User login
Predictors of Long-Term Opioid Use After Opioid Initiation at Discharge From Medical and Surgical Hospitalizations
While patients may be newly exposed to opioids during medical and surgical hospitalization and the prescription of opioids at discharge is common,1-5 prescribers of opioids at discharge may not intend to initiate long-term opioid (LTO) use. By understanding the frequency of progression to LTO use, hospitalists can better balance postdischarge pain treatment and the risk for unintended LTO initiation.
Estimates of LTO use rates following hospital discharge in selected populations1,2,4-6 have varied depending on the population studied and the method of defining LTO use.7 Rates of LTO use following incident opioid prescription have not been directly compared at medical versus surgical discharge or compared with initiation in the ambulatory setting. We present the rates of LTO use following incident opioid exposure at surgical discharge and medical discharge and identify the factors associated with LTO use following surgical and medical discharge.
METHODS
Data Sources
Veterans Health Administration (VHA) data were obtained through the Austin Information Technology Center for fiscal years (FYs) 2003 through 2012 (Austin, Texas). Decision support system national data extracts were used to identify prescription-dispensing events, and inpatient and outpatient medical SAS data sets were used to identify diagnostic codes. The study was approved by the University of Iowa Institutional Review Board and the Iowa City Veterans Affairs (VA) Health Care System Research and Development Committee.
Patients
We included all patients with an outpatient opioid prescription during FY 2011 that was preceded by a 1-year opioid-free period.7 Patients with broadly accepted indications for LTO use (eg, metastatic cancer, palliative care, or opioid-dependence treatment) were excluded.7
Opioid Exposure
We included all outpatient prescription fills for noninjectable dosage forms of butorphanol, fentanyl, hydrocodone, hydromorphone, levorphanol, meperidine, methadone, morphine, oxycodone, oxymorphone, pentazocine, and tramadol. Consistent with the Centers for Disease Control and Prevention and VA/Department of Defense guidelines, LTO use was defined conceptually as regular use for >90 days. Operationalizing this definition to pharmacy refill data was established by using a cabinet supply methodology,7 which allows for the construction of episodes of continuous medication therapy by estimating the medication supply available to a patient for each day during a defined period based on the pattern of observed refills. LTO use was defined as an episode of continuous opioid supply for >90 days and beginning within 30 days of the initial prescription. While some studies have defined LTO use based on onset within 1 year following surgery,5 the requirement for onset within 30 days of initiation was applied to more strongly tie the association of developing LTO use with the discharge event and minimize various forms of bias that are introduced with extended follow-up periods.
Clinical Characteristics
Patients were classified as being medical discharges, surgical discharges, or outpatient initiators. Patients with an opioid index date within 2 days following discharge were designated based on discharge bed section; additionally, if patients had a surgical bed section during hospitalization, they were assigned as surgical discharges. Demographic, diagnosis, and medication exposure variables that were previously associated with LTO use were selected.8,9 Substance use disorder, chronic pain, anxiety disorder, and depressive disorder were based on International Classification of Diseases, 9th Revision (ICD-9) codes in the preceding year. The use of concurrent benzodiazepines, skeletal muscle relaxants, and antidepressants were determined at opioid initiation.10 Rural or urban residence was assigned by using the Rural-Urban Commuting Area Codes system and mapped with the zip code of a veteran’s residence.11
Analysis
Bivariate and multivariable relationships were determined by using logistic regression. The multivariable model considered all pairwise interaction terms between inpatient service (surgery versus medicine) and each of the variables in the model. Statistically significant interaction terms (P < .05) were retained, and all others were omitted from the final model. The main effects for variables that were involved in a significant interaction term were not reported in the final multivariable model; instead, we created fully specified multivariable models for surgery service and medicine service and reported odds ratios (ORs) for the main effects. All analyses were conducted by using SAS version 9.4 (SAS Institute Inc, Cary, North Carolina).
RESULTS

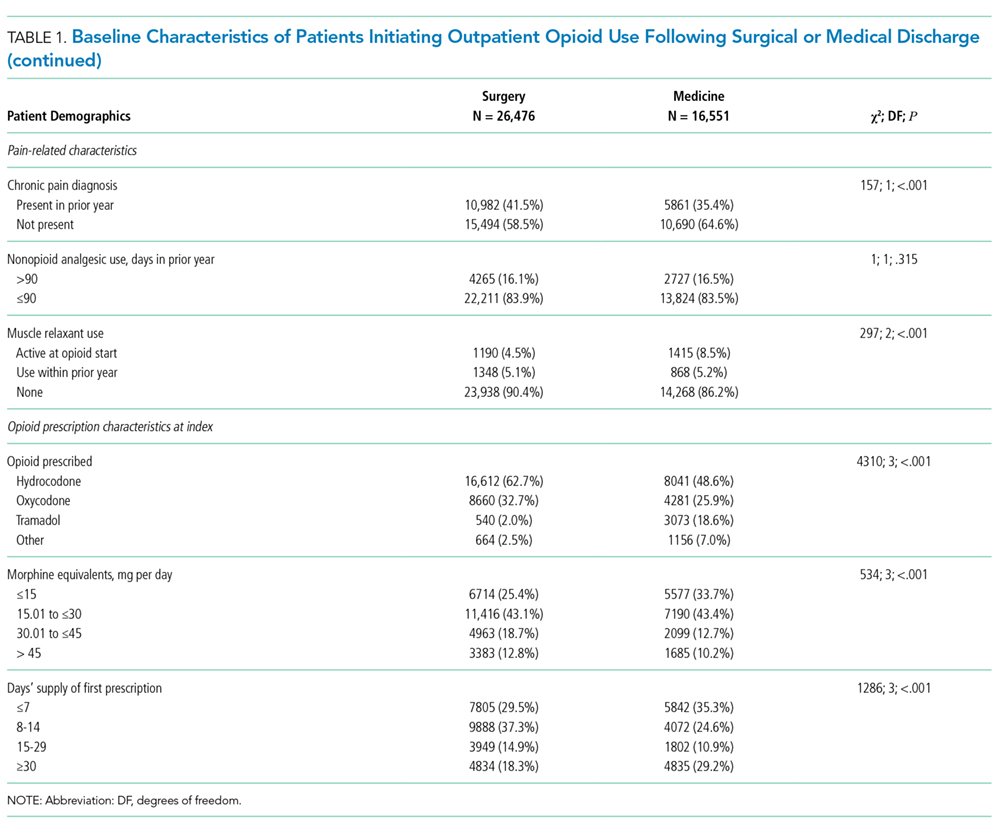
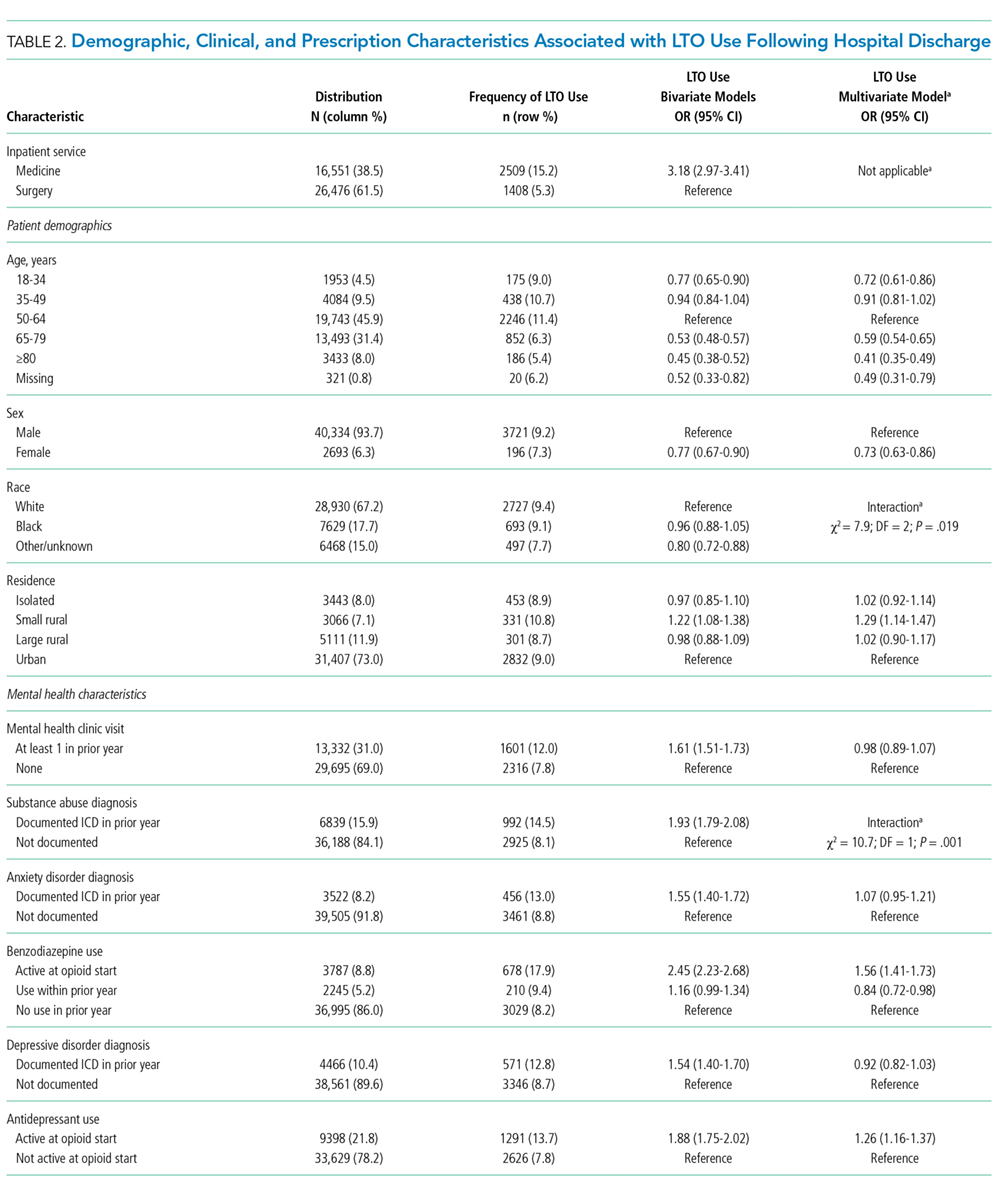
Days’ supply was associated with LTO use in a dose-dependent fashion relative to the reference category of ≤7 days: OR of 1.24 (95% CI, 1.12-1.37) for 8 to 14 days; OR of 1.56 (95% CI, 1.39-1.76) for 15 to 29 days; and OR of 2.59 (95% CI, 2.35-2.86) for 30 days (Table 2). LTO risk was higher among patients with an estimated dose of ≥15 morphine equivalents per day (MED) compared with those with doses of <15 equivalents (OR = 1.11; 95% CI, 1.02-1.21); patients who received >45 MED were at the greatest risk (OR = 1.70; 95% CI, 1.49-1.94).
DISCUSSION

The observation that subsequent LTO use occurs more frequently in discharged medical patients than surgical patients is consistent with the findings of Calcaterra et al.1 that among patients with no surgery versus surgery during hospitalization, opioid receipt at discharge resulted in a higher adjusted OR (7.24 for no surgery versus 3.40 for surgery) for chronic opioid use at 1 year. One explanation for this finding may be an artifact of cohort selection in the study design: patients with prior opioid use are excluded from the cohort, and prior use may be more common among surgical patients presenting for elective inpatient surgery for painful conditions. Previous work suggests that opioid use preoperatively is a robust predictor of postoperative use, and rates of LTO use are low among patients without preoperative opioid exposure.6
Demographic characteristics associated with persistent opioid receipt were similar to those previously reported.5,8,9 The inclusion of medication classes indicated in the treatment of mental health or pain conditions (ie, antidepressants, benzodiazepines, muscle relaxants, and nonopioid analgesics) resulted in diagnoses based on ICD-9 codes being no longer associated with LTO use. Severity or activity of illness, preferences regarding pharmacologic or nonpharmacologic treatment and undiagnosed or undocumented pain-comorbid conditions may all contribute to this finding. Future work studying opioid-related outcomes should include variables that reflect pharmacologic management of comorbid diagnoses in the cohort development or analytic design.
The strongest risk factors were potentially modifiable: days’ supply, dose, and concurrent medications. The measures of opioid quantity supplied are associated with subsequent ongoing use and are consistent with recent work based on prescription drug–monitoring data in a single state14 and in a nationally representative sample.15 That this relationship persists following hospital discharge, a scenario in which LTO use is unlikely to be initiated by a provider (who would be expected to subsequently titrate or monitor therapy), further supports the potential to curtail unintended LTO use through judicious early prescribing decisions.
We assessed only opioids that were supplied through a VA pharmacy, which may lead to the misclassification of patients as opioid naive for inclusion and an underestimation of the rate of opioid use following discharge. It is possible that differences in the rates of non-VA pharmacy use differ in medical and surgical populations in a nonrandom way. This study was performed in a large, integrated health system and may not be generalizable outside the VA system, where more discontinuities between hospital and ambulatory care may exist.
CONCLUSION
The initiation of LTO use at discharge is more common in veterans who are discharged from medical than surgical hospitalizations, likely reflecting differences in the patient population, pain conditions, and discharge prescribing decisions. While patient characteristics are associated with LTO use, the strongest associations are with increasing index dose and days’ supply; both represent potentially modifiable prescriber behaviors. These findings support policy changes and other efforts to minimize dose and days supplied when short-term use is intended as a means to address the current opioid epidemic.
Acknowledgments
The work reported here was supported by the Department of Veterans Affairs Office of Academic Affiliations and Office of Research and Development (Dr. Mosher and Dr. Hofmeyer), and Health Services Research and Development Service (HSR&D) through the Comprehensive Access and Delivery Research and Evaluation Center (CIN 13-412) and a Career Development Award (CDA 10-017; Dr. Lund).
Disclosures
The authors report no conflict of interest in regard to this study. The authors had full access to and take full responsibility for the integrity of the data. All analyses were conducted by using SAS version 9.2 (SAS Institute Inc, Cary, NC). This manuscript is not under review elsewhere, and there is no prior publication of the manuscript contents. The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs. The study was approved by the University of Iowa Institutional Review Board and the Iowa City Healthcare System Research and Development Committee.
1. Calcaterra SL, Yamashita TE, Min SJ, Keniston A, Frank JW, Binswanger IA. Opioid Prescribing at Hospital Discharge Contributes to Chronic Opioid Use. J Gen Intern Med. 2016;31(5):478-485. PubMed
2. Raebel MA, Newcomer SR, Reifler LM, et al. Chronic use of opioid medications before and after bariatric surgery. JAMA. 2013;310(13):1369-1376. PubMed
3. Mosher HJ, Jiang L, Vaughan Sarrazin MS, Cram P, Kaboli PJ, Vander Weg MW. Prevalence and characteristics of hospitalized adults on chronic opioid therapy. J Hosp Med. 2014;9(2):82-87. PubMed
4. Holman JE, Stoddard GJ, Higgins TF. Rates of prescription opiate use before and after injury in patients with orthopaedic trauma and the risk factors for prolonged opiate use. J Bone Joint Surg Am. 2013;95(12):1075-1080.
5. Sun EC, Darnall BD, Baker LC, Mackey S. Incidence of and Risk Factors for Chronic Opioid Use Among Opioid-Naive Patients in the Postoperative Period. JAMA Intern Med. 2016;176(9):1286-1293. PubMed
6. Goesling J, Moser SE, Zaidi B, et al. Trends and predictors of opioid use after total knee and total hip arthroplasty. Pain. 2016;157(6):1259-1265. PubMed
7. Mosher HJ, Richardson KK, Lund BC. The 1-Year Treatment Course of New Opioid Recipients in Veterans Health Administration. Pain Med. 2016. [Epub ahead of print]. PubMed
8. Sullivan MD, Edlund MJ, Fan MY, Devries A, Brennan Braden J, Martin BC. Risks for possible and probable opioid misuse among recipients of chronic opioid therapy in commercial and medicaid insurance plans: The TROUP Study. Pain. 2010;150(2):332-339. PubMed
9. Seal KH, Shi Y, Cohen G, et al. Association of mental health disorders with prescription opioids and high-risk opioid use in US veterans of Iraq and Afghanistan. JAMA. 2012;307(9):940-947. PubMed
10. Mosher HJ, Richardson KK, Lund BC. Sedative Prescriptions Are Common at Opioid Initiation: An Observational Study in the Veterans Health Administration. Pain Med. 2017. [Epub ahead of print]. PubMed
11. Lund BC, Abrams TE, Bernardy NC, Alexander B, Friedman MJ. Benzodiazepine prescribing variation and clinical uncertainty in treating posttraumatic stress disorder. Psychiatr Serv. 2013;64(1):21-27. PubMed
12. Brummett CM, Waljee JF, Goesling J, et al. New Persistent Opioid Use After Minor and Major Surgical Procedures in US Adults. JAMA Surg. 2017;152(6):e170504. PubMed
13. Mellbye A, Karlstad O, Skurtveit S, Borchgrevink PC, Fredheim OM. The duration and course of opioid therapy in patients with chronic non-malignant pain. Acta Anaesthesiol Scand. 2016;60(1):128-137. PubMed
14. Deyo RA, Hallvik SE, Hildebran C, et al. Association Between Initial Opioid Prescribing Patterns and Subsequent Long-Term Use Among Opioid-Naive Patients: A Statewide Retrospective Cohort Study. J Gen Intern Med. 2017;32(1):21-27. PubMed
15. Shah A, Hayes CJ, Martin BC. Factors Influencing Long-Term Opioid Use Among Opioid Naive Patients: An Examination of Initial Prescription Characteristics and Pain Etiologies. J Pain. 2017;18(11):1374-1383. PubMed
While patients may be newly exposed to opioids during medical and surgical hospitalization and the prescription of opioids at discharge is common,1-5 prescribers of opioids at discharge may not intend to initiate long-term opioid (LTO) use. By understanding the frequency of progression to LTO use, hospitalists can better balance postdischarge pain treatment and the risk for unintended LTO initiation.
Estimates of LTO use rates following hospital discharge in selected populations1,2,4-6 have varied depending on the population studied and the method of defining LTO use.7 Rates of LTO use following incident opioid prescription have not been directly compared at medical versus surgical discharge or compared with initiation in the ambulatory setting. We present the rates of LTO use following incident opioid exposure at surgical discharge and medical discharge and identify the factors associated with LTO use following surgical and medical discharge.
METHODS
Data Sources
Veterans Health Administration (VHA) data were obtained through the Austin Information Technology Center for fiscal years (FYs) 2003 through 2012 (Austin, Texas). Decision support system national data extracts were used to identify prescription-dispensing events, and inpatient and outpatient medical SAS data sets were used to identify diagnostic codes. The study was approved by the University of Iowa Institutional Review Board and the Iowa City Veterans Affairs (VA) Health Care System Research and Development Committee.
Patients
We included all patients with an outpatient opioid prescription during FY 2011 that was preceded by a 1-year opioid-free period.7 Patients with broadly accepted indications for LTO use (eg, metastatic cancer, palliative care, or opioid-dependence treatment) were excluded.7
Opioid Exposure
We included all outpatient prescription fills for noninjectable dosage forms of butorphanol, fentanyl, hydrocodone, hydromorphone, levorphanol, meperidine, methadone, morphine, oxycodone, oxymorphone, pentazocine, and tramadol. Consistent with the Centers for Disease Control and Prevention and VA/Department of Defense guidelines, LTO use was defined conceptually as regular use for >90 days. Operationalizing this definition to pharmacy refill data was established by using a cabinet supply methodology,7 which allows for the construction of episodes of continuous medication therapy by estimating the medication supply available to a patient for each day during a defined period based on the pattern of observed refills. LTO use was defined as an episode of continuous opioid supply for >90 days and beginning within 30 days of the initial prescription. While some studies have defined LTO use based on onset within 1 year following surgery,5 the requirement for onset within 30 days of initiation was applied to more strongly tie the association of developing LTO use with the discharge event and minimize various forms of bias that are introduced with extended follow-up periods.
