User login
Fingolimod cuts pediatric MS relapse rate more than interferon beta-1a
Pediatric patients with relapsing remitting multiple sclerosis (MS) had fewer relapses after receiving the oral drug fingolimod when compared with patients who received intramuscular interferon beta-1a in the randomized, double-blind PARADIGMS study, suggesting that the sphingosine-1-phosphate receptor modulator could offer a new treatment option to patients younger than 18 years.
A new agent in this patient population is particularly important because most children and adolescents have the relapsing remitting form of the disease, and generally experience a relapse rate that is two to three times higher than that seen in people with adult-onset multiple sclerosis.

The phase 3 PARADIGMS study is the first international controlled trial to evaluate the safety and efficacy of fingolimod in pediatric and adolescent patients. Dr. Chitnis and her colleagues randomized 215 participants aged 10-17 years to up to 0.5 mg/day of fingolimod based on body weight or to a once-a-week intramuscular injection of 30 mcg of interferon beta-1a. The trial lasted 2 years and was followed by an open-label extension for an additional 5 years.
The annualized relapse rate was the primary endpoint. The fingolimod group experienced 25 relapses in 180 patient-years, compared with 120 relapses in 163 patient-years in the interferon beta-1a group.
MRI findings and outcomes associated with relapse were secondary endpoints. The researchers found that, compared with the interferon beta-1a group, patients randomized to fingolimod had fewer lesions identified on MRI: There was a 53% annualized reduction in new or newly enlarged T2 lesions and 66% decrease in gadolinium-enhancing T1 lesions.
“These results indicate that fingolimod is more effective than the current standard of care, beta-interferon, in patients aged 10-17 and is a consideration for treatment in teenagers,” said Dr. Chitnis, director of the Partners Pediatric MS Center at the MassGeneral Hospital for Children, Boston. “The overall safety profile was reasonable, and there were no new major adverse events observed in comparison to adult studies.”
Participants were primarily Caucasian (92%) and female (62%). The mean age of each group at randomization was similar: 15.2 years in the fingolimod group and 15.4 years in the interferon beta-1a group. Disease duration since onset of first symptom was shorter in the fingolimod patients, a mean of 1.9 years, compared with a mean 2.4 years in the interferon beta-1a patients. At baseline, patients in both groups reported a mean 1.5 relapses in the previous year and a median Expanded Disability Status Scale (EDSS) score of 1.5.
To be included in the study, the children and teenagers had to have an EDSS score of 0 to 5.5; one or more relapses in the past year or two relapses in the previous two years; or MRI evidence of one or more gadolinium-enhancing lesions in the 6 months prior to trial randomization.
Fingolimod (Gilenya) is approved for the first-line treatment of relapsing forms of MS in adults in the United States. It is not yet FDA-approved for treatment of pediatric patients.
Dr. Chitnis said she was somewhat surprised by the strength of the findings in the PARADIGMS study. “These results showed very strong efficacy in young patients. However, as this study was being conducted, our group looked in more detail at the young adult subpopulation in the pivotal fingolimod adult studies, there was an improved effect in younger adults [those younger than 20 or younger than 30], compared to the entire group. Thus, one could extrapolate that the effects in adolescents would follow and show even greater efficacy.”
The study was sponsored by Novartis, the maker of fingolimod. Dr. Chitnis and nearly all of her coauthors disclosed financial ties to Novartis. Three authors are employees of Novartis.
SOURCE: Chitnis T et al. ACTRIMS Forum 2018, Abstract P025.
Pediatric patients with relapsing remitting multiple sclerosis (MS) had fewer relapses after receiving the oral drug fingolimod when compared with patients who received intramuscular interferon beta-1a in the randomized, double-blind PARADIGMS study, suggesting that the sphingosine-1-phosphate receptor modulator could offer a new treatment option to patients younger than 18 years.
A new agent in this patient population is particularly important because most children and adolescents have the relapsing remitting form of the disease, and generally experience a relapse rate that is two to three times higher than that seen in people with adult-onset multiple sclerosis.
The phase 3 PARADIGMS study is the first international controlled trial to evaluate the safety and efficacy of fingolimod in pediatric and adolescent patients. Dr. Chitnis and her colleagues randomized 215 participants aged 10-17 years to up to 0.5 mg/day of fingolimod based on body weight or to a once-a-week intramuscular injection of 30 mcg of interferon beta-1a. The trial lasted 2 years and was followed by an open-label extension for an additional 5 years.
The annualized relapse rate was the primary endpoint. The fingolimod group experienced 25 relapses in 180 patient-years, compared with 120 relapses in 163 patient-years in the interferon beta-1a group.
MRI findings and outcomes associated with relapse were secondary endpoints. The researchers found that, compared with the interferon beta-1a group, patients randomized to fingolimod had fewer lesions identified on MRI: There was a 53% annualized reduction in new or newly enlarged T2 lesions and 66% decrease in gadolinium-enhancing T1 lesions.
“These results indicate that fingolimod is more effective than the current standard of care, beta-interferon, in patients aged 10-17 and is a consideration for treatment in teenagers,” said Dr. Chitnis, director of the Partners Pediatric MS Center at the MassGeneral Hospital for Children, Boston. “The overall safety profile was reasonable, and there were no new major adverse events observed in comparison to adult studies.”
Participants were primarily Caucasian (92%) and female (62%). The mean age of each group at randomization was similar: 15.2 years in the fingolimod group and 15.4 years in the interferon beta-1a group. Disease duration since onset of first symptom was shorter in the fingolimod patients, a mean of 1.9 years, compared with a mean 2.4 years in the interferon beta-1a patients. At baseline, patients in both groups reported a mean 1.5 relapses in the previous year and a median Expanded Disability Status Scale (EDSS) score of 1.5.
To be included in the study, the children and teenagers had to have an EDSS score of 0 to 5.5; one or more relapses in the past year or two relapses in the previous two years; or MRI evidence of one or more gadolinium-enhancing lesions in the 6 months prior to trial randomization.
Fingolimod (Gilenya) is approved for the first-line treatment of relapsing forms of MS in adults in the United States. It is not yet FDA-approved for treatment of pediatric patients.
Dr. Chitnis said she was somewhat surprised by the strength of the findings in the PARADIGMS study. “These results showed very strong efficacy in young patients. However, as this study was being conducted, our group looked in more detail at the young adult subpopulation in the pivotal fingolimod adult studies, there was an improved effect in younger adults [those younger than 20 or younger than 30], compared to the entire group. Thus, one could extrapolate that the effects in adolescents would follow and show even greater efficacy.”
The study was sponsored by Novartis, the maker of fingolimod. Dr. Chitnis and nearly all of her coauthors disclosed financial ties to Novartis. Three authors are employees of Novartis.
SOURCE: Chitnis T et al. ACTRIMS Forum 2018, Abstract P025.
Pediatric patients with relapsing remitting multiple sclerosis (MS) had fewer relapses after receiving the oral drug fingolimod when compared with patients who received intramuscular interferon beta-1a in the randomized, double-blind PARADIGMS study, suggesting that the sphingosine-1-phosphate receptor modulator could offer a new treatment option to patients younger than 18 years.
A new agent in this patient population is particularly important because most children and adolescents have the relapsing remitting form of the disease, and generally experience a relapse rate that is two to three times higher than that seen in people with adult-onset multiple sclerosis.
The phase 3 PARADIGMS study is the first international controlled trial to evaluate the safety and efficacy of fingolimod in pediatric and adolescent patients. Dr. Chitnis and her colleagues randomized 215 participants aged 10-17 years to up to 0.5 mg/day of fingolimod based on body weight or to a once-a-week intramuscular injection of 30 mcg of interferon beta-1a. The trial lasted 2 years and was followed by an open-label extension for an additional 5 years.
The annualized relapse rate was the primary endpoint. The fingolimod group experienced 25 relapses in 180 patient-years, compared with 120 relapses in 163 patient-years in the interferon beta-1a group.
MRI findings and outcomes associated with relapse were secondary endpoints. The researchers found that, compared with the interferon beta-1a group, patients randomized to fingolimod had fewer lesions identified on MRI: There was a 53% annualized reduction in new or newly enlarged T2 lesions and 66% decrease in gadolinium-enhancing T1 lesions.
“These results indicate that fingolimod is more effective than the current standard of care, beta-interferon, in patients aged 10-17 and is a consideration for treatment in teenagers,” said Dr. Chitnis, director of the Partners Pediatric MS Center at the MassGeneral Hospital for Children, Boston. “The overall safety profile was reasonable, and there were no new major adverse events observed in comparison to adult studies.”
Participants were primarily Caucasian (92%) and female (62%). The mean age of each group at randomization was similar: 15.2 years in the fingolimod group and 15.4 years in the interferon beta-1a group. Disease duration since onset of first symptom was shorter in the fingolimod patients, a mean of 1.9 years, compared with a mean 2.4 years in the interferon beta-1a patients. At baseline, patients in both groups reported a mean 1.5 relapses in the previous year and a median Expanded Disability Status Scale (EDSS) score of 1.5.
To be included in the study, the children and teenagers had to have an EDSS score of 0 to 5.5; one or more relapses in the past year or two relapses in the previous two years; or MRI evidence of one or more gadolinium-enhancing lesions in the 6 months prior to trial randomization.
Fingolimod (Gilenya) is approved for the first-line treatment of relapsing forms of MS in adults in the United States. It is not yet FDA-approved for treatment of pediatric patients.
Dr. Chitnis said she was somewhat surprised by the strength of the findings in the PARADIGMS study. “These results showed very strong efficacy in young patients. However, as this study was being conducted, our group looked in more detail at the young adult subpopulation in the pivotal fingolimod adult studies, there was an improved effect in younger adults [those younger than 20 or younger than 30], compared to the entire group. Thus, one could extrapolate that the effects in adolescents would follow and show even greater efficacy.”
The study was sponsored by Novartis, the maker of fingolimod. Dr. Chitnis and nearly all of her coauthors disclosed financial ties to Novartis. Three authors are employees of Novartis.
SOURCE: Chitnis T et al. ACTRIMS Forum 2018, Abstract P025.
FROM ACTRIMS FORUM 2018
Key clinical point:
Major finding: The fingolimod group experienced 25 relapses in 180 patient-years, compared with 120 relapses in 163 patient-years in the interferon beta-1a group.
Study details: International, randomized, double-blind, parallel-group study of 215 people aged 10-17 years.
Disclosures: The study was sponsored by Novartis, the maker of fingolimod. Dr. Chitnis and nearly all of her coauthors disclosed financial ties to Novartis. Three authors are employees of Novartis.
Source: Chitnis T et al. ACTRIMS Forum 2018, Abstract P025.
Days of Therapy Avoided: A Novel Method for Measuring the Impact of an Antimicrobial Stewardship Program to Stop Antibiotics
A proposed metric to quantify the impact of an antimicrobial stewardship program (ASP) is using changes in the antibiotic days of therapy (DOT) per 1000 patient-days, which is the total number of days any dose of an antibiotic is administered during a specified time period, standardized by the number of patient-days.1 Although DOT is useful for comparing antibiotic use among hospitals or time periods, this metric is a composite result of an ASP’s often multifaceted approach to improving antibiotic use. Thus, DOT provides a loose estimate of the direct impact of specific ASP activities and does not quantify the amount of antibiotics directly avoided or direct cost savings on the patient level. To ameliorate this, we reviewed our institution’s ASP prospective audit and feedback (PAF) and applied a novel metric, days of therapy avoided (DOTA), to calculate the number of antibiotic days avoided that directly result from our ASP’s actions targeting antibiotic stoppage. From DOTA, we also calculate attributable cost savings.
METHODS
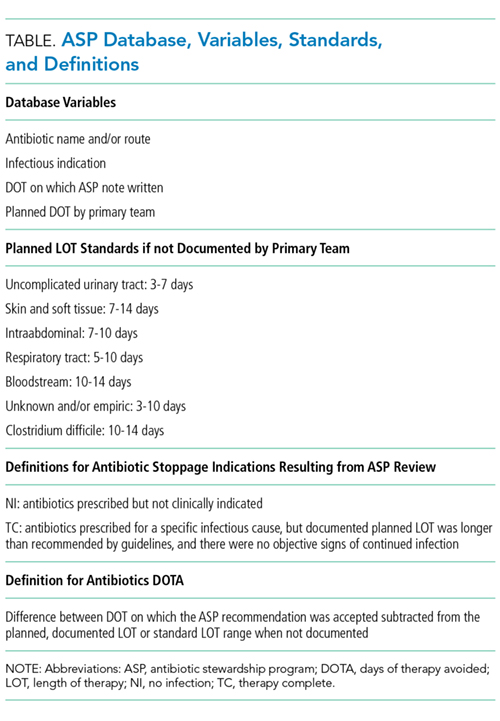
To quantify the direct impact of PAF, DOTA (Table) was calculated. Antibiotic costs avoided were calculated by multiplying the average wholesale price (AWP) per day (range: $0.44-$534; mean: $67.85) by DOTA. This calculation was done twice under 2 assumptions: that PAF led to the prevention of (1) 1 more day of antibiotic prescription and (2) the remainder of the documented or assumed LOT.
RESULTS
Over 4 years, the ASP made 1594 interventions to stop antibiotics. Accepted interventions totaled 1151 (72%): 513 (44.5%) for NI and 638 (55.4%) for TC, involving 431 and 575 unique patients, respectively. Nearly half (45.8%) of the NI interventions targeted asymptomatic bacteriuria, whereas respiratory tract infections were the most common (42.2%) indication for the TC intervention.
Under the most conservative assumption that each accepted PAF recommendation avoided 1 day of unnecessary antibiotics, we estimated a total of 1151 DOTA; 690 (59.9%) were intravenous antibiotics. The average DOT on which the PAF note was written was 3.07 ± 1.69 for NI and 6.38 ± 2.73 for TC. A planned LOT was documented for only 36.7% of the courses. On the basis of documented or assumed LOT, we estimate that the NI and TC interventions led to between 1077 and 2826 DOTA and between 397 and 1598 DOTA, respectively. Potential fluoroquinolone DOTA ranged from 300 to 1126; for third- and fourth-generation cephalosporins, there were 314 to 1017 DOTA.
Using the conservative estimate of 1151 DOTA, the costs avoided totaled $16,700, which includes $10,700 for intravenous antibiotics. When the AWP per day of each antibiotic was applied to the remaining LOTs avoided, the maximum potential cost savings was $67,100. Additional cost savings may have been realized if indirect expenses, such as pharmacy preparation and nursing administration time or costs of medical supplies, were evaluated.
CONCLUSION
We investigated DOTA as a measure of the direct patient-level and intervention-specific impact of an ASP’s PAF. DOTA may be useful for ASPs with limited access to an electronic record or electronically generated DOT reports because DOTA and cost savings can be tracked manually and prospectively with each accepted intervention. DOTA can also help ASPs identify which clinical conditions are responsible for the most antibiotic overuse, and thus may benefit from the development of clinical treatment guidelines. We found that the highest yield areas for DOTA were targeting asymptomatic bacteriuria (NI) and respiratory infections (TC). In doing so, these have also succeeded in reducing high-risk, broad-spectrum antimicrobials, such as fluoroquinolones and advanced-generation cephalosporins. Further research is needed to assess if DOTA correlates with other ASP metrics and clinical outcomes; however, current evidence supports that reducing unnecessary antibiotic use is fundamental to reducing antibiotic resistance and adverse events.10
The limitations of measuring DOTA include time consumption, particularly if not collected prospectively. However, we make several conclusions. ASP PAF stopping antibiotics was well accepted and reduced antibiotic use. Second, calculating DOTA requires little technology and only knowledge of the planned LOT and drug costs. DOTA also identifies which infectious indications to focus PAF efforts on and gain the greatest impact. Overall, DOTA is a simple, useful, and promising measurement of the direct antibiotic and economic impacts of specific ASP PAF and warrants further investigation as an ASP metric.
Acknowledgments
The authors thank the patients and RGH staff, particularly the departments of infectious diseases, pharmacy, and internal medicine, for their support.
Disclosure
The authors declare no conflicts of interest. This study was previously presented in poster form at the Society for Healthcare Epidemiology of America Spring Conference in St. Louis, Missouri (March 29-31, 2017).
1. Moehring RW, Anderson DJ, Cochran RL, Hicks LA, Srinivasan A, Dodds-Ashley ES. Structured Taskforce of Experts Working at Reliable Standards for Stewardship Panel. Expert consensus on metrics to assess the impact of patient-level antimicrobial stewardship interventions in acute-care settings. Clin Infect Dis. 2016;64(3):377-383. PubMed
2. Gupta K, Hooton TM, Naber KG, et al. International clinical practice guidelines for the treatment of acute uncomplicated cystitis and pyelonephritis in women: a 2010 update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clin Infect Dis. 2011;52(5):e103-e120. PubMed
3. Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59(2):e10-e52. PubMed
4. Lipsky BA, Berendt AR, Cornia PB, et al. 2012 Infectious Diseases Society of America clinical practice guideline for the diagnosis and treatment of diabetic foot infections. Clin Infect Dis. 2012;54(12):e132-e173. PubMed
5. Solomkin JS, Mazuski JE, Bradley JS, et al. Diagnosis and management of complicated intraabdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin Infect Dis. 2010;50(2):133-164. PubMed
6. Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis. 2007;44(Supplement 2):S27-S72. PubMed
7. American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171(4):388-416. PubMed
8. Havey TC, Fowler RA, Daneman N. Duration of antibiotic therapy for bacteremia: a systematic review and meta-analysis. Crit Care. 2011;15(6):R267. PubMed
9. Cohen SH, Gerding DN, Johnson S, Kelly CP. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455. PubMed
10. Llewelyn MJ, Fitzpatrick JM, Darwin E, et al. The antibiotic course has had its day. BMJ 2017;358:j3418. PubMed
A proposed metric to quantify the impact of an antimicrobial stewardship program (ASP) is using changes in the antibiotic days of therapy (DOT) per 1000 patient-days, which is the total number of days any dose of an antibiotic is administered during a specified time period, standardized by the number of patient-days.1 Although DOT is useful for comparing antibiotic use among hospitals or time periods, this metric is a composite result of an ASP’s often multifaceted approach to improving antibiotic use. Thus, DOT provides a loose estimate of the direct impact of specific ASP activities and does not quantify the amount of antibiotics directly avoided or direct cost savings on the patient level. To ameliorate this, we reviewed our institution’s ASP prospective audit and feedback (PAF) and applied a novel metric, days of therapy avoided (DOTA), to calculate the number of antibiotic days avoided that directly result from our ASP’s actions targeting antibiotic stoppage. From DOTA, we also calculate attributable cost savings.
METHODS
To quantify the direct impact of PAF, DOTA (Table) was calculated. Antibiotic costs avoided were calculated by multiplying the average wholesale price (AWP) per day (range: $0.44-$534; mean: $67.85) by DOTA. This calculation was done twice under 2 assumptions: that PAF led to the prevention of (1) 1 more day of antibiotic prescription and (2) the remainder of the documented or assumed LOT.
RESULTS
Over 4 years, the ASP made 1594 interventions to stop antibiotics. Accepted interventions totaled 1151 (72%): 513 (44.5%) for NI and 638 (55.4%) for TC, involving 431 and 575 unique patients, respectively. Nearly half (45.8%) of the NI interventions targeted asymptomatic bacteriuria, whereas respiratory tract infections were the most common (42.2%) indication for the TC intervention.
Under the most conservative assumption that each accepted PAF recommendation avoided 1 day of unnecessary antibiotics, we estimated a total of 1151 DOTA; 690 (59.9%) were intravenous antibiotics. The average DOT on which the PAF note was written was 3.07 ± 1.69 for NI and 6.38 ± 2.73 for TC. A planned LOT was documented for only 36.7% of the courses. On the basis of documented or assumed LOT, we estimate that the NI and TC interventions led to between 1077 and 2826 DOTA and between 397 and 1598 DOTA, respectively. Potential fluoroquinolone DOTA ranged from 300 to 1126; for third- and fourth-generation cephalosporins, there were 314 to 1017 DOTA.
Using the conservative estimate of 1151 DOTA, the costs avoided totaled $16,700, which includes $10,700 for intravenous antibiotics. When the AWP per day of each antibiotic was applied to the remaining LOTs avoided, the maximum potential cost savings was $67,100. Additional cost savings may have been realized if indirect expenses, such as pharmacy preparation and nursing administration time or costs of medical supplies, were evaluated.
CONCLUSION
We investigated DOTA as a measure of the direct patient-level and intervention-specific impact of an ASP’s PAF. DOTA may be useful for ASPs with limited access to an electronic record or electronically generated DOT reports because DOTA and cost savings can be tracked manually and prospectively with each accepted intervention. DOTA can also help ASPs identify which clinical conditions are responsible for the most antibiotic overuse, and thus may benefit from the development of clinical treatment guidelines. We found that the highest yield areas for DOTA were targeting asymptomatic bacteriuria (NI) and respiratory infections (TC). In doing so, these have also succeeded in reducing high-risk, broad-spectrum antimicrobials, such as fluoroquinolones and advanced-generation cephalosporins. Further research is needed to assess if DOTA correlates with other ASP metrics and clinical outcomes; however, current evidence supports that reducing unnecessary antibiotic use is fundamental to reducing antibiotic resistance and adverse events.10
The limitations of measuring DOTA include time consumption, particularly if not collected prospectively. However, we make several conclusions. ASP PAF stopping antibiotics was well accepted and reduced antibiotic use. Second, calculating DOTA requires little technology and only knowledge of the planned LOT and drug costs. DOTA also identifies which infectious indications to focus PAF efforts on and gain the greatest impact. Overall, DOTA is a simple, useful, and promising measurement of the direct antibiotic and economic impacts of specific ASP PAF and warrants further investigation as an ASP metric.
Acknowledgments
The authors thank the patients and RGH staff, particularly the departments of infectious diseases, pharmacy, and internal medicine, for their support.
Disclosure
The authors declare no conflicts of interest. This study was previously presented in poster form at the Society for Healthcare Epidemiology of America Spring Conference in St. Louis, Missouri (March 29-31, 2017).
A proposed metric to quantify the impact of an antimicrobial stewardship program (ASP) is using changes in the antibiotic days of therapy (DOT) per 1000 patient-days, which is the total number of days any dose of an antibiotic is administered during a specified time period, standardized by the number of patient-days.1 Although DOT is useful for comparing antibiotic use among hospitals or time periods, this metric is a composite result of an ASP’s often multifaceted approach to improving antibiotic use. Thus, DOT provides a loose estimate of the direct impact of specific ASP activities and does not quantify the amount of antibiotics directly avoided or direct cost savings on the patient level. To ameliorate this, we reviewed our institution’s ASP prospective audit and feedback (PAF) and applied a novel metric, days of therapy avoided (DOTA), to calculate the number of antibiotic days avoided that directly result from our ASP’s actions targeting antibiotic stoppage. From DOTA, we also calculate attributable cost savings.
METHODS
To quantify the direct impact of PAF, DOTA (Table) was calculated. Antibiotic costs avoided were calculated by multiplying the average wholesale price (AWP) per day (range: $0.44-$534; mean: $67.85) by DOTA. This calculation was done twice under 2 assumptions: that PAF led to the prevention of (1) 1 more day of antibiotic prescription and (2) the remainder of the documented or assumed LOT.
RESULTS
Over 4 years, the ASP made 1594 interventions to stop antibiotics. Accepted interventions totaled 1151 (72%): 513 (44.5%) for NI and 638 (55.4%) for TC, involving 431 and 575 unique patients, respectively. Nearly half (45.8%) of the NI interventions targeted asymptomatic bacteriuria, whereas respiratory tract infections were the most common (42.2%) indication for the TC intervention.
Under the most conservative assumption that each accepted PAF recommendation avoided 1 day of unnecessary antibiotics, we estimated a total of 1151 DOTA; 690 (59.9%) were intravenous antibiotics. The average DOT on which the PAF note was written was 3.07 ± 1.69 for NI and 6.38 ± 2.73 for TC. A planned LOT was documented for only 36.7% of the courses. On the basis of documented or assumed LOT, we estimate that the NI and TC interventions led to between 1077 and 2826 DOTA and between 397 and 1598 DOTA, respectively. Potential fluoroquinolone DOTA ranged from 300 to 1126; for third- and fourth-generation cephalosporins, there were 314 to 1017 DOTA.
Using the conservative estimate of 1151 DOTA, the costs avoided totaled $16,700, which includes $10,700 for intravenous antibiotics. When the AWP per day of each antibiotic was applied to the remaining LOTs avoided, the maximum potential cost savings was $67,100. Additional cost savings may have been realized if indirect expenses, such as pharmacy preparation and nursing administration time or costs of medical supplies, were evaluated.
CONCLUSION
We investigated DOTA as a measure of the direct patient-level and intervention-specific impact of an ASP’s PAF. DOTA may be useful for ASPs with limited access to an electronic record or electronically generated DOT reports because DOTA and cost savings can be tracked manually and prospectively with each accepted intervention. DOTA can also help ASPs identify which clinical conditions are responsible for the most antibiotic overuse, and thus may benefit from the development of clinical treatment guidelines. We found that the highest yield areas for DOTA were targeting asymptomatic bacteriuria (NI) and respiratory infections (TC). In doing so, these have also succeeded in reducing high-risk, broad-spectrum antimicrobials, such as fluoroquinolones and advanced-generation cephalosporins. Further research is needed to assess if DOTA correlates with other ASP metrics and clinical outcomes; however, current evidence supports that reducing unnecessary antibiotic use is fundamental to reducing antibiotic resistance and adverse events.10
The limitations of measuring DOTA include time consumption, particularly if not collected prospectively. However, we make several conclusions. ASP PAF stopping antibiotics was well accepted and reduced antibiotic use. Second, calculating DOTA requires little technology and only knowledge of the planned LOT and drug costs. DOTA also identifies which infectious indications to focus PAF efforts on and gain the greatest impact. Overall, DOTA is a simple, useful, and promising measurement of the direct antibiotic and economic impacts of specific ASP PAF and warrants further investigation as an ASP metric.
Acknowledgments
The authors thank the patients and RGH staff, particularly the departments of infectious diseases, pharmacy, and internal medicine, for their support.
Disclosure
The authors declare no conflicts of interest. This study was previously presented in poster form at the Society for Healthcare Epidemiology of America Spring Conference in St. Louis, Missouri (March 29-31, 2017).
1. Moehring RW, Anderson DJ, Cochran RL, Hicks LA, Srinivasan A, Dodds-Ashley ES. Structured Taskforce of Experts Working at Reliable Standards for Stewardship Panel. Expert consensus on metrics to assess the impact of patient-level antimicrobial stewardship interventions in acute-care settings. Clin Infect Dis. 2016;64(3):377-383. PubMed
2. Gupta K, Hooton TM, Naber KG, et al. International clinical practice guidelines for the treatment of acute uncomplicated cystitis and pyelonephritis in women: a 2010 update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clin Infect Dis. 2011;52(5):e103-e120. PubMed
3. Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59(2):e10-e52. PubMed
4. Lipsky BA, Berendt AR, Cornia PB, et al. 2012 Infectious Diseases Society of America clinical practice guideline for the diagnosis and treatment of diabetic foot infections. Clin Infect Dis. 2012;54(12):e132-e173. PubMed
5. Solomkin JS, Mazuski JE, Bradley JS, et al. Diagnosis and management of complicated intraabdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin Infect Dis. 2010;50(2):133-164. PubMed
6. Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis. 2007;44(Supplement 2):S27-S72. PubMed
7. American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171(4):388-416. PubMed
8. Havey TC, Fowler RA, Daneman N. Duration of antibiotic therapy for bacteremia: a systematic review and meta-analysis. Crit Care. 2011;15(6):R267. PubMed
9. Cohen SH, Gerding DN, Johnson S, Kelly CP. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455. PubMed
10. Llewelyn MJ, Fitzpatrick JM, Darwin E, et al. The antibiotic course has had its day. BMJ 2017;358:j3418. PubMed
1. Moehring RW, Anderson DJ, Cochran RL, Hicks LA, Srinivasan A, Dodds-Ashley ES. Structured Taskforce of Experts Working at Reliable Standards for Stewardship Panel. Expert consensus on metrics to assess the impact of patient-level antimicrobial stewardship interventions in acute-care settings. Clin Infect Dis. 2016;64(3):377-383. PubMed
2. Gupta K, Hooton TM, Naber KG, et al. International clinical practice guidelines for the treatment of acute uncomplicated cystitis and pyelonephritis in women: a 2010 update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clin Infect Dis. 2011;52(5):e103-e120. PubMed
3. Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59(2):e10-e52. PubMed
4. Lipsky BA, Berendt AR, Cornia PB, et al. 2012 Infectious Diseases Society of America clinical practice guideline for the diagnosis and treatment of diabetic foot infections. Clin Infect Dis. 2012;54(12):e132-e173. PubMed
5. Solomkin JS, Mazuski JE, Bradley JS, et al. Diagnosis and management of complicated intraabdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin Infect Dis. 2010;50(2):133-164. PubMed
6. Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis. 2007;44(Supplement 2):S27-S72. PubMed
7. American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171(4):388-416. PubMed
8. Havey TC, Fowler RA, Daneman N. Duration of antibiotic therapy for bacteremia: a systematic review and meta-analysis. Crit Care. 2011;15(6):R267. PubMed
9. Cohen SH, Gerding DN, Johnson S, Kelly CP. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455. PubMed
10. Llewelyn MJ, Fitzpatrick JM, Darwin E, et al. The antibiotic course has had its day. BMJ 2017;358:j3418. PubMed
© 2018 Society of Hospital Medicine
Immunotherapy-Induced Colitis: An Emerging Problem for the Hospitalist
Immune checkpoint inhibitors (ICIs), a form of immunotherapy, have changed the management of cancer since their introduction in 2011.1 They were initially tested on melanoma.2 Their use in the advanced stages of the disease demonstrated a 2-year survival of 18% compared with 5% by using other therapies.3 Similar results were observed in nonsmall cell lung carcinoma (NSCLC); the overall survival benefit was 3 months with the use of ICIs compared with traditional chemotherapy (42% and 24% at 1 year, respectively).4 Antitumor activity has also been seen in the treatment of other malignancies, including renal cell carcinoma,5 bladder carcinoma,6,7 head and neck carcinoma,8 colorectal cancer,9 Hodgkin lymphoma,10 and, more recently, hepatocellular carcinoma.11 The use of ICIs has also been linked to serious complications.12 Although the skin, kidneys, lungs, and endocrine and nervous systems may be affected, complications of the gastrointestinal (GI) tract are frequent and can be life-threatening.12-16 We performed a thorough review of the literature to familiarize hospitalists with the mechanism of action and uses of ICIs, the clinical presentation of their GI toxicity, and the current recommendations regarding diagnosis and treatment.
CASE PRESENTATION
A 66-year-old man was admitted to our institution with a 1-week history of severe, diffuse abdominal pain and profuse watery diarrhea. He reported having more than 8 watery bowel movements per day and denied fever, recent travel, ill contacts, or ingestion of undercooked food. He had a history of metastatic melanoma and was undergoing treatment with both nivolumab and ipilimumab; the drugs were started 6 weeks prior to presentation. Physical examination revealed a heart rate of 110 beats/minute while supine and 123 beats/minute while standing, blood pressure of 112/69 mm Hg while supine and 92/62 mm Hg while standing, and a temperature of 37.2°C. He was in mild distress and had dry oral mucosa. Abdominal examination revealed hyperactive bowel sounds and mild diffuse abdominal tenderness with no guarding or rebound. His extremities were cool, but peripheral pulses were present. Initial laboratory results included a hemoglobin level of 15.3 g/dL (range 12.0-16.0 mg/dL), white blood cell count 14.2 × 109/L (range 4.5-11.0 × 109/L), and platelet count 236 × 109/L (range 150-400 × 109/L); other test results included a sodium level of 130 mmol/L (range 135-145 mmol/L), potassium 2.3 mmol/L (range 3.5-5.5 mmol/L), serum creatinine 2.2 mg/dL (range 0.8-1.3 mg/dL), blood urea nitrogen 72 mg/dL (range 8-21 mg/dL), and serum venous lactate 5.9 mmol/L (range 0.9-1.7 mmol/L).
MECHANISM OF ACTION AND USES OF ICIS
T-cell lymphocytes play a pivotal role in acquired immunity, but their function requires an appropriate balance between stimulatory and inhibitory signals to prevent autoimmunity.17 Immune checkpoint molecules are used by the immune system to assist with this balance.18 Although several of these molecules exist, the cytotoxic T-lymphocyte antigen-4 (CTLA-4) and programmed cell death-1 (PD-1) are among the most widely studied.12
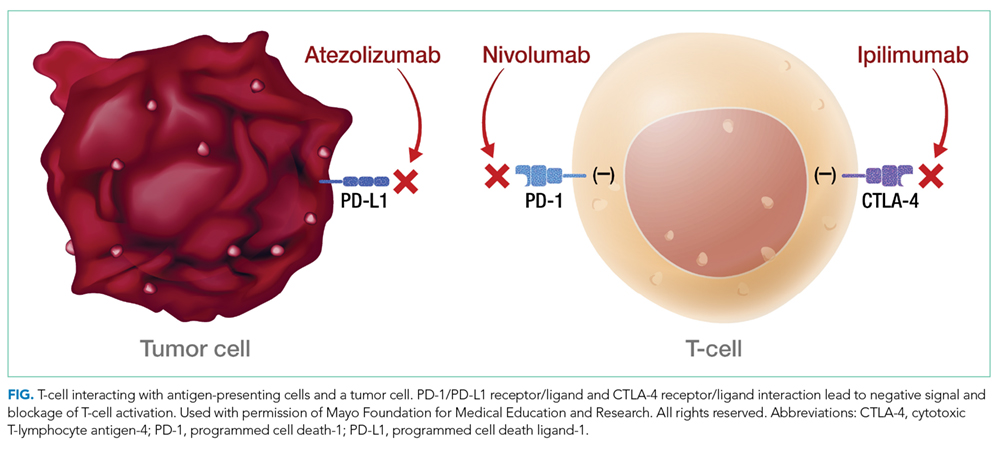
Ipilimumab is a monoclonal antibody directed against CTLA-4.24 After demonstrating survival benefits in patients with unresectable and metastatic melanoma, ipilimumab was the first ICI approved for use by the US Food and Drug Administration (FDA).1,3 Another monoclonal antibody directed against CTLA-4, tremelimumab, is not currently approved for use by the FDA.
TOXIC PROFILE
Because of the sustained T-cell activation, ICIs have been associated with autoimmune-like toxicities known as immune-related adverse events (irAEs).19,31 Because the PD-1/PD-L1 pathway is more tumor-specific than the CTLA-4 pathway,21-23 there is a higher incidence of serious irAEs seen with ipilimumab, reported to be around 27%.18,22 Furthermore, the risk of developing irAEs is dose-dependent and can increase up to 55% when anti-CTLA-4 are used with other ICIs such as nivolumab.13,32-34
The skin and GI tract are the most commonly involved organs.14-16 Skin is affected in 50% of patients receiving ipilimumab and 40% of patients on nivolumab or pembrolizumab, often in the form of a rash or pruritus.12,35-37 The rash is often described as faintly erythematous, reticular, and maculopapular and typically affects the trunk and extremities.38 Importantly, these events usually occur within the first 2 weeks of treatment, and fewer than 5% are severe.12,36,39 A higher percentage of severe adverse events occurs in the GI tract, with a reported incidence of 12%.3,14,36,39
CLINICAL PRESENTATION
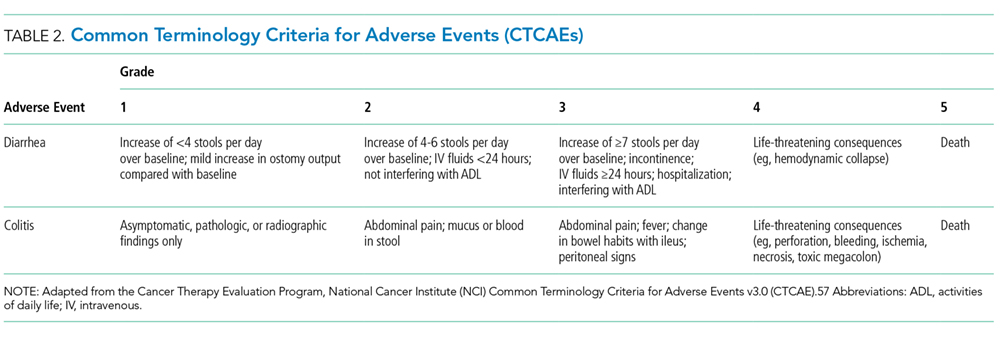
Colitis, defined by either the presence of symptoms or radiologic findings suggestive of inflammation, occurs less often than diarrhea alone, with a reported incidence of 2.3%.37,43 This incidence increases to almost 12% when anti-CTLA-4 and anti-PD-1/PD-L1 are combined.32 Colitis symptoms include abdominal pain (20%), nausea and vomiting (15%), fever (12%), and, less often, bloody diarrhea or rectal bleeding.19,20 Colitis severity is graded according to the CTCAE (Table 2).42 Most patients have mild colitis (grade 1 or 2).19 The risk for developing severe colitis (grade 3 or higher) is almost 10 times higher with the use of anti-CTLA-4 compared with anti-PD-1/PD-L1 agents.43 Patients with severe disease are at risk of developing life-threatening complications, such as ileus, toxic megacolon, bowel ischemia, necrosis, or even perforation, which has been reported in up to 5% of patients with colitis because of ipilimumab.13,17
CASE APPROACH STRATEGY
Based on the patient’s symptoms, physical findings, and temporal relationship to ICI therapy, he was believed to have immune-mediated colitis. Stool studies, including those looking for ova and parasites, C
DIAGNOSIS
In a patient undergoing ICI treatment who has diarrhea, the initial assessment should exclude C. difficile and Salmonella by stool culture, PCR, or pathogenic antigens.19 Cytomegalovirus reactivation should also be considered. Immune-mediated colitis and infection can coexist; thus, a positive infectious etiology does not rule out the presence of immune colitis or vice versa.44 Fecal calprotectin, a marker of neutrophil-associated inflammation, is nonspecific for ICI-induced colitis; however, it may help to distinguish inflammatory from noninflammatory diarrhea.33,45
No clear guideline exists for the use of abdominal imaging. Some experts suggest using computed tomography in patients with severe, persistent, or progressive symptoms in order to exclude bowel obstruction, toxic megacolon, or perforation.19,46
In patients with typical symptoms, and after infectious etiologies are ruled out, empiric use of corticosteroids can be initiated without an endoscopic evaluation, which is not necessary to establish a diagnosis and rarely changes management.12,37,47 In patients with atypical presentations or for whom the diagnosis remains in question, endoscopic evaluation with biopsies may be required. Macroscopic findings may be similar to those seen with inflammatory bowel disease (IBD), including erythema, edema, ulceration, granularity, or loss of vascular pattern. Although immune-mediated colitis affects the descending colon more often than IBD, this feature and any macroscopic findings are insufficient to make this distinction.20,36 Furthermore, the lack of macroscopic abnormalities does not rule out immune-mediated colitis.20
When endoscopic biopsies are obtained, histologic findings for anti-CTLA-4 medications (eg, ipilimumab) usually follow 3 patterns: neutrophilic infiltrate (46%), lymphocytic infiltrate (15%), and mixed infiltrate (38%).41 Other findings include crypt abscesses and tissue destruction.20 No biopsy-specific pattern has been described with anti-PD-1/PD-L1 medications, such as nivolumab or pembrolizumab.18 A normal colonic tissue does not exclude the presence of an irAE, as cases of isolated ileitis48 or enteritis49 without colitis can also occur.
CASE MANAGEMENT STRATEGY
The patient was started on intravenous (IV) methylprednisolone 2 mg/kg twice a day. After 48 hours, he still had more than 7 episodes of diarrhea per day, so he was treated with 1 dose of infliximab 5 mg/kg without stopping corticosteroids. Within 72 hours, the patient’s abdominal pain improved and his diarrhea stopped. He was discharged on an 8-week taper of prednisone starting at 1 mg/kg/day, pneumocystis pneumonia (PCP) prophylaxis was started, and ICI therapy was discontinued indefinitely.
MANAGEMENT OF COLITIS
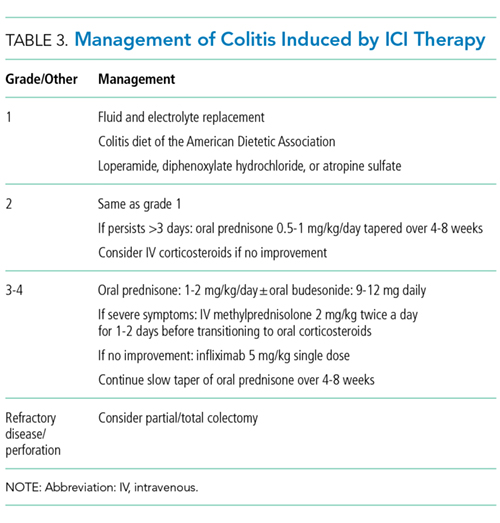
Management of grade 1 and 2 colitis is mainly supportive, consisting of fluid and electrolyte replacement, the American Dietetic Association colitis diet, and antimotility agents, such as loperamide, oral diphenoxylate hydrochloride, or atropine sulfate.36,37 Persistent grade 2 symptoms (lasting >3 days), should prompt initiation of 0.5 to 1 mg/kg/day of oral prednisone or an equivalent.19 If symptoms do not improve with oral corticosteroids, patient hospitalization for IV corticosteroids should be considered.37 Importantly, opioids and antidiarrheals may mask the pain and severity of symptoms and, therefore, should be used cautiously.19
Patients with grade 3 and 4 colitis (≥7 stools per day, severe abdominal pain, or complications) require the use of systemic corticosteroids at a dose of 1 to 2 mg/kg/day of prednisone or an equivalent.15 Patients who fail to respond to prednisone alone may benefit from the addition of oral budesonide at a dose of 9 to 12 mg/day.50 In severe cases of colitis, hospitalization may be necessary for IV hydration, electrolyte replacement, and IV methylprednisolone at a starting dose of 2 mg/kg twice a day for 1 to 2 days before transitioning to oral corticosteroids.12,15 Though improvement is usually noted within the first 2 weeks of treatment, prednisone should be slowly tapered over a period of 4 to 8 weeks to ensure complete healing and prevent relapse.20,36 Patients who receive an equivalent dose of prednisone 20 mg daily during a period of 4 weeks or more should receive PCP prophylaxis.51 Some patients fail to respond to IV corticosteroids despite adequate dosing. Many of these patients have severe disease, possibly because of delayed recognition and initiation of treatment.19 As with IBD, the addition of infliximab to corticosteroids at 5 mg/kg as a single dose is usually successful for this population subset.52-54 Although a response is seen within 1 to 3 days,41 some patients benefit from an additional dose of infliximab 2 weeks after the initial dose.19 If sepsis or perforation is suspected at any point, corticosteroids or infliximab should be avoided and antibiotics should be started immediately.15,19 Patients with a medically unresponsive disease may require partial or complete colectomy.20 The use of prophylactic budesonide to prevent diarrhea or colitis has not been proven effective and should not be used.55 Despite complications, mortality from colitis has markedly decreased given the increased awareness of this adverse event, reduction in the time to recognition and treatment, and increased adherence to corticosteroids.12
Treating physicians may be delayed in starting appropriate therapy because patients are concerned that using corticosteroids will negatively impact immunotherapy efficacy. Current evidence shows that the use of temporary immunosuppression to treat irAEs does not affect overall survival, efficacy, or time to treatment failure of the ICI.12,56 Restarting ICI therapy is a complex decision and should always be individualized. In grade 1 and 2 colitis, ICI therapy is typically restarted after symptoms have improved.5 In grade 3 and 4 colitis, ICI therapy is often permanently discontinued.20
CONCLUSION
ICIs have not only increased our understanding of the biology of cancer, but they have also improved survival in advanced stages of malignancies like melanoma, NSCLC, and renal cell carcinoma. The expanding use of these medications increases the likelihood that healthcare providers will encounter patients experiencing their adverse events.
Immune-mediated GI adverse events include a wide range of symptoms, from mild diarrhea to severe colitis complicated by perforation and death. Diagnosis requires exclusion of an infectious process. Early recognition and treatment with corticosteroids or another immunosuppressant such as infliximab hastens recovery and decreases complications and mortality. Treatment should be started within 5 days of symptom onset. Corticosteroids should be slowly tapered for no less than 4 weeks to prevent relapse and PCP prophylaxis administered in appropriate patients. Restarting ICI therapy may be considered in cases of mild colitis, but in severe cases, ICI therapy is usually discontinued.
Disclosure
Julian Marin-Acevedo, Dana Harris, and M. Caroline Burton have no conflicts of interest or funding sources to declare.
1. Ledford H. Melanoma drug wins US approval. Nature. 2011;471(7340):561. PubMed
2. Ribas A. Clinical development of the anti-CTLA-4 antibody tremelimumab. Semin Oncol. 2010;37(5):450-454. PubMed
3. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723. PubMed
4. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373(2):123-135. PubMed
5. Motzer RJ, Rini BI, McDermott DF, et al. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J Clin Oncol. 2015;33(13):1430-1437. PubMed
6. Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558-562. PubMed
7. Massard C, Gordon MS, Sharma S, et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti-Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J Clin Oncol. 2016;34(26):3119-3125. PubMed
8. Ferris RL, Blumenschein G Jr, Fayette J, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375(19):1856-1867. PubMed
9. Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509-2520. PubMed
10. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311-319. PubMed
11. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088)2492-2502. PubMed
12. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the Immune-Related Adverse Effects of Immune Checkpoint Inhibitors: A Review. JAMA Oncol. 2016;2(10):1346-1353. PubMed
13. Heinzerling L, Goldinger SM. A review of serious adverse effects under treatment with checkpoint inhibitors. Curr Opin Oncol. 2017;29(2):136-144. PubMed
14. Kahler KC, Hauschild A. Treatment and side effect management of CTLA-4 antibody therapy in metastatic melanoma. J Dtsch Dermatol Ges. 2011;9(4):277-286. PubMed
15. Weber JS, Postow M, Lao CD, Schadendorf D. Management of Adverse Events Following Treatment With Anti-Programmed Death-1 Agents. Oncologist. 2016;21(10):1230-1240. PubMed
16. Bertrand A, Kostine M, Barnetche T, Truchetet ME, Schaeverbeke T. Immune related adverse events associated with anti-CTLA-4 antibodies: systematic review and meta-analysis. BMC Med. 2015;13:211-224. PubMed
17. Abdel-Wahab N, Shah M, Suarez-Almazor ME. Adverse Events Associated with Immune Checkpoint Blockade in Patients with Cancer: A Systematic Review of Case Reports. PLoS One. 2016;11(7):e0160221. doi:10.1371/journal.pone.0160221 PubMed
18. Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26(12):2375-2391. PubMed
19. Gupta A, De Felice KM, Loftus EV Jr, Khanna S. Systematic review: colitis associated with anti-CTLA-4 therapy. Aliment Pharmacol Ther. 2015;42(4):406-417. PubMed
20. Pernot S, Ramtohul T, Taieb J. Checkpoint inhibitors and gastrointestinal immune-related adverse events. Curr Opin Oncol. 2016;28(4):264-268. PubMed
21. Kamata T, Suzuki A, Mise N, et al. Blockade of programmed death-1/programmed death ligand pathway enhances the antitumor immunity of human invariant natural killer T cells. Cancer Immunol Immunother. 2016;65(12):1477-1489. PubMed
22. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252-264. PubMed
23. Velu V, Titanji K, Zhu B, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458(7235):206-210. PubMed
24. Phan GQ, Yang JC, Sherry RM, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2003;100(14):8372-8377. PubMed
25. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Atezolizumab BLA 761041 approval letter (urothelial carcinoma). https://www.genentech-access.com/content/dam/gene/accesssolutions/brands/tecentriq/Appeals%20Tips/TECENTRIQ-FDA-Approval-Letter-Metastatic-Urothelial-Carcinoma-First-Line-Therapy.pdf. Accessed September 30, 2017.
26. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Imfinzi (durvalumab) approval letter. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/761069Orig1s000ltr.pdf. Accessed September 30, 2017.
27. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Bavencio (avelumab) accelerated approval letter - urothelial carcinoma. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/761078Orig1s000ltr.pdf. Accessed May 16, 2017.
28. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Atezolizumab BLA 761041 approval letter (NSCLC).
29. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Bavencio (avelumab) approval letter - Merkel cell carcinoma. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/761049Orig1s000ltr.pdf. Accessed April 27, 2017.
30. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Atezolizumab BLA 761041 approval letter. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761034Orig1s000Approv.pdf. Accessed April 6, 2017.
31. Voskens CJ, Goldinger SM, Loquai C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8(1):e53745. doi:10.1371/journal.pone.0053745. PubMed
32. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373(1):23-34. PubMed
33. Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139-148. PubMed
34. Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res. 2015;4(5):560-575. PubMed
35. Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697. PubMed
36. Kahler KC, Hassel JC, Heinzerling L, et al. Management of side effects of immune checkpoint blockade by anti-CTLA-4 and anti-PD-1 antibodies in metastatic melanoma. J Dtsch Dermatol Ges. 2016;14(7):662-681. PubMed
37. Postow MA. Managing immune checkpoint-blocking antibody side effects. Am Soc Clin Oncol Educ Book. 2015:76-83. PubMed
38. Lacouture ME, Wolchok JD, Yosipovitch G, Kahler KC, Busam KJ, Hauschild A. Ipilimumab in patients with cancer and the management of dermatologic adverse events. J Am Acad Dermatol. 2014;71(1):161-169. PubMed
39. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521-2532. PubMed
40. Weber J. Ipilimumab: controversies in its development, utility and autoimmune adverse events. Cancer Immunol Immunother. 2009;58(5):823-830. PubMed
41. Beck KE, Blansfield JA, Tran KQ, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol. 2006;24(15):2283-2289. PubMed
42. Cancer Therapy Evaluation Program, National Cancer Institute (NCI). Common terminology criteria for adverse events v3.0 (CTCAE). https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed April 9, 2017.
43. De Velasco G, Je Y, Bosse D, et al. Comprehensive Meta-analysis of Key Immune-Related Adverse Events from CTLA-4 and PD-1/PD-L1 Inhibitors in Cancer Patients. Cancer Immunol Res. 2017;5(4):312-318. PubMed
44. McCutcheon JL, McClain CM, Puzanov I, Smith TA. Infectious Colitis Associated With Ipilimumab Therapy. Gastroenterology Res. 2014;7(1):28-31. PubMed
45. Berman D, Parker SM, Siegel J, et al. Blockade of cytotoxic T-lymphocyte antigen-4 by ipilimumab results in dysregulation of gastrointestinal immunity in patients with advanced melanoma. Cancer Immun. 2010;10:11-20. PubMed
46. Reynolds K, Ananthakrishnan A, Dougan M, Bardia A. Immune-Related Adverse Events (irAEs) in Cancer Patients. In: McKean SC, Ross JJ, Dressler DD, Scheurer DB, eds. Principles and Practice of Hospital Medicine. 2nd ed. New York: McGraw-Hill Education; 2017.
47. Garcia-Neuer M, Marmarelis ME, Jangi SR, et al. Diagnostic Comparison of CT Scans and Colonoscopy for Immune-Related Colitis in Ipilimumab-Treated Advanced Melanoma Patients. Cancer Immunol Res. 2017;5(4):286-291. PubMed
48. Venditti O, De Lisi D, Caricato M, et al. Ipilimumab and immune-mediated adverse events: a case report of anti-CTLA4 induced ileitis. BMC Cancer. 2015;15:87-91. PubMed
49. Messmer M, Upreti S, Tarabishy Y, et al. Ipilimumab-Induced Enteritis without Colitis: A New Challenge. Case Rep Oncol. 2016;9(3):705-713. PubMed
50. De Felice KM, Gupta A, Rakshit S, et al. Ipilimumab-induced colitis in patients with metastatic melanoma. Melanoma Res. 2015;25(4):321-327. PubMed
51. Baden LR, Swaminathan S, Angarone M, et al. Prevention and Treatment of Cancer-Related Infections, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Newt. 2017;14(7):882-913. PubMed
52. Minor DR, Chin K, Kashani-Sabet M. Infliximab in the treatment of anti-CTLA4 antibody (ipilimumab) induced immune-related colitis. Cancer Biother Radiopharm. 2009;24(3):321-325. PubMed
53. Merrill SP, Reynolds P, Kalra A, Biehl J, Vandivier RW, Mueller SW. Early administration of infliximab for severe ipilimumab-related diarrhea in a critically ill patient. Ann Pharmacother. 2014;48(6):806-810. PubMed
54. Pages C, Gornet JM, Monsel G, et al. Ipilimumab-induced acute severe colitis treated by infliximab. Melanoma Res. 2013;23(3):227-230. PubMed
55. Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15(17):5591-5598. PubMed
56. Horvat TZ, Adel NG, Dung TO, et al. Immune-Related Adverse Events, Need for Systemic Immunosuppression, and Effects on Survival and Time to Treatment Failure in Patients With Melanoma Treated With Ipilimumab at Memorial Sloan Kettering Cancer Center. J Clin Oncol. 2015;33(28):3193-3198. PubMed
57. Cancer Therapy Evaluation Program, National Cancer Institute (NCI). Common terminology criteria for adverse events v3.0 (CTCAE). https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed April 9, 2017.
Immune checkpoint inhibitors (ICIs), a form of immunotherapy, have changed the management of cancer since their introduction in 2011.1 They were initially tested on melanoma.2 Their use in the advanced stages of the disease demonstrated a 2-year survival of 18% compared with 5% by using other therapies.3 Similar results were observed in nonsmall cell lung carcinoma (NSCLC); the overall survival benefit was 3 months with the use of ICIs compared with traditional chemotherapy (42% and 24% at 1 year, respectively).4 Antitumor activity has also been seen in the treatment of other malignancies, including renal cell carcinoma,5 bladder carcinoma,6,7 head and neck carcinoma,8 colorectal cancer,9 Hodgkin lymphoma,10 and, more recently, hepatocellular carcinoma.11 The use of ICIs has also been linked to serious complications.12 Although the skin, kidneys, lungs, and endocrine and nervous systems may be affected, complications of the gastrointestinal (GI) tract are frequent and can be life-threatening.12-16 We performed a thorough review of the literature to familiarize hospitalists with the mechanism of action and uses of ICIs, the clinical presentation of their GI toxicity, and the current recommendations regarding diagnosis and treatment.
CASE PRESENTATION
A 66-year-old man was admitted to our institution with a 1-week history of severe, diffuse abdominal pain and profuse watery diarrhea. He reported having more than 8 watery bowel movements per day and denied fever, recent travel, ill contacts, or ingestion of undercooked food. He had a history of metastatic melanoma and was undergoing treatment with both nivolumab and ipilimumab; the drugs were started 6 weeks prior to presentation. Physical examination revealed a heart rate of 110 beats/minute while supine and 123 beats/minute while standing, blood pressure of 112/69 mm Hg while supine and 92/62 mm Hg while standing, and a temperature of 37.2°C. He was in mild distress and had dry oral mucosa. Abdominal examination revealed hyperactive bowel sounds and mild diffuse abdominal tenderness with no guarding or rebound. His extremities were cool, but peripheral pulses were present. Initial laboratory results included a hemoglobin level of 15.3 g/dL (range 12.0-16.0 mg/dL), white blood cell count 14.2 × 109/L (range 4.5-11.0 × 109/L), and platelet count 236 × 109/L (range 150-400 × 109/L); other test results included a sodium level of 130 mmol/L (range 135-145 mmol/L), potassium 2.3 mmol/L (range 3.5-5.5 mmol/L), serum creatinine 2.2 mg/dL (range 0.8-1.3 mg/dL), blood urea nitrogen 72 mg/dL (range 8-21 mg/dL), and serum venous lactate 5.9 mmol/L (range 0.9-1.7 mmol/L).
MECHANISM OF ACTION AND USES OF ICIS
T-cell lymphocytes play a pivotal role in acquired immunity, but their function requires an appropriate balance between stimulatory and inhibitory signals to prevent autoimmunity.17 Immune checkpoint molecules are used by the immune system to assist with this balance.18 Although several of these molecules exist, the cytotoxic T-lymphocyte antigen-4 (CTLA-4) and programmed cell death-1 (PD-1) are among the most widely studied.12
Ipilimumab is a monoclonal antibody directed against CTLA-4.24 After demonstrating survival benefits in patients with unresectable and metastatic melanoma, ipilimumab was the first ICI approved for use by the US Food and Drug Administration (FDA).1,3 Another monoclonal antibody directed against CTLA-4, tremelimumab, is not currently approved for use by the FDA.
TOXIC PROFILE
Because of the sustained T-cell activation, ICIs have been associated with autoimmune-like toxicities known as immune-related adverse events (irAEs).19,31 Because the PD-1/PD-L1 pathway is more tumor-specific than the CTLA-4 pathway,21-23 there is a higher incidence of serious irAEs seen with ipilimumab, reported to be around 27%.18,22 Furthermore, the risk of developing irAEs is dose-dependent and can increase up to 55% when anti-CTLA-4 are used with other ICIs such as nivolumab.13,32-34
The skin and GI tract are the most commonly involved organs.14-16 Skin is affected in 50% of patients receiving ipilimumab and 40% of patients on nivolumab or pembrolizumab, often in the form of a rash or pruritus.12,35-37 The rash is often described as faintly erythematous, reticular, and maculopapular and typically affects the trunk and extremities.38 Importantly, these events usually occur within the first 2 weeks of treatment, and fewer than 5% are severe.12,36,39 A higher percentage of severe adverse events occurs in the GI tract, with a reported incidence of 12%.3,14,36,39
CLINICAL PRESENTATION
Colitis, defined by either the presence of symptoms or radiologic findings suggestive of inflammation, occurs less often than diarrhea alone, with a reported incidence of 2.3%.37,43 This incidence increases to almost 12% when anti-CTLA-4 and anti-PD-1/PD-L1 are combined.32 Colitis symptoms include abdominal pain (20%), nausea and vomiting (15%), fever (12%), and, less often, bloody diarrhea or rectal bleeding.19,20 Colitis severity is graded according to the CTCAE (Table 2).42 Most patients have mild colitis (grade 1 or 2).19 The risk for developing severe colitis (grade 3 or higher) is almost 10 times higher with the use of anti-CTLA-4 compared with anti-PD-1/PD-L1 agents.43 Patients with severe disease are at risk of developing life-threatening complications, such as ileus, toxic megacolon, bowel ischemia, necrosis, or even perforation, which has been reported in up to 5% of patients with colitis because of ipilimumab.13,17
CASE APPROACH STRATEGY
Based on the patient’s symptoms, physical findings, and temporal relationship to ICI therapy, he was believed to have immune-mediated colitis. Stool studies, including those looking for ova and parasites, C
DIAGNOSIS
In a patient undergoing ICI treatment who has diarrhea, the initial assessment should exclude C. difficile and Salmonella by stool culture, PCR, or pathogenic antigens.19 Cytomegalovirus reactivation should also be considered. Immune-mediated colitis and infection can coexist; thus, a positive infectious etiology does not rule out the presence of immune colitis or vice versa.44 Fecal calprotectin, a marker of neutrophil-associated inflammation, is nonspecific for ICI-induced colitis; however, it may help to distinguish inflammatory from noninflammatory diarrhea.33,45
No clear guideline exists for the use of abdominal imaging. Some experts suggest using computed tomography in patients with severe, persistent, or progressive symptoms in order to exclude bowel obstruction, toxic megacolon, or perforation.19,46
In patients with typical symptoms, and after infectious etiologies are ruled out, empiric use of corticosteroids can be initiated without an endoscopic evaluation, which is not necessary to establish a diagnosis and rarely changes management.12,37,47 In patients with atypical presentations or for whom the diagnosis remains in question, endoscopic evaluation with biopsies may be required. Macroscopic findings may be similar to those seen with inflammatory bowel disease (IBD), including erythema, edema, ulceration, granularity, or loss of vascular pattern. Although immune-mediated colitis affects the descending colon more often than IBD, this feature and any macroscopic findings are insufficient to make this distinction.20,36 Furthermore, the lack of macroscopic abnormalities does not rule out immune-mediated colitis.20
When endoscopic biopsies are obtained, histologic findings for anti-CTLA-4 medications (eg, ipilimumab) usually follow 3 patterns: neutrophilic infiltrate (46%), lymphocytic infiltrate (15%), and mixed infiltrate (38%).41 Other findings include crypt abscesses and tissue destruction.20 No biopsy-specific pattern has been described with anti-PD-1/PD-L1 medications, such as nivolumab or pembrolizumab.18 A normal colonic tissue does not exclude the presence of an irAE, as cases of isolated ileitis48 or enteritis49 without colitis can also occur.
CASE MANAGEMENT STRATEGY
The patient was started on intravenous (IV) methylprednisolone 2 mg/kg twice a day. After 48 hours, he still had more than 7 episodes of diarrhea per day, so he was treated with 1 dose of infliximab 5 mg/kg without stopping corticosteroids. Within 72 hours, the patient’s abdominal pain improved and his diarrhea stopped. He was discharged on an 8-week taper of prednisone starting at 1 mg/kg/day, pneumocystis pneumonia (PCP) prophylaxis was started, and ICI therapy was discontinued indefinitely.
MANAGEMENT OF COLITIS
Management of grade 1 and 2 colitis is mainly supportive, consisting of fluid and electrolyte replacement, the American Dietetic Association colitis diet, and antimotility agents, such as loperamide, oral diphenoxylate hydrochloride, or atropine sulfate.36,37 Persistent grade 2 symptoms (lasting >3 days), should prompt initiation of 0.5 to 1 mg/kg/day of oral prednisone or an equivalent.19 If symptoms do not improve with oral corticosteroids, patient hospitalization for IV corticosteroids should be considered.37 Importantly, opioids and antidiarrheals may mask the pain and severity of symptoms and, therefore, should be used cautiously.19
Patients with grade 3 and 4 colitis (≥7 stools per day, severe abdominal pain, or complications) require the use of systemic corticosteroids at a dose of 1 to 2 mg/kg/day of prednisone or an equivalent.15 Patients who fail to respond to prednisone alone may benefit from the addition of oral budesonide at a dose of 9 to 12 mg/day.50 In severe cases of colitis, hospitalization may be necessary for IV hydration, electrolyte replacement, and IV methylprednisolone at a starting dose of 2 mg/kg twice a day for 1 to 2 days before transitioning to oral corticosteroids.12,15 Though improvement is usually noted within the first 2 weeks of treatment, prednisone should be slowly tapered over a period of 4 to 8 weeks to ensure complete healing and prevent relapse.20,36 Patients who receive an equivalent dose of prednisone 20 mg daily during a period of 4 weeks or more should receive PCP prophylaxis.51 Some patients fail to respond to IV corticosteroids despite adequate dosing. Many of these patients have severe disease, possibly because of delayed recognition and initiation of treatment.19 As with IBD, the addition of infliximab to corticosteroids at 5 mg/kg as a single dose is usually successful for this population subset.52-54 Although a response is seen within 1 to 3 days,41 some patients benefit from an additional dose of infliximab 2 weeks after the initial dose.19 If sepsis or perforation is suspected at any point, corticosteroids or infliximab should be avoided and antibiotics should be started immediately.15,19 Patients with a medically unresponsive disease may require partial or complete colectomy.20 The use of prophylactic budesonide to prevent diarrhea or colitis has not been proven effective and should not be used.55 Despite complications, mortality from colitis has markedly decreased given the increased awareness of this adverse event, reduction in the time to recognition and treatment, and increased adherence to corticosteroids.12
Treating physicians may be delayed in starting appropriate therapy because patients are concerned that using corticosteroids will negatively impact immunotherapy efficacy. Current evidence shows that the use of temporary immunosuppression to treat irAEs does not affect overall survival, efficacy, or time to treatment failure of the ICI.12,56 Restarting ICI therapy is a complex decision and should always be individualized. In grade 1 and 2 colitis, ICI therapy is typically restarted after symptoms have improved.5 In grade 3 and 4 colitis, ICI therapy is often permanently discontinued.20
CONCLUSION
ICIs have not only increased our understanding of the biology of cancer, but they have also improved survival in advanced stages of malignancies like melanoma, NSCLC, and renal cell carcinoma. The expanding use of these medications increases the likelihood that healthcare providers will encounter patients experiencing their adverse events.
Immune-mediated GI adverse events include a wide range of symptoms, from mild diarrhea to severe colitis complicated by perforation and death. Diagnosis requires exclusion of an infectious process. Early recognition and treatment with corticosteroids or another immunosuppressant such as infliximab hastens recovery and decreases complications and mortality. Treatment should be started within 5 days of symptom onset. Corticosteroids should be slowly tapered for no less than 4 weeks to prevent relapse and PCP prophylaxis administered in appropriate patients. Restarting ICI therapy may be considered in cases of mild colitis, but in severe cases, ICI therapy is usually discontinued.
Disclosure
Julian Marin-Acevedo, Dana Harris, and M. Caroline Burton have no conflicts of interest or funding sources to declare.
Immune checkpoint inhibitors (ICIs), a form of immunotherapy, have changed the management of cancer since their introduction in 2011.1 They were initially tested on melanoma.2 Their use in the advanced stages of the disease demonstrated a 2-year survival of 18% compared with 5% by using other therapies.3 Similar results were observed in nonsmall cell lung carcinoma (NSCLC); the overall survival benefit was 3 months with the use of ICIs compared with traditional chemotherapy (42% and 24% at 1 year, respectively).4 Antitumor activity has also been seen in the treatment of other malignancies, including renal cell carcinoma,5 bladder carcinoma,6,7 head and neck carcinoma,8 colorectal cancer,9 Hodgkin lymphoma,10 and, more recently, hepatocellular carcinoma.11 The use of ICIs has also been linked to serious complications.12 Although the skin, kidneys, lungs, and endocrine and nervous systems may be affected, complications of the gastrointestinal (GI) tract are frequent and can be life-threatening.12-16 We performed a thorough review of the literature to familiarize hospitalists with the mechanism of action and uses of ICIs, the clinical presentation of their GI toxicity, and the current recommendations regarding diagnosis and treatment.
CASE PRESENTATION
A 66-year-old man was admitted to our institution with a 1-week history of severe, diffuse abdominal pain and profuse watery diarrhea. He reported having more than 8 watery bowel movements per day and denied fever, recent travel, ill contacts, or ingestion of undercooked food. He had a history of metastatic melanoma and was undergoing treatment with both nivolumab and ipilimumab; the drugs were started 6 weeks prior to presentation. Physical examination revealed a heart rate of 110 beats/minute while supine and 123 beats/minute while standing, blood pressure of 112/69 mm Hg while supine and 92/62 mm Hg while standing, and a temperature of 37.2°C. He was in mild distress and had dry oral mucosa. Abdominal examination revealed hyperactive bowel sounds and mild diffuse abdominal tenderness with no guarding or rebound. His extremities were cool, but peripheral pulses were present. Initial laboratory results included a hemoglobin level of 15.3 g/dL (range 12.0-16.0 mg/dL), white blood cell count 14.2 × 109/L (range 4.5-11.0 × 109/L), and platelet count 236 × 109/L (range 150-400 × 109/L); other test results included a sodium level of 130 mmol/L (range 135-145 mmol/L), potassium 2.3 mmol/L (range 3.5-5.5 mmol/L), serum creatinine 2.2 mg/dL (range 0.8-1.3 mg/dL), blood urea nitrogen 72 mg/dL (range 8-21 mg/dL), and serum venous lactate 5.9 mmol/L (range 0.9-1.7 mmol/L).
MECHANISM OF ACTION AND USES OF ICIS
T-cell lymphocytes play a pivotal role in acquired immunity, but their function requires an appropriate balance between stimulatory and inhibitory signals to prevent autoimmunity.17 Immune checkpoint molecules are used by the immune system to assist with this balance.18 Although several of these molecules exist, the cytotoxic T-lymphocyte antigen-4 (CTLA-4) and programmed cell death-1 (PD-1) are among the most widely studied.12
Ipilimumab is a monoclonal antibody directed against CTLA-4.24 After demonstrating survival benefits in patients with unresectable and metastatic melanoma, ipilimumab was the first ICI approved for use by the US Food and Drug Administration (FDA).1,3 Another monoclonal antibody directed against CTLA-4, tremelimumab, is not currently approved for use by the FDA.
TOXIC PROFILE
Because of the sustained T-cell activation, ICIs have been associated with autoimmune-like toxicities known as immune-related adverse events (irAEs).19,31 Because the PD-1/PD-L1 pathway is more tumor-specific than the CTLA-4 pathway,21-23 there is a higher incidence of serious irAEs seen with ipilimumab, reported to be around 27%.18,22 Furthermore, the risk of developing irAEs is dose-dependent and can increase up to 55% when anti-CTLA-4 are used with other ICIs such as nivolumab.13,32-34
The skin and GI tract are the most commonly involved organs.14-16 Skin is affected in 50% of patients receiving ipilimumab and 40% of patients on nivolumab or pembrolizumab, often in the form of a rash or pruritus.12,35-37 The rash is often described as faintly erythematous, reticular, and maculopapular and typically affects the trunk and extremities.38 Importantly, these events usually occur within the first 2 weeks of treatment, and fewer than 5% are severe.12,36,39 A higher percentage of severe adverse events occurs in the GI tract, with a reported incidence of 12%.3,14,36,39
CLINICAL PRESENTATION
Colitis, defined by either the presence of symptoms or radiologic findings suggestive of inflammation, occurs less often than diarrhea alone, with a reported incidence of 2.3%.37,43 This incidence increases to almost 12% when anti-CTLA-4 and anti-PD-1/PD-L1 are combined.32 Colitis symptoms include abdominal pain (20%), nausea and vomiting (15%), fever (12%), and, less often, bloody diarrhea or rectal bleeding.19,20 Colitis severity is graded according to the CTCAE (Table 2).42 Most patients have mild colitis (grade 1 or 2).19 The risk for developing severe colitis (grade 3 or higher) is almost 10 times higher with the use of anti-CTLA-4 compared with anti-PD-1/PD-L1 agents.43 Patients with severe disease are at risk of developing life-threatening complications, such as ileus, toxic megacolon, bowel ischemia, necrosis, or even perforation, which has been reported in up to 5% of patients with colitis because of ipilimumab.13,17
CASE APPROACH STRATEGY
Based on the patient’s symptoms, physical findings, and temporal relationship to ICI therapy, he was believed to have immune-mediated colitis. Stool studies, including those looking for ova and parasites, C
DIAGNOSIS
In a patient undergoing ICI treatment who has diarrhea, the initial assessment should exclude C. difficile and Salmonella by stool culture, PCR, or pathogenic antigens.19 Cytomegalovirus reactivation should also be considered. Immune-mediated colitis and infection can coexist; thus, a positive infectious etiology does not rule out the presence of immune colitis or vice versa.44 Fecal calprotectin, a marker of neutrophil-associated inflammation, is nonspecific for ICI-induced colitis; however, it may help to distinguish inflammatory from noninflammatory diarrhea.33,45
No clear guideline exists for the use of abdominal imaging. Some experts suggest using computed tomography in patients with severe, persistent, or progressive symptoms in order to exclude bowel obstruction, toxic megacolon, or perforation.19,46
In patients with typical symptoms, and after infectious etiologies are ruled out, empiric use of corticosteroids can be initiated without an endoscopic evaluation, which is not necessary to establish a diagnosis and rarely changes management.12,37,47 In patients with atypical presentations or for whom the diagnosis remains in question, endoscopic evaluation with biopsies may be required. Macroscopic findings may be similar to those seen with inflammatory bowel disease (IBD), including erythema, edema, ulceration, granularity, or loss of vascular pattern. Although immune-mediated colitis affects the descending colon more often than IBD, this feature and any macroscopic findings are insufficient to make this distinction.20,36 Furthermore, the lack of macroscopic abnormalities does not rule out immune-mediated colitis.20
When endoscopic biopsies are obtained, histologic findings for anti-CTLA-4 medications (eg, ipilimumab) usually follow 3 patterns: neutrophilic infiltrate (46%), lymphocytic infiltrate (15%), and mixed infiltrate (38%).41 Other findings include crypt abscesses and tissue destruction.20 No biopsy-specific pattern has been described with anti-PD-1/PD-L1 medications, such as nivolumab or pembrolizumab.18 A normal colonic tissue does not exclude the presence of an irAE, as cases of isolated ileitis48 or enteritis49 without colitis can also occur.
CASE MANAGEMENT STRATEGY
The patient was started on intravenous (IV) methylprednisolone 2 mg/kg twice a day. After 48 hours, he still had more than 7 episodes of diarrhea per day, so he was treated with 1 dose of infliximab 5 mg/kg without stopping corticosteroids. Within 72 hours, the patient’s abdominal pain improved and his diarrhea stopped. He was discharged on an 8-week taper of prednisone starting at 1 mg/kg/day, pneumocystis pneumonia (PCP) prophylaxis was started, and ICI therapy was discontinued indefinitely.
MANAGEMENT OF COLITIS
Management of grade 1 and 2 colitis is mainly supportive, consisting of fluid and electrolyte replacement, the American Dietetic Association colitis diet, and antimotility agents, such as loperamide, oral diphenoxylate hydrochloride, or atropine sulfate.36,37 Persistent grade 2 symptoms (lasting >3 days), should prompt initiation of 0.5 to 1 mg/kg/day of oral prednisone or an equivalent.19 If symptoms do not improve with oral corticosteroids, patient hospitalization for IV corticosteroids should be considered.37 Importantly, opioids and antidiarrheals may mask the pain and severity of symptoms and, therefore, should be used cautiously.19
Patients with grade 3 and 4 colitis (≥7 stools per day, severe abdominal pain, or complications) require the use of systemic corticosteroids at a dose of 1 to 2 mg/kg/day of prednisone or an equivalent.15 Patients who fail to respond to prednisone alone may benefit from the addition of oral budesonide at a dose of 9 to 12 mg/day.50 In severe cases of colitis, hospitalization may be necessary for IV hydration, electrolyte replacement, and IV methylprednisolone at a starting dose of 2 mg/kg twice a day for 1 to 2 days before transitioning to oral corticosteroids.12,15 Though improvement is usually noted within the first 2 weeks of treatment, prednisone should be slowly tapered over a period of 4 to 8 weeks to ensure complete healing and prevent relapse.20,36 Patients who receive an equivalent dose of prednisone 20 mg daily during a period of 4 weeks or more should receive PCP prophylaxis.51 Some patients fail to respond to IV corticosteroids despite adequate dosing. Many of these patients have severe disease, possibly because of delayed recognition and initiation of treatment.19 As with IBD, the addition of infliximab to corticosteroids at 5 mg/kg as a single dose is usually successful for this population subset.52-54 Although a response is seen within 1 to 3 days,41 some patients benefit from an additional dose of infliximab 2 weeks after the initial dose.19 If sepsis or perforation is suspected at any point, corticosteroids or infliximab should be avoided and antibiotics should be started immediately.15,19 Patients with a medically unresponsive disease may require partial or complete colectomy.20 The use of prophylactic budesonide to prevent diarrhea or colitis has not been proven effective and should not be used.55 Despite complications, mortality from colitis has markedly decreased given the increased awareness of this adverse event, reduction in the time to recognition and treatment, and increased adherence to corticosteroids.12
Treating physicians may be delayed in starting appropriate therapy because patients are concerned that using corticosteroids will negatively impact immunotherapy efficacy. Current evidence shows that the use of temporary immunosuppression to treat irAEs does not affect overall survival, efficacy, or time to treatment failure of the ICI.12,56 Restarting ICI therapy is a complex decision and should always be individualized. In grade 1 and 2 colitis, ICI therapy is typically restarted after symptoms have improved.5 In grade 3 and 4 colitis, ICI therapy is often permanently discontinued.20
CONCLUSION
ICIs have not only increased our understanding of the biology of cancer, but they have also improved survival in advanced stages of malignancies like melanoma, NSCLC, and renal cell carcinoma. The expanding use of these medications increases the likelihood that healthcare providers will encounter patients experiencing their adverse events.
Immune-mediated GI adverse events include a wide range of symptoms, from mild diarrhea to severe colitis complicated by perforation and death. Diagnosis requires exclusion of an infectious process. Early recognition and treatment with corticosteroids or another immunosuppressant such as infliximab hastens recovery and decreases complications and mortality. Treatment should be started within 5 days of symptom onset. Corticosteroids should be slowly tapered for no less than 4 weeks to prevent relapse and PCP prophylaxis administered in appropriate patients. Restarting ICI therapy may be considered in cases of mild colitis, but in severe cases, ICI therapy is usually discontinued.
Disclosure
Julian Marin-Acevedo, Dana Harris, and M. Caroline Burton have no conflicts of interest or funding sources to declare.
1. Ledford H. Melanoma drug wins US approval. Nature. 2011;471(7340):561. PubMed
2. Ribas A. Clinical development of the anti-CTLA-4 antibody tremelimumab. Semin Oncol. 2010;37(5):450-454. PubMed
3. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723. PubMed
4. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373(2):123-135. PubMed
5. Motzer RJ, Rini BI, McDermott DF, et al. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J Clin Oncol. 2015;33(13):1430-1437. PubMed
6. Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558-562. PubMed
7. Massard C, Gordon MS, Sharma S, et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti-Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J Clin Oncol. 2016;34(26):3119-3125. PubMed
8. Ferris RL, Blumenschein G Jr, Fayette J, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375(19):1856-1867. PubMed
9. Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509-2520. PubMed
10. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311-319. PubMed
11. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088)2492-2502. PubMed
12. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the Immune-Related Adverse Effects of Immune Checkpoint Inhibitors: A Review. JAMA Oncol. 2016;2(10):1346-1353. PubMed
13. Heinzerling L, Goldinger SM. A review of serious adverse effects under treatment with checkpoint inhibitors. Curr Opin Oncol. 2017;29(2):136-144. PubMed
14. Kahler KC, Hauschild A. Treatment and side effect management of CTLA-4 antibody therapy in metastatic melanoma. J Dtsch Dermatol Ges. 2011;9(4):277-286. PubMed
15. Weber JS, Postow M, Lao CD, Schadendorf D. Management of Adverse Events Following Treatment With Anti-Programmed Death-1 Agents. Oncologist. 2016;21(10):1230-1240. PubMed
16. Bertrand A, Kostine M, Barnetche T, Truchetet ME, Schaeverbeke T. Immune related adverse events associated with anti-CTLA-4 antibodies: systematic review and meta-analysis. BMC Med. 2015;13:211-224. PubMed
17. Abdel-Wahab N, Shah M, Suarez-Almazor ME. Adverse Events Associated with Immune Checkpoint Blockade in Patients with Cancer: A Systematic Review of Case Reports. PLoS One. 2016;11(7):e0160221. doi:10.1371/journal.pone.0160221 PubMed
18. Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26(12):2375-2391. PubMed
19. Gupta A, De Felice KM, Loftus EV Jr, Khanna S. Systematic review: colitis associated with anti-CTLA-4 therapy. Aliment Pharmacol Ther. 2015;42(4):406-417. PubMed
20. Pernot S, Ramtohul T, Taieb J. Checkpoint inhibitors and gastrointestinal immune-related adverse events. Curr Opin Oncol. 2016;28(4):264-268. PubMed
21. Kamata T, Suzuki A, Mise N, et al. Blockade of programmed death-1/programmed death ligand pathway enhances the antitumor immunity of human invariant natural killer T cells. Cancer Immunol Immunother. 2016;65(12):1477-1489. PubMed
22. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252-264. PubMed
23. Velu V, Titanji K, Zhu B, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458(7235):206-210. PubMed
24. Phan GQ, Yang JC, Sherry RM, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2003;100(14):8372-8377. PubMed
25. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Atezolizumab BLA 761041 approval letter (urothelial carcinoma). https://www.genentech-access.com/content/dam/gene/accesssolutions/brands/tecentriq/Appeals%20Tips/TECENTRIQ-FDA-Approval-Letter-Metastatic-Urothelial-Carcinoma-First-Line-Therapy.pdf. Accessed September 30, 2017.
26. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Imfinzi (durvalumab) approval letter. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/761069Orig1s000ltr.pdf. Accessed September 30, 2017.
27. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Bavencio (avelumab) accelerated approval letter - urothelial carcinoma. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/761078Orig1s000ltr.pdf. Accessed May 16, 2017.
28. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Atezolizumab BLA 761041 approval letter (NSCLC).
29. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Bavencio (avelumab) approval letter - Merkel cell carcinoma. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/761049Orig1s000ltr.pdf. Accessed April 27, 2017.
30. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Atezolizumab BLA 761041 approval letter. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761034Orig1s000Approv.pdf. Accessed April 6, 2017.
31. Voskens CJ, Goldinger SM, Loquai C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8(1):e53745. doi:10.1371/journal.pone.0053745. PubMed
32. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373(1):23-34. PubMed
33. Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139-148. PubMed
34. Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res. 2015;4(5):560-575. PubMed
35. Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697. PubMed
36. Kahler KC, Hassel JC, Heinzerling L, et al. Management of side effects of immune checkpoint blockade by anti-CTLA-4 and anti-PD-1 antibodies in metastatic melanoma. J Dtsch Dermatol Ges. 2016;14(7):662-681. PubMed
37. Postow MA. Managing immune checkpoint-blocking antibody side effects. Am Soc Clin Oncol Educ Book. 2015:76-83. PubMed
38. Lacouture ME, Wolchok JD, Yosipovitch G, Kahler KC, Busam KJ, Hauschild A. Ipilimumab in patients with cancer and the management of dermatologic adverse events. J Am Acad Dermatol. 2014;71(1):161-169. PubMed
39. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521-2532. PubMed
40. Weber J. Ipilimumab: controversies in its development, utility and autoimmune adverse events. Cancer Immunol Immunother. 2009;58(5):823-830. PubMed
41. Beck KE, Blansfield JA, Tran KQ, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol. 2006;24(15):2283-2289. PubMed
42. Cancer Therapy Evaluation Program, National Cancer Institute (NCI). Common terminology criteria for adverse events v3.0 (CTCAE). https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed April 9, 2017.
43. De Velasco G, Je Y, Bosse D, et al. Comprehensive Meta-analysis of Key Immune-Related Adverse Events from CTLA-4 and PD-1/PD-L1 Inhibitors in Cancer Patients. Cancer Immunol Res. 2017;5(4):312-318. PubMed
44. McCutcheon JL, McClain CM, Puzanov I, Smith TA. Infectious Colitis Associated With Ipilimumab Therapy. Gastroenterology Res. 2014;7(1):28-31. PubMed
45. Berman D, Parker SM, Siegel J, et al. Blockade of cytotoxic T-lymphocyte antigen-4 by ipilimumab results in dysregulation of gastrointestinal immunity in patients with advanced melanoma. Cancer Immun. 2010;10:11-20. PubMed
46. Reynolds K, Ananthakrishnan A, Dougan M, Bardia A. Immune-Related Adverse Events (irAEs) in Cancer Patients. In: McKean SC, Ross JJ, Dressler DD, Scheurer DB, eds. Principles and Practice of Hospital Medicine. 2nd ed. New York: McGraw-Hill Education; 2017.
47. Garcia-Neuer M, Marmarelis ME, Jangi SR, et al. Diagnostic Comparison of CT Scans and Colonoscopy for Immune-Related Colitis in Ipilimumab-Treated Advanced Melanoma Patients. Cancer Immunol Res. 2017;5(4):286-291. PubMed
48. Venditti O, De Lisi D, Caricato M, et al. Ipilimumab and immune-mediated adverse events: a case report of anti-CTLA4 induced ileitis. BMC Cancer. 2015;15:87-91. PubMed
49. Messmer M, Upreti S, Tarabishy Y, et al. Ipilimumab-Induced Enteritis without Colitis: A New Challenge. Case Rep Oncol. 2016;9(3):705-713. PubMed
50. De Felice KM, Gupta A, Rakshit S, et al. Ipilimumab-induced colitis in patients with metastatic melanoma. Melanoma Res. 2015;25(4):321-327. PubMed
51. Baden LR, Swaminathan S, Angarone M, et al. Prevention and Treatment of Cancer-Related Infections, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Newt. 2017;14(7):882-913. PubMed
52. Minor DR, Chin K, Kashani-Sabet M. Infliximab in the treatment of anti-CTLA4 antibody (ipilimumab) induced immune-related colitis. Cancer Biother Radiopharm. 2009;24(3):321-325. PubMed
53. Merrill SP, Reynolds P, Kalra A, Biehl J, Vandivier RW, Mueller SW. Early administration of infliximab for severe ipilimumab-related diarrhea in a critically ill patient. Ann Pharmacother. 2014;48(6):806-810. PubMed
54. Pages C, Gornet JM, Monsel G, et al. Ipilimumab-induced acute severe colitis treated by infliximab. Melanoma Res. 2013;23(3):227-230. PubMed
55. Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15(17):5591-5598. PubMed
56. Horvat TZ, Adel NG, Dung TO, et al. Immune-Related Adverse Events, Need for Systemic Immunosuppression, and Effects on Survival and Time to Treatment Failure in Patients With Melanoma Treated With Ipilimumab at Memorial Sloan Kettering Cancer Center. J Clin Oncol. 2015;33(28):3193-3198. PubMed
57. Cancer Therapy Evaluation Program, National Cancer Institute (NCI). Common terminology criteria for adverse events v3.0 (CTCAE). https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed April 9, 2017.
1. Ledford H. Melanoma drug wins US approval. Nature. 2011;471(7340):561. PubMed
2. Ribas A. Clinical development of the anti-CTLA-4 antibody tremelimumab. Semin Oncol. 2010;37(5):450-454. PubMed
3. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723. PubMed
4. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373(2):123-135. PubMed
5. Motzer RJ, Rini BI, McDermott DF, et al. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J Clin Oncol. 2015;33(13):1430-1437. PubMed
6. Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558-562. PubMed
7. Massard C, Gordon MS, Sharma S, et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti-Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J Clin Oncol. 2016;34(26):3119-3125. PubMed
8. Ferris RL, Blumenschein G Jr, Fayette J, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375(19):1856-1867. PubMed
9. Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509-2520. PubMed
10. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311-319. PubMed
11. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088)2492-2502. PubMed
12. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the Immune-Related Adverse Effects of Immune Checkpoint Inhibitors: A Review. JAMA Oncol. 2016;2(10):1346-1353. PubMed
13. Heinzerling L, Goldinger SM. A review of serious adverse effects under treatment with checkpoint inhibitors. Curr Opin Oncol. 2017;29(2):136-144. PubMed
14. Kahler KC, Hauschild A. Treatment and side effect management of CTLA-4 antibody therapy in metastatic melanoma. J Dtsch Dermatol Ges. 2011;9(4):277-286. PubMed
15. Weber JS, Postow M, Lao CD, Schadendorf D. Management of Adverse Events Following Treatment With Anti-Programmed Death-1 Agents. Oncologist. 2016;21(10):1230-1240. PubMed
16. Bertrand A, Kostine M, Barnetche T, Truchetet ME, Schaeverbeke T. Immune related adverse events associated with anti-CTLA-4 antibodies: systematic review and meta-analysis. BMC Med. 2015;13:211-224. PubMed
17. Abdel-Wahab N, Shah M, Suarez-Almazor ME. Adverse Events Associated with Immune Checkpoint Blockade in Patients with Cancer: A Systematic Review of Case Reports. PLoS One. 2016;11(7):e0160221. doi:10.1371/journal.pone.0160221 PubMed
18. Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26(12):2375-2391. PubMed
19. Gupta A, De Felice KM, Loftus EV Jr, Khanna S. Systematic review: colitis associated with anti-CTLA-4 therapy. Aliment Pharmacol Ther. 2015;42(4):406-417. PubMed
20. Pernot S, Ramtohul T, Taieb J. Checkpoint inhibitors and gastrointestinal immune-related adverse events. Curr Opin Oncol. 2016;28(4):264-268. PubMed
21. Kamata T, Suzuki A, Mise N, et al. Blockade of programmed death-1/programmed death ligand pathway enhances the antitumor immunity of human invariant natural killer T cells. Cancer Immunol Immunother. 2016;65(12):1477-1489. PubMed
22. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252-264. PubMed
23. Velu V, Titanji K, Zhu B, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458(7235):206-210. PubMed
24. Phan GQ, Yang JC, Sherry RM, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2003;100(14):8372-8377. PubMed
25. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Atezolizumab BLA 761041 approval letter (urothelial carcinoma). https://www.genentech-access.com/content/dam/gene/accesssolutions/brands/tecentriq/Appeals%20Tips/TECENTRIQ-FDA-Approval-Letter-Metastatic-Urothelial-Carcinoma-First-Line-Therapy.pdf. Accessed September 30, 2017.
26. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Imfinzi (durvalumab) approval letter. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/761069Orig1s000ltr.pdf. Accessed September 30, 2017.
27. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Bavencio (avelumab) accelerated approval letter - urothelial carcinoma. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/761078Orig1s000ltr.pdf. Accessed May 16, 2017.
28. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Atezolizumab BLA 761041 approval letter (NSCLC).
29. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Bavencio (avelumab) approval letter - Merkel cell carcinoma. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/761049Orig1s000ltr.pdf. Accessed April 27, 2017.
30. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. Atezolizumab BLA 761041 approval letter. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761034Orig1s000Approv.pdf. Accessed April 6, 2017.
31. Voskens CJ, Goldinger SM, Loquai C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8(1):e53745. doi:10.1371/journal.pone.0053745. PubMed
32. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373(1):23-34. PubMed
33. Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139-148. PubMed
34. Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res. 2015;4(5):560-575. PubMed
35. Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697. PubMed
36. Kahler KC, Hassel JC, Heinzerling L, et al. Management of side effects of immune checkpoint blockade by anti-CTLA-4 and anti-PD-1 antibodies in metastatic melanoma. J Dtsch Dermatol Ges. 2016;14(7):662-681. PubMed
37. Postow MA. Managing immune checkpoint-blocking antibody side effects. Am Soc Clin Oncol Educ Book. 2015:76-83. PubMed
38. Lacouture ME, Wolchok JD, Yosipovitch G, Kahler KC, Busam KJ, Hauschild A. Ipilimumab in patients with cancer and the management of dermatologic adverse events. J Am Acad Dermatol. 2014;71(1):161-169. PubMed
39. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521-2532. PubMed
40. Weber J. Ipilimumab: controversies in its development, utility and autoimmune adverse events. Cancer Immunol Immunother. 2009;58(5):823-830. PubMed
41. Beck KE, Blansfield JA, Tran KQ, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol. 2006;24(15):2283-2289. PubMed
42. Cancer Therapy Evaluation Program, National Cancer Institute (NCI). Common terminology criteria for adverse events v3.0 (CTCAE). https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed April 9, 2017.
43. De Velasco G, Je Y, Bosse D, et al. Comprehensive Meta-analysis of Key Immune-Related Adverse Events from CTLA-4 and PD-1/PD-L1 Inhibitors in Cancer Patients. Cancer Immunol Res. 2017;5(4):312-318. PubMed
44. McCutcheon JL, McClain CM, Puzanov I, Smith TA. Infectious Colitis Associated With Ipilimumab Therapy. Gastroenterology Res. 2014;7(1):28-31. PubMed
45. Berman D, Parker SM, Siegel J, et al. Blockade of cytotoxic T-lymphocyte antigen-4 by ipilimumab results in dysregulation of gastrointestinal immunity in patients with advanced melanoma. Cancer Immun. 2010;10:11-20. PubMed
46. Reynolds K, Ananthakrishnan A, Dougan M, Bardia A. Immune-Related Adverse Events (irAEs) in Cancer Patients. In: McKean SC, Ross JJ, Dressler DD, Scheurer DB, eds. Principles and Practice of Hospital Medicine. 2nd ed. New York: McGraw-Hill Education; 2017.
47. Garcia-Neuer M, Marmarelis ME, Jangi SR, et al. Diagnostic Comparison of CT Scans and Colonoscopy for Immune-Related Colitis in Ipilimumab-Treated Advanced Melanoma Patients. Cancer Immunol Res. 2017;5(4):286-291. PubMed
48. Venditti O, De Lisi D, Caricato M, et al. Ipilimumab and immune-mediated adverse events: a case report of anti-CTLA4 induced ileitis. BMC Cancer. 2015;15:87-91. PubMed
49. Messmer M, Upreti S, Tarabishy Y, et al. Ipilimumab-Induced Enteritis without Colitis: A New Challenge. Case Rep Oncol. 2016;9(3):705-713. PubMed
50. De Felice KM, Gupta A, Rakshit S, et al. Ipilimumab-induced colitis in patients with metastatic melanoma. Melanoma Res. 2015;25(4):321-327. PubMed
51. Baden LR, Swaminathan S, Angarone M, et al. Prevention and Treatment of Cancer-Related Infections, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Newt. 2017;14(7):882-913. PubMed
52. Minor DR, Chin K, Kashani-Sabet M. Infliximab in the treatment of anti-CTLA4 antibody (ipilimumab) induced immune-related colitis. Cancer Biother Radiopharm. 2009;24(3):321-325. PubMed
53. Merrill SP, Reynolds P, Kalra A, Biehl J, Vandivier RW, Mueller SW. Early administration of infliximab for severe ipilimumab-related diarrhea in a critically ill patient. Ann Pharmacother. 2014;48(6):806-810. PubMed
54. Pages C, Gornet JM, Monsel G, et al. Ipilimumab-induced acute severe colitis treated by infliximab. Melanoma Res. 2013;23(3):227-230. PubMed
55. Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15(17):5591-5598. PubMed
56. Horvat TZ, Adel NG, Dung TO, et al. Immune-Related Adverse Events, Need for Systemic Immunosuppression, and Effects on Survival and Time to Treatment Failure in Patients With Melanoma Treated With Ipilimumab at Memorial Sloan Kettering Cancer Center. J Clin Oncol. 2015;33(28):3193-3198. PubMed
57. Cancer Therapy Evaluation Program, National Cancer Institute (NCI). Common terminology criteria for adverse events v3.0 (CTCAE). https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed April 9, 2017.
© 2018 Society of Hospital Medicine
Poor Adherence to Risk Stratification Guidelines Results in Overuse of Venous Thromboembolism Prophylaxis in Hospitalized Older Adults
Venous thromboembolism (VTE) prophylaxis is an important consideration for every older adult admitted to the hospital1 but should not be prescribed to all patients. Use of anticoagulants (specifically low-molecular-weight heparin, low-dose unfractionated heparin, and fondaparinux) when not medically indicated may be harmful, especially for older adults who on average have more chronic conditions,1 take more potentially interacting medications,2 and have higher risks of bleeding.3 The American College of Chest Physicians (ACCP) Ninth Edition Guidelines for Antithrombotic Therapy and Prevention of Thrombosis explicitly recommend a risk-stratification approach using the Padua Prediction Score (PPS) to select those patients most likely to benefit from VTE prophylaxis.4,5 This study aimed to describe the use of risk stratification and pharmacologic VTE prophylaxis use in a population of medically ill, hospitalized older patients.
METHODS
We conducted a retrospective cohort study using data from patients aged 70 years or older admitted to Duke University Hospital general medicine services between January 1, 2014, to December 31, 2014. The PPS variables, 11 in total, are each weighed and sum to a score that stratifies patients into either high or low risk for VTE occurrence.5 Manual chart abstraction was performed using the electronic health record (EHR) to determine each patient’s PPS, inpatient pharmacologic VTE prophylaxis use, and contraindications to VTE prophylaxis. Descriptive statistics are presented for the important confounders/covariates, VTE risk, and VTE prophylaxis use.
RESULTS
Of the total eligible cohort (N = 1399), 400 patients were randomly selected for manual chart review; 89 of these patients were not eligible because they were on anticoagulation upon admission, leaving n = 311 patients in the analytic sample. Mean age for the sample was 80.6 years (standard deviation [SD]: 7.3); 42% were male and 34% were African American, and median length of stay was 4.0 days. The overall mean PPS for the sample was 3.6 (SD 1.8), resulting in 59% (n = 182) defined as “low risk.” Reasons for admission, median length of stay, and aspirin use did not differ between the risk groups.
Pharmacological VTE prophylaxis was present in 74% (134 out of 182) of low-risk patients and 71% (92 out of 129) of high-risk patients (Figure). In both low- and high-risk patients who received pharmacological VTE prophylaxis, over 90% had the therapy initiated within 24 hours of admission, and it was continued for over 60% of their hospital days.
DISCUSSION
We found no association between PPS and use of anticoagulants for VTE prophylaxis, suggesting that risk stratification is not being used to guide clinical decision-making. There are several barriers to implementing guideline directed use of VTE risk stratification. First, there is a lack of consensus on which VTE risk assessment tool is best to use with medically ill, hospitalized patients. While the ACCP Ninth Edition Guidelines support the use of the PPS, the American College of Physicians does not recommend a specific tool for VTE risk assessment.5,6 Although other risk stratification tools exist, concordance between these tools has not been well studied.7 Second, manual calculation of the PPS can be cumbersome, error prone, and disruptive to the clinical workflow. Automated data extraction leveraging existing structured data elements in the EHR may be particularly attractive to many health systems striving to use EHRs to improve care. Designing and testing automatically populated VTE risk stratification tools may facilitate translation of evidence-based guidelines into routine clinical practice. Lastly, a key barrier is clinician education and awareness about these tools. Adding risk stratification tools to admission order sets is one way to increase clinician awareness and has been shown to decrease inappropriate VTE prophylaxis use.8 High-quality studies that use implementation science to promote uptake and efficacy of risk stratification tools into clinical practice are urgently needed.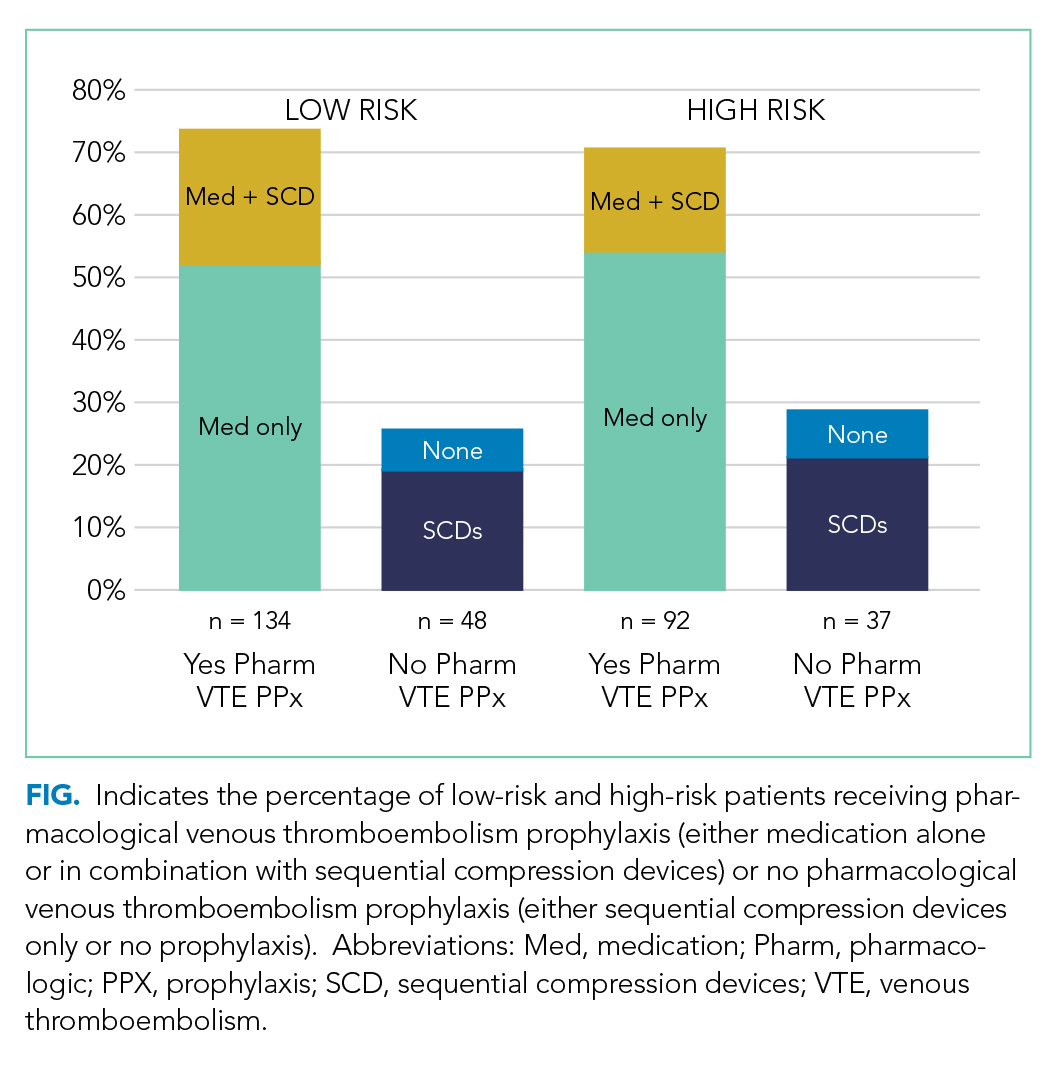
Our study has several limitations. First, this was a single-site study at an academic center, which may limit generalizability of the findings. However, our design enabled us to look at other specific patient-level data that is typically not available in larger databases. Second, determination of PPS is limited to data available in the EHR, resulting in measurement error and possibly the underreporting of risk factors. Finally, due to feasibility and the low probability of VTE, we did not collect data on long-term VTE outcome and were unable to determine the impact that inappropriate VTE prophylaxis use has in low-risk hospitalized older adults.
In summary, we found poor adherence to risk stratification guidelines among medically ill, hospitalized older adults, resulting in overuse of anticoagulants for VTE prophylaxis. Automating risk stratification tools and incorporating results into order sets may ensure that adequate prophylaxis is used for patients who need it, while minimizing excess prophylaxis in those who do not.
Acknowledgments
The authors would like to thank Shenglan Li from Research Triangle Institute for her assistance in the data programming and database creation.
Disclosure
The authors have no conflicts of interest to report. This study was funded by the National Institute on Aging (NIA) GEMSSTAR Award (NIA R03AG048007) and the Duke Older Americans Independence Center (NIA P30 AG028716–01). This work was also supported by the Duke University Internal Medicine Chair’s Award, the Duke University Hartford Center of Excellence, and the Center of Innovation for Health Services Research in Primary Care (CIN 13-410) at the Durham VA Health Care System. This work was conducted while Dr. Pavon was supported by the T. Franklin Williams Scholars Program. Dr. Colón-Emeric is supported by K24 AG049077-01A1. The funding sources had no role in the design and conduct of the study; analysis or interpretation of the data; preparation or final approval of the manuscript before publication, and decision to submit the manuscript for publication. Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of Duke University or the Department of Veterans Affairs.
1. Kniffin WD, Baron JA, Barrett J, Birkmeyer JD, Anderson FA. The epidemiology of diagnosed pulmonary embolism and deep venous thrombosis in the elderly. Arch Intern Med. 1994;154(8):861-866. PubMed
2. Pasina L, Djade CD, Nobili A, et al. Drug-drug interactions in a cohort of hospitalized elderly patients. Pharmacoepidemiol Drug Saf. 2013; 22(10):1054-1060. PubMed
3. Campbell NR, Hull RD, Brant R, Hogan DB, Pineo GF, Raskob GE. Aging and heparin-related bleeding. Arch Intern Med. 1996;156(8):857-860. PubMed
4. Gould MK, Garcia DA, Wren SM, et al. Prevention of VTE in nonorthopedic surgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e227S-e277S. PubMed
5. Barbar S, Noventa F, Rossetto V, et al. A risk assessment model for the identification of hospitalized medical patients at risk for venous thromboembolism: the Padua Prediction Score. J Thromb Haemost. 2010;8(11):2450-2457. PubMed
6. Qaseem A, Chou R, Humphrey LL, Starkey M, Shekelle P. Venous thromboembolism prophylaxis in hospitalized patients: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2011;155(9):625-632. PubMed
7. Stuck AK, Spirk D, Schaudt J, Kucher N. Risk assessment models for venous thromboembolism in acutely ill medical patients. A systematic review. Thromb Haemost. 2017;117(4):801-808. PubMed
8. Khanna R, Vittinghoff E, Maselli J, Auerbach A. Unintended consequences of a standard admission order set on venous thromboembolism prophylaxis and patient outcomes. J Gen Intern Med. 2012;27(3):318-324. PubMed
Venous thromboembolism (VTE) prophylaxis is an important consideration for every older adult admitted to the hospital1 but should not be prescribed to all patients. Use of anticoagulants (specifically low-molecular-weight heparin, low-dose unfractionated heparin, and fondaparinux) when not medically indicated may be harmful, especially for older adults who on average have more chronic conditions,1 take more potentially interacting medications,2 and have higher risks of bleeding.3 The American College of Chest Physicians (ACCP) Ninth Edition Guidelines for Antithrombotic Therapy and Prevention of Thrombosis explicitly recommend a risk-stratification approach using the Padua Prediction Score (PPS) to select those patients most likely to benefit from VTE prophylaxis.4,5 This study aimed to describe the use of risk stratification and pharmacologic VTE prophylaxis use in a population of medically ill, hospitalized older patients.
METHODS
We conducted a retrospective cohort study using data from patients aged 70 years or older admitted to Duke University Hospital general medicine services between January 1, 2014, to December 31, 2014. The PPS variables, 11 in total, are each weighed and sum to a score that stratifies patients into either high or low risk for VTE occurrence.5 Manual chart abstraction was performed using the electronic health record (EHR) to determine each patient’s PPS, inpatient pharmacologic VTE prophylaxis use, and contraindications to VTE prophylaxis. Descriptive statistics are presented for the important confounders/covariates, VTE risk, and VTE prophylaxis use.
RESULTS
Of the total eligible cohort (N = 1399), 400 patients were randomly selected for manual chart review; 89 of these patients were not eligible because they were on anticoagulation upon admission, leaving n = 311 patients in the analytic sample. Mean age for the sample was 80.6 years (standard deviation [SD]: 7.3); 42% were male and 34% were African American, and median length of stay was 4.0 days. The overall mean PPS for the sample was 3.6 (SD 1.8), resulting in 59% (n = 182) defined as “low risk.” Reasons for admission, median length of stay, and aspirin use did not differ between the risk groups.
Pharmacological VTE prophylaxis was present in 74% (134 out of 182) of low-risk patients and 71% (92 out of 129) of high-risk patients (Figure). In both low- and high-risk patients who received pharmacological VTE prophylaxis, over 90% had the therapy initiated within 24 hours of admission, and it was continued for over 60% of their hospital days.
DISCUSSION
We found no association between PPS and use of anticoagulants for VTE prophylaxis, suggesting that risk stratification is not being used to guide clinical decision-making. There are several barriers to implementing guideline directed use of VTE risk stratification. First, there is a lack of consensus on which VTE risk assessment tool is best to use with medically ill, hospitalized patients. While the ACCP Ninth Edition Guidelines support the use of the PPS, the American College of Physicians does not recommend a specific tool for VTE risk assessment.5,6 Although other risk stratification tools exist, concordance between these tools has not been well studied.7 Second, manual calculation of the PPS can be cumbersome, error prone, and disruptive to the clinical workflow. Automated data extraction leveraging existing structured data elements in the EHR may be particularly attractive to many health systems striving to use EHRs to improve care. Designing and testing automatically populated VTE risk stratification tools may facilitate translation of evidence-based guidelines into routine clinical practice. Lastly, a key barrier is clinician education and awareness about these tools. Adding risk stratification tools to admission order sets is one way to increase clinician awareness and has been shown to decrease inappropriate VTE prophylaxis use.8 High-quality studies that use implementation science to promote uptake and efficacy of risk stratification tools into clinical practice are urgently needed.
