User login
Effective Management and Counseling of Patients with Recurrent Bacterial Vaginosis
Click Here to Read Supplement.

Bacterial vaginosis (BV) affects women worldwide and recurrent BV can be a frustrating condition for both patients and providers. In this new supplement, expert Khady Diouf, MD, discusses her treatment approach and suggested counseling for patients with recurrent BV.
Click Here to Read Supplement.
Bacterial vaginosis (BV) affects women worldwide and recurrent BV can be a frustrating condition for both patients and providers. In this new supplement, expert Khady Diouf, MD, discusses her treatment approach and suggested counseling for patients with recurrent BV.
Click Here to Read Supplement.
Bacterial vaginosis (BV) affects women worldwide and recurrent BV can be a frustrating condition for both patients and providers. In this new supplement, expert Khady Diouf, MD, discusses her treatment approach and suggested counseling for patients with recurrent BV.

Preterm infant GER is a normal phenomenon
Treatment of gastroesophageal reflux (GER) in preterm infants with traditional treatments, such as body positioning, and newer treatments with pharmacologic agents appear to be ineffective, and pharmacologic agents in particular may cause significant harm, according to a clinical report by the American Academy of Pediatrics Committee on Fetus and Newborn.
“I think that probably the most important point for any physician, including neonatologists, is that the committee concluded on the basis of the evidence that Eric Eichenwald, MD, lead author of the committee’s clinical report and chief of neonatology at Children’s Hospital of Philadelphia, said in an interview. “So really the bottom line of the clinical report is watchful waiting, conservative management, and patience is the most important approach to a baby that you think is suffering from reflux.”
Pharmacologic management
The committee members focused on four categories of pharmacologic interventions in their report in Pediatrics.
Prokinetic (promotility) agents, such as metoclopramide, domperidone, and erythromycin, are widely used in treating symptoms of GER in older infants and appear to improve gastric emptying, reduce regurgitation, and enhance lower esophageal sphincter tone, but they do not appear to reduce GER symptoms in preterm infants. In addition to not being effective in these infants, there is also a potential for significant adverse events, including cardiac arrhythmia and neurologic side effects. Another common pharmacologic treatment is the use of sodium alginate in combination with sodium bicarbonate. In the presence of gastric acid, sodium alginate precipitates as a gel that forms a physical barrier that protects the gastric mucosa. When sodium bicarbonate is added, a carbon dioxide foam forms that is less harmful to the esophagus than GER-related fluids. While this combination treatment has reduced the number of acidic GER exposures and esophageal acid exposure in preterm infants in small studies, the long-term safety has not been evaluated in this populations.
Histamine2 (H2) blockers, like famotidine and ranitidine, also are commonly prescribed to treat preterm infant gastroesophageal reflux. H2 blockers compete with H2 for the histamine receptors of the parietal cells, which causes a decrease in hydrochloric acid and a subsequent increase in intragastric pH. These are often prescribed on the premise that GER symptoms are secondary to acid reflux in the lower esophagus, but there is no research on the efficacy of H2 blockers on the symptom profile of GER in preterm infants. This class of drugs also has been linked with an increased risk of necrotizing enterocolitis and a higher incidence of late-onset infections and death. This is thought to be caused by alteration of the intestinal microbiome, according to the clinical report.
Proton pump inhibitors (PPIs) are another treatment for reducing acid secretion by the parietal cells, but are largely ineffective in relieving clinical signs of GER in preterm infants. PPIs also have been associated with a higher risk of bacterial overgrowth, gastroenteritis, and community-acquired pneumonia in older children. It is theorized that, because of the acid mitigating effects of PPIs, they will have the potential for adverse effects similar to those seen with H2 blockers, although this has not been investigated.
Traditional treatments
Dr. Eichenwald also was quick to point out that even traditional methods of treating preterm infant GER are not particularly effective.
“Some of the conservative approaches that have been advocated include head-up position and different ways of side-lying to enhance emptying of the stomach after feeding. And none of those have been shown to reduce clinically appreciated signs of reflux in preterm infants. If anything – in term babies – some of those positions have been shown to increase the amount of reflux,” he said in an interview.
“I think that the other important point to make about this is that there are many signs that clinicians attribute to reflux in preterm babies, which include wakefulness, irritability, arching after a feeding. And none of those behaviors have been shown to be associated with reflux when it’s critically examined using either a pH Probe or multichannel impedance monitoring. And therefore the treatments to try to decrease reflux don’t really have an effect on those behaviors either.”
Parental concern
Treating a pediatric issue is not as simple as diagnosis and treatment. Often, parents are justifiably concerned about their children. Dr. Eichenwald sees educating parents as an important facet of treating GER in preterm infants.
“Quite honestly I think that there’s some projection on the part of adults who say, ‘I know how I feel when I have heartburn, which is the adult equivalent of reflux, and the baby must be experiencing the same thing, and that’s why they’re acting uncomfortable,’ ” suggested Dr. Eichenwald. “I think that it’s important for clinicians to educate families that a lot of the signs that we typically have attributed to gastroesophageal reflux are not really related to it.”
With both traditional and pharmacological interventions failing to treat preterm infant GER, Dr. Eichenwald believes that the most effective treatment could be patiently waiting. “I think that the important thing to stress is that reflux is a normal physiologic phenomenon. It rarely causes pathology in preterm infants, and therefore, in treating it, you’re not treating any pathology. You should just be patient and it will likely just go away on its own.”
Dr. Eichenwald has no potential conflicts of interest or external funding to report.
SOURCE: Eichenwald E et al. Pediatrics. 2018 June. doi: 10.1542/peds.2018-1061 .
Treatment of gastroesophageal reflux (GER) in preterm infants with traditional treatments, such as body positioning, and newer treatments with pharmacologic agents appear to be ineffective, and pharmacologic agents in particular may cause significant harm, according to a clinical report by the American Academy of Pediatrics Committee on Fetus and Newborn.
“I think that probably the most important point for any physician, including neonatologists, is that the committee concluded on the basis of the evidence that Eric Eichenwald, MD, lead author of the committee’s clinical report and chief of neonatology at Children’s Hospital of Philadelphia, said in an interview. “So really the bottom line of the clinical report is watchful waiting, conservative management, and patience is the most important approach to a baby that you think is suffering from reflux.”
Pharmacologic management
The committee members focused on four categories of pharmacologic interventions in their report in Pediatrics.
Prokinetic (promotility) agents, such as metoclopramide, domperidone, and erythromycin, are widely used in treating symptoms of GER in older infants and appear to improve gastric emptying, reduce regurgitation, and enhance lower esophageal sphincter tone, but they do not appear to reduce GER symptoms in preterm infants. In addition to not being effective in these infants, there is also a potential for significant adverse events, including cardiac arrhythmia and neurologic side effects. Another common pharmacologic treatment is the use of sodium alginate in combination with sodium bicarbonate. In the presence of gastric acid, sodium alginate precipitates as a gel that forms a physical barrier that protects the gastric mucosa. When sodium bicarbonate is added, a carbon dioxide foam forms that is less harmful to the esophagus than GER-related fluids. While this combination treatment has reduced the number of acidic GER exposures and esophageal acid exposure in preterm infants in small studies, the long-term safety has not been evaluated in this populations.
Histamine2 (H2) blockers, like famotidine and ranitidine, also are commonly prescribed to treat preterm infant gastroesophageal reflux. H2 blockers compete with H2 for the histamine receptors of the parietal cells, which causes a decrease in hydrochloric acid and a subsequent increase in intragastric pH. These are often prescribed on the premise that GER symptoms are secondary to acid reflux in the lower esophagus, but there is no research on the efficacy of H2 blockers on the symptom profile of GER in preterm infants. This class of drugs also has been linked with an increased risk of necrotizing enterocolitis and a higher incidence of late-onset infections and death. This is thought to be caused by alteration of the intestinal microbiome, according to the clinical report.
Proton pump inhibitors (PPIs) are another treatment for reducing acid secretion by the parietal cells, but are largely ineffective in relieving clinical signs of GER in preterm infants. PPIs also have been associated with a higher risk of bacterial overgrowth, gastroenteritis, and community-acquired pneumonia in older children. It is theorized that, because of the acid mitigating effects of PPIs, they will have the potential for adverse effects similar to those seen with H2 blockers, although this has not been investigated.
Traditional treatments
Dr. Eichenwald also was quick to point out that even traditional methods of treating preterm infant GER are not particularly effective.
“Some of the conservative approaches that have been advocated include head-up position and different ways of side-lying to enhance emptying of the stomach after feeding. And none of those have been shown to reduce clinically appreciated signs of reflux in preterm infants. If anything – in term babies – some of those positions have been shown to increase the amount of reflux,” he said in an interview.
“I think that the other important point to make about this is that there are many signs that clinicians attribute to reflux in preterm babies, which include wakefulness, irritability, arching after a feeding. And none of those behaviors have been shown to be associated with reflux when it’s critically examined using either a pH Probe or multichannel impedance monitoring. And therefore the treatments to try to decrease reflux don’t really have an effect on those behaviors either.”
Parental concern
Treating a pediatric issue is not as simple as diagnosis and treatment. Often, parents are justifiably concerned about their children. Dr. Eichenwald sees educating parents as an important facet of treating GER in preterm infants.
“Quite honestly I think that there’s some projection on the part of adults who say, ‘I know how I feel when I have heartburn, which is the adult equivalent of reflux, and the baby must be experiencing the same thing, and that’s why they’re acting uncomfortable,’ ” suggested Dr. Eichenwald. “I think that it’s important for clinicians to educate families that a lot of the signs that we typically have attributed to gastroesophageal reflux are not really related to it.”
With both traditional and pharmacological interventions failing to treat preterm infant GER, Dr. Eichenwald believes that the most effective treatment could be patiently waiting. “I think that the important thing to stress is that reflux is a normal physiologic phenomenon. It rarely causes pathology in preterm infants, and therefore, in treating it, you’re not treating any pathology. You should just be patient and it will likely just go away on its own.”
Dr. Eichenwald has no potential conflicts of interest or external funding to report.
SOURCE: Eichenwald E et al. Pediatrics. 2018 June. doi: 10.1542/peds.2018-1061 .
Treatment of gastroesophageal reflux (GER) in preterm infants with traditional treatments, such as body positioning, and newer treatments with pharmacologic agents appear to be ineffective, and pharmacologic agents in particular may cause significant harm, according to a clinical report by the American Academy of Pediatrics Committee on Fetus and Newborn.
“I think that probably the most important point for any physician, including neonatologists, is that the committee concluded on the basis of the evidence that Eric Eichenwald, MD, lead author of the committee’s clinical report and chief of neonatology at Children’s Hospital of Philadelphia, said in an interview. “So really the bottom line of the clinical report is watchful waiting, conservative management, and patience is the most important approach to a baby that you think is suffering from reflux.”
Pharmacologic management
The committee members focused on four categories of pharmacologic interventions in their report in Pediatrics.
Prokinetic (promotility) agents, such as metoclopramide, domperidone, and erythromycin, are widely used in treating symptoms of GER in older infants and appear to improve gastric emptying, reduce regurgitation, and enhance lower esophageal sphincter tone, but they do not appear to reduce GER symptoms in preterm infants. In addition to not being effective in these infants, there is also a potential for significant adverse events, including cardiac arrhythmia and neurologic side effects. Another common pharmacologic treatment is the use of sodium alginate in combination with sodium bicarbonate. In the presence of gastric acid, sodium alginate precipitates as a gel that forms a physical barrier that protects the gastric mucosa. When sodium bicarbonate is added, a carbon dioxide foam forms that is less harmful to the esophagus than GER-related fluids. While this combination treatment has reduced the number of acidic GER exposures and esophageal acid exposure in preterm infants in small studies, the long-term safety has not been evaluated in this populations.
Histamine2 (H2) blockers, like famotidine and ranitidine, also are commonly prescribed to treat preterm infant gastroesophageal reflux. H2 blockers compete with H2 for the histamine receptors of the parietal cells, which causes a decrease in hydrochloric acid and a subsequent increase in intragastric pH. These are often prescribed on the premise that GER symptoms are secondary to acid reflux in the lower esophagus, but there is no research on the efficacy of H2 blockers on the symptom profile of GER in preterm infants. This class of drugs also has been linked with an increased risk of necrotizing enterocolitis and a higher incidence of late-onset infections and death. This is thought to be caused by alteration of the intestinal microbiome, according to the clinical report.
Proton pump inhibitors (PPIs) are another treatment for reducing acid secretion by the parietal cells, but are largely ineffective in relieving clinical signs of GER in preterm infants. PPIs also have been associated with a higher risk of bacterial overgrowth, gastroenteritis, and community-acquired pneumonia in older children. It is theorized that, because of the acid mitigating effects of PPIs, they will have the potential for adverse effects similar to those seen with H2 blockers, although this has not been investigated.
Traditional treatments
Dr. Eichenwald also was quick to point out that even traditional methods of treating preterm infant GER are not particularly effective.
“Some of the conservative approaches that have been advocated include head-up position and different ways of side-lying to enhance emptying of the stomach after feeding. And none of those have been shown to reduce clinically appreciated signs of reflux in preterm infants. If anything – in term babies – some of those positions have been shown to increase the amount of reflux,” he said in an interview.
“I think that the other important point to make about this is that there are many signs that clinicians attribute to reflux in preterm babies, which include wakefulness, irritability, arching after a feeding. And none of those behaviors have been shown to be associated with reflux when it’s critically examined using either a pH Probe or multichannel impedance monitoring. And therefore the treatments to try to decrease reflux don’t really have an effect on those behaviors either.”
Parental concern
Treating a pediatric issue is not as simple as diagnosis and treatment. Often, parents are justifiably concerned about their children. Dr. Eichenwald sees educating parents as an important facet of treating GER in preterm infants.
“Quite honestly I think that there’s some projection on the part of adults who say, ‘I know how I feel when I have heartburn, which is the adult equivalent of reflux, and the baby must be experiencing the same thing, and that’s why they’re acting uncomfortable,’ ” suggested Dr. Eichenwald. “I think that it’s important for clinicians to educate families that a lot of the signs that we typically have attributed to gastroesophageal reflux are not really related to it.”
With both traditional and pharmacological interventions failing to treat preterm infant GER, Dr. Eichenwald believes that the most effective treatment could be patiently waiting. “I think that the important thing to stress is that reflux is a normal physiologic phenomenon. It rarely causes pathology in preterm infants, and therefore, in treating it, you’re not treating any pathology. You should just be patient and it will likely just go away on its own.”
Dr. Eichenwald has no potential conflicts of interest or external funding to report.
SOURCE: Eichenwald E et al. Pediatrics. 2018 June. doi: 10.1542/peds.2018-1061 .
FROM PEDIATRICS
Impulse control disorders in Parkinson’s patients may be higher than thought
Nearly half of patients with Parkinson’s disease who were taking dopamine agonist treatment experienced impulse control disorders over a follow-up of 5 years, according to recently published results of a longitudinal study.
The 5-year cumulative incidence of impulse control disorders was approximately 45% in the study, which included 411 patients with a high prevalence of dopamine agonist use and disease duration of 5 years or less at baseline.
