User login
The Association Between Sleep and Seizure Types
Patients with non-acquired focal epilepsy are more likely to experience seizures while asleep, when compared to patients with generalized epilepsy. An analysis of nearly 1400 patients enrolled in the Epilepsy Phenome/Genome Project also revealed that these sleep/wake patterns applied to both convulsive and nonconvulsive seizures. The study further found that seizures occurring within an hour of awakening were more likely to happen in patients with generalized epilepsy, for both convulsive and nonconvulsive seizures. The researchers also discovered that the timing of seizures in first degree relatives predicted the timing of seizures in the proband, suggesting a genetic underpinning to the correlations.
Winawer MR, Shih J, Beck ES, Hunter JE, Epstein MP; EPGP Investigators. Genetic effects on sleep/wake variation on seizures. Epilepsia. 2016;57(4):557-665.
Patients with non-acquired focal epilepsy are more likely to experience seizures while asleep, when compared to patients with generalized epilepsy. An analysis of nearly 1400 patients enrolled in the Epilepsy Phenome/Genome Project also revealed that these sleep/wake patterns applied to both convulsive and nonconvulsive seizures. The study further found that seizures occurring within an hour of awakening were more likely to happen in patients with generalized epilepsy, for both convulsive and nonconvulsive seizures. The researchers also discovered that the timing of seizures in first degree relatives predicted the timing of seizures in the proband, suggesting a genetic underpinning to the correlations.
Winawer MR, Shih J, Beck ES, Hunter JE, Epstein MP; EPGP Investigators. Genetic effects on sleep/wake variation on seizures. Epilepsia. 2016;57(4):557-665.
Patients with non-acquired focal epilepsy are more likely to experience seizures while asleep, when compared to patients with generalized epilepsy. An analysis of nearly 1400 patients enrolled in the Epilepsy Phenome/Genome Project also revealed that these sleep/wake patterns applied to both convulsive and nonconvulsive seizures. The study further found that seizures occurring within an hour of awakening were more likely to happen in patients with generalized epilepsy, for both convulsive and nonconvulsive seizures. The researchers also discovered that the timing of seizures in first degree relatives predicted the timing of seizures in the proband, suggesting a genetic underpinning to the correlations.
Winawer MR, Shih J, Beck ES, Hunter JE, Epstein MP; EPGP Investigators. Genetic effects on sleep/wake variation on seizures. Epilepsia. 2016;57(4):557-665.
The Stigma Attached to Epilepsy is Alive and Unwell
Misconceptions about epilepsy abound in the Western world. An analysis of English language publications revealed that many people have “socially exclusionary attitudes” toward persons with epilepsy, are ignorant about proper treatment, and tend to overgeneralize about people with epilepsy in a way that stigmatizes them. The literature review also found that intervention studies have been effective in improving attitudes about the disease but concluded that “many were targeted to healthcare and education settings, were time intensive, and impractical for broad general population implementation.”
Herrman LK, Welter E, Berg AT, et al. Epilepsy misconceptions and stigma reduction: current status in Western countries. Epilepsy Behav. 2016;60:165-173.
Misconceptions about epilepsy abound in the Western world. An analysis of English language publications revealed that many people have “socially exclusionary attitudes” toward persons with epilepsy, are ignorant about proper treatment, and tend to overgeneralize about people with epilepsy in a way that stigmatizes them. The literature review also found that intervention studies have been effective in improving attitudes about the disease but concluded that “many were targeted to healthcare and education settings, were time intensive, and impractical for broad general population implementation.”
Herrman LK, Welter E, Berg AT, et al. Epilepsy misconceptions and stigma reduction: current status in Western countries. Epilepsy Behav. 2016;60:165-173.
Misconceptions about epilepsy abound in the Western world. An analysis of English language publications revealed that many people have “socially exclusionary attitudes” toward persons with epilepsy, are ignorant about proper treatment, and tend to overgeneralize about people with epilepsy in a way that stigmatizes them. The literature review also found that intervention studies have been effective in improving attitudes about the disease but concluded that “many were targeted to healthcare and education settings, were time intensive, and impractical for broad general population implementation.”
Herrman LK, Welter E, Berg AT, et al. Epilepsy misconceptions and stigma reduction: current status in Western countries. Epilepsy Behav. 2016;60:165-173.
Wilson disease
To the Editor: We read the IM Board Review article by Hanouneh et al in the February issue of the Journal with great interest.1 The authors described an interesting case of a young woman presenting with what initially seemed to be jaundice of acute onset, with rapid progression to acute encephalopathy and worsening liver failure. The patient was eventually diagnosed with fulminant Wilson disease and, thankfully, underwent successful liver transplant. We thank the authors for their in-depth review of the common causes of acute liver failure, the general approach to management, and the tailored treatment of Wilson disease in such settings.
However, we believe that several aspects merit further attention. First, on initial presentation and investigation, it would have been important to consider cholestatic hepatobiliary pathologic processes (eg, choledocholithiasis, cholangitis, primary biliary cirrhosis, primary sclerosing cholangitis), given the characteristic liver panel results.
Second, the authors rightly pointed out that hemolytic anemia is common in patients with acute liver failure secondary to Wilson disease. However, it is important to keep in mind that additional testing should include Coombs testing (typically negative in Wilson disease) and examination of the peripheral smear to exclude other etiologies, since such conditions as thrombotic thrombocytopenic purpura may present with multiorgan failure as well.2
Third, the authors report that Kayser-Fleischer rings are pathognomonic for Wilson disease. However, many reports in peer-reviewed medical journals suggest that this may not be the case and the overall clinical picture should be
considered.3
Fourth, while the authors focus their attention on liver transplant, several other treatments deserve mentioning. We agree that liver transplant is considered the only lifesaving treatment. But in certain situations, molecular absorbent recirculation systems and hemodialysis may provide temporary support while awaiting transportation to a liver transplant center or actual liver transplant.4
- Hanouneh MA, Garber A, Tavill AS, Zein NN, Hanouneh IA. A tale of two sisters with liver disease. Cleve Clin J Med 2016; 83:109–115.
- Nguyen TC, Cruz MA, Carcillo JA. Thrombocytopenia-associated multiple organ failure and acute kidney injury. Crit Care Clin 2015; 31:661–674.
- Frommer D, Morris J, Sherlock S, Abrams J, Newman S. Kayser-Fleischer-like rings in patients without Wilson’s disease. Gastroenterology 1977; 72:1331–1335.
- Hamlyn AN, Gollan JL, Douglas AP, Sherlock S. Fulminant Wilson’s disease with haemolysis and renal failure: copper studies and assessment of dialysis regimens. Br Med J 1977; 2:660–663.
To the Editor: We read the IM Board Review article by Hanouneh et al in the February issue of the Journal with great interest.1 The authors described an interesting case of a young woman presenting with what initially seemed to be jaundice of acute onset, with rapid progression to acute encephalopathy and worsening liver failure. The patient was eventually diagnosed with fulminant Wilson disease and, thankfully, underwent successful liver transplant. We thank the authors for their in-depth review of the common causes of acute liver failure, the general approach to management, and the tailored treatment of Wilson disease in such settings.
However, we believe that several aspects merit further attention. First, on initial presentation and investigation, it would have been important to consider cholestatic hepatobiliary pathologic processes (eg, choledocholithiasis, cholangitis, primary biliary cirrhosis, primary sclerosing cholangitis), given the characteristic liver panel results.
Second, the authors rightly pointed out that hemolytic anemia is common in patients with acute liver failure secondary to Wilson disease. However, it is important to keep in mind that additional testing should include Coombs testing (typically negative in Wilson disease) and examination of the peripheral smear to exclude other etiologies, since such conditions as thrombotic thrombocytopenic purpura may present with multiorgan failure as well.2
Third, the authors report that Kayser-Fleischer rings are pathognomonic for Wilson disease. However, many reports in peer-reviewed medical journals suggest that this may not be the case and the overall clinical picture should be
considered.3
Fourth, while the authors focus their attention on liver transplant, several other treatments deserve mentioning. We agree that liver transplant is considered the only lifesaving treatment. But in certain situations, molecular absorbent recirculation systems and hemodialysis may provide temporary support while awaiting transportation to a liver transplant center or actual liver transplant.4
To the Editor: We read the IM Board Review article by Hanouneh et al in the February issue of the Journal with great interest.1 The authors described an interesting case of a young woman presenting with what initially seemed to be jaundice of acute onset, with rapid progression to acute encephalopathy and worsening liver failure. The patient was eventually diagnosed with fulminant Wilson disease and, thankfully, underwent successful liver transplant. We thank the authors for their in-depth review of the common causes of acute liver failure, the general approach to management, and the tailored treatment of Wilson disease in such settings.
However, we believe that several aspects merit further attention. First, on initial presentation and investigation, it would have been important to consider cholestatic hepatobiliary pathologic processes (eg, choledocholithiasis, cholangitis, primary biliary cirrhosis, primary sclerosing cholangitis), given the characteristic liver panel results.
Second, the authors rightly pointed out that hemolytic anemia is common in patients with acute liver failure secondary to Wilson disease. However, it is important to keep in mind that additional testing should include Coombs testing (typically negative in Wilson disease) and examination of the peripheral smear to exclude other etiologies, since such conditions as thrombotic thrombocytopenic purpura may present with multiorgan failure as well.2
Third, the authors report that Kayser-Fleischer rings are pathognomonic for Wilson disease. However, many reports in peer-reviewed medical journals suggest that this may not be the case and the overall clinical picture should be
considered.3
Fourth, while the authors focus their attention on liver transplant, several other treatments deserve mentioning. We agree that liver transplant is considered the only lifesaving treatment. But in certain situations, molecular absorbent recirculation systems and hemodialysis may provide temporary support while awaiting transportation to a liver transplant center or actual liver transplant.4
- Hanouneh MA, Garber A, Tavill AS, Zein NN, Hanouneh IA. A tale of two sisters with liver disease. Cleve Clin J Med 2016; 83:109–115.
- Nguyen TC, Cruz MA, Carcillo JA. Thrombocytopenia-associated multiple organ failure and acute kidney injury. Crit Care Clin 2015; 31:661–674.
- Frommer D, Morris J, Sherlock S, Abrams J, Newman S. Kayser-Fleischer-like rings in patients without Wilson’s disease. Gastroenterology 1977; 72:1331–1335.
- Hamlyn AN, Gollan JL, Douglas AP, Sherlock S. Fulminant Wilson’s disease with haemolysis and renal failure: copper studies and assessment of dialysis regimens. Br Med J 1977; 2:660–663.
- Hanouneh MA, Garber A, Tavill AS, Zein NN, Hanouneh IA. A tale of two sisters with liver disease. Cleve Clin J Med 2016; 83:109–115.
- Nguyen TC, Cruz MA, Carcillo JA. Thrombocytopenia-associated multiple organ failure and acute kidney injury. Crit Care Clin 2015; 31:661–674.
- Frommer D, Morris J, Sherlock S, Abrams J, Newman S. Kayser-Fleischer-like rings in patients without Wilson’s disease. Gastroenterology 1977; 72:1331–1335.
- Hamlyn AN, Gollan JL, Douglas AP, Sherlock S. Fulminant Wilson’s disease with haemolysis and renal failure: copper studies and assessment of dialysis regimens. Br Med J 1977; 2:660–663.
In reply: Wilson disease
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
What’s the most effective topical Tx for scalp psoriasis?
Single-agent therapy with a very potent or potent topical corticosteroid appears more effective than other topical agents, including vitamin D3 analogues, for treating scalp psoriasis (strength of recommendation [SOR]: A, systematic reviews of randomized controlled trials [RCTs]).
Combined therapy with a vitamin D3 analogue and a potent topical corticosteroid may be slightly more effective than monotherapy with either agent (SOR: B, systematic reviews of RCTs with inconsistent results).
Evidence summary
A 2013 meta-analysis of 26 RCTs with 8020 patients evaluated topical treatments for scalp psoriasis as part of a subanalysis of a larger Cochrane review of psoriasis therapy.1 Only 20 studies reported the severity of disease: 13 studies looked at moderate to severe scalp psoriasis and the others examined mild to severe disease.
Results were reported as standardized mean differences (SMD) and also converted to a 6-point global improvement scale created by the authors to provide a combined endpoint of provider- or patient-assessed improvement in symptoms such as redness, thickness, and scaling. Higher scores indicate more improvement.
Compared with placebo, the very potent corticosteroid clobetasol propionate improved psoriasis by 1.9 points on the 6-point scale (4 trials, 788 patients; SMD= −1.6; 95% confidence interval [CI], −1.8 to −1.3). The potent steroid betamethasone diproprionate improved symptoms by 1.3 points compared with placebo (2 trials, 712 patients; SMD= −1.1; 95% CI, −1.3 to −0.90).
The topical corticosteroids clobetasol, betamethasone diproprionate, and betamethasone valerate improved symptoms more than the vitamin D3 analogue calcipotriol in head-to-head trials. The corticosteroid improvement scores exceeded calcipotriol scores by 0.5 points (1 trial, 151 patients; SMD=0.37; 95% CI, 0.05-0.69), 0.6 points (1 trial, 1676 patients; SMD=0.48; 95% CI, 0.32-0.64), and 0.5 points (1 trial, 510 patients; SMD=0.37; 95% CI, 0.20-0.55), respectively.
Combination therapy with a vitamin D3 analogue and a corticosteroid yielded approximately 0.2 points of improvement over corticosteroid alone (6 trials, 2444 patients; SMD= −0.18; 95% CI, −0.26 to −0.10). Four trials of combination therapy (2581 patients) resulted in 0.5 to 1.2 points of improvement compared with vitamin D3 analogues alone (SMD=0.64; 95% CI, 0.44-0.84). Specific strengths and dosing regimens weren’t reported.
The Cochrane systematic review, using the same outcome reporting methods, provided data on the vitamin D3 analogue calcipotriol compared with placebo for treating scalp psoriasis.2 Calcipotriol resulted in 0.9 points of improvement on the 6-point global improvement scale (2 trials, 457 patients; SMD= −0.72; 95% CI, −1.3 to −0.16).
Very potent corticosteroids show a better response than potent agents
In 2013, a meta-analysis of 13 placebo-controlled RCTs (5640 patients) evaluated topical therapies for scalp psoriasis licensed in the United Kingdom. This meta-analysis included the same placebo-controlled studies as the Cochrane review but added one study published after the search date of the review.3
The outcome reporting was different from the Cochrane review. The primary outcome was percentage of patients with at least moderate scalp psoriasis who achieved clear or nearly clear status on provider assessment scales. All treatments were compared to twice-daily placebo with a response rate of 11%.
Very potent steroids had response rates of 78% for twice-daily application (risk ratio [RR]=7.0; 95% CI, 5.6-8.0) and 69% for once-daily application (RR=6.2; 95% CI, 3.0-8.3). The combination of a vitamin D3 analogue and a potent corticosteroid showed a response rate of 64% (RR=5.7; 95% CI, 2.4-8.0) whereas response rates for potent corticosteroids alone were 57% (RR=5.0; 95% CI, 1.6-7.8) for once-daily application and 49% (RR=4.4; 95% CI, 2.2-6.7) for twice-daily administration. The authors suggested patient satisfaction at using once daily vs twice daily application as a possible explanation for the difference in response rate.
Vitamin D3 analogues showed response rates of approximately 34%, which is nonsignificant for once-daily application (RR=3.1; 95% CI, 0.71-6.6) but significant for twice-daily administration (RR=3.1; 95% CI, 1.3-5.9). Exact numbers of studies and participants, as well as specific agents and preparation information, were not included.
1. Mason AR, Mason JM, Cork MJ, et al. Topical treatments for chronic plaque psoriasis of the scalp: a systematic review. Br J Dermatol. 2013;169:519-527.
2. Mason A, Mason J, Cork M, et al. Topical treatments for chronic plaque psoriasis: an abridged Cochrane systematic review. J Am Acad Dermatol. 2013; 69:799-807.
3. Samarasekera EJ, Sawyer L, Wonderling D, et al. Topical therapies for the treatment of plaque psoriasis: systematic review and network meta-analyses. Br J Dermatol. 2013;168:954-967.
Single-agent therapy with a very potent or potent topical corticosteroid appears more effective than other topical agents, including vitamin D3 analogues, for treating scalp psoriasis (strength of recommendation [SOR]: A, systematic reviews of randomized controlled trials [RCTs]).
Combined therapy with a vitamin D3 analogue and a potent topical corticosteroid may be slightly more effective than monotherapy with either agent (SOR: B, systematic reviews of RCTs with inconsistent results).
Evidence summary
A 2013 meta-analysis of 26 RCTs with 8020 patients evaluated topical treatments for scalp psoriasis as part of a subanalysis of a larger Cochrane review of psoriasis therapy.1 Only 20 studies reported the severity of disease: 13 studies looked at moderate to severe scalp psoriasis and the others examined mild to severe disease.
Results were reported as standardized mean differences (SMD) and also converted to a 6-point global improvement scale created by the authors to provide a combined endpoint of provider- or patient-assessed improvement in symptoms such as redness, thickness, and scaling. Higher scores indicate more improvement.
Compared with placebo, the very potent corticosteroid clobetasol propionate improved psoriasis by 1.9 points on the 6-point scale (4 trials, 788 patients; SMD= −1.6; 95% confidence interval [CI], −1.8 to −1.3). The potent steroid betamethasone diproprionate improved symptoms by 1.3 points compared with placebo (2 trials, 712 patients; SMD= −1.1; 95% CI, −1.3 to −0.90).
The topical corticosteroids clobetasol, betamethasone diproprionate, and betamethasone valerate improved symptoms more than the vitamin D3 analogue calcipotriol in head-to-head trials. The corticosteroid improvement scores exceeded calcipotriol scores by 0.5 points (1 trial, 151 patients; SMD=0.37; 95% CI, 0.05-0.69), 0.6 points (1 trial, 1676 patients; SMD=0.48; 95% CI, 0.32-0.64), and 0.5 points (1 trial, 510 patients; SMD=0.37; 95% CI, 0.20-0.55), respectively.
Combination therapy with a vitamin D3 analogue and a corticosteroid yielded approximately 0.2 points of improvement over corticosteroid alone (6 trials, 2444 patients; SMD= −0.18; 95% CI, −0.26 to −0.10). Four trials of combination therapy (2581 patients) resulted in 0.5 to 1.2 points of improvement compared with vitamin D3 analogues alone (SMD=0.64; 95% CI, 0.44-0.84). Specific strengths and dosing regimens weren’t reported.
The Cochrane systematic review, using the same outcome reporting methods, provided data on the vitamin D3 analogue calcipotriol compared with placebo for treating scalp psoriasis.2 Calcipotriol resulted in 0.9 points of improvement on the 6-point global improvement scale (2 trials, 457 patients; SMD= −0.72; 95% CI, −1.3 to −0.16).
Very potent corticosteroids show a better response than potent agents
In 2013, a meta-analysis of 13 placebo-controlled RCTs (5640 patients) evaluated topical therapies for scalp psoriasis licensed in the United Kingdom. This meta-analysis included the same placebo-controlled studies as the Cochrane review but added one study published after the search date of the review.3
The outcome reporting was different from the Cochrane review. The primary outcome was percentage of patients with at least moderate scalp psoriasis who achieved clear or nearly clear status on provider assessment scales. All treatments were compared to twice-daily placebo with a response rate of 11%.
Very potent steroids had response rates of 78% for twice-daily application (risk ratio [RR]=7.0; 95% CI, 5.6-8.0) and 69% for once-daily application (RR=6.2; 95% CI, 3.0-8.3). The combination of a vitamin D3 analogue and a potent corticosteroid showed a response rate of 64% (RR=5.7; 95% CI, 2.4-8.0) whereas response rates for potent corticosteroids alone were 57% (RR=5.0; 95% CI, 1.6-7.8) for once-daily application and 49% (RR=4.4; 95% CI, 2.2-6.7) for twice-daily administration. The authors suggested patient satisfaction at using once daily vs twice daily application as a possible explanation for the difference in response rate.
Vitamin D3 analogues showed response rates of approximately 34%, which is nonsignificant for once-daily application (RR=3.1; 95% CI, 0.71-6.6) but significant for twice-daily administration (RR=3.1; 95% CI, 1.3-5.9). Exact numbers of studies and participants, as well as specific agents and preparation information, were not included.
Single-agent therapy with a very potent or potent topical corticosteroid appears more effective than other topical agents, including vitamin D3 analogues, for treating scalp psoriasis (strength of recommendation [SOR]: A, systematic reviews of randomized controlled trials [RCTs]).
Combined therapy with a vitamin D3 analogue and a potent topical corticosteroid may be slightly more effective than monotherapy with either agent (SOR: B, systematic reviews of RCTs with inconsistent results).
Evidence summary
A 2013 meta-analysis of 26 RCTs with 8020 patients evaluated topical treatments for scalp psoriasis as part of a subanalysis of a larger Cochrane review of psoriasis therapy.1 Only 20 studies reported the severity of disease: 13 studies looked at moderate to severe scalp psoriasis and the others examined mild to severe disease.
Results were reported as standardized mean differences (SMD) and also converted to a 6-point global improvement scale created by the authors to provide a combined endpoint of provider- or patient-assessed improvement in symptoms such as redness, thickness, and scaling. Higher scores indicate more improvement.
Compared with placebo, the very potent corticosteroid clobetasol propionate improved psoriasis by 1.9 points on the 6-point scale (4 trials, 788 patients; SMD= −1.6; 95% confidence interval [CI], −1.8 to −1.3). The potent steroid betamethasone diproprionate improved symptoms by 1.3 points compared with placebo (2 trials, 712 patients; SMD= −1.1; 95% CI, −1.3 to −0.90).
The topical corticosteroids clobetasol, betamethasone diproprionate, and betamethasone valerate improved symptoms more than the vitamin D3 analogue calcipotriol in head-to-head trials. The corticosteroid improvement scores exceeded calcipotriol scores by 0.5 points (1 trial, 151 patients; SMD=0.37; 95% CI, 0.05-0.69), 0.6 points (1 trial, 1676 patients; SMD=0.48; 95% CI, 0.32-0.64), and 0.5 points (1 trial, 510 patients; SMD=0.37; 95% CI, 0.20-0.55), respectively.
Combination therapy with a vitamin D3 analogue and a corticosteroid yielded approximately 0.2 points of improvement over corticosteroid alone (6 trials, 2444 patients; SMD= −0.18; 95% CI, −0.26 to −0.10). Four trials of combination therapy (2581 patients) resulted in 0.5 to 1.2 points of improvement compared with vitamin D3 analogues alone (SMD=0.64; 95% CI, 0.44-0.84). Specific strengths and dosing regimens weren’t reported.
The Cochrane systematic review, using the same outcome reporting methods, provided data on the vitamin D3 analogue calcipotriol compared with placebo for treating scalp psoriasis.2 Calcipotriol resulted in 0.9 points of improvement on the 6-point global improvement scale (2 trials, 457 patients; SMD= −0.72; 95% CI, −1.3 to −0.16).
Very potent corticosteroids show a better response than potent agents
In 2013, a meta-analysis of 13 placebo-controlled RCTs (5640 patients) evaluated topical therapies for scalp psoriasis licensed in the United Kingdom. This meta-analysis included the same placebo-controlled studies as the Cochrane review but added one study published after the search date of the review.3
The outcome reporting was different from the Cochrane review. The primary outcome was percentage of patients with at least moderate scalp psoriasis who achieved clear or nearly clear status on provider assessment scales. All treatments were compared to twice-daily placebo with a response rate of 11%.
Very potent steroids had response rates of 78% for twice-daily application (risk ratio [RR]=7.0; 95% CI, 5.6-8.0) and 69% for once-daily application (RR=6.2; 95% CI, 3.0-8.3). The combination of a vitamin D3 analogue and a potent corticosteroid showed a response rate of 64% (RR=5.7; 95% CI, 2.4-8.0) whereas response rates for potent corticosteroids alone were 57% (RR=5.0; 95% CI, 1.6-7.8) for once-daily application and 49% (RR=4.4; 95% CI, 2.2-6.7) for twice-daily administration. The authors suggested patient satisfaction at using once daily vs twice daily application as a possible explanation for the difference in response rate.
Vitamin D3 analogues showed response rates of approximately 34%, which is nonsignificant for once-daily application (RR=3.1; 95% CI, 0.71-6.6) but significant for twice-daily administration (RR=3.1; 95% CI, 1.3-5.9). Exact numbers of studies and participants, as well as specific agents and preparation information, were not included.
1. Mason AR, Mason JM, Cork MJ, et al. Topical treatments for chronic plaque psoriasis of the scalp: a systematic review. Br J Dermatol. 2013;169:519-527.
2. Mason A, Mason J, Cork M, et al. Topical treatments for chronic plaque psoriasis: an abridged Cochrane systematic review. J Am Acad Dermatol. 2013; 69:799-807.
3. Samarasekera EJ, Sawyer L, Wonderling D, et al. Topical therapies for the treatment of plaque psoriasis: systematic review and network meta-analyses. Br J Dermatol. 2013;168:954-967.
1. Mason AR, Mason JM, Cork MJ, et al. Topical treatments for chronic plaque psoriasis of the scalp: a systematic review. Br J Dermatol. 2013;169:519-527.
2. Mason A, Mason J, Cork M, et al. Topical treatments for chronic plaque psoriasis: an abridged Cochrane systematic review. J Am Acad Dermatol. 2013; 69:799-807.
3. Samarasekera EJ, Sawyer L, Wonderling D, et al. Topical therapies for the treatment of plaque psoriasis: systematic review and network meta-analyses. Br J Dermatol. 2013;168:954-967.
Evidence-based answers from the Family Physicians Inquiries Network
Patients With Epilepsy Have an Internet Disadvantage That May Impede Self-Management
Persons with epilepsy are less likely to use the Internet compared with the general public. The recent CDC study that arrived at that conclusion suggested that this disparity may put patients with epilepsy at a disadvantage because it limits their access to online tools that can optimize their self-care and improve their quality of life. The study was based on data from the 2013 National Health Interview Survey, which confirmed that the disparity existed in all three age groups analyzed: 18-44 years, 45-59 years, 60 years and older.
US Centers for Disease Control and Prevention Epilepsy Program. Internet use and looking up information online in adults with epilepsy varies by epilepsy status — 2013 National Health Interview Survey. Epilepsy Behav. 2016;54:47-49.
Persons with epilepsy are less likely to use the Internet compared with the general public. The recent CDC study that arrived at that conclusion suggested that this disparity may put patients with epilepsy at a disadvantage because it limits their access to online tools that can optimize their self-care and improve their quality of life. The study was based on data from the 2013 National Health Interview Survey, which confirmed that the disparity existed in all three age groups analyzed: 18-44 years, 45-59 years, 60 years and older.
US Centers for Disease Control and Prevention Epilepsy Program. Internet use and looking up information online in adults with epilepsy varies by epilepsy status — 2013 National Health Interview Survey. Epilepsy Behav. 2016;54:47-49.
Persons with epilepsy are less likely to use the Internet compared with the general public. The recent CDC study that arrived at that conclusion suggested that this disparity may put patients with epilepsy at a disadvantage because it limits their access to online tools that can optimize their self-care and improve their quality of life. The study was based on data from the 2013 National Health Interview Survey, which confirmed that the disparity existed in all three age groups analyzed: 18-44 years, 45-59 years, 60 years and older.
US Centers for Disease Control and Prevention Epilepsy Program. Internet use and looking up information online in adults with epilepsy varies by epilepsy status — 2013 National Health Interview Survey. Epilepsy Behav. 2016;54:47-49.
Do novel oral anticoagulants safely prevent stroke in patients with nonvalvular A-fib?
Yes. Dabigatran, rivaroxaban, and apixaban are safe and effective compared with warfarin for preventing stroke in patients with nonvalvular atrial fibrillation. These novel oral anticoagulants (NOACs) are noninferior in reducing the number of strokes and systemic emboli and in lowering all-cause mortality while not increasing major bleeding complications and hemorrhagic events (strength of recommendation: A, consistent meta-analyses of randomized controlled trials [RCTs]).
Evidence summary
A 2014 meta-analysis of 4 RCTs including 71,683 patients with nonvalvular atrial fibrillation evaluated the NOACs dabigatran, rivaroxaban, apixaban, and edoxaban, for efficacy and safety compared with warfarin.1 The RCTs analyzed 42,411 patients receiving NOACs and 29,272 patients receiving warfarin. All trials were designed to show noninferiority. Selection criteria for RCTs included all phase 3 trials of available NOACs (edoxaban isn’t available in the United States). Median follow-up was 1.8 to 2.8 years.
Pooled data demonstrated that NOACs were noninferior to warfarin in preventing stroke or systemic embolism (relative risk [RR]=0.81; 95% confidence interval [CI], 0.73-0.91; number needed to treat [NNT]=147). The main benefit was derived from the relatively large decrease in the rate of hemorrhagic stroke (RR=0.49; 95% CI, 0.38-0.64; NNT=97) compared with warfarin. All-cause mortality was lower with NOACs as well (RR=0.90; 95% CI, 0.85-0.95; NNT=128).
A significant increase in gastrointestinal bleeding occurred with NOACs compared with warfarin (RR=1.3; 95% CI, 1.1-1.6; number needed to harm=185), but NOACs were associated with a decrease in intracranial hemorrhage similar to the reduction in hemorrhagic stroke (RR=0.48; 95% CI, 0.39-0.59; NNT=132).
NOACs show no significant difference in bleeding complications vs warfarin
A 2013 meta-analysis of 5 RCTs including 51,895 patients with nonvalvular atrial fibrillation compared the efficacy and safety of the NOACs dabigatran, rivaroxaban, apixaban, and ximelagatran, with the efficacy and safety of warfarin.2 This review included the 3 studies of dabigatran, rivaroxaban, and apixaban from the previously described review, as well as 2 trials of ximelagatran that were not included in the other review (presumably because ximelagatran was no longer available owing to liver toxicity). This review didn’t include the study of edoxaban that was published after the search dates of the literature review.
All trials were designed to show noninferiority. Selection criteria included a study population of at least 3000 patients and use of intention-to-treat analysis. Only 3 of the trials were double-blinded, and 2 were open-label. Mean follow-up was 16 months; median was 24 months.
NOACs were noninferior to vitamin K antagonists in the rate of stroke or systemic embolism (RR=0.82; 95% CI, 0.69-0.98; NNT=200), the rate of death from any cause (RR=0.91; 95% CI, 0.85-0.96; NNT=145), and the rate of hemorrhagic strokes (RR=0.51; 95% CI, 0.41-0.64). NOACs showed no significant difference in major bleeding compared with warfarin (RR=0.83; 95% CI, 0.69-1.0), and were noninferior for minor bleeding (RR=0.88; 95% CI, 0.80-0.97). There was no difference in ischemic stroke (RR=0.87; 95% CI, 0.75-1.06) and major noncerebral bleeding (RR=0.88; 95% CI, 0.73-1.08).
The ACCP weighs in
The American College of Chest Physicians’ 2012 clinical practice guidelines for antithrombotic therapy for atrial fibrillation recommend dabigatran 150 mg twice daily rather than adjusted-dose warfarin therapy for patients with nonvalvular atrial fibrillation requiring thromboembolism prophylaxis (Grade 2B, weak recommendation based on RCTs with important limitations).3
1. Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383:955-962.
2. Dogliotti A, Paolasso E, Giugliano RP, et al. Novel oral anticoagulants in atrial fibrillation: a meta-analysis of large, randomized, controlled trials vs warfarin. Clin Cardiol. 2013;36:61-67.
3. You JJ, Singer DE, Howard PA, et al. Antithrombotic therapy for atrial fibrillation: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e531S-e575S.
Yes. Dabigatran, rivaroxaban, and apixaban are safe and effective compared with warfarin for preventing stroke in patients with nonvalvular atrial fibrillation. These novel oral anticoagulants (NOACs) are noninferior in reducing the number of strokes and systemic emboli and in lowering all-cause mortality while not increasing major bleeding complications and hemorrhagic events (strength of recommendation: A, consistent meta-analyses of randomized controlled trials [RCTs]).
Evidence summary
A 2014 meta-analysis of 4 RCTs including 71,683 patients with nonvalvular atrial fibrillation evaluated the NOACs dabigatran, rivaroxaban, apixaban, and edoxaban, for efficacy and safety compared with warfarin.1 The RCTs analyzed 42,411 patients receiving NOACs and 29,272 patients receiving warfarin. All trials were designed to show noninferiority. Selection criteria for RCTs included all phase 3 trials of available NOACs (edoxaban isn’t available in the United States). Median follow-up was 1.8 to 2.8 years.
Pooled data demonstrated that NOACs were noninferior to warfarin in preventing stroke or systemic embolism (relative risk [RR]=0.81; 95% confidence interval [CI], 0.73-0.91; number needed to treat [NNT]=147). The main benefit was derived from the relatively large decrease in the rate of hemorrhagic stroke (RR=0.49; 95% CI, 0.38-0.64; NNT=97) compared with warfarin. All-cause mortality was lower with NOACs as well (RR=0.90; 95% CI, 0.85-0.95; NNT=128).
A significant increase in gastrointestinal bleeding occurred with NOACs compared with warfarin (RR=1.3; 95% CI, 1.1-1.6; number needed to harm=185), but NOACs were associated with a decrease in intracranial hemorrhage similar to the reduction in hemorrhagic stroke (RR=0.48; 95% CI, 0.39-0.59; NNT=132).
NOACs show no significant difference in bleeding complications vs warfarin
A 2013 meta-analysis of 5 RCTs including 51,895 patients with nonvalvular atrial fibrillation compared the efficacy and safety of the NOACs dabigatran, rivaroxaban, apixaban, and ximelagatran, with the efficacy and safety of warfarin.2 This review included the 3 studies of dabigatran, rivaroxaban, and apixaban from the previously described review, as well as 2 trials of ximelagatran that were not included in the other review (presumably because ximelagatran was no longer available owing to liver toxicity). This review didn’t include the study of edoxaban that was published after the search dates of the literature review.
All trials were designed to show noninferiority. Selection criteria included a study population of at least 3000 patients and use of intention-to-treat analysis. Only 3 of the trials were double-blinded, and 2 were open-label. Mean follow-up was 16 months; median was 24 months.
NOACs were noninferior to vitamin K antagonists in the rate of stroke or systemic embolism (RR=0.82; 95% CI, 0.69-0.98; NNT=200), the rate of death from any cause (RR=0.91; 95% CI, 0.85-0.96; NNT=145), and the rate of hemorrhagic strokes (RR=0.51; 95% CI, 0.41-0.64). NOACs showed no significant difference in major bleeding compared with warfarin (RR=0.83; 95% CI, 0.69-1.0), and were noninferior for minor bleeding (RR=0.88; 95% CI, 0.80-0.97). There was no difference in ischemic stroke (RR=0.87; 95% CI, 0.75-1.06) and major noncerebral bleeding (RR=0.88; 95% CI, 0.73-1.08).
The ACCP weighs in
The American College of Chest Physicians’ 2012 clinical practice guidelines for antithrombotic therapy for atrial fibrillation recommend dabigatran 150 mg twice daily rather than adjusted-dose warfarin therapy for patients with nonvalvular atrial fibrillation requiring thromboembolism prophylaxis (Grade 2B, weak recommendation based on RCTs with important limitations).3
Yes. Dabigatran, rivaroxaban, and apixaban are safe and effective compared with warfarin for preventing stroke in patients with nonvalvular atrial fibrillation. These novel oral anticoagulants (NOACs) are noninferior in reducing the number of strokes and systemic emboli and in lowering all-cause mortality while not increasing major bleeding complications and hemorrhagic events (strength of recommendation: A, consistent meta-analyses of randomized controlled trials [RCTs]).
Evidence summary
A 2014 meta-analysis of 4 RCTs including 71,683 patients with nonvalvular atrial fibrillation evaluated the NOACs dabigatran, rivaroxaban, apixaban, and edoxaban, for efficacy and safety compared with warfarin.1 The RCTs analyzed 42,411 patients receiving NOACs and 29,272 patients receiving warfarin. All trials were designed to show noninferiority. Selection criteria for RCTs included all phase 3 trials of available NOACs (edoxaban isn’t available in the United States). Median follow-up was 1.8 to 2.8 years.
Pooled data demonstrated that NOACs were noninferior to warfarin in preventing stroke or systemic embolism (relative risk [RR]=0.81; 95% confidence interval [CI], 0.73-0.91; number needed to treat [NNT]=147). The main benefit was derived from the relatively large decrease in the rate of hemorrhagic stroke (RR=0.49; 95% CI, 0.38-0.64; NNT=97) compared with warfarin. All-cause mortality was lower with NOACs as well (RR=0.90; 95% CI, 0.85-0.95; NNT=128).
A significant increase in gastrointestinal bleeding occurred with NOACs compared with warfarin (RR=1.3; 95% CI, 1.1-1.6; number needed to harm=185), but NOACs were associated with a decrease in intracranial hemorrhage similar to the reduction in hemorrhagic stroke (RR=0.48; 95% CI, 0.39-0.59; NNT=132).
NOACs show no significant difference in bleeding complications vs warfarin
A 2013 meta-analysis of 5 RCTs including 51,895 patients with nonvalvular atrial fibrillation compared the efficacy and safety of the NOACs dabigatran, rivaroxaban, apixaban, and ximelagatran, with the efficacy and safety of warfarin.2 This review included the 3 studies of dabigatran, rivaroxaban, and apixaban from the previously described review, as well as 2 trials of ximelagatran that were not included in the other review (presumably because ximelagatran was no longer available owing to liver toxicity). This review didn’t include the study of edoxaban that was published after the search dates of the literature review.
All trials were designed to show noninferiority. Selection criteria included a study population of at least 3000 patients and use of intention-to-treat analysis. Only 3 of the trials were double-blinded, and 2 were open-label. Mean follow-up was 16 months; median was 24 months.
NOACs were noninferior to vitamin K antagonists in the rate of stroke or systemic embolism (RR=0.82; 95% CI, 0.69-0.98; NNT=200), the rate of death from any cause (RR=0.91; 95% CI, 0.85-0.96; NNT=145), and the rate of hemorrhagic strokes (RR=0.51; 95% CI, 0.41-0.64). NOACs showed no significant difference in major bleeding compared with warfarin (RR=0.83; 95% CI, 0.69-1.0), and were noninferior for minor bleeding (RR=0.88; 95% CI, 0.80-0.97). There was no difference in ischemic stroke (RR=0.87; 95% CI, 0.75-1.06) and major noncerebral bleeding (RR=0.88; 95% CI, 0.73-1.08).
The ACCP weighs in
The American College of Chest Physicians’ 2012 clinical practice guidelines for antithrombotic therapy for atrial fibrillation recommend dabigatran 150 mg twice daily rather than adjusted-dose warfarin therapy for patients with nonvalvular atrial fibrillation requiring thromboembolism prophylaxis (Grade 2B, weak recommendation based on RCTs with important limitations).3
1. Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383:955-962.
2. Dogliotti A, Paolasso E, Giugliano RP, et al. Novel oral anticoagulants in atrial fibrillation: a meta-analysis of large, randomized, controlled trials vs warfarin. Clin Cardiol. 2013;36:61-67.
3. You JJ, Singer DE, Howard PA, et al. Antithrombotic therapy for atrial fibrillation: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e531S-e575S.
1. Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383:955-962.
2. Dogliotti A, Paolasso E, Giugliano RP, et al. Novel oral anticoagulants in atrial fibrillation: a meta-analysis of large, randomized, controlled trials vs warfarin. Clin Cardiol. 2013;36:61-67.
3. You JJ, Singer DE, Howard PA, et al. Antithrombotic therapy for atrial fibrillation: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e531S-e575S.
Evidence-based answers from the Family Physicians Inquiries Network
Management of adnexal masses in pregnancy
Roughly 1%-2% of pregnancies are complicated by an adnexal mass, and prenatal ultrasound for fetal evaluation has detected more asymptomatic ovarian masses as a result.
The differential diagnosis for adnexal mass is broad and includes follicular or corpus luteum cysts, mature teratoma, theca lutein cyst, hydrosalpinx, endometrioma, cystadenoma, pedunculated leiomyoma, luteoma, as well as malignant neoplasms of epithelial, germ cell, and sex cord–stromal origin (J Ultrasound Med. 2004 Jun;23[6]:805-19). Most masses will be benign neoplasms, with a fraction identified as malignancies.

In 2013, Baser et al. performed a retrospective study of 151 women who underwent surgery of an adnexal mass at time of cesarean delivery. Of the 151 cases reviewed, 148 (98%) of the masses were benign (Int J Gynaecol Obstet. 2013 Nov;123[2]:124-6). Additionally, if the patient presents with pain, diagnoses such as ectopic pregnancy, heterotopic pregnancy, degenerating fibroid, and torsion should also be considered.
Diagnostic evaluation and management
The majority of adnexal masses identified in pregnancy are benign simple cysts measuring less than 5 cm in diameter. Approximately 70% of cystic masses detected in the first trimester will spontaneously resolve by the second trimester (Clin Obstet Gynecol. 2006 Sep;49[3]:492-505). However, for some masses, surgical resection is warranted.
Masses present after the first trimester and that are (1) greater than 10cm in diameter or (2) are solid or contain solid and cystic areas or have septated or papillary areas, are generally managed surgically as these features increase the risk of malignancy or complications such as adnexal torsion, rupture, or labor dystocia (Gynecol Oncol. 2006 May;101(2):315-21).

Adnexal masses without these features often resolve during pregnancy and can be expectantly managed (Obstet Gynecol. 2005 May;105[5 Pt 1]:1098-103). The optimal time for surgical intervention is after the first trimester as organogenesis is largely complete, therefore minimizing the risk of drug-induced teratogenesis, and any necessary cystectomy or oophorectomy will not disrupt the required progesterone production of the corpus luteum as this has been replaced by the placenta.
Preoperative assessment
For most cases, imaging with ultrasound is adequate for preoperative evaluation; however, in some cases, further imaging is needed for appropriate characterization of the mass. In this situation, further imaging with MRI is preferred as this modality has good resolution for visualization of soft tissue pathology and does not expose the patient and fetus to ionizing radiation. Of note, Gadolinium-based contrast should be avoided as effects have not been well established in pregnancy (AJR Am J Roentgenol. 2008 Aug;191[2]:364-70).
If there is concern for malignancy during pregnancy, drawing serum tumor markers preoperatively is typically not suggested. Oncofetal antigens, including alpha fetoprotein (AFP), human chorionic gonadotropin (hCG), carcinoembryonic antigen (CEA), and cancer antigen 125 (CA-125), are elevated during gestation, making them poor markers for malignancy. If malignancy is ultimately diagnosed, then tumor markers can be obtained immediately postoperatively.
Surgical approach and prognosis
If there is low suspicion for malignancy, a laparoscopic approach is preferable and reasonable at all stages of pregnancy, although early second trimester is ideal. Entry at Palmer’s point in the left upper quadrant versus the umbilicus is preferable in order to minimize risk of uterine injury.
If malignancy is suspected, maximum exposure should be obtained with a midline vertical incision. Peritoneal washings should be obtained on immediate entry of the peritoneal cavity, and the contralateral ovary should also be adequately examined along with a general abdominopelvic survey. If the mass demonstrates concerning features, such as solid features or presence of ascites, then the specimen should be sent for intraoperative frozen pathology, and the pathologist should be made aware of the concurrent pregnancy. If malignancy is confirmed on frozen pathology, a full staging procedure should be performed and a gynecologic oncologist consulted.
Roughly three-quarters of invasive ovarian cancers diagnosed in pregnancy are early stage disease, and the 5-year survival of ovarian cancers associated with pregnancy is between 72% and 90% (Int J Gynecol Cancer. 2006 Jan-Feb;16[1]:8-15).
In a retrospective cohort study of 101 pregnant women, 31% of adnexal masses resected in pregnant women greater than 14 weeks gestation were teratomas. In total, 23% of masses were luteal cysts. Less commonly, patients were diagnosed with serous cystadenoma (14%), endometrioma (8%), mucinous cystadenoma (7%), benign cyst (6%), tumor of low malignant potential (5%), and paratubal cyst (3%).
In this study, approximately half of the women underwent minimally invasive surgery and half had surgery via laparotomy. There were more complications in the women undergoing laparotomy (ileus) and there were no differences between the groups with regards to pregnancy and neonatal outcomes (J Minim Invasive Gynecol. 2011 Nov-Dec;18[6]:720-5).
In general, characteristics that are favorable for spontaneous resolution include masses that are simple in nature by ultrasound and less than 5 cm to 6 cm in diameter.
For women with simple-appearing masses on ultrasound, reimaging can occur during the remainder of the pregnancy at the discretion of the physician or during the postpartum period. All women should be provided with torsion and rupture precautions during the pregnancy (Am J Obstet Gynecol. 2011 Aug;205[2]:97-102). For women with more concerning features on ultrasound, referral to a gynecologic oncologist is warranted. If the decision for surgical management is made, minimally invasive surgery should be strongly considered due to minimal maternal and perinatal morbidity.
Dr. Staley is a resident physician in the department of obstetrics and gynecology at the University of North Carolina at Chapel Hill. Dr. Gehrig is professor and director of gynecologic oncology at the university. They reported having no relevant financial disclosures.
Roughly 1%-2% of pregnancies are complicated by an adnexal mass, and prenatal ultrasound for fetal evaluation has detected more asymptomatic ovarian masses as a result.
The differential diagnosis for adnexal mass is broad and includes follicular or corpus luteum cysts, mature teratoma, theca lutein cyst, hydrosalpinx, endometrioma, cystadenoma, pedunculated leiomyoma, luteoma, as well as malignant neoplasms of epithelial, germ cell, and sex cord–stromal origin (J Ultrasound Med. 2004 Jun;23[6]:805-19). Most masses will be benign neoplasms, with a fraction identified as malignancies.
In 2013, Baser et al. performed a retrospective study of 151 women who underwent surgery of an adnexal mass at time of cesarean delivery. Of the 151 cases reviewed, 148 (98%) of the masses were benign (Int J Gynaecol Obstet. 2013 Nov;123[2]:124-6). Additionally, if the patient presents with pain, diagnoses such as ectopic pregnancy, heterotopic pregnancy, degenerating fibroid, and torsion should also be considered.
Diagnostic evaluation and management
The majority of adnexal masses identified in pregnancy are benign simple cysts measuring less than 5 cm in diameter. Approximately 70% of cystic masses detected in the first trimester will spontaneously resolve by the second trimester (Clin Obstet Gynecol. 2006 Sep;49[3]:492-505). However, for some masses, surgical resection is warranted.
Masses present after the first trimester and that are (1) greater than 10cm in diameter or (2) are solid or contain solid and cystic areas or have septated or papillary areas, are generally managed surgically as these features increase the risk of malignancy or complications such as adnexal torsion, rupture, or labor dystocia (Gynecol Oncol. 2006 May;101(2):315-21).
Adnexal masses without these features often resolve during pregnancy and can be expectantly managed (Obstet Gynecol. 2005 May;105[5 Pt 1]:1098-103). The optimal time for surgical intervention is after the first trimester as organogenesis is largely complete, therefore minimizing the risk of drug-induced teratogenesis, and any necessary cystectomy or oophorectomy will not disrupt the required progesterone production of the corpus luteum as this has been replaced by the placenta.
Preoperative assessment
For most cases, imaging with ultrasound is adequate for preoperative evaluation; however, in some cases, further imaging is needed for appropriate characterization of the mass. In this situation, further imaging with MRI is preferred as this modality has good resolution for visualization of soft tissue pathology and does not expose the patient and fetus to ionizing radiation. Of note, Gadolinium-based contrast should be avoided as effects have not been well established in pregnancy (AJR Am J Roentgenol. 2008 Aug;191[2]:364-70).
If there is concern for malignancy during pregnancy, drawing serum tumor markers preoperatively is typically not suggested. Oncofetal antigens, including alpha fetoprotein (AFP), human chorionic gonadotropin (hCG), carcinoembryonic antigen (CEA), and cancer antigen 125 (CA-125), are elevated during gestation, making them poor markers for malignancy. If malignancy is ultimately diagnosed, then tumor markers can be obtained immediately postoperatively.
Surgical approach and prognosis
If there is low suspicion for malignancy, a laparoscopic approach is preferable and reasonable at all stages of pregnancy, although early second trimester is ideal. Entry at Palmer’s point in the left upper quadrant versus the umbilicus is preferable in order to minimize risk of uterine injury.
If malignancy is suspected, maximum exposure should be obtained with a midline vertical incision. Peritoneal washings should be obtained on immediate entry of the peritoneal cavity, and the contralateral ovary should also be adequately examined along with a general abdominopelvic survey. If the mass demonstrates concerning features, such as solid features or presence of ascites, then the specimen should be sent for intraoperative frozen pathology, and the pathologist should be made aware of the concurrent pregnancy. If malignancy is confirmed on frozen pathology, a full staging procedure should be performed and a gynecologic oncologist consulted.
Roughly three-quarters of invasive ovarian cancers diagnosed in pregnancy are early stage disease, and the 5-year survival of ovarian cancers associated with pregnancy is between 72% and 90% (Int J Gynecol Cancer. 2006 Jan-Feb;16[1]:8-15).
In a retrospective cohort study of 101 pregnant women, 31% of adnexal masses resected in pregnant women greater than 14 weeks gestation were teratomas. In total, 23% of masses were luteal cysts. Less commonly, patients were diagnosed with serous cystadenoma (14%), endometrioma (8%), mucinous cystadenoma (7%), benign cyst (6%), tumor of low malignant potential (5%), and paratubal cyst (3%).
In this study, approximately half of the women underwent minimally invasive surgery and half had surgery via laparotomy. There were more complications in the women undergoing laparotomy (ileus) and there were no differences between the groups with regards to pregnancy and neonatal outcomes (J Minim Invasive Gynecol. 2011 Nov-Dec;18[6]:720-5).
In general, characteristics that are favorable for spontaneous resolution include masses that are simple in nature by ultrasound and less than 5 cm to 6 cm in diameter.
For women with simple-appearing masses on ultrasound, reimaging can occur during the remainder of the pregnancy at the discretion of the physician or during the postpartum period. All women should be provided with torsion and rupture precautions during the pregnancy (Am J Obstet Gynecol. 2011 Aug;205[2]:97-102). For women with more concerning features on ultrasound, referral to a gynecologic oncologist is warranted. If the decision for surgical management is made, minimally invasive surgery should be strongly considered due to minimal maternal and perinatal morbidity.
Dr. Staley is a resident physician in the department of obstetrics and gynecology at the University of North Carolina at Chapel Hill. Dr. Gehrig is professor and director of gynecologic oncology at the university. They reported having no relevant financial disclosures.
Roughly 1%-2% of pregnancies are complicated by an adnexal mass, and prenatal ultrasound for fetal evaluation has detected more asymptomatic ovarian masses as a result.
The differential diagnosis for adnexal mass is broad and includes follicular or corpus luteum cysts, mature teratoma, theca lutein cyst, hydrosalpinx, endometrioma, cystadenoma, pedunculated leiomyoma, luteoma, as well as malignant neoplasms of epithelial, germ cell, and sex cord–stromal origin (J Ultrasound Med. 2004 Jun;23[6]:805-19). Most masses will be benign neoplasms, with a fraction identified as malignancies.
In 2013, Baser et al. performed a retrospective study of 151 women who underwent surgery of an adnexal mass at time of cesarean delivery. Of the 151 cases reviewed, 148 (98%) of the masses were benign (Int J Gynaecol Obstet. 2013 Nov;123[2]:124-6). Additionally, if the patient presents with pain, diagnoses such as ectopic pregnancy, heterotopic pregnancy, degenerating fibroid, and torsion should also be considered.
Diagnostic evaluation and management
The majority of adnexal masses identified in pregnancy are benign simple cysts measuring less than 5 cm in diameter. Approximately 70% of cystic masses detected in the first trimester will spontaneously resolve by the second trimester (Clin Obstet Gynecol. 2006 Sep;49[3]:492-505). However, for some masses, surgical resection is warranted.
Masses present after the first trimester and that are (1) greater than 10cm in diameter or (2) are solid or contain solid and cystic areas or have septated or papillary areas, are generally managed surgically as these features increase the risk of malignancy or complications such as adnexal torsion, rupture, or labor dystocia (Gynecol Oncol. 2006 May;101(2):315-21).
Adnexal masses without these features often resolve during pregnancy and can be expectantly managed (Obstet Gynecol. 2005 May;105[5 Pt 1]:1098-103). The optimal time for surgical intervention is after the first trimester as organogenesis is largely complete, therefore minimizing the risk of drug-induced teratogenesis, and any necessary cystectomy or oophorectomy will not disrupt the required progesterone production of the corpus luteum as this has been replaced by the placenta.
Preoperative assessment
For most cases, imaging with ultrasound is adequate for preoperative evaluation; however, in some cases, further imaging is needed for appropriate characterization of the mass. In this situation, further imaging with MRI is preferred as this modality has good resolution for visualization of soft tissue pathology and does not expose the patient and fetus to ionizing radiation. Of note, Gadolinium-based contrast should be avoided as effects have not been well established in pregnancy (AJR Am J Roentgenol. 2008 Aug;191[2]:364-70).
If there is concern for malignancy during pregnancy, drawing serum tumor markers preoperatively is typically not suggested. Oncofetal antigens, including alpha fetoprotein (AFP), human chorionic gonadotropin (hCG), carcinoembryonic antigen (CEA), and cancer antigen 125 (CA-125), are elevated during gestation, making them poor markers for malignancy. If malignancy is ultimately diagnosed, then tumor markers can be obtained immediately postoperatively.
Surgical approach and prognosis
If there is low suspicion for malignancy, a laparoscopic approach is preferable and reasonable at all stages of pregnancy, although early second trimester is ideal. Entry at Palmer’s point in the left upper quadrant versus the umbilicus is preferable in order to minimize risk of uterine injury.
If malignancy is suspected, maximum exposure should be obtained with a midline vertical incision. Peritoneal washings should be obtained on immediate entry of the peritoneal cavity, and the contralateral ovary should also be adequately examined along with a general abdominopelvic survey. If the mass demonstrates concerning features, such as solid features or presence of ascites, then the specimen should be sent for intraoperative frozen pathology, and the pathologist should be made aware of the concurrent pregnancy. If malignancy is confirmed on frozen pathology, a full staging procedure should be performed and a gynecologic oncologist consulted.
Roughly three-quarters of invasive ovarian cancers diagnosed in pregnancy are early stage disease, and the 5-year survival of ovarian cancers associated with pregnancy is between 72% and 90% (Int J Gynecol Cancer. 2006 Jan-Feb;16[1]:8-15).
In a retrospective cohort study of 101 pregnant women, 31% of adnexal masses resected in pregnant women greater than 14 weeks gestation were teratomas. In total, 23% of masses were luteal cysts. Less commonly, patients were diagnosed with serous cystadenoma (14%), endometrioma (8%), mucinous cystadenoma (7%), benign cyst (6%), tumor of low malignant potential (5%), and paratubal cyst (3%).
In this study, approximately half of the women underwent minimally invasive surgery and half had surgery via laparotomy. There were more complications in the women undergoing laparotomy (ileus) and there were no differences between the groups with regards to pregnancy and neonatal outcomes (J Minim Invasive Gynecol. 2011 Nov-Dec;18[6]:720-5).
In general, characteristics that are favorable for spontaneous resolution include masses that are simple in nature by ultrasound and less than 5 cm to 6 cm in diameter.
For women with simple-appearing masses on ultrasound, reimaging can occur during the remainder of the pregnancy at the discretion of the physician or during the postpartum period. All women should be provided with torsion and rupture precautions during the pregnancy (Am J Obstet Gynecol. 2011 Aug;205[2]:97-102). For women with more concerning features on ultrasound, referral to a gynecologic oncologist is warranted. If the decision for surgical management is made, minimally invasive surgery should be strongly considered due to minimal maternal and perinatal morbidity.
Dr. Staley is a resident physician in the department of obstetrics and gynecology at the University of North Carolina at Chapel Hill. Dr. Gehrig is professor and director of gynecologic oncology at the university. They reported having no relevant financial disclosures.

Navigating pneumococcal vaccination in adults
STREPTOCOCCUS PNEUMONIAE (the “pneumococcus”) causes a variety of clinical syndromes that range from otitis media to bacteremia, meningitis, and pneumonia. Hardest hit are immunocompromised people and those at the extremes of age. Therefore, preventing disease through pneumococcal vaccination is very important in these groups.
This review summarizes the current guidelines from the Advisory Committee on Immunization Practices (ACIP) of the US Centers for Disease Control and Prevention (CDC) for pneumococcal immunization in adults.
STRIKES THE VERY YOUNG, VERY OLD, AND IMMUNOCOMPROMISED
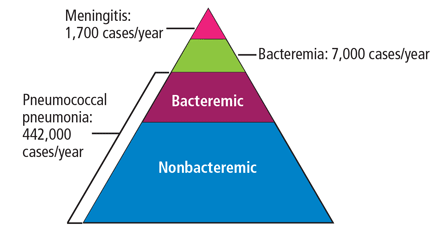
Invasive pneumococcal disease is defined as infection in which S pneumoniae can be found in a normally sterile site such as the cerebrospinal fluid or blood, and it includes bacteremic pneumonia.1 By far the most common type of pneumococcal disease is pneumonia, followed by bacteremia and meningitis (Figure 1);2,3 about 25% of patients with pneumococcal pneumonia also have bacteremia.2
Invasive pneumococcal disease most often occurs in children age 2 and younger, adults age 65 and older, and people who are immunocompromised. In 2010, the incidence was 3.8 per 100,000 in people ages 18 to 34 but was 10 times higher in the elderly and those with compromised immunity.1
Even now that vaccines are available, invasive pneumococcal disease continues to cause 4,000 deaths per year in the United States.1
TWO INACTIVATED VACCINES
S pneumoniae is a gram-positive coccus with an outer capsule composed of polysaccharides that protect the bacterium from being ingested and killed by host phagocytic cells. Some 91 serotypes of this organism have been identified on the basis of genetic differences in capsular polysaccharide composition.
Currently, two inactivated vaccines are available that elicit antibody responses to the most common pneumococcal serotypes that infect humans.
- PPSV23 (pneumococcal polysaccharide vaccine-23, or Pneumovax 23) contains purified capsular polysaccharides from 23 pneumococcal serotypes.
- PCV13 (pneumococcal conjugate vaccine-13, or Prevnar 13) contains purified capsular polysaccharides from 13 serotypes that are covalently bound to (conjugated with) a carrier protein.
PPSV23 AND PCV13 ARE NOT THE SAME
Apart from the number of serotypes covered, the two vaccines differ in important ways. Both of them elicit a B-cell-mediated immune response, but only PCV13 produces a T-cell-dependent response, which is essential for maturation of the B-cell response and development of immune memory.
PPSV23 generally provides 3 to 5 years of immunity, and repeat doses do not offer additive or “boosted” protection. It is ineffective in children under 2 years of age.
Pneumococcal conjugate vaccine has been available since 2000 for children starting at 2 months of age. Since then it has directly reduced the incidence of invasive pneumococcal disease in children and indirectly in adults. The impact on pneumococcal disease rates in adults has probably been related to reduction in rates of pneumococcal nasopharyngeal carriage in children, another unique benefit of conjugated vaccines.3
In December 2011, the US Food and Drug Administration (FDA) approved PCV13 for adults on the basis of immunologic studies and anticipation that clinical efficacy would be similar to that observed in children.
HOW EFFECTIVE ARE THEY?
The efficacy and safety of PPSV23 and PCV13 have been studied in a variety of patient populations. Though antibody responses to PCV13 were similar to or better than those with PPSV23, no studies of specific correlations between immunologic responses and disease outcomes are available.4,5
In large studies in healthy adults, both vaccines reduced the incidence of invasive pneumococcal disease. A study in more than 47,000 adults age 65 and older showed a significant reduction in pneumococcal bacteremia (hazard ratio 0.56, 95% confidence interval 0.33–0.93) in those who received PPSV23 compared with those who received placebo.6 However, PPSV23 was not effective in preventing nonbacteremic and noninvasive pneumococcal community-acquired pneumonia when all bacterial serotypes were considered.6
In a placebo-controlled trial in more than 84,000 people age 65 and older, PCV13 prevented both nonbacteremic and bacteremic community-acquired pneumococcal pneumonia due to serotypes included in the vaccine (relative risk reduction 45%, P < .007) and overall invasive pneumococcal disease due to serotypes included in the vaccine (relative risk reduction 70%, P < .001).7
Both vaccines have also demonstrated efficacy in immunocompromised adults. Several studies showed an equivalent or superior antibody response to a seven-valent pneumococcal conjugate vaccine (PCV7, which has been replaced by PCV13) compared with PPSV23 in adults with human immunodeficiency virus (HIV) infection.8,9 While specific clinical studies of the efficacy of PCV13 among immunocompromised people are not available, a study of vaccination with PCV7 in 496 people in Malawi, of whom 88% were infected with HIV, found that the vaccine was effective in preventing invasive pneumococcal disease (hazard ratio 26%, 95% confidence interval 0.10–0.70).10
AT-RISK PATIENT POPULATIONS
Since both PPSV23 and PCV13 are approved for use in adults, it is important to understand appropriate indications for their use. The ACIP recommends pneumococcal vaccination in adults at an increased risk of invasive pneumococcal disease: ie, people age 65 and older, at-risk people ages 19 to 64, and people who are immunocompromised or asplenic.
A more robust antibody response has been shown with PCV13 compared to PPSV23 in healthy people.5 Of note, when PPSV23 is given before PCV13, there is a diminished immune response to PCV13.11,12 Therefore, unvaccinated people who will receive both PCV13 and PPSV23 should be given the conjugate vaccine PCV13 first. (See Commonly asked questions.)

ADULTS AGE 65 AND OLDER: ONE DOSE EACH OF PCV13 AND PPSV23
Before September 2014, the ACIP recommended one dose of PPSV23 for adults age 65 and older to prevent invasive pneumococcal disease.13 With evidence that PCV13 also produces an antibody response and is clinically effective against pneumococcal pneumonia in older people, the ACIP now recommends that all adults age 65 and older receive one dose of PCV13 and one dose of PPSV23.3,14
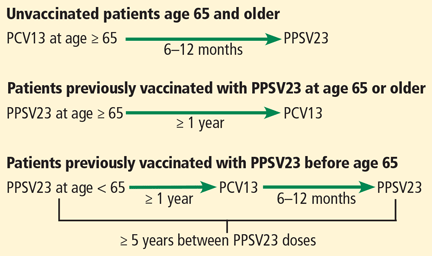
Based on antibody studies, the ACIP recommends giving PCV13 first and PPSV23 12 months after.11,12 Patients who received PPSV23 at age 65 or older should receive PCV13 at least 1 year after PPSV23 (Figure 2).3,14 Patients who had previously received one dose of PPSV23 before age 65 who are now age 65 or older should receive one dose of PCV13 at least 1 year after PPSV23 and an additional dose of PPSV23 at least 5 years after the first dose of PPSV23 and at least 1 year after the dose of PCV13.3 Patients who received a dose of PCV13 before age 65 do not need an additional dose after age 65.
The Centers for Medicare and Medicaid Services have updated the reimbursement for pneumococcal vaccines to include both PCV13 and PPSV23. Patients can receive one dose of pneumococcal vaccine followed by a different, second pneumococcal vaccine at least 11 full months after the month in which the first pneumococcal vaccine was administered.15
AT-RISK PATIENTS AGES 19 TO 64
Before 2012, the ACIP recommended that patients at risk, including immunocompromised patients and those without a spleen, with cerebrospinal fluid leaks, or with cochlear implants, receive only PPSV23 before age 65.13 In 2010, 50% of cases of invasive pneumococcal disease in immunocompromised adults were due to serotypes contained in PCV13.16 Additionally, according to CDC data from 2013, in adults ages 19 to 64 at risk of pneumococcal disease, only 21.2% had received pneumococcal vaccine.17 With information on epidemiology, safety, and efficacy, as well as expanded FDA approval of PCV13 for adults in 2011, the ACIP updated its guidelines for pneumococcal immunization of adults with immunocompromising conditions in October 2012.16 The updated guidelines now include giving PCV13 to adults at increased risk of invasive pneumococcal disease.16
Adults under age 65 at risk of invasive pneumococcal disease can be further divided into those who are immunocompetent with comorbid conditions, and those with cochlear implants or cerebrospinal fluid leak (Table 1).16
Patients with cochlear implants or cerebrospinal fluid leaks should receive one dose of PCV13 followed by one dose of PPSV23 8 weeks later. If PPSV23 is given first in this group, PCV13 can be given 1 year later.
Immunocompetent patients with comorbid conditions, including cigarette smoking, chronic heart, liver, or lung disease, asthma, cirrhosis, and diabetes mellitus, should receive one dose of PPSV23 before age 65 (Table 1).16
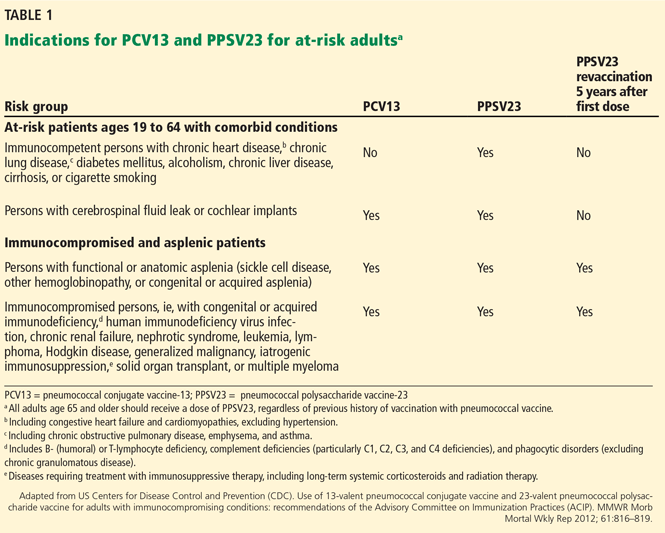
IMMUNOCOMPROMISED AND ASPLENIC PATIENTS
Immunocompromised patients at risk for invasive pneumococcal disease include patients with functional or anatomic asplenia or immunocompromising conditions such as HIV infection, chronic renal failure, generalized malignancy, solid organ transplant, iatrogenic immunosuppression (eg, due to corticosteroid therapy), and other immunocompromising conditions.16 Patients on corticosteroid therapy are considered immunosuppressed if they take 20 mg or more of prednisone daily (or an equivalent corticosteroid dose) for at least 14 days.16 These immunocompromised patients should receive one dose of PCV13, followed by a PPSV23 dose 8 weeks later and a second PPSV23 dose 5 years after the first.16
The time between vaccinations is also important. If PCV13 is given first, PPSV23 can be given after at least 8 weeks. If PPSV23 is given first, PCV13 should be given after 12 months. The time between PPSV23 doses is 5 years (Figure 3).16
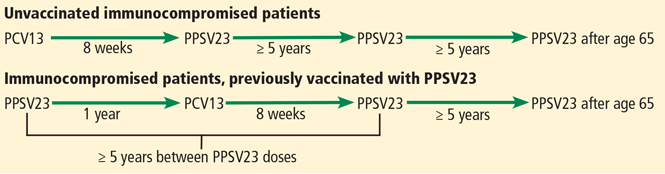
ADDRESSING BARRIERS TO PNEUMOCOCCAL VACCINATION
In 2013, only 59.7% of adults age 65 and older and 21.1% of younger, at-risk adults with immunocompromising conditions had received pneumococcal vaccination.17 Healthcare providers have the opportunity to improve pneumococcal vaccination rates. The National Foundation for Infectious Diseases (www.nfid.org) summarized challenges in vaccinating at-risk patients and recommended strategies to overcome barriers.18
Challenges include the cost of vaccine coverage, limited time (with competing priorities during office appointments or hospitalizations), patient refusal, and knowledge gaps.
Strategies to overcome barriers include incorporating vaccination into protocols and procedures; educating healthcare providers and patients about pneumococcal disease, vaccines, costs, and reimbursement; engaging nonclinical staff members; and monitoring local vaccination rates. However, the most important factor affecting whether adults are vaccinated is whether the healthcare provider recommends it.
AN OPPORTUNITY TO IMPROVE
In the last 30 years, great strides have been made in recognizing and preventing pneumococcal disease, but challenges remain. Adherence to the new ACIP guidelines for pneumococcal vaccination in immunocompromised, at risk and elderly patients is important in reducing invasive pneumococcal disease.
Healthcare providers have the opportunity to improve pneumococcal vaccination rates at outpatient appointments to decrease the burden of invasive pneumococcal disease in at-risk populations. A comprehensive understanding of the guideline recommendations for pneumococcal vaccination can aid the provider in identifying patients who are eligible for vaccination.
Adult pneumococcal immunization rates are low due to missed opportunities. Healthcare providers can improve these rates by viewing every patient encounter as a chance to provide vaccination.
- Centers for Disease Control and Prevention (CDC). Active Bacterial Core surveillance report (ABCs). ABCs Report: Streptococcus pneumoniae, 2010. www.cdc.gov/abcs/reports-findings/survreports/spneu10-orig.html. Accessed May 13, 2016.
- Said MA, Johnson, HL, Nonyane BA, et al. Estimating the burden of pneumococcal pneumonia among adults: a systematic review and meta-analysis of diagnostic techniques. Plos One. 2013;8:e60273.
- Tomczyk S, Bennett NM, Stoecker C, et al; Centers for Disease Control and Prevention (CDC). Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine among adults aged ≥ 65 years: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep. 2014;63:822–825.
- Crum-Cianflone NF, Huppler Hullsiek K, Roediger M, et al; Infectious Disease Clinical Research Program HIV Working Group. A randomized clinical trial comparing revaccination with pneumococcal conjugate vaccine to polysaccharide vaccine among HIV-infected adults. J Infect Dis. 2010:202:1114–1125.
- Jackson LA, Gurtman A, van Cleeff M, et al. Immunogenicity and safety of a 13-valent pneumococcal conjugate vaccine compared to a 23-valent pneumococcal polysaccharide vaccine in pneumococcal vaccine-naïve adults. Vaccine. 2013;31:3577–3584.
- Jackson LA, Neuzil KM, Yu O, et al; Vaccine Safety Datalink. Effectiveness of pneumococcal polysaccharide vaccine in older adults. N Engl J Med. 2003;348:1747–1755.
- Bonten M, Huijts S, Bolkenbaas M, et al. Polysaccharide conjugate vaccine against pneumococcal pneumonia in adults. N Engl J Med. 2015;372:1114–1125.
- Lesprit P, Pedrono G, Molina JM, et al; ANRS 114-Pneumovac Study Group. Immunological efficacy of a prime-boosted pneumococcal vaccination in HIV-infected adults. AIDS. 2007;21:2425–2434.
- Feikin DR, Elie CM, Goetz MB, et al. Randomized trial of the quantitative and functional antibody responses to a 7-valent pneumococcal conjugate vaccine and/or 23-valent polysaccharide vaccine among HIV-infected adults. Vaccine. 2001;20:545–553.
- French N, Gordon SB, Mwalukomo T, et al. A trial of a 7-valent pneumococcal conjugate vaccine in HIV-infected adults. N Engl J Med. 2010;362:812–822.
- Jackson LA, Gurtman A, Rice K, et al. Immunogenicity and safety of a 13-valent pneumococcal conjugate vaccine in adults 70 years of age and older previously vaccinated with 23-valent pneumococcal polysaccharide vaccine. Vaccine. 2013;31:3585–3593.
- Greenberg RN, Gurtman A, French RW, et al. Sequential administration of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine in pneumococcal vaccine-naïve adults 60-64 years of age. Vaccine. 2014;32:2364–2374.
- Centers for Disease Control and Prevention (CDC); Advisory Committee on Immunization Practices. Updated recommendations for prevention of invasive pneumococcal disease among adults using the 23-valent pneumococcal polysaccharide vaccine (PPSV23). MMWR Morb Mortal Wkly Rep. 2010:59:1102–1106.
- Kobayashi M, Bennett NM, Gierke R, et al. Centers for Disease Control and Prevention (CDC). Intervals between PCV13 and PPSV23; Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morbid Mortal Wkly Rep. 2015;64:944-947.
- Department of Health and Human Services; Centers for Medicare and Medicaid Services. Modifications to Medicare Part B coverage of pneumococcal vaccinations. www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/Downloads/MM9051.pdf. Accessed May 13, 2016.
- Centers for Disease Control and Prevention (CDC). Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine for adults with immunocompromising conditions: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep. 2012;61:816–819.
- Williams WW, Lu PJ, O’Halloran A, et al; Centers for Disease Control and Prevention (CDC). Noninfluenza vaccination coverage among adults - United States, 2013. MMWR Morb Mortal Wkly Rep. 2015;64:95–102.
- Rehm SJ, File TM, Metersky M, Nichol KL, Schaffner W; National Foundation for Infectious Diseases Pneumococcal Disease Advisory Board. Identifying barriers to adult pneumococcal vaccination: an NFID task force meeting. Postgrad Med. 2012;124:71–79.
- Centers for Disease Control and Prevention (CDC). Vaccines and immunizations. PCV13 (pneumococcal conjugate) vaccine. Recommendations, scenarios and Q&As for healthcare professionals about PCV13 for adults. www.cdc.gov/vaccines/vpd-vac/pneumo/vac-PCV13-adults.htm. Accessed May 13, 2016.
- Harpaz R, Ortega-Sanchez IR, Seward JF; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep. 2008;57:1–30.
- Immunization Action Coalition. Ask the experts: diseases & vaccines. Pneumococcal vaccines (PCV13 and PPSV23). www.immunize.org/askexperts/experts_pneumococcal_vaccines.asp. Accessed May 13, 2016.
STREPTOCOCCUS PNEUMONIAE (the “pneumococcus”) causes a variety of clinical syndromes that range from otitis media to bacteremia, meningitis, and pneumonia. Hardest hit are immunocompromised people and those at the extremes of age. Therefore, preventing disease through pneumococcal vaccination is very important in these groups.
This review summarizes the current guidelines from the Advisory Committee on Immunization Practices (ACIP) of the US Centers for Disease Control and Prevention (CDC) for pneumococcal immunization in adults.
STRIKES THE VERY YOUNG, VERY OLD, AND IMMUNOCOMPROMISED
Invasive pneumococcal disease is defined as infection in which S pneumoniae can be found in a normally sterile site such as the cerebrospinal fluid or blood, and it includes bacteremic pneumonia.1 By far the most common type of pneumococcal disease is pneumonia, followed by bacteremia and meningitis (Figure 1);2,3 about 25% of patients with pneumococcal pneumonia also have bacteremia.2
Invasive pneumococcal disease most often occurs in children age 2 and younger, adults age 65 and older, and people who are immunocompromised. In 2010, the incidence was 3.8 per 100,000 in people ages 18 to 34 but was 10 times higher in the elderly and those with compromised immunity.1
Even now that vaccines are available, invasive pneumococcal disease continues to cause 4,000 deaths per year in the United States.1
TWO INACTIVATED VACCINES
S pneumoniae is a gram-positive coccus with an outer capsule composed of polysaccharides that protect the bacterium from being ingested and killed by host phagocytic cells. Some 91 serotypes of this organism have been identified on the basis of genetic differences in capsular polysaccharide composition.
Currently, two inactivated vaccines are available that elicit antibody responses to the most common pneumococcal serotypes that infect humans.
- PPSV23 (pneumococcal polysaccharide vaccine-23, or Pneumovax 23) contains purified capsular polysaccharides from 23 pneumococcal serotypes.
- PCV13 (pneumococcal conjugate vaccine-13, or Prevnar 13) contains purified capsular polysaccharides from 13 serotypes that are covalently bound to (conjugated with) a carrier protein.
PPSV23 AND PCV13 ARE NOT THE SAME
Apart from the number of serotypes covered, the two vaccines differ in important ways. Both of them elicit a B-cell-mediated immune response, but only PCV13 produces a T-cell-dependent response, which is essential for maturation of the B-cell response and development of immune memory.
PPSV23 generally provides 3 to 5 years of immunity, and repeat doses do not offer additive or “boosted” protection. It is ineffective in children under 2 years of age.
Pneumococcal conjugate vaccine has been available since 2000 for children starting at 2 months of age. Since then it has directly reduced the incidence of invasive pneumococcal disease in children and indirectly in adults. The impact on pneumococcal disease rates in adults has probably been related to reduction in rates of pneumococcal nasopharyngeal carriage in children, another unique benefit of conjugated vaccines.3
In December 2011, the US Food and Drug Administration (FDA) approved PCV13 for adults on the basis of immunologic studies and anticipation that clinical efficacy would be similar to that observed in children.
HOW EFFECTIVE ARE THEY?
The efficacy and safety of PPSV23 and PCV13 have been studied in a variety of patient populations. Though antibody responses to PCV13 were similar to or better than those with PPSV23, no studies of specific correlations between immunologic responses and disease outcomes are available.4,5
In large studies in healthy adults, both vaccines reduced the incidence of invasive pneumococcal disease. A study in more than 47,000 adults age 65 and older showed a significant reduction in pneumococcal bacteremia (hazard ratio 0.56, 95% confidence interval 0.33–0.93) in those who received PPSV23 compared with those who received placebo.6 However, PPSV23 was not effective in preventing nonbacteremic and noninvasive pneumococcal community-acquired pneumonia when all bacterial serotypes were considered.6
In a placebo-controlled trial in more than 84,000 people age 65 and older, PCV13 prevented both nonbacteremic and bacteremic community-acquired pneumococcal pneumonia due to serotypes included in the vaccine (relative risk reduction 45%, P < .007) and overall invasive pneumococcal disease due to serotypes included in the vaccine (relative risk reduction 70%, P < .001).7
Both vaccines have also demonstrated efficacy in immunocompromised adults. Several studies showed an equivalent or superior antibody response to a seven-valent pneumococcal conjugate vaccine (PCV7, which has been replaced by PCV13) compared with PPSV23 in adults with human immunodeficiency virus (HIV) infection.8,9 While specific clinical studies of the efficacy of PCV13 among immunocompromised people are not available, a study of vaccination with PCV7 in 496 people in Malawi, of whom 88% were infected with HIV, found that the vaccine was effective in preventing invasive pneumococcal disease (hazard ratio 26%, 95% confidence interval 0.10–0.70).10
AT-RISK PATIENT POPULATIONS
Since both PPSV23 and PCV13 are approved for use in adults, it is important to understand appropriate indications for their use. The ACIP recommends pneumococcal vaccination in adults at an increased risk of invasive pneumococcal disease: ie, people age 65 and older, at-risk people ages 19 to 64, and people who are immunocompromised or asplenic.
A more robust antibody response has been shown with PCV13 compared to PPSV23 in healthy people.5 Of note, when PPSV23 is given before PCV13, there is a diminished immune response to PCV13.11,12 Therefore, unvaccinated people who will receive both PCV13 and PPSV23 should be given the conjugate vaccine PCV13 first. (See Commonly asked questions.)
ADULTS AGE 65 AND OLDER: ONE DOSE EACH OF PCV13 AND PPSV23
Before September 2014, the ACIP recommended one dose of PPSV23 for adults age 65 and older to prevent invasive pneumococcal disease.13 With evidence that PCV13 also produces an antibody response and is clinically effective against pneumococcal pneumonia in older people, the ACIP now recommends that all adults age 65 and older receive one dose of PCV13 and one dose of PPSV23.3,14
Based on antibody studies, the ACIP recommends giving PCV13 first and PPSV23 12 months after.11,12 Patients who received PPSV23 at age 65 or older should receive PCV13 at least 1 year after PPSV23 (Figure 2).3,14 Patients who had previously received one dose of PPSV23 before age 65 who are now age 65 or older should receive one dose of PCV13 at least 1 year after PPSV23 and an additional dose of PPSV23 at least 5 years after the first dose of PPSV23 and at least 1 year after the dose of PCV13.3 Patients who received a dose of PCV13 before age 65 do not need an additional dose after age 65.
The Centers for Medicare and Medicaid Services have updated the reimbursement for pneumococcal vaccines to include both PCV13 and PPSV23. Patients can receive one dose of pneumococcal vaccine followed by a different, second pneumococcal vaccine at least 11 full months after the month in which the first pneumococcal vaccine was administered.15
AT-RISK PATIENTS AGES 19 TO 64
Before 2012, the ACIP recommended that patients at risk, including immunocompromised patients and those without a spleen, with cerebrospinal fluid leaks, or with cochlear implants, receive only PPSV23 before age 65.13 In 2010, 50% of cases of invasive pneumococcal disease in immunocompromised adults were due to serotypes contained in PCV13.16 Additionally, according to CDC data from 2013, in adults ages 19 to 64 at risk of pneumococcal disease, only 21.2% had received pneumococcal vaccine.17 With information on epidemiology, safety, and efficacy, as well as expanded FDA approval of PCV13 for adults in 2011, the ACIP updated its guidelines for pneumococcal immunization of adults with immunocompromising conditions in October 2012.16 The updated guidelines now include giving PCV13 to adults at increased risk of invasive pneumococcal disease.16
Adults under age 65 at risk of invasive pneumococcal disease can be further divided into those who are immunocompetent with comorbid conditions, and those with cochlear implants or cerebrospinal fluid leak (Table 1).16
Patients with cochlear implants or cerebrospinal fluid leaks should receive one dose of PCV13 followed by one dose of PPSV23 8 weeks later. If PPSV23 is given first in this group, PCV13 can be given 1 year later.
Immunocompetent patients with comorbid conditions, including cigarette smoking, chronic heart, liver, or lung disease, asthma, cirrhosis, and diabetes mellitus, should receive one dose of PPSV23 before age 65 (Table 1).16
IMMUNOCOMPROMISED AND ASPLENIC PATIENTS
Immunocompromised patients at risk for invasive pneumococcal disease include patients with functional or anatomic asplenia or immunocompromising conditions such as HIV infection, chronic renal failure, generalized malignancy, solid organ transplant, iatrogenic immunosuppression (eg, due to corticosteroid therapy), and other immunocompromising conditions.16 Patients on corticosteroid therapy are considered immunosuppressed if they take 20 mg or more of prednisone daily (or an equivalent corticosteroid dose) for at least 14 days.16 These immunocompromised patients should receive one dose of PCV13, followed by a PPSV23 dose 8 weeks later and a second PPSV23 dose 5 years after the first.16
The time between vaccinations is also important. If PCV13 is given first, PPSV23 can be given after at least 8 weeks. If PPSV23 is given first, PCV13 should be given after 12 months. The time between PPSV23 doses is 5 years (Figure 3).16
ADDRESSING BARRIERS TO PNEUMOCOCCAL VACCINATION
In 2013, only 59.7% of adults age 65 and older and 21.1% of younger, at-risk adults with immunocompromising conditions had received pneumococcal vaccination.17 Healthcare providers have the opportunity to improve pneumococcal vaccination rates. The National Foundation for Infectious Diseases (www.nfid.org) summarized challenges in vaccinating at-risk patients and recommended strategies to overcome barriers.18
Challenges include the cost of vaccine coverage, limited time (with competing priorities during office appointments or hospitalizations), patient refusal, and knowledge gaps.
Strategies to overcome barriers include incorporating vaccination into protocols and procedures; educating healthcare providers and patients about pneumococcal disease, vaccines, costs, and reimbursement; engaging nonclinical staff members; and monitoring local vaccination rates. However, the most important factor affecting whether adults are vaccinated is whether the healthcare provider recommends it.
AN OPPORTUNITY TO IMPROVE
In the last 30 years, great strides have been made in recognizing and preventing pneumococcal disease, but challenges remain. Adherence to the new ACIP guidelines for pneumococcal vaccination in immunocompromised, at risk and elderly patients is important in reducing invasive pneumococcal disease.
Healthcare providers have the opportunity to improve pneumococcal vaccination rates at outpatient appointments to decrease the burden of invasive pneumococcal disease in at-risk populations. A comprehensive understanding of the guideline recommendations for pneumococcal vaccination can aid the provider in identifying patients who are eligible for vaccination.
Adult pneumococcal immunization rates are low due to missed opportunities. Healthcare providers can improve these rates by viewing every patient encounter as a chance to provide vaccination.
STREPTOCOCCUS PNEUMONIAE (the “pneumococcus”) causes a variety of clinical syndromes that range from otitis media to bacteremia, meningitis, and pneumonia. Hardest hit are immunocompromised people and those at the extremes of age. Therefore, preventing disease through pneumococcal vaccination is very important in these groups.
This review summarizes the current guidelines from the Advisory Committee on Immunization Practices (ACIP) of the US Centers for Disease Control and Prevention (CDC) for pneumococcal immunization in adults.
STRIKES THE VERY YOUNG, VERY OLD, AND IMMUNOCOMPROMISED
Invasive pneumococcal disease is defined as infection in which S pneumoniae can be found in a normally sterile site such as the cerebrospinal fluid or blood, and it includes bacteremic pneumonia.1 By far the most common type of pneumococcal disease is pneumonia, followed by bacteremia and meningitis (Figure 1);2,3 about 25% of patients with pneumococcal pneumonia also have bacteremia.2
Invasive pneumococcal disease most often occurs in children age 2 and younger, adults age 65 and older, and people who are immunocompromised. In 2010, the incidence was 3.8 per 100,000 in people ages 18 to 34 but was 10 times higher in the elderly and those with compromised immunity.1
Even now that vaccines are available, invasive pneumococcal disease continues to cause 4,000 deaths per year in the United States.1
TWO INACTIVATED VACCINES
S pneumoniae is a gram-positive coccus with an outer capsule composed of polysaccharides that protect the bacterium from being ingested and killed by host phagocytic cells. Some 91 serotypes of this organism have been identified on the basis of genetic differences in capsular polysaccharide composition.
Currently, two inactivated vaccines are available that elicit antibody responses to the most common pneumococcal serotypes that infect humans.
- PPSV23 (pneumococcal polysaccharide vaccine-23, or Pneumovax 23) contains purified capsular polysaccharides from 23 pneumococcal serotypes.
- PCV13 (pneumococcal conjugate vaccine-13, or Prevnar 13) contains purified capsular polysaccharides from 13 serotypes that are covalently bound to (conjugated with) a carrier protein.
PPSV23 AND PCV13 ARE NOT THE SAME
Apart from the number of serotypes covered, the two vaccines differ in important ways. Both of them elicit a B-cell-mediated immune response, but only PCV13 produces a T-cell-dependent response, which is essential for maturation of the B-cell response and development of immune memory.
PPSV23 generally provides 3 to 5 years of immunity, and repeat doses do not offer additive or “boosted” protection. It is ineffective in children under 2 years of age.
Pneumococcal conjugate vaccine has been available since 2000 for children starting at 2 months of age. Since then it has directly reduced the incidence of invasive pneumococcal disease in children and indirectly in adults. The impact on pneumococcal disease rates in adults has probably been related to reduction in rates of pneumococcal nasopharyngeal carriage in children, another unique benefit of conjugated vaccines.3
In December 2011, the US Food and Drug Administration (FDA) approved PCV13 for adults on the basis of immunologic studies and anticipation that clinical efficacy would be similar to that observed in children.
HOW EFFECTIVE ARE THEY?
The efficacy and safety of PPSV23 and PCV13 have been studied in a variety of patient populations. Though antibody responses to PCV13 were similar to or better than those with PPSV23, no studies of specific correlations between immunologic responses and disease outcomes are available.4,5
In large studies in healthy adults, both vaccines reduced the incidence of invasive pneumococcal disease. A study in more than 47,000 adults age 65 and older showed a significant reduction in pneumococcal bacteremia (hazard ratio 0.56, 95% confidence interval 0.33–0.93) in those who received PPSV23 compared with those who received placebo.6 However, PPSV23 was not effective in preventing nonbacteremic and noninvasive pneumococcal community-acquired pneumonia when all bacterial serotypes were considered.6
In a placebo-controlled trial in more than 84,000 people age 65 and older, PCV13 prevented both nonbacteremic and bacteremic community-acquired pneumococcal pneumonia due to serotypes included in the vaccine (relative risk reduction 45%, P < .007) and overall invasive pneumococcal disease due to serotypes included in the vaccine (relative risk reduction 70%, P < .001).7
Both vaccines have also demonstrated efficacy in immunocompromised adults. Several studies showed an equivalent or superior antibody response to a seven-valent pneumococcal conjugate vaccine (PCV7, which has been replaced by PCV13) compared with PPSV23 in adults with human immunodeficiency virus (HIV) infection.8,9 While specific clinical studies of the efficacy of PCV13 among immunocompromised people are not available, a study of vaccination with PCV7 in 496 people in Malawi, of whom 88% were infected with HIV, found that the vaccine was effective in preventing invasive pneumococcal disease (hazard ratio 26%, 95% confidence interval 0.10–0.70).10
AT-RISK PATIENT POPULATIONS
Since both PPSV23 and PCV13 are approved for use in adults, it is important to understand appropriate indications for their use. The ACIP recommends pneumococcal vaccination in adults at an increased risk of invasive pneumococcal disease: ie, people age 65 and older, at-risk people ages 19 to 64, and people who are immunocompromised or asplenic.
A more robust antibody response has been shown with PCV13 compared to PPSV23 in healthy people.5 Of note, when PPSV23 is given before PCV13, there is a diminished immune response to PCV13.11,12 Therefore, unvaccinated people who will receive both PCV13 and PPSV23 should be given the conjugate vaccine PCV13 first. (See Commonly asked questions.)
ADULTS AGE 65 AND OLDER: ONE DOSE EACH OF PCV13 AND PPSV23
Before September 2014, the ACIP recommended one dose of PPSV23 for adults age 65 and older to prevent invasive pneumococcal disease.13 With evidence that PCV13 also produces an antibody response and is clinically effective against pneumococcal pneumonia in older people, the ACIP now recommends that all adults age 65 and older receive one dose of PCV13 and one dose of PPSV23.3,14
Based on antibody studies, the ACIP recommends giving PCV13 first and PPSV23 12 months after.11,12 Patients who received PPSV23 at age 65 or older should receive PCV13 at least 1 year after PPSV23 (Figure 2).3,14 Patients who had previously received one dose of PPSV23 before age 65 who are now age 65 or older should receive one dose of PCV13 at least 1 year after PPSV23 and an additional dose of PPSV23 at least 5 years after the first dose of PPSV23 and at least 1 year after the dose of PCV13.3 Patients who received a dose of PCV13 before age 65 do not need an additional dose after age 65.
The Centers for Medicare and Medicaid Services have updated the reimbursement for pneumococcal vaccines to include both PCV13 and PPSV23. Patients can receive one dose of pneumococcal vaccine followed by a different, second pneumococcal vaccine at least 11 full months after the month in which the first pneumococcal vaccine was administered.15
AT-RISK PATIENTS AGES 19 TO 64
Before 2012, the ACIP recommended that patients at risk, including immunocompromised patients and those without a spleen, with cerebrospinal fluid leaks, or with cochlear implants, receive only PPSV23 before age 65.13 In 2010, 50% of cases of invasive pneumococcal disease in immunocompromised adults were due to serotypes contained in PCV13.16 Additionally, according to CDC data from 2013, in adults ages 19 to 64 at risk of pneumococcal disease, only 21.2% had received pneumococcal vaccine.17 With information on epidemiology, safety, and efficacy, as well as expanded FDA approval of PCV13 for adults in 2011, the ACIP updated its guidelines for pneumococcal immunization of adults with immunocompromising conditions in October 2012.16 The updated guidelines now include giving PCV13 to adults at increased risk of invasive pneumococcal disease.16
Adults under age 65 at risk of invasive pneumococcal disease can be further divided into those who are immunocompetent with comorbid conditions, and those with cochlear implants or cerebrospinal fluid leak (Table 1).16
Patients with cochlear implants or cerebrospinal fluid leaks should receive one dose of PCV13 followed by one dose of PPSV23 8 weeks later. If PPSV23 is given first in this group, PCV13 can be given 1 year later.
Immunocompetent patients with comorbid conditions, including cigarette smoking, chronic heart, liver, or lung disease, asthma, cirrhosis, and diabetes mellitus, should receive one dose of PPSV23 before age 65 (Table 1).16
IMMUNOCOMPROMISED AND ASPLENIC PATIENTS
Immunocompromised patients at risk for invasive pneumococcal disease include patients with functional or anatomic asplenia or immunocompromising conditions such as HIV infection, chronic renal failure, generalized malignancy, solid organ transplant, iatrogenic immunosuppression (eg, due to corticosteroid therapy), and other immunocompromising conditions.16 Patients on corticosteroid therapy are considered immunosuppressed if they take 20 mg or more of prednisone daily (or an equivalent corticosteroid dose) for at least 14 days.16 These immunocompromised patients should receive one dose of PCV13, followed by a PPSV23 dose 8 weeks later and a second PPSV23 dose 5 years after the first.16
The time between vaccinations is also important. If PCV13 is given first, PPSV23 can be given after at least 8 weeks. If PPSV23 is given first, PCV13 should be given after 12 months. The time between PPSV23 doses is 5 years (Figure 3).16
ADDRESSING BARRIERS TO PNEUMOCOCCAL VACCINATION
In 2013, only 59.7% of adults age 65 and older and 21.1% of younger, at-risk adults with immunocompromising conditions had received pneumococcal vaccination.17 Healthcare providers have the opportunity to improve pneumococcal vaccination rates. The National Foundation for Infectious Diseases (www.nfid.org) summarized challenges in vaccinating at-risk patients and recommended strategies to overcome barriers.18
Challenges include the cost of vaccine coverage, limited time (with competing priorities during office appointments or hospitalizations), patient refusal, and knowledge gaps.
Strategies to overcome barriers include incorporating vaccination into protocols and procedures; educating healthcare providers and patients about pneumococcal disease, vaccines, costs, and reimbursement; engaging nonclinical staff members; and monitoring local vaccination rates. However, the most important factor affecting whether adults are vaccinated is whether the healthcare provider recommends it.
AN OPPORTUNITY TO IMPROVE
In the last 30 years, great strides have been made in recognizing and preventing pneumococcal disease, but challenges remain. Adherence to the new ACIP guidelines for pneumococcal vaccination in immunocompromised, at risk and elderly patients is important in reducing invasive pneumococcal disease.
Healthcare providers have the opportunity to improve pneumococcal vaccination rates at outpatient appointments to decrease the burden of invasive pneumococcal disease in at-risk populations. A comprehensive understanding of the guideline recommendations for pneumococcal vaccination can aid the provider in identifying patients who are eligible for vaccination.
Adult pneumococcal immunization rates are low due to missed opportunities. Healthcare providers can improve these rates by viewing every patient encounter as a chance to provide vaccination.
- Centers for Disease Control and Prevention (CDC). Active Bacterial Core surveillance report (ABCs). ABCs Report: Streptococcus pneumoniae, 2010. www.cdc.gov/abcs/reports-findings/survreports/spneu10-orig.html. Accessed May 13, 2016.
- Said MA, Johnson, HL, Nonyane BA, et al. Estimating the burden of pneumococcal pneumonia among adults: a systematic review and meta-analysis of diagnostic techniques. Plos One. 2013;8:e60273.
- Tomczyk S, Bennett NM, Stoecker C, et al; Centers for Disease Control and Prevention (CDC). Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine among adults aged ≥ 65 years: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep. 2014;63:822–825.
- Crum-Cianflone NF, Huppler Hullsiek K, Roediger M, et al; Infectious Disease Clinical Research Program HIV Working Group. A randomized clinical trial comparing revaccination with pneumococcal conjugate vaccine to polysaccharide vaccine among HIV-infected adults. J Infect Dis. 2010:202:1114–1125.
- Jackson LA, Gurtman A, van Cleeff M, et al. Immunogenicity and safety of a 13-valent pneumococcal conjugate vaccine compared to a 23-valent pneumococcal polysaccharide vaccine in pneumococcal vaccine-naïve adults. Vaccine. 2013;31:3577–3584.
- Jackson LA, Neuzil KM, Yu O, et al; Vaccine Safety Datalink. Effectiveness of pneumococcal polysaccharide vaccine in older adults. N Engl J Med. 2003;348:1747–1755.
- Bonten M, Huijts S, Bolkenbaas M, et al. Polysaccharide conjugate vaccine against pneumococcal pneumonia in adults. N Engl J Med. 2015;372:1114–1125.
- Lesprit P, Pedrono G, Molina JM, et al; ANRS 114-Pneumovac Study Group. Immunological efficacy of a prime-boosted pneumococcal vaccination in HIV-infected adults. AIDS. 2007;21:2425–2434.
- Feikin DR, Elie CM, Goetz MB, et al. Randomized trial of the quantitative and functional antibody responses to a 7-valent pneumococcal conjugate vaccine and/or 23-valent polysaccharide vaccine among HIV-infected adults. Vaccine. 2001;20:545–553.
- French N, Gordon SB, Mwalukomo T, et al. A trial of a 7-valent pneumococcal conjugate vaccine in HIV-infected adults. N Engl J Med. 2010;362:812–822.
- Jackson LA, Gurtman A, Rice K, et al. Immunogenicity and safety of a 13-valent pneumococcal conjugate vaccine in adults 70 years of age and older previously vaccinated with 23-valent pneumococcal polysaccharide vaccine. Vaccine. 2013;31:3585–3593.
- Greenberg RN, Gurtman A, French RW, et al. Sequential administration of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine in pneumococcal vaccine-naïve adults 60-64 years of age. Vaccine. 2014;32:2364–2374.
- Centers for Disease Control and Prevention (CDC); Advisory Committee on Immunization Practices. Updated recommendations for prevention of invasive pneumococcal disease among adults using the 23-valent pneumococcal polysaccharide vaccine (PPSV23). MMWR Morb Mortal Wkly Rep. 2010:59:1102–1106.
- Kobayashi M, Bennett NM, Gierke R, et al. Centers for Disease Control and Prevention (CDC). Intervals between PCV13 and PPSV23; Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morbid Mortal Wkly Rep. 2015;64:944-947.
- Department of Health and Human Services; Centers for Medicare and Medicaid Services. Modifications to Medicare Part B coverage of pneumococcal vaccinations. www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/Downloads/MM9051.pdf. Accessed May 13, 2016.
- Centers for Disease Control and Prevention (CDC). Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine for adults with immunocompromising conditions: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep. 2012;61:816–819.
- Williams WW, Lu PJ, O’Halloran A, et al; Centers for Disease Control and Prevention (CDC). Noninfluenza vaccination coverage among adults - United States, 2013. MMWR Morb Mortal Wkly Rep. 2015;64:95–102.
- Rehm SJ, File TM, Metersky M, Nichol KL, Schaffner W; National Foundation for Infectious Diseases Pneumococcal Disease Advisory Board. Identifying barriers to adult pneumococcal vaccination: an NFID task force meeting. Postgrad Med. 2012;124:71–79.
- Centers for Disease Control and Prevention (CDC). Vaccines and immunizations. PCV13 (pneumococcal conjugate) vaccine. Recommendations, scenarios and Q&As for healthcare professionals about PCV13 for adults. www.cdc.gov/vaccines/vpd-vac/pneumo/vac-PCV13-adults.htm. Accessed May 13, 2016.
- Harpaz R, Ortega-Sanchez IR, Seward JF; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep. 2008;57:1–30.
- Immunization Action Coalition. Ask the experts: diseases & vaccines. Pneumococcal vaccines (PCV13 and PPSV23). www.immunize.org/askexperts/experts_pneumococcal_vaccines.asp. Accessed May 13, 2016.
- Centers for Disease Control and Prevention (CDC). Active Bacterial Core surveillance report (ABCs). ABCs Report: Streptococcus pneumoniae, 2010. www.cdc.gov/abcs/reports-findings/survreports/spneu10-orig.html. Accessed May 13, 2016.
- Said MA, Johnson, HL, Nonyane BA, et al. Estimating the burden of pneumococcal pneumonia among adults: a systematic review and meta-analysis of diagnostic techniques. Plos One. 2013;8:e60273.
- Tomczyk S, Bennett NM, Stoecker C, et al; Centers for Disease Control and Prevention (CDC). Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine among adults aged ≥ 65 years: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep. 2014;63:822–825.
- Crum-Cianflone NF, Huppler Hullsiek K, Roediger M, et al; Infectious Disease Clinical Research Program HIV Working Group. A randomized clinical trial comparing revaccination with pneumococcal conjugate vaccine to polysaccharide vaccine among HIV-infected adults. J Infect Dis. 2010:202:1114–1125.
- Jackson LA, Gurtman A, van Cleeff M, et al. Immunogenicity and safety of a 13-valent pneumococcal conjugate vaccine compared to a 23-valent pneumococcal polysaccharide vaccine in pneumococcal vaccine-naïve adults. Vaccine. 2013;31:3577–3584.
- Jackson LA, Neuzil KM, Yu O, et al; Vaccine Safety Datalink. Effectiveness of pneumococcal polysaccharide vaccine in older adults. N Engl J Med. 2003;348:1747–1755.
- Bonten M, Huijts S, Bolkenbaas M, et al. Polysaccharide conjugate vaccine against pneumococcal pneumonia in adults. N Engl J Med. 2015;372:1114–1125.
- Lesprit P, Pedrono G, Molina JM, et al; ANRS 114-Pneumovac Study Group. Immunological efficacy of a prime-boosted pneumococcal vaccination in HIV-infected adults. AIDS. 2007;21:2425–2434.
- Feikin DR, Elie CM, Goetz MB, et al. Randomized trial of the quantitative and functional antibody responses to a 7-valent pneumococcal conjugate vaccine and/or 23-valent polysaccharide vaccine among HIV-infected adults. Vaccine. 2001;20:545–553.
- French N, Gordon SB, Mwalukomo T, et al. A trial of a 7-valent pneumococcal conjugate vaccine in HIV-infected adults. N Engl J Med. 2010;362:812–822.
- Jackson LA, Gurtman A, Rice K, et al. Immunogenicity and safety of a 13-valent pneumococcal conjugate vaccine in adults 70 years of age and older previously vaccinated with 23-valent pneumococcal polysaccharide vaccine. Vaccine. 2013;31:3585–3593.
- Greenberg RN, Gurtman A, French RW, et al. Sequential administration of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine in pneumococcal vaccine-naïve adults 60-64 years of age. Vaccine. 2014;32:2364–2374.
- Centers for Disease Control and Prevention (CDC); Advisory Committee on Immunization Practices. Updated recommendations for prevention of invasive pneumococcal disease among adults using the 23-valent pneumococcal polysaccharide vaccine (PPSV23). MMWR Morb Mortal Wkly Rep. 2010:59:1102–1106.
- Kobayashi M, Bennett NM, Gierke R, et al. Centers for Disease Control and Prevention (CDC). Intervals between PCV13 and PPSV23; Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morbid Mortal Wkly Rep. 2015;64:944-947.
- Department of Health and Human Services; Centers for Medicare and Medicaid Services. Modifications to Medicare Part B coverage of pneumococcal vaccinations. www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/Downloads/MM9051.pdf. Accessed May 13, 2016.
- Centers for Disease Control and Prevention (CDC). Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine for adults with immunocompromising conditions: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep. 2012;61:816–819.
- Williams WW, Lu PJ, O’Halloran A, et al; Centers for Disease Control and Prevention (CDC). Noninfluenza vaccination coverage among adults - United States, 2013. MMWR Morb Mortal Wkly Rep. 2015;64:95–102.
- Rehm SJ, File TM, Metersky M, Nichol KL, Schaffner W; National Foundation for Infectious Diseases Pneumococcal Disease Advisory Board. Identifying barriers to adult pneumococcal vaccination: an NFID task force meeting. Postgrad Med. 2012;124:71–79.
- Centers for Disease Control and Prevention (CDC). Vaccines and immunizations. PCV13 (pneumococcal conjugate) vaccine. Recommendations, scenarios and Q&As for healthcare professionals about PCV13 for adults. www.cdc.gov/vaccines/vpd-vac/pneumo/vac-PCV13-adults.htm. Accessed May 13, 2016.
- Harpaz R, Ortega-Sanchez IR, Seward JF; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep. 2008;57:1–30.
- Immunization Action Coalition. Ask the experts: diseases & vaccines. Pneumococcal vaccines (PCV13 and PPSV23). www.immunize.org/askexperts/experts_pneumococcal_vaccines.asp. Accessed May 13, 2016.
KEY POINTS
- At highest risk of invasive pneumococcal disease are people who are immunocompromised, very young, or very old.
- Pneumococcal polysaccharide vaccine-23 (PPSV23) covers more serotypes of S pneumoniae than pneumococcal conjugate vaccine-13 (PCV13), but the latter induces a stronger antibody response.
- The combination of both vaccines in sequence produces a better antibody response than either vaccine alone.
- The Advisory Committee on Immunization Practices now recommends that immunocompromised and asplenic adults who need pneumococcal vaccination receive both vaccines, preferably PCV13 first, followed by PPSV23 8 weeks later. Those who have already received PPSV23 can receive PCV13 after at least 1 year has passed.
- People with asplenia or immunocompromising conditions should receive a second dose of PPSV23 at least 5 years after the first dose.
- Vaccination schedules and information are available from the US Centers for Disease Control and Prevention at www.cdc.gov.
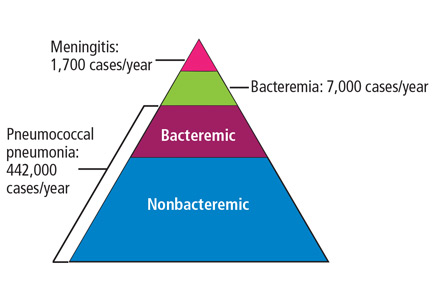
Register for Hospital Medicine 2017

- Reenergize and focus your practice with the latest research, best practices, and newest innovations in the field that can immediately be applied to improving patient care.
- Learn from the “best of the best,” including nationally renowned leaders in the field of hospital medicine.
- Connect and collaborate with a vibrant community of hospital medicine professionals.
It’s not too early to register! Sign up now and save at www.hospitalmedicine2017.org.
- Reenergize and focus your practice with the latest research, best practices, and newest innovations in the field that can immediately be applied to improving patient care.
- Learn from the “best of the best,” including nationally renowned leaders in the field of hospital medicine.
- Connect and collaborate with a vibrant community of hospital medicine professionals.
It’s not too early to register! Sign up now and save at www.hospitalmedicine2017.org.
- Reenergize and focus your practice with the latest research, best practices, and newest innovations in the field that can immediately be applied to improving patient care.
- Learn from the “best of the best,” including nationally renowned leaders in the field of hospital medicine.
- Connect and collaborate with a vibrant community of hospital medicine professionals.
It’s not too early to register! Sign up now and save at www.hospitalmedicine2017.org.
