User login
Parkinsonism and Vitamin C Deficiency
Vitamin C (ascorbic acid) deficiency is known to affect brain function and is associated with parkinsonism.1 In 1752, James Lind, MD, described emotional and behavioral changes that herald the onset of scurvy and precede hemorrhagic findings.2 The World Health Organization (WHO) today refers to this stage as latent scurvy.3 The 2 case studies that follow present examples of patients with vitamin C deficiencies whose parkinsonism responded robustly to vitamin C replacement. These cases suggest that vitamin C deficiency may be a treatable cause of parkinsonism.
Case 1
Mr. A, a 60-year-old white male, was admitted to the Medicine Service for alcohol detoxification. The patient had a history of alcohol dependence, alcohol withdrawal seizures, tobacco dependence, and hyperlipidemia. He took no medications as an outpatient. On admission Mr. A’s body mass index (BMI) was 27.2. An initial examination revealed a marked resting tremor of the patient’s right hand with cogwheeling, which had not been present in examinations conducted in the previous 3 years. Mr. A had no prior history of a tremor. He had no cerebellar findings and no evidence of asterixis or of tremulousness associated with high-output cardiac states, such as de Musset sign.
Mr. A reported he had experienced the tremor for a month and that it had been worsening. He also was having difficulty using his dominant right hand, for routine daily activities. Mr. A was oriented, and his short-term memory was intact. He was ill-appearing, irritable with psychomotor slowing, and did not wish to rise from his bed. He had no gingival or periungual bleeding and did not bruise easily. He had no corkscrew hairs. The patient was started on no medications known to cause extrapyramidal symptoms (EPS).
In the hospital, the tremor persisted unabated for 2 days. On the third day, Mr. A was started on 1,000 mg vitamin C IV twice daily. He received a total of 2,000 mg IV that day, but the IV fell out, and he refused its replacement. Several hours later, Mr. A stated that he felt much better, got out of bed, and asked to go outside to smoke. The author noted complete resolution of the right hand tremor and cogwheeling 20 hours after starting the vitamin C IV. Mr. A refused a repeat serum vitamin C assay.
Laboratory studies initially revealed that Mr. A had hyponatremia with a serum sodium of 121 mmol/L (normal range: 133 to 145 mmol/L) as well as hypokalemia with a serum potassium of 3.2 mmol/L (normal range: 3.5 to 5.0 mmol/L). He was hypoosmolar, with a serum osmolality of 276 mOsm/kg (normal range: 278 to 305 mOsm/kg). His vitamin C level was low at 0.2 mg/dL (normal range: 0.4 to 2.0 mg/dL). Mr. A also had a serum vitamin C level drawn 2 years prior that showed no symptoms of EPS, and at that time, the reading was 0.7 mg/dL. At admission to Medicine Services, Mr. A had a serum alcohol level of 211 mg/dL. Neuroimaging revealed diffuse cerebral and cerebellar volume loss.
Normal laboratory results included serum levels of vitamin B12, red cell folate, homocysteine, methylmalonic acid, free and total carnitine, alkaline phosphatase, manganese, and zinc. A urine drug screen was negative.
Case 2
Mr. B, a 69-year-old black male, was admitted to the hospital for depression complicated by alcohol dependence. He also had tobacco dependence, type 2 diabetes mellitus, hypertension, and gout. The patient’s BMI at admission was 16.1. Mr. B appeared ill, was worried about his health, and remained recumbent unless asked to move. He reported that his right hand had begun to shake at rest in the month prior to admission. The tremor made it difficult for him to drink. He pointed out stains on his hospital gurney from an attempt to drink orange juice prior to being assessed.
A physical examination revealed a distinct resting tremor with cogwheeling of the right hand; there was no other evidence of EPS, nor was there evidence of cognitive, cerebellar, or skin abnormalities, such as hemorrhages or corkscrew hairs. Asterixis was absent as was evidence of a high-output cardiac state that might produce a tremor, such as de Musset sign. A serum vitamin C level was obtained and returned at 0.0 mg/dL. A head computed tomography scan obtained the next day revealed mild cerebellar volume loss. A serum alkaline phosphatase level was elevated slightly at 136 U/L (normal range: 42 to 113 U/L). Normal serum values were returned for zinc, vitamins B12 and folate, rapid plasma reagin, sodium, and serum osmolality. A urine drug screen was negative, and serum alcohol level was < 5.0 mg/dL.
Mr. B took no medications expected to cause EPS. He received no micronutrient replacement until the day after admission when he began receiving oral vitamin C 1,000 mg twice a day. After receiving 3 doses, Mr. B’s resting tremor and cogwheeling completely resolved. He noticed he had stopped shaking and could now drink without spilling fluids. He also got out of bed and began interacting with others. Mr. B said he felt he was “doing well.” A repeat serum vitamin C level was 0.2 mg/dL on that day. The improvement was sustained over 3 days, and Mr. B was discharged to home.
Discussion
Both Mr. A and Mr. B presented with a typical picture of latent scurvy and the additional finding of parkinsonism. These cases are important for 2 reasons. First, the swift and full response of these patients’ parkinsonism to vitamin C replacement underscores the importance of considering a vitamin C deficiency when confronted with EPS. And second, both patients lacked signs of bleeding or of impaired collagen synthesis. This differs from the classic presentation of scurvy as a disorder primarily of collagen metabolism.4
Lind described the onset of scurvy as one in which striking emotional and behavior changes developed and later were followed by abnormal bleeding and even death.2 These early changes also were recognized by Shapter in 1847.5 Furthermore, the evidence that exists about the time-course of scurvy’s development suggests that neuropsychiatric findings precede the hemorrhagic.6 Indeed, classic skin findings, such as petechiae or corkscrew hairs, may develop years after the onset of neuropsychiatric changes.7,8
Despite WHO characterizing it as latent scurvy, the distinct syndromal presentation of hypovitaminosis C with parkinsonism along with the rapid response to vitamin C replacement argues for its recognition as a distinct clinical entity and not just a prelude to the hemorrhagic state. To assist in recognizing neuropsychiatric scurvy, the author suggests the operationalized approach described in Table 1.9

Pathophysiology
Vitamin C has an intimate role in the normal functioning of the basal ganglia. It is involved in the synthesis of catechecholamines, the regulation of the release and postsynaptic activities of various neurotransmitters, and managing the oxyradical toxicity of aerobic metabolism. Table 2 outlines some of the normal brain functions of vitamin C and the potential consequences of inadequate central vitamin C.9,10 Risk factors for vitamin C deficiency include those affecting the uptake, response to, and elimination of this vitamin (Table 3).11-14
The potential role of alcohol use by both patients also warrants mention. Current data suggest a nonlinear relationship between alcohol use and neurotoxicity. Epidemiologic data show that moderate alcohol consumption protects against the development of such neurodegenerative processes as Parkinson disease and Alzheimer disease.15,16 But the cases here reflect excessive use of alcohol. In this situation, a variety of progressive insults, such as those caused by oxyradical toxicity as well as malnutrition may foster the development of basal ganglia dysfunction.17
Measuring Deficiency
A deficiency of vitamin C may be determined in several ways. The most frequently used laboratory measure of vitamin C status is the serum vitamin C level. This level is included in the WHO’s recommendations for diagnosis.3 However, this assay is limited because when facing total body depletion, the kidneys may restrict the elimination of vitamin C and tend to maintain serum vitamin C levels even as target tissue levels fall. An interesting example of this is the 0.2 mg/dL value that each patient registered. In Mr. A’s case, this reflected a systemic deficit of vitamin C, while in Mr. B’s case it correlated with the onset of effective repletion of body’s stores.
A fall in urinary output of vitamin C is another marker of hypovitaminosis C. When available, this laboratory test can be used with the serum level to assess total body stores of vitamin C. Lymphocytes, neutrophils, and platelets also store vitamin C. These target tissues tend to saturate when the oral intake ranges between 100 mg to 200 mg a day. This is the same point at which serum vitamin C levels peak and level off in normal, healthy adults.18,19 Once again, the limited availability of target-tissue assays puts these studies out of reach for most clinicians.
No evaluation is complete without some assurance of what the disease is not. Deficiencies of biotin, zinc, folate, and B12 all may affect the function of the basal ganglia.20 The biotin deficiencies literature is particularly robust. Biotin deficiencies affecting basal ganglia function are best known as inherited disorders of metabolism.21 Manganese intoxication also may present as a movement disorder.22
Treatment
Treatment of neuropsychiatric scurvy has relied on IV administration of vitamin C. Although the bioavailability of oral vitamin C among healthy adult volunteers is nearly complete up to about 200 mg a day, a patient with neuropsychiatric scurvy may need substantially more than that amount to accommodate total body deficiencies and increased demands.23 The IV route allows serum vitamin C levels up to 100 times higher than by the oral route.24 Mr. B is, in fact, the first person reported in the literature with neuropsychiatric scurvy to respond to oral vitamin C replacement alone. Once repletion of vitamin C is complete, it is useful to consider a maintenance replacement dose based on a patient’s risk factors and needs.
A healthy adult should ingest about 120 mg of vitamin C daily. Smokers and pregnant women may require more, but this recommendation was intended to address their needs as well.25 Many commercial multivitamins use the old recommended daily allowance of 60 mg, so it may be safest to recommend specifically a vitamin C tablet with at least 120 mg when ordering vitamin C replacement.
Tight control of the serum vitamin C concentration means that little additional vitamin C will be taken up by the gut beyond 200 mg orally a day, which helps minimize any concerns about long-term toxicity. It takes several weeks to deplete vitamin C from the human body when vitamin C is removed from the diet, so a patient with a previously treated deficiency of vitamin C should wait a month before a repeat serum vitamin C level measurement.
The half life of vitamin C is normally ≤ 2 hours. When renal function is intact, vitamin C in excess of immediate need is lost through renal filtration. Toxicity is rare under these conditions.26 When vitamin C toxicity has been reported, it has occurred in the setting of prolonged supplementation, usually when a patient already experienced a renal injury. The main toxicities attributed to vitamin C are oxalate crystal formation with subsequent renal injury and exacerbation of glucose 6-phosphate dehydrogenase deficiency (G6PD).24
Oxalate formation due to vitamin C replacement is uncommon, but patients with preexisting calcium oxalate stones may be at risk for further stone formation when they receive additional vitamin C.27 This is most likely to occur when treatment with parenteral vitamin C is prolonged, which is not typical for patients with neuropsychiatric scurvy who tend to respond rapidly to vitamin C replenishment. Reports of acute hemolytic episodes among patient with G6PD deficiency receiving vitamin C exist, although these cases are rare.28 Furthermore, some authors advocate for the use of ascorbic acid to treat methemoglobinemia associated with G6PD deficiency, when methylene blue is not available.29 It may be reasonable to begin treatment with oral vitamin C for patients with NPS and G6PD deficiency. This is equivalent to a low-dose form of vitamin C replacement and may help avoid the theoretically pro-oxidant effects of larger, IV doses of vitamin C.30
Conclusion
The recent discovery of movement disorders in scurvy has enlarged the picture of vitamin C deficiency. The cases here demonstrate how hypovitaminosis C with central nervous system manifestations may be identified and treated. This relationship fits well within the established basic science and clinical framework for scurvy, and the clinical implications for scurvy remain in many ways unchanged. First, malnutrition must be considered even when a patient’s habitus suggests he is well fed. Also, it is more likely to see scurvy without all of the classic findings than an end-stage case of the disease.31 In the right clinical setting, it is reasonable to think of a vitamin C deficiency before the patient develops bleeding gums and corkscrew hairs. And as is typical of vitamin deficiencies, the treatment of a vitamin C deficiency usually results in swift improvement. Finally, for those who treat movement disorders or who prescribe agents such as antipsychotics that may cause movement disorders, it is important to recognize vitamin C deficiency as another potential explanation for EPS.
1. Ide K, Yamada H, Umegaki K, et al. Lymphocyte vitamin C levels as potential biomarker for progression of Parkinson’s disease. Nutrition. 2015;31(2):406-408.
2. Lind J. The diagnostics, or symptoms. A Treatise on the Scurvy, in Three Parts. 3rd ed. London: S. Crowder, D. Wilson and G. Nicholls, T. Cadell, T. Becket and Co., G. Pearch, and Woodfall; 1772:98-129.
3. World Health Organization. Scurvy and its prevention and control in major emergencies. http://whqlibdoc.who.int/hq/1999/WHO_NHD_99.11.pdf. Published 1999. Accessed July 6, 2017.
4. Sasseville D. Scurvy: curse and cure in New France. JAMA Dermatol. 2015;151(4):431.
5. Shapter T. On the recent occurrence of scurvy in Exeter and the neighbourhood. Prov Med Surg J. 1847;11(11):281-285.
6. Kinsman RA, Hood J. Some behavioral effects of ascorbic acid deficiency. Am J Clin Nutr. 1971;24(4):455-464.
7. DeSantis J. Scurvy and psychiatric symptoms. Perspect Psychiatr Care. 1993;29(1):18-22.
8. Walter JF. Scurvy resulting from a self-imposed diet. West J Med. 1979;130(2):177-179.
9. Brown TM. Neuropsychiatric scurvy. Psychosomatics. 2015;56(1):12-20.
10. Feuerstein TJ, Weinheimer G, Lang G, Ginap T, Rossner R. Inhibition by ascorbic acid of NMDA-evoked acetylcholine release in rabbit caudate nucleus. Naunyn Schmiedebergs Arch Pharmacol. 1993;348(5):549-551.
11. Kim J, Kwon J, Noh G, Lee SS. The effects of elimination diet on nutritional status in subjects with atopic dermatitis. Nutr Res Pract. 2013;7(6):488-494.
12. Langlois M, Duprez D, Delanghe J, De Buyzere M, Clement DL. Serum vitamin C concentration is low in peripheral arterial disease and is associated with inflammation and severity of atherosclerosis. Circulation. 2001;103(14):1863-1868.
13. Nappe TM, Pacelli AM, Katz K. An atypical case of methemoglobinemia due to self-administered benzocaine. Case Rep Emerg Med. 2015;2015:670979.
14. Wright AD, Stevens E, Ali M, Carroll DW, Brown TM. The neuropsychiatry of scurvy. Psychosomatics. 2014;55(2):179-185.
15. Bate C, Williams A. Ethanol protects cultured neurons against amyloid-β and α-synuclein-induced synapse damage. Neuropharmacology. 2011;61(8):1406-1412.
16. Vasanthi HR, Parameswari RP, DeLeiris J, Das DK. Health benefits of wine and alcohol from neuroprotection to heart health. Front Biosci (Elite Ed). 2012;4:1505-1512.
17. Vaglini F, Viaggi C, Piro V, et al. Acetaldehyde and parkinsonism: role of CYP450 2E1. Front Behav Neurosci. 2013;7:71.
18. Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci USA. 2001;98(17):9842-9846.
19. Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv Nutr. 2011;2(2):78-88.
20. Quiroga MJ, Carroll DW, Brown TM. Ascorbate- and zinc-responsive parkinsonism. Ann Pharmacother. 2014;48(11):1515-1520.
21. Tabarki B, Al-Shafi S, Al-Shahwan S, et al. Biotin-responsive basal ganglia disease revisited: clinical, radiologic, and genetic findings. Neurology. 2013;80(3):261-267.
22. Tuschl K, Mills PB, Clayton PT. Manganese and the brain. Int Rev Neurobiol. 2013;110:277-312.
23. Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci USA. 1996;93(8):3704-3709.
24. Wilson MK, Baguley BC, Wall C, Jameson MB, Findlay MP. Review of high-dose intravenous vitamin C as an anticancer agent. Asia Pac J Clin Oncol. 2014;10(1):22-37.
25. Carr AC, Frei B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am J Clin Nutr. 1999;69(6):1086-1107.
26. Nielsen TK, Højgaard M, Andersen JT, Poulsen HE, Lykkesfeldt J, Mikines KJ. Elimination of ascorbic acid after high-dose infusion in prostate cancer patients: a pharmacokinetic evaluation. Basic Clin Pharmacol Toxicol. 2015;116(4):343-348.
27. Baxmann AC, De O G Mendonça C, Heilberg IP. Effect of vitamin C supplements on urinary oxalate and pH in calcium stone-forming patients. Kidney Int. 2003;63(3):1066-1071.
28. Huang YC, Chang TK, Fu YC, Jan SL. C for colored urine: acute hemolysis induced by high-dose ascorbic acid. Clin Toxicol (Phila). 2014;52(9):984.
29. Rino PB, Scolnik D, Fustiñana A, Mitelpunkt A, Glatstein M. Ascorbic acid for the treatment of methemoglobinemia: the experience of a large tertiary care pediatric hospital. Am J Ther. 2014;21(4):240-243.
30. Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012;1826(2):443-457.
31. Fouron JC, Chicoine L. Le scorbut: aspects particuliers de l’association rachitisme-scorbut. Can Med Assoc J. 1962;86(26):1191-1196.
Vitamin C (ascorbic acid) deficiency is known to affect brain function and is associated with parkinsonism.1 In 1752, James Lind, MD, described emotional and behavioral changes that herald the onset of scurvy and precede hemorrhagic findings.2 The World Health Organization (WHO) today refers to this stage as latent scurvy.3 The 2 case studies that follow present examples of patients with vitamin C deficiencies whose parkinsonism responded robustly to vitamin C replacement. These cases suggest that vitamin C deficiency may be a treatable cause of parkinsonism.
Case 1
Mr. A, a 60-year-old white male, was admitted to the Medicine Service for alcohol detoxification. The patient had a history of alcohol dependence, alcohol withdrawal seizures, tobacco dependence, and hyperlipidemia. He took no medications as an outpatient. On admission Mr. A’s body mass index (BMI) was 27.2. An initial examination revealed a marked resting tremor of the patient’s right hand with cogwheeling, which had not been present in examinations conducted in the previous 3 years. Mr. A had no prior history of a tremor. He had no cerebellar findings and no evidence of asterixis or of tremulousness associated with high-output cardiac states, such as de Musset sign.
Mr. A reported he had experienced the tremor for a month and that it had been worsening. He also was having difficulty using his dominant right hand, for routine daily activities. Mr. A was oriented, and his short-term memory was intact. He was ill-appearing, irritable with psychomotor slowing, and did not wish to rise from his bed. He had no gingival or periungual bleeding and did not bruise easily. He had no corkscrew hairs. The patient was started on no medications known to cause extrapyramidal symptoms (EPS).
In the hospital, the tremor persisted unabated for 2 days. On the third day, Mr. A was started on 1,000 mg vitamin C IV twice daily. He received a total of 2,000 mg IV that day, but the IV fell out, and he refused its replacement. Several hours later, Mr. A stated that he felt much better, got out of bed, and asked to go outside to smoke. The author noted complete resolution of the right hand tremor and cogwheeling 20 hours after starting the vitamin C IV. Mr. A refused a repeat serum vitamin C assay.
Laboratory studies initially revealed that Mr. A had hyponatremia with a serum sodium of 121 mmol/L (normal range: 133 to 145 mmol/L) as well as hypokalemia with a serum potassium of 3.2 mmol/L (normal range: 3.5 to 5.0 mmol/L). He was hypoosmolar, with a serum osmolality of 276 mOsm/kg (normal range: 278 to 305 mOsm/kg). His vitamin C level was low at 0.2 mg/dL (normal range: 0.4 to 2.0 mg/dL). Mr. A also had a serum vitamin C level drawn 2 years prior that showed no symptoms of EPS, and at that time, the reading was 0.7 mg/dL. At admission to Medicine Services, Mr. A had a serum alcohol level of 211 mg/dL. Neuroimaging revealed diffuse cerebral and cerebellar volume loss.
Normal laboratory results included serum levels of vitamin B12, red cell folate, homocysteine, methylmalonic acid, free and total carnitine, alkaline phosphatase, manganese, and zinc. A urine drug screen was negative.
Case 2
Mr. B, a 69-year-old black male, was admitted to the hospital for depression complicated by alcohol dependence. He also had tobacco dependence, type 2 diabetes mellitus, hypertension, and gout. The patient’s BMI at admission was 16.1. Mr. B appeared ill, was worried about his health, and remained recumbent unless asked to move. He reported that his right hand had begun to shake at rest in the month prior to admission. The tremor made it difficult for him to drink. He pointed out stains on his hospital gurney from an attempt to drink orange juice prior to being assessed.
A physical examination revealed a distinct resting tremor with cogwheeling of the right hand; there was no other evidence of EPS, nor was there evidence of cognitive, cerebellar, or skin abnormalities, such as hemorrhages or corkscrew hairs. Asterixis was absent as was evidence of a high-output cardiac state that might produce a tremor, such as de Musset sign. A serum vitamin C level was obtained and returned at 0.0 mg/dL. A head computed tomography scan obtained the next day revealed mild cerebellar volume loss. A serum alkaline phosphatase level was elevated slightly at 136 U/L (normal range: 42 to 113 U/L). Normal serum values were returned for zinc, vitamins B12 and folate, rapid plasma reagin, sodium, and serum osmolality. A urine drug screen was negative, and serum alcohol level was < 5.0 mg/dL.
Mr. B took no medications expected to cause EPS. He received no micronutrient replacement until the day after admission when he began receiving oral vitamin C 1,000 mg twice a day. After receiving 3 doses, Mr. B’s resting tremor and cogwheeling completely resolved. He noticed he had stopped shaking and could now drink without spilling fluids. He also got out of bed and began interacting with others. Mr. B said he felt he was “doing well.” A repeat serum vitamin C level was 0.2 mg/dL on that day. The improvement was sustained over 3 days, and Mr. B was discharged to home.
Discussion
Both Mr. A and Mr. B presented with a typical picture of latent scurvy and the additional finding of parkinsonism. These cases are important for 2 reasons. First, the swift and full response of these patients’ parkinsonism to vitamin C replacement underscores the importance of considering a vitamin C deficiency when confronted with EPS. And second, both patients lacked signs of bleeding or of impaired collagen synthesis. This differs from the classic presentation of scurvy as a disorder primarily of collagen metabolism.4
Lind described the onset of scurvy as one in which striking emotional and behavior changes developed and later were followed by abnormal bleeding and even death.2 These early changes also were recognized by Shapter in 1847.5 Furthermore, the evidence that exists about the time-course of scurvy’s development suggests that neuropsychiatric findings precede the hemorrhagic.6 Indeed, classic skin findings, such as petechiae or corkscrew hairs, may develop years after the onset of neuropsychiatric changes.7,8
Despite WHO characterizing it as latent scurvy, the distinct syndromal presentation of hypovitaminosis C with parkinsonism along with the rapid response to vitamin C replacement argues for its recognition as a distinct clinical entity and not just a prelude to the hemorrhagic state. To assist in recognizing neuropsychiatric scurvy, the author suggests the operationalized approach described in Table 1.9
Pathophysiology
Vitamin C has an intimate role in the normal functioning of the basal ganglia. It is involved in the synthesis of catechecholamines, the regulation of the release and postsynaptic activities of various neurotransmitters, and managing the oxyradical toxicity of aerobic metabolism. Table 2 outlines some of the normal brain functions of vitamin C and the potential consequences of inadequate central vitamin C.9,10 Risk factors for vitamin C deficiency include those affecting the uptake, response to, and elimination of this vitamin (Table 3).11-14
The potential role of alcohol use by both patients also warrants mention. Current data suggest a nonlinear relationship between alcohol use and neurotoxicity. Epidemiologic data show that moderate alcohol consumption protects against the development of such neurodegenerative processes as Parkinson disease and Alzheimer disease.15,16 But the cases here reflect excessive use of alcohol. In this situation, a variety of progressive insults, such as those caused by oxyradical toxicity as well as malnutrition may foster the development of basal ganglia dysfunction.17
Measuring Deficiency
A deficiency of vitamin C may be determined in several ways. The most frequently used laboratory measure of vitamin C status is the serum vitamin C level. This level is included in the WHO’s recommendations for diagnosis.3 However, this assay is limited because when facing total body depletion, the kidneys may restrict the elimination of vitamin C and tend to maintain serum vitamin C levels even as target tissue levels fall. An interesting example of this is the 0.2 mg/dL value that each patient registered. In Mr. A’s case, this reflected a systemic deficit of vitamin C, while in Mr. B’s case it correlated with the onset of effective repletion of body’s stores.
A fall in urinary output of vitamin C is another marker of hypovitaminosis C. When available, this laboratory test can be used with the serum level to assess total body stores of vitamin C. Lymphocytes, neutrophils, and platelets also store vitamin C. These target tissues tend to saturate when the oral intake ranges between 100 mg to 200 mg a day. This is the same point at which serum vitamin C levels peak and level off in normal, healthy adults.18,19 Once again, the limited availability of target-tissue assays puts these studies out of reach for most clinicians.
No evaluation is complete without some assurance of what the disease is not. Deficiencies of biotin, zinc, folate, and B12 all may affect the function of the basal ganglia.20 The biotin deficiencies literature is particularly robust. Biotin deficiencies affecting basal ganglia function are best known as inherited disorders of metabolism.21 Manganese intoxication also may present as a movement disorder.22
Treatment
Treatment of neuropsychiatric scurvy has relied on IV administration of vitamin C. Although the bioavailability of oral vitamin C among healthy adult volunteers is nearly complete up to about 200 mg a day, a patient with neuropsychiatric scurvy may need substantially more than that amount to accommodate total body deficiencies and increased demands.23 The IV route allows serum vitamin C levels up to 100 times higher than by the oral route.24 Mr. B is, in fact, the first person reported in the literature with neuropsychiatric scurvy to respond to oral vitamin C replacement alone. Once repletion of vitamin C is complete, it is useful to consider a maintenance replacement dose based on a patient’s risk factors and needs.
A healthy adult should ingest about 120 mg of vitamin C daily. Smokers and pregnant women may require more, but this recommendation was intended to address their needs as well.25 Many commercial multivitamins use the old recommended daily allowance of 60 mg, so it may be safest to recommend specifically a vitamin C tablet with at least 120 mg when ordering vitamin C replacement.
Tight control of the serum vitamin C concentration means that little additional vitamin C will be taken up by the gut beyond 200 mg orally a day, which helps minimize any concerns about long-term toxicity. It takes several weeks to deplete vitamin C from the human body when vitamin C is removed from the diet, so a patient with a previously treated deficiency of vitamin C should wait a month before a repeat serum vitamin C level measurement.
The half life of vitamin C is normally ≤ 2 hours. When renal function is intact, vitamin C in excess of immediate need is lost through renal filtration. Toxicity is rare under these conditions.26 When vitamin C toxicity has been reported, it has occurred in the setting of prolonged supplementation, usually when a patient already experienced a renal injury. The main toxicities attributed to vitamin C are oxalate crystal formation with subsequent renal injury and exacerbation of glucose 6-phosphate dehydrogenase deficiency (G6PD).24
Oxalate formation due to vitamin C replacement is uncommon, but patients with preexisting calcium oxalate stones may be at risk for further stone formation when they receive additional vitamin C.27 This is most likely to occur when treatment with parenteral vitamin C is prolonged, which is not typical for patients with neuropsychiatric scurvy who tend to respond rapidly to vitamin C replenishment. Reports of acute hemolytic episodes among patient with G6PD deficiency receiving vitamin C exist, although these cases are rare.28 Furthermore, some authors advocate for the use of ascorbic acid to treat methemoglobinemia associated with G6PD deficiency, when methylene blue is not available.29 It may be reasonable to begin treatment with oral vitamin C for patients with NPS and G6PD deficiency. This is equivalent to a low-dose form of vitamin C replacement and may help avoid the theoretically pro-oxidant effects of larger, IV doses of vitamin C.30
Conclusion
The recent discovery of movement disorders in scurvy has enlarged the picture of vitamin C deficiency. The cases here demonstrate how hypovitaminosis C with central nervous system manifestations may be identified and treated. This relationship fits well within the established basic science and clinical framework for scurvy, and the clinical implications for scurvy remain in many ways unchanged. First, malnutrition must be considered even when a patient’s habitus suggests he is well fed. Also, it is more likely to see scurvy without all of the classic findings than an end-stage case of the disease.31 In the right clinical setting, it is reasonable to think of a vitamin C deficiency before the patient develops bleeding gums and corkscrew hairs. And as is typical of vitamin deficiencies, the treatment of a vitamin C deficiency usually results in swift improvement. Finally, for those who treat movement disorders or who prescribe agents such as antipsychotics that may cause movement disorders, it is important to recognize vitamin C deficiency as another potential explanation for EPS.
Vitamin C (ascorbic acid) deficiency is known to affect brain function and is associated with parkinsonism.1 In 1752, James Lind, MD, described emotional and behavioral changes that herald the onset of scurvy and precede hemorrhagic findings.2 The World Health Organization (WHO) today refers to this stage as latent scurvy.3 The 2 case studies that follow present examples of patients with vitamin C deficiencies whose parkinsonism responded robustly to vitamin C replacement. These cases suggest that vitamin C deficiency may be a treatable cause of parkinsonism.
Case 1
Mr. A, a 60-year-old white male, was admitted to the Medicine Service for alcohol detoxification. The patient had a history of alcohol dependence, alcohol withdrawal seizures, tobacco dependence, and hyperlipidemia. He took no medications as an outpatient. On admission Mr. A’s body mass index (BMI) was 27.2. An initial examination revealed a marked resting tremor of the patient’s right hand with cogwheeling, which had not been present in examinations conducted in the previous 3 years. Mr. A had no prior history of a tremor. He had no cerebellar findings and no evidence of asterixis or of tremulousness associated with high-output cardiac states, such as de Musset sign.
Mr. A reported he had experienced the tremor for a month and that it had been worsening. He also was having difficulty using his dominant right hand, for routine daily activities. Mr. A was oriented, and his short-term memory was intact. He was ill-appearing, irritable with psychomotor slowing, and did not wish to rise from his bed. He had no gingival or periungual bleeding and did not bruise easily. He had no corkscrew hairs. The patient was started on no medications known to cause extrapyramidal symptoms (EPS).
In the hospital, the tremor persisted unabated for 2 days. On the third day, Mr. A was started on 1,000 mg vitamin C IV twice daily. He received a total of 2,000 mg IV that day, but the IV fell out, and he refused its replacement. Several hours later, Mr. A stated that he felt much better, got out of bed, and asked to go outside to smoke. The author noted complete resolution of the right hand tremor and cogwheeling 20 hours after starting the vitamin C IV. Mr. A refused a repeat serum vitamin C assay.
Laboratory studies initially revealed that Mr. A had hyponatremia with a serum sodium of 121 mmol/L (normal range: 133 to 145 mmol/L) as well as hypokalemia with a serum potassium of 3.2 mmol/L (normal range: 3.5 to 5.0 mmol/L). He was hypoosmolar, with a serum osmolality of 276 mOsm/kg (normal range: 278 to 305 mOsm/kg). His vitamin C level was low at 0.2 mg/dL (normal range: 0.4 to 2.0 mg/dL). Mr. A also had a serum vitamin C level drawn 2 years prior that showed no symptoms of EPS, and at that time, the reading was 0.7 mg/dL. At admission to Medicine Services, Mr. A had a serum alcohol level of 211 mg/dL. Neuroimaging revealed diffuse cerebral and cerebellar volume loss.
Normal laboratory results included serum levels of vitamin B12, red cell folate, homocysteine, methylmalonic acid, free and total carnitine, alkaline phosphatase, manganese, and zinc. A urine drug screen was negative.
Case 2
Mr. B, a 69-year-old black male, was admitted to the hospital for depression complicated by alcohol dependence. He also had tobacco dependence, type 2 diabetes mellitus, hypertension, and gout. The patient’s BMI at admission was 16.1. Mr. B appeared ill, was worried about his health, and remained recumbent unless asked to move. He reported that his right hand had begun to shake at rest in the month prior to admission. The tremor made it difficult for him to drink. He pointed out stains on his hospital gurney from an attempt to drink orange juice prior to being assessed.
A physical examination revealed a distinct resting tremor with cogwheeling of the right hand; there was no other evidence of EPS, nor was there evidence of cognitive, cerebellar, or skin abnormalities, such as hemorrhages or corkscrew hairs. Asterixis was absent as was evidence of a high-output cardiac state that might produce a tremor, such as de Musset sign. A serum vitamin C level was obtained and returned at 0.0 mg/dL. A head computed tomography scan obtained the next day revealed mild cerebellar volume loss. A serum alkaline phosphatase level was elevated slightly at 136 U/L (normal range: 42 to 113 U/L). Normal serum values were returned for zinc, vitamins B12 and folate, rapid plasma reagin, sodium, and serum osmolality. A urine drug screen was negative, and serum alcohol level was < 5.0 mg/dL.
Mr. B took no medications expected to cause EPS. He received no micronutrient replacement until the day after admission when he began receiving oral vitamin C 1,000 mg twice a day. After receiving 3 doses, Mr. B’s resting tremor and cogwheeling completely resolved. He noticed he had stopped shaking and could now drink without spilling fluids. He also got out of bed and began interacting with others. Mr. B said he felt he was “doing well.” A repeat serum vitamin C level was 0.2 mg/dL on that day. The improvement was sustained over 3 days, and Mr. B was discharged to home.
Discussion
Both Mr. A and Mr. B presented with a typical picture of latent scurvy and the additional finding of parkinsonism. These cases are important for 2 reasons. First, the swift and full response of these patients’ parkinsonism to vitamin C replacement underscores the importance of considering a vitamin C deficiency when confronted with EPS. And second, both patients lacked signs of bleeding or of impaired collagen synthesis. This differs from the classic presentation of scurvy as a disorder primarily of collagen metabolism.4
Lind described the onset of scurvy as one in which striking emotional and behavior changes developed and later were followed by abnormal bleeding and even death.2 These early changes also were recognized by Shapter in 1847.5 Furthermore, the evidence that exists about the time-course of scurvy’s development suggests that neuropsychiatric findings precede the hemorrhagic.6 Indeed, classic skin findings, such as petechiae or corkscrew hairs, may develop years after the onset of neuropsychiatric changes.7,8
Despite WHO characterizing it as latent scurvy, the distinct syndromal presentation of hypovitaminosis C with parkinsonism along with the rapid response to vitamin C replacement argues for its recognition as a distinct clinical entity and not just a prelude to the hemorrhagic state. To assist in recognizing neuropsychiatric scurvy, the author suggests the operationalized approach described in Table 1.9
Pathophysiology
Vitamin C has an intimate role in the normal functioning of the basal ganglia. It is involved in the synthesis of catechecholamines, the regulation of the release and postsynaptic activities of various neurotransmitters, and managing the oxyradical toxicity of aerobic metabolism. Table 2 outlines some of the normal brain functions of vitamin C and the potential consequences of inadequate central vitamin C.9,10 Risk factors for vitamin C deficiency include those affecting the uptake, response to, and elimination of this vitamin (Table 3).11-14
The potential role of alcohol use by both patients also warrants mention. Current data suggest a nonlinear relationship between alcohol use and neurotoxicity. Epidemiologic data show that moderate alcohol consumption protects against the development of such neurodegenerative processes as Parkinson disease and Alzheimer disease.15,16 But the cases here reflect excessive use of alcohol. In this situation, a variety of progressive insults, such as those caused by oxyradical toxicity as well as malnutrition may foster the development of basal ganglia dysfunction.17
Measuring Deficiency
A deficiency of vitamin C may be determined in several ways. The most frequently used laboratory measure of vitamin C status is the serum vitamin C level. This level is included in the WHO’s recommendations for diagnosis.3 However, this assay is limited because when facing total body depletion, the kidneys may restrict the elimination of vitamin C and tend to maintain serum vitamin C levels even as target tissue levels fall. An interesting example of this is the 0.2 mg/dL value that each patient registered. In Mr. A’s case, this reflected a systemic deficit of vitamin C, while in Mr. B’s case it correlated with the onset of effective repletion of body’s stores.
A fall in urinary output of vitamin C is another marker of hypovitaminosis C. When available, this laboratory test can be used with the serum level to assess total body stores of vitamin C. Lymphocytes, neutrophils, and platelets also store vitamin C. These target tissues tend to saturate when the oral intake ranges between 100 mg to 200 mg a day. This is the same point at which serum vitamin C levels peak and level off in normal, healthy adults.18,19 Once again, the limited availability of target-tissue assays puts these studies out of reach for most clinicians.
No evaluation is complete without some assurance of what the disease is not. Deficiencies of biotin, zinc, folate, and B12 all may affect the function of the basal ganglia.20 The biotin deficiencies literature is particularly robust. Biotin deficiencies affecting basal ganglia function are best known as inherited disorders of metabolism.21 Manganese intoxication also may present as a movement disorder.22
Treatment
Treatment of neuropsychiatric scurvy has relied on IV administration of vitamin C. Although the bioavailability of oral vitamin C among healthy adult volunteers is nearly complete up to about 200 mg a day, a patient with neuropsychiatric scurvy may need substantially more than that amount to accommodate total body deficiencies and increased demands.23 The IV route allows serum vitamin C levels up to 100 times higher than by the oral route.24 Mr. B is, in fact, the first person reported in the literature with neuropsychiatric scurvy to respond to oral vitamin C replacement alone. Once repletion of vitamin C is complete, it is useful to consider a maintenance replacement dose based on a patient’s risk factors and needs.
A healthy adult should ingest about 120 mg of vitamin C daily. Smokers and pregnant women may require more, but this recommendation was intended to address their needs as well.25 Many commercial multivitamins use the old recommended daily allowance of 60 mg, so it may be safest to recommend specifically a vitamin C tablet with at least 120 mg when ordering vitamin C replacement.
Tight control of the serum vitamin C concentration means that little additional vitamin C will be taken up by the gut beyond 200 mg orally a day, which helps minimize any concerns about long-term toxicity. It takes several weeks to deplete vitamin C from the human body when vitamin C is removed from the diet, so a patient with a previously treated deficiency of vitamin C should wait a month before a repeat serum vitamin C level measurement.
The half life of vitamin C is normally ≤ 2 hours. When renal function is intact, vitamin C in excess of immediate need is lost through renal filtration. Toxicity is rare under these conditions.26 When vitamin C toxicity has been reported, it has occurred in the setting of prolonged supplementation, usually when a patient already experienced a renal injury. The main toxicities attributed to vitamin C are oxalate crystal formation with subsequent renal injury and exacerbation of glucose 6-phosphate dehydrogenase deficiency (G6PD).24
Oxalate formation due to vitamin C replacement is uncommon, but patients with preexisting calcium oxalate stones may be at risk for further stone formation when they receive additional vitamin C.27 This is most likely to occur when treatment with parenteral vitamin C is prolonged, which is not typical for patients with neuropsychiatric scurvy who tend to respond rapidly to vitamin C replenishment. Reports of acute hemolytic episodes among patient with G6PD deficiency receiving vitamin C exist, although these cases are rare.28 Furthermore, some authors advocate for the use of ascorbic acid to treat methemoglobinemia associated with G6PD deficiency, when methylene blue is not available.29 It may be reasonable to begin treatment with oral vitamin C for patients with NPS and G6PD deficiency. This is equivalent to a low-dose form of vitamin C replacement and may help avoid the theoretically pro-oxidant effects of larger, IV doses of vitamin C.30
Conclusion
The recent discovery of movement disorders in scurvy has enlarged the picture of vitamin C deficiency. The cases here demonstrate how hypovitaminosis C with central nervous system manifestations may be identified and treated. This relationship fits well within the established basic science and clinical framework for scurvy, and the clinical implications for scurvy remain in many ways unchanged. First, malnutrition must be considered even when a patient’s habitus suggests he is well fed. Also, it is more likely to see scurvy without all of the classic findings than an end-stage case of the disease.31 In the right clinical setting, it is reasonable to think of a vitamin C deficiency before the patient develops bleeding gums and corkscrew hairs. And as is typical of vitamin deficiencies, the treatment of a vitamin C deficiency usually results in swift improvement. Finally, for those who treat movement disorders or who prescribe agents such as antipsychotics that may cause movement disorders, it is important to recognize vitamin C deficiency as another potential explanation for EPS.
1. Ide K, Yamada H, Umegaki K, et al. Lymphocyte vitamin C levels as potential biomarker for progression of Parkinson’s disease. Nutrition. 2015;31(2):406-408.
2. Lind J. The diagnostics, or symptoms. A Treatise on the Scurvy, in Three Parts. 3rd ed. London: S. Crowder, D. Wilson and G. Nicholls, T. Cadell, T. Becket and Co., G. Pearch, and Woodfall; 1772:98-129.
3. World Health Organization. Scurvy and its prevention and control in major emergencies. http://whqlibdoc.who.int/hq/1999/WHO_NHD_99.11.pdf. Published 1999. Accessed July 6, 2017.
4. Sasseville D. Scurvy: curse and cure in New France. JAMA Dermatol. 2015;151(4):431.
5. Shapter T. On the recent occurrence of scurvy in Exeter and the neighbourhood. Prov Med Surg J. 1847;11(11):281-285.
6. Kinsman RA, Hood J. Some behavioral effects of ascorbic acid deficiency. Am J Clin Nutr. 1971;24(4):455-464.
7. DeSantis J. Scurvy and psychiatric symptoms. Perspect Psychiatr Care. 1993;29(1):18-22.
8. Walter JF. Scurvy resulting from a self-imposed diet. West J Med. 1979;130(2):177-179.
9. Brown TM. Neuropsychiatric scurvy. Psychosomatics. 2015;56(1):12-20.
10. Feuerstein TJ, Weinheimer G, Lang G, Ginap T, Rossner R. Inhibition by ascorbic acid of NMDA-evoked acetylcholine release in rabbit caudate nucleus. Naunyn Schmiedebergs Arch Pharmacol. 1993;348(5):549-551.
11. Kim J, Kwon J, Noh G, Lee SS. The effects of elimination diet on nutritional status in subjects with atopic dermatitis. Nutr Res Pract. 2013;7(6):488-494.
12. Langlois M, Duprez D, Delanghe J, De Buyzere M, Clement DL. Serum vitamin C concentration is low in peripheral arterial disease and is associated with inflammation and severity of atherosclerosis. Circulation. 2001;103(14):1863-1868.
13. Nappe TM, Pacelli AM, Katz K. An atypical case of methemoglobinemia due to self-administered benzocaine. Case Rep Emerg Med. 2015;2015:670979.
14. Wright AD, Stevens E, Ali M, Carroll DW, Brown TM. The neuropsychiatry of scurvy. Psychosomatics. 2014;55(2):179-185.
15. Bate C, Williams A. Ethanol protects cultured neurons against amyloid-β and α-synuclein-induced synapse damage. Neuropharmacology. 2011;61(8):1406-1412.
16. Vasanthi HR, Parameswari RP, DeLeiris J, Das DK. Health benefits of wine and alcohol from neuroprotection to heart health. Front Biosci (Elite Ed). 2012;4:1505-1512.
17. Vaglini F, Viaggi C, Piro V, et al. Acetaldehyde and parkinsonism: role of CYP450 2E1. Front Behav Neurosci. 2013;7:71.
18. Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci USA. 2001;98(17):9842-9846.
19. Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv Nutr. 2011;2(2):78-88.
20. Quiroga MJ, Carroll DW, Brown TM. Ascorbate- and zinc-responsive parkinsonism. Ann Pharmacother. 2014;48(11):1515-1520.
21. Tabarki B, Al-Shafi S, Al-Shahwan S, et al. Biotin-responsive basal ganglia disease revisited: clinical, radiologic, and genetic findings. Neurology. 2013;80(3):261-267.
22. Tuschl K, Mills PB, Clayton PT. Manganese and the brain. Int Rev Neurobiol. 2013;110:277-312.
23. Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci USA. 1996;93(8):3704-3709.
24. Wilson MK, Baguley BC, Wall C, Jameson MB, Findlay MP. Review of high-dose intravenous vitamin C as an anticancer agent. Asia Pac J Clin Oncol. 2014;10(1):22-37.
25. Carr AC, Frei B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am J Clin Nutr. 1999;69(6):1086-1107.
26. Nielsen TK, Højgaard M, Andersen JT, Poulsen HE, Lykkesfeldt J, Mikines KJ. Elimination of ascorbic acid after high-dose infusion in prostate cancer patients: a pharmacokinetic evaluation. Basic Clin Pharmacol Toxicol. 2015;116(4):343-348.
27. Baxmann AC, De O G Mendonça C, Heilberg IP. Effect of vitamin C supplements on urinary oxalate and pH in calcium stone-forming patients. Kidney Int. 2003;63(3):1066-1071.
28. Huang YC, Chang TK, Fu YC, Jan SL. C for colored urine: acute hemolysis induced by high-dose ascorbic acid. Clin Toxicol (Phila). 2014;52(9):984.
29. Rino PB, Scolnik D, Fustiñana A, Mitelpunkt A, Glatstein M. Ascorbic acid for the treatment of methemoglobinemia: the experience of a large tertiary care pediatric hospital. Am J Ther. 2014;21(4):240-243.
30. Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012;1826(2):443-457.
31. Fouron JC, Chicoine L. Le scorbut: aspects particuliers de l’association rachitisme-scorbut. Can Med Assoc J. 1962;86(26):1191-1196.
1. Ide K, Yamada H, Umegaki K, et al. Lymphocyte vitamin C levels as potential biomarker for progression of Parkinson’s disease. Nutrition. 2015;31(2):406-408.
2. Lind J. The diagnostics, or symptoms. A Treatise on the Scurvy, in Three Parts. 3rd ed. London: S. Crowder, D. Wilson and G. Nicholls, T. Cadell, T. Becket and Co., G. Pearch, and Woodfall; 1772:98-129.
3. World Health Organization. Scurvy and its prevention and control in major emergencies. http://whqlibdoc.who.int/hq/1999/WHO_NHD_99.11.pdf. Published 1999. Accessed July 6, 2017.
4. Sasseville D. Scurvy: curse and cure in New France. JAMA Dermatol. 2015;151(4):431.
5. Shapter T. On the recent occurrence of scurvy in Exeter and the neighbourhood. Prov Med Surg J. 1847;11(11):281-285.
6. Kinsman RA, Hood J. Some behavioral effects of ascorbic acid deficiency. Am J Clin Nutr. 1971;24(4):455-464.
7. DeSantis J. Scurvy and psychiatric symptoms. Perspect Psychiatr Care. 1993;29(1):18-22.
8. Walter JF. Scurvy resulting from a self-imposed diet. West J Med. 1979;130(2):177-179.
9. Brown TM. Neuropsychiatric scurvy. Psychosomatics. 2015;56(1):12-20.
10. Feuerstein TJ, Weinheimer G, Lang G, Ginap T, Rossner R. Inhibition by ascorbic acid of NMDA-evoked acetylcholine release in rabbit caudate nucleus. Naunyn Schmiedebergs Arch Pharmacol. 1993;348(5):549-551.
11. Kim J, Kwon J, Noh G, Lee SS. The effects of elimination diet on nutritional status in subjects with atopic dermatitis. Nutr Res Pract. 2013;7(6):488-494.
12. Langlois M, Duprez D, Delanghe J, De Buyzere M, Clement DL. Serum vitamin C concentration is low in peripheral arterial disease and is associated with inflammation and severity of atherosclerosis. Circulation. 2001;103(14):1863-1868.
13. Nappe TM, Pacelli AM, Katz K. An atypical case of methemoglobinemia due to self-administered benzocaine. Case Rep Emerg Med. 2015;2015:670979.
14. Wright AD, Stevens E, Ali M, Carroll DW, Brown TM. The neuropsychiatry of scurvy. Psychosomatics. 2014;55(2):179-185.
15. Bate C, Williams A. Ethanol protects cultured neurons against amyloid-β and α-synuclein-induced synapse damage. Neuropharmacology. 2011;61(8):1406-1412.
16. Vasanthi HR, Parameswari RP, DeLeiris J, Das DK. Health benefits of wine and alcohol from neuroprotection to heart health. Front Biosci (Elite Ed). 2012;4:1505-1512.
17. Vaglini F, Viaggi C, Piro V, et al. Acetaldehyde and parkinsonism: role of CYP450 2E1. Front Behav Neurosci. 2013;7:71.
18. Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci USA. 2001;98(17):9842-9846.
19. Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration-function approach yields pharmacology and therapeutic discoveries. Adv Nutr. 2011;2(2):78-88.
20. Quiroga MJ, Carroll DW, Brown TM. Ascorbate- and zinc-responsive parkinsonism. Ann Pharmacother. 2014;48(11):1515-1520.
21. Tabarki B, Al-Shafi S, Al-Shahwan S, et al. Biotin-responsive basal ganglia disease revisited: clinical, radiologic, and genetic findings. Neurology. 2013;80(3):261-267.
22. Tuschl K, Mills PB, Clayton PT. Manganese and the brain. Int Rev Neurobiol. 2013;110:277-312.
23. Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci USA. 1996;93(8):3704-3709.
24. Wilson MK, Baguley BC, Wall C, Jameson MB, Findlay MP. Review of high-dose intravenous vitamin C as an anticancer agent. Asia Pac J Clin Oncol. 2014;10(1):22-37.
25. Carr AC, Frei B. Toward a new recommended dietary allowance for vitamin C based on antioxidant and health effects in humans. Am J Clin Nutr. 1999;69(6):1086-1107.
26. Nielsen TK, Højgaard M, Andersen JT, Poulsen HE, Lykkesfeldt J, Mikines KJ. Elimination of ascorbic acid after high-dose infusion in prostate cancer patients: a pharmacokinetic evaluation. Basic Clin Pharmacol Toxicol. 2015;116(4):343-348.