Our study has several limitations. First, this was a single-site study at an academic center, which may limit generalizability of the findings. However, our design enabled us to look at other specific patient-level data that is typically not available in larger databases. Second, determination of PPS is limited to data available in the EHR, resulting in measurement error and possibly the underreporting of risk factors. Finally, due to feasibility and the low probability of VTE, we did not collect data on long-term VTE outcome and were unable to determine the impact that inappropriate VTE prophylaxis use has in low-risk hospitalized older adults.
In summary, we found poor adherence to risk stratification guidelines among medically ill, hospitalized older adults, resulting in overuse of anticoagulants for VTE prophylaxis. Automating risk stratification tools and incorporating results into order sets may ensure that adequate prophylaxis is used for patients who need it, while minimizing excess prophylaxis in those who do not.
Acknowledgments
The authors would like to thank Shenglan Li from Research Triangle Institute for her assistance in the data programming and database creation.
Disclosure
The authors have no conflicts of interest to report. This study was funded by the National Institute on Aging (NIA) GEMSSTAR Award (NIA R03AG048007) and the Duke Older Americans Independence Center (NIA P30 AG028716–01). This work was also supported by the Duke University Internal Medicine Chair’s Award, the Duke University Hartford Center of Excellence, and the Center of Innovation for Health Services Research in Primary Care (CIN 13-410) at the Durham VA Health Care System. This work was conducted while Dr. Pavon was supported by the T. Franklin Williams Scholars Program. Dr. Colón-Emeric is supported by K24 AG049077-01A1. The funding sources had no role in the design and conduct of the study; analysis or interpretation of the data; preparation or final approval of the manuscript before publication, and decision to submit the manuscript for publication. Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of Duke University or the Department of Veterans Affairs.
Venous thromboembolism (VTE) prophylaxis is an important consideration for every older adult admitted to the hospital1 but should not be prescribed to all patients. Use of anticoagulants (specifically low-molecular-weight heparin, low-dose unfractionated heparin, and fondaparinux) when not medically indicated may be harmful, especially for older adults who on average have more chronic conditions,1 take more potentially interacting medications,2 and have higher risks of bleeding.3 The American College of Chest Physicians (ACCP) Ninth Edition Guidelines for Antithrombotic Therapy and Prevention of Thrombosis explicitly recommend a risk-stratification approach using the Padua Prediction Score (PPS) to select those patients most likely to benefit from VTE prophylaxis.4,5 This study aimed to describe the use of risk stratification and pharmacologic VTE prophylaxis use in a population of medically ill, hospitalized older patients.
METHODS
We conducted a retrospective cohort study using data from patients aged 70 years or older admitted to Duke University Hospital general medicine services between January 1, 2014, to December 31, 2014. The PPS variables, 11 in total, are each weighed and sum to a score that stratifies patients into either high or low risk for VTE occurrence.5 Manual chart abstraction was performed using the electronic health record (EHR) to determine each patient’s PPS, inpatient pharmacologic VTE prophylaxis use, and contraindications to VTE prophylaxis. Descriptive statistics are presented for the important confounders/covariates, VTE risk, and VTE prophylaxis use.
RESULTS
Of the total eligible cohort (N = 1399), 400 patients were randomly selected for manual chart review; 89 of these patients were not eligible because they were on anticoagulation upon admission, leaving n = 311 patients in the analytic sample. Mean age for the sample was 80.6 years (standard deviation [SD]: 7.3); 42% were male and 34% were African American, and median length of stay was 4.0 days. The overall mean PPS for the sample was 3.6 (SD 1.8), resulting in 59% (n = 182) defined as “low risk.” Reasons for admission, median length of stay, and aspirin use did not differ between the risk groups.
Pharmacological VTE prophylaxis was present in 74% (134 out of 182) of low-risk patients and 71% (92 out of 129) of high-risk patients (Figure). In both low- and high-risk patients who received pharmacological VTE prophylaxis, over 90% had the therapy initiated within 24 hours of admission, and it was continued for over 60% of their hospital days.
DISCUSSION
We found no association between PPS and use of anticoagulants for VTE prophylaxis, suggesting that risk stratification is not being used to guide clinical decision-making. There are several barriers to implementing guideline directed use of VTE risk stratification. First, there is a lack of consensus on which VTE risk assessment tool is best to use with medically ill, hospitalized patients. While the ACCP Ninth Edition Guidelines support the use of the PPS, the American College of Physicians does not recommend a specific tool for VTE risk assessment.5,6 Although other risk stratification tools exist, concordance between these tools has not been well studied.7 Second, manual calculation of the PPS can be cumbersome, error prone, and disruptive to the clinical workflow. Automated data extraction leveraging existing structured data elements in the EHR may be particularly attractive to many health systems striving to use EHRs to improve care. Designing and testing automatically populated VTE risk stratification tools may facilitate translation of evidence-based guidelines into routine clinical practice. Lastly, a key barrier is clinician education and awareness about these tools. Adding risk stratification tools to admission order sets is one way to increase clinician awareness and has been shown to decrease inappropriate VTE prophylaxis use.8 High-quality studies that use implementation science to promote uptake and efficacy of risk stratification tools into clinical practice are urgently needed.
Our study has several limitations. First, this was a single-site study at an academic center, which may limit generalizability of the findings. However, our design enabled us to look at other specific patient-level data that is typically not available in larger databases. Second, determination of PPS is limited to data available in the EHR, resulting in measurement error and possibly the underreporting of risk factors. Finally, due to feasibility and the low probability of VTE, we did not collect data on long-term VTE outcome and were unable to determine the impact that inappropriate VTE prophylaxis use has in low-risk hospitalized older adults.
In summary, we found poor adherence to risk stratification guidelines among medically ill, hospitalized older adults, resulting in overuse of anticoagulants for VTE prophylaxis. Automating risk stratification tools and incorporating results into order sets may ensure that adequate prophylaxis is used for patients who need it, while minimizing excess prophylaxis in those who do not.
Acknowledgments
The authors would like to thank Shenglan Li from Research Triangle Institute for her assistance in the data programming and database creation.
Disclosure
The authors have no conflicts of interest to report. This study was funded by the National Institute on Aging (NIA) GEMSSTAR Award (NIA R03AG048007) and the Duke Older Americans Independence Center (NIA P30 AG028716–01). This work was also supported by the Duke University Internal Medicine Chair’s Award, the Duke University Hartford Center of Excellence, and the Center of Innovation for Health Services Research in Primary Care (CIN 13-410) at the Durham VA Health Care System. This work was conducted while Dr. Pavon was supported by the T. Franklin Williams Scholars Program. Dr. Colón-Emeric is supported by K24 AG049077-01A1. The funding sources had no role in the design and conduct of the study; analysis or interpretation of the data; preparation or final approval of the manuscript before publication, and decision to submit the manuscript for publication. Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of Duke University or the Department of Veterans Affairs.
1. Kniffin WD, Baron JA, Barrett J, Birkmeyer JD, Anderson FA. The epidemiology of diagnosed pulmonary embolism and deep venous thrombosis in the elderly. Arch Intern Med. 1994;154(8):861-866. PubMed
2. Pasina L, Djade CD, Nobili A, et al. Drug-drug interactions in a cohort of hospitalized elderly patients. Pharmacoepidemiol Drug Saf. 2013; 22(10):1054-1060. PubMed
3. Campbell NR, Hull RD, Brant R, Hogan DB, Pineo GF, Raskob GE. Aging and heparin-related bleeding. Arch Intern Med. 1996;156(8):857-860. PubMed
4. Gould MK, Garcia DA, Wren SM, et al. Prevention of VTE in nonorthopedic surgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e227S-e277S. PubMed
5. Barbar S, Noventa F, Rossetto V, et al. A risk assessment model for the identification of hospitalized medical patients at risk for venous thromboembolism: the Padua Prediction Score. J Thromb Haemost. 2010;8(11):2450-2457. PubMed
6. Qaseem A, Chou R, Humphrey LL, Starkey M, Shekelle P. Venous thromboembolism prophylaxis in hospitalized patients: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2011;155(9):625-632. PubMed
7. Stuck AK, Spirk D, Schaudt J, Kucher N. Risk assessment models for venous thromboembolism in acutely ill medical patients. A systematic review. Thromb Haemost. 2017;117(4):801-808. PubMed
8. Khanna R, Vittinghoff E, Maselli J, Auerbach A. Unintended consequences of a standard admission order set on venous thromboembolism prophylaxis and patient outcomes. J Gen Intern Med. 2012;27(3):318-324. PubMed
1. Kniffin WD, Baron JA, Barrett J, Birkmeyer JD, Anderson FA. The epidemiology of diagnosed pulmonary embolism and deep venous thrombosis in the elderly. Arch Intern Med. 1994;154(8):861-866. PubMed
2. Pasina L, Djade CD, Nobili A, et al. Drug-drug interactions in a cohort of hospitalized elderly patients. Pharmacoepidemiol Drug Saf. 2013; 22(10):1054-1060. PubMed
3. Campbell NR, Hull RD, Brant R, Hogan DB, Pineo GF, Raskob GE. Aging and heparin-related bleeding. Arch Intern Med. 1996;156(8):857-860. PubMed
4. Gould MK, Garcia DA, Wren SM, et al. Prevention of VTE in nonorthopedic surgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e227S-e277S. PubMed
5. Barbar S, Noventa F, Rossetto V, et al. A risk assessment model for the identification of hospitalized medical patients at risk for venous thromboembolism: the Padua Prediction Score. J Thromb Haemost. 2010;8(11):2450-2457. PubMed
6. Qaseem A, Chou R, Humphrey LL, Starkey M, Shekelle P. Venous thromboembolism prophylaxis in hospitalized patients: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2011;155(9):625-632. PubMed
7. Stuck AK, Spirk D, Schaudt J, Kucher N. Risk assessment models for venous thromboembolism in acutely ill medical patients. A systematic review. Thromb Haemost. 2017;117(4):801-808. PubMed
8. Khanna R, Vittinghoff E, Maselli J, Auerbach A. Unintended consequences of a standard admission order set on venous thromboembolism prophylaxis and patient outcomes. J Gen Intern Med. 2012;27(3):318-324. PubMed
© 2018 Society of Hospital Medicine
Shared Decision-Making During Inpatient Rounds: Opportunities for Improvement in Patient Engagement and Communication
The ethos of medicine has shifted from paternalistic, physician-driven care to patient autonomy and engagement, in which the physician shares information and advises.1-3 Although there are ethical, legal, and practical reasons to respect patient preferences,1-4 patient engagement also fosters quality and safety5 and may improve clinical outcomes.5-8 Patients whose preferences are respected are more likely to trust their doctor, feel empowered, and adhere to treatments.9
Providers may partner with patients through shared decision-making (SDM).10,11 Several SDM models describe the process of providers and patients balancing evidence, preferences and context to arrive at a clinical decision.12-15 The National Academy of Medicine and the American Academy of Pediatrics has called for more SDM,16,17 including when clinical evidence is limited,2 equally beneficial options exist,18 clinical stakes are high,19 and even with deferential patients.20 Despite its value, SDM does not reliably occur21,22 and SDM training is often unavailable.4 Clinical decision tools, patient education aids, and various training interventions have shown promising, although inconsistent results.23, 24
Little is known about SDM in inpatient settings where unique patient, clinician, and environmental factors may influence SDM. This study describes the quality and possible predictors of inpatient SDM during attending rounds in 4 academic training settings. Although SDM may occur anytime during a hospitalization, attending rounds present a valuable opportunity for SDM observation given their centrality to inpatient care and teaching.25,26 Because attending physicians bear ultimate responsibility for patient management, we examined whether SDM performance varies among attendings within each service. In addition, we tested the hypothesis that service-level, team-level, and patient-level features explain variation in SDM quality more than individual attending physicians. Finally, we compared peer-observer perspectives of SDM behaviors with patient and/or guardian perspectives.
METHODS
Study Design and Setting
This cross-sectional, observational study examined the diversity of SDM practice within and between 4 inpatient services during attending rounds, including the internal medicine and pediatrics services at Stanford University and the University of California, San Francisco (UCSF). Both institutions provide quaternary care to diverse patient populations with approximately half enrolled in Medicare and/or Medicaid.
One institution had 42 internal medicine (Med-1) and 15 pediatric hospitalists (Peds-1) compared to 8 internal medicine (Med-2) and 12 pediatric hospitalists (Peds-2) at the second location. Both pediatric services used family-centered rounds that included discussions between the patients’ families and the whole team. One medicine service used a similar rounding model that did not necessarily involve the patients’ families. In contrast, the smaller medicine service typically began rounds by discussing all patients in a conference room and then visiting select patients afterwards.
From August 2014 to November 2014, peer observers gathered data on team SDM behaviors during attending rounds. After the rounding team departed, nonphysician interviewers surveyed consenting patients’ (or guardians’) views of the SDM experience, yielding paired evaluations for a subset of SDM encounters. Institutional review board approval was obtained from Stanford University and UCSF.
Participants and Inclusion Criteria
Attending physicians were hospitalists who supervised rounds at least 1 month per year, and did not include those conducting the study. All provided verbal assent to be observed on 3 days within a 7-day period. While team composition varied as needed (eg, to include the nurse, pharmacist, interpreter, etc), we restricted study observations to those teams with an attending and at least one learner (eg, resident, intern, medical student) to capture the influence of attending physicians in their training role. Because services vary in number of attendings on staff, rounds assigned per attending, and patients per round, it was not possible to enroll equal sample sizes per service in the study.
Nonintensive care unit patients who were deemed medically stable by the team were eligible for peer observation and participation in a subsequent patient interview once during the study period. Pediatric patients were invited for an interview if they were between 13 and 21 years old and had the option of having a parent or guardian present; if the pediatric patients were less than 13 years old or they were not interested in being interviewed, then their parents or guardians were invited to be interviewed. Interpreters were on rounds, and thus, non-English participants were able to participate in the peer observations, but could not participate in patient interviews because interpreters were not available during afternoons for study purposes. Consent was obtained from all participating patients and/or guardians.
Data Collection
Round and Patient Characteristics
Peer observers recorded rounding, team, and patient characteristics using a standardized form. Rounding data included date, attending name, duration of rounds, and patient census. Patient level data included the decision(s) discussed, the seniority of the clinician leading the discussion, team composition, minutes spent discussing the patient (both with the patient and/or guardian and total time), hospitalization week, and patient’s primary language. Additional patient data obtained from electronic health records included age, gender, race, ethnicity, date of admission, and admitting diagnosis.
SDM Measures
Peer-observed SDM behaviors were quantified per patient encounter using the 9-item Rochester Participatory Decision-Making Scale (RPAD), with credit given for SDM behaviors exhibited by anyone on the rounding team (team-level metric).27 Each item was scored on a 3-point scale (0 = absent, 0.5 = partial, and 1 = present) for a maximum of 9 points, with higher scores indicating higher-quality SDM (Peer-RPAD Score). We created semistructured patient interview guides by adapting each RPAD item into layperson language (Patient-RPAD Score) and adding open-ended questions to assess the patient experience.
Peer-Observer Training
Eight peer-observers (7 hospitalists and 1 palliative care physician) were trained to perform RPAD ratings using videos of patient encounters. Initially, raters viewed videos together and discussed ratings for each RPAD item. The observers incorporated behavioral anchors and clinical examples into the development of an RPAD rating guide, which they subsequently used to independently score 4 videos from an online medical communication library.28 These scores were discussed to resolve any differences before 4 additional videos were independently viewed, scored, and compared. Interrater reliability was achieved when the standard deviation of summed SDM scores across raters was less than 1 for all 4 videos.
Patient Interviewers
Interviewers were English-speaking volunteers without formal medical training. They were educated in hospital etiquette by a physician and in administering patient interviews through peer-to-peer role playing and an observation and feedback interview with at least 1 patient.
Data Analysis
The analysis set included every unique patient with whom a medical decision was made by an eligible clinical team. To account for the nested study design (patient-level scores within rounds, rounds within attending, and attendings within service), we used mixed-effects models to estimate mean (summary or item) RPAD score by levels of fixed covariate(s). The models included random effects accounting for attending-level and round-level correlations among scores via variance components, and allowing the attending-level random effect to differ by service. Analyses were performed using SAS version 9.4 (SAS Institute Inc, Cary, NC). We used descriptive statistics to summarize round- and patient-level characteristics.
SDM Variation by Attending and Service
Box plots were used to summarize raw patient-level, Peer-RPAD scores by service and attending. By using the methods described above, we estimated the mean score overall and by service. In both models, we examined the statistical significance of service-specific variation in attending-level random effects by using likelihood-ratio test (LRT) to compare models.
SDM Variation by Round and Patient Characteristics
We used the models described above to identify covariates associated with Peer-RPAD scores. We fit univariate models separately for each covariate, then fit 2 multivariable models, including (1) all covariates and (2) all effects significant in either model at P ≤ .20 according to F tests. For uniformity of presentation, we express continuous covariates categorically; however, we report P values based on continuous versions. Means generated by the multivariable models were calculated at the mean values of all other covariates in model.
Patient-Level RPAD Data
A subsample of patients completed semistructured interviews with analogous RPAD questions. To identify possible selection bias in the full sample, we summarized response rates by service and patient language and modeled Peer-RPAD scores by interview response status. Among responders, we estimated the mean Peer-RPAD and Patient-RPAD scores and their paired differences and correlations, testing for non-zero correlations via the Spearman rank test.
RESULTS
All Patient Encounters
![]()
The median duration of rounding sessions was 1.8 hours, median patient census was 9, and median patient encounter was 13 minutes. The duration of rounds and minutes per patient were longest at Med-2 and shortest at Peds-1. See Table 1 for other team characteristics.
Peer Evaluations of SDM Encounters
Characteristics of Patients
![]()
Teams spent a median of 13 minutes per SDM encounter, which was not higher than the round median. SDM topics discussed included 47% treatment, 15% diagnostic, 30% both treatment and diagnostic, and 7% other.
Variation in SDM Quality Among Attending Physicians
Overall Peer-RPAD Scores were normally distributed. After adjusting for the nested study design, the overall mean (standard error) score was 4.16 (0.11). Score variability among attendings differed significantly by service (LRT P = .0067). For example, raw scores were lower and more variable among attending physicians at Med-2 than other among attendings in other services (see Appendix Figure in Supporting Information). However, when service was included in the model as a fixed effect, mean scores varied significantly, from 3.0 at Med-2 to 4.7 at Med-1 (P < .0001), but the random variation among attendings no longer differed significantly by service (P = .13). This finding supports the hypothesis that service-level influences are stronger than influences of individual attending physicians, that is, that variation between services exceeded variation among attendings within service.
Aspects of SDM That Are More Prevalent on Rounds
![]()
Rounds and Patient Characteristics Associated With Peer-RPAD Scores
![]()
Patient-RPAD Results: Dissimilar Perspectives of Patients and/or Guardians and Physician Observers
![]()
Among responders, mean Patient-RPAD scores were 6.8 to 7.1 for medicine services and 7.6 to 7.8 for pediatric services (P = .01). The overall mean Patient-RPAD score, 7.46, was significantly greater than the paired Peer-RPAD score by 3.5 (P = .011); however, correlations were not statistically significantly different from 0 (by service, each P > .12).
To understand drivers of the differences between Peer-RPAD and Patient-RPAD scores, we analyzed findings by item. Each mean patient-item score exceeded its peer counterpart (P ≤ .01; panel B of Figure). Peer-item scores fell below 33% on 2 items (Items 9 and 4) and only exceeded 67% on 2 items (Items 1 and 6), whereas patient-item scores ranged from 60% (Item 8) to 97% (Item 7). Three paired differences exceeded 50% (Items 9, 4, and 7) and 3 were below 20% (Items 6, 8 and 1), underlying the lack of correlation between peer and patient scores.
DISCUSSION
In this multisite study of SDM during inpatient attending rounds, SDM quality, specific SDM behaviors, and factors contributing to SDM were identified. Our study found an adjusted overall Peer-RPAD Score of 4.4 out of 9, and found the following 3 SDM elements most needing improvement according to trained peer observers: (1) “Checking understanding of the patient’s perspective”, (2) “Examining barriers to follow-through with the treatment plan”, and (3) “Asking if the patient has questions.” Areas of strength included explaining the clinical issue or nature of the decision and matching medical language to the patient’s level of understanding, with each rated highly by both peer-observers and patients. Broadly speaking, physicians were skillful in delivering information to patients but failed to solicit input from patients. Characteristics associated with increased SDM in the multivariate analysis included the following: service, patient gender, timing of rounds during patient’s hospital stay, and amount of time rounding with each patient.
Patients similarly found that physicians could improve their abilities to elicit information from patients and families, noting the 3 lowest patient-rated SDM elements were as follows: (1) asking open-ended questions, (2) discussing alternatives or uncertainties, and (3) discussing barriers to treatment plan follow through. Overall, patients and guardians perceived the quantity and quality of SDM on rounds more favorably than peer observers, which is consistent with other studies of patient perceptions of communication. 29-31 It is possible that patient ratings are more influenced by demand characteristics, fear of negatively impacting their patient-provider relationships, and conflation of overall satisfaction with quality of communication.32 This difference in patient perception of SDM is worthy of further study.
Prior work has revealed that SDM may occur infrequently during inpatient rounds.11 This study further elucidates specific SDM behaviors used along with univariate and multivariate modeling to explore possible contributing factors. The strengths and weaknesses found were similar at all 4 services and the influence of the service was more important than variability across attendings. This study’s findings are similar to a study by Shields et al.,33 in which the findings in a geographically different outpatient setting 10 years earlier suggesting global and enduring challenges to SDM. To our knowledge, this is the first published study to characterize inpatient SDM behaviors and may serve as the basis for future interventions.
Although the item-level components were ranked similarly across services, on average the summary Peer-RPAD score was lowest at Med-2, where we observed high variability within and between attendings, and was highest at Med-1, where variability was low. Med-2 carried the highest caseload and held the longest rounds, while Med-1 carried the lowest caseload, suggesting that modifiable burdens may hamper SDM performance. Prior studies suggest that patients are often selected based on teaching opportunities, immediate medical need and being newly admitted.34 The high scores at Med-1 may reflect that service’s prediscussion of patients during card-flipping rounds or their selection of which patients to round on as a team. Consistent with prior studies29,35 of SDM and the family-centered rounding model, which includes the involvement of nurses, respiratory therapists, pharmacists, case managers, social workers, and interpreters on rounds, both pediatrics services showed higher SDM scores.
In contrast to prior studies,34,36 team size and number of learners did not affect SDM performance, nor did decision type. Despite teams having up to 17 members, 8 learners, and 14 complex patients, SDM scores did not vary significantly by team. Nonetheless, trends were in the directions expected: Scores tended to decrease as the team size or the percentage of trainees grew, and increased with the seniority of the presenting physician. Interestingly, SDM performance decreased with round-average minutes per patient, which may be measuring on-going intensity across cases that leads to exhaustion. Statistically significant patient factors for increased SDM included longer duration of patient encounters, second week of hospital stay, and female patient gender. Although we anticipated that the high number of decisions made early in hospitalization would facilitate higher SDM scores, continuity and stronger patient-provider relationships may enhance SDM.36 We report service-specific team and patient characteristics, in addition to SDM findings in anticipation that some readers will identify with 1 service more than others.
This study has several important limitations. First, our peer observers were not blinded and primarily observed encounters at their own site. To minimize bias, observers periodically rated videos to recalibrate RPAD scoring. Second, additional SDM conversations with a patient and/or guardian may have occurred outside of rounds and were not captured, and poor patient recall may have affected Patient-RPAD scores despite interviewer prompts and timeliness of interviews within 12 hours of rounds. Third, there might have been a selection bias for the one service who selected a smaller number of patients to see, compared with the three other services that performed bedside rounds on all patients. It is possible that attending physicians selected patients who were deemed most able to have SDM conversations, thus affecting RPAD scores on that service. Fourth, study services had fewer patients on average than other academic hospitals (median 9, range 3-14), which might limit its generalizability. Last, as in any observational study, there is always the possibility of the Hawthorne effect. However, neither teams nor patients knew the study objectives.
Nevertheless, important findings emerged through the use of RPAD Scores to evaluate inpatient SDM practices. In particular, we found that to increase SDM quality in inpatient settings, practitioners should (1) check their understanding of the patient’s perspective, (2) examine barriers to follow-through with the treatment plan, and (3) ask if the patient has questions. Variation among services remained very influential after adjusting for team and patient characteristics, which suggests that “climate” or service culture should be targeted by an intervention, rather than individual attendings or subgroups defined by team or patient characteristics. Notably, team size, number of learners, patient census, and type of decision being made did not affect SDM performance, suggesting that even large, busy services can perform SDM if properly trained.
Acknowledgments
The authors thank the patients, families, pediatric and internal medicine residents, and hospitalists at Stanford School of Medicine and University of California, San Francisco School of Medicine for their participation in this study. We would also like to thank the student volunteers who collected patient perspectives on the encounters.
Disclosure
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by an NIH/NCCIH grant R25 AT006573.
1. Braddock CH. The emerging importance and relevance of shared decision making to clinical practice. Med Decis Mak. 2010;30(5 Suppl):5S-7S. doi:10.1177/0272989X10381344. PubMed
2. Braddock CH. Supporting shared decision making when clinical evidence is low. Med Care Res Rev MCRR. 2013;70(1 Suppl):129S-140S. doi:10.1177/1077558712460280. PubMed
3. Elwyn G, Tilburt J, Montori V. The ethical imperative for shared decision-making. Eur J Pers Centered Healthc. 2013;1(1):129-131. doi:10.5750/ejpch.v1i1.645.
4. Stiggelbout AM, Pieterse AH, De Haes JCJM. Shared decision making: Concepts, evidence, and practice. Patient Educ Couns. 2015;98(10):1172-1179. doi:10.1016/j.pec.2015.06.022. PubMed
5. Stacey D, Légaré F, Col NF, et al. Decision aids for people facing health treatment or screening decisions. Cochrane Database Syst Rev. 2014;(10):CD001431. doi:10.1002/14651858.CD001431.pub4. PubMed
6. Wilson SR, Strub P, Buist AS, et al. Shared treatment decision making improves adherence and outcomes in poorly controlled asthma. Am J Respir Crit Care Med. 2010;181(6):566-577. doi:10.1164/rccm.200906-0907OC. PubMed
7. Parchman ML, Zeber JE, Palmer RF. Participatory decision making, patient activation, medication adherence, and intermediate clinical outcomes in type 2 diabetes: a STARNet study. Ann Fam Med. 2010;8(5):410-417. doi:10.1370/afm.1161. PubMed
8. Weiner SJ, Schwartz A, Sharma G, et al. Patient-centered decision making and health care outcomes: an observational study. Ann Intern Med. 2013;158(8):573-579. doi:10.7326/0003-4819-158-8-201304160-00001. PubMed
9. Butterworth JE, Campbell JL. Older patients and their GPs: shared decision making in enhancing trust. Br J Gen Pract. 2014;64(628):e709-e718. doi:10.3399/bjgp14X682297. PubMed
10. Barry MJ, Edgman-Levitan S. Shared decision making--pinnacle of patient-centered care. N Engl J Med. 2012;366(9):780-781. doi:10.1056/NEJMp1109283. PubMed
11. Satterfield JM, Bereknyei S, Hilton JF, et al. The prevalence of social and behavioral topics and related educational opportunities during attending rounds. Acad Med J Assoc Am Med Coll. 2014;89(11):1548-1557. doi:10.1097/ACM.0000000000000483. PubMed
12. Charles C, Gafni A, Whelan T. Shared decision-making in the medical encounter: what does it mean? (or it takes at least two to tango). Soc Sci Med. 1997;44(5):681-692. PubMed
13. Elwyn G, Frosch D, Thomson R, et al. Shared decision making: a model for clinical practice. J Gen Intern Med. 2012;27(10):1361-1367. doi:10.1007/s11606-012-2077-6. PubMed
14. Légaré F, St-Jacques S, Gagnon S, et al. Prenatal screening for Down syndrome: a survey of willingness in women and family physicians to engage in shared decision-making. Prenat Diagn. 2011;31(4):319-326. doi:10.1002/pd.2624. PubMed
15. Satterfield JM, Spring B, Brownson RC, et al. Toward a Transdisciplinary Model of Evidence-Based Practice. Milbank Q. 2009;87(2):368-390. PubMed
16. National Academy of Medicine. Crossing the quality chasm: a new health system for the 21st century. https://www.nationalacademies.org/hmd/~/media/Files/Report%20Files/2001/Crossing-the-Quality-Chasm/Quality%20Chasm%202001%20%20report%20brief.pdf. Accessed on November 30, 2016.
17. Adams RC, Levy SE, Council on Children with Disabilities. Shared Decision-Making and Children with Disabilities: Pathways to Consensus. Pediatrics. 2017; 139(6):1-9. PubMed
18. Müller-Engelmann M, Keller H, Donner-Banzhoff N, Krones T. Shared decision making in medicine: The influence of situational treatment factors. Patient Educ Couns. 2011;82(2):240-246. doi:10.1016/j.pec.2010.04.028. PubMed
19. Whitney SN. A New Model of Medical Decisions: Exploring the Limits of Shared Decision Making. Med Decis Making. 2003;23(4):275-280. doi:10.1177/0272989X03256006. PubMed
20. Kehl KL, Landrum MB, Arora NK, et al. Association of Actual and Preferred Decision Roles With Patient-Reported Quality of Care: Shared Decision Making in Cancer Care. JAMA Oncol. 2015;1(1):50-58. doi:10.1001/jamaoncol.2014.112. PubMed
21. Couët N, Desroches S, Robitaille H, et al. Assessments of the extent to which health-care providers involve patients in decision making: a systematic review of studies using the OPTION instrument. Health Expect Int J Public Particip Health Care Health Policy. 2015;18(4):542-561. doi:10.1111/hex.12054. PubMed
22. Fowler FJ, Gerstein BS, Barry MJ. How patient centered are medical decisions?: Results of a national survey. JAMA Intern Med. 2013;173(13):1215-1221. doi:10.1001/jamainternmed.2013.6172. PubMed
23. Légaré F, Stacey D, Turcotte S, et al. Interventions for improving the adoption of shared decision making by healthcare professionals. Cochrane Database Syst Rev. 2014;(9):CD006732. doi:10.1002/14651858.CD006732.pub3. PubMed
24. Stacey D, Bennett CL, Barry MJ, et al. Decision aids for people facing health treatment or screening decisions. Cochrane Database Syst Rev. 2011;(10):CD001431. doi:10.1002/14651858.CD001431.pub3. PubMed
25. Di Francesco L, Pistoria MJ, Auerbach AD, Nardino RJ, Holmboe ES. Internal medicine training in the inpatient setting. A review of published educational interventions. J Gen Intern Med. 2005;20(12):1173-1180. doi:10.1111/j.1525-1497.2005.00250.x. PubMed
26. Janicik RW, Fletcher KE. Teaching at the bedside: a new model. Med Teach. 2003;25(2):127-130. PubMed
27. Shields CG, Franks P, Fiscella K, Meldrum S, Epstein RM. Rochester Participatory Decision-Making Scale (RPAD): reliability and validity. Ann Fam Med. 2005;3(5):436-442. doi:10.1370/afm.305. PubMed
28. DocCom - enhancing competence in healthcare communication. https://webcampus.drexelmed.edu/doccom/user/. Accessed on November 30, 2016.
29. Bailey SM, Hendricks-Muñoz KD, Mally P. Parental influence on clinical management during neonatal intensive care: a survey of US neonatologists. J Matern Fetal Neonatal Med. 2013;26(12):1239-1244. doi:10.3109/14767058.2013.776531. PubMed
30. Janz NK, Wren PA, Copeland LA, Lowery JC, Goldfarb SL, Wilkins EG. Patient-physician concordance: preferences, perceptions, and factors influencing the breast cancer surgical decision. J Clin Oncol. 2004;22(15):3091-3098. doi:10.1200/JCO.2004.09.069. PubMed
31. Schoenborn NL, Cayea D, McNabney M, Ray A, Boyd C. Prognosis communication with older patients with multimorbidity: Assessment after an educational intervention. Gerontol Geriatr Educ. 2016;38(4):471-481. doi:10.1080/02701960.2015.1115983. PubMed
32. Lipkin M. Shared decision making. JAMA Intern Med. 2013;173(13):1204-1205. doi:10.1001/jamainternmed.2013.6248. PubMed
33. Gonzalo JD, Heist BS, Duffy BL, et al. The art of bedside rounds: a multi-center qualitative study of strategies used by experienced bedside teachers. J Gen Intern Med. 2013;28(3):412-420. doi:10.1007/s11606-012-2259-2. PubMed
34. Rosen P, Stenger E, Bochkoris M, Hannon MJ, Kwoh CK. Family-centered multidisciplinary rounds enhance the team approach in pediatrics. Pediatrics. 2009;123(4):e603-e608. doi:10.1542/peds.2008-2238. PubMed
35. Harrison R, Allen E. Teaching internal medicine residents in the new era. Inpatient attending with duty-hour regulations. J Gen Intern Med. 2006;21(5):447-452. doi:10.1111/j.1525-1497.2006.00425.x. PubMed
36. Smith SK, Dixon A, Trevena L, Nutbeam D, McCaffery KJ. Exploring patient involvement in healthcare decision making across different education and functional health literacy groups. Soc Sci Med 1982. 2009;69(12):1805-1812. doi:10.1016/j.socscimed.2009.09.056. PubMed
The ethos of medicine has shifted from paternalistic, physician-driven care to patient autonomy and engagement, in which the physician shares information and advises.1-3 Although there are ethical, legal, and practical reasons to respect patient preferences,1-4 patient engagement also fosters quality and safety5 and may improve clinical outcomes.5-8 Patients whose preferences are respected are more likely to trust their doctor, feel empowered, and adhere to treatments.9
Providers may partner with patients through shared decision-making (SDM).10,11 Several SDM models describe the process of providers and patients balancing evidence, preferences and context to arrive at a clinical decision.12-15 The National Academy of Medicine and the American Academy of Pediatrics has called for more SDM,16,17 including when clinical evidence is limited,2 equally beneficial options exist,18 clinical stakes are high,19 and even with deferential patients.20 Despite its value, SDM does not reliably occur21,22 and SDM training is often unavailable.4 Clinical decision tools, patient education aids, and various training interventions have shown promising, although inconsistent results.23, 24
Little is known about SDM in inpatient settings where unique patient, clinician, and environmental factors may influence SDM. This study describes the quality and possible predictors of inpatient SDM during attending rounds in 4 academic training settings. Although SDM may occur anytime during a hospitalization, attending rounds present a valuable opportunity for SDM observation given their centrality to inpatient care and teaching.25,26 Because attending physicians bear ultimate responsibility for patient management, we examined whether SDM performance varies among attendings within each service. In addition, we tested the hypothesis that service-level, team-level, and patient-level features explain variation in SDM quality more than individual attending physicians. Finally, we compared peer-observer perspectives of SDM behaviors with patient and/or guardian perspectives.
METHODS
Study Design and Setting
This cross-sectional, observational study examined the diversity of SDM practice within and between 4 inpatient services during attending rounds, including the internal medicine and pediatrics services at Stanford University and the University of California, San Francisco (UCSF). Both institutions provide quaternary care to diverse patient populations with approximately half enrolled in Medicare and/or Medicaid.
One institution had 42 internal medicine (Med-1) and 15 pediatric hospitalists (Peds-1) compared to 8 internal medicine (Med-2) and 12 pediatric hospitalists (Peds-2) at the second location. Both pediatric services used family-centered rounds that included discussions between the patients’ families and the whole team. One medicine service used a similar rounding model that did not necessarily involve the patients’ families. In contrast, the smaller medicine service typically began rounds by discussing all patients in a conference room and then visiting select patients afterwards.
From August 2014 to November 2014, peer observers gathered data on team SDM behaviors during attending rounds. After the rounding team departed, nonphysician interviewers surveyed consenting patients’ (or guardians’) views of the SDM experience, yielding paired evaluations for a subset of SDM encounters. Institutional review board approval was obtained from Stanford University and UCSF.
Participants and Inclusion Criteria
Attending physicians were hospitalists who supervised rounds at least 1 month per year, and did not include those conducting the study. All provided verbal assent to be observed on 3 days within a 7-day period. While team composition varied as needed (eg, to include the nurse, pharmacist, interpreter, etc), we restricted study observations to those teams with an attending and at least one learner (eg, resident, intern, medical student) to capture the influence of attending physicians in their training role. Because services vary in number of attendings on staff, rounds assigned per attending, and patients per round, it was not possible to enroll equal sample sizes per service in the study.
Nonintensive care unit patients who were deemed medically stable by the team were eligible for peer observation and participation in a subsequent patient interview once during the study period. Pediatric patients were invited for an interview if they were between 13 and 21 years old and had the option of having a parent or guardian present; if the pediatric patients were less than 13 years old or they were not interested in being interviewed, then their parents or guardians were invited to be interviewed. Interpreters were on rounds, and thus, non-English participants were able to participate in the peer observations, but could not participate in patient interviews because interpreters were not available during afternoons for study purposes. Consent was obtained from all participating patients and/or guardians.
Data Collection
Round and Patient Characteristics
Peer observers recorded rounding, team, and patient characteristics using a standardized form. Rounding data included date, attending name, duration of rounds, and patient census. Patient level data included the decision(s) discussed, the seniority of the clinician leading the discussion, team composition, minutes spent discussing the patient (both with the patient and/or guardian and total time), hospitalization week, and patient’s primary language. Additional patient data obtained from electronic health records included age, gender, race, ethnicity, date of admission, and admitting diagnosis.
SDM Measures
Peer-observed SDM behaviors were quantified per patient encounter using the 9-item Rochester Participatory Decision-Making Scale (RPAD), with credit given for SDM behaviors exhibited by anyone on the rounding team (team-level metric).27 Each item was scored on a 3-point scale (0 = absent, 0.5 = partial, and 1 = present) for a maximum of 9 points, with higher scores indicating higher-quality SDM (Peer-RPAD Score). We created semistructured patient interview guides by adapting each RPAD item into layperson language (Patient-RPAD Score) and adding open-ended questions to assess the patient experience.
Peer-Observer Training
Eight peer-observers (7 hospitalists and 1 palliative care physician) were trained to perform RPAD ratings using videos of patient encounters. Initially, raters viewed videos together and discussed ratings for each RPAD item. The observers incorporated behavioral anchors and clinical examples into the development of an RPAD rating guide, which they subsequently used to independently score 4 videos from an online medical communication library.28 These scores were discussed to resolve any differences before 4 additional videos were independently viewed, scored, and compared. Interrater reliability was achieved when the standard deviation of summed SDM scores across raters was less than 1 for all 4 videos.
Patient Interviewers
Interviewers were English-speaking volunteers without formal medical training. They were educated in hospital etiquette by a physician and in administering patient interviews through peer-to-peer role playing and an observation and feedback interview with at least 1 patient.
Data Analysis
The analysis set included every unique patient with whom a medical decision was made by an eligible clinical team. To account for the nested study design (patient-level scores within rounds, rounds within attending, and attendings within service), we used mixed-effects models to estimate mean (summary or item) RPAD score by levels of fixed covariate(s). The models included random effects accounting for attending-level and round-level correlations among scores via variance components, and allowing the attending-level random effect to differ by service. Analyses were performed using SAS version 9.4 (SAS Institute Inc, Cary, NC). We used descriptive statistics to summarize round- and patient-level characteristics.
SDM Variation by Attending and Service
Box plots were used to summarize raw patient-level, Peer-RPAD scores by service and attending. By using the methods described above, we estimated the mean score overall and by service. In both models, we examined the statistical significance of service-specific variation in attending-level random effects by using likelihood-ratio test (LRT) to compare models.
SDM Variation by Round and Patient Characteristics
We used the models described above to identify covariates associated with Peer-RPAD scores. We fit univariate models separately for each covariate, then fit 2 multivariable models, including (1) all covariates and (2) all effects significant in either model at P ≤ .20 according to F tests. For uniformity of presentation, we express continuous covariates categorically; however, we report P values based on continuous versions. Means generated by the multivariable models were calculated at the mean values of all other covariates in model.
Patient-Level RPAD Data
A subsample of patients completed semistructured interviews with analogous RPAD questions. To identify possible selection bias in the full sample, we summarized response rates by service and patient language and modeled Peer-RPAD scores by interview response status. Among responders, we estimated the mean Peer-RPAD and Patient-RPAD scores and their paired differences and correlations, testing for non-zero correlations via the Spearman rank test.
RESULTS
All Patient Encounters
![]()
The median duration of rounding sessions was 1.8 hours, median patient census was 9, and median patient encounter was 13 minutes. The duration of rounds and minutes per patient were longest at Med-2 and shortest at Peds-1. See Table 1 for other team characteristics.
Peer Evaluations of SDM Encounters
Characteristics of Patients
![]()
Teams spent a median of 13 minutes per SDM encounter, which was not higher than the round median. SDM topics discussed included 47% treatment, 15% diagnostic, 30% both treatment and diagnostic, and 7% other.
Variation in SDM Quality Among Attending Physicians
Overall Peer-RPAD Scores were normally distributed. After adjusting for the nested study design, the overall mean (standard error) score was 4.16 (0.11). Score variability among attendings differed significantly by service (LRT P = .0067). For example, raw scores were lower and more variable among attending physicians at Med-2 than other among attendings in other services (see Appendix Figure in Supporting Information). However, when service was included in the model as a fixed effect, mean scores varied significantly, from 3.0 at Med-2 to 4.7 at Med-1 (P < .0001), but the random variation among attendings no longer differed significantly by service (P = .13). This finding supports the hypothesis that service-level influences are stronger than influences of individual attending physicians, that is, that variation between services exceeded variation among attendings within service.
Aspects of SDM That Are More Prevalent on Rounds
![]()
Rounds and Patient Characteristics Associated With Peer-RPAD Scores
![]()
Patient-RPAD Results: Dissimilar Perspectives of Patients and/or Guardians and Physician Observers
![]()
Among responders, mean Patient-RPAD scores were 6.8 to 7.1 for medicine services and 7.6 to 7.8 for pediatric services (P = .01). The overall mean Patient-RPAD score, 7.46, was significantly greater than the paired Peer-RPAD score by 3.5 (P = .011); however, correlations were not statistically significantly different from 0 (by service, each P > .12).
To understand drivers of the differences between Peer-RPAD and Patient-RPAD scores, we analyzed findings by item. Each mean patient-item score exceeded its peer counterpart (P ≤ .01; panel B of Figure). Peer-item scores fell below 33% on 2 items (Items 9 and 4) and only exceeded 67% on 2 items (Items 1 and 6), whereas patient-item scores ranged from 60% (Item 8) to 97% (Item 7). Three paired differences exceeded 50% (Items 9, 4, and 7) and 3 were below 20% (Items 6, 8 and 1), underlying the lack of correlation between peer and patient scores.
DISCUSSION
In this multisite study of SDM during inpatient attending rounds, SDM quality, specific SDM behaviors, and factors contributing to SDM were identified. Our study found an adjusted overall Peer-RPAD Score of 4.4 out of 9, and found the following 3 SDM elements most needing improvement according to trained peer observers: (1) “Checking understanding of the patient’s perspective”, (2) “Examining barriers to follow-through with the treatment plan”, and (3) “Asking if the patient has questions.” Areas of strength included explaining the clinical issue or nature of the decision and matching medical language to the patient’s level of understanding, with each rated highly by both peer-observers and patients. Broadly speaking, physicians were skillful in delivering information to patients but failed to solicit input from patients. Characteristics associated with increased SDM in the multivariate analysis included the following: service, patient gender, timing of rounds during patient’s hospital stay, and amount of time rounding with each patient.
Patients similarly found that physicians could improve their abilities to elicit information from patients and families, noting the 3 lowest patient-rated SDM elements were as follows: (1) asking open-ended questions, (2) discussing alternatives or uncertainties, and (3) discussing barriers to treatment plan follow through. Overall, patients and guardians perceived the quantity and quality of SDM on rounds more favorably than peer observers, which is consistent with other studies of patient perceptions of communication. 29-31 It is possible that patient ratings are more influenced by demand characteristics, fear of negatively impacting their patient-provider relationships, and conflation of overall satisfaction with quality of communication.32 This difference in patient perception of SDM is worthy of further study.
Prior work has revealed that SDM may occur infrequently during inpatient rounds.11 This study further elucidates specific SDM behaviors used along with univariate and multivariate modeling to explore possible contributing factors. The strengths and weaknesses found were similar at all 4 services and the influence of the service was more important than variability across attendings. This study’s findings are similar to a study by Shields et al.,33 in which the findings in a geographically different outpatient setting 10 years earlier suggesting global and enduring challenges to SDM. To our knowledge, this is the first published study to characterize inpatient SDM behaviors and may serve as the basis for future interventions.
Although the item-level components were ranked similarly across services, on average the summary Peer-RPAD score was lowest at Med-2, where we observed high variability within and between attendings, and was highest at Med-1, where variability was low. Med-2 carried the highest caseload and held the longest rounds, while Med-1 carried the lowest caseload, suggesting that modifiable burdens may hamper SDM performance. Prior studies suggest that patients are often selected based on teaching opportunities, immediate medical need and being newly admitted.34 The high scores at Med-1 may reflect that service’s prediscussion of patients during card-flipping rounds or their selection of which patients to round on as a team. Consistent with prior studies29,35 of SDM and the family-centered rounding model, which includes the involvement of nurses, respiratory therapists, pharmacists, case managers, social workers, and interpreters on rounds, both pediatrics services showed higher SDM scores.
In contrast to prior studies,34,36 team size and number of learners did not affect SDM performance, nor did decision type. Despite teams having up to 17 members, 8 learners, and 14 complex patients, SDM scores did not vary significantly by team. Nonetheless, trends were in the directions expected: Scores tended to decrease as the team size or the percentage of trainees grew, and increased with the seniority of the presenting physician. Interestingly, SDM performance decreased with round-average minutes per patient, which may be measuring on-going intensity across cases that leads to exhaustion. Statistically significant patient factors for increased SDM included longer duration of patient encounters, second week of hospital stay, and female patient gender. Although we anticipated that the high number of decisions made early in hospitalization would facilitate higher SDM scores, continuity and stronger patient-provider relationships may enhance SDM.36 We report service-specific team and patient characteristics, in addition to SDM findings in anticipation that some readers will identify with 1 service more than others.
This study has several important limitations. First, our peer observers were not blinded and primarily observed encounters at their own site. To minimize bias, observers periodically rated videos to recalibrate RPAD scoring. Second, additional SDM conversations with a patient and/or guardian may have occurred outside of rounds and were not captured, and poor patient recall may have affected Patient-RPAD scores despite interviewer prompts and timeliness of interviews within 12 hours of rounds. Third, there might have been a selection bias for the one service who selected a smaller number of patients to see, compared with the three other services that performed bedside rounds on all patients. It is possible that attending physicians selected patients who were deemed most able to have SDM conversations, thus affecting RPAD scores on that service. Fourth, study services had fewer patients on average than other academic hospitals (median 9, range 3-14), which might limit its generalizability. Last, as in any observational study, there is always the possibility of the Hawthorne effect. However, neither teams nor patients knew the study objectives.
Nevertheless, important findings emerged through the use of RPAD Scores to evaluate inpatient SDM practices. In particular, we found that to increase SDM quality in inpatient settings, practitioners should (1) check their understanding of the patient’s perspective, (2) examine barriers to follow-through with the treatment plan, and (3) ask if the patient has questions. Variation among services remained very influential after adjusting for team and patient characteristics, which suggests that “climate” or service culture should be targeted by an intervention, rather than individual attendings or subgroups defined by team or patient characteristics. Notably, team size, number of learners, patient census, and type of decision being made did not affect SDM performance, suggesting that even large, busy services can perform SDM if properly trained.
Acknowledgments
The authors thank the patients, families, pediatric and internal medicine residents, and hospitalists at Stanford School of Medicine and University of California, San Francisco School of Medicine for their participation in this study. We would also like to thank the student volunteers who collected patient perspectives on the encounters.
Disclosure
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by an NIH/NCCIH grant R25 AT006573.
The ethos of medicine has shifted from paternalistic, physician-driven care to patient autonomy and engagement, in which the physician shares information and advises.1-3 Although there are ethical, legal, and practical reasons to respect patient preferences,1-4 patient engagement also fosters quality and safety5 and may improve clinical outcomes.5-8 Patients whose preferences are respected are more likely to trust their doctor, feel empowered, and adhere to treatments.9
Providers may partner with patients through shared decision-making (SDM).10,11 Several SDM models describe the process of providers and patients balancing evidence, preferences and context to arrive at a clinical decision.12-15 The National Academy of Medicine and the American Academy of Pediatrics has called for more SDM,16,17 including when clinical evidence is limited,2 equally beneficial options exist,18 clinical stakes are high,19 and even with deferential patients.20 Despite its value, SDM does not reliably occur21,22 and SDM training is often unavailable.4 Clinical decision tools, patient education aids, and various training interventions have shown promising, although inconsistent results.23, 24
Little is known about SDM in inpatient settings where unique patient, clinician, and environmental factors may influence SDM. This study describes the quality and possible predictors of inpatient SDM during attending rounds in 4 academic training settings. Although SDM may occur anytime during a hospitalization, attending rounds present a valuable opportunity for SDM observation given their centrality to inpatient care and teaching.25,26 Because attending physicians bear ultimate responsibility for patient management, we examined whether SDM performance varies among attendings within each service. In addition, we tested the hypothesis that service-level, team-level, and patient-level features explain variation in SDM quality more than individual attending physicians. Finally, we compared peer-observer perspectives of SDM behaviors with patient and/or guardian perspectives.
METHODS
Study Design and Setting
This cross-sectional, observational study examined the diversity of SDM practice within and between 4 inpatient services during attending rounds, including the internal medicine and pediatrics services at Stanford University and the University of California, San Francisco (UCSF). Both institutions provide quaternary care to diverse patient populations with approximately half enrolled in Medicare and/or Medicaid.
One institution had 42 internal medicine (Med-1) and 15 pediatric hospitalists (Peds-1) compared to 8 internal medicine (Med-2) and 12 pediatric hospitalists (Peds-2) at the second location. Both pediatric services used family-centered rounds that included discussions between the patients’ families and the whole team. One medicine service used a similar rounding model that did not necessarily involve the patients’ families. In contrast, the smaller medicine service typically began rounds by discussing all patients in a conference room and then visiting select patients afterwards.
From August 2014 to November 2014, peer observers gathered data on team SDM behaviors during attending rounds. After the rounding team departed, nonphysician interviewers surveyed consenting patients’ (or guardians’) views of the SDM experience, yielding paired evaluations for a subset of SDM encounters. Institutional review board approval was obtained from Stanford University and UCSF.
Participants and Inclusion Criteria
Attending physicians were hospitalists who supervised rounds at least 1 month per year, and did not include those conducting the study. All provided verbal assent to be observed on 3 days within a 7-day period. While team composition varied as needed (eg, to include the nurse, pharmacist, interpreter, etc), we restricted study observations to those teams with an attending and at least one learner (eg, resident, intern, medical student) to capture the influence of attending physicians in their training role. Because services vary in number of attendings on staff, rounds assigned per attending, and patients per round, it was not possible to enroll equal sample sizes per service in the study.
Nonintensive care unit patients who were deemed medically stable by the team were eligible for peer observation and participation in a subsequent patient interview once during the study period. Pediatric patients were invited for an interview if they were between 13 and 21 years old and had the option of having a parent or guardian present; if the pediatric patients were less than 13 years old or they were not interested in being interviewed, then their parents or guardians were invited to be interviewed. Interpreters were on rounds, and thus, non-English participants were able to participate in the peer observations, but could not participate in patient interviews because interpreters were not available during afternoons for study purposes. Consent was obtained from all participating patients and/or guardians.
Data Collection
Round and Patient Characteristics
Peer observers recorded rounding, team, and patient characteristics using a standardized form. Rounding data included date, attending name, duration of rounds, and patient census. Patient level data included the decision(s) discussed, the seniority of the clinician leading the discussion, team composition, minutes spent discussing the patient (both with the patient and/or guardian and total time), hospitalization week, and patient’s primary language. Additional patient data obtained from electronic health records included age, gender, race, ethnicity, date of admission, and admitting diagnosis.
SDM Measures
Peer-observed SDM behaviors were quantified per patient encounter using the 9-item Rochester Participatory Decision-Making Scale (RPAD), with credit given for SDM behaviors exhibited by anyone on the rounding team (team-level metric).27 Each item was scored on a 3-point scale (0 = absent, 0.5 = partial, and 1 = present) for a maximum of 9 points, with higher scores indicating higher-quality SDM (Peer-RPAD Score). We created semistructured patient interview guides by adapting each RPAD item into layperson language (Patient-RPAD Score) and adding open-ended questions to assess the patient experience.
Peer-Observer Training
Eight peer-observers (7 hospitalists and 1 palliative care physician) were trained to perform RPAD ratings using videos of patient encounters. Initially, raters viewed videos together and discussed ratings for each RPAD item. The observers incorporated behavioral anchors and clinical examples into the development of an RPAD rating guide, which they subsequently used to independently score 4 videos from an online medical communication library.28 These scores were discussed to resolve any differences before 4 additional videos were independently viewed, scored, and compared. Interrater reliability was achieved when the standard deviation of summed SDM scores across raters was less than 1 for all 4 videos.
Patient Interviewers
Interviewers were English-speaking volunteers without formal medical training. They were educated in hospital etiquette by a physician and in administering patient interviews through peer-to-peer role playing and an observation and feedback interview with at least 1 patient.
Data Analysis
The analysis set included every unique patient with whom a medical decision was made by an eligible clinical team. To account for the nested study design (patient-level scores within rounds, rounds within attending, and attendings within service), we used mixed-effects models to estimate mean (summary or item) RPAD score by levels of fixed covariate(s). The models included random effects accounting for attending-level and round-level correlations among scores via variance components, and allowing the attending-level random effect to differ by service. Analyses were performed using SAS version 9.4 (SAS Institute Inc, Cary, NC). We used descriptive statistics to summarize round- and patient-level characteristics.
