User login
More Hospitals to Be Replaced by FSEDs
If an ED is considered the “front door” to the hospital, how do we regard a free-standing emergency department (FSED) with no hospital attached to it? Fueled by continued hospital closures in the face of steadily increasing demands for emergency care, FSEDs are now replacing hospitals in previously well-served urban areas in addition to serving rural areas lacking alternative facilities.
According to The New York Times (http://nyti.ms/1TB8Z44), since 2000, 19 New York City hospitals “have either closed or overhauled how they operate.” As this issue of Emergency Medicine went to press, plans had been announced to replace Manhattan’s Beth Israel and Brooklyn’s Wyckoff Heights hospitals with FSEDs and expanded outpatient facilities. These hospitals and many others that have recently closed, including St Vincent’s (2010) and the Long Island College Hospital (2014), had been part of the health care landscape in New York for over 125 years.
What do FSEDs mean for emergency medicine (EM) and emergency physicians (EPs), and are they safe alternatives to traditional hospital-based EDs? Newer technologies and treatments, coupled with steadily increasing pressures to reduce inpatient stays, razor-thin hospital operating margins, and the refusal of state and local governments to bail out financially failing hospitals, have created a disconnect between the increasing need for emergency care and the decreasing number of inpatient beds.
On one end of the EM patient care spectrum, urgent care centers (UCCs) and retail pharmacy clinics—collectively referred to as “convenient care” centers—are rapidly proliferating to offer care to those with urgent, episodic, and relatively minor medical and surgical problems. (See “Urgent Care and the Urgent Need for Care” at http://bit.ly/1OSrHSA). With little or no regulatory oversight, convenient care centers staffed by EPs, family practitioners, internists, NPs, and PAs, offer extended hour care—but not 24/7 care—to anyone with adequate health insurance or the ability to pay for the care.
On the other end of the EM patient care spectrum are the FSEDs, now divided into two types: satellite EDs of nearby hospitals, and “FS”-FSEDs with no direct hospital connections. Almost all FSEDs receive 911 ambulances, are staffed at all times by trained and certified EPs and registered nurses (RNs) provide acute care and stabilization consistent with the standards for hospital-based EDs, and are open 24/7—a hallmark that distinguishes EDs from UCCs. FSEDs code and bill both for facility and provider services in the same way hospital-based EDs do. Although organized EM has enthusiastically embraced and endorsed FSEDs, its position on UCCs has been decidedly mixed.
Are FSEDs safe for patients requiring emergency care? The lack of uniform definitions and federal and state regulatory requirements make it difficult to gather and interpret meaningful clinical data on FSEDs and convenient care centers. But a well-equipped FSED, served by state-of-the-art pre- and inter-facility ambulances, and staffed by qualified EPs and RNs, should provide a safe alternative to hospital-based EDs for almost all patients in need of emergency care—especially when no hospital-based ED is available.
Specialty designations of qualifying area hospitals such as “Level I trauma center” will minimize but not completely eliminate bad outcomes of cases where even seconds may make the difference between life and death. In the end though, the real question may be is an FSED better than no ED at all?
Ideally, a hospital-based ED should be the epicenter of a network of both satellite convenient care centers and FSEDs, coordinating services, providing management and staffing for all parts of the network, and arranging safe, appropriate intranetwork ambulance transport.
Should you think that FSEDs are a new phenomenon, you might be surprised to discover that in 1875, after New York Hospital (now part of New York Presbyterian) closed its original lower Manhattan site to move further uptown, it opened a “House of Relief” in its old neighborhood that contained an emergency treatment center, an operating room, an isolation area, a dispensary, a reception area, examination rooms, an ambulance entrance, and wards to observe and treat patients until they could be safely transported to the new main hospital. FSEDs served 19th-century patients well, and in the 21st century may serve as a reminder that sometimes even in medicine, “everything old is new again!” (See http://bit.ly/1NSPlDG.)
Editor’s Note: Portions of this editorial were previously published in Emergency Medicine.
If an ED is considered the “front door” to the hospital, how do we regard a free-standing emergency department (FSED) with no hospital attached to it? Fueled by continued hospital closures in the face of steadily increasing demands for emergency care, FSEDs are now replacing hospitals in previously well-served urban areas in addition to serving rural areas lacking alternative facilities.
According to The New York Times (http://nyti.ms/1TB8Z44), since 2000, 19 New York City hospitals “have either closed or overhauled how they operate.” As this issue of Emergency Medicine went to press, plans had been announced to replace Manhattan’s Beth Israel and Brooklyn’s Wyckoff Heights hospitals with FSEDs and expanded outpatient facilities. These hospitals and many others that have recently closed, including St Vincent’s (2010) and the Long Island College Hospital (2014), had been part of the health care landscape in New York for over 125 years.
What do FSEDs mean for emergency medicine (EM) and emergency physicians (EPs), and are they safe alternatives to traditional hospital-based EDs? Newer technologies and treatments, coupled with steadily increasing pressures to reduce inpatient stays, razor-thin hospital operating margins, and the refusal of state and local governments to bail out financially failing hospitals, have created a disconnect between the increasing need for emergency care and the decreasing number of inpatient beds.
On one end of the EM patient care spectrum, urgent care centers (UCCs) and retail pharmacy clinics—collectively referred to as “convenient care” centers—are rapidly proliferating to offer care to those with urgent, episodic, and relatively minor medical and surgical problems. (See “Urgent Care and the Urgent Need for Care” at http://bit.ly/1OSrHSA). With little or no regulatory oversight, convenient care centers staffed by EPs, family practitioners, internists, NPs, and PAs, offer extended hour care—but not 24/7 care—to anyone with adequate health insurance or the ability to pay for the care.
On the other end of the EM patient care spectrum are the FSEDs, now divided into two types: satellite EDs of nearby hospitals, and “FS”-FSEDs with no direct hospital connections. Almost all FSEDs receive 911 ambulances, are staffed at all times by trained and certified EPs and registered nurses (RNs) provide acute care and stabilization consistent with the standards for hospital-based EDs, and are open 24/7—a hallmark that distinguishes EDs from UCCs. FSEDs code and bill both for facility and provider services in the same way hospital-based EDs do. Although organized EM has enthusiastically embraced and endorsed FSEDs, its position on UCCs has been decidedly mixed.
Are FSEDs safe for patients requiring emergency care? The lack of uniform definitions and federal and state regulatory requirements make it difficult to gather and interpret meaningful clinical data on FSEDs and convenient care centers. But a well-equipped FSED, served by state-of-the-art pre- and inter-facility ambulances, and staffed by qualified EPs and RNs, should provide a safe alternative to hospital-based EDs for almost all patients in need of emergency care—especially when no hospital-based ED is available.
Specialty designations of qualifying area hospitals such as “Level I trauma center” will minimize but not completely eliminate bad outcomes of cases where even seconds may make the difference between life and death. In the end though, the real question may be is an FSED better than no ED at all?
Ideally, a hospital-based ED should be the epicenter of a network of both satellite convenient care centers and FSEDs, coordinating services, providing management and staffing for all parts of the network, and arranging safe, appropriate intranetwork ambulance transport.
Should you think that FSEDs are a new phenomenon, you might be surprised to discover that in 1875, after New York Hospital (now part of New York Presbyterian) closed its original lower Manhattan site to move further uptown, it opened a “House of Relief” in its old neighborhood that contained an emergency treatment center, an operating room, an isolation area, a dispensary, a reception area, examination rooms, an ambulance entrance, and wards to observe and treat patients until they could be safely transported to the new main hospital. FSEDs served 19th-century patients well, and in the 21st century may serve as a reminder that sometimes even in medicine, “everything old is new again!” (See http://bit.ly/1NSPlDG.)
Editor’s Note: Portions of this editorial were previously published in Emergency Medicine.
If an ED is considered the “front door” to the hospital, how do we regard a free-standing emergency department (FSED) with no hospital attached to it? Fueled by continued hospital closures in the face of steadily increasing demands for emergency care, FSEDs are now replacing hospitals in previously well-served urban areas in addition to serving rural areas lacking alternative facilities.
According to The New York Times (http://nyti.ms/1TB8Z44), since 2000, 19 New York City hospitals “have either closed or overhauled how they operate.” As this issue of Emergency Medicine went to press, plans had been announced to replace Manhattan’s Beth Israel and Brooklyn’s Wyckoff Heights hospitals with FSEDs and expanded outpatient facilities. These hospitals and many others that have recently closed, including St Vincent’s (2010) and the Long Island College Hospital (2014), had been part of the health care landscape in New York for over 125 years.
What do FSEDs mean for emergency medicine (EM) and emergency physicians (EPs), and are they safe alternatives to traditional hospital-based EDs? Newer technologies and treatments, coupled with steadily increasing pressures to reduce inpatient stays, razor-thin hospital operating margins, and the refusal of state and local governments to bail out financially failing hospitals, have created a disconnect between the increasing need for emergency care and the decreasing number of inpatient beds.
On one end of the EM patient care spectrum, urgent care centers (UCCs) and retail pharmacy clinics—collectively referred to as “convenient care” centers—are rapidly proliferating to offer care to those with urgent, episodic, and relatively minor medical and surgical problems. (See “Urgent Care and the Urgent Need for Care” at http://bit.ly/1OSrHSA). With little or no regulatory oversight, convenient care centers staffed by EPs, family practitioners, internists, NPs, and PAs, offer extended hour care—but not 24/7 care—to anyone with adequate health insurance or the ability to pay for the care.
On the other end of the EM patient care spectrum are the FSEDs, now divided into two types: satellite EDs of nearby hospitals, and “FS”-FSEDs with no direct hospital connections. Almost all FSEDs receive 911 ambulances, are staffed at all times by trained and certified EPs and registered nurses (RNs) provide acute care and stabilization consistent with the standards for hospital-based EDs, and are open 24/7—a hallmark that distinguishes EDs from UCCs. FSEDs code and bill both for facility and provider services in the same way hospital-based EDs do. Although organized EM has enthusiastically embraced and endorsed FSEDs, its position on UCCs has been decidedly mixed.
Are FSEDs safe for patients requiring emergency care? The lack of uniform definitions and federal and state regulatory requirements make it difficult to gather and interpret meaningful clinical data on FSEDs and convenient care centers. But a well-equipped FSED, served by state-of-the-art pre- and inter-facility ambulances, and staffed by qualified EPs and RNs, should provide a safe alternative to hospital-based EDs for almost all patients in need of emergency care—especially when no hospital-based ED is available.
Specialty designations of qualifying area hospitals such as “Level I trauma center” will minimize but not completely eliminate bad outcomes of cases where even seconds may make the difference between life and death. In the end though, the real question may be is an FSED better than no ED at all?
Ideally, a hospital-based ED should be the epicenter of a network of both satellite convenient care centers and FSEDs, coordinating services, providing management and staffing for all parts of the network, and arranging safe, appropriate intranetwork ambulance transport.
Should you think that FSEDs are a new phenomenon, you might be surprised to discover that in 1875, after New York Hospital (now part of New York Presbyterian) closed its original lower Manhattan site to move further uptown, it opened a “House of Relief” in its old neighborhood that contained an emergency treatment center, an operating room, an isolation area, a dispensary, a reception area, examination rooms, an ambulance entrance, and wards to observe and treat patients until they could be safely transported to the new main hospital. FSEDs served 19th-century patients well, and in the 21st century may serve as a reminder that sometimes even in medicine, “everything old is new again!” (See http://bit.ly/1NSPlDG.)
Editor’s Note: Portions of this editorial were previously published in Emergency Medicine.