27. Baxmann AC, De O G Mendonça C, Heilberg IP. Effect of vitamin C supplements on urinary oxalate and pH in calcium stone-forming patients. Kidney Int. 2003;63(3):1066-1071.
28. Huang YC, Chang TK, Fu YC, Jan SL. C for colored urine: acute hemolysis induced by high-dose ascorbic acid. Clin Toxicol (Phila). 2014;52(9):984.
29. Rino PB, Scolnik D, Fustiñana A, Mitelpunkt A, Glatstein M. Ascorbic acid for the treatment of methemoglobinemia: the experience of a large tertiary care pediatric hospital. Am J Ther. 2014;21(4):240-243.
30. Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012;1826(2):443-457.
31. Fouron JC, Chicoine L. Le scorbut: aspects particuliers de l’association rachitisme-scorbut. Can Med Assoc J. 1962;86(26):1191-1196.
Testing the Limits of Dual Antiplatelet Treatment for PCI Patients
What’s the right duration of dual antiplatelet therapy (DAPT) for patients who have had drug-eluting stents implanted? According to researchers from Sungkyunkwan University in Seoul, Korea, long-term clinical outcomes are similar whether patients receive therapy for less than or longer than 12 months.
The researchers conducted a retrospective study of 512 patients who had undergone percutaneous coronary intervention (PCI) for coronary chronic total occlusion (CTO) and were event free at 12 months. They separated the patients into 2 groups: 199 received aspirin and clopidogrel for ≤ 12 months, and 313 for > 12 months. The primary outcome was major adverse cardiac and cerebrovascular event (MACCE) during follow-up. Median follow-up was 64 months.
Related: Statins for the Physically Fit: Do They Help or Hurt?
No significant differences were seen between the groups in the incidence of MACCE: 21.6% of the patients in the ≤ 12-month group and 17.6% of the patients in the > 12-month group developed MACCE. After propensity matching, moderate or severe bleeding rates were also similar (1.6% in the shorter duration group and 2.2% in the longer duration group).
The researchers note that previously published data showed that longer duration DAPT was not associated with improved clinical outcomes in patients with CTO, although other subsets of complex PCI, such as longer stent length, bifurcation stenting, or multiple stents showed better clinical outcomes. To the best of their knowledge, the researchers add, theirs is the first study to directly compare DAPT durations in patients with CTO-PCI.
Related: A Heart Failure Management Program Using Shared Medical Appointments
Source:
Lee SH, Yang JH, Choi SH, et al. PLoS One. 2017;12(5):e0176737.
doi: 10.1371/journal.pone.0176737.
What’s the right duration of dual antiplatelet therapy (DAPT) for patients who have had drug-eluting stents implanted? According to researchers from Sungkyunkwan University in Seoul, Korea, long-term clinical outcomes are similar whether patients receive therapy for less than or longer than 12 months.
The researchers conducted a retrospective study of 512 patients who had undergone percutaneous coronary intervention (PCI) for coronary chronic total occlusion (CTO) and were event free at 12 months. They separated the patients into 2 groups: 199 received aspirin and clopidogrel for ≤ 12 months, and 313 for > 12 months. The primary outcome was major adverse cardiac and cerebrovascular event (MACCE) during follow-up. Median follow-up was 64 months.
Related: Statins for the Physically Fit: Do They Help or Hurt?
No significant differences were seen between the groups in the incidence of MACCE: 21.6% of the patients in the ≤ 12-month group and 17.6% of the patients in the > 12-month group developed MACCE. After propensity matching, moderate or severe bleeding rates were also similar (1.6% in the shorter duration group and 2.2% in the longer duration group).
The researchers note that previously published data showed that longer duration DAPT was not associated with improved clinical outcomes in patients with CTO, although other subsets of complex PCI, such as longer stent length, bifurcation stenting, or multiple stents showed better clinical outcomes. To the best of their knowledge, the researchers add, theirs is the first study to directly compare DAPT durations in patients with CTO-PCI.
Related: A Heart Failure Management Program Using Shared Medical Appointments
Source:
Lee SH, Yang JH, Choi SH, et al. PLoS One. 2017;12(5):e0176737.
doi: 10.1371/journal.pone.0176737.
What’s the right duration of dual antiplatelet therapy (DAPT) for patients who have had drug-eluting stents implanted? According to researchers from Sungkyunkwan University in Seoul, Korea, long-term clinical outcomes are similar whether patients receive therapy for less than or longer than 12 months.
The researchers conducted a retrospective study of 512 patients who had undergone percutaneous coronary intervention (PCI) for coronary chronic total occlusion (CTO) and were event free at 12 months. They separated the patients into 2 groups: 199 received aspirin and clopidogrel for ≤ 12 months, and 313 for > 12 months. The primary outcome was major adverse cardiac and cerebrovascular event (MACCE) during follow-up. Median follow-up was 64 months.
Related: Statins for the Physically Fit: Do They Help or Hurt?
No significant differences were seen between the groups in the incidence of MACCE: 21.6% of the patients in the ≤ 12-month group and 17.6% of the patients in the > 12-month group developed MACCE. After propensity matching, moderate or severe bleeding rates were also similar (1.6% in the shorter duration group and 2.2% in the longer duration group).
The researchers note that previously published data showed that longer duration DAPT was not associated with improved clinical outcomes in patients with CTO, although other subsets of complex PCI, such as longer stent length, bifurcation stenting, or multiple stents showed better clinical outcomes. To the best of their knowledge, the researchers add, theirs is the first study to directly compare DAPT durations in patients with CTO-PCI.
Related: A Heart Failure Management Program Using Shared Medical Appointments
Source:
Lee SH, Yang JH, Choi SH, et al. PLoS One. 2017;12(5):e0176737.
doi: 10.1371/journal.pone.0176737.
Sleep Apnea on the Rise Among Veterans
Male veterans who reported mild-to-moderate psychological distress in the previous year had 61% higher odds of experiencing sleep apnea, according to a California State University study. Those with serious psychological distress had 138% higher odds. The average prevalence of sleep apnea was 5.9%, but the proportions rose from 3.7% in 2005 to 8.1% in 2014.
The researchers analyzed data from the 2005-2014 National Survey on Drug Use and Health. They cite other research that found the age-adjusted prevalence of sleep apnea among U.S. veterans increased almost 6-fold from 2000 to 2010. They also point to an evaluation of veterans of Operation Enduring Freedom, Operation Iraqi Freedom, and Operation New Dawn that found that 69.2% of 159 veterans screened were at high risk for obstructive sleep apnea.
An even stronger risk factor was asthma. Veterans with a past-year diagnosis of asthma had 256% higher odds of experiencing sleep apnea than among those without asthma. The researchers note that men and women may be asymptomatic when they are recruited but develop asthma due to environmental factors during deployment. The researchers urge more and better screening regardless of whether the service member had asthma symptoms during recruitment.
Their study is unique, the researchers say, in that it demonstrates a putative relationship between sleep apnea and mental illness. They suggest multidisciplinary interventions, including peer-support strategies to improve veterans’ mental health and community-based resources to help improve access to health care. Above all, the researchers urge more rigorous screening of sleep apnea and better sleep apnea treatment for veterans.
Male veterans who reported mild-to-moderate psychological distress in the previous year had 61% higher odds of experiencing sleep apnea, according to a California State University study. Those with serious psychological distress had 138% higher odds. The average prevalence of sleep apnea was 5.9%, but the proportions rose from 3.7% in 2005 to 8.1% in 2014.
The researchers analyzed data from the 2005-2014 National Survey on Drug Use and Health. They cite other research that found the age-adjusted prevalence of sleep apnea among U.S. veterans increased almost 6-fold from 2000 to 2010. They also point to an evaluation of veterans of Operation Enduring Freedom, Operation Iraqi Freedom, and Operation New Dawn that found that 69.2% of 159 veterans screened were at high risk for obstructive sleep apnea.
An even stronger risk factor was asthma. Veterans with a past-year diagnosis of asthma had 256% higher odds of experiencing sleep apnea than among those without asthma. The researchers note that men and women may be asymptomatic when they are recruited but develop asthma due to environmental factors during deployment. The researchers urge more and better screening regardless of whether the service member had asthma symptoms during recruitment.
Their study is unique, the researchers say, in that it demonstrates a putative relationship between sleep apnea and mental illness. They suggest multidisciplinary interventions, including peer-support strategies to improve veterans’ mental health and community-based resources to help improve access to health care. Above all, the researchers urge more rigorous screening of sleep apnea and better sleep apnea treatment for veterans.
Male veterans who reported mild-to-moderate psychological distress in the previous year had 61% higher odds of experiencing sleep apnea, according to a California State University study. Those with serious psychological distress had 138% higher odds. The average prevalence of sleep apnea was 5.9%, but the proportions rose from 3.7% in 2005 to 8.1% in 2014.
The researchers analyzed data from the 2005-2014 National Survey on Drug Use and Health. They cite other research that found the age-adjusted prevalence of sleep apnea among U.S. veterans increased almost 6-fold from 2000 to 2010. They also point to an evaluation of veterans of Operation Enduring Freedom, Operation Iraqi Freedom, and Operation New Dawn that found that 69.2% of 159 veterans screened were at high risk for obstructive sleep apnea.
An even stronger risk factor was asthma. Veterans with a past-year diagnosis of asthma had 256% higher odds of experiencing sleep apnea than among those without asthma. The researchers note that men and women may be asymptomatic when they are recruited but develop asthma due to environmental factors during deployment. The researchers urge more and better screening regardless of whether the service member had asthma symptoms during recruitment.
Their study is unique, the researchers say, in that it demonstrates a putative relationship between sleep apnea and mental illness. They suggest multidisciplinary interventions, including peer-support strategies to improve veterans’ mental health and community-based resources to help improve access to health care. Above all, the researchers urge more rigorous screening of sleep apnea and better sleep apnea treatment for veterans.
Glucose metabolism deemed key to platelet survival
New research published in Cell Reports suggests platelets are highly reliant on their ability to metabolize glucose.
Researchers studied mice lacking proteins that platelets use to import glucose—glucose transporter (GLUT) 1 and GLUT3.
These mice had lower platelet counts and their platelets had shorter life spans than platelets in wild-type mice.
“We found that glucose metabolism is very critical across the entire life cycle of platelets—from production, to what they do in the body, to how they get cleared from the body,” said E. Dale Abel, MBBS, PhD, of the University of Iowa in Iowa City.
For this study, Dr Abel and his colleagues studied genetically engineered mouse models that were missing GLUT1 and GLUT3 or GLUT3 alone and observed how platelet formation, function, and clearance were affected.
Mice missing glucose transporter proteins did produce platelets, and the platelets’ mitochondria metabolized other substances in place of glucose. However, the mice had platelet counts that were lower than normal.
The researchers were able to pinpoint 2 causes for the low platelet count in mice lacking GLUT1 and GLUT3—fewer platelets being produced and increased clearance of platelets.
The team tested megakaryocytes’ ability to generate new platelets by depleting the blood of platelets and observing the subsequent recovery, which was lower than normal.
They also tested the megakaryocytes in culture, stimulating them to create new platelets, and found the generation of new platelets was defective.
“Clearly, we show that there’s an obligate need for glucose to bud platelets off from the bone marrow,” Dr Abel said.
In addition, the team observed that young platelets functioned normally, even in the absence of glucose. But as they aged, the platelets were cleared from the circulation earlier than normal because they were being destroyed.
“We identified a new mechanism of necrosis by which the absence of glucose leads to the cleavage of a protein called calpain, which marks them for this necrotic pathway,” Dr Abel said. “If we treated the animals with a calpain inhibitor, we could reduce the increased platelet clearance.”
The team also sought to determine whether platelets could exist and function when mitochondria metabolism is halted.
They injected the mice with oligomycin, which inhibits mitochondrial metabolism. In the mice deficient in GLUT1 and GLUT3, platelet counts dropped to 0 within about 30 minutes. The same effect did not occur in wild-type mice.
“This work defined a very important role for metabolism in how platelets leave the bone marrow, how they come into circulation, and how they stay in circulation,” Dr Abel said. “It could even have implications for why platelets have to be used within a certain period of time when they’re donated at the blood bank—the fresher the better.” ![]()
New research published in Cell Reports suggests platelets are highly reliant on their ability to metabolize glucose.
Researchers studied mice lacking proteins that platelets use to import glucose—glucose transporter (GLUT) 1 and GLUT3.
These mice had lower platelet counts and their platelets had shorter life spans than platelets in wild-type mice.
“We found that glucose metabolism is very critical across the entire life cycle of platelets—from production, to what they do in the body, to how they get cleared from the body,” said E. Dale Abel, MBBS, PhD, of the University of Iowa in Iowa City.
For this study, Dr Abel and his colleagues studied genetically engineered mouse models that were missing GLUT1 and GLUT3 or GLUT3 alone and observed how platelet formation, function, and clearance were affected.
Mice missing glucose transporter proteins did produce platelets, and the platelets’ mitochondria metabolized other substances in place of glucose. However, the mice had platelet counts that were lower than normal.
The researchers were able to pinpoint 2 causes for the low platelet count in mice lacking GLUT1 and GLUT3—fewer platelets being produced and increased clearance of platelets.
The team tested megakaryocytes’ ability to generate new platelets by depleting the blood of platelets and observing the subsequent recovery, which was lower than normal.
They also tested the megakaryocytes in culture, stimulating them to create new platelets, and found the generation of new platelets was defective.
“Clearly, we show that there’s an obligate need for glucose to bud platelets off from the bone marrow,” Dr Abel said.
In addition, the team observed that young platelets functioned normally, even in the absence of glucose. But as they aged, the platelets were cleared from the circulation earlier than normal because they were being destroyed.
“We identified a new mechanism of necrosis by which the absence of glucose leads to the cleavage of a protein called calpain, which marks them for this necrotic pathway,” Dr Abel said. “If we treated the animals with a calpain inhibitor, we could reduce the increased platelet clearance.”
The team also sought to determine whether platelets could exist and function when mitochondria metabolism is halted.
They injected the mice with oligomycin, which inhibits mitochondrial metabolism. In the mice deficient in GLUT1 and GLUT3, platelet counts dropped to 0 within about 30 minutes. The same effect did not occur in wild-type mice.
“This work defined a very important role for metabolism in how platelets leave the bone marrow, how they come into circulation, and how they stay in circulation,” Dr Abel said. “It could even have implications for why platelets have to be used within a certain period of time when they’re donated at the blood bank—the fresher the better.” ![]()
New research published in Cell Reports suggests platelets are highly reliant on their ability to metabolize glucose.
Researchers studied mice lacking proteins that platelets use to import glucose—glucose transporter (GLUT) 1 and GLUT3.
These mice had lower platelet counts and their platelets had shorter life spans than platelets in wild-type mice.
“We found that glucose metabolism is very critical across the entire life cycle of platelets—from production, to what they do in the body, to how they get cleared from the body,” said E. Dale Abel, MBBS, PhD, of the University of Iowa in Iowa City.
For this study, Dr Abel and his colleagues studied genetically engineered mouse models that were missing GLUT1 and GLUT3 or GLUT3 alone and observed how platelet formation, function, and clearance were affected.
Mice missing glucose transporter proteins did produce platelets, and the platelets’ mitochondria metabolized other substances in place of glucose. However, the mice had platelet counts that were lower than normal.
The researchers were able to pinpoint 2 causes for the low platelet count in mice lacking GLUT1 and GLUT3—fewer platelets being produced and increased clearance of platelets.
The team tested megakaryocytes’ ability to generate new platelets by depleting the blood of platelets and observing the subsequent recovery, which was lower than normal.
They also tested the megakaryocytes in culture, stimulating them to create new platelets, and found the generation of new platelets was defective.
“Clearly, we show that there’s an obligate need for glucose to bud platelets off from the bone marrow,” Dr Abel said.
In addition, the team observed that young platelets functioned normally, even in the absence of glucose. But as they aged, the platelets were cleared from the circulation earlier than normal because they were being destroyed.
“We identified a new mechanism of necrosis by which the absence of glucose leads to the cleavage of a protein called calpain, which marks them for this necrotic pathway,” Dr Abel said. “If we treated the animals with a calpain inhibitor, we could reduce the increased platelet clearance.”
The team also sought to determine whether platelets could exist and function when mitochondria metabolism is halted.
They injected the mice with oligomycin, which inhibits mitochondrial metabolism. In the mice deficient in GLUT1 and GLUT3, platelet counts dropped to 0 within about 30 minutes. The same effect did not occur in wild-type mice.
“This work defined a very important role for metabolism in how platelets leave the bone marrow, how they come into circulation, and how they stay in circulation,” Dr Abel said. “It could even have implications for why platelets have to be used within a certain period of time when they’re donated at the blood bank—the fresher the better.” ![]()
Team characterizes RIMs in childhood cancer survivors

Neuroscientists say they have uncovered genetic differences between radiation-induced meningiomas (RIMs) and sporadic meningiomas (SMs).
Their work suggests RIMs have a different “mutational landscape” from SMs, a finding that may have “significant therapeutic implications” for childhood cancer survivors who undergo cranial radiation.
Gelareh Zadeh, MD, PhD, of the University of Toronto in Ontario, Canada, and her colleagues reported these findings in Nature Communications.
“Radiation-induced meningiomas appear the same [as SMs] on MRI and pathology, feel the same during surgery, and look the same under the operating microscope,” Dr Zadeh said.
“What’s different is they are more aggressive, tend to recur in multiples, and invade the brain, causing significant morbidity and limitations (or impairments) for individuals who survive following childhood radiation. By understanding the biology, the goal is to identify a therapeutic strategy that could be implemented early on after childhood radiation to prevent the formation of these tumors in the first place.”
To better understand the biology, Dr Zadeh and her colleagues analyzed 31 RIMs. This included 18 tumors from 16 patients with childhood cancers, 9 with leukemia.
The researchers found NF2 gene rearrangements in 12 of the RIMs and noted that such rearrangements have not been observed in SMs.
On the other hand, recurrent mutations characteristic of SMs—AKT1, KLF4, TRAF7, and SMO—were not found in the RIMs.
The researchers also noted that, overall, chromosomal aberrations in RIMs were more complex than those observed in SMs. And combined loss of chromosomes 1p and 22q was common in RIMs (16/18).
“Our research identified a specific rearrangement involving the NF2 gene that causes radiation-induced meningiomas,” said Kenneth Aldape, MD, of University Health Network in Toronto.
“But there are likely other genetic rearrangements that are occurring as a result of that radiation-induced DNA damage. So one of the next steps is to identify what the radiation is doing to the DNA of the meninges.”
“In addition, identifying the subset of childhood cancer patients who are at highest risk to develop meningioma is critical so that they could be followed closely for early detection and management.” ![]()
Neuroscientists say they have uncovered genetic differences between radiation-induced meningiomas (RIMs) and sporadic meningiomas (SMs).
Their work suggests RIMs have a different “mutational landscape” from SMs, a finding that may have “significant therapeutic implications” for childhood cancer survivors who undergo cranial radiation.
Gelareh Zadeh, MD, PhD, of the University of Toronto in Ontario, Canada, and her colleagues reported these findings in Nature Communications.
“Radiation-induced meningiomas appear the same [as SMs] on MRI and pathology, feel the same during surgery, and look the same under the operating microscope,” Dr Zadeh said.
“What’s different is they are more aggressive, tend to recur in multiples, and invade the brain, causing significant morbidity and limitations (or impairments) for individuals who survive following childhood radiation. By understanding the biology, the goal is to identify a therapeutic strategy that could be implemented early on after childhood radiation to prevent the formation of these tumors in the first place.”
To better understand the biology, Dr Zadeh and her colleagues analyzed 31 RIMs. This included 18 tumors from 16 patients with childhood cancers, 9 with leukemia.
The researchers found NF2 gene rearrangements in 12 of the RIMs and noted that such rearrangements have not been observed in SMs.
On the other hand, recurrent mutations characteristic of SMs—AKT1, KLF4, TRAF7, and SMO—were not found in the RIMs.
The researchers also noted that, overall, chromosomal aberrations in RIMs were more complex than those observed in SMs. And combined loss of chromosomes 1p and 22q was common in RIMs (16/18).
“Our research identified a specific rearrangement involving the NF2 gene that causes radiation-induced meningiomas,” said Kenneth Aldape, MD, of University Health Network in Toronto.
“But there are likely other genetic rearrangements that are occurring as a result of that radiation-induced DNA damage. So one of the next steps is to identify what the radiation is doing to the DNA of the meninges.”
“In addition, identifying the subset of childhood cancer patients who are at highest risk to develop meningioma is critical so that they could be followed closely for early detection and management.” ![]()
Neuroscientists say they have uncovered genetic differences between radiation-induced meningiomas (RIMs) and sporadic meningiomas (SMs).
Their work suggests RIMs have a different “mutational landscape” from SMs, a finding that may have “significant therapeutic implications” for childhood cancer survivors who undergo cranial radiation.
Gelareh Zadeh, MD, PhD, of the University of Toronto in Ontario, Canada, and her colleagues reported these findings in Nature Communications.
“Radiation-induced meningiomas appear the same [as SMs] on MRI and pathology, feel the same during surgery, and look the same under the operating microscope,” Dr Zadeh said.
“What’s different is they are more aggressive, tend to recur in multiples, and invade the brain, causing significant morbidity and limitations (or impairments) for individuals who survive following childhood radiation. By understanding the biology, the goal is to identify a therapeutic strategy that could be implemented early on after childhood radiation to prevent the formation of these tumors in the first place.”
To better understand the biology, Dr Zadeh and her colleagues analyzed 31 RIMs. This included 18 tumors from 16 patients with childhood cancers, 9 with leukemia.
The researchers found NF2 gene rearrangements in 12 of the RIMs and noted that such rearrangements have not been observed in SMs.
On the other hand, recurrent mutations characteristic of SMs—AKT1, KLF4, TRAF7, and SMO—were not found in the RIMs.
The researchers also noted that, overall, chromosomal aberrations in RIMs were more complex than those observed in SMs. And combined loss of chromosomes 1p and 22q was common in RIMs (16/18).
“Our research identified a specific rearrangement involving the NF2 gene that causes radiation-induced meningiomas,” said Kenneth Aldape, MD, of University Health Network in Toronto.
“But there are likely other genetic rearrangements that are occurring as a result of that radiation-induced DNA damage. So one of the next steps is to identify what the radiation is doing to the DNA of the meninges.”
“In addition, identifying the subset of childhood cancer patients who are at highest risk to develop meningioma is critical so that they could be followed closely for early detection and management.” ![]()
Drug granted priority review, breakthrough designation for ECD

The US Food and Drug Administration (FDA) has granted priority review and breakthrough therapy designation to vemurafenib (Zelboraf®) as a treatment for Erdheim-Chester disease (ECD) with BRAF V600 mutation.
Vemurafenib is a kinase inhibitor designed to inhibit some mutated forms of BRAF.
The drug is already FDA-approved to treat patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test.
The FDA is expected to make a decision on the approval of vemurafenib in ECD by December 7, 2017.
The supplemental new drug application for vemurafenib in this indication is supported by data from the phase 2 VE-BASKET study.
VE-BASKET was designed to investigate the use of vemurafenib in patients with BRAF V600 mutation-positive diseases, including ECD.
Final results for the 22 people with ECD showed a best overall response rate of 54.5%. The median duration of response, progression-free survival, and overall survival were not reached at a median follow-up of 26.6 months.
The most common adverse events were joint pain, rash, hair loss, change in heart rhythm, fatigue, skin tags, diarrhea, and thickening of the skin. The most common grade 3 or higher adverse events were new skin cancers, high blood pressure, rash, and joint pain.
Initial results from this study were published in NEJM in August 2015.
About priority review
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need. ![]()
The US Food and Drug Administration (FDA) has granted priority review and breakthrough therapy designation to vemurafenib (Zelboraf®) as a treatment for Erdheim-Chester disease (ECD) with BRAF V600 mutation.
Vemurafenib is a kinase inhibitor designed to inhibit some mutated forms of BRAF.
The drug is already FDA-approved to treat patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test.
The FDA is expected to make a decision on the approval of vemurafenib in ECD by December 7, 2017.
The supplemental new drug application for vemurafenib in this indication is supported by data from the phase 2 VE-BASKET study.
VE-BASKET was designed to investigate the use of vemurafenib in patients with BRAF V600 mutation-positive diseases, including ECD.
Final results for the 22 people with ECD showed a best overall response rate of 54.5%. The median duration of response, progression-free survival, and overall survival were not reached at a median follow-up of 26.6 months.
The most common adverse events were joint pain, rash, hair loss, change in heart rhythm, fatigue, skin tags, diarrhea, and thickening of the skin. The most common grade 3 or higher adverse events were new skin cancers, high blood pressure, rash, and joint pain.
Initial results from this study were published in NEJM in August 2015.
About priority review
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need. ![]()
The US Food and Drug Administration (FDA) has granted priority review and breakthrough therapy designation to vemurafenib (Zelboraf®) as a treatment for Erdheim-Chester disease (ECD) with BRAF V600 mutation.
Vemurafenib is a kinase inhibitor designed to inhibit some mutated forms of BRAF.
The drug is already FDA-approved to treat patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test.
The FDA is expected to make a decision on the approval of vemurafenib in ECD by December 7, 2017.
The supplemental new drug application for vemurafenib in this indication is supported by data from the phase 2 VE-BASKET study.
VE-BASKET was designed to investigate the use of vemurafenib in patients with BRAF V600 mutation-positive diseases, including ECD.
Final results for the 22 people with ECD showed a best overall response rate of 54.5%. The median duration of response, progression-free survival, and overall survival were not reached at a median follow-up of 26.6 months.
The most common adverse events were joint pain, rash, hair loss, change in heart rhythm, fatigue, skin tags, diarrhea, and thickening of the skin. The most common grade 3 or higher adverse events were new skin cancers, high blood pressure, rash, and joint pain.
Initial results from this study were published in NEJM in August 2015.
About priority review
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need. ![]()
FDA issues EUA for Zika virus test

The US Food and Drug Administration (FDA) has issued an emergency use authorization (EUA) for Thermo Fisher Scientific’s TaqPath Zika Virus Kit.
This means the TaqPath Zika Virus Kit is authorized for the qualitative detection of RNA from Zika virus and diagnosis of Zika virus infection in human serum and urine (collected alongside a patient-matched serum specimen) from individuals meeting the US Centers for Disease Control and Prevention’s criteria for testing.
The EUA does not mean the TaqPath Zika Virus Kit is FDA cleared or approved.
An EUA allows for the use of unapproved medical products or unapproved uses of approved medical products in an emergency. The products must be used to diagnose, treat, or prevent serious or life-threatening conditions caused by chemical, biological, radiological, or nuclear threat agents, when there are no adequate alternatives.
The EUA for the TaqPath Zika Virus Kit means the test is only authorized as long as circumstances exist to justify the emergency use of in vitro diagnostics for the detection of Zika virus, unless the authorization is terminated or revoked sooner.
Testing using the TaqPath Zika Virus Kit is authorized to be conducted by laboratories in the US that are certified under the Clinical Laboratory Improvement Amendments of 1988, 42 U.S.C. § 263a, to perform high complexity tests, or by similarly qualified non-US laboratories.
More information on the TaqPath Zika Virus Kit and other Zika tests granted EUAs can be found on the FDA’s EUA page. ![]()
The US Food and Drug Administration (FDA) has issued an emergency use authorization (EUA) for Thermo Fisher Scientific’s TaqPath Zika Virus Kit.
This means the TaqPath Zika Virus Kit is authorized for the qualitative detection of RNA from Zika virus and diagnosis of Zika virus infection in human serum and urine (collected alongside a patient-matched serum specimen) from individuals meeting the US Centers for Disease Control and Prevention’s criteria for testing.
The EUA does not mean the TaqPath Zika Virus Kit is FDA cleared or approved.
An EUA allows for the use of unapproved medical products or unapproved uses of approved medical products in an emergency. The products must be used to diagnose, treat, or prevent serious or life-threatening conditions caused by chemical, biological, radiological, or nuclear threat agents, when there are no adequate alternatives.
The EUA for the TaqPath Zika Virus Kit means the test is only authorized as long as circumstances exist to justify the emergency use of in vitro diagnostics for the detection of Zika virus, unless the authorization is terminated or revoked sooner.
Testing using the TaqPath Zika Virus Kit is authorized to be conducted by laboratories in the US that are certified under the Clinical Laboratory Improvement Amendments of 1988, 42 U.S.C. § 263a, to perform high complexity tests, or by similarly qualified non-US laboratories.
More information on the TaqPath Zika Virus Kit and other Zika tests granted EUAs can be found on the FDA’s EUA page. ![]()
The US Food and Drug Administration (FDA) has issued an emergency use authorization (EUA) for Thermo Fisher Scientific’s TaqPath Zika Virus Kit.
This means the TaqPath Zika Virus Kit is authorized for the qualitative detection of RNA from Zika virus and diagnosis of Zika virus infection in human serum and urine (collected alongside a patient-matched serum specimen) from individuals meeting the US Centers for Disease Control and Prevention’s criteria for testing.
The EUA does not mean the TaqPath Zika Virus Kit is FDA cleared or approved.
An EUA allows for the use of unapproved medical products or unapproved uses of approved medical products in an emergency. The products must be used to diagnose, treat, or prevent serious or life-threatening conditions caused by chemical, biological, radiological, or nuclear threat agents, when there are no adequate alternatives.
The EUA for the TaqPath Zika Virus Kit means the test is only authorized as long as circumstances exist to justify the emergency use of in vitro diagnostics for the detection of Zika virus, unless the authorization is terminated or revoked sooner.
Testing using the TaqPath Zika Virus Kit is authorized to be conducted by laboratories in the US that are certified under the Clinical Laboratory Improvement Amendments of 1988, 42 U.S.C. § 263a, to perform high complexity tests, or by similarly qualified non-US laboratories.
More information on the TaqPath Zika Virus Kit and other Zika tests granted EUAs can be found on the FDA’s EUA page. ![]()
Small study advances noninvasive ICP monitoring
A device that noninvasively measures intracranial pressure (ICP) had a sensitivity of 75% and a specificity of 89%, compared with standard invasive monitoring, based on the results of an industry-sponsored study of 14 patients with traumatic brain injury or subarachnoid hemorrhage.
“This study provides the first clinical data on the accuracy of the HS-1000 noninvasive ICP monitor, which uses advanced signal analysis algorithms to evaluate properties of acoustic signals traveling through the brain,” wrote Oliver Ganslandt, MD, of Klinikum Stuttgart (Germany) and his associates. The findings were published online Aug. 8 in the Journal of Neurosurgery.
The noninvasive and invasive measurements produced more than 2,500 parallel ICP data points. Notably, each of the two methods produced the same number of data points. Readings averaged 10 (standard deviation, 6.1) mm Hg with invasive monitoring and 9.5 (SD, 4.7) mm Hg for noninvasive monitoring with the HS-1000. Compared with invasive ICP monitoring, the HS-1000 had a sensitivity of 75% and a specificity of 89% at an arbitrary cutoff of at least 17 mm Hg. Linear regression showed a “strong positive relationship between the [noninvasive and invasive] measurements,” the investigators said. In all, 63% of paired data points fell within 3 mm Hg of each other, and 85% fell within 5 mm Hg of each other. A receiver operating characteristic area under the curve analysis of the two methods generated an area under the curve of almost 90%.
The study did not include children or patients who were pregnant or had ear disease or ear injuries, rhinorrhea or otorrhea, or skull defects, the researchers said. If the HS-1000 holds up in other ongoing studies (NCT02284217, NCT02773901), physicians might be able to use it to decide if patients needs invasive ICP monitoring, they added. Use of the noninvasive method could also help prevent infections and other morbidity associated with invasive ICP monitoring in both neurocritical intensive care units and low-resource settings, they said.
HeadSense Medical sponsored the study. The researchers had no relevant disclosures.
A device that noninvasively measures intracranial pressure (ICP) had a sensitivity of 75% and a specificity of 89%, compared with standard invasive monitoring, based on the results of an industry-sponsored study of 14 patients with traumatic brain injury or subarachnoid hemorrhage.
“This study provides the first clinical data on the accuracy of the HS-1000 noninvasive ICP monitor, which uses advanced signal analysis algorithms to evaluate properties of acoustic signals traveling through the brain,” wrote Oliver Ganslandt, MD, of Klinikum Stuttgart (Germany) and his associates. The findings were published online Aug. 8 in the Journal of Neurosurgery.
