User login
Gene therapy accepted into PRIME program
![]()
Photo by Chad McNeeley
An investigational gene therapy known as LentiGlobin BB305 has been accepted into the European Medicines Agency’s (EMA’s) Priority Medicines (PRIME) program as a treatment for patients with transfusion-dependent beta-thalassemia (TDT).
LentiGlobin BB305 is created by inserting a functional human beta-globin gene into a patient’s hematopoietic stem cells ex vivo. The cells are then returned to the patient via transplant.
The goal of the EMA’s PRIME program is to accelerate the development of therapies that target unmet medical needs. The program provides enhanced EMA support and increased interaction to developers, in order to optimize development plans and speed regulatory evaluations to potentially bring these therapies to patients more quickly.
To be accepted for PRIME, a therapy must demonstrate potential to benefit patients with unmet medical need through early clinical or nonclinical data.
Bluebird bio, the company developing LentiGlobin BB305, is also participating in the EMA’s Adaptive Pathways pilot program.
Like PRIME, the Adaptive Pathways program aims to expedite patient access to therapies with the potential to treat serious conditions with unmet need. It uses the existing European Union (EU) regulatory framework for medicines, including conditional approval.
“PRIME designation will allow bluebird bio to further improve our communication with European regulators as we continue to refine our evidence generation plan in the context of adaptive biomedical innovation,” said David Davidson, MD, chief medical officer of bluebird bio.
“Overall, we believe this will enable us to accelerate development of LentiGlobin drug product for patients with transfusion-dependent beta-thalassemia, a life-shortening disease with significant unmet medical need.”
“Earlier this year, we completed enrollment in the Northstar (HGB-204) global clinical study of LentiGlobin drug product in patients with TDT, which, along with the supporting HGB-205 study, will form the basis of our eventual application for conditional approval in the EU under the Adaptive Pathways pilot program.” ![]()
![]()
Photo by Chad McNeeley
An investigational gene therapy known as LentiGlobin BB305 has been accepted into the European Medicines Agency’s (EMA’s) Priority Medicines (PRIME) program as a treatment for patients with transfusion-dependent beta-thalassemia (TDT).
LentiGlobin BB305 is created by inserting a functional human beta-globin gene into a patient’s hematopoietic stem cells ex vivo. The cells are then returned to the patient via transplant.
The goal of the EMA’s PRIME program is to accelerate the development of therapies that target unmet medical needs. The program provides enhanced EMA support and increased interaction to developers, in order to optimize development plans and speed regulatory evaluations to potentially bring these therapies to patients more quickly.
To be accepted for PRIME, a therapy must demonstrate potential to benefit patients with unmet medical need through early clinical or nonclinical data.
Bluebird bio, the company developing LentiGlobin BB305, is also participating in the EMA’s Adaptive Pathways pilot program.
Like PRIME, the Adaptive Pathways program aims to expedite patient access to therapies with the potential to treat serious conditions with unmet need. It uses the existing European Union (EU) regulatory framework for medicines, including conditional approval.
“PRIME designation will allow bluebird bio to further improve our communication with European regulators as we continue to refine our evidence generation plan in the context of adaptive biomedical innovation,” said David Davidson, MD, chief medical officer of bluebird bio.
“Overall, we believe this will enable us to accelerate development of LentiGlobin drug product for patients with transfusion-dependent beta-thalassemia, a life-shortening disease with significant unmet medical need.”
“Earlier this year, we completed enrollment in the Northstar (HGB-204) global clinical study of LentiGlobin drug product in patients with TDT, which, along with the supporting HGB-205 study, will form the basis of our eventual application for conditional approval in the EU under the Adaptive Pathways pilot program.” ![]()
![]()
Photo by Chad McNeeley
An investigational gene therapy known as LentiGlobin BB305 has been accepted into the European Medicines Agency’s (EMA’s) Priority Medicines (PRIME) program as a treatment for patients with transfusion-dependent beta-thalassemia (TDT).
LentiGlobin BB305 is created by inserting a functional human beta-globin gene into a patient’s hematopoietic stem cells ex vivo. The cells are then returned to the patient via transplant.
The goal of the EMA’s PRIME program is to accelerate the development of therapies that target unmet medical needs. The program provides enhanced EMA support and increased interaction to developers, in order to optimize development plans and speed regulatory evaluations to potentially bring these therapies to patients more quickly.
To be accepted for PRIME, a therapy must demonstrate potential to benefit patients with unmet medical need through early clinical or nonclinical data.
Bluebird bio, the company developing LentiGlobin BB305, is also participating in the EMA’s Adaptive Pathways pilot program.
Like PRIME, the Adaptive Pathways program aims to expedite patient access to therapies with the potential to treat serious conditions with unmet need. It uses the existing European Union (EU) regulatory framework for medicines, including conditional approval.
“PRIME designation will allow bluebird bio to further improve our communication with European regulators as we continue to refine our evidence generation plan in the context of adaptive biomedical innovation,” said David Davidson, MD, chief medical officer of bluebird bio.
“Overall, we believe this will enable us to accelerate development of LentiGlobin drug product for patients with transfusion-dependent beta-thalassemia, a life-shortening disease with significant unmet medical need.”
“Earlier this year, we completed enrollment in the Northstar (HGB-204) global clinical study of LentiGlobin drug product in patients with TDT, which, along with the supporting HGB-205 study, will form the basis of our eventual application for conditional approval in the EU under the Adaptive Pathways pilot program.” ![]()
Compound could treat resistant malaria
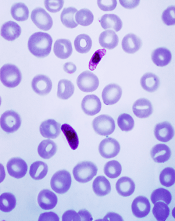
of the P falciparum parasite
Image by Mae Melvin/CDC
A novel compound could be effective against malaria infections that are resistant to currently available antimalarial drugs, according to researchers.
In a phase 2 study, the compound, KAF156, proved active against Plasmodium vivax and Plasmodium falciparum malaria.
KAF156 was able to clear both blood and liver stages of malaria parasites, including artemisinin-resistant parasites.
Most of the patients in this study had at least 1 adverse event (AE). However, most were grade 1, and there were no grade 4 or serious AEs.
Results from this study were published in NEJM. The trial was funded, in part, by Novartis, the company developing KAF156.
KAF156 is the first compound from a novel class of drugs called imidazolopiperazines. The drugs’ mechanism of action is still being characterized, but it may be related to a previously uncharacterized gene—Plasmodium falciparum cyclic amine resistance locus (Pfcarl).
From March to August 2013, researchers conducted a phase 2 trial of KAF156 at 5 centers in Thailand and Vietnam.
Twenty-one of the patients in this study had acute, uncomplicated malaria—11 with P vivax and 10 with P falciparum malaria. They received multiple doses of KAF156—400 mg once daily for 3 days.
Twenty-two of the patients studied had uncomplicated, P falciparum malaria and received a single dose of KAF156 at 800 mg.
All patients were assessed for fever and parasite clearance as well as AEs. Patients in the single-dose cohort were also followed for 28 days to assess the cure rate.
Two patients were excluded from the efficacy analysis. One was a P vivax patient receiving the 400 mg dose who turned out to have a mixed infection. And the other was a P falciparum patient who vomited repeatedly after receiving 800 mg of KAF156 (after 3 attempts at dosing).
Efficacy
In the multiple-dose cohorts, the median time to fever clearance after receiving KAF156 was 14 hours (range, 4 to 30) in patients with P vivax malaria and 6 hours (range, 4 to 24) in patients with P falciparum malaria. In the single-dose cohort, the median time to fever clearance was 4 hours (range, 4 to 66).
In the multiple-dose cohorts, the median time to parasite clearance was 24 hours (range, 16 to 36) in patients with P vivax and 45 hours (range, 36 to 66) in patients with P falciparum.
In the single-dose cohort, the median time to parasite clearance was 49 hours (range, 16 to 68). One of these patients had parasite clearance at 66 hours but an asymptomatic recurrence at 84 hours. In all of the other patients, parasitemia cleared.
During follow-up of the single-dose cohort, 1 patient had reinfection, and 7 had recrudescent infections.
Safety
Most patients had at least 1 AE, although none were serious. Seventy-two percent of patients had grade 1 AEs, 35% had grade 2 AEs, and 14% had grade 3 AEs.
Sixty percent of AEs were considered related to treatment. In the multiple-dose cohorts, 14 of the 31 AEs (45%) reported in 7 patients (1 with P vivax and 6 with P falciparum malaria) were considered drug-related. All 22 P falciparum patients in the single-dose cohort had at least 1 AE that was considered drug-related.
The most common AEs were sinus bradycardia, thrombocytopenia, hypokalemia, anemia, and hyperbilirubinemia.
Two patients experienced vomiting of grade 2 or higher. One of these patients discontinued treatment because of repeated vomiting after the 800 mg dose (mentioned above). ![]()
of the P falciparum parasite
Image by Mae Melvin/CDC
A novel compound could be effective against malaria infections that are resistant to currently available antimalarial drugs, according to researchers.
In a phase 2 study, the compound, KAF156, proved active against Plasmodium vivax and Plasmodium falciparum malaria.
KAF156 was able to clear both blood and liver stages of malaria parasites, including artemisinin-resistant parasites.
Most of the patients in this study had at least 1 adverse event (AE). However, most were grade 1, and there were no grade 4 or serious AEs.
Results from this study were published in NEJM. The trial was funded, in part, by Novartis, the company developing KAF156.
KAF156 is the first compound from a novel class of drugs called imidazolopiperazines. The drugs’ mechanism of action is still being characterized, but it may be related to a previously uncharacterized gene—Plasmodium falciparum cyclic amine resistance locus (Pfcarl).
From March to August 2013, researchers conducted a phase 2 trial of KAF156 at 5 centers in Thailand and Vietnam.
Twenty-one of the patients in this study had acute, uncomplicated malaria—11 with P vivax and 10 with P falciparum malaria. They received multiple doses of KAF156—400 mg once daily for 3 days.
Twenty-two of the patients studied had uncomplicated, P falciparum malaria and received a single dose of KAF156 at 800 mg.
All patients were assessed for fever and parasite clearance as well as AEs. Patients in the single-dose cohort were also followed for 28 days to assess the cure rate.
Two patients were excluded from the efficacy analysis. One was a P vivax patient receiving the 400 mg dose who turned out to have a mixed infection. And the other was a P falciparum patient who vomited repeatedly after receiving 800 mg of KAF156 (after 3 attempts at dosing).
Efficacy
In the multiple-dose cohorts, the median time to fever clearance after receiving KAF156 was 14 hours (range, 4 to 30) in patients with P vivax malaria and 6 hours (range, 4 to 24) in patients with P falciparum malaria. In the single-dose cohort, the median time to fever clearance was 4 hours (range, 4 to 66).
In the multiple-dose cohorts, the median time to parasite clearance was 24 hours (range, 16 to 36) in patients with P vivax and 45 hours (range, 36 to 66) in patients with P falciparum.
In the single-dose cohort, the median time to parasite clearance was 49 hours (range, 16 to 68). One of these patients had parasite clearance at 66 hours but an asymptomatic recurrence at 84 hours. In all of the other patients, parasitemia cleared.
During follow-up of the single-dose cohort, 1 patient had reinfection, and 7 had recrudescent infections.
Safety
Most patients had at least 1 AE, although none were serious. Seventy-two percent of patients had grade 1 AEs, 35% had grade 2 AEs, and 14% had grade 3 AEs.
Sixty percent of AEs were considered related to treatment. In the multiple-dose cohorts, 14 of the 31 AEs (45%) reported in 7 patients (1 with P vivax and 6 with P falciparum malaria) were considered drug-related. All 22 P falciparum patients in the single-dose cohort had at least 1 AE that was considered drug-related.
The most common AEs were sinus bradycardia, thrombocytopenia, hypokalemia, anemia, and hyperbilirubinemia.
Two patients experienced vomiting of grade 2 or higher. One of these patients discontinued treatment because of repeated vomiting after the 800 mg dose (mentioned above). ![]()
of the P falciparum parasite
Image by Mae Melvin/CDC
A novel compound could be effective against malaria infections that are resistant to currently available antimalarial drugs, according to researchers.
In a phase 2 study, the compound, KAF156, proved active against Plasmodium vivax and Plasmodium falciparum malaria.
KAF156 was able to clear both blood and liver stages of malaria parasites, including artemisinin-resistant parasites.
Most of the patients in this study had at least 1 adverse event (AE). However, most were grade 1, and there were no grade 4 or serious AEs.
Results from this study were published in NEJM. The trial was funded, in part, by Novartis, the company developing KAF156.
KAF156 is the first compound from a novel class of drugs called imidazolopiperazines. The drugs’ mechanism of action is still being characterized, but it may be related to a previously uncharacterized gene—Plasmodium falciparum cyclic amine resistance locus (Pfcarl).
From March to August 2013, researchers conducted a phase 2 trial of KAF156 at 5 centers in Thailand and Vietnam.
Twenty-one of the patients in this study had acute, uncomplicated malaria—11 with P vivax and 10 with P falciparum malaria. They received multiple doses of KAF156—400 mg once daily for 3 days.
Twenty-two of the patients studied had uncomplicated, P falciparum malaria and received a single dose of KAF156 at 800 mg.
All patients were assessed for fever and parasite clearance as well as AEs. Patients in the single-dose cohort were also followed for 28 days to assess the cure rate.
Two patients were excluded from the efficacy analysis. One was a P vivax patient receiving the 400 mg dose who turned out to have a mixed infection. And the other was a P falciparum patient who vomited repeatedly after receiving 800 mg of KAF156 (after 3 attempts at dosing).
Efficacy
In the multiple-dose cohorts, the median time to fever clearance after receiving KAF156 was 14 hours (range, 4 to 30) in patients with P vivax malaria and 6 hours (range, 4 to 24) in patients with P falciparum malaria. In the single-dose cohort, the median time to fever clearance was 4 hours (range, 4 to 66).
In the multiple-dose cohorts, the median time to parasite clearance was 24 hours (range, 16 to 36) in patients with P vivax and 45 hours (range, 36 to 66) in patients with P falciparum.
In the single-dose cohort, the median time to parasite clearance was 49 hours (range, 16 to 68). One of these patients had parasite clearance at 66 hours but an asymptomatic recurrence at 84 hours. In all of the other patients, parasitemia cleared.
During follow-up of the single-dose cohort, 1 patient had reinfection, and 7 had recrudescent infections.
Safety
Most patients had at least 1 AE, although none were serious. Seventy-two percent of patients had grade 1 AEs, 35% had grade 2 AEs, and 14% had grade 3 AEs.
Sixty percent of AEs were considered related to treatment. In the multiple-dose cohorts, 14 of the 31 AEs (45%) reported in 7 patients (1 with P vivax and 6 with P falciparum malaria) were considered drug-related. All 22 P falciparum patients in the single-dose cohort had at least 1 AE that was considered drug-related.
The most common AEs were sinus bradycardia, thrombocytopenia, hypokalemia, anemia, and hyperbilirubinemia.
Two patients experienced vomiting of grade 2 or higher. One of these patients discontinued treatment because of repeated vomiting after the 800 mg dose (mentioned above). ![]()
Of Cell Phones & Skin Damage
A 54-year-old man is seen for evaluation of various skin problems, none of which he believes to be serious. His concern primarily stems from his job of 30 years, which keeps him outdoors most days with minimal protection from the sun.
In addition to the generally weathered skin on his sun-exposed arms and face, his wife has noticed redness around his neck and chest that is slowly worsening with time.
He denies any discomfort with the changes in his neck skin. He has a history of diabetes and smoking.
EXAMINATION
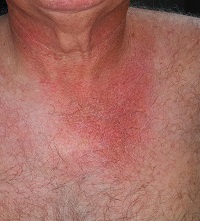
There is well-defined redness around his neck, especially in the front, that spills down onto his chest. Strangely, instead of matching the scoop neckline of the t-shirts he always wears, the chest redness is asymmetrical. It veers off to the patient’s left, sparing the midline chest. The erythema is partially blanchable with digital pressure but totally macular with no increased warmth.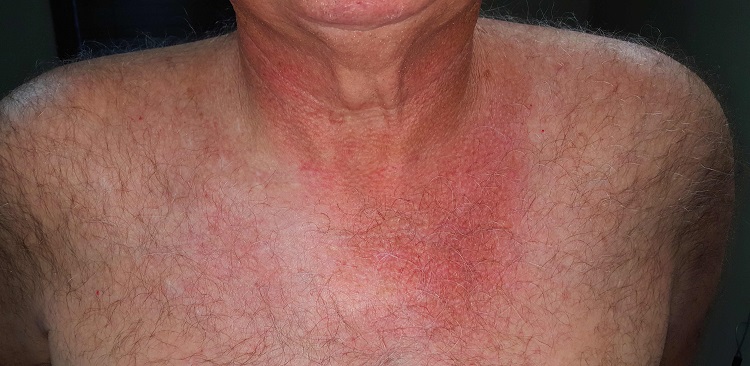
What is the diagnosis?
DISCUSSION
This patient’s condition is poikiloderma of Civatte (PC), an increase in surface vasculature etched into the skin by chronic overexposure to the sun. PC usually follows a very predictable pattern, mirroring the patient’s usual attire. This made the asymmetry in this patient’s case striking and puzzling. Speculation included uneven sunscreen application or use of clothing such as overalls, which tend to shift during wearing, but the cause remained a mystery—that is, until the patient got dressed.
After putting his shirt back on, he placed his large, heavy cell phone in the left pocket, effectively pulling his shirt down and to the left. This exposed more skin on that side, covering up the right side of the chest. He confirmed that he carried the cell phone in his shirt pocket constantly during his waking hours, at home and on the job.
This conundrum needed to be solved in order to rule out other possible conditions. There’s no rule that prevents a person from having two adjacent diagnoses at the same time, such as PC and Bowen disease (a superficial form of squamous cell carcinoma). In this case, the patient’s skin changes were all consistent with one diagnosis.
TAKE-HOME LEARNING POINTS
• Poikiloderma of Civatte (PC) is the term given to a particular pattern of chronic overexposure to UV rays, typically affecting the neck and upper chest.
• PC is often caused by occupational sun exposure.
• Unless treated with a laser, PC changes are permanent but harmless. They do, however, identify the patient as being at risk for sun-caused skin cancer.
• Atypical presentations of what appear to be common problems may need a bit of investigation to rule out other items in the differential.
A 54-year-old man is seen for evaluation of various skin problems, none of which he believes to be serious. His concern primarily stems from his job of 30 years, which keeps him outdoors most days with minimal protection from the sun.
In addition to the generally weathered skin on his sun-exposed arms and face, his wife has noticed redness around his neck and chest that is slowly worsening with time.
He denies any discomfort with the changes in his neck skin. He has a history of diabetes and smoking.
EXAMINATION
There is well-defined redness around his neck, especially in the front, that spills down onto his chest. Strangely, instead of matching the scoop neckline of the t-shirts he always wears, the chest redness is asymmetrical. It veers off to the patient’s left, sparing the midline chest. The erythema is partially blanchable with digital pressure but totally macular with no increased warmth.
What is the diagnosis?
DISCUSSION
This patient’s condition is poikiloderma of Civatte (PC), an increase in surface vasculature etched into the skin by chronic overexposure to the sun. PC usually follows a very predictable pattern, mirroring the patient’s usual attire. This made the asymmetry in this patient’s case striking and puzzling. Speculation included uneven sunscreen application or use of clothing such as overalls, which tend to shift during wearing, but the cause remained a mystery—that is, until the patient got dressed.
After putting his shirt back on, he placed his large, heavy cell phone in the left pocket, effectively pulling his shirt down and to the left. This exposed more skin on that side, covering up the right side of the chest. He confirmed that he carried the cell phone in his shirt pocket constantly during his waking hours, at home and on the job.
This conundrum needed to be solved in order to rule out other possible conditions. There’s no rule that prevents a person from having two adjacent diagnoses at the same time, such as PC and Bowen disease (a superficial form of squamous cell carcinoma). In this case, the patient’s skin changes were all consistent with one diagnosis.
TAKE-HOME LEARNING POINTS
• Poikiloderma of Civatte (PC) is the term given to a particular pattern of chronic overexposure to UV rays, typically affecting the neck and upper chest.
• PC is often caused by occupational sun exposure.
• Unless treated with a laser, PC changes are permanent but harmless. They do, however, identify the patient as being at risk for sun-caused skin cancer.
• Atypical presentations of what appear to be common problems may need a bit of investigation to rule out other items in the differential.
A 54-year-old man is seen for evaluation of various skin problems, none of which he believes to be serious. His concern primarily stems from his job of 30 years, which keeps him outdoors most days with minimal protection from the sun.
In addition to the generally weathered skin on his sun-exposed arms and face, his wife has noticed redness around his neck and chest that is slowly worsening with time.
He denies any discomfort with the changes in his neck skin. He has a history of diabetes and smoking.
EXAMINATION
There is well-defined redness around his neck, especially in the front, that spills down onto his chest. Strangely, instead of matching the scoop neckline of the t-shirts he always wears, the chest redness is asymmetrical. It veers off to the patient’s left, sparing the midline chest. The erythema is partially blanchable with digital pressure but totally macular with no increased warmth.
What is the diagnosis?
DISCUSSION
This patient’s condition is poikiloderma of Civatte (PC), an increase in surface vasculature etched into the skin by chronic overexposure to the sun. PC usually follows a very predictable pattern, mirroring the patient’s usual attire. This made the asymmetry in this patient’s case striking and puzzling. Speculation included uneven sunscreen application or use of clothing such as overalls, which tend to shift during wearing, but the cause remained a mystery—that is, until the patient got dressed.
After putting his shirt back on, he placed his large, heavy cell phone in the left pocket, effectively pulling his shirt down and to the left. This exposed more skin on that side, covering up the right side of the chest. He confirmed that he carried the cell phone in his shirt pocket constantly during his waking hours, at home and on the job.
This conundrum needed to be solved in order to rule out other possible conditions. There’s no rule that prevents a person from having two adjacent diagnoses at the same time, such as PC and Bowen disease (a superficial form of squamous cell carcinoma). In this case, the patient’s skin changes were all consistent with one diagnosis.
TAKE-HOME LEARNING POINTS
• Poikiloderma of Civatte (PC) is the term given to a particular pattern of chronic overexposure to UV rays, typically affecting the neck and upper chest.
• PC is often caused by occupational sun exposure.
• Unless treated with a laser, PC changes are permanent but harmless. They do, however, identify the patient as being at risk for sun-caused skin cancer.
• Atypical presentations of what appear to be common problems may need a bit of investigation to rule out other items in the differential.
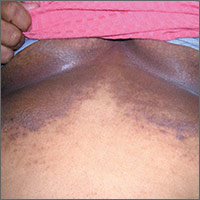
Dark rash under breasts
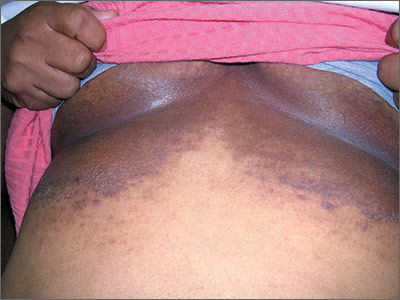
The family physician (FP) suspected that the patient had some type of fungal infection under her breasts. The FP scraped the edge and performed a potassium hydroxide (KOH) preparation that showed pseudohyphae and budding yeast forms, indicating a candida infection. (See video on how to perform a KOH preparation.) The FP also noted that there appeared to be some satellite lesions around the edge of the hyperpigmented area. Under the left breast was a small fissure and some white coloration at the skinfold. The differential diagnosis included tinea corporis and inverse psoriasis.
The FP prescribed topical clotrimazole cream 1% over the counter to be applied twice daily until at least one week after the rash resolved. While nystatin would be another option, it required a prescription and would not cover any possible dermatophyte in the area. The FP also explained that the dark pigmentation is the result of a longstanding inflammatory process related to the infection and would not go away immediately with treatment. In fact, the postinflammatory hyperpigmentation might remain for life.
The FP also spent time counseling the patient on an improved diet, increased exercise, and adherence to her diabetes medicines. A follow-up appointment for one month was set to review the patient's general health and to see if the candida infection resolved.
In cases where intertriginous fungal infections are treated without KOH or fungal culture confirmation, be suspicious for inverse psoriasis if the patient does not respond to antifungal medications. With such a large area involved, an oral antifungal such as fluconazole might occasionally be needed. Terbinafine is not an effective medicine for cutaneous candidiasis. If psoriasis is suspected, it helps to look for clues, such as nail pitting or cutaneous involvement with plaques on the elbows, knees, or scalp. If all else fails, a punch biopsy can be used to determine the correct diagnosis.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Candidiasis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:777-781.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The family physician (FP) suspected that the patient had some type of fungal infection under her breasts. The FP scraped the edge and performed a potassium hydroxide (KOH) preparation that showed pseudohyphae and budding yeast forms, indicating a candida infection. (See video on how to perform a KOH preparation.) The FP also noted that there appeared to be some satellite lesions around the edge of the hyperpigmented area. Under the left breast was a small fissure and some white coloration at the skinfold. The differential diagnosis included tinea corporis and inverse psoriasis.
The FP prescribed topical clotrimazole cream 1% over the counter to be applied twice daily until at least one week after the rash resolved. While nystatin would be another option, it required a prescription and would not cover any possible dermatophyte in the area. The FP also explained that the dark pigmentation is the result of a longstanding inflammatory process related to the infection and would not go away immediately with treatment. In fact, the postinflammatory hyperpigmentation might remain for life.
The FP also spent time counseling the patient on an improved diet, increased exercise, and adherence to her diabetes medicines. A follow-up appointment for one month was set to review the patient's general health and to see if the candida infection resolved.
In cases where intertriginous fungal infections are treated without KOH or fungal culture confirmation, be suspicious for inverse psoriasis if the patient does not respond to antifungal medications. With such a large area involved, an oral antifungal such as fluconazole might occasionally be needed. Terbinafine is not an effective medicine for cutaneous candidiasis. If psoriasis is suspected, it helps to look for clues, such as nail pitting or cutaneous involvement with plaques on the elbows, knees, or scalp. If all else fails, a punch biopsy can be used to determine the correct diagnosis.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Candidiasis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:777-781.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The family physician (FP) suspected that the patient had some type of fungal infection under her breasts. The FP scraped the edge and performed a potassium hydroxide (KOH) preparation that showed pseudohyphae and budding yeast forms, indicating a candida infection. (See video on how to perform a KOH preparation.) The FP also noted that there appeared to be some satellite lesions around the edge of the hyperpigmented area. Under the left breast was a small fissure and some white coloration at the skinfold. The differential diagnosis included tinea corporis and inverse psoriasis.
The FP prescribed topical clotrimazole cream 1% over the counter to be applied twice daily until at least one week after the rash resolved. While nystatin would be another option, it required a prescription and would not cover any possible dermatophyte in the area. The FP also explained that the dark pigmentation is the result of a longstanding inflammatory process related to the infection and would not go away immediately with treatment. In fact, the postinflammatory hyperpigmentation might remain for life.
The FP also spent time counseling the patient on an improved diet, increased exercise, and adherence to her diabetes medicines. A follow-up appointment for one month was set to review the patient's general health and to see if the candida infection resolved.
In cases where intertriginous fungal infections are treated without KOH or fungal culture confirmation, be suspicious for inverse psoriasis if the patient does not respond to antifungal medications. With such a large area involved, an oral antifungal such as fluconazole might occasionally be needed. Terbinafine is not an effective medicine for cutaneous candidiasis. If psoriasis is suspected, it helps to look for clues, such as nail pitting or cutaneous involvement with plaques on the elbows, knees, or scalp. If all else fails, a punch biopsy can be used to determine the correct diagnosis.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Candidiasis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:777-781.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
ICS/LABA exacerbation benefit outweighs pneumonia risk
LONDON – The benefit of a fixed-dose inhaled corticosteroid (ICS) and long-acting beta-agonist (LABA) combination in reducing exacerbations of chronic obstructive pulmonary disease (COPD) far outweighed any risk for pneumonia in a post hoc analysis of the 48-week FORWARD study.
Although there were 13 extra pneumonia events when a fixed-dose combination of beclometasone diproprionate and formoterol fumarate (Foster, Chiesi Farmaceutici SpA) was used as compared to formoterol fumarate alone, there were 123 fewer moderate to severe COPD exacerbations over a 342-day analysis period.
“Analysis of pneumonia and exacerbation cumulative number of events shows that the number of incident pneumonia remains very small relative to that of moderate to severe exacerbations,” Massimo Corradi, MD, of the University of Parma (Italy), reported at the annual congress of the European Respiratory Society.
Dr. Corradi added that the new analysis confirms that the ICS/LABA combination has a “positive risk-benefit balance over LABA monotherapy, supporting [the argument that] the benefits of adding an ICS to a bronchodilator significantly outweigh potential risks.”
The FORWARD study was a two-arm trial designed to compare the efficacy and safety of fixed-dose treatment with beclometasone diproprionate and formoterol fumarate versus formoterol fumarate alone in 1,199 patients with severe COPD.
For inclusion in the study, patients had to have a post-bronchodilator forced expiratory volume in 1 second below 50% of predicted and a forced vital capacity ratio of less than 0.7. They also had to have a smoking history of 10 pack-years or more, and a history of at least one COPD exacerbation in the previous 12 months that had required treatment or hospitalization (Eur Respir J. 2013;41[1]:12-7).
After a 2-week run-in period, where all patients received a 24-mcg dose of formoterol fumarate, patients were randomized to continue treatment with formoterol fumarate or to receive the fixed-dose combination of beclometasone diproprionate 400 mcg and beclometasone diproprionate 24 mcg for 48 weeks.
A total of 1,186 patients, most of whom were male (69%) with a mean age of 64 years, formed the intention-to-treat population.
Published results (Respir Med. 2014;108[8]:1153-62) showed that the combination of the ICS beclometasone diproprionate and the LABA formoterol fumarate (Chiesi Farmaceutici SpA) was associated with a 28% reduction in the annual rate of moderate to severe exacerbations versus the LABA alone.
The adjusted rate of exacerbations per patient per year was 0.80 in patients treated with the ICS/LABA combination versus 1.12 for those treated with just the LABA, with an adjusted rate ratio of 0.72 (P less than .001).
The published data also showed that pneumonia was reported by 23 patients (3.8%) treated with the ICS/LABA and by 11 (1.8%) treated with the LABA only.
For the new analysis, Dr. Corradi and his coinvestigators looked at the cases of pneumonia and COPD exacerbations in more detail, plotting out the cumulative number of events over time and also characterizing the types of pneumonia in more detail.
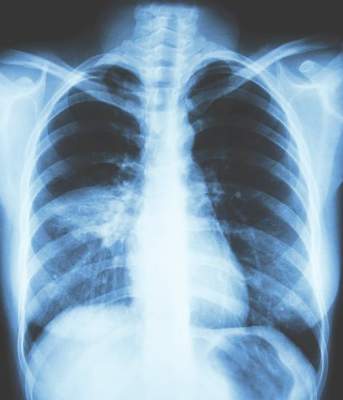
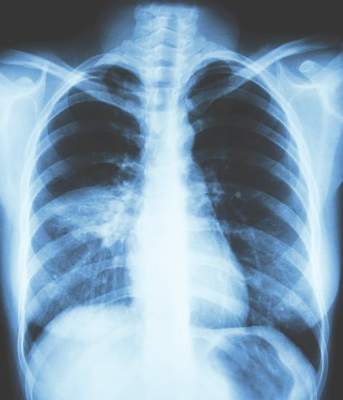
All patients had a chest x-ray to confirm the presence of pneumonia, he said, noting that overall there were 35 cases of pneumonia, 24 occurring in patients treated with the fixed-dose beclometasone diproprionate and formoterol fumarate combination and 11 in patients treated only with formoterol fumarate.
Of these cases, 25 required in-hospital treatment – 16 patients in the ICS/LABA arm and 9 in the LABA-only arm. There were three instances of patients acquiring pneumonia in hospital – two in the ICS/LABA and one in the LABA-only arm.
There were also two fatal cases of pneumonia – one in each treatment group. Neither were thought to be related to either of the treatments.
These findings are in line with a recent review of the use of ICS for COPD by the European Medicines Agency (EMA/488280/2016), which noted that “overall the benefits of inhaled corticosteroid medicines in treating COPD continue to outweigh their risks and there should be no change to the way in which these medicines are used.”
The European Medicines Agency advised that patients and clinicians need to “be alert for signs and symptoms of pneumonia, bearing in mind that the clinical features of pneumonia overlap with those of a worsening (exacerbation) of the underlying disease.”
Dr. Corradi has received speaker fees from Chiesi Farmaceutici SpA, which funded the FORWARD study, and his coauthors are employees of the company.
LONDON – The benefit of a fixed-dose inhaled corticosteroid (ICS) and long-acting beta-agonist (LABA) combination in reducing exacerbations of chronic obstructive pulmonary disease (COPD) far outweighed any risk for pneumonia in a post hoc analysis of the 48-week FORWARD study.
Although there were 13 extra pneumonia events when a fixed-dose combination of beclometasone diproprionate and formoterol fumarate (Foster, Chiesi Farmaceutici SpA) was used as compared to formoterol fumarate alone, there were 123 fewer moderate to severe COPD exacerbations over a 342-day analysis period.
“Analysis of pneumonia and exacerbation cumulative number of events shows that the number of incident pneumonia remains very small relative to that of moderate to severe exacerbations,” Massimo Corradi, MD, of the University of Parma (Italy), reported at the annual congress of the European Respiratory Society.
Dr. Corradi added that the new analysis confirms that the ICS/LABA combination has a “positive risk-benefit balance over LABA monotherapy, supporting [the argument that] the benefits of adding an ICS to a bronchodilator significantly outweigh potential risks.”
The FORWARD study was a two-arm trial designed to compare the efficacy and safety of fixed-dose treatment with beclometasone diproprionate and formoterol fumarate versus formoterol fumarate alone in 1,199 patients with severe COPD.
For inclusion in the study, patients had to have a post-bronchodilator forced expiratory volume in 1 second below 50% of predicted and a forced vital capacity ratio of less than 0.7. They also had to have a smoking history of 10 pack-years or more, and a history of at least one COPD exacerbation in the previous 12 months that had required treatment or hospitalization (Eur Respir J. 2013;41[1]:12-7).
After a 2-week run-in period, where all patients received a 24-mcg dose of formoterol fumarate, patients were randomized to continue treatment with formoterol fumarate or to receive the fixed-dose combination of beclometasone diproprionate 400 mcg and beclometasone diproprionate 24 mcg for 48 weeks.
A total of 1,186 patients, most of whom were male (69%) with a mean age of 64 years, formed the intention-to-treat population.
Published results (Respir Med. 2014;108[8]:1153-62) showed that the combination of the ICS beclometasone diproprionate and the LABA formoterol fumarate (Chiesi Farmaceutici SpA) was associated with a 28% reduction in the annual rate of moderate to severe exacerbations versus the LABA alone.
The adjusted rate of exacerbations per patient per year was 0.80 in patients treated with the ICS/LABA combination versus 1.12 for those treated with just the LABA, with an adjusted rate ratio of 0.72 (P less than .001).
The published data also showed that pneumonia was reported by 23 patients (3.8%) treated with the ICS/LABA and by 11 (1.8%) treated with the LABA only.
For the new analysis, Dr. Corradi and his coinvestigators looked at the cases of pneumonia and COPD exacerbations in more detail, plotting out the cumulative number of events over time and also characterizing the types of pneumonia in more detail.
All patients had a chest x-ray to confirm the presence of pneumonia, he said, noting that overall there were 35 cases of pneumonia, 24 occurring in patients treated with the fixed-dose beclometasone diproprionate and formoterol fumarate combination and 11 in patients treated only with formoterol fumarate.
Of these cases, 25 required in-hospital treatment – 16 patients in the ICS/LABA arm and 9 in the LABA-only arm. There were three instances of patients acquiring pneumonia in hospital – two in the ICS/LABA and one in the LABA-only arm.
There were also two fatal cases of pneumonia – one in each treatment group. Neither were thought to be related to either of the treatments.
These findings are in line with a recent review of the use of ICS for COPD by the European Medicines Agency (EMA/488280/2016), which noted that “overall the benefits of inhaled corticosteroid medicines in treating COPD continue to outweigh their risks and there should be no change to the way in which these medicines are used.”
The European Medicines Agency advised that patients and clinicians need to “be alert for signs and symptoms of pneumonia, bearing in mind that the clinical features of pneumonia overlap with those of a worsening (exacerbation) of the underlying disease.”
Dr. Corradi has received speaker fees from Chiesi Farmaceutici SpA, which funded the FORWARD study, and his coauthors are employees of the company.
LONDON – The benefit of a fixed-dose inhaled corticosteroid (ICS) and long-acting beta-agonist (LABA) combination in reducing exacerbations of chronic obstructive pulmonary disease (COPD) far outweighed any risk for pneumonia in a post hoc analysis of the 48-week FORWARD study.
Although there were 13 extra pneumonia events when a fixed-dose combination of beclometasone diproprionate and formoterol fumarate (Foster, Chiesi Farmaceutici SpA) was used as compared to formoterol fumarate alone, there were 123 fewer moderate to severe COPD exacerbations over a 342-day analysis period.
“Analysis of pneumonia and exacerbation cumulative number of events shows that the number of incident pneumonia remains very small relative to that of moderate to severe exacerbations,” Massimo Corradi, MD, of the University of Parma (Italy), reported at the annual congress of the European Respiratory Society.
Dr. Corradi added that the new analysis confirms that the ICS/LABA combination has a “positive risk-benefit balance over LABA monotherapy, supporting [the argument that] the benefits of adding an ICS to a bronchodilator significantly outweigh potential risks.”
The FORWARD study was a two-arm trial designed to compare the efficacy and safety of fixed-dose treatment with beclometasone diproprionate and formoterol fumarate versus formoterol fumarate alone in 1,199 patients with severe COPD.
For inclusion in the study, patients had to have a post-bronchodilator forced expiratory volume in 1 second below 50% of predicted and a forced vital capacity ratio of less than 0.7. They also had to have a smoking history of 10 pack-years or more, and a history of at least one COPD exacerbation in the previous 12 months that had required treatment or hospitalization (Eur Respir J. 2013;41[1]:12-7).
After a 2-week run-in period, where all patients received a 24-mcg dose of formoterol fumarate, patients were randomized to continue treatment with formoterol fumarate or to receive the fixed-dose combination of beclometasone diproprionate 400 mcg and beclometasone diproprionate 24 mcg for 48 weeks.
A total of 1,186 patients, most of whom were male (69%) with a mean age of 64 years, formed the intention-to-treat population.
Published results (Respir Med. 2014;108[8]:1153-62) showed that the combination of the ICS beclometasone diproprionate and the LABA formoterol fumarate (Chiesi Farmaceutici SpA) was associated with a 28% reduction in the annual rate of moderate to severe exacerbations versus the LABA alone.
The adjusted rate of exacerbations per patient per year was 0.80 in patients treated with the ICS/LABA combination versus 1.12 for those treated with just the LABA, with an adjusted rate ratio of 0.72 (P less than .001).
The published data also showed that pneumonia was reported by 23 patients (3.8%) treated with the ICS/LABA and by 11 (1.8%) treated with the LABA only.
For the new analysis, Dr. Corradi and his coinvestigators looked at the cases of pneumonia and COPD exacerbations in more detail, plotting out the cumulative number of events over time and also characterizing the types of pneumonia in more detail.
All patients had a chest x-ray to confirm the presence of pneumonia, he said, noting that overall there were 35 cases of pneumonia, 24 occurring in patients treated with the fixed-dose beclometasone diproprionate and formoterol fumarate combination and 11 in patients treated only with formoterol fumarate.
Of these cases, 25 required in-hospital treatment – 16 patients in the ICS/LABA arm and 9 in the LABA-only arm. There were three instances of patients acquiring pneumonia in hospital – two in the ICS/LABA and one in the LABA-only arm.
There were also two fatal cases of pneumonia – one in each treatment group. Neither were thought to be related to either of the treatments.
These findings are in line with a recent review of the use of ICS for COPD by the European Medicines Agency (EMA/488280/2016), which noted that “overall the benefits of inhaled corticosteroid medicines in treating COPD continue to outweigh their risks and there should be no change to the way in which these medicines are used.”
The European Medicines Agency advised that patients and clinicians need to “be alert for signs and symptoms of pneumonia, bearing in mind that the clinical features of pneumonia overlap with those of a worsening (exacerbation) of the underlying disease.”
Dr. Corradi has received speaker fees from Chiesi Farmaceutici SpA, which funded the FORWARD study, and his coauthors are employees of the company.
AT THE ERS CONGRESS 2016
Key clinical point: The risk for pneumonia associated with drugs containing inhaled corticosteroids (ICS) is outweighed by the reduction in chronic obstructive pulmonary disease (COPD) exacerbations that can be achieved.
Major finding: There were 13 extra pneumonia events but 123 fewer COPD exacerbations when a fixed dose ICS/long-acting beta-agonist (LABA) combination was used versus a LABA alone.
Data source: Post hoc analysis of the FORWARD study, a multicenter, randomized, double-blind, active-controlled 48-week trial of a fixed-dose ICS/LABA combination versus LABA for reducing exacerbations in 1,186 patients with COPD.
Disclosures: Dr. Corradi has received speaker fees from Chiesi Farmaceutici SpA, which funded the FORWARD study, and his coauthors are employees of the company.
How Do Diet, Exercise, and Supplements Affect Parkinson’s Disease Progression?

Prior studies have found that people who consume green tea, coffee, and blueberries and avoid dairy may have a lower risk of Parkinson’s disease. Whether nutrition is associated with rate of disease progression in patients with Parkinson’s disease, however, is not known.
To evaluate whether diet, exercise, and supplements are associated with rate of Parkinson’s disease progression, Laurie Mischley, ND, PhD, MPH, Assistant Research Scientist at Bastyr University Research Institute in Kenmore, Washington, and Richard Lau, MPH, a PhD student in the College of Public Health and Human Sciences at Oregon State University in Corvalis conducted an Internet-based natural history study. A total of 1,024 patients participated in the study. Participants had a mean age of 60.7 and had been diagnosed with Parkinson’s disease for an average of 6.7 years.
The researchers used the Patient-Reported Outcomes in Parkinson’s Disease (PRO-PD) scale to assess Parkinson’s disease severity. Disease progression was defined as PRO-PD score adjusted for age and years since diagnosis. They used baseline food frequency questionnaires to quantify dietary intake in the cross-sectional analysis.
Fresh fruit, fresh vegetables, nuts and seeds, olive oil, fish (non-fried), wine, eggs, and fresh herbs were associated with a statistically significant improvement in PRO-PD score, the researchers said. Fried foods, beef, diet soda, canned fruits, and canned vegetables were associated with more severe disease. Dairy consumption was not associated with disease severity.
Of the supplements and pharmaceuticals studied, oral glutathione, rasagiline, and coenzyme Q10 were associated with improved PRO-PD scores, whereas iron was associated with more severe disease. The effect of melatonin was not significant, however, when the researchers considered poor sleep.The researchers observed a dose response curve with exercise. Exercising at least 30 minutes daily was associated with the greatest reduction in disease severity.
“Whether iron, fried foods, diet soda, or canned goods provide environmental insults that accelerate disease progression warrants immediate attention,” the researchers concluded. “This pragmatic natural history study offers the first evidence base for prescribing lifestyle modification (beyond exercise) to patients with Parkinson’s disease. Patients should be empowered to know that they can make choices that affect outcomes."
—Jake Remaly
Prior studies have found that people who consume green tea, coffee, and blueberries and avoid dairy may have a lower risk of Parkinson’s disease. Whether nutrition is associated with rate of disease progression in patients with Parkinson’s disease, however, is not known.
To evaluate whether diet, exercise, and supplements are associated with rate of Parkinson’s disease progression, Laurie Mischley, ND, PhD, MPH, Assistant Research Scientist at Bastyr University Research Institute in Kenmore, Washington, and Richard Lau, MPH, a PhD student in the College of Public Health and Human Sciences at Oregon State University in Corvalis conducted an Internet-based natural history study. A total of 1,024 patients participated in the study. Participants had a mean age of 60.7 and had been diagnosed with Parkinson’s disease for an average of 6.7 years.
The researchers used the Patient-Reported Outcomes in Parkinson’s Disease (PRO-PD) scale to assess Parkinson’s disease severity. Disease progression was defined as PRO-PD score adjusted for age and years since diagnosis. They used baseline food frequency questionnaires to quantify dietary intake in the cross-sectional analysis.
Fresh fruit, fresh vegetables, nuts and seeds, olive oil, fish (non-fried), wine, eggs, and fresh herbs were associated with a statistically significant improvement in PRO-PD score, the researchers said. Fried foods, beef, diet soda, canned fruits, and canned vegetables were associated with more severe disease. Dairy consumption was not associated with disease severity.
Of the supplements and pharmaceuticals studied, oral glutathione, rasagiline, and coenzyme Q10 were associated with improved PRO-PD scores, whereas iron was associated with more severe disease. The effect of melatonin was not significant, however, when the researchers considered poor sleep.The researchers observed a dose response curve with exercise. Exercising at least 30 minutes daily was associated with the greatest reduction in disease severity.
“Whether iron, fried foods, diet soda, or canned goods provide environmental insults that accelerate disease progression warrants immediate attention,” the researchers concluded. “This pragmatic natural history study offers the first evidence base for prescribing lifestyle modification (beyond exercise) to patients with Parkinson’s disease. Patients should be empowered to know that they can make choices that affect outcomes."
—Jake Remaly
Prior studies have found that people who consume green tea, coffee, and blueberries and avoid dairy may have a lower risk of Parkinson’s disease. Whether nutrition is associated with rate of disease progression in patients with Parkinson’s disease, however, is not known.
To evaluate whether diet, exercise, and supplements are associated with rate of Parkinson’s disease progression, Laurie Mischley, ND, PhD, MPH, Assistant Research Scientist at Bastyr University Research Institute in Kenmore, Washington, and Richard Lau, MPH, a PhD student in the College of Public Health and Human Sciences at Oregon State University in Corvalis conducted an Internet-based natural history study. A total of 1,024 patients participated in the study. Participants had a mean age of 60.7 and had been diagnosed with Parkinson’s disease for an average of 6.7 years.
The researchers used the Patient-Reported Outcomes in Parkinson’s Disease (PRO-PD) scale to assess Parkinson’s disease severity. Disease progression was defined as PRO-PD score adjusted for age and years since diagnosis. They used baseline food frequency questionnaires to quantify dietary intake in the cross-sectional analysis.
Fresh fruit, fresh vegetables, nuts and seeds, olive oil, fish (non-fried), wine, eggs, and fresh herbs were associated with a statistically significant improvement in PRO-PD score, the researchers said. Fried foods, beef, diet soda, canned fruits, and canned vegetables were associated with more severe disease. Dairy consumption was not associated with disease severity.
Of the supplements and pharmaceuticals studied, oral glutathione, rasagiline, and coenzyme Q10 were associated with improved PRO-PD scores, whereas iron was associated with more severe disease. The effect of melatonin was not significant, however, when the researchers considered poor sleep.The researchers observed a dose response curve with exercise. Exercising at least 30 minutes daily was associated with the greatest reduction in disease severity.
“Whether iron, fried foods, diet soda, or canned goods provide environmental insults that accelerate disease progression warrants immediate attention,” the researchers concluded. “This pragmatic natural history study offers the first evidence base for prescribing lifestyle modification (beyond exercise) to patients with Parkinson’s disease. Patients should be empowered to know that they can make choices that affect outcomes."
—Jake Remaly
Can Treating Neuroinflammation in REM Sleep Behavior Disorder Delay Parkinson’s Disease Onset?
PORTLAND, OR—In patients with idiopathic REM sleep behavior disorder, microglial activation is increased in the substantia nigra, compared with controls, and microglial activation correlates with putamenal dopaminergic dysfunction, according to research presented at the Fourth World Parkinson Congress. These findings suggest that “anti-inflammatory agents could possibly delay progression to a manifest synucleinopathy in subjects with idiopathic REM sleep behavior disorder,” researchers said.
Longitudinal studies have found that patients with idiopathic REM sleep behavior disorder have an increased risk of Parkinson’s disease and related Lewy body disorders. “This implies that, in idiopathic REM sleep behavior disorder, the underlying pathology of developing neurodegenerative disorders can be investigated years prior to the development of manifest symptoms,” said Morten Gersel Stokholm, MD, a researcher in the Department of Clinical Medicine at Aarhus University and the Department of Nuclear Medicine & PET-Centre at Aarhus University Hospital, Denmark, and his research colleagues.

Chronic activation of microglial cells may have a detrimental effect on neurons and contribute to the development of neurodegenerative disorders. Studies have found that uptake of an in vivo marker of microglial activation, 11C-PK11195, is increased in Parkinson’s disease and other neurodegenerative disorders.
To investigate the in vivo occurrence of neuroinflammation in the brains of patients with idiopathic REM sleep behavior disorder and neuroinflammation’s temporal relationship with striatal dopamine dysfunction, Dr. Stokholm and colleagues conducted a multitracer PET study of patients with idiopathic REM sleep behavior disorder.
The investigators enrolled 15 patients with polysomnography-confirmed idiopathic REM sleep behavior disorder at Aarhus University Hospital and Hospital Clínic de Barcelona. They also enrolled 19 matched controls. Participants underwent two PET scans with 18F-DOPA and 11C- PK11195 and a structural T1 MRI scan. Parametric images of specific tracer uptake (ie, F-dopa Ki-values and PK11195 binding potential) were constructed at voxel level using Patlak graphical analysis and a supervised cluster-analysis with compartmental modeling, respectively. A region of interest analysis was performed on a priori defined regions.
Compared with controls, patients with idiopathic REM sleep behavior disorder showed significantly reduced 18F-DOPA tracer uptake in the substantia nigra. Patients with higher substantia nigra11C-PK11195 binding also had increased binding in the ipsilateral putamen. Patients with more severe reductions in putaminal 18F-DOPA uptake had significantly higher 11C-PK11195 binding in the putamen and substantia nigra.
—Jake Remaly
PORTLAND, OR—In patients with idiopathic REM sleep behavior disorder, microglial activation is increased in the substantia nigra, compared with controls, and microglial activation correlates with putamenal dopaminergic dysfunction, according to research presented at the Fourth World Parkinson Congress. These findings suggest that “anti-inflammatory agents could possibly delay progression to a manifest synucleinopathy in subjects with idiopathic REM sleep behavior disorder,” researchers said.
Longitudinal studies have found that patients with idiopathic REM sleep behavior disorder have an increased risk of Parkinson’s disease and related Lewy body disorders. “This implies that, in idiopathic REM sleep behavior disorder, the underlying pathology of developing neurodegenerative disorders can be investigated years prior to the development of manifest symptoms,” said Morten Gersel Stokholm, MD, a researcher in the Department of Clinical Medicine at Aarhus University and the Department of Nuclear Medicine & PET-Centre at Aarhus University Hospital, Denmark, and his research colleagues.
Chronic activation of microglial cells may have a detrimental effect on neurons and contribute to the development of neurodegenerative disorders. Studies have found that uptake of an in vivo marker of microglial activation, 11C-PK11195, is increased in Parkinson’s disease and other neurodegenerative disorders.
To investigate the in vivo occurrence of neuroinflammation in the brains of patients with idiopathic REM sleep behavior disorder and neuroinflammation’s temporal relationship with striatal dopamine dysfunction, Dr. Stokholm and colleagues conducted a multitracer PET study of patients with idiopathic REM sleep behavior disorder.
The investigators enrolled 15 patients with polysomnography-confirmed idiopathic REM sleep behavior disorder at Aarhus University Hospital and Hospital Clínic de Barcelona. They also enrolled 19 matched controls. Participants underwent two PET scans with 18F-DOPA and 11C- PK11195 and a structural T1 MRI scan. Parametric images of specific tracer uptake (ie, F-dopa Ki-values and PK11195 binding potential) were constructed at voxel level using Patlak graphical analysis and a supervised cluster-analysis with compartmental modeling, respectively. A region of interest analysis was performed on a priori defined regions.
Compared with controls, patients with idiopathic REM sleep behavior disorder showed significantly reduced 18F-DOPA tracer uptake in the substantia nigra. Patients with higher substantia nigra11C-PK11195 binding also had increased binding in the ipsilateral putamen. Patients with more severe reductions in putaminal 18F-DOPA uptake had significantly higher 11C-PK11195 binding in the putamen and substantia nigra.
—Jake Remaly
PORTLAND, OR—In patients with idiopathic REM sleep behavior disorder, microglial activation is increased in the substantia nigra, compared with controls, and microglial activation correlates with putamenal dopaminergic dysfunction, according to research presented at the Fourth World Parkinson Congress. These findings suggest that “anti-inflammatory agents could possibly delay progression to a manifest synucleinopathy in subjects with idiopathic REM sleep behavior disorder,” researchers said.
Longitudinal studies have found that patients with idiopathic REM sleep behavior disorder have an increased risk of Parkinson’s disease and related Lewy body disorders. “This implies that, in idiopathic REM sleep behavior disorder, the underlying pathology of developing neurodegenerative disorders can be investigated years prior to the development of manifest symptoms,” said Morten Gersel Stokholm, MD, a researcher in the Department of Clinical Medicine at Aarhus University and the Department of Nuclear Medicine & PET-Centre at Aarhus University Hospital, Denmark, and his research colleagues.
Chronic activation of microglial cells may have a detrimental effect on neurons and contribute to the development of neurodegenerative disorders. Studies have found that uptake of an in vivo marker of microglial activation, 11C-PK11195, is increased in Parkinson’s disease and other neurodegenerative disorders.
To investigate the in vivo occurrence of neuroinflammation in the brains of patients with idiopathic REM sleep behavior disorder and neuroinflammation’s temporal relationship with striatal dopamine dysfunction, Dr. Stokholm and colleagues conducted a multitracer PET study of patients with idiopathic REM sleep behavior disorder.
The investigators enrolled 15 patients with polysomnography-confirmed idiopathic REM sleep behavior disorder at Aarhus University Hospital and Hospital Clínic de Barcelona. They also enrolled 19 matched controls. Participants underwent two PET scans with 18F-DOPA and 11C- PK11195 and a structural T1 MRI scan. Parametric images of specific tracer uptake (ie, F-dopa Ki-values and PK11195 binding potential) were constructed at voxel level using Patlak graphical analysis and a supervised cluster-analysis with compartmental modeling, respectively. A region of interest analysis was performed on a priori defined regions.
Compared with controls, patients with idiopathic REM sleep behavior disorder showed significantly reduced 18F-DOPA tracer uptake in the substantia nigra. Patients with higher substantia nigra11C-PK11195 binding also had increased binding in the ipsilateral putamen. Patients with more severe reductions in putaminal 18F-DOPA uptake had significantly higher 11C-PK11195 binding in the putamen and substantia nigra.
—Jake Remaly
NOACs outpace warfarin for afib anticoagulation
The NOAC revolution has happened.
New oral anticoagulants (NOACs) now claim the majority of the oral anticoagulant market in patients with new atrial fibrillation, wresting the advantage from warfarin in the U.S. and globally. This is the promise NOACs held even while still in development, the potential to whittle warfarin use down to a shadow of what it was. Despite a sputtering reception when the first NOAC, dabigatran (Pradaxa), came onto the U.S. market in late 2010, the four NOACs (also apixaban [Eliquis], edoxaban [Savaysa], and rivaroxaban [Xarelto]) now available in the United States and elsewhere gradually gained traction and today they collectively form the most widely used oral anticoagulant option for patients newly diagnosed with atrial fibrillation.

Data reported in August at the annual congress of the European Society of Cardiology showed that in Denmark during the first half of 2015, NOACs accounted for 73% of oral anticoagulant prescriptions for patients with newly diagnosed atrial fibrillation, Kasper Gadsboll, MD, reported. He and his associates studied NOAC and warfarin use in a total of 108,410 atrial fibrillation patients newly diagnosed starting in 2005. Through 2010, warfarin was the only option, but starting in early 2011 when the first NOAC became available in Denmark, use of the class rose sharply with a corresponding plummet in warfarin prescriptions. Not only did the NOACs largely supplant warfarin during 2011-2015, but they also powered an overall rise in the percentage of atrial fibrillation patients treated with an oral anticoagulant, boosting the rate by a relative 75% between the end of 2009 and mid-2015.
“People were reluctant to start an oral anticoagulant because they feared bleeding. Now with NOACs they are not as fearful,” said Dr. Gadsboll, a cardiology researcher at Gentofte Hospital in Hellerup, Denmark. He also noted that in Denmark the national health system pays for prescribed drugs, so the increased direct cost for NOAC treatment in place of warfarin is covered by the Danish government.

“NOAC uptake is gathering steam,” commented Stuart J. Connolly, MD, an electrophysiologist and professor of medicine at McMaster University in Hamilton, Ontario. “When the government pays for it, people prescribe it more, but increased NOAC use is happening everywhere,” said Dr. Connolly, who led major trials that assessed dabigatran and apixaban in atrial fibrillation patients. “I’ve seen Canadian data that show warfarin use is falling and NOAC use is increasing. People are more willing to prescribe NOACs because they are safer,” he said in an interview.
The most current U.S. data I found came from the IMS Health National Disease and Therapeutic Index in 2014, based on a survey of a representative sample of about 4,800 U.S. office-based physicians. The results showed that the NOAC share of oral anticoagulant use in patients who had office visits for atrial fibrillation jumped from 6% of patients in early 2011 to roughly half of all atrial fibrillation patients, 48%, during 2014 (Am J Med. 2015 Dec;128[12]:1300-5). That trajectory makes it likely that by now NOACs have a clear lead.
It’s a similar story in several European countries, such as in Belgium where NOACs now account for about 70% of new prescriptions for atrial fibrillation patients, commented Freek W.A. Verheugt, MD. In the Netherlands, however, NOACs have not taken off, in large part because a popular and entrenched thrombosis clinic system exists that employs thousands of Dutch workers, thereby making choice of an anticoagulant drug a social and political question as well as a medical one, he said. “Warfarin is still indicated for patients with an artificial heart valve or poor kidney function,” noted Dr. Verheugt, professor of cardiology at the University of Nijmegen in the Netherlands, “but a majority of atrial fibrillation patients will eventually change to a NOAC. Even when treatment with warfarin has a good time-in-therapeutic-range, NOACs are still better. They are so much safer.”
On Twitter @mitchelzoler
The NOAC revolution has happened.
New oral anticoagulants (NOACs) now claim the majority of the oral anticoagulant market in patients with new atrial fibrillation, wresting the advantage from warfarin in the U.S. and globally. This is the promise NOACs held even while still in development, the potential to whittle warfarin use down to a shadow of what it was. Despite a sputtering reception when the first NOAC, dabigatran (Pradaxa), came onto the U.S. market in late 2010, the four NOACs (also apixaban [Eliquis], edoxaban [Savaysa], and rivaroxaban [Xarelto]) now available in the United States and elsewhere gradually gained traction and today they collectively form the most widely used oral anticoagulant option for patients newly diagnosed with atrial fibrillation.
Data reported in August at the annual congress of the European Society of Cardiology showed that in Denmark during the first half of 2015, NOACs accounted for 73% of oral anticoagulant prescriptions for patients with newly diagnosed atrial fibrillation, Kasper Gadsboll, MD, reported. He and his associates studied NOAC and warfarin use in a total of 108,410 atrial fibrillation patients newly diagnosed starting in 2005. Through 2010, warfarin was the only option, but starting in early 2011 when the first NOAC became available in Denmark, use of the class rose sharply with a corresponding plummet in warfarin prescriptions. Not only did the NOACs largely supplant warfarin during 2011-2015, but they also powered an overall rise in the percentage of atrial fibrillation patients treated with an oral anticoagulant, boosting the rate by a relative 75% between the end of 2009 and mid-2015.
“People were reluctant to start an oral anticoagulant because they feared bleeding. Now with NOACs they are not as fearful,” said Dr. Gadsboll, a cardiology researcher at Gentofte Hospital in Hellerup, Denmark. He also noted that in Denmark the national health system pays for prescribed drugs, so the increased direct cost for NOAC treatment in place of warfarin is covered by the Danish government.
“NOAC uptake is gathering steam,” commented Stuart J. Connolly, MD, an electrophysiologist and professor of medicine at McMaster University in Hamilton, Ontario. “When the government pays for it, people prescribe it more, but increased NOAC use is happening everywhere,” said Dr. Connolly, who led major trials that assessed dabigatran and apixaban in atrial fibrillation patients. “I’ve seen Canadian data that show warfarin use is falling and NOAC use is increasing. People are more willing to prescribe NOACs because they are safer,” he said in an interview.
The most current U.S. data I found came from the IMS Health National Disease and Therapeutic Index in 2014, based on a survey of a representative sample of about 4,800 U.S. office-based physicians. The results showed that the NOAC share of oral anticoagulant use in patients who had office visits for atrial fibrillation jumped from 6% of patients in early 2011 to roughly half of all atrial fibrillation patients, 48%, during 2014 (Am J Med. 2015 Dec;128[12]:1300-5). That trajectory makes it likely that by now NOACs have a clear lead.
It’s a similar story in several European countries, such as in Belgium where NOACs now account for about 70% of new prescriptions for atrial fibrillation patients, commented Freek W.A. Verheugt, MD. In the Netherlands, however, NOACs have not taken off, in large part because a popular and entrenched thrombosis clinic system exists that employs thousands of Dutch workers, thereby making choice of an anticoagulant drug a social and political question as well as a medical one, he said. “Warfarin is still indicated for patients with an artificial heart valve or poor kidney function,” noted Dr. Verheugt, professor of cardiology at the University of Nijmegen in the Netherlands, “but a majority of atrial fibrillation patients will eventually change to a NOAC. Even when treatment with warfarin has a good time-in-therapeutic-range, NOACs are still better. They are so much safer.”
On Twitter @mitchelzoler
The NOAC revolution has happened.
New oral anticoagulants (NOACs) now claim the majority of the oral anticoagulant market in patients with new atrial fibrillation, wresting the advantage from warfarin in the U.S. and globally. This is the promise NOACs held even while still in development, the potential to whittle warfarin use down to a shadow of what it was. Despite a sputtering reception when the first NOAC, dabigatran (Pradaxa), came onto the U.S. market in late 2010, the four NOACs (also apixaban [Eliquis], edoxaban [Savaysa], and rivaroxaban [Xarelto]) now available in the United States and elsewhere gradually gained traction and today they collectively form the most widely used oral anticoagulant option for patients newly diagnosed with atrial fibrillation.
Data reported in August at the annual congress of the European Society of Cardiology showed that in Denmark during the first half of 2015, NOACs accounted for 73% of oral anticoagulant prescriptions for patients with newly diagnosed atrial fibrillation, Kasper Gadsboll, MD, reported. He and his associates studied NOAC and warfarin use in a total of 108,410 atrial fibrillation patients newly diagnosed starting in 2005. Through 2010, warfarin was the only option, but starting in early 2011 when the first NOAC became available in Denmark, use of the class rose sharply with a corresponding plummet in warfarin prescriptions. Not only did the NOACs largely supplant warfarin during 2011-2015, but they also powered an overall rise in the percentage of atrial fibrillation patients treated with an oral anticoagulant, boosting the rate by a relative 75% between the end of 2009 and mid-2015.
“People were reluctant to start an oral anticoagulant because they feared bleeding. Now with NOACs they are not as fearful,” said Dr. Gadsboll, a cardiology researcher at Gentofte Hospital in Hellerup, Denmark. He also noted that in Denmark the national health system pays for prescribed drugs, so the increased direct cost for NOAC treatment in place of warfarin is covered by the Danish government.
“NOAC uptake is gathering steam,” commented Stuart J. Connolly, MD, an electrophysiologist and professor of medicine at McMaster University in Hamilton, Ontario. “When the government pays for it, people prescribe it more, but increased NOAC use is happening everywhere,” said Dr. Connolly, who led major trials that assessed dabigatran and apixaban in atrial fibrillation patients. “I’ve seen Canadian data that show warfarin use is falling and NOAC use is increasing. People are more willing to prescribe NOACs because they are safer,” he said in an interview.
The most current U.S. data I found came from the IMS Health National Disease and Therapeutic Index in 2014, based on a survey of a representative sample of about 4,800 U.S. office-based physicians. The results showed that the NOAC share of oral anticoagulant use in patients who had office visits for atrial fibrillation jumped from 6% of patients in early 2011 to roughly half of all atrial fibrillation patients, 48%, during 2014 (Am J Med. 2015 Dec;128[12]:1300-5). That trajectory makes it likely that by now NOACs have a clear lead.
It’s a similar story in several European countries, such as in Belgium where NOACs now account for about 70% of new prescriptions for atrial fibrillation patients, commented Freek W.A. Verheugt, MD. In the Netherlands, however, NOACs have not taken off, in large part because a popular and entrenched thrombosis clinic system exists that employs thousands of Dutch workers, thereby making choice of an anticoagulant drug a social and political question as well as a medical one, he said. “Warfarin is still indicated for patients with an artificial heart valve or poor kidney function,” noted Dr. Verheugt, professor of cardiology at the University of Nijmegen in the Netherlands, “but a majority of atrial fibrillation patients will eventually change to a NOAC. Even when treatment with warfarin has a good time-in-therapeutic-range, NOACs are still better. They are so much safer.”
On Twitter @mitchelzoler

Direct Immunofluorescence Staining Patterns in Blistering Disorders
Review the PDF of the fact sheet on Direct Immunofluorescence Staining Patterns in Blistering Disorders with board-relevant, easy-to-review material. This fact sheet reviews the dermatologic conditions that typically have positive immunofluorescence staining patterns.
Practice Questions
1. Which autoimmune blistering disease shows deposition of immunoglobulin on the floor of salt-split skin?
a. BP
b. dermatitis herpetiformis
c. epidermolysis bullosa acquisita
d. paraneoplastic pemphigus
e. PV
2. What medicine is commonly implicated in drug-induced pemphigus?
a. acetaminophen
b. amoxicillin
c. naproxen
d. penicillamine
e. penicillin
3. Which autoimmune blistering disease predominantly shows deposition of IgG on DIF?
a. dermatitis herpetiformis
b. IgA pemphigus
c. linear IgA bullous dermatosis
d. paraneoplastic pemphigus
e. porphyria cutanea tarda
4. Which of the following diseases has a negative direct immunofluorescence?
a. dermatitis herpetiformis
b. herpes gestationis
c. pemphigus vulgaris
d. porphyria cutanea tarda
e. transient acantholytic dermatosis
5. Which of the following diseases shows a linear deposition of IgG and C3 along the dermoepidermal junction?
a. CP
b. IgA pemphigus
c. PF
d. porphyria cutanea tarda
e. PV
Answers to practice questions provided on next page
Practice Question Answers
1. Which autoimmune blistering disease shows deposition of immunoglobulin on the floor of salt-split skin?
a. BP
b. dermatitis herpetiformis
c. epidermolysis bullosa acquisita
d. paraneoplastic pemphigus
e. PV
2. What medicine is commonly implicated in drug-induced pemphigus?
a. acetaminophen
b. amoxicillin
c. naproxen
d. penicillamine
e. penicillin
3. Which autoimmune blistering disease predominantly shows deposition of IgG on DIF?
a. dermatitis herpetiformis
b. IgA pemphigus
c. linear IgA bullous dermatosis
d. paraneoplastic pemphigus
e. porphyria cutanea tarda
4. Which of the following diseases has a negative direct immunofluorescence?
a. dermatitis herpetiformis
b. herpes gestationis
c. pemphigus vulgaris
d. porphyria cutanea tarda
e. transient acantholytic dermatosis
5. Which of the following diseases shows a linear deposition of IgG and C3 along the dermoepidermal junction?
a. CP
b. IgA pemphigus
c. PF
d. porphyria cutanea tarda
e. PV
Review the PDF of the fact sheet on Direct Immunofluorescence Staining Patterns in Blistering Disorders with board-relevant, easy-to-review material. This fact sheet reviews the dermatologic conditions that typically have positive immunofluorescence staining patterns.
Practice Questions
1. Which autoimmune blistering disease shows deposition of immunoglobulin on the floor of salt-split skin?
a. BP
b. dermatitis herpetiformis
c. epidermolysis bullosa acquisita
d. paraneoplastic pemphigus
e. PV
2. What medicine is commonly implicated in drug-induced pemphigus?
a. acetaminophen
b. amoxicillin
c. naproxen
d. penicillamine
e. penicillin
3. Which autoimmune blistering disease predominantly shows deposition of IgG on DIF?
a. dermatitis herpetiformis
b. IgA pemphigus
c. linear IgA bullous dermatosis
d. paraneoplastic pemphigus
e. porphyria cutanea tarda
4. Which of the following diseases has a negative direct immunofluorescence?
a. dermatitis herpetiformis
b. herpes gestationis
c. pemphigus vulgaris
d. porphyria cutanea tarda
e. transient acantholytic dermatosis
5. Which of the following diseases shows a linear deposition of IgG and C3 along the dermoepidermal junction?
a. CP
b. IgA pemphigus
c. PF
d. porphyria cutanea tarda
e. PV
Answers to practice questions provided on next page
Practice Question Answers
1. Which autoimmune blistering disease shows deposition of immunoglobulin on the floor of salt-split skin?
a. BP
b. dermatitis herpetiformis
c. epidermolysis bullosa acquisita
d. paraneoplastic pemphigus
e. PV
2. What medicine is commonly implicated in drug-induced pemphigus?
a. acetaminophen
b. amoxicillin
c. naproxen
d. penicillamine
e. penicillin
3. Which autoimmune blistering disease predominantly shows deposition of IgG on DIF?
a. dermatitis herpetiformis
b. IgA pemphigus
c. linear IgA bullous dermatosis
d. paraneoplastic pemphigus
e. porphyria cutanea tarda
4. Which of the following diseases has a negative direct immunofluorescence?
a. dermatitis herpetiformis
b. herpes gestationis
c. pemphigus vulgaris
d. porphyria cutanea tarda
e. transient acantholytic dermatosis
5. Which of the following diseases shows a linear deposition of IgG and C3 along the dermoepidermal junction?
a. CP
b. IgA pemphigus
c. PF
d. porphyria cutanea tarda
e. PV
Review the PDF of the fact sheet on Direct Immunofluorescence Staining Patterns in Blistering Disorders with board-relevant, easy-to-review material. This fact sheet reviews the dermatologic conditions that typically have positive immunofluorescence staining patterns.
Practice Questions
1. Which autoimmune blistering disease shows deposition of immunoglobulin on the floor of salt-split skin?
a. BP
b. dermatitis herpetiformis
c. epidermolysis bullosa acquisita
d. paraneoplastic pemphigus
e. PV
2. What medicine is commonly implicated in drug-induced pemphigus?
a. acetaminophen
b. amoxicillin
c. naproxen
d. penicillamine
e. penicillin
3. Which autoimmune blistering disease predominantly shows deposition of IgG on DIF?
a. dermatitis herpetiformis
b. IgA pemphigus
c. linear IgA bullous dermatosis
d. paraneoplastic pemphigus
e. porphyria cutanea tarda
4. Which of the following diseases has a negative direct immunofluorescence?
a. dermatitis herpetiformis
b. herpes gestationis
c. pemphigus vulgaris
d. porphyria cutanea tarda
e. transient acantholytic dermatosis
5. Which of the following diseases shows a linear deposition of IgG and C3 along the dermoepidermal junction?
a. CP
b. IgA pemphigus
c. PF
d. porphyria cutanea tarda
e. PV
Answers to practice questions provided on next page
Practice Question Answers
1. Which autoimmune blistering disease shows deposition of immunoglobulin on the floor of salt-split skin?
a. BP
b. dermatitis herpetiformis
c. epidermolysis bullosa acquisita
d. paraneoplastic pemphigus
e. PV
2. What medicine is commonly implicated in drug-induced pemphigus?
a. acetaminophen
b. amoxicillin
c. naproxen
d. penicillamine
e. penicillin
3. Which autoimmune blistering disease predominantly shows deposition of IgG on DIF?
a. dermatitis herpetiformis
b. IgA pemphigus
c. linear IgA bullous dermatosis
d. paraneoplastic pemphigus
e. porphyria cutanea tarda
4. Which of the following diseases has a negative direct immunofluorescence?
a. dermatitis herpetiformis
b. herpes gestationis
c. pemphigus vulgaris
d. porphyria cutanea tarda
e. transient acantholytic dermatosis
5. Which of the following diseases shows a linear deposition of IgG and C3 along the dermoepidermal junction?
a. CP
b. IgA pemphigus
c. PF
d. porphyria cutanea tarda
e. PV

CardioMEMS shows real-world heart failure benefit
ORLANDO – Pulmonary artery pressure monitoring using an implanted device was even more effective for controlling pulmonary artery pressures in 2,000 real-world U.S. heart failure patients than it was in the pivotal trial that led to the device’s regulatory approval.
Data from the first 2,000 U.S. heart failure patients to receive the CardioMEMS pulmonary artery (PA) pressure monitoring device and have at least 6 months of follow-up data since the device received Food and Drug Administration approval in 2014 showed that cumulative PA pressure reductions in these patients during the first 6 months of use averaged 434 mm Hg per patient when compared with their baseline PA pressure when they first received the device. This was nearly threefold better than the average 150–mm Hg cumulative reduction in PA pressure per patient during 6 months of use seen in the CHAMPION trial, Dr. William T. Abraham reported at the annual scientific meeting of the Heart Failure Society of America.

Although this analysis of data from the registry maintained for U.S. patients who receive the CardioMEMS device does not yet include information on how these patients fared clinically, and specifically how often they required rehospitalization for heart failure, the strikingly high level of PA pressure control seen in the first 2,000 U.S. patients bodes well for what the clinical findings will show once they are available.
“In our experience with PA pressure monitoring, there is almost a linear relationship between reduced PA pressures and reduced numbers of events” in the form of rehospitalizations for heart failure, said Dr. Abraham, professor of medicine and director of cardiovascular medicine at Ohio State University in Columbus. Once data on outcomes are analyzed for the registry patients, “I think they will be even better than they were in the trial,” he said in an interview.
The PA pressure data in these initial patients “are very important because they tell us that in general use, clinicians – many of whom are at community hospitals – are very capable of using the CardioMEMS data to control patient pressures, and in CHAMPION we showed that there is a relationship between controlled pressures and improved outcomes,” he said. The findings also help allay a key concern about the potential benefit from implanting a device to monitor PA pressure, which is that clinicians must respond to the information and tweak a patient’s diuretic and vasodilator treatments in order for pressure monitoring to have an effect on heart failure outcomes.
“These data clearly refute that concern,” Dr. Abraham said.
He expressed some surprise that PA pressure control with monitoring was so much more effective in real-world use than in the CHAMPION pivotal trial. “In the trial, it was a paradigm shift to manage heart failure patients based on their PA pressures and not according to their symptoms,” he said. With CardioMEMS pressure monitoring, clinicians are supposed to treat high PA pressure with dose adjustments even if the patient feels okay. The new data suggest that clinicians now using the device “have gotten the message that if you don’t do something with the data the patients won’t improve.”
The registry patients came from 47 states and 427 unique physicians who worked in a range of settings including large and small centers, and academic and nonacademic community centers. The patients averaged 70 years, 40% were women, a third had a left ventricular ejection fraction at or above 40%, and their average PA pressure at the time they had their device implanted was 34.9 mm Hg. This pressure was notably higher than the average 31.6 mm Hg pressure among patients enrolled in CHAMPION, a fact that also helps explain why the registry patients received a larger pressure-reduction benefit: They started from a higher level than the trial patients, and during follow-up, their achieved pressures were always compared back to their high baseline pressures.
The registry patients were also substantially older than the trial patients, who had averaged 62 years, and the registry included substantially more women and more patients with higher ejection fractions. Dr. Abraham did not report data on their New York Heart Association class at entry, but labeling for CardioMEMS specifies that patients should have class III heart failure as well as a recent heart failure hospitalization.
Dr. Abraham’s analysis also showed that the greatest degree of PA pressure control occurred in the patients who began device-based treatment with the highest PA pressures. Nearly half the 2,000 registry patients had an entry PA pressure at or above 35 mm Hg, and over a period of 6 months, they averaged a cumulative 876–mm Hg reduction in their PA pressure relative to their baseline level. The third of patients who began with a PA pressure of 25-34 mm Hg had an average 169–mm Hg cumulative pressure reduction over the 6 month period, and the 18% of patients who began with a PA pressure of less than 25 mm Hg actually had an average cumulative increase in the PA pressure of 163 mm Hg. Target PA pressures are usually in the normal range of 18-25 mm Hg.
The analyses also showed that the impact of PA pressure monitoring on pressure was roughly similar regardless of the left ventricular ejection fraction patients had at baseline, and regardless of their sex.
The registry data were collected by St. Jude, the company that markets the CardioMEMS device. Dr. Abraham is a consultant to St. Jude and was lead investigator for the CHAMPION pivotal trial.
On Twitter @mitchelzoler
ORLANDO – Pulmonary artery pressure monitoring using an implanted device was even more effective for controlling pulmonary artery pressures in 2,000 real-world U.S. heart failure patients than it was in the pivotal trial that led to the device’s regulatory approval.
Data from the first 2,000 U.S. heart failure patients to receive the CardioMEMS pulmonary artery (PA) pressure monitoring device and have at least 6 months of follow-up data since the device received Food and Drug Administration approval in 2014 showed that cumulative PA pressure reductions in these patients during the first 6 months of use averaged 434 mm Hg per patient when compared with their baseline PA pressure when they first received the device. This was nearly threefold better than the average 150–mm Hg cumulative reduction in PA pressure per patient during 6 months of use seen in the CHAMPION trial, Dr. William T. Abraham reported at the annual scientific meeting of the Heart Failure Society of America.
Although this analysis of data from the registry maintained for U.S. patients who receive the CardioMEMS device does not yet include information on how these patients fared clinically, and specifically how often they required rehospitalization for heart failure, the strikingly high level of PA pressure control seen in the first 2,000 U.S. patients bodes well for what the clinical findings will show once they are available.
“In our experience with PA pressure monitoring, there is almost a linear relationship between reduced PA pressures and reduced numbers of events” in the form of rehospitalizations for heart failure, said Dr. Abraham, professor of medicine and director of cardiovascular medicine at Ohio State University in Columbus. Once data on outcomes are analyzed for the registry patients, “I think they will be even better than they were in the trial,” he said in an interview.
The PA pressure data in these initial patients “are very important because they tell us that in general use, clinicians – many of whom are at community hospitals – are very capable of using the CardioMEMS data to control patient pressures, and in CHAMPION we showed that there is a relationship between controlled pressures and improved outcomes,” he said. The findings also help allay a key concern about the potential benefit from implanting a device to monitor PA pressure, which is that clinicians must respond to the information and tweak a patient’s diuretic and vasodilator treatments in order for pressure monitoring to have an effect on heart failure outcomes.
“These data clearly refute that concern,” Dr. Abraham said.
He expressed some surprise that PA pressure control with monitoring was so much more effective in real-world use than in the CHAMPION pivotal trial. “In the trial, it was a paradigm shift to manage heart failure patients based on their PA pressures and not according to their symptoms,” he said. With CardioMEMS pressure monitoring, clinicians are supposed to treat high PA pressure with dose adjustments even if the patient feels okay. The new data suggest that clinicians now using the device “have gotten the message that if you don’t do something with the data the patients won’t improve.”
The registry patients came from 47 states and 427 unique physicians who worked in a range of settings including large and small centers, and academic and nonacademic community centers. The patients averaged 70 years, 40% were women, a third had a left ventricular ejection fraction at or above 40%, and their average PA pressure at the time they had their device implanted was 34.9 mm Hg. This pressure was notably higher than the average 31.6 mm Hg pressure among patients enrolled in CHAMPION, a fact that also helps explain why the registry patients received a larger pressure-reduction benefit: They started from a higher level than the trial patients, and during follow-up, their achieved pressures were always compared back to their high baseline pressures.
The registry patients were also substantially older than the trial patients, who had averaged 62 years, and the registry included substantially more women and more patients with higher ejection fractions. Dr. Abraham did not report data on their New York Heart Association class at entry, but labeling for CardioMEMS specifies that patients should have class III heart failure as well as a recent heart failure hospitalization.
Dr. Abraham’s analysis also showed that the greatest degree of PA pressure control occurred in the patients who began device-based treatment with the highest PA pressures. Nearly half the 2,000 registry patients had an entry PA pressure at or above 35 mm Hg, and over a period of 6 months, they averaged a cumulative 876–mm Hg reduction in their PA pressure relative to their baseline level. The third of patients who began with a PA pressure of 25-34 mm Hg had an average 169–mm Hg cumulative pressure reduction over the 6 month period, and the 18% of patients who began with a PA pressure of less than 25 mm Hg actually had an average cumulative increase in the PA pressure of 163 mm Hg. Target PA pressures are usually in the normal range of 18-25 mm Hg.
The analyses also showed that the impact of PA pressure monitoring on pressure was roughly similar regardless of the left ventricular ejection fraction patients had at baseline, and regardless of their sex.
The registry data were collected by St. Jude, the company that markets the CardioMEMS device. Dr. Abraham is a consultant to St. Jude and was lead investigator for the CHAMPION pivotal trial.
On Twitter @mitchelzoler
ORLANDO – Pulmonary artery pressure monitoring using an implanted device was even more effective for controlling pulmonary artery pressures in 2,000 real-world U.S. heart failure patients than it was in the pivotal trial that led to the device’s regulatory approval.
Data from the first 2,000 U.S. heart failure patients to receive the CardioMEMS pulmonary artery (PA) pressure monitoring device and have at least 6 months of follow-up data since the device received Food and Drug Administration approval in 2014 showed that cumulative PA pressure reductions in these patients during the first 6 months of use averaged 434 mm Hg per patient when compared with their baseline PA pressure when they first received the device. This was nearly threefold better than the average 150–mm Hg cumulative reduction in PA pressure per patient during 6 months of use seen in the CHAMPION trial, Dr. William T. Abraham reported at the annual scientific meeting of the Heart Failure Society of America.
Although this analysis of data from the registry maintained for U.S. patients who receive the CardioMEMS device does not yet include information on how these patients fared clinically, and specifically how often they required rehospitalization for heart failure, the strikingly high level of PA pressure control seen in the first 2,000 U.S. patients bodes well for what the clinical findings will show once they are available.
“In our experience with PA pressure monitoring, there is almost a linear relationship between reduced PA pressures and reduced numbers of events” in the form of rehospitalizations for heart failure, said Dr. Abraham, professor of medicine and director of cardiovascular medicine at Ohio State University in Columbus. Once data on outcomes are analyzed for the registry patients, “I think they will be even better than they were in the trial,” he said in an interview.
The PA pressure data in these initial patients “are very important because they tell us that in general use, clinicians – many of whom are at community hospitals – are very capable of using the CardioMEMS data to control patient pressures, and in CHAMPION we showed that there is a relationship between controlled pressures and improved outcomes,” he said. The findings also help allay a key concern about the potential benefit from implanting a device to monitor PA pressure, which is that clinicians must respond to the information and tweak a patient’s diuretic and vasodilator treatments in order for pressure monitoring to have an effect on heart failure outcomes.
“These data clearly refute that concern,” Dr. Abraham said.
He expressed some surprise that PA pressure control with monitoring was so much more effective in real-world use than in the CHAMPION pivotal trial. “In the trial, it was a paradigm shift to manage heart failure patients based on their PA pressures and not according to their symptoms,” he said. With CardioMEMS pressure monitoring, clinicians are supposed to treat high PA pressure with dose adjustments even if the patient feels okay. The new data suggest that clinicians now using the device “have gotten the message that if you don’t do something with the data the patients won’t improve.”
The registry patients came from 47 states and 427 unique physicians who worked in a range of settings including large and small centers, and academic and nonacademic community centers. The patients averaged 70 years, 40% were women, a third had a left ventricular ejection fraction at or above 40%, and their average PA pressure at the time they had their device implanted was 34.9 mm Hg. This pressure was notably higher than the average 31.6 mm Hg pressure among patients enrolled in CHAMPION, a fact that also helps explain why the registry patients received a larger pressure-reduction benefit: They started from a higher level than the trial patients, and during follow-up, their achieved pressures were always compared back to their high baseline pressures.
The registry patients were also substantially older than the trial patients, who had averaged 62 years, and the registry included substantially more women and more patients with higher ejection fractions. Dr. Abraham did not report data on their New York Heart Association class at entry, but labeling for CardioMEMS specifies that patients should have class III heart failure as well as a recent heart failure hospitalization.
Dr. Abraham’s analysis also showed that the greatest degree of PA pressure control occurred in the patients who began device-based treatment with the highest PA pressures. Nearly half the 2,000 registry patients had an entry PA pressure at or above 35 mm Hg, and over a period of 6 months, they averaged a cumulative 876–mm Hg reduction in their PA pressure relative to their baseline level. The third of patients who began with a PA pressure of 25-34 mm Hg had an average 169–mm Hg cumulative pressure reduction over the 6 month period, and the 18% of patients who began with a PA pressure of less than 25 mm Hg actually had an average cumulative increase in the PA pressure of 163 mm Hg. Target PA pressures are usually in the normal range of 18-25 mm Hg.
The analyses also showed that the impact of PA pressure monitoring on pressure was roughly similar regardless of the left ventricular ejection fraction patients had at baseline, and regardless of their sex.
The registry data were collected by St. Jude, the company that markets the CardioMEMS device. Dr. Abraham is a consultant to St. Jude and was lead investigator for the CHAMPION pivotal trial.
On Twitter @mitchelzoler
AT THE HFSA ANNUAL SCIENTIFIC MEETING
Key clinical point: Registry data from the first 2,000 U.S. heart failure patients who received an implanted pulmonary artery pressure monitor showed a level of pressure control over a period of 6 months nearly triple that seen in the pivotal trial.
Major finding: Cumulative pulmonary artery pressure reductions averaged 434 mm Hg over a period of 6 months, compared with an average reduction of 150 mm Hg in the pivotal trial.
Data source: The first 2,000 U.S. patients who received a CardioMEMS device and were followed for at least 6 months.
Disclosures: The registry data were collected by St. Jude, the company that markets the CardioMEMS device. Dr. Abraham is a consultant to St. Jude and was lead investigator for the CHAMPION pivotal trial.
