User login
Global Health Hospitalists Share a Passion for Their Work

Global health hospitalists are passionate about their work. The Hospitalist asked them to expand on the reasons they choose this work.
“Working in Haiti has been the most compelling work in my life,” says Michelle Morse, MD, MPH, an instructor in medicine at Harvard Medical School and deputy chief medical officer for Partners in Health (PIH) in Boston. She has worked with the Navajo Nation in conjunction with PIH’s Community Outreach and Patient Empowerment (COPE) program. The sharing of information is “bi-directional,” Dr. Morse says.

Her Haitian colleagues, she says, have developed “transformative” systems improvements, and she’s found that her own diagnostic and physical exam skills have strengthened because of her work abroad.
“You really have to think bigger than your group of patients and bigger than your community, and think about the whole system to make things better around the world,” she says. “I think that is a fundamental part of becoming a physician.”
UCSF clinical fellow Varun Verma, MD, says he was tired of working in “fragmented volunteer assignments” with relief organizations. Three-month clinical rotations, in which he essentially functions as a teaching attending, have solved the “filling in” feeling he’d grown weary of.
“Here at St. Thérèse Hospital [in Hinche, Haiti], they do not need us to take care of patients on a moment-to-moment basis. There are Haitian clinicians for that,” he says. “Part of our job is to do medical teaching of residents and try to involve everyone in quality improvement projects. It’s sometimes challenging discussing best practices of managing conditions, given the resources at hand, but I find that the Haitian doctors are always interested in learning how we do things in the U.S.”
Evan Lyon, MD, assistant professor of medicine in the section of hospital medicine, supervises clinical fellows in the department of medicine at the University of Chicago. He believes hospitalists who take on global health assignments gain a deeper appreciation for assessing patients’ social histories.
“There’s no better way to deepen your learning of physical exam and history-taking skills than to be out here on the edge and have to rely on those skills,” he says. “Back in the states, you might order an echocardiogram before you listen to the patient’s heart. I think all of us have a different relationship to labs, testing, and X-rays when we return. But the deepest influence for me has been around understanding patients’ social histories and their social context, which is a neglected piece of American medicine.”

Sharing resources and knowledge is what drives Marwa Shoeb MD, MS, assistant professor in the division of hospital medicine at UCSF. “I see this as an extension of our daily work,” she says. “We are just taking it to a different context.”
Gretchen Henkel is a freelance writer in southern California.
Global health hospitalists are passionate about their work. The Hospitalist asked them to expand on the reasons they choose this work.
“Working in Haiti has been the most compelling work in my life,” says Michelle Morse, MD, MPH, an instructor in medicine at Harvard Medical School and deputy chief medical officer for Partners in Health (PIH) in Boston. She has worked with the Navajo Nation in conjunction with PIH’s Community Outreach and Patient Empowerment (COPE) program. The sharing of information is “bi-directional,” Dr. Morse says.
Her Haitian colleagues, she says, have developed “transformative” systems improvements, and she’s found that her own diagnostic and physical exam skills have strengthened because of her work abroad.
“You really have to think bigger than your group of patients and bigger than your community, and think about the whole system to make things better around the world,” she says. “I think that is a fundamental part of becoming a physician.”
UCSF clinical fellow Varun Verma, MD, says he was tired of working in “fragmented volunteer assignments” with relief organizations. Three-month clinical rotations, in which he essentially functions as a teaching attending, have solved the “filling in” feeling he’d grown weary of.
“Here at St. Thérèse Hospital [in Hinche, Haiti], they do not need us to take care of patients on a moment-to-moment basis. There are Haitian clinicians for that,” he says. “Part of our job is to do medical teaching of residents and try to involve everyone in quality improvement projects. It’s sometimes challenging discussing best practices of managing conditions, given the resources at hand, but I find that the Haitian doctors are always interested in learning how we do things in the U.S.”
Evan Lyon, MD, assistant professor of medicine in the section of hospital medicine, supervises clinical fellows in the department of medicine at the University of Chicago. He believes hospitalists who take on global health assignments gain a deeper appreciation for assessing patients’ social histories.
“There’s no better way to deepen your learning of physical exam and history-taking skills than to be out here on the edge and have to rely on those skills,” he says. “Back in the states, you might order an echocardiogram before you listen to the patient’s heart. I think all of us have a different relationship to labs, testing, and X-rays when we return. But the deepest influence for me has been around understanding patients’ social histories and their social context, which is a neglected piece of American medicine.”
Sharing resources and knowledge is what drives Marwa Shoeb MD, MS, assistant professor in the division of hospital medicine at UCSF. “I see this as an extension of our daily work,” she says. “We are just taking it to a different context.”
Gretchen Henkel is a freelance writer in southern California.
Global health hospitalists are passionate about their work. The Hospitalist asked them to expand on the reasons they choose this work.
“Working in Haiti has been the most compelling work in my life,” says Michelle Morse, MD, MPH, an instructor in medicine at Harvard Medical School and deputy chief medical officer for Partners in Health (PIH) in Boston. She has worked with the Navajo Nation in conjunction with PIH’s Community Outreach and Patient Empowerment (COPE) program. The sharing of information is “bi-directional,” Dr. Morse says.
Her Haitian colleagues, she says, have developed “transformative” systems improvements, and she’s found that her own diagnostic and physical exam skills have strengthened because of her work abroad.
“You really have to think bigger than your group of patients and bigger than your community, and think about the whole system to make things better around the world,” she says. “I think that is a fundamental part of becoming a physician.”
UCSF clinical fellow Varun Verma, MD, says he was tired of working in “fragmented volunteer assignments” with relief organizations. Three-month clinical rotations, in which he essentially functions as a teaching attending, have solved the “filling in” feeling he’d grown weary of.
“Here at St. Thérèse Hospital [in Hinche, Haiti], they do not need us to take care of patients on a moment-to-moment basis. There are Haitian clinicians for that,” he says. “Part of our job is to do medical teaching of residents and try to involve everyone in quality improvement projects. It’s sometimes challenging discussing best practices of managing conditions, given the resources at hand, but I find that the Haitian doctors are always interested in learning how we do things in the U.S.”
Evan Lyon, MD, assistant professor of medicine in the section of hospital medicine, supervises clinical fellows in the department of medicine at the University of Chicago. He believes hospitalists who take on global health assignments gain a deeper appreciation for assessing patients’ social histories.
“There’s no better way to deepen your learning of physical exam and history-taking skills than to be out here on the edge and have to rely on those skills,” he says. “Back in the states, you might order an echocardiogram before you listen to the patient’s heart. I think all of us have a different relationship to labs, testing, and X-rays when we return. But the deepest influence for me has been around understanding patients’ social histories and their social context, which is a neglected piece of American medicine.”
Sharing resources and knowledge is what drives Marwa Shoeb MD, MS, assistant professor in the division of hospital medicine at UCSF. “I see this as an extension of our daily work,” she says. “We are just taking it to a different context.”
Gretchen Henkel is a freelance writer in southern California.
Brett Hendel-Paterson, MD, Discusses Advantages of Needs Assessments
Listen to more of our interview with Dr. Hendel-Paterson, as he discusses the advantages of a good needs assessment.
Listen to more of our interview with Dr. Hendel-Paterson, as he discusses the advantages of a good needs assessment.
Listen to more of our interview with Dr. Hendel-Paterson, as he discusses the advantages of a good needs assessment.
What Patients Undergoing Gastrointestinal Endoscopic Procedures Should Receive Antibiotic Prophylaxis?
Case
You are asked to admit two patients. The first is a 75-year-old male with a prosthetic aortic valve on warfarin who presents with bright red blood per rectum and is scheduled for colonoscopy. The second patient is a 35-year-old female with biliary obstruction due to choledocholithiasis; she is afebrile with normal vital signs and no leukocytosis. She underwent endoscopic retrograde cholangiopancreatography (ERCP), which did not resolve her biliary obstruction. Should you prescribe prophylactic antibiotics for either patient?
Overview
Providers are often confused regarding which patients undergoing gastrointestinal (GI) endoscopic procedures should receive antibiotic prophylaxis. To answer this question, it is important to understand the goal of prophylactic antibiotics. Are we trying to prevent infective endocarditis or a localized infection?
There are few large, prospective, randomized controlled trials that have examined the need for antibiotic prophylaxis with GI endoscopic procedures. Guidelines from professional societies are mainly based on expert opinion, evidence from retrospective case studies, and meta-analysis reviews.
Review of the Data
Infective endocarditis resulting from GI endoscopy has been a concern of physicians for decades. The American Heart Association (AHA) first published its recommendations for antibiotic prophylaxis of GI tract procedures in 1965. The most recent antibacterial prophylaxis guidelines, published in 2007, have simplified recommendations and greatly scaled back the indications for antibiotics. The new guidelines conclude that frequent bacteremia from daily activities is more likely to precipitate endocarditis than a single dental, GI, or genitourinary tract procedure.1
The American Society for Gastrointestinal Endoscopy (ASGE) reports that 14.2 million colonoscopies, 2.8 million flexible sigmoidoscopies, and nearly as many upper endoscopies are performed in the U.S. each year, but only 15 cases of endocarditis have been reported with a temporal association to a procedure.2
The British Society of Gastroenterology (BSG) found, after reviewing the histories of patients with infective endocarditis from 1983 through 2006, that there is not enough evidence to warrant antibiotic prophylaxis prior to endoscopy. They noted less than one case of endocarditis after GI endoscopy per year as well as significant variation in the time interval between the procedure and symptoms. The BSG also recognized that antibiotic prophylaxis does not always protect against infection and that clinical factors unrelated to the endoscopy may play a significant role in the development of endocarditis.3
Upper GI Endoscopy, Colonoscopy with Biopsy, and Esophageal Dilatation. Administering antibiotics to prevent infective endocarditis is not recommended for patients undergoing routine procedures such as endoscopy with biopsy and colonoscopy with polypectomy. Likewise, patients with a history of prosthetic heart valves, valve repair with prosthetic material, endocarditis, congenital heart disease, or cardiac transplant with valvulopathy do not need prophylactic antibiotics before GI endoscopic procedures. However, for patients who are being treated for an active GI infection, antibiotic coverage for enterococcus may be warranted given the increased risk of developing endocarditis. The AHA acknowledges there are no published studies to support the efficacy of antibiotics to prevent enterococcal endocarditis in patients in this clinical setting.1
Unlike routine endoscopy, esophageal dilation is associated with an increased rate of bacteremia (12%-100%).4 Streptococcus viridans has been found in blood cultures up to 79% of the time after esophageal dilation.5 Patients with malignant strictures have higher rates of bacteremia than those with benign strictures (52.9% versus 15.7%). Patients treated with multiple passes with the esophageal dilator compared to those treated with a single dilation have a higher risk of bacteremia.6 All patients undergoing esophageal stricture dilation should receive pre-procedural prophylactic antibiotics.7
Patients with bleeding esophageal varices also have high rates of bacteremia. Up to 20% of patients with cirrhosis and GI bleeding on admission develop an infection within 48 hours of presentation.8 There is evidence that the bacteremia may actually be related to the variceal bleeding rather than the procedure.9 Patients with bleeding esophageal varices treated with antibiotics have improved outcomes, including a decrease in mortality.10 Therefore, all patients with bleeding esophageal varices should be placed on antibiotic therapy regardless of whether an endoscopic intervention is planned.
Percutaneous Endoscopic Gastrostomy (PEG) Placement. Prophylactic antibiotics are recommended before placement of a PEG. The indication for prophylactic antibiotics is to prevent a gastrostomy site infection, not infective endocarditis. Gastrostomy site infection is unfortunately a fairly common infection, affecting 4% to 30% of patients who undergo PEG tube placement. There is significant evidence that antibiotics are beneficial in preventing peristomal infections. A meta-analysis showed that only eight patients need to be treated with prophylactic antibiotics to prevent a single peristomal infection.11 Since these infections are believed to be caused by contamination from the oropharynx, physicians should consider prophylaxis against pathogens from the oral flora.12
More recently, it has been noted that methicillin-resistant Staphylococcus aureus (MRSA) is increasingly cultured from infection sites.13 In centers with endemic MRSA, patients should be screened and then undergo decontamination prior to the PEG placement in positive cases.
Endoscopic Ultrasound with Fine Needle Aspiration (EUS-FNA). Antibiotic prophylaxis before EUS-FNA of a solid lesion in an organ is generally thought to be unnecessary because the risk of bacteremia with this procedure is low, comparable to routine GI endoscopy with biopsy. The recommendation for prophylactic antibiotics before biopsy of a cystic lesion is different. There is concern that puncturing cystic lesions may create a new infected fluid collection.2 A systematic review of more than 10,000 patients undergoing EUS-FNA with a full range of target organs revealed that, overall, 11.2% of patients experienced a fever and 4.7% of patients had a peri-procedural infection. While it was not possible in this study to determine which patients received prophylactic antibiotics, 93.7% of patients with pancreatic cystic lesions were reported to have been treated with antibiotics.14
A separate, single-center, retrospective trial produced different results. This study examined a population of 253 patients who underwent 266 EUS-FNA of pancreatic cysts and found that prophylactic antibiotics were associated with more adverse events and were not protective for the 3% of the patients with infectious symptoms.15 Despite the conflicting data, guidelines at this time recommend prophylactic antibiotics before drainage of a sterile pancreatic fluid collection that communicates with the pancreatic duct and also for aspiration of cystic lesions along the GI tract and the mediastinum.2
Endoscopic Retrograde Cholangiopancreatography (ERCP). In patients undergoing ERCP, the routine use of prophylactic antibiotics has not been found to be effective in decreasing the risk of post-procedure cholangitis.16 Guidelines recommend the use of prophylactic antibiotics only in those patients in which the ERCP may not completely resolve the biliary obstruction.2 In these patients, the thought is that ERCP can precipitate infection by disturbing bacteria already present in the biliary tree, especially with increased intrabiliary pressure at the time of contrast dye injection.17
Patients with incomplete biliary drainage, including those with primary sclerosing cholangitis (PSC), hilar cholangiocarcinoma, persistent biliary that were not extracted, and strictures that continue to obstruct despite attempted intervention, are thought to be at elevated risk of developing cholangitis post-ERCP. These patients should be placed on prophylactic antibiotics at the time of the procedure to cover biliary flora such as enteric gram negatives and enterococci. Antibiotics should be continued until the biliary obstruction is resolved.2
Additional Populations to Consider. Previously, the International Society for Peritoneal Dialysis recommended that patients on peritoneal dialysis receive prophylactic antibiotics and empty their abdomen of dialysate prior to colonoscopy. This recommendation has been removed from the 2010 guidelines.18 There is also no indication that patients with synthetic vascular grafts or cardiac devices should receive prophylactic antibiotics prior to routine GI endoscopy.19 The American Academy of Orthopaedic Surgeons no longer recommends that patients with joint replacements receive antibiotic prophylaxis prior to GI endoscopy.20
Back to the Case
The older gentleman with a prosthetic valve undergoing colonoscopy should not receive prophylactic antibiotics, because even in the setting of valvulopathy, colonoscopy does not pose a significant risk for infective endocarditis. The young patient with severe choledocholithiasis should be placed on prophylactic antibiotics because she has continued biliary obstruction, which could result in a cholangitis after ERCP.
Bottom Line
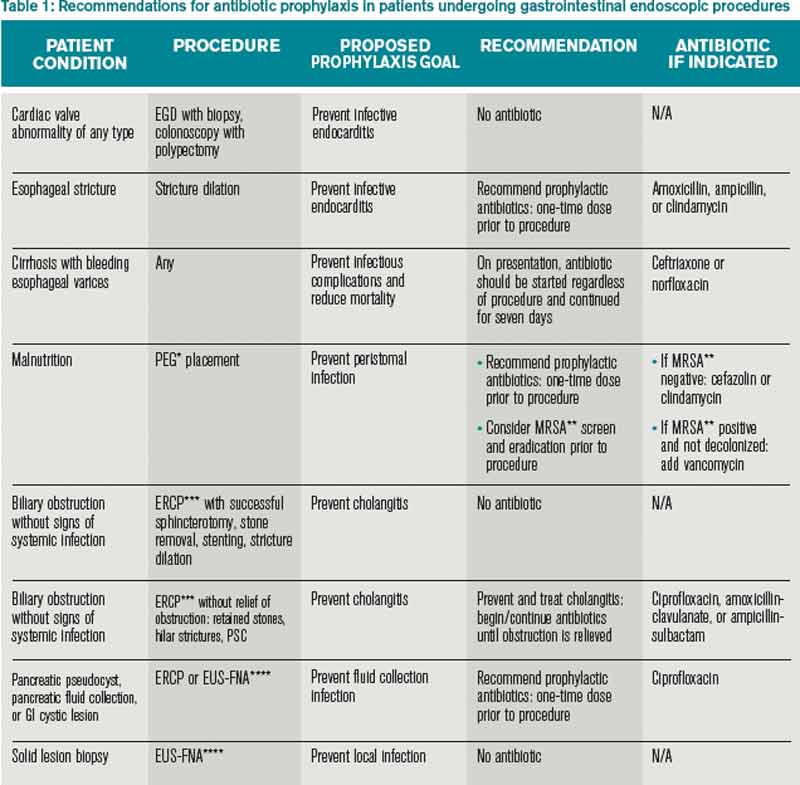
Prophylactic antibiotics are not recommended for any patient undergoing routine endoscopy or colonoscopy. They are indicated for patients with bleeding esophageal varices and for patients who undergo esophageal stricture dilation, PEG placement, or pseudocyst or cyst drainage, and those with continued biliary obstruction undergoing ERCP as summarized in Table 1.
Drs. Ritter, Jupiter, Carbo, and Li are hospitalists at Beth Israel Deaconess Medical Center and Harvard Medical School faculty in Boston.
References
- Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007;116(15):1736-1754.
- Banerjee S, Shen B, Baron TH, et al. Antibiotic prophylaxis for GI endoscopy. Gastrointest Endosc. 2008;67(6):791-798.
- Allison MC, Sandoe JA, Tighe R, Simpson IA, Hall RJ, Elliott TS. Antibiotic prophylaxis in gastrointestinal endoscopy. Gut. 2009;58(6):869-880.
- Nelson DB. Infectious disease complications of GI endoscopy: Part I, endogenous infections. Gastrointest Endosc. 2003;57(4):546-556.
- Zuccaro G Jr., Richter JE, Rice TW, et al. Viridans streptococcal bacteremia after esophageal stricture dilation. Gastrointest Endosc. 1998;48(6):568-573.
- Nelson DB, Sanderson SJ, Azar MM. Bacteremia with esophageal dilation. Gastrointest Endosc.1998;48(6):563-567.
- Hirota WK, Petersen K, Baron TH, et al. Guidelines for antibiotic prophylaxis for GI endoscopy. Gastrointest Endosc. 2003;58(4):475-482.
- Ho H, Zuckerman MJ, Wassem C. A prospective controlled study of the risk of bacteremia in emergency sclerotherapy of esophageal varices. Gastroenterology. 1991;101(6):1642-1648.
- Rolando N, Gimson A, Philpott-Howard J, et al. Infectious sequelae after endoscopic sclerotherapy of oesophageal varices: Role of antibiotic prophylaxis. J Hepatol. 1993;18(3):290-294.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology. 2007;46(3):922-938.
- Jafri NS, Mahid SS, Minor KS, Idstein SR, Hornung CA, Galandiuk S. Meta-analysis: Antibiotic prophylaxis to prevent peristomal infection following percutaneous endoscopic gastrostomy. Aliment Pharmacol Ther. 2007;25(6):647-656.
- Chuang CH, Hung KH, Chen JR, et al. Airway infection predisposes to peristomal infection after percutaneous endoscopic gastrostomy with high concordance between sputum and wound isolates. J Gastrointest Surg. 2010;14(1):45-51.
- Chaudhary KA, Smith OJ, Cuddy PG, Clarkston WK. PEG site infections: The emergence of methicillin resistant Staphylococcus aureus as a major pathogen. Am J Gastroenterol. 2002;97(7):1713-1716.
- Wang KX, Ben QW, Jin ZD, et al. Assessment of morbidity and mortality associated with EUS-guided FNA: A systematic review. Gastrointest Endosc. 2011;73(2):283-290.
- Guarner-Argente C, Shah P, Buchner A, Ahmad NA, Kochman ML, Ginsberg GG. Use of antimicrobials for EUS-guided FNA of pancreatic cysts: A retrospective, comparative analysis. Gastrointest Endosc. 2011;74(1):81-86.
- Bai Y, Gao F, Gao J, Zou DW, Li ZS. Prophylactic antibiotics cannot prevent endoscopic retrograde cholangiopancreatography-induced cholangitis: A meta-analysis. Pancreas. 2009;38(2):126-130.
- Cotton PB, Connor P, Rawls E, Romagnuolo J. Infection after ERCP, and antibiotic prophylaxis: A sequential quality-improvement approach over 11 years. Gastrointest Endosc. 2008;67(3):471-475.
- Li PK, Szeto CC, Piraino B, et al. Peritoneal dialysis-related infections recommendations: 2010 update. Perit Dial Int. 2010;30(4):393-423.
- Baddour LM, Bettmann MA, Bolger AF, et al. Nonvalvular cardiovascular device-related infections. Circulation. 2003;108(16):2015-2031.
- Rethman MP, Watters W III, Abt E, et al. The American Academy of Orthopaedic Surgeons and the American Dental Association clinical practice guideline on the prevention of orthopaedic implant infection in patients undergoing dental procedures. J Bone Joint Surg Am. 2013;95(8):745-747.
Case
You are asked to admit two patients. The first is a 75-year-old male with a prosthetic aortic valve on warfarin who presents with bright red blood per rectum and is scheduled for colonoscopy. The second patient is a 35-year-old female with biliary obstruction due to choledocholithiasis; she is afebrile with normal vital signs and no leukocytosis. She underwent endoscopic retrograde cholangiopancreatography (ERCP), which did not resolve her biliary obstruction. Should you prescribe prophylactic antibiotics for either patient?
Overview
Providers are often confused regarding which patients undergoing gastrointestinal (GI) endoscopic procedures should receive antibiotic prophylaxis. To answer this question, it is important to understand the goal of prophylactic antibiotics. Are we trying to prevent infective endocarditis or a localized infection?
There are few large, prospective, randomized controlled trials that have examined the need for antibiotic prophylaxis with GI endoscopic procedures. Guidelines from professional societies are mainly based on expert opinion, evidence from retrospective case studies, and meta-analysis reviews.
Review of the Data
Infective endocarditis resulting from GI endoscopy has been a concern of physicians for decades. The American Heart Association (AHA) first published its recommendations for antibiotic prophylaxis of GI tract procedures in 1965. The most recent antibacterial prophylaxis guidelines, published in 2007, have simplified recommendations and greatly scaled back the indications for antibiotics. The new guidelines conclude that frequent bacteremia from daily activities is more likely to precipitate endocarditis than a single dental, GI, or genitourinary tract procedure.1
The American Society for Gastrointestinal Endoscopy (ASGE) reports that 14.2 million colonoscopies, 2.8 million flexible sigmoidoscopies, and nearly as many upper endoscopies are performed in the U.S. each year, but only 15 cases of endocarditis have been reported with a temporal association to a procedure.2
The British Society of Gastroenterology (BSG) found, after reviewing the histories of patients with infective endocarditis from 1983 through 2006, that there is not enough evidence to warrant antibiotic prophylaxis prior to endoscopy. They noted less than one case of endocarditis after GI endoscopy per year as well as significant variation in the time interval between the procedure and symptoms. The BSG also recognized that antibiotic prophylaxis does not always protect against infection and that clinical factors unrelated to the endoscopy may play a significant role in the development of endocarditis.3
Upper GI Endoscopy, Colonoscopy with Biopsy, and Esophageal Dilatation. Administering antibiotics to prevent infective endocarditis is not recommended for patients undergoing routine procedures such as endoscopy with biopsy and colonoscopy with polypectomy. Likewise, patients with a history of prosthetic heart valves, valve repair with prosthetic material, endocarditis, congenital heart disease, or cardiac transplant with valvulopathy do not need prophylactic antibiotics before GI endoscopic procedures. However, for patients who are being treated for an active GI infection, antibiotic coverage for enterococcus may be warranted given the increased risk of developing endocarditis. The AHA acknowledges there are no published studies to support the efficacy of antibiotics to prevent enterococcal endocarditis in patients in this clinical setting.1
Unlike routine endoscopy, esophageal dilation is associated with an increased rate of bacteremia (12%-100%).4 Streptococcus viridans has been found in blood cultures up to 79% of the time after esophageal dilation.5 Patients with malignant strictures have higher rates of bacteremia than those with benign strictures (52.9% versus 15.7%). Patients treated with multiple passes with the esophageal dilator compared to those treated with a single dilation have a higher risk of bacteremia.6 All patients undergoing esophageal stricture dilation should receive pre-procedural prophylactic antibiotics.7
Patients with bleeding esophageal varices also have high rates of bacteremia. Up to 20% of patients with cirrhosis and GI bleeding on admission develop an infection within 48 hours of presentation.8 There is evidence that the bacteremia may actually be related to the variceal bleeding rather than the procedure.9 Patients with bleeding esophageal varices treated with antibiotics have improved outcomes, including a decrease in mortality.10 Therefore, all patients with bleeding esophageal varices should be placed on antibiotic therapy regardless of whether an endoscopic intervention is planned.
Percutaneous Endoscopic Gastrostomy (PEG) Placement. Prophylactic antibiotics are recommended before placement of a PEG. The indication for prophylactic antibiotics is to prevent a gastrostomy site infection, not infective endocarditis. Gastrostomy site infection is unfortunately a fairly common infection, affecting 4% to 30% of patients who undergo PEG tube placement. There is significant evidence that antibiotics are beneficial in preventing peristomal infections. A meta-analysis showed that only eight patients need to be treated with prophylactic antibiotics to prevent a single peristomal infection.11 Since these infections are believed to be caused by contamination from the oropharynx, physicians should consider prophylaxis against pathogens from the oral flora.12
More recently, it has been noted that methicillin-resistant Staphylococcus aureus (MRSA) is increasingly cultured from infection sites.13 In centers with endemic MRSA, patients should be screened and then undergo decontamination prior to the PEG placement in positive cases.
Endoscopic Ultrasound with Fine Needle Aspiration (EUS-FNA). Antibiotic prophylaxis before EUS-FNA of a solid lesion in an organ is generally thought to be unnecessary because the risk of bacteremia with this procedure is low, comparable to routine GI endoscopy with biopsy. The recommendation for prophylactic antibiotics before biopsy of a cystic lesion is different. There is concern that puncturing cystic lesions may create a new infected fluid collection.2 A systematic review of more than 10,000 patients undergoing EUS-FNA with a full range of target organs revealed that, overall, 11.2% of patients experienced a fever and 4.7% of patients had a peri-procedural infection. While it was not possible in this study to determine which patients received prophylactic antibiotics, 93.7% of patients with pancreatic cystic lesions were reported to have been treated with antibiotics.14
A separate, single-center, retrospective trial produced different results. This study examined a population of 253 patients who underwent 266 EUS-FNA of pancreatic cysts and found that prophylactic antibiotics were associated with more adverse events and were not protective for the 3% of the patients with infectious symptoms.15 Despite the conflicting data, guidelines at this time recommend prophylactic antibiotics before drainage of a sterile pancreatic fluid collection that communicates with the pancreatic duct and also for aspiration of cystic lesions along the GI tract and the mediastinum.2
Endoscopic Retrograde Cholangiopancreatography (ERCP). In patients undergoing ERCP, the routine use of prophylactic antibiotics has not been found to be effective in decreasing the risk of post-procedure cholangitis.16 Guidelines recommend the use of prophylactic antibiotics only in those patients in which the ERCP may not completely resolve the biliary obstruction.2 In these patients, the thought is that ERCP can precipitate infection by disturbing bacteria already present in the biliary tree, especially with increased intrabiliary pressure at the time of contrast dye injection.17
Patients with incomplete biliary drainage, including those with primary sclerosing cholangitis (PSC), hilar cholangiocarcinoma, persistent biliary that were not extracted, and strictures that continue to obstruct despite attempted intervention, are thought to be at elevated risk of developing cholangitis post-ERCP. These patients should be placed on prophylactic antibiotics at the time of the procedure to cover biliary flora such as enteric gram negatives and enterococci. Antibiotics should be continued until the biliary obstruction is resolved.2
Additional Populations to Consider. Previously, the International Society for Peritoneal Dialysis recommended that patients on peritoneal dialysis receive prophylactic antibiotics and empty their abdomen of dialysate prior to colonoscopy. This recommendation has been removed from the 2010 guidelines.18 There is also no indication that patients with synthetic vascular grafts or cardiac devices should receive prophylactic antibiotics prior to routine GI endoscopy.19 The American Academy of Orthopaedic Surgeons no longer recommends that patients with joint replacements receive antibiotic prophylaxis prior to GI endoscopy.20
Back to the Case
The older gentleman with a prosthetic valve undergoing colonoscopy should not receive prophylactic antibiotics, because even in the setting of valvulopathy, colonoscopy does not pose a significant risk for infective endocarditis. The young patient with severe choledocholithiasis should be placed on prophylactic antibiotics because she has continued biliary obstruction, which could result in a cholangitis after ERCP.
Bottom Line
Prophylactic antibiotics are not recommended for any patient undergoing routine endoscopy or colonoscopy. They are indicated for patients with bleeding esophageal varices and for patients who undergo esophageal stricture dilation, PEG placement, or pseudocyst or cyst drainage, and those with continued biliary obstruction undergoing ERCP as summarized in Table 1.
Drs. Ritter, Jupiter, Carbo, and Li are hospitalists at Beth Israel Deaconess Medical Center and Harvard Medical School faculty in Boston.
References
- Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007;116(15):1736-1754.
- Banerjee S, Shen B, Baron TH, et al. Antibiotic prophylaxis for GI endoscopy. Gastrointest Endosc. 2008;67(6):791-798.
- Allison MC, Sandoe JA, Tighe R, Simpson IA, Hall RJ, Elliott TS. Antibiotic prophylaxis in gastrointestinal endoscopy. Gut. 2009;58(6):869-880.
- Nelson DB. Infectious disease complications of GI endoscopy: Part I, endogenous infections. Gastrointest Endosc. 2003;57(4):546-556.
- Zuccaro G Jr., Richter JE, Rice TW, et al. Viridans streptococcal bacteremia after esophageal stricture dilation. Gastrointest Endosc. 1998;48(6):568-573.
- Nelson DB, Sanderson SJ, Azar MM. Bacteremia with esophageal dilation. Gastrointest Endosc.1998;48(6):563-567.
- Hirota WK, Petersen K, Baron TH, et al. Guidelines for antibiotic prophylaxis for GI endoscopy. Gastrointest Endosc. 2003;58(4):475-482.
- Ho H, Zuckerman MJ, Wassem C. A prospective controlled study of the risk of bacteremia in emergency sclerotherapy of esophageal varices. Gastroenterology. 1991;101(6):1642-1648.
- Rolando N, Gimson A, Philpott-Howard J, et al. Infectious sequelae after endoscopic sclerotherapy of oesophageal varices: Role of antibiotic prophylaxis. J Hepatol. 1993;18(3):290-294.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology. 2007;46(3):922-938.
- Jafri NS, Mahid SS, Minor KS, Idstein SR, Hornung CA, Galandiuk S. Meta-analysis: Antibiotic prophylaxis to prevent peristomal infection following percutaneous endoscopic gastrostomy. Aliment Pharmacol Ther. 2007;25(6):647-656.
- Chuang CH, Hung KH, Chen JR, et al. Airway infection predisposes to peristomal infection after percutaneous endoscopic gastrostomy with high concordance between sputum and wound isolates. J Gastrointest Surg. 2010;14(1):45-51.
- Chaudhary KA, Smith OJ, Cuddy PG, Clarkston WK. PEG site infections: The emergence of methicillin resistant Staphylococcus aureus as a major pathogen. Am J Gastroenterol. 2002;97(7):1713-1716.
- Wang KX, Ben QW, Jin ZD, et al. Assessment of morbidity and mortality associated with EUS-guided FNA: A systematic review. Gastrointest Endosc. 2011;73(2):283-290.
- Guarner-Argente C, Shah P, Buchner A, Ahmad NA, Kochman ML, Ginsberg GG. Use of antimicrobials for EUS-guided FNA of pancreatic cysts: A retrospective, comparative analysis. Gastrointest Endosc. 2011;74(1):81-86.
- Bai Y, Gao F, Gao J, Zou DW, Li ZS. Prophylactic antibiotics cannot prevent endoscopic retrograde cholangiopancreatography-induced cholangitis: A meta-analysis. Pancreas. 2009;38(2):126-130.
- Cotton PB, Connor P, Rawls E, Romagnuolo J. Infection after ERCP, and antibiotic prophylaxis: A sequential quality-improvement approach over 11 years. Gastrointest Endosc. 2008;67(3):471-475.
- Li PK, Szeto CC, Piraino B, et al. Peritoneal dialysis-related infections recommendations: 2010 update. Perit Dial Int. 2010;30(4):393-423.
- Baddour LM, Bettmann MA, Bolger AF, et al. Nonvalvular cardiovascular device-related infections. Circulation. 2003;108(16):2015-2031.
- Rethman MP, Watters W III, Abt E, et al. The American Academy of Orthopaedic Surgeons and the American Dental Association clinical practice guideline on the prevention of orthopaedic implant infection in patients undergoing dental procedures. J Bone Joint Surg Am. 2013;95(8):745-747.
Case
You are asked to admit two patients. The first is a 75-year-old male with a prosthetic aortic valve on warfarin who presents with bright red blood per rectum and is scheduled for colonoscopy. The second patient is a 35-year-old female with biliary obstruction due to choledocholithiasis; she is afebrile with normal vital signs and no leukocytosis. She underwent endoscopic retrograde cholangiopancreatography (ERCP), which did not resolve her biliary obstruction. Should you prescribe prophylactic antibiotics for either patient?
Overview
Providers are often confused regarding which patients undergoing gastrointestinal (GI) endoscopic procedures should receive antibiotic prophylaxis. To answer this question, it is important to understand the goal of prophylactic antibiotics. Are we trying to prevent infective endocarditis or a localized infection?
There are few large, prospective, randomized controlled trials that have examined the need for antibiotic prophylaxis with GI endoscopic procedures. Guidelines from professional societies are mainly based on expert opinion, evidence from retrospective case studies, and meta-analysis reviews.
Review of the Data
Infective endocarditis resulting from GI endoscopy has been a concern of physicians for decades. The American Heart Association (AHA) first published its recommendations for antibiotic prophylaxis of GI tract procedures in 1965. The most recent antibacterial prophylaxis guidelines, published in 2007, have simplified recommendations and greatly scaled back the indications for antibiotics. The new guidelines conclude that frequent bacteremia from daily activities is more likely to precipitate endocarditis than a single dental, GI, or genitourinary tract procedure.1
The American Society for Gastrointestinal Endoscopy (ASGE) reports that 14.2 million colonoscopies, 2.8 million flexible sigmoidoscopies, and nearly as many upper endoscopies are performed in the U.S. each year, but only 15 cases of endocarditis have been reported with a temporal association to a procedure.2
The British Society of Gastroenterology (BSG) found, after reviewing the histories of patients with infective endocarditis from 1983 through 2006, that there is not enough evidence to warrant antibiotic prophylaxis prior to endoscopy. They noted less than one case of endocarditis after GI endoscopy per year as well as significant variation in the time interval between the procedure and symptoms. The BSG also recognized that antibiotic prophylaxis does not always protect against infection and that clinical factors unrelated to the endoscopy may play a significant role in the development of endocarditis.3
Upper GI Endoscopy, Colonoscopy with Biopsy, and Esophageal Dilatation. Administering antibiotics to prevent infective endocarditis is not recommended for patients undergoing routine procedures such as endoscopy with biopsy and colonoscopy with polypectomy. Likewise, patients with a history of prosthetic heart valves, valve repair with prosthetic material, endocarditis, congenital heart disease, or cardiac transplant with valvulopathy do not need prophylactic antibiotics before GI endoscopic procedures. However, for patients who are being treated for an active GI infection, antibiotic coverage for enterococcus may be warranted given the increased risk of developing endocarditis. The AHA acknowledges there are no published studies to support the efficacy of antibiotics to prevent enterococcal endocarditis in patients in this clinical setting.1
Unlike routine endoscopy, esophageal dilation is associated with an increased rate of bacteremia (12%-100%).4 Streptococcus viridans has been found in blood cultures up to 79% of the time after esophageal dilation.5 Patients with malignant strictures have higher rates of bacteremia than those with benign strictures (52.9% versus 15.7%). Patients treated with multiple passes with the esophageal dilator compared to those treated with a single dilation have a higher risk of bacteremia.6 All patients undergoing esophageal stricture dilation should receive pre-procedural prophylactic antibiotics.7
Patients with bleeding esophageal varices also have high rates of bacteremia. Up to 20% of patients with cirrhosis and GI bleeding on admission develop an infection within 48 hours of presentation.8 There is evidence that the bacteremia may actually be related to the variceal bleeding rather than the procedure.9 Patients with bleeding esophageal varices treated with antibiotics have improved outcomes, including a decrease in mortality.10 Therefore, all patients with bleeding esophageal varices should be placed on antibiotic therapy regardless of whether an endoscopic intervention is planned.
Percutaneous Endoscopic Gastrostomy (PEG) Placement. Prophylactic antibiotics are recommended before placement of a PEG. The indication for prophylactic antibiotics is to prevent a gastrostomy site infection, not infective endocarditis. Gastrostomy site infection is unfortunately a fairly common infection, affecting 4% to 30% of patients who undergo PEG tube placement. There is significant evidence that antibiotics are beneficial in preventing peristomal infections. A meta-analysis showed that only eight patients need to be treated with prophylactic antibiotics to prevent a single peristomal infection.11 Since these infections are believed to be caused by contamination from the oropharynx, physicians should consider prophylaxis against pathogens from the oral flora.12
More recently, it has been noted that methicillin-resistant Staphylococcus aureus (MRSA) is increasingly cultured from infection sites.13 In centers with endemic MRSA, patients should be screened and then undergo decontamination prior to the PEG placement in positive cases.
Endoscopic Ultrasound with Fine Needle Aspiration (EUS-FNA). Antibiotic prophylaxis before EUS-FNA of a solid lesion in an organ is generally thought to be unnecessary because the risk of bacteremia with this procedure is low, comparable to routine GI endoscopy with biopsy. The recommendation for prophylactic antibiotics before biopsy of a cystic lesion is different. There is concern that puncturing cystic lesions may create a new infected fluid collection.2 A systematic review of more than 10,000 patients undergoing EUS-FNA with a full range of target organs revealed that, overall, 11.2% of patients experienced a fever and 4.7% of patients had a peri-procedural infection. While it was not possible in this study to determine which patients received prophylactic antibiotics, 93.7% of patients with pancreatic cystic lesions were reported to have been treated with antibiotics.14
A separate, single-center, retrospective trial produced different results. This study examined a population of 253 patients who underwent 266 EUS-FNA of pancreatic cysts and found that prophylactic antibiotics were associated with more adverse events and were not protective for the 3% of the patients with infectious symptoms.15 Despite the conflicting data, guidelines at this time recommend prophylactic antibiotics before drainage of a sterile pancreatic fluid collection that communicates with the pancreatic duct and also for aspiration of cystic lesions along the GI tract and the mediastinum.2
Endoscopic Retrograde Cholangiopancreatography (ERCP). In patients undergoing ERCP, the routine use of prophylactic antibiotics has not been found to be effective in decreasing the risk of post-procedure cholangitis.16 Guidelines recommend the use of prophylactic antibiotics only in those patients in which the ERCP may not completely resolve the biliary obstruction.2 In these patients, the thought is that ERCP can precipitate infection by disturbing bacteria already present in the biliary tree, especially with increased intrabiliary pressure at the time of contrast dye injection.17
Patients with incomplete biliary drainage, including those with primary sclerosing cholangitis (PSC), hilar cholangiocarcinoma, persistent biliary that were not extracted, and strictures that continue to obstruct despite attempted intervention, are thought to be at elevated risk of developing cholangitis post-ERCP. These patients should be placed on prophylactic antibiotics at the time of the procedure to cover biliary flora such as enteric gram negatives and enterococci. Antibiotics should be continued until the biliary obstruction is resolved.2
Additional Populations to Consider. Previously, the International Society for Peritoneal Dialysis recommended that patients on peritoneal dialysis receive prophylactic antibiotics and empty their abdomen of dialysate prior to colonoscopy. This recommendation has been removed from the 2010 guidelines.18 There is also no indication that patients with synthetic vascular grafts or cardiac devices should receive prophylactic antibiotics prior to routine GI endoscopy.19 The American Academy of Orthopaedic Surgeons no longer recommends that patients with joint replacements receive antibiotic prophylaxis prior to GI endoscopy.20
Back to the Case
The older gentleman with a prosthetic valve undergoing colonoscopy should not receive prophylactic antibiotics, because even in the setting of valvulopathy, colonoscopy does not pose a significant risk for infective endocarditis. The young patient with severe choledocholithiasis should be placed on prophylactic antibiotics because she has continued biliary obstruction, which could result in a cholangitis after ERCP.
Bottom Line
Prophylactic antibiotics are not recommended for any patient undergoing routine endoscopy or colonoscopy. They are indicated for patients with bleeding esophageal varices and for patients who undergo esophageal stricture dilation, PEG placement, or pseudocyst or cyst drainage, and those with continued biliary obstruction undergoing ERCP as summarized in Table 1.
Drs. Ritter, Jupiter, Carbo, and Li are hospitalists at Beth Israel Deaconess Medical Center and Harvard Medical School faculty in Boston.
References
- Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007;116(15):1736-1754.
- Banerjee S, Shen B, Baron TH, et al. Antibiotic prophylaxis for GI endoscopy. Gastrointest Endosc. 2008;67(6):791-798.
- Allison MC, Sandoe JA, Tighe R, Simpson IA, Hall RJ, Elliott TS. Antibiotic prophylaxis in gastrointestinal endoscopy. Gut. 2009;58(6):869-880.
- Nelson DB. Infectious disease complications of GI endoscopy: Part I, endogenous infections. Gastrointest Endosc. 2003;57(4):546-556.
- Zuccaro G Jr., Richter JE, Rice TW, et al. Viridans streptococcal bacteremia after esophageal stricture dilation. Gastrointest Endosc. 1998;48(6):568-573.
- Nelson DB, Sanderson SJ, Azar MM. Bacteremia with esophageal dilation. Gastrointest Endosc.1998;48(6):563-567.
- Hirota WK, Petersen K, Baron TH, et al. Guidelines for antibiotic prophylaxis for GI endoscopy. Gastrointest Endosc. 2003;58(4):475-482.
- Ho H, Zuckerman MJ, Wassem C. A prospective controlled study of the risk of bacteremia in emergency sclerotherapy of esophageal varices. Gastroenterology. 1991;101(6):1642-1648.
- Rolando N, Gimson A, Philpott-Howard J, et al. Infectious sequelae after endoscopic sclerotherapy of oesophageal varices: Role of antibiotic prophylaxis. J Hepatol. 1993;18(3):290-294.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology. 2007;46(3):922-938.
- Jafri NS, Mahid SS, Minor KS, Idstein SR, Hornung CA, Galandiuk S. Meta-analysis: Antibiotic prophylaxis to prevent peristomal infection following percutaneous endoscopic gastrostomy. Aliment Pharmacol Ther. 2007;25(6):647-656.
- Chuang CH, Hung KH, Chen JR, et al. Airway infection predisposes to peristomal infection after percutaneous endoscopic gastrostomy with high concordance between sputum and wound isolates. J Gastrointest Surg. 2010;14(1):45-51.
- Chaudhary KA, Smith OJ, Cuddy PG, Clarkston WK. PEG site infections: The emergence of methicillin resistant Staphylococcus aureus as a major pathogen. Am J Gastroenterol. 2002;97(7):1713-1716.
- Wang KX, Ben QW, Jin ZD, et al. Assessment of morbidity and mortality associated with EUS-guided FNA: A systematic review. Gastrointest Endosc. 2011;73(2):283-290.
- Guarner-Argente C, Shah P, Buchner A, Ahmad NA, Kochman ML, Ginsberg GG. Use of antimicrobials for EUS-guided FNA of pancreatic cysts: A retrospective, comparative analysis. Gastrointest Endosc. 2011;74(1):81-86.
- Bai Y, Gao F, Gao J, Zou DW, Li ZS. Prophylactic antibiotics cannot prevent endoscopic retrograde cholangiopancreatography-induced cholangitis: A meta-analysis. Pancreas. 2009;38(2):126-130.
- Cotton PB, Connor P, Rawls E, Romagnuolo J. Infection after ERCP, and antibiotic prophylaxis: A sequential quality-improvement approach over 11 years. Gastrointest Endosc. 2008;67(3):471-475.
- Li PK, Szeto CC, Piraino B, et al. Peritoneal dialysis-related infections recommendations: 2010 update. Perit Dial Int. 2010;30(4):393-423.
- Baddour LM, Bettmann MA, Bolger AF, et al. Nonvalvular cardiovascular device-related infections. Circulation. 2003;108(16):2015-2031.
- Rethman MP, Watters W III, Abt E, et al. The American Academy of Orthopaedic Surgeons and the American Dental Association clinical practice guideline on the prevention of orthopaedic implant infection in patients undergoing dental procedures. J Bone Joint Surg Am. 2013;95(8):745-747.
HM14 to Feature Free Wi-Fi
At today’s meetings and conferences, wireless Internet access is a necessity for hospitalists who need to stay in touch with home and work, network with other conference-goers, and receive updates through the HM14 at Hand app. HM14 attendees will enjoy free Wi-Fi at the Mandalay Bay Convention Center in Las Vegas. To access, jot down the username (HM14) and the access code (hospitalist14), and log in with all your devices when you arrive.
Wi-Fi at HM14
Username: HM14
Access Code: hospitalist14
At today’s meetings and conferences, wireless Internet access is a necessity for hospitalists who need to stay in touch with home and work, network with other conference-goers, and receive updates through the HM14 at Hand app. HM14 attendees will enjoy free Wi-Fi at the Mandalay Bay Convention Center in Las Vegas. To access, jot down the username (HM14) and the access code (hospitalist14), and log in with all your devices when you arrive.
Wi-Fi at HM14
Username: HM14
Access Code: hospitalist14
At today’s meetings and conferences, wireless Internet access is a necessity for hospitalists who need to stay in touch with home and work, network with other conference-goers, and receive updates through the HM14 at Hand app. HM14 attendees will enjoy free Wi-Fi at the Mandalay Bay Convention Center in Las Vegas. To access, jot down the username (HM14) and the access code (hospitalist14), and log in with all your devices when you arrive.
Wi-Fi at HM14
Username: HM14
Access Code: hospitalist14
Problem Solving In Multi-Site Hospital Medicine Groups


Serving as the lead physician for a hospital medicine group (HMG) makes for challenging work. And the challenges and complexity only increase for anyone who serves as the physician leader for multiple practice sites in the same hospital system. In my November 2013 column on multi-site HMG leaders, I listed a few of the tricky issues they face and will mention a few more here.
Large-Small Friction
Unfortunately, tension between hospitalists at the big hospital and doctors at the small, “feeder” hospitals seems pretty common, and I think it’s due largely to high stress and a wide variation in workload, neither of which are in our direct control. At facilities where there is significant tension, I’m impressed by how vigorously the hospitalists at both the small and large hospitals argue that their own site faces the most stress and challenges. (This is a little like the endless debate about who works harder, those who work with residents and those who don’t.)
The hospitalists at the small site point out that they work with little or no subspecialty help and might even have to take night call from home while working during the day. Those at the big hospital say they are the ones with the very large scope of clinical practice and that, rather than making their life easier, the presence of lots of subspecialists makes for additional work coordinating care and communicating with all parties.
Where it exists, this tension is most evident during a transfer from one of the small hospitals to the large one. After all, one of the reasons to form a system of hospitals is so that nearly all patient needs can be met at one of the facilities in the system. Yet, for many reasons, the hospitalists at the large hospital are—sometimes—not as receptive to transfers as might be ideal. They might be short staffed or facing a high census or an unusually high number of admissions from their own ED. Or, perhaps, they’re concerned that the subspecialty services for which the patient is being transferred (e.g. to be scoped by a GI doctor) won’t be as helpful or prompt as needed. Or maybe they’ve felt “burned” by their colleagues at the small hospital for past transfers that didn’t seem necessary.
The result can be that the doctors at the smaller hospital complain that the “mother ship” hospitalists often are unfriendly and unreceptive to transfer requests. Although there may not be a definitive “cure” for this issue, there are several ways to help address the problem.
- In my last column, I mentioned the value of one or more in-person meetings between those who tend to be on the sending and receiving end of transfers, to establish some criteria regarding transfers that are appropriate and review the process of requesting a transfer and making the associated arrangements. In most cases there will be value in the parties meeting routinely—perhaps two to four times annually—to review how the system is working and address any difficulties.
- Periodic social meetings among the hospitalists at each site will help to form relationships that can make it less likely that any conversation about transfers will go in an unhelpful direction. Things can be very different when the people on each end of the phone call know each other personally.
- Record the phone calls between those seeking and accepting/declining each transfer. Scott Rissmiller, MD, the lead hospitalist for the 17 practice sites in Carolinas Healthcare, has said that having underperforming doctors listen to recordings of their phone calls about transfers has, in most cases he’s been involved with, proven to be a very effective way to encourage improvement.
Shared Staffing
The small hospitals in many systems sometimes struggle to find a way to provide economical night coverage. Hospitals below a certain size find it very difficult to justify a separate, in-house night provider. Some hospital systems have had success sharing night staffing, with the large hospital’s night hospitalist, nurse practitioner, or physician assistant providing telephone coverage for “cross cover” issues that arise after hours.
For example, when a nurse at the small hospital needs to contact a night hospitalist, staff will page the provider at the big hospital, and, in many cases, the issue can be managed effectively by phone. This works best when both hospitals are on the same electronic medical record, so that the responding provider can look through the record as needed.
The hospitalist at the small hospital typically stays on back-up call and is contacted if bedside attention is required.
Or, if the large and small hospitals are a short drive apart, the night hospitalist at the large facility might make the short drive to the small hospital when needed. In the case of emergencies (i.e., a code blue), the in-house night ED physician is relied on as the first responder.
Dr. Nelson has been a practicing hospitalist since 1988. He is co-founder and past president of SHM, and principal in Nelson Flores Hospital Medicine Consultants. He is co-director for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. Write to him at [email protected].
Serving as the lead physician for a hospital medicine group (HMG) makes for challenging work. And the challenges and complexity only increase for anyone who serves as the physician leader for multiple practice sites in the same hospital system. In my November 2013 column on multi-site HMG leaders, I listed a few of the tricky issues they face and will mention a few more here.
Large-Small Friction
Unfortunately, tension between hospitalists at the big hospital and doctors at the small, “feeder” hospitals seems pretty common, and I think it’s due largely to high stress and a wide variation in workload, neither of which are in our direct control. At facilities where there is significant tension, I’m impressed by how vigorously the hospitalists at both the small and large hospitals argue that their own site faces the most stress and challenges. (This is a little like the endless debate about who works harder, those who work with residents and those who don’t.)
The hospitalists at the small site point out that they work with little or no subspecialty help and might even have to take night call from home while working during the day. Those at the big hospital say they are the ones with the very large scope of clinical practice and that, rather than making their life easier, the presence of lots of subspecialists makes for additional work coordinating care and communicating with all parties.
Where it exists, this tension is most evident during a transfer from one of the small hospitals to the large one. After all, one of the reasons to form a system of hospitals is so that nearly all patient needs can be met at one of the facilities in the system. Yet, for many reasons, the hospitalists at the large hospital are—sometimes—not as receptive to transfers as might be ideal. They might be short staffed or facing a high census or an unusually high number of admissions from their own ED. Or, perhaps, they’re concerned that the subspecialty services for which the patient is being transferred (e.g. to be scoped by a GI doctor) won’t be as helpful or prompt as needed. Or maybe they’ve felt “burned” by their colleagues at the small hospital for past transfers that didn’t seem necessary.
The result can be that the doctors at the smaller hospital complain that the “mother ship” hospitalists often are unfriendly and unreceptive to transfer requests. Although there may not be a definitive “cure” for this issue, there are several ways to help address the problem.
- In my last column, I mentioned the value of one or more in-person meetings between those who tend to be on the sending and receiving end of transfers, to establish some criteria regarding transfers that are appropriate and review the process of requesting a transfer and making the associated arrangements. In most cases there will be value in the parties meeting routinely—perhaps two to four times annually—to review how the system is working and address any difficulties.
- Periodic social meetings among the hospitalists at each site will help to form relationships that can make it less likely that any conversation about transfers will go in an unhelpful direction. Things can be very different when the people on each end of the phone call know each other personally.
- Record the phone calls between those seeking and accepting/declining each transfer. Scott Rissmiller, MD, the lead hospitalist for the 17 practice sites in Carolinas Healthcare, has said that having underperforming doctors listen to recordings of their phone calls about transfers has, in most cases he’s been involved with, proven to be a very effective way to encourage improvement.
Shared Staffing
The small hospitals in many systems sometimes struggle to find a way to provide economical night coverage. Hospitals below a certain size find it very difficult to justify a separate, in-house night provider. Some hospital systems have had success sharing night staffing, with the large hospital’s night hospitalist, nurse practitioner, or physician assistant providing telephone coverage for “cross cover” issues that arise after hours.
For example, when a nurse at the small hospital needs to contact a night hospitalist, staff will page the provider at the big hospital, and, in many cases, the issue can be managed effectively by phone. This works best when both hospitals are on the same electronic medical record, so that the responding provider can look through the record as needed.
The hospitalist at the small hospital typically stays on back-up call and is contacted if bedside attention is required.
Or, if the large and small hospitals are a short drive apart, the night hospitalist at the large facility might make the short drive to the small hospital when needed. In the case of emergencies (i.e., a code blue), the in-house night ED physician is relied on as the first responder.
Dr. Nelson has been a practicing hospitalist since 1988. He is co-founder and past president of SHM, and principal in Nelson Flores Hospital Medicine Consultants. He is co-director for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. Write to him at [email protected].
Serving as the lead physician for a hospital medicine group (HMG) makes for challenging work. And the challenges and complexity only increase for anyone who serves as the physician leader for multiple practice sites in the same hospital system. In my November 2013 column on multi-site HMG leaders, I listed a few of the tricky issues they face and will mention a few more here.
Large-Small Friction
Unfortunately, tension between hospitalists at the big hospital and doctors at the small, “feeder” hospitals seems pretty common, and I think it’s due largely to high stress and a wide variation in workload, neither of which are in our direct control. At facilities where there is significant tension, I’m impressed by how vigorously the hospitalists at both the small and large hospitals argue that their own site faces the most stress and challenges. (This is a little like the endless debate about who works harder, those who work with residents and those who don’t.)
The hospitalists at the small site point out that they work with little or no subspecialty help and might even have to take night call from home while working during the day. Those at the big hospital say they are the ones with the very large scope of clinical practice and that, rather than making their life easier, the presence of lots of subspecialists makes for additional work coordinating care and communicating with all parties.
Where it exists, this tension is most evident during a transfer from one of the small hospitals to the large one. After all, one of the reasons to form a system of hospitals is so that nearly all patient needs can be met at one of the facilities in the system. Yet, for many reasons, the hospitalists at the large hospital are—sometimes—not as receptive to transfers as might be ideal. They might be short staffed or facing a high census or an unusually high number of admissions from their own ED. Or, perhaps, they’re concerned that the subspecialty services for which the patient is being transferred (e.g. to be scoped by a GI doctor) won’t be as helpful or prompt as needed. Or maybe they’ve felt “burned” by their colleagues at the small hospital for past transfers that didn’t seem necessary.
The result can be that the doctors at the smaller hospital complain that the “mother ship” hospitalists often are unfriendly and unreceptive to transfer requests. Although there may not be a definitive “cure” for this issue, there are several ways to help address the problem.
- In my last column, I mentioned the value of one or more in-person meetings between those who tend to be on the sending and receiving end of transfers, to establish some criteria regarding transfers that are appropriate and review the process of requesting a transfer and making the associated arrangements. In most cases there will be value in the parties meeting routinely—perhaps two to four times annually—to review how the system is working and address any difficulties.
- Periodic social meetings among the hospitalists at each site will help to form relationships that can make it less likely that any conversation about transfers will go in an unhelpful direction. Things can be very different when the people on each end of the phone call know each other personally.
- Record the phone calls between those seeking and accepting/declining each transfer. Scott Rissmiller, MD, the lead hospitalist for the 17 practice sites in Carolinas Healthcare, has said that having underperforming doctors listen to recordings of their phone calls about transfers has, in most cases he’s been involved with, proven to be a very effective way to encourage improvement.
Shared Staffing
The small hospitals in many systems sometimes struggle to find a way to provide economical night coverage. Hospitals below a certain size find it very difficult to justify a separate, in-house night provider. Some hospital systems have had success sharing night staffing, with the large hospital’s night hospitalist, nurse practitioner, or physician assistant providing telephone coverage for “cross cover” issues that arise after hours.
For example, when a nurse at the small hospital needs to contact a night hospitalist, staff will page the provider at the big hospital, and, in many cases, the issue can be managed effectively by phone. This works best when both hospitals are on the same electronic medical record, so that the responding provider can look through the record as needed.
The hospitalist at the small hospital typically stays on back-up call and is contacted if bedside attention is required.
Or, if the large and small hospitals are a short drive apart, the night hospitalist at the large facility might make the short drive to the small hospital when needed. In the case of emergencies (i.e., a code blue), the in-house night ED physician is relied on as the first responder.
Dr. Nelson has been a practicing hospitalist since 1988. He is co-founder and past president of SHM, and principal in Nelson Flores Hospital Medicine Consultants. He is co-director for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. Write to him at [email protected].
Federal Grant Extends Anti-Infection Initiative
The American Hospital Association’s Health Research and Educational Trust (HRET) recently obtained a grant from the federal Agency for Healthcare Research and Quality to expand CUSP, the Comprehensive Unit-based Safety Program for reducing catheter-associated urinary tract infections (CAUTI) and other healthcare-associated infections, to nursing homes and skilled nursing facilities nationwide.
CUSP has posted a 40% reduction in central line-associated bloodstream infections (CLABSI) in 1,000 participating hospitals by providing education and support and an evidence-based protocol. The grant will be administered by HRET in partnership with others, including the University of Michigan Health System, the Association for Professionals in Infection Control and Epidemiology, and SHM.
Meanwhile, a study published in the American Journal of Infection Control found that rates of catheter-associated urinary tract infections in adult patients given urinary catheter placements dropped nationwide to 5.3% in 2010 from 9.4% in 2001.3 The retrospective analysis of data from the National Hospital Discharge Survey found that CAUTI-related mortality and associated length of hospital stay also declined during the same period.
Larry Beresford is a freelance writer in Alameda, Calif.
The American Hospital Association’s Health Research and Educational Trust (HRET) recently obtained a grant from the federal Agency for Healthcare Research and Quality to expand CUSP, the Comprehensive Unit-based Safety Program for reducing catheter-associated urinary tract infections (CAUTI) and other healthcare-associated infections, to nursing homes and skilled nursing facilities nationwide.
CUSP has posted a 40% reduction in central line-associated bloodstream infections (CLABSI) in 1,000 participating hospitals by providing education and support and an evidence-based protocol. The grant will be administered by HRET in partnership with others, including the University of Michigan Health System, the Association for Professionals in Infection Control and Epidemiology, and SHM.
Meanwhile, a study published in the American Journal of Infection Control found that rates of catheter-associated urinary tract infections in adult patients given urinary catheter placements dropped nationwide to 5.3% in 2010 from 9.4% in 2001.3 The retrospective analysis of data from the National Hospital Discharge Survey found that CAUTI-related mortality and associated length of hospital stay also declined during the same period.
Larry Beresford is a freelance writer in Alameda, Calif.
The American Hospital Association’s Health Research and Educational Trust (HRET) recently obtained a grant from the federal Agency for Healthcare Research and Quality to expand CUSP, the Comprehensive Unit-based Safety Program for reducing catheter-associated urinary tract infections (CAUTI) and other healthcare-associated infections, to nursing homes and skilled nursing facilities nationwide.
CUSP has posted a 40% reduction in central line-associated bloodstream infections (CLABSI) in 1,000 participating hospitals by providing education and support and an evidence-based protocol. The grant will be administered by HRET in partnership with others, including the University of Michigan Health System, the Association for Professionals in Infection Control and Epidemiology, and SHM.
Meanwhile, a study published in the American Journal of Infection Control found that rates of catheter-associated urinary tract infections in adult patients given urinary catheter placements dropped nationwide to 5.3% in 2010 from 9.4% in 2001.3 The retrospective analysis of data from the National Hospital Discharge Survey found that CAUTI-related mortality and associated length of hospital stay also declined during the same period.
Larry Beresford is a freelance writer in Alameda, Calif.
Patient Activation Measure Tool Helps Patients Avoid Hospital Readmissions
–Dr. Hibbard
A recent article in the Journal of Internal Medicine draws a strong link between readmission rates and the degree to which patients are activated—possessing the knowledge, skills, and confidence to manage their own health post-discharge.2 Co-author Judith Hibbard, DrPh, professor of health policy at the University of Oregon, is the lead inventor of the Patient Activation Measure (PAM), an eight-item tool that assigns patients to one of four levels of activation.
In a sample of 700 patients discharged from Boston Medical Center, those with the lowest levels of activation had 1.75 times the risk of 30-day readmissions, more ED visits, and greater utilization of health services, even after adjusting for severity of illness and demographics.
“Contrary to what some may assume, patients who demonstrate a lower level of activation do not fall into any specific racial, economic, or educational demographic,” Dr. Hibbard says, adding that providers should not expect to be able to reliably judge their patients’ ability to self-manage outside of the hospital. “We know that people who measure low tend to have little confidence in their ability to manage their own health. They feel overwhelmed, show poor problem-solving skills, don’t understand what professionals are telling them, and, as a result, may not pay close attention.”
Dr. Hibbard says higher activation scores reflect greater focus on personal health and the effort to monitor it—with more confidence.
The take-home message for hospitalists, she says, is to understand the importance of their patients’ activation level and to tailor interventions accordingly.
“Those with low activation may need more support,” such as post-discharge home visits instead of just a phone call. Low-activation patients should not be overwhelmed with information but should instead be given just a few prioritized key points, combined with the use of reinforcing communications techniques such as teach-back.
“Someone should sit with them and help negotiate their health behaviors,” she adds. “That’s how they get more activated. It doesn’t have to be a doctor going through these things. But just using the clinical lens to understand your patients is not enough.”
Larry Beresford is a freelance writer in Alameda, Calif.
–Dr. Hibbard
A recent article in the Journal of Internal Medicine draws a strong link between readmission rates and the degree to which patients are activated—possessing the knowledge, skills, and confidence to manage their own health post-discharge.2 Co-author Judith Hibbard, DrPh, professor of health policy at the University of Oregon, is the lead inventor of the Patient Activation Measure (PAM), an eight-item tool that assigns patients to one of four levels of activation.
In a sample of 700 patients discharged from Boston Medical Center, those with the lowest levels of activation had 1.75 times the risk of 30-day readmissions, more ED visits, and greater utilization of health services, even after adjusting for severity of illness and demographics.
“Contrary to what some may assume, patients who demonstrate a lower level of activation do not fall into any specific racial, economic, or educational demographic,” Dr. Hibbard says, adding that providers should not expect to be able to reliably judge their patients’ ability to self-manage outside of the hospital. “We know that people who measure low tend to have little confidence in their ability to manage their own health. They feel overwhelmed, show poor problem-solving skills, don’t understand what professionals are telling them, and, as a result, may not pay close attention.”
Dr. Hibbard says higher activation scores reflect greater focus on personal health and the effort to monitor it—with more confidence.
The take-home message for hospitalists, she says, is to understand the importance of their patients’ activation level and to tailor interventions accordingly.
“Those with low activation may need more support,” such as post-discharge home visits instead of just a phone call. Low-activation patients should not be overwhelmed with information but should instead be given just a few prioritized key points, combined with the use of reinforcing communications techniques such as teach-back.
“Someone should sit with them and help negotiate their health behaviors,” she adds. “That’s how they get more activated. It doesn’t have to be a doctor going through these things. But just using the clinical lens to understand your patients is not enough.”
Larry Beresford is a freelance writer in Alameda, Calif.
–Dr. Hibbard
A recent article in the Journal of Internal Medicine draws a strong link between readmission rates and the degree to which patients are activated—possessing the knowledge, skills, and confidence to manage their own health post-discharge.2 Co-author Judith Hibbard, DrPh, professor of health policy at the University of Oregon, is the lead inventor of the Patient Activation Measure (PAM), an eight-item tool that assigns patients to one of four levels of activation.
In a sample of 700 patients discharged from Boston Medical Center, those with the lowest levels of activation had 1.75 times the risk of 30-day readmissions, more ED visits, and greater utilization of health services, even after adjusting for severity of illness and demographics.
“Contrary to what some may assume, patients who demonstrate a lower level of activation do not fall into any specific racial, economic, or educational demographic,” Dr. Hibbard says, adding that providers should not expect to be able to reliably judge their patients’ ability to self-manage outside of the hospital. “We know that people who measure low tend to have little confidence in their ability to manage their own health. They feel overwhelmed, show poor problem-solving skills, don’t understand what professionals are telling them, and, as a result, may not pay close attention.”
Dr. Hibbard says higher activation scores reflect greater focus on personal health and the effort to monitor it—with more confidence.
The take-home message for hospitalists, she says, is to understand the importance of their patients’ activation level and to tailor interventions accordingly.
“Those with low activation may need more support,” such as post-discharge home visits instead of just a phone call. Low-activation patients should not be overwhelmed with information but should instead be given just a few prioritized key points, combined with the use of reinforcing communications techniques such as teach-back.
“Someone should sit with them and help negotiate their health behaviors,” she adds. “That’s how they get more activated. It doesn’t have to be a doctor going through these things. But just using the clinical lens to understand your patients is not enough.”
Larry Beresford is a freelance writer in Alameda, Calif.
Hospitalists Use Online Game to Identify, Manage Sepsis
Teaching trainees to identify and manage sepsis using an online game known as “Septris” earned hospitalists at Stanford University Medical Center in Palo Alto, Calif., a Research, Innovation, and Clinical Vignette category award at HM13.1
“We took third-year medical students and residents in medicine, surgery, and emergency medicine—people who would be sepsis first responders on the floor—and gave them pre- and post-tests that documented improvements in both attitudes and knowledge,” says lead author Lisa Shieh, MD, PhD, Stanford’s medical director of quality in the department of medicine. All participants said they enjoyed playing the game, she reported.
Septris was developed by a multidisciplinary group of physicians, educational technology specialists, and programmers at Stanford. The game offers a case-based interactive learning environment drawn from evidence-based treatment algorithms. Players make treatment decisions and watch as the patient outcome rises or declines. The game’s rapid pace underscores the importance of early diagnosis and treatment.
“We tried to make our game as engaging and real-life as possible,” Dr. Shieh says.
The Stanford team is in touch with the Society of Critical Care Medicine’s Surviving Sepsis Campaign (www.survivingsepsis.org) and with other medical groups internationally. Thousands of players have accessed the game online for free (http://cme.stanford.edu/septris/game/SepsisTetris.html), with a nominal fee for CME credit. It is best played on an iPad or iPhone, Dr. Shieh says.
Larry Beresford is a freelance writer in Alameda, Calif.
Teaching trainees to identify and manage sepsis using an online game known as “Septris” earned hospitalists at Stanford University Medical Center in Palo Alto, Calif., a Research, Innovation, and Clinical Vignette category award at HM13.1
“We took third-year medical students and residents in medicine, surgery, and emergency medicine—people who would be sepsis first responders on the floor—and gave them pre- and post-tests that documented improvements in both attitudes and knowledge,” says lead author Lisa Shieh, MD, PhD, Stanford’s medical director of quality in the department of medicine. All participants said they enjoyed playing the game, she reported.
Septris was developed by a multidisciplinary group of physicians, educational technology specialists, and programmers at Stanford. The game offers a case-based interactive learning environment drawn from evidence-based treatment algorithms. Players make treatment decisions and watch as the patient outcome rises or declines. The game’s rapid pace underscores the importance of early diagnosis and treatment.
“We tried to make our game as engaging and real-life as possible,” Dr. Shieh says.
The Stanford team is in touch with the Society of Critical Care Medicine’s Surviving Sepsis Campaign (www.survivingsepsis.org) and with other medical groups internationally. Thousands of players have accessed the game online for free (http://cme.stanford.edu/septris/game/SepsisTetris.html), with a nominal fee for CME credit. It is best played on an iPad or iPhone, Dr. Shieh says.
Larry Beresford is a freelance writer in Alameda, Calif.
Teaching trainees to identify and manage sepsis using an online game known as “Septris” earned hospitalists at Stanford University Medical Center in Palo Alto, Calif., a Research, Innovation, and Clinical Vignette category award at HM13.1
“We took third-year medical students and residents in medicine, surgery, and emergency medicine—people who would be sepsis first responders on the floor—and gave them pre- and post-tests that documented improvements in both attitudes and knowledge,” says lead author Lisa Shieh, MD, PhD, Stanford’s medical director of quality in the department of medicine. All participants said they enjoyed playing the game, she reported.
Septris was developed by a multidisciplinary group of physicians, educational technology specialists, and programmers at Stanford. The game offers a case-based interactive learning environment drawn from evidence-based treatment algorithms. Players make treatment decisions and watch as the patient outcome rises or declines. The game’s rapid pace underscores the importance of early diagnosis and treatment.
“We tried to make our game as engaging and real-life as possible,” Dr. Shieh says.
The Stanford team is in touch with the Society of Critical Care Medicine’s Surviving Sepsis Campaign (www.survivingsepsis.org) and with other medical groups internationally. Thousands of players have accessed the game online for free (http://cme.stanford.edu/septris/game/SepsisTetris.html), with a nominal fee for CME credit. It is best played on an iPad or iPhone, Dr. Shieh says.
Larry Beresford is a freelance writer in Alameda, Calif.
Should Unaffiliated Physicians Have Infusion Privileges?

“Infusion Privileges” a Simple Answer to Complex Issue
I have a couple of questions based on the following scenario: hospital infusion center treating patients referred by physicians who are not members of hospital staff and don’t have hospital privileges. Since they are not credentialed at the hospital, they cannot give orders for infusion treatment for their patients. And they are not interested in applying for membership and hospital privileges. First, is it OK for the referring physicians to talk to our hospitalist of the day and give an infusion treatment order? Second, what CPT code would the hospitalist use for just writing an infusion treatment order—and can they bill the service?
—Glena Loyola
Dr. Hospitalist responds:
The alternate site infusion therapy market has exploded in the U.S. in the past 25 years. Most of this surge has been driven by increased emphasis on cost containment and the desires of patients to resume their usual lifestyles while recovering from illness. Most recent estimates show that these services represent approximately $9-$11 billion a year. Although the cost is substantial, it is far lower than the cost of inpatient treatment.
Many hospitals have infusion centers, both as revenue-generating ventures and to provide a service for their patients without admitting them to the hospital. Initially, most centers focused on oncologic medications; most now provide a variety of infusion services and therapies. Having clinical staff, prescribing physicians, and pharmacists under the same roof, or in the same healthcare system, should lead to better communication, which is key when administering these specialty drugs. The center at my hospital is of average size, and it seems there are at least one or two medical emergencies there every month. I can imagine the wasted time and lives lost in situations where a full cadre of emergency staff was not immediately available.
The processes and procedures developed by hospital administrators to allow physicians to administer these medications are highly variable. When the centers first came on the scene, most of the prescribing physicians were practicing oncologists and active members of the medical staff. While oncologists still make up the largest group utilizing these centers, rheumatologists, cardiologists, and endocrinologists also are active participants. As these clinicians have aged, and as the services, as well as the variety of infusions, have expanded, hospitals have needed alternate staffing models to keep up.
My CMO created specific “infusion privileges” for health system physicians working on alternate campuses. This privilege allows them to write for the medications but does not give them core privileges like most courtesy staff designations. There is no associated hospital call or ED coverage requirement, and no quality monitoring is needed with this “special” designation. We did consider having our hospitalist write the orders for these docs, but there were many reasons not to go that route—most importantly the logistics and our current HM program’s bandwidth.
The situation you describe, in which physicians call in and give infusion orders to another physician/hospitalist, is the one I believe is most fraught with problems. The potential for prescribing error is very high. Plus, the multiple downstream opportunities for the patient’s care to be compromised are myriad. Because the consequences of a medication error with many of these infusions can be catastrophic, most institutions (including ours) limit who can prescribe them to those specializing in that field. Many also require physicians to use computerized physician order entry, which has been shown to reduce medication errors, for these agents.
The billing requirements for infusion centers and prescribers are very complex and were last globally consolidated in May 2004. CMS annually updates using National Correct Coding Initiative Edits, with which most coders are familiar. The CPT code is tied to the infusion or type of infusion that is given and even incorporates the amount of time it takes to administer. Prior to 2004, the codes incorporated practice expense as well as malpractice relative value units (RVUs), but zero physician RVUs. Since then, a lot has changed. Although a physician can usually bill for services using E&M codes, most require face-to-face time to be allowable. If you would like to bill independently as a prescriber for your services, I recommend you sit down with your coders and decide if it’s feasible.
“Infusion Privileges” a Simple Answer to Complex Issue
I have a couple of questions based on the following scenario: hospital infusion center treating patients referred by physicians who are not members of hospital staff and don’t have hospital privileges. Since they are not credentialed at the hospital, they cannot give orders for infusion treatment for their patients. And they are not interested in applying for membership and hospital privileges. First, is it OK for the referring physicians to talk to our hospitalist of the day and give an infusion treatment order? Second, what CPT code would the hospitalist use for just writing an infusion treatment order—and can they bill the service?
—Glena Loyola
Dr. Hospitalist responds:
The alternate site infusion therapy market has exploded in the U.S. in the past 25 years. Most of this surge has been driven by increased emphasis on cost containment and the desires of patients to resume their usual lifestyles while recovering from illness. Most recent estimates show that these services represent approximately $9-$11 billion a year. Although the cost is substantial, it is far lower than the cost of inpatient treatment.
Many hospitals have infusion centers, both as revenue-generating ventures and to provide a service for their patients without admitting them to the hospital. Initially, most centers focused on oncologic medications; most now provide a variety of infusion services and therapies. Having clinical staff, prescribing physicians, and pharmacists under the same roof, or in the same healthcare system, should lead to better communication, which is key when administering these specialty drugs. The center at my hospital is of average size, and it seems there are at least one or two medical emergencies there every month. I can imagine the wasted time and lives lost in situations where a full cadre of emergency staff was not immediately available.
The processes and procedures developed by hospital administrators to allow physicians to administer these medications are highly variable. When the centers first came on the scene, most of the prescribing physicians were practicing oncologists and active members of the medical staff. While oncologists still make up the largest group utilizing these centers, rheumatologists, cardiologists, and endocrinologists also are active participants. As these clinicians have aged, and as the services, as well as the variety of infusions, have expanded, hospitals have needed alternate staffing models to keep up.
My CMO created specific “infusion privileges” for health system physicians working on alternate campuses. This privilege allows them to write for the medications but does not give them core privileges like most courtesy staff designations. There is no associated hospital call or ED coverage requirement, and no quality monitoring is needed with this “special” designation. We did consider having our hospitalist write the orders for these docs, but there were many reasons not to go that route—most importantly the logistics and our current HM program’s bandwidth.
The situation you describe, in which physicians call in and give infusion orders to another physician/hospitalist, is the one I believe is most fraught with problems. The potential for prescribing error is very high. Plus, the multiple downstream opportunities for the patient’s care to be compromised are myriad. Because the consequences of a medication error with many of these infusions can be catastrophic, most institutions (including ours) limit who can prescribe them to those specializing in that field. Many also require physicians to use computerized physician order entry, which has been shown to reduce medication errors, for these agents.
The billing requirements for infusion centers and prescribers are very complex and were last globally consolidated in May 2004. CMS annually updates using National Correct Coding Initiative Edits, with which most coders are familiar. The CPT code is tied to the infusion or type of infusion that is given and even incorporates the amount of time it takes to administer. Prior to 2004, the codes incorporated practice expense as well as malpractice relative value units (RVUs), but zero physician RVUs. Since then, a lot has changed. Although a physician can usually bill for services using E&M codes, most require face-to-face time to be allowable. If you would like to bill independently as a prescriber for your services, I recommend you sit down with your coders and decide if it’s feasible.
“Infusion Privileges” a Simple Answer to Complex Issue
I have a couple of questions based on the following scenario: hospital infusion center treating patients referred by physicians who are not members of hospital staff and don’t have hospital privileges. Since they are not credentialed at the hospital, they cannot give orders for infusion treatment for their patients. And they are not interested in applying for membership and hospital privileges. First, is it OK for the referring physicians to talk to our hospitalist of the day and give an infusion treatment order? Second, what CPT code would the hospitalist use for just writing an infusion treatment order—and can they bill the service?
—Glena Loyola
Dr. Hospitalist responds:
The alternate site infusion therapy market has exploded in the U.S. in the past 25 years. Most of this surge has been driven by increased emphasis on cost containment and the desires of patients to resume their usual lifestyles while recovering from illness. Most recent estimates show that these services represent approximately $9-$11 billion a year. Although the cost is substantial, it is far lower than the cost of inpatient treatment.
Many hospitals have infusion centers, both as revenue-generating ventures and to provide a service for their patients without admitting them to the hospital. Initially, most centers focused on oncologic medications; most now provide a variety of infusion services and therapies. Having clinical staff, prescribing physicians, and pharmacists under the same roof, or in the same healthcare system, should lead to better communication, which is key when administering these specialty drugs. The center at my hospital is of average size, and it seems there are at least one or two medical emergencies there every month. I can imagine the wasted time and lives lost in situations where a full cadre of emergency staff was not immediately available.
The processes and procedures developed by hospital administrators to allow physicians to administer these medications are highly variable. When the centers first came on the scene, most of the prescribing physicians were practicing oncologists and active members of the medical staff. While oncologists still make up the largest group utilizing these centers, rheumatologists, cardiologists, and endocrinologists also are active participants. As these clinicians have aged, and as the services, as well as the variety of infusions, have expanded, hospitals have needed alternate staffing models to keep up.
My CMO created specific “infusion privileges” for health system physicians working on alternate campuses. This privilege allows them to write for the medications but does not give them core privileges like most courtesy staff designations. There is no associated hospital call or ED coverage requirement, and no quality monitoring is needed with this “special” designation. We did consider having our hospitalist write the orders for these docs, but there were many reasons not to go that route—most importantly the logistics and our current HM program’s bandwidth.
The situation you describe, in which physicians call in and give infusion orders to another physician/hospitalist, is the one I believe is most fraught with problems. The potential for prescribing error is very high. Plus, the multiple downstream opportunities for the patient’s care to be compromised are myriad. Because the consequences of a medication error with many of these infusions can be catastrophic, most institutions (including ours) limit who can prescribe them to those specializing in that field. Many also require physicians to use computerized physician order entry, which has been shown to reduce medication errors, for these agents.
The billing requirements for infusion centers and prescribers are very complex and were last globally consolidated in May 2004. CMS annually updates using National Correct Coding Initiative Edits, with which most coders are familiar. The CPT code is tied to the infusion or type of infusion that is given and even incorporates the amount of time it takes to administer. Prior to 2004, the codes incorporated practice expense as well as malpractice relative value units (RVUs), but zero physician RVUs. Since then, a lot has changed. Although a physician can usually bill for services using E&M codes, most require face-to-face time to be allowable. If you would like to bill independently as a prescriber for your services, I recommend you sit down with your coders and decide if it’s feasible.
Hospitalist Reviews on Treatments for Acute Asthma, Stroke, Healthcare-Associated Pneumonia, and More

In This Edition
Literature At A Glance
A guide to this month’s studies
- ICU pressures improve transfers to the floor
- Morbidity, mortality rates high for respiratory syncytial virus infections
- Antibiotic algorithm can guide therapy in healthcare-associated pneumonia
- Three-month dual antiplatelet therapy for zotarolimus-eluting stents
- De-escalating antibiotics in sepsis
- New oral anticoagulants increase GI bleed risk
- Single vs. dual antiplatelet therapy after stroke
- Endoscopic vs. surgical cystogastrostomy for pancreatic pseudocyst drainage
- Long-term cognitive impairment after critical illness
- Holding chambers vs. nebulizers for acute asthma
ICU Pressures Improve Transfers to the Floor
Clinical question: Does ICU strain negatively affect the outcomes of patients transferred to the floor?
Background: With healthcare costs increasing and critical care staff shortages projected, ICUs will have to operate under increasing strain. This may influence decisions on discharging patients from ICUs and could affect patient outcomes.
Study design: Retrospective cohort study.
Setting: One hundred fifty-five ICUs in the United States.
Synopsis: Using the Project IMPACT database, 200,730 adult patients from 107 different hospitals were evaluated in times of ICU strain, determined by the current census, new admissions, and acuity level. Outcomes measured were initial ICU length of stay (LOS), readmission within 72 hours, in-hospital mortality rates, and post-ICU discharge LOS.
Increases of the strain variables from the fifth to the 95th percentiles resulted in a 6.3-hour reduction in ICU LOS, a 2.0-hour decrease in post-ICU discharge LOS, and a 1.0% increase in probability of ICU readmission within 72 hours. Mortality rates during the hospital stay and odds of being discharged home showed no significant change. This study was limited because the ICUs participating were not randomly chosen, outcomes of patients transferred to other hospitals were not measured, and no post-hospital data was collected, so no long-term outcomes could be measured.
Bottom line: ICU bed pressures prompt physicians to allocate ICU resources more efficiently without changing short-term patient outcomes.
Citation: Wagner J, Gabler NB, Ratcliffe SJ, Brown SE, Strom BL, Halpern SD. Outcomes among patients discharged from busy intensive care units. Ann Intern Med. 2013;159(7):447-455.
Adults Hospitalized for Respiratory Syncytial Virus Infections Have High Morbidity, Mortality Rates
Clinical question: What are the complications and outcomes of respiratory syncytial virus (RSV) infection in adults requiring hospitalization?
Background: RSV is a common cause of lower respiratory tract infection in infants and young children, leading to hospitalization and even death. RSV has been estimated to affect 3%-10% of adults annually, generally causing mild disease. However, the outcomes of adults with more severe disease are not fully known.
Study design: Retrospective cohort study.
Setting: Three acute care, public hospitals in Hong Kong.
Synopsis: All adult patients hospitalized with laboratory-confirmed RSV infection were included during the defined time period. The main outcome measure was all-cause death, with secondary outcome measures of development of acute respiratory failure requiring ventilator support and total duration of hospitalization among survivors. Additionally, the cohort of RSV patients was compared to patients admitted with seasonal influenza during this same time frame. Patients with pandemic 2009 H1N1 infection were not included.
Of patients with RSV, pneumonia was found in 42.3%, bacterial superinfection in 12.5%, and cardiovascular complications in 14.3%. Additionally, 11.1% developed respiratory failure requiring ventilator support. All-cause mortality at 30 days and 60 days was 9.1% and 11.9%, respectively, with pneumonia the most common cause of death. Use of systemic corticosteroids did not improve survival. When the RSV cohort was compared to the influenza cohort, the patients were similar in age, but the RSV patients were more likely to have underlying chronic lung disease and major systemic co-morbidities. The rate of survival and duration of hospitalization were not significantly different.
Bottom line: RSV infection is an underappreciated cause of lower tract respiratory infection in adults; severe infections that require hospitalization have rates of mortality similar to seasonal influenza. Further research on treatment or immunization is needed.
Citation: Lee N, Lui GC, Wong KT, et al. High morbidity and mortality in adults hospitalized for respiratory syncytial virus infections. Clin Infect Dis. 2013;57(8):1069-1077.
Antibiotic Algorithm Can Guide Therapy in Healthcare-Associated Pneumonia
Clinical question: Can an algorithm based on risk for multidrug-resistant (MDR) organisms and illness severity guide antibiotic selection in healthcare-associated pneumonia (HCAP)?
Background: The 2005 American Thoracic Society/Infectious Diseases Society of America (ATS/IDSA) guidelines identify patients with HCAP as those with recent contact with a healthcare environment, including nursing homes and hemodialysis; however, previous studies have shown that not all patients with healthcare contact have equal risk for MDR organisms.
Study design: Prospective cohort study.
Setting: Japan, multi-center.
Synopsis: Of the 445 enrolled patients, 124 were diagnosed with community-acquired pneumonia (CAP) and 321 with HCAP. Patients with HCAP were classified based on severity of illness or MDR pathogen risk factors (immune suppression, hospitalization within the last 90 days, poor functional status, and antibiotics within the past six months). Patients with low risk (0-1 factors) for MDR organisms were treated for CAP, and patients with high risk (≥2 factors) or moderate risk (≥1 factor) for severe illness were treated for HCAP.
HCAP patients had a higher 30-day mortality rate (13.7% vs. 5.6%, P=0.017), but mortality rate was less in the patients at low risk for MDR pathogens (8.6% vs. 18.2%, P=0.012). Of the HCAP patients, only 7.1% received inappropriate therapy (pathogen resistant to initial antibiotic regimen), and treatment failure was 19.3%.
Appropriateness of initial empiric therapy was determined not to be a mortality risk; however, this trial might be limited by its location, because Japan appears to have fewer MDR pathogens than the U.S.
Bottom line: A treatment algorithm based on risk for MDR organisms and severity of illness can be used to guide empiric antibiotic therapy in patients with HCAP, and, ideally, to reduce excessive use of broad-spectrum antibiotics.
Citation: Maruyama T, Fujisawa T, Okuno M, et al. A new strategy for healthcare-associated pneumonia: a 2-year prospective multicenter cohort study using risk factors for multidrug-resistant pathogens to select initial empiric therapy. Clin Infect Dis. 2013;57(10):1373-1383.
Three-Month Dual Antiplatelet Therapy for Zotarolimus-Eluting Stents
Clinical question: Is short-term, dual antiplatelet therapy noninferior to long-term therapy in zotarolimus-eluting stents?
Background: Current guidelines recommend long-term (>12 months) dual antiplatelet therapy after the placement of drug-eluting stents. The optimal therapy duration in second-generation drug-eluting stents has not been studied; moreover, some studies with multiple drug-eluting stents have suggested no added benefit from long-term therapy.
Study design: Randomized controlled trial.
Setting: Brazil, multi-center.
Synopsis: Researchers randomized 3,211 patients with stable coronary artery disease (CAD) or low-risk acute coronary syndrome (ACS) undergoing intervention with zotarolimus-eluting stents to short-term (three months) or long-term (12 months) dual antiplatelet therapy. Exclusion criteria included ST-elevation myocardial infarction (STEMI), previous drug-eluting stent, scheduled elective surgery within 12 months, or contraindication to aspirin or clopidogrel. Primary endpoints were a composite of death from any cause, MI, stroke, or major bleeding. Secondary endpoints were stent thrombosis, target lesion revascularization, adverse cardiac event, and any bleed.
At one-year follow-up, the short-term group had similar primary (6.0% vs. 5.8%) and secondary (8.3% vs. 7.4%) outcomes compared to the long-term. The short-term group’s noninferiority also was seen in several key subgroups.
This study included patients with stable CAD or low-risk ACS and cannot be generalized to higher-risk patients. Results for zotarolimus-eluting stents cannot be generalized to other second-generation drug-eluting stents.
Bottom line: Zotarolimus-eluting stents, followed by three months of dual antiplatelet therapy, were noninferior to 12 months of therapy in patients with stable CAD or low-risk ACS.
Citation: Feres F, Costa RA, Abizaid A, et al. Three vs. twelve months of dual antiplatelet therapy after zotarolimus-eluting stents: the OPTIMIZE randomized trial. JAMA. 2013;310(23):2510-2522.
De-Escalating Antibiotics in Sepsis
Clinical question: Does tailoring antibiotics based on known pathogens impact mortality for patients with severe sepsis or shock?
Background: In patients with sepsis, the use of early empiric antibiotics reduces morbidity and mortality. De-escalation therapy refers to narrowing the broad-spectrum antibiotics once the pathogen and sensitivities are known; however, no randomized controlled studies have assessed the impact of this therapy on critically ill patients.
Study design: Prospective observational study.
Setting: Academic hospital ICU in Spain.
Synopsis: From January 2008 to May 2012, 628 adult patients were treated empirically with broad-spectrum antibiotics. De-escalation was applied to 219 patients (34.9%). Outcomes measured were ICU mortality, hospital mortality, and 90-day mortality in patients who received de-escalation therapy, patients whose antibiotics were not changed, and patients for whom antibiotics were escalated.
The in-hospital mortality rate was 27.4% in patients who were de-escalated, 32.6% in the unchanged group, and 42.9% in the escalation group. ICU and 90-day mortality were lower in the de-escalation group. De-escalation was more commonly used in medical than in surgical patients.
This study is limited because it is not a randomized controlled study and was single-centered, so it might only be applicable on the larger scale. Also, multi-drug resistant organisms were not evaluated.
Overall, it is safe to narrow empiric antibiotics in severe sepsis and shock when the pathogen and sensitivities are known.
Bottom line: De-escalation of antibiotics in severe sepsis and septic shock is associated with a lower mortality.
Citation: Garnacho-Montero J, Gutierrez-Pizarraya A, Escoresca-Ortega A, et al. De-escalation of empirical therapy is associated with lower mortality in patients with severe sepsis and septic shock. Intensive Care Med. 2014;40(1):32-40.
New Oral Anticoagulants Increase GI Bleed Risk
Clinical question: Do thrombin and factor Xa inhibitors increase the risk of gastrointestinal (GI) bleeding when compared to vitamin K antagonists and heparins?
Background: New oral anticoagulants (thrombin and factor Xa inhibitors) are available and being used with increased frequency due to equal efficacy and ease of administration. Some studies indicate a higher risk of GI bleeding with these agents. Further evaluation is needed, because no reversal therapy is available.
Study design: Systematic review and meta-analysis.
Setting: Data from MEDLINE, Embase, and the Cochrane Library.
Synopsis: More than 150,000 patients from 43 randomized controlled trials were evaluated for risk of GI bleed when treated with new anticoagulants versus traditional therapy. Patients were treated for one of the following: embolism prevention from atrial fibrillation, venous thromboembolism (VTE) prophylaxis post orthopedic surgery, VTE prophylaxis of medical patients, acute VTE, and acute coronary syndrome (ACS). Use of aspirin or NSAIDs was discouraged but not documented. The odds ratio for GI bleeding with use of the new anticoagulants was 1.45, with a number needed to harm of 500. Evaluation of subgroups revealed increased GI bleed risk in patients treated for ACS and acute thrombosis versus prophylaxis. Post-surgical patients had the lowest risk.
This study was limited by the heterogeneity and differing primary outcomes (mostly efficacy rather than safety) of the included trials. Studies excluded high-risk patients, which the authors estimate to be 25%-40% of actual patients. More studies need to be done that include high-risk patients and focus on GI bleed as a primary outcome.
Bottom line: The new anticoagulants tend to have a higher incidence of GI bleed than traditional therapy, but this varies based on indication of therapy and needs further evaluation to clarify risk.
Citation: Holster IL, Valkhoff VE, Kuipers EJ, Tjwa ET. New oral anticoagulants increase risk for gastrointestinal bleeding: a systematic review and meta-analysis. Gastroenterology. 2013;145(1):105-112.
Single vs. Dual Antiplatelet Therapy after Stroke
Clinical question: Is dual antiplatelet therapy more beneficial or harmful than monotherapy after ischemic stroke?
Background: It is recommended that patients with ischemic stroke or transient ischemic attack (TIA) receive lifelong antiplatelet therapy; however, there have been insufficient studies evaluating the long-term safety of dual antiplatelet therapy.
Study design: Meta-analysis of randomized controlled trials (RCTs)
Setting: Data from PubMed, Embase, and the Cochrane Central Register of Controlled Trials.
Synopsis: Data from seven RCTs, including 39,574 patients with recent TIA or ischemic stroke, were reviewed. Comparisons were made regarding occurrence of intracranial hemorrhage (ICH) and recurrent stroke between patients receiving dual antiplatelet therapy and those receiving aspirin or clopidogrel monotherapy. All patients were treated for at least one year.
There was no difference in recurrent stroke or ICH between patients on dual antiplatelet therapy versus aspirin monotherapy. Patients treated with dual antiplatelet therapy did have a 46% increased risk of ICH without any additional protective benefit for recurrent stroke or TIA when compared with patients on clopidogrel monotherapy.
This information should not be applied in the acute setting, given the high risk of stroke after TIA or ischemic stroke. One major limitation of this study was that the individual trials used different combinations of dual antiplatelet therapy.
Bottom line: The risk of recurrent stroke or TIA after dual antiplatelet therapy and after monotherapy with aspirin or clopidogrel is equal, but the risk of ICH compared to clopidogrel monotherapy is increased.
Citation: Lee M, Saver JL, Hong KS, Rao NM, Wu YL, Ovbiagele B. Risk-benefit profile of long-term dual- versus single-antiplatelet therapy among patients with ischemic stroke: a systematic review and meta-analysis. Ann Intern Med. 2013;159(7):463-470.
Endoscopic vs. Surgical Cystogastrostomy for Pancreatic Pseudocyst Drainage
Clinical question: How does endoscopic cystogastrostomy for pancreatic pseudocyst drainage compare to the standard surgical approach?
Background: Pancreatic pseudocysts are a common complication of pancreatitis and necessitate decompression when they are accompanied by pain, infection, or obstruction. Decompression of the pseudocyst can be accomplished using either endoscopic or surgical cystogastrostomy.
Study design: Open-label, single-center, randomized trial.
Setting: Single-center U.S. hospital.
Synopsis: A total of 40 patients were randomly equalized to both treatment arms; 20 patients underwent endoscopic and 20 patients underwent surgical cystogastrostomy. Zero patients in the endoscopic therapy had a pseudocyst recurrence, compared with one patient treated surgically. Length of stay (LOS) and cost were lower for the endoscopic group compared to the surgical group (two days vs. six days, P<0.001, $7,011 vs. $15,052, P=0.003).
This study is limited due to several factors. First, patients with pancreatic necrosis were excluded; had these patients been included, the complication rates and LOS would have been higher. Second, cost difference cannot be generalized across the U.S., because Medicare payments are based on provider types and regions.
Bottom line: Endoscopic cystogastrostomy for pancreatic pseudocyst is equal to the standard surgical therapy and results in decreased LOS and reduced costs.
Citation: Varadarajulu S, Bang JY, Sutton BS, Trevino JM, Christein JD, Wilcox CM. Equal efficacy of endoscopic and surgical cystogastrostomy for pancreatic pseudocyst drainage in a randomized trial. Gastroenterology. 2013;145(3):583-590.
Long-Term Cognitive Impairment after Critical Illness
Clinical question: Are a longer duration of delirium and higher doses of sedatives associated with cognitive impairment in the hospital?
Background: Survivors of critical illness are at risk for prolonged cognitive dysfunction. Delirium (and factors associated with delirium, namely sedative and analgesic medications) has been implicated in cognitive dysfunction.
Study design: Prospective cohort study.
Setting: Multi-center, academic, and acute care hospitals.
Synopsis: The study examined 821 adults admitted to the ICU with respiratory failure, cardiogenic shock, or septic shock. Patients excluded were those with pre-existing cognitive impairment, those with psychotic disorders, and those for whom follow-up would not be possible. Two risk factors measured were duration of delirium and use of sedative/analgesics. Delirium was assessed at three and 12 months using the CAM-ICU algorithm in the ICU by trained psychology professionals who were unaware of the patients’ in-hospital course.
At three months, 40% of patients had global cognition scores that were 1.5 standard deviations (SD) below population mean (similar to traumatic brain injury), and 26% had scores two SD below population mean (similar to mild Alzheimer’s). At 12 months, 34% had scores similar to traumatic brain injury patients, and 24% had scores similar to mild Alzheimer’s. A longer duration of delirium was associated with worse global cognition at three and 12 months. Use of sedatives/analgesics was not associated with cognitive impairment.
Bottom line: Critically ill patients in the ICU who experience a longer duration of delirium are at risk of long-term cognitive impairments lasting 12 months.
Citation: Pandharipande PP, Girard TD, Jackson JC, et al. Long-term cognitive impairment after critical illness. N Engl J Med. 2013;369(14):1306-1316.
Holding Chambers (Spacers) vs. Nebulizers for Acute Asthma
Clinical question: Are beta-2 agonists as effective when administered through a holding chamber (spacer) as they are when administered by a nebulizer?
Background: During an acute asthma attack, beta-2 agonists must be delivered to the peripheral airways. There has been considerable controversy regarding the use of a spacer compared with a nebulizer. Aside from admission rates and length of stay, factors taken into account include cost, maintenance of nebulizer machines, and infection control (potential of cross-infection via nebulizers).
Study design: Meta-analysis review of randomized controlled trials (RCTs).
Setting: Multi-centered, worldwide studies from community setting and EDs.
Synopsis: In 39 studies of patients with an acute asthma attack (selected from Cochrane Airways Group Specialized Register), the hospital admission rates did not differ on the basis of delivery method in 729 adults (risk ratio=0.94, confidence interval 0.61-1.43) or in 1,897 children (risk ratio=0.71, confidence interval 0.47-1.08). Secondary outcomes included the duration of time in the ED and the duration of hospital admission. Time spent in the ED varied for adults but was shorter for children with spacers (based on three studies). Duration of hospital admission also did not differ when modes of delivery were compared.
Bottom line: Providing beta-2 agonists using nebulizers during an acute asthma attack is not more effective than administration using a spacer.
Citation: Cates CJ, Welsh EJ, Rowe BH. Holding chambers (spacers) versus nebulisers for beta-agonist treatment of acute asthma. Cochrane Database Syst Rev. 2013;9:CD000052.
In This Edition
Literature At A Glance
A guide to this month’s studies
- ICU pressures improve transfers to the floor
- Morbidity, mortality rates high for respiratory syncytial virus infections
- Antibiotic algorithm can guide therapy in healthcare-associated pneumonia
- Three-month dual antiplatelet therapy for zotarolimus-eluting stents
- De-escalating antibiotics in sepsis
- New oral anticoagulants increase GI bleed risk
- Single vs. dual antiplatelet therapy after stroke
- Endoscopic vs. surgical cystogastrostomy for pancreatic pseudocyst drainage
- Long-term cognitive impairment after critical illness
- Holding chambers vs. nebulizers for acute asthma
ICU Pressures Improve Transfers to the Floor
Clinical question: Does ICU strain negatively affect the outcomes of patients transferred to the floor?
Background: With healthcare costs increasing and critical care staff shortages projected, ICUs will have to operate under increasing strain. This may influence decisions on discharging patients from ICUs and could affect patient outcomes.
Study design: Retrospective cohort study.
Setting: One hundred fifty-five ICUs in the United States.
Synopsis: Using the Project IMPACT database, 200,730 adult patients from 107 different hospitals were evaluated in times of ICU strain, determined by the current census, new admissions, and acuity level. Outcomes measured were initial ICU length of stay (LOS), readmission within 72 hours, in-hospital mortality rates, and post-ICU discharge LOS.
Increases of the strain variables from the fifth to the 95th percentiles resulted in a 6.3-hour reduction in ICU LOS, a 2.0-hour decrease in post-ICU discharge LOS, and a 1.0% increase in probability of ICU readmission within 72 hours. Mortality rates during the hospital stay and odds of being discharged home showed no significant change. This study was limited because the ICUs participating were not randomly chosen, outcomes of patients transferred to other hospitals were not measured, and no post-hospital data was collected, so no long-term outcomes could be measured.
Bottom line: ICU bed pressures prompt physicians to allocate ICU resources more efficiently without changing short-term patient outcomes.
Citation: Wagner J, Gabler NB, Ratcliffe SJ, Brown SE, Strom BL, Halpern SD. Outcomes among patients discharged from busy intensive care units. Ann Intern Med. 2013;159(7):447-455.
Adults Hospitalized for Respiratory Syncytial Virus Infections Have High Morbidity, Mortality Rates
Clinical question: What are the complications and outcomes of respiratory syncytial virus (RSV) infection in adults requiring hospitalization?
Background: RSV is a common cause of lower respiratory tract infection in infants and young children, leading to hospitalization and even death. RSV has been estimated to affect 3%-10% of adults annually, generally causing mild disease. However, the outcomes of adults with more severe disease are not fully known.
Study design: Retrospective cohort study.
Setting: Three acute care, public hospitals in Hong Kong.
Synopsis: All adult patients hospitalized with laboratory-confirmed RSV infection were included during the defined time period. The main outcome measure was all-cause death, with secondary outcome measures of development of acute respiratory failure requiring ventilator support and total duration of hospitalization among survivors. Additionally, the cohort of RSV patients was compared to patients admitted with seasonal influenza during this same time frame. Patients with pandemic 2009 H1N1 infection were not included.
Of patients with RSV, pneumonia was found in 42.3%, bacterial superinfection in 12.5%, and cardiovascular complications in 14.3%. Additionally, 11.1% developed respiratory failure requiring ventilator support. All-cause mortality at 30 days and 60 days was 9.1% and 11.9%, respectively, with pneumonia the most common cause of death. Use of systemic corticosteroids did not improve survival. When the RSV cohort was compared to the influenza cohort, the patients were similar in age, but the RSV patients were more likely to have underlying chronic lung disease and major systemic co-morbidities. The rate of survival and duration of hospitalization were not significantly different.
Bottom line: RSV infection is an underappreciated cause of lower tract respiratory infection in adults; severe infections that require hospitalization have rates of mortality similar to seasonal influenza. Further research on treatment or immunization is needed.
Citation: Lee N, Lui GC, Wong KT, et al. High morbidity and mortality in adults hospitalized for respiratory syncytial virus infections. Clin Infect Dis. 2013;57(8):1069-1077.
Antibiotic Algorithm Can Guide Therapy in Healthcare-Associated Pneumonia
Clinical question: Can an algorithm based on risk for multidrug-resistant (MDR) organisms and illness severity guide antibiotic selection in healthcare-associated pneumonia (HCAP)?
Background: The 2005 American Thoracic Society/Infectious Diseases Society of America (ATS/IDSA) guidelines identify patients with HCAP as those with recent contact with a healthcare environment, including nursing homes and hemodialysis; however, previous studies have shown that not all patients with healthcare contact have equal risk for MDR organisms.
Study design: Prospective cohort study.
Setting: Japan, multi-center.
Synopsis: Of the 445 enrolled patients, 124 were diagnosed with community-acquired pneumonia (CAP) and 321 with HCAP. Patients with HCAP were classified based on severity of illness or MDR pathogen risk factors (immune suppression, hospitalization within the last 90 days, poor functional status, and antibiotics within the past six months). Patients with low risk (0-1 factors) for MDR organisms were treated for CAP, and patients with high risk (≥2 factors) or moderate risk (≥1 factor) for severe illness were treated for HCAP.
HCAP patients had a higher 30-day mortality rate (13.7% vs. 5.6%, P=0.017), but mortality rate was less in the patients at low risk for MDR pathogens (8.6% vs. 18.2%, P=0.012). Of the HCAP patients, only 7.1% received inappropriate therapy (pathogen resistant to initial antibiotic regimen), and treatment failure was 19.3%.
Appropriateness of initial empiric therapy was determined not to be a mortality risk; however, this trial might be limited by its location, because Japan appears to have fewer MDR pathogens than the U.S.
Bottom line: A treatment algorithm based on risk for MDR organisms and severity of illness can be used to guide empiric antibiotic therapy in patients with HCAP, and, ideally, to reduce excessive use of broad-spectrum antibiotics.
Citation: Maruyama T, Fujisawa T, Okuno M, et al. A new strategy for healthcare-associated pneumonia: a 2-year prospective multicenter cohort study using risk factors for multidrug-resistant pathogens to select initial empiric therapy. Clin Infect Dis. 2013;57(10):1373-1383.
Three-Month Dual Antiplatelet Therapy for Zotarolimus-Eluting Stents
Clinical question: Is short-term, dual antiplatelet therapy noninferior to long-term therapy in zotarolimus-eluting stents?
Background: Current guidelines recommend long-term (>12 months) dual antiplatelet therapy after the placement of drug-eluting stents. The optimal therapy duration in second-generation drug-eluting stents has not been studied; moreover, some studies with multiple drug-eluting stents have suggested no added benefit from long-term therapy.
Study design: Randomized controlled trial.
Setting: Brazil, multi-center.
Synopsis: Researchers randomized 3,211 patients with stable coronary artery disease (CAD) or low-risk acute coronary syndrome (ACS) undergoing intervention with zotarolimus-eluting stents to short-term (three months) or long-term (12 months) dual antiplatelet therapy. Exclusion criteria included ST-elevation myocardial infarction (STEMI), previous drug-eluting stent, scheduled elective surgery within 12 months, or contraindication to aspirin or clopidogrel. Primary endpoints were a composite of death from any cause, MI, stroke, or major bleeding. Secondary endpoints were stent thrombosis, target lesion revascularization, adverse cardiac event, and any bleed.
At one-year follow-up, the short-term group had similar primary (6.0% vs. 5.8%) and secondary (8.3% vs. 7.4%) outcomes compared to the long-term. The short-term group’s noninferiority also was seen in several key subgroups.
This study included patients with stable CAD or low-risk ACS and cannot be generalized to higher-risk patients. Results for zotarolimus-eluting stents cannot be generalized to other second-generation drug-eluting stents.
Bottom line: Zotarolimus-eluting stents, followed by three months of dual antiplatelet therapy, were noninferior to 12 months of therapy in patients with stable CAD or low-risk ACS.
Citation: Feres F, Costa RA, Abizaid A, et al. Three vs. twelve months of dual antiplatelet therapy after zotarolimus-eluting stents: the OPTIMIZE randomized trial. JAMA. 2013;310(23):2510-2522.
De-Escalating Antibiotics in Sepsis
Clinical question: Does tailoring antibiotics based on known pathogens impact mortality for patients with severe sepsis or shock?
Background: In patients with sepsis, the use of early empiric antibiotics reduces morbidity and mortality. De-escalation therapy refers to narrowing the broad-spectrum antibiotics once the pathogen and sensitivities are known; however, no randomized controlled studies have assessed the impact of this therapy on critically ill patients.
Study design: Prospective observational study.
Setting: Academic hospital ICU in Spain.
Synopsis: From January 2008 to May 2012, 628 adult patients were treated empirically with broad-spectrum antibiotics. De-escalation was applied to 219 patients (34.9%). Outcomes measured were ICU mortality, hospital mortality, and 90-day mortality in patients who received de-escalation therapy, patients whose antibiotics were not changed, and patients for whom antibiotics were escalated.
The in-hospital mortality rate was 27.4% in patients who were de-escalated, 32.6% in the unchanged group, and 42.9% in the escalation group. ICU and 90-day mortality were lower in the de-escalation group. De-escalation was more commonly used in medical than in surgical patients.
This study is limited because it is not a randomized controlled study and was single-centered, so it might only be applicable on the larger scale. Also, multi-drug resistant organisms were not evaluated.
Overall, it is safe to narrow empiric antibiotics in severe sepsis and shock when the pathogen and sensitivities are known.
Bottom line: De-escalation of antibiotics in severe sepsis and septic shock is associated with a lower mortality.
Citation: Garnacho-Montero J, Gutierrez-Pizarraya A, Escoresca-Ortega A, et al. De-escalation of empirical therapy is associated with lower mortality in patients with severe sepsis and septic shock. Intensive Care Med. 2014;40(1):32-40.
New Oral Anticoagulants Increase GI Bleed Risk
Clinical question: Do thrombin and factor Xa inhibitors increase the risk of gastrointestinal (GI) bleeding when compared to vitamin K antagonists and heparins?
Background: New oral anticoagulants (thrombin and factor Xa inhibitors) are available and being used with increased frequency due to equal efficacy and ease of administration. Some studies indicate a higher risk of GI bleeding with these agents. Further evaluation is needed, because no reversal therapy is available.
Study design: Systematic review and meta-analysis.
Setting: Data from MEDLINE, Embase, and the Cochrane Library.
Synopsis: More than 150,000 patients from 43 randomized controlled trials were evaluated for risk of GI bleed when treated with new anticoagulants versus traditional therapy. Patients were treated for one of the following: embolism prevention from atrial fibrillation, venous thromboembolism (VTE) prophylaxis post orthopedic surgery, VTE prophylaxis of medical patients, acute VTE, and acute coronary syndrome (ACS). Use of aspirin or NSAIDs was discouraged but not documented. The odds ratio for GI bleeding with use of the new anticoagulants was 1.45, with a number needed to harm of 500. Evaluation of subgroups revealed increased GI bleed risk in patients treated for ACS and acute thrombosis versus prophylaxis. Post-surgical patients had the lowest risk.
This study was limited by the heterogeneity and differing primary outcomes (mostly efficacy rather than safety) of the included trials. Studies excluded high-risk patients, which the authors estimate to be 25%-40% of actual patients. More studies need to be done that include high-risk patients and focus on GI bleed as a primary outcome.
Bottom line: The new anticoagulants tend to have a higher incidence of GI bleed than traditional therapy, but this varies based on indication of therapy and needs further evaluation to clarify risk.
Citation: Holster IL, Valkhoff VE, Kuipers EJ, Tjwa ET. New oral anticoagulants increase risk for gastrointestinal bleeding: a systematic review and meta-analysis. Gastroenterology. 2013;145(1):105-112.
Single vs. Dual Antiplatelet Therapy after Stroke
Clinical question: Is dual antiplatelet therapy more beneficial or harmful than monotherapy after ischemic stroke?
Background: It is recommended that patients with ischemic stroke or transient ischemic attack (TIA) receive lifelong antiplatelet therapy; however, there have been insufficient studies evaluating the long-term safety of dual antiplatelet therapy.
Study design: Meta-analysis of randomized controlled trials (RCTs)
Setting: Data from PubMed, Embase, and the Cochrane Central Register of Controlled Trials.
Synopsis: Data from seven RCTs, including 39,574 patients with recent TIA or ischemic stroke, were reviewed. Comparisons were made regarding occurrence of intracranial hemorrhage (ICH) and recurrent stroke between patients receiving dual antiplatelet therapy and those receiving aspirin or clopidogrel monotherapy. All patients were treated for at least one year.
There was no difference in recurrent stroke or ICH between patients on dual antiplatelet therapy versus aspirin monotherapy. Patients treated with dual antiplatelet therapy did have a 46% increased risk of ICH without any additional protective benefit for recurrent stroke or TIA when compared with patients on clopidogrel monotherapy.
This information should not be applied in the acute setting, given the high risk of stroke after TIA or ischemic stroke. One major limitation of this study was that the individual trials used different combinations of dual antiplatelet therapy.
Bottom line: The risk of recurrent stroke or TIA after dual antiplatelet therapy and after monotherapy with aspirin or clopidogrel is equal, but the risk of ICH compared to clopidogrel monotherapy is increased.
Citation: Lee M, Saver JL, Hong KS, Rao NM, Wu YL, Ovbiagele B. Risk-benefit profile of long-term dual- versus single-antiplatelet therapy among patients with ischemic stroke: a systematic review and meta-analysis. Ann Intern Med. 2013;159(7):463-470.
Endoscopic vs. Surgical Cystogastrostomy for Pancreatic Pseudocyst Drainage
Clinical question: How does endoscopic cystogastrostomy for pancreatic pseudocyst drainage compare to the standard surgical approach?
Background: Pancreatic pseudocysts are a common complication of pancreatitis and necessitate decompression when they are accompanied by pain, infection, or obstruction. Decompression of the pseudocyst can be accomplished using either endoscopic or surgical cystogastrostomy.
Study design: Open-label, single-center, randomized trial.
Setting: Single-center U.S. hospital.
Synopsis: A total of 40 patients were randomly equalized to both treatment arms; 20 patients underwent endoscopic and 20 patients underwent surgical cystogastrostomy. Zero patients in the endoscopic therapy had a pseudocyst recurrence, compared with one patient treated surgically. Length of stay (LOS) and cost were lower for the endoscopic group compared to the surgical group (two days vs. six days, P<0.001, $7,011 vs. $15,052, P=0.003).
This study is limited due to several factors. First, patients with pancreatic necrosis were excluded; had these patients been included, the complication rates and LOS would have been higher. Second, cost difference cannot be generalized across the U.S., because Medicare payments are based on provider types and regions.
Bottom line: Endoscopic cystogastrostomy for pancreatic pseudocyst is equal to the standard surgical therapy and results in decreased LOS and reduced costs.
Citation: Varadarajulu S, Bang JY, Sutton BS, Trevino JM, Christein JD, Wilcox CM. Equal efficacy of endoscopic and surgical cystogastrostomy for pancreatic pseudocyst drainage in a randomized trial. Gastroenterology. 2013;145(3):583-590.
Long-Term Cognitive Impairment after Critical Illness
Clinical question: Are a longer duration of delirium and higher doses of sedatives associated with cognitive impairment in the hospital?
Background: Survivors of critical illness are at risk for prolonged cognitive dysfunction. Delirium (and factors associated with delirium, namely sedative and analgesic medications) has been implicated in cognitive dysfunction.
Study design: Prospective cohort study.
Setting: Multi-center, academic, and acute care hospitals.
Synopsis: The study examined 821 adults admitted to the ICU with respiratory failure, cardiogenic shock, or septic shock. Patients excluded were those with pre-existing cognitive impairment, those with psychotic disorders, and those for whom follow-up would not be possible. Two risk factors measured were duration of delirium and use of sedative/analgesics. Delirium was assessed at three and 12 months using the CAM-ICU algorithm in the ICU by trained psychology professionals who were unaware of the patients’ in-hospital course.
At three months, 40% of patients had global cognition scores that were 1.5 standard deviations (SD) below population mean (similar to traumatic brain injury), and 26% had scores two SD below population mean (similar to mild Alzheimer’s). At 12 months, 34% had scores similar to traumatic brain injury patients, and 24% had scores similar to mild Alzheimer’s. A longer duration of delirium was associated with worse global cognition at three and 12 months. Use of sedatives/analgesics was not associated with cognitive impairment.
Bottom line: Critically ill patients in the ICU who experience a longer duration of delirium are at risk of long-term cognitive impairments lasting 12 months.
Citation: Pandharipande PP, Girard TD, Jackson JC, et al. Long-term cognitive impairment after critical illness. N Engl J Med. 2013;369(14):1306-1316.
Holding Chambers (Spacers) vs. Nebulizers for Acute Asthma
Clinical question: Are beta-2 agonists as effective when administered through a holding chamber (spacer) as they are when administered by a nebulizer?
Background: During an acute asthma attack, beta-2 agonists must be delivered to the peripheral airways. There has been considerable controversy regarding the use of a spacer compared with a nebulizer. Aside from admission rates and length of stay, factors taken into account include cost, maintenance of nebulizer machines, and infection control (potential of cross-infection via nebulizers).
Study design: Meta-analysis review of randomized controlled trials (RCTs).
Setting: Multi-centered, worldwide studies from community setting and EDs.
Synopsis: In 39 studies of patients with an acute asthma attack (selected from Cochrane Airways Group Specialized Register), the hospital admission rates did not differ on the basis of delivery method in 729 adults (risk ratio=0.94, confidence interval 0.61-1.43) or in 1,897 children (risk ratio=0.71, confidence interval 0.47-1.08). Secondary outcomes included the duration of time in the ED and the duration of hospital admission. Time spent in the ED varied for adults but was shorter for children with spacers (based on three studies). Duration of hospital admission also did not differ when modes of delivery were compared.
Bottom line: Providing beta-2 agonists using nebulizers during an acute asthma attack is not more effective than administration using a spacer.
Citation: Cates CJ, Welsh EJ, Rowe BH. Holding chambers (spacers) versus nebulisers for beta-agonist treatment of acute asthma. Cochrane Database Syst Rev. 2013;9:CD000052.
In This Edition
Literature At A Glance
A guide to this month’s studies
- ICU pressures improve transfers to the floor
- Morbidity, mortality rates high for respiratory syncytial virus infections
- Antibiotic algorithm can guide therapy in healthcare-associated pneumonia
- Three-month dual antiplatelet therapy for zotarolimus-eluting stents
- De-escalating antibiotics in sepsis
- New oral anticoagulants increase GI bleed risk
- Single vs. dual antiplatelet therapy after stroke
- Endoscopic vs. surgical cystogastrostomy for pancreatic pseudocyst drainage
- Long-term cognitive impairment after critical illness
- Holding chambers vs. nebulizers for acute asthma
ICU Pressures Improve Transfers to the Floor
Clinical question: Does ICU strain negatively affect the outcomes of patients transferred to the floor?
Background: With healthcare costs increasing and critical care staff shortages projected, ICUs will have to operate under increasing strain. This may influence decisions on discharging patients from ICUs and could affect patient outcomes.
Study design: Retrospective cohort study.
Setting: One hundred fifty-five ICUs in the United States.
Synopsis: Using the Project IMPACT database, 200,730 adult patients from 107 different hospitals were evaluated in times of ICU strain, determined by the current census, new admissions, and acuity level. Outcomes measured were initial ICU length of stay (LOS), readmission within 72 hours, in-hospital mortality rates, and post-ICU discharge LOS.
Increases of the strain variables from the fifth to the 95th percentiles resulted in a 6.3-hour reduction in ICU LOS, a 2.0-hour decrease in post-ICU discharge LOS, and a 1.0% increase in probability of ICU readmission within 72 hours. Mortality rates during the hospital stay and odds of being discharged home showed no significant change. This study was limited because the ICUs participating were not randomly chosen, outcomes of patients transferred to other hospitals were not measured, and no post-hospital data was collected, so no long-term outcomes could be measured.
Bottom line: ICU bed pressures prompt physicians to allocate ICU resources more efficiently without changing short-term patient outcomes.
Citation: Wagner J, Gabler NB, Ratcliffe SJ, Brown SE, Strom BL, Halpern SD. Outcomes among patients discharged from busy intensive care units. Ann Intern Med. 2013;159(7):447-455.
Adults Hospitalized for Respiratory Syncytial Virus Infections Have High Morbidity, Mortality Rates
Clinical question: What are the complications and outcomes of respiratory syncytial virus (RSV) infection in adults requiring hospitalization?
Background: RSV is a common cause of lower respiratory tract infection in infants and young children, leading to hospitalization and even death. RSV has been estimated to affect 3%-10% of adults annually, generally causing mild disease. However, the outcomes of adults with more severe disease are not fully known.
Study design: Retrospective cohort study.
Setting: Three acute care, public hospitals in Hong Kong.
Synopsis: All adult patients hospitalized with laboratory-confirmed RSV infection were included during the defined time period. The main outcome measure was all-cause death, with secondary outcome measures of development of acute respiratory failure requiring ventilator support and total duration of hospitalization among survivors. Additionally, the cohort of RSV patients was compared to patients admitted with seasonal influenza during this same time frame. Patients with pandemic 2009 H1N1 infection were not included.
Of patients with RSV, pneumonia was found in 42.3%, bacterial superinfection in 12.5%, and cardiovascular complications in 14.3%. Additionally, 11.1% developed respiratory failure requiring ventilator support. All-cause mortality at 30 days and 60 days was 9.1% and 11.9%, respectively, with pneumonia the most common cause of death. Use of systemic corticosteroids did not improve survival. When the RSV cohort was compared to the influenza cohort, the patients were similar in age, but the RSV patients were more likely to have underlying chronic lung disease and major systemic co-morbidities. The rate of survival and duration of hospitalization were not significantly different.
Bottom line: RSV infection is an underappreciated cause of lower tract respiratory infection in adults; severe infections that require hospitalization have rates of mortality similar to seasonal influenza. Further research on treatment or immunization is needed.
Citation: Lee N, Lui GC, Wong KT, et al. High morbidity and mortality in adults hospitalized for respiratory syncytial virus infections. Clin Infect Dis. 2013;57(8):1069-1077.
Antibiotic Algorithm Can Guide Therapy in Healthcare-Associated Pneumonia
Clinical question: Can an algorithm based on risk for multidrug-resistant (MDR) organisms and illness severity guide antibiotic selection in healthcare-associated pneumonia (HCAP)?
Background: The 2005 American Thoracic Society/Infectious Diseases Society of America (ATS/IDSA) guidelines identify patients with HCAP as those with recent contact with a healthcare environment, including nursing homes and hemodialysis; however, previous studies have shown that not all patients with healthcare contact have equal risk for MDR organisms.
Study design: Prospective cohort study.
Setting: Japan, multi-center.
Synopsis: Of the 445 enrolled patients, 124 were diagnosed with community-acquired pneumonia (CAP) and 321 with HCAP. Patients with HCAP were classified based on severity of illness or MDR pathogen risk factors (immune suppression, hospitalization within the last 90 days, poor functional status, and antibiotics within the past six months). Patients with low risk (0-1 factors) for MDR organisms were treated for CAP, and patients with high risk (≥2 factors) or moderate risk (≥1 factor) for severe illness were treated for HCAP.
HCAP patients had a higher 30-day mortality rate (13.7% vs. 5.6%, P=0.017), but mortality rate was less in the patients at low risk for MDR pathogens (8.6% vs. 18.2%, P=0.012). Of the HCAP patients, only 7.1% received inappropriate therapy (pathogen resistant to initial antibiotic regimen), and treatment failure was 19.3%.
Appropriateness of initial empiric therapy was determined not to be a mortality risk; however, this trial might be limited by its location, because Japan appears to have fewer MDR pathogens than the U.S.
Bottom line: A treatment algorithm based on risk for MDR organisms and severity of illness can be used to guide empiric antibiotic therapy in patients with HCAP, and, ideally, to reduce excessive use of broad-spectrum antibiotics.
Citation: Maruyama T, Fujisawa T, Okuno M, et al. A new strategy for healthcare-associated pneumonia: a 2-year prospective multicenter cohort study using risk factors for multidrug-resistant pathogens to select initial empiric therapy. Clin Infect Dis. 2013;57(10):1373-1383.
Three-Month Dual Antiplatelet Therapy for Zotarolimus-Eluting Stents
Clinical question: Is short-term, dual antiplatelet therapy noninferior to long-term therapy in zotarolimus-eluting stents?
Background: Current guidelines recommend long-term (>12 months) dual antiplatelet therapy after the placement of drug-eluting stents. The optimal therapy duration in second-generation drug-eluting stents has not been studied; moreover, some studies with multiple drug-eluting stents have suggested no added benefit from long-term therapy.
Study design: Randomized controlled trial.
Setting: Brazil, multi-center.
Synopsis: Researchers randomized 3,211 patients with stable coronary artery disease (CAD) or low-risk acute coronary syndrome (ACS) undergoing intervention with zotarolimus-eluting stents to short-term (three months) or long-term (12 months) dual antiplatelet therapy. Exclusion criteria included ST-elevation myocardial infarction (STEMI), previous drug-eluting stent, scheduled elective surgery within 12 months, or contraindication to aspirin or clopidogrel. Primary endpoints were a composite of death from any cause, MI, stroke, or major bleeding. Secondary endpoints were stent thrombosis, target lesion revascularization, adverse cardiac event, and any bleed.
At one-year follow-up, the short-term group had similar primary (6.0% vs. 5.8%) and secondary (8.3% vs. 7.4%) outcomes compared to the long-term. The short-term group’s noninferiority also was seen in several key subgroups.
This study included patients with stable CAD or low-risk ACS and cannot be generalized to higher-risk patients. Results for zotarolimus-eluting stents cannot be generalized to other second-generation drug-eluting stents.
Bottom line: Zotarolimus-eluting stents, followed by three months of dual antiplatelet therapy, were noninferior to 12 months of therapy in patients with stable CAD or low-risk ACS.
Citation: Feres F, Costa RA, Abizaid A, et al. Three vs. twelve months of dual antiplatelet therapy after zotarolimus-eluting stents: the OPTIMIZE randomized trial. JAMA. 2013;310(23):2510-2522.
De-Escalating Antibiotics in Sepsis
Clinical question: Does tailoring antibiotics based on known pathogens impact mortality for patients with severe sepsis or shock?
Background: In patients with sepsis, the use of early empiric antibiotics reduces morbidity and mortality. De-escalation therapy refers to narrowing the broad-spectrum antibiotics once the pathogen and sensitivities are known; however, no randomized controlled studies have assessed the impact of this therapy on critically ill patients.
Study design: Prospective observational study.
Setting: Academic hospital ICU in Spain.
Synopsis: From January 2008 to May 2012, 628 adult patients were treated empirically with broad-spectrum antibiotics. De-escalation was applied to 219 patients (34.9%). Outcomes measured were ICU mortality, hospital mortality, and 90-day mortality in patients who received de-escalation therapy, patients whose antibiotics were not changed, and patients for whom antibiotics were escalated.
The in-hospital mortality rate was 27.4% in patients who were de-escalated, 32.6% in the unchanged group, and 42.9% in the escalation group. ICU and 90-day mortality were lower in the de-escalation group. De-escalation was more commonly used in medical than in surgical patients.
This study is limited because it is not a randomized controlled study and was single-centered, so it might only be applicable on the larger scale. Also, multi-drug resistant organisms were not evaluated.
Overall, it is safe to narrow empiric antibiotics in severe sepsis and shock when the pathogen and sensitivities are known.
Bottom line: De-escalation of antibiotics in severe sepsis and septic shock is associated with a lower mortality.
Citation: Garnacho-Montero J, Gutierrez-Pizarraya A, Escoresca-Ortega A, et al. De-escalation of empirical therapy is associated with lower mortality in patients with severe sepsis and septic shock. Intensive Care Med. 2014;40(1):32-40.
New Oral Anticoagulants Increase GI Bleed Risk
Clinical question: Do thrombin and factor Xa inhibitors increase the risk of gastrointestinal (GI) bleeding when compared to vitamin K antagonists and heparins?
Background: New oral anticoagulants (thrombin and factor Xa inhibitors) are available and being used with increased frequency due to equal efficacy and ease of administration. Some studies indicate a higher risk of GI bleeding with these agents. Further evaluation is needed, because no reversal therapy is available.
Study design: Systematic review and meta-analysis.
Setting: Data from MEDLINE, Embase, and the Cochrane Library.
Synopsis: More than 150,000 patients from 43 randomized controlled trials were evaluated for risk of GI bleed when treated with new anticoagulants versus traditional therapy. Patients were treated for one of the following: embolism prevention from atrial fibrillation, venous thromboembolism (VTE) prophylaxis post orthopedic surgery, VTE prophylaxis of medical patients, acute VTE, and acute coronary syndrome (ACS). Use of aspirin or NSAIDs was discouraged but not documented. The odds ratio for GI bleeding with use of the new anticoagulants was 1.45, with a number needed to harm of 500. Evaluation of subgroups revealed increased GI bleed risk in patients treated for ACS and acute thrombosis versus prophylaxis. Post-surgical patients had the lowest risk.
This study was limited by the heterogeneity and differing primary outcomes (mostly efficacy rather than safety) of the included trials. Studies excluded high-risk patients, which the authors estimate to be 25%-40% of actual patients. More studies need to be done that include high-risk patients and focus on GI bleed as a primary outcome.
Bottom line: The new anticoagulants tend to have a higher incidence of GI bleed than traditional therapy, but this varies based on indication of therapy and needs further evaluation to clarify risk.
Citation: Holster IL, Valkhoff VE, Kuipers EJ, Tjwa ET. New oral anticoagulants increase risk for gastrointestinal bleeding: a systematic review and meta-analysis. Gastroenterology. 2013;145(1):105-112.
Single vs. Dual Antiplatelet Therapy after Stroke
Clinical question: Is dual antiplatelet therapy more beneficial or harmful than monotherapy after ischemic stroke?
Background: It is recommended that patients with ischemic stroke or transient ischemic attack (TIA) receive lifelong antiplatelet therapy; however, there have been insufficient studies evaluating the long-term safety of dual antiplatelet therapy.
Study design: Meta-analysis of randomized controlled trials (RCTs)
Setting: Data from PubMed, Embase, and the Cochrane Central Register of Controlled Trials.
Synopsis: Data from seven RCTs, including 39,574 patients with recent TIA or ischemic stroke, were reviewed. Comparisons were made regarding occurrence of intracranial hemorrhage (ICH) and recurrent stroke between patients receiving dual antiplatelet therapy and those receiving aspirin or clopidogrel monotherapy. All patients were treated for at least one year.
There was no difference in recurrent stroke or ICH between patients on dual antiplatelet therapy versus aspirin monotherapy. Patients treated with dual antiplatelet therapy did have a 46% increased risk of ICH without any additional protective benefit for recurrent stroke or TIA when compared with patients on clopidogrel monotherapy.
This information should not be applied in the acute setting, given the high risk of stroke after TIA or ischemic stroke. One major limitation of this study was that the individual trials used different combinations of dual antiplatelet therapy.
Bottom line: The risk of recurrent stroke or TIA after dual antiplatelet therapy and after monotherapy with aspirin or clopidogrel is equal, but the risk of ICH compared to clopidogrel monotherapy is increased.
Citation: Lee M, Saver JL, Hong KS, Rao NM, Wu YL, Ovbiagele B. Risk-benefit profile of long-term dual- versus single-antiplatelet therapy among patients with ischemic stroke: a systematic review and meta-analysis. Ann Intern Med. 2013;159(7):463-470.
Endoscopic vs. Surgical Cystogastrostomy for Pancreatic Pseudocyst Drainage
Clinical question: How does endoscopic cystogastrostomy for pancreatic pseudocyst drainage compare to the standard surgical approach?
Background: Pancreatic pseudocysts are a common complication of pancreatitis and necessitate decompression when they are accompanied by pain, infection, or obstruction. Decompression of the pseudocyst can be accomplished using either endoscopic or surgical cystogastrostomy.
Study design: Open-label, single-center, randomized trial.
Setting: Single-center U.S. hospital.
Synopsis: A total of 40 patients were randomly equalized to both treatment arms; 20 patients underwent endoscopic and 20 patients underwent surgical cystogastrostomy. Zero patients in the endoscopic therapy had a pseudocyst recurrence, compared with one patient treated surgically. Length of stay (LOS) and cost were lower for the endoscopic group compared to the surgical group (two days vs. six days, P<0.001, $7,011 vs. $15,052, P=0.003).
This study is limited due to several factors. First, patients with pancreatic necrosis were excluded; had these patients been included, the complication rates and LOS would have been higher. Second, cost difference cannot be generalized across the U.S., because Medicare payments are based on provider types and regions.
Bottom line: Endoscopic cystogastrostomy for pancreatic pseudocyst is equal to the standard surgical therapy and results in decreased LOS and reduced costs.
Citation: Varadarajulu S, Bang JY, Sutton BS, Trevino JM, Christein JD, Wilcox CM. Equal efficacy of endoscopic and surgical cystogastrostomy for pancreatic pseudocyst drainage in a randomized trial. Gastroenterology. 2013;145(3):583-590.
Long-Term Cognitive Impairment after Critical Illness
Clinical question: Are a longer duration of delirium and higher doses of sedatives associated with cognitive impairment in the hospital?
Background: Survivors of critical illness are at risk for prolonged cognitive dysfunction. Delirium (and factors associated with delirium, namely sedative and analgesic medications) has been implicated in cognitive dysfunction.
Study design: Prospective cohort study.
Setting: Multi-center, academic, and acute care hospitals.
Synopsis: The study examined 821 adults admitted to the ICU with respiratory failure, cardiogenic shock, or septic shock. Patients excluded were those with pre-existing cognitive impairment, those with psychotic disorders, and those for whom follow-up would not be possible. Two risk factors measured were duration of delirium and use of sedative/analgesics. Delirium was assessed at three and 12 months using the CAM-ICU algorithm in the ICU by trained psychology professionals who were unaware of the patients’ in-hospital course.
At three months, 40% of patients had global cognition scores that were 1.5 standard deviations (SD) below population mean (similar to traumatic brain injury), and 26% had scores two SD below population mean (similar to mild Alzheimer’s). At 12 months, 34% had scores similar to traumatic brain injury patients, and 24% had scores similar to mild Alzheimer’s. A longer duration of delirium was associated with worse global cognition at three and 12 months. Use of sedatives/analgesics was not associated with cognitive impairment.
Bottom line: Critically ill patients in the ICU who experience a longer duration of delirium are at risk of long-term cognitive impairments lasting 12 months.
Citation: Pandharipande PP, Girard TD, Jackson JC, et al. Long-term cognitive impairment after critical illness. N Engl J Med. 2013;369(14):1306-1316.
Holding Chambers (Spacers) vs. Nebulizers for Acute Asthma
Clinical question: Are beta-2 agonists as effective when administered through a holding chamber (spacer) as they are when administered by a nebulizer?
Background: During an acute asthma attack, beta-2 agonists must be delivered to the peripheral airways. There has been considerable controversy regarding the use of a spacer compared with a nebulizer. Aside from admission rates and length of stay, factors taken into account include cost, maintenance of nebulizer machines, and infection control (potential of cross-infection via nebulizers).
Study design: Meta-analysis review of randomized controlled trials (RCTs).
Setting: Multi-centered, worldwide studies from community setting and EDs.
Synopsis: In 39 studies of patients with an acute asthma attack (selected from Cochrane Airways Group Specialized Register), the hospital admission rates did not differ on the basis of delivery method in 729 adults (risk ratio=0.94, confidence interval 0.61-1.43) or in 1,897 children (risk ratio=0.71, confidence interval 0.47-1.08). Secondary outcomes included the duration of time in the ED and the duration of hospital admission. Time spent in the ED varied for adults but was shorter for children with spacers (based on three studies). Duration of hospital admission also did not differ when modes of delivery were compared.
Bottom line: Providing beta-2 agonists using nebulizers during an acute asthma attack is not more effective than administration using a spacer.
Citation: Cates CJ, Welsh EJ, Rowe BH. Holding chambers (spacers) versus nebulisers for beta-agonist treatment of acute asthma. Cochrane Database Syst Rev. 2013;9:CD000052.