Clinical Characteristics
Patients were classified as being medical discharges, surgical discharges, or outpatient initiators. Patients with an opioid index date within 2 days following discharge were designated based on discharge bed section; additionally, if patients had a surgical bed section during hospitalization, they were assigned as surgical discharges. Demographic, diagnosis, and medication exposure variables that were previously associated with LTO use were selected.8,9 Substance use disorder, chronic pain, anxiety disorder, and depressive disorder were based on International Classification of Diseases, 9th Revision (ICD-9) codes in the preceding year. The use of concurrent benzodiazepines, skeletal muscle relaxants, and antidepressants were determined at opioid initiation.10 Rural or urban residence was assigned by using the Rural-Urban Commuting Area Codes system and mapped with the zip code of a veteran’s residence.11
Analysis
Bivariate and multivariable relationships were determined by using logistic regression. The multivariable model considered all pairwise interaction terms between inpatient service (surgery versus medicine) and each of the variables in the model. Statistically significant interaction terms (P < .05) were retained, and all others were omitted from the final model. The main effects for variables that were involved in a significant interaction term were not reported in the final multivariable model; instead, we created fully specified multivariable models for surgery service and medicine service and reported odds ratios (ORs) for the main effects. All analyses were conducted by using SAS version 9.4 (SAS Institute Inc, Cary, North Carolina).
RESULTS
Days’ supply was associated with LTO use in a dose-dependent fashion relative to the reference category of ≤7 days: OR of 1.24 (95% CI, 1.12-1.37) for 8 to 14 days; OR of 1.56 (95% CI, 1.39-1.76) for 15 to 29 days; and OR of 2.59 (95% CI, 2.35-2.86) for 30 days (Table 2). LTO risk was higher among patients with an estimated dose of ≥15 morphine equivalents per day (MED) compared with those with doses of <15 equivalents (OR = 1.11; 95% CI, 1.02-1.21); patients who received >45 MED were at the greatest risk (OR = 1.70; 95% CI, 1.49-1.94).
DISCUSSION
The observation that subsequent LTO use occurs more frequently in discharged medical patients than surgical patients is consistent with the findings of Calcaterra et al.1 that among patients with no surgery versus surgery during hospitalization, opioid receipt at discharge resulted in a higher adjusted OR (7.24 for no surgery versus 3.40 for surgery) for chronic opioid use at 1 year. One explanation for this finding may be an artifact of cohort selection in the study design: patients with prior opioid use are excluded from the cohort, and prior use may be more common among surgical patients presenting for elective inpatient surgery for painful conditions. Previous work suggests that opioid use preoperatively is a robust predictor of postoperative use, and rates of LTO use are low among patients without preoperative opioid exposure.6
Demographic characteristics associated with persistent opioid receipt were similar to those previously reported.5,8,9 The inclusion of medication classes indicated in the treatment of mental health or pain conditions (ie, antidepressants, benzodiazepines, muscle relaxants, and nonopioid analgesics) resulted in diagnoses based on ICD-9 codes being no longer associated with LTO use. Severity or activity of illness, preferences regarding pharmacologic or nonpharmacologic treatment and undiagnosed or undocumented pain-comorbid conditions may all contribute to this finding. Future work studying opioid-related outcomes should include variables that reflect pharmacologic management of comorbid diagnoses in the cohort development or analytic design.
The strongest risk factors were potentially modifiable: days’ supply, dose, and concurrent medications. The measures of opioid quantity supplied are associated with subsequent ongoing use and are consistent with recent work based on prescription drug–monitoring data in a single state14 and in a nationally representative sample.15 That this relationship persists following hospital discharge, a scenario in which LTO use is unlikely to be initiated by a provider (who would be expected to subsequently titrate or monitor therapy), further supports the potential to curtail unintended LTO use through judicious early prescribing decisions.
We assessed only opioids that were supplied through a VA pharmacy, which may lead to the misclassification of patients as opioid naive for inclusion and an underestimation of the rate of opioid use following discharge. It is possible that differences in the rates of non-VA pharmacy use differ in medical and surgical populations in a nonrandom way. This study was performed in a large, integrated health system and may not be generalizable outside the VA system, where more discontinuities between hospital and ambulatory care may exist.
CONCLUSION
The initiation of LTO use at discharge is more common in veterans who are discharged from medical than surgical hospitalizations, likely reflecting differences in the patient population, pain conditions, and discharge prescribing decisions. While patient characteristics are associated with LTO use, the strongest associations are with increasing index dose and days’ supply; both represent potentially modifiable prescriber behaviors. These findings support policy changes and other efforts to minimize dose and days supplied when short-term use is intended as a means to address the current opioid epidemic.
Acknowledgments
The work reported here was supported by the Department of Veterans Affairs Office of Academic Affiliations and Office of Research and Development (Dr. Mosher and Dr. Hofmeyer), and Health Services Research and Development Service (HSR&D) through the Comprehensive Access and Delivery Research and Evaluation Center (CIN 13-412) and a Career Development Award (CDA 10-017; Dr. Lund).
Disclosures
The authors report no conflict of interest in regard to this study. The authors had full access to and take full responsibility for the integrity of the data. All analyses were conducted by using SAS version 9.2 (SAS Institute Inc, Cary, NC). This manuscript is not under review elsewhere, and there is no prior publication of the manuscript contents. The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs. The study was approved by the University of Iowa Institutional Review Board and the Iowa City Healthcare System Research and Development Committee.
While patients may be newly exposed to opioids during medical and surgical hospitalization and the prescription of opioids at discharge is common,1-5 prescribers of opioids at discharge may not intend to initiate long-term opioid (LTO) use. By understanding the frequency of progression to LTO use, hospitalists can better balance postdischarge pain treatment and the risk for unintended LTO initiation.
Estimates of LTO use rates following hospital discharge in selected populations1,2,4-6 have varied depending on the population studied and the method of defining LTO use.7 Rates of LTO use following incident opioid prescription have not been directly compared at medical versus surgical discharge or compared with initiation in the ambulatory setting. We present the rates of LTO use following incident opioid exposure at surgical discharge and medical discharge and identify the factors associated with LTO use following surgical and medical discharge.
METHODS
Data Sources
Veterans Health Administration (VHA) data were obtained through the Austin Information Technology Center for fiscal years (FYs) 2003 through 2012 (Austin, Texas). Decision support system national data extracts were used to identify prescription-dispensing events, and inpatient and outpatient medical SAS data sets were used to identify diagnostic codes. The study was approved by the University of Iowa Institutional Review Board and the Iowa City Veterans Affairs (VA) Health Care System Research and Development Committee.
Patients
We included all patients with an outpatient opioid prescription during FY 2011 that was preceded by a 1-year opioid-free period.7 Patients with broadly accepted indications for LTO use (eg, metastatic cancer, palliative care, or opioid-dependence treatment) were excluded.7
Opioid Exposure
We included all outpatient prescription fills for noninjectable dosage forms of butorphanol, fentanyl, hydrocodone, hydromorphone, levorphanol, meperidine, methadone, morphine, oxycodone, oxymorphone, pentazocine, and tramadol. Consistent with the Centers for Disease Control and Prevention and VA/Department of Defense guidelines, LTO use was defined conceptually as regular use for >90 days. Operationalizing this definition to pharmacy refill data was established by using a cabinet supply methodology,7 which allows for the construction of episodes of continuous medication therapy by estimating the medication supply available to a patient for each day during a defined period based on the pattern of observed refills. LTO use was defined as an episode of continuous opioid supply for >90 days and beginning within 30 days of the initial prescription. While some studies have defined LTO use based on onset within 1 year following surgery,5 the requirement for onset within 30 days of initiation was applied to more strongly tie the association of developing LTO use with the discharge event and minimize various forms of bias that are introduced with extended follow-up periods.
Clinical Characteristics
Patients were classified as being medical discharges, surgical discharges, or outpatient initiators. Patients with an opioid index date within 2 days following discharge were designated based on discharge bed section; additionally, if patients had a surgical bed section during hospitalization, they were assigned as surgical discharges. Demographic, diagnosis, and medication exposure variables that were previously associated with LTO use were selected.8,9 Substance use disorder, chronic pain, anxiety disorder, and depressive disorder were based on International Classification of Diseases, 9th Revision (ICD-9) codes in the preceding year. The use of concurrent benzodiazepines, skeletal muscle relaxants, and antidepressants were determined at opioid initiation.10 Rural or urban residence was assigned by using the Rural-Urban Commuting Area Codes system and mapped with the zip code of a veteran’s residence.11
Analysis
Bivariate and multivariable relationships were determined by using logistic regression. The multivariable model considered all pairwise interaction terms between inpatient service (surgery versus medicine) and each of the variables in the model. Statistically significant interaction terms (P < .05) were retained, and all others were omitted from the final model. The main effects for variables that were involved in a significant interaction term were not reported in the final multivariable model; instead, we created fully specified multivariable models for surgery service and medicine service and reported odds ratios (ORs) for the main effects. All analyses were conducted by using SAS version 9.4 (SAS Institute Inc, Cary, North Carolina).
RESULTS
Days’ supply was associated with LTO use in a dose-dependent fashion relative to the reference category of ≤7 days: OR of 1.24 (95% CI, 1.12-1.37) for 8 to 14 days; OR of 1.56 (95% CI, 1.39-1.76) for 15 to 29 days; and OR of 2.59 (95% CI, 2.35-2.86) for 30 days (Table 2). LTO risk was higher among patients with an estimated dose of ≥15 morphine equivalents per day (MED) compared with those with doses of <15 equivalents (OR = 1.11; 95% CI, 1.02-1.21); patients who received >45 MED were at the greatest risk (OR = 1.70; 95% CI, 1.49-1.94).
DISCUSSION
The observation that subsequent LTO use occurs more frequently in discharged medical patients than surgical patients is consistent with the findings of Calcaterra et al.1 that among patients with no surgery versus surgery during hospitalization, opioid receipt at discharge resulted in a higher adjusted OR (7.24 for no surgery versus 3.40 for surgery) for chronic opioid use at 1 year. One explanation for this finding may be an artifact of cohort selection in the study design: patients with prior opioid use are excluded from the cohort, and prior use may be more common among surgical patients presenting for elective inpatient surgery for painful conditions. Previous work suggests that opioid use preoperatively is a robust predictor of postoperative use, and rates of LTO use are low among patients without preoperative opioid exposure.6
Demographic characteristics associated with persistent opioid receipt were similar to those previously reported.5,8,9 The inclusion of medication classes indicated in the treatment of mental health or pain conditions (ie, antidepressants, benzodiazepines, muscle relaxants, and nonopioid analgesics) resulted in diagnoses based on ICD-9 codes being no longer associated with LTO use. Severity or activity of illness, preferences regarding pharmacologic or nonpharmacologic treatment and undiagnosed or undocumented pain-comorbid conditions may all contribute to this finding. Future work studying opioid-related outcomes should include variables that reflect pharmacologic management of comorbid diagnoses in the cohort development or analytic design.
The strongest risk factors were potentially modifiable: days’ supply, dose, and concurrent medications. The measures of opioid quantity supplied are associated with subsequent ongoing use and are consistent with recent work based on prescription drug–monitoring data in a single state14 and in a nationally representative sample.15 That this relationship persists following hospital discharge, a scenario in which LTO use is unlikely to be initiated by a provider (who would be expected to subsequently titrate or monitor therapy), further supports the potential to curtail unintended LTO use through judicious early prescribing decisions.
We assessed only opioids that were supplied through a VA pharmacy, which may lead to the misclassification of patients as opioid naive for inclusion and an underestimation of the rate of opioid use following discharge. It is possible that differences in the rates of non-VA pharmacy use differ in medical and surgical populations in a nonrandom way. This study was performed in a large, integrated health system and may not be generalizable outside the VA system, where more discontinuities between hospital and ambulatory care may exist.
CONCLUSION
The initiation of LTO use at discharge is more common in veterans who are discharged from medical than surgical hospitalizations, likely reflecting differences in the patient population, pain conditions, and discharge prescribing decisions. While patient characteristics are associated with LTO use, the strongest associations are with increasing index dose and days’ supply; both represent potentially modifiable prescriber behaviors. These findings support policy changes and other efforts to minimize dose and days supplied when short-term use is intended as a means to address the current opioid epidemic.
Acknowledgments
The work reported here was supported by the Department of Veterans Affairs Office of Academic Affiliations and Office of Research and Development (Dr. Mosher and Dr. Hofmeyer), and Health Services Research and Development Service (HSR&D) through the Comprehensive Access and Delivery Research and Evaluation Center (CIN 13-412) and a Career Development Award (CDA 10-017; Dr. Lund).
Disclosures
The authors report no conflict of interest in regard to this study. The authors had full access to and take full responsibility for the integrity of the data. All analyses were conducted by using SAS version 9.2 (SAS Institute Inc, Cary, NC). This manuscript is not under review elsewhere, and there is no prior publication of the manuscript contents. The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs. The study was approved by the University of Iowa Institutional Review Board and the Iowa City Healthcare System Research and Development Committee.
1. Calcaterra SL, Yamashita TE, Min SJ, Keniston A, Frank JW, Binswanger IA. Opioid Prescribing at Hospital Discharge Contributes to Chronic Opioid Use. J Gen Intern Med. 2016;31(5):478-485. PubMed
2. Raebel MA, Newcomer SR, Reifler LM, et al. Chronic use of opioid medications before and after bariatric surgery. JAMA. 2013;310(13):1369-1376. PubMed
3. Mosher HJ, Jiang L, Vaughan Sarrazin MS, Cram P, Kaboli PJ, Vander Weg MW. Prevalence and characteristics of hospitalized adults on chronic opioid therapy. J Hosp Med. 2014;9(2):82-87. PubMed
4. Holman JE, Stoddard GJ, Higgins TF. Rates of prescription opiate use before and after injury in patients with orthopaedic trauma and the risk factors for prolonged opiate use. J Bone Joint Surg Am. 2013;95(12):1075-1080.
5. Sun EC, Darnall BD, Baker LC, Mackey S. Incidence of and Risk Factors for Chronic Opioid Use Among Opioid-Naive Patients in the Postoperative Period. JAMA Intern Med. 2016;176(9):1286-1293. PubMed
6. Goesling J, Moser SE, Zaidi B, et al. Trends and predictors of opioid use after total knee and total hip arthroplasty. Pain. 2016;157(6):1259-1265. PubMed
7. Mosher HJ, Richardson KK, Lund BC. The 1-Year Treatment Course of New Opioid Recipients in Veterans Health Administration. Pain Med. 2016. [Epub ahead of print]. PubMed
8. Sullivan MD, Edlund MJ, Fan MY, Devries A, Brennan Braden J, Martin BC. Risks for possible and probable opioid misuse among recipients of chronic opioid therapy in commercial and medicaid insurance plans: The TROUP Study. Pain. 2010;150(2):332-339. PubMed
9. Seal KH, Shi Y, Cohen G, et al. Association of mental health disorders with prescription opioids and high-risk opioid use in US veterans of Iraq and Afghanistan. JAMA. 2012;307(9):940-947. PubMed
10. Mosher HJ, Richardson KK, Lund BC. Sedative Prescriptions Are Common at Opioid Initiation: An Observational Study in the Veterans Health Administration. Pain Med. 2017. [Epub ahead of print]. PubMed
11. Lund BC, Abrams TE, Bernardy NC, Alexander B, Friedman MJ. Benzodiazepine prescribing variation and clinical uncertainty in treating posttraumatic stress disorder. Psychiatr Serv. 2013;64(1):21-27. PubMed
12. Brummett CM, Waljee JF, Goesling J, et al. New Persistent Opioid Use After Minor and Major Surgical Procedures in US Adults. JAMA Surg. 2017;152(6):e170504. PubMed
13. Mellbye A, Karlstad O, Skurtveit S, Borchgrevink PC, Fredheim OM. The duration and course of opioid therapy in patients with chronic non-malignant pain. Acta Anaesthesiol Scand. 2016;60(1):128-137. PubMed
14. Deyo RA, Hallvik SE, Hildebran C, et al. Association Between Initial Opioid Prescribing Patterns and Subsequent Long-Term Use Among Opioid-Naive Patients: A Statewide Retrospective Cohort Study. J Gen Intern Med. 2017;32(1):21-27. PubMed
15. Shah A, Hayes CJ, Martin BC. Factors Influencing Long-Term Opioid Use Among Opioid Naive Patients: An Examination of Initial Prescription Characteristics and Pain Etiologies. J Pain. 2017;18(11):1374-1383. PubMed
1. Calcaterra SL, Yamashita TE, Min SJ, Keniston A, Frank JW, Binswanger IA. Opioid Prescribing at Hospital Discharge Contributes to Chronic Opioid Use. J Gen Intern Med. 2016;31(5):478-485. PubMed
2. Raebel MA, Newcomer SR, Reifler LM, et al. Chronic use of opioid medications before and after bariatric surgery. JAMA. 2013;310(13):1369-1376. PubMed
3. Mosher HJ, Jiang L, Vaughan Sarrazin MS, Cram P, Kaboli PJ, Vander Weg MW. Prevalence and characteristics of hospitalized adults on chronic opioid therapy. J Hosp Med. 2014;9(2):82-87. PubMed
4. Holman JE, Stoddard GJ, Higgins TF. Rates of prescription opiate use before and after injury in patients with orthopaedic trauma and the risk factors for prolonged opiate use. J Bone Joint Surg Am. 2013;95(12):1075-1080.
5. Sun EC, Darnall BD, Baker LC, Mackey S. Incidence of and Risk Factors for Chronic Opioid Use Among Opioid-Naive Patients in the Postoperative Period. JAMA Intern Med. 2016;176(9):1286-1293. PubMed
6. Goesling J, Moser SE, Zaidi B, et al. Trends and predictors of opioid use after total knee and total hip arthroplasty. Pain. 2016;157(6):1259-1265. PubMed
7. Mosher HJ, Richardson KK, Lund BC. The 1-Year Treatment Course of New Opioid Recipients in Veterans Health Administration. Pain Med. 2016. [Epub ahead of print]. PubMed
8. Sullivan MD, Edlund MJ, Fan MY, Devries A, Brennan Braden J, Martin BC. Risks for possible and probable opioid misuse among recipients of chronic opioid therapy in commercial and medicaid insurance plans: The TROUP Study. Pain. 2010;150(2):332-339. PubMed
9. Seal KH, Shi Y, Cohen G, et al. Association of mental health disorders with prescription opioids and high-risk opioid use in US veterans of Iraq and Afghanistan. JAMA. 2012;307(9):940-947. PubMed
10. Mosher HJ, Richardson KK, Lund BC. Sedative Prescriptions Are Common at Opioid Initiation: An Observational Study in the Veterans Health Administration. Pain Med. 2017. [Epub ahead of print]. PubMed
11. Lund BC, Abrams TE, Bernardy NC, Alexander B, Friedman MJ. Benzodiazepine prescribing variation and clinical uncertainty in treating posttraumatic stress disorder. Psychiatr Serv. 2013;64(1):21-27. PubMed
12. Brummett CM, Waljee JF, Goesling J, et al. New Persistent Opioid Use After Minor and Major Surgical Procedures in US Adults. JAMA Surg. 2017;152(6):e170504. PubMed
13. Mellbye A, Karlstad O, Skurtveit S, Borchgrevink PC, Fredheim OM. The duration and course of opioid therapy in patients with chronic non-malignant pain. Acta Anaesthesiol Scand. 2016;60(1):128-137. PubMed
14. Deyo RA, Hallvik SE, Hildebran C, et al. Association Between Initial Opioid Prescribing Patterns and Subsequent Long-Term Use Among Opioid-Naive Patients: A Statewide Retrospective Cohort Study. J Gen Intern Med. 2017;32(1):21-27. PubMed
15. Shah A, Hayes CJ, Martin BC. Factors Influencing Long-Term Opioid Use Among Opioid Naive Patients: An Examination of Initial Prescription Characteristics and Pain Etiologies. J Pain. 2017;18(11):1374-1383. PubMed
Engaging skeptical parents
While every day seems to bring extraordinary new advances in science – robotic surgery, individually targeted medications, and even gene therapy – there are many people who currently approach the science of medicine with skepticism.
