User login
Genetic studies link JIA subtypes to adult diseases, show uniqueness of systemic disease
Two new studies of the genetic relationships between the seven designated categories of juvenile idiopathic arthritis (JIA) provide compelling support for reclassification of the categories, particularly for systemic JIA, and give evidence that some of the categories have clear equivalents in the realm of adult-onset diseases.
The International League of Associations for Rheumatology (ILAR) classification system (J Rheumatol. 2004;31:390-2) that defined the seven juvenile idiopathic arthritis (JIA) categories – systemic arthritis, oligoarticular arthritis, rheumatoid factor (RF)–negative polyarticular arthritis, RF-positive polyarticular arthritis, psoriatic arthritis (PsA), enthesitis-related arthritis (ERA), and undifferentiated arthritis – is problematic because the long-term outcome and response to treatment varies not only between subtypes but also within the subtypes, suggesting that these subgroups do not yet represent uniform groups of patients,” Wendy Thomson, PhD, professor of genetic epidemiology at the University of Manchester (England) and a senior author on both studies, explained in an interview.

“Despite the differences between the subtypes, current treatments of this disease often involve using the same drugs across all subtypes of JIA,” said Dr. Thomson, who is also deputy director of the Arthritis Research UK Centre for Genetics and Genomics at the university. “Understanding the genetic basis of the subtypes of this disease could help understand the cause of this disease and identify more appropriate treatment options [because] the current classification is largely based on clinical data and it is proposed that the addition of genetic data could improve classification.”
Interrelationships between JIA categories and adult disease
The first of these studies found that particular alleles in the human leukocyte antigen (HLA) region that have been associated with the different JIA categories strongly correlate some of the categories with one another, and that each JIA category potentially has an adult-onset counterpart based on shared HLA associations. It is the largest investigation of association of the HLA region with JIA and its categories to date, according to the researchers (Ann Rheum Dis. 2016 Dec 20. doi: 10.1136/annrheumdis-2016-210025).
In particular, the investigators demonstrated for the first time that RF-negative polyarticular JIA and oligoarticular JIA are genetically similar in their HLA associations. They also reported that RF-positive polyarthritis shares an association with adult seropositive rheumatoid arthritis and that combined data for oligoarthritis and RF-negative polyarthritis share the same association with adult seronegative RA. In addition, the researchers generated support for genetic associations between the particular JIA categories that are most commonly thought to have adult counterparts, such as ERA and adult ankylosing spondylitis (AS) as well as juvenile PsA and adult PsA.
Dr. Thomson and her coinvestigators used ImmunoChip genotype array data for 191,494 single nucleotide polymorphism (SNP) markers from the HLA region of 5,737 JIA patients and 16,403 controls in the United States, United Kingdom, Canada, Norway, and Germany to impute classical HLA alleles as well as specific amino acid positions within HLA alleles that may play an important functional role. After quality-control measures for the data were put in place, comparisons between 5,043 JIA cases and 14,390 controls showed that oligoarthritis and RF-negative polyarthritis were the most common and homogeneous JIA categories investigated (2,409 and 1,525 patients, respectively), and that they share a significant association across the HLA spectrum, meaning that they are genetically similar. When combined, the data from these two JIA categories correspond with seronegative rheumatoid arthritis in adults, while RF-positive polyarthritis on its own has an association with seropositive rheumatoid arthritis involving histidine at position 13 of the HLA-DRB1 allele. As expected, the most significant association between the ERA category and AS was for HLA-B*27. For juvenile PsA, no associations reached genome-wide level of significance, although HLA-C*0602 was modestly associated with juvenile PsA, and it is known to be associated with adult-onset PsA and is the primary HLA association in psoriasis.
“The results of this study have important implications for understanding disease pathogenesis, etiology, and potential future therapeutic strategies for JIA categories,” Dr. Thomson and her coauthors wrote, adding that “heterogeneity of JIA remains a key challenge to pediatric rheumatologists; however, these results may inform the debate on classification and help define a more biological-driven and molecular-driven classification system.”
Uniqueness of systemic JIA
In the second of the two studies, investigators from many different childhood arthritis study groups focused on systemic JIA (sJIA), also known in the past as Still’s disease. According to the authors, it is the first large-scale genomic study of sJIA ever published (Ann Rheum Dis. 2016 Dec 7. doi: 10.1136/annrheumdis-2016-210324).
“[sJIA] is characterized by prominent systemic inflammation and has a rare adult-onset counterpart; and undifferentiated arthritis includes arthritis that does not fit into any single category,” the authors wrote, adding that the “unique clinical characteristics of sJIA suggest that it is distinct from other forms of JIA, leading to the contention by some that sJIA should be separated from other forms of JIA and labeled as an autoinflammatory disease.”
The investigators carried out SNP genotyping on 982 children with sJIA in the United States, United Kingdom, Germany, Turkey, Italy, Brazil, Argentina, Canada, and Spain, along with 431 healthy children without sJIA who came from the same countries. A total of 7,579 control patients also underwent genotyping. Ultimately, 770 sJIA and 6,947 control subjects completed the study. sJIA was then compared with other JIA subtypes by using weighted genetic risk scores.
Comparison of the data sets for sJIA with those for the combined categories of RF-negative polyarticular JIA and oligoarticular JIA, as well as for the individual category of RF-positive polyarticular JIA, showed that sJIA has a distinct genetic architecture that separates it from those JIA categories, providing further evidence that sJIA should be classified separately from other forms of JIA and, potentially, treated differently as well.
According to Dr. Thomson, the finding “provides important evidence that sJIA should be considered a unique disease with its own specific disease mechanisms.” Dr. Thomson explained that “knowing more about the genetic risk factors of this disease might give a greater understanding of the disease processes involved in this condition and ultimately lead to novel therapies for this severely disabling disease.”
Funding for the first study was provided by the Wellcome Trust, the National Institutes of Health, the Doris Duke Charitable Foundation, the Medical Research Council, the Canadian Institutes of Health Research, the Liaison Committee between the Central Norway Regional Health Authority and the Norwegian University of Science and Technology, the Juvenile Diabetes Research Foundation International, and the Texas Scottish Rite Hospital for Children. Funding for the second study was provided by intramural research programs of the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Human Genome Research Institute, individual NIH grants, and grants from charitable foundations, advocacy organizations, and the governments of individual researchers’ countries. Dr. Thomson did not report any relevant financial disclosures; however, a number of coauthors reported potential conflicts of interest.
These two excellent multi-institutional genetic studies demonstrate once and for all the error promulgated by the International League of Associations for Rheumatology (ILAR) criteria formulated in the 1990s. Despite contrary voices, the ILAR committee grouped all childhood arthritis that was not due to sepsis or trauma as “juvenile idiopathic arthritis.” And despite the lack of clarity of the subtypes and the many cases that are “unclassifiable,” pediatric rheumatologists have continued to use these criteria in both drug trials and scientific studies.

Thomas Lehman, MD, is chief of the division of pediatric rheumatology at the Hospital for Special Surgery in New York. He has no relevant financial disclosures.
These two excellent multi-institutional genetic studies demonstrate once and for all the error promulgated by the International League of Associations for Rheumatology (ILAR) criteria formulated in the 1990s. Despite contrary voices, the ILAR committee grouped all childhood arthritis that was not due to sepsis or trauma as “juvenile idiopathic arthritis.” And despite the lack of clarity of the subtypes and the many cases that are “unclassifiable,” pediatric rheumatologists have continued to use these criteria in both drug trials and scientific studies.
Thomas Lehman, MD, is chief of the division of pediatric rheumatology at the Hospital for Special Surgery in New York. He has no relevant financial disclosures.
These two excellent multi-institutional genetic studies demonstrate once and for all the error promulgated by the International League of Associations for Rheumatology (ILAR) criteria formulated in the 1990s. Despite contrary voices, the ILAR committee grouped all childhood arthritis that was not due to sepsis or trauma as “juvenile idiopathic arthritis.” And despite the lack of clarity of the subtypes and the many cases that are “unclassifiable,” pediatric rheumatologists have continued to use these criteria in both drug trials and scientific studies.
Thomas Lehman, MD, is chief of the division of pediatric rheumatology at the Hospital for Special Surgery in New York. He has no relevant financial disclosures.
Two new studies of the genetic relationships between the seven designated categories of juvenile idiopathic arthritis (JIA) provide compelling support for reclassification of the categories, particularly for systemic JIA, and give evidence that some of the categories have clear equivalents in the realm of adult-onset diseases.
The International League of Associations for Rheumatology (ILAR) classification system (J Rheumatol. 2004;31:390-2) that defined the seven juvenile idiopathic arthritis (JIA) categories – systemic arthritis, oligoarticular arthritis, rheumatoid factor (RF)–negative polyarticular arthritis, RF-positive polyarticular arthritis, psoriatic arthritis (PsA), enthesitis-related arthritis (ERA), and undifferentiated arthritis – is problematic because the long-term outcome and response to treatment varies not only between subtypes but also within the subtypes, suggesting that these subgroups do not yet represent uniform groups of patients,” Wendy Thomson, PhD, professor of genetic epidemiology at the University of Manchester (England) and a senior author on both studies, explained in an interview.
“Despite the differences between the subtypes, current treatments of this disease often involve using the same drugs across all subtypes of JIA,” said Dr. Thomson, who is also deputy director of the Arthritis Research UK Centre for Genetics and Genomics at the university. “Understanding the genetic basis of the subtypes of this disease could help understand the cause of this disease and identify more appropriate treatment options [because] the current classification is largely based on clinical data and it is proposed that the addition of genetic data could improve classification.”
Interrelationships between JIA categories and adult disease
The first of these studies found that particular alleles in the human leukocyte antigen (HLA) region that have been associated with the different JIA categories strongly correlate some of the categories with one another, and that each JIA category potentially has an adult-onset counterpart based on shared HLA associations. It is the largest investigation of association of the HLA region with JIA and its categories to date, according to the researchers (Ann Rheum Dis. 2016 Dec 20. doi: 10.1136/annrheumdis-2016-210025).
In particular, the investigators demonstrated for the first time that RF-negative polyarticular JIA and oligoarticular JIA are genetically similar in their HLA associations. They also reported that RF-positive polyarthritis shares an association with adult seropositive rheumatoid arthritis and that combined data for oligoarthritis and RF-negative polyarthritis share the same association with adult seronegative RA. In addition, the researchers generated support for genetic associations between the particular JIA categories that are most commonly thought to have adult counterparts, such as ERA and adult ankylosing spondylitis (AS) as well as juvenile PsA and adult PsA.
Dr. Thomson and her coinvestigators used ImmunoChip genotype array data for 191,494 single nucleotide polymorphism (SNP) markers from the HLA region of 5,737 JIA patients and 16,403 controls in the United States, United Kingdom, Canada, Norway, and Germany to impute classical HLA alleles as well as specific amino acid positions within HLA alleles that may play an important functional role. After quality-control measures for the data were put in place, comparisons between 5,043 JIA cases and 14,390 controls showed that oligoarthritis and RF-negative polyarthritis were the most common and homogeneous JIA categories investigated (2,409 and 1,525 patients, respectively), and that they share a significant association across the HLA spectrum, meaning that they are genetically similar. When combined, the data from these two JIA categories correspond with seronegative rheumatoid arthritis in adults, while RF-positive polyarthritis on its own has an association with seropositive rheumatoid arthritis involving histidine at position 13 of the HLA-DRB1 allele. As expected, the most significant association between the ERA category and AS was for HLA-B*27. For juvenile PsA, no associations reached genome-wide level of significance, although HLA-C*0602 was modestly associated with juvenile PsA, and it is known to be associated with adult-onset PsA and is the primary HLA association in psoriasis.
“The results of this study have important implications for understanding disease pathogenesis, etiology, and potential future therapeutic strategies for JIA categories,” Dr. Thomson and her coauthors wrote, adding that “heterogeneity of JIA remains a key challenge to pediatric rheumatologists; however, these results may inform the debate on classification and help define a more biological-driven and molecular-driven classification system.”
Uniqueness of systemic JIA
In the second of the two studies, investigators from many different childhood arthritis study groups focused on systemic JIA (sJIA), also known in the past as Still’s disease. According to the authors, it is the first large-scale genomic study of sJIA ever published (Ann Rheum Dis. 2016 Dec 7. doi: 10.1136/annrheumdis-2016-210324).
“[sJIA] is characterized by prominent systemic inflammation and has a rare adult-onset counterpart; and undifferentiated arthritis includes arthritis that does not fit into any single category,” the authors wrote, adding that the “unique clinical characteristics of sJIA suggest that it is distinct from other forms of JIA, leading to the contention by some that sJIA should be separated from other forms of JIA and labeled as an autoinflammatory disease.”
The investigators carried out SNP genotyping on 982 children with sJIA in the United States, United Kingdom, Germany, Turkey, Italy, Brazil, Argentina, Canada, and Spain, along with 431 healthy children without sJIA who came from the same countries. A total of 7,579 control patients also underwent genotyping. Ultimately, 770 sJIA and 6,947 control subjects completed the study. sJIA was then compared with other JIA subtypes by using weighted genetic risk scores.
Comparison of the data sets for sJIA with those for the combined categories of RF-negative polyarticular JIA and oligoarticular JIA, as well as for the individual category of RF-positive polyarticular JIA, showed that sJIA has a distinct genetic architecture that separates it from those JIA categories, providing further evidence that sJIA should be classified separately from other forms of JIA and, potentially, treated differently as well.
According to Dr. Thomson, the finding “provides important evidence that sJIA should be considered a unique disease with its own specific disease mechanisms.” Dr. Thomson explained that “knowing more about the genetic risk factors of this disease might give a greater understanding of the disease processes involved in this condition and ultimately lead to novel therapies for this severely disabling disease.”
Funding for the first study was provided by the Wellcome Trust, the National Institutes of Health, the Doris Duke Charitable Foundation, the Medical Research Council, the Canadian Institutes of Health Research, the Liaison Committee between the Central Norway Regional Health Authority and the Norwegian University of Science and Technology, the Juvenile Diabetes Research Foundation International, and the Texas Scottish Rite Hospital for Children. Funding for the second study was provided by intramural research programs of the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Human Genome Research Institute, individual NIH grants, and grants from charitable foundations, advocacy organizations, and the governments of individual researchers’ countries. Dr. Thomson did not report any relevant financial disclosures; however, a number of coauthors reported potential conflicts of interest.
Two new studies of the genetic relationships between the seven designated categories of juvenile idiopathic arthritis (JIA) provide compelling support for reclassification of the categories, particularly for systemic JIA, and give evidence that some of the categories have clear equivalents in the realm of adult-onset diseases.
The International League of Associations for Rheumatology (ILAR) classification system (J Rheumatol. 2004;31:390-2) that defined the seven juvenile idiopathic arthritis (JIA) categories – systemic arthritis, oligoarticular arthritis, rheumatoid factor (RF)–negative polyarticular arthritis, RF-positive polyarticular arthritis, psoriatic arthritis (PsA), enthesitis-related arthritis (ERA), and undifferentiated arthritis – is problematic because the long-term outcome and response to treatment varies not only between subtypes but also within the subtypes, suggesting that these subgroups do not yet represent uniform groups of patients,” Wendy Thomson, PhD, professor of genetic epidemiology at the University of Manchester (England) and a senior author on both studies, explained in an interview.
“Despite the differences between the subtypes, current treatments of this disease often involve using the same drugs across all subtypes of JIA,” said Dr. Thomson, who is also deputy director of the Arthritis Research UK Centre for Genetics and Genomics at the university. “Understanding the genetic basis of the subtypes of this disease could help understand the cause of this disease and identify more appropriate treatment options [because] the current classification is largely based on clinical data and it is proposed that the addition of genetic data could improve classification.”
Interrelationships between JIA categories and adult disease
The first of these studies found that particular alleles in the human leukocyte antigen (HLA) region that have been associated with the different JIA categories strongly correlate some of the categories with one another, and that each JIA category potentially has an adult-onset counterpart based on shared HLA associations. It is the largest investigation of association of the HLA region with JIA and its categories to date, according to the researchers (Ann Rheum Dis. 2016 Dec 20. doi: 10.1136/annrheumdis-2016-210025).
In particular, the investigators demonstrated for the first time that RF-negative polyarticular JIA and oligoarticular JIA are genetically similar in their HLA associations. They also reported that RF-positive polyarthritis shares an association with adult seropositive rheumatoid arthritis and that combined data for oligoarthritis and RF-negative polyarthritis share the same association with adult seronegative RA. In addition, the researchers generated support for genetic associations between the particular JIA categories that are most commonly thought to have adult counterparts, such as ERA and adult ankylosing spondylitis (AS) as well as juvenile PsA and adult PsA.
Dr. Thomson and her coinvestigators used ImmunoChip genotype array data for 191,494 single nucleotide polymorphism (SNP) markers from the HLA region of 5,737 JIA patients and 16,403 controls in the United States, United Kingdom, Canada, Norway, and Germany to impute classical HLA alleles as well as specific amino acid positions within HLA alleles that may play an important functional role. After quality-control measures for the data were put in place, comparisons between 5,043 JIA cases and 14,390 controls showed that oligoarthritis and RF-negative polyarthritis were the most common and homogeneous JIA categories investigated (2,409 and 1,525 patients, respectively), and that they share a significant association across the HLA spectrum, meaning that they are genetically similar. When combined, the data from these two JIA categories correspond with seronegative rheumatoid arthritis in adults, while RF-positive polyarthritis on its own has an association with seropositive rheumatoid arthritis involving histidine at position 13 of the HLA-DRB1 allele. As expected, the most significant association between the ERA category and AS was for HLA-B*27. For juvenile PsA, no associations reached genome-wide level of significance, although HLA-C*0602 was modestly associated with juvenile PsA, and it is known to be associated with adult-onset PsA and is the primary HLA association in psoriasis.
“The results of this study have important implications for understanding disease pathogenesis, etiology, and potential future therapeutic strategies for JIA categories,” Dr. Thomson and her coauthors wrote, adding that “heterogeneity of JIA remains a key challenge to pediatric rheumatologists; however, these results may inform the debate on classification and help define a more biological-driven and molecular-driven classification system.”
Uniqueness of systemic JIA
In the second of the two studies, investigators from many different childhood arthritis study groups focused on systemic JIA (sJIA), also known in the past as Still’s disease. According to the authors, it is the first large-scale genomic study of sJIA ever published (Ann Rheum Dis. 2016 Dec 7. doi: 10.1136/annrheumdis-2016-210324).
“[sJIA] is characterized by prominent systemic inflammation and has a rare adult-onset counterpart; and undifferentiated arthritis includes arthritis that does not fit into any single category,” the authors wrote, adding that the “unique clinical characteristics of sJIA suggest that it is distinct from other forms of JIA, leading to the contention by some that sJIA should be separated from other forms of JIA and labeled as an autoinflammatory disease.”
The investigators carried out SNP genotyping on 982 children with sJIA in the United States, United Kingdom, Germany, Turkey, Italy, Brazil, Argentina, Canada, and Spain, along with 431 healthy children without sJIA who came from the same countries. A total of 7,579 control patients also underwent genotyping. Ultimately, 770 sJIA and 6,947 control subjects completed the study. sJIA was then compared with other JIA subtypes by using weighted genetic risk scores.
Comparison of the data sets for sJIA with those for the combined categories of RF-negative polyarticular JIA and oligoarticular JIA, as well as for the individual category of RF-positive polyarticular JIA, showed that sJIA has a distinct genetic architecture that separates it from those JIA categories, providing further evidence that sJIA should be classified separately from other forms of JIA and, potentially, treated differently as well.
According to Dr. Thomson, the finding “provides important evidence that sJIA should be considered a unique disease with its own specific disease mechanisms.” Dr. Thomson explained that “knowing more about the genetic risk factors of this disease might give a greater understanding of the disease processes involved in this condition and ultimately lead to novel therapies for this severely disabling disease.”
Funding for the first study was provided by the Wellcome Trust, the National Institutes of Health, the Doris Duke Charitable Foundation, the Medical Research Council, the Canadian Institutes of Health Research, the Liaison Committee between the Central Norway Regional Health Authority and the Norwegian University of Science and Technology, the Juvenile Diabetes Research Foundation International, and the Texas Scottish Rite Hospital for Children. Funding for the second study was provided by intramural research programs of the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Human Genome Research Institute, individual NIH grants, and grants from charitable foundations, advocacy organizations, and the governments of individual researchers’ countries. Dr. Thomson did not report any relevant financial disclosures; however, a number of coauthors reported potential conflicts of interest.
FROM ANNALS OF THE RHEUMATIC DISEASES
Rituximab vanquished MRD in mantle cell lymphoma
SAN DIEGO – Rituximab can at least temporarily vanquish minimal residual disease (MRD) in mantle cell lymphoma (MCL) patients who relapse after induction therapy and autologous stem cell transplantation (ASCT), researchers reported at the annual meeting of the American Society of Hematology.
Of 58 patients whose MCL relapsed after induction therapy and ASCT, 82% converted back to an MRD-negative state after receiving 4 weekly doses of rituximab (375 mg/m2), Arne Kolstad, MD, PhD, and his associates. The data “strongly suggest that preemptive rituximab treatment delayed clinical relapse in MCL,” they wrote in their abstract. They recommended molecular and clinical monitoring after ASCT, not only “as an alternative to maintenance therapy for all MCL patients” but to identify MRD-positive candidates for clinical trials.
The study was an analysis of the Nordic Lymphoma Group phase II MCL2 and MCL3 trials (NTC 00514475), in which patients received six alternating cycles of R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab) and R-Ara-C (rituximab-cytarabine). followed by high-dose ASCT. In MCL3, responders who fell short of complete remission also received intensification with yttium-90 ibritumomab tiuxetan (0.4 mCi/kg) 1 week before treatment with BEAM/C (carmustine, etoposide, cytarabine, and melphalan or cyclophosphamide). Patients were evaluated 2-3 months after completing ASCT, and then every 6 months for 5 years or until relapse. Survivors were followed for a median of 8.5 years, noted Dr. Kolstad, who is with Oslo University Hospital in Norway.
Among 183 patients who underwent polymerase chain reaction–based testing for markers of MRD, median time to molecular relapse was 55 months. However, the relapse-free survival curve did not plateau – patients in all risk groups continued to relapse 5-10 years after undergoing ASCT, the researchers said. “Hence, it is fair to consider MCL as a chronic incurable lymphoma entity, and novel approaches will be necessary to change the natural course of this disease,” they wrote.
After controlling for potential confounders, significant predictors of molecular relapse included high MCL international prognostic index at diagnosis (hazard ratio, 1.9; 95% confidence interval 1.4-2.7; P = .0001) and detection of MRD before patients underwent ASCT (HR, 2.5; 95% CI, 1.5-4.1; P = .0005). Minimal residual disease predicted clinical relapse and shorter survival (P less than .001 for both associations). In contrast, the 86 patients who remained in continuous molecular remission had a 76% chance of having at least a 10-year clinical remission, the investigators said.
Minimal residual disease was assessed by testing bone marrow and blood samples with combined standard nested and quantitative real-time polymerase chain reaction (PCR) for Bcl-1 or IgH rearrangement. They defined molecular relapse as conversion from a negative to a positive result on standard nested PCR, or, for patients who were MRD positive after ASCT, as a more than fivefold rise in real-time quantitative PCR levels in two consecutive bone marrow samples.
Oslo University sponsored the trials. Dr. Kolstad reported ties to Nordic Nanovector, Bayer Schering Pharma, Merck, and Roche.
SAN DIEGO – Rituximab can at least temporarily vanquish minimal residual disease (MRD) in mantle cell lymphoma (MCL) patients who relapse after induction therapy and autologous stem cell transplantation (ASCT), researchers reported at the annual meeting of the American Society of Hematology.
Of 58 patients whose MCL relapsed after induction therapy and ASCT, 82% converted back to an MRD-negative state after receiving 4 weekly doses of rituximab (375 mg/m2), Arne Kolstad, MD, PhD, and his associates. The data “strongly suggest that preemptive rituximab treatment delayed clinical relapse in MCL,” they wrote in their abstract. They recommended molecular and clinical monitoring after ASCT, not only “as an alternative to maintenance therapy for all MCL patients” but to identify MRD-positive candidates for clinical trials.
The study was an analysis of the Nordic Lymphoma Group phase II MCL2 and MCL3 trials (NTC 00514475), in which patients received six alternating cycles of R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab) and R-Ara-C (rituximab-cytarabine). followed by high-dose ASCT. In MCL3, responders who fell short of complete remission also received intensification with yttium-90 ibritumomab tiuxetan (0.4 mCi/kg) 1 week before treatment with BEAM/C (carmustine, etoposide, cytarabine, and melphalan or cyclophosphamide). Patients were evaluated 2-3 months after completing ASCT, and then every 6 months for 5 years or until relapse. Survivors were followed for a median of 8.5 years, noted Dr. Kolstad, who is with Oslo University Hospital in Norway.
Among 183 patients who underwent polymerase chain reaction–based testing for markers of MRD, median time to molecular relapse was 55 months. However, the relapse-free survival curve did not plateau – patients in all risk groups continued to relapse 5-10 years after undergoing ASCT, the researchers said. “Hence, it is fair to consider MCL as a chronic incurable lymphoma entity, and novel approaches will be necessary to change the natural course of this disease,” they wrote.
After controlling for potential confounders, significant predictors of molecular relapse included high MCL international prognostic index at diagnosis (hazard ratio, 1.9; 95% confidence interval 1.4-2.7; P = .0001) and detection of MRD before patients underwent ASCT (HR, 2.5; 95% CI, 1.5-4.1; P = .0005). Minimal residual disease predicted clinical relapse and shorter survival (P less than .001 for both associations). In contrast, the 86 patients who remained in continuous molecular remission had a 76% chance of having at least a 10-year clinical remission, the investigators said.
Minimal residual disease was assessed by testing bone marrow and blood samples with combined standard nested and quantitative real-time polymerase chain reaction (PCR) for Bcl-1 or IgH rearrangement. They defined molecular relapse as conversion from a negative to a positive result on standard nested PCR, or, for patients who were MRD positive after ASCT, as a more than fivefold rise in real-time quantitative PCR levels in two consecutive bone marrow samples.
Oslo University sponsored the trials. Dr. Kolstad reported ties to Nordic Nanovector, Bayer Schering Pharma, Merck, and Roche.
SAN DIEGO – Rituximab can at least temporarily vanquish minimal residual disease (MRD) in mantle cell lymphoma (MCL) patients who relapse after induction therapy and autologous stem cell transplantation (ASCT), researchers reported at the annual meeting of the American Society of Hematology.
Of 58 patients whose MCL relapsed after induction therapy and ASCT, 82% converted back to an MRD-negative state after receiving 4 weekly doses of rituximab (375 mg/m2), Arne Kolstad, MD, PhD, and his associates. The data “strongly suggest that preemptive rituximab treatment delayed clinical relapse in MCL,” they wrote in their abstract. They recommended molecular and clinical monitoring after ASCT, not only “as an alternative to maintenance therapy for all MCL patients” but to identify MRD-positive candidates for clinical trials.
The study was an analysis of the Nordic Lymphoma Group phase II MCL2 and MCL3 trials (NTC 00514475), in which patients received six alternating cycles of R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab) and R-Ara-C (rituximab-cytarabine). followed by high-dose ASCT. In MCL3, responders who fell short of complete remission also received intensification with yttium-90 ibritumomab tiuxetan (0.4 mCi/kg) 1 week before treatment with BEAM/C (carmustine, etoposide, cytarabine, and melphalan or cyclophosphamide). Patients were evaluated 2-3 months after completing ASCT, and then every 6 months for 5 years or until relapse. Survivors were followed for a median of 8.5 years, noted Dr. Kolstad, who is with Oslo University Hospital in Norway.
Among 183 patients who underwent polymerase chain reaction–based testing for markers of MRD, median time to molecular relapse was 55 months. However, the relapse-free survival curve did not plateau – patients in all risk groups continued to relapse 5-10 years after undergoing ASCT, the researchers said. “Hence, it is fair to consider MCL as a chronic incurable lymphoma entity, and novel approaches will be necessary to change the natural course of this disease,” they wrote.
After controlling for potential confounders, significant predictors of molecular relapse included high MCL international prognostic index at diagnosis (hazard ratio, 1.9; 95% confidence interval 1.4-2.7; P = .0001) and detection of MRD before patients underwent ASCT (HR, 2.5; 95% CI, 1.5-4.1; P = .0005). Minimal residual disease predicted clinical relapse and shorter survival (P less than .001 for both associations). In contrast, the 86 patients who remained in continuous molecular remission had a 76% chance of having at least a 10-year clinical remission, the investigators said.
Minimal residual disease was assessed by testing bone marrow and blood samples with combined standard nested and quantitative real-time polymerase chain reaction (PCR) for Bcl-1 or IgH rearrangement. They defined molecular relapse as conversion from a negative to a positive result on standard nested PCR, or, for patients who were MRD positive after ASCT, as a more than fivefold rise in real-time quantitative PCR levels in two consecutive bone marrow samples.
Oslo University sponsored the trials. Dr. Kolstad reported ties to Nordic Nanovector, Bayer Schering Pharma, Merck, and Roche.
AT ASH 2016
Key clinical point:
Major finding: Among 58 patients who relapsed after induction therapy and autologous stem cell transplantation, 82% converted back to an MRD-negative state with 4 weekly doses of rituximab (375 mg/m2).
Data source: A study of 183 patients with mantle cell lymphoma from the Nordic MCL2 and MCL3 trials.
Disclosures: Oslo University sponsored the trials. Dr. Kolstad reported ties to Nordic Nanovector, Bayer Schering Pharma, Merck, and Roche.
LMWH trumps unfractionated heparin in reducing posttrauma thrombotic events
HOLLYWOOD, FLA. – Low-molecular-weight heparin (LMWH) decreased the risk of venous thromboembolism in trauma patients significantly more than did unfractionated heparin, a large state database review has found.
It also was associated with a 37% decrease in overall mortality, compared with unfractionated heparin, Benjamin Jacobs, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.

He extracted data describing thromboembolism prophylaxis among 37,868 trauma patients included in the Michigan Trauma Quality Improvement Program from 2012 to 2014. The patients were treated at 23 hospitals around the state. They received either unfractionated or LMWH as their only clot-preventing protocol.
The primary outcomes of the study were reductions in the risk of venous thromboembolism (VTE), deep vein thrombosis (DVT), pulmonary thrombosis (PT), and mortality.
LMWH was given at either 40 mg every day or 30 mg twice a day. The comparator was unfractionated heparin at 5,000 U either two or three times a day.
The preferred method was LMWH, which 83% of patients received, compared with 17% who got the unfractionated heparin. Most patients who got LMWH received the 40 mg/day dose (70%). Most who got unfractionated heparin received 5,000 U three times a day (87%).
Both types of heparin reduced the risk of all thromboembolic outcomes, and both doses of LMWH significantly reduced the risks. However, the 40 mg/day dose was significantly more effective than the twice-daily 30-mg dose in reducing the risk of VTE and DVT. Risk reductions for PT and mortality were not significantly different between the doses.
Overall, compared with unfractionated heparin, LMWH decreased the risk of VTE by 33%; of PT by 48%; and of DVT by 27%. It also conferred a significant mortality benefit, reducing the risk of death by 37%, compared with the unfractionated type
When Dr. Jacobs grouped the patients according to Injury Severity Score (ISS), he saw a consistently higher benefit among patients with lower scores. For example, LMWH significantly reduced the risk of PT by 59% in patients with an ISS of 5-14. In those with an ISS of 25 or higher, the drug was associated with a 20% increased risk, although that wasn’t statistically significant.
There was a similar finding in DVT. LMWH reduced the risk by 18% in those with an ISS of 5-15, and by 50% among those with an score of 16-24 – both significant reductions. Among those with an ISS of at least 25, the risk was 18% higher, although, again, it was not a significant finding.
Curiously, the mortality benefit was stronger among sicker patients. The benefit was nonsignificant among those with an ISS of less than 25 but for those above 25, the mortality risk reduction was a significant 45%.
Dr Jacobs had no financial disclosures.
[email protected]
On Twitter @alz_gal
HOLLYWOOD, FLA. – Low-molecular-weight heparin (LMWH) decreased the risk of venous thromboembolism in trauma patients significantly more than did unfractionated heparin, a large state database review has found.
It also was associated with a 37% decrease in overall mortality, compared with unfractionated heparin, Benjamin Jacobs, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
He extracted data describing thromboembolism prophylaxis among 37,868 trauma patients included in the Michigan Trauma Quality Improvement Program from 2012 to 2014. The patients were treated at 23 hospitals around the state. They received either unfractionated or LMWH as their only clot-preventing protocol.
The primary outcomes of the study were reductions in the risk of venous thromboembolism (VTE), deep vein thrombosis (DVT), pulmonary thrombosis (PT), and mortality.
LMWH was given at either 40 mg every day or 30 mg twice a day. The comparator was unfractionated heparin at 5,000 U either two or three times a day.
The preferred method was LMWH, which 83% of patients received, compared with 17% who got the unfractionated heparin. Most patients who got LMWH received the 40 mg/day dose (70%). Most who got unfractionated heparin received 5,000 U three times a day (87%).
Both types of heparin reduced the risk of all thromboembolic outcomes, and both doses of LMWH significantly reduced the risks. However, the 40 mg/day dose was significantly more effective than the twice-daily 30-mg dose in reducing the risk of VTE and DVT. Risk reductions for PT and mortality were not significantly different between the doses.
Overall, compared with unfractionated heparin, LMWH decreased the risk of VTE by 33%; of PT by 48%; and of DVT by 27%. It also conferred a significant mortality benefit, reducing the risk of death by 37%, compared with the unfractionated type
When Dr. Jacobs grouped the patients according to Injury Severity Score (ISS), he saw a consistently higher benefit among patients with lower scores. For example, LMWH significantly reduced the risk of PT by 59% in patients with an ISS of 5-14. In those with an ISS of 25 or higher, the drug was associated with a 20% increased risk, although that wasn’t statistically significant.
There was a similar finding in DVT. LMWH reduced the risk by 18% in those with an ISS of 5-15, and by 50% among those with an score of 16-24 – both significant reductions. Among those with an ISS of at least 25, the risk was 18% higher, although, again, it was not a significant finding.
Curiously, the mortality benefit was stronger among sicker patients. The benefit was nonsignificant among those with an ISS of less than 25 but for those above 25, the mortality risk reduction was a significant 45%.
Dr Jacobs had no financial disclosures.
[email protected]
On Twitter @alz_gal
HOLLYWOOD, FLA. – Low-molecular-weight heparin (LMWH) decreased the risk of venous thromboembolism in trauma patients significantly more than did unfractionated heparin, a large state database review has found.
It also was associated with a 37% decrease in overall mortality, compared with unfractionated heparin, Benjamin Jacobs, MD, said at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
He extracted data describing thromboembolism prophylaxis among 37,868 trauma patients included in the Michigan Trauma Quality Improvement Program from 2012 to 2014. The patients were treated at 23 hospitals around the state. They received either unfractionated or LMWH as their only clot-preventing protocol.
The primary outcomes of the study were reductions in the risk of venous thromboembolism (VTE), deep vein thrombosis (DVT), pulmonary thrombosis (PT), and mortality.
LMWH was given at either 40 mg every day or 30 mg twice a day. The comparator was unfractionated heparin at 5,000 U either two or three times a day.
The preferred method was LMWH, which 83% of patients received, compared with 17% who got the unfractionated heparin. Most patients who got LMWH received the 40 mg/day dose (70%). Most who got unfractionated heparin received 5,000 U three times a day (87%).
Both types of heparin reduced the risk of all thromboembolic outcomes, and both doses of LMWH significantly reduced the risks. However, the 40 mg/day dose was significantly more effective than the twice-daily 30-mg dose in reducing the risk of VTE and DVT. Risk reductions for PT and mortality were not significantly different between the doses.
Overall, compared with unfractionated heparin, LMWH decreased the risk of VTE by 33%; of PT by 48%; and of DVT by 27%. It also conferred a significant mortality benefit, reducing the risk of death by 37%, compared with the unfractionated type
When Dr. Jacobs grouped the patients according to Injury Severity Score (ISS), he saw a consistently higher benefit among patients with lower scores. For example, LMWH significantly reduced the risk of PT by 59% in patients with an ISS of 5-14. In those with an ISS of 25 or higher, the drug was associated with a 20% increased risk, although that wasn’t statistically significant.
There was a similar finding in DVT. LMWH reduced the risk by 18% in those with an ISS of 5-15, and by 50% among those with an score of 16-24 – both significant reductions. Among those with an ISS of at least 25, the risk was 18% higher, although, again, it was not a significant finding.
Curiously, the mortality benefit was stronger among sicker patients. The benefit was nonsignificant among those with an ISS of less than 25 but for those above 25, the mortality risk reduction was a significant 45%.
Dr Jacobs had no financial disclosures.
[email protected]
On Twitter @alz_gal
AT THE EAST ANNUAL SCIENTIFIC ASSEMBLY
Key clinical point:
Major finding: Overall mortality was reduced by 37% with LMWH, compared with unfractionated heparin.
Data source: The review comprised 37,868 patients included in the Michigan Trauma Quality Improvement Program.
Disclosures: Dr. Jacobs had no financial disclosures.
NAS linked to poor and deteriorating school performance
A neonatal diagnostic code of neonatal abstinence syndrome (NAS) is strongly associated with poor and deteriorating school performance, according to Ju Lee Oei, MD, of the University of New South Wales, Sydney, and her associates.
In a study of 604,829 children born in 2000-2006 in New South Wales, linkage rates were similar between matched controls (77.6% of 4,330) and other NSW children (77.4% of 598,265; P = .83) but were significantly lower in children with NAS (75.6% of 2,234; P = .03). The controls were matched for gender,gestation, and socioeconomic status.
The children with NAS had significantly lower scores than the controls and other NSW children in every grade and every domain of testing (reading, writing, numeracy, spelling, and grammar/punctuation). By grade 7, 38% of children with NAS did not meet National Minimum Standard (NMS) in one or more domains (versus 18.4% of controls and 14.5% of other NSW children). The mean serial composite scores also were lower in children with NAS from grades 3 to 7, compared with the other two groups; the difference was progressive, the investigators said. And by grade 7, the scores for children with NAS were lower than other children’s scores in grade 5.
It was noted that children with NAS, indigenous status (adjusted odds ratio, 1.7), male gender (aOR, 1.3), and having a primary parent without a grade 9 education (aOR, 1.3) increased the risk of failure to meet NMS. Overall, NAS (aOR, 2.5), indigenous status (aOR, 2.2), male gender (aOR, 1.3), and prematurity (less than 37 weeks’ gestation [aOR, 1.2]) increased the risk of failure to meet NMS.
“To date these are the only data demonstrating long-term school outcomes for children with a history of NAS. Similar data for children born from the current opioid epidemic gripping much of the Northern Hemisphere, assuming linkage is possible, will be available only in 7-10 years,” researchers concluded. “Although this study was conducted in Australia, the high risk of poor academic performance in this vulnerable group of children is applicable to all countries, and strategies to address this risk and prevent poor adult outcomes and intergenerational vulnerability must be urgently addressed.”
Read the full study in Pediatrics (doi: 10.1542/peds.2016-2651).
A neonatal diagnostic code of neonatal abstinence syndrome (NAS) is strongly associated with poor and deteriorating school performance, according to Ju Lee Oei, MD, of the University of New South Wales, Sydney, and her associates.
In a study of 604,829 children born in 2000-2006 in New South Wales, linkage rates were similar between matched controls (77.6% of 4,330) and other NSW children (77.4% of 598,265; P = .83) but were significantly lower in children with NAS (75.6% of 2,234; P = .03). The controls were matched for gender,gestation, and socioeconomic status.
The children with NAS had significantly lower scores than the controls and other NSW children in every grade and every domain of testing (reading, writing, numeracy, spelling, and grammar/punctuation). By grade 7, 38% of children with NAS did not meet National Minimum Standard (NMS) in one or more domains (versus 18.4% of controls and 14.5% of other NSW children). The mean serial composite scores also were lower in children with NAS from grades 3 to 7, compared with the other two groups; the difference was progressive, the investigators said. And by grade 7, the scores for children with NAS were lower than other children’s scores in grade 5.
It was noted that children with NAS, indigenous status (adjusted odds ratio, 1.7), male gender (aOR, 1.3), and having a primary parent without a grade 9 education (aOR, 1.3) increased the risk of failure to meet NMS. Overall, NAS (aOR, 2.5), indigenous status (aOR, 2.2), male gender (aOR, 1.3), and prematurity (less than 37 weeks’ gestation [aOR, 1.2]) increased the risk of failure to meet NMS.
“To date these are the only data demonstrating long-term school outcomes for children with a history of NAS. Similar data for children born from the current opioid epidemic gripping much of the Northern Hemisphere, assuming linkage is possible, will be available only in 7-10 years,” researchers concluded. “Although this study was conducted in Australia, the high risk of poor academic performance in this vulnerable group of children is applicable to all countries, and strategies to address this risk and prevent poor adult outcomes and intergenerational vulnerability must be urgently addressed.”
Read the full study in Pediatrics (doi: 10.1542/peds.2016-2651).
A neonatal diagnostic code of neonatal abstinence syndrome (NAS) is strongly associated with poor and deteriorating school performance, according to Ju Lee Oei, MD, of the University of New South Wales, Sydney, and her associates.
In a study of 604,829 children born in 2000-2006 in New South Wales, linkage rates were similar between matched controls (77.6% of 4,330) and other NSW children (77.4% of 598,265; P = .83) but were significantly lower in children with NAS (75.6% of 2,234; P = .03). The controls were matched for gender,gestation, and socioeconomic status.
The children with NAS had significantly lower scores than the controls and other NSW children in every grade and every domain of testing (reading, writing, numeracy, spelling, and grammar/punctuation). By grade 7, 38% of children with NAS did not meet National Minimum Standard (NMS) in one or more domains (versus 18.4% of controls and 14.5% of other NSW children). The mean serial composite scores also were lower in children with NAS from grades 3 to 7, compared with the other two groups; the difference was progressive, the investigators said. And by grade 7, the scores for children with NAS were lower than other children’s scores in grade 5.
It was noted that children with NAS, indigenous status (adjusted odds ratio, 1.7), male gender (aOR, 1.3), and having a primary parent without a grade 9 education (aOR, 1.3) increased the risk of failure to meet NMS. Overall, NAS (aOR, 2.5), indigenous status (aOR, 2.2), male gender (aOR, 1.3), and prematurity (less than 37 weeks’ gestation [aOR, 1.2]) increased the risk of failure to meet NMS.
“To date these are the only data demonstrating long-term school outcomes for children with a history of NAS. Similar data for children born from the current opioid epidemic gripping much of the Northern Hemisphere, assuming linkage is possible, will be available only in 7-10 years,” researchers concluded. “Although this study was conducted in Australia, the high risk of poor academic performance in this vulnerable group of children is applicable to all countries, and strategies to address this risk and prevent poor adult outcomes and intergenerational vulnerability must be urgently addressed.”
Read the full study in Pediatrics (doi: 10.1542/peds.2016-2651).
FROM PEDIATRICS
Bank could help docs identify optimal AML treatment, team says

Photo courtesy of
University Hospital Ulm
Research published in Nature Genetics suggests a knowledge bank
can reveal the optimal treatment for patients with acute
myeloid leukemia (AML), although more research is needed before such
banks can be used in the clinic.
Researchers built a knowledge
bank using data from 1540 AML patients enrolled in clinical
trials in Germany and Austria.
The bank includes information on genetic features, treatment, and outcomes for each patient.
The researchers used this information to develop models that could predict a patient’s likelihood of remission, relapse, and mortality.
The team then validated those results using data from patients in The Cancer Genome Atlas.
The researchers estimate that up to 1 in 3 AML patients would be prescribed a different treatment regimen if physicians used the knowledge bank approach rather than current practice.
“The knowledge bank approach makes far more detailed and accurate predictions about the likely future course of a patient with AML than what we can make in the clinic at the moment,” said study author Peter Campbell, PhD, of the Wellcome Trust Sanger Institute in Hinxton, UK.
“Current guides use a simple set of rules based on only a few genetic findings. For any given patient, using the new tool, we can compare the likely future outcomes under a transplant route versus a standard chemotherapy route. This means that we can make a treatment choice that is personally tailored to the unique features of that particular patient.”
However, the researchers said the knowledge bank approach requires further testing before it can be used to prescribe treatment in AML clinics.
“Our analysis reveals that knowledge banks of up to 10,000 patients would be needed to obtain the precision needed for routine clinical application,” said study author Moritz Gerstung, PhD, of the European Bioinformatics Institute in Hinxton, UK.
“Building knowledge banks is not easy,” added author Hartmut Döhner, MD, of the University of Ulm in Germany. “To get accurate treatment predictions, you need data from thousands of patients and all tumor types.”
“Furthermore, such knowledge banks will need continuous updating as new therapies become approved and available. As genetic testing enters routine clinical practice, there is an opportunity to learn from patients undergoing care in our health systems. Our paper gives the first real evidence that the approach is worthwhile, how it could be used, and what the scale needs to be.” ![]()
Photo courtesy of
University Hospital Ulm
Research published in Nature Genetics suggests a knowledge bank
can reveal the optimal treatment for patients with acute
myeloid leukemia (AML), although more research is needed before such
banks can be used in the clinic.
Researchers built a knowledge
bank using data from 1540 AML patients enrolled in clinical
trials in Germany and Austria.
The bank includes information on genetic features, treatment, and outcomes for each patient.
The researchers used this information to develop models that could predict a patient’s likelihood of remission, relapse, and mortality.
The team then validated those results using data from patients in The Cancer Genome Atlas.
The researchers estimate that up to 1 in 3 AML patients would be prescribed a different treatment regimen if physicians used the knowledge bank approach rather than current practice.
“The knowledge bank approach makes far more detailed and accurate predictions about the likely future course of a patient with AML than what we can make in the clinic at the moment,” said study author Peter Campbell, PhD, of the Wellcome Trust Sanger Institute in Hinxton, UK.
“Current guides use a simple set of rules based on only a few genetic findings. For any given patient, using the new tool, we can compare the likely future outcomes under a transplant route versus a standard chemotherapy route. This means that we can make a treatment choice that is personally tailored to the unique features of that particular patient.”
However, the researchers said the knowledge bank approach requires further testing before it can be used to prescribe treatment in AML clinics.
“Our analysis reveals that knowledge banks of up to 10,000 patients would be needed to obtain the precision needed for routine clinical application,” said study author Moritz Gerstung, PhD, of the European Bioinformatics Institute in Hinxton, UK.
“Building knowledge banks is not easy,” added author Hartmut Döhner, MD, of the University of Ulm in Germany. “To get accurate treatment predictions, you need data from thousands of patients and all tumor types.”
“Furthermore, such knowledge banks will need continuous updating as new therapies become approved and available. As genetic testing enters routine clinical practice, there is an opportunity to learn from patients undergoing care in our health systems. Our paper gives the first real evidence that the approach is worthwhile, how it could be used, and what the scale needs to be.” ![]()
Photo courtesy of
University Hospital Ulm
Research published in Nature Genetics suggests a knowledge bank
can reveal the optimal treatment for patients with acute
myeloid leukemia (AML), although more research is needed before such
banks can be used in the clinic.
Researchers built a knowledge
bank using data from 1540 AML patients enrolled in clinical
trials in Germany and Austria.
The bank includes information on genetic features, treatment, and outcomes for each patient.
The researchers used this information to develop models that could predict a patient’s likelihood of remission, relapse, and mortality.
The team then validated those results using data from patients in The Cancer Genome Atlas.
The researchers estimate that up to 1 in 3 AML patients would be prescribed a different treatment regimen if physicians used the knowledge bank approach rather than current practice.
“The knowledge bank approach makes far more detailed and accurate predictions about the likely future course of a patient with AML than what we can make in the clinic at the moment,” said study author Peter Campbell, PhD, of the Wellcome Trust Sanger Institute in Hinxton, UK.
“Current guides use a simple set of rules based on only a few genetic findings. For any given patient, using the new tool, we can compare the likely future outcomes under a transplant route versus a standard chemotherapy route. This means that we can make a treatment choice that is personally tailored to the unique features of that particular patient.”
However, the researchers said the knowledge bank approach requires further testing before it can be used to prescribe treatment in AML clinics.
“Our analysis reveals that knowledge banks of up to 10,000 patients would be needed to obtain the precision needed for routine clinical application,” said study author Moritz Gerstung, PhD, of the European Bioinformatics Institute in Hinxton, UK.
“Building knowledge banks is not easy,” added author Hartmut Döhner, MD, of the University of Ulm in Germany. “To get accurate treatment predictions, you need data from thousands of patients and all tumor types.”
“Furthermore, such knowledge banks will need continuous updating as new therapies become approved and available. As genetic testing enters routine clinical practice, there is an opportunity to learn from patients undergoing care in our health systems. Our paper gives the first real evidence that the approach is worthwhile, how it could be used, and what the scale needs to be.” ![]()
Get to Know the Trump Administration VA Secretary Nominee
A little over a year ago David J. Shulkin, MD, was approved by Senate to take over the position of under secretary for health at the VA, after being nominated by President Obama in March 2015. Since his appointment, Shulkin has strongly voiced the importance of improving VA wait times, providing veterans with the utmost quality of care, and championing the VA health care system for its ability to provide services that the private sector cannot.
Here is a collection of articles published by Federal Practitioner featuring David Shulkin, MD, including exclusive interviews and an editorial written by the VA Secretary Nominee himself.
- Shulkin Nominated to Replace McDonald at VA
- Why VA Health Care Is Different
- Shulkin: VA "Not a Political Issue”
- Shulkin Addresses APRN Rule, Health Care Vacancies, and Access
- A New View for the VA
- Nearly 20,000 Comment on Controversial APRN Rule
- McDonald and Shulkin Lay Out Strategies for Eliminating VA Wait Times in 2016
- "Call to Action" on Veteran Suicide Yields Policy Shifts
- New VA Under Secretary of Health Focuses on Access, Employee Engagement
- Senate Confirms New VA Under Secretary for Health
A little over a year ago David J. Shulkin, MD, was approved by Senate to take over the position of under secretary for health at the VA, after being nominated by President Obama in March 2015. Since his appointment, Shulkin has strongly voiced the importance of improving VA wait times, providing veterans with the utmost quality of care, and championing the VA health care system for its ability to provide services that the private sector cannot.
Here is a collection of articles published by Federal Practitioner featuring David Shulkin, MD, including exclusive interviews and an editorial written by the VA Secretary Nominee himself.
- Shulkin Nominated to Replace McDonald at VA
- Why VA Health Care Is Different
- Shulkin: VA "Not a Political Issue”
- Shulkin Addresses APRN Rule, Health Care Vacancies, and Access
- A New View for the VA
- Nearly 20,000 Comment on Controversial APRN Rule
- McDonald and Shulkin Lay Out Strategies for Eliminating VA Wait Times in 2016
- "Call to Action" on Veteran Suicide Yields Policy Shifts
- New VA Under Secretary of Health Focuses on Access, Employee Engagement
- Senate Confirms New VA Under Secretary for Health
A little over a year ago David J. Shulkin, MD, was approved by Senate to take over the position of under secretary for health at the VA, after being nominated by President Obama in March 2015. Since his appointment, Shulkin has strongly voiced the importance of improving VA wait times, providing veterans with the utmost quality of care, and championing the VA health care system for its ability to provide services that the private sector cannot.
Here is a collection of articles published by Federal Practitioner featuring David Shulkin, MD, including exclusive interviews and an editorial written by the VA Secretary Nominee himself.
- Shulkin Nominated to Replace McDonald at VA
- Why VA Health Care Is Different
- Shulkin: VA "Not a Political Issue”
- Shulkin Addresses APRN Rule, Health Care Vacancies, and Access
- A New View for the VA
- Nearly 20,000 Comment on Controversial APRN Rule
- McDonald and Shulkin Lay Out Strategies for Eliminating VA Wait Times in 2016
- "Call to Action" on Veteran Suicide Yields Policy Shifts
- New VA Under Secretary of Health Focuses on Access, Employee Engagement
- Senate Confirms New VA Under Secretary for Health

SHM Practice Administrators’ Mentor Program benefits both parties
Editor’s note: Each month, SHM puts the spotlight on our most active members and explores how they are making substantial contributions to hospital medicine. Visit www.hospitalmedicine.org/getinvolved for more information on how you can lend your expertise and help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Alessandra G. Cornelio, MPH, the acquisition manager at Hartford Healthcare Medical Group in Connecticut. Ms. Cornelio is an active member of SHM’s Practice Administrators Committee. She developed and now directs the Practice Administrators’ Mentor Program.

Answer: I was finishing my internship at the Middlesex Hospital Cancer Center. I was interested in hospital administration and learning more about the inpatient side of health care. I chose to work within hospital medicine because I wanted to help build a team of compassionate doctors who could provide an excellent patient experience while maintaining an environment with safe, high-quality care.
To complement my career goals, SHM helped my professional growth by exposing me to the variety of topics and issues that practice administrators deal with regularly in their practices. I was also able to review and learn from the many resources available on the SHM website, such as white papers and articles, which were extremely useful for a new administrator.
Q: What prompted you to join the Practice Administrators Committee? What are some of the most impactful projects the committee is currently working on?
A: Within my first year of being a practice administrator, I attended a practice administrators’ forum at the SHM annual meeting in Washington. I found that the information was relevant to my daily functions as an administrator, and I was also able to meet and share ideas with other practice administrators from throughout the country. Down the line, I learned that SHM needed new members for the Practice Administrators Committee. I wanted to become more involved in a meaningful way, so I decided to apply.
The Practice Administrators Committee is a hardworking committee that takes on many meaningful projects. Most recently, the team has been working on developing a more user-friendly website for practice administrators, and a subgroup of the committee has cross-referenced “The Key Principles and Characteristics of an Effective Hospital Medicine Group” with existing resources, which will prove valuable to all administrators in the final product.
Q: Can you discuss how you began leading the work group for the Practice Administrators’ Mentor Program and how it has evolved since its inception?
A: As part of the committee’s initiative to help fellow practice administrators, we formed a subcommittee to begin developing a mentor program. (Former SHM staffer) Joseph Miller and I worked together to create an appropriate program model through research and brainstorming. We also utilized the HMX Practice Administrators Community to ask fellow practice administrators what they would expect from a mentor program and if they would participate. There was a strong favorable response rate, and we were able to implement a pilot program.
We implemented two different tracks for the program – the buddy system track and the career development track. The buddy system track is for those of any level of expertise or experience who are more interested in short-term assistance or in need of a sounding board. The career development track is a more traditional approach, matching a seasoned practice administrator with a less experienced practice administrator.
The program was designed to have annual cohorts, with the Practice Administrators Committee members as mentors. There is a detailed application process to ensure that each mentee is matched with an appropriate mentor, based on their interests and needs. We provide an orientation webinar to both parties before kicking off the relationship to present program expectations. The pilot program used this model, and comments from 6-month and annual evaluations showed tremendous satisfaction with the structure and value of this program.
There were approximately 16 pairs during the pilot year, and the following year, we grew to almost 20 pairs. Our goal as a committee is to maintain this program year after year, and in order to expand, we’ll need more than just the committee members to volunteer as mentors. There are so many talented practice administrators, and it would be wonderful to fold them into this gratifying program to pay it forward.
Many mentors, including myself, found value in acting as a mentor. I learned from my mentees as well as made connections and friendships with other professionals in the field.
Q: Given your intimate involvement, how have you seen the Practice Administrators’ Mentor Program benefit both the mentors and the mentees? Can you provide any specific examples?
A: Mentees are able to connect with seasoned mentors and can ask specific questions about career development and any issues they may be experiencing. Mentors are able to share their experiences and pass along important and valuable lessons learned to mentees. I served as a mentor, even though I did not yet consider myself a qualified candidate. However, I found that I was more equipped than I had realized, and I was able to assist my mentee with many aspects of career development (i.e., resume building, discussions with the C-suite, etc.).
My mentee was a practice coordinator who had only been in hospital medicine for 1 year. She had little experience hiring hospitalists, so this was a major area that we worked on together during our yearlong connection. I introduced her to collaborating with her HR department when posting positions, as well as working with permanent placement agencies. Her service was also undergoing a change in leadership, which can be difficult for any service line to experience. We discussed ways in which she could present important information to the new medical director that would produce a meaningful conversation.
In turn, my mentee introduced me to new online resources and was able to connect me with the manager of her practice, who assisted me with streamlining the payroll structure in my practice. I truly enjoyed my experience developing and participating in the program.
Felicia Steele is SHM’s communications coordinator.
Learn more about how you can benefit from the Practice Administrators’ Mentor program via the SHM website.
Editor’s note: Each month, SHM puts the spotlight on our most active members and explores how they are making substantial contributions to hospital medicine. Visit www.hospitalmedicine.org/getinvolved for more information on how you can lend your expertise and help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Alessandra G. Cornelio, MPH, the acquisition manager at Hartford Healthcare Medical Group in Connecticut. Ms. Cornelio is an active member of SHM’s Practice Administrators Committee. She developed and now directs the Practice Administrators’ Mentor Program.
Answer: I was finishing my internship at the Middlesex Hospital Cancer Center. I was interested in hospital administration and learning more about the inpatient side of health care. I chose to work within hospital medicine because I wanted to help build a team of compassionate doctors who could provide an excellent patient experience while maintaining an environment with safe, high-quality care.
To complement my career goals, SHM helped my professional growth by exposing me to the variety of topics and issues that practice administrators deal with regularly in their practices. I was also able to review and learn from the many resources available on the SHM website, such as white papers and articles, which were extremely useful for a new administrator.
Q: What prompted you to join the Practice Administrators Committee? What are some of the most impactful projects the committee is currently working on?
A: Within my first year of being a practice administrator, I attended a practice administrators’ forum at the SHM annual meeting in Washington. I found that the information was relevant to my daily functions as an administrator, and I was also able to meet and share ideas with other practice administrators from throughout the country. Down the line, I learned that SHM needed new members for the Practice Administrators Committee. I wanted to become more involved in a meaningful way, so I decided to apply.
The Practice Administrators Committee is a hardworking committee that takes on many meaningful projects. Most recently, the team has been working on developing a more user-friendly website for practice administrators, and a subgroup of the committee has cross-referenced “The Key Principles and Characteristics of an Effective Hospital Medicine Group” with existing resources, which will prove valuable to all administrators in the final product.
Q: Can you discuss how you began leading the work group for the Practice Administrators’ Mentor Program and how it has evolved since its inception?
A: As part of the committee’s initiative to help fellow practice administrators, we formed a subcommittee to begin developing a mentor program. (Former SHM staffer) Joseph Miller and I worked together to create an appropriate program model through research and brainstorming. We also utilized the HMX Practice Administrators Community to ask fellow practice administrators what they would expect from a mentor program and if they would participate. There was a strong favorable response rate, and we were able to implement a pilot program.
We implemented two different tracks for the program – the buddy system track and the career development track. The buddy system track is for those of any level of expertise or experience who are more interested in short-term assistance or in need of a sounding board. The career development track is a more traditional approach, matching a seasoned practice administrator with a less experienced practice administrator.
The program was designed to have annual cohorts, with the Practice Administrators Committee members as mentors. There is a detailed application process to ensure that each mentee is matched with an appropriate mentor, based on their interests and needs. We provide an orientation webinar to both parties before kicking off the relationship to present program expectations. The pilot program used this model, and comments from 6-month and annual evaluations showed tremendous satisfaction with the structure and value of this program.
There were approximately 16 pairs during the pilot year, and the following year, we grew to almost 20 pairs. Our goal as a committee is to maintain this program year after year, and in order to expand, we’ll need more than just the committee members to volunteer as mentors. There are so many talented practice administrators, and it would be wonderful to fold them into this gratifying program to pay it forward.
Many mentors, including myself, found value in acting as a mentor. I learned from my mentees as well as made connections and friendships with other professionals in the field.
Q: Given your intimate involvement, how have you seen the Practice Administrators’ Mentor Program benefit both the mentors and the mentees? Can you provide any specific examples?
A: Mentees are able to connect with seasoned mentors and can ask specific questions about career development and any issues they may be experiencing. Mentors are able to share their experiences and pass along important and valuable lessons learned to mentees. I served as a mentor, even though I did not yet consider myself a qualified candidate. However, I found that I was more equipped than I had realized, and I was able to assist my mentee with many aspects of career development (i.e., resume building, discussions with the C-suite, etc.).
My mentee was a practice coordinator who had only been in hospital medicine for 1 year. She had little experience hiring hospitalists, so this was a major area that we worked on together during our yearlong connection. I introduced her to collaborating with her HR department when posting positions, as well as working with permanent placement agencies. Her service was also undergoing a change in leadership, which can be difficult for any service line to experience. We discussed ways in which she could present important information to the new medical director that would produce a meaningful conversation.
In turn, my mentee introduced me to new online resources and was able to connect me with the manager of her practice, who assisted me with streamlining the payroll structure in my practice. I truly enjoyed my experience developing and participating in the program.
Felicia Steele is SHM’s communications coordinator.
Learn more about how you can benefit from the Practice Administrators’ Mentor program via the SHM website.
Editor’s note: Each month, SHM puts the spotlight on our most active members and explores how they are making substantial contributions to hospital medicine. Visit www.hospitalmedicine.org/getinvolved for more information on how you can lend your expertise and help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Alessandra G. Cornelio, MPH, the acquisition manager at Hartford Healthcare Medical Group in Connecticut. Ms. Cornelio is an active member of SHM’s Practice Administrators Committee. She developed and now directs the Practice Administrators’ Mentor Program.
Answer: I was finishing my internship at the Middlesex Hospital Cancer Center. I was interested in hospital administration and learning more about the inpatient side of health care. I chose to work within hospital medicine because I wanted to help build a team of compassionate doctors who could provide an excellent patient experience while maintaining an environment with safe, high-quality care.
To complement my career goals, SHM helped my professional growth by exposing me to the variety of topics and issues that practice administrators deal with regularly in their practices. I was also able to review and learn from the many resources available on the SHM website, such as white papers and articles, which were extremely useful for a new administrator.
Q: What prompted you to join the Practice Administrators Committee? What are some of the most impactful projects the committee is currently working on?
A: Within my first year of being a practice administrator, I attended a practice administrators’ forum at the SHM annual meeting in Washington. I found that the information was relevant to my daily functions as an administrator, and I was also able to meet and share ideas with other practice administrators from throughout the country. Down the line, I learned that SHM needed new members for the Practice Administrators Committee. I wanted to become more involved in a meaningful way, so I decided to apply.
The Practice Administrators Committee is a hardworking committee that takes on many meaningful projects. Most recently, the team has been working on developing a more user-friendly website for practice administrators, and a subgroup of the committee has cross-referenced “The Key Principles and Characteristics of an Effective Hospital Medicine Group” with existing resources, which will prove valuable to all administrators in the final product.
Q: Can you discuss how you began leading the work group for the Practice Administrators’ Mentor Program and how it has evolved since its inception?
A: As part of the committee’s initiative to help fellow practice administrators, we formed a subcommittee to begin developing a mentor program. (Former SHM staffer) Joseph Miller and I worked together to create an appropriate program model through research and brainstorming. We also utilized the HMX Practice Administrators Community to ask fellow practice administrators what they would expect from a mentor program and if they would participate. There was a strong favorable response rate, and we were able to implement a pilot program.
We implemented two different tracks for the program – the buddy system track and the career development track. The buddy system track is for those of any level of expertise or experience who are more interested in short-term assistance or in need of a sounding board. The career development track is a more traditional approach, matching a seasoned practice administrator with a less experienced practice administrator.
The program was designed to have annual cohorts, with the Practice Administrators Committee members as mentors. There is a detailed application process to ensure that each mentee is matched with an appropriate mentor, based on their interests and needs. We provide an orientation webinar to both parties before kicking off the relationship to present program expectations. The pilot program used this model, and comments from 6-month and annual evaluations showed tremendous satisfaction with the structure and value of this program.
There were approximately 16 pairs during the pilot year, and the following year, we grew to almost 20 pairs. Our goal as a committee is to maintain this program year after year, and in order to expand, we’ll need more than just the committee members to volunteer as mentors. There are so many talented practice administrators, and it would be wonderful to fold them into this gratifying program to pay it forward.
Many mentors, including myself, found value in acting as a mentor. I learned from my mentees as well as made connections and friendships with other professionals in the field.
Q: Given your intimate involvement, how have you seen the Practice Administrators’ Mentor Program benefit both the mentors and the mentees? Can you provide any specific examples?
A: Mentees are able to connect with seasoned mentors and can ask specific questions about career development and any issues they may be experiencing. Mentors are able to share their experiences and pass along important and valuable lessons learned to mentees. I served as a mentor, even though I did not yet consider myself a qualified candidate. However, I found that I was more equipped than I had realized, and I was able to assist my mentee with many aspects of career development (i.e., resume building, discussions with the C-suite, etc.).
My mentee was a practice coordinator who had only been in hospital medicine for 1 year. She had little experience hiring hospitalists, so this was a major area that we worked on together during our yearlong connection. I introduced her to collaborating with her HR department when posting positions, as well as working with permanent placement agencies. Her service was also undergoing a change in leadership, which can be difficult for any service line to experience. We discussed ways in which she could present important information to the new medical director that would produce a meaningful conversation.
In turn, my mentee introduced me to new online resources and was able to connect me with the manager of her practice, who assisted me with streamlining the payroll structure in my practice. I truly enjoyed my experience developing and participating in the program.
Felicia Steele is SHM’s communications coordinator.
Learn more about how you can benefit from the Practice Administrators’ Mentor program via the SHM website.
HAIs in kids are most common among infants, study shows
Photo by Bertrand Devouard
Data from hospitals in 29 European countries suggest that healthcare-associated infections (HAIs) reported in children occur most often in infants younger than 12 months of age.
And the prevalence of infection is highest for children in intensive care units (ICUs).
Walter Zingg, MD, of Imperial College London in the UK, and his colleagues conducted this research and detailed the results in The Lancet Infectious Diseases.
The researchers analyzed data from the European Centre for Disease Prevention and Control (ECDC) point prevalence survey of HAIs and antimicrobial use in European acute care hospitals.
The data encompassed 17,273 children and adolescents (ages 0 to 18) treated at 1149 hospitals in 29 European countries* from May 2011 to November 2012.
During that time, there were 770 infections reported in 726 children and adolescents.
The researchers found that most HAIs (77%) occurred in infants younger than 12 months. And the prevalence of infections was highest in pediatric ICUs and neonatal ICUs—15.5% and 10.7%, respectively.
Bloodstream infections were the most common type (45%), followed by lower respiratory tract infections (22%); gastrointestinal infections (8%); eye, ear, nose, and throat infections (7%); urinary tract infections (5%); and surgical-site infections (4%).
Analyses suggested that independent risk factors for infection included being younger than 12 months, having a fatal disease, a prolonged length of hospital stay, and receiving treatment with invasive medical devices.
The researchers said a pan-European program is urgently needed to prevent and reduce the unacceptably high rates of HAIs in children in Europe, with a focus in neonatal and pediatric ICUs and addressing the issues related to healthcare-associated bloodstream infections.
The team said this is the largest multinational study describing HAIs in children thus far.
A second point prevalence survey is ongoing in Europe, and its results are expected to be published by the ECDC after 2017. ![]()
*Austria, Belgium, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Netherlands, Norway, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, and the UK.
Photo by Bertrand Devouard
Data from hospitals in 29 European countries suggest that healthcare-associated infections (HAIs) reported in children occur most often in infants younger than 12 months of age.
And the prevalence of infection is highest for children in intensive care units (ICUs).
Walter Zingg, MD, of Imperial College London in the UK, and his colleagues conducted this research and detailed the results in The Lancet Infectious Diseases.
The researchers analyzed data from the European Centre for Disease Prevention and Control (ECDC) point prevalence survey of HAIs and antimicrobial use in European acute care hospitals.
The data encompassed 17,273 children and adolescents (ages 0 to 18) treated at 1149 hospitals in 29 European countries* from May 2011 to November 2012.
During that time, there were 770 infections reported in 726 children and adolescents.
The researchers found that most HAIs (77%) occurred in infants younger than 12 months. And the prevalence of infections was highest in pediatric ICUs and neonatal ICUs—15.5% and 10.7%, respectively.
Bloodstream infections were the most common type (45%), followed by lower respiratory tract infections (22%); gastrointestinal infections (8%); eye, ear, nose, and throat infections (7%); urinary tract infections (5%); and surgical-site infections (4%).
Analyses suggested that independent risk factors for infection included being younger than 12 months, having a fatal disease, a prolonged length of hospital stay, and receiving treatment with invasive medical devices.
The researchers said a pan-European program is urgently needed to prevent and reduce the unacceptably high rates of HAIs in children in Europe, with a focus in neonatal and pediatric ICUs and addressing the issues related to healthcare-associated bloodstream infections.
The team said this is the largest multinational study describing HAIs in children thus far.
A second point prevalence survey is ongoing in Europe, and its results are expected to be published by the ECDC after 2017. ![]()
*Austria, Belgium, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Netherlands, Norway, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, and the UK.
Photo by Bertrand Devouard
Data from hospitals in 29 European countries suggest that healthcare-associated infections (HAIs) reported in children occur most often in infants younger than 12 months of age.
And the prevalence of infection is highest for children in intensive care units (ICUs).
Walter Zingg, MD, of Imperial College London in the UK, and his colleagues conducted this research and detailed the results in The Lancet Infectious Diseases.
The researchers analyzed data from the European Centre for Disease Prevention and Control (ECDC) point prevalence survey of HAIs and antimicrobial use in European acute care hospitals.
The data encompassed 17,273 children and adolescents (ages 0 to 18) treated at 1149 hospitals in 29 European countries* from May 2011 to November 2012.
During that time, there were 770 infections reported in 726 children and adolescents.
The researchers found that most HAIs (77%) occurred in infants younger than 12 months. And the prevalence of infections was highest in pediatric ICUs and neonatal ICUs—15.5% and 10.7%, respectively.
Bloodstream infections were the most common type (45%), followed by lower respiratory tract infections (22%); gastrointestinal infections (8%); eye, ear, nose, and throat infections (7%); urinary tract infections (5%); and surgical-site infections (4%).
Analyses suggested that independent risk factors for infection included being younger than 12 months, having a fatal disease, a prolonged length of hospital stay, and receiving treatment with invasive medical devices.
The researchers said a pan-European program is urgently needed to prevent and reduce the unacceptably high rates of HAIs in children in Europe, with a focus in neonatal and pediatric ICUs and addressing the issues related to healthcare-associated bloodstream infections.
The team said this is the largest multinational study describing HAIs in children thus far.
A second point prevalence survey is ongoing in Europe, and its results are expected to be published by the ECDC after 2017. ![]()
*Austria, Belgium, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Netherlands, Norway, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, and the UK.
Decentralized vs Centralized Pharmacist Treatment of Patients With Atrial Fibrillation Managed With Direct Oral Anticoagulants
In the U.S. about 2.7 to 6.1 million people have atrial fibrillation (AF).1 This condition affects the rhythm of the heart, causes blood in the heart to become stagnant, and puts patients at high risk for developing a systemic embolism, particularly a stroke.1 Recent studies have shown that AF accounts for at least 15% of all strokes in the U.S. and 36% of strokes in people aged > 80 years.2
For patients aged > 60 years, the gold standard of long-term anticoagulation for reducing the risk of stroke has been oral vitamin K antagonist (warfarin) therapy.2 Although overwhelming evidence exists that supports the use of warfarin in these patients, warfarin is a narrow therapeutic index medication that requires frequent laboratory monitoring of international normalized ratio (INR) for dose titration guidance. There is also strong evidence that pharmacist-run anticoagulation clinics have improved patient-centered outcomes in patients prescribed warfarin.3-5
Direct oral anticoagulants (DOACs) are recently approved oral medications used as alternatives to warfarin for anticoagulation in AF. Direct oral anticoagulants do not require INR monitoring or any laboratory test for efficacy. In 2010, the FDA approved the first DOAC, dabigatran, for use in patients with AF. In 2011, rivaroxaban received approval for the same indication. One potential drawback of these new agents relative to warfarin is the lack of availability of a reversal agent that can be used in the event of a life-threatening bleeding event. Dabigatran is the only DOAC with an FDA-approved available reversal agent. In both 2011 and 2012, dabigatran, warfarin, and other anticoagulants topped the Institute for Safe Medicine Practice list of suspect drugs related to adverse events (AEs). These data prompted the Joint Commission to incorporate anticoagulation into the 2017 National Hospital Patient Safety Goals to improve patient outcomes and reduce harm from use of anticoagulants.6
In early 2011, the VHA produced national guidance on the treatment of patients who receive DOACs; this guidance was updated most recently in September 2016.7 Patients who were receiving DOACs at the Ralph H. Johnson VAMC (RHJVAMC) were initially monitored by 12 primary care pharmacists at the main hospital or at community-based outpatient clinics (CBOCs). Ambulatory care pharmacists at RHJVAMC work under a scope of practice to prescribe and adjust certain classes of medications to provide the highest level of care to more than 65,000 veterans in South Carolina and Georgia. Historically at RHJVAMC, warfarin has been the anticoagulant most commonly used for AF, though dabigatran and rivaroxaban have gained in popularity after being added to the national VA formulary.
In November 2012, for better monitoring of patient outcomes, improved efficiency of the primary care pharmacist clinics, and increased access to care in these clinics, treatment of patients prescribed DOACs was shifted to a centralized model that involved 3 anticoagulation clinical pharmacy specialists.
Centralized pharmacy services have a small number of core team members in a specific service for a particular disease, which reduces the number of different pharmacists a patient could talk to for management of a particular condition. Centralized pharmacy services allow for streamlining anticoagulation management to a small group of individual pharmacists considered specialists in anticoagulation. This shift in management to centralized anticoagulation services was supported at RHJVAMC by findings from a study of a pharmacist-run centralized anticoagulation clinic: Patients treated by the centralized clinic were 39% less likely to experience an anticoagulation therapy complication.8
Protocol for dabigatran follow-up and monitoring at RHJVAMC was developed by clinical and supervisory pharmacy staff, to align with national VA guidance. When a provider determines a patient is a candidate for dabigatran, an outpatient consultation is entered for the clinical pharmacy specialist to review the appropriateness of the patient selection for therapy. If the patient is eligible for therapy, the pharmacist contacts the patient to set up an initial visit to confirm selection and to provide the first dabigatran prescription and counseling. For assessments, with specific emphasis on adherence and AE monitoring, the patient is contacted 2 weeks, 1 month, 3 months, and every 6 months after the initial appointment.
Although most of the literature supports pharmacist-managed anticoagulation for patients who receive warfarin, DOACs have become more integrated into practice and more evaluated. Evidence supports pharmacists' interventions on evaluation of patient education and dosing, but there is conflicting evidence regarding pharmacists' impact on adherence after 3 months of therapy.9,10 In a larger VA study of the impact of dabigatran adherence on patient-centered outcomes, patients were mostly nonadherent to prescribed dosing.11 These studies support the need for improved adherence in patients prescribed DOACs and the need for further investigation of pharmacists' roles in improving patient outcomes.
Methods
This single-center, retrospective anticoagulant-use evaluation covered 2 study periods between November 1, 2011 and October 31, 2013. Study approval was obtained from the institutional review board of the Medical University of South Carolina and the research and development committee of RHJVAMC. The study population consisted of veterans who had a diagnosis of AF and received at least 3 outpatient prescription fills of a 30-day supply of dabigatran at RHJVAMC during either or both of the study periods. Patients were excluded if they were pregnant or planning to become pregnant or were incarcerated at any time during the study period. Dabigatran was selected because it was the first DOAC added to the local VA formulary before the start of this study.
Patients who met the inclusion criteria were separated into 2 groups based on the dates of their prescription fills. The precentralization group included patients treated by primary care pharmacists from November 1, 2011 to October 31, 2012; the postcentralization group included patients treated by anticoagulation clinical pharmacy specialists from November 1, 2012 to October 31, 2013. In each group, patients were followed for 1 year during their respective study period. For analysis, patients were included in both study periods if they received at least 3 fills of dabigatran during each period.
Medication possession ratio (MPR), which was used to measure the primary endpoint of adherence, is defined as the proportion of days a patient had dabigatran. The MPR denominator is the total number of days between the first and last prescription refill dates within the 52-week study period; the numerator is calculated by summing the days' supply for all but the last filling of the medication during each respective period. Nonadherence was defined as an MPR < 0.8 (or 80%), which has been used to define poor adherence in the literature.12 The authors calculated all patients' mean MPRs and compared them to determine statistical significance by repeated-measures linear regression. Descriptive statistics on proportion of patients in each study group with MPR < 0.8 were examined. Last, the authors performed a comparative subanalysis of median MPRs to determine whether there was an adherence difference between patients initially started on dabigatran at RHJVAMC and patients who were started on dabigatran before receiving it at RHJVAMC.
The secondary focus of this study was safety outcomes, including any bleeding event or thromboembolism within either study period. A bleeding event was defined as any major or minor bleeding event recognized through ICD-9 codes or any bleeding recorded in the patient's chart and noted during chart review, as well as any serum hemoglobin (Hgb) level decrease of ≥ to 2 g/dL during the study period. Thromboembolism was defined as a thromboembolism recognized through ICD-9 codes or any thromboembolism noted during chart review. Descriptive statistics were reported for this outcome, and a chi-square test was used to compare bleeding events between groups to determine significance.
The tertiary focus of this study was clinical efficiency as determined by number of primary care pharmacist visits during each study period. Primary care pharmacist visits were included for all primary care pharmacists in primary care clinics at the main hospital and in all 6 CBOCs.
For statistical analysis α was set at 0.05, and P < .05 was considered statistically significant. SAS Enterprise Guide software (Cary, North Carolina) was used for all statistical analyses.
Results
An initial data pull was completed from the RHJVAMC prescription records database for patients who had ≥ 3 prescriptions of dabigatran filled for treatment of AF during the study period, which yielded 65 unique patients. There were 34 patients in the precentralization group and 55 patients in the postcentralization group. Twenty-four unique patients were included in both study groups.
Mean MPR was 1.01 (range, 0.59-1.41) for the precentralization study period and 0.96 (range, 0.33-1.36) for the postcentralization period (Table 1). The difference was not statistically significant (P = .91). Number of patients considered nonadherent (MPR < 0.8) was 3 (8.82%) in the precentralization group and 8 (14.6%) in the postcentralization group.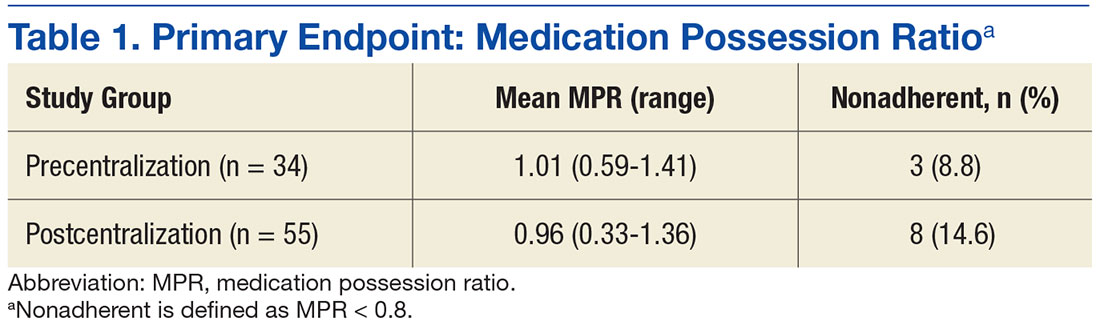
The primary endpoint subanalysis compared the median MPRs for the patients initially started on dabigatran at RHJVAMC (de novo starts) and the patients who were started on dabigatran before receiving it at RHJVAMC (prior starts). In each group, number and percentage of patients determined to be nonadherent by MPR were evaluated as well. De novo patients received initial assessment, counseling, and a dabigatran prescription from RHJVAMC pharmacists before or during the study period, and prior patients were initially prescribed dabigatran at another VA facility or at a non-VA facility (Table 2).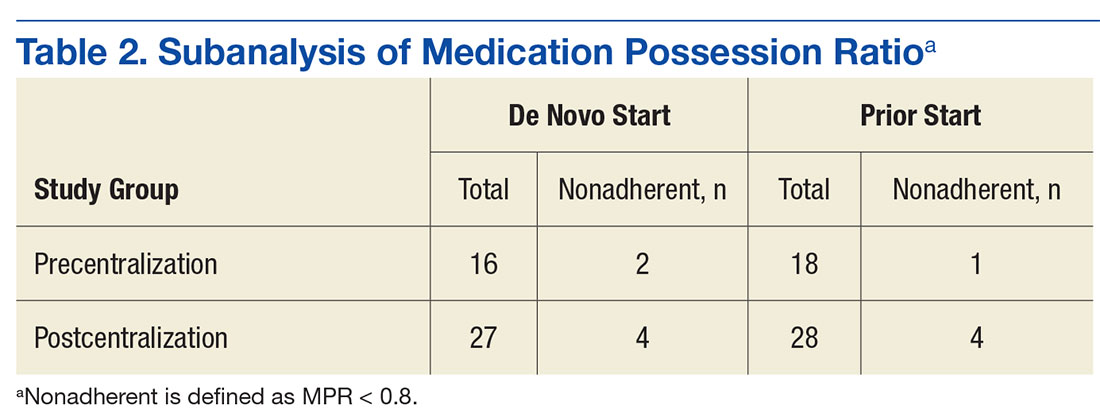
Regarding safety outcomes (secondary endpoint), a bleeding event was identified in 6 (17.7%) of the precentralization patients and 7 (12.7%) of the postcentralization patients. Of the 6 precentralization events, 1 was a case of hemoptysis, 1 was a hematoma on the forehead, 1 was a lower gastrointestinal bleed (unconfirmed), 1 was retinal hemorrhaging (noted by ophthalmologist), and 2 were serum Hgb level decreases of more than 2 g/dL (neither patient required transfusion of packed red blood cells). Of the 7 postcentralization events, 1 was persistent hematochezia caused by hemorrhoids, 1 was hematuria, 1 was a hematoma, 1 was an upper gastrointestinal bleed (required blood transfusion), and 4 were serum Hgb level decreases of more than 2 g/dL (1 of the 4 required transfusion). No precentralization patient had any evidence of thromboembolism during the study period; 1 postcentralization patient had a superficial venous thromboembolism near a hematoma on the elbow.
Discussion
In this single-center, retrospective medication-use evaluation, the authors found a high rate of adherence to dabigatran before and after centralization of outpatient DOAC management by pharmacists. There was no statistically significant difference in bleeding events between the study periods, but primary care pharmacist visits increased by 108% from precentralization to postcentralization. Although the primary outcome findings did not refute the study's null hypothesis, results support implementing centralized pharmacist DOAC management to maintain a high rate of adherence and a low incidence of adverse outcomes and providing more primary care pharmacist services to increase access to care for other chronic diseases.
Although there was no statistically significant difference in adherence rates between study periods, the 2 groups' rates were higher than the national average of 72%, as calculated by the proportion-of-days-covered (PDC) equation (median, 74%) in a 2015 large-scale study of site-level adherence in more than 5,000 VA patients.13 The authors' findings support that study's significant finding of a high rate of adherence to pharmacist-provided dabigatran treatment. This study's adherence rate also was higher than the median PDC rate reported in a 2014 study that focused on dabigatran adherence: 94% (mean, 84%; SD, 22%).11
The RHJVAMC follows national VA guidance on pharmacist follow-up for patients who receive DOACs. This follow-up focuses on frequent counseling over the first 6 months of de novo DOAC treatment and on monitoring and assessing adherence and AEs. Although there is less laboratory monitoring for DOAC treatment than for treatment with vitamin K antagonists (eg, warfarin), telephone monitoring as described in this study has been associated with a high adherence rate and minimization of AEs. The 2014 study with the 94% median PDC rate also showed an association of decreased adherence and increased harm, including combined all-cause mortality and stroke (hazard ratio, 1.13; 95% confidence interval [CI], 1.07-1.19 per 10% decrease in PDC rate).11
This study's subanalysis revealed no difference in adherence between patients initially started on dabigatran at RHJVAMC and patients who were started on dabigatran before receiving it at RHJVAMC. Each group had a high rate of adherence. Shore and colleagues found that most of the VA sites they surveyed (22/41) had anticoagulation clinics monitoring patients who were prescribed dabigatran.13 Pharmacist-led monitoring of adherence and AEs led to increased adherence to dabigatran treatment (relative risk, 1.25; 95% CI, 1.11-1.41), which was the standard of care at RHJVAMC throughout their entire study. Many of these factors may explain the very high rate of adherence found in the present study, specifically in comparison to previously reported national averages.
