User login
Tracking the Decline of AAA Repair in the United States
Has the advent of branched-fenestrated aortic endografts increased the number of abdominal aortic aneurysm (AAA) repairs due to patients who otherwise would not be offered open repair or infrarenal endovascular repair (EVAR)?
This is the question addressed by Dr. Bjoern D. Suckow, who will present the results of a study that he and his colleagues at the Dartmouth-Hitchcock Medical Center performed to evaluate trends in open AAA repair, EVAR, and branched-fenestrated EVAR (BEVAR) for AAA in Medicare beneficiaries from 2003 to 2013. They used a 100% sample of Medicare Part B claims to determine the number of each of these repair types and to compare them annually over the study period.
Between 2003 and 2013, the total number of AAA repairs significantly declined by 13% from 27,352 to 23,835, after a peak number of 29,924 repairs performed in 2005. The number of open AAA repairs significantly and steadily declined by 78%, from 15,385 in 2003 to 3,315 in 2013. In contrast, while the number of EVARs significantly increased from 11,967 in 2003 to 20,845 in 2008, that number has since declined a total of 13% to only 18,098 repairs in 2013, although this was not a significant difference.
Prior to 2011, there were no BEVAR cases among Medicare beneficiaries, according to Dr. Suckow. However, the number of BEVAR cases continuously and significantly rose more than 500% from 401 procedures in 2011 to 2,422 procedures in 2013. Over this same time period, the number of open repairs and the number of EVARs declined by 2,499 and 2,415 cases, respectively .
“The total number of AAA repairs in the Unites States has been declining annually since 2005,” said Dr. Suckow. “Our study shows that the continuous increase in BEVAR cases performed among Medicare patients over the last 3 years has not reversed the steady decline in overall numbers of AAA repairs, because of a concomitant decline in both open aneurysm repair and EVAR.”
Their results indicated that BEVAR was not likely responsible for the overall decline in open repair, as this trend was found to be present over a much longer time period, according to Dr. Suckow. Similarly, the rapid rise in BEVAR cases occurred when there was an almost equal decline in EVAR case numbers.
“These trends suggest a potential decline in the incidence of AAA in the U.S. Medicare population, with shifts in the type of repair based on available technology and perhaps patient preference,” said Dr. Suckow. “Future efforts are needed to determine the characteristics of patients treated with BEVAR as well as the long-term effectiveness of these new treatments,” he concluded.
Friday: Plenary Session 4
8:00 A.M. - 9:30 A.M.
Potomac Ballroom A/B
Has the advent of branched-fenestrated aortic endografts increased the number of abdominal aortic aneurysm (AAA) repairs due to patients who otherwise would not be offered open repair or infrarenal endovascular repair (EVAR)?
This is the question addressed by Dr. Bjoern D. Suckow, who will present the results of a study that he and his colleagues at the Dartmouth-Hitchcock Medical Center performed to evaluate trends in open AAA repair, EVAR, and branched-fenestrated EVAR (BEVAR) for AAA in Medicare beneficiaries from 2003 to 2013. They used a 100% sample of Medicare Part B claims to determine the number of each of these repair types and to compare them annually over the study period.
Between 2003 and 2013, the total number of AAA repairs significantly declined by 13% from 27,352 to 23,835, after a peak number of 29,924 repairs performed in 2005. The number of open AAA repairs significantly and steadily declined by 78%, from 15,385 in 2003 to 3,315 in 2013. In contrast, while the number of EVARs significantly increased from 11,967 in 2003 to 20,845 in 2008, that number has since declined a total of 13% to only 18,098 repairs in 2013, although this was not a significant difference.
Prior to 2011, there were no BEVAR cases among Medicare beneficiaries, according to Dr. Suckow. However, the number of BEVAR cases continuously and significantly rose more than 500% from 401 procedures in 2011 to 2,422 procedures in 2013. Over this same time period, the number of open repairs and the number of EVARs declined by 2,499 and 2,415 cases, respectively .
“The total number of AAA repairs in the Unites States has been declining annually since 2005,” said Dr. Suckow. “Our study shows that the continuous increase in BEVAR cases performed among Medicare patients over the last 3 years has not reversed the steady decline in overall numbers of AAA repairs, because of a concomitant decline in both open aneurysm repair and EVAR.”
Their results indicated that BEVAR was not likely responsible for the overall decline in open repair, as this trend was found to be present over a much longer time period, according to Dr. Suckow. Similarly, the rapid rise in BEVAR cases occurred when there was an almost equal decline in EVAR case numbers.
“These trends suggest a potential decline in the incidence of AAA in the U.S. Medicare population, with shifts in the type of repair based on available technology and perhaps patient preference,” said Dr. Suckow. “Future efforts are needed to determine the characteristics of patients treated with BEVAR as well as the long-term effectiveness of these new treatments,” he concluded.
Friday: Plenary Session 4
8:00 A.M. - 9:30 A.M.
Potomac Ballroom A/B
Has the advent of branched-fenestrated aortic endografts increased the number of abdominal aortic aneurysm (AAA) repairs due to patients who otherwise would not be offered open repair or infrarenal endovascular repair (EVAR)?
This is the question addressed by Dr. Bjoern D. Suckow, who will present the results of a study that he and his colleagues at the Dartmouth-Hitchcock Medical Center performed to evaluate trends in open AAA repair, EVAR, and branched-fenestrated EVAR (BEVAR) for AAA in Medicare beneficiaries from 2003 to 2013. They used a 100% sample of Medicare Part B claims to determine the number of each of these repair types and to compare them annually over the study period.
Between 2003 and 2013, the total number of AAA repairs significantly declined by 13% from 27,352 to 23,835, after a peak number of 29,924 repairs performed in 2005. The number of open AAA repairs significantly and steadily declined by 78%, from 15,385 in 2003 to 3,315 in 2013. In contrast, while the number of EVARs significantly increased from 11,967 in 2003 to 20,845 in 2008, that number has since declined a total of 13% to only 18,098 repairs in 2013, although this was not a significant difference.
Prior to 2011, there were no BEVAR cases among Medicare beneficiaries, according to Dr. Suckow. However, the number of BEVAR cases continuously and significantly rose more than 500% from 401 procedures in 2011 to 2,422 procedures in 2013. Over this same time period, the number of open repairs and the number of EVARs declined by 2,499 and 2,415 cases, respectively .
“The total number of AAA repairs in the Unites States has been declining annually since 2005,” said Dr. Suckow. “Our study shows that the continuous increase in BEVAR cases performed among Medicare patients over the last 3 years has not reversed the steady decline in overall numbers of AAA repairs, because of a concomitant decline in both open aneurysm repair and EVAR.”
Their results indicated that BEVAR was not likely responsible for the overall decline in open repair, as this trend was found to be present over a much longer time period, according to Dr. Suckow. Similarly, the rapid rise in BEVAR cases occurred when there was an almost equal decline in EVAR case numbers.
“These trends suggest a potential decline in the incidence of AAA in the U.S. Medicare population, with shifts in the type of repair based on available technology and perhaps patient preference,” said Dr. Suckow. “Future efforts are needed to determine the characteristics of patients treated with BEVAR as well as the long-term effectiveness of these new treatments,” he concluded.
Friday: Plenary Session 4
8:00 A.M. - 9:30 A.M.
Potomac Ballroom A/B

Learn Radiation Safety During Vascular Surgery
Practical tips for protecting your vascular surgery patients and yourselves from radiation exposure will be featured as part of this morning’s session “Radiation Safety for the Vascular Surgeon.”
“Since endovascular procedures became an essential part of the vascular field about 25 years ago, minimally invasive, radiation-guided procedures have become more sophisticated, more involved and more frequent while requiring better resolution,” said session co-moderator Dr. Peter Schneider of Kaiser Permanente Medical Group in Honolulu. “All of this means more radiation for the patient and for the physician. Going forward, as our field continues to develop, the likelihood is high that these requirements will continue and will intensify and that the radiation risk to patients and providers will increase.
“Radiation is a necessary tool in our ability to help our patients,” Dr. Schneider added. “Yet radiation has negative effects, such as radiation-induced cancers and cataracts. We need to do everything we can to minimize those negative effects.”
A good 60%-70% of vascular procedures involve endovascular techniques that utilize radiation, said session co-moderator Dr. Amy Reed of Rush University Medical Center in Chicago. “Even though your patient may be exposed multiple times in the future through CAT scans or cardiac catheterizations, you as the surgeon are going to be doing this for decades – so the long-term exposure to radiation is a concern. Usually it goes hand in hand that if you’re protecting the patient, you’re protecting yourself as well.”
There are a number of measures vascular surgeons can take to minimize radiation exposure, the moderators said. “These include simple things like the use of radiation filters during imaging, decreasing frame rates to the lowest level that gives the imaging required, being careful about maintaining distance from the image intensifier during fluoroscopy and use of radiation shielding,” Dr. Schneider said.
Additional steps can include collimating or focusing the view and changing table positions, Dr. Reed noted. Good radiation safety techniques are especially important for residency program directors, she added: “Trainees stand closer to the radiation, so it’s important to have good technique. Just like universal precautions measures such as wearing gloves and washing hands, trainees are going to learn radiation safety by emulating you.”
During the session, six expert clinicians knowledgeable about the effects of radiation will share their wisdom. Dr. Christopher Carsten of Greenville Health System in South Carolina will cover what is known about the clinically allowable limits of radiation to the provider and to the patient; Dr. Sunita Srivastava of the Cleveland Clinic will provide a basic understanding of the mechanism of the biological effects of radiation; Dr. Melissa Kirkwood of the University of Texas Southwestern Medical Center in Dallas will offer practical tips to the vascular surgeon for decreasing radiation; Dr. Audra Duncan of the University of Western Ontario will discuss the need for diagnostic tests or radiation-guided procedures in patients who are pregnant; Dr. Mark Farber of the University of North Carolina will discuss new technologies in radiation monitoring; and Dr. Lois Killewich of the University of Texas Medical Branch in Galveston will cover governmental regulations associated with the clinical use of radiation.
“We’re really trying to increase the awareness of radiation safety,” Dr. Reed said. “It’s not to go over all of the things radiation can cause – we know that – but what’s on the horizon that might be helpful for you, and what tips you can take back for protection if you’re not currently using them.”
Friday Breakfast Session B4
6:30 A.M. - 8:00 A.M.
Potomac Ballroom C
Practical tips for protecting your vascular surgery patients and yourselves from radiation exposure will be featured as part of this morning’s session “Radiation Safety for the Vascular Surgeon.”
“Since endovascular procedures became an essential part of the vascular field about 25 years ago, minimally invasive, radiation-guided procedures have become more sophisticated, more involved and more frequent while requiring better resolution,” said session co-moderator Dr. Peter Schneider of Kaiser Permanente Medical Group in Honolulu. “All of this means more radiation for the patient and for the physician. Going forward, as our field continues to develop, the likelihood is high that these requirements will continue and will intensify and that the radiation risk to patients and providers will increase.
“Radiation is a necessary tool in our ability to help our patients,” Dr. Schneider added. “Yet radiation has negative effects, such as radiation-induced cancers and cataracts. We need to do everything we can to minimize those negative effects.”
A good 60%-70% of vascular procedures involve endovascular techniques that utilize radiation, said session co-moderator Dr. Amy Reed of Rush University Medical Center in Chicago. “Even though your patient may be exposed multiple times in the future through CAT scans or cardiac catheterizations, you as the surgeon are going to be doing this for decades – so the long-term exposure to radiation is a concern. Usually it goes hand in hand that if you’re protecting the patient, you’re protecting yourself as well.”
There are a number of measures vascular surgeons can take to minimize radiation exposure, the moderators said. “These include simple things like the use of radiation filters during imaging, decreasing frame rates to the lowest level that gives the imaging required, being careful about maintaining distance from the image intensifier during fluoroscopy and use of radiation shielding,” Dr. Schneider said.
Additional steps can include collimating or focusing the view and changing table positions, Dr. Reed noted. Good radiation safety techniques are especially important for residency program directors, she added: “Trainees stand closer to the radiation, so it’s important to have good technique. Just like universal precautions measures such as wearing gloves and washing hands, trainees are going to learn radiation safety by emulating you.”
During the session, six expert clinicians knowledgeable about the effects of radiation will share their wisdom. Dr. Christopher Carsten of Greenville Health System in South Carolina will cover what is known about the clinically allowable limits of radiation to the provider and to the patient; Dr. Sunita Srivastava of the Cleveland Clinic will provide a basic understanding of the mechanism of the biological effects of radiation; Dr. Melissa Kirkwood of the University of Texas Southwestern Medical Center in Dallas will offer practical tips to the vascular surgeon for decreasing radiation; Dr. Audra Duncan of the University of Western Ontario will discuss the need for diagnostic tests or radiation-guided procedures in patients who are pregnant; Dr. Mark Farber of the University of North Carolina will discuss new technologies in radiation monitoring; and Dr. Lois Killewich of the University of Texas Medical Branch in Galveston will cover governmental regulations associated with the clinical use of radiation.
“We’re really trying to increase the awareness of radiation safety,” Dr. Reed said. “It’s not to go over all of the things radiation can cause – we know that – but what’s on the horizon that might be helpful for you, and what tips you can take back for protection if you’re not currently using them.”
Friday Breakfast Session B4
6:30 A.M. - 8:00 A.M.
Potomac Ballroom C
Practical tips for protecting your vascular surgery patients and yourselves from radiation exposure will be featured as part of this morning’s session “Radiation Safety for the Vascular Surgeon.”
“Since endovascular procedures became an essential part of the vascular field about 25 years ago, minimally invasive, radiation-guided procedures have become more sophisticated, more involved and more frequent while requiring better resolution,” said session co-moderator Dr. Peter Schneider of Kaiser Permanente Medical Group in Honolulu. “All of this means more radiation for the patient and for the physician. Going forward, as our field continues to develop, the likelihood is high that these requirements will continue and will intensify and that the radiation risk to patients and providers will increase.
“Radiation is a necessary tool in our ability to help our patients,” Dr. Schneider added. “Yet radiation has negative effects, such as radiation-induced cancers and cataracts. We need to do everything we can to minimize those negative effects.”
A good 60%-70% of vascular procedures involve endovascular techniques that utilize radiation, said session co-moderator Dr. Amy Reed of Rush University Medical Center in Chicago. “Even though your patient may be exposed multiple times in the future through CAT scans or cardiac catheterizations, you as the surgeon are going to be doing this for decades – so the long-term exposure to radiation is a concern. Usually it goes hand in hand that if you’re protecting the patient, you’re protecting yourself as well.”
There are a number of measures vascular surgeons can take to minimize radiation exposure, the moderators said. “These include simple things like the use of radiation filters during imaging, decreasing frame rates to the lowest level that gives the imaging required, being careful about maintaining distance from the image intensifier during fluoroscopy and use of radiation shielding,” Dr. Schneider said.
Additional steps can include collimating or focusing the view and changing table positions, Dr. Reed noted. Good radiation safety techniques are especially important for residency program directors, she added: “Trainees stand closer to the radiation, so it’s important to have good technique. Just like universal precautions measures such as wearing gloves and washing hands, trainees are going to learn radiation safety by emulating you.”
During the session, six expert clinicians knowledgeable about the effects of radiation will share their wisdom. Dr. Christopher Carsten of Greenville Health System in South Carolina will cover what is known about the clinically allowable limits of radiation to the provider and to the patient; Dr. Sunita Srivastava of the Cleveland Clinic will provide a basic understanding of the mechanism of the biological effects of radiation; Dr. Melissa Kirkwood of the University of Texas Southwestern Medical Center in Dallas will offer practical tips to the vascular surgeon for decreasing radiation; Dr. Audra Duncan of the University of Western Ontario will discuss the need for diagnostic tests or radiation-guided procedures in patients who are pregnant; Dr. Mark Farber of the University of North Carolina will discuss new technologies in radiation monitoring; and Dr. Lois Killewich of the University of Texas Medical Branch in Galveston will cover governmental regulations associated with the clinical use of radiation.
“We’re really trying to increase the awareness of radiation safety,” Dr. Reed said. “It’s not to go over all of the things radiation can cause – we know that – but what’s on the horizon that might be helpful for you, and what tips you can take back for protection if you’re not currently using them.”
Friday Breakfast Session B4
6:30 A.M. - 8:00 A.M.
Potomac Ballroom C

Enjoy a ‘Capitol’ Performance with the Capitol Steps
First, combine a Washington, D.C. venue and an upcoming and hotly debated presidential election. Add a date shortly before the GOP and Democratic national conventions.
What do you get? The perfect time and place for a political comedy show performed by the “Capitol Steps,” a group that’s been poking fun at politics, politicians and the headlines of the day since the Reagan administration.
The group will perform its particular brand of satirical humor – song parodies, standup comedy and more – at a special presentation for all VAM attendees from 8:30 to 9:30 p.m., Friday, in Potomac Ballroom A/B. The event is open at no charge to all registered attendees, guests and exhibitors.
No topic is safe, whether it’s the FBI’s fight earlier this year with Apple over a locked iPhone to Bill Clinton commenting on Hillary to Donald Trump and Sarah Palin. Perhaps the audience will hear Bernie Sanders sing a show tune or hear about “Deleter of the Facts.”
The group got its start in 1981. Senate staffers, planning entertainment for a Christmas party, created song parodies and skits from the headlines at the time and created the Capitol Steps in the process. The group has recorded more than 30 albums (the latest is “What to Expect When You’re Electing”) and has been featured on national television and radio.
“We are thrilled to offer the humor of the Capitol Steps to our members and guests,” said SVS President Dr. Bruce A. Perler, who was instrumental in bringing the “Steps” to VAM. “I am certain that, no matter what a particular member’s politics, in this most ‘unusual’ of presidential election campaigns, the Capitol Steps will provide plenty of laughs and remarkable satirical insights.”
First, combine a Washington, D.C. venue and an upcoming and hotly debated presidential election. Add a date shortly before the GOP and Democratic national conventions.
What do you get? The perfect time and place for a political comedy show performed by the “Capitol Steps,” a group that’s been poking fun at politics, politicians and the headlines of the day since the Reagan administration.
The group will perform its particular brand of satirical humor – song parodies, standup comedy and more – at a special presentation for all VAM attendees from 8:30 to 9:30 p.m., Friday, in Potomac Ballroom A/B. The event is open at no charge to all registered attendees, guests and exhibitors.
No topic is safe, whether it’s the FBI’s fight earlier this year with Apple over a locked iPhone to Bill Clinton commenting on Hillary to Donald Trump and Sarah Palin. Perhaps the audience will hear Bernie Sanders sing a show tune or hear about “Deleter of the Facts.”
The group got its start in 1981. Senate staffers, planning entertainment for a Christmas party, created song parodies and skits from the headlines at the time and created the Capitol Steps in the process. The group has recorded more than 30 albums (the latest is “What to Expect When You’re Electing”) and has been featured on national television and radio.
“We are thrilled to offer the humor of the Capitol Steps to our members and guests,” said SVS President Dr. Bruce A. Perler, who was instrumental in bringing the “Steps” to VAM. “I am certain that, no matter what a particular member’s politics, in this most ‘unusual’ of presidential election campaigns, the Capitol Steps will provide plenty of laughs and remarkable satirical insights.”
First, combine a Washington, D.C. venue and an upcoming and hotly debated presidential election. Add a date shortly before the GOP and Democratic national conventions.
What do you get? The perfect time and place for a political comedy show performed by the “Capitol Steps,” a group that’s been poking fun at politics, politicians and the headlines of the day since the Reagan administration.
The group will perform its particular brand of satirical humor – song parodies, standup comedy and more – at a special presentation for all VAM attendees from 8:30 to 9:30 p.m., Friday, in Potomac Ballroom A/B. The event is open at no charge to all registered attendees, guests and exhibitors.
No topic is safe, whether it’s the FBI’s fight earlier this year with Apple over a locked iPhone to Bill Clinton commenting on Hillary to Donald Trump and Sarah Palin. Perhaps the audience will hear Bernie Sanders sing a show tune or hear about “Deleter of the Facts.”
The group got its start in 1981. Senate staffers, planning entertainment for a Christmas party, created song parodies and skits from the headlines at the time and created the Capitol Steps in the process. The group has recorded more than 30 albums (the latest is “What to Expect When You’re Electing”) and has been featured on national television and radio.
“We are thrilled to offer the humor of the Capitol Steps to our members and guests,” said SVS President Dr. Bruce A. Perler, who was instrumental in bringing the “Steps” to VAM. “I am certain that, no matter what a particular member’s politics, in this most ‘unusual’ of presidential election campaigns, the Capitol Steps will provide plenty of laughs and remarkable satirical insights.”

Presidentially Speaking
Dr. Bruce Perler, Johns Hopkins Hospital, will give his SVS Presidential Address Friday after a short introduction by President-Elect Dr. Ronald Fairman.
Friday: 11 a.m. – 12:15 p.m. Potomac Ballroom A/B
Dr. Bruce Perler, Johns Hopkins Hospital, will give his SVS Presidential Address Friday after a short introduction by President-Elect Dr. Ronald Fairman.
Friday: 11 a.m. – 12:15 p.m. Potomac Ballroom A/B
Dr. Bruce Perler, Johns Hopkins Hospital, will give his SVS Presidential Address Friday after a short introduction by President-Elect Dr. Ronald Fairman.
Friday: 11 a.m. – 12:15 p.m. Potomac Ballroom A/B

Are You Going to the Fair? A Guide to What You Need to Know
There’s no Ferris Wheel or tilt-a-whirl at this fair – just useful information on 73 vascular training programs and the chance for aspiring vascular surgeons to find a good fit for their futures.
Programs are arranged on the floor by the types of offered paradigms: 0+5, 5+2 as well as those offering both training pathways. The configuration, introduced in 2015, makes it easier for attendees to locate the programs in which they are specifically interested.
This year residents/students will receive more data – such as if the program is academic- or community practice-based – to help them better locate the programs in which they are most interested in visiting in the time allotted. The change was made as the result of attendee feedback from earlier fairs.
The Residency Fair is the perfect way for residents and students to network and make connections. Program directors, faculty and current trainees from the 73 institutions will be on hand to talk with interested attendees.
More than 200 students and residents who have an active interest in vascular surgery are expected to participate.
A directory of all participating programs was distributed to students and residents earlier and also will be available at the fair. V
Friday: 5:00 P.M. - 6:30 P.M.
Exhibit Hall D
There’s no Ferris Wheel or tilt-a-whirl at this fair – just useful information on 73 vascular training programs and the chance for aspiring vascular surgeons to find a good fit for their futures.