SDM Variation by Attending and Service
Box plots were used to summarize raw patient-level, Peer-RPAD scores by service and attending. By using the methods described above, we estimated the mean score overall and by service. In both models, we examined the statistical significance of service-specific variation in attending-level random effects by using likelihood-ratio test (LRT) to compare models.
SDM Variation by Round and Patient Characteristics
We used the models described above to identify covariates associated with Peer-RPAD scores. We fit univariate models separately for each covariate, then fit 2 multivariable models, including (1) all covariates and (2) all effects significant in either model at P ≤ .20 according to F tests. For uniformity of presentation, we express continuous covariates categorically; however, we report P values based on continuous versions. Means generated by the multivariable models were calculated at the mean values of all other covariates in model.
Patient-Level RPAD Data
A subsample of patients completed semistructured interviews with analogous RPAD questions. To identify possible selection bias in the full sample, we summarized response rates by service and patient language and modeled Peer-RPAD scores by interview response status. Among responders, we estimated the mean Peer-RPAD and Patient-RPAD scores and their paired differences and correlations, testing for non-zero correlations via the Spearman rank test.
RESULTS
All Patient Encounters
![]()
The median duration of rounding sessions was 1.8 hours, median patient census was 9, and median patient encounter was 13 minutes. The duration of rounds and minutes per patient were longest at Med-2 and shortest at Peds-1. See Table 1 for other team characteristics.
Peer Evaluations of SDM Encounters
Characteristics of Patients
![]()
Teams spent a median of 13 minutes per SDM encounter, which was not higher than the round median. SDM topics discussed included 47% treatment, 15% diagnostic, 30% both treatment and diagnostic, and 7% other.
Variation in SDM Quality Among Attending Physicians
Overall Peer-RPAD Scores were normally distributed. After adjusting for the nested study design, the overall mean (standard error) score was 4.16 (0.11). Score variability among attendings differed significantly by service (LRT P = .0067). For example, raw scores were lower and more variable among attending physicians at Med-2 than other among attendings in other services (see Appendix Figure in Supporting Information). However, when service was included in the model as a fixed effect, mean scores varied significantly, from 3.0 at Med-2 to 4.7 at Med-1 (P < .0001), but the random variation among attendings no longer differed significantly by service (P = .13). This finding supports the hypothesis that service-level influences are stronger than influences of individual attending physicians, that is, that variation between services exceeded variation among attendings within service.
Aspects of SDM That Are More Prevalent on Rounds
![]()
Rounds and Patient Characteristics Associated With Peer-RPAD Scores
![]()
Patient-RPAD Results: Dissimilar Perspectives of Patients and/or Guardians and Physician Observers
![]()
Among responders, mean Patient-RPAD scores were 6.8 to 7.1 for medicine services and 7.6 to 7.8 for pediatric services (P = .01). The overall mean Patient-RPAD score, 7.46, was significantly greater than the paired Peer-RPAD score by 3.5 (P = .011); however, correlations were not statistically significantly different from 0 (by service, each P > .12).
To understand drivers of the differences between Peer-RPAD and Patient-RPAD scores, we analyzed findings by item. Each mean patient-item score exceeded its peer counterpart (P ≤ .01; panel B of Figure). Peer-item scores fell below 33% on 2 items (Items 9 and 4) and only exceeded 67% on 2 items (Items 1 and 6), whereas patient-item scores ranged from 60% (Item 8) to 97% (Item 7). Three paired differences exceeded 50% (Items 9, 4, and 7) and 3 were below 20% (Items 6, 8 and 1), underlying the lack of correlation between peer and patient scores.
DISCUSSION
In this multisite study of SDM during inpatient attending rounds, SDM quality, specific SDM behaviors, and factors contributing to SDM were identified. Our study found an adjusted overall Peer-RPAD Score of 4.4 out of 9, and found the following 3 SDM elements most needing improvement according to trained peer observers: (1) “Checking understanding of the patient’s perspective”, (2) “Examining barriers to follow-through with the treatment plan”, and (3) “Asking if the patient has questions.” Areas of strength included explaining the clinical issue or nature of the decision and matching medical language to the patient’s level of understanding, with each rated highly by both peer-observers and patients. Broadly speaking, physicians were skillful in delivering information to patients but failed to solicit input from patients. Characteristics associated with increased SDM in the multivariate analysis included the following: service, patient gender, timing of rounds during patient’s hospital stay, and amount of time rounding with each patient.
Patients similarly found that physicians could improve their abilities to elicit information from patients and families, noting the 3 lowest patient-rated SDM elements were as follows: (1) asking open-ended questions, (2) discussing alternatives or uncertainties, and (3) discussing barriers to treatment plan follow through. Overall, patients and guardians perceived the quantity and quality of SDM on rounds more favorably than peer observers, which is consistent with other studies of patient perceptions of communication. 29-31 It is possible that patient ratings are more influenced by demand characteristics, fear of negatively impacting their patient-provider relationships, and conflation of overall satisfaction with quality of communication.32 This difference in patient perception of SDM is worthy of further study.
Prior work has revealed that SDM may occur infrequently during inpatient rounds.11 This study further elucidates specific SDM behaviors used along with univariate and multivariate modeling to explore possible contributing factors. The strengths and weaknesses found were similar at all 4 services and the influence of the service was more important than variability across attendings. This study’s findings are similar to a study by Shields et al.,33 in which the findings in a geographically different outpatient setting 10 years earlier suggesting global and enduring challenges to SDM. To our knowledge, this is the first published study to characterize inpatient SDM behaviors and may serve as the basis for future interventions.
Although the item-level components were ranked similarly across services, on average the summary Peer-RPAD score was lowest at Med-2, where we observed high variability within and between attendings, and was highest at Med-1, where variability was low. Med-2 carried the highest caseload and held the longest rounds, while Med-1 carried the lowest caseload, suggesting that modifiable burdens may hamper SDM performance. Prior studies suggest that patients are often selected based on teaching opportunities, immediate medical need and being newly admitted.34 The high scores at Med-1 may reflect that service’s prediscussion of patients during card-flipping rounds or their selection of which patients to round on as a team. Consistent with prior studies29,35 of SDM and the family-centered rounding model, which includes the involvement of nurses, respiratory therapists, pharmacists, case managers, social workers, and interpreters on rounds, both pediatrics services showed higher SDM scores.
In contrast to prior studies,34,36 team size and number of learners did not affect SDM performance, nor did decision type. Despite teams having up to 17 members, 8 learners, and 14 complex patients, SDM scores did not vary significantly by team. Nonetheless, trends were in the directions expected: Scores tended to decrease as the team size or the percentage of trainees grew, and increased with the seniority of the presenting physician. Interestingly, SDM performance decreased with round-average minutes per patient, which may be measuring on-going intensity across cases that leads to exhaustion. Statistically significant patient factors for increased SDM included longer duration of patient encounters, second week of hospital stay, and female patient gender. Although we anticipated that the high number of decisions made early in hospitalization would facilitate higher SDM scores, continuity and stronger patient-provider relationships may enhance SDM.36 We report service-specific team and patient characteristics, in addition to SDM findings in anticipation that some readers will identify with 1 service more than others.
This study has several important limitations. First, our peer observers were not blinded and primarily observed encounters at their own site. To minimize bias, observers periodically rated videos to recalibrate RPAD scoring. Second, additional SDM conversations with a patient and/or guardian may have occurred outside of rounds and were not captured, and poor patient recall may have affected Patient-RPAD scores despite interviewer prompts and timeliness of interviews within 12 hours of rounds. Third, there might have been a selection bias for the one service who selected a smaller number of patients to see, compared with the three other services that performed bedside rounds on all patients. It is possible that attending physicians selected patients who were deemed most able to have SDM conversations, thus affecting RPAD scores on that service. Fourth, study services had fewer patients on average than other academic hospitals (median 9, range 3-14), which might limit its generalizability. Last, as in any observational study, there is always the possibility of the Hawthorne effect. However, neither teams nor patients knew the study objectives.
Nevertheless, important findings emerged through the use of RPAD Scores to evaluate inpatient SDM practices. In particular, we found that to increase SDM quality in inpatient settings, practitioners should (1) check their understanding of the patient’s perspective, (2) examine barriers to follow-through with the treatment plan, and (3) ask if the patient has questions. Variation among services remained very influential after adjusting for team and patient characteristics, which suggests that “climate” or service culture should be targeted by an intervention, rather than individual attendings or subgroups defined by team or patient characteristics. Notably, team size, number of learners, patient census, and type of decision being made did not affect SDM performance, suggesting that even large, busy services can perform SDM if properly trained.
Acknowledgments
The authors thank the patients, families, pediatric and internal medicine residents, and hospitalists at Stanford School of Medicine and University of California, San Francisco School of Medicine for their participation in this study. We would also like to thank the student volunteers who collected patient perspectives on the encounters.
Disclosure
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by an NIH/NCCIH grant R25 AT006573.
1. Braddock CH. The emerging importance and relevance of shared decision making to clinical practice. Med Decis Mak. 2010;30(5 Suppl):5S-7S. doi:10.1177/0272989X10381344. PubMed
2. Braddock CH. Supporting shared decision making when clinical evidence is low. Med Care Res Rev MCRR. 2013;70(1 Suppl):129S-140S. doi:10.1177/1077558712460280. PubMed
3. Elwyn G, Tilburt J, Montori V. The ethical imperative for shared decision-making. Eur J Pers Centered Healthc. 2013;1(1):129-131. doi:10.5750/ejpch.v1i1.645.
4. Stiggelbout AM, Pieterse AH, De Haes JCJM. Shared decision making: Concepts, evidence, and practice. Patient Educ Couns. 2015;98(10):1172-1179. doi:10.1016/j.pec.2015.06.022. PubMed
5. Stacey D, Légaré F, Col NF, et al. Decision aids for people facing health treatment or screening decisions. Cochrane Database Syst Rev. 2014;(10):CD001431. doi:10.1002/14651858.CD001431.pub4. PubMed
6. Wilson SR, Strub P, Buist AS, et al. Shared treatment decision making improves adherence and outcomes in poorly controlled asthma. Am J Respir Crit Care Med. 2010;181(6):566-577. doi:10.1164/rccm.200906-0907OC. PubMed
7. Parchman ML, Zeber JE, Palmer RF. Participatory decision making, patient activation, medication adherence, and intermediate clinical outcomes in type 2 diabetes: a STARNet study. Ann Fam Med. 2010;8(5):410-417. doi:10.1370/afm.1161. PubMed
8. Weiner SJ, Schwartz A, Sharma G, et al. Patient-centered decision making and health care outcomes: an observational study. Ann Intern Med. 2013;158(8):573-579. doi:10.7326/0003-4819-158-8-201304160-00001. PubMed
9. Butterworth JE, Campbell JL. Older patients and their GPs: shared decision making in enhancing trust. Br J Gen Pract. 2014;64(628):e709-e718. doi:10.3399/bjgp14X682297. PubMed
10. Barry MJ, Edgman-Levitan S. Shared decision making--pinnacle of patient-centered care. N Engl J Med. 2012;366(9):780-781. doi:10.1056/NEJMp1109283. PubMed
11. Satterfield JM, Bereknyei S, Hilton JF, et al. The prevalence of social and behavioral topics and related educational opportunities during attending rounds. Acad Med J Assoc Am Med Coll. 2014;89(11):1548-1557. doi:10.1097/ACM.0000000000000483. PubMed
12. Charles C, Gafni A, Whelan T. Shared decision-making in the medical encounter: what does it mean? (or it takes at least two to tango). Soc Sci Med. 1997;44(5):681-692. PubMed
13. Elwyn G, Frosch D, Thomson R, et al. Shared decision making: a model for clinical practice. J Gen Intern Med. 2012;27(10):1361-1367. doi:10.1007/s11606-012-2077-6. PubMed
14. Légaré F, St-Jacques S, Gagnon S, et al. Prenatal screening for Down syndrome: a survey of willingness in women and family physicians to engage in shared decision-making. Prenat Diagn. 2011;31(4):319-326. doi:10.1002/pd.2624. PubMed
15. Satterfield JM, Spring B, Brownson RC, et al. Toward a Transdisciplinary Model of Evidence-Based Practice. Milbank Q. 2009;87(2):368-390. PubMed
16. National Academy of Medicine. Crossing the quality chasm: a new health system for the 21st century. https://www.nationalacademies.org/hmd/~/media/Files/Report%20Files/2001/Crossing-the-Quality-Chasm/Quality%20Chasm%202001%20%20report%20brief.pdf. Accessed on November 30, 2016.
17. Adams RC, Levy SE, Council on Children with Disabilities. Shared Decision-Making and Children with Disabilities: Pathways to Consensus. Pediatrics. 2017; 139(6):1-9. PubMed
18. Müller-Engelmann M, Keller H, Donner-Banzhoff N, Krones T. Shared decision making in medicine: The influence of situational treatment factors. Patient Educ Couns. 2011;82(2):240-246. doi:10.1016/j.pec.2010.04.028. PubMed
19. Whitney SN. A New Model of Medical Decisions: Exploring the Limits of Shared Decision Making. Med Decis Making. 2003;23(4):275-280. doi:10.1177/0272989X03256006. PubMed
20. Kehl KL, Landrum MB, Arora NK, et al. Association of Actual and Preferred Decision Roles With Patient-Reported Quality of Care: Shared Decision Making in Cancer Care. JAMA Oncol. 2015;1(1):50-58. doi:10.1001/jamaoncol.2014.112. PubMed
21. Couët N, Desroches S, Robitaille H, et al. Assessments of the extent to which health-care providers involve patients in decision making: a systematic review of studies using the OPTION instrument. Health Expect Int J Public Particip Health Care Health Policy. 2015;18(4):542-561. doi:10.1111/hex.12054. PubMed
22. Fowler FJ, Gerstein BS, Barry MJ. How patient centered are medical decisions?: Results of a national survey. JAMA Intern Med. 2013;173(13):1215-1221. doi:10.1001/jamainternmed.2013.6172. PubMed
23. Légaré F, Stacey D, Turcotte S, et al. Interventions for improving the adoption of shared decision making by healthcare professionals. Cochrane Database Syst Rev. 2014;(9):CD006732. doi:10.1002/14651858.CD006732.pub3. PubMed
24. Stacey D, Bennett CL, Barry MJ, et al. Decision aids for people facing health treatment or screening decisions. Cochrane Database Syst Rev. 2011;(10):CD001431. doi:10.1002/14651858.CD001431.pub3. PubMed
25. Di Francesco L, Pistoria MJ, Auerbach AD, Nardino RJ, Holmboe ES. Internal medicine training in the inpatient setting. A review of published educational interventions. J Gen Intern Med. 2005;20(12):1173-1180. doi:10.1111/j.1525-1497.2005.00250.x. PubMed
26. Janicik RW, Fletcher KE. Teaching at the bedside: a new model. Med Teach. 2003;25(2):127-130. PubMed
27. Shields CG, Franks P, Fiscella K, Meldrum S, Epstein RM. Rochester Participatory Decision-Making Scale (RPAD): reliability and validity. Ann Fam Med. 2005;3(5):436-442. doi:10.1370/afm.305. PubMed
28. DocCom - enhancing competence in healthcare communication. https://webcampus.drexelmed.edu/doccom/user/. Accessed on November 30, 2016.
29. Bailey SM, Hendricks-Muñoz KD, Mally P. Parental influence on clinical management during neonatal intensive care: a survey of US neonatologists. J Matern Fetal Neonatal Med. 2013;26(12):1239-1244. doi:10.3109/14767058.2013.776531. PubMed
30. Janz NK, Wren PA, Copeland LA, Lowery JC, Goldfarb SL, Wilkins EG. Patient-physician concordance: preferences, perceptions, and factors influencing the breast cancer surgical decision. J Clin Oncol. 2004;22(15):3091-3098. doi:10.1200/JCO.2004.09.069. PubMed
31. Schoenborn NL, Cayea D, McNabney M, Ray A, Boyd C. Prognosis communication with older patients with multimorbidity: Assessment after an educational intervention. Gerontol Geriatr Educ. 2016;38(4):471-481. doi:10.1080/02701960.2015.1115983. PubMed
32. Lipkin M. Shared decision making. JAMA Intern Med. 2013;173(13):1204-1205. doi:10.1001/jamainternmed.2013.6248. PubMed
33. Gonzalo JD, Heist BS, Duffy BL, et al. The art of bedside rounds: a multi-center qualitative study of strategies used by experienced bedside teachers. J Gen Intern Med. 2013;28(3):412-420. doi:10.1007/s11606-012-2259-2. PubMed
34. Rosen P, Stenger E, Bochkoris M, Hannon MJ, Kwoh CK. Family-centered multidisciplinary rounds enhance the team approach in pediatrics. Pediatrics. 2009;123(4):e603-e608. doi:10.1542/peds.2008-2238. PubMed
35. Harrison R, Allen E. Teaching internal medicine residents in the new era. Inpatient attending with duty-hour regulations. J Gen Intern Med. 2006;21(5):447-452. doi:10.1111/j.1525-1497.2006.00425.x. PubMed
36. Smith SK, Dixon A, Trevena L, Nutbeam D, McCaffery KJ. Exploring patient involvement in healthcare decision making across different education and functional health literacy groups. Soc Sci Med 1982. 2009;69(12):1805-1812. doi:10.1016/j.socscimed.2009.09.056. PubMed
1. Braddock CH. The emerging importance and relevance of shared decision making to clinical practice. Med Decis Mak. 2010;30(5 Suppl):5S-7S. doi:10.1177/0272989X10381344. PubMed
2. Braddock CH. Supporting shared decision making when clinical evidence is low. Med Care Res Rev MCRR. 2013;70(1 Suppl):129S-140S. doi:10.1177/1077558712460280. PubMed
3. Elwyn G, Tilburt J, Montori V. The ethical imperative for shared decision-making. Eur J Pers Centered Healthc. 2013;1(1):129-131. doi:10.5750/ejpch.v1i1.645.
4. Stiggelbout AM, Pieterse AH, De Haes JCJM. Shared decision making: Concepts, evidence, and practice. Patient Educ Couns. 2015;98(10):1172-1179. doi:10.1016/j.pec.2015.06.022. PubMed
5. Stacey D, Légaré F, Col NF, et al. Decision aids for people facing health treatment or screening decisions. Cochrane Database Syst Rev. 2014;(10):CD001431. doi:10.1002/14651858.CD001431.pub4. PubMed
6. Wilson SR, Strub P, Buist AS, et al. Shared treatment decision making improves adherence and outcomes in poorly controlled asthma. Am J Respir Crit Care Med. 2010;181(6):566-577. doi:10.1164/rccm.200906-0907OC. PubMed
7. Parchman ML, Zeber JE, Palmer RF. Participatory decision making, patient activation, medication adherence, and intermediate clinical outcomes in type 2 diabetes: a STARNet study. Ann Fam Med. 2010;8(5):410-417. doi:10.1370/afm.1161. PubMed
8. Weiner SJ, Schwartz A, Sharma G, et al. Patient-centered decision making and health care outcomes: an observational study. Ann Intern Med. 2013;158(8):573-579. doi:10.7326/0003-4819-158-8-201304160-00001. PubMed
9. Butterworth JE, Campbell JL. Older patients and their GPs: shared decision making in enhancing trust. Br J Gen Pract. 2014;64(628):e709-e718. doi:10.3399/bjgp14X682297. PubMed
10. Barry MJ, Edgman-Levitan S. Shared decision making--pinnacle of patient-centered care. N Engl J Med. 2012;366(9):780-781. doi:10.1056/NEJMp1109283. PubMed
11. Satterfield JM, Bereknyei S, Hilton JF, et al. The prevalence of social and behavioral topics and related educational opportunities during attending rounds. Acad Med J Assoc Am Med Coll. 2014;89(11):1548-1557. doi:10.1097/ACM.0000000000000483. PubMed
12. Charles C, Gafni A, Whelan T. Shared decision-making in the medical encounter: what does it mean? (or it takes at least two to tango). Soc Sci Med. 1997;44(5):681-692. PubMed
13. Elwyn G, Frosch D, Thomson R, et al. Shared decision making: a model for clinical practice. J Gen Intern Med. 2012;27(10):1361-1367. doi:10.1007/s11606-012-2077-6. PubMed
14. Légaré F, St-Jacques S, Gagnon S, et al. Prenatal screening for Down syndrome: a survey of willingness in women and family physicians to engage in shared decision-making. Prenat Diagn. 2011;31(4):319-326. doi:10.1002/pd.2624. PubMed
15. Satterfield JM, Spring B, Brownson RC, et al. Toward a Transdisciplinary Model of Evidence-Based Practice. Milbank Q. 2009;87(2):368-390. PubMed
16. National Academy of Medicine. Crossing the quality chasm: a new health system for the 21st century. https://www.nationalacademies.org/hmd/~/media/Files/Report%20Files/2001/Crossing-the-Quality-Chasm/Quality%20Chasm%202001%20%20report%20brief.pdf. Accessed on November 30, 2016.
17. Adams RC, Levy SE, Council on Children with Disabilities. Shared Decision-Making and Children with Disabilities: Pathways to Consensus. Pediatrics. 2017; 139(6):1-9. PubMed
18. Müller-Engelmann M, Keller H, Donner-Banzhoff N, Krones T. Shared decision making in medicine: The influence of situational treatment factors. Patient Educ Couns. 2011;82(2):240-246. doi:10.1016/j.pec.2010.04.028. PubMed
19. Whitney SN. A New Model of Medical Decisions: Exploring the Limits of Shared Decision Making. Med Decis Making. 2003;23(4):275-280. doi:10.1177/0272989X03256006. PubMed
20. Kehl KL, Landrum MB, Arora NK, et al. Association of Actual and Preferred Decision Roles With Patient-Reported Quality of Care: Shared Decision Making in Cancer Care. JAMA Oncol. 2015;1(1):50-58. doi:10.1001/jamaoncol.2014.112. PubMed
21. Couët N, Desroches S, Robitaille H, et al. Assessments of the extent to which health-care providers involve patients in decision making: a systematic review of studies using the OPTION instrument. Health Expect Int J Public Particip Health Care Health Policy. 2015;18(4):542-561. doi:10.1111/hex.12054. PubMed
22. Fowler FJ, Gerstein BS, Barry MJ. How patient centered are medical decisions?: Results of a national survey. JAMA Intern Med. 2013;173(13):1215-1221. doi:10.1001/jamainternmed.2013.6172. PubMed
23. Légaré F, Stacey D, Turcotte S, et al. Interventions for improving the adoption of shared decision making by healthcare professionals. Cochrane Database Syst Rev. 2014;(9):CD006732. doi:10.1002/14651858.CD006732.pub3. PubMed
24. Stacey D, Bennett CL, Barry MJ, et al. Decision aids for people facing health treatment or screening decisions. Cochrane Database Syst Rev. 2011;(10):CD001431. doi:10.1002/14651858.CD001431.pub3. PubMed
25. Di Francesco L, Pistoria MJ, Auerbach AD, Nardino RJ, Holmboe ES. Internal medicine training in the inpatient setting. A review of published educational interventions. J Gen Intern Med. 2005;20(12):1173-1180. doi:10.1111/j.1525-1497.2005.00250.x. PubMed
26. Janicik RW, Fletcher KE. Teaching at the bedside: a new model. Med Teach. 2003;25(2):127-130. PubMed
27. Shields CG, Franks P, Fiscella K, Meldrum S, Epstein RM. Rochester Participatory Decision-Making Scale (RPAD): reliability and validity. Ann Fam Med. 2005;3(5):436-442. doi:10.1370/afm.305. PubMed
28. DocCom - enhancing competence in healthcare communication. https://webcampus.drexelmed.edu/doccom/user/. Accessed on November 30, 2016.
29. Bailey SM, Hendricks-Muñoz KD, Mally P. Parental influence on clinical management during neonatal intensive care: a survey of US neonatologists. J Matern Fetal Neonatal Med. 2013;26(12):1239-1244. doi:10.3109/14767058.2013.776531. PubMed
30. Janz NK, Wren PA, Copeland LA, Lowery JC, Goldfarb SL, Wilkins EG. Patient-physician concordance: preferences, perceptions, and factors influencing the breast cancer surgical decision. J Clin Oncol. 2004;22(15):3091-3098. doi:10.1200/JCO.2004.09.069. PubMed
31. Schoenborn NL, Cayea D, McNabney M, Ray A, Boyd C. Prognosis communication with older patients with multimorbidity: Assessment after an educational intervention. Gerontol Geriatr Educ. 2016;38(4):471-481. doi:10.1080/02701960.2015.1115983. PubMed
32. Lipkin M. Shared decision making. JAMA Intern Med. 2013;173(13):1204-1205. doi:10.1001/jamainternmed.2013.6248. PubMed
33. Gonzalo JD, Heist BS, Duffy BL, et al. The art of bedside rounds: a multi-center qualitative study of strategies used by experienced bedside teachers. J Gen Intern Med. 2013;28(3):412-420. doi:10.1007/s11606-012-2259-2. PubMed
34. Rosen P, Stenger E, Bochkoris M, Hannon MJ, Kwoh CK. Family-centered multidisciplinary rounds enhance the team approach in pediatrics. Pediatrics. 2009;123(4):e603-e608. doi:10.1542/peds.2008-2238. PubMed
35. Harrison R, Allen E. Teaching internal medicine residents in the new era. Inpatient attending with duty-hour regulations. J Gen Intern Med. 2006;21(5):447-452. doi:10.1111/j.1525-1497.2006.00425.x. PubMed
36. Smith SK, Dixon A, Trevena L, Nutbeam D, McCaffery KJ. Exploring patient involvement in healthcare decision making across different education and functional health literacy groups. Soc Sci Med 1982. 2009;69(12):1805-1812. doi:10.1016/j.socscimed.2009.09.056. PubMed
© 2018 Society of Hospital Medicine
Iron deficiency anemia: Disease heterogeneity and the rapid evolution of traditional diagnosis and treatment paradigms

Release Date: February 1, 2018
Expiration Date: January 31, 2019
Note: This activity is no longer available for credit
Agenda
Classification and Causes of Iron Deficiency and Iron Deficiency Anemia (duration 14:30)
Maureen M. Achebe, MD, MPH
Clinical Director, Non-Malignant Hematology Clinic
Dana-Farber Cancer Institute
Assistant Professor of Medicine
Harvard Medical School
Boston, MA
Diagnostic Approaches to Iron Deficiency Anemia:
Conventional Tests or Newer Assessments? (duration 13:00)
Thomas DeLoughery, MD, MACP
Professor of Medicine
OHSU School of Medicine
Portland, OR
Treatment: From Oral Iron Supplementation to Intravenous Therapy
(duration 21:00)
Michael Auerbach, MD, FACP
Clinical Professor of Medicine
Georgetown University School of Medicine
Washington, DC
Provided by:
![]()
Original activity supported by an educational grant from: Luitpold Pharmaceuticals, Inc.
Learning Objectives
After completing the activity, clinicians will be better able to:
- Describe iron metabolism and the mechanism of iron deficiency and iron deficiency anemia
- Differentiate the causes of iron deficiency and iron deficiency anemia and confounding factors
- Evaluate the different approaches to diagnose iron deficiency and iron deficiency anemia
- Discuss the indications for intravenous iron therapy, the various formulations, and the benefits and risk of each
Target Audience
Hematologists, oncologists, and allied healthcare professionals who manage patients with iron deficiency anemia
Statement of Need
Iron deficiency (ID) and iron deficiency anemia (IDA) are common conditions worldwide, with 1.6 billion people in both developing and developed countries affected by these conditions, which can have serious consequences. Clinicians can readily diagnose ID and IDA in a healthy individual with a single cause of anemia. However, “explosive knowledge” of iron metabolism over the last 20 years has made it increasingly more difficult for them to determine the correct diagnosis and treatment [Camaschella, Blood Rev 2017]. This leaves clinicians uncertain as to the appropriate tests to order, how to interpret the results, what treatment to use, and at what dose. This activity is designed to clarify these issues and help the healthcare team provide optimal care for patients.
Disclosures
Maureen Achebe, MD (Presenter)
Consulting fees: Luitpold Pharmaceuticals, Inc., AMAG Pharmaceuticals, Inc., Syros Pharmaceuticals, Inc.
Michael Auerbach, MD (Course Director and Presenter)
Consulting fee: AMAG Pharmaceuticals, Inc., American Regent Luitpold, Pharmacosmos
Contracted research (Data management only): AMAG Pharmaceuticals, Pharmacosmos
Thomas DeLoughery, MD (Presenter)
No relevant financial relationships with a commercial interest.
Permissions
Maureen Achebe presentation
- Slide 14: Functional Iron Deficiency
Republished with permission of the American Society of Hematology, from Bruganara C, et al. Red blood cell regeneration induced by subcutaneous recombinant erythropoietin: iron-deficient erythropoiesis in iron-replete subjects. Blood 1993; 81(4):956-64; permission conveyed through Copyright Clearance Center, Inc.
- Slide 22: Prevalence of Anemia by Cr and GFR in a Pre-dialysis Population
McClellan W, et al. The prevalence of anemia in patients with chronic kidney disease. Curr Med Res Opin 2004;20(9):1501-10, reprinted by permission of the publisher (Taylor & Francis Ltd, http://www.tandfonline.com)
Michael Auerbach presentation
- Slide 9: Once vs Twice Daily Dosing
Reprinted from Lancet Haematol, Vol 4, No 11, Stoffel NU, Cercamondi CI, Brittenham G, et al. Iron absorption from oral iron supplements given on consecutive versus alternate days and as single morning doses versus twice-daily split dosing in iron-depleted women: two open-label, randomised controlled trials, e524-e533, © 2017, with permission from Elsevier.
- Slide 22: Iron Deficiency in Infancy Alters Neural Correlates of Recognition Memory at 10 Years: Key Study Results
Reprinted from J Pediatr, Vol 160, Congdon EL, Westerlund A, Algarin CR, et al. Iron deficiency in infancy is associated with altered neural correlates of recognition memory at 10 years, pp 1027-1033, © 2012, with permission from Elsevier.
Thomas DeLoughery presentation
- Slide 12: [No title]
From N Engl J Med, Lipschitz DA, Cook, JD, Finch CA, A clinical evaluation of serum ferritin as an index of iron stores, Vol 290, pages 1213-1216, © 1974 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
- Slide 15: Most Women Have Low Iron Stores
Reproduced with permission from JAMA, 1967, 199(12):897-900. Copyright© 1967 American Medical Association. All rights reserved
- Slide 16: Age and Ferritin
Reprinted from Am Heart J, Vol 140, Zacharski LR, Ornstein, DL, Woloshin S, et al. Association of age, sex, and race with body iron stores in adults: analysis of NHANES III data, pp 98-104, © 2000, with permission from Elsevier.
Disclaimer
The content and views presented in this educational activity are those of the authors and do not necessarily reflect those of Hemedicus, the supporter, or Frontline Medical Communications. This material is prepared based upon a review of multiple sources of information, but it is not exhaustive of the subject matter. Therefore, healthcare professionals and other individuals should review and consider other publications and materials on the subject matter before relying solely upon the information contained within this educational activity.
Release Date: February 1, 2018
Expiration Date: January 31, 2019
Note: This activity is no longer available for credit
Agenda
Classification and Causes of Iron Deficiency and Iron Deficiency Anemia (duration 14:30)
Maureen M. Achebe, MD, MPH
Clinical Director, Non-Malignant Hematology Clinic
Dana-Farber Cancer Institute
Assistant Professor of Medicine
Harvard Medical School
Boston, MA
Diagnostic Approaches to Iron Deficiency Anemia:
Conventional Tests or Newer Assessments? (duration 13:00)
Thomas DeLoughery, MD, MACP
Professor of Medicine
OHSU School of Medicine
Portland, OR
Treatment: From Oral Iron Supplementation to Intravenous Therapy
(duration 21:00)
Michael Auerbach, MD, FACP
Clinical Professor of Medicine
Georgetown University School of Medicine
Washington, DC
Provided by:
![]()
Original activity supported by an educational grant from: Luitpold Pharmaceuticals, Inc.
Learning Objectives
After completing the activity, clinicians will be better able to:
- Describe iron metabolism and the mechanism of iron deficiency and iron deficiency anemia
- Differentiate the causes of iron deficiency and iron deficiency anemia and confounding factors
- Evaluate the different approaches to diagnose iron deficiency and iron deficiency anemia
- Discuss the indications for intravenous iron therapy, the various formulations, and the benefits and risk of each
Target Audience
Hematologists, oncologists, and allied healthcare professionals who manage patients with iron deficiency anemia
Statement of Need
Iron deficiency (ID) and iron deficiency anemia (IDA) are common conditions worldwide, with 1.6 billion people in both developing and developed countries affected by these conditions, which can have serious consequences. Clinicians can readily diagnose ID and IDA in a healthy individual with a single cause of anemia. However, “explosive knowledge” of iron metabolism over the last 20 years has made it increasingly more difficult for them to determine the correct diagnosis and treatment [Camaschella, Blood Rev 2017]. This leaves clinicians uncertain as to the appropriate tests to order, how to interpret the results, what treatment to use, and at what dose. This activity is designed to clarify these issues and help the healthcare team provide optimal care for patients.
Disclosures
Maureen Achebe, MD (Presenter)
Consulting fees: Luitpold Pharmaceuticals, Inc., AMAG Pharmaceuticals, Inc., Syros Pharmaceuticals, Inc.
Michael Auerbach, MD (Course Director and Presenter)
Consulting fee: AMAG Pharmaceuticals, Inc., American Regent Luitpold, Pharmacosmos
Contracted research (Data management only): AMAG Pharmaceuticals, Pharmacosmos
Thomas DeLoughery, MD (Presenter)
No relevant financial relationships with a commercial interest.
Permissions
Maureen Achebe presentation
- Slide 14: Functional Iron Deficiency
Republished with permission of the American Society of Hematology, from Bruganara C, et al. Red blood cell regeneration induced by subcutaneous recombinant erythropoietin: iron-deficient erythropoiesis in iron-replete subjects. Blood 1993; 81(4):956-64; permission conveyed through Copyright Clearance Center, Inc.
- Slide 22: Prevalence of Anemia by Cr and GFR in a Pre-dialysis Population
McClellan W, et al. The prevalence of anemia in patients with chronic kidney disease. Curr Med Res Opin 2004;20(9):1501-10, reprinted by permission of the publisher (Taylor & Francis Ltd, http://www.tandfonline.com)
Michael Auerbach presentation
- Slide 9: Once vs Twice Daily Dosing
Reprinted from Lancet Haematol, Vol 4, No 11, Stoffel NU, Cercamondi CI, Brittenham G, et al. Iron absorption from oral iron supplements given on consecutive versus alternate days and as single morning doses versus twice-daily split dosing in iron-depleted women: two open-label, randomised controlled trials, e524-e533, © 2017, with permission from Elsevier.
- Slide 22: Iron Deficiency in Infancy Alters Neural Correlates of Recognition Memory at 10 Years: Key Study Results
Reprinted from J Pediatr, Vol 160, Congdon EL, Westerlund A, Algarin CR, et al. Iron deficiency in infancy is associated with altered neural correlates of recognition memory at 10 years, pp 1027-1033, © 2012, with permission from Elsevier.
Thomas DeLoughery presentation
- Slide 12: [No title]
From N Engl J Med, Lipschitz DA, Cook, JD, Finch CA, A clinical evaluation of serum ferritin as an index of iron stores, Vol 290, pages 1213-1216, © 1974 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
- Slide 15: Most Women Have Low Iron Stores
Reproduced with permission from JAMA, 1967, 199(12):897-900. Copyright© 1967 American Medical Association. All rights reserved
- Slide 16: Age and Ferritin
Reprinted from Am Heart J, Vol 140, Zacharski LR, Ornstein, DL, Woloshin S, et al. Association of age, sex, and race with body iron stores in adults: analysis of NHANES III data, pp 98-104, © 2000, with permission from Elsevier.
Disclaimer
The content and views presented in this educational activity are those of the authors and do not necessarily reflect those of Hemedicus, the supporter, or Frontline Medical Communications. This material is prepared based upon a review of multiple sources of information, but it is not exhaustive of the subject matter. Therefore, healthcare professionals and other individuals should review and consider other publications and materials on the subject matter before relying solely upon the information contained within this educational activity.
Release Date: February 1, 2018
Expiration Date: January 31, 2019
Note: This activity is no longer available for credit
Agenda
Classification and Causes of Iron Deficiency and Iron Deficiency Anemia (duration 14:30)
Maureen M. Achebe, MD, MPH
Clinical Director, Non-Malignant Hematology Clinic
Dana-Farber Cancer Institute
Assistant Professor of Medicine
Harvard Medical School
Boston, MA
Diagnostic Approaches to Iron Deficiency Anemia:
Conventional Tests or Newer Assessments? (duration 13:00)
Thomas DeLoughery, MD, MACP
Professor of Medicine
OHSU School of Medicine
Portland, OR
Treatment: From Oral Iron Supplementation to Intravenous Therapy
(duration 21:00)
Michael Auerbach, MD, FACP
Clinical Professor of Medicine
Georgetown University School of Medicine
Washington, DC
Provided by:
![]()
Original activity supported by an educational grant from: Luitpold Pharmaceuticals, Inc.
Learning Objectives
After completing the activity, clinicians will be better able to:
- Describe iron metabolism and the mechanism of iron deficiency and iron deficiency anemia
- Differentiate the causes of iron deficiency and iron deficiency anemia and confounding factors
- Evaluate the different approaches to diagnose iron deficiency and iron deficiency anemia
- Discuss the indications for intravenous iron therapy, the various formulations, and the benefits and risk of each
Target Audience
Hematologists, oncologists, and allied healthcare professionals who manage patients with iron deficiency anemia
Statement of Need
Iron deficiency (ID) and iron deficiency anemia (IDA) are common conditions worldwide, with 1.6 billion people in both developing and developed countries affected by these conditions, which can have serious consequences. Clinicians can readily diagnose ID and IDA in a healthy individual with a single cause of anemia. However, “explosive knowledge” of iron metabolism over the last 20 years has made it increasingly more difficult for them to determine the correct diagnosis and treatment [Camaschella, Blood Rev 2017]. This leaves clinicians uncertain as to the appropriate tests to order, how to interpret the results, what treatment to use, and at what dose. This activity is designed to clarify these issues and help the healthcare team provide optimal care for patients.
Disclosures
Maureen Achebe, MD (Presenter)
Consulting fees: Luitpold Pharmaceuticals, Inc., AMAG Pharmaceuticals, Inc., Syros Pharmaceuticals, Inc.
Michael Auerbach, MD (Course Director and Presenter)
Consulting fee: AMAG Pharmaceuticals, Inc., American Regent Luitpold, Pharmacosmos
Contracted research (Data management only): AMAG Pharmaceuticals, Pharmacosmos
Thomas DeLoughery, MD (Presenter)
No relevant financial relationships with a commercial interest.
Permissions
Maureen Achebe presentation
- Slide 14: Functional Iron Deficiency
Republished with permission of the American Society of Hematology, from Bruganara C, et al. Red blood cell regeneration induced by subcutaneous recombinant erythropoietin: iron-deficient erythropoiesis in iron-replete subjects. Blood 1993; 81(4):956-64; permission conveyed through Copyright Clearance Center, Inc.
- Slide 22: Prevalence of Anemia by Cr and GFR in a Pre-dialysis Population
McClellan W, et al. The prevalence of anemia in patients with chronic kidney disease. Curr Med Res Opin 2004;20(9):1501-10, reprinted by permission of the publisher (Taylor & Francis Ltd, http://www.tandfonline.com)
Michael Auerbach presentation
- Slide 9: Once vs Twice Daily Dosing
Reprinted from Lancet Haematol, Vol 4, No 11, Stoffel NU, Cercamondi CI, Brittenham G, et al. Iron absorption from oral iron supplements given on consecutive versus alternate days and as single morning doses versus twice-daily split dosing in iron-depleted women: two open-label, randomised controlled trials, e524-e533, © 2017, with permission from Elsevier.
- Slide 22: Iron Deficiency in Infancy Alters Neural Correlates of Recognition Memory at 10 Years: Key Study Results
Reprinted from J Pediatr, Vol 160, Congdon EL, Westerlund A, Algarin CR, et al. Iron deficiency in infancy is associated with altered neural correlates of recognition memory at 10 years, pp 1027-1033, © 2012, with permission from Elsevier.
Thomas DeLoughery presentation
- Slide 12: [No title]
From N Engl J Med, Lipschitz DA, Cook, JD, Finch CA, A clinical evaluation of serum ferritin as an index of iron stores, Vol 290, pages 1213-1216, © 1974 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
- Slide 15: Most Women Have Low Iron Stores
Reproduced with permission from JAMA, 1967, 199(12):897-900. Copyright© 1967 American Medical Association. All rights reserved
- Slide 16: Age and Ferritin
Reprinted from Am Heart J, Vol 140, Zacharski LR, Ornstein, DL, Woloshin S, et al. Association of age, sex, and race with body iron stores in adults: analysis of NHANES III data, pp 98-104, © 2000, with permission from Elsevier.
Disclaimer
The content and views presented in this educational activity are those of the authors and do not necessarily reflect those of Hemedicus, the supporter, or Frontline Medical Communications. This material is prepared based upon a review of multiple sources of information, but it is not exhaustive of the subject matter. Therefore, healthcare professionals and other individuals should review and consider other publications and materials on the subject matter before relying solely upon the information contained within this educational activity.
Brown spot on back

The dermatoscope revealed a fine reticular network around a central scar, which confirmed a diagnosis of dermatofibroma. A dermatofibroma is a benign fibrohistiocytic tumor found in the mid dermis, composed of a mixture of fibroblastic and histiocytic cells. It represents a fibrous reaction triggered by trauma, a viral infection, or an insect bite. Many dermatofibromas have a hyperpigmented halo around a central hypopigmented fibrous scar.
Dermoscopy is a very useful diagnostic technique for dermatofibroma. The most common pattern found is a peripheral reticular pigment network with a central hypopigmented stellate area.
No treatment is necessary unless the diagnosis is uncertain or symptoms warrant it. Dermatofibromas can be removed using punch excision for small lesions or an elliptical (fusiform) excision down to the subcutaneous fat for larger lesions. Cryotherapy is one option to shrink the lesion, but the cure rate is low and lesions may regrow.
The patient was relieved that the lesion was not cancer and opted to leave it be, as it was not bothering him.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Smith M. Usatine R. Dermatofibroma. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 935-939.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The dermatoscope revealed a fine reticular network around a central scar, which confirmed a diagnosis of dermatofibroma. A dermatofibroma is a benign fibrohistiocytic tumor found in the mid dermis, composed of a mixture of fibroblastic and histiocytic cells. It represents a fibrous reaction triggered by trauma, a viral infection, or an insect bite. Many dermatofibromas have a hyperpigmented halo around a central hypopigmented fibrous scar.
Dermoscopy is a very useful diagnostic technique for dermatofibroma. The most common pattern found is a peripheral reticular pigment network with a central hypopigmented stellate area.
No treatment is necessary unless the diagnosis is uncertain or symptoms warrant it. Dermatofibromas can be removed using punch excision for small lesions or an elliptical (fusiform) excision down to the subcutaneous fat for larger lesions. Cryotherapy is one option to shrink the lesion, but the cure rate is low and lesions may regrow.
The patient was relieved that the lesion was not cancer and opted to leave it be, as it was not bothering him.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Smith M. Usatine R. Dermatofibroma. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 935-939.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The dermatoscope revealed a fine reticular network around a central scar, which confirmed a diagnosis of dermatofibroma. A dermatofibroma is a benign fibrohistiocytic tumor found in the mid dermis, composed of a mixture of fibroblastic and histiocytic cells. It represents a fibrous reaction triggered by trauma, a viral infection, or an insect bite. Many dermatofibromas have a hyperpigmented halo around a central hypopigmented fibrous scar.
Dermoscopy is a very useful diagnostic technique for dermatofibroma. The most common pattern found is a peripheral reticular pigment network with a central hypopigmented stellate area.
No treatment is necessary unless the diagnosis is uncertain or symptoms warrant it. Dermatofibromas can be removed using punch excision for small lesions or an elliptical (fusiform) excision down to the subcutaneous fat for larger lesions. Cryotherapy is one option to shrink the lesion, but the cure rate is low and lesions may regrow.
The patient was relieved that the lesion was not cancer and opted to leave it be, as it was not bothering him.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Smith M. Usatine R. Dermatofibroma. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 935-939.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Transient neurologic syndromes: A diagnostic approach
Many patients present to their primary care physicians, urgent care centers, and emergency rooms because of neurologic symptoms lasting seconds to hours. Their problems can be a cause for concern and a challenge to diagnose, as in many cases their symptoms have returned to baseline by the time of evaluation. Referral to a neurologist may not be practical for all of them, particularly given that a consultation may take a long time to obtain.
Understanding the causes of transient neurologic syndromes and their phenomenology may help the clinician diagnose, triage, and treat such conditions effectively.
Here, we outline several transient neurologic syndromes—transient ischemic attack (TIA), migraine with aura, partial seizures, hypoglycemic encephalopathy, hyperventilation syndrome, transient global amnesia, narcolepsy, parasomnias, and some rarer conditions— focusing on their diagnostic elements. Others, such as drug-induced transient neurologic syndromes, vertigo, and dizziness, have been well discussed elsewhere.1–3
THE BIG 3: TIA, MIGRAINE, SEIZURES
A 45-year-old woman with a history of tobacco use and headaches presents to the emergency department with a 4-month history of episodic numbness and tingling of her right arm and face. She reports a prodromal state of anxiety and irritability 24 to 48 hours before symptom onset.
The sensory symptoms begin on her face and gradually progress down the arm and eventually to her fingers. They fully resolve within 2 hours without sequelae. Family members have noted some “slurred speech” during the episodes, and the episodes are occasionally preceded by a unilateral, throbbing headache that improves with rest.
What are the possible causes of her symptoms?
Transient ischemic attack
If a patient reports transient neurologic symptoms and has vascular risk factors, TIA is often the default diagnostic consideration. The risk of stroke is 9.9% in the 2 days after a TIA, 13.4% at 30 days, and 17.3% at 90 days.4 Rapid recognition offers a crucial period to minimize the possibility of permanent impairment. Interventions include modifying risk factors (hypertension, diabetes, and smoking) and starting an antiplatelet drug, an anticoagulant drug, or both, and possibly a statin.
It can be difficult to determine if this workup needs to be completed in the inpatient or outpatient setting. There is no clear consensus, but the ultimate goal is timely evaluation (within 24 to 48 hours). The ABCD2 (Age, Blood pressure, Clinical features, Duration of symptoms, and Diabetes) risk factor calculator was developed to help triage patients, though it has limitations.5,6
One should assess a patient’s history of a possible TIA in a stepwise fashion. First, analyze the patient’s age and demographics for known vascular risk factors or central embolic sources (eg, atrial fibrillation). Then consider the symptoms. TIA symptoms have rapid onset, usually within seconds7; symptoms with a more gradual crescendo suggest a nonvascular cause.8 TIA manifestations should resolve within 1 hour, and most studies suggest symptom resolution within 10 minutes is specific for a TIA.9–11 TIA symptoms are negative neurologic phenomena that denote a loss of function, such as loss of vision, motor weakness, or sensory numbness.
Symptoms should also correlate with a defined vascular territory:
- The middle cerebral artery is commonly involved; its blockage is associated with aphasia, weakness of the face and arm, and homonymous visual field impairment (loss of one-half of the visual fields in both eyes)
- Blockage in the posterior circulation generally causes symptoms localized to the brainstem, cerebellum, and occipital cortex. The symptoms are usually grouped together as the “5Ds”: dizziness, diplopia, dysarthria, dysphagia, and dystaxia/ataxia. Brainstem involvement classically produces “crossed” findings, with ipsilateral cranial findings and contralateral motor or sensory findings.
- Lacunar strokes involve the subcortical white matter and produce typical patterns including pure motor or sensory syndromes.
Loss of consciousness is rarely a symptom of TIA and should suggest another etiology.
The definition of TIA has evolved from an operational one, ie, symptoms lasting less than 24 hours, to a tissue-based one, ie, focal cerebral ischemia not associated with permanent cerebral infarction.12 Though imperfect, this pathophysiology should help reinforce the most common features of TIA, including a sudden onset of negative symptoms that are localized to a defined vascular territory.13,14
Migraine with aura
Migraine with aura is common in patients ages 25 to 55 who have a long-standing history of headache. The pathophysiologic mechanism of an aura is believed to be a disseminating wave of cortical depression, which is a self-propagating wave of neural depression and then activation. Ultimately, this leads to a cascade of inflammatory and pain signals, resulting in a headache.
This background helps explain the positive (superimposed) symptoms associated with the aura. Positive symptoms are produced by excessive neuronal discharges stimulating the visual (flashing lights, zigzag lines), sensory (paresthesias), or motor (limb movements) pathways.
Common symptoms associated with aura include visual disturbances such as scintillating scotoma (a blind spot), sensory changes such as tingling, or auditory disruption with tinnitus. Symptoms may evolve over the course of 5 to 20 minutes, first affecting vision and then other senses. In contrast, in a TIA, symptoms usually begin simultaneously and are confined to a vascular territory.7,15 Symptoms of an aura usually resolve within an hour, but there is evidence showing a substantial number of patients have an aura lasting much longer.16 Focal weakness is uncommon during an aura but is reported in specific migraine conditions such as hemiplegic migraine and migraine with unilateral motor symptoms. The vast majority of patients experience other neurologic symptoms during this prodrome.17,18
The prodromal period (2 to 48 hours leading up to the onset of migraine) is a commonly overlooked feature of migraine.19 Common symptoms during this time include fatigue, mood change, and gastrointestinal symptoms.20 One study demonstrated that patients generally had good intuition concerning these nonspecific prodromal symptoms and could predict the onset of migraine 72% of the time.21
In addition, a myriad of possible triggers and exacerbating factors can be identified (and sometimes avoided) such as visual stimuli, weather changes, nitrates, sleep disturbances, menstruation, foods, and stressors.22
Although headache is often the cardinal manifestation of migraine, some patients experience aura without headache—acephalgic migraine.23 This can be a diagnostic challenge, especially in an older population with multiple vascular risk factors. New-onset acephalgic migraine may be a cause for concern but is not uncommon and is not associated with a significantly increased risk of stroke.24 Focusing on the character of the neurologic symptoms in regard to timing, progression, and resolution will help differentiate this disease from other transient neurologic syndromes.25
Partial seizure
Partial seizure produces a diverse range of stereotypical symptoms due to focal abnormal neuronal activation. The aberrant electrical firing generates positive symptoms involving the motor, sensory, or visual pathway. A history of trauma, neurosurgical intervention, central nervous system infection, stroke, or other seizure foci can suggest this diagnosis. Other prodromal clues include abdominal discomfort, sense of detachment, déjà vu, or jamais vu.26
During a seizure, there may be a progression of positive symptoms similar to what happens in migraine aura, because both represent cortical spread and depression.
Involvement of the motor pathway may produce tonic (stiffening) or clonic (twitching) movement. Other common motor abnormalities include automatisms such as lip smacking, chewing, and hand gestures (picking, fidgeting, fumbling).27
Epileptic discharges in the sensory cortex commonly cause paresthesias or distortion of a sensory input. Visual symptoms may be more complex. In occipital epilepsy, circular phenomena with a colored pattern are common, which contrasts with the photopsia (flashes of light) or fortification (a bright zigzag of lines resembling a fort) seen in migraines.28
Autonomic or somatosensory symptoms can also occur.
Todd paralysis, also called transient postictal paralysis, occurs in only 13% of seizures but can linger for 0.5 to 36 hours.29,30 This weakness is most pronounced within the affected region after a partial seizure.
In general, focal seizures are often stereotyped with positive neurologic features, usually last a few minutes, and resolve fully. These episodes may cause an arrest in activity but not usually loss of consciousness unless the epileptic discharge secondarily generalizes into the adjacent hemisphere.
A common differential diagnosis encountered during an epilepsy workup is psychogenic nonepileptic seizures. Nonepileptic seizures consist of transient, abnormal movements, sensation, or cognition but lack ictal electroencephalographic changes. This is a specifically challenging patient population, with high healthcare utilization and high risk for iatrogenic harm. In addition, on average, diagnosis can take years to establish and usually requires referral to a tertiary care facility.31,32
The big 3: Back to our patient
Our patient’s vascular risk factors, transient symptoms, and language involvement support the diagnosis of TIA. A feature that points away from the diagnosis of TIA is the gradual onset of positive neurologic symptoms. This pattern is not consistent with neuronal ischemia.
Also, our patient had a repetitive, stereotypical pattern of symptoms, which supports including partial seizures in the differential diagnosis. On the other hand, her lack of risk factors for seizure (a history of febrile seizures, developmental delay, trauma, or infection) would make this diagnosis less likely. Also pointing away from the diagnosis of seizures are her lack of typical prodromal symptoms, the length of the events, and the postevent headache.

Table 1 summarizes the clinical findings associated with TIA, migraine, and partial seizure.
EPISODES OF CONFUSION
A 35-year-old woman with a history of depression, anxiety, and poorly controlled type 1 diabetes presents to the clinic after several weeks of episodes of confusion, usually accompanied by paresthesias in both hands, dizziness, and palpitations. In each episode, soon after the symptoms began, she had painful cramps in her hand. The symptoms fully resolved within 10 minutes without sequelae.
Questioned further, the patient describes the confusion as a “mental haze” but denies frank disorientation. She has not kept a log of her blood sugar levels but has not noticed a temporal relationship with regard to her meals or insulin injections.
What are the possible causes of these episodes?
Hypoglycemic encephalopathy
Hypoglycemia is common in most people with diabetes, who have been reported to suffer from 62 to 320 severe hypoglycemic episodes in their lifetime.33,34 The neurologic consequences can be devastating in these severe cases.