There was a strong association between dopamine agonist use and impulse control disorders in the study, which was conducted by Jean-Christophe Corvol, MD, of Publique Hôpitaux de Paris and his coinvestigators.
Impulse disorders increased in incidence with both duration and dose of dopamine agonists and resolved progressively after discontinuation of those agents, the investigators reported online June 20 in Neurology. The investigators used item 1.6 of part I of the Movement Disorder Society Unified Parkinson’s Disease Rating Scale to determine the presence of an impulse control disorder.
“Given the high cumulative incidence of impulse control disorders in patients with Parkinson’s disease, these adverse effects should be carefully monitored in patients ever treated with dopamine agonists,” Dr. Corvol and his coauthors wrote.
The results came from the ongoing Drug Interaction With Genes in Parkinson’s Disease (DIGPD) study, a longitudinal cohort study including Parkinson’s disease patients consecutively recruited between 2009 and 2013 at eight French hospitals. All patients had no more than 5 years of disease duration at recruitment, and follow-up included annual evaluations by movement disorder specialists.
At baseline, the majority of patients (302, or 73.5%) had taken dopamine agonists within the past 12 months.
Over the course of 5 years, the prevalence of impulse control disorders increased from 19.7% at baseline to 32.8%, Dr. Corvol and his colleagues reported.
Among 306 patients with no impulse control disorders at baseline, 94 developed one, for a 5-year cumulative incidence of 46.1%, they added. Only 4 of the 94 new cases occurred in patients who never used dopamine agonists.
Dopamine agonist use in the previous 12 months was associated with a 2.23-fold higher prevalence of impulse control disorders (P less than .001), with prevalence increasing along with average daily dose and cumulative dose duration over that time period, according to the investigators.
These findings suggests tools are needed to screen for impulse control disorders and identify high-risk patients, they said.
“Further studies are needed to understand the mechanisms involved in the relation between [dopamine agonists] and [impulse control disorders], in particular the role of apathy, anxiety, and depression,” they added.
The study was funded by grants from the French Ministry of Health and from the French Drug Agency. Dr. Corvol and many of his colleagues reported financial disclosures with many pharmaceutical companies.
SOURCE: Corvol J-C et al. Neurology. 2018 Jun 20. doi: 10.1212/WNL.0000000000005816.
Data from the study by Dr. Corvol and colleagues are robust and suggest neurologists may be “missing the boat or even harming patients” by underestimating the adverse effects associated with dopamine agonists, the authors of an editorial wrote.
The 5-year incidence of impulse control disorders approaching 50% suggests they are even more common than previously reported, according to editorial authors Laura S. Boylan, MD, and Vladimir S. Kostic, MD, PhD.Compulsive gambling, shopping, eating, sexual behaviors and other impulse control disorders at their worst can ruin finances, disrupt families, and have legal implications, Dr. Boylan and Dr. Kostic said in their editorial.
Neurologists are “often uncomfortable” with psychiatric disorders, they added, even though they are the ones managing movement disorder medications.
There is an absence of high-quality evidence on how to treat impulse control disorders, though one common approach, switching to levodopa, is in the wheelhouse of neurologists. However, “levodopaphobia” persists in some circles despite evidence debunking the notion that the medication is neurotoxic, according to Dr. Boylan and Dr. Kostic.
“Practice change in medicine, as in other areas, can be like turning a cruise ship,” they wrote, “but this study may help give a little push to the boat and, we hope, promote further basic and clinical research on nonmotor aspects of PD and other movement disorders.”
Dr. Boylan is with Essentia Health, Duluth, Minn., Albany-Stratton VA Medical Center, Albany, N.Y., and Bellevue Hospital/New York University. Dr. Kosticis with the Institute of Neurology CCS, School of Medicine University of Belgrade (Serbia). Dr. Kostic reported receiving speaker honoraria from Novartis, Teva, and Salveo. Their editorial accompanied Dr. Corvol and colleagues’ report (Neurology. 2018 Jun 20. doi: 10.1212/WNL.0000000000005806 ).
Data from the study by Dr. Corvol and colleagues are robust and suggest neurologists may be “missing the boat or even harming patients” by underestimating the adverse effects associated with dopamine agonists, the authors of an editorial wrote.
The 5-year incidence of impulse control disorders approaching 50% suggests they are even more common than previously reported, according to editorial authors Laura S. Boylan, MD, and Vladimir S. Kostic, MD, PhD.Compulsive gambling, shopping, eating, sexual behaviors and other impulse control disorders at their worst can ruin finances, disrupt families, and have legal implications, Dr. Boylan and Dr. Kostic said in their editorial.
Neurologists are “often uncomfortable” with psychiatric disorders, they added, even though they are the ones managing movement disorder medications.
There is an absence of high-quality evidence on how to treat impulse control disorders, though one common approach, switching to levodopa, is in the wheelhouse of neurologists. However, “levodopaphobia” persists in some circles despite evidence debunking the notion that the medication is neurotoxic, according to Dr. Boylan and Dr. Kostic.
“Practice change in medicine, as in other areas, can be like turning a cruise ship,” they wrote, “but this study may help give a little push to the boat and, we hope, promote further basic and clinical research on nonmotor aspects of PD and other movement disorders.”
Dr. Boylan is with Essentia Health, Duluth, Minn., Albany-Stratton VA Medical Center, Albany, N.Y., and Bellevue Hospital/New York University. Dr. Kosticis with the Institute of Neurology CCS, School of Medicine University of Belgrade (Serbia). Dr. Kostic reported receiving speaker honoraria from Novartis, Teva, and Salveo. Their editorial accompanied Dr. Corvol and colleagues’ report (Neurology. 2018 Jun 20. doi: 10.1212/WNL.0000000000005806 ).
Data from the study by Dr. Corvol and colleagues are robust and suggest neurologists may be “missing the boat or even harming patients” by underestimating the adverse effects associated with dopamine agonists, the authors of an editorial wrote.
The 5-year incidence of impulse control disorders approaching 50% suggests they are even more common than previously reported, according to editorial authors Laura S. Boylan, MD, and Vladimir S. Kostic, MD, PhD.Compulsive gambling, shopping, eating, sexual behaviors and other impulse control disorders at their worst can ruin finances, disrupt families, and have legal implications, Dr. Boylan and Dr. Kostic said in their editorial.
Neurologists are “often uncomfortable” with psychiatric disorders, they added, even though they are the ones managing movement disorder medications.
There is an absence of high-quality evidence on how to treat impulse control disorders, though one common approach, switching to levodopa, is in the wheelhouse of neurologists. However, “levodopaphobia” persists in some circles despite evidence debunking the notion that the medication is neurotoxic, according to Dr. Boylan and Dr. Kostic.
“Practice change in medicine, as in other areas, can be like turning a cruise ship,” they wrote, “but this study may help give a little push to the boat and, we hope, promote further basic and clinical research on nonmotor aspects of PD and other movement disorders.”
Dr. Boylan is with Essentia Health, Duluth, Minn., Albany-Stratton VA Medical Center, Albany, N.Y., and Bellevue Hospital/New York University. Dr. Kosticis with the Institute of Neurology CCS, School of Medicine University of Belgrade (Serbia). Dr. Kostic reported receiving speaker honoraria from Novartis, Teva, and Salveo. Their editorial accompanied Dr. Corvol and colleagues’ report (Neurology. 2018 Jun 20. doi: 10.1212/WNL.0000000000005806 ).
Nearly half of patients with Parkinson’s disease who were taking dopamine agonist treatment experienced impulse control disorders over a follow-up of 5 years, according to recently published results of a longitudinal study.
The 5-year cumulative incidence of impulse control disorders was approximately 45% in the study, which included 411 patients with a high prevalence of dopamine agonist use and disease duration of 5 years or less at baseline.
There was a strong association between dopamine agonist use and impulse control disorders in the study, which was conducted by Jean-Christophe Corvol, MD, of Publique Hôpitaux de Paris and his coinvestigators.
Impulse disorders increased in incidence with both duration and dose of dopamine agonists and resolved progressively after discontinuation of those agents, the investigators reported online June 20 in Neurology. The investigators used item 1.6 of part I of the Movement Disorder Society Unified Parkinson’s Disease Rating Scale to determine the presence of an impulse control disorder.
“Given the high cumulative incidence of impulse control disorders in patients with Parkinson’s disease, these adverse effects should be carefully monitored in patients ever treated with dopamine agonists,” Dr. Corvol and his coauthors wrote.
The results came from the ongoing Drug Interaction With Genes in Parkinson’s Disease (DIGPD) study, a longitudinal cohort study including Parkinson’s disease patients consecutively recruited between 2009 and 2013 at eight French hospitals. All patients had no more than 5 years of disease duration at recruitment, and follow-up included annual evaluations by movement disorder specialists.
At baseline, the majority of patients (302, or 73.5%) had taken dopamine agonists within the past 12 months.
Over the course of 5 years, the prevalence of impulse control disorders increased from 19.7% at baseline to 32.8%, Dr. Corvol and his colleagues reported.
Among 306 patients with no impulse control disorders at baseline, 94 developed one, for a 5-year cumulative incidence of 46.1%, they added. Only 4 of the 94 new cases occurred in patients who never used dopamine agonists.
Dopamine agonist use in the previous 12 months was associated with a 2.23-fold higher prevalence of impulse control disorders (P less than .001), with prevalence increasing along with average daily dose and cumulative dose duration over that time period, according to the investigators.
These findings suggests tools are needed to screen for impulse control disorders and identify high-risk patients, they said.
“Further studies are needed to understand the mechanisms involved in the relation between [dopamine agonists] and [impulse control disorders], in particular the role of apathy, anxiety, and depression,” they added.
The study was funded by grants from the French Ministry of Health and from the French Drug Agency. Dr. Corvol and many of his colleagues reported financial disclosures with many pharmaceutical companies.
SOURCE: Corvol J-C et al. Neurology. 2018 Jun 20. doi: 10.1212/WNL.0000000000005816.
Nearly half of patients with Parkinson’s disease who were taking dopamine agonist treatment experienced impulse control disorders over a follow-up of 5 years, according to recently published results of a longitudinal study.
The 5-year cumulative incidence of impulse control disorders was approximately 45% in the study, which included 411 patients with a high prevalence of dopamine agonist use and disease duration of 5 years or less at baseline.
There was a strong association between dopamine agonist use and impulse control disorders in the study, which was conducted by Jean-Christophe Corvol, MD, of Publique Hôpitaux de Paris and his coinvestigators.
Impulse disorders increased in incidence with both duration and dose of dopamine agonists and resolved progressively after discontinuation of those agents, the investigators reported online June 20 in Neurology. The investigators used item 1.6 of part I of the Movement Disorder Society Unified Parkinson’s Disease Rating Scale to determine the presence of an impulse control disorder.
“Given the high cumulative incidence of impulse control disorders in patients with Parkinson’s disease, these adverse effects should be carefully monitored in patients ever treated with dopamine agonists,” Dr. Corvol and his coauthors wrote.
The results came from the ongoing Drug Interaction With Genes in Parkinson’s Disease (DIGPD) study, a longitudinal cohort study including Parkinson’s disease patients consecutively recruited between 2009 and 2013 at eight French hospitals. All patients had no more than 5 years of disease duration at recruitment, and follow-up included annual evaluations by movement disorder specialists.
At baseline, the majority of patients (302, or 73.5%) had taken dopamine agonists within the past 12 months.
Over the course of 5 years, the prevalence of impulse control disorders increased from 19.7% at baseline to 32.8%, Dr. Corvol and his colleagues reported.
Among 306 patients with no impulse control disorders at baseline, 94 developed one, for a 5-year cumulative incidence of 46.1%, they added. Only 4 of the 94 new cases occurred in patients who never used dopamine agonists.
Dopamine agonist use in the previous 12 months was associated with a 2.23-fold higher prevalence of impulse control disorders (P less than .001), with prevalence increasing along with average daily dose and cumulative dose duration over that time period, according to the investigators.
These findings suggests tools are needed to screen for impulse control disorders and identify high-risk patients, they said.
“Further studies are needed to understand the mechanisms involved in the relation between [dopamine agonists] and [impulse control disorders], in particular the role of apathy, anxiety, and depression,” they added.
The study was funded by grants from the French Ministry of Health and from the French Drug Agency. Dr. Corvol and many of his colleagues reported financial disclosures with many pharmaceutical companies.
SOURCE: Corvol J-C et al. Neurology. 2018 Jun 20. doi: 10.1212/WNL.0000000000005816.
FROM NEUROLOGY
Key clinical point: Nearly half of Parkinson’s disease patients reported having an impulse control disorder during a 5-year period, the great majority of whom were receiving dopamine agonist treatment.
Major finding: The 5-year cumulative incidence of impulse control disorders was approximately 45%, with increased risk correlating with dose and duration of dopamine agonist treatment.
Study details: Analysis of a multicenter, longitudinal cohort including 5 years of follow-up on 411 consecutive patients with Parkinson’s disease and a disease duration of 5 years or less at baseline.
Disclosures: The study was funded by grants from the French Ministry of Health and from the French Drug Agency. Many of the authors reported financial disclosures with many pharmaceutical companies.
Source: Corvol J-C et al. Neurology. 2018 Jun 20. doi: 10.1212/WNL.0000000000005816.
Neurology Board Review: Epilepsy
Click here to read Neurology Board Review: Epilepsy
Neurology Board Review: Epilepsy is a resource developed by leading clinical educators for studying for board certification and maintenance of certification exams.

After reading the article, Click Here to Access the Board Review Questions
About the Authors
Shavonne L. Massey, MD
Clinical Instructor
Departments of Neurology and Pediatrics
Children’s Hospital of Philadelphia
University of Pennsylvania
Philadelphia, Pennsylvania
Hannah C. Glass, MDCM, MAS
Associate Professor
Departments of Neurology, Pediatrics, and
Epidemiology & Biostatistics
University of California, San Francisco
San Francisco, California
Click here to read Neurology Board Review: Epilepsy
After reading the article, Click Here to Access the Board Review Questions
Click here to read Neurology Board Review: Epilepsy
Neurology Board Review: Epilepsy is a resource developed by leading clinical educators for studying for board certification and maintenance of certification exams.
After reading the article, Click Here to Access the Board Review Questions
About the Authors
Shavonne L. Massey, MD
Clinical Instructor
Departments of Neurology and Pediatrics
Children’s Hospital of Philadelphia
University of Pennsylvania
Philadelphia, Pennsylvania
Hannah C. Glass, MDCM, MAS
Associate Professor
Departments of Neurology, Pediatrics, and
Epidemiology & Biostatistics
University of California, San Francisco
San Francisco, California
Click here to read Neurology Board Review: Epilepsy
After reading the article, Click Here to Access the Board Review Questions
Click here to read Neurology Board Review: Epilepsy
Neurology Board Review: Epilepsy is a resource developed by leading clinical educators for studying for board certification and maintenance of certification exams.
