors and the interferon-driven chemokine MCP-3; a prevention trial is now underway, she noted.
Beyond genetics
Genetics are just one piece of the precision medicine puzzle, and other areas of investigation that may help to divide patients into subgroups for more precise treatment include genomics, soluble mediators, and immunophenotyping, Dr. James said.
“It may be that we need different pieces of all of these things to help guide our treatment in lupus patients,” she said.
Longitudinal clinical and blood transcriptional profiling of patients in the Dallas Pediatric SLE cohort, for example, identified a molecular classification system for SLE patients. The analysis of 972 samples from 158 SLE patients and 48 healthy controls, which were collected for up to 4 years, showed that an interferon response signature was present in 784 of the samples.
The investigators found that a plasmablast signature, which is found more in African-American patients than in other populations, best correlates with disease activity and that a neutrophil-related signature is associated with progression to active lupus nephritis (Cell. 2016;165[3]:551-65).
“This is something that will potentially be helpful [in the clinic], and we need to test this in the adult population,” Dr. James said.
The investigators also were able to stratify patients, based on individual immunoprofiling, into seven major groups based on molecular correlates. They concluded that such stratification could help improve the outcomes of clinical trials in SLE.
In another study, researchers looked at longitudinal gene expression in SLE patients by stratifying each of two independent sets of patients (a pediatric cohort and an adult cohort) into three clinically differentiated disease clusters defined by mechanisms of disease progression (Arthritis Rheumatol. Dec 2018;70[12]:2025-35).
The clusters included one showing a correlation between the percentage of neutrophils and disease activity progression, one showing a correlation between the percentage of lymphocytes and disease activity progression, and a third for which the percentage of neutrophils correlated to a lesser degree with disease activity but was functionally more heterogeneous. Patients in the two neutrophil‐driven clusters had an increased risk of developing proliferative nephritis.
The results have implications for treatment, trial design, and understanding of disease etiology, the investigators concluded.
“This may help us in the future as we think about which medicine to start patients on, and which medicines to start patients on first,” Dr. James said.
It is clear that precision medicine will play an increasingly important role in rheumatology, Dr. James said, when considering the context of other findings in recent years, such as those from studies looking at soluble mediators of inflammation associated with disease flare, as well as those that involved extensive immunophenotyping and showed widely divergent transcriptional patterns based on ancestral backgrounds. Other research, such as the BOLD (Biomarkers of Lupus Disease) study, looked at various mechanisms of disease flare.
Numerous types of personalized therapies are being considered in rheumatology, ranging from expanded regulatory T cells to chimeric antigen receptor T cell therapy to risk profiling for disease prevention, just to name a few. Going forward it will be important to perform more systems biology analyses to assemble precision medicine–related data that can inform clinical diagnosis, prognosis, and therapy selection and optimization, she said.
The future of personalized therapies in rheumatology will require more input from rheumatologists on large-scale precision medicine projects such as the National Institutes of Health’s All of Us Research Project and the Million Veteran Program, as well as other similar programs of major health systems, she noted, adding that different types of training and interaction with molecular pathologists, genetic counselors, health coaches, and other key players also are needed.
Dr. James reported having no relevant disclosures.
Advances in precision medicine present enormous opportunity for rheumatology, but optimizing its benefits requires more input from the specialty and a sharper focus on related training for rheumatologists, according to Judith A. James, MD, PhD.
Precision medicine is getting a great deal of attention and is an exciting area, but it is already widely used in the field; think treat-to-target in rheumatoid arthritis, autoantibody testing for patient stratification across various conditions, and individual monitoring and dose escalation to achieve optimal uric acid levels in gout patients, Dr. James, professor of medicine and associate vice provost of clinical and translational science at the University of Oklahoma, Oklahoma City, said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
“We have historically ... actually had the highest number of FDA approved biomarker tests in rheumatology compared to all other specialties until this last couple of years where we’re starting to see this explosion of genetic testing in oncology – and we’ve been doing genetic testing,” she said.
However, there is a great deal more work to be done.
“We still have a long way to go to go to get the right drug at the right dose at the right time in the right patient in order to optimize outcomes in all of these diseases that we are responsible for as rheumatologists,” she said.
The fields of oncology and hematology have been intensely focused on precision medicine – the development of unique therapies based on specific genetic abnormalities in an individual’s tumor – and this focus is apparent in practice patterns: A recent survey of 132 medical oncologists and hematologists/oncologists showed that nearly 90% had ordered DNA sequencing, about 65% do so monthly, and 25% do so weekly.
“Those numbers are just going to continue to climb, and I think will see this in other disciplines as well,” she said.
The possibilities for improved outcomes in rheumatologic conditions using tailored treatments based on individual characteristics are practically limitless, she said, noting the heterogeneity of many rheumatologic conditions.
This is particularly true for systemic lupus erythematosus (SLE) patients, she said.
Identifying patient subsets based on organ involvement, demographics, and biomarkers, for example, could lead to personalized treatments with different doses, routes of administration, and concurrent medications, she explained.
Genetics in SLE
Dr. James highlighted the role of genetics and the value of precision medicine in the SLE setting in a large transancestral association study published in 2017. The investigators analyzed Immunochip genotype data from 27,574 SLE cases and controls and identified 58 distinct non–human leukocyte antigen (HLA) regions in Americans with European ancestry, 9 in those with African ancestry, and 16 in those with Hispanic ancestry. The investigators found that these non-HLA regions included 24 novel to SLE, and in their analysis the researchers were able to refine association signals in previously established regions, extend associations to additional ancestries, and reveal a complex multigenic effect just outside of the HLA region (Nature Commun. 2017;8:16021).
The findings led to a “cumulative hit hypothesis” for autoimmune disease, and help to clarify genetic architecture and ethnic disparities in SLE, they concluded.
“So we now have over a hundred genetic regions that have been associated with lupus, compared to healthy controls,” Dr. James said.
A frustration with genetic data such as these, however, is the challenge of “getting it into the clinic,” she noted.
“I think that looking at individual [single nuclear polymorphisms] is probably not what we’re going to be doing, but we’re seeing a lot of interest in the idea of genetic load,” she said, explaining that it may soon be possible to use genetic load information to evaluate patient risk.
A recent study at her institution looked at lupus risk from another angle: She and her colleagues recontacted family members from Oklahoma Lupus Genetics studies to look more closely at which blood relatives of SLE patients transitioned to SLE, and what factors were associated with that transition when compared with relatives who remained unaffected (Arthritis Rheumatol. 2017;69[3]:630-42).
Among the findings was a higher risk of transitioning among family members with both a positive antinuclear antibody test and a baseline Connective Tissue Disease Screening Questionnaire score indicative of connective tissue disease.
“We also found, of course, biomarkers, or blood markers, that helped us identify the individuals who were at the highest risk of transitioning, so we think a blood test might really be helpful,” she said.
That study also suggested that there may be ways to intervene in SLE patients’ relatives at increased risk for also developing lupus. For example, those who transitioned had increased levels of soluble tumor necrosis factor receptors and the interferon-driven chemokine MCP-3; a prevention trial is now underway, she noted.
Beyond genetics
Genetics are just one piece of the precision medicine puzzle, and other areas of investigation that may help to divide patients into subgroups for more precise treatment include genomics, soluble mediators, and immunophenotyping, Dr. James said.
“It may be that we need different pieces of all of these things to help guide our treatment in lupus patients,” she said.
Longitudinal clinical and blood transcriptional profiling of patients in the Dallas Pediatric SLE cohort, for example, identified a molecular classification system for SLE patients. The analysis of 972 samples from 158 SLE patients and 48 healthy controls, which were collected for up to 4 years, showed that an interferon response signature was present in 784 of the samples.
The investigators found that a plasmablast signature, which is found more in African-American patients than in other populations, best correlates with disease activity and that a neutrophil-related signature is associated with progression to active lupus nephritis (Cell. 2016;165[3]:551-65).
“This is something that will potentially be helpful [in the clinic], and we need to test this in the adult population,” Dr. James said.
The investigators also were able to stratify patients, based on individual immunoprofiling, into seven major groups based on molecular correlates. They concluded that such stratification could help improve the outcomes of clinical trials in SLE.
In another study, researchers looked at longitudinal gene expression in SLE patients by stratifying each of two independent sets of patients (a pediatric cohort and an adult cohort) into three clinically differentiated disease clusters defined by mechanisms of disease progression (Arthritis Rheumatol. Dec 2018;70[12]:2025-35).
The clusters included one showing a correlation between the percentage of neutrophils and disease activity progression, one showing a correlation between the percentage of lymphocytes and disease activity progression, and a third for which the percentage of neutrophils correlated to a lesser degree with disease activity but was functionally more heterogeneous. Patients in the two neutrophil‐driven clusters had an increased risk of developing proliferative nephritis.
The results have implications for treatment, trial design, and understanding of disease etiology, the investigators concluded.
“This may help us in the future as we think about which medicine to start patients on, and which medicines to start patients on first,” Dr. James said.
It is clear that precision medicine will play an increasingly important role in rheumatology, Dr. James said, when considering the context of other findings in recent years, such as those from studies looking at soluble mediators of inflammation associated with disease flare, as well as those that involved extensive immunophenotyping and showed widely divergent transcriptional patterns based on ancestral backgrounds. Other research, such as the BOLD (Biomarkers of Lupus Disease) study, looked at various mechanisms of disease flare.
Numerous types of personalized therapies are being considered in rheumatology, ranging from expanded regulatory T cells to chimeric antigen receptor T cell therapy to risk profiling for disease prevention, just to name a few. Going forward it will be important to perform more systems biology analyses to assemble precision medicine–related data that can inform clinical diagnosis, prognosis, and therapy selection and optimization, she said.
The future of personalized therapies in rheumatology will require more input from rheumatologists on large-scale precision medicine projects such as the National Institutes of Health’s All of Us Research Project and the Million Veteran Program, as well as other similar programs of major health systems, she noted, adding that different types of training and interaction with molecular pathologists, genetic counselors, health coaches, and other key players also are needed.
Dr. James reported having no relevant disclosures.
EXPERT ANALYSIS FROM THE WINTER RHEUMATOLOGY SYMPOSIUM
Will microneedling enhance the impact of photodynamic therapy?
ORLANDO –
Dr. Spencer, who practices in St. Petersburg, Fla., and is cochair of the conference, gave attendees a roundup of what’s new in adjuncts and delivery methods for photodynamic therapy (PDT). Among the updates is the promise of PDT delivered by means of an ultrashort incubation time of 10-20 minutes, followed by prolonged blue light exposure time of 1 hour. “The idea is that the enzymatic conversion is occurring during the light exposure,” Dr. Spencer said, adding that reports of this approach are mostly anecdotal.
A variation on the ultrashort incubation adds microneedling, he said. In one recent study, 33 patients who had facial actinic keratoses (AKs) were randomized to 10 or 20 minutes of incubation after application of aminolevulinic acid (ALA), followed by 1,000 seconds of exposure to blue light. However, in this split-face study, participants each had one side of their faces treated with microneedling and the other half with a sham treatment before ALA was applied.
Those who had the shorter incubation time had 43% of AKs cleared on the side that received microneedling, compared with 38% on the sham side. For those who received 20 minutes of ALA incubation, rates were higher, with 76% AK clearance on the treated side and 58% on the sham side. “Patients reported that the procedure was virtually painless on both sides,” said Dr. Spencer.
Though the addition of microneedling to PDT is a newer trend, there’s one that’s been a mainstay in Europe for some time: daylight PDT. He cited a review article published in 2016, which identified 17 studies on the use of daylight PDT (Dermatol Surg. 2016 Mar;42[3]:286-95).
Advantages of daylight PDT, he said, include less time in the office for patients and “supposedly less pain.” European protocols vary, but most use methyl aminolevulinate, which he said is a “little more lipophilic than ALA,” with incubation times ranging from 0 to 30 minutes. Exposure time is also variable, but will usually range from 1.5 to 2.5 hours. Most patients receive just one treatment, but some protocols will include up to three treatments.
Overall, studies show a range from 46% to almost 90% complete response rates when AKs are treated with daylight PDT. One study that looked at daylight PDT for small basal cell carcinomas showed that 94% of patients had clinical clearance of their lesions after two treatment sessions; however, the recurrence rate at 12 months post therapy was 21%, Dr. Spencer said.
He shared results of a recent head-to-head study of conventional and daylight PDT; conducted in Greece, the study enrolled patients with “high sun exposure” and used a split-face design.
Of the 46 patients who received MAL on both sides of their faces, response rates were similar at both 3 and 12 months, with slightly numerically higher clearance rates for conventional versus daylight PDT. The 3-month clearance rate for conventional PDT was 80.6%, compared with 78.0% for daylight PDT. At 12 months, the respective clearance rates were 73.7% and 71.8% (J Eur Acad Dermatol Venereol. 2018 Apr;32[4]:595-600). However, “significantly less pain was reported with daylight PDT,” Dr. Spencer said.
Daylight PDT hasn’t caught on the United States. Physicians have concern about the lack of control of UV dosing, and, he pointed out, “this, of course, is not billable.”
Dr. Spencer reported that he serves on the speakers bureau for Genentech.
ORLANDO –
Dr. Spencer, who practices in St. Petersburg, Fla., and is cochair of the conference, gave attendees a roundup of what’s new in adjuncts and delivery methods for photodynamic therapy (PDT). Among the updates is the promise of PDT delivered by means of an ultrashort incubation time of 10-20 minutes, followed by prolonged blue light exposure time of 1 hour. “The idea is that the enzymatic conversion is occurring during the light exposure,” Dr. Spencer said, adding that reports of this approach are mostly anecdotal.
A variation on the ultrashort incubation adds microneedling, he said. In one recent study, 33 patients who had facial actinic keratoses (AKs) were randomized to 10 or 20 minutes of incubation after application of aminolevulinic acid (ALA), followed by 1,000 seconds of exposure to blue light. However, in this split-face study, participants each had one side of their faces treated with microneedling and the other half with a sham treatment before ALA was applied.
Those who had the shorter incubation time had 43% of AKs cleared on the side that received microneedling, compared with 38% on the sham side. For those who received 20 minutes of ALA incubation, rates were higher, with 76% AK clearance on the treated side and 58% on the sham side. “Patients reported that the procedure was virtually painless on both sides,” said Dr. Spencer.
Though the addition of microneedling to PDT is a newer trend, there’s one that’s been a mainstay in Europe for some time: daylight PDT. He cited a review article published in 2016, which identified 17 studies on the use of daylight PDT (Dermatol Surg. 2016 Mar;42[3]:286-95).
Advantages of daylight PDT, he said, include less time in the office for patients and “supposedly less pain.” European protocols vary, but most use methyl aminolevulinate, which he said is a “little more lipophilic than ALA,” with incubation times ranging from 0 to 30 minutes. Exposure time is also variable, but will usually range from 1.5 to 2.5 hours. Most patients receive just one treatment, but some protocols will include up to three treatments.
Overall, studies show a range from 46% to almost 90% complete response rates when AKs are treated with daylight PDT. One study that looked at daylight PDT for small basal cell carcinomas showed that 94% of patients had clinical clearance of their lesions after two treatment sessions; however, the recurrence rate at 12 months post therapy was 21%, Dr. Spencer said.
He shared results of a recent head-to-head study of conventional and daylight PDT; conducted in Greece, the study enrolled patients with “high sun exposure” and used a split-face design.
Of the 46 patients who received MAL on both sides of their faces, response rates were similar at both 3 and 12 months, with slightly numerically higher clearance rates for conventional versus daylight PDT. The 3-month clearance rate for conventional PDT was 80.6%, compared with 78.0% for daylight PDT. At 12 months, the respective clearance rates were 73.7% and 71.8% (J Eur Acad Dermatol Venereol. 2018 Apr;32[4]:595-600). However, “significantly less pain was reported with daylight PDT,” Dr. Spencer said.
Daylight PDT hasn’t caught on the United States. Physicians have concern about the lack of control of UV dosing, and, he pointed out, “this, of course, is not billable.”
Dr. Spencer reported that he serves on the speakers bureau for Genentech.
ORLANDO –
Dr. Spencer, who practices in St. Petersburg, Fla., and is cochair of the conference, gave attendees a roundup of what’s new in adjuncts and delivery methods for photodynamic therapy (PDT). Among the updates is the promise of PDT delivered by means of an ultrashort incubation time of 10-20 minutes, followed by prolonged blue light exposure time of 1 hour. “The idea is that the enzymatic conversion is occurring during the light exposure,” Dr. Spencer said, adding that reports of this approach are mostly anecdotal.
A variation on the ultrashort incubation adds microneedling, he said. In one recent study, 33 patients who had facial actinic keratoses (AKs) were randomized to 10 or 20 minutes of incubation after application of aminolevulinic acid (ALA), followed by 1,000 seconds of exposure to blue light. However, in this split-face study, participants each had one side of their faces treated with microneedling and the other half with a sham treatment before ALA was applied.
Those who had the shorter incubation time had 43% of AKs cleared on the side that received microneedling, compared with 38% on the sham side. For those who received 20 minutes of ALA incubation, rates were higher, with 76% AK clearance on the treated side and 58% on the sham side. “Patients reported that the procedure was virtually painless on both sides,” said Dr. Spencer.
Though the addition of microneedling to PDT is a newer trend, there’s one that’s been a mainstay in Europe for some time: daylight PDT. He cited a review article published in 2016, which identified 17 studies on the use of daylight PDT (Dermatol Surg. 2016 Mar;42[3]:286-95).
Advantages of daylight PDT, he said, include less time in the office for patients and “supposedly less pain.” European protocols vary, but most use methyl aminolevulinate, which he said is a “little more lipophilic than ALA,” with incubation times ranging from 0 to 30 minutes. Exposure time is also variable, but will usually range from 1.5 to 2.5 hours. Most patients receive just one treatment, but some protocols will include up to three treatments.
Overall, studies show a range from 46% to almost 90% complete response rates when AKs are treated with daylight PDT. One study that looked at daylight PDT for small basal cell carcinomas showed that 94% of patients had clinical clearance of their lesions after two treatment sessions; however, the recurrence rate at 12 months post therapy was 21%, Dr. Spencer said.
He shared results of a recent head-to-head study of conventional and daylight PDT; conducted in Greece, the study enrolled patients with “high sun exposure” and used a split-face design.
Of the 46 patients who received MAL on both sides of their faces, response rates were similar at both 3 and 12 months, with slightly numerically higher clearance rates for conventional versus daylight PDT. The 3-month clearance rate for conventional PDT was 80.6%, compared with 78.0% for daylight PDT. At 12 months, the respective clearance rates were 73.7% and 71.8% (J Eur Acad Dermatol Venereol. 2018 Apr;32[4]:595-600). However, “significantly less pain was reported with daylight PDT,” Dr. Spencer said.
Daylight PDT hasn’t caught on the United States. Physicians have concern about the lack of control of UV dosing, and, he pointed out, “this, of course, is not billable.”
Dr. Spencer reported that he serves on the speakers bureau for Genentech.
EXPERT ANALYSIS FROM ODAC 2019
AF ablation referral
Also today, a theory of relativity for rosacea patients, long-term opioid use is substantial in elderly adults prior to total joint replacement, and pediatricians get more guidance to be proactive on the use of e-cigarettes among youth.
Amazon Alexa
Apple Podcasts
Google Podcasts
Spotify
Also today, a theory of relativity for rosacea patients, long-term opioid use is substantial in elderly adults prior to total joint replacement, and pediatricians get more guidance to be proactive on the use of e-cigarettes among youth.
Amazon Alexa
Apple Podcasts
Google Podcasts
Spotify
Also today, a theory of relativity for rosacea patients, long-term opioid use is substantial in elderly adults prior to total joint replacement, and pediatricians get more guidance to be proactive on the use of e-cigarettes among youth.
Amazon Alexa
Apple Podcasts
Google Podcasts
Spotify

Timeout or not?
However, when it comes to timeout, child behavior specialists have failed to reach consensus. In a recent Washington Post article, Claire Gillespie quotes several experts who feel that timeout is ineffective at best and damaging and dangerous at its worst. (Timeouts are a dated and ineffective parenting strategy. So what’s a good alternative? Washington Post, Nov. 29, 2019.)

How do you feel about timeouts? Do you think they are effective? Do you think that brief periods of isolation in a home setting will increase a child’s anxiety? Will the threat of isolation create long-lasting psychological harm? Or do you believe that properly done timeout can be a safe consequence when a child misbehaves?
The disagreement seems to be another one of those issues of apples and oranges. Do I believe that solitary confinement in a prison or chained to a metal cot in the basement of mentally deranged and obsessive parent will leave psychological scars? Of course I do. But, do I believe that a few minutes alone in a child’s own room in a home in which her parents frequently express their affection will cause any harm? Not for a moment. It’s not so much where the child is. It’s where she isn’t. Of course, she doesn’t want to be isolated from the family and that sends a powerful but not harmful message. A big hug and a kiss at the end of the timeout wipes the slate clear.
Some critics believe that timeout should be condemned because it is a punishment. Here again, it’s a case of semantics. Punishments in my mind are inhumane, “a pound of flesh” or “an eye for an eye” response. A well-done timeout is a harmless consequence and one that particularly makes sense when the misbehavior has been or is creating an unpleasant atmosphere in the family.
Other critics will claim that timeouts aren’t an effective deterrent. Correct! They aren’t meant to be a deterrent. A detailed discussion, more likely a lecture, about the misbehavior before and even immediately after a timeout is a waste of time. If timeouts are a deterrent it is because of their safety. Parents will be more likely to use them as a consequence, and most importantly to follow up on their threats. A parent whose words can be believed is his or her own best deterrent.
Finally, many parents who have tried timeouts will claim that they don’t work. This is true if they were talking about deterrent value. Maybe the timeouts have been too long or too short. About 30-60 seconds after the child stops crying may be enough. However, if the parents mean that the child wouldn’t stay in timeout in his room, then they have not taken the difficult final step. If the parent doesn’t have the stamina to keep walking the child back into his room, then it is time to put a latch on the door. Whoops. ... I may have lost some of you who up to this point have been nodding agreement along with my rationale. I know, I know it smacks of prison. It may be used only once or twice, but it will remain as a tangible reminder that sometimes enough is enough. Frequent trips into the room to help the child self-calm make it clear he hasn’t been abandoned.
It’s hard to provide a fully nuanced argument for including timeout in the consequence arsenal in 500 words. I’m eager to hear how you feel on the subject. I can take the heat.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
However, when it comes to timeout, child behavior specialists have failed to reach consensus. In a recent Washington Post article, Claire Gillespie quotes several experts who feel that timeout is ineffective at best and damaging and dangerous at its worst. (Timeouts are a dated and ineffective parenting strategy. So what’s a good alternative? Washington Post, Nov. 29, 2019.)
How do you feel about timeouts? Do you think they are effective? Do you think that brief periods of isolation in a home setting will increase a child’s anxiety? Will the threat of isolation create long-lasting psychological harm? Or do you believe that properly done timeout can be a safe consequence when a child misbehaves?
The disagreement seems to be another one of those issues of apples and oranges. Do I believe that solitary confinement in a prison or chained to a metal cot in the basement of mentally deranged and obsessive parent will leave psychological scars? Of course I do. But, do I believe that a few minutes alone in a child’s own room in a home in which her parents frequently express their affection will cause any harm? Not for a moment. It’s not so much where the child is. It’s where she isn’t. Of course, she doesn’t want to be isolated from the family and that sends a powerful but not harmful message. A big hug and a kiss at the end of the timeout wipes the slate clear.
Some critics believe that timeout should be condemned because it is a punishment. Here again, it’s a case of semantics. Punishments in my mind are inhumane, “a pound of flesh” or “an eye for an eye” response. A well-done timeout is a harmless consequence and one that particularly makes sense when the misbehavior has been or is creating an unpleasant atmosphere in the family.
Other critics will claim that timeouts aren’t an effective deterrent. Correct! They aren’t meant to be a deterrent. A detailed discussion, more likely a lecture, about the misbehavior before and even immediately after a timeout is a waste of time. If timeouts are a deterrent it is because of their safety. Parents will be more likely to use them as a consequence, and most importantly to follow up on their threats. A parent whose words can be believed is his or her own best deterrent.
Finally, many parents who have tried timeouts will claim that they don’t work. This is true if they were talking about deterrent value. Maybe the timeouts have been too long or too short. About 30-60 seconds after the child stops crying may be enough. However, if the parents mean that the child wouldn’t stay in timeout in his room, then they have not taken the difficult final step. If the parent doesn’t have the stamina to keep walking the child back into his room, then it is time to put a latch on the door. Whoops. ... I may have lost some of you who up to this point have been nodding agreement along with my rationale. I know, I know it smacks of prison. It may be used only once or twice, but it will remain as a tangible reminder that sometimes enough is enough. Frequent trips into the room to help the child self-calm make it clear he hasn’t been abandoned.
It’s hard to provide a fully nuanced argument for including timeout in the consequence arsenal in 500 words. I’m eager to hear how you feel on the subject. I can take the heat.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
However, when it comes to timeout, child behavior specialists have failed to reach consensus. In a recent Washington Post article, Claire Gillespie quotes several experts who feel that timeout is ineffective at best and damaging and dangerous at its worst. (Timeouts are a dated and ineffective parenting strategy. So what’s a good alternative? Washington Post, Nov. 29, 2019.)
How do you feel about timeouts? Do you think they are effective? Do you think that brief periods of isolation in a home setting will increase a child’s anxiety? Will the threat of isolation create long-lasting psychological harm? Or do you believe that properly done timeout can be a safe consequence when a child misbehaves?
The disagreement seems to be another one of those issues of apples and oranges. Do I believe that solitary confinement in a prison or chained to a metal cot in the basement of mentally deranged and obsessive parent will leave psychological scars? Of course I do. But, do I believe that a few minutes alone in a child’s own room in a home in which her parents frequently express their affection will cause any harm? Not for a moment. It’s not so much where the child is. It’s where she isn’t. Of course, she doesn’t want to be isolated from the family and that sends a powerful but not harmful message. A big hug and a kiss at the end of the timeout wipes the slate clear.
Some critics believe that timeout should be condemned because it is a punishment. Here again, it’s a case of semantics. Punishments in my mind are inhumane, “a pound of flesh” or “an eye for an eye” response. A well-done timeout is a harmless consequence and one that particularly makes sense when the misbehavior has been or is creating an unpleasant atmosphere in the family.
Other critics will claim that timeouts aren’t an effective deterrent. Correct! They aren’t meant to be a deterrent. A detailed discussion, more likely a lecture, about the misbehavior before and even immediately after a timeout is a waste of time. If timeouts are a deterrent it is because of their safety. Parents will be more likely to use them as a consequence, and most importantly to follow up on their threats. A parent whose words can be believed is his or her own best deterrent.
Finally, many parents who have tried timeouts will claim that they don’t work. This is true if they were talking about deterrent value. Maybe the timeouts have been too long or too short. About 30-60 seconds after the child stops crying may be enough. However, if the parents mean that the child wouldn’t stay in timeout in his room, then they have not taken the difficult final step. If the parent doesn’t have the stamina to keep walking the child back into his room, then it is time to put a latch on the door. Whoops. ... I may have lost some of you who up to this point have been nodding agreement along with my rationale. I know, I know it smacks of prison. It may be used only once or twice, but it will remain as a tangible reminder that sometimes enough is enough. Frequent trips into the room to help the child self-calm make it clear he hasn’t been abandoned.
It’s hard to provide a fully nuanced argument for including timeout in the consequence arsenal in 500 words. I’m eager to hear how you feel on the subject. I can take the heat.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
SPRINT MIND published: Extension trial to add 2 years’ follow-up
A new iteration of the SPRINT MIND hypertension trial will seek to prove conclusively the original study’s tantalizing suggestion: that intensive blood pressure control decreases the risk of developing mild cognitive impairment (MCI) and, eventually, dementia.

SPRINT MIND 2.0 will re-recruit SPRINT MIND subjects and enable another follow-up cognitive assessment and other clinical tests as they remain on their standard of care blood pressure regimen. It is largely funded by an $800,000 grant from the Alzheimer’s Association.
Initially released last July at the Alzheimer’s Association International Conference, the results of the SPRINT MIND have now appeared online in JAMA. Although it failed to meet its primary endpoint of reducing dementia incidence, the study did score on two secondary endpoints. Patients who reduced their systolic blood pressure to less than 120 mm Hg were 19% less likely to develop MCI and 17% less likely to be diagnosed with all-cause dementia than were those who achieved a hypertension target of less than 140 mm Hg.
The secondary results, and positive movement in the primary results, sparked excitement in the dementia research community last summer. They have suggested that the median 5-year follow-up just wasn’t long enough to show any significant effects on dementia, which can take years to fully manifest. Adding 2 more years with SPRINT MIND 2.0 should be long enough to discern those benefits, if indeed they exist.
“SPRINT MIND 2.0 and the work leading up to it offers genuine, concrete hope,” Maria C. Carrillo, PhD, chief science officer for the Alzheimer’s Association, said in a press statement. “MCI is a known risk factor for dementia, and everyone who experiences dementia passes through MCI. When you prevent new cases of MCI, you are preventing new cases of dementia. The Alzheimer’s Association finds these data to be compelling and is committed to getting clarity and certainty on the dementia outcome by following participants for a longer period of time.”
The study strengthens the new and energetic push to find ways to prevent dementia, which has proven itself intractable in every drug study to date.
“This study is in line with where the field of dementia research is going: preventing memory loss earlier,” said Laurie Ryan, PhD, chief of the dementias of aging branch in the National Institute on Aging. “Much like we have research-based interventions for heart health and cancer prevention, we hope to have guidance based on this and subsequent studies that will more definitively show how to slow or even stop dementia well before symptoms appear.”
NIA director Richard J. Hodes, MD, agreed.
“Dementia continues to be a large public health challenge, and based on the primary results of this study, we still have yet to find an intervention strategy proven to reduce the risk of dementia,” he said in a press statement. “Nevertheless, the secondary results showing that intensive lowering of blood pressure may reduce risk for MCI, a known risk factor for dementia, gives us additional avenues to explore on the path to prevention.”
SPRINT MIND was a substudy of the Systolic Blood Pressure Intervention Trial (SPRINT). It compared two strategies for managing hypertension in older adults. The intensive strategy had a target of less than 120 mm Hg, while standard care had a target of less than 140 mm Hg. SPRINT showed that more intensive blood pressure control produced a 25% reduction in the composite primary composite endpoint of cardiovascular events, stroke, and cardiovascular death. The intensive arm was so successful that SPRINT helped inform the 2017 high blood pressure clinical guidelines from the American Heart Association and American College of Cardiology.

The SPRINT MIND substudy, headed by Jeff D. Williamson, MD, of Wake Forest University, Winston-Salem, NC, asked whether intensive management had any effect on probable all-cause dementia or MCI, as well as imaging evidence of changes in white matter lesions and brain volume. It followed patients for up to 7 years and comprised 9,361 SPRINT subjects at least 50 years old (mean, 68 years) with at least one cardiovascular risk factor. Nearly a third (30%) were black, and 10% Hispanic. The primary outcome was incident probable dementia. Secondary outcomes were MCI and a composite of MCI and/or probable dementia. About a third had a SBP of 132 mm Hg or less, another third had a systolic pressure of 132-145 mm Hg, and the remainder had a systolic pressure greater than 145 mm Hg.
Physicians could use their choice of antihypertensive treatments. The study protocol encouraged, but did not mandate, thiazide-type diuretics as a first-line agent, followed by loop diuretics and beta-adrenergic blockers. Chlorthalidone was encouraged as the primary thiazide-type diuretic, and amlodipine as the preferred calcium-channel blocker.
The interventions did successfully control blood pressure, with a significant difference between the treatment groups. The mean SBP was 121.6 mm Hg in the intensive therapy group and 134.8 mm Hg in the standard group – a statistically significant difference of 13.3 mm Hg.
Dementia developed in 149 in the aggressive control group and 176 in the standard group – a nonsignificant difference of 17% (hazard ratio, 0.83). MCI developed in 287 in the intensive group and 353 in the standard treatment group. This amounted to a statistically significant 19% reduction. There was also a significant 15% reduction in the composite outcome of MCI or probable dementia in favor of intensive treatment.
As evidenced by the Alzheimer’s Association grant, dementia researchers chose to focus on SPRINT MIND’s positive secondary endpoints. At the AAIC meeting, Dr. Williamson even suggested that antihypertensive medications could be seen as disease-modifying agents for cognitive decline. Data support his claim: No dementia intervention yet tested has approached this level of success.
“I think we can say this is the first disease-modifying strategy to reduce the risk of MCI,” Dr. Williamson said during a press briefing. And although the primary endpoint – the 17% relative risk reduction for probable all-cause dementia – didn’t meet statistical significance, “It’s comforting to see that the benefit went in the same direction and was of the same magnitude..”
SOURCE: Williamson JD et al. JAMA 2019 Jan 28. doi:10.1001/jama.2018.21442.
SPRINT MIND offers hope that a very achievable blood pressure goal can dramatically alter the trajectory from mild cognitive impairment to dementia, Kristine Yaffe, MD, wrote in an accompanying editorial. But at this point, it’s impossible to make specific clinical recommendations.

Additionally it is not possible, right now, to know which hypertension treatment regimens were most effective in improved cognitive outcomes.
“Information necessary to compare the effects of classes of antihypertensive agents on cognitive outcomes is also not provided. SPRINT used a quasi-pragmatic approach with suggestions for treatment choice, but practitioners approached SBP control individually, and most participants were taking multiple drugs.”
Nevertheless, the positive secondary findings and the encouraging trajectory on dementia risk should fix blood pressure management squarely into a cornerstone of dementia prevention algorithms.
“The SPRINT MIND study may not be the final approach for prevention of AD or other cognitive impairment, but it represents a major leap forward in what has emerged as a marathon journey.”
Dr. Kristine Yaffe is professor of psychiatry, neurology and epidemiology and the Roy and Marie Scola Endowed Chair at the University of California, San Francisco.
SPRINT MIND offers hope that a very achievable blood pressure goal can dramatically alter the trajectory from mild cognitive impairment to dementia, Kristine Yaffe, MD, wrote in an accompanying editorial. But at this point, it’s impossible to make specific clinical recommendations.
Additionally it is not possible, right now, to know which hypertension treatment regimens were most effective in improved cognitive outcomes.
“Information necessary to compare the effects of classes of antihypertensive agents on cognitive outcomes is also not provided. SPRINT used a quasi-pragmatic approach with suggestions for treatment choice, but practitioners approached SBP control individually, and most participants were taking multiple drugs.”
Nevertheless, the positive secondary findings and the encouraging trajectory on dementia risk should fix blood pressure management squarely into a cornerstone of dementia prevention algorithms.
“The SPRINT MIND study may not be the final approach for prevention of AD or other cognitive impairment, but it represents a major leap forward in what has emerged as a marathon journey.”
Dr. Kristine Yaffe is professor of psychiatry, neurology and epidemiology and the Roy and Marie Scola Endowed Chair at the University of California, San Francisco.
SPRINT MIND offers hope that a very achievable blood pressure goal can dramatically alter the trajectory from mild cognitive impairment to dementia, Kristine Yaffe, MD, wrote in an accompanying editorial. But at this point, it’s impossible to make specific clinical recommendations.
Additionally it is not possible, right now, to know which hypertension treatment regimens were most effective in improved cognitive outcomes.
“Information necessary to compare the effects of classes of antihypertensive agents on cognitive outcomes is also not provided. SPRINT used a quasi-pragmatic approach with suggestions for treatment choice, but practitioners approached SBP control individually, and most participants were taking multiple drugs.”
Nevertheless, the positive secondary findings and the encouraging trajectory on dementia risk should fix blood pressure management squarely into a cornerstone of dementia prevention algorithms.
“The SPRINT MIND study may not be the final approach for prevention of AD or other cognitive impairment, but it represents a major leap forward in what has emerged as a marathon journey.”
Dr. Kristine Yaffe is professor of psychiatry, neurology and epidemiology and the Roy and Marie Scola Endowed Chair at the University of California, San Francisco.
A new iteration of the SPRINT MIND hypertension trial will seek to prove conclusively the original study’s tantalizing suggestion: that intensive blood pressure control decreases the risk of developing mild cognitive impairment (MCI) and, eventually, dementia.
SPRINT MIND 2.0 will re-recruit SPRINT MIND subjects and enable another follow-up cognitive assessment and other clinical tests as they remain on their standard of care blood pressure regimen. It is largely funded by an $800,000 grant from the Alzheimer’s Association.
Initially released last July at the Alzheimer’s Association International Conference, the results of the SPRINT MIND have now appeared online in JAMA. Although it failed to meet its primary endpoint of reducing dementia incidence, the study did score on two secondary endpoints. Patients who reduced their systolic blood pressure to less than 120 mm Hg were 19% less likely to develop MCI and 17% less likely to be diagnosed with all-cause dementia than were those who achieved a hypertension target of less than 140 mm Hg.
The secondary results, and positive movement in the primary results, sparked excitement in the dementia research community last summer. They have suggested that the median 5-year follow-up just wasn’t long enough to show any significant effects on dementia, which can take years to fully manifest. Adding 2 more years with SPRINT MIND 2.0 should be long enough to discern those benefits, if indeed they exist.
“SPRINT MIND 2.0 and the work leading up to it offers genuine, concrete hope,” Maria C. Carrillo, PhD, chief science officer for the Alzheimer’s Association, said in a press statement. “MCI is a known risk factor for dementia, and everyone who experiences dementia passes through MCI. When you prevent new cases of MCI, you are preventing new cases of dementia. The Alzheimer’s Association finds these data to be compelling and is committed to getting clarity and certainty on the dementia outcome by following participants for a longer period of time.”
The study strengthens the new and energetic push to find ways to prevent dementia, which has proven itself intractable in every drug study to date.
“This study is in line with where the field of dementia research is going: preventing memory loss earlier,” said Laurie Ryan, PhD, chief of the dementias of aging branch in the National Institute on Aging. “Much like we have research-based interventions for heart health and cancer prevention, we hope to have guidance based on this and subsequent studies that will more definitively show how to slow or even stop dementia well before symptoms appear.”
NIA director Richard J. Hodes, MD, agreed.
“Dementia continues to be a large public health challenge, and based on the primary results of this study, we still have yet to find an intervention strategy proven to reduce the risk of dementia,” he said in a press statement. “Nevertheless, the secondary results showing that intensive lowering of blood pressure may reduce risk for MCI, a known risk factor for dementia, gives us additional avenues to explore on the path to prevention.”
SPRINT MIND was a substudy of the Systolic Blood Pressure Intervention Trial (SPRINT). It compared two strategies for managing hypertension in older adults. The intensive strategy had a target of less than 120 mm Hg, while standard care had a target of less than 140 mm Hg. SPRINT showed that more intensive blood pressure control produced a 25% reduction in the composite primary composite endpoint of cardiovascular events, stroke, and cardiovascular death. The intensive arm was so successful that SPRINT helped inform the 2017 high blood pressure clinical guidelines from the American Heart Association and American College of Cardiology.
The SPRINT MIND substudy, headed by Jeff D. Williamson, MD, of Wake Forest University, Winston-Salem, NC, asked whether intensive management had any effect on probable all-cause dementia or MCI, as well as imaging evidence of changes in white matter lesions and brain volume. It followed patients for up to 7 years and comprised 9,361 SPRINT subjects at least 50 years old (mean, 68 years) with at least one cardiovascular risk factor. Nearly a third (30%) were black, and 10% Hispanic. The primary outcome was incident probable dementia. Secondary outcomes were MCI and a composite of MCI and/or probable dementia. About a third had a SBP of 132 mm Hg or less, another third had a systolic pressure of 132-145 mm Hg, and the remainder had a systolic pressure greater than 145 mm Hg.
Physicians could use their choice of antihypertensive treatments. The study protocol encouraged, but did not mandate, thiazide-type diuretics as a first-line agent, followed by loop diuretics and beta-adrenergic blockers. Chlorthalidone was encouraged as the primary thiazide-type diuretic, and amlodipine as the preferred calcium-channel blocker.
The interventions did successfully control blood pressure, with a significant difference between the treatment groups. The mean SBP was 121.6 mm Hg in the intensive therapy group and 134.8 mm Hg in the standard group – a statistically significant difference of 13.3 mm Hg.
Dementia developed in 149 in the aggressive control group and 176 in the standard group – a nonsignificant difference of 17% (hazard ratio, 0.83). MCI developed in 287 in the intensive group and 353 in the standard treatment group. This amounted to a statistically significant 19% reduction. There was also a significant 15% reduction in the composite outcome of MCI or probable dementia in favor of intensive treatment.
As evidenced by the Alzheimer’s Association grant, dementia researchers chose to focus on SPRINT MIND’s positive secondary endpoints. At the AAIC meeting, Dr. Williamson even suggested that antihypertensive medications could be seen as disease-modifying agents for cognitive decline. Data support his claim: No dementia intervention yet tested has approached this level of success.
“I think we can say this is the first disease-modifying strategy to reduce the risk of MCI,” Dr. Williamson said during a press briefing. And although the primary endpoint – the 17% relative risk reduction for probable all-cause dementia – didn’t meet statistical significance, “It’s comforting to see that the benefit went in the same direction and was of the same magnitude..”
SOURCE: Williamson JD et al. JAMA 2019 Jan 28. doi:10.1001/jama.2018.21442.
A new iteration of the SPRINT MIND hypertension trial will seek to prove conclusively the original study’s tantalizing suggestion: that intensive blood pressure control decreases the risk of developing mild cognitive impairment (MCI) and, eventually, dementia.
SPRINT MIND 2.0 will re-recruit SPRINT MIND subjects and enable another follow-up cognitive assessment and other clinical tests as they remain on their standard of care blood pressure regimen. It is largely funded by an $800,000 grant from the Alzheimer’s Association.
Initially released last July at the Alzheimer’s Association International Conference, the results of the SPRINT MIND have now appeared online in JAMA. Although it failed to meet its primary endpoint of reducing dementia incidence, the study did score on two secondary endpoints. Patients who reduced their systolic blood pressure to less than 120 mm Hg were 19% less likely to develop MCI and 17% less likely to be diagnosed with all-cause dementia than were those who achieved a hypertension target of less than 140 mm Hg.
The secondary results, and positive movement in the primary results, sparked excitement in the dementia research community last summer. They have suggested that the median 5-year follow-up just wasn’t long enough to show any significant effects on dementia, which can take years to fully manifest. Adding 2 more years with SPRINT MIND 2.0 should be long enough to discern those benefits, if indeed they exist.
“SPRINT MIND 2.0 and the work leading up to it offers genuine, concrete hope,” Maria C. Carrillo, PhD, chief science officer for the Alzheimer’s Association, said in a press statement. “MCI is a known risk factor for dementia, and everyone who experiences dementia passes through MCI. When you prevent new cases of MCI, you are preventing new cases of dementia. The Alzheimer’s Association finds these data to be compelling and is committed to getting clarity and certainty on the dementia outcome by following participants for a longer period of time.”
The study strengthens the new and energetic push to find ways to prevent dementia, which has proven itself intractable in every drug study to date.
“This study is in line with where the field of dementia research is going: preventing memory loss earlier,” said Laurie Ryan, PhD, chief of the dementias of aging branch in the National Institute on Aging. “Much like we have research-based interventions for heart health and cancer prevention, we hope to have guidance based on this and subsequent studies that will more definitively show how to slow or even stop dementia well before symptoms appear.”
NIA director Richard J. Hodes, MD, agreed.
“Dementia continues to be a large public health challenge, and based on the primary results of this study, we still have yet to find an intervention strategy proven to reduce the risk of dementia,” he said in a press statement. “Nevertheless, the secondary results showing that intensive lowering of blood pressure may reduce risk for MCI, a known risk factor for dementia, gives us additional avenues to explore on the path to prevention.”
SPRINT MIND was a substudy of the Systolic Blood Pressure Intervention Trial (SPRINT). It compared two strategies for managing hypertension in older adults. The intensive strategy had a target of less than 120 mm Hg, while standard care had a target of less than 140 mm Hg. SPRINT showed that more intensive blood pressure control produced a 25% reduction in the composite primary composite endpoint of cardiovascular events, stroke, and cardiovascular death. The intensive arm was so successful that SPRINT helped inform the 2017 high blood pressure clinical guidelines from the American Heart Association and American College of Cardiology.
The SPRINT MIND substudy, headed by Jeff D. Williamson, MD, of Wake Forest University, Winston-Salem, NC, asked whether intensive management had any effect on probable all-cause dementia or MCI, as well as imaging evidence of changes in white matter lesions and brain volume. It followed patients for up to 7 years and comprised 9,361 SPRINT subjects at least 50 years old (mean, 68 years) with at least one cardiovascular risk factor. Nearly a third (30%) were black, and 10% Hispanic. The primary outcome was incident probable dementia. Secondary outcomes were MCI and a composite of MCI and/or probable dementia. About a third had a SBP of 132 mm Hg or less, another third had a systolic pressure of 132-145 mm Hg, and the remainder had a systolic pressure greater than 145 mm Hg.
Physicians could use their choice of antihypertensive treatments. The study protocol encouraged, but did not mandate, thiazide-type diuretics as a first-line agent, followed by loop diuretics and beta-adrenergic blockers. Chlorthalidone was encouraged as the primary thiazide-type diuretic, and amlodipine as the preferred calcium-channel blocker.
The interventions did successfully control blood pressure, with a significant difference between the treatment groups. The mean SBP was 121.6 mm Hg in the intensive therapy group and 134.8 mm Hg in the standard group – a statistically significant difference of 13.3 mm Hg.
Dementia developed in 149 in the aggressive control group and 176 in the standard group – a nonsignificant difference of 17% (hazard ratio, 0.83). MCI developed in 287 in the intensive group and 353 in the standard treatment group. This amounted to a statistically significant 19% reduction. There was also a significant 15% reduction in the composite outcome of MCI or probable dementia in favor of intensive treatment.
As evidenced by the Alzheimer’s Association grant, dementia researchers chose to focus on SPRINT MIND’s positive secondary endpoints. At the AAIC meeting, Dr. Williamson even suggested that antihypertensive medications could be seen as disease-modifying agents for cognitive decline. Data support his claim: No dementia intervention yet tested has approached this level of success.
“I think we can say this is the first disease-modifying strategy to reduce the risk of MCI,” Dr. Williamson said during a press briefing. And although the primary endpoint – the 17% relative risk reduction for probable all-cause dementia – didn’t meet statistical significance, “It’s comforting to see that the benefit went in the same direction and was of the same magnitude..”
SOURCE: Williamson JD et al. JAMA 2019 Jan 28. doi:10.1001/jama.2018.21442.
FROM JAMA
Key clinical point: Keeping systolic blood pressure lower than 120 mm Hg did not significantly reduce the risk of all-cause dementia in patients with hypertension, but it did lower the risk of mild cognitive impairment and probable dementia.
Major finding: The intensively treated group had a nonsignificant 17% lower risk of dementia, and significant reductions in the risk of MCI (19%) and probable dementia (15%).
Study details: SPRINT MIND was a substudy of the SPRINT antihypertension trial.
Source: Williamson JD et al. JAMA 2019 Jan 28. doi:10.1001/jama.2018.21442.
When sweaty palms are more than just sweaty palms
ORLANDO – When you extend your hand to a new patient, and he reflexively wipes his palm before shaking hands, be alert. It’s possible you’re seeing primary hyperhidrosis, a condition that’s both more common and more disabling than once thought.

“Looking at the biology of sweating, normally, it’s a good thing – we need it to survive. However, hyperhidrosis is too much of a good thing – it’s an excess of what is needed for normal biology,” said Adam Friedman, MD, speaking at the Orlando Dermatology Aesthetic and Clinical Conference.
Recent data, he pointed out, show that hyperhidrosis is more prevalent than previously thought – about 4.8% of individuals may have the condition, with about half having axillary hyperhidrosis. Symptoms peak in early adulthood, with adults aged 18-54 most affected. “These are the prime working years,” he said.
About 2% of teens are affected, and many adults report that symptoms began before they were 12 years old. Hand hyperhidrosis is a factor for computer and electronic device work, sports, and even handling paper and pencils, noted Dr. Friedman, professor of dermatology at George Washington University, Washington.
“Does it affect quality of life? Yes. We have data to support the impact. The adverse impact is actually greater than that of eczema and psoriasis,” he said, adding that patients won’t always bring up their concerns about sweating. “Often, it’s the patient who apologizes for having sweaty palms or who sticks to the paper on the exam table. It’s worth asking these patients if they are bothered by excessive sweating.”
Is it hyperhidrosis?
A 2016 paper defined hyperhidrosis as “a condition that involves chronic excessive sweating of the underarms, hands, feet, face, groin, or other bodily areas, which is much more than what is normal, and occurs regardless of temperature, exercise, or situation, and may have an impact on quality of life” (Arch Dermatol Res. 2016 Dec;308[10]:743-9). The amount of sweating can be four to five times that seen in healthy controls.
Other clues that excess sweating might be hyperhidrosis? General hyperhidrosis is a secondary syndrome that can be caused by a variety of conditions including endocrine and metabolic disorders and malignancies. Drugs and toxins can also cause generalized excessive sweating.
Focal hyperhidrosis can be primary idiopathic disease; some neuropathies and certain spinal diseases and spinal cord injury can also cause focal hyperhidrosis, though not usually in the axillary/palmar/plantar distribution seen in primary hyperhidrosis.
Before settling on primary hyperhidrosis, the history and exam should also account for other possibilities in the differential: social anxiety disorder, eccrine nevus, gustatory sweating, Frey syndrome, and impaired evaporation could all account for excess sweating, which is also a postsurgical phenomenon for some patients.
Diagnostic criteria call for “focal, visible, excessive sweating” persisting for at least 6 months with no apparent cause. Additionally, patients must have at least two of the additional following criteria: sweating that is bilateral and symmetric, occurs at least once weekly, impairs daily activities, and starts before age 25 years, as well as a positive family history of hyperhidrosis and cessation of sweating during sleep.
The last point is critical, Dr. Friedman said. “If you sweat a lot at night, it’s not hyperhidrosis!”
Though gravimetric evaluation is used in hyperhidrosis research, the history and exam are really where the diagnosis is made in practice, he noted. The Hyperhidrosis Disease Severity Scale is a brief, useful clinical tool that asks patients to peg the extent to which their sweating interferes with daily life.
Topical treatments to try
Topical antiperspirants and other topical agents are a logical place to start and may be required as part of step therapy by insurers. Many patients will already have tried clinical strength over-the-counter antiperspirants containing aluminum zirconium trichlorohydrex, but these products rate low in patient satisfaction among those with primary hyperhidrosis.
Prescription aluminum salts can be compounded to various strengths, with 10%-20% concentration appropriate for axillae and 30%-40% a good strength for palms and soles, according to Dr. Friedman. All of these agents work by precipitating out solids that form a shallow plug in sweat ducts, slowing the flow of perspiration.
Pearls for topical treatment include the need for the product to be on the skin for 6-8 hours overnight. “Remember hyperhidrosis patients do not sweat at night,” so this is the time when the occlusive plugs can form. Then residue can be washed off in the morning, and patients can apply a deodorant. “I remind my patients that antiperspirants are for sweating, and deodorants are for odor,” said Dr. Friedman. These products can damage fabric, and they can be irritating, a problem addressed with low-potency topical steroids.
Topical regimens don’t need to be adjusted for pediatric patients, said Dr. Friedman.
Iontophoresis has been around since the 1950s, is effective, has few side effects, and is considered first-line treatment for severe palmar and plantar hyperhidrosis. But he said there’s one big rub: time. To be effective, patients need 20-30 minutes of application of 15-20 milliamperes of current 3-4 times weekly, not a schedule that works for most patients or practitioners, Dr. Friedman noted.
A treatment recently approved by the Food and Drug Administration for primary axillary hyperhidrosis is a topical anticholinergic, glycopyrronium tosylate, applied with wipes impregnated with glycopyrronium solution. This product significantly outperformed placebo in two clinical trials, with up to 64% of users meeting the primary endpoint of improving by at least 4 points on the Axillary Sweating Daily Diary (ASDD) scale. This product significantly outperformed placebo in two clinical trials, with 53% and 66% of users meeting the primary endpoint, improvement of at least 4 points from baseline in the weekly mean ASDD Item #2. It was approved in those aged 9 years and older.
“You can use this in kids, but you need to educate the kid and the parent or adult,” he said. “This is the last thing you do before bed, after brushing your teeth and after washing your face.”
Patients should apply one swipe to the clean skin of each underarm, and then wash their hands thoroughly. Clinical trials saw a greater proportion of off-target effects such as dry eyes and mouth and mydriasis in the active arm; unilateral mydriasis was more common than bilateral, underscoring the importance of hand washing as this was probably secondary transfer from hands to face during sleep, said Dr. Friedman. Patients can expect results in 2-3 weeks, and doses can be held as needed for anticholinergic side effects.
Systemic choices are limited
There are no FDA-approved systemic agents for hyperhidrosis, and the literature holds only case reports or small series, Dr. Friedman pointed out.
Though systemic treatment may be more effective in generalized hyperhidrosis and for patients with dysautonomia-associated hyperhidrosis, glycopyrrolate is a logical choice if a systemic anticholinergic is desired. A starting dose of 1 mg twice daily can be titrated for effect to about 6 mg daily. Though off-target effects may be a dose-limiting factor, glycopyrrolate is not very lipid soluble, so it penetrates the blood-brain barrier relatively poorly, he said.
Oxybutynin is available in many forms, including a slow-release tablet that permits once-daily dosing. Starting at 5-10 mg daily is a good idea, but dosing may need to be increased to as high as 20 mg daily to be effective. However, patients will often experience “major side effects” with oxybutynin, including significant xerostomia, constipation, blurred vision, and difficulty urinating.
For children, small studies have seen improvement with glycopyrrolate at an average dose of about 2 mg/day. Oxybutynin, which has been extensively studied in the pediatric population, was also effective, but central nervous system adverse events were common.
For some, beta-adrenergic blockade can be an extremely valuable tool, said Dr. Friedman. When sweating is linked to social phobia or performance anxiety, 10-20 mg of atenolol about an hour before the performance or public appearance can make a big difference. Bradycardia, atrioventricular block, and asthma are all contraindications, and the usual precautions should be taken with a host of other comorbidities, he noted.
It’s a good idea to check resting blood pressure and heart rate and take body mass into consideration, and adjust the dose downward appropriately. A key pearl: “Have them do a test run at home, to make sure they don’t keel over on the podium!” said Dr. Friedman.
Botulinum toxin tips and tricks
Botulinum toxin can be very effective and works directly by blocking acetylcholine release at the junction of the sympathetic sudomotor neuron and the sweat gland.
Before treatment, make sure the patient prepares correctly by abstaining from over-the-counter deodorants or antiperspirants, and resting without exertion or drinking hot beverages for about 30 minutes before the procedure.
To ascertain the follicular outline of the area to be injected, the iodine starch test can be used: Paint the axilla with iodine, allow it to dry, and then dust corn starch over the area. The follicular outline is mapped by the purple-blue reaction of the starch and iodine in the presence of moisture from perspiration, Dr. Friedman said.
Applying topical analgesia 30 minutes prior to the procedure helps with patient discomfort with axillary injections. When it comes time to inject, a shallow approach with the bevel side up works well, with a goal of blanketing the field identified by the iodine starch test with small aliquots of toxin placed 1-2 centimeters apart, said Dr. Friedman. However, for patients who might have tattoos that extend to the axillary area, “Avoid the ink!”
Patients will start to see improvement within 2-4 days, and although the literature says a toxin treatment can last 6-9 months, Dr. Friedman said he sees patients coming back in 4-5 months.
Obtaining botulinum toxin can be done in one of two ways: the “buy and bill” approach has the dermatologist purchasing the medication, using CPT 64650 and J code J0585 – “Remember the units!” said Dr. Friedman, because reimbursement will be based on the volume of toxin purchased.. This route may be cheaper for the patient because it avoids a medication copay. The physician obtains preauthorization for both the medication and procedure with this strategy.
The other route is to have the provider prescribe botulinum toxin and the patient purchase it at a regular or specialty pharmacy. In this case, the pharmacist obtains precertification for the medication, but the physician still needs to be precertified – and bill – for the injection procedure itself. This scenario is less risky for the physician but may trigger two separate copays for the patient.
Botulinum toxin can be effective for up to 90% of patients, but at a cost: Without insurance reimbursement, treatments can cost in the neighborhood of $1,500.
A good resource for patients and clinicians is the International Hyperhidrosis Society’s website (sweathelp.org), said Dr. Friedman.
Dr. Friedman disclosed relationships with multiple pharmaceutical and cosmetic companies, including Dermira, which markets topical glycopyrronium tosylate as Qbrexza.
ORLANDO – When you extend your hand to a new patient, and he reflexively wipes his palm before shaking hands, be alert. It’s possible you’re seeing primary hyperhidrosis, a condition that’s both more common and more disabling than once thought.
“Looking at the biology of sweating, normally, it’s a good thing – we need it to survive. However, hyperhidrosis is too much of a good thing – it’s an excess of what is needed for normal biology,” said Adam Friedman, MD, speaking at the Orlando Dermatology Aesthetic and Clinical Conference.
Recent data, he pointed out, show that hyperhidrosis is more prevalent than previously thought – about 4.8% of individuals may have the condition, with about half having axillary hyperhidrosis. Symptoms peak in early adulthood, with adults aged 18-54 most affected. “These are the prime working years,” he said.
About 2% of teens are affected, and many adults report that symptoms began before they were 12 years old. Hand hyperhidrosis is a factor for computer and electronic device work, sports, and even handling paper and pencils, noted Dr. Friedman, professor of dermatology at George Washington University, Washington.
“Does it affect quality of life? Yes. We have data to support the impact. The adverse impact is actually greater than that of eczema and psoriasis,” he said, adding that patients won’t always bring up their concerns about sweating. “Often, it’s the patient who apologizes for having sweaty palms or who sticks to the paper on the exam table. It’s worth asking these patients if they are bothered by excessive sweating.”
Is it hyperhidrosis?
A 2016 paper defined hyperhidrosis as “a condition that involves chronic excessive sweating of the underarms, hands, feet, face, groin, or other bodily areas, which is much more than what is normal, and occurs regardless of temperature, exercise, or situation, and may have an impact on quality of life” (Arch Dermatol Res. 2016 Dec;308[10]:743-9). The amount of sweating can be four to five times that seen in healthy controls.
Other clues that excess sweating might be hyperhidrosis? General hyperhidrosis is a secondary syndrome that can be caused by a variety of conditions including endocrine and metabolic disorders and malignancies. Drugs and toxins can also cause generalized excessive sweating.
Focal hyperhidrosis can be primary idiopathic disease; some neuropathies and certain spinal diseases and spinal cord injury can also cause focal hyperhidrosis, though not usually in the axillary/palmar/plantar distribution seen in primary hyperhidrosis.
Before settling on primary hyperhidrosis, the history and exam should also account for other possibilities in the differential: social anxiety disorder, eccrine nevus, gustatory sweating, Frey syndrome, and impaired evaporation could all account for excess sweating, which is also a postsurgical phenomenon for some patients.
Diagnostic criteria call for “focal, visible, excessive sweating” persisting for at least 6 months with no apparent cause. Additionally, patients must have at least two of the additional following criteria: sweating that is bilateral and symmetric, occurs at least once weekly, impairs daily activities, and starts before age 25 years, as well as a positive family history of hyperhidrosis and cessation of sweating during sleep.
The last point is critical, Dr. Friedman said. “If you sweat a lot at night, it’s not hyperhidrosis!”
Though gravimetric evaluation is used in hyperhidrosis research, the history and exam are really where the diagnosis is made in practice, he noted. The Hyperhidrosis Disease Severity Scale is a brief, useful clinical tool that asks patients to peg the extent to which their sweating interferes with daily life.
Topical treatments to try
Topical antiperspirants and other topical agents are a logical place to start and may be required as part of step therapy by insurers. Many patients will already have tried clinical strength over-the-counter antiperspirants containing aluminum zirconium trichlorohydrex, but these products rate low in patient satisfaction among those with primary hyperhidrosis.
Prescription aluminum salts can be compounded to various strengths, with 10%-20% concentration appropriate for axillae and 30%-40% a good strength for palms and soles, according to Dr. Friedman. All of these agents work by precipitating out solids that form a shallow plug in sweat ducts, slowing the flow of perspiration.
Pearls for topical treatment include the need for the product to be on the skin for 6-8 hours overnight. “Remember hyperhidrosis patients do not sweat at night,” so this is the time when the occlusive plugs can form. Then residue can be washed off in the morning, and patients can apply a deodorant. “I remind my patients that antiperspirants are for sweating, and deodorants are for odor,” said Dr. Friedman. These products can damage fabric, and they can be irritating, a problem addressed with low-potency topical steroids.
Topical regimens don’t need to be adjusted for pediatric patients, said Dr. Friedman.
Iontophoresis has been around since the 1950s, is effective, has few side effects, and is considered first-line treatment for severe palmar and plantar hyperhidrosis. But he said there’s one big rub: time. To be effective, patients need 20-30 minutes of application of 15-20 milliamperes of current 3-4 times weekly, not a schedule that works for most patients or practitioners, Dr. Friedman noted.
A treatment recently approved by the Food and Drug Administration for primary axillary hyperhidrosis is a topical anticholinergic, glycopyrronium tosylate, applied with wipes impregnated with glycopyrronium solution. This product significantly outperformed placebo in two clinical trials, with up to 64% of users meeting the primary endpoint of improving by at least 4 points on the Axillary Sweating Daily Diary (ASDD) scale. This product significantly outperformed placebo in two clinical trials, with 53% and 66% of users meeting the primary endpoint, improvement of at least 4 points from baseline in the weekly mean ASDD Item #2. It was approved in those aged 9 years and older.
“You can use this in kids, but you need to educate the kid and the parent or adult,” he said. “This is the last thing you do before bed, after brushing your teeth and after washing your face.”
Patients should apply one swipe to the clean skin of each underarm, and then wash their hands thoroughly. Clinical trials saw a greater proportion of off-target effects such as dry eyes and mouth and mydriasis in the active arm; unilateral mydriasis was more common than bilateral, underscoring the importance of hand washing as this was probably secondary transfer from hands to face during sleep, said Dr. Friedman. Patients can expect results in 2-3 weeks, and doses can be held as needed for anticholinergic side effects.
Systemic choices are limited
There are no FDA-approved systemic agents for hyperhidrosis, and the literature holds only case reports or small series, Dr. Friedman pointed out.
Though systemic treatment may be more effective in generalized hyperhidrosis and for patients with dysautonomia-associated hyperhidrosis, glycopyrrolate is a logical choice if a systemic anticholinergic is desired. A starting dose of 1 mg twice daily can be titrated for effect to about 6 mg daily. Though off-target effects may be a dose-limiting factor, glycopyrrolate is not very lipid soluble, so it penetrates the blood-brain barrier relatively poorly, he said.
Oxybutynin is available in many forms, including a slow-release tablet that permits once-daily dosing. Starting at 5-10 mg daily is a good idea, but dosing may need to be increased to as high as 20 mg daily to be effective. However, patients will often experience “major side effects” with oxybutynin, including significant xerostomia, constipation, blurred vision, and difficulty urinating.
For children, small studies have seen improvement with glycopyrrolate at an average dose of about 2 mg/day. Oxybutynin, which has been extensively studied in the pediatric population, was also effective, but central nervous system adverse events were common.
For some, beta-adrenergic blockade can be an extremely valuable tool, said Dr. Friedman. When sweating is linked to social phobia or performance anxiety, 10-20 mg of atenolol about an hour before the performance or public appearance can make a big difference. Bradycardia, atrioventricular block, and asthma are all contraindications, and the usual precautions should be taken with a host of other comorbidities, he noted.
It’s a good idea to check resting blood pressure and heart rate and take body mass into consideration, and adjust the dose downward appropriately. A key pearl: “Have them do a test run at home, to make sure they don’t keel over on the podium!” said Dr. Friedman.
Botulinum toxin tips and tricks
Botulinum toxin can be very effective and works directly by blocking acetylcholine release at the junction of the sympathetic sudomotor neuron and the sweat gland.
Before treatment, make sure the patient prepares correctly by abstaining from over-the-counter deodorants or antiperspirants, and resting without exertion or drinking hot beverages for about 30 minutes before the procedure.
To ascertain the follicular outline of the area to be injected, the iodine starch test can be used: Paint the axilla with iodine, allow it to dry, and then dust corn starch over the area. The follicular outline is mapped by the purple-blue reaction of the starch and iodine in the presence of moisture from perspiration, Dr. Friedman said.
Applying topical analgesia 30 minutes prior to the procedure helps with patient discomfort with axillary injections. When it comes time to inject, a shallow approach with the bevel side up works well, with a goal of blanketing the field identified by the iodine starch test with small aliquots of toxin placed 1-2 centimeters apart, said Dr. Friedman. However, for patients who might have tattoos that extend to the axillary area, “Avoid the ink!”
Patients will start to see improvement within 2-4 days, and although the literature says a toxin treatment can last 6-9 months, Dr. Friedman said he sees patients coming back in 4-5 months.
Obtaining botulinum toxin can be done in one of two ways: the “buy and bill” approach has the dermatologist purchasing the medication, using CPT 64650 and J code J0585 – “Remember the units!” said Dr. Friedman, because reimbursement will be based on the volume of toxin purchased.. This route may be cheaper for the patient because it avoids a medication copay. The physician obtains preauthorization for both the medication and procedure with this strategy.
The other route is to have the provider prescribe botulinum toxin and the patient purchase it at a regular or specialty pharmacy. In this case, the pharmacist obtains precertification for the medication, but the physician still needs to be precertified – and bill – for the injection procedure itself. This scenario is less risky for the physician but may trigger two separate copays for the patient.
Botulinum toxin can be effective for up to 90% of patients, but at a cost: Without insurance reimbursement, treatments can cost in the neighborhood of $1,500.
A good resource for patients and clinicians is the International Hyperhidrosis Society’s website (sweathelp.org), said Dr. Friedman.
Dr. Friedman disclosed relationships with multiple pharmaceutical and cosmetic companies, including Dermira, which markets topical glycopyrronium tosylate as Qbrexza.
ORLANDO – When you extend your hand to a new patient, and he reflexively wipes his palm before shaking hands, be alert. It’s possible you’re seeing primary hyperhidrosis, a condition that’s both more common and more disabling than once thought.
“Looking at the biology of sweating, normally, it’s a good thing – we need it to survive. However, hyperhidrosis is too much of a good thing – it’s an excess of what is needed for normal biology,” said Adam Friedman, MD, speaking at the Orlando Dermatology Aesthetic and Clinical Conference.
Recent data, he pointed out, show that hyperhidrosis is more prevalent than previously thought – about 4.8% of individuals may have the condition, with about half having axillary hyperhidrosis. Symptoms peak in early adulthood, with adults aged 18-54 most affected. “These are the prime working years,” he said.
About 2% of teens are affected, and many adults report that symptoms began before they were 12 years old. Hand hyperhidrosis is a factor for computer and electronic device work, sports, and even handling paper and pencils, noted Dr. Friedman, professor of dermatology at George Washington University, Washington.
“Does it affect quality of life? Yes. We have data to support the impact. The adverse impact is actually greater than that of eczema and psoriasis,” he said, adding that patients won’t always bring up their concerns about sweating. “Often, it’s the patient who apologizes for having sweaty palms or who sticks to the paper on the exam table. It’s worth asking these patients if they are bothered by excessive sweating.”
Is it hyperhidrosis?
A 2016 paper defined hyperhidrosis as “a condition that involves chronic excessive sweating of the underarms, hands, feet, face, groin, or other bodily areas, which is much more than what is normal, and occurs regardless of temperature, exercise, or situation, and may have an impact on quality of life” (Arch Dermatol Res. 2016 Dec;308[10]:743-9). The amount of sweating can be four to five times that seen in healthy controls.
Other clues that excess sweating might be hyperhidrosis? General hyperhidrosis is a secondary syndrome that can be caused by a variety of conditions including endocrine and metabolic disorders and malignancies. Drugs and toxins can also cause generalized excessive sweating.
Focal hyperhidrosis can be primary idiopathic disease; some neuropathies and certain spinal diseases and spinal cord injury can also cause focal hyperhidrosis, though not usually in the axillary/palmar/plantar distribution seen in primary hyperhidrosis.
Before settling on primary hyperhidrosis, the history and exam should also account for other possibilities in the differential: social anxiety disorder, eccrine nevus, gustatory sweating, Frey syndrome, and impaired evaporation could all account for excess sweating, which is also a postsurgical phenomenon for some patients.
Diagnostic criteria call for “focal, visible, excessive sweating” persisting for at least 6 months with no apparent cause. Additionally, patients must have at least two of the additional following criteria: sweating that is bilateral and symmetric, occurs at least once weekly, impairs daily activities, and starts before age 25 years, as well as a positive family history of hyperhidrosis and cessation of sweating during sleep.
The last point is critical, Dr. Friedman said. “If you sweat a lot at night, it’s not hyperhidrosis!”
Though gravimetric evaluation is used in hyperhidrosis research, the history and exam are really where the diagnosis is made in practice, he noted. The Hyperhidrosis Disease Severity Scale is a brief, useful clinical tool that asks patients to peg the extent to which their sweating interferes with daily life.
Topical treatments to try
Topical antiperspirants and other topical agents are a logical place to start and may be required as part of step therapy by insurers. Many patients will already have tried clinical strength over-the-counter antiperspirants containing aluminum zirconium trichlorohydrex, but these products rate low in patient satisfaction among those with primary hyperhidrosis.
Prescription aluminum salts can be compounded to various strengths, with 10%-20% concentration appropriate for axillae and 30%-40% a good strength for palms and soles, according to Dr. Friedman. All of these agents work by precipitating out solids that form a shallow plug in sweat ducts, slowing the flow of perspiration.
Pearls for topical treatment include the need for the product to be on the skin for 6-8 hours overnight. “Remember hyperhidrosis patients do not sweat at night,” so this is the time when the occlusive plugs can form. Then residue can be washed off in the morning, and patients can apply a deodorant. “I remind my patients that antiperspirants are for sweating, and deodorants are for odor,” said Dr. Friedman. These products can damage fabric, and they can be irritating, a problem addressed with low-potency topical steroids.
Topical regimens don’t need to be adjusted for pediatric patients, said Dr. Friedman.
Iontophoresis has been around since the 1950s, is effective, has few side effects, and is considered first-line treatment for severe palmar and plantar hyperhidrosis. But he said there’s one big rub: time. To be effective, patients need 20-30 minutes of application of 15-20 milliamperes of current 3-4 times weekly, not a schedule that works for most patients or practitioners, Dr. Friedman noted.
A treatment recently approved by the Food and Drug Administration for primary axillary hyperhidrosis is a topical anticholinergic, glycopyrronium tosylate, applied with wipes impregnated with glycopyrronium solution. This product significantly outperformed placebo in two clinical trials, with up to 64% of users meeting the primary endpoint of improving by at least 4 points on the Axillary Sweating Daily Diary (ASDD) scale. This product significantly outperformed placebo in two clinical trials, with 53% and 66% of users meeting the primary endpoint, improvement of at least 4 points from baseline in the weekly mean ASDD Item #2. It was approved in those aged 9 years and older.
“You can use this in kids, but you need to educate the kid and the parent or adult,” he said. “This is the last thing you do before bed, after brushing your teeth and after washing your face.”
Patients should apply one swipe to the clean skin of each underarm, and then wash their hands thoroughly. Clinical trials saw a greater proportion of off-target effects such as dry eyes and mouth and mydriasis in the active arm; unilateral mydriasis was more common than bilateral, underscoring the importance of hand washing as this was probably secondary transfer from hands to face during sleep, said Dr. Friedman. Patients can expect results in 2-3 weeks, and doses can be held as needed for anticholinergic side effects.
Systemic choices are limited
There are no FDA-approved systemic agents for hyperhidrosis, and the literature holds only case reports or small series, Dr. Friedman pointed out.
Though systemic treatment may be more effective in generalized hyperhidrosis and for patients with dysautonomia-associated hyperhidrosis, glycopyrrolate is a logical choice if a systemic anticholinergic is desired. A starting dose of 1 mg twice daily can be titrated for effect to about 6 mg daily. Though off-target effects may be a dose-limiting factor, glycopyrrolate is not very lipid soluble, so it penetrates the blood-brain barrier relatively poorly, he said.
Oxybutynin is available in many forms, including a slow-release tablet that permits once-daily dosing. Starting at 5-10 mg daily is a good idea, but dosing may need to be increased to as high as 20 mg daily to be effective. However, patients will often experience “major side effects” with oxybutynin, including significant xerostomia, constipation, blurred vision, and difficulty urinating.
For children, small studies have seen improvement with glycopyrrolate at an average dose of about 2 mg/day. Oxybutynin, which has been extensively studied in the pediatric population, was also effective, but central nervous system adverse events were common.
For some, beta-adrenergic blockade can be an extremely valuable tool, said Dr. Friedman. When sweating is linked to social phobia or performance anxiety, 10-20 mg of atenolol about an hour before the performance or public appearance can make a big difference. Bradycardia, atrioventricular block, and asthma are all contraindications, and the usual precautions should be taken with a host of other comorbidities, he noted.
It’s a good idea to check resting blood pressure and heart rate and take body mass into consideration, and adjust the dose downward appropriately. A key pearl: “Have them do a test run at home, to make sure they don’t keel over on the podium!” said Dr. Friedman.
Botulinum toxin tips and tricks
Botulinum toxin can be very effective and works directly by blocking acetylcholine release at the junction of the sympathetic sudomotor neuron and the sweat gland.
Before treatment, make sure the patient prepares correctly by abstaining from over-the-counter deodorants or antiperspirants, and resting without exertion or drinking hot beverages for about 30 minutes before the procedure.
To ascertain the follicular outline of the area to be injected, the iodine starch test can be used: Paint the axilla with iodine, allow it to dry, and then dust corn starch over the area. The follicular outline is mapped by the purple-blue reaction of the starch and iodine in the presence of moisture from perspiration, Dr. Friedman said.
Applying topical analgesia 30 minutes prior to the procedure helps with patient discomfort with axillary injections. When it comes time to inject, a shallow approach with the bevel side up works well, with a goal of blanketing the field identified by the iodine starch test with small aliquots of toxin placed 1-2 centimeters apart, said Dr. Friedman. However, for patients who might have tattoos that extend to the axillary area, “Avoid the ink!”
Patients will start to see improvement within 2-4 days, and although the literature says a toxin treatment can last 6-9 months, Dr. Friedman said he sees patients coming back in 4-5 months.
Obtaining botulinum toxin can be done in one of two ways: the “buy and bill” approach has the dermatologist purchasing the medication, using CPT 64650 and J code J0585 – “Remember the units!” said Dr. Friedman, because reimbursement will be based on the volume of toxin purchased.. This route may be cheaper for the patient because it avoids a medication copay. The physician obtains preauthorization for both the medication and procedure with this strategy.
The other route is to have the provider prescribe botulinum toxin and the patient purchase it at a regular or specialty pharmacy. In this case, the pharmacist obtains precertification for the medication, but the physician still needs to be precertified – and bill – for the injection procedure itself. This scenario is less risky for the physician but may trigger two separate copays for the patient.
Botulinum toxin can be effective for up to 90% of patients, but at a cost: Without insurance reimbursement, treatments can cost in the neighborhood of $1,500.
A good resource for patients and clinicians is the International Hyperhidrosis Society’s website (sweathelp.org), said Dr. Friedman.
Dr. Friedman disclosed relationships with multiple pharmaceutical and cosmetic companies, including Dermira, which markets topical glycopyrronium tosylate as Qbrexza.
EXPERT ANALYSIS FROM ODAC 2019
Writing an effective cover letter
You have run your experiments, analyzed your data, and finished your manuscript, but now you are asked to write a cover letter for journal submission. How do you effectively convey your message in a cover letter? To understand how to gain the editors’ support for your paper, let us first discuss the role of this letter.

The cover letter is your first communication with the editors. As this serves as your first impression, you want to send a clear and concise message that highlights the novelty, validity, and significance of your manuscript. You also want to state why your manuscript would be a good fit for the journal. Keeping this letter concise and ideally to one page allows the editors to quickly review the highlights of your manuscript. Here are 10 steps we believe are important to follow when writing an effective cover letter (Table 1). We have listed examples to accompany each step; note that our examples are for illustrative purposes only.
1) Address the editor(s) formally by name in your cover letter
This information is found on the journal’s website and in the journal. Common mistakes we have seen is reporting the wrong editor(s) for submission or omitting an introduction. Though this cover letter is not published, it reflects poorly on the authors if there are incorrect editors listed or misspelled words.
Example:
Richard M. Peek, Jr., MD, and Douglas A. Corley, MD, PhD, MPH
Editors in Chief, Gastroenterology
Dear Drs. Peek and Corley:
2) Manuscript title and authors
Bolding your manuscript and listing the authors are essential parts of the cover letter.

Example:
We are submitting our original article entitled “Endoscopic and medical management of acute food impaction: A double-blinded randomized control trial” by Naik R and Inadomi J.
3) Methods
We recommend dedicating one sentence to your methods. Take this time to highlight key features if appropriate, such as novel techniques, prospective enrollment, and randomized, controlled studies, which are all generally viewed as stronger study designs than retrospective studies. Make sure you are truthful in your claims. Mistakes we have seen include making claims the group is the “first” or the “largest,” but our review of the literature disproves these statements. These are false claims and raise flags regarding the integrity of the science.

In this pragmatic, randomized, double-blind placebo-controlled study, our primary aim was to calculate the efficacy of endoscopic dilation and oral steroids to reduce recurrent dysphagia in individuals aged 20-49 years with eosinophilic esophagitis admitted with acute food impaction.
4) Results and key findings
We recommend dedicating a sentence to the key finding of this study. This allows the editors in chief to identify which associate editor should handle the manuscript and highlights the importance of the study, which will determine whether the associate editor will send your paper for external review.
Example:
We found that combined endoscopic dilation and oral steroid therapy at index food impaction improved quality of life and need for repeat dilation measured at 1-year follow-up, compared with sham dilation and placebo.
5) Novelty
The editors want to know the degree to which your paper is unique among other publications in the field. We recommend emphasizing the novelty of your study as compared with other published work. The focus of this should be how your manuscript adds to the literature and serves to advance the science in this field.
Example:
Our study is unique because we enrolled patients in a pragmatic fashion at time of admission for food impaction to the emergency department. This design allows implementation of our intervention in most clinical settings, where we expect our findings to translate broadly into reduced hospital admissions and repeat endoscopic interventions, as well as improved quality of life as documented by patient-reported outcomes.
6) Submission to other journals
Certain manuscripts are only part of the entire study, which can be because of interim results or prespecified secondary endpoints. It is important to state whether this manuscript or any part has been published elsewhere or parts of the study are in submission elsewhere.
Example:
Neither the entire paper nor any part of its content has been published or has been accepted elsewhere. This work has not been submitted to any other journal, and in case of acceptance of the manuscript, the copyright is transferred to Gastroenterology.
7) Journal fit
Not all journals have the same readership or focus. When submitting to the journals, please highlight why this manuscript fits this journal and its readership. When able, also recommend what section of the journal this manuscript should go to for review. This allows for expedited review process and shows you understand the journal’s readership and categories.
Example:
We believe our study will be of great interest to readers of Gastroenterology and suggest the manuscript section: “Clinical: Alimentary Tract.”
8) Influence of sponsors and conflicts of interest
Given the concern for potential conflict of interests in study design, data collection, or analysis, we recommend listing any important conflicts of interest or study sponsors. The entire list of conflicts of interest is typically included elsewhere, but a sponsored study or direct conflict should be listed to prevent any perceived influence from sponsors that may limit data integrity.
Example:
The study was supported by RDN-Gastro, a nonprofit private organization aimed to promote clinical and translational research in eosinophilic esophagitis. The RDN-Gastro organization receives support from the National Institutes of Health and the Emergency Department Eosinophilic Esophagitis Association.
9) Suggestions for editors and reviewers
It is helpful to suggest potential associate editors to handle your manuscript. Similarly, suggesting potential reviewers (and providing their email addresses) who are experts in the field and understand your research topic can enhance the quality of reviews. Soliciting reviewers who you know may seem like a recipe for friendly reviews; however, this is a flawed assumption because sometimes these reviewers are more rigorous in their comments.
Example:
Preferred Associate Editor: John M. Inadomi.
Preferred Reviewer: Rishi Naik ([email protected]); expertise in esophageal motility.
10) Do Not Review list (optional)
Unfortunately, not all review processes for all journals are created equal. For particular topics, you may wish to ensure your data are not compromised through the review process. Some journals will have the option for selecting reviewers who you prefer not review your manuscript and this request should be respected in the review process.
Example:
Nonpreferred reviewers: Rishi D. Naik (Nashville, TN).
In summary, the cover letter should be viewed as an opportunity to highlight the significance of your manuscript and reasons why your manuscript should be published in the journal. Highlighting the innovation, validity, and importance of your manuscript using this systematic approach will allow the editors and reviewers to more effectively evaluate your paper. Being truthful about potential conflicts of interest and sponsorships will also be appreciated by the editors. Good luck with your future submissions!
Dr. Naik is a gastroenterology fellow, department of gastroenterology, Vanderbilt University, Nashville, Tenn., as well as fellow editor of Gastroenterology. Dr. Inadomi is the Cyrus E. Rubin Chair, professor of medicine and head, division of gastroenterology, University of Washington, Seattle, as well as associate editor of Gastroenterology. Dr. Naik and Dr. Inadomi have no conflicts of interest to disclose.
You have run your experiments, analyzed your data, and finished your manuscript, but now you are asked to write a cover letter for journal submission. How do you effectively convey your message in a cover letter? To understand how to gain the editors’ support for your paper, let us first discuss the role of this letter.
The cover letter is your first communication with the editors. As this serves as your first impression, you want to send a clear and concise message that highlights the novelty, validity, and significance of your manuscript. You also want to state why your manuscript would be a good fit for the journal. Keeping this letter concise and ideally to one page allows the editors to quickly review the highlights of your manuscript. Here are 10 steps we believe are important to follow when writing an effective cover letter (Table 1). We have listed examples to accompany each step; note that our examples are for illustrative purposes only.
1) Address the editor(s) formally by name in your cover letter
This information is found on the journal’s website and in the journal. Common mistakes we have seen is reporting the wrong editor(s) for submission or omitting an introduction. Though this cover letter is not published, it reflects poorly on the authors if there are incorrect editors listed or misspelled words.
Example:
Richard M. Peek, Jr., MD, and Douglas A. Corley, MD, PhD, MPH
Editors in Chief, Gastroenterology
Dear Drs. Peek and Corley:
2) Manuscript title and authors
Bolding your manuscript and listing the authors are essential parts of the cover letter.
Example:
We are submitting our original article entitled “Endoscopic and medical management of acute food impaction: A double-blinded randomized control trial” by Naik R and Inadomi J.
3) Methods
We recommend dedicating one sentence to your methods. Take this time to highlight key features if appropriate, such as novel techniques, prospective enrollment, and randomized, controlled studies, which are all generally viewed as stronger study designs than retrospective studies. Make sure you are truthful in your claims. Mistakes we have seen include making claims the group is the “first” or the “largest,” but our review of the literature disproves these statements. These are false claims and raise flags regarding the integrity of the science.
In this pragmatic, randomized, double-blind placebo-controlled study, our primary aim was to calculate the efficacy of endoscopic dilation and oral steroids to reduce recurrent dysphagia in individuals aged 20-49 years with eosinophilic esophagitis admitted with acute food impaction.
4) Results and key findings
We recommend dedicating a sentence to the key finding of this study. This allows the editors in chief to identify which associate editor should handle the manuscript and highlights the importance of the study, which will determine whether the associate editor will send your paper for external review.
Example:
We found that combined endoscopic dilation and oral steroid therapy at index food impaction improved quality of life and need for repeat dilation measured at 1-year follow-up, compared with sham dilation and placebo.
5) Novelty
The editors want to know the degree to which your paper is unique among other publications in the field. We recommend emphasizing the novelty of your study as compared with other published work. The focus of this should be how your manuscript adds to the literature and serves to advance the science in this field.
Example:
Our study is unique because we enrolled patients in a pragmatic fashion at time of admission for food impaction to the emergency department. This design allows implementation of our intervention in most clinical settings, where we expect our findings to translate broadly into reduced hospital admissions and repeat endoscopic interventions, as well as improved quality of life as documented by patient-reported outcomes.
6) Submission to other journals
Certain manuscripts are only part of the entire study, which can be because of interim results or prespecified secondary endpoints. It is important to state whether this manuscript or any part has been published elsewhere or parts of the study are in submission elsewhere.
Example:
Neither the entire paper nor any part of its content has been published or has been accepted elsewhere. This work has not been submitted to any other journal, and in case of acceptance of the manuscript, the copyright is transferred to Gastroenterology.
7) Journal fit
Not all journals have the same readership or focus. When submitting to the journals, please highlight why this manuscript fits this journal and its readership. When able, also recommend what section of the journal this manuscript should go to for review. This allows for expedited review process and shows you understand the journal’s readership and categories.
Example:
We believe our study will be of great interest to readers of Gastroenterology and suggest the manuscript section: “Clinical: Alimentary Tract.”
8) Influence of sponsors and conflicts of interest
Given the concern for potential conflict of interests in study design, data collection, or analysis, we recommend listing any important conflicts of interest or study sponsors. The entire list of conflicts of interest is typically included elsewhere, but a sponsored study or direct conflict should be listed to prevent any perceived influence from sponsors that may limit data integrity.
Example:
The study was supported by RDN-Gastro, a nonprofit private organization aimed to promote clinical and translational research in eosinophilic esophagitis. The RDN-Gastro organization receives support from the National Institutes of Health and the Emergency Department Eosinophilic Esophagitis Association.
9) Suggestions for editors and reviewers
It is helpful to suggest potential associate editors to handle your manuscript. Similarly, suggesting potential reviewers (and providing their email addresses) who are experts in the field and understand your research topic can enhance the quality of reviews. Soliciting reviewers who you know may seem like a recipe for friendly reviews; however, this is a flawed assumption because sometimes these reviewers are more rigorous in their comments.
Example:
Preferred Associate Editor: John M. Inadomi.
Preferred Reviewer: Rishi Naik ([email protected]); expertise in esophageal motility.
10) Do Not Review list (optional)
Unfortunately, not all review processes for all journals are created equal. For particular topics, you may wish to ensure your data are not compromised through the review process. Some journals will have the option for selecting reviewers who you prefer not review your manuscript and this request should be respected in the review process.
Example:
Nonpreferred reviewers: Rishi D. Naik (Nashville, TN).
In summary, the cover letter should be viewed as an opportunity to highlight the significance of your manuscript and reasons why your manuscript should be published in the journal. Highlighting the innovation, validity, and importance of your manuscript using this systematic approach will allow the editors and reviewers to more effectively evaluate your paper. Being truthful about potential conflicts of interest and sponsorships will also be appreciated by the editors. Good luck with your future submissions!
Dr. Naik is a gastroenterology fellow, department of gastroenterology, Vanderbilt University, Nashville, Tenn., as well as fellow editor of Gastroenterology. Dr. Inadomi is the Cyrus E. Rubin Chair, professor of medicine and head, division of gastroenterology, University of Washington, Seattle, as well as associate editor of Gastroenterology. Dr. Naik and Dr. Inadomi have no conflicts of interest to disclose.
You have run your experiments, analyzed your data, and finished your manuscript, but now you are asked to write a cover letter for journal submission. How do you effectively convey your message in a cover letter? To understand how to gain the editors’ support for your paper, let us first discuss the role of this letter.
The cover letter is your first communication with the editors. As this serves as your first impression, you want to send a clear and concise message that highlights the novelty, validity, and significance of your manuscript. You also want to state why your manuscript would be a good fit for the journal. Keeping this letter concise and ideally to one page allows the editors to quickly review the highlights of your manuscript. Here are 10 steps we believe are important to follow when writing an effective cover letter (Table 1). We have listed examples to accompany each step; note that our examples are for illustrative purposes only.
1) Address the editor(s) formally by name in your cover letter
This information is found on the journal’s website and in the journal. Common mistakes we have seen is reporting the wrong editor(s) for submission or omitting an introduction. Though this cover letter is not published, it reflects poorly on the authors if there are incorrect editors listed or misspelled words.
Example:
Richard M. Peek, Jr., MD, and Douglas A. Corley, MD, PhD, MPH
Editors in Chief, Gastroenterology
Dear Drs. Peek and Corley:
2) Manuscript title and authors
Bolding your manuscript and listing the authors are essential parts of the cover letter.
Example:
We are submitting our original article entitled “Endoscopic and medical management of acute food impaction: A double-blinded randomized control trial” by Naik R and Inadomi J.
3) Methods
We recommend dedicating one sentence to your methods. Take this time to highlight key features if appropriate, such as novel techniques, prospective enrollment, and randomized, controlled studies, which are all generally viewed as stronger study designs than retrospective studies. Make sure you are truthful in your claims. Mistakes we have seen include making claims the group is the “first” or the “largest,” but our review of the literature disproves these statements. These are false claims and raise flags regarding the integrity of the science.
In this pragmatic, randomized, double-blind placebo-controlled study, our primary aim was to calculate the efficacy of endoscopic dilation and oral steroids to reduce recurrent dysphagia in individuals aged 20-49 years with eosinophilic esophagitis admitted with acute food impaction.
4) Results and key findings
We recommend dedicating a sentence to the key finding of this study. This allows the editors in chief to identify which associate editor should handle the manuscript and highlights the importance of the study, which will determine whether the associate editor will send your paper for external review.
Example:
We found that combined endoscopic dilation and oral steroid therapy at index food impaction improved quality of life and need for repeat dilation measured at 1-year follow-up, compared with sham dilation and placebo.
5) Novelty
The editors want to know the degree to which your paper is unique among other publications in the field. We recommend emphasizing the novelty of your study as compared with other published work. The focus of this should be how your manuscript adds to the literature and serves to advance the science in this field.
Example:
Our study is unique because we enrolled patients in a pragmatic fashion at time of admission for food impaction to the emergency department. This design allows implementation of our intervention in most clinical settings, where we expect our findings to translate broadly into reduced hospital admissions and repeat endoscopic interventions, as well as improved quality of life as documented by patient-reported outcomes.
6) Submission to other journals
Certain manuscripts are only part of the entire study, which can be because of interim results or prespecified secondary endpoints. It is important to state whether this manuscript or any part has been published elsewhere or parts of the study are in submission elsewhere.
Example:
Neither the entire paper nor any part of its content has been published or has been accepted elsewhere. This work has not been submitted to any other journal, and in case of acceptance of the manuscript, the copyright is transferred to Gastroenterology.
7) Journal fit
Not all journals have the same readership or focus. When submitting to the journals, please highlight why this manuscript fits this journal and its readership. When able, also recommend what section of the journal this manuscript should go to for review. This allows for expedited review process and shows you understand the journal’s readership and categories.
Example:
We believe our study will be of great interest to readers of Gastroenterology and suggest the manuscript section: “Clinical: Alimentary Tract.”
8) Influence of sponsors and conflicts of interest
Given the concern for potential conflict of interests in study design, data collection, or analysis, we recommend listing any important conflicts of interest or study sponsors. The entire list of conflicts of interest is typically included elsewhere, but a sponsored study or direct conflict should be listed to prevent any perceived influence from sponsors that may limit data integrity.
Example:
The study was supported by RDN-Gastro, a nonprofit private organization aimed to promote clinical and translational research in eosinophilic esophagitis. The RDN-Gastro organization receives support from the National Institutes of Health and the Emergency Department Eosinophilic Esophagitis Association.
9) Suggestions for editors and reviewers
It is helpful to suggest potential associate editors to handle your manuscript. Similarly, suggesting potential reviewers (and providing their email addresses) who are experts in the field and understand your research topic can enhance the quality of reviews. Soliciting reviewers who you know may seem like a recipe for friendly reviews; however, this is a flawed assumption because sometimes these reviewers are more rigorous in their comments.
Example:
Preferred Associate Editor: John M. Inadomi.
Preferred Reviewer: Rishi Naik ([email protected]); expertise in esophageal motility.
10) Do Not Review list (optional)
Unfortunately, not all review processes for all journals are created equal. For particular topics, you may wish to ensure your data are not compromised through the review process. Some journals will have the option for selecting reviewers who you prefer not review your manuscript and this request should be respected in the review process.
Example:
Nonpreferred reviewers: Rishi D. Naik (Nashville, TN).
In summary, the cover letter should be viewed as an opportunity to highlight the significance of your manuscript and reasons why your manuscript should be published in the journal. Highlighting the innovation, validity, and importance of your manuscript using this systematic approach will allow the editors and reviewers to more effectively evaluate your paper. Being truthful about potential conflicts of interest and sponsorships will also be appreciated by the editors. Good luck with your future submissions!
Dr. Naik is a gastroenterology fellow, department of gastroenterology, Vanderbilt University, Nashville, Tenn., as well as fellow editor of Gastroenterology. Dr. Inadomi is the Cyrus E. Rubin Chair, professor of medicine and head, division of gastroenterology, University of Washington, Seattle, as well as associate editor of Gastroenterology. Dr. Naik and Dr. Inadomi have no conflicts of interest to disclose.
