User login
Stroke Prevention in Patients With Silent Cerebrovascular Disease
Two decades of epidemiologic research show that silent cerebrovascular disease is common and associated with an increased risk of stroke and dementia. To summarize evidence for the diagnosis and management of silent cerebrovascular disease to prevent stroke, the Stroke Council of the American Heart Association (AHA) convened a writing committee to evaluate existing evidence, to discuss clinical considerations, and to offer suggestions for future research on stroke prevention. The committee focused on patients with three cardinal manifestations of silent cerebrovascular disease: silent brain infarcts, MRI white matter hyperintensities of presumed vascular origin, and cerebral microbleeds. The committee’s report, which was published as a Scientific Statement from the AHA and the American Stroke Association (ASA), appeared online ahead of print December 15, 2016, in Stroke. The American Academy of Neurology affirmed the value of the AHA/ASA statement as an educational tool for neurologists.
The AHA/ASA writing committee found strong and consistent evidence that silent cerebrovascular disease is a common problem of aging and that silent brain infarcts and white matter hyperintensities are strongly associated with future symptomatic stroke risk, entailing a two- to threefold increase in relative risk, even after controlling for vascular risk factors such as hypertension. Among patients with cerebral microbleeds, evidence suggests that those treated with thrombolysis for acute ischemic stroke have a moderately increased risk of symptomatic intracranial hemorrhage, but little prospective evidence indicates the risk of symptomatic hemorrhage in patients who receive anticoagulation, said lead author and chair of the writing committee, Eric E. Smith, MD, MPH, Associate Professor of Neurology and the Katthy Taylor Chair in Vascular Dementia in the Department of Clinical Neurosciences at the University of Calgary, Alberta, and Hotchkiss Brain Institute. There were no randomized controlled trials targeted specifically to stroke prevention in participants with silent cerebrovascular disease.

According to the AHA/ASA Scientific Statement, primary stroke prevention is indicated in patients with silent brain infarcts, white matter hyperintensities, or microbleeds. The AHA/ASA statement further advises that accurate and reliable radiologic reporting using consensus terms understood by radiologists and nonradiologists alike will be essential to identify patients with silent cerebrovascular disease for appropriate treatment. Adoption of standard terms and definitions for silent cerebrovascular disease, as provided by prior AHA/ASA statements and by a consensus group, may facilitate diagnosis and communication of findings from radiologists to clinicians.
—Glenn S. Williams
Suggested Reading
Smith EE, Saposnik G, Biessels GJ, et al. Prevention of stroke in patients with silent cerebrovascular disease: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2016 December 15 [Epub ahead of print].
Two decades of epidemiologic research show that silent cerebrovascular disease is common and associated with an increased risk of stroke and dementia. To summarize evidence for the diagnosis and management of silent cerebrovascular disease to prevent stroke, the Stroke Council of the American Heart Association (AHA) convened a writing committee to evaluate existing evidence, to discuss clinical considerations, and to offer suggestions for future research on stroke prevention. The committee focused on patients with three cardinal manifestations of silent cerebrovascular disease: silent brain infarcts, MRI white matter hyperintensities of presumed vascular origin, and cerebral microbleeds. The committee’s report, which was published as a Scientific Statement from the AHA and the American Stroke Association (ASA), appeared online ahead of print December 15, 2016, in Stroke. The American Academy of Neurology affirmed the value of the AHA/ASA statement as an educational tool for neurologists.
The AHA/ASA writing committee found strong and consistent evidence that silent cerebrovascular disease is a common problem of aging and that silent brain infarcts and white matter hyperintensities are strongly associated with future symptomatic stroke risk, entailing a two- to threefold increase in relative risk, even after controlling for vascular risk factors such as hypertension. Among patients with cerebral microbleeds, evidence suggests that those treated with thrombolysis for acute ischemic stroke have a moderately increased risk of symptomatic intracranial hemorrhage, but little prospective evidence indicates the risk of symptomatic hemorrhage in patients who receive anticoagulation, said lead author and chair of the writing committee, Eric E. Smith, MD, MPH, Associate Professor of Neurology and the Katthy Taylor Chair in Vascular Dementia in the Department of Clinical Neurosciences at the University of Calgary, Alberta, and Hotchkiss Brain Institute. There were no randomized controlled trials targeted specifically to stroke prevention in participants with silent cerebrovascular disease.
According to the AHA/ASA Scientific Statement, primary stroke prevention is indicated in patients with silent brain infarcts, white matter hyperintensities, or microbleeds. The AHA/ASA statement further advises that accurate and reliable radiologic reporting using consensus terms understood by radiologists and nonradiologists alike will be essential to identify patients with silent cerebrovascular disease for appropriate treatment. Adoption of standard terms and definitions for silent cerebrovascular disease, as provided by prior AHA/ASA statements and by a consensus group, may facilitate diagnosis and communication of findings from radiologists to clinicians.
—Glenn S. Williams
Suggested Reading
Smith EE, Saposnik G, Biessels GJ, et al. Prevention of stroke in patients with silent cerebrovascular disease: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2016 December 15 [Epub ahead of print].
Two decades of epidemiologic research show that silent cerebrovascular disease is common and associated with an increased risk of stroke and dementia. To summarize evidence for the diagnosis and management of silent cerebrovascular disease to prevent stroke, the Stroke Council of the American Heart Association (AHA) convened a writing committee to evaluate existing evidence, to discuss clinical considerations, and to offer suggestions for future research on stroke prevention. The committee focused on patients with three cardinal manifestations of silent cerebrovascular disease: silent brain infarcts, MRI white matter hyperintensities of presumed vascular origin, and cerebral microbleeds. The committee’s report, which was published as a Scientific Statement from the AHA and the American Stroke Association (ASA), appeared online ahead of print December 15, 2016, in Stroke. The American Academy of Neurology affirmed the value of the AHA/ASA statement as an educational tool for neurologists.
The AHA/ASA writing committee found strong and consistent evidence that silent cerebrovascular disease is a common problem of aging and that silent brain infarcts and white matter hyperintensities are strongly associated with future symptomatic stroke risk, entailing a two- to threefold increase in relative risk, even after controlling for vascular risk factors such as hypertension. Among patients with cerebral microbleeds, evidence suggests that those treated with thrombolysis for acute ischemic stroke have a moderately increased risk of symptomatic intracranial hemorrhage, but little prospective evidence indicates the risk of symptomatic hemorrhage in patients who receive anticoagulation, said lead author and chair of the writing committee, Eric E. Smith, MD, MPH, Associate Professor of Neurology and the Katthy Taylor Chair in Vascular Dementia in the Department of Clinical Neurosciences at the University of Calgary, Alberta, and Hotchkiss Brain Institute. There were no randomized controlled trials targeted specifically to stroke prevention in participants with silent cerebrovascular disease.
According to the AHA/ASA Scientific Statement, primary stroke prevention is indicated in patients with silent brain infarcts, white matter hyperintensities, or microbleeds. The AHA/ASA statement further advises that accurate and reliable radiologic reporting using consensus terms understood by radiologists and nonradiologists alike will be essential to identify patients with silent cerebrovascular disease for appropriate treatment. Adoption of standard terms and definitions for silent cerebrovascular disease, as provided by prior AHA/ASA statements and by a consensus group, may facilitate diagnosis and communication of findings from radiologists to clinicians.
—Glenn S. Williams
Suggested Reading
Smith EE, Saposnik G, Biessels GJ, et al. Prevention of stroke in patients with silent cerebrovascular disease: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2016 December 15 [Epub ahead of print].
Latuda receives FDA approval for adolescent schizophrenia treatment
Lurasidone HCl, marketed as Latuda, has received approval from the Food and Drug Administration for the treatment of schizophrenia in adolescents aged 13-17 years old, according to a press release from Sunovion Pharmaceuticals.
FDA approval of Latuda is based on results of a 6-week study in which adolescents with schizophrenia received either 40 mg of lurasidone per day, 80 mg per day, or a placebo. Patients who received lurasidone HCI showed statistical and clinical improvements in schizophrenia symptoms, compared with the placebo group. The drug was well tolerated with no significant adverse events reported.![]()
“The impact on development and poor prognosis frequently associated with schizophrenia that begins in adolescence underscores the need for treatment that is both well tolerated and effective,” Robert L. Findling, MD, study investigator, and director of child and adolescent psychiatry at Johns Hopkins University, Baltimore, said in the press release. “The availability of Latuda provides health care providers with an important new option for helping adolescents with this illness that is chronic and severely disabling,” said Dr. Findling, also vice president of psychiatric services and research at the Kennedy Krieger Institute, Baltimore.
Find the full press release on the Sunovion Pharmaceuticals website.
Lurasidone HCl, marketed as Latuda, has received approval from the Food and Drug Administration for the treatment of schizophrenia in adolescents aged 13-17 years old, according to a press release from Sunovion Pharmaceuticals.
FDA approval of Latuda is based on results of a 6-week study in which adolescents with schizophrenia received either 40 mg of lurasidone per day, 80 mg per day, or a placebo. Patients who received lurasidone HCI showed statistical and clinical improvements in schizophrenia symptoms, compared with the placebo group. The drug was well tolerated with no significant adverse events reported.![]()
“The impact on development and poor prognosis frequently associated with schizophrenia that begins in adolescence underscores the need for treatment that is both well tolerated and effective,” Robert L. Findling, MD, study investigator, and director of child and adolescent psychiatry at Johns Hopkins University, Baltimore, said in the press release. “The availability of Latuda provides health care providers with an important new option for helping adolescents with this illness that is chronic and severely disabling,” said Dr. Findling, also vice president of psychiatric services and research at the Kennedy Krieger Institute, Baltimore.
Find the full press release on the Sunovion Pharmaceuticals website.
Lurasidone HCl, marketed as Latuda, has received approval from the Food and Drug Administration for the treatment of schizophrenia in adolescents aged 13-17 years old, according to a press release from Sunovion Pharmaceuticals.
FDA approval of Latuda is based on results of a 6-week study in which adolescents with schizophrenia received either 40 mg of lurasidone per day, 80 mg per day, or a placebo. Patients who received lurasidone HCI showed statistical and clinical improvements in schizophrenia symptoms, compared with the placebo group. The drug was well tolerated with no significant adverse events reported.![]()
“The impact on development and poor prognosis frequently associated with schizophrenia that begins in adolescence underscores the need for treatment that is both well tolerated and effective,” Robert L. Findling, MD, study investigator, and director of child and adolescent psychiatry at Johns Hopkins University, Baltimore, said in the press release. “The availability of Latuda provides health care providers with an important new option for helping adolescents with this illness that is chronic and severely disabling,” said Dr. Findling, also vice president of psychiatric services and research at the Kennedy Krieger Institute, Baltimore.
Find the full press release on the Sunovion Pharmaceuticals website.
Recreational marijuana use should not rule out ADHD stimulant treatment
SAN FRANCISCO – “If one is going to say ‘you need 6 months of abstinence from cannabis before I am going to treat your ADHD,’ that’s absurd. If [kids] are stoned all the time, no, but if it’s intermittent, it’s really not a factor,” according to James J. McGough, MD, director of the Attention Deficit Disorder Clinic at the University of California, Los Angeles.
There’s no evidence that ongoing stimulant treatment increases the risk of substance abuse, and “we must admit to ourselves that substance use among adolescents and young adults is normative. It happens,” even among well-adapted kids. “Think of your own past histories,” he said to an audience of psychiatrists and other medical professionals at a psychopharmacology update held by the American Academy of Child and Adolescent Psychiatry.

“You have” to deal with substance abuse that’s ruining lives, but otherwise, “I’m comfortable giving a prescription under some circumstances even if I know [patients] are using drugs. It’s the extent and type of [drug use] that informs if we should prescribe or not,” said Dr. McGough, also a professor of clinical psychiatry at the university, but be careful to “really document what you’re doing.”
Misuse and diversion is mostly related to performance enhancement, especially at competitive universities, or when grades head south. Children and young adults in those situations, as well as smokers, drinkers, and those with conduct disorders, “are the ones you worry about.” If a Stanford medical school student at age 27 suddenly develops a horrible problem focusing, with no past ADHD history, “I’m a little suspicious,” he said, noting that people who malinger “tend to go off the scale in terms of endorsing symptoms.”
If misuse and abuse are a concern, extended-release stimulants, as well as nonstimulants, are better options than immediate-release formulations.
Audience members also were curious about alternative approaches for ADHD, but the news wasn’t very good.
For now, there’s no brain scan, test, or lab measurement that reliably detects ADHD. Even with neuropsychiatric testing, only half of children and adolescents will have executive-functioning deficits. The problem with computerized tests, meanwhile, is that a lot of children and adolescents with ADHD have no problem focusing on computer games, so they aren’t helpful for diagnosis.
“The gold standard for ADHD assessment remains a good interview,” and documenting DSM-5 criteria, he said.
As for alternative treatments, “what studies have shown is that if you are highly invested in the treatment and not blinded” to it, “you tend to think you do better. In good studies where they look at teachers who don’t know what’s going on, it doesn’t pan out,” he said. Metanalysis of neurofeedback hasn’t shown effect, nor have computer games to train focus; kids will get better at them with practice, but they haven’t been shown to improve ADHD.
The only exception is omega-3, but the effect size is barely measurable. “If you can get the kid so swallow those fish oil things, go for it, or they can eat salmon,” Dr. McGough said.
In short, medications remain the best option for treating the core symptoms of ADHD, first with stimulants then, as needed, nonstimulants.
But “you are not meeting your patients’ needs if you are only prescribing medications,” he said. Psychosocial education, family behavioral therapy, and school interventions – even things as simple as sitting toward the front of the class and having a little more time on tests – are critical for overall improvement.
It’s also important to let patients and families know that “they don’t have to be ashamed of this,” and the patients need to know why they are taking their medication, he said.
Too often, children are started on a low dose of medication and told to come back in a month. “That’s a waste of time, because it’s a whole month when the person is probably inadequately treated,” Dr. McGough said.
Instead, he has patients titrate up at first, with advice to cut back or stop and call if there are problems. The patient returns after 3 weeks, and the most effective, best-tolerated dose is selected for treatment.
Dr. McGough disclosed relationships with Purdue, Shire, Tris, Sunovion, NeuroSigma, and Neurovance.
SAN FRANCISCO – “If one is going to say ‘you need 6 months of abstinence from cannabis before I am going to treat your ADHD,’ that’s absurd. If [kids] are stoned all the time, no, but if it’s intermittent, it’s really not a factor,” according to James J. McGough, MD, director of the Attention Deficit Disorder Clinic at the University of California, Los Angeles.
There’s no evidence that ongoing stimulant treatment increases the risk of substance abuse, and “we must admit to ourselves that substance use among adolescents and young adults is normative. It happens,” even among well-adapted kids. “Think of your own past histories,” he said to an audience of psychiatrists and other medical professionals at a psychopharmacology update held by the American Academy of Child and Adolescent Psychiatry.
“You have” to deal with substance abuse that’s ruining lives, but otherwise, “I’m comfortable giving a prescription under some circumstances even if I know [patients] are using drugs. It’s the extent and type of [drug use] that informs if we should prescribe or not,” said Dr. McGough, also a professor of clinical psychiatry at the university, but be careful to “really document what you’re doing.”
Misuse and diversion is mostly related to performance enhancement, especially at competitive universities, or when grades head south. Children and young adults in those situations, as well as smokers, drinkers, and those with conduct disorders, “are the ones you worry about.” If a Stanford medical school student at age 27 suddenly develops a horrible problem focusing, with no past ADHD history, “I’m a little suspicious,” he said, noting that people who malinger “tend to go off the scale in terms of endorsing symptoms.”
If misuse and abuse are a concern, extended-release stimulants, as well as nonstimulants, are better options than immediate-release formulations.
Audience members also were curious about alternative approaches for ADHD, but the news wasn’t very good.
For now, there’s no brain scan, test, or lab measurement that reliably detects ADHD. Even with neuropsychiatric testing, only half of children and adolescents will have executive-functioning deficits. The problem with computerized tests, meanwhile, is that a lot of children and adolescents with ADHD have no problem focusing on computer games, so they aren’t helpful for diagnosis.
“The gold standard for ADHD assessment remains a good interview,” and documenting DSM-5 criteria, he said.
As for alternative treatments, “what studies have shown is that if you are highly invested in the treatment and not blinded” to it, “you tend to think you do better. In good studies where they look at teachers who don’t know what’s going on, it doesn’t pan out,” he said. Metanalysis of neurofeedback hasn’t shown effect, nor have computer games to train focus; kids will get better at them with practice, but they haven’t been shown to improve ADHD.
The only exception is omega-3, but the effect size is barely measurable. “If you can get the kid so swallow those fish oil things, go for it, or they can eat salmon,” Dr. McGough said.
In short, medications remain the best option for treating the core symptoms of ADHD, first with stimulants then, as needed, nonstimulants.
But “you are not meeting your patients’ needs if you are only prescribing medications,” he said. Psychosocial education, family behavioral therapy, and school interventions – even things as simple as sitting toward the front of the class and having a little more time on tests – are critical for overall improvement.
It’s also important to let patients and families know that “they don’t have to be ashamed of this,” and the patients need to know why they are taking their medication, he said.
Too often, children are started on a low dose of medication and told to come back in a month. “That’s a waste of time, because it’s a whole month when the person is probably inadequately treated,” Dr. McGough said.
Instead, he has patients titrate up at first, with advice to cut back or stop and call if there are problems. The patient returns after 3 weeks, and the most effective, best-tolerated dose is selected for treatment.
Dr. McGough disclosed relationships with Purdue, Shire, Tris, Sunovion, NeuroSigma, and Neurovance.
SAN FRANCISCO – “If one is going to say ‘you need 6 months of abstinence from cannabis before I am going to treat your ADHD,’ that’s absurd. If [kids] are stoned all the time, no, but if it’s intermittent, it’s really not a factor,” according to James J. McGough, MD, director of the Attention Deficit Disorder Clinic at the University of California, Los Angeles.
There’s no evidence that ongoing stimulant treatment increases the risk of substance abuse, and “we must admit to ourselves that substance use among adolescents and young adults is normative. It happens,” even among well-adapted kids. “Think of your own past histories,” he said to an audience of psychiatrists and other medical professionals at a psychopharmacology update held by the American Academy of Child and Adolescent Psychiatry.
“You have” to deal with substance abuse that’s ruining lives, but otherwise, “I’m comfortable giving a prescription under some circumstances even if I know [patients] are using drugs. It’s the extent and type of [drug use] that informs if we should prescribe or not,” said Dr. McGough, also a professor of clinical psychiatry at the university, but be careful to “really document what you’re doing.”
Misuse and diversion is mostly related to performance enhancement, especially at competitive universities, or when grades head south. Children and young adults in those situations, as well as smokers, drinkers, and those with conduct disorders, “are the ones you worry about.” If a Stanford medical school student at age 27 suddenly develops a horrible problem focusing, with no past ADHD history, “I’m a little suspicious,” he said, noting that people who malinger “tend to go off the scale in terms of endorsing symptoms.”
If misuse and abuse are a concern, extended-release stimulants, as well as nonstimulants, are better options than immediate-release formulations.
Audience members also were curious about alternative approaches for ADHD, but the news wasn’t very good.
For now, there’s no brain scan, test, or lab measurement that reliably detects ADHD. Even with neuropsychiatric testing, only half of children and adolescents will have executive-functioning deficits. The problem with computerized tests, meanwhile, is that a lot of children and adolescents with ADHD have no problem focusing on computer games, so they aren’t helpful for diagnosis.
“The gold standard for ADHD assessment remains a good interview,” and documenting DSM-5 criteria, he said.
As for alternative treatments, “what studies have shown is that if you are highly invested in the treatment and not blinded” to it, “you tend to think you do better. In good studies where they look at teachers who don’t know what’s going on, it doesn’t pan out,” he said. Metanalysis of neurofeedback hasn’t shown effect, nor have computer games to train focus; kids will get better at them with practice, but they haven’t been shown to improve ADHD.
The only exception is omega-3, but the effect size is barely measurable. “If you can get the kid so swallow those fish oil things, go for it, or they can eat salmon,” Dr. McGough said.
In short, medications remain the best option for treating the core symptoms of ADHD, first with stimulants then, as needed, nonstimulants.
But “you are not meeting your patients’ needs if you are only prescribing medications,” he said. Psychosocial education, family behavioral therapy, and school interventions – even things as simple as sitting toward the front of the class and having a little more time on tests – are critical for overall improvement.
It’s also important to let patients and families know that “they don’t have to be ashamed of this,” and the patients need to know why they are taking their medication, he said.
Too often, children are started on a low dose of medication and told to come back in a month. “That’s a waste of time, because it’s a whole month when the person is probably inadequately treated,” Dr. McGough said.
Instead, he has patients titrate up at first, with advice to cut back or stop and call if there are problems. The patient returns after 3 weeks, and the most effective, best-tolerated dose is selected for treatment.
Dr. McGough disclosed relationships with Purdue, Shire, Tris, Sunovion, NeuroSigma, and Neurovance.
VIDEO: Pattern recognition provides clues to rheumatologic diagnoses
WAILEA, HAWAII – Several patterns can provide clues to diagnoses of rheumatologic diseases, according to Daniel E. Furst, MD, professor of rheumatology, University of Washington, Seattle.
Dermatology and rheumatology share the use of pattern recognition when making a diagnosis, he said in a video interview at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation. An important pattern is the idea of polyarticular versus monoarticular disease. Rheumatoid arthritis, osteoarthritis, autoimmune diseases, and gout and crystalline diseases, he said, encompass “about 80% of the cases with polyarticular arthritis.”
Another pattern to consider is symmetry versus asymmetry. Osteoarthritis and gout will be asymmetric, while autoimmune diseases and RA will be symmetrical, Dr. Furst pointed out. Also consider areas of involvement, such as upper and lower parts of the body. “Lower tends to be more gout and more OA,” while upper-extremity involvement is more likely to be autoimmune diseases and RA, he said.
“You don’t have to be Einstein to be a rheumatologist; just remember some simple stuff,” added Dr. Furst, who also discusses the use of lab tests in the interview.
Dr. Furst is also professor emeritus, University of California, Los Angeles, and is affiliated with the University of Florence (Italy) Medical School.
He disclosed financial relationships with companies including AbbVie, Actelion, Amgen, BMS, Cytori, Genentech/Roche, Novartis, and Pfizer.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WAILEA, HAWAII – Several patterns can provide clues to diagnoses of rheumatologic diseases, according to Daniel E. Furst, MD, professor of rheumatology, University of Washington, Seattle.
Dermatology and rheumatology share the use of pattern recognition when making a diagnosis, he said in a video interview at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation. An important pattern is the idea of polyarticular versus monoarticular disease. Rheumatoid arthritis, osteoarthritis, autoimmune diseases, and gout and crystalline diseases, he said, encompass “about 80% of the cases with polyarticular arthritis.”
Another pattern to consider is symmetry versus asymmetry. Osteoarthritis and gout will be asymmetric, while autoimmune diseases and RA will be symmetrical, Dr. Furst pointed out. Also consider areas of involvement, such as upper and lower parts of the body. “Lower tends to be more gout and more OA,” while upper-extremity involvement is more likely to be autoimmune diseases and RA, he said.
“You don’t have to be Einstein to be a rheumatologist; just remember some simple stuff,” added Dr. Furst, who also discusses the use of lab tests in the interview.
Dr. Furst is also professor emeritus, University of California, Los Angeles, and is affiliated with the University of Florence (Italy) Medical School.
He disclosed financial relationships with companies including AbbVie, Actelion, Amgen, BMS, Cytori, Genentech/Roche, Novartis, and Pfizer.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WAILEA, HAWAII – Several patterns can provide clues to diagnoses of rheumatologic diseases, according to Daniel E. Furst, MD, professor of rheumatology, University of Washington, Seattle.
Dermatology and rheumatology share the use of pattern recognition when making a diagnosis, he said in a video interview at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation. An important pattern is the idea of polyarticular versus monoarticular disease. Rheumatoid arthritis, osteoarthritis, autoimmune diseases, and gout and crystalline diseases, he said, encompass “about 80% of the cases with polyarticular arthritis.”
Another pattern to consider is symmetry versus asymmetry. Osteoarthritis and gout will be asymmetric, while autoimmune diseases and RA will be symmetrical, Dr. Furst pointed out. Also consider areas of involvement, such as upper and lower parts of the body. “Lower tends to be more gout and more OA,” while upper-extremity involvement is more likely to be autoimmune diseases and RA, he said.
“You don’t have to be Einstein to be a rheumatologist; just remember some simple stuff,” added Dr. Furst, who also discusses the use of lab tests in the interview.
Dr. Furst is also professor emeritus, University of California, Los Angeles, and is affiliated with the University of Florence (Italy) Medical School.
He disclosed financial relationships with companies including AbbVie, Actelion, Amgen, BMS, Cytori, Genentech/Roche, Novartis, and Pfizer.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EXPERT ANALYSIS FROM SDEF HAWAII DERMATOLOGY SEMINAR

BOS beat placebo for eosinophilic esophagitis
Budesonide oral suspension (BOS) was safe and significantly outperformed placebo on validated measures of eosinophilic esophagitis, according to a first-in-kind, multicenter, randomized, double-blind, phase II trial presented in the March issue of Gastroenterology (doi: 10.1053/j.gastro.2016.11.021).
The novel topical corticosteroid formulation yielded a significant histologic response and was associated with 3 fewer days of dysphagia over 2 weeks compared with placebo, reported Evan S. Dellon, MD, MPH, of University of North Carolina, Chapel Hill, and his associates. “There were no unexpected safety signals, and compliance with medication was high, suggesting that this formulation can be reliably used,” they wrote. Their findings earned BOS (SHP621) an FDA Breakthrough Therapy Designation in June 2016. Although corticosteroids are first-line therapy for eosinophilic esophagitis, symptom response in other studies has been mixed, and the Food and Drug Administration had approved neither fluticasone nor budesonide for this disease, the researchers noted. They formulated BOS to adhere better to the esophageal mucosa in order to enhance esophageal delivery while decreasing unwanted pulmonary deposition.
For the study, they randomly assigned 93 patients aged 11-40 years with eosinophilic esophagitis to receive either placebo or 2 mg BOS twice daily. By week 12, Dysphagia Symptom Questionnaire scores had fallen by 14.3 points with BOS group and by 7.5 points with placebo (P = .001). Endoscopic severity scores dropped by 3.8 points with BOS and rose by 0.4 points with placebo (P less than .0001). Rates of histologic response were 39% and 3%, respectively (P less than .0001). Nonresponders averaged 10 kg more body weight than responders, and had been diagnosed about 21 months earlier (average disease duration, 46 months and 25 months, respectively).
Rates of reported adverse effects were similar with BOS (47%) and placebo (50%). Individual rates of nasopharyngitis, upper respiratory infections, and oropharyngeal pain also were comparable between groups, but one patient stopped BOS after developing dyspnea, nausea, and vomiting that were considered treatment related. Esophageal candidiasis developed in two BOS recipients – a rate similar rate to that in a prior study of BOS (Clin Gastroenterol Hepatol. 2015 Jan 13. doi: 10.1016/j.cgh.2014.05.02), and a lower percentage than in other studies of topical steroids for eosinophilic esophagitis, according to the researchers. Morning cortisol levels were similar between groups, and there were no adverse laboratory effects, they added.
Patients in this trial had severe symptoms and histology and were highly compliant with treatment. They filled out at least 70% of their symptom diary, had at least 15 eosinophils per high-power frame from at least two esophageal levels on screening endoscopy, and reported at least 4 days of dysphagia during the second half of a 4-week, blinded placebo run-in period. Researchers should consider using these strict inclusion criteria in future trials of eosinophilic esophagitis, especially because previous studies have failed to show a treatment benefit for topical steroid therapy, the investigators noted.
Meritage Pharma, which is now a part of the Shire group, makes budesonide oral suspension and sponsored the study. Dr. Dellon disclosed ties to Meritage, Receptos, Regeneron, Aptalis, Banner Life Sciences, Novartis, and Roche. All five coinvestigators disclosed ties to industry, including Meritage, Shire, Receptos, Regeneron, and Biogen Idec.
Budesonide oral suspension (BOS) was safe and significantly outperformed placebo on validated measures of eosinophilic esophagitis, according to a first-in-kind, multicenter, randomized, double-blind, phase II trial presented in the March issue of Gastroenterology (doi: 10.1053/j.gastro.2016.11.021).
The novel topical corticosteroid formulation yielded a significant histologic response and was associated with 3 fewer days of dysphagia over 2 weeks compared with placebo, reported Evan S. Dellon, MD, MPH, of University of North Carolina, Chapel Hill, and his associates. “There were no unexpected safety signals, and compliance with medication was high, suggesting that this formulation can be reliably used,” they wrote. Their findings earned BOS (SHP621) an FDA Breakthrough Therapy Designation in June 2016. Although corticosteroids are first-line therapy for eosinophilic esophagitis, symptom response in other studies has been mixed, and the Food and Drug Administration had approved neither fluticasone nor budesonide for this disease, the researchers noted. They formulated BOS to adhere better to the esophageal mucosa in order to enhance esophageal delivery while decreasing unwanted pulmonary deposition.
For the study, they randomly assigned 93 patients aged 11-40 years with eosinophilic esophagitis to receive either placebo or 2 mg BOS twice daily. By week 12, Dysphagia Symptom Questionnaire scores had fallen by 14.3 points with BOS group and by 7.5 points with placebo (P = .001). Endoscopic severity scores dropped by 3.8 points with BOS and rose by 0.4 points with placebo (P less than .0001). Rates of histologic response were 39% and 3%, respectively (P less than .0001). Nonresponders averaged 10 kg more body weight than responders, and had been diagnosed about 21 months earlier (average disease duration, 46 months and 25 months, respectively).
Rates of reported adverse effects were similar with BOS (47%) and placebo (50%). Individual rates of nasopharyngitis, upper respiratory infections, and oropharyngeal pain also were comparable between groups, but one patient stopped BOS after developing dyspnea, nausea, and vomiting that were considered treatment related. Esophageal candidiasis developed in two BOS recipients – a rate similar rate to that in a prior study of BOS (Clin Gastroenterol Hepatol. 2015 Jan 13. doi: 10.1016/j.cgh.2014.05.02), and a lower percentage than in other studies of topical steroids for eosinophilic esophagitis, according to the researchers. Morning cortisol levels were similar between groups, and there were no adverse laboratory effects, they added.
Patients in this trial had severe symptoms and histology and were highly compliant with treatment. They filled out at least 70% of their symptom diary, had at least 15 eosinophils per high-power frame from at least two esophageal levels on screening endoscopy, and reported at least 4 days of dysphagia during the second half of a 4-week, blinded placebo run-in period. Researchers should consider using these strict inclusion criteria in future trials of eosinophilic esophagitis, especially because previous studies have failed to show a treatment benefit for topical steroid therapy, the investigators noted.
Meritage Pharma, which is now a part of the Shire group, makes budesonide oral suspension and sponsored the study. Dr. Dellon disclosed ties to Meritage, Receptos, Regeneron, Aptalis, Banner Life Sciences, Novartis, and Roche. All five coinvestigators disclosed ties to industry, including Meritage, Shire, Receptos, Regeneron, and Biogen Idec.
Budesonide oral suspension (BOS) was safe and significantly outperformed placebo on validated measures of eosinophilic esophagitis, according to a first-in-kind, multicenter, randomized, double-blind, phase II trial presented in the March issue of Gastroenterology (doi: 10.1053/j.gastro.2016.11.021).
The novel topical corticosteroid formulation yielded a significant histologic response and was associated with 3 fewer days of dysphagia over 2 weeks compared with placebo, reported Evan S. Dellon, MD, MPH, of University of North Carolina, Chapel Hill, and his associates. “There were no unexpected safety signals, and compliance with medication was high, suggesting that this formulation can be reliably used,” they wrote. Their findings earned BOS (SHP621) an FDA Breakthrough Therapy Designation in June 2016. Although corticosteroids are first-line therapy for eosinophilic esophagitis, symptom response in other studies has been mixed, and the Food and Drug Administration had approved neither fluticasone nor budesonide for this disease, the researchers noted. They formulated BOS to adhere better to the esophageal mucosa in order to enhance esophageal delivery while decreasing unwanted pulmonary deposition.
For the study, they randomly assigned 93 patients aged 11-40 years with eosinophilic esophagitis to receive either placebo or 2 mg BOS twice daily. By week 12, Dysphagia Symptom Questionnaire scores had fallen by 14.3 points with BOS group and by 7.5 points with placebo (P = .001). Endoscopic severity scores dropped by 3.8 points with BOS and rose by 0.4 points with placebo (P less than .0001). Rates of histologic response were 39% and 3%, respectively (P less than .0001). Nonresponders averaged 10 kg more body weight than responders, and had been diagnosed about 21 months earlier (average disease duration, 46 months and 25 months, respectively).
Rates of reported adverse effects were similar with BOS (47%) and placebo (50%). Individual rates of nasopharyngitis, upper respiratory infections, and oropharyngeal pain also were comparable between groups, but one patient stopped BOS after developing dyspnea, nausea, and vomiting that were considered treatment related. Esophageal candidiasis developed in two BOS recipients – a rate similar rate to that in a prior study of BOS (Clin Gastroenterol Hepatol. 2015 Jan 13. doi: 10.1016/j.cgh.2014.05.02), and a lower percentage than in other studies of topical steroids for eosinophilic esophagitis, according to the researchers. Morning cortisol levels were similar between groups, and there were no adverse laboratory effects, they added.
Patients in this trial had severe symptoms and histology and were highly compliant with treatment. They filled out at least 70% of their symptom diary, had at least 15 eosinophils per high-power frame from at least two esophageal levels on screening endoscopy, and reported at least 4 days of dysphagia during the second half of a 4-week, blinded placebo run-in period. Researchers should consider using these strict inclusion criteria in future trials of eosinophilic esophagitis, especially because previous studies have failed to show a treatment benefit for topical steroid therapy, the investigators noted.
Meritage Pharma, which is now a part of the Shire group, makes budesonide oral suspension and sponsored the study. Dr. Dellon disclosed ties to Meritage, Receptos, Regeneron, Aptalis, Banner Life Sciences, Novartis, and Roche. All five coinvestigators disclosed ties to industry, including Meritage, Shire, Receptos, Regeneron, and Biogen Idec.
FROM GASTROENTEROLOGY
Key clinical point: Budesonide oral suspension (BOS) (2 mg twice daily) was safe and significantly outperformed placebo on validated measures of eosinophilic esophagitis.
Major finding: Dysphagia Symptom Questionnaire scores decreased by 14.3 points with BOS and by 7.5 points with placebo (P = .001). Endoscopic severity scores decreased by 3.8 points and rose by 0.4 points, respectively (P less than .0001).
Data source: A 12-week, double-blind, placebo-controlled, parallel-group, phase II trial of 93 adolescents and adults with eosinophilic esophagitis.
Disclosures: Meritage Pharma, which is now a part of the Shire group, makes budesonide oral suspension and sponsored the study. Dr. Dellon disclosed ties to Meritage, Receptos, Regeneron, Aptalis, Banner Life Sciences, Novartis, and Roche. All five coinvestigators disclosed ties to industry, including Meritage, Shire, Receptos, Regeneron, and Biogen Idec.
Can a Sigma-1 Agonist Stabilize Cognition and Function in Alzheimer’s Disease?
SAN DIEGO—A novel Alzheimer’s disease drug candidate appeared to stabilize cognition and function over 57 weeks in a small, early-phase, open-label trial.
Patients with mild to moderate Alzheimer’s disease who took ANAVEX 2-73, an agonist of the sigma-1 receptor, experienced virtually no decline on the Mini-Mental State Examination (MMSE) and Alzheimer’s Disease Cooperative
ANAVEX 2-73 (Anavex Life Sciences, New York) also conferred an unexpected benefit upon subjects with insomnia. “Any patient who scored on the insomnia measure [of the Hamilton Depression Rating Scale] at baseline had no sleep disturbance at all by weeks 12 and 26,” Stephen Macfarlane, MBBS, said at the Clinical Trials on Alzheimer’s Disease conference.

The findings must be interpreted cautiously. The phase IIa study was designed to assess safety and tolerability; cognitive and functional end points were secondary. It comprised 32 patients at baseline, 25 of whom completed both a five-week, randomized, dose-finding, crossover trial and a 52-week, open-label, extension study. There was no placebo comparator. Instead, researchers used three different sets of historical control data.
Investigators will continue to treat and follow the extension study cohort, and plan to launch a placebo-controlled study in 2017, said Dr. Macfarlane, Head of Clinical Governance for the Dementia Centre in Melbourne.
The five-week, randomized, dose-finding, crossover trial started one group of patients on 30 mg/day or 50 mg/day oral ANAVEX 2-73 for 11 days after an initial two-day, single-dose, pharmacokinetic analysis, followed by an 11-day washout period, and then 11 days of 3 mg/day or 5 mg/day intravenously. A second group first received 11 days of 3 mg/day or 5 mg/day ANAVEX 2-73 intravenously after an initial 2-day, single-dose, pharmacokinetic analysis, followed by an 11-day washout period, and then 30 mg/day or 50 mg/day oral ANAVEX 2-73 for 11 days. This trial was followed by a 52-week, open-label, extension trial of 10-50 mg/day orally, titrating each patient to the maximum tolerated dose. The extension phase was originally planned to last six months, but patients and caregivers wanted to continue on the medication, so the company extended it to 12 months. It is ongoing.
An Attractive Drug Target
The sigma-1 receptor targeted by ANAVEX 2-73 is found on neurons and glia in many areas of the CNS. It modulates a number of processes implicated in neurodegenerative diseases, including glutamate and calcium activity, reaction to oxidative stress, and mitochondrial function. There is some evidence that sigma-1 receptor activation can induce neuronal regrowth and functional recovery after stroke.
The sigma-1 receptor also appears to play a role in helping cells clear misfolded proteins—a pathway that makes it an attractive drug target in Alzheimer’s disease, as well as other neurodegenerative diseases with aberrant proteins, such as Parkinson’s and Huntington’s diseases.