Optical Imaging to Detect Lentigo Maligna

In an article published online on January 26 in the Journal of the American Academy of Dermatology, my colleagues and I (Menge et al) reported on the use of reflectance confocal microscopy (RCM) for challenging facial lesions. We studied the diagnosis of lentigo maligna (LM) based on RCM versus the histopathologic diagnosis after biopsy.
In this study 17 patients were seen for evaluation of known or suspected LM at Memorial Sloan Kettering Cancer Center (New York, New York). Among these patients, a total of 63 sites on the skin were evaluated using RCM and a presumptive diagnosis was made. These sites were then biopsied to compare the diagnosis using RCM with that made by histopathology. When LM was present as determined by biopsy, RCM also was able to detect it 100% of the time (sensitivity). When LM was absent as determined by biopsy, RCM also indicated it was absent 71% of the time (specificity).
What’s the issue?
Lentigo maligna is a form of melanoma in situ occurring on sun-damaged skin. It can be quite subtle to detect clinically and therefore may go undiagnosed for a while. Lentigo maligna also has been shown to have notable subclinical extension with which traditional surgical margins for truncal melanoma may be too narrow to clear LM on the head and neck. Therefore, presurgical consultation may be difficult due to the amorphous borders. Random blind biopsies also are discouraged because of sampling error.
Additionally, repetitive biopsies over time, which may be frequently needed in individuals with heavy sun exposure, can be costly and cause adverse effects.
This study showed the usefulness and reliability of using RCM for challenging facial lesions that are suspicious for LM. The sensitivity and specificity of RCM in this study indicated that this technology performs well in detecting LM when present; however, false-positives were noted in this study. False-positives included pigmented actinic keratosis and melanocytosis. Dermatologists who are advanced in RCM technology and interpretation also were utilized in this study. More research is needed to understand how to best utilize this technology, but overall the ability of RCM to accurately identify LM without biopsy represents an exciting new development in how dermatologists can better diagnose, manage, and treat melanoma.
How will you adopt advances in cutaneous noninvasive imaging?
In an article published online on January 26 in the Journal of the American Academy of Dermatology, my colleagues and I (Menge et al) reported on the use of reflectance confocal microscopy (RCM) for challenging facial lesions. We studied the diagnosis of lentigo maligna (LM) based on RCM versus the histopathologic diagnosis after biopsy.
In this study 17 patients were seen for evaluation of known or suspected LM at Memorial Sloan Kettering Cancer Center (New York, New York). Among these patients, a total of 63 sites on the skin were evaluated using RCM and a presumptive diagnosis was made. These sites were then biopsied to compare the diagnosis using RCM with that made by histopathology. When LM was present as determined by biopsy, RCM also was able to detect it 100% of the time (sensitivity). When LM was absent as determined by biopsy, RCM also indicated it was absent 71% of the time (specificity).
What’s the issue?
Lentigo maligna is a form of melanoma in situ occurring on sun-damaged skin. It can be quite subtle to detect clinically and therefore may go undiagnosed for a while. Lentigo maligna also has been shown to have notable subclinical extension with which traditional surgical margins for truncal melanoma may be too narrow to clear LM on the head and neck. Therefore, presurgical consultation may be difficult due to the amorphous borders. Random blind biopsies also are discouraged because of sampling error.
Additionally, repetitive biopsies over time, which may be frequently needed in individuals with heavy sun exposure, can be costly and cause adverse effects.
This study showed the usefulness and reliability of using RCM for challenging facial lesions that are suspicious for LM. The sensitivity and specificity of RCM in this study indicated that this technology performs well in detecting LM when present; however, false-positives were noted in this study. False-positives included pigmented actinic keratosis and melanocytosis. Dermatologists who are advanced in RCM technology and interpretation also were utilized in this study. More research is needed to understand how to best utilize this technology, but overall the ability of RCM to accurately identify LM without biopsy represents an exciting new development in how dermatologists can better diagnose, manage, and treat melanoma.
How will you adopt advances in cutaneous noninvasive imaging?
In an article published online on January 26 in the Journal of the American Academy of Dermatology, my colleagues and I (Menge et al) reported on the use of reflectance confocal microscopy (RCM) for challenging facial lesions. We studied the diagnosis of lentigo maligna (LM) based on RCM versus the histopathologic diagnosis after biopsy.
In this study 17 patients were seen for evaluation of known or suspected LM at Memorial Sloan Kettering Cancer Center (New York, New York). Among these patients, a total of 63 sites on the skin were evaluated using RCM and a presumptive diagnosis was made. These sites were then biopsied to compare the diagnosis using RCM with that made by histopathology. When LM was present as determined by biopsy, RCM also was able to detect it 100% of the time (sensitivity). When LM was absent as determined by biopsy, RCM also indicated it was absent 71% of the time (specificity).
What’s the issue?
Lentigo maligna is a form of melanoma in situ occurring on sun-damaged skin. It can be quite subtle to detect clinically and therefore may go undiagnosed for a while. Lentigo maligna also has been shown to have notable subclinical extension with which traditional surgical margins for truncal melanoma may be too narrow to clear LM on the head and neck. Therefore, presurgical consultation may be difficult due to the amorphous borders. Random blind biopsies also are discouraged because of sampling error.
Additionally, repetitive biopsies over time, which may be frequently needed in individuals with heavy sun exposure, can be costly and cause adverse effects.
This study showed the usefulness and reliability of using RCM for challenging facial lesions that are suspicious for LM. The sensitivity and specificity of RCM in this study indicated that this technology performs well in detecting LM when present; however, false-positives were noted in this study. False-positives included pigmented actinic keratosis and melanocytosis. Dermatologists who are advanced in RCM technology and interpretation also were utilized in this study. More research is needed to understand how to best utilize this technology, but overall the ability of RCM to accurately identify LM without biopsy represents an exciting new development in how dermatologists can better diagnose, manage, and treat melanoma.
How will you adopt advances in cutaneous noninvasive imaging?

Spontaneous Repigmentation of Silvery Hair in an Infant With Congenital Hydrops Fetalis and Hypoproteinemia
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
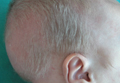
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
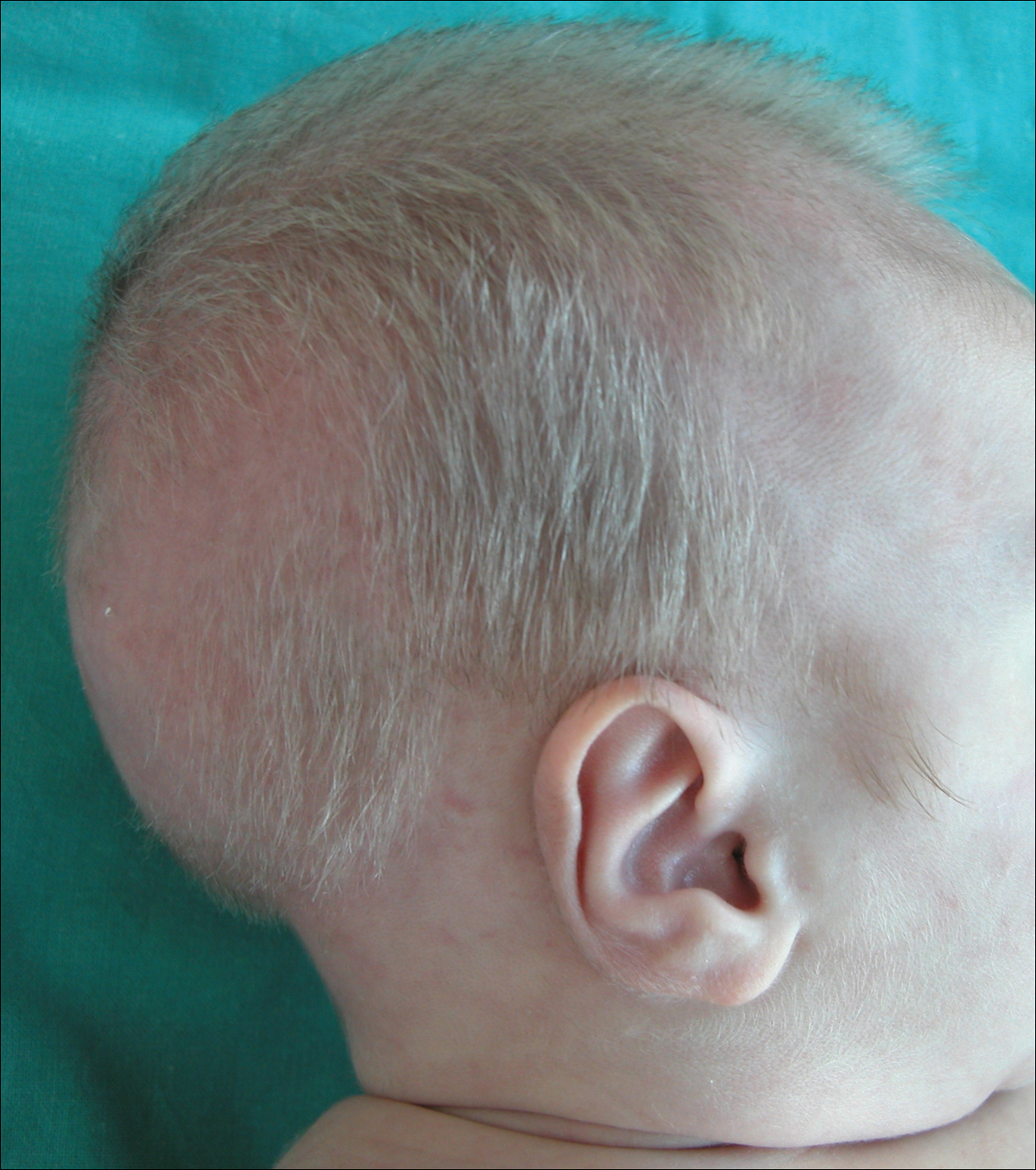
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
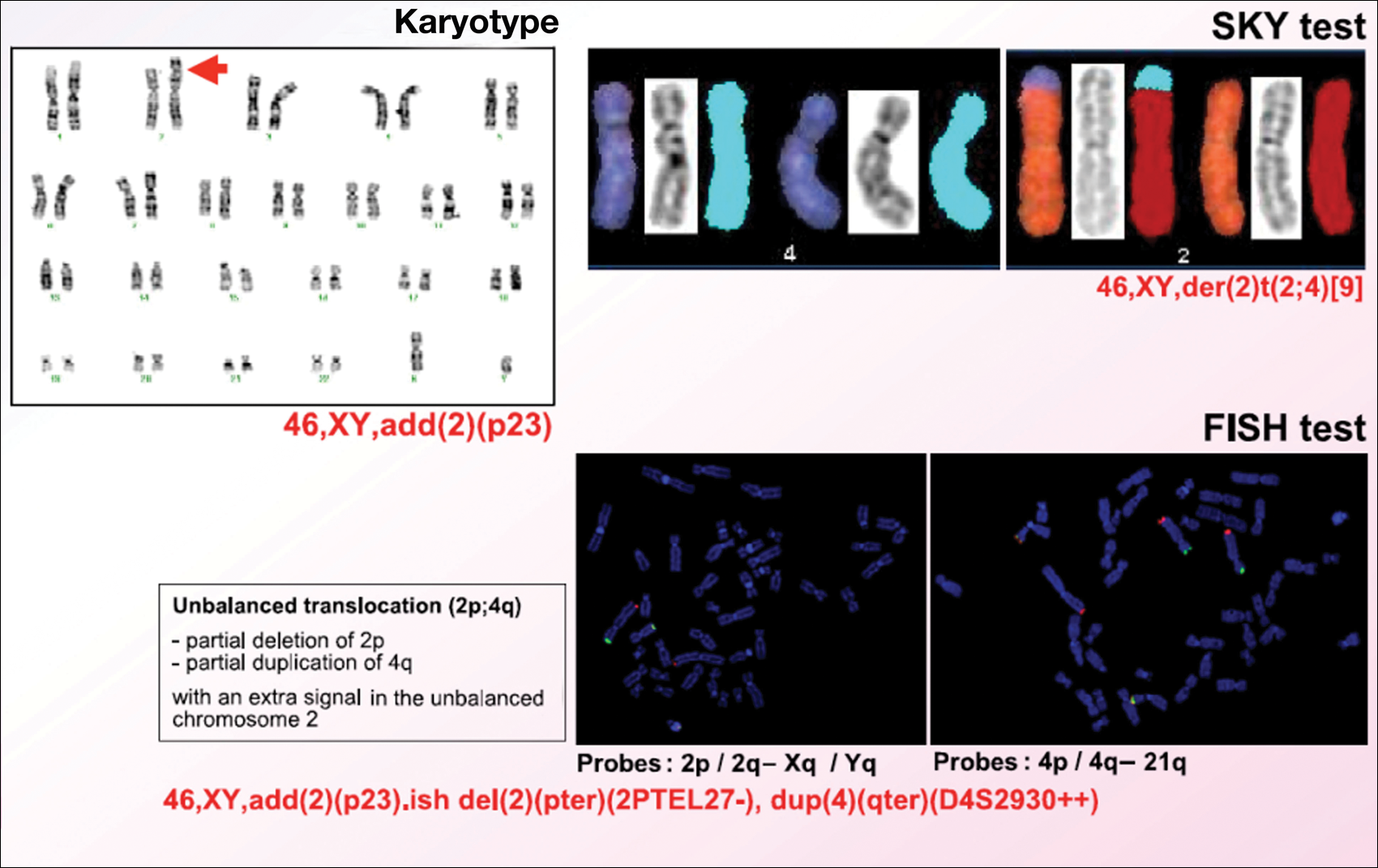
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.


Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
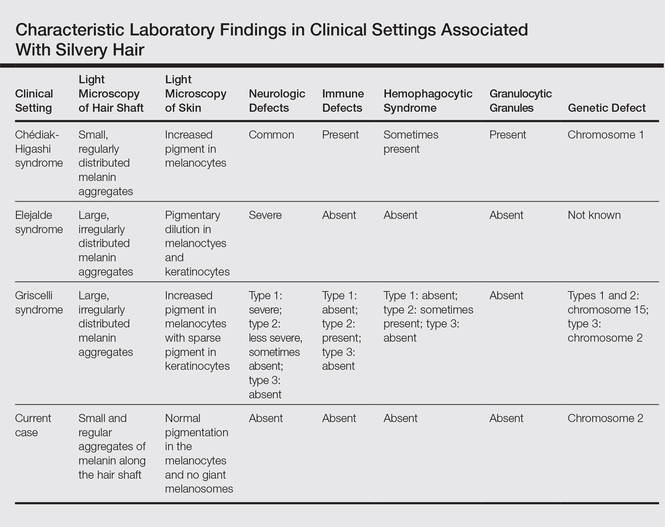
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
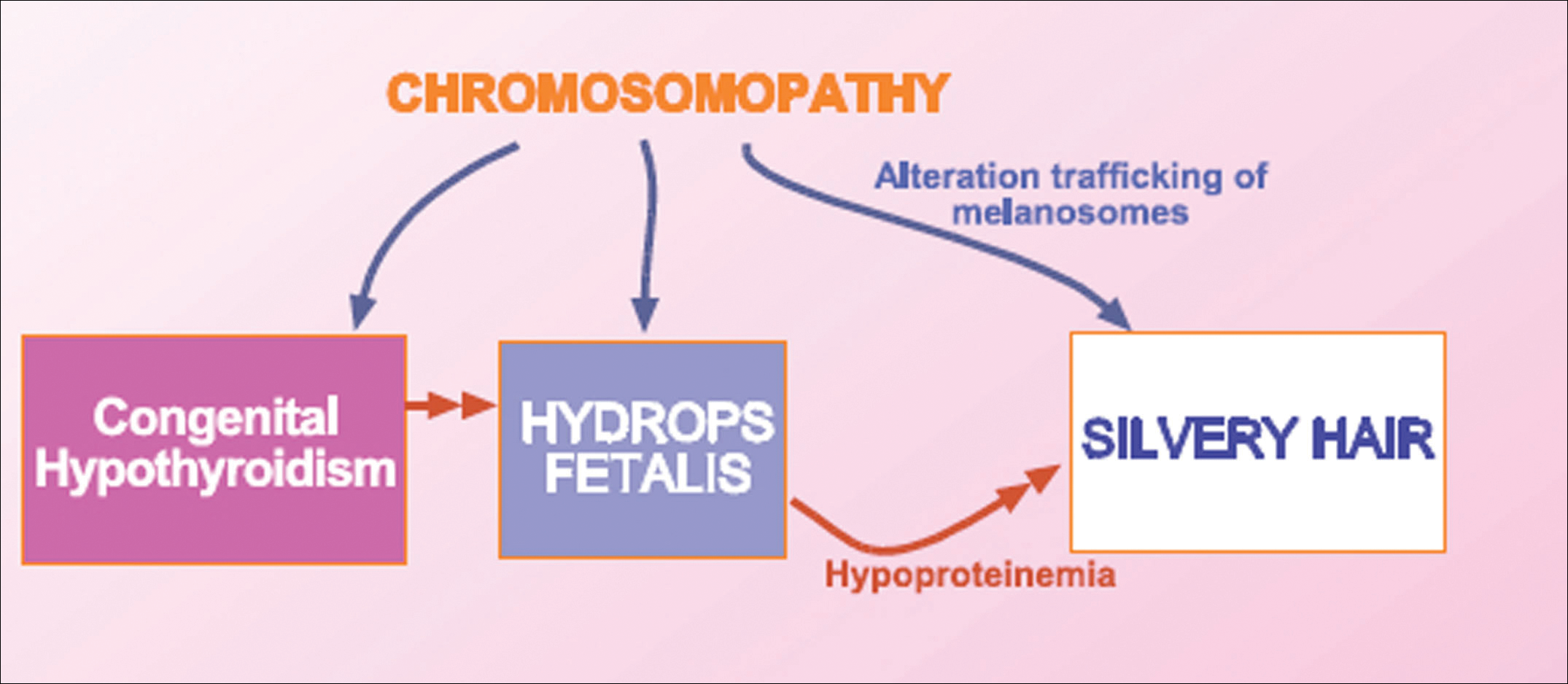
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.
Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.
Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
Practice Points
- Silvery hair is characteristic of 3 rare autosomal-recessive disorders: Chédiak-Higashi syndrome, Elejalde syndrome, and Griscelli syndrome.
- Hypopigmentation is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes.
- Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
Coldiron Truth: Beware the state pharmacy board
What does the pharmacy board have to do with me? I’m a physician, regulated by the state medical board. Well heads up. If regulations coming your way are adopted, you will have the additional privilege of being licensed, inspected by, and financially supporting your state pharmacy board.
How did all this happen? In 2012, a compounding pharmacy inadequately sterilized multiple lots of methylprednisolone, which were sold around the country and used for intrathecal injections. As a result, 753 patients developed fungal infections, including 386 cases of meningitis, and 64 of them died. The owner of the pharmacy and the head pharmacist are up on second degree murder charges.

But what does this have to do with you?
After a media bonfire, a congressional hearing complete with the taking of the fifth amendment, and a major rewrite of pharmacy regulations with increased scrutiny and oversight, the State of Ohio Board of Pharmacy rushed to adopt rules before reasonable regulations could be worked out by the Food and Drug Administration, the American Medical Association, and the Federation of State Medical Boards. The Ohio board of pharmacy adopted the U.S. Pharmacopeial Convention (USP) regulations, written for compounding pharmacies, and applied them to physicians’ offices.
In an overreaching bureaucratic coup de grace, any practitioners who reconstitute any drug in their offices is considered to be a compounding pharmacy, ordered to pay compounding pharmacy registration fees ($112 yearly), and to undergo the same inspections as compounding pharmacies. You can’t be too safe, you know, and all those registration fees (totaling about $2 million per year in Ohio alone) will decrease what would have been an onerous registration and inspection expense for true compounding pharmacies.
This is the reality we are facing in Ohio, and this situation may soon be “coming to a theater near you.” I understand that several pharmacy boards in other states are preparing to roll out similar regulations.
As a kicker, if the product you reconstitute is preservative free (botulinum toxin anyone?), you must use it or dispose of it within one hour. Yes, one hour. If you dilute bleomycin or 5-fluorouracil (5-FU), you must install an outside vented laminar flow hood, and wear level 5 hazmat gear while drawing it up.
Is this situation insane or what? As the result of a pharmacy in Massachusetts that skirted existing regulations and sold contaminated drugs that killed patients, doctors now need more regulations, licensing, inspections, and fees?
The real problem here, of course, is not the $112 fee. It will be the loss of many drugs and therapies that can be used inexpensively in the office, but will now either be unavailable to patients or available at a greatly increased cost. I pointed this situation out at a pharmacy board meeting, and they helpfully responded that I can have my friendly local pharmacist compound any drug I need in a specific strength and unit dose. Who is going to pay for this? I can make diclofenac or 5-FU cream in my office for less than $20. Instead, it will cost over $700 at the pharmacy! Further, making something fiscally impossible, like installing a laminar flow hood, is not different that denying it outright. I consider this to be restraint of trade.
Don’t allow yourselves to be compromised as Ohio physicians have been. You must be vigilant. Attend the public hearings and testify. In Ohio, the hearings were held over the Christmas holidays. Guess what? No one came to the hearings! You must show up and complain. Loudly. You must point out how patients are going to be hurt, not helped, by these rules. You must point out the superb safety record of physicians when using in-office pharmaceuticals. You must alert your neurology, ophthalmology, gynecology, and urology colleagues to the problem since they all use neurotoxins, too. The primary care doctors all reconstitute drugs (think antibiotics) for office use, too.
These efforts are also part of a larger campaign to give pharmacists a larger clinical role in patient care. If pharmacists license you, if they inspect your office, how can you oppose them when they want clinical privileges?
The fix is to enact a moratorium on regulations until the FDA rules come out. These will be more reasonable than the rules issued by the USP. Another fix is a legislative change that instructs that physicians, not pharmacists, will define what is considered to be a dangerous drug.
It is time to be alert, vigilant, and outspoken. You must do this to preserve your ability to do what is best for patients, to be able to deliver care in an expeditious, efficient, and cost-effective manner. This is what being physician is all about! Keep your state board of pharmacy off your license and out of your office.
Dr. Coldiron is a past president of the American Academy of Dermatology. He is currently in private practice, but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of more than 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics. Reach him at [email protected].
What does the pharmacy board have to do with me? I’m a physician, regulated by the state medical board. Well heads up. If regulations coming your way are adopted, you will have the additional privilege of being licensed, inspected by, and financially supporting your state pharmacy board.
How did all this happen? In 2012, a compounding pharmacy inadequately sterilized multiple lots of methylprednisolone, which were sold around the country and used for intrathecal injections. As a result, 753 patients developed fungal infections, including 386 cases of meningitis, and 64 of them died. The owner of the pharmacy and the head pharmacist are up on second degree murder charges.
But what does this have to do with you?
After a media bonfire, a congressional hearing complete with the taking of the fifth amendment, and a major rewrite of pharmacy regulations with increased scrutiny and oversight, the State of Ohio Board of Pharmacy rushed to adopt rules before reasonable regulations could be worked out by the Food and Drug Administration, the American Medical Association, and the Federation of State Medical Boards. The Ohio board of pharmacy adopted the U.S. Pharmacopeial Convention (USP) regulations, written for compounding pharmacies, and applied them to physicians’ offices.
In an overreaching bureaucratic coup de grace, any practitioners who reconstitute any drug in their offices is considered to be a compounding pharmacy, ordered to pay compounding pharmacy registration fees ($112 yearly), and to undergo the same inspections as compounding pharmacies. You can’t be too safe, you know, and all those registration fees (totaling about $2 million per year in Ohio alone) will decrease what would have been an onerous registration and inspection expense for true compounding pharmacies.
This is the reality we are facing in Ohio, and this situation may soon be “coming to a theater near you.” I understand that several pharmacy boards in other states are preparing to roll out similar regulations.
As a kicker, if the product you reconstitute is preservative free (botulinum toxin anyone?), you must use it or dispose of it within one hour. Yes, one hour. If you dilute bleomycin or 5-fluorouracil (5-FU), you must install an outside vented laminar flow hood, and wear level 5 hazmat gear while drawing it up.
Is this situation insane or what? As the result of a pharmacy in Massachusetts that skirted existing regulations and sold contaminated drugs that killed patients, doctors now need more regulations, licensing, inspections, and fees?
The real problem here, of course, is not the $112 fee. It will be the loss of many drugs and therapies that can be used inexpensively in the office, but will now either be unavailable to patients or available at a greatly increased cost. I pointed this situation out at a pharmacy board meeting, and they helpfully responded that I can have my friendly local pharmacist compound any drug I need in a specific strength and unit dose. Who is going to pay for this? I can make diclofenac or 5-FU cream in my office for less than $20. Instead, it will cost over $700 at the pharmacy! Further, making something fiscally impossible, like installing a laminar flow hood, is not different that denying it outright. I consider this to be restraint of trade.
Don’t allow yourselves to be compromised as Ohio physicians have been. You must be vigilant. Attend the public hearings and testify. In Ohio, the hearings were held over the Christmas holidays. Guess what? No one came to the hearings! You must show up and complain. Loudly. You must point out how patients are going to be hurt, not helped, by these rules. You must point out the superb safety record of physicians when using in-office pharmaceuticals. You must alert your neurology, ophthalmology, gynecology, and urology colleagues to the problem since they all use neurotoxins, too. The primary care doctors all reconstitute drugs (think antibiotics) for office use, too.
These efforts are also part of a larger campaign to give pharmacists a larger clinical role in patient care. If pharmacists license you, if they inspect your office, how can you oppose them when they want clinical privileges?
The fix is to enact a moratorium on regulations until the FDA rules come out. These will be more reasonable than the rules issued by the USP. Another fix is a legislative change that instructs that physicians, not pharmacists, will define what is considered to be a dangerous drug.
It is time to be alert, vigilant, and outspoken. You must do this to preserve your ability to do what is best for patients, to be able to deliver care in an expeditious, efficient, and cost-effective manner. This is what being physician is all about! Keep your state board of pharmacy off your license and out of your office.
Dr. Coldiron is a past president of the American Academy of Dermatology. He is currently in private practice, but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of more than 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics. Reach him at [email protected].
What does the pharmacy board have to do with me? I’m a physician, regulated by the state medical board. Well heads up. If regulations coming your way are adopted, you will have the additional privilege of being licensed, inspected by, and financially supporting your state pharmacy board.
How did all this happen? In 2012, a compounding pharmacy inadequately sterilized multiple lots of methylprednisolone, which were sold around the country and used for intrathecal injections. As a result, 753 patients developed fungal infections, including 386 cases of meningitis, and 64 of them died. The owner of the pharmacy and the head pharmacist are up on second degree murder charges.
But what does this have to do with you?
After a media bonfire, a congressional hearing complete with the taking of the fifth amendment, and a major rewrite of pharmacy regulations with increased scrutiny and oversight, the State of Ohio Board of Pharmacy rushed to adopt rules before reasonable regulations could be worked out by the Food and Drug Administration, the American Medical Association, and the Federation of State Medical Boards. The Ohio board of pharmacy adopted the U.S. Pharmacopeial Convention (USP) regulations, written for compounding pharmacies, and applied them to physicians’ offices.
In an overreaching bureaucratic coup de grace, any practitioners who reconstitute any drug in their offices is considered to be a compounding pharmacy, ordered to pay compounding pharmacy registration fees ($112 yearly), and to undergo the same inspections as compounding pharmacies. You can’t be too safe, you know, and all those registration fees (totaling about $2 million per year in Ohio alone) will decrease what would have been an onerous registration and inspection expense for true compounding pharmacies.
This is the reality we are facing in Ohio, and this situation may soon be “coming to a theater near you.” I understand that several pharmacy boards in other states are preparing to roll out similar regulations.
As a kicker, if the product you reconstitute is preservative free (botulinum toxin anyone?), you must use it or dispose of it within one hour. Yes, one hour. If you dilute bleomycin or 5-fluorouracil (5-FU), you must install an outside vented laminar flow hood, and wear level 5 hazmat gear while drawing it up.
Is this situation insane or what? As the result of a pharmacy in Massachusetts that skirted existing regulations and sold contaminated drugs that killed patients, doctors now need more regulations, licensing, inspections, and fees?
The real problem here, of course, is not the $112 fee. It will be the loss of many drugs and therapies that can be used inexpensively in the office, but will now either be unavailable to patients or available at a greatly increased cost. I pointed this situation out at a pharmacy board meeting, and they helpfully responded that I can have my friendly local pharmacist compound any drug I need in a specific strength and unit dose. Who is going to pay for this? I can make diclofenac or 5-FU cream in my office for less than $20. Instead, it will cost over $700 at the pharmacy! Further, making something fiscally impossible, like installing a laminar flow hood, is not different that denying it outright. I consider this to be restraint of trade.
Don’t allow yourselves to be compromised as Ohio physicians have been. You must be vigilant. Attend the public hearings and testify. In Ohio, the hearings were held over the Christmas holidays. Guess what? No one came to the hearings! You must show up and complain. Loudly. You must point out how patients are going to be hurt, not helped, by these rules. You must point out the superb safety record of physicians when using in-office pharmaceuticals. You must alert your neurology, ophthalmology, gynecology, and urology colleagues to the problem since they all use neurotoxins, too. The primary care doctors all reconstitute drugs (think antibiotics) for office use, too.
These efforts are also part of a larger campaign to give pharmacists a larger clinical role in patient care. If pharmacists license you, if they inspect your office, how can you oppose them when they want clinical privileges?
The fix is to enact a moratorium on regulations until the FDA rules come out. These will be more reasonable than the rules issued by the USP. Another fix is a legislative change that instructs that physicians, not pharmacists, will define what is considered to be a dangerous drug.
It is time to be alert, vigilant, and outspoken. You must do this to preserve your ability to do what is best for patients, to be able to deliver care in an expeditious, efficient, and cost-effective manner. This is what being physician is all about! Keep your state board of pharmacy off your license and out of your office.
Dr. Coldiron is a past president of the American Academy of Dermatology. He is currently in private practice, but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of more than 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics. Reach him at [email protected].

Experts present first recommendations for treating acne fulminans
SCOTTSDALE, ARIZ. – Patients with acne fulminans should first begin corticosteroid monotherapy before adding isotretinoin, according to the first evidence-based consensus recommendations on this disease.
Antibiotics should not be used as first line treatment or monotherapy for acne fulminans, according to the experts who drafted the recommendations. Acne fulminans is an uncommon, understudied, and severe disorder that “typically manifests as an explosive worsening and ulceration of skin lesions, and can be associated with fever, bone pain, and other systemic symptoms,” noted panel co-chairs Dr. Andrea Zaenglein, professor of dermatology and pediatric dermatology, Pennsylvania State University, Hershey, and Dr. Sheila Friedlander, professor of medicine and pediatrics, University of California, San Diego, together with their associates.
The recommendations – based on a full literature review, a 5-hour audioconference, and two rounds of surveys to achieve consensus on these topics – were summarized in a poster presented at the annual meeting of the Society for Investigative Dermatology.
Until now, there have been no clear guidelines on the pathogenesis, treatment, and prevention of acne fulminans, according to the panelists.
Consensus definitions
“Acne fulminans is not just an extreme form of acne, but rather, a distinct, likely auto-inflammatory disorder,” the experts stated. Affected patients typically have an “abrupt, dramatic flare of inflammatory acne, with erosions, and with or without crusts, hemorrhagic nodules/plaques, and systemic findings.”
Systemic involvement is uncommon, but when present, includes fever, malaise, bone pain, arthralgias, erythema nodosum, and leukocytosis, they noted. Some patients also have anemia, an elevated erythrocyte sedimentation rate, and an increased C-reactive protein level. Radiography typically reveals osteolytic lesions of the sternum, clavicles, sacroiliac joints, and hips.
Acne fulminans is most often triggered by isotretinoin therapy, but can occur without it, the panel said. Isotretinoin-induced acne fulminans can have systemic involvement, but usually does not.
Corticosteroids
Patients should start corticosteroid monotherapy at a dose of 0.5 to 1.0 mg per kg per day, according to the recommendations. Patients with systemic involvement should receive steroids for at least 4 weeks, and other patients should continue steroids for at least 2 weeks and until all lesions have healed.
Oral corticosteroids should be tapered slowly over about 4 to 8 weeks, first by halving the dose to a physiologic dose each week, and then by dosing every other day for 2 weeks. Topical corticosteroids also can be used for eroded sites with granulation tissue, they noted.
Isotretinoin
Ironically, isotretinoin is both a treatment and a potential trigger of acne fulminans, and the recommendations included detailed guidance on its use.
Patients should wait at least 2 weeks after crusting resolves before starting isotretinoin, and should overlap isotretinoin with corticosteroids for at least 4 weeks, the experts emphasized. They recommended starting isotretinoin at 0.1 mg per kg per day, and waiting at least 2 months to increase this dose. Because patients clear at different rates, there is no universal optimal cumulative dose of isotretinoin, they noted.
If patients on isotretinoin develop flare, crusts, and erosions, they should halt treatment and either start corticosteroids, or increase the steroid dose to 1.0 mg per kg. If crusts and erosions persist, the panelists recommended considering cyclosporine, biologics, or dapsone.
If hemorrhagic crusts or erosions resolve after stopping isotretinoin, it can be restarted at the initial dose of 0.1 mg per kg and overlapped with steroids for 4 weeks.
If flares, crusts, and erosions begin when patients on isotretinoin are tapering corticosteroids, then steroids should be continued without tapering, isotretinoin should be stopped temporarily, and it should be restarted at 0.1 mg per kg after crusts and erosions have healed. This dose should be continued for 4 weeks, and then slowly increased as tolerated.
Antibiotics
Antibiotics are not useful as monotherapy for first-line therapy for acne fulminans, according to the recommendations. But to avoid isotretinoin-induced acne fulminans, the experts often pretreat patients with oral antibiotics before starting isotretinoin, and may continue oral antibiotics during the initial phase of isotretinoin treatment. However, there have been no prospective studies supporting this approach, they noted.
Other treatment considerations
Acne fulminans lesions should not be debrided, the experts emphasized. Acne fulminans associated with SAPHO (synovitis, acne, pustulosis, hyperostosis, and osteitis), PAPA (pyogenic arthritis, pyoderma gangrenosum, and acne), and PAPASH (pyogenic arthritis, pyoderma gangrenosum, acne, and hidradenitis suppurativa) has responded successfully to tumor necrosis factor–alpha inhibitors and interleukin-1 receptor antagonists, they noted. Pulsed dye laser also has been used to successfully treat acne fulminans, particularly when there is associated granulation tissue, they stated.
Reducing the risk of pseudotumor cerebri syndrome
Several drugs used to treat acne fulminans have been linked to pseudotumor cerebri syndrome, the experts cautioned. “Among the tetracyclines, minocycline carries the highest risk,” they noted. Both isotretinoin and corticosteroids have been linked to pseudotumor cerebri syndrome, but clinicians can reduce this risk by avoiding an abrupt steroid taper, they emphasized.
The panel included physicians from the University of California, San Diego; Pennsylvania State University, Hershey; State University of New York, Brooklyn; Cornell University, New York; Touro University California, Vallejo; the University of California, San Francisco; the University of Pennsylvania, Philadelphia; and Jefferson Medical College, Philadelphia. They did not disclose conflicts of interest.
SCOTTSDALE, ARIZ. – Patients with acne fulminans should first begin corticosteroid monotherapy before adding isotretinoin, according to the first evidence-based consensus recommendations on this disease.
Antibiotics should not be used as first line treatment or monotherapy for acne fulminans, according to the experts who drafted the recommendations. Acne fulminans is an uncommon, understudied, and severe disorder that “typically manifests as an explosive worsening and ulceration of skin lesions, and can be associated with fever, bone pain, and other systemic symptoms,” noted panel co-chairs Dr. Andrea Zaenglein, professor of dermatology and pediatric dermatology, Pennsylvania State University, Hershey, and Dr. Sheila Friedlander, professor of medicine and pediatrics, University of California, San Diego, together with their associates.
The recommendations – based on a full literature review, a 5-hour audioconference, and two rounds of surveys to achieve consensus on these topics – were summarized in a poster presented at the annual meeting of the Society for Investigative Dermatology.
Until now, there have been no clear guidelines on the pathogenesis, treatment, and prevention of acne fulminans, according to the panelists.
Consensus definitions
“Acne fulminans is not just an extreme form of acne, but rather, a distinct, likely auto-inflammatory disorder,” the experts stated. Affected patients typically have an “abrupt, dramatic flare of inflammatory acne, with erosions, and with or without crusts, hemorrhagic nodules/plaques, and systemic findings.”
Systemic involvement is uncommon, but when present, includes fever, malaise, bone pain, arthralgias, erythema nodosum, and leukocytosis, they noted. Some patients also have anemia, an elevated erythrocyte sedimentation rate, and an increased C-reactive protein level. Radiography typically reveals osteolytic lesions of the sternum, clavicles, sacroiliac joints, and hips.
Acne fulminans is most often triggered by isotretinoin therapy, but can occur without it, the panel said. Isotretinoin-induced acne fulminans can have systemic involvement, but usually does not.
Corticosteroids
Patients should start corticosteroid monotherapy at a dose of 0.5 to 1.0 mg per kg per day, according to the recommendations. Patients with systemic involvement should receive steroids for at least 4 weeks, and other patients should continue steroids for at least 2 weeks and until all lesions have healed.
Oral corticosteroids should be tapered slowly over about 4 to 8 weeks, first by halving the dose to a physiologic dose each week, and then by dosing every other day for 2 weeks. Topical corticosteroids also can be used for eroded sites with granulation tissue, they noted.
Isotretinoin
Ironically, isotretinoin is both a treatment and a potential trigger of acne fulminans, and the recommendations included detailed guidance on its use.
Patients should wait at least 2 weeks after crusting resolves before starting isotretinoin, and should overlap isotretinoin with corticosteroids for at least 4 weeks, the experts emphasized. They recommended starting isotretinoin at 0.1 mg per kg per day, and waiting at least 2 months to increase this dose. Because patients clear at different rates, there is no universal optimal cumulative dose of isotretinoin, they noted.
If patients on isotretinoin develop flare, crusts, and erosions, they should halt treatment and either start corticosteroids, or increase the steroid dose to 1.0 mg per kg. If crusts and erosions persist, the panelists recommended considering cyclosporine, biologics, or dapsone.
If hemorrhagic crusts or erosions resolve after stopping isotretinoin, it can be restarted at the initial dose of 0.1 mg per kg and overlapped with steroids for 4 weeks.
If flares, crusts, and erosions begin when patients on isotretinoin are tapering corticosteroids, then steroids should be continued without tapering, isotretinoin should be stopped temporarily, and it should be restarted at 0.1 mg per kg after crusts and erosions have healed. This dose should be continued for 4 weeks, and then slowly increased as tolerated.
Antibiotics
Antibiotics are not useful as monotherapy for first-line therapy for acne fulminans, according to the recommendations. But to avoid isotretinoin-induced acne fulminans, the experts often pretreat patients with oral antibiotics before starting isotretinoin, and may continue oral antibiotics during the initial phase of isotretinoin treatment. However, there have been no prospective studies supporting this approach, they noted.
Other treatment considerations
Acne fulminans lesions should not be debrided, the experts emphasized. Acne fulminans associated with SAPHO (synovitis, acne, pustulosis, hyperostosis, and osteitis), PAPA (pyogenic arthritis, pyoderma gangrenosum, and acne), and PAPASH (pyogenic arthritis, pyoderma gangrenosum, acne, and hidradenitis suppurativa) has responded successfully to tumor necrosis factor–alpha inhibitors and interleukin-1 receptor antagonists, they noted. Pulsed dye laser also has been used to successfully treat acne fulminans, particularly when there is associated granulation tissue, they stated.
Reducing the risk of pseudotumor cerebri syndrome
Several drugs used to treat acne fulminans have been linked to pseudotumor cerebri syndrome, the experts cautioned. “Among the tetracyclines, minocycline carries the highest risk,” they noted. Both isotretinoin and corticosteroids have been linked to pseudotumor cerebri syndrome, but clinicians can reduce this risk by avoiding an abrupt steroid taper, they emphasized.
The panel included physicians from the University of California, San Diego; Pennsylvania State University, Hershey; State University of New York, Brooklyn; Cornell University, New York; Touro University California, Vallejo; the University of California, San Francisco; the University of Pennsylvania, Philadelphia; and Jefferson Medical College, Philadelphia. They did not disclose conflicts of interest.
SCOTTSDALE, ARIZ. – Patients with acne fulminans should first begin corticosteroid monotherapy before adding isotretinoin, according to the first evidence-based consensus recommendations on this disease.
Antibiotics should not be used as first line treatment or monotherapy for acne fulminans, according to the experts who drafted the recommendations. Acne fulminans is an uncommon, understudied, and severe disorder that “typically manifests as an explosive worsening and ulceration of skin lesions, and can be associated with fever, bone pain, and other systemic symptoms,” noted panel co-chairs Dr. Andrea Zaenglein, professor of dermatology and pediatric dermatology, Pennsylvania State University, Hershey, and Dr. Sheila Friedlander, professor of medicine and pediatrics, University of California, San Diego, together with their associates.
The recommendations – based on a full literature review, a 5-hour audioconference, and two rounds of surveys to achieve consensus on these topics – were summarized in a poster presented at the annual meeting of the Society for Investigative Dermatology.
Until now, there have been no clear guidelines on the pathogenesis, treatment, and prevention of acne fulminans, according to the panelists.
Consensus definitions
“Acne fulminans is not just an extreme form of acne, but rather, a distinct, likely auto-inflammatory disorder,” the experts stated. Affected patients typically have an “abrupt, dramatic flare of inflammatory acne, with erosions, and with or without crusts, hemorrhagic nodules/plaques, and systemic findings.”
Systemic involvement is uncommon, but when present, includes fever, malaise, bone pain, arthralgias, erythema nodosum, and leukocytosis, they noted. Some patients also have anemia, an elevated erythrocyte sedimentation rate, and an increased C-reactive protein level. Radiography typically reveals osteolytic lesions of the sternum, clavicles, sacroiliac joints, and hips.
Acne fulminans is most often triggered by isotretinoin therapy, but can occur without it, the panel said. Isotretinoin-induced acne fulminans can have systemic involvement, but usually does not.
Corticosteroids
Patients should start corticosteroid monotherapy at a dose of 0.5 to 1.0 mg per kg per day, according to the recommendations. Patients with systemic involvement should receive steroids for at least 4 weeks, and other patients should continue steroids for at least 2 weeks and until all lesions have healed.
Oral corticosteroids should be tapered slowly over about 4 to 8 weeks, first by halving the dose to a physiologic dose each week, and then by dosing every other day for 2 weeks. Topical corticosteroids also can be used for eroded sites with granulation tissue, they noted.
Isotretinoin
Ironically, isotretinoin is both a treatment and a potential trigger of acne fulminans, and the recommendations included detailed guidance on its use.
Patients should wait at least 2 weeks after crusting resolves before starting isotretinoin, and should overlap isotretinoin with corticosteroids for at least 4 weeks, the experts emphasized. They recommended starting isotretinoin at 0.1 mg per kg per day, and waiting at least 2 months to increase this dose. Because patients clear at different rates, there is no universal optimal cumulative dose of isotretinoin, they noted.
If patients on isotretinoin develop flare, crusts, and erosions, they should halt treatment and either start corticosteroids, or increase the steroid dose to 1.0 mg per kg. If crusts and erosions persist, the panelists recommended considering cyclosporine, biologics, or dapsone.
If hemorrhagic crusts or erosions resolve after stopping isotretinoin, it can be restarted at the initial dose of 0.1 mg per kg and overlapped with steroids for 4 weeks.
If flares, crusts, and erosions begin when patients on isotretinoin are tapering corticosteroids, then steroids should be continued without tapering, isotretinoin should be stopped temporarily, and it should be restarted at 0.1 mg per kg after crusts and erosions have healed. This dose should be continued for 4 weeks, and then slowly increased as tolerated.
Antibiotics
Antibiotics are not useful as monotherapy for first-line therapy for acne fulminans, according to the recommendations. But to avoid isotretinoin-induced acne fulminans, the experts often pretreat patients with oral antibiotics before starting isotretinoin, and may continue oral antibiotics during the initial phase of isotretinoin treatment. However, there have been no prospective studies supporting this approach, they noted.
Other treatment considerations
Acne fulminans lesions should not be debrided, the experts emphasized. Acne fulminans associated with SAPHO (synovitis, acne, pustulosis, hyperostosis, and osteitis), PAPA (pyogenic arthritis, pyoderma gangrenosum, and acne), and PAPASH (pyogenic arthritis, pyoderma gangrenosum, acne, and hidradenitis suppurativa) has responded successfully to tumor necrosis factor–alpha inhibitors and interleukin-1 receptor antagonists, they noted. Pulsed dye laser also has been used to successfully treat acne fulminans, particularly when there is associated granulation tissue, they stated.
Reducing the risk of pseudotumor cerebri syndrome
Several drugs used to treat acne fulminans have been linked to pseudotumor cerebri syndrome, the experts cautioned. “Among the tetracyclines, minocycline carries the highest risk,” they noted. Both isotretinoin and corticosteroids have been linked to pseudotumor cerebri syndrome, but clinicians can reduce this risk by avoiding an abrupt steroid taper, they emphasized.
The panel included physicians from the University of California, San Diego; Pennsylvania State University, Hershey; State University of New York, Brooklyn; Cornell University, New York; Touro University California, Vallejo; the University of California, San Francisco; the University of Pennsylvania, Philadelphia; and Jefferson Medical College, Philadelphia. They did not disclose conflicts of interest.
AT THE 2016 SID ANNUAL MEETING
Mobile App: VAM Info at Your Fingertips
At the Vascular Annual Meeting, there’s no need to cart around a lot of paper. Instead, the Mobile App puts all the information you need – abstracts, exhibitor map, schedules and more – within fingertip reach, to keep you more organized than ever.
Quick tips for using the Mobile App:

Create a personal schedule. Bookmark potential sessions in the program tab by clinking the small calendar icon the right for each session of interest. Reminders will pop up 10 minutes before the start time, including session details. The information will be pinned to the top of your activity feed to help you stay organized.
Review abstracts and take notes. The index contains all abstracts with author names and affiliations, presentation time, location, and a link to view the full abstract online. All index information is searchable. Take notes on each abstract within the app and export those notes to an email. Create a list of favorite abstracts by bookmarking those you want to refer to again.
Share. Share your thoughts in the app’s activity feed. Let all your friends know what you’re up to by linking your social media accounts to the app so you can post in the app and your social media feed at the same time!
The VAM Meeting App Is ...
Comprehensive: It includes all meeting content, including abstracts.
Searchable: Quickly locate sessions, abstracts, speakers and more.
Interactive: Network with colleagues, share photos and rate programs.
Visit vsweb.org/mobileapp to download the app.
At the Vascular Annual Meeting, there’s no need to cart around a lot of paper. Instead, the Mobile App puts all the information you need – abstracts, exhibitor map, schedules and more – within fingertip reach, to keep you more organized than ever.
Quick tips for using the Mobile App:
Create a personal schedule. Bookmark potential sessions in the program tab by clinking the small calendar icon the right for each session of interest. Reminders will pop up 10 minutes before the start time, including session details. The information will be pinned to the top of your activity feed to help you stay organized.
Review abstracts and take notes. The index contains all abstracts with author names and affiliations, presentation time, location, and a link to view the full abstract online. All index information is searchable. Take notes on each abstract within the app and export those notes to an email. Create a list of favorite abstracts by bookmarking those you want to refer to again.
Share. Share your thoughts in the app’s activity feed. Let all your friends know what you’re up to by linking your social media accounts to the app so you can post in the app and your social media feed at the same time!
The VAM Meeting App Is ...
Comprehensive: It includes all meeting content, including abstracts.
Searchable: Quickly locate sessions, abstracts, speakers and more.
Interactive: Network with colleagues, share photos and rate programs.
Visit vsweb.org/mobileapp to download the app.
At the Vascular Annual Meeting, there’s no need to cart around a lot of paper. Instead, the Mobile App puts all the information you need – abstracts, exhibitor map, schedules and more – within fingertip reach, to keep you more organized than ever.
Quick tips for using the Mobile App:
Create a personal schedule. Bookmark potential sessions in the program tab by clinking the small calendar icon the right for each session of interest. Reminders will pop up 10 minutes before the start time, including session details. The information will be pinned to the top of your activity feed to help you stay organized.
Review abstracts and take notes. The index contains all abstracts with author names and affiliations, presentation time, location, and a link to view the full abstract online. All index information is searchable. Take notes on each abstract within the app and export those notes to an email. Create a list of favorite abstracts by bookmarking those you want to refer to again.
Share. Share your thoughts in the app’s activity feed. Let all your friends know what you’re up to by linking your social media accounts to the app so you can post in the app and your social media feed at the same time!
The VAM Meeting App Is ...
Comprehensive: It includes all meeting content, including abstracts.
Searchable: Quickly locate sessions, abstracts, speakers and more.
Interactive: Network with colleagues, share photos and rate programs.
Visit vsweb.org/mobileapp to download the app.

Enhancing Mobility Reduces Length of Stay
Clinical question: Can a nurse-driven early ambulation program aimed at all hospitalized adults increase patient mobility and decrease length of stay?

Background: Many adults experience decline of functional abilities during their hospitalization. Interventions to increase early mobilization of patients in the ICU have been associated with decreased length of stay, decreased costs, and improved patient satisfaction. Previous studies of interventions in non-ICU patients have used specialized staff or have targeted select patient populations.
Study design: Before-after cohort study.
Setting: Patients admitted to two general medical units at a single large academic hospital.
Synopsis: The authors implemented a 12-month multidisciplinary quality improvement project in 3,352 patients, with the goal of mobilizing patients three times per day. Additional goals included consistently documenting daily mobility, setting daily goals to increase activity, and standardizing the description of mobility across disciplines. Ambulation, documentation, and goal setting were assigned to regular nursing staff and targeted at each of the patients admitted to these units during the study period. Highest level of mobility was documented using a locally derived simple eight-point ordinal scale. Daily documentation rate of mobility averaged 85% over the 12 months of the project. Comparing the four-month study period at the beginning of the project implementation to the four-month period after implementation, more patients ambulated (70% versus 43%), patients with improved mobility scores increased from 32% to 45%, and length of stay declined by 0.40 days. All of these differences were statistically significant. There was no increase in falls with injury.
Bottom line: A nurse-driven early mobility program aimed at all patients admitted to general medical services may improve mobility and decrease length of stay.
Citation: Hoyer EH, Friedman M, Lavezza A, et al. Promoting mobility and reducing length of stay in hospitalized general medicine patients: a quality-improvement project [published online ahead of print February 5, 2016]. J Hosp Med. doi:10.1002/jhm.2546.
Short Take
Prednisolone is Equivalent to NSAIDs in the Treatment of Acute Gout
In a multicenter, double-blind, randomized equivalence trial of 416 patients presenting to the emergency department with symptoms of acute gout, treatment with prednisolone was equivalent to indomethacin for pain treatment without any difference in adverse events.
Citation: Rainer TH, Cheng CH, Janssens HJEM, et al. Oral prednisolone in the treatment of acute gout: a pragmatic, multicenter, double-blind, randomized trial. Ann Intern Med. 2016;164(7):464-471. doi:10.7326/M14-2070.
Clinical question: Can a nurse-driven early ambulation program aimed at all hospitalized adults increase patient mobility and decrease length of stay?
Background: Many adults experience decline of functional abilities during their hospitalization. Interventions to increase early mobilization of patients in the ICU have been associated with decreased length of stay, decreased costs, and improved patient satisfaction. Previous studies of interventions in non-ICU patients have used specialized staff or have targeted select patient populations.
Study design: Before-after cohort study.
Setting: Patients admitted to two general medical units at a single large academic hospital.
Synopsis: The authors implemented a 12-month multidisciplinary quality improvement project in 3,352 patients, with the goal of mobilizing patients three times per day. Additional goals included consistently documenting daily mobility, setting daily goals to increase activity, and standardizing the description of mobility across disciplines. Ambulation, documentation, and goal setting were assigned to regular nursing staff and targeted at each of the patients admitted to these units during the study period. Highest level of mobility was documented using a locally derived simple eight-point ordinal scale. Daily documentation rate of mobility averaged 85% over the 12 months of the project. Comparing the four-month study period at the beginning of the project implementation to the four-month period after implementation, more patients ambulated (70% versus 43%), patients with improved mobility scores increased from 32% to 45%, and length of stay declined by 0.40 days. All of these differences were statistically significant. There was no increase in falls with injury.
Bottom line: A nurse-driven early mobility program aimed at all patients admitted to general medical services may improve mobility and decrease length of stay.
Citation: Hoyer EH, Friedman M, Lavezza A, et al. Promoting mobility and reducing length of stay in hospitalized general medicine patients: a quality-improvement project [published online ahead of print February 5, 2016]. J Hosp Med. doi:10.1002/jhm.2546.
Short Take
Prednisolone is Equivalent to NSAIDs in the Treatment of Acute Gout
In a multicenter, double-blind, randomized equivalence trial of 416 patients presenting to the emergency department with symptoms of acute gout, treatment with prednisolone was equivalent to indomethacin for pain treatment without any difference in adverse events.
Citation: Rainer TH, Cheng CH, Janssens HJEM, et al. Oral prednisolone in the treatment of acute gout: a pragmatic, multicenter, double-blind, randomized trial. Ann Intern Med. 2016;164(7):464-471. doi:10.7326/M14-2070.
Clinical question: Can a nurse-driven early ambulation program aimed at all hospitalized adults increase patient mobility and decrease length of stay?
Background: Many adults experience decline of functional abilities during their hospitalization. Interventions to increase early mobilization of patients in the ICU have been associated with decreased length of stay, decreased costs, and improved patient satisfaction. Previous studies of interventions in non-ICU patients have used specialized staff or have targeted select patient populations.
Study design: Before-after cohort study.
Setting: Patients admitted to two general medical units at a single large academic hospital.
Synopsis: The authors implemented a 12-month multidisciplinary quality improvement project in 3,352 patients, with the goal of mobilizing patients three times per day. Additional goals included consistently documenting daily mobility, setting daily goals to increase activity, and standardizing the description of mobility across disciplines. Ambulation, documentation, and goal setting were assigned to regular nursing staff and targeted at each of the patients admitted to these units during the study period. Highest level of mobility was documented using a locally derived simple eight-point ordinal scale. Daily documentation rate of mobility averaged 85% over the 12 months of the project. Comparing the four-month study period at the beginning of the project implementation to the four-month period after implementation, more patients ambulated (70% versus 43%), patients with improved mobility scores increased from 32% to 45%, and length of stay declined by 0.40 days. All of these differences were statistically significant. There was no increase in falls with injury.
Bottom line: A nurse-driven early mobility program aimed at all patients admitted to general medical services may improve mobility and decrease length of stay.
Citation: Hoyer EH, Friedman M, Lavezza A, et al. Promoting mobility and reducing length of stay in hospitalized general medicine patients: a quality-improvement project [published online ahead of print February 5, 2016]. J Hosp Med. doi:10.1002/jhm.2546.
Short Take
Prednisolone is Equivalent to NSAIDs in the Treatment of Acute Gout
In a multicenter, double-blind, randomized equivalence trial of 416 patients presenting to the emergency department with symptoms of acute gout, treatment with prednisolone was equivalent to indomethacin for pain treatment without any difference in adverse events.
Citation: Rainer TH, Cheng CH, Janssens HJEM, et al. Oral prednisolone in the treatment of acute gout: a pragmatic, multicenter, double-blind, randomized trial. Ann Intern Med. 2016;164(7):464-471. doi:10.7326/M14-2070.

Can Sepsis Be Better Defined?
Clinical question: Given advances in the understanding and treatment of sepsis, can sepsis be better defined?
Background: Definitions of sepsis and septic shock were last revised in 2001. The current definitions are based on a constellation of clinical signs and symptoms in a patient with suspected infection. Recent studies suggest that the definitions have low sensitivity and specificity, and they do not correlate well with patient outcomes.
Study design: Consensus guidelines.
Setting: Task force of 19 critical care, infectious disease, surgical, and pulmonary specialists convened in 2014 by the European Society of Intensive Care Medicine and the Society of Critical Care Medicine.
Synopsis: The task force recommended that sepsis be defined as life-threatening organ dysfunction caused by a dysregulated host response to infection and that it be identified by a change of more than one point in the Sequential Organ Failure Assessment (SOFA) score. This score incorporates the Glasgow Coma Scale, mean arterial blood pressure (MAP), PaO2/FiO2, platelet count, creatinine, and bilirubin. Septic shock is defined as a subset of sepsis with profound circulatory, cellular, and metabolic abnormalities, and it’s identified by serum lactate level >2 mmol/L and vasopressor requirement to maintain a MAP of ≥65 mm Hg in the absence of hypovolemia. These new definitions have higher sensitivity and specificity and can predict mortality more accurately. Patients with these definitions of sepsis and septic shock have in-hospital mortality >10% and >40%, respectively. The presence of two or more quick SOFA (qSOFA) elements (altered mentation, systolic blood pressure ≤100 mm Hg, and respiratory rate ≥22/min) identifies adult patients with suspected infection who need more extensive laboratory testing to exclude sepsis.
Bottom line: Defining sepsis now requires more laboratory testing but provides more diagnostic consistency and more accurately predicts outcomes.
Citation: Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801-810. doi:10.1001/jama.2016.0287.
Clinical question: Given advances in the understanding and treatment of sepsis, can sepsis be better defined?
Background: Definitions of sepsis and septic shock were last revised in 2001. The current definitions are based on a constellation of clinical signs and symptoms in a patient with suspected infection. Recent studies suggest that the definitions have low sensitivity and specificity, and they do not correlate well with patient outcomes.
Study design: Consensus guidelines.
Setting: Task force of 19 critical care, infectious disease, surgical, and pulmonary specialists convened in 2014 by the European Society of Intensive Care Medicine and the Society of Critical Care Medicine.
Synopsis: The task force recommended that sepsis be defined as life-threatening organ dysfunction caused by a dysregulated host response to infection and that it be identified by a change of more than one point in the Sequential Organ Failure Assessment (SOFA) score. This score incorporates the Glasgow Coma Scale, mean arterial blood pressure (MAP), PaO2/FiO2, platelet count, creatinine, and bilirubin. Septic shock is defined as a subset of sepsis with profound circulatory, cellular, and metabolic abnormalities, and it’s identified by serum lactate level >2 mmol/L and vasopressor requirement to maintain a MAP of ≥65 mm Hg in the absence of hypovolemia. These new definitions have higher sensitivity and specificity and can predict mortality more accurately. Patients with these definitions of sepsis and septic shock have in-hospital mortality >10% and >40%, respectively. The presence of two or more quick SOFA (qSOFA) elements (altered mentation, systolic blood pressure ≤100 mm Hg, and respiratory rate ≥22/min) identifies adult patients with suspected infection who need more extensive laboratory testing to exclude sepsis.
Bottom line: Defining sepsis now requires more laboratory testing but provides more diagnostic consistency and more accurately predicts outcomes.
Citation: Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801-810. doi:10.1001/jama.2016.0287.
Clinical question: Given advances in the understanding and treatment of sepsis, can sepsis be better defined?
Background: Definitions of sepsis and septic shock were last revised in 2001. The current definitions are based on a constellation of clinical signs and symptoms in a patient with suspected infection. Recent studies suggest that the definitions have low sensitivity and specificity, and they do not correlate well with patient outcomes.
Study design: Consensus guidelines.
Setting: Task force of 19 critical care, infectious disease, surgical, and pulmonary specialists convened in 2014 by the European Society of Intensive Care Medicine and the Society of Critical Care Medicine.
Synopsis: The task force recommended that sepsis be defined as life-threatening organ dysfunction caused by a dysregulated host response to infection and that it be identified by a change of more than one point in the Sequential Organ Failure Assessment (SOFA) score. This score incorporates the Glasgow Coma Scale, mean arterial blood pressure (MAP), PaO2/FiO2, platelet count, creatinine, and bilirubin. Septic shock is defined as a subset of sepsis with profound circulatory, cellular, and metabolic abnormalities, and it’s identified by serum lactate level >2 mmol/L and vasopressor requirement to maintain a MAP of ≥65 mm Hg in the absence of hypovolemia. These new definitions have higher sensitivity and specificity and can predict mortality more accurately. Patients with these definitions of sepsis and septic shock have in-hospital mortality >10% and >40%, respectively. The presence of two or more quick SOFA (qSOFA) elements (altered mentation, systolic blood pressure ≤100 mm Hg, and respiratory rate ≥22/min) identifies adult patients with suspected infection who need more extensive laboratory testing to exclude sepsis.
Bottom line: Defining sepsis now requires more laboratory testing but provides more diagnostic consistency and more accurately predicts outcomes.
Citation: Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801-810. doi:10.1001/jama.2016.0287.
Team develops new approach to programming T cells
Using mouse models, researchers have developed a new cellular programming approach to create alloreactive T cells they say eliminate leukemic cells without causing graft-versus-host disease (GVHD).
They created the T cells using the donor key immune cell. When used in allogeneic hematopoietic stem cell transplantation and anti-leukemia therapy, the new approach reduced the toxicities that cause GVHD while preserving the anti-leukemia activity of the immune cell.
“This approach will be useful in the future when developing novel methods for immunotherapy,” said Yi Zhang, MD, PhD, of Temple University in Philadelphia, Pennsylvania.
Dr Zhang and colleagues took murine bone marrow using Flt3 ligand and Toll-like receptor agonists to produce δ-like ligand 4-positive dendritic cells (Dll4hiDCs). When the dendritic cells were stimulated, CD4+ naïve T cells underwent effector differentiation and produced high levels of IFN-γ and IL-17 in vitro.
The team then transferred the allogeneic Dll4hiDC-induced T cells into the mice. The cells did not induce severe GVHD and preserved anti-leukemic activity, “significantly improving the survival of leukemic mice undergoing allogeneic HSCT,” they said.
They noted that the IFN-γ was important for Dll4hiDC programming in reducing the GVHD toxicities of alloreactive T cells. When the researchers transferred unstimulated T cells into mice, 5 of 8 mice died from GVHD and 3 of 8 died with tumor. Those that received Dll4hiDC-induced T cells did not develop GVHD.
They also emphasized that this platform does not require transfection with viral vectors, which has limitations of safety and efficiency.
“This system will not only be useful for reducing GvHD,” Dr Zhang said, “but can also be used in the identification of T cells for the improvement of other types of immunotherapy for advanced cancer.”
The team published this research in Blood. ![]()
Using mouse models, researchers have developed a new cellular programming approach to create alloreactive T cells they say eliminate leukemic cells without causing graft-versus-host disease (GVHD).
They created the T cells using the donor key immune cell. When used in allogeneic hematopoietic stem cell transplantation and anti-leukemia therapy, the new approach reduced the toxicities that cause GVHD while preserving the anti-leukemia activity of the immune cell.
“This approach will be useful in the future when developing novel methods for immunotherapy,” said Yi Zhang, MD, PhD, of Temple University in Philadelphia, Pennsylvania.
Dr Zhang and colleagues took murine bone marrow using Flt3 ligand and Toll-like receptor agonists to produce δ-like ligand 4-positive dendritic cells (Dll4hiDCs). When the dendritic cells were stimulated, CD4+ naïve T cells underwent effector differentiation and produced high levels of IFN-γ and IL-17 in vitro.
The team then transferred the allogeneic Dll4hiDC-induced T cells into the mice. The cells did not induce severe GVHD and preserved anti-leukemic activity, “significantly improving the survival of leukemic mice undergoing allogeneic HSCT,” they said.
They noted that the IFN-γ was important for Dll4hiDC programming in reducing the GVHD toxicities of alloreactive T cells. When the researchers transferred unstimulated T cells into mice, 5 of 8 mice died from GVHD and 3 of 8 died with tumor. Those that received Dll4hiDC-induced T cells did not develop GVHD.
They also emphasized that this platform does not require transfection with viral vectors, which has limitations of safety and efficiency.
“This system will not only be useful for reducing GvHD,” Dr Zhang said, “but can also be used in the identification of T cells for the improvement of other types of immunotherapy for advanced cancer.”
The team published this research in Blood. ![]()
Using mouse models, researchers have developed a new cellular programming approach to create alloreactive T cells they say eliminate leukemic cells without causing graft-versus-host disease (GVHD).
They created the T cells using the donor key immune cell. When used in allogeneic hematopoietic stem cell transplantation and anti-leukemia therapy, the new approach reduced the toxicities that cause GVHD while preserving the anti-leukemia activity of the immune cell.
“This approach will be useful in the future when developing novel methods for immunotherapy,” said Yi Zhang, MD, PhD, of Temple University in Philadelphia, Pennsylvania.
Dr Zhang and colleagues took murine bone marrow using Flt3 ligand and Toll-like receptor agonists to produce δ-like ligand 4-positive dendritic cells (Dll4hiDCs). When the dendritic cells were stimulated, CD4+ naïve T cells underwent effector differentiation and produced high levels of IFN-γ and IL-17 in vitro.
The team then transferred the allogeneic Dll4hiDC-induced T cells into the mice. The cells did not induce severe GVHD and preserved anti-leukemic activity, “significantly improving the survival of leukemic mice undergoing allogeneic HSCT,” they said.
They noted that the IFN-γ was important for Dll4hiDC programming in reducing the GVHD toxicities of alloreactive T cells. When the researchers transferred unstimulated T cells into mice, 5 of 8 mice died from GVHD and 3 of 8 died with tumor. Those that received Dll4hiDC-induced T cells did not develop GVHD.
They also emphasized that this platform does not require transfection with viral vectors, which has limitations of safety and efficiency.
“This system will not only be useful for reducing GvHD,” Dr Zhang said, “but can also be used in the identification of T cells for the improvement of other types of immunotherapy for advanced cancer.”
The team published this research in Blood. ![]()
Young HCT survivors have increased risk for frailty
for transplant
Photo credit: Chad McNeeley
Frailty among young adult hematopoietic cell transplant (HCT) survivors is high and approaches that of a community-based elderly population, according to results of the Bone Marrow Transplant Survivor Study.
Of the 998 HCT participants, frailty exceeded 8%, and they were 8.4 times more likely to be frail than their siblings.
Investigators defined frailty as exhibiting 3 or more of the following traits: clinically underweight, exhaustion, low energy expenditure, slow walking speed, and muscle weakness.
Because HCT recipients are exposed to high-intensity chemotherapy, radiation, and immunosuppression at points before, during, and after transplant, the investigators set out to determine whether non-elderly HCT recipients who have survived 2 years or more after transplant were at a higher risk of frailty compared with a sibling comparator group.
Smita Bhatia, MD, of the University of Alabama at Birmingham, and colleagues conducted the study of HCT survivors between the ages of 18 and 64 and compared the results to a sibling control group. The authors also looked at the subsequent mortality of HCT survivors.
They reported their findings in JAMA Oncology.
The 998 HCT survivors who participated in the study received their transplants at City of Hope in Duarte, California, or at the University of Minnesota in Minneapolis, between 1974 and 1998. The survivors and the 297 siblings completed questionnaires between February 1999 and June 2005.
Demographics
The HCT survivors were a mean age of 42.5 years, and 911 (93%) had health insurance coverage.
This was comparable to the sibling controls, who were a mean age of 43.8 years (P=0.09) and 279 (95%) had health insurance coverage.
However, more siblings were female (64%), non-Hispanic white (88%), college graduates (56%), and 92% had annual household incomes of $20,000 or more.
In the HCT survivor group, 46% were female (P<0.001), 81% non-Hispanic white (P=0.004), 49% college graduates (P<0.001), and 80% had annual incomes of $20,000 or more (P<0.001).
HCT survivors were a mean age of 33.8 years when they had their transplants and the interval between HCT and participation in the study was 8.7 years.
Hematologic malignancies were the major diagnoses leading to HCT. Twenty-three percent had a primary diagnosis of chronic myeloid leukemia, 24% had acute myeloid leukemia, 19% had non-Hodgkin lymphoma, 10% had acute lymphoblastic leukemia, and 9% had Hodgkin lymphoma.
Seventy-seven percent of the HCT survivors had total body irradiation, and 300 of the 562 who received allogeneic transplants had chronic GVHD, with 24% of them reporting active GVHD at the time they participated in the study.
Frailty
Only 2 siblings (0.7%) considered themselves frail compared to 84 (8.4%) HCT survivors.
More survivors were underweight and reported low energy expenditure compared to the sibling group, but the differences were not statistically significant, P=0.26 and P=0.14, respectively.
However, significantly more survivors reported exhaustion (P<0.001), slowness (P<0.001), and weakness (P<0.001) compared to the sibling group,
The investigators then adjusted the data for age at study participation, sex, race/ethnicity, education, household income, health insurance, presence of grades 3 or 4 chronic health conditions, and transplant institution. They then found the HCT survivors to be 8.35 times more likely to be frail than their siblings (P=0.003).
HCT survivors with low annual incomes (P=0.03), less than a college education (P=0.002), with grades 3 or 4 chronic health conditions (P=0.02), with multiple myeloma (P=0.05), or with resolved chronic (P=0.04) or active chronic GVHD (P<0.001) were more likely to be frail compared to the other HCT survivors.
Mortality
The investigators followed the patients for a median of 10.3 years from the time participants completed the survey. At that time, 182 (18%) patients had died.
The 10-year cumulative all-cause mortality was 39.3% for patients with frailty and 14.7% for patients without frailty (P<0.001).
The 10-year cumulative relapse-related mortality was 15.5% among frail HCT patients and 4.5% for non-frail HCT patients.
And the 10-year cumulative non-relapse mortality was also higher among frail HCT recipients, 23.9% compared to 10.2% of the non-frail HCT recipients (P<0.001).
Multivariate analysis revealed that frailty was associated with a 2.76-fold increase in death. The variables included age at study participation, sex, presence of grades 3 to 4 chronic health conditions, primary diagnosis, annual household income, and risk of relapse at transplant.
The investigators concluded that the therapies transplant patients undergo and post-transplant complications constitute a substantial stressor, placing HCT survivors at risk for frailty and premature aging.
“These findings demonstrate the need for interventions,” they added, “including personalized assessments and multidisciplinary efforts targeting both pre-frail and frail individuals to improve outcomes.” ![]()
for transplant
Photo credit: Chad McNeeley
Frailty among young adult hematopoietic cell transplant (HCT) survivors is high and approaches that of a community-based elderly population, according to results of the Bone Marrow Transplant Survivor Study.
Of the 998 HCT participants, frailty exceeded 8%, and they were 8.4 times more likely to be frail than their siblings.
Investigators defined frailty as exhibiting 3 or more of the following traits: clinically underweight, exhaustion, low energy expenditure, slow walking speed, and muscle weakness.
Because HCT recipients are exposed to high-intensity chemotherapy, radiation, and immunosuppression at points before, during, and after transplant, the investigators set out to determine whether non-elderly HCT recipients who have survived 2 years or more after transplant were at a higher risk of frailty compared with a sibling comparator group.
Smita Bhatia, MD, of the University of Alabama at Birmingham, and colleagues conducted the study of HCT survivors between the ages of 18 and 64 and compared the results to a sibling control group. The authors also looked at the subsequent mortality of HCT survivors.
They reported their findings in JAMA Oncology.
The 998 HCT survivors who participated in the study received their transplants at City of Hope in Duarte, California, or at the University of Minnesota in Minneapolis, between 1974 and 1998. The survivors and the 297 siblings completed questionnaires between February 1999 and June 2005.
Demographics
The HCT survivors were a mean age of 42.5 years, and 911 (93%) had health insurance coverage.
This was comparable to the sibling controls, who were a mean age of 43.8 years (P=0.09) and 279 (95%) had health insurance coverage.
However, more siblings were female (64%), non-Hispanic white (88%), college graduates (56%), and 92% had annual household incomes of $20,000 or more.
In the HCT survivor group, 46% were female (P<0.001), 81% non-Hispanic white (P=0.004), 49% college graduates (P<0.001), and 80% had annual incomes of $20,000 or more (P<0.001).
HCT survivors were a mean age of 33.8 years when they had their transplants and the interval between HCT and participation in the study was 8.7 years.
Hematologic malignancies were the major diagnoses leading to HCT. Twenty-three percent had a primary diagnosis of chronic myeloid leukemia, 24% had acute myeloid leukemia, 19% had non-Hodgkin lymphoma, 10% had acute lymphoblastic leukemia, and 9% had Hodgkin lymphoma.
Seventy-seven percent of the HCT survivors had total body irradiation, and 300 of the 562 who received allogeneic transplants had chronic GVHD, with 24% of them reporting active GVHD at the time they participated in the study.
Frailty
Only 2 siblings (0.7%) considered themselves frail compared to 84 (8.4%) HCT survivors.
More survivors were underweight and reported low energy expenditure compared to the sibling group, but the differences were not statistically significant, P=0.26 and P=0.14, respectively.
However, significantly more survivors reported exhaustion (P<0.001), slowness (P<0.001), and weakness (P<0.001) compared to the sibling group,
The investigators then adjusted the data for age at study participation, sex, race/ethnicity, education, household income, health insurance, presence of grades 3 or 4 chronic health conditions, and transplant institution. They then found the HCT survivors to be 8.35 times more likely to be frail than their siblings (P=0.003).
HCT survivors with low annual incomes (P=0.03), less than a college education (P=0.002), with grades 3 or 4 chronic health conditions (P=0.02), with multiple myeloma (P=0.05), or with resolved chronic (P=0.04) or active chronic GVHD (P<0.001) were more likely to be frail compared to the other HCT survivors.
Mortality
The investigators followed the patients for a median of 10.3 years from the time participants completed the survey. At that time, 182 (18%) patients had died.
The 10-year cumulative all-cause mortality was 39.3% for patients with frailty and 14.7% for patients without frailty (P<0.001).
The 10-year cumulative relapse-related mortality was 15.5% among frail HCT patients and 4.5% for non-frail HCT patients.
And the 10-year cumulative non-relapse mortality was also higher among frail HCT recipients, 23.9% compared to 10.2% of the non-frail HCT recipients (P<0.001).
Multivariate analysis revealed that frailty was associated with a 2.76-fold increase in death. The variables included age at study participation, sex, presence of grades 3 to 4 chronic health conditions, primary diagnosis, annual household income, and risk of relapse at transplant.
The investigators concluded that the therapies transplant patients undergo and post-transplant complications constitute a substantial stressor, placing HCT survivors at risk for frailty and premature aging.
“These findings demonstrate the need for interventions,” they added, “including personalized assessments and multidisciplinary efforts targeting both pre-frail and frail individuals to improve outcomes.” ![]()
for transplant
Photo credit: Chad McNeeley
Frailty among young adult hematopoietic cell transplant (HCT) survivors is high and approaches that of a community-based elderly population, according to results of the Bone Marrow Transplant Survivor Study.
Of the 998 HCT participants, frailty exceeded 8%, and they were 8.4 times more likely to be frail than their siblings.
Investigators defined frailty as exhibiting 3 or more of the following traits: clinically underweight, exhaustion, low energy expenditure, slow walking speed, and muscle weakness.
Because HCT recipients are exposed to high-intensity chemotherapy, radiation, and immunosuppression at points before, during, and after transplant, the investigators set out to determine whether non-elderly HCT recipients who have survived 2 years or more after transplant were at a higher risk of frailty compared with a sibling comparator group.
Smita Bhatia, MD, of the University of Alabama at Birmingham, and colleagues conducted the study of HCT survivors between the ages of 18 and 64 and compared the results to a sibling control group. The authors also looked at the subsequent mortality of HCT survivors.
They reported their findings in JAMA Oncology.
The 998 HCT survivors who participated in the study received their transplants at City of Hope in Duarte, California, or at the University of Minnesota in Minneapolis, between 1974 and 1998. The survivors and the 297 siblings completed questionnaires between February 1999 and June 2005.
Demographics
The HCT survivors were a mean age of 42.5 years, and 911 (93%) had health insurance coverage.
This was comparable to the sibling controls, who were a mean age of 43.8 years (P=0.09) and 279 (95%) had health insurance coverage.
However, more siblings were female (64%), non-Hispanic white (88%), college graduates (56%), and 92% had annual household incomes of $20,000 or more.
In the HCT survivor group, 46% were female (P<0.001), 81% non-Hispanic white (P=0.004), 49% college graduates (P<0.001), and 80% had annual incomes of $20,000 or more (P<0.001).
HCT survivors were a mean age of 33.8 years when they had their transplants and the interval between HCT and participation in the study was 8.7 years.
Hematologic malignancies were the major diagnoses leading to HCT. Twenty-three percent had a primary diagnosis of chronic myeloid leukemia, 24% had acute myeloid leukemia, 19% had non-Hodgkin lymphoma, 10% had acute lymphoblastic leukemia, and 9% had Hodgkin lymphoma.
Seventy-seven percent of the HCT survivors had total body irradiation, and 300 of the 562 who received allogeneic transplants had chronic GVHD, with 24% of them reporting active GVHD at the time they participated in the study.
Frailty
Only 2 siblings (0.7%) considered themselves frail compared to 84 (8.4%) HCT survivors.
More survivors were underweight and reported low energy expenditure compared to the sibling group, but the differences were not statistically significant, P=0.26 and P=0.14, respectively.
However, significantly more survivors reported exhaustion (P<0.001), slowness (P<0.001), and weakness (P<0.001) compared to the sibling group,
The investigators then adjusted the data for age at study participation, sex, race/ethnicity, education, household income, health insurance, presence of grades 3 or 4 chronic health conditions, and transplant institution. They then found the HCT survivors to be 8.35 times more likely to be frail than their siblings (P=0.003).
HCT survivors with low annual incomes (P=0.03), less than a college education (P=0.002), with grades 3 or 4 chronic health conditions (P=0.02), with multiple myeloma (P=0.05), or with resolved chronic (P=0.04) or active chronic GVHD (P<0.001) were more likely to be frail compared to the other HCT survivors.
Mortality
The investigators followed the patients for a median of 10.3 years from the time participants completed the survey. At that time, 182 (18%) patients had died.
The 10-year cumulative all-cause mortality was 39.3% for patients with frailty and 14.7% for patients without frailty (P<0.001).
The 10-year cumulative relapse-related mortality was 15.5% among frail HCT patients and 4.5% for non-frail HCT patients.
And the 10-year cumulative non-relapse mortality was also higher among frail HCT recipients, 23.9% compared to 10.2% of the non-frail HCT recipients (P<0.001).
Multivariate analysis revealed that frailty was associated with a 2.76-fold increase in death. The variables included age at study participation, sex, presence of grades 3 to 4 chronic health conditions, primary diagnosis, annual household income, and risk of relapse at transplant.
The investigators concluded that the therapies transplant patients undergo and post-transplant complications constitute a substantial stressor, placing HCT survivors at risk for frailty and premature aging.
“These findings demonstrate the need for interventions,” they added, “including personalized assessments and multidisciplinary efforts targeting both pre-frail and frail individuals to improve outcomes.” ![]()