The noninvasive and invasive measurements produced more than 2,500 parallel ICP data points. Notably, each of the two methods produced the same number of data points. Readings averaged 10 (standard deviation, 6.1) mm Hg with invasive monitoring and 9.5 (SD, 4.7) mm Hg for noninvasive monitoring with the HS-1000. Compared with invasive ICP monitoring, the HS-1000 had a sensitivity of 75% and a specificity of 89% at an arbitrary cutoff of at least 17 mm Hg. Linear regression showed a “strong positive relationship between the [noninvasive and invasive] measurements,” the investigators said. In all, 63% of paired data points fell within 3 mm Hg of each other, and 85% fell within 5 mm Hg of each other. A receiver operating characteristic area under the curve analysis of the two methods generated an area under the curve of almost 90%.
The study did not include children or patients who were pregnant or had ear disease or ear injuries, rhinorrhea or otorrhea, or skull defects, the researchers said. If the HS-1000 holds up in other ongoing studies (NCT02284217, NCT02773901), physicians might be able to use it to decide if patients needs invasive ICP monitoring, they added. Use of the noninvasive method could also help prevent infections and other morbidity associated with invasive ICP monitoring in both neurocritical intensive care units and low-resource settings, they said.
HeadSense Medical sponsored the study. The researchers had no relevant disclosures.
A device that noninvasively measures intracranial pressure (ICP) had a sensitivity of 75% and a specificity of 89%, compared with standard invasive monitoring, based on the results of an industry-sponsored study of 14 patients with traumatic brain injury or subarachnoid hemorrhage.
“This study provides the first clinical data on the accuracy of the HS-1000 noninvasive ICP monitor, which uses advanced signal analysis algorithms to evaluate properties of acoustic signals traveling through the brain,” wrote Oliver Ganslandt, MD, of Klinikum Stuttgart (Germany) and his associates. The findings were published online Aug. 8 in the Journal of Neurosurgery.
The noninvasive and invasive measurements produced more than 2,500 parallel ICP data points. Notably, each of the two methods produced the same number of data points. Readings averaged 10 (standard deviation, 6.1) mm Hg with invasive monitoring and 9.5 (SD, 4.7) mm Hg for noninvasive monitoring with the HS-1000. Compared with invasive ICP monitoring, the HS-1000 had a sensitivity of 75% and a specificity of 89% at an arbitrary cutoff of at least 17 mm Hg. Linear regression showed a “strong positive relationship between the [noninvasive and invasive] measurements,” the investigators said. In all, 63% of paired data points fell within 3 mm Hg of each other, and 85% fell within 5 mm Hg of each other. A receiver operating characteristic area under the curve analysis of the two methods generated an area under the curve of almost 90%.
The study did not include children or patients who were pregnant or had ear disease or ear injuries, rhinorrhea or otorrhea, or skull defects, the researchers said. If the HS-1000 holds up in other ongoing studies (NCT02284217, NCT02773901), physicians might be able to use it to decide if patients needs invasive ICP monitoring, they added. Use of the noninvasive method could also help prevent infections and other morbidity associated with invasive ICP monitoring in both neurocritical intensive care units and low-resource settings, they said.
HeadSense Medical sponsored the study. The researchers had no relevant disclosures.
FROM THE JOURNAL OF NEUROSURGERY
Key clinical point: A noninvasive device that measures intracranial pressure generated data that was comparable with standard invasive methods.
Major finding: Sensitivity was 75%, and specificity was 89%, compared with standard invasive monitoring at an arbitrary cutoff of at least 17 mm Hg.
Data source: Noninvasive and invasive intracranial pressure monitoring of 14 patients with traumatic brain injury or subarachnoid hemorrhage.
Disclosures: HeadSense Medical sponsored the study. The researchers had no relevant disclosures.

Prescribe This Combined OC With CV Safety in Mind

A 28-year-old woman presents to your office for a routine health maintenance exam. She is currently using an oral contraceptive containing desogestrel and ethinyl estradiol for contraception and is inquiring about a refill for the coming year. What would you recommend?
When choosing a combined oral contraceptive (COC) for a pa
In general, when compared with nonusers, women who use COCs have a two- to four-fold increase in risk for venous thromboembolism (VTE) and an increased risk for myocardial infarction (MI) and stroke.2,3 More specifically, higher doses of estrogen combined with the progesterones gestodene, desogestrel, and levonorgestrel, are associated with a higher risk for VTE.2-6
In 2012, the European Medicines Agency warned that COCs containing drospirenone were associated with a higher risk for VTE than other preparations, despite similar estrogen content.7 The FDA produced a similar statement that same year, recommending that providers carefully consider the risks and benefits before prescribing contraceptives containing drospirenone.8
The risks for ischemic stroke and MI have not been clearly established for varying doses of estrogen and different progesterones. This large observational study fills that informational gap by providing risk estimates for the various COC options.
STUDY SUMMARY
One COC comes out ahead
The authors used an observational cohort model to determine the effects of different doses of estrogen combined with different progesterones in COCs on the risks for pulmonary embolism (PE), ischemic stroke, and MI.1 Data were collected from the French national health insurance database and the French national hospital discharge database.9,10 The study included nearly 5 million women ages 15 to 49, living in France, who had at least one prescription filled for COCs between July 2010 and September 2012.
The investigators calculated the absolute and relative risks for first PE, ischemic stroke, and MI in women using COC formulations containing either low-dose estrogen (20 µg) or high-dose estrogen (30-40 µg) combined with one of five progesterones (norethisterone, norgestrel, levonorgestrel, desogestrel, gestodene). The relative risk (RR) was adjusted for confounding factors, including age, complimentary universal health insurance, socioeconomic status, hypertension, diabetes, and consultation with a gynecologist in the previous year.
The absolute risk per 100,000 woman-years for all COC use was 33 for PE, 19 for ischemic stroke, and 7 for MI, with a composite risk of 60. The RRs for low-dose estrogen vs high-dose estrogen were 0.75 for PE, 0.82 for ischemic stroke, and 0.56 for MI. The absolute risk reduction (ARR) with low-dose estrogen vs high-dose estrogen was 14/100,000 person-years of use; the number needed to harm (NNH) was 7,143.
Compared with levonorgestrel, desogestrel and gestodene were associated with higher RRs for PE but not arterial events (2.16 for desogestrel and 1.63 for gestodene). For PE, the ARR with levonorgestrel compared to desogestrel and gestodene, respectively, was 19/100,000 and 12/100,000 person-years of use (NNH, 5,263 and 8,333, respectively). The authors concluded that for the same progesterone, using a lower dose of estrogen decreases risk for PE, ischemic stroke, and MI, and that oral contraceptives containing levonorgestrel and low-dose estrogen resulted in the lowest overall risks for PE and arterial thromboembolism.
WHAT’S NEW?
Low-dose estrogen + levonorgestrel confer lowest risk
Prior studies have shown that COCs increase the risk for PE and may also increase the risks for ischemic stroke and MI.3,11 Studies have also suggested that a higher dose of estrogen in COCs is associated with an increased risk for VTE.11,12 This study shows that 20 µg of estrogen combined with levonorgestrel is associated with the lowest risks for PE, MI, and ischemic stroke.
CAVEATS
Cohort study, no start date, incomplete tobacco use data
This is an observational cohort study, so it is subject to confounding factors and biases. It does, however, include a very large population, which improves validity. The study did not account for COC start date, which may be confounding because the risk for VTE is highest in the first three months to one year of COC use.12 Data on tobacco use, a significant independent risk factor for arterial but not venous thromboembolism, was incomplete; however, in other studies, it has only marginally affected outcomes.3,13
CHALLENGES TO IMPLEMENTATION
Increased vaginal spotting
One potential challenge to implementing this practice changer may be the increased rate of vaginal spotting associated with low-dose estrogen. COCs containing 20 µg of estrogen are associated with spotting in approximately two-thirds of menstrual cycles over the course of a year.14 That said, women may prefer to endure the spotting in light of the improved safety profile of a lower-dose estrogen pill.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2017. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2017;66[7]:454-456).
1. Weill A, Dalichampt M, Raguideau F, et al. Low dose oestrogen combined oral contraception and risk of pulmonary embolism, stroke, and myocardial infarction in five million French women: cohort study. BMJ. 2016;353:i2002.
2. Lidegaard Ø, Løkkegaard E, Svendsen AL, et al. Hormonal contraception and risk of venous thromboembolism: national follow-up study. BMJ. 2009;339:b2890.
3. Lidegaard Ø, Løkkegaard E, Jensen A, et al. Thrombotic stroke and myocardial infarction with hormonal contraception. N Engl J Med. 2012;366:2257-2266.
4. Stegeman BH, de Bastos M, Rosendaal FR, et al. Different combined oral contraceptives and the risk of venous thrombosis: systematic review and network meta-analysis. BMJ. 2013;347:f5298.
5. FDA. Combined hormonal contraceptives (CHCs) and the risk of cardiovascular disease endpoints. www.fda.gov/downloads/drugs/drugsafety/ucm277384. Accessed July 5, 2017.
6. Seeger JD, Loughlin J, Eng PM, et al. Risk of thromboembolism in women taking ethinyl estradiol/drospirenone and other oral contraceptives. Obstet Gynecol. 2007;110:587-593.
7. European Medicines Agency. PhVWP monthly report on safety concerns, guidelines and general matters. 2012. www.ema.europa.eu/docs/en_GB/document_library/Report/2012/01/WC500121387.pdf. Accessed July 5, 2017.
8. FDA. FDA Drug Safety Communication: Updated information about the risk of blood clots in women taking birth control pills containing drospirenone. 2012. www.fda.gov/Drugs/DrugSafety/ucm299305.htm. Accessed July 5, 2017.
9. Tuppin P, de Roquefeuil L, Weill A, et al. French national health insurance information system and the permanent beneficiaries sample. Rev Epidemiol Sante Publique. 2010;58:286-290.
10. Moulis G, Lapeyre-Mestre M, Palmaro A, et al. French health insurance databases: what interest for medical research? Rev Med Interne. 2015;36:411-417.
11. Farmer RD, Lawrenson RA, Thompson CR, et al. Population-based study of risk of venous thromboembolism associated with various oral contraceptives. Lancet. 1997;349:83-88.
12. Lidegaard Ø, Nielsen LH, Skovlund CW, et al. Risk of venous thromboembolism from use of oral contraceptives containing different progestogens and oestrogen doses: Danish cohort study, 2001-9. BMJ. 2011;343:d6423.
13. Zhang G, Xu X, Su W, et al. Smoking and risk of venous thromboembolism: a systematic review. Southeast Asian J Trop Med Public Health. 2014;45:736-745.
14. Akerlund M, Røde A, Westergaard J. Comparative profiles of reliability, cycle control and side effects of two oral contraceptive formulations containing 150 micrograms desogestrel and either 30 micrograms or 20 micrograms ethinyl oestradiol. Br J Obstet Gynaecol. 1993;100:832-838.
A 28-year-old woman presents to your office for a routine health maintenance exam. She is currently using an oral contraceptive containing desogestrel and ethinyl estradiol for contraception and is inquiring about a refill for the coming year. What would you recommend?
When choosing a combined oral contraceptive (COC) for a pa
In general, when compared with nonusers, women who use COCs have a two- to four-fold increase in risk for venous thromboembolism (VTE) and an increased risk for myocardial infarction (MI) and stroke.2,3 More specifically, higher doses of estrogen combined with the progesterones gestodene, desogestrel, and levonorgestrel, are associated with a higher risk for VTE.2-6
In 2012, the European Medicines Agency warned that COCs containing drospirenone were associated with a higher risk for VTE than other preparations, despite similar estrogen content.7 The FDA produced a similar statement that same year, recommending that providers carefully consider the risks and benefits before prescribing contraceptives containing drospirenone.8
The risks for ischemic stroke and MI have not been clearly established for varying doses of estrogen and different progesterones. This large observational study fills that informational gap by providing risk estimates for the various COC options.
STUDY SUMMARY
One COC comes out ahead
The authors used an observational cohort model to determine the effects of different doses of estrogen combined with different progesterones in COCs on the risks for pulmonary embolism (PE), ischemic stroke, and MI.1 Data were collected from the French national health insurance database and the French national hospital discharge database.9,10 The study included nearly 5 million women ages 15 to 49, living in France, who had at least one prescription filled for COCs between July 2010 and September 2012.
The investigators calculated the absolute and relative risks for first PE, ischemic stroke, and MI in women using COC formulations containing either low-dose estrogen (20 µg) or high-dose estrogen (30-40 µg) combined with one of five progesterones (norethisterone, norgestrel, levonorgestrel, desogestrel, gestodene). The relative risk (RR) was adjusted for confounding factors, including age, complimentary universal health insurance, socioeconomic status, hypertension, diabetes, and consultation with a gynecologist in the previous year.
The absolute risk per 100,000 woman-years for all COC use was 33 for PE, 19 for ischemic stroke, and 7 for MI, with a composite risk of 60. The RRs for low-dose estrogen vs high-dose estrogen were 0.75 for PE, 0.82 for ischemic stroke, and 0.56 for MI. The absolute risk reduction (ARR) with low-dose estrogen vs high-dose estrogen was 14/100,000 person-years of use; the number needed to harm (NNH) was 7,143.
Compared with levonorgestrel, desogestrel and gestodene were associated with higher RRs for PE but not arterial events (2.16 for desogestrel and 1.63 for gestodene). For PE, the ARR with levonorgestrel compared to desogestrel and gestodene, respectively, was 19/100,000 and 12/100,000 person-years of use (NNH, 5,263 and 8,333, respectively). The authors concluded that for the same progesterone, using a lower dose of estrogen decreases risk for PE, ischemic stroke, and MI, and that oral contraceptives containing levonorgestrel and low-dose estrogen resulted in the lowest overall risks for PE and arterial thromboembolism.
WHAT’S NEW?
Low-dose estrogen + levonorgestrel confer lowest risk
Prior studies have shown that COCs increase the risk for PE and may also increase the risks for ischemic stroke and MI.3,11 Studies have also suggested that a higher dose of estrogen in COCs is associated with an increased risk for VTE.11,12 This study shows that 20 µg of estrogen combined with levonorgestrel is associated with the lowest risks for PE, MI, and ischemic stroke.
CAVEATS
Cohort study, no start date, incomplete tobacco use data
This is an observational cohort study, so it is subject to confounding factors and biases. It does, however, include a very large population, which improves validity. The study did not account for COC start date, which may be confounding because the risk for VTE is highest in the first three months to one year of COC use.12 Data on tobacco use, a significant independent risk factor for arterial but not venous thromboembolism, was incomplete; however, in other studies, it has only marginally affected outcomes.3,13
CHALLENGES TO IMPLEMENTATION
Increased vaginal spotting
One potential challenge to implementing this practice changer may be the increased rate of vaginal spotting associated with low-dose estrogen. COCs containing 20 µg of estrogen are associated with spotting in approximately two-thirds of menstrual cycles over the course of a year.14 That said, women may prefer to endure the spotting in light of the improved safety profile of a lower-dose estrogen pill.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2017. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2017;66[7]:454-456).
A 28-year-old woman presents to your office for a routine health maintenance exam. She is currently using an oral contraceptive containing desogestrel and ethinyl estradiol for contraception and is inquiring about a refill for the coming year. What would you recommend?
When choosing a combined oral contraceptive (COC) for a pa
In general, when compared with nonusers, women who use COCs have a two- to four-fold increase in risk for venous thromboembolism (VTE) and an increased risk for myocardial infarction (MI) and stroke.2,3 More specifically, higher doses of estrogen combined with the progesterones gestodene, desogestrel, and levonorgestrel, are associated with a higher risk for VTE.2-6
In 2012, the European Medicines Agency warned that COCs containing drospirenone were associated with a higher risk for VTE than other preparations, despite similar estrogen content.7 The FDA produced a similar statement that same year, recommending that providers carefully consider the risks and benefits before prescribing contraceptives containing drospirenone.8
The risks for ischemic stroke and MI have not been clearly established for varying doses of estrogen and different progesterones. This large observational study fills that informational gap by providing risk estimates for the various COC options.
STUDY SUMMARY
One COC comes out ahead
The authors used an observational cohort model to determine the effects of different doses of estrogen combined with different progesterones in COCs on the risks for pulmonary embolism (PE), ischemic stroke, and MI.1 Data were collected from the French national health insurance database and the French national hospital discharge database.9,10 The study included nearly 5 million women ages 15 to 49, living in France, who had at least one prescription filled for COCs between July 2010 and September 2012.
The investigators calculated the absolute and relative risks for first PE, ischemic stroke, and MI in women using COC formulations containing either low-dose estrogen (20 µg) or high-dose estrogen (30-40 µg) combined with one of five progesterones (norethisterone, norgestrel, levonorgestrel, desogestrel, gestodene). The relative risk (RR) was adjusted for confounding factors, including age, complimentary universal health insurance, socioeconomic status, hypertension, diabetes, and consultation with a gynecologist in the previous year.
