User login
How to Initiate a Choosing Wisely Project in Your Hospital
Securing institutional support is the first step in any Choosing Wisely project. Evaluate your hospital in the areas covered by Choosing Wisely recommendations. How does the number of transfusions occurring in your hospital compare to national averages? What about number of catheter days and frequency of CAUTIs?
Once you choose an area for improvement, present a strong business case, strongly supported by data and the best projections you can muster, to your hospital’s stakeholders to prove that your proposed initiative will positively impact healthcare costs, quality, and patient safety. Secure institutional support to hold meetings and set specific goals.
And, remember, starting a Choosing Wisely project takes time and commitment. There must be a period of deliberate infrastructure building in which protocols that facilitate Choosing Wisely practices are put in place and metrics for measuring their success are established. Only when this foundation is properly laid can the initiatives be truly and perceptively effective.
Securing institutional support is the first step in any Choosing Wisely project. Evaluate your hospital in the areas covered by Choosing Wisely recommendations. How does the number of transfusions occurring in your hospital compare to national averages? What about number of catheter days and frequency of CAUTIs?
Once you choose an area for improvement, present a strong business case, strongly supported by data and the best projections you can muster, to your hospital’s stakeholders to prove that your proposed initiative will positively impact healthcare costs, quality, and patient safety. Secure institutional support to hold meetings and set specific goals.
And, remember, starting a Choosing Wisely project takes time and commitment. There must be a period of deliberate infrastructure building in which protocols that facilitate Choosing Wisely practices are put in place and metrics for measuring their success are established. Only when this foundation is properly laid can the initiatives be truly and perceptively effective.
Securing institutional support is the first step in any Choosing Wisely project. Evaluate your hospital in the areas covered by Choosing Wisely recommendations. How does the number of transfusions occurring in your hospital compare to national averages? What about number of catheter days and frequency of CAUTIs?
Once you choose an area for improvement, present a strong business case, strongly supported by data and the best projections you can muster, to your hospital’s stakeholders to prove that your proposed initiative will positively impact healthcare costs, quality, and patient safety. Secure institutional support to hold meetings and set specific goals.
And, remember, starting a Choosing Wisely project takes time and commitment. There must be a period of deliberate infrastructure building in which protocols that facilitate Choosing Wisely practices are put in place and metrics for measuring their success are established. Only when this foundation is properly laid can the initiatives be truly and perceptively effective.
ICD-10 Medical Coding System Likely to Improve Documentation, Reimbursement

ICD-10 is the system that will replace ICD-9 for all parties covered by the Health Insurance Portability and Accountability Act (HIPAA). ICD-10 contains a code set used for inpatient procedural reporting and a code set used for diagnosis reporting. Physicians billing for professional services will only be affected when reporting diagnoses codes on their claims, but both physician and hospital selection of ICD-10 codes relies heavily on physician documentation. Therefore, documentation must be scrutinized. The most widely noted impact ICD-10 will have on documentation is increased specificity, with enhanced reporting of the patient’s presenting problem(s). Expanding from a pool of 14,000 3/5-digit codes to 69,000 7-digit codes, and accommodating this change, are daunting tasks. These anticipated burdens make it hard for physicians to recognize the positive effects ICD-10 may create, such as:1
- Better clinical decisions as better data is documented, collected, and evaluated;
- Improved protocol and clinical pathway design for various health conditions;
- Improved public health reporting and tracking of illnesses and severity over time;
- Better definition of patient conditions, providing improved matching of professional resources and care teams and increasing communications between providers;
- Support in practice transition to risk-sharing models with more precise data for patients and populations;
- Provision of clear objective data for credentialing and privileges, and support for professional Maintenance of Certification reporting across specialties;
- Better documentation of patient complexity and level of care, supporting reimbursement and measures for quality and efficiency reporting; and
- Reduction in audit risk exposure by encouraging the use of diagnosis codes with a greater degree of specificity as supported by the clinical documentation.
With the Oct. 1 implementation date rapidly approaching, physicians need to ask themselves, “Am I prepared?”
Getting Started
Everyone has a role and responsibility in transitioning to ICD-10. Active participation by all involved parties guarantees a more successful outcome. Practice administration must ensure that each aspect of implementation is reviewed and appropriately addressed. If not already done, immediate steps should be taken to verify the products and services that affect implementation. These include:
- Payer mix and related contracts: Entities not covered by HIPAA (e.g. workers’ compensation and auto insurance companies) may choose not to implement ICD-10. Since ICD-9 will no longer be maintained post-ICD-10 implementation, it is in the best interest of non-covered entities to use the new coding system.2 For payers who are required to transition to ICD-10, it is important to identify whether patient eligibility, claim processing, and/or payment timelines will be affected, as well as fee schedules or capitated rates.
- Vendor readiness: Physician groups may use a variety of vendors to assist with different aspects of the revenue cycle, including an electronic health record (e.g. documenting services and transmitting physician orders/prescriptions); a practice management system (e.g. scheduling and registering patients); a billing service (e.g. processing patient claims and payments); and a clearinghouse (e.g. verifying patient eligibility and obtaining authorizations). Know when software and/or hardware upgrades are available and if there are additional upgrade fees. Identify vendors that provide support services, training, and tools or templates to ease the transition. Most importantly, inquire about a testing period for products and applications to ensure functionality and adequate feedback on use of the system(s).
- Internal coding and billing resources. Identify physicians and staff who use ICD-9 codes and need to know ICD-10 codes in order to fulfill their responsibilities. Both physicians and staff can assist in identifying common clinical scenarios and the most frequently used ICD-9 codes, in order to develop a list of common ICD-10 specialty codes. Payer coverage policies currently include ICD-10 codes for provider review and comparison. Revise current forms/templates that include diagnosis codes to reflect this updated information. Schedule ICD-10 training for clinicians, office managers, billers, coders, and other key staff. Coding professionals recommend that training take place approximately six months prior to the ICD-10 compliance deadline.3 Training sessions are available from consultants, professional societies, payers, and other entities. Cost varies depending upon the type and length of training. CMS provides some free services, but in-depth training or certification for at least one practice member should be considered.
Once training is completed, dual coding is an option. Dual coding is the process by which both ICD-9 codes and ICD-10 codes are selected during the coding process. Some practices rely on independent selection of each code, while others rely on the General Equivalence Mappings (GEMs). GEMs were developed to assist industry migration to ICD-10. They are intended to be used primarily for translations of code lists or code tables used by an application or other coded data when codes in one code set are the only source of information; they are not intended as a substitution for direct use of ICD-9-CM and ICD-10-CM/PCS.4 Manual coding enhances coding efficiency and also identifies physician documentation deficiencies. Dual coding should begin as soon as possible, prior to October 1.
End-to-end testing is an opportunity to submit test claims to CMS with ICD-10 codes; providers will receive a remittance advice that explains the adjudication of the claims.5 This testing is limited to a small group of providers who were required to register in April, and its final week is July 20-24.
Provide Feedback
The importance of feedback is often understated. Many physician practices do not have the time to plan ahead and, as a result, find themselves in a reactive rather than proactive role. Over the next couple of months, find the time and resources to audit physician documentation based on ICD-10 criteria. Ask yourself whether or not the information contains enough specificity to select the best possible code, or does code selection default to an “unspecified” code?
Avoid “unspecified” codes when possible in preparation for payer policy revisions that are aimed at reducing or eliminating these types of codes. If the documentation lacks detail, educate physicians on the missing elements.
Review ICD-10 code sets with physicians to improve their understanding of the new system. For example, diabetes mellitus is identified in ICD-9 as one category (250.xx), with digits to specify Type I or Type II, controlled vs. uncontrolled, with or without complications. ICD-10 separates diabetes into categories of Type I (E10) or Type II (E11), with subcategories to identify complications and affected body systems, thereby expanding the volume of codes and corresponding documentation criteria.6
Post-implementation feedback will become even more important. Monitor claim denials for invalid codes and medical necessity issues (i.e., valid codes not included for coverage). If the medical necessity denials are a result of inaccurate code selection related to insufficient documentation details, provider education will be crucial in resolving these errors. Continuing education to strengthen and update staff skills is imperative.
CMS has developed many tools and resources to promote a successful transition and assess your ICD-10 preparedness. Physician practices can develop an “action plan,” learn basic ICD-10 concepts, and much more.
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
References
- Centers for Medicare and Medicaid Services. Road to 10: the small physician practice’s route to ICD-10? Accessed June 6, 2015.
- Department of Health and Human Services. Centers for Medicare and Medicaid Services. Medicare Learning Network: ICD-10-CM/PCS Myths and Facts. Accessed June 6, 2015.
- Centers for Medicare and Medicaid Services. ICD-10: ICD-10 Basics for Medical Practices. Accessed June 6, 2015.
- American Health Information Management Association (AHIMA). Putting the ICD-10-CM/PCS GEMs into practice. Accessed June 6, 2015.
- Novitas Solutions. Medicare JL, Part B. ICD-10 Implementation. Accessed June 6, 2015.
- Centers for Medicare and Medicaid Services. ICD-10 Coding and Diabetes. Accessed June 6, 2015.
ICD-10 is the system that will replace ICD-9 for all parties covered by the Health Insurance Portability and Accountability Act (HIPAA). ICD-10 contains a code set used for inpatient procedural reporting and a code set used for diagnosis reporting. Physicians billing for professional services will only be affected when reporting diagnoses codes on their claims, but both physician and hospital selection of ICD-10 codes relies heavily on physician documentation. Therefore, documentation must be scrutinized. The most widely noted impact ICD-10 will have on documentation is increased specificity, with enhanced reporting of the patient’s presenting problem(s). Expanding from a pool of 14,000 3/5-digit codes to 69,000 7-digit codes, and accommodating this change, are daunting tasks. These anticipated burdens make it hard for physicians to recognize the positive effects ICD-10 may create, such as:1
- Better clinical decisions as better data is documented, collected, and evaluated;
- Improved protocol and clinical pathway design for various health conditions;
- Improved public health reporting and tracking of illnesses and severity over time;
- Better definition of patient conditions, providing improved matching of professional resources and care teams and increasing communications between providers;
- Support in practice transition to risk-sharing models with more precise data for patients and populations;
- Provision of clear objective data for credentialing and privileges, and support for professional Maintenance of Certification reporting across specialties;
- Better documentation of patient complexity and level of care, supporting reimbursement and measures for quality and efficiency reporting; and
- Reduction in audit risk exposure by encouraging the use of diagnosis codes with a greater degree of specificity as supported by the clinical documentation.
With the Oct. 1 implementation date rapidly approaching, physicians need to ask themselves, “Am I prepared?”
Getting Started
Everyone has a role and responsibility in transitioning to ICD-10. Active participation by all involved parties guarantees a more successful outcome. Practice administration must ensure that each aspect of implementation is reviewed and appropriately addressed. If not already done, immediate steps should be taken to verify the products and services that affect implementation. These include:
- Payer mix and related contracts: Entities not covered by HIPAA (e.g. workers’ compensation and auto insurance companies) may choose not to implement ICD-10. Since ICD-9 will no longer be maintained post-ICD-10 implementation, it is in the best interest of non-covered entities to use the new coding system.2 For payers who are required to transition to ICD-10, it is important to identify whether patient eligibility, claim processing, and/or payment timelines will be affected, as well as fee schedules or capitated rates.
- Vendor readiness: Physician groups may use a variety of vendors to assist with different aspects of the revenue cycle, including an electronic health record (e.g. documenting services and transmitting physician orders/prescriptions); a practice management system (e.g. scheduling and registering patients); a billing service (e.g. processing patient claims and payments); and a clearinghouse (e.g. verifying patient eligibility and obtaining authorizations). Know when software and/or hardware upgrades are available and if there are additional upgrade fees. Identify vendors that provide support services, training, and tools or templates to ease the transition. Most importantly, inquire about a testing period for products and applications to ensure functionality and adequate feedback on use of the system(s).
- Internal coding and billing resources. Identify physicians and staff who use ICD-9 codes and need to know ICD-10 codes in order to fulfill their responsibilities. Both physicians and staff can assist in identifying common clinical scenarios and the most frequently used ICD-9 codes, in order to develop a list of common ICD-10 specialty codes. Payer coverage policies currently include ICD-10 codes for provider review and comparison. Revise current forms/templates that include diagnosis codes to reflect this updated information. Schedule ICD-10 training for clinicians, office managers, billers, coders, and other key staff. Coding professionals recommend that training take place approximately six months prior to the ICD-10 compliance deadline.3 Training sessions are available from consultants, professional societies, payers, and other entities. Cost varies depending upon the type and length of training. CMS provides some free services, but in-depth training or certification for at least one practice member should be considered.
Once training is completed, dual coding is an option. Dual coding is the process by which both ICD-9 codes and ICD-10 codes are selected during the coding process. Some practices rely on independent selection of each code, while others rely on the General Equivalence Mappings (GEMs). GEMs were developed to assist industry migration to ICD-10. They are intended to be used primarily for translations of code lists or code tables used by an application or other coded data when codes in one code set are the only source of information; they are not intended as a substitution for direct use of ICD-9-CM and ICD-10-CM/PCS.4 Manual coding enhances coding efficiency and also identifies physician documentation deficiencies. Dual coding should begin as soon as possible, prior to October 1.
End-to-end testing is an opportunity to submit test claims to CMS with ICD-10 codes; providers will receive a remittance advice that explains the adjudication of the claims.5 This testing is limited to a small group of providers who were required to register in April, and its final week is July 20-24.
Provide Feedback
The importance of feedback is often understated. Many physician practices do not have the time to plan ahead and, as a result, find themselves in a reactive rather than proactive role. Over the next couple of months, find the time and resources to audit physician documentation based on ICD-10 criteria. Ask yourself whether or not the information contains enough specificity to select the best possible code, or does code selection default to an “unspecified” code?
Avoid “unspecified” codes when possible in preparation for payer policy revisions that are aimed at reducing or eliminating these types of codes. If the documentation lacks detail, educate physicians on the missing elements.
Review ICD-10 code sets with physicians to improve their understanding of the new system. For example, diabetes mellitus is identified in ICD-9 as one category (250.xx), with digits to specify Type I or Type II, controlled vs. uncontrolled, with or without complications. ICD-10 separates diabetes into categories of Type I (E10) or Type II (E11), with subcategories to identify complications and affected body systems, thereby expanding the volume of codes and corresponding documentation criteria.6
Post-implementation feedback will become even more important. Monitor claim denials for invalid codes and medical necessity issues (i.e., valid codes not included for coverage). If the medical necessity denials are a result of inaccurate code selection related to insufficient documentation details, provider education will be crucial in resolving these errors. Continuing education to strengthen and update staff skills is imperative.
CMS has developed many tools and resources to promote a successful transition and assess your ICD-10 preparedness. Physician practices can develop an “action plan,” learn basic ICD-10 concepts, and much more.
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
References
- Centers for Medicare and Medicaid Services. Road to 10: the small physician practice’s route to ICD-10? Accessed June 6, 2015.
- Department of Health and Human Services. Centers for Medicare and Medicaid Services. Medicare Learning Network: ICD-10-CM/PCS Myths and Facts. Accessed June 6, 2015.
- Centers for Medicare and Medicaid Services. ICD-10: ICD-10 Basics for Medical Practices. Accessed June 6, 2015.
- American Health Information Management Association (AHIMA). Putting the ICD-10-CM/PCS GEMs into practice. Accessed June 6, 2015.
- Novitas Solutions. Medicare JL, Part B. ICD-10 Implementation. Accessed June 6, 2015.
- Centers for Medicare and Medicaid Services. ICD-10 Coding and Diabetes. Accessed June 6, 2015.
ICD-10 is the system that will replace ICD-9 for all parties covered by the Health Insurance Portability and Accountability Act (HIPAA). ICD-10 contains a code set used for inpatient procedural reporting and a code set used for diagnosis reporting. Physicians billing for professional services will only be affected when reporting diagnoses codes on their claims, but both physician and hospital selection of ICD-10 codes relies heavily on physician documentation. Therefore, documentation must be scrutinized. The most widely noted impact ICD-10 will have on documentation is increased specificity, with enhanced reporting of the patient’s presenting problem(s). Expanding from a pool of 14,000 3/5-digit codes to 69,000 7-digit codes, and accommodating this change, are daunting tasks. These anticipated burdens make it hard for physicians to recognize the positive effects ICD-10 may create, such as:1
- Better clinical decisions as better data is documented, collected, and evaluated;
- Improved protocol and clinical pathway design for various health conditions;
- Improved public health reporting and tracking of illnesses and severity over time;
- Better definition of patient conditions, providing improved matching of professional resources and care teams and increasing communications between providers;
- Support in practice transition to risk-sharing models with more precise data for patients and populations;
- Provision of clear objective data for credentialing and privileges, and support for professional Maintenance of Certification reporting across specialties;
- Better documentation of patient complexity and level of care, supporting reimbursement and measures for quality and efficiency reporting; and
- Reduction in audit risk exposure by encouraging the use of diagnosis codes with a greater degree of specificity as supported by the clinical documentation.
With the Oct. 1 implementation date rapidly approaching, physicians need to ask themselves, “Am I prepared?”
Getting Started
Everyone has a role and responsibility in transitioning to ICD-10. Active participation by all involved parties guarantees a more successful outcome. Practice administration must ensure that each aspect of implementation is reviewed and appropriately addressed. If not already done, immediate steps should be taken to verify the products and services that affect implementation. These include:
- Payer mix and related contracts: Entities not covered by HIPAA (e.g. workers’ compensation and auto insurance companies) may choose not to implement ICD-10. Since ICD-9 will no longer be maintained post-ICD-10 implementation, it is in the best interest of non-covered entities to use the new coding system.2 For payers who are required to transition to ICD-10, it is important to identify whether patient eligibility, claim processing, and/or payment timelines will be affected, as well as fee schedules or capitated rates.
- Vendor readiness: Physician groups may use a variety of vendors to assist with different aspects of the revenue cycle, including an electronic health record (e.g. documenting services and transmitting physician orders/prescriptions); a practice management system (e.g. scheduling and registering patients); a billing service (e.g. processing patient claims and payments); and a clearinghouse (e.g. verifying patient eligibility and obtaining authorizations). Know when software and/or hardware upgrades are available and if there are additional upgrade fees. Identify vendors that provide support services, training, and tools or templates to ease the transition. Most importantly, inquire about a testing period for products and applications to ensure functionality and adequate feedback on use of the system(s).
- Internal coding and billing resources. Identify physicians and staff who use ICD-9 codes and need to know ICD-10 codes in order to fulfill their responsibilities. Both physicians and staff can assist in identifying common clinical scenarios and the most frequently used ICD-9 codes, in order to develop a list of common ICD-10 specialty codes. Payer coverage policies currently include ICD-10 codes for provider review and comparison. Revise current forms/templates that include diagnosis codes to reflect this updated information. Schedule ICD-10 training for clinicians, office managers, billers, coders, and other key staff. Coding professionals recommend that training take place approximately six months prior to the ICD-10 compliance deadline.3 Training sessions are available from consultants, professional societies, payers, and other entities. Cost varies depending upon the type and length of training. CMS provides some free services, but in-depth training or certification for at least one practice member should be considered.
Once training is completed, dual coding is an option. Dual coding is the process by which both ICD-9 codes and ICD-10 codes are selected during the coding process. Some practices rely on independent selection of each code, while others rely on the General Equivalence Mappings (GEMs). GEMs were developed to assist industry migration to ICD-10. They are intended to be used primarily for translations of code lists or code tables used by an application or other coded data when codes in one code set are the only source of information; they are not intended as a substitution for direct use of ICD-9-CM and ICD-10-CM/PCS.4 Manual coding enhances coding efficiency and also identifies physician documentation deficiencies. Dual coding should begin as soon as possible, prior to October 1.
End-to-end testing is an opportunity to submit test claims to CMS with ICD-10 codes; providers will receive a remittance advice that explains the adjudication of the claims.5 This testing is limited to a small group of providers who were required to register in April, and its final week is July 20-24.
Provide Feedback
The importance of feedback is often understated. Many physician practices do not have the time to plan ahead and, as a result, find themselves in a reactive rather than proactive role. Over the next couple of months, find the time and resources to audit physician documentation based on ICD-10 criteria. Ask yourself whether or not the information contains enough specificity to select the best possible code, or does code selection default to an “unspecified” code?
Avoid “unspecified” codes when possible in preparation for payer policy revisions that are aimed at reducing or eliminating these types of codes. If the documentation lacks detail, educate physicians on the missing elements.
Review ICD-10 code sets with physicians to improve their understanding of the new system. For example, diabetes mellitus is identified in ICD-9 as one category (250.xx), with digits to specify Type I or Type II, controlled vs. uncontrolled, with or without complications. ICD-10 separates diabetes into categories of Type I (E10) or Type II (E11), with subcategories to identify complications and affected body systems, thereby expanding the volume of codes and corresponding documentation criteria.6
Post-implementation feedback will become even more important. Monitor claim denials for invalid codes and medical necessity issues (i.e., valid codes not included for coverage). If the medical necessity denials are a result of inaccurate code selection related to insufficient documentation details, provider education will be crucial in resolving these errors. Continuing education to strengthen and update staff skills is imperative.
CMS has developed many tools and resources to promote a successful transition and assess your ICD-10 preparedness. Physician practices can develop an “action plan,” learn basic ICD-10 concepts, and much more.
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
References
- Centers for Medicare and Medicaid Services. Road to 10: the small physician practice’s route to ICD-10? Accessed June 6, 2015.
- Department of Health and Human Services. Centers for Medicare and Medicaid Services. Medicare Learning Network: ICD-10-CM/PCS Myths and Facts. Accessed June 6, 2015.
- Centers for Medicare and Medicaid Services. ICD-10: ICD-10 Basics for Medical Practices. Accessed June 6, 2015.
- American Health Information Management Association (AHIMA). Putting the ICD-10-CM/PCS GEMs into practice. Accessed June 6, 2015.
- Novitas Solutions. Medicare JL, Part B. ICD-10 Implementation. Accessed June 6, 2015.
- Centers for Medicare and Medicaid Services. ICD-10 Coding and Diabetes. Accessed June 6, 2015.
Post-Acute Patient Care Offers Opportunities for Non-Physicians
More than in the inpatient setting, post-acute care offers opportunities for nurse practitioners and physician assistants to play important clinical and administrative roles. Physician assistant (PA) Edwin Lopez, PA-C, is chief of an eight-member HM group—four doctors, four PAs—that provides coverage at St. Elizabeth, a rural critical access hospital in Enumclaw, Wash., population 10,669, and in the 80-bed SNF located across the street. Lopez was recruited to establish the HM group “in the shadow of Mt. Rainier” about eight years ago, at a time when the hospital’s parent, CHI Franciscan Health System, was trying to rebuild its quality and reputation while planning a new building.
He succeeded, dramatically improving its performance on HCAHPS surveys and other metrics; however, hospital readmissions then emerged as an issue.
“I began to realize, with our little facility’s large population of elderly patients with multiple chronic problems—typically the highest cohort for readmissions—all the gains we had made could be lost if we didn’t do something about this problem,” Lopez says. “I ran the numbers and found that the nursing home across the street readmitted 35% of patients discharged from our hospital.”
It took a year to get the larger system’s approval, but Lopez’s hospitalist group manages all the patients transferred to the nearby nursing home, with daily visits by the doctor and/or PA on duty.
“We started the program in January 2014, and, in one year, readmissions went from 35% down to 7%,” he says. “We developed culturally from the very beginning as a PA/MD collaborative model. The doctor doesn’t need to see the more routine patients with more common conditions but instead is freed up to focus on higher-acuity, more complex patients.”
In Dr. Tollman’s opinion, physician extenders “own” the post-acute realm, because of the demand for their care.
“There just aren’t going to be enough doctors for all of the patients who need to be seen,” he says, “and the amount of money for this care isn’t enough for these facilities to employ groups of doctors.”
Emily Rosenbaum, PA-C, works for Northwest Community Healthcare in Arlington Heights, Ill. She is the lone PA working with eight physician hospitalists. Much of her work is in a rehabilitation facility across the street from Northwest Community Hospital.
“I see all of the new admissions, although under my scope of practice I can’t bill for the initial visit. But I do the follow-ups, see patients that have been in rehab for 30 days or less, and put out [clinical] fires in the facility,” she says.
Rosenbaum works at the hospital part of her day taking care of acute patients, then works with the hospitalist assigned to the rehabilitation facility.
“It’s easier for me to go back and forth and keep my finger on the patients’ pulse,” she says. “As there are more demands on doctors on the acute side, it’s natural for the NP and PA to step up and take a larger role on the post-acute side.”
More than in the inpatient setting, post-acute care offers opportunities for nurse practitioners and physician assistants to play important clinical and administrative roles. Physician assistant (PA) Edwin Lopez, PA-C, is chief of an eight-member HM group—four doctors, four PAs—that provides coverage at St. Elizabeth, a rural critical access hospital in Enumclaw, Wash., population 10,669, and in the 80-bed SNF located across the street. Lopez was recruited to establish the HM group “in the shadow of Mt. Rainier” about eight years ago, at a time when the hospital’s parent, CHI Franciscan Health System, was trying to rebuild its quality and reputation while planning a new building.
He succeeded, dramatically improving its performance on HCAHPS surveys and other metrics; however, hospital readmissions then emerged as an issue.
“I began to realize, with our little facility’s large population of elderly patients with multiple chronic problems—typically the highest cohort for readmissions—all the gains we had made could be lost if we didn’t do something about this problem,” Lopez says. “I ran the numbers and found that the nursing home across the street readmitted 35% of patients discharged from our hospital.”
It took a year to get the larger system’s approval, but Lopez’s hospitalist group manages all the patients transferred to the nearby nursing home, with daily visits by the doctor and/or PA on duty.
“We started the program in January 2014, and, in one year, readmissions went from 35% down to 7%,” he says. “We developed culturally from the very beginning as a PA/MD collaborative model. The doctor doesn’t need to see the more routine patients with more common conditions but instead is freed up to focus on higher-acuity, more complex patients.”
In Dr. Tollman’s opinion, physician extenders “own” the post-acute realm, because of the demand for their care.
“There just aren’t going to be enough doctors for all of the patients who need to be seen,” he says, “and the amount of money for this care isn’t enough for these facilities to employ groups of doctors.”
Emily Rosenbaum, PA-C, works for Northwest Community Healthcare in Arlington Heights, Ill. She is the lone PA working with eight physician hospitalists. Much of her work is in a rehabilitation facility across the street from Northwest Community Hospital.
“I see all of the new admissions, although under my scope of practice I can’t bill for the initial visit. But I do the follow-ups, see patients that have been in rehab for 30 days or less, and put out [clinical] fires in the facility,” she says.
Rosenbaum works at the hospital part of her day taking care of acute patients, then works with the hospitalist assigned to the rehabilitation facility.
“It’s easier for me to go back and forth and keep my finger on the patients’ pulse,” she says. “As there are more demands on doctors on the acute side, it’s natural for the NP and PA to step up and take a larger role on the post-acute side.”
More than in the inpatient setting, post-acute care offers opportunities for nurse practitioners and physician assistants to play important clinical and administrative roles. Physician assistant (PA) Edwin Lopez, PA-C, is chief of an eight-member HM group—four doctors, four PAs—that provides coverage at St. Elizabeth, a rural critical access hospital in Enumclaw, Wash., population 10,669, and in the 80-bed SNF located across the street. Lopez was recruited to establish the HM group “in the shadow of Mt. Rainier” about eight years ago, at a time when the hospital’s parent, CHI Franciscan Health System, was trying to rebuild its quality and reputation while planning a new building.
He succeeded, dramatically improving its performance on HCAHPS surveys and other metrics; however, hospital readmissions then emerged as an issue.
“I began to realize, with our little facility’s large population of elderly patients with multiple chronic problems—typically the highest cohort for readmissions—all the gains we had made could be lost if we didn’t do something about this problem,” Lopez says. “I ran the numbers and found that the nursing home across the street readmitted 35% of patients discharged from our hospital.”
It took a year to get the larger system’s approval, but Lopez’s hospitalist group manages all the patients transferred to the nearby nursing home, with daily visits by the doctor and/or PA on duty.
“We started the program in January 2014, and, in one year, readmissions went from 35% down to 7%,” he says. “We developed culturally from the very beginning as a PA/MD collaborative model. The doctor doesn’t need to see the more routine patients with more common conditions but instead is freed up to focus on higher-acuity, more complex patients.”
In Dr. Tollman’s opinion, physician extenders “own” the post-acute realm, because of the demand for their care.
“There just aren’t going to be enough doctors for all of the patients who need to be seen,” he says, “and the amount of money for this care isn’t enough for these facilities to employ groups of doctors.”
Emily Rosenbaum, PA-C, works for Northwest Community Healthcare in Arlington Heights, Ill. She is the lone PA working with eight physician hospitalists. Much of her work is in a rehabilitation facility across the street from Northwest Community Hospital.
“I see all of the new admissions, although under my scope of practice I can’t bill for the initial visit. But I do the follow-ups, see patients that have been in rehab for 30 days or less, and put out [clinical] fires in the facility,” she says.
Rosenbaum works at the hospital part of her day taking care of acute patients, then works with the hospitalist assigned to the rehabilitation facility.
“It’s easier for me to go back and forth and keep my finger on the patients’ pulse,” she says. “As there are more demands on doctors on the acute side, it’s natural for the NP and PA to step up and take a larger role on the post-acute side.”
Post-Acute Patient Care New Frontier for Hospitalists

In spite of all the gadgets and technologies available to hospital-based physicians nowadays, Jerome Wilborn, MD, FCCP, sees a much simpler symbol of patient care. Dr. Wilborn, national medical director for post-acute care services at IPC Healthcare, Inc., in North Hollywood, Calif., believes stethoscopes are key to post-acute patient care, and hospitalists are the ones “redefining” the practice. It’s not just a metaphor for working in settings that lack access to the specialists, equipment, and other resources of the acute-care hospital, he explains. A stethoscope, he says, reflects on the physicians’ clinical acumen and listening skills.
“Acute-care doctors need to understand that in the post-acute setting, it’s not about ordering labs. It is important to talk to the family,” he says. “Over the next 14 to 30 days, you can really dig into relationships with patients, optimize their medical care, reduce poly-pharmacy, and even prevent readmissions.”
These are among the sickest of patients, with multiple co-morbidities and limitations in activities of daily living (ADLs), Dr. Wilborn notes.
“Many internists and hospitalists who come to the nursing home are astounded by the clinical acuity of the patients and don’t appreciate how, even without the hospital treatment armamentarium they are used to, they can still make a big difference in the care,” he explains. But the key, he adds, is for doctors to go into the facility often enough to have an impact, with regularly scheduled presence and a commitment to standardizing the care.

For hospitalists who are more accustomed to the high-intensity, fast-paced world of the acute hospital, post-acute care may not seem very sexy.
“But that’s changing, along with the medical landscape,” Dr. Wilborn says. “For those who can appreciate the opportunity to build relationships with patients and to practice more independently, it could be a great place to change your career trajectory and have an immediate impact on the quality of patient care.”
What Is the Post-Acute Space?
Although post-acute care could refer broadly to all settings for care following the patient’s discharge from the hospital, including home care, three post-acute settings, defined by their licensure, are more likely to involve physicians such as hospitalists:
- The inpatient rehabilitation facility (IRF) , which is a freestanding rehabilitation unit or hospital inside an acute hospital for patients who need rehabilitation care in order to function effectively and are medically stable and able to participate in rehabilitation therapies;
- The long-term acute-care hospital (LTACH), a hospital that specializes in treatment and recovery of medical patients who require prolonged lengths of stay, typically measured in weeks;
- The The skilled nursing facility (SNF), which focuses on the health, social, and personal needs of chronically ill or disabled patients, either for rehabilitation stays of two weeks to a month or longer stays for chronic illness.
Although hospital medicine began as a practice specific to the inpatient setting, increasing numbers of hospitalists are spending at least part of their working lives outside of the hospital, visiting patients in post-acute settings. IPC is just one of the many national hospitalist management companies, medical groups, and hospital-employed practices that are defining new roles for their physicians, nurse practitioners, and physician assistants in these settings.
The presence of hospitalists in post-acute care is growing, according to the Society of Hospital Medicine, with 25.4% of adult hospital medicine groups in its most recent survey saying that they see patients in post-acute care facilities.1 In response to this trend, SHM in 2012 impaneled the Post-Acute Care Task Force, chaired by Sean Muldoon, MD, MPH, senior vice president and chief medical officer of Kindred Healthcare’s Hospital Division, Louisville, Ky. The task force was formed to help SHM members explore post-acute care and learn about what to expect. The task force developed a toolbox and a transitions quality improvement toolkit, and a new white paper, “Primer for Hospitalists on Skilled Nursing Facilities.”

“Why does it matter to hospitalists?” Dr. Muldoon asks, rhetorically. “Everything in terms of acuity is being pushed down to lower-level settings. If hospitalists think they will only do hospital work in the future, well, they will miss much of inpatient care because of the shift from the hospital to other hospital-like settings.”
At HM15 in late March, the task force outlined its agenda for the coming year, including promotion of its toolkit, development of a web-based CME seminar, and creation of a web-based reference repository.
Scott Rissmiller, MD, chief hospitalist at the 43-hospital Carolinas Healthcare System in Charlotte, N.C., says the transformation now taking place in post-acute care is more than just hospitalists doing some or all of their work in long-term care facilities. Post-acute care is becoming less of a side job for moonlighting hospitalists, with more of a focus on integrated care. Dr. Rissmiller, a member of SHM’s Multi-Site Hospitalist Group Task Force, says hospitalists are bringing to the post-acute arena the same standardization, accountability, and quality improvement the field has brought to hospitals across the country.
For Carolinas Healthcare and other multi-site hospitalist groups, the goal is to elevate the quality of care in LTACHs and other long-term care settings.
“It’s upping the game in post-acute care. It’s looking at the whole continuum of care from a systems perspective, improving handoffs and transitions,” Dr. Rissmiller says. “For years, we tried to improve communication with post-acute providers. It wasn’t until we started partnering with these facilities that we started to see changes.”
Dr. Rissmiller believes the best practice is to have one cohesive team caring for patients in both hospital and post-acute settings, under the leadership of the hospitalist group. The goal is to ensure that patients go to the proper level of care—and only for as long as they need to be there—using the system’s resources correctly.
Not every member of the hospitalist group will go to post-acute care facilities, while others will choose to specialize in that setting, he says, “but they meet every month with their acute care counterparts to work on improving care.”

What Should We Call This?
The amount of medical care being provided by hospitalists in post-acute facilities is growing, experts say, inclusive of physician assistants and nurse practitioners working as part of hospitalist groups. As many as 30% of SHM members are involved in post-acute care, according to the latest SHM survey, with large management groups like IPC, TeamHealth, and Tacoma, Wash.-based Sound Physicians, expanding rapidly in this area.1
“I don’t think that hospitalists have taken over from PCPs in post-acute care in general, but they make up a significant physician cohort,” Dr. Wilborn says.
IPC has a presence today in more than 1,700 post-acute care facilities, with 20% of its physicians working in both acute and post-acute care, more than 2,800 affiliated clinicians, and a third of the company’s revenue coming from the post-acute space, Dr. Wilborn notes.
Interestingly, what to call these providers seems to be a problem.
“Hospitalist groups are employing people who, strictly speaking, aren’t really hospitalists, although the post-acute setting marries up very well with the hospitalist model, mindset, and historical leadership role,” Dr. Rissmiller says. “It requires a different skill set.”
He prefers the term “post-acute specialist.”
Others refer to these providers as “SNF-ists,” although that word doesn’t exactly roll off the tongue, nor does it convey the scope of post-acute care.
“If hospitalists are doctors who round in acute-care hospitals, the parallel term for doctors who round in post-acute facilities is not well established,” Dr. Wilborn says. “It’s site-specific care. I call them post-acute care providers. This certainly is a specialty, for a lot of different reasons. It’s post-acute care medicine, and the hospitalist term isn’t going to stick.”
Scott Sears, MD, FACP, chief clinical officer of Tacoma, Wash.-based Sound Physicians, which has physicians deployed in roughly 100 post-acute settings, labels his providers “transitional care physicians.” Most of them are dedicated full time to post-acute care.
“Our main source of transitional care doctors are former hospitalists who are interested in more than a three-to-seven day relationship with their patients,” Dr. Sears says.
“It’s almost an art in itself,” he adds. “That’s why it’s not in the patient’s best interest to have a doctor who just dabbles in post-acute care. That’s where the dedicated provider with a passion and vision for the work is so valuable. We also have more success with people who have more experience.”
General Medicine PC, a Novi, Mich.-based company of physicians, NPs, and PAs who specialize in treating geriatric and chronically ill patients in long-term care settings, calls itself “the post-hospitalist company.” It claims its primary customers tend to be payers and managed care systems.
“We use the term post-hospitalist, and we are the country’s largest provider of post-hospitalist services,” explains CEO Thomas Prose, MD, MPH, MBA, who founded the company in 1983. “We tailor services to the needs of each accountable care organization, hospital, and integrated health system we contract with, in order to improve patient care and reduce hospital readmissions, ED visits, and overall spending.”
The company has posted readmissions rates lower than 95% of the industry, with higher quality metrics, he says, adding that the majority of General Medicine’s physicians are not transitioning hospitalists but doctors who were drawn to geriatrics and long-term care settings from the outset.
What’s Driving the Post-Acute Space?
A significant portion of healthcare expenditures is in post-acute care, and that money hasn’t always been well spent, experts interviewed for this article emphasized. Without adequate physician involvement in their care, many of these patients would be sent back to the hospital for complications that might have been managed outside of the hospital. Often it is payers, managed care plans, health systems, medical groups, and other risk-bearing entities that are driving the growth of physician involvement in post-acute care—just as insurers had a role in pushing the early growth of hospital medicine—and accountable care organizations (ACOs) are more often acting and contracting like payers.
In fact, spending on post-acute care overall, not just the physician’s role, is growing rapidly enough to attract the concerns of policymakers, reflected in a recent hearing by the U.S. House Energy and Commerce Committee that found drastic variations in payment rates across settings, with overall Medicare spending of $59 billion on post-acute care in 2013.2 The Bundling and Coordinating Post-Acute Care Act, a bill first introduced last year by Rep. David McKinley (D-W. Va.) and reintroduced in 2015 as HR1458, aims to address these growing costs while preserving patient choice by requiring a single bundled payment for post-acute care services under Medicare parts A and B.
Bundled payment is definitely coming, Dr. Rissmiller says, and will fuel the move to inpatient-outpatient partnerships.
“A lot of this work is in preparation for ACOs and bundled payments, even if the new models are not yet dominant in the marketplace.”
Bundling payment for an episode of care, including the hospital stay and all of the post-acute follow-up, will be a game-changer, Dr. Wilborn adds. CMS is now testing bundled payment models and, by 2017, he says they will be an established fact in nursing homes, with half of their reimbursement coming from some kind of bundle.
James Tollman, MD, FHM, heads a small HM group, Essex Inpatient Physicians, which he started in 2007 in Boxford, Mass. Essex includes full- and part-time physicians and physician extenders and has contracts with several hospitals, but Dr. Tollman estimates that 95% of the practice is in post-acute care.
“For us, as a small group in the current environment, it’s a good idea to diversify,” Dr. Tollman says. “It’s important to be flexible and have a foot in many venues.
“I view myself as a hospitalist by personality and history,” he adds. But experience working in post-acute care enables physicians to view the hospital in perspective—as part of the larger continuum of care and not the center of the universe.
Dr. Tollman says risk contracts with ACOs are the new frontier for hospitalists in post-acute care.
“Who manages the money is an important question,” he says. “The quality metrics are still poorly aligned with what the SNF-ist does. Right now, we’re entering contracts with three different ACO-type exchanges. None of them have really figured out what we are about, and we don’t have much leverage yet.”
Larry Beresford is a freelance writer in Alameda, Calif.
References
- Society of Hospital Medicine. 2014 State of Hospital Medicine report. Philadelphia, Pa.; Society of Hospital Medicine; 2014.
- Firth S. House panel considers medicare reform for post-acute care. Medpage Today. April 18, 2015. Accessed June 10, 2015.
In spite of all the gadgets and technologies available to hospital-based physicians nowadays, Jerome Wilborn, MD, FCCP, sees a much simpler symbol of patient care. Dr. Wilborn, national medical director for post-acute care services at IPC Healthcare, Inc., in North Hollywood, Calif., believes stethoscopes are key to post-acute patient care, and hospitalists are the ones “redefining” the practice. It’s not just a metaphor for working in settings that lack access to the specialists, equipment, and other resources of the acute-care hospital, he explains. A stethoscope, he says, reflects on the physicians’ clinical acumen and listening skills.
“Acute-care doctors need to understand that in the post-acute setting, it’s not about ordering labs. It is important to talk to the family,” he says. “Over the next 14 to 30 days, you can really dig into relationships with patients, optimize their medical care, reduce poly-pharmacy, and even prevent readmissions.”
These are among the sickest of patients, with multiple co-morbidities and limitations in activities of daily living (ADLs), Dr. Wilborn notes.
“Many internists and hospitalists who come to the nursing home are astounded by the clinical acuity of the patients and don’t appreciate how, even without the hospital treatment armamentarium they are used to, they can still make a big difference in the care,” he explains. But the key, he adds, is for doctors to go into the facility often enough to have an impact, with regularly scheduled presence and a commitment to standardizing the care.
For hospitalists who are more accustomed to the high-intensity, fast-paced world of the acute hospital, post-acute care may not seem very sexy.
“But that’s changing, along with the medical landscape,” Dr. Wilborn says. “For those who can appreciate the opportunity to build relationships with patients and to practice more independently, it could be a great place to change your career trajectory and have an immediate impact on the quality of patient care.”
What Is the Post-Acute Space?
Although post-acute care could refer broadly to all settings for care following the patient’s discharge from the hospital, including home care, three post-acute settings, defined by their licensure, are more likely to involve physicians such as hospitalists:
- The inpatient rehabilitation facility (IRF) , which is a freestanding rehabilitation unit or hospital inside an acute hospital for patients who need rehabilitation care in order to function effectively and are medically stable and able to participate in rehabilitation therapies;
- The long-term acute-care hospital (LTACH), a hospital that specializes in treatment and recovery of medical patients who require prolonged lengths of stay, typically measured in weeks;
- The The skilled nursing facility (SNF), which focuses on the health, social, and personal needs of chronically ill or disabled patients, either for rehabilitation stays of two weeks to a month or longer stays for chronic illness.
Although hospital medicine began as a practice specific to the inpatient setting, increasing numbers of hospitalists are spending at least part of their working lives outside of the hospital, visiting patients in post-acute settings. IPC is just one of the many national hospitalist management companies, medical groups, and hospital-employed practices that are defining new roles for their physicians, nurse practitioners, and physician assistants in these settings.
The presence of hospitalists in post-acute care is growing, according to the Society of Hospital Medicine, with 25.4% of adult hospital medicine groups in its most recent survey saying that they see patients in post-acute care facilities.1 In response to this trend, SHM in 2012 impaneled the Post-Acute Care Task Force, chaired by Sean Muldoon, MD, MPH, senior vice president and chief medical officer of Kindred Healthcare’s Hospital Division, Louisville, Ky. The task force was formed to help SHM members explore post-acute care and learn about what to expect. The task force developed a toolbox and a transitions quality improvement toolkit, and a new white paper, “Primer for Hospitalists on Skilled Nursing Facilities.”
“Why does it matter to hospitalists?” Dr. Muldoon asks, rhetorically. “Everything in terms of acuity is being pushed down to lower-level settings. If hospitalists think they will only do hospital work in the future, well, they will miss much of inpatient care because of the shift from the hospital to other hospital-like settings.”
At HM15 in late March, the task force outlined its agenda for the coming year, including promotion of its toolkit, development of a web-based CME seminar, and creation of a web-based reference repository.
Scott Rissmiller, MD, chief hospitalist at the 43-hospital Carolinas Healthcare System in Charlotte, N.C., says the transformation now taking place in post-acute care is more than just hospitalists doing some or all of their work in long-term care facilities. Post-acute care is becoming less of a side job for moonlighting hospitalists, with more of a focus on integrated care. Dr. Rissmiller, a member of SHM’s Multi-Site Hospitalist Group Task Force, says hospitalists are bringing to the post-acute arena the same standardization, accountability, and quality improvement the field has brought to hospitals across the country.
For Carolinas Healthcare and other multi-site hospitalist groups, the goal is to elevate the quality of care in LTACHs and other long-term care settings.
“It’s upping the game in post-acute care. It’s looking at the whole continuum of care from a systems perspective, improving handoffs and transitions,” Dr. Rissmiller says. “For years, we tried to improve communication with post-acute providers. It wasn’t until we started partnering with these facilities that we started to see changes.”
Dr. Rissmiller believes the best practice is to have one cohesive team caring for patients in both hospital and post-acute settings, under the leadership of the hospitalist group. The goal is to ensure that patients go to the proper level of care—and only for as long as they need to be there—using the system’s resources correctly.
Not every member of the hospitalist group will go to post-acute care facilities, while others will choose to specialize in that setting, he says, “but they meet every month with their acute care counterparts to work on improving care.”
What Should We Call This?
The amount of medical care being provided by hospitalists in post-acute facilities is growing, experts say, inclusive of physician assistants and nurse practitioners working as part of hospitalist groups. As many as 30% of SHM members are involved in post-acute care, according to the latest SHM survey, with large management groups like IPC, TeamHealth, and Tacoma, Wash.-based Sound Physicians, expanding rapidly in this area.1
“I don’t think that hospitalists have taken over from PCPs in post-acute care in general, but they make up a significant physician cohort,” Dr. Wilborn says.
IPC has a presence today in more than 1,700 post-acute care facilities, with 20% of its physicians working in both acute and post-acute care, more than 2,800 affiliated clinicians, and a third of the company’s revenue coming from the post-acute space, Dr. Wilborn notes.
Interestingly, what to call these providers seems to be a problem.
“Hospitalist groups are employing people who, strictly speaking, aren’t really hospitalists, although the post-acute setting marries up very well with the hospitalist model, mindset, and historical leadership role,” Dr. Rissmiller says. “It requires a different skill set.”
He prefers the term “post-acute specialist.”
Others refer to these providers as “SNF-ists,” although that word doesn’t exactly roll off the tongue, nor does it convey the scope of post-acute care.
“If hospitalists are doctors who round in acute-care hospitals, the parallel term for doctors who round in post-acute facilities is not well established,” Dr. Wilborn says. “It’s site-specific care. I call them post-acute care providers. This certainly is a specialty, for a lot of different reasons. It’s post-acute care medicine, and the hospitalist term isn’t going to stick.”
Scott Sears, MD, FACP, chief clinical officer of Tacoma, Wash.-based Sound Physicians, which has physicians deployed in roughly 100 post-acute settings, labels his providers “transitional care physicians.” Most of them are dedicated full time to post-acute care.
“Our main source of transitional care doctors are former hospitalists who are interested in more than a three-to-seven day relationship with their patients,” Dr. Sears says.
“It’s almost an art in itself,” he adds. “That’s why it’s not in the patient’s best interest to have a doctor who just dabbles in post-acute care. That’s where the dedicated provider with a passion and vision for the work is so valuable. We also have more success with people who have more experience.”
General Medicine PC, a Novi, Mich.-based company of physicians, NPs, and PAs who specialize in treating geriatric and chronically ill patients in long-term care settings, calls itself “the post-hospitalist company.” It claims its primary customers tend to be payers and managed care systems.
“We use the term post-hospitalist, and we are the country’s largest provider of post-hospitalist services,” explains CEO Thomas Prose, MD, MPH, MBA, who founded the company in 1983. “We tailor services to the needs of each accountable care organization, hospital, and integrated health system we contract with, in order to improve patient care and reduce hospital readmissions, ED visits, and overall spending.”
The company has posted readmissions rates lower than 95% of the industry, with higher quality metrics, he says, adding that the majority of General Medicine’s physicians are not transitioning hospitalists but doctors who were drawn to geriatrics and long-term care settings from the outset.
What’s Driving the Post-Acute Space?
A significant portion of healthcare expenditures is in post-acute care, and that money hasn’t always been well spent, experts interviewed for this article emphasized. Without adequate physician involvement in their care, many of these patients would be sent back to the hospital for complications that might have been managed outside of the hospital. Often it is payers, managed care plans, health systems, medical groups, and other risk-bearing entities that are driving the growth of physician involvement in post-acute care—just as insurers had a role in pushing the early growth of hospital medicine—and accountable care organizations (ACOs) are more often acting and contracting like payers.
In fact, spending on post-acute care overall, not just the physician’s role, is growing rapidly enough to attract the concerns of policymakers, reflected in a recent hearing by the U.S. House Energy and Commerce Committee that found drastic variations in payment rates across settings, with overall Medicare spending of $59 billion on post-acute care in 2013.2 The Bundling and Coordinating Post-Acute Care Act, a bill first introduced last year by Rep. David McKinley (D-W. Va.) and reintroduced in 2015 as HR1458, aims to address these growing costs while preserving patient choice by requiring a single bundled payment for post-acute care services under Medicare parts A and B.
Bundled payment is definitely coming, Dr. Rissmiller says, and will fuel the move to inpatient-outpatient partnerships.
“A lot of this work is in preparation for ACOs and bundled payments, even if the new models are not yet dominant in the marketplace.”
Bundling payment for an episode of care, including the hospital stay and all of the post-acute follow-up, will be a game-changer, Dr. Wilborn adds. CMS is now testing bundled payment models and, by 2017, he says they will be an established fact in nursing homes, with half of their reimbursement coming from some kind of bundle.
James Tollman, MD, FHM, heads a small HM group, Essex Inpatient Physicians, which he started in 2007 in Boxford, Mass. Essex includes full- and part-time physicians and physician extenders and has contracts with several hospitals, but Dr. Tollman estimates that 95% of the practice is in post-acute care.
“For us, as a small group in the current environment, it’s a good idea to diversify,” Dr. Tollman says. “It’s important to be flexible and have a foot in many venues.
“I view myself as a hospitalist by personality and history,” he adds. But experience working in post-acute care enables physicians to view the hospital in perspective—as part of the larger continuum of care and not the center of the universe.
Dr. Tollman says risk contracts with ACOs are the new frontier for hospitalists in post-acute care.
“Who manages the money is an important question,” he says. “The quality metrics are still poorly aligned with what the SNF-ist does. Right now, we’re entering contracts with three different ACO-type exchanges. None of them have really figured out what we are about, and we don’t have much leverage yet.”
Larry Beresford is a freelance writer in Alameda, Calif.
References
- Society of Hospital Medicine. 2014 State of Hospital Medicine report. Philadelphia, Pa.; Society of Hospital Medicine; 2014.
- Firth S. House panel considers medicare reform for post-acute care. Medpage Today. April 18, 2015. Accessed June 10, 2015.
In spite of all the gadgets and technologies available to hospital-based physicians nowadays, Jerome Wilborn, MD, FCCP, sees a much simpler symbol of patient care. Dr. Wilborn, national medical director for post-acute care services at IPC Healthcare, Inc., in North Hollywood, Calif., believes stethoscopes are key to post-acute patient care, and hospitalists are the ones “redefining” the practice. It’s not just a metaphor for working in settings that lack access to the specialists, equipment, and other resources of the acute-care hospital, he explains. A stethoscope, he says, reflects on the physicians’ clinical acumen and listening skills.
“Acute-care doctors need to understand that in the post-acute setting, it’s not about ordering labs. It is important to talk to the family,” he says. “Over the next 14 to 30 days, you can really dig into relationships with patients, optimize their medical care, reduce poly-pharmacy, and even prevent readmissions.”
These are among the sickest of patients, with multiple co-morbidities and limitations in activities of daily living (ADLs), Dr. Wilborn notes.
“Many internists and hospitalists who come to the nursing home are astounded by the clinical acuity of the patients and don’t appreciate how, even without the hospital treatment armamentarium they are used to, they can still make a big difference in the care,” he explains. But the key, he adds, is for doctors to go into the facility often enough to have an impact, with regularly scheduled presence and a commitment to standardizing the care.
For hospitalists who are more accustomed to the high-intensity, fast-paced world of the acute hospital, post-acute care may not seem very sexy.
“But that’s changing, along with the medical landscape,” Dr. Wilborn says. “For those who can appreciate the opportunity to build relationships with patients and to practice more independently, it could be a great place to change your career trajectory and have an immediate impact on the quality of patient care.”
What Is the Post-Acute Space?
Although post-acute care could refer broadly to all settings for care following the patient’s discharge from the hospital, including home care, three post-acute settings, defined by their licensure, are more likely to involve physicians such as hospitalists:
- The inpatient rehabilitation facility (IRF) , which is a freestanding rehabilitation unit or hospital inside an acute hospital for patients who need rehabilitation care in order to function effectively and are medically stable and able to participate in rehabilitation therapies;
- The long-term acute-care hospital (LTACH), a hospital that specializes in treatment and recovery of medical patients who require prolonged lengths of stay, typically measured in weeks;
- The The skilled nursing facility (SNF), which focuses on the health, social, and personal needs of chronically ill or disabled patients, either for rehabilitation stays of two weeks to a month or longer stays for chronic illness.
Although hospital medicine began as a practice specific to the inpatient setting, increasing numbers of hospitalists are spending at least part of their working lives outside of the hospital, visiting patients in post-acute settings. IPC is just one of the many national hospitalist management companies, medical groups, and hospital-employed practices that are defining new roles for their physicians, nurse practitioners, and physician assistants in these settings.
The presence of hospitalists in post-acute care is growing, according to the Society of Hospital Medicine, with 25.4% of adult hospital medicine groups in its most recent survey saying that they see patients in post-acute care facilities.1 In response to this trend, SHM in 2012 impaneled the Post-Acute Care Task Force, chaired by Sean Muldoon, MD, MPH, senior vice president and chief medical officer of Kindred Healthcare’s Hospital Division, Louisville, Ky. The task force was formed to help SHM members explore post-acute care and learn about what to expect. The task force developed a toolbox and a transitions quality improvement toolkit, and a new white paper, “Primer for Hospitalists on Skilled Nursing Facilities.”
“Why does it matter to hospitalists?” Dr. Muldoon asks, rhetorically. “Everything in terms of acuity is being pushed down to lower-level settings. If hospitalists think they will only do hospital work in the future, well, they will miss much of inpatient care because of the shift from the hospital to other hospital-like settings.”
At HM15 in late March, the task force outlined its agenda for the coming year, including promotion of its toolkit, development of a web-based CME seminar, and creation of a web-based reference repository.
Scott Rissmiller, MD, chief hospitalist at the 43-hospital Carolinas Healthcare System in Charlotte, N.C., says the transformation now taking place in post-acute care is more than just hospitalists doing some or all of their work in long-term care facilities. Post-acute care is becoming less of a side job for moonlighting hospitalists, with more of a focus on integrated care. Dr. Rissmiller, a member of SHM’s Multi-Site Hospitalist Group Task Force, says hospitalists are bringing to the post-acute arena the same standardization, accountability, and quality improvement the field has brought to hospitals across the country.
For Carolinas Healthcare and other multi-site hospitalist groups, the goal is to elevate the quality of care in LTACHs and other long-term care settings.
“It’s upping the game in post-acute care. It’s looking at the whole continuum of care from a systems perspective, improving handoffs and transitions,” Dr. Rissmiller says. “For years, we tried to improve communication with post-acute providers. It wasn’t until we started partnering with these facilities that we started to see changes.”
Dr. Rissmiller believes the best practice is to have one cohesive team caring for patients in both hospital and post-acute settings, under the leadership of the hospitalist group. The goal is to ensure that patients go to the proper level of care—and only for as long as they need to be there—using the system’s resources correctly.
Not every member of the hospitalist group will go to post-acute care facilities, while others will choose to specialize in that setting, he says, “but they meet every month with their acute care counterparts to work on improving care.”
What Should We Call This?
The amount of medical care being provided by hospitalists in post-acute facilities is growing, experts say, inclusive of physician assistants and nurse practitioners working as part of hospitalist groups. As many as 30% of SHM members are involved in post-acute care, according to the latest SHM survey, with large management groups like IPC, TeamHealth, and Tacoma, Wash.-based Sound Physicians, expanding rapidly in this area.1
“I don’t think that hospitalists have taken over from PCPs in post-acute care in general, but they make up a significant physician cohort,” Dr. Wilborn says.
IPC has a presence today in more than 1,700 post-acute care facilities, with 20% of its physicians working in both acute and post-acute care, more than 2,800 affiliated clinicians, and a third of the company’s revenue coming from the post-acute space, Dr. Wilborn notes.
Interestingly, what to call these providers seems to be a problem.
“Hospitalist groups are employing people who, strictly speaking, aren’t really hospitalists, although the post-acute setting marries up very well with the hospitalist model, mindset, and historical leadership role,” Dr. Rissmiller says. “It requires a different skill set.”
He prefers the term “post-acute specialist.”
Others refer to these providers as “SNF-ists,” although that word doesn’t exactly roll off the tongue, nor does it convey the scope of post-acute care.
“If hospitalists are doctors who round in acute-care hospitals, the parallel term for doctors who round in post-acute facilities is not well established,” Dr. Wilborn says. “It’s site-specific care. I call them post-acute care providers. This certainly is a specialty, for a lot of different reasons. It’s post-acute care medicine, and the hospitalist term isn’t going to stick.”
Scott Sears, MD, FACP, chief clinical officer of Tacoma, Wash.-based Sound Physicians, which has physicians deployed in roughly 100 post-acute settings, labels his providers “transitional care physicians.” Most of them are dedicated full time to post-acute care.
“Our main source of transitional care doctors are former hospitalists who are interested in more than a three-to-seven day relationship with their patients,” Dr. Sears says.
“It’s almost an art in itself,” he adds. “That’s why it’s not in the patient’s best interest to have a doctor who just dabbles in post-acute care. That’s where the dedicated provider with a passion and vision for the work is so valuable. We also have more success with people who have more experience.”
General Medicine PC, a Novi, Mich.-based company of physicians, NPs, and PAs who specialize in treating geriatric and chronically ill patients in long-term care settings, calls itself “the post-hospitalist company.” It claims its primary customers tend to be payers and managed care systems.
“We use the term post-hospitalist, and we are the country’s largest provider of post-hospitalist services,” explains CEO Thomas Prose, MD, MPH, MBA, who founded the company in 1983. “We tailor services to the needs of each accountable care organization, hospital, and integrated health system we contract with, in order to improve patient care and reduce hospital readmissions, ED visits, and overall spending.”
The company has posted readmissions rates lower than 95% of the industry, with higher quality metrics, he says, adding that the majority of General Medicine’s physicians are not transitioning hospitalists but doctors who were drawn to geriatrics and long-term care settings from the outset.
What’s Driving the Post-Acute Space?
A significant portion of healthcare expenditures is in post-acute care, and that money hasn’t always been well spent, experts interviewed for this article emphasized. Without adequate physician involvement in their care, many of these patients would be sent back to the hospital for complications that might have been managed outside of the hospital. Often it is payers, managed care plans, health systems, medical groups, and other risk-bearing entities that are driving the growth of physician involvement in post-acute care—just as insurers had a role in pushing the early growth of hospital medicine—and accountable care organizations (ACOs) are more often acting and contracting like payers.
In fact, spending on post-acute care overall, not just the physician’s role, is growing rapidly enough to attract the concerns of policymakers, reflected in a recent hearing by the U.S. House Energy and Commerce Committee that found drastic variations in payment rates across settings, with overall Medicare spending of $59 billion on post-acute care in 2013.2 The Bundling and Coordinating Post-Acute Care Act, a bill first introduced last year by Rep. David McKinley (D-W. Va.) and reintroduced in 2015 as HR1458, aims to address these growing costs while preserving patient choice by requiring a single bundled payment for post-acute care services under Medicare parts A and B.
Bundled payment is definitely coming, Dr. Rissmiller says, and will fuel the move to inpatient-outpatient partnerships.
“A lot of this work is in preparation for ACOs and bundled payments, even if the new models are not yet dominant in the marketplace.”
Bundling payment for an episode of care, including the hospital stay and all of the post-acute follow-up, will be a game-changer, Dr. Wilborn adds. CMS is now testing bundled payment models and, by 2017, he says they will be an established fact in nursing homes, with half of their reimbursement coming from some kind of bundle.
James Tollman, MD, FHM, heads a small HM group, Essex Inpatient Physicians, which he started in 2007 in Boxford, Mass. Essex includes full- and part-time physicians and physician extenders and has contracts with several hospitals, but Dr. Tollman estimates that 95% of the practice is in post-acute care.
“For us, as a small group in the current environment, it’s a good idea to diversify,” Dr. Tollman says. “It’s important to be flexible and have a foot in many venues.
“I view myself as a hospitalist by personality and history,” he adds. But experience working in post-acute care enables physicians to view the hospital in perspective—as part of the larger continuum of care and not the center of the universe.
Dr. Tollman says risk contracts with ACOs are the new frontier for hospitalists in post-acute care.
“Who manages the money is an important question,” he says. “The quality metrics are still poorly aligned with what the SNF-ist does. Right now, we’re entering contracts with three different ACO-type exchanges. None of them have really figured out what we are about, and we don’t have much leverage yet.”
Larry Beresford is a freelance writer in Alameda, Calif.
References
- Society of Hospital Medicine. 2014 State of Hospital Medicine report. Philadelphia, Pa.; Society of Hospital Medicine; 2014.
- Firth S. House panel considers medicare reform for post-acute care. Medpage Today. April 18, 2015. Accessed June 10, 2015.
11 Things Gastroenterologists Think Hospitalists Need to Know

So many symptoms that are staples of gastroenterology—chest pain, nausea, diarrhea—are mainstay causes for hospitalization that it might be worth fine-tuning how well you handle patients with gastroenterology disorders.
The Hospitalist asked several gastroenterologists for their guidance on better care and their suggestions for correcting some common mistakes that they encounter. Here are their tips:
1 Fluid resuscitation is crucial for pancreatitis patients.
It’s very important to rehydrate these patients within the first 24 hours, because those who remain underhydrated can have a worse prognosis, says Robert Coben, MD, academic coordinator for the Gastroentestinal Fellowship Program at Thomas Jefferson University Hospital in Philadelphia. On occasion, physicians are reluctant to give extra fluids to these patients, he says, particularly if they have heart failure or suffer kidney problems. A 70-kg patient should be receiving about 200 cc per hour, he notes.
“Sometimes we’ll walk in the room and they’re getting 80 cc an hour,” Dr. Coben says.
“These patients…need to be flooded with fluids,” says Rajeev Jain, MD, chief of gastroenterology at Presbyterian Hospital of Dallas, partner at Texas Digestive Disease Consultants, and chair of the Practice Management and Economics Committee of the American Gastroenterological Association. “We’re talking sometimes liters and liters of IV normal saline or lactated Ringer’s (solution) in a 24-hour period.”1
Marcelo Vela, MD, a gastroenterologist and hepatologist at the Mayo Clinic in Scottsdale, Ariz., and an associate editor with Clinical Gastroenterology and Hepatology, says Ringer’s solution is a better choice than normal saline.2
“If you’re going to start IV fluids on somebody who’s coming in with acute pancreatitis, Ringer’s solution has been shown to be superior to saline in randomized controlled trials,” Dr. Vela says. “It reduces systemic inflammation.”
2 Gastrointestinal bleeding decisions
When inpatients have gastrointestinal bleeding, the hospitalist often has to assess its severity and make the call on whether a patient needs the ICU.
“The most important thing for that is obviously the vital signs,” says John Pandolfino, MD, chief of the division of medicine–gastroenterology and hepatology at Northwestern University Feinberg School of Medicine in Chicago. “If people are tachycardic and they’re not responding to hydration and blood transfusion, that usually means it’s a pretty active bleed and they need to go to the intensive care unit. If you have somebody who’s GI bleeding and they’re coagulopathic (i.e., they’re on anti-coagulation because they have a valve and they need anti-coagulation or they have cancer or bad cardiovascular disease), those are the people that you should have a low threshold for sending to the intensive care unit with a GI bleed; those are the people who are at a very high mortality [risk].”
He added that those with an ulcer, with a visible vessel, are at a high-risk of a rebleed and should spend some time in the ICU.
“Those people should be evaluated in the intensive care unit for at least 24 hours, maybe even 72 hours,” Dr. Pandolfino says, “and they should have IV PPI [proton pump inhibitor] therapy.”
3 Endoscopy has very low yield for diagnosis of reflux.
“Endoscopy has good yield for mucosa abnormalities on inspection of the esophageal mucosa, but it does not give you a diagnosis of reflux, especially on patients who have already been treated with a PPI,” says Prakash Gyawali, MD, MRCP, professor of gastroenterology at Washington University in St. Louis, Mo. “So, usually, in those settings, obtaining a consult or trying to decide exactly what you’re looking for to explain the symptoms has better yield than an endoscopy.”
Sometimes a pH study is needed, but that has to be planned, because patients may have to be taken off of a PPI in advance. That means those studies are not easy to set up in the hospital and might best be arranged by the gastroenterologist, Dr. Gyawali says.
4 In cases of acute diarrhea, order a stool sample right away.
That will help guide care from the gastroenterologist, if and when the gastroenterologist is called in, Dr. Pandolfino says.
“One of the things that is frustrating for the gastroenterologist is that we get called initially, but really the hospitalist should be getting stool studies, and they should have at least a very good idea of what they need from us,” he says. “Because, really, endoscopy is not usually needed very often in diagnosis of acute diarrhea.”
Broad-range stool studies, a good history and physical, and examining labs for a possible chronic inflammatory process or anemia are good ways to begin to assess patients with diarrhea, he says. Clostridium difficile colitis has to be considered as well, Dr. Pandolfino says.
Endoscopy is more helpful in evaluating acute diarrhea in those with bloody diarrhea suspected of having inflammatory bowel disease or an infectious diarrhea but on whom cultures have come back negative. For those with compromised immune systems, endoscopy could be done earlier, as well.
“So for us, I think we really need to get into the picture a little bit after the patient has been brought into the hospital and the stool study is negative, unless they’re an immune-compromised patient,” Dr. Pandolfino says.
5 When—and how—to test the stool.
If a patient develops diarrhea while already in the hospital, the only stool test needed is C. diff.
“They shouldn’t be developing a viral diarrhea, they shouldn’t be developing an infectious diarrhea—let’s say, from E. coli or Salmonella—unless they literally developed it a couple hours after getting in,” Dr. Jain says. “It should either be C. diff or a side effect of some medication. … We don’t need to spend the extra money, which is of low-value care to send for OVA and parasites, or bacterial pathogens and so forth.”
Dr. Jain says he thinks such testing is being done more appropriately of late.
“But I still will see multiple stool tests sent on somebody who’s been in the hospital for a week and then develops diarrhea,” he explains.
6 Gastroenterologists do not need to be consulted for every C. diff infection.
“I think that we really should get involved when patients are either not responding or when they’re very ill,” Dr. Pandolfino says.
Hospitalists should consider whether patients are on antibiotics or a PPI and whether or not they need to stay on those medications. Also, medications that slow motility (i.e., loperamide) should be avoided, if possible.
“We don’t want it to linger,” Dr. Pandolfino says. “One of the basic mechanisms of how we get rid of pathogens is to expel them with diarrhea.
“But you certainly don’t want the patient to be uncomfortable to the point where they’re having 20 to 30 bowel movements a day.”
In non-severe patients who have some diarrhea, abdominal pain, nausea, and vomiting—but are able to keep food down—the specialists might not have to be called in, and patients can just be treated with oral metronidazole or oral vancomycin fairly simply, he says.
For those who seem severely ill with a dilating colon, are in nearly a septic state, and have very severe diarrhea, the gastroenterologist should probably be called in, he says.

7 For patients with a possible GI bleed and black stools, do an exam before calling in the gastroenterologist.
The exam should determine what the stool color is and whether it is heme-positive, and the patient’s blood count should be evaluated, Dr. Coben says.
Frequently, “the consultant’s the one who ends up doing the rectal exam and checking that,” he says. “Sometimes we find that [the patient is] really not bleeding,” and it was just a case of a hospitalist taking the patient’s word that they were bleeding.
Being on iron therapy or taking Pepto-Bismol can turn the stool black, and the stool might not really be black; colors can sometimes be open to interpretation.
But he cautions that colon cancer could be the cause for a GI bleed.
“We’ve had it happen a few times where this occurred,” he says. “The patient was discharged, and they really didn’t get proper follow-up, and it ended up that they had a colon cancer. It kind of delayed that diagnosis. So I think you have to be aware of [the fact that], especially in somebody over the age of 40 or 50, if they have an iron-deficiency, anemia, or heme-positive stool, the first thing you really need to exclude is a colon cancer.”
8 Minimize CT scans in early evaluation and management of acute pancreatitis patients.
“The reason for that is they tend to be intravascularly volume-depleted,” Dr. Jain says. “So [with] the IV contrast, there’s an increased risk of developing kidney failure. It’s also been associated with increased risk of necrotizing pancreatitis.”
Dr. Jain notes that when he is consulted on this kind of patient, he will order a sonogram. If it doesn’t show gallstones, and there’s no clear reason for the pancreatitis, he will want a CT scan, but he will wait a few days until the patient is fluid-resuscitated.
Dr. Jain says that this is a problem more often seen in the ED—where 90% of patients with acute pancreatitis have already gotten the CT scan—and less so among hospitalists, but it’s still worth the reminder.
A CT scan right away is justified to rule out something such as a perforation, but not in the case of classic symptoms of acute pancreatitis, he adds.
9 Actively bleeding patients?
It’s smart to give patients a unit of packed red blood cells more quickly if they’re actively bleeding. Even over just one hour is OK, Dr. Jain says.
“Even if they have underlying heart failure, if they’re volume-depleted, they need that volume,” Dr. Jain explains. “Sometimes you’ll see that it takes eight hours to get two units of blood in. That’s inadequate.”
Another point worth a reminder, he says: Two large-bore peripheral IV’s are “much, much better” than a central or PICC line to deliver IV fluid resuscitation.

“Sometimes hospitalists order barium studies because these can be done the same day, whereas with endoscopy, patients need to be put on a schedule and have to be NPO [nothing by mouth] overnight.” —Prakash Gyawali, MD, MRCP
10 Don’t be too quick to order barium studies, especially in patients with dysphagia.
“The problem with that is it takes much longer to do an endoscopy if barium is put in the esophagus, because we usually wait until the barium clears,” Dr. Gyawali says. “And inpatient evaluation of new-onset dysphagia should be endoscopy first, and not barium, because biopsies need to be taken to rule out eosinophilic esophagitis.
“Sometimes hospitalists order barium studies because these can be done the same day, whereas with endoscopy, patients need to be put on a schedule and have to be NPO [nothing by mouth] overnight.”
11 Gastric-emptying studies should be outpatient.
Gastric-emptying studies are often better done when patients are not in a hospital, experts say, because they might be on medications that would interfere with the study.
“A common issue, not just among hospitalists but also gastroenterologists, is that patients may be on a bunch of medicines that would affect stomach-emptying while they are in-house for some other problem,” Dr. Gyawali says. “A lot of times, these patients with pain may get narcotics. They may be on medicines to prevent them from throwing up. … And all of these will slow down gastric emptying.”
With an abnormal test result, “you are left to decide whether that is a real abnormality or whether the medicines the patients were on impacted the abnormality.”
If emptying was significantly prolonged, the test may have some value, “but then it probably will need to be corroborated with symptoms and with endoscopic findings.”
Thomas Collins is a freelance writer in South Florida.
References
- Warndorf MG, Kurtzman JT, Bartel MJ, et al. Early fluid resuscitation reduces morbidity among patients with acute pancreatitis. Clin Gastroenterol Hepatol. 2011;9(8):705-709.
- Wu BU, Hwang JQ, Gardner TH, et al. Lactated Ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol. 2011;9(8):710-717.
So many symptoms that are staples of gastroenterology—chest pain, nausea, diarrhea—are mainstay causes for hospitalization that it might be worth fine-tuning how well you handle patients with gastroenterology disorders.
The Hospitalist asked several gastroenterologists for their guidance on better care and their suggestions for correcting some common mistakes that they encounter. Here are their tips:
1 Fluid resuscitation is crucial for pancreatitis patients.
It’s very important to rehydrate these patients within the first 24 hours, because those who remain underhydrated can have a worse prognosis, says Robert Coben, MD, academic coordinator for the Gastroentestinal Fellowship Program at Thomas Jefferson University Hospital in Philadelphia. On occasion, physicians are reluctant to give extra fluids to these patients, he says, particularly if they have heart failure or suffer kidney problems. A 70-kg patient should be receiving about 200 cc per hour, he notes.
“Sometimes we’ll walk in the room and they’re getting 80 cc an hour,” Dr. Coben says.
“These patients…need to be flooded with fluids,” says Rajeev Jain, MD, chief of gastroenterology at Presbyterian Hospital of Dallas, partner at Texas Digestive Disease Consultants, and chair of the Practice Management and Economics Committee of the American Gastroenterological Association. “We’re talking sometimes liters and liters of IV normal saline or lactated Ringer’s (solution) in a 24-hour period.”1
Marcelo Vela, MD, a gastroenterologist and hepatologist at the Mayo Clinic in Scottsdale, Ariz., and an associate editor with Clinical Gastroenterology and Hepatology, says Ringer’s solution is a better choice than normal saline.2
“If you’re going to start IV fluids on somebody who’s coming in with acute pancreatitis, Ringer’s solution has been shown to be superior to saline in randomized controlled trials,” Dr. Vela says. “It reduces systemic inflammation.”
2 Gastrointestinal bleeding decisions
When inpatients have gastrointestinal bleeding, the hospitalist often has to assess its severity and make the call on whether a patient needs the ICU.
“The most important thing for that is obviously the vital signs,” says John Pandolfino, MD, chief of the division of medicine–gastroenterology and hepatology at Northwestern University Feinberg School of Medicine in Chicago. “If people are tachycardic and they’re not responding to hydration and blood transfusion, that usually means it’s a pretty active bleed and they need to go to the intensive care unit. If you have somebody who’s GI bleeding and they’re coagulopathic (i.e., they’re on anti-coagulation because they have a valve and they need anti-coagulation or they have cancer or bad cardiovascular disease), those are the people that you should have a low threshold for sending to the intensive care unit with a GI bleed; those are the people who are at a very high mortality [risk].”
He added that those with an ulcer, with a visible vessel, are at a high-risk of a rebleed and should spend some time in the ICU.
“Those people should be evaluated in the intensive care unit for at least 24 hours, maybe even 72 hours,” Dr. Pandolfino says, “and they should have IV PPI [proton pump inhibitor] therapy.”
3 Endoscopy has very low yield for diagnosis of reflux.
“Endoscopy has good yield for mucosa abnormalities on inspection of the esophageal mucosa, but it does not give you a diagnosis of reflux, especially on patients who have already been treated with a PPI,” says Prakash Gyawali, MD, MRCP, professor of gastroenterology at Washington University in St. Louis, Mo. “So, usually, in those settings, obtaining a consult or trying to decide exactly what you’re looking for to explain the symptoms has better yield than an endoscopy.”
Sometimes a pH study is needed, but that has to be planned, because patients may have to be taken off of a PPI in advance. That means those studies are not easy to set up in the hospital and might best be arranged by the gastroenterologist, Dr. Gyawali says.
4 In cases of acute diarrhea, order a stool sample right away.
That will help guide care from the gastroenterologist, if and when the gastroenterologist is called in, Dr. Pandolfino says.
“One of the things that is frustrating for the gastroenterologist is that we get called initially, but really the hospitalist should be getting stool studies, and they should have at least a very good idea of what they need from us,” he says. “Because, really, endoscopy is not usually needed very often in diagnosis of acute diarrhea.”
Broad-range stool studies, a good history and physical, and examining labs for a possible chronic inflammatory process or anemia are good ways to begin to assess patients with diarrhea, he says. Clostridium difficile colitis has to be considered as well, Dr. Pandolfino says.
Endoscopy is more helpful in evaluating acute diarrhea in those with bloody diarrhea suspected of having inflammatory bowel disease or an infectious diarrhea but on whom cultures have come back negative. For those with compromised immune systems, endoscopy could be done earlier, as well.
“So for us, I think we really need to get into the picture a little bit after the patient has been brought into the hospital and the stool study is negative, unless they’re an immune-compromised patient,” Dr. Pandolfino says.
5 When—and how—to test the stool.
If a patient develops diarrhea while already in the hospital, the only stool test needed is C. diff.
“They shouldn’t be developing a viral diarrhea, they shouldn’t be developing an infectious diarrhea—let’s say, from E. coli or Salmonella—unless they literally developed it a couple hours after getting in,” Dr. Jain says. “It should either be C. diff or a side effect of some medication. … We don’t need to spend the extra money, which is of low-value care to send for OVA and parasites, or bacterial pathogens and so forth.”
Dr. Jain says he thinks such testing is being done more appropriately of late.
“But I still will see multiple stool tests sent on somebody who’s been in the hospital for a week and then develops diarrhea,” he explains.
6 Gastroenterologists do not need to be consulted for every C. diff infection.
“I think that we really should get involved when patients are either not responding or when they’re very ill,” Dr. Pandolfino says.
Hospitalists should consider whether patients are on antibiotics or a PPI and whether or not they need to stay on those medications. Also, medications that slow motility (i.e., loperamide) should be avoided, if possible.
“We don’t want it to linger,” Dr. Pandolfino says. “One of the basic mechanisms of how we get rid of pathogens is to expel them with diarrhea.
“But you certainly don’t want the patient to be uncomfortable to the point where they’re having 20 to 30 bowel movements a day.”
In non-severe patients who have some diarrhea, abdominal pain, nausea, and vomiting—but are able to keep food down—the specialists might not have to be called in, and patients can just be treated with oral metronidazole or oral vancomycin fairly simply, he says.
For those who seem severely ill with a dilating colon, are in nearly a septic state, and have very severe diarrhea, the gastroenterologist should probably be called in, he says.
7 For patients with a possible GI bleed and black stools, do an exam before calling in the gastroenterologist.
The exam should determine what the stool color is and whether it is heme-positive, and the patient’s blood count should be evaluated, Dr. Coben says.
Frequently, “the consultant’s the one who ends up doing the rectal exam and checking that,” he says. “Sometimes we find that [the patient is] really not bleeding,” and it was just a case of a hospitalist taking the patient’s word that they were bleeding.
Being on iron therapy or taking Pepto-Bismol can turn the stool black, and the stool might not really be black; colors can sometimes be open to interpretation.
But he cautions that colon cancer could be the cause for a GI bleed.
“We’ve had it happen a few times where this occurred,” he says. “The patient was discharged, and they really didn’t get proper follow-up, and it ended up that they had a colon cancer. It kind of delayed that diagnosis. So I think you have to be aware of [the fact that], especially in somebody over the age of 40 or 50, if they have an iron-deficiency, anemia, or heme-positive stool, the first thing you really need to exclude is a colon cancer.”
8 Minimize CT scans in early evaluation and management of acute pancreatitis patients.
“The reason for that is they tend to be intravascularly volume-depleted,” Dr. Jain says. “So [with] the IV contrast, there’s an increased risk of developing kidney failure. It’s also been associated with increased risk of necrotizing pancreatitis.”
Dr. Jain notes that when he is consulted on this kind of patient, he will order a sonogram. If it doesn’t show gallstones, and there’s no clear reason for the pancreatitis, he will want a CT scan, but he will wait a few days until the patient is fluid-resuscitated.
Dr. Jain says that this is a problem more often seen in the ED—where 90% of patients with acute pancreatitis have already gotten the CT scan—and less so among hospitalists, but it’s still worth the reminder.
A CT scan right away is justified to rule out something such as a perforation, but not in the case of classic symptoms of acute pancreatitis, he adds.
9 Actively bleeding patients?
It’s smart to give patients a unit of packed red blood cells more quickly if they’re actively bleeding. Even over just one hour is OK, Dr. Jain says.
“Even if they have underlying heart failure, if they’re volume-depleted, they need that volume,” Dr. Jain explains. “Sometimes you’ll see that it takes eight hours to get two units of blood in. That’s inadequate.”
Another point worth a reminder, he says: Two large-bore peripheral IV’s are “much, much better” than a central or PICC line to deliver IV fluid resuscitation.
“Sometimes hospitalists order barium studies because these can be done the same day, whereas with endoscopy, patients need to be put on a schedule and have to be NPO [nothing by mouth] overnight.” —Prakash Gyawali, MD, MRCP
10 Don’t be too quick to order barium studies, especially in patients with dysphagia.
“The problem with that is it takes much longer to do an endoscopy if barium is put in the esophagus, because we usually wait until the barium clears,” Dr. Gyawali says. “And inpatient evaluation of new-onset dysphagia should be endoscopy first, and not barium, because biopsies need to be taken to rule out eosinophilic esophagitis.
“Sometimes hospitalists order barium studies because these can be done the same day, whereas with endoscopy, patients need to be put on a schedule and have to be NPO [nothing by mouth] overnight.”
11 Gastric-emptying studies should be outpatient.
Gastric-emptying studies are often better done when patients are not in a hospital, experts say, because they might be on medications that would interfere with the study.
“A common issue, not just among hospitalists but also gastroenterologists, is that patients may be on a bunch of medicines that would affect stomach-emptying while they are in-house for some other problem,” Dr. Gyawali says. “A lot of times, these patients with pain may get narcotics. They may be on medicines to prevent them from throwing up. … And all of these will slow down gastric emptying.”
With an abnormal test result, “you are left to decide whether that is a real abnormality or whether the medicines the patients were on impacted the abnormality.”
If emptying was significantly prolonged, the test may have some value, “but then it probably will need to be corroborated with symptoms and with endoscopic findings.”
Thomas Collins is a freelance writer in South Florida.
References
- Warndorf MG, Kurtzman JT, Bartel MJ, et al. Early fluid resuscitation reduces morbidity among patients with acute pancreatitis. Clin Gastroenterol Hepatol. 2011;9(8):705-709.
- Wu BU, Hwang JQ, Gardner TH, et al. Lactated Ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol. 2011;9(8):710-717.
So many symptoms that are staples of gastroenterology—chest pain, nausea, diarrhea—are mainstay causes for hospitalization that it might be worth fine-tuning how well you handle patients with gastroenterology disorders.
The Hospitalist asked several gastroenterologists for their guidance on better care and their suggestions for correcting some common mistakes that they encounter. Here are their tips:
1 Fluid resuscitation is crucial for pancreatitis patients.
It’s very important to rehydrate these patients within the first 24 hours, because those who remain underhydrated can have a worse prognosis, says Robert Coben, MD, academic coordinator for the Gastroentestinal Fellowship Program at Thomas Jefferson University Hospital in Philadelphia. On occasion, physicians are reluctant to give extra fluids to these patients, he says, particularly if they have heart failure or suffer kidney problems. A 70-kg patient should be receiving about 200 cc per hour, he notes.
“Sometimes we’ll walk in the room and they’re getting 80 cc an hour,” Dr. Coben says.
“These patients…need to be flooded with fluids,” says Rajeev Jain, MD, chief of gastroenterology at Presbyterian Hospital of Dallas, partner at Texas Digestive Disease Consultants, and chair of the Practice Management and Economics Committee of the American Gastroenterological Association. “We’re talking sometimes liters and liters of IV normal saline or lactated Ringer’s (solution) in a 24-hour period.”1
Marcelo Vela, MD, a gastroenterologist and hepatologist at the Mayo Clinic in Scottsdale, Ariz., and an associate editor with Clinical Gastroenterology and Hepatology, says Ringer’s solution is a better choice than normal saline.2
“If you’re going to start IV fluids on somebody who’s coming in with acute pancreatitis, Ringer’s solution has been shown to be superior to saline in randomized controlled trials,” Dr. Vela says. “It reduces systemic inflammation.”
2 Gastrointestinal bleeding decisions
When inpatients have gastrointestinal bleeding, the hospitalist often has to assess its severity and make the call on whether a patient needs the ICU.
“The most important thing for that is obviously the vital signs,” says John Pandolfino, MD, chief of the division of medicine–gastroenterology and hepatology at Northwestern University Feinberg School of Medicine in Chicago. “If people are tachycardic and they’re not responding to hydration and blood transfusion, that usually means it’s a pretty active bleed and they need to go to the intensive care unit. If you have somebody who’s GI bleeding and they’re coagulopathic (i.e., they’re on anti-coagulation because they have a valve and they need anti-coagulation or they have cancer or bad cardiovascular disease), those are the people that you should have a low threshold for sending to the intensive care unit with a GI bleed; those are the people who are at a very high mortality [risk].”
He added that those with an ulcer, with a visible vessel, are at a high-risk of a rebleed and should spend some time in the ICU.
“Those people should be evaluated in the intensive care unit for at least 24 hours, maybe even 72 hours,” Dr. Pandolfino says, “and they should have IV PPI [proton pump inhibitor] therapy.”
3 Endoscopy has very low yield for diagnosis of reflux.
“Endoscopy has good yield for mucosa abnormalities on inspection of the esophageal mucosa, but it does not give you a diagnosis of reflux, especially on patients who have already been treated with a PPI,” says Prakash Gyawali, MD, MRCP, professor of gastroenterology at Washington University in St. Louis, Mo. “So, usually, in those settings, obtaining a consult or trying to decide exactly what you’re looking for to explain the symptoms has better yield than an endoscopy.”
Sometimes a pH study is needed, but that has to be planned, because patients may have to be taken off of a PPI in advance. That means those studies are not easy to set up in the hospital and might best be arranged by the gastroenterologist, Dr. Gyawali says.
4 In cases of acute diarrhea, order a stool sample right away.
That will help guide care from the gastroenterologist, if and when the gastroenterologist is called in, Dr. Pandolfino says.
“One of the things that is frustrating for the gastroenterologist is that we get called initially, but really the hospitalist should be getting stool studies, and they should have at least a very good idea of what they need from us,” he says. “Because, really, endoscopy is not usually needed very often in diagnosis of acute diarrhea.”
Broad-range stool studies, a good history and physical, and examining labs for a possible chronic inflammatory process or anemia are good ways to begin to assess patients with diarrhea, he says. Clostridium difficile colitis has to be considered as well, Dr. Pandolfino says.
Endoscopy is more helpful in evaluating acute diarrhea in those with bloody diarrhea suspected of having inflammatory bowel disease or an infectious diarrhea but on whom cultures have come back negative. For those with compromised immune systems, endoscopy could be done earlier, as well.
“So for us, I think we really need to get into the picture a little bit after the patient has been brought into the hospital and the stool study is negative, unless they’re an immune-compromised patient,” Dr. Pandolfino says.
5 When—and how—to test the stool.
If a patient develops diarrhea while already in the hospital, the only stool test needed is C. diff.
“They shouldn’t be developing a viral diarrhea, they shouldn’t be developing an infectious diarrhea—let’s say, from E. coli or Salmonella—unless they literally developed it a couple hours after getting in,” Dr. Jain says. “It should either be C. diff or a side effect of some medication. … We don’t need to spend the extra money, which is of low-value care to send for OVA and parasites, or bacterial pathogens and so forth.”
Dr. Jain says he thinks such testing is being done more appropriately of late.
“But I still will see multiple stool tests sent on somebody who’s been in the hospital for a week and then develops diarrhea,” he explains.
6 Gastroenterologists do not need to be consulted for every C. diff infection.
“I think that we really should get involved when patients are either not responding or when they’re very ill,” Dr. Pandolfino says.
Hospitalists should consider whether patients are on antibiotics or a PPI and whether or not they need to stay on those medications. Also, medications that slow motility (i.e., loperamide) should be avoided, if possible.
“We don’t want it to linger,” Dr. Pandolfino says. “One of the basic mechanisms of how we get rid of pathogens is to expel them with diarrhea.
“But you certainly don’t want the patient to be uncomfortable to the point where they’re having 20 to 30 bowel movements a day.”
In non-severe patients who have some diarrhea, abdominal pain, nausea, and vomiting—but are able to keep food down—the specialists might not have to be called in, and patients can just be treated with oral metronidazole or oral vancomycin fairly simply, he says.
For those who seem severely ill with a dilating colon, are in nearly a septic state, and have very severe diarrhea, the gastroenterologist should probably be called in, he says.
7 For patients with a possible GI bleed and black stools, do an exam before calling in the gastroenterologist.
The exam should determine what the stool color is and whether it is heme-positive, and the patient’s blood count should be evaluated, Dr. Coben says.
Frequently, “the consultant’s the one who ends up doing the rectal exam and checking that,” he says. “Sometimes we find that [the patient is] really not bleeding,” and it was just a case of a hospitalist taking the patient’s word that they were bleeding.
Being on iron therapy or taking Pepto-Bismol can turn the stool black, and the stool might not really be black; colors can sometimes be open to interpretation.
But he cautions that colon cancer could be the cause for a GI bleed.
“We’ve had it happen a few times where this occurred,” he says. “The patient was discharged, and they really didn’t get proper follow-up, and it ended up that they had a colon cancer. It kind of delayed that diagnosis. So I think you have to be aware of [the fact that], especially in somebody over the age of 40 or 50, if they have an iron-deficiency, anemia, or heme-positive stool, the first thing you really need to exclude is a colon cancer.”
8 Minimize CT scans in early evaluation and management of acute pancreatitis patients.
“The reason for that is they tend to be intravascularly volume-depleted,” Dr. Jain says. “So [with] the IV contrast, there’s an increased risk of developing kidney failure. It’s also been associated with increased risk of necrotizing pancreatitis.”
Dr. Jain notes that when he is consulted on this kind of patient, he will order a sonogram. If it doesn’t show gallstones, and there’s no clear reason for the pancreatitis, he will want a CT scan, but he will wait a few days until the patient is fluid-resuscitated.
Dr. Jain says that this is a problem more often seen in the ED—where 90% of patients with acute pancreatitis have already gotten the CT scan—and less so among hospitalists, but it’s still worth the reminder.
A CT scan right away is justified to rule out something such as a perforation, but not in the case of classic symptoms of acute pancreatitis, he adds.
9 Actively bleeding patients?
It’s smart to give patients a unit of packed red blood cells more quickly if they’re actively bleeding. Even over just one hour is OK, Dr. Jain says.
“Even if they have underlying heart failure, if they’re volume-depleted, they need that volume,” Dr. Jain explains. “Sometimes you’ll see that it takes eight hours to get two units of blood in. That’s inadequate.”
Another point worth a reminder, he says: Two large-bore peripheral IV’s are “much, much better” than a central or PICC line to deliver IV fluid resuscitation.
“Sometimes hospitalists order barium studies because these can be done the same day, whereas with endoscopy, patients need to be put on a schedule and have to be NPO [nothing by mouth] overnight.” —Prakash Gyawali, MD, MRCP
10 Don’t be too quick to order barium studies, especially in patients with dysphagia.
“The problem with that is it takes much longer to do an endoscopy if barium is put in the esophagus, because we usually wait until the barium clears,” Dr. Gyawali says. “And inpatient evaluation of new-onset dysphagia should be endoscopy first, and not barium, because biopsies need to be taken to rule out eosinophilic esophagitis.
“Sometimes hospitalists order barium studies because these can be done the same day, whereas with endoscopy, patients need to be put on a schedule and have to be NPO [nothing by mouth] overnight.”
11 Gastric-emptying studies should be outpatient.
Gastric-emptying studies are often better done when patients are not in a hospital, experts say, because they might be on medications that would interfere with the study.
“A common issue, not just among hospitalists but also gastroenterologists, is that patients may be on a bunch of medicines that would affect stomach-emptying while they are in-house for some other problem,” Dr. Gyawali says. “A lot of times, these patients with pain may get narcotics. They may be on medicines to prevent them from throwing up. … And all of these will slow down gastric emptying.”
With an abnormal test result, “you are left to decide whether that is a real abnormality or whether the medicines the patients were on impacted the abnormality.”
If emptying was significantly prolonged, the test may have some value, “but then it probably will need to be corroborated with symptoms and with endoscopic findings.”
Thomas Collins is a freelance writer in South Florida.
References
- Warndorf MG, Kurtzman JT, Bartel MJ, et al. Early fluid resuscitation reduces morbidity among patients with acute pancreatitis. Clin Gastroenterol Hepatol. 2011;9(8):705-709.
- Wu BU, Hwang JQ, Gardner TH, et al. Lactated Ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol. 2011;9(8):710-717.
Hospitalist Brian Chen, MD, Marries Medicine With Music

Most young boys dream about scoring touchdowns or being a superhero, combatting villains or evil aliens. When Brian Chen, MD, was young, he wanted superpowers, too, but of a different sort. He wanted to become a concert violinist.
Now a hospitalist at St. Joseph Mercy Ann Arbor Hospital in Michigan, Dr. Chen recalls watching Itzhak Perlman, the superstar virtuoso of the violin, perform on the PBS show “Great Performances” when he was just five years old. Entranced by Perlman’s genius, he vowed to master the violin. After years of study, practice, and commitment, Dr. Chen has not quite reached Perlman’s status but has achieved more in his musical career than some musicians ever dream of.
Musical Gifts
After watching “Great Performances,” Dr. Chen begged his parents for a violin. At the time, he was learning to play piano.
“My parents were smart about having me start with piano first,” he says, explaining that the piano helped him learn the basics of reading music and distinguishing tones. “With piano, you press a key that produces a sound you intended. With the violin, there’s no such luck. It requires precise placement of fingers. It was sort of something my parents saw that I could eventually [play] once I got past learning the basics.”
Dr. Chen received his first violin when he was six years old. By then, he had already been playing the piano for a year. His excitement quickly turned into frustration, however. Why couldn’t he play like Perlman?
Instead of admitting defeat, he became that much more determined. Throughout middle school and high school, he took piano and violin lessons and practiced both instruments for several hours each day after school. He joined the high school orchestra and the Dayton (Ohio) Philharmonic Youth Orchestra. Both gave free public concerts, which exposed Dr. Chen to the art of performing at a tender age.
He says the violin appeals to him partly because it is a difficult instrument to learn. Not everyone can play it. But, once mastered, he believes “its voice is the most beautiful and expressive, even more than piano.”
Likewise, he says violinists can play stronger and richer tones with a little vibrato, which adds yet another dimension to the instrument.
Like most teenagers, Dr. Chen was active, trying out for sports like his school’s swim team, but sports did not offer anywhere near the same satisfaction that he received from playing music. His musical ability was critiqued at state performances, where he scored high marks in the areas of technique, creativity, performance, and interpretation.
It wasn’t until he attended Cornell University in Ithaca, N.Y., that the piano took a back seat to the violin. He joined the university’s orchestra, chamber ensembles, and quartets.
“With the violin, I had more opportunity to meet new people, as opposed to playing the piano,” he says. “I had no misconception [about choosing] music as a career, because I felt I wasn’t good enough to be a professional musician.”
But opinions are a lot like music—subjective. Thanks to its unique talent, the university’s orchestra was invited to perform a public concert at Carnegie Hall.
“[It] was a once-in-a-lifetime [experience],” Dr. Chen says, adding that some student musicians at Cornell who share similar interests—music and medicine—recently contacted him to serve as their mentor. “Whether I could get back to that that level of perfection, in terms of playing, is certainly worth attempting, [and it would be] a lot of fun trying.”
Perfect Match
According to Dr. Chen, medicine and music are complementary.
“Medicine and music can be inexorably connected in more ways than one,” he says. “Learning music early on certainly set a tone and established a certain discipline that I rely on every day as a physician.
“Even the process of how you learn music—the repetition, the constant trying to obtain perfection—you can also find in medicine.”
He adds that music and medicine possess similar qualities in terms of their duality. They both offer structure and opportunities for creative expression. Doctors keep track of all the minutia to form a big picture. So do musicians, whose musical notes are combined into a song or symphony. To be a really good doctor or musician, he says, people need to excel at both the creative and technical aspects.
Dr. Chen still practices the violin, roughly an hour each day. Among his favorite pieces are those composed during the romantic period of classical music—generally between the 18th and early 19th centuries.
“You can play musical notes [that are] technically precise, but unless you add a certain creativity to make the music beautiful, then the music doesn’t carry any meaning,” he says.
His music is appreciated by many of his peers when he performs at staff meetings or concerts as part of the hospital’s orchestra. But performing has stirred up a new passion—to pursue other performance venues. He says a handful of exceptional musicians in the hospital’s orchestra are members of the Ann Arbor Symphony Orchestra. Dr. Chen hopes to audition for the orchestra within the next two years, after polishing a new classical piece he is working on—Mendelssohn’s Concerto in E minor.
Until then, he’s considering playing his violin in the hospital’s lobby. Since music is medicine, performing mini concerts throughout the year may help minimize patients’ pain or ease the anxiety experienced by family members.
Not to mention that it also helps Dr. Chen maintain balance in his own life. At this point, he has no plans to sacrifice one career for the other.
“Music has been able to help me get in touch with my human side, nourish and nurture it,” Dr. Chen says. “Music, by all means, helps give equilibrium, so I can stay a complete individual. That’s how I function best, whether as a musician or physician.”
Carol Patton is a freelance writer in Las Vegas.
Most young boys dream about scoring touchdowns or being a superhero, combatting villains or evil aliens. When Brian Chen, MD, was young, he wanted superpowers, too, but of a different sort. He wanted to become a concert violinist.
Now a hospitalist at St. Joseph Mercy Ann Arbor Hospital in Michigan, Dr. Chen recalls watching Itzhak Perlman, the superstar virtuoso of the violin, perform on the PBS show “Great Performances” when he was just five years old. Entranced by Perlman’s genius, he vowed to master the violin. After years of study, practice, and commitment, Dr. Chen has not quite reached Perlman’s status but has achieved more in his musical career than some musicians ever dream of.
Musical Gifts
After watching “Great Performances,” Dr. Chen begged his parents for a violin. At the time, he was learning to play piano.
“My parents were smart about having me start with piano first,” he says, explaining that the piano helped him learn the basics of reading music and distinguishing tones. “With piano, you press a key that produces a sound you intended. With the violin, there’s no such luck. It requires precise placement of fingers. It was sort of something my parents saw that I could eventually [play] once I got past learning the basics.”
Dr. Chen received his first violin when he was six years old. By then, he had already been playing the piano for a year. His excitement quickly turned into frustration, however. Why couldn’t he play like Perlman?
Instead of admitting defeat, he became that much more determined. Throughout middle school and high school, he took piano and violin lessons and practiced both instruments for several hours each day after school. He joined the high school orchestra and the Dayton (Ohio) Philharmonic Youth Orchestra. Both gave free public concerts, which exposed Dr. Chen to the art of performing at a tender age.
He says the violin appeals to him partly because it is a difficult instrument to learn. Not everyone can play it. But, once mastered, he believes “its voice is the most beautiful and expressive, even more than piano.”
Likewise, he says violinists can play stronger and richer tones with a little vibrato, which adds yet another dimension to the instrument.
Like most teenagers, Dr. Chen was active, trying out for sports like his school’s swim team, but sports did not offer anywhere near the same satisfaction that he received from playing music. His musical ability was critiqued at state performances, where he scored high marks in the areas of technique, creativity, performance, and interpretation.
It wasn’t until he attended Cornell University in Ithaca, N.Y., that the piano took a back seat to the violin. He joined the university’s orchestra, chamber ensembles, and quartets.
“With the violin, I had more opportunity to meet new people, as opposed to playing the piano,” he says. “I had no misconception [about choosing] music as a career, because I felt I wasn’t good enough to be a professional musician.”
But opinions are a lot like music—subjective. Thanks to its unique talent, the university’s orchestra was invited to perform a public concert at Carnegie Hall.
“[It] was a once-in-a-lifetime [experience],” Dr. Chen says, adding that some student musicians at Cornell who share similar interests—music and medicine—recently contacted him to serve as their mentor. “Whether I could get back to that that level of perfection, in terms of playing, is certainly worth attempting, [and it would be] a lot of fun trying.”
Perfect Match
According to Dr. Chen, medicine and music are complementary.
“Medicine and music can be inexorably connected in more ways than one,” he says. “Learning music early on certainly set a tone and established a certain discipline that I rely on every day as a physician.
“Even the process of how you learn music—the repetition, the constant trying to obtain perfection—you can also find in medicine.”
He adds that music and medicine possess similar qualities in terms of their duality. They both offer structure and opportunities for creative expression. Doctors keep track of all the minutia to form a big picture. So do musicians, whose musical notes are combined into a song or symphony. To be a really good doctor or musician, he says, people need to excel at both the creative and technical aspects.
Dr. Chen still practices the violin, roughly an hour each day. Among his favorite pieces are those composed during the romantic period of classical music—generally between the 18th and early 19th centuries.
“You can play musical notes [that are] technically precise, but unless you add a certain creativity to make the music beautiful, then the music doesn’t carry any meaning,” he says.
His music is appreciated by many of his peers when he performs at staff meetings or concerts as part of the hospital’s orchestra. But performing has stirred up a new passion—to pursue other performance venues. He says a handful of exceptional musicians in the hospital’s orchestra are members of the Ann Arbor Symphony Orchestra. Dr. Chen hopes to audition for the orchestra within the next two years, after polishing a new classical piece he is working on—Mendelssohn’s Concerto in E minor.
Until then, he’s considering playing his violin in the hospital’s lobby. Since music is medicine, performing mini concerts throughout the year may help minimize patients’ pain or ease the anxiety experienced by family members.
Not to mention that it also helps Dr. Chen maintain balance in his own life. At this point, he has no plans to sacrifice one career for the other.
“Music has been able to help me get in touch with my human side, nourish and nurture it,” Dr. Chen says. “Music, by all means, helps give equilibrium, so I can stay a complete individual. That’s how I function best, whether as a musician or physician.”
Carol Patton is a freelance writer in Las Vegas.
Most young boys dream about scoring touchdowns or being a superhero, combatting villains or evil aliens. When Brian Chen, MD, was young, he wanted superpowers, too, but of a different sort. He wanted to become a concert violinist.
Now a hospitalist at St. Joseph Mercy Ann Arbor Hospital in Michigan, Dr. Chen recalls watching Itzhak Perlman, the superstar virtuoso of the violin, perform on the PBS show “Great Performances” when he was just five years old. Entranced by Perlman’s genius, he vowed to master the violin. After years of study, practice, and commitment, Dr. Chen has not quite reached Perlman’s status but has achieved more in his musical career than some musicians ever dream of.
Musical Gifts
After watching “Great Performances,” Dr. Chen begged his parents for a violin. At the time, he was learning to play piano.
“My parents were smart about having me start with piano first,” he says, explaining that the piano helped him learn the basics of reading music and distinguishing tones. “With piano, you press a key that produces a sound you intended. With the violin, there’s no such luck. It requires precise placement of fingers. It was sort of something my parents saw that I could eventually [play] once I got past learning the basics.”
Dr. Chen received his first violin when he was six years old. By then, he had already been playing the piano for a year. His excitement quickly turned into frustration, however. Why couldn’t he play like Perlman?
Instead of admitting defeat, he became that much more determined. Throughout middle school and high school, he took piano and violin lessons and practiced both instruments for several hours each day after school. He joined the high school orchestra and the Dayton (Ohio) Philharmonic Youth Orchestra. Both gave free public concerts, which exposed Dr. Chen to the art of performing at a tender age.
He says the violin appeals to him partly because it is a difficult instrument to learn. Not everyone can play it. But, once mastered, he believes “its voice is the most beautiful and expressive, even more than piano.”
Likewise, he says violinists can play stronger and richer tones with a little vibrato, which adds yet another dimension to the instrument.
Like most teenagers, Dr. Chen was active, trying out for sports like his school’s swim team, but sports did not offer anywhere near the same satisfaction that he received from playing music. His musical ability was critiqued at state performances, where he scored high marks in the areas of technique, creativity, performance, and interpretation.
It wasn’t until he attended Cornell University in Ithaca, N.Y., that the piano took a back seat to the violin. He joined the university’s orchestra, chamber ensembles, and quartets.
“With the violin, I had more opportunity to meet new people, as opposed to playing the piano,” he says. “I had no misconception [about choosing] music as a career, because I felt I wasn’t good enough to be a professional musician.”
But opinions are a lot like music—subjective. Thanks to its unique talent, the university’s orchestra was invited to perform a public concert at Carnegie Hall.
“[It] was a once-in-a-lifetime [experience],” Dr. Chen says, adding that some student musicians at Cornell who share similar interests—music and medicine—recently contacted him to serve as their mentor. “Whether I could get back to that that level of perfection, in terms of playing, is certainly worth attempting, [and it would be] a lot of fun trying.”
Perfect Match
According to Dr. Chen, medicine and music are complementary.
“Medicine and music can be inexorably connected in more ways than one,” he says. “Learning music early on certainly set a tone and established a certain discipline that I rely on every day as a physician.
“Even the process of how you learn music—the repetition, the constant trying to obtain perfection—you can also find in medicine.”
He adds that music and medicine possess similar qualities in terms of their duality. They both offer structure and opportunities for creative expression. Doctors keep track of all the minutia to form a big picture. So do musicians, whose musical notes are combined into a song or symphony. To be a really good doctor or musician, he says, people need to excel at both the creative and technical aspects.
Dr. Chen still practices the violin, roughly an hour each day. Among his favorite pieces are those composed during the romantic period of classical music—generally between the 18th and early 19th centuries.
“You can play musical notes [that are] technically precise, but unless you add a certain creativity to make the music beautiful, then the music doesn’t carry any meaning,” he says.
His music is appreciated by many of his peers when he performs at staff meetings or concerts as part of the hospital’s orchestra. But performing has stirred up a new passion—to pursue other performance venues. He says a handful of exceptional musicians in the hospital’s orchestra are members of the Ann Arbor Symphony Orchestra. Dr. Chen hopes to audition for the orchestra within the next two years, after polishing a new classical piece he is working on—Mendelssohn’s Concerto in E minor.
Until then, he’s considering playing his violin in the hospital’s lobby. Since music is medicine, performing mini concerts throughout the year may help minimize patients’ pain or ease the anxiety experienced by family members.
Not to mention that it also helps Dr. Chen maintain balance in his own life. At this point, he has no plans to sacrifice one career for the other.
“Music has been able to help me get in touch with my human side, nourish and nurture it,” Dr. Chen says. “Music, by all means, helps give equilibrium, so I can stay a complete individual. That’s how I function best, whether as a musician or physician.”
Carol Patton is a freelance writer in Las Vegas.
Medicare Rankings Favor Small, For-Profit Hospitals

In April, the Centers for Medicare and Medicaid Services (CMS) publicly revealed for the first time which hospitals achieved five stars and which had room for improvement based on patient experience per the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey.
Although these measures are not new, this year CMS assembled the star ratings from HCAHPS survey results and made them available on its consumer-facing website, in an effort to increase transparency.
The decision has not been met without controversy, particularly given the fact that just 251 hospitals out of more than 3,500 received five stars, and only two major teaching hospitals achieved the highest rating. Some professional groups, like the American Hospital Association (AHA), which issued a statement the day CMS released its ratings, believe the rankings risk “oversimplifying the complexity of quality care or misinterpreting what is important to a particular patient, especially since patients seek care for many different reasons.”
Others argue that there is a disconnect between what hospital leaders perceive as important drivers of patient experience and what patients really want. For instance, a 2013 Harvard Business Review article cites a 2012 survey in which C-suite leaders suggested new facilities, private rooms, on-demand food, bedside electronics, and more amenities were necessary to improve patient experience in the hospital.1
“I am surprised at how much controversy there is on this,” says Ashish Jha, MD, MPH, hospitalist at the VA Boston Healthcare System and professor of health policy at the Harvard T.H. Chan School of Public Health. “Modestly good evidence suggests that hospitals that do well on patient experience scores are also the hospitals that have better patient outcomes on more hard measures, like mortality and evidence-based guidelines.”
Dr. Jha cites a February 2015 study, published in the Journal of Hospital Medicine, in which patients were moved from one clinical building to a newer one with more patient-centered features.2 The care team remained the same. The study concluded that patients were able to differentiate between satisfactory clinical care and their surroundings, and that clinical care had a greater impact on patient experience than any other factor.
“Was your pain controlled adequately? Were people responsive to your needs? Were you treated with dignity and respect?” Dr. Jha says. “I think it’s disrespectful to say patients can’t tell the difference between high thread-count bed sheets and being treated with disrespect.”
The HCAHPS survey, Dr. Jha notes, reflects important aspects of healthcare that only patients can report. It encompasses 11 measures that gauge, for example, how well patients felt nurses and physicians communicated with them. It also asks patients to provide an overall hospital rank on a 10-point scale (counting only those that receive a nine or 10), according to Kaiser Health News, and to rate the cleanliness and quietness of the rooms.
Hospitals must send surveys to a random sample of adult patients monthly, including those not on Medicare, within six weeks of discharge, and Inpatient Prospective Payment System hospitals should collect at least 300 surveys every four years, CMS says.
“I think one of the most important things for a hospital to understand is [that] the methodology behind creating the star ratings and the way CMS structures the ratings does make it challenging to achieve the very highest score,” says Akin Demehin, AHA senior associate director of policy.
While CMS applies adjustments to account for sampling methods and patient characteristics of hospitals, an analysis by Dr. Jha’s team showed significant disparities between the rankings of large, academic medical centers and those of small, for-profit hospitals, as well as a substantial difference between hospitals that provide for the greatest number of poor patients and those that serve the fewest.3
However, he writes on his blog, "An Ounce of Evidence," that survey methodology is not the problem and that he believes star ratings are a good idea. Although some hospitals might find themselves at score cut-offs—a one-point difference can translate to a full star change—it’s a “small price to pay to make data more accessible to patients,” he writes.
“There is pretty good evidence hospitals are paying attention, and one that gets a one or two-star rating may be motivated to be better,” Dr. Jha says.
“Every hospital is interested in this because it’s part of value-based purchasing,” says Trina Dorrah, MD, MPH, a hospitalist and director of quality at Baylor Scott & White Health in Round Rock, Texas.
Dr. Dorrah has authored two books focused on patient experience, and she suggests simple ways hospitals can work toward improving their HCAHPS scores, and potentially their star ratings, from having nurses round with physicians to installing communication-facilitating whiteboards in every room.
Her hospital also awards bonuses to the hospitalist group for achieving set goals. Some hospitalist programs around the country are also adding questions to their surveys to link individual providers to patient rankings, she said, though many also do it in aggregate, because linking patients to individual physicians can get “very messy.”
CMS advises caution in interpreting star rankings, acknowledging that they are not the only valuable measures of care quality. Despite the concern over the contextual value of the new rankings, Demehin says AHA supports use of the HCAHPS survey and the value of patient experience measures and believes they should be consulted in conjunction with other quality improvement efforts.
“When I’m really sick and I go to the hospital, I want to be treated with dignity and respect and I want my pain treated quickly, but I also want to survive and not develop an infection,” Dr. Jha says. “That’s obviously not in the star ratings.”
Kelly April Tyrrell is a freelance writer in Madison, Wis.
References
- Merlino J, Raman A. Understanding the drivers of the patient experience. Harvard Business Review. Sept. 17, 2013. Accessed May 14, 2015.
- Siddiqui Z, Zuccarelli R, Durkin N, Wu AW, Brotman DJ. Changes in patient satisfaction related to hospital renovation: Experience with a new clinical building. J Hosp Med. 2015;10(3):165-171.
- Jha A. Finding the stars of hospital care in the U.S. An Ounce of Evidence blog. April 20, 2015. Accessed June 4, 2015.
In April, the Centers for Medicare and Medicaid Services (CMS) publicly revealed for the first time which hospitals achieved five stars and which had room for improvement based on patient experience per the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey.
Although these measures are not new, this year CMS assembled the star ratings from HCAHPS survey results and made them available on its consumer-facing website, in an effort to increase transparency.
The decision has not been met without controversy, particularly given the fact that just 251 hospitals out of more than 3,500 received five stars, and only two major teaching hospitals achieved the highest rating. Some professional groups, like the American Hospital Association (AHA), which issued a statement the day CMS released its ratings, believe the rankings risk “oversimplifying the complexity of quality care or misinterpreting what is important to a particular patient, especially since patients seek care for many different reasons.”
Others argue that there is a disconnect between what hospital leaders perceive as important drivers of patient experience and what patients really want. For instance, a 2013 Harvard Business Review article cites a 2012 survey in which C-suite leaders suggested new facilities, private rooms, on-demand food, bedside electronics, and more amenities were necessary to improve patient experience in the hospital.1
“I am surprised at how much controversy there is on this,” says Ashish Jha, MD, MPH, hospitalist at the VA Boston Healthcare System and professor of health policy at the Harvard T.H. Chan School of Public Health. “Modestly good evidence suggests that hospitals that do well on patient experience scores are also the hospitals that have better patient outcomes on more hard measures, like mortality and evidence-based guidelines.”
Dr. Jha cites a February 2015 study, published in the Journal of Hospital Medicine, in which patients were moved from one clinical building to a newer one with more patient-centered features.2 The care team remained the same. The study concluded that patients were able to differentiate between satisfactory clinical care and their surroundings, and that clinical care had a greater impact on patient experience than any other factor.
“Was your pain controlled adequately? Were people responsive to your needs? Were you treated with dignity and respect?” Dr. Jha says. “I think it’s disrespectful to say patients can’t tell the difference between high thread-count bed sheets and being treated with disrespect.”
The HCAHPS survey, Dr. Jha notes, reflects important aspects of healthcare that only patients can report. It encompasses 11 measures that gauge, for example, how well patients felt nurses and physicians communicated with them. It also asks patients to provide an overall hospital rank on a 10-point scale (counting only those that receive a nine or 10), according to Kaiser Health News, and to rate the cleanliness and quietness of the rooms.
Hospitals must send surveys to a random sample of adult patients monthly, including those not on Medicare, within six weeks of discharge, and Inpatient Prospective Payment System hospitals should collect at least 300 surveys every four years, CMS says.
“I think one of the most important things for a hospital to understand is [that] the methodology behind creating the star ratings and the way CMS structures the ratings does make it challenging to achieve the very highest score,” says Akin Demehin, AHA senior associate director of policy.
While CMS applies adjustments to account for sampling methods and patient characteristics of hospitals, an analysis by Dr. Jha’s team showed significant disparities between the rankings of large, academic medical centers and those of small, for-profit hospitals, as well as a substantial difference between hospitals that provide for the greatest number of poor patients and those that serve the fewest.3
However, he writes on his blog, "An Ounce of Evidence," that survey methodology is not the problem and that he believes star ratings are a good idea. Although some hospitals might find themselves at score cut-offs—a one-point difference can translate to a full star change—it’s a “small price to pay to make data more accessible to patients,” he writes.
“There is pretty good evidence hospitals are paying attention, and one that gets a one or two-star rating may be motivated to be better,” Dr. Jha says.
“Every hospital is interested in this because it’s part of value-based purchasing,” says Trina Dorrah, MD, MPH, a hospitalist and director of quality at Baylor Scott & White Health in Round Rock, Texas.
Dr. Dorrah has authored two books focused on patient experience, and she suggests simple ways hospitals can work toward improving their HCAHPS scores, and potentially their star ratings, from having nurses round with physicians to installing communication-facilitating whiteboards in every room.
Her hospital also awards bonuses to the hospitalist group for achieving set goals. Some hospitalist programs around the country are also adding questions to their surveys to link individual providers to patient rankings, she said, though many also do it in aggregate, because linking patients to individual physicians can get “very messy.”
CMS advises caution in interpreting star rankings, acknowledging that they are not the only valuable measures of care quality. Despite the concern over the contextual value of the new rankings, Demehin says AHA supports use of the HCAHPS survey and the value of patient experience measures and believes they should be consulted in conjunction with other quality improvement efforts.
“When I’m really sick and I go to the hospital, I want to be treated with dignity and respect and I want my pain treated quickly, but I also want to survive and not develop an infection,” Dr. Jha says. “That’s obviously not in the star ratings.”
Kelly April Tyrrell is a freelance writer in Madison, Wis.
References
- Merlino J, Raman A. Understanding the drivers of the patient experience. Harvard Business Review. Sept. 17, 2013. Accessed May 14, 2015.
- Siddiqui Z, Zuccarelli R, Durkin N, Wu AW, Brotman DJ. Changes in patient satisfaction related to hospital renovation: Experience with a new clinical building. J Hosp Med. 2015;10(3):165-171.
- Jha A. Finding the stars of hospital care in the U.S. An Ounce of Evidence blog. April 20, 2015. Accessed June 4, 2015.
In April, the Centers for Medicare and Medicaid Services (CMS) publicly revealed for the first time which hospitals achieved five stars and which had room for improvement based on patient experience per the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey.
Although these measures are not new, this year CMS assembled the star ratings from HCAHPS survey results and made them available on its consumer-facing website, in an effort to increase transparency.
The decision has not been met without controversy, particularly given the fact that just 251 hospitals out of more than 3,500 received five stars, and only two major teaching hospitals achieved the highest rating. Some professional groups, like the American Hospital Association (AHA), which issued a statement the day CMS released its ratings, believe the rankings risk “oversimplifying the complexity of quality care or misinterpreting what is important to a particular patient, especially since patients seek care for many different reasons.”
Others argue that there is a disconnect between what hospital leaders perceive as important drivers of patient experience and what patients really want. For instance, a 2013 Harvard Business Review article cites a 2012 survey in which C-suite leaders suggested new facilities, private rooms, on-demand food, bedside electronics, and more amenities were necessary to improve patient experience in the hospital.1
“I am surprised at how much controversy there is on this,” says Ashish Jha, MD, MPH, hospitalist at the VA Boston Healthcare System and professor of health policy at the Harvard T.H. Chan School of Public Health. “Modestly good evidence suggests that hospitals that do well on patient experience scores are also the hospitals that have better patient outcomes on more hard measures, like mortality and evidence-based guidelines.”
Dr. Jha cites a February 2015 study, published in the Journal of Hospital Medicine, in which patients were moved from one clinical building to a newer one with more patient-centered features.2 The care team remained the same. The study concluded that patients were able to differentiate between satisfactory clinical care and their surroundings, and that clinical care had a greater impact on patient experience than any other factor.
“Was your pain controlled adequately? Were people responsive to your needs? Were you treated with dignity and respect?” Dr. Jha says. “I think it’s disrespectful to say patients can’t tell the difference between high thread-count bed sheets and being treated with disrespect.”
The HCAHPS survey, Dr. Jha notes, reflects important aspects of healthcare that only patients can report. It encompasses 11 measures that gauge, for example, how well patients felt nurses and physicians communicated with them. It also asks patients to provide an overall hospital rank on a 10-point scale (counting only those that receive a nine or 10), according to Kaiser Health News, and to rate the cleanliness and quietness of the rooms.
Hospitals must send surveys to a random sample of adult patients monthly, including those not on Medicare, within six weeks of discharge, and Inpatient Prospective Payment System hospitals should collect at least 300 surveys every four years, CMS says.
“I think one of the most important things for a hospital to understand is [that] the methodology behind creating the star ratings and the way CMS structures the ratings does make it challenging to achieve the very highest score,” says Akin Demehin, AHA senior associate director of policy.
While CMS applies adjustments to account for sampling methods and patient characteristics of hospitals, an analysis by Dr. Jha’s team showed significant disparities between the rankings of large, academic medical centers and those of small, for-profit hospitals, as well as a substantial difference between hospitals that provide for the greatest number of poor patients and those that serve the fewest.3
However, he writes on his blog, "An Ounce of Evidence," that survey methodology is not the problem and that he believes star ratings are a good idea. Although some hospitals might find themselves at score cut-offs—a one-point difference can translate to a full star change—it’s a “small price to pay to make data more accessible to patients,” he writes.
“There is pretty good evidence hospitals are paying attention, and one that gets a one or two-star rating may be motivated to be better,” Dr. Jha says.
“Every hospital is interested in this because it’s part of value-based purchasing,” says Trina Dorrah, MD, MPH, a hospitalist and director of quality at Baylor Scott & White Health in Round Rock, Texas.
Dr. Dorrah has authored two books focused on patient experience, and she suggests simple ways hospitals can work toward improving their HCAHPS scores, and potentially their star ratings, from having nurses round with physicians to installing communication-facilitating whiteboards in every room.
Her hospital also awards bonuses to the hospitalist group for achieving set goals. Some hospitalist programs around the country are also adding questions to their surveys to link individual providers to patient rankings, she said, though many also do it in aggregate, because linking patients to individual physicians can get “very messy.”
CMS advises caution in interpreting star rankings, acknowledging that they are not the only valuable measures of care quality. Despite the concern over the contextual value of the new rankings, Demehin says AHA supports use of the HCAHPS survey and the value of patient experience measures and believes they should be consulted in conjunction with other quality improvement efforts.
“When I’m really sick and I go to the hospital, I want to be treated with dignity and respect and I want my pain treated quickly, but I also want to survive and not develop an infection,” Dr. Jha says. “That’s obviously not in the star ratings.”
Kelly April Tyrrell is a freelance writer in Madison, Wis.
References
- Merlino J, Raman A. Understanding the drivers of the patient experience. Harvard Business Review. Sept. 17, 2013. Accessed May 14, 2015.
- Siddiqui Z, Zuccarelli R, Durkin N, Wu AW, Brotman DJ. Changes in patient satisfaction related to hospital renovation: Experience with a new clinical building. J Hosp Med. 2015;10(3):165-171.
- Jha A. Finding the stars of hospital care in the U.S. An Ounce of Evidence blog. April 20, 2015. Accessed June 4, 2015.
The Hospital Leader Explores Ways Hospitalists Can Tackle Healthcare Costs
EDITOR’S NOTE: A version of this article originally appeared on Medpage Today on May 5, 2015.
By now, we have all heard the stories about unconscionable medical bills causing financial harm for patients. We have read about more Americans than ever before on high-deductible health insurance plans. Some of us have even helped our parents navigate the deceptively simple-looking bronze, silver, and gold tiers of the insurance exchanges, weighing the gamble of increasingly unaffordable monthly premiums against catastrophically high deductibles and out-of-pocket costs.

We have accepted the fact that healthcare costs are out of control and causing real constraints on every level, from individuals to communities to businesses to states to our nation.
OK, but now what are we supposed to do about it?
“Remarkably, given the importance of this issue, until now, we lacked a roadmap to attack it,” wrote Bob Wachter, MD, in the foreword to our new book, Understanding Value-Based Healthcare. “Now we have one.”
For starters, we can supply a pipeline for change by embedding the principles of value-based care in the apprenticeship of health professional education. Recently, the leaders of the soon-to-open Dell Medical School at University of Texas-Austin articulated their plan to build their entire curriculum around teaching students to root out waste and to care for the health of the community. This is the clearest example that medical educators are taking seriously the calls to action coming from the Accreditation Council for Graduate Medical Education (ACGME), the Association of American Medical Colleges (AAMC), the American College of Physicians (ACP), and other leaders to address healthcare value in training very seriously. However, the front lines are not waiting for new medical schools to open up or for massive curricular overhauls. The Second Annual Teaching Value and Choosing Wisely Challenge that we organized resulted in 80 submissions spanning the country (and Canada). The authors, including five students, 30 residents or fellows, and at least 41 faculty members, described their bright ideas and innovations for integrating healthcare value into education.
Education is fundamental but will not be enough. We must also practice what we preach. Practicing clinicians can deflate medical bills for their patients by advocating for appropriate care, considering patient affordability in customizing treatments, and leading local initiatives to improve value of care.
Clinicians can advocate for appropriate care by avoiding low-value services at the point of care. Specific targets for improving appropriate resource utilization may be identified from resources such as Choosing Wisely lists, guidelines, and appropriateness criteria. Physicians will need to understand the true risks and benefits of recommended therapies and learn ways to communicate this balance with patients.
Patient affordability is increasingly important, with more patients now facing astronomically high out-of-pocket bills, even for simple medical treatments or procedures. A December 2014 CBS/New York Times poll found that 80% of Americans now think their doctor should discuss the cost of recommended medical treatment with them ahead of time. Clinicians can screen their patients for financial harm and can help them navigate the trade-offs of lower cost options. Physicians should seek to provide high-value prescribing, which entails recommending the simplest medication regimen that minimizes physical and financial risk to the patient while achieving the best outcome. In other words, decreasing either cost, complexity, or risk of medications can improve value—and clinicians should aim to improve all three simultaneously.
In addition to reducing waste and considering patient affordability, clinicians are ideal leaders of local value initiatives, whether they accomplish this by running value improvement projects or by launching formal high-value care programs. Our framework to guide value improvement project design is “COST”: culture, oversight accountability, system support, and training. This approach leverages principles from implementation science to ensure that value improvement projects successfully provide multi-pronged tactics for overcoming the many barriers to high-value care delivery. At some locations across the country, individual efforts have matured into entire groups dedicated to designing and implementing value-improvement initiatives, including the UCSF High Value Care Committee, the Johns Hopkins High-Value Care Committee, Johns Hopkins Bayview Physicians for Responsible Ordering (PRO), and “High-Value Carolina” in North Carolina.
Health professionals are faced with a responsibility to help deflate medical bills. To achieve this goal, clinicians can advocate for appropriate care, consider patient affordability, and lead local value improvement initiatives. For those ready to tackle this challenge, we elaborate on and explain some of these necessary tools in our book, Understanding Value-Based Healthcare.
The Hospital Leader

Hot on HMX
![]()
• Patient satisfaction • Geographic rounding • LACE Score
To share your own perspective on these topics—or start a new conversation.
EDITOR’S NOTE: A version of this article originally appeared on Medpage Today on May 5, 2015.
By now, we have all heard the stories about unconscionable medical bills causing financial harm for patients. We have read about more Americans than ever before on high-deductible health insurance plans. Some of us have even helped our parents navigate the deceptively simple-looking bronze, silver, and gold tiers of the insurance exchanges, weighing the gamble of increasingly unaffordable monthly premiums against catastrophically high deductibles and out-of-pocket costs.
We have accepted the fact that healthcare costs are out of control and causing real constraints on every level, from individuals to communities to businesses to states to our nation.
OK, but now what are we supposed to do about it?
“Remarkably, given the importance of this issue, until now, we lacked a roadmap to attack it,” wrote Bob Wachter, MD, in the foreword to our new book, Understanding Value-Based Healthcare. “Now we have one.”
For starters, we can supply a pipeline for change by embedding the principles of value-based care in the apprenticeship of health professional education. Recently, the leaders of the soon-to-open Dell Medical School at University of Texas-Austin articulated their plan to build their entire curriculum around teaching students to root out waste and to care for the health of the community. This is the clearest example that medical educators are taking seriously the calls to action coming from the Accreditation Council for Graduate Medical Education (ACGME), the Association of American Medical Colleges (AAMC), the American College of Physicians (ACP), and other leaders to address healthcare value in training very seriously. However, the front lines are not waiting for new medical schools to open up or for massive curricular overhauls. The Second Annual Teaching Value and Choosing Wisely Challenge that we organized resulted in 80 submissions spanning the country (and Canada). The authors, including five students, 30 residents or fellows, and at least 41 faculty members, described their bright ideas and innovations for integrating healthcare value into education.
Education is fundamental but will not be enough. We must also practice what we preach. Practicing clinicians can deflate medical bills for their patients by advocating for appropriate care, considering patient affordability in customizing treatments, and leading local initiatives to improve value of care.
Clinicians can advocate for appropriate care by avoiding low-value services at the point of care. Specific targets for improving appropriate resource utilization may be identified from resources such as Choosing Wisely lists, guidelines, and appropriateness criteria. Physicians will need to understand the true risks and benefits of recommended therapies and learn ways to communicate this balance with patients.
Patient affordability is increasingly important, with more patients now facing astronomically high out-of-pocket bills, even for simple medical treatments or procedures. A December 2014 CBS/New York Times poll found that 80% of Americans now think their doctor should discuss the cost of recommended medical treatment with them ahead of time. Clinicians can screen their patients for financial harm and can help them navigate the trade-offs of lower cost options. Physicians should seek to provide high-value prescribing, which entails recommending the simplest medication regimen that minimizes physical and financial risk to the patient while achieving the best outcome. In other words, decreasing either cost, complexity, or risk of medications can improve value—and clinicians should aim to improve all three simultaneously.
In addition to reducing waste and considering patient affordability, clinicians are ideal leaders of local value initiatives, whether they accomplish this by running value improvement projects or by launching formal high-value care programs. Our framework to guide value improvement project design is “COST”: culture, oversight accountability, system support, and training. This approach leverages principles from implementation science to ensure that value improvement projects successfully provide multi-pronged tactics for overcoming the many barriers to high-value care delivery. At some locations across the country, individual efforts have matured into entire groups dedicated to designing and implementing value-improvement initiatives, including the UCSF High Value Care Committee, the Johns Hopkins High-Value Care Committee, Johns Hopkins Bayview Physicians for Responsible Ordering (PRO), and “High-Value Carolina” in North Carolina.
Health professionals are faced with a responsibility to help deflate medical bills. To achieve this goal, clinicians can advocate for appropriate care, consider patient affordability, and lead local value improvement initiatives. For those ready to tackle this challenge, we elaborate on and explain some of these necessary tools in our book, Understanding Value-Based Healthcare.
The Hospital Leader
Hot on HMX
![]()
• Patient satisfaction • Geographic rounding • LACE Score
To share your own perspective on these topics—or start a new conversation.
EDITOR’S NOTE: A version of this article originally appeared on Medpage Today on May 5, 2015.
By now, we have all heard the stories about unconscionable medical bills causing financial harm for patients. We have read about more Americans than ever before on high-deductible health insurance plans. Some of us have even helped our parents navigate the deceptively simple-looking bronze, silver, and gold tiers of the insurance exchanges, weighing the gamble of increasingly unaffordable monthly premiums against catastrophically high deductibles and out-of-pocket costs.
We have accepted the fact that healthcare costs are out of control and causing real constraints on every level, from individuals to communities to businesses to states to our nation.
OK, but now what are we supposed to do about it?
“Remarkably, given the importance of this issue, until now, we lacked a roadmap to attack it,” wrote Bob Wachter, MD, in the foreword to our new book, Understanding Value-Based Healthcare. “Now we have one.”
For starters, we can supply a pipeline for change by embedding the principles of value-based care in the apprenticeship of health professional education. Recently, the leaders of the soon-to-open Dell Medical School at University of Texas-Austin articulated their plan to build their entire curriculum around teaching students to root out waste and to care for the health of the community. This is the clearest example that medical educators are taking seriously the calls to action coming from the Accreditation Council for Graduate Medical Education (ACGME), the Association of American Medical Colleges (AAMC), the American College of Physicians (ACP), and other leaders to address healthcare value in training very seriously. However, the front lines are not waiting for new medical schools to open up or for massive curricular overhauls. The Second Annual Teaching Value and Choosing Wisely Challenge that we organized resulted in 80 submissions spanning the country (and Canada). The authors, including five students, 30 residents or fellows, and at least 41 faculty members, described their bright ideas and innovations for integrating healthcare value into education.
Education is fundamental but will not be enough. We must also practice what we preach. Practicing clinicians can deflate medical bills for their patients by advocating for appropriate care, considering patient affordability in customizing treatments, and leading local initiatives to improve value of care.
Clinicians can advocate for appropriate care by avoiding low-value services at the point of care. Specific targets for improving appropriate resource utilization may be identified from resources such as Choosing Wisely lists, guidelines, and appropriateness criteria. Physicians will need to understand the true risks and benefits of recommended therapies and learn ways to communicate this balance with patients.
Patient affordability is increasingly important, with more patients now facing astronomically high out-of-pocket bills, even for simple medical treatments or procedures. A December 2014 CBS/New York Times poll found that 80% of Americans now think their doctor should discuss the cost of recommended medical treatment with them ahead of time. Clinicians can screen their patients for financial harm and can help them navigate the trade-offs of lower cost options. Physicians should seek to provide high-value prescribing, which entails recommending the simplest medication regimen that minimizes physical and financial risk to the patient while achieving the best outcome. In other words, decreasing either cost, complexity, or risk of medications can improve value—and clinicians should aim to improve all three simultaneously.
In addition to reducing waste and considering patient affordability, clinicians are ideal leaders of local value initiatives, whether they accomplish this by running value improvement projects or by launching formal high-value care programs. Our framework to guide value improvement project design is “COST”: culture, oversight accountability, system support, and training. This approach leverages principles from implementation science to ensure that value improvement projects successfully provide multi-pronged tactics for overcoming the many barriers to high-value care delivery. At some locations across the country, individual efforts have matured into entire groups dedicated to designing and implementing value-improvement initiatives, including the UCSF High Value Care Committee, the Johns Hopkins High-Value Care Committee, Johns Hopkins Bayview Physicians for Responsible Ordering (PRO), and “High-Value Carolina” in North Carolina.
Health professionals are faced with a responsibility to help deflate medical bills. To achieve this goal, clinicians can advocate for appropriate care, consider patient affordability, and lead local value improvement initiatives. For those ready to tackle this challenge, we elaborate on and explain some of these necessary tools in our book, Understanding Value-Based Healthcare.
The Hospital Leader
Hot on HMX
![]()
• Patient satisfaction • Geographic rounding • LACE Score
To share your own perspective on these topics—or start a new conversation.
SGR Repeal: What It Means for Hospitalists
On April 16, President Obama signed into law a bipartisan, bicameral piece of legislation that not only fully repealed the sustainable growth rate (SGR) but also permanently eliminated the recurring threat of physician payment cuts in Medicare.
Along with the SGR repeal, the Medicare Access and CHIP Reauthorization Act, or MACRA, institutes the Merit-based Incentive Payment System (MIPS). Starting in 2019, the MIPS will consolidate all of Medicare’s current quality reporting programs: the Physician Quality Reporting System (PQRS), the Value-Based Payment Modifier (VBPM) Program, and the meaningful use (MU) requirements, and will restructure their associated penalties.
Under current law, however, physicians are still required to participate in PQRS, VBPM, and MU, or face their associated penalties until the MIPS is fully implemented in 2019. MACRA also incentivizes the adoption of alternative payment models (APMs). APMs are broadly defined within the law as models that involve both upside and downside financial risk (e.g. ACOs or bundled payments) or patient-centered medical homes, provided they improve quality without increasing costs or lower costs without decreasing quality. Those participating in and deriving substantial revenue from an approved APM will not only be exempt from reporting within the MIPS, but will also receive an automatic 5% bonus in their Medicare billing.
Pay-for-performance programs lack relevant quality metrics and are structured in ways that do not account for the realities of providing inpatient care, which increasingly result in headaches for hospitalists. MACRA has the potential to alleviate this burden and reshape the way in which hospitalists are measured.
SHM worked closely with key Congressional committees, as they were developing the SGR repeal legislation, to include flexible language that could better align quality measures for hospitalists. As a result, buried deep in the text of MACRA lies a two-sentence section that makes this goal possible. The law authorizes the “use [of] measures used for a payment system other than for physicians, such as measures for inpatient hospitals, for the purposes of the performance categories [quality and resource use].” Permitting the use of measures from other payment systems allows hospitalists to have the opportunity to align their quality and resource use performance with that of their institutions. As this alignment is not allowed under current law, it brings new potential to level the playing field and increase the relevance of hospitalist quality reporting in the future.
SHM has been pressing CMS to pursue this concept for the last three years, and MACRA finally gives CMS clear authority to move ahead.
The law is not overly specific, so it is not exactly clear how this provision will be implemented. SHM will remain vigilant, working with CMS to ensure that the MIPS-related regulations set the stage for more fair assessment of hospitalists when the MIPS goes into effect in 2019.
Although hospitalists face an uphill battle in terms of current PQRS reporting, the flexibility contained in MACRA provides an important first step toward a better pathway to reporting quality measures that are fair and relevant for hospitalists.
Ellen Boyer is SHM’s government relations project coordinator.
On April 16, President Obama signed into law a bipartisan, bicameral piece of legislation that not only fully repealed the sustainable growth rate (SGR) but also permanently eliminated the recurring threat of physician payment cuts in Medicare.
Along with the SGR repeal, the Medicare Access and CHIP Reauthorization Act, or MACRA, institutes the Merit-based Incentive Payment System (MIPS). Starting in 2019, the MIPS will consolidate all of Medicare’s current quality reporting programs: the Physician Quality Reporting System (PQRS), the Value-Based Payment Modifier (VBPM) Program, and the meaningful use (MU) requirements, and will restructure their associated penalties.
Under current law, however, physicians are still required to participate in PQRS, VBPM, and MU, or face their associated penalties until the MIPS is fully implemented in 2019. MACRA also incentivizes the adoption of alternative payment models (APMs). APMs are broadly defined within the law as models that involve both upside and downside financial risk (e.g. ACOs or bundled payments) or patient-centered medical homes, provided they improve quality without increasing costs or lower costs without decreasing quality. Those participating in and deriving substantial revenue from an approved APM will not only be exempt from reporting within the MIPS, but will also receive an automatic 5% bonus in their Medicare billing.
Pay-for-performance programs lack relevant quality metrics and are structured in ways that do not account for the realities of providing inpatient care, which increasingly result in headaches for hospitalists. MACRA has the potential to alleviate this burden and reshape the way in which hospitalists are measured.
SHM worked closely with key Congressional committees, as they were developing the SGR repeal legislation, to include flexible language that could better align quality measures for hospitalists. As a result, buried deep in the text of MACRA lies a two-sentence section that makes this goal possible. The law authorizes the “use [of] measures used for a payment system other than for physicians, such as measures for inpatient hospitals, for the purposes of the performance categories [quality and resource use].” Permitting the use of measures from other payment systems allows hospitalists to have the opportunity to align their quality and resource use performance with that of their institutions. As this alignment is not allowed under current law, it brings new potential to level the playing field and increase the relevance of hospitalist quality reporting in the future.
SHM has been pressing CMS to pursue this concept for the last three years, and MACRA finally gives CMS clear authority to move ahead.
The law is not overly specific, so it is not exactly clear how this provision will be implemented. SHM will remain vigilant, working with CMS to ensure that the MIPS-related regulations set the stage for more fair assessment of hospitalists when the MIPS goes into effect in 2019.
Although hospitalists face an uphill battle in terms of current PQRS reporting, the flexibility contained in MACRA provides an important first step toward a better pathway to reporting quality measures that are fair and relevant for hospitalists.
Ellen Boyer is SHM’s government relations project coordinator.
On April 16, President Obama signed into law a bipartisan, bicameral piece of legislation that not only fully repealed the sustainable growth rate (SGR) but also permanently eliminated the recurring threat of physician payment cuts in Medicare.
Along with the SGR repeal, the Medicare Access and CHIP Reauthorization Act, or MACRA, institutes the Merit-based Incentive Payment System (MIPS). Starting in 2019, the MIPS will consolidate all of Medicare’s current quality reporting programs: the Physician Quality Reporting System (PQRS), the Value-Based Payment Modifier (VBPM) Program, and the meaningful use (MU) requirements, and will restructure their associated penalties.
Under current law, however, physicians are still required to participate in PQRS, VBPM, and MU, or face their associated penalties until the MIPS is fully implemented in 2019. MACRA also incentivizes the adoption of alternative payment models (APMs). APMs are broadly defined within the law as models that involve both upside and downside financial risk (e.g. ACOs or bundled payments) or patient-centered medical homes, provided they improve quality without increasing costs or lower costs without decreasing quality. Those participating in and deriving substantial revenue from an approved APM will not only be exempt from reporting within the MIPS, but will also receive an automatic 5% bonus in their Medicare billing.
Pay-for-performance programs lack relevant quality metrics and are structured in ways that do not account for the realities of providing inpatient care, which increasingly result in headaches for hospitalists. MACRA has the potential to alleviate this burden and reshape the way in which hospitalists are measured.
SHM worked closely with key Congressional committees, as they were developing the SGR repeal legislation, to include flexible language that could better align quality measures for hospitalists. As a result, buried deep in the text of MACRA lies a two-sentence section that makes this goal possible. The law authorizes the “use [of] measures used for a payment system other than for physicians, such as measures for inpatient hospitals, for the purposes of the performance categories [quality and resource use].” Permitting the use of measures from other payment systems allows hospitalists to have the opportunity to align their quality and resource use performance with that of their institutions. As this alignment is not allowed under current law, it brings new potential to level the playing field and increase the relevance of hospitalist quality reporting in the future.
SHM has been pressing CMS to pursue this concept for the last three years, and MACRA finally gives CMS clear authority to move ahead.
The law is not overly specific, so it is not exactly clear how this provision will be implemented. SHM will remain vigilant, working with CMS to ensure that the MIPS-related regulations set the stage for more fair assessment of hospitalists when the MIPS goes into effect in 2019.
Although hospitalists face an uphill battle in terms of current PQRS reporting, the flexibility contained in MACRA provides an important first step toward a better pathway to reporting quality measures that are fair and relevant for hospitalists.
Ellen Boyer is SHM’s government relations project coordinator.
Hospitalists Play Key Role Developing Electronic Health Records
If you are a hospitalist, you know this to be true: Electronic health records (EHRs) run more smoothly because of our input. According to the 2014 State of Hospital Medicine (SOHM) report, hospitalists played a significant role in implementing their hospitals’ EHR systems; 34% of respondent hospital medicine groups (HMGs) serving adults only develop order sets, protocols, and decision support, while 28.3% of HMGs serve as super users. As super users, hospitalists are role models who help other physicians work successfully with the computerized systems and set the tone for acceptance within their medical staff and the larger hospital staff.

When EHRs were implemented a decade ago at Allina Health, a 14-hospital system headquartered in Minneapolis, hospitalists acted as super users and were able to interface closely with IT staff, quickly fixing computer troubles that physicians encountered. We partnered with IT to help them prioritize their work by letting them know which computer issues involved patient safety, which were high frequency, irksome issues, and which EHR quirks could be queued farther down the line for repair.
Although successful EHR implementation is a mammoth accomplishment, it is just the tip of the iceberg. Once a system is implemented, an unending amount of work is needed to make sure EHRs deliver, through data analysis and standardization of care, all the efficiency, safety, and cost containment we hoped for at their inception.
When hospitalists at Allina were asked to be the first group of physicians to convert to professional fee billing using the EHR, we found that we needed to create a check-and-balance reporting system to ensure that our charges were indeed being captured. All of our health system physicians now use the hospitalist-developed reporting system.
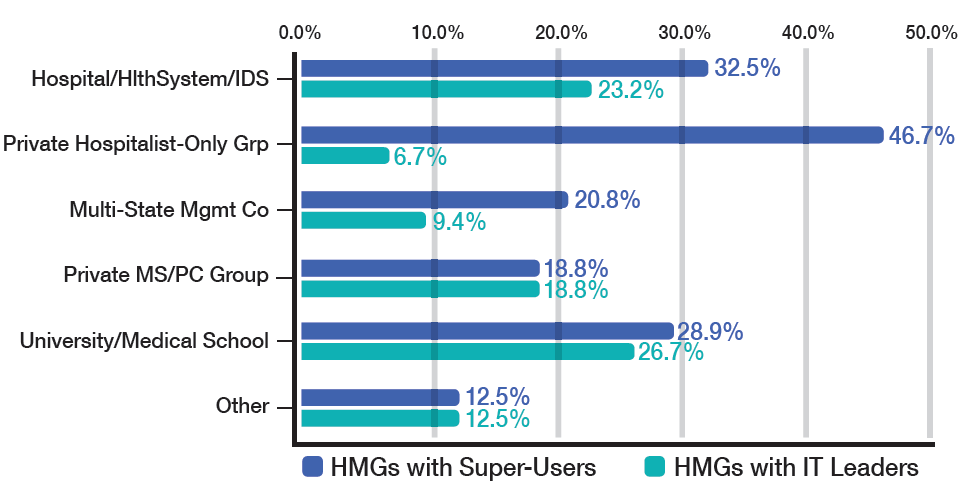
Hospitalists in our system also forged the way to getting meaningful data out of the EHR system. When we first implemented our EHR, we found it difficult to get hospitalist-specific data. We worked closely with data analytics staff to develop a hospitalist “flag” that would differentiate hospitalists from other physicians providing inpatient care.
Because hospitalists are involved with patients through all phases of their care, from admission to discharge, they encounter firsthand the benefits and glitches of EHRs. By the very nature of our work, we are positioned to help improve EHRs. Hospitalist input ranges from informal feedback to the IT department to participation in focused work groups dedicated to specific aspects of improving patient care. Not surprisingly, 18.4% of adults-only HMGs who participated in the last SOHM survey have hospitalists serving in an IT leadership role for their organizations.
When looked at by ownership/employment models (see Table 1), this number is higher for the “University, Medical School, or Faculty Practice Plan” (26.7%) and for the “Hospital, Health System, or Integrated Delivery System” (23.2%) and lower for the “Private Local/Regional Hospitalist-Only Medical Groups” (6.7%).
Interestingly, the “Private Local/Regional Hospitalist-Only” employment model has the highest percentage of super users (46.7%).
Although most hospitalists across the nation spend a majority of their time interfacing with EHRs, 4.5% of HMGs in the most recent SOHM responded that their hospitals or health systems have not yet begun EHR implementation. I’m guessing the hospitalists in those organizations have a lot of work ahead of them in the next year or two!
Dr. Stephan is a hospitalist at Abbott Northwestern Hospital in Minneapolis and a member of the SHM Practice Analysis Committee.
If you are a hospitalist, you know this to be true: Electronic health records (EHRs) run more smoothly because of our input. According to the 2014 State of Hospital Medicine (SOHM) report, hospitalists played a significant role in implementing their hospitals’ EHR systems; 34% of respondent hospital medicine groups (HMGs) serving adults only develop order sets, protocols, and decision support, while 28.3% of HMGs serve as super users. As super users, hospitalists are role models who help other physicians work successfully with the computerized systems and set the tone for acceptance within their medical staff and the larger hospital staff.
When EHRs were implemented a decade ago at Allina Health, a 14-hospital system headquartered in Minneapolis, hospitalists acted as super users and were able to interface closely with IT staff, quickly fixing computer troubles that physicians encountered. We partnered with IT to help them prioritize their work by letting them know which computer issues involved patient safety, which were high frequency, irksome issues, and which EHR quirks could be queued farther down the line for repair.
Although successful EHR implementation is a mammoth accomplishment, it is just the tip of the iceberg. Once a system is implemented, an unending amount of work is needed to make sure EHRs deliver, through data analysis and standardization of care, all the efficiency, safety, and cost containment we hoped for at their inception.
When hospitalists at Allina were asked to be the first group of physicians to convert to professional fee billing using the EHR, we found that we needed to create a check-and-balance reporting system to ensure that our charges were indeed being captured. All of our health system physicians now use the hospitalist-developed reporting system.
Hospitalists in our system also forged the way to getting meaningful data out of the EHR system. When we first implemented our EHR, we found it difficult to get hospitalist-specific data. We worked closely with data analytics staff to develop a hospitalist “flag” that would differentiate hospitalists from other physicians providing inpatient care.
Because hospitalists are involved with patients through all phases of their care, from admission to discharge, they encounter firsthand the benefits and glitches of EHRs. By the very nature of our work, we are positioned to help improve EHRs. Hospitalist input ranges from informal feedback to the IT department to participation in focused work groups dedicated to specific aspects of improving patient care. Not surprisingly, 18.4% of adults-only HMGs who participated in the last SOHM survey have hospitalists serving in an IT leadership role for their organizations.
When looked at by ownership/employment models (see Table 1), this number is higher for the “University, Medical School, or Faculty Practice Plan” (26.7%) and for the “Hospital, Health System, or Integrated Delivery System” (23.2%) and lower for the “Private Local/Regional Hospitalist-Only Medical Groups” (6.7%).
Interestingly, the “Private Local/Regional Hospitalist-Only” employment model has the highest percentage of super users (46.7%).
Although most hospitalists across the nation spend a majority of their time interfacing with EHRs, 4.5% of HMGs in the most recent SOHM responded that their hospitals or health systems have not yet begun EHR implementation. I’m guessing the hospitalists in those organizations have a lot of work ahead of them in the next year or two!
Dr. Stephan is a hospitalist at Abbott Northwestern Hospital in Minneapolis and a member of the SHM Practice Analysis Committee.
If you are a hospitalist, you know this to be true: Electronic health records (EHRs) run more smoothly because of our input. According to the 2014 State of Hospital Medicine (SOHM) report, hospitalists played a significant role in implementing their hospitals’ EHR systems; 34% of respondent hospital medicine groups (HMGs) serving adults only develop order sets, protocols, and decision support, while 28.3% of HMGs serve as super users. As super users, hospitalists are role models who help other physicians work successfully with the computerized systems and set the tone for acceptance within their medical staff and the larger hospital staff.
When EHRs were implemented a decade ago at Allina Health, a 14-hospital system headquartered in Minneapolis, hospitalists acted as super users and were able to interface closely with IT staff, quickly fixing computer troubles that physicians encountered. We partnered with IT to help them prioritize their work by letting them know which computer issues involved patient safety, which were high frequency, irksome issues, and which EHR quirks could be queued farther down the line for repair.
Although successful EHR implementation is a mammoth accomplishment, it is just the tip of the iceberg. Once a system is implemented, an unending amount of work is needed to make sure EHRs deliver, through data analysis and standardization of care, all the efficiency, safety, and cost containment we hoped for at their inception.
When hospitalists at Allina were asked to be the first group of physicians to convert to professional fee billing using the EHR, we found that we needed to create a check-and-balance reporting system to ensure that our charges were indeed being captured. All of our health system physicians now use the hospitalist-developed reporting system.
Hospitalists in our system also forged the way to getting meaningful data out of the EHR system. When we first implemented our EHR, we found it difficult to get hospitalist-specific data. We worked closely with data analytics staff to develop a hospitalist “flag” that would differentiate hospitalists from other physicians providing inpatient care.
Because hospitalists are involved with patients through all phases of their care, from admission to discharge, they encounter firsthand the benefits and glitches of EHRs. By the very nature of our work, we are positioned to help improve EHRs. Hospitalist input ranges from informal feedback to the IT department to participation in focused work groups dedicated to specific aspects of improving patient care. Not surprisingly, 18.4% of adults-only HMGs who participated in the last SOHM survey have hospitalists serving in an IT leadership role for their organizations.
When looked at by ownership/employment models (see Table 1), this number is higher for the “University, Medical School, or Faculty Practice Plan” (26.7%) and for the “Hospital, Health System, or Integrated Delivery System” (23.2%) and lower for the “Private Local/Regional Hospitalist-Only Medical Groups” (6.7%).
Interestingly, the “Private Local/Regional Hospitalist-Only” employment model has the highest percentage of super users (46.7%).
Although most hospitalists across the nation spend a majority of their time interfacing with EHRs, 4.5% of HMGs in the most recent SOHM responded that their hospitals or health systems have not yet begun EHR implementation. I’m guessing the hospitalists in those organizations have a lot of work ahead of them in the next year or two!
Dr. Stephan is a hospitalist at Abbott Northwestern Hospital in Minneapolis and a member of the SHM Practice Analysis Committee.