User login
Less education tied to more anxiety
Adults with less than a high school education are more than twice as likely as are those with high school degrees to have reported an anxiety disorder in the past year, according to the National Survey on Drug Use and Health’s 2008 to 2012 Mental Health Surveillance Study (MHSS), published June 2.
The data identified anxiety disorders in 13% of non–high school graduates vs. 5% of high school graduates. Adults with at least a college degree had the lowest rates of past year anxiety (4.3%). The MHSS estimated that approximately 13 million adults in the United States had at least one anxiety disorder within the past year.
“Although the MHSS results cannot be used to determine whether anxiety stopped people from finishing high school, having an anxiety disorder can lower the odds of graduating from high school and the odds of attending college,” the researchers wrote. The findings emphasize the need to support people with anxiety to help them remain in school and succeed, they added.
The findings were published in the Center for Behavioral Health Statistics and Quality Report. Read the full study here.
Adults with less than a high school education are more than twice as likely as are those with high school degrees to have reported an anxiety disorder in the past year, according to the National Survey on Drug Use and Health’s 2008 to 2012 Mental Health Surveillance Study (MHSS), published June 2.
The data identified anxiety disorders in 13% of non–high school graduates vs. 5% of high school graduates. Adults with at least a college degree had the lowest rates of past year anxiety (4.3%). The MHSS estimated that approximately 13 million adults in the United States had at least one anxiety disorder within the past year.
“Although the MHSS results cannot be used to determine whether anxiety stopped people from finishing high school, having an anxiety disorder can lower the odds of graduating from high school and the odds of attending college,” the researchers wrote. The findings emphasize the need to support people with anxiety to help them remain in school and succeed, they added.
The findings were published in the Center for Behavioral Health Statistics and Quality Report. Read the full study here.
Adults with less than a high school education are more than twice as likely as are those with high school degrees to have reported an anxiety disorder in the past year, according to the National Survey on Drug Use and Health’s 2008 to 2012 Mental Health Surveillance Study (MHSS), published June 2.
The data identified anxiety disorders in 13% of non–high school graduates vs. 5% of high school graduates. Adults with at least a college degree had the lowest rates of past year anxiety (4.3%). The MHSS estimated that approximately 13 million adults in the United States had at least one anxiety disorder within the past year.
“Although the MHSS results cannot be used to determine whether anxiety stopped people from finishing high school, having an anxiety disorder can lower the odds of graduating from high school and the odds of attending college,” the researchers wrote. The findings emphasize the need to support people with anxiety to help them remain in school and succeed, they added.
The findings were published in the Center for Behavioral Health Statistics and Quality Report. Read the full study here.
FROM THE CBHSQ REPORT
Necrolytic Migratory Erythema With Recalcitrant Dermatitis as the Only Presenting Symptom
To the Editor:
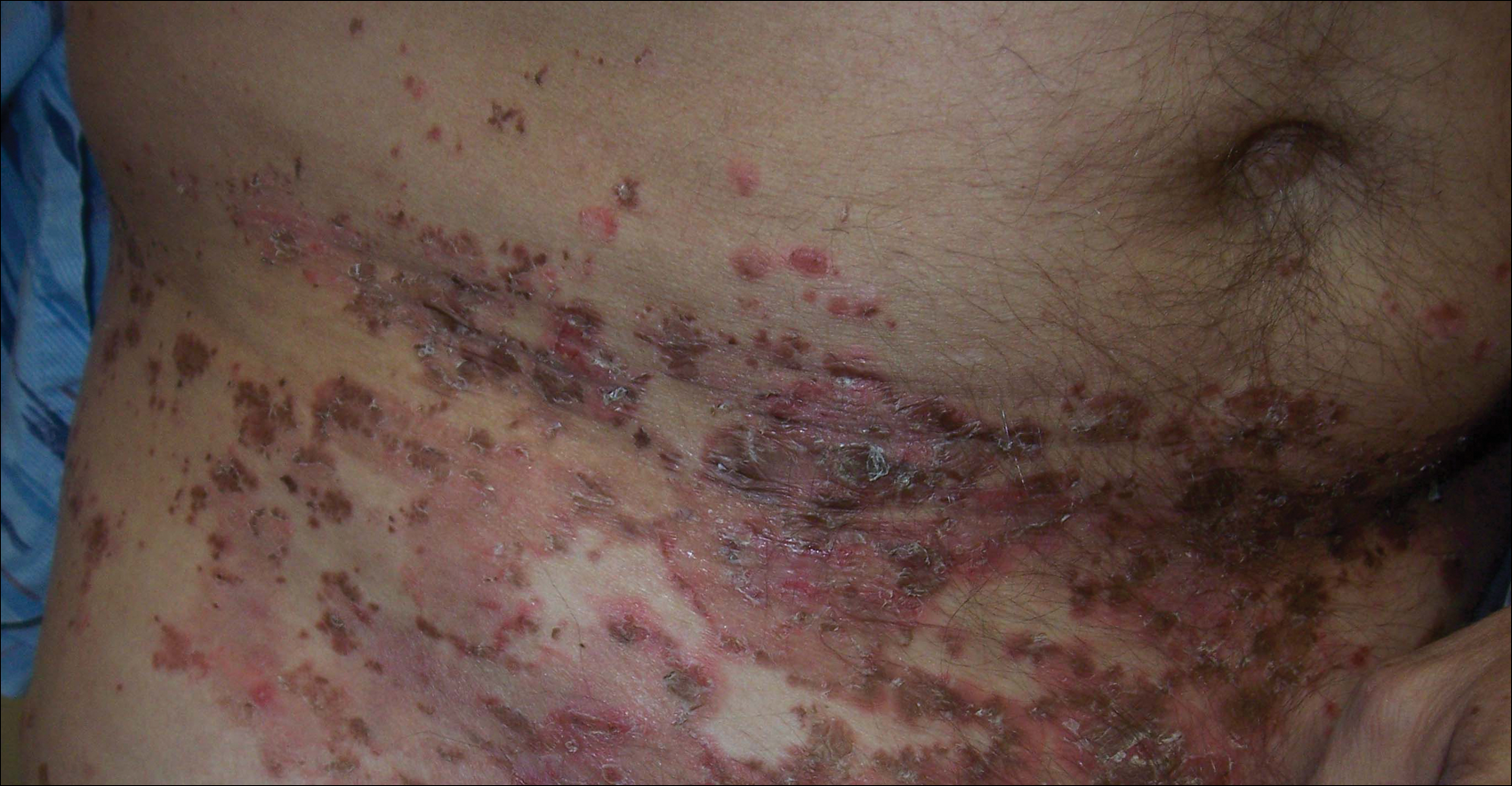
A 52-year-old man presented with recalcitrant dermatitis of 6 years’ duration. He was otherwise in excellent health. On initial presentation, physical examination revealed symmetrical, erythematous, blanching plaques with areas of erosions and overlying hemorrhagic crust on the eyebrows, scalp, back, dorsal aspects of the hands, axillae, abdomen (Figure), buttocks, groin, scrotum, pubis, and lower legs. Some areas showed slight necrosis. He denied any fevers, chills, night sweats, cough, chest pain, shortness of breath, dizziness, lightheadedness, weight loss, or appetite change.
Throughout the disease course the patient had visited numerous dermatologists seeking treatment. He had response to higher doses of oral prednisone (80 mg taper), but the condition would recur at the end of an extended taper. Treatment with narrowband UVB, mycophenolate mofetil, methotrexate, acitretin, topical clobetasol, and topical pimecrolimus provided no relief. Eventually he was placed on azathioprine 100 mg twice daily, which led to near-complete resolution. Outbreaks continued every few months and required courses of prednisone.
Multiple biopsies over the years revealed subacute spongiotic or psoriasiform dermatitis. At multiple visits it was noted that during flares there were areas of crusting and mild necrosis, which led to an extensive biochemical investigation. The glucagon level was markedly elevated at 630 ng/L (reference range, 40–130 ng/L), as was insulin at 71 μIU/mL (reference range, 6–27 μIU/mL). Complete blood cell counts over the disease course showed mild normochromic normocytic anemia. The abnormal laboratory findings led to computed tomography of the abdomen, which revealed a mass in the body of the pancreas measuring 3×3.8 cm. After computed tomography, the patient underwent a laparoscopic distal pancreatectomy and splenectomy. Histologic examination revealed a well-differentiated pancreatic endocrine tumor (glucagonoma) confined to the pancreas. After the surgery, the patient’s rash resolved within a few days and he discontinued all medications.
Diagnosis of glucagonomas often is delayed due to their rarity and lack of classical signs and symptoms. The distribution of the lesions seen in necrolytic migratory erythema (NME) usually involves the inguinal crease, perineum, lower extremities, buttocks, and other intertriginous areas.1 Our patient had involvement in the typical distribution but also had involvement of the scalp, face, and upper body. The typical histology for NME is crusted psoriasiform dermatitis with a tendency for the upper epidermis to have necrosis and a vacuolated pale epidermis.2 Our patient’s histologic findings were less specific showing epidermal spongiosis with a scant lymphocytic infiltrate and at times acanthosis. The lack of classical skin findings and histology delayed diagnosis. In more than 50% of patients, metastasis has already occurred by the time the patient is diagnosed.3 Treatment is aimed at complete removal of the pancreatic tumor, which typically leads to a rapid improvement in symptoms. For patients unable to undergo surgery, chemotherapy agents and octreotide are used; unfortunately, symptoms may persist.4 The response to azathioprine in our patient suggests it is a possible alternate therapy for those with persistent NME.
This patient highlights the difficulty of diagnosing a glucagonoma when the only clinical manifestation may be NME. Moreover, skin biopsies that can sometimes be diagnostic may be nonspecific. This patient also shows a potential benefit of azathioprine in the treatment of NME.
- Shi W, Liao W, Mei X, et al. Necrolytic migratory erythema associated with glucagonoma syndrome [published online June 7, 2010]. J Clin Oncol. 2010;28:e329-e331.
- Rapini RP. Practical Dermatopathology. London, England: Elsevier Mosby; 2005.
- Oberg K, Eriksson B. Endocrine tumors of the pancreas. Best Pract Res Clin Gastroenterol. 2005;19:753-781.
- Wermers RA, Fatourechi V, Wynne AG, et al. The glucagonoma syndrome: clinical and pathologic features in 21 patients. Medicine (Baltimore). 1996;72:53-63.
To the Editor:
A 52-year-old man presented with recalcitrant dermatitis of 6 years’ duration. He was otherwise in excellent health. On initial presentation, physical examination revealed symmetrical, erythematous, blanching plaques with areas of erosions and overlying hemorrhagic crust on the eyebrows, scalp, back, dorsal aspects of the hands, axillae, abdomen (Figure), buttocks, groin, scrotum, pubis, and lower legs. Some areas showed slight necrosis. He denied any fevers, chills, night sweats, cough, chest pain, shortness of breath, dizziness, lightheadedness, weight loss, or appetite change.
Throughout the disease course the patient had visited numerous dermatologists seeking treatment. He had response to higher doses of oral prednisone (80 mg taper), but the condition would recur at the end of an extended taper. Treatment with narrowband UVB, mycophenolate mofetil, methotrexate, acitretin, topical clobetasol, and topical pimecrolimus provided no relief. Eventually he was placed on azathioprine 100 mg twice daily, which led to near-complete resolution. Outbreaks continued every few months and required courses of prednisone.
Multiple biopsies over the years revealed subacute spongiotic or psoriasiform dermatitis. At multiple visits it was noted that during flares there were areas of crusting and mild necrosis, which led to an extensive biochemical investigation. The glucagon level was markedly elevated at 630 ng/L (reference range, 40–130 ng/L), as was insulin at 71 μIU/mL (reference range, 6–27 μIU/mL). Complete blood cell counts over the disease course showed mild normochromic normocytic anemia. The abnormal laboratory findings led to computed tomography of the abdomen, which revealed a mass in the body of the pancreas measuring 3×3.8 cm. After computed tomography, the patient underwent a laparoscopic distal pancreatectomy and splenectomy. Histologic examination revealed a well-differentiated pancreatic endocrine tumor (glucagonoma) confined to the pancreas. After the surgery, the patient’s rash resolved within a few days and he discontinued all medications.
Diagnosis of glucagonomas often is delayed due to their rarity and lack of classical signs and symptoms. The distribution of the lesions seen in necrolytic migratory erythema (NME) usually involves the inguinal crease, perineum, lower extremities, buttocks, and other intertriginous areas.1 Our patient had involvement in the typical distribution but also had involvement of the scalp, face, and upper body. The typical histology for NME is crusted psoriasiform dermatitis with a tendency for the upper epidermis to have necrosis and a vacuolated pale epidermis.2 Our patient’s histologic findings were less specific showing epidermal spongiosis with a scant lymphocytic infiltrate and at times acanthosis. The lack of classical skin findings and histology delayed diagnosis. In more than 50% of patients, metastasis has already occurred by the time the patient is diagnosed.3 Treatment is aimed at complete removal of the pancreatic tumor, which typically leads to a rapid improvement in symptoms. For patients unable to undergo surgery, chemotherapy agents and octreotide are used; unfortunately, symptoms may persist.4 The response to azathioprine in our patient suggests it is a possible alternate therapy for those with persistent NME.
This patient highlights the difficulty of diagnosing a glucagonoma when the only clinical manifestation may be NME. Moreover, skin biopsies that can sometimes be diagnostic may be nonspecific. This patient also shows a potential benefit of azathioprine in the treatment of NME.
To the Editor:
A 52-year-old man presented with recalcitrant dermatitis of 6 years’ duration. He was otherwise in excellent health. On initial presentation, physical examination revealed symmetrical, erythematous, blanching plaques with areas of erosions and overlying hemorrhagic crust on the eyebrows, scalp, back, dorsal aspects of the hands, axillae, abdomen (Figure), buttocks, groin, scrotum, pubis, and lower legs. Some areas showed slight necrosis. He denied any fevers, chills, night sweats, cough, chest pain, shortness of breath, dizziness, lightheadedness, weight loss, or appetite change.
Throughout the disease course the patient had visited numerous dermatologists seeking treatment. He had response to higher doses of oral prednisone (80 mg taper), but the condition would recur at the end of an extended taper. Treatment with narrowband UVB, mycophenolate mofetil, methotrexate, acitretin, topical clobetasol, and topical pimecrolimus provided no relief. Eventually he was placed on azathioprine 100 mg twice daily, which led to near-complete resolution. Outbreaks continued every few months and required courses of prednisone.
Multiple biopsies over the years revealed subacute spongiotic or psoriasiform dermatitis. At multiple visits it was noted that during flares there were areas of crusting and mild necrosis, which led to an extensive biochemical investigation. The glucagon level was markedly elevated at 630 ng/L (reference range, 40–130 ng/L), as was insulin at 71 μIU/mL (reference range, 6–27 μIU/mL). Complete blood cell counts over the disease course showed mild normochromic normocytic anemia. The abnormal laboratory findings led to computed tomography of the abdomen, which revealed a mass in the body of the pancreas measuring 3×3.8 cm. After computed tomography, the patient underwent a laparoscopic distal pancreatectomy and splenectomy. Histologic examination revealed a well-differentiated pancreatic endocrine tumor (glucagonoma) confined to the pancreas. After the surgery, the patient’s rash resolved within a few days and he discontinued all medications.
Diagnosis of glucagonomas often is delayed due to their rarity and lack of classical signs and symptoms. The distribution of the lesions seen in necrolytic migratory erythema (NME) usually involves the inguinal crease, perineum, lower extremities, buttocks, and other intertriginous areas.1 Our patient had involvement in the typical distribution but also had involvement of the scalp, face, and upper body. The typical histology for NME is crusted psoriasiform dermatitis with a tendency for the upper epidermis to have necrosis and a vacuolated pale epidermis.2 Our patient’s histologic findings were less specific showing epidermal spongiosis with a scant lymphocytic infiltrate and at times acanthosis. The lack of classical skin findings and histology delayed diagnosis. In more than 50% of patients, metastasis has already occurred by the time the patient is diagnosed.3 Treatment is aimed at complete removal of the pancreatic tumor, which typically leads to a rapid improvement in symptoms. For patients unable to undergo surgery, chemotherapy agents and octreotide are used; unfortunately, symptoms may persist.4 The response to azathioprine in our patient suggests it is a possible alternate therapy for those with persistent NME.
This patient highlights the difficulty of diagnosing a glucagonoma when the only clinical manifestation may be NME. Moreover, skin biopsies that can sometimes be diagnostic may be nonspecific. This patient also shows a potential benefit of azathioprine in the treatment of NME.
- Shi W, Liao W, Mei X, et al. Necrolytic migratory erythema associated with glucagonoma syndrome [published online June 7, 2010]. J Clin Oncol. 2010;28:e329-e331.
- Rapini RP. Practical Dermatopathology. London, England: Elsevier Mosby; 2005.
- Oberg K, Eriksson B. Endocrine tumors of the pancreas. Best Pract Res Clin Gastroenterol. 2005;19:753-781.
- Wermers RA, Fatourechi V, Wynne AG, et al. The glucagonoma syndrome: clinical and pathologic features in 21 patients. Medicine (Baltimore). 1996;72:53-63.
- Shi W, Liao W, Mei X, et al. Necrolytic migratory erythema associated with glucagonoma syndrome [published online June 7, 2010]. J Clin Oncol. 2010;28:e329-e331.
- Rapini RP. Practical Dermatopathology. London, England: Elsevier Mosby; 2005.
- Oberg K, Eriksson B. Endocrine tumors of the pancreas. Best Pract Res Clin Gastroenterol. 2005;19:753-781.
- Wermers RA, Fatourechi V, Wynne AG, et al. The glucagonoma syndrome: clinical and pathologic features in 21 patients. Medicine (Baltimore). 1996;72:53-63.
Practice Points
- Recalcitrant dermatitis may be a symptom of internal malignancy.
- Glucagon levels are helpful in identifying glucagonomas of the pancreas.
- Although surgical excision is the preferred treatment of glucagonomas, azathioprine also can control dermatitis associated with necrolytic migratory erythema.
Sell skin care products to protect your patients
The ethics behind selling skin care products to patients has been hotly debated within the field of cosmetic dermatology for several decades. In 15 years of practice, I have come to the conclusion that patients want you to and need you to because otherwise they are easily taken advantage of. Other physicians are doing it but we – the dermatologists – are the most qualified to offer skin care advice. This article will discuss the reasons that you need to get over the ethical dilemma and offer skin care to your patients.

Using the correct skin care regimen for the face and body will improve outcomes
Whether a patient suffers from acne, rosacea, melasma, psoriasis, eczema, contact dermatitis, or even tinea versicolor, using the proper skin care regimen will improve outcomes by affecting the skin barrier, pH, hydration level, and function of the keratinocytes and fibroblasts. In fact, every personal care product that touches the skin has an impact on skin health. For example, if a patient uses a detergent-laden bar soap, the skin barrier will be impaired, which can cause them to react to allergens and irritants. Personal care products can affect the skin pH; this is shown to play a role in Malassezia colonization in atopic dermatitis patients (J Clin Med. 2015;4[6]:1217-28). As dermatologists, we know better than anyone that daily use of SPF improves skin health and lowers the risk of postinflammatory pigmentation. We all agree that patients should cleanse the skin and apply a SPF every day. Giving them guidance about which to choose is very important.
Giving the patient exact instructions will lead to improved compliance
Why should recommending skin care products be perceived differently than prescribing a prescription medication? We should prescribe to our patients in writing the exact skin care regimen they should use for their face or body to ensure that they understand the directions. I have been surprised by patients who have said, “I did not know I was supposed to wash my cleanser off,” or “I wash my face with hand soap.” We can help them by educating them and giving them specific instructions. Improved education and communication results in increased compliance. When you do surgery on a wound, you probably tell them to apply a topical antibiotic ointment, but do you direct them to what cleanser to use or tell them which SPF to use on the stitched wound? Providing written instructions for all dermatologic disorders and postprocedure care is necessary to improve compliance and outcomes.
Combine cosmeceuticals, prescription medications, and medical procedures
You (unlike the cosmetic counter salesperson) have the ability to combine cosmeceuticals with prescription medications and medical procedures. In fact, selling your patients the right skin care products to use after a procedure saves them a trip to the store and ensures that they use the correct products. Of course it makes sense that patients getting toxins and fillers should use a retinoid to improve skin aging; however, many general dermatologic diseases would improve with the proper skin care. For example, do you use biologics for psoriasis? Using the proper skin care to regulate skin pH and improve the skin barrier may help prevent colonization of yeast, fungus, and bacteria. The same is true for atopic patients. Do you use liquid nitrogen? Studies show that using a retinoid before a procedure speeds healing. Skin care goes way beyond wrinkles and dark circles under the eyes, so if you are not prescribing the patient an exact regimen, you are not maximizing outcomes.
I don’t have time to talk to my patients about skin care
The missing piece is that most of us don’t have the time to spend discussing skin care. This is where using a standardized scientific methodology is crucial. I developed and use a skin typing methodology in my office and have seen improved physician/patient relationships and increased patient satisfaction resulting in a significant amount of referrals. We also have noted decreased call backs and fewer adverse events from products because the patients have a better understanding of how to properly apply the cosmeceuticals and prescription products. The best part is, it does not add any time onto the patient visit when standardized methodologies are properly adopted.
What if I still do not feel comfortable profiting from the sale of skin care products?
First you need to realize that time is money and you are saving the patient the cost in time it would have taken them to go to a store, park, and shop for the correct product. I have seen data presented from several companies that show that patients usually spend a large amount of money on skin care products after they see their dermatologist. Without guidance, they will likely buy the incorrect products. If they buy the wrong product, you save them the hassle of having to make another office visit and the aggravation of the side effects from the incorrect product. These are often of poor quality or not appropriate for their skin issues. Counterfeit products are rampant on the Internet and many new companies tout worthless products with stem cells and other nonsense. Only you can help your patients make sure that money is spent on the proper products.
Conclusion
Do you really want someone else giving your patients skin care advice? Your patients deserve to have someone with your insights, knowledge, compassion, and honesty help them achieve optimal skin health through use of the proper cosmeceuticals and prescription medications. It is up to you and your staff to save your patients from falling prey to persuasive salespeople with no scientific knowledge or concern for long-term skin health.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever. Dr. Baumann also developed and owns the Baumann Skin Type Solution skin typing systems and related products.
The ethics behind selling skin care products to patients has been hotly debated within the field of cosmetic dermatology for several decades. In 15 years of practice, I have come to the conclusion that patients want you to and need you to because otherwise they are easily taken advantage of. Other physicians are doing it but we – the dermatologists – are the most qualified to offer skin care advice. This article will discuss the reasons that you need to get over the ethical dilemma and offer skin care to your patients.
Using the correct skin care regimen for the face and body will improve outcomes
Whether a patient suffers from acne, rosacea, melasma, psoriasis, eczema, contact dermatitis, or even tinea versicolor, using the proper skin care regimen will improve outcomes by affecting the skin barrier, pH, hydration level, and function of the keratinocytes and fibroblasts. In fact, every personal care product that touches the skin has an impact on skin health. For example, if a patient uses a detergent-laden bar soap, the skin barrier will be impaired, which can cause them to react to allergens and irritants. Personal care products can affect the skin pH; this is shown to play a role in Malassezia colonization in atopic dermatitis patients (J Clin Med. 2015;4[6]:1217-28). As dermatologists, we know better than anyone that daily use of SPF improves skin health and lowers the risk of postinflammatory pigmentation. We all agree that patients should cleanse the skin and apply a SPF every day. Giving them guidance about which to choose is very important.
Giving the patient exact instructions will lead to improved compliance
Why should recommending skin care products be perceived differently than prescribing a prescription medication? We should prescribe to our patients in writing the exact skin care regimen they should use for their face or body to ensure that they understand the directions. I have been surprised by patients who have said, “I did not know I was supposed to wash my cleanser off,” or “I wash my face with hand soap.” We can help them by educating them and giving them specific instructions. Improved education and communication results in increased compliance. When you do surgery on a wound, you probably tell them to apply a topical antibiotic ointment, but do you direct them to what cleanser to use or tell them which SPF to use on the stitched wound? Providing written instructions for all dermatologic disorders and postprocedure care is necessary to improve compliance and outcomes.
Combine cosmeceuticals, prescription medications, and medical procedures
You (unlike the cosmetic counter salesperson) have the ability to combine cosmeceuticals with prescription medications and medical procedures. In fact, selling your patients the right skin care products to use after a procedure saves them a trip to the store and ensures that they use the correct products. Of course it makes sense that patients getting toxins and fillers should use a retinoid to improve skin aging; however, many general dermatologic diseases would improve with the proper skin care. For example, do you use biologics for psoriasis? Using the proper skin care to regulate skin pH and improve the skin barrier may help prevent colonization of yeast, fungus, and bacteria. The same is true for atopic patients. Do you use liquid nitrogen? Studies show that using a retinoid before a procedure speeds healing. Skin care goes way beyond wrinkles and dark circles under the eyes, so if you are not prescribing the patient an exact regimen, you are not maximizing outcomes.
I don’t have time to talk to my patients about skin care
The missing piece is that most of us don’t have the time to spend discussing skin care. This is where using a standardized scientific methodology is crucial. I developed and use a skin typing methodology in my office and have seen improved physician/patient relationships and increased patient satisfaction resulting in a significant amount of referrals. We also have noted decreased call backs and fewer adverse events from products because the patients have a better understanding of how to properly apply the cosmeceuticals and prescription products. The best part is, it does not add any time onto the patient visit when standardized methodologies are properly adopted.
What if I still do not feel comfortable profiting from the sale of skin care products?
First you need to realize that time is money and you are saving the patient the cost in time it would have taken them to go to a store, park, and shop for the correct product. I have seen data presented from several companies that show that patients usually spend a large amount of money on skin care products after they see their dermatologist. Without guidance, they will likely buy the incorrect products. If they buy the wrong product, you save them the hassle of having to make another office visit and the aggravation of the side effects from the incorrect product. These are often of poor quality or not appropriate for their skin issues. Counterfeit products are rampant on the Internet and many new companies tout worthless products with stem cells and other nonsense. Only you can help your patients make sure that money is spent on the proper products.
Conclusion
Do you really want someone else giving your patients skin care advice? Your patients deserve to have someone with your insights, knowledge, compassion, and honesty help them achieve optimal skin health through use of the proper cosmeceuticals and prescription medications. It is up to you and your staff to save your patients from falling prey to persuasive salespeople with no scientific knowledge or concern for long-term skin health.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever. Dr. Baumann also developed and owns the Baumann Skin Type Solution skin typing systems and related products.
The ethics behind selling skin care products to patients has been hotly debated within the field of cosmetic dermatology for several decades. In 15 years of practice, I have come to the conclusion that patients want you to and need you to because otherwise they are easily taken advantage of. Other physicians are doing it but we – the dermatologists – are the most qualified to offer skin care advice. This article will discuss the reasons that you need to get over the ethical dilemma and offer skin care to your patients.
Using the correct skin care regimen for the face and body will improve outcomes
Whether a patient suffers from acne, rosacea, melasma, psoriasis, eczema, contact dermatitis, or even tinea versicolor, using the proper skin care regimen will improve outcomes by affecting the skin barrier, pH, hydration level, and function of the keratinocytes and fibroblasts. In fact, every personal care product that touches the skin has an impact on skin health. For example, if a patient uses a detergent-laden bar soap, the skin barrier will be impaired, which can cause them to react to allergens and irritants. Personal care products can affect the skin pH; this is shown to play a role in Malassezia colonization in atopic dermatitis patients (J Clin Med. 2015;4[6]:1217-28). As dermatologists, we know better than anyone that daily use of SPF improves skin health and lowers the risk of postinflammatory pigmentation. We all agree that patients should cleanse the skin and apply a SPF every day. Giving them guidance about which to choose is very important.
Giving the patient exact instructions will lead to improved compliance
Why should recommending skin care products be perceived differently than prescribing a prescription medication? We should prescribe to our patients in writing the exact skin care regimen they should use for their face or body to ensure that they understand the directions. I have been surprised by patients who have said, “I did not know I was supposed to wash my cleanser off,” or “I wash my face with hand soap.” We can help them by educating them and giving them specific instructions. Improved education and communication results in increased compliance. When you do surgery on a wound, you probably tell them to apply a topical antibiotic ointment, but do you direct them to what cleanser to use or tell them which SPF to use on the stitched wound? Providing written instructions for all dermatologic disorders and postprocedure care is necessary to improve compliance and outcomes.
Combine cosmeceuticals, prescription medications, and medical procedures
You (unlike the cosmetic counter salesperson) have the ability to combine cosmeceuticals with prescription medications and medical procedures. In fact, selling your patients the right skin care products to use after a procedure saves them a trip to the store and ensures that they use the correct products. Of course it makes sense that patients getting toxins and fillers should use a retinoid to improve skin aging; however, many general dermatologic diseases would improve with the proper skin care. For example, do you use biologics for psoriasis? Using the proper skin care to regulate skin pH and improve the skin barrier may help prevent colonization of yeast, fungus, and bacteria. The same is true for atopic patients. Do you use liquid nitrogen? Studies show that using a retinoid before a procedure speeds healing. Skin care goes way beyond wrinkles and dark circles under the eyes, so if you are not prescribing the patient an exact regimen, you are not maximizing outcomes.
I don’t have time to talk to my patients about skin care
The missing piece is that most of us don’t have the time to spend discussing skin care. This is where using a standardized scientific methodology is crucial. I developed and use a skin typing methodology in my office and have seen improved physician/patient relationships and increased patient satisfaction resulting in a significant amount of referrals. We also have noted decreased call backs and fewer adverse events from products because the patients have a better understanding of how to properly apply the cosmeceuticals and prescription products. The best part is, it does not add any time onto the patient visit when standardized methodologies are properly adopted.
What if I still do not feel comfortable profiting from the sale of skin care products?
First you need to realize that time is money and you are saving the patient the cost in time it would have taken them to go to a store, park, and shop for the correct product. I have seen data presented from several companies that show that patients usually spend a large amount of money on skin care products after they see their dermatologist. Without guidance, they will likely buy the incorrect products. If they buy the wrong product, you save them the hassle of having to make another office visit and the aggravation of the side effects from the incorrect product. These are often of poor quality or not appropriate for their skin issues. Counterfeit products are rampant on the Internet and many new companies tout worthless products with stem cells and other nonsense. Only you can help your patients make sure that money is spent on the proper products.
Conclusion
Do you really want someone else giving your patients skin care advice? Your patients deserve to have someone with your insights, knowledge, compassion, and honesty help them achieve optimal skin health through use of the proper cosmeceuticals and prescription medications. It is up to you and your staff to save your patients from falling prey to persuasive salespeople with no scientific knowledge or concern for long-term skin health.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever. Dr. Baumann also developed and owns the Baumann Skin Type Solution skin typing systems and related products.
• Most skin care products that patients buy are not appropriate for their skin issues.
• Dermatologists have the most knowledge and insights to prescribe skin care.
• Giving specific skin care instructions helps improve communication.
• Increased communication improves outcomes.

Targeting vagal activity could improve breast cancer survival
ATLANTA – Vagal activity predicts survival in patients with metastatic or recurrent breast cancer, a study showed.
The findings are intriguing, given that vagal activity is modifiable, according to Dr. David Spiegel, Willson Professor of Psychiatry and Behavioral Sciences and director of the center on stress and health at Stanford (Calif.) University.

The study, conducted by Dr. Spiegel and his colleagues, is one of several that together are beginning to elucidate the connections among sleep, stress, and vagal tone, and the effects these factors have on cancer outcomes. For example, in one earlier study of metastatic breast cancer patients, the group demonstrated that good sleep efficiency predicted longer survival (Sleep. 2014 May 1;37[5]:837-42).
“There’s something about sleep that we think has an effect on disease progression,” Dr. Spiegel said at the annual meeting of the American Psychiatric Association.
In another study, the team showed that breast cancer patients who slept better at night had better vagal tone the following morning.
“We all kind of know that a problem that has been keeping you from getting to sleep or worrying you a lot the night before suddenly seems more soluble in the morning after you’ve had a good night’s sleep. You’re better able to self-soothe in the morning,” he said, noting the importance of this evidence that “sleep improves vagal tone.”
Heart rate variability is a good measure of vagal tone, vagal activity, and the ability to self-soothe, he explained, noting that heart rate variability also predicts longer survival with cardiac disease; it seems to reduce the risk of fatal arrhythmias, and also predicts recovery from myocardial infarction.
Others have suggested that it might have an effect on cancer, and there seems to be a link between vagal activity and inflammatory processes, Dr. Spiegel said.
“There is reason to think that poor heart rate variability might be associated with cancer progression as well, and that’s what we wanted to study in a group of metastatic cancer patients,” he said.
Dr. Spiegel and his colleagues measured high-frequency heart rate variability (HF-HRV), which appears to be the best measure of parasympathetic tone, has been associated with longer survival in humans and animals, and is related to immune system functioning.
“We hypothesized that higher heart rate variability would predict longer survival in patients with MRBC [metastatic or recurrent breast cancer],” he said.
In 87 patients with metastases to bone, skin, or viscera who underwent a variety of stress measures, including a 5-minute resting baseline electrocardiogram, 43 had higher HF-HRV, and 44 had lower HF-HRV. Higher baseline HF-HRV did, indeed, predict significantly longer survival (hazard ratio, 0.75).
“The main hypothesis was confirmed – that patients with better vagal tone, higher high-frequency heart rate variability had significantly longer survival over the ensuing 7 years, compared with the patients who had poorer heart rate variability, poorer vagal tone,” he said (Psychosom Med. 2015;77[4]:346-55).
Visceral metastasis status and baseline heart rate both were related to HF-HRV and survival, and the combination of HF-HRV and heart rate further improved survival prediction (HR, 0.64), he noted.
“This is basically coactivation of higher parasympathetic and lower sympathetic activity related to longer survival,” he explained.
Reconstructive surgery, the presence of visceral metastases, and sleep efficiency each were found to be associated with heart rate variability; thus several analyses were conducted “to try to disentangle these relationships and determine what the major variables were that predicted survival,” Dr. Spiegel said.
“It turns out that heart rate variability and visceral metastases were significantly related, and heart rate variability did not predict survival,” he said, explaining that those with visceral metastases (and therefore, cancer with a much poorer prognosis) died sooner, but heart rate variability didn’t make much of a difference. “Where we saw the heart rate variability effect was among those with better prognosis.”
A combination measure of high heart rate variability (“a pretty pure measure of vagal activity, not sympathetic activity”) and low heart rate (“more driven by the sympathetic adrenal-medullary system”) is an even stronger predictor of overall survival, he said.
This suggests that autonomic nervous system variables play a strong role in predicting overall cancer survival, Dr. Spiegel said, noting that depression was not a confounder.
“This is important, because we have, in other studies, found that depression is associated with lower heart rate variability, as you might expect,” he said. In fact, depression has been found to predict shorter survival in cancer patients over a period of 10 years. In an earlier study, cancer patients with worsening depression in the first year died sooner than those with depression that improved during the first year (J Clin Oncol. 2011;29[4]:413-20).
“The median survival difference was about 2 years, so this is not a trivial difference in overall survival,” he said, stressing that the finding is based on more chronic and severe depression.
Other studies have demonstrated relationships between circadian rhythm disruption and cancer survival. In one such study involving patients with metastatic colorectal cancer, circadian rhythm/rest activity cycle (more activity during the day, more rest at night) was associated with better quality of life and predicted survival.
“This has been shown now in several cancers, and it’s clear that a combination of higher activity and better sleep predict longer survival with different kinds of cancers,” Dr. Spiegel said, explaining that a hallmark of a healthy hypothalamic-pituitary-adrenal axis is good diurnal variation of cortisol with high levels in the morning and declining levels throughout the day.
Women with metastatic breast cancer and poorer survival tended to have flat or increasing levels of cortisol throughout the day, he said, adding that the same was true in a study of patients with lung cancer, and that there is evidence that those among them with flatter, more abnormal cortisol patterns throughout the day also have shorter survival.
Animal studies suggest that cortisol might directly suppress the activity of tumor suppressor genes, he explained, noting that there is also increasing evidence of autonomic dysregulation effects on inflammatory processes associated with tumor growth.
“Some basic research on this in animal models shows that if you block adrenergic arousal, you can block the growth of blood vessels from tumors. This has led some people to look at the use of antiadrenergic drugs like the beta-blocker propranolol, and it turned out – to everyone’s surprise – that breast cancer patients who happened to be on beta-blockers for hypertension actually lived longer than those who didn’t,” he said.
This adds to the growing evidence that dysregulation in the sympathetic and parasympathetic systems have effects on survival, he said.
A look at another factor related to sleep disruption – bedtime misalignment – showed that patients who adhere to their preferred sleep pattern, and who are therefore sleeping better, had a difference in disease-free interval; those whose bedtime was misaligned had a shorter time between diagnosis and disease recurrence (Chronobiol Int. 2014 Mar 31[2]214-21).
“Disease-free interval is a very strong predictor of ultimate overall survival, so there seems to be another relationship here between circadian disruption and disease progression in breast cancer,” Dr. Spiegel said.
Taken together, these findings demonstrate a strong association between vagal activity and survival in patients with metastatic or recurrent breast cancer, extending the known predictive window of HF-HRV beyond palliative care to cancer, Dr. Spiegel said.
“Vagal activity can be altered through behavioral, pharmacological, and surgical interventions and thus may be a promising target for increasing survival in patients with metastatic cancer,” he said.
Dr. Spiegel’s studies were funded by the National Cancer Institute and the National Institute on Aging.
ATLANTA – Vagal activity predicts survival in patients with metastatic or recurrent breast cancer, a study showed.
The findings are intriguing, given that vagal activity is modifiable, according to Dr. David Spiegel, Willson Professor of Psychiatry and Behavioral Sciences and director of the center on stress and health at Stanford (Calif.) University.
The study, conducted by Dr. Spiegel and his colleagues, is one of several that together are beginning to elucidate the connections among sleep, stress, and vagal tone, and the effects these factors have on cancer outcomes. For example, in one earlier study of metastatic breast cancer patients, the group demonstrated that good sleep efficiency predicted longer survival (Sleep. 2014 May 1;37[5]:837-42).
“There’s something about sleep that we think has an effect on disease progression,” Dr. Spiegel said at the annual meeting of the American Psychiatric Association.
In another study, the team showed that breast cancer patients who slept better at night had better vagal tone the following morning.
“We all kind of know that a problem that has been keeping you from getting to sleep or worrying you a lot the night before suddenly seems more soluble in the morning after you’ve had a good night’s sleep. You’re better able to self-soothe in the morning,” he said, noting the importance of this evidence that “sleep improves vagal tone.”
Heart rate variability is a good measure of vagal tone, vagal activity, and the ability to self-soothe, he explained, noting that heart rate variability also predicts longer survival with cardiac disease; it seems to reduce the risk of fatal arrhythmias, and also predicts recovery from myocardial infarction.
Others have suggested that it might have an effect on cancer, and there seems to be a link between vagal activity and inflammatory processes, Dr. Spiegel said.
“There is reason to think that poor heart rate variability might be associated with cancer progression as well, and that’s what we wanted to study in a group of metastatic cancer patients,” he said.
Dr. Spiegel and his colleagues measured high-frequency heart rate variability (HF-HRV), which appears to be the best measure of parasympathetic tone, has been associated with longer survival in humans and animals, and is related to immune system functioning.
“We hypothesized that higher heart rate variability would predict longer survival in patients with MRBC [metastatic or recurrent breast cancer],” he said.
In 87 patients with metastases to bone, skin, or viscera who underwent a variety of stress measures, including a 5-minute resting baseline electrocardiogram, 43 had higher HF-HRV, and 44 had lower HF-HRV. Higher baseline HF-HRV did, indeed, predict significantly longer survival (hazard ratio, 0.75).
“The main hypothesis was confirmed – that patients with better vagal tone, higher high-frequency heart rate variability had significantly longer survival over the ensuing 7 years, compared with the patients who had poorer heart rate variability, poorer vagal tone,” he said (Psychosom Med. 2015;77[4]:346-55).
Visceral metastasis status and baseline heart rate both were related to HF-HRV and survival, and the combination of HF-HRV and heart rate further improved survival prediction (HR, 0.64), he noted.
“This is basically coactivation of higher parasympathetic and lower sympathetic activity related to longer survival,” he explained.
Reconstructive surgery, the presence of visceral metastases, and sleep efficiency each were found to be associated with heart rate variability; thus several analyses were conducted “to try to disentangle these relationships and determine what the major variables were that predicted survival,” Dr. Spiegel said.
“It turns out that heart rate variability and visceral metastases were significantly related, and heart rate variability did not predict survival,” he said, explaining that those with visceral metastases (and therefore, cancer with a much poorer prognosis) died sooner, but heart rate variability didn’t make much of a difference. “Where we saw the heart rate variability effect was among those with better prognosis.”
A combination measure of high heart rate variability (“a pretty pure measure of vagal activity, not sympathetic activity”) and low heart rate (“more driven by the sympathetic adrenal-medullary system”) is an even stronger predictor of overall survival, he said.
This suggests that autonomic nervous system variables play a strong role in predicting overall cancer survival, Dr. Spiegel said, noting that depression was not a confounder.
“This is important, because we have, in other studies, found that depression is associated with lower heart rate variability, as you might expect,” he said. In fact, depression has been found to predict shorter survival in cancer patients over a period of 10 years. In an earlier study, cancer patients with worsening depression in the first year died sooner than those with depression that improved during the first year (J Clin Oncol. 2011;29[4]:413-20).
“The median survival difference was about 2 years, so this is not a trivial difference in overall survival,” he said, stressing that the finding is based on more chronic and severe depression.
Other studies have demonstrated relationships between circadian rhythm disruption and cancer survival. In one such study involving patients with metastatic colorectal cancer, circadian rhythm/rest activity cycle (more activity during the day, more rest at night) was associated with better quality of life and predicted survival.
“This has been shown now in several cancers, and it’s clear that a combination of higher activity and better sleep predict longer survival with different kinds of cancers,” Dr. Spiegel said, explaining that a hallmark of a healthy hypothalamic-pituitary-adrenal axis is good diurnal variation of cortisol with high levels in the morning and declining levels throughout the day.
Women with metastatic breast cancer and poorer survival tended to have flat or increasing levels of cortisol throughout the day, he said, adding that the same was true in a study of patients with lung cancer, and that there is evidence that those among them with flatter, more abnormal cortisol patterns throughout the day also have shorter survival.
Animal studies suggest that cortisol might directly suppress the activity of tumor suppressor genes, he explained, noting that there is also increasing evidence of autonomic dysregulation effects on inflammatory processes associated with tumor growth.
“Some basic research on this in animal models shows that if you block adrenergic arousal, you can block the growth of blood vessels from tumors. This has led some people to look at the use of antiadrenergic drugs like the beta-blocker propranolol, and it turned out – to everyone’s surprise – that breast cancer patients who happened to be on beta-blockers for hypertension actually lived longer than those who didn’t,” he said.
This adds to the growing evidence that dysregulation in the sympathetic and parasympathetic systems have effects on survival, he said.
A look at another factor related to sleep disruption – bedtime misalignment – showed that patients who adhere to their preferred sleep pattern, and who are therefore sleeping better, had a difference in disease-free interval; those whose bedtime was misaligned had a shorter time between diagnosis and disease recurrence (Chronobiol Int. 2014 Mar 31[2]214-21).
“Disease-free interval is a very strong predictor of ultimate overall survival, so there seems to be another relationship here between circadian disruption and disease progression in breast cancer,” Dr. Spiegel said.
Taken together, these findings demonstrate a strong association between vagal activity and survival in patients with metastatic or recurrent breast cancer, extending the known predictive window of HF-HRV beyond palliative care to cancer, Dr. Spiegel said.
“Vagal activity can be altered through behavioral, pharmacological, and surgical interventions and thus may be a promising target for increasing survival in patients with metastatic cancer,” he said.
Dr. Spiegel’s studies were funded by the National Cancer Institute and the National Institute on Aging.
ATLANTA – Vagal activity predicts survival in patients with metastatic or recurrent breast cancer, a study showed.
The findings are intriguing, given that vagal activity is modifiable, according to Dr. David Spiegel, Willson Professor of Psychiatry and Behavioral Sciences and director of the center on stress and health at Stanford (Calif.) University.
The study, conducted by Dr. Spiegel and his colleagues, is one of several that together are beginning to elucidate the connections among sleep, stress, and vagal tone, and the effects these factors have on cancer outcomes. For example, in one earlier study of metastatic breast cancer patients, the group demonstrated that good sleep efficiency predicted longer survival (Sleep. 2014 May 1;37[5]:837-42).
“There’s something about sleep that we think has an effect on disease progression,” Dr. Spiegel said at the annual meeting of the American Psychiatric Association.
In another study, the team showed that breast cancer patients who slept better at night had better vagal tone the following morning.
“We all kind of know that a problem that has been keeping you from getting to sleep or worrying you a lot the night before suddenly seems more soluble in the morning after you’ve had a good night’s sleep. You’re better able to self-soothe in the morning,” he said, noting the importance of this evidence that “sleep improves vagal tone.”
Heart rate variability is a good measure of vagal tone, vagal activity, and the ability to self-soothe, he explained, noting that heart rate variability also predicts longer survival with cardiac disease; it seems to reduce the risk of fatal arrhythmias, and also predicts recovery from myocardial infarction.
Others have suggested that it might have an effect on cancer, and there seems to be a link between vagal activity and inflammatory processes, Dr. Spiegel said.
“There is reason to think that poor heart rate variability might be associated with cancer progression as well, and that’s what we wanted to study in a group of metastatic cancer patients,” he said.
Dr. Spiegel and his colleagues measured high-frequency heart rate variability (HF-HRV), which appears to be the best measure of parasympathetic tone, has been associated with longer survival in humans and animals, and is related to immune system functioning.
“We hypothesized that higher heart rate variability would predict longer survival in patients with MRBC [metastatic or recurrent breast cancer],” he said.
In 87 patients with metastases to bone, skin, or viscera who underwent a variety of stress measures, including a 5-minute resting baseline electrocardiogram, 43 had higher HF-HRV, and 44 had lower HF-HRV. Higher baseline HF-HRV did, indeed, predict significantly longer survival (hazard ratio, 0.75).
“The main hypothesis was confirmed – that patients with better vagal tone, higher high-frequency heart rate variability had significantly longer survival over the ensuing 7 years, compared with the patients who had poorer heart rate variability, poorer vagal tone,” he said (Psychosom Med. 2015;77[4]:346-55).
Visceral metastasis status and baseline heart rate both were related to HF-HRV and survival, and the combination of HF-HRV and heart rate further improved survival prediction (HR, 0.64), he noted.
“This is basically coactivation of higher parasympathetic and lower sympathetic activity related to longer survival,” he explained.
Reconstructive surgery, the presence of visceral metastases, and sleep efficiency each were found to be associated with heart rate variability; thus several analyses were conducted “to try to disentangle these relationships and determine what the major variables were that predicted survival,” Dr. Spiegel said.
“It turns out that heart rate variability and visceral metastases were significantly related, and heart rate variability did not predict survival,” he said, explaining that those with visceral metastases (and therefore, cancer with a much poorer prognosis) died sooner, but heart rate variability didn’t make much of a difference. “Where we saw the heart rate variability effect was among those with better prognosis.”
A combination measure of high heart rate variability (“a pretty pure measure of vagal activity, not sympathetic activity”) and low heart rate (“more driven by the sympathetic adrenal-medullary system”) is an even stronger predictor of overall survival, he said.
This suggests that autonomic nervous system variables play a strong role in predicting overall cancer survival, Dr. Spiegel said, noting that depression was not a confounder.
“This is important, because we have, in other studies, found that depression is associated with lower heart rate variability, as you might expect,” he said. In fact, depression has been found to predict shorter survival in cancer patients over a period of 10 years. In an earlier study, cancer patients with worsening depression in the first year died sooner than those with depression that improved during the first year (J Clin Oncol. 2011;29[4]:413-20).
“The median survival difference was about 2 years, so this is not a trivial difference in overall survival,” he said, stressing that the finding is based on more chronic and severe depression.
Other studies have demonstrated relationships between circadian rhythm disruption and cancer survival. In one such study involving patients with metastatic colorectal cancer, circadian rhythm/rest activity cycle (more activity during the day, more rest at night) was associated with better quality of life and predicted survival.
“This has been shown now in several cancers, and it’s clear that a combination of higher activity and better sleep predict longer survival with different kinds of cancers,” Dr. Spiegel said, explaining that a hallmark of a healthy hypothalamic-pituitary-adrenal axis is good diurnal variation of cortisol with high levels in the morning and declining levels throughout the day.
Women with metastatic breast cancer and poorer survival tended to have flat or increasing levels of cortisol throughout the day, he said, adding that the same was true in a study of patients with lung cancer, and that there is evidence that those among them with flatter, more abnormal cortisol patterns throughout the day also have shorter survival.
Animal studies suggest that cortisol might directly suppress the activity of tumor suppressor genes, he explained, noting that there is also increasing evidence of autonomic dysregulation effects on inflammatory processes associated with tumor growth.
“Some basic research on this in animal models shows that if you block adrenergic arousal, you can block the growth of blood vessels from tumors. This has led some people to look at the use of antiadrenergic drugs like the beta-blocker propranolol, and it turned out – to everyone’s surprise – that breast cancer patients who happened to be on beta-blockers for hypertension actually lived longer than those who didn’t,” he said.
This adds to the growing evidence that dysregulation in the sympathetic and parasympathetic systems have effects on survival, he said.
A look at another factor related to sleep disruption – bedtime misalignment – showed that patients who adhere to their preferred sleep pattern, and who are therefore sleeping better, had a difference in disease-free interval; those whose bedtime was misaligned had a shorter time between diagnosis and disease recurrence (Chronobiol Int. 2014 Mar 31[2]214-21).
“Disease-free interval is a very strong predictor of ultimate overall survival, so there seems to be another relationship here between circadian disruption and disease progression in breast cancer,” Dr. Spiegel said.
Taken together, these findings demonstrate a strong association between vagal activity and survival in patients with metastatic or recurrent breast cancer, extending the known predictive window of HF-HRV beyond palliative care to cancer, Dr. Spiegel said.
“Vagal activity can be altered through behavioral, pharmacological, and surgical interventions and thus may be a promising target for increasing survival in patients with metastatic cancer,” he said.
Dr. Spiegel’s studies were funded by the National Cancer Institute and the National Institute on Aging.
AT THE APA ANNUAL MEETING
Key clinical point: Vagal activity predicted survival in patients with metastatic or recurrent breast cancer.
Major finding: Higher baseline HF-HRV predicted significantly longer survival (hazard ratio, 0.75).
Data source: A study of 87 patients with metastatic or recurrent breast cancer.
Disclosures: Dr. Spiegel’s studies were funded by the National Cancer Institute and the National Institute on Aging.

Book offers even-handed, scholarly treatment of AA
Several years ago, the British psychoanalyst Enid Balint suggested that people might not necessarily emerge from a group experience less neurotic or psychotic, but invariably, they were more mature.1 Her suggestion is consistent with my clinical experience. Some of the most admirable and mature patients with whom I have worked have been individuals who participated regularly in 12-step meetings as part of their treatment and recovery.
In this book, “What is Alcoholics Anonymous? A Path From Addiction to Recovery,” (Oxford University Press, 2016), Dr. Marc Galanter, a distinguished scholar and clinician, provides a basis to understand how this occurs. He has devoted a better part of his career to the study and treatment of addictive disorders with a special interest in how 12-step groups curtail the unbridled drinking of alcoholic individuals and stimulate a process for their growth and recovery.

Despite the volumes written about AA, Dr. Galanter maintains that there are few if any scholarly accounts that explain how it works, and how it benefits those who attend. He believes that this book will help the alcoholic who wonders whether AA is for him or her, as it will guide stymied family members and friends who wonder what help to offer, as well as health professionals who need a “coherent and objective sense of what the fellowship is about.”
Dr. Galanter invites the reader to witness the changes he and others have observed that occur in the lives of alcoholics with their encounters and immersion in AA. Allowance is made that not all who try the program benefit or continue, but for those who do, the change and help of AA, as Dr. Galanter repeatedly provides compelling data and touching examples, are transformative.
He offers that an informed appreciation of what AA is about, and most importantly, guides alcoholic individuals to understand how AA can help them, as well as assist their family and friends.

The book also provides health professionals and the public an awareness of essential aspects of the program that meet the needs of alcoholic individuals. Dr. Galanter benefits the reader by providing a brief background on the beginnings of AA, how it governs itself, and the different pathways by which its participants achieve recovery. Helpful chapters addressing controversies such as the God concept and whether alcoholism is a disease are balanced and illuminating, as are the chapters that review the 12 steps and the process of engagement.
He also provides a balanced explanation of the spiritual elements of the program, the different form they take, and how the program helps those who can draw on those elements. In addition, Dr. Galanter reviews the evidence that AA changes the brain, and finally, he concludes with a scholarly consideration and formulation on the effectiveness of AA. His even-handed, scholarly assessment of these issues is refreshing and welcome, given recent polarizations and controversies about the effectiveness AA.
As Dr. Galanter and others see and understand the culture, it is a bottom-up, democratic one in which the guides, and wisdom for sobriety and recovery for its members are garnered from each other and not from authorities on high. To his credit, Dr. Galanter avoids doctrinaire views on rules for participation, such as number and frequency of meetings (in contrast to rigid and strident rules advocated by some members), but describes flexible and alternative ways individuals participate and benefit. Dr. Galanter brings the reader to this experience with his own erudite and investigative accounts, but just as compelling, through the absorbing words and experiences of the many who have experienced the help and wisdom of the program.
I find little to criticize about this book on the merits. It is lucid and well written, and it will be instructive for all who take it up. Dr. Galanter clearly succeeds in getting to the main audiences he targets, namely those who wonder whether AA is for them, for family and friends who worry about those with the condition, and for clinicians who want guidance in how and why the program works. But ultimately, the book will profit anyone who wants a better insight into how AA achieves its successes.
References
1. Int J Psychoanal. 1972;53[1]61-5.
Dr. Khantzian is a professor of psychiatry, Harvard Medical School, Boston, and past president of the American Academy of Addiction Psychiatry.
Several years ago, the British psychoanalyst Enid Balint suggested that people might not necessarily emerge from a group experience less neurotic or psychotic, but invariably, they were more mature.1 Her suggestion is consistent with my clinical experience. Some of the most admirable and mature patients with whom I have worked have been individuals who participated regularly in 12-step meetings as part of their treatment and recovery.
In this book, “What is Alcoholics Anonymous? A Path From Addiction to Recovery,” (Oxford University Press, 2016), Dr. Marc Galanter, a distinguished scholar and clinician, provides a basis to understand how this occurs. He has devoted a better part of his career to the study and treatment of addictive disorders with a special interest in how 12-step groups curtail the unbridled drinking of alcoholic individuals and stimulate a process for their growth and recovery.
Despite the volumes written about AA, Dr. Galanter maintains that there are few if any scholarly accounts that explain how it works, and how it benefits those who attend. He believes that this book will help the alcoholic who wonders whether AA is for him or her, as it will guide stymied family members and friends who wonder what help to offer, as well as health professionals who need a “coherent and objective sense of what the fellowship is about.”
Dr. Galanter invites the reader to witness the changes he and others have observed that occur in the lives of alcoholics with their encounters and immersion in AA. Allowance is made that not all who try the program benefit or continue, but for those who do, the change and help of AA, as Dr. Galanter repeatedly provides compelling data and touching examples, are transformative.
He offers that an informed appreciation of what AA is about, and most importantly, guides alcoholic individuals to understand how AA can help them, as well as assist their family and friends.
The book also provides health professionals and the public an awareness of essential aspects of the program that meet the needs of alcoholic individuals. Dr. Galanter benefits the reader by providing a brief background on the beginnings of AA, how it governs itself, and the different pathways by which its participants achieve recovery. Helpful chapters addressing controversies such as the God concept and whether alcoholism is a disease are balanced and illuminating, as are the chapters that review the 12 steps and the process of engagement.
He also provides a balanced explanation of the spiritual elements of the program, the different form they take, and how the program helps those who can draw on those elements. In addition, Dr. Galanter reviews the evidence that AA changes the brain, and finally, he concludes with a scholarly consideration and formulation on the effectiveness of AA. His even-handed, scholarly assessment of these issues is refreshing and welcome, given recent polarizations and controversies about the effectiveness AA.
As Dr. Galanter and others see and understand the culture, it is a bottom-up, democratic one in which the guides, and wisdom for sobriety and recovery for its members are garnered from each other and not from authorities on high. To his credit, Dr. Galanter avoids doctrinaire views on rules for participation, such as number and frequency of meetings (in contrast to rigid and strident rules advocated by some members), but describes flexible and alternative ways individuals participate and benefit. Dr. Galanter brings the reader to this experience with his own erudite and investigative accounts, but just as compelling, through the absorbing words and experiences of the many who have experienced the help and wisdom of the program.
I find little to criticize about this book on the merits. It is lucid and well written, and it will be instructive for all who take it up. Dr. Galanter clearly succeeds in getting to the main audiences he targets, namely those who wonder whether AA is for them, for family and friends who worry about those with the condition, and for clinicians who want guidance in how and why the program works. But ultimately, the book will profit anyone who wants a better insight into how AA achieves its successes.
References
1. Int J Psychoanal. 1972;53[1]61-5.
Dr. Khantzian is a professor of psychiatry, Harvard Medical School, Boston, and past president of the American Academy of Addiction Psychiatry.
Several years ago, the British psychoanalyst Enid Balint suggested that people might not necessarily emerge from a group experience less neurotic or psychotic, but invariably, they were more mature.1 Her suggestion is consistent with my clinical experience. Some of the most admirable and mature patients with whom I have worked have been individuals who participated regularly in 12-step meetings as part of their treatment and recovery.
In this book, “What is Alcoholics Anonymous? A Path From Addiction to Recovery,” (Oxford University Press, 2016), Dr. Marc Galanter, a distinguished scholar and clinician, provides a basis to understand how this occurs. He has devoted a better part of his career to the study and treatment of addictive disorders with a special interest in how 12-step groups curtail the unbridled drinking of alcoholic individuals and stimulate a process for their growth and recovery.
Despite the volumes written about AA, Dr. Galanter maintains that there are few if any scholarly accounts that explain how it works, and how it benefits those who attend. He believes that this book will help the alcoholic who wonders whether AA is for him or her, as it will guide stymied family members and friends who wonder what help to offer, as well as health professionals who need a “coherent and objective sense of what the fellowship is about.”
Dr. Galanter invites the reader to witness the changes he and others have observed that occur in the lives of alcoholics with their encounters and immersion in AA. Allowance is made that not all who try the program benefit or continue, but for those who do, the change and help of AA, as Dr. Galanter repeatedly provides compelling data and touching examples, are transformative.
He offers that an informed appreciation of what AA is about, and most importantly, guides alcoholic individuals to understand how AA can help them, as well as assist their family and friends.
The book also provides health professionals and the public an awareness of essential aspects of the program that meet the needs of alcoholic individuals. Dr. Galanter benefits the reader by providing a brief background on the beginnings of AA, how it governs itself, and the different pathways by which its participants achieve recovery. Helpful chapters addressing controversies such as the God concept and whether alcoholism is a disease are balanced and illuminating, as are the chapters that review the 12 steps and the process of engagement.
He also provides a balanced explanation of the spiritual elements of the program, the different form they take, and how the program helps those who can draw on those elements. In addition, Dr. Galanter reviews the evidence that AA changes the brain, and finally, he concludes with a scholarly consideration and formulation on the effectiveness of AA. His even-handed, scholarly assessment of these issues is refreshing and welcome, given recent polarizations and controversies about the effectiveness AA.
As Dr. Galanter and others see and understand the culture, it is a bottom-up, democratic one in which the guides, and wisdom for sobriety and recovery for its members are garnered from each other and not from authorities on high. To his credit, Dr. Galanter avoids doctrinaire views on rules for participation, such as number and frequency of meetings (in contrast to rigid and strident rules advocated by some members), but describes flexible and alternative ways individuals participate and benefit. Dr. Galanter brings the reader to this experience with his own erudite and investigative accounts, but just as compelling, through the absorbing words and experiences of the many who have experienced the help and wisdom of the program.
I find little to criticize about this book on the merits. It is lucid and well written, and it will be instructive for all who take it up. Dr. Galanter clearly succeeds in getting to the main audiences he targets, namely those who wonder whether AA is for them, for family and friends who worry about those with the condition, and for clinicians who want guidance in how and why the program works. But ultimately, the book will profit anyone who wants a better insight into how AA achieves its successes.
References
1. Int J Psychoanal. 1972;53[1]61-5.
Dr. Khantzian is a professor of psychiatry, Harvard Medical School, Boston, and past president of the American Academy of Addiction Psychiatry.

Hungry and obese
Does it seem strange to you that while on one hand we hear from multiple sources that a troubling number of adults and children are going hungry, but on the other hand data from the Centers for Disease Control and Prevention indicate that self reports of obesity by adults in 2014 in different ethnic groups ranged from 27% to 38% and approximately 17% of children and adolescents aged 2-19 years are obese per 2011-2012 data?
One might guess that this situation is simply that too many Americans can afford to overeat and their numbers overwhelm the data from a smaller segment of the population who are underweight because they can’t afford to feed themselves.
But this isn’t the case at all, because the obesity rates in our poorest counties are nearly 12% above the national median (“Obesity: The New Hunger” by Robert Paarlberg, Ph.D., The Wall Street Journal, May 10, 2016). So the question is why do we have so many overweight adults and children if we also have a hunger problem? The answer is very complicated and even more complicated because of how we define hunger.
While obesity is relatively easy to measure with a scale and a tape measure, hunger is a perception that is difficult, if not impossible, to quantify. Possibly in an attempt to create clarity for people like me who are confused by the coexistence of hunger and obesity, there has been a trend toward replacing “hunger” with the more techno-sounding buzz words, “food insecurity.”
According to Dr. Paarlberg, an adjunct professor of public policy at Harvard University, Cambridge, Mass., and author of “The United States of Excess: Gluttony and the Dark Side of American Exceptionalism” (New York, N.Y.: Oxford University Press, 2015), the United States Department of Agriculture calculates our national food insecurity quotient by way of an annual survey of a sample of households. Family members are asked questions such as whether “they had failed to eat or worried about running out of food for lack of money at any time in the previous 12 months.”

There are many reasons why a survey respondent might be concerned that he or she wouldn’t have enough to eat on a given day. It could have been poor planning on the part of the head of the household or a consequence of family chaos. And we have to assume that in some cases, it is simply because there wasn’t enough money to buy food that day. If it were only a matter of money, the solution would be easy. We could simply provide economically challenged families with more money to buy more food, but that is already being done through programs such as the Supplemental Nutrition Assistance Program (often referred to as food stamps or SNAP). But the coexistence of obesity and hunger suggests to me that more food isn’t the answer.
Part of the problem is that “food” is too broadly defined. Some foods are more likely to contribute to obesity than others, and some foods satiate more quickly than others. While some restrictions have been built into the SNAP program to encourage participants to eat a healthier diet, the fact that soda and candy can be bought with food stamps is a serious error that must be corrected. It may be time to take a harder look at tightening other guidelines to make the subsidized diet healthier.
Unfortunately, the last step in the process occurs in the home. A diet that discourages obesity often includes fresh fruits and vegetables that can be expensive and may not be appealing to a family accustomed to calorie-dense foods. And a healthy diet often requires preparation skills and time, both of which economically challenged families may not have.
All of this makes me wonder whether we should stop worrying so much about hunger in America and shift the focus of our nutritional support programs more toward obesity prevention. Of course, that is easy to say for someone like myself who is lean and can always find something in the refrigerator to eat. But let’s remember that while starvation and obesity can kill, hunger doesn’t.
The problem is that “hunger” and the less emotionally charged term “food insecurity” are potent motivators for legislators who control the funding of our critical nutritional support programs. It still makes sense politically to continue to talk about eliminating hunger. But we need to craft our programs so that they address the larger problem of obesity.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
Does it seem strange to you that while on one hand we hear from multiple sources that a troubling number of adults and children are going hungry, but on the other hand data from the Centers for Disease Control and Prevention indicate that self reports of obesity by adults in 2014 in different ethnic groups ranged from 27% to 38% and approximately 17% of children and adolescents aged 2-19 years are obese per 2011-2012 data?
One might guess that this situation is simply that too many Americans can afford to overeat and their numbers overwhelm the data from a smaller segment of the population who are underweight because they can’t afford to feed themselves.
But this isn’t the case at all, because the obesity rates in our poorest counties are nearly 12% above the national median (“Obesity: The New Hunger” by Robert Paarlberg, Ph.D., The Wall Street Journal, May 10, 2016). So the question is why do we have so many overweight adults and children if we also have a hunger problem? The answer is very complicated and even more complicated because of how we define hunger.
While obesity is relatively easy to measure with a scale and a tape measure, hunger is a perception that is difficult, if not impossible, to quantify. Possibly in an attempt to create clarity for people like me who are confused by the coexistence of hunger and obesity, there has been a trend toward replacing “hunger” with the more techno-sounding buzz words, “food insecurity.”
According to Dr. Paarlberg, an adjunct professor of public policy at Harvard University, Cambridge, Mass., and author of “The United States of Excess: Gluttony and the Dark Side of American Exceptionalism” (New York, N.Y.: Oxford University Press, 2015), the United States Department of Agriculture calculates our national food insecurity quotient by way of an annual survey of a sample of households. Family members are asked questions such as whether “they had failed to eat or worried about running out of food for lack of money at any time in the previous 12 months.”
There are many reasons why a survey respondent might be concerned that he or she wouldn’t have enough to eat on a given day. It could have been poor planning on the part of the head of the household or a consequence of family chaos. And we have to assume that in some cases, it is simply because there wasn’t enough money to buy food that day. If it were only a matter of money, the solution would be easy. We could simply provide economically challenged families with more money to buy more food, but that is already being done through programs such as the Supplemental Nutrition Assistance Program (often referred to as food stamps or SNAP). But the coexistence of obesity and hunger suggests to me that more food isn’t the answer.
Part of the problem is that “food” is too broadly defined. Some foods are more likely to contribute to obesity than others, and some foods satiate more quickly than others. While some restrictions have been built into the SNAP program to encourage participants to eat a healthier diet, the fact that soda and candy can be bought with food stamps is a serious error that must be corrected. It may be time to take a harder look at tightening other guidelines to make the subsidized diet healthier.
Unfortunately, the last step in the process occurs in the home. A diet that discourages obesity often includes fresh fruits and vegetables that can be expensive and may not be appealing to a family accustomed to calorie-dense foods. And a healthy diet often requires preparation skills and time, both of which economically challenged families may not have.
All of this makes me wonder whether we should stop worrying so much about hunger in America and shift the focus of our nutritional support programs more toward obesity prevention. Of course, that is easy to say for someone like myself who is lean and can always find something in the refrigerator to eat. But let’s remember that while starvation and obesity can kill, hunger doesn’t.
The problem is that “hunger” and the less emotionally charged term “food insecurity” are potent motivators for legislators who control the funding of our critical nutritional support programs. It still makes sense politically to continue to talk about eliminating hunger. But we need to craft our programs so that they address the larger problem of obesity.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
Does it seem strange to you that while on one hand we hear from multiple sources that a troubling number of adults and children are going hungry, but on the other hand data from the Centers for Disease Control and Prevention indicate that self reports of obesity by adults in 2014 in different ethnic groups ranged from 27% to 38% and approximately 17% of children and adolescents aged 2-19 years are obese per 2011-2012 data?
One might guess that this situation is simply that too many Americans can afford to overeat and their numbers overwhelm the data from a smaller segment of the population who are underweight because they can’t afford to feed themselves.
But this isn’t the case at all, because the obesity rates in our poorest counties are nearly 12% above the national median (“Obesity: The New Hunger” by Robert Paarlberg, Ph.D., The Wall Street Journal, May 10, 2016). So the question is why do we have so many overweight adults and children if we also have a hunger problem? The answer is very complicated and even more complicated because of how we define hunger.
While obesity is relatively easy to measure with a scale and a tape measure, hunger is a perception that is difficult, if not impossible, to quantify. Possibly in an attempt to create clarity for people like me who are confused by the coexistence of hunger and obesity, there has been a trend toward replacing “hunger” with the more techno-sounding buzz words, “food insecurity.”
According to Dr. Paarlberg, an adjunct professor of public policy at Harvard University, Cambridge, Mass., and author of “The United States of Excess: Gluttony and the Dark Side of American Exceptionalism” (New York, N.Y.: Oxford University Press, 2015), the United States Department of Agriculture calculates our national food insecurity quotient by way of an annual survey of a sample of households. Family members are asked questions such as whether “they had failed to eat or worried about running out of food for lack of money at any time in the previous 12 months.”
There are many reasons why a survey respondent might be concerned that he or she wouldn’t have enough to eat on a given day. It could have been poor planning on the part of the head of the household or a consequence of family chaos. And we have to assume that in some cases, it is simply because there wasn’t enough money to buy food that day. If it were only a matter of money, the solution would be easy. We could simply provide economically challenged families with more money to buy more food, but that is already being done through programs such as the Supplemental Nutrition Assistance Program (often referred to as food stamps or SNAP). But the coexistence of obesity and hunger suggests to me that more food isn’t the answer.
Part of the problem is that “food” is too broadly defined. Some foods are more likely to contribute to obesity than others, and some foods satiate more quickly than others. While some restrictions have been built into the SNAP program to encourage participants to eat a healthier diet, the fact that soda and candy can be bought with food stamps is a serious error that must be corrected. It may be time to take a harder look at tightening other guidelines to make the subsidized diet healthier.
Unfortunately, the last step in the process occurs in the home. A diet that discourages obesity often includes fresh fruits and vegetables that can be expensive and may not be appealing to a family accustomed to calorie-dense foods. And a healthy diet often requires preparation skills and time, both of which economically challenged families may not have.
All of this makes me wonder whether we should stop worrying so much about hunger in America and shift the focus of our nutritional support programs more toward obesity prevention. Of course, that is easy to say for someone like myself who is lean and can always find something in the refrigerator to eat. But let’s remember that while starvation and obesity can kill, hunger doesn’t.
The problem is that “hunger” and the less emotionally charged term “food insecurity” are potent motivators for legislators who control the funding of our critical nutritional support programs. It still makes sense politically to continue to talk about eliminating hunger. But we need to craft our programs so that they address the larger problem of obesity.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”

People With HIV Are Less Likely To Get Cancer Treatment
We’ve made great progress treating people who are infected with HIV, but if they get cancer they’re less likely to get the care they need, a recent study found.
Researchers examined treatment for a variety of cancers, including upper gastrointestinal tract, colorectal, prostate, lung, head and neck, cervix, breast, anal and two blood cancers. With the exception of anal cancer, treatment rates differed significantly between HIV-infected people and those who weren’t infected, according to the study.
For example, 33 percent of patients with HIV and lung cancer failed to receive any treatment for the cancer compared with 14 percent of those who weren’t infected. Similarly, 44 percent of people who were HIV positive didn’t receive treatment for upper GI cancer versus 18 percent of those who weren’t infected with HIV. Twenty-four percent of men with prostate cancer who were HIV positive didn’t get treatment compared with 7 percent of non-HIV infected men.
Cancer treatment was defined as radiation, chemotherapy and/or surgery.
“To have made such great strides with treating HIV only to have them succumb to cancer is devastating,” said Dr. Gita Suneja, a radiation oncologist at the University of Utah’s Huntsman Cancer Institute in Salt Lake City and the lead author of the study. It was published online this month in the journal Cancer.
The study used the National Cancer Data Base to analyze treatment for adults younger than 65 who were diagnosed with any of the 10 most common cancers to affect HIV patients between 2003 and 2011. The study included 10,265 HIV-infected adults and 2.2 million without HIV.
The data base, which is sponsored by the American Cancer Society and the American College of Surgeons, captures roughly 70 percent of newly diagnosed cancer cases in the United States.
The study noted that more than a third of the patients with HIV had stage 4 cancer — cancer that has metastasized — when they were diagnosed, while only 19 percent of those without HIV did.
Improvements in antiretroviral therapy to treat HIV have helped reduce the incidence of cancers such as Kaposi sarcoma that are closely linked to AIDS, but rates for other cancers often associated with normal aging have increased among HIV patients. In addition, people with HIV have a higher incidence of some lifestyle-related cancers, such as lung cancer, which could be linked to higher rates of smoking. Cancer is now the second most common cause of death among HIV-infected people, behind AIDS-related causes.
HIV patients are more likely to be uninsured or underinsured, and lack of coverage can affect access to cancer care. But having insurance didn’t eliminate the problem: privately insured people with HIV were significantly more likely to be untreated for many cancers than were privately insured people without HIV, the study found.
“We know that people with Medicaid or who are uninsured receive subpar cancer treatment, and that’s a big public health issue,” said Suneja. “But even factoring that in, HIV-infected people are still less likely to receive cancer treatment. That means there are other drivers that we couldn’t measure in the study.”
Disparities in cancer treatment could exist for several reasons. For one thing, for most cancers there are no national treatment guidelines for HIV-infected patients, Suneja said. One of the few exceptions is anal cancer, the only cancer for which the study found little discrepancy in treatment among HIV-infected and non-infected patients. According to the research, the difference among those not receiving treatment was 4.8 percent for HIV patients versus 3.1 percent for others.
For other cancers, “the oncologist may pause and ask, ‘Does the HIV infection mean they shouldn’t get standard cancer treatment?’” Suneja added.
This story was produced by Kaiser Health News, which publishes California Healthline, a service of the California Health Care Foundation.
Please contact Kaiser Health News to send comments or ideas for future topics for the Insuring Your Health column.
We’ve made great progress treating people who are infected with HIV, but if they get cancer they’re less likely to get the care they need, a recent study found.
Researchers examined treatment for a variety of cancers, including upper gastrointestinal tract, colorectal, prostate, lung, head and neck, cervix, breast, anal and two blood cancers. With the exception of anal cancer, treatment rates differed significantly between HIV-infected people and those who weren’t infected, according to the study.
For example, 33 percent of patients with HIV and lung cancer failed to receive any treatment for the cancer compared with 14 percent of those who weren’t infected. Similarly, 44 percent of people who were HIV positive didn’t receive treatment for upper GI cancer versus 18 percent of those who weren’t infected with HIV. Twenty-four percent of men with prostate cancer who were HIV positive didn’t get treatment compared with 7 percent of non-HIV infected men.
Cancer treatment was defined as radiation, chemotherapy and/or surgery.
“To have made such great strides with treating HIV only to have them succumb to cancer is devastating,” said Dr. Gita Suneja, a radiation oncologist at the University of Utah’s Huntsman Cancer Institute in Salt Lake City and the lead author of the study. It was published online this month in the journal Cancer.
The study used the National Cancer Data Base to analyze treatment for adults younger than 65 who were diagnosed with any of the 10 most common cancers to affect HIV patients between 2003 and 2011. The study included 10,265 HIV-infected adults and 2.2 million without HIV.
The data base, which is sponsored by the American Cancer Society and the American College of Surgeons, captures roughly 70 percent of newly diagnosed cancer cases in the United States.
The study noted that more than a third of the patients with HIV had stage 4 cancer — cancer that has metastasized — when they were diagnosed, while only 19 percent of those without HIV did.
Improvements in antiretroviral therapy to treat HIV have helped reduce the incidence of cancers such as Kaposi sarcoma that are closely linked to AIDS, but rates for other cancers often associated with normal aging have increased among HIV patients. In addition, people with HIV have a higher incidence of some lifestyle-related cancers, such as lung cancer, which could be linked to higher rates of smoking. Cancer is now the second most common cause of death among HIV-infected people, behind AIDS-related causes.
HIV patients are more likely to be uninsured or underinsured, and lack of coverage can affect access to cancer care. But having insurance didn’t eliminate the problem: privately insured people with HIV were significantly more likely to be untreated for many cancers than were privately insured people without HIV, the study found.
“We know that people with Medicaid or who are uninsured receive subpar cancer treatment, and that’s a big public health issue,” said Suneja. “But even factoring that in, HIV-infected people are still less likely to receive cancer treatment. That means there are other drivers that we couldn’t measure in the study.”
Disparities in cancer treatment could exist for several reasons. For one thing, for most cancers there are no national treatment guidelines for HIV-infected patients, Suneja said. One of the few exceptions is anal cancer, the only cancer for which the study found little discrepancy in treatment among HIV-infected and non-infected patients. According to the research, the difference among those not receiving treatment was 4.8 percent for HIV patients versus 3.1 percent for others.
For other cancers, “the oncologist may pause and ask, ‘Does the HIV infection mean they shouldn’t get standard cancer treatment?’” Suneja added.
This story was produced by Kaiser Health News, which publishes California Healthline, a service of the California Health Care Foundation.
Please contact Kaiser Health News to send comments or ideas for future topics for the Insuring Your Health column.
We’ve made great progress treating people who are infected with HIV, but if they get cancer they’re less likely to get the care they need, a recent study found.
Researchers examined treatment for a variety of cancers, including upper gastrointestinal tract, colorectal, prostate, lung, head and neck, cervix, breast, anal and two blood cancers. With the exception of anal cancer, treatment rates differed significantly between HIV-infected people and those who weren’t infected, according to the study.
For example, 33 percent of patients with HIV and lung cancer failed to receive any treatment for the cancer compared with 14 percent of those who weren’t infected. Similarly, 44 percent of people who were HIV positive didn’t receive treatment for upper GI cancer versus 18 percent of those who weren’t infected with HIV. Twenty-four percent of men with prostate cancer who were HIV positive didn’t get treatment compared with 7 percent of non-HIV infected men.
Cancer treatment was defined as radiation, chemotherapy and/or surgery.
“To have made such great strides with treating HIV only to have them succumb to cancer is devastating,” said Dr. Gita Suneja, a radiation oncologist at the University of Utah’s Huntsman Cancer Institute in Salt Lake City and the lead author of the study. It was published online this month in the journal Cancer.
The study used the National Cancer Data Base to analyze treatment for adults younger than 65 who were diagnosed with any of the 10 most common cancers to affect HIV patients between 2003 and 2011. The study included 10,265 HIV-infected adults and 2.2 million without HIV.
The data base, which is sponsored by the American Cancer Society and the American College of Surgeons, captures roughly 70 percent of newly diagnosed cancer cases in the United States.
The study noted that more than a third of the patients with HIV had stage 4 cancer — cancer that has metastasized — when they were diagnosed, while only 19 percent of those without HIV did.
Improvements in antiretroviral therapy to treat HIV have helped reduce the incidence of cancers such as Kaposi sarcoma that are closely linked to AIDS, but rates for other cancers often associated with normal aging have increased among HIV patients. In addition, people with HIV have a higher incidence of some lifestyle-related cancers, such as lung cancer, which could be linked to higher rates of smoking. Cancer is now the second most common cause of death among HIV-infected people, behind AIDS-related causes.
HIV patients are more likely to be uninsured or underinsured, and lack of coverage can affect access to cancer care. But having insurance didn’t eliminate the problem: privately insured people with HIV were significantly more likely to be untreated for many cancers than were privately insured people without HIV, the study found.
“We know that people with Medicaid or who are uninsured receive subpar cancer treatment, and that’s a big public health issue,” said Suneja. “But even factoring that in, HIV-infected people are still less likely to receive cancer treatment. That means there are other drivers that we couldn’t measure in the study.”
Disparities in cancer treatment could exist for several reasons. For one thing, for most cancers there are no national treatment guidelines for HIV-infected patients, Suneja said. One of the few exceptions is anal cancer, the only cancer for which the study found little discrepancy in treatment among HIV-infected and non-infected patients. According to the research, the difference among those not receiving treatment was 4.8 percent for HIV patients versus 3.1 percent for others.
For other cancers, “the oncologist may pause and ask, ‘Does the HIV infection mean they shouldn’t get standard cancer treatment?’” Suneja added.
This story was produced by Kaiser Health News, which publishes California Healthline, a service of the California Health Care Foundation.
Please contact Kaiser Health News to send comments or ideas for future topics for the Insuring Your Health column.
VA to Reexamine 24,000 Veterans for TBI
More than 24,000 veterans who received examinations but were not diagnosed with traumatic brain injuries (TBIs) will be eligible for new medical examinations, the VA has announced. Due to confusing guidance documents, the original examinations were not conducted by a psychiatrist, physiatrist, neurosurgeon, or neurologist as mandated by VA policy. The 24,000 veterans may be eligible for additional benefits and service-connected compensation based on the results of the new examinations.
“Traumatic Brain Injury is a signature injury in veterans returning from the conflicts in Iraq and Afghanistan, and VA is proud to be an organization that sets the bar high for supporting these, and all, veterans,” said Secretary of Veterans Affairs Robert McDonald in a statement. “Providing support for veterans suffering from a TBI is a priority and a privilege, and we must make certain they receive a just and fair rating for their disabilities.”
The current VA policy dates to 2007 and requires that a specialist complete a TBI examination when VA does not have a prior diagnosis. However, given the rapidly changing science around TBI since 2007, the VA has issued multiple additional guidance documents. These additional guidance documents, the VA notes, “created confusion regarding the policy.”
“We let these veterans down,” Secretary McDonald said. “That is why we are taking every step necessary to grant equitable relief to those affected to ensure they receive the full benefits to which they are entitled.”
Veterans will not be required to submit new claims and the VA has pledged to contact the identified patients to offer them a new examination. According to the VA > 13,000 veterans are already receiving 10% or higher service-connected compensation benefits for TBI.
More than 24,000 veterans who received examinations but were not diagnosed with traumatic brain injuries (TBIs) will be eligible for new medical examinations, the VA has announced. Due to confusing guidance documents, the original examinations were not conducted by a psychiatrist, physiatrist, neurosurgeon, or neurologist as mandated by VA policy. The 24,000 veterans may be eligible for additional benefits and service-connected compensation based on the results of the new examinations.
“Traumatic Brain Injury is a signature injury in veterans returning from the conflicts in Iraq and Afghanistan, and VA is proud to be an organization that sets the bar high for supporting these, and all, veterans,” said Secretary of Veterans Affairs Robert McDonald in a statement. “Providing support for veterans suffering from a TBI is a priority and a privilege, and we must make certain they receive a just and fair rating for their disabilities.”
The current VA policy dates to 2007 and requires that a specialist complete a TBI examination when VA does not have a prior diagnosis. However, given the rapidly changing science around TBI since 2007, the VA has issued multiple additional guidance documents. These additional guidance documents, the VA notes, “created confusion regarding the policy.”
“We let these veterans down,” Secretary McDonald said. “That is why we are taking every step necessary to grant equitable relief to those affected to ensure they receive the full benefits to which they are entitled.”
Veterans will not be required to submit new claims and the VA has pledged to contact the identified patients to offer them a new examination. According to the VA > 13,000 veterans are already receiving 10% or higher service-connected compensation benefits for TBI.
More than 24,000 veterans who received examinations but were not diagnosed with traumatic brain injuries (TBIs) will be eligible for new medical examinations, the VA has announced. Due to confusing guidance documents, the original examinations were not conducted by a psychiatrist, physiatrist, neurosurgeon, or neurologist as mandated by VA policy. The 24,000 veterans may be eligible for additional benefits and service-connected compensation based on the results of the new examinations.
“Traumatic Brain Injury is a signature injury in veterans returning from the conflicts in Iraq and Afghanistan, and VA is proud to be an organization that sets the bar high for supporting these, and all, veterans,” said Secretary of Veterans Affairs Robert McDonald in a statement. “Providing support for veterans suffering from a TBI is a priority and a privilege, and we must make certain they receive a just and fair rating for their disabilities.”
The current VA policy dates to 2007 and requires that a specialist complete a TBI examination when VA does not have a prior diagnosis. However, given the rapidly changing science around TBI since 2007, the VA has issued multiple additional guidance documents. These additional guidance documents, the VA notes, “created confusion regarding the policy.”
“We let these veterans down,” Secretary McDonald said. “That is why we are taking every step necessary to grant equitable relief to those affected to ensure they receive the full benefits to which they are entitled.”
Veterans will not be required to submit new claims and the VA has pledged to contact the identified patients to offer them a new examination. According to the VA > 13,000 veterans are already receiving 10% or higher service-connected compensation benefits for TBI.
Two-step UTI screening cuts catheterization rate in half
After implementation of a quality improvement initiative to more effectively screen febrile children for urinary tract infections (UTIs) in the emergency department, catheterization rates dropped from 63% to 30% over a 6-month period, a study found.
The sustained drop prevented more than 350 young children from catheterization without increasing revisit rates or missing UTIs in the 39% of children who were followed in the care network. This was in a study that compared catheterization rates in 1,520 children aged 6-24 months in the year before the intervention and 828 children in the 6 months during the intervention.
“Although urine catheterization remains the gold standard in diagnosing UTIs, it is an invasive procedure that may be avoided in most patients who are being screened,” wrote Dr. Jane M. Lavelle of Children’s Hospital of Philadelphia (CHOP) and her associates. UTI screening by this method can be “painful, time consuming, and costly,” they added (Pediatrics. 2016 June 2. doi: 10.1542/peds.2015-3023).

An alternative method to automatic catheterization is a two-step process already included as an option in American Academy of Pediatrics guidelines: Instead of collecting urine through catheterization just once for screening and culture, an emergency department first noninvasively collects urine with a urine bag for screening in those indicated with evidence-based risk factors, and then catheterizes only those who screen positive (Pediatrics. 2011;128[3]:595-610).
“Due to the predictive models’ higher sensitivity than specificity for screening, most urine samples will have a negative screen for pyuria or bacteriuria by urine dipstick or microscopy,” the authors wrote.
At baseline, CHOP’s ED was screening 63% of febrile children under age 24 months using catheterization, but screens were most commonly negative and only 4.3% had positive cultures. The authors therefore initiated a switch to the two-step method as a pilot run in one ED area before educating all ED personnel and expanding to the full department in the second month, using three cycles of Plan-Do-Study-Act protocol.
Children aged 6-24 months comprised approximately 20% of the ED’s more than 90,000 annual patients, and about 22% of these children presented with fever as the primary concern. Children with a history of genitourinary problems or immune deficiency were excluded.
The pilot ran in an “urgent care section of ED where there are typically more children with less complex medical histories and where ‘fever’ is a common complaint,” the investigators said. The staff completed a learning module with assessment and then received in-person and visual reminders of the procedure. Nurse feedback was then used to develop a nursing-specific educational module before expanding the intervention to all ED areas.
While 69% of 828 febrile young children still underwent screening during the 6-month intervention period, only 16% still underwent urethral catheterization as the initial screening step, typically because of strong clinical indications for a UTI. Another 14% underwent catheterization only after a positive urine screen from an initial noninvasive urine collection or because of an inability to get an adequate urine specimen with the bag. The reduction in catheterization dropped to 55% within 2 weeks of the intervention’s start and spread to other hospital departments. The drop to a 30% catheterization rate remained throughout 18 additional months of monitoring.
“Through online education modules, staff meetings, printed and EHR reminders, family involvement, team review of weekly data, individual and group feedback, and nurse scripting, the ED was able to achieve our aim of reducing catheterization rates among febrile young children ages 6-24 months by half over a 6-month period with sustained results,” Dr. Lavelle and her associates reported.
The research did not use external funding, and the researchers reported they had no financial disclosures.
When children are febrile and younger than 24 months old and they go to a pediatric emergency department, a pretty big work-up can be anticipated, taking several hours and including a test for possible UTI.

|
Dr. Michael E. Pichichero |
In this report from investigators at Children’s Hospital of Philadelphia, we learn the results of a study undertaken to reduce the frequency of bladder catheterization by using a urine bag collection as a screening tool. It worked! Before the study, 63% of 1,520 young children got catheterized, and 4.3% had a UTI. With the screening method, only those with a positive screen for a possible UTI (16% of 828) proceeded to be catheterized, reducing catheterization rates significantly. Only 4.4% of the 69% of children screened by either method had a UTI. So by screening with a bag urine specimen first, many of the children who would have been catheterized did not undergo the procedure.
A hidden gem of information in the study was the length of a visit to the emergency department. The kids were there an average of 4.7 hours whether the provider waited for the bag urine and then catheterized or went straight to the procedure! If only they had gone to their primary care provider, I wonder how much time could have been saved?
Dr. Michael E. Pichichero, a specialist in pediatric infectious diseases, is director of the Research Institute, Rochester (N.Y.) General Hospital. He is also a pediatrician at Legacy Pediatrics in Rochester. Dr. Pichichero said he had no relevant financial disclosures.
When children are febrile and younger than 24 months old and they go to a pediatric emergency department, a pretty big work-up can be anticipated, taking several hours and including a test for possible UTI.
|
|
Dr. Michael E. Pichichero |
In this report from investigators at Children’s Hospital of Philadelphia, we learn the results of a study undertaken to reduce the frequency of bladder catheterization by using a urine bag collection as a screening tool. It worked! Before the study, 63% of 1,520 young children got catheterized, and 4.3% had a UTI. With the screening method, only those with a positive screen for a possible UTI (16% of 828) proceeded to be catheterized, reducing catheterization rates significantly. Only 4.4% of the 69% of children screened by either method had a UTI. So by screening with a bag urine specimen first, many of the children who would have been catheterized did not undergo the procedure.
A hidden gem of information in the study was the length of a visit to the emergency department. The kids were there an average of 4.7 hours whether the provider waited for the bag urine and then catheterized or went straight to the procedure! If only they had gone to their primary care provider, I wonder how much time could have been saved?
Dr. Michael E. Pichichero, a specialist in pediatric infectious diseases, is director of the Research Institute, Rochester (N.Y.) General Hospital. He is also a pediatrician at Legacy Pediatrics in Rochester. Dr. Pichichero said he had no relevant financial disclosures.
When children are febrile and younger than 24 months old and they go to a pediatric emergency department, a pretty big work-up can be anticipated, taking several hours and including a test for possible UTI.
|
|
Dr. Michael E. Pichichero |
In this report from investigators at Children’s Hospital of Philadelphia, we learn the results of a study undertaken to reduce the frequency of bladder catheterization by using a urine bag collection as a screening tool. It worked! Before the study, 63% of 1,520 young children got catheterized, and 4.3% had a UTI. With the screening method, only those with a positive screen for a possible UTI (16% of 828) proceeded to be catheterized, reducing catheterization rates significantly. Only 4.4% of the 69% of children screened by either method had a UTI. So by screening with a bag urine specimen first, many of the children who would have been catheterized did not undergo the procedure.
A hidden gem of information in the study was the length of a visit to the emergency department. The kids were there an average of 4.7 hours whether the provider waited for the bag urine and then catheterized or went straight to the procedure! If only they had gone to their primary care provider, I wonder how much time could have been saved?
Dr. Michael E. Pichichero, a specialist in pediatric infectious diseases, is director of the Research Institute, Rochester (N.Y.) General Hospital. He is also a pediatrician at Legacy Pediatrics in Rochester. Dr. Pichichero said he had no relevant financial disclosures.
After implementation of a quality improvement initiative to more effectively screen febrile children for urinary tract infections (UTIs) in the emergency department, catheterization rates dropped from 63% to 30% over a 6-month period, a study found.
The sustained drop prevented more than 350 young children from catheterization without increasing revisit rates or missing UTIs in the 39% of children who were followed in the care network. This was in a study that compared catheterization rates in 1,520 children aged 6-24 months in the year before the intervention and 828 children in the 6 months during the intervention.
“Although urine catheterization remains the gold standard in diagnosing UTIs, it is an invasive procedure that may be avoided in most patients who are being screened,” wrote Dr. Jane M. Lavelle of Children’s Hospital of Philadelphia (CHOP) and her associates. UTI screening by this method can be “painful, time consuming, and costly,” they added (Pediatrics. 2016 June 2. doi: 10.1542/peds.2015-3023).
An alternative method to automatic catheterization is a two-step process already included as an option in American Academy of Pediatrics guidelines: Instead of collecting urine through catheterization just once for screening and culture, an emergency department first noninvasively collects urine with a urine bag for screening in those indicated with evidence-based risk factors, and then catheterizes only those who screen positive (Pediatrics. 2011;128[3]:595-610).
“Due to the predictive models’ higher sensitivity than specificity for screening, most urine samples will have a negative screen for pyuria or bacteriuria by urine dipstick or microscopy,” the authors wrote.
At baseline, CHOP’s ED was screening 63% of febrile children under age 24 months using catheterization, but screens were most commonly negative and only 4.3% had positive cultures. The authors therefore initiated a switch to the two-step method as a pilot run in one ED area before educating all ED personnel and expanding to the full department in the second month, using three cycles of Plan-Do-Study-Act protocol.
Children aged 6-24 months comprised approximately 20% of the ED’s more than 90,000 annual patients, and about 22% of these children presented with fever as the primary concern. Children with a history of genitourinary problems or immune deficiency were excluded.
The pilot ran in an “urgent care section of ED where there are typically more children with less complex medical histories and where ‘fever’ is a common complaint,” the investigators said. The staff completed a learning module with assessment and then received in-person and visual reminders of the procedure. Nurse feedback was then used to develop a nursing-specific educational module before expanding the intervention to all ED areas.
While 69% of 828 febrile young children still underwent screening during the 6-month intervention period, only 16% still underwent urethral catheterization as the initial screening step, typically because of strong clinical indications for a UTI. Another 14% underwent catheterization only after a positive urine screen from an initial noninvasive urine collection or because of an inability to get an adequate urine specimen with the bag. The reduction in catheterization dropped to 55% within 2 weeks of the intervention’s start and spread to other hospital departments. The drop to a 30% catheterization rate remained throughout 18 additional months of monitoring.
“Through online education modules, staff meetings, printed and EHR reminders, family involvement, team review of weekly data, individual and group feedback, and nurse scripting, the ED was able to achieve our aim of reducing catheterization rates among febrile young children ages 6-24 months by half over a 6-month period with sustained results,” Dr. Lavelle and her associates reported.
The research did not use external funding, and the researchers reported they had no financial disclosures.
After implementation of a quality improvement initiative to more effectively screen febrile children for urinary tract infections (UTIs) in the emergency department, catheterization rates dropped from 63% to 30% over a 6-month period, a study found.
The sustained drop prevented more than 350 young children from catheterization without increasing revisit rates or missing UTIs in the 39% of children who were followed in the care network. This was in a study that compared catheterization rates in 1,520 children aged 6-24 months in the year before the intervention and 828 children in the 6 months during the intervention.
“Although urine catheterization remains the gold standard in diagnosing UTIs, it is an invasive procedure that may be avoided in most patients who are being screened,” wrote Dr. Jane M. Lavelle of Children’s Hospital of Philadelphia (CHOP) and her associates. UTI screening by this method can be “painful, time consuming, and costly,” they added (Pediatrics. 2016 June 2. doi: 10.1542/peds.2015-3023).
An alternative method to automatic catheterization is a two-step process already included as an option in American Academy of Pediatrics guidelines: Instead of collecting urine through catheterization just once for screening and culture, an emergency department first noninvasively collects urine with a urine bag for screening in those indicated with evidence-based risk factors, and then catheterizes only those who screen positive (Pediatrics. 2011;128[3]:595-610).
“Due to the predictive models’ higher sensitivity than specificity for screening, most urine samples will have a negative screen for pyuria or bacteriuria by urine dipstick or microscopy,” the authors wrote.
At baseline, CHOP’s ED was screening 63% of febrile children under age 24 months using catheterization, but screens were most commonly negative and only 4.3% had positive cultures. The authors therefore initiated a switch to the two-step method as a pilot run in one ED area before educating all ED personnel and expanding to the full department in the second month, using three cycles of Plan-Do-Study-Act protocol.
Children aged 6-24 months comprised approximately 20% of the ED’s more than 90,000 annual patients, and about 22% of these children presented with fever as the primary concern. Children with a history of genitourinary problems or immune deficiency were excluded.
The pilot ran in an “urgent care section of ED where there are typically more children with less complex medical histories and where ‘fever’ is a common complaint,” the investigators said. The staff completed a learning module with assessment and then received in-person and visual reminders of the procedure. Nurse feedback was then used to develop a nursing-specific educational module before expanding the intervention to all ED areas.
While 69% of 828 febrile young children still underwent screening during the 6-month intervention period, only 16% still underwent urethral catheterization as the initial screening step, typically because of strong clinical indications for a UTI. Another 14% underwent catheterization only after a positive urine screen from an initial noninvasive urine collection or because of an inability to get an adequate urine specimen with the bag. The reduction in catheterization dropped to 55% within 2 weeks of the intervention’s start and spread to other hospital departments. The drop to a 30% catheterization rate remained throughout 18 additional months of monitoring.
“Through online education modules, staff meetings, printed and EHR reminders, family involvement, team review of weekly data, individual and group feedback, and nurse scripting, the ED was able to achieve our aim of reducing catheterization rates among febrile young children ages 6-24 months by half over a 6-month period with sustained results,” Dr. Lavelle and her associates reported.
The research did not use external funding, and the researchers reported they had no financial disclosures.
FROM PEDIATRICS
Key clinical point: Use of a two-step UTI screening process reduced unnecessary catheterizations by half.
Major finding: The ED catheterization rate in febrile children suspected of a UTI dropped from 63% to 30% over a 6-month period.
Data source: An assessment of a quality improvement initiative in the Children’s Hospital of Philadelphia ED that compared catheterization rates in 1,520 children aged 6-24 months in the year before the intervention and 828 children in the 6 months during the intervention.
Disclosures: The research did not use external funding, and the researchers reported they had no financial disclosures.

Maternal vaccination against pertussis can protect premature infants
Maternal immunization in the early third trimester (from 28 weeks’ gestation) may protect premature infants from pertussis, study results found.
This was the finding of an observational substudy of a larger multicenter, randomized, controlled vaccination trial of premature infants (the PUNS trial), which compared pertussis antibody concentrations before and after primary immunization in premature infants born to mothers who were or were not vaccinated with Repevax. Dr. Alison Kent of St George’s, University of London, and colleagues assessed the levels of the five vaccine antigens present in the maternal combination Repevax vaccine (pertussis toxoid, filamentous hemagglutinin, fimbriae types 2 and 3, diphtheria toxoid, tetanus toxoid, and inactivated poliovirus) in premature infants born to mothers who either received or did not receive Repevax from 28 weeks’ gestation. Antigen quantifications were conducted in these premature infants at approximately 2, 5, and 12 months of age.

Thirty-one (19%) of the 160 premature infants in the substudy were born to mothers who had been vaccinated. Two months after their premature birth, infants born to vaccinated mothers had significantly higher concentrations of all five measured antigens, compared with those born to unvaccinated mothers (all P values less than .001). Although fewer infants were sampled at 5 months of age, significantly higher concentrations of filamentous hemagglutinin and diphtheria toxoid were still found in those born to vaccinated mothers (both P = .003). Data collected at the 12-month assessment indicated that only tetanus antibody concentrations remained significantly higher in those born to vaccinated mothers (P = .015). A positive correlation between the number of days from maternal vaccination to delivery was found for all measured antigens, with the exception of fimbriae types 2 and 3.
“The emergency introduction of a maternal immunization program to control a national pertussis outbreak serendipitously provided an opportunity to assess antibody concentrations to maternal vaccine antigens in premature infants,” Dr. Kent and associates noted in Pediatrics (June 2016 doi: 10.1542/peds.2015-3854). This unexpected opportunity resulted in evidence supporting a protective effect against pertussis in the early lives of infants born prematurely to mothers immunized in their early third trimester.
Pfizer and the National Institute for Health Research Clinical Research Network funded the study. Professor Heath and Dr. Ladhani disclosed conducting studies on behalf of St George’s, University of London funded by vaccine manufacturers without receiving personal payments or travel support. The other authors reported no conflicts of interest.
Maternal immunization in the early third trimester (from 28 weeks’ gestation) may protect premature infants from pertussis, study results found.
This was the finding of an observational substudy of a larger multicenter, randomized, controlled vaccination trial of premature infants (the PUNS trial), which compared pertussis antibody concentrations before and after primary immunization in premature infants born to mothers who were or were not vaccinated with Repevax. Dr. Alison Kent of St George’s, University of London, and colleagues assessed the levels of the five vaccine antigens present in the maternal combination Repevax vaccine (pertussis toxoid, filamentous hemagglutinin, fimbriae types 2 and 3, diphtheria toxoid, tetanus toxoid, and inactivated poliovirus) in premature infants born to mothers who either received or did not receive Repevax from 28 weeks’ gestation. Antigen quantifications were conducted in these premature infants at approximately 2, 5, and 12 months of age.
Thirty-one (19%) of the 160 premature infants in the substudy were born to mothers who had been vaccinated. Two months after their premature birth, infants born to vaccinated mothers had significantly higher concentrations of all five measured antigens, compared with those born to unvaccinated mothers (all P values less than .001). Although fewer infants were sampled at 5 months of age, significantly higher concentrations of filamentous hemagglutinin and diphtheria toxoid were still found in those born to vaccinated mothers (both P = .003). Data collected at the 12-month assessment indicated that only tetanus antibody concentrations remained significantly higher in those born to vaccinated mothers (P = .015). A positive correlation between the number of days from maternal vaccination to delivery was found for all measured antigens, with the exception of fimbriae types 2 and 3.
“The emergency introduction of a maternal immunization program to control a national pertussis outbreak serendipitously provided an opportunity to assess antibody concentrations to maternal vaccine antigens in premature infants,” Dr. Kent and associates noted in Pediatrics (June 2016 doi: 10.1542/peds.2015-3854). This unexpected opportunity resulted in evidence supporting a protective effect against pertussis in the early lives of infants born prematurely to mothers immunized in their early third trimester.
Pfizer and the National Institute for Health Research Clinical Research Network funded the study. Professor Heath and Dr. Ladhani disclosed conducting studies on behalf of St George’s, University of London funded by vaccine manufacturers without receiving personal payments or travel support. The other authors reported no conflicts of interest.
Maternal immunization in the early third trimester (from 28 weeks’ gestation) may protect premature infants from pertussis, study results found.
This was the finding of an observational substudy of a larger multicenter, randomized, controlled vaccination trial of premature infants (the PUNS trial), which compared pertussis antibody concentrations before and after primary immunization in premature infants born to mothers who were or were not vaccinated with Repevax. Dr. Alison Kent of St George’s, University of London, and colleagues assessed the levels of the five vaccine antigens present in the maternal combination Repevax vaccine (pertussis toxoid, filamentous hemagglutinin, fimbriae types 2 and 3, diphtheria toxoid, tetanus toxoid, and inactivated poliovirus) in premature infants born to mothers who either received or did not receive Repevax from 28 weeks’ gestation. Antigen quantifications were conducted in these premature infants at approximately 2, 5, and 12 months of age.
Thirty-one (19%) of the 160 premature infants in the substudy were born to mothers who had been vaccinated. Two months after their premature birth, infants born to vaccinated mothers had significantly higher concentrations of all five measured antigens, compared with those born to unvaccinated mothers (all P values less than .001). Although fewer infants were sampled at 5 months of age, significantly higher concentrations of filamentous hemagglutinin and diphtheria toxoid were still found in those born to vaccinated mothers (both P = .003). Data collected at the 12-month assessment indicated that only tetanus antibody concentrations remained significantly higher in those born to vaccinated mothers (P = .015). A positive correlation between the number of days from maternal vaccination to delivery was found for all measured antigens, with the exception of fimbriae types 2 and 3.
“The emergency introduction of a maternal immunization program to control a national pertussis outbreak serendipitously provided an opportunity to assess antibody concentrations to maternal vaccine antigens in premature infants,” Dr. Kent and associates noted in Pediatrics (June 2016 doi: 10.1542/peds.2015-3854). This unexpected opportunity resulted in evidence supporting a protective effect against pertussis in the early lives of infants born prematurely to mothers immunized in their early third trimester.
Pfizer and the National Institute for Health Research Clinical Research Network funded the study. Professor Heath and Dr. Ladhani disclosed conducting studies on behalf of St George’s, University of London funded by vaccine manufacturers without receiving personal payments or travel support. The other authors reported no conflicts of interest.
FROM PEDIATRICS
