User login
Team discovers oncogenic driver of T-ALL
Preclinical research suggests the TOX protein is an oncogenic driver of T-cell acute lymphoblastic leukemia (T-ALL).
Results indicate that TOX may be expressed in as many as 95% of human T-ALL cases, and the protein is required for the cancer’s growth and persistence.
“A major role for TOX in T-ALL is to elicit defects in DNA repair, leading to genetic changes that drive normal cells into cancer,” said study author David Langenau, PhD, of Massachusetts General Hospital in Boston.
“TOX then continues to be expressed within leukemic cells and is required for continued tumor growth. That means that, if we can successfully target TOX with small molecules in the future, the 95% of T-ALL patients whose tumors express TOX would have new treatment options for this aggressive leukemia.”
Dr Langenau and his colleagues described this new role for TOX in Cancer Discovery.
The team noted that T-ALL has several molecular subtypes, many of which are driven by common oncogenes such as MYC and NOTCH. However, evidence has suggested the cancer’s initiation is likely driven by aberrations in DNA repair.
To identify genes that might help drive T-ALL, the researchers performed a transgenic screen in zebrafish.
The team found that TOX collaborates with known oncogene pathways to transform T-cell precursors into leukemia cells by altering DNA repair and then expanding the population of transformed cells.
In human T-ALL cells, TOX was shown to suppress non-homologous end joining (NHEJ) repair, a pathway required for repairing double-strand DNA breaks that, when disrupted, is known to cause errant DNA repair and genomic instability.
Nearly all of the human T-ALL samples the researchers tested were found to express TOX. And TOX proved essential for the proliferation and survival of T-ALL.
Dr Langenau explained that TOX is known to have important roles in the development and maturation of several types of immune cells, yet its roles in leukemia initiation and genomic instability were not described until this work.
TOX belongs to a group of proteins known to regulate the configuration or expression of genes by binding to DNA molecules, yet its mechanism in T-ALL—blocking NHEJ repair by binding to DNA repair proteins rather than directly to DNA—was totally unexpected.
The researchers believe that, in addition to better understanding how TOX regulates the continued growth of T-ALL, it will be important to determine whether related proteins have similar molecular functions in other cancers. ![]()
Preclinical research suggests the TOX protein is an oncogenic driver of T-cell acute lymphoblastic leukemia (T-ALL).
Results indicate that TOX may be expressed in as many as 95% of human T-ALL cases, and the protein is required for the cancer’s growth and persistence.
“A major role for TOX in T-ALL is to elicit defects in DNA repair, leading to genetic changes that drive normal cells into cancer,” said study author David Langenau, PhD, of Massachusetts General Hospital in Boston.
“TOX then continues to be expressed within leukemic cells and is required for continued tumor growth. That means that, if we can successfully target TOX with small molecules in the future, the 95% of T-ALL patients whose tumors express TOX would have new treatment options for this aggressive leukemia.”
Dr Langenau and his colleagues described this new role for TOX in Cancer Discovery.
The team noted that T-ALL has several molecular subtypes, many of which are driven by common oncogenes such as MYC and NOTCH. However, evidence has suggested the cancer’s initiation is likely driven by aberrations in DNA repair.
To identify genes that might help drive T-ALL, the researchers performed a transgenic screen in zebrafish.
The team found that TOX collaborates with known oncogene pathways to transform T-cell precursors into leukemia cells by altering DNA repair and then expanding the population of transformed cells.
In human T-ALL cells, TOX was shown to suppress non-homologous end joining (NHEJ) repair, a pathway required for repairing double-strand DNA breaks that, when disrupted, is known to cause errant DNA repair and genomic instability.
Nearly all of the human T-ALL samples the researchers tested were found to express TOX. And TOX proved essential for the proliferation and survival of T-ALL.
Dr Langenau explained that TOX is known to have important roles in the development and maturation of several types of immune cells, yet its roles in leukemia initiation and genomic instability were not described until this work.
TOX belongs to a group of proteins known to regulate the configuration or expression of genes by binding to DNA molecules, yet its mechanism in T-ALL—blocking NHEJ repair by binding to DNA repair proteins rather than directly to DNA—was totally unexpected.
The researchers believe that, in addition to better understanding how TOX regulates the continued growth of T-ALL, it will be important to determine whether related proteins have similar molecular functions in other cancers. ![]()
Preclinical research suggests the TOX protein is an oncogenic driver of T-cell acute lymphoblastic leukemia (T-ALL).
Results indicate that TOX may be expressed in as many as 95% of human T-ALL cases, and the protein is required for the cancer’s growth and persistence.
“A major role for TOX in T-ALL is to elicit defects in DNA repair, leading to genetic changes that drive normal cells into cancer,” said study author David Langenau, PhD, of Massachusetts General Hospital in Boston.
“TOX then continues to be expressed within leukemic cells and is required for continued tumor growth. That means that, if we can successfully target TOX with small molecules in the future, the 95% of T-ALL patients whose tumors express TOX would have new treatment options for this aggressive leukemia.”
Dr Langenau and his colleagues described this new role for TOX in Cancer Discovery.
The team noted that T-ALL has several molecular subtypes, many of which are driven by common oncogenes such as MYC and NOTCH. However, evidence has suggested the cancer’s initiation is likely driven by aberrations in DNA repair.
To identify genes that might help drive T-ALL, the researchers performed a transgenic screen in zebrafish.
The team found that TOX collaborates with known oncogene pathways to transform T-cell precursors into leukemia cells by altering DNA repair and then expanding the population of transformed cells.
In human T-ALL cells, TOX was shown to suppress non-homologous end joining (NHEJ) repair, a pathway required for repairing double-strand DNA breaks that, when disrupted, is known to cause errant DNA repair and genomic instability.
Nearly all of the human T-ALL samples the researchers tested were found to express TOX. And TOX proved essential for the proliferation and survival of T-ALL.
Dr Langenau explained that TOX is known to have important roles in the development and maturation of several types of immune cells, yet its roles in leukemia initiation and genomic instability were not described until this work.
TOX belongs to a group of proteins known to regulate the configuration or expression of genes by binding to DNA molecules, yet its mechanism in T-ALL—blocking NHEJ repair by binding to DNA repair proteins rather than directly to DNA—was totally unexpected.
The researchers believe that, in addition to better understanding how TOX regulates the continued growth of T-ALL, it will be important to determine whether related proteins have similar molecular functions in other cancers. ![]()
Some cancers linked to weight are on the rise in the US

Cancers associated with being overweight or obese accounted for about 40% of all cancers diagnosed in the US in 2014, according to the latest Vital Signs report by the US Centers for Disease Control and Prevention.
The International Agency for Research on Cancer has identified 13 cancers associated with overweight and obesity—multiple myeloma (MM), meningioma, adenocarcinoma of the esophagus, and thyroid, postmenopausal breast, gallbladder, stomach, liver, pancreatic, kidney, ovarian, uterine, and colorectal cancers.
For the Vital Signs report, researchers reviewed US data from 2005 to 2014 to determine trends for cancers associated with being overweight (having a body mass index [BMI] of 25 to 29.9 kg/m2) or obese (having a BMI of 30 kg/m2 and higher).
In 2014, roughly 631,000 people were diagnosed with cancers associated with overweight and obesity, which represents 40% of all cancers diagnosed.
Fifty-five percent of all cancers diagnosed in women and 24% of those diagnosed in men were associated with overweight and obesity.
Incidence rates of the 13 cancers combined were highest in non-Hispanic blacks, followed by non-Hispanic whites, American Indians/Alaska Natives, Hispanics, and Asians/Pacific Islanders.
Incidence over time
Overall, the incidence of cancers associated with overweight and obesity decreased 2% from 2005 to 2014.
However, when colorectal cancer was excluded, the incidence of the other 12 cancers combined increased 7% from 2005 to 2014. The incidence of colorectal cancer decreased 23% during that time. Researchers said this was due, in large part, to screening.
The incidence of cancers not associated with overweight and obesity decreased 13% from 2005 to 2014.
The incidence of MM increased over the period studied, but it was not a significant increase. The incidence rate of MM was 5.6 per 100,000 persons (age-adjusted to the 2000 US standard population) in 2005 and 6.0 per 100,000 persons in 2014.
