User login
Use of isolation in juvenile detention centers
Isolation in juvenile detention centers persists despite ample data demonstrating the traumatizing consequences to youth who often already have been traumatized. I recently visited a detention center as part of my pediatric residency’s advocacy rotation. There, I learned that youth are kept in isolation for several days, with a vague definition by staff on the limit of “several days.” Multiple words were used for confinement, the most stunning and horrific of which was “segregation” – “He got into a fight and was placed in segregation.” While in isolation or segregation – whatever it is called – mental illness and posttraumatic stress disorder are exacerbated. Youth do not participate in school classes, and they are barred from the daily hour of physical activity. In extreme cases, they go from complete isolation one day to complete freedom the next.
The vibe word in the facility I visited was “evidence-based strategies,” stressed by the new administration. But evidence-based strategies do not include isolation. They include educating staff about the pervasive effects of trauma in children. They include communication interventions, conflict resolution, and the implementation of rewards such as extra visitation, computer time, or the use of an adolescent’s own personal hygiene items or clothing. They include knowledge of the adolescent brain, and how the use of isolation in juvenile centers has led to increased suicide rates in those children.

Lindsay M. Hayes, author of the National Center on Institutions and Alternatives’ 2004 report “Juvenile Suicide in Confinement: A National Study,” wrote: ”Although room confinement remains a staple in most juvenile facilities, it is a sanction that can have deadly consequences. … More than 50% of all youths’ suicides in juvenile facilities occurred while young people were isolated alone in their rooms, and … more than 60% of young people who committed suicide in custody had a history of being held in isolation.”
The United Nations has called on all countries to absolutely prohibit solitary confinement for juveniles, as has the American Academy of Child and Adolescent Psychiatry. Thus, extreme isolation should not be another tool for juvenile detention centers.
Currently, 20 states have banned solitary confinement in juvenile detention facilities. The major barriers are from staff, who state it would remove a tool, put staff in danger, and allow youth to run the facilities. None of these has been shown to be true. Some juvenile detention centers have changed the traditional meaning of isolation – youth will have a minimum of 8 hours away from isolation when confined for a day or longer. “During that 8 hours, they have the opportunity to talk to and be in the company of staff,” said Adam Schwartz, a lawyer for the American Civil Liberties Union of Illinois, whose lawsuit drastically limited solitary confinement practices in Illinois’s juvenile detention centers. The policy also requires that inmates in isolation continue to receive education and mental health services.
There is a human dignity that not even detainees deserve to lose. President Obama has discussed this, as has the 2012 Report of the Attorney General’s National Task Force on Children Exposed to Violence, which concluded: “Nowhere is the damaging impact of incarceration on vulnerable children more obvious than when it involves solitary confinement.” I am writing this article to raise awareness about this underreported problem in hopes that new legislation will lead to change that is in the best interest of our children.
Dr. Raffa is in postgraduate year 2 in her pediatric residency at Vanderbilt Children’s Hospital in Nashville, Tenn.
Isolation in juvenile detention centers persists despite ample data demonstrating the traumatizing consequences to youth who often already have been traumatized. I recently visited a detention center as part of my pediatric residency’s advocacy rotation. There, I learned that youth are kept in isolation for several days, with a vague definition by staff on the limit of “several days.” Multiple words were used for confinement, the most stunning and horrific of which was “segregation” – “He got into a fight and was placed in segregation.” While in isolation or segregation – whatever it is called – mental illness and posttraumatic stress disorder are exacerbated. Youth do not participate in school classes, and they are barred from the daily hour of physical activity. In extreme cases, they go from complete isolation one day to complete freedom the next.
The vibe word in the facility I visited was “evidence-based strategies,” stressed by the new administration. But evidence-based strategies do not include isolation. They include educating staff about the pervasive effects of trauma in children. They include communication interventions, conflict resolution, and the implementation of rewards such as extra visitation, computer time, or the use of an adolescent’s own personal hygiene items or clothing. They include knowledge of the adolescent brain, and how the use of isolation in juvenile centers has led to increased suicide rates in those children.
Lindsay M. Hayes, author of the National Center on Institutions and Alternatives’ 2004 report “Juvenile Suicide in Confinement: A National Study,” wrote: ”Although room confinement remains a staple in most juvenile facilities, it is a sanction that can have deadly consequences. … More than 50% of all youths’ suicides in juvenile facilities occurred while young people were isolated alone in their rooms, and … more than 60% of young people who committed suicide in custody had a history of being held in isolation.”
The United Nations has called on all countries to absolutely prohibit solitary confinement for juveniles, as has the American Academy of Child and Adolescent Psychiatry. Thus, extreme isolation should not be another tool for juvenile detention centers.
Currently, 20 states have banned solitary confinement in juvenile detention facilities. The major barriers are from staff, who state it would remove a tool, put staff in danger, and allow youth to run the facilities. None of these has been shown to be true. Some juvenile detention centers have changed the traditional meaning of isolation – youth will have a minimum of 8 hours away from isolation when confined for a day or longer. “During that 8 hours, they have the opportunity to talk to and be in the company of staff,” said Adam Schwartz, a lawyer for the American Civil Liberties Union of Illinois, whose lawsuit drastically limited solitary confinement practices in Illinois’s juvenile detention centers. The policy also requires that inmates in isolation continue to receive education and mental health services.
There is a human dignity that not even detainees deserve to lose. President Obama has discussed this, as has the 2012 Report of the Attorney General’s National Task Force on Children Exposed to Violence, which concluded: “Nowhere is the damaging impact of incarceration on vulnerable children more obvious than when it involves solitary confinement.” I am writing this article to raise awareness about this underreported problem in hopes that new legislation will lead to change that is in the best interest of our children.
Dr. Raffa is in postgraduate year 2 in her pediatric residency at Vanderbilt Children’s Hospital in Nashville, Tenn.
Isolation in juvenile detention centers persists despite ample data demonstrating the traumatizing consequences to youth who often already have been traumatized. I recently visited a detention center as part of my pediatric residency’s advocacy rotation. There, I learned that youth are kept in isolation for several days, with a vague definition by staff on the limit of “several days.” Multiple words were used for confinement, the most stunning and horrific of which was “segregation” – “He got into a fight and was placed in segregation.” While in isolation or segregation – whatever it is called – mental illness and posttraumatic stress disorder are exacerbated. Youth do not participate in school classes, and they are barred from the daily hour of physical activity. In extreme cases, they go from complete isolation one day to complete freedom the next.
The vibe word in the facility I visited was “evidence-based strategies,” stressed by the new administration. But evidence-based strategies do not include isolation. They include educating staff about the pervasive effects of trauma in children. They include communication interventions, conflict resolution, and the implementation of rewards such as extra visitation, computer time, or the use of an adolescent’s own personal hygiene items or clothing. They include knowledge of the adolescent brain, and how the use of isolation in juvenile centers has led to increased suicide rates in those children.
Lindsay M. Hayes, author of the National Center on Institutions and Alternatives’ 2004 report “Juvenile Suicide in Confinement: A National Study,” wrote: ”Although room confinement remains a staple in most juvenile facilities, it is a sanction that can have deadly consequences. … More than 50% of all youths’ suicides in juvenile facilities occurred while young people were isolated alone in their rooms, and … more than 60% of young people who committed suicide in custody had a history of being held in isolation.”
The United Nations has called on all countries to absolutely prohibit solitary confinement for juveniles, as has the American Academy of Child and Adolescent Psychiatry. Thus, extreme isolation should not be another tool for juvenile detention centers.
Currently, 20 states have banned solitary confinement in juvenile detention facilities. The major barriers are from staff, who state it would remove a tool, put staff in danger, and allow youth to run the facilities. None of these has been shown to be true. Some juvenile detention centers have changed the traditional meaning of isolation – youth will have a minimum of 8 hours away from isolation when confined for a day or longer. “During that 8 hours, they have the opportunity to talk to and be in the company of staff,” said Adam Schwartz, a lawyer for the American Civil Liberties Union of Illinois, whose lawsuit drastically limited solitary confinement practices in Illinois’s juvenile detention centers. The policy also requires that inmates in isolation continue to receive education and mental health services.
There is a human dignity that not even detainees deserve to lose. President Obama has discussed this, as has the 2012 Report of the Attorney General’s National Task Force on Children Exposed to Violence, which concluded: “Nowhere is the damaging impact of incarceration on vulnerable children more obvious than when it involves solitary confinement.” I am writing this article to raise awareness about this underreported problem in hopes that new legislation will lead to change that is in the best interest of our children.
Dr. Raffa is in postgraduate year 2 in her pediatric residency at Vanderbilt Children’s Hospital in Nashville, Tenn.

AHA: Three measures risk stratify acute heart failure
ORLANDO – Three simple, routinely collected measurements together provide a lot of insight into the risk faced by community-dwelling patients hospitalized for acute decompensated heart failure, according to data collected from 3,628 patients at one U.S. center.
The three measures are blood urea nitrogen (BUN), systolic blood pressure, and serum creatinine. Using dichotomous cutoffs first calculated a decade ago, these three parameters distinguish up to an eightfold range of postdischarge mortality during the 30 or 90 days following an index hospitalization, and up to a fourfold range of risk for rehospitalization for heart failure during the ensuing 30 or 90 days, Dr. Sithu Win said at the American Heart Association scientific sessions.

Applying this three-measure assessment to patients hospitalized with acute decompensated heart failure “may guide care-transition planning and promote efficient allocation of limited resources,” said Dr. Win, a cardiologist at the Mayo Clinic in Rochester, Minn. The next step is to try to figure out the best way to use this risk prognostication in routine practice, he added.
Researchers published the original analysis that identified BUN, systolic BP, and serum creatinine as key prognostic measures in 2005 using data taken from more than 65,000 U.S. heart failure patients enrolled in ADHERE (Acute Decompensated Heart Failure National Registry) (JAMA. 2005 Feb 2;293[5]:572-80). Using a classification and regression tree analysis, the 2005 study verified dichotomous cutoffs for these three parameters that identified patients at highest risk for in-hospital mortality.
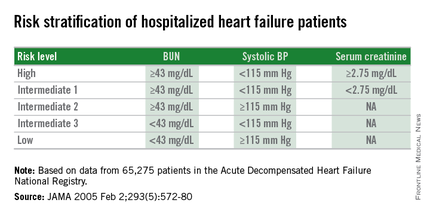
The 2005 study prioritized the application of these cutoffs to define in-hospital mortality risk: first BUN, then the systolic BP criterion, and lastly the serum creatinine criterion. This resulted in five risk levels: Highest-risk patients had a BUN of at least 43 mg/dL, a systolic BP of less than 115 mm Hg, and a serum creatinine of at least 2.75 mg/dL. Lowest-risk patients had a BUN of less than 43 mg/dL and a systolic BP of more than 115 mm Hg. (In lower-risk patients, serum-creatinine level dropped out as a risk determinant.) The analysis also created three categories of patients with intermediate risk based on various combinations of the three measures.
The new study run by Dr. Win and his associates evaluated how this risk-assessment tool developed to predict in-hospital mortality performed for predicting event rates among community-based heart failure patients who had a total of 5,918 hospitalizations for acute decompensated heart failure at the Mayo Clinic during 2000-2013. They averaged 78 years old, half were women, and 48% had heart failure with preserved ejection fraction.
The risk-level distribution of the 3,628 Mayo patients closely matched the pattern seen in the original ADHERE registry: 63% were low risk, 17% were at intermediate level 3 (the lowest risk level in the intermediate range), 13% were at intermediate level 2, 5% at intermediate level 1, and 2% were categorized as high risk.
For 30-day mortality post hospitalization, patients at the highest risk level had a mortality rate eightfold higher than did the lowest-risk patients, those rated intermediate level 1 had a fivefold higher mortality rate, intermediate level 2 patients had a threefold higher rate, and those at intermediate 3 had a 50% higher rate, Dr. Win reported. During the 90 days after discharge, mortality rates relative to the lowest risk level ranged from a sixfold higher rate among the highest-risk patients to a 50% higher rate among patients with an intermediate 3 designation.
Analysis of rehospitalizations for heart failure showed that, by 30 days after hospitalization, the readmission rate ran threefold higher in the highest-risk patients, compared with those at the lowest risk and fourfold higher among those at intermediate risk level 1. Heart failure readmissions by 90 days following the index hospitalization ran threefold higher for both the highest-risk patients as well as those at intermediate level 1, compared with the patients at lowest risk.
The new analyses also showed that roughly similar risk patterns occurred regardless of whether patients had heart failure with reduced or preserved ejection fraction during their index hospitalization, although the relatively increased rate of 30-day mortality with a worse risk profile was most dramatic among patients with reduced ejection fraction. Age, sex, and comorbidity severity did not have a marked effect on the relationships between event rates and risk levels, Dr. Win said.
Dr. Win had no disclosures.
On Twitter @mitchelzoler
ORLANDO – Three simple, routinely collected measurements together provide a lot of insight into the risk faced by community-dwelling patients hospitalized for acute decompensated heart failure, according to data collected from 3,628 patients at one U.S. center.
The three measures are blood urea nitrogen (BUN), systolic blood pressure, and serum creatinine. Using dichotomous cutoffs first calculated a decade ago, these three parameters distinguish up to an eightfold range of postdischarge mortality during the 30 or 90 days following an index hospitalization, and up to a fourfold range of risk for rehospitalization for heart failure during the ensuing 30 or 90 days, Dr. Sithu Win said at the American Heart Association scientific sessions.
Applying this three-measure assessment to patients hospitalized with acute decompensated heart failure “may guide care-transition planning and promote efficient allocation of limited resources,” said Dr. Win, a cardiologist at the Mayo Clinic in Rochester, Minn. The next step is to try to figure out the best way to use this risk prognostication in routine practice, he added.
Researchers published the original analysis that identified BUN, systolic BP, and serum creatinine as key prognostic measures in 2005 using data taken from more than 65,000 U.S. heart failure patients enrolled in ADHERE (Acute Decompensated Heart Failure National Registry) (JAMA. 2005 Feb 2;293[5]:572-80). Using a classification and regression tree analysis, the 2005 study verified dichotomous cutoffs for these three parameters that identified patients at highest risk for in-hospital mortality.
The 2005 study prioritized the application of these cutoffs to define in-hospital mortality risk: first BUN, then the systolic BP criterion, and lastly the serum creatinine criterion. This resulted in five risk levels: Highest-risk patients had a BUN of at least 43 mg/dL, a systolic BP of less than 115 mm Hg, and a serum creatinine of at least 2.75 mg/dL. Lowest-risk patients had a BUN of less than 43 mg/dL and a systolic BP of more than 115 mm Hg. (In lower-risk patients, serum-creatinine level dropped out as a risk determinant.) The analysis also created three categories of patients with intermediate risk based on various combinations of the three measures.
The new study run by Dr. Win and his associates evaluated how this risk-assessment tool developed to predict in-hospital mortality performed for predicting event rates among community-based heart failure patients who had a total of 5,918 hospitalizations for acute decompensated heart failure at the Mayo Clinic during 2000-2013. They averaged 78 years old, half were women, and 48% had heart failure with preserved ejection fraction.
The risk-level distribution of the 3,628 Mayo patients closely matched the pattern seen in the original ADHERE registry: 63% were low risk, 17% were at intermediate level 3 (the lowest risk level in the intermediate range), 13% were at intermediate level 2, 5% at intermediate level 1, and 2% were categorized as high risk.
For 30-day mortality post hospitalization, patients at the highest risk level had a mortality rate eightfold higher than did the lowest-risk patients, those rated intermediate level 1 had a fivefold higher mortality rate, intermediate level 2 patients had a threefold higher rate, and those at intermediate 3 had a 50% higher rate, Dr. Win reported. During the 90 days after discharge, mortality rates relative to the lowest risk level ranged from a sixfold higher rate among the highest-risk patients to a 50% higher rate among patients with an intermediate 3 designation.
Analysis of rehospitalizations for heart failure showed that, by 30 days after hospitalization, the readmission rate ran threefold higher in the highest-risk patients, compared with those at the lowest risk and fourfold higher among those at intermediate risk level 1. Heart failure readmissions by 90 days following the index hospitalization ran threefold higher for both the highest-risk patients as well as those at intermediate level 1, compared with the patients at lowest risk.
The new analyses also showed that roughly similar risk patterns occurred regardless of whether patients had heart failure with reduced or preserved ejection fraction during their index hospitalization, although the relatively increased rate of 30-day mortality with a worse risk profile was most dramatic among patients with reduced ejection fraction. Age, sex, and comorbidity severity did not have a marked effect on the relationships between event rates and risk levels, Dr. Win said.
Dr. Win had no disclosures.
On Twitter @mitchelzoler
ORLANDO – Three simple, routinely collected measurements together provide a lot of insight into the risk faced by community-dwelling patients hospitalized for acute decompensated heart failure, according to data collected from 3,628 patients at one U.S. center.
The three measures are blood urea nitrogen (BUN), systolic blood pressure, and serum creatinine. Using dichotomous cutoffs first calculated a decade ago, these three parameters distinguish up to an eightfold range of postdischarge mortality during the 30 or 90 days following an index hospitalization, and up to a fourfold range of risk for rehospitalization for heart failure during the ensuing 30 or 90 days, Dr. Sithu Win said at the American Heart Association scientific sessions.
Applying this three-measure assessment to patients hospitalized with acute decompensated heart failure “may guide care-transition planning and promote efficient allocation of limited resources,” said Dr. Win, a cardiologist at the Mayo Clinic in Rochester, Minn. The next step is to try to figure out the best way to use this risk prognostication in routine practice, he added.
Researchers published the original analysis that identified BUN, systolic BP, and serum creatinine as key prognostic measures in 2005 using data taken from more than 65,000 U.S. heart failure patients enrolled in ADHERE (Acute Decompensated Heart Failure National Registry) (JAMA. 2005 Feb 2;293[5]:572-80). Using a classification and regression tree analysis, the 2005 study verified dichotomous cutoffs for these three parameters that identified patients at highest risk for in-hospital mortality.
The 2005 study prioritized the application of these cutoffs to define in-hospital mortality risk: first BUN, then the systolic BP criterion, and lastly the serum creatinine criterion. This resulted in five risk levels: Highest-risk patients had a BUN of at least 43 mg/dL, a systolic BP of less than 115 mm Hg, and a serum creatinine of at least 2.75 mg/dL. Lowest-risk patients had a BUN of less than 43 mg/dL and a systolic BP of more than 115 mm Hg. (In lower-risk patients, serum-creatinine level dropped out as a risk determinant.) The analysis also created three categories of patients with intermediate risk based on various combinations of the three measures.
The new study run by Dr. Win and his associates evaluated how this risk-assessment tool developed to predict in-hospital mortality performed for predicting event rates among community-based heart failure patients who had a total of 5,918 hospitalizations for acute decompensated heart failure at the Mayo Clinic during 2000-2013. They averaged 78 years old, half were women, and 48% had heart failure with preserved ejection fraction.
The risk-level distribution of the 3,628 Mayo patients closely matched the pattern seen in the original ADHERE registry: 63% were low risk, 17% were at intermediate level 3 (the lowest risk level in the intermediate range), 13% were at intermediate level 2, 5% at intermediate level 1, and 2% were categorized as high risk.
For 30-day mortality post hospitalization, patients at the highest risk level had a mortality rate eightfold higher than did the lowest-risk patients, those rated intermediate level 1 had a fivefold higher mortality rate, intermediate level 2 patients had a threefold higher rate, and those at intermediate 3 had a 50% higher rate, Dr. Win reported. During the 90 days after discharge, mortality rates relative to the lowest risk level ranged from a sixfold higher rate among the highest-risk patients to a 50% higher rate among patients with an intermediate 3 designation.
Analysis of rehospitalizations for heart failure showed that, by 30 days after hospitalization, the readmission rate ran threefold higher in the highest-risk patients, compared with those at the lowest risk and fourfold higher among those at intermediate risk level 1. Heart failure readmissions by 90 days following the index hospitalization ran threefold higher for both the highest-risk patients as well as those at intermediate level 1, compared with the patients at lowest risk.
The new analyses also showed that roughly similar risk patterns occurred regardless of whether patients had heart failure with reduced or preserved ejection fraction during their index hospitalization, although the relatively increased rate of 30-day mortality with a worse risk profile was most dramatic among patients with reduced ejection fraction. Age, sex, and comorbidity severity did not have a marked effect on the relationships between event rates and risk levels, Dr. Win said.
Dr. Win had no disclosures.
On Twitter @mitchelzoler
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: Dichotomous cutoffs for BUN, systolic BP, and serum creatinine together robustly risk stratified patients hospitalized for acute decompensated heart failure.
Major finding: Three baseline measures together stratified patients over an eightfold range for 30-day mortality following hospital discharge.
Data source: Application of the risk-stratification formula to 3,628 heart failure patients hospitalized at one U.S. center during 2000-2013.
Disclosures: Dr. Win had no disclosures.

Electronic Health Records Key Driver of Physician Burnout
Half of U.S. physicians are experiencing some of the symptoms of burnout, with even higher rates for general internists. Implementation of the electronic health record (EHR) has been cited as the biggest driver of physician job dissatisfaction, said Christine Sinsky, MD, a former hospitalist and currently vice president of professional satisfaction at the American Medical Association (AMA), to attendees at the 19th Management of the Hospitalized Patient Conference, presented by the University of California-San Francisco.1
Dr. Sinsky deemed physician discontent “the canary in the coal mine” for a dysfunctional healthcare system. After visiting 23 high-functioning medical teams, Dr. Sinsky said she had found that 70% to 80% of physician work output could be considered waste, defined as work that doesn’t need to be done and doesn’t add value to the patient. The AMA, she said, has made a commitment to addressing physicians’ dissatisfaction and burnout.
Dr. Sinsky offered a number of suggestions for physicians and the larger system. Among them was the idea of medical teams employing a documentation specialist, or scribe, to accompany physicians on patient rounds to help with the clerical tasks that divert physicians from patient care. She also cited David Reuben, MD, a gerontologist at UCLA whose JAMA Internal Medicine study documented his training of physician “practice partners,” often medical or nursing students, who help queue up orders in the EHR, and the improved patient satisfaction that resulted.2
“Be bold,” she advised hospitalists. “The patient care delivery modes of the future can’t be met with staffing models from the past.”
Larry Beresford is a freelance writer in Alameda, Calif.
References
- Friedberg M, Chen PG, Van Busum KR, et al. Factors affecting physician professional satisfaction and their implications for patient care, health systems, and health policy. 2013. Accessed November 3, 2015.
- Reuben DB, Knudsen J, Senelick W, Glazier E, Koretz BK. The effect of a physician partner program on physician efficiency and patient satisfaction. JAMA Intern Med. 2014;174(7):1190-1193.
Half of U.S. physicians are experiencing some of the symptoms of burnout, with even higher rates for general internists. Implementation of the electronic health record (EHR) has been cited as the biggest driver of physician job dissatisfaction, said Christine Sinsky, MD, a former hospitalist and currently vice president of professional satisfaction at the American Medical Association (AMA), to attendees at the 19th Management of the Hospitalized Patient Conference, presented by the University of California-San Francisco.1
Dr. Sinsky deemed physician discontent “the canary in the coal mine” for a dysfunctional healthcare system. After visiting 23 high-functioning medical teams, Dr. Sinsky said she had found that 70% to 80% of physician work output could be considered waste, defined as work that doesn’t need to be done and doesn’t add value to the patient. The AMA, she said, has made a commitment to addressing physicians’ dissatisfaction and burnout.
Dr. Sinsky offered a number of suggestions for physicians and the larger system. Among them was the idea of medical teams employing a documentation specialist, or scribe, to accompany physicians on patient rounds to help with the clerical tasks that divert physicians from patient care. She also cited David Reuben, MD, a gerontologist at UCLA whose JAMA Internal Medicine study documented his training of physician “practice partners,” often medical or nursing students, who help queue up orders in the EHR, and the improved patient satisfaction that resulted.2
“Be bold,” she advised hospitalists. “The patient care delivery modes of the future can’t be met with staffing models from the past.”
Larry Beresford is a freelance writer in Alameda, Calif.
References
- Friedberg M, Chen PG, Van Busum KR, et al. Factors affecting physician professional satisfaction and their implications for patient care, health systems, and health policy. 2013. Accessed November 3, 2015.
- Reuben DB, Knudsen J, Senelick W, Glazier E, Koretz BK. The effect of a physician partner program on physician efficiency and patient satisfaction. JAMA Intern Med. 2014;174(7):1190-1193.
Half of U.S. physicians are experiencing some of the symptoms of burnout, with even higher rates for general internists. Implementation of the electronic health record (EHR) has been cited as the biggest driver of physician job dissatisfaction, said Christine Sinsky, MD, a former hospitalist and currently vice president of professional satisfaction at the American Medical Association (AMA), to attendees at the 19th Management of the Hospitalized Patient Conference, presented by the University of California-San Francisco.1
Dr. Sinsky deemed physician discontent “the canary in the coal mine” for a dysfunctional healthcare system. After visiting 23 high-functioning medical teams, Dr. Sinsky said she had found that 70% to 80% of physician work output could be considered waste, defined as work that doesn’t need to be done and doesn’t add value to the patient. The AMA, she said, has made a commitment to addressing physicians’ dissatisfaction and burnout.
Dr. Sinsky offered a number of suggestions for physicians and the larger system. Among them was the idea of medical teams employing a documentation specialist, or scribe, to accompany physicians on patient rounds to help with the clerical tasks that divert physicians from patient care. She also cited David Reuben, MD, a gerontologist at UCLA whose JAMA Internal Medicine study documented his training of physician “practice partners,” often medical or nursing students, who help queue up orders in the EHR, and the improved patient satisfaction that resulted.2
“Be bold,” she advised hospitalists. “The patient care delivery modes of the future can’t be met with staffing models from the past.”
Larry Beresford is a freelance writer in Alameda, Calif.
References
- Friedberg M, Chen PG, Van Busum KR, et al. Factors affecting physician professional satisfaction and their implications for patient care, health systems, and health policy. 2013. Accessed November 3, 2015.
- Reuben DB, Knudsen J, Senelick W, Glazier E, Koretz BK. The effect of a physician partner program on physician efficiency and patient satisfaction. JAMA Intern Med. 2014;174(7):1190-1193.
Claims Data Misclassify Cardiovascular Disease Event Rates
NEW YORK - Diagnostic codes from administrative claims data underestimate cardiovascular disease event rates, researchers report.
"Increasingly, the diagnostic codes from administrative claims data are being used to measure clinical outcomes," Dr. Bruce M. Psaty from the University of Washington, Seattle, said by email. "The methods of using only claims data as outcomes nonetheless influence the results. Methods that seek to avoid misclassification tend to underestimate event rates, and methods that attempt to include all events tend to include misclassified events such as non-event hospitalizations as part of the outcome."
Dr. Psaty's team used data from the Cardiovascular Health Study (CHS) to evaluate the degree of both misclassification and underestimation of event rates for cardiovascular disease outcomes identified solely from claims data compared with those identified through active surveillance.
An ICD9 code of 410 in the first position had a 90.6% positive predictive value (PPV) for MI, but this code only identified 53.8% of the incident MIs ascertained by active surveillance. Inclusion of this code as a secondary diagnosis identified an additional 16.6% of MI events.
Similarly, main stroke codes in the first position had an 80.4% PPV for stroke, but identified only 63.8% of the incident stroke events. For heart failure, main diagnostic codes had a high PPV of 93.2%, but identified only 27.2% of heart failure events.
Estimates of disease incidence differed markedly according to whether the incidence rates were determined by CHS surveillance, a first-position diagnostic code, or a diagnostic code in any position.
In general, misclassified events in the administrative claims data appeared to have little effect on the magnitude of associations for most cardiovascular disease risk factors, the researchers report in Circulation, online Nov. 4.
"No study is perfect, and some incomplete identification of events in a study is common," Dr. Psaty explained. "The effect depends on both the type of study and the degree of incompleteness. In a clinical trial, if there is no bias, the relative-risk comparison between the two groups remains valid even if some of the events were missed. If, on the other hand, the goal is the development of a model for the prediction of event rates to decide on whether to start a cholesterol lowering therapy, the incomplete identification of events introduces a bias in the model that is directly related to the degree of incompleteness in the identification of events."
"Events data collection should be appropriate for the study," Dr. Psaty concluded. "Published studies need to provide sufficient detail so that readers can judge whether the methods were indeed fit for purpose."
NEW YORK - Diagnostic codes from administrative claims data underestimate cardiovascular disease event rates, researchers report.
"Increasingly, the diagnostic codes from administrative claims data are being used to measure clinical outcomes," Dr. Bruce M. Psaty from the University of Washington, Seattle, said by email. "The methods of using only claims data as outcomes nonetheless influence the results. Methods that seek to avoid misclassification tend to underestimate event rates, and methods that attempt to include all events tend to include misclassified events such as non-event hospitalizations as part of the outcome."
Dr. Psaty's team used data from the Cardiovascular Health Study (CHS) to evaluate the degree of both misclassification and underestimation of event rates for cardiovascular disease outcomes identified solely from claims data compared with those identified through active surveillance.
An ICD9 code of 410 in the first position had a 90.6% positive predictive value (PPV) for MI, but this code only identified 53.8% of the incident MIs ascertained by active surveillance. Inclusion of this code as a secondary diagnosis identified an additional 16.6% of MI events.
Similarly, main stroke codes in the first position had an 80.4% PPV for stroke, but identified only 63.8% of the incident stroke events. For heart failure, main diagnostic codes had a high PPV of 93.2%, but identified only 27.2% of heart failure events.
Estimates of disease incidence differed markedly according to whether the incidence rates were determined by CHS surveillance, a first-position diagnostic code, or a diagnostic code in any position.
In general, misclassified events in the administrative claims data appeared to have little effect on the magnitude of associations for most cardiovascular disease risk factors, the researchers report in Circulation, online Nov. 4.
"No study is perfect, and some incomplete identification of events in a study is common," Dr. Psaty explained. "The effect depends on both the type of study and the degree of incompleteness. In a clinical trial, if there is no bias, the relative-risk comparison between the two groups remains valid even if some of the events were missed. If, on the other hand, the goal is the development of a model for the prediction of event rates to decide on whether to start a cholesterol lowering therapy, the incomplete identification of events introduces a bias in the model that is directly related to the degree of incompleteness in the identification of events."
"Events data collection should be appropriate for the study," Dr. Psaty concluded. "Published studies need to provide sufficient detail so that readers can judge whether the methods were indeed fit for purpose."
NEW YORK - Diagnostic codes from administrative claims data underestimate cardiovascular disease event rates, researchers report.
"Increasingly, the diagnostic codes from administrative claims data are being used to measure clinical outcomes," Dr. Bruce M. Psaty from the University of Washington, Seattle, said by email. "The methods of using only claims data as outcomes nonetheless influence the results. Methods that seek to avoid misclassification tend to underestimate event rates, and methods that attempt to include all events tend to include misclassified events such as non-event hospitalizations as part of the outcome."
Dr. Psaty's team used data from the Cardiovascular Health Study (CHS) to evaluate the degree of both misclassification and underestimation of event rates for cardiovascular disease outcomes identified solely from claims data compared with those identified through active surveillance.
An ICD9 code of 410 in the first position had a 90.6% positive predictive value (PPV) for MI, but this code only identified 53.8% of the incident MIs ascertained by active surveillance. Inclusion of this code as a secondary diagnosis identified an additional 16.6% of MI events.
Similarly, main stroke codes in the first position had an 80.4% PPV for stroke, but identified only 63.8% of the incident stroke events. For heart failure, main diagnostic codes had a high PPV of 93.2%, but identified only 27.2% of heart failure events.
Estimates of disease incidence differed markedly according to whether the incidence rates were determined by CHS surveillance, a first-position diagnostic code, or a diagnostic code in any position.
In general, misclassified events in the administrative claims data appeared to have little effect on the magnitude of associations for most cardiovascular disease risk factors, the researchers report in Circulation, online Nov. 4.
"No study is perfect, and some incomplete identification of events in a study is common," Dr. Psaty explained. "The effect depends on both the type of study and the degree of incompleteness. In a clinical trial, if there is no bias, the relative-risk comparison between the two groups remains valid even if some of the events were missed. If, on the other hand, the goal is the development of a model for the prediction of event rates to decide on whether to start a cholesterol lowering therapy, the incomplete identification of events introduces a bias in the model that is directly related to the degree of incompleteness in the identification of events."
"Events data collection should be appropriate for the study," Dr. Psaty concluded. "Published studies need to provide sufficient detail so that readers can judge whether the methods were indeed fit for purpose."
Movers and Shakers in Hospital Medicine December 2015
John Carlile, MD, is the new medical director of pediatric hospitalist and intensivist services at Summerville (S.C.) Medical Center. Most recently, Dr. Carlile worked as a pediatric critical care specialist at Cox Medical Center in Springfield, Mo.


Elaine McElveen, RN, has been named director of the hospitalist program at McLeod Medical Center Dillon in Dillon, S.C. In her new role, McElveen also will oversee the case management department. She has been a practicing nurse for the McLeod Health system for 28 years.

Christine Wentt, PA, an adult hospitalist physician assistant, has been inducted into the VIP Woman of the Year Circle for 2015-2016 by the National Association of Professional Women (NAPW). Wentt works for Physicians Inpatient Care Specialists (MDICS), a private hospitalist group in Glen Burnie, Md. Wentt also serves as an adjunct professor in Drexel University’s physician assistant program.
Robert Zurcher, MD, has been appointed chief medical officer for emergency medicine at HNI Healthcare (formerly Hospitalists Now, Inc.), based in Austin, Texas. Dr. Zurcher comes to HNI Healthcare from IPC Healthcare, where he served as the administrative director for their Northwest Region. Dr. Zurcher has 27 years of practice experience and has founded and managed physician services companies in both hospital medicine and emergency medicine.
Business Moves
North Hollywood, Calif.-based IPC Healthcare recently announced its acquisition of Hospital Medicine Consultants, LLC, in Elgin, Ill., a locally owned practice serving several hospitals in the Chicago suburbs. IPC staffs hospitalist providers in more than 400 hospitals across the country.
The Schumacher Group, a private hospitalist and emergency medicine provider, announced the implementation of a new telehospitalist program at Abrom Kaplan Memorial and Acadia General Hospitals, both in Lafayette, La. The program will allow physicians to provide overnight care to the two facilities from a remote location. The Schumacher Group was founded in 1994 and currently serves more than four million patients per year.

Mike O’Neal is a freelance writer in New York City.
In Memoriam

Dr. Ayantuga completed his PhD thesis at Oxford University in less than three years, during which time he received the Bishop Frazer Prize for excellence in research in organic chemistry. Following his tenure at Oxford, Dr. Ayantuga obtained his medical degree from Cambridge University in 1994.
As his memorial site notes, “he continued until his death, to demonstrate dedication to quality and process involvement, illustrating commitment to active engagement in lifelong learning.”
John Carlile, MD, is the new medical director of pediatric hospitalist and intensivist services at Summerville (S.C.) Medical Center. Most recently, Dr. Carlile worked as a pediatric critical care specialist at Cox Medical Center in Springfield, Mo.
Elaine McElveen, RN, has been named director of the hospitalist program at McLeod Medical Center Dillon in Dillon, S.C. In her new role, McElveen also will oversee the case management department. She has been a practicing nurse for the McLeod Health system for 28 years.
Christine Wentt, PA, an adult hospitalist physician assistant, has been inducted into the VIP Woman of the Year Circle for 2015-2016 by the National Association of Professional Women (NAPW). Wentt works for Physicians Inpatient Care Specialists (MDICS), a private hospitalist group in Glen Burnie, Md. Wentt also serves as an adjunct professor in Drexel University’s physician assistant program.
Robert Zurcher, MD, has been appointed chief medical officer for emergency medicine at HNI Healthcare (formerly Hospitalists Now, Inc.), based in Austin, Texas. Dr. Zurcher comes to HNI Healthcare from IPC Healthcare, where he served as the administrative director for their Northwest Region. Dr. Zurcher has 27 years of practice experience and has founded and managed physician services companies in both hospital medicine and emergency medicine.
Business Moves
North Hollywood, Calif.-based IPC Healthcare recently announced its acquisition of Hospital Medicine Consultants, LLC, in Elgin, Ill., a locally owned practice serving several hospitals in the Chicago suburbs. IPC staffs hospitalist providers in more than 400 hospitals across the country.
The Schumacher Group, a private hospitalist and emergency medicine provider, announced the implementation of a new telehospitalist program at Abrom Kaplan Memorial and Acadia General Hospitals, both in Lafayette, La. The program will allow physicians to provide overnight care to the two facilities from a remote location. The Schumacher Group was founded in 1994 and currently serves more than four million patients per year.
Mike O’Neal is a freelance writer in New York City.
In Memoriam
Dr. Ayantuga completed his PhD thesis at Oxford University in less than three years, during which time he received the Bishop Frazer Prize for excellence in research in organic chemistry. Following his tenure at Oxford, Dr. Ayantuga obtained his medical degree from Cambridge University in 1994.
As his memorial site notes, “he continued until his death, to demonstrate dedication to quality and process involvement, illustrating commitment to active engagement in lifelong learning.”
John Carlile, MD, is the new medical director of pediatric hospitalist and intensivist services at Summerville (S.C.) Medical Center. Most recently, Dr. Carlile worked as a pediatric critical care specialist at Cox Medical Center in Springfield, Mo.
Elaine McElveen, RN, has been named director of the hospitalist program at McLeod Medical Center Dillon in Dillon, S.C. In her new role, McElveen also will oversee the case management department. She has been a practicing nurse for the McLeod Health system for 28 years.
Christine Wentt, PA, an adult hospitalist physician assistant, has been inducted into the VIP Woman of the Year Circle for 2015-2016 by the National Association of Professional Women (NAPW). Wentt works for Physicians Inpatient Care Specialists (MDICS), a private hospitalist group in Glen Burnie, Md. Wentt also serves as an adjunct professor in Drexel University’s physician assistant program.
Robert Zurcher, MD, has been appointed chief medical officer for emergency medicine at HNI Healthcare (formerly Hospitalists Now, Inc.), based in Austin, Texas. Dr. Zurcher comes to HNI Healthcare from IPC Healthcare, where he served as the administrative director for their Northwest Region. Dr. Zurcher has 27 years of practice experience and has founded and managed physician services companies in both hospital medicine and emergency medicine.
Business Moves
North Hollywood, Calif.-based IPC Healthcare recently announced its acquisition of Hospital Medicine Consultants, LLC, in Elgin, Ill., a locally owned practice serving several hospitals in the Chicago suburbs. IPC staffs hospitalist providers in more than 400 hospitals across the country.
The Schumacher Group, a private hospitalist and emergency medicine provider, announced the implementation of a new telehospitalist program at Abrom Kaplan Memorial and Acadia General Hospitals, both in Lafayette, La. The program will allow physicians to provide overnight care to the two facilities from a remote location. The Schumacher Group was founded in 1994 and currently serves more than four million patients per year.
Mike O’Neal is a freelance writer in New York City.
In Memoriam
Dr. Ayantuga completed his PhD thesis at Oxford University in less than three years, during which time he received the Bishop Frazer Prize for excellence in research in organic chemistry. Following his tenure at Oxford, Dr. Ayantuga obtained his medical degree from Cambridge University in 1994.
As his memorial site notes, “he continued until his death, to demonstrate dedication to quality and process involvement, illustrating commitment to active engagement in lifelong learning.”
Healthcare Policies Affecting Hospitalists, Hospitalized Patients Advance in 2015

Laws and regulations regularly impact hospitalists and hospitalized patients, which is why SHM’s Advocacy Department and Public Policy Committee (PPC) work on behalf of SHM membership, the hospital medicine movement, and its patients.
This year has seen significant positive movement within several key policy areas for all of these groups. Some of these issues have made headlines, while others happened under the radar, but SHM and its members have played an important role within each of them.
In general, observation care and related issues have received increased attention from lawmakers in no small part as a result of SHM’s efforts in this area. Early in the summer, the Centers for Medicare and Medicaid Services (CMS) responded to SHM’s efforts by proposing changes to the two-midnight rule—softening yet still retaining the rule. SHM commented positively on the proposed changes but signaled to both CMS and Congress that still more needs to be done to address problems inherent within observation policy.
As CMS and Congress work on a long-term solution to issues related to observation stays, SHM will play an important role in both proposing solutions and providing feedback for potential changes.
In July 2015, CMS proposed to pay for advance care planning (ACP). This proposal, assuming it is finalized at press time, is one that SHM has advocated for and supported for almost two years. Hospitalists should consider it a victory. The PPC, SHM members, and staff have consistently advocated for improved ACP policies in urging Medicare to recognize the value in ACP and reimburse for these critical services.
Hospitalists have also played a major part in the advocacy efforts on the Congressional level, dispelling misconceptions surrounding ACP by educating members of Congress and their staff and explaining the importance of these conversations within the healthcare system. Consistent advocacy and hospitalist involvement have led to a positive response that has been a long time coming.
These are just a few examples of areas in which SHM has seen tangible accomplishments, but there are also issues that have yet to play out and require SHM’s efforts to remain consistent in the coming years.
One of these issues is a hospitalist-specific problem within the meaningful use program. Hospital-based physicians are exempt from meaningful use and associated penalties based on their percentages of inpatient services; however, due to the way the law was written and changes in the healthcare landscape since passage, an unintended consequence has arisen.
Hospitalists who care for significant numbers of “observation” and skilled nursing facility patients (coded as outpatient services) do not qualify as hospital-based under the law and are finding themselves subject to unfair penalties. In response to SHM advocacy, CMS provided a limited hardship exception for hospitalists in 2015 and extended it into 2016. This exception saved numerous hospitalist groups an estimated $2,000 to $3,000 per hospitalist per year.
The temporary exception was limited in scope, however, and a legislative fix is needed to ensure a permanent solution. SHM will continue to meet with legislators to discuss this issue and garner interest in what we hope will result in a permanent resolution to this issue.
Finally, passage of the Medicare Access and CHIP Reauthorization Act (MACRA) in April 2015 was one of the most important laws impacting physicians to pass in years. While it finally repealed the broken sustainable growth rate, its passage was just the beginning of SHM’s advocacy efforts on the legislation.
MACRA will fundamentally change the way in which physicians are compensated and will accelerate the transition away from fee-for-service (FFS) by encouraging alternative payment model (APM) participation; however, many details remain unclear and are in need of hospitalist-specific clarification. SHM has already begun engaging with federal agencies and lawmakers as the regulations for MACRA are developed, including initiating multi-stakeholder conversations about problems facing facility-based providers in pay-for-performance programs.
SHM’s Performance Measurement and Reporting Committee has been working closely with the PPC to devise concrete proposals that will allow physician/hospital alignment within mandated quality reporting programs, and the PPC has launched an APM workgroup that is exploring the most effective avenues for hospitalists to move away from FFS and take advantage of incentives that will be available under MACRA.
Successful advocacy efforts often take time, persistence, and most importantly, patience; these victories demonstrate that endurance pays off.
SHM is clearly making headway on behalf of hospitalists and patients, and will build off of the momentum that these victories have generated. As SHM Public Policy Committee chair, Ron Greeno, stated after SHM’s most recent victories, “There are times when we all ask if our efforts are all worth it, and the clear message that we have heard in the past few weeks is a resounding ‘Yes!’”
Success does take time, however, and help from SHM members is a critical part of the equation. Your voice does make a difference.
To stay up to date and get involved with SHM’s advocacy efforts, connect with SHM’s Grassroots Network at www.hospitalmedicine.org/grassroots, and join the policy discussions in the Advocacy and Public Policy community on HMX.
Josh Boswell is SHM’s director of government relations.
Laws and regulations regularly impact hospitalists and hospitalized patients, which is why SHM’s Advocacy Department and Public Policy Committee (PPC) work on behalf of SHM membership, the hospital medicine movement, and its patients.
This year has seen significant positive movement within several key policy areas for all of these groups. Some of these issues have made headlines, while others happened under the radar, but SHM and its members have played an important role within each of them.
In general, observation care and related issues have received increased attention from lawmakers in no small part as a result of SHM’s efforts in this area. Early in the summer, the Centers for Medicare and Medicaid Services (CMS) responded to SHM’s efforts by proposing changes to the two-midnight rule—softening yet still retaining the rule. SHM commented positively on the proposed changes but signaled to both CMS and Congress that still more needs to be done to address problems inherent within observation policy.
As CMS and Congress work on a long-term solution to issues related to observation stays, SHM will play an important role in both proposing solutions and providing feedback for potential changes.
In July 2015, CMS proposed to pay for advance care planning (ACP). This proposal, assuming it is finalized at press time, is one that SHM has advocated for and supported for almost two years. Hospitalists should consider it a victory. The PPC, SHM members, and staff have consistently advocated for improved ACP policies in urging Medicare to recognize the value in ACP and reimburse for these critical services.
Hospitalists have also played a major part in the advocacy efforts on the Congressional level, dispelling misconceptions surrounding ACP by educating members of Congress and their staff and explaining the importance of these conversations within the healthcare system. Consistent advocacy and hospitalist involvement have led to a positive response that has been a long time coming.
These are just a few examples of areas in which SHM has seen tangible accomplishments, but there are also issues that have yet to play out and require SHM’s efforts to remain consistent in the coming years.
One of these issues is a hospitalist-specific problem within the meaningful use program. Hospital-based physicians are exempt from meaningful use and associated penalties based on their percentages of inpatient services; however, due to the way the law was written and changes in the healthcare landscape since passage, an unintended consequence has arisen.
Hospitalists who care for significant numbers of “observation” and skilled nursing facility patients (coded as outpatient services) do not qualify as hospital-based under the law and are finding themselves subject to unfair penalties. In response to SHM advocacy, CMS provided a limited hardship exception for hospitalists in 2015 and extended it into 2016. This exception saved numerous hospitalist groups an estimated $2,000 to $3,000 per hospitalist per year.
The temporary exception was limited in scope, however, and a legislative fix is needed to ensure a permanent solution. SHM will continue to meet with legislators to discuss this issue and garner interest in what we hope will result in a permanent resolution to this issue.
Finally, passage of the Medicare Access and CHIP Reauthorization Act (MACRA) in April 2015 was one of the most important laws impacting physicians to pass in years. While it finally repealed the broken sustainable growth rate, its passage was just the beginning of SHM’s advocacy efforts on the legislation.
MACRA will fundamentally change the way in which physicians are compensated and will accelerate the transition away from fee-for-service (FFS) by encouraging alternative payment model (APM) participation; however, many details remain unclear and are in need of hospitalist-specific clarification. SHM has already begun engaging with federal agencies and lawmakers as the regulations for MACRA are developed, including initiating multi-stakeholder conversations about problems facing facility-based providers in pay-for-performance programs.
SHM’s Performance Measurement and Reporting Committee has been working closely with the PPC to devise concrete proposals that will allow physician/hospital alignment within mandated quality reporting programs, and the PPC has launched an APM workgroup that is exploring the most effective avenues for hospitalists to move away from FFS and take advantage of incentives that will be available under MACRA.
Successful advocacy efforts often take time, persistence, and most importantly, patience; these victories demonstrate that endurance pays off.
SHM is clearly making headway on behalf of hospitalists and patients, and will build off of the momentum that these victories have generated. As SHM Public Policy Committee chair, Ron Greeno, stated after SHM’s most recent victories, “There are times when we all ask if our efforts are all worth it, and the clear message that we have heard in the past few weeks is a resounding ‘Yes!’”
Success does take time, however, and help from SHM members is a critical part of the equation. Your voice does make a difference.
To stay up to date and get involved with SHM’s advocacy efforts, connect with SHM’s Grassroots Network at www.hospitalmedicine.org/grassroots, and join the policy discussions in the Advocacy and Public Policy community on HMX.
Josh Boswell is SHM’s director of government relations.
Laws and regulations regularly impact hospitalists and hospitalized patients, which is why SHM’s Advocacy Department and Public Policy Committee (PPC) work on behalf of SHM membership, the hospital medicine movement, and its patients.
This year has seen significant positive movement within several key policy areas for all of these groups. Some of these issues have made headlines, while others happened under the radar, but SHM and its members have played an important role within each of them.
In general, observation care and related issues have received increased attention from lawmakers in no small part as a result of SHM’s efforts in this area. Early in the summer, the Centers for Medicare and Medicaid Services (CMS) responded to SHM’s efforts by proposing changes to the two-midnight rule—softening yet still retaining the rule. SHM commented positively on the proposed changes but signaled to both CMS and Congress that still more needs to be done to address problems inherent within observation policy.
As CMS and Congress work on a long-term solution to issues related to observation stays, SHM will play an important role in both proposing solutions and providing feedback for potential changes.
In July 2015, CMS proposed to pay for advance care planning (ACP). This proposal, assuming it is finalized at press time, is one that SHM has advocated for and supported for almost two years. Hospitalists should consider it a victory. The PPC, SHM members, and staff have consistently advocated for improved ACP policies in urging Medicare to recognize the value in ACP and reimburse for these critical services.
Hospitalists have also played a major part in the advocacy efforts on the Congressional level, dispelling misconceptions surrounding ACP by educating members of Congress and their staff and explaining the importance of these conversations within the healthcare system. Consistent advocacy and hospitalist involvement have led to a positive response that has been a long time coming.
These are just a few examples of areas in which SHM has seen tangible accomplishments, but there are also issues that have yet to play out and require SHM’s efforts to remain consistent in the coming years.
One of these issues is a hospitalist-specific problem within the meaningful use program. Hospital-based physicians are exempt from meaningful use and associated penalties based on their percentages of inpatient services; however, due to the way the law was written and changes in the healthcare landscape since passage, an unintended consequence has arisen.
Hospitalists who care for significant numbers of “observation” and skilled nursing facility patients (coded as outpatient services) do not qualify as hospital-based under the law and are finding themselves subject to unfair penalties. In response to SHM advocacy, CMS provided a limited hardship exception for hospitalists in 2015 and extended it into 2016. This exception saved numerous hospitalist groups an estimated $2,000 to $3,000 per hospitalist per year.
The temporary exception was limited in scope, however, and a legislative fix is needed to ensure a permanent solution. SHM will continue to meet with legislators to discuss this issue and garner interest in what we hope will result in a permanent resolution to this issue.
Finally, passage of the Medicare Access and CHIP Reauthorization Act (MACRA) in April 2015 was one of the most important laws impacting physicians to pass in years. While it finally repealed the broken sustainable growth rate, its passage was just the beginning of SHM’s advocacy efforts on the legislation.
MACRA will fundamentally change the way in which physicians are compensated and will accelerate the transition away from fee-for-service (FFS) by encouraging alternative payment model (APM) participation; however, many details remain unclear and are in need of hospitalist-specific clarification. SHM has already begun engaging with federal agencies and lawmakers as the regulations for MACRA are developed, including initiating multi-stakeholder conversations about problems facing facility-based providers in pay-for-performance programs.
SHM’s Performance Measurement and Reporting Committee has been working closely with the PPC to devise concrete proposals that will allow physician/hospital alignment within mandated quality reporting programs, and the PPC has launched an APM workgroup that is exploring the most effective avenues for hospitalists to move away from FFS and take advantage of incentives that will be available under MACRA.
Successful advocacy efforts often take time, persistence, and most importantly, patience; these victories demonstrate that endurance pays off.
SHM is clearly making headway on behalf of hospitalists and patients, and will build off of the momentum that these victories have generated. As SHM Public Policy Committee chair, Ron Greeno, stated after SHM’s most recent victories, “There are times when we all ask if our efforts are all worth it, and the clear message that we have heard in the past few weeks is a resounding ‘Yes!’”
Success does take time, however, and help from SHM members is a critical part of the equation. Your voice does make a difference.
To stay up to date and get involved with SHM’s advocacy efforts, connect with SHM’s Grassroots Network at www.hospitalmedicine.org/grassroots, and join the policy discussions in the Advocacy and Public Policy community on HMX.
Josh Boswell is SHM’s director of government relations.
Gray matter of first-degree bipolar-patient relatives same as general population
Unaffected first-degree relatives of bipolar disorder patients show no differences in gray-matter volume compared with other healthy adults, Dr. Fabiano G. Nery of the University of São Paulo and colleagues reported.
Investigators took magnetic resonance images of the brains of 25 patients with bipolar disorder, 23 unaffected relatives, and 27 healthy controls recruited from outpatient facilities at the university and the local community. The total gray-matter volume from images was 646.64 mL plus or minus 71.87 among bipolar disorder patients, 645.97 mL plus or minus 48.20 in unaffected relatives, and 637.87 mL plus or minus 62.50 in healthy controls, indicating no significant differences. Bipolar disorder patients, however, had reduced gray-matter volumes in the bilateral thalamus, compared with healthy controls.
This finding was present after controlling for possible confounding effects of age and gender, suggesting that the thalamus “may be involved in the neurocircuitry responsible for the clinical manifestations of” bipolar disorder, they wrote.
“These results suggest that there is no structural endophenotype for [bipolar disorder] and support the role of the thalamus in the pathophysiology of” bipolar disorder, the authors noted.
Read the article in Psychiatry Research: Neuroimaging (doi: 10.1016/j.pscychresns.2015.09.005).
Unaffected first-degree relatives of bipolar disorder patients show no differences in gray-matter volume compared with other healthy adults, Dr. Fabiano G. Nery of the University of São Paulo and colleagues reported.
Investigators took magnetic resonance images of the brains of 25 patients with bipolar disorder, 23 unaffected relatives, and 27 healthy controls recruited from outpatient facilities at the university and the local community. The total gray-matter volume from images was 646.64 mL plus or minus 71.87 among bipolar disorder patients, 645.97 mL plus or minus 48.20 in unaffected relatives, and 637.87 mL plus or minus 62.50 in healthy controls, indicating no significant differences. Bipolar disorder patients, however, had reduced gray-matter volumes in the bilateral thalamus, compared with healthy controls.
This finding was present after controlling for possible confounding effects of age and gender, suggesting that the thalamus “may be involved in the neurocircuitry responsible for the clinical manifestations of” bipolar disorder, they wrote.
“These results suggest that there is no structural endophenotype for [bipolar disorder] and support the role of the thalamus in the pathophysiology of” bipolar disorder, the authors noted.
Read the article in Psychiatry Research: Neuroimaging (doi: 10.1016/j.pscychresns.2015.09.005).
Unaffected first-degree relatives of bipolar disorder patients show no differences in gray-matter volume compared with other healthy adults, Dr. Fabiano G. Nery of the University of São Paulo and colleagues reported.
Investigators took magnetic resonance images of the brains of 25 patients with bipolar disorder, 23 unaffected relatives, and 27 healthy controls recruited from outpatient facilities at the university and the local community. The total gray-matter volume from images was 646.64 mL plus or minus 71.87 among bipolar disorder patients, 645.97 mL plus or minus 48.20 in unaffected relatives, and 637.87 mL plus or minus 62.50 in healthy controls, indicating no significant differences. Bipolar disorder patients, however, had reduced gray-matter volumes in the bilateral thalamus, compared with healthy controls.
This finding was present after controlling for possible confounding effects of age and gender, suggesting that the thalamus “may be involved in the neurocircuitry responsible for the clinical manifestations of” bipolar disorder, they wrote.
“These results suggest that there is no structural endophenotype for [bipolar disorder] and support the role of the thalamus in the pathophysiology of” bipolar disorder, the authors noted.
Read the article in Psychiatry Research: Neuroimaging (doi: 10.1016/j.pscychresns.2015.09.005).
FROM PSYCHIATRY RESEARCH: NEUROIMAGING
Top Trends at Society of Hospital Medicine for 2016

Ready or not, 2016 is almost here, and SHM is gearing up for another year jam packed with exciting, enriching opportunities for hospitalists and their teams. Here are 10 things to have on your radar as we head into the new year.
1 Hospital Medicine 2016: March 6–9, 2016
This year’s annual meeting in San Diego promises to be the biggest yet, with new tracks in Co-Management/Perioperative Medicine, Health IT for Hospitalists, and Post-Acute Care—and opportunities to connect and collaborate with a vibrant community of hospital medicine professionals from around the nation. Register before early bird rates end on Jan. 11, 2016!
2 The Year of the Hospitalist
In celebration of the 20-year anniversary of the coining of the word “hospitalist,” SHM is preparing for a yearlong series of special events, contests, and opportunities. Follow SHM on Twitter at @SHMLive, and visit Hospital Medicine for the latest news!
3 Get Engaged with Public Policy
Healthcare legislation is constantly evolving, and hospitalists play an important role in advocating for hospitalized patients and the hospital medicine movement. SHM is an active voice in many conversations on policy development and reform. Sign up for the Grassroots Network now to stay updated on developments in healthcare policy, share your experiences with healthcare programs, and even participate in policy forums.
4 Fight the Resistance Campaign to Promote Antibiotic Stewardship
SHM is partnering with the CDC to change hospital culture and, in turn, change antibiotic prescription behaviors to prevent antibiotic resistance. Learn how you can be a part of the campaign, and download the campaign posters—featuring striking designs inspired by 1940s propaganda posters.
5 2016 State of Hospital Medicine Survey
The 2016 State of Hospital Medicine survey will take place January through March, with the release of the report scheduled for September. The survey consists of comprehensive, current information crucial to understanding the hospital medicine landscape and making better decisions in the hospital. Visit SHM’s website to find out how you can participate.
6 Expansion of the Quality Improvement Portfolio
SHM’s Center for Hospital Innovation and Improvement continues to develop guides, toolkits, and programs to meet the evolving needs of hospital-based clinicians and improve the care of hospitalized patients. New additions to the portfolio include resources for VTE, chronic heart failure, delirium, anemia, and end-of-life care.
7 SHM Student Hospitalist Scholar Grant Applications
Student members of SHM could be eligible to apply for an SHM Student Hospitalist Scholar Grant, including funding to complete scholarly work with an active SHM mentor in a project related to patient safety, quality improvement, or other hospital medicine-related fields. The deadline to apply is Feb. 15, 2016.
8 Innovative Additions to SHM’s Digital Learning Offerings
In addition to SHM SPARK, an MOC preparation tool for the Focused Practice in Hospital Medicine exam, and SHMConsults, online modules for consultative and perioperative medicine, look for new SHM Learning Portal activities in 2016, like “Management of Postoperative Atrial Fibrillation,” “Managing Pain in Postoperative Patients: What the Hospitalist Needs to Know,” and “Perioperative Bridging of Anticoagulant Theory.”
9 Get #SHeMpowered on Social Media
Have a success story to share about how SHM helped you advance your career or enhance patient care in your hospital? Maybe you improved your clinical skills at the annual meeting or improved care transitions with Project BOOST? Shout it from the rooftops—tweet @SHMLive and use the hashtag #SHeMpowered. If you haven’t followed SHM on Twitter, head over to @SHMLive.
10 “Are You Number 15,000?”
This is a question you want to say “yes” to! SHM is poised to welcome its 15,000th member in early 2016 as part of the Year of the Hospitalist. The 15,000th member will receive an assortment of exciting prizes, including complimentary registration to Hospital Medicine 2016 in San Diego. Know someone who is interested in joining? Spread the word!
Brett Radler is SHM’s communications coordinator.
Ready or not, 2016 is almost here, and SHM is gearing up for another year jam packed with exciting, enriching opportunities for hospitalists and their teams. Here are 10 things to have on your radar as we head into the new year.
1 Hospital Medicine 2016: March 6–9, 2016
This year’s annual meeting in San Diego promises to be the biggest yet, with new tracks in Co-Management/Perioperative Medicine, Health IT for Hospitalists, and Post-Acute Care—and opportunities to connect and collaborate with a vibrant community of hospital medicine professionals from around the nation. Register before early bird rates end on Jan. 11, 2016!
2 The Year of the Hospitalist
In celebration of the 20-year anniversary of the coining of the word “hospitalist,” SHM is preparing for a yearlong series of special events, contests, and opportunities. Follow SHM on Twitter at @SHMLive, and visit Hospital Medicine for the latest news!
3 Get Engaged with Public Policy
Healthcare legislation is constantly evolving, and hospitalists play an important role in advocating for hospitalized patients and the hospital medicine movement. SHM is an active voice in many conversations on policy development and reform. Sign up for the Grassroots Network now to stay updated on developments in healthcare policy, share your experiences with healthcare programs, and even participate in policy forums.
4 Fight the Resistance Campaign to Promote Antibiotic Stewardship
SHM is partnering with the CDC to change hospital culture and, in turn, change antibiotic prescription behaviors to prevent antibiotic resistance. Learn how you can be a part of the campaign, and download the campaign posters—featuring striking designs inspired by 1940s propaganda posters.
5 2016 State of Hospital Medicine Survey
The 2016 State of Hospital Medicine survey will take place January through March, with the release of the report scheduled for September. The survey consists of comprehensive, current information crucial to understanding the hospital medicine landscape and making better decisions in the hospital. Visit SHM’s website to find out how you can participate.
6 Expansion of the Quality Improvement Portfolio
SHM’s Center for Hospital Innovation and Improvement continues to develop guides, toolkits, and programs to meet the evolving needs of hospital-based clinicians and improve the care of hospitalized patients. New additions to the portfolio include resources for VTE, chronic heart failure, delirium, anemia, and end-of-life care.
7 SHM Student Hospitalist Scholar Grant Applications
Student members of SHM could be eligible to apply for an SHM Student Hospitalist Scholar Grant, including funding to complete scholarly work with an active SHM mentor in a project related to patient safety, quality improvement, or other hospital medicine-related fields. The deadline to apply is Feb. 15, 2016.
8 Innovative Additions to SHM’s Digital Learning Offerings
In addition to SHM SPARK, an MOC preparation tool for the Focused Practice in Hospital Medicine exam, and SHMConsults, online modules for consultative and perioperative medicine, look for new SHM Learning Portal activities in 2016, like “Management of Postoperative Atrial Fibrillation,” “Managing Pain in Postoperative Patients: What the Hospitalist Needs to Know,” and “Perioperative Bridging of Anticoagulant Theory.”
9 Get #SHeMpowered on Social Media
Have a success story to share about how SHM helped you advance your career or enhance patient care in your hospital? Maybe you improved your clinical skills at the annual meeting or improved care transitions with Project BOOST? Shout it from the rooftops—tweet @SHMLive and use the hashtag #SHeMpowered. If you haven’t followed SHM on Twitter, head over to @SHMLive.
10 “Are You Number 15,000?”
This is a question you want to say “yes” to! SHM is poised to welcome its 15,000th member in early 2016 as part of the Year of the Hospitalist. The 15,000th member will receive an assortment of exciting prizes, including complimentary registration to Hospital Medicine 2016 in San Diego. Know someone who is interested in joining? Spread the word!
Brett Radler is SHM’s communications coordinator.
Ready or not, 2016 is almost here, and SHM is gearing up for another year jam packed with exciting, enriching opportunities for hospitalists and their teams. Here are 10 things to have on your radar as we head into the new year.
1 Hospital Medicine 2016: March 6–9, 2016
This year’s annual meeting in San Diego promises to be the biggest yet, with new tracks in Co-Management/Perioperative Medicine, Health IT for Hospitalists, and Post-Acute Care—and opportunities to connect and collaborate with a vibrant community of hospital medicine professionals from around the nation. Register before early bird rates end on Jan. 11, 2016!
2 The Year of the Hospitalist
In celebration of the 20-year anniversary of the coining of the word “hospitalist,” SHM is preparing for a yearlong series of special events, contests, and opportunities. Follow SHM on Twitter at @SHMLive, and visit Hospital Medicine for the latest news!
3 Get Engaged with Public Policy
Healthcare legislation is constantly evolving, and hospitalists play an important role in advocating for hospitalized patients and the hospital medicine movement. SHM is an active voice in many conversations on policy development and reform. Sign up for the Grassroots Network now to stay updated on developments in healthcare policy, share your experiences with healthcare programs, and even participate in policy forums.
4 Fight the Resistance Campaign to Promote Antibiotic Stewardship
SHM is partnering with the CDC to change hospital culture and, in turn, change antibiotic prescription behaviors to prevent antibiotic resistance. Learn how you can be a part of the campaign, and download the campaign posters—featuring striking designs inspired by 1940s propaganda posters.
5 2016 State of Hospital Medicine Survey
The 2016 State of Hospital Medicine survey will take place January through March, with the release of the report scheduled for September. The survey consists of comprehensive, current information crucial to understanding the hospital medicine landscape and making better decisions in the hospital. Visit SHM’s website to find out how you can participate.
6 Expansion of the Quality Improvement Portfolio
SHM’s Center for Hospital Innovation and Improvement continues to develop guides, toolkits, and programs to meet the evolving needs of hospital-based clinicians and improve the care of hospitalized patients. New additions to the portfolio include resources for VTE, chronic heart failure, delirium, anemia, and end-of-life care.
7 SHM Student Hospitalist Scholar Grant Applications
Student members of SHM could be eligible to apply for an SHM Student Hospitalist Scholar Grant, including funding to complete scholarly work with an active SHM mentor in a project related to patient safety, quality improvement, or other hospital medicine-related fields. The deadline to apply is Feb. 15, 2016.
8 Innovative Additions to SHM’s Digital Learning Offerings
In addition to SHM SPARK, an MOC preparation tool for the Focused Practice in Hospital Medicine exam, and SHMConsults, online modules for consultative and perioperative medicine, look for new SHM Learning Portal activities in 2016, like “Management of Postoperative Atrial Fibrillation,” “Managing Pain in Postoperative Patients: What the Hospitalist Needs to Know,” and “Perioperative Bridging of Anticoagulant Theory.”
9 Get #SHeMpowered on Social Media
Have a success story to share about how SHM helped you advance your career or enhance patient care in your hospital? Maybe you improved your clinical skills at the annual meeting or improved care transitions with Project BOOST? Shout it from the rooftops—tweet @SHMLive and use the hashtag #SHeMpowered. If you haven’t followed SHM on Twitter, head over to @SHMLive.
10 “Are You Number 15,000?”
This is a question you want to say “yes” to! SHM is poised to welcome its 15,000th member in early 2016 as part of the Year of the Hospitalist. The 15,000th member will receive an assortment of exciting prizes, including complimentary registration to Hospital Medicine 2016 in San Diego. Know someone who is interested in joining? Spread the word!
Brett Radler is SHM’s communications coordinator.
ICD-10 Flexibility Helps Transition to New Coding Systems, Principles, Payer Policy Requirements
Effective October 1, providers submit claims with ICD-10-CM codes. As they adapt to this new system, physicians, clinical staff, and billers should be relying on feedback from each other to achieve a successful transition. On July 6, the Centers for Medicare and Medicaid Services (CMS), in conjunction with the AMA, issued a letter to the provider community offering ICD-10-CM guidance. The joint announcement and guidance regarding ICD-10 flexibilities minimizes the anxiety that often accompanies change and clarifies a few key points about claim scrutiny.1
According to the correspondence, “CMS is releasing additional guidance that will allow for flexibility in the claims auditing and quality reporting process as the medical community gains experience using the new ICD-10 code set.”1 The guidance specifies the flexibility that will be used during the first 12 months of ICD-10-CM use.
This “flexibility” is an opportunity and should not be disregarded. Physician practices can effectively use this time to become accustomed to the ICD-10-CM system, correct coding principles, and payer policy requirements. Internal audit and review processes should increase in order to correct or confirm appropriate coding and claim submission.
Valid Codes
Medicare review contractors are instructed “not to deny physician or other practitioner claims billed under the Part B physician fee schedule through either automated medical review or complex medical review based solely on the specificity of the ICD-10 diagnosis code as long as the physician/practitioner used a valid code from the right family.”2 This “flexibility” will only occur for the first 12 months of ICD-10-CM implementation; the ultimate goal is for providers to assign the correct diagnosis code and the appropriate level of specificity after one year.
The “family code” allowance should not be confused with provision of an incomplete or truncated diagnosis code; these types of codes will always result in claim denial. The ICD-10-CM code presented on the claim form must be carried out to the highest character available for that particular code.
For example, an initial encounter involving an infected peripherally inserted central catheter (PICC) is reported with ICD-10-CM T80.212A (local infection due to central venous catheter). An individual unfamiliar with ICD-10-CM nomenclature may not realize that the seventh extension character of the code is required to carry the code out to its highest level of specificity. If T880.212 is mistakenly reported because the encounter detail (i.e., initial encounter [A], subsequent encounter [D], or sequela [S]) was not documented or provided to the biller, the payers’ claim edit system will identify this as a truncated or invalid diagnosis and reject the claim. Therefore, the code is required to be complete. The “flexibility” refers to reporting the code that best reflects the documented condition. As long as the reported code comes from the same family of codes and is valid, the claim cannot be denied.
Code Families
Code families are “codes within a category [that] are clinically related and provide differences in capturing specific information on the type of condition.”3 Upon review, Medicare will allow ICD-10-CM codes from the same code family to be reported on the claim without penalty if the most accurate code is not selected.
For example, a patient with COPD with acute exacerbation is admitted to the hospital. During the 12-month “flexibility” period, the claim could include J44.9 (COPD, unspecified) without being considered erroneous. The most appropriate code, however, is J44.1 (COPD with acute exacerbation). During the course of the hospitalization, if the physician determines that the COPD exacerbation was caused by an acute lower respiratory infection, J44.0 (COPD with acute lower respiratory infection) is the best option.
The provider goal for this flexibility period is to identify all of the “unspecified codes” used on their claims, review the documentation, and determine the most appropriate code. The practice staff assigned to this task would then provide feedback to the physicians to enhance their future reporting strategies. Although “unspecified” codes are often reported by default, physicians and staff should attempt to reduce usage of this code type unless the patient’s condition is unable to be further specified or categorized at a given point in time.
For example, it would not be acceptable to report R10.8 (unspecified abdominal pain) when a more specific diagnosis code can be easily determined by patient history or exam findings (e.g. right upper quadrant abdominal pain, R10.11).
Affected Claims
As previously stated, “Medicare review contractors will not deny physician or other practitioner claims billed under the Part B physician fee schedule through either automated medical review or complex medical record review.”3 The review contractors included are as follows:
- Medicare Administrative Contractors (MACs) process claims submitted by physicians, hospitals, and other healthcare professionals and submit payment to those providers according to Medicare rules and regulations (including identifying and correcting underpayments and overpayments);
- Recovery Auditors (RACs) review claims to identify potential underpayments and overpayments in Medicare fee-for-service, as part of the Recovery Audit Program;
- Zone Program Integrity Contractors (ZPICs) perform investigations that are unique and tailored to the specific circumstances and occur only in situations where there is potential fraud and take appropriate corrective actions; and
- Supplemental Medical Review Contractor (SMRCs) conduct nationwide medical review as directed by CMS (including identifying underpayments and overpayments).4
This instruction applies to claims that are typically selected for review due to the ICD-10-CM code used on the claim but does not affect claims that are selected for review for other reasons (e.g. modifier 25 [separately identifiable visit performed on the same day as another procedure or service], unbundling, service-specific current procedural terminology code). If a claim is selected for one of these other reasons and does not meet the corresponding criterion, the service will be denied. This instruction also excludes claims for services that correspond to an existing local coverage determination (LCD) or national coverage determination (NCD).
For example, an esophagogastroduodenoscopy (EGD) is not considered “medically necessary” when reported with R10.8 (unspecified abdominal pain) and would be denied. EGD requires a more specific diagnosis (e.g. right upper quadrant abdominal pain, R10.11) per Medicare LCD.
Non-Medicare Payer Considerations
Most payers that are required to convert to ICD-10-CM have also provided some guidance about claim submission. Although most do not address the audit and review process, payers will follow some basic principles:
- Claims submitted with service dates on or after October 1 must use ICD-10-CM codes.
- Claims submitted with service dates prior to October 1 must use ICD-9-CM codes; this includes claims that are initially submitted after October 1 or require correction and resubmission after October 1.
- Physician claims will be held to medical necessity guidelines identified by specific ICD-10-CM codes represented in existing payer policies.
- General equivalence mappings (GEMs) should only be used as a starting point to convert large databases and large code lists from ICD-9 to ICD-10. Many ICD-9-CM codes do not crosswalk directly to an ICD-10-CM code. Physician and staff should continue to use the ICD-10-CM coding books and resources to determine the most accurate code selection.
- “Unspecified” codes are only for use when the information in the medical record is insufficient to assign a more specific code.5,6,7
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is on the faculty of SHM’s inpatient coding course.
References
- Centers for Medicare and Medicaid Services. CMS and AMA announce efforts to help providers get ready for ICD-10. July 6, 2015. Accessed October 3, 2015.
- Centers for Medicare and Medicaid Services. CMS and AMA announce efforts to help providers get ready for ICD-10: frequently asked questions. Accessed October 3, 2015.
- Centers for Medicare and Medicaid Services. Clarifying questions and answers related to the July 6, 2015 CMS/AMA joint announcement and guidance regarding ICD-10 flexibilities. Accessed October 3, 2015.
- Centers for Medicare and Medicaid Services. Medicare Learning Network: Medicare claim review programs. May 2015. Accessed October 3, 2015.
- Aetna. Preparation for ICD-10-CM: frequently asked questions. Accessed October 3, 2015.
- Independence Blue Cross. Transition to ICD-10: frequently asked questions. Accessed October 3, 2015.
- Cigna. Ready, Set, Switch: Know Your ICD-10 Codes. Accessed November 16, 2015.
Effective October 1, providers submit claims with ICD-10-CM codes. As they adapt to this new system, physicians, clinical staff, and billers should be relying on feedback from each other to achieve a successful transition. On July 6, the Centers for Medicare and Medicaid Services (CMS), in conjunction with the AMA, issued a letter to the provider community offering ICD-10-CM guidance. The joint announcement and guidance regarding ICD-10 flexibilities minimizes the anxiety that often accompanies change and clarifies a few key points about claim scrutiny.1
According to the correspondence, “CMS is releasing additional guidance that will allow for flexibility in the claims auditing and quality reporting process as the medical community gains experience using the new ICD-10 code set.”1 The guidance specifies the flexibility that will be used during the first 12 months of ICD-10-CM use.
This “flexibility” is an opportunity and should not be disregarded. Physician practices can effectively use this time to become accustomed to the ICD-10-CM system, correct coding principles, and payer policy requirements. Internal audit and review processes should increase in order to correct or confirm appropriate coding and claim submission.
Valid Codes
Medicare review contractors are instructed “not to deny physician or other practitioner claims billed under the Part B physician fee schedule through either automated medical review or complex medical review based solely on the specificity of the ICD-10 diagnosis code as long as the physician/practitioner used a valid code from the right family.”2 This “flexibility” will only occur for the first 12 months of ICD-10-CM implementation; the ultimate goal is for providers to assign the correct diagnosis code and the appropriate level of specificity after one year.
The “family code” allowance should not be confused with provision of an incomplete or truncated diagnosis code; these types of codes will always result in claim denial. The ICD-10-CM code presented on the claim form must be carried out to the highest character available for that particular code.
For example, an initial encounter involving an infected peripherally inserted central catheter (PICC) is reported with ICD-10-CM T80.212A (local infection due to central venous catheter). An individual unfamiliar with ICD-10-CM nomenclature may not realize that the seventh extension character of the code is required to carry the code out to its highest level of specificity. If T880.212 is mistakenly reported because the encounter detail (i.e., initial encounter [A], subsequent encounter [D], or sequela [S]) was not documented or provided to the biller, the payers’ claim edit system will identify this as a truncated or invalid diagnosis and reject the claim. Therefore, the code is required to be complete. The “flexibility” refers to reporting the code that best reflects the documented condition. As long as the reported code comes from the same family of codes and is valid, the claim cannot be denied.
Code Families
Code families are “codes within a category [that] are clinically related and provide differences in capturing specific information on the type of condition.”3 Upon review, Medicare will allow ICD-10-CM codes from the same code family to be reported on the claim without penalty if the most accurate code is not selected.
For example, a patient with COPD with acute exacerbation is admitted to the hospital. During the 12-month “flexibility” period, the claim could include J44.9 (COPD, unspecified) without being considered erroneous. The most appropriate code, however, is J44.1 (COPD with acute exacerbation). During the course of the hospitalization, if the physician determines that the COPD exacerbation was caused by an acute lower respiratory infection, J44.0 (COPD with acute lower respiratory infection) is the best option.
The provider goal for this flexibility period is to identify all of the “unspecified codes” used on their claims, review the documentation, and determine the most appropriate code. The practice staff assigned to this task would then provide feedback to the physicians to enhance their future reporting strategies. Although “unspecified” codes are often reported by default, physicians and staff should attempt to reduce usage of this code type unless the patient’s condition is unable to be further specified or categorized at a given point in time.
For example, it would not be acceptable to report R10.8 (unspecified abdominal pain) when a more specific diagnosis code can be easily determined by patient history or exam findings (e.g. right upper quadrant abdominal pain, R10.11).
Affected Claims
As previously stated, “Medicare review contractors will not deny physician or other practitioner claims billed under the Part B physician fee schedule through either automated medical review or complex medical record review.”3 The review contractors included are as follows:
- Medicare Administrative Contractors (MACs) process claims submitted by physicians, hospitals, and other healthcare professionals and submit payment to those providers according to Medicare rules and regulations (including identifying and correcting underpayments and overpayments);
- Recovery Auditors (RACs) review claims to identify potential underpayments and overpayments in Medicare fee-for-service, as part of the Recovery Audit Program;
- Zone Program Integrity Contractors (ZPICs) perform investigations that are unique and tailored to the specific circumstances and occur only in situations where there is potential fraud and take appropriate corrective actions; and
- Supplemental Medical Review Contractor (SMRCs) conduct nationwide medical review as directed by CMS (including identifying underpayments and overpayments).4
This instruction applies to claims that are typically selected for review due to the ICD-10-CM code used on the claim but does not affect claims that are selected for review for other reasons (e.g. modifier 25 [separately identifiable visit performed on the same day as another procedure or service], unbundling, service-specific current procedural terminology code). If a claim is selected for one of these other reasons and does not meet the corresponding criterion, the service will be denied. This instruction also excludes claims for services that correspond to an existing local coverage determination (LCD) or national coverage determination (NCD).
For example, an esophagogastroduodenoscopy (EGD) is not considered “medically necessary” when reported with R10.8 (unspecified abdominal pain) and would be denied. EGD requires a more specific diagnosis (e.g. right upper quadrant abdominal pain, R10.11) per Medicare LCD.
Non-Medicare Payer Considerations
Most payers that are required to convert to ICD-10-CM have also provided some guidance about claim submission. Although most do not address the audit and review process, payers will follow some basic principles:
- Claims submitted with service dates on or after October 1 must use ICD-10-CM codes.
- Claims submitted with service dates prior to October 1 must use ICD-9-CM codes; this includes claims that are initially submitted after October 1 or require correction and resubmission after October 1.
- Physician claims will be held to medical necessity guidelines identified by specific ICD-10-CM codes represented in existing payer policies.
- General equivalence mappings (GEMs) should only be used as a starting point to convert large databases and large code lists from ICD-9 to ICD-10. Many ICD-9-CM codes do not crosswalk directly to an ICD-10-CM code. Physician and staff should continue to use the ICD-10-CM coding books and resources to determine the most accurate code selection.
- “Unspecified” codes are only for use when the information in the medical record is insufficient to assign a more specific code.5,6,7
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is on the faculty of SHM’s inpatient coding course.
References
- Centers for Medicare and Medicaid Services. CMS and AMA announce efforts to help providers get ready for ICD-10. July 6, 2015. Accessed October 3, 2015.
- Centers for Medicare and Medicaid Services. CMS and AMA announce efforts to help providers get ready for ICD-10: frequently asked questions. Accessed October 3, 2015.
- Centers for Medicare and Medicaid Services. Clarifying questions and answers related to the July 6, 2015 CMS/AMA joint announcement and guidance regarding ICD-10 flexibilities. Accessed October 3, 2015.
- Centers for Medicare and Medicaid Services. Medicare Learning Network: Medicare claim review programs. May 2015. Accessed October 3, 2015.
- Aetna. Preparation for ICD-10-CM: frequently asked questions. Accessed October 3, 2015.
- Independence Blue Cross. Transition to ICD-10: frequently asked questions. Accessed October 3, 2015.
- Cigna. Ready, Set, Switch: Know Your ICD-10 Codes. Accessed November 16, 2015.
Effective October 1, providers submit claims with ICD-10-CM codes. As they adapt to this new system, physicians, clinical staff, and billers should be relying on feedback from each other to achieve a successful transition. On July 6, the Centers for Medicare and Medicaid Services (CMS), in conjunction with the AMA, issued a letter to the provider community offering ICD-10-CM guidance. The joint announcement and guidance regarding ICD-10 flexibilities minimizes the anxiety that often accompanies change and clarifies a few key points about claim scrutiny.1
According to the correspondence, “CMS is releasing additional guidance that will allow for flexibility in the claims auditing and quality reporting process as the medical community gains experience using the new ICD-10 code set.”1 The guidance specifies the flexibility that will be used during the first 12 months of ICD-10-CM use.
This “flexibility” is an opportunity and should not be disregarded. Physician practices can effectively use this time to become accustomed to the ICD-10-CM system, correct coding principles, and payer policy requirements. Internal audit and review processes should increase in order to correct or confirm appropriate coding and claim submission.
Valid Codes
Medicare review contractors are instructed “not to deny physician or other practitioner claims billed under the Part B physician fee schedule through either automated medical review or complex medical review based solely on the specificity of the ICD-10 diagnosis code as long as the physician/practitioner used a valid code from the right family.”2 This “flexibility” will only occur for the first 12 months of ICD-10-CM implementation; the ultimate goal is for providers to assign the correct diagnosis code and the appropriate level of specificity after one year.
The “family code” allowance should not be confused with provision of an incomplete or truncated diagnosis code; these types of codes will always result in claim denial. The ICD-10-CM code presented on the claim form must be carried out to the highest character available for that particular code.
For example, an initial encounter involving an infected peripherally inserted central catheter (PICC) is reported with ICD-10-CM T80.212A (local infection due to central venous catheter). An individual unfamiliar with ICD-10-CM nomenclature may not realize that the seventh extension character of the code is required to carry the code out to its highest level of specificity. If T880.212 is mistakenly reported because the encounter detail (i.e., initial encounter [A], subsequent encounter [D], or sequela [S]) was not documented or provided to the biller, the payers’ claim edit system will identify this as a truncated or invalid diagnosis and reject the claim. Therefore, the code is required to be complete. The “flexibility” refers to reporting the code that best reflects the documented condition. As long as the reported code comes from the same family of codes and is valid, the claim cannot be denied.
Code Families
Code families are “codes within a category [that] are clinically related and provide differences in capturing specific information on the type of condition.”3 Upon review, Medicare will allow ICD-10-CM codes from the same code family to be reported on the claim without penalty if the most accurate code is not selected.
For example, a patient with COPD with acute exacerbation is admitted to the hospital. During the 12-month “flexibility” period, the claim could include J44.9 (COPD, unspecified) without being considered erroneous. The most appropriate code, however, is J44.1 (COPD with acute exacerbation). During the course of the hospitalization, if the physician determines that the COPD exacerbation was caused by an acute lower respiratory infection, J44.0 (COPD with acute lower respiratory infection) is the best option.
The provider goal for this flexibility period is to identify all of the “unspecified codes” used on their claims, review the documentation, and determine the most appropriate code. The practice staff assigned to this task would then provide feedback to the physicians to enhance their future reporting strategies. Although “unspecified” codes are often reported by default, physicians and staff should attempt to reduce usage of this code type unless the patient’s condition is unable to be further specified or categorized at a given point in time.
For example, it would not be acceptable to report R10.8 (unspecified abdominal pain) when a more specific diagnosis code can be easily determined by patient history or exam findings (e.g. right upper quadrant abdominal pain, R10.11).
Affected Claims
As previously stated, “Medicare review contractors will not deny physician or other practitioner claims billed under the Part B physician fee schedule through either automated medical review or complex medical record review.”3 The review contractors included are as follows:
- Medicare Administrative Contractors (MACs) process claims submitted by physicians, hospitals, and other healthcare professionals and submit payment to those providers according to Medicare rules and regulations (including identifying and correcting underpayments and overpayments);
- Recovery Auditors (RACs) review claims to identify potential underpayments and overpayments in Medicare fee-for-service, as part of the Recovery Audit Program;
- Zone Program Integrity Contractors (ZPICs) perform investigations that are unique and tailored to the specific circumstances and occur only in situations where there is potential fraud and take appropriate corrective actions; and
- Supplemental Medical Review Contractor (SMRCs) conduct nationwide medical review as directed by CMS (including identifying underpayments and overpayments).4
This instruction applies to claims that are typically selected for review due to the ICD-10-CM code used on the claim but does not affect claims that are selected for review for other reasons (e.g. modifier 25 [separately identifiable visit performed on the same day as another procedure or service], unbundling, service-specific current procedural terminology code). If a claim is selected for one of these other reasons and does not meet the corresponding criterion, the service will be denied. This instruction also excludes claims for services that correspond to an existing local coverage determination (LCD) or national coverage determination (NCD).
For example, an esophagogastroduodenoscopy (EGD) is not considered “medically necessary” when reported with R10.8 (unspecified abdominal pain) and would be denied. EGD requires a more specific diagnosis (e.g. right upper quadrant abdominal pain, R10.11) per Medicare LCD.
Non-Medicare Payer Considerations
Most payers that are required to convert to ICD-10-CM have also provided some guidance about claim submission. Although most do not address the audit and review process, payers will follow some basic principles:
- Claims submitted with service dates on or after October 1 must use ICD-10-CM codes.
- Claims submitted with service dates prior to October 1 must use ICD-9-CM codes; this includes claims that are initially submitted after October 1 or require correction and resubmission after October 1.
- Physician claims will be held to medical necessity guidelines identified by specific ICD-10-CM codes represented in existing payer policies.
- General equivalence mappings (GEMs) should only be used as a starting point to convert large databases and large code lists from ICD-9 to ICD-10. Many ICD-9-CM codes do not crosswalk directly to an ICD-10-CM code. Physician and staff should continue to use the ICD-10-CM coding books and resources to determine the most accurate code selection.
- “Unspecified” codes are only for use when the information in the medical record is insufficient to assign a more specific code.5,6,7
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is on the faculty of SHM’s inpatient coding course.
References
- Centers for Medicare and Medicaid Services. CMS and AMA announce efforts to help providers get ready for ICD-10. July 6, 2015. Accessed October 3, 2015.
- Centers for Medicare and Medicaid Services. CMS and AMA announce efforts to help providers get ready for ICD-10: frequently asked questions. Accessed October 3, 2015.
- Centers for Medicare and Medicaid Services. Clarifying questions and answers related to the July 6, 2015 CMS/AMA joint announcement and guidance regarding ICD-10 flexibilities. Accessed October 3, 2015.
- Centers for Medicare and Medicaid Services. Medicare Learning Network: Medicare claim review programs. May 2015. Accessed October 3, 2015.
- Aetna. Preparation for ICD-10-CM: frequently asked questions. Accessed October 3, 2015.
- Independence Blue Cross. Transition to ICD-10: frequently asked questions. Accessed October 3, 2015.
- Cigna. Ready, Set, Switch: Know Your ICD-10 Codes. Accessed November 16, 2015.
CPAP, oral devices reduced blood pressure in sleep apnea
Continuous positive airway pressure (CPAP) and mandibular advancement devices (MADs) both achieved similar reductions in blood pressure in individuals with obstructive sleep apnea, compared with inactive controls.
In a systematic review and meta-analysis of 51 studies involving 4,888 patients, researchers found that CPAP use was associated with a significant mean systolic blood pressure reduction of 2.5 mm Hg and mean diastolic reduction of 2 mm Hg, compared with inactive controls. Each 1-hour increase in mean CPAP use was associated with a significant additional 1.5 mm Hg systolic and 0.9 mm Hg diastolic blood pressure reduction.
Similarly, MADs were associated with a significant 2.1 mm Hg reduction in systolic pressure and 1.9 mm Hg reduction in diastolic pressure, compared with inactive controls.
“This is partly in contrast to a previous meta-analysis, which did not find a beneficial association with MADs, perhaps due to including only two [randomized controlled trials] and thus having inadequate power to detect a difference,” wrote Daniel J. Bratton, Ph.D., of the department of pulmonology, University Hospital, Zurich, and coauthors (JAMA. 2015 Dec 1;314:2280-93).
Overall, the authors found no significant differences between CPAP and MADs in the associated changes in systolic or diastolic blood pressure, although they noted that CPAP showed the strongest association with systolic blood pressure reductions.
The Swiss National Science Foundation and the University of Zurich supported the study. The authors declared no conflicts of interest.
Continuous positive airway pressure (CPAP) and mandibular advancement devices (MADs) both achieved similar reductions in blood pressure in individuals with obstructive sleep apnea, compared with inactive controls.
In a systematic review and meta-analysis of 51 studies involving 4,888 patients, researchers found that CPAP use was associated with a significant mean systolic blood pressure reduction of 2.5 mm Hg and mean diastolic reduction of 2 mm Hg, compared with inactive controls. Each 1-hour increase in mean CPAP use was associated with a significant additional 1.5 mm Hg systolic and 0.9 mm Hg diastolic blood pressure reduction.
Similarly, MADs were associated with a significant 2.1 mm Hg reduction in systolic pressure and 1.9 mm Hg reduction in diastolic pressure, compared with inactive controls.
“This is partly in contrast to a previous meta-analysis, which did not find a beneficial association with MADs, perhaps due to including only two [randomized controlled trials] and thus having inadequate power to detect a difference,” wrote Daniel J. Bratton, Ph.D., of the department of pulmonology, University Hospital, Zurich, and coauthors (JAMA. 2015 Dec 1;314:2280-93).
Overall, the authors found no significant differences between CPAP and MADs in the associated changes in systolic or diastolic blood pressure, although they noted that CPAP showed the strongest association with systolic blood pressure reductions.
The Swiss National Science Foundation and the University of Zurich supported the study. The authors declared no conflicts of interest.
Continuous positive airway pressure (CPAP) and mandibular advancement devices (MADs) both achieved similar reductions in blood pressure in individuals with obstructive sleep apnea, compared with inactive controls.
In a systematic review and meta-analysis of 51 studies involving 4,888 patients, researchers found that CPAP use was associated with a significant mean systolic blood pressure reduction of 2.5 mm Hg and mean diastolic reduction of 2 mm Hg, compared with inactive controls. Each 1-hour increase in mean CPAP use was associated with a significant additional 1.5 mm Hg systolic and 0.9 mm Hg diastolic blood pressure reduction.
Similarly, MADs were associated with a significant 2.1 mm Hg reduction in systolic pressure and 1.9 mm Hg reduction in diastolic pressure, compared with inactive controls.
“This is partly in contrast to a previous meta-analysis, which did not find a beneficial association with MADs, perhaps due to including only two [randomized controlled trials] and thus having inadequate power to detect a difference,” wrote Daniel J. Bratton, Ph.D., of the department of pulmonology, University Hospital, Zurich, and coauthors (JAMA. 2015 Dec 1;314:2280-93).
Overall, the authors found no significant differences between CPAP and MADs in the associated changes in systolic or diastolic blood pressure, although they noted that CPAP showed the strongest association with systolic blood pressure reductions.
The Swiss National Science Foundation and the University of Zurich supported the study. The authors declared no conflicts of interest.
FROM JAMA
Key clinical point: Continuous positive airway pressure and mandibular advancement devices both achieve similar reductions in blood pressure in individuals with obstructive sleep apnea.
Major finding: CPAP use was associated with a mean systolic blood pressure reduction of 2.5 mm Hg, and MADs were associated with a 2.1 mm Hg reduction, compared with inactive controls.
Data source: A systematic review and meta-analysis of 51 studies involving 4,888 patients.
Disclosures: The Swiss National Science Foundation and the University of Zurich supported the study. The authors declared no conflicts of interest.