The absolute risk per 100,000 woman-years for all COC use was 33 for PE, 19 for ischemic stroke, and 7 for MI, with a composite risk of 60. The RRs for low-dose estrogen vs high-dose estrogen were 0.75 for PE, 0.82 for ischemic stroke, and 0.56 for MI. The absolute risk reduction (ARR) with low-dose estrogen vs high-dose estrogen was 14/100,000 person-years of use; the number needed to harm (NNH) was 7,143.
Compared with levonorgestrel, desogestrel and gestodene were associated with higher RRs for PE but not arterial events (2.16 for desogestrel and 1.63 for gestodene). For PE, the ARR with levonorgestrel compared to desogestrel and gestodene, respectively, was 19/100,000 and 12/100,000 person-years of use (NNH, 5,263 and 8,333, respectively). The authors concluded that for the same progesterone, using a lower dose of estrogen decreases risk for PE, ischemic stroke, and MI, and that oral contraceptives containing levonorgestrel and low-dose estrogen resulted in the lowest overall risks for PE and arterial thromboembolism.
WHAT’S NEW?
Low-dose estrogen + levonorgestrel confer lowest risk
Prior studies have shown that COCs increase the risk for PE and may also increase the risks for ischemic stroke and MI.3,11 Studies have also suggested that a higher dose of estrogen in COCs is associated with an increased risk for VTE.11,12 This study shows that 20 µg of estrogen combined with levonorgestrel is associated with the lowest risks for PE, MI, and ischemic stroke.
CAVEATS
Cohort study, no start date, incomplete tobacco use data
This is an observational cohort study, so it is subject to confounding factors and biases. It does, however, include a very large population, which improves validity. The study did not account for COC start date, which may be confounding because the risk for VTE is highest in the first three months to one year of COC use.12 Data on tobacco use, a significant independent risk factor for arterial but not venous thromboembolism, was incomplete; however, in other studies, it has only marginally affected outcomes.3,13
CHALLENGES TO IMPLEMENTATION
Increased vaginal spotting
One potential challenge to implementing this practice changer may be the increased rate of vaginal spotting associated with low-dose estrogen. COCs containing 20 µg of estrogen are associated with spotting in approximately two-thirds of menstrual cycles over the course of a year.14 That said, women may prefer to endure the spotting in light of the improved safety profile of a lower-dose estrogen pill.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2017. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2017;66[7]:454-456).
1. Weill A, Dalichampt M, Raguideau F, et al. Low dose oestrogen combined oral contraception and risk of pulmonary embolism, stroke, and myocardial infarction in five million French women: cohort study. BMJ. 2016;353:i2002.
2. Lidegaard Ø, Løkkegaard E, Svendsen AL, et al. Hormonal contraception and risk of venous thromboembolism: national follow-up study. BMJ. 2009;339:b2890.
3. Lidegaard Ø, Løkkegaard E, Jensen A, et al. Thrombotic stroke and myocardial infarction with hormonal contraception. N Engl J Med. 2012;366:2257-2266.
4. Stegeman BH, de Bastos M, Rosendaal FR, et al. Different combined oral contraceptives and the risk of venous thrombosis: systematic review and network meta-analysis. BMJ. 2013;347:f5298.
5. FDA. Combined hormonal contraceptives (CHCs) and the risk of cardiovascular disease endpoints. www.fda.gov/downloads/drugs/drugsafety/ucm277384. Accessed July 5, 2017.
6. Seeger JD, Loughlin J, Eng PM, et al. Risk of thromboembolism in women taking ethinyl estradiol/drospirenone and other oral contraceptives. Obstet Gynecol. 2007;110:587-593.
7. European Medicines Agency. PhVWP monthly report on safety concerns, guidelines and general matters. 2012. www.ema.europa.eu/docs/en_GB/document_library/Report/2012/01/WC500121387.pdf. Accessed July 5, 2017.
8. FDA. FDA Drug Safety Communication: Updated information about the risk of blood clots in women taking birth control pills containing drospirenone. 2012. www.fda.gov/Drugs/DrugSafety/ucm299305.htm. Accessed July 5, 2017.
9. Tuppin P, de Roquefeuil L, Weill A, et al. French national health insurance information system and the permanent beneficiaries sample. Rev Epidemiol Sante Publique. 2010;58:286-290.
10. Moulis G, Lapeyre-Mestre M, Palmaro A, et al. French health insurance databases: what interest for medical research? Rev Med Interne. 2015;36:411-417.
11. Farmer RD, Lawrenson RA, Thompson CR, et al. Population-based study of risk of venous thromboembolism associated with various oral contraceptives. Lancet. 1997;349:83-88.
12. Lidegaard Ø, Nielsen LH, Skovlund CW, et al. Risk of venous thromboembolism from use of oral contraceptives containing different progestogens and oestrogen doses: Danish cohort study, 2001-9. BMJ. 2011;343:d6423.
13. Zhang G, Xu X, Su W, et al. Smoking and risk of venous thromboembolism: a systematic review. Southeast Asian J Trop Med Public Health. 2014;45:736-745.
14. Akerlund M, Røde A, Westergaard J. Comparative profiles of reliability, cycle control and side effects of two oral contraceptive formulations containing 150 micrograms desogestrel and either 30 micrograms or 20 micrograms ethinyl oestradiol. Br J Obstet Gynaecol. 1993;100:832-838.
1. Weill A, Dalichampt M, Raguideau F, et al. Low dose oestrogen combined oral contraception and risk of pulmonary embolism, stroke, and myocardial infarction in five million French women: cohort study. BMJ. 2016;353:i2002.
2. Lidegaard Ø, Løkkegaard E, Svendsen AL, et al. Hormonal contraception and risk of venous thromboembolism: national follow-up study. BMJ. 2009;339:b2890.
3. Lidegaard Ø, Løkkegaard E, Jensen A, et al. Thrombotic stroke and myocardial infarction with hormonal contraception. N Engl J Med. 2012;366:2257-2266.
4. Stegeman BH, de Bastos M, Rosendaal FR, et al. Different combined oral contraceptives and the risk of venous thrombosis: systematic review and network meta-analysis. BMJ. 2013;347:f5298.
5. FDA. Combined hormonal contraceptives (CHCs) and the risk of cardiovascular disease endpoints. www.fda.gov/downloads/drugs/drugsafety/ucm277384. Accessed July 5, 2017.
6. Seeger JD, Loughlin J, Eng PM, et al. Risk of thromboembolism in women taking ethinyl estradiol/drospirenone and other oral contraceptives. Obstet Gynecol. 2007;110:587-593.
7. European Medicines Agency. PhVWP monthly report on safety concerns, guidelines and general matters. 2012. www.ema.europa.eu/docs/en_GB/document_library/Report/2012/01/WC500121387.pdf. Accessed July 5, 2017.
8. FDA. FDA Drug Safety Communication: Updated information about the risk of blood clots in women taking birth control pills containing drospirenone. 2012. www.fda.gov/Drugs/DrugSafety/ucm299305.htm. Accessed July 5, 2017.
9. Tuppin P, de Roquefeuil L, Weill A, et al. French national health insurance information system and the permanent beneficiaries sample. Rev Epidemiol Sante Publique. 2010;58:286-290.
10. Moulis G, Lapeyre-Mestre M, Palmaro A, et al. French health insurance databases: what interest for medical research? Rev Med Interne. 2015;36:411-417.
11. Farmer RD, Lawrenson RA, Thompson CR, et al. Population-based study of risk of venous thromboembolism associated with various oral contraceptives. Lancet. 1997;349:83-88.
12. Lidegaard Ø, Nielsen LH, Skovlund CW, et al. Risk of venous thromboembolism from use of oral contraceptives containing different progestogens and oestrogen doses: Danish cohort study, 2001-9. BMJ. 2011;343:d6423.
13. Zhang G, Xu X, Su W, et al. Smoking and risk of venous thromboembolism: a systematic review. Southeast Asian J Trop Med Public Health. 2014;45:736-745.
14. Akerlund M, Røde A, Westergaard J. Comparative profiles of reliability, cycle control and side effects of two oral contraceptive formulations containing 150 micrograms desogestrel and either 30 micrograms or 20 micrograms ethinyl oestradiol. Br J Obstet Gynaecol. 1993;100:832-838.
Thyroid Storm: Early Management and Prevention
A 73-year-old man is transported to the emergency department (ED) by ambulance for nausea, vomiting, diarrhea, and weakness of three days’ duration. Earlier today, he presented to his primary care provider with these symptoms and was found to be hypotensive; he was advised to go to the ED but instead went home against medical advice.
The patient’s medical history is significant for type 2 diabetes, stage 3b chronic kidney disease, dyslipidemia, hypertension, coronary artery disease, and benign prostatic hyperplasia. He has undergone stent placement and triple coronary artery bypass graft surgery. His medication list includes insulin glargine, glimepiride, liraglutide, atorvastatin, benazepril, carvedilol, amlodipine, clopidogrel, and tamsulosin.
Upon admission, the patient has a pulse of 98 beats/min; temperature, 98.2°F; respiratory rate, 18 breaths/min-1; and PO2, 98 mm Hg. An ECG, chest radiograph, and CT (without contrast) of the head, chest, and abdomen are all within normal limits. Lab evaluation is significant for severe thyrotoxicosis (see Table 1).
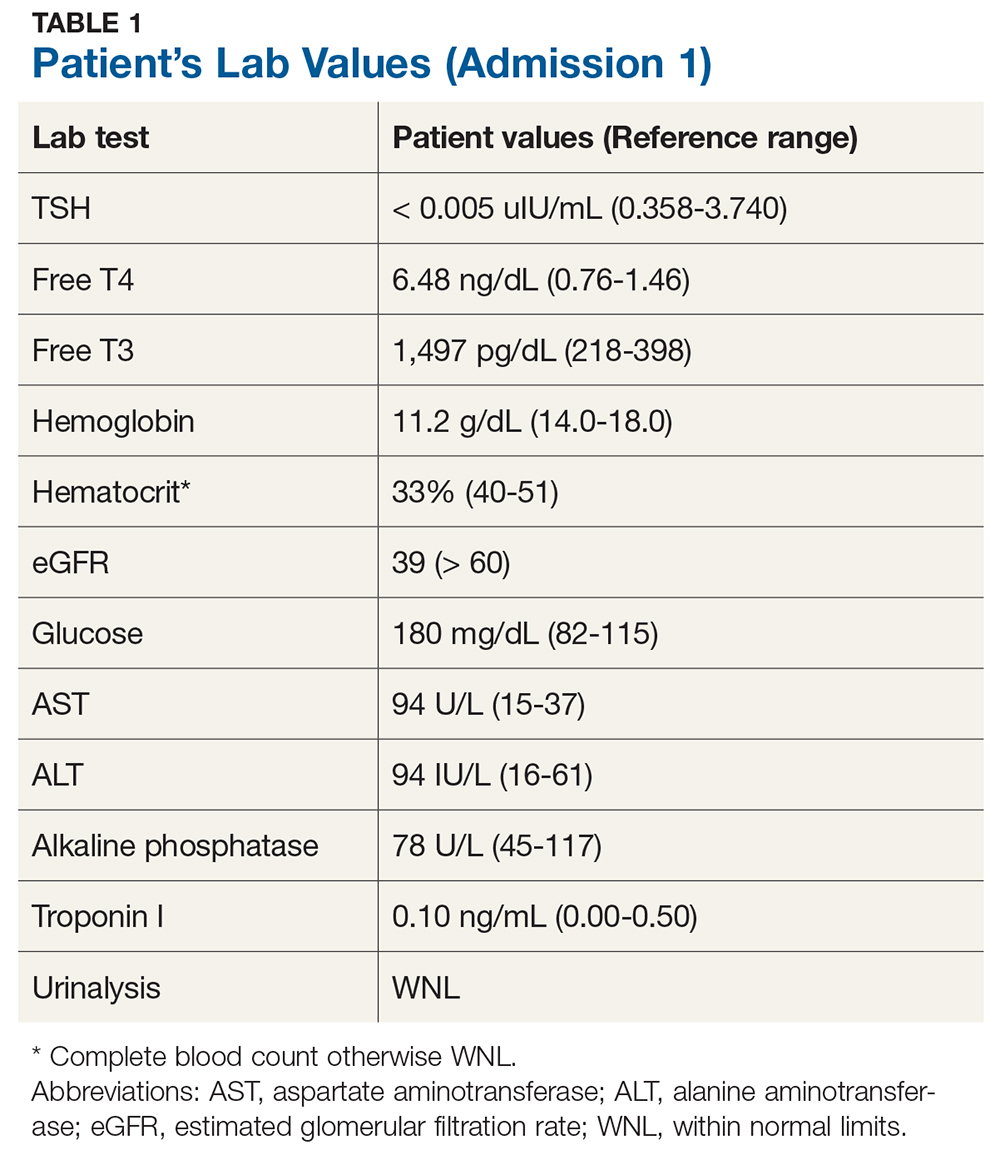
Endocrinology consult is requested. Further testing yields the following findings
- Thyroid-stimulating immunoglobulin: 309% (reference range, < 30%)
- Nuclear medicine thyroid scan with uptake: 6-hour uptake of 70.3% (10%-25%) and 24-hour uptake, 81.8% (15%-35%)
- Homogeneous radiotracer uptake within the thyroid gland: no evidence of hot or cold nodules
- Thyroid ultrasound: bilateral enlarged heterogeneous gland and multiple subcentimeter nodules (largest measuring 6 × 7 mm)
These results confirm a diagnosis of Graves’ disease. Treatment options, including antithyroid medications, radioactive iodine ablation (RAI), and surgery, are discussed. The patient is treated with RAI therapy (10 mCi) and discharged from the hospital.
Six days later, however, he returns to the ED with severe intermittent dizziness and lightheadedness of two hours’ duration, new-onset atrial fibrillation (A-fib), and mild shortness of breath. His vital signs include a pulse of 116 beats/min; temperature, 98.1°F; respiratory rate, 18 breaths/min-1, blood pressure, 154/88 mm Hg; and PO2, 100 mm Hg.
His lab values include
- TSH < 0.005 uIU/mL
- Free T4, 8.01 ng/dL
- Free T3, 3,701 pg/dL
- eGFR, 60 mL/min/1.73 m2
Cardiology consult is requested. A pacemaker is placed for bradycardia-tachycardia syndrome, and the patient is put on rivaroxaban for stroke prevention.
The endocrinologist suspects post-RAI thyroiditis or ineffective RAI treatment. The patient is started on methimazole (10 mg bid), and his carvedilol is replaced with metoprolol (50 mg bid).
Two weeks postdischarge, the patient returns to the office. Although he says he’s doing better, he seems uneasy and agitated and has a pulse of 120 beats/min. His methimazole and metoprolol are increased (to 10 mg tid and 50 mg tid, respectively).
Another two weeks later, lab results still show elevated thyroid levels—now with increased enzyme levels on liver function testing. The patient reports worsening dizziness and shortness of breath. He is sent back to hospital and admitted for inpatient management, with urgent surgical consult for thyroidectomy. Total thyroidectomy is successfully performed, and the final pathology report shows a benign goiter.
DISCUSSION
Thyroid storm is an extreme form of thyrotoxicosis with an associated mortality rate of 8% to 25%.1 When thyroid hormone levels are elevated, adrenaline receptors are upregulated—but, while it is possible for persistent thyrotoxicosis to progress to thyroid storm on its own, a surge of adrenaline is usually needed. Most cases are triggered by acute stressors (ie, myocardial infarction, surgery, anesthesia, labor and delivery) in the context of underlying thyrotoxicosis.1
Diagnosis of thyroid storm is made clinically in patients who are thyrotoxic and present with systemic decompensation (ie, altered mental status, cardiovascular dysfunction, hyperpyrexia). Although no universally accepted criteria currently exist, the Burch-Wartofsky Point Scale (BWPS; see Table 2) can be used to assess disease severity and guide the extent of treatment and monitoring.2 However, this measure should not replace clinical judgment—the distinction between compensated thyrotoxicosis and decompensating thyrotoxicosis (thyroid storm) should be made by sound but prompt clinical assessment.
Once thyroid storm is suspected, aggressive treatment should be implemented to improve the systemic thyrotoxic state. Propylthiouracil (PTU) is preferred over methimazole, as it blocks T4 to T3 conversion in addition to blocking new hormone synthesis. Propranolol is the best choice of ß-blocker because it also blocks T4 to T3 conversion and controls cardiac rhythm.
Iodine can rapidly block new hormone synthesis and release; it is often used to reduce thyroid hormone levels prior to emergency thyroid surgery. However, it should be given at least one hour after a dose of PTU. Hydrocortisone is given prophylactically for relative adrenal insufficiency (due to rapid cortisol clearance during thyrotoxic state); it may block T4 to T3 conversion as well. Volume resuscitation, respiratory care, temperature control (eg, antipyretics, cooling blankets), and nutritional support should also be incorporated, ideally in the intensive care unit (ICU). During or after thyroid storm management, treatment of the precipitating event/illness and of hyperthyroidism should be initiated to prevent recurrence.1
The patient’s initial BWPS was 30 (gastrointestinal [GI] score 10 + central nervous system [CNS] score 10 + without precipitating factor 10), which put him in the “impending storm” category. At his second ED visit, his BWPS was 40 (cardiovascular score 10 + A-fib 10 + GI score 10 + CNS score 10 + precipitating factor [RAI ablation] score 0)—still in the “impending storm” category but certainly indicating a worsened state.
RAI for hyperthyroidism can transiently increase thyroid hormone levels due to inflammation of the gland. To prevent exacerbation of the thyrotoxic state, pretreatment with methimazole should be considered in patients with risk factors (eg, older age, cardiovascular complications, cerebrovascular disease, pulmonary disease, renal failure, infection, trauma, and poorly controlled diabetes). Patients should also be placed on ß-blockers prior to treatment, in anticipation of a transient rise in thyroid hormone levels.
Due to this patient’s age, severity of thyrotoxicosis, and multiple risk factors, strong consideration should have been given to pretreating him with antithyroid medication and a ß-blocker before definitive treatment was given. This would have potentially averted his subsequent hospital visits and urgent need for thyroidectomy.
CONCLUSION
Thyroid storm is an uncommon but serious medical condition with a high mortality rate. Prompt recognition and an aggressive multimodal treatment approach, ideally in the ICU, are paramount to stabilize patients and seek definitive treatment.
1. Ross DS, Burch HB, Cooper DS, et al. 2016 American Thyroid Association guidelines for diagnosis and management of hyperthyroidism and other causes of thyrotoxicosis. Thyroid. 2016;26(10):1343-1421.
2. Burch HB, Wartofsky L. Life-threatening thyrotoxicosis: thyroid storm. Endocrinol Metab Clin North Am. 1993; 22(2):263-277.
Clinician Reviews in partnership with

Ji Hyun Chun (CJ) practices at OptumCare Medical Group in Orange County, California; he is President of the American Society of Endocrine PAs and a member of the Clinician Reviews editorial board.
Clinician Reviews in partnership with
Ji Hyun Chun (CJ) practices at OptumCare Medical Group in Orange County, California; he is President of the American Society of Endocrine PAs and a member of the Clinician Reviews editorial board.
Clinician Reviews in partnership with
Ji Hyun Chun (CJ) practices at OptumCare Medical Group in Orange County, California; he is President of the American Society of Endocrine PAs and a member of the Clinician Reviews editorial board.
A 73-year-old man is transported to the emergency department (ED) by ambulance for nausea, vomiting, diarrhea, and weakness of three days’ duration. Earlier today, he presented to his primary care provider with these symptoms and was found to be hypotensive; he was advised to go to the ED but instead went home against medical advice.
The patient’s medical history is significant for type 2 diabetes, stage 3b chronic kidney disease, dyslipidemia, hypertension, coronary artery disease, and benign prostatic hyperplasia. He has undergone stent placement and triple coronary artery bypass graft surgery. His medication list includes insulin glargine, glimepiride, liraglutide, atorvastatin, benazepril, carvedilol, amlodipine, clopidogrel, and tamsulosin.
Upon admission, the patient has a pulse of 98 beats/min; temperature, 98.2°F; respiratory rate, 18 breaths/min-1; and PO2, 98 mm Hg. An ECG, chest radiograph, and CT (without contrast) of the head, chest, and abdomen are all within normal limits. Lab evaluation is significant for severe thyrotoxicosis (see Table 1).
Endocrinology consult is requested. Further testing yields the following findings
- Thyroid-stimulating immunoglobulin: 309% (reference range, < 30%)
- Nuclear medicine thyroid scan with uptake: 6-hour uptake of 70.3% (10%-25%) and 24-hour uptake, 81.8% (15%-35%)
- Homogeneous radiotracer uptake within the thyroid gland: no evidence of hot or cold nodules
- Thyroid ultrasound: bilateral enlarged heterogeneous gland and multiple subcentimeter nodules (largest measuring 6 × 7 mm)
These results confirm a diagnosis of Graves’ disease. Treatment options, including antithyroid medications, radioactive iodine ablation (RAI), and surgery, are discussed. The patient is treated with RAI therapy (10 mCi) and discharged from the hospital.
Six days later, however, he returns to the ED with severe intermittent dizziness and lightheadedness of two hours’ duration, new-onset atrial fibrillation (A-fib), and mild shortness of breath. His vital signs include a pulse of 116 beats/min; temperature, 98.1°F; respiratory rate, 18 breaths/min-1, blood pressure, 154/88 mm Hg; and PO2, 100 mm Hg.
His lab values include
- TSH < 0.005 uIU/mL
- Free T4, 8.01 ng/dL
- Free T3, 3,701 pg/dL
- eGFR, 60 mL/min/1.73 m2
Cardiology consult is requested. A pacemaker is placed for bradycardia-tachycardia syndrome, and the patient is put on rivaroxaban for stroke prevention.
The endocrinologist suspects post-RAI thyroiditis or ineffective RAI treatment. The patient is started on methimazole (10 mg bid), and his carvedilol is replaced with metoprolol (50 mg bid).
Two weeks postdischarge, the patient returns to the office. Although he says he’s doing better, he seems uneasy and agitated and has a pulse of 120 beats/min. His methimazole and metoprolol are increased (to 10 mg tid and 50 mg tid, respectively).
Another two weeks later, lab results still show elevated thyroid levels—now with increased enzyme levels on liver function testing. The patient reports worsening dizziness and shortness of breath. He is sent back to hospital and admitted for inpatient management, with urgent surgical consult for thyroidectomy. Total thyroidectomy is successfully performed, and the final pathology report shows a benign goiter.
DISCUSSION
Thyroid storm is an extreme form of thyrotoxicosis with an associated mortality rate of 8% to 25%.1 When thyroid hormone levels are elevated, adrenaline receptors are upregulated—but, while it is possible for persistent thyrotoxicosis to progress to thyroid storm on its own, a surge of adrenaline is usually needed. Most cases are triggered by acute stressors (ie, myocardial infarction, surgery, anesthesia, labor and delivery) in the context of underlying thyrotoxicosis.1
Diagnosis of thyroid storm is made clinically in patients who are thyrotoxic and present with systemic decompensation (ie, altered mental status, cardiovascular dysfunction, hyperpyrexia). Although no universally accepted criteria currently exist, the Burch-Wartofsky Point Scale (BWPS; see Table 2) can be used to assess disease severity and guide the extent of treatment and monitoring.2 However, this measure should not replace clinical judgment—the distinction between compensated thyrotoxicosis and decompensating thyrotoxicosis (thyroid storm) should be made by sound but prompt clinical assessment.
Once thyroid storm is suspected, aggressive treatment should be implemented to improve the systemic thyrotoxic state. Propylthiouracil (PTU) is preferred over methimazole, as it blocks T4 to T3 conversion in addition to blocking new hormone synthesis. Propranolol is the best choice of ß-blocker because it also blocks T4 to T3 conversion and controls cardiac rhythm.
Iodine can rapidly block new hormone synthesis and release; it is often used to reduce thyroid hormone levels prior to emergency thyroid surgery. However, it should be given at least one hour after a dose of PTU. Hydrocortisone is given prophylactically for relative adrenal insufficiency (due to rapid cortisol clearance during thyrotoxic state); it may block T4 to T3 conversion as well. Volume resuscitation, respiratory care, temperature control (eg, antipyretics, cooling blankets), and nutritional support should also be incorporated, ideally in the intensive care unit (ICU). During or after thyroid storm management, treatment of the precipitating event/illness and of hyperthyroidism should be initiated to prevent recurrence.1
The patient’s initial BWPS was 30 (gastrointestinal [GI] score 10 + central nervous system [CNS] score 10 + without precipitating factor 10), which put him in the “impending storm” category. At his second ED visit, his BWPS was 40 (cardiovascular score 10 + A-fib 10 + GI score 10 + CNS score 10 + precipitating factor [RAI ablation] score 0)—still in the “impending storm” category but certainly indicating a worsened state.
RAI for hyperthyroidism can transiently increase thyroid hormone levels due to inflammation of the gland. To prevent exacerbation of the thyrotoxic state, pretreatment with methimazole should be considered in patients with risk factors (eg, older age, cardiovascular complications, cerebrovascular disease, pulmonary disease, renal failure, infection, trauma, and poorly controlled diabetes). Patients should also be placed on ß-blockers prior to treatment, in anticipation of a transient rise in thyroid hormone levels.
Due to this patient’s age, severity of thyrotoxicosis, and multiple risk factors, strong consideration should have been given to pretreating him with antithyroid medication and a ß-blocker before definitive treatment was given. This would have potentially averted his subsequent hospital visits and urgent need for thyroidectomy.
CONCLUSION
Thyroid storm is an uncommon but serious medical condition with a high mortality rate. Prompt recognition and an aggressive multimodal treatment approach, ideally in the ICU, are paramount to stabilize patients and seek definitive treatment.
A 73-year-old man is transported to the emergency department (ED) by ambulance for nausea, vomiting, diarrhea, and weakness of three days’ duration. Earlier today, he presented to his primary care provider with these symptoms and was found to be hypotensive; he was advised to go to the ED but instead went home against medical advice.
The patient’s medical history is significant for type 2 diabetes, stage 3b chronic kidney disease, dyslipidemia, hypertension, coronary artery disease, and benign prostatic hyperplasia. He has undergone stent placement and triple coronary artery bypass graft surgery. His medication list includes insulin glargine, glimepiride, liraglutide, atorvastatin, benazepril, carvedilol, amlodipine, clopidogrel, and tamsulosin.
Upon admission, the patient has a pulse of 98 beats/min; temperature, 98.2°F; respiratory rate, 18 breaths/min-1; and PO2, 98 mm Hg. An ECG, chest radiograph, and CT (without contrast) of the head, chest, and abdomen are all within normal limits. Lab evaluation is significant for severe thyrotoxicosis (see Table 1).
Endocrinology consult is requested. Further testing yields the following findings
- Thyroid-stimulating immunoglobulin: 309% (reference range, < 30%)
- Nuclear medicine thyroid scan with uptake: 6-hour uptake of 70.3% (10%-25%) and 24-hour uptake, 81.8% (15%-35%)
- Homogeneous radiotracer uptake within the thyroid gland: no evidence of hot or cold nodules
- Thyroid ultrasound: bilateral enlarged heterogeneous gland and multiple subcentimeter nodules (largest measuring 6 × 7 mm)
These results confirm a diagnosis of Graves’ disease. Treatment options, including antithyroid medications, radioactive iodine ablation (RAI), and surgery, are discussed. The patient is treated with RAI therapy (10 mCi) and discharged from the hospital.
Six days later, however, he returns to the ED with severe intermittent dizziness and lightheadedness of two hours’ duration, new-onset atrial fibrillation (A-fib), and mild shortness of breath. His vital signs include a pulse of 116 beats/min; temperature, 98.1°F; respiratory rate, 18 breaths/min-1, blood pressure, 154/88 mm Hg; and PO2, 100 mm Hg.
His lab values include
- TSH < 0.005 uIU/mL
- Free T4, 8.01 ng/dL
- Free T3, 3,701 pg/dL
- eGFR, 60 mL/min/1.73 m2
Cardiology consult is requested. A pacemaker is placed for bradycardia-tachycardia syndrome, and the patient is put on rivaroxaban for stroke prevention.
The endocrinologist suspects post-RAI thyroiditis or ineffective RAI treatment. The patient is started on methimazole (10 mg bid), and his carvedilol is replaced with metoprolol (50 mg bid).
Two weeks postdischarge, the patient returns to the office. Although he says he’s doing better, he seems uneasy and agitated and has a pulse of 120 beats/min. His methimazole and metoprolol are increased (to 10 mg tid and 50 mg tid, respectively).
Another two weeks later, lab results still show elevated thyroid levels—now with increased enzyme levels on liver function testing. The patient reports worsening dizziness and shortness of breath. He is sent back to hospital and admitted for inpatient management, with urgent surgical consult for thyroidectomy. Total thyroidectomy is successfully performed, and the final pathology report shows a benign goiter.
DISCUSSION
Thyroid storm is an extreme form of thyrotoxicosis with an associated mortality rate of 8% to 25%.1 When thyroid hormone levels are elevated, adrenaline receptors are upregulated—but, while it is possible for persistent thyrotoxicosis to progress to thyroid storm on its own, a surge of adrenaline is usually needed. Most cases are triggered by acute stressors (ie, myocardial infarction, surgery, anesthesia, labor and delivery) in the context of underlying thyrotoxicosis.1
Diagnosis of thyroid storm is made clinically in patients who are thyrotoxic and present with systemic decompensation (ie, altered mental status, cardiovascular dysfunction, hyperpyrexia). Although no universally accepted criteria currently exist, the Burch-Wartofsky Point Scale (BWPS; see Table 2) can be used to assess disease severity and guide the extent of treatment and monitoring.2 However, this measure should not replace clinical judgment—the distinction between compensated thyrotoxicosis and decompensating thyrotoxicosis (thyroid storm) should be made by sound but prompt clinical assessment.
Once thyroid storm is suspected, aggressive treatment should be implemented to improve the systemic thyrotoxic state. Propylthiouracil (PTU) is preferred over methimazole, as it blocks T4 to T3 conversion in addition to blocking new hormone synthesis. Propranolol is the best choice of ß-blocker because it also blocks T4 to T3 conversion and controls cardiac rhythm.
Iodine can rapidly block new hormone synthesis and release; it is often used to reduce thyroid hormone levels prior to emergency thyroid surgery. However, it should be given at least one hour after a dose of PTU. Hydrocortisone is given prophylactically for relative adrenal insufficiency (due to rapid cortisol clearance during thyrotoxic state); it may block T4 to T3 conversion as well. Volume resuscitation, respiratory care, temperature control (eg, antipyretics, cooling blankets), and nutritional support should also be incorporated, ideally in the intensive care unit (ICU). During or after thyroid storm management, treatment of the precipitating event/illness and of hyperthyroidism should be initiated to prevent recurrence.1
The patient’s initial BWPS was 30 (gastrointestinal [GI] score 10 + central nervous system [CNS] score 10 + without precipitating factor 10), which put him in the “impending storm” category. At his second ED visit, his BWPS was 40 (cardiovascular score 10 + A-fib 10 + GI score 10 + CNS score 10 + precipitating factor [RAI ablation] score 0)—still in the “impending storm” category but certainly indicating a worsened state.
RAI for hyperthyroidism can transiently increase thyroid hormone levels due to inflammation of the gland. To prevent exacerbation of the thyrotoxic state, pretreatment with methimazole should be considered in patients with risk factors (eg, older age, cardiovascular complications, cerebrovascular disease, pulmonary disease, renal failure, infection, trauma, and poorly controlled diabetes). Patients should also be placed on ß-blockers prior to treatment, in anticipation of a transient rise in thyroid hormone levels.
Due to this patient’s age, severity of thyrotoxicosis, and multiple risk factors, strong consideration should have been given to pretreating him with antithyroid medication and a ß-blocker before definitive treatment was given. This would have potentially averted his subsequent hospital visits and urgent need for thyroidectomy.
CONCLUSION
Thyroid storm is an uncommon but serious medical condition with a high mortality rate. Prompt recognition and an aggressive multimodal treatment approach, ideally in the ICU, are paramount to stabilize patients and seek definitive treatment.
1. Ross DS, Burch HB, Cooper DS, et al. 2016 American Thyroid Association guidelines for diagnosis and management of hyperthyroidism and other causes of thyrotoxicosis. Thyroid. 2016;26(10):1343-1421.
2. Burch HB, Wartofsky L. Life-threatening thyrotoxicosis: thyroid storm. Endocrinol Metab Clin North Am. 1993; 22(2):263-277.
1. Ross DS, Burch HB, Cooper DS, et al. 2016 American Thyroid Association guidelines for diagnosis and management of hyperthyroidism and other causes of thyrotoxicosis. Thyroid. 2016;26(10):1343-1421.
2. Burch HB, Wartofsky L. Life-threatening thyrotoxicosis: thyroid storm. Endocrinol Metab Clin North Am. 1993; 22(2):263-277.