After reading the article, Click Here to Access the Board Review Questions
About the Authors
Shavonne L. Massey, MD
Clinical Instructor
Departments of Neurology and Pediatrics
Children’s Hospital of Philadelphia
University of Pennsylvania
Philadelphia, Pennsylvania
Hannah C. Glass, MDCM, MAS
Associate Professor
Departments of Neurology, Pediatrics, and
Epidemiology & Biostatistics
University of California, San Francisco
San Francisco, California
Click here to read Neurology Board Review: Epilepsy
After reading the article, Click Here to Access the Board Review Questions

Clinical trials to look for at ADA 2018
More than 2,000 abstracts will be presented at the annual scientific sessions of the American Diabetes Association in Orlando, from basic science studies to clinical trials. Maureen A. Gannon, PhD, who chairs the Scientific Sessions Meeting Planning Committee, highlighted several as being the most relevant to clinical practice.
TEDDY at 13

VADT at 15
Final follow-up data from Veterans Administration Diabetes Trial will be presented on Sunday, June 24, at 4:30 p.m. The trial randomized nearly 2,000 military veterans with poor glycemic control to a mean of 5.6 years of intensive glycemic therapy versus standard treatment, with a goal of lowering HbA1c below 8%.
RISE
Restoring Insulin Secretion (RISE) comprises three intervention trials, two in adults and one in adolescents. The trials are studying whether aggressive glucose lowering will lead to recovery of beta-cell function can be sustained after withdrawal of treatment. Initial results from the adolescent trial will be reported on Monday, June 25, at 2:15 p.m.
SGLT inhibition in type 1 diabetes
Presenters in this session, on Tuesday, June 26 at 10:15 a.m., will provide trial results an insights on a regulatory pathway for sodium-glucose cotransporter (SGLT)-1 and -2 inhibitors in type 1 diabetes patients. Julio Rosenstock, MD, who will present the latest data on empagliflozin from the EASE (Empagliflozin as Adjunctive to InSulin thErapy) trial program, said, “This symposium brings together the lead investigators from the three major competitors that are pursuing approval of a SGLT inhibitor for type 1 diabetes. They will report top-level data that will eventually be submitted to regulators.”
DIY technology
This symposium on Saturday at 1:45 pm, The Diabetes Do-It-Yourself Revolution, will explore the evolving, DIY revolution in diabetes, in which patients are upending traditional treatment pathways and closing their own insulin delivery loop.
“I’m excited about the variety we have in the program this year,” said Dr. Gannon, professor of medicine in the division of diabetes, endocrinology and metabolism; molecular physiology and biophysics; and cell and developmental biology at Vanderbilt University, Nashville, Tenn. “This is the place for cutting-edge information for anybody who is involved in diabetes research or patient care.”
More than 2,000 abstracts will be presented at the annual scientific sessions of the American Diabetes Association in Orlando, from basic science studies to clinical trials. Maureen A. Gannon, PhD, who chairs the Scientific Sessions Meeting Planning Committee, highlighted several as being the most relevant to clinical practice.
TEDDY at 13
VADT at 15
Final follow-up data from Veterans Administration Diabetes Trial will be presented on Sunday, June 24, at 4:30 p.m. The trial randomized nearly 2,000 military veterans with poor glycemic control to a mean of 5.6 years of intensive glycemic therapy versus standard treatment, with a goal of lowering HbA1c below 8%.
RISE
Restoring Insulin Secretion (RISE) comprises three intervention trials, two in adults and one in adolescents. The trials are studying whether aggressive glucose lowering will lead to recovery of beta-cell function can be sustained after withdrawal of treatment. Initial results from the adolescent trial will be reported on Monday, June 25, at 2:15 p.m.
SGLT inhibition in type 1 diabetes
Presenters in this session, on Tuesday, June 26 at 10:15 a.m., will provide trial results an insights on a regulatory pathway for sodium-glucose cotransporter (SGLT)-1 and -2 inhibitors in type 1 diabetes patients. Julio Rosenstock, MD, who will present the latest data on empagliflozin from the EASE (Empagliflozin as Adjunctive to InSulin thErapy) trial program, said, “This symposium brings together the lead investigators from the three major competitors that are pursuing approval of a SGLT inhibitor for type 1 diabetes. They will report top-level data that will eventually be submitted to regulators.”
DIY technology
This symposium on Saturday at 1:45 pm, The Diabetes Do-It-Yourself Revolution, will explore the evolving, DIY revolution in diabetes, in which patients are upending traditional treatment pathways and closing their own insulin delivery loop.
“I’m excited about the variety we have in the program this year,” said Dr. Gannon, professor of medicine in the division of diabetes, endocrinology and metabolism; molecular physiology and biophysics; and cell and developmental biology at Vanderbilt University, Nashville, Tenn. “This is the place for cutting-edge information for anybody who is involved in diabetes research or patient care.”
More than 2,000 abstracts will be presented at the annual scientific sessions of the American Diabetes Association in Orlando, from basic science studies to clinical trials. Maureen A. Gannon, PhD, who chairs the Scientific Sessions Meeting Planning Committee, highlighted several as being the most relevant to clinical practice.
TEDDY at 13
VADT at 15
Final follow-up data from Veterans Administration Diabetes Trial will be presented on Sunday, June 24, at 4:30 p.m. The trial randomized nearly 2,000 military veterans with poor glycemic control to a mean of 5.6 years of intensive glycemic therapy versus standard treatment, with a goal of lowering HbA1c below 8%.
RISE
Restoring Insulin Secretion (RISE) comprises three intervention trials, two in adults and one in adolescents. The trials are studying whether aggressive glucose lowering will lead to recovery of beta-cell function can be sustained after withdrawal of treatment. Initial results from the adolescent trial will be reported on Monday, June 25, at 2:15 p.m.
SGLT inhibition in type 1 diabetes
Presenters in this session, on Tuesday, June 26 at 10:15 a.m., will provide trial results an insights on a regulatory pathway for sodium-glucose cotransporter (SGLT)-1 and -2 inhibitors in type 1 diabetes patients. Julio Rosenstock, MD, who will present the latest data on empagliflozin from the EASE (Empagliflozin as Adjunctive to InSulin thErapy) trial program, said, “This symposium brings together the lead investigators from the three major competitors that are pursuing approval of a SGLT inhibitor for type 1 diabetes. They will report top-level data that will eventually be submitted to regulators.”
DIY technology
This symposium on Saturday at 1:45 pm, The Diabetes Do-It-Yourself Revolution, will explore the evolving, DIY revolution in diabetes, in which patients are upending traditional treatment pathways and closing their own insulin delivery loop.
“I’m excited about the variety we have in the program this year,” said Dr. Gannon, professor of medicine in the division of diabetes, endocrinology and metabolism; molecular physiology and biophysics; and cell and developmental biology at Vanderbilt University, Nashville, Tenn. “This is the place for cutting-edge information for anybody who is involved in diabetes research or patient care.”
Free Composite Serratus Anterior-Latissimus-Rib Flaps for Acute One-Stage Reconstruction of Gustilo IIIB Tibia Fractures
ABSTRACT
Gustilo IIIB injuries of the tibia with segmental bone loss continue to be a difficult reconstructive problem. The serratus anterior-latissimus-rib (SALR) composite flap consists of bone and muscle; this flap can provide soft tissue coverage and vascularized bone in a single surgical procedure. The purpose of this study is to describe the use of the SALR flap for the treatment of a large open tibia fracture with segmental bone loss, with a specific focus on postoperative complications, limb salvage, and time to union.
We reviewed the medical records of patients undergoing an SALR flap (n = 5) for the treatment of Gustilo Type IIIB tibia fractures within 1 month of injury. We compared the mechanism of injury, injury severity score, time from injury to free tissue transfer, complications, and time to radiographic and clinical union.
All patients were male, with a mean age of 25 years. On average, patients underwent free tissue transfer within 1 week of injury. The average time to radiographic union was 7 months. Two patients underwent reoperation. There were no graft failures.
Free SALR flaps can be a useful option for the treatment of high-energy tibia fractures with extensive soft tissue and bone loss. These flaps provide immediate osseous and soft tissue reconstruction with an acceptable complication profile.
Reconstruction of the lower extremity following Gustilo’s grade IIIB injuries is difficult due to loss of both combined soft tissue and segmental bone loss. Since these injuries necessitate the need for soft tissue flap coverage along with vascularized bone grafting, free fibula flaps have classically been used for reconstruction.1-3 In the setting of bilateral lower extremity injury, the contralateral fibula is often not appropriate to harvest and transfer; therefore, other sources of vascularized bone grafts must be utilized including vascularized iliac crest and rib.1-5 The vascularized iliac crest graft is insufficient to provide the bony reconstruction of bone defects >6 cm to 7 cm and does not have a reliable skin paddle.4 In contrast, free composite serratus anterior-latissimus-rib (SALR) flaps can provide both long segments of vascularized bone and abundant soft tissue coverage for large segmental defects.1-5
Continue to: Free fibula grafts have been considered...
Free fibula grafts have been considered the gold standard for the reconstruction of large (>6 cm) bone defects.6 In cases of “mangled extremities,” bone defects are associated with large soft tissue defects, which require either single-stage surgery consisting of 2 separate free flaps (ie, free fibula and free latissimus) or a 2-stage procedure where the soft tissue reconstruction precedes the bone reconstruction.2,7-9 Unlike free fibula and latissimus flaps, composite SALR flaps provide both osseous reconstruction and soft tissue in 1 flap supplied by a single vascular pedicle; unfortunately, outcomes using this flap for large Gustilo IIIB injuries are limited.1-5 The purpose of this study is to examine the use of free composite SALR flaps for soft tissue coverage in cases of Gustilo IIIB injuries with large soft tissue and bony deficits. This study specifically examines time to union, need for reoperation, and graft failure following the use of these flaps.
MATERIALS AND METHODS
Following approval from our Institutional Review Board, we retrospectively reviewed the medical records of patients undergoing a free composite SALR flap (n = 5) for the treatment of a severe open tibia fracture within 1 month of injury. All patients sustained open injuries classified as IIIB on the Gustilo-Anderson scale.10 Medical records were examined for the mechanism of injury (MOI), injury severity score (ISS), time from injury to free tissue transfer, medical comorbidities, surgical complications, and time to radiographic and clinical union. Radiographic union was determined by the presence of bridging bone on 3 of 4 of cortices on plain film radiographs.
All patients were male (n = 5), with a mean age of 25 years (range, 19-30 years) at the time of injury (Table).
Table. Demographics and Outcomes of Patients Undergoing Free Tissue Transfer
| Free Serratus Anterior-Latissimus-Rib Flaps |
Age (Mean ± SEM) | 23 ± 2 years |
Males | 5 |
Females | 0 |
Tobacco Use | 2 |
Body Mass Index (Mean ± SEM) | 26.2 ± 0.9 kg/m2 |
Injury Severity Score (Mean ± SEM) | 18 ± 5 |
Time to Tissue Transfer (Mean ± SEM) | 1 ± 0.3 weeks |
Time to Boney Union (Mean ± SEM) | 7 ± 0.7 months |
Time Non-Weight-Bearing (Mean ± SEM) | 5 ± 0.5 months |
The MOI included motorcycle collisions (n = 2), pedestrian struck by car (n = 1), motor vehicle collisions (n = 1), and direct blow to the leg (n = 1). The mean ISS of the cohort was 18 (range, 10-34) (Table). On average, patients underwent free tissue transfer within 1 week (range, 3 days to 2 weeks) from the time of injury. Patients in this cohort were followed clinically for a mean of 4 years (range, 1-6 years) after surgery. Patients were non-weight-bearing for an average of 5 months (range, 4-6 months) following their reconstructions.
RESULTS
All flaps survived. The mean time to radiographic and clinical union was 7 months (range, 6-9 months). Two patients underwent reoperation. One patient underwent a bone grafting procedure for a delayed union at 6 months postoperative, and 1 patient underwent irrigation and débridement of superficial soft tissue infection. Donor site complications occurred in 2 patients, including chronic rib pain (n = 1) and a pleural effusion requiring drainage (n = 1). At the last follow-up, all ribs had incorporated, and all patients were weight-bearing as tolerated on the limb.
CASE EXAMPLE
A 22-year-old male smoker was transferred to our facility after a motor vehicle accident with bilateral tibia fractures, 1 closed and 1 open with significant bone loss (Figures 1A, 1B). 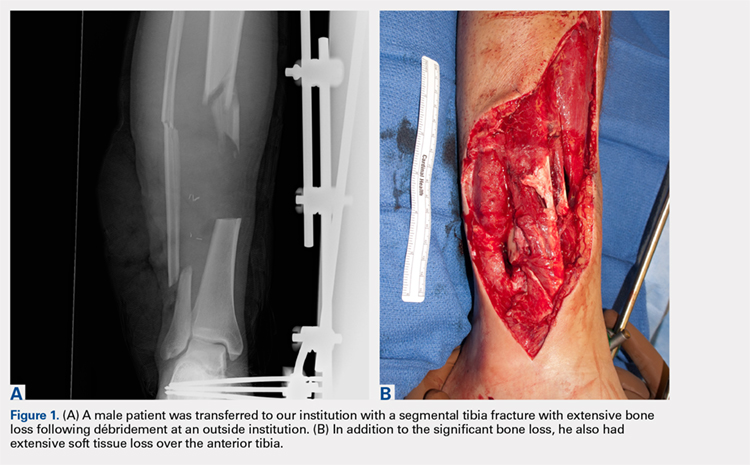
Continue to: Surgical Technique...
SURGICAL TECHNIQUE
The patient is placed in the lateral decubitus position during the procedure. A 2-team approach is used for dissection of the flap and preparation of recipient vessels to decrease operative time. A J-shaped incision is started on the chest at the mid-axillary line and extended just over the fifth and sixth rib. The incision can be made to fall into the intermammary crease in a woman to hide the scar. The dissection begins by exposing the anterior border of the latissimus muscle (Figure 2A). Next, the latissimus is dissected to reveal the thoracodorsal vessels (Figure 2B). At this level, the thoracodorsal vessel can be traced into the axilla. The branch going into the fifth, sixth, and lower slips of the serratus are dissected. The long thoracic nerve and the thoracodorsal nerve are preserved during the dissection (Figure 2C). The fifth, sixth, and seventh slips of the serratus are preferentially included in the dissection while leaving the most superior slips of the serratus to preserve scapular stability. Dissection begins by identifying 2 adjacent rib sections of the fifth and sixth or sixth and seventh ribs. The defect in the lower extremity determines the length of rib harvested. The serratus slips are then divided anteriorly over the chest wall. The dissection is extended to the intercostal spaces of the fourth and fifth ribs. The supraperiosteal dissection is performed at the anterior margin of the rib (Figure 2D). 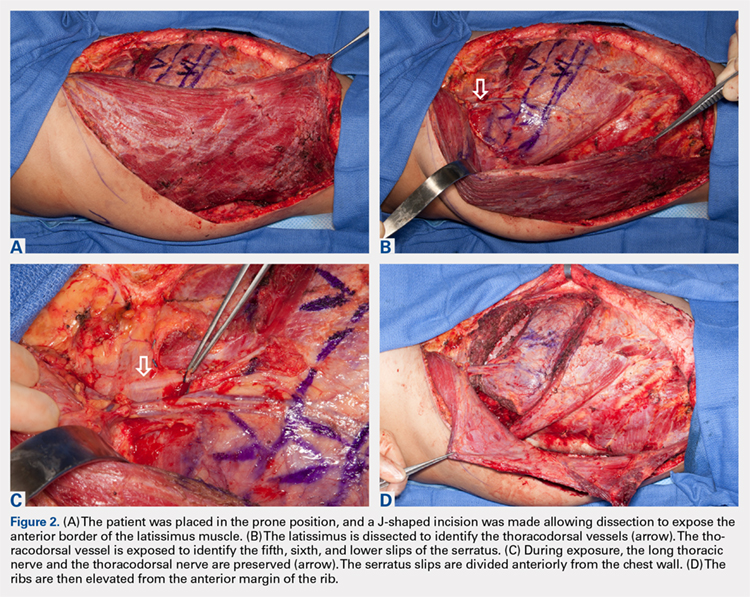

Continue to: Following the surgical procedure...
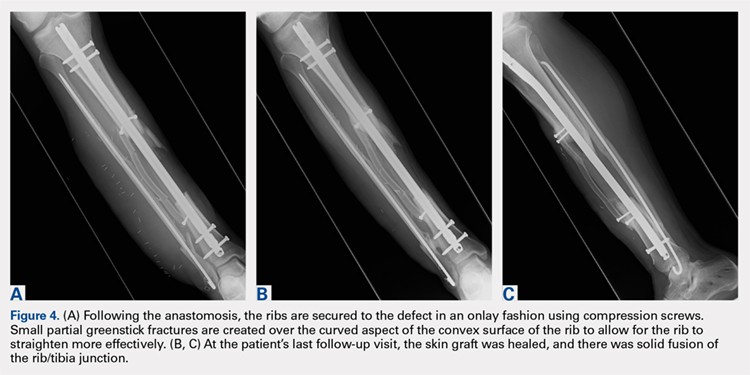
Following the surgical procedure, patients are made non-weight-bearing on the operative extremity until signs of healing are apparent on radiographs. In this case, at the patients’ last follow-up visit, the skin graft was healed, and there was solid fusion of the rib/tibia junction (Figures 4B, 4C).
DISCUSSION
High-energy open injuries to the lower extremities are devastating injuries, with a high rate of late amputation and poor functional outcomes.11-13 Vascularized bone grafting provides both essential osteoinductive and osteoconductive properties to segmental bone defects in areas with inadequate soft tissue coverage, particularly in the setting of >6 cm of bone loss.4,14 The results of this study show that acute reconstruction of the lower limb with a composite vascularized SALR graft is a reliable procedure with an acceptable complication profile.
The timing of soft tissue coverage should be performed as soon as the patient is medically stable enough to undergo a reconstructive procedure, ideally within 7 to 10 days; and this timetable has been shown to decrease rates of infection and free flap failure.15-19 Early coverage provides both control of the soft-tissue envelope and reduces the risk of losing bone.1 Unlike the timing of coverage, the staging of the procedure is controversial. Proponents of the 2-stage free tissue (soft tissue followed by bony flap) transfer feel that although the tissue may not be infected at the time of coverage, it is contaminated with bacteria at the time of bone reconstruction, and as such is at high risk for both infection and complications.20 Unlike 2-stage procedures, single-stage coverage provides immediate soft tissue coverage, as well as bony support. This reduces the time to bony union and negates the need for repeated surgery in a mangled extremity where secondary surgery is complicated by both scar tissue and altered anatomy.1,2 Furthermore, it has been shown that there is no difference in the rates of infection when performing a single-stage compared with a 2-stage procedure.9 In this study, SALR flaps were typically performed within 2 weeks following an injury as a single procedure. We feel this resulted in the low number of complications in the SALR group.
Unlike free fibulas, rib flaps are easily pedicled with an associated soft-tissue flap due to their blood supply, making them ideal for 1-stage reconstruction. The rib has a dual blood supply: 1 from the posterior intercostal artery, and the other, an abundant periosteal blood supply, from the serratus anterior muscle.4 The blood supply to the serratus anterior comes from the thoracodorsal artery, and usually provides 14 cm of a large-caliber pedicle, making it a reliable flap for soft tissue reconstruction.21,22 Another unique feature of the blood supply to this flap is the amount of soft tissue available for both harvest and transfer; larger portions of serratus muscle and latissimus muscle can be harvested if necessary to cover the soft tissue defect.4
Comminuted tibias with segmental bone loss are difficult to manage since they are associated with bony as well as soft tissue defects.1,12,13,23 These injuries are ideal candidates for a single-stage reconstruction using a vascularized SALR flap. In our series, the use of an SALR flap resulted ultimately in a 100% union and limb salvage rate, with no flap failures and a low complication profile. Unlike the SALR, free fibular flaps must be transferred along with a separate latissimus dorsi flap to provide enough soft tissue coverage necessary for reconstructing large Gustilo IIIB injuries, which could increase the risk of flap failure. Since ribs are composed of membranous bone and have a similar cross-sectional area to both metacarpal and metatarsals, there are concerns regarding the biomechanical properties of ribs for weight-bearing.4,22,24-26 To compensate for this relatively small cross-sectional area, 2 ribs (either consecutive or alternative) are frequently harvested.1,4,5,23 Previous studies examining the use of ribs for bony reconstruction have frequently supplemented the rib reconstruction to the tibia using screws and external fixation alone.1,4,5,23 In our series, all SALR grafts were supported with the use of an intramedullary nail (n = 3) or locked plating (n = 1). The use of this supplemental fixation of the SALR graft allowed our patients to return to full weight-bearing (mean, 6 months) much earlier than the length of time cited in previous reports (12 months) examining these injuries.1,4,5,23
Continue to: There are several limitations...
There are several limitations to this study. The small sample size and retrospective nature of the study limits the amount of data we are able to collect from the medical record and places obvious constraints on the analysis. Although all these procedures were performed at 1 institution, multiple providers were involved in the reconstruction of these injuries, and there is no standard protocol for their treatment. Similarly, although other forms of extremity reconstruction were used during this time period, there was no standard protocol that could serve as a comparator for patients who underwent an SALR compared with other reconstructive procedures.
Overall, SALR grafts provide an excellent option for 1-stage reconstruction of severe, open lower extremity injuries. In this series we noted a 100% graft success rate with an acceptable complication profile.
This paper will be judged for the Resident Writer’s Award.
1. Yazar S, Lin CH, Wei FC. One-stage reconstruction of composite bone and soft-tissue defects in traumatic lower extremities. Plast Reconstr Surg. 2004;114(6):1457-1466. doi:10.1097/01.PRS.0000138811.88807.65.
2. Lin CH, Wei FC, Chen HC, Chuang DC. Outcome comparison in traumatic lower-extremity reconstruction by using various composite vascularized bone transplantation. Plast Reconstr Surg. 1999;104(4):984-992. doi:10.1097/00006534-199909040-00013.
3. Tu YK, Yen CY, Yeh WL, Wang IC, Wang KC, Ueng SW. Reconstruction of posttraumatic long bone defect with free vascularized bone graft: good outcome in 48 patients with 6 years' follow-up. Acta Orthopaedica Scandinavica. 2001;72(4):359-364. doi:10.1080/000164701753542014.
4. Lin CH, Wei FC, Levin LS, Su JI, Fan KF, Yeh WL, Hsu DT. Free composite serratus anterior and rib flaps for tibial composite bone and soft-tissue defect. Plast Reconstr Surg. 1997;99(6):1656-1665. Doi:10.1097/00006534-199705000-00028.
5. Georgescu AV, Ignatiadis I, Ileana M, Irina C, Filip A, Olariu R. Long-term results after muscle-rib flap transfer for reconstruction of composite limb defects. Microsurgery. 2011;31(3):218-222. doi:10.1002/micr.20857.
6. Moran CG, Wood MB. Vascularized bone autografts. Orthop Rev. 1993;22(2):187-197. doi:10.1097/01241398-199307000-00031.
7. Banic A, Hertel R. Double vascularized fibulas for reconstruction of large tibial defects. J Reconstr Microsurg. 1993;9(6):421-428. doi:10.1055/s-2007-1006751.
8. Malizos KN, Nunley JA, Goldner RD, Urbaniak JR, Harrelson JM. Free vascularized fibula in traumatic long bone defects and in limb salvaging following tumor resection: comparative study. Microsurgery. 1993;14(6):368-374. doi:10.1002/micr.1920140603.
9. Peat BG, Liggins DF. Microvascular soft tissue reconstruction for acute tibial fractures--late complications and the role of bone grafting. Ann Plast Surg. 1990;24(6):517-520.
10. Gustilo RB, Anderson JT. Prevention of infection in the treatment of one thousand and twenty-five open fractures of long bones: retrospective and prospective analyses. J Bone Joint Surg Am. 1976;58(4):453-458.
11. Gustilo RB, Mendoza RM, Williams DN. Problems in the management of type III (severe) open fractures: a new classification of type III open fractures. J Trauma. 1984;24(8):742-746. doi:10.1097/00005373-198408000-00009.
12. Bosse MJ, MacKenzie EJ, Kellam JF, et al. An analysis of outcomes of reconstruction or amputation after leg-threatening injuries. NEJM. 2002;347(24):1924-1931. doi:10.1056/NEJMoa012604.
13. MacKenzie EJ, Bosse MJ, Pollak AN, et al. Long-term persistence of disability following severe lower-limb trauma. Results of a seven-year follow-up. J Bone Joint Surg Am. 2005;87(8):1801-1809. doi:10.2106/JBJS.E.00032.
14. Bieber EJ, Wood MB. Bone reconstruction. Clin Plast Surg. 1986;13(4):645-655.
15. Melvin JS, Dombroski DG, Torbert JT, Kovach SJ, Esterhai JL, Mehta S. Open tibial shaft fractures: II. Definitive management and limb salvage. J Am Acad Orthop Surg. 2010;18(2):108-117. doi:10.5435/00124635-201002000-00005.
16. Godina M. Early microsurgical reconstruction of complex trauma of the extremities. Plast Reconstr Surg. 1986;78(3):285-292. doi:10.1055/s-2006-944324.
17. Gopal S, Majumder S, Batchelor AG, Knight SL, De Boer P, Smith RM. Fix and flap: the radical orthopaedic and plastic treatment of severe open fractures of the tibia. J Bone Joint Surg Br. 2000;82(7):959-966. doi:10.1302/0301-620X.82B7.0820959.
18. Fischer MD, Gustilo RB, Varecka TF. The timing of flap coverage, bone-grafting, and intramedullary nailing in patients who have a fracture of the tibial shaft with extensive soft-tissue injury. J Bone Joint Surg Am. 1991;73(9):1316-1322. doi:10.2106/00004623-199173090-00005.
19. Tielinen L, Lindahl JE, Tukiainen EJ. Acute unreamed intramedullary nailing and soft tissue reconstruction with muscle flaps for the treatment of severe open tibial shaft fractures. Injury. 2007;38(8):906-912. doi:10.1016/j.injury.2007.02.052.
20. Yaremchuk MJ, Brumback RJ, Manson PN, Burgess AR, Poka A, Weiland AJ. Acute and definitive management of traumatic osteocutaneous defects of the lower extremity. Plast Reconstr Surg. 1987;80(1):1-14. doi:10.1097/00006534-198707000-00002.
21. Ueng WN, Chuang CC, Shih CH. Double-rib composite free transfer to reconstruct a single-spared lower extremity defect. J Trauma. 1995;38(2):210-212.
22. Bruck JC, Bier J, Kistler D. The serratus anterior osteocutaneous free flap. J Reconstr Microsurg. 1990;6(3):209-213. doi:10.1055/s-2007-1006820.
23. Lin CH, Yazar S. Revisiting the serratus anterior rib flap for composite tibial defects. Plast Reconstr Surg. 2004;114(7):1871-1877. doi:10.1097/01.PRS.0000142767.13493.63.
24. Hui KC, Zhang F, Lineaweaver WC, Moon W, Buncke GM, Buncke HJ. Serratus anterior-rib composite flap: anatomic studies and clinical application to hand reconstruction. Ann Plast Surg. 1999;42(2):132-136. doi:10.1097/00000637-199902000-00004.
25. Buncke HJ, Furnas DW, Gordon L, Achauer BM. Free osteocutaneous flap from a rib to the tibia. Plast Reconstr Surg. 1977;59(6):799-804. doi:10.1097/00006534-197706000-00002.
26. Nusbickel FR, Dell PC, Mcandrew MP, Moore MM. Vascularized autografts for reconstruction of skeletal defects following lower extremity trauma. A review. Clin Orthop Relat Res. 1989;(243):65-70.
ABSTRACT
Gustilo IIIB injuries of the tibia with segmental bone loss continue to be a difficult reconstructive problem. The serratus anterior-latissimus-rib (SALR) composite flap consists of bone and muscle; this flap can provide soft tissue coverage and vascularized bone in a single surgical procedure. The purpose of this study is to describe the use of the SALR flap for the treatment of a large open tibia fracture with segmental bone loss, with a specific focus on postoperative complications, limb salvage, and time to union.
We reviewed the medical records of patients undergoing an SALR flap (n = 5) for the treatment of Gustilo Type IIIB tibia fractures within 1 month of injury. We compared the mechanism of injury, injury severity score, time from injury to free tissue transfer, complications, and time to radiographic and clinical union.
All patients were male, with a mean age of 25 years. On average, patients underwent free tissue transfer within 1 week of injury. The average time to radiographic union was 7 months. Two patients underwent reoperation. There were no graft failures.
Free SALR flaps can be a useful option for the treatment of high-energy tibia fractures with extensive soft tissue and bone loss. These flaps provide immediate osseous and soft tissue reconstruction with an acceptable complication profile.
Reconstruction of the lower extremity following Gustilo’s grade IIIB injuries is difficult due to loss of both combined soft tissue and segmental bone loss. Since these injuries necessitate the need for soft tissue flap coverage along with vascularized bone grafting, free fibula flaps have classically been used for reconstruction.1-3 In the setting of bilateral lower extremity injury, the contralateral fibula is often not appropriate to harvest and transfer; therefore, other sources of vascularized bone grafts must be utilized including vascularized iliac crest and rib.1-5 The vascularized iliac crest graft is insufficient to provide the bony reconstruction of bone defects >6 cm to 7 cm and does not have a reliable skin paddle.4 In contrast, free composite serratus anterior-latissimus-rib (SALR) flaps can provide both long segments of vascularized bone and abundant soft tissue coverage for large segmental defects.1-5
Continue to: Free fibula grafts have been considered...
Free fibula grafts have been considered the gold standard for the reconstruction of large (>6 cm) bone defects.6 In cases of “mangled extremities,” bone defects are associated with large soft tissue defects, which require either single-stage surgery consisting of 2 separate free flaps (ie, free fibula and free latissimus) or a 2-stage procedure where the soft tissue reconstruction precedes the bone reconstruction.2,7-9 Unlike free fibula and latissimus flaps, composite SALR flaps provide both osseous reconstruction and soft tissue in 1 flap supplied by a single vascular pedicle; unfortunately, outcomes using this flap for large Gustilo IIIB injuries are limited.1-5 The purpose of this study is to examine the use of free composite SALR flaps for soft tissue coverage in cases of Gustilo IIIB injuries with large soft tissue and bony deficits. This study specifically examines time to union, need for reoperation, and graft failure following the use of these flaps.
MATERIALS AND METHODS
Following approval from our Institutional Review Board, we retrospectively reviewed the medical records of patients undergoing a free composite SALR flap (n = 5) for the treatment of a severe open tibia fracture within 1 month of injury. All patients sustained open injuries classified as IIIB on the Gustilo-Anderson scale.10 Medical records were examined for the mechanism of injury (MOI), injury severity score (ISS), time from injury to free tissue transfer, medical comorbidities, surgical complications, and time to radiographic and clinical union. Radiographic union was determined by the presence of bridging bone on 3 of 4 of cortices on plain film radiographs.
All patients were male (n = 5), with a mean age of 25 years (range, 19-30 years) at the time of injury (Table).
Table. Demographics and Outcomes of Patients Undergoing Free Tissue Transfer
| Free Serratus Anterior-Latissimus-Rib Flaps |
Age (Mean ± SEM) | 23 ± 2 years |
Males | 5 |
Females | 0 |
Tobacco Use | 2 |
Body Mass Index (Mean ± SEM) | 26.2 ± 0.9 kg/m2 |
Injury Severity Score (Mean ± SEM) | 18 ± 5 |
Time to Tissue Transfer (Mean ± SEM) | 1 ± 0.3 weeks |
Time to Boney Union (Mean ± SEM) | 7 ± 0.7 months |
Time Non-Weight-Bearing (Mean ± SEM) | 5 ± 0.5 months |
The MOI included motorcycle collisions (n = 2), pedestrian struck by car (n = 1), motor vehicle collisions (n = 1), and direct blow to the leg (n = 1). The mean ISS of the cohort was 18 (range, 10-34) (Table). On average, patients underwent free tissue transfer within 1 week (range, 3 days to 2 weeks) from the time of injury. Patients in this cohort were followed clinically for a mean of 4 years (range, 1-6 years) after surgery. Patients were non-weight-bearing for an average of 5 months (range, 4-6 months) following their reconstructions.
RESULTS
All flaps survived. The mean time to radiographic and clinical union was 7 months (range, 6-9 months). Two patients underwent reoperation. One patient underwent a bone grafting procedure for a delayed union at 6 months postoperative, and 1 patient underwent irrigation and débridement of superficial soft tissue infection. Donor site complications occurred in 2 patients, including chronic rib pain (n = 1) and a pleural effusion requiring drainage (n = 1). At the last follow-up, all ribs had incorporated, and all patients were weight-bearing as tolerated on the limb.
CASE EXAMPLE
A 22-year-old male smoker was transferred to our facility after a motor vehicle accident with bilateral tibia fractures, 1 closed and 1 open with significant bone loss (Figures 1A, 1B).
Continue to: Surgical Technique...
SURGICAL TECHNIQUE
The patient is placed in the lateral decubitus position during the procedure. A 2-team approach is used for dissection of the flap and preparation of recipient vessels to decrease operative time. A J-shaped incision is started on the chest at the mid-axillary line and extended just over the fifth and sixth rib. The incision can be made to fall into the intermammary crease in a woman to hide the scar. The dissection begins by exposing the anterior border of the latissimus muscle (Figure 2A). Next, the latissimus is dissected to reveal the thoracodorsal vessels (Figure 2B). At this level, the thoracodorsal vessel can be traced into the axilla. The branch going into the fifth, sixth, and lower slips of the serratus are dissected. The long thoracic nerve and the thoracodorsal nerve are preserved during the dissection (Figure 2C). The fifth, sixth, and seventh slips of the serratus are preferentially included in the dissection while leaving the most superior slips of the serratus to preserve scapular stability. Dissection begins by identifying 2 adjacent rib sections of the fifth and sixth or sixth and seventh ribs. The defect in the lower extremity determines the length of rib harvested. The serratus slips are then divided anteriorly over the chest wall. The dissection is extended to the intercostal spaces of the fourth and fifth ribs. The supraperiosteal dissection is performed at the anterior margin of the rib (Figure 2D).
Continue to: Following the surgical procedure...
Following the surgical procedure, patients are made non-weight-bearing on the operative extremity until signs of healing are apparent on radiographs. In this case, at the patients’ last follow-up visit, the skin graft was healed, and there was solid fusion of the rib/tibia junction (Figures 4B, 4C).
DISCUSSION
High-energy open injuries to the lower extremities are devastating injuries, with a high rate of late amputation and poor functional outcomes.11-13 Vascularized bone grafting provides both essential osteoinductive and osteoconductive properties to segmental bone defects in areas with inadequate soft tissue coverage, particularly in the setting of >6 cm of bone loss.4,14 The results of this study show that acute reconstruction of the lower limb with a composite vascularized SALR graft is a reliable procedure with an acceptable complication profile.
The timing of soft tissue coverage should be performed as soon as the patient is medically stable enough to undergo a reconstructive procedure, ideally within 7 to 10 days; and this timetable has been shown to decrease rates of infection and free flap failure.15-19 Early coverage provides both control of the soft-tissue envelope and reduces the risk of losing bone.1 Unlike the timing of coverage, the staging of the procedure is controversial. Proponents of the 2-stage free tissue (soft tissue followed by bony flap) transfer feel that although the tissue may not be infected at the time of coverage, it is contaminated with bacteria at the time of bone reconstruction, and as such is at high risk for both infection and complications.20 Unlike 2-stage procedures, single-stage coverage provides immediate soft tissue coverage, as well as bony support. This reduces the time to bony union and negates the need for repeated surgery in a mangled extremity where secondary surgery is complicated by both scar tissue and altered anatomy.1,2 Furthermore, it has been shown that there is no difference in the rates of infection when performing a single-stage compared with a 2-stage procedure.9 In this study, SALR flaps were typically performed within 2 weeks following an injury as a single procedure. We feel this resulted in the low number of complications in the SALR group.
Unlike free fibulas, rib flaps are easily pedicled with an associated soft-tissue flap due to their blood supply, making them ideal for 1-stage reconstruction. The rib has a dual blood supply: 1 from the posterior intercostal artery, and the other, an abundant periosteal blood supply, from the serratus anterior muscle.4 The blood supply to the serratus anterior comes from the thoracodorsal artery, and usually provides 14 cm of a large-caliber pedicle, making it a reliable flap for soft tissue reconstruction.21,22 Another unique feature of the blood supply to this flap is the amount of soft tissue available for both harvest and transfer; larger portions of serratus muscle and latissimus muscle can be harvested if necessary to cover the soft tissue defect.4
Comminuted tibias with segmental bone loss are difficult to manage since they are associated with bony as well as soft tissue defects.1,12,13,23 These injuries are ideal candidates for a single-stage reconstruction using a vascularized SALR flap. In our series, the use of an SALR flap resulted ultimately in a 100% union and limb salvage rate, with no flap failures and a low complication profile. Unlike the SALR, free fibular flaps must be transferred along with a separate latissimus dorsi flap to provide enough soft tissue coverage necessary for reconstructing large Gustilo IIIB injuries, which could increase the risk of flap failure. Since ribs are composed of membranous bone and have a similar cross-sectional area to both metacarpal and metatarsals, there are concerns regarding the biomechanical properties of ribs for weight-bearing.4,22,24-26 To compensate for this relatively small cross-sectional area, 2 ribs (either consecutive or alternative) are frequently harvested.1,4,5,23 Previous studies examining the use of ribs for bony reconstruction have frequently supplemented the rib reconstruction to the tibia using screws and external fixation alone.1,4,5,23 In our series, all SALR grafts were supported with the use of an intramedullary nail (n = 3) or locked plating (n = 1). The use of this supplemental fixation of the SALR graft allowed our patients to return to full weight-bearing (mean, 6 months) much earlier than the length of time cited in previous reports (12 months) examining these injuries.1,4,5,23
Continue to: There are several limitations...
There are several limitations to this study. The small sample size and retrospective nature of the study limits the amount of data we are able to collect from the medical record and places obvious constraints on the analysis. Although all these procedures were performed at 1 institution, multiple providers were involved in the reconstruction of these injuries, and there is no standard protocol for their treatment. Similarly, although other forms of extremity reconstruction were used during this time period, there was no standard protocol that could serve as a comparator for patients who underwent an SALR compared with other reconstructive procedures.
Overall, SALR grafts provide an excellent option for 1-stage reconstruction of severe, open lower extremity injuries. In this series we noted a 100% graft success rate with an acceptable complication profile.
This paper will be judged for the Resident Writer’s Award.
ABSTRACT
Gustilo IIIB injuries of the tibia with segmental bone loss continue to be a difficult reconstructive problem. The serratus anterior-latissimus-rib (SALR) composite flap consists of bone and muscle; this flap can provide soft tissue coverage and vascularized bone in a single surgical procedure. The purpose of this study is to describe the use of the SALR flap for the treatment of a large open tibia fracture with segmental bone loss, with a specific focus on postoperative complications, limb salvage, and time to union.
We reviewed the medical records of patients undergoing an SALR flap (n = 5) for the treatment of Gustilo Type IIIB tibia fractures within 1 month of injury. We compared the mechanism of injury, injury severity score, time from injury to free tissue transfer, complications, and time to radiographic and clinical union.
All patients were male, with a mean age of 25 years. On average, patients underwent free tissue transfer within 1 week of injury. The average time to radiographic union was 7 months. Two patients underwent reoperation. There were no graft failures.
Free SALR flaps can be a useful option for the treatment of high-energy tibia fractures with extensive soft tissue and bone loss. These flaps provide immediate osseous and soft tissue reconstruction with an acceptable complication profile.
Reconstruction of the lower extremity following Gustilo’s grade IIIB injuries is difficult due to loss of both combined soft tissue and segmental bone loss. Since these injuries necessitate the need for soft tissue flap coverage along with vascularized bone grafting, free fibula flaps have classically been used for reconstruction.1-3 In the setting of bilateral lower extremity injury, the contralateral fibula is often not appropriate to harvest and transfer; therefore, other sources of vascularized bone grafts must be utilized including vascularized iliac crest and rib.1-5 The vascularized iliac crest graft is insufficient to provide the bony reconstruction of bone defects >6 cm to 7 cm and does not have a reliable skin paddle.4 In contrast, free composite serratus anterior-latissimus-rib (SALR) flaps can provide both long segments of vascularized bone and abundant soft tissue coverage for large segmental defects.1-5
Continue to: Free fibula grafts have been considered...
Free fibula grafts have been considered the gold standard for the reconstruction of large (>6 cm) bone defects.6 In cases of “mangled extremities,” bone defects are associated with large soft tissue defects, which require either single-stage surgery consisting of 2 separate free flaps (ie, free fibula and free latissimus) or a 2-stage procedure where the soft tissue reconstruction precedes the bone reconstruction.2,7-9 Unlike free fibula and latissimus flaps, composite SALR flaps provide both osseous reconstruction and soft tissue in 1 flap supplied by a single vascular pedicle; unfortunately, outcomes using this flap for large Gustilo IIIB injuries are limited.1-5 The purpose of this study is to examine the use of free composite SALR flaps for soft tissue coverage in cases of Gustilo IIIB injuries with large soft tissue and bony deficits. This study specifically examines time to union, need for reoperation, and graft failure following the use of these flaps.
MATERIALS AND METHODS
Following approval from our Institutional Review Board, we retrospectively reviewed the medical records of patients undergoing a free composite SALR flap (n = 5) for the treatment of a severe open tibia fracture within 1 month of injury. All patients sustained open injuries classified as IIIB on the Gustilo-Anderson scale.10 Medical records were examined for the mechanism of injury (MOI), injury severity score (ISS), time from injury to free tissue transfer, medical comorbidities, surgical complications, and time to radiographic and clinical union. Radiographic union was determined by the presence of bridging bone on 3 of 4 of cortices on plain film radiographs.
All patients were male (n = 5), with a mean age of 25 years (range, 19-30 years) at the time of injury (Table).
Table. Demographics and Outcomes of Patients Undergoing Free Tissue Transfer
| Free Serratus Anterior-Latissimus-Rib Flaps |
Age (Mean ± SEM) | 23 ± 2 years |
Males | 5 |
Females | 0 |
Tobacco Use | 2 |
Body Mass Index (Mean ± SEM) | 26.2 ± 0.9 kg/m2 |
Injury Severity Score (Mean ± SEM) | 18 ± 5 |
Time to Tissue Transfer (Mean ± SEM) | 1 ± 0.3 weeks |
Time to Boney Union (Mean ± SEM) | 7 ± 0.7 months |
Time Non-Weight-Bearing (Mean ± SEM) | 5 ± 0.5 months |
The MOI included motorcycle collisions (n = 2), pedestrian struck by car (n = 1), motor vehicle collisions (n = 1), and direct blow to the leg (n = 1). The mean ISS of the cohort was 18 (range, 10-34) (Table). On average, patients underwent free tissue transfer within 1 week (range, 3 days to 2 weeks) from the time of injury. Patients in this cohort were followed clinically for a mean of 4 years (range, 1-6 years) after surgery. Patients were non-weight-bearing for an average of 5 months (range, 4-6 months) following their reconstructions.
RESULTS
All flaps survived. The mean time to radiographic and clinical union was 7 months (range, 6-9 months). Two patients underwent reoperation. One patient underwent a bone grafting procedure for a delayed union at 6 months postoperative, and 1 patient underwent irrigation and débridement of superficial soft tissue infection. Donor site complications occurred in 2 patients, including chronic rib pain (n = 1) and a pleural effusion requiring drainage (n = 1). At the last follow-up, all ribs had incorporated, and all patients were weight-bearing as tolerated on the limb.
CASE EXAMPLE
A 22-year-old male smoker was transferred to our facility after a motor vehicle accident with bilateral tibia fractures, 1 closed and 1 open with significant bone loss (Figures 1A, 1B).
Continue to: Surgical Technique...
SURGICAL TECHNIQUE
The patient is placed in the lateral decubitus position during the procedure. A 2-team approach is used for dissection of the flap and preparation of recipient vessels to decrease operative time. A J-shaped incision is started on the chest at the mid-axillary line and extended just over the fifth and sixth rib. The incision can be made to fall into the intermammary crease in a woman to hide the scar. The dissection begins by exposing the anterior border of the latissimus muscle (Figure 2A). Next, the latissimus is dissected to reveal the thoracodorsal vessels (Figure 2B). At this level, the thoracodorsal vessel can be traced into the axilla. The branch going into the fifth, sixth, and lower slips of the serratus are dissected. The long thoracic nerve and the thoracodorsal nerve are preserved during the dissection (Figure 2C). The fifth, sixth, and seventh slips of the serratus are preferentially included in the dissection while leaving the most superior slips of the serratus to preserve scapular stability. Dissection begins by identifying 2 adjacent rib sections of the fifth and sixth or sixth and seventh ribs. The defect in the lower extremity determines the length of rib harvested. The serratus slips are then divided anteriorly over the chest wall. The dissection is extended to the intercostal spaces of the fourth and fifth ribs. The supraperiosteal dissection is performed at the anterior margin of the rib (Figure 2D).
