User login
Help Bolster Your Skills at Leadership Academy 2016
A successful hospitalist program requires strong leadership from the unit to the C-suite. SHM’s Leadership Academy (www.shmleadershipacademy.org) prepares clinical and academic leaders with vital skills traditionally not taught in medical school or typical residency programs. This year’s meeting will be held from October 24 to 27 at Disney’s BoardWalk Inn in Lake Buena Vista, Fla. Courses offered include:
- Leadership Foundations: Evaluate your personal leadership strengths and weaknesses, understand key hospital drivers, and more.
- Advanced Leadership: Influential Management: Learn the skills needed to drive culture change through specific leadership behaviors and actions as well as financial storytelling.
(Prerequisite: Leadership Foundations or an advanced management degree upon course director approval.)
- Advanced Leadership: Mastering Teamwork: Learn to critically assess program growth opportunities, lead and motivate teams, and design effective communication strategies. (Prerequisite: Leadership Foundations or an advanced management degree upon course director approval.)
A successful hospitalist program requires strong leadership from the unit to the C-suite. SHM’s Leadership Academy (www.shmleadershipacademy.org) prepares clinical and academic leaders with vital skills traditionally not taught in medical school or typical residency programs. This year’s meeting will be held from October 24 to 27 at Disney’s BoardWalk Inn in Lake Buena Vista, Fla. Courses offered include:
- Leadership Foundations: Evaluate your personal leadership strengths and weaknesses, understand key hospital drivers, and more.
- Advanced Leadership: Influential Management: Learn the skills needed to drive culture change through specific leadership behaviors and actions as well as financial storytelling.
(Prerequisite: Leadership Foundations or an advanced management degree upon course director approval.)
- Advanced Leadership: Mastering Teamwork: Learn to critically assess program growth opportunities, lead and motivate teams, and design effective communication strategies. (Prerequisite: Leadership Foundations or an advanced management degree upon course director approval.)
A successful hospitalist program requires strong leadership from the unit to the C-suite. SHM’s Leadership Academy (www.shmleadershipacademy.org) prepares clinical and academic leaders with vital skills traditionally not taught in medical school or typical residency programs. This year’s meeting will be held from October 24 to 27 at Disney’s BoardWalk Inn in Lake Buena Vista, Fla. Courses offered include:
- Leadership Foundations: Evaluate your personal leadership strengths and weaknesses, understand key hospital drivers, and more.
- Advanced Leadership: Influential Management: Learn the skills needed to drive culture change through specific leadership behaviors and actions as well as financial storytelling.
(Prerequisite: Leadership Foundations or an advanced management degree upon course director approval.)
- Advanced Leadership: Mastering Teamwork: Learn to critically assess program growth opportunities, lead and motivate teams, and design effective communication strategies. (Prerequisite: Leadership Foundations or an advanced management degree upon course director approval.)
Negative RT-PCR result doesn’t exclude Zika infection
Photo by Jeremy L. Grisham
The Centers for Disease Control and Prevention (CDC) has issued an interim guidance on how to interpret results of the Zika virus antibody test.
Over the past few weeks, the FDA has authorized the use of new Zika tests, including RealStar® Zika Virus RT-PCR Kit U.S., Zika Virus RNA Qualitative Real-Time RT-PCR test, and the Trioplex Real-time RT-PCR Assay.
The CDC is now updating its guidance, since a negative real time reverse transcription-polymerase chain reaction (rRT-PCR) test does not necessarily rule out Zika infection.
In these cases, the CDC recommends immunoglobulin (Ig) M and neutralizing antibody testing, which can identify additional recent Zika virus infections.
The Zika antibody test, however, is difficult to interpret because of cross-reactivity with other flaviviruses. Zika is a mosquito-borne flavivirus that is closely related to dengue, West Nile, Japanese encephalitis, and yellow fever viruses.
The cross-reactivity can preclude identification of the specific infecting virus, particularly if a person was previously infected with or vaccinated against a related flavivirus. And appropriate clinical management is dependent upon proper identification of the virus.
If IgM test results are positive, equivocal, or inconclusive, the CDC recommends performing a plaque reduction neutralization test (PRNT) to confirm the diagnosis.
However, in people who have been previously infected with or vaccinated against a related flavivirus, even a 4-fold higher titer by PRNT may not be able to discriminate between anti-Zika virus antibodies and cross-reacting antibodies.
Therefore, the CDC now recommends an even more conservative approach to interpreting PRNT results to reduce the possibility of missing the diagnosis of either Zika or dengue virus infection.
The US Food and Drug Administration issued in February an Emergency Use Authorization for the CDC Zika IgM Antibody Capture Enzyme-Linked Immunosorbent Assay (Zika MAC-ELISA) for antibody testing.
It is used to detect Zika virus IgM antibodies in serum or cerebrospinal fluid from people with suspect Zika virus disease.
Presumptive positive results should be confirmed with PRNT against Zika, dengue, and other flaviviruses.
Equivocal and inconclusive results that are not resolved by re-testing should also have PRNT performed to rule out false-positive results.
For more information on interpretation of the Zika virus antibody test results, see the CDC’s Interim Guidance published as part of the Morbidity and Mortality Weekly Report for 31 May 2016. ![]()
Photo by Jeremy L. Grisham
The Centers for Disease Control and Prevention (CDC) has issued an interim guidance on how to interpret results of the Zika virus antibody test.
Over the past few weeks, the FDA has authorized the use of new Zika tests, including RealStar® Zika Virus RT-PCR Kit U.S., Zika Virus RNA Qualitative Real-Time RT-PCR test, and the Trioplex Real-time RT-PCR Assay.
The CDC is now updating its guidance, since a negative real time reverse transcription-polymerase chain reaction (rRT-PCR) test does not necessarily rule out Zika infection.
In these cases, the CDC recommends immunoglobulin (Ig) M and neutralizing antibody testing, which can identify additional recent Zika virus infections.
The Zika antibody test, however, is difficult to interpret because of cross-reactivity with other flaviviruses. Zika is a mosquito-borne flavivirus that is closely related to dengue, West Nile, Japanese encephalitis, and yellow fever viruses.
The cross-reactivity can preclude identification of the specific infecting virus, particularly if a person was previously infected with or vaccinated against a related flavivirus. And appropriate clinical management is dependent upon proper identification of the virus.
If IgM test results are positive, equivocal, or inconclusive, the CDC recommends performing a plaque reduction neutralization test (PRNT) to confirm the diagnosis.
However, in people who have been previously infected with or vaccinated against a related flavivirus, even a 4-fold higher titer by PRNT may not be able to discriminate between anti-Zika virus antibodies and cross-reacting antibodies.
Therefore, the CDC now recommends an even more conservative approach to interpreting PRNT results to reduce the possibility of missing the diagnosis of either Zika or dengue virus infection.
The US Food and Drug Administration issued in February an Emergency Use Authorization for the CDC Zika IgM Antibody Capture Enzyme-Linked Immunosorbent Assay (Zika MAC-ELISA) for antibody testing.
It is used to detect Zika virus IgM antibodies in serum or cerebrospinal fluid from people with suspect Zika virus disease.
Presumptive positive results should be confirmed with PRNT against Zika, dengue, and other flaviviruses.
Equivocal and inconclusive results that are not resolved by re-testing should also have PRNT performed to rule out false-positive results.
For more information on interpretation of the Zika virus antibody test results, see the CDC’s Interim Guidance published as part of the Morbidity and Mortality Weekly Report for 31 May 2016. ![]()
Photo by Jeremy L. Grisham
The Centers for Disease Control and Prevention (CDC) has issued an interim guidance on how to interpret results of the Zika virus antibody test.
Over the past few weeks, the FDA has authorized the use of new Zika tests, including RealStar® Zika Virus RT-PCR Kit U.S., Zika Virus RNA Qualitative Real-Time RT-PCR test, and the Trioplex Real-time RT-PCR Assay.
The CDC is now updating its guidance, since a negative real time reverse transcription-polymerase chain reaction (rRT-PCR) test does not necessarily rule out Zika infection.
In these cases, the CDC recommends immunoglobulin (Ig) M and neutralizing antibody testing, which can identify additional recent Zika virus infections.
The Zika antibody test, however, is difficult to interpret because of cross-reactivity with other flaviviruses. Zika is a mosquito-borne flavivirus that is closely related to dengue, West Nile, Japanese encephalitis, and yellow fever viruses.
The cross-reactivity can preclude identification of the specific infecting virus, particularly if a person was previously infected with or vaccinated against a related flavivirus. And appropriate clinical management is dependent upon proper identification of the virus.
If IgM test results are positive, equivocal, or inconclusive, the CDC recommends performing a plaque reduction neutralization test (PRNT) to confirm the diagnosis.
However, in people who have been previously infected with or vaccinated against a related flavivirus, even a 4-fold higher titer by PRNT may not be able to discriminate between anti-Zika virus antibodies and cross-reacting antibodies.
Therefore, the CDC now recommends an even more conservative approach to interpreting PRNT results to reduce the possibility of missing the diagnosis of either Zika or dengue virus infection.
The US Food and Drug Administration issued in February an Emergency Use Authorization for the CDC Zika IgM Antibody Capture Enzyme-Linked Immunosorbent Assay (Zika MAC-ELISA) for antibody testing.
It is used to detect Zika virus IgM antibodies in serum or cerebrospinal fluid from people with suspect Zika virus disease.
Presumptive positive results should be confirmed with PRNT against Zika, dengue, and other flaviviruses.
Equivocal and inconclusive results that are not resolved by re-testing should also have PRNT performed to rule out false-positive results.
For more information on interpretation of the Zika virus antibody test results, see the CDC’s Interim Guidance published as part of the Morbidity and Mortality Weekly Report for 31 May 2016. ![]()
Blood test may aid diagnosis of HELLP

Photo by Nina Matthews
A blood test developed to diagnose a rare genetic blood cell disorder, atypical hemolytic uremic syndrome (aHUS), may also aid in the diagnosis of HELLP syndrome, a life-threatening high blood pressure condition that affects 1% of all pregnant women.
The study, based on blood samples from a small number of women, suggests that aHUS has similar underlying biochemistry to HELLP, which affects hemolysis, elevates liver enzymes, and causes a low platelet count. Both conditions have over activation of the alternative pathway of complement.
At present, no diagnostic blood or biomarker test exists to diagnose HELLP, which is thought to be a severe form of preeclampsia. The condition is diagnosed only by its symptoms.
Senior study author Robert Brodsky, MD, of Johns Hopkins, and his team developed the modified Ham test to diagnose aHUS, a genetic disorder in which abnormal blood clots form in small blood vessels in the kidneys. They published that work last year in Blood.
The two conditions share a number of traits, such as hemolysis, elevated liver enzymes, a low platelet count, kidney dysfunction, high blood pressure, and seizures. This led the investigators to believe that the modified Ham test could also help identify women with HELLP syndrome.
"The clinical implications from an obstetric point of view are potentially huge," said lead study author Arthur Vaught, MD, also of Johns Hopkins. "If this works, we can reduce pre-term deliveries, stays in the neonatal intensive care unit, and other complications for mothers and their babies."
The team analyzed serum samples from 14 women with classic or atypical HELLP syndrome, 7 with severe preeclampsia, 11 women with normal pregnancies, and 8 healthy nonpregnant women. All pregnant women were at least 23 weeks’ gestation, the point at which HELLP symptoms start to arise.
The team evaluated patient sera using terminal product of complement activation (C5b-9). They observed that women with classic or atypical HELLP had increased complement activation compared to nonpregnant controls.
Women with classic HELLP had an average cell killing of 34.3% compared with 26% in women with atypical HELLP, 5% in women with normal pregnancies, and3.3% in women who were not pregnant.
The investigators then added eculizumab to HELLP sera to see whether the agent could inhibit complement activation. They found that mixing HELLP serum with eculizumab-containing serum significantly decreased cell killing compared with HELLP serum alone.
The kill rate in women with classic or atypical HELLP decreased from 34% to 5% with eculizumab.
Eculizumab (Soliris), manufactured by Alexion Pharmaceuticals, is a monoclonal antibody approved by the US Food and Drug Administration for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) and aHUS.
Further investigation is required to confirm these findings, but thus far, the investigators believe the modified Ham assay may assist in diagnosing the HELLP syndrome and corroborate its relationship to aHUS.
The current study by Vaught et al is published in Experimental Hematology. ![]()
Photo by Nina Matthews
A blood test developed to diagnose a rare genetic blood cell disorder, atypical hemolytic uremic syndrome (aHUS), may also aid in the diagnosis of HELLP syndrome, a life-threatening high blood pressure condition that affects 1% of all pregnant women.
The study, based on blood samples from a small number of women, suggests that aHUS has similar underlying biochemistry to HELLP, which affects hemolysis, elevates liver enzymes, and causes a low platelet count. Both conditions have over activation of the alternative pathway of complement.
At present, no diagnostic blood or biomarker test exists to diagnose HELLP, which is thought to be a severe form of preeclampsia. The condition is diagnosed only by its symptoms.
Senior study author Robert Brodsky, MD, of Johns Hopkins, and his team developed the modified Ham test to diagnose aHUS, a genetic disorder in which abnormal blood clots form in small blood vessels in the kidneys. They published that work last year in Blood.
The two conditions share a number of traits, such as hemolysis, elevated liver enzymes, a low platelet count, kidney dysfunction, high blood pressure, and seizures. This led the investigators to believe that the modified Ham test could also help identify women with HELLP syndrome.
"The clinical implications from an obstetric point of view are potentially huge," said lead study author Arthur Vaught, MD, also of Johns Hopkins. "If this works, we can reduce pre-term deliveries, stays in the neonatal intensive care unit, and other complications for mothers and their babies."
The team analyzed serum samples from 14 women with classic or atypical HELLP syndrome, 7 with severe preeclampsia, 11 women with normal pregnancies, and 8 healthy nonpregnant women. All pregnant women were at least 23 weeks’ gestation, the point at which HELLP symptoms start to arise.
The team evaluated patient sera using terminal product of complement activation (C5b-9). They observed that women with classic or atypical HELLP had increased complement activation compared to nonpregnant controls.
Women with classic HELLP had an average cell killing of 34.3% compared with 26% in women with atypical HELLP, 5% in women with normal pregnancies, and3.3% in women who were not pregnant.
The investigators then added eculizumab to HELLP sera to see whether the agent could inhibit complement activation. They found that mixing HELLP serum with eculizumab-containing serum significantly decreased cell killing compared with HELLP serum alone.
The kill rate in women with classic or atypical HELLP decreased from 34% to 5% with eculizumab.
Eculizumab (Soliris), manufactured by Alexion Pharmaceuticals, is a monoclonal antibody approved by the US Food and Drug Administration for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) and aHUS.
Further investigation is required to confirm these findings, but thus far, the investigators believe the modified Ham assay may assist in diagnosing the HELLP syndrome and corroborate its relationship to aHUS.
The current study by Vaught et al is published in Experimental Hematology. ![]()
Photo by Nina Matthews
A blood test developed to diagnose a rare genetic blood cell disorder, atypical hemolytic uremic syndrome (aHUS), may also aid in the diagnosis of HELLP syndrome, a life-threatening high blood pressure condition that affects 1% of all pregnant women.
The study, based on blood samples from a small number of women, suggests that aHUS has similar underlying biochemistry to HELLP, which affects hemolysis, elevates liver enzymes, and causes a low platelet count. Both conditions have over activation of the alternative pathway of complement.
At present, no diagnostic blood or biomarker test exists to diagnose HELLP, which is thought to be a severe form of preeclampsia. The condition is diagnosed only by its symptoms.
Senior study author Robert Brodsky, MD, of Johns Hopkins, and his team developed the modified Ham test to diagnose aHUS, a genetic disorder in which abnormal blood clots form in small blood vessels in the kidneys. They published that work last year in Blood.
The two conditions share a number of traits, such as hemolysis, elevated liver enzymes, a low platelet count, kidney dysfunction, high blood pressure, and seizures. This led the investigators to believe that the modified Ham test could also help identify women with HELLP syndrome.
"The clinical implications from an obstetric point of view are potentially huge," said lead study author Arthur Vaught, MD, also of Johns Hopkins. "If this works, we can reduce pre-term deliveries, stays in the neonatal intensive care unit, and other complications for mothers and their babies."
The team analyzed serum samples from 14 women with classic or atypical HELLP syndrome, 7 with severe preeclampsia, 11 women with normal pregnancies, and 8 healthy nonpregnant women. All pregnant women were at least 23 weeks’ gestation, the point at which HELLP symptoms start to arise.
The team evaluated patient sera using terminal product of complement activation (C5b-9). They observed that women with classic or atypical HELLP had increased complement activation compared to nonpregnant controls.
Women with classic HELLP had an average cell killing of 34.3% compared with 26% in women with atypical HELLP, 5% in women with normal pregnancies, and3.3% in women who were not pregnant.
The investigators then added eculizumab to HELLP sera to see whether the agent could inhibit complement activation. They found that mixing HELLP serum with eculizumab-containing serum significantly decreased cell killing compared with HELLP serum alone.
The kill rate in women with classic or atypical HELLP decreased from 34% to 5% with eculizumab.
Eculizumab (Soliris), manufactured by Alexion Pharmaceuticals, is a monoclonal antibody approved by the US Food and Drug Administration for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) and aHUS.
Further investigation is required to confirm these findings, but thus far, the investigators believe the modified Ham assay may assist in diagnosing the HELLP syndrome and corroborate its relationship to aHUS.
The current study by Vaught et al is published in Experimental Hematology. ![]()
June 2016 Digital Edition
Table of Contents
- Discrimination, Dignity, and Duty
- Survival After Long-Term Residence in an Intensive Care Unit
- Disease-Modifying Therapies in Multiple Sclerosis: Overview and Treatment Considerations
- The Relationship Between Male Patients’ Antihypertensive Medication Beliefs and Erectile Function
- A Physician With Thigh Pain
- Shared Medical Appointments for Glycemic Management in Rural Veterans

Table of Contents
- Discrimination, Dignity, and Duty
- Survival After Long-Term Residence in an Intensive Care Unit
- Disease-Modifying Therapies in Multiple Sclerosis: Overview and Treatment Considerations
- The Relationship Between Male Patients’ Antihypertensive Medication Beliefs and Erectile Function
- A Physician With Thigh Pain
- Shared Medical Appointments for Glycemic Management in Rural Veterans
Table of Contents
- Discrimination, Dignity, and Duty
- Survival After Long-Term Residence in an Intensive Care Unit
- Disease-Modifying Therapies in Multiple Sclerosis: Overview and Treatment Considerations
- The Relationship Between Male Patients’ Antihypertensive Medication Beliefs and Erectile Function
- A Physician With Thigh Pain
- Shared Medical Appointments for Glycemic Management in Rural Veterans

HCV regimen found safe, effective in patients with severe renal disease
A 12-week regimen achieved sustained viral response for 90% of patients with genotype 1 hepatitis C virus (HCV) infection and stage 4 or 5 chronic kidney disease (CKD), researchers reported in the April issue of Gastroenterology.
“The regimen is well tolerated, though ribavirin use may require a reduction or interruption to manage anemia,” said Dr. Paul Pockros at Scripps Clinic and Scripps Translational Science Institute in La Jolla, Calif., and his associates. The second phase of the study will evaluate the regimen in treatment-experienced CKD patients and those with compensated cirrhosis, they said.

The regimen contained ombitasvir, paritaprevir, ritonavir, and dasabuvir.
Between 8% and 44% of hemodialysis patients are HCV positive, and CKD is known to heighten the risk of HCV-associated cirrhosis, hepatocellular carcinoma, and liver-related death, the researchers noted. While sofosbuvir is cleared renally, ombitasvir, paritaprevir, ritonavir, and dasabuvir undergo hepatic metabolism and needed no dose adjustment in phase I studies of patients with mild, moderate, or severe renal impairment. To further investigate the safety and efficacy of these direct-acting antivirals in patients with severe kidney disease, the researchers performed a single-arm, open-label, multicenter study of 20 treatment-naive, noncirrhotic, HCV-infected adults with stage 4 CKD (estimated glomerular filtration rate, 15-30 mL/min per 1.73 m2) or stage 5 (eGFR, less than 15 mL/min per 1.73 m2 or requiring hemodialysis). Patients received once-daily ombitasvir (25 mg), paritaprevir (150 mg), and ritonavir (100 mg) plus dasabuvir (250 mg) for 12 weeks. The 13 patients with genotype 1a infections also received once-daily ribavirin (200 mg). Most patients were black men with stage 5 CKD, and 14 were on hemodialysis, the researchers said (Gastroenterology. 2016 Apr 16. doi: 10.1053/j.gastro.2016.02.078).
All 20 patients completed treatment, and 18 (90%) achieved sustained viral response (SVR) at posttreatment week 12 (SVR12; 95% confidence interval, 70%-97%). No patients developed hepatic decompensation, the researchers said. The most common adverse effects were anemia (45%), fatigue (35%), diarrhea (25%), and nausea (25%). Anemia developed only in patients receiving ribavirin and was more pronounced than in phase III studies of this regimen, the researchers said. Hemoglobin levels dropped an average of 1.38 plus or minus 1.54 g/dL among patients who received ribavirin, compared with 0.02 plus or minus 0.9 g/dL among patients who did not receive ribavirin. There was one case of grade 3 anemia related to incorrect dosing of ribavirin; the lowest measured hemoglobin level was 7.0 g/dL, which improved to more than 10 g/dL after stopping ribavirin and starting erythropoietin treatment. This patient also achieved SVR12. The other eight patients who developed anemia also stopped ribavirin, although three were able to resume it after their hemoglobin levels improved.
Of the two patients who did not achieve SVR12, one relapsed 4 weeks after treatment, and one died of cardiac arrest 14 days after treatment. The patient who died had a history of hypertension; his hemoglobin level was stable (9-11 g/dL) during the last 6 weeks of treatment, and was 10 g/dL at admission, suggesting that ribavirin-induced anemia did not cause the cardiac event, the investigators said.
“The results of this study are important for hepatologists, gastroenterologists, and infectious disease specialists who are accustomed to treating HCV-infected patients with DAA [direct-acting antiviral] therapy but who may not yet have seen sufficient data to initiate DAA therapy in patients with ESRD [end-stage renal disease],” the researchers concluded. “Nephrologists, who may not be accustomed to treating HCV, should also be aware that treatment options may now be available that can help prevent the end-stage sequelae of HCV. How treatment of HCV infection affects early or intermediate stages of CKD and how achievement of SVR impacts strategies for kidney transplantation in patients with ESRD require more study.”
AbbVie makes the regimen and sponsored the study. Dr. Pockros and six coinvestigators disclosed financial relationships with AbbVie and numerous other pharmaceutical companies. Seven coinvestigators reported being employed by AbbVie.
A 12-week regimen achieved sustained viral response for 90% of patients with genotype 1 hepatitis C virus (HCV) infection and stage 4 or 5 chronic kidney disease (CKD), researchers reported in the April issue of Gastroenterology.
“The regimen is well tolerated, though ribavirin use may require a reduction or interruption to manage anemia,” said Dr. Paul Pockros at Scripps Clinic and Scripps Translational Science Institute in La Jolla, Calif., and his associates. The second phase of the study will evaluate the regimen in treatment-experienced CKD patients and those with compensated cirrhosis, they said.
The regimen contained ombitasvir, paritaprevir, ritonavir, and dasabuvir.
Between 8% and 44% of hemodialysis patients are HCV positive, and CKD is known to heighten the risk of HCV-associated cirrhosis, hepatocellular carcinoma, and liver-related death, the researchers noted. While sofosbuvir is cleared renally, ombitasvir, paritaprevir, ritonavir, and dasabuvir undergo hepatic metabolism and needed no dose adjustment in phase I studies of patients with mild, moderate, or severe renal impairment. To further investigate the safety and efficacy of these direct-acting antivirals in patients with severe kidney disease, the researchers performed a single-arm, open-label, multicenter study of 20 treatment-naive, noncirrhotic, HCV-infected adults with stage 4 CKD (estimated glomerular filtration rate, 15-30 mL/min per 1.73 m2) or stage 5 (eGFR, less than 15 mL/min per 1.73 m2 or requiring hemodialysis). Patients received once-daily ombitasvir (25 mg), paritaprevir (150 mg), and ritonavir (100 mg) plus dasabuvir (250 mg) for 12 weeks. The 13 patients with genotype 1a infections also received once-daily ribavirin (200 mg). Most patients were black men with stage 5 CKD, and 14 were on hemodialysis, the researchers said (Gastroenterology. 2016 Apr 16. doi: 10.1053/j.gastro.2016.02.078).
All 20 patients completed treatment, and 18 (90%) achieved sustained viral response (SVR) at posttreatment week 12 (SVR12; 95% confidence interval, 70%-97%). No patients developed hepatic decompensation, the researchers said. The most common adverse effects were anemia (45%), fatigue (35%), diarrhea (25%), and nausea (25%). Anemia developed only in patients receiving ribavirin and was more pronounced than in phase III studies of this regimen, the researchers said. Hemoglobin levels dropped an average of 1.38 plus or minus 1.54 g/dL among patients who received ribavirin, compared with 0.02 plus or minus 0.9 g/dL among patients who did not receive ribavirin. There was one case of grade 3 anemia related to incorrect dosing of ribavirin; the lowest measured hemoglobin level was 7.0 g/dL, which improved to more than 10 g/dL after stopping ribavirin and starting erythropoietin treatment. This patient also achieved SVR12. The other eight patients who developed anemia also stopped ribavirin, although three were able to resume it after their hemoglobin levels improved.
Of the two patients who did not achieve SVR12, one relapsed 4 weeks after treatment, and one died of cardiac arrest 14 days after treatment. The patient who died had a history of hypertension; his hemoglobin level was stable (9-11 g/dL) during the last 6 weeks of treatment, and was 10 g/dL at admission, suggesting that ribavirin-induced anemia did not cause the cardiac event, the investigators said.
“The results of this study are important for hepatologists, gastroenterologists, and infectious disease specialists who are accustomed to treating HCV-infected patients with DAA [direct-acting antiviral] therapy but who may not yet have seen sufficient data to initiate DAA therapy in patients with ESRD [end-stage renal disease],” the researchers concluded. “Nephrologists, who may not be accustomed to treating HCV, should also be aware that treatment options may now be available that can help prevent the end-stage sequelae of HCV. How treatment of HCV infection affects early or intermediate stages of CKD and how achievement of SVR impacts strategies for kidney transplantation in patients with ESRD require more study.”
AbbVie makes the regimen and sponsored the study. Dr. Pockros and six coinvestigators disclosed financial relationships with AbbVie and numerous other pharmaceutical companies. Seven coinvestigators reported being employed by AbbVie.
A 12-week regimen achieved sustained viral response for 90% of patients with genotype 1 hepatitis C virus (HCV) infection and stage 4 or 5 chronic kidney disease (CKD), researchers reported in the April issue of Gastroenterology.
“The regimen is well tolerated, though ribavirin use may require a reduction or interruption to manage anemia,” said Dr. Paul Pockros at Scripps Clinic and Scripps Translational Science Institute in La Jolla, Calif., and his associates. The second phase of the study will evaluate the regimen in treatment-experienced CKD patients and those with compensated cirrhosis, they said.
The regimen contained ombitasvir, paritaprevir, ritonavir, and dasabuvir.
Between 8% and 44% of hemodialysis patients are HCV positive, and CKD is known to heighten the risk of HCV-associated cirrhosis, hepatocellular carcinoma, and liver-related death, the researchers noted. While sofosbuvir is cleared renally, ombitasvir, paritaprevir, ritonavir, and dasabuvir undergo hepatic metabolism and needed no dose adjustment in phase I studies of patients with mild, moderate, or severe renal impairment. To further investigate the safety and efficacy of these direct-acting antivirals in patients with severe kidney disease, the researchers performed a single-arm, open-label, multicenter study of 20 treatment-naive, noncirrhotic, HCV-infected adults with stage 4 CKD (estimated glomerular filtration rate, 15-30 mL/min per 1.73 m2) or stage 5 (eGFR, less than 15 mL/min per 1.73 m2 or requiring hemodialysis). Patients received once-daily ombitasvir (25 mg), paritaprevir (150 mg), and ritonavir (100 mg) plus dasabuvir (250 mg) for 12 weeks. The 13 patients with genotype 1a infections also received once-daily ribavirin (200 mg). Most patients were black men with stage 5 CKD, and 14 were on hemodialysis, the researchers said (Gastroenterology. 2016 Apr 16. doi: 10.1053/j.gastro.2016.02.078).
All 20 patients completed treatment, and 18 (90%) achieved sustained viral response (SVR) at posttreatment week 12 (SVR12; 95% confidence interval, 70%-97%). No patients developed hepatic decompensation, the researchers said. The most common adverse effects were anemia (45%), fatigue (35%), diarrhea (25%), and nausea (25%). Anemia developed only in patients receiving ribavirin and was more pronounced than in phase III studies of this regimen, the researchers said. Hemoglobin levels dropped an average of 1.38 plus or minus 1.54 g/dL among patients who received ribavirin, compared with 0.02 plus or minus 0.9 g/dL among patients who did not receive ribavirin. There was one case of grade 3 anemia related to incorrect dosing of ribavirin; the lowest measured hemoglobin level was 7.0 g/dL, which improved to more than 10 g/dL after stopping ribavirin and starting erythropoietin treatment. This patient also achieved SVR12. The other eight patients who developed anemia also stopped ribavirin, although three were able to resume it after their hemoglobin levels improved.
Of the two patients who did not achieve SVR12, one relapsed 4 weeks after treatment, and one died of cardiac arrest 14 days after treatment. The patient who died had a history of hypertension; his hemoglobin level was stable (9-11 g/dL) during the last 6 weeks of treatment, and was 10 g/dL at admission, suggesting that ribavirin-induced anemia did not cause the cardiac event, the investigators said.
“The results of this study are important for hepatologists, gastroenterologists, and infectious disease specialists who are accustomed to treating HCV-infected patients with DAA [direct-acting antiviral] therapy but who may not yet have seen sufficient data to initiate DAA therapy in patients with ESRD [end-stage renal disease],” the researchers concluded. “Nephrologists, who may not be accustomed to treating HCV, should also be aware that treatment options may now be available that can help prevent the end-stage sequelae of HCV. How treatment of HCV infection affects early or intermediate stages of CKD and how achievement of SVR impacts strategies for kidney transplantation in patients with ESRD require more study.”
AbbVie makes the regimen and sponsored the study. Dr. Pockros and six coinvestigators disclosed financial relationships with AbbVie and numerous other pharmaceutical companies. Seven coinvestigators reported being employed by AbbVie.
FROM GASTROENTEROLOGY
Key clinical point: Twelve weeks of ombitasvir, paritaprevir, ritonavir, and dasabuvir cured 90% of patients with hepatitis C virus infection and severe or end-stage renal disease.
Major finding: The rate of SVR12 was 90% (95% CI, 70%-97%).
Data source: A single-arm, open-label, multicenter trial of 20 noncirrhotic genotype 1 HCV-infected adults with stage 4 or 5 chronic kidney disease.
Disclosures: AbbVie makes the regimen and sponsored the study. Dr. Pockros and six coinvestigators disclosed financial relationships with AbbVie and numerous other pharmaceutical companies. Seven coinvestigators reported being employed by AbbVie.
VIDEO: Asymptomatic pancreatic cysts rarely became malignant
Only 1% of adults with asymptomatic neoplastic pancreatic cysts developed invasive pancreatic adenocarcinoma after more than 5 years of follow-up, according to a multicenter retrospective study reported in the June issue of Clinical Gastroenterology and Hepatology.
Furthermore, there were no malignant conversions among patients lacking American Gastroenterological Association high-risk features – that is, mural nodules, dilated pancreatic ducts, or cysts measuring more than 3 cm, said Dr. Wilson Kwong at the University of California San Diego Health Sciences in La Jolla. “There is a very low risk of malignant transformation of asymptomatic neoplastic pancreatic cysts after 5 years,” he and his associates wrote.
Up to 20% of cross-sectional imaging studies reveal incidental pancreatic cysts, the researchers noted. Cysts with neoplastic features are recommended for indefinite surveillance, even though there is little or no data on their natural history and malignant potential beyond 5- 10 years, they added. Therefore, they studied 310 patients who underwent endoscopic ultrasound of pancreatic cysts at an academic medical center, a Veterans’ Affairs hospital, and two community health care systems in California between 2002 and 2010. The most common age at enrollment was 66 years, 60% of patients were women, and the median follow-up period was 87 months (range, 60 to 189 months). A total of 90% of patients were followed for 5-10 years, while 10% were followed for more than 10 years (Clin Gastroenterol Hepatol. 2016 Feb 10. doi: 10.1016/j.cgh.2015.11.013).
Source: American Gastroenterological Association
In all, three patients developed invasive pancreatic malignancies after 6, 8, and 11 years of follow-up, for an overall conversion rate of 1%. Conversion rates by subgroup were 0% for patients with no high-risk AGA features, 1% (one case) for patients with one high-risk feature, and 15% (two cases) for patients with two high-risk features. “Because the risk of malignant transformation beyond 5 years is lower than the 1.4% mortality risk of pancreatic resection at high-volume centers, the argument can be made that discontinuing surveillance after 5 years is justified,” the researchers said. Specifically, surveillance could be discontinued after 5 years for neoplastic pancreatic cysts with up to one high-risk feature, particularly if patients have significant comorbidities that increase their risk of imminent death from other causes, they added. In contrast, healthy patients in their 60s and 70s might benefit from long-term surveillance given their longer life expectancy, they said. “Among patients with two high-risk features who remain surgically fit, discussion of surgery or surveillance beyond 5 years should be considered,” they emphasized.
A total of two patients developed high-grade dysplasia – a risk factor for invasive pancreatic cancer – but even so, the aggregate rate of cancer and high-grade dysplasia was 1.6%, only slightly higher than the fatality rate associated with pancreatic resection, the researchers noted. By excluding patients with recent acute pancreatitis (because of the likelihood of pseudocysts), they might have inadvertently excluded “a small number” of patients with pancreatic intraductal papillary mucinous neoplasms, they added.
The University of California San Diego Health Care System supported the study. The investigators had no disclosures.
Kwong et al. present important data demonstrating a low risk of malignant transformation for pancreas cysts followed for more than 5 years, which is similar to the risk of surgical resection. Mortality from nonpancreatic causes was found to be eightfold higher than mortality from pancreatic cancer. The goal of pancreas cyst surveillance is to prevent death from pancreatic cancer, currently accomplished by identifying high-risk cysts for surgical resection. When evaluating the utility of surveillance, patient and cyst characteristics can be considered.
Elderly patients with multiple comorbidities are unlikely to benefit from long-term surveillance as they may be poor surgical candidates and are unlikely to die from the malignant progression of a pancreas cyst. Healthy patients with a family history of pancreatic cancer and/or identifiable genetic risk factors, however, may benefit from long-term surveillance. Although demonstrated to be infrequent, cysts that have been stable for 5-10 years rarely may progress to cancer. The presence of more than one high-risk cyst feature increased the risk of progression from approximately 1% to 15%. The study of larger groups of cysts with morphologic high-risk features is required. The addition of molecular and genetic cyst and patient features has the potential to assist in risk stratification.
Clarifying which cysts and patients are likely to benefit from surveillance and resection is of increasing importance as high-resolution, cross-sectional imaging identifies greater numbers of pancreas cysts.
Dr. Harry R. Aslanian, AGAF, is director, Advanced Endoscopy Fellowship, and associate professor, Yale University, New Haven, Conn. He is a consultant for Boston Scientific and Olympus.
Kwong et al. present important data demonstrating a low risk of malignant transformation for pancreas cysts followed for more than 5 years, which is similar to the risk of surgical resection. Mortality from nonpancreatic causes was found to be eightfold higher than mortality from pancreatic cancer. The goal of pancreas cyst surveillance is to prevent death from pancreatic cancer, currently accomplished by identifying high-risk cysts for surgical resection. When evaluating the utility of surveillance, patient and cyst characteristics can be considered.
Elderly patients with multiple comorbidities are unlikely to benefit from long-term surveillance as they may be poor surgical candidates and are unlikely to die from the malignant progression of a pancreas cyst. Healthy patients with a family history of pancreatic cancer and/or identifiable genetic risk factors, however, may benefit from long-term surveillance. Although demonstrated to be infrequent, cysts that have been stable for 5-10 years rarely may progress to cancer. The presence of more than one high-risk cyst feature increased the risk of progression from approximately 1% to 15%. The study of larger groups of cysts with morphologic high-risk features is required. The addition of molecular and genetic cyst and patient features has the potential to assist in risk stratification.
Clarifying which cysts and patients are likely to benefit from surveillance and resection is of increasing importance as high-resolution, cross-sectional imaging identifies greater numbers of pancreas cysts.
Dr. Harry R. Aslanian, AGAF, is director, Advanced Endoscopy Fellowship, and associate professor, Yale University, New Haven, Conn. He is a consultant for Boston Scientific and Olympus.
Kwong et al. present important data demonstrating a low risk of malignant transformation for pancreas cysts followed for more than 5 years, which is similar to the risk of surgical resection. Mortality from nonpancreatic causes was found to be eightfold higher than mortality from pancreatic cancer. The goal of pancreas cyst surveillance is to prevent death from pancreatic cancer, currently accomplished by identifying high-risk cysts for surgical resection. When evaluating the utility of surveillance, patient and cyst characteristics can be considered.
Elderly patients with multiple comorbidities are unlikely to benefit from long-term surveillance as they may be poor surgical candidates and are unlikely to die from the malignant progression of a pancreas cyst. Healthy patients with a family history of pancreatic cancer and/or identifiable genetic risk factors, however, may benefit from long-term surveillance. Although demonstrated to be infrequent, cysts that have been stable for 5-10 years rarely may progress to cancer. The presence of more than one high-risk cyst feature increased the risk of progression from approximately 1% to 15%. The study of larger groups of cysts with morphologic high-risk features is required. The addition of molecular and genetic cyst and patient features has the potential to assist in risk stratification.
Clarifying which cysts and patients are likely to benefit from surveillance and resection is of increasing importance as high-resolution, cross-sectional imaging identifies greater numbers of pancreas cysts.
Dr. Harry R. Aslanian, AGAF, is director, Advanced Endoscopy Fellowship, and associate professor, Yale University, New Haven, Conn. He is a consultant for Boston Scientific and Olympus.
Only 1% of adults with asymptomatic neoplastic pancreatic cysts developed invasive pancreatic adenocarcinoma after more than 5 years of follow-up, according to a multicenter retrospective study reported in the June issue of Clinical Gastroenterology and Hepatology.
Furthermore, there were no malignant conversions among patients lacking American Gastroenterological Association high-risk features – that is, mural nodules, dilated pancreatic ducts, or cysts measuring more than 3 cm, said Dr. Wilson Kwong at the University of California San Diego Health Sciences in La Jolla. “There is a very low risk of malignant transformation of asymptomatic neoplastic pancreatic cysts after 5 years,” he and his associates wrote.
Up to 20% of cross-sectional imaging studies reveal incidental pancreatic cysts, the researchers noted. Cysts with neoplastic features are recommended for indefinite surveillance, even though there is little or no data on their natural history and malignant potential beyond 5- 10 years, they added. Therefore, they studied 310 patients who underwent endoscopic ultrasound of pancreatic cysts at an academic medical center, a Veterans’ Affairs hospital, and two community health care systems in California between 2002 and 2010. The most common age at enrollment was 66 years, 60% of patients were women, and the median follow-up period was 87 months (range, 60 to 189 months). A total of 90% of patients were followed for 5-10 years, while 10% were followed for more than 10 years (Clin Gastroenterol Hepatol. 2016 Feb 10. doi: 10.1016/j.cgh.2015.11.013).
Source: American Gastroenterological Association
In all, three patients developed invasive pancreatic malignancies after 6, 8, and 11 years of follow-up, for an overall conversion rate of 1%. Conversion rates by subgroup were 0% for patients with no high-risk AGA features, 1% (one case) for patients with one high-risk feature, and 15% (two cases) for patients with two high-risk features. “Because the risk of malignant transformation beyond 5 years is lower than the 1.4% mortality risk of pancreatic resection at high-volume centers, the argument can be made that discontinuing surveillance after 5 years is justified,” the researchers said. Specifically, surveillance could be discontinued after 5 years for neoplastic pancreatic cysts with up to one high-risk feature, particularly if patients have significant comorbidities that increase their risk of imminent death from other causes, they added. In contrast, healthy patients in their 60s and 70s might benefit from long-term surveillance given their longer life expectancy, they said. “Among patients with two high-risk features who remain surgically fit, discussion of surgery or surveillance beyond 5 years should be considered,” they emphasized.
A total of two patients developed high-grade dysplasia – a risk factor for invasive pancreatic cancer – but even so, the aggregate rate of cancer and high-grade dysplasia was 1.6%, only slightly higher than the fatality rate associated with pancreatic resection, the researchers noted. By excluding patients with recent acute pancreatitis (because of the likelihood of pseudocysts), they might have inadvertently excluded “a small number” of patients with pancreatic intraductal papillary mucinous neoplasms, they added.
The University of California San Diego Health Care System supported the study. The investigators had no disclosures.
Only 1% of adults with asymptomatic neoplastic pancreatic cysts developed invasive pancreatic adenocarcinoma after more than 5 years of follow-up, according to a multicenter retrospective study reported in the June issue of Clinical Gastroenterology and Hepatology.
Furthermore, there were no malignant conversions among patients lacking American Gastroenterological Association high-risk features – that is, mural nodules, dilated pancreatic ducts, or cysts measuring more than 3 cm, said Dr. Wilson Kwong at the University of California San Diego Health Sciences in La Jolla. “There is a very low risk of malignant transformation of asymptomatic neoplastic pancreatic cysts after 5 years,” he and his associates wrote.
Up to 20% of cross-sectional imaging studies reveal incidental pancreatic cysts, the researchers noted. Cysts with neoplastic features are recommended for indefinite surveillance, even though there is little or no data on their natural history and malignant potential beyond 5- 10 years, they added. Therefore, they studied 310 patients who underwent endoscopic ultrasound of pancreatic cysts at an academic medical center, a Veterans’ Affairs hospital, and two community health care systems in California between 2002 and 2010. The most common age at enrollment was 66 years, 60% of patients were women, and the median follow-up period was 87 months (range, 60 to 189 months). A total of 90% of patients were followed for 5-10 years, while 10% were followed for more than 10 years (Clin Gastroenterol Hepatol. 2016 Feb 10. doi: 10.1016/j.cgh.2015.11.013).
Source: American Gastroenterological Association
In all, three patients developed invasive pancreatic malignancies after 6, 8, and 11 years of follow-up, for an overall conversion rate of 1%. Conversion rates by subgroup were 0% for patients with no high-risk AGA features, 1% (one case) for patients with one high-risk feature, and 15% (two cases) for patients with two high-risk features. “Because the risk of malignant transformation beyond 5 years is lower than the 1.4% mortality risk of pancreatic resection at high-volume centers, the argument can be made that discontinuing surveillance after 5 years is justified,” the researchers said. Specifically, surveillance could be discontinued after 5 years for neoplastic pancreatic cysts with up to one high-risk feature, particularly if patients have significant comorbidities that increase their risk of imminent death from other causes, they added. In contrast, healthy patients in their 60s and 70s might benefit from long-term surveillance given their longer life expectancy, they said. “Among patients with two high-risk features who remain surgically fit, discussion of surgery or surveillance beyond 5 years should be considered,” they emphasized.
A total of two patients developed high-grade dysplasia – a risk factor for invasive pancreatic cancer – but even so, the aggregate rate of cancer and high-grade dysplasia was 1.6%, only slightly higher than the fatality rate associated with pancreatic resection, the researchers noted. By excluding patients with recent acute pancreatitis (because of the likelihood of pseudocysts), they might have inadvertently excluded “a small number” of patients with pancreatic intraductal papillary mucinous neoplasms, they added.
The University of California San Diego Health Care System supported the study. The investigators had no disclosures.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: Asymptomatic neoplastic pancreatic cysts rarely become malignant, especially in the absence of multiple American Gastroenterological Association high-risk features.
Major finding: Only 1% of patients developed invasive pancreatic adenocarcinoma after more than 5 years of surveillance.
Data source: A multicenter retrospective study of 310 patients who underwent endoscopic ultrasound evaluations of pancreatic cysts.
Disclosures: The University of California San Diego Health Care System supported the study. The investigators had no disclosures.
VIDEO: High sensitivity–CRP, IL-6 predicted inflammatory bowel disease
Women with high circulating levels of interleukin-6 and high-sensitivity C-reactive protein were at significantly greater risk of inflammatory bowel disease (IBD) compared with those testing in the lowest quintiles, according to a prospective nested case-control study.
The findings point to a preclinical state in IBD, in which patients are not yet symptomatic but have positive serologic markers, as occurs in rheumatoid arthritis and systemic lupus erythematosus, said Dr. Paul Lochhead at Massachusetts General Hospital in Boston, and his associates. “To our knowledge, no previous study has examined prediagnostic inflammatory markers in relation to IBD risk,” the investigators added. “Characterizing preclinical inflammation in IBD could give insights into the natural history of [Crohn’s disease] and [ulcerative colitis], and might help identify potential windows for early therapeutic or preventive interventions in high-risk individuals.”
SOURCE: American Gastroenterological Society
The study included 83 patients with Crohn’s disease, 90 patients with ulcerative colitis, and 344 matched controls. Patients were from two national prospective cohort studies – the Nurses’ Health Study, which includes female nurses aged 35-55 years at enrollment, and the Nurses’ Health Study II, which includes female nurses aged 24-42 years at enrollment. Both studies are ongoing, with follow-up rates exceeding 90%. To assemble the cohort, the researchers extracted questionnaire data and then obtained medical records for blinded review. They confirmed diagnoses of Crohn’s disease and ulcerative colitis using standard case definitions, they said (Clin Gastroenterol Hepatol. 2016 Feb 13. doi: 10.1016/j.cgh.2016.01.016).
Participants testing in the highest quintiles for circulating hs-CRP and IL-6 were at greater risk of Crohn’s disease and ulcerative colitis than were those in the lowest quintiles, even after accounting for age, smoking status, body mass index, oral contraceptive use, and cumulative physical activity. For IL-6, odds ratios were 4.7 for Crohn’s disease (95% confidence interval; 1.9-11.5), and 3.4 for ulcerative colitis (95% CI; 1.4-8.2). For hs-CRP, odds ratios were 2.8 for Crohn’s disease (95% CI; 1.15-6.9) and 1.8 for ulcerative colitis (95% CI; 0.8-4.0). The longest interval between testing and diagnosis of IBD was 20 years, Crohn’s disease patients were diagnosed within 10 years, and patients testing in the upper quintile for the inflammatory markers were diagnosed an average of 10.6 years later, the researchers said.
Study participants tended to be in their early 50s when first tested, which exceeds the typical age of Crohn’s disease and ulcerative colitis onset and might limit the generalizability of the findings, the investigators said. They tried to eliminate confounding from undiagnosed baseline IBD by excluding participants diagnosed within 2 years of blood collection, they added. “The differences in overall median inflammatory marker levels between cases and control subjects in our study were small; however, differences of similar magnitude have been reported between groups with disparate outcomes in studies of cardiovascular disease,” they noted. “Moreover, when comparing extreme quintiles of median inflammatory marker levels, where risk of [Crohn’s disease] or [ulcerative colitis] was most evident, the differences were more substantial.”
The study was funded by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Crohn’s and Colitis Foundation of America, and the American Gastroenterological Association. Dr. Lochhead had no disclosures. Two coinvestigators disclosed relationships with Exact Sciences, AbbVie, Cubist Pharmaceuticals, Bayer Healthcare, Pfizer, and Pozen.
Women with high circulating levels of interleukin-6 and high-sensitivity C-reactive protein were at significantly greater risk of inflammatory bowel disease (IBD) compared with those testing in the lowest quintiles, according to a prospective nested case-control study.
The findings point to a preclinical state in IBD, in which patients are not yet symptomatic but have positive serologic markers, as occurs in rheumatoid arthritis and systemic lupus erythematosus, said Dr. Paul Lochhead at Massachusetts General Hospital in Boston, and his associates. “To our knowledge, no previous study has examined prediagnostic inflammatory markers in relation to IBD risk,” the investigators added. “Characterizing preclinical inflammation in IBD could give insights into the natural history of [Crohn’s disease] and [ulcerative colitis], and might help identify potential windows for early therapeutic or preventive interventions in high-risk individuals.”
SOURCE: American Gastroenterological Society
The study included 83 patients with Crohn’s disease, 90 patients with ulcerative colitis, and 344 matched controls. Patients were from two national prospective cohort studies – the Nurses’ Health Study, which includes female nurses aged 35-55 years at enrollment, and the Nurses’ Health Study II, which includes female nurses aged 24-42 years at enrollment. Both studies are ongoing, with follow-up rates exceeding 90%. To assemble the cohort, the researchers extracted questionnaire data and then obtained medical records for blinded review. They confirmed diagnoses of Crohn’s disease and ulcerative colitis using standard case definitions, they said (Clin Gastroenterol Hepatol. 2016 Feb 13. doi: 10.1016/j.cgh.2016.01.016).
Participants testing in the highest quintiles for circulating hs-CRP and IL-6 were at greater risk of Crohn’s disease and ulcerative colitis than were those in the lowest quintiles, even after accounting for age, smoking status, body mass index, oral contraceptive use, and cumulative physical activity. For IL-6, odds ratios were 4.7 for Crohn’s disease (95% confidence interval; 1.9-11.5), and 3.4 for ulcerative colitis (95% CI; 1.4-8.2). For hs-CRP, odds ratios were 2.8 for Crohn’s disease (95% CI; 1.15-6.9) and 1.8 for ulcerative colitis (95% CI; 0.8-4.0). The longest interval between testing and diagnosis of IBD was 20 years, Crohn’s disease patients were diagnosed within 10 years, and patients testing in the upper quintile for the inflammatory markers were diagnosed an average of 10.6 years later, the researchers said.
Study participants tended to be in their early 50s when first tested, which exceeds the typical age of Crohn’s disease and ulcerative colitis onset and might limit the generalizability of the findings, the investigators said. They tried to eliminate confounding from undiagnosed baseline IBD by excluding participants diagnosed within 2 years of blood collection, they added. “The differences in overall median inflammatory marker levels between cases and control subjects in our study were small; however, differences of similar magnitude have been reported between groups with disparate outcomes in studies of cardiovascular disease,” they noted. “Moreover, when comparing extreme quintiles of median inflammatory marker levels, where risk of [Crohn’s disease] or [ulcerative colitis] was most evident, the differences were more substantial.”
The study was funded by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Crohn’s and Colitis Foundation of America, and the American Gastroenterological Association. Dr. Lochhead had no disclosures. Two coinvestigators disclosed relationships with Exact Sciences, AbbVie, Cubist Pharmaceuticals, Bayer Healthcare, Pfizer, and Pozen.
Women with high circulating levels of interleukin-6 and high-sensitivity C-reactive protein were at significantly greater risk of inflammatory bowel disease (IBD) compared with those testing in the lowest quintiles, according to a prospective nested case-control study.
The findings point to a preclinical state in IBD, in which patients are not yet symptomatic but have positive serologic markers, as occurs in rheumatoid arthritis and systemic lupus erythematosus, said Dr. Paul Lochhead at Massachusetts General Hospital in Boston, and his associates. “To our knowledge, no previous study has examined prediagnostic inflammatory markers in relation to IBD risk,” the investigators added. “Characterizing preclinical inflammation in IBD could give insights into the natural history of [Crohn’s disease] and [ulcerative colitis], and might help identify potential windows for early therapeutic or preventive interventions in high-risk individuals.”
SOURCE: American Gastroenterological Society
The study included 83 patients with Crohn’s disease, 90 patients with ulcerative colitis, and 344 matched controls. Patients were from two national prospective cohort studies – the Nurses’ Health Study, which includes female nurses aged 35-55 years at enrollment, and the Nurses’ Health Study II, which includes female nurses aged 24-42 years at enrollment. Both studies are ongoing, with follow-up rates exceeding 90%. To assemble the cohort, the researchers extracted questionnaire data and then obtained medical records for blinded review. They confirmed diagnoses of Crohn’s disease and ulcerative colitis using standard case definitions, they said (Clin Gastroenterol Hepatol. 2016 Feb 13. doi: 10.1016/j.cgh.2016.01.016).
Participants testing in the highest quintiles for circulating hs-CRP and IL-6 were at greater risk of Crohn’s disease and ulcerative colitis than were those in the lowest quintiles, even after accounting for age, smoking status, body mass index, oral contraceptive use, and cumulative physical activity. For IL-6, odds ratios were 4.7 for Crohn’s disease (95% confidence interval; 1.9-11.5), and 3.4 for ulcerative colitis (95% CI; 1.4-8.2). For hs-CRP, odds ratios were 2.8 for Crohn’s disease (95% CI; 1.15-6.9) and 1.8 for ulcerative colitis (95% CI; 0.8-4.0). The longest interval between testing and diagnosis of IBD was 20 years, Crohn’s disease patients were diagnosed within 10 years, and patients testing in the upper quintile for the inflammatory markers were diagnosed an average of 10.6 years later, the researchers said.
Study participants tended to be in their early 50s when first tested, which exceeds the typical age of Crohn’s disease and ulcerative colitis onset and might limit the generalizability of the findings, the investigators said. They tried to eliminate confounding from undiagnosed baseline IBD by excluding participants diagnosed within 2 years of blood collection, they added. “The differences in overall median inflammatory marker levels between cases and control subjects in our study were small; however, differences of similar magnitude have been reported between groups with disparate outcomes in studies of cardiovascular disease,” they noted. “Moreover, when comparing extreme quintiles of median inflammatory marker levels, where risk of [Crohn’s disease] or [ulcerative colitis] was most evident, the differences were more substantial.”
The study was funded by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Crohn’s and Colitis Foundation of America, and the American Gastroenterological Association. Dr. Lochhead had no disclosures. Two coinvestigators disclosed relationships with Exact Sciences, AbbVie, Cubist Pharmaceuticals, Bayer Healthcare, Pfizer, and Pozen.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: Patients might have subclinical inflammation for several years before developing inflammatory bowel disease.
Major finding: Participants testing in the highest quintiles for circulating high-sensitivity C-reactive protein and interleukin-6 were at greater risk of Crohn’s disease and ulcerative colitis, compared with individuals testing in the lowest quintiles for each marker, with estimated odds ratios of 1.8, 2.8, 3.4, and 4.7.
Data source: A prospective nested case-control study of female nurses, including 83 with Crohn’s disease, 90 with ulcerative colitis, and 344 matched controls.
Disclosures: The study was funded by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Crohn’s and Colitis Foundation of America, and the American Gastroenterological Association. Dr. Lochhead had no disclosures. Two coinvestigators disclosed relationships with Exact Sciences, AbbVie, Cubist Pharmaceuticals, Bayer Healthcare, Pfizer, and Pozen.

Combination OCs tied to increased risk of surgery for Crohn’s
Women with Crohn’s disease who were prescribed combination oral contraceptive pills for more than 3 years were 68% more likely to need gastrointestinal surgery than patients who did not use oral contraceptives, according to a national prospective cohort study reported in the June issue of Gastroenterology.
“Our data suggest the importance of carefully evaluating contraceptive options among women with established Crohn’s disease. Future studies should focus on mechanisms by which oral contraceptive use alters risk and progression,” said Dr. Hamed Khalili of Harvard Medical School in Boston and his associates at Harvard and Karolinska Institutet, Solna, Sweden.
Several studies have linked OC exposure to Crohn’s disease itself. But past studies of OCs and Crohn’s disease progression were small, retrospective, or did not adequately ascertain OC exposure, Dr. Khalili and his associates said. To help fill this gap, they identified 4,036 women with Crohn’s disease aged 16-51 years through the Swedish National Patient Register, and ascertained OC exposure by analyzing Sweden’s national prescription database (Gastroenterology. 2016 Feb 23. doi: 10.1053/j.gastro.2016.02.041).
During a median follow-up period of 58 months, 482 patients (12%) underwent surgery related to Crohn’s disease, the researchers said. Use of OCs was associated with surgery, but the link only reached statistical significance among women prescribed combination (estrogen-containing) regimens for more than 3 years (adjusted hazard ratio, 1.68; 95% confidence interval, 1.06-2.67) or for more than 900 doses (aHR, 1.60; 95% CI, 1.1-2.34). For each additional year that combination OCs were prescribed, surgery risk rose by nearly 30% (aHR, 1.29; 95% CI, 1.05-1.57). Thus, one extra surgery was needed for every 83 patients who received combination OCs for at least 1 year, said the investigators. Progestin-only prescriptions did not increase the likelihood of needing surgery, and there was no link between current or prior OC exposure and the chances of being prescribed steroids, they noted.
Only one other study has linked OC exposure with Crohn’s disease progression, and it included only 158 patients followed for just a year, Dr. Khalili and his associates said. Exactly how estrogen exposure might trigger Crohn’s disease progression is unclear, but OCs have been linked to changes in intestinal barrier function, increased humoral immunity, and modulation of testosterone levels, which in turn affects cytokine function, they added. “Regardless of the potential mechanism, the effect of OCs on Crohn’s disease progression appears to be related to consistent and long-term use of these medications. Similar patterns of associations have also been reported with other chronic illnesses, such as breast cancer and cardiovascular diseases,” said the researchers. Current OC use itself might not have predicted surgery in the study because about one in four women in Sweden stop taking OCs or switch to a nonhormonal form within 6 months of being prescribed them, they added.
The work was funded by the Crohn’s and Colitis Foundation of America, the National Institute of Diabetes and Digestive and Kidney Diseases, the American Gastroenterological Association, and the American College of Gastroenterology. Dr. Khalili reported receiving consulting fees from Abbvie. One coinvestigator reported consulting relationships with Bayer Healthcare, Pfizer, and Pozen. The other investigators had no disclosures.
Women with Crohn’s disease who were prescribed combination oral contraceptive pills for more than 3 years were 68% more likely to need gastrointestinal surgery than patients who did not use oral contraceptives, according to a national prospective cohort study reported in the June issue of Gastroenterology.
“Our data suggest the importance of carefully evaluating contraceptive options among women with established Crohn’s disease. Future studies should focus on mechanisms by which oral contraceptive use alters risk and progression,” said Dr. Hamed Khalili of Harvard Medical School in Boston and his associates at Harvard and Karolinska Institutet, Solna, Sweden.
Several studies have linked OC exposure to Crohn’s disease itself. But past studies of OCs and Crohn’s disease progression were small, retrospective, or did not adequately ascertain OC exposure, Dr. Khalili and his associates said. To help fill this gap, they identified 4,036 women with Crohn’s disease aged 16-51 years through the Swedish National Patient Register, and ascertained OC exposure by analyzing Sweden’s national prescription database (Gastroenterology. 2016 Feb 23. doi: 10.1053/j.gastro.2016.02.041).
During a median follow-up period of 58 months, 482 patients (12%) underwent surgery related to Crohn’s disease, the researchers said. Use of OCs was associated with surgery, but the link only reached statistical significance among women prescribed combination (estrogen-containing) regimens for more than 3 years (adjusted hazard ratio, 1.68; 95% confidence interval, 1.06-2.67) or for more than 900 doses (aHR, 1.60; 95% CI, 1.1-2.34). For each additional year that combination OCs were prescribed, surgery risk rose by nearly 30% (aHR, 1.29; 95% CI, 1.05-1.57). Thus, one extra surgery was needed for every 83 patients who received combination OCs for at least 1 year, said the investigators. Progestin-only prescriptions did not increase the likelihood of needing surgery, and there was no link between current or prior OC exposure and the chances of being prescribed steroids, they noted.
Only one other study has linked OC exposure with Crohn’s disease progression, and it included only 158 patients followed for just a year, Dr. Khalili and his associates said. Exactly how estrogen exposure might trigger Crohn’s disease progression is unclear, but OCs have been linked to changes in intestinal barrier function, increased humoral immunity, and modulation of testosterone levels, which in turn affects cytokine function, they added. “Regardless of the potential mechanism, the effect of OCs on Crohn’s disease progression appears to be related to consistent and long-term use of these medications. Similar patterns of associations have also been reported with other chronic illnesses, such as breast cancer and cardiovascular diseases,” said the researchers. Current OC use itself might not have predicted surgery in the study because about one in four women in Sweden stop taking OCs or switch to a nonhormonal form within 6 months of being prescribed them, they added.
The work was funded by the Crohn’s and Colitis Foundation of America, the National Institute of Diabetes and Digestive and Kidney Diseases, the American Gastroenterological Association, and the American College of Gastroenterology. Dr. Khalili reported receiving consulting fees from Abbvie. One coinvestigator reported consulting relationships with Bayer Healthcare, Pfizer, and Pozen. The other investigators had no disclosures.
Women with Crohn’s disease who were prescribed combination oral contraceptive pills for more than 3 years were 68% more likely to need gastrointestinal surgery than patients who did not use oral contraceptives, according to a national prospective cohort study reported in the June issue of Gastroenterology.
“Our data suggest the importance of carefully evaluating contraceptive options among women with established Crohn’s disease. Future studies should focus on mechanisms by which oral contraceptive use alters risk and progression,” said Dr. Hamed Khalili of Harvard Medical School in Boston and his associates at Harvard and Karolinska Institutet, Solna, Sweden.
Several studies have linked OC exposure to Crohn’s disease itself. But past studies of OCs and Crohn’s disease progression were small, retrospective, or did not adequately ascertain OC exposure, Dr. Khalili and his associates said. To help fill this gap, they identified 4,036 women with Crohn’s disease aged 16-51 years through the Swedish National Patient Register, and ascertained OC exposure by analyzing Sweden’s national prescription database (Gastroenterology. 2016 Feb 23. doi: 10.1053/j.gastro.2016.02.041).
During a median follow-up period of 58 months, 482 patients (12%) underwent surgery related to Crohn’s disease, the researchers said. Use of OCs was associated with surgery, but the link only reached statistical significance among women prescribed combination (estrogen-containing) regimens for more than 3 years (adjusted hazard ratio, 1.68; 95% confidence interval, 1.06-2.67) or for more than 900 doses (aHR, 1.60; 95% CI, 1.1-2.34). For each additional year that combination OCs were prescribed, surgery risk rose by nearly 30% (aHR, 1.29; 95% CI, 1.05-1.57). Thus, one extra surgery was needed for every 83 patients who received combination OCs for at least 1 year, said the investigators. Progestin-only prescriptions did not increase the likelihood of needing surgery, and there was no link between current or prior OC exposure and the chances of being prescribed steroids, they noted.
Only one other study has linked OC exposure with Crohn’s disease progression, and it included only 158 patients followed for just a year, Dr. Khalili and his associates said. Exactly how estrogen exposure might trigger Crohn’s disease progression is unclear, but OCs have been linked to changes in intestinal barrier function, increased humoral immunity, and modulation of testosterone levels, which in turn affects cytokine function, they added. “Regardless of the potential mechanism, the effect of OCs on Crohn’s disease progression appears to be related to consistent and long-term use of these medications. Similar patterns of associations have also been reported with other chronic illnesses, such as breast cancer and cardiovascular diseases,” said the researchers. Current OC use itself might not have predicted surgery in the study because about one in four women in Sweden stop taking OCs or switch to a nonhormonal form within 6 months of being prescribed them, they added.
The work was funded by the Crohn’s and Colitis Foundation of America, the National Institute of Diabetes and Digestive and Kidney Diseases, the American Gastroenterological Association, and the American College of Gastroenterology. Dr. Khalili reported receiving consulting fees from Abbvie. One coinvestigator reported consulting relationships with Bayer Healthcare, Pfizer, and Pozen. The other investigators had no disclosures.
FROM GASTROENTEROLOGY
Key clinical point: Long-term use of combination oral contraceptives significantly increased the risk of surgery among women with Crohn’s disease.
Major finding: Women who used combination OCs for more than 3 years were 68% more likely to need surgery than were nonusers.
Data source: A prospective national registry study of 4,036 women with Crohn’s disease.
Disclosures: The study was funded by the Crohn’s and Colitis Foundation of America, the National Institute of Diabetes and Digestive and Kidney Diseases, the American Gastroenterological Association, and the American College of Gastroenterology. Dr. Khalili reported receiving consulting fees from Abbvie. One coinvestigator reported consulting relationships with Bayer Healthcare, Pfizer, and Pozen. The other investigators had no disclosures.

In Middle of Trip, Woman Falls
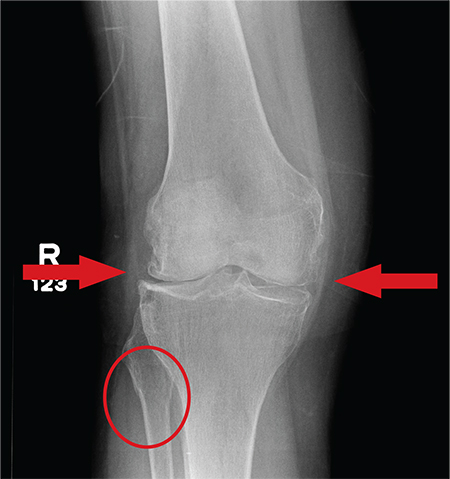
Answer
The radiograph has several findings, one of which is a nondisplaced proximal fibula fracture. In addition, there is a moderate suprapatellar joint effusion. The patient also has fairly advanced tricompartment degenerative arthrosis. (To review, the tricompartment comprises all three anatomic areas of the knee: the patellofemoral, lateral tibiofemoral, and medial tibiofemoral joints.)
Answer
The radiograph has several findings, one of which is a nondisplaced proximal fibula fracture. In addition, there is a moderate suprapatellar joint effusion. The patient also has fairly advanced tricompartment degenerative arthrosis. (To review, the tricompartment comprises all three anatomic areas of the knee: the patellofemoral, lateral tibiofemoral, and medial tibiofemoral joints.)
Answer
The radiograph has several findings, one of which is a nondisplaced proximal fibula fracture. In addition, there is a moderate suprapatellar joint effusion. The patient also has fairly advanced tricompartment degenerative arthrosis. (To review, the tricompartment comprises all three anatomic areas of the knee: the patellofemoral, lateral tibiofemoral, and medial tibiofemoral joints.)
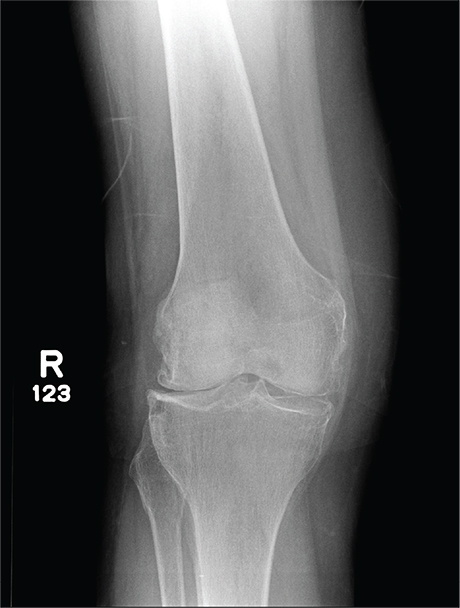
A 70-year-old woman presents to your emergency department for evaluation of right knee pain secondary to a fall. She and her husband, in the process of driving from Florida to their home in California, stopped for the night in your town. The patient states that shortly after getting up this morning, she tripped, lost her balance, and fell. All her weight landed on her right knee; she says it is now “extremely painful” to bear weight on that leg. She also twisted her right ankle, causing additional discomfort. Her medical history is significant for hypertension, which is controlled by medication. On physical exam, you note an elderly female who is uncomfortable but in no obvious distress. Inspection of her right knee shows no obvious deformity but a moderate amount of swelling. The patient has limited range of motion secondary to the swelling. She also has moderate tenderness circumferentially around the knee. There is additional swelling and mild bruising on both the medial and lateral aspects of the right ankle. You obtain a radiograph of the right knee. What is your impression?
Moles: Their Role in Skin Cancer Diagnosis
ANSWER
The correct answer is none of the above (choice “d”). These lesions are all intradermal nevi, which have little, if any, risk for malignant transformation. Deeper nevi are considered quite safe, unless significant change has occurred. Despite the unlikelihood, however, it is risky to declare a 0% chance of skin cancer.
DISCUSSION
Slow growth and increased prominence are not the kinds of changes to look for in skin lesions. Rather, look for marked asymmetry (eg, the growth of a new, darker, macular component) or other change in color or consistency.
Hairs on these lesions are quite normal and are actually reassuring in confirming their benign nature. Skin cancers seldom support hair growth.
Most melanomas don’t come from moles. Instead, they are “de novo” lesions, literally coming from nothing, out of clear skin. It is true that the more moles someone has, the greater his or her risk for skin cancer, though not necessarily in one of the moles. When melanomas do develop from nevi (a collection of a certain type of melanocyte), this usually occurs in superficial types, such as compound or junctional nevi. From an objective standpoint, in this patient’s case, family history means nothing.
What does matter is to pay as much attention to the owner as to the lesion. The more fair-skinned and sun-damaged (freckles, blue eyes, red hair) the patient is, the more worrisome a lesion can be.
This patient had none of those traits, and she will likely have one of her lesions surgically excised to ensure she’s satisfied with the resulting scar. Of course, the tissue sample will be sent for pathologic examination, as any specimen should be.
ANSWER
The correct answer is none of the above (choice “d”). These lesions are all intradermal nevi, which have little, if any, risk for malignant transformation. Deeper nevi are considered quite safe, unless significant change has occurred. Despite the unlikelihood, however, it is risky to declare a 0% chance of skin cancer.
DISCUSSION
Slow growth and increased prominence are not the kinds of changes to look for in skin lesions. Rather, look for marked asymmetry (eg, the growth of a new, darker, macular component) or other change in color or consistency.
Hairs on these lesions are quite normal and are actually reassuring in confirming their benign nature. Skin cancers seldom support hair growth.
Most melanomas don’t come from moles. Instead, they are “de novo” lesions, literally coming from nothing, out of clear skin. It is true that the more moles someone has, the greater his or her risk for skin cancer, though not necessarily in one of the moles. When melanomas do develop from nevi (a collection of a certain type of melanocyte), this usually occurs in superficial types, such as compound or junctional nevi. From an objective standpoint, in this patient’s case, family history means nothing.
What does matter is to pay as much attention to the owner as to the lesion. The more fair-skinned and sun-damaged (freckles, blue eyes, red hair) the patient is, the more worrisome a lesion can be.
This patient had none of those traits, and she will likely have one of her lesions surgically excised to ensure she’s satisfied with the resulting scar. Of course, the tissue sample will be sent for pathologic examination, as any specimen should be.
ANSWER
The correct answer is none of the above (choice “d”). These lesions are all intradermal nevi, which have little, if any, risk for malignant transformation. Deeper nevi are considered quite safe, unless significant change has occurred. Despite the unlikelihood, however, it is risky to declare a 0% chance of skin cancer.
DISCUSSION
Slow growth and increased prominence are not the kinds of changes to look for in skin lesions. Rather, look for marked asymmetry (eg, the growth of a new, darker, macular component) or other change in color or consistency.
Hairs on these lesions are quite normal and are actually reassuring in confirming their benign nature. Skin cancers seldom support hair growth.
Most melanomas don’t come from moles. Instead, they are “de novo” lesions, literally coming from nothing, out of clear skin. It is true that the more moles someone has, the greater his or her risk for skin cancer, though not necessarily in one of the moles. When melanomas do develop from nevi (a collection of a certain type of melanocyte), this usually occurs in superficial types, such as compound or junctional nevi. From an objective standpoint, in this patient’s case, family history means nothing.
What does matter is to pay as much attention to the owner as to the lesion. The more fair-skinned and sun-damaged (freckles, blue eyes, red hair) the patient is, the more worrisome a lesion can be.
This patient had none of those traits, and she will likely have one of her lesions surgically excised to ensure she’s satisfied with the resulting scar. Of course, the tissue sample will be sent for pathologic examination, as any specimen should be.
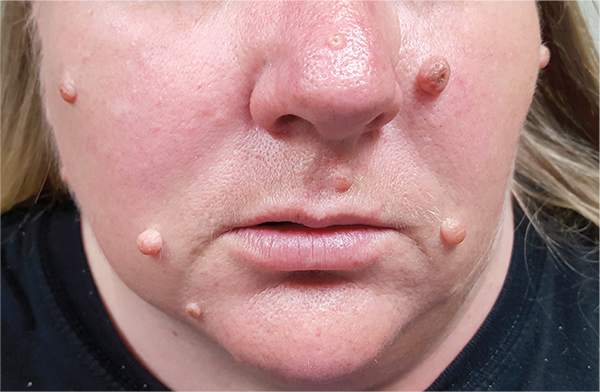
A 39-year-old woman self-refers for evaluation of moles she’s had on her face “all her life.” They have become more prominent with age, and many now have hairs growing in them. They are often traumatized by contact with fingernails or clothing. The patient worries that they might “turn into cancer” the way her grandfather’s moles did. The patient looks her stated age, is moderately overweight, and has more than her share of moles (some of which exceed 6 mm in diameter.) For the most part, they are skin-colored, and several are hair-bearing. Further questioning reveals that her moles manifested during puberty and have not been present “all her life.” Her type II skin is otherwise unremarkable and free of sun damage.