Programs are arranged on the floor by the types of offered paradigms: 0+5, 5+2 as well as those offering both training pathways. The configuration, introduced in 2015, makes it easier for attendees to locate the programs in which they are specifically interested.
This year residents/students will receive more data – such as if the program is academic- or community practice-based – to help them better locate the programs in which they are most interested in visiting in the time allotted. The change was made as the result of attendee feedback from earlier fairs.
The Residency Fair is the perfect way for residents and students to network and make connections. Program directors, faculty and current trainees from the 73 institutions will be on hand to talk with interested attendees.
More than 200 students and residents who have an active interest in vascular surgery are expected to participate.
A directory of all participating programs was distributed to students and residents earlier and also will be available at the fair. V
Friday: 5:00 P.M. - 6:30 P.M.
Exhibit Hall D
There’s no Ferris Wheel or tilt-a-whirl at this fair – just useful information on 73 vascular training programs and the chance for aspiring vascular surgeons to find a good fit for their futures.
Programs are arranged on the floor by the types of offered paradigms: 0+5, 5+2 as well as those offering both training pathways. The configuration, introduced in 2015, makes it easier for attendees to locate the programs in which they are specifically interested.
This year residents/students will receive more data – such as if the program is academic- or community practice-based – to help them better locate the programs in which they are most interested in visiting in the time allotted. The change was made as the result of attendee feedback from earlier fairs.
The Residency Fair is the perfect way for residents and students to network and make connections. Program directors, faculty and current trainees from the 73 institutions will be on hand to talk with interested attendees.
More than 200 students and residents who have an active interest in vascular surgery are expected to participate.
A directory of all participating programs was distributed to students and residents earlier and also will be available at the fair. V
Friday: 5:00 P.M. - 6:30 P.M.
Exhibit Hall D

Study finds most antidepressants ineffective or harmful in children, adolescents
Most antidepressants prescribed to children and adolescents with acute major depression might not be nearly as effective as they are believed to be – and one might be harmful, according to a retrospective review published June 8 and including more than 5,000 participants.
“Our analysis found robust evidence to suggest a significantly increased risk for suicidality for young people given venlafaxine,” wrote Dr. Andrea Cipriani, Xinyu Zhou, Ph.D., and their colleagues. “Unfortunately, due to the absence of reliable data on suicidality for many antidepressants, it was not possible to comprehensively assess the risk of suicidality for all drugs.”
The review looked at 34 double-blind, randomized, controlled trials investigating at least one of 14 major drugs typically prescribed as antidepressants for pediatric patients. In addition to venlafaxine, the researchers looked at amitriptyline, citalopram, clomipramine, desipramine, duloxetine, escitalopram, fluoxetine, imipramine, mirtazapine, nefazodone, nortriptyline, paroxetine, and sertraline (Lancet. 2016 Jun 8. doi: 10.1016/S0140-6736[16]30385-3).
All trials were published before May 31, 2015, and were found in the PubMed, Cochrane Library, Web of Science, Embase, CINAHL, PsycINFO, and LiLACS databases, as well as regulatory agencies’ websites, and international registers for published and unpublished trials. Trials either compared one or more drugs against one another, or one or more drugs against a placebo.
Fluoxetine was the only drug found to be significantly more effective than placebo, and it also was found to be significantly more effective than nortriptyline. In addition, fluoxetine proved better tolerated than imipramine and duloxetine. “However, the clinical interpretation of these findings is limited not only by the uncertainty around these estimates, but also by the potential bias due to selective reporting and the small number of trials,” said Dr. Cipriani of the University of Oxford (England) and Dr. Zhou of the First Affiliated Hospital of Chongqing Medical University in China.
Regardless of which treatment clinicians choose, children and adolescents prescribed antidepressants should be monitored closely. Clinical guidelines for young people with major depression recommend psychotherapy, particularly cognitive-behavioral therapy or interpersonal therapy as first-line interventions, and “fluoxetine should be considered only for those patients with moderate to severe depression who do not have access to psychotherapy or have not responded to nonpharmacological interventions,” the researchers said.
The study was funded by the National Basic Research Program of China. The authors did not report any relevant financial disclosures.
The study by Dr. Andrea Cipriani, Xinyu Zhou, Ph.D., and their colleagues has “disturbing implications for clinical practice” in that it concludes that the risk-benefit profile of antidepressants in the acute treatment of major depression in children and adolescents “does not seem to offer a clear advantage” for these young patients, Dr. Jon Jureidini wrote in an accompanying editorial (Lancet. 2016 Jun 8. doi: 10.1016/S0140-6736[16]30385-2).
For clinicians, the implications are that every decision about whether and what to prescribe requires a calculation on the part of the clinician that is both complex and intuitive, he wrote.
“With research evidence as an important part of that calculation, we now know that we need to make a conscious correction for favorable misrepresentation of outcomes in published and unpublished study reports,” Dr. Jureidini wrote. “Only if the discounted benefit outweighs the boosted harm should the treatment be prescribed. For antidepressants in adolescents, this equation will rarely favor prescribing; in younger children, almost never.”
Dr. Jureidini is a research leader for the Robinson Research Institute at the University of Adelaide in North Adelaide, Australia.
The study by Dr. Andrea Cipriani, Xinyu Zhou, Ph.D., and their colleagues has “disturbing implications for clinical practice” in that it concludes that the risk-benefit profile of antidepressants in the acute treatment of major depression in children and adolescents “does not seem to offer a clear advantage” for these young patients, Dr. Jon Jureidini wrote in an accompanying editorial (Lancet. 2016 Jun 8. doi: 10.1016/S0140-6736[16]30385-2).
For clinicians, the implications are that every decision about whether and what to prescribe requires a calculation on the part of the clinician that is both complex and intuitive, he wrote.
“With research evidence as an important part of that calculation, we now know that we need to make a conscious correction for favorable misrepresentation of outcomes in published and unpublished study reports,” Dr. Jureidini wrote. “Only if the discounted benefit outweighs the boosted harm should the treatment be prescribed. For antidepressants in adolescents, this equation will rarely favor prescribing; in younger children, almost never.”
Dr. Jureidini is a research leader for the Robinson Research Institute at the University of Adelaide in North Adelaide, Australia.
The study by Dr. Andrea Cipriani, Xinyu Zhou, Ph.D., and their colleagues has “disturbing implications for clinical practice” in that it concludes that the risk-benefit profile of antidepressants in the acute treatment of major depression in children and adolescents “does not seem to offer a clear advantage” for these young patients, Dr. Jon Jureidini wrote in an accompanying editorial (Lancet. 2016 Jun 8. doi: 10.1016/S0140-6736[16]30385-2).
For clinicians, the implications are that every decision about whether and what to prescribe requires a calculation on the part of the clinician that is both complex and intuitive, he wrote.
“With research evidence as an important part of that calculation, we now know that we need to make a conscious correction for favorable misrepresentation of outcomes in published and unpublished study reports,” Dr. Jureidini wrote. “Only if the discounted benefit outweighs the boosted harm should the treatment be prescribed. For antidepressants in adolescents, this equation will rarely favor prescribing; in younger children, almost never.”
Dr. Jureidini is a research leader for the Robinson Research Institute at the University of Adelaide in North Adelaide, Australia.
Most antidepressants prescribed to children and adolescents with acute major depression might not be nearly as effective as they are believed to be – and one might be harmful, according to a retrospective review published June 8 and including more than 5,000 participants.
“Our analysis found robust evidence to suggest a significantly increased risk for suicidality for young people given venlafaxine,” wrote Dr. Andrea Cipriani, Xinyu Zhou, Ph.D., and their colleagues. “Unfortunately, due to the absence of reliable data on suicidality for many antidepressants, it was not possible to comprehensively assess the risk of suicidality for all drugs.”
The review looked at 34 double-blind, randomized, controlled trials investigating at least one of 14 major drugs typically prescribed as antidepressants for pediatric patients. In addition to venlafaxine, the researchers looked at amitriptyline, citalopram, clomipramine, desipramine, duloxetine, escitalopram, fluoxetine, imipramine, mirtazapine, nefazodone, nortriptyline, paroxetine, and sertraline (Lancet. 2016 Jun 8. doi: 10.1016/S0140-6736[16]30385-3).
All trials were published before May 31, 2015, and were found in the PubMed, Cochrane Library, Web of Science, Embase, CINAHL, PsycINFO, and LiLACS databases, as well as regulatory agencies’ websites, and international registers for published and unpublished trials. Trials either compared one or more drugs against one another, or one or more drugs against a placebo.
Fluoxetine was the only drug found to be significantly more effective than placebo, and it also was found to be significantly more effective than nortriptyline. In addition, fluoxetine proved better tolerated than imipramine and duloxetine. “However, the clinical interpretation of these findings is limited not only by the uncertainty around these estimates, but also by the potential bias due to selective reporting and the small number of trials,” said Dr. Cipriani of the University of Oxford (England) and Dr. Zhou of the First Affiliated Hospital of Chongqing Medical University in China.
Regardless of which treatment clinicians choose, children and adolescents prescribed antidepressants should be monitored closely. Clinical guidelines for young people with major depression recommend psychotherapy, particularly cognitive-behavioral therapy or interpersonal therapy as first-line interventions, and “fluoxetine should be considered only for those patients with moderate to severe depression who do not have access to psychotherapy or have not responded to nonpharmacological interventions,” the researchers said.
The study was funded by the National Basic Research Program of China. The authors did not report any relevant financial disclosures.
Most antidepressants prescribed to children and adolescents with acute major depression might not be nearly as effective as they are believed to be – and one might be harmful, according to a retrospective review published June 8 and including more than 5,000 participants.
“Our analysis found robust evidence to suggest a significantly increased risk for suicidality for young people given venlafaxine,” wrote Dr. Andrea Cipriani, Xinyu Zhou, Ph.D., and their colleagues. “Unfortunately, due to the absence of reliable data on suicidality for many antidepressants, it was not possible to comprehensively assess the risk of suicidality for all drugs.”
The review looked at 34 double-blind, randomized, controlled trials investigating at least one of 14 major drugs typically prescribed as antidepressants for pediatric patients. In addition to venlafaxine, the researchers looked at amitriptyline, citalopram, clomipramine, desipramine, duloxetine, escitalopram, fluoxetine, imipramine, mirtazapine, nefazodone, nortriptyline, paroxetine, and sertraline (Lancet. 2016 Jun 8. doi: 10.1016/S0140-6736[16]30385-3).
All trials were published before May 31, 2015, and were found in the PubMed, Cochrane Library, Web of Science, Embase, CINAHL, PsycINFO, and LiLACS databases, as well as regulatory agencies’ websites, and international registers for published and unpublished trials. Trials either compared one or more drugs against one another, or one or more drugs against a placebo.
Fluoxetine was the only drug found to be significantly more effective than placebo, and it also was found to be significantly more effective than nortriptyline. In addition, fluoxetine proved better tolerated than imipramine and duloxetine. “However, the clinical interpretation of these findings is limited not only by the uncertainty around these estimates, but also by the potential bias due to selective reporting and the small number of trials,” said Dr. Cipriani of the University of Oxford (England) and Dr. Zhou of the First Affiliated Hospital of Chongqing Medical University in China.
Regardless of which treatment clinicians choose, children and adolescents prescribed antidepressants should be monitored closely. Clinical guidelines for young people with major depression recommend psychotherapy, particularly cognitive-behavioral therapy or interpersonal therapy as first-line interventions, and “fluoxetine should be considered only for those patients with moderate to severe depression who do not have access to psychotherapy or have not responded to nonpharmacological interventions,” the researchers said.
The study was funded by the National Basic Research Program of China. The authors did not report any relevant financial disclosures.
FROM THE LANCET
Key clinical point: The majority of antidepressant drugs prescribed to children and adolescents are ineffective, with only fluoxetine showing significant improvement.
Major finding: Fluoxetine showed the most promising results among all 14 antidepressants studied.
Data source: Retrospective review of 34 studies with 5,260 participants investigating 14 distinct drugs used to treat depression in children and adolescents.
Disclosures: The study was funded by the National Basic Research Program of China. Authors did not report any relevant financial disclosures.
Postpartum glycemic screening rates are low for women with GDM history
Most women with a history of gestational diabetes mellitus are not receiving recommended glycemic screenings in their first postpartum year. And screening rates vary based on geography, race, and use of antiglycemic medication in pregnancy, according to the results of a study published in Obstetrics & Gynecology.
Currently, the American College of Obstetricians and Gynecologists recommends screening all women with gestational diabetes mellitus (GDM) at 6-12 weeks postpartum with either fasting plasma glucose (FPG) or a 75-g 2-hour oral glucose tolerance test (OGTT). A hemoglobin A1c (HBA1c) is not recommended in the early postpartum period.

Dr. Emma Morton Eggleston of Harvard Pilgrim Health Care Institute in Boston and her colleagues performed a retrospective analysis of medical records from a large U.S. health plan database to determine the rates of glycemic screenings – 75-g OGTT, HBA1c only, FPG only, or HbA1c plus FPG – in women with a history of GDM who were enrolled in the health plan from 2000-2012.
Rates were also measured for specific geographic regions, races/ethnicities, and patient clinical characteristics, including comorbidity in or before pregnancy, a visit to a nutritionist or diabetes educator during pregnancy, a visit to an endocrinologist during pregnancy, and the use of any antiglycemic agent during pregnancy (Obstet Gynecol. 2016;128:159-67. doi: 10.1097/AOG.0000000000001467).
Of all 447,556 women continuously enrolled in the health plan for 1 year before and after delivery, 32,253 (7.2%) had a history of GDM. The majority of women (76.1%) did not receive any of the glycemic screening tests in their first postpartum year.
The rates of these recommended tests were found to be low in general, although improvements in rates were noted between 2001 and 2011 for all but FPG alone, which declined from 7% within 12 weeks postpartum to 2% (adjusted odds ratio, 0.2). Conversely, the rate of receiving a 75-g OGTT within 12 weeks postpartum increased from 3% to 8% (adjusted OR, 3.2).
Geography was a predictor of postpartum screening. Women who lived in the West were the most likely to receive any screening within 12 weeks (18%) and at 1 year (31%). Among those who were screened, women in the West were most likely to receive a 75-g OGTT within 12 weeks (36%), compared with women in the Northeast (19%) and South (18%).
Race also played a role. Black women were the least likely to receive a 75-g OGTT and the most likely to receive HbA1c alone, even though this group has the highest rates of conversion to type 2 diabetes.
The strongest predictor of screening was the use of antiglycemic medication during pregnancy, according to the study. Women on antiglycemic medication in pregnancy (21%) were twice as likely to receive any type of screening, compared with women who were not on medication. Women who saw a nutritionist or diabetes educator during pregnancy, as well as those who saw an endocrinologist, were also more likely to receive any type of glycemic screening.
“Whether at the level of health system or population, quality improvement efforts must identify effective means of postpartum screening that are feasible for both women and health care providers and based on risk factors rather than geography or disparities in care,” the researchers wrote.
The researchers received grant support from the National Institutes of Health and the Centers for Disease Control and Prevention. No potential conflicts of interest were reported.
Most women with a history of gestational diabetes mellitus are not receiving recommended glycemic screenings in their first postpartum year. And screening rates vary based on geography, race, and use of antiglycemic medication in pregnancy, according to the results of a study published in Obstetrics & Gynecology.
Currently, the American College of Obstetricians and Gynecologists recommends screening all women with gestational diabetes mellitus (GDM) at 6-12 weeks postpartum with either fasting plasma glucose (FPG) or a 75-g 2-hour oral glucose tolerance test (OGTT). A hemoglobin A1c (HBA1c) is not recommended in the early postpartum period.
Dr. Emma Morton Eggleston of Harvard Pilgrim Health Care Institute in Boston and her colleagues performed a retrospective analysis of medical records from a large U.S. health plan database to determine the rates of glycemic screenings – 75-g OGTT, HBA1c only, FPG only, or HbA1c plus FPG – in women with a history of GDM who were enrolled in the health plan from 2000-2012.
Rates were also measured for specific geographic regions, races/ethnicities, and patient clinical characteristics, including comorbidity in or before pregnancy, a visit to a nutritionist or diabetes educator during pregnancy, a visit to an endocrinologist during pregnancy, and the use of any antiglycemic agent during pregnancy (Obstet Gynecol. 2016;128:159-67. doi: 10.1097/AOG.0000000000001467).
Of all 447,556 women continuously enrolled in the health plan for 1 year before and after delivery, 32,253 (7.2%) had a history of GDM. The majority of women (76.1%) did not receive any of the glycemic screening tests in their first postpartum year.
The rates of these recommended tests were found to be low in general, although improvements in rates were noted between 2001 and 2011 for all but FPG alone, which declined from 7% within 12 weeks postpartum to 2% (adjusted odds ratio, 0.2). Conversely, the rate of receiving a 75-g OGTT within 12 weeks postpartum increased from 3% to 8% (adjusted OR, 3.2).
Geography was a predictor of postpartum screening. Women who lived in the West were the most likely to receive any screening within 12 weeks (18%) and at 1 year (31%). Among those who were screened, women in the West were most likely to receive a 75-g OGTT within 12 weeks (36%), compared with women in the Northeast (19%) and South (18%).
Race also played a role. Black women were the least likely to receive a 75-g OGTT and the most likely to receive HbA1c alone, even though this group has the highest rates of conversion to type 2 diabetes.
The strongest predictor of screening was the use of antiglycemic medication during pregnancy, according to the study. Women on antiglycemic medication in pregnancy (21%) were twice as likely to receive any type of screening, compared with women who were not on medication. Women who saw a nutritionist or diabetes educator during pregnancy, as well as those who saw an endocrinologist, were also more likely to receive any type of glycemic screening.
“Whether at the level of health system or population, quality improvement efforts must identify effective means of postpartum screening that are feasible for both women and health care providers and based on risk factors rather than geography or disparities in care,” the researchers wrote.
The researchers received grant support from the National Institutes of Health and the Centers for Disease Control and Prevention. No potential conflicts of interest were reported.
Most women with a history of gestational diabetes mellitus are not receiving recommended glycemic screenings in their first postpartum year. And screening rates vary based on geography, race, and use of antiglycemic medication in pregnancy, according to the results of a study published in Obstetrics & Gynecology.
Currently, the American College of Obstetricians and Gynecologists recommends screening all women with gestational diabetes mellitus (GDM) at 6-12 weeks postpartum with either fasting plasma glucose (FPG) or a 75-g 2-hour oral glucose tolerance test (OGTT). A hemoglobin A1c (HBA1c) is not recommended in the early postpartum period.
Dr. Emma Morton Eggleston of Harvard Pilgrim Health Care Institute in Boston and her colleagues performed a retrospective analysis of medical records from a large U.S. health plan database to determine the rates of glycemic screenings – 75-g OGTT, HBA1c only, FPG only, or HbA1c plus FPG – in women with a history of GDM who were enrolled in the health plan from 2000-2012.
Rates were also measured for specific geographic regions, races/ethnicities, and patient clinical characteristics, including comorbidity in or before pregnancy, a visit to a nutritionist or diabetes educator during pregnancy, a visit to an endocrinologist during pregnancy, and the use of any antiglycemic agent during pregnancy (Obstet Gynecol. 2016;128:159-67. doi: 10.1097/AOG.0000000000001467).
Of all 447,556 women continuously enrolled in the health plan for 1 year before and after delivery, 32,253 (7.2%) had a history of GDM. The majority of women (76.1%) did not receive any of the glycemic screening tests in their first postpartum year.
The rates of these recommended tests were found to be low in general, although improvements in rates were noted between 2001 and 2011 for all but FPG alone, which declined from 7% within 12 weeks postpartum to 2% (adjusted odds ratio, 0.2). Conversely, the rate of receiving a 75-g OGTT within 12 weeks postpartum increased from 3% to 8% (adjusted OR, 3.2).
Geography was a predictor of postpartum screening. Women who lived in the West were the most likely to receive any screening within 12 weeks (18%) and at 1 year (31%). Among those who were screened, women in the West were most likely to receive a 75-g OGTT within 12 weeks (36%), compared with women in the Northeast (19%) and South (18%).
Race also played a role. Black women were the least likely to receive a 75-g OGTT and the most likely to receive HbA1c alone, even though this group has the highest rates of conversion to type 2 diabetes.
The strongest predictor of screening was the use of antiglycemic medication during pregnancy, according to the study. Women on antiglycemic medication in pregnancy (21%) were twice as likely to receive any type of screening, compared with women who were not on medication. Women who saw a nutritionist or diabetes educator during pregnancy, as well as those who saw an endocrinologist, were also more likely to receive any type of glycemic screening.
“Whether at the level of health system or population, quality improvement efforts must identify effective means of postpartum screening that are feasible for both women and health care providers and based on risk factors rather than geography or disparities in care,” the researchers wrote.
The researchers received grant support from the National Institutes of Health and the Centers for Disease Control and Prevention. No potential conflicts of interest were reported.
FROM OBSTETRICS & GYNECOLOGY
Key clinical point: Glycemic screening rates for women who had gestational diabetes mellitus are generally low and vary based on race, geography, and medication treatment in pregnancy.
Major finding: More than three-quarters of women with a history of gestational diabetes mellitus did not receive a glycemic screen in their first year postpartum.
Data sources: A retrospective analysis of medical records from women enrolled in a large, U.S. commercial health plan from 2000-2012.
Disclosures: The researchers received grant support from the National Institutes of Health and the Centers for Disease Control and Prevention. No potential conflicts of interest were reported.

Gyn. oncologists are in demand for robotic hysterectomy
SAN DIEGO – The demand for gynecologic oncologists to perform robotic hysterectomies – even for benign indications – has increased to the point that additional fellowship training spots will be necessary to meet the need, Dr. Kayla M. Wishall said at the annual meeting of the Society of Gynecologic Oncology.
More and more patients want their hysterectomies performed robotically. They find the high-quality optics and minimally invasive nature of the robotic procedure appealing – smaller incisions, less blood loss, shorter hospital stay, and faster recovery. And gynecologic oncologists are getting an increasing number of referrals because of their special expertise in robotic surgery and extensive experience with higher-risk patients, explained Dr. Wishall, a gynecologic oncologist at Hahnemann University Hospital/Drexel University in Philadelphia.
“This trend will likely tax the limited resources of gynecologic oncologists,” she added.
Another possible reason for the growing demand for gynecologic oncologist–performed robotic hysterectomies is that these subspecialists achieve better outcomes than gynecologists who do robotic hysterectomies, at least according to the findings of a retrospective study performed by Dr. Wishall, which included all of the 468 robotic hysterectomies performed at a large academic medical center in a recent 5-year period.
Gynecologic oncologists performed 64 (16.5%) of the 387 robotic hysterectomies done for benign indications. All told, gynecologists did 254 of the robotic hysterectomies; gynecologic oncologists performed 214.
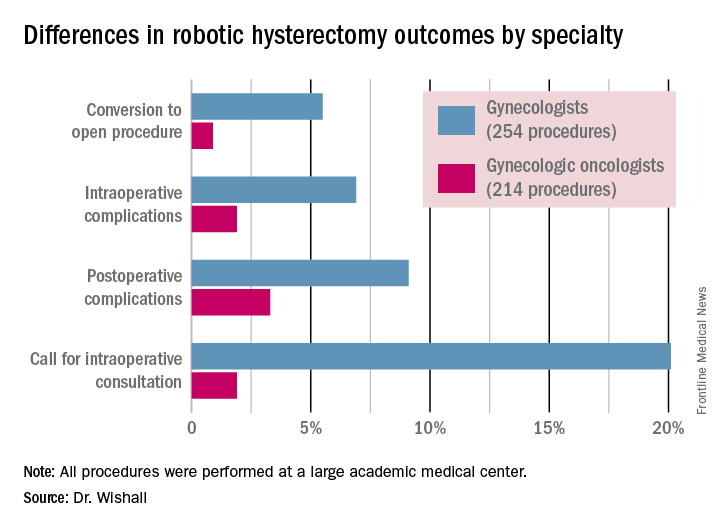
Even though patients referred to gynecologic oncologists for these procedures were older, heavier, more likely to have had previous abdominal surgery, more often members of racial minorities, and had a higher prevalence of cardiac comorbidities, they experienced significantly fewer intra- and postoperative complications than patients whose robotic hysterectomies were performed by gynecologists, Dr. Wishall reported.
The combined intraoperative and postoperative complication rate for robotic hysterectomies performed by gynecologic oncologists was 5.2%, compared with 16% for gynecologists. But the rate of cardiac comorbidities, for instance, was 36.4% among patients seeing gynecologic oncologists, compared with 23.6% among those seeing gynecologists.
Moreover, gynecologists were about 10-fold more likely than gynecologic oncologists to call for an intraoperative consultation and sixfold more likely to convert their robotic hysterectomy to an open procedure. Their average operating room time was about 40% longer (244 minutes versus 171 minutes), too, in this single-center experience.
Dr. Wishall reported having no financial conflicts related to her study, which was conducted free of commercial support.
I read this article initially with amusement and then with outrage and disdain. This article summarizes the single-site, retrospective study by Dr. Kayla Wishall at the annual meeting of the Society of Gynecologic Oncology. Not only is this nonscience, but nonsensical science. As a single center retrospective study, conclusions must be suspect.

|
Dr. Charles E. Miller |
The comparison numbers of the two groups are small. While confounders would appear to be greater in the oncology group, we know nothing about the difficulty of the surgeries themselves – size of uterus, adnexal disease, endometriosis, pelvic adhesions, etc. Oftentimes, gynecologic oncologists dealing with endometrial carcinoma are going to face a less difficult challenge than a generalist dealing with an 18-weeks–size uterus in a woman who has undergone three prior C-sections, an open myomectomy, or stage IV endometriosis.
We are also not privy to the experience of the surgeons involved; that is, the number of procedures performed by each surgeon in the compared groups. It is certainly well known that complications decrease with surgeon experience. In a multicenter analysis by Peter Lim et al., looking at robotic assisted hysterectomies performed by high-volume surgeons (60 or more prior procedures), the intraoperative complication rate was only 0.7% and the postoperative complication rate 6.3% (Int J Gynaecol Obstet. 2016 Jun;133[3]:359-64).
As a benign gynecologist who has been performing minimally invasive gynecologic surgery for 30 years and more recently, robotic surgery, I am shocked with the tenor of this study, as it would imply that unless someone is boarded in gynecologic oncology, he or she should not be performing robotic hysterectomies.
I would advise Dr. Wishall to reevaluate her surgeon population and look at the impact of experience as well as procedure difficultly. I am absolutely sure that she will find that many of the surgeons with excellent outcomes will be generalists, who are well experienced in robotic hysterectomy.
Dr. Charles E. Miller is a clinical associate professor at the University of Illinois at Chicago, and a reproductive endocrinologist and minimally invasive gynecologic surgeon in private practice in Naperville and Schaumburg, Ill. He reported having no financial disclosures relevant to this article.
I read this article initially with amusement and then with outrage and disdain. This article summarizes the single-site, retrospective study by Dr. Kayla Wishall at the annual meeting of the Society of Gynecologic Oncology. Not only is this nonscience, but nonsensical science. As a single center retrospective study, conclusions must be suspect.
|
|
Dr. Charles E. Miller |
The comparison numbers of the two groups are small. While confounders would appear to be greater in the oncology group, we know nothing about the difficulty of the surgeries themselves – size of uterus, adnexal disease, endometriosis, pelvic adhesions, etc. Oftentimes, gynecologic oncologists dealing with endometrial carcinoma are going to face a less difficult challenge than a generalist dealing with an 18-weeks–size uterus in a woman who has undergone three prior C-sections, an open myomectomy, or stage IV endometriosis.
We are also not privy to the experience of the surgeons involved; that is, the number of procedures performed by each surgeon in the compared groups. It is certainly well known that complications decrease with surgeon experience. In a multicenter analysis by Peter Lim et al., looking at robotic assisted hysterectomies performed by high-volume surgeons (60 or more prior procedures), the intraoperative complication rate was only 0.7% and the postoperative complication rate 6.3% (Int J Gynaecol Obstet. 2016 Jun;133[3]:359-64).
As a benign gynecologist who has been performing minimally invasive gynecologic surgery for 30 years and more recently, robotic surgery, I am shocked with the tenor of this study, as it would imply that unless someone is boarded in gynecologic oncology, he or she should not be performing robotic hysterectomies.
I would advise Dr. Wishall to reevaluate her surgeon population and look at the impact of experience as well as procedure difficultly. I am absolutely sure that she will find that many of the surgeons with excellent outcomes will be generalists, who are well experienced in robotic hysterectomy.
Dr. Charles E. Miller is a clinical associate professor at the University of Illinois at Chicago, and a reproductive endocrinologist and minimally invasive gynecologic surgeon in private practice in Naperville and Schaumburg, Ill. He reported having no financial disclosures relevant to this article.
I read this article initially with amusement and then with outrage and disdain. This article summarizes the single-site, retrospective study by Dr. Kayla Wishall at the annual meeting of the Society of Gynecologic Oncology. Not only is this nonscience, but nonsensical science. As a single center retrospective study, conclusions must be suspect.
|
|
Dr. Charles E. Miller |
The comparison numbers of the two groups are small. While confounders would appear to be greater in the oncology group, we know nothing about the difficulty of the surgeries themselves – size of uterus, adnexal disease, endometriosis, pelvic adhesions, etc. Oftentimes, gynecologic oncologists dealing with endometrial carcinoma are going to face a less difficult challenge than a generalist dealing with an 18-weeks–size uterus in a woman who has undergone three prior C-sections, an open myomectomy, or stage IV endometriosis.
We are also not privy to the experience of the surgeons involved; that is, the number of procedures performed by each surgeon in the compared groups. It is certainly well known that complications decrease with surgeon experience. In a multicenter analysis by Peter Lim et al., looking at robotic assisted hysterectomies performed by high-volume surgeons (60 or more prior procedures), the intraoperative complication rate was only 0.7% and the postoperative complication rate 6.3% (Int J Gynaecol Obstet. 2016 Jun;133[3]:359-64).
As a benign gynecologist who has been performing minimally invasive gynecologic surgery for 30 years and more recently, robotic surgery, I am shocked with the tenor of this study, as it would imply that unless someone is boarded in gynecologic oncology, he or she should not be performing robotic hysterectomies.
I would advise Dr. Wishall to reevaluate her surgeon population and look at the impact of experience as well as procedure difficultly. I am absolutely sure that she will find that many of the surgeons with excellent outcomes will be generalists, who are well experienced in robotic hysterectomy.
Dr. Charles E. Miller is a clinical associate professor at the University of Illinois at Chicago, and a reproductive endocrinologist and minimally invasive gynecologic surgeon in private practice in Naperville and Schaumburg, Ill. He reported having no financial disclosures relevant to this article.
SAN DIEGO – The demand for gynecologic oncologists to perform robotic hysterectomies – even for benign indications – has increased to the point that additional fellowship training spots will be necessary to meet the need, Dr. Kayla M. Wishall said at the annual meeting of the Society of Gynecologic Oncology.
More and more patients want their hysterectomies performed robotically. They find the high-quality optics and minimally invasive nature of the robotic procedure appealing – smaller incisions, less blood loss, shorter hospital stay, and faster recovery. And gynecologic oncologists are getting an increasing number of referrals because of their special expertise in robotic surgery and extensive experience with higher-risk patients, explained Dr. Wishall, a gynecologic oncologist at Hahnemann University Hospital/Drexel University in Philadelphia.
“This trend will likely tax the limited resources of gynecologic oncologists,” she added.
Another possible reason for the growing demand for gynecologic oncologist–performed robotic hysterectomies is that these subspecialists achieve better outcomes than gynecologists who do robotic hysterectomies, at least according to the findings of a retrospective study performed by Dr. Wishall, which included all of the 468 robotic hysterectomies performed at a large academic medical center in a recent 5-year period.
Gynecologic oncologists performed 64 (16.5%) of the 387 robotic hysterectomies done for benign indications. All told, gynecologists did 254 of the robotic hysterectomies; gynecologic oncologists performed 214.
Even though patients referred to gynecologic oncologists for these procedures were older, heavier, more likely to have had previous abdominal surgery, more often members of racial minorities, and had a higher prevalence of cardiac comorbidities, they experienced significantly fewer intra- and postoperative complications than patients whose robotic hysterectomies were performed by gynecologists, Dr. Wishall reported.
The combined intraoperative and postoperative complication rate for robotic hysterectomies performed by gynecologic oncologists was 5.2%, compared with 16% for gynecologists. But the rate of cardiac comorbidities, for instance, was 36.4% among patients seeing gynecologic oncologists, compared with 23.6% among those seeing gynecologists.
Moreover, gynecologists were about 10-fold more likely than gynecologic oncologists to call for an intraoperative consultation and sixfold more likely to convert their robotic hysterectomy to an open procedure. Their average operating room time was about 40% longer (244 minutes versus 171 minutes), too, in this single-center experience.
Dr. Wishall reported having no financial conflicts related to her study, which was conducted free of commercial support.
SAN DIEGO – The demand for gynecologic oncologists to perform robotic hysterectomies – even for benign indications – has increased to the point that additional fellowship training spots will be necessary to meet the need, Dr. Kayla M. Wishall said at the annual meeting of the Society of Gynecologic Oncology.
More and more patients want their hysterectomies performed robotically. They find the high-quality optics and minimally invasive nature of the robotic procedure appealing – smaller incisions, less blood loss, shorter hospital stay, and faster recovery. And gynecologic oncologists are getting an increasing number of referrals because of their special expertise in robotic surgery and extensive experience with higher-risk patients, explained Dr. Wishall, a gynecologic oncologist at Hahnemann University Hospital/Drexel University in Philadelphia.
“This trend will likely tax the limited resources of gynecologic oncologists,” she added.
Another possible reason for the growing demand for gynecologic oncologist–performed robotic hysterectomies is that these subspecialists achieve better outcomes than gynecologists who do robotic hysterectomies, at least according to the findings of a retrospective study performed by Dr. Wishall, which included all of the 468 robotic hysterectomies performed at a large academic medical center in a recent 5-year period.
Gynecologic oncologists performed 64 (16.5%) of the 387 robotic hysterectomies done for benign indications. All told, gynecologists did 254 of the robotic hysterectomies; gynecologic oncologists performed 214.
Even though patients referred to gynecologic oncologists for these procedures were older, heavier, more likely to have had previous abdominal surgery, more often members of racial minorities, and had a higher prevalence of cardiac comorbidities, they experienced significantly fewer intra- and postoperative complications than patients whose robotic hysterectomies were performed by gynecologists, Dr. Wishall reported.
The combined intraoperative and postoperative complication rate for robotic hysterectomies performed by gynecologic oncologists was 5.2%, compared with 16% for gynecologists. But the rate of cardiac comorbidities, for instance, was 36.4% among patients seeing gynecologic oncologists, compared with 23.6% among those seeing gynecologists.
Moreover, gynecologists were about 10-fold more likely than gynecologic oncologists to call for an intraoperative consultation and sixfold more likely to convert their robotic hysterectomy to an open procedure. Their average operating room time was about 40% longer (244 minutes versus 171 minutes), too, in this single-center experience.
Dr. Wishall reported having no financial conflicts related to her study, which was conducted free of commercial support.
AT THE ANNUAL MEETING ON WOMEN’S CANCER
Key clinical point: Gynecologic oncologists achieved better robotic hysterectomy outcomes than gynecologists despite challenging referrals.
Major finding: The combined intraoperative and postoperative complication rate for robotic hysterectomies performed by gynecologic oncologists was 5.2%, compared with 16% for gynecologists.
Data source: A retrospective observational study conducted at a single center included 254 women whose robotic hysterectomies were performed by gynecologists and 214 done by gynecologic oncologists.
Disclosures: Dr. Wishall reported having no financial conflicts related to the study, which was conducted free of commercial support.
Marijuana use disorders in adolescents on the decline
New findings show that marijuana use disorders are on the decline among U.S. adolescents, according to Richard A. Grucza, Ph.D., and his associates.
They examined National Survey on Drug Use and Health data for 2002-2013 from 216,852 adolescents aged 12-17 years. They divided the data into two age groups: 12-14 and 15-17.
The prevalence of past-year marijuana use of the adolescents declined steadily from 15.8% in 2002 to 12.5% in 2007. After that it began to climb, peaking at 14.2% to 14.3% in 2010 and 2011. Then it dropped back to 13.2% in 2013. The decline was significant for both age groups, with no significant difference in trends between the groups (ages 12-14 years, odds ratio, 0.978 per year; ages 15-17 years, OR, 0.987).

In examining risk factors and protective factors, the researchers observed significant negative trends for the three risk factors and significant positive trends for four of the six protective factors. The two protective factors that decreased over time were drug education and religious commitment. “Thus, seven of the nine risk/protective factors changed over time in a manner that might partially explain the downward trend in the prevalence of marijuana use disorders,” they noted.
“Our findings underscore the importance of adolescent mental health in conferring resilience to risk for substance use disorders,” the researchers concluded. “There are one or more environmental factors – yet to be identified – that may be changing over time in a manner that leads to both lower risk for marijuana use disorders and for other behavioral problems.”
Read the study in the Journal of the American Academy of Child & Adolescent Psychiatry (doi: 10.1016/j.jaac.2016.04.002).
New findings show that marijuana use disorders are on the decline among U.S. adolescents, according to Richard A. Grucza, Ph.D., and his associates.
They examined National Survey on Drug Use and Health data for 2002-2013 from 216,852 adolescents aged 12-17 years. They divided the data into two age groups: 12-14 and 15-17.
The prevalence of past-year marijuana use of the adolescents declined steadily from 15.8% in 2002 to 12.5% in 2007. After that it began to climb, peaking at 14.2% to 14.3% in 2010 and 2011. Then it dropped back to 13.2% in 2013. The decline was significant for both age groups, with no significant difference in trends between the groups (ages 12-14 years, odds ratio, 0.978 per year; ages 15-17 years, OR, 0.987).
In examining risk factors and protective factors, the researchers observed significant negative trends for the three risk factors and significant positive trends for four of the six protective factors. The two protective factors that decreased over time were drug education and religious commitment. “Thus, seven of the nine risk/protective factors changed over time in a manner that might partially explain the downward trend in the prevalence of marijuana use disorders,” they noted.
“Our findings underscore the importance of adolescent mental health in conferring resilience to risk for substance use disorders,” the researchers concluded. “There are one or more environmental factors – yet to be identified – that may be changing over time in a manner that leads to both lower risk for marijuana use disorders and for other behavioral problems.”
Read the study in the Journal of the American Academy of Child & Adolescent Psychiatry (doi: 10.1016/j.jaac.2016.04.002).
New findings show that marijuana use disorders are on the decline among U.S. adolescents, according to Richard A. Grucza, Ph.D., and his associates.
They examined National Survey on Drug Use and Health data for 2002-2013 from 216,852 adolescents aged 12-17 years. They divided the data into two age groups: 12-14 and 15-17.
The prevalence of past-year marijuana use of the adolescents declined steadily from 15.8% in 2002 to 12.5% in 2007. After that it began to climb, peaking at 14.2% to 14.3% in 2010 and 2011. Then it dropped back to 13.2% in 2013. The decline was significant for both age groups, with no significant difference in trends between the groups (ages 12-14 years, odds ratio, 0.978 per year; ages 15-17 years, OR, 0.987).
In examining risk factors and protective factors, the researchers observed significant negative trends for the three risk factors and significant positive trends for four of the six protective factors. The two protective factors that decreased over time were drug education and religious commitment. “Thus, seven of the nine risk/protective factors changed over time in a manner that might partially explain the downward trend in the prevalence of marijuana use disorders,” they noted.
“Our findings underscore the importance of adolescent mental health in conferring resilience to risk for substance use disorders,” the researchers concluded. “There are one or more environmental factors – yet to be identified – that may be changing over time in a manner that leads to both lower risk for marijuana use disorders and for other behavioral problems.”
Read the study in the Journal of the American Academy of Child & Adolescent Psychiatry (doi: 10.1016/j.jaac.2016.04.002).
FROM JOURNAL OF THE AMERICAN ACADEMY OF CHILD & ADOLESCENT PSYCHIATRY

Management Challenges in Sarcoidosis
From the New Cross Hospital, Wolverhampton, UK.
Abstract
- Objective: To discuss the management of sarcoidosis.
- Methods: Review of the literature.
- Results: Sarcoidosis is a challenging multisystem disorder of uncertain etiology characterized by granulomatous inflammation in the affected organs. Treatment is dependent on the severity of disease and organ involvement at the time of diagnosis. Glucocorticoids have traditionally been considered first-line pharmacologic treatment; however, a significant proportion of patients do not require drug treatment due to the propensity toward spontaneous disease remission. Treated patients who fail to respond to corticosteroids or develop significant adverse effects can be offered a second-line agent, eg, methotrexate. Anti-TNF therapy may be considered as a treatment option in carefully selected patients with refractory disease after discussion of potential adverse effects followed by close monitoring at a specialist center.
- Conclusion: Further research into therapeutic options is likely to unveil novel agents with different mechanisms of action and better safety profiles than those seen with currently available immunosuppressive regimens.
Sarcoidosis is a multisystem disorder of uncertain etiology characterized by granulomatous inflammation in the affected organs. The diagnosis of sarcoidosis is best supported by histological evidence of noncaseating granuloma formation. It is a disease with a generally good prognosis; less than 5% patients die from the disease, with cause of death usually secondary to respiratory failure or cardiac or neurologic involvement. This review aims to discuss the management of sarcoidosis with a special emphasis on the management challenges resulting from the myriad clinical manifestations and potential complications seen in this chronic multisystem disease.
Case Study
Initial Presentation
A 40-year-old African-American man presents to his primary care physician with symptoms of fatigue, dry cough, exertional breathlessness, dry and painful eyes, generalized arthralgia, and multiple skin lesions for 3 months. He has a history of essential hypertension and is a former smoker with 10 pack-year history. He is not on any regular medications. Examination reveals bilateral cervical lymphadenopathy and multiple skin lesions on trunk. The rest of the systemic examination (including respiratory and cardiovascular system) is normal.