Continue to: Following the surgical procedure...
Following the surgical procedure, patients are made non-weight-bearing on the operative extremity until signs of healing are apparent on radiographs. In this case, at the patients’ last follow-up visit, the skin graft was healed, and there was solid fusion of the rib/tibia junction (Figures 4B, 4C).
DISCUSSION
High-energy open injuries to the lower extremities are devastating injuries, with a high rate of late amputation and poor functional outcomes.11-13 Vascularized bone grafting provides both essential osteoinductive and osteoconductive properties to segmental bone defects in areas with inadequate soft tissue coverage, particularly in the setting of >6 cm of bone loss.4,14 The results of this study show that acute reconstruction of the lower limb with a composite vascularized SALR graft is a reliable procedure with an acceptable complication profile.
The timing of soft tissue coverage should be performed as soon as the patient is medically stable enough to undergo a reconstructive procedure, ideally within 7 to 10 days; and this timetable has been shown to decrease rates of infection and free flap failure.15-19 Early coverage provides both control of the soft-tissue envelope and reduces the risk of losing bone.1 Unlike the timing of coverage, the staging of the procedure is controversial. Proponents of the 2-stage free tissue (soft tissue followed by bony flap) transfer feel that although the tissue may not be infected at the time of coverage, it is contaminated with bacteria at the time of bone reconstruction, and as such is at high risk for both infection and complications.20 Unlike 2-stage procedures, single-stage coverage provides immediate soft tissue coverage, as well as bony support. This reduces the time to bony union and negates the need for repeated surgery in a mangled extremity where secondary surgery is complicated by both scar tissue and altered anatomy.1,2 Furthermore, it has been shown that there is no difference in the rates of infection when performing a single-stage compared with a 2-stage procedure.9 In this study, SALR flaps were typically performed within 2 weeks following an injury as a single procedure. We feel this resulted in the low number of complications in the SALR group.
Unlike free fibulas, rib flaps are easily pedicled with an associated soft-tissue flap due to their blood supply, making them ideal for 1-stage reconstruction. The rib has a dual blood supply: 1 from the posterior intercostal artery, and the other, an abundant periosteal blood supply, from the serratus anterior muscle.4 The blood supply to the serratus anterior comes from the thoracodorsal artery, and usually provides 14 cm of a large-caliber pedicle, making it a reliable flap for soft tissue reconstruction.21,22 Another unique feature of the blood supply to this flap is the amount of soft tissue available for both harvest and transfer; larger portions of serratus muscle and latissimus muscle can be harvested if necessary to cover the soft tissue defect.4
Comminuted tibias with segmental bone loss are difficult to manage since they are associated with bony as well as soft tissue defects.1,12,13,23 These injuries are ideal candidates for a single-stage reconstruction using a vascularized SALR flap. In our series, the use of an SALR flap resulted ultimately in a 100% union and limb salvage rate, with no flap failures and a low complication profile. Unlike the SALR, free fibular flaps must be transferred along with a separate latissimus dorsi flap to provide enough soft tissue coverage necessary for reconstructing large Gustilo IIIB injuries, which could increase the risk of flap failure. Since ribs are composed of membranous bone and have a similar cross-sectional area to both metacarpal and metatarsals, there are concerns regarding the biomechanical properties of ribs for weight-bearing.4,22,24-26 To compensate for this relatively small cross-sectional area, 2 ribs (either consecutive or alternative) are frequently harvested.1,4,5,23 Previous studies examining the use of ribs for bony reconstruction have frequently supplemented the rib reconstruction to the tibia using screws and external fixation alone.1,4,5,23 In our series, all SALR grafts were supported with the use of an intramedullary nail (n = 3) or locked plating (n = 1). The use of this supplemental fixation of the SALR graft allowed our patients to return to full weight-bearing (mean, 6 months) much earlier than the length of time cited in previous reports (12 months) examining these injuries.1,4,5,23
Continue to: There are several limitations...
There are several limitations to this study. The small sample size and retrospective nature of the study limits the amount of data we are able to collect from the medical record and places obvious constraints on the analysis. Although all these procedures were performed at 1 institution, multiple providers were involved in the reconstruction of these injuries, and there is no standard protocol for their treatment. Similarly, although other forms of extremity reconstruction were used during this time period, there was no standard protocol that could serve as a comparator for patients who underwent an SALR compared with other reconstructive procedures.
Overall, SALR grafts provide an excellent option for 1-stage reconstruction of severe, open lower extremity injuries. In this series we noted a 100% graft success rate with an acceptable complication profile.
This paper will be judged for the Resident Writer’s Award.
1. Yazar S, Lin CH, Wei FC. One-stage reconstruction of composite bone and soft-tissue defects in traumatic lower extremities. Plast Reconstr Surg. 2004;114(6):1457-1466. doi:10.1097/01.PRS.0000138811.88807.65.
2. Lin CH, Wei FC, Chen HC, Chuang DC. Outcome comparison in traumatic lower-extremity reconstruction by using various composite vascularized bone transplantation. Plast Reconstr Surg. 1999;104(4):984-992. doi:10.1097/00006534-199909040-00013.
3. Tu YK, Yen CY, Yeh WL, Wang IC, Wang KC, Ueng SW. Reconstruction of posttraumatic long bone defect with free vascularized bone graft: good outcome in 48 patients with 6 years' follow-up. Acta Orthopaedica Scandinavica. 2001;72(4):359-364. doi:10.1080/000164701753542014.
4. Lin CH, Wei FC, Levin LS, Su JI, Fan KF, Yeh WL, Hsu DT. Free composite serratus anterior and rib flaps for tibial composite bone and soft-tissue defect. Plast Reconstr Surg. 1997;99(6):1656-1665. Doi:10.1097/00006534-199705000-00028.
5. Georgescu AV, Ignatiadis I, Ileana M, Irina C, Filip A, Olariu R. Long-term results after muscle-rib flap transfer for reconstruction of composite limb defects. Microsurgery. 2011;31(3):218-222. doi:10.1002/micr.20857.
6. Moran CG, Wood MB. Vascularized bone autografts. Orthop Rev. 1993;22(2):187-197. doi:10.1097/01241398-199307000-00031.
7. Banic A, Hertel R. Double vascularized fibulas for reconstruction of large tibial defects. J Reconstr Microsurg. 1993;9(6):421-428. doi:10.1055/s-2007-1006751.
8. Malizos KN, Nunley JA, Goldner RD, Urbaniak JR, Harrelson JM. Free vascularized fibula in traumatic long bone defects and in limb salvaging following tumor resection: comparative study. Microsurgery. 1993;14(6):368-374. doi:10.1002/micr.1920140603.
9. Peat BG, Liggins DF. Microvascular soft tissue reconstruction for acute tibial fractures--late complications and the role of bone grafting. Ann Plast Surg. 1990;24(6):517-520.
10. Gustilo RB, Anderson JT. Prevention of infection in the treatment of one thousand and twenty-five open fractures of long bones: retrospective and prospective analyses. J Bone Joint Surg Am. 1976;58(4):453-458.
11. Gustilo RB, Mendoza RM, Williams DN. Problems in the management of type III (severe) open fractures: a new classification of type III open fractures. J Trauma. 1984;24(8):742-746. doi:10.1097/00005373-198408000-00009.
12. Bosse MJ, MacKenzie EJ, Kellam JF, et al. An analysis of outcomes of reconstruction or amputation after leg-threatening injuries. NEJM. 2002;347(24):1924-1931. doi:10.1056/NEJMoa012604.
13. MacKenzie EJ, Bosse MJ, Pollak AN, et al. Long-term persistence of disability following severe lower-limb trauma. Results of a seven-year follow-up. J Bone Joint Surg Am. 2005;87(8):1801-1809. doi:10.2106/JBJS.E.00032.
14. Bieber EJ, Wood MB. Bone reconstruction. Clin Plast Surg. 1986;13(4):645-655.
15. Melvin JS, Dombroski DG, Torbert JT, Kovach SJ, Esterhai JL, Mehta S. Open tibial shaft fractures: II. Definitive management and limb salvage. J Am Acad Orthop Surg. 2010;18(2):108-117. doi:10.5435/00124635-201002000-00005.
16. Godina M. Early microsurgical reconstruction of complex trauma of the extremities. Plast Reconstr Surg. 1986;78(3):285-292. doi:10.1055/s-2006-944324.
17. Gopal S, Majumder S, Batchelor AG, Knight SL, De Boer P, Smith RM. Fix and flap: the radical orthopaedic and plastic treatment of severe open fractures of the tibia. J Bone Joint Surg Br. 2000;82(7):959-966. doi:10.1302/0301-620X.82B7.0820959.
18. Fischer MD, Gustilo RB, Varecka TF. The timing of flap coverage, bone-grafting, and intramedullary nailing in patients who have a fracture of the tibial shaft with extensive soft-tissue injury. J Bone Joint Surg Am. 1991;73(9):1316-1322. doi:10.2106/00004623-199173090-00005.
19. Tielinen L, Lindahl JE, Tukiainen EJ. Acute unreamed intramedullary nailing and soft tissue reconstruction with muscle flaps for the treatment of severe open tibial shaft fractures. Injury. 2007;38(8):906-912. doi:10.1016/j.injury.2007.02.052.
20. Yaremchuk MJ, Brumback RJ, Manson PN, Burgess AR, Poka A, Weiland AJ. Acute and definitive management of traumatic osteocutaneous defects of the lower extremity. Plast Reconstr Surg. 1987;80(1):1-14. doi:10.1097/00006534-198707000-00002.
21. Ueng WN, Chuang CC, Shih CH. Double-rib composite free transfer to reconstruct a single-spared lower extremity defect. J Trauma. 1995;38(2):210-212.
22. Bruck JC, Bier J, Kistler D. The serratus anterior osteocutaneous free flap. J Reconstr Microsurg. 1990;6(3):209-213. doi:10.1055/s-2007-1006820.
23. Lin CH, Yazar S. Revisiting the serratus anterior rib flap for composite tibial defects. Plast Reconstr Surg. 2004;114(7):1871-1877. doi:10.1097/01.PRS.0000142767.13493.63.
24. Hui KC, Zhang F, Lineaweaver WC, Moon W, Buncke GM, Buncke HJ. Serratus anterior-rib composite flap: anatomic studies and clinical application to hand reconstruction. Ann Plast Surg. 1999;42(2):132-136. doi:10.1097/00000637-199902000-00004.
25. Buncke HJ, Furnas DW, Gordon L, Achauer BM. Free osteocutaneous flap from a rib to the tibia. Plast Reconstr Surg. 1977;59(6):799-804. doi:10.1097/00006534-197706000-00002.
26. Nusbickel FR, Dell PC, Mcandrew MP, Moore MM. Vascularized autografts for reconstruction of skeletal defects following lower extremity trauma. A review. Clin Orthop Relat Res. 1989;(243):65-70.
1. Yazar S, Lin CH, Wei FC. One-stage reconstruction of composite bone and soft-tissue defects in traumatic lower extremities. Plast Reconstr Surg. 2004;114(6):1457-1466. doi:10.1097/01.PRS.0000138811.88807.65.
2. Lin CH, Wei FC, Chen HC, Chuang DC. Outcome comparison in traumatic lower-extremity reconstruction by using various composite vascularized bone transplantation. Plast Reconstr Surg. 1999;104(4):984-992. doi:10.1097/00006534-199909040-00013.
3. Tu YK, Yen CY, Yeh WL, Wang IC, Wang KC, Ueng SW. Reconstruction of posttraumatic long bone defect with free vascularized bone graft: good outcome in 48 patients with 6 years' follow-up. Acta Orthopaedica Scandinavica. 2001;72(4):359-364. doi:10.1080/000164701753542014.
4. Lin CH, Wei FC, Levin LS, Su JI, Fan KF, Yeh WL, Hsu DT. Free composite serratus anterior and rib flaps for tibial composite bone and soft-tissue defect. Plast Reconstr Surg. 1997;99(6):1656-1665. Doi:10.1097/00006534-199705000-00028.
5. Georgescu AV, Ignatiadis I, Ileana M, Irina C, Filip A, Olariu R. Long-term results after muscle-rib flap transfer for reconstruction of composite limb defects. Microsurgery. 2011;31(3):218-222. doi:10.1002/micr.20857.
6. Moran CG, Wood MB. Vascularized bone autografts. Orthop Rev. 1993;22(2):187-197. doi:10.1097/01241398-199307000-00031.
7. Banic A, Hertel R. Double vascularized fibulas for reconstruction of large tibial defects. J Reconstr Microsurg. 1993;9(6):421-428. doi:10.1055/s-2007-1006751.
8. Malizos KN, Nunley JA, Goldner RD, Urbaniak JR, Harrelson JM. Free vascularized fibula in traumatic long bone defects and in limb salvaging following tumor resection: comparative study. Microsurgery. 1993;14(6):368-374. doi:10.1002/micr.1920140603.
9. Peat BG, Liggins DF. Microvascular soft tissue reconstruction for acute tibial fractures--late complications and the role of bone grafting. Ann Plast Surg. 1990;24(6):517-520.
10. Gustilo RB, Anderson JT. Prevention of infection in the treatment of one thousand and twenty-five open fractures of long bones: retrospective and prospective analyses. J Bone Joint Surg Am. 1976;58(4):453-458.
11. Gustilo RB, Mendoza RM, Williams DN. Problems in the management of type III (severe) open fractures: a new classification of type III open fractures. J Trauma. 1984;24(8):742-746. doi:10.1097/00005373-198408000-00009.
12. Bosse MJ, MacKenzie EJ, Kellam JF, et al. An analysis of outcomes of reconstruction or amputation after leg-threatening injuries. NEJM. 2002;347(24):1924-1931. doi:10.1056/NEJMoa012604.
13. MacKenzie EJ, Bosse MJ, Pollak AN, et al. Long-term persistence of disability following severe lower-limb trauma. Results of a seven-year follow-up. J Bone Joint Surg Am. 2005;87(8):1801-1809. doi:10.2106/JBJS.E.00032.
14. Bieber EJ, Wood MB. Bone reconstruction. Clin Plast Surg. 1986;13(4):645-655.
15. Melvin JS, Dombroski DG, Torbert JT, Kovach SJ, Esterhai JL, Mehta S. Open tibial shaft fractures: II. Definitive management and limb salvage. J Am Acad Orthop Surg. 2010;18(2):108-117. doi:10.5435/00124635-201002000-00005.
16. Godina M. Early microsurgical reconstruction of complex trauma of the extremities. Plast Reconstr Surg. 1986;78(3):285-292. doi:10.1055/s-2006-944324.
17. Gopal S, Majumder S, Batchelor AG, Knight SL, De Boer P, Smith RM. Fix and flap: the radical orthopaedic and plastic treatment of severe open fractures of the tibia. J Bone Joint Surg Br. 2000;82(7):959-966. doi:10.1302/0301-620X.82B7.0820959.
18. Fischer MD, Gustilo RB, Varecka TF. The timing of flap coverage, bone-grafting, and intramedullary nailing in patients who have a fracture of the tibial shaft with extensive soft-tissue injury. J Bone Joint Surg Am. 1991;73(9):1316-1322. doi:10.2106/00004623-199173090-00005.
19. Tielinen L, Lindahl JE, Tukiainen EJ. Acute unreamed intramedullary nailing and soft tissue reconstruction with muscle flaps for the treatment of severe open tibial shaft fractures. Injury. 2007;38(8):906-912. doi:10.1016/j.injury.2007.02.052.
20. Yaremchuk MJ, Brumback RJ, Manson PN, Burgess AR, Poka A, Weiland AJ. Acute and definitive management of traumatic osteocutaneous defects of the lower extremity. Plast Reconstr Surg. 1987;80(1):1-14. doi:10.1097/00006534-198707000-00002.
21. Ueng WN, Chuang CC, Shih CH. Double-rib composite free transfer to reconstruct a single-spared lower extremity defect. J Trauma. 1995;38(2):210-212.
22. Bruck JC, Bier J, Kistler D. The serratus anterior osteocutaneous free flap. J Reconstr Microsurg. 1990;6(3):209-213. doi:10.1055/s-2007-1006820.
23. Lin CH, Yazar S. Revisiting the serratus anterior rib flap for composite tibial defects. Plast Reconstr Surg. 2004;114(7):1871-1877. doi:10.1097/01.PRS.0000142767.13493.63.
24. Hui KC, Zhang F, Lineaweaver WC, Moon W, Buncke GM, Buncke HJ. Serratus anterior-rib composite flap: anatomic studies and clinical application to hand reconstruction. Ann Plast Surg. 1999;42(2):132-136. doi:10.1097/00000637-199902000-00004.
25. Buncke HJ, Furnas DW, Gordon L, Achauer BM. Free osteocutaneous flap from a rib to the tibia. Plast Reconstr Surg. 1977;59(6):799-804. doi:10.1097/00006534-197706000-00002.
26. Nusbickel FR, Dell PC, Mcandrew MP, Moore MM. Vascularized autografts for reconstruction of skeletal defects following lower extremity trauma. A review. Clin Orthop Relat Res. 1989;(243):65-70.
TAKE-HOME POINTS
- Gustilo IIIB injuries with segmental bone loss can be difficult to treat with conventional means.
- Vascularized bone grafts are beneficial for reconstructing bone defects >5 cm.
- The SALR composite flap consists of bone and muscle.
- This flap can provide soft tissue coverage and vascularized bone in a single surgical procedure.
- In this study, the use of the SALR composite flap was capable of healing large segmental bony defects at an average of 7 months.
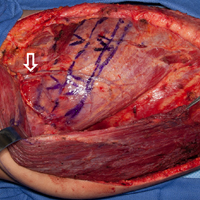
Cosmetic procedures show continued growth
and two declining, according to the American Society of Plastic Surgeons.
The gainers were the three most popular procedures: OnabotulinumtoxinA injections topped the list with 7.23 million anatomic sites injected – an increase of 2% over 2016 – followed by injection of soft tissue fillers with 2.69 million procedures (up 3%) and chemical peels with 1.38 million procedures (an increase of 1%), the ASPS said in its 2017 Plastic Surgery Statistics Report.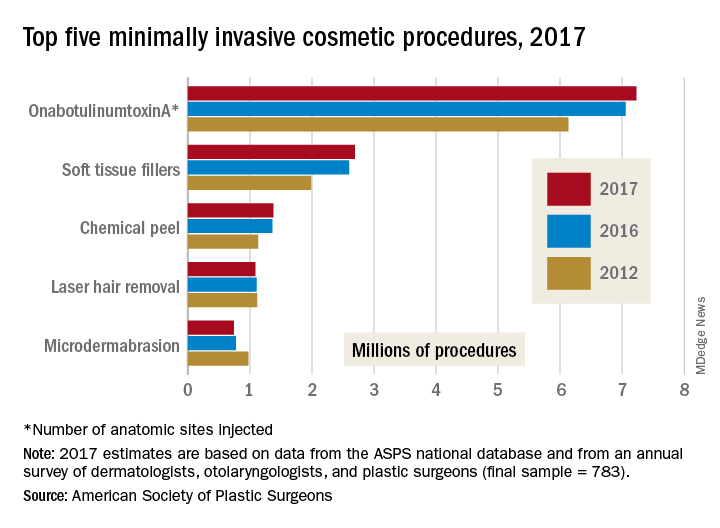
The two decliners among the top five were laser hair removal, which dropped 2% to 1.09 million procedures, and microdermabrasion, which continued a long-term decline by falling 4% to 740,000 procedures in 2017, the ASPS reported.
The minimally invasive cosmetic sector as a whole was up by 2% last year, bringing the number of total procedures to 15.7 million. Cosmetic surgical procedures were up by 1% from 2016 to 2017, reaching a total of 1.79 million. The five most popular cosmetic surgeries were breast augmentation (300,000 performed), liposuction (246,000), rhinoplasty (219,000), blepharoplasty (210,000), and abdominoplasty (130,000), according to the ASPS Tracking Operations and Outcomes for Plastic Surgeons database and an annual survey of board-certified dermatologists, otolaryngologists, and plastic surgeons (final sample = 783).
and two declining, according to the American Society of Plastic Surgeons.
The gainers were the three most popular procedures: OnabotulinumtoxinA injections topped the list with 7.23 million anatomic sites injected – an increase of 2% over 2016 – followed by injection of soft tissue fillers with 2.69 million procedures (up 3%) and chemical peels with 1.38 million procedures (an increase of 1%), the ASPS said in its 2017 Plastic Surgery Statistics Report.
The two decliners among the top five were laser hair removal, which dropped 2% to 1.09 million procedures, and microdermabrasion, which continued a long-term decline by falling 4% to 740,000 procedures in 2017, the ASPS reported.
The minimally invasive cosmetic sector as a whole was up by 2% last year, bringing the number of total procedures to 15.7 million. Cosmetic surgical procedures were up by 1% from 2016 to 2017, reaching a total of 1.79 million. The five most popular cosmetic surgeries were breast augmentation (300,000 performed), liposuction (246,000), rhinoplasty (219,000), blepharoplasty (210,000), and abdominoplasty (130,000), according to the ASPS Tracking Operations and Outcomes for Plastic Surgeons database and an annual survey of board-certified dermatologists, otolaryngologists, and plastic surgeons (final sample = 783).
and two declining, according to the American Society of Plastic Surgeons.
The gainers were the three most popular procedures: OnabotulinumtoxinA injections topped the list with 7.23 million anatomic sites injected – an increase of 2% over 2016 – followed by injection of soft tissue fillers with 2.69 million procedures (up 3%) and chemical peels with 1.38 million procedures (an increase of 1%), the ASPS said in its 2017 Plastic Surgery Statistics Report.
The two decliners among the top five were laser hair removal, which dropped 2% to 1.09 million procedures, and microdermabrasion, which continued a long-term decline by falling 4% to 740,000 procedures in 2017, the ASPS reported.
The minimally invasive cosmetic sector as a whole was up by 2% last year, bringing the number of total procedures to 15.7 million. Cosmetic surgical procedures were up by 1% from 2016 to 2017, reaching a total of 1.79 million. The five most popular cosmetic surgeries were breast augmentation (300,000 performed), liposuction (246,000), rhinoplasty (219,000), blepharoplasty (210,000), and abdominoplasty (130,000), according to the ASPS Tracking Operations and Outcomes for Plastic Surgeons database and an annual survey of board-certified dermatologists, otolaryngologists, and plastic surgeons (final sample = 783).
MI risk prediction after noncardiac surgery simplified
ORLANDO – The risk of perioperative MI or death associated with noncardiac surgery is vanishingly low in patients free of diabetes, hypertension, and smoking, Tanya Wilcox, MD, reported at the annual meeting of the American College of Cardiology.
How small is the risk? A mere 1 in 1,000, according to her analysis of more than 3.8 million major noncardiac surgeries in the American College of Surgeons National Surgical Quality Improvement Program database for 2009-2015, according to Dr. Wilcox of New York University.
Physicians are frequently asked by surgeons to clear patients for noncardiac surgery in terms of cardiovascular risk. Because current risk scores are complex, aren’t amenable to rapid bedside calculations, and may entail cardiac stress testing, Dr. Wilcox decided it was worth assessing the impact of three straightforward cardiovascular risk factors – current smoking and treatment for hypertension or diabetes – on 30-day postoperative MI-free survival. For this purpose she turned to the National Surgical Quality Improvement Program database, a validated, risk-adjusted, outcomes-based program to measure and improve the quality of surgical care utilizing data from 250 U.S. surgical centers.
Of the 3,817,113 patients who underwent major noncardiac surgery, 1,586,020 (42%) of them had none of the three cardiovascular risk factors of interest, 1,541,846 (40%) had one, 643,424 (17%) had two, and 45,823, or 1.2%, had all three. The patients’ mean age was 57, 75% were white, and 57% were women. About half of all patients underwent various operations within the realm of general surgery; next most frequent were orthopedic procedures, accounting for 18% of total noncardiac surgery. Of note, only 23% of patients with zero risk factors were American Society of Anesthesiologists Class 3-5, compared with 51% of those with one cardiovascular risk factor, 76% with two, and 71% with all three.
The incidence of acute MI or death within 30 days of noncardiac surgery climbed in stepwise fashion according to a patient’s risk factor burden. In a multivariate analysis adjusted for age, race, and gender, patients with any one of the cardiovascular risk factors had a 30-day risk of acute MI or death that was 1.52 times greater than those with no risk factors, patients with two risk factors were at 2.4-fold increased risk, and those with all three were at 3.63-fold greater risk than those with none. The degree of increased risk associated with any single risk factor ranged from 1.47-fold for hypertension to 1.94-fold for smoking.
“Further study is needed to determine whether aggressive risk factor modifications in the form of blood pressure control, glycemic control, and smoking cessation could reduce the incidence of postoperative MI,” Dr. Wilcox observed.
She reported having no financial conflicts regarding her study.
ORLANDO – The risk of perioperative MI or death associated with noncardiac surgery is vanishingly low in patients free of diabetes, hypertension, and smoking, Tanya Wilcox, MD, reported at the annual meeting of the American College of Cardiology.
How small is the risk? A mere 1 in 1,000, according to her analysis of more than 3.8 million major noncardiac surgeries in the American College of Surgeons National Surgical Quality Improvement Program database for 2009-2015, according to Dr. Wilcox of New York University.
Physicians are frequently asked by surgeons to clear patients for noncardiac surgery in terms of cardiovascular risk. Because current risk scores are complex, aren’t amenable to rapid bedside calculations, and may entail cardiac stress testing, Dr. Wilcox decided it was worth assessing the impact of three straightforward cardiovascular risk factors – current smoking and treatment for hypertension or diabetes – on 30-day postoperative MI-free survival. For this purpose she turned to the National Surgical Quality Improvement Program database, a validated, risk-adjusted, outcomes-based program to measure and improve the quality of surgical care utilizing data from 250 U.S. surgical centers.
Of the 3,817,113 patients who underwent major noncardiac surgery, 1,586,020 (42%) of them had none of the three cardiovascular risk factors of interest, 1,541,846 (40%) had one, 643,424 (17%) had two, and 45,823, or 1.2%, had all three. The patients’ mean age was 57, 75% were white, and 57% were women. About half of all patients underwent various operations within the realm of general surgery; next most frequent were orthopedic procedures, accounting for 18% of total noncardiac surgery. Of note, only 23% of patients with zero risk factors were American Society of Anesthesiologists Class 3-5, compared with 51% of those with one cardiovascular risk factor, 76% with two, and 71% with all three.
The incidence of acute MI or death within 30 days of noncardiac surgery climbed in stepwise fashion according to a patient’s risk factor burden. In a multivariate analysis adjusted for age, race, and gender, patients with any one of the cardiovascular risk factors had a 30-day risk of acute MI or death that was 1.52 times greater than those with no risk factors, patients with two risk factors were at 2.4-fold increased risk, and those with all three were at 3.63-fold greater risk than those with none. The degree of increased risk associated with any single risk factor ranged from 1.47-fold for hypertension to 1.94-fold for smoking.
“Further study is needed to determine whether aggressive risk factor modifications in the form of blood pressure control, glycemic control, and smoking cessation could reduce the incidence of postoperative MI,” Dr. Wilcox observed.
She reported having no financial conflicts regarding her study.
ORLANDO – The risk of perioperative MI or death associated with noncardiac surgery is vanishingly low in patients free of diabetes, hypertension, and smoking, Tanya Wilcox, MD, reported at the annual meeting of the American College of Cardiology.
How small is the risk? A mere 1 in 1,000, according to her analysis of more than 3.8 million major noncardiac surgeries in the American College of Surgeons National Surgical Quality Improvement Program database for 2009-2015, according to Dr. Wilcox of New York University.
Physicians are frequently asked by surgeons to clear patients for noncardiac surgery in terms of cardiovascular risk. Because current risk scores are complex, aren’t amenable to rapid bedside calculations, and may entail cardiac stress testing, Dr. Wilcox decided it was worth assessing the impact of three straightforward cardiovascular risk factors – current smoking and treatment for hypertension or diabetes – on 30-day postoperative MI-free survival. For this purpose she turned to the National Surgical Quality Improvement Program database, a validated, risk-adjusted, outcomes-based program to measure and improve the quality of surgical care utilizing data from 250 U.S. surgical centers.
Of the 3,817,113 patients who underwent major noncardiac surgery, 1,586,020 (42%) of them had none of the three cardiovascular risk factors of interest, 1,541,846 (40%) had one, 643,424 (17%) had two, and 45,823, or 1.2%, had all three. The patients’ mean age was 57, 75% were white, and 57% were women. About half of all patients underwent various operations within the realm of general surgery; next most frequent were orthopedic procedures, accounting for 18% of total noncardiac surgery. Of note, only 23% of patients with zero risk factors were American Society of Anesthesiologists Class 3-5, compared with 51% of those with one cardiovascular risk factor, 76% with two, and 71% with all three.
The incidence of acute MI or death within 30 days of noncardiac surgery climbed in stepwise fashion according to a patient’s risk factor burden. In a multivariate analysis adjusted for age, race, and gender, patients with any one of the cardiovascular risk factors had a 30-day risk of acute MI or death that was 1.52 times greater than those with no risk factors, patients with two risk factors were at 2.4-fold increased risk, and those with all three were at 3.63-fold greater risk than those with none. The degree of increased risk associated with any single risk factor ranged from 1.47-fold for hypertension to 1.94-fold for smoking.
“Further study is needed to determine whether aggressive risk factor modifications in the form of blood pressure control, glycemic control, and smoking cessation could reduce the incidence of postoperative MI,” Dr. Wilcox observed.
She reported having no financial conflicts regarding her study.
REPORTING FROM ACC 2018
Key clinical point: Noncardiac surgery patients can breathe easier regarding perioperative cardiovascular risk provided they don’t smoke and aren’t hypertensive or diabetic.
Major finding: .
Study details: This was a retrospective analysis of more than 3.8 million noncardiac surgeries contained in the American College of Surgeons National Surgical Quality Improvement Program database for 2009-2015.
Disclosures: The study presenter reported having no financial conflicts.
Canakinumab cut gout attacks in CANTOS
AMSTERDAM – in an exploratory, post hoc analysis of data collected from more than 10,000 patients in the CANTOS multicenter, randomized trial.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
While this result is only a hypothesis-generating suggestion that blocking interleukin (IL)-1 beta can have a significant impact on the frequency of gout flares, it serves as a proof-of-concept that IL-1 beta blockade is a potentially clinically meaningful strategy for future efforts to block gout attacks, Daniel H. Solomon, MD, said at the European Congress of Rheumatology.
“IL-1 beta is incredibly important in the inflammation associated with gout. Gout is considered by many to be the canonical IL-1 beta disease,” and hence it was important to examine the impact that treatment with the IL-1 beta blocker canakinumab had on gout in the CANTOS trial, Dr. Solomon explained in a video interview.
The answer was that treatment with canakinumab was linked with a roughly 50% reduction in gout flares in the total study group. The same reduction was seen in both the subgroups of patients with and without a history of gout. The effect was seen across all three subgroups of patients, based on their baseline serum urate levels including those with normal, elevated, or very elevated levels and across all the other prespecified subgroups including divisions based on sex, age, baseline body mass index, and baseline level of high-sensitivity C-reactive protein (hsCRP).
It’s also unclear that canakinumab (Ilaris) is the best type of IL-1 beta blocking drug to use for prevention of gout flares. In CANTOS, this expensive drug was administered subcutaneously every 3 months. A more appropriate agent might be an oral, small-molecule drug that blocks IL-1 beta. Several examples of this type of agent are currently in clinical development, said Dr. Solomon, a professor of medicine at Harvard Medical School and a rheumatologist at Brigham and Women’s Hospital, both in Boston.
CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcome Study) randomized 10,061 patients with a history of MI and a hsCRP level of at least 2 mg/L at centers in 39 countries. The study’s primary endpoint was the combined rate of cardiovascular death, MI, or stroke, and canakinumab treatment at the 150-mg dosage level linked with a 15% relative reduction in this endpoint, compared with placebo in this secondary-prevention study (N Engl J Med. 2017 Sept 21;377[12]:1119-31). The study also randomized patients to either of two other canakinumab dosages, 50 mg or 300 mg, administered every 3 months, and, while each of these produced reductions in the primary endpoint relative to placebo, the 150-mg dosage had the largest effect. In the gout analysis reported by Dr. Solomon, the three different canakinumab dosages produced somewhat different levels of gout-flare reductions, but, generally, the effect was similar across the three treatment groups.
In the total study population, regardless of gout history, treatment with 50 mg, 150 mg, and 300 mg canakinumab every 3 months was linked with a reduction in gout attacks of 46%, 57%, and 53%, respectively, compared with placebo-treated patients, Dr. Solomon reported. The three dosages also uniformly produced significantly drops in serum levels of hsCRP, compared with placebo, but canakinumab treatment had no impact on serum urate levels, indicating that the gout-reducing effects of the drug did not occur via a mechanism that involved serum urate.
Because CANTOS exclusively enrolled patients with established coronary disease, the new analysis could not address whether IL-1 beta blockade would also be an effective strategy for reducing gout flares in people without cardiovascular disease, Dr. Solomon cautioned. Although it probably would, he said. He also stressed that treatment with an IL-1 blocking drug should not be seen as a substitute for appropriate urate-lowering treatment in patients with elevated levels of serum urate.
SOURCE: Solomon DH et al. Ann Rheum Dis. 2018;77(Suppl 2):56. Abstract OP0014.
AMSTERDAM – in an exploratory, post hoc analysis of data collected from more than 10,000 patients in the CANTOS multicenter, randomized trial.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
While this result is only a hypothesis-generating suggestion that blocking interleukin (IL)-1 beta can have a significant impact on the frequency of gout flares, it serves as a proof-of-concept that IL-1 beta blockade is a potentially clinically meaningful strategy for future efforts to block gout attacks, Daniel H. Solomon, MD, said at the European Congress of Rheumatology.
“IL-1 beta is incredibly important in the inflammation associated with gout. Gout is considered by many to be the canonical IL-1 beta disease,” and hence it was important to examine the impact that treatment with the IL-1 beta blocker canakinumab had on gout in the CANTOS trial, Dr. Solomon explained in a video interview.
The answer was that treatment with canakinumab was linked with a roughly 50% reduction in gout flares in the total study group. The same reduction was seen in both the subgroups of patients with and without a history of gout. The effect was seen across all three subgroups of patients, based on their baseline serum urate levels including those with normal, elevated, or very elevated levels and across all the other prespecified subgroups including divisions based on sex, age, baseline body mass index, and baseline level of high-sensitivity C-reactive protein (hsCRP).
It’s also unclear that canakinumab (Ilaris) is the best type of IL-1 beta blocking drug to use for prevention of gout flares. In CANTOS, this expensive drug was administered subcutaneously every 3 months. A more appropriate agent might be an oral, small-molecule drug that blocks IL-1 beta. Several examples of this type of agent are currently in clinical development, said Dr. Solomon, a professor of medicine at Harvard Medical School and a rheumatologist at Brigham and Women’s Hospital, both in Boston.
CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcome Study) randomized 10,061 patients with a history of MI and a hsCRP level of at least 2 mg/L at centers in 39 countries. The study’s primary endpoint was the combined rate of cardiovascular death, MI, or stroke, and canakinumab treatment at the 150-mg dosage level linked with a 15% relative reduction in this endpoint, compared with placebo in this secondary-prevention study (N Engl J Med. 2017 Sept 21;377[12]:1119-31). The study also randomized patients to either of two other canakinumab dosages, 50 mg or 300 mg, administered every 3 months, and, while each of these produced reductions in the primary endpoint relative to placebo, the 150-mg dosage had the largest effect. In the gout analysis reported by Dr. Solomon, the three different canakinumab dosages produced somewhat different levels of gout-flare reductions, but, generally, the effect was similar across the three treatment groups.
In the total study population, regardless of gout history, treatment with 50 mg, 150 mg, and 300 mg canakinumab every 3 months was linked with a reduction in gout attacks of 46%, 57%, and 53%, respectively, compared with placebo-treated patients, Dr. Solomon reported. The three dosages also uniformly produced significantly drops in serum levels of hsCRP, compared with placebo, but canakinumab treatment had no impact on serum urate levels, indicating that the gout-reducing effects of the drug did not occur via a mechanism that involved serum urate.
Because CANTOS exclusively enrolled patients with established coronary disease, the new analysis could not address whether IL-1 beta blockade would also be an effective strategy for reducing gout flares in people without cardiovascular disease, Dr. Solomon cautioned. Although it probably would, he said. He also stressed that treatment with an IL-1 blocking drug should not be seen as a substitute for appropriate urate-lowering treatment in patients with elevated levels of serum urate.
SOURCE: Solomon DH et al. Ann Rheum Dis. 2018;77(Suppl 2):56. Abstract OP0014.
AMSTERDAM – in an exploratory, post hoc analysis of data collected from more than 10,000 patients in the CANTOS multicenter, randomized trial.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
While this result is only a hypothesis-generating suggestion that blocking interleukin (IL)-1 beta can have a significant impact on the frequency of gout flares, it serves as a proof-of-concept that IL-1 beta blockade is a potentially clinically meaningful strategy for future efforts to block gout attacks, Daniel H. Solomon, MD, said at the European Congress of Rheumatology.
“IL-1 beta is incredibly important in the inflammation associated with gout. Gout is considered by many to be the canonical IL-1 beta disease,” and hence it was important to examine the impact that treatment with the IL-1 beta blocker canakinumab had on gout in the CANTOS trial, Dr. Solomon explained in a video interview.
The answer was that treatment with canakinumab was linked with a roughly 50% reduction in gout flares in the total study group. The same reduction was seen in both the subgroups of patients with and without a history of gout. The effect was seen across all three subgroups of patients, based on their baseline serum urate levels including those with normal, elevated, or very elevated levels and across all the other prespecified subgroups including divisions based on sex, age, baseline body mass index, and baseline level of high-sensitivity C-reactive protein (hsCRP).
It’s also unclear that canakinumab (Ilaris) is the best type of IL-1 beta blocking drug to use for prevention of gout flares. In CANTOS, this expensive drug was administered subcutaneously every 3 months. A more appropriate agent might be an oral, small-molecule drug that blocks IL-1 beta. Several examples of this type of agent are currently in clinical development, said Dr. Solomon, a professor of medicine at Harvard Medical School and a rheumatologist at Brigham and Women’s Hospital, both in Boston.
CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcome Study) randomized 10,061 patients with a history of MI and a hsCRP level of at least 2 mg/L at centers in 39 countries. The study’s primary endpoint was the combined rate of cardiovascular death, MI, or stroke, and canakinumab treatment at the 150-mg dosage level linked with a 15% relative reduction in this endpoint, compared with placebo in this secondary-prevention study (N Engl J Med. 2017 Sept 21;377[12]:1119-31). The study also randomized patients to either of two other canakinumab dosages, 50 mg or 300 mg, administered every 3 months, and, while each of these produced reductions in the primary endpoint relative to placebo, the 150-mg dosage had the largest effect. In the gout analysis reported by Dr. Solomon, the three different canakinumab dosages produced somewhat different levels of gout-flare reductions, but, generally, the effect was similar across the three treatment groups.
In the total study population, regardless of gout history, treatment with 50 mg, 150 mg, and 300 mg canakinumab every 3 months was linked with a reduction in gout attacks of 46%, 57%, and 53%, respectively, compared with placebo-treated patients, Dr. Solomon reported. The three dosages also uniformly produced significantly drops in serum levels of hsCRP, compared with placebo, but canakinumab treatment had no impact on serum urate levels, indicating that the gout-reducing effects of the drug did not occur via a mechanism that involved serum urate.
Because CANTOS exclusively enrolled patients with established coronary disease, the new analysis could not address whether IL-1 beta blockade would also be an effective strategy for reducing gout flares in people without cardiovascular disease, Dr. Solomon cautioned. Although it probably would, he said. He also stressed that treatment with an IL-1 blocking drug should not be seen as a substitute for appropriate urate-lowering treatment in patients with elevated levels of serum urate.
SOURCE: Solomon DH et al. Ann Rheum Dis. 2018;77(Suppl 2):56. Abstract OP0014.
REPORTING FROM THE EULAR 2018 CONGRESS
Key clinical point: IL-1 blockade seems to be an effective way to cut the incidence of gout attacks.
Major finding: IL-1 blockade with canakinumab was linked with about a 50% cut in gout flares, compared with placebo.
Study details: CANTOS, a multicenter, randomized trial with 10,061 patients.
Disclosures: CANTOS was funded by Novartis, the company that markets canakinumab. Dr. Solomon has no relationships with Novartis. Brigham and Women’s Hospital, the center at which he works, has received research funding from Amgen, Bristol-Myers Squibb, Genentech, and Pfizer for studies that Dr. Solomon has helped direct.
Source: Solomon DH et al. Ann Rheum Dis. 2018;77(Suppl 2):56. Abstract OP0014.

Creating a digital pill
Technology battles medication noncompliance
Hospitalists and other physicians have long struggled with medication noncompliance, which can lead to sicker patients and higher rates of readmittance, and costs some $100-$289 billion a year.
There is a growing field of digital devices being developed to address this problem. The Food and Drug Administration has just approved the newest one: a medication with a sensor embedded that can tell doctors if, and when, patients take their medicine, according to an article in the New York Times.1 It’s expected to become available in 2018.
The digital medication is a version of the antipsychotic Abilify. Patients who agree to take it will sign consent forms allowing their doctors (and up to four other people) to receive electronic data showing the date and time pills are ingested.
The sensor, created by Proteus Digital Health, contains copper, magnesium, and silicon, all said to be safe ingredients found in foods. The electrical signal is created when stomach fluids contact the sensor; a patch worn on the rib cage detects that signal and sends the message.
Other companies are joining the race to create digital medication technologies; these are being tested in medications for patients with conditions including heart disease, diabetes, and HIV infection. Some researchers predict the technology might have applications for monitoring the opioid intake of postsurgical patients or patients in medication clinical trials.
Reference
1. Belluck P. “First Digital Pill Approved to Worries About Biomedical ‘Big Brother.’ ” New York Times. Nov 13, 2017.
Technology battles medication noncompliance
Technology battles medication noncompliance
Hospitalists and other physicians have long struggled with medication noncompliance, which can lead to sicker patients and higher rates of readmittance, and costs some $100-$289 billion a year.
There is a growing field of digital devices being developed to address this problem. The Food and Drug Administration has just approved the newest one: a medication with a sensor embedded that can tell doctors if, and when, patients take their medicine, according to an article in the New York Times.1 It’s expected to become available in 2018.
The digital medication is a version of the antipsychotic Abilify. Patients who agree to take it will sign consent forms allowing their doctors (and up to four other people) to receive electronic data showing the date and time pills are ingested.
The sensor, created by Proteus Digital Health, contains copper, magnesium, and silicon, all said to be safe ingredients found in foods. The electrical signal is created when stomach fluids contact the sensor; a patch worn on the rib cage detects that signal and sends the message.
Other companies are joining the race to create digital medication technologies; these are being tested in medications for patients with conditions including heart disease, diabetes, and HIV infection. Some researchers predict the technology might have applications for monitoring the opioid intake of postsurgical patients or patients in medication clinical trials.
Reference
1. Belluck P. “First Digital Pill Approved to Worries About Biomedical ‘Big Brother.’ ” New York Times. Nov 13, 2017.
Hospitalists and other physicians have long struggled with medication noncompliance, which can lead to sicker patients and higher rates of readmittance, and costs some $100-$289 billion a year.
There is a growing field of digital devices being developed to address this problem. The Food and Drug Administration has just approved the newest one: a medication with a sensor embedded that can tell doctors if, and when, patients take their medicine, according to an article in the New York Times.1 It’s expected to become available in 2018.
The digital medication is a version of the antipsychotic Abilify. Patients who agree to take it will sign consent forms allowing their doctors (and up to four other people) to receive electronic data showing the date and time pills are ingested.
The sensor, created by Proteus Digital Health, contains copper, magnesium, and silicon, all said to be safe ingredients found in foods. The electrical signal is created when stomach fluids contact the sensor; a patch worn on the rib cage detects that signal and sends the message.
Other companies are joining the race to create digital medication technologies; these are being tested in medications for patients with conditions including heart disease, diabetes, and HIV infection. Some researchers predict the technology might have applications for monitoring the opioid intake of postsurgical patients or patients in medication clinical trials.
Reference
1. Belluck P. “First Digital Pill Approved to Worries About Biomedical ‘Big Brother.’ ” New York Times. Nov 13, 2017.