During mild to moderate drops in the glucose level, generalized symptoms stem from sympathetic activation. These include generalized anxiety, tremor, palpitations, and sweating. Focal symptoms such as unilateral weakness have also been reported.35,36
Unfortunately, people with long-standing diabetes have a blunted response to epinephrine that reduces their sensitivity to hypoglycemia, placing them at high risk of permanent neurologic damage. This can lead to seizures and coma, as the hypoglycemia has a greater effect on cortical and subcortical structures (highly metabolic areas) than on the brainstem. Thus, respiratory and cardiovascular function is maintained but cerebral function is abnormal. If this state is prolonged, brain death can occur.37,38
Hyperventilation syndrome
Hyperventilation syndrome is not well characterized. Most think of it as synonymous with an underlying psychopathology, but there is evidence to suggest it can occur without underlying anxiety.
There is no clear mechanism, but it is hypothesized that diminished carbon dioxide levels lead to cerebral vasoconstriction. This may lead to reduced cerebral blood flow, causing dizziness, lightheadedness, or vertigo.39 Appendicular symptoms including paresthesias, carpopedal spasm, or tetany have been core features since the syndrome was first described in the early 1900s.40
Though the disorder has rather nonspecific features, it can be easily reproduced in the clinical setting by asking the patient to breathe deeply and rapidly. This can help confirm the underlying diagnosis and also reassure the patient that the underlying pathology is not life-threatening and that he or she has some control over the disease.
Transient global amnesia
Transient global amnesia usually strikes older patients (50 to 70 years old) in the setting of an acute physical or emotional stressor. There is also a correlation between transient global amnesia and migraine, with studies showing migraineurs are at higher risk than the general population.41 Despite common clinical concerns, there is no relationship between transient global amnesia and stroke.42
Transient global amnesia is defined by acute transient anterograde amnesia (coding of new memories). To try to reorient themselves, patients will repeatedly ask questions such as “What day is it?” or “Why are we here?” Retrograde memories, especially long-standing ones, are usually well preserved. The patient’s cognition is otherwise intact, and there are no other focal neurologic symptoms. The event usually lasts 2 to 24 hours and resolves without sequelae.43,44 Afterward, patients remember the event only poorly, which supports the notion that they cannot code new memories.
Confusional episodes: Discussion
Evaluating confusional episodes can be time-consuming and vexing. The subjective nature of the symptoms and the vast differential diagnosis can be overwhelming. Subtle clinical details can help formulate an appropriate evaluation.
Hypoglycemia can produce bizarre neurologic symptoms. Most cases of hypoglycemia produce an exaggerated sympathetic response, though this is blunted in people with longstanding diabetes. In addition, there should be a temporal association with meals, insulin doses, or both.
Transient global amnesia usually occurs with acute stressors and produces a confusional state. These episodes rarely recur, and the patient cannot provide much history regarding the episode secondary to the anterograde amnesia.

Table 2 summarizes the clinical findings associated with hypoglycemic encephalopathy, hyperventilation syndrome, and transient global amnesia.
Back to our patient
In our patient, the likely diagnosis is hyperventilation syndrome, even though we don’t know if her respiratory rate is increased during attacks. Some patients lack awareness of their breathing or are too distracted by the vague symptoms to have insight into the true cause. The cramps and contractions in the hands are a specific feature of the disease and can be accompanied by confusion.
SLEEP DISORDERS
A 17-year-old boy with a history of depression and anxiety presents to his pediatrician because he has had difficulty staying awake in school over the past year. His sleepiness has gradually worsened over the last few months and has taken a toll on his grades, leading to discord in his family. Over the past month he has had some difficulty holding his head up during arguments with friends. He does not lose consciousness during these events but is described as “unresponsive.” He describes vivid dreams when going to sleep that have startled him awake at times. His family history is positive for somnambulism on his father’s side.
Does this patient have a sleep disorder, and if so, which one?
Narcolepsy
Narcolepsy is defined by excessive daytime sleepiness, cataplexy, hypnagogic hallucination, and sleep paralysis. It is more common in men but its prevalence varies widely by geographic region, supporting an underlying interplay between genetics and environment.45
Sleep attacks or excessive daytime sleepiness are the cardinal features of narcolepsy. The dissociation between the sleep-wake cycle is evident with rapid transition into rapid-eye-movement (REM) sleep during these sleep attacks. This results in a “refreshing nap” that commonly involves vivid dreams. These episodes occur about 3 to 5 times per day, varying in duration from a few minutes to hours.46
Cataplexy is very specific feature of narcolepsy. Triggered by strong emotion, the body loses skeletal muscle tone except for the diaphragm and ocular muscles. The patient does not lose consciousness and remains aware of his or her environment. Of note, the loss of tone does not need to be dramatic. The hypotonia can manifest as jaw-dropping or head-nodding. The paralysis is related to prolonged REM atonia and impaired transition from sleep to wakefulness.47 Hypnagogic hallucination and sleep paralysis can occur, together with vivid visual hallucinations.
Parasomnias: Somnambulism and night terrors
Most non-REM parasomnias occur in childhood and diminish in adulthood. Two of the most common disorders are sleepwalking (somnambulism) and night terrors. Both are characterized by arousal from slow-wave sleep and are commonly associated with sedating medication, sleep deprivation, or psychopathology.
In somnambulism, patients exhibit complex motor behavior without interaction with their environment. Most have little recollection of the event.48 Sleep terrors produce a more intense reaction. The patient erupts out of sleep with profound terror, confusion, and autonomic changes. Interestingly, the patient can normally fall right back into sleep after the event.49–51
Back to our patient
Excessive daytime sleepiness and generalized fatigue are commonly encountered in outpatients. They can be frustrating because in many cases, no clear etiology can be discovered.52
This patient has several risk factors for parasomnias. His history of anxiety and depression in the setting of recent stressors sets the stage for night terrors. In addition, like many patients with parasomnias, he has a family history of sleep disorders. His vivid dreams make night terrors possible, but without the stark sympathetic activation it is a less likely diagnosis. It also does not account for the other symptoms he describes.
Our patient’s excessive daytime sleepiness interfering with daily activities, cataplexy, and hypnagogic hallucinations support the diagnosis of narcolepsy. This case highlights the variable weakness experienced during a cataplexy attack. It can range from a simple head droop to complete paralysis. Subtle findings require specific probing by the clinician. Patients with narcolepsy typically present in their late teens to early adulthood, but the cataplexy attacks may develop later in the disease course.

Table 3 summarizes the clinical findings associated with night terrors, somnambulism, and narcolepsy.
RARE CAUSES OF TRANSIENT NEUROLOGIC SYMPTOMS
Transient (paroxysmal) neurologic events in multiple sclerosis
A less well-known phenomenon in multiple sclerosis is termed “transient” (paroxysmal) neurologic events. These are typically stereotyped episodes lasting seconds, occurring sometimes hundreds of times a day. They are thought to arise from spontaneous electrical activity in an area of demyelination (ephaptic transmission), creating a wide range of symptoms. Some common events include positive sensory symptoms, alteration of the motor system such as spasms, or brainstem symptoms.53
Channelopathy
Two prototypical channelopathies are hyperkalemic and hypokalemic periodic paralysis. They are rare conditions, usually inherited in an autosomal dominant pattern.54 Both produce episodic, flaccid weakness in the setting of activity or other stressors (fasting, pregnancy, an emotionally charged episode). The attacks last a few minutes to hours and affect proximal skeletal muscles, with very little respiratory or bulbar involvement.
Hyperkalemic periodic paralysis is also associated with myotonia, which is the inability to voluntarily relax after stimulation. This can be evident after shaking a patient’s hand, as he or she would be unable to release because of the sustained activation. The myotonia is evident between attacks and may help cue a physician to the diagnosis even if the weakness has abated.55
As the name implies, potassium levels can vary during the attack, though hyperkalemic periodic paralysis can be seen with normal levels of serum potassium. The underlying pathology is tied to a voltage-gated sodium channel or calcium channel necessary for action potential generation.56
Paroxysmal dyskinesias
Paroxysmal dyskinesias encompass a rare group of movement disorders characterized by attacks without alterations in consciousness. Patients have reported dystonic, choreoathetotic, or ballistic movements. The attacks can be triggered by stress, eating, or even other types of movements. Most reported cases have a strong family history and are inherited in an autosomal dominant pattern. The exact pathophysiology is unclear. When paroxysmal dyskinesia was initially discovered, many thought it was a form of epilepsy, but the lack of electroencephalographic changes and postictal events argues against this etiology.
Transient focal neurologic episodes in cerebral amyloid angiopathy
Cerebral amyloid angiopathy is a degenerative condition in which amyloid is deposited in cerebral vessels, making them friable and at risk of bleeding. Most patients have no symptoms whatsoever, and the diagnosis is made by magnetic resonance imaging. Small microbleeds are common, but lobar intraparenchymal hemorrhage is the most feared complication.
Transient focal neurologic episodes, sometimes termed “amyloid spells,” are recurrent, stereotyped neurologic events that are spurred by cortical superficial siderosis (deposition of iron). Unfortunately, these events are difficult to characterize by their clinical morphology. The events can involve the visual, motor, and sensory pathways with both positive and negative symptoms, making the diagnosis difficult without imaging. These events may precede a symptomatic intraparenchymal hemorrhage, offering a unique window to reconsider the decision to continue an antiplatelet or anticoagulant drug.57,58
- Vuadens P, Regli F. Drug-induced neurological complications in a hospital cohort. Schweiz Med Wochenschr 1995; 125:1625–1633. French.
- Hanley K, O’Dowd T, Considine N. A systematic review of vertigo in primary care. Br J Gen Pract 2001; 51:666–671.
- Brignole M. Diagnosis and treatment of syncope. Heart 2007; 93:130–136.
- Giles MF, Rothwell PM. Risk of stroke early after transient ischaemic attack: a systematic review and meta-analysis. Lancet Neurol 2007; 6:1063–1072.
- Johnston SC, Rothwell PM, Nguyen-Huynh MN, et al. Validation and refinement of scores to predict very early stroke risk after transient ischaemic attack. Lancet 2007; 369:283–292.
- Wardlaw JM, Brazzelli M, Chappell FM, et al. ABCD2 score and secondary stroke prevention: meta-analysis and effect per 1,000 patients triaged. Neurology 2015; 85:373–380.
- Nadarajan V, Perry RJ, Johnson J, Werring DJ. Transient ischaemic attacks: mimics and chameleons. Pract Neurol 2014; 14:23–31.
- Prabhakaran S, Silver AJ, Warrior L, McClenathan B, Lee VH. Misdiagnosis of transient ischemic attacks in the emergency room. Cerebrovasc Dis 2008; 26:630–635.
- Sorensen AG, Ay H. Transient ischemic attack: definition, diagnosis, and risk stratification. Neuroimaging Clin N Am 2011; 21:303–313.
- Kimura K, Minematsu K, Yasaka M, Wada K, Yamaguchi T. The duration of symptoms in transient ischemic attack. Neurology 1999; 52:976–980.
- Lewandowski CA, Rao CP, Silver B. Transient ischemic attack: definitions and clinical presentations. Ann Emerg Med 2008; 52:S7–S16.
- Easton JD, Saver JL, Albers GW, et al; American Heart Association; American Stroke Association Stroke Council; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Interdisciplinary Council on Peripheral Vascular Disease. Definition and evaluation of transient ischemic attack: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association Stroke Council; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; and the Interdisciplinary Council on Peripheral Vascular Disease. The American Academy of Neurology affirms the value of this statement as an educational tool for neurologists. Stroke 2009; 40:2276–2293.
- Bos MJ, van Rijn MJ, Witteman JC, Hofman A, Koudstaal PJ, Breteler MM. Incidence and prognosis of transient neurological attacks. JAMA 2007; 298:2877–2885.
- van Rooij FG, Vermeer SE, Goraj BM, et al. Diffusion-weighted imaging in transient neurological attacks. Ann Neurol 2015; 78:1005–1010.
- Silberstein SD, Young WB. Migraine aura and prodrome. Semin Neurol 1995; 15:175–182.
- Viana M, Sprenger T, Andelova M, Goadsby PJ. The typical duration of migraine aura: a systematic review. Cephalalgia 2013; 33:483–490.
- Young WB, Gangal KS, Aponte RJ, Kaiser RS. Migraine with unilateral motor symptoms: a case-control study. J Neurol Neurosurg Psychiatry 2007; 78:600–604.
- Thomsen LL, Eriksen MK, Roemer SF, Andersen I, Olesen J, Russell MB. A population-based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain 2002; 125:1379–1391.
- Buzzi MG, Cologno D, Formisano R, Rossi P. Prodromes and the early phase of the migraine attack: therapeutic relevance. Funct Neurol 2005; 20:179–183.
- Kelman L. The premonitory symptoms (prodrome): a tertiary care study of 893 migraineurs. Headache 2004; 44:865–872.
- Giffin NJ, Ruggiero L, Lipton RB, et al. Premonitory symptoms in migraine: an electronic diary study. Neurology 2003; 60:935–940.
- Martin VT, Behbehani MM. Toward a rational understanding of migraine trigger factors. Med Clin North Am 2001; 85:911–941.
- Naeije G, Gaspard N, Legros B, Mavroudakis N, Pandolfo M. Transient CNS deficits and migrainous auras in individuals without a history of headache. Headache 2014; 54:493–499.
- Tuna MA, Mehta Z, Rothwell PM; Stroke Prevention Research Unit, Neuroscience Department, John Radcliffe Hospital, Oxford University. Stroke risk after a first late–onset migraine–like transient neurological attack (TNA): Oxford vascular study TNA cohort. J Neurol Neurosurg Psychiatry 2013; 84:e2.
- Fisher CM. Late-life migraine accompaniments—further experience. Stroke 1986; 17:1033–1042.
- Walker HK, Hall WD, Hurst JW. Clinical methods: the history, physical, and laboratory examinations. 3rd ed. Boston, MA: Butterworths; 1990.
- Wyllie E, Rothner AD, Luders H. Partial seizures in children: clinical features, medical treatment, and surgical considerations. Pediatr Clin North Am 1989; 36:343–364.
- Panayiotopoulos CP. Visual phenomena and headache in occipital epilepsy: a review, a systematic study and differentiation from migraine. Epileptic Disord 1999; 1:205–216.
- Gallmetzer P, Leutmezer F, Serles W, Assem-Hilger E, Spatt J, Baumgartner C. Postictal paresis in focal epilepsies—incidence, duration, and causes: a video-EEG monitoring study. Neurology 2004; 62:2160–2164.
- Rolak LA, Rutecki P, Ashizawa T, Harati Y. Clinical features of Todd’s post-epileptic paralysis. J Neurol Neurosurg Psychiatry 1992; 55:63–64.
- Reuber M, Elger CE. Psychogenic nonepileptic seizures: review and update. Epilepsy Behav 2003; 4:205–216.
- LaFrance WC Jr, Baird GL, Barry JJ, et al; NES Treatment Trial (NEST-T) Consortium. Multicenter pilot treatment trial for psychogenic nonepileptic seizures: a randomized clinical trial. JAMA Psychiatry 2014; 71:997–1005.
- UK Hypoglycaemia Study Group. Risk of hypoglycaemia in types 1 and 2 diabetes: effects of treatment modalities and their duration. Diabetologia 2007; 50:1140–1147.
- Cryer PE, Axelrod L, Grossman AB, et al; Endocrine Society. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2009; 94:709–728.
- Yoshino T, Meguro S, Soeda Y, Itoh A, Kawai T, Itoh H. A case of hypoglycemic hemiparesis and literature review. Ups J Med Sci 2012; 117:347–351.
- Lee SH, Kang CD, Kim SS, et al. Lateralization of hypoglycemic encephalopathy: evidence of a mechanism of selective vulnerability. J Clin Neurol 2010; 6:104–108.
- Siegel GJ, Agranoff BW. Basic neurochemistry: molecular, cellular, and medical aspects. 6th ed. Philadelphia, PA: Lippincott-Williams & Wilkins; 1999.
- Holstein A, Plaschke A, Egberts EH. Clinical characterisation of severe hypoglycaemia—a prospective population-based study. Exp Clin Endocrinol Diabetes 2003; 111:364–369.
- Raichle ME, Plum F. Hyperventilation and cerebral blood flow. Stroke 1972; 3:566–575.
- Kerr WJ, Gliebe PA, Dalton JW. Physical phenomena associated with anxiety states: the hyperventilation syndrome. Cal West Med 1938; 48:12–16.
- Lin KH, Chen YT, Fuh JL, et al. Migraine is associated with a higher risk of transient global amnesia: a nationwide cohort study. Eur J Neurol 2014; 21:718–724.
- Arena JE, Brown RD, Mandrekar J, Rabinstein AA. Long-term outcome in patients with transient global amnesia: a population-based study. Mayo Clin Proc 2017; 92:399–405.
- Arena JE, Rabinstein AA. Transient global amnesia. Mayo Clin Proc 2015; 90:264–272.
- Bartsch T, Butler C. Transient amnesic syndromes. Nat Rev Neurol 2013; 9:86–97.
- Scammell TE. Narcolepsy. N Engl J Med 2015; 373:2654–2662.
- Ahmed I, Thorpy M. Clinical features, diagnosis and treatment of narcolepsy. Clin Chest Med 2010; 31:371–381.
- Leschziner G. Narcolepsy: a clinical review. Pract Neurol 2014; 14:323–331.
- Hughes JR. A review of sleepwalking (somnambulism): the enigma of neurophysiology and polysomnography with differential diagnosis of complex partial seizures. Epilepsy Behav 2007; 11:483–491.
- Gremmo M, Blanchi I, Costa B, et al. An abilitative approach to the premature infant in neonatal intensive care unit (NICU). J Perinat Med 1994; 22(suppl 1):102–105.
- Howell MJ. Parasomnias: an updated review. Neurotherapeutics 2012; 9:753–775.
- Giglio P, Undevia N, Spire JP. The primary parasomnias. A review for neurologists. Neurologist 2005; 11:90–97.
- Viner R, Christie D. Fatigue and somatic symptoms. BMJ 2005; 330:1012–1015.
- Rae-Grant AD. Unusual symptoms and syndromes in multiple sclerosis. Continuum (Minneap Minn) 2013; 19:992–1006.
- Fontaine B. Periodic paralysis. Adv Genet 2008; 63:3–23.
- Jurkat-Rott K, Lehmann-Horn F. Paroxysmal muscle weakness: the familial periodic paralyses. J Neurol 2006; 253:1391–1398.
- Lehmann-Horn F, Jurkat-Rott K, Rudel R. Periodic paralysis: understanding channelopathies. Curr Neurol Neurosci Rep 2002; 2:61–69.
- Katoh M, Yoshino M, Asaoka K, et al. A restricted subarachnoid hemorrhage in the cortical sulcus in cerebral amyloid angiopathy: could it be a warning sign? Surg Neurol 2007; 68:457–460.
- Charidimou A, Peeters A, Fox Z, et al. Spectrum of transient focal neurological episodes in cerebral amyloid angiopathy: multicentre magnetic resonance imaging cohort study and meta-analysis. Stroke 2012; 43:2324–2330.
Many patients present to their primary care physicians, urgent care centers, and emergency rooms because of neurologic symptoms lasting seconds to hours. Their problems can be a cause for concern and a challenge to diagnose, as in many cases their symptoms have returned to baseline by the time of evaluation. Referral to a neurologist may not be practical for all of them, particularly given that a consultation may take a long time to obtain.
Understanding the causes of transient neurologic syndromes and their phenomenology may help the clinician diagnose, triage, and treat such conditions effectively.
Here, we outline several transient neurologic syndromes—transient ischemic attack (TIA), migraine with aura, partial seizures, hypoglycemic encephalopathy, hyperventilation syndrome, transient global amnesia, narcolepsy, parasomnias, and some rarer conditions— focusing on their diagnostic elements. Others, such as drug-induced transient neurologic syndromes, vertigo, and dizziness, have been well discussed elsewhere.1–3
THE BIG 3: TIA, MIGRAINE, SEIZURES
A 45-year-old woman with a history of tobacco use and headaches presents to the emergency department with a 4-month history of episodic numbness and tingling of her right arm and face. She reports a prodromal state of anxiety and irritability 24 to 48 hours before symptom onset.
The sensory symptoms begin on her face and gradually progress down the arm and eventually to her fingers. They fully resolve within 2 hours without sequelae. Family members have noted some “slurred speech” during the episodes, and the episodes are occasionally preceded by a unilateral, throbbing headache that improves with rest.
What are the possible causes of her symptoms?
Transient ischemic attack
If a patient reports transient neurologic symptoms and has vascular risk factors, TIA is often the default diagnostic consideration. The risk of stroke is 9.9% in the 2 days after a TIA, 13.4% at 30 days, and 17.3% at 90 days.4 Rapid recognition offers a crucial period to minimize the possibility of permanent impairment. Interventions include modifying risk factors (hypertension, diabetes, and smoking) and starting an antiplatelet drug, an anticoagulant drug, or both, and possibly a statin.
It can be difficult to determine if this workup needs to be completed in the inpatient or outpatient setting. There is no clear consensus, but the ultimate goal is timely evaluation (within 24 to 48 hours). The ABCD2 (Age, Blood pressure, Clinical features, Duration of symptoms, and Diabetes) risk factor calculator was developed to help triage patients, though it has limitations.5,6
One should assess a patient’s history of a possible TIA in a stepwise fashion. First, analyze the patient’s age and demographics for known vascular risk factors or central embolic sources (eg, atrial fibrillation). Then consider the symptoms. TIA symptoms have rapid onset, usually within seconds7; symptoms with a more gradual crescendo suggest a nonvascular cause.8 TIA manifestations should resolve within 1 hour, and most studies suggest symptom resolution within 10 minutes is specific for a TIA.9–11 TIA symptoms are negative neurologic phenomena that denote a loss of function, such as loss of vision, motor weakness, or sensory numbness.
Symptoms should also correlate with a defined vascular territory:
- The middle cerebral artery is commonly involved; its blockage is associated with aphasia, weakness of the face and arm, and homonymous visual field impairment (loss of one-half of the visual fields in both eyes)
- Blockage in the posterior circulation generally causes symptoms localized to the brainstem, cerebellum, and occipital cortex. The symptoms are usually grouped together as the “5Ds”: dizziness, diplopia, dysarthria, dysphagia, and dystaxia/ataxia. Brainstem involvement classically produces “crossed” findings, with ipsilateral cranial findings and contralateral motor or sensory findings.
- Lacunar strokes involve the subcortical white matter and produce typical patterns including pure motor or sensory syndromes.
Loss of consciousness is rarely a symptom of TIA and should suggest another etiology.
The definition of TIA has evolved from an operational one, ie, symptoms lasting less than 24 hours, to a tissue-based one, ie, focal cerebral ischemia not associated with permanent cerebral infarction.12 Though imperfect, this pathophysiology should help reinforce the most common features of TIA, including a sudden onset of negative symptoms that are localized to a defined vascular territory.13,14
Migraine with aura
Migraine with aura is common in patients ages 25 to 55 who have a long-standing history of headache. The pathophysiologic mechanism of an aura is believed to be a disseminating wave of cortical depression, which is a self-propagating wave of neural depression and then activation. Ultimately, this leads to a cascade of inflammatory and pain signals, resulting in a headache.
This background helps explain the positive (superimposed) symptoms associated with the aura. Positive symptoms are produced by excessive neuronal discharges stimulating the visual (flashing lights, zigzag lines), sensory (paresthesias), or motor (limb movements) pathways.
Common symptoms associated with aura include visual disturbances such as scintillating scotoma (a blind spot), sensory changes such as tingling, or auditory disruption with tinnitus. Symptoms may evolve over the course of 5 to 20 minutes, first affecting vision and then other senses. In contrast, in a TIA, symptoms usually begin simultaneously and are confined to a vascular territory.7,15 Symptoms of an aura usually resolve within an hour, but there is evidence showing a substantial number of patients have an aura lasting much longer.16 Focal weakness is uncommon during an aura but is reported in specific migraine conditions such as hemiplegic migraine and migraine with unilateral motor symptoms. The vast majority of patients experience other neurologic symptoms during this prodrome.17,18
The prodromal period (2 to 48 hours leading up to the onset of migraine) is a commonly overlooked feature of migraine.19 Common symptoms during this time include fatigue, mood change, and gastrointestinal symptoms.20 One study demonstrated that patients generally had good intuition concerning these nonspecific prodromal symptoms and could predict the onset of migraine 72% of the time.21
In addition, a myriad of possible triggers and exacerbating factors can be identified (and sometimes avoided) such as visual stimuli, weather changes, nitrates, sleep disturbances, menstruation, foods, and stressors.22
Although headache is often the cardinal manifestation of migraine, some patients experience aura without headache—acephalgic migraine.23 This can be a diagnostic challenge, especially in an older population with multiple vascular risk factors. New-onset acephalgic migraine may be a cause for concern but is not uncommon and is not associated with a significantly increased risk of stroke.24 Focusing on the character of the neurologic symptoms in regard to timing, progression, and resolution will help differentiate this disease from other transient neurologic syndromes.25
Partial seizure
Partial seizure produces a diverse range of stereotypical symptoms due to focal abnormal neuronal activation. The aberrant electrical firing generates positive symptoms involving the motor, sensory, or visual pathway. A history of trauma, neurosurgical intervention, central nervous system infection, stroke, or other seizure foci can suggest this diagnosis. Other prodromal clues include abdominal discomfort, sense of detachment, déjà vu, or jamais vu.26
During a seizure, there may be a progression of positive symptoms similar to what happens in migraine aura, because both represent cortical spread and depression.
Involvement of the motor pathway may produce tonic (stiffening) or clonic (twitching) movement. Other common motor abnormalities include automatisms such as lip smacking, chewing, and hand gestures (picking, fidgeting, fumbling).27
Epileptic discharges in the sensory cortex commonly cause paresthesias or distortion of a sensory input. Visual symptoms may be more complex. In occipital epilepsy, circular phenomena with a colored pattern are common, which contrasts with the photopsia (flashes of light) or fortification (a bright zigzag of lines resembling a fort) seen in migraines.28
Autonomic or somatosensory symptoms can also occur.
Todd paralysis, also called transient postictal paralysis, occurs in only 13% of seizures but can linger for 0.5 to 36 hours.29,30 This weakness is most pronounced within the affected region after a partial seizure.
In general, focal seizures are often stereotyped with positive neurologic features, usually last a few minutes, and resolve fully. These episodes may cause an arrest in activity but not usually loss of consciousness unless the epileptic discharge secondarily generalizes into the adjacent hemisphere.
A common differential diagnosis encountered during an epilepsy workup is psychogenic nonepileptic seizures. Nonepileptic seizures consist of transient, abnormal movements, sensation, or cognition but lack ictal electroencephalographic changes. This is a specifically challenging patient population, with high healthcare utilization and high risk for iatrogenic harm. In addition, on average, diagnosis can take years to establish and usually requires referral to a tertiary care facility.31,32
The big 3: Back to our patient
Our patient’s vascular risk factors, transient symptoms, and language involvement support the diagnosis of TIA. A feature that points away from the diagnosis of TIA is the gradual onset of positive neurologic symptoms. This pattern is not consistent with neuronal ischemia.
Also, our patient had a repetitive, stereotypical pattern of symptoms, which supports including partial seizures in the differential diagnosis. On the other hand, her lack of risk factors for seizure (a history of febrile seizures, developmental delay, trauma, or infection) would make this diagnosis less likely. Also pointing away from the diagnosis of seizures are her lack of typical prodromal symptoms, the length of the events, and the postevent headache.
Table 1 summarizes the clinical findings associated with TIA, migraine, and partial seizure.
EPISODES OF CONFUSION
A 35-year-old woman with a history of depression, anxiety, and poorly controlled type 1 diabetes presents to the clinic after several weeks of episodes of confusion, usually accompanied by paresthesias in both hands, dizziness, and palpitations. In each episode, soon after the symptoms began, she had painful cramps in her hand. The symptoms fully resolved within 10 minutes without sequelae.
Questioned further, the patient describes the confusion as a “mental haze” but denies frank disorientation. She has not kept a log of her blood sugar levels but has not noticed a temporal relationship with regard to her meals or insulin injections.
What are the possible causes of these episodes?
Hypoglycemic encephalopathy
Hypoglycemia is common in most people with diabetes, who have been reported to suffer from 62 to 320 severe hypoglycemic episodes in their lifetime.33,34 The neurologic consequences can be devastating in these severe cases.
During mild to moderate drops in the glucose level, generalized symptoms stem from sympathetic activation. These include generalized anxiety, tremor, palpitations, and sweating. Focal symptoms such as unilateral weakness have also been reported.35,36
Unfortunately, people with long-standing diabetes have a blunted response to epinephrine that reduces their sensitivity to hypoglycemia, placing them at high risk of permanent neurologic damage. This can lead to seizures and coma, as the hypoglycemia has a greater effect on cortical and subcortical structures (highly metabolic areas) than on the brainstem. Thus, respiratory and cardiovascular function is maintained but cerebral function is abnormal. If this state is prolonged, brain death can occur.37,38
Hyperventilation syndrome
Hyperventilation syndrome is not well characterized. Most think of it as synonymous with an underlying psychopathology, but there is evidence to suggest it can occur without underlying anxiety.
There is no clear mechanism, but it is hypothesized that diminished carbon dioxide levels lead to cerebral vasoconstriction. This may lead to reduced cerebral blood flow, causing dizziness, lightheadedness, or vertigo.39 Appendicular symptoms including paresthesias, carpopedal spasm, or tetany have been core features since the syndrome was first described in the early 1900s.40
Though the disorder has rather nonspecific features, it can be easily reproduced in the clinical setting by asking the patient to breathe deeply and rapidly. This can help confirm the underlying diagnosis and also reassure the patient that the underlying pathology is not life-threatening and that he or she has some control over the disease.
Transient global amnesia
Transient global amnesia usually strikes older patients (50 to 70 years old) in the setting of an acute physical or emotional stressor. There is also a correlation between transient global amnesia and migraine, with studies showing migraineurs are at higher risk than the general population.41 Despite common clinical concerns, there is no relationship between transient global amnesia and stroke.42
Transient global amnesia is defined by acute transient anterograde amnesia (coding of new memories). To try to reorient themselves, patients will repeatedly ask questions such as “What day is it?” or “Why are we here?” Retrograde memories, especially long-standing ones, are usually well preserved. The patient’s cognition is otherwise intact, and there are no other focal neurologic symptoms. The event usually lasts 2 to 24 hours and resolves without sequelae.43,44 Afterward, patients remember the event only poorly, which supports the notion that they cannot code new memories.
Confusional episodes: Discussion
Evaluating confusional episodes can be time-consuming and vexing. The subjective nature of the symptoms and the vast differential diagnosis can be overwhelming. Subtle clinical details can help formulate an appropriate evaluation.
Hypoglycemia can produce bizarre neurologic symptoms. Most cases of hypoglycemia produce an exaggerated sympathetic response, though this is blunted in people with longstanding diabetes. In addition, there should be a temporal association with meals, insulin doses, or both.
Transient global amnesia usually occurs with acute stressors and produces a confusional state. These episodes rarely recur, and the patient cannot provide much history regarding the episode secondary to the anterograde amnesia.
Table 2 summarizes the clinical findings associated with hypoglycemic encephalopathy, hyperventilation syndrome, and transient global amnesia.
Back to our patient
In our patient, the likely diagnosis is hyperventilation syndrome, even though we don’t know if her respiratory rate is increased during attacks. Some patients lack awareness of their breathing or are too distracted by the vague symptoms to have insight into the true cause. The cramps and contractions in the hands are a specific feature of the disease and can be accompanied by confusion.
SLEEP DISORDERS
A 17-year-old boy with a history of depression and anxiety presents to his pediatrician because he has had difficulty staying awake in school over the past year. His sleepiness has gradually worsened over the last few months and has taken a toll on his grades, leading to discord in his family. Over the past month he has had some difficulty holding his head up during arguments with friends. He does not lose consciousness during these events but is described as “unresponsive.” He describes vivid dreams when going to sleep that have startled him awake at times. His family history is positive for somnambulism on his father’s side.
Does this patient have a sleep disorder, and if so, which one?
Narcolepsy
Narcolepsy is defined by excessive daytime sleepiness, cataplexy, hypnagogic hallucination, and sleep paralysis. It is more common in men but its prevalence varies widely by geographic region, supporting an underlying interplay between genetics and environment.45
Sleep attacks or excessive daytime sleepiness are the cardinal features of narcolepsy. The dissociation between the sleep-wake cycle is evident with rapid transition into rapid-eye-movement (REM) sleep during these sleep attacks. This results in a “refreshing nap” that commonly involves vivid dreams. These episodes occur about 3 to 5 times per day, varying in duration from a few minutes to hours.46
Cataplexy is very specific feature of narcolepsy. Triggered by strong emotion, the body loses skeletal muscle tone except for the diaphragm and ocular muscles. The patient does not lose consciousness and remains aware of his or her environment. Of note, the loss of tone does not need to be dramatic. The hypotonia can manifest as jaw-dropping or head-nodding. The paralysis is related to prolonged REM atonia and impaired transition from sleep to wakefulness.47 Hypnagogic hallucination and sleep paralysis can occur, together with vivid visual hallucinations.
Parasomnias: Somnambulism and night terrors
Most non-REM parasomnias occur in childhood and diminish in adulthood. Two of the most common disorders are sleepwalking (somnambulism) and night terrors. Both are characterized by arousal from slow-wave sleep and are commonly associated with sedating medication, sleep deprivation, or psychopathology.
In somnambulism, patients exhibit complex motor behavior without interaction with their environment. Most have little recollection of the event.48 Sleep terrors produce a more intense reaction. The patient erupts out of sleep with profound terror, confusion, and autonomic changes. Interestingly, the patient can normally fall right back into sleep after the event.49–51
Back to our patient
Excessive daytime sleepiness and generalized fatigue are commonly encountered in outpatients. They can be frustrating because in many cases, no clear etiology can be discovered.52
This patient has several risk factors for parasomnias. His history of anxiety and depression in the setting of recent stressors sets the stage for night terrors. In addition, like many patients with parasomnias, he has a family history of sleep disorders. His vivid dreams make night terrors possible, but without the stark sympathetic activation it is a less likely diagnosis. It also does not account for the other symptoms he describes.
Our patient’s excessive daytime sleepiness interfering with daily activities, cataplexy, and hypnagogic hallucinations support the diagnosis of narcolepsy. This case highlights the variable weakness experienced during a cataplexy attack. It can range from a simple head droop to complete paralysis. Subtle findings require specific probing by the clinician. Patients with narcolepsy typically present in their late teens to early adulthood, but the cataplexy attacks may develop later in the disease course.
Table 3 summarizes the clinical findings associated with night terrors, somnambulism, and narcolepsy.
RARE CAUSES OF TRANSIENT NEUROLOGIC SYMPTOMS
Transient (paroxysmal) neurologic events in multiple sclerosis
A less well-known phenomenon in multiple sclerosis is termed “transient” (paroxysmal) neurologic events. These are typically stereotyped episodes lasting seconds, occurring sometimes hundreds of times a day. They are thought to arise from spontaneous electrical activity in an area of demyelination (ephaptic transmission), creating a wide range of symptoms. Some common events include positive sensory symptoms, alteration of the motor system such as spasms, or brainstem symptoms.53
Channelopathy
Two prototypical channelopathies are hyperkalemic and hypokalemic periodic paralysis. They are rare conditions, usually inherited in an autosomal dominant pattern.54 Both produce episodic, flaccid weakness in the setting of activity or other stressors (fasting, pregnancy, an emotionally charged episode). The attacks last a few minutes to hours and affect proximal skeletal muscles, with very little respiratory or bulbar involvement.
Hyperkalemic periodic paralysis is also associated with myotonia, which is the inability to voluntarily relax after stimulation. This can be evident after shaking a patient’s hand, as he or she would be unable to release because of the sustained activation. The myotonia is evident between attacks and may help cue a physician to the diagnosis even if the weakness has abated.55
As the name implies, potassium levels can vary during the attack, though hyperkalemic periodic paralysis can be seen with normal levels of serum potassium. The underlying pathology is tied to a voltage-gated sodium channel or calcium channel necessary for action potential generation.56
Paroxysmal dyskinesias
Paroxysmal dyskinesias encompass a rare group of movement disorders characterized by attacks without alterations in consciousness. Patients have reported dystonic, choreoathetotic, or ballistic movements. The attacks can be triggered by stress, eating, or even other types of movements. Most reported cases have a strong family history and are inherited in an autosomal dominant pattern. The exact pathophysiology is unclear. When paroxysmal dyskinesia was initially discovered, many thought it was a form of epilepsy, but the lack of electroencephalographic changes and postictal events argues against this etiology.
Transient focal neurologic episodes in cerebral amyloid angiopathy
Cerebral amyloid angiopathy is a degenerative condition in which amyloid is deposited in cerebral vessels, making them friable and at risk of bleeding. Most patients have no symptoms whatsoever, and the diagnosis is made by magnetic resonance imaging. Small microbleeds are common, but lobar intraparenchymal hemorrhage is the most feared complication.
Transient focal neurologic episodes, sometimes termed “amyloid spells,” are recurrent, stereotyped neurologic events that are spurred by cortical superficial siderosis (deposition of iron). Unfortunately, these events are difficult to characterize by their clinical morphology. The events can involve the visual, motor, and sensory pathways with both positive and negative symptoms, making the diagnosis difficult without imaging. These events may precede a symptomatic intraparenchymal hemorrhage, offering a unique window to reconsider the decision to continue an antiplatelet or anticoagulant drug.57,58
Many patients present to their primary care physicians, urgent care centers, and emergency rooms because of neurologic symptoms lasting seconds to hours. Their problems can be a cause for concern and a challenge to diagnose, as in many cases their symptoms have returned to baseline by the time of evaluation. Referral to a neurologist may not be practical for all of them, particularly given that a consultation may take a long time to obtain.
Understanding the causes of transient neurologic syndromes and their phenomenology may help the clinician diagnose, triage, and treat such conditions effectively.
Here, we outline several transient neurologic syndromes—transient ischemic attack (TIA), migraine with aura, partial seizures, hypoglycemic encephalopathy, hyperventilation syndrome, transient global amnesia, narcolepsy, parasomnias, and some rarer conditions— focusing on their diagnostic elements. Others, such as drug-induced transient neurologic syndromes, vertigo, and dizziness, have been well discussed elsewhere.1–3
THE BIG 3: TIA, MIGRAINE, SEIZURES
A 45-year-old woman with a history of tobacco use and headaches presents to the emergency department with a 4-month history of episodic numbness and tingling of her right arm and face. She reports a prodromal state of anxiety and irritability 24 to 48 hours before symptom onset.
The sensory symptoms begin on her face and gradually progress down the arm and eventually to her fingers. They fully resolve within 2 hours without sequelae. Family members have noted some “slurred speech” during the episodes, and the episodes are occasionally preceded by a unilateral, throbbing headache that improves with rest.
What are the possible causes of her symptoms?
Transient ischemic attack
If a patient reports transient neurologic symptoms and has vascular risk factors, TIA is often the default diagnostic consideration. The risk of stroke is 9.9% in the 2 days after a TIA, 13.4% at 30 days, and 17.3% at 90 days.4 Rapid recognition offers a crucial period to minimize the possibility of permanent impairment. Interventions include modifying risk factors (hypertension, diabetes, and smoking) and starting an antiplatelet drug, an anticoagulant drug, or both, and possibly a statin.
It can be difficult to determine if this workup needs to be completed in the inpatient or outpatient setting. There is no clear consensus, but the ultimate goal is timely evaluation (within 24 to 48 hours). The ABCD2 (Age, Blood pressure, Clinical features, Duration of symptoms, and Diabetes) risk factor calculator was developed to help triage patients, though it has limitations.5,6
One should assess a patient’s history of a possible TIA in a stepwise fashion. First, analyze the patient’s age and demographics for known vascular risk factors or central embolic sources (eg, atrial fibrillation). Then consider the symptoms. TIA symptoms have rapid onset, usually within seconds7; symptoms with a more gradual crescendo suggest a nonvascular cause.8 TIA manifestations should resolve within 1 hour, and most studies suggest symptom resolution within 10 minutes is specific for a TIA.9–11 TIA symptoms are negative neurologic phenomena that denote a loss of function, such as loss of vision, motor weakness, or sensory numbness.
Symptoms should also correlate with a defined vascular territory:
- The middle cerebral artery is commonly involved; its blockage is associated with aphasia, weakness of the face and arm, and homonymous visual field impairment (loss of one-half of the visual fields in both eyes)
- Blockage in the posterior circulation generally causes symptoms localized to the brainstem, cerebellum, and occipital cortex. The symptoms are usually grouped together as the “5Ds”: dizziness, diplopia, dysarthria, dysphagia, and dystaxia/ataxia. Brainstem involvement classically produces “crossed” findings, with ipsilateral cranial findings and contralateral motor or sensory findings.
- Lacunar strokes involve the subcortical white matter and produce typical patterns including pure motor or sensory syndromes.
Loss of consciousness is rarely a symptom of TIA and should suggest another etiology.
The definition of TIA has evolved from an operational one, ie, symptoms lasting less than 24 hours, to a tissue-based one, ie, focal cerebral ischemia not associated with permanent cerebral infarction.12 Though imperfect, this pathophysiology should help reinforce the most common features of TIA, including a sudden onset of negative symptoms that are localized to a defined vascular territory.13,14
Migraine with aura
Migraine with aura is common in patients ages 25 to 55 who have a long-standing history of headache. The pathophysiologic mechanism of an aura is believed to be a disseminating wave of cortical depression, which is a self-propagating wave of neural depression and then activation. Ultimately, this leads to a cascade of inflammatory and pain signals, resulting in a headache.
This background helps explain the positive (superimposed) symptoms associated with the aura. Positive symptoms are produced by excessive neuronal discharges stimulating the visual (flashing lights, zigzag lines), sensory (paresthesias), or motor (limb movements) pathways.
Common symptoms associated with aura include visual disturbances such as scintillating scotoma (a blind spot), sensory changes such as tingling, or auditory disruption with tinnitus. Symptoms may evolve over the course of 5 to 20 minutes, first affecting vision and then other senses. In contrast, in a TIA, symptoms usually begin simultaneously and are confined to a vascular territory.7,15 Symptoms of an aura usually resolve within an hour, but there is evidence showing a substantial number of patients have an aura lasting much longer.16 Focal weakness is uncommon during an aura but is reported in specific migraine conditions such as hemiplegic migraine and migraine with unilateral motor symptoms. The vast majority of patients experience other neurologic symptoms during this prodrome.17,18
The prodromal period (2 to 48 hours leading up to the onset of migraine) is a commonly overlooked feature of migraine.19 Common symptoms during this time include fatigue, mood change, and gastrointestinal symptoms.20 One study demonstrated that patients generally had good intuition concerning these nonspecific prodromal symptoms and could predict the onset of migraine 72% of the time.21
In addition, a myriad of possible triggers and exacerbating factors can be identified (and sometimes avoided) such as visual stimuli, weather changes, nitrates, sleep disturbances, menstruation, foods, and stressors.22
Although headache is often the cardinal manifestation of migraine, some patients experience aura without headache—acephalgic migraine.23 This can be a diagnostic challenge, especially in an older population with multiple vascular risk factors. New-onset acephalgic migraine may be a cause for concern but is not uncommon and is not associated with a significantly increased risk of stroke.24 Focusing on the character of the neurologic symptoms in regard to timing, progression, and resolution will help differentiate this disease from other transient neurologic syndromes.25
Partial seizure
Partial seizure produces a diverse range of stereotypical symptoms due to focal abnormal neuronal activation. The aberrant electrical firing generates positive symptoms involving the motor, sensory, or visual pathway. A history of trauma, neurosurgical intervention, central nervous system infection, stroke, or other seizure foci can suggest this diagnosis. Other prodromal clues include abdominal discomfort, sense of detachment, déjà vu, or jamais vu.26
During a seizure, there may be a progression of positive symptoms similar to what happens in migraine aura, because both represent cortical spread and depression.
Involvement of the motor pathway may produce tonic (stiffening) or clonic (twitching) movement. Other common motor abnormalities include automatisms such as lip smacking, chewing, and hand gestures (picking, fidgeting, fumbling).27
Epileptic discharges in the sensory cortex commonly cause paresthesias or distortion of a sensory input. Visual symptoms may be more complex. In occipital epilepsy, circular phenomena with a colored pattern are common, which contrasts with the photopsia (flashes of light) or fortification (a bright zigzag of lines resembling a fort) seen in migraines.28
Autonomic or somatosensory symptoms can also occur.
Todd paralysis, also called transient postictal paralysis, occurs in only 13% of seizures but can linger for 0.5 to 36 hours.29,30 This weakness is most pronounced within the affected region after a partial seizure.
In general, focal seizures are often stereotyped with positive neurologic features, usually last a few minutes, and resolve fully. These episodes may cause an arrest in activity but not usually loss of consciousness unless the epileptic discharge secondarily generalizes into the adjacent hemisphere.
A common differential diagnosis encountered during an epilepsy workup is psychogenic nonepileptic seizures. Nonepileptic seizures consist of transient, abnormal movements, sensation, or cognition but lack ictal electroencephalographic changes. This is a specifically challenging patient population, with high healthcare utilization and high risk for iatrogenic harm. In addition, on average, diagnosis can take years to establish and usually requires referral to a tertiary care facility.31,32
The big 3: Back to our patient
Our patient’s vascular risk factors, transient symptoms, and language involvement support the diagnosis of TIA. A feature that points away from the diagnosis of TIA is the gradual onset of positive neurologic symptoms. This pattern is not consistent with neuronal ischemia.
Also, our patient had a repetitive, stereotypical pattern of symptoms, which supports including partial seizures in the differential diagnosis. On the other hand, her lack of risk factors for seizure (a history of febrile seizures, developmental delay, trauma, or infection) would make this diagnosis less likely. Also pointing away from the diagnosis of seizures are her lack of typical prodromal symptoms, the length of the events, and the postevent headache.
Table 1 summarizes the clinical findings associated with TIA, migraine, and partial seizure.
EPISODES OF CONFUSION
A 35-year-old woman with a history of depression, anxiety, and poorly controlled type 1 diabetes presents to the clinic after several weeks of episodes of confusion, usually accompanied by paresthesias in both hands, dizziness, and palpitations. In each episode, soon after the symptoms began, she had painful cramps in her hand. The symptoms fully resolved within 10 minutes without sequelae.
Questioned further, the patient describes the confusion as a “mental haze” but denies frank disorientation. She has not kept a log of her blood sugar levels but has not noticed a temporal relationship with regard to her meals or insulin injections.
What are the possible causes of these episodes?
Hypoglycemic encephalopathy
Hypoglycemia is common in most people with diabetes, who have been reported to suffer from 62 to 320 severe hypoglycemic episodes in their lifetime.33,34 The neurologic consequences can be devastating in these severe cases.
During mild to moderate drops in the glucose level, generalized symptoms stem from sympathetic activation. These include generalized anxiety, tremor, palpitations, and sweating. Focal symptoms such as unilateral weakness have also been reported.35,36
Unfortunately, people with long-standing diabetes have a blunted response to epinephrine that reduces their sensitivity to hypoglycemia, placing them at high risk of permanent neurologic damage. This can lead to seizures and coma, as the hypoglycemia has a greater effect on cortical and subcortical structures (highly metabolic areas) than on the brainstem. Thus, respiratory and cardiovascular function is maintained but cerebral function is abnormal. If this state is prolonged, brain death can occur.37,38
Hyperventilation syndrome
Hyperventilation syndrome is not well characterized. Most think of it as synonymous with an underlying psychopathology, but there is evidence to suggest it can occur without underlying anxiety.
There is no clear mechanism, but it is hypothesized that diminished carbon dioxide levels lead to cerebral vasoconstriction. This may lead to reduced cerebral blood flow, causing dizziness, lightheadedness, or vertigo.39 Appendicular symptoms including paresthesias, carpopedal spasm, or tetany have been core features since the syndrome was first described in the early 1900s.40
Though the disorder has rather nonspecific features, it can be easily reproduced in the clinical setting by asking the patient to breathe deeply and rapidly. This can help confirm the underlying diagnosis and also reassure the patient that the underlying pathology is not life-threatening and that he or she has some control over the disease.
Transient global amnesia
Transient global amnesia usually strikes older patients (50 to 70 years old) in the setting of an acute physical or emotional stressor. There is also a correlation between transient global amnesia and migraine, with studies showing migraineurs are at higher risk than the general population.41 Despite common clinical concerns, there is no relationship between transient global amnesia and stroke.42
Transient global amnesia is defined by acute transient anterograde amnesia (coding of new memories). To try to reorient themselves, patients will repeatedly ask questions such as “What day is it?” or “Why are we here?” Retrograde memories, especially long-standing ones, are usually well preserved. The patient’s cognition is otherwise intact, and there are no other focal neurologic symptoms. The event usually lasts 2 to 24 hours and resolves without sequelae.43,44 Afterward, patients remember the event only poorly, which supports the notion that they cannot code new memories.
Confusional episodes: Discussion
Evaluating confusional episodes can be time-consuming and vexing. The subjective nature of the symptoms and the vast differential diagnosis can be overwhelming. Subtle clinical details can help formulate an appropriate evaluation.
Hypoglycemia can produce bizarre neurologic symptoms. Most cases of hypoglycemia produce an exaggerated sympathetic response, though this is blunted in people with longstanding diabetes. In addition, there should be a temporal association with meals, insulin doses, or both.
Transient global amnesia usually occurs with acute stressors and produces a confusional state. These episodes rarely recur, and the patient cannot provide much history regarding the episode secondary to the anterograde amnesia.
Table 2 summarizes the clinical findings associated with hypoglycemic encephalopathy, hyperventilation syndrome, and transient global amnesia.
Back to our patient
In our patient, the likely diagnosis is hyperventilation syndrome, even though we don’t know if her respiratory rate is increased during attacks. Some patients lack awareness of their breathing or are too distracted by the vague symptoms to have insight into the true cause. The cramps and contractions in the hands are a specific feature of the disease and can be accompanied by confusion.
SLEEP DISORDERS
A 17-year-old boy with a history of depression and anxiety presents to his pediatrician because he has had difficulty staying awake in school over the past year. His sleepiness has gradually worsened over the last few months and has taken a toll on his grades, leading to discord in his family. Over the past month he has had some difficulty holding his head up during arguments with friends. He does not lose consciousness during these events but is described as “unresponsive.” He describes vivid dreams when going to sleep that have startled him awake at times. His family history is positive for somnambulism on his father’s side.
Does this patient have a sleep disorder, and if so, which one?
Narcolepsy
Narcolepsy is defined by excessive daytime sleepiness, cataplexy, hypnagogic hallucination, and sleep paralysis. It is more common in men but its prevalence varies widely by geographic region, supporting an underlying interplay between genetics and environment.45
Sleep attacks or excessive daytime sleepiness are the cardinal features of narcolepsy. The dissociation between the sleep-wake cycle is evident with rapid transition into rapid-eye-movement (REM) sleep during these sleep attacks. This results in a “refreshing nap” that commonly involves vivid dreams. These episodes occur about 3 to 5 times per day, varying in duration from a few minutes to hours.46
Cataplexy is very specific feature of narcolepsy. Triggered by strong emotion, the body loses skeletal muscle tone except for the diaphragm and ocular muscles. The patient does not lose consciousness and remains aware of his or her environment. Of note, the loss of tone does not need to be dramatic. The hypotonia can manifest as jaw-dropping or head-nodding. The paralysis is related to prolonged REM atonia and impaired transition from sleep to wakefulness.47 Hypnagogic hallucination and sleep paralysis can occur, together with vivid visual hallucinations.
Parasomnias: Somnambulism and night terrors
Most non-REM parasomnias occur in childhood and diminish in adulthood. Two of the most common disorders are sleepwalking (somnambulism) and night terrors. Both are characterized by arousal from slow-wave sleep and are commonly associated with sedating medication, sleep deprivation, or psychopathology.
In somnambulism, patients exhibit complex motor behavior without interaction with their environment. Most have little recollection of the event.48 Sleep terrors produce a more intense reaction. The patient erupts out of sleep with profound terror, confusion, and autonomic changes. Interestingly, the patient can normally fall right back into sleep after the event.49–51
Back to our patient
Excessive daytime sleepiness and generalized fatigue are commonly encountered in outpatients. They can be frustrating because in many cases, no clear etiology can be discovered.52
This patient has several risk factors for parasomnias. His history of anxiety and depression in the setting of recent stressors sets the stage for night terrors. In addition, like many patients with parasomnias, he has a family history of sleep disorders. His vivid dreams make night terrors possible, but without the stark sympathetic activation it is a less likely diagnosis. It also does not account for the other symptoms he describes.
Our patient’s excessive daytime sleepiness interfering with daily activities, cataplexy, and hypnagogic hallucinations support the diagnosis of narcolepsy. This case highlights the variable weakness experienced during a cataplexy attack. It can range from a simple head droop to complete paralysis. Subtle findings require specific probing by the clinician. Patients with narcolepsy typically present in their late teens to early adulthood, but the cataplexy attacks may develop later in the disease course.
Table 3 summarizes the clinical findings associated with night terrors, somnambulism, and narcolepsy.
RARE CAUSES OF TRANSIENT NEUROLOGIC SYMPTOMS
Transient (paroxysmal) neurologic events in multiple sclerosis
A less well-known phenomenon in multiple sclerosis is termed “transient” (paroxysmal) neurologic events. These are typically stereotyped episodes lasting seconds, occurring sometimes hundreds of times a day. They are thought to arise from spontaneous electrical activity in an area of demyelination (ephaptic transmission), creating a wide range of symptoms. Some common events include positive sensory symptoms, alteration of the motor system such as spasms, or brainstem symptoms.53
Channelopathy
Two prototypical channelopathies are hyperkalemic and hypokalemic periodic paralysis. They are rare conditions, usually inherited in an autosomal dominant pattern.54 Both produce episodic, flaccid weakness in the setting of activity or other stressors (fasting, pregnancy, an emotionally charged episode). The attacks last a few minutes to hours and affect proximal skeletal muscles, with very little respiratory or bulbar involvement.