While it is the right of legally competent adults in a free society to chose how best to care for their own health, to explore holistic or alternative therapies, or avoid medicine altogether, it is more complex when they are skeptical of accepted medical practice in managing the health of their children. For those parents who trust you enough to bring their children to you for care but remain skeptical of vaccines or other treatments, you have an opportunity to work with that trust and engage in a discussion so that they might reconsider their position on valuable and even life-saving treatments for their children.
In each of these cases, launching into an enthusiastic explanation of the advanced statistics that underpin your recommendation is unlikely to bridge the gap. Instead, you want to start with these parents by being curious. Resist the urge to tell, and listen instead. What is their understanding of the problem you are treating or preventing? What have they heard or read about the treatment or test in question? What do they most fear is going to happen to their child if they do or do not accept your recommendation? Are there specific events (with their child or with the health care system) that have informed this fear?
Respectfully listening to their experiences, thoughts, and feelings goes a long way toward building a trusting alliance. It can help overcome feelings of distrust or defensiveness around authority figures. And it models the thoughtful, respectful give and take that are essential to a healthy collaboration between pediatrician and parents.

Once you have information about what they think and some about how they think and make decisions, you then can offer your perspective. “You are the expert on your child, what I bring to this equation is experience with (this problem) and with assessing the scientific evidence that guides treatments in medicine. It is true that treatments often change as we learn more, but here is what the evidence currently supports.”
After learning something about how they think, you might offer more data or more warm acknowledgment of how difficult it can be to make medical decisions for your children with imperfect information. Be humble while also being accurate about your level of confidence in a recommendation. Humility is important because it is easy for parents to feel insecure and condescended to. You understand their greatest fear, now let them know what your greatest worry is for their child should they forgo a recommended treatment. Explaining all of this with humility and warmth makes it more likely that the parents will take in the facts you are trying to share with them and not be derailed by suspicion, defensiveness, or insecurity.
Make building an alliance with the parents your top priority. This does not mean that you do not offer your best recommendation for their child. Rather, it means that, if they still decline recommended treatment, you treat them with respect and invest your time in explaining what they should be watching or monitoring their child for without recommended treatment. Building trust is a long game. If you patiently stick with parents even when it’s not easy, they may be ready to trust you with a subsequent decision when the stakes are even higher.

Of course, all this thoughtful communication takes a lot of time! You may learn to block off more time for certain families. It also can be helpful to have these conversations as a team. If you and your nurse or social worker can meet with parents together, then some of the listening and learning can be done by the nurse or social worker alone, so that everyone’s time might be managed more efficiently. And managing skeptical parents as a team also can help to prevent frustration or burnout. It will not always succeed, but in some cases, your investment will pay off in a trusting alliance, mutual respect, and healthy patients.
Dr. Swick is an attending psychiatrist in the division of child psychiatry at Massachusetts General Hospital, Boston, and director of the Parenting at a Challenging Time (PACT) Program at the Vernon Cancer Center at Newton Wellesley Hospital, also in Boston. Dr. Jellinek is professor emeritus of psychiatry and pediatrics at Harvard Medical School, Boston. Email them at [email protected].
While every day seems to bring extraordinary new advances in science – robotic surgery, individually targeted medications, and even gene therapy – there are many people who currently approach the science of medicine with skepticism.
While it is the right of legally competent adults in a free society to chose how best to care for their own health, to explore holistic or alternative therapies, or avoid medicine altogether, it is more complex when they are skeptical of accepted medical practice in managing the health of their children. For those parents who trust you enough to bring their children to you for care but remain skeptical of vaccines or other treatments, you have an opportunity to work with that trust and engage in a discussion so that they might reconsider their position on valuable and even life-saving treatments for their children.
In each of these cases, launching into an enthusiastic explanation of the advanced statistics that underpin your recommendation is unlikely to bridge the gap. Instead, you want to start with these parents by being curious. Resist the urge to tell, and listen instead. What is their understanding of the problem you are treating or preventing? What have they heard or read about the treatment or test in question? What do they most fear is going to happen to their child if they do or do not accept your recommendation? Are there specific events (with their child or with the health care system) that have informed this fear?
Respectfully listening to their experiences, thoughts, and feelings goes a long way toward building a trusting alliance. It can help overcome feelings of distrust or defensiveness around authority figures. And it models the thoughtful, respectful give and take that are essential to a healthy collaboration between pediatrician and parents.
Once you have information about what they think and some about how they think and make decisions, you then can offer your perspective. “You are the expert on your child, what I bring to this equation is experience with (this problem) and with assessing the scientific evidence that guides treatments in medicine. It is true that treatments often change as we learn more, but here is what the evidence currently supports.”
After learning something about how they think, you might offer more data or more warm acknowledgment of how difficult it can be to make medical decisions for your children with imperfect information. Be humble while also being accurate about your level of confidence in a recommendation. Humility is important because it is easy for parents to feel insecure and condescended to. You understand their greatest fear, now let them know what your greatest worry is for their child should they forgo a recommended treatment. Explaining all of this with humility and warmth makes it more likely that the parents will take in the facts you are trying to share with them and not be derailed by suspicion, defensiveness, or insecurity.
Make building an alliance with the parents your top priority. This does not mean that you do not offer your best recommendation for their child. Rather, it means that, if they still decline recommended treatment, you treat them with respect and invest your time in explaining what they should be watching or monitoring their child for without recommended treatment. Building trust is a long game. If you patiently stick with parents even when it’s not easy, they may be ready to trust you with a subsequent decision when the stakes are even higher.
Of course, all this thoughtful communication takes a lot of time! You may learn to block off more time for certain families. It also can be helpful to have these conversations as a team. If you and your nurse or social worker can meet with parents together, then some of the listening and learning can be done by the nurse or social worker alone, so that everyone’s time might be managed more efficiently. And managing skeptical parents as a team also can help to prevent frustration or burnout. It will not always succeed, but in some cases, your investment will pay off in a trusting alliance, mutual respect, and healthy patients.
Dr. Swick is an attending psychiatrist in the division of child psychiatry at Massachusetts General Hospital, Boston, and director of the Parenting at a Challenging Time (PACT) Program at the Vernon Cancer Center at Newton Wellesley Hospital, also in Boston. Dr. Jellinek is professor emeritus of psychiatry and pediatrics at Harvard Medical School, Boston. Email them at [email protected].
While every day seems to bring extraordinary new advances in science – robotic surgery, individually targeted medications, and even gene therapy – there are many people who currently approach the science of medicine with skepticism.
While it is the right of legally competent adults in a free society to chose how best to care for their own health, to explore holistic or alternative therapies, or avoid medicine altogether, it is more complex when they are skeptical of accepted medical practice in managing the health of their children. For those parents who trust you enough to bring their children to you for care but remain skeptical of vaccines or other treatments, you have an opportunity to work with that trust and engage in a discussion so that they might reconsider their position on valuable and even life-saving treatments for their children.
In each of these cases, launching into an enthusiastic explanation of the advanced statistics that underpin your recommendation is unlikely to bridge the gap. Instead, you want to start with these parents by being curious. Resist the urge to tell, and listen instead. What is their understanding of the problem you are treating or preventing? What have they heard or read about the treatment or test in question? What do they most fear is going to happen to their child if they do or do not accept your recommendation? Are there specific events (with their child or with the health care system) that have informed this fear?
Respectfully listening to their experiences, thoughts, and feelings goes a long way toward building a trusting alliance. It can help overcome feelings of distrust or defensiveness around authority figures. And it models the thoughtful, respectful give and take that are essential to a healthy collaboration between pediatrician and parents.
Once you have information about what they think and some about how they think and make decisions, you then can offer your perspective. “You are the expert on your child, what I bring to this equation is experience with (this problem) and with assessing the scientific evidence that guides treatments in medicine. It is true that treatments often change as we learn more, but here is what the evidence currently supports.”
After learning something about how they think, you might offer more data or more warm acknowledgment of how difficult it can be to make medical decisions for your children with imperfect information. Be humble while also being accurate about your level of confidence in a recommendation. Humility is important because it is easy for parents to feel insecure and condescended to. You understand their greatest fear, now let them know what your greatest worry is for their child should they forgo a recommended treatment. Explaining all of this with humility and warmth makes it more likely that the parents will take in the facts you are trying to share with them and not be derailed by suspicion, defensiveness, or insecurity.
Make building an alliance with the parents your top priority. This does not mean that you do not offer your best recommendation for their child. Rather, it means that, if they still decline recommended treatment, you treat them with respect and invest your time in explaining what they should be watching or monitoring their child for without recommended treatment. Building trust is a long game. If you patiently stick with parents even when it’s not easy, they may be ready to trust you with a subsequent decision when the stakes are even higher.
Of course, all this thoughtful communication takes a lot of time! You may learn to block off more time for certain families. It also can be helpful to have these conversations as a team. If you and your nurse or social worker can meet with parents together, then some of the listening and learning can be done by the nurse or social worker alone, so that everyone’s time might be managed more efficiently. And managing skeptical parents as a team also can help to prevent frustration or burnout. It will not always succeed, but in some cases, your investment will pay off in a trusting alliance, mutual respect, and healthy patients.
Dr. Swick is an attending psychiatrist in the division of child psychiatry at Massachusetts General Hospital, Boston, and director of the Parenting at a Challenging Time (PACT) Program at the Vernon Cancer Center at Newton Wellesley Hospital, also in Boston. Dr. Jellinek is professor emeritus of psychiatry and pediatrics at Harvard Medical School, Boston. Email them at [email protected].
Oral SGLT-2 inhibitor reduced liver fat in diabetics with NAFLD
CHICAGO – and improved ALT in patients with nonalcoholic fatty liver disease (NAFLD) and type 2 diabetes mellitus, according to a study presented at the annual meeting of the Endocrine Society.
As insulin resistance is the mechanism for NAFLD development, this new addition to the list of drugs on offer to patients with diabetes could help decrease the chance of developing metabolic syndrome and cardiovascular disease.
“SGLT-2 inhibitors are newer antidiabetic agents that reduce blood glucose by promoting urinary glucose excretion,” said presenter Mohammad Shafi Kuchay, MD, DM, an endocrinologist at Medanta The Medicity, Gurugram, India. “NAFLD, which also increases the risk of type 2 diabetes, often responds to strategies that improve hyperglycemia.”
Dr. Kuchay and fellow investigators conducted a small, 20-week randomized controlled trial of 42 patients with type 2 diabetes and NAFLD.
Patients in the test group were mostly male and on average 50 years old, with baseline AST, ALT, and gamma-glutamyltransferase scores of 44.6 U/L, 64.3 U/L, and 65.8 U/L, respectively. Those randomized to the control group had similar characteristics.
After adding 10 mg of empagliflozin to their diabetes regimen, liver fat density in test patients decreased from 16.2% to 11.3% (P less than or equal to .0001). The drop stands in sharp contrast to the control group, which decreased from 16.4% to 15.5% (P = .054). Measurement of liver fat density was made by MRI-derived proton density fat fraction (MRI-PDFF). This method has higher sensitivity for detecting changes in liver fat, compared with histology, explained Dr. Kuchay.
When broken down by individual liver fat, 25% of patients in the control group increased in liver fat, 50% had no significant change, and 25% decreased in liver fat, according to Dr. Kuchay.
In comparison, 77% of patients in the empagliflozin group had a decrease in liver fat, 23% had no change, and no patients saw an increase in liver fat.
When comparing levels of hemoglobin A1c between the two groups, both had a similarly significant reduction of around 2%, which Dr. Kuchay attributes to deliberate intervention by investigators.
Further studies will need to be conducted regarding the long-term effects of this treatment; however, using SGLT-2 to reduce liver fat could be a boon to preventing more serious liver diseases, concluded Dr. Kuchay.
“There are studies in which liver fat reduction led to improvement in inflammation and fibrosis,” said Dr. Kuchay in response to a question from the audience. “Because liver fat accumulation is the first inhibitor in the pathogenesis of more severe forms of liver disease, reducing liver fat should help improve patient outcomes.”
Dr. Kuchay reported no relevant financial disclosures.
Source: M. Kuchay et al. ENDO 2018, Abstract OR27-2.
CHICAGO – and improved ALT in patients with nonalcoholic fatty liver disease (NAFLD) and type 2 diabetes mellitus, according to a study presented at the annual meeting of the Endocrine Society.
As insulin resistance is the mechanism for NAFLD development, this new addition to the list of drugs on offer to patients with diabetes could help decrease the chance of developing metabolic syndrome and cardiovascular disease.
“SGLT-2 inhibitors are newer antidiabetic agents that reduce blood glucose by promoting urinary glucose excretion,” said presenter Mohammad Shafi Kuchay, MD, DM, an endocrinologist at Medanta The Medicity, Gurugram, India. “NAFLD, which also increases the risk of type 2 diabetes, often responds to strategies that improve hyperglycemia.”
Dr. Kuchay and fellow investigators conducted a small, 20-week randomized controlled trial of 42 patients with type 2 diabetes and NAFLD.
Patients in the test group were mostly male and on average 50 years old, with baseline AST, ALT, and gamma-glutamyltransferase scores of 44.6 U/L, 64.3 U/L, and 65.8 U/L, respectively. Those randomized to the control group had similar characteristics.
After adding 10 mg of empagliflozin to their diabetes regimen, liver fat density in test patients decreased from 16.2% to 11.3% (P less than or equal to .0001). The drop stands in sharp contrast to the control group, which decreased from 16.4% to 15.5% (P = .054). Measurement of liver fat density was made by MRI-derived proton density fat fraction (MRI-PDFF). This method has higher sensitivity for detecting changes in liver fat, compared with histology, explained Dr. Kuchay.
When broken down by individual liver fat, 25% of patients in the control group increased in liver fat, 50% had no significant change, and 25% decreased in liver fat, according to Dr. Kuchay.
In comparison, 77% of patients in the empagliflozin group had a decrease in liver fat, 23% had no change, and no patients saw an increase in liver fat.
When comparing levels of hemoglobin A1c between the two groups, both had a similarly significant reduction of around 2%, which Dr. Kuchay attributes to deliberate intervention by investigators.
Further studies will need to be conducted regarding the long-term effects of this treatment; however, using SGLT-2 to reduce liver fat could be a boon to preventing more serious liver diseases, concluded Dr. Kuchay.
“There are studies in which liver fat reduction led to improvement in inflammation and fibrosis,” said Dr. Kuchay in response to a question from the audience. “Because liver fat accumulation is the first inhibitor in the pathogenesis of more severe forms of liver disease, reducing liver fat should help improve patient outcomes.”
Dr. Kuchay reported no relevant financial disclosures.
Source: M. Kuchay et al. ENDO 2018, Abstract OR27-2.
CHICAGO – and improved ALT in patients with nonalcoholic fatty liver disease (NAFLD) and type 2 diabetes mellitus, according to a study presented at the annual meeting of the Endocrine Society.
As insulin resistance is the mechanism for NAFLD development, this new addition to the list of drugs on offer to patients with diabetes could help decrease the chance of developing metabolic syndrome and cardiovascular disease.
“SGLT-2 inhibitors are newer antidiabetic agents that reduce blood glucose by promoting urinary glucose excretion,” said presenter Mohammad Shafi Kuchay, MD, DM, an endocrinologist at Medanta The Medicity, Gurugram, India. “NAFLD, which also increases the risk of type 2 diabetes, often responds to strategies that improve hyperglycemia.”
Dr. Kuchay and fellow investigators conducted a small, 20-week randomized controlled trial of 42 patients with type 2 diabetes and NAFLD.
Patients in the test group were mostly male and on average 50 years old, with baseline AST, ALT, and gamma-glutamyltransferase scores of 44.6 U/L, 64.3 U/L, and 65.8 U/L, respectively. Those randomized to the control group had similar characteristics.
After adding 10 mg of empagliflozin to their diabetes regimen, liver fat density in test patients decreased from 16.2% to 11.3% (P less than or equal to .0001). The drop stands in sharp contrast to the control group, which decreased from 16.4% to 15.5% (P = .054). Measurement of liver fat density was made by MRI-derived proton density fat fraction (MRI-PDFF). This method has higher sensitivity for detecting changes in liver fat, compared with histology, explained Dr. Kuchay.
When broken down by individual liver fat, 25% of patients in the control group increased in liver fat, 50% had no significant change, and 25% decreased in liver fat, according to Dr. Kuchay.
In comparison, 77% of patients in the empagliflozin group had a decrease in liver fat, 23% had no change, and no patients saw an increase in liver fat.
When comparing levels of hemoglobin A1c between the two groups, both had a similarly significant reduction of around 2%, which Dr. Kuchay attributes to deliberate intervention by investigators.
Further studies will need to be conducted regarding the long-term effects of this treatment; however, using SGLT-2 to reduce liver fat could be a boon to preventing more serious liver diseases, concluded Dr. Kuchay.
“There are studies in which liver fat reduction led to improvement in inflammation and fibrosis,” said Dr. Kuchay in response to a question from the audience. “Because liver fat accumulation is the first inhibitor in the pathogenesis of more severe forms of liver disease, reducing liver fat should help improve patient outcomes.”
Dr. Kuchay reported no relevant financial disclosures.
Source: M. Kuchay et al. ENDO 2018, Abstract OR27-2.
REPORTING FROM ENDO 2018
Key clinical point: Empagliflozin reduced liver fat in patients with NAFLD and type 2 diabetes.
Major finding: MRI-PDFF in test patients decreased from 16.2% to 11.3% (P less than or equal to .0001), compared with control patients, who saw a decrease from 19.4% to 15.5% (P = .057)
Data source: Prospective, randomized, controlled trial of 60 patients with type 2 diabetes and NAFLD.
Disclosures: Dr. Kuchay reported no relevant financial disclosures.
Source: Kuchay M et al. ENDO 2018, Abstract OR27-2.
The Use of Bolus-Dose Vasopressors in the Emergency Department
The use of bolus-dose vasopressors in anesthesiology and other areas of critical care medicine is well known. This common medical intervention, however, is not often employed in emergency medicine (EM). Bolus-dose vasopressors are defined as the administration of small bolus doses of vasopressor agents, such as epinephrine or phenylephrine, to patients with compromised perfusion who continue to have a pulse (ie, these patients are not in cardiac arrest). This intervention is considered as a temporizing measure for transient hypotension or as a bridge to more definitive therapy.
Clinical Application
Bolus-dose vasopressive therapy is also referred to as push-dose pressor (PDP) therapy—a term coined by Weingart.1-3 Theoretically, any vasopressor could be used in a mini-dose, bolus fashion, though in current clinical practice, anesthesiologists primarily employ ephedrine, epinephrine, and phenylephrine. Two of these agents are likely more appropriate for the ED, including epinephrine and phenylephrine. Both of these agents have a short half-life and therefore an abbreviated period of effect. In addition, dosing and related administration of epinephrine and phenylephrine is relatively straightforward. Moreover, most emergency physicians and nurses are quite familiar with both agents.
With respect to ephedrine, due to its longer half-life, complex dosing regimen, and associated higher-incidence of cardiovascular (CV) complications, its use is likely not appropriate in the ED as a bolus-dose vasopressor.
Epinephrine and Phenylephrine
Epinephrine is a potent sympathomimetic agent with alpha- and beta-receptor activity. In addition to its vasopressor effects, epinephrine is also an inotropic and chronotropic agent, increasing cardiac output, heart rate (HR), and systemic vascular resistance, which can markedly improve perfusion. Epinephrine also can be given to patients with hypoperfusion and/or shock due to low-cardiac output with or without vasodilation, lacking significant tachycardia.
Phenylephrine is a pure alpha agonist and therefore does not appreciably affect cardiac output and HR, but does significantly increase systemic vascular resistance and thus systemic perfusion. Phenylephrine can be used to treat patients with hypoperfusion and/or shock states due to vasodilation with coexistent, significant tachycardia.