In preclinical testing, ANAVEX 2-73 seemed to enhance cognition in wild-type and Alzheimer’s disease–model mice.
The mean age of patients in the extension study was 71. The median MMSE score was 20.5. Most patients (78%) were taking a stable dose of an acetylcholinesterase inhibitor. During the extension phase, they were titrated to the maximum tolerated dose; 14 mg was the minimum dose necessary to achieve a therapeutic effect and keep the MMSE stable.
The primary end points were safety, tolerability, and pharmacokinetics. The exploratory measures included the P300 electroencephalogram, MMSE score, the Computerized Cogstate Alzheimer’s Battery, and the ADCS-ADL. The Hamilton Depression (HAM-D) Scale was also employed as a neuropsychiatric symptom measure.
The cohort had low baseline depression scores, with a mean score of 2 on the HAM-D. By the study’s end, it had decreased to a mean of 1 point. Patients also reported improvements in their ability to work or do other activities, and in anxiety, agitation, hypochondriasis, and insight.
Electrophysiologic Measures
The P300 wave amplitude showed a small initial increase from about 6 microvolts to 7 microvolts by four weeks, and then returned to about 6 microvolts until about week 32. Thereafter, it steadily improved to about 8 microvolts by 57 weeks—a level usually seen in healthy age-matched controls. There was a significant separation from the P300 decline seen in a matched historical Alzheimer’s disease cohort, which decreased to about 4 microvolts over a 52-week period while patients were taking donepezil.
Researchers used a second historical control group to assess changes on the Computerized Cogstate Alzheimer’s Battery. All subjects in a large Australian prospective cohort study, called AIBL (Australian Imaging, Biomarkers & Lifestyle Flagship Study of Ageing), were taking standard of care Alzheimer’s drugs. Compared with that cohort, the ANAVEX 2-73 group experienced benefits in processing speed, attention, and working memory, which became statistically significant at week 31 and continued to improve.
At 57 weeks, patients’ mean MMSE score was near the baseline mean score of 20. ADCS-ADL scores declined slightly, from a mean of about 70 to approximately 65.
Finally, the investigators used another historical cohort to assess projected cognitive and functional benefit. Compared with a pooled, placebo-arm, cohort study conducted by the Alzheimer Disease Cooperative Study Group over 12 months, ANAVEX 2-73 would have been associated with 1.8-point benefit on the MMSE and a 4-point benefit on the ADCS-ADL.
“The MMSE declined 45% less and the ADCS-ADL declined 56% less than what we would have expected from the historical control data,” Dr. Macfarlane said. “This is not only statistically significant, but clearly clinically meaningful for patients.”
Nearly all patients (98%) experienced an adverse event. Most events were mild, transitory dizziness or headache; 76% of the events were grade 1, and 2% were grade 2. There were no serious adverse events. Three subjects dropped out of the trial because of adverse events (ie, delirium, dizziness, and a combination of confusion, disorientation, and lethargy). There were no problematic interactions between the study drug and any standard of care Alzheimer’s disease medications.
—Michelle G. Sullivan
SAN DIEGO—A novel Alzheimer’s disease drug candidate appeared to stabilize cognition and function over 57 weeks in a small, early-phase, open-label trial.
Patients with mild to moderate Alzheimer’s disease who took ANAVEX 2-73, an agonist of the sigma-1 receptor, experienced virtually no decline on the Mini-Mental State Examination (MMSE) and Alzheimer’s Disease Cooperative
ANAVEX 2-73 (Anavex Life Sciences, New York) also conferred an unexpected benefit upon subjects with insomnia. “Any patient who scored on the insomnia measure [of the Hamilton Depression Rating Scale] at baseline had no sleep disturbance at all by weeks 12 and 26,” Stephen Macfarlane, MBBS, said at the Clinical Trials on Alzheimer’s Disease conference.
The findings must be interpreted cautiously. The phase IIa study was designed to assess safety and tolerability; cognitive and functional end points were secondary. It comprised 32 patients at baseline, 25 of whom completed both a five-week, randomized, dose-finding, crossover trial and a 52-week, open-label, extension study. There was no placebo comparator. Instead, researchers used three different sets of historical control data.
Investigators will continue to treat and follow the extension study cohort, and plan to launch a placebo-controlled study in 2017, said Dr. Macfarlane, Head of Clinical Governance for the Dementia Centre in Melbourne.
The five-week, randomized, dose-finding, crossover trial started one group of patients on 30 mg/day or 50 mg/day oral ANAVEX 2-73 for 11 days after an initial two-day, single-dose, pharmacokinetic analysis, followed by an 11-day washout period, and then 11 days of 3 mg/day or 5 mg/day intravenously. A second group first received 11 days of 3 mg/day or 5 mg/day ANAVEX 2-73 intravenously after an initial 2-day, single-dose, pharmacokinetic analysis, followed by an 11-day washout period, and then 30 mg/day or 50 mg/day oral ANAVEX 2-73 for 11 days. This trial was followed by a 52-week, open-label, extension trial of 10-50 mg/day orally, titrating each patient to the maximum tolerated dose. The extension phase was originally planned to last six months, but patients and caregivers wanted to continue on the medication, so the company extended it to 12 months. It is ongoing.
An Attractive Drug Target
The sigma-1 receptor targeted by ANAVEX 2-73 is found on neurons and glia in many areas of the CNS. It modulates a number of processes implicated in neurodegenerative diseases, including glutamate and calcium activity, reaction to oxidative stress, and mitochondrial function. There is some evidence that sigma-1 receptor activation can induce neuronal regrowth and functional recovery after stroke.
The sigma-1 receptor also appears to play a role in helping cells clear misfolded proteins—a pathway that makes it an attractive drug target in Alzheimer’s disease, as well as other neurodegenerative diseases with aberrant proteins, such as Parkinson’s and Huntington’s diseases.
In preclinical testing, ANAVEX 2-73 seemed to enhance cognition in wild-type and Alzheimer’s disease–model mice.
The mean age of patients in the extension study was 71. The median MMSE score was 20.5. Most patients (78%) were taking a stable dose of an acetylcholinesterase inhibitor. During the extension phase, they were titrated to the maximum tolerated dose; 14 mg was the minimum dose necessary to achieve a therapeutic effect and keep the MMSE stable.
The primary end points were safety, tolerability, and pharmacokinetics. The exploratory measures included the P300 electroencephalogram, MMSE score, the Computerized Cogstate Alzheimer’s Battery, and the ADCS-ADL. The Hamilton Depression (HAM-D) Scale was also employed as a neuropsychiatric symptom measure.
The cohort had low baseline depression scores, with a mean score of 2 on the HAM-D. By the study’s end, it had decreased to a mean of 1 point. Patients also reported improvements in their ability to work or do other activities, and in anxiety, agitation, hypochondriasis, and insight.
Electrophysiologic Measures
The P300 wave amplitude showed a small initial increase from about 6 microvolts to 7 microvolts by four weeks, and then returned to about 6 microvolts until about week 32. Thereafter, it steadily improved to about 8 microvolts by 57 weeks—a level usually seen in healthy age-matched controls. There was a significant separation from the P300 decline seen in a matched historical Alzheimer’s disease cohort, which decreased to about 4 microvolts over a 52-week period while patients were taking donepezil.
Researchers used a second historical control group to assess changes on the Computerized Cogstate Alzheimer’s Battery. All subjects in a large Australian prospective cohort study, called AIBL (Australian Imaging, Biomarkers & Lifestyle Flagship Study of Ageing), were taking standard of care Alzheimer’s drugs. Compared with that cohort, the ANAVEX 2-73 group experienced benefits in processing speed, attention, and working memory, which became statistically significant at week 31 and continued to improve.
At 57 weeks, patients’ mean MMSE score was near the baseline mean score of 20. ADCS-ADL scores declined slightly, from a mean of about 70 to approximately 65.
Finally, the investigators used another historical cohort to assess projected cognitive and functional benefit. Compared with a pooled, placebo-arm, cohort study conducted by the Alzheimer Disease Cooperative Study Group over 12 months, ANAVEX 2-73 would have been associated with 1.8-point benefit on the MMSE and a 4-point benefit on the ADCS-ADL.
“The MMSE declined 45% less and the ADCS-ADL declined 56% less than what we would have expected from the historical control data,” Dr. Macfarlane said. “This is not only statistically significant, but clearly clinically meaningful for patients.”
Nearly all patients (98%) experienced an adverse event. Most events were mild, transitory dizziness or headache; 76% of the events were grade 1, and 2% were grade 2. There were no serious adverse events. Three subjects dropped out of the trial because of adverse events (ie, delirium, dizziness, and a combination of confusion, disorientation, and lethargy). There were no problematic interactions between the study drug and any standard of care Alzheimer’s disease medications.
—Michelle G. Sullivan
SAN DIEGO—A novel Alzheimer’s disease drug candidate appeared to stabilize cognition and function over 57 weeks in a small, early-phase, open-label trial.
Patients with mild to moderate Alzheimer’s disease who took ANAVEX 2-73, an agonist of the sigma-1 receptor, experienced virtually no decline on the Mini-Mental State Examination (MMSE) and Alzheimer’s Disease Cooperative
ANAVEX 2-73 (Anavex Life Sciences, New York) also conferred an unexpected benefit upon subjects with insomnia. “Any patient who scored on the insomnia measure [of the Hamilton Depression Rating Scale] at baseline had no sleep disturbance at all by weeks 12 and 26,” Stephen Macfarlane, MBBS, said at the Clinical Trials on Alzheimer’s Disease conference.
The findings must be interpreted cautiously. The phase IIa study was designed to assess safety and tolerability; cognitive and functional end points were secondary. It comprised 32 patients at baseline, 25 of whom completed both a five-week, randomized, dose-finding, crossover trial and a 52-week, open-label, extension study. There was no placebo comparator. Instead, researchers used three different sets of historical control data.
Investigators will continue to treat and follow the extension study cohort, and plan to launch a placebo-controlled study in 2017, said Dr. Macfarlane, Head of Clinical Governance for the Dementia Centre in Melbourne.
The five-week, randomized, dose-finding, crossover trial started one group of patients on 30 mg/day or 50 mg/day oral ANAVEX 2-73 for 11 days after an initial two-day, single-dose, pharmacokinetic analysis, followed by an 11-day washout period, and then 11 days of 3 mg/day or 5 mg/day intravenously. A second group first received 11 days of 3 mg/day or 5 mg/day ANAVEX 2-73 intravenously after an initial 2-day, single-dose, pharmacokinetic analysis, followed by an 11-day washout period, and then 30 mg/day or 50 mg/day oral ANAVEX 2-73 for 11 days. This trial was followed by a 52-week, open-label, extension trial of 10-50 mg/day orally, titrating each patient to the maximum tolerated dose. The extension phase was originally planned to last six months, but patients and caregivers wanted to continue on the medication, so the company extended it to 12 months. It is ongoing.
An Attractive Drug Target
The sigma-1 receptor targeted by ANAVEX 2-73 is found on neurons and glia in many areas of the CNS. It modulates a number of processes implicated in neurodegenerative diseases, including glutamate and calcium activity, reaction to oxidative stress, and mitochondrial function. There is some evidence that sigma-1 receptor activation can induce neuronal regrowth and functional recovery after stroke.
The sigma-1 receptor also appears to play a role in helping cells clear misfolded proteins—a pathway that makes it an attractive drug target in Alzheimer’s disease, as well as other neurodegenerative diseases with aberrant proteins, such as Parkinson’s and Huntington’s diseases.
In preclinical testing, ANAVEX 2-73 seemed to enhance cognition in wild-type and Alzheimer’s disease–model mice.
The mean age of patients in the extension study was 71. The median MMSE score was 20.5. Most patients (78%) were taking a stable dose of an acetylcholinesterase inhibitor. During the extension phase, they were titrated to the maximum tolerated dose; 14 mg was the minimum dose necessary to achieve a therapeutic effect and keep the MMSE stable.
The primary end points were safety, tolerability, and pharmacokinetics. The exploratory measures included the P300 electroencephalogram, MMSE score, the Computerized Cogstate Alzheimer’s Battery, and the ADCS-ADL. The Hamilton Depression (HAM-D) Scale was also employed as a neuropsychiatric symptom measure.
The cohort had low baseline depression scores, with a mean score of 2 on the HAM-D. By the study’s end, it had decreased to a mean of 1 point. Patients also reported improvements in their ability to work or do other activities, and in anxiety, agitation, hypochondriasis, and insight.
Electrophysiologic Measures
The P300 wave amplitude showed a small initial increase from about 6 microvolts to 7 microvolts by four weeks, and then returned to about 6 microvolts until about week 32. Thereafter, it steadily improved to about 8 microvolts by 57 weeks—a level usually seen in healthy age-matched controls. There was a significant separation from the P300 decline seen in a matched historical Alzheimer’s disease cohort, which decreased to about 4 microvolts over a 52-week period while patients were taking donepezil.
Researchers used a second historical control group to assess changes on the Computerized Cogstate Alzheimer’s Battery. All subjects in a large Australian prospective cohort study, called AIBL (Australian Imaging, Biomarkers & Lifestyle Flagship Study of Ageing), were taking standard of care Alzheimer’s drugs. Compared with that cohort, the ANAVEX 2-73 group experienced benefits in processing speed, attention, and working memory, which became statistically significant at week 31 and continued to improve.
At 57 weeks, patients’ mean MMSE score was near the baseline mean score of 20. ADCS-ADL scores declined slightly, from a mean of about 70 to approximately 65.
Finally, the investigators used another historical cohort to assess projected cognitive and functional benefit. Compared with a pooled, placebo-arm, cohort study conducted by the Alzheimer Disease Cooperative Study Group over 12 months, ANAVEX 2-73 would have been associated with 1.8-point benefit on the MMSE and a 4-point benefit on the ADCS-ADL.
“The MMSE declined 45% less and the ADCS-ADL declined 56% less than what we would have expected from the historical control data,” Dr. Macfarlane said. “This is not only statistically significant, but clearly clinically meaningful for patients.”
Nearly all patients (98%) experienced an adverse event. Most events were mild, transitory dizziness or headache; 76% of the events were grade 1, and 2% were grade 2. There were no serious adverse events. Three subjects dropped out of the trial because of adverse events (ie, delirium, dizziness, and a combination of confusion, disorientation, and lethargy). There were no problematic interactions between the study drug and any standard of care Alzheimer’s disease medications.
—Michelle G. Sullivan

Clinical Guidelines: ADA 2017 Standards of Medical Care in Diabetes
In 2012, 29.1 million Americans, or 9.3% of the population, had diabetes. Of this number, 21 million were diagnosed, and 8.1 million were undiagnosed. Each year almost 1.5 million Americans receive a new diagnosis of diabetes. The management of diabetes relies upon excellent primary care. Each year the American Diabetes Association reviews new evidence and publishes an updated Standards of Care in the January issue of Diabetes Care. Here we give a short overview of the guidelines with emphasis on fundamentals and changes in the standards over the past year.
Self-management education and support, nutrition therapy, and physical activity
All patients should participate in ongoing diabetes self-management education (DSME) to facilitate the knowledge, skills, and abilities necessary to obtain optimal self-care and incorporate the needs, goals, and life experiences of the person with diabetes as they face new challenges throughout a lifetime of diabetes.
In addition, each patient should receive individualized medical nutrition therapy (MNT) provided by a registered dietitian with knowledge regarding diabetes-specific MNT. Most patients should increase aerobic physical activity to 150 min/week. Providers should encourage patients to reduce the amount of time spent sedentary by briefly standing, walking, or performing other light physical activities every 30 minutes.
Glycemic targets
A reasonable hemoglobin A1c goal for many diabetic nonpregnant adults is less than 7%. A less stringent goal under 8% may be appropriate for patients with a history of severe hypoglycemia, limited life expectancy, advanced microvascular and macrovascular complications, and extensive comorbid conditions. HbA1c measurements should be done at diagnosis and routinely to monitor glycemic control. To aid in achieving glycemic targets, self-monitoring blood glucose (SMBG) allows patients to evaluate their individual response to therapy. Integrating SMBG data into diabetes management can help guide MNT, adjust medications, determine physical activity requirements, and prevent hypoglycemia. Individuals at risk for hypoglycemia should be asked about symptomatic and asymptomatic hypoglycemia at each encounter and counseled regarding treatment of hypoglycemic events.

Obesity management
There is strong and consistent evidence that obesity management may be beneficial in the treatment of type 2 diabetes. For overweight and obese patients with type 2 diabetes, interventions should be high intensity (more than 16 sessions in 6 months) and focus on diet, physical activity, and behavioral therapy designed to achieve a greater than 5% weight loss (energy deficit of 500-750 kcal/day).
For select patients, weight loss medications may be effective as adjuncts to lifestyle changes. When choosing additional pharmacologic interventions to improve glycemic control in overweight or obese patients, providers should use medications that promote weight loss or are weight neutral including metformin, sodium-glucose cotransporter-2 (SGLT-2) inhibitors, glucagon-like peptide-1 (GLP-1) agonists, and dipeptidyl peptidase-4 inhibitors (DPP-4) versus those that cause weight gain such as insulin secretagogues, thiazolidinediones, and insulin.
Metabolic surgery should be recommended to patients with type 2 diabetes and body mass index above 40 kg/m2 (BMI above 37.5 kg/m2 in Asian Americans), regardless of adequate glycemic control and for patients with BMI above 35 kg/m2 (more than 32.5 kg/m2 in Asian Americans) without adequate glycemic control despite lifestyle modifications and optimal medical therapy. Metabolic surgery should be considered for appropriate candidates with BMIs as low as 30 if hyperglycemia is inadequately controlled despite optical medical control by either oral or injectable medications.
CV disease and risk management: BP, lipids, antiplatelet therapy, and glycemic medication management
Atherosclerotic cardiovascular disease is the leading cause of morbidity and mortality for individuals with diabetes. Screening for atherosclerotic cardiovascular disease is not recommended; rather, the emphasis is on careful risk factor management.
If systolic blood pressure (SBP) is confirmed to be above 140 mm Hg and/or the diastolic blood pressure (DBP) is confirmed to be above 90 mm Hg, pharmacologic therapy should be initiated. A meta-analysis of randomized trials of adults with type 2 diabetes comparing intensive blood pressure targets (upper limit of 130 mm Hg SBP and 80 mm Hg DBP) with standard targets (upper limit of 140-160 mm Hg SBP and 85-100 mm Hg DBP) found no significant reduction in mortality or nonfatal MI. There was a statistically significant, 35% relative risk (RR) reduction in stroke with intensive targets, but intensive targets were associated with an increased risk for adverse events such as hypotension and syncope. Recommendations suggest that antihypertension treatment in adults with diabetes without albuminuria should include any of the classes of medication demonstrated to reduce cardiovascular events in patients with diabetes, such as ACE inhibitors, angiotensin receptor blockers (ARBs), thiazide-like diuretics, or dihydropyridine calcium-channel blockers. ACE inhibitors and ARBs continue to be recommended as first-line medications for the treatment of hypertension in patients with diabetes and elevated urine albumin/creatinine ratios (above 30 mg/g creatinine). The standards also suggest consideration of administering one or more antihypertensive medications at bedtime, which may improve cardiovascular outcomes.
For patients aged 40-75 years who have diabetes without additional atherosclerotic CV disease risk factors, a moderate-intensity statin should be considered. If there are additional cardiovascular risk factors, then a high-intensity statin should be considered. For patients who are younger than 40 years of age and have diabetes with additional atherosclerotic CV disease risk factors, a less strong recommendation is to consider using moderate-intensity or high-intensity statins. For patients older than 75 years with diabetes without additional atherosclerotic CV disease risk factors, consider using moderate-intensity statin therapy; high-intensity statin therapy may be considered in older adults with risk factors for atherosclerotic cardiovascular disease.
Both women and men who are at least 50 years old and have diabetes with at least one additional cardiovascular risk factor should consider taking daily aspirin therapy (75-162 mg/day) if they do not have any risk for excessive bleeding.
In patients with long-standing suboptimally controlled type 2 diabetes and established atherosclerotic CV disease, empagliflozin or liraglutide should be considered as they have been shown to reduce cardiovascular and all-cause mortality when added to standard care.

Microvascular disease and foot care
Large prospective studies have demonstrated that optimized glucose control can reduce the onset and progression of diabetic microvascular complications. Diabetic kidney disease occurs in about 20%-40% of persons with diabetes. Annual screening includes estimated glomerular filtration rate and spot urine albumin-to-creatinine ratio. Treatment includes ACE inhibitors or ARBs in addition to a target blood pressure of under 140/90 mm Hg.
Diabetic retinopathy screening includes a dilated eye exam by an eye care professional. Treatment includes laser photocoagulation therapy for high risk nonproliferative retinopathy or proliferative retinopathy, or intravitreal injections of antivascular endothelial growth factor agents for central-involved diabetic macular edema.
Diabetic peripheral neuropathy screening includes annual 10-g monofilament and 128-HZ tuning fork vibration sensation. Medications for painful diabetic neuropathy may include gabapentin, pregabalin, duloxetine, and other agents.
Neuropathy and vascular disease are contributors to diabetic foot ulcers and amputation. A comprehensive foot examination along with appropriate risk factor oriented history to include neuropathic and vascular components (pulses, claudication) should be performed annually, while all patients with diabetes should have their feet checked at every visit.
Older adults
Prioritizing treatment goals in older adults is important in this heterogeneous population. Cardiovascular risk factor treatment is likely to be beneficial.
In setting HbA1c goals, functional status, and comorbid conditions should be considered. Metformin can still be a first-line agent for many older adults with type 2 diabetes, with consideration to renal status (creatinine clearance above 30 mL/min per 1.73 m2) and heart failure. DPP-4s have few side effects and low risk of hypoglycemia. GLP-1 receptor agonists have a low risk of hypoglycemia but may be associated with GI side effects and weight loss. SGLT-2 inhibitors have a low risk of hypoglycemia, and attention should be paid to renal thresholds for use. Thiazolidinediones should be used cautiously in those with heart failure or at elevated fracture risk. Sulfonylureas should be used cautiously because of their elevated risk of hypoglycemia. When used, a short-acting sulfonylurea – such as glipizide – is preferred, as long-acting sulfonylureas are contraindicated because of even greater hypoglycemic risk. Single-injection basal insulin may be appropriate for many with ease of use and efficacy.
The bottom line
Diabetes is a rapidly changing field and each year the American Diabetes Association updates the Standards of Medical Care document to be consistent with the latest evidence. Highlights of the standards include emphasis on diabetes self-management education, individualized glycemic goal setting, obesity management, setting blood pressure targets to less than 140/90 mm Hg, as well as statins and daily aspirin for most people with diabetes. In addition, ADA now recommends the use of specific antihyperglycemic medications to reduce cardiovascular and all-cause mortality in patients with diabetes and established cardiovascular disease.
Reference
American Diabetes Association Standards of Medical Care in Diabetes – 2017. Diabetes Care 2017; 40 (sup 1):S1-S138
Dr. Skolnik is professor of family and community medicine, Temple University School of Medicine, Philadelphia, and associate director, Family Medicine Residency Program, Abington-Jefferson Health, Abington, Pa. Dr. Johnson is associate professor at the University of North Dakota School of Medicine and Health Sciences, and practices at the Altru Diabetes Center, Grand Forks. Ms. Neuman practices at St. Mark’s Hospital, Salt Lake City.
In 2012, 29.1 million Americans, or 9.3% of the population, had diabetes. Of this number, 21 million were diagnosed, and 8.1 million were undiagnosed. Each year almost 1.5 million Americans receive a new diagnosis of diabetes. The management of diabetes relies upon excellent primary care. Each year the American Diabetes Association reviews new evidence and publishes an updated Standards of Care in the January issue of Diabetes Care. Here we give a short overview of the guidelines with emphasis on fundamentals and changes in the standards over the past year.
Self-management education and support, nutrition therapy, and physical activity
All patients should participate in ongoing diabetes self-management education (DSME) to facilitate the knowledge, skills, and abilities necessary to obtain optimal self-care and incorporate the needs, goals, and life experiences of the person with diabetes as they face new challenges throughout a lifetime of diabetes.
In addition, each patient should receive individualized medical nutrition therapy (MNT) provided by a registered dietitian with knowledge regarding diabetes-specific MNT. Most patients should increase aerobic physical activity to 150 min/week. Providers should encourage patients to reduce the amount of time spent sedentary by briefly standing, walking, or performing other light physical activities every 30 minutes.
Glycemic targets
A reasonable hemoglobin A1c goal for many diabetic nonpregnant adults is less than 7%. A less stringent goal under 8% may be appropriate for patients with a history of severe hypoglycemia, limited life expectancy, advanced microvascular and macrovascular complications, and extensive comorbid conditions. HbA1c measurements should be done at diagnosis and routinely to monitor glycemic control. To aid in achieving glycemic targets, self-monitoring blood glucose (SMBG) allows patients to evaluate their individual response to therapy. Integrating SMBG data into diabetes management can help guide MNT, adjust medications, determine physical activity requirements, and prevent hypoglycemia. Individuals at risk for hypoglycemia should be asked about symptomatic and asymptomatic hypoglycemia at each encounter and counseled regarding treatment of hypoglycemic events.
Obesity management
There is strong and consistent evidence that obesity management may be beneficial in the treatment of type 2 diabetes. For overweight and obese patients with type 2 diabetes, interventions should be high intensity (more than 16 sessions in 6 months) and focus on diet, physical activity, and behavioral therapy designed to achieve a greater than 5% weight loss (energy deficit of 500-750 kcal/day).
For select patients, weight loss medications may be effective as adjuncts to lifestyle changes. When choosing additional pharmacologic interventions to improve glycemic control in overweight or obese patients, providers should use medications that promote weight loss or are weight neutral including metformin, sodium-glucose cotransporter-2 (SGLT-2) inhibitors, glucagon-like peptide-1 (GLP-1) agonists, and dipeptidyl peptidase-4 inhibitors (DPP-4) versus those that cause weight gain such as insulin secretagogues, thiazolidinediones, and insulin.
Metabolic surgery should be recommended to patients with type 2 diabetes and body mass index above 40 kg/m2 (BMI above 37.5 kg/m2 in Asian Americans), regardless of adequate glycemic control and for patients with BMI above 35 kg/m2 (more than 32.5 kg/m2 in Asian Americans) without adequate glycemic control despite lifestyle modifications and optimal medical therapy. Metabolic surgery should be considered for appropriate candidates with BMIs as low as 30 if hyperglycemia is inadequately controlled despite optical medical control by either oral or injectable medications.
CV disease and risk management: BP, lipids, antiplatelet therapy, and glycemic medication management
Atherosclerotic cardiovascular disease is the leading cause of morbidity and mortality for individuals with diabetes. Screening for atherosclerotic cardiovascular disease is not recommended; rather, the emphasis is on careful risk factor management.
If systolic blood pressure (SBP) is confirmed to be above 140 mm Hg and/or the diastolic blood pressure (DBP) is confirmed to be above 90 mm Hg, pharmacologic therapy should be initiated. A meta-analysis of randomized trials of adults with type 2 diabetes comparing intensive blood pressure targets (upper limit of 130 mm Hg SBP and 80 mm Hg DBP) with standard targets (upper limit of 140-160 mm Hg SBP and 85-100 mm Hg DBP) found no significant reduction in mortality or nonfatal MI. There was a statistically significant, 35% relative risk (RR) reduction in stroke with intensive targets, but intensive targets were associated with an increased risk for adverse events such as hypotension and syncope. Recommendations suggest that antihypertension treatment in adults with diabetes without albuminuria should include any of the classes of medication demonstrated to reduce cardiovascular events in patients with diabetes, such as ACE inhibitors, angiotensin receptor blockers (ARBs), thiazide-like diuretics, or dihydropyridine calcium-channel blockers. ACE inhibitors and ARBs continue to be recommended as first-line medications for the treatment of hypertension in patients with diabetes and elevated urine albumin/creatinine ratios (above 30 mg/g creatinine). The standards also suggest consideration of administering one or more antihypertensive medications at bedtime, which may improve cardiovascular outcomes.
For patients aged 40-75 years who have diabetes without additional atherosclerotic CV disease risk factors, a moderate-intensity statin should be considered. If there are additional cardiovascular risk factors, then a high-intensity statin should be considered. For patients who are younger than 40 years of age and have diabetes with additional atherosclerotic CV disease risk factors, a less strong recommendation is to consider using moderate-intensity or high-intensity statins. For patients older than 75 years with diabetes without additional atherosclerotic CV disease risk factors, consider using moderate-intensity statin therapy; high-intensity statin therapy may be considered in older adults with risk factors for atherosclerotic cardiovascular disease.
Both women and men who are at least 50 years old and have diabetes with at least one additional cardiovascular risk factor should consider taking daily aspirin therapy (75-162 mg/day) if they do not have any risk for excessive bleeding.
In patients with long-standing suboptimally controlled type 2 diabetes and established atherosclerotic CV disease, empagliflozin or liraglutide should be considered as they have been shown to reduce cardiovascular and all-cause mortality when added to standard care.
Microvascular disease and foot care
Large prospective studies have demonstrated that optimized glucose control can reduce the onset and progression of diabetic microvascular complications. Diabetic kidney disease occurs in about 20%-40% of persons with diabetes. Annual screening includes estimated glomerular filtration rate and spot urine albumin-to-creatinine ratio. Treatment includes ACE inhibitors or ARBs in addition to a target blood pressure of under 140/90 mm Hg.
Diabetic retinopathy screening includes a dilated eye exam by an eye care professional. Treatment includes laser photocoagulation therapy for high risk nonproliferative retinopathy or proliferative retinopathy, or intravitreal injections of antivascular endothelial growth factor agents for central-involved diabetic macular edema.
Diabetic peripheral neuropathy screening includes annual 10-g monofilament and 128-HZ tuning fork vibration sensation. Medications for painful diabetic neuropathy may include gabapentin, pregabalin, duloxetine, and other agents.
Neuropathy and vascular disease are contributors to diabetic foot ulcers and amputation. A comprehensive foot examination along with appropriate risk factor oriented history to include neuropathic and vascular components (pulses, claudication) should be performed annually, while all patients with diabetes should have their feet checked at every visit.
Older adults
Prioritizing treatment goals in older adults is important in this heterogeneous population. Cardiovascular risk factor treatment is likely to be beneficial.
In setting HbA1c goals, functional status, and comorbid conditions should be considered. Metformin can still be a first-line agent for many older adults with type 2 diabetes, with consideration to renal status (creatinine clearance above 30 mL/min per 1.73 m2) and heart failure. DPP-4s have few side effects and low risk of hypoglycemia. GLP-1 receptor agonists have a low risk of hypoglycemia but may be associated with GI side effects and weight loss. SGLT-2 inhibitors have a low risk of hypoglycemia, and attention should be paid to renal thresholds for use. Thiazolidinediones should be used cautiously in those with heart failure or at elevated fracture risk. Sulfonylureas should be used cautiously because of their elevated risk of hypoglycemia. When used, a short-acting sulfonylurea – such as glipizide – is preferred, as long-acting sulfonylureas are contraindicated because of even greater hypoglycemic risk. Single-injection basal insulin may be appropriate for many with ease of use and efficacy.
The bottom line
Diabetes is a rapidly changing field and each year the American Diabetes Association updates the Standards of Medical Care document to be consistent with the latest evidence. Highlights of the standards include emphasis on diabetes self-management education, individualized glycemic goal setting, obesity management, setting blood pressure targets to less than 140/90 mm Hg, as well as statins and daily aspirin for most people with diabetes. In addition, ADA now recommends the use of specific antihyperglycemic medications to reduce cardiovascular and all-cause mortality in patients with diabetes and established cardiovascular disease.
Reference
American Diabetes Association Standards of Medical Care in Diabetes – 2017. Diabetes Care 2017; 40 (sup 1):S1-S138
Dr. Skolnik is professor of family and community medicine, Temple University School of Medicine, Philadelphia, and associate director, Family Medicine Residency Program, Abington-Jefferson Health, Abington, Pa. Dr. Johnson is associate professor at the University of North Dakota School of Medicine and Health Sciences, and practices at the Altru Diabetes Center, Grand Forks. Ms. Neuman practices at St. Mark’s Hospital, Salt Lake City.
In 2012, 29.1 million Americans, or 9.3% of the population, had diabetes. Of this number, 21 million were diagnosed, and 8.1 million were undiagnosed. Each year almost 1.5 million Americans receive a new diagnosis of diabetes. The management of diabetes relies upon excellent primary care. Each year the American Diabetes Association reviews new evidence and publishes an updated Standards of Care in the January issue of Diabetes Care. Here we give a short overview of the guidelines with emphasis on fundamentals and changes in the standards over the past year.
Self-management education and support, nutrition therapy, and physical activity
All patients should participate in ongoing diabetes self-management education (DSME) to facilitate the knowledge, skills, and abilities necessary to obtain optimal self-care and incorporate the needs, goals, and life experiences of the person with diabetes as they face new challenges throughout a lifetime of diabetes.
In addition, each patient should receive individualized medical nutrition therapy (MNT) provided by a registered dietitian with knowledge regarding diabetes-specific MNT. Most patients should increase aerobic physical activity to 150 min/week. Providers should encourage patients to reduce the amount of time spent sedentary by briefly standing, walking, or performing other light physical activities every 30 minutes.
Glycemic targets
A reasonable hemoglobin A1c goal for many diabetic nonpregnant adults is less than 7%. A less stringent goal under 8% may be appropriate for patients with a history of severe hypoglycemia, limited life expectancy, advanced microvascular and macrovascular complications, and extensive comorbid conditions. HbA1c measurements should be done at diagnosis and routinely to monitor glycemic control. To aid in achieving glycemic targets, self-monitoring blood glucose (SMBG) allows patients to evaluate their individual response to therapy. Integrating SMBG data into diabetes management can help guide MNT, adjust medications, determine physical activity requirements, and prevent hypoglycemia. Individuals at risk for hypoglycemia should be asked about symptomatic and asymptomatic hypoglycemia at each encounter and counseled regarding treatment of hypoglycemic events.
Obesity management
There is strong and consistent evidence that obesity management may be beneficial in the treatment of type 2 diabetes. For overweight and obese patients with type 2 diabetes, interventions should be high intensity (more than 16 sessions in 6 months) and focus on diet, physical activity, and behavioral therapy designed to achieve a greater than 5% weight loss (energy deficit of 500-750 kcal/day).
For select patients, weight loss medications may be effective as adjuncts to lifestyle changes. When choosing additional pharmacologic interventions to improve glycemic control in overweight or obese patients, providers should use medications that promote weight loss or are weight neutral including metformin, sodium-glucose cotransporter-2 (SGLT-2) inhibitors, glucagon-like peptide-1 (GLP-1) agonists, and dipeptidyl peptidase-4 inhibitors (DPP-4) versus those that cause weight gain such as insulin secretagogues, thiazolidinediones, and insulin.
Metabolic surgery should be recommended to patients with type 2 diabetes and body mass index above 40 kg/m2 (BMI above 37.5 kg/m2 in Asian Americans), regardless of adequate glycemic control and for patients with BMI above 35 kg/m2 (more than 32.5 kg/m2 in Asian Americans) without adequate glycemic control despite lifestyle modifications and optimal medical therapy. Metabolic surgery should be considered for appropriate candidates with BMIs as low as 30 if hyperglycemia is inadequately controlled despite optical medical control by either oral or injectable medications.
CV disease and risk management: BP, lipids, antiplatelet therapy, and glycemic medication management
Atherosclerotic cardiovascular disease is the leading cause of morbidity and mortality for individuals with diabetes. Screening for atherosclerotic cardiovascular disease is not recommended; rather, the emphasis is on careful risk factor management.
If systolic blood pressure (SBP) is confirmed to be above 140 mm Hg and/or the diastolic blood pressure (DBP) is confirmed to be above 90 mm Hg, pharmacologic therapy should be initiated. A meta-analysis of randomized trials of adults with type 2 diabetes comparing intensive blood pressure targets (upper limit of 130 mm Hg SBP and 80 mm Hg DBP) with standard targets (upper limit of 140-160 mm Hg SBP and 85-100 mm Hg DBP) found no significant reduction in mortality or nonfatal MI. There was a statistically significant, 35% relative risk (RR) reduction in stroke with intensive targets, but intensive targets were associated with an increased risk for adverse events such as hypotension and syncope. Recommendations suggest that antihypertension treatment in adults with diabetes without albuminuria should include any of the classes of medication demonstrated to reduce cardiovascular events in patients with diabetes, such as ACE inhibitors, angiotensin receptor blockers (ARBs), thiazide-like diuretics, or dihydropyridine calcium-channel blockers. ACE inhibitors and ARBs continue to be recommended as first-line medications for the treatment of hypertension in patients with diabetes and elevated urine albumin/creatinine ratios (above 30 mg/g creatinine). The standards also suggest consideration of administering one or more antihypertensive medications at bedtime, which may improve cardiovascular outcomes.
For patients aged 40-75 years who have diabetes without additional atherosclerotic CV disease risk factors, a moderate-intensity statin should be considered. If there are additional cardiovascular risk factors, then a high-intensity statin should be considered. For patients who are younger than 40 years of age and have diabetes with additional atherosclerotic CV disease risk factors, a less strong recommendation is to consider using moderate-intensity or high-intensity statins. For patients older than 75 years with diabetes without additional atherosclerotic CV disease risk factors, consider using moderate-intensity statin therapy; high-intensity statin therapy may be considered in older adults with risk factors for atherosclerotic cardiovascular disease.
Both women and men who are at least 50 years old and have diabetes with at least one additional cardiovascular risk factor should consider taking daily aspirin therapy (75-162 mg/day) if they do not have any risk for excessive bleeding.
In patients with long-standing suboptimally controlled type 2 diabetes and established atherosclerotic CV disease, empagliflozin or liraglutide should be considered as they have been shown to reduce cardiovascular and all-cause mortality when added to standard care.
Microvascular disease and foot care
Large prospective studies have demonstrated that optimized glucose control can reduce the onset and progression of diabetic microvascular complications. Diabetic kidney disease occurs in about 20%-40% of persons with diabetes. Annual screening includes estimated glomerular filtration rate and spot urine albumin-to-creatinine ratio. Treatment includes ACE inhibitors or ARBs in addition to a target blood pressure of under 140/90 mm Hg.
Diabetic retinopathy screening includes a dilated eye exam by an eye care professional. Treatment includes laser photocoagulation therapy for high risk nonproliferative retinopathy or proliferative retinopathy, or intravitreal injections of antivascular endothelial growth factor agents for central-involved diabetic macular edema.
Diabetic peripheral neuropathy screening includes annual 10-g monofilament and 128-HZ tuning fork vibration sensation. Medications for painful diabetic neuropathy may include gabapentin, pregabalin, duloxetine, and other agents.
Neuropathy and vascular disease are contributors to diabetic foot ulcers and amputation. A comprehensive foot examination along with appropriate risk factor oriented history to include neuropathic and vascular components (pulses, claudication) should be performed annually, while all patients with diabetes should have their feet checked at every visit.
Older adults
Prioritizing treatment goals in older adults is important in this heterogeneous population. Cardiovascular risk factor treatment is likely to be beneficial.
In setting HbA1c goals, functional status, and comorbid conditions should be considered. Metformin can still be a first-line agent for many older adults with type 2 diabetes, with consideration to renal status (creatinine clearance above 30 mL/min per 1.73 m2) and heart failure. DPP-4s have few side effects and low risk of hypoglycemia. GLP-1 receptor agonists have a low risk of hypoglycemia but may be associated with GI side effects and weight loss. SGLT-2 inhibitors have a low risk of hypoglycemia, and attention should be paid to renal thresholds for use. Thiazolidinediones should be used cautiously in those with heart failure or at elevated fracture risk. Sulfonylureas should be used cautiously because of their elevated risk of hypoglycemia. When used, a short-acting sulfonylurea – such as glipizide – is preferred, as long-acting sulfonylureas are contraindicated because of even greater hypoglycemic risk. Single-injection basal insulin may be appropriate for many with ease of use and efficacy.
The bottom line
Diabetes is a rapidly changing field and each year the American Diabetes Association updates the Standards of Medical Care document to be consistent with the latest evidence. Highlights of the standards include emphasis on diabetes self-management education, individualized glycemic goal setting, obesity management, setting blood pressure targets to less than 140/90 mm Hg, as well as statins and daily aspirin for most people with diabetes. In addition, ADA now recommends the use of specific antihyperglycemic medications to reduce cardiovascular and all-cause mortality in patients with diabetes and established cardiovascular disease.
Reference
American Diabetes Association Standards of Medical Care in Diabetes – 2017. Diabetes Care 2017; 40 (sup 1):S1-S138
Dr. Skolnik is professor of family and community medicine, Temple University School of Medicine, Philadelphia, and associate director, Family Medicine Residency Program, Abington-Jefferson Health, Abington, Pa. Dr. Johnson is associate professor at the University of North Dakota School of Medicine and Health Sciences, and practices at the Altru Diabetes Center, Grand Forks. Ms. Neuman practices at St. Mark’s Hospital, Salt Lake City.

Underdosing of Lorazepam in Children Is Associated With Increased Seizure Duration
HOUSTON—The first dose of lorazepam, when administered as a first-line antiepileptic drug (AED) for pediatric refractory status epilepticus, frequently is underdosed, and doses lower than 0.1 mg/kg are associated with increased seizure duration, according to research presented at the 70th Annual Meeting of the American Epilepsy Society (AES).
The results emphasize the importance of following AES status epilepticus guidelines, which call for lorazepam dosing of 0.1 mg/kg, said Dmitry Tchapyjnikov, MD, of Duke University School of Medicine in Durham, North Carolina, and colleagues.

“There is high variability in lorazepam dosing when used in the treatment of status epilepticus, but little is known about how this dosing variability affects seizure duration,” the researchers said. The investigators analyzed data from a multicenter prospective observational cohort of pediatric patients admitted with refractory status epilepticus (ie, status epilepticus did not resolve after two or more AEDs) between 2011 and 2016. The data were compiled by the Pediatric Status Epilepticus Research Group.
Researchers grouped patients by those who received a lower dose ( < 0.05 mg/kg), medium dose (0.05 mg/kg to < 0.1 mg/kg), and higher dose ( ≥ 0.1 mg/kg) of lorazepam. They used Cox proportional hazards models to assess the association between lorazepam dose and time to seizure resolution, adjusting for age, sex, presumed seizure cause, seizure duration prior to lorazepam administration, home AED use, prior neurologic conditions, and study site.
A total of 103 patients were included in the analysis. Patients had a median age of 4.5, and 48% were female. Lorazepam was administered at a median of 20 minutes after seizure onset. Twenty-eight percent of patients received a lower dose, 43% a medium dose, and 29% a higher dose. Individuals in the higher dose group were significantly more likely to experience seizure resolution sooner than patients in the medium and lower dose groups, with hazard ratios of 1.62 and 2.49, respectively. Median time to total seizure resolution following lorazepam administration was 93 minutes in the higher dose group, 160 minutes in the medium dose group, and 350 minutes in the lower dose group.
Among patients who had convulsive seizures, those in the higher dose group were more likely to experience convulsive seizure resolution sooner than those in the lower dose group (hazard ratio, 1.89). Median time to convulsive seizure resolution was 67 minutes in the higher dose group and 120 minutes in the lower dose group.
—Jake Remaly
HOUSTON—The first dose of lorazepam, when administered as a first-line antiepileptic drug (AED) for pediatric refractory status epilepticus, frequently is underdosed, and doses lower than 0.1 mg/kg are associated with increased seizure duration, according to research presented at the 70th Annual Meeting of the American Epilepsy Society (AES).
The results emphasize the importance of following AES status epilepticus guidelines, which call for lorazepam dosing of 0.1 mg/kg, said Dmitry Tchapyjnikov, MD, of Duke University School of Medicine in Durham, North Carolina, and colleagues.
“There is high variability in lorazepam dosing when used in the treatment of status epilepticus, but little is known about how this dosing variability affects seizure duration,” the researchers said. The investigators analyzed data from a multicenter prospective observational cohort of pediatric patients admitted with refractory status epilepticus (ie, status epilepticus did not resolve after two or more AEDs) between 2011 and 2016. The data were compiled by the Pediatric Status Epilepticus Research Group.
Researchers grouped patients by those who received a lower dose ( < 0.05 mg/kg), medium dose (0.05 mg/kg to < 0.1 mg/kg), and higher dose ( ≥ 0.1 mg/kg) of lorazepam. They used Cox proportional hazards models to assess the association between lorazepam dose and time to seizure resolution, adjusting for age, sex, presumed seizure cause, seizure duration prior to lorazepam administration, home AED use, prior neurologic conditions, and study site.
A total of 103 patients were included in the analysis. Patients had a median age of 4.5, and 48% were female. Lorazepam was administered at a median of 20 minutes after seizure onset. Twenty-eight percent of patients received a lower dose, 43% a medium dose, and 29% a higher dose. Individuals in the higher dose group were significantly more likely to experience seizure resolution sooner than patients in the medium and lower dose groups, with hazard ratios of 1.62 and 2.49, respectively. Median time to total seizure resolution following lorazepam administration was 93 minutes in the higher dose group, 160 minutes in the medium dose group, and 350 minutes in the lower dose group.
Among patients who had convulsive seizures, those in the higher dose group were more likely to experience convulsive seizure resolution sooner than those in the lower dose group (hazard ratio, 1.89). Median time to convulsive seizure resolution was 67 minutes in the higher dose group and 120 minutes in the lower dose group.
—Jake Remaly
HOUSTON—The first dose of lorazepam, when administered as a first-line antiepileptic drug (AED) for pediatric refractory status epilepticus, frequently is underdosed, and doses lower than 0.1 mg/kg are associated with increased seizure duration, according to research presented at the 70th Annual Meeting of the American Epilepsy Society (AES).
The results emphasize the importance of following AES status epilepticus guidelines, which call for lorazepam dosing of 0.1 mg/kg, said Dmitry Tchapyjnikov, MD, of Duke University School of Medicine in Durham, North Carolina, and colleagues.
“There is high variability in lorazepam dosing when used in the treatment of status epilepticus, but little is known about how this dosing variability affects seizure duration,” the researchers said. The investigators analyzed data from a multicenter prospective observational cohort of pediatric patients admitted with refractory status epilepticus (ie, status epilepticus did not resolve after two or more AEDs) between 2011 and 2016. The data were compiled by the Pediatric Status Epilepticus Research Group.
Researchers grouped patients by those who received a lower dose ( < 0.05 mg/kg), medium dose (0.05 mg/kg to < 0.1 mg/kg), and higher dose ( ≥ 0.1 mg/kg) of lorazepam. They used Cox proportional hazards models to assess the association between lorazepam dose and time to seizure resolution, adjusting for age, sex, presumed seizure cause, seizure duration prior to lorazepam administration, home AED use, prior neurologic conditions, and study site.
A total of 103 patients were included in the analysis. Patients had a median age of 4.5, and 48% were female. Lorazepam was administered at a median of 20 minutes after seizure onset. Twenty-eight percent of patients received a lower dose, 43% a medium dose, and 29% a higher dose. Individuals in the higher dose group were significantly more likely to experience seizure resolution sooner than patients in the medium and lower dose groups, with hazard ratios of 1.62 and 2.49, respectively. Median time to total seizure resolution following lorazepam administration was 93 minutes in the higher dose group, 160 minutes in the medium dose group, and 350 minutes in the lower dose group.
Among patients who had convulsive seizures, those in the higher dose group were more likely to experience convulsive seizure resolution sooner than those in the lower dose group (hazard ratio, 1.89). Median time to convulsive seizure resolution was 67 minutes in the higher dose group and 120 minutes in the lower dose group.
—Jake Remaly
7 Myomectomy myths debunked
Fibroids are extremely common and can be detected in 60% of African American women and 40% of white women by age 35. By age 50, more than 80% of African American women and almost 70% of white women have fibroids. Although most women with fibroids are relatively asymptomatic, women who have bothersome symptoms, such as heavy menstrual bleeding, urinary frequency, pelvic or abdominal pressure, or pain, account for nearly 30% of all gynecologic admissions in the United States. The cost of fibroid-related care, including surgery, hospital admissions, outpatient visits, and medications, is estimated at $4 to $9 billion per year.1 In addition, each woman seeking treatment for fibroid-related symptoms incurs an expense of $4,500 to $30,000 for lost work or disability every year.1
Many treatment options, including medical therapy and noninvasive procedures, are now available for women with symptomatic fibroids. For women who require surgical treatment, however, hysterectomy is often recommended. Fibroid-related hysterectomy currently accounts for 45% of all hysterectomies, or approximately 195,700 per year. Although the American College of Obstetricians and Gynecologists (ACOG) clinical management guidelines state that myomectomy is a safe and effective alternative to hysterectomy for treatment of women with symptomatic fibroids, only 30,000 myomectomies (abdominal, laparoscopic, and robotic-assisted approaches) are performed each year.2 Why is this? One reason may be that, although many women wish to have uterus-preserving treatment, they often feel that doctors are too quick to recommend hysterectomy as the first—and sometimes only—treatment option for fibroids.3
CASE: Woman with fibroids seeks alternative to hysterectomy
A 42-year-old woman (G2P2) presents for a third opinion regarding her heavy menstrual bleeding and known uterine fibroids. She does not want to have any more children, but she wishes to avoid a hysterectomy. Both her regular gynecologist and the second gynecologist she consulted recommended hysterectomy as the first, and only, treatment option. Physical examination reveals a 16-week-sized uterus, and ultrasonography shows at least 6 fibroids, 2 of which impinge on the uterine cavity. The patient’s other gynecologists advised her that a myomectomy would be a “bloody operation,” would leave her uterus looking like Swiss cheese, and is not appropriate for women who have completed childbearing.
The patient asks if myomectomy could be considered in her situation. How would you advise her regarding myomectomy as an alternative to hysterectomy?
Organ conservation is important
In 1931, prominent British gynecologic surgeon Victor Bonney said, “Since cure without deformity or loss of function must ever be surgery’s highest ideal, the general proposition that myomectomy is a greater surgical achievement is incontestable.”4 As current hysterectomy and myomectomy rates indicate, however, we are not attempting organ conservation very often.
Other specialties almost never remove an entire organ for benign growths. Using breast cancer surgery as an admirable paradigm, consider that in the early 20th century the standard treatment for breast cancer was a Halsted radical mastectomy with axial lymphadenectomy. By the 1930s, this disfiguring operation was replaced by simple mastectomy and radiation, and by the 1970s, by lumpectomy and lymphadenectomy. Currently, lumpectomy and sentinel node sampling is the standard of care for early stage breast cancer. This is an excellent example of “minimally invasive surgery,” a term fostered by gynecologists. And, these organ-preservingsurgeries are performed for women with cancer, not a benign condition like fibroids.
Although our approach to hysterectomy has evolved with the increasing use of laparoscopic or robotic assistance, removal of the entire uterus nevertheless remains the surgical goal. I think this narrow view of surgical options is a disservice to our patients.
Many of us were taught that myomectomy was associated with more complications and more blood loss than hysterectomy. We were taught that the uterus had no function other than childbearing and that removing the uterus had no adverse health effects. The dogma suggested that myomectomy preserved a uterus that looked like Swiss cheese and would not heal properly and that the risk of fibroid recurrence was high. These beliefs, however, are myths, which are discussed and debunked below. In second and third installments for this series on myomectomy, I present steps for successful abdominal and laparoscopic technique.
Read myths on hysterectomy, myomectomy, and fibroids
MYTH #1: Hysterectomy is safer than myomectomy
Myomectomy is performed within the confines of the uterus and myometrium, with only infrequent occasion to operate near the ureters, uterine vessels, bowel, or bladder. Therefore, it should not be surprising that studies show that fewer complications occur with myomectomy than with hysterectomy.
A retrospective review of 197 women who had myomectomy and 197 women who underwent hysterectomy with similar uterine size (14 vs 15 weeks) reported that 13% (n = 26) of women in the hysterectomy group experienced complications, including 1 bladder injury, 1 ureteral injury, and 3 bowel injuries; 8 women had an ileus and 6 women had a pelvic abscess.5 Only 5% (n = 11) of the myomectomy patients had complications, including 1 bladder injury; 2 women had reoperation for small bowel obstruction, and 6 women had an ileus. The risks of febrile morbidity, unintended surgical procedure, life-threatening events, and rehospitalization were similar for both groups.
Authors of a recent systematic review of 6 studies, which included 1,520 women with uterine size up to 18 weeks, found higher rates of visceral injury and longer hospital stays for women who had a hysterectomy compared with those who had a myomectomy (TABLE 1).6
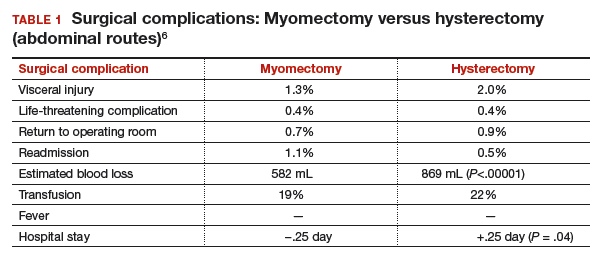
MYTH #2: Myomectomy is associated with more surgical blood loss than hysterectomy
In the previously cited study of 197 women treated with myomectomy and 197 women treated with hysterectomy, the estimated blood loss was greater in the hysterectomy group (484 mL) than in the myomectomy group (227 mL). When uterine size was corrected for, blood loss was no greater for myomectomy than for hysterectomy.5 The risk of hemorrhage (>500 mL blood loss) was greater in the hysterectomy group (14.2% vs 9.6%). Authors of the recent meta-analysis also found that the rate of transfusion was higher in the hysterectomy cohort. Tourniquets, misoprostol, vasopressin, and tranexamic acid all have been shown to significantly decrease surgical blood loss. (These treatments will be discussed in the next installment of this article series.)
MYTH #3: A uterus will look like Swiss cheese after a myomectomy
The uterus heals remarkably well after myomectomy. Three months following laparoscopic myomectomy, 3-dimensional Doppler ultrasonography demonstrated complete myometrial healing and normal blood flow to the uterus.7 In a study of women undergoing abdominal myomectomy, follow-up magnetic resonance imaging (MRI) with gadolinium showed complete healing of the myometrium and normal myometrial perfusion by 3 months.8 This study also found that, after removal of 65 g to 380 g of fibroids, the uterine volume 3 months after surgery was 65 mL, essentially equivalent to the normal volume of a uterus without fibroids (57 mL).8 See FIGURE for MRI scans of the uterus before and after myomectomy.
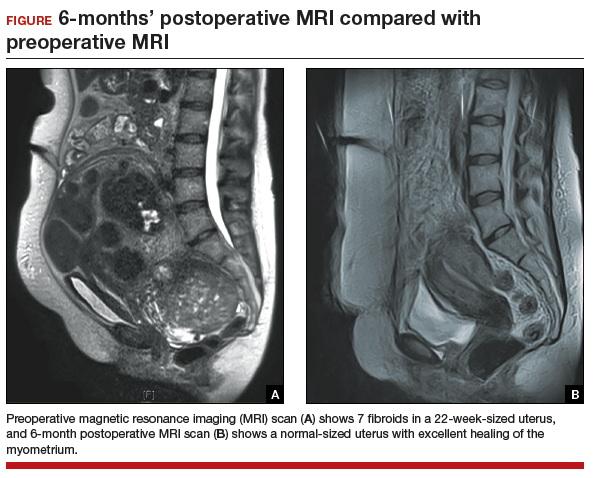
MYTH #4: Fibroids will just grow back after myomectomy
Once a fibroid is completely removed surgically, it does not grow back. The risk of new fibroid growth depends on the number of fibroids originally removed and the amount of time until menopause, when fibroids reduce in size and symptoms usually resolve. Given that the prevalence of fibroids is nearly 80% by age 50, studies measuring the detection of new fibroid growth of 1 cm on ultrasound imaging overstate the problem.9 What is likely a more important consideration for women is whether, following myomectomy, they will need another procedure for new fibroid-related symptoms.
Results of a meta-analysis of 872 women in 7 studies with 10- to 25-year follow-up indicated that 89% of women did not require another surgery.10 In another study, authors found that, over an average follow-up of 7.6 years, a second surgery occurred in 11% of the women who had 1 fibroid initially removed and for 26% of women who had multiple fibroids initially removed.11 In another study of 92 women who had either abdominal or laparoscopic myomectomy after age 45and who were followed for an average of 30 months, only 1 woman (1%) required a hysterectomy for fibroid-related symptoms.12 That patient had growth of a fibroid that was present but was not removed at her initial laparoscopic myomectomy.
Read myths 5–7 on ovarian conservation, fibroid growth, and symptom improvement
MYTH #5: Hysterectomy with ovarian conservation does not change hormone levels
Following hysterectomy with ovarian conservation, some women begin menopause earlier than age-matched women who have not undergone any surgery.13 Hysterectomy with ovarian conservation prior to age 50 has been associated with a significant increase in the risk of coronary heart disease, stroke, and heart failure.14 In a prospective longitudinal study, antimüllerian hormone (AMH) levels were persistently decreased following hysterectomy despite ovarian conservation.15 However, 3 months after myomectomy, no such changes in AMH levels were seen (TABLE 2).15

Early natural menopause has been associated with an increase in cardiovascular disease and death, and bilateral oophorectomy has been associated with increased risks of cardiovascular disease, all-cause mortality, lung cancer, colon cancer, anxiety, and depression. Although taking estrogen might obviate these adverse health effects, the majority of women who receive a prescription for estrogen following surgery are no longer taking it 5 years later.
MYTH #6: Fibroid growth in a premenopausal patient means cancer may be present
While most fibroids grow slowly, rapid growth of benign fibroids is very common. Using computerized analysis of a group of 72 women having serial MRI scans, investigators found that 34% of benign fibroids increased more than 20% in volume over 6 months.16 In premenopausal women, “rapid uterine growth” almost never indicates presence of uterine sarcoma. One study reported only 1 sarcoma among 371 women operated on for rapid growth of presumed fibroids.17 Using current criteria from the World Health Organization to determine the pathologic diagnosis, however, that 1 woman was determined to have had an atypical leiomyoma. Therefore, the prevalence of leiomyosarcoma in that study approached zero. In addition, in the 198 women who had a 6-week increase in uterine size over 1 year (one published definition of rapid growth), no sarcomas were found.17
Because of recent concern about leiomyosarcoma and morcellation of fibroids, some gynecologists have reverted to advising women that growing fibroids might be cancer and that hysterectomy is recommended. However, there is no evidence that fibroid growth is a sign of leiomyosarcoma in premenopausal women. Leiomyosarcoma should strongly be considered in a postmenopausal woman on no hormone therapy who has growth of a presumed fibroid.
MYTH #7: Myomectomy will not improve symptoms
Fibroid-related symptoms can be significant; women who undergo hysterectomy because of fibroid-related symptoms have significantly worse scores on the 36-Item Short-Form Survey (SF-36) quality-of-life questionnaire than women diagnosed with hypertension, heart disease, chronic lung disease, or arthritis.18
For women with fibroid-related symptoms, myomectomy has been shown to improve quality of life. A study of 72 women showed that SF-36 scores improved significantly following myomectomy (TABLE 3, page 48).19 In another study that used the European Quality of Life Five-Dimension Scale and Visual Analog Scale, 95 women had significant improvement in quality of life (P<.001) following laparoscopic myomectomy.20
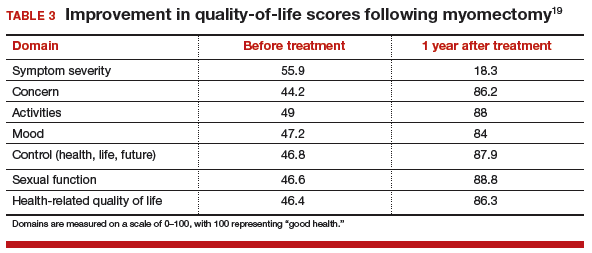
For some women, hysterectomy may have an impact on emotional quality of life. Some women report decreased sexual desire after hysterectomy. They worry that partners will see them as “not whole” and less desirable. Some women expect that hysterectomy will lead to depression, crying, lack of sexual desire, and vaginal dryness.21 No such changes have been reported for women having myomectomy.
CASE Continued: Third consult leads patient to schedule surgical procedure
After reviewing the patient’s symptoms, examination, and ultrasound results, we advise the patient that abdominal myomectomy is indeed appropriate and feasible in her case. She schedules surgery for the following month.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Cardozo ER, Clark AD, Banks NK, Henne MB, Stegmann BJ, Segars JH. The estimated annual cost of leiomyomata in the United States. Am J Obstet Gynecol. 2012;206(3):211.e1–e9.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins–Gynecology. ACOG Practice Bulletin No. 96: alternatives to hysterectomy in the management of leiomyomas. Obstet Gynecol. 2008;112(2 pt 1):387–400.
- Borah BJ, Nicholson WK, Bradley L, Stewart EA. The impact of uterine leiomyomas: a national survey of affected women. Am J Obstet Gynecol. 2013;209(4):319.e1–e20.
- Bonney V. The technique and results of myomectomy. Lancet. 1931;217(5604):171-177.
- Sawin SW, Pilevsky ND, Berlin JA, Barnhart KT. Comparability of perioperative morbidity between abdominal myomectomy and hysterectomy for women with uterine leiomyomas. Am J Obstet Gynecol. 2000;183(6):1448–1455.
- Pundir J, Walawalkar R, Seshadri S, Khalaf Y, El-Toukhy T. Perioperative morbidity associated with abdominal myomectomy compared with total abdominal hysterectomy for uterine fibroids. J Obstet Gynecol. 2013;33(7):655–662.
- Chang WC, Chang DY, Huang SC, et al. Use of three-dimensional ultrasonography in the evaluation of uterine perfusion and healing after laparoscopic myomectomy. Fertil Steril. 2009;92(3):1110–1115.
- Tsuji S, Takahashi K, Imaoka I, Sugimura K, Miyazaki K, Noda Y. MRI evaluation of the uterine structure after myomectomy. Gynecol Obstet Invest. 2006;61(2):106–110.
- Sudik R, Husch K, Steller J, Daume E. Fertility and pregnancy outcome after myomectomy in sterility patients. Eur J Obstet Gynecol Reprod Biol. 1996;65(2):209–214.
- Fauconnier A, Chapron C, Babaki-Fard K, Dubuisson JB. Recurrence of leiomyomata after myomectomy. Hum Reprod Update. 2000;6(6):595–602.
- Malone, LJ. Myomectomy: recurrence after removal of solitary and multiple myomas. Obstet Gynecol. 1969;34(2):200–203.
- Kim DH, Kim ML, Song T, Kim MK, Yoon BS, Seong SJ. Is myomectomy in women aged 45 years and older an effective option? Eur J Obstet Gynecol Reprod Biol. 2014;177:57–60.
- Farquhar CM, Sadler L, Harvey SA, Stewart AW. The association of hysterectomy and menopause: a prospective cohort study. BJOG. 2005;112(7):956–962.
- Ingelsson E, Lundholm C, Johansson AL, Altman D. Hysterectomy and risk of cardiovascular disease: a population-based cohort study. Eur Heart J. 2011;32(6):745–750.
- Wang HY, Quan S, Zhang RL, et al. Comparison of serum anti-Mullerian hormone levels following hysterectomy and myomectomy for benign gynaecological conditions. Eur J Obstet Gynecol Reprod Biol. 2013;171(2):368–371.
- Peddada SD, Laughlin SK, Miner K, et al. Growth of uterine leiomyomata among premenopausal black and white women. Proc Natl Acad Sci. 2008;105(50):19887–19892.
- Parker W, Fu YS, Berek JS. Uterine sarcoma in patients operated on for presumed leiomyoma and rapidly growing leiomyoma. Obstet Gynecol. 1994;83(3):414–418.
- Rowe MK, Kanouse DE, Mittman BS, Bernstein SJ. Quality of life among women undergoing hysterectomies. Obstet Gynecol. 1999;93(6):915–921.
- Dilek S, Ertunc D, Tok EC, Cimen R, Doruk A. The effect of myomectomy on health-related quality of life of women with myoma uteri. J Obstet Gynaecol Res. 2010;36(2):364–369.
- Radosa JC, Radosa CG, Mavrova R, et al. Postoperative quality of life and sexual function in premenopausal women undergoing laparoscopic myomectomy for symptomatic fibroids: a prospective observational cohort study. PLoS One. 2016;29;11(11):e0166659.
- Groff JY, Mullen PD, Byrd T, Shelton AJ, Lees E, Goode J. Decision making, beliefs, and attitudes toward hysterectomy: a focus group study with medically underserved women in Texas. J Womens Health Gend Based Med. 2000;9(suppl 2):39S–50S.

Dr. Parker is Director of Minimally Invasive Gynecologic Surgery at Santa Monica-UCLA Medical Center, Santa Monica, California. He is a past president of AAGL.
The author reports no financial relationships relevant to this article.
Dr. Parker is Director of Minimally Invasive Gynecologic Surgery at Santa Monica-UCLA Medical Center, Santa Monica, California. He is a past president of AAGL.
The author reports no financial relationships relevant to this article.
Dr. Parker is Director of Minimally Invasive Gynecologic Surgery at Santa Monica-UCLA Medical Center, Santa Monica, California. He is a past president of AAGL.
The author reports no financial relationships relevant to this article.
Fibroids are extremely common and can be detected in 60% of African American women and 40% of white women by age 35. By age 50, more than 80% of African American women and almost 70% of white women have fibroids. Although most women with fibroids are relatively asymptomatic, women who have bothersome symptoms, such as heavy menstrual bleeding, urinary frequency, pelvic or abdominal pressure, or pain, account for nearly 30% of all gynecologic admissions in the United States. The cost of fibroid-related care, including surgery, hospital admissions, outpatient visits, and medications, is estimated at $4 to $9 billion per year.1 In addition, each woman seeking treatment for fibroid-related symptoms incurs an expense of $4,500 to $30,000 for lost work or disability every year.1
Many treatment options, including medical therapy and noninvasive procedures, are now available for women with symptomatic fibroids. For women who require surgical treatment, however, hysterectomy is often recommended. Fibroid-related hysterectomy currently accounts for 45% of all hysterectomies, or approximately 195,700 per year. Although the American College of Obstetricians and Gynecologists (ACOG) clinical management guidelines state that myomectomy is a safe and effective alternative to hysterectomy for treatment of women with symptomatic fibroids, only 30,000 myomectomies (abdominal, laparoscopic, and robotic-assisted approaches) are performed each year.2 Why is this? One reason may be that, although many women wish to have uterus-preserving treatment, they often feel that doctors are too quick to recommend hysterectomy as the first—and sometimes only—treatment option for fibroids.3
CASE: Woman with fibroids seeks alternative to hysterectomy
A 42-year-old woman (G2P2) presents for a third opinion regarding her heavy menstrual bleeding and known uterine fibroids. She does not want to have any more children, but she wishes to avoid a hysterectomy. Both her regular gynecologist and the second gynecologist she consulted recommended hysterectomy as the first, and only, treatment option. Physical examination reveals a 16-week-sized uterus, and ultrasonography shows at least 6 fibroids, 2 of which impinge on the uterine cavity. The patient’s other gynecologists advised her that a myomectomy would be a “bloody operation,” would leave her uterus looking like Swiss cheese, and is not appropriate for women who have completed childbearing.
The patient asks if myomectomy could be considered in her situation. How would you advise her regarding myomectomy as an alternative to hysterectomy?
Organ conservation is important
In 1931, prominent British gynecologic surgeon Victor Bonney said, “Since cure without deformity or loss of function must ever be surgery’s highest ideal, the general proposition that myomectomy is a greater surgical achievement is incontestable.”4 As current hysterectomy and myomectomy rates indicate, however, we are not attempting organ conservation very often.
Other specialties almost never remove an entire organ for benign growths. Using breast cancer surgery as an admirable paradigm, consider that in the early 20th century the standard treatment for breast cancer was a Halsted radical mastectomy with axial lymphadenectomy. By the 1930s, this disfiguring operation was replaced by simple mastectomy and radiation, and by the 1970s, by lumpectomy and lymphadenectomy. Currently, lumpectomy and sentinel node sampling is the standard of care for early stage breast cancer. This is an excellent example of “minimally invasive surgery,” a term fostered by gynecologists. And, these organ-preservingsurgeries are performed for women with cancer, not a benign condition like fibroids.
Although our approach to hysterectomy has evolved with the increasing use of laparoscopic or robotic assistance, removal of the entire uterus nevertheless remains the surgical goal. I think this narrow view of surgical options is a disservice to our patients.
Many of us were taught that myomectomy was associated with more complications and more blood loss than hysterectomy. We were taught that the uterus had no function other than childbearing and that removing the uterus had no adverse health effects. The dogma suggested that myomectomy preserved a uterus that looked like Swiss cheese and would not heal properly and that the risk of fibroid recurrence was high. These beliefs, however, are myths, which are discussed and debunked below. In second and third installments for this series on myomectomy, I present steps for successful abdominal and laparoscopic technique.
Read myths on hysterectomy, myomectomy, and fibroids
MYTH #1: Hysterectomy is safer than myomectomy
Myomectomy is performed within the confines of the uterus and myometrium, with only infrequent occasion to operate near the ureters, uterine vessels, bowel, or bladder. Therefore, it should not be surprising that studies show that fewer complications occur with myomectomy than with hysterectomy.
A retrospective review of 197 women who had myomectomy and 197 women who underwent hysterectomy with similar uterine size (14 vs 15 weeks) reported that 13% (n = 26) of women in the hysterectomy group experienced complications, including 1 bladder injury, 1 ureteral injury, and 3 bowel injuries; 8 women had an ileus and 6 women had a pelvic abscess.5 Only 5% (n = 11) of the myomectomy patients had complications, including 1 bladder injury; 2 women had reoperation for small bowel obstruction, and 6 women had an ileus. The risks of febrile morbidity, unintended surgical procedure, life-threatening events, and rehospitalization were similar for both groups.
Authors of a recent systematic review of 6 studies, which included 1,520 women with uterine size up to 18 weeks, found higher rates of visceral injury and longer hospital stays for women who had a hysterectomy compared with those who had a myomectomy (TABLE 1).6
MYTH #2: Myomectomy is associated with more surgical blood loss than hysterectomy
In the previously cited study of 197 women treated with myomectomy and 197 women treated with hysterectomy, the estimated blood loss was greater in the hysterectomy group (484 mL) than in the myomectomy group (227 mL). When uterine size was corrected for, blood loss was no greater for myomectomy than for hysterectomy.5 The risk of hemorrhage (>500 mL blood loss) was greater in the hysterectomy group (14.2% vs 9.6%). Authors of the recent meta-analysis also found that the rate of transfusion was higher in the hysterectomy cohort. Tourniquets, misoprostol, vasopressin, and tranexamic acid all have been shown to significantly decrease surgical blood loss. (These treatments will be discussed in the next installment of this article series.)
MYTH #3: A uterus will look like Swiss cheese after a myomectomy
The uterus heals remarkably well after myomectomy. Three months following laparoscopic myomectomy, 3-dimensional Doppler ultrasonography demonstrated complete myometrial healing and normal blood flow to the uterus.7 In a study of women undergoing abdominal myomectomy, follow-up magnetic resonance imaging (MRI) with gadolinium showed complete healing of the myometrium and normal myometrial perfusion by 3 months.8 This study also found that, after removal of 65 g to 380 g of fibroids, the uterine volume 3 months after surgery was 65 mL, essentially equivalent to the normal volume of a uterus without fibroids (57 mL).8 See FIGURE for MRI scans of the uterus before and after myomectomy.
MYTH #4: Fibroids will just grow back after myomectomy
Once a fibroid is completely removed surgically, it does not grow back. The risk of new fibroid growth depends on the number of fibroids originally removed and the amount of time until menopause, when fibroids reduce in size and symptoms usually resolve. Given that the prevalence of fibroids is nearly 80% by age 50, studies measuring the detection of new fibroid growth of 1 cm on ultrasound imaging overstate the problem.9 What is likely a more important consideration for women is whether, following myomectomy, they will need another procedure for new fibroid-related symptoms.
Results of a meta-analysis of 872 women in 7 studies with 10- to 25-year follow-up indicated that 89% of women did not require another surgery.10 In another study, authors found that, over an average follow-up of 7.6 years, a second surgery occurred in 11% of the women who had 1 fibroid initially removed and for 26% of women who had multiple fibroids initially removed.11 In another study of 92 women who had either abdominal or laparoscopic myomectomy after age 45and who were followed for an average of 30 months, only 1 woman (1%) required a hysterectomy for fibroid-related symptoms.12 That patient had growth of a fibroid that was present but was not removed at her initial laparoscopic myomectomy.
Read myths 5–7 on ovarian conservation, fibroid growth, and symptom improvement
MYTH #5: Hysterectomy with ovarian conservation does not change hormone levels
Following hysterectomy with ovarian conservation, some women begin menopause earlier than age-matched women who have not undergone any surgery.13 Hysterectomy with ovarian conservation prior to age 50 has been associated with a significant increase in the risk of coronary heart disease, stroke, and heart failure.14 In a prospective longitudinal study, antimüllerian hormone (AMH) levels were persistently decreased following hysterectomy despite ovarian conservation.15 However, 3 months after myomectomy, no such changes in AMH levels were seen (TABLE 2).15
Early natural menopause has been associated with an increase in cardiovascular disease and death, and bilateral oophorectomy has been associated with increased risks of cardiovascular disease, all-cause mortality, lung cancer, colon cancer, anxiety, and depression. Although taking estrogen might obviate these adverse health effects, the majority of women who receive a prescription for estrogen following surgery are no longer taking it 5 years later.
MYTH #6: Fibroid growth in a premenopausal patient means cancer may be present
While most fibroids grow slowly, rapid growth of benign fibroids is very common. Using computerized analysis of a group of 72 women having serial MRI scans, investigators found that 34% of benign fibroids increased more than 20% in volume over 6 months.16 In premenopausal women, “rapid uterine growth” almost never indicates presence of uterine sarcoma. One study reported only 1 sarcoma among 371 women operated on for rapid growth of presumed fibroids.17 Using current criteria from the World Health Organization to determine the pathologic diagnosis, however, that 1 woman was determined to have had an atypical leiomyoma. Therefore, the prevalence of leiomyosarcoma in that study approached zero. In addition, in the 198 women who had a 6-week increase in uterine size over 1 year (one published definition of rapid growth), no sarcomas were found.17
Because of recent concern about leiomyosarcoma and morcellation of fibroids, some gynecologists have reverted to advising women that growing fibroids might be cancer and that hysterectomy is recommended. However, there is no evidence that fibroid growth is a sign of leiomyosarcoma in premenopausal women. Leiomyosarcoma should strongly be considered in a postmenopausal woman on no hormone therapy who has growth of a presumed fibroid.
MYTH #7: Myomectomy will not improve symptoms
Fibroid-related symptoms can be significant; women who undergo hysterectomy because of fibroid-related symptoms have significantly worse scores on the 36-Item Short-Form Survey (SF-36) quality-of-life questionnaire than women diagnosed with hypertension, heart disease, chronic lung disease, or arthritis.18
For women with fibroid-related symptoms, myomectomy has been shown to improve quality of life. A study of 72 women showed that SF-36 scores improved significantly following myomectomy (TABLE 3, page 48).19 In another study that used the European Quality of Life Five-Dimension Scale and Visual Analog Scale, 95 women had significant improvement in quality of life (P<.001) following laparoscopic myomectomy.20
For some women, hysterectomy may have an impact on emotional quality of life. Some women report decreased sexual desire after hysterectomy. They worry that partners will see them as “not whole” and less desirable. Some women expect that hysterectomy will lead to depression, crying, lack of sexual desire, and vaginal dryness.21 No such changes have been reported for women having myomectomy.
CASE Continued: Third consult leads patient to schedule surgical procedure
After reviewing the patient’s symptoms, examination, and ultrasound results, we advise the patient that abdominal myomectomy is indeed appropriate and feasible in her case. She schedules surgery for the following month.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Fibroids are extremely common and can be detected in 60% of African American women and 40% of white women by age 35. By age 50, more than 80% of African American women and almost 70% of white women have fibroids. Although most women with fibroids are relatively asymptomatic, women who have bothersome symptoms, such as heavy menstrual bleeding, urinary frequency, pelvic or abdominal pressure, or pain, account for nearly 30% of all gynecologic admissions in the United States. The cost of fibroid-related care, including surgery, hospital admissions, outpatient visits, and medications, is estimated at $4 to $9 billion per year.1 In addition, each woman seeking treatment for fibroid-related symptoms incurs an expense of $4,500 to $30,000 for lost work or disability every year.1
Many treatment options, including medical therapy and noninvasive procedures, are now available for women with symptomatic fibroids. For women who require surgical treatment, however, hysterectomy is often recommended. Fibroid-related hysterectomy currently accounts for 45% of all hysterectomies, or approximately 195,700 per year. Although the American College of Obstetricians and Gynecologists (ACOG) clinical management guidelines state that myomectomy is a safe and effective alternative to hysterectomy for treatment of women with symptomatic fibroids, only 30,000 myomectomies (abdominal, laparoscopic, and robotic-assisted approaches) are performed each year.2 Why is this? One reason may be that, although many women wish to have uterus-preserving treatment, they often feel that doctors are too quick to recommend hysterectomy as the first—and sometimes only—treatment option for fibroids.3
CASE: Woman with fibroids seeks alternative to hysterectomy
A 42-year-old woman (G2P2) presents for a third opinion regarding her heavy menstrual bleeding and known uterine fibroids. She does not want to have any more children, but she wishes to avoid a hysterectomy. Both her regular gynecologist and the second gynecologist she consulted recommended hysterectomy as the first, and only, treatment option. Physical examination reveals a 16-week-sized uterus, and ultrasonography shows at least 6 fibroids, 2 of which impinge on the uterine cavity. The patient’s other gynecologists advised her that a myomectomy would be a “bloody operation,” would leave her uterus looking like Swiss cheese, and is not appropriate for women who have completed childbearing.
The patient asks if myomectomy could be considered in her situation. How would you advise her regarding myomectomy as an alternative to hysterectomy?
Organ conservation is important
In 1931, prominent British gynecologic surgeon Victor Bonney said, “Since cure without deformity or loss of function must ever be surgery’s highest ideal, the general proposition that myomectomy is a greater surgical achievement is incontestable.”4 As current hysterectomy and myomectomy rates indicate, however, we are not attempting organ conservation very often.
Other specialties almost never remove an entire organ for benign growths. Using breast cancer surgery as an admirable paradigm, consider that in the early 20th century the standard treatment for breast cancer was a Halsted radical mastectomy with axial lymphadenectomy. By the 1930s, this disfiguring operation was replaced by simple mastectomy and radiation, and by the 1970s, by lumpectomy and lymphadenectomy. Currently, lumpectomy and sentinel node sampling is the standard of care for early stage breast cancer. This is an excellent example of “minimally invasive surgery,” a term fostered by gynecologists. And, these organ-preservingsurgeries are performed for women with cancer, not a benign condition like fibroids.
Although our approach to hysterectomy has evolved with the increasing use of laparoscopic or robotic assistance, removal of the entire uterus nevertheless remains the surgical goal. I think this narrow view of surgical options is a disservice to our patients.
Many of us were taught that myomectomy was associated with more complications and more blood loss than hysterectomy. We were taught that the uterus had no function other than childbearing and that removing the uterus had no adverse health effects. The dogma suggested that myomectomy preserved a uterus that looked like Swiss cheese and would not heal properly and that the risk of fibroid recurrence was high. These beliefs, however, are myths, which are discussed and debunked below. In second and third installments for this series on myomectomy, I present steps for successful abdominal and laparoscopic technique.
Read myths on hysterectomy, myomectomy, and fibroids
MYTH #1: Hysterectomy is safer than myomectomy
Myomectomy is performed within the confines of the uterus and myometrium, with only infrequent occasion to operate near the ureters, uterine vessels, bowel, or bladder. Therefore, it should not be surprising that studies show that fewer complications occur with myomectomy than with hysterectomy.
A retrospective review of 197 women who had myomectomy and 197 women who underwent hysterectomy with similar uterine size (14 vs 15 weeks) reported that 13% (n = 26) of women in the hysterectomy group experienced complications, including 1 bladder injury, 1 ureteral injury, and 3 bowel injuries; 8 women had an ileus and 6 women had a pelvic abscess.5 Only 5% (n = 11) of the myomectomy patients had complications, including 1 bladder injury; 2 women had reoperation for small bowel obstruction, and 6 women had an ileus. The risks of febrile morbidity, unintended surgical procedure, life-threatening events, and rehospitalization were similar for both groups.
Authors of a recent systematic review of 6 studies, which included 1,520 women with uterine size up to 18 weeks, found higher rates of visceral injury and longer hospital stays for women who had a hysterectomy compared with those who had a myomectomy (TABLE 1).6
MYTH #2: Myomectomy is associated with more surgical blood loss than hysterectomy
In the previously cited study of 197 women treated with myomectomy and 197 women treated with hysterectomy, the estimated blood loss was greater in the hysterectomy group (484 mL) than in the myomectomy group (227 mL). When uterine size was corrected for, blood loss was no greater for myomectomy than for hysterectomy.5 The risk of hemorrhage (>500 mL blood loss) was greater in the hysterectomy group (14.2% vs 9.6%). Authors of the recent meta-analysis also found that the rate of transfusion was higher in the hysterectomy cohort. Tourniquets, misoprostol, vasopressin, and tranexamic acid all have been shown to significantly decrease surgical blood loss. (These treatments will be discussed in the next installment of this article series.)
MYTH #3: A uterus will look like Swiss cheese after a myomectomy
The uterus heals remarkably well after myomectomy. Three months following laparoscopic myomectomy, 3-dimensional Doppler ultrasonography demonstrated complete myometrial healing and normal blood flow to the uterus.7 In a study of women undergoing abdominal myomectomy, follow-up magnetic resonance imaging (MRI) with gadolinium showed complete healing of the myometrium and normal myometrial perfusion by 3 months.8 This study also found that, after removal of 65 g to 380 g of fibroids, the uterine volume 3 months after surgery was 65 mL, essentially equivalent to the normal volume of a uterus without fibroids (57 mL).8 See FIGURE for MRI scans of the uterus before and after myomectomy.
MYTH #4: Fibroids will just grow back after myomectomy
Once a fibroid is completely removed surgically, it does not grow back. The risk of new fibroid growth depends on the number of fibroids originally removed and the amount of time until menopause, when fibroids reduce in size and symptoms usually resolve. Given that the prevalence of fibroids is nearly 80% by age 50, studies measuring the detection of new fibroid growth of 1 cm on ultrasound imaging overstate the problem.9 What is likely a more important consideration for women is whether, following myomectomy, they will need another procedure for new fibroid-related symptoms.
Results of a meta-analysis of 872 women in 7 studies with 10- to 25-year follow-up indicated that 89% of women did not require another surgery.10 In another study, authors found that, over an average follow-up of 7.6 years, a second surgery occurred in 11% of the women who had 1 fibroid initially removed and for 26% of women who had multiple fibroids initially removed.11 In another study of 92 women who had either abdominal or laparoscopic myomectomy after age 45and who were followed for an average of 30 months, only 1 woman (1%) required a hysterectomy for fibroid-related symptoms.12 That patient had growth of a fibroid that was present but was not removed at her initial laparoscopic myomectomy.
Read myths 5–7 on ovarian conservation, fibroid growth, and symptom improvement
MYTH #5: Hysterectomy with ovarian conservation does not change hormone levels
Following hysterectomy with ovarian conservation, some women begin menopause earlier than age-matched women who have not undergone any surgery.13 Hysterectomy with ovarian conservation prior to age 50 has been associated with a significant increase in the risk of coronary heart disease, stroke, and heart failure.14 In a prospective longitudinal study, antimüllerian hormone (AMH) levels were persistently decreased following hysterectomy despite ovarian conservation.15 However, 3 months after myomectomy, no such changes in AMH levels were seen (TABLE 2).15
Early natural menopause has been associated with an increase in cardiovascular disease and death, and bilateral oophorectomy has been associated with increased risks of cardiovascular disease, all-cause mortality, lung cancer, colon cancer, anxiety, and depression. Although taking estrogen might obviate these adverse health effects, the majority of women who receive a prescription for estrogen following surgery are no longer taking it 5 years later.
MYTH #6: Fibroid growth in a premenopausal patient means cancer may be present
While most fibroids grow slowly, rapid growth of benign fibroids is very common. Using computerized analysis of a group of 72 women having serial MRI scans, investigators found that 34% of benign fibroids increased more than 20% in volume over 6 months.16 In premenopausal women, “rapid uterine growth” almost never indicates presence of uterine sarcoma. One study reported only 1 sarcoma among 371 women operated on for rapid growth of presumed fibroids.17 Using current criteria from the World Health Organization to determine the pathologic diagnosis, however, that 1 woman was determined to have had an atypical leiomyoma. Therefore, the prevalence of leiomyosarcoma in that study approached zero. In addition, in the 198 women who had a 6-week increase in uterine size over 1 year (one published definition of rapid growth), no sarcomas were found.17
Because of recent concern about leiomyosarcoma and morcellation of fibroids, some gynecologists have reverted to advising women that growing fibroids might be cancer and that hysterectomy is recommended. However, there is no evidence that fibroid growth is a sign of leiomyosarcoma in premenopausal women. Leiomyosarcoma should strongly be considered in a postmenopausal woman on no hormone therapy who has growth of a presumed fibroid.
MYTH #7: Myomectomy will not improve symptoms
Fibroid-related symptoms can be significant; women who undergo hysterectomy because of fibroid-related symptoms have significantly worse scores on the 36-Item Short-Form Survey (SF-36) quality-of-life questionnaire than women diagnosed with hypertension, heart disease, chronic lung disease, or arthritis.18
For women with fibroid-related symptoms, myomectomy has been shown to improve quality of life. A study of 72 women showed that SF-36 scores improved significantly following myomectomy (TABLE 3, page 48).19 In another study that used the European Quality of Life Five-Dimension Scale and Visual Analog Scale, 95 women had significant improvement in quality of life (P<.001) following laparoscopic myomectomy.20
For some women, hysterectomy may have an impact on emotional quality of life. Some women report decreased sexual desire after hysterectomy. They worry that partners will see them as “not whole” and less desirable. Some women expect that hysterectomy will lead to depression, crying, lack of sexual desire, and vaginal dryness.21 No such changes have been reported for women having myomectomy.
CASE Continued: Third consult leads patient to schedule surgical procedure
After reviewing the patient’s symptoms, examination, and ultrasound results, we advise the patient that abdominal myomectomy is indeed appropriate and feasible in her case. She schedules surgery for the following month.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Cardozo ER, Clark AD, Banks NK, Henne MB, Stegmann BJ, Segars JH. The estimated annual cost of leiomyomata in the United States. Am J Obstet Gynecol. 2012;206(3):211.e1–e9.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins–Gynecology. ACOG Practice Bulletin No. 96: alternatives to hysterectomy in the management of leiomyomas. Obstet Gynecol. 2008;112(2 pt 1):387–400.
- Borah BJ, Nicholson WK, Bradley L, Stewart EA. The impact of uterine leiomyomas: a national survey of affected women. Am J Obstet Gynecol. 2013;209(4):319.e1–e20.
- Bonney V. The technique and results of myomectomy. Lancet. 1931;217(5604):171-177.
- Sawin SW, Pilevsky ND, Berlin JA, Barnhart KT. Comparability of perioperative morbidity between abdominal myomectomy and hysterectomy for women with uterine leiomyomas. Am J Obstet Gynecol. 2000;183(6):1448–1455.
- Pundir J, Walawalkar R, Seshadri S, Khalaf Y, El-Toukhy T. Perioperative morbidity associated with abdominal myomectomy compared with total abdominal hysterectomy for uterine fibroids. J Obstet Gynecol. 2013;33(7):655–662.
- Chang WC, Chang DY, Huang SC, et al. Use of three-dimensional ultrasonography in the evaluation of uterine perfusion and healing after laparoscopic myomectomy. Fertil Steril. 2009;92(3):1110–1115.
- Tsuji S, Takahashi K, Imaoka I, Sugimura K, Miyazaki K, Noda Y. MRI evaluation of the uterine structure after myomectomy. Gynecol Obstet Invest. 2006;61(2):106–110.
- Sudik R, Husch K, Steller J, Daume E. Fertility and pregnancy outcome after myomectomy in sterility patients. Eur J Obstet Gynecol Reprod Biol. 1996;65(2):209–214.
- Fauconnier A, Chapron C, Babaki-Fard K, Dubuisson JB. Recurrence of leiomyomata after myomectomy. Hum Reprod Update. 2000;6(6):595–602.
- Malone, LJ. Myomectomy: recurrence after removal of solitary and multiple myomas. Obstet Gynecol. 1969;34(2):200–203.
- Kim DH, Kim ML, Song T, Kim MK, Yoon BS, Seong SJ. Is myomectomy in women aged 45 years and older an effective option? Eur J Obstet Gynecol Reprod Biol. 2014;177:57–60.
- Farquhar CM, Sadler L, Harvey SA, Stewart AW. The association of hysterectomy and menopause: a prospective cohort study. BJOG. 2005;112(7):956–962.
- Ingelsson E, Lundholm C, Johansson AL, Altman D. Hysterectomy and risk of cardiovascular disease: a population-based cohort study. Eur Heart J. 2011;32(6):745–750.
- Wang HY, Quan S, Zhang RL, et al. Comparison of serum anti-Mullerian hormone levels following hysterectomy and myomectomy for benign gynaecological conditions. Eur J Obstet Gynecol Reprod Biol. 2013;171(2):368–371.
- Peddada SD, Laughlin SK, Miner K, et al. Growth of uterine leiomyomata among premenopausal black and white women. Proc Natl Acad Sci. 2008;105(50):19887–19892.
- Parker W, Fu YS, Berek JS. Uterine sarcoma in patients operated on for presumed leiomyoma and rapidly growing leiomyoma. Obstet Gynecol. 1994;83(3):414–418.
- Rowe MK, Kanouse DE, Mittman BS, Bernstein SJ. Quality of life among women undergoing hysterectomies. Obstet Gynecol. 1999;93(6):915–921.
- Dilek S, Ertunc D, Tok EC, Cimen R, Doruk A. The effect of myomectomy on health-related quality of life of women with myoma uteri. J Obstet Gynaecol Res. 2010;36(2):364–369.
- Radosa JC, Radosa CG, Mavrova R, et al. Postoperative quality of life and sexual function in premenopausal women undergoing laparoscopic myomectomy for symptomatic fibroids: a prospective observational cohort study. PLoS One. 2016;29;11(11):e0166659.
- Groff JY, Mullen PD, Byrd T, Shelton AJ, Lees E, Goode J. Decision making, beliefs, and attitudes toward hysterectomy: a focus group study with medically underserved women in Texas. J Womens Health Gend Based Med. 2000;9(suppl 2):39S–50S.
- Cardozo ER, Clark AD, Banks NK, Henne MB, Stegmann BJ, Segars JH. The estimated annual cost of leiomyomata in the United States. Am J Obstet Gynecol. 2012;206(3):211.e1–e9.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins–Gynecology. ACOG Practice Bulletin No. 96: alternatives to hysterectomy in the management of leiomyomas. Obstet Gynecol. 2008;112(2 pt 1):387–400.
- Borah BJ, Nicholson WK, Bradley L, Stewart EA. The impact of uterine leiomyomas: a national survey of affected women. Am J Obstet Gynecol. 2013;209(4):319.e1–e20.
- Bonney V. The technique and results of myomectomy. Lancet. 1931;217(5604):171-177.
- Sawin SW, Pilevsky ND, Berlin JA, Barnhart KT. Comparability of perioperative morbidity between abdominal myomectomy and hysterectomy for women with uterine leiomyomas. Am J Obstet Gynecol. 2000;183(6):1448–1455.
- Pundir J, Walawalkar R, Seshadri S, Khalaf Y, El-Toukhy T. Perioperative morbidity associated with abdominal myomectomy compared with total abdominal hysterectomy for uterine fibroids. J Obstet Gynecol. 2013;33(7):655–662.
- Chang WC, Chang DY, Huang SC, et al. Use of three-dimensional ultrasonography in the evaluation of uterine perfusion and healing after laparoscopic myomectomy. Fertil Steril. 2009;92(3):1110–1115.
- Tsuji S, Takahashi K, Imaoka I, Sugimura K, Miyazaki K, Noda Y. MRI evaluation of the uterine structure after myomectomy. Gynecol Obstet Invest. 2006;61(2):106–110.
- Sudik R, Husch K, Steller J, Daume E. Fertility and pregnancy outcome after myomectomy in sterility patients. Eur J Obstet Gynecol Reprod Biol. 1996;65(2):209–214.
- Fauconnier A, Chapron C, Babaki-Fard K, Dubuisson JB. Recurrence of leiomyomata after myomectomy. Hum Reprod Update. 2000;6(6):595–602.
- Malone, LJ. Myomectomy: recurrence after removal of solitary and multiple myomas. Obstet Gynecol. 1969;34(2):200–203.
- Kim DH, Kim ML, Song T, Kim MK, Yoon BS, Seong SJ. Is myomectomy in women aged 45 years and older an effective option? Eur J Obstet Gynecol Reprod Biol. 2014;177:57–60.
- Farquhar CM, Sadler L, Harvey SA, Stewart AW. The association of hysterectomy and menopause: a prospective cohort study. BJOG. 2005;112(7):956–962.
- Ingelsson E, Lundholm C, Johansson AL, Altman D. Hysterectomy and risk of cardiovascular disease: a population-based cohort study. Eur Heart J. 2011;32(6):745–750.
- Wang HY, Quan S, Zhang RL, et al. Comparison of serum anti-Mullerian hormone levels following hysterectomy and myomectomy for benign gynaecological conditions. Eur J Obstet Gynecol Reprod Biol. 2013;171(2):368–371.
- Peddada SD, Laughlin SK, Miner K, et al. Growth of uterine leiomyomata among premenopausal black and white women. Proc Natl Acad Sci. 2008;105(50):19887–19892.
- Parker W, Fu YS, Berek JS. Uterine sarcoma in patients operated on for presumed leiomyoma and rapidly growing leiomyoma. Obstet Gynecol. 1994;83(3):414–418.
- Rowe MK, Kanouse DE, Mittman BS, Bernstein SJ. Quality of life among women undergoing hysterectomies. Obstet Gynecol. 1999;93(6):915–921.
- Dilek S, Ertunc D, Tok EC, Cimen R, Doruk A. The effect of myomectomy on health-related quality of life of women with myoma uteri. J Obstet Gynaecol Res. 2010;36(2):364–369.
- Radosa JC, Radosa CG, Mavrova R, et al. Postoperative quality of life and sexual function in premenopausal women undergoing laparoscopic myomectomy for symptomatic fibroids: a prospective observational cohort study. PLoS One. 2016;29;11(11):e0166659.