Hyperkalemic periodic paralysis is also associated with myotonia, which is the inability to voluntarily relax after stimulation. This can be evident after shaking a patient’s hand, as he or she would be unable to release because of the sustained activation. The myotonia is evident between attacks and may help cue a physician to the diagnosis even if the weakness has abated.55
As the name implies, potassium levels can vary during the attack, though hyperkalemic periodic paralysis can be seen with normal levels of serum potassium. The underlying pathology is tied to a voltage-gated sodium channel or calcium channel necessary for action potential generation.56
Paroxysmal dyskinesias
Paroxysmal dyskinesias encompass a rare group of movement disorders characterized by attacks without alterations in consciousness. Patients have reported dystonic, choreoathetotic, or ballistic movements. The attacks can be triggered by stress, eating, or even other types of movements. Most reported cases have a strong family history and are inherited in an autosomal dominant pattern. The exact pathophysiology is unclear. When paroxysmal dyskinesia was initially discovered, many thought it was a form of epilepsy, but the lack of electroencephalographic changes and postictal events argues against this etiology.
Transient focal neurologic episodes in cerebral amyloid angiopathy
Cerebral amyloid angiopathy is a degenerative condition in which amyloid is deposited in cerebral vessels, making them friable and at risk of bleeding. Most patients have no symptoms whatsoever, and the diagnosis is made by magnetic resonance imaging. Small microbleeds are common, but lobar intraparenchymal hemorrhage is the most feared complication.
Transient focal neurologic episodes, sometimes termed “amyloid spells,” are recurrent, stereotyped neurologic events that are spurred by cortical superficial siderosis (deposition of iron). Unfortunately, these events are difficult to characterize by their clinical morphology. The events can involve the visual, motor, and sensory pathways with both positive and negative symptoms, making the diagnosis difficult without imaging. These events may precede a symptomatic intraparenchymal hemorrhage, offering a unique window to reconsider the decision to continue an antiplatelet or anticoagulant drug.57,58
- Vuadens P, Regli F. Drug-induced neurological complications in a hospital cohort. Schweiz Med Wochenschr 1995; 125:1625–1633. French.
- Hanley K, O’Dowd T, Considine N. A systematic review of vertigo in primary care. Br J Gen Pract 2001; 51:666–671.
- Brignole M. Diagnosis and treatment of syncope. Heart 2007; 93:130–136.
- Giles MF, Rothwell PM. Risk of stroke early after transient ischaemic attack: a systematic review and meta-analysis. Lancet Neurol 2007; 6:1063–1072.
- Johnston SC, Rothwell PM, Nguyen-Huynh MN, et al. Validation and refinement of scores to predict very early stroke risk after transient ischaemic attack. Lancet 2007; 369:283–292.
- Wardlaw JM, Brazzelli M, Chappell FM, et al. ABCD2 score and secondary stroke prevention: meta-analysis and effect per 1,000 patients triaged. Neurology 2015; 85:373–380.
- Nadarajan V, Perry RJ, Johnson J, Werring DJ. Transient ischaemic attacks: mimics and chameleons. Pract Neurol 2014; 14:23–31.
- Prabhakaran S, Silver AJ, Warrior L, McClenathan B, Lee VH. Misdiagnosis of transient ischemic attacks in the emergency room. Cerebrovasc Dis 2008; 26:630–635.
- Sorensen AG, Ay H. Transient ischemic attack: definition, diagnosis, and risk stratification. Neuroimaging Clin N Am 2011; 21:303–313.
- Kimura K, Minematsu K, Yasaka M, Wada K, Yamaguchi T. The duration of symptoms in transient ischemic attack. Neurology 1999; 52:976–980.
- Lewandowski CA, Rao CP, Silver B. Transient ischemic attack: definitions and clinical presentations. Ann Emerg Med 2008; 52:S7–S16.
- Easton JD, Saver JL, Albers GW, et al; American Heart Association; American Stroke Association Stroke Council; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Interdisciplinary Council on Peripheral Vascular Disease. Definition and evaluation of transient ischemic attack: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association Stroke Council; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; and the Interdisciplinary Council on Peripheral Vascular Disease. The American Academy of Neurology affirms the value of this statement as an educational tool for neurologists. Stroke 2009; 40:2276–2293.
- Bos MJ, van Rijn MJ, Witteman JC, Hofman A, Koudstaal PJ, Breteler MM. Incidence and prognosis of transient neurological attacks. JAMA 2007; 298:2877–2885.
- van Rooij FG, Vermeer SE, Goraj BM, et al. Diffusion-weighted imaging in transient neurological attacks. Ann Neurol 2015; 78:1005–1010.
- Silberstein SD, Young WB. Migraine aura and prodrome. Semin Neurol 1995; 15:175–182.
- Viana M, Sprenger T, Andelova M, Goadsby PJ. The typical duration of migraine aura: a systematic review. Cephalalgia 2013; 33:483–490.
- Young WB, Gangal KS, Aponte RJ, Kaiser RS. Migraine with unilateral motor symptoms: a case-control study. J Neurol Neurosurg Psychiatry 2007; 78:600–604.
- Thomsen LL, Eriksen MK, Roemer SF, Andersen I, Olesen J, Russell MB. A population-based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain 2002; 125:1379–1391.
- Buzzi MG, Cologno D, Formisano R, Rossi P. Prodromes and the early phase of the migraine attack: therapeutic relevance. Funct Neurol 2005; 20:179–183.
- Kelman L. The premonitory symptoms (prodrome): a tertiary care study of 893 migraineurs. Headache 2004; 44:865–872.
- Giffin NJ, Ruggiero L, Lipton RB, et al. Premonitory symptoms in migraine: an electronic diary study. Neurology 2003; 60:935–940.
- Martin VT, Behbehani MM. Toward a rational understanding of migraine trigger factors. Med Clin North Am 2001; 85:911–941.
- Naeije G, Gaspard N, Legros B, Mavroudakis N, Pandolfo M. Transient CNS deficits and migrainous auras in individuals without a history of headache. Headache 2014; 54:493–499.
- Tuna MA, Mehta Z, Rothwell PM; Stroke Prevention Research Unit, Neuroscience Department, John Radcliffe Hospital, Oxford University. Stroke risk after a first late–onset migraine–like transient neurological attack (TNA): Oxford vascular study TNA cohort. J Neurol Neurosurg Psychiatry 2013; 84:e2.
- Fisher CM. Late-life migraine accompaniments—further experience. Stroke 1986; 17:1033–1042.
- Walker HK, Hall WD, Hurst JW. Clinical methods: the history, physical, and laboratory examinations. 3rd ed. Boston, MA: Butterworths; 1990.
- Wyllie E, Rothner AD, Luders H. Partial seizures in children: clinical features, medical treatment, and surgical considerations. Pediatr Clin North Am 1989; 36:343–364.
- Panayiotopoulos CP. Visual phenomena and headache in occipital epilepsy: a review, a systematic study and differentiation from migraine. Epileptic Disord 1999; 1:205–216.
- Gallmetzer P, Leutmezer F, Serles W, Assem-Hilger E, Spatt J, Baumgartner C. Postictal paresis in focal epilepsies—incidence, duration, and causes: a video-EEG monitoring study. Neurology 2004; 62:2160–2164.
- Rolak LA, Rutecki P, Ashizawa T, Harati Y. Clinical features of Todd’s post-epileptic paralysis. J Neurol Neurosurg Psychiatry 1992; 55:63–64.
- Reuber M, Elger CE. Psychogenic nonepileptic seizures: review and update. Epilepsy Behav 2003; 4:205–216.
- LaFrance WC Jr, Baird GL, Barry JJ, et al; NES Treatment Trial (NEST-T) Consortium. Multicenter pilot treatment trial for psychogenic nonepileptic seizures: a randomized clinical trial. JAMA Psychiatry 2014; 71:997–1005.
- UK Hypoglycaemia Study Group. Risk of hypoglycaemia in types 1 and 2 diabetes: effects of treatment modalities and their duration. Diabetologia 2007; 50:1140–1147.
- Cryer PE, Axelrod L, Grossman AB, et al; Endocrine Society. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2009; 94:709–728.
- Yoshino T, Meguro S, Soeda Y, Itoh A, Kawai T, Itoh H. A case of hypoglycemic hemiparesis and literature review. Ups J Med Sci 2012; 117:347–351.
- Lee SH, Kang CD, Kim SS, et al. Lateralization of hypoglycemic encephalopathy: evidence of a mechanism of selective vulnerability. J Clin Neurol 2010; 6:104–108.
- Siegel GJ, Agranoff BW. Basic neurochemistry: molecular, cellular, and medical aspects. 6th ed. Philadelphia, PA: Lippincott-Williams & Wilkins; 1999.
- Holstein A, Plaschke A, Egberts EH. Clinical characterisation of severe hypoglycaemia—a prospective population-based study. Exp Clin Endocrinol Diabetes 2003; 111:364–369.
- Raichle ME, Plum F. Hyperventilation and cerebral blood flow. Stroke 1972; 3:566–575.
- Kerr WJ, Gliebe PA, Dalton JW. Physical phenomena associated with anxiety states: the hyperventilation syndrome. Cal West Med 1938; 48:12–16.
- Lin KH, Chen YT, Fuh JL, et al. Migraine is associated with a higher risk of transient global amnesia: a nationwide cohort study. Eur J Neurol 2014; 21:718–724.
- Arena JE, Brown RD, Mandrekar J, Rabinstein AA. Long-term outcome in patients with transient global amnesia: a population-based study. Mayo Clin Proc 2017; 92:399–405.
- Arena JE, Rabinstein AA. Transient global amnesia. Mayo Clin Proc 2015; 90:264–272.
- Bartsch T, Butler C. Transient amnesic syndromes. Nat Rev Neurol 2013; 9:86–97.
- Scammell TE. Narcolepsy. N Engl J Med 2015; 373:2654–2662.
- Ahmed I, Thorpy M. Clinical features, diagnosis and treatment of narcolepsy. Clin Chest Med 2010; 31:371–381.
- Leschziner G. Narcolepsy: a clinical review. Pract Neurol 2014; 14:323–331.
- Hughes JR. A review of sleepwalking (somnambulism): the enigma of neurophysiology and polysomnography with differential diagnosis of complex partial seizures. Epilepsy Behav 2007; 11:483–491.
- Gremmo M, Blanchi I, Costa B, et al. An abilitative approach to the premature infant in neonatal intensive care unit (NICU). J Perinat Med 1994; 22(suppl 1):102–105.
- Howell MJ. Parasomnias: an updated review. Neurotherapeutics 2012; 9:753–775.
- Giglio P, Undevia N, Spire JP. The primary parasomnias. A review for neurologists. Neurologist 2005; 11:90–97.
- Viner R, Christie D. Fatigue and somatic symptoms. BMJ 2005; 330:1012–1015.
- Rae-Grant AD. Unusual symptoms and syndromes in multiple sclerosis. Continuum (Minneap Minn) 2013; 19:992–1006.
- Fontaine B. Periodic paralysis. Adv Genet 2008; 63:3–23.
- Jurkat-Rott K, Lehmann-Horn F. Paroxysmal muscle weakness: the familial periodic paralyses. J Neurol 2006; 253:1391–1398.
- Lehmann-Horn F, Jurkat-Rott K, Rudel R. Periodic paralysis: understanding channelopathies. Curr Neurol Neurosci Rep 2002; 2:61–69.
- Katoh M, Yoshino M, Asaoka K, et al. A restricted subarachnoid hemorrhage in the cortical sulcus in cerebral amyloid angiopathy: could it be a warning sign? Surg Neurol 2007; 68:457–460.
- Charidimou A, Peeters A, Fox Z, et al. Spectrum of transient focal neurological episodes in cerebral amyloid angiopathy: multicentre magnetic resonance imaging cohort study and meta-analysis. Stroke 2012; 43:2324–2330.
- Vuadens P, Regli F. Drug-induced neurological complications in a hospital cohort. Schweiz Med Wochenschr 1995; 125:1625–1633. French.
- Hanley K, O’Dowd T, Considine N. A systematic review of vertigo in primary care. Br J Gen Pract 2001; 51:666–671.
- Brignole M. Diagnosis and treatment of syncope. Heart 2007; 93:130–136.
- Giles MF, Rothwell PM. Risk of stroke early after transient ischaemic attack: a systematic review and meta-analysis. Lancet Neurol 2007; 6:1063–1072.
- Johnston SC, Rothwell PM, Nguyen-Huynh MN, et al. Validation and refinement of scores to predict very early stroke risk after transient ischaemic attack. Lancet 2007; 369:283–292.
- Wardlaw JM, Brazzelli M, Chappell FM, et al. ABCD2 score and secondary stroke prevention: meta-analysis and effect per 1,000 patients triaged. Neurology 2015; 85:373–380.
- Nadarajan V, Perry RJ, Johnson J, Werring DJ. Transient ischaemic attacks: mimics and chameleons. Pract Neurol 2014; 14:23–31.
- Prabhakaran S, Silver AJ, Warrior L, McClenathan B, Lee VH. Misdiagnosis of transient ischemic attacks in the emergency room. Cerebrovasc Dis 2008; 26:630–635.
- Sorensen AG, Ay H. Transient ischemic attack: definition, diagnosis, and risk stratification. Neuroimaging Clin N Am 2011; 21:303–313.
- Kimura K, Minematsu K, Yasaka M, Wada K, Yamaguchi T. The duration of symptoms in transient ischemic attack. Neurology 1999; 52:976–980.
- Lewandowski CA, Rao CP, Silver B. Transient ischemic attack: definitions and clinical presentations. Ann Emerg Med 2008; 52:S7–S16.
- Easton JD, Saver JL, Albers GW, et al; American Heart Association; American Stroke Association Stroke Council; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Interdisciplinary Council on Peripheral Vascular Disease. Definition and evaluation of transient ischemic attack: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association Stroke Council; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; and the Interdisciplinary Council on Peripheral Vascular Disease. The American Academy of Neurology affirms the value of this statement as an educational tool for neurologists. Stroke 2009; 40:2276–2293.
- Bos MJ, van Rijn MJ, Witteman JC, Hofman A, Koudstaal PJ, Breteler MM. Incidence and prognosis of transient neurological attacks. JAMA 2007; 298:2877–2885.
- van Rooij FG, Vermeer SE, Goraj BM, et al. Diffusion-weighted imaging in transient neurological attacks. Ann Neurol 2015; 78:1005–1010.
- Silberstein SD, Young WB. Migraine aura and prodrome. Semin Neurol 1995; 15:175–182.
- Viana M, Sprenger T, Andelova M, Goadsby PJ. The typical duration of migraine aura: a systematic review. Cephalalgia 2013; 33:483–490.
- Young WB, Gangal KS, Aponte RJ, Kaiser RS. Migraine with unilateral motor symptoms: a case-control study. J Neurol Neurosurg Psychiatry 2007; 78:600–604.
- Thomsen LL, Eriksen MK, Roemer SF, Andersen I, Olesen J, Russell MB. A population-based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain 2002; 125:1379–1391.
- Buzzi MG, Cologno D, Formisano R, Rossi P. Prodromes and the early phase of the migraine attack: therapeutic relevance. Funct Neurol 2005; 20:179–183.
- Kelman L. The premonitory symptoms (prodrome): a tertiary care study of 893 migraineurs. Headache 2004; 44:865–872.
- Giffin NJ, Ruggiero L, Lipton RB, et al. Premonitory symptoms in migraine: an electronic diary study. Neurology 2003; 60:935–940.
- Martin VT, Behbehani MM. Toward a rational understanding of migraine trigger factors. Med Clin North Am 2001; 85:911–941.
- Naeije G, Gaspard N, Legros B, Mavroudakis N, Pandolfo M. Transient CNS deficits and migrainous auras in individuals without a history of headache. Headache 2014; 54:493–499.
- Tuna MA, Mehta Z, Rothwell PM; Stroke Prevention Research Unit, Neuroscience Department, John Radcliffe Hospital, Oxford University. Stroke risk after a first late–onset migraine–like transient neurological attack (TNA): Oxford vascular study TNA cohort. J Neurol Neurosurg Psychiatry 2013; 84:e2.
- Fisher CM. Late-life migraine accompaniments—further experience. Stroke 1986; 17:1033–1042.
- Walker HK, Hall WD, Hurst JW. Clinical methods: the history, physical, and laboratory examinations. 3rd ed. Boston, MA: Butterworths; 1990.
- Wyllie E, Rothner AD, Luders H. Partial seizures in children: clinical features, medical treatment, and surgical considerations. Pediatr Clin North Am 1989; 36:343–364.
- Panayiotopoulos CP. Visual phenomena and headache in occipital epilepsy: a review, a systematic study and differentiation from migraine. Epileptic Disord 1999; 1:205–216.
- Gallmetzer P, Leutmezer F, Serles W, Assem-Hilger E, Spatt J, Baumgartner C. Postictal paresis in focal epilepsies—incidence, duration, and causes: a video-EEG monitoring study. Neurology 2004; 62:2160–2164.
- Rolak LA, Rutecki P, Ashizawa T, Harati Y. Clinical features of Todd’s post-epileptic paralysis. J Neurol Neurosurg Psychiatry 1992; 55:63–64.
- Reuber M, Elger CE. Psychogenic nonepileptic seizures: review and update. Epilepsy Behav 2003; 4:205–216.
- LaFrance WC Jr, Baird GL, Barry JJ, et al; NES Treatment Trial (NEST-T) Consortium. Multicenter pilot treatment trial for psychogenic nonepileptic seizures: a randomized clinical trial. JAMA Psychiatry 2014; 71:997–1005.
- UK Hypoglycaemia Study Group. Risk of hypoglycaemia in types 1 and 2 diabetes: effects of treatment modalities and their duration. Diabetologia 2007; 50:1140–1147.
- Cryer PE, Axelrod L, Grossman AB, et al; Endocrine Society. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2009; 94:709–728.
- Yoshino T, Meguro S, Soeda Y, Itoh A, Kawai T, Itoh H. A case of hypoglycemic hemiparesis and literature review. Ups J Med Sci 2012; 117:347–351.
- Lee SH, Kang CD, Kim SS, et al. Lateralization of hypoglycemic encephalopathy: evidence of a mechanism of selective vulnerability. J Clin Neurol 2010; 6:104–108.
- Siegel GJ, Agranoff BW. Basic neurochemistry: molecular, cellular, and medical aspects. 6th ed. Philadelphia, PA: Lippincott-Williams & Wilkins; 1999.
- Holstein A, Plaschke A, Egberts EH. Clinical characterisation of severe hypoglycaemia—a prospective population-based study. Exp Clin Endocrinol Diabetes 2003; 111:364–369.
- Raichle ME, Plum F. Hyperventilation and cerebral blood flow. Stroke 1972; 3:566–575.
- Kerr WJ, Gliebe PA, Dalton JW. Physical phenomena associated with anxiety states: the hyperventilation syndrome. Cal West Med 1938; 48:12–16.
- Lin KH, Chen YT, Fuh JL, et al. Migraine is associated with a higher risk of transient global amnesia: a nationwide cohort study. Eur J Neurol 2014; 21:718–724.
- Arena JE, Brown RD, Mandrekar J, Rabinstein AA. Long-term outcome in patients with transient global amnesia: a population-based study. Mayo Clin Proc 2017; 92:399–405.
- Arena JE, Rabinstein AA. Transient global amnesia. Mayo Clin Proc 2015; 90:264–272.
- Bartsch T, Butler C. Transient amnesic syndromes. Nat Rev Neurol 2013; 9:86–97.
- Scammell TE. Narcolepsy. N Engl J Med 2015; 373:2654–2662.
- Ahmed I, Thorpy M. Clinical features, diagnosis and treatment of narcolepsy. Clin Chest Med 2010; 31:371–381.
- Leschziner G. Narcolepsy: a clinical review. Pract Neurol 2014; 14:323–331.
- Hughes JR. A review of sleepwalking (somnambulism): the enigma of neurophysiology and polysomnography with differential diagnosis of complex partial seizures. Epilepsy Behav 2007; 11:483–491.
- Gremmo M, Blanchi I, Costa B, et al. An abilitative approach to the premature infant in neonatal intensive care unit (NICU). J Perinat Med 1994; 22(suppl 1):102–105.
- Howell MJ. Parasomnias: an updated review. Neurotherapeutics 2012; 9:753–775.
- Giglio P, Undevia N, Spire JP. The primary parasomnias. A review for neurologists. Neurologist 2005; 11:90–97.
- Viner R, Christie D. Fatigue and somatic symptoms. BMJ 2005; 330:1012–1015.
- Rae-Grant AD. Unusual symptoms and syndromes in multiple sclerosis. Continuum (Minneap Minn) 2013; 19:992–1006.
- Fontaine B. Periodic paralysis. Adv Genet 2008; 63:3–23.
- Jurkat-Rott K, Lehmann-Horn F. Paroxysmal muscle weakness: the familial periodic paralyses. J Neurol 2006; 253:1391–1398.
- Lehmann-Horn F, Jurkat-Rott K, Rudel R. Periodic paralysis: understanding channelopathies. Curr Neurol Neurosci Rep 2002; 2:61–69.
- Katoh M, Yoshino M, Asaoka K, et al. A restricted subarachnoid hemorrhage in the cortical sulcus in cerebral amyloid angiopathy: could it be a warning sign? Surg Neurol 2007; 68:457–460.
- Charidimou A, Peeters A, Fox Z, et al. Spectrum of transient focal neurological episodes in cerebral amyloid angiopathy: multicentre magnetic resonance imaging cohort study and meta-analysis. Stroke 2012; 43:2324–2330.
KEY POINTS
- Transient ischemic attack, migraine aura, and partial seizures are common and often can be differentiated by their distinctive symptoms.
- Episodes of confusion in a patient with diabetes raise the possibility of hypoglycemic encephalopathy; other possibilities include hyperventilation syndrome and transient global amnesia.
- Daytime sleepiness in a young patient may be due to narcolepsy or parasomnias.
Kidney transplant: New opportunities and challenges
Much has improved in renal transplantation over the past 20 years. The focus has shifted to using stronger immunotherapy rather than trying to minimize it. There has been increasing recognition of infection and ways to prevent and treat it. Induction therapy now has greater emphasis so that maintenance therapy can be eased, with the aim of reducing long-term toxicity. Perhaps the biggest change is the practice of screening for donor-specific antibodies at the time of transplant so that predictable problems can be prevented or better handled if they occur. Such advances have helped patients directly and by extending the life of their transplanted organs.
LONGER SURVIVAL
As early as the 1990s, it was recognized that kidney transplant offers a survival advantage for patients with end-stage renal disease over maintenance on dialysis.1 Although the risk of death is higher immediately after transplant, within a few months it becomes much lower than for patients on dialysis. Survival varies according to the health of the patient and the quality of the transplanted organ.
In general, patients who obtain the greatest benefit from transplants in terms of years of life gained are those with diabetes, especially those who are younger. Those ages 20 to 39 live about 8 years on dialysis vs 25 years after transplant.
CONTRAINDICATIONS TO TRANSPLANT

There are multiple contraindications to a solitary kidney transplant (Table 1), including smoking. Most transplant centers require that smokers quit before transplant. Long-standing smokers almost double their risk of a cardiac event after transplant and double their rate of malignancy. Active smoking at the time of transplant is associated with twice the risk of death by 10 years after transplant compared with that of nonsmokers.2 Cotinine testing can detect whether a patient is an active smoker.
WAITING-LIST CONSIDERATIONS
Organs are scarce
The number of patients on the kidney waiting list has increased rapidly in the last few decades, while the number of transplants performed each year has remained about the same. In 2016, about 100,000 patients were on the list, but only about 19,000 transplants were performed.3 Wait times, especially for deceased-donor organs, have increased to about 6 years, varying by blood type and geographic region.
Waiting-list placement
Placement on the waiting list for a deceased-donor kidney transplant occurs when a patient has an estimated glomerular filtration rate (GFR) of 20 mL/min/1.73 m2 or less, although referral to the list can be made earlier. Early listing remains advantageous, as total time on the list will be counted before starting dialysis. “Preemptive transplant” means the patient had no dialysis before transplant; this applies to about 10% of transplant recipients. These patients tend to fare the best and are usually recipients of a living-donor organ.
Most patients do not receive a transplant until the GFR is less than 15 mL/min/1.73 m2.
Since 2014, wait time has been measured from the beginning of dialysis rather than the date of waiting-list placement in patients who are listed after starting dialysis therapy. This approach is more fair but sometimes introduces problems. A patient who did not previously know about the list may suddenly jump to the head of the line after 10 years of dialysis, by which time comorbidities associated with long-term dialysis make the patient less likely to gain as much benefit from a transplant as people lower on the list. Time on dialysis, or “dialysis vintage,” predicts patient and kidney survival after transplant, with reduced survival associated with increasing time on dialysis.4
Shorter wait for a suboptimal kidney
The aging population has increased the number of older patients being listed for transplant, presenting multiple challenges. Patients age 65 or older have a 50% chance of dying before they receive a transplant during a 5-year wait.
A patient may shorten the wait by joining the list for a suboptimal organ. All deceased-donor organs are given a Kidney Donor Profile Index score, which predicts the longevity of an organ after transplant. The score is determined by donor age, kidney function based on the serum creatinine at the time of death, and other donor factors.
A kidney with a score higher than 85% is likely to function longer than only 15% of available kidneys. Patients who receive a kidney with that score have a longer period of risk of death soon after transplant and a slightly higher risk of death in the long term than patients who receive a healthier kidney, although on average they still do better than patients on dialysis.5
Older patients should be encouraged to sign up for both the regular waiting list and the suboptimal kidney waiting list to reduce the risk of dying before they get a kidney.
LIVING-DONOR ORGAN TRANSPLANT
Many advantages
Living-donor organ transplant is associated with a better survival rate than deceased-donor organ transplant, and the advantage becomes greater over time. At 1 year, patient survival is more than 90% in both groups, but by 5 years about 80% of patients with a living-donor organ are still alive vs only about 65% of patients with a deceased-donor organ.
The waiting time for a living-donor transplant may be only weeks to months, rather than years. Because increasing time on dialysis predicts worse patient and graft survival after transplant, the shorter wait time is a big advantage. In addition, because the donor and recipient are typically in adjacent operating rooms, the organ sustains less ischemic damage. In general, the kidney quality is better from healthy donors, resulting in superior function early on and longer graft survival by an average of 4 years. If the living donor is related to the recipient, human leukocyte antigen matching also tends to be better and predicts better outcomes.
Special challenges
Opting for a living-donor organ also entails special challenges. In addition to the ethical issues surrounding living-donor organ donation, an appropriate donor must be found. Donors must be highly motivated and pass physical, laboratory, and psychological evaluations.
For older patients, if the donor is a spouse or close friend, he or she is also likely to be older, making the organ less viable than one from a younger person. Even an adult child may not be an ideal donor if there is a family propensity to kidney disease, such as diabetic nephropathy. No test is available to determine the risk for future diabetes, but it is known to run in families.
POTENT IMMUNOSUPPRESSION
Induction therapy
Induction therapy with antithymocyte globulin or basiliximab provides intense immunosuppression to prevent acute rejection during the early posttransplant period.
Antithymocyte globulin is a potent agent that contains antibodies directed at T cells, B cells, neutrophils, platelets, adhesion molecules, and complement. It binds T cells and removes them from circulation by opsonization in splenic and lymphoid tissue. The immunosuppressive effect is sustained for at least 2 to 3 months after a series of injections (dosage 1.5 mg/kg/day, usually for 4 to 10 doses). Antithymocyte globulin is also used to treat acute rejection, especially high-grade rejection for which steroid therapy is likely to be insufficient.
Basiliximab consists of antibodies to the interleukin 2 (IL-2) receptor of T cells. Binding to T cells prevents their activation rather than removing them from circulation. The drug prevents rejection, with 30% relative reduction in early studies compared with placebo. However, it is ineffective in reversing established rejection. Dosage is 20 mg at day 0 and day 4, which provides receptor saturation for 30 to 45 days.
Basiliximab is also sometimes used off-label for patients who need to discontinue a calcineurin inhibitor (ie, tacrolimus or cyclosporine). In such cases, normal therapy is put on hold while basiliximab is given for 1 or 2 doses. Case series have been reported for this use, particularly for patients with a heart and liver transplant who develop acute kidney injury while hospitalized.6,7
Antithymocyte globulin is more effective but also more risky. Brennan et al8 randomized 278 transplant recipients to either antithymocyte globulin or basiliximab. Patients in the antithymocyte globulin group had a 16% rejection rate vs 26% in the basiliximab group.
Antithymocyte globulin therapy is associated with multiple adverse effects, including fever and chills, pulmonary edema, and long-standing immunosuppressive effects such as increased risk of lymphoma and cytomegalovirus (CMV) infection. Basiliximab side-effect profiles are similar to those of placebo.
Maintenance therapy
The calcineurin inhibitors cyclosporine and tacrolimus remain the standard of care in kidney transplant despite multiple drug interactions and side effects that include renal toxicity and fibrosis. Cyclosporine and tacrolimus both bind intracellular immunophilins and thereby prevent transcription of IL-2 and production of T cells. The drugs work similarly but have different binding sites. Cyclosporine has largely been replaced by tacrolimus because its reliability of dosing and higher potency are associated with lower rejection rates.
Tacrolimus is typically given twice daily (1–6 mg/dose). Twelve-hour trough levels are followed (target: 8–12 ng/mL early on, then 5–8 ng/mL after 3 months posttransplant). Side effects include hypertension and hypercholesterolemia, but less so than with cyclosporine. On the other hand, hyperglycemia tends to be worse with tacrolimus than with cyclosporine, and combining tacrolimus with steroids frequently leads to diabetes. Tacrolimus can also cause acute and chronic renal failure, especially at high drug levels, as well as neurotoxicity, tremors, and hair loss.

Cyclosporine, tacrolimus, and sirolimus (not a calcineurin inhibitor) are metabolized through the same cytochrome P450 pathway (CYP3A4), so they have common drug interactions (Table 2).
Mycophenolate mofetil is typically used as an adjunct therapy (500–1,000 mg twice daily). It is also used for other kidney diseases before transplant, including lupus nephritis. Transplanted kidney rejection rates with mycophenolate mofetil with steroids are about 40%, so the drug is not potent enough to be used without a calcineurin inhibitor.
Side effects include gastrointestinal toxicity in up to 20% of patients, and leukopenia, which is associated with viral infections.
CORONARY ARTERY DISEASE IS COMMON WITH DIALYSIS
Coronary artery disease is highly associated with end-stage kidney disease and occurs in as many as 85% of older patients with diabetes on dialysis. Although patients with end-stage kidney disease tend to have more numerous and severe atherosclerotic lesions compared with the general population, justifying aggressive management, cardiac care tends to be conservative in patients on dialysis.9
Death from acute myocardial infarction occurs in about 20% to 30% of patients on dialysis vs about 2% of patients with normal renal function. Five years after myocardial infarction, survival is only about 30% in patients on dialysis.9
There are many explanations for excess coronary artery disease in patients on dialysis. In addition to the traditional cardiovascular risk factors of diabetes, hypertension, and preexisting coronary artery disease, patients are in a proinflammatory uremic state and have high levels of phosphorus and fibroblast growth factor 23 that contribute to vascular calcification. Almost all patients have high homocysteine levels and hemodynamic instability, particularly if they are on hemodialysis.
Pretransplant evaluation for heart disease
Patients on the kidney transplant waiting list are screened aggressively for heart disease. A history of myocardial infarction usually results in removal from the list. All patients have an initial electrocardiogram and echocardiogram. Thallium or echocardiographic stress testing is used for patients who are age 50 and older, have diabetes, or have had dialysis for many years. Patients with evidence of ischemia undergo catheterization.
Patients are also screened with computed tomography before transplant. Because the kidney is typically anastomosed to the iliac artery and vein, heavy calcification of the iliac artery can make the procedure too difficult to perform.
Reduced long-term risk of myocardial infarction after transplant
Kasiske et al10 analyzed data from more than 50,000 patients from the US Renal Data System and found that, for about the first year after transplant, patients who underwent kidney transplant were more likely to have a myocardial infarction than those on dialysis. After that, they fared better than patients who remained on dialysis. Those with a living-donor transplant were less likely at all times to have a myocardial infarction than those with a deceased-donor transplant. By 3 years after transplant, the relative risk of having a myocardial infarction was 0.89 for deceased-donor organ recipients and 0.69 for living-donor recipients compared with patients on the waiting list.10
INFECTIOUS COMPLICATIONS IN KIDNEY RECIPIENTS

Kidney recipients are prone to many common and uncommon infections (Table 3). All potential recipients are tested pretransplant for hepatitis B, hepatitis C, human immunodeficiency virus, syphilis, and tuberculosis. A positive result does not necessarily rule out transplant.
The following viral serology tests are also done before transplant:
Epstein-Barr virus (antibodies are positive in about 90% of adults)
CMV (about 70% of adults are seropositive)
Varicella zoster (seronegative patients should be given live-attenuated varicella vaccine).
Risk of transmission of these viruses relates to the serostatus of the donor and recipient before transplant. If a donor is positive for viral antibodies but the recipient is not (a so-called “mismatch”), risk is higher after transplant.
Hepatitis C
Patients with hepatitis C fare better if they get a transplant than if they remain on dialysis, although their posttransplant course is worse compared with transplant patients who do not have hepatitis. Some patients develop accelerated liver disease after kidney transplant. Hepatitis C-related kidney disease—membranous proliferative glomerulonephritis—also occurs, as do comorbidities such as diabetes.
Careful evaluation is warranted before transplant, including liver imaging, alpha-fetoprotein testing, and liver biopsy to evaluate for hepatocellular carcinoma. A patient with advanced fibrosis or cirrhosis may not be a candidate for kidney transplant alone but could possibly receive a combined kidney and liver transplant.
There is a need to determine the best time to treat hepatitis C infection. Patients with advanced liver disease or hepatitis C-related kidney disease would likely benefit from early treatment. However, delaying treatment could shorten the wait time for a deceased-donor organ positive for hepatitis C. Transplant candidates with active hepatitis C are uniquely considered to accept hepatitis C-positive kidneys, which are often discarded, and may only wait weeks for such a transplant. The shortened kidney survival associated with a hepatitis C-positive kidney may no longer be true with the new antiviral hepatitis C therapy, which has been shown to be effective post-transplant.
Hepatitis B
No cure is available for hepatitis B infection, but it can be well controlled with antiviral therapy. Patients with hepatitis B infection may be candidates for transplant, but they should be stable on antiviral therapy (lamivudine, entecavir, or tenofovir) to eliminate the viral load before transplant, and therapy should be continued afterward. Liver imaging, alpha-fetoprotein levels, and biopsy are recommended for evaluation. All hepatitis B- negative patients should be vaccinated before transplant.
Organs from living or deceased donors that test positive for hepatitis B core antibody, indicating prior exposure, can be considered for transplant in a patient who tests positive for hepatitis B surface antibody, indicating successful vaccination or prior exposure in the recipient. But donors must have negative surface antigen and polymerase chain reaction (PCR) tests that indicate no active hepatitis B infection.
Cytomegalovirus
CMV typically does not appear until prophylactic therapy is stopped. Classic symptoms are fever, leukopenia, and diarrhea. Infection can involve any organ, and patients may present with hepatitis, pancreatitis or, less commonly, pneumonitis.
Patients who are negative for CMV before transplant and receive a donor-positive organ are at the highest risk. Patients who are CMV IgG-positive are considered to be at intermediate risk, regardless of the donor status. Patients who are negative for CMV and receive a donor-negative organ are at the lowest risk and do not need prophylaxis with valganciclovir.
CMV infection is diagnosed by PCR testing of the blood or immunostaining in tissue biopsy. Occasionally, blood testing is negative in the face of tissue-based disease.
BK virus
BK is a polyoma virus and a common virus associated with kidney transplant. Viremia is seen in about 18% of patients, whereas actual kidney disease associated with a higher level of virus is seen in fewer than 10% of patients. Most people are exposed to BK virus, often in childhood, and it can remain indolent in the bladder and uroepithelium.
Patients can develop BK nephropathy after exposure to transplant immunosuppression.11 Posttransplant monitoring protocols typically include PCR testing for BK virus at 1, 3, 6, and 12 months. No agent has been identified to specifically treat BK virus. The general strategy is to minimize immunosuppressive therapy by reducing or eliminating mycophenolate mofetil. Fortunately, BK virus does not tend to recur, and patients can have a low-level viremia (< 10,000 copies/mL) persisting over months or even years but often without clinical consequences.
The appearance of BK virus on biopsy can mimic acute rejection. Before BK viral nephropathy was a recognized entity, patients would have been diagnosed with acute rejection and may have been put on high-dose steroids, which would have worsened the BK infection.
Posttransplant lymphoproliferative disorder
Posttransplant lymphoproliferative disorder is most often associated with Epstein-Barr virus and usually involves a large, diffuse B-cell lymphoma. Burkitt lymphoma and plasma cell neoplasms also can occur less commonly.
The condition is about 30 times more common in patients after transplant than in the general population, and it is the third most common malignancy in transplant patients after skin and cervical cancers. About 80% of the cases occur early after transplant, within the first year.
Patients typically have a marked elevation in viral load of Epstein-Barr virus, although a negative viral load does not rule it out. A patient who is serologically negative for Epstein-Barr virus receiving a donor-positive kidney is at highest risk; this situation is most often seen in the pediatric population. Potent induction therapies (eg, antilymphocyte antibody therapy) are also associated with posttransplant lymphoproliferative disorder.
Patients typically present with fever of unknown origin with no localizing signs or symptoms. Mass lesions can be challenging to find; positron emission tomography may be helpful. The culprit is usually a focal mass, ulcer (especially in the gastrointestinal tract), or infiltrate (commonly localized to the allograft). Multifocal or disseminated disease can also occur, including lymphoma or with central nervous system, gastrointestinal, or pulmonary involvement.
Biopsy of the affected site is required for histopathology and Epstein-Barr virus markers. PCR blood testing is often positive for Epstein-Barr virus.
Typical antiviral therapy does not eliminate Epstein-Barr virus. In early polyclonal viral proliferation, the first goal is to reduce immunosuppressive therapy. Rituximab alone may also help in polymorphic cases. With disease that is clearly monomorphic and has transformed to a true malignancy, cytotoxic chemotherapy is also required. “R-CHOP,” a combination therapy consisting of rituximab with cyclophosphamide, doxorubicin, vincristine, and prednisone, is usually used. Radiation therapy may help in some cases.
Cryptococcal infection
Previously seen in patients with acquired immune deficiency syndrome, cryptococcal infection is now most commonly encountered in patients with solid-organ transplants. Vilchez et al12 found a 1% incidence in a series of more than 5,000 patients who had received an organ transplant.
Immunosuppression likely conveys risk, but because cryptococcal infection is acquired, environmental exposure also plays a role. It tends to appear more than 6 months after transplant, indicating that its cause is a primary infection by spore inhalation rather than by reactivation or transmission from the donor organ.13 Bird exposure is a risk factor for cryptococcal infection. One case identified the same strain of Cryptococcus in a kidney transplant recipient and the family’s pet cockatoo.14
Cryptococcal infection typically starts as pneumonia, which may be subclinical. The infection can then disseminate, with meningitis presenting with headache and mental status changes being the most concerning complication. The death rate is about 50% in most series of patients with meningitis. Skin and soft-tissue manifestations may also occur in 10% to 15% of cases and can be nodular, ulcerative, or cellulitic.
More than 75% of fungal infections requiring hospitalization in US patients who have undergone transplant are attributed to either Candida, Aspergillus, or Cryptococcus species.15 Risk of fungal infection is increased with diabetes, duration of pretransplant dialysis, tacrolimus therapy, or rejection treatment.
- Wolfe RA, Ashby VB, Milford EL, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med 1999; 341:1725–1730.
- Kasiske BL, Klinger D. Cigarette smoking in renal transplant recipients. J Am Soc Nephrol 2000; 11:753–759.
- United Network for Organ Sharing. Transplant trends. https://transplantpro.org/technology/transplant-trends/#waitlists_by_organ. Accessed December 13, 2017.
- Meier-Kriesche HU, Kaplan B. Waiting time on dialysis as the strongest modifiable risk factor for renal transplant outcomes: a paired donor kidney analysis. Transplantation 2002; 74:1377–1381.
- Ojo AO, Hanson JA, Meier-Kriesche H, et al. Survival in recipients of marginal cadaveric donor kidneys compared with other recipients and wait-listed transplant candidates. J Am Soc Nephrol 2001; 12:589–597.
- Alonso P. Sanchez-Lazaro I, Almenar L, et al. Use of a “CNI holidays” strategy in acute renal dysfunction late after heart transplant. Report of two cases. Heart Int 2014; 9:74–77.
- Cantarovich M, Metrakos P, Giannetti N, Cecere R, Barkun J, Tchervenkov J. Anti-CD25 monoclonal antibody coverage allows for calcineurin inhibitor “holiday” in solid organ transplant patients with acute renal dysfunction. Transplantation 2002; 73:1169–1172.
- Brennan DC, Daller JA, Lake KD, Cibrik D, Del Castillo D; Thymoglobulin Induction Study Group. Rabbit antithymocyte globulin versus basiliximab in renal transplantation. N Engl J Med 2006; 355:1967–1977.
- McCullough PA. Evaluation and treatment of coronary artery disease in patients with end-stage renal disease. Kidney Int 2005; 67:S51–S58.
- Kasiske BL, Maclean JR, Snyder JJ. Acute myocardial infarction and kidney transplantation. J Am Soc Nephrol 2006; 17:900–907.
- Bohl DL, Storch GA, Ryschkewitsch C, et al. Donor origin of BK virus in renal transplantation and role of HLA C7 in susceptibility to sustained BK viremia. Am J Transplant 2005; 5:2213–2221.
- Vilchez RA, Fung J, Kusne S. Cryptococcosis in organ transplant recipients: an overview. Am J Transplant 2002; 2:575–580.
- Vilchez R, Shapiro R, McCurry K, et al. Longitudinal study of cryptococcosis in adult solid-organ transplant recipients. Transpl Int 2003; 16:336–340.
- Nosanchuk JD, Shoham S, Fries BC, Shapiro DS, Levitz SM, Casadevall A. Evidence of zoonotic transmission of Cryptococcus neoformans from a pet cockatoo to an immunocompromised patient. Ann Intern Med 2000; 132:205–208.
- Abbott KC, Hypolite I, Poropatich RK, et al. Hospitalizations for fungal infections after renal transplantation in the United States. Transpl Infect Dis 2001; 3:203–211.
Much has improved in renal transplantation over the past 20 years. The focus has shifted to using stronger immunotherapy rather than trying to minimize it. There has been increasing recognition of infection and ways to prevent and treat it. Induction therapy now has greater emphasis so that maintenance therapy can be eased, with the aim of reducing long-term toxicity. Perhaps the biggest change is the practice of screening for donor-specific antibodies at the time of transplant so that predictable problems can be prevented or better handled if they occur. Such advances have helped patients directly and by extending the life of their transplanted organs.
LONGER SURVIVAL
As early as the 1990s, it was recognized that kidney transplant offers a survival advantage for patients with end-stage renal disease over maintenance on dialysis.1 Although the risk of death is higher immediately after transplant, within a few months it becomes much lower than for patients on dialysis. Survival varies according to the health of the patient and the quality of the transplanted organ.
In general, patients who obtain the greatest benefit from transplants in terms of years of life gained are those with diabetes, especially those who are younger. Those ages 20 to 39 live about 8 years on dialysis vs 25 years after transplant.
CONTRAINDICATIONS TO TRANSPLANT
There are multiple contraindications to a solitary kidney transplant (Table 1), including smoking. Most transplant centers require that smokers quit before transplant. Long-standing smokers almost double their risk of a cardiac event after transplant and double their rate of malignancy. Active smoking at the time of transplant is associated with twice the risk of death by 10 years after transplant compared with that of nonsmokers.2 Cotinine testing can detect whether a patient is an active smoker.
WAITING-LIST CONSIDERATIONS
Organs are scarce
The number of patients on the kidney waiting list has increased rapidly in the last few decades, while the number of transplants performed each year has remained about the same. In 2016, about 100,000 patients were on the list, but only about 19,000 transplants were performed.3 Wait times, especially for deceased-donor organs, have increased to about 6 years, varying by blood type and geographic region.
Waiting-list placement
Placement on the waiting list for a deceased-donor kidney transplant occurs when a patient has an estimated glomerular filtration rate (GFR) of 20 mL/min/1.73 m2 or less, although referral to the list can be made earlier. Early listing remains advantageous, as total time on the list will be counted before starting dialysis. “Preemptive transplant” means the patient had no dialysis before transplant; this applies to about 10% of transplant recipients. These patients tend to fare the best and are usually recipients of a living-donor organ.
Most patients do not receive a transplant until the GFR is less than 15 mL/min/1.73 m2.
Since 2014, wait time has been measured from the beginning of dialysis rather than the date of waiting-list placement in patients who are listed after starting dialysis therapy. This approach is more fair but sometimes introduces problems. A patient who did not previously know about the list may suddenly jump to the head of the line after 10 years of dialysis, by which time comorbidities associated with long-term dialysis make the patient less likely to gain as much benefit from a transplant as people lower on the list. Time on dialysis, or “dialysis vintage,” predicts patient and kidney survival after transplant, with reduced survival associated with increasing time on dialysis.4
Shorter wait for a suboptimal kidney
The aging population has increased the number of older patients being listed for transplant, presenting multiple challenges. Patients age 65 or older have a 50% chance of dying before they receive a transplant during a 5-year wait.
A patient may shorten the wait by joining the list for a suboptimal organ. All deceased-donor organs are given a Kidney Donor Profile Index score, which predicts the longevity of an organ after transplant. The score is determined by donor age, kidney function based on the serum creatinine at the time of death, and other donor factors.
A kidney with a score higher than 85% is likely to function longer than only 15% of available kidneys. Patients who receive a kidney with that score have a longer period of risk of death soon after transplant and a slightly higher risk of death in the long term than patients who receive a healthier kidney, although on average they still do better than patients on dialysis.5
Older patients should be encouraged to sign up for both the regular waiting list and the suboptimal kidney waiting list to reduce the risk of dying before they get a kidney.
LIVING-DONOR ORGAN TRANSPLANT
Many advantages
Living-donor organ transplant is associated with a better survival rate than deceased-donor organ transplant, and the advantage becomes greater over time. At 1 year, patient survival is more than 90% in both groups, but by 5 years about 80% of patients with a living-donor organ are still alive vs only about 65% of patients with a deceased-donor organ.
The waiting time for a living-donor transplant may be only weeks to months, rather than years. Because increasing time on dialysis predicts worse patient and graft survival after transplant, the shorter wait time is a big advantage. In addition, because the donor and recipient are typically in adjacent operating rooms, the organ sustains less ischemic damage. In general, the kidney quality is better from healthy donors, resulting in superior function early on and longer graft survival by an average of 4 years. If the living donor is related to the recipient, human leukocyte antigen matching also tends to be better and predicts better outcomes.
Special challenges
Opting for a living-donor organ also entails special challenges. In addition to the ethical issues surrounding living-donor organ donation, an appropriate donor must be found. Donors must be highly motivated and pass physical, laboratory, and psychological evaluations.
For older patients, if the donor is a spouse or close friend, he or she is also likely to be older, making the organ less viable than one from a younger person. Even an adult child may not be an ideal donor if there is a family propensity to kidney disease, such as diabetic nephropathy. No test is available to determine the risk for future diabetes, but it is known to run in families.
POTENT IMMUNOSUPPRESSION
Induction therapy
Induction therapy with antithymocyte globulin or basiliximab provides intense immunosuppression to prevent acute rejection during the early posttransplant period.
Antithymocyte globulin is a potent agent that contains antibodies directed at T cells, B cells, neutrophils, platelets, adhesion molecules, and complement. It binds T cells and removes them from circulation by opsonization in splenic and lymphoid tissue. The immunosuppressive effect is sustained for at least 2 to 3 months after a series of injections (dosage 1.5 mg/kg/day, usually for 4 to 10 doses). Antithymocyte globulin is also used to treat acute rejection, especially high-grade rejection for which steroid therapy is likely to be insufficient.
Basiliximab consists of antibodies to the interleukin 2 (IL-2) receptor of T cells. Binding to T cells prevents their activation rather than removing them from circulation. The drug prevents rejection, with 30% relative reduction in early studies compared with placebo. However, it is ineffective in reversing established rejection. Dosage is 20 mg at day 0 and day 4, which provides receptor saturation for 30 to 45 days.
Basiliximab is also sometimes used off-label for patients who need to discontinue a calcineurin inhibitor (ie, tacrolimus or cyclosporine). In such cases, normal therapy is put on hold while basiliximab is given for 1 or 2 doses. Case series have been reported for this use, particularly for patients with a heart and liver transplant who develop acute kidney injury while hospitalized.6,7
Antithymocyte globulin is more effective but also more risky. Brennan et al8 randomized 278 transplant recipients to either antithymocyte globulin or basiliximab. Patients in the antithymocyte globulin group had a 16% rejection rate vs 26% in the basiliximab group.
Antithymocyte globulin therapy is associated with multiple adverse effects, including fever and chills, pulmonary edema, and long-standing immunosuppressive effects such as increased risk of lymphoma and cytomegalovirus (CMV) infection. Basiliximab side-effect profiles are similar to those of placebo.
Maintenance therapy
The calcineurin inhibitors cyclosporine and tacrolimus remain the standard of care in kidney transplant despite multiple drug interactions and side effects that include renal toxicity and fibrosis. Cyclosporine and tacrolimus both bind intracellular immunophilins and thereby prevent transcription of IL-2 and production of T cells. The drugs work similarly but have different binding sites. Cyclosporine has largely been replaced by tacrolimus because its reliability of dosing and higher potency are associated with lower rejection rates.
Tacrolimus is typically given twice daily (1–6 mg/dose). Twelve-hour trough levels are followed (target: 8–12 ng/mL early on, then 5–8 ng/mL after 3 months posttransplant). Side effects include hypertension and hypercholesterolemia, but less so than with cyclosporine. On the other hand, hyperglycemia tends to be worse with tacrolimus than with cyclosporine, and combining tacrolimus with steroids frequently leads to diabetes. Tacrolimus can also cause acute and chronic renal failure, especially at high drug levels, as well as neurotoxicity, tremors, and hair loss.
Cyclosporine, tacrolimus, and sirolimus (not a calcineurin inhibitor) are metabolized through the same cytochrome P450 pathway (CYP3A4), so they have common drug interactions (Table 2).
Mycophenolate mofetil is typically used as an adjunct therapy (500–1,000 mg twice daily). It is also used for other kidney diseases before transplant, including lupus nephritis. Transplanted kidney rejection rates with mycophenolate mofetil with steroids are about 40%, so the drug is not potent enough to be used without a calcineurin inhibitor.
Side effects include gastrointestinal toxicity in up to 20% of patients, and leukopenia, which is associated with viral infections.
CORONARY ARTERY DISEASE IS COMMON WITH DIALYSIS
Coronary artery disease is highly associated with end-stage kidney disease and occurs in as many as 85% of older patients with diabetes on dialysis. Although patients with end-stage kidney disease tend to have more numerous and severe atherosclerotic lesions compared with the general population, justifying aggressive management, cardiac care tends to be conservative in patients on dialysis.9
Death from acute myocardial infarction occurs in about 20% to 30% of patients on dialysis vs about 2% of patients with normal renal function. Five years after myocardial infarction, survival is only about 30% in patients on dialysis.9
There are many explanations for excess coronary artery disease in patients on dialysis. In addition to the traditional cardiovascular risk factors of diabetes, hypertension, and preexisting coronary artery disease, patients are in a proinflammatory uremic state and have high levels of phosphorus and fibroblast growth factor 23 that contribute to vascular calcification. Almost all patients have high homocysteine levels and hemodynamic instability, particularly if they are on hemodialysis.
Pretransplant evaluation for heart disease
Patients on the kidney transplant waiting list are screened aggressively for heart disease. A history of myocardial infarction usually results in removal from the list. All patients have an initial electrocardiogram and echocardiogram. Thallium or echocardiographic stress testing is used for patients who are age 50 and older, have diabetes, or have had dialysis for many years. Patients with evidence of ischemia undergo catheterization.
Patients are also screened with computed tomography before transplant. Because the kidney is typically anastomosed to the iliac artery and vein, heavy calcification of the iliac artery can make the procedure too difficult to perform.
Reduced long-term risk of myocardial infarction after transplant
Kasiske et al10 analyzed data from more than 50,000 patients from the US Renal Data System and found that, for about the first year after transplant, patients who underwent kidney transplant were more likely to have a myocardial infarction than those on dialysis. After that, they fared better than patients who remained on dialysis. Those with a living-donor transplant were less likely at all times to have a myocardial infarction than those with a deceased-donor transplant. By 3 years after transplant, the relative risk of having a myocardial infarction was 0.89 for deceased-donor organ recipients and 0.69 for living-donor recipients compared with patients on the waiting list.10
INFECTIOUS COMPLICATIONS IN KIDNEY RECIPIENTS
Kidney recipients are prone to many common and uncommon infections (Table 3). All potential recipients are tested pretransplant for hepatitis B, hepatitis C, human immunodeficiency virus, syphilis, and tuberculosis. A positive result does not necessarily rule out transplant.
The following viral serology tests are also done before transplant:
Epstein-Barr virus (antibodies are positive in about 90% of adults)
CMV (about 70% of adults are seropositive)
Varicella zoster (seronegative patients should be given live-attenuated varicella vaccine).
Risk of transmission of these viruses relates to the serostatus of the donor and recipient before transplant. If a donor is positive for viral antibodies but the recipient is not (a so-called “mismatch”), risk is higher after transplant.
Hepatitis C
Patients with hepatitis C fare better if they get a transplant than if they remain on dialysis, although their posttransplant course is worse compared with transplant patients who do not have hepatitis. Some patients develop accelerated liver disease after kidney transplant. Hepatitis C-related kidney disease—membranous proliferative glomerulonephritis—also occurs, as do comorbidities such as diabetes.