Preparation and Administration
The preparation and dosing of push-dose epinephrine and phenylephrine are not particularly complex. Many clinicians recommend the pre-mixed, manufacturer-prepared agents for PDP therapy. These premixed formulations not only facilitate administration, but also reduce the chance of a preparation error that can result in incorrect dosing.3-5 If pre-mixed formulations are not available, clinicians can readily prepare epinephrine and phenylephrine for PDP use.
Push-Dose Epinephrine. Clinicians can prepare epinephrine for push-dose administration as follows:1-3
- Obtain 1 mL of epinephrine 1:10,000 (ie, 0.1 mg/mL or 100 mcg/mL);
- Obtain a 10 mL syringe of normal saline and remove 1 mL;
- Inject the 1 mL of epinephrine 1:10,000 (100 mcg/mL) into this syringe containing 9 mL of normal saline; and
- Result: 10 mL of epinephrine (10 mcg/mL), with each 1 mL of this solution containing 10 mcg of epinephrine.
Administration of push-dose epinephrine (10 mcg/mL) produces effect within 1 minute of use with a duration of approximately 5 to 10 minutes. Dosing at this concentration ranges from 0.5 to 2.0 mL every 2 to 5 minutes, delivering 5 to 20 mcg.1-3Push-Dose Phenylephrine. To prepare phenylephrine for push-dose administration, clinicians may use the following approach:1-3
- Obtain 1 mL of phenylephrine (10 mg/mL concentration);
- Inject this 1 mL of phenylephrine (10 mg/mL) into a 100 mL bag of normal saline; and
- Result: 100 mL of phenylephrine (100 mcg/mL), with each 1 mL of this solution containing 100 mcg of phenylephrine.
Administration of push-dose phenylephrine (100 mcg/mL) produces effect within 1 minute of use with a duration of approximately 10 to 20 minutes. Dosing at this concentration ranges from 0.5 to 2.0 mL every 2 to 5 minutes, delivering 50 to 200 mcg.1-3Alternative Push-Dose Preparations for Phenylephrine. Two other methods of preparing phenylephrine for bolus-dose administration include the following: (1) the addition of phenylephrine 20 mg to a bag of 250 cc of normal saline, resulting in an 80 mcg/mL concentration; and/or (2) phenylephrine (20 mg) is commercially available for continuous infusion in a 250 mL bag of normal saline, yielding the same concentration of 80 mcg/mL; in either case, medication can be drawn up and administered. Dosing at this concentration ranges from 0.5 to 2.5 mL every 2 to 5 minutes, delivering 40 to 200 mcg. Lastly, phenylephrine is also commercially available in pre-made mixtures, specifically manufactured for bolus-dose therapy.
Indications
Both epinephrine and phenylephrine can be considered in the management of significant transient or sustained hypoperfusion. Although the definition of significant hypotension is complex, Brunauer et al6 have suggested that a mean arterial pressure (MAP) of approximately 35 mm Hg is associated with a significant risk of CV collapse. Of course, a MAP of 40 to 50 mm Hg is also very concerning clinically, with significant risk of deterioration and CV collapse.
Procedural events, such as conscious sedation or rapid sequence intubation (RSI), can produce significant hypotension; PDP can rapidly correct hypotension. In other clinical scenarios in which sustained hypotension is likely and not transient (eg, sepsis with shock), PDP can be used as a bridge to definitive care (eg, volume replacement, continuous vasopressor infusion). It is important to note, however, that PDP administration must occur in conjunction with or after the patient has received other appropriate therapies such as a normal saline bolus and continuous vasopressor infusions. Push-dose pressors are not a replacement for these proven interventions, but rather are an important augmentation to these therapies.
Emergency Medicine Literature
As previously noted, the literature base describing and supporting the clinical use of PDP in EM is extremely limited. The few articles that comprise this literature base address significant hypotension in periendotracheal intubation intervention, post-return of spontaneous circulation (ROSC) management, and shock management with preload augmentation.7-9In addition, there are several articles in the literature that address safety concerns surrounding the use of PDP in the ED.4,5
Panchal et al10 investigated the use of phenylephrine in hypotensive patients undergoing RSI-assisted endotracheal intubation. The authors performed a 1-year retrospective review of hypotensive patients managed with endotracheal intubation for a range of clinical conditions that required clinical care intervention. In this study, 20 of the 119 patients received phenylephrine in the peri-intubation period. A range of clinical conditions requiring critical care intervention were encountered; in addition, almost three-quarters of these patients were receiving at least one other vasopressor infusion. Further differences were seen in the timing of PDP administration. In those patients receiving bolus-dose phenylephrine, blood pressure (BP) improved without change in HR. Panchal et al10 concluded that while push-dose phenylephrine improved hemodynamic status, there was significant variation among clinicians regarding dosing, timing of use, and overall clinical situation The significant variation in PDP management in this study was noted to be a potential source of medical error, thus increasing the chance of adverse clinical event.
Push-dose pressor therapy can be employed for significant hypotension while more definitive therapy is being readied and applied. For instance, patients with significant hypotension requiring continuous vasopressor infusion can be managed with PDP while appropriate venous access is established, intravenous fluids are administered, and medications are prepared. The immediate period after resuscitation from cardiac arrest can be complicated by shock of many types. In fact, hypotension following ROSC in the cardiac arrest patient is not uncommon and has been identified as a risk issue associated with poor outcome. Prompt treatment of this altered perfusion may improve outcome. Gottlieb8 described three patients with ROSC after cardiac arrest. All three patients experienced significant, sustained hypotension with systolic blood pressure reading in the 50 to 60 mm Hg range; bolus-dose epinephrine was administered with significant improvement in the hemodynamic status while central venous access was established.
In a related clinical scenario, Schwartz et al9 considered the impact of PDP on central venous line (CVL) placement with continuous vasopressor infusion. In this ED study, although patients experienced an increase in BP, this impact was transient with approximately half of these individuals ultimately requiring CVL. In addition, serious adverse effect was noted more commonly in the phenylephrine-treated patients with “reactive” hypertension and ventricular tachycardia occurring in study patients.
Patient-Safety Considerations
In addition to the limited literature base supporting PDP use in the ED, another major significant issue focuses on safety concerns and adverse effects. Extremely limited data is available describing adverse events related to ED-administered PDP. Extrapolating from other EM and critical care administrations of peripheral epinephrine, both local and systemic adverse effects have been reported.11,12 The range of adverse events noted in these studies are considerable, including local skin and soft-tissue injury (necrosis), end-organ tissue ischemia (eg, digits, tip of nose), acute hypertension, cardiac ischemic events, and left ventricular (LV) dysfunction.11,12
When comparing peripheral infusion with central infusion, the risk of extravasation with resultant local tissue injury is markedly greater with peripheral vasopressor administration. In a systematic review of this issue, Loubani and Green11 noted that such local adverse events were much more commonly associated with peripheral administration.
In another report of vasopressor use in the ED, Kanwar et al12 described apparent confusion with epinephrine dosing and route of administration, resulting in very significant, systemic CV maladies, including severe elevations in BP, acute LV dysfunction, and chest pain associated with ST segment elevation.
It must be stressed that the publications by Loubani and Green11 and Kanwar et al12 described peripheral vasopressor administration: neither study included PDP therapy. Therefore, as previously noted, the aforementioned statements are extrapolated from when applied to PDP strategy.
Acquisto et al4 describe several errors in medication administration of PDP in the ED and other critical care areas of the hospital. In this report, all treating physicians were present at the patients’ bedside, either administering the medication or directly supervising its use. Agents involved included epinephrine and phenylephrine, delivered at exceedingly high doses. In their study, the authors noted several issues which they believe contributed to medication errors, including heterogeneity of pathology treated in these patients, apparent “earlier-than-appropriate” use of vasopressors (ie, prior to giving an appropriate fluid bolus), and medication preparation at the bedside by clinicians who may not possess the experience and training to mix these agents.
From a patient-safety perspective, Holden et al5 noted the potential for dosing error with significant adverse medical consequence related to PDP, as well as several contributing issues. First, they highlight the lack of a solid literature base to support administration of PDP in the ED and the development of decision-making guidelines for use in the ED. They also observed an inconsistency in approach to patient selection, medication choice, agent preparation, dosing, and other therapies. As seen in the Acquisto et al4 report, the patient-care scenarios are high risk and quite dynamic.
Conclusion
Bolus-dose vasopressor therapy is a potentially very useful treatment in the ED and other emergency/critical care settings. However, despite its benefits in treating patients in shock or with hypoperfusion, PDP is not widely used in EM due to the lack of studies, reviews, and guidelines in the literature to support its use in the ED. Such a literature base is required to provide an appropriate, safe means of patient selection, medication choice, dosing, and administration. Continued educational and research efforts are needed to more fully explore the use of PDP therapy in the ED.
When used correctly and appropriately, PDP has promise to be an important aid in the management of shock in the ED. Although bolus-dose therapy is appropriate for select clinical scenarios involving significant shock states which have the potential for progression to complete CV collapse without timely therapy, it is an adjunct to, not a replacement for commonly employed and medically indicated therapies such as crystalloid bolus or continuous vasopressor infusions.
1. Weingart S. EMCrit podcast 6—push-dose pressors. EMCrit RACC Web site. July 2009. https://emcrit.org/racc/bolus-dose-pressors. Accessed March 12, 2018.
2. Weingart S. EMCrit podcast 205—push-dose pressors update. EMCrit RACC Web site. August 2017. https://emcrit.org/racc/push-dose-pressor-update/. March 12, 2018.
3. Weingart S. Push-dose pressors for immediate blood pressure control. Clin Exp Emerg Med. 2015;2(2):131-132. doi:10.15441/ceem.15.010.
4. Acquisto NM, Bodkin RP, Johnstone C. Medication errors with push dose pressors in the emergency department and intensive care units. Am J Emerg Med. 2017;35(12):1964-1965. doi:10.1016/j.ajem.2017.06.013.
5. Holden D, Ramich J, Timm E, Pauze D, Lesar T. Safety considerations and guideline-based safe use recommendations for “bolus-dose” vasopressors in the emergency department. Ann Emerg Med. 2018;71(1):83-92. doi:10.1016/j.annemergmed.2017.04.021.
6. Brunauer A, Koköfer A, Bataar O, Gradwohl-Matis I, Dankl D, Dünser MW. The arterial blood pressure associated with terminal cardiovascular collapse in critically ill patients: a retrospective cohort study. Crit Care. 2014;18(6):719. doi:10.1186/s13054-014-0719-2.
7. Panchal AR, Satyanarayan A, Bahadir JD, Hays D, Mosier J. Efficacy of bolus-dose phenylephrine for peri-intubation hypotension. J Emerg Med. 2015;49(4):488-494. doi:10.1016/j.jemermed.2015.04.033.
8. Gottlieb M. Bolus dose of epinephrine for refractory post-arrest hypotension. Can J Emerg Med. 2017;409:1-5. doi:10.1017/cem.2016.409.
9. Schwartz MB, Ferreira JA, Aaronson PM. The impact of push-dose phenylephrine use on subsequent preload expansion in the ED setting. Am J Emerg Med. 2016;34(12):2419-2422. doi:10.1016/j.ajem.2016.09.041.
10. Panchal AR, Satyanarayan A, Bahadir JD, Hays D, Mosier J. Efficacy of bolus-dose phenylephrine for peri-intubation hypotension. J Emerg Med. 2015;49(4):488-494. doi:10.1016/j.jemermed.2015.04.033.
11. Loubani OM, Green RS. A systematic review of extravasation and local tissue injury from administration of vasopressors through peripheral intravenous catheters and central venous catheters. J Crit Care. 2015;30:653.e9-e17.
12. Kanwar M, Irvin CB, Frank JJ, et al. Confusion about epinephrine dosing leading to iatrogenic overdose: A life-threatening problem with a potential solution. Ann Emerg Med. 2010;55:341-344.
The use of bolus-dose vasopressors in anesthesiology and other areas of critical care medicine is well known. This common medical intervention, however, is not often employed in emergency medicine (EM). Bolus-dose vasopressors are defined as the administration of small bolus doses of vasopressor agents, such as epinephrine or phenylephrine, to patients with compromised perfusion who continue to have a pulse (ie, these patients are not in cardiac arrest). This intervention is considered as a temporizing measure for transient hypotension or as a bridge to more definitive therapy.
Clinical Application
Bolus-dose vasopressive therapy is also referred to as push-dose pressor (PDP) therapy—a term coined by Weingart.1-3 Theoretically, any vasopressor could be used in a mini-dose, bolus fashion, though in current clinical practice, anesthesiologists primarily employ ephedrine, epinephrine, and phenylephrine. Two of these agents are likely more appropriate for the ED, including epinephrine and phenylephrine. Both of these agents have a short half-life and therefore an abbreviated period of effect. In addition, dosing and related administration of epinephrine and phenylephrine is relatively straightforward. Moreover, most emergency physicians and nurses are quite familiar with both agents.
With respect to ephedrine, due to its longer half-life, complex dosing regimen, and associated higher-incidence of cardiovascular (CV) complications, its use is likely not appropriate in the ED as a bolus-dose vasopressor.
Epinephrine and Phenylephrine
Epinephrine is a potent sympathomimetic agent with alpha- and beta-receptor activity. In addition to its vasopressor effects, epinephrine is also an inotropic and chronotropic agent, increasing cardiac output, heart rate (HR), and systemic vascular resistance, which can markedly improve perfusion. Epinephrine also can be given to patients with hypoperfusion and/or shock due to low-cardiac output with or without vasodilation, lacking significant tachycardia.
Phenylephrine is a pure alpha agonist and therefore does not appreciably affect cardiac output and HR, but does significantly increase systemic vascular resistance and thus systemic perfusion. Phenylephrine can be used to treat patients with hypoperfusion and/or shock states due to vasodilation with coexistent, significant tachycardia.
Preparation and Administration
The preparation and dosing of push-dose epinephrine and phenylephrine are not particularly complex. Many clinicians recommend the pre-mixed, manufacturer-prepared agents for PDP therapy. These premixed formulations not only facilitate administration, but also reduce the chance of a preparation error that can result in incorrect dosing.3-5 If pre-mixed formulations are not available, clinicians can readily prepare epinephrine and phenylephrine for PDP use.
Push-Dose Epinephrine. Clinicians can prepare epinephrine for push-dose administration as follows:1-3
- Obtain 1 mL of epinephrine 1:10,000 (ie, 0.1 mg/mL or 100 mcg/mL);
- Obtain a 10 mL syringe of normal saline and remove 1 mL;
- Inject the 1 mL of epinephrine 1:10,000 (100 mcg/mL) into this syringe containing 9 mL of normal saline; and
- Result: 10 mL of epinephrine (10 mcg/mL), with each 1 mL of this solution containing 10 mcg of epinephrine.
Administration of push-dose epinephrine (10 mcg/mL) produces effect within 1 minute of use with a duration of approximately 5 to 10 minutes. Dosing at this concentration ranges from 0.5 to 2.0 mL every 2 to 5 minutes, delivering 5 to 20 mcg.1-3Push-Dose Phenylephrine. To prepare phenylephrine for push-dose administration, clinicians may use the following approach:1-3
- Obtain 1 mL of phenylephrine (10 mg/mL concentration);
- Inject this 1 mL of phenylephrine (10 mg/mL) into a 100 mL bag of normal saline; and
- Result: 100 mL of phenylephrine (100 mcg/mL), with each 1 mL of this solution containing 100 mcg of phenylephrine.
Administration of push-dose phenylephrine (100 mcg/mL) produces effect within 1 minute of use with a duration of approximately 10 to 20 minutes. Dosing at this concentration ranges from 0.5 to 2.0 mL every 2 to 5 minutes, delivering 50 to 200 mcg.1-3Alternative Push-Dose Preparations for Phenylephrine. Two other methods of preparing phenylephrine for bolus-dose administration include the following: (1) the addition of phenylephrine 20 mg to a bag of 250 cc of normal saline, resulting in an 80 mcg/mL concentration; and/or (2) phenylephrine (20 mg) is commercially available for continuous infusion in a 250 mL bag of normal saline, yielding the same concentration of 80 mcg/mL; in either case, medication can be drawn up and administered. Dosing at this concentration ranges from 0.5 to 2.5 mL every 2 to 5 minutes, delivering 40 to 200 mcg. Lastly, phenylephrine is also commercially available in pre-made mixtures, specifically manufactured for bolus-dose therapy.
Indications
Both epinephrine and phenylephrine can be considered in the management of significant transient or sustained hypoperfusion. Although the definition of significant hypotension is complex, Brunauer et al6 have suggested that a mean arterial pressure (MAP) of approximately 35 mm Hg is associated with a significant risk of CV collapse. Of course, a MAP of 40 to 50 mm Hg is also very concerning clinically, with significant risk of deterioration and CV collapse.
Procedural events, such as conscious sedation or rapid sequence intubation (RSI), can produce significant hypotension; PDP can rapidly correct hypotension. In other clinical scenarios in which sustained hypotension is likely and not transient (eg, sepsis with shock), PDP can be used as a bridge to definitive care (eg, volume replacement, continuous vasopressor infusion). It is important to note, however, that PDP administration must occur in conjunction with or after the patient has received other appropriate therapies such as a normal saline bolus and continuous vasopressor infusions. Push-dose pressors are not a replacement for these proven interventions, but rather are an important augmentation to these therapies.
Emergency Medicine Literature
As previously noted, the literature base describing and supporting the clinical use of PDP in EM is extremely limited. The few articles that comprise this literature base address significant hypotension in periendotracheal intubation intervention, post-return of spontaneous circulation (ROSC) management, and shock management with preload augmentation.7-9In addition, there are several articles in the literature that address safety concerns surrounding the use of PDP in the ED.4,5
Panchal et al10 investigated the use of phenylephrine in hypotensive patients undergoing RSI-assisted endotracheal intubation. The authors performed a 1-year retrospective review of hypotensive patients managed with endotracheal intubation for a range of clinical conditions that required clinical care intervention. In this study, 20 of the 119 patients received phenylephrine in the peri-intubation period. A range of clinical conditions requiring critical care intervention were encountered; in addition, almost three-quarters of these patients were receiving at least one other vasopressor infusion. Further differences were seen in the timing of PDP administration. In those patients receiving bolus-dose phenylephrine, blood pressure (BP) improved without change in HR. Panchal et al10 concluded that while push-dose phenylephrine improved hemodynamic status, there was significant variation among clinicians regarding dosing, timing of use, and overall clinical situation The significant variation in PDP management in this study was noted to be a potential source of medical error, thus increasing the chance of adverse clinical event.
Push-dose pressor therapy can be employed for significant hypotension while more definitive therapy is being readied and applied. For instance, patients with significant hypotension requiring continuous vasopressor infusion can be managed with PDP while appropriate venous access is established, intravenous fluids are administered, and medications are prepared. The immediate period after resuscitation from cardiac arrest can be complicated by shock of many types. In fact, hypotension following ROSC in the cardiac arrest patient is not uncommon and has been identified as a risk issue associated with poor outcome. Prompt treatment of this altered perfusion may improve outcome. Gottlieb8 described three patients with ROSC after cardiac arrest. All three patients experienced significant, sustained hypotension with systolic blood pressure reading in the 50 to 60 mm Hg range; bolus-dose epinephrine was administered with significant improvement in the hemodynamic status while central venous access was established.
In a related clinical scenario, Schwartz et al9 considered the impact of PDP on central venous line (CVL) placement with continuous vasopressor infusion. In this ED study, although patients experienced an increase in BP, this impact was transient with approximately half of these individuals ultimately requiring CVL. In addition, serious adverse effect was noted more commonly in the phenylephrine-treated patients with “reactive” hypertension and ventricular tachycardia occurring in study patients.
Patient-Safety Considerations
In addition to the limited literature base supporting PDP use in the ED, another major significant issue focuses on safety concerns and adverse effects. Extremely limited data is available describing adverse events related to ED-administered PDP. Extrapolating from other EM and critical care administrations of peripheral epinephrine, both local and systemic adverse effects have been reported.11,12 The range of adverse events noted in these studies are considerable, including local skin and soft-tissue injury (necrosis), end-organ tissue ischemia (eg, digits, tip of nose), acute hypertension, cardiac ischemic events, and left ventricular (LV) dysfunction.11,12
When comparing peripheral infusion with central infusion, the risk of extravasation with resultant local tissue injury is markedly greater with peripheral vasopressor administration. In a systematic review of this issue, Loubani and Green11 noted that such local adverse events were much more commonly associated with peripheral administration.
In another report of vasopressor use in the ED, Kanwar et al12 described apparent confusion with epinephrine dosing and route of administration, resulting in very significant, systemic CV maladies, including severe elevations in BP, acute LV dysfunction, and chest pain associated with ST segment elevation.
It must be stressed that the publications by Loubani and Green11 and Kanwar et al12 described peripheral vasopressor administration: neither study included PDP therapy. Therefore, as previously noted, the aforementioned statements are extrapolated from when applied to PDP strategy.
Acquisto et al4 describe several errors in medication administration of PDP in the ED and other critical care areas of the hospital. In this report, all treating physicians were present at the patients’ bedside, either administering the medication or directly supervising its use. Agents involved included epinephrine and phenylephrine, delivered at exceedingly high doses. In their study, the authors noted several issues which they believe contributed to medication errors, including heterogeneity of pathology treated in these patients, apparent “earlier-than-appropriate” use of vasopressors (ie, prior to giving an appropriate fluid bolus), and medication preparation at the bedside by clinicians who may not possess the experience and training to mix these agents.
From a patient-safety perspective, Holden et al5 noted the potential for dosing error with significant adverse medical consequence related to PDP, as well as several contributing issues. First, they highlight the lack of a solid literature base to support administration of PDP in the ED and the development of decision-making guidelines for use in the ED. They also observed an inconsistency in approach to patient selection, medication choice, agent preparation, dosing, and other therapies. As seen in the Acquisto et al4 report, the patient-care scenarios are high risk and quite dynamic.
Conclusion
Bolus-dose vasopressor therapy is a potentially very useful treatment in the ED and other emergency/critical care settings. However, despite its benefits in treating patients in shock or with hypoperfusion, PDP is not widely used in EM due to the lack of studies, reviews, and guidelines in the literature to support its use in the ED. Such a literature base is required to provide an appropriate, safe means of patient selection, medication choice, dosing, and administration. Continued educational and research efforts are needed to more fully explore the use of PDP therapy in the ED.
When used correctly and appropriately, PDP has promise to be an important aid in the management of shock in the ED. Although bolus-dose therapy is appropriate for select clinical scenarios involving significant shock states which have the potential for progression to complete CV collapse without timely therapy, it is an adjunct to, not a replacement for commonly employed and medically indicated therapies such as crystalloid bolus or continuous vasopressor infusions.
The use of bolus-dose vasopressors in anesthesiology and other areas of critical care medicine is well known. This common medical intervention, however, is not often employed in emergency medicine (EM). Bolus-dose vasopressors are defined as the administration of small bolus doses of vasopressor agents, such as epinephrine or phenylephrine, to patients with compromised perfusion who continue to have a pulse (ie, these patients are not in cardiac arrest). This intervention is considered as a temporizing measure for transient hypotension or as a bridge to more definitive therapy.
Clinical Application
Bolus-dose vasopressive therapy is also referred to as push-dose pressor (PDP) therapy—a term coined by Weingart.1-3 Theoretically, any vasopressor could be used in a mini-dose, bolus fashion, though in current clinical practice, anesthesiologists primarily employ ephedrine, epinephrine, and phenylephrine. Two of these agents are likely more appropriate for the ED, including epinephrine and phenylephrine. Both of these agents have a short half-life and therefore an abbreviated period of effect. In addition, dosing and related administration of epinephrine and phenylephrine is relatively straightforward. Moreover, most emergency physicians and nurses are quite familiar with both agents.
With respect to ephedrine, due to its longer half-life, complex dosing regimen, and associated higher-incidence of cardiovascular (CV) complications, its use is likely not appropriate in the ED as a bolus-dose vasopressor.
Epinephrine and Phenylephrine
Epinephrine is a potent sympathomimetic agent with alpha- and beta-receptor activity. In addition to its vasopressor effects, epinephrine is also an inotropic and chronotropic agent, increasing cardiac output, heart rate (HR), and systemic vascular resistance, which can markedly improve perfusion. Epinephrine also can be given to patients with hypoperfusion and/or shock due to low-cardiac output with or without vasodilation, lacking significant tachycardia.
Phenylephrine is a pure alpha agonist and therefore does not appreciably affect cardiac output and HR, but does significantly increase systemic vascular resistance and thus systemic perfusion. Phenylephrine can be used to treat patients with hypoperfusion and/or shock states due to vasodilation with coexistent, significant tachycardia.
Preparation and Administration
The preparation and dosing of push-dose epinephrine and phenylephrine are not particularly complex. Many clinicians recommend the pre-mixed, manufacturer-prepared agents for PDP therapy. These premixed formulations not only facilitate administration, but also reduce the chance of a preparation error that can result in incorrect dosing.3-5 If pre-mixed formulations are not available, clinicians can readily prepare epinephrine and phenylephrine for PDP use.
Push-Dose Epinephrine. Clinicians can prepare epinephrine for push-dose administration as follows:1-3
- Obtain 1 mL of epinephrine 1:10,000 (ie, 0.1 mg/mL or 100 mcg/mL);
- Obtain a 10 mL syringe of normal saline and remove 1 mL;
- Inject the 1 mL of epinephrine 1:10,000 (100 mcg/mL) into this syringe containing 9 mL of normal saline; and
- Result: 10 mL of epinephrine (10 mcg/mL), with each 1 mL of this solution containing 10 mcg of epinephrine.
Administration of push-dose epinephrine (10 mcg/mL) produces effect within 1 minute of use with a duration of approximately 5 to 10 minutes. Dosing at this concentration ranges from 0.5 to 2.0 mL every 2 to 5 minutes, delivering 5 to 20 mcg.1-3Push-Dose Phenylephrine. To prepare phenylephrine for push-dose administration, clinicians may use the following approach:1-3
- Obtain 1 mL of phenylephrine (10 mg/mL concentration);
- Inject this 1 mL of phenylephrine (10 mg/mL) into a 100 mL bag of normal saline; and
- Result: 100 mL of phenylephrine (100 mcg/mL), with each 1 mL of this solution containing 100 mcg of phenylephrine.
Administration of push-dose phenylephrine (100 mcg/mL) produces effect within 1 minute of use with a duration of approximately 10 to 20 minutes. Dosing at this concentration ranges from 0.5 to 2.0 mL every 2 to 5 minutes, delivering 50 to 200 mcg.1-3Alternative Push-Dose Preparations for Phenylephrine. Two other methods of preparing phenylephrine for bolus-dose administration include the following: (1) the addition of phenylephrine 20 mg to a bag of 250 cc of normal saline, resulting in an 80 mcg/mL concentration; and/or (2) phenylephrine (20 mg) is commercially available for continuous infusion in a 250 mL bag of normal saline, yielding the same concentration of 80 mcg/mL; in either case, medication can be drawn up and administered. Dosing at this concentration ranges from 0.5 to 2.5 mL every 2 to 5 minutes, delivering 40 to 200 mcg. Lastly, phenylephrine is also commercially available in pre-made mixtures, specifically manufactured for bolus-dose therapy.
Indications
Both epinephrine and phenylephrine can be considered in the management of significant transient or sustained hypoperfusion. Although the definition of significant hypotension is complex, Brunauer et al6 have suggested that a mean arterial pressure (MAP) of approximately 35 mm Hg is associated with a significant risk of CV collapse. Of course, a MAP of 40 to 50 mm Hg is also very concerning clinically, with significant risk of deterioration and CV collapse.
Procedural events, such as conscious sedation or rapid sequence intubation (RSI), can produce significant hypotension; PDP can rapidly correct hypotension. In other clinical scenarios in which sustained hypotension is likely and not transient (eg, sepsis with shock), PDP can be used as a bridge to definitive care (eg, volume replacement, continuous vasopressor infusion). It is important to note, however, that PDP administration must occur in conjunction with or after the patient has received other appropriate therapies such as a normal saline bolus and continuous vasopressor infusions. Push-dose pressors are not a replacement for these proven interventions, but rather are an important augmentation to these therapies.
Emergency Medicine Literature
As previously noted, the literature base describing and supporting the clinical use of PDP in EM is extremely limited. The few articles that comprise this literature base address significant hypotension in periendotracheal intubation intervention, post-return of spontaneous circulation (ROSC) management, and shock management with preload augmentation.7-9In addition, there are several articles in the literature that address safety concerns surrounding the use of PDP in the ED.4,5
Panchal et al10 investigated the use of phenylephrine in hypotensive patients undergoing RSI-assisted endotracheal intubation. The authors performed a 1-year retrospective review of hypotensive patients managed with endotracheal intubation for a range of clinical conditions that required clinical care intervention. In this study, 20 of the 119 patients received phenylephrine in the peri-intubation period. A range of clinical conditions requiring critical care intervention were encountered; in addition, almost three-quarters of these patients were receiving at least one other vasopressor infusion. Further differences were seen in the timing of PDP administration. In those patients receiving bolus-dose phenylephrine, blood pressure (BP) improved without change in HR. Panchal et al10 concluded that while push-dose phenylephrine improved hemodynamic status, there was significant variation among clinicians regarding dosing, timing of use, and overall clinical situation The significant variation in PDP management in this study was noted to be a potential source of medical error, thus increasing the chance of adverse clinical event.
Push-dose pressor therapy can be employed for significant hypotension while more definitive therapy is being readied and applied. For instance, patients with significant hypotension requiring continuous vasopressor infusion can be managed with PDP while appropriate venous access is established, intravenous fluids are administered, and medications are prepared. The immediate period after resuscitation from cardiac arrest can be complicated by shock of many types. In fact, hypotension following ROSC in the cardiac arrest patient is not uncommon and has been identified as a risk issue associated with poor outcome. Prompt treatment of this altered perfusion may improve outcome. Gottlieb8 described three patients with ROSC after cardiac arrest. All three patients experienced significant, sustained hypotension with systolic blood pressure reading in the 50 to 60 mm Hg range; bolus-dose epinephrine was administered with significant improvement in the hemodynamic status while central venous access was established.
In a related clinical scenario, Schwartz et al9 considered the impact of PDP on central venous line (CVL) placement with continuous vasopressor infusion. In this ED study, although patients experienced an increase in BP, this impact was transient with approximately half of these individuals ultimately requiring CVL. In addition, serious adverse effect was noted more commonly in the phenylephrine-treated patients with “reactive” hypertension and ventricular tachycardia occurring in study patients.
Patient-Safety Considerations
In addition to the limited literature base supporting PDP use in the ED, another major significant issue focuses on safety concerns and adverse effects. Extremely limited data is available describing adverse events related to ED-administered PDP. Extrapolating from other EM and critical care administrations of peripheral epinephrine, both local and systemic adverse effects have been reported.11,12 The range of adverse events noted in these studies are considerable, including local skin and soft-tissue injury (necrosis), end-organ tissue ischemia (eg, digits, tip of nose), acute hypertension, cardiac ischemic events, and left ventricular (LV) dysfunction.11,12
When comparing peripheral infusion with central infusion, the risk of extravasation with resultant local tissue injury is markedly greater with peripheral vasopressor administration. In a systematic review of this issue, Loubani and Green11 noted that such local adverse events were much more commonly associated with peripheral administration.
In another report of vasopressor use in the ED, Kanwar et al12 described apparent confusion with epinephrine dosing and route of administration, resulting in very significant, systemic CV maladies, including severe elevations in BP, acute LV dysfunction, and chest pain associated with ST segment elevation.
It must be stressed that the publications by Loubani and Green11 and Kanwar et al12 described peripheral vasopressor administration: neither study included PDP therapy. Therefore, as previously noted, the aforementioned statements are extrapolated from when applied to PDP strategy.
Acquisto et al4 describe several errors in medication administration of PDP in the ED and other critical care areas of the hospital. In this report, all treating physicians were present at the patients’ bedside, either administering the medication or directly supervising its use. Agents involved included epinephrine and phenylephrine, delivered at exceedingly high doses. In their study, the authors noted several issues which they believe contributed to medication errors, including heterogeneity of pathology treated in these patients, apparent “earlier-than-appropriate” use of vasopressors (ie, prior to giving an appropriate fluid bolus), and medication preparation at the bedside by clinicians who may not possess the experience and training to mix these agents.
From a patient-safety perspective, Holden et al5 noted the potential for dosing error with significant adverse medical consequence related to PDP, as well as several contributing issues. First, they highlight the lack of a solid literature base to support administration of PDP in the ED and the development of decision-making guidelines for use in the ED. They also observed an inconsistency in approach to patient selection, medication choice, agent preparation, dosing, and other therapies. As seen in the Acquisto et al4 report, the patient-care scenarios are high risk and quite dynamic.
Conclusion
Bolus-dose vasopressor therapy is a potentially very useful treatment in the ED and other emergency/critical care settings. However, despite its benefits in treating patients in shock or with hypoperfusion, PDP is not widely used in EM due to the lack of studies, reviews, and guidelines in the literature to support its use in the ED. Such a literature base is required to provide an appropriate, safe means of patient selection, medication choice, dosing, and administration. Continued educational and research efforts are needed to more fully explore the use of PDP therapy in the ED.
When used correctly and appropriately, PDP has promise to be an important aid in the management of shock in the ED. Although bolus-dose therapy is appropriate for select clinical scenarios involving significant shock states which have the potential for progression to complete CV collapse without timely therapy, it is an adjunct to, not a replacement for commonly employed and medically indicated therapies such as crystalloid bolus or continuous vasopressor infusions.
1. Weingart S. EMCrit podcast 6—push-dose pressors. EMCrit RACC Web site. July 2009. https://emcrit.org/racc/bolus-dose-pressors. Accessed March 12, 2018.
2. Weingart S. EMCrit podcast 205—push-dose pressors update. EMCrit RACC Web site. August 2017. https://emcrit.org/racc/push-dose-pressor-update/. March 12, 2018.
3. Weingart S. Push-dose pressors for immediate blood pressure control. Clin Exp Emerg Med. 2015;2(2):131-132. doi:10.15441/ceem.15.010.
4. Acquisto NM, Bodkin RP, Johnstone C. Medication errors with push dose pressors in the emergency department and intensive care units. Am J Emerg Med. 2017;35(12):1964-1965. doi:10.1016/j.ajem.2017.06.013.
5. Holden D, Ramich J, Timm E, Pauze D, Lesar T. Safety considerations and guideline-based safe use recommendations for “bolus-dose” vasopressors in the emergency department. Ann Emerg Med. 2018;71(1):83-92. doi:10.1016/j.annemergmed.2017.04.021.
6. Brunauer A, Koköfer A, Bataar O, Gradwohl-Matis I, Dankl D, Dünser MW. The arterial blood pressure associated with terminal cardiovascular collapse in critically ill patients: a retrospective cohort study. Crit Care. 2014;18(6):719. doi:10.1186/s13054-014-0719-2.
7. Panchal AR, Satyanarayan A, Bahadir JD, Hays D, Mosier J. Efficacy of bolus-dose phenylephrine for peri-intubation hypotension. J Emerg Med. 2015;49(4):488-494. doi:10.1016/j.jemermed.2015.04.033.
8. Gottlieb M. Bolus dose of epinephrine for refractory post-arrest hypotension. Can J Emerg Med. 2017;409:1-5. doi:10.1017/cem.2016.409.
9. Schwartz MB, Ferreira JA, Aaronson PM. The impact of push-dose phenylephrine use on subsequent preload expansion in the ED setting. Am J Emerg Med. 2016;34(12):2419-2422. doi:10.1016/j.ajem.2016.09.041.
10. Panchal AR, Satyanarayan A, Bahadir JD, Hays D, Mosier J. Efficacy of bolus-dose phenylephrine for peri-intubation hypotension. J Emerg Med. 2015;49(4):488-494. doi:10.1016/j.jemermed.2015.04.033.
11. Loubani OM, Green RS. A systematic review of extravasation and local tissue injury from administration of vasopressors through peripheral intravenous catheters and central venous catheters. J Crit Care. 2015;30:653.e9-e17.
12. Kanwar M, Irvin CB, Frank JJ, et al. Confusion about epinephrine dosing leading to iatrogenic overdose: A life-threatening problem with a potential solution. Ann Emerg Med. 2010;55:341-344.
1. Weingart S. EMCrit podcast 6—push-dose pressors. EMCrit RACC Web site. July 2009. https://emcrit.org/racc/bolus-dose-pressors. Accessed March 12, 2018.
2. Weingart S. EMCrit podcast 205—push-dose pressors update. EMCrit RACC Web site. August 2017. https://emcrit.org/racc/push-dose-pressor-update/. March 12, 2018.
3. Weingart S. Push-dose pressors for immediate blood pressure control. Clin Exp Emerg Med. 2015;2(2):131-132. doi:10.15441/ceem.15.010.
4. Acquisto NM, Bodkin RP, Johnstone C. Medication errors with push dose pressors in the emergency department and intensive care units. Am J Emerg Med. 2017;35(12):1964-1965. doi:10.1016/j.ajem.2017.06.013.
5. Holden D, Ramich J, Timm E, Pauze D, Lesar T. Safety considerations and guideline-based safe use recommendations for “bolus-dose” vasopressors in the emergency department. Ann Emerg Med. 2018;71(1):83-92. doi:10.1016/j.annemergmed.2017.04.021.
6. Brunauer A, Koköfer A, Bataar O, Gradwohl-Matis I, Dankl D, Dünser MW. The arterial blood pressure associated with terminal cardiovascular collapse in critically ill patients: a retrospective cohort study. Crit Care. 2014;18(6):719. doi:10.1186/s13054-014-0719-2.
7. Panchal AR, Satyanarayan A, Bahadir JD, Hays D, Mosier J. Efficacy of bolus-dose phenylephrine for peri-intubation hypotension. J Emerg Med. 2015;49(4):488-494. doi:10.1016/j.jemermed.2015.04.033.
8. Gottlieb M. Bolus dose of epinephrine for refractory post-arrest hypotension. Can J Emerg Med. 2017;409:1-5. doi:10.1017/cem.2016.409.
9. Schwartz MB, Ferreira JA, Aaronson PM. The impact of push-dose phenylephrine use on subsequent preload expansion in the ED setting. Am J Emerg Med. 2016;34(12):2419-2422. doi:10.1016/j.ajem.2016.09.041.
10. Panchal AR, Satyanarayan A, Bahadir JD, Hays D, Mosier J. Efficacy of bolus-dose phenylephrine for peri-intubation hypotension. J Emerg Med. 2015;49(4):488-494. doi:10.1016/j.jemermed.2015.04.033.
11. Loubani OM, Green RS. A systematic review of extravasation and local tissue injury from administration of vasopressors through peripheral intravenous catheters and central venous catheters. J Crit Care. 2015;30:653.e9-e17.
12. Kanwar M, Irvin CB, Frank JJ, et al. Confusion about epinephrine dosing leading to iatrogenic overdose: A life-threatening problem with a potential solution. Ann Emerg Med. 2010;55:341-344.
Transgender trauma patients: What surgeons need to know
The likelihood that a is increasing every year.
The number of patients who self-identify as transgender and who have undergone both medical and/or surgical gender-affirming treatment is on the rise. The trend has accelerated since private insurers, Medicare, and Medicaid are now covering some of the costs (JAMA Surg. 2018 Feb 28. doi: 10.1001/jamasurg.2017.6231).

Lead author Samuel Mandell, MD, FACS, a trauma surgeon at the University of Washington Harborview Medical Center, Seattle, and his colleagues quote an estimate of 1 million transgender people in the United States. These individuals, many of whom have experienced gender dysphoria, in addition to stigma and negative psychosocial sequelae, may or may not have sought medical treatment. Medical interventions range from hormonal treatments to craniofacial plastic surgery and/or genital surgery.
“As transgender patients are more likely to be victims of assault and intimate partner violence or suicide, they are at increased risk for traumatic injury,” Dr. Mandell and coauthors said. More than 60% of the transgender population has been subjected to assault and more than 40% have attempted suicide. A recent study found that 42% of transgender individuals had a history on nonsuicidal self-injury (Psychiatr Clin North Am. 2017;40:41-50). The research team based their recommendations on managing transgender trauma patients on their own experience, and suggest some topics for future research
The authors searched the MEDLINE database for articles with the key words “trauma” or “injury” and “transgender/transsexual,” in addition to “surgery” and “transgender.” While the search yielded 388 articles, only 6 were relevant to acute care surgery or physical trauma/injury in the transgender population. “No articles were identified that addressed trauma/injury from the perspective of caring for the injured transgender patient,” Dr. Mandell and coauthors said.
The researchers recommend that the trauma surgeon begin if possible by working to establish patient-provider trust. “During surgical consultation, it is important to be aware that any transgender patient may have limited or negative interactions with general health care providers due to the significant discrimination this population faces,” the investigators wrote. Among the steps they suggest for the initial encounter with transgender patients are respectful questions about gender identity, asking what name they prefer, as well as what pronoun should be used.
Privacy concerns can be of particular sensitivity. “Care must be taken to maintain privacy for the patient, as others outside of the hospital may not know they are transgender. Consultation with the patient’s primary care provider may be beneficial to determine the extent of gender-affirmation and the patient’s disclosure to family and friends,” the investigator advised. In addition, the clinician needs to establish which if any nonmedical interventions the transgender patients has had. These may include nonprescription hormone therapy and silicone injections.
The encounter should include an evaluation for injury to genitalia. “Transgender patients may have significant dysphoria associated with their preoperative genitals,” Dr. Mandell and his coauthors said. In these cases, “involvement of providers experienced with examination of transgender patients should be sought, if possible.” These patients should be screened for potential abuse by a companion or self-injury, the investigators suggested.
Dr. Mandell and his coauthors also discussed some of the nuances of trauma care for this population. For example, transgender women may need a smaller endotracheal tube for establishing an airway as intubation to avoid damaging surgically altered vocal chords. Other craniofacial alterations can get in the way of establishing an airway. Clinicians also should keep in mind the increased likelihood of a venous thromboembolism from estrogen hormone therapy in immobilized transgender patients in the trauma setting. Implants and surgical alterations can add a layer of complexity to reading images. Anatomical rearrangement can make catheterization challenging.
Dr. Mandell and his coauthors concluded, “Further research is needed on the appropriate management of cross-gender hormones, dosing of medications and nutrition, and the special considerations for injury patterns and risks in transgender patients. Development of a system for quickly determining the state of gender-affirmation of the patient in regards to hormone therapy, surgeries, and social aspects may prove beneficial to providers in the setting of trauma, but involvement of the transgender population in the development of any such system is crucial.”
Eileen M. Bulger, MD, FACS, Chair ACS Committee on Trauma and one of the coauthors of the study, views the findings as potentially useful to meet the training deficit on transgender trauma issues. “As trauma surgeons, we strive to provide optimal care by attending to the physical, psychological, and social needs of our patients. This review raises awareness of critical issues to consider when caring for transgender patients and should be included in our educational programs for trauma fellowship training and used as a resource to raise awareness in our trauma centers.”
Dr. Mandell and his coauthors reported having no financial disclosures.
Education on the care of transgender and gender nonbinary population is lacking in both medical schools as well as surgical residencies, and it is often left to individual surgeons to seek out their own training. Unfortunately, this leaves many uncertain how to ask a patient about his/her/their history without making the patient uncomfortable. If we don’t ask the right questions, some patients may not disclose information that could be very detrimental to their care. Documentation in EHRs can be made difficult if the software doesn’t include transgender female, transgender male, and gender nonbinary options in addition to the binary choice of female or male. This can contribute to the misgendering and distress of the patient.
Asking which pronouns a transgender individual uses can be a big first step because it allows that person know that you are being respectful. Be prepared for pronouns you may not be used to: Some may use she/her or he/his, and some may use they/their, ze/hir, ze/zir or xe/xyr. It is important to have appropriate registration forms, gender neutral bathrooms, and respect and discretion from every individual provider for all of our patients. Providers should seek out education and training so that the patients aren’t forced to do the educating themselves. As trauma and acute care surgeons, we are used to caring for a diverse patient population with many unique needs. However, we don’t know enough about the trauma and surgery risks in the transgender and gender nonbinary population as only a limited research has been done. Studies such as this by Dr. Mandell et al. are encouraging and hopefully more will follow.
Andrea Long, MD, is an acute care surgeon and an assistant clinical professor at University of San Francisco, Fresno.
Education on the care of transgender and gender nonbinary population is lacking in both medical schools as well as surgical residencies, and it is often left to individual surgeons to seek out their own training. Unfortunately, this leaves many uncertain how to ask a patient about his/her/their history without making the patient uncomfortable. If we don’t ask the right questions, some patients may not disclose information that could be very detrimental to their care. Documentation in EHRs can be made difficult if the software doesn’t include transgender female, transgender male, and gender nonbinary options in addition to the binary choice of female or male. This can contribute to the misgendering and distress of the patient.
Asking which pronouns a transgender individual uses can be a big first step because it allows that person know that you are being respectful. Be prepared for pronouns you may not be used to: Some may use she/her or he/his, and some may use they/their, ze/hir, ze/zir or xe/xyr. It is important to have appropriate registration forms, gender neutral bathrooms, and respect and discretion from every individual provider for all of our patients. Providers should seek out education and training so that the patients aren’t forced to do the educating themselves. As trauma and acute care surgeons, we are used to caring for a diverse patient population with many unique needs. However, we don’t know enough about the trauma and surgery risks in the transgender and gender nonbinary population as only a limited research has been done. Studies such as this by Dr. Mandell et al. are encouraging and hopefully more will follow.
Andrea Long, MD, is an acute care surgeon and an assistant clinical professor at University of San Francisco, Fresno.
Education on the care of transgender and gender nonbinary population is lacking in both medical schools as well as surgical residencies, and it is often left to individual surgeons to seek out their own training. Unfortunately, this leaves many uncertain how to ask a patient about his/her/their history without making the patient uncomfortable. If we don’t ask the right questions, some patients may not disclose information that could be very detrimental to their care. Documentation in EHRs can be made difficult if the software doesn’t include transgender female, transgender male, and gender nonbinary options in addition to the binary choice of female or male. This can contribute to the misgendering and distress of the patient.
Asking which pronouns a transgender individual uses can be a big first step because it allows that person know that you are being respectful. Be prepared for pronouns you may not be used to: Some may use she/her or he/his, and some may use they/their, ze/hir, ze/zir or xe/xyr. It is important to have appropriate registration forms, gender neutral bathrooms, and respect and discretion from every individual provider for all of our patients. Providers should seek out education and training so that the patients aren’t forced to do the educating themselves. As trauma and acute care surgeons, we are used to caring for a diverse patient population with many unique needs. However, we don’t know enough about the trauma and surgery risks in the transgender and gender nonbinary population as only a limited research has been done. Studies such as this by Dr. Mandell et al. are encouraging and hopefully more will follow.
Andrea Long, MD, is an acute care surgeon and an assistant clinical professor at University of San Francisco, Fresno.
The likelihood that a is increasing every year.
The number of patients who self-identify as transgender and who have undergone both medical and/or surgical gender-affirming treatment is on the rise. The trend has accelerated since private insurers, Medicare, and Medicaid are now covering some of the costs (JAMA Surg. 2018 Feb 28. doi: 10.1001/jamasurg.2017.6231).
Lead author Samuel Mandell, MD, FACS, a trauma surgeon at the University of Washington Harborview Medical Center, Seattle, and his colleagues quote an estimate of 1 million transgender people in the United States. These individuals, many of whom have experienced gender dysphoria, in addition to stigma and negative psychosocial sequelae, may or may not have sought medical treatment. Medical interventions range from hormonal treatments to craniofacial plastic surgery and/or genital surgery.
“As transgender patients are more likely to be victims of assault and intimate partner violence or suicide, they are at increased risk for traumatic injury,” Dr. Mandell and coauthors said. More than 60% of the transgender population has been subjected to assault and more than 40% have attempted suicide. A recent study found that 42% of transgender individuals had a history on nonsuicidal self-injury (Psychiatr Clin North Am. 2017;40:41-50). The research team based their recommendations on managing transgender trauma patients on their own experience, and suggest some topics for future research
The authors searched the MEDLINE database for articles with the key words “trauma” or “injury” and “transgender/transsexual,” in addition to “surgery” and “transgender.” While the search yielded 388 articles, only 6 were relevant to acute care surgery or physical trauma/injury in the transgender population. “No articles were identified that addressed trauma/injury from the perspective of caring for the injured transgender patient,” Dr. Mandell and coauthors said.
The researchers recommend that the trauma surgeon begin if possible by working to establish patient-provider trust. “During surgical consultation, it is important to be aware that any transgender patient may have limited or negative interactions with general health care providers due to the significant discrimination this population faces,” the investigators wrote. Among the steps they suggest for the initial encounter with transgender patients are respectful questions about gender identity, asking what name they prefer, as well as what pronoun should be used.
Privacy concerns can be of particular sensitivity. “Care must be taken to maintain privacy for the patient, as others outside of the hospital may not know they are transgender. Consultation with the patient’s primary care provider may be beneficial to determine the extent of gender-affirmation and the patient’s disclosure to family and friends,” the investigator advised. In addition, the clinician needs to establish which if any nonmedical interventions the transgender patients has had. These may include nonprescription hormone therapy and silicone injections.
The encounter should include an evaluation for injury to genitalia. “Transgender patients may have significant dysphoria associated with their preoperative genitals,” Dr. Mandell and his coauthors said. In these cases, “involvement of providers experienced with examination of transgender patients should be sought, if possible.” These patients should be screened for potential abuse by a companion or self-injury, the investigators suggested.
Dr. Mandell and his coauthors also discussed some of the nuances of trauma care for this population. For example, transgender women may need a smaller endotracheal tube for establishing an airway as intubation to avoid damaging surgically altered vocal chords. Other craniofacial alterations can get in the way of establishing an airway. Clinicians also should keep in mind the increased likelihood of a venous thromboembolism from estrogen hormone therapy in immobilized transgender patients in the trauma setting. Implants and surgical alterations can add a layer of complexity to reading images. Anatomical rearrangement can make catheterization challenging.
Dr. Mandell and his coauthors concluded, “Further research is needed on the appropriate management of cross-gender hormones, dosing of medications and nutrition, and the special considerations for injury patterns and risks in transgender patients. Development of a system for quickly determining the state of gender-affirmation of the patient in regards to hormone therapy, surgeries, and social aspects may prove beneficial to providers in the setting of trauma, but involvement of the transgender population in the development of any such system is crucial.”
Eileen M. Bulger, MD, FACS, Chair ACS Committee on Trauma and one of the coauthors of the study, views the findings as potentially useful to meet the training deficit on transgender trauma issues. “As trauma surgeons, we strive to provide optimal care by attending to the physical, psychological, and social needs of our patients. This review raises awareness of critical issues to consider when caring for transgender patients and should be included in our educational programs for trauma fellowship training and used as a resource to raise awareness in our trauma centers.”
Dr. Mandell and his coauthors reported having no financial disclosures.
The likelihood that a is increasing every year.
The number of patients who self-identify as transgender and who have undergone both medical and/or surgical gender-affirming treatment is on the rise. The trend has accelerated since private insurers, Medicare, and Medicaid are now covering some of the costs (JAMA Surg. 2018 Feb 28. doi: 10.1001/jamasurg.2017.6231).
Lead author Samuel Mandell, MD, FACS, a trauma surgeon at the University of Washington Harborview Medical Center, Seattle, and his colleagues quote an estimate of 1 million transgender people in the United States. These individuals, many of whom have experienced gender dysphoria, in addition to stigma and negative psychosocial sequelae, may or may not have sought medical treatment. Medical interventions range from hormonal treatments to craniofacial plastic surgery and/or genital surgery.
“As transgender patients are more likely to be victims of assault and intimate partner violence or suicide, they are at increased risk for traumatic injury,” Dr. Mandell and coauthors said. More than 60% of the transgender population has been subjected to assault and more than 40% have attempted suicide. A recent study found that 42% of transgender individuals had a history on nonsuicidal self-injury (Psychiatr Clin North Am. 2017;40:41-50). The research team based their recommendations on managing transgender trauma patients on their own experience, and suggest some topics for future research
The authors searched the MEDLINE database for articles with the key words “trauma” or “injury” and “transgender/transsexual,” in addition to “surgery” and “transgender.” While the search yielded 388 articles, only 6 were relevant to acute care surgery or physical trauma/injury in the transgender population. “No articles were identified that addressed trauma/injury from the perspective of caring for the injured transgender patient,” Dr. Mandell and coauthors said.
The researchers recommend that the trauma surgeon begin if possible by working to establish patient-provider trust. “During surgical consultation, it is important to be aware that any transgender patient may have limited or negative interactions with general health care providers due to the significant discrimination this population faces,” the investigators wrote. Among the steps they suggest for the initial encounter with transgender patients are respectful questions about gender identity, asking what name they prefer, as well as what pronoun should be used.
Privacy concerns can be of particular sensitivity. “Care must be taken to maintain privacy for the patient, as others outside of the hospital may not know they are transgender. Consultation with the patient’s primary care provider may be beneficial to determine the extent of gender-affirmation and the patient’s disclosure to family and friends,” the investigator advised. In addition, the clinician needs to establish which if any nonmedical interventions the transgender patients has had. These may include nonprescription hormone therapy and silicone injections.
The encounter should include an evaluation for injury to genitalia. “Transgender patients may have significant dysphoria associated with their preoperative genitals,” Dr. Mandell and his coauthors said. In these cases, “involvement of providers experienced with examination of transgender patients should be sought, if possible.” These patients should be screened for potential abuse by a companion or self-injury, the investigators suggested.
Dr. Mandell and his coauthors also discussed some of the nuances of trauma care for this population. For example, transgender women may need a smaller endotracheal tube for establishing an airway as intubation to avoid damaging surgically altered vocal chords. Other craniofacial alterations can get in the way of establishing an airway. Clinicians also should keep in mind the increased likelihood of a venous thromboembolism from estrogen hormone therapy in immobilized transgender patients in the trauma setting. Implants and surgical alterations can add a layer of complexity to reading images. Anatomical rearrangement can make catheterization challenging.
Dr. Mandell and his coauthors concluded, “Further research is needed on the appropriate management of cross-gender hormones, dosing of medications and nutrition, and the special considerations for injury patterns and risks in transgender patients. Development of a system for quickly determining the state of gender-affirmation of the patient in regards to hormone therapy, surgeries, and social aspects may prove beneficial to providers in the setting of trauma, but involvement of the transgender population in the development of any such system is crucial.”
Eileen M. Bulger, MD, FACS, Chair ACS Committee on Trauma and one of the coauthors of the study, views the findings as potentially useful to meet the training deficit on transgender trauma issues. “As trauma surgeons, we strive to provide optimal care by attending to the physical, psychological, and social needs of our patients. This review raises awareness of critical issues to consider when caring for transgender patients and should be included in our educational programs for trauma fellowship training and used as a resource to raise awareness in our trauma centers.”
Dr. Mandell and his coauthors reported having no financial disclosures.
FROM THE JOURNAL OF TRAUMA AND ACUTE CARE SURGERY
Over one-third report financial burden from breast cancer treatment
CHICAGO – Women who have treatment for breast cancer seldom talk about the costs of care with their medical team, but a study out of Duke University has found that more than one-third reported having a financial burden from their breast cancer treatment, even among women with health insurance, according to a report presented at the Society of Surgical Oncology Annual Cancer Symposium.
“The financial harm associated with cancer treatment is now known as ‘financial toxicity,’ ” Rachel A. Greenup, MD, MPH, said in reporting the results of an 88-item survey completed by 654 adult women who had treatment for breast cancer. The women were recruited through the Army of Women of the Dr. Susan Love Research Foundation and The Sister’s Network of North Carolina, an African-American breast cancer survivors’ organization.
Overall, 69% of survey respondents had private insurance and 26% had Medicare. Of the patients surveyed, 94% had breast cancer surgery: 40.6% lumpectomy, 23.7% mastectomy, and 29.7% bilateral mastectomy; 34% also had breast reconstruction. Among those surveyed, 43% reported considering costs in their treatment decision. Of these, 29% considered costs when making surgical treatment decisions, including 14% who reported that costs were “extremely” important.
Despite the high levels of insurance coverage, 35% of the study participants reported a financial burden resulting from cancer treatment, ranging from “somewhat” burdensome to “catastrophic.” The median out-of-pocket cost for the study participants was $4,000, and 5% exceeded $40,000 in such costs, Dr. Greenup said. “The risk of financial harm and increased out-of-pocket costs to patients differed by surgery type,” with higher financial burdens seen in women who underwent bilateral mastectomy.
Cost was one of many factors survey participants reported considering when making surgical treatment decisions, but the most important factors were the opinions and advice of the medical team and the individual patient’s fear of recurrence. However, in lower-income women, cost factored more significantly in decision making. “In a subset of women who reported an annual income of $45,000 a year or less, cost of treatment gained importance and, interestingly, became more important than many variables we routinely discuss – for example, appearance of the breast, sexuality, avoiding radiation, and breast preservation,” Dr. Greenup said. “An income of $74,000 a year was the tipping point at which women reported incorporating costs into their cancer treatment decisions.”
She added that younger, minority women who did not have Medicare coverage were more likely to consider costs in breast cancer treatment decisions.
Most women surveyed (79%) said they preferred to know their out-of-pocket costs before they begin treatment, Dr. Greenup said, “and 40% believed that we as physicians should be considering out-of-pocket costs while making medical decisions.” However, 78% of those surveyed said they never discussed costs with their cancer team – despite American Society of Clinical Oncologists guidelines, she pointed out – and 35% said their treatment costs were higher than expected.
Dr. Greenup described the study population as “well engaged … with good insurance and strong educational background that likely does not reflect the general population.” The results may not be generalizable. “We expect that in a general cohort of women, our findings would be even more exaggerated,” she said.
The study points out the need to better understand how cost transparency may affect breast cancer treatment decisions, Dr. Greenup said. “As eligible women with breast cancer choose between surgical options, it’s important that we consider the potential risk of financial harm as we guide them through these difficult treatment decisions,” she said.
Dr. Greenup and her study coauthors reported having no financial disclosures.
SOURCE: Greenup RA. SSO 2018, Abstract No. 24.
CHICAGO – Women who have treatment for breast cancer seldom talk about the costs of care with their medical team, but a study out of Duke University has found that more than one-third reported having a financial burden from their breast cancer treatment, even among women with health insurance, according to a report presented at the Society of Surgical Oncology Annual Cancer Symposium.
“The financial harm associated with cancer treatment is now known as ‘financial toxicity,’ ” Rachel A. Greenup, MD, MPH, said in reporting the results of an 88-item survey completed by 654 adult women who had treatment for breast cancer. The women were recruited through the Army of Women of the Dr. Susan Love Research Foundation and The Sister’s Network of North Carolina, an African-American breast cancer survivors’ organization.
Overall, 69% of survey respondents had private insurance and 26% had Medicare. Of the patients surveyed, 94% had breast cancer surgery: 40.6% lumpectomy, 23.7% mastectomy, and 29.7% bilateral mastectomy; 34% also had breast reconstruction. Among those surveyed, 43% reported considering costs in their treatment decision. Of these, 29% considered costs when making surgical treatment decisions, including 14% who reported that costs were “extremely” important.
Despite the high levels of insurance coverage, 35% of the study participants reported a financial burden resulting from cancer treatment, ranging from “somewhat” burdensome to “catastrophic.” The median out-of-pocket cost for the study participants was $4,000, and 5% exceeded $40,000 in such costs, Dr. Greenup said. “The risk of financial harm and increased out-of-pocket costs to patients differed by surgery type,” with higher financial burdens seen in women who underwent bilateral mastectomy.
Cost was one of many factors survey participants reported considering when making surgical treatment decisions, but the most important factors were the opinions and advice of the medical team and the individual patient’s fear of recurrence. However, in lower-income women, cost factored more significantly in decision making. “In a subset of women who reported an annual income of $45,000 a year or less, cost of treatment gained importance and, interestingly, became more important than many variables we routinely discuss – for example, appearance of the breast, sexuality, avoiding radiation, and breast preservation,” Dr. Greenup said. “An income of $74,000 a year was the tipping point at which women reported incorporating costs into their cancer treatment decisions.”
She added that younger, minority women who did not have Medicare coverage were more likely to consider costs in breast cancer treatment decisions.
Most women surveyed (79%) said they preferred to know their out-of-pocket costs before they begin treatment, Dr. Greenup said, “and 40% believed that we as physicians should be considering out-of-pocket costs while making medical decisions.” However, 78% of those surveyed said they never discussed costs with their cancer team – despite American Society of Clinical Oncologists guidelines, she pointed out – and 35% said their treatment costs were higher than expected.
Dr. Greenup described the study population as “well engaged … with good insurance and strong educational background that likely does not reflect the general population.” The results may not be generalizable. “We expect that in a general cohort of women, our findings would be even more exaggerated,” she said.
The study points out the need to better understand how cost transparency may affect breast cancer treatment decisions, Dr. Greenup said. “As eligible women with breast cancer choose between surgical options, it’s important that we consider the potential risk of financial harm as we guide them through these difficult treatment decisions,” she said.
Dr. Greenup and her study coauthors reported having no financial disclosures.
SOURCE: Greenup RA. SSO 2018, Abstract No. 24.
CHICAGO – Women who have treatment for breast cancer seldom talk about the costs of care with their medical team, but a study out of Duke University has found that more than one-third reported having a financial burden from their breast cancer treatment, even among women with health insurance, according to a report presented at the Society of Surgical Oncology Annual Cancer Symposium.
“The financial harm associated with cancer treatment is now known as ‘financial toxicity,’ ” Rachel A. Greenup, MD, MPH, said in reporting the results of an 88-item survey completed by 654 adult women who had treatment for breast cancer. The women were recruited through the Army of Women of the Dr. Susan Love Research Foundation and The Sister’s Network of North Carolina, an African-American breast cancer survivors’ organization.
Overall, 69% of survey respondents had private insurance and 26% had Medicare. Of the patients surveyed, 94% had breast cancer surgery: 40.6% lumpectomy, 23.7% mastectomy, and 29.7% bilateral mastectomy; 34% also had breast reconstruction. Among those surveyed, 43% reported considering costs in their treatment decision. Of these, 29% considered costs when making surgical treatment decisions, including 14% who reported that costs were “extremely” important.
Despite the high levels of insurance coverage, 35% of the study participants reported a financial burden resulting from cancer treatment, ranging from “somewhat” burdensome to “catastrophic.” The median out-of-pocket cost for the study participants was $4,000, and 5% exceeded $40,000 in such costs, Dr. Greenup said. “The risk of financial harm and increased out-of-pocket costs to patients differed by surgery type,” with higher financial burdens seen in women who underwent bilateral mastectomy.
Cost was one of many factors survey participants reported considering when making surgical treatment decisions, but the most important factors were the opinions and advice of the medical team and the individual patient’s fear of recurrence. However, in lower-income women, cost factored more significantly in decision making. “In a subset of women who reported an annual income of $45,000 a year or less, cost of treatment gained importance and, interestingly, became more important than many variables we routinely discuss – for example, appearance of the breast, sexuality, avoiding radiation, and breast preservation,” Dr. Greenup said. “An income of $74,000 a year was the tipping point at which women reported incorporating costs into their cancer treatment decisions.”
She added that younger, minority women who did not have Medicare coverage were more likely to consider costs in breast cancer treatment decisions.
Most women surveyed (79%) said they preferred to know their out-of-pocket costs before they begin treatment, Dr. Greenup said, “and 40% believed that we as physicians should be considering out-of-pocket costs while making medical decisions.” However, 78% of those surveyed said they never discussed costs with their cancer team – despite American Society of Clinical Oncologists guidelines, she pointed out – and 35% said their treatment costs were higher than expected.
Dr. Greenup described the study population as “well engaged … with good insurance and strong educational background that likely does not reflect the general population.” The results may not be generalizable. “We expect that in a general cohort of women, our findings would be even more exaggerated,” she said.
The study points out the need to better understand how cost transparency may affect breast cancer treatment decisions, Dr. Greenup said. “As eligible women with breast cancer choose between surgical options, it’s important that we consider the potential risk of financial harm as we guide them through these difficult treatment decisions,” she said.
Dr. Greenup and her study coauthors reported having no financial disclosures.
SOURCE: Greenup RA. SSO 2018, Abstract No. 24.
REPORTING FROM SSO 2018
Key clinical point: Treatment costs are important to many women with breast cancer, although most report not having cost discussions with their physicians.
Major finding: Despite the high levels of insurance coverage, 35% of study participants reported a financial burden resulting from cancer treatment, ranging from “somewhat” burdensome to “catastrophic.”
Study details: An 88-item survey completed by 654 adult women who had treatment for breast cancer.
Disclosures: Dr. Greenup and her coauthors reported having no financial disclosures.
Source: Greenup RA. SSO 2018, Abstract No. 24.
FDA advisors recommend lofexidine for opioid withdrawal
SILVER SPRING, MD. – Members of the Food and Drug Administration Psychopharmacologic Drugs Advisory Committee voted 11 to 1 to recommend approval of lofexidine as the first nonopioid treatment option for the symptomatic treatment of opioid withdrawal.
Opioid withdrawal symptoms are the largest barrier to discontinuing opioid use, according to Louis Baxter, MD, executive medical director of the Professional Assistance Program in Princeton, N.J., who presented on behalf of U.S. WorldMeds, which plans to market lofexidine as Lucemyra.

Lofexidine, a selective alpha2-adrenergic receptor agonist that regulates norepinephrine release has been approved for management of opioid withdrawal in the United Kingdom since 1992.
The advisory committee voted to recommend lofexidine on the strength of the results of two randomized, double-blind, and placebo controlled phase 3 studies on the safety and efficacy of lofexidine for symptomatic treatment of opioid withdrawal between days 1 through 7. One study randomized 264 patients to lofexidine (134) or placebo (130), with patients in the treatment arm received 3.2 mg of lofexidine on days 1-5, then placebo until day 7. The second study randomized 603 patients to three groups, comparing high dose (3.2 mg/day) and low dose (2.4 mg/day) regimens of lofexidine to placebo; patients in the treatment arms took four smaller doses of lofexidine throughout the day to achieve the cumulative dose.
Researchers enrolled heavy users of short-acting opioids; heroin was the predominant agent. Both studies were conducted in the scenario of abrupt withdrawal, or the most intense withdrawal situation.
Symptomatic benefit was measured using the Short Opiate Withdrawal Scale of Gossop (SOWS-Gossop), a patient reported outcome. Patients were asked to rank their symptoms as none, mild, moderate or severe across measures like feeling sick, stomach cramps, and heart pounding among other symptoms.
Lofexidine increased completion of withdrawal treatment compared to placebo. Patients in the first study had a 5-day completion rate of 53%, compared to 35% for the placebo group. Researchers observed similar results in the 7-day completion rates the second study, with low and high dose completion rates of 42% and 40%, respectively, both of which were much higher than placebo (28%).
Lofexidine also reduced patient withdrawal symptoms, according to SOWS-Gossop scores during peak withdrawal. In the first study, SOWS-Gossop scores were 2-4 points lower in the lofexidine group compared to placebo. Similarly, the scores were significantly better in both lofexidine groups in the second study, compared to placebo, particularly on days 1 to 4. Decreasing withdrawal symptoms during this period is particularly important because this is the most vulnerable window for patient dropout, briefing documents from US WorldMeds.
Several notable adverse events occurred during the study, particularly at higher doses of lofexidine. The risk of bradycardia and hypotension are prominent in patients taking lofexidine, but these are risks associated with this class of drug, according to Mark Pirner, MD, senior medical director at US WorldMeds, who noted that “the lower dose, if that’s what ultimately gets approved [by the FDA], is safe and effective too.”
Development of lofexidine was conducted in collaboration with the National Institute on Drug Abuse and the FDA, according to briefing documents from US WorldMeds.
The Prescription Drug User Fee Act (PDUFA) for lofexidine is May 26; FDA actions on new drug applications often occur at near the PDUFA date.
The FDA is not obligated to follow the recommendations of its advisory committees.
SILVER SPRING, MD. – Members of the Food and Drug Administration Psychopharmacologic Drugs Advisory Committee voted 11 to 1 to recommend approval of lofexidine as the first nonopioid treatment option for the symptomatic treatment of opioid withdrawal.
Opioid withdrawal symptoms are the largest barrier to discontinuing opioid use, according to Louis Baxter, MD, executive medical director of the Professional Assistance Program in Princeton, N.J., who presented on behalf of U.S. WorldMeds, which plans to market lofexidine as Lucemyra.
Lofexidine, a selective alpha2-adrenergic receptor agonist that regulates norepinephrine release has been approved for management of opioid withdrawal in the United Kingdom since 1992.
The advisory committee voted to recommend lofexidine on the strength of the results of two randomized, double-blind, and placebo controlled phase 3 studies on the safety and efficacy of lofexidine for symptomatic treatment of opioid withdrawal between days 1 through 7. One study randomized 264 patients to lofexidine (134) or placebo (130), with patients in the treatment arm received 3.2 mg of lofexidine on days 1-5, then placebo until day 7. The second study randomized 603 patients to three groups, comparing high dose (3.2 mg/day) and low dose (2.4 mg/day) regimens of lofexidine to placebo; patients in the treatment arms took four smaller doses of lofexidine throughout the day to achieve the cumulative dose.
Researchers enrolled heavy users of short-acting opioids; heroin was the predominant agent. Both studies were conducted in the scenario of abrupt withdrawal, or the most intense withdrawal situation.
Symptomatic benefit was measured using the Short Opiate Withdrawal Scale of Gossop (SOWS-Gossop), a patient reported outcome. Patients were asked to rank their symptoms as none, mild, moderate or severe across measures like feeling sick, stomach cramps, and heart pounding among other symptoms.
Lofexidine increased completion of withdrawal treatment compared to placebo. Patients in the first study had a 5-day completion rate of 53%, compared to 35% for the placebo group. Researchers observed similar results in the 7-day completion rates the second study, with low and high dose completion rates of 42% and 40%, respectively, both of which were much higher than placebo (28%).
Lofexidine also reduced patient withdrawal symptoms, according to SOWS-Gossop scores during peak withdrawal. In the first study, SOWS-Gossop scores were 2-4 points lower in the lofexidine group compared to placebo. Similarly, the scores were significantly better in both lofexidine groups in the second study, compared to placebo, particularly on days 1 to 4. Decreasing withdrawal symptoms during this period is particularly important because this is the most vulnerable window for patient dropout, briefing documents from US WorldMeds.
Several notable adverse events occurred during the study, particularly at higher doses of lofexidine. The risk of bradycardia and hypotension are prominent in patients taking lofexidine, but these are risks associated with this class of drug, according to Mark Pirner, MD, senior medical director at US WorldMeds, who noted that “the lower dose, if that’s what ultimately gets approved [by the FDA], is safe and effective too.”
Development of lofexidine was conducted in collaboration with the National Institute on Drug Abuse and the FDA, according to briefing documents from US WorldMeds.
The Prescription Drug User Fee Act (PDUFA) for lofexidine is May 26; FDA actions on new drug applications often occur at near the PDUFA date.
The FDA is not obligated to follow the recommendations of its advisory committees.
SILVER SPRING, MD. – Members of the Food and Drug Administration Psychopharmacologic Drugs Advisory Committee voted 11 to 1 to recommend approval of lofexidine as the first nonopioid treatment option for the symptomatic treatment of opioid withdrawal.
Opioid withdrawal symptoms are the largest barrier to discontinuing opioid use, according to Louis Baxter, MD, executive medical director of the Professional Assistance Program in Princeton, N.J., who presented on behalf of U.S. WorldMeds, which plans to market lofexidine as Lucemyra.
Lofexidine, a selective alpha2-adrenergic receptor agonist that regulates norepinephrine release has been approved for management of opioid withdrawal in the United Kingdom since 1992.
The advisory committee voted to recommend lofexidine on the strength of the results of two randomized, double-blind, and placebo controlled phase 3 studies on the safety and efficacy of lofexidine for symptomatic treatment of opioid withdrawal between days 1 through 7. One study randomized 264 patients to lofexidine (134) or placebo (130), with patients in the treatment arm received 3.2 mg of lofexidine on days 1-5, then placebo until day 7. The second study randomized 603 patients to three groups, comparing high dose (3.2 mg/day) and low dose (2.4 mg/day) regimens of lofexidine to placebo; patients in the treatment arms took four smaller doses of lofexidine throughout the day to achieve the cumulative dose.
Researchers enrolled heavy users of short-acting opioids; heroin was the predominant agent. Both studies were conducted in the scenario of abrupt withdrawal, or the most intense withdrawal situation.
Symptomatic benefit was measured using the Short Opiate Withdrawal Scale of Gossop (SOWS-Gossop), a patient reported outcome. Patients were asked to rank their symptoms as none, mild, moderate or severe across measures like feeling sick, stomach cramps, and heart pounding among other symptoms.
Lofexidine increased completion of withdrawal treatment compared to placebo. Patients in the first study had a 5-day completion rate of 53%, compared to 35% for the placebo group. Researchers observed similar results in the 7-day completion rates the second study, with low and high dose completion rates of 42% and 40%, respectively, both of which were much higher than placebo (28%).
Lofexidine also reduced patient withdrawal symptoms, according to SOWS-Gossop scores during peak withdrawal. In the first study, SOWS-Gossop scores were 2-4 points lower in the lofexidine group compared to placebo. Similarly, the scores were significantly better in both lofexidine groups in the second study, compared to placebo, particularly on days 1 to 4. Decreasing withdrawal symptoms during this period is particularly important because this is the most vulnerable window for patient dropout, briefing documents from US WorldMeds.
Several notable adverse events occurred during the study, particularly at higher doses of lofexidine. The risk of bradycardia and hypotension are prominent in patients taking lofexidine, but these are risks associated with this class of drug, according to Mark Pirner, MD, senior medical director at US WorldMeds, who noted that “the lower dose, if that’s what ultimately gets approved [by the FDA], is safe and effective too.”
Development of lofexidine was conducted in collaboration with the National Institute on Drug Abuse and the FDA, according to briefing documents from US WorldMeds.
The Prescription Drug User Fee Act (PDUFA) for lofexidine is May 26; FDA actions on new drug applications often occur at near the PDUFA date.
The FDA is not obligated to follow the recommendations of its advisory committees.
REPORTING FROM AN FDA ADVISORY COMMITTEE MEETING

From the Washington Office: An opportunity to address policymakers on the concerns of Fellows
On March 15, 2018, I had the opportunity to present on behalf of the ACS at a roundtable discussion on Capitol Hill to members of the House Ways and Means Committee on the topic of Medicare red tape relief
The roundtable provided members of this key committee of jurisdiction over Medicare policy the opportunity to hear from representatives from a variety of health care professional organizations on how Congress can improve Medicare to work more effectively and efficiently for both patients and providers. Each group was allotted just three minutes for their presentation. A summary of my presentation is included below:
E/M Documentation Guidelines
The ACS has significant concerns regarding Evaluation and Management (E/M) Documentation Guidelines. Though CMS created the E/M documentation guidelines 23 years ago with the laudable goal of adding structure to the various levels of E/M services, and in an effort to create a sense of equivalency of E/M services across the multitude of specialties, ACS believes the time has come to re-examine and revise these guidelines to be more appropriate in the modern digital information era.

Again, the primary goal of all medical record documentation is to provide an accurate, chronologic record of patient care. That said, the medical record also serves other important goals including communication between providers, data exchange to facilitate clinical decisions, and a legal document. The payment-focused E/M documentation guidelines do not serve any of these objectives.
There must be some level of trust of the provider by the payers. Physicians should have the ability to meet the primary goal of the medical record without being required to repeatedly enter the same information. If a family history is recorded on Monday, there should be no requirement to re-record it on Thursday unless something cogent changes in the interim. ACS believes that documentation should focus on the minimum data elements needed to establish an accurate chronologic record of patient care.
The ACS is prepared to assist in an effort to explore ways to revise the current paper-based E/M documentation guidelines such that they more efficiently and accurately document patient care information in the modern digital era.
Meaningful Measurement of Surgical Quality
I also addressed concerns relative to the meaningful measurement of surgical quality. Despite having expended significant human and financial resources toward helping Fellows succeed in MIPS, the College is becoming increasingly concerned that MIPS is not actually measuring surgical quality, and therefore, is not a quality program for surgery and serves primarily as a payment program.
As evidence, the most recent quality metric data available (from the 2015 Physician Quality Reporting System) show that many of the CMS quality measures reported by surgeons have little to do with improving the quality of the actual surgical care provided to patients. For general surgeons, the two most commonly reported measures were the documentation of a patient’s medications in the medical record and tobacco use screening. While no one would deny the importance of either of these activities, neither is of much real value in the effort to measure the quality of surgical care provided. In another, perhaps even more illustrative example, one of the most common quality measures reported by urologists was inquiring of their patients whether they had received a pneumovax. This obviously has little to do with why one would see an urologist, much less the quality of care provided.
As an organization, the ACS and its members are absolutely dedicated to improving the quality of care they provide to their patients. However, the quality measures forming the basis of the assessment of their care must first be relevant to the surgical care they provide, and second be achievable. Fellows are increasingly expressing concerns about the burdens imposed by the Quality component of MIPS and believe their efforts to participate do little to meaningfully measure the quality of surgical care they provide. I asked that the Ways and Means Committee hold a hearing specifically addressing issues relative to the Quality component of MIPS.
Standardizing Electronic Prior Authorization for Safe Prescribing Act
I expressed ACS’ support for the Standardizing Electronic Prior Authorization for Safe Prescribing Act, H.R. 4841, which would allow for electronic prior authorization under Medicare Part D and allow for the creation of technical standards for the electronic transmission of prior authorization. While the College believes this legislation is a good first step for electronic prior authorization, I asked that the scope of the legislation be expanded to include all medical services, supplies, and prescription drugs covered by the Medicare program, and also requested prior authorization policies be standardized across all insurers and that prior authorization requests, decisions, and appeals processes be automated through uniform electronic transaction portals for all services and supplies.
As evidence, I provided data from a 2017 ACS survey of nearly 300 surgeons and their staff, which indicated that, on average, a medical practice receives approximately 37 prior authorization requests per provider, per week. Action on these requests requires 25 hours to complete. This exorbitant expenditure of time and resources required for prior authorization is largely due to a lack of automated prior authorization processes that integrate with current electronic health record systems. The ACS is committed to working with the bill’s sponsors and the Ways and Means Committee toward a goal of swift passage.
Questions from and discussion with members of the Ways and Means Committee were truncated because of votes on the House floor. We look forward to continuing the dialogue in the coming weeks when the roundtable is reconvened.
Until next month ….
On March 15, 2018, I had the opportunity to present on behalf of the ACS at a roundtable discussion on Capitol Hill to members of the House Ways and Means Committee on the topic of Medicare red tape relief
The roundtable provided members of this key committee of jurisdiction over Medicare policy the opportunity to hear from representatives from a variety of health care professional organizations on how Congress can improve Medicare to work more effectively and efficiently for both patients and providers. Each group was allotted just three minutes for their presentation. A summary of my presentation is included below:
E/M Documentation Guidelines
The ACS has significant concerns regarding Evaluation and Management (E/M) Documentation Guidelines. Though CMS created the E/M documentation guidelines 23 years ago with the laudable goal of adding structure to the various levels of E/M services, and in an effort to create a sense of equivalency of E/M services across the multitude of specialties, ACS believes the time has come to re-examine and revise these guidelines to be more appropriate in the modern digital information era.
