User login
Common Hair Disorders
Review the PDF of the fact sheet on common hair disorders with board-relevant, easy-to-review material. This fact sheet reviews information about the most common hair disorders, including clinical and histopathological features, trichoscopy, and management of these diseases.
Practice Questions
1. A 40-year-old woman presents to the clinic with a burning sensation and tenderness on the scalp. At physical examination you notice erythematous papules and pustules on the vertex scalp. The most likely diagnosis is:
a. alopecia areata
b. CCSA
c. folliculitis decalvans
d. lichen planopilaris
e. traction alopecia
2. A 60-year-old woman presents with receding hair loss on the frontal and bitemporal scalp. She has noticed hair loss on her eyebrows. She has a history of oral ulcers. On physical examination there is mild erythema and perifollicular scales on the frontal hairline. A hair pull test is positive in this area. The most likely diagnosis is:
a. androgenetic alopecia
b. chronic cutaneous lupus erythematosus
c. frontal fibrosing alopecia
d. telogen effluvium
e. trichotillomania
3. A 5-year-old girl with a history of seasonal allergies and eczema presents with recurrent patchy hair loss on the scalp of 6 months’ duration. Her mother has noticed rapidly progressive hair loss affecting the whole scalp. On trichoscopy, you find yellow dots, broken hairs, and tapering hairs. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. telogen effluvium
d. traction alopecia
e. trichotillomania
4. A 30-year-old white woman with history of obsessive-compulsive disorder presents to the clinic with hair loss for the last 3 years. She says she has noticed worsening of the hair loss when she is under stress. She also bites her nails. On physical examination you identify an irregular patch of alopecia with broken hairs on the occipital scalp. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. lichen planopilaris
d. traction alopecia
e. trichotillomania
5. A 45-year-old black woman who has a family history of hair loss in her mother presents with tenderness and burning sensation on the vertex scalp. She reports the hair loss was worse after she got a hair relaxer 6 months prior. She uses braids on her scalp and she has not had a relaxer since then. The most likely diagnosis is:
a. CCSA
b. chronic cutaneous lupus erythematosus
c. folliculitis decalvans
d. lichen planopilaris
e. trichotillomania
Answers to practice questions provided on next page
Practice Question Answers
1. A 40-year-old woman presents to the clinic with a burning sensation and tenderness on the scalp. At physical examination you notice erythematous papules and pustules on the vertex scalp. The most likely diagnosis is:
a. alopecia areata
b. CCSA
c. folliculitis decalvans
d. lichen planopilaris
e. traction alopecia
2. A 60-year-old woman presents with receding hair loss on the frontal and bitemporal scalp. She has noticed hair loss on her eyebrows. She has a history of oral ulcers. On physical examination there is mild erythema and perifollicular scales on the frontal hairline. A hair pull test is positive in this area. The most likely diagnosis is:
a. androgenetic alopecia
b. chronic cutaneous lupus erythematosus
c. frontal fibrosing alopecia
d. telogen effluvium
e. trichotillomania
3. A 5-year-old girl with a history of seasonal allergies and eczema presents with recurrent patchy hair loss on the scalp of 6 months’ duration. Her mother has noticed rapidly progressive hair loss affecting the whole scalp. On trichoscopy, you find yellow dots, broken hairs, and tapering hairs. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. telogen effluvium
d. traction alopecia
e. trichotillomania
4. A 30-year-old white woman with history of obsessive-compulsive disorder presents to the clinic with hair loss for the last 3 years. She says she has noticed worsening of the hair loss when she is under stress. She also bites her nails. On physical examination you identify an irregular patch of alopecia with broken hairs on the occipital scalp. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. lichen planopilaris
d. traction alopecia
e. trichotillomania
5. A 45-year-old black woman who has a family history of hair loss in her mother presents with tenderness and burning sensation on the vertex scalp. She reports the hair loss was worse after she got a hair relaxer 6 months prior. She uses braids on her scalp and she has not had a relaxer since then. The most likely diagnosis is:
a. CCSA
b. chronic cutaneous lupus erythematosus
c. folliculitis decalvans
d. lichen planopilaris
e. trichotillomania
Review the PDF of the fact sheet on common hair disorders with board-relevant, easy-to-review material. This fact sheet reviews information about the most common hair disorders, including clinical and histopathological features, trichoscopy, and management of these diseases.
Practice Questions
1. A 40-year-old woman presents to the clinic with a burning sensation and tenderness on the scalp. At physical examination you notice erythematous papules and pustules on the vertex scalp. The most likely diagnosis is:
a. alopecia areata
b. CCSA
c. folliculitis decalvans
d. lichen planopilaris
e. traction alopecia
2. A 60-year-old woman presents with receding hair loss on the frontal and bitemporal scalp. She has noticed hair loss on her eyebrows. She has a history of oral ulcers. On physical examination there is mild erythema and perifollicular scales on the frontal hairline. A hair pull test is positive in this area. The most likely diagnosis is:
a. androgenetic alopecia
b. chronic cutaneous lupus erythematosus
c. frontal fibrosing alopecia
d. telogen effluvium
e. trichotillomania
3. A 5-year-old girl with a history of seasonal allergies and eczema presents with recurrent patchy hair loss on the scalp of 6 months’ duration. Her mother has noticed rapidly progressive hair loss affecting the whole scalp. On trichoscopy, you find yellow dots, broken hairs, and tapering hairs. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. telogen effluvium
d. traction alopecia
e. trichotillomania
4. A 30-year-old white woman with history of obsessive-compulsive disorder presents to the clinic with hair loss for the last 3 years. She says she has noticed worsening of the hair loss when she is under stress. She also bites her nails. On physical examination you identify an irregular patch of alopecia with broken hairs on the occipital scalp. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. lichen planopilaris
d. traction alopecia
e. trichotillomania
5. A 45-year-old black woman who has a family history of hair loss in her mother presents with tenderness and burning sensation on the vertex scalp. She reports the hair loss was worse after she got a hair relaxer 6 months prior. She uses braids on her scalp and she has not had a relaxer since then. The most likely diagnosis is:
a. CCSA
b. chronic cutaneous lupus erythematosus
c. folliculitis decalvans
d. lichen planopilaris
e. trichotillomania
Answers to practice questions provided on next page
Practice Question Answers
1. A 40-year-old woman presents to the clinic with a burning sensation and tenderness on the scalp. At physical examination you notice erythematous papules and pustules on the vertex scalp. The most likely diagnosis is:
a. alopecia areata
b. CCSA
c. folliculitis decalvans
d. lichen planopilaris
e. traction alopecia
2. A 60-year-old woman presents with receding hair loss on the frontal and bitemporal scalp. She has noticed hair loss on her eyebrows. She has a history of oral ulcers. On physical examination there is mild erythema and perifollicular scales on the frontal hairline. A hair pull test is positive in this area. The most likely diagnosis is:
a. androgenetic alopecia
b. chronic cutaneous lupus erythematosus
c. frontal fibrosing alopecia
d. telogen effluvium
e. trichotillomania
3. A 5-year-old girl with a history of seasonal allergies and eczema presents with recurrent patchy hair loss on the scalp of 6 months’ duration. Her mother has noticed rapidly progressive hair loss affecting the whole scalp. On trichoscopy, you find yellow dots, broken hairs, and tapering hairs. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. telogen effluvium
d. traction alopecia
e. trichotillomania
4. A 30-year-old white woman with history of obsessive-compulsive disorder presents to the clinic with hair loss for the last 3 years. She says she has noticed worsening of the hair loss when she is under stress. She also bites her nails. On physical examination you identify an irregular patch of alopecia with broken hairs on the occipital scalp. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. lichen planopilaris
d. traction alopecia
e. trichotillomania
5. A 45-year-old black woman who has a family history of hair loss in her mother presents with tenderness and burning sensation on the vertex scalp. She reports the hair loss was worse after she got a hair relaxer 6 months prior. She uses braids on her scalp and she has not had a relaxer since then. The most likely diagnosis is:
a. CCSA
b. chronic cutaneous lupus erythematosus
c. folliculitis decalvans
d. lichen planopilaris
e. trichotillomania
Review the PDF of the fact sheet on common hair disorders with board-relevant, easy-to-review material. This fact sheet reviews information about the most common hair disorders, including clinical and histopathological features, trichoscopy, and management of these diseases.
Practice Questions
1. A 40-year-old woman presents to the clinic with a burning sensation and tenderness on the scalp. At physical examination you notice erythematous papules and pustules on the vertex scalp. The most likely diagnosis is:
a. alopecia areata
b. CCSA
c. folliculitis decalvans
d. lichen planopilaris
e. traction alopecia
2. A 60-year-old woman presents with receding hair loss on the frontal and bitemporal scalp. She has noticed hair loss on her eyebrows. She has a history of oral ulcers. On physical examination there is mild erythema and perifollicular scales on the frontal hairline. A hair pull test is positive in this area. The most likely diagnosis is:
a. androgenetic alopecia
b. chronic cutaneous lupus erythematosus
c. frontal fibrosing alopecia
d. telogen effluvium
e. trichotillomania
3. A 5-year-old girl with a history of seasonal allergies and eczema presents with recurrent patchy hair loss on the scalp of 6 months’ duration. Her mother has noticed rapidly progressive hair loss affecting the whole scalp. On trichoscopy, you find yellow dots, broken hairs, and tapering hairs. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. telogen effluvium
d. traction alopecia
e. trichotillomania
4. A 30-year-old white woman with history of obsessive-compulsive disorder presents to the clinic with hair loss for the last 3 years. She says she has noticed worsening of the hair loss when she is under stress. She also bites her nails. On physical examination you identify an irregular patch of alopecia with broken hairs on the occipital scalp. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. lichen planopilaris
d. traction alopecia
e. trichotillomania
5. A 45-year-old black woman who has a family history of hair loss in her mother presents with tenderness and burning sensation on the vertex scalp. She reports the hair loss was worse after she got a hair relaxer 6 months prior. She uses braids on her scalp and she has not had a relaxer since then. The most likely diagnosis is:
a. CCSA
b. chronic cutaneous lupus erythematosus
c. folliculitis decalvans
d. lichen planopilaris
e. trichotillomania
Answers to practice questions provided on next page
Practice Question Answers
1. A 40-year-old woman presents to the clinic with a burning sensation and tenderness on the scalp. At physical examination you notice erythematous papules and pustules on the vertex scalp. The most likely diagnosis is:
a. alopecia areata
b. CCSA
c. folliculitis decalvans
d. lichen planopilaris
e. traction alopecia
2. A 60-year-old woman presents with receding hair loss on the frontal and bitemporal scalp. She has noticed hair loss on her eyebrows. She has a history of oral ulcers. On physical examination there is mild erythema and perifollicular scales on the frontal hairline. A hair pull test is positive in this area. The most likely diagnosis is:
a. androgenetic alopecia
b. chronic cutaneous lupus erythematosus
c. frontal fibrosing alopecia
d. telogen effluvium
e. trichotillomania
3. A 5-year-old girl with a history of seasonal allergies and eczema presents with recurrent patchy hair loss on the scalp of 6 months’ duration. Her mother has noticed rapidly progressive hair loss affecting the whole scalp. On trichoscopy, you find yellow dots, broken hairs, and tapering hairs. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. telogen effluvium
d. traction alopecia
e. trichotillomania
4. A 30-year-old white woman with history of obsessive-compulsive disorder presents to the clinic with hair loss for the last 3 years. She says she has noticed worsening of the hair loss when she is under stress. She also bites her nails. On physical examination you identify an irregular patch of alopecia with broken hairs on the occipital scalp. The most likely diagnosis is:
a. alopecia areata
b. androgenetic alopecia
c. lichen planopilaris
d. traction alopecia
e. trichotillomania
5. A 45-year-old black woman who has a family history of hair loss in her mother presents with tenderness and burning sensation on the vertex scalp. She reports the hair loss was worse after she got a hair relaxer 6 months prior. She uses braids on her scalp and she has not had a relaxer since then. The most likely diagnosis is:
a. CCSA
b. chronic cutaneous lupus erythematosus
c. folliculitis decalvans
d. lichen planopilaris
e. trichotillomania

Dermoscopy Update and Noninvasive Imaging Devices for Skin Cancer: Report From the Mount Sinai Winter Symposium
At the 19th Annual Mount Sinai Winter Symposium, Dr. Orit Markowitz provided an update on dermoscopy as a first-line noninvasive imaging modality for skin cancer screening and diagnosis along with reflectance confocal microscopy and dynamic optical coherence tomography. She explained how noninvasive imaging offers a more complete picture of lesions along with what is seen clinically and on pathology and discussed how it can help catch aggressive melanomas and other skin cancers at earlier stages. For these reasons, she emphasized that increased use of dermoscopy can be used to justify the need for regular skin cancer screenings. Finally, she discussed how noninvasive imaging can be used to guide dermatologists in performing optimal biposies of suspicious lesions.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
At the 19th Annual Mount Sinai Winter Symposium, Dr. Orit Markowitz provided an update on dermoscopy as a first-line noninvasive imaging modality for skin cancer screening and diagnosis along with reflectance confocal microscopy and dynamic optical coherence tomography. She explained how noninvasive imaging offers a more complete picture of lesions along with what is seen clinically and on pathology and discussed how it can help catch aggressive melanomas and other skin cancers at earlier stages. For these reasons, she emphasized that increased use of dermoscopy can be used to justify the need for regular skin cancer screenings. Finally, she discussed how noninvasive imaging can be used to guide dermatologists in performing optimal biposies of suspicious lesions.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
At the 19th Annual Mount Sinai Winter Symposium, Dr. Orit Markowitz provided an update on dermoscopy as a first-line noninvasive imaging modality for skin cancer screening and diagnosis along with reflectance confocal microscopy and dynamic optical coherence tomography. She explained how noninvasive imaging offers a more complete picture of lesions along with what is seen clinically and on pathology and discussed how it can help catch aggressive melanomas and other skin cancers at earlier stages. For these reasons, she emphasized that increased use of dermoscopy can be used to justify the need for regular skin cancer screenings. Finally, she discussed how noninvasive imaging can be used to guide dermatologists in performing optimal biposies of suspicious lesions.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Trump HHS nominee could curb regulations, reshape health insurance
Opinions are mixed on what the nominations of Rep. Tom Price (R-Ga.) as Secretary of Health & Human Services will mean for medicine and health care.
An orthopedic surgeon and six-term congressman, Dr. Price is an outspoken critic of the Affordable Care Act and has sponsored or cosponsored numerous bills to replace it. President-elect Trump called Rep. Price “a renowned physician” who has “earned a reputation for being a tireless problem solver and the go-to expert on health care policy,” according to a statement.
Not everyone agrees.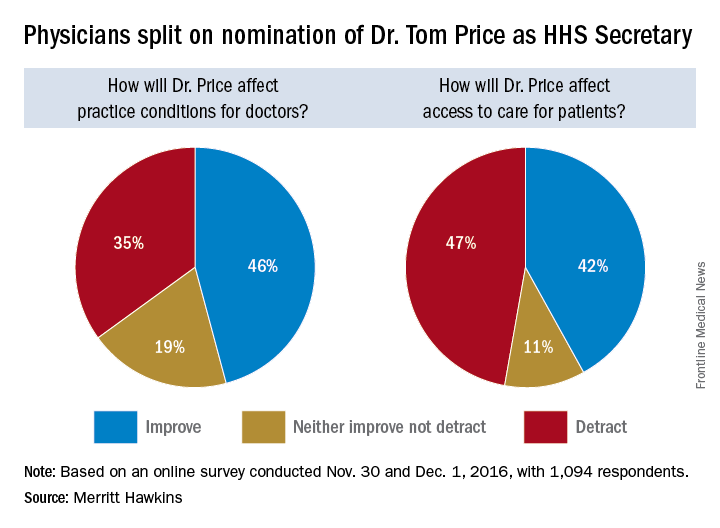
But Adam Gaffney, MD, a pulmonologist at the Cambridge (Mass.) Health Alliance, said physicians’ ability to care for their patients would be compromised if Rep. Price succeeds with many of his proposals, such as the privatization of Medicare and block grants for Medicaid.
“If these reforms go through, we’re going to see the insurance protections of our patients get worse,” said Dr. Gaffney, a board member for Physicians for a National Health Program, which advocates for a single-payer health care system. “If [his] agenda is successful, I think it’s going to have a detrimental impact on our ability to provide the care that our patients need.”
ACA repeal, malpractice reform
In the House, Rep. Price has introduced the Empowering Patients First Act, legislation, which would allow doctors to opt out of Medicare and enter into private contracts with Medicare patients. The bill is seen by many as a potential blueprint for Trump administration health reform. Rep. Price is also a proponent of malpractice reform that would make it tougher for patients to sue doctors and would lower liability insurance premiums.
The Empowering Patients First Act would repeal the ACA and offer tax credits for the purchase of individual and family health insurance policies. It would also create incentives for patients to contribute to health savings accounts, offer state grants to subsidize coverage for high-risk patients, and authorize businesses to cover members through association health plans.
The American Medical Association praised Rep. Price’s nomination, expressing support for ability to lead HHS.
“Dr. Price has been a leader in the development of health policies to advance patient choice and market-based solutions as well as reduce excessive regulatory burdens that diminish time devoted to patient care and increase costs,” AMA Board of Trustees Chair Patrice A. Harris, MD, said in a statement.
The American College of Surgeons' Executive Director, David B. Hoyt, MD, FACS, issued a supportive statement about the nomination of Dr. Price. "“Dr. Price is a stalwart champion for patients and their surgeons, and the ACS looks forward to working with him on key issues, such as the implementation of the Medicare Access and CHIP Reauthorization Act,” said Dr. Hoyt in a statement. “The ACS encourages the Senate to swiftly confirm Dr. Price’s nomination as Secretary of HHS."
But thousands of physicians disagree. Rep. Price’s proposals on Medicaid and Medicare threaten to harm vulnerable patients and limit access to healthcare, according to an open letter to the AMA published on Medium and credited to Clinician Action Network, a nonpartisan group that supports evidence-based policies. The group was started in opposition to the nomination of Rep. Price.
“We cannot support the dismantling of Medicaid, which has helped 15 million Americans gain health coverage since 2014,” the letter states. “We oppose Dr. Price’s proposals to reduce funding for the Children’s Health Insurance Program, a critical mechanism by which poor children access preventative care.”
Value-based payment or fee for service?
Rep. Price’s experience as a physician fuels his efforts to reduce burdensome regulations for doctors and enhance care efficiency, according to one of his predecessors, Louis W. Sullivan, MD. If confirmed, Rep. Price will become the third physician to be HHS secretary; Dr. Sullivan served in the George H.W. Bush administration and Otis R. Bowen, MD, served in the Reagan administration.
“He is very much aware of the challenges that physicians face in trying to delivery care,” said Dr. Sullivan. “I know that he’ll be working to reduce regulation when feasible so that the cost and delays that some regulatory issues present will hopefully be relieved,”
Some of those regulatory modifications could affect value-based care programs, Dr. Rodriguez said. Rep. Price has been critical of the move from fee for service to quality-based care and has opposed some corresponding programs, such as bundled payment initiatives. Rep. Price and members of the GOP Doctors Caucus wrote to Centers for Medicare & Medicaid Services in October to protest the regulations to implement the Medicare Access and CHIP Reauthorization Act of 2015 (MACRA) as too burdensome for smaller practices and calling for flexibility in quality reporting.
Rep. Price voted for passage of MACRA.
“He has been cautious about some of the changes that are being promoted in health care,” Dr. Rodriguez said. “He could slow that down – the processes being put in place. That might delay the impact those systems have in bringing about the improved quality that we want. [This would be] enormous, given the amount of work that we’ve been doing.”
A fair medical liability system also is a priority for Rep. Price, Dr. Sullivan said. His Empowering Patients First bill would require collaboration between HHS and physician associations to develop best practice guidelines that would provide a litigation safe harbor to physicians who practiced in accordance with the standards.
“I know that he will be working to develop strategies to reduce litigation in the health space,” Dr. Sullivan said in an interview. “That is one of the challenges that adds to health care costs, adds tension, and enhances an adversarial relationship between physicians and patients.”
But Dr. Gaffney said that he believes Rep. Price’s views on reproductive rights and gay marriage are regressive and that his agenda regarding health policy issues is bad for medicine.
“The overall [theme] of that agenda can be summed up as ‘take from the poor and sick and give to the rich,’ ” Dr. Gaffney said in an interview. “I think the financing of this [new health reform] system will be much more aggressive, and the result will be greater health care inequity.”
Rep. Price also has supported a ban on federal funding for Planned Parenthood, calling some of their practices barbaric. He has also voted to prohibit the importation of prescription drugs by nonsanctioned importers and has voted to repeal the medical device excise tax.
[email protected]
On Twitter @legal_med
Opinions are mixed on what the nominations of Rep. Tom Price (R-Ga.) as Secretary of Health & Human Services will mean for medicine and health care.
An orthopedic surgeon and six-term congressman, Dr. Price is an outspoken critic of the Affordable Care Act and has sponsored or cosponsored numerous bills to replace it. President-elect Trump called Rep. Price “a renowned physician” who has “earned a reputation for being a tireless problem solver and the go-to expert on health care policy,” according to a statement.
Not everyone agrees.
But Adam Gaffney, MD, a pulmonologist at the Cambridge (Mass.) Health Alliance, said physicians’ ability to care for their patients would be compromised if Rep. Price succeeds with many of his proposals, such as the privatization of Medicare and block grants for Medicaid.
“If these reforms go through, we’re going to see the insurance protections of our patients get worse,” said Dr. Gaffney, a board member for Physicians for a National Health Program, which advocates for a single-payer health care system. “If [his] agenda is successful, I think it’s going to have a detrimental impact on our ability to provide the care that our patients need.”
ACA repeal, malpractice reform
In the House, Rep. Price has introduced the Empowering Patients First Act, legislation, which would allow doctors to opt out of Medicare and enter into private contracts with Medicare patients. The bill is seen by many as a potential blueprint for Trump administration health reform. Rep. Price is also a proponent of malpractice reform that would make it tougher for patients to sue doctors and would lower liability insurance premiums.
The Empowering Patients First Act would repeal the ACA and offer tax credits for the purchase of individual and family health insurance policies. It would also create incentives for patients to contribute to health savings accounts, offer state grants to subsidize coverage for high-risk patients, and authorize businesses to cover members through association health plans.
The American Medical Association praised Rep. Price’s nomination, expressing support for ability to lead HHS.
“Dr. Price has been a leader in the development of health policies to advance patient choice and market-based solutions as well as reduce excessive regulatory burdens that diminish time devoted to patient care and increase costs,” AMA Board of Trustees Chair Patrice A. Harris, MD, said in a statement.
The American College of Surgeons' Executive Director, David B. Hoyt, MD, FACS, issued a supportive statement about the nomination of Dr. Price. "“Dr. Price is a stalwart champion for patients and their surgeons, and the ACS looks forward to working with him on key issues, such as the implementation of the Medicare Access and CHIP Reauthorization Act,” said Dr. Hoyt in a statement. “The ACS encourages the Senate to swiftly confirm Dr. Price’s nomination as Secretary of HHS."
But thousands of physicians disagree. Rep. Price’s proposals on Medicaid and Medicare threaten to harm vulnerable patients and limit access to healthcare, according to an open letter to the AMA published on Medium and credited to Clinician Action Network, a nonpartisan group that supports evidence-based policies. The group was started in opposition to the nomination of Rep. Price.
“We cannot support the dismantling of Medicaid, which has helped 15 million Americans gain health coverage since 2014,” the letter states. “We oppose Dr. Price’s proposals to reduce funding for the Children’s Health Insurance Program, a critical mechanism by which poor children access preventative care.”
Value-based payment or fee for service?
Rep. Price’s experience as a physician fuels his efforts to reduce burdensome regulations for doctors and enhance care efficiency, according to one of his predecessors, Louis W. Sullivan, MD. If confirmed, Rep. Price will become the third physician to be HHS secretary; Dr. Sullivan served in the George H.W. Bush administration and Otis R. Bowen, MD, served in the Reagan administration.
“He is very much aware of the challenges that physicians face in trying to delivery care,” said Dr. Sullivan. “I know that he’ll be working to reduce regulation when feasible so that the cost and delays that some regulatory issues present will hopefully be relieved,”
Some of those regulatory modifications could affect value-based care programs, Dr. Rodriguez said. Rep. Price has been critical of the move from fee for service to quality-based care and has opposed some corresponding programs, such as bundled payment initiatives. Rep. Price and members of the GOP Doctors Caucus wrote to Centers for Medicare & Medicaid Services in October to protest the regulations to implement the Medicare Access and CHIP Reauthorization Act of 2015 (MACRA) as too burdensome for smaller practices and calling for flexibility in quality reporting.
Rep. Price voted for passage of MACRA.
“He has been cautious about some of the changes that are being promoted in health care,” Dr. Rodriguez said. “He could slow that down – the processes being put in place. That might delay the impact those systems have in bringing about the improved quality that we want. [This would be] enormous, given the amount of work that we’ve been doing.”
A fair medical liability system also is a priority for Rep. Price, Dr. Sullivan said. His Empowering Patients First bill would require collaboration between HHS and physician associations to develop best practice guidelines that would provide a litigation safe harbor to physicians who practiced in accordance with the standards.
“I know that he will be working to develop strategies to reduce litigation in the health space,” Dr. Sullivan said in an interview. “That is one of the challenges that adds to health care costs, adds tension, and enhances an adversarial relationship between physicians and patients.”
But Dr. Gaffney said that he believes Rep. Price’s views on reproductive rights and gay marriage are regressive and that his agenda regarding health policy issues is bad for medicine.
“The overall [theme] of that agenda can be summed up as ‘take from the poor and sick and give to the rich,’ ” Dr. Gaffney said in an interview. “I think the financing of this [new health reform] system will be much more aggressive, and the result will be greater health care inequity.”
Rep. Price also has supported a ban on federal funding for Planned Parenthood, calling some of their practices barbaric. He has also voted to prohibit the importation of prescription drugs by nonsanctioned importers and has voted to repeal the medical device excise tax.
[email protected]
On Twitter @legal_med
Opinions are mixed on what the nominations of Rep. Tom Price (R-Ga.) as Secretary of Health & Human Services will mean for medicine and health care.
An orthopedic surgeon and six-term congressman, Dr. Price is an outspoken critic of the Affordable Care Act and has sponsored or cosponsored numerous bills to replace it. President-elect Trump called Rep. Price “a renowned physician” who has “earned a reputation for being a tireless problem solver and the go-to expert on health care policy,” according to a statement.
Not everyone agrees.
But Adam Gaffney, MD, a pulmonologist at the Cambridge (Mass.) Health Alliance, said physicians’ ability to care for their patients would be compromised if Rep. Price succeeds with many of his proposals, such as the privatization of Medicare and block grants for Medicaid.
“If these reforms go through, we’re going to see the insurance protections of our patients get worse,” said Dr. Gaffney, a board member for Physicians for a National Health Program, which advocates for a single-payer health care system. “If [his] agenda is successful, I think it’s going to have a detrimental impact on our ability to provide the care that our patients need.”
ACA repeal, malpractice reform
In the House, Rep. Price has introduced the Empowering Patients First Act, legislation, which would allow doctors to opt out of Medicare and enter into private contracts with Medicare patients. The bill is seen by many as a potential blueprint for Trump administration health reform. Rep. Price is also a proponent of malpractice reform that would make it tougher for patients to sue doctors and would lower liability insurance premiums.
The Empowering Patients First Act would repeal the ACA and offer tax credits for the purchase of individual and family health insurance policies. It would also create incentives for patients to contribute to health savings accounts, offer state grants to subsidize coverage for high-risk patients, and authorize businesses to cover members through association health plans.
The American Medical Association praised Rep. Price’s nomination, expressing support for ability to lead HHS.
“Dr. Price has been a leader in the development of health policies to advance patient choice and market-based solutions as well as reduce excessive regulatory burdens that diminish time devoted to patient care and increase costs,” AMA Board of Trustees Chair Patrice A. Harris, MD, said in a statement.
The American College of Surgeons' Executive Director, David B. Hoyt, MD, FACS, issued a supportive statement about the nomination of Dr. Price. "“Dr. Price is a stalwart champion for patients and their surgeons, and the ACS looks forward to working with him on key issues, such as the implementation of the Medicare Access and CHIP Reauthorization Act,” said Dr. Hoyt in a statement. “The ACS encourages the Senate to swiftly confirm Dr. Price’s nomination as Secretary of HHS."
But thousands of physicians disagree. Rep. Price’s proposals on Medicaid and Medicare threaten to harm vulnerable patients and limit access to healthcare, according to an open letter to the AMA published on Medium and credited to Clinician Action Network, a nonpartisan group that supports evidence-based policies. The group was started in opposition to the nomination of Rep. Price.
“We cannot support the dismantling of Medicaid, which has helped 15 million Americans gain health coverage since 2014,” the letter states. “We oppose Dr. Price’s proposals to reduce funding for the Children’s Health Insurance Program, a critical mechanism by which poor children access preventative care.”
Value-based payment or fee for service?
Rep. Price’s experience as a physician fuels his efforts to reduce burdensome regulations for doctors and enhance care efficiency, according to one of his predecessors, Louis W. Sullivan, MD. If confirmed, Rep. Price will become the third physician to be HHS secretary; Dr. Sullivan served in the George H.W. Bush administration and Otis R. Bowen, MD, served in the Reagan administration.
“He is very much aware of the challenges that physicians face in trying to delivery care,” said Dr. Sullivan. “I know that he’ll be working to reduce regulation when feasible so that the cost and delays that some regulatory issues present will hopefully be relieved,”
Some of those regulatory modifications could affect value-based care programs, Dr. Rodriguez said. Rep. Price has been critical of the move from fee for service to quality-based care and has opposed some corresponding programs, such as bundled payment initiatives. Rep. Price and members of the GOP Doctors Caucus wrote to Centers for Medicare & Medicaid Services in October to protest the regulations to implement the Medicare Access and CHIP Reauthorization Act of 2015 (MACRA) as too burdensome for smaller practices and calling for flexibility in quality reporting.
Rep. Price voted for passage of MACRA.
“He has been cautious about some of the changes that are being promoted in health care,” Dr. Rodriguez said. “He could slow that down – the processes being put in place. That might delay the impact those systems have in bringing about the improved quality that we want. [This would be] enormous, given the amount of work that we’ve been doing.”
A fair medical liability system also is a priority for Rep. Price, Dr. Sullivan said. His Empowering Patients First bill would require collaboration between HHS and physician associations to develop best practice guidelines that would provide a litigation safe harbor to physicians who practiced in accordance with the standards.
“I know that he will be working to develop strategies to reduce litigation in the health space,” Dr. Sullivan said in an interview. “That is one of the challenges that adds to health care costs, adds tension, and enhances an adversarial relationship between physicians and patients.”
But Dr. Gaffney said that he believes Rep. Price’s views on reproductive rights and gay marriage are regressive and that his agenda regarding health policy issues is bad for medicine.
“The overall [theme] of that agenda can be summed up as ‘take from the poor and sick and give to the rich,’ ” Dr. Gaffney said in an interview. “I think the financing of this [new health reform] system will be much more aggressive, and the result will be greater health care inequity.”
Rep. Price also has supported a ban on federal funding for Planned Parenthood, calling some of their practices barbaric. He has also voted to prohibit the importation of prescription drugs by nonsanctioned importers and has voted to repeal the medical device excise tax.
[email protected]
On Twitter @legal_med
Dermoscopy Pearls: Report From the Mount Sinai Winter Symposium
At the 19th Annual Mount Sinai Winter Symposium, Dr. Orit Markowitz addressed some common questions physicians have about dermoscopy, including what kind of dermatoscope to buy, how to incorporate dermoscopy into a dermatology practice, and how to efficiently perform skin examinations using a dermatoscope. She also emphasized the importance of attending courses and workshops to learn how to utilize dermoscopy and other noninvasive imaging devices effectively.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
At the 19th Annual Mount Sinai Winter Symposium, Dr. Orit Markowitz addressed some common questions physicians have about dermoscopy, including what kind of dermatoscope to buy, how to incorporate dermoscopy into a dermatology practice, and how to efficiently perform skin examinations using a dermatoscope. She also emphasized the importance of attending courses and workshops to learn how to utilize dermoscopy and other noninvasive imaging devices effectively.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
At the 19th Annual Mount Sinai Winter Symposium, Dr. Orit Markowitz addressed some common questions physicians have about dermoscopy, including what kind of dermatoscope to buy, how to incorporate dermoscopy into a dermatology practice, and how to efficiently perform skin examinations using a dermatoscope. She also emphasized the importance of attending courses and workshops to learn how to utilize dermoscopy and other noninvasive imaging devices effectively.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Aspirin use linked to increased ICH in trauma patients
WAIKOLOA, HAWAII – Among a group of anticoagulated trauma patients, those on aspirin had the highest rate and risk of intracranial hemorrhage (ICH), while those on novel oral anticoagulants were not at higher risk for ICH, ICH progression, or death, a multicenter study found.
“The number of patients on warfarin and antiplatelet agents has significantly increased over time,” Leslie Kobayashi, MD, said at the annual meeting of the American Association for the Surgery of Trauma. “These oral antithrombotic agents have been associated with poor outcomes following traumatic injury, including increased rates of intracranial hemorrhage, increased progression of intracranial hemorrhage, and increased mortality.”

In a prospective, multicenter observational study conducted by the AAST’s Multi-institutional Trials Committee, Dr. Kobayashi and her associates set out identify injury patterns and outcomes in trauma patients taking the NOAs, and to test their hypothesis that patients taking NOAs would have higher rates of ICH, ICH progression, and death, compared with patients taking traditional oral anticoagulant therapies (OATs). Patients were included if they were admitted to the trauma service on warfarin, aspirin, clopidogrel, dabigatran, apixaban, or rivaroxaban. Pregnant patients, prisoners, and minors were excluded from the study. Data collected included demographics, mechanism of injury, vitals on admission, injuries/injury severity scores, labs, interventions, and reversal agents used such as vitamin K, prothrombin complexes, dialysis, and transfusion of fresh frozen plasma (FFP). Outcomes studied included ICH, ICH progression, and death.
In all, 16 Level 1 trauma centers enrolled 1,847 patients over a 2-year period. Their average age was 75 years, 46% were female, 77% were white, their median Injury Severity Score (ISS) was 9, and 99% sustained a blunt mechanism of trauma. The top two causes of injury were falls (71%) and motor vehicle crashes (15%). One-third of patients (33%) were on warfarin, while the remainder were on aspirin (26%), clopidogrel (24%), NOAs (10%), and 7% took multiple or other agents.
The mechanism of injury pattern was similar between patients taking NOAs and those taking OATs, with the exception of patients on aspirin being significantly less likely to have sustained a fall. Patients on aspirin also had a significantly higher median ISS. “Patients on NOAs presented more frequently in shock as defined by a systolic blood pressure of less than 90 mmHg, but this was not associated with increased need for packed red blood cell transfusion, bleeding requiring an intervention, need for surgical procedure, hospital LOS, complications, or death,” Dr. Kobayashi said.
About 30% of all patients studied underwent an attempt at reversal. The types of agents used to reverse the patients differed depending on drug agent, with antiplatelet patients more frequently getting platelets, and patients on warfarin more frequently receiving FFP, vitamin K, and prothrombin complex. “Interestingly, patients on the anti-Xa inhibitors more frequently received prothrombin complex as well,” she said. “This likely reflects some of the recent literature which suggests that there may be a therapeutic benefit to using prothrombin complex in patients taking the oral anti-Xa inhibitors but not in patients on dabigatran.”
Overall, bleeding, need for surgical procedure, need for neurosurgical procedure, complications, length of stay, and death were similar between those on NOAs and those on OATs. However, the rate of ICH was significantly higher in patients on aspirin. “What is even more surprising is that 89% of the patients in the aspirin-only group were on an 81-mg baby aspirin rather than the larger 325-mg dose,” Dr. Kobayashi said. This difference was significant on univariate analysis and was retained after multivariate logistic regression adjusted for differences between populations, with an OR for aspirin of 1.7 and a P value of .024. “This is not to suggest that patients on aspirin are doing markedly worse, compared to their counterparts, but I think most of us would have assumed that aspirin patients would have done better,” she commented. “I think we’ve definitively shown that is not the case.” Other independent predictors of ICH were advanced age (OR, 1.02), Asian race (OR, 3.1), ISS of 10 or greater (OR, 2.2), and a Glasgow coma score (GCS) of 8 or less (OR, 5.6).
Despite their increased risk for ICH, patients on aspirin were significantly less likely to undergo an attempt at reversal with any type of agent, at 16% with a P value of less than .001, on univariate analysis. “This was significantly lower than all other medications and was retained after multivariate logistic regression, with an OR of 0.3 and a P value of less than .001,” she said.
Progression of ICH did not differ by medication group. Other independent predictors included intraparenchymal location of hemorrhage (OR, 2.2), need for a neurosurgical procedure (OR, 5.1), an attempt at reversal (OR, 2.3) and a GCS of 8 or lower at admission (OR, 4.3). Similarly, multivariate analysis of death showed no significant differences between the different medication groups. Independent predictors included advanced age (OR, 1.06), GCS of 8 or less (OR, 13), progression of head injury (OR, 10), bleeding (OR, 2.3), and complications (OR, 2.1).
Dr. Kobayashi acknowledged that the study’s observational design is a limitation, as well as the fact that it lacked a control group of age-matched patients who were not taking anticoagulants. “Additionally, we had a relatively low number of patients on NOAs, at only 10% of the study population,” she said. “Lastly, there is potential for enrollment bias as all sites involved in this study were level one trauma centers.” She reported having no financial disclosures.
WAIKOLOA, HAWAII – Among a group of anticoagulated trauma patients, those on aspirin had the highest rate and risk of intracranial hemorrhage (ICH), while those on novel oral anticoagulants were not at higher risk for ICH, ICH progression, or death, a multicenter study found.
“The number of patients on warfarin and antiplatelet agents has significantly increased over time,” Leslie Kobayashi, MD, said at the annual meeting of the American Association for the Surgery of Trauma. “These oral antithrombotic agents have been associated with poor outcomes following traumatic injury, including increased rates of intracranial hemorrhage, increased progression of intracranial hemorrhage, and increased mortality.”
In a prospective, multicenter observational study conducted by the AAST’s Multi-institutional Trials Committee, Dr. Kobayashi and her associates set out identify injury patterns and outcomes in trauma patients taking the NOAs, and to test their hypothesis that patients taking NOAs would have higher rates of ICH, ICH progression, and death, compared with patients taking traditional oral anticoagulant therapies (OATs). Patients were included if they were admitted to the trauma service on warfarin, aspirin, clopidogrel, dabigatran, apixaban, or rivaroxaban. Pregnant patients, prisoners, and minors were excluded from the study. Data collected included demographics, mechanism of injury, vitals on admission, injuries/injury severity scores, labs, interventions, and reversal agents used such as vitamin K, prothrombin complexes, dialysis, and transfusion of fresh frozen plasma (FFP). Outcomes studied included ICH, ICH progression, and death.
In all, 16 Level 1 trauma centers enrolled 1,847 patients over a 2-year period. Their average age was 75 years, 46% were female, 77% were white, their median Injury Severity Score (ISS) was 9, and 99% sustained a blunt mechanism of trauma. The top two causes of injury were falls (71%) and motor vehicle crashes (15%). One-third of patients (33%) were on warfarin, while the remainder were on aspirin (26%), clopidogrel (24%), NOAs (10%), and 7% took multiple or other agents.
The mechanism of injury pattern was similar between patients taking NOAs and those taking OATs, with the exception of patients on aspirin being significantly less likely to have sustained a fall. Patients on aspirin also had a significantly higher median ISS. “Patients on NOAs presented more frequently in shock as defined by a systolic blood pressure of less than 90 mmHg, but this was not associated with increased need for packed red blood cell transfusion, bleeding requiring an intervention, need for surgical procedure, hospital LOS, complications, or death,” Dr. Kobayashi said.
About 30% of all patients studied underwent an attempt at reversal. The types of agents used to reverse the patients differed depending on drug agent, with antiplatelet patients more frequently getting platelets, and patients on warfarin more frequently receiving FFP, vitamin K, and prothrombin complex. “Interestingly, patients on the anti-Xa inhibitors more frequently received prothrombin complex as well,” she said. “This likely reflects some of the recent literature which suggests that there may be a therapeutic benefit to using prothrombin complex in patients taking the oral anti-Xa inhibitors but not in patients on dabigatran.”
Overall, bleeding, need for surgical procedure, need for neurosurgical procedure, complications, length of stay, and death were similar between those on NOAs and those on OATs. However, the rate of ICH was significantly higher in patients on aspirin. “What is even more surprising is that 89% of the patients in the aspirin-only group were on an 81-mg baby aspirin rather than the larger 325-mg dose,” Dr. Kobayashi said. This difference was significant on univariate analysis and was retained after multivariate logistic regression adjusted for differences between populations, with an OR for aspirin of 1.7 and a P value of .024. “This is not to suggest that patients on aspirin are doing markedly worse, compared to their counterparts, but I think most of us would have assumed that aspirin patients would have done better,” she commented. “I think we’ve definitively shown that is not the case.” Other independent predictors of ICH were advanced age (OR, 1.02), Asian race (OR, 3.1), ISS of 10 or greater (OR, 2.2), and a Glasgow coma score (GCS) of 8 or less (OR, 5.6).
Despite their increased risk for ICH, patients on aspirin were significantly less likely to undergo an attempt at reversal with any type of agent, at 16% with a P value of less than .001, on univariate analysis. “This was significantly lower than all other medications and was retained after multivariate logistic regression, with an OR of 0.3 and a P value of less than .001,” she said.
Progression of ICH did not differ by medication group. Other independent predictors included intraparenchymal location of hemorrhage (OR, 2.2), need for a neurosurgical procedure (OR, 5.1), an attempt at reversal (OR, 2.3) and a GCS of 8 or lower at admission (OR, 4.3). Similarly, multivariate analysis of death showed no significant differences between the different medication groups. Independent predictors included advanced age (OR, 1.06), GCS of 8 or less (OR, 13), progression of head injury (OR, 10), bleeding (OR, 2.3), and complications (OR, 2.1).
Dr. Kobayashi acknowledged that the study’s observational design is a limitation, as well as the fact that it lacked a control group of age-matched patients who were not taking anticoagulants. “Additionally, we had a relatively low number of patients on NOAs, at only 10% of the study population,” she said. “Lastly, there is potential for enrollment bias as all sites involved in this study were level one trauma centers.” She reported having no financial disclosures.
WAIKOLOA, HAWAII – Among a group of anticoagulated trauma patients, those on aspirin had the highest rate and risk of intracranial hemorrhage (ICH), while those on novel oral anticoagulants were not at higher risk for ICH, ICH progression, or death, a multicenter study found.
“The number of patients on warfarin and antiplatelet agents has significantly increased over time,” Leslie Kobayashi, MD, said at the annual meeting of the American Association for the Surgery of Trauma. “These oral antithrombotic agents have been associated with poor outcomes following traumatic injury, including increased rates of intracranial hemorrhage, increased progression of intracranial hemorrhage, and increased mortality.”
In a prospective, multicenter observational study conducted by the AAST’s Multi-institutional Trials Committee, Dr. Kobayashi and her associates set out identify injury patterns and outcomes in trauma patients taking the NOAs, and to test their hypothesis that patients taking NOAs would have higher rates of ICH, ICH progression, and death, compared with patients taking traditional oral anticoagulant therapies (OATs). Patients were included if they were admitted to the trauma service on warfarin, aspirin, clopidogrel, dabigatran, apixaban, or rivaroxaban. Pregnant patients, prisoners, and minors were excluded from the study. Data collected included demographics, mechanism of injury, vitals on admission, injuries/injury severity scores, labs, interventions, and reversal agents used such as vitamin K, prothrombin complexes, dialysis, and transfusion of fresh frozen plasma (FFP). Outcomes studied included ICH, ICH progression, and death.
In all, 16 Level 1 trauma centers enrolled 1,847 patients over a 2-year period. Their average age was 75 years, 46% were female, 77% were white, their median Injury Severity Score (ISS) was 9, and 99% sustained a blunt mechanism of trauma. The top two causes of injury were falls (71%) and motor vehicle crashes (15%). One-third of patients (33%) were on warfarin, while the remainder were on aspirin (26%), clopidogrel (24%), NOAs (10%), and 7% took multiple or other agents.
The mechanism of injury pattern was similar between patients taking NOAs and those taking OATs, with the exception of patients on aspirin being significantly less likely to have sustained a fall. Patients on aspirin also had a significantly higher median ISS. “Patients on NOAs presented more frequently in shock as defined by a systolic blood pressure of less than 90 mmHg, but this was not associated with increased need for packed red blood cell transfusion, bleeding requiring an intervention, need for surgical procedure, hospital LOS, complications, or death,” Dr. Kobayashi said.
About 30% of all patients studied underwent an attempt at reversal. The types of agents used to reverse the patients differed depending on drug agent, with antiplatelet patients more frequently getting platelets, and patients on warfarin more frequently receiving FFP, vitamin K, and prothrombin complex. “Interestingly, patients on the anti-Xa inhibitors more frequently received prothrombin complex as well,” she said. “This likely reflects some of the recent literature which suggests that there may be a therapeutic benefit to using prothrombin complex in patients taking the oral anti-Xa inhibitors but not in patients on dabigatran.”
Overall, bleeding, need for surgical procedure, need for neurosurgical procedure, complications, length of stay, and death were similar between those on NOAs and those on OATs. However, the rate of ICH was significantly higher in patients on aspirin. “What is even more surprising is that 89% of the patients in the aspirin-only group were on an 81-mg baby aspirin rather than the larger 325-mg dose,” Dr. Kobayashi said. This difference was significant on univariate analysis and was retained after multivariate logistic regression adjusted for differences between populations, with an OR for aspirin of 1.7 and a P value of .024. “This is not to suggest that patients on aspirin are doing markedly worse, compared to their counterparts, but I think most of us would have assumed that aspirin patients would have done better,” she commented. “I think we’ve definitively shown that is not the case.” Other independent predictors of ICH were advanced age (OR, 1.02), Asian race (OR, 3.1), ISS of 10 or greater (OR, 2.2), and a Glasgow coma score (GCS) of 8 or less (OR, 5.6).
Despite their increased risk for ICH, patients on aspirin were significantly less likely to undergo an attempt at reversal with any type of agent, at 16% with a P value of less than .001, on univariate analysis. “This was significantly lower than all other medications and was retained after multivariate logistic regression, with an OR of 0.3 and a P value of less than .001,” she said.
Progression of ICH did not differ by medication group. Other independent predictors included intraparenchymal location of hemorrhage (OR, 2.2), need for a neurosurgical procedure (OR, 5.1), an attempt at reversal (OR, 2.3) and a GCS of 8 or lower at admission (OR, 4.3). Similarly, multivariate analysis of death showed no significant differences between the different medication groups. Independent predictors included advanced age (OR, 1.06), GCS of 8 or less (OR, 13), progression of head injury (OR, 10), bleeding (OR, 2.3), and complications (OR, 2.1).
Dr. Kobayashi acknowledged that the study’s observational design is a limitation, as well as the fact that it lacked a control group of age-matched patients who were not taking anticoagulants. “Additionally, we had a relatively low number of patients on NOAs, at only 10% of the study population,” she said. “Lastly, there is potential for enrollment bias as all sites involved in this study were level one trauma centers.” She reported having no financial disclosures.
AT THE AAST ANNUAL MEETING
Key clinical point:
Major finding: The rate of ICH was significantly higher in patients on aspirin, compared with those on novel oral anticoagulant therapies (OR, 1.7; P = .024).
Data source: A prospective evaluation of 1,847 patients treated at 16 level one trauma centers over a 2-year period.
Disclosures: Dr. Kobayashi reported having no financial disclosures.
HIV research update: Early November 2016
A great volume of HIV and AIDS research enters the medical literature every month. It’s difficult to monitor everything, so here’s a quick look at some notable news items and journal articles published over the past few weeks.
HIV-infected U.S. smokers aged 40 years lose more than 6 years of life expectancy from smoking, according to a computer simulation study, possibly outweighing the loss from HIV infection itself.
A study published in JAIDS provides insight into a novel mechanism of ritonavir-induced insulin resistance involving proinflammatory properties of heme oxygenase-1 (HO-1).
Antiretroviral therapy with efavirenz, emtricitabine, and tenofovir disoproxil fumarate (TDF) appeared to have a more favorable renal safety profile than did TDF administered with a protease inhibitor or cobicistat, according to a study in HIV Clinical Trials.
Researchers said the first study to describe the dynamics of the biomarker of cerebrospinal fluid YKL-40 in two groups of HIV-infected individuals, before and after combination antiretroviral therapy, has demonstrated the value of the marker in understanding HIV neuropathogenesis.
A study of HIV testing among ethnic minority adolescents in New Jersey underscored the importance of developing multifaceted HIV/AIDS prevention protocols that provide direct education and skill-building activities, leverage peer education as a means to disseminate health-related information, and deliver broad-based prevention messaging that is culturally tailored and gender specific.
HIV-infected adults in a contemporary, high-resource setting have poor dietary patterns, a recent study found, and alcohol use was associated with worse gut integrity and increased inflammation, while other aspects of diet were not.
A study in BMC Infectious Diseases found immune recovery comparable in primary and chronic HIV infection, whereas differences in absolute counts and proportions of CD4+ T cell subpopulations were found between primary HIV infection and late presenters supporting early initiation of combination antiretroviral therapy.
HIV/HCV coinfection leads to a significant increase in plasma HCV RNA, according to a study in Pathogens and Global Health.
A Canadian study identified elevated rates of intentional and unintentional injury among people living with HIV.
Partner services for persons with acute and early HIV infection (AEH) within 30 days of diagnosis represents an effective tool to find HIV unaware persons, a recent study found, including those with AEH who are at greatest risk of HIV transmission.
Maternal tenofovir use was not associated with lower length or head circumference of HIV-exposed uninfected infants at 2 years of age, a recent study revealed, but may be related to greater weight among those exposed to combination antiretroviral therapy early in pregnancy.
Cryptococcal meningitis may be considered one of the causes of acute vision loss in pregnant/postpartum HIV-positive females, according to a study in BMC Infectious Diseases.
According to a study in JAIDS, red cell distribution width remains a powerful marker of cardiovascular disease in the context of the inflammatory milieu that accompanies HIV infection.
HIV infection moderates the association between vascular remodeling and neurocognitive function but not the association between pulse pressure and neurocognitive function, a recent study revealed.
HIV viral blips on therapy are associated with subsequent viral rebound on stopping antiretroviral therapy among individuals treated in primary HIV infection, according to a new study.
Adipocytokine dysregulation seems to be related to metabolic syndrome in HIV-infected children, according to a study in the Pediatric Infectious Disease Journal.
[email protected]
On Twitter @richpizzi
A great volume of HIV and AIDS research enters the medical literature every month. It’s difficult to monitor everything, so here’s a quick look at some notable news items and journal articles published over the past few weeks.
HIV-infected U.S. smokers aged 40 years lose more than 6 years of life expectancy from smoking, according to a computer simulation study, possibly outweighing the loss from HIV infection itself.
A study published in JAIDS provides insight into a novel mechanism of ritonavir-induced insulin resistance involving proinflammatory properties of heme oxygenase-1 (HO-1).
Antiretroviral therapy with efavirenz, emtricitabine, and tenofovir disoproxil fumarate (TDF) appeared to have a more favorable renal safety profile than did TDF administered with a protease inhibitor or cobicistat, according to a study in HIV Clinical Trials.
Researchers said the first study to describe the dynamics of the biomarker of cerebrospinal fluid YKL-40 in two groups of HIV-infected individuals, before and after combination antiretroviral therapy, has demonstrated the value of the marker in understanding HIV neuropathogenesis.
A study of HIV testing among ethnic minority adolescents in New Jersey underscored the importance of developing multifaceted HIV/AIDS prevention protocols that provide direct education and skill-building activities, leverage peer education as a means to disseminate health-related information, and deliver broad-based prevention messaging that is culturally tailored and gender specific.
HIV-infected adults in a contemporary, high-resource setting have poor dietary patterns, a recent study found, and alcohol use was associated with worse gut integrity and increased inflammation, while other aspects of diet were not.
A study in BMC Infectious Diseases found immune recovery comparable in primary and chronic HIV infection, whereas differences in absolute counts and proportions of CD4+ T cell subpopulations were found between primary HIV infection and late presenters supporting early initiation of combination antiretroviral therapy.
HIV/HCV coinfection leads to a significant increase in plasma HCV RNA, according to a study in Pathogens and Global Health.
A Canadian study identified elevated rates of intentional and unintentional injury among people living with HIV.
Partner services for persons with acute and early HIV infection (AEH) within 30 days of diagnosis represents an effective tool to find HIV unaware persons, a recent study found, including those with AEH who are at greatest risk of HIV transmission.
Maternal tenofovir use was not associated with lower length or head circumference of HIV-exposed uninfected infants at 2 years of age, a recent study revealed, but may be related to greater weight among those exposed to combination antiretroviral therapy early in pregnancy.
Cryptococcal meningitis may be considered one of the causes of acute vision loss in pregnant/postpartum HIV-positive females, according to a study in BMC Infectious Diseases.
According to a study in JAIDS, red cell distribution width remains a powerful marker of cardiovascular disease in the context of the inflammatory milieu that accompanies HIV infection.
HIV infection moderates the association between vascular remodeling and neurocognitive function but not the association between pulse pressure and neurocognitive function, a recent study revealed.
HIV viral blips on therapy are associated with subsequent viral rebound on stopping antiretroviral therapy among individuals treated in primary HIV infection, according to a new study.
Adipocytokine dysregulation seems to be related to metabolic syndrome in HIV-infected children, according to a study in the Pediatric Infectious Disease Journal.
[email protected]
On Twitter @richpizzi
A great volume of HIV and AIDS research enters the medical literature every month. It’s difficult to monitor everything, so here’s a quick look at some notable news items and journal articles published over the past few weeks.
HIV-infected U.S. smokers aged 40 years lose more than 6 years of life expectancy from smoking, according to a computer simulation study, possibly outweighing the loss from HIV infection itself.
A study published in JAIDS provides insight into a novel mechanism of ritonavir-induced insulin resistance involving proinflammatory properties of heme oxygenase-1 (HO-1).
Antiretroviral therapy with efavirenz, emtricitabine, and tenofovir disoproxil fumarate (TDF) appeared to have a more favorable renal safety profile than did TDF administered with a protease inhibitor or cobicistat, according to a study in HIV Clinical Trials.
Researchers said the first study to describe the dynamics of the biomarker of cerebrospinal fluid YKL-40 in two groups of HIV-infected individuals, before and after combination antiretroviral therapy, has demonstrated the value of the marker in understanding HIV neuropathogenesis.
A study of HIV testing among ethnic minority adolescents in New Jersey underscored the importance of developing multifaceted HIV/AIDS prevention protocols that provide direct education and skill-building activities, leverage peer education as a means to disseminate health-related information, and deliver broad-based prevention messaging that is culturally tailored and gender specific.
HIV-infected adults in a contemporary, high-resource setting have poor dietary patterns, a recent study found, and alcohol use was associated with worse gut integrity and increased inflammation, while other aspects of diet were not.
A study in BMC Infectious Diseases found immune recovery comparable in primary and chronic HIV infection, whereas differences in absolute counts and proportions of CD4+ T cell subpopulations were found between primary HIV infection and late presenters supporting early initiation of combination antiretroviral therapy.