Careful evaluation is warranted before transplant, including liver imaging, alpha-fetoprotein testing, and liver biopsy to evaluate for hepatocellular carcinoma. A patient with advanced fibrosis or cirrhosis may not be a candidate for kidney transplant alone but could possibly receive a combined kidney and liver transplant.
There is a need to determine the best time to treat hepatitis C infection. Patients with advanced liver disease or hepatitis C-related kidney disease would likely benefit from early treatment. However, delaying treatment could shorten the wait time for a deceased-donor organ positive for hepatitis C. Transplant candidates with active hepatitis C are uniquely considered to accept hepatitis C-positive kidneys, which are often discarded, and may only wait weeks for such a transplant. The shortened kidney survival associated with a hepatitis C-positive kidney may no longer be true with the new antiviral hepatitis C therapy, which has been shown to be effective post-transplant.
Hepatitis B
No cure is available for hepatitis B infection, but it can be well controlled with antiviral therapy. Patients with hepatitis B infection may be candidates for transplant, but they should be stable on antiviral therapy (lamivudine, entecavir, or tenofovir) to eliminate the viral load before transplant, and therapy should be continued afterward. Liver imaging, alpha-fetoprotein levels, and biopsy are recommended for evaluation. All hepatitis B- negative patients should be vaccinated before transplant.
Organs from living or deceased donors that test positive for hepatitis B core antibody, indicating prior exposure, can be considered for transplant in a patient who tests positive for hepatitis B surface antibody, indicating successful vaccination or prior exposure in the recipient. But donors must have negative surface antigen and polymerase chain reaction (PCR) tests that indicate no active hepatitis B infection.
Cytomegalovirus
CMV typically does not appear until prophylactic therapy is stopped. Classic symptoms are fever, leukopenia, and diarrhea. Infection can involve any organ, and patients may present with hepatitis, pancreatitis or, less commonly, pneumonitis.
Patients who are negative for CMV before transplant and receive a donor-positive organ are at the highest risk. Patients who are CMV IgG-positive are considered to be at intermediate risk, regardless of the donor status. Patients who are negative for CMV and receive a donor-negative organ are at the lowest risk and do not need prophylaxis with valganciclovir.
CMV infection is diagnosed by PCR testing of the blood or immunostaining in tissue biopsy. Occasionally, blood testing is negative in the face of tissue-based disease.
BK virus
BK is a polyoma virus and a common virus associated with kidney transplant. Viremia is seen in about 18% of patients, whereas actual kidney disease associated with a higher level of virus is seen in fewer than 10% of patients. Most people are exposed to BK virus, often in childhood, and it can remain indolent in the bladder and uroepithelium.
Patients can develop BK nephropathy after exposure to transplant immunosuppression.11 Posttransplant monitoring protocols typically include PCR testing for BK virus at 1, 3, 6, and 12 months. No agent has been identified to specifically treat BK virus. The general strategy is to minimize immunosuppressive therapy by reducing or eliminating mycophenolate mofetil. Fortunately, BK virus does not tend to recur, and patients can have a low-level viremia (< 10,000 copies/mL) persisting over months or even years but often without clinical consequences.
The appearance of BK virus on biopsy can mimic acute rejection. Before BK viral nephropathy was a recognized entity, patients would have been diagnosed with acute rejection and may have been put on high-dose steroids, which would have worsened the BK infection.
Posttransplant lymphoproliferative disorder
Posttransplant lymphoproliferative disorder is most often associated with Epstein-Barr virus and usually involves a large, diffuse B-cell lymphoma. Burkitt lymphoma and plasma cell neoplasms also can occur less commonly.
The condition is about 30 times more common in patients after transplant than in the general population, and it is the third most common malignancy in transplant patients after skin and cervical cancers. About 80% of the cases occur early after transplant, within the first year.
Patients typically have a marked elevation in viral load of Epstein-Barr virus, although a negative viral load does not rule it out. A patient who is serologically negative for Epstein-Barr virus receiving a donor-positive kidney is at highest risk; this situation is most often seen in the pediatric population. Potent induction therapies (eg, antilymphocyte antibody therapy) are also associated with posttransplant lymphoproliferative disorder.
Patients typically present with fever of unknown origin with no localizing signs or symptoms. Mass lesions can be challenging to find; positron emission tomography may be helpful. The culprit is usually a focal mass, ulcer (especially in the gastrointestinal tract), or infiltrate (commonly localized to the allograft). Multifocal or disseminated disease can also occur, including lymphoma or with central nervous system, gastrointestinal, or pulmonary involvement.
Biopsy of the affected site is required for histopathology and Epstein-Barr virus markers. PCR blood testing is often positive for Epstein-Barr virus.
Typical antiviral therapy does not eliminate Epstein-Barr virus. In early polyclonal viral proliferation, the first goal is to reduce immunosuppressive therapy. Rituximab alone may also help in polymorphic cases. With disease that is clearly monomorphic and has transformed to a true malignancy, cytotoxic chemotherapy is also required. “R-CHOP,” a combination therapy consisting of rituximab with cyclophosphamide, doxorubicin, vincristine, and prednisone, is usually used. Radiation therapy may help in some cases.
Cryptococcal infection
Previously seen in patients with acquired immune deficiency syndrome, cryptococcal infection is now most commonly encountered in patients with solid-organ transplants. Vilchez et al12 found a 1% incidence in a series of more than 5,000 patients who had received an organ transplant.
Immunosuppression likely conveys risk, but because cryptococcal infection is acquired, environmental exposure also plays a role. It tends to appear more than 6 months after transplant, indicating that its cause is a primary infection by spore inhalation rather than by reactivation or transmission from the donor organ.13 Bird exposure is a risk factor for cryptococcal infection. One case identified the same strain of Cryptococcus in a kidney transplant recipient and the family’s pet cockatoo.14
Cryptococcal infection typically starts as pneumonia, which may be subclinical. The infection can then disseminate, with meningitis presenting with headache and mental status changes being the most concerning complication. The death rate is about 50% in most series of patients with meningitis. Skin and soft-tissue manifestations may also occur in 10% to 15% of cases and can be nodular, ulcerative, or cellulitic.
More than 75% of fungal infections requiring hospitalization in US patients who have undergone transplant are attributed to either Candida, Aspergillus, or Cryptococcus species.15 Risk of fungal infection is increased with diabetes, duration of pretransplant dialysis, tacrolimus therapy, or rejection treatment.
Much has improved in renal transplantation over the past 20 years. The focus has shifted to using stronger immunotherapy rather than trying to minimize it. There has been increasing recognition of infection and ways to prevent and treat it. Induction therapy now has greater emphasis so that maintenance therapy can be eased, with the aim of reducing long-term toxicity. Perhaps the biggest change is the practice of screening for donor-specific antibodies at the time of transplant so that predictable problems can be prevented or better handled if they occur. Such advances have helped patients directly and by extending the life of their transplanted organs.
LONGER SURVIVAL
As early as the 1990s, it was recognized that kidney transplant offers a survival advantage for patients with end-stage renal disease over maintenance on dialysis.1 Although the risk of death is higher immediately after transplant, within a few months it becomes much lower than for patients on dialysis. Survival varies according to the health of the patient and the quality of the transplanted organ.
In general, patients who obtain the greatest benefit from transplants in terms of years of life gained are those with diabetes, especially those who are younger. Those ages 20 to 39 live about 8 years on dialysis vs 25 years after transplant.
CONTRAINDICATIONS TO TRANSPLANT
There are multiple contraindications to a solitary kidney transplant (Table 1), including smoking. Most transplant centers require that smokers quit before transplant. Long-standing smokers almost double their risk of a cardiac event after transplant and double their rate of malignancy. Active smoking at the time of transplant is associated with twice the risk of death by 10 years after transplant compared with that of nonsmokers.2 Cotinine testing can detect whether a patient is an active smoker.
WAITING-LIST CONSIDERATIONS
Organs are scarce
The number of patients on the kidney waiting list has increased rapidly in the last few decades, while the number of transplants performed each year has remained about the same. In 2016, about 100,000 patients were on the list, but only about 19,000 transplants were performed.3 Wait times, especially for deceased-donor organs, have increased to about 6 years, varying by blood type and geographic region.
Waiting-list placement
Placement on the waiting list for a deceased-donor kidney transplant occurs when a patient has an estimated glomerular filtration rate (GFR) of 20 mL/min/1.73 m2 or less, although referral to the list can be made earlier. Early listing remains advantageous, as total time on the list will be counted before starting dialysis. “Preemptive transplant” means the patient had no dialysis before transplant; this applies to about 10% of transplant recipients. These patients tend to fare the best and are usually recipients of a living-donor organ.
Most patients do not receive a transplant until the GFR is less than 15 mL/min/1.73 m2.
Since 2014, wait time has been measured from the beginning of dialysis rather than the date of waiting-list placement in patients who are listed after starting dialysis therapy. This approach is more fair but sometimes introduces problems. A patient who did not previously know about the list may suddenly jump to the head of the line after 10 years of dialysis, by which time comorbidities associated with long-term dialysis make the patient less likely to gain as much benefit from a transplant as people lower on the list. Time on dialysis, or “dialysis vintage,” predicts patient and kidney survival after transplant, with reduced survival associated with increasing time on dialysis.4
Shorter wait for a suboptimal kidney
The aging population has increased the number of older patients being listed for transplant, presenting multiple challenges. Patients age 65 or older have a 50% chance of dying before they receive a transplant during a 5-year wait.
A patient may shorten the wait by joining the list for a suboptimal organ. All deceased-donor organs are given a Kidney Donor Profile Index score, which predicts the longevity of an organ after transplant. The score is determined by donor age, kidney function based on the serum creatinine at the time of death, and other donor factors.
A kidney with a score higher than 85% is likely to function longer than only 15% of available kidneys. Patients who receive a kidney with that score have a longer period of risk of death soon after transplant and a slightly higher risk of death in the long term than patients who receive a healthier kidney, although on average they still do better than patients on dialysis.5
Older patients should be encouraged to sign up for both the regular waiting list and the suboptimal kidney waiting list to reduce the risk of dying before they get a kidney.
LIVING-DONOR ORGAN TRANSPLANT
Many advantages
Living-donor organ transplant is associated with a better survival rate than deceased-donor organ transplant, and the advantage becomes greater over time. At 1 year, patient survival is more than 90% in both groups, but by 5 years about 80% of patients with a living-donor organ are still alive vs only about 65% of patients with a deceased-donor organ.
The waiting time for a living-donor transplant may be only weeks to months, rather than years. Because increasing time on dialysis predicts worse patient and graft survival after transplant, the shorter wait time is a big advantage. In addition, because the donor and recipient are typically in adjacent operating rooms, the organ sustains less ischemic damage. In general, the kidney quality is better from healthy donors, resulting in superior function early on and longer graft survival by an average of 4 years. If the living donor is related to the recipient, human leukocyte antigen matching also tends to be better and predicts better outcomes.
Special challenges
Opting for a living-donor organ also entails special challenges. In addition to the ethical issues surrounding living-donor organ donation, an appropriate donor must be found. Donors must be highly motivated and pass physical, laboratory, and psychological evaluations.
For older patients, if the donor is a spouse or close friend, he or she is also likely to be older, making the organ less viable than one from a younger person. Even an adult child may not be an ideal donor if there is a family propensity to kidney disease, such as diabetic nephropathy. No test is available to determine the risk for future diabetes, but it is known to run in families.
POTENT IMMUNOSUPPRESSION
Induction therapy
Induction therapy with antithymocyte globulin or basiliximab provides intense immunosuppression to prevent acute rejection during the early posttransplant period.
Antithymocyte globulin is a potent agent that contains antibodies directed at T cells, B cells, neutrophils, platelets, adhesion molecules, and complement. It binds T cells and removes them from circulation by opsonization in splenic and lymphoid tissue. The immunosuppressive effect is sustained for at least 2 to 3 months after a series of injections (dosage 1.5 mg/kg/day, usually for 4 to 10 doses). Antithymocyte globulin is also used to treat acute rejection, especially high-grade rejection for which steroid therapy is likely to be insufficient.
Basiliximab consists of antibodies to the interleukin 2 (IL-2) receptor of T cells. Binding to T cells prevents their activation rather than removing them from circulation. The drug prevents rejection, with 30% relative reduction in early studies compared with placebo. However, it is ineffective in reversing established rejection. Dosage is 20 mg at day 0 and day 4, which provides receptor saturation for 30 to 45 days.
Basiliximab is also sometimes used off-label for patients who need to discontinue a calcineurin inhibitor (ie, tacrolimus or cyclosporine). In such cases, normal therapy is put on hold while basiliximab is given for 1 or 2 doses. Case series have been reported for this use, particularly for patients with a heart and liver transplant who develop acute kidney injury while hospitalized.6,7
Antithymocyte globulin is more effective but also more risky. Brennan et al8 randomized 278 transplant recipients to either antithymocyte globulin or basiliximab. Patients in the antithymocyte globulin group had a 16% rejection rate vs 26% in the basiliximab group.
Antithymocyte globulin therapy is associated with multiple adverse effects, including fever and chills, pulmonary edema, and long-standing immunosuppressive effects such as increased risk of lymphoma and cytomegalovirus (CMV) infection. Basiliximab side-effect profiles are similar to those of placebo.
Maintenance therapy
The calcineurin inhibitors cyclosporine and tacrolimus remain the standard of care in kidney transplant despite multiple drug interactions and side effects that include renal toxicity and fibrosis. Cyclosporine and tacrolimus both bind intracellular immunophilins and thereby prevent transcription of IL-2 and production of T cells. The drugs work similarly but have different binding sites. Cyclosporine has largely been replaced by tacrolimus because its reliability of dosing and higher potency are associated with lower rejection rates.
Tacrolimus is typically given twice daily (1–6 mg/dose). Twelve-hour trough levels are followed (target: 8–12 ng/mL early on, then 5–8 ng/mL after 3 months posttransplant). Side effects include hypertension and hypercholesterolemia, but less so than with cyclosporine. On the other hand, hyperglycemia tends to be worse with tacrolimus than with cyclosporine, and combining tacrolimus with steroids frequently leads to diabetes. Tacrolimus can also cause acute and chronic renal failure, especially at high drug levels, as well as neurotoxicity, tremors, and hair loss.
Cyclosporine, tacrolimus, and sirolimus (not a calcineurin inhibitor) are metabolized through the same cytochrome P450 pathway (CYP3A4), so they have common drug interactions (Table 2).
Mycophenolate mofetil is typically used as an adjunct therapy (500–1,000 mg twice daily). It is also used for other kidney diseases before transplant, including lupus nephritis. Transplanted kidney rejection rates with mycophenolate mofetil with steroids are about 40%, so the drug is not potent enough to be used without a calcineurin inhibitor.
Side effects include gastrointestinal toxicity in up to 20% of patients, and leukopenia, which is associated with viral infections.
CORONARY ARTERY DISEASE IS COMMON WITH DIALYSIS
Coronary artery disease is highly associated with end-stage kidney disease and occurs in as many as 85% of older patients with diabetes on dialysis. Although patients with end-stage kidney disease tend to have more numerous and severe atherosclerotic lesions compared with the general population, justifying aggressive management, cardiac care tends to be conservative in patients on dialysis.9
Death from acute myocardial infarction occurs in about 20% to 30% of patients on dialysis vs about 2% of patients with normal renal function. Five years after myocardial infarction, survival is only about 30% in patients on dialysis.9
There are many explanations for excess coronary artery disease in patients on dialysis. In addition to the traditional cardiovascular risk factors of diabetes, hypertension, and preexisting coronary artery disease, patients are in a proinflammatory uremic state and have high levels of phosphorus and fibroblast growth factor 23 that contribute to vascular calcification. Almost all patients have high homocysteine levels and hemodynamic instability, particularly if they are on hemodialysis.
Pretransplant evaluation for heart disease
Patients on the kidney transplant waiting list are screened aggressively for heart disease. A history of myocardial infarction usually results in removal from the list. All patients have an initial electrocardiogram and echocardiogram. Thallium or echocardiographic stress testing is used for patients who are age 50 and older, have diabetes, or have had dialysis for many years. Patients with evidence of ischemia undergo catheterization.
Patients are also screened with computed tomography before transplant. Because the kidney is typically anastomosed to the iliac artery and vein, heavy calcification of the iliac artery can make the procedure too difficult to perform.
Reduced long-term risk of myocardial infarction after transplant
Kasiske et al10 analyzed data from more than 50,000 patients from the US Renal Data System and found that, for about the first year after transplant, patients who underwent kidney transplant were more likely to have a myocardial infarction than those on dialysis. After that, they fared better than patients who remained on dialysis. Those with a living-donor transplant were less likely at all times to have a myocardial infarction than those with a deceased-donor transplant. By 3 years after transplant, the relative risk of having a myocardial infarction was 0.89 for deceased-donor organ recipients and 0.69 for living-donor recipients compared with patients on the waiting list.10
INFECTIOUS COMPLICATIONS IN KIDNEY RECIPIENTS
Kidney recipients are prone to many common and uncommon infections (Table 3). All potential recipients are tested pretransplant for hepatitis B, hepatitis C, human immunodeficiency virus, syphilis, and tuberculosis. A positive result does not necessarily rule out transplant.
The following viral serology tests are also done before transplant:
Epstein-Barr virus (antibodies are positive in about 90% of adults)
CMV (about 70% of adults are seropositive)
Varicella zoster (seronegative patients should be given live-attenuated varicella vaccine).
Risk of transmission of these viruses relates to the serostatus of the donor and recipient before transplant. If a donor is positive for viral antibodies but the recipient is not (a so-called “mismatch”), risk is higher after transplant.
Hepatitis C
Patients with hepatitis C fare better if they get a transplant than if they remain on dialysis, although their posttransplant course is worse compared with transplant patients who do not have hepatitis. Some patients develop accelerated liver disease after kidney transplant. Hepatitis C-related kidney disease—membranous proliferative glomerulonephritis—also occurs, as do comorbidities such as diabetes.
Careful evaluation is warranted before transplant, including liver imaging, alpha-fetoprotein testing, and liver biopsy to evaluate for hepatocellular carcinoma. A patient with advanced fibrosis or cirrhosis may not be a candidate for kidney transplant alone but could possibly receive a combined kidney and liver transplant.
There is a need to determine the best time to treat hepatitis C infection. Patients with advanced liver disease or hepatitis C-related kidney disease would likely benefit from early treatment. However, delaying treatment could shorten the wait time for a deceased-donor organ positive for hepatitis C. Transplant candidates with active hepatitis C are uniquely considered to accept hepatitis C-positive kidneys, which are often discarded, and may only wait weeks for such a transplant. The shortened kidney survival associated with a hepatitis C-positive kidney may no longer be true with the new antiviral hepatitis C therapy, which has been shown to be effective post-transplant.
Hepatitis B
No cure is available for hepatitis B infection, but it can be well controlled with antiviral therapy. Patients with hepatitis B infection may be candidates for transplant, but they should be stable on antiviral therapy (lamivudine, entecavir, or tenofovir) to eliminate the viral load before transplant, and therapy should be continued afterward. Liver imaging, alpha-fetoprotein levels, and biopsy are recommended for evaluation. All hepatitis B- negative patients should be vaccinated before transplant.
Organs from living or deceased donors that test positive for hepatitis B core antibody, indicating prior exposure, can be considered for transplant in a patient who tests positive for hepatitis B surface antibody, indicating successful vaccination or prior exposure in the recipient. But donors must have negative surface antigen and polymerase chain reaction (PCR) tests that indicate no active hepatitis B infection.
Cytomegalovirus
CMV typically does not appear until prophylactic therapy is stopped. Classic symptoms are fever, leukopenia, and diarrhea. Infection can involve any organ, and patients may present with hepatitis, pancreatitis or, less commonly, pneumonitis.
Patients who are negative for CMV before transplant and receive a donor-positive organ are at the highest risk. Patients who are CMV IgG-positive are considered to be at intermediate risk, regardless of the donor status. Patients who are negative for CMV and receive a donor-negative organ are at the lowest risk and do not need prophylaxis with valganciclovir.
CMV infection is diagnosed by PCR testing of the blood or immunostaining in tissue biopsy. Occasionally, blood testing is negative in the face of tissue-based disease.
BK virus
BK is a polyoma virus and a common virus associated with kidney transplant. Viremia is seen in about 18% of patients, whereas actual kidney disease associated with a higher level of virus is seen in fewer than 10% of patients. Most people are exposed to BK virus, often in childhood, and it can remain indolent in the bladder and uroepithelium.
Patients can develop BK nephropathy after exposure to transplant immunosuppression.11 Posttransplant monitoring protocols typically include PCR testing for BK virus at 1, 3, 6, and 12 months. No agent has been identified to specifically treat BK virus. The general strategy is to minimize immunosuppressive therapy by reducing or eliminating mycophenolate mofetil. Fortunately, BK virus does not tend to recur, and patients can have a low-level viremia (< 10,000 copies/mL) persisting over months or even years but often without clinical consequences.
The appearance of BK virus on biopsy can mimic acute rejection. Before BK viral nephropathy was a recognized entity, patients would have been diagnosed with acute rejection and may have been put on high-dose steroids, which would have worsened the BK infection.
Posttransplant lymphoproliferative disorder
Posttransplant lymphoproliferative disorder is most often associated with Epstein-Barr virus and usually involves a large, diffuse B-cell lymphoma. Burkitt lymphoma and plasma cell neoplasms also can occur less commonly.
The condition is about 30 times more common in patients after transplant than in the general population, and it is the third most common malignancy in transplant patients after skin and cervical cancers. About 80% of the cases occur early after transplant, within the first year.
Patients typically have a marked elevation in viral load of Epstein-Barr virus, although a negative viral load does not rule it out. A patient who is serologically negative for Epstein-Barr virus receiving a donor-positive kidney is at highest risk; this situation is most often seen in the pediatric population. Potent induction therapies (eg, antilymphocyte antibody therapy) are also associated with posttransplant lymphoproliferative disorder.
Patients typically present with fever of unknown origin with no localizing signs or symptoms. Mass lesions can be challenging to find; positron emission tomography may be helpful. The culprit is usually a focal mass, ulcer (especially in the gastrointestinal tract), or infiltrate (commonly localized to the allograft). Multifocal or disseminated disease can also occur, including lymphoma or with central nervous system, gastrointestinal, or pulmonary involvement.
Biopsy of the affected site is required for histopathology and Epstein-Barr virus markers. PCR blood testing is often positive for Epstein-Barr virus.
Typical antiviral therapy does not eliminate Epstein-Barr virus. In early polyclonal viral proliferation, the first goal is to reduce immunosuppressive therapy. Rituximab alone may also help in polymorphic cases. With disease that is clearly monomorphic and has transformed to a true malignancy, cytotoxic chemotherapy is also required. “R-CHOP,” a combination therapy consisting of rituximab with cyclophosphamide, doxorubicin, vincristine, and prednisone, is usually used. Radiation therapy may help in some cases.
Cryptococcal infection
Previously seen in patients with acquired immune deficiency syndrome, cryptococcal infection is now most commonly encountered in patients with solid-organ transplants. Vilchez et al12 found a 1% incidence in a series of more than 5,000 patients who had received an organ transplant.
Immunosuppression likely conveys risk, but because cryptococcal infection is acquired, environmental exposure also plays a role. It tends to appear more than 6 months after transplant, indicating that its cause is a primary infection by spore inhalation rather than by reactivation or transmission from the donor organ.13 Bird exposure is a risk factor for cryptococcal infection. One case identified the same strain of Cryptococcus in a kidney transplant recipient and the family’s pet cockatoo.14
Cryptococcal infection typically starts as pneumonia, which may be subclinical. The infection can then disseminate, with meningitis presenting with headache and mental status changes being the most concerning complication. The death rate is about 50% in most series of patients with meningitis. Skin and soft-tissue manifestations may also occur in 10% to 15% of cases and can be nodular, ulcerative, or cellulitic.
More than 75% of fungal infections requiring hospitalization in US patients who have undergone transplant are attributed to either Candida, Aspergillus, or Cryptococcus species.15 Risk of fungal infection is increased with diabetes, duration of pretransplant dialysis, tacrolimus therapy, or rejection treatment.
- Wolfe RA, Ashby VB, Milford EL, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med 1999; 341:1725–1730.
- Kasiske BL, Klinger D. Cigarette smoking in renal transplant recipients. J Am Soc Nephrol 2000; 11:753–759.
- United Network for Organ Sharing. Transplant trends. https://transplantpro.org/technology/transplant-trends/#waitlists_by_organ. Accessed December 13, 2017.
- Meier-Kriesche HU, Kaplan B. Waiting time on dialysis as the strongest modifiable risk factor for renal transplant outcomes: a paired donor kidney analysis. Transplantation 2002; 74:1377–1381.
- Ojo AO, Hanson JA, Meier-Kriesche H, et al. Survival in recipients of marginal cadaveric donor kidneys compared with other recipients and wait-listed transplant candidates. J Am Soc Nephrol 2001; 12:589–597.
- Alonso P. Sanchez-Lazaro I, Almenar L, et al. Use of a “CNI holidays” strategy in acute renal dysfunction late after heart transplant. Report of two cases. Heart Int 2014; 9:74–77.
- Cantarovich M, Metrakos P, Giannetti N, Cecere R, Barkun J, Tchervenkov J. Anti-CD25 monoclonal antibody coverage allows for calcineurin inhibitor “holiday” in solid organ transplant patients with acute renal dysfunction. Transplantation 2002; 73:1169–1172.
- Brennan DC, Daller JA, Lake KD, Cibrik D, Del Castillo D; Thymoglobulin Induction Study Group. Rabbit antithymocyte globulin versus basiliximab in renal transplantation. N Engl J Med 2006; 355:1967–1977.
- McCullough PA. Evaluation and treatment of coronary artery disease in patients with end-stage renal disease. Kidney Int 2005; 67:S51–S58.
- Kasiske BL, Maclean JR, Snyder JJ. Acute myocardial infarction and kidney transplantation. J Am Soc Nephrol 2006; 17:900–907.
- Bohl DL, Storch GA, Ryschkewitsch C, et al. Donor origin of BK virus in renal transplantation and role of HLA C7 in susceptibility to sustained BK viremia. Am J Transplant 2005; 5:2213–2221.
- Vilchez RA, Fung J, Kusne S. Cryptococcosis in organ transplant recipients: an overview. Am J Transplant 2002; 2:575–580.
- Vilchez R, Shapiro R, McCurry K, et al. Longitudinal study of cryptococcosis in adult solid-organ transplant recipients. Transpl Int 2003; 16:336–340.
- Nosanchuk JD, Shoham S, Fries BC, Shapiro DS, Levitz SM, Casadevall A. Evidence of zoonotic transmission of Cryptococcus neoformans from a pet cockatoo to an immunocompromised patient. Ann Intern Med 2000; 132:205–208.
- Abbott KC, Hypolite I, Poropatich RK, et al. Hospitalizations for fungal infections after renal transplantation in the United States. Transpl Infect Dis 2001; 3:203–211.
- Wolfe RA, Ashby VB, Milford EL, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med 1999; 341:1725–1730.
- Kasiske BL, Klinger D. Cigarette smoking in renal transplant recipients. J Am Soc Nephrol 2000; 11:753–759.
- United Network for Organ Sharing. Transplant trends. https://transplantpro.org/technology/transplant-trends/#waitlists_by_organ. Accessed December 13, 2017.
- Meier-Kriesche HU, Kaplan B. Waiting time on dialysis as the strongest modifiable risk factor for renal transplant outcomes: a paired donor kidney analysis. Transplantation 2002; 74:1377–1381.
- Ojo AO, Hanson JA, Meier-Kriesche H, et al. Survival in recipients of marginal cadaveric donor kidneys compared with other recipients and wait-listed transplant candidates. J Am Soc Nephrol 2001; 12:589–597.
- Alonso P. Sanchez-Lazaro I, Almenar L, et al. Use of a “CNI holidays” strategy in acute renal dysfunction late after heart transplant. Report of two cases. Heart Int 2014; 9:74–77.
- Cantarovich M, Metrakos P, Giannetti N, Cecere R, Barkun J, Tchervenkov J. Anti-CD25 monoclonal antibody coverage allows for calcineurin inhibitor “holiday” in solid organ transplant patients with acute renal dysfunction. Transplantation 2002; 73:1169–1172.
- Brennan DC, Daller JA, Lake KD, Cibrik D, Del Castillo D; Thymoglobulin Induction Study Group. Rabbit antithymocyte globulin versus basiliximab in renal transplantation. N Engl J Med 2006; 355:1967–1977.
- McCullough PA. Evaluation and treatment of coronary artery disease in patients with end-stage renal disease. Kidney Int 2005; 67:S51–S58.
- Kasiske BL, Maclean JR, Snyder JJ. Acute myocardial infarction and kidney transplantation. J Am Soc Nephrol 2006; 17:900–907.
- Bohl DL, Storch GA, Ryschkewitsch C, et al. Donor origin of BK virus in renal transplantation and role of HLA C7 in susceptibility to sustained BK viremia. Am J Transplant 2005; 5:2213–2221.
- Vilchez RA, Fung J, Kusne S. Cryptococcosis in organ transplant recipients: an overview. Am J Transplant 2002; 2:575–580.
- Vilchez R, Shapiro R, McCurry K, et al. Longitudinal study of cryptococcosis in adult solid-organ transplant recipients. Transpl Int 2003; 16:336–340.
- Nosanchuk JD, Shoham S, Fries BC, Shapiro DS, Levitz SM, Casadevall A. Evidence of zoonotic transmission of Cryptococcus neoformans from a pet cockatoo to an immunocompromised patient. Ann Intern Med 2000; 132:205–208.
- Abbott KC, Hypolite I, Poropatich RK, et al. Hospitalizations for fungal infections after renal transplantation in the United States. Transpl Infect Dis 2001; 3:203–211.
KEY POINTS
- Kidney transplant improves survival and long-term outcomes in patients with renal failure.
- Before transplant, patients should be carefully evaluated for cardiovascular and infectious disease risk.
- Potent immunosuppression is required to maintain a successful kidney transplant.
- After transplant, patients must be monitored for recurrent disease, side effects of immunosuppression, and opportunistic infections.
PCI for stable angina: A missed opportunity for shared decision-making
Multiple randomized controlled trials have compared percutaneous coronary intervention (PCI) vs optimal medical therapy for patients with chronic stable angina. All have consistently shown that PCI does not reduce the risk of death or even myocardial infarction (MI) but that it may relieve angina temporarily. Nevertheless, PCI is still commonly performed for patients with stable coronary disease, often in the absence of angina, and patients mistakenly believe the procedure is life-saving. Cardiologists may not be aware of patients’ misperceptions, or worse, may encourage them. In either case, if patients do not understand the benefits of the procedure, they cannot give informed consent.
This article reviews the pathophysiology of coronary artery disease, evidence from clinical trials of the value of PCI for chronic stable angina, patient and physician perceptions of PCI, and ways to promote patient-centered, shared decision-making.
CLINICAL CASE: EXERTIONAL ANGINA
While climbing 4 flights of stairs, a 55-year-old man noticed tightness in his chest, which lasted for 5 minutes and resolved spontaneously. Several weeks later, when visiting his primary care physician, he mentioned the episode. He had had no symptoms in the interim, but the physician ordered an exercise stress test.
Six minutes into a standard Bruce protocol, the patient experienced the same chest tightness, accompanied by 1-mm ST-segment depressions in leads II, III, and aVF. He was then referred to a cardiologist, who recommended catheterization.
Catheterization demonstrated a 95% stenosis of the right coronary artery with nonsignificant stenoses of the left anterior descending and circumflex arteries. A drug-eluting stent was placed in the right coronary artery, with no residual stenosis.
Did this intervention likely prevent an MI and perhaps save the man’s life?
HOW MYOCARDIAL INFARCTION HAPPENS
Understanding the pathogenesis of MI is critical to having realistic expectations of the benefits of stent placement.
Doctors often describe coronary atherosclerosis as a plumbing problem, where deposits of cholesterol and fat build up in arterial walls, clogging the pipes and eventually causing a heart attack. This analogy, which has been around since the 1950s, is easy to for patients to grasp and has been popularized in the press and internalized by the public—as one patient with a 95% stenosis put it, “I was 95% dead.” In that model, angioplasty and stenting can resolve the blockage and “fix” the problem, much as a plumber can clear your pipes with a Roto-Rooter.
Despite the visual appeal of this model,1 it doesn’t accurately convey what we know about the pathophysiology of coronary artery disease. Instead of a gradual buildup of fatty deposits, low-density lipoprotein cholesterol particles infiltrate arterial walls and trigger an inflammatory reaction as they are engulfed by macrophages, leading to a cascade of cytokines and recruitment of more inflammatory cells.2 This immune response can eventually cause the rupture of the plaque’s fibrous cap, triggering thrombosis and infarction, often at a site of insignificant stenosis.
In this new model, coronary artery disease is primarily a problem of inflammation distributed throughout the vasculature, rather than a mechanical problem localized to the site of a significant stenosis.
Significant stenosis does not equal unstable plaque
Not all plaques are equally likely to rupture. Stable plaques tend to be long-standing and calcified, with a thick fibrous cap. A stable plaque causing a 95% stenosis may cause symptoms with exertion, but it is unlikely to cause infarction.3 Conversely, rupture-prone plaques may cause little stenosis, but a large and dangerous plaque may be lurking beneath the thin fibrous cap.
Relying on angiography can be misleading. Treating all significant stenoses improves blood flow, but does not reduce the risk of infarction, because infarction most often occurs in areas where the lumen is not obstructed. A plaque causing only 30% stenosis can suddenly rupture, causing thrombosis and complete occlusion.
The current model explains why PCI is no better than optimal medical therapy (ie, risk factor modification, antiplatelet therapy with aspirin, and a statin). Diet, exercise, smoking cessation, and statins target inflammatory processes and lower low-density lipoprotein cholesterol levels, while aspirin prevents platelet aggregation, among other likely actions.
The model also explains why coronary artery bypass grafting reduces the risk of MI and death in patients with left main or 3-vessel disease. A patient with generalized coronary artery disease has multiple lesions, many of which do not cause significant stenoses. PCI corrects only a single stenosis, whereas coronary artery bypass grafting circumvents all the vulnerable plaques in a vessel.
THE LANDMARK COURAGE TRIAL
Published in 2007, the Clinical Outcomes Utilizing Revascularization and Aggressive Drug Evaluation (COURAGE) trial4 randomized more than 2,000 patients to receive either optimal medical therapy plus PCI or optimal medical therapy alone. The primary outcome was a composite of death from any cause and nonfatal MI. Patients were followed for at least 3 years, and some for as long as 7 years.
There was an initial small upward spike in the primary outcome in the PCI arm due to periprocedural events. By 5 years, the outcomes of the 2 arms converged and then stayed the same for up to 15 years.5 The authors concluded that PCI conferred no benefit over optimal medical therapy in the risk of death or MI.
Some doctors dismiss the study because of its stringent entry criteria—of 35,539 patients assessed, only 3,071 met the eligibility criteria. However, the entry criteria were meant to identify patients most likely to benefit from PCI. Many patients who undergo PCI today would not have qualified for the study because they lack objective evidence of ischemia.6 To enroll, patients needed a proximal stenosis of at least 70% and objective evidence of ischemia or a coronary stenosis of more than 80% and classic angina. Exclusion criteria disqualified few patients: Canadian Cardiovascular Society class IV angina (ie, angina evoked from minimal activity or at rest); a markedly positive stress test (substantial ST-segment depression or hypotension during stage I of the Bruce protocol); refractory heart failure or cardiogenic shock; an ejection fraction of less than 30%; revascularization within the past 6 months; and coronary anatomy unsuitable for PCI.
OTHER TRIALS SUPPORT COURAGE FINDINGS
Although COURAGE was hailed as a landmark trial, it largely supported the results of previous studies. A meta-analysis of PCI vs optimal medical therapy published in 2005 found no significant differences in death, cardiac death, MI, or nonfatal MI.7 MI was actually slightly more common in the PCI group due to the increased risk of MI during the periprocedural period.
Nor has the evidence from COURAGE discouraged additional studies of the same topic. Despite consistent findings that fit with our understanding of coronary disease as inflammation, we continue to conduct studies aimed at addressing significant stenosis, as if that was the problem. Thus, there have been studies of angioplasty alone, followed by studies of bare-metal stents and then drug-eluting stents.
In 2009, Trikalinos et al published a review of 61 randomized controlled trials comprising more than 25,000 patients with stable coronary disease and comparing medical therapy and angioplasty in its various forms over the previous 20 years.8 In all direct and indirect comparisons of PCI and medical therapy, there were no improvements in rates of death or MI.
Even so, the studies continue. The most recent “improvement” was the addition of fractional flow reserve, which served as the inclusion criterion for the Fractional Flow Reserve versus Angiography for Multivessel Evaluation 2 (FAME 2) trial.9 In that study, patients with at least 1 stenosis with a fractional flow reserve less than 0.80 were randomized to PCI plus medical therapy or to medical therapy alone. The primary end point was a composite of death from any cause, MI, and urgent revascularization. Unfortunately, the study was stopped early when the primary end point was met due to a reduction in the need for urgent revascularization. There was no reduction in the rate of MI (hazard ratio 1.05, 95% confidence interval 0.51–2.19).
The reduction in urgent revascularization has also been shown consistently in past studies, but this is the weakest outcome measure because it does not equate to a reduction in the rate of MI. There is no demonstrable harm to putting off stent placement, even in functionally significant arteries, and most patients do not require a stent, even in the future.
In summary, the primary benefit of getting a stent now is a reduced likelihood of needing one later.
PCI MAY IMPROVE ANGINA FASTER
Another important finding of the COURAGE trial was that PCI improved symptoms more than optimal medical therapy.10 This is not surprising, because angina is often a direct result of a significant stenosis. What was unexpected was that even after PCI, most patients were not symptom-free. At 1 month, significantly more PCI patients were angina-free (42%) than were medical patients (33%). This translates into an absolute risk reduction of 9% or a number needed to treat of 11 to prevent 1 case of angina.
Patients in both groups improved over time, and after 3 years, the difference between the 2 groups was no longer significant: 59% in the PCI group vs 56% in the medical therapy group were angina-free.
A more recent study has raised the possibility that the improvement in angina with PCI is primarily a placebo effect. Researchers in the United Kingdom randomized patients with stable angina and at least a 70% stenosis of one vessel to either PCI or sham PCI, in which they threaded the catheter but did not deploy the stent.11 All patients received aggressive antianginal therapy before the procedure. At 6 weeks, there was improvement in angina in both groups, but no statistically significant difference between them in either exercise time or angina. Approximately half the patients in each group improved by at least 1 grade on the Canadian Cardiovascular Society angina classification, and more than 20% improved 2 grades.
This finding is not without precedent. Ligation of the internal mammary arteries, a popular treatment for angina in the 1950s, often provided dramatic relief of symptoms, until it was proven to be no better than a sham operation.12,13 More recently, a placebo-controlled trial of percutaneous laser myocardial revascularization also failed to show improvement over a sham treatment, despite promising results from a phase 1 trial.14 Together, these studies emphasize the subjective nature of angina as an outcome and call into question the routine use of PCI to relieve it.
PCI ENTAILS RISK
PCI entails a small but not inconsequential risk. During the procedure, 2% of patients develop bleeding or blood vessel damage, and another 1% die or have an MI or a stroke. In the first year after stent placement, 3% of patients have a bleeding event from the antiplatelet therapy needed for the stent, and an additional 2% develop a clot in the stent that leads to MI.15
INFORMED CONSENT IS CRITICAL
As demonstrated above, for patients with stable angina, the only evidence-based benefit of PCI over optimal medical therapy is that symptoms may respond faster. At the same time, there are costs and risks associated with the procedure. Because symptoms are subjective, patients should play a key role in deciding whether PCI is appropriate for them.
The American Medical Association states that a physician providing any treatment or procedure should disclose and discuss with patients the risks and benefits. Unfortunately, a substantial body of evidence demonstrates that this is not occurring in practice.
Patients and cardiologists have conflicting beliefs about PCI
Studies over the past 20 years demonstrate that patients with chronic stable angina consistently overestimate the benefits of PCI, with 71% to 88% believing that it will reduce their chance of death.16–19 Patients also understand that PCI can relieve their symptoms, though no study seems to have assessed the perceived magnitude of this benefit.
In contrast, when cardiologists were asked about the benefits their patients could expect from PCI, only 20% said that it would reduce mortality and 25% said it would prevent MI.18 These are still surprisingly high percentages, since the study was conducted after the COURAGE trial.
Nevertheless, these differences in perception show that cardiologists fail to successfully communicate the benefits of the procedure to their patients. Without complete information, patients cannot make informed decisions.
Cardiologists’ reasons for performing PCI
If PCI cannot improve hard outcomes like MI or death in stable coronary disease, why do cardiologists continue to perform it so frequently?
Soon after the COURAGE trial, Lin et al conducted focus groups with cardiologists to find out.20 Some said that they doubted the clinical trial evidence, given the reduction in the cardiac mortality rate over the past 30 years. Others remarked that their overriding goal is to stamp out ischemia, and that once a lesion is found by catheterization, one must proceed with PCI. This has been termed the “oculostenotic reflex,” ie, the interventionist sees coronary artery disease and immediately places a stent.
Atreya et al found objective evidence of this practice.21 In a 2016 study of 207 patients with obstructive lesions amenable to PCI, the only factors associated with medical management were those that increased the risk of the procedure: age, chronic kidney disease, distal location of the lesion, and type C lesions (the most difficult ones to treat by PCI). More important, evidence of ischemia, presence of angina, and being on optimal medical therapy or maximal antianginal therapy were not associated with PCI.
When surveyed, cardiologists offered reasons similar to those identified by Lin et al, including a positive stress test (70%) and significant myocardium at risk (50%).18 Optimal medical therapy failure was cited less often (40%). Over 30% identified relief of chest pain for patients who were not prescribed optimal medical therapy. Another 30% said that patient anxiety contributed to their decision, but patients who reported anxiety were not more likely to get PCI than those who did not.
True informed consent rarely occurs
Surveys of patients and recordings of doctor visits suggest that doctors often discuss the risks of the procedure but rarely accurately describe the benefits or mention alternative treatments, including optimal medical therapy.
Fowler et al22 surveyed 472 Medicare patients who had undergone PCI in the past year about their consent discussion, particularly regarding alternative options. Only 6% of patients recalled discussing medication as a serious option with their doctor.
In 2 published studies,23,24 we analyzed recorded conversations between doctors and patients in which angiography and PCI were discussed.
In a qualitative assessment of how cardiologists presented the rationale for PCI to patients,23 we observed that cardiologists gave an accurate presentation of the benefits in only 5% of cases. In 13% of the conversations the benefits were explicitly overstated (eg, “If you don’t do it [angiogram/PCI], what could happen? Well, you could…have a heart attack involving that area which can lead to a sudden death”). In another 35% of cases, physicians offered an implicit overstatement of the benefit by saying they could “fix” the problem (eg, “So that’s where we start thinking, Well maybe we better try to fix that [blockage]”), without specifically stating that fixing the problem would offer any benefit. Patients were left to fill in the blanks. Conversations frequently focused on the rationale for performing PCI (eg, ischemia on a stress test) and a description of the procedure, rather than on the risks and benefits.
In a quantitative study of the same data set, we assessed how often physicians addressed the 7 elements of informed decision-making as defined by Braddock et al.24
- Explaining the patient’s role in decision-making (ie, that the patient has a choice to make) was present in half of the conversations. Sometimes a doctor would simply say, “The next step is to perform catheterization.”
- Discussion of clinical issues (eg, having a blockage, stress test results) was performed in almost every case, demonstrating physicians’ comfort with that element.
- Discussing treatment alternatives occurred in only 1 in 4 conversations. This was more frequent than previously reported, and appeared most often when patients expressed hesitancy about proceeding to PCI.
- Discussing pros and cons of the alternatives was done in 42%.
- Uncertainty about the procedure (eg, that it might not relieve the angina) was expressed in only 10% of conversations.
- Assessment of patient understanding was done in 65% of cases. This included even minimal efforts (eg, “Do you have any questions?”). More advanced methods such as teach-back were never used.
- Exploration of patient preferences (eg, asking patients which treatment they prefer, or attempting to understand how angina affects a patient’s life) the final element, occurred in 73% of conversations.
Only 3% of the conversations contained all 7 elements. Even using a more relaxed definition of 3 critical elements (ie, discussing clinical issues, treatment alternatives, and pros and cons), only 13% of conversations included them all.
Discussion affects decisions
Informed decision-making is not only important because of its ethical implications. Offering patients more information was associated with their choosing not to have PCI. The probability of a patient undergoing PCI was negatively associated with 3 specific elements of informed decision-making. Patients were less likely to choose PCI if the patient’s role in decision-making was discussed (61% vs 86% chose PCI, P < .03); if alternatives were discussed (31% vs 89% chose PCI, P < .01); and if uncertainties were discussed (17% vs 80% chose PCI, P < .01).
There was also a linear relationship between the total number of elements discussed and the probability of choosing PCI: it ranged from 100% of patients choosing PCI when just 1 element was present to 3% of patients choosing PCI when all 7 elements were present. The relationship is not entirely causal, since doctors were more likely to talk about alternatives and risks if patients hesitated and raised questions. Cautious patients received more information.
From these observational studies, we know that physicians do not generally communicate the benefits of PCI, and patients make incorrect assumptions about the benefits they can expect. We know that those who receive more information are less likely to choose PCI, but what would happen if patients were randomly assigned to receive complete information?
An online survey
We conducted an online survey of more than 1,000 participants over age 50 who had never undergone PCI, asking them to imagine visiting a cardiologist after having a positive stress test for stable chest pain.25 Three intervention groups read different scenarios couched as information provided by their cardiologist:
- The “standard care” group received no specific information about the effects of PCI on the risk of myocardial infarction
- The “specific information” group was specifically told that PCI does not reduce the risk of myocardial infarction
- The “explanatory information” group was told how medications work and why PCI does not reduce the risk of myocardial infarction.
All 3 groups received information about the risks of PCI, its role in reducing angina, and the risks and benefits of optimal medical therapy.
After reading their scenario, all participants completed an identical questionnaire, which asked if they would opt for PCI, medical therapy, or both. Overall, 55% chose PCI, ranging from 70% in the standard care group to 46% in the group given explanatory information. Rates in the specific-information and explanatory-information groups were not statistically different from each other, but both were significantly different from that in the standard-care group. Interestingly, the more information patients were given about PCI, the more likely they were to choose optimal medical therapy.
After reading the scenario, participants were also asked if PCI would “prevent a heart attack.” Of those who received standard care, 71% endorsed that belief, which is remarkably similar to studies of real patients who have received standard care. In contrast, only 39% of those given specific information and 31% given explanatory information held that belief. Moreover, the belief that PCI prevented MI was the strongest predictor of choosing PCI (odds ratio 5.82, 95% confidence interval 4.13–8.26).25
Interestingly, 52% of the standard care group falsely remembered that the doctor had told them that PCI would prevent an MI, even though the doctor said nothing about it one way or the other. It appears that participants were projecting their own beliefs onto the encounter. This highlights the importance of providing full information to patients who are considering this procedure.
TOWARD SHARED DECISION-MAKING
Shared decision-making is a process in which physicians enter into a partnership with a patient, offer information, elicit the patient’s preferences, and then come to a decision in concert with the patient.
Although many decisions can and should involve elements of shared decision-making, the decision to proceed with PCI for stable angina is particularly well-suited to shared decision-making. This is because the benefit of PCI depends on the value a patient attaches to being free of angina sooner. Since there is no difference in the risk of MI or death, the patient must decide if the risks of the procedure and the inconvenience of taking dual antiplatelet therapy are worth the benefit of improving symptoms faster. Presumably, patients who have more severe symptoms or experienced side effects from antianginal therapy would be more likely to choose PCI.
Despite having substantial experience educating patients, most physicians are unfamiliar with the process of shared decision-making. In particular, the process of eliciting preferences is often overlooked.
To address this issue, researchers at the Mayo Clinic developed a decision aid that compares PCI plus optimal medical therapy vs optimal medical therapy alone in an easily understandable information card.15 On one side, the 2 options are clearly stated, with the magnitude of symptom improvement over time graphically illustrated and the statement, “NO DIFFERENCE in heart attack or death,” prominently displayed. The back of the card discusses the risks of each option in easily understood tables.
The decision aid was compared with standard care in a randomized trial involving patients who were referred for catheterization and possible PCI.26 The decision aid improved patients’ overall knowledge about PCI. In particular, 60% of those who used the decision aid knew that PCI did not prevent death or MI vs 40% of usual-care patients—results similar to those of the online experiment.
Interestingly, the decision about whether to undergo PCI did not differ significantly between the 2 groups, although there was a trend toward more patients in the decision-aid group choosing medical therapy alone (53%) vs the standard-care patients (39%).
To understand why the decision aid did not make more of a difference, the investigators performed qualitative interviews of the cardiologists in the study.27 One theme was the timing of the intervention. Patients using the decision aid had already been referred for catheterization, and some felt the process should have occurred earlier. Engaging in shared decision-making with a general cardiologist before referral could help to improve the quality of patient decisions.
Cardiologists also noted the difficulty in changing their work flow to incorporate the decision aid. Although some embraced the idea of shared decision-making, others were concerned that many patients could not participate, and there was confusion about the difference between an educational tool, which could be used by a patient alone, and a decision aid, which is meant to generate discussion between the doctor and patient. Some expressed interest in using the tool in the future.
These findings serve to emphasize that providing information alone is not enough. If the physician does not “buy in” to the idea of shared decision-making, it will not occur.
PRACTICE IMPLICATIONS
Based on the pathophysiology of coronary artery disease and the results of multiple randomized controlled trials, it is evident that PCI does not prevent heart attacks in patients with chronic stable angina. However, most patients who undergo PCI are unaware of this and therefore do not truly give informed consent. In the absence of explicit information to the contrary, most patients with stable angina assume that PCI prevents MI and thus are biased toward choosing PCI.
Even minimal amounts of explicit information can partially overcome that bias and influence decision-making. In particular, explaining why PCI does not prevent MI was the most effective means of overcoming the bias.
To this end, shared decision aids may help physicians to engage in shared decision-making. Shared decision-making is most likely to occur if physicians are trained in the concept of shared decision-making, are committed to practicing it, and can fit it into their work flow. Ideally, this would occur in the office of a general cardiologist before referral for PCI.
For those practicing in accountable-care organizations, Medicare has recently introduced the shared decision-making model for 6 preference-sensitive conditions, including stable ischemic heart disease. Participants in this program will have the opportunity to receive payments for shared decision-making services and to share in any savings that result from reduced use of resources. Use of these tools holds the promise for providing more patient-centered care at lower cost.
- Jones DS. Visions of a cure. Visualization, clinical trials, and controversies in cardiac therapeutics, 1968–1998. Isis 2000; 91:504–541.
- Hansson G. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 2005; 352:1685–1695.
- Stone GW, Maehara A, Lansky AJ, et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med 2011; 364:226–235.
- Boden WE, O’Rourke RA, Teo KK, et al. Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med 2007; 356:1503–1516.
- Sedlis SP, Hartigan PM, Teo KK, et al. Effect of PCI on long-term survival in patients with stable ischemic heart disease. N Engl J Med 2015; 373:1937–1946.
- Lin GA, Dudley RA, Lucas FL, Malenka DJ, Vittinghoff E, Redberg RF. Frequency of stress testing to document ischemia prior to elective percutaneous coronary intervention. JAMA 2008; 300:1765–1773.
- Katritsis DG, Ioannidis JP. Percutaneous coronary intervention versus conservative therapy in nonacute coronary artery disease: a meta-analysis. Circulation 2005; 111:2906–2912.
- Trikalinos TA, Alsheikh-Ali AA, Tatsioni A, Nallamothu BK, Kent DM. Percutaneous coronary interventions for non-acute coronary artery disease: a quantitative 20-year synopsis and a network meta-analysis. Lancet 2009; 373:911–918.
- De Bruyne B, Pijls NHJ, Kalesan B, et al. Fractional flow reserve–guided PCI versus medical therapy in stable coronary disease. N Engl J Med 2012; 367:991–1001.
- Weintraub WS, Spertus JA, Kolm P, et al. Effect of PCI on quality of life in patients with stable coronary disease. N Engl J Med 2008; 359:677–687.
- Al-Lamee R, Thompson D, Dehbi H-M, et al, on behalf of the ORBITA Investigators. Percutaneous coronary intervention in stable angina (ORBITA): a double-blind, randomised controlled trial. Lancet. Published online November 2, 2017. http://dx.doi.org/10.1016/S0140-6736(17)32714-9. Accessed November 10, 2017.
- Cobb LA, Thomas GI, Dillard DH, et al. An evaluation of internal mammary-artery ligation by a double-blind technic. N Engl J Med 1959; 260:1115–1118.
- Dimond EG, Fittle F, Crockett JE. Comparison of internal mammary artery ligation and sham operation for angina pectoris. Am J Cardiol 1960; 5:483-486.
- Leon MB, Kornowski R, Downey WE, et al. A blinded, randomized placebo-controlled trial of percutaneous laser myocardial revascularization to improve angina symptoms in patients with severe coronary disease. J Am Coll Cardiol 2005; 46:1812–1819.
- Coylewright M, Shepel K, Leblanc A, et al. Shared decision making in patients with stable coronary artery disease: PCI choice. PLoS One 2012; 7:e49827.
- Holmboe ES, Fiellin DA, Cusanelli E, Remetz M, Krumholz HM. Perceptions of benefit and risk of patients undergoing first-time elective percutaneous coronary revascularization. J Gen Intern Med 2000; 15:632–637.
- Kee F, McDonald P, Gaffney B. Risks and benefits of coronary angioplasty: the patients perspective: a preliminary study. Qual Health Care 1997; 6:131–139.
- Rothberg MB, Sivalingam SK, Ashraf J, et al. Patients’ and cardiologists’ perceptions of the benefits of percutaneous coronary intervention for stable coronary disease. Ann Intern Med 2010; 153:307–313.
- Whittle J, Conigliaro J, Good CB, Kelley ME, Skanderson M. Understanding of the benefits of coronary revascularization procedures among patients who are offered such procedures. Am Heart J 2007; 154:662–668.
- Lin GA, Dudley RA, Redberg RF. Cardiologists’ use of percutaneous coronary interventions for stable coronary artery disease. Arch Intern Med 2007; 167:1604–1609.
- Atreya AR, Sivalingam SK, Arora S, et al. Predictors of medical management in patients undergoing elective cardiac catheterization for chronic ischemic heart disease. Clin Cardiol 2016; 39:207–214.
- Fowler FJ Jr, Gallagher PM, Bynum JP, Barry MJ, Lucas FL, Skinner JS. Decision-making process reported by Medicare patients who had coronary artery stenting or surgery for prostate cancer. J Gen Intern Med 2012; 27:911–916.
- Goff SL, Mazor KM, Ting HH, Kleppel R, Rothberg MB. How cardiologists present the benefits of percutaneous coronary interventions to patients with stable angina: a qualitative analysis. JAMA Intern Med 2014; 174:1614–1621.
- Braddock CH 3rd, Edwards KA, Hasenberg NM, Laidley TL, Levinson W. Informed decision making in outpatient practice: time to get back to basics. JAMA 1999; 282:2313–2320.
- Rothberg MB, Scherer L, Kashef MA, et al. The effect of information presentation on beliefs about the benefits of elective percutaneous coronary intervention. JAMA Intern Med 2014; 174:1623–1629.
- Coylewright M, Dick S, Zmolek B, et al. PCI choice decision aid for stable coronary artery disease: a randomized trial. Circ Cardiovasc Qual Outcomes 2016; 9:767–776.
- Coylewright M, O’Neill ES, Dick S, Grande SW. PCI choice: cardiovascular clinicians’ perceptions of shared decision making in stable coronary artery disease. Patient Educ Couns 2017; 100:1136–1143.
Multiple randomized controlled trials have compared percutaneous coronary intervention (PCI) vs optimal medical therapy for patients with chronic stable angina. All have consistently shown that PCI does not reduce the risk of death or even myocardial infarction (MI) but that it may relieve angina temporarily. Nevertheless, PCI is still commonly performed for patients with stable coronary disease, often in the absence of angina, and patients mistakenly believe the procedure is life-saving. Cardiologists may not be aware of patients’ misperceptions, or worse, may encourage them. In either case, if patients do not understand the benefits of the procedure, they cannot give informed consent.
This article reviews the pathophysiology of coronary artery disease, evidence from clinical trials of the value of PCI for chronic stable angina, patient and physician perceptions of PCI, and ways to promote patient-centered, shared decision-making.
CLINICAL CASE: EXERTIONAL ANGINA
While climbing 4 flights of stairs, a 55-year-old man noticed tightness in his chest, which lasted for 5 minutes and resolved spontaneously. Several weeks later, when visiting his primary care physician, he mentioned the episode. He had had no symptoms in the interim, but the physician ordered an exercise stress test.
Six minutes into a standard Bruce protocol, the patient experienced the same chest tightness, accompanied by 1-mm ST-segment depressions in leads II, III, and aVF. He was then referred to a cardiologist, who recommended catheterization.
Catheterization demonstrated a 95% stenosis of the right coronary artery with nonsignificant stenoses of the left anterior descending and circumflex arteries. A drug-eluting stent was placed in the right coronary artery, with no residual stenosis.
Did this intervention likely prevent an MI and perhaps save the man’s life?
HOW MYOCARDIAL INFARCTION HAPPENS
Understanding the pathogenesis of MI is critical to having realistic expectations of the benefits of stent placement.
Doctors often describe coronary atherosclerosis as a plumbing problem, where deposits of cholesterol and fat build up in arterial walls, clogging the pipes and eventually causing a heart attack. This analogy, which has been around since the 1950s, is easy to for patients to grasp and has been popularized in the press and internalized by the public—as one patient with a 95% stenosis put it, “I was 95% dead.” In that model, angioplasty and stenting can resolve the blockage and “fix” the problem, much as a plumber can clear your pipes with a Roto-Rooter.
Despite the visual appeal of this model,1 it doesn’t accurately convey what we know about the pathophysiology of coronary artery disease. Instead of a gradual buildup of fatty deposits, low-density lipoprotein cholesterol particles infiltrate arterial walls and trigger an inflammatory reaction as they are engulfed by macrophages, leading to a cascade of cytokines and recruitment of more inflammatory cells.2 This immune response can eventually cause the rupture of the plaque’s fibrous cap, triggering thrombosis and infarction, often at a site of insignificant stenosis.
In this new model, coronary artery disease is primarily a problem of inflammation distributed throughout the vasculature, rather than a mechanical problem localized to the site of a significant stenosis.
Significant stenosis does not equal unstable plaque
Not all plaques are equally likely to rupture. Stable plaques tend to be long-standing and calcified, with a thick fibrous cap. A stable plaque causing a 95% stenosis may cause symptoms with exertion, but it is unlikely to cause infarction.3 Conversely, rupture-prone plaques may cause little stenosis, but a large and dangerous plaque may be lurking beneath the thin fibrous cap.
Relying on angiography can be misleading. Treating all significant stenoses improves blood flow, but does not reduce the risk of infarction, because infarction most often occurs in areas where the lumen is not obstructed. A plaque causing only 30% stenosis can suddenly rupture, causing thrombosis and complete occlusion.
The current model explains why PCI is no better than optimal medical therapy (ie, risk factor modification, antiplatelet therapy with aspirin, and a statin). Diet, exercise, smoking cessation, and statins target inflammatory processes and lower low-density lipoprotein cholesterol levels, while aspirin prevents platelet aggregation, among other likely actions.
The model also explains why coronary artery bypass grafting reduces the risk of MI and death in patients with left main or 3-vessel disease. A patient with generalized coronary artery disease has multiple lesions, many of which do not cause significant stenoses. PCI corrects only a single stenosis, whereas coronary artery bypass grafting circumvents all the vulnerable plaques in a vessel.
THE LANDMARK COURAGE TRIAL
Published in 2007, the Clinical Outcomes Utilizing Revascularization and Aggressive Drug Evaluation (COURAGE) trial4 randomized more than 2,000 patients to receive either optimal medical therapy plus PCI or optimal medical therapy alone. The primary outcome was a composite of death from any cause and nonfatal MI. Patients were followed for at least 3 years, and some for as long as 7 years.
There was an initial small upward spike in the primary outcome in the PCI arm due to periprocedural events. By 5 years, the outcomes of the 2 arms converged and then stayed the same for up to 15 years.5 The authors concluded that PCI conferred no benefit over optimal medical therapy in the risk of death or MI.
Some doctors dismiss the study because of its stringent entry criteria—of 35,539 patients assessed, only 3,071 met the eligibility criteria. However, the entry criteria were meant to identify patients most likely to benefit from PCI. Many patients who undergo PCI today would not have qualified for the study because they lack objective evidence of ischemia.6 To enroll, patients needed a proximal stenosis of at least 70% and objective evidence of ischemia or a coronary stenosis of more than 80% and classic angina. Exclusion criteria disqualified few patients: Canadian Cardiovascular Society class IV angina (ie, angina evoked from minimal activity or at rest); a markedly positive stress test (substantial ST-segment depression or hypotension during stage I of the Bruce protocol); refractory heart failure or cardiogenic shock; an ejection fraction of less than 30%; revascularization within the past 6 months; and coronary anatomy unsuitable for PCI.
OTHER TRIALS SUPPORT COURAGE FINDINGS
Although COURAGE was hailed as a landmark trial, it largely supported the results of previous studies. A meta-analysis of PCI vs optimal medical therapy published in 2005 found no significant differences in death, cardiac death, MI, or nonfatal MI.7 MI was actually slightly more common in the PCI group due to the increased risk of MI during the periprocedural period.
Nor has the evidence from COURAGE discouraged additional studies of the same topic. Despite consistent findings that fit with our understanding of coronary disease as inflammation, we continue to conduct studies aimed at addressing significant stenosis, as if that was the problem. Thus, there have been studies of angioplasty alone, followed by studies of bare-metal stents and then drug-eluting stents.
In 2009, Trikalinos et al published a review of 61 randomized controlled trials comprising more than 25,000 patients with stable coronary disease and comparing medical therapy and angioplasty in its various forms over the previous 20 years.8 In all direct and indirect comparisons of PCI and medical therapy, there were no improvements in rates of death or MI.
Even so, the studies continue. The most recent “improvement” was the addition of fractional flow reserve, which served as the inclusion criterion for the Fractional Flow Reserve versus Angiography for Multivessel Evaluation 2 (FAME 2) trial.9 In that study, patients with at least 1 stenosis with a fractional flow reserve less than 0.80 were randomized to PCI plus medical therapy or to medical therapy alone. The primary end point was a composite of death from any cause, MI, and urgent revascularization. Unfortunately, the study was stopped early when the primary end point was met due to a reduction in the need for urgent revascularization. There was no reduction in the rate of MI (hazard ratio 1.05, 95% confidence interval 0.51–2.19).
The reduction in urgent revascularization has also been shown consistently in past studies, but this is the weakest outcome measure because it does not equate to a reduction in the rate of MI. There is no demonstrable harm to putting off stent placement, even in functionally significant arteries, and most patients do not require a stent, even in the future.
In summary, the primary benefit of getting a stent now is a reduced likelihood of needing one later.
PCI MAY IMPROVE ANGINA FASTER
Another important finding of the COURAGE trial was that PCI improved symptoms more than optimal medical therapy.10 This is not surprising, because angina is often a direct result of a significant stenosis. What was unexpected was that even after PCI, most patients were not symptom-free. At 1 month, significantly more PCI patients were angina-free (42%) than were medical patients (33%). This translates into an absolute risk reduction of 9% or a number needed to treat of 11 to prevent 1 case of angina.
Patients in both groups improved over time, and after 3 years, the difference between the 2 groups was no longer significant: 59% in the PCI group vs 56% in the medical therapy group were angina-free.
A more recent study has raised the possibility that the improvement in angina with PCI is primarily a placebo effect. Researchers in the United Kingdom randomized patients with stable angina and at least a 70% stenosis of one vessel to either PCI or sham PCI, in which they threaded the catheter but did not deploy the stent.11 All patients received aggressive antianginal therapy before the procedure. At 6 weeks, there was improvement in angina in both groups, but no statistically significant difference between them in either exercise time or angina. Approximately half the patients in each group improved by at least 1 grade on the Canadian Cardiovascular Society angina classification, and more than 20% improved 2 grades.
This finding is not without precedent. Ligation of the internal mammary arteries, a popular treatment for angina in the 1950s, often provided dramatic relief of symptoms, until it was proven to be no better than a sham operation.12,13 More recently, a placebo-controlled trial of percutaneous laser myocardial revascularization also failed to show improvement over a sham treatment, despite promising results from a phase 1 trial.14 Together, these studies emphasize the subjective nature of angina as an outcome and call into question the routine use of PCI to relieve it.
PCI ENTAILS RISK
PCI entails a small but not inconsequential risk. During the procedure, 2% of patients develop bleeding or blood vessel damage, and another 1% die or have an MI or a stroke. In the first year after stent placement, 3% of patients have a bleeding event from the antiplatelet therapy needed for the stent, and an additional 2% develop a clot in the stent that leads to MI.15
INFORMED CONSENT IS CRITICAL
As demonstrated above, for patients with stable angina, the only evidence-based benefit of PCI over optimal medical therapy is that symptoms may respond faster. At the same time, there are costs and risks associated with the procedure. Because symptoms are subjective, patients should play a key role in deciding whether PCI is appropriate for them.
The American Medical Association states that a physician providing any treatment or procedure should disclose and discuss with patients the risks and benefits. Unfortunately, a substantial body of evidence demonstrates that this is not occurring in practice.
Patients and cardiologists have conflicting beliefs about PCI
Studies over the past 20 years demonstrate that patients with chronic stable angina consistently overestimate the benefits of PCI, with 71% to 88% believing that it will reduce their chance of death.16–19 Patients also understand that PCI can relieve their symptoms, though no study seems to have assessed the perceived magnitude of this benefit.
In contrast, when cardiologists were asked about the benefits their patients could expect from PCI, only 20% said that it would reduce mortality and 25% said it would prevent MI.18 These are still surprisingly high percentages, since the study was conducted after the COURAGE trial.
Nevertheless, these differences in perception show that cardiologists fail to successfully communicate the benefits of the procedure to their patients. Without complete information, patients cannot make informed decisions.
Cardiologists’ reasons for performing PCI
If PCI cannot improve hard outcomes like MI or death in stable coronary disease, why do cardiologists continue to perform it so frequently?
Soon after the COURAGE trial, Lin et al conducted focus groups with cardiologists to find out.20 Some said that they doubted the clinical trial evidence, given the reduction in the cardiac mortality rate over the past 30 years. Others remarked that their overriding goal is to stamp out ischemia, and that once a lesion is found by catheterization, one must proceed with PCI. This has been termed the “oculostenotic reflex,” ie, the interventionist sees coronary artery disease and immediately places a stent.
Atreya et al found objective evidence of this practice.21 In a 2016 study of 207 patients with obstructive lesions amenable to PCI, the only factors associated with medical management were those that increased the risk of the procedure: age, chronic kidney disease, distal location of the lesion, and type C lesions (the most difficult ones to treat by PCI). More important, evidence of ischemia, presence of angina, and being on optimal medical therapy or maximal antianginal therapy were not associated with PCI.
When surveyed, cardiologists offered reasons similar to those identified by Lin et al, including a positive stress test (70%) and significant myocardium at risk (50%).18 Optimal medical therapy failure was cited less often (40%). Over 30% identified relief of chest pain for patients who were not prescribed optimal medical therapy. Another 30% said that patient anxiety contributed to their decision, but patients who reported anxiety were not more likely to get PCI than those who did not.
True informed consent rarely occurs
Surveys of patients and recordings of doctor visits suggest that doctors often discuss the risks of the procedure but rarely accurately describe the benefits or mention alternative treatments, including optimal medical therapy.
Fowler et al22 surveyed 472 Medicare patients who had undergone PCI in the past year about their consent discussion, particularly regarding alternative options. Only 6% of patients recalled discussing medication as a serious option with their doctor.
In 2 published studies,23,24 we analyzed recorded conversations between doctors and patients in which angiography and PCI were discussed.
In a qualitative assessment of how cardiologists presented the rationale for PCI to patients,23 we observed that cardiologists gave an accurate presentation of the benefits in only 5% of cases. In 13% of the conversations the benefits were explicitly overstated (eg, “If you don’t do it [angiogram/PCI], what could happen? Well, you could…have a heart attack involving that area which can lead to a sudden death”). In another 35% of cases, physicians offered an implicit overstatement of the benefit by saying they could “fix” the problem (eg, “So that’s where we start thinking, Well maybe we better try to fix that [blockage]”), without specifically stating that fixing the problem would offer any benefit. Patients were left to fill in the blanks. Conversations frequently focused on the rationale for performing PCI (eg, ischemia on a stress test) and a description of the procedure, rather than on the risks and benefits.
In a quantitative study of the same data set, we assessed how often physicians addressed the 7 elements of informed decision-making as defined by Braddock et al.24
- Explaining the patient’s role in decision-making (ie, that the patient has a choice to make) was present in half of the conversations. Sometimes a doctor would simply say, “The next step is to perform catheterization.”
- Discussion of clinical issues (eg, having a blockage, stress test results) was performed in almost every case, demonstrating physicians’ comfort with that element.
- Discussing treatment alternatives occurred in only 1 in 4 conversations. This was more frequent than previously reported, and appeared most often when patients expressed hesitancy about proceeding to PCI.
- Discussing pros and cons of the alternatives was done in 42%.
- Uncertainty about the procedure (eg, that it might not relieve the angina) was expressed in only 10% of conversations.
- Assessment of patient understanding was done in 65% of cases. This included even minimal efforts (eg, “Do you have any questions?”). More advanced methods such as teach-back were never used.
- Exploration of patient preferences (eg, asking patients which treatment they prefer, or attempting to understand how angina affects a patient’s life) the final element, occurred in 73% of conversations.
Only 3% of the conversations contained all 7 elements. Even using a more relaxed definition of 3 critical elements (ie, discussing clinical issues, treatment alternatives, and pros and cons), only 13% of conversations included them all.
Discussion affects decisions
Informed decision-making is not only important because of its ethical implications. Offering patients more information was associated with their choosing not to have PCI. The probability of a patient undergoing PCI was negatively associated with 3 specific elements of informed decision-making. Patients were less likely to choose PCI if the patient’s role in decision-making was discussed (61% vs 86% chose PCI, P < .03); if alternatives were discussed (31% vs 89% chose PCI, P < .01); and if uncertainties were discussed (17% vs 80% chose PCI, P < .01).
There was also a linear relationship between the total number of elements discussed and the probability of choosing PCI: it ranged from 100% of patients choosing PCI when just 1 element was present to 3% of patients choosing PCI when all 7 elements were present. The relationship is not entirely causal, since doctors were more likely to talk about alternatives and risks if patients hesitated and raised questions. Cautious patients received more information.
From these observational studies, we know that physicians do not generally communicate the benefits of PCI, and patients make incorrect assumptions about the benefits they can expect. We know that those who receive more information are less likely to choose PCI, but what would happen if patients were randomly assigned to receive complete information?
An online survey
We conducted an online survey of more than 1,000 participants over age 50 who had never undergone PCI, asking them to imagine visiting a cardiologist after having a positive stress test for stable chest pain.25 Three intervention groups read different scenarios couched as information provided by their cardiologist:
- The “standard care” group received no specific information about the effects of PCI on the risk of myocardial infarction
- The “specific information” group was specifically told that PCI does not reduce the risk of myocardial infarction
- The “explanatory information” group was told how medications work and why PCI does not reduce the risk of myocardial infarction.
All 3 groups received information about the risks of PCI, its role in reducing angina, and the risks and benefits of optimal medical therapy.
After reading their scenario, all participants completed an identical questionnaire, which asked if they would opt for PCI, medical therapy, or both. Overall, 55% chose PCI, ranging from 70% in the standard care group to 46% in the group given explanatory information. Rates in the specific-information and explanatory-information groups were not statistically different from each other, but both were significantly different from that in the standard-care group. Interestingly, the more information patients were given about PCI, the more likely they were to choose optimal medical therapy.
After reading the scenario, participants were also asked if PCI would “prevent a heart attack.” Of those who received standard care, 71% endorsed that belief, which is remarkably similar to studies of real patients who have received standard care. In contrast, only 39% of those given specific information and 31% given explanatory information held that belief. Moreover, the belief that PCI prevented MI was the strongest predictor of choosing PCI (odds ratio 5.82, 95% confidence interval 4.13–8.26).25
Interestingly, 52% of the standard care group falsely remembered that the doctor had told them that PCI would prevent an MI, even though the doctor said nothing about it one way or the other. It appears that participants were projecting their own beliefs onto the encounter. This highlights the importance of providing full information to patients who are considering this procedure.
TOWARD SHARED DECISION-MAKING
Shared decision-making is a process in which physicians enter into a partnership with a patient, offer information, elicit the patient’s preferences, and then come to a decision in concert with the patient.
Although many decisions can and should involve elements of shared decision-making, the decision to proceed with PCI for stable angina is particularly well-suited to shared decision-making. This is because the benefit of PCI depends on the value a patient attaches to being free of angina sooner. Since there is no difference in the risk of MI or death, the patient must decide if the risks of the procedure and the inconvenience of taking dual antiplatelet therapy are worth the benefit of improving symptoms faster. Presumably, patients who have more severe symptoms or experienced side effects from antianginal therapy would be more likely to choose PCI.
Despite having substantial experience educating patients, most physicians are unfamiliar with the process of shared decision-making. In particular, the process of eliciting preferences is often overlooked.
To address this issue, researchers at the Mayo Clinic developed a decision aid that compares PCI plus optimal medical therapy vs optimal medical therapy alone in an easily understandable information card.15 On one side, the 2 options are clearly stated, with the magnitude of symptom improvement over time graphically illustrated and the statement, “NO DIFFERENCE in heart attack or death,” prominently displayed. The back of the card discusses the risks of each option in easily understood tables.
The decision aid was compared with standard care in a randomized trial involving patients who were referred for catheterization and possible PCI.26 The decision aid improved patients’ overall knowledge about PCI. In particular, 60% of those who used the decision aid knew that PCI did not prevent death or MI vs 40% of usual-care patients—results similar to those of the online experiment.
Interestingly, the decision about whether to undergo PCI did not differ significantly between the 2 groups, although there was a trend toward more patients in the decision-aid group choosing medical therapy alone (53%) vs the standard-care patients (39%).
To understand why the decision aid did not make more of a difference, the investigators performed qualitative interviews of the cardiologists in the study.27 One theme was the timing of the intervention. Patients using the decision aid had already been referred for catheterization, and some felt the process should have occurred earlier. Engaging in shared decision-making with a general cardiologist before referral could help to improve the quality of patient decisions.
Cardiologists also noted the difficulty in changing their work flow to incorporate the decision aid. Although some embraced the idea of shared decision-making, others were concerned that many patients could not participate, and there was confusion about the difference between an educational tool, which could be used by a patient alone, and a decision aid, which is meant to generate discussion between the doctor and patient. Some expressed interest in using the tool in the future.
These findings serve to emphasize that providing information alone is not enough. If the physician does not “buy in” to the idea of shared decision-making, it will not occur.
PRACTICE IMPLICATIONS
Based on the pathophysiology of coronary artery disease and the results of multiple randomized controlled trials, it is evident that PCI does not prevent heart attacks in patients with chronic stable angina. However, most patients who undergo PCI are unaware of this and therefore do not truly give informed consent. In the absence of explicit information to the contrary, most patients with stable angina assume that PCI prevents MI and thus are biased toward choosing PCI.
Even minimal amounts of explicit information can partially overcome that bias and influence decision-making. In particular, explaining why PCI does not prevent MI was the most effective means of overcoming the bias.
To this end, shared decision aids may help physicians to engage in shared decision-making. Shared decision-making is most likely to occur if physicians are trained in the concept of shared decision-making, are committed to practicing it, and can fit it into their work flow. Ideally, this would occur in the office of a general cardiologist before referral for PCI.
For those practicing in accountable-care organizations, Medicare has recently introduced the shared decision-making model for 6 preference-sensitive conditions, including stable ischemic heart disease. Participants in this program will have the opportunity to receive payments for shared decision-making services and to share in any savings that result from reduced use of resources. Use of these tools holds the promise for providing more patient-centered care at lower cost.
Multiple randomized controlled trials have compared percutaneous coronary intervention (PCI) vs optimal medical therapy for patients with chronic stable angina. All have consistently shown that PCI does not reduce the risk of death or even myocardial infarction (MI) but that it may relieve angina temporarily. Nevertheless, PCI is still commonly performed for patients with stable coronary disease, often in the absence of angina, and patients mistakenly believe the procedure is life-saving. Cardiologists may not be aware of patients’ misperceptions, or worse, may encourage them. In either case, if patients do not understand the benefits of the procedure, they cannot give informed consent.
This article reviews the pathophysiology of coronary artery disease, evidence from clinical trials of the value of PCI for chronic stable angina, patient and physician perceptions of PCI, and ways to promote patient-centered, shared decision-making.
CLINICAL CASE: EXERTIONAL ANGINA
While climbing 4 flights of stairs, a 55-year-old man noticed tightness in his chest, which lasted for 5 minutes and resolved spontaneously. Several weeks later, when visiting his primary care physician, he mentioned the episode. He had had no symptoms in the interim, but the physician ordered an exercise stress test.
Six minutes into a standard Bruce protocol, the patient experienced the same chest tightness, accompanied by 1-mm ST-segment depressions in leads II, III, and aVF. He was then referred to a cardiologist, who recommended catheterization.
Catheterization demonstrated a 95% stenosis of the right coronary artery with nonsignificant stenoses of the left anterior descending and circumflex arteries. A drug-eluting stent was placed in the right coronary artery, with no residual stenosis.
Did this intervention likely prevent an MI and perhaps save the man’s life?
HOW MYOCARDIAL INFARCTION HAPPENS
Understanding the pathogenesis of MI is critical to having realistic expectations of the benefits of stent placement.
Doctors often describe coronary atherosclerosis as a plumbing problem, where deposits of cholesterol and fat build up in arterial walls, clogging the pipes and eventually causing a heart attack. This analogy, which has been around since the 1950s, is easy to for patients to grasp and has been popularized in the press and internalized by the public—as one patient with a 95% stenosis put it, “I was 95% dead.” In that model, angioplasty and stenting can resolve the blockage and “fix” the problem, much as a plumber can clear your pipes with a Roto-Rooter.
Despite the visual appeal of this model,1 it doesn’t accurately convey what we know about the pathophysiology of coronary artery disease. Instead of a gradual buildup of fatty deposits, low-density lipoprotein cholesterol particles infiltrate arterial walls and trigger an inflammatory reaction as they are engulfed by macrophages, leading to a cascade of cytokines and recruitment of more inflammatory cells.2 This immune response can eventually cause the rupture of the plaque’s fibrous cap, triggering thrombosis and infarction, often at a site of insignificant stenosis.
In this new model, coronary artery disease is primarily a problem of inflammation distributed throughout the vasculature, rather than a mechanical problem localized to the site of a significant stenosis.
Significant stenosis does not equal unstable plaque
Not all plaques are equally likely to rupture. Stable plaques tend to be long-standing and calcified, with a thick fibrous cap. A stable plaque causing a 95% stenosis may cause symptoms with exertion, but it is unlikely to cause infarction.3 Conversely, rupture-prone plaques may cause little stenosis, but a large and dangerous plaque may be lurking beneath the thin fibrous cap.
Relying on angiography can be misleading. Treating all significant stenoses improves blood flow, but does not reduce the risk of infarction, because infarction most often occurs in areas where the lumen is not obstructed. A plaque causing only 30% stenosis can suddenly rupture, causing thrombosis and complete occlusion.
The current model explains why PCI is no better than optimal medical therapy (ie, risk factor modification, antiplatelet therapy with aspirin, and a statin). Diet, exercise, smoking cessation, and statins target inflammatory processes and lower low-density lipoprotein cholesterol levels, while aspirin prevents platelet aggregation, among other likely actions.
The model also explains why coronary artery bypass grafting reduces the risk of MI and death in patients with left main or 3-vessel disease. A patient with generalized coronary artery disease has multiple lesions, many of which do not cause significant stenoses. PCI corrects only a single stenosis, whereas coronary artery bypass grafting circumvents all the vulnerable plaques in a vessel.
THE LANDMARK COURAGE TRIAL
Published in 2007, the Clinical Outcomes Utilizing Revascularization and Aggressive Drug Evaluation (COURAGE) trial4 randomized more than 2,000 patients to receive either optimal medical therapy plus PCI or optimal medical therapy alone. The primary outcome was a composite of death from any cause and nonfatal MI. Patients were followed for at least 3 years, and some for as long as 7 years.
There was an initial small upward spike in the primary outcome in the PCI arm due to periprocedural events. By 5 years, the outcomes of the 2 arms converged and then stayed the same for up to 15 years.5 The authors concluded that PCI conferred no benefit over optimal medical therapy in the risk of death or MI.
Some doctors dismiss the study because of its stringent entry criteria—of 35,539 patients assessed, only 3,071 met the eligibility criteria. However, the entry criteria were meant to identify patients most likely to benefit from PCI. Many patients who undergo PCI today would not have qualified for the study because they lack objective evidence of ischemia.6 To enroll, patients needed a proximal stenosis of at least 70% and objective evidence of ischemia or a coronary stenosis of more than 80% and classic angina. Exclusion criteria disqualified few patients: Canadian Cardiovascular Society class IV angina (ie, angina evoked from minimal activity or at rest); a markedly positive stress test (substantial ST-segment depression or hypotension during stage I of the Bruce protocol); refractory heart failure or cardiogenic shock; an ejection fraction of less than 30%; revascularization within the past 6 months; and coronary anatomy unsuitable for PCI.
OTHER TRIALS SUPPORT COURAGE FINDINGS
Although COURAGE was hailed as a landmark trial, it largely supported the results of previous studies. A meta-analysis of PCI vs optimal medical therapy published in 2005 found no significant differences in death, cardiac death, MI, or nonfatal MI.7 MI was actually slightly more common in the PCI group due to the increased risk of MI during the periprocedural period.
Nor has the evidence from COURAGE discouraged additional studies of the same topic. Despite consistent findings that fit with our understanding of coronary disease as inflammation, we continue to conduct studies aimed at addressing significant stenosis, as if that was the problem. Thus, there have been studies of angioplasty alone, followed by studies of bare-metal stents and then drug-eluting stents.
In 2009, Trikalinos et al published a review of 61 randomized controlled trials comprising more than 25,000 patients with stable coronary disease and comparing medical therapy and angioplasty in its various forms over the previous 20 years.8 In all direct and indirect comparisons of PCI and medical therapy, there were no improvements in rates of death or MI.
Even so, the studies continue. The most recent “improvement” was the addition of fractional flow reserve, which served as the inclusion criterion for the Fractional Flow Reserve versus Angiography for Multivessel Evaluation 2 (FAME 2) trial.9 In that study, patients with at least 1 stenosis with a fractional flow reserve less than 0.80 were randomized to PCI plus medical therapy or to medical therapy alone. The primary end point was a composite of death from any cause, MI, and urgent revascularization. Unfortunately, the study was stopped early when the primary end point was met due to a reduction in the need for urgent revascularization. There was no reduction in the rate of MI (hazard ratio 1.05, 95% confidence interval 0.51–2.19).
The reduction in urgent revascularization has also been shown consistently in past studies, but this is the weakest outcome measure because it does not equate to a reduction in the rate of MI. There is no demonstrable harm to putting off stent placement, even in functionally significant arteries, and most patients do not require a stent, even in the future.
In summary, the primary benefit of getting a stent now is a reduced likelihood of needing one later.
PCI MAY IMPROVE ANGINA FASTER
Another important finding of the COURAGE trial was that PCI improved symptoms more than optimal medical therapy.10 This is not surprising, because angina is often a direct result of a significant stenosis. What was unexpected was that even after PCI, most patients were not symptom-free. At 1 month, significantly more PCI patients were angina-free (42%) than were medical patients (33%). This translates into an absolute risk reduction of 9% or a number needed to treat of 11 to prevent 1 case of angina.
Patients in both groups improved over time, and after 3 years, the difference between the 2 groups was no longer significant: 59% in the PCI group vs 56% in the medical therapy group were angina-free.
A more recent study has raised the possibility that the improvement in angina with PCI is primarily a placebo effect. Researchers in the United Kingdom randomized patients with stable angina and at least a 70% stenosis of one vessel to either PCI or sham PCI, in which they threaded the catheter but did not deploy the stent.11 All patients received aggressive antianginal therapy before the procedure. At 6 weeks, there was improvement in angina in both groups, but no statistically significant difference between them in either exercise time or angina. Approximately half the patients in each group improved by at least 1 grade on the Canadian Cardiovascular Society angina classification, and more than 20% improved 2 grades.
This finding is not without precedent. Ligation of the internal mammary arteries, a popular treatment for angina in the 1950s, often provided dramatic relief of symptoms, until it was proven to be no better than a sham operation.12,13 More recently, a placebo-controlled trial of percutaneous laser myocardial revascularization also failed to show improvement over a sham treatment, despite promising results from a phase 1 trial.14 Together, these studies emphasize the subjective nature of angina as an outcome and call into question the routine use of PCI to relieve it.
PCI ENTAILS RISK
PCI entails a small but not inconsequential risk. During the procedure, 2% of patients develop bleeding or blood vessel damage, and another 1% die or have an MI or a stroke. In the first year after stent placement, 3% of patients have a bleeding event from the antiplatelet therapy needed for the stent, and an additional 2% develop a clot in the stent that leads to MI.15
INFORMED CONSENT IS CRITICAL
As demonstrated above, for patients with stable angina, the only evidence-based benefit of PCI over optimal medical therapy is that symptoms may respond faster. At the same time, there are costs and risks associated with the procedure. Because symptoms are subjective, patients should play a key role in deciding whether PCI is appropriate for them.
The American Medical Association states that a physician providing any treatment or procedure should disclose and discuss with patients the risks and benefits. Unfortunately, a substantial body of evidence demonstrates that this is not occurring in practice.
Patients and cardiologists have conflicting beliefs about PCI
Studies over the past 20 years demonstrate that patients with chronic stable angina consistently overestimate the benefits of PCI, with 71% to 88% believing that it will reduce their chance of death.16–19 Patients also understand that PCI can relieve their symptoms, though no study seems to have assessed the perceived magnitude of this benefit.
In contrast, when cardiologists were asked about the benefits their patients could expect from PCI, only 20% said that it would reduce mortality and 25% said it would prevent MI.18 These are still surprisingly high percentages, since the study was conducted after the COURAGE trial.
Nevertheless, these differences in perception show that cardiologists fail to successfully communicate the benefits of the procedure to their patients. Without complete information, patients cannot make informed decisions.
Cardiologists’ reasons for performing PCI
If PCI cannot improve hard outcomes like MI or death in stable coronary disease, why do cardiologists continue to perform it so frequently?
Soon after the COURAGE trial, Lin et al conducted focus groups with cardiologists to find out.20 Some said that they doubted the clinical trial evidence, given the reduction in the cardiac mortality rate over the past 30 years. Others remarked that their overriding goal is to stamp out ischemia, and that once a lesion is found by catheterization, one must proceed with PCI. This has been termed the “oculostenotic reflex,” ie, the interventionist sees coronary artery disease and immediately places a stent.
Atreya et al found objective evidence of this practice.21 In a 2016 study of 207 patients with obstructive lesions amenable to PCI, the only factors associated with medical management were those that increased the risk of the procedure: age, chronic kidney disease, distal location of the lesion, and type C lesions (the most difficult ones to treat by PCI). More important, evidence of ischemia, presence of angina, and being on optimal medical therapy or maximal antianginal therapy were not associated with PCI.
When surveyed, cardiologists offered reasons similar to those identified by Lin et al, including a positive stress test (70%) and significant myocardium at risk (50%).18 Optimal medical therapy failure was cited less often (40%). Over 30% identified relief of chest pain for patients who were not prescribed optimal medical therapy. Another 30% said that patient anxiety contributed to their decision, but patients who reported anxiety were not more likely to get PCI than those who did not.
True informed consent rarely occurs
Surveys of patients and recordings of doctor visits suggest that doctors often discuss the risks of the procedure but rarely accurately describe the benefits or mention alternative treatments, including optimal medical therapy.
Fowler et al22 surveyed 472 Medicare patients who had undergone PCI in the past year about their consent discussion, particularly regarding alternative options. Only 6% of patients recalled discussing medication as a serious option with their doctor.
In 2 published studies,23,24 we analyzed recorded conversations between doctors and patients in which angiography and PCI were discussed.
In a qualitative assessment of how cardiologists presented the rationale for PCI to patients,23 we observed that cardiologists gave an accurate presentation of the benefits in only 5% of cases. In 13% of the conversations the benefits were explicitly overstated (eg, “If you don’t do it [angiogram/PCI], what could happen? Well, you could…have a heart attack involving that area which can lead to a sudden death”). In another 35% of cases, physicians offered an implicit overstatement of the benefit by saying they could “fix” the problem (eg, “So that’s where we start thinking, Well maybe we better try to fix that [blockage]”), without specifically stating that fixing the problem would offer any benefit. Patients were left to fill in the blanks. Conversations frequently focused on the rationale for performing PCI (eg, ischemia on a stress test) and a description of the procedure, rather than on the risks and benefits.
In a quantitative study of the same data set, we assessed how often physicians addressed the 7 elements of informed decision-making as defined by Braddock et al.24
- Explaining the patient’s role in decision-making (ie, that the patient has a choice to make) was present in half of the conversations. Sometimes a doctor would simply say, “The next step is to perform catheterization.”
- Discussion of clinical issues (eg, having a blockage, stress test results) was performed in almost every case, demonstrating physicians’ comfort with that element.
- Discussing treatment alternatives occurred in only 1 in 4 conversations. This was more frequent than previously reported, and appeared most often when patients expressed hesitancy about proceeding to PCI.
- Discussing pros and cons of the alternatives was done in 42%.
- Uncertainty about the procedure (eg, that it might not relieve the angina) was expressed in only 10% of conversations.
- Assessment of patient understanding was done in 65% of cases. This included even minimal efforts (eg, “Do you have any questions?”). More advanced methods such as teach-back were never used.
- Exploration of patient preferences (eg, asking patients which treatment they prefer, or attempting to understand how angina affects a patient’s life) the final element, occurred in 73% of conversations.
Only 3% of the conversations contained all 7 elements. Even using a more relaxed definition of 3 critical elements (ie, discussing clinical issues, treatment alternatives, and pros and cons), only 13% of conversations included them all.
Discussion affects decisions
Informed decision-making is not only important because of its ethical implications. Offering patients more information was associated with their choosing not to have PCI. The probability of a patient undergoing PCI was negatively associated with 3 specific elements of informed decision-making. Patients were less likely to choose PCI if the patient’s role in decision-making was discussed (61% vs 86% chose PCI, P < .03); if alternatives were discussed (31% vs 89% chose PCI, P < .01); and if uncertainties were discussed (17% vs 80% chose PCI, P < .01).
There was also a linear relationship between the total number of elements discussed and the probability of choosing PCI: it ranged from 100% of patients choosing PCI when just 1 element was present to 3% of patients choosing PCI when all 7 elements were present. The relationship is not entirely causal, since doctors were more likely to talk about alternatives and risks if patients hesitated and raised questions. Cautious patients received more information.
From these observational studies, we know that physicians do not generally communicate the benefits of PCI, and patients make incorrect assumptions about the benefits they can expect. We know that those who receive more information are less likely to choose PCI, but what would happen if patients were randomly assigned to receive complete information?
An online survey
We conducted an online survey of more than 1,000 participants over age 50 who had never undergone PCI, asking them to imagine visiting a cardiologist after having a positive stress test for stable chest pain.25 Three intervention groups read different scenarios couched as information provided by their cardiologist:
- The “standard care” group received no specific information about the effects of PCI on the risk of myocardial infarction
- The “specific information” group was specifically told that PCI does not reduce the risk of myocardial infarction
- The “explanatory information” group was told how medications work and why PCI does not reduce the risk of myocardial infarction.
All 3 groups received information about the risks of PCI, its role in reducing angina, and the risks and benefits of optimal medical therapy.
After reading their scenario, all participants completed an identical questionnaire, which asked if they would opt for PCI, medical therapy, or both. Overall, 55% chose PCI, ranging from 70% in the standard care group to 46% in the group given explanatory information. Rates in the specific-information and explanatory-information groups were not statistically different from each other, but both were significantly different from that in the standard-care group. Interestingly, the more information patients were given about PCI, the more likely they were to choose optimal medical therapy.
After reading the scenario, participants were also asked if PCI would “prevent a heart attack.” Of those who received standard care, 71% endorsed that belief, which is remarkably similar to studies of real patients who have received standard care. In contrast, only 39% of those given specific information and 31% given explanatory information held that belief. Moreover, the belief that PCI prevented MI was the strongest predictor of choosing PCI (odds ratio 5.82, 95% confidence interval 4.13–8.26).25
Interestingly, 52% of the standard care group falsely remembered that the doctor had told them that PCI would prevent an MI, even though the doctor said nothing about it one way or the other. It appears that participants were projecting their own beliefs onto the encounter. This highlights the importance of providing full information to patients who are considering this procedure.
TOWARD SHARED DECISION-MAKING
Shared decision-making is a process in which physicians enter into a partnership with a patient, offer information, elicit the patient’s preferences, and then come to a decision in concert with the patient.
Although many decisions can and should involve elements of shared decision-making, the decision to proceed with PCI for stable angina is particularly well-suited to shared decision-making. This is because the benefit of PCI depends on the value a patient attaches to being free of angina sooner. Since there is no difference in the risk of MI or death, the patient must decide if the risks of the procedure and the inconvenience of taking dual antiplatelet therapy are worth the benefit of improving symptoms faster. Presumably, patients who have more severe symptoms or experienced side effects from antianginal therapy would be more likely to choose PCI.
Despite having substantial experience educating patients, most physicians are unfamiliar with the process of shared decision-making. In particular, the process of eliciting preferences is often overlooked.
To address this issue, researchers at the Mayo Clinic developed a decision aid that compares PCI plus optimal medical therapy vs optimal medical therapy alone in an easily understandable information card.15 On one side, the 2 options are clearly stated, with the magnitude of symptom improvement over time graphically illustrated and the statement, “NO DIFFERENCE in heart attack or death,” prominently displayed. The back of the card discusses the risks of each option in easily understood tables.
The decision aid was compared with standard care in a randomized trial involving patients who were referred for catheterization and possible PCI.26 The decision aid improved patients’ overall knowledge about PCI. In particular, 60% of those who used the decision aid knew that PCI did not prevent death or MI vs 40% of usual-care patients—results similar to those of the online experiment.
Interestingly, the decision about whether to undergo PCI did not differ significantly between the 2 groups, although there was a trend toward more patients in the decision-aid group choosing medical therapy alone (53%) vs the standard-care patients (39%).
To understand why the decision aid did not make more of a difference, the investigators performed qualitative interviews of the cardiologists in the study.27 One theme was the timing of the intervention. Patients using the decision aid had already been referred for catheterization, and some felt the process should have occurred earlier. Engaging in shared decision-making with a general cardiologist before referral could help to improve the quality of patient decisions.
Cardiologists also noted the difficulty in changing their work flow to incorporate the decision aid. Although some embraced the idea of shared decision-making, others were concerned that many patients could not participate, and there was confusion about the difference between an educational tool, which could be used by a patient alone, and a decision aid, which is meant to generate discussion between the doctor and patient. Some expressed interest in using the tool in the future.
These findings serve to emphasize that providing information alone is not enough. If the physician does not “buy in” to the idea of shared decision-making, it will not occur.
PRACTICE IMPLICATIONS
Based on the pathophysiology of coronary artery disease and the results of multiple randomized controlled trials, it is evident that PCI does not prevent heart attacks in patients with chronic stable angina. However, most patients who undergo PCI are unaware of this and therefore do not truly give informed consent. In the absence of explicit information to the contrary, most patients with stable angina assume that PCI prevents MI and thus are biased toward choosing PCI.
Even minimal amounts of explicit information can partially overcome that bias and influence decision-making. In particular, explaining why PCI does not prevent MI was the most effective means of overcoming the bias.
To this end, shared decision aids may help physicians to engage in shared decision-making. Shared decision-making is most likely to occur if physicians are trained in the concept of shared decision-making, are committed to practicing it, and can fit it into their work flow. Ideally, this would occur in the office of a general cardiologist before referral for PCI.
For those practicing in accountable-care organizations, Medicare has recently introduced the shared decision-making model for 6 preference-sensitive conditions, including stable ischemic heart disease. Participants in this program will have the opportunity to receive payments for shared decision-making services and to share in any savings that result from reduced use of resources. Use of these tools holds the promise for providing more patient-centered care at lower cost.
- Jones DS. Visions of a cure. Visualization, clinical trials, and controversies in cardiac therapeutics, 1968–1998. Isis 2000; 91:504–541.
- Hansson G. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 2005; 352:1685–1695.
- Stone GW, Maehara A, Lansky AJ, et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med 2011; 364:226–235.
- Boden WE, O’Rourke RA, Teo KK, et al. Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med 2007; 356:1503–1516.
- Sedlis SP, Hartigan PM, Teo KK, et al. Effect of PCI on long-term survival in patients with stable ischemic heart disease. N Engl J Med 2015; 373:1937–1946.
- Lin GA, Dudley RA, Lucas FL, Malenka DJ, Vittinghoff E, Redberg RF. Frequency of stress testing to document ischemia prior to elective percutaneous coronary intervention. JAMA 2008; 300:1765–1773.
- Katritsis DG, Ioannidis JP. Percutaneous coronary intervention versus conservative therapy in nonacute coronary artery disease: a meta-analysis. Circulation 2005; 111:2906–2912.
- Trikalinos TA, Alsheikh-Ali AA, Tatsioni A, Nallamothu BK, Kent DM. Percutaneous coronary interventions for non-acute coronary artery disease: a quantitative 20-year synopsis and a network meta-analysis. Lancet 2009; 373:911–918.
- De Bruyne B, Pijls NHJ, Kalesan B, et al. Fractional flow reserve–guided PCI versus medical therapy in stable coronary disease. N Engl J Med 2012; 367:991–1001.
- Weintraub WS, Spertus JA, Kolm P, et al. Effect of PCI on quality of life in patients with stable coronary disease. N Engl J Med 2008; 359:677–687.
- Al-Lamee R, Thompson D, Dehbi H-M, et al, on behalf of the ORBITA Investigators. Percutaneous coronary intervention in stable angina (ORBITA): a double-blind, randomised controlled trial. Lancet. Published online November 2, 2017. http://dx.doi.org/10.1016/S0140-6736(17)32714-9. Accessed November 10, 2017.
- Cobb LA, Thomas GI, Dillard DH, et al. An evaluation of internal mammary-artery ligation by a double-blind technic. N Engl J Med 1959; 260:1115–1118.
- Dimond EG, Fittle F, Crockett JE. Comparison of internal mammary artery ligation and sham operation for angina pectoris. Am J Cardiol 1960; 5:483-486.
- Leon MB, Kornowski R, Downey WE, et al. A blinded, randomized placebo-controlled trial of percutaneous laser myocardial revascularization to improve angina symptoms in patients with severe coronary disease. J Am Coll Cardiol 2005; 46:1812–1819.
- Coylewright M, Shepel K, Leblanc A, et al. Shared decision making in patients with stable coronary artery disease: PCI choice. PLoS One 2012; 7:e49827.
- Holmboe ES, Fiellin DA, Cusanelli E, Remetz M, Krumholz HM. Perceptions of benefit and risk of patients undergoing first-time elective percutaneous coronary revascularization. J Gen Intern Med 2000; 15:632–637.
- Kee F, McDonald P, Gaffney B. Risks and benefits of coronary angioplasty: the patients perspective: a preliminary study. Qual Health Care 1997; 6:131–139.
- Rothberg MB, Sivalingam SK, Ashraf J, et al. Patients’ and cardiologists’ perceptions of the benefits of percutaneous coronary intervention for stable coronary disease. Ann Intern Med 2010; 153:307–313.
- Whittle J, Conigliaro J, Good CB, Kelley ME, Skanderson M. Understanding of the benefits of coronary revascularization procedures among patients who are offered such procedures. Am Heart J 2007; 154:662–668.
- Lin GA, Dudley RA, Redberg RF. Cardiologists’ use of percutaneous coronary interventions for stable coronary artery disease. Arch Intern Med 2007; 167:1604–1609.
- Atreya AR, Sivalingam SK, Arora S, et al. Predictors of medical management in patients undergoing elective cardiac catheterization for chronic ischemic heart disease. Clin Cardiol 2016; 39:207–214.
- Fowler FJ Jr, Gallagher PM, Bynum JP, Barry MJ, Lucas FL, Skinner JS. Decision-making process reported by Medicare patients who had coronary artery stenting or surgery for prostate cancer. J Gen Intern Med 2012; 27:911–916.
- Goff SL, Mazor KM, Ting HH, Kleppel R, Rothberg MB. How cardiologists present the benefits of percutaneous coronary interventions to patients with stable angina: a qualitative analysis. JAMA Intern Med 2014; 174:1614–1621.
- Braddock CH 3rd, Edwards KA, Hasenberg NM, Laidley TL, Levinson W. Informed decision making in outpatient practice: time to get back to basics. JAMA 1999; 282:2313–2320.
- Rothberg MB, Scherer L, Kashef MA, et al. The effect of information presentation on beliefs about the benefits of elective percutaneous coronary intervention. JAMA Intern Med 2014; 174:1623–1629.
- Coylewright M, Dick S, Zmolek B, et al. PCI choice decision aid for stable coronary artery disease: a randomized trial. Circ Cardiovasc Qual Outcomes 2016; 9:767–776.
- Coylewright M, O’Neill ES, Dick S, Grande SW. PCI choice: cardiovascular clinicians’ perceptions of shared decision making in stable coronary artery disease. Patient Educ Couns 2017; 100:1136–1143.
- Jones DS. Visions of a cure. Visualization, clinical trials, and controversies in cardiac therapeutics, 1968–1998. Isis 2000; 91:504–541.
- Hansson G. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 2005; 352:1685–1695.
- Stone GW, Maehara A, Lansky AJ, et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med 2011; 364:226–235.
- Boden WE, O’Rourke RA, Teo KK, et al. Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med 2007; 356:1503–1516.
- Sedlis SP, Hartigan PM, Teo KK, et al. Effect of PCI on long-term survival in patients with stable ischemic heart disease. N Engl J Med 2015; 373:1937–1946.
- Lin GA, Dudley RA, Lucas FL, Malenka DJ, Vittinghoff E, Redberg RF. Frequency of stress testing to document ischemia prior to elective percutaneous coronary intervention. JAMA 2008; 300:1765–1773.
- Katritsis DG, Ioannidis JP. Percutaneous coronary intervention versus conservative therapy in nonacute coronary artery disease: a meta-analysis. Circulation 2005; 111:2906–2912.
- Trikalinos TA, Alsheikh-Ali AA, Tatsioni A, Nallamothu BK, Kent DM. Percutaneous coronary interventions for non-acute coronary artery disease: a quantitative 20-year synopsis and a network meta-analysis. Lancet 2009; 373:911–918.
- De Bruyne B, Pijls NHJ, Kalesan B, et al. Fractional flow reserve–guided PCI versus medical therapy in stable coronary disease. N Engl J Med 2012; 367:991–1001.
- Weintraub WS, Spertus JA, Kolm P, et al. Effect of PCI on quality of life in patients with stable coronary disease. N Engl J Med 2008; 359:677–687.
- Al-Lamee R, Thompson D, Dehbi H-M, et al, on behalf of the ORBITA Investigators. Percutaneous coronary intervention in stable angina (ORBITA): a double-blind, randomised controlled trial. Lancet. Published online November 2, 2017. http://dx.doi.org/10.1016/S0140-6736(17)32714-9. Accessed November 10, 2017.
- Cobb LA, Thomas GI, Dillard DH, et al. An evaluation of internal mammary-artery ligation by a double-blind technic. N Engl J Med 1959; 260:1115–1118.
- Dimond EG, Fittle F, Crockett JE. Comparison of internal mammary artery ligation and sham operation for angina pectoris. Am J Cardiol 1960; 5:483-486.
- Leon MB, Kornowski R, Downey WE, et al. A blinded, randomized placebo-controlled trial of percutaneous laser myocardial revascularization to improve angina symptoms in patients with severe coronary disease. J Am Coll Cardiol 2005; 46:1812–1819.
- Coylewright M, Shepel K, Leblanc A, et al. Shared decision making in patients with stable coronary artery disease: PCI choice. PLoS One 2012; 7:e49827.
- Holmboe ES, Fiellin DA, Cusanelli E, Remetz M, Krumholz HM. Perceptions of benefit and risk of patients undergoing first-time elective percutaneous coronary revascularization. J Gen Intern Med 2000; 15:632–637.
- Kee F, McDonald P, Gaffney B. Risks and benefits of coronary angioplasty: the patients perspective: a preliminary study. Qual Health Care 1997; 6:131–139.
- Rothberg MB, Sivalingam SK, Ashraf J, et al. Patients’ and cardiologists’ perceptions of the benefits of percutaneous coronary intervention for stable coronary disease. Ann Intern Med 2010; 153:307–313.
- Whittle J, Conigliaro J, Good CB, Kelley ME, Skanderson M. Understanding of the benefits of coronary revascularization procedures among patients who are offered such procedures. Am Heart J 2007; 154:662–668.
- Lin GA, Dudley RA, Redberg RF. Cardiologists’ use of percutaneous coronary interventions for stable coronary artery disease. Arch Intern Med 2007; 167:1604–1609.
- Atreya AR, Sivalingam SK, Arora S, et al. Predictors of medical management in patients undergoing elective cardiac catheterization for chronic ischemic heart disease. Clin Cardiol 2016; 39:207–214.
- Fowler FJ Jr, Gallagher PM, Bynum JP, Barry MJ, Lucas FL, Skinner JS. Decision-making process reported by Medicare patients who had coronary artery stenting or surgery for prostate cancer. J Gen Intern Med 2012; 27:911–916.
- Goff SL, Mazor KM, Ting HH, Kleppel R, Rothberg MB. How cardiologists present the benefits of percutaneous coronary interventions to patients with stable angina: a qualitative analysis. JAMA Intern Med 2014; 174:1614–1621.
- Braddock CH 3rd, Edwards KA, Hasenberg NM, Laidley TL, Levinson W. Informed decision making in outpatient practice: time to get back to basics. JAMA 1999; 282:2313–2320.
- Rothberg MB, Scherer L, Kashef MA, et al. The effect of information presentation on beliefs about the benefits of elective percutaneous coronary intervention. JAMA Intern Med 2014; 174:1623–1629.
- Coylewright M, Dick S, Zmolek B, et al. PCI choice decision aid for stable coronary artery disease: a randomized trial. Circ Cardiovasc Qual Outcomes 2016; 9:767–776.
- Coylewright M, O’Neill ES, Dick S, Grande SW. PCI choice: cardiovascular clinicians’ perceptions of shared decision making in stable coronary artery disease. Patient Educ Couns 2017; 100:1136–1143.
KEY POINTS
- For patients with stable angina pectoris, PCI does not prevent myocardial infarction or death.
- Optimal medical therapy with aspirin and a statin can reduce the risk of myocardial infarction and should be recommended for all patients with stable angina, regardless of whether they undergo PCI.
- PCI improves symptoms of angina faster than medical therapy alone, but more than half of patients will be free of angina in about 2 years with either option.
- In the absence of information to the contrary, most patients and some doctors assume that PCI is life-saving and are biased towards choosing it. As a result, patients are rarely able to give true informed consent to undergo PCI.