Workup
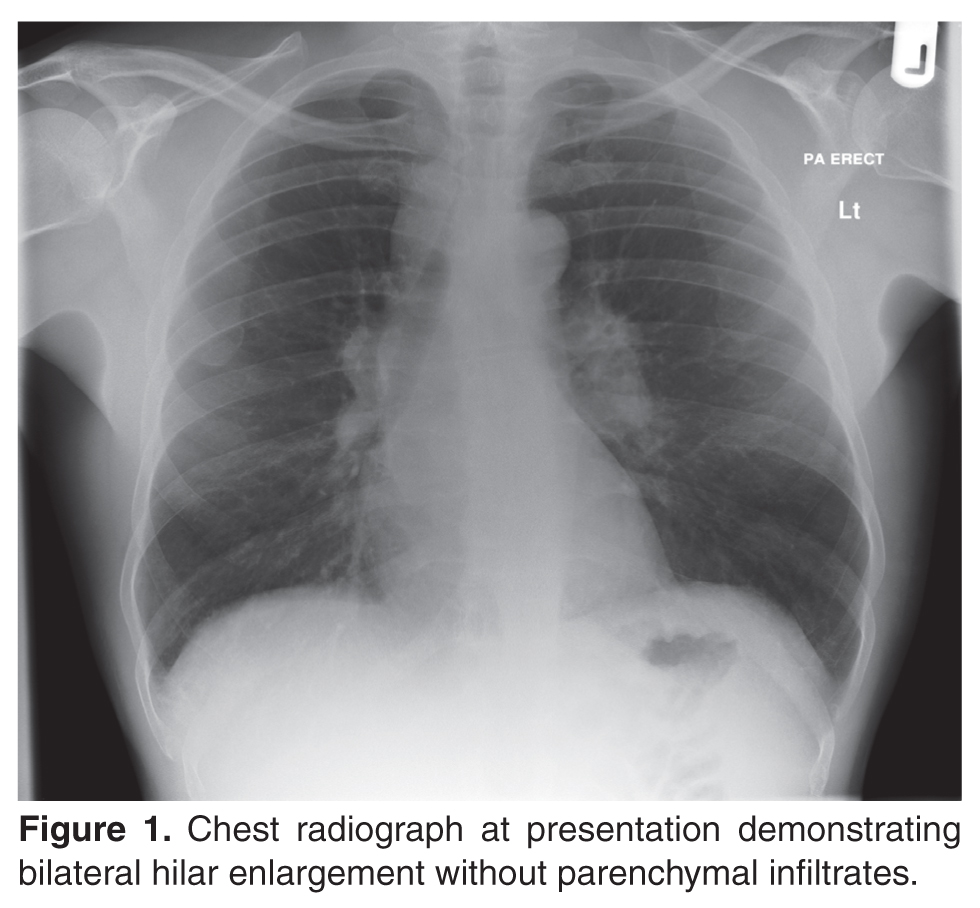
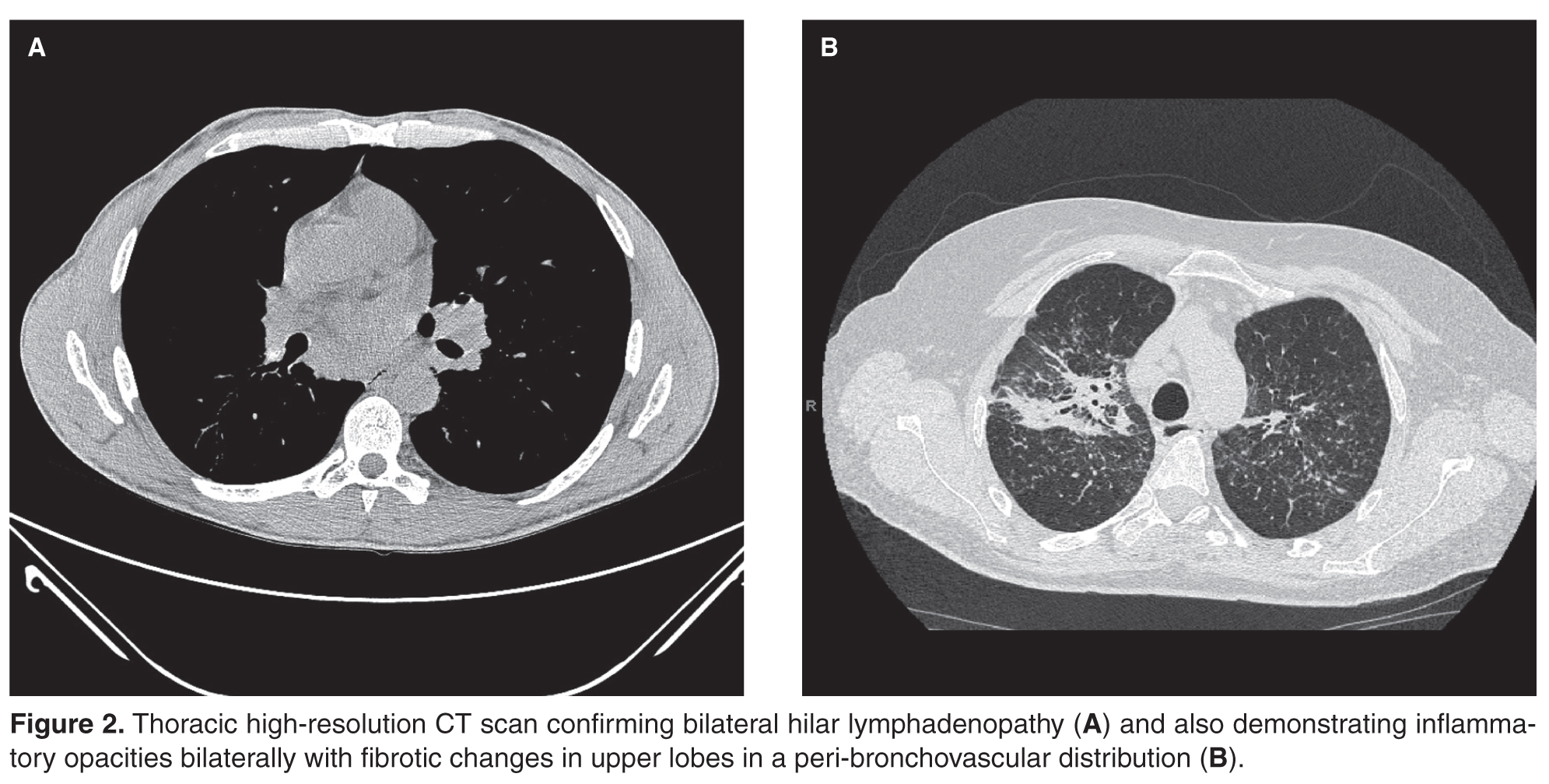
The patient’s initial bloodwork showed a mild degree of lymphopenia (1.1 × 109/L, normal range 1.5–4.5). Other bloodwork results, bone profile and immunology screen (including ANA, rheumatoid factor, immunoglobulins, and extractable nuclear antigen antibodies) were negative. Angiotensin-converting enzyme (ACE) was elevated at 149 U/L (normal range 5–58), while serum calcium and vitamin D levels (including vitamin D3) were normal. The findings on CT scan along with the biochemical profile suggest a plausible diagnosis of sarcoidosis.
What is the next step in the workup to establish the suspected diagnosis?
The radiological findings of hilar lymphadenopathy are not confirmatory. There are a number of entities in the differential diagnoses, including tuberculosis, malignancy, lymphoma, and other granulomatous disorders such as histoplasmosis, schistosomiasis, and blastomycosis. It is important to obtain histological evidence before a definitive diagnosis of sarcoidosis can be made. This is necessary as management differs for each of the diagnostic categories mentioned above. Furthermore, diagnostic confirmation would be helpful later in the disease course if the patient develops any associated complications such as pulmonary hypertension or respiratory failure and/or need for lung transplant assessment.
As this patient had palpable cervical lymphadenopathy, an ultrasound guided biopsy of the lymph node was obtained. The histological examination demonstrated evidence of noncaseating granulomas that were well formed and highly consistent with the suspected clinical and radiological diagnosis of sarcoidosis. In addition, the special stains for acid-fast bacilli and other infections including fungi were negative.
Our practice is to evaluate patients with suspected sarcoidosis with neck ultrasound and tru-cut biopsy of cervical lymph nodes (if appropriate) as a first-line investigation, as it is less invasive than bronchoscopy/endoscopic ultrasound–guided transbronchial needle aspiration (EBUS-TBNA) or thoracoscopic lung biopsy. The diagnostic yield of EBUS-TBNA has been evaluated in a number of studies with variable results [1–5]. A large multicenter randomized clinical trial [5] of 304 patients investigated the diagnostic yield of endosonography (endobronchial and esophageal ultrasound) in comparison with bronchoscopy with transbronchial biopsy (TBB) and endobronchial biopsy. The study cohort was made up of patients with stage I/II sarcoidosis. The results showed that endosonography had a higher diagnostic yield to detect granulomas (80% vs 53%; P < 0.001) and there were no serious adverse events related to endoscopy. Hence, ultrasound-guided endoscopic procedures are becoming common first-line investigations for sarcoidosis in the absence of other readily identifiable biopsy sites such as peripheral lymph nodes in the cervical area.
What should be done about the cutaneous and ophthalmologic symptoms in this patient?
As sarcoidosis commonly involves eyes and skin (after pulmonary involvement, which is seen in 90% of cases), the patient was referred to ophthalmology and dermatology departments for further evaluation. These assessments confirmed him to have bilateral uveitis and skin involvement with granulomatous inflammation consistent with ocular and cutaneous sarcoidosis respectively. Hence, the diagnosis of multisystem sarcoidosis was made. At this stage, the patient also mentioned symptoms of intermittent palpitations for 3 months’ duration and feeling of missing a beat, so an urgent cardiological evaluation was undertaken that showed him to have ectopic beats on Holter monitoring. However, his trans-thoracic echocardiogram and a cardiac MRI scan were normal with good biventricular function, excluding cardiac sarcoidosis as a cause of his palpitations. Cardiac involvement with sarcoidosis is clinically apparent in only 5% of cases and presents as cardiomyopathy and or cardiac arrhythmias (both tachy and bradyarrythmias). As cardiac involvement with sarcoid granulomas is usually patchy, endomyocardial biopsy has a limited diagnostic yield of < 20% [6]. In this particular case, endomyocardial biopsy was not attempted in view of normal cardiac MR and echocardiography as well as no significant cardiac dysrythmia on holter monitoring.
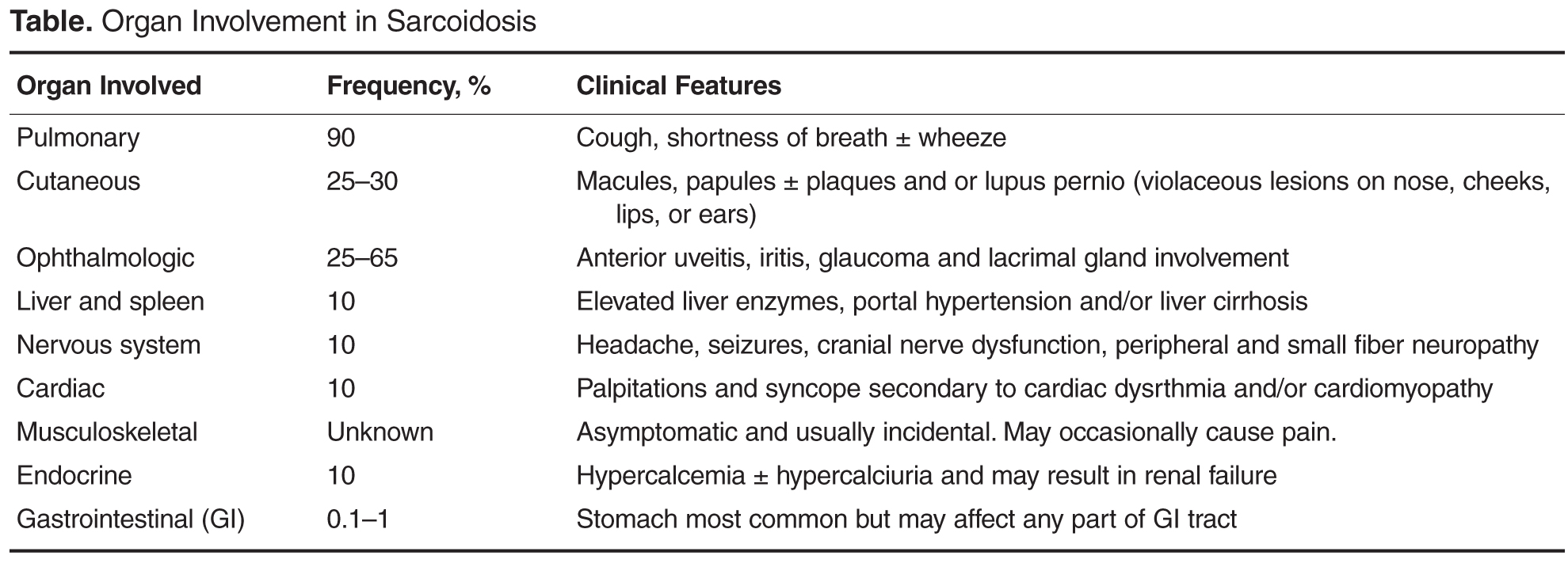
Case Continued
The patient was started on corticosteroid eye drops and steroid ointment for his ophthalmologic and cutaneous sarcoidosis, respectively, and symptoms gradually improved over the next few months. As the patient was symptomatic with cough and breathlessness and there was evidence of reduction in FVC (along with reduced DLco), a trial of oral corticosteroids was considered to treat the pulmonary sarcoidosis.
What is first-line pharmacological treatment for sarcoidosis and when is it indicated?
Treatment of sarcoidosis is dependent upon the severity of disease and organ involvement at the time of the diagnosis. Glucocorticoids have traditionally been considered first-line pharmacological agents in selective cases, as a significant proportion of patients do not require drug treatment due to the propensity for spontaneous remission. Furthermore, sarcoidosis remains stable without anti-inflammatory/immunosuppressive therapies in a majority of patients. A number of clinical trials have evaluated the value of corticosteroids in the management of sarcoidosis, with variable outcomes [10–16]. The disease tends to be severe in patients of African descent compared with patients from other racial backgrounds. The European cohort of sarcoidosis patients generally have milder disease with less propensity for vital organ involvement such as cardiac or central nervous system (CNS) disease.
The decision to initiate corticosteroids for sarcoidosis is not a straightforward one as there is variability in symptom presentation, disease severity, and response to corticosteroids. We initiate first-line therapy with oral prednisolone in the following circumstances:
- Evidence of pulmonary impairment (forced vital capacity FVC < 80% predicted) with or without reduction in gas transfer (DLco) along with respiratory symptoms of cough, chest pain, and/or breathlessness (as seen in the case patient)
- Vital organ involvement such as cardiac, ophthalmic (such as panuveitis) or CNS sarcoidosis once confirmed by respective investigations
- Selective cases of sarcoid-associated pulmonary hypertension (SAPH) along with close liaison with pulmonary hypertension specialists
We recommend an initial starting dose of 20 to 40 mg of prednisolone for a period of 1 to 3 months, followed by maintenance dose of 10 mg or less for a further 6 to 9 months, aiming for a total duration of treatment of 12 months. However, the duration may vary depending on the response and any associated adverse effects with corticosteroids. It is usual practice to supplement with calcium and vitamin D when beginning patients on oral corticosteroids due to the potential risk of osteoporosis. However, this may result in significant hypercalcemia, which itself may be an endocrine manifestation of sarcoidosis. Hence, we recommend monitoring serum calcium during treatment and supplement vitamin D in patients who are vitamin D–deficient [17]. Furthermore, serum vitamin 1,25(OH)2 vitamin D3 has been demonstrated to the best available test to evaluate vitamin D status in sarcoidosis [18].
Case Continued
What are the preferred pharmacological agents for second-line treatment in sarcoidosis?
Alternative immunosuppression should be considered in the following circumstances in patients diagnosed with sarcoidosis:
- Failure or less than optimal response to oral corticosteroids
- Use as a steroid-sparing agent in patients requiring high doses of steroids for symptomatic control
- Failure to tolerate corticosteroids due to significant adverse effects such as excessive weight gain, steroid-induced psychosis, osteoporosis, and worsening diabetic control
A small trial of 11 patients examined azathioprine as a steroid-sparing agent and found it as an acceptable immunosuppressive agent for that purpose [19]. It was associated with good safety profile and adherence to treatment was 82% (9 out of 11 patients). However, the small sample size makes it difficult to draw firm conclusions from the findings of this study. In another trial evaluating methotrexate as a steroid-sparing agent in the first year after the diagnosis of sarcoidosis, Baughman and colleagues [20] reported that methotrexate is an attractive alternative to other immunosuppressive agents in term of a steroid sparer. In this double-blind randomized controlled trial (RCT), 15 patients were studied with at least 6 months of treatment with methotrexate vs placebo. There was a significantly reduced dosage of prednisolone observed in methotrexate group. However, the difference was not significant when data were analyzed for all patients, including the dropouts. More recently, a large international retrospective cohort study of 200 patients with sarcoidosis demonstrated that both methotrexate and azathioprine have similar efficacy and steroid-sparing capacity in sarcoidosis [21]. However, infection rates were significantly higher in the azathioprine group as compared to methotrexate (34.6% vs. 18.1%, P = 0.01). Hence methotrexate should be considered as a preferred second-line agent in sarcoidosis after detailed discussion about potential side effects.
Case Continued
The patient was started on methotrexate after discussion about the potential adverse effects of bone marrow suppression, hepatotoxicity, and pneumonitis. He was screened for latent tuberculosis and viral hepatitis prior to starting methotrexate. The dosage was 7.5 mg per week along with folic acid once a week. We gradually increase the dose in increments of 2.5 mg every 2 weeks with a view to reach 15 mg every week as maintenance therapy. In severely obese patients, a dose of up to 20 mg weekly is occasionally considered if 15 mg is suboptimal after careful clinical assessment.
The patient failed to make significant progress after being on methotrexate for a period of 6 months and lung function tests continued to demonstrate a persistent decline with symptomatic worsening of dyspnea and cough.
What are treatment options in refractory sarcoidosis?
Options to consider in the setting of refractory sarcoidosis are leflunomide, hydroxychloroquine, or combination therapy of methotrexate and leflunomide. Leflunomide has been shown to be of similar efficacy to methotrexate as demonstrated by a retrospective analysis of 32 patients treated with the drug in a tertiary care center [22]. Complete or partial response was noted in 12 of 17 patients treated solely with leflunomide and 13 of 15 treated in conjunction with methotrexate. Hence, combination therapy has been suggested as a viable option for these patients who fail to respond to initial glucocorticoid agents and alternative immunosuppressive drugs, as combination therapy may enhance efficacy with reduced toxicity if considered in a rational manner after careful selection of patients [23].
How should the symptom of fatigue be addressed?
The patient had ongoing fatigue during the treatment period with corticosteroids and alternative immunosuppressants. Fatigue, noted in a majority of patients with sarcoidosis [24], is one of the commonest symptoms of sarcoidosis and one of the most difficult to treat. We recommend excluding alternative etiologies of fatigue when confronted with this symptom and evaluating for associated comorbidities such as thyroid dysfunction, vitamin D deficiency, and hypoadrenalism. Extra-pulmonary sarcoidosis seems to be associated with fatigue in comparison to sarcoidosis restricted to the pulmonary system [25]. Furthermore, there is a paucity of good quality data on the benefit of pharmacological intervention for treatment of fatigue. A small double-blind randomized study of 10 patients demonstrated a positive impact of treatment with dexmethylphenidate hydrochloride (d-MPH) for a period of 8 weeks [26]. However, the small sample and lack of long-term outcomes data make it difficult to draw firm conclusions based on the findings of this study and larger randomized trials are warranted to investigate this important aspect of sarcoidosis before recommending a pharmacological agent routinely for this disabling symptom.
What are the pharmacological agents for cutaneous sarcoidosis?
Corticosteroids (local and or systemic) are the mainstay of treatment in cutaneous sarcoidosis. However, patients who fail to respond to these agents or develop significant adverse effects should be offered second-line agents in the form of hydroxychloroquine/chloroquine, methotrexate, or leflunomide. It is important to acknowledge that the evidence of benefit for these agents is derived from uncontrolled studies [27–29]. Anti-malarial agents are usually well tolerated; patients do require a baseline ophthalmological assessment and subsequent periodic examinations to monitor for any ocular toxicity associated with their use. Leflunomide is also a second-line option in cutaneous disease and is associated with lesser toxicity than methotrexate [22]. More recently, topical tacrolimus has shown promising results in isolated case reports [30–33]. Hence, it may be considered a treatment option in refractory cutaneous sarcoidosis.
Is there a role for anti-TNF therapy in the management of sarcoidosis? Should it be considered for the case patient?
Tumour necrosis factor-α (TNF-α) is implicated in the pathogenesis of sarcoidosis and is believed to have a significant role in the inflammatory processes in sarcoidosis. It is released from alveolar macrophages of patients with active disease and is involved in formation and maintenance of granulomas in the lung tissue. There has been significant progress in therapeutic options alternative to traditional immunosuppressants such as corticosteroids and TNF-α inhibition has been on the horizon as a treatment strategy for few years. It may be considered as a steroid-sparing agent or a third-line treatment option in refractory cases.
The initial evidence of benefit of anti-TNF therapy comes from case reports and small case series [34,35]. However, there have been 2 RCTs published so far invest-igating infliximab in sarcoidosis [36,37]. Baughman and colleagues [36] evaluated 138 patients with chronic pulmonary sarcoidosis in a double-blind study. There was a statistically significant improvement in FVC after 24 weeks of therapy with infliximab as compared to placebo (2.5% increase in mean FVC % predicted from baseline, P = 0.038). However, there was no improvement in any of the secondary outcome variables including SGRQ (St George’s Respiratory Questionnaire), 6-minute walk distance, and dyspnea scores. The benefit seemed to be more pronounced in the severe disease category on post-hoc analysis. The clinical significance of these findings are however unclear and it is difficult to draw firm conclusions based on the findings of this study alone.
The other RCT exploring the role of infliximab in sarcoidosis was conducted by Rossman et al [37]. This multi-center phase II study was conducted to evaluate the safety and tolerability of the drug in active pulmonary sarcoidosis (stage II to IV). The trial had a small number of participants with only 19 patients and failed to show a significant improvement in lung function after 6 weeks of treatment. Furthermore, 4 patients developed serious adverse events after treatment. Doty and colleagues [34] retrospectively analysed 10 patients treated with infliximab for refractory sarcoidosis and found subjective and objective evidence of improvement in all patients. This therapy resulted in reduction of corticosteroid dosage in 83% of cases (5 out of 6). However, adverse reactions were noted in 3 cases, including development of angioimmunoblastic lymphoma in one case. A retrospective study of 16 consecutive unselected cases of refractory sarcoidosis by Chapelon-Abric and colleagues [38] demonstrated a positive response in a majority of cases. However 38% of patients experienced a relapse. Furthermore, 44% of patients (7 out of 16) had infectious complications associated with anti-TNF therapy. Finally, infliximab may be used successfully in treating severe small fibre neuropathy [39] and should be considered in refractory cases where neuropathy is associated with autonomic dysfunction.
In summary, based on the findings of the aforementioned trials and case series, anti-TNF therapy may be considered as a treatment option in carefully selected patients after discussion of the potential adverse effects, followed by close monitoring at a specialist center for the management of sarcoidosis. Furthermore, anti-TNF therapy should not delay referral for lung transplant assessment, particularly when disease progression is relatively rapid. Duration of treatment and timing of thoracic imaging after anti-TNF therapy is subject to debate and should be individualized in close collaboration with a specialist center.
Case Continued
What is the role of newer biologics in the treatment of sarcoidosis? And what other therapies are on the horizon?
Although sarcoidosis is a T cell–mediated disease, humoral immunity has been implicated in sarcoidosis [40] and B cell depletion by rituximab (anti-CD 20+chimeric monoclonal antibody) has been successfully utilized in T cell–mediated diseases such as rheumatoid arthritis. Rituximab has been studied in phase I/II trial [41] in patients with refractory pulmonary sarcoidosis. The response to rituximab was inconsistent in these patients, with only a small group demonstrating > 5% improvement in FVC or walking distance. Hence, further studies are required to demonstrate a significant clinical benefit of this treatment in refractory sarcoidosis and potentially identify the characteristics of patients that may respond to B-cell depletion.
Adalimumab is another biologic agent that may be a potential option in refractory sarcoidosis as demonstrated in an open-label study of 11 patients [42]. This small trial of 52 weeks’ duration showed that adalimumab is well tolerated and may be considered in refractory pulmonary sarcoidosis when other treatment options have been exhausted.
Acthar had been used to treat pulmonary sarcoidosis in 1950s and there has been recent interest in evaluating the value of acthar gel therapy in the management of sarcoidosis. Baughman and colleagues [43] carried out a retrospective analysis of 47 patients with advanced sarcoidosis treated with acthar gel. The results showed that there was an objective improvement in approximately a third of patients receiving at least 3 months of treatment. Thirty-six percent of patients (n = 17) managed to reduce their oral corticosteroid dosage by more than 50% while on this therapy. However, a significant proportion of patients were unable to take ≥ 3 months of treatment, suggesting poor tolerance/adherence. The utility of acthar gel therapy would need to be examined in larger prospective randomized trial before it can be recommended for a treatment option in advanced disease.