- Groff JY, Mullen PD, Byrd T, Shelton AJ, Lees E, Goode J. Decision making, beliefs, and attitudes toward hysterectomy: a focus group study with medically underserved women in Texas. J Womens Health Gend Based Med. 2000;9(suppl 2):39S–50S.
Acute Plasma Tau Predicts Prolonged Return to Play After Concussion
Among collegiate athletes, elevated plasma tau concentrations within six hours after a sport-related concussion predict a prolonged recovery, according to research published online ahead of print January 6 in Neurology. This finding suggests that tau levels may help to determine when athletes should return to play. Variability of tau concentrations across athletes and the effect of physical exertion on plasma tau may complicate the use of the biomarker for concussion management, however.
Approximately 3.8 million sport-related concussions occur each year in the United States, but no biomarkers are known to predict recovery and an athlete’s readiness to return to play. Postconcussive symptoms typically resolve within 10 days in about half of collegiate athletes, but symptoms are chronic in a subset of patients. Shahim et al found that plasma tau elevations predicted a return to play of more than 10 days in professional ice hockey players in Sweden.
Diagnosing Sport-Related Concussion
Jessica Gill, RN, PhD, an investigator with the National Institute of Nursing Research at the NIH, and colleagues conducted a study to determine whether changes in plasma tau after sport-related concussion relate to return to play in men and women collegiate athletes. The researchers included students with concussion, as well as athlete and nonathlete controls. The athletes participated in various National Collegiate Athletic Association (NCAA) division I and III contact sports (ie, football, soccer, basketball, hockey, and lacrosse).

Between 2009 and 2014, 632 athletes underwent plasma sampling and cognitive testing prior to the sports seasons and were followed prospectively for a diagnosis of sport-related concussion. Sport-related concussions were witnessed by an on-field certified athletic trainer and met the Sport Concussion Assessment Tool 2 definition of concussion.Investigators collected blood samples from athletes with concussion and athlete controls at six hours, 24 hours, 72 hours, and seven days after a concussion. Nonathlete controls had blood draws at an unrelated time point. Investigators measured total tau using an ultrasensitive immunoassay.
Return to play for each athlete was determined by athletic trainers or team physicians. They followed NCAA guidelines, which recommend that athletes be asymptomatic at rest and as they progressively resume activity before returning to play.
A total of 46 athletes were diagnosed with a sport-related concussion. Concussions occurred between 19 days and 218 days after baseline assessments (mean, 92.3 days). Thirty-seven athletes without concussion served as athlete controls. Athletes with and without concussion did not differ significantly in sport played, history of sport-related concussion, or other demographic features. A control group of 21 healthy nonathletes was demographically similar to the athlete groups.Return to play information was available for 41 of the athletes with concussion. Athletes who took more than 10 days to recover were considered to have a long return to play (23 athletes). Those who recovered in less than 10 days had a short return to play (18 athletes). The mean return to play duration was 21.68 days (range, two days to 263 days). Five athletes had a return to play duration of 30 days or more. Approximately 39% returned to play in less than 10 days. There were no significant differences in sport played or history of concussion among those with long return to play versus short return to play. Women made up 61% of the long return to play group and 28% of the short return to play group.
Tau Measurements
Compared with nonathletes, athletes had significantly higher mean tau concentrations at baseline and all other time points. The longitudinal pattern of tau differed significantly between athletes with and without concussion. Athletes with concussion had significantly lower mean total tau at 24 hours (6.06 pg/mL vs 7.89 pg/mL) and 72 hours (5.19 pg/mL vs 6.94 pg/mL), compared with athlete controls.
Athletes with a long return to play had higher tau concentrations overall, after controlling for sex, than those with a short return to play. The differences were statistically significant at six hours (10.98 pg/mL vs 7.02 pg/mL), 24 hours (7.19 pg/mL vs 4.08 pg/mL), and 72 hours (6.29 pg/mL vs 3.94 pg/mL).
Mean change in tau from baseline also significantly differed between the return to play groups. Athletes with long return to play had a mean increase of 2.26 pg/mL at six hours postconcussion, compared with a mean reduction of 1.19 pg/mL in the short return to play group, after controlling for sex. Area under the curve (AUC) analyses revealed that higher total tau six hours post concussion and change in tau from baseline to six hours post concussion predicted long return to play (AUC of 0.81 and 0.80, respectively). Higher total tau at 72 hours postconcussion also was a significant predictor of long return to play (AUC, 0.82).
“These findings suggest that changes in total tau within six hours of a sport-related concussion may provide vital information about return to play decisions, and may serve to mitigate the negative consequences of returning to play prematurely,” Dr. Gill and colleagues said. Preclinical models link insufficient recovery time from a mild traumatic brain injury (mTBI) to greater neuropathology following a subsequent mTBI, including pathology that overlaps with that of chronic traumatic encephalopathy.
Lower levels of tau in athletes with concussion, compared with athletes without concussion, at 24 hours and 72 hours “may be due to the effects of physical exertion on tau,” the researchers said. Limitations of the study include the relatively small sample size within subanalyses of long and short return to play.
More Research Is Needed
“While normally measured in CSF, tau measured in blood could provide the opportunity to assess neurologic injury shortly after concussion, as well as facilitate monitoring of recovery over time,” said Barbara B. Bendlin, PhD, Associate Professor of Medicine and Public Health at the University of Wisconsin–Madison, and Michael Makdissi, MBBS, PhD, research fellow at the Florey Institute of Neuroscience and Mental Health and Adjunct Associate Professor of Rehabilitation, Nutrition, and Sport at the La Trobe Sport and Exercise Medicine Research Centre in Australia, in an accompanying editorial.
However, differences in plasma tau levels between athletes and nonathletes; lower plasma tau levels at 24 hours and 72 hours post concussion in athletes with concussion, compared with nonconcussed teammates; variability across players; and fluctuations in plasma tau levels over time in general may complicate the use of the biomarker in concussion management. In addition, tau in plasma may reflect CNS and peripheral nervous system origins.
“This study and others conducted in the sports setting open the door for further evaluation and possible future implementation of blood-based biomarkers for evaluation of concussion,” they said. “Nevertheless, more work is needed before blood-based biomarkers can be used for management of sport-related concussion.”
—Jake Remaly
Suggested Reading
Bendlin BB, Makdissi M. Blood-based biomarkers for evaluating sport-related concussion: Back in the game. Neurology. 2017 Jan 6 [Epub ahead of print].
Gill J, Merchant-Borna K, Jeromin A, et al. Acute plasma tau relates to prolonged return to play after concussion. Neurology. 2017 Jan 6 [Epub ahead of print].
Shahim P, Tegner Y, Wilson DH, et al. Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol. 2014;71(6):684-692.
Among collegiate athletes, elevated plasma tau concentrations within six hours after a sport-related concussion predict a prolonged recovery, according to research published online ahead of print January 6 in Neurology. This finding suggests that tau levels may help to determine when athletes should return to play. Variability of tau concentrations across athletes and the effect of physical exertion on plasma tau may complicate the use of the biomarker for concussion management, however.
Approximately 3.8 million sport-related concussions occur each year in the United States, but no biomarkers are known to predict recovery and an athlete’s readiness to return to play. Postconcussive symptoms typically resolve within 10 days in about half of collegiate athletes, but symptoms are chronic in a subset of patients. Shahim et al found that plasma tau elevations predicted a return to play of more than 10 days in professional ice hockey players in Sweden.
Diagnosing Sport-Related Concussion
Jessica Gill, RN, PhD, an investigator with the National Institute of Nursing Research at the NIH, and colleagues conducted a study to determine whether changes in plasma tau after sport-related concussion relate to return to play in men and women collegiate athletes. The researchers included students with concussion, as well as athlete and nonathlete controls. The athletes participated in various National Collegiate Athletic Association (NCAA) division I and III contact sports (ie, football, soccer, basketball, hockey, and lacrosse).
Between 2009 and 2014, 632 athletes underwent plasma sampling and cognitive testing prior to the sports seasons and were followed prospectively for a diagnosis of sport-related concussion. Sport-related concussions were witnessed by an on-field certified athletic trainer and met the Sport Concussion Assessment Tool 2 definition of concussion.Investigators collected blood samples from athletes with concussion and athlete controls at six hours, 24 hours, 72 hours, and seven days after a concussion. Nonathlete controls had blood draws at an unrelated time point. Investigators measured total tau using an ultrasensitive immunoassay.
Return to play for each athlete was determined by athletic trainers or team physicians. They followed NCAA guidelines, which recommend that athletes be asymptomatic at rest and as they progressively resume activity before returning to play.
A total of 46 athletes were diagnosed with a sport-related concussion. Concussions occurred between 19 days and 218 days after baseline assessments (mean, 92.3 days). Thirty-seven athletes without concussion served as athlete controls. Athletes with and without concussion did not differ significantly in sport played, history of sport-related concussion, or other demographic features. A control group of 21 healthy nonathletes was demographically similar to the athlete groups.Return to play information was available for 41 of the athletes with concussion. Athletes who took more than 10 days to recover were considered to have a long return to play (23 athletes). Those who recovered in less than 10 days had a short return to play (18 athletes). The mean return to play duration was 21.68 days (range, two days to 263 days). Five athletes had a return to play duration of 30 days or more. Approximately 39% returned to play in less than 10 days. There were no significant differences in sport played or history of concussion among those with long return to play versus short return to play. Women made up 61% of the long return to play group and 28% of the short return to play group.
Tau Measurements
Compared with nonathletes, athletes had significantly higher mean tau concentrations at baseline and all other time points. The longitudinal pattern of tau differed significantly between athletes with and without concussion. Athletes with concussion had significantly lower mean total tau at 24 hours (6.06 pg/mL vs 7.89 pg/mL) and 72 hours (5.19 pg/mL vs 6.94 pg/mL), compared with athlete controls.
Athletes with a long return to play had higher tau concentrations overall, after controlling for sex, than those with a short return to play. The differences were statistically significant at six hours (10.98 pg/mL vs 7.02 pg/mL), 24 hours (7.19 pg/mL vs 4.08 pg/mL), and 72 hours (6.29 pg/mL vs 3.94 pg/mL).
Mean change in tau from baseline also significantly differed between the return to play groups. Athletes with long return to play had a mean increase of 2.26 pg/mL at six hours postconcussion, compared with a mean reduction of 1.19 pg/mL in the short return to play group, after controlling for sex. Area under the curve (AUC) analyses revealed that higher total tau six hours post concussion and change in tau from baseline to six hours post concussion predicted long return to play (AUC of 0.81 and 0.80, respectively). Higher total tau at 72 hours postconcussion also was a significant predictor of long return to play (AUC, 0.82).
“These findings suggest that changes in total tau within six hours of a sport-related concussion may provide vital information about return to play decisions, and may serve to mitigate the negative consequences of returning to play prematurely,” Dr. Gill and colleagues said. Preclinical models link insufficient recovery time from a mild traumatic brain injury (mTBI) to greater neuropathology following a subsequent mTBI, including pathology that overlaps with that of chronic traumatic encephalopathy.
Lower levels of tau in athletes with concussion, compared with athletes without concussion, at 24 hours and 72 hours “may be due to the effects of physical exertion on tau,” the researchers said. Limitations of the study include the relatively small sample size within subanalyses of long and short return to play.
More Research Is Needed
“While normally measured in CSF, tau measured in blood could provide the opportunity to assess neurologic injury shortly after concussion, as well as facilitate monitoring of recovery over time,” said Barbara B. Bendlin, PhD, Associate Professor of Medicine and Public Health at the University of Wisconsin–Madison, and Michael Makdissi, MBBS, PhD, research fellow at the Florey Institute of Neuroscience and Mental Health and Adjunct Associate Professor of Rehabilitation, Nutrition, and Sport at the La Trobe Sport and Exercise Medicine Research Centre in Australia, in an accompanying editorial.
However, differences in plasma tau levels between athletes and nonathletes; lower plasma tau levels at 24 hours and 72 hours post concussion in athletes with concussion, compared with nonconcussed teammates; variability across players; and fluctuations in plasma tau levels over time in general may complicate the use of the biomarker in concussion management. In addition, tau in plasma may reflect CNS and peripheral nervous system origins.
“This study and others conducted in the sports setting open the door for further evaluation and possible future implementation of blood-based biomarkers for evaluation of concussion,” they said. “Nevertheless, more work is needed before blood-based biomarkers can be used for management of sport-related concussion.”
—Jake Remaly
Suggested Reading
Bendlin BB, Makdissi M. Blood-based biomarkers for evaluating sport-related concussion: Back in the game. Neurology. 2017 Jan 6 [Epub ahead of print].
Gill J, Merchant-Borna K, Jeromin A, et al. Acute plasma tau relates to prolonged return to play after concussion. Neurology. 2017 Jan 6 [Epub ahead of print].
Shahim P, Tegner Y, Wilson DH, et al. Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol. 2014;71(6):684-692.
Among collegiate athletes, elevated plasma tau concentrations within six hours after a sport-related concussion predict a prolonged recovery, according to research published online ahead of print January 6 in Neurology. This finding suggests that tau levels may help to determine when athletes should return to play. Variability of tau concentrations across athletes and the effect of physical exertion on plasma tau may complicate the use of the biomarker for concussion management, however.
Approximately 3.8 million sport-related concussions occur each year in the United States, but no biomarkers are known to predict recovery and an athlete’s readiness to return to play. Postconcussive symptoms typically resolve within 10 days in about half of collegiate athletes, but symptoms are chronic in a subset of patients. Shahim et al found that plasma tau elevations predicted a return to play of more than 10 days in professional ice hockey players in Sweden.
Diagnosing Sport-Related Concussion
Jessica Gill, RN, PhD, an investigator with the National Institute of Nursing Research at the NIH, and colleagues conducted a study to determine whether changes in plasma tau after sport-related concussion relate to return to play in men and women collegiate athletes. The researchers included students with concussion, as well as athlete and nonathlete controls. The athletes participated in various National Collegiate Athletic Association (NCAA) division I and III contact sports (ie, football, soccer, basketball, hockey, and lacrosse).
Between 2009 and 2014, 632 athletes underwent plasma sampling and cognitive testing prior to the sports seasons and were followed prospectively for a diagnosis of sport-related concussion. Sport-related concussions were witnessed by an on-field certified athletic trainer and met the Sport Concussion Assessment Tool 2 definition of concussion.Investigators collected blood samples from athletes with concussion and athlete controls at six hours, 24 hours, 72 hours, and seven days after a concussion. Nonathlete controls had blood draws at an unrelated time point. Investigators measured total tau using an ultrasensitive immunoassay.
Return to play for each athlete was determined by athletic trainers or team physicians. They followed NCAA guidelines, which recommend that athletes be asymptomatic at rest and as they progressively resume activity before returning to play.
A total of 46 athletes were diagnosed with a sport-related concussion. Concussions occurred between 19 days and 218 days after baseline assessments (mean, 92.3 days). Thirty-seven athletes without concussion served as athlete controls. Athletes with and without concussion did not differ significantly in sport played, history of sport-related concussion, or other demographic features. A control group of 21 healthy nonathletes was demographically similar to the athlete groups.Return to play information was available for 41 of the athletes with concussion. Athletes who took more than 10 days to recover were considered to have a long return to play (23 athletes). Those who recovered in less than 10 days had a short return to play (18 athletes). The mean return to play duration was 21.68 days (range, two days to 263 days). Five athletes had a return to play duration of 30 days or more. Approximately 39% returned to play in less than 10 days. There were no significant differences in sport played or history of concussion among those with long return to play versus short return to play. Women made up 61% of the long return to play group and 28% of the short return to play group.
Tau Measurements
Compared with nonathletes, athletes had significantly higher mean tau concentrations at baseline and all other time points. The longitudinal pattern of tau differed significantly between athletes with and without concussion. Athletes with concussion had significantly lower mean total tau at 24 hours (6.06 pg/mL vs 7.89 pg/mL) and 72 hours (5.19 pg/mL vs 6.94 pg/mL), compared with athlete controls.
Athletes with a long return to play had higher tau concentrations overall, after controlling for sex, than those with a short return to play. The differences were statistically significant at six hours (10.98 pg/mL vs 7.02 pg/mL), 24 hours (7.19 pg/mL vs 4.08 pg/mL), and 72 hours (6.29 pg/mL vs 3.94 pg/mL).
Mean change in tau from baseline also significantly differed between the return to play groups. Athletes with long return to play had a mean increase of 2.26 pg/mL at six hours postconcussion, compared with a mean reduction of 1.19 pg/mL in the short return to play group, after controlling for sex. Area under the curve (AUC) analyses revealed that higher total tau six hours post concussion and change in tau from baseline to six hours post concussion predicted long return to play (AUC of 0.81 and 0.80, respectively). Higher total tau at 72 hours postconcussion also was a significant predictor of long return to play (AUC, 0.82).
“These findings suggest that changes in total tau within six hours of a sport-related concussion may provide vital information about return to play decisions, and may serve to mitigate the negative consequences of returning to play prematurely,” Dr. Gill and colleagues said. Preclinical models link insufficient recovery time from a mild traumatic brain injury (mTBI) to greater neuropathology following a subsequent mTBI, including pathology that overlaps with that of chronic traumatic encephalopathy.
Lower levels of tau in athletes with concussion, compared with athletes without concussion, at 24 hours and 72 hours “may be due to the effects of physical exertion on tau,” the researchers said. Limitations of the study include the relatively small sample size within subanalyses of long and short return to play.
More Research Is Needed
“While normally measured in CSF, tau measured in blood could provide the opportunity to assess neurologic injury shortly after concussion, as well as facilitate monitoring of recovery over time,” said Barbara B. Bendlin, PhD, Associate Professor of Medicine and Public Health at the University of Wisconsin–Madison, and Michael Makdissi, MBBS, PhD, research fellow at the Florey Institute of Neuroscience and Mental Health and Adjunct Associate Professor of Rehabilitation, Nutrition, and Sport at the La Trobe Sport and Exercise Medicine Research Centre in Australia, in an accompanying editorial.
However, differences in plasma tau levels between athletes and nonathletes; lower plasma tau levels at 24 hours and 72 hours post concussion in athletes with concussion, compared with nonconcussed teammates; variability across players; and fluctuations in plasma tau levels over time in general may complicate the use of the biomarker in concussion management. In addition, tau in plasma may reflect CNS and peripheral nervous system origins.
“This study and others conducted in the sports setting open the door for further evaluation and possible future implementation of blood-based biomarkers for evaluation of concussion,” they said. “Nevertheless, more work is needed before blood-based biomarkers can be used for management of sport-related concussion.”
—Jake Remaly
Suggested Reading
Bendlin BB, Makdissi M. Blood-based biomarkers for evaluating sport-related concussion: Back in the game. Neurology. 2017 Jan 6 [Epub ahead of print].
Gill J, Merchant-Borna K, Jeromin A, et al. Acute plasma tau relates to prolonged return to play after concussion. Neurology. 2017 Jan 6 [Epub ahead of print].
Shahim P, Tegner Y, Wilson DH, et al. Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol. 2014;71(6):684-692.