Again, the primary goal of all medical record documentation is to provide an accurate, chronologic record of patient care. That said, the medical record also serves other important goals including communication between providers, data exchange to facilitate clinical decisions, and a legal document. The payment-focused E/M documentation guidelines do not serve any of these objectives.
There must be some level of trust of the provider by the payers. Physicians should have the ability to meet the primary goal of the medical record without being required to repeatedly enter the same information. If a family history is recorded on Monday, there should be no requirement to re-record it on Thursday unless something cogent changes in the interim. ACS believes that documentation should focus on the minimum data elements needed to establish an accurate chronologic record of patient care.
The ACS is prepared to assist in an effort to explore ways to revise the current paper-based E/M documentation guidelines such that they more efficiently and accurately document patient care information in the modern digital era.
Meaningful Measurement of Surgical Quality
I also addressed concerns relative to the meaningful measurement of surgical quality. Despite having expended significant human and financial resources toward helping Fellows succeed in MIPS, the College is becoming increasingly concerned that MIPS is not actually measuring surgical quality, and therefore, is not a quality program for surgery and serves primarily as a payment program.
As evidence, the most recent quality metric data available (from the 2015 Physician Quality Reporting System) show that many of the CMS quality measures reported by surgeons have little to do with improving the quality of the actual surgical care provided to patients. For general surgeons, the two most commonly reported measures were the documentation of a patient’s medications in the medical record and tobacco use screening. While no one would deny the importance of either of these activities, neither is of much real value in the effort to measure the quality of surgical care provided. In another, perhaps even more illustrative example, one of the most common quality measures reported by urologists was inquiring of their patients whether they had received a pneumovax. This obviously has little to do with why one would see an urologist, much less the quality of care provided.
As an organization, the ACS and its members are absolutely dedicated to improving the quality of care they provide to their patients. However, the quality measures forming the basis of the assessment of their care must first be relevant to the surgical care they provide, and second be achievable. Fellows are increasingly expressing concerns about the burdens imposed by the Quality component of MIPS and believe their efforts to participate do little to meaningfully measure the quality of surgical care they provide. I asked that the Ways and Means Committee hold a hearing specifically addressing issues relative to the Quality component of MIPS.
Standardizing Electronic Prior Authorization for Safe Prescribing Act
I expressed ACS’ support for the Standardizing Electronic Prior Authorization for Safe Prescribing Act, H.R. 4841, which would allow for electronic prior authorization under Medicare Part D and allow for the creation of technical standards for the electronic transmission of prior authorization. While the College believes this legislation is a good first step for electronic prior authorization, I asked that the scope of the legislation be expanded to include all medical services, supplies, and prescription drugs covered by the Medicare program, and also requested prior authorization policies be standardized across all insurers and that prior authorization requests, decisions, and appeals processes be automated through uniform electronic transaction portals for all services and supplies.
As evidence, I provided data from a 2017 ACS survey of nearly 300 surgeons and their staff, which indicated that, on average, a medical practice receives approximately 37 prior authorization requests per provider, per week. Action on these requests requires 25 hours to complete. This exorbitant expenditure of time and resources required for prior authorization is largely due to a lack of automated prior authorization processes that integrate with current electronic health record systems. The ACS is committed to working with the bill’s sponsors and the Ways and Means Committee toward a goal of swift passage.
Questions from and discussion with members of the Ways and Means Committee were truncated because of votes on the House floor. We look forward to continuing the dialogue in the coming weeks when the roundtable is reconvened.
Until next month ….
On March 15, 2018, I had the opportunity to present on behalf of the ACS at a roundtable discussion on Capitol Hill to members of the House Ways and Means Committee on the topic of Medicare red tape relief
The roundtable provided members of this key committee of jurisdiction over Medicare policy the opportunity to hear from representatives from a variety of health care professional organizations on how Congress can improve Medicare to work more effectively and efficiently for both patients and providers. Each group was allotted just three minutes for their presentation. A summary of my presentation is included below:
E/M Documentation Guidelines
The ACS has significant concerns regarding Evaluation and Management (E/M) Documentation Guidelines. Though CMS created the E/M documentation guidelines 23 years ago with the laudable goal of adding structure to the various levels of E/M services, and in an effort to create a sense of equivalency of E/M services across the multitude of specialties, ACS believes the time has come to re-examine and revise these guidelines to be more appropriate in the modern digital information era.
Again, the primary goal of all medical record documentation is to provide an accurate, chronologic record of patient care. That said, the medical record also serves other important goals including communication between providers, data exchange to facilitate clinical decisions, and a legal document. The payment-focused E/M documentation guidelines do not serve any of these objectives.
There must be some level of trust of the provider by the payers. Physicians should have the ability to meet the primary goal of the medical record without being required to repeatedly enter the same information. If a family history is recorded on Monday, there should be no requirement to re-record it on Thursday unless something cogent changes in the interim. ACS believes that documentation should focus on the minimum data elements needed to establish an accurate chronologic record of patient care.
The ACS is prepared to assist in an effort to explore ways to revise the current paper-based E/M documentation guidelines such that they more efficiently and accurately document patient care information in the modern digital era.
Meaningful Measurement of Surgical Quality
I also addressed concerns relative to the meaningful measurement of surgical quality. Despite having expended significant human and financial resources toward helping Fellows succeed in MIPS, the College is becoming increasingly concerned that MIPS is not actually measuring surgical quality, and therefore, is not a quality program for surgery and serves primarily as a payment program.
As evidence, the most recent quality metric data available (from the 2015 Physician Quality Reporting System) show that many of the CMS quality measures reported by surgeons have little to do with improving the quality of the actual surgical care provided to patients. For general surgeons, the two most commonly reported measures were the documentation of a patient’s medications in the medical record and tobacco use screening. While no one would deny the importance of either of these activities, neither is of much real value in the effort to measure the quality of surgical care provided. In another, perhaps even more illustrative example, one of the most common quality measures reported by urologists was inquiring of their patients whether they had received a pneumovax. This obviously has little to do with why one would see an urologist, much less the quality of care provided.
As an organization, the ACS and its members are absolutely dedicated to improving the quality of care they provide to their patients. However, the quality measures forming the basis of the assessment of their care must first be relevant to the surgical care they provide, and second be achievable. Fellows are increasingly expressing concerns about the burdens imposed by the Quality component of MIPS and believe their efforts to participate do little to meaningfully measure the quality of surgical care they provide. I asked that the Ways and Means Committee hold a hearing specifically addressing issues relative to the Quality component of MIPS.
Standardizing Electronic Prior Authorization for Safe Prescribing Act
I expressed ACS’ support for the Standardizing Electronic Prior Authorization for Safe Prescribing Act, H.R. 4841, which would allow for electronic prior authorization under Medicare Part D and allow for the creation of technical standards for the electronic transmission of prior authorization. While the College believes this legislation is a good first step for electronic prior authorization, I asked that the scope of the legislation be expanded to include all medical services, supplies, and prescription drugs covered by the Medicare program, and also requested prior authorization policies be standardized across all insurers and that prior authorization requests, decisions, and appeals processes be automated through uniform electronic transaction portals for all services and supplies.
As evidence, I provided data from a 2017 ACS survey of nearly 300 surgeons and their staff, which indicated that, on average, a medical practice receives approximately 37 prior authorization requests per provider, per week. Action on these requests requires 25 hours to complete. This exorbitant expenditure of time and resources required for prior authorization is largely due to a lack of automated prior authorization processes that integrate with current electronic health record systems. The ACS is committed to working with the bill’s sponsors and the Ways and Means Committee toward a goal of swift passage.
Questions from and discussion with members of the Ways and Means Committee were truncated because of votes on the House floor. We look forward to continuing the dialogue in the coming weeks when the roundtable is reconvened.
Until next month ….
VIDEO: Pelvic radiation surpasses brachytherapy/chemo for early endometrial cancer
NEW ORLEANS – Pelvic radiation was as effective for producing recurrence-free survival as vaginal cuff brachytherapy plus chemotherapy but with less acute toxicity and fewer local recurrences in women with high-risk stage I or stage II endometrial cancer in a multicenter, randomized trial with 601 patients.
These findings should result in wider use of pelvic radiation as the preferred treatment for these patients, Marcus E. Randall, MD, said at the annual meeting of the Society of Gynecologic Oncology. “It will change practice,” he predicted.
Dr. Randall and his colleagues from the Gynecologic Oncology Group (which recently became part of NRG Oncology) designed the trial, GOG-0249, to address recent interest in using vaginal cuff brachytherapy plus chemotherapy with carboplatin and paclitaxel as an alternative to the more standard approach of pelvic radiation for treating women with either high-risk stage I or stage II endometrial cancers. Clinicians had considered the brachytherapy plus chemotherapy approach a reasonable option by “extrapolating from studies with advanced” endometrial cancer, but with no direct evidence to support this alternative, Dr. Randall explained in a video interview.
To generate evidence, the researchers enrolled 601 patients at several participating U.S. centers and followed them for a median of 53 months (4.4 years), with 259 patients treated and followed in the pelvic radiation arm and 268 patients treated and followed in the brachytherapy plus chemotherapy arm. Clinicians administered the complete planned treatment regimen to 91% of patients assigned to pelvic radiation and to 87% of those assigned to brachytherapy plus chemotherapy. Three quarters of enrolled patients had high-risk stage I disease, and the entire study group averaged about 62 years old.
The trial’s primary endpoint was recurrence-free survival, which occurred in 78% of the pelvic radiation patients and 79% of brachytherapy plus chemotherapy patients after 5 years when analyzed on an intention-to-treat basis. The two subgroups also showed similar rates of overall survival during follow-up.
Although the two treatments produced essentially identical outcomes for the primary result, they showed two important differences on secondary outcomes, reported Dr. Randall, professor and chair of radiation medicine at the University of Kentucky in Lexington.
Acute adverse effects rated as grade 3 severity or higher occurred in 11% of the pelvic radiation patients and in 64% of the brachytherapy plus chemotherapy patients, although late toxicities occurred at similar rates (13% and 12%, respectively) in the two subgroups.
Local pelvic and para-aortic nodal recurrences occurred in 4% of the pelvic radiation patients and in 9% of the brachytherapy plus chemotherapy patients, a 53% relative risk reduction with pelvic radiation. The difference in the nodal recurrences was apparent within the first year of follow-up, and the difference in rates continued to steadily widen over time after that. However the rates of both vaginal and distant recurrences were very similar in the two treatment arms. Distant recurrences were the most common type of treatment failure, occurring in about 18% of patients in both subgroups during complete follow-up.
“Pelvic radiation therapy remains an appropriate and preferable treatment for high-risk, early stage endometrial carcinoma,” Dr. Randall concluded.
SOURCE: Randall ME et al. SGO 2018.
NEW ORLEANS – Pelvic radiation was as effective for producing recurrence-free survival as vaginal cuff brachytherapy plus chemotherapy but with less acute toxicity and fewer local recurrences in women with high-risk stage I or stage II endometrial cancer in a multicenter, randomized trial with 601 patients.
These findings should result in wider use of pelvic radiation as the preferred treatment for these patients, Marcus E. Randall, MD, said at the annual meeting of the Society of Gynecologic Oncology. “It will change practice,” he predicted.
Dr. Randall and his colleagues from the Gynecologic Oncology Group (which recently became part of NRG Oncology) designed the trial, GOG-0249, to address recent interest in using vaginal cuff brachytherapy plus chemotherapy with carboplatin and paclitaxel as an alternative to the more standard approach of pelvic radiation for treating women with either high-risk stage I or stage II endometrial cancers. Clinicians had considered the brachytherapy plus chemotherapy approach a reasonable option by “extrapolating from studies with advanced” endometrial cancer, but with no direct evidence to support this alternative, Dr. Randall explained in a video interview.
To generate evidence, the researchers enrolled 601 patients at several participating U.S. centers and followed them for a median of 53 months (4.4 years), with 259 patients treated and followed in the pelvic radiation arm and 268 patients treated and followed in the brachytherapy plus chemotherapy arm. Clinicians administered the complete planned treatment regimen to 91% of patients assigned to pelvic radiation and to 87% of those assigned to brachytherapy plus chemotherapy. Three quarters of enrolled patients had high-risk stage I disease, and the entire study group averaged about 62 years old.
The trial’s primary endpoint was recurrence-free survival, which occurred in 78% of the pelvic radiation patients and 79% of brachytherapy plus chemotherapy patients after 5 years when analyzed on an intention-to-treat basis. The two subgroups also showed similar rates of overall survival during follow-up.
Although the two treatments produced essentially identical outcomes for the primary result, they showed two important differences on secondary outcomes, reported Dr. Randall, professor and chair of radiation medicine at the University of Kentucky in Lexington.
Acute adverse effects rated as grade 3 severity or higher occurred in 11% of the pelvic radiation patients and in 64% of the brachytherapy plus chemotherapy patients, although late toxicities occurred at similar rates (13% and 12%, respectively) in the two subgroups.
Local pelvic and para-aortic nodal recurrences occurred in 4% of the pelvic radiation patients and in 9% of the brachytherapy plus chemotherapy patients, a 53% relative risk reduction with pelvic radiation. The difference in the nodal recurrences was apparent within the first year of follow-up, and the difference in rates continued to steadily widen over time after that. However the rates of both vaginal and distant recurrences were very similar in the two treatment arms. Distant recurrences were the most common type of treatment failure, occurring in about 18% of patients in both subgroups during complete follow-up.
“Pelvic radiation therapy remains an appropriate and preferable treatment for high-risk, early stage endometrial carcinoma,” Dr. Randall concluded.
SOURCE: Randall ME et al. SGO 2018.
NEW ORLEANS – Pelvic radiation was as effective for producing recurrence-free survival as vaginal cuff brachytherapy plus chemotherapy but with less acute toxicity and fewer local recurrences in women with high-risk stage I or stage II endometrial cancer in a multicenter, randomized trial with 601 patients.
These findings should result in wider use of pelvic radiation as the preferred treatment for these patients, Marcus E. Randall, MD, said at the annual meeting of the Society of Gynecologic Oncology. “It will change practice,” he predicted.
Dr. Randall and his colleagues from the Gynecologic Oncology Group (which recently became part of NRG Oncology) designed the trial, GOG-0249, to address recent interest in using vaginal cuff brachytherapy plus chemotherapy with carboplatin and paclitaxel as an alternative to the more standard approach of pelvic radiation for treating women with either high-risk stage I or stage II endometrial cancers. Clinicians had considered the brachytherapy plus chemotherapy approach a reasonable option by “extrapolating from studies with advanced” endometrial cancer, but with no direct evidence to support this alternative, Dr. Randall explained in a video interview.
To generate evidence, the researchers enrolled 601 patients at several participating U.S. centers and followed them for a median of 53 months (4.4 years), with 259 patients treated and followed in the pelvic radiation arm and 268 patients treated and followed in the brachytherapy plus chemotherapy arm. Clinicians administered the complete planned treatment regimen to 91% of patients assigned to pelvic radiation and to 87% of those assigned to brachytherapy plus chemotherapy. Three quarters of enrolled patients had high-risk stage I disease, and the entire study group averaged about 62 years old.
The trial’s primary endpoint was recurrence-free survival, which occurred in 78% of the pelvic radiation patients and 79% of brachytherapy plus chemotherapy patients after 5 years when analyzed on an intention-to-treat basis. The two subgroups also showed similar rates of overall survival during follow-up.
Although the two treatments produced essentially identical outcomes for the primary result, they showed two important differences on secondary outcomes, reported Dr. Randall, professor and chair of radiation medicine at the University of Kentucky in Lexington.
Acute adverse effects rated as grade 3 severity or higher occurred in 11% of the pelvic radiation patients and in 64% of the brachytherapy plus chemotherapy patients, although late toxicities occurred at similar rates (13% and 12%, respectively) in the two subgroups.
Local pelvic and para-aortic nodal recurrences occurred in 4% of the pelvic radiation patients and in 9% of the brachytherapy plus chemotherapy patients, a 53% relative risk reduction with pelvic radiation. The difference in the nodal recurrences was apparent within the first year of follow-up, and the difference in rates continued to steadily widen over time after that. However the rates of both vaginal and distant recurrences were very similar in the two treatment arms. Distant recurrences were the most common type of treatment failure, occurring in about 18% of patients in both subgroups during complete follow-up.
“Pelvic radiation therapy remains an appropriate and preferable treatment for high-risk, early stage endometrial carcinoma,” Dr. Randall concluded.
SOURCE: Randall ME et al. SGO 2018.
REPORTING FROM SGO 2018
Key clinical point:
Major finding: Acute, higher-grade toxicities occurred in 11% of pelvic radiation patients and 64% of brachytherapy/chemotherapy patients.
Study details: GOG-0249, a multicenter, randomized phase III trial with 601 patients.
Disclosures: GOG-0249 had no commercial funding. Dr. Randall had no disclosures.
Source: Randall ME et al. SGO 2018.

Diabetes does its part to increase health care costs
, which was enough to make it “the most costly chronic illness in the country,” the American Diabetes Association said.
The estimated total economic burden of diabetes went from an inflation-adjusted estimate of $261 billion in 2012 to $327 billion – $237 billion in direct medical costs and $90 billion in indirect costs such as absenteeism, reduced productivity, and premature mortality – in 2017, according to a new report from the ADA published in Diabetes Care.
“One of every four health care dollars is incurred by someone with diagnosed diabetes, and one of every seven health care dollars is spent directly treating diabetes and its complications,” the ADA said in a written statement.
The study used data from a large number of sources, including the American Community Survey, the OptumInsight de-identified Normative Health Information database, the Medical Expenditure Panel Survey, and the Medicare 5% sample Standard Analytical Files. All cost estimates were extrapolated to the 2017 U.S. population and adjusted to 2017 dollars.
SOURCE: Diabetes Care. 2018 Mar 22. doi: 10.2337/dci18-0007.
, which was enough to make it “the most costly chronic illness in the country,” the American Diabetes Association said.
The estimated total economic burden of diabetes went from an inflation-adjusted estimate of $261 billion in 2012 to $327 billion – $237 billion in direct medical costs and $90 billion in indirect costs such as absenteeism, reduced productivity, and premature mortality – in 2017, according to a new report from the ADA published in Diabetes Care.
“One of every four health care dollars is incurred by someone with diagnosed diabetes, and one of every seven health care dollars is spent directly treating diabetes and its complications,” the ADA said in a written statement.
The study used data from a large number of sources, including the American Community Survey, the OptumInsight de-identified Normative Health Information database, the Medical Expenditure Panel Survey, and the Medicare 5% sample Standard Analytical Files. All cost estimates were extrapolated to the 2017 U.S. population and adjusted to 2017 dollars.
SOURCE: Diabetes Care. 2018 Mar 22. doi: 10.2337/dci18-0007.
, which was enough to make it “the most costly chronic illness in the country,” the American Diabetes Association said.
The estimated total economic burden of diabetes went from an inflation-adjusted estimate of $261 billion in 2012 to $327 billion – $237 billion in direct medical costs and $90 billion in indirect costs such as absenteeism, reduced productivity, and premature mortality – in 2017, according to a new report from the ADA published in Diabetes Care.
“One of every four health care dollars is incurred by someone with diagnosed diabetes, and one of every seven health care dollars is spent directly treating diabetes and its complications,” the ADA said in a written statement.
The study used data from a large number of sources, including the American Community Survey, the OptumInsight de-identified Normative Health Information database, the Medical Expenditure Panel Survey, and the Medicare 5% sample Standard Analytical Files. All cost estimates were extrapolated to the 2017 U.S. population and adjusted to 2017 dollars.
SOURCE: Diabetes Care. 2018 Mar 22. doi: 10.2337/dci18-0007.
FROM DIABETES CARE