In addition, the authors found no statistically significant difference in bleeding outcomes between the precentralization and postcentralization groups. Their incidence of bleeding was similar to the 16.6% rate reported in the package insert for dabigatran.14 Furthermore, the safety outcomes were similar for both groups in this study, which may be attributable to the quality of patient care provided by all RHJVAMC pharmacists, particularly in the setting of dabigatran management.
Many studies have found an association between dabigatran use and an increased rate of bleeding, particularly gastrointestinal, as demonstrated in several patients in this study. Evidence of these clinically significant AEs further supports pharmacists' close monitoring to detect these AEs and working with patients' providers to determine whether an alternative anticoagulant should be used.
A significant finding of this study regarding centralization of DOAC management by pharmacists was the increased number of primary care pharmacist visits. By streamlining all anticoagulant services to anticoagulation clinical pharmacy specialists, primary care pharmacists were able to care for more veterans and increase access to care without adding staff. The centralized anticoagulation pharmacists were volunteers who held other positions within the department; they did not have to be replaced when they became anticoagulation providers. This workload reallocation helped the RHJVAMC pharmacy department increase access to care.
Limitations
This study had several potential limitations. First, MPR, a widely studied common tool for assessing adherence, has been criticized for often being imprecise when used with short study periods.12 Another commonly used adherence measure is PDC rate, which has been reported in several large-scale studies of dabigatran therapy. The authors selected MPR for the present study because MPR calculation is more practical in the patient population and because MPR and PDC rate are predicted to yield similar results in assessments of adherence to a single medication.12 It also should be noted that both MPR and PDC rate are surrogate markers for adherence and assume adherence based on the availability of medication to the patient. Assessing adherence in a retrospective study is a challenge, as more reliable adherence assessment--for example, with use of pill counts or blister packs--is not possible. This study's retrospective design was another potential limitation, as an active intervention was not used.
In addition, this study had a small sample, likely attributable to the addition of dabigatran to the VA national formulary just months before the start of the study period. Furthermore, this study was not powered to detect significant differences in safety or efficacy outcomes. Other potential study limitations included having national VA guidance regarding follow-up periods and dabigatran prescription quantity limits during both study periods. Also, there was some potential for pharmacist-initiated refills at follow-up visits, which could falsely increase MPR. Last, the study analyzed only 1 DOAC and not the entire class of medications.
Conclusion
Centralizing DOAC management by clinical pharmacy specialists at a single VA facility helped maintain high rates of dabigatran adherence, above the national average, and low rates of adverse outcomes were maintained in both study groups. In addition, centralization of anticoagulation services improved access to care through an increase in primary care pharmacist visits without the addition of staff. Centralization of DOAC management by pharmacists is a viable option for maintaining high rates of adherence and low rates of adverse outcomes in facilities where the goal is to achieve clinical efficiency.
1. January CT, Wann LS, Alpert JS, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society [published correction appears in J Am Coll Cardiol. 2014;64(21):2305-2307. J Am Coll Cardiol. 2014;64(21):e1-e76.
2. Reiffel JA. New versus traditional approaches to oral anticoagulation in patients with atrial fibrillation. Am J Med. 2014;127(4):e15.
3. Locke C, Ravnan SL, Patel R, Uchizono JA. Reduction in warfarin adverse events requiring patient hospitalization after implementation of pharmacist-managed anticoagulation service. Pharmacotherapy. 2005;25(5):685-689.
4. Poon IO, Lal L, Brown EN, Braun UK. The impact of pharmacist-managed oral anticoagulation therapy in older veterans. J Clin Pharm Ther. 2007;32(1):21-29.
5. Chiquette E, Amato MG, Bussey HI. Comparison of an anticoagulation clinic with usual medical care. Arch Intern Med. 1998;158(15):1641-1647.
6. The Joint Commission. National patient safety goals. https://www.jointcommission.org/as sets/1/6/2017_NPSG_HAP_ER.pdf. Published 2016. Accessed December 6, 2016.
7. Department of Veterans Affairs Pharmacy Benefits Management Services, Medical Advisory Panel, and VISN Pharmacist Executives. Direct oral anticoagulants (DOACs) (formerly called TSOACs) dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis): Criteria for Use for Stroke Prevention in nonvalvular atrial fibrillation (AF) and Edoxaban (SAVAYSA). http://www.pbm.va.gov/PBM/clinicalguidance/criteriaforuse/Anticoagulants_Direct_Oral_DOACs_CFU_and_Algorithm_for_Nonvalvular_Atrial_Fibrillation_Sep_2016.pdf. Updated September 2016. Accessed December 6, 2016.
8. Witt DM, Sadler MA, Shanahan RL, Mazzoli G, Tillman DJ. Effect of a centralized clinical pharmacy anticoagulation service on the outcomes of anticoagulation therapy. Chest. 2005;127(5):1515-1522.
9. Chan LL, Crumpler WL, Jacobson AK. Implementation of pharmacist-managed anticoagulation in patients receiving newer anticoagulants. Am J Health Syst Pharm. 2013;70(15):1285-1286, 1288.
10. Lee PY, Han SY, Miyahara RK. Adherence and outcomes of patients treated with dabigatran: pharmacist-managed anticoagulation clinic versus usual care. Am J Health Syst Pharm. 2013;70(13):1154-1161.
11. Shore S, Carey EP, Turakhia MP, et al. Adherence to dabigatran therapy and longitudinal patient outcomes: insights from the Veterans Health Administration. Am Heart J. 2014;167(6):810-817.
12. Martin BC, Wiley-Exley EK, Richards S, Domino ME, Carey TS, Sleath BL. Contrasting measures of adherence with simple drug use, medication switching and therapeutic duplication. Ann Pharmacother. 2009;43(1):36-44.
13. Shore S, Ho PM, Lambert-Kerzner A, et al. Site-level variation in and practices associated with dabigatran adherence. JAMA. 2015;313(14):1443-1450.
14. Pradaxa [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals; 2015.
In the U.S. about 2.7 to 6.1 million people have atrial fibrillation (AF).1 This condition affects the rhythm of the heart, causes blood in the heart to become stagnant, and puts patients at high risk for developing a systemic embolism, particularly a stroke.1 Recent studies have shown that AF accounts for at least 15% of all strokes in the U.S. and 36% of strokes in people aged > 80 years.2
For patients aged > 60 years, the gold standard of long-term anticoagulation for reducing the risk of stroke has been oral vitamin K antagonist (warfarin) therapy.2 Although overwhelming evidence exists that supports the use of warfarin in these patients, warfarin is a narrow therapeutic index medication that requires frequent laboratory monitoring of international normalized ratio (INR) for dose titration guidance. There is also strong evidence that pharmacist-run anticoagulation clinics have improved patient-centered outcomes in patients prescribed warfarin.3-5
Direct oral anticoagulants (DOACs) are recently approved oral medications used as alternatives to warfarin for anticoagulation in AF. Direct oral anticoagulants do not require INR monitoring or any laboratory test for efficacy. In 2010, the FDA approved the first DOAC, dabigatran, for use in patients with AF. In 2011, rivaroxaban received approval for the same indication. One potential drawback of these new agents relative to warfarin is the lack of availability of a reversal agent that can be used in the event of a life-threatening bleeding event. Dabigatran is the only DOAC with an FDA-approved available reversal agent. In both 2011 and 2012, dabigatran, warfarin, and other anticoagulants topped the Institute for Safe Medicine Practice list of suspect drugs related to adverse events (AEs). These data prompted the Joint Commission to incorporate anticoagulation into the 2017 National Hospital Patient Safety Goals to improve patient outcomes and reduce harm from use of anticoagulants.6
In early 2011, the VHA produced national guidance on the treatment of patients who receive DOACs; this guidance was updated most recently in September 2016.7 Patients who were receiving DOACs at the Ralph H. Johnson VAMC (RHJVAMC) were initially monitored by 12 primary care pharmacists at the main hospital or at community-based outpatient clinics (CBOCs). Ambulatory care pharmacists at RHJVAMC work under a scope of practice to prescribe and adjust certain classes of medications to provide the highest level of care to more than 65,000 veterans in South Carolina and Georgia. Historically at RHJVAMC, warfarin has been the anticoagulant most commonly used for AF, though dabigatran and rivaroxaban have gained in popularity after being added to the national VA formulary.
In November 2012, for better monitoring of patient outcomes, improved efficiency of the primary care pharmacist clinics, and increased access to care in these clinics, treatment of patients prescribed DOACs was shifted to a centralized model that involved 3 anticoagulation clinical pharmacy specialists.
Centralized pharmacy services have a small number of core team members in a specific service for a particular disease, which reduces the number of different pharmacists a patient could talk to for management of a particular condition. Centralized pharmacy services allow for streamlining anticoagulation management to a small group of individual pharmacists considered specialists in anticoagulation. This shift in management to centralized anticoagulation services was supported at RHJVAMC by findings from a study of a pharmacist-run centralized anticoagulation clinic: Patients treated by the centralized clinic were 39% less likely to experience an anticoagulation therapy complication.8
Protocol for dabigatran follow-up and monitoring at RHJVAMC was developed by clinical and supervisory pharmacy staff, to align with national VA guidance. When a provider determines a patient is a candidate for dabigatran, an outpatient consultation is entered for the clinical pharmacy specialist to review the appropriateness of the patient selection for therapy. If the patient is eligible for therapy, the pharmacist contacts the patient to set up an initial visit to confirm selection and to provide the first dabigatran prescription and counseling. For assessments, with specific emphasis on adherence and AE monitoring, the patient is contacted 2 weeks, 1 month, 3 months, and every 6 months after the initial appointment.
Although most of the literature supports pharmacist-managed anticoagulation for patients who receive warfarin, DOACs have become more integrated into practice and more evaluated. Evidence supports pharmacists' interventions on evaluation of patient education and dosing, but there is conflicting evidence regarding pharmacists' impact on adherence after 3 months of therapy.9,10 In a larger VA study of the impact of dabigatran adherence on patient-centered outcomes, patients were mostly nonadherent to prescribed dosing.11 These studies support the need for improved adherence in patients prescribed DOACs and the need for further investigation of pharmacists' roles in improving patient outcomes.
Methods
This single-center, retrospective anticoagulant-use evaluation covered 2 study periods between November 1, 2011 and October 31, 2013. Study approval was obtained from the institutional review board of the Medical University of South Carolina and the research and development committee of RHJVAMC. The study population consisted of veterans who had a diagnosis of AF and received at least 3 outpatient prescription fills of a 30-day supply of dabigatran at RHJVAMC during either or both of the study periods. Patients were excluded if they were pregnant or planning to become pregnant or were incarcerated at any time during the study period. Dabigatran was selected because it was the first DOAC added to the local VA formulary before the start of this study.
Patients who met the inclusion criteria were separated into 2 groups based on the dates of their prescription fills. The precentralization group included patients treated by primary care pharmacists from November 1, 2011 to October 31, 2012; the postcentralization group included patients treated by anticoagulation clinical pharmacy specialists from November 1, 2012 to October 31, 2013. In each group, patients were followed for 1 year during their respective study period. For analysis, patients were included in both study periods if they received at least 3 fills of dabigatran during each period.
Medication possession ratio (MPR), which was used to measure the primary endpoint of adherence, is defined as the proportion of days a patient had dabigatran. The MPR denominator is the total number of days between the first and last prescription refill dates within the 52-week study period; the numerator is calculated by summing the days' supply for all but the last filling of the medication during each respective period. Nonadherence was defined as an MPR < 0.8 (or 80%), which has been used to define poor adherence in the literature.12 The authors calculated all patients' mean MPRs and compared them to determine statistical significance by repeated-measures linear regression. Descriptive statistics on proportion of patients in each study group with MPR < 0.8 were examined. Last, the authors performed a comparative subanalysis of median MPRs to determine whether there was an adherence difference between patients initially started on dabigatran at RHJVAMC and patients who were started on dabigatran before receiving it at RHJVAMC.
The secondary focus of this study was safety outcomes, including any bleeding event or thromboembolism within either study period. A bleeding event was defined as any major or minor bleeding event recognized through ICD-9 codes or any bleeding recorded in the patient's chart and noted during chart review, as well as any serum hemoglobin (Hgb) level decrease of ≥ to 2 g/dL during the study period. Thromboembolism was defined as a thromboembolism recognized through ICD-9 codes or any thromboembolism noted during chart review. Descriptive statistics were reported for this outcome, and a chi-square test was used to compare bleeding events between groups to determine significance.
The tertiary focus of this study was clinical efficiency as determined by number of primary care pharmacist visits during each study period. Primary care pharmacist visits were included for all primary care pharmacists in primary care clinics at the main hospital and in all 6 CBOCs.
For statistical analysis α was set at 0.05, and P < .05 was considered statistically significant. SAS Enterprise Guide software (Cary, North Carolina) was used for all statistical analyses.
Results
An initial data pull was completed from the RHJVAMC prescription records database for patients who had ≥ 3 prescriptions of dabigatran filled for treatment of AF during the study period, which yielded 65 unique patients. There were 34 patients in the precentralization group and 55 patients in the postcentralization group. Twenty-four unique patients were included in both study groups.
Mean MPR was 1.01 (range, 0.59-1.41) for the precentralization study period and 0.96 (range, 0.33-1.36) for the postcentralization period (Table 1). The difference was not statistically significant (P = .91). Number of patients considered nonadherent (MPR < 0.8) was 3 (8.82%) in the precentralization group and 8 (14.6%) in the postcentralization group.
The primary endpoint subanalysis compared the median MPRs for the patients initially started on dabigatran at RHJVAMC (de novo starts) and the patients who were started on dabigatran before receiving it at RHJVAMC (prior starts). In each group, number and percentage of patients determined to be nonadherent by MPR were evaluated as well. De novo patients received initial assessment, counseling, and a dabigatran prescription from RHJVAMC pharmacists before or during the study period, and prior patients were initially prescribed dabigatran at another VA facility or at a non-VA facility (Table 2).
Regarding safety outcomes (secondary endpoint), a bleeding event was identified in 6 (17.7%) of the precentralization patients and 7 (12.7%) of the postcentralization patients. Of the 6 precentralization events, 1 was a case of hemoptysis, 1 was a hematoma on the forehead, 1 was a lower gastrointestinal bleed (unconfirmed), 1 was retinal hemorrhaging (noted by ophthalmologist), and 2 were serum Hgb level decreases of more than 2 g/dL (neither patient required transfusion of packed red blood cells). Of the 7 postcentralization events, 1 was persistent hematochezia caused by hemorrhoids, 1 was hematuria, 1 was a hematoma, 1 was an upper gastrointestinal bleed (required blood transfusion), and 4 were serum Hgb level decreases of more than 2 g/dL (1 of the 4 required transfusion). No precentralization patient had any evidence of thromboembolism during the study period; 1 postcentralization patient had a superficial venous thromboembolism near a hematoma on the elbow.
Discussion
In this single-center, retrospective medication-use evaluation, the authors found a high rate of adherence to dabigatran before and after centralization of outpatient DOAC management by pharmacists. There was no statistically significant difference in bleeding events between the study periods, but primary care pharmacist visits increased by 108% from precentralization to postcentralization. Although the primary outcome findings did not refute the study's null hypothesis, results support implementing centralized pharmacist DOAC management to maintain a high rate of adherence and a low incidence of adverse outcomes and providing more primary care pharmacist services to increase access to care for other chronic diseases.
Although there was no statistically significant difference in adherence rates between study periods, the 2 groups' rates were higher than the national average of 72%, as calculated by the proportion-of-days-covered (PDC) equation (median, 74%) in a 2015 large-scale study of site-level adherence in more than 5,000 VA patients.13 The authors' findings support that study's significant finding of a high rate of adherence to pharmacist-provided dabigatran treatment. This study's adherence rate also was higher than the median PDC rate reported in a 2014 study that focused on dabigatran adherence: 94% (mean, 84%; SD, 22%).11
The RHJVAMC follows national VA guidance on pharmacist follow-up for patients who receive DOACs. This follow-up focuses on frequent counseling over the first 6 months of de novo DOAC treatment and on monitoring and assessing adherence and AEs. Although there is less laboratory monitoring for DOAC treatment than for treatment with vitamin K antagonists (eg, warfarin), telephone monitoring as described in this study has been associated with a high adherence rate and minimization of AEs. The 2014 study with the 94% median PDC rate also showed an association of decreased adherence and increased harm, including combined all-cause mortality and stroke (hazard ratio, 1.13; 95% confidence interval [CI], 1.07-1.19 per 10% decrease in PDC rate).11
This study's subanalysis revealed no difference in adherence between patients initially started on dabigatran at RHJVAMC and patients who were started on dabigatran before receiving it at RHJVAMC. Each group had a high rate of adherence. Shore and colleagues found that most of the VA sites they surveyed (22/41) had anticoagulation clinics monitoring patients who were prescribed dabigatran.13 Pharmacist-led monitoring of adherence and AEs led to increased adherence to dabigatran treatment (relative risk, 1.25; 95% CI, 1.11-1.41), which was the standard of care at RHJVAMC throughout their entire study. Many of these factors may explain the very high rate of adherence found in the present study, specifically in comparison to previously reported national averages.
In addition, the authors found no statistically significant difference in bleeding outcomes between the precentralization and postcentralization groups. Their incidence of bleeding was similar to the 16.6% rate reported in the package insert for dabigatran.14 Furthermore, the safety outcomes were similar for both groups in this study, which may be attributable to the quality of patient care provided by all RHJVAMC pharmacists, particularly in the setting of dabigatran management.
Many studies have found an association between dabigatran use and an increased rate of bleeding, particularly gastrointestinal, as demonstrated in several patients in this study. Evidence of these clinically significant AEs further supports pharmacists' close monitoring to detect these AEs and working with patients' providers to determine whether an alternative anticoagulant should be used.
A significant finding of this study regarding centralization of DOAC management by pharmacists was the increased number of primary care pharmacist visits. By streamlining all anticoagulant services to anticoagulation clinical pharmacy specialists, primary care pharmacists were able to care for more veterans and increase access to care without adding staff. The centralized anticoagulation pharmacists were volunteers who held other positions within the department; they did not have to be replaced when they became anticoagulation providers. This workload reallocation helped the RHJVAMC pharmacy department increase access to care.
Limitations
This study had several potential limitations. First, MPR, a widely studied common tool for assessing adherence, has been criticized for often being imprecise when used with short study periods.12 Another commonly used adherence measure is PDC rate, which has been reported in several large-scale studies of dabigatran therapy. The authors selected MPR for the present study because MPR calculation is more practical in the patient population and because MPR and PDC rate are predicted to yield similar results in assessments of adherence to a single medication.12 It also should be noted that both MPR and PDC rate are surrogate markers for adherence and assume adherence based on the availability of medication to the patient. Assessing adherence in a retrospective study is a challenge, as more reliable adherence assessment--for example, with use of pill counts or blister packs--is not possible. This study's retrospective design was another potential limitation, as an active intervention was not used.
In addition, this study had a small sample, likely attributable to the addition of dabigatran to the VA national formulary just months before the start of the study period. Furthermore, this study was not powered to detect significant differences in safety or efficacy outcomes. Other potential study limitations included having national VA guidance regarding follow-up periods and dabigatran prescription quantity limits during both study periods. Also, there was some potential for pharmacist-initiated refills at follow-up visits, which could falsely increase MPR. Last, the study analyzed only 1 DOAC and not the entire class of medications.
Conclusion
Centralizing DOAC management by clinical pharmacy specialists at a single VA facility helped maintain high rates of dabigatran adherence, above the national average, and low rates of adverse outcomes were maintained in both study groups. In addition, centralization of anticoagulation services improved access to care through an increase in primary care pharmacist visits without the addition of staff. Centralization of DOAC management by pharmacists is a viable option for maintaining high rates of adherence and low rates of adverse outcomes in facilities where the goal is to achieve clinical efficiency.
In the U.S. about 2.7 to 6.1 million people have atrial fibrillation (AF).1 This condition affects the rhythm of the heart, causes blood in the heart to become stagnant, and puts patients at high risk for developing a systemic embolism, particularly a stroke.1 Recent studies have shown that AF accounts for at least 15% of all strokes in the U.S. and 36% of strokes in people aged > 80 years.2
For patients aged > 60 years, the gold standard of long-term anticoagulation for reducing the risk of stroke has been oral vitamin K antagonist (warfarin) therapy.2 Although overwhelming evidence exists that supports the use of warfarin in these patients, warfarin is a narrow therapeutic index medication that requires frequent laboratory monitoring of international normalized ratio (INR) for dose titration guidance. There is also strong evidence that pharmacist-run anticoagulation clinics have improved patient-centered outcomes in patients prescribed warfarin.3-5
Direct oral anticoagulants (DOACs) are recently approved oral medications used as alternatives to warfarin for anticoagulation in AF. Direct oral anticoagulants do not require INR monitoring or any laboratory test for efficacy. In 2010, the FDA approved the first DOAC, dabigatran, for use in patients with AF. In 2011, rivaroxaban received approval for the same indication. One potential drawback of these new agents relative to warfarin is the lack of availability of a reversal agent that can be used in the event of a life-threatening bleeding event. Dabigatran is the only DOAC with an FDA-approved available reversal agent. In both 2011 and 2012, dabigatran, warfarin, and other anticoagulants topped the Institute for Safe Medicine Practice list of suspect drugs related to adverse events (AEs). These data prompted the Joint Commission to incorporate anticoagulation into the 2017 National Hospital Patient Safety Goals to improve patient outcomes and reduce harm from use of anticoagulants.6
In early 2011, the VHA produced national guidance on the treatment of patients who receive DOACs; this guidance was updated most recently in September 2016.7 Patients who were receiving DOACs at the Ralph H. Johnson VAMC (RHJVAMC) were initially monitored by 12 primary care pharmacists at the main hospital or at community-based outpatient clinics (CBOCs). Ambulatory care pharmacists at RHJVAMC work under a scope of practice to prescribe and adjust certain classes of medications to provide the highest level of care to more than 65,000 veterans in South Carolina and Georgia. Historically at RHJVAMC, warfarin has been the anticoagulant most commonly used for AF, though dabigatran and rivaroxaban have gained in popularity after being added to the national VA formulary.
In November 2012, for better monitoring of patient outcomes, improved efficiency of the primary care pharmacist clinics, and increased access to care in these clinics, treatment of patients prescribed DOACs was shifted to a centralized model that involved 3 anticoagulation clinical pharmacy specialists.
Centralized pharmacy services have a small number of core team members in a specific service for a particular disease, which reduces the number of different pharmacists a patient could talk to for management of a particular condition. Centralized pharmacy services allow for streamlining anticoagulation management to a small group of individual pharmacists considered specialists in anticoagulation. This shift in management to centralized anticoagulation services was supported at RHJVAMC by findings from a study of a pharmacist-run centralized anticoagulation clinic: Patients treated by the centralized clinic were 39% less likely to experience an anticoagulation therapy complication.8
Protocol for dabigatran follow-up and monitoring at RHJVAMC was developed by clinical and supervisory pharmacy staff, to align with national VA guidance. When a provider determines a patient is a candidate for dabigatran, an outpatient consultation is entered for the clinical pharmacy specialist to review the appropriateness of the patient selection for therapy. If the patient is eligible for therapy, the pharmacist contacts the patient to set up an initial visit to confirm selection and to provide the first dabigatran prescription and counseling. For assessments, with specific emphasis on adherence and AE monitoring, the patient is contacted 2 weeks, 1 month, 3 months, and every 6 months after the initial appointment.
Although most of the literature supports pharmacist-managed anticoagulation for patients who receive warfarin, DOACs have become more integrated into practice and more evaluated. Evidence supports pharmacists' interventions on evaluation of patient education and dosing, but there is conflicting evidence regarding pharmacists' impact on adherence after 3 months of therapy.9,10 In a larger VA study of the impact of dabigatran adherence on patient-centered outcomes, patients were mostly nonadherent to prescribed dosing.11 These studies support the need for improved adherence in patients prescribed DOACs and the need for further investigation of pharmacists' roles in improving patient outcomes.
Methods
This single-center, retrospective anticoagulant-use evaluation covered 2 study periods between November 1, 2011 and October 31, 2013. Study approval was obtained from the institutional review board of the Medical University of South Carolina and the research and development committee of RHJVAMC. The study population consisted of veterans who had a diagnosis of AF and received at least 3 outpatient prescription fills of a 30-day supply of dabigatran at RHJVAMC during either or both of the study periods. Patients were excluded if they were pregnant or planning to become pregnant or were incarcerated at any time during the study period. Dabigatran was selected because it was the first DOAC added to the local VA formulary before the start of this study.
Patients who met the inclusion criteria were separated into 2 groups based on the dates of their prescription fills. The precentralization group included patients treated by primary care pharmacists from November 1, 2011 to October 31, 2012; the postcentralization group included patients treated by anticoagulation clinical pharmacy specialists from November 1, 2012 to October 31, 2013. In each group, patients were followed for 1 year during their respective study period. For analysis, patients were included in both study periods if they received at least 3 fills of dabigatran during each period.
Medication possession ratio (MPR), which was used to measure the primary endpoint of adherence, is defined as the proportion of days a patient had dabigatran. The MPR denominator is the total number of days between the first and last prescription refill dates within the 52-week study period; the numerator is calculated by summing the days' supply for all but the last filling of the medication during each respective period. Nonadherence was defined as an MPR < 0.8 (or 80%), which has been used to define poor adherence in the literature.12 The authors calculated all patients' mean MPRs and compared them to determine statistical significance by repeated-measures linear regression. Descriptive statistics on proportion of patients in each study group with MPR < 0.8 were examined. Last, the authors performed a comparative subanalysis of median MPRs to determine whether there was an adherence difference between patients initially started on dabigatran at RHJVAMC and patients who were started on dabigatran before receiving it at RHJVAMC.
The secondary focus of this study was safety outcomes, including any bleeding event or thromboembolism within either study period. A bleeding event was defined as any major or minor bleeding event recognized through ICD-9 codes or any bleeding recorded in the patient's chart and noted during chart review, as well as any serum hemoglobin (Hgb) level decrease of ≥ to 2 g/dL during the study period. Thromboembolism was defined as a thromboembolism recognized through ICD-9 codes or any thromboembolism noted during chart review. Descriptive statistics were reported for this outcome, and a chi-square test was used to compare bleeding events between groups to determine significance.
The tertiary focus of this study was clinical efficiency as determined by number of primary care pharmacist visits during each study period. Primary care pharmacist visits were included for all primary care pharmacists in primary care clinics at the main hospital and in all 6 CBOCs.
For statistical analysis α was set at 0.05, and P < .05 was considered statistically significant. SAS Enterprise Guide software (Cary, North Carolina) was used for all statistical analyses.
Results
An initial data pull was completed from the RHJVAMC prescription records database for patients who had ≥ 3 prescriptions of dabigatran filled for treatment of AF during the study period, which yielded 65 unique patients. There were 34 patients in the precentralization group and 55 patients in the postcentralization group. Twenty-four unique patients were included in both study groups.
Mean MPR was 1.01 (range, 0.59-1.41) for the precentralization study period and 0.96 (range, 0.33-1.36) for the postcentralization period (Table 1). The difference was not statistically significant (P = .91). Number of patients considered nonadherent (MPR < 0.8) was 3 (8.82%) in the precentralization group and 8 (14.6%) in the postcentralization group.
The primary endpoint subanalysis compared the median MPRs for the patients initially started on dabigatran at RHJVAMC (de novo starts) and the patients who were started on dabigatran before receiving it at RHJVAMC (prior starts). In each group, number and percentage of patients determined to be nonadherent by MPR were evaluated as well. De novo patients received initial assessment, counseling, and a dabigatran prescription from RHJVAMC pharmacists before or during the study period, and prior patients were initially prescribed dabigatran at another VA facility or at a non-VA facility (Table 2).
Regarding safety outcomes (secondary endpoint), a bleeding event was identified in 6 (17.7%) of the precentralization patients and 7 (12.7%) of the postcentralization patients. Of the 6 precentralization events, 1 was a case of hemoptysis, 1 was a hematoma on the forehead, 1 was a lower gastrointestinal bleed (unconfirmed), 1 was retinal hemorrhaging (noted by ophthalmologist), and 2 were serum Hgb level decreases of more than 2 g/dL (neither patient required transfusion of packed red blood cells). Of the 7 postcentralization events, 1 was persistent hematochezia caused by hemorrhoids, 1 was hematuria, 1 was a hematoma, 1 was an upper gastrointestinal bleed (required blood transfusion), and 4 were serum Hgb level decreases of more than 2 g/dL (1 of the 4 required transfusion). No precentralization patient had any evidence of thromboembolism during the study period; 1 postcentralization patient had a superficial venous thromboembolism near a hematoma on the elbow.
Discussion
In this single-center, retrospective medication-use evaluation, the authors found a high rate of adherence to dabigatran before and after centralization of outpatient DOAC management by pharmacists. There was no statistically significant difference in bleeding events between the study periods, but primary care pharmacist visits increased by 108% from precentralization to postcentralization. Although the primary outcome findings did not refute the study's null hypothesis, results support implementing centralized pharmacist DOAC management to maintain a high rate of adherence and a low incidence of adverse outcomes and providing more primary care pharmacist services to increase access to care for other chronic diseases.
Although there was no statistically significant difference in adherence rates between study periods, the 2 groups' rates were higher than the national average of 72%, as calculated by the proportion-of-days-covered (PDC) equation (median, 74%) in a 2015 large-scale study of site-level adherence in more than 5,000 VA patients.13 The authors' findings support that study's significant finding of a high rate of adherence to pharmacist-provided dabigatran treatment. This study's adherence rate also was higher than the median PDC rate reported in a 2014 study that focused on dabigatran adherence: 94% (mean, 84%; SD, 22%).11
The RHJVAMC follows national VA guidance on pharmacist follow-up for patients who receive DOACs. This follow-up focuses on frequent counseling over the first 6 months of de novo DOAC treatment and on monitoring and assessing adherence and AEs. Although there is less laboratory monitoring for DOAC treatment than for treatment with vitamin K antagonists (eg, warfarin), telephone monitoring as described in this study has been associated with a high adherence rate and minimization of AEs. The 2014 study with the 94% median PDC rate also showed an association of decreased adherence and increased harm, including combined all-cause mortality and stroke (hazard ratio, 1.13; 95% confidence interval [CI], 1.07-1.19 per 10% decrease in PDC rate).11
This study's subanalysis revealed no difference in adherence between patients initially started on dabigatran at RHJVAMC and patients who were started on dabigatran before receiving it at RHJVAMC. Each group had a high rate of adherence. Shore and colleagues found that most of the VA sites they surveyed (22/41) had anticoagulation clinics monitoring patients who were prescribed dabigatran.13 Pharmacist-led monitoring of adherence and AEs led to increased adherence to dabigatran treatment (relative risk, 1.25; 95% CI, 1.11-1.41), which was the standard of care at RHJVAMC throughout their entire study. Many of these factors may explain the very high rate of adherence found in the present study, specifically in comparison to previously reported national averages.
In addition, the authors found no statistically significant difference in bleeding outcomes between the precentralization and postcentralization groups. Their incidence of bleeding was similar to the 16.6% rate reported in the package insert for dabigatran.14 Furthermore, the safety outcomes were similar for both groups in this study, which may be attributable to the quality of patient care provided by all RHJVAMC pharmacists, particularly in the setting of dabigatran management.
Many studies have found an association between dabigatran use and an increased rate of bleeding, particularly gastrointestinal, as demonstrated in several patients in this study. Evidence of these clinically significant AEs further supports pharmacists' close monitoring to detect these AEs and working with patients' providers to determine whether an alternative anticoagulant should be used.
A significant finding of this study regarding centralization of DOAC management by pharmacists was the increased number of primary care pharmacist visits. By streamlining all anticoagulant services to anticoagulation clinical pharmacy specialists, primary care pharmacists were able to care for more veterans and increase access to care without adding staff. The centralized anticoagulation pharmacists were volunteers who held other positions within the department; they did not have to be replaced when they became anticoagulation providers. This workload reallocation helped the RHJVAMC pharmacy department increase access to care.
Limitations
This study had several potential limitations. First, MPR, a widely studied common tool for assessing adherence, has been criticized for often being imprecise when used with short study periods.12 Another commonly used adherence measure is PDC rate, which has been reported in several large-scale studies of dabigatran therapy. The authors selected MPR for the present study because MPR calculation is more practical in the patient population and because MPR and PDC rate are predicted to yield similar results in assessments of adherence to a single medication.12 It also should be noted that both MPR and PDC rate are surrogate markers for adherence and assume adherence based on the availability of medication to the patient. Assessing adherence in a retrospective study is a challenge, as more reliable adherence assessment--for example, with use of pill counts or blister packs--is not possible. This study's retrospective design was another potential limitation, as an active intervention was not used.
In addition, this study had a small sample, likely attributable to the addition of dabigatran to the VA national formulary just months before the start of the study period. Furthermore, this study was not powered to detect significant differences in safety or efficacy outcomes. Other potential study limitations included having national VA guidance regarding follow-up periods and dabigatran prescription quantity limits during both study periods. Also, there was some potential for pharmacist-initiated refills at follow-up visits, which could falsely increase MPR. Last, the study analyzed only 1 DOAC and not the entire class of medications.
Conclusion
Centralizing DOAC management by clinical pharmacy specialists at a single VA facility helped maintain high rates of dabigatran adherence, above the national average, and low rates of adverse outcomes were maintained in both study groups. In addition, centralization of anticoagulation services improved access to care through an increase in primary care pharmacist visits without the addition of staff. Centralization of DOAC management by pharmacists is a viable option for maintaining high rates of adherence and low rates of adverse outcomes in facilities where the goal is to achieve clinical efficiency.
1. January CT, Wann LS, Alpert JS, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society [published correction appears in J Am Coll Cardiol. 2014;64(21):2305-2307. J Am Coll Cardiol. 2014;64(21):e1-e76.
2. Reiffel JA. New versus traditional approaches to oral anticoagulation in patients with atrial fibrillation. Am J Med. 2014;127(4):e15.
3. Locke C, Ravnan SL, Patel R, Uchizono JA. Reduction in warfarin adverse events requiring patient hospitalization after implementation of pharmacist-managed anticoagulation service. Pharmacotherapy. 2005;25(5):685-689.
4. Poon IO, Lal L, Brown EN, Braun UK. The impact of pharmacist-managed oral anticoagulation therapy in older veterans. J Clin Pharm Ther. 2007;32(1):21-29.
5. Chiquette E, Amato MG, Bussey HI. Comparison of an anticoagulation clinic with usual medical care. Arch Intern Med. 1998;158(15):1641-1647.
6. The Joint Commission. National patient safety goals. https://www.jointcommission.org/as sets/1/6/2017_NPSG_HAP_ER.pdf. Published 2016. Accessed December 6, 2016.
7. Department of Veterans Affairs Pharmacy Benefits Management Services, Medical Advisory Panel, and VISN Pharmacist Executives. Direct oral anticoagulants (DOACs) (formerly called TSOACs) dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis): Criteria for Use for Stroke Prevention in nonvalvular atrial fibrillation (AF) and Edoxaban (SAVAYSA). http://www.pbm.va.gov/PBM/clinicalguidance/criteriaforuse/Anticoagulants_Direct_Oral_DOACs_CFU_and_Algorithm_for_Nonvalvular_Atrial_Fibrillation_Sep_2016.pdf. Updated September 2016. Accessed December 6, 2016.
8. Witt DM, Sadler MA, Shanahan RL, Mazzoli G, Tillman DJ. Effect of a centralized clinical pharmacy anticoagulation service on the outcomes of anticoagulation therapy. Chest. 2005;127(5):1515-1522.
9. Chan LL, Crumpler WL, Jacobson AK. Implementation of pharmacist-managed anticoagulation in patients receiving newer anticoagulants. Am J Health Syst Pharm. 2013;70(15):1285-1286, 1288.
10. Lee PY, Han SY, Miyahara RK. Adherence and outcomes of patients treated with dabigatran: pharmacist-managed anticoagulation clinic versus usual care. Am J Health Syst Pharm. 2013;70(13):1154-1161.
11. Shore S, Carey EP, Turakhia MP, et al. Adherence to dabigatran therapy and longitudinal patient outcomes: insights from the Veterans Health Administration. Am Heart J. 2014;167(6):810-817.
12. Martin BC, Wiley-Exley EK, Richards S, Domino ME, Carey TS, Sleath BL. Contrasting measures of adherence with simple drug use, medication switching and therapeutic duplication. Ann Pharmacother. 2009;43(1):36-44.
13. Shore S, Ho PM, Lambert-Kerzner A, et al. Site-level variation in and practices associated with dabigatran adherence. JAMA. 2015;313(14):1443-1450.
14. Pradaxa [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals; 2015.
1. January CT, Wann LS, Alpert JS, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society [published correction appears in J Am Coll Cardiol. 2014;64(21):2305-2307. J Am Coll Cardiol. 2014;64(21):e1-e76.
2. Reiffel JA. New versus traditional approaches to oral anticoagulation in patients with atrial fibrillation. Am J Med. 2014;127(4):e15.
3. Locke C, Ravnan SL, Patel R, Uchizono JA. Reduction in warfarin adverse events requiring patient hospitalization after implementation of pharmacist-managed anticoagulation service. Pharmacotherapy. 2005;25(5):685-689.
4. Poon IO, Lal L, Brown EN, Braun UK. The impact of pharmacist-managed oral anticoagulation therapy in older veterans. J Clin Pharm Ther. 2007;32(1):21-29.
5. Chiquette E, Amato MG, Bussey HI. Comparison of an anticoagulation clinic with usual medical care. Arch Intern Med. 1998;158(15):1641-1647.
6. The Joint Commission. National patient safety goals. https://www.jointcommission.org/as sets/1/6/2017_NPSG_HAP_ER.pdf. Published 2016. Accessed December 6, 2016.
7. Department of Veterans Affairs Pharmacy Benefits Management Services, Medical Advisory Panel, and VISN Pharmacist Executives. Direct oral anticoagulants (DOACs) (formerly called TSOACs) dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis): Criteria for Use for Stroke Prevention in nonvalvular atrial fibrillation (AF) and Edoxaban (SAVAYSA). http://www.pbm.va.gov/PBM/clinicalguidance/criteriaforuse/Anticoagulants_Direct_Oral_DOACs_CFU_and_Algorithm_for_Nonvalvular_Atrial_Fibrillation_Sep_2016.pdf. Updated September 2016. Accessed December 6, 2016.
8. Witt DM, Sadler MA, Shanahan RL, Mazzoli G, Tillman DJ. Effect of a centralized clinical pharmacy anticoagulation service on the outcomes of anticoagulation therapy. Chest. 2005;127(5):1515-1522.
9. Chan LL, Crumpler WL, Jacobson AK. Implementation of pharmacist-managed anticoagulation in patients receiving newer anticoagulants. Am J Health Syst Pharm. 2013;70(15):1285-1286, 1288.
10. Lee PY, Han SY, Miyahara RK. Adherence and outcomes of patients treated with dabigatran: pharmacist-managed anticoagulation clinic versus usual care. Am J Health Syst Pharm. 2013;70(13):1154-1161.
11. Shore S, Carey EP, Turakhia MP, et al. Adherence to dabigatran therapy and longitudinal patient outcomes: insights from the Veterans Health Administration. Am Heart J. 2014;167(6):810-817.
12. Martin BC, Wiley-Exley EK, Richards S, Domino ME, Carey TS, Sleath BL. Contrasting measures of adherence with simple drug use, medication switching and therapeutic duplication. Ann Pharmacother. 2009;43(1):36-44.
13. Shore S, Ho PM, Lambert-Kerzner A, et al. Site-level variation in and practices associated with dabigatran adherence. JAMA. 2015;313(14):1443-1450.
14. Pradaxa [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals; 2015.
Shingles Strikes With a Vengeance
1. A 16-year-old girl presents with a rash manifesting several weeks ago that became enlarged and more symptomatic after she applied hydrogen peroxide and scrubbed with antibacterial soap. Large, annular, honey-colored crusts are focally located around the left eye, and faint pinkness is noted peripherally around the lesions. Modest but palpable adenopathy is detected in the pretragal and submental nodal areas.
Diagnosis: Impetigo has also been called impetiginized dermatitis because it almost always starts with minor breaks in the skin as a result of conditions such as eczema, acne, contact dermatitis, or insect bite. Thus provided with access to deeper portions of the epithelial surface, bacterial organisms that normally cause no problems on intact skin are able to create the minor but annoying condition we call impetigo. Rarely associated with morbidity, it tends to resolve in two to three weeks at most, even without treatment.
For more information, see “Is It Ringworm, Herpes— Or Something Else Entirely?” Clinician Reviews. 2014;24(11):8-9.
2. A 38-year-old man presents with an itchy, blistery rash that usually appears in the summer, getting worse each year. The lesions are collections of vesicles with faint underlying erythema that crisscross his legs in linear configurations. Smaller but similar lesions are scattered over his arms and trunk.
Diagnosis: The rash produced by poison ivy exposure can be severe and can last six weeks or more without treatment. Poison ivy is not contagious, cannot be spread by scratching, and (despite its name) is not poisonous in any way. The number of poison ivy plants has doubled in the past 50 years and is expected to double again within 20 years. The potency of the plant’s allergen is also expected to increase. The patient (height, 6’3”; weight, > 300 lb) was treated with a 60-mg IM injection of triamcinolone, a two-week, 40-mg taper of prednisone, and twice-daily application of betamethasone cream. This, of course, followed a discussion of the risks versus benefits of such a course of action.
For more information, see “He Tried So Hard to Avoid It … .” Clinician Reviews. 2015 July;25(7):W2.
3. For several months, a 69-year-old woman has had a rash around her eyes. It is terribly symptomatic, burning and itching regardless of any type of OTC treatment. She finally requests referral to dermatology from her primary care provider.
Diagnosis: Eyelid dermatitis, or irritant contact dermatitis, is an extremely common complaint, and this patient’s history is quite typical: The worse the problem gets, the more attempts the patient makes to relieve symptoms.
When this patient presented to dermatology, she was applying six different products (all OTC) to the affected areas. None helped, and in fact, most seemed to worsen the problem. Even if one had helped, she would never have known which. But desperation drives patients to do irrational things, especially when the problem is out in the open for the whole world to see.
For more information, see “The Eyes Have It, and It Itches Like Crazy.” Clinician Reviews. 2015;25(9):W1.
4. A 58-year-old man seeks care for burning in his right eye and a skin eruption on his forehead and scalp with progressive worsening over the past 10 days. The patient has decreased vision in his right eye, as well as fever, chills, photophobia, and headache. A physical exam reveals vesicles on an erythematous base on his right scalp, forehead, upper and lower eyelids, dorsum of his nose, and cheek distributed along the ophthalmic branch of the trigeminal nerve.
Diagnosis: Herpes zoster ophthalmicus, confirmed by an ophthalmologic exam. This serious condition has been linked to reactivation of the varicella-zoster virus (VZV) within the trigeminal ganglion. Primary infection with VZV results in varicella (chickenpox), whereas reactivation of a latent VZV infection within the sensory ganglia is known as herpes zoster.
For more information, see “Painful rash on face.” J Fam Pract. 2015;64(11):E1-E3.
RELATED ARTICLE
Jacobsen E, Hull CE. “Herpes Zoster Infection.” Clinician Reviews. 2013;23(8):42-49.
1. A 16-year-old girl presents with a rash manifesting several weeks ago that became enlarged and more symptomatic after she applied hydrogen peroxide and scrubbed with antibacterial soap. Large, annular, honey-colored crusts are focally located around the left eye, and faint pinkness is noted peripherally around the lesions. Modest but palpable adenopathy is detected in the pretragal and submental nodal areas.
Diagnosis: Impetigo has also been called impetiginized dermatitis because it almost always starts with minor breaks in the skin as a result of conditions such as eczema, acne, contact dermatitis, or insect bite. Thus provided with access to deeper portions of the epithelial surface, bacterial organisms that normally cause no problems on intact skin are able to create the minor but annoying condition we call impetigo. Rarely associated with morbidity, it tends to resolve in two to three weeks at most, even without treatment.
For more information, see “Is It Ringworm, Herpes— Or Something Else Entirely?” Clinician Reviews. 2014;24(11):8-9.
2. A 38-year-old man presents with an itchy, blistery rash that usually appears in the summer, getting worse each year. The lesions are collections of vesicles with faint underlying erythema that crisscross his legs in linear configurations. Smaller but similar lesions are scattered over his arms and trunk.
Diagnosis: The rash produced by poison ivy exposure can be severe and can last six weeks or more without treatment. Poison ivy is not contagious, cannot be spread by scratching, and (despite its name) is not poisonous in any way. The number of poison ivy plants has doubled in the past 50 years and is expected to double again within 20 years. The potency of the plant’s allergen is also expected to increase. The patient (height, 6’3”; weight, > 300 lb) was treated with a 60-mg IM injection of triamcinolone, a two-week, 40-mg taper of prednisone, and twice-daily application of betamethasone cream. This, of course, followed a discussion of the risks versus benefits of such a course of action.
For more information, see “He Tried So Hard to Avoid It … .” Clinician Reviews. 2015 July;25(7):W2.
3. For several months, a 69-year-old woman has had a rash around her eyes. It is terribly symptomatic, burning and itching regardless of any type of OTC treatment. She finally requests referral to dermatology from her primary care provider.
Diagnosis: Eyelid dermatitis, or irritant contact dermatitis, is an extremely common complaint, and this patient’s history is quite typical: The worse the problem gets, the more attempts the patient makes to relieve symptoms.
When this patient presented to dermatology, she was applying six different products (all OTC) to the affected areas. None helped, and in fact, most seemed to worsen the problem. Even if one had helped, she would never have known which. But desperation drives patients to do irrational things, especially when the problem is out in the open for the whole world to see.
For more information, see “The Eyes Have It, and It Itches Like Crazy.” Clinician Reviews. 2015;25(9):W1.
4. A 58-year-old man seeks care for burning in his right eye and a skin eruption on his forehead and scalp with progressive worsening over the past 10 days. The patient has decreased vision in his right eye, as well as fever, chills, photophobia, and headache. A physical exam reveals vesicles on an erythematous base on his right scalp, forehead, upper and lower eyelids, dorsum of his nose, and cheek distributed along the ophthalmic branch of the trigeminal nerve.
Diagnosis: Herpes zoster ophthalmicus, confirmed by an ophthalmologic exam. This serious condition has been linked to reactivation of the varicella-zoster virus (VZV) within the trigeminal ganglion. Primary infection with VZV results in varicella (chickenpox), whereas reactivation of a latent VZV infection within the sensory ganglia is known as herpes zoster.
For more information, see “Painful rash on face.” J Fam Pract. 2015;64(11):E1-E3.
RELATED ARTICLE
Jacobsen E, Hull CE. “Herpes Zoster Infection.” Clinician Reviews. 2013;23(8):42-49.
1. A 16-year-old girl presents with a rash manifesting several weeks ago that became enlarged and more symptomatic after she applied hydrogen peroxide and scrubbed with antibacterial soap. Large, annular, honey-colored crusts are focally located around the left eye, and faint pinkness is noted peripherally around the lesions. Modest but palpable adenopathy is detected in the pretragal and submental nodal areas.
Diagnosis: Impetigo has also been called impetiginized dermatitis because it almost always starts with minor breaks in the skin as a result of conditions such as eczema, acne, contact dermatitis, or insect bite. Thus provided with access to deeper portions of the epithelial surface, bacterial organisms that normally cause no problems on intact skin are able to create the minor but annoying condition we call impetigo. Rarely associated with morbidity, it tends to resolve in two to three weeks at most, even without treatment.
For more information, see “Is It Ringworm, Herpes— Or Something Else Entirely?” Clinician Reviews. 2014;24(11):8-9.
2. A 38-year-old man presents with an itchy, blistery rash that usually appears in the summer, getting worse each year. The lesions are collections of vesicles with faint underlying erythema that crisscross his legs in linear configurations. Smaller but similar lesions are scattered over his arms and trunk.
Diagnosis: The rash produced by poison ivy exposure can be severe and can last six weeks or more without treatment. Poison ivy is not contagious, cannot be spread by scratching, and (despite its name) is not poisonous in any way. The number of poison ivy plants has doubled in the past 50 years and is expected to double again within 20 years. The potency of the plant’s allergen is also expected to increase. The patient (height, 6’3”; weight, > 300 lb) was treated with a 60-mg IM injection of triamcinolone, a two-week, 40-mg taper of prednisone, and twice-daily application of betamethasone cream. This, of course, followed a discussion of the risks versus benefits of such a course of action.
For more information, see “He Tried So Hard to Avoid It … .” Clinician Reviews. 2015 July;25(7):W2.
3. For several months, a 69-year-old woman has had a rash around her eyes. It is terribly symptomatic, burning and itching regardless of any type of OTC treatment. She finally requests referral to dermatology from her primary care provider.
Diagnosis: Eyelid dermatitis, or irritant contact dermatitis, is an extremely common complaint, and this patient’s history is quite typical: The worse the problem gets, the more attempts the patient makes to relieve symptoms.
When this patient presented to dermatology, she was applying six different products (all OTC) to the affected areas. None helped, and in fact, most seemed to worsen the problem. Even if one had helped, she would never have known which. But desperation drives patients to do irrational things, especially when the problem is out in the open for the whole world to see.
For more information, see “The Eyes Have It, and It Itches Like Crazy.” Clinician Reviews. 2015;25(9):W1.
4. A 58-year-old man seeks care for burning in his right eye and a skin eruption on his forehead and scalp with progressive worsening over the past 10 days. The patient has decreased vision in his right eye, as well as fever, chills, photophobia, and headache. A physical exam reveals vesicles on an erythematous base on his right scalp, forehead, upper and lower eyelids, dorsum of his nose, and cheek distributed along the ophthalmic branch of the trigeminal nerve.
Diagnosis: Herpes zoster ophthalmicus, confirmed by an ophthalmologic exam. This serious condition has been linked to reactivation of the varicella-zoster virus (VZV) within the trigeminal ganglion. Primary infection with VZV results in varicella (chickenpox), whereas reactivation of a latent VZV infection within the sensory ganglia is known as herpes zoster.
For more information, see “Painful rash on face.” J Fam Pract. 2015;64(11):E1-E3.
RELATED ARTICLE
Jacobsen E, Hull CE. “Herpes Zoster Infection.” Clinician Reviews. 2013;23(8):42-49.