HIV/HCV coinfection leads to a significant increase in plasma HCV RNA, according to a study in Pathogens and Global Health.
A Canadian study identified elevated rates of intentional and unintentional injury among people living with HIV.
Partner services for persons with acute and early HIV infection (AEH) within 30 days of diagnosis represents an effective tool to find HIV unaware persons, a recent study found, including those with AEH who are at greatest risk of HIV transmission.
Maternal tenofovir use was not associated with lower length or head circumference of HIV-exposed uninfected infants at 2 years of age, a recent study revealed, but may be related to greater weight among those exposed to combination antiretroviral therapy early in pregnancy.
Cryptococcal meningitis may be considered one of the causes of acute vision loss in pregnant/postpartum HIV-positive females, according to a study in BMC Infectious Diseases.
According to a study in JAIDS, red cell distribution width remains a powerful marker of cardiovascular disease in the context of the inflammatory milieu that accompanies HIV infection.
HIV infection moderates the association between vascular remodeling and neurocognitive function but not the association between pulse pressure and neurocognitive function, a recent study revealed.
HIV viral blips on therapy are associated with subsequent viral rebound on stopping antiretroviral therapy among individuals treated in primary HIV infection, according to a new study.
Adipocytokine dysregulation seems to be related to metabolic syndrome in HIV-infected children, according to a study in the Pediatric Infectious Disease Journal.
[email protected]
On Twitter @richpizzi
Preventing weight gain after smoking cessation
About three-quarters of current cigarette smokers want to quit, 40% will attempt to quit annually, and 90% of self-initiated attempts will be unsuccessful. Mean weight gain after smoking cessation may be as much as 13 pounds at 1 year, and 21 pounds over 5 years.
Population data suggest that more than one-half of women and one-third of men with a previous attempt to quit smoking report that weight gain was one of the primary reasons for relapse back to smoking.

Lorcaserin is a 5-HT2c (serotonin) receptor agonist FDA-approved for weight loss. Varenicline is the most effective monotherapy for smoking cessation and targets the alpha-4 beta-2 nicotinic acetylcholine receptor.
Ryan Hurt, MD, and his colleagues recently completed a pilot clinical trial evaluating the potential efficacy of combining varenicline and lorcaserin for the prevention of PCWG in obese and overweight smokers (Nicotine Tob Res. 2016 Nov 16. doi: 10.1093/ntr/ntw304).
In this study, 20 smokers with a body mass index of 27-40 kg/m2 received varenicline and lorcaserin for 12 weeks.
Fifty percent of subjects were abstinent from smoking at 12 weeks, among whom weight gain was only +1.1 ± 3.9 kg (90% confidence interval, –0.9 to +3.1). The most-common side effect of the combination was sleep disturbance, reported by five patients.
The study was limited by the small sample size and the absence of a control group or placebo.
As clinicians, we frequently employ combination therapy in chronic diseases such as diabetes and hypertension when single-agent therapy is ineffective. By combining drugs with different therapeutic targets, we can achieve our treatment goals.
In tobacco dependence treatment, we use combination pharmacotherapy for heavier smokers or for those who have tried and failed to quit previously. Interestingly, lorcaserin has been demonstrated in another pilot study to increase smoking cessation rates by itself.
The combination of lorcaserin and varenicline holds promise for the treatment of tobacco dependence by attacking tobacco dependence through two different mechanisms and preventing PCWG, which may prevent relapse back to smoking.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article.
About three-quarters of current cigarette smokers want to quit, 40% will attempt to quit annually, and 90% of self-initiated attempts will be unsuccessful. Mean weight gain after smoking cessation may be as much as 13 pounds at 1 year, and 21 pounds over 5 years.
Population data suggest that more than one-half of women and one-third of men with a previous attempt to quit smoking report that weight gain was one of the primary reasons for relapse back to smoking.
Lorcaserin is a 5-HT2c (serotonin) receptor agonist FDA-approved for weight loss. Varenicline is the most effective monotherapy for smoking cessation and targets the alpha-4 beta-2 nicotinic acetylcholine receptor.
Ryan Hurt, MD, and his colleagues recently completed a pilot clinical trial evaluating the potential efficacy of combining varenicline and lorcaserin for the prevention of PCWG in obese and overweight smokers (Nicotine Tob Res. 2016 Nov 16. doi: 10.1093/ntr/ntw304).
In this study, 20 smokers with a body mass index of 27-40 kg/m2 received varenicline and lorcaserin for 12 weeks.
Fifty percent of subjects were abstinent from smoking at 12 weeks, among whom weight gain was only +1.1 ± 3.9 kg (90% confidence interval, –0.9 to +3.1). The most-common side effect of the combination was sleep disturbance, reported by five patients.
The study was limited by the small sample size and the absence of a control group or placebo.
As clinicians, we frequently employ combination therapy in chronic diseases such as diabetes and hypertension when single-agent therapy is ineffective. By combining drugs with different therapeutic targets, we can achieve our treatment goals.
In tobacco dependence treatment, we use combination pharmacotherapy for heavier smokers or for those who have tried and failed to quit previously. Interestingly, lorcaserin has been demonstrated in another pilot study to increase smoking cessation rates by itself.
The combination of lorcaserin and varenicline holds promise for the treatment of tobacco dependence by attacking tobacco dependence through two different mechanisms and preventing PCWG, which may prevent relapse back to smoking.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article.
About three-quarters of current cigarette smokers want to quit, 40% will attempt to quit annually, and 90% of self-initiated attempts will be unsuccessful. Mean weight gain after smoking cessation may be as much as 13 pounds at 1 year, and 21 pounds over 5 years.
Population data suggest that more than one-half of women and one-third of men with a previous attempt to quit smoking report that weight gain was one of the primary reasons for relapse back to smoking.
Lorcaserin is a 5-HT2c (serotonin) receptor agonist FDA-approved for weight loss. Varenicline is the most effective monotherapy for smoking cessation and targets the alpha-4 beta-2 nicotinic acetylcholine receptor.
Ryan Hurt, MD, and his colleagues recently completed a pilot clinical trial evaluating the potential efficacy of combining varenicline and lorcaserin for the prevention of PCWG in obese and overweight smokers (Nicotine Tob Res. 2016 Nov 16. doi: 10.1093/ntr/ntw304).
In this study, 20 smokers with a body mass index of 27-40 kg/m2 received varenicline and lorcaserin for 12 weeks.
Fifty percent of subjects were abstinent from smoking at 12 weeks, among whom weight gain was only +1.1 ± 3.9 kg (90% confidence interval, –0.9 to +3.1). The most-common side effect of the combination was sleep disturbance, reported by five patients.
The study was limited by the small sample size and the absence of a control group or placebo.
As clinicians, we frequently employ combination therapy in chronic diseases such as diabetes and hypertension when single-agent therapy is ineffective. By combining drugs with different therapeutic targets, we can achieve our treatment goals.
In tobacco dependence treatment, we use combination pharmacotherapy for heavier smokers or for those who have tried and failed to quit previously. Interestingly, lorcaserin has been demonstrated in another pilot study to increase smoking cessation rates by itself.
The combination of lorcaserin and varenicline holds promise for the treatment of tobacco dependence by attacking tobacco dependence through two different mechanisms and preventing PCWG, which may prevent relapse back to smoking.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article.
Second transplant, consolidation don’t add benefit in upfront multiple myeloma therapy
SAN DIEGO – It took a clinical trial with a byzantine design to prove it, but neither posttransplant consolidation therapy nor second transplant offered any additional survival benefits to patients with multiple myeloma, including patients with high-risk disease who were treated with an upfront thalidomide analogue and a proteasome inhibitor, followed by stem cell transplant and lenalidomide maintenance.
Among 758 patients with multiple myeloma who underwent standard induction therapy, followed by melphalan conditioning and autologous stem cell transplant (ASCT), there were no differences in either progression-free survival (PFS) or overall survival (OS) among patients assigned to follow-on therapy with either lenalidomide (Revlimid) maintenance alone; consolidation therapy with four cycles of lenalidomide (Revlimid), bortezomib (Velcade), and dexamethasone (RVD), followed by lenalidomide maintenance; or second transplant, followed by lenalidomide maintenance, reported Edward A. Stadtmauer, MD, coleader of the hematologic malignancies program at the Abramson Cancer Center, and chief of the section of hematologic malignancies, University of Pennsylvania, Philadelphia.

Investigators in the STAMINA (Stem Cell Transplant With Lenalidomide Maintenance in Patients With Multiple Myeloma) trial (BMT CTN 0702) hypothesized that the use of thalidomide analogues and proteasome inhibitors used in first-line therapy, consolidation, and long-term maintenance after high-dose melphalan and ASCT would improve survival, compared with a second ASCT.
To test this idea, they enrolled 758 patients and randomized them to one of the three aforementioned posttransplant strategies prior to transplant conditioning with high-dose melphalan (200 mg/m2) and ASCT.
Roughly 25% of patients in each treatment arm had high-risk disease, defined as beta2 microglobulin levels greater than 5.5 mg/L, high-risk cytogenetics, and deletion 13 detected by standard cytogenetics only. The remaining patients in each arm had standard-risk disease.
Slightly more than half of patients received RVD upfront; about 13% received cyclophosphamide, bortezomib, and dexamethasone (CyBorD); roughly 10% received lenalidomide dexamethasone; 12% were treated with bortezomib/dexamethasone; and about 8% received other, unspecified combinations.
At a median follow-up time of 37.8 months, the PFS rate, which was the primary endpoint, was 56.5% for the second transplant arm, 56.7% for the RVD arm, and 52.2% for the maintenance-only arm. The differences were not statistically significant.
Similarly, there were no among-arm differences in PFS for patients with standard-risk disease (60.9%, 59.5%, and 55.9%) or for those with high-risk myeloma (42.2%, 48.3%, and 40.2%)
Overall survival, a secondary endpoint, was also not significantly different among the groups, at 82%, 85.7%, and 83.4%, respectively.
Encouragingly, however, despite lower PFS rates, patients with high-risk disease had high OS rates, with 79.6% of patients in the double-transplant arm, 77.5% of those in the RVD consolidation arm, and 79.5% of those in the lenalidomide maintenance-alone arm still alive at 38 months.
Secondary malignancies occurred among 5.1% of patients overall: 14 in the dual-transplant arm, 15 in the consolidation arm, and 10 in the maintenance-only arm. The most frequently reported second malignancies were leukemia, which occurred in 3 of 14 patients with second cancers after second transplant and in 9 of 15 patients with second cancers after consolidation, and solid tumors, which occurred most frequently among second cancers in the maintenance arm.
The investigators are continuing to parse the data by study arm to see whether response assessment correlates with outcomes and with complete remissions. They also plan to examine minimal residual disease via flow cytometry and sequencing, and to obtain long-term data on survival, toxicities, and second primary malignancies.
The trial was funded by the National Institutes of Health with support from Celgene and Millennium/Takeda. Dr. Stadtmauer disclosed consulting for Takeda and travel expenses from Celgene.
SAN DIEGO – It took a clinical trial with a byzantine design to prove it, but neither posttransplant consolidation therapy nor second transplant offered any additional survival benefits to patients with multiple myeloma, including patients with high-risk disease who were treated with an upfront thalidomide analogue and a proteasome inhibitor, followed by stem cell transplant and lenalidomide maintenance.
Among 758 patients with multiple myeloma who underwent standard induction therapy, followed by melphalan conditioning and autologous stem cell transplant (ASCT), there were no differences in either progression-free survival (PFS) or overall survival (OS) among patients assigned to follow-on therapy with either lenalidomide (Revlimid) maintenance alone; consolidation therapy with four cycles of lenalidomide (Revlimid), bortezomib (Velcade), and dexamethasone (RVD), followed by lenalidomide maintenance; or second transplant, followed by lenalidomide maintenance, reported Edward A. Stadtmauer, MD, coleader of the hematologic malignancies program at the Abramson Cancer Center, and chief of the section of hematologic malignancies, University of Pennsylvania, Philadelphia.
Investigators in the STAMINA (Stem Cell Transplant With Lenalidomide Maintenance in Patients With Multiple Myeloma) trial (BMT CTN 0702) hypothesized that the use of thalidomide analogues and proteasome inhibitors used in first-line therapy, consolidation, and long-term maintenance after high-dose melphalan and ASCT would improve survival, compared with a second ASCT.
To test this idea, they enrolled 758 patients and randomized them to one of the three aforementioned posttransplant strategies prior to transplant conditioning with high-dose melphalan (200 mg/m2) and ASCT.
Roughly 25% of patients in each treatment arm had high-risk disease, defined as beta2 microglobulin levels greater than 5.5 mg/L, high-risk cytogenetics, and deletion 13 detected by standard cytogenetics only. The remaining patients in each arm had standard-risk disease.
Slightly more than half of patients received RVD upfront; about 13% received cyclophosphamide, bortezomib, and dexamethasone (CyBorD); roughly 10% received lenalidomide dexamethasone; 12% were treated with bortezomib/dexamethasone; and about 8% received other, unspecified combinations.
At a median follow-up time of 37.8 months, the PFS rate, which was the primary endpoint, was 56.5% for the second transplant arm, 56.7% for the RVD arm, and 52.2% for the maintenance-only arm. The differences were not statistically significant.
Similarly, there were no among-arm differences in PFS for patients with standard-risk disease (60.9%, 59.5%, and 55.9%) or for those with high-risk myeloma (42.2%, 48.3%, and 40.2%)
Overall survival, a secondary endpoint, was also not significantly different among the groups, at 82%, 85.7%, and 83.4%, respectively.
Encouragingly, however, despite lower PFS rates, patients with high-risk disease had high OS rates, with 79.6% of patients in the double-transplant arm, 77.5% of those in the RVD consolidation arm, and 79.5% of those in the lenalidomide maintenance-alone arm still alive at 38 months.
Secondary malignancies occurred among 5.1% of patients overall: 14 in the dual-transplant arm, 15 in the consolidation arm, and 10 in the maintenance-only arm. The most frequently reported second malignancies were leukemia, which occurred in 3 of 14 patients with second cancers after second transplant and in 9 of 15 patients with second cancers after consolidation, and solid tumors, which occurred most frequently among second cancers in the maintenance arm.
The investigators are continuing to parse the data by study arm to see whether response assessment correlates with outcomes and with complete remissions. They also plan to examine minimal residual disease via flow cytometry and sequencing, and to obtain long-term data on survival, toxicities, and second primary malignancies.
The trial was funded by the National Institutes of Health with support from Celgene and Millennium/Takeda. Dr. Stadtmauer disclosed consulting for Takeda and travel expenses from Celgene.
SAN DIEGO – It took a clinical trial with a byzantine design to prove it, but neither posttransplant consolidation therapy nor second transplant offered any additional survival benefits to patients with multiple myeloma, including patients with high-risk disease who were treated with an upfront thalidomide analogue and a proteasome inhibitor, followed by stem cell transplant and lenalidomide maintenance.
Among 758 patients with multiple myeloma who underwent standard induction therapy, followed by melphalan conditioning and autologous stem cell transplant (ASCT), there were no differences in either progression-free survival (PFS) or overall survival (OS) among patients assigned to follow-on therapy with either lenalidomide (Revlimid) maintenance alone; consolidation therapy with four cycles of lenalidomide (Revlimid), bortezomib (Velcade), and dexamethasone (RVD), followed by lenalidomide maintenance; or second transplant, followed by lenalidomide maintenance, reported Edward A. Stadtmauer, MD, coleader of the hematologic malignancies program at the Abramson Cancer Center, and chief of the section of hematologic malignancies, University of Pennsylvania, Philadelphia.
Investigators in the STAMINA (Stem Cell Transplant With Lenalidomide Maintenance in Patients With Multiple Myeloma) trial (BMT CTN 0702) hypothesized that the use of thalidomide analogues and proteasome inhibitors used in first-line therapy, consolidation, and long-term maintenance after high-dose melphalan and ASCT would improve survival, compared with a second ASCT.
To test this idea, they enrolled 758 patients and randomized them to one of the three aforementioned posttransplant strategies prior to transplant conditioning with high-dose melphalan (200 mg/m2) and ASCT.
Roughly 25% of patients in each treatment arm had high-risk disease, defined as beta2 microglobulin levels greater than 5.5 mg/L, high-risk cytogenetics, and deletion 13 detected by standard cytogenetics only. The remaining patients in each arm had standard-risk disease.
Slightly more than half of patients received RVD upfront; about 13% received cyclophosphamide, bortezomib, and dexamethasone (CyBorD); roughly 10% received lenalidomide dexamethasone; 12% were treated with bortezomib/dexamethasone; and about 8% received other, unspecified combinations.
At a median follow-up time of 37.8 months, the PFS rate, which was the primary endpoint, was 56.5% for the second transplant arm, 56.7% for the RVD arm, and 52.2% for the maintenance-only arm. The differences were not statistically significant.
Similarly, there were no among-arm differences in PFS for patients with standard-risk disease (60.9%, 59.5%, and 55.9%) or for those with high-risk myeloma (42.2%, 48.3%, and 40.2%)
Overall survival, a secondary endpoint, was also not significantly different among the groups, at 82%, 85.7%, and 83.4%, respectively.
Encouragingly, however, despite lower PFS rates, patients with high-risk disease had high OS rates, with 79.6% of patients in the double-transplant arm, 77.5% of those in the RVD consolidation arm, and 79.5% of those in the lenalidomide maintenance-alone arm still alive at 38 months.
Secondary malignancies occurred among 5.1% of patients overall: 14 in the dual-transplant arm, 15 in the consolidation arm, and 10 in the maintenance-only arm. The most frequently reported second malignancies were leukemia, which occurred in 3 of 14 patients with second cancers after second transplant and in 9 of 15 patients with second cancers after consolidation, and solid tumors, which occurred most frequently among second cancers in the maintenance arm.
The investigators are continuing to parse the data by study arm to see whether response assessment correlates with outcomes and with complete remissions. They also plan to examine minimal residual disease via flow cytometry and sequencing, and to obtain long-term data on survival, toxicities, and second primary malignancies.
The trial was funded by the National Institutes of Health with support from Celgene and Millennium/Takeda. Dr. Stadtmauer disclosed consulting for Takeda and travel expenses from Celgene.
AT ASH 2016
Key clinical point: Three posttransplant strategies for patients with previously untreated myeloma were comparably effective.
Major finding: There were no differences in PFS or OS among patients treated with upfront therapy and transplant followed by either second transplant, consolidation, or lenalidomide maintenance alone.
Data source: Randomized prospective trial of 758 patients with multiple myeloma treated with a thalidomide analogue, proteasome inhibitor, and autologous stem cell transplant.
Disclosures: The trial was funded by the National Institutes of Health with support from Celgene and Millennium/Takeda. Dr. Stadtmauer disclosed consulting for Takeda and travel expenses from Celgene.
The war on pain
When your peer group is dominated by folks in their early 70s, conversations at dinner parties and lobster bakes invariably morph into storytelling competitions between the survivors of recent hospitalizations and medical procedures. I try to redirect this tedious and repetitive chatter with a topic from my standard collection of conversation re-starters that includes “How about those Red Sox?” and “How’s your granddaughter’s soccer season going?” But sadly I am not always successful.
Often embedded in these tales of medical misadventure are stories of unfortunate experiences with pain medications. Sometimes the story includes a description of how prescribed pain medication created symptoms that were far worse than the pain it was intended to treat. Vomiting, constipation, and “feeling goofy” are high on the list of complaints.

These caches of unused opioids, many of which were never needed in the first place, are evidence of why our health care has become so expensive, and also represent the seeds from which the addiction epidemic has grown. Ironically, they also are collateral damage from an unsuccessful and sometimes misguided war on pain.
It isn’t clear exactly when or where the war on pain began, but I’m sure those who fired the first shots were understandably concerned that many patients with incurable and terminal conditions were suffering needlessly because their pain was being under-treated. Coincidently came the realization that the sooner we could get postoperative patients on their feet and taking deep breaths, the fewer complications we would see. And the more adequately we treated their pain, the sooner we could get those patients moving and breathing optimally.
In a good faith effort to be more “scientific” about pain management, patients were asked to rate their pain and smiley face charts appeared. Unfortunately, somewhere along the line came the mantra that not only should no patient’s pain go unmeasured, but no patient’s pain should go unmedicated.
The federal government entered the war when the Centers for Medicare & Medicaid Services issued the directive that hospitals ask patients who were being discharged if their pain had been well controlled and how often did the hospital staff do what they could to ease their pain? The answers to these questions, along with others, was collected and used in assessing a hospital’s quality of care and determining its level of reimbursement.
So far, there is insufficient data to determine how frequently this directive on pain management induced hospitals to over-prescribe medication, but it certainly hasn’t been associated with a decline in opioid abuse. It is reasonable to suspect that this salvo by the government has resulted in some collateral damage as it encouraged a steady flow of unused and unnecessary prescription narcotics out of the hospital and on to the streets.
The good news is that there has been enough concern voiced about the unintended effect of these pain management questions that the CMS has decided to eliminate financial pressure clinicians might feel to over-prescribe medications by withdrawing the questions from the patient discharge questionnaire.
The bad news is that we continue to fight the war on pain with a limited arsenal. As long as clinicians simply believe that no pain should go unmedicated, they will continue to miss opportunities to use other modalities such as counseling, physical therapy, and education that can be effective without the risk of collateral damage. Instead of asking the patient (who may not know the answer), we should be asking ourselves if we have been doing everything we could to help the patient deal with his pain. The answer is often not written on prescription pads.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
When your peer group is dominated by folks in their early 70s, conversations at dinner parties and lobster bakes invariably morph into storytelling competitions between the survivors of recent hospitalizations and medical procedures. I try to redirect this tedious and repetitive chatter with a topic from my standard collection of conversation re-starters that includes “How about those Red Sox?” and “How’s your granddaughter’s soccer season going?” But sadly I am not always successful.
Often embedded in these tales of medical misadventure are stories of unfortunate experiences with pain medications. Sometimes the story includes a description of how prescribed pain medication created symptoms that were far worse than the pain it was intended to treat. Vomiting, constipation, and “feeling goofy” are high on the list of complaints.
These caches of unused opioids, many of which were never needed in the first place, are evidence of why our health care has become so expensive, and also represent the seeds from which the addiction epidemic has grown. Ironically, they also are collateral damage from an unsuccessful and sometimes misguided war on pain.
It isn’t clear exactly when or where the war on pain began, but I’m sure those who fired the first shots were understandably concerned that many patients with incurable and terminal conditions were suffering needlessly because their pain was being under-treated. Coincidently came the realization that the sooner we could get postoperative patients on their feet and taking deep breaths, the fewer complications we would see. And the more adequately we treated their pain, the sooner we could get those patients moving and breathing optimally.
In a good faith effort to be more “scientific” about pain management, patients were asked to rate their pain and smiley face charts appeared. Unfortunately, somewhere along the line came the mantra that not only should no patient’s pain go unmeasured, but no patient’s pain should go unmedicated.
The federal government entered the war when the Centers for Medicare & Medicaid Services issued the directive that hospitals ask patients who were being discharged if their pain had been well controlled and how often did the hospital staff do what they could to ease their pain? The answers to these questions, along with others, was collected and used in assessing a hospital’s quality of care and determining its level of reimbursement.
So far, there is insufficient data to determine how frequently this directive on pain management induced hospitals to over-prescribe medication, but it certainly hasn’t been associated with a decline in opioid abuse. It is reasonable to suspect that this salvo by the government has resulted in some collateral damage as it encouraged a steady flow of unused and unnecessary prescription narcotics out of the hospital and on to the streets.
The good news is that there has been enough concern voiced about the unintended effect of these pain management questions that the CMS has decided to eliminate financial pressure clinicians might feel to over-prescribe medications by withdrawing the questions from the patient discharge questionnaire.
The bad news is that we continue to fight the war on pain with a limited arsenal. As long as clinicians simply believe that no pain should go unmedicated, they will continue to miss opportunities to use other modalities such as counseling, physical therapy, and education that can be effective without the risk of collateral damage. Instead of asking the patient (who may not know the answer), we should be asking ourselves if we have been doing everything we could to help the patient deal with his pain. The answer is often not written on prescription pads.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
When your peer group is dominated by folks in their early 70s, conversations at dinner parties and lobster bakes invariably morph into storytelling competitions between the survivors of recent hospitalizations and medical procedures. I try to redirect this tedious and repetitive chatter with a topic from my standard collection of conversation re-starters that includes “How about those Red Sox?” and “How’s your granddaughter’s soccer season going?” But sadly I am not always successful.
Often embedded in these tales of medical misadventure are stories of unfortunate experiences with pain medications. Sometimes the story includes a description of how prescribed pain medication created symptoms that were far worse than the pain it was intended to treat. Vomiting, constipation, and “feeling goofy” are high on the list of complaints.
These caches of unused opioids, many of which were never needed in the first place, are evidence of why our health care has become so expensive, and also represent the seeds from which the addiction epidemic has grown. Ironically, they also are collateral damage from an unsuccessful and sometimes misguided war on pain.
It isn’t clear exactly when or where the war on pain began, but I’m sure those who fired the first shots were understandably concerned that many patients with incurable and terminal conditions were suffering needlessly because their pain was being under-treated. Coincidently came the realization that the sooner we could get postoperative patients on their feet and taking deep breaths, the fewer complications we would see. And the more adequately we treated their pain, the sooner we could get those patients moving and breathing optimally.
In a good faith effort to be more “scientific” about pain management, patients were asked to rate their pain and smiley face charts appeared. Unfortunately, somewhere along the line came the mantra that not only should no patient’s pain go unmeasured, but no patient’s pain should go unmedicated.
The federal government entered the war when the Centers for Medicare & Medicaid Services issued the directive that hospitals ask patients who were being discharged if their pain had been well controlled and how often did the hospital staff do what they could to ease their pain? The answers to these questions, along with others, was collected and used in assessing a hospital’s quality of care and determining its level of reimbursement.
So far, there is insufficient data to determine how frequently this directive on pain management induced hospitals to over-prescribe medication, but it certainly hasn’t been associated with a decline in opioid abuse. It is reasonable to suspect that this salvo by the government has resulted in some collateral damage as it encouraged a steady flow of unused and unnecessary prescription narcotics out of the hospital and on to the streets.
The good news is that there has been enough concern voiced about the unintended effect of these pain management questions that the CMS has decided to eliminate financial pressure clinicians might feel to over-prescribe medications by withdrawing the questions from the patient discharge questionnaire.
The bad news is that we continue to fight the war on pain with a limited arsenal. As long as clinicians simply believe that no pain should go unmedicated, they will continue to miss opportunities to use other modalities such as counseling, physical therapy, and education that can be effective without the risk of collateral damage. Instead of asking the patient (who may not know the answer), we should be asking ourselves if we have been doing everything we could to help the patient deal with his pain. The answer is often not written on prescription pads.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
VIDEO: Ankylosing spondylitis problems outside the joints strike more women than men
WASHINGTON – Women are almost twice as likely as men to develop extra-articular manifestations of ankylosing spondylitis such as uveitis and inflammatory bowel disease, according to an analysis of patients in the Ankylosing Spondylitis Registry of Ireland.
Each of those manifestations exerts its own difficulties upon patients over and above the inflammatory back pain of the underlying disease, Gillian Fitzgerald, MD, said at the annual meeting of the American College of Rheumatology. Many patients can develop several of these separate manifestations – a circumstance that seriously affects their quality of life.
The findings of the large registry study were a bit surprising, she said during presentation of the study at a press briefing, as ankylosing spondylitis is generally thought to affect largely men. “However, this isn’t the case,” said Dr. Fitzgerald of St. James’s Hospital, Dublin. “Recent studies show that women can be affected as often as men are.”
In light of those findings, Dr. Fitzgerald and her coauthors wanted to further define the gender differences, especially with regard to extra-articular manifestations.
They accessed data on 564 patients in the registry, which was established in 2013. The majority of patients (78%) were men; the mean age was 47 years. Patients had a mean disease duration of nearly 21 years. For almost half that time (9 years) they had remained undiagnosed, Dr. Fitzgerald added. They had a mean age of about 47 years, and 78% fulfilled the modified New York criteria for ankylosing spondylitis.
Overall, extra-articular manifestations were common, with 35% having uveitis, 18% psoriasis, and 10% inflammatory bowel disease.
Uveitis was significantly more common among women (47% vs. 32%) and among those with disease duration of more than 10 years (40% vs. 22% with less than 10 years).
Inflammatory bowel disease was also significantly more common among women (16.5% vs. 8%). It wasn’t related to disease duration, but it was related to elevated baseline C-reactive protein, peptic ulcer disease, and osteoporosis.
In a multivariate regression analysis, women were 70% more likely to experience an extra-articular manifestation of the disease than were men (hazard ratio, 1.7). Having the disease for more than 10 years more than doubled the risk of an extra-articular manifestation (HR, 2.4).
Dr. Fitzgerald discussed the study’s findings in a video interview at the meeting. She had no financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
[email protected]
On Twitter @alz_gal
WASHINGTON – Women are almost twice as likely as men to develop extra-articular manifestations of ankylosing spondylitis such as uveitis and inflammatory bowel disease, according to an analysis of patients in the Ankylosing Spondylitis Registry of Ireland.
Each of those manifestations exerts its own difficulties upon patients over and above the inflammatory back pain of the underlying disease, Gillian Fitzgerald, MD, said at the annual meeting of the American College of Rheumatology. Many patients can develop several of these separate manifestations – a circumstance that seriously affects their quality of life.
The findings of the large registry study were a bit surprising, she said during presentation of the study at a press briefing, as ankylosing spondylitis is generally thought to affect largely men. “However, this isn’t the case,” said Dr. Fitzgerald of St. James’s Hospital, Dublin. “Recent studies show that women can be affected as often as men are.”
In light of those findings, Dr. Fitzgerald and her coauthors wanted to further define the gender differences, especially with regard to extra-articular manifestations.
They accessed data on 564 patients in the registry, which was established in 2013. The majority of patients (78%) were men; the mean age was 47 years. Patients had a mean disease duration of nearly 21 years. For almost half that time (9 years) they had remained undiagnosed, Dr. Fitzgerald added. They had a mean age of about 47 years, and 78% fulfilled the modified New York criteria for ankylosing spondylitis.
Overall, extra-articular manifestations were common, with 35% having uveitis, 18% psoriasis, and 10% inflammatory bowel disease.
Uveitis was significantly more common among women (47% vs. 32%) and among those with disease duration of more than 10 years (40% vs. 22% with less than 10 years).
Inflammatory bowel disease was also significantly more common among women (16.5% vs. 8%). It wasn’t related to disease duration, but it was related to elevated baseline C-reactive protein, peptic ulcer disease, and osteoporosis.
In a multivariate regression analysis, women were 70% more likely to experience an extra-articular manifestation of the disease than were men (hazard ratio, 1.7). Having the disease for more than 10 years more than doubled the risk of an extra-articular manifestation (HR, 2.4).
Dr. Fitzgerald discussed the study’s findings in a video interview at the meeting. She had no financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
[email protected]
On Twitter @alz_gal
WASHINGTON – Women are almost twice as likely as men to develop extra-articular manifestations of ankylosing spondylitis such as uveitis and inflammatory bowel disease, according to an analysis of patients in the Ankylosing Spondylitis Registry of Ireland.
Each of those manifestations exerts its own difficulties upon patients over and above the inflammatory back pain of the underlying disease, Gillian Fitzgerald, MD, said at the annual meeting of the American College of Rheumatology. Many patients can develop several of these separate manifestations – a circumstance that seriously affects their quality of life.
The findings of the large registry study were a bit surprising, she said during presentation of the study at a press briefing, as ankylosing spondylitis is generally thought to affect largely men. “However, this isn’t the case,” said Dr. Fitzgerald of St. James’s Hospital, Dublin. “Recent studies show that women can be affected as often as men are.”
In light of those findings, Dr. Fitzgerald and her coauthors wanted to further define the gender differences, especially with regard to extra-articular manifestations.
They accessed data on 564 patients in the registry, which was established in 2013. The majority of patients (78%) were men; the mean age was 47 years. Patients had a mean disease duration of nearly 21 years. For almost half that time (9 years) they had remained undiagnosed, Dr. Fitzgerald added. They had a mean age of about 47 years, and 78% fulfilled the modified New York criteria for ankylosing spondylitis.
Overall, extra-articular manifestations were common, with 35% having uveitis, 18% psoriasis, and 10% inflammatory bowel disease.
Uveitis was significantly more common among women (47% vs. 32%) and among those with disease duration of more than 10 years (40% vs. 22% with less than 10 years).
Inflammatory bowel disease was also significantly more common among women (16.5% vs. 8%). It wasn’t related to disease duration, but it was related to elevated baseline C-reactive protein, peptic ulcer disease, and osteoporosis.
In a multivariate regression analysis, women were 70% more likely to experience an extra-articular manifestation of the disease than were men (hazard ratio, 1.7). Having the disease for more than 10 years more than doubled the risk of an extra-articular manifestation (HR, 2.4).
Dr. Fitzgerald discussed the study’s findings in a video interview at the meeting. She had no financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
[email protected]
On Twitter @alz_gal
AT THE ACR ANNUAL MEETING
Key clinical point:
Major finding: Women were 70% more likely than men to develop an extra-articular manifestation of the disease.
Data source: The registry study comprised 564 patients.
Disclosures: Dr. Fitzgerald had no financial disclosures.