Should pneumocystis pneumonia (PCP) prophylaxis be considered in sarcoidosis?
We do not routinely consider PCP prophylaxis in all sarcoidosis patients. However, it should be considered in the following clinical situations:
- Failure to reduce oral corticosteroid dose to less than 20 mg of prednisolone daily
- Concomitant use of anti-TNF agents with relatively high maintenance dose of oral corticosteroids (≥ 20 mg per day)
- Significant comorbid condition in association with sarcoidosis resulting in significant level of immunosuppression
As our patient did not fall in any of the above categories, we did not offer him PCP prophylaxis. However, clinicians treating patients with sarcoidosis with strong immunosuppressants should be aware of this potential complication and be vigilant about discussing prophylaxis with trimethoprim-sulphamethoxazole, which is the first-line agent for this purpose.
Is there a role of anti-tuberculous treatment in sarcoidosis?
Both mycobacterium tuberculosis (MTB) and non-mycobateria (NTM) have been implicated in sarcoidosis [44] and it is challenging to differentiate tuberculosis (TB) from sarcoidosis in certain clinical situations, such as when dealing with patients from countries with a high incidence of TB. The association of mycobacterium with sarcoidosis was explored in 2 recent trials of concomitant use of levofloxacin, ethambutol, azithromycin, and rifampin in cutaneous [45] and pulmonary sarcoidosis [46]. Drake and colleagues evaluated 15 chronic pulmonary sarcoidosis patients in an open-label trial to investigate if the combination of these drugs was associated with improvement in pulmonary sarcoidosis. The patients who completed 8 weeks of treatment had improvement in FVC at both 4 and 8 weeks of treatment. However, only 8 patients could complete 8 weeks of therapy, with significant adverse events. The small sample size limits our ability to draw meaningful conclusions and larger randomized trials are warranted to investigate this approach in sarcoidosis management.
Are there any valid serum biomarkers for sarcoidosis?
A biomarker is defined as a compound easily measurable in serum, urine, or other body fluids that can be used as indicator of presence and/or severity of particular disease state. Moreover, it helps in evaluation of effectiveness of drug therapy and useful to monitor the disease longitudinally. Unfortunately, there is no ideal serum biomarker in sarcoidosis. The most widely evaluated marker is serum angiotensin-converting enzyme (ACE). It has been found to be elevated in three quarters of patients with sarcoidosis [47]. However, it has poor diagnostic utility due to limited sensitivity and specificity. Furthermore, levels of serum ACE are reduced in patients taking ACE inhibitors and hence measurement of ACE levels in patients taking ACE inhibitors may lead to inaccurate interpretations [48].
A number of other serum biomarkers have been evaluated in sarcoidosis. Gungor and colleagues [49] evaluated 48 patients with sarcoidosis and 20 healthy controls. The biomarkers measured were ACE, adenosine deaminase (ADA), total IgE, serum amyloid-A (SAA), soluble interleukin-2 receptor (sIL2R), and C-reactive protein (CRP). This study showed that SAA was significantly elevated in sarcoidosis as compared to controls (P < 0.001). Furthermore, sIL2R levels were raised in extra-pulmonary sarcoidosis (P < 0.014). In another study, Grutters and co-workers [50] demonstrated elevated levels of sIL2R in 47 patients with active sarcoidosis. However, the levels did not correlate with radiolographic or physiological outcomes or response to treatment. Hence, the utility of these biomarkers in clinical practice is questionable and larger longitudinal studies are required to demonstrate the actual benefit of these biomarkers in everyday clinical practice.
There is a complex relationship between vitamin D and calcium metabolism and risk of osteoporosis in sarcoidosis. The value of active vitamin D metabolite 1, 25-dihydroxy vitamin D in relation to the degree of sarcoidosis disease chronicity was evaluated in a study of 59 patients with sarcoidosis [51]. It was noted that increased levels of 1,25 vitamin D were associated with increased risk of chronic phenotype and the need to require repeated treatments with immunosuppressive agents. Hence, serum levels of 1,25 vitamin D may have a prognostic value in sarcoidosis. It is important to note that both vitamin D deficiency as well as vitamin D excess can result in osteoporosis [52], and there is a risk of bone fragility and fractures in patients who may need long-term oral corticosteroid therapy. Hence, an optimal level of vitamin D is crucial; we recommend a value of serum 25(OH) vitamin D between 10 and 20 ng/mL as evidenced by a cross-sectional analysis of 142 consecutive patients with biopsy-proven sarcoidosis [53]. The above range of 25(OH) vitamin D was associated with higher bone mineral density and values above 20 ng/mL resulted in increased risk of fractures, demonstrating the need to keep vitamin D levels of these patients to be lower than those recommended for general population.
When should a patient with sarcoidosis be referred for lung transplantation?
Lung transplantation is a treatment option in advanced/end-stage disease after pharmacologic treatments have been exhausted and there is no evidence of progressive or severe extrapulmonary disease. The most critical decision regarding transplantation in pulmonary sarcoidosis is the timing of the referral and close liaison with the transplant center to ensure the maximal chance of success with lung transplantation. We recommend considering transplant referral when % predicted FVC approaches a value of 50% or less with or without significant pulmonary hypertension. Single lung transplant is appropriate for the majority of patients with sarcoidosis. However, bilateral transplant should be considered in bilateral mycetomas and bilateral bronchiectasis. Arcasoy and colleagues [54] analysed 43 patients listed for transplantation and found the following factors associated with increased risk of mortality:
- pulmonary hypertension
- hypoxia
- low cardiac output
- elevated right atrial pressure
It is important to note that pulmonary function parameters have not been found to be predictive of mortality in sarcoidosis [54,55]. Although granuloma recurrence in transplanted lung has been observed, it is a rare to have organ failure secondary to recurrence, and lung transplant should be considered in refractory cases of pulmonary sarcoidosis in the absence of contraindications and close liaison with the transplant center at the earliest opportunity is recommended.
What is the optimal duration of clinical follow-up in sarcoidosis?
There is no consensus on appropriate follow-up duration once a diagnosis of sarcoidosis is confirmed, and it is dependent on physician preference, vital organ involvement, disease progression, need for pharmacological treatment, and availability of resources. The American Thoracic Society statement on sarcoidosis suggested at least 3 years of follow-up after corticosteroid treatment is completed irrespective of radiographic stage [56]. Patients with serious extrapulmonary symptoms would require longer-term follow-up and a recent single-center Japanese study of corticosteroid-naive patients showed that the number of organs involved at the outset dictates the cumulative risk of subsequent progression of sarcoidosis [57]. This study of 150 patients with sarcoidosis with a median follow-up of 7.7 years demonstrated significantly increased risk of progression when there were > 3 organ systems involved as compared to ≤ 3 involved. These corticosteroid-naive patients may require longer follow-up (up to 10 years) than previously thought. However, the findings of this study would require confirmation in multi-center and multi-ethnic cohort of sarcoidosis patients.
Summary
Sarcoidosis presents as a fascinating and challenging disease with myriad clinical, radiological, and pathological manifestations. Despite extensive research in the last decade to increase our understanding of the mechanisms of disease evolution and progression, the underlying etiology remains unidentified. The treatment paradigm has changed over the last 10 years with the introduction of biological agents such as infliximab and adalimumab [42]; these anti-TNF drugs offer a treatment option in refractory cases of sarcoidosis with or without corticosteroids and other anti-metabolites. Methotrexate is the preferred first-line immunosuppressive agent after corticosteroids and has a safer adverse effect profile in comparison to azathioprine. Larger randomized studies to evaluate the efficacy of antimycobacterial agents and newer biologics are warranted before making strong recommendations for the use of these drugs, as there are significant potential toxic effects associated with their use.
Corresponding author: Dr. Ahmed Fahim, Dept. of Respiratory Medicine, McHale Centre New Cross Hospital, Wolverhampton, UK WV10 0QP, [email protected].
Financial disclosures: None.
1. Kitamura AY, Takiguchi K, Kurosu N, et al. Feasibility of cytological diagnosis of sarcoidosis with endobronchial US-guided transbronchial aspiration. Sarcoidosis Vasc Diffuse Lung Dis 2012;29:82–9.
2. Chee A, Khalil M, Stather DR, et al. Cytologic assessment of endobronchial ultrasound-guided transbronchial needle aspirates in sarcoidosis. J Bronchology Interv Pulmonol 2012;19:24–8.
3. Oki M, Saka H, Kitagawa C, et al. Prospective study of endobronchial ultrasound-guided transbronchial needle aspiration of lymph nodes versus transbronchial lung biopsy of lung tissue for diagnosis of sarcoidosis. J Thorac Cardiovasc Surg 2012;143:1324–9.
4. Plit ML, Havryk AP, Hodgson A, et al. Rapid cytological analysis of endobronchial ultrasound-guided aspirates in sarcoidosis. Eur Respir J 2013;42:1302-8.
5. von Bartheld MB, Dekkers OM, Szlubowski A, et al. Endosonography vs conventional bronchoscopy for the diagnosis of sarcoidosis: the GRANULOMA randomized clinical trial. JAMA 2013;309:2457–64.
6. Uemura A, Morimoto S, Hiramitsu S, et al. Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J 1999;138:299–302.
7. Ishimaru S, Tsujino I, Sakaue S, et al. Combination of 18F-fluoro-2-deoxyglucose positron emission tomography and magnetic resonance imaging in assessing cardiac sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2005;22:234–5.
8. Youssef G, Leung E, Mylonas I, et al. The use of 18F-FDG PET in the diagnosis of cardiac sarcoidosis: a systematic review and metaanalysis including the Ontario experience. J Nucl Med 2012;53:241–8.
9. Blankstein R, Osborne M, Naya M, et al. Cardiac positron emission tomography enhances prognostic assessments of patients with suspected cardiac sarcoidosis. J Am Coll Cardiol 2014;63:329–36.
10. Baughman RP, Nunes H, Sweiss NJ, Lower EE. Established and experimental medical therapy of pulmonary sarcoidosis. Eur Respir J 2013;41:1424–8.
11. James DG, Carstairs LS, Trowell J, Sharma OP. Treatment of sarcoidosis. Report of a controlled therapeutic trial. Lancet 1967;2:526–8.
12. Pietinalho A, Tukiainen P, Haahtela T, et al. Oral prednisolone followed by inhaled budesonide in newly diagnosed pulmonary sarcoidosis: a double-blind, placebo-controlled multicenter study. Finnish Pulmonary Sarcoidosis Study Group. Chest 1999;116:424–31.
13. Eule H, Roth I, Ehrke I, Weinecke W. Corticosteroid therapy of intrathoracic sarcoidosis stages I and II--results of a controlled clinical trial. Z Erkr Atmungsorgane 1977;149:142–7.
14. Israel HL, Fouts DW, Beggs RA. A controlled trial of prednisone treatment of sarcoidosis. Am Rev Respir Dis 1973;107:609–14.
15. Zaki MH, Lyons HA, Leilop L, Huang CT. Corticosteroid therapy in sarcoidosis. A five-year, controlled follow-up study. N Y State J Med 1987;87:496–9.
16. Gibson GJ, Prescott RJ, Muers MF, et al. British Thoracic Society Sarcoidosis study: effects of long term corticosteroid treatment. Thorax 1996;51:238–47.
17. Kamphuis LS, Bonte-Mineur F, van Laar JA, et al. Calcium and vitamin D in sarcoidosis: is supplementation safe? J Bone Miner Res 2014;29:2498–503.
18. Baughman RP, Janovcik J, Ray M, et al. Calcium and vitamin D metabolism in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:113–20.
19. Muller-Quernheim J, Kienast K, Held M, et al. Treatment of chronic sarcoidosis with an azathioprine/prednisolone regimen. Eur Respir J 1999;14:1117–22.
20. Baughman RP, Winget DB, Lower EE. Methotrexate is steroid sparing in acute sarcoidosis: results of a double blind, randomized trial. Sarcoidosis Vasc Diffuse Lung Dis 2000;17:60–6.
21. Vorselaars AD, Wuyts WA, Vorselaars VM, et al. Methotrexate versus azathioprine in second line therapy of sarcoidosis. Chest 2013.
22. Baughman RP, Lower EE. Leflunomide for chronic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2004;21:43–8.
23. Baughman RP, Ohmichi M, Lower EE. Combination therapy for sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2001;18:133–7.
24. Drent M, Lower EE, De Vries J. Sarcoidosis-associated fatigue. Eur Respir J 2012;40:255–63.
25. Fleischer M, Hinz A, Brahler E, et al. Factors associated with fatigue in sarcoidosis. Respir Care 2014;59:1086–94.
26. Lower EE, Harman S, Baughman RP. Double-blind, randomized trial of dexmethylphenidate hydrochloride for the treatment of sarcoidosis-associated fatigue. Chest 2008;133:1189–95.
27. Veien NK, Brodthagen H. Cutaneous sarcoidosis treated with methotrexate. Br J Dermatol 1977;97:213–6.
28. Lower EE, Baughman RP. Prolonged use of methotrexate for sarcoidosis. Arch Intern Med 1995;155:846–51.
29. Kaye O, Palazzo E, Grossin M, et al. Low-dose methotrexate: an effective corticosteroid-sparing agent in the musculoskeletal manifestations of sarcoidosis. Br J Rheumatol 1995;34:642–4.
30. Green CM. Topical tacrolimus for the treatment of cutaneous sarcoidosis. Clin Exp Dermatol 2007:32:457–8.
31. Vano-Galvan S, Fernandez-Guarino M, Carmona LP, et al. Lichenoid type of cutaneous sarcoidosis: great response to topical tacrolimus. Eur J Dermatol 2008;18:89–90.
32. Gutzmer R, Volker B, Kapp A, Werfel T. [Successful topical treatment of cutaneous sarcoidosis with tacrolimus]. Hautarzt 2003;54:1193–7.
33. Katoh N, Mihara H, Yasuno H. Cutaneous sarcoidosis successfully treated with topical tacrolimus. Br J Dermatol 2002;147:154–6.
34. Doty JD, Mazur JE, Judson MA. Treatment of sarcoidosis with infliximab. Chest 2005;127:1064–71.
35. Berrios I, Jun-O’Connell A, Ghiran S, Ionete C. A case of neurosarcoidosis secondary to treatment of etanercept and review of the literature. BMJ Case Rep 2015 Jul 6;2015.
36. Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med 2006;174:795–802.
37. Rossman MD, Newman LS, Baughman RP, et al. A double-blinded, randomized, placebo-controlled trial of infliximab in subjects with active pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2006;23:201–8.
38. Chapelon-Abric D, Saadoun D, Biard L. Long-term outcome of infliximab in severe chronic and refractory systemic sarcoidosis: a report of 16 cases. Clin Exp Rheumatol 2015;33:509–15.
39. Hoitsma E, Faber CG, Santen-Hoeufft M, et al. Improvement of small fiber neuropathy in a sarcoidosis patient after treatment with infliximab. Sarcoidosis Vasc Diffuse Lung Dis 2006;23:73–7.
40. Lee NS, Barber L, Kanchwala A, et al. Low levels of NF-kappaB/p65 mark anergic CD4+ T cells and correlate with disease severity in sarcoidosis. Clin Vaccine Immunol 2011;18:223–34.
41. Sweiss NJ, Lower EE, Mirsaeidi M, et al. Rituximab in the treatment of refractory pulmonary sarcoidosis. Eur Respir J 2014;43:1525–8.
42. Sweiss NJ, Noth I, Mirsaeidi M, et al. Efficacy Results of a 52-week trial of adalimumab in the treatment of refractory sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:46–54.
43. Baughman RP, Barney JB, O’Hare L, Lower EE. A retrospective pilot study examining the use of Acthar gel in sarcoidosis patients. Respir Med 2016;110:66–72.
44. Mangiapan G, Hance AJ. Mycobacteria and sarcoidosis: an overview and summary of recent molecular biological data. Sarcoidosis 1995;12:20-37.
45. Drake WP, Oswald-Richter K, Richmond BW, et al. Oral antimycobacterial therapy in chronic cutaneous sarcoidosis: a randomized, single-masked, placebo-controlled study. JAMA Dermatol 2013;149:1040–9.
46. Drake WP, Richmond BW, Oswald-Richter K, et al. Effects of broad-spectrum antimycobacterial therapy on chronic pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:201–11.
47. Studdy PR, Bird R. Serum angiotensin converting enzyme in sarcoidosis--its value in present clinical practice. Ann Clin Biochem 1989;26 ( Pt 1):13–8.
48. Krasowski MD, Savage J, Ehlers A, et al. Ordering of the serum angiotensin-converting enzyme test in patients receiving angiotensin-converting enzyme inhibitor therapy: an avoidable but common error. Chest 2015;148:1447–53.
49. Gungor S, Ozseker F, Yalcinsoy M. Conventional markers in determination of activity of sarcoidosis. Int Immunopharmacol 2015;25:174–9.
50. Grutters JC, Fellrath JM, Mulder L, et al. Serum soluble interleukin-2 receptor measurement in patients with sarcoidosis: a clinical evaluation. Chest 2003;124:186–95.
51. Kavathia D, Buckley JD, Rao D, et al. Elevated 1, 25-dihydroxyvitamin D levels are associated with protracted treatment in sarcoidosis. Respir Med 2010;104:564–70.
52. Baughman RP, Lower EE. Goldilocks, vitamin D and sarcoidosis. Arthritis Res Ther 2014;16:111.
53. Saidenberg-Kermanac’h N, Semerano L, Nunes H, et al. Bone fragility in sarcoidosis and relationships with calcium metabolism disorders: a cross sectional study on 142 patients. Arthritis Res Ther 2014;16:R78.
54. Arcasoy SM, Christie JD, Pochettino A, et al. Characteristics and outcomes of patients with sarcoidosis listed for lung transplantation. Chest 2001;120:873–80.
55. Shorr AF, Davies DB, Nathan SD. Predicting mortality in patients with sarcoidosis awaiting lung transplantation. Chest 2003;124:922–8.
56. Hunninghake GW, Costabel U, Ando M, et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis 1999;16:149–73.
57. Inoue Y, Inui N, Hashimoto D, et al. Cumulative incidence and predictors of progression in corticosteroid-naive patients with sarcoidosis. PLoS One 2015;10:e0143371.
From the New Cross Hospital, Wolverhampton, UK.
Abstract
- Objective: To discuss the management of sarcoidosis.
- Methods: Review of the literature.
- Results: Sarcoidosis is a challenging multisystem disorder of uncertain etiology characterized by granulomatous inflammation in the affected organs. Treatment is dependent on the severity of disease and organ involvement at the time of diagnosis. Glucocorticoids have traditionally been considered first-line pharmacologic treatment; however, a significant proportion of patients do not require drug treatment due to the propensity toward spontaneous disease remission. Treated patients who fail to respond to corticosteroids or develop significant adverse effects can be offered a second-line agent, eg, methotrexate. Anti-TNF therapy may be considered as a treatment option in carefully selected patients with refractory disease after discussion of potential adverse effects followed by close monitoring at a specialist center.
- Conclusion: Further research into therapeutic options is likely to unveil novel agents with different mechanisms of action and better safety profiles than those seen with currently available immunosuppressive regimens.
Sarcoidosis is a multisystem disorder of uncertain etiology characterized by granulomatous inflammation in the affected organs. The diagnosis of sarcoidosis is best supported by histological evidence of noncaseating granuloma formation. It is a disease with a generally good prognosis; less than 5% patients die from the disease, with cause of death usually secondary to respiratory failure or cardiac or neurologic involvement. This review aims to discuss the management of sarcoidosis with a special emphasis on the management challenges resulting from the myriad clinical manifestations and potential complications seen in this chronic multisystem disease.
Case Study
Initial Presentation
A 40-year-old African-American man presents to his primary care physician with symptoms of fatigue, dry cough, exertional breathlessness, dry and painful eyes, generalized arthralgia, and multiple skin lesions for 3 months. He has a history of essential hypertension and is a former smoker with 10 pack-year history. He is not on any regular medications. Examination reveals bilateral cervical lymphadenopathy and multiple skin lesions on trunk. The rest of the systemic examination (including respiratory and cardiovascular system) is normal.
Workup
The patient’s initial bloodwork showed a mild degree of lymphopenia (1.1 × 109/L, normal range 1.5–4.5). Other bloodwork results, bone profile and immunology screen (including ANA, rheumatoid factor, immunoglobulins, and extractable nuclear antigen antibodies) were negative. Angiotensin-converting enzyme (ACE) was elevated at 149 U/L (normal range 5–58), while serum calcium and vitamin D levels (including vitamin D3) were normal. The findings on CT scan along with the biochemical profile suggest a plausible diagnosis of sarcoidosis.
What is the next step in the workup to establish the suspected diagnosis?
The radiological findings of hilar lymphadenopathy are not confirmatory. There are a number of entities in the differential diagnoses, including tuberculosis, malignancy, lymphoma, and other granulomatous disorders such as histoplasmosis, schistosomiasis, and blastomycosis. It is important to obtain histological evidence before a definitive diagnosis of sarcoidosis can be made. This is necessary as management differs for each of the diagnostic categories mentioned above. Furthermore, diagnostic confirmation would be helpful later in the disease course if the patient develops any associated complications such as pulmonary hypertension or respiratory failure and/or need for lung transplant assessment.
As this patient had palpable cervical lymphadenopathy, an ultrasound guided biopsy of the lymph node was obtained. The histological examination demonstrated evidence of noncaseating granulomas that were well formed and highly consistent with the suspected clinical and radiological diagnosis of sarcoidosis. In addition, the special stains for acid-fast bacilli and other infections including fungi were negative.
Our practice is to evaluate patients with suspected sarcoidosis with neck ultrasound and tru-cut biopsy of cervical lymph nodes (if appropriate) as a first-line investigation, as it is less invasive than bronchoscopy/endoscopic ultrasound–guided transbronchial needle aspiration (EBUS-TBNA) or thoracoscopic lung biopsy. The diagnostic yield of EBUS-TBNA has been evaluated in a number of studies with variable results [1–5]. A large multicenter randomized clinical trial [5] of 304 patients investigated the diagnostic yield of endosonography (endobronchial and esophageal ultrasound) in comparison with bronchoscopy with transbronchial biopsy (TBB) and endobronchial biopsy. The study cohort was made up of patients with stage I/II sarcoidosis. The results showed that endosonography had a higher diagnostic yield to detect granulomas (80% vs 53%; P < 0.001) and there were no serious adverse events related to endoscopy. Hence, ultrasound-guided endoscopic procedures are becoming common first-line investigations for sarcoidosis in the absence of other readily identifiable biopsy sites such as peripheral lymph nodes in the cervical area.
What should be done about the cutaneous and ophthalmologic symptoms in this patient?
As sarcoidosis commonly involves eyes and skin (after pulmonary involvement, which is seen in 90% of cases), the patient was referred to ophthalmology and dermatology departments for further evaluation. These assessments confirmed him to have bilateral uveitis and skin involvement with granulomatous inflammation consistent with ocular and cutaneous sarcoidosis respectively. Hence, the diagnosis of multisystem sarcoidosis was made. At this stage, the patient also mentioned symptoms of intermittent palpitations for 3 months’ duration and feeling of missing a beat, so an urgent cardiological evaluation was undertaken that showed him to have ectopic beats on Holter monitoring. However, his trans-thoracic echocardiogram and a cardiac MRI scan were normal with good biventricular function, excluding cardiac sarcoidosis as a cause of his palpitations. Cardiac involvement with sarcoidosis is clinically apparent in only 5% of cases and presents as cardiomyopathy and or cardiac arrhythmias (both tachy and bradyarrythmias). As cardiac involvement with sarcoid granulomas is usually patchy, endomyocardial biopsy has a limited diagnostic yield of < 20% [6]. In this particular case, endomyocardial biopsy was not attempted in view of normal cardiac MR and echocardiography as well as no significant cardiac dysrythmia on holter monitoring.
Case Continued
The patient was started on corticosteroid eye drops and steroid ointment for his ophthalmologic and cutaneous sarcoidosis, respectively, and symptoms gradually improved over the next few months. As the patient was symptomatic with cough and breathlessness and there was evidence of reduction in FVC (along with reduced DLco), a trial of oral corticosteroids was considered to treat the pulmonary sarcoidosis.
What is first-line pharmacological treatment for sarcoidosis and when is it indicated?
Treatment of sarcoidosis is dependent upon the severity of disease and organ involvement at the time of the diagnosis. Glucocorticoids have traditionally been considered first-line pharmacological agents in selective cases, as a significant proportion of patients do not require drug treatment due to the propensity for spontaneous remission. Furthermore, sarcoidosis remains stable without anti-inflammatory/immunosuppressive therapies in a majority of patients. A number of clinical trials have evaluated the value of corticosteroids in the management of sarcoidosis, with variable outcomes [10–16]. The disease tends to be severe in patients of African descent compared with patients from other racial backgrounds. The European cohort of sarcoidosis patients generally have milder disease with less propensity for vital organ involvement such as cardiac or central nervous system (CNS) disease.
The decision to initiate corticosteroids for sarcoidosis is not a straightforward one as there is variability in symptom presentation, disease severity, and response to corticosteroids. We initiate first-line therapy with oral prednisolone in the following circumstances:
- Evidence of pulmonary impairment (forced vital capacity FVC < 80% predicted) with or without reduction in gas transfer (DLco) along with respiratory symptoms of cough, chest pain, and/or breathlessness (as seen in the case patient)
- Vital organ involvement such as cardiac, ophthalmic (such as panuveitis) or CNS sarcoidosis once confirmed by respective investigations
- Selective cases of sarcoid-associated pulmonary hypertension (SAPH) along with close liaison with pulmonary hypertension specialists
We recommend an initial starting dose of 20 to 40 mg of prednisolone for a period of 1 to 3 months, followed by maintenance dose of 10 mg or less for a further 6 to 9 months, aiming for a total duration of treatment of 12 months. However, the duration may vary depending on the response and any associated adverse effects with corticosteroids. It is usual practice to supplement with calcium and vitamin D when beginning patients on oral corticosteroids due to the potential risk of osteoporosis. However, this may result in significant hypercalcemia, which itself may be an endocrine manifestation of sarcoidosis. Hence, we recommend monitoring serum calcium during treatment and supplement vitamin D in patients who are vitamin D–deficient [17]. Furthermore, serum vitamin 1,25(OH)2 vitamin D3 has been demonstrated to the best available test to evaluate vitamin D status in sarcoidosis [18].
Case Continued
What are the preferred pharmacological agents for second-line treatment in sarcoidosis?
Alternative immunosuppression should be considered in the following circumstances in patients diagnosed with sarcoidosis:
- Failure or less than optimal response to oral corticosteroids
- Use as a steroid-sparing agent in patients requiring high doses of steroids for symptomatic control
- Failure to tolerate corticosteroids due to significant adverse effects such as excessive weight gain, steroid-induced psychosis, osteoporosis, and worsening diabetic control
A small trial of 11 patients examined azathioprine as a steroid-sparing agent and found it as an acceptable immunosuppressive agent for that purpose [19]. It was associated with good safety profile and adherence to treatment was 82% (9 out of 11 patients). However, the small sample size makes it difficult to draw firm conclusions from the findings of this study. In another trial evaluating methotrexate as a steroid-sparing agent in the first year after the diagnosis of sarcoidosis, Baughman and colleagues [20] reported that methotrexate is an attractive alternative to other immunosuppressive agents in term of a steroid sparer. In this double-blind randomized controlled trial (RCT), 15 patients were studied with at least 6 months of treatment with methotrexate vs placebo. There was a significantly reduced dosage of prednisolone observed in methotrexate group. However, the difference was not significant when data were analyzed for all patients, including the dropouts. More recently, a large international retrospective cohort study of 200 patients with sarcoidosis demonstrated that both methotrexate and azathioprine have similar efficacy and steroid-sparing capacity in sarcoidosis [21]. However, infection rates were significantly higher in the azathioprine group as compared to methotrexate (34.6% vs. 18.1%, P = 0.01). Hence methotrexate should be considered as a preferred second-line agent in sarcoidosis after detailed discussion about potential side effects.
Case Continued
The patient was started on methotrexate after discussion about the potential adverse effects of bone marrow suppression, hepatotoxicity, and pneumonitis. He was screened for latent tuberculosis and viral hepatitis prior to starting methotrexate. The dosage was 7.5 mg per week along with folic acid once a week. We gradually increase the dose in increments of 2.5 mg every 2 weeks with a view to reach 15 mg every week as maintenance therapy. In severely obese patients, a dose of up to 20 mg weekly is occasionally considered if 15 mg is suboptimal after careful clinical assessment.
The patient failed to make significant progress after being on methotrexate for a period of 6 months and lung function tests continued to demonstrate a persistent decline with symptomatic worsening of dyspnea and cough.
What are treatment options in refractory sarcoidosis?
Options to consider in the setting of refractory sarcoidosis are leflunomide, hydroxychloroquine, or combination therapy of methotrexate and leflunomide. Leflunomide has been shown to be of similar efficacy to methotrexate as demonstrated by a retrospective analysis of 32 patients treated with the drug in a tertiary care center [22]. Complete or partial response was noted in 12 of 17 patients treated solely with leflunomide and 13 of 15 treated in conjunction with methotrexate. Hence, combination therapy has been suggested as a viable option for these patients who fail to respond to initial glucocorticoid agents and alternative immunosuppressive drugs, as combination therapy may enhance efficacy with reduced toxicity if considered in a rational manner after careful selection of patients [23].
How should the symptom of fatigue be addressed?
The patient had ongoing fatigue during the treatment period with corticosteroids and alternative immunosuppressants. Fatigue, noted in a majority of patients with sarcoidosis [24], is one of the commonest symptoms of sarcoidosis and one of the most difficult to treat. We recommend excluding alternative etiologies of fatigue when confronted with this symptom and evaluating for associated comorbidities such as thyroid dysfunction, vitamin D deficiency, and hypoadrenalism. Extra-pulmonary sarcoidosis seems to be associated with fatigue in comparison to sarcoidosis restricted to the pulmonary system [25]. Furthermore, there is a paucity of good quality data on the benefit of pharmacological intervention for treatment of fatigue. A small double-blind randomized study of 10 patients demonstrated a positive impact of treatment with dexmethylphenidate hydrochloride (d-MPH) for a period of 8 weeks [26]. However, the small sample and lack of long-term outcomes data make it difficult to draw firm conclusions based on the findings of this study and larger randomized trials are warranted to investigate this important aspect of sarcoidosis before recommending a pharmacological agent routinely for this disabling symptom.
What are the pharmacological agents for cutaneous sarcoidosis?
Corticosteroids (local and or systemic) are the mainstay of treatment in cutaneous sarcoidosis. However, patients who fail to respond to these agents or develop significant adverse effects should be offered second-line agents in the form of hydroxychloroquine/chloroquine, methotrexate, or leflunomide. It is important to acknowledge that the evidence of benefit for these agents is derived from uncontrolled studies [27–29]. Anti-malarial agents are usually well tolerated; patients do require a baseline ophthalmological assessment and subsequent periodic examinations to monitor for any ocular toxicity associated with their use. Leflunomide is also a second-line option in cutaneous disease and is associated with lesser toxicity than methotrexate [22]. More recently, topical tacrolimus has shown promising results in isolated case reports [30–33]. Hence, it may be considered a treatment option in refractory cutaneous sarcoidosis.
Is there a role for anti-TNF therapy in the management of sarcoidosis? Should it be considered for the case patient?
Tumour necrosis factor-α (TNF-α) is implicated in the pathogenesis of sarcoidosis and is believed to have a significant role in the inflammatory processes in sarcoidosis. It is released from alveolar macrophages of patients with active disease and is involved in formation and maintenance of granulomas in the lung tissue. There has been significant progress in therapeutic options alternative to traditional immunosuppressants such as corticosteroids and TNF-α inhibition has been on the horizon as a treatment strategy for few years. It may be considered as a steroid-sparing agent or a third-line treatment option in refractory cases.
The initial evidence of benefit of anti-TNF therapy comes from case reports and small case series [34,35]. However, there have been 2 RCTs published so far invest-igating infliximab in sarcoidosis [36,37]. Baughman and colleagues [36] evaluated 138 patients with chronic pulmonary sarcoidosis in a double-blind study. There was a statistically significant improvement in FVC after 24 weeks of therapy with infliximab as compared to placebo (2.5% increase in mean FVC % predicted from baseline, P = 0.038). However, there was no improvement in any of the secondary outcome variables including SGRQ (St George’s Respiratory Questionnaire), 6-minute walk distance, and dyspnea scores. The benefit seemed to be more pronounced in the severe disease category on post-hoc analysis. The clinical significance of these findings are however unclear and it is difficult to draw firm conclusions based on the findings of this study alone.
The other RCT exploring the role of infliximab in sarcoidosis was conducted by Rossman et al [37]. This multi-center phase II study was conducted to evaluate the safety and tolerability of the drug in active pulmonary sarcoidosis (stage II to IV). The trial had a small number of participants with only 19 patients and failed to show a significant improvement in lung function after 6 weeks of treatment. Furthermore, 4 patients developed serious adverse events after treatment. Doty and colleagues [34] retrospectively analysed 10 patients treated with infliximab for refractory sarcoidosis and found subjective and objective evidence of improvement in all patients. This therapy resulted in reduction of corticosteroid dosage in 83% of cases (5 out of 6). However, adverse reactions were noted in 3 cases, including development of angioimmunoblastic lymphoma in one case. A retrospective study of 16 consecutive unselected cases of refractory sarcoidosis by Chapelon-Abric and colleagues [38] demonstrated a positive response in a majority of cases. However 38% of patients experienced a relapse. Furthermore, 44% of patients (7 out of 16) had infectious complications associated with anti-TNF therapy. Finally, infliximab may be used successfully in treating severe small fibre neuropathy [39] and should be considered in refractory cases where neuropathy is associated with autonomic dysfunction.
In summary, based on the findings of the aforementioned trials and case series, anti-TNF therapy may be considered as a treatment option in carefully selected patients after discussion of the potential adverse effects, followed by close monitoring at a specialist center for the management of sarcoidosis. Furthermore, anti-TNF therapy should not delay referral for lung transplant assessment, particularly when disease progression is relatively rapid. Duration of treatment and timing of thoracic imaging after anti-TNF therapy is subject to debate and should be individualized in close collaboration with a specialist center.
Case Continued
What is the role of newer biologics in the treatment of sarcoidosis? And what other therapies are on the horizon?
Although sarcoidosis is a T cell–mediated disease, humoral immunity has been implicated in sarcoidosis [40] and B cell depletion by rituximab (anti-CD 20+chimeric monoclonal antibody) has been successfully utilized in T cell–mediated diseases such as rheumatoid arthritis. Rituximab has been studied in phase I/II trial [41] in patients with refractory pulmonary sarcoidosis. The response to rituximab was inconsistent in these patients, with only a small group demonstrating > 5% improvement in FVC or walking distance. Hence, further studies are required to demonstrate a significant clinical benefit of this treatment in refractory sarcoidosis and potentially identify the characteristics of patients that may respond to B-cell depletion.
Adalimumab is another biologic agent that may be a potential option in refractory sarcoidosis as demonstrated in an open-label study of 11 patients [42]. This small trial of 52 weeks’ duration showed that adalimumab is well tolerated and may be considered in refractory pulmonary sarcoidosis when other treatment options have been exhausted.
Acthar had been used to treat pulmonary sarcoidosis in 1950s and there has been recent interest in evaluating the value of acthar gel therapy in the management of sarcoidosis. Baughman and colleagues [43] carried out a retrospective analysis of 47 patients with advanced sarcoidosis treated with acthar gel. The results showed that there was an objective improvement in approximately a third of patients receiving at least 3 months of treatment. Thirty-six percent of patients (n = 17) managed to reduce their oral corticosteroid dosage by more than 50% while on this therapy. However, a significant proportion of patients were unable to take ≥ 3 months of treatment, suggesting poor tolerance/adherence. The utility of acthar gel therapy would need to be examined in larger prospective randomized trial before it can be recommended for a treatment option in advanced disease.
Should pneumocystis pneumonia (PCP) prophylaxis be considered in sarcoidosis?
We do not routinely consider PCP prophylaxis in all sarcoidosis patients. However, it should be considered in the following clinical situations:
- Failure to reduce oral corticosteroid dose to less than 20 mg of prednisolone daily
- Concomitant use of anti-TNF agents with relatively high maintenance dose of oral corticosteroids (≥ 20 mg per day)
- Significant comorbid condition in association with sarcoidosis resulting in significant level of immunosuppression
As our patient did not fall in any of the above categories, we did not offer him PCP prophylaxis. However, clinicians treating patients with sarcoidosis with strong immunosuppressants should be aware of this potential complication and be vigilant about discussing prophylaxis with trimethoprim-sulphamethoxazole, which is the first-line agent for this purpose.
Is there a role of anti-tuberculous treatment in sarcoidosis?
Both mycobacterium tuberculosis (MTB) and non-mycobateria (NTM) have been implicated in sarcoidosis [44] and it is challenging to differentiate tuberculosis (TB) from sarcoidosis in certain clinical situations, such as when dealing with patients from countries with a high incidence of TB. The association of mycobacterium with sarcoidosis was explored in 2 recent trials of concomitant use of levofloxacin, ethambutol, azithromycin, and rifampin in cutaneous [45] and pulmonary sarcoidosis [46]. Drake and colleagues evaluated 15 chronic pulmonary sarcoidosis patients in an open-label trial to investigate if the combination of these drugs was associated with improvement in pulmonary sarcoidosis. The patients who completed 8 weeks of treatment had improvement in FVC at both 4 and 8 weeks of treatment. However, only 8 patients could complete 8 weeks of therapy, with significant adverse events. The small sample size limits our ability to draw meaningful conclusions and larger randomized trials are warranted to investigate this approach in sarcoidosis management.
Are there any valid serum biomarkers for sarcoidosis?
A biomarker is defined as a compound easily measurable in serum, urine, or other body fluids that can be used as indicator of presence and/or severity of particular disease state. Moreover, it helps in evaluation of effectiveness of drug therapy and useful to monitor the disease longitudinally. Unfortunately, there is no ideal serum biomarker in sarcoidosis. The most widely evaluated marker is serum angiotensin-converting enzyme (ACE). It has been found to be elevated in three quarters of patients with sarcoidosis [47]. However, it has poor diagnostic utility due to limited sensitivity and specificity. Furthermore, levels of serum ACE are reduced in patients taking ACE inhibitors and hence measurement of ACE levels in patients taking ACE inhibitors may lead to inaccurate interpretations [48].
A number of other serum biomarkers have been evaluated in sarcoidosis. Gungor and colleagues [49] evaluated 48 patients with sarcoidosis and 20 healthy controls. The biomarkers measured were ACE, adenosine deaminase (ADA), total IgE, serum amyloid-A (SAA), soluble interleukin-2 receptor (sIL2R), and C-reactive protein (CRP). This study showed that SAA was significantly elevated in sarcoidosis as compared to controls (P < 0.001). Furthermore, sIL2R levels were raised in extra-pulmonary sarcoidosis (P < 0.014). In another study, Grutters and co-workers [50] demonstrated elevated levels of sIL2R in 47 patients with active sarcoidosis. However, the levels did not correlate with radiolographic or physiological outcomes or response to treatment. Hence, the utility of these biomarkers in clinical practice is questionable and larger longitudinal studies are required to demonstrate the actual benefit of these biomarkers in everyday clinical practice.
There is a complex relationship between vitamin D and calcium metabolism and risk of osteoporosis in sarcoidosis. The value of active vitamin D metabolite 1, 25-dihydroxy vitamin D in relation to the degree of sarcoidosis disease chronicity was evaluated in a study of 59 patients with sarcoidosis [51]. It was noted that increased levels of 1,25 vitamin D were associated with increased risk of chronic phenotype and the need to require repeated treatments with immunosuppressive agents. Hence, serum levels of 1,25 vitamin D may have a prognostic value in sarcoidosis. It is important to note that both vitamin D deficiency as well as vitamin D excess can result in osteoporosis [52], and there is a risk of bone fragility and fractures in patients who may need long-term oral corticosteroid therapy. Hence, an optimal level of vitamin D is crucial; we recommend a value of serum 25(OH) vitamin D between 10 and 20 ng/mL as evidenced by a cross-sectional analysis of 142 consecutive patients with biopsy-proven sarcoidosis [53]. The above range of 25(OH) vitamin D was associated with higher bone mineral density and values above 20 ng/mL resulted in increased risk of fractures, demonstrating the need to keep vitamin D levels of these patients to be lower than those recommended for general population.
When should a patient with sarcoidosis be referred for lung transplantation?
Lung transplantation is a treatment option in advanced/end-stage disease after pharmacologic treatments have been exhausted and there is no evidence of progressive or severe extrapulmonary disease. The most critical decision regarding transplantation in pulmonary sarcoidosis is the timing of the referral and close liaison with the transplant center to ensure the maximal chance of success with lung transplantation. We recommend considering transplant referral when % predicted FVC approaches a value of 50% or less with or without significant pulmonary hypertension. Single lung transplant is appropriate for the majority of patients with sarcoidosis. However, bilateral transplant should be considered in bilateral mycetomas and bilateral bronchiectasis. Arcasoy and colleagues [54] analysed 43 patients listed for transplantation and found the following factors associated with increased risk of mortality:
- pulmonary hypertension
- hypoxia
- low cardiac output
- elevated right atrial pressure
It is important to note that pulmonary function parameters have not been found to be predictive of mortality in sarcoidosis [54,55]. Although granuloma recurrence in transplanted lung has been observed, it is a rare to have organ failure secondary to recurrence, and lung transplant should be considered in refractory cases of pulmonary sarcoidosis in the absence of contraindications and close liaison with the transplant center at the earliest opportunity is recommended.
What is the optimal duration of clinical follow-up in sarcoidosis?
There is no consensus on appropriate follow-up duration once a diagnosis of sarcoidosis is confirmed, and it is dependent on physician preference, vital organ involvement, disease progression, need for pharmacological treatment, and availability of resources. The American Thoracic Society statement on sarcoidosis suggested at least 3 years of follow-up after corticosteroid treatment is completed irrespective of radiographic stage [56]. Patients with serious extrapulmonary symptoms would require longer-term follow-up and a recent single-center Japanese study of corticosteroid-naive patients showed that the number of organs involved at the outset dictates the cumulative risk of subsequent progression of sarcoidosis [57]. This study of 150 patients with sarcoidosis with a median follow-up of 7.7 years demonstrated significantly increased risk of progression when there were > 3 organ systems involved as compared to ≤ 3 involved. These corticosteroid-naive patients may require longer follow-up (up to 10 years) than previously thought. However, the findings of this study would require confirmation in multi-center and multi-ethnic cohort of sarcoidosis patients.
Summary
Sarcoidosis presents as a fascinating and challenging disease with myriad clinical, radiological, and pathological manifestations. Despite extensive research in the last decade to increase our understanding of the mechanisms of disease evolution and progression, the underlying etiology remains unidentified. The treatment paradigm has changed over the last 10 years with the introduction of biological agents such as infliximab and adalimumab [42]; these anti-TNF drugs offer a treatment option in refractory cases of sarcoidosis with or without corticosteroids and other anti-metabolites. Methotrexate is the preferred first-line immunosuppressive agent after corticosteroids and has a safer adverse effect profile in comparison to azathioprine. Larger randomized studies to evaluate the efficacy of antimycobacterial agents and newer biologics are warranted before making strong recommendations for the use of these drugs, as there are significant potential toxic effects associated with their use.
Corresponding author: Dr. Ahmed Fahim, Dept. of Respiratory Medicine, McHale Centre New Cross Hospital, Wolverhampton, UK WV10 0QP, [email protected].
Financial disclosures: None.
From the New Cross Hospital, Wolverhampton, UK.
Abstract
- Objective: To discuss the management of sarcoidosis.
- Methods: Review of the literature.
- Results: Sarcoidosis is a challenging multisystem disorder of uncertain etiology characterized by granulomatous inflammation in the affected organs. Treatment is dependent on the severity of disease and organ involvement at the time of diagnosis. Glucocorticoids have traditionally been considered first-line pharmacologic treatment; however, a significant proportion of patients do not require drug treatment due to the propensity toward spontaneous disease remission. Treated patients who fail to respond to corticosteroids or develop significant adverse effects can be offered a second-line agent, eg, methotrexate. Anti-TNF therapy may be considered as a treatment option in carefully selected patients with refractory disease after discussion of potential adverse effects followed by close monitoring at a specialist center.
- Conclusion: Further research into therapeutic options is likely to unveil novel agents with different mechanisms of action and better safety profiles than those seen with currently available immunosuppressive regimens.
Sarcoidosis is a multisystem disorder of uncertain etiology characterized by granulomatous inflammation in the affected organs. The diagnosis of sarcoidosis is best supported by histological evidence of noncaseating granuloma formation. It is a disease with a generally good prognosis; less than 5% patients die from the disease, with cause of death usually secondary to respiratory failure or cardiac or neurologic involvement. This review aims to discuss the management of sarcoidosis with a special emphasis on the management challenges resulting from the myriad clinical manifestations and potential complications seen in this chronic multisystem disease.
Case Study
Initial Presentation
A 40-year-old African-American man presents to his primary care physician with symptoms of fatigue, dry cough, exertional breathlessness, dry and painful eyes, generalized arthralgia, and multiple skin lesions for 3 months. He has a history of essential hypertension and is a former smoker with 10 pack-year history. He is not on any regular medications. Examination reveals bilateral cervical lymphadenopathy and multiple skin lesions on trunk. The rest of the systemic examination (including respiratory and cardiovascular system) is normal.
Workup
The patient’s initial bloodwork showed a mild degree of lymphopenia (1.1 × 109/L, normal range 1.5–4.5). Other bloodwork results, bone profile and immunology screen (including ANA, rheumatoid factor, immunoglobulins, and extractable nuclear antigen antibodies) were negative. Angiotensin-converting enzyme (ACE) was elevated at 149 U/L (normal range 5–58), while serum calcium and vitamin D levels (including vitamin D3) were normal. The findings on CT scan along with the biochemical profile suggest a plausible diagnosis of sarcoidosis.
What is the next step in the workup to establish the suspected diagnosis?
The radiological findings of hilar lymphadenopathy are not confirmatory. There are a number of entities in the differential diagnoses, including tuberculosis, malignancy, lymphoma, and other granulomatous disorders such as histoplasmosis, schistosomiasis, and blastomycosis. It is important to obtain histological evidence before a definitive diagnosis of sarcoidosis can be made. This is necessary as management differs for each of the diagnostic categories mentioned above. Furthermore, diagnostic confirmation would be helpful later in the disease course if the patient develops any associated complications such as pulmonary hypertension or respiratory failure and/or need for lung transplant assessment.
As this patient had palpable cervical lymphadenopathy, an ultrasound guided biopsy of the lymph node was obtained. The histological examination demonstrated evidence of noncaseating granulomas that were well formed and highly consistent with the suspected clinical and radiological diagnosis of sarcoidosis. In addition, the special stains for acid-fast bacilli and other infections including fungi were negative.
Our practice is to evaluate patients with suspected sarcoidosis with neck ultrasound and tru-cut biopsy of cervical lymph nodes (if appropriate) as a first-line investigation, as it is less invasive than bronchoscopy/endoscopic ultrasound–guided transbronchial needle aspiration (EBUS-TBNA) or thoracoscopic lung biopsy. The diagnostic yield of EBUS-TBNA has been evaluated in a number of studies with variable results [1–5]. A large multicenter randomized clinical trial [5] of 304 patients investigated the diagnostic yield of endosonography (endobronchial and esophageal ultrasound) in comparison with bronchoscopy with transbronchial biopsy (TBB) and endobronchial biopsy. The study cohort was made up of patients with stage I/II sarcoidosis. The results showed that endosonography had a higher diagnostic yield to detect granulomas (80% vs 53%; P < 0.001) and there were no serious adverse events related to endoscopy. Hence, ultrasound-guided endoscopic procedures are becoming common first-line investigations for sarcoidosis in the absence of other readily identifiable biopsy sites such as peripheral lymph nodes in the cervical area.
What should be done about the cutaneous and ophthalmologic symptoms in this patient?
As sarcoidosis commonly involves eyes and skin (after pulmonary involvement, which is seen in 90% of cases), the patient was referred to ophthalmology and dermatology departments for further evaluation. These assessments confirmed him to have bilateral uveitis and skin involvement with granulomatous inflammation consistent with ocular and cutaneous sarcoidosis respectively. Hence, the diagnosis of multisystem sarcoidosis was made. At this stage, the patient also mentioned symptoms of intermittent palpitations for 3 months’ duration and feeling of missing a beat, so an urgent cardiological evaluation was undertaken that showed him to have ectopic beats on Holter monitoring. However, his trans-thoracic echocardiogram and a cardiac MRI scan were normal with good biventricular function, excluding cardiac sarcoidosis as a cause of his palpitations. Cardiac involvement with sarcoidosis is clinically apparent in only 5% of cases and presents as cardiomyopathy and or cardiac arrhythmias (both tachy and bradyarrythmias). As cardiac involvement with sarcoid granulomas is usually patchy, endomyocardial biopsy has a limited diagnostic yield of < 20% [6]. In this particular case, endomyocardial biopsy was not attempted in view of normal cardiac MR and echocardiography as well as no significant cardiac dysrythmia on holter monitoring.
Case Continued
The patient was started on corticosteroid eye drops and steroid ointment for his ophthalmologic and cutaneous sarcoidosis, respectively, and symptoms gradually improved over the next few months. As the patient was symptomatic with cough and breathlessness and there was evidence of reduction in FVC (along with reduced DLco), a trial of oral corticosteroids was considered to treat the pulmonary sarcoidosis.
What is first-line pharmacological treatment for sarcoidosis and when is it indicated?
Treatment of sarcoidosis is dependent upon the severity of disease and organ involvement at the time of the diagnosis. Glucocorticoids have traditionally been considered first-line pharmacological agents in selective cases, as a significant proportion of patients do not require drug treatment due to the propensity for spontaneous remission. Furthermore, sarcoidosis remains stable without anti-inflammatory/immunosuppressive therapies in a majority of patients. A number of clinical trials have evaluated the value of corticosteroids in the management of sarcoidosis, with variable outcomes [10–16]. The disease tends to be severe in patients of African descent compared with patients from other racial backgrounds. The European cohort of sarcoidosis patients generally have milder disease with less propensity for vital organ involvement such as cardiac or central nervous system (CNS) disease.
The decision to initiate corticosteroids for sarcoidosis is not a straightforward one as there is variability in symptom presentation, disease severity, and response to corticosteroids. We initiate first-line therapy with oral prednisolone in the following circumstances:
- Evidence of pulmonary impairment (forced vital capacity FVC < 80% predicted) with or without reduction in gas transfer (DLco) along with respiratory symptoms of cough, chest pain, and/or breathlessness (as seen in the case patient)
- Vital organ involvement such as cardiac, ophthalmic (such as panuveitis) or CNS sarcoidosis once confirmed by respective investigations
- Selective cases of sarcoid-associated pulmonary hypertension (SAPH) along with close liaison with pulmonary hypertension specialists
We recommend an initial starting dose of 20 to 40 mg of prednisolone for a period of 1 to 3 months, followed by maintenance dose of 10 mg or less for a further 6 to 9 months, aiming for a total duration of treatment of 12 months. However, the duration may vary depending on the response and any associated adverse effects with corticosteroids. It is usual practice to supplement with calcium and vitamin D when beginning patients on oral corticosteroids due to the potential risk of osteoporosis. However, this may result in significant hypercalcemia, which itself may be an endocrine manifestation of sarcoidosis. Hence, we recommend monitoring serum calcium during treatment and supplement vitamin D in patients who are vitamin D–deficient [17]. Furthermore, serum vitamin 1,25(OH)2 vitamin D3 has been demonstrated to the best available test to evaluate vitamin D status in sarcoidosis [18].
Case Continued
What are the preferred pharmacological agents for second-line treatment in sarcoidosis?
Alternative immunosuppression should be considered in the following circumstances in patients diagnosed with sarcoidosis:
- Failure or less than optimal response to oral corticosteroids
- Use as a steroid-sparing agent in patients requiring high doses of steroids for symptomatic control
- Failure to tolerate corticosteroids due to significant adverse effects such as excessive weight gain, steroid-induced psychosis, osteoporosis, and worsening diabetic control
A small trial of 11 patients examined azathioprine as a steroid-sparing agent and found it as an acceptable immunosuppressive agent for that purpose [19]. It was associated with good safety profile and adherence to treatment was 82% (9 out of 11 patients). However, the small sample size makes it difficult to draw firm conclusions from the findings of this study. In another trial evaluating methotrexate as a steroid-sparing agent in the first year after the diagnosis of sarcoidosis, Baughman and colleagues [20] reported that methotrexate is an attractive alternative to other immunosuppressive agents in term of a steroid sparer. In this double-blind randomized controlled trial (RCT), 15 patients were studied with at least 6 months of treatment with methotrexate vs placebo. There was a significantly reduced dosage of prednisolone observed in methotrexate group. However, the difference was not significant when data were analyzed for all patients, including the dropouts. More recently, a large international retrospective cohort study of 200 patients with sarcoidosis demonstrated that both methotrexate and azathioprine have similar efficacy and steroid-sparing capacity in sarcoidosis [21]. However, infection rates were significantly higher in the azathioprine group as compared to methotrexate (34.6% vs. 18.1%, P = 0.01). Hence methotrexate should be considered as a preferred second-line agent in sarcoidosis after detailed discussion about potential side effects.
Case Continued
The patient was started on methotrexate after discussion about the potential adverse effects of bone marrow suppression, hepatotoxicity, and pneumonitis. He was screened for latent tuberculosis and viral hepatitis prior to starting methotrexate. The dosage was 7.5 mg per week along with folic acid once a week. We gradually increase the dose in increments of 2.5 mg every 2 weeks with a view to reach 15 mg every week as maintenance therapy. In severely obese patients, a dose of up to 20 mg weekly is occasionally considered if 15 mg is suboptimal after careful clinical assessment.
The patient failed to make significant progress after being on methotrexate for a period of 6 months and lung function tests continued to demonstrate a persistent decline with symptomatic worsening of dyspnea and cough.
What are treatment options in refractory sarcoidosis?
Options to consider in the setting of refractory sarcoidosis are leflunomide, hydroxychloroquine, or combination therapy of methotrexate and leflunomide. Leflunomide has been shown to be of similar efficacy to methotrexate as demonstrated by a retrospective analysis of 32 patients treated with the drug in a tertiary care center [22]. Complete or partial response was noted in 12 of 17 patients treated solely with leflunomide and 13 of 15 treated in conjunction with methotrexate. Hence, combination therapy has been suggested as a viable option for these patients who fail to respond to initial glucocorticoid agents and alternative immunosuppressive drugs, as combination therapy may enhance efficacy with reduced toxicity if considered in a rational manner after careful selection of patients [23].
How should the symptom of fatigue be addressed?
The patient had ongoing fatigue during the treatment period with corticosteroids and alternative immunosuppressants. Fatigue, noted in a majority of patients with sarcoidosis [24], is one of the commonest symptoms of sarcoidosis and one of the most difficult to treat. We recommend excluding alternative etiologies of fatigue when confronted with this symptom and evaluating for associated comorbidities such as thyroid dysfunction, vitamin D deficiency, and hypoadrenalism. Extra-pulmonary sarcoidosis seems to be associated with fatigue in comparison to sarcoidosis restricted to the pulmonary system [25]. Furthermore, there is a paucity of good quality data on the benefit of pharmacological intervention for treatment of fatigue. A small double-blind randomized study of 10 patients demonstrated a positive impact of treatment with dexmethylphenidate hydrochloride (d-MPH) for a period of 8 weeks [26]. However, the small sample and lack of long-term outcomes data make it difficult to draw firm conclusions based on the findings of this study and larger randomized trials are warranted to investigate this important aspect of sarcoidosis before recommending a pharmacological agent routinely for this disabling symptom.
What are the pharmacological agents for cutaneous sarcoidosis?
Corticosteroids (local and or systemic) are the mainstay of treatment in cutaneous sarcoidosis. However, patients who fail to respond to these agents or develop significant adverse effects should be offered second-line agents in the form of hydroxychloroquine/chloroquine, methotrexate, or leflunomide. It is important to acknowledge that the evidence of benefit for these agents is derived from uncontrolled studies [27–29]. Anti-malarial agents are usually well tolerated; patients do require a baseline ophthalmological assessment and subsequent periodic examinations to monitor for any ocular toxicity associated with their use. Leflunomide is also a second-line option in cutaneous disease and is associated with lesser toxicity than methotrexate [22]. More recently, topical tacrolimus has shown promising results in isolated case reports [30–33]. Hence, it may be considered a treatment option in refractory cutaneous sarcoidosis.
Is there a role for anti-TNF therapy in the management of sarcoidosis? Should it be considered for the case patient?
Tumour necrosis factor-α (TNF-α) is implicated in the pathogenesis of sarcoidosis and is believed to have a significant role in the inflammatory processes in sarcoidosis. It is released from alveolar macrophages of patients with active disease and is involved in formation and maintenance of granulomas in the lung tissue. There has been significant progress in therapeutic options alternative to traditional immunosuppressants such as corticosteroids and TNF-α inhibition has been on the horizon as a treatment strategy for few years. It may be considered as a steroid-sparing agent or a third-line treatment option in refractory cases.
The initial evidence of benefit of anti-TNF therapy comes from case reports and small case series [34,35]. However, there have been 2 RCTs published so far invest-igating infliximab in sarcoidosis [36,37]. Baughman and colleagues [36] evaluated 138 patients with chronic pulmonary sarcoidosis in a double-blind study. There was a statistically significant improvement in FVC after 24 weeks of therapy with infliximab as compared to placebo (2.5% increase in mean FVC % predicted from baseline, P = 0.038). However, there was no improvement in any of the secondary outcome variables including SGRQ (St George’s Respiratory Questionnaire), 6-minute walk distance, and dyspnea scores. The benefit seemed to be more pronounced in the severe disease category on post-hoc analysis. The clinical significance of these findings are however unclear and it is difficult to draw firm conclusions based on the findings of this study alone.
The other RCT exploring the role of infliximab in sarcoidosis was conducted by Rossman et al [37]. This multi-center phase II study was conducted to evaluate the safety and tolerability of the drug in active pulmonary sarcoidosis (stage II to IV). The trial had a small number of participants with only 19 patients and failed to show a significant improvement in lung function after 6 weeks of treatment. Furthermore, 4 patients developed serious adverse events after treatment. Doty and colleagues [34] retrospectively analysed 10 patients treated with infliximab for refractory sarcoidosis and found subjective and objective evidence of improvement in all patients. This therapy resulted in reduction of corticosteroid dosage in 83% of cases (5 out of 6). However, adverse reactions were noted in 3 cases, including development of angioimmunoblastic lymphoma in one case. A retrospective study of 16 consecutive unselected cases of refractory sarcoidosis by Chapelon-Abric and colleagues [38] demonstrated a positive response in a majority of cases. However 38% of patients experienced a relapse. Furthermore, 44% of patients (7 out of 16) had infectious complications associated with anti-TNF therapy. Finally, infliximab may be used successfully in treating severe small fibre neuropathy [39] and should be considered in refractory cases where neuropathy is associated with autonomic dysfunction.
In summary, based on the findings of the aforementioned trials and case series, anti-TNF therapy may be considered as a treatment option in carefully selected patients after discussion of the potential adverse effects, followed by close monitoring at a specialist center for the management of sarcoidosis. Furthermore, anti-TNF therapy should not delay referral for lung transplant assessment, particularly when disease progression is relatively rapid. Duration of treatment and timing of thoracic imaging after anti-TNF therapy is subject to debate and should be individualized in close collaboration with a specialist center.
Case Continued
What is the role of newer biologics in the treatment of sarcoidosis? And what other therapies are on the horizon?
Although sarcoidosis is a T cell–mediated disease, humoral immunity has been implicated in sarcoidosis [40] and B cell depletion by rituximab (anti-CD 20+chimeric monoclonal antibody) has been successfully utilized in T cell–mediated diseases such as rheumatoid arthritis. Rituximab has been studied in phase I/II trial [41] in patients with refractory pulmonary sarcoidosis. The response to rituximab was inconsistent in these patients, with only a small group demonstrating > 5% improvement in FVC or walking distance. Hence, further studies are required to demonstrate a significant clinical benefit of this treatment in refractory sarcoidosis and potentially identify the characteristics of patients that may respond to B-cell depletion.
Adalimumab is another biologic agent that may be a potential option in refractory sarcoidosis as demonstrated in an open-label study of 11 patients [42]. This small trial of 52 weeks’ duration showed that adalimumab is well tolerated and may be considered in refractory pulmonary sarcoidosis when other treatment options have been exhausted.
Acthar had been used to treat pulmonary sarcoidosis in 1950s and there has been recent interest in evaluating the value of acthar gel therapy in the management of sarcoidosis. Baughman and colleagues [43] carried out a retrospective analysis of 47 patients with advanced sarcoidosis treated with acthar gel. The results showed that there was an objective improvement in approximately a third of patients receiving at least 3 months of treatment. Thirty-six percent of patients (n = 17) managed to reduce their oral corticosteroid dosage by more than 50% while on this therapy. However, a significant proportion of patients were unable to take ≥ 3 months of treatment, suggesting poor tolerance/adherence. The utility of acthar gel therapy would need to be examined in larger prospective randomized trial before it can be recommended for a treatment option in advanced disease.
Should pneumocystis pneumonia (PCP) prophylaxis be considered in sarcoidosis?
We do not routinely consider PCP prophylaxis in all sarcoidosis patients. However, it should be considered in the following clinical situations:
- Failure to reduce oral corticosteroid dose to less than 20 mg of prednisolone daily
- Concomitant use of anti-TNF agents with relatively high maintenance dose of oral corticosteroids (≥ 20 mg per day)
- Significant comorbid condition in association with sarcoidosis resulting in significant level of immunosuppression
As our patient did not fall in any of the above categories, we did not offer him PCP prophylaxis. However, clinicians treating patients with sarcoidosis with strong immunosuppressants should be aware of this potential complication and be vigilant about discussing prophylaxis with trimethoprim-sulphamethoxazole, which is the first-line agent for this purpose.
Is there a role of anti-tuberculous treatment in sarcoidosis?
Both mycobacterium tuberculosis (MTB) and non-mycobateria (NTM) have been implicated in sarcoidosis [44] and it is challenging to differentiate tuberculosis (TB) from sarcoidosis in certain clinical situations, such as when dealing with patients from countries with a high incidence of TB. The association of mycobacterium with sarcoidosis was explored in 2 recent trials of concomitant use of levofloxacin, ethambutol, azithromycin, and rifampin in cutaneous [45] and pulmonary sarcoidosis [46]. Drake and colleagues evaluated 15 chronic pulmonary sarcoidosis patients in an open-label trial to investigate if the combination of these drugs was associated with improvement in pulmonary sarcoidosis. The patients who completed 8 weeks of treatment had improvement in FVC at both 4 and 8 weeks of treatment. However, only 8 patients could complete 8 weeks of therapy, with significant adverse events. The small sample size limits our ability to draw meaningful conclusions and larger randomized trials are warranted to investigate this approach in sarcoidosis management.
Are there any valid serum biomarkers for sarcoidosis?
A biomarker is defined as a compound easily measurable in serum, urine, or other body fluids that can be used as indicator of presence and/or severity of particular disease state. Moreover, it helps in evaluation of effectiveness of drug therapy and useful to monitor the disease longitudinally. Unfortunately, there is no ideal serum biomarker in sarcoidosis. The most widely evaluated marker is serum angiotensin-converting enzyme (ACE). It has been found to be elevated in three quarters of patients with sarcoidosis [47]. However, it has poor diagnostic utility due to limited sensitivity and specificity. Furthermore, levels of serum ACE are reduced in patients taking ACE inhibitors and hence measurement of ACE levels in patients taking ACE inhibitors may lead to inaccurate interpretations [48].
A number of other serum biomarkers have been evaluated in sarcoidosis. Gungor and colleagues [49] evaluated 48 patients with sarcoidosis and 20 healthy controls. The biomarkers measured were ACE, adenosine deaminase (ADA), total IgE, serum amyloid-A (SAA), soluble interleukin-2 receptor (sIL2R), and C-reactive protein (CRP). This study showed that SAA was significantly elevated in sarcoidosis as compared to controls (P < 0.001). Furthermore, sIL2R levels were raised in extra-pulmonary sarcoidosis (P < 0.014). In another study, Grutters and co-workers [50] demonstrated elevated levels of sIL2R in 47 patients with active sarcoidosis. However, the levels did not correlate with radiolographic or physiological outcomes or response to treatment. Hence, the utility of these biomarkers in clinical practice is questionable and larger longitudinal studies are required to demonstrate the actual benefit of these biomarkers in everyday clinical practice.
There is a complex relationship between vitamin D and calcium metabolism and risk of osteoporosis in sarcoidosis. The value of active vitamin D metabolite 1, 25-dihydroxy vitamin D in relation to the degree of sarcoidosis disease chronicity was evaluated in a study of 59 patients with sarcoidosis [51]. It was noted that increased levels of 1,25 vitamin D were associated with increased risk of chronic phenotype and the need to require repeated treatments with immunosuppressive agents. Hence, serum levels of 1,25 vitamin D may have a prognostic value in sarcoidosis. It is important to note that both vitamin D deficiency as well as vitamin D excess can result in osteoporosis [52], and there is a risk of bone fragility and fractures in patients who may need long-term oral corticosteroid therapy. Hence, an optimal level of vitamin D is crucial; we recommend a value of serum 25(OH) vitamin D between 10 and 20 ng/mL as evidenced by a cross-sectional analysis of 142 consecutive patients with biopsy-proven sarcoidosis [53]. The above range of 25(OH) vitamin D was associated with higher bone mineral density and values above 20 ng/mL resulted in increased risk of fractures, demonstrating the need to keep vitamin D levels of these patients to be lower than those recommended for general population.
When should a patient with sarcoidosis be referred for lung transplantation?
Lung transplantation is a treatment option in advanced/end-stage disease after pharmacologic treatments have been exhausted and there is no evidence of progressive or severe extrapulmonary disease. The most critical decision regarding transplantation in pulmonary sarcoidosis is the timing of the referral and close liaison with the transplant center to ensure the maximal chance of success with lung transplantation. We recommend considering transplant referral when % predicted FVC approaches a value of 50% or less with or without significant pulmonary hypertension. Single lung transplant is appropriate for the majority of patients with sarcoidosis. However, bilateral transplant should be considered in bilateral mycetomas and bilateral bronchiectasis. Arcasoy and colleagues [54] analysed 43 patients listed for transplantation and found the following factors associated with increased risk of mortality:
- pulmonary hypertension
- hypoxia
- low cardiac output
- elevated right atrial pressure
It is important to note that pulmonary function parameters have not been found to be predictive of mortality in sarcoidosis [54,55]. Although granuloma recurrence in transplanted lung has been observed, it is a rare to have organ failure secondary to recurrence, and lung transplant should be considered in refractory cases of pulmonary sarcoidosis in the absence of contraindications and close liaison with the transplant center at the earliest opportunity is recommended.
What is the optimal duration of clinical follow-up in sarcoidosis?
There is no consensus on appropriate follow-up duration once a diagnosis of sarcoidosis is confirmed, and it is dependent on physician preference, vital organ involvement, disease progression, need for pharmacological treatment, and availability of resources. The American Thoracic Society statement on sarcoidosis suggested at least 3 years of follow-up after corticosteroid treatment is completed irrespective of radiographic stage [56]. Patients with serious extrapulmonary symptoms would require longer-term follow-up and a recent single-center Japanese study of corticosteroid-naive patients showed that the number of organs involved at the outset dictates the cumulative risk of subsequent progression of sarcoidosis [57]. This study of 150 patients with sarcoidosis with a median follow-up of 7.7 years demonstrated significantly increased risk of progression when there were > 3 organ systems involved as compared to ≤ 3 involved. These corticosteroid-naive patients may require longer follow-up (up to 10 years) than previously thought. However, the findings of this study would require confirmation in multi-center and multi-ethnic cohort of sarcoidosis patients.
Summary
Sarcoidosis presents as a fascinating and challenging disease with myriad clinical, radiological, and pathological manifestations. Despite extensive research in the last decade to increase our understanding of the mechanisms of disease evolution and progression, the underlying etiology remains unidentified. The treatment paradigm has changed over the last 10 years with the introduction of biological agents such as infliximab and adalimumab [42]; these anti-TNF drugs offer a treatment option in refractory cases of sarcoidosis with or without corticosteroids and other anti-metabolites. Methotrexate is the preferred first-line immunosuppressive agent after corticosteroids and has a safer adverse effect profile in comparison to azathioprine. Larger randomized studies to evaluate the efficacy of antimycobacterial agents and newer biologics are warranted before making strong recommendations for the use of these drugs, as there are significant potential toxic effects associated with their use.
Corresponding author: Dr. Ahmed Fahim, Dept. of Respiratory Medicine, McHale Centre New Cross Hospital, Wolverhampton, UK WV10 0QP, [email protected].
Financial disclosures: None.
1. Kitamura AY, Takiguchi K, Kurosu N, et al. Feasibility of cytological diagnosis of sarcoidosis with endobronchial US-guided transbronchial aspiration. Sarcoidosis Vasc Diffuse Lung Dis 2012;29:82–9.
2. Chee A, Khalil M, Stather DR, et al. Cytologic assessment of endobronchial ultrasound-guided transbronchial needle aspirates in sarcoidosis. J Bronchology Interv Pulmonol 2012;19:24–8.
3. Oki M, Saka H, Kitagawa C, et al. Prospective study of endobronchial ultrasound-guided transbronchial needle aspiration of lymph nodes versus transbronchial lung biopsy of lung tissue for diagnosis of sarcoidosis. J Thorac Cardiovasc Surg 2012;143:1324–9.
4. Plit ML, Havryk AP, Hodgson A, et al. Rapid cytological analysis of endobronchial ultrasound-guided aspirates in sarcoidosis. Eur Respir J 2013;42:1302-8.
5. von Bartheld MB, Dekkers OM, Szlubowski A, et al. Endosonography vs conventional bronchoscopy for the diagnosis of sarcoidosis: the GRANULOMA randomized clinical trial. JAMA 2013;309:2457–64.
6. Uemura A, Morimoto S, Hiramitsu S, et al. Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J 1999;138:299–302.
7. Ishimaru S, Tsujino I, Sakaue S, et al. Combination of 18F-fluoro-2-deoxyglucose positron emission tomography and magnetic resonance imaging in assessing cardiac sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2005;22:234–5.
8. Youssef G, Leung E, Mylonas I, et al. The use of 18F-FDG PET in the diagnosis of cardiac sarcoidosis: a systematic review and metaanalysis including the Ontario experience. J Nucl Med 2012;53:241–8.
9. Blankstein R, Osborne M, Naya M, et al. Cardiac positron emission tomography enhances prognostic assessments of patients with suspected cardiac sarcoidosis. J Am Coll Cardiol 2014;63:329–36.
10. Baughman RP, Nunes H, Sweiss NJ, Lower EE. Established and experimental medical therapy of pulmonary sarcoidosis. Eur Respir J 2013;41:1424–8.
11. James DG, Carstairs LS, Trowell J, Sharma OP. Treatment of sarcoidosis. Report of a controlled therapeutic trial. Lancet 1967;2:526–8.
12. Pietinalho A, Tukiainen P, Haahtela T, et al. Oral prednisolone followed by inhaled budesonide in newly diagnosed pulmonary sarcoidosis: a double-blind, placebo-controlled multicenter study. Finnish Pulmonary Sarcoidosis Study Group. Chest 1999;116:424–31.
13. Eule H, Roth I, Ehrke I, Weinecke W. Corticosteroid therapy of intrathoracic sarcoidosis stages I and II--results of a controlled clinical trial. Z Erkr Atmungsorgane 1977;149:142–7.
14. Israel HL, Fouts DW, Beggs RA. A controlled trial of prednisone treatment of sarcoidosis. Am Rev Respir Dis 1973;107:609–14.
15. Zaki MH, Lyons HA, Leilop L, Huang CT. Corticosteroid therapy in sarcoidosis. A five-year, controlled follow-up study. N Y State J Med 1987;87:496–9.
16. Gibson GJ, Prescott RJ, Muers MF, et al. British Thoracic Society Sarcoidosis study: effects of long term corticosteroid treatment. Thorax 1996;51:238–47.
17. Kamphuis LS, Bonte-Mineur F, van Laar JA, et al. Calcium and vitamin D in sarcoidosis: is supplementation safe? J Bone Miner Res 2014;29:2498–503.
18. Baughman RP, Janovcik J, Ray M, et al. Calcium and vitamin D metabolism in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:113–20.
19. Muller-Quernheim J, Kienast K, Held M, et al. Treatment of chronic sarcoidosis with an azathioprine/prednisolone regimen. Eur Respir J 1999;14:1117–22.
20. Baughman RP, Winget DB, Lower EE. Methotrexate is steroid sparing in acute sarcoidosis: results of a double blind, randomized trial. Sarcoidosis Vasc Diffuse Lung Dis 2000;17:60–6.
21. Vorselaars AD, Wuyts WA, Vorselaars VM, et al. Methotrexate versus azathioprine in second line therapy of sarcoidosis. Chest 2013.
22. Baughman RP, Lower EE. Leflunomide for chronic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2004;21:43–8.
23. Baughman RP, Ohmichi M, Lower EE. Combination therapy for sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2001;18:133–7.
24. Drent M, Lower EE, De Vries J. Sarcoidosis-associated fatigue. Eur Respir J 2012;40:255–63.
25. Fleischer M, Hinz A, Brahler E, et al. Factors associated with fatigue in sarcoidosis. Respir Care 2014;59:1086–94.
26. Lower EE, Harman S, Baughman RP. Double-blind, randomized trial of dexmethylphenidate hydrochloride for the treatment of sarcoidosis-associated fatigue. Chest 2008;133:1189–95.
27. Veien NK, Brodthagen H. Cutaneous sarcoidosis treated with methotrexate. Br J Dermatol 1977;97:213–6.
28. Lower EE, Baughman RP. Prolonged use of methotrexate for sarcoidosis. Arch Intern Med 1995;155:846–51.
29. Kaye O, Palazzo E, Grossin M, et al. Low-dose methotrexate: an effective corticosteroid-sparing agent in the musculoskeletal manifestations of sarcoidosis. Br J Rheumatol 1995;34:642–4.
30. Green CM. Topical tacrolimus for the treatment of cutaneous sarcoidosis. Clin Exp Dermatol 2007:32:457–8.
31. Vano-Galvan S, Fernandez-Guarino M, Carmona LP, et al. Lichenoid type of cutaneous sarcoidosis: great response to topical tacrolimus. Eur J Dermatol 2008;18:89–90.
32. Gutzmer R, Volker B, Kapp A, Werfel T. [Successful topical treatment of cutaneous sarcoidosis with tacrolimus]. Hautarzt 2003;54:1193–7.
33. Katoh N, Mihara H, Yasuno H. Cutaneous sarcoidosis successfully treated with topical tacrolimus. Br J Dermatol 2002;147:154–6.
34. Doty JD, Mazur JE, Judson MA. Treatment of sarcoidosis with infliximab. Chest 2005;127:1064–71.
35. Berrios I, Jun-O’Connell A, Ghiran S, Ionete C. A case of neurosarcoidosis secondary to treatment of etanercept and review of the literature. BMJ Case Rep 2015 Jul 6;2015.
36. Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med 2006;174:795–802.
37. Rossman MD, Newman LS, Baughman RP, et al. A double-blinded, randomized, placebo-controlled trial of infliximab in subjects with active pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2006;23:201–8.
38. Chapelon-Abric D, Saadoun D, Biard L. Long-term outcome of infliximab in severe chronic and refractory systemic sarcoidosis: a report of 16 cases. Clin Exp Rheumatol 2015;33:509–15.
39. Hoitsma E, Faber CG, Santen-Hoeufft M, et al. Improvement of small fiber neuropathy in a sarcoidosis patient after treatment with infliximab. Sarcoidosis Vasc Diffuse Lung Dis 2006;23:73–7.
40. Lee NS, Barber L, Kanchwala A, et al. Low levels of NF-kappaB/p65 mark anergic CD4+ T cells and correlate with disease severity in sarcoidosis. Clin Vaccine Immunol 2011;18:223–34.
41. Sweiss NJ, Lower EE, Mirsaeidi M, et al. Rituximab in the treatment of refractory pulmonary sarcoidosis. Eur Respir J 2014;43:1525–8.
42. Sweiss NJ, Noth I, Mirsaeidi M, et al. Efficacy Results of a 52-week trial of adalimumab in the treatment of refractory sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:46–54.
43. Baughman RP, Barney JB, O’Hare L, Lower EE. A retrospective pilot study examining the use of Acthar gel in sarcoidosis patients. Respir Med 2016;110:66–72.
44. Mangiapan G, Hance AJ. Mycobacteria and sarcoidosis: an overview and summary of recent molecular biological data. Sarcoidosis 1995;12:20-37.
45. Drake WP, Oswald-Richter K, Richmond BW, et al. Oral antimycobacterial therapy in chronic cutaneous sarcoidosis: a randomized, single-masked, placebo-controlled study. JAMA Dermatol 2013;149:1040–9.
46. Drake WP, Richmond BW, Oswald-Richter K, et al. Effects of broad-spectrum antimycobacterial therapy on chronic pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:201–11.
47. Studdy PR, Bird R. Serum angiotensin converting enzyme in sarcoidosis--its value in present clinical practice. Ann Clin Biochem 1989;26 ( Pt 1):13–8.
48. Krasowski MD, Savage J, Ehlers A, et al. Ordering of the serum angiotensin-converting enzyme test in patients receiving angiotensin-converting enzyme inhibitor therapy: an avoidable but common error. Chest 2015;148:1447–53.
49. Gungor S, Ozseker F, Yalcinsoy M. Conventional markers in determination of activity of sarcoidosis. Int Immunopharmacol 2015;25:174–9.
50. Grutters JC, Fellrath JM, Mulder L, et al. Serum soluble interleukin-2 receptor measurement in patients with sarcoidosis: a clinical evaluation. Chest 2003;124:186–95.
51. Kavathia D, Buckley JD, Rao D, et al. Elevated 1, 25-dihydroxyvitamin D levels are associated with protracted treatment in sarcoidosis. Respir Med 2010;104:564–70.
52. Baughman RP, Lower EE. Goldilocks, vitamin D and sarcoidosis. Arthritis Res Ther 2014;16:111.
53. Saidenberg-Kermanac’h N, Semerano L, Nunes H, et al. Bone fragility in sarcoidosis and relationships with calcium metabolism disorders: a cross sectional study on 142 patients. Arthritis Res Ther 2014;16:R78.
54. Arcasoy SM, Christie JD, Pochettino A, et al. Characteristics and outcomes of patients with sarcoidosis listed for lung transplantation. Chest 2001;120:873–80.
55. Shorr AF, Davies DB, Nathan SD. Predicting mortality in patients with sarcoidosis awaiting lung transplantation. Chest 2003;124:922–8.
56. Hunninghake GW, Costabel U, Ando M, et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis 1999;16:149–73.
57. Inoue Y, Inui N, Hashimoto D, et al. Cumulative incidence and predictors of progression in corticosteroid-naive patients with sarcoidosis. PLoS One 2015;10:e0143371.
1. Kitamura AY, Takiguchi K, Kurosu N, et al. Feasibility of cytological diagnosis of sarcoidosis with endobronchial US-guided transbronchial aspiration. Sarcoidosis Vasc Diffuse Lung Dis 2012;29:82–9.
2. Chee A, Khalil M, Stather DR, et al. Cytologic assessment of endobronchial ultrasound-guided transbronchial needle aspirates in sarcoidosis. J Bronchology Interv Pulmonol 2012;19:24–8.
3. Oki M, Saka H, Kitagawa C, et al. Prospective study of endobronchial ultrasound-guided transbronchial needle aspiration of lymph nodes versus transbronchial lung biopsy of lung tissue for diagnosis of sarcoidosis. J Thorac Cardiovasc Surg 2012;143:1324–9.
4. Plit ML, Havryk AP, Hodgson A, et al. Rapid cytological analysis of endobronchial ultrasound-guided aspirates in sarcoidosis. Eur Respir J 2013;42:1302-8.
5. von Bartheld MB, Dekkers OM, Szlubowski A, et al. Endosonography vs conventional bronchoscopy for the diagnosis of sarcoidosis: the GRANULOMA randomized clinical trial. JAMA 2013;309:2457–64.
6. Uemura A, Morimoto S, Hiramitsu S, et al. Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J 1999;138:299–302.
7. Ishimaru S, Tsujino I, Sakaue S, et al. Combination of 18F-fluoro-2-deoxyglucose positron emission tomography and magnetic resonance imaging in assessing cardiac sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2005;22:234–5.
8. Youssef G, Leung E, Mylonas I, et al. The use of 18F-FDG PET in the diagnosis of cardiac sarcoidosis: a systematic review and metaanalysis including the Ontario experience. J Nucl Med 2012;53:241–8.
9. Blankstein R, Osborne M, Naya M, et al. Cardiac positron emission tomography enhances prognostic assessments of patients with suspected cardiac sarcoidosis. J Am Coll Cardiol 2014;63:329–36.
10. Baughman RP, Nunes H, Sweiss NJ, Lower EE. Established and experimental medical therapy of pulmonary sarcoidosis. Eur Respir J 2013;41:1424–8.
11. James DG, Carstairs LS, Trowell J, Sharma OP. Treatment of sarcoidosis. Report of a controlled therapeutic trial. Lancet 1967;2:526–8.
12. Pietinalho A, Tukiainen P, Haahtela T, et al. Oral prednisolone followed by inhaled budesonide in newly diagnosed pulmonary sarcoidosis: a double-blind, placebo-controlled multicenter study. Finnish Pulmonary Sarcoidosis Study Group. Chest 1999;116:424–31.
13. Eule H, Roth I, Ehrke I, Weinecke W. Corticosteroid therapy of intrathoracic sarcoidosis stages I and II--results of a controlled clinical trial. Z Erkr Atmungsorgane 1977;149:142–7.
14. Israel HL, Fouts DW, Beggs RA. A controlled trial of prednisone treatment of sarcoidosis. Am Rev Respir Dis 1973;107:609–14.
15. Zaki MH, Lyons HA, Leilop L, Huang CT. Corticosteroid therapy in sarcoidosis. A five-year, controlled follow-up study. N Y State J Med 1987;87:496–9.
16. Gibson GJ, Prescott RJ, Muers MF, et al. British Thoracic Society Sarcoidosis study: effects of long term corticosteroid treatment. Thorax 1996;51:238–47.
17. Kamphuis LS, Bonte-Mineur F, van Laar JA, et al. Calcium and vitamin D in sarcoidosis: is supplementation safe? J Bone Miner Res 2014;29:2498–503.
18. Baughman RP, Janovcik J, Ray M, et al. Calcium and vitamin D metabolism in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:113–20.
19. Muller-Quernheim J, Kienast K, Held M, et al. Treatment of chronic sarcoidosis with an azathioprine/prednisolone regimen. Eur Respir J 1999;14:1117–22.
20. Baughman RP, Winget DB, Lower EE. Methotrexate is steroid sparing in acute sarcoidosis: results of a double blind, randomized trial. Sarcoidosis Vasc Diffuse Lung Dis 2000;17:60–6.
21. Vorselaars AD, Wuyts WA, Vorselaars VM, et al. Methotrexate versus azathioprine in second line therapy of sarcoidosis. Chest 2013.
22. Baughman RP, Lower EE. Leflunomide for chronic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2004;21:43–8.
23. Baughman RP, Ohmichi M, Lower EE. Combination therapy for sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2001;18:133–7.
24. Drent M, Lower EE, De Vries J. Sarcoidosis-associated fatigue. Eur Respir J 2012;40:255–63.
25. Fleischer M, Hinz A, Brahler E, et al. Factors associated with fatigue in sarcoidosis. Respir Care 2014;59:1086–94.
26. Lower EE, Harman S, Baughman RP. Double-blind, randomized trial of dexmethylphenidate hydrochloride for the treatment of sarcoidosis-associated fatigue. Chest 2008;133:1189–95.
27. Veien NK, Brodthagen H. Cutaneous sarcoidosis treated with methotrexate. Br J Dermatol 1977;97:213–6.
28. Lower EE, Baughman RP. Prolonged use of methotrexate for sarcoidosis. Arch Intern Med 1995;155:846–51.
29. Kaye O, Palazzo E, Grossin M, et al. Low-dose methotrexate: an effective corticosteroid-sparing agent in the musculoskeletal manifestations of sarcoidosis. Br J Rheumatol 1995;34:642–4.
30. Green CM. Topical tacrolimus for the treatment of cutaneous sarcoidosis. Clin Exp Dermatol 2007:32:457–8.
31. Vano-Galvan S, Fernandez-Guarino M, Carmona LP, et al. Lichenoid type of cutaneous sarcoidosis: great response to topical tacrolimus. Eur J Dermatol 2008;18:89–90.
32. Gutzmer R, Volker B, Kapp A, Werfel T. [Successful topical treatment of cutaneous sarcoidosis with tacrolimus]. Hautarzt 2003;54:1193–7.
33. Katoh N, Mihara H, Yasuno H. Cutaneous sarcoidosis successfully treated with topical tacrolimus. Br J Dermatol 2002;147:154–6.
34. Doty JD, Mazur JE, Judson MA. Treatment of sarcoidosis with infliximab. Chest 2005;127:1064–71.
35. Berrios I, Jun-O’Connell A, Ghiran S, Ionete C. A case of neurosarcoidosis secondary to treatment of etanercept and review of the literature. BMJ Case Rep 2015 Jul 6;2015.
36. Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med 2006;174:795–802.
37. Rossman MD, Newman LS, Baughman RP, et al. A double-blinded, randomized, placebo-controlled trial of infliximab in subjects with active pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2006;23:201–8.
38. Chapelon-Abric D, Saadoun D, Biard L. Long-term outcome of infliximab in severe chronic and refractory systemic sarcoidosis: a report of 16 cases. Clin Exp Rheumatol 2015;33:509–15.
39. Hoitsma E, Faber CG, Santen-Hoeufft M, et al. Improvement of small fiber neuropathy in a sarcoidosis patient after treatment with infliximab. Sarcoidosis Vasc Diffuse Lung Dis 2006;23:73–7.
40. Lee NS, Barber L, Kanchwala A, et al. Low levels of NF-kappaB/p65 mark anergic CD4+ T cells and correlate with disease severity in sarcoidosis. Clin Vaccine Immunol 2011;18:223–34.
41. Sweiss NJ, Lower EE, Mirsaeidi M, et al. Rituximab in the treatment of refractory pulmonary sarcoidosis. Eur Respir J 2014;43:1525–8.
42. Sweiss NJ, Noth I, Mirsaeidi M, et al. Efficacy Results of a 52-week trial of adalimumab in the treatment of refractory sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:46–54.
43. Baughman RP, Barney JB, O’Hare L, Lower EE. A retrospective pilot study examining the use of Acthar gel in sarcoidosis patients. Respir Med 2016;110:66–72.
44. Mangiapan G, Hance AJ. Mycobacteria and sarcoidosis: an overview and summary of recent molecular biological data. Sarcoidosis 1995;12:20-37.
45. Drake WP, Oswald-Richter K, Richmond BW, et al. Oral antimycobacterial therapy in chronic cutaneous sarcoidosis: a randomized, single-masked, placebo-controlled study. JAMA Dermatol 2013;149:1040–9.
46. Drake WP, Richmond BW, Oswald-Richter K, et al. Effects of broad-spectrum antimycobacterial therapy on chronic pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:201–11.
47. Studdy PR, Bird R. Serum angiotensin converting enzyme in sarcoidosis--its value in present clinical practice. Ann Clin Biochem 1989;26 ( Pt 1):13–8.
48. Krasowski MD, Savage J, Ehlers A, et al. Ordering of the serum angiotensin-converting enzyme test in patients receiving angiotensin-converting enzyme inhibitor therapy: an avoidable but common error. Chest 2015;148:1447–53.
49. Gungor S, Ozseker F, Yalcinsoy M. Conventional markers in determination of activity of sarcoidosis. Int Immunopharmacol 2015;25:174–9.
50. Grutters JC, Fellrath JM, Mulder L, et al. Serum soluble interleukin-2 receptor measurement in patients with sarcoidosis: a clinical evaluation. Chest 2003;124:186–95.
51. Kavathia D, Buckley JD, Rao D, et al. Elevated 1, 25-dihydroxyvitamin D levels are associated with protracted treatment in sarcoidosis. Respir Med 2010;104:564–70.
52. Baughman RP, Lower EE. Goldilocks, vitamin D and sarcoidosis. Arthritis Res Ther 2014;16:111.
53. Saidenberg-Kermanac’h N, Semerano L, Nunes H, et al. Bone fragility in sarcoidosis and relationships with calcium metabolism disorders: a cross sectional study on 142 patients. Arthritis Res Ther 2014;16:R78.
54. Arcasoy SM, Christie JD, Pochettino A, et al. Characteristics and outcomes of patients with sarcoidosis listed for lung transplantation. Chest 2001;120:873–80.
55. Shorr AF, Davies DB, Nathan SD. Predicting mortality in patients with sarcoidosis awaiting lung transplantation. Chest 2003;124:922–8.
56. Hunninghake GW, Costabel U, Ando M, et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis 1999;16:149–73.
57. Inoue Y, Inui N, Hashimoto D, et al. Cumulative incidence and predictors of progression in corticosteroid-naive patients with sarcoidosis. PLoS One 2015;10:e0143371.