So overall, there was an 8% increase in MM incidence rate from 2005 to 2014, or a 1.1% average annual increase. There was a 2% increase in the risk of MM per 1 kg/m2 increase in BMI.
Like MM, 3 other cancers had fairly stable incidence rates over the study period—adenocarcinoma of the esophagus, gallbladder cancer, and postmenopausal breast cancer.
However, incidence rates increased significantly each year for thyroid cancer (4.0% per year), liver cancer (2.9%), gastric cardia cancer (1.2%), endometrial cancer (1.1%), pancreatic cancer (0.8%), and kidney cancer (0.7%).
And incidence rates decreased significantly each year for meningioma (-3.8%), colorectal cancer (-2.9%), and ovarian cancer (-2.0%). ![]()
Cancers associated with being overweight or obese accounted for about 40% of all cancers diagnosed in the US in 2014, according to the latest Vital Signs report by the US Centers for Disease Control and Prevention.
The International Agency for Research on Cancer has identified 13 cancers associated with overweight and obesity—multiple myeloma (MM), meningioma, adenocarcinoma of the esophagus, and thyroid, postmenopausal breast, gallbladder, stomach, liver, pancreatic, kidney, ovarian, uterine, and colorectal cancers.
For the Vital Signs report, researchers reviewed US data from 2005 to 2014 to determine trends for cancers associated with being overweight (having a body mass index [BMI] of 25 to 29.9 kg/m2) or obese (having a BMI of 30 kg/m2 and higher).
In 2014, roughly 631,000 people were diagnosed with cancers associated with overweight and obesity, which represents 40% of all cancers diagnosed.
Fifty-five percent of all cancers diagnosed in women and 24% of those diagnosed in men were associated with overweight and obesity.
Incidence rates of the 13 cancers combined were highest in non-Hispanic blacks, followed by non-Hispanic whites, American Indians/Alaska Natives, Hispanics, and Asians/Pacific Islanders.
Incidence over time
Overall, the incidence of cancers associated with overweight and obesity decreased 2% from 2005 to 2014.
However, when colorectal cancer was excluded, the incidence of the other 12 cancers combined increased 7% from 2005 to 2014. The incidence of colorectal cancer decreased 23% during that time. Researchers said this was due, in large part, to screening.
The incidence of cancers not associated with overweight and obesity decreased 13% from 2005 to 2014.
The incidence of MM increased over the period studied, but it was not a significant increase. The incidence rate of MM was 5.6 per 100,000 persons (age-adjusted to the 2000 US standard population) in 2005 and 6.0 per 100,000 persons in 2014.
So overall, there was an 8% increase in MM incidence rate from 2005 to 2014, or a 1.1% average annual increase. There was a 2% increase in the risk of MM per 1 kg/m2 increase in BMI.
Like MM, 3 other cancers had fairly stable incidence rates over the study period—adenocarcinoma of the esophagus, gallbladder cancer, and postmenopausal breast cancer.
However, incidence rates increased significantly each year for thyroid cancer (4.0% per year), liver cancer (2.9%), gastric cardia cancer (1.2%), endometrial cancer (1.1%), pancreatic cancer (0.8%), and kidney cancer (0.7%).
And incidence rates decreased significantly each year for meningioma (-3.8%), colorectal cancer (-2.9%), and ovarian cancer (-2.0%). ![]()
Cancers associated with being overweight or obese accounted for about 40% of all cancers diagnosed in the US in 2014, according to the latest Vital Signs report by the US Centers for Disease Control and Prevention.
The International Agency for Research on Cancer has identified 13 cancers associated with overweight and obesity—multiple myeloma (MM), meningioma, adenocarcinoma of the esophagus, and thyroid, postmenopausal breast, gallbladder, stomach, liver, pancreatic, kidney, ovarian, uterine, and colorectal cancers.
For the Vital Signs report, researchers reviewed US data from 2005 to 2014 to determine trends for cancers associated with being overweight (having a body mass index [BMI] of 25 to 29.9 kg/m2) or obese (having a BMI of 30 kg/m2 and higher).
In 2014, roughly 631,000 people were diagnosed with cancers associated with overweight and obesity, which represents 40% of all cancers diagnosed.
Fifty-five percent of all cancers diagnosed in women and 24% of those diagnosed in men were associated with overweight and obesity.
Incidence rates of the 13 cancers combined were highest in non-Hispanic blacks, followed by non-Hispanic whites, American Indians/Alaska Natives, Hispanics, and Asians/Pacific Islanders.
Incidence over time
Overall, the incidence of cancers associated with overweight and obesity decreased 2% from 2005 to 2014.
However, when colorectal cancer was excluded, the incidence of the other 12 cancers combined increased 7% from 2005 to 2014. The incidence of colorectal cancer decreased 23% during that time. Researchers said this was due, in large part, to screening.
The incidence of cancers not associated with overweight and obesity decreased 13% from 2005 to 2014.
The incidence of MM increased over the period studied, but it was not a significant increase. The incidence rate of MM was 5.6 per 100,000 persons (age-adjusted to the 2000 US standard population) in 2005 and 6.0 per 100,000 persons in 2014.
So overall, there was an 8% increase in MM incidence rate from 2005 to 2014, or a 1.1% average annual increase. There was a 2% increase in the risk of MM per 1 kg/m2 increase in BMI.
Like MM, 3 other cancers had fairly stable incidence rates over the study period—adenocarcinoma of the esophagus, gallbladder cancer, and postmenopausal breast cancer.
However, incidence rates increased significantly each year for thyroid cancer (4.0% per year), liver cancer (2.9%), gastric cardia cancer (1.2%), endometrial cancer (1.1%), pancreatic cancer (0.8%), and kidney cancer (0.7%).
And incidence rates decreased significantly each year for meningioma (-3.8%), colorectal cancer (-2.9%), and ovarian cancer (-2.0%). ![]()
Device could improve anemia detection

A new microfluidic device could improve testing for anemia, according to researchers.
The team said the device can detect the level of hemoglobin in a whole blood sample using optical absorbance.
The device is portable, requires only a few drops of blood for analysis, and eliminates the need for chemical preparations.
In addition, researchers believe the device could be integrated with other microfluidic approaches to blood analysis.
The researchers described their device in AIP Advances.
The team noted that blood analyzers currently on the market measure hemoglobin via hemolysis. This requires hands-on expertise to prepare and run a sample, limiting the ability to monitor anemia in many parts of the world.
“The most exciting aspect to this analyzer is that it uses whole blood and does not require the additional steps and reagents to prepare a sample,” said study author Nathan Sniadecki, PhD, of the University of Washington in Seattle.
“You just run blood into the channel, and that’s it,” added Nikita Taparia, a doctoral candidate in Sniadecki’s lab. “It can be used anywhere.”
The analyzer takes advantage of the optical properties of blood, such as absorption and scattering, to measure hemoglobin concentration. Anemic blood transmits more light than normal blood, so the severity of anemia can be measured as a ratio of transmitted to original light intensity.
To simulate anemia, the researchers diluted blood samples with a buffer solution. The blood analyzer was effective at predicting cases of moderate to severe anemia, defined as less than 10 g/dL of hemoglobin in a sample. The analyzer did not produce any false-negative results.
The optical density of samples did not increase linearly, so a higher concentration of hemoglobin defines the upper limit of detection for the device.
The researchers said the device could be integrated with other microfluidic devices to analyze whole blood samples in parallel to diagnose anemia and other underlying factors that could contribute to the condition.
The team said such an integrated diagnostic tool would “aid the global health community in their continued surveillance of anemia and its etiology in high-risk subpopulations.” ![]()
A new microfluidic device could improve testing for anemia, according to researchers.
The team said the device can detect the level of hemoglobin in a whole blood sample using optical absorbance.
The device is portable, requires only a few drops of blood for analysis, and eliminates the need for chemical preparations.
In addition, researchers believe the device could be integrated with other microfluidic approaches to blood analysis.
The researchers described their device in AIP Advances.
The team noted that blood analyzers currently on the market measure hemoglobin via hemolysis. This requires hands-on expertise to prepare and run a sample, limiting the ability to monitor anemia in many parts of the world.
“The most exciting aspect to this analyzer is that it uses whole blood and does not require the additional steps and reagents to prepare a sample,” said study author Nathan Sniadecki, PhD, of the University of Washington in Seattle.
“You just run blood into the channel, and that’s it,” added Nikita Taparia, a doctoral candidate in Sniadecki’s lab. “It can be used anywhere.”
The analyzer takes advantage of the optical properties of blood, such as absorption and scattering, to measure hemoglobin concentration. Anemic blood transmits more light than normal blood, so the severity of anemia can be measured as a ratio of transmitted to original light intensity.
To simulate anemia, the researchers diluted blood samples with a buffer solution. The blood analyzer was effective at predicting cases of moderate to severe anemia, defined as less than 10 g/dL of hemoglobin in a sample. The analyzer did not produce any false-negative results.
The optical density of samples did not increase linearly, so a higher concentration of hemoglobin defines the upper limit of detection for the device.
The researchers said the device could be integrated with other microfluidic devices to analyze whole blood samples in parallel to diagnose anemia and other underlying factors that could contribute to the condition.
The team said such an integrated diagnostic tool would “aid the global health community in their continued surveillance of anemia and its etiology in high-risk subpopulations.” ![]()
A new microfluidic device could improve testing for anemia, according to researchers.
The team said the device can detect the level of hemoglobin in a whole blood sample using optical absorbance.
The device is portable, requires only a few drops of blood for analysis, and eliminates the need for chemical preparations.
In addition, researchers believe the device could be integrated with other microfluidic approaches to blood analysis.
The researchers described their device in AIP Advances.
The team noted that blood analyzers currently on the market measure hemoglobin via hemolysis. This requires hands-on expertise to prepare and run a sample, limiting the ability to monitor anemia in many parts of the world.
“The most exciting aspect to this analyzer is that it uses whole blood and does not require the additional steps and reagents to prepare a sample,” said study author Nathan Sniadecki, PhD, of the University of Washington in Seattle.
“You just run blood into the channel, and that’s it,” added Nikita Taparia, a doctoral candidate in Sniadecki’s lab. “It can be used anywhere.”
The analyzer takes advantage of the optical properties of blood, such as absorption and scattering, to measure hemoglobin concentration. Anemic blood transmits more light than normal blood, so the severity of anemia can be measured as a ratio of transmitted to original light intensity.
To simulate anemia, the researchers diluted blood samples with a buffer solution. The blood analyzer was effective at predicting cases of moderate to severe anemia, defined as less than 10 g/dL of hemoglobin in a sample. The analyzer did not produce any false-negative results.
The optical density of samples did not increase linearly, so a higher concentration of hemoglobin defines the upper limit of detection for the device.
The researchers said the device could be integrated with other microfluidic devices to analyze whole blood samples in parallel to diagnose anemia and other underlying factors that could contribute to the condition.
The team said such an integrated diagnostic tool would “aid the global health community in their continued surveillance of anemia and its etiology in high-risk subpopulations.” ![]()
Researchers win Nobel Prize for developing cryo-EM

Three researchers have received the Nobel Prize in Chemistry 2017 for the development of cryo-electron microscopy (EM), which has simplified and improved the imaging of biomolecules.
Cryo-EM is a method used to image frozen biological molecules without the use of structure-altering dyes or fixatives or the need to coax the molecules into crystalline form.
This provides a simpler way to generate images of molecules in their normal states and greater understanding of biological function. It also aids the development of pharmaceuticals.
For developing cryo-EM, this year’s Nobel Prize in Chemistry* was awarded to:
- Jacques Dubochet, PhD, of University of Lausanne in Switzerland
- Joachim Frank, PhD, of Columbia University in New York, New York
- Richard Henderson, PhD, of MRC Laboratory of Molecular Biology in Cambridge, UK.
About the work
Electron microscopes were long believed to be suitable only for imaging dead matter because the electron beam destroys biological material.
However, in 1990, Dr Henderson succeeded in using an electron microscope to generate a 3-dimensional image of a protein at atomic resolution. This breakthrough proved the technology’s potential.
Dr Frank made the technology generally applicable. Between 1975 and 1986, he developed an image processing method in which the electron microscope’s fuzzy, 2-dimensional images are analyzed and merged to reveal a sharp, 3-dimensional structure.
Dr Dubochet added water to the mix. Liquid water evaporates in the electron microscope’s vacuum, which makes the biomolecules collapse.
In the early 1980s, Dr Dubochet succeeded in vitrifying water. He cooled water so rapidly that it solidified in its liquid form around a biological sample, allowing the biomolecules to retain their natural shape even in a vacuum.
Following these discoveries, the electron microscope’s every nut and bolt have been optimized. The desired atomic resolution was reached in 2013, and researchers can now routinely produce 3-dimensional structures of biomolecules.
In the past few years, the scientific literature has been filled with images of everything from proteins that cause antibiotic resistance to the surface of the Zika virus. ![]()
*The prize amount is 9 million Swedish krona to be shared equally among the Laureates.
Three researchers have received the Nobel Prize in Chemistry 2017 for the development of cryo-electron microscopy (EM), which has simplified and improved the imaging of biomolecules.
Cryo-EM is a method used to image frozen biological molecules without the use of structure-altering dyes or fixatives or the need to coax the molecules into crystalline form.
This provides a simpler way to generate images of molecules in their normal states and greater understanding of biological function. It also aids the development of pharmaceuticals.
For developing cryo-EM, this year’s Nobel Prize in Chemistry* was awarded to:
- Jacques Dubochet, PhD, of University of Lausanne in Switzerland
- Joachim Frank, PhD, of Columbia University in New York, New York
- Richard Henderson, PhD, of MRC Laboratory of Molecular Biology in Cambridge, UK.
About the work
Electron microscopes were long believed to be suitable only for imaging dead matter because the electron beam destroys biological material.
However, in 1990, Dr Henderson succeeded in using an electron microscope to generate a 3-dimensional image of a protein at atomic resolution. This breakthrough proved the technology’s potential.
Dr Frank made the technology generally applicable. Between 1975 and 1986, he developed an image processing method in which the electron microscope’s fuzzy, 2-dimensional images are analyzed and merged to reveal a sharp, 3-dimensional structure.
Dr Dubochet added water to the mix. Liquid water evaporates in the electron microscope’s vacuum, which makes the biomolecules collapse.
In the early 1980s, Dr Dubochet succeeded in vitrifying water. He cooled water so rapidly that it solidified in its liquid form around a biological sample, allowing the biomolecules to retain their natural shape even in a vacuum.
Following these discoveries, the electron microscope’s every nut and bolt have been optimized. The desired atomic resolution was reached in 2013, and researchers can now routinely produce 3-dimensional structures of biomolecules.
In the past few years, the scientific literature has been filled with images of everything from proteins that cause antibiotic resistance to the surface of the Zika virus. ![]()
*The prize amount is 9 million Swedish krona to be shared equally among the Laureates.
Three researchers have received the Nobel Prize in Chemistry 2017 for the development of cryo-electron microscopy (EM), which has simplified and improved the imaging of biomolecules.
Cryo-EM is a method used to image frozen biological molecules without the use of structure-altering dyes or fixatives or the need to coax the molecules into crystalline form.
This provides a simpler way to generate images of molecules in their normal states and greater understanding of biological function. It also aids the development of pharmaceuticals.
For developing cryo-EM, this year’s Nobel Prize in Chemistry* was awarded to:
- Jacques Dubochet, PhD, of University of Lausanne in Switzerland
- Joachim Frank, PhD, of Columbia University in New York, New York
- Richard Henderson, PhD, of MRC Laboratory of Molecular Biology in Cambridge, UK.
About the work
Electron microscopes were long believed to be suitable only for imaging dead matter because the electron beam destroys biological material.
However, in 1990, Dr Henderson succeeded in using an electron microscope to generate a 3-dimensional image of a protein at atomic resolution. This breakthrough proved the technology’s potential.
Dr Frank made the technology generally applicable. Between 1975 and 1986, he developed an image processing method in which the electron microscope’s fuzzy, 2-dimensional images are analyzed and merged to reveal a sharp, 3-dimensional structure.
Dr Dubochet added water to the mix. Liquid water evaporates in the electron microscope’s vacuum, which makes the biomolecules collapse.
In the early 1980s, Dr Dubochet succeeded in vitrifying water. He cooled water so rapidly that it solidified in its liquid form around a biological sample, allowing the biomolecules to retain their natural shape even in a vacuum.
Following these discoveries, the electron microscope’s every nut and bolt have been optimized. The desired atomic resolution was reached in 2013, and researchers can now routinely produce 3-dimensional structures of biomolecules.
In the past few years, the scientific literature has been filled with images of everything from proteins that cause antibiotic resistance to the surface of the Zika virus. ![]()
*The prize amount is 9 million Swedish krona to be shared equally among the Laureates.
Rash on back and wrists
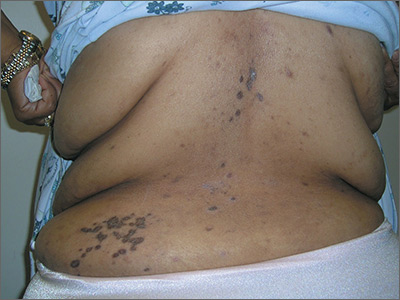
The FP was aware that HCTZ might precipitate lichen planus or create a lichenoid drug reaction, so he performed a 4-mm punch biopsy and asked the patient to stop taking her HCTZ while the biopsy results were pending. (The patient also had several of the 5 Ps of lichen planus: planar, polygonal, pruritic, papular, and purple lesions. She didn’t have any papular lesions and the lesions she did have were more brown than purple because of her dark skin color.)
The FP prescribed topical fluocinonide 0.05% ointment to be applied twice daily to the affected areas until the biopsy results were available. The FP also increased her lisinopril dose, hoping to keep her BP under control.
Biopsy results came back as probable lichen planus and possible lichenoid drug reaction. As treatment for each condition would be the same, it didn’t matter that the pathologist couldn’t be more specific.
On the patient’s second visit for suture removal and discussion of the biopsy results, she said she was feeling better. Her BP was still under control, so the FP decided to continue the treatment plan. At follow-up one month later, the pruritus had resolved completely and the skin lesions were no longer palpable. There was postinflammatory hyperpigmentation at the sites of the lesions, but the patient was not concerned about this because most of it was on her back.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Kraft RL, Usatine R. Lichen planus. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 901-909.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP was aware that HCTZ might precipitate lichen planus or create a lichenoid drug reaction, so he performed a 4-mm punch biopsy and asked the patient to stop taking her HCTZ while the biopsy results were pending. (The patient also had several of the 5 Ps of lichen planus: planar, polygonal, pruritic, papular, and purple lesions. She didn’t have any papular lesions and the lesions she did have were more brown than purple because of her dark skin color.)
The FP prescribed topical fluocinonide 0.05% ointment to be applied twice daily to the affected areas until the biopsy results were available. The FP also increased her lisinopril dose, hoping to keep her BP under control.
Biopsy results came back as probable lichen planus and possible lichenoid drug reaction. As treatment for each condition would be the same, it didn’t matter that the pathologist couldn’t be more specific.
On the patient’s second visit for suture removal and discussion of the biopsy results, she said she was feeling better. Her BP was still under control, so the FP decided to continue the treatment plan. At follow-up one month later, the pruritus had resolved completely and the skin lesions were no longer palpable. There was postinflammatory hyperpigmentation at the sites of the lesions, but the patient was not concerned about this because most of it was on her back.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Kraft RL, Usatine R. Lichen planus. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 901-909.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP was aware that HCTZ might precipitate lichen planus or create a lichenoid drug reaction, so he performed a 4-mm punch biopsy and asked the patient to stop taking her HCTZ while the biopsy results were pending. (The patient also had several of the 5 Ps of lichen planus: planar, polygonal, pruritic, papular, and purple lesions. She didn’t have any papular lesions and the lesions she did have were more brown than purple because of her dark skin color.)
The FP prescribed topical fluocinonide 0.05% ointment to be applied twice daily to the affected areas until the biopsy results were available. The FP also increased her lisinopril dose, hoping to keep her BP under control.
Biopsy results came back as probable lichen planus and possible lichenoid drug reaction. As treatment for each condition would be the same, it didn’t matter that the pathologist couldn’t be more specific.
On the patient’s second visit for suture removal and discussion of the biopsy results, she said she was feeling better. Her BP was still under control, so the FP decided to continue the treatment plan. At follow-up one month later, the pruritus had resolved completely and the skin lesions were no longer palpable. There was postinflammatory hyperpigmentation at the sites of the lesions, but the patient was not concerned about this because most of it was on her back.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Kraft RL, Usatine R. Lichen planus. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 901-909.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Cosmetic Corner: Dermatologists Weigh in on Postprocedural Makeup
To improve patient care and outcomes, leading dermatologists offered their recommendations on postprocedural makeup. Consideration must be given to:
- Dual Action Redness Relief
PCA Skin
“This product is great immediately after laser treatment or filler/botulinum toxin injections to reduce postprocedural redness.”— Gary Goldenberg, MD, New York, New York
- Isdinceutics Skin Drops
ISDIN
“This product is great to reduce or camouflage postprocedural bruising or redness.”—Gary Goldenberg, MD, New York, New York
- Oxygenating Foundation
Oxygenetix
“This is my favorite postprocedural makeup. Originally designed for burn victims, this makeup has botanicals, SPF, and is water resistant and soothing.”—Jeannette Graf, MD, Great Neck, New York
- Quick-Fix Concealer Stick
Dermablend
“This product is customized to match your patient’s skin type. It’s great at covering up purpura postprocedure.”—Shari Lipner, MD, PhD, New York, New York
“I love Dermablend because it can essentially camouflage anything postprocedure, getting patients back to work or their social activities.”— Jerome Potozkin, MD, Danville, California
Cutis invites readers to send us their recommendations. Pigment corrector, lip plumper, moisturizers for men, and wet skin moisturizers will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on postprocedural makeup. Consideration must be given to:
- Dual Action Redness Relief
PCA Skin
“This product is great immediately after laser treatment or filler/botulinum toxin injections to reduce postprocedural redness.”— Gary Goldenberg, MD, New York, New York
- Isdinceutics Skin Drops
ISDIN
“This product is great to reduce or camouflage postprocedural bruising or redness.”—Gary Goldenberg, MD, New York, New York
- Oxygenating Foundation
Oxygenetix
“This is my favorite postprocedural makeup. Originally designed for burn victims, this makeup has botanicals, SPF, and is water resistant and soothing.”—Jeannette Graf, MD, Great Neck, New York
- Quick-Fix Concealer Stick
Dermablend
“This product is customized to match your patient’s skin type. It’s great at covering up purpura postprocedure.”—Shari Lipner, MD, PhD, New York, New York
“I love Dermablend because it can essentially camouflage anything postprocedure, getting patients back to work or their social activities.”— Jerome Potozkin, MD, Danville, California
Cutis invites readers to send us their recommendations. Pigment corrector, lip plumper, moisturizers for men, and wet skin moisturizers will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on postprocedural makeup. Consideration must be given to:
- Dual Action Redness Relief
PCA Skin
“This product is great immediately after laser treatment or filler/botulinum toxin injections to reduce postprocedural redness.”— Gary Goldenberg, MD, New York, New York
- Isdinceutics Skin Drops
ISDIN
“This product is great to reduce or camouflage postprocedural bruising or redness.”—Gary Goldenberg, MD, New York, New York
- Oxygenating Foundation
Oxygenetix
“This is my favorite postprocedural makeup. Originally designed for burn victims, this makeup has botanicals, SPF, and is water resistant and soothing.”—Jeannette Graf, MD, Great Neck, New York
- Quick-Fix Concealer Stick
Dermablend
“This product is customized to match your patient’s skin type. It’s great at covering up purpura postprocedure.”—Shari Lipner, MD, PhD, New York, New York
“I love Dermablend because it can essentially camouflage anything postprocedure, getting patients back to work or their social activities.”— Jerome Potozkin, MD, Danville, California
Cutis invites readers to send us their recommendations. Pigment corrector, lip plumper, moisturizers for men, and wet skin moisturizers will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.

When Epilepsy Does Not Respond to Treatment

How to apply SPRINT findings to elderly patients
SAN FRANCISCO – The benefit of lowering blood pressure exceeded the potential for harm, even among the most frail elderly, in SPRINT, but it’s important to remember who was excluded from the trial when using the findings in the clinic, according to Mark Supiano, MD.
SPRINT (Systolic Blood Pressure Intervention Trial) excluded people with histories of stroke, diabetes, heart failure, and chronic kidney disease with a markedly reduced glomerular filtration rate. People living in nursing homes, assisted living centers, and those with prevalent dementia were also excluded, as were individuals with a standing systolic pressure below 110 mm Hg (N Engl J Med. 2015 Nov 26;373:2103-16).
Even with those exclusions, however, the 2,636 patients in SPRINT who were 75 years and older “were not a super healthy group of older people,” Dr. Supiano said at the joint scientific sessions of the American Heart Association Council on Hypertension, AHA Council on Kidney in Cardiovascular Disease, and American Society of Hypertension.
They were at high risk for cardiovascular disease (CVD), with a median 10-year Framingham risk score of almost 25%. More than a quarter had gait speeds below 0.8 m/sec, and almost a third were classified as frail. Many had mild cognitive impairment at baseline.
In the United States, Dr. Supiano and his colleagues estimate that there are almost 6 million similar people 75 years or older with hypertension who would likely achieve the same benefits from hypertension control as elderly subjects in the trial. “As a geriatrician, there are very few things that I can offer patients 75 years and older that will have a profound improvement in their overall mortality.” Blood pressure control is one of them, said Dr. Supiano, chief of geriatrics at the University of Utah, Salt Lake City, and a SPRINT investigator.
In SPRINT, intensive treatment to systolic pressure below 120 mm Hg showed greater benefit for patients 75 years and older than it did for younger patients, even among the frail, with a 34% reduction in fatal and nonfatal CVD events versus patients treated to below 140 mm Hg, and a 33% lower rate of death from any cause.
It should be no surprise that older patients had greater benefit from tighter control, because elderly patients have “a greater CVD risk. There’s more bang for the buck” with blood pressure lowering in an older population. “Overall, benefits exceed the potential for harm, even among the frailest older patients,” Dr. Supiano said.
“A systemic target of less than 140 mm Hg is, I believe, appropriate for most healthy people age 60 and older. A benefit-based systemic target of less than 120 mm Hg may be appropriate for those at higher CVD risk.” Among patients 60-75 years old, that would include those with a Framingham score above 15%. Among patients older than age 75 with an elevated CVD risk, treatment to below 120 mm Hg makes sense if it aligns with patient’s goals of care, Dr. Supiano said.
The 120–mm Hg target in SPRINT was associated with a greater incidence of some transient side effects in the elderly, including hypotension, syncope, acute kidney injury, and electrolyte imbalance, but not a higher risk of serious adverse events or injurious falls.
There were concerns raised at the joint sessions about the effect of blood pressure lowering on the cognitive function of older people. Dr. Supiano noted that the cognitive outcomes in SPRINT, as well as outcomes in patients with chronic kidney disease, have not yet been released, but are expected soon.
Dr. Supiano had no relevant disclosures.
SAN FRANCISCO – The benefit of lowering blood pressure exceeded the potential for harm, even among the most frail elderly, in SPRINT, but it’s important to remember who was excluded from the trial when using the findings in the clinic, according to Mark Supiano, MD.
SPRINT (Systolic Blood Pressure Intervention Trial) excluded people with histories of stroke, diabetes, heart failure, and chronic kidney disease with a markedly reduced glomerular filtration rate. People living in nursing homes, assisted living centers, and those with prevalent dementia were also excluded, as were individuals with a standing systolic pressure below 110 mm Hg (N Engl J Med. 2015 Nov 26;373:2103-16).
Even with those exclusions, however, the 2,636 patients in SPRINT who were 75 years and older “were not a super healthy group of older people,” Dr. Supiano said at the joint scientific sessions of the American Heart Association Council on Hypertension, AHA Council on Kidney in Cardiovascular Disease, and American Society of Hypertension.
They were at high risk for cardiovascular disease (CVD), with a median 10-year Framingham risk score of almost 25%. More than a quarter had gait speeds below 0.8 m/sec, and almost a third were classified as frail. Many had mild cognitive impairment at baseline.
In the United States, Dr. Supiano and his colleagues estimate that there are almost 6 million similar people 75 years or older with hypertension who would likely achieve the same benefits from hypertension control as elderly subjects in the trial. “As a geriatrician, there are very few things that I can offer patients 75 years and older that will have a profound improvement in their overall mortality.” Blood pressure control is one of them, said Dr. Supiano, chief of geriatrics at the University of Utah, Salt Lake City, and a SPRINT investigator.
In SPRINT, intensive treatment to systolic pressure below 120 mm Hg showed greater benefit for patients 75 years and older than it did for younger patients, even among the frail, with a 34% reduction in fatal and nonfatal CVD events versus patients treated to below 140 mm Hg, and a 33% lower rate of death from any cause.
It should be no surprise that older patients had greater benefit from tighter control, because elderly patients have “a greater CVD risk. There’s more bang for the buck” with blood pressure lowering in an older population. “Overall, benefits exceed the potential for harm, even among the frailest older patients,” Dr. Supiano said.
“A systemic target of less than 140 mm Hg is, I believe, appropriate for most healthy people age 60 and older. A benefit-based systemic target of less than 120 mm Hg may be appropriate for those at higher CVD risk.” Among patients 60-75 years old, that would include those with a Framingham score above 15%. Among patients older than age 75 with an elevated CVD risk, treatment to below 120 mm Hg makes sense if it aligns with patient’s goals of care, Dr. Supiano said.
The 120–mm Hg target in SPRINT was associated with a greater incidence of some transient side effects in the elderly, including hypotension, syncope, acute kidney injury, and electrolyte imbalance, but not a higher risk of serious adverse events or injurious falls.
There were concerns raised at the joint sessions about the effect of blood pressure lowering on the cognitive function of older people. Dr. Supiano noted that the cognitive outcomes in SPRINT, as well as outcomes in patients with chronic kidney disease, have not yet been released, but are expected soon.
Dr. Supiano had no relevant disclosures.
SAN FRANCISCO – The benefit of lowering blood pressure exceeded the potential for harm, even among the most frail elderly, in SPRINT, but it’s important to remember who was excluded from the trial when using the findings in the clinic, according to Mark Supiano, MD.
SPRINT (Systolic Blood Pressure Intervention Trial) excluded people with histories of stroke, diabetes, heart failure, and chronic kidney disease with a markedly reduced glomerular filtration rate. People living in nursing homes, assisted living centers, and those with prevalent dementia were also excluded, as were individuals with a standing systolic pressure below 110 mm Hg (N Engl J Med. 2015 Nov 26;373:2103-16).
Even with those exclusions, however, the 2,636 patients in SPRINT who were 75 years and older “were not a super healthy group of older people,” Dr. Supiano said at the joint scientific sessions of the American Heart Association Council on Hypertension, AHA Council on Kidney in Cardiovascular Disease, and American Society of Hypertension.
They were at high risk for cardiovascular disease (CVD), with a median 10-year Framingham risk score of almost 25%. More than a quarter had gait speeds below 0.8 m/sec, and almost a third were classified as frail. Many had mild cognitive impairment at baseline.
In the United States, Dr. Supiano and his colleagues estimate that there are almost 6 million similar people 75 years or older with hypertension who would likely achieve the same benefits from hypertension control as elderly subjects in the trial. “As a geriatrician, there are very few things that I can offer patients 75 years and older that will have a profound improvement in their overall mortality.” Blood pressure control is one of them, said Dr. Supiano, chief of geriatrics at the University of Utah, Salt Lake City, and a SPRINT investigator.
In SPRINT, intensive treatment to systolic pressure below 120 mm Hg showed greater benefit for patients 75 years and older than it did for younger patients, even among the frail, with a 34% reduction in fatal and nonfatal CVD events versus patients treated to below 140 mm Hg, and a 33% lower rate of death from any cause.
It should be no surprise that older patients had greater benefit from tighter control, because elderly patients have “a greater CVD risk. There’s more bang for the buck” with blood pressure lowering in an older population. “Overall, benefits exceed the potential for harm, even among the frailest older patients,” Dr. Supiano said.
“A systemic target of less than 140 mm Hg is, I believe, appropriate for most healthy people age 60 and older. A benefit-based systemic target of less than 120 mm Hg may be appropriate for those at higher CVD risk.” Among patients 60-75 years old, that would include those with a Framingham score above 15%. Among patients older than age 75 with an elevated CVD risk, treatment to below 120 mm Hg makes sense if it aligns with patient’s goals of care, Dr. Supiano said.
The 120–mm Hg target in SPRINT was associated with a greater incidence of some transient side effects in the elderly, including hypotension, syncope, acute kidney injury, and electrolyte imbalance, but not a higher risk of serious adverse events or injurious falls.
There were concerns raised at the joint sessions about the effect of blood pressure lowering on the cognitive function of older people. Dr. Supiano noted that the cognitive outcomes in SPRINT, as well as outcomes in patients with chronic kidney disease, have not yet been released, but are expected soon.
Dr. Supiano had no relevant disclosures.
EXPERT ANALYSIS FROM JOINT HYPERTENSION 2017
Prehospital tourniquets in civilian settings significantly decreased mortality
BALTIMORE – Prehospital tourniquet use on injured civilians in trauma situations was associated with a nearly sixfold decrease in mortality, according to a study presented at the annual meeting of the American Association for Surgery of Trauma.
While tourniquets have been an effective tool in military settings, data on successful applications in civilian settings have been scarce.
Dr. Teixeira and his coinvestigators conducted a multicenter, retrospective study of 1,026 peripheral–vascular injury patients admitted to level I trauma centers between January 2011 and December 2016. Among the patients studied, 181 (17.6%) received a tourniquet prior to hospital admission.
A majority of tourniquets were applied to the limbs, with the most common application sites on the arm (49%) and the thigh (29%).Tourniquets were held in place for an average 77 minutes.
Of the patients in the study, 98 (9.6%) underwent an amputation; 35 of these patients had received a tourniquet.
After adjusting for confounding factors, such as age and mechanism of injury, investigators found patients who received tourniquets were nearly six times more likely to survive than were their nontourniquet counterparts (odds ratio, 5.86; 95% confidence interval, 1.41-24.47; P = .015).
While the overall mortality rate among those with a tourniquet – compared with those without a tourniquet – was significantly lower, the comparative mortality rate among amputee patients was not significant, which investigators hypothesized could be because of the smaller number of patients in this subgroup.
Additionally, patients who did not receive a tourniquet had lower injury severity scores, had better vital signs, and needed less blood, according to investigators.
The findings of this study mirror what many military medical professionals have historically, and adamantly, supported, according to discussant Jay J. Doucet, MD, FACS, medical director for the surgical intensive care unit at the University of California San Diego Medical Center and a former combat surgeon.
“The medical lessons on our battlefields that hold such great promise have to be carefully relearned, brought home, and fearlessly applied here,” said Dr. Doucet. “I have yet to meet an employed military surgeon who does not believe the tourniquet is an indispensable tool.” While Dr. Doucet did acknowledge the benefit of tourniquets outside military use and addressed the need for increased implementation among civilian hospitals, he did pose a query about the mortality rate that investigators had found.
“The no-tourniquet group has an adjusted odds of death at a rate that is 5.86 times higher, yet they had better vitals, needed less blood, had lower [injury severity scores], had less head injury, fewer traumatic amputations, and fewer complications,” said Dr. Doucet. “So why do they die?”
Investigators were not able to pinpoint the cause of death among patients because of the limitations of their study; however, Dr. Teixeira and his colleagues were able to determine the presence of cardiac complications, pulmonary complications, and acute kidney injury, none of which had a significantly different presence between the two study groups.
The data gathered from this study are strong enough to support the use of tourniquets in civilian situations, asserted Dr. Teixeira, which means the next hurdle is to integrate it into the health system.
“What’s important from our perspective as leaders of this issue is what we are doing to increase the rate [of tourniquet use],” said Dr. Teixeira. “I think one of the important things is the Stop the Bleed program, [in which] we are actually teaching the Austin police department, and we are trying to increase the use of the tourniquet and demonstrate its importance.”
Investigators reported no relevant financial disclosures.
[email protected]
On Twitter @eaztweets
BALTIMORE – Prehospital tourniquet use on injured civilians in trauma situations was associated with a nearly sixfold decrease in mortality, according to a study presented at the annual meeting of the American Association for Surgery of Trauma.
While tourniquets have been an effective tool in military settings, data on successful applications in civilian settings have been scarce.
Dr. Teixeira and his coinvestigators conducted a multicenter, retrospective study of 1,026 peripheral–vascular injury patients admitted to level I trauma centers between January 2011 and December 2016. Among the patients studied, 181 (17.6%) received a tourniquet prior to hospital admission.
A majority of tourniquets were applied to the limbs, with the most common application sites on the arm (49%) and the thigh (29%).Tourniquets were held in place for an average 77 minutes.
Of the patients in the study, 98 (9.6%) underwent an amputation; 35 of these patients had received a tourniquet.
After adjusting for confounding factors, such as age and mechanism of injury, investigators found patients who received tourniquets were nearly six times more likely to survive than were their nontourniquet counterparts (odds ratio, 5.86; 95% confidence interval, 1.41-24.47; P = .015).
While the overall mortality rate among those with a tourniquet – compared with those without a tourniquet – was significantly lower, the comparative mortality rate among amputee patients was not significant, which investigators hypothesized could be because of the smaller number of patients in this subgroup.
Additionally, patients who did not receive a tourniquet had lower injury severity scores, had better vital signs, and needed less blood, according to investigators.
The findings of this study mirror what many military medical professionals have historically, and adamantly, supported, according to discussant Jay J. Doucet, MD, FACS, medical director for the surgical intensive care unit at the University of California San Diego Medical Center and a former combat surgeon.
“The medical lessons on our battlefields that hold such great promise have to be carefully relearned, brought home, and fearlessly applied here,” said Dr. Doucet. “I have yet to meet an employed military surgeon who does not believe the tourniquet is an indispensable tool.” While Dr. Doucet did acknowledge the benefit of tourniquets outside military use and addressed the need for increased implementation among civilian hospitals, he did pose a query about the mortality rate that investigators had found.
“The no-tourniquet group has an adjusted odds of death at a rate that is 5.86 times higher, yet they had better vitals, needed less blood, had lower [injury severity scores], had less head injury, fewer traumatic amputations, and fewer complications,” said Dr. Doucet. “So why do they die?”
Investigators were not able to pinpoint the cause of death among patients because of the limitations of their study; however, Dr. Teixeira and his colleagues were able to determine the presence of cardiac complications, pulmonary complications, and acute kidney injury, none of which had a significantly different presence between the two study groups.
The data gathered from this study are strong enough to support the use of tourniquets in civilian situations, asserted Dr. Teixeira, which means the next hurdle is to integrate it into the health system.
“What’s important from our perspective as leaders of this issue is what we are doing to increase the rate [of tourniquet use],” said Dr. Teixeira. “I think one of the important things is the Stop the Bleed program, [in which] we are actually teaching the Austin police department, and we are trying to increase the use of the tourniquet and demonstrate its importance.”
Investigators reported no relevant financial disclosures.
[email protected]
On Twitter @eaztweets
BALTIMORE – Prehospital tourniquet use on injured civilians in trauma situations was associated with a nearly sixfold decrease in mortality, according to a study presented at the annual meeting of the American Association for Surgery of Trauma.
While tourniquets have been an effective tool in military settings, data on successful applications in civilian settings have been scarce.
Dr. Teixeira and his coinvestigators conducted a multicenter, retrospective study of 1,026 peripheral–vascular injury patients admitted to level I trauma centers between January 2011 and December 2016. Among the patients studied, 181 (17.6%) received a tourniquet prior to hospital admission.
A majority of tourniquets were applied to the limbs, with the most common application sites on the arm (49%) and the thigh (29%).Tourniquets were held in place for an average 77 minutes.
Of the patients in the study, 98 (9.6%) underwent an amputation; 35 of these patients had received a tourniquet.
After adjusting for confounding factors, such as age and mechanism of injury, investigators found patients who received tourniquets were nearly six times more likely to survive than were their nontourniquet counterparts (odds ratio, 5.86; 95% confidence interval, 1.41-24.47; P = .015).
While the overall mortality rate among those with a tourniquet – compared with those without a tourniquet – was significantly lower, the comparative mortality rate among amputee patients was not significant, which investigators hypothesized could be because of the smaller number of patients in this subgroup.
Additionally, patients who did not receive a tourniquet had lower injury severity scores, had better vital signs, and needed less blood, according to investigators.
The findings of this study mirror what many military medical professionals have historically, and adamantly, supported, according to discussant Jay J. Doucet, MD, FACS, medical director for the surgical intensive care unit at the University of California San Diego Medical Center and a former combat surgeon.
“The medical lessons on our battlefields that hold such great promise have to be carefully relearned, brought home, and fearlessly applied here,” said Dr. Doucet. “I have yet to meet an employed military surgeon who does not believe the tourniquet is an indispensable tool.” While Dr. Doucet did acknowledge the benefit of tourniquets outside military use and addressed the need for increased implementation among civilian hospitals, he did pose a query about the mortality rate that investigators had found.
“The no-tourniquet group has an adjusted odds of death at a rate that is 5.86 times higher, yet they had better vitals, needed less blood, had lower [injury severity scores], had less head injury, fewer traumatic amputations, and fewer complications,” said Dr. Doucet. “So why do they die?”
Investigators were not able to pinpoint the cause of death among patients because of the limitations of their study; however, Dr. Teixeira and his colleagues were able to determine the presence of cardiac complications, pulmonary complications, and acute kidney injury, none of which had a significantly different presence between the two study groups.
The data gathered from this study are strong enough to support the use of tourniquets in civilian situations, asserted Dr. Teixeira, which means the next hurdle is to integrate it into the health system.
“What’s important from our perspective as leaders of this issue is what we are doing to increase the rate [of tourniquet use],” said Dr. Teixeira. “I think one of the important things is the Stop the Bleed program, [in which] we are actually teaching the Austin police department, and we are trying to increase the use of the tourniquet and demonstrate its importance.”
Investigators reported no relevant financial disclosures.
[email protected]
On Twitter @eaztweets
At the AAST Annual Meeting
Key clinical point:
Major finding: Patients who were given a prehospital tourniquet were associated with a survival odds ratio nearly sixfold higher than those without (odds ratio, 5.86; 95% confidence interval, 1.41-24.47; P = .015).
Data source: Multicenter retrospective study of 1,026 patients with peripheral vascular injuries admitted to a level I trauma facility between January 2011 and December 2016.
Disclosures: Investigators reported no relevant financial disclosures.
Five takeaways from Congress’ failure to extend funding for children’s coverage
Congress finally seems ready to take action on the Children’s Health Insurance Program after funding lapsed Sept. 30.
Before the deadline, lawmakers were busy grappling with the failed repeal of the Affordable Care Act.
CHIP covers 9 million children nationwide. But until Congress renews CHIP, states are cut off from additional federal funding that helps lower- and middle-income families.
CHIP, which has enjoyed broad bipartisan support, helps lower- and middle-income families that otherwise earn too much to be eligible for Medicaid. Besides children, it covers 370,000 pregnant women a year. Like Medicaid, CHIP is traditionally paid for with state and federal funds, but the federal government covers most of the cost.
Though current authorization for spending has expired, states can use some of their unspent federal CHIP money. Still, several states are expected to run out of money before the end of 2017, and most of the rest will run out by next summer. CHIP has been in this fix only one other time since it was established in 1997. In 2007, CHIP went weeks without funding authorization from Congress.
Here’s a quick look at what may lie ahead for the program.
1. Will children lose coverage because Congress missed the deadline?
They could eventually, but not immediately. A few states facing the most immediate threat – including California and Arizona – have enough funding to last only until the end of the year.
No states have yet announced plans to freeze enrollment or alert families about any potential end in coverage. But if Congress fails to renew funding quickly, some states may begin taking steps to unwind the program in the next few weeks.
2. What are states doing in reaction to Congress missing the deadline?
Most states are doing little except reaching into their unspent federal funds.
However, Minnesota was among those most imperiled because it had spent all its funds. State officials said Tuesday that the federal Centers for Medicare & Medicaid Services (CMS) was giving Minnesota $3.6 million from unspent national funds to cover CHIP this month.
Emily Piper, commissioner of the Minnesota Department of Human Services, reported in a newspaper commentary in September that her state’s funds would be exhausted by the end of that month.
Even without the last-minute infusion of funding from CMS, most of the children covered by CHIP would have continued to receive care under the state’s Medicaid program, but Minnesota would get fewer federal dollars for each child, according to Piper’s commentary. However, she added, those most at risk are the 1,700 pregnant women covered by CHIP, because they wouldn’t be eligible for Medicaid.
Utah has notified CMS that it plans to discontinue its CHIP program by the end of the year unless it receives more federal money. About 19,000 children are in the state’s CHIP program, state officials say. So far, though, the state said it is not moving to suspend service or enrollment or alert enrollees about any possible changes.
Nevada officials said if funding is not extended it might have to freeze enrollment on Nov. 1 and end coverage by Nov. 30.
California, which has 1.3 million children covered by CHIP, has the highest enrollment of any state running out of funding this year. But, so far, it’s continuing business as usual.
“We estimate that we have available CHIP funding at least through December 2017,” said Tony Cava, spokesman for California Department of Health Services. “Our CHIP program is open for enrollment and continues to operate normally.”
Oregon said it has enough CHIP funding to last through October for its program that covers 98,000 children.
3. When is Congress likely to act?
The Senate Finance and the House Energy and Commerce committees have scheduled votes Wednesday on legislation to extend CHIP funding. If both approve their individual bills, floor votes could come quickly, and then both houses would need to resolve any differences.
Senate Finance Committee Chairman Orrin Hatch (R-Utah) and the committee’s ranking Democrat, Sen. Ron Wyden of Oregon, announced an agreement in mid-September to renew CHIP funding. Under the proposed deal, federal CHIP funding would drop by 23 percentage points starting in by 2020, returning to its pre-Affordable Care Act levels. The agreement would extend the life of the CHIP program through 2022.
Hatch and Wyden did not provide any details on how they would pay for the CHIP extension.
The House Energy and Commerce Committee posted its bill just before midnight Monday. It mirrors the Senate Finance plan by extending funding for CHIP for five years and gradually phasing down the 23-percentage-point funding increase provided under Affordable Care Act over the next two years.
4. If CHIP is so popular among Republicans and Democrats, why hasn’t Congress renewed the program yet?
The funding renewal was not a priority among Republican leaders, who have spent most of this year trying to replace the Affordable Care Act and dramatically overhaul the Medicaid program. Some in Congress also thought the Sept. 30 deadline was squishy since states could extend their existing funds beyond that.
5. Who benefits from CHIP?
While CHIP income eligibility levels vary by state, about 90 percent of children covered are in families earning 200 percent of poverty or less ($40,840 for a family of three). CHIP covers children up to age 19. States have the option to cover pregnant women, and 18 plus the District of Columbia do so.
The program is known by different names in different states such as Hoosier Healthwise in Indiana and PeachCare for Kids in Georgia.
For families that move out of Medicaid as their incomes rise, CHIP is an affordable option that ensures continued coverage for their children. Many states operate their CHIP programs as part of Medicaid.
KHN’s coverage of children’s health care issues is supported in part by a grant from The Heising-Simons Foundation.
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
Congress finally seems ready to take action on the Children’s Health Insurance Program after funding lapsed Sept. 30.
Before the deadline, lawmakers were busy grappling with the failed repeal of the Affordable Care Act.
CHIP covers 9 million children nationwide. But until Congress renews CHIP, states are cut off from additional federal funding that helps lower- and middle-income families.
CHIP, which has enjoyed broad bipartisan support, helps lower- and middle-income families that otherwise earn too much to be eligible for Medicaid. Besides children, it covers 370,000 pregnant women a year. Like Medicaid, CHIP is traditionally paid for with state and federal funds, but the federal government covers most of the cost.
Though current authorization for spending has expired, states can use some of their unspent federal CHIP money. Still, several states are expected to run out of money before the end of 2017, and most of the rest will run out by next summer. CHIP has been in this fix only one other time since it was established in 1997. In 2007, CHIP went weeks without funding authorization from Congress.
Here’s a quick look at what may lie ahead for the program.
1. Will children lose coverage because Congress missed the deadline?
They could eventually, but not immediately. A few states facing the most immediate threat – including California and Arizona – have enough funding to last only until the end of the year.
No states have yet announced plans to freeze enrollment or alert families about any potential end in coverage. But if Congress fails to renew funding quickly, some states may begin taking steps to unwind the program in the next few weeks.
2. What are states doing in reaction to Congress missing the deadline?
Most states are doing little except reaching into their unspent federal funds.
However, Minnesota was among those most imperiled because it had spent all its funds. State officials said Tuesday that the federal Centers for Medicare & Medicaid Services (CMS) was giving Minnesota $3.6 million from unspent national funds to cover CHIP this month.
Emily Piper, commissioner of the Minnesota Department of Human Services, reported in a newspaper commentary in September that her state’s funds would be exhausted by the end of that month.
Even without the last-minute infusion of funding from CMS, most of the children covered by CHIP would have continued to receive care under the state’s Medicaid program, but Minnesota would get fewer federal dollars for each child, according to Piper’s commentary. However, she added, those most at risk are the 1,700 pregnant women covered by CHIP, because they wouldn’t be eligible for Medicaid.
Utah has notified CMS that it plans to discontinue its CHIP program by the end of the year unless it receives more federal money. About 19,000 children are in the state’s CHIP program, state officials say. So far, though, the state said it is not moving to suspend service or enrollment or alert enrollees about any possible changes.
Nevada officials said if funding is not extended it might have to freeze enrollment on Nov. 1 and end coverage by Nov. 30.
California, which has 1.3 million children covered by CHIP, has the highest enrollment of any state running out of funding this year. But, so far, it’s continuing business as usual.
“We estimate that we have available CHIP funding at least through December 2017,” said Tony Cava, spokesman for California Department of Health Services. “Our CHIP program is open for enrollment and continues to operate normally.”
Oregon said it has enough CHIP funding to last through October for its program that covers 98,000 children.
3. When is Congress likely to act?
The Senate Finance and the House Energy and Commerce committees have scheduled votes Wednesday on legislation to extend CHIP funding. If both approve their individual bills, floor votes could come quickly, and then both houses would need to resolve any differences.
Senate Finance Committee Chairman Orrin Hatch (R-Utah) and the committee’s ranking Democrat, Sen. Ron Wyden of Oregon, announced an agreement in mid-September to renew CHIP funding. Under the proposed deal, federal CHIP funding would drop by 23 percentage points starting in by 2020, returning to its pre-Affordable Care Act levels. The agreement would extend the life of the CHIP program through 2022.
Hatch and Wyden did not provide any details on how they would pay for the CHIP extension.
The House Energy and Commerce Committee posted its bill just before midnight Monday. It mirrors the Senate Finance plan by extending funding for CHIP for five years and gradually phasing down the 23-percentage-point funding increase provided under Affordable Care Act over the next two years.
4. If CHIP is so popular among Republicans and Democrats, why hasn’t Congress renewed the program yet?
The funding renewal was not a priority among Republican leaders, who have spent most of this year trying to replace the Affordable Care Act and dramatically overhaul the Medicaid program. Some in Congress also thought the Sept. 30 deadline was squishy since states could extend their existing funds beyond that.
5. Who benefits from CHIP?
While CHIP income eligibility levels vary by state, about 90 percent of children covered are in families earning 200 percent of poverty or less ($40,840 for a family of three). CHIP covers children up to age 19. States have the option to cover pregnant women, and 18 plus the District of Columbia do so.
The program is known by different names in different states such as Hoosier Healthwise in Indiana and PeachCare for Kids in Georgia.
For families that move out of Medicaid as their incomes rise, CHIP is an affordable option that ensures continued coverage for their children. Many states operate their CHIP programs as part of Medicaid.
KHN’s coverage of children’s health care issues is supported in part by a grant from The Heising-Simons Foundation.
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.
Congress finally seems ready to take action on the Children’s Health Insurance Program after funding lapsed Sept. 30.
Before the deadline, lawmakers were busy grappling with the failed repeal of the Affordable Care Act.
CHIP covers 9 million children nationwide. But until Congress renews CHIP, states are cut off from additional federal funding that helps lower- and middle-income families.
CHIP, which has enjoyed broad bipartisan support, helps lower- and middle-income families that otherwise earn too much to be eligible for Medicaid. Besides children, it covers 370,000 pregnant women a year. Like Medicaid, CHIP is traditionally paid for with state and federal funds, but the federal government covers most of the cost.
Though current authorization for spending has expired, states can use some of their unspent federal CHIP money. Still, several states are expected to run out of money before the end of 2017, and most of the rest will run out by next summer. CHIP has been in this fix only one other time since it was established in 1997. In 2007, CHIP went weeks without funding authorization from Congress.
Here’s a quick look at what may lie ahead for the program.
1. Will children lose coverage because Congress missed the deadline?
They could eventually, but not immediately. A few states facing the most immediate threat – including California and Arizona – have enough funding to last only until the end of the year.
No states have yet announced plans to freeze enrollment or alert families about any potential end in coverage. But if Congress fails to renew funding quickly, some states may begin taking steps to unwind the program in the next few weeks.
2. What are states doing in reaction to Congress missing the deadline?
Most states are doing little except reaching into their unspent federal funds.
However, Minnesota was among those most imperiled because it had spent all its funds. State officials said Tuesday that the federal Centers for Medicare & Medicaid Services (CMS) was giving Minnesota $3.6 million from unspent national funds to cover CHIP this month.
Emily Piper, commissioner of the Minnesota Department of Human Services, reported in a newspaper commentary in September that her state’s funds would be exhausted by the end of that month.
Even without the last-minute infusion of funding from CMS, most of the children covered by CHIP would have continued to receive care under the state’s Medicaid program, but Minnesota would get fewer federal dollars for each child, according to Piper’s commentary. However, she added, those most at risk are the 1,700 pregnant women covered by CHIP, because they wouldn’t be eligible for Medicaid.
Utah has notified CMS that it plans to discontinue its CHIP program by the end of the year unless it receives more federal money. About 19,000 children are in the state’s CHIP program, state officials say. So far, though, the state said it is not moving to suspend service or enrollment or alert enrollees about any possible changes.
Nevada officials said if funding is not extended it might have to freeze enrollment on Nov. 1 and end coverage by Nov. 30.
California, which has 1.3 million children covered by CHIP, has the highest enrollment of any state running out of funding this year. But, so far, it’s continuing business as usual.
“We estimate that we have available CHIP funding at least through December 2017,” said Tony Cava, spokesman for California Department of Health Services. “Our CHIP program is open for enrollment and continues to operate normally.”
Oregon said it has enough CHIP funding to last through October for its program that covers 98,000 children.
3. When is Congress likely to act?
The Senate Finance and the House Energy and Commerce committees have scheduled votes Wednesday on legislation to extend CHIP funding. If both approve their individual bills, floor votes could come quickly, and then both houses would need to resolve any differences.
Senate Finance Committee Chairman Orrin Hatch (R-Utah) and the committee’s ranking Democrat, Sen. Ron Wyden of Oregon, announced an agreement in mid-September to renew CHIP funding. Under the proposed deal, federal CHIP funding would drop by 23 percentage points starting in by 2020, returning to its pre-Affordable Care Act levels. The agreement would extend the life of the CHIP program through 2022.
Hatch and Wyden did not provide any details on how they would pay for the CHIP extension.
The House Energy and Commerce Committee posted its bill just before midnight Monday. It mirrors the Senate Finance plan by extending funding for CHIP for five years and gradually phasing down the 23-percentage-point funding increase provided under Affordable Care Act over the next two years.
4. If CHIP is so popular among Republicans and Democrats, why hasn’t Congress renewed the program yet?
The funding renewal was not a priority among Republican leaders, who have spent most of this year trying to replace the Affordable Care Act and dramatically overhaul the Medicaid program. Some in Congress also thought the Sept. 30 deadline was squishy since states could extend their existing funds beyond that.
5. Who benefits from CHIP?
While CHIP income eligibility levels vary by state, about 90 percent of children covered are in families earning 200 percent of poverty or less ($40,840 for a family of three). CHIP covers children up to age 19. States have the option to cover pregnant women, and 18 plus the District of Columbia do so.
The program is known by different names in different states such as Hoosier Healthwise in Indiana and PeachCare for Kids in Georgia.
For families that move out of Medicaid as their incomes rise, CHIP is an affordable option that ensures continued coverage for their children. Many states operate their CHIP programs as part of Medicaid.
KHN’s coverage of children’s health care issues is supported in part by a grant from The Heising-Simons Foundation.
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation.