User login
FDA says heparin contamination was likely intentional
The contaminant found in heparin was most likely introduced intentionally, according to the US Food and Drug Administration.
In a Senate hearing on April 15, FDA Commissioner Andrew von Eschenbach said the FDA suspects the contaminant, oversulfated chondroitin sulfate, was introduced to increase profits.
However, it is unclear where in the chain of production the contaminant was added and, therefore, who would be responsible.
Baxter International Inc. and Scientific Protein Laboratories Inc. (SPL), suppliers of heparin, say the contaminant was not added in their factories.
Baxter said it has been seeking access to consolidators and workshops in China that handled the crude material before it went to SPL. According to spokeswoman Erin Gardiner, Baxter has not determined how or why the contaminant was introduced.
SPL has issued a press release claiming its lack of involvement in the contamination.
The company said, “Based upon testing and reports from around the world, it is clear that the contamination occurred on a widespread basis earlier in the Chinese heparin raw material supply chain, before those materials reached Changzhou SPL and SPL.”
Baxter began recalling heparin January 17 of this year, after receiving reports of allergic reactions and deaths resulting from use of the drug. Recalls of heparin have continued since that time.
In mid-March, the FDA released the news that a contaminant was found in crude lots of heparin at a Chinese processing plant. The substance was identified as over-sulfated chondroitin sulfate, which mimics heparin and is cheaper to produce than real heparin.
Earlier this month, the FDA said a total of 62 people have died since January as a result of heparin use. ![]()
The contaminant found in heparin was most likely introduced intentionally, according to the US Food and Drug Administration.
In a Senate hearing on April 15, FDA Commissioner Andrew von Eschenbach said the FDA suspects the contaminant, oversulfated chondroitin sulfate, was introduced to increase profits.
However, it is unclear where in the chain of production the contaminant was added and, therefore, who would be responsible.
Baxter International Inc. and Scientific Protein Laboratories Inc. (SPL), suppliers of heparin, say the contaminant was not added in their factories.
Baxter said it has been seeking access to consolidators and workshops in China that handled the crude material before it went to SPL. According to spokeswoman Erin Gardiner, Baxter has not determined how or why the contaminant was introduced.
SPL has issued a press release claiming its lack of involvement in the contamination.
The company said, “Based upon testing and reports from around the world, it is clear that the contamination occurred on a widespread basis earlier in the Chinese heparin raw material supply chain, before those materials reached Changzhou SPL and SPL.”
Baxter began recalling heparin January 17 of this year, after receiving reports of allergic reactions and deaths resulting from use of the drug. Recalls of heparin have continued since that time.
In mid-March, the FDA released the news that a contaminant was found in crude lots of heparin at a Chinese processing plant. The substance was identified as over-sulfated chondroitin sulfate, which mimics heparin and is cheaper to produce than real heparin.
Earlier this month, the FDA said a total of 62 people have died since January as a result of heparin use. ![]()
The contaminant found in heparin was most likely introduced intentionally, according to the US Food and Drug Administration.
In a Senate hearing on April 15, FDA Commissioner Andrew von Eschenbach said the FDA suspects the contaminant, oversulfated chondroitin sulfate, was introduced to increase profits.
However, it is unclear where in the chain of production the contaminant was added and, therefore, who would be responsible.
Baxter International Inc. and Scientific Protein Laboratories Inc. (SPL), suppliers of heparin, say the contaminant was not added in their factories.
Baxter said it has been seeking access to consolidators and workshops in China that handled the crude material before it went to SPL. According to spokeswoman Erin Gardiner, Baxter has not determined how or why the contaminant was introduced.
SPL has issued a press release claiming its lack of involvement in the contamination.
The company said, “Based upon testing and reports from around the world, it is clear that the contamination occurred on a widespread basis earlier in the Chinese heparin raw material supply chain, before those materials reached Changzhou SPL and SPL.”
Baxter began recalling heparin January 17 of this year, after receiving reports of allergic reactions and deaths resulting from use of the drug. Recalls of heparin have continued since that time.
In mid-March, the FDA released the news that a contaminant was found in crude lots of heparin at a Chinese processing plant. The substance was identified as over-sulfated chondroitin sulfate, which mimics heparin and is cheaper to produce than real heparin.
Earlier this month, the FDA said a total of 62 people have died since January as a result of heparin use. ![]()
When Crisis Comes
One hospitalist spent three weeks without a break treating victims of Hurricane Katrina in 2005. Another couldn’t get to work when the I-35W bridge collapsed in Minneapolis on Aug. 1, 2007, but there were enough physicians on hand for that tragedy and fewer victims to treat than feared.
Yet another shudders when he recalls treating victims of an 89-car pile-up caused by a dust storm in southern Idaho.
Not all hospitalists have been in the trenches treating victims of disasters. But two emerging trends likely will put hospitalists on the front lines of preparing for disasters and treating victims.
The first is the increasing recognition that there are many threats to the safety of the public, including terrorism, natural disasters, disease outbreaks, and criminal acts like the mass killings a year ago at Virginia Tech in Blacksburg.
The second is the rapidly expanding role hospitalists have in caring for critically ill and injured patients.
“Hospitalists will be a key,” says Timothy Close, senior safety officer for the University of Colorado Hospital in Denver and chairman of its emergency management committee. “Because of their understanding of all hospital services and treatments, they can handle a multitude of clinical roles. Facilities should deploy hospitalists’ understanding of the organization to facilitate patient care.”
Close, who has 15 years of experience in planning and preparedness, urges organizations to implement plans “that are realistic and doable based on local resources and conditions.” He also urges facilities to conduct emergency drills and have hospitalists participate.
He has dealt with crises wrought by fires, workplace violence, severe weather, and abductions, but adds it is important to remember that “you never know what’s going to happen.”
Close helped treat the victims of the dust storm pile-up. “It was caused by an unfortunate series of events,” he says. “A new land owner plowed during a dry time, and when the winds came it was catastrophic. The cars ran right into the dust cloud with zero visibility.”
Prepare for the Unseen
Lisa Kirkland, MD, a hospitalist at the Mayo Clinic in Rochester, Minn., agrees disaster planning should be local in the sense of preparing for specific events. Tornadoes are the most likely weather-related crisis to occur in Rochester, she says, and the area is not a prime terrorism target.
Yet disasters don’t have to happen suddenly or involve mass casualties. “A disaster is anything that overwhelms the usual system,” she says. “Putting a community under quarantine during an outbreak of influenza or bird flu, for example, could require the initiation of disaster plans since staff couldn’t get to hospitals.”
In this sort of scenario, like during the SARS outbreak in Toronto in 2003, patient care would be largely medical, rather than surgical, so hospitalists would be key providers of treatment, Dr. Kirkland says.
Hospitalists would also be key in maintaining effective communication, internally and with the outside world because of their thorough knowledge of hospital services, she adds.
Some 75 miles away in Minneapolis, many victims of the I-35W bridge collapse were taken to Hennepin County Medical Center (HCMC). Glen Varns, MD, hospitalist program leader at HCMC, was unable to get to work because he lives on the other side of the bridge. But he says hospitalists played a critical role in dealing with the crisis.
“Since our hospitalists are most familiar with the inner workings of the facility, they played a huge role in determining who needed to be hospitalized and where in the hospital they would best be treated,” he says. “This included reviewing the existing patient census when the collapse happened so we could discharge and transfer inpatients appropriately to ensure that the hospital was in the best position to deal with the collapse victims.”
Because the bridge collapsed during the early evening, there was plenty of staff on-hand to treat the victims, including residents who worked hand-in-hand with hospitalists in making admission and transfer decisions.
Challenge for Hospitalists
In smaller facilities where there are no residents, or in small emergency departments (ED) and intensive-care units, hospitalists will and should have even more critical roles in handling disasters and planning for them, Dr. Varns says.
He believes all hospitalists—but especially those in small, nonteaching facilities—should get triage training. “Hospitalists have a very broad skill set—especially with increasing responsibility for co-management of surgical cases—but they should develop triage skills,” says Dr. Varns, who suggests hospitalists take a two or three-day advanced trauma life support course.
Steven B. Deitelzweig, MD, FACP, system chairman, department of hospital medicine and vice president of medical affairs for the Ochsner Health System in the New Orleans area, agrees.
“I think the folks who are closest to guiding the care should be offering input into triage decisions,” he says. “Hospitalists can be invaluable in doing triage of inpatients. They provide objective detailed information.”
Dr. Deitelzweig, who experienced the three-week lock-down following Katrina, suggests hospitalist groups create a system of prioritizing evacuation of patients—including what kind of support they’ll need.
He believes hospitalists will be invaluable during crises because they are “front-line decision-makers, along with ED physicians and intensivists.” Hospitalists should be on disaster-preparedness committees and a key part of communication during an actual crisis, he urges.
“Communication is critical during a crisis—and hospitalists know their systems,” he continues, noting that Ochsner has out-of-state cell phones, satellite phones, ham radios, spectral light phones, radio frequency antennas in secure places, and more.
In addition to equipment and supplies, hospitalists need to be prepared to do whatever is needed in a crisis, Dr. Deitelzweig says. “In a disaster, you might have to do a procedure usually done by a specialist—with supervision—to extend that person,” he says. “You also may have to go past the physician role. That’s where leadership shows. Our CEO served food in the cafeteria during Katrina. During a disaster, you have to be a flat organization and just do what needs to be done. That gives emotional support to everyone.”
Still, the need to prepare before a disaster cannot be overemphasized, he says.
Ochsner now has two teams of pre-selected physicians, including hospitalists, dedicated to working through specific types of crises. Having the list of essential personnel online at all times is intended to prevent last-minute scurrying around to find the right people, he says.
In addition, providing balanced scheduling—especially in long-lasting crisis situations like Katrina—is important, says Dr. Deitelzweig. “Timing for release must be included, and having more staff on hand than necessary can help alleviate stress,” he advises.
Lessons of Katrina
Neal Axon, MD, an assistant professor at the Medical University of South Carolina, says he and his colleagues learned from those who went through Katrina as they prepared for the most likely disaster in Charleston: a severe hurricane.
Dr. Axon, a senior hospitalist in his group, says the facility has a system that generates e-mail, pages, text messages, and cell phone calls to keep hospital staff informed about potential crises. He also says the preparedness plan provides for relief of staff working for extended periods.
In addition, the hospital has trailers and inflatable tents to extend its facilities if there is a surge in patients. It also has a facility to provide decontamination for exposure to chemicals and radiation.
Brian Bossard, MD, director of Inpatient Physician Associates and medical staff quality designee at BryanLGH Medical Center in Lincoln Neb., says preparedness plans should be tested and updated regularly—especially the systems used to call in staff.
Dr. Bossard strongly believes hospitalists should be involved in disaster planning: “Every day hospitalists work hospital systems. We have a broad scope and perspective. That’s what you need in a disaster.” TH
Karla Feuer is a journalist based in New York.
One hospitalist spent three weeks without a break treating victims of Hurricane Katrina in 2005. Another couldn’t get to work when the I-35W bridge collapsed in Minneapolis on Aug. 1, 2007, but there were enough physicians on hand for that tragedy and fewer victims to treat than feared.
Yet another shudders when he recalls treating victims of an 89-car pile-up caused by a dust storm in southern Idaho.
Not all hospitalists have been in the trenches treating victims of disasters. But two emerging trends likely will put hospitalists on the front lines of preparing for disasters and treating victims.
The first is the increasing recognition that there are many threats to the safety of the public, including terrorism, natural disasters, disease outbreaks, and criminal acts like the mass killings a year ago at Virginia Tech in Blacksburg.
The second is the rapidly expanding role hospitalists have in caring for critically ill and injured patients.
“Hospitalists will be a key,” says Timothy Close, senior safety officer for the University of Colorado Hospital in Denver and chairman of its emergency management committee. “Because of their understanding of all hospital services and treatments, they can handle a multitude of clinical roles. Facilities should deploy hospitalists’ understanding of the organization to facilitate patient care.”
Close, who has 15 years of experience in planning and preparedness, urges organizations to implement plans “that are realistic and doable based on local resources and conditions.” He also urges facilities to conduct emergency drills and have hospitalists participate.
He has dealt with crises wrought by fires, workplace violence, severe weather, and abductions, but adds it is important to remember that “you never know what’s going to happen.”
Close helped treat the victims of the dust storm pile-up. “It was caused by an unfortunate series of events,” he says. “A new land owner plowed during a dry time, and when the winds came it was catastrophic. The cars ran right into the dust cloud with zero visibility.”
Prepare for the Unseen
Lisa Kirkland, MD, a hospitalist at the Mayo Clinic in Rochester, Minn., agrees disaster planning should be local in the sense of preparing for specific events. Tornadoes are the most likely weather-related crisis to occur in Rochester, she says, and the area is not a prime terrorism target.
Yet disasters don’t have to happen suddenly or involve mass casualties. “A disaster is anything that overwhelms the usual system,” she says. “Putting a community under quarantine during an outbreak of influenza or bird flu, for example, could require the initiation of disaster plans since staff couldn’t get to hospitals.”
In this sort of scenario, like during the SARS outbreak in Toronto in 2003, patient care would be largely medical, rather than surgical, so hospitalists would be key providers of treatment, Dr. Kirkland says.
Hospitalists would also be key in maintaining effective communication, internally and with the outside world because of their thorough knowledge of hospital services, she adds.
Some 75 miles away in Minneapolis, many victims of the I-35W bridge collapse were taken to Hennepin County Medical Center (HCMC). Glen Varns, MD, hospitalist program leader at HCMC, was unable to get to work because he lives on the other side of the bridge. But he says hospitalists played a critical role in dealing with the crisis.
“Since our hospitalists are most familiar with the inner workings of the facility, they played a huge role in determining who needed to be hospitalized and where in the hospital they would best be treated,” he says. “This included reviewing the existing patient census when the collapse happened so we could discharge and transfer inpatients appropriately to ensure that the hospital was in the best position to deal with the collapse victims.”
Because the bridge collapsed during the early evening, there was plenty of staff on-hand to treat the victims, including residents who worked hand-in-hand with hospitalists in making admission and transfer decisions.
Challenge for Hospitalists
In smaller facilities where there are no residents, or in small emergency departments (ED) and intensive-care units, hospitalists will and should have even more critical roles in handling disasters and planning for them, Dr. Varns says.
He believes all hospitalists—but especially those in small, nonteaching facilities—should get triage training. “Hospitalists have a very broad skill set—especially with increasing responsibility for co-management of surgical cases—but they should develop triage skills,” says Dr. Varns, who suggests hospitalists take a two or three-day advanced trauma life support course.
Steven B. Deitelzweig, MD, FACP, system chairman, department of hospital medicine and vice president of medical affairs for the Ochsner Health System in the New Orleans area, agrees.
“I think the folks who are closest to guiding the care should be offering input into triage decisions,” he says. “Hospitalists can be invaluable in doing triage of inpatients. They provide objective detailed information.”
Dr. Deitelzweig, who experienced the three-week lock-down following Katrina, suggests hospitalist groups create a system of prioritizing evacuation of patients—including what kind of support they’ll need.
He believes hospitalists will be invaluable during crises because they are “front-line decision-makers, along with ED physicians and intensivists.” Hospitalists should be on disaster-preparedness committees and a key part of communication during an actual crisis, he urges.
“Communication is critical during a crisis—and hospitalists know their systems,” he continues, noting that Ochsner has out-of-state cell phones, satellite phones, ham radios, spectral light phones, radio frequency antennas in secure places, and more.
In addition to equipment and supplies, hospitalists need to be prepared to do whatever is needed in a crisis, Dr. Deitelzweig says. “In a disaster, you might have to do a procedure usually done by a specialist—with supervision—to extend that person,” he says. “You also may have to go past the physician role. That’s where leadership shows. Our CEO served food in the cafeteria during Katrina. During a disaster, you have to be a flat organization and just do what needs to be done. That gives emotional support to everyone.”
Still, the need to prepare before a disaster cannot be overemphasized, he says.
Ochsner now has two teams of pre-selected physicians, including hospitalists, dedicated to working through specific types of crises. Having the list of essential personnel online at all times is intended to prevent last-minute scurrying around to find the right people, he says.
In addition, providing balanced scheduling—especially in long-lasting crisis situations like Katrina—is important, says Dr. Deitelzweig. “Timing for release must be included, and having more staff on hand than necessary can help alleviate stress,” he advises.
Lessons of Katrina
Neal Axon, MD, an assistant professor at the Medical University of South Carolina, says he and his colleagues learned from those who went through Katrina as they prepared for the most likely disaster in Charleston: a severe hurricane.
Dr. Axon, a senior hospitalist in his group, says the facility has a system that generates e-mail, pages, text messages, and cell phone calls to keep hospital staff informed about potential crises. He also says the preparedness plan provides for relief of staff working for extended periods.
In addition, the hospital has trailers and inflatable tents to extend its facilities if there is a surge in patients. It also has a facility to provide decontamination for exposure to chemicals and radiation.
Brian Bossard, MD, director of Inpatient Physician Associates and medical staff quality designee at BryanLGH Medical Center in Lincoln Neb., says preparedness plans should be tested and updated regularly—especially the systems used to call in staff.
Dr. Bossard strongly believes hospitalists should be involved in disaster planning: “Every day hospitalists work hospital systems. We have a broad scope and perspective. That’s what you need in a disaster.” TH
Karla Feuer is a journalist based in New York.
One hospitalist spent three weeks without a break treating victims of Hurricane Katrina in 2005. Another couldn’t get to work when the I-35W bridge collapsed in Minneapolis on Aug. 1, 2007, but there were enough physicians on hand for that tragedy and fewer victims to treat than feared.
Yet another shudders when he recalls treating victims of an 89-car pile-up caused by a dust storm in southern Idaho.
Not all hospitalists have been in the trenches treating victims of disasters. But two emerging trends likely will put hospitalists on the front lines of preparing for disasters and treating victims.
The first is the increasing recognition that there are many threats to the safety of the public, including terrorism, natural disasters, disease outbreaks, and criminal acts like the mass killings a year ago at Virginia Tech in Blacksburg.
The second is the rapidly expanding role hospitalists have in caring for critically ill and injured patients.
“Hospitalists will be a key,” says Timothy Close, senior safety officer for the University of Colorado Hospital in Denver and chairman of its emergency management committee. “Because of their understanding of all hospital services and treatments, they can handle a multitude of clinical roles. Facilities should deploy hospitalists’ understanding of the organization to facilitate patient care.”
Close, who has 15 years of experience in planning and preparedness, urges organizations to implement plans “that are realistic and doable based on local resources and conditions.” He also urges facilities to conduct emergency drills and have hospitalists participate.
He has dealt with crises wrought by fires, workplace violence, severe weather, and abductions, but adds it is important to remember that “you never know what’s going to happen.”
Close helped treat the victims of the dust storm pile-up. “It was caused by an unfortunate series of events,” he says. “A new land owner plowed during a dry time, and when the winds came it was catastrophic. The cars ran right into the dust cloud with zero visibility.”
Prepare for the Unseen
Lisa Kirkland, MD, a hospitalist at the Mayo Clinic in Rochester, Minn., agrees disaster planning should be local in the sense of preparing for specific events. Tornadoes are the most likely weather-related crisis to occur in Rochester, she says, and the area is not a prime terrorism target.
Yet disasters don’t have to happen suddenly or involve mass casualties. “A disaster is anything that overwhelms the usual system,” she says. “Putting a community under quarantine during an outbreak of influenza or bird flu, for example, could require the initiation of disaster plans since staff couldn’t get to hospitals.”
In this sort of scenario, like during the SARS outbreak in Toronto in 2003, patient care would be largely medical, rather than surgical, so hospitalists would be key providers of treatment, Dr. Kirkland says.
Hospitalists would also be key in maintaining effective communication, internally and with the outside world because of their thorough knowledge of hospital services, she adds.
Some 75 miles away in Minneapolis, many victims of the I-35W bridge collapse were taken to Hennepin County Medical Center (HCMC). Glen Varns, MD, hospitalist program leader at HCMC, was unable to get to work because he lives on the other side of the bridge. But he says hospitalists played a critical role in dealing with the crisis.
“Since our hospitalists are most familiar with the inner workings of the facility, they played a huge role in determining who needed to be hospitalized and where in the hospital they would best be treated,” he says. “This included reviewing the existing patient census when the collapse happened so we could discharge and transfer inpatients appropriately to ensure that the hospital was in the best position to deal with the collapse victims.”
Because the bridge collapsed during the early evening, there was plenty of staff on-hand to treat the victims, including residents who worked hand-in-hand with hospitalists in making admission and transfer decisions.
Challenge for Hospitalists
In smaller facilities where there are no residents, or in small emergency departments (ED) and intensive-care units, hospitalists will and should have even more critical roles in handling disasters and planning for them, Dr. Varns says.
He believes all hospitalists—but especially those in small, nonteaching facilities—should get triage training. “Hospitalists have a very broad skill set—especially with increasing responsibility for co-management of surgical cases—but they should develop triage skills,” says Dr. Varns, who suggests hospitalists take a two or three-day advanced trauma life support course.
Steven B. Deitelzweig, MD, FACP, system chairman, department of hospital medicine and vice president of medical affairs for the Ochsner Health System in the New Orleans area, agrees.
“I think the folks who are closest to guiding the care should be offering input into triage decisions,” he says. “Hospitalists can be invaluable in doing triage of inpatients. They provide objective detailed information.”
Dr. Deitelzweig, who experienced the three-week lock-down following Katrina, suggests hospitalist groups create a system of prioritizing evacuation of patients—including what kind of support they’ll need.
He believes hospitalists will be invaluable during crises because they are “front-line decision-makers, along with ED physicians and intensivists.” Hospitalists should be on disaster-preparedness committees and a key part of communication during an actual crisis, he urges.
“Communication is critical during a crisis—and hospitalists know their systems,” he continues, noting that Ochsner has out-of-state cell phones, satellite phones, ham radios, spectral light phones, radio frequency antennas in secure places, and more.
In addition to equipment and supplies, hospitalists need to be prepared to do whatever is needed in a crisis, Dr. Deitelzweig says. “In a disaster, you might have to do a procedure usually done by a specialist—with supervision—to extend that person,” he says. “You also may have to go past the physician role. That’s where leadership shows. Our CEO served food in the cafeteria during Katrina. During a disaster, you have to be a flat organization and just do what needs to be done. That gives emotional support to everyone.”
Still, the need to prepare before a disaster cannot be overemphasized, he says.
Ochsner now has two teams of pre-selected physicians, including hospitalists, dedicated to working through specific types of crises. Having the list of essential personnel online at all times is intended to prevent last-minute scurrying around to find the right people, he says.
In addition, providing balanced scheduling—especially in long-lasting crisis situations like Katrina—is important, says Dr. Deitelzweig. “Timing for release must be included, and having more staff on hand than necessary can help alleviate stress,” he advises.
Lessons of Katrina
Neal Axon, MD, an assistant professor at the Medical University of South Carolina, says he and his colleagues learned from those who went through Katrina as they prepared for the most likely disaster in Charleston: a severe hurricane.
Dr. Axon, a senior hospitalist in his group, says the facility has a system that generates e-mail, pages, text messages, and cell phone calls to keep hospital staff informed about potential crises. He also says the preparedness plan provides for relief of staff working for extended periods.
In addition, the hospital has trailers and inflatable tents to extend its facilities if there is a surge in patients. It also has a facility to provide decontamination for exposure to chemicals and radiation.
Brian Bossard, MD, director of Inpatient Physician Associates and medical staff quality designee at BryanLGH Medical Center in Lincoln Neb., says preparedness plans should be tested and updated regularly—especially the systems used to call in staff.
Dr. Bossard strongly believes hospitalists should be involved in disaster planning: “Every day hospitalists work hospital systems. We have a broad scope and perspective. That’s what you need in a disaster.” TH
Karla Feuer is a journalist based in New York.
Manage Your Work Flow
As a soon-to-be-attending hospitalist, you’ll shortly be on your own directing patient care. According to SHM data, you will see 12 to 18 patients per day, if not more. You understand the medicine, but how can you optimize your day to make it home in time? Here’s how you can direct your workday more efficiently.
1) Organize: This should come as no surprise. This crucial skill, reiterated during residency training, will prove invaluable as a practicing hospitalist. It certainly helps to maintain a structured and accurate daily census. Keeping a list of things to do handy and refreshing that list keeps you from having to rethink or reread your notes. On some occasions, doing things rather than writing them down to complete later can be faster. Whether you utilize handheld PCs or note cards, find a method that works for you.
2) Plan your day: If you know you are going to have a busy day, accept it. Start the day with a positive attitude and know you have to keep moving and can’t get stuck on trivial things. See your sickest patients first, or the ones you know will require a lot of time. Also, see your potential discharges as early in the day as possible to optimize the discharge process and pinpoint potential problems. If you are the attending on a teaching service, spend time with your resident to go over the structure of a typical day.
3) Consolidate: If you have patients in different areas in the hospital, start with the areas where you have the most patients—especially the sickest ones. See all the patients in proximity to each other. Avoid running between the computer, the chart, and the patient’s room for every patient. Lump some of these tasks together and avoid losing time.
4) Avoid hold music: Instead of paging people and waiting for a call back, send a text message to increase your efficiency. Try to contact (and wait for) someone to call you back while you are doing something productive—like writing a note. You can also make other work-related calls, such as to families and consultants, during your commute to or from work. This saves you some time when you are in the hospital.
5) Delegate: A lot of new hospitalists have difficulty relinquishing control—similar to when they made the transition from intern to resident. As an academic attending, don’t micromanage. Rather, attend to the global issues and problems that might need a greater degree of attending involvement, such as challenging family situations. This requires a certain degree of trust in your resident.
Assign specific responsibilities to members of your team (residents, interns, and medical students) and go over their roles. If you are not at an academic institution, you can still delegate tasks like procedures (central lines, spinal taps, thoracentesis, and paracentesis). There are other specialists in the hospital who perform these procedures more frequently and more efficiently than you.
6) Give yourself a time limit: To improve efficiency, some people find it helpful to give themselves a time limit to get their work done. Making this time limit practical may help get you home at reasonable hour. Also, learn to gracefully extract yourself from chatty patients, family, or colleagues if time is short.
7) Document efficiently: When rounding on patients, make sure the note is written when you see the patient—then move on. You can always come back for an addendum if needed. Group your note writing as much as possible on each floor. When admitting or discharging a patient, do all documentation at once, including notes and orders. This way you don’t waste time getting back to information you have in front of you. If there is a history and physical available on a new admission or consult, print it out and use it as a template during the patient interview. It helps to confirm details with the patient and fill in gaps. If time permits, prepare discharge papers and prescriptions in advance of anticipated discharges to save time on the day of discharge.
8) Define inpatient vs. outpatient management: Differentiate between important inpatient workup and evaluation that can be performed on an outpatient basis to save time and reduce length of stay. When patients can safely leave the hospital to continue work-ups and follow-ups with their primary care providers and specialists, you gain more time the following day—when you are no longer rounding on them.
9) Schedule a time to see family members: Conversations with family members are usually more productive if those times are scheduled. If possible, schedule them after you have seen a bulk of your patients to avoid feeling pressured to cut the meeting short. It is important to know who the family spokesperson is for large families so you can refer other family members to them and avoid multiple call-backs.
10) Develop and maintain good relationships: Your cordial interaction with various hospital department staff (nursing, case management, social work, radiology, and physical therapy to name a few) will help facilitate the inpatient care plan. It certainly helps not to have to wait two or more days to have a diagnostic test performed or assessment made. Sustaining a healthy working relationship promotes an understanding of your expectations for inpatient care.
11) Advocate for constructive change: Much inefficiency is systems based. Thinking about what interferes with your effectiveness in your system and suggesting changes can help a lot. For example, if your institution is going to switch to an electronic medical record, it certainly helps for you or a member of your hospitalist team to get involved in the implementation. Many hospitals are invested in quality improvement—and hospitalists are and should be at the forefront of this change.
As a hospitalist, you can initiate changes within your practice and your hospital’s system to keep things efficient. Making those adjustments sometimes takes time—but they’re well worth the effort.
Meanwhile, maximizing your efficiency can help promote patient throughput, enhance patient satisfaction, improve quality of care and increase job satisfaction. TH
Dr. Magnet is a hospitalist at the Singing River Hospital System on the Gulf Coast of Mississippi and a member of SHM’s Young Physician Committee.
As a soon-to-be-attending hospitalist, you’ll shortly be on your own directing patient care. According to SHM data, you will see 12 to 18 patients per day, if not more. You understand the medicine, but how can you optimize your day to make it home in time? Here’s how you can direct your workday more efficiently.
1) Organize: This should come as no surprise. This crucial skill, reiterated during residency training, will prove invaluable as a practicing hospitalist. It certainly helps to maintain a structured and accurate daily census. Keeping a list of things to do handy and refreshing that list keeps you from having to rethink or reread your notes. On some occasions, doing things rather than writing them down to complete later can be faster. Whether you utilize handheld PCs or note cards, find a method that works for you.
2) Plan your day: If you know you are going to have a busy day, accept it. Start the day with a positive attitude and know you have to keep moving and can’t get stuck on trivial things. See your sickest patients first, or the ones you know will require a lot of time. Also, see your potential discharges as early in the day as possible to optimize the discharge process and pinpoint potential problems. If you are the attending on a teaching service, spend time with your resident to go over the structure of a typical day.
3) Consolidate: If you have patients in different areas in the hospital, start with the areas where you have the most patients—especially the sickest ones. See all the patients in proximity to each other. Avoid running between the computer, the chart, and the patient’s room for every patient. Lump some of these tasks together and avoid losing time.
4) Avoid hold music: Instead of paging people and waiting for a call back, send a text message to increase your efficiency. Try to contact (and wait for) someone to call you back while you are doing something productive—like writing a note. You can also make other work-related calls, such as to families and consultants, during your commute to or from work. This saves you some time when you are in the hospital.
5) Delegate: A lot of new hospitalists have difficulty relinquishing control—similar to when they made the transition from intern to resident. As an academic attending, don’t micromanage. Rather, attend to the global issues and problems that might need a greater degree of attending involvement, such as challenging family situations. This requires a certain degree of trust in your resident.
Assign specific responsibilities to members of your team (residents, interns, and medical students) and go over their roles. If you are not at an academic institution, you can still delegate tasks like procedures (central lines, spinal taps, thoracentesis, and paracentesis). There are other specialists in the hospital who perform these procedures more frequently and more efficiently than you.
6) Give yourself a time limit: To improve efficiency, some people find it helpful to give themselves a time limit to get their work done. Making this time limit practical may help get you home at reasonable hour. Also, learn to gracefully extract yourself from chatty patients, family, or colleagues if time is short.
7) Document efficiently: When rounding on patients, make sure the note is written when you see the patient—then move on. You can always come back for an addendum if needed. Group your note writing as much as possible on each floor. When admitting or discharging a patient, do all documentation at once, including notes and orders. This way you don’t waste time getting back to information you have in front of you. If there is a history and physical available on a new admission or consult, print it out and use it as a template during the patient interview. It helps to confirm details with the patient and fill in gaps. If time permits, prepare discharge papers and prescriptions in advance of anticipated discharges to save time on the day of discharge.
8) Define inpatient vs. outpatient management: Differentiate between important inpatient workup and evaluation that can be performed on an outpatient basis to save time and reduce length of stay. When patients can safely leave the hospital to continue work-ups and follow-ups with their primary care providers and specialists, you gain more time the following day—when you are no longer rounding on them.
9) Schedule a time to see family members: Conversations with family members are usually more productive if those times are scheduled. If possible, schedule them after you have seen a bulk of your patients to avoid feeling pressured to cut the meeting short. It is important to know who the family spokesperson is for large families so you can refer other family members to them and avoid multiple call-backs.
10) Develop and maintain good relationships: Your cordial interaction with various hospital department staff (nursing, case management, social work, radiology, and physical therapy to name a few) will help facilitate the inpatient care plan. It certainly helps not to have to wait two or more days to have a diagnostic test performed or assessment made. Sustaining a healthy working relationship promotes an understanding of your expectations for inpatient care.
11) Advocate for constructive change: Much inefficiency is systems based. Thinking about what interferes with your effectiveness in your system and suggesting changes can help a lot. For example, if your institution is going to switch to an electronic medical record, it certainly helps for you or a member of your hospitalist team to get involved in the implementation. Many hospitals are invested in quality improvement—and hospitalists are and should be at the forefront of this change.
As a hospitalist, you can initiate changes within your practice and your hospital’s system to keep things efficient. Making those adjustments sometimes takes time—but they’re well worth the effort.
Meanwhile, maximizing your efficiency can help promote patient throughput, enhance patient satisfaction, improve quality of care and increase job satisfaction. TH
Dr. Magnet is a hospitalist at the Singing River Hospital System on the Gulf Coast of Mississippi and a member of SHM’s Young Physician Committee.
As a soon-to-be-attending hospitalist, you’ll shortly be on your own directing patient care. According to SHM data, you will see 12 to 18 patients per day, if not more. You understand the medicine, but how can you optimize your day to make it home in time? Here’s how you can direct your workday more efficiently.
1) Organize: This should come as no surprise. This crucial skill, reiterated during residency training, will prove invaluable as a practicing hospitalist. It certainly helps to maintain a structured and accurate daily census. Keeping a list of things to do handy and refreshing that list keeps you from having to rethink or reread your notes. On some occasions, doing things rather than writing them down to complete later can be faster. Whether you utilize handheld PCs or note cards, find a method that works for you.
2) Plan your day: If you know you are going to have a busy day, accept it. Start the day with a positive attitude and know you have to keep moving and can’t get stuck on trivial things. See your sickest patients first, or the ones you know will require a lot of time. Also, see your potential discharges as early in the day as possible to optimize the discharge process and pinpoint potential problems. If you are the attending on a teaching service, spend time with your resident to go over the structure of a typical day.
3) Consolidate: If you have patients in different areas in the hospital, start with the areas where you have the most patients—especially the sickest ones. See all the patients in proximity to each other. Avoid running between the computer, the chart, and the patient’s room for every patient. Lump some of these tasks together and avoid losing time.
4) Avoid hold music: Instead of paging people and waiting for a call back, send a text message to increase your efficiency. Try to contact (and wait for) someone to call you back while you are doing something productive—like writing a note. You can also make other work-related calls, such as to families and consultants, during your commute to or from work. This saves you some time when you are in the hospital.
5) Delegate: A lot of new hospitalists have difficulty relinquishing control—similar to when they made the transition from intern to resident. As an academic attending, don’t micromanage. Rather, attend to the global issues and problems that might need a greater degree of attending involvement, such as challenging family situations. This requires a certain degree of trust in your resident.
Assign specific responsibilities to members of your team (residents, interns, and medical students) and go over their roles. If you are not at an academic institution, you can still delegate tasks like procedures (central lines, spinal taps, thoracentesis, and paracentesis). There are other specialists in the hospital who perform these procedures more frequently and more efficiently than you.
6) Give yourself a time limit: To improve efficiency, some people find it helpful to give themselves a time limit to get their work done. Making this time limit practical may help get you home at reasonable hour. Also, learn to gracefully extract yourself from chatty patients, family, or colleagues if time is short.
7) Document efficiently: When rounding on patients, make sure the note is written when you see the patient—then move on. You can always come back for an addendum if needed. Group your note writing as much as possible on each floor. When admitting or discharging a patient, do all documentation at once, including notes and orders. This way you don’t waste time getting back to information you have in front of you. If there is a history and physical available on a new admission or consult, print it out and use it as a template during the patient interview. It helps to confirm details with the patient and fill in gaps. If time permits, prepare discharge papers and prescriptions in advance of anticipated discharges to save time on the day of discharge.
8) Define inpatient vs. outpatient management: Differentiate between important inpatient workup and evaluation that can be performed on an outpatient basis to save time and reduce length of stay. When patients can safely leave the hospital to continue work-ups and follow-ups with their primary care providers and specialists, you gain more time the following day—when you are no longer rounding on them.
9) Schedule a time to see family members: Conversations with family members are usually more productive if those times are scheduled. If possible, schedule them after you have seen a bulk of your patients to avoid feeling pressured to cut the meeting short. It is important to know who the family spokesperson is for large families so you can refer other family members to them and avoid multiple call-backs.
10) Develop and maintain good relationships: Your cordial interaction with various hospital department staff (nursing, case management, social work, radiology, and physical therapy to name a few) will help facilitate the inpatient care plan. It certainly helps not to have to wait two or more days to have a diagnostic test performed or assessment made. Sustaining a healthy working relationship promotes an understanding of your expectations for inpatient care.
11) Advocate for constructive change: Much inefficiency is systems based. Thinking about what interferes with your effectiveness in your system and suggesting changes can help a lot. For example, if your institution is going to switch to an electronic medical record, it certainly helps for you or a member of your hospitalist team to get involved in the implementation. Many hospitals are invested in quality improvement—and hospitalists are and should be at the forefront of this change.
As a hospitalist, you can initiate changes within your practice and your hospital’s system to keep things efficient. Making those adjustments sometimes takes time—but they’re well worth the effort.
Meanwhile, maximizing your efficiency can help promote patient throughput, enhance patient satisfaction, improve quality of care and increase job satisfaction. TH
Dr. Magnet is a hospitalist at the Singing River Hospital System on the Gulf Coast of Mississippi and a member of SHM’s Young Physician Committee.
Is That Your Patient?
How many times have you been asked a medical question outside the hospital? Undoubtedly, it happens too many times to count.
An acquaintance asks about a strange pain; you look at a rash on your neighbor’s son; you guide a nurse when she can’t reach a patient’s physician; a colleague asks for a curbside consult; or you provide medical advice over the phone to another provider while on-call at the hospital. When do any of the people in these situations become your patient?
Unfortunately, there is no easy answer. Legally, the question of whether a physician-patient relationship is created is determined on a case-by-case basis. As a general rule, if a physician undertakes to treat or provide medical care, a physician-patient relationship exists and the physician contracts to exercise reasonable skill in providing the care.
Implied Relationships
Absent an express agreement to enter a physician-patient relationship, the law may imply a relationship based on conduct that demonstrates consent to a relationship. A patient demonstrates consent by seeking medical services. Consent may also be implied when, for example, a patient needs emergency care, services are provided at the request of a treating physician, or treatment is mandated by a court.
Physicians consent to a relationship by diagnosing, treating, or otherwise providing care. A physician can also consent simply because of a working arrangement with a hospital or other entity—such as an agreement to accept assignment of patients.
In determining whether a physician-patient relationship has been created, consider the absence or existence of affirmative acts by a physician. For example, when a physician receives a call from a patient’s treating physician and the two physicians discuss the patient, the conversation might not create a physician-patient relationship if the consulting physician does not expressly provide an opinion. This is because there is no affirmative action upon which a court can imply a duty. Likewise, an on-call doctor does not create a physician-patient relationship simply by being on-call when she does not see, treat, or participate in the care of a patient.
Conversely, acts sufficient to create a physician-patient relationship exist when an on-call or consulting physician offers advice, provides treatment, or discharges a patient. Notably, an implied duty can be inferred even if the physician has not had direct contact with a patient if the court determines the physician’s conduct has interfered with a patient’s interests—thereby entitling the patient to legal protection.
Duties of Physicians
Even absent a physician-patient relationship, the law can impose general duties on physicians. Physicians have a duty to use reasonable care in regard to affirmative conduct when it is foreseeable that another might be injured.
For example, the Colorado Supreme Court found that an anesthesiologist owed a general duty to hospital patients who were not his patients when the physician’s failure to properly dispose of medication exposed patients to a foreseeable risk of harm.
The court has also found that a physician retained by defendants in a personal injury lawsuit owed a duty of reasonable care when subjecting the plaintiff to medical examinations. Similarly, the court concluded that an independent medical examiner could be liable for any injury the examiner causes during an examination, even though the examiner does not owe a duty to accurately diagnose the patient.
Is That Your Patient?
Ultimately, a physician-patient relationship and its corresponding duties arise when reasonable people would recognize a duty and agree that it exists. You must analyze your conduct and interactions, including your:
- Communication with patients or nonpatients (including e-mail or conversations in passing);
- On-call status;
- Agreements with facilities, a service, or other providers to accept patients;
- Degree of responsibility for a given patient’s care;
- Charges or fee discussion;
- Affirmative acts of care or treatment as distinguished from examination solely for the benefit of a third party;
- Initiation of contact with the patient or patient’s family;
- Referral from another physician or non-physician; and
- Consultations with other physicians, either formal or informal and whether different areas of expertise are involved or specific care or advise is given.
Ultimate determination of whether a physician-patient relationship exists is fact-specific—and no single fact is definitive. The above factors may guide you in assessing the nature of your interactions with patients and your attendant responsibilities. TH
Patrick O’Rourke works in the Office of University Counsel, Department of Litigation, University of Colorado, Denver.
How many times have you been asked a medical question outside the hospital? Undoubtedly, it happens too many times to count.
An acquaintance asks about a strange pain; you look at a rash on your neighbor’s son; you guide a nurse when she can’t reach a patient’s physician; a colleague asks for a curbside consult; or you provide medical advice over the phone to another provider while on-call at the hospital. When do any of the people in these situations become your patient?
Unfortunately, there is no easy answer. Legally, the question of whether a physician-patient relationship is created is determined on a case-by-case basis. As a general rule, if a physician undertakes to treat or provide medical care, a physician-patient relationship exists and the physician contracts to exercise reasonable skill in providing the care.
Implied Relationships
Absent an express agreement to enter a physician-patient relationship, the law may imply a relationship based on conduct that demonstrates consent to a relationship. A patient demonstrates consent by seeking medical services. Consent may also be implied when, for example, a patient needs emergency care, services are provided at the request of a treating physician, or treatment is mandated by a court.
Physicians consent to a relationship by diagnosing, treating, or otherwise providing care. A physician can also consent simply because of a working arrangement with a hospital or other entity—such as an agreement to accept assignment of patients.
In determining whether a physician-patient relationship has been created, consider the absence or existence of affirmative acts by a physician. For example, when a physician receives a call from a patient’s treating physician and the two physicians discuss the patient, the conversation might not create a physician-patient relationship if the consulting physician does not expressly provide an opinion. This is because there is no affirmative action upon which a court can imply a duty. Likewise, an on-call doctor does not create a physician-patient relationship simply by being on-call when she does not see, treat, or participate in the care of a patient.
Conversely, acts sufficient to create a physician-patient relationship exist when an on-call or consulting physician offers advice, provides treatment, or discharges a patient. Notably, an implied duty can be inferred even if the physician has not had direct contact with a patient if the court determines the physician’s conduct has interfered with a patient’s interests—thereby entitling the patient to legal protection.
Duties of Physicians
Even absent a physician-patient relationship, the law can impose general duties on physicians. Physicians have a duty to use reasonable care in regard to affirmative conduct when it is foreseeable that another might be injured.
For example, the Colorado Supreme Court found that an anesthesiologist owed a general duty to hospital patients who were not his patients when the physician’s failure to properly dispose of medication exposed patients to a foreseeable risk of harm.
The court has also found that a physician retained by defendants in a personal injury lawsuit owed a duty of reasonable care when subjecting the plaintiff to medical examinations. Similarly, the court concluded that an independent medical examiner could be liable for any injury the examiner causes during an examination, even though the examiner does not owe a duty to accurately diagnose the patient.
Is That Your Patient?
Ultimately, a physician-patient relationship and its corresponding duties arise when reasonable people would recognize a duty and agree that it exists. You must analyze your conduct and interactions, including your:
- Communication with patients or nonpatients (including e-mail or conversations in passing);
- On-call status;
- Agreements with facilities, a service, or other providers to accept patients;
- Degree of responsibility for a given patient’s care;
- Charges or fee discussion;
- Affirmative acts of care or treatment as distinguished from examination solely for the benefit of a third party;
- Initiation of contact with the patient or patient’s family;
- Referral from another physician or non-physician; and
- Consultations with other physicians, either formal or informal and whether different areas of expertise are involved or specific care or advise is given.
Ultimate determination of whether a physician-patient relationship exists is fact-specific—and no single fact is definitive. The above factors may guide you in assessing the nature of your interactions with patients and your attendant responsibilities. TH
Patrick O’Rourke works in the Office of University Counsel, Department of Litigation, University of Colorado, Denver.
How many times have you been asked a medical question outside the hospital? Undoubtedly, it happens too many times to count.
An acquaintance asks about a strange pain; you look at a rash on your neighbor’s son; you guide a nurse when she can’t reach a patient’s physician; a colleague asks for a curbside consult; or you provide medical advice over the phone to another provider while on-call at the hospital. When do any of the people in these situations become your patient?
Unfortunately, there is no easy answer. Legally, the question of whether a physician-patient relationship is created is determined on a case-by-case basis. As a general rule, if a physician undertakes to treat or provide medical care, a physician-patient relationship exists and the physician contracts to exercise reasonable skill in providing the care.
Implied Relationships
Absent an express agreement to enter a physician-patient relationship, the law may imply a relationship based on conduct that demonstrates consent to a relationship. A patient demonstrates consent by seeking medical services. Consent may also be implied when, for example, a patient needs emergency care, services are provided at the request of a treating physician, or treatment is mandated by a court.
Physicians consent to a relationship by diagnosing, treating, or otherwise providing care. A physician can also consent simply because of a working arrangement with a hospital or other entity—such as an agreement to accept assignment of patients.
In determining whether a physician-patient relationship has been created, consider the absence or existence of affirmative acts by a physician. For example, when a physician receives a call from a patient’s treating physician and the two physicians discuss the patient, the conversation might not create a physician-patient relationship if the consulting physician does not expressly provide an opinion. This is because there is no affirmative action upon which a court can imply a duty. Likewise, an on-call doctor does not create a physician-patient relationship simply by being on-call when she does not see, treat, or participate in the care of a patient.
Conversely, acts sufficient to create a physician-patient relationship exist when an on-call or consulting physician offers advice, provides treatment, or discharges a patient. Notably, an implied duty can be inferred even if the physician has not had direct contact with a patient if the court determines the physician’s conduct has interfered with a patient’s interests—thereby entitling the patient to legal protection.
Duties of Physicians
Even absent a physician-patient relationship, the law can impose general duties on physicians. Physicians have a duty to use reasonable care in regard to affirmative conduct when it is foreseeable that another might be injured.
For example, the Colorado Supreme Court found that an anesthesiologist owed a general duty to hospital patients who were not his patients when the physician’s failure to properly dispose of medication exposed patients to a foreseeable risk of harm.
The court has also found that a physician retained by defendants in a personal injury lawsuit owed a duty of reasonable care when subjecting the plaintiff to medical examinations. Similarly, the court concluded that an independent medical examiner could be liable for any injury the examiner causes during an examination, even though the examiner does not owe a duty to accurately diagnose the patient.
Is That Your Patient?
Ultimately, a physician-patient relationship and its corresponding duties arise when reasonable people would recognize a duty and agree that it exists. You must analyze your conduct and interactions, including your:
- Communication with patients or nonpatients (including e-mail or conversations in passing);
- On-call status;
- Agreements with facilities, a service, or other providers to accept patients;
- Degree of responsibility for a given patient’s care;
- Charges or fee discussion;
- Affirmative acts of care or treatment as distinguished from examination solely for the benefit of a third party;
- Initiation of contact with the patient or patient’s family;
- Referral from another physician or non-physician; and
- Consultations with other physicians, either formal or informal and whether different areas of expertise are involved or specific care or advise is given.
Ultimate determination of whether a physician-patient relationship exists is fact-specific—and no single fact is definitive. The above factors may guide you in assessing the nature of your interactions with patients and your attendant responsibilities. TH
Patrick O’Rourke works in the Office of University Counsel, Department of Litigation, University of Colorado, Denver.
Document Patient History
Documentation in the medical record serves many purposes: communication among healthcare professionals, evidence of patient care, and justification for provider claims.
Although these three aspects of documentation are intertwined, the first two prevent physicians from paying settlements involving malpractice allegations, while the last one assists in obtaining appropriate reimbursement for services rendered. This is the first of a three-part series that will focus on claim reporting and outline the documentation guidelines set forth by the Centers for Medicare and Medicaid Services (CMS) in conjunction with the American Medical Association (AMA).
1995, 1997 Guidelines
Two sets of documentation guidelines are in place, referred to as the 1995 and 1997 guidelines. Increased criticism of the ambiguity in the 1995 guidelines from auditors and providers inspired development of the 1997 guidelines.
While the 1997 guidelines were intended to create a more objective and unified approach to documentation, the level of specificity required brought criticism and frustration. But while the physician community balked, most auditors praised these efforts.
To satisfy all parties and allow physicians to document as they prefer, both sets of guidelines remain. Physicians can document according to either style, and auditors are obligated to review provider records against both sets of guidelines, selecting the final visit level with the set that best supports provider documentation.
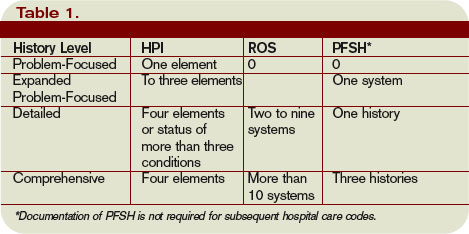
Elements of History
Chief complaint (CC): The CC is the reason for the visit as stated in the patient’s own words. This must be present for each encounter, and should reference a specific condition or complaint (e.g., patient complains of abdominal pain).
History of present illness (HPI): This is a description of the present illness as it developed. It is typically formatted and documented with reference to location, quality, severity, timing, context, modifying factors, and associated signs/symptoms as related to the chief complaint. The HPI may be classified as brief (a comment on fewer than HPI elements) or extended (a comment on more than four HPI elements). Sample documentation of an extended HPI is: “The patient has intermittent (duration), sharp (quality) pain in the right upper quadrant (location) without associated nausea, vomiting, or diarrhea (associated signs/symptoms).”
The 1997 guidelines offer an alternate format for documenting the HPI. In contrast to the standard method above, the physician may list and status the patient’s chronic or inactive conditions. An extended HPI consists of the status of at least three chronic or inactive conditions (e.g., “Diabetes controlled by oral medication; extrinsic asthma without acute exacerbation in past six months; hypertension stable with pressures ranging from 130-140/80-90”). Failing to document the status negates the opportunity for the physician to receive HPI credit. Instead, he will receive credit for a past medical history.
The HPI should never be documented by ancillary staff (e.g., registered nurse, medical assistant, students). HPI might be documented by residents (e.g., residents, fellows, interns) or nonphysician providers (nurse practitioners and physician assistants) when utilizing the Teaching Physician Rules or Split-Shared Billing Rules, respectively (teaching Physician Rules and Split-Shared Billing Rules will be addressed in an upcoming issue).
Review of systems (ROS): This is a series of questions used to elicit information about additional signs, symptoms, or problems currently or previously experienced by the patient:
- Constitutional;
- Eyes; ears, nose, mouth, throat;
- Cardiovascular;
- Respiratory;
- Gastrointestinal;
- Genitourinary;
- Musculoskeletal;
- Integumentary (including skin and/or breast);
- Neurological;
- Psychiatric;
- Endocrine;
- Hematologic/lymphatic; and
- Allergic/immunologic.
The ROS may be classified as brief (a comment on one system), expanded (a comment on two to nine systems), or complete (a comment on more than 10 systems).
Documentation of a complete ROS (more than 10 systems) can occur in two ways:
- The physician can individually document each system. For example: “No fever/chills (constitutional) or blurred vision (eyes); no chest pain (cardiovascular); shortness of breath (respiratory); or belly pain (gastrointestinal); etc.”; or
- The physician can document the positive findings and pertinent negative findings related to the chief complaint, along with a comment that “all other systems are negative.” This latter statement is not accepted by all local Medicare contractors.
Information involving the ROS can be documented by anyone, including the patient. If documented by someone else (e.g., a medical student) other than residents under the Teaching Physician Rules or nonphysician providers under the Split-Shared Billing Rules, the physician should reference the documented ROS in his progress note. Re-documentation of the ROS is not necessary unless a revision is required.
Past, family, and social history (PFSH): Documentation of PFSH involves data obtained about the patient’s previous illness or medical conditions/therapies, family occurrences with illness, and relevant patient activities. The PFSH can be classified as pertinent (a comment on one history) or complete (a comment in each of the three histories). Documentation that exemplifies a complete PFSH is: “Patient currently on Prilosec 20 mg daily; family history of Barrett’s esophagus; no tobacco or alcohol use.”
As with ROS, the PFSH can be documented by anyone, including the patient. If documented by someone else (e.g., a medical student) other than residents under the Teaching Physician Rules or nonphysician providers under the Split-Shared Billing Rules, the physician should reference the documented PFSH in his progress note. Re-documentation of the PFSH is not necessary unless a revision is required. It is important to note that while documentation of the PFSH is required when billing higher level consultations (99254-99255) or initial inpatient care (99221-99223), it is not required when reporting subsequent hospital care services (99231-99233).

Levels of History
There are four levels of history, determined by the number of elements documented in the progress note (see Table 1, p. 21). The physician must meet all the requirements in a specific level of history before assigning it.
If all of the required elements in a given history level are not documented, the level assigned is that of the least documented element. For example, physician documentation may include four HPI elements and a complete PFSH, yet only eight ROS. The physician can only receive credit for a detailed history. If the physician submitted a claim for 99222 (initial hospital care requiring a comprehensive history, a comprehensive exam, and moderate-complexity decision making), documentation would not support the reported service due to the underdocumented ROS. Deficiencies in the ROS and family history are the most common physician documentation errors involving the history component.
A specific level of history is associated with each type of physician encounter, and must be documented accordingly (see Table 2, right). The most common visit categories provided by hospitalists that include documentation requirements for history are initial inpatient consultations, initial hospital care, subsequent hospital care, and initial observation care. Other visit categories, such as critical care and discharge day management, have neither associated levels of history nor documentation requirements for historical elements. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
Documentation in the medical record serves many purposes: communication among healthcare professionals, evidence of patient care, and justification for provider claims.
Although these three aspects of documentation are intertwined, the first two prevent physicians from paying settlements involving malpractice allegations, while the last one assists in obtaining appropriate reimbursement for services rendered. This is the first of a three-part series that will focus on claim reporting and outline the documentation guidelines set forth by the Centers for Medicare and Medicaid Services (CMS) in conjunction with the American Medical Association (AMA).
1995, 1997 Guidelines
Two sets of documentation guidelines are in place, referred to as the 1995 and 1997 guidelines. Increased criticism of the ambiguity in the 1995 guidelines from auditors and providers inspired development of the 1997 guidelines.
While the 1997 guidelines were intended to create a more objective and unified approach to documentation, the level of specificity required brought criticism and frustration. But while the physician community balked, most auditors praised these efforts.
To satisfy all parties and allow physicians to document as they prefer, both sets of guidelines remain. Physicians can document according to either style, and auditors are obligated to review provider records against both sets of guidelines, selecting the final visit level with the set that best supports provider documentation.
Elements of History
Chief complaint (CC): The CC is the reason for the visit as stated in the patient’s own words. This must be present for each encounter, and should reference a specific condition or complaint (e.g., patient complains of abdominal pain).
History of present illness (HPI): This is a description of the present illness as it developed. It is typically formatted and documented with reference to location, quality, severity, timing, context, modifying factors, and associated signs/symptoms as related to the chief complaint. The HPI may be classified as brief (a comment on fewer than HPI elements) or extended (a comment on more than four HPI elements). Sample documentation of an extended HPI is: “The patient has intermittent (duration), sharp (quality) pain in the right upper quadrant (location) without associated nausea, vomiting, or diarrhea (associated signs/symptoms).”
The 1997 guidelines offer an alternate format for documenting the HPI. In contrast to the standard method above, the physician may list and status the patient’s chronic or inactive conditions. An extended HPI consists of the status of at least three chronic or inactive conditions (e.g., “Diabetes controlled by oral medication; extrinsic asthma without acute exacerbation in past six months; hypertension stable with pressures ranging from 130-140/80-90”). Failing to document the status negates the opportunity for the physician to receive HPI credit. Instead, he will receive credit for a past medical history.
The HPI should never be documented by ancillary staff (e.g., registered nurse, medical assistant, students). HPI might be documented by residents (e.g., residents, fellows, interns) or nonphysician providers (nurse practitioners and physician assistants) when utilizing the Teaching Physician Rules or Split-Shared Billing Rules, respectively (teaching Physician Rules and Split-Shared Billing Rules will be addressed in an upcoming issue).
Review of systems (ROS): This is a series of questions used to elicit information about additional signs, symptoms, or problems currently or previously experienced by the patient:
- Constitutional;
- Eyes; ears, nose, mouth, throat;
- Cardiovascular;
- Respiratory;
- Gastrointestinal;
- Genitourinary;
- Musculoskeletal;
- Integumentary (including skin and/or breast);
- Neurological;
- Psychiatric;
- Endocrine;
- Hematologic/lymphatic; and
- Allergic/immunologic.
The ROS may be classified as brief (a comment on one system), expanded (a comment on two to nine systems), or complete (a comment on more than 10 systems).
Documentation of a complete ROS (more than 10 systems) can occur in two ways:
- The physician can individually document each system. For example: “No fever/chills (constitutional) or blurred vision (eyes); no chest pain (cardiovascular); shortness of breath (respiratory); or belly pain (gastrointestinal); etc.”; or
- The physician can document the positive findings and pertinent negative findings related to the chief complaint, along with a comment that “all other systems are negative.” This latter statement is not accepted by all local Medicare contractors.
Information involving the ROS can be documented by anyone, including the patient. If documented by someone else (e.g., a medical student) other than residents under the Teaching Physician Rules or nonphysician providers under the Split-Shared Billing Rules, the physician should reference the documented ROS in his progress note. Re-documentation of the ROS is not necessary unless a revision is required.
Past, family, and social history (PFSH): Documentation of PFSH involves data obtained about the patient’s previous illness or medical conditions/therapies, family occurrences with illness, and relevant patient activities. The PFSH can be classified as pertinent (a comment on one history) or complete (a comment in each of the three histories). Documentation that exemplifies a complete PFSH is: “Patient currently on Prilosec 20 mg daily; family history of Barrett’s esophagus; no tobacco or alcohol use.”
As with ROS, the PFSH can be documented by anyone, including the patient. If documented by someone else (e.g., a medical student) other than residents under the Teaching Physician Rules or nonphysician providers under the Split-Shared Billing Rules, the physician should reference the documented PFSH in his progress note. Re-documentation of the PFSH is not necessary unless a revision is required. It is important to note that while documentation of the PFSH is required when billing higher level consultations (99254-99255) or initial inpatient care (99221-99223), it is not required when reporting subsequent hospital care services (99231-99233).
Levels of History
There are four levels of history, determined by the number of elements documented in the progress note (see Table 1, p. 21). The physician must meet all the requirements in a specific level of history before assigning it.
If all of the required elements in a given history level are not documented, the level assigned is that of the least documented element. For example, physician documentation may include four HPI elements and a complete PFSH, yet only eight ROS. The physician can only receive credit for a detailed history. If the physician submitted a claim for 99222 (initial hospital care requiring a comprehensive history, a comprehensive exam, and moderate-complexity decision making), documentation would not support the reported service due to the underdocumented ROS. Deficiencies in the ROS and family history are the most common physician documentation errors involving the history component.
A specific level of history is associated with each type of physician encounter, and must be documented accordingly (see Table 2, right). The most common visit categories provided by hospitalists that include documentation requirements for history are initial inpatient consultations, initial hospital care, subsequent hospital care, and initial observation care. Other visit categories, such as critical care and discharge day management, have neither associated levels of history nor documentation requirements for historical elements. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
Documentation in the medical record serves many purposes: communication among healthcare professionals, evidence of patient care, and justification for provider claims.
Although these three aspects of documentation are intertwined, the first two prevent physicians from paying settlements involving malpractice allegations, while the last one assists in obtaining appropriate reimbursement for services rendered. This is the first of a three-part series that will focus on claim reporting and outline the documentation guidelines set forth by the Centers for Medicare and Medicaid Services (CMS) in conjunction with the American Medical Association (AMA).
1995, 1997 Guidelines
Two sets of documentation guidelines are in place, referred to as the 1995 and 1997 guidelines. Increased criticism of the ambiguity in the 1995 guidelines from auditors and providers inspired development of the 1997 guidelines.
While the 1997 guidelines were intended to create a more objective and unified approach to documentation, the level of specificity required brought criticism and frustration. But while the physician community balked, most auditors praised these efforts.
To satisfy all parties and allow physicians to document as they prefer, both sets of guidelines remain. Physicians can document according to either style, and auditors are obligated to review provider records against both sets of guidelines, selecting the final visit level with the set that best supports provider documentation.
Elements of History
Chief complaint (CC): The CC is the reason for the visit as stated in the patient’s own words. This must be present for each encounter, and should reference a specific condition or complaint (e.g., patient complains of abdominal pain).
History of present illness (HPI): This is a description of the present illness as it developed. It is typically formatted and documented with reference to location, quality, severity, timing, context, modifying factors, and associated signs/symptoms as related to the chief complaint. The HPI may be classified as brief (a comment on fewer than HPI elements) or extended (a comment on more than four HPI elements). Sample documentation of an extended HPI is: “The patient has intermittent (duration), sharp (quality) pain in the right upper quadrant (location) without associated nausea, vomiting, or diarrhea (associated signs/symptoms).”
The 1997 guidelines offer an alternate format for documenting the HPI. In contrast to the standard method above, the physician may list and status the patient’s chronic or inactive conditions. An extended HPI consists of the status of at least three chronic or inactive conditions (e.g., “Diabetes controlled by oral medication; extrinsic asthma without acute exacerbation in past six months; hypertension stable with pressures ranging from 130-140/80-90”). Failing to document the status negates the opportunity for the physician to receive HPI credit. Instead, he will receive credit for a past medical history.
The HPI should never be documented by ancillary staff (e.g., registered nurse, medical assistant, students). HPI might be documented by residents (e.g., residents, fellows, interns) or nonphysician providers (nurse practitioners and physician assistants) when utilizing the Teaching Physician Rules or Split-Shared Billing Rules, respectively (teaching Physician Rules and Split-Shared Billing Rules will be addressed in an upcoming issue).
Review of systems (ROS): This is a series of questions used to elicit information about additional signs, symptoms, or problems currently or previously experienced by the patient:
- Constitutional;
- Eyes; ears, nose, mouth, throat;
- Cardiovascular;
- Respiratory;
- Gastrointestinal;
- Genitourinary;
- Musculoskeletal;
- Integumentary (including skin and/or breast);
- Neurological;
- Psychiatric;
- Endocrine;
- Hematologic/lymphatic; and
- Allergic/immunologic.
The ROS may be classified as brief (a comment on one system), expanded (a comment on two to nine systems), or complete (a comment on more than 10 systems).
Documentation of a complete ROS (more than 10 systems) can occur in two ways:
- The physician can individually document each system. For example: “No fever/chills (constitutional) or blurred vision (eyes); no chest pain (cardiovascular); shortness of breath (respiratory); or belly pain (gastrointestinal); etc.”; or
- The physician can document the positive findings and pertinent negative findings related to the chief complaint, along with a comment that “all other systems are negative.” This latter statement is not accepted by all local Medicare contractors.
Information involving the ROS can be documented by anyone, including the patient. If documented by someone else (e.g., a medical student) other than residents under the Teaching Physician Rules or nonphysician providers under the Split-Shared Billing Rules, the physician should reference the documented ROS in his progress note. Re-documentation of the ROS is not necessary unless a revision is required.
Past, family, and social history (PFSH): Documentation of PFSH involves data obtained about the patient’s previous illness or medical conditions/therapies, family occurrences with illness, and relevant patient activities. The PFSH can be classified as pertinent (a comment on one history) or complete (a comment in each of the three histories). Documentation that exemplifies a complete PFSH is: “Patient currently on Prilosec 20 mg daily; family history of Barrett’s esophagus; no tobacco or alcohol use.”
As with ROS, the PFSH can be documented by anyone, including the patient. If documented by someone else (e.g., a medical student) other than residents under the Teaching Physician Rules or nonphysician providers under the Split-Shared Billing Rules, the physician should reference the documented PFSH in his progress note. Re-documentation of the PFSH is not necessary unless a revision is required. It is important to note that while documentation of the PFSH is required when billing higher level consultations (99254-99255) or initial inpatient care (99221-99223), it is not required when reporting subsequent hospital care services (99231-99233).
Levels of History
There are four levels of history, determined by the number of elements documented in the progress note (see Table 1, p. 21). The physician must meet all the requirements in a specific level of history before assigning it.
If all of the required elements in a given history level are not documented, the level assigned is that of the least documented element. For example, physician documentation may include four HPI elements and a complete PFSH, yet only eight ROS. The physician can only receive credit for a detailed history. If the physician submitted a claim for 99222 (initial hospital care requiring a comprehensive history, a comprehensive exam, and moderate-complexity decision making), documentation would not support the reported service due to the underdocumented ROS. Deficiencies in the ROS and family history are the most common physician documentation errors involving the history component.
A specific level of history is associated with each type of physician encounter, and must be documented accordingly (see Table 2, right). The most common visit categories provided by hospitalists that include documentation requirements for history are initial inpatient consultations, initial hospital care, subsequent hospital care, and initial observation care. Other visit categories, such as critical care and discharge day management, have neither associated levels of history nor documentation requirements for historical elements. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is also on the faculty of SHM’s inpatient coding course.
Report on PQRI
The current pay-for-reporting program from the Centers for Medicare and Medicaid (CMS) seems tailor-made for hospitalists. Here’s a look at the voluntary Physician Quality Reporting Initiative (PQRI) program, and why and how hospitalists are—and are not—participating.
CMS has revised the reporting program that began as a six-month trial in 2007. The current PQRI runs the full calendar year for 2008 and includes 119 quality measures—11 of which hospitalists can report on. Detailed specifications for the measures are available on the CMS Web site at www.cms.hhs.gov.
The earnings in this pay-for-reporting program remain the same as 2007: Physicians who successfully report on measures can earn a bonus payment equal to 1.5% of their total Medicare-allowed charges. Some hospitalists have collected their bonus for participating in the 2007 trial; it’s likely more will participate this year.
CMS has yet to release data on participation in the 2007 PQRI trial or this year’s initiative. However, SHM has urged hospitalists to participate, and many are. During a national, SHM-sponsored conference call with CMS in summer 2007, approximately 20% of the 160 hospitalists participating in the call responded to a follow-up survey. Almost half of all respondents indicated they planned to participate in PQRI reporting.
“That percentage comes from a select group of hospitalists who were highly interested in the PQRI,” points out Patrick J. Torcson, MD, MMM, FACP, director of hospital medicine at St. Tammany Parish Hospital in Covington, La.
Unlike many specialists, hospitalists are finding reporting to be a straightforward process. “For hospitalists, PQRI reporting on specific measures harmonizes nicely with workflow,” says Dr. Torcson. “Most applicable measures take place during admission or discharge. Documentation and reporting for PQRI can take place during these times.”
Report on Reporting
At St. Tammany, Dr. Torcson’s eight-hospitalist team is participating in PQRI. Although you need only to report on three measures to qualify for a bonus payment from the program, “we’re actually reporting on the full list of [hospitalist-applicable] measures,” Dr. Torcson says. It’s up to each St. Tammany hospitalist to remember to report on the 11 measures.
“Support for [reporting] really comes down to physician memory,” says Dr. Torcson. “Long term, this is going to have to be part of an electronic system, with decision support and billing capability from an electronic health record.”
In spite of the added step of PQRI reporting, Dr. Torcson says, “we’ve had an enthusiastic response from our hospitalists.” The payoff for the hospital medicine program and the hospital is yet to be seen. “You hope that PQRI performance reporting will result in improved quality of care,” henotes.
But many physicians—including hospitalists—are not participating in PQRI.
“It comes down to different practice models,” explains Dr. Torcson. “But for many physicians, a major reason not to participate is that they’re taking a wait-and-see approach. They’re waiting to see if this is just the latest flavor of the month, and think it’s not worth investing time and effort until it proves otherwise.”
Gregory B. Seymann, MD, associate clinical professor, University of California, San Diego (UCSD) School of Medicine, Division of Hospital Medicine, is a member of SHM’s Public Policy Committee and says he was disappointed his group is unable to participate in PQRI.
“I work for UCSD, where our hospitalist group is one of many, many subspecialty groups that work out of our hospital,” he explains “We do a lot of QI work, and we were certainly interested in participating in PQRI.” However, the hospital uses an electronic billing system incompatible with reporting on the measures. The software could be upgraded for about $15,000, says Dr. Seymann, but hospital administration sees no return on the investment.
“The cost wouldn’t match the increase in revenues because besides hospital medicine, there aren’t a lot of other subspecialties that would be interested in participating,” explains Dr. Seymann. “As much as I wanted our group to participate, I can’t fully fault UCSD on this decision on business grounds. They want to see some stability in [the decision to continue PQRI] before they invest.”
In the meantime, the orthopedics group at UCSD has invested in reporting. They are tracking PQRI measures on paper and reporting to CMS, and they’ll ultimately be able to show the administration whether the bonus per physician might add up to the cost of the necessary billing-system upgrade.
Beyond 2008
Everyone involved—not just UCSD—is asking: Is PQRI here to stay? That decision rests with federal lawmakers. At the end of this year, Congress must vote on whether to extend the program—and no one can guarantee whether that will happen.
“The chairs of the Senate Finance Committee have been tremendously supportive of the PQRI,” says Dr. Torcson. “There is a lot of political will behind this right now. [PQRI supporters in Congress] want better quality in healthcare for better pay.”
This year’s election will have a major impact on this decision: “A change in administration will definitely factor in,” warns Dr. Torcson. “The 2008 Medicare Physician Payment Update seemed to divide along party lines. Republicans were somewhat supportive, and Democrats didn’t seem to support it. It’s not quite that simple, but that was a general pattern.”
The best advice for physicians invested or interested in investing in PQRI is to keep an eye on the November election results and the Senate Finance Committee to find out what 2009 and beyond will look like for PQRI or other CMS pay-for-reporting initiatives.
Too Late to Participate?
Although the PQRI began Jan. 1, there is no enrollment process; physicians can start reporting any time during the year. However, participants reporting on three measures report in at least 80% of the instances in which those measures are reportable—that means all year—in order to qualify for a bonus. If you begin reporting this far into the year, you’re not likely to reach that threshold and earn your bonus.
“Starting late in the year could affect reaching that threshold, but it’s never too late to start the practice and process of reporting,” says Dr. Torcson. “You can still make that commitment to performance reporting. Even if you don’t get the 1.5% bonus, you get the benefit of getting started in the important practice of performance reporting.”
Read more about the PQRI on SHM’s Web site (www.hospitalmedicine.org). TH
Jane Jerrard has written for The Hospitalist since 2005.
The current pay-for-reporting program from the Centers for Medicare and Medicaid (CMS) seems tailor-made for hospitalists. Here’s a look at the voluntary Physician Quality Reporting Initiative (PQRI) program, and why and how hospitalists are—and are not—participating.
CMS has revised the reporting program that began as a six-month trial in 2007. The current PQRI runs the full calendar year for 2008 and includes 119 quality measures—11 of which hospitalists can report on. Detailed specifications for the measures are available on the CMS Web site at www.cms.hhs.gov.
The earnings in this pay-for-reporting program remain the same as 2007: Physicians who successfully report on measures can earn a bonus payment equal to 1.5% of their total Medicare-allowed charges. Some hospitalists have collected their bonus for participating in the 2007 trial; it’s likely more will participate this year.
CMS has yet to release data on participation in the 2007 PQRI trial or this year’s initiative. However, SHM has urged hospitalists to participate, and many are. During a national, SHM-sponsored conference call with CMS in summer 2007, approximately 20% of the 160 hospitalists participating in the call responded to a follow-up survey. Almost half of all respondents indicated they planned to participate in PQRI reporting.
“That percentage comes from a select group of hospitalists who were highly interested in the PQRI,” points out Patrick J. Torcson, MD, MMM, FACP, director of hospital medicine at St. Tammany Parish Hospital in Covington, La.
Unlike many specialists, hospitalists are finding reporting to be a straightforward process. “For hospitalists, PQRI reporting on specific measures harmonizes nicely with workflow,” says Dr. Torcson. “Most applicable measures take place during admission or discharge. Documentation and reporting for PQRI can take place during these times.”
Report on Reporting
At St. Tammany, Dr. Torcson’s eight-hospitalist team is participating in PQRI. Although you need only to report on three measures to qualify for a bonus payment from the program, “we’re actually reporting on the full list of [hospitalist-applicable] measures,” Dr. Torcson says. It’s up to each St. Tammany hospitalist to remember to report on the 11 measures.
“Support for [reporting] really comes down to physician memory,” says Dr. Torcson. “Long term, this is going to have to be part of an electronic system, with decision support and billing capability from an electronic health record.”
In spite of the added step of PQRI reporting, Dr. Torcson says, “we’ve had an enthusiastic response from our hospitalists.” The payoff for the hospital medicine program and the hospital is yet to be seen. “You hope that PQRI performance reporting will result in improved quality of care,” henotes.
But many physicians—including hospitalists—are not participating in PQRI.
“It comes down to different practice models,” explains Dr. Torcson. “But for many physicians, a major reason not to participate is that they’re taking a wait-and-see approach. They’re waiting to see if this is just the latest flavor of the month, and think it’s not worth investing time and effort until it proves otherwise.”
Gregory B. Seymann, MD, associate clinical professor, University of California, San Diego (UCSD) School of Medicine, Division of Hospital Medicine, is a member of SHM’s Public Policy Committee and says he was disappointed his group is unable to participate in PQRI.
“I work for UCSD, where our hospitalist group is one of many, many subspecialty groups that work out of our hospital,” he explains “We do a lot of QI work, and we were certainly interested in participating in PQRI.” However, the hospital uses an electronic billing system incompatible with reporting on the measures. The software could be upgraded for about $15,000, says Dr. Seymann, but hospital administration sees no return on the investment.
“The cost wouldn’t match the increase in revenues because besides hospital medicine, there aren’t a lot of other subspecialties that would be interested in participating,” explains Dr. Seymann. “As much as I wanted our group to participate, I can’t fully fault UCSD on this decision on business grounds. They want to see some stability in [the decision to continue PQRI] before they invest.”
In the meantime, the orthopedics group at UCSD has invested in reporting. They are tracking PQRI measures on paper and reporting to CMS, and they’ll ultimately be able to show the administration whether the bonus per physician might add up to the cost of the necessary billing-system upgrade.
Beyond 2008
Everyone involved—not just UCSD—is asking: Is PQRI here to stay? That decision rests with federal lawmakers. At the end of this year, Congress must vote on whether to extend the program—and no one can guarantee whether that will happen.
“The chairs of the Senate Finance Committee have been tremendously supportive of the PQRI,” says Dr. Torcson. “There is a lot of political will behind this right now. [PQRI supporters in Congress] want better quality in healthcare for better pay.”
This year’s election will have a major impact on this decision: “A change in administration will definitely factor in,” warns Dr. Torcson. “The 2008 Medicare Physician Payment Update seemed to divide along party lines. Republicans were somewhat supportive, and Democrats didn’t seem to support it. It’s not quite that simple, but that was a general pattern.”
The best advice for physicians invested or interested in investing in PQRI is to keep an eye on the November election results and the Senate Finance Committee to find out what 2009 and beyond will look like for PQRI or other CMS pay-for-reporting initiatives.
Too Late to Participate?
Although the PQRI began Jan. 1, there is no enrollment process; physicians can start reporting any time during the year. However, participants reporting on three measures report in at least 80% of the instances in which those measures are reportable—that means all year—in order to qualify for a bonus. If you begin reporting this far into the year, you’re not likely to reach that threshold and earn your bonus.
“Starting late in the year could affect reaching that threshold, but it’s never too late to start the practice and process of reporting,” says Dr. Torcson. “You can still make that commitment to performance reporting. Even if you don’t get the 1.5% bonus, you get the benefit of getting started in the important practice of performance reporting.”
Read more about the PQRI on SHM’s Web site (www.hospitalmedicine.org). TH
Jane Jerrard has written for The Hospitalist since 2005.
The current pay-for-reporting program from the Centers for Medicare and Medicaid (CMS) seems tailor-made for hospitalists. Here’s a look at the voluntary Physician Quality Reporting Initiative (PQRI) program, and why and how hospitalists are—and are not—participating.
CMS has revised the reporting program that began as a six-month trial in 2007. The current PQRI runs the full calendar year for 2008 and includes 119 quality measures—11 of which hospitalists can report on. Detailed specifications for the measures are available on the CMS Web site at www.cms.hhs.gov.
The earnings in this pay-for-reporting program remain the same as 2007: Physicians who successfully report on measures can earn a bonus payment equal to 1.5% of their total Medicare-allowed charges. Some hospitalists have collected their bonus for participating in the 2007 trial; it’s likely more will participate this year.
CMS has yet to release data on participation in the 2007 PQRI trial or this year’s initiative. However, SHM has urged hospitalists to participate, and many are. During a national, SHM-sponsored conference call with CMS in summer 2007, approximately 20% of the 160 hospitalists participating in the call responded to a follow-up survey. Almost half of all respondents indicated they planned to participate in PQRI reporting.
“That percentage comes from a select group of hospitalists who were highly interested in the PQRI,” points out Patrick J. Torcson, MD, MMM, FACP, director of hospital medicine at St. Tammany Parish Hospital in Covington, La.
Unlike many specialists, hospitalists are finding reporting to be a straightforward process. “For hospitalists, PQRI reporting on specific measures harmonizes nicely with workflow,” says Dr. Torcson. “Most applicable measures take place during admission or discharge. Documentation and reporting for PQRI can take place during these times.”
Report on Reporting
At St. Tammany, Dr. Torcson’s eight-hospitalist team is participating in PQRI. Although you need only to report on three measures to qualify for a bonus payment from the program, “we’re actually reporting on the full list of [hospitalist-applicable] measures,” Dr. Torcson says. It’s up to each St. Tammany hospitalist to remember to report on the 11 measures.
“Support for [reporting] really comes down to physician memory,” says Dr. Torcson. “Long term, this is going to have to be part of an electronic system, with decision support and billing capability from an electronic health record.”
In spite of the added step of PQRI reporting, Dr. Torcson says, “we’ve had an enthusiastic response from our hospitalists.” The payoff for the hospital medicine program and the hospital is yet to be seen. “You hope that PQRI performance reporting will result in improved quality of care,” henotes.
But many physicians—including hospitalists—are not participating in PQRI.
“It comes down to different practice models,” explains Dr. Torcson. “But for many physicians, a major reason not to participate is that they’re taking a wait-and-see approach. They’re waiting to see if this is just the latest flavor of the month, and think it’s not worth investing time and effort until it proves otherwise.”
Gregory B. Seymann, MD, associate clinical professor, University of California, San Diego (UCSD) School of Medicine, Division of Hospital Medicine, is a member of SHM’s Public Policy Committee and says he was disappointed his group is unable to participate in PQRI.
“I work for UCSD, where our hospitalist group is one of many, many subspecialty groups that work out of our hospital,” he explains “We do a lot of QI work, and we were certainly interested in participating in PQRI.” However, the hospital uses an electronic billing system incompatible with reporting on the measures. The software could be upgraded for about $15,000, says Dr. Seymann, but hospital administration sees no return on the investment.
“The cost wouldn’t match the increase in revenues because besides hospital medicine, there aren’t a lot of other subspecialties that would be interested in participating,” explains Dr. Seymann. “As much as I wanted our group to participate, I can’t fully fault UCSD on this decision on business grounds. They want to see some stability in [the decision to continue PQRI] before they invest.”
In the meantime, the orthopedics group at UCSD has invested in reporting. They are tracking PQRI measures on paper and reporting to CMS, and they’ll ultimately be able to show the administration whether the bonus per physician might add up to the cost of the necessary billing-system upgrade.
Beyond 2008
Everyone involved—not just UCSD—is asking: Is PQRI here to stay? That decision rests with federal lawmakers. At the end of this year, Congress must vote on whether to extend the program—and no one can guarantee whether that will happen.
“The chairs of the Senate Finance Committee have been tremendously supportive of the PQRI,” says Dr. Torcson. “There is a lot of political will behind this right now. [PQRI supporters in Congress] want better quality in healthcare for better pay.”
This year’s election will have a major impact on this decision: “A change in administration will definitely factor in,” warns Dr. Torcson. “The 2008 Medicare Physician Payment Update seemed to divide along party lines. Republicans were somewhat supportive, and Democrats didn’t seem to support it. It’s not quite that simple, but that was a general pattern.”
The best advice for physicians invested or interested in investing in PQRI is to keep an eye on the November election results and the Senate Finance Committee to find out what 2009 and beyond will look like for PQRI or other CMS pay-for-reporting initiatives.
Too Late to Participate?
Although the PQRI began Jan. 1, there is no enrollment process; physicians can start reporting any time during the year. However, participants reporting on three measures report in at least 80% of the instances in which those measures are reportable—that means all year—in order to qualify for a bonus. If you begin reporting this far into the year, you’re not likely to reach that threshold and earn your bonus.
“Starting late in the year could affect reaching that threshold, but it’s never too late to start the practice and process of reporting,” says Dr. Torcson. “You can still make that commitment to performance reporting. Even if you don’t get the 1.5% bonus, you get the benefit of getting started in the important practice of performance reporting.”
Read more about the PQRI on SHM’s Web site (www.hospitalmedicine.org). TH
Jane Jerrard has written for The Hospitalist since 2005.
Speak Up
By putting a little time and effort into your presentation skills, you can become more persuasive and effective in your day-to-day job—and even advance your career and reputation.
For hospitalists, with their often-heavy committee load and frequent formal or informal teaching conversations, addressing groups is part of the job.
“At the end of the day, hospitalists are advocates—whether for quality improvement or patient-care issues,” says Jeffrey Wiese, MD, FACP, associate professor of medicine at Tulane University Health Sciences Center in New Orleans, associate chairman of medicine, director of the Tulane Internal Medicine Residency Program, and associate director of student programs, internal medicine. “And most of their advocacy efforts [are] going to be person-to-person, verbal discussions, where their passion and conviction can come through.”
Even if you’re never asked to present at a national meeting, you are likely to address a lot of committees, teams, and task forces in your career.
“It’s important to realize that people’s time is valuable in committee meetings,” stresses Dr. Wiese. “You have to be able to speak clearly, concisely, and to the point to make your case effectively.”
Learn by Listening
If you haven’t had much experience addressing groups or you feel your presentation skills are lacking, there are simple steps to become comfortable—even accomplished—at speaking.
“Most effective speakers are partly born but mostly made,” says Robert Wachter, MD, co-founder of SHM, frequent keynote speaker and professor and associate chairman of the Department of Medicine at the University of California, San Francisco.
Becoming an effective speaker may require formal training, perhaps from a course or a book. But one step every aspiring speaker can easily take is to listen to other speakers—a lot of them.
While working on his own presentation skills, Dr. Wachter says: “I learned to be a shameless mimic and thief. Even now, when I hear a good lecture, I always ask myself what that person did really well, and can I do that, too. And when I hear a crummy speaker, I wonder what I would tell them to them improve.”
Dr. Wiese does the same thing. “My strategy is to learn from every talk I sit in on,” he says. “Watch how the speaker is performing—not just at medical meetings, but also on TV. In this election year there are a lot of opportunities to listen to speeches. Note good speakers’ cadence, pitch and tone, and borrow from them.”
Simple Secrets
Effective speaking is built on some basic tenets. “There are fundamental skills that most speakers don’t use—you’d be surprised how basic these skills are,” says Dr. Wiese. These basics include:
Practice makes perfect: No matter how confident you are of your material, practice. Whether you’ll teach, speak to a quality-improvement committee or address a national group, make an outline and run through your speech. “There’s no talk I give without at least sitting down an hour beforehand to think through what I’m going to say,” says Dr. Wiese.
Give it all you’ve got: “When you’re asked to address a group, you have to convince yourself that this is the most important talk you’ve ever given,” stresses Dr. Wiese. “Your belief in this will give you the passion and commitment to your topic that comes out in how you speak.”
Start strong: Getting your audience’s interest and attention immediately is crucial.
“Engaging the audience successfully in the first one to three minutes is unbelievably important because unless you get them to care enough to listen at the outset, you’ve lost them for the rest of the talk,” he says. He believes only about one in 100 speakers do this well. “I assume the audience is not really with me and that I need to actively engage them—and I make sure they know enough to care about the topic. I start with the reasonable assumption that I know more and care more about my topic than they do. Make sure you give them enough background to get them started.”
Fledgling speakers can try capturing their audience’s attention by starting with a joke, story, dramatic anecdote, or shocking data. Starting your presentation with a bang, says Dr. Wachter, “is a learnable skill, and it’s a lot easier when you’re addressing a small group of people you know.”
Spice up dry information: If you’re stuck with a topic you fear is too boring to engage, find a “hook” to draw the audience in. Dr. Wachter suggests, “When you explain facts, use analogy and metaphors, and use graphics only when appropriate,” he suggests.
Find your voice: A tricky thing for new speakers is controlling their voice and using it to maintain interest. Avoid using a monotone—a common effect of reading from notes or slides.
“It’s important to work on your cadence and on the pitch and tone of your voice,” advises Dr. Wiese. “I think speaking is similar to music. Music has rest notes for a reason: to augment what you just said and to set up what you’re about to say. Try replacing the “ums” and “uhs” you use while you’re thinking about what to say next with silence. The audience will be riveted.”
Go easy on the PowerPoint: Don’t rely on your slides or flipchart to influence or engage your audience. Make eye contact with individuals and in a small group; touch a shoulder or two. “The truth is that most people use PowerPoint slides because they didn’t practice their talk,” says Dr. Wiese. “Turn away from your slides and talk person to person—you’ll be much more compelling.”
Speaking Opportunities
For an ambitious hospitalist, opportunities are abundant. “Find the residency director at the nearest program and tell them you’d like to give a conference for free,” Dr. Wiese recommends. “I guarantee this will get you 20 or 30 offers.”
He says national and regional organizations are great opportunities to get involved. “All it really takes is to attend the meetings, find the people doing the talks and tell them that you want an opportunity to hone your speaking skills,” he notes.
If you’re convinced that practicing your speaking skills will help you influence committees, enhance your reputation and improve your career possibilities, then take Dr. Wiese’s advice and get ready to launch your speaking career. TH
Jane Jerrard writes “Public Policy” for The Hospitalist.
By putting a little time and effort into your presentation skills, you can become more persuasive and effective in your day-to-day job—and even advance your career and reputation.
For hospitalists, with their often-heavy committee load and frequent formal or informal teaching conversations, addressing groups is part of the job.
“At the end of the day, hospitalists are advocates—whether for quality improvement or patient-care issues,” says Jeffrey Wiese, MD, FACP, associate professor of medicine at Tulane University Health Sciences Center in New Orleans, associate chairman of medicine, director of the Tulane Internal Medicine Residency Program, and associate director of student programs, internal medicine. “And most of their advocacy efforts [are] going to be person-to-person, verbal discussions, where their passion and conviction can come through.”
Even if you’re never asked to present at a national meeting, you are likely to address a lot of committees, teams, and task forces in your career.
“It’s important to realize that people’s time is valuable in committee meetings,” stresses Dr. Wiese. “You have to be able to speak clearly, concisely, and to the point to make your case effectively.”
Learn by Listening
If you haven’t had much experience addressing groups or you feel your presentation skills are lacking, there are simple steps to become comfortable—even accomplished—at speaking.
“Most effective speakers are partly born but mostly made,” says Robert Wachter, MD, co-founder of SHM, frequent keynote speaker and professor and associate chairman of the Department of Medicine at the University of California, San Francisco.
Becoming an effective speaker may require formal training, perhaps from a course or a book. But one step every aspiring speaker can easily take is to listen to other speakers—a lot of them.
While working on his own presentation skills, Dr. Wachter says: “I learned to be a shameless mimic and thief. Even now, when I hear a good lecture, I always ask myself what that person did really well, and can I do that, too. And when I hear a crummy speaker, I wonder what I would tell them to them improve.”
Dr. Wiese does the same thing. “My strategy is to learn from every talk I sit in on,” he says. “Watch how the speaker is performing—not just at medical meetings, but also on TV. In this election year there are a lot of opportunities to listen to speeches. Note good speakers’ cadence, pitch and tone, and borrow from them.”
Simple Secrets
Effective speaking is built on some basic tenets. “There are fundamental skills that most speakers don’t use—you’d be surprised how basic these skills are,” says Dr. Wiese. These basics include:
Practice makes perfect: No matter how confident you are of your material, practice. Whether you’ll teach, speak to a quality-improvement committee or address a national group, make an outline and run through your speech. “There’s no talk I give without at least sitting down an hour beforehand to think through what I’m going to say,” says Dr. Wiese.
Give it all you’ve got: “When you’re asked to address a group, you have to convince yourself that this is the most important talk you’ve ever given,” stresses Dr. Wiese. “Your belief in this will give you the passion and commitment to your topic that comes out in how you speak.”
Start strong: Getting your audience’s interest and attention immediately is crucial.
“Engaging the audience successfully in the first one to three minutes is unbelievably important because unless you get them to care enough to listen at the outset, you’ve lost them for the rest of the talk,” he says. He believes only about one in 100 speakers do this well. “I assume the audience is not really with me and that I need to actively engage them—and I make sure they know enough to care about the topic. I start with the reasonable assumption that I know more and care more about my topic than they do. Make sure you give them enough background to get them started.”
Fledgling speakers can try capturing their audience’s attention by starting with a joke, story, dramatic anecdote, or shocking data. Starting your presentation with a bang, says Dr. Wachter, “is a learnable skill, and it’s a lot easier when you’re addressing a small group of people you know.”
Spice up dry information: If you’re stuck with a topic you fear is too boring to engage, find a “hook” to draw the audience in. Dr. Wachter suggests, “When you explain facts, use analogy and metaphors, and use graphics only when appropriate,” he suggests.
Find your voice: A tricky thing for new speakers is controlling their voice and using it to maintain interest. Avoid using a monotone—a common effect of reading from notes or slides.
“It’s important to work on your cadence and on the pitch and tone of your voice,” advises Dr. Wiese. “I think speaking is similar to music. Music has rest notes for a reason: to augment what you just said and to set up what you’re about to say. Try replacing the “ums” and “uhs” you use while you’re thinking about what to say next with silence. The audience will be riveted.”
Go easy on the PowerPoint: Don’t rely on your slides or flipchart to influence or engage your audience. Make eye contact with individuals and in a small group; touch a shoulder or two. “The truth is that most people use PowerPoint slides because they didn’t practice their talk,” says Dr. Wiese. “Turn away from your slides and talk person to person—you’ll be much more compelling.”
Speaking Opportunities
For an ambitious hospitalist, opportunities are abundant. “Find the residency director at the nearest program and tell them you’d like to give a conference for free,” Dr. Wiese recommends. “I guarantee this will get you 20 or 30 offers.”
He says national and regional organizations are great opportunities to get involved. “All it really takes is to attend the meetings, find the people doing the talks and tell them that you want an opportunity to hone your speaking skills,” he notes.
If you’re convinced that practicing your speaking skills will help you influence committees, enhance your reputation and improve your career possibilities, then take Dr. Wiese’s advice and get ready to launch your speaking career. TH
Jane Jerrard writes “Public Policy” for The Hospitalist.
By putting a little time and effort into your presentation skills, you can become more persuasive and effective in your day-to-day job—and even advance your career and reputation.
For hospitalists, with their often-heavy committee load and frequent formal or informal teaching conversations, addressing groups is part of the job.
“At the end of the day, hospitalists are advocates—whether for quality improvement or patient-care issues,” says Jeffrey Wiese, MD, FACP, associate professor of medicine at Tulane University Health Sciences Center in New Orleans, associate chairman of medicine, director of the Tulane Internal Medicine Residency Program, and associate director of student programs, internal medicine. “And most of their advocacy efforts [are] going to be person-to-person, verbal discussions, where their passion and conviction can come through.”
Even if you’re never asked to present at a national meeting, you are likely to address a lot of committees, teams, and task forces in your career.
“It’s important to realize that people’s time is valuable in committee meetings,” stresses Dr. Wiese. “You have to be able to speak clearly, concisely, and to the point to make your case effectively.”
Learn by Listening
If you haven’t had much experience addressing groups or you feel your presentation skills are lacking, there are simple steps to become comfortable—even accomplished—at speaking.
“Most effective speakers are partly born but mostly made,” says Robert Wachter, MD, co-founder of SHM, frequent keynote speaker and professor and associate chairman of the Department of Medicine at the University of California, San Francisco.
Becoming an effective speaker may require formal training, perhaps from a course or a book. But one step every aspiring speaker can easily take is to listen to other speakers—a lot of them.
While working on his own presentation skills, Dr. Wachter says: “I learned to be a shameless mimic and thief. Even now, when I hear a good lecture, I always ask myself what that person did really well, and can I do that, too. And when I hear a crummy speaker, I wonder what I would tell them to them improve.”
Dr. Wiese does the same thing. “My strategy is to learn from every talk I sit in on,” he says. “Watch how the speaker is performing—not just at medical meetings, but also on TV. In this election year there are a lot of opportunities to listen to speeches. Note good speakers’ cadence, pitch and tone, and borrow from them.”
Simple Secrets
Effective speaking is built on some basic tenets. “There are fundamental skills that most speakers don’t use—you’d be surprised how basic these skills are,” says Dr. Wiese. These basics include:
Practice makes perfect: No matter how confident you are of your material, practice. Whether you’ll teach, speak to a quality-improvement committee or address a national group, make an outline and run through your speech. “There’s no talk I give without at least sitting down an hour beforehand to think through what I’m going to say,” says Dr. Wiese.
Give it all you’ve got: “When you’re asked to address a group, you have to convince yourself that this is the most important talk you’ve ever given,” stresses Dr. Wiese. “Your belief in this will give you the passion and commitment to your topic that comes out in how you speak.”
Start strong: Getting your audience’s interest and attention immediately is crucial.
“Engaging the audience successfully in the first one to three minutes is unbelievably important because unless you get them to care enough to listen at the outset, you’ve lost them for the rest of the talk,” he says. He believes only about one in 100 speakers do this well. “I assume the audience is not really with me and that I need to actively engage them—and I make sure they know enough to care about the topic. I start with the reasonable assumption that I know more and care more about my topic than they do. Make sure you give them enough background to get them started.”
Fledgling speakers can try capturing their audience’s attention by starting with a joke, story, dramatic anecdote, or shocking data. Starting your presentation with a bang, says Dr. Wachter, “is a learnable skill, and it’s a lot easier when you’re addressing a small group of people you know.”
Spice up dry information: If you’re stuck with a topic you fear is too boring to engage, find a “hook” to draw the audience in. Dr. Wachter suggests, “When you explain facts, use analogy and metaphors, and use graphics only when appropriate,” he suggests.
Find your voice: A tricky thing for new speakers is controlling their voice and using it to maintain interest. Avoid using a monotone—a common effect of reading from notes or slides.
“It’s important to work on your cadence and on the pitch and tone of your voice,” advises Dr. Wiese. “I think speaking is similar to music. Music has rest notes for a reason: to augment what you just said and to set up what you’re about to say. Try replacing the “ums” and “uhs” you use while you’re thinking about what to say next with silence. The audience will be riveted.”
Go easy on the PowerPoint: Don’t rely on your slides or flipchart to influence or engage your audience. Make eye contact with individuals and in a small group; touch a shoulder or two. “The truth is that most people use PowerPoint slides because they didn’t practice their talk,” says Dr. Wiese. “Turn away from your slides and talk person to person—you’ll be much more compelling.”
Speaking Opportunities
For an ambitious hospitalist, opportunities are abundant. “Find the residency director at the nearest program and tell them you’d like to give a conference for free,” Dr. Wiese recommends. “I guarantee this will get you 20 or 30 offers.”
He says national and regional organizations are great opportunities to get involved. “All it really takes is to attend the meetings, find the people doing the talks and tell them that you want an opportunity to hone your speaking skills,” he notes.
If you’re convinced that practicing your speaking skills will help you influence committees, enhance your reputation and improve your career possibilities, then take Dr. Wiese’s advice and get ready to launch your speaking career. TH
Jane Jerrard writes “Public Policy” for The Hospitalist.
Drug Misuse Varies
Elderly inpatients’ risk of receiving potentially inappropriate medication (PIM) varies widely depending on where in the country they’re hospitalized and the specialty of their attending physicians, according to a study in the March-April edition of the Journal of Hospital Medicine.
Hospitalists may be encouraged by the fact that they, along with geriatricians, internists, and family physicians, were less likely than cardiologists to prescribe PIMs. Still, the major take-home message of the study is to “examine your individual practice and think about whether it’s appropriate to prescribe these medications,” says lead author Michael Rothberg, MD, assistant professor of medicine at Tufts University School of Medicine in Boston.
PIM use was highest in hospitals in the South. There, 55% of elderly patients received at least one PIM, compared with 34% of patients in Northeastern hospitals, where PIM use was lowest. The exact reason for this discrepancy is not known, but Dr. Rothberg hypothesizes that “we tend to prescribe like people in our hospital and like people in our region.” In other words, “it has to do with learning from the people around us.”
Most interesting to him is the wide variation in prescribing practices among individual doctors—even within the same specialty. “The decision to prescribe a drug is based on the individual provider and has to do with how you as a doctor feel about these drugs,” he explains. Although nearly half of all of the patients had received at least one PIM, there were seven hospitals in which those drugs never were prescribed. Somehow, “they found a way to care for people without [those medications],” he points out.
PIM use has been examined among elderly outpatients and nursing home residents, but only a handful of small studies have looked at the problem in hospital inpatients, says Dr. Rothberg. He and his coauthors used data from hospitals across the United States participating in Perspective, a database developed by Charlotte, N.C.-based Premier to measure quality and healthcare utilization.
The survey included patients 65 years or older admitted between Sept. 1, 2002, and June 30, 2005. Their principal diagnoses were acute myocardial infarction, chronic obstructive pulmonary disease, chest pain, community acquired pneumonia, congestive heart failure, ischemic stroke, or urinary tract infection. Surgical patients were excluded. Using the 2002 update of the Beers criteria for PIM use in older adults, the authors identified the total number of PIMs administered to each patient during his or her hospital stay. They further classified each PIM as high- or low-severity, based on the expert consensus expressed in the 1997 update of the Beers criteria.
Data were available on 493,971 patients from 384 hospitals. Of those individuals, 49% received at least one PIM, and 6% received three or more. Thirty-eight percent of patients received at least one PIM with a high severity rating.
The three agents most likely to be prescribed were promethazine, diphenhydramine, and propoxyphene—probably because these drugs treat the problems most commonly encountered in hospitals, such as allergies, sleep problems, nausea, and pain, Dr. Rothberg says.
Hospital region emerged as the most important predictor of PIM use. Compared with patients in the Midwest, patients in the South had an odds ratio of 1.63 of receiving a high-severity PIM. The odds ratio for patients in the West was 1.43. Patients in the Northeast had an odds ratio of 0.85.
The median rate of prescribing high-severity PIMs was lowest among geriatricians, at 24%. Rates among hospitalists, internists, and family physicians were 33% to 36%. Cardiologists had the highest rate: 48% prescribed at least one high-severity PIM.
Interestingly, older patient age also was associated with a lower risk of PIM use. Of patients 85 or older, 42% received at least one PIM, compared with 53% of patients age 65 to 74 (p<0.0001). This suggests that “doctors are aware that the older patients are more frail and vulnerable” and take extra care to avoid prescribing PIMs to people in that age range, Dr. Rothberg says. A diagnosis of stroke or chronic obstructive pulmonary disease also was associated with a lower risk of receiving a PIM—further evidence that “doctors were, to some extent, taking patient factors into account” when prescribing medication.
PIM use among inpatients, as reported in this study, far exceeds the rates published for elders dwelling in the community or in nursing homes, writes Daniel S. Budnitz, MD, MPH, in an editorial accompanying the study.
The wide variation in prescribing practices means each facility must monitor its use of PIMs, just as individual hospitals monitor antibiotic use and resistance, advises Dr. Budnitz, a medical officer in the Division of Healthcare Quality Promotion at the Centers for Disease Control and Prevention. He also points out that the evidence that PIMs cause clinically significant adverse events is “weak and based largely on observational studies with inconsistent results.” The drugs in the Beers criteria are “potentially” inappropriate, he says, but some centers have recategorized them as “ ‘always avoid’ medications, ‘rarely acceptable’ medications, and medications which, indeed, have ‘some indications’ for use in older adults.” Thus, some variation among hospitals may be acceptable.
Rather than concentrate on the Beers criteria, hospitalists should focus “on identifying and mitigating the most common and most severe adverse drug events occurring in their hospitals,” such as bleeding from anticoagulants, hypoglycemic events from insulin, and oversedation from opioid analgesics, Dr. Budnitz points out. TH
Norra MacReady is a medical writer based in California.
Elderly inpatients’ risk of receiving potentially inappropriate medication (PIM) varies widely depending on where in the country they’re hospitalized and the specialty of their attending physicians, according to a study in the March-April edition of the Journal of Hospital Medicine.
Hospitalists may be encouraged by the fact that they, along with geriatricians, internists, and family physicians, were less likely than cardiologists to prescribe PIMs. Still, the major take-home message of the study is to “examine your individual practice and think about whether it’s appropriate to prescribe these medications,” says lead author Michael Rothberg, MD, assistant professor of medicine at Tufts University School of Medicine in Boston.
PIM use was highest in hospitals in the South. There, 55% of elderly patients received at least one PIM, compared with 34% of patients in Northeastern hospitals, where PIM use was lowest. The exact reason for this discrepancy is not known, but Dr. Rothberg hypothesizes that “we tend to prescribe like people in our hospital and like people in our region.” In other words, “it has to do with learning from the people around us.”
Most interesting to him is the wide variation in prescribing practices among individual doctors—even within the same specialty. “The decision to prescribe a drug is based on the individual provider and has to do with how you as a doctor feel about these drugs,” he explains. Although nearly half of all of the patients had received at least one PIM, there were seven hospitals in which those drugs never were prescribed. Somehow, “they found a way to care for people without [those medications],” he points out.
PIM use has been examined among elderly outpatients and nursing home residents, but only a handful of small studies have looked at the problem in hospital inpatients, says Dr. Rothberg. He and his coauthors used data from hospitals across the United States participating in Perspective, a database developed by Charlotte, N.C.-based Premier to measure quality and healthcare utilization.
The survey included patients 65 years or older admitted between Sept. 1, 2002, and June 30, 2005. Their principal diagnoses were acute myocardial infarction, chronic obstructive pulmonary disease, chest pain, community acquired pneumonia, congestive heart failure, ischemic stroke, or urinary tract infection. Surgical patients were excluded. Using the 2002 update of the Beers criteria for PIM use in older adults, the authors identified the total number of PIMs administered to each patient during his or her hospital stay. They further classified each PIM as high- or low-severity, based on the expert consensus expressed in the 1997 update of the Beers criteria.
Data were available on 493,971 patients from 384 hospitals. Of those individuals, 49% received at least one PIM, and 6% received three or more. Thirty-eight percent of patients received at least one PIM with a high severity rating.
The three agents most likely to be prescribed were promethazine, diphenhydramine, and propoxyphene—probably because these drugs treat the problems most commonly encountered in hospitals, such as allergies, sleep problems, nausea, and pain, Dr. Rothberg says.
Hospital region emerged as the most important predictor of PIM use. Compared with patients in the Midwest, patients in the South had an odds ratio of 1.63 of receiving a high-severity PIM. The odds ratio for patients in the West was 1.43. Patients in the Northeast had an odds ratio of 0.85.
The median rate of prescribing high-severity PIMs was lowest among geriatricians, at 24%. Rates among hospitalists, internists, and family physicians were 33% to 36%. Cardiologists had the highest rate: 48% prescribed at least one high-severity PIM.
Interestingly, older patient age also was associated with a lower risk of PIM use. Of patients 85 or older, 42% received at least one PIM, compared with 53% of patients age 65 to 74 (p<0.0001). This suggests that “doctors are aware that the older patients are more frail and vulnerable” and take extra care to avoid prescribing PIMs to people in that age range, Dr. Rothberg says. A diagnosis of stroke or chronic obstructive pulmonary disease also was associated with a lower risk of receiving a PIM—further evidence that “doctors were, to some extent, taking patient factors into account” when prescribing medication.
PIM use among inpatients, as reported in this study, far exceeds the rates published for elders dwelling in the community or in nursing homes, writes Daniel S. Budnitz, MD, MPH, in an editorial accompanying the study.
The wide variation in prescribing practices means each facility must monitor its use of PIMs, just as individual hospitals monitor antibiotic use and resistance, advises Dr. Budnitz, a medical officer in the Division of Healthcare Quality Promotion at the Centers for Disease Control and Prevention. He also points out that the evidence that PIMs cause clinically significant adverse events is “weak and based largely on observational studies with inconsistent results.” The drugs in the Beers criteria are “potentially” inappropriate, he says, but some centers have recategorized them as “ ‘always avoid’ medications, ‘rarely acceptable’ medications, and medications which, indeed, have ‘some indications’ for use in older adults.” Thus, some variation among hospitals may be acceptable.
Rather than concentrate on the Beers criteria, hospitalists should focus “on identifying and mitigating the most common and most severe adverse drug events occurring in their hospitals,” such as bleeding from anticoagulants, hypoglycemic events from insulin, and oversedation from opioid analgesics, Dr. Budnitz points out. TH
Norra MacReady is a medical writer based in California.
Elderly inpatients’ risk of receiving potentially inappropriate medication (PIM) varies widely depending on where in the country they’re hospitalized and the specialty of their attending physicians, according to a study in the March-April edition of the Journal of Hospital Medicine.
Hospitalists may be encouraged by the fact that they, along with geriatricians, internists, and family physicians, were less likely than cardiologists to prescribe PIMs. Still, the major take-home message of the study is to “examine your individual practice and think about whether it’s appropriate to prescribe these medications,” says lead author Michael Rothberg, MD, assistant professor of medicine at Tufts University School of Medicine in Boston.
PIM use was highest in hospitals in the South. There, 55% of elderly patients received at least one PIM, compared with 34% of patients in Northeastern hospitals, where PIM use was lowest. The exact reason for this discrepancy is not known, but Dr. Rothberg hypothesizes that “we tend to prescribe like people in our hospital and like people in our region.” In other words, “it has to do with learning from the people around us.”
Most interesting to him is the wide variation in prescribing practices among individual doctors—even within the same specialty. “The decision to prescribe a drug is based on the individual provider and has to do with how you as a doctor feel about these drugs,” he explains. Although nearly half of all of the patients had received at least one PIM, there were seven hospitals in which those drugs never were prescribed. Somehow, “they found a way to care for people without [those medications],” he points out.
PIM use has been examined among elderly outpatients and nursing home residents, but only a handful of small studies have looked at the problem in hospital inpatients, says Dr. Rothberg. He and his coauthors used data from hospitals across the United States participating in Perspective, a database developed by Charlotte, N.C.-based Premier to measure quality and healthcare utilization.
The survey included patients 65 years or older admitted between Sept. 1, 2002, and June 30, 2005. Their principal diagnoses were acute myocardial infarction, chronic obstructive pulmonary disease, chest pain, community acquired pneumonia, congestive heart failure, ischemic stroke, or urinary tract infection. Surgical patients were excluded. Using the 2002 update of the Beers criteria for PIM use in older adults, the authors identified the total number of PIMs administered to each patient during his or her hospital stay. They further classified each PIM as high- or low-severity, based on the expert consensus expressed in the 1997 update of the Beers criteria.
Data were available on 493,971 patients from 384 hospitals. Of those individuals, 49% received at least one PIM, and 6% received three or more. Thirty-eight percent of patients received at least one PIM with a high severity rating.
The three agents most likely to be prescribed were promethazine, diphenhydramine, and propoxyphene—probably because these drugs treat the problems most commonly encountered in hospitals, such as allergies, sleep problems, nausea, and pain, Dr. Rothberg says.
Hospital region emerged as the most important predictor of PIM use. Compared with patients in the Midwest, patients in the South had an odds ratio of 1.63 of receiving a high-severity PIM. The odds ratio for patients in the West was 1.43. Patients in the Northeast had an odds ratio of 0.85.
The median rate of prescribing high-severity PIMs was lowest among geriatricians, at 24%. Rates among hospitalists, internists, and family physicians were 33% to 36%. Cardiologists had the highest rate: 48% prescribed at least one high-severity PIM.
Interestingly, older patient age also was associated with a lower risk of PIM use. Of patients 85 or older, 42% received at least one PIM, compared with 53% of patients age 65 to 74 (p<0.0001). This suggests that “doctors are aware that the older patients are more frail and vulnerable” and take extra care to avoid prescribing PIMs to people in that age range, Dr. Rothberg says. A diagnosis of stroke or chronic obstructive pulmonary disease also was associated with a lower risk of receiving a PIM—further evidence that “doctors were, to some extent, taking patient factors into account” when prescribing medication.
PIM use among inpatients, as reported in this study, far exceeds the rates published for elders dwelling in the community or in nursing homes, writes Daniel S. Budnitz, MD, MPH, in an editorial accompanying the study.
The wide variation in prescribing practices means each facility must monitor its use of PIMs, just as individual hospitals monitor antibiotic use and resistance, advises Dr. Budnitz, a medical officer in the Division of Healthcare Quality Promotion at the Centers for Disease Control and Prevention. He also points out that the evidence that PIMs cause clinically significant adverse events is “weak and based largely on observational studies with inconsistent results.” The drugs in the Beers criteria are “potentially” inappropriate, he says, but some centers have recategorized them as “ ‘always avoid’ medications, ‘rarely acceptable’ medications, and medications which, indeed, have ‘some indications’ for use in older adults.” Thus, some variation among hospitals may be acceptable.
Rather than concentrate on the Beers criteria, hospitalists should focus “on identifying and mitigating the most common and most severe adverse drug events occurring in their hospitals,” such as bleeding from anticoagulants, hypoglycemic events from insulin, and oversedation from opioid analgesics, Dr. Budnitz points out. TH
Norra MacReady is a medical writer based in California.
Vital VTE Interventions
Venous thromboembolism (VTE) affects more than 2 million Americans every year.1 Pulmonary embolism (PE) is one of the most common preventable causes of in-hospital deaths in the United States. Clinical manifestations of PE may be the first indication the patient has a VTE, and fatal PEs occur in at least 75% of hospitalized medical patients. More than 300,000 patients die from PE each year—an estimated incidence of 10%. This makes VTE prevention a top patient-safety goal in hospitals.2,3
Thromboprophylaxis can be accomplished with unfractionated heparin (UFH), low-molecular-weight heparin (LMWH; e.g., enoxaparin, dalteparin, tinzaparin) or heparinoid, or a selective factor Xa inhibitor (e.g., fondaparinux).4 For long-term treatment, oral warfarin is often used. Doses and duration of prophylaxis and treatment regimens vary.
Current guidelines should be reviewed for specific recommendations. Two current guidelines are the American College of Chest Physicians (ACCP) Seventh Conference on the Prevention of VTE and the American Society of Clinical Oncology (ASCO) Guideline for VTE prophylaxis and treatment in oncology patients. Although guidelines are available, thromboprophylaxis continues to baffle many healthcare providers. There are many advantages to thromboprophylaxis including the prevention of significant morbidity, prevention of PE, decreases in resource consumption, and decreases in the long-term clinical and economic sequelae.
The ACCP notes that most surgical patients will require thromboprophylaxis. Contraindications need to be evaluated prior to antithrombotic/anticoagulant use. Additionally, all trauma patients with at least one VTE risk factor should receive thromboprophylaxis. Acutely ill patients hospitalized with congestive heart failure or severe respiratory distress or who are confined to bed and have one or more additional risk factors, should receive VTE prophylaxis. Additionally, most patients upon admission to an intensive-care unit should be assessed for VTE risk and receive thromboprophylaxis as required.
VTE is a major complication in up to 20% of cancer patients, with hospitalized oncology patients and those undergoing treatment at the highest risk. Some of the newer drug treatments used in these patients have higher VTE rates (e.g., bevacizumab, thalidomide, lenalidomide). These patients need to be carefully evaluated for VTE prophylaxis and closely monitored.5
Generally, in hospitalized patients with cancer, VTE prophylaxis should be considered with UFH, LMWH, or fondaparinux, in the absence of bleeding or other contraindications to anticoagulation. Relative contraindications to anticoagulation include (but are not limited to):
- Active uncontrolled bleeding;
- Active cerebrovascular hemorrhage;
- Dissecting or cerebral aneurysm;
- Bacterial endocarditis;
- Pericarditis;
- Active peptic or gastrointestinal ulceration;
- Severe uncontrolled or malignant hypertension;
- Severe head trauma;
- Pregnancy (warfarin contraindication);
- Heparin-induced thrombocytopenia (heparin, LMWH); and
- Epidural catheter placement.
These same contraindications can be applied to the non-oncology patient, as well.
An important aspect of VTE management is the “Clinical Practice Guideline from the American Academy of Family Physicians and the American College of Physicians on the Diagnosis of VTE from the Annals of Family Medicine.” Consult this for a review of diagnostic tests for VTE.
Thromboprophylaxis is a necessity in a number of at-risk hospitalized patients. Knowing which patients will benefit, and the contraindications for use, will improve patient outcomes. Consult current guidelines for diagnosis recommendations as well as agents of choice, dosing regimens, and therapy duration. TH
Michele B. Kaufman is registered pharmacist based in New York City.
References
- DVT: Assess Your Patients’ Risk, Take Preventive Measures. ASHP Foundation Discoveries, Summer 2007;19(1):1,5. Available at www.ashpfoundation.org/MainMenuCategories/AboutUs/Newsletter/DiscoveriesSummer2007.aspx. Last accessed Nov. 26, 2007.
- Geertz WH, Pineo Graham F, Heit JA et al. Prevention of venous thromboembolism: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:338-400.
- Wein L, Wein S, Haas SJ, et al. Pharmacological venous thromboembolism prophylaxis in hospitalized medical patients, a meta-analysis of randomized controlled trials. Arch Intern Med. 2007;167:1476-1486.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol. 2007;25(34): 5490–5505.
- Qaseem A, Snow V, Barry P for the Joint American Academy of Family Physicians/American College of Physicians Panel on Deep Vein Thrombosis/Pulmonary Embolism. Current diagnosis of venous thromboembolism in primary care: a clinical practice guideline from the American Academy of Family Physicians and the American College of Physicians. Ann Fam Med. 2007;5:57-62.
Venous thromboembolism (VTE) affects more than 2 million Americans every year.1 Pulmonary embolism (PE) is one of the most common preventable causes of in-hospital deaths in the United States. Clinical manifestations of PE may be the first indication the patient has a VTE, and fatal PEs occur in at least 75% of hospitalized medical patients. More than 300,000 patients die from PE each year—an estimated incidence of 10%. This makes VTE prevention a top patient-safety goal in hospitals.2,3
Thromboprophylaxis can be accomplished with unfractionated heparin (UFH), low-molecular-weight heparin (LMWH; e.g., enoxaparin, dalteparin, tinzaparin) or heparinoid, or a selective factor Xa inhibitor (e.g., fondaparinux).4 For long-term treatment, oral warfarin is often used. Doses and duration of prophylaxis and treatment regimens vary.
Current guidelines should be reviewed for specific recommendations. Two current guidelines are the American College of Chest Physicians (ACCP) Seventh Conference on the Prevention of VTE and the American Society of Clinical Oncology (ASCO) Guideline for VTE prophylaxis and treatment in oncology patients. Although guidelines are available, thromboprophylaxis continues to baffle many healthcare providers. There are many advantages to thromboprophylaxis including the prevention of significant morbidity, prevention of PE, decreases in resource consumption, and decreases in the long-term clinical and economic sequelae.
The ACCP notes that most surgical patients will require thromboprophylaxis. Contraindications need to be evaluated prior to antithrombotic/anticoagulant use. Additionally, all trauma patients with at least one VTE risk factor should receive thromboprophylaxis. Acutely ill patients hospitalized with congestive heart failure or severe respiratory distress or who are confined to bed and have one or more additional risk factors, should receive VTE prophylaxis. Additionally, most patients upon admission to an intensive-care unit should be assessed for VTE risk and receive thromboprophylaxis as required.
VTE is a major complication in up to 20% of cancer patients, with hospitalized oncology patients and those undergoing treatment at the highest risk. Some of the newer drug treatments used in these patients have higher VTE rates (e.g., bevacizumab, thalidomide, lenalidomide). These patients need to be carefully evaluated for VTE prophylaxis and closely monitored.5
Generally, in hospitalized patients with cancer, VTE prophylaxis should be considered with UFH, LMWH, or fondaparinux, in the absence of bleeding or other contraindications to anticoagulation. Relative contraindications to anticoagulation include (but are not limited to):
- Active uncontrolled bleeding;
- Active cerebrovascular hemorrhage;
- Dissecting or cerebral aneurysm;
- Bacterial endocarditis;
- Pericarditis;
- Active peptic or gastrointestinal ulceration;
- Severe uncontrolled or malignant hypertension;
- Severe head trauma;
- Pregnancy (warfarin contraindication);
- Heparin-induced thrombocytopenia (heparin, LMWH); and
- Epidural catheter placement.
These same contraindications can be applied to the non-oncology patient, as well.
An important aspect of VTE management is the “Clinical Practice Guideline from the American Academy of Family Physicians and the American College of Physicians on the Diagnosis of VTE from the Annals of Family Medicine.” Consult this for a review of diagnostic tests for VTE.
Thromboprophylaxis is a necessity in a number of at-risk hospitalized patients. Knowing which patients will benefit, and the contraindications for use, will improve patient outcomes. Consult current guidelines for diagnosis recommendations as well as agents of choice, dosing regimens, and therapy duration. TH
Michele B. Kaufman is registered pharmacist based in New York City.
References
- DVT: Assess Your Patients’ Risk, Take Preventive Measures. ASHP Foundation Discoveries, Summer 2007;19(1):1,5. Available at www.ashpfoundation.org/MainMenuCategories/AboutUs/Newsletter/DiscoveriesSummer2007.aspx. Last accessed Nov. 26, 2007.
- Geertz WH, Pineo Graham F, Heit JA et al. Prevention of venous thromboembolism: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:338-400.
- Wein L, Wein S, Haas SJ, et al. Pharmacological venous thromboembolism prophylaxis in hospitalized medical patients, a meta-analysis of randomized controlled trials. Arch Intern Med. 2007;167:1476-1486.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol. 2007;25(34): 5490–5505.
- Qaseem A, Snow V, Barry P for the Joint American Academy of Family Physicians/American College of Physicians Panel on Deep Vein Thrombosis/Pulmonary Embolism. Current diagnosis of venous thromboembolism in primary care: a clinical practice guideline from the American Academy of Family Physicians and the American College of Physicians. Ann Fam Med. 2007;5:57-62.
Venous thromboembolism (VTE) affects more than 2 million Americans every year.1 Pulmonary embolism (PE) is one of the most common preventable causes of in-hospital deaths in the United States. Clinical manifestations of PE may be the first indication the patient has a VTE, and fatal PEs occur in at least 75% of hospitalized medical patients. More than 300,000 patients die from PE each year—an estimated incidence of 10%. This makes VTE prevention a top patient-safety goal in hospitals.2,3
Thromboprophylaxis can be accomplished with unfractionated heparin (UFH), low-molecular-weight heparin (LMWH; e.g., enoxaparin, dalteparin, tinzaparin) or heparinoid, or a selective factor Xa inhibitor (e.g., fondaparinux).4 For long-term treatment, oral warfarin is often used. Doses and duration of prophylaxis and treatment regimens vary.
Current guidelines should be reviewed for specific recommendations. Two current guidelines are the American College of Chest Physicians (ACCP) Seventh Conference on the Prevention of VTE and the American Society of Clinical Oncology (ASCO) Guideline for VTE prophylaxis and treatment in oncology patients. Although guidelines are available, thromboprophylaxis continues to baffle many healthcare providers. There are many advantages to thromboprophylaxis including the prevention of significant morbidity, prevention of PE, decreases in resource consumption, and decreases in the long-term clinical and economic sequelae.
The ACCP notes that most surgical patients will require thromboprophylaxis. Contraindications need to be evaluated prior to antithrombotic/anticoagulant use. Additionally, all trauma patients with at least one VTE risk factor should receive thromboprophylaxis. Acutely ill patients hospitalized with congestive heart failure or severe respiratory distress or who are confined to bed and have one or more additional risk factors, should receive VTE prophylaxis. Additionally, most patients upon admission to an intensive-care unit should be assessed for VTE risk and receive thromboprophylaxis as required.
VTE is a major complication in up to 20% of cancer patients, with hospitalized oncology patients and those undergoing treatment at the highest risk. Some of the newer drug treatments used in these patients have higher VTE rates (e.g., bevacizumab, thalidomide, lenalidomide). These patients need to be carefully evaluated for VTE prophylaxis and closely monitored.5
Generally, in hospitalized patients with cancer, VTE prophylaxis should be considered with UFH, LMWH, or fondaparinux, in the absence of bleeding or other contraindications to anticoagulation. Relative contraindications to anticoagulation include (but are not limited to):
- Active uncontrolled bleeding;
- Active cerebrovascular hemorrhage;
- Dissecting or cerebral aneurysm;
- Bacterial endocarditis;
- Pericarditis;
- Active peptic or gastrointestinal ulceration;
- Severe uncontrolled or malignant hypertension;
- Severe head trauma;
- Pregnancy (warfarin contraindication);
- Heparin-induced thrombocytopenia (heparin, LMWH); and
- Epidural catheter placement.
These same contraindications can be applied to the non-oncology patient, as well.
An important aspect of VTE management is the “Clinical Practice Guideline from the American Academy of Family Physicians and the American College of Physicians on the Diagnosis of VTE from the Annals of Family Medicine.” Consult this for a review of diagnostic tests for VTE.
Thromboprophylaxis is a necessity in a number of at-risk hospitalized patients. Knowing which patients will benefit, and the contraindications for use, will improve patient outcomes. Consult current guidelines for diagnosis recommendations as well as agents of choice, dosing regimens, and therapy duration. TH
Michele B. Kaufman is registered pharmacist based in New York City.
References
- DVT: Assess Your Patients’ Risk, Take Preventive Measures. ASHP Foundation Discoveries, Summer 2007;19(1):1,5. Available at www.ashpfoundation.org/MainMenuCategories/AboutUs/Newsletter/DiscoveriesSummer2007.aspx. Last accessed Nov. 26, 2007.
- Geertz WH, Pineo Graham F, Heit JA et al. Prevention of venous thromboembolism: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:338-400.
- Wein L, Wein S, Haas SJ, et al. Pharmacological venous thromboembolism prophylaxis in hospitalized medical patients, a meta-analysis of randomized controlled trials. Arch Intern Med. 2007;167:1476-1486.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol. 2007;25(34): 5490–5505.
- Qaseem A, Snow V, Barry P for the Joint American Academy of Family Physicians/American College of Physicians Panel on Deep Vein Thrombosis/Pulmonary Embolism. Current diagnosis of venous thromboembolism in primary care: a clinical practice guideline from the American Academy of Family Physicians and the American College of Physicians. Ann Fam Med. 2007;5:57-62.
In the Literature
Literature at a Glance
A guide to this month’s abstracts
- Steroids reduce mortality only in patients with confirmed bacterial meningitis.
- Probiotics can be useful in the treatment of acute diarrhea in children.
- CT pulmonary angiography is not inferior to V/Q scanning for exclusion of PE.
- Hospitalist care results in shorter LOS compared with care by traditional general internists and family practice physicians.
- The early risk of stroke after TIA is approximately 15% to 20% at 90 days after the sentinel event.
- Different anti-thrombotic strategies produce no difference in outcomes of early acute coronary syndromes.
- The risk of fatal PE is highest in the first year after medication is stopped.
- Beers criteria medications are associated with fewer ED visits by elderly patients compared with warfarin, digoxin, and insulin.
Do Steroids Affect the Outcome in Patients with Meningitis?
Background: Pyogenic (bacterial) meningitis has high morbidity and mortality. Studies suggest some benefit of steroids in children but provide limited evidence for adult use.
Study design: Intention-to-treat, randomized control trial.
Setting: Single hospital in Vietnam.
Synopsis: Of 435 patients older than 14 with suspected meningitis all received lumbar puncture with randomization to IV dexamethasone or placebo for four days. Results showed 69% of patients had definite meningitis, 28.3% were probable, and 2.8% had an alternative diagnosis based on culture results.
The primary outcome was death after one month, which did not differ among groups (risk ratio [RR] 0.79, confidence interval [CI] 0.45-1.39).
Predefined subgroup analysis of patients with definitive meningitis showed a significant reduction in mortality at one month (RR 0.43, CI 0.2-0.94) and death/disability at six months (odds ratio [OR] 0.56, CI 0.32-0.98).
In patients with probable meningitis, those who received steroids demonstrated a trend toward harm (OR 2.65, CI 0.73-9.63).
Probable versus definite meningitis was determined retrospectively based on cultures. The most common isolate was Streptococcus suis.
Bottom line: This study provides some evidence for using steroids in adults with confirmed bacterial meningitis. Clinical application is limited by bacterial epidemiology and the difficulty of prospectively separating patients who would benefit from those who might be harmed.
Citation: Nguyen TH, Tran TH, Thwaites G, et. al. Dexamethasone in Vietnamese adolescents and adults with bacterial meningitis. N Engl J Med. 2007;357:2431-2439.
Which Probiotic Preparations Best Reduce the Duration of Acute Diarrhea in Children?
Background: Probiotics have been suggested as an adjunctive therapy to reduce the severity and duration of acute diarrhea in children. However, there are no clear data to suggest if specific probiotic agents are superior to others.
Study design: Prospective single-blind, randomized, controlled trial.
Setting: Outpatient primary care in Naples, Italy.
Synopsis: This study compared five commercially available probiotic preparations (mix of Lactobacillus delbrueckii var bulgaricus/Streptococcus thermophilus/L. acidophilus/ Bifido-bacterium bifidum; L. rhamnosus strain GG; Saccharomyces boulardii; Bacillus clausii; or Enterococcus faecium SF68) and a control group in the treatment of outpatient acute diarrhea in 571 children age 3 months to 36 months.
The primary outcomes were the duration of diarrhea and the number and consistency of stools. The groups receiving Lactobacillus GG and the mixture had a shorter total duration of diarrhea (78.5 and 70 hours, respectively), decreased total number of stools, and improved stool consistency when compared with the control (115.5 hours). The other therapies showed no improvement over the control group. These data report on products commercially available in Italy, which may differ greatly from products available locally.
Bottom line: Probiotic preparations for the treatment of acute diarrhea in children should be chosen based on effectiveness data.
Citation: Canani RB, Cirillo P, Terrin G, et al. Probiotics for treatment of acute diarrhoea in children: randomised clinical trial of five different preparations. BMJ 2007;335:340-345.
Is CTPA a Reliable Alternative to V/Q Scan for Diagnosing PE?
Background: Computed tomography pulmonary angiogram (CTPA) has replaced ventilation/perfusion (V/Q) scanning at many hospitals as the test of choice for ruling out pulmonary embolism (PE). But limited clinical data compare CTPA with V/Q scanning in those suspected of having venous thromboembolism (VTE).
Study design: Randomized, investigator blinded, controlled trial.
Setting: The emergency departments (ED), inpatient wards, and outpatient clinics of five academic centers.
Synopsis: In the study, 1,411 patients were enrolled from five medical centers. Of 694 patients randomized to CTPA, 133 (19.2%) were diagnosed with VTE in the initial evaluation period, while 101 of 712 patients (14.2%) receiving a V/Q scan were diagnosed with VTE.
Patients not initially diagnosed with VTE were monitored. At three-month follow-up, 0.4% of the CTPA group and 1.0% of the V/Q group had a diagnosed VTE.
The overall rate of VTE found in the initial diagnostic period was significantly greater in patients randomized to CTPA (19.2% vs. 14.2%; difference, 5.0%; 95% CI; 1.1% to 8.9% p=.01). This suggests CTPA has a higher false positive rate or detects clinically insignificant thrombi.
Bottom line: CTPA was not inferior to V/Q scanning for excluding clinically meaningful PE, but CTPA diagnosed about 30% more patients with VTE than did V/Q scanning.
Citation: Anderson DR, Kahn SR, Rodger MA, et al. Computed tomographic pulmonary angiography vs. ventilation-perfusion lung scanning in patients with suspected pulmonary embolism: a randomized controlled trial. JAMA. 2007;298(23):2743-2753.
Does the Hospitalist Model Improve Length of Stay, Quality, and Cost of Care?
Background: The hospitalist model, with increased physician availability and expertise but greater discontinuity of care, is becoming more prevalent in U.S. medicine. What little is known about how this model will affect patient care is derived from a number of small studies.
Study design: Retrospective cohort study.
Setting: 45 small to midsize, predominantly nonteaching hospitals throughout the U.S.
Synopsis: Using the Premier Healthcare Informatics database, this study examined information on 76,926 patients admitted for seven common diagnoses to one of three services: hospitalist, general internist, or family physician. Analysis showed that patients on a hospitalist service had a 0.4-day shorter length of stay (p<0.001) compared with those on a general internist or family physician service.
The cost to patients cared for by a hospitalist was lower than the cost of family physicians ($125 less, p=0.33) and internists ($268 less, p=0.02). There was no difference found in death rate or 14-day readmission rate among the three services.
Given the retrospective design of this study, no causal relationship can be deduced. This study is further limited by its lack of specific data on the physicians categorized into one of the three groups solely by administrative data. The authors had concerns that the biases inherent to the retrospective nature of their work accounted for the significant difference found between hospitalists and internists.
Bottom line: The hospitalist model is associated with modest improvements in length of stay as compared with traditional inpatient approaches.
Citation: Lindenauer PK, Rothberg MB, Pekow PS, et. al. Outcomes of care by hospitalists, general internists, and family physicians. N Engl J Med. 2007;357:2589-2600.
What Is the Stroke Risk Soon after TIA, and What Factors Drive the Variability of Previous Findings?
Background: Many studies have attempted to estimate the risk of stroke in the early period after a transient ischemic attack (TIA). These studies vary widely in their calculation of the estimated risk. Further, the clinical and methodological factors underlying this variability are unclear.
Study design: Systematic review and meta-analysis.
Setting: Community and hospital.
Synopsis: Searching the Cochrane review database, MEDLINE, EMBASE, CINAHL, and BIOSIS, 11 studies from 1973 to 2006 were included for meta-analysis, selected from 694 potential candidate studies identified on initial screening. The studies ranged in size from 62 to 2,285 patients.
The pooled estimate of risk for stroke following TIA was found to be 3.5%, 8%, and 9.2% at two, 30, and 90 days following TIA, respectively. However, there was significant heterogeneity for all periods considered (p<0.001).
Outcome ascertainment was identified as a major source of methodological heterogeneity. When risk of stroke at follow-up was determined by passive ascertainment (e.g., administrative documentation) the early risk of stroke was 3.1% two days after TIA, 6.4% at 30 days, and 8.7% at 90 days. But active ascertainment (e.g., direct, personal contact with study participants) determined stroke risk to be 9.9%, 13.4%, and 17.4% at two, 30, and 90 days after TIA, respectively.
Bottom line: Based on analysis of completed studies that included directly observed follow-up of study participants, the early risk of stroke after TIA is approximately 15% to 20% at 90 days following the sentinel event.
Citation: Wu CM, McLaughlin K, Lorenzetti DL, Hill MD, Manns BJ, Ghali WA. Early risk of stroke after transient ischemic attack. Arch Intern Med. 2007;167:2417-2422.
What Is the 1-year Ischemia and Mortality Rate for Three Anti-thrombotic Therapies for Early Invasive Management of ACS?
Background: Early interventional or surgical revascularization has improved morbidity and mortality in patients with acute coronary syndrome (ACS). The optimal anti-thrombotic regimen to reduce late ischemic and death rates has not been determined.
Study design: Prospective, open-label randomized control trial.
Setting: 450 academic and community-based institutions in 17 countries.
Synopsis: A total of 13,819 patients were enrolled between August 2003 and December 2005. They were assigned to heparin plus glycoprotein (GP) IIb/IIIa inhibitors (n=4,603), bivalirudin (Angiomax) plus IIb/IIIa inhibitors (n=4,604), or bivalirudin monotherapy (n=4,612).
For patients receiving GP IIb/IIIa inhibitors, a 2x2 factorial design assigned half the heparin and bivalirudin groups to routine upstream GP inhibitor administration (4,605 patients). The other half received selective GP IIb/IIIa inhibitors administration if PCI was indicated (4,602 patients).
At one year, there was no statistically significant difference in ischemia or mortality rate among the three therapy groups. No difference in ischemia rate was detected between the two GP IIb/IIIa inhibitor utilization strategies.
Since the hypotheses and the power for the one-year analysis in this trial were not prospectively determined, the results are considered to be exploratory and hypothesis generating.
Bottom line: At one year, there is no statistically significant difference in ischemia or mortality rate for the three antithrombotic regiments and the two glycoprotein utilization strategies.
Citation: Stone GW, Ware JH, Bertrand ME, et. al. Antithrombotic strategies in patients with acute coronary syndromes undergoing early invasive management. One-year results from the ACUITY trial. JAMA 2007;298:2497-2505.
What Is the PE Risk after Discontinuing Anticoagulation in Patients with Symptomatic VTE?
Background: The natural history of patients with symptomatic VTE who have completed anticoagulation is not well understood.
Study design: Inception cohort using pooled data from a prospective cohort study and one arm of an open-label randomized trial.
Setting: Academic medical centers in Canada, Sweden, and Italy.
Synopsis: Using pooled data from two previous studies, 2,052 patients with a first diagnosis of symptomatic VTE (lower-extremity deep-vein thrombosis [DVT], PE, or both) were evaluated for fatal PE after a standard course of therapy (mean of six months) with a vitamin K antagonist.
Patients were followed for up to 120 months. The investigators found an annual event risk of 0.19-0.49 per 100 person-years for fatal PE. Patients with prolonged immobility, active cancer, and thrombophilia were excluded, as were those with recurrent acute DVT.
Secondary analysis revealed an incidence of any fatal, definite or probable PE within the first year of discontinuing therapy of 0.35%-0.81%.
After the first year, the annual event risk ranged from 0.15-0.40 events per 100 person-years. Patients with advanced age, idiopathic VTE as well as those presenting with PE had higher rates of fatal PE.
Bottom line: There is a real though small (less than 1%) risk of fatal PE in the first year following discontinuation of anticoagulation for the first VTE episode. The optimal course of treatment for patients with idiopathic VTE is yet to be determined.
Citation: Douketis JD, Gu CS, Schulman S, et al. The risk for fatal pulmonary embolism after discontinuing anticoagulant therapy for venous thromboembolism. Ann Intern Med. 2007;147(11):766-774.
Do the Beers Criteria Predict ED Visits Associated with Adverse Drug Events?
Background: Adverse drug events are common in the elderly. The Beers criteria are a consensus-based list of 41 medications that are considered inappropriate for use in older adults and often lead to poor outcomes.
Study design: Retrospective medical record review and data analysis.
Setting: Three nationally representative, U.S. public health surveillance systems: the National Electronic Injury Surveillance System-Cooperative Adverse Drug Event Surveillance System (NEISS-CADES), 2004-2005; the National Ambulatory Medical Care Survey (NAMCS), 2004; and National Hospital Ambulatory Medical Care Survey (NHAMCS), 2004.
Synopsis: Using data collected from ED visits at 58 hospitals in the NEISS-CADES system, this study estimated that 177,504 visits for adverse drug events occur annually in the United States. Only 8.8% of such visits were attributable to the 41 medications included in the Beers criteria. Three drug classes (anticoagulant and antiplatelet agents, antidiabetic agents, and narrow therapeutic index agents) accounted for nearly half of all such ED visits. Warfarin (17.3%), insulin (13%), and digoxin (3.2%) were the most commonly implicated medications, collectively accounting for 33% of visits (CI, 27.8% to 38.7%).
This study suggests that because of the common use and high risk of adverse events associated with these three drugs, interventions targeting their use may prevent ED visits for adverse drug events in the elderly, compared with interventions aimed at reducing the use of medications identified in the Beers criteria.
This study only included adverse drug events identified in the ED and relied on the diagnosis and documentation of such events by the ED physician.
Bottom line: Beers criteria medications, although considered inappropriate for use in the elderly, were associated with significantly fewer ED visits for adverse events compared with warfarin, digoxin, and insulin.
Citation: Budnitz DS, Shehab N, Kegler SR, et. al. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med. 2007;147:755-765. TH
Literature at a Glance
A guide to this month’s abstracts
- Steroids reduce mortality only in patients with confirmed bacterial meningitis.
- Probiotics can be useful in the treatment of acute diarrhea in children.
- CT pulmonary angiography is not inferior to V/Q scanning for exclusion of PE.
- Hospitalist care results in shorter LOS compared with care by traditional general internists and family practice physicians.
- The early risk of stroke after TIA is approximately 15% to 20% at 90 days after the sentinel event.
- Different anti-thrombotic strategies produce no difference in outcomes of early acute coronary syndromes.
- The risk of fatal PE is highest in the first year after medication is stopped.
- Beers criteria medications are associated with fewer ED visits by elderly patients compared with warfarin, digoxin, and insulin.
Do Steroids Affect the Outcome in Patients with Meningitis?
Background: Pyogenic (bacterial) meningitis has high morbidity and mortality. Studies suggest some benefit of steroids in children but provide limited evidence for adult use.
Study design: Intention-to-treat, randomized control trial.
Setting: Single hospital in Vietnam.
Synopsis: Of 435 patients older than 14 with suspected meningitis all received lumbar puncture with randomization to IV dexamethasone or placebo for four days. Results showed 69% of patients had definite meningitis, 28.3% were probable, and 2.8% had an alternative diagnosis based on culture results.
The primary outcome was death after one month, which did not differ among groups (risk ratio [RR] 0.79, confidence interval [CI] 0.45-1.39).
Predefined subgroup analysis of patients with definitive meningitis showed a significant reduction in mortality at one month (RR 0.43, CI 0.2-0.94) and death/disability at six months (odds ratio [OR] 0.56, CI 0.32-0.98).
In patients with probable meningitis, those who received steroids demonstrated a trend toward harm (OR 2.65, CI 0.73-9.63).
Probable versus definite meningitis was determined retrospectively based on cultures. The most common isolate was Streptococcus suis.
Bottom line: This study provides some evidence for using steroids in adults with confirmed bacterial meningitis. Clinical application is limited by bacterial epidemiology and the difficulty of prospectively separating patients who would benefit from those who might be harmed.
Citation: Nguyen TH, Tran TH, Thwaites G, et. al. Dexamethasone in Vietnamese adolescents and adults with bacterial meningitis. N Engl J Med. 2007;357:2431-2439.
Which Probiotic Preparations Best Reduce the Duration of Acute Diarrhea in Children?
Background: Probiotics have been suggested as an adjunctive therapy to reduce the severity and duration of acute diarrhea in children. However, there are no clear data to suggest if specific probiotic agents are superior to others.
Study design: Prospective single-blind, randomized, controlled trial.
Setting: Outpatient primary care in Naples, Italy.
Synopsis: This study compared five commercially available probiotic preparations (mix of Lactobacillus delbrueckii var bulgaricus/Streptococcus thermophilus/L. acidophilus/ Bifido-bacterium bifidum; L. rhamnosus strain GG; Saccharomyces boulardii; Bacillus clausii; or Enterococcus faecium SF68) and a control group in the treatment of outpatient acute diarrhea in 571 children age 3 months to 36 months.
The primary outcomes were the duration of diarrhea and the number and consistency of stools. The groups receiving Lactobacillus GG and the mixture had a shorter total duration of diarrhea (78.5 and 70 hours, respectively), decreased total number of stools, and improved stool consistency when compared with the control (115.5 hours). The other therapies showed no improvement over the control group. These data report on products commercially available in Italy, which may differ greatly from products available locally.
Bottom line: Probiotic preparations for the treatment of acute diarrhea in children should be chosen based on effectiveness data.
Citation: Canani RB, Cirillo P, Terrin G, et al. Probiotics for treatment of acute diarrhoea in children: randomised clinical trial of five different preparations. BMJ 2007;335:340-345.
Is CTPA a Reliable Alternative to V/Q Scan for Diagnosing PE?
Background: Computed tomography pulmonary angiogram (CTPA) has replaced ventilation/perfusion (V/Q) scanning at many hospitals as the test of choice for ruling out pulmonary embolism (PE). But limited clinical data compare CTPA with V/Q scanning in those suspected of having venous thromboembolism (VTE).
Study design: Randomized, investigator blinded, controlled trial.
Setting: The emergency departments (ED), inpatient wards, and outpatient clinics of five academic centers.
Synopsis: In the study, 1,411 patients were enrolled from five medical centers. Of 694 patients randomized to CTPA, 133 (19.2%) were diagnosed with VTE in the initial evaluation period, while 101 of 712 patients (14.2%) receiving a V/Q scan were diagnosed with VTE.
Patients not initially diagnosed with VTE were monitored. At three-month follow-up, 0.4% of the CTPA group and 1.0% of the V/Q group had a diagnosed VTE.
The overall rate of VTE found in the initial diagnostic period was significantly greater in patients randomized to CTPA (19.2% vs. 14.2%; difference, 5.0%; 95% CI; 1.1% to 8.9% p=.01). This suggests CTPA has a higher false positive rate or detects clinically insignificant thrombi.
Bottom line: CTPA was not inferior to V/Q scanning for excluding clinically meaningful PE, but CTPA diagnosed about 30% more patients with VTE than did V/Q scanning.
Citation: Anderson DR, Kahn SR, Rodger MA, et al. Computed tomographic pulmonary angiography vs. ventilation-perfusion lung scanning in patients with suspected pulmonary embolism: a randomized controlled trial. JAMA. 2007;298(23):2743-2753.
Does the Hospitalist Model Improve Length of Stay, Quality, and Cost of Care?
Background: The hospitalist model, with increased physician availability and expertise but greater discontinuity of care, is becoming more prevalent in U.S. medicine. What little is known about how this model will affect patient care is derived from a number of small studies.
Study design: Retrospective cohort study.
Setting: 45 small to midsize, predominantly nonteaching hospitals throughout the U.S.
Synopsis: Using the Premier Healthcare Informatics database, this study examined information on 76,926 patients admitted for seven common diagnoses to one of three services: hospitalist, general internist, or family physician. Analysis showed that patients on a hospitalist service had a 0.4-day shorter length of stay (p<0.001) compared with those on a general internist or family physician service.
The cost to patients cared for by a hospitalist was lower than the cost of family physicians ($125 less, p=0.33) and internists ($268 less, p=0.02). There was no difference found in death rate or 14-day readmission rate among the three services.
Given the retrospective design of this study, no causal relationship can be deduced. This study is further limited by its lack of specific data on the physicians categorized into one of the three groups solely by administrative data. The authors had concerns that the biases inherent to the retrospective nature of their work accounted for the significant difference found between hospitalists and internists.
Bottom line: The hospitalist model is associated with modest improvements in length of stay as compared with traditional inpatient approaches.
Citation: Lindenauer PK, Rothberg MB, Pekow PS, et. al. Outcomes of care by hospitalists, general internists, and family physicians. N Engl J Med. 2007;357:2589-2600.
What Is the Stroke Risk Soon after TIA, and What Factors Drive the Variability of Previous Findings?
Background: Many studies have attempted to estimate the risk of stroke in the early period after a transient ischemic attack (TIA). These studies vary widely in their calculation of the estimated risk. Further, the clinical and methodological factors underlying this variability are unclear.
Study design: Systematic review and meta-analysis.
Setting: Community and hospital.
Synopsis: Searching the Cochrane review database, MEDLINE, EMBASE, CINAHL, and BIOSIS, 11 studies from 1973 to 2006 were included for meta-analysis, selected from 694 potential candidate studies identified on initial screening. The studies ranged in size from 62 to 2,285 patients.
The pooled estimate of risk for stroke following TIA was found to be 3.5%, 8%, and 9.2% at two, 30, and 90 days following TIA, respectively. However, there was significant heterogeneity for all periods considered (p<0.001).
Outcome ascertainment was identified as a major source of methodological heterogeneity. When risk of stroke at follow-up was determined by passive ascertainment (e.g., administrative documentation) the early risk of stroke was 3.1% two days after TIA, 6.4% at 30 days, and 8.7% at 90 days. But active ascertainment (e.g., direct, personal contact with study participants) determined stroke risk to be 9.9%, 13.4%, and 17.4% at two, 30, and 90 days after TIA, respectively.
Bottom line: Based on analysis of completed studies that included directly observed follow-up of study participants, the early risk of stroke after TIA is approximately 15% to 20% at 90 days following the sentinel event.
Citation: Wu CM, McLaughlin K, Lorenzetti DL, Hill MD, Manns BJ, Ghali WA. Early risk of stroke after transient ischemic attack. Arch Intern Med. 2007;167:2417-2422.
What Is the 1-year Ischemia and Mortality Rate for Three Anti-thrombotic Therapies for Early Invasive Management of ACS?
Background: Early interventional or surgical revascularization has improved morbidity and mortality in patients with acute coronary syndrome (ACS). The optimal anti-thrombotic regimen to reduce late ischemic and death rates has not been determined.
Study design: Prospective, open-label randomized control trial.
Setting: 450 academic and community-based institutions in 17 countries.
Synopsis: A total of 13,819 patients were enrolled between August 2003 and December 2005. They were assigned to heparin plus glycoprotein (GP) IIb/IIIa inhibitors (n=4,603), bivalirudin (Angiomax) plus IIb/IIIa inhibitors (n=4,604), or bivalirudin monotherapy (n=4,612).
For patients receiving GP IIb/IIIa inhibitors, a 2x2 factorial design assigned half the heparin and bivalirudin groups to routine upstream GP inhibitor administration (4,605 patients). The other half received selective GP IIb/IIIa inhibitors administration if PCI was indicated (4,602 patients).
At one year, there was no statistically significant difference in ischemia or mortality rate among the three therapy groups. No difference in ischemia rate was detected between the two GP IIb/IIIa inhibitor utilization strategies.
Since the hypotheses and the power for the one-year analysis in this trial were not prospectively determined, the results are considered to be exploratory and hypothesis generating.
Bottom line: At one year, there is no statistically significant difference in ischemia or mortality rate for the three antithrombotic regiments and the two glycoprotein utilization strategies.
Citation: Stone GW, Ware JH, Bertrand ME, et. al. Antithrombotic strategies in patients with acute coronary syndromes undergoing early invasive management. One-year results from the ACUITY trial. JAMA 2007;298:2497-2505.
What Is the PE Risk after Discontinuing Anticoagulation in Patients with Symptomatic VTE?
Background: The natural history of patients with symptomatic VTE who have completed anticoagulation is not well understood.
Study design: Inception cohort using pooled data from a prospective cohort study and one arm of an open-label randomized trial.
Setting: Academic medical centers in Canada, Sweden, and Italy.
Synopsis: Using pooled data from two previous studies, 2,052 patients with a first diagnosis of symptomatic VTE (lower-extremity deep-vein thrombosis [DVT], PE, or both) were evaluated for fatal PE after a standard course of therapy (mean of six months) with a vitamin K antagonist.
Patients were followed for up to 120 months. The investigators found an annual event risk of 0.19-0.49 per 100 person-years for fatal PE. Patients with prolonged immobility, active cancer, and thrombophilia were excluded, as were those with recurrent acute DVT.
Secondary analysis revealed an incidence of any fatal, definite or probable PE within the first year of discontinuing therapy of 0.35%-0.81%.
After the first year, the annual event risk ranged from 0.15-0.40 events per 100 person-years. Patients with advanced age, idiopathic VTE as well as those presenting with PE had higher rates of fatal PE.
Bottom line: There is a real though small (less than 1%) risk of fatal PE in the first year following discontinuation of anticoagulation for the first VTE episode. The optimal course of treatment for patients with idiopathic VTE is yet to be determined.
Citation: Douketis JD, Gu CS, Schulman S, et al. The risk for fatal pulmonary embolism after discontinuing anticoagulant therapy for venous thromboembolism. Ann Intern Med. 2007;147(11):766-774.
Do the Beers Criteria Predict ED Visits Associated with Adverse Drug Events?
Background: Adverse drug events are common in the elderly. The Beers criteria are a consensus-based list of 41 medications that are considered inappropriate for use in older adults and often lead to poor outcomes.
Study design: Retrospective medical record review and data analysis.
Setting: Three nationally representative, U.S. public health surveillance systems: the National Electronic Injury Surveillance System-Cooperative Adverse Drug Event Surveillance System (NEISS-CADES), 2004-2005; the National Ambulatory Medical Care Survey (NAMCS), 2004; and National Hospital Ambulatory Medical Care Survey (NHAMCS), 2004.
Synopsis: Using data collected from ED visits at 58 hospitals in the NEISS-CADES system, this study estimated that 177,504 visits for adverse drug events occur annually in the United States. Only 8.8% of such visits were attributable to the 41 medications included in the Beers criteria. Three drug classes (anticoagulant and antiplatelet agents, antidiabetic agents, and narrow therapeutic index agents) accounted for nearly half of all such ED visits. Warfarin (17.3%), insulin (13%), and digoxin (3.2%) were the most commonly implicated medications, collectively accounting for 33% of visits (CI, 27.8% to 38.7%).
This study suggests that because of the common use and high risk of adverse events associated with these three drugs, interventions targeting their use may prevent ED visits for adverse drug events in the elderly, compared with interventions aimed at reducing the use of medications identified in the Beers criteria.
This study only included adverse drug events identified in the ED and relied on the diagnosis and documentation of such events by the ED physician.
Bottom line: Beers criteria medications, although considered inappropriate for use in the elderly, were associated with significantly fewer ED visits for adverse events compared with warfarin, digoxin, and insulin.
Citation: Budnitz DS, Shehab N, Kegler SR, et. al. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med. 2007;147:755-765. TH
Literature at a Glance
A guide to this month’s abstracts
- Steroids reduce mortality only in patients with confirmed bacterial meningitis.
- Probiotics can be useful in the treatment of acute diarrhea in children.
- CT pulmonary angiography is not inferior to V/Q scanning for exclusion of PE.
- Hospitalist care results in shorter LOS compared with care by traditional general internists and family practice physicians.
- The early risk of stroke after TIA is approximately 15% to 20% at 90 days after the sentinel event.
- Different anti-thrombotic strategies produce no difference in outcomes of early acute coronary syndromes.
- The risk of fatal PE is highest in the first year after medication is stopped.
- Beers criteria medications are associated with fewer ED visits by elderly patients compared with warfarin, digoxin, and insulin.
Do Steroids Affect the Outcome in Patients with Meningitis?
Background: Pyogenic (bacterial) meningitis has high morbidity and mortality. Studies suggest some benefit of steroids in children but provide limited evidence for adult use.
Study design: Intention-to-treat, randomized control trial.
Setting: Single hospital in Vietnam.
Synopsis: Of 435 patients older than 14 with suspected meningitis all received lumbar puncture with randomization to IV dexamethasone or placebo for four days. Results showed 69% of patients had definite meningitis, 28.3% were probable, and 2.8% had an alternative diagnosis based on culture results.
The primary outcome was death after one month, which did not differ among groups (risk ratio [RR] 0.79, confidence interval [CI] 0.45-1.39).
Predefined subgroup analysis of patients with definitive meningitis showed a significant reduction in mortality at one month (RR 0.43, CI 0.2-0.94) and death/disability at six months (odds ratio [OR] 0.56, CI 0.32-0.98).
In patients with probable meningitis, those who received steroids demonstrated a trend toward harm (OR 2.65, CI 0.73-9.63).
Probable versus definite meningitis was determined retrospectively based on cultures. The most common isolate was Streptococcus suis.
Bottom line: This study provides some evidence for using steroids in adults with confirmed bacterial meningitis. Clinical application is limited by bacterial epidemiology and the difficulty of prospectively separating patients who would benefit from those who might be harmed.
Citation: Nguyen TH, Tran TH, Thwaites G, et. al. Dexamethasone in Vietnamese adolescents and adults with bacterial meningitis. N Engl J Med. 2007;357:2431-2439.
Which Probiotic Preparations Best Reduce the Duration of Acute Diarrhea in Children?
Background: Probiotics have been suggested as an adjunctive therapy to reduce the severity and duration of acute diarrhea in children. However, there are no clear data to suggest if specific probiotic agents are superior to others.
Study design: Prospective single-blind, randomized, controlled trial.
Setting: Outpatient primary care in Naples, Italy.
Synopsis: This study compared five commercially available probiotic preparations (mix of Lactobacillus delbrueckii var bulgaricus/Streptococcus thermophilus/L. acidophilus/ Bifido-bacterium bifidum; L. rhamnosus strain GG; Saccharomyces boulardii; Bacillus clausii; or Enterococcus faecium SF68) and a control group in the treatment of outpatient acute diarrhea in 571 children age 3 months to 36 months.
The primary outcomes were the duration of diarrhea and the number and consistency of stools. The groups receiving Lactobacillus GG and the mixture had a shorter total duration of diarrhea (78.5 and 70 hours, respectively), decreased total number of stools, and improved stool consistency when compared with the control (115.5 hours). The other therapies showed no improvement over the control group. These data report on products commercially available in Italy, which may differ greatly from products available locally.
Bottom line: Probiotic preparations for the treatment of acute diarrhea in children should be chosen based on effectiveness data.
Citation: Canani RB, Cirillo P, Terrin G, et al. Probiotics for treatment of acute diarrhoea in children: randomised clinical trial of five different preparations. BMJ 2007;335:340-345.
Is CTPA a Reliable Alternative to V/Q Scan for Diagnosing PE?
Background: Computed tomography pulmonary angiogram (CTPA) has replaced ventilation/perfusion (V/Q) scanning at many hospitals as the test of choice for ruling out pulmonary embolism (PE). But limited clinical data compare CTPA with V/Q scanning in those suspected of having venous thromboembolism (VTE).
Study design: Randomized, investigator blinded, controlled trial.
Setting: The emergency departments (ED), inpatient wards, and outpatient clinics of five academic centers.
Synopsis: In the study, 1,411 patients were enrolled from five medical centers. Of 694 patients randomized to CTPA, 133 (19.2%) were diagnosed with VTE in the initial evaluation period, while 101 of 712 patients (14.2%) receiving a V/Q scan were diagnosed with VTE.
Patients not initially diagnosed with VTE were monitored. At three-month follow-up, 0.4% of the CTPA group and 1.0% of the V/Q group had a diagnosed VTE.
The overall rate of VTE found in the initial diagnostic period was significantly greater in patients randomized to CTPA (19.2% vs. 14.2%; difference, 5.0%; 95% CI; 1.1% to 8.9% p=.01). This suggests CTPA has a higher false positive rate or detects clinically insignificant thrombi.
Bottom line: CTPA was not inferior to V/Q scanning for excluding clinically meaningful PE, but CTPA diagnosed about 30% more patients with VTE than did V/Q scanning.
Citation: Anderson DR, Kahn SR, Rodger MA, et al. Computed tomographic pulmonary angiography vs. ventilation-perfusion lung scanning in patients with suspected pulmonary embolism: a randomized controlled trial. JAMA. 2007;298(23):2743-2753.
Does the Hospitalist Model Improve Length of Stay, Quality, and Cost of Care?
Background: The hospitalist model, with increased physician availability and expertise but greater discontinuity of care, is becoming more prevalent in U.S. medicine. What little is known about how this model will affect patient care is derived from a number of small studies.
Study design: Retrospective cohort study.
Setting: 45 small to midsize, predominantly nonteaching hospitals throughout the U.S.
Synopsis: Using the Premier Healthcare Informatics database, this study examined information on 76,926 patients admitted for seven common diagnoses to one of three services: hospitalist, general internist, or family physician. Analysis showed that patients on a hospitalist service had a 0.4-day shorter length of stay (p<0.001) compared with those on a general internist or family physician service.
The cost to patients cared for by a hospitalist was lower than the cost of family physicians ($125 less, p=0.33) and internists ($268 less, p=0.02). There was no difference found in death rate or 14-day readmission rate among the three services.
Given the retrospective design of this study, no causal relationship can be deduced. This study is further limited by its lack of specific data on the physicians categorized into one of the three groups solely by administrative data. The authors had concerns that the biases inherent to the retrospective nature of their work accounted for the significant difference found between hospitalists and internists.
Bottom line: The hospitalist model is associated with modest improvements in length of stay as compared with traditional inpatient approaches.
Citation: Lindenauer PK, Rothberg MB, Pekow PS, et. al. Outcomes of care by hospitalists, general internists, and family physicians. N Engl J Med. 2007;357:2589-2600.
What Is the Stroke Risk Soon after TIA, and What Factors Drive the Variability of Previous Findings?
Background: Many studies have attempted to estimate the risk of stroke in the early period after a transient ischemic attack (TIA). These studies vary widely in their calculation of the estimated risk. Further, the clinical and methodological factors underlying this variability are unclear.
Study design: Systematic review and meta-analysis.
Setting: Community and hospital.
Synopsis: Searching the Cochrane review database, MEDLINE, EMBASE, CINAHL, and BIOSIS, 11 studies from 1973 to 2006 were included for meta-analysis, selected from 694 potential candidate studies identified on initial screening. The studies ranged in size from 62 to 2,285 patients.
The pooled estimate of risk for stroke following TIA was found to be 3.5%, 8%, and 9.2% at two, 30, and 90 days following TIA, respectively. However, there was significant heterogeneity for all periods considered (p<0.001).
Outcome ascertainment was identified as a major source of methodological heterogeneity. When risk of stroke at follow-up was determined by passive ascertainment (e.g., administrative documentation) the early risk of stroke was 3.1% two days after TIA, 6.4% at 30 days, and 8.7% at 90 days. But active ascertainment (e.g., direct, personal contact with study participants) determined stroke risk to be 9.9%, 13.4%, and 17.4% at two, 30, and 90 days after TIA, respectively.
Bottom line: Based on analysis of completed studies that included directly observed follow-up of study participants, the early risk of stroke after TIA is approximately 15% to 20% at 90 days following the sentinel event.
Citation: Wu CM, McLaughlin K, Lorenzetti DL, Hill MD, Manns BJ, Ghali WA. Early risk of stroke after transient ischemic attack. Arch Intern Med. 2007;167:2417-2422.
What Is the 1-year Ischemia and Mortality Rate for Three Anti-thrombotic Therapies for Early Invasive Management of ACS?
Background: Early interventional or surgical revascularization has improved morbidity and mortality in patients with acute coronary syndrome (ACS). The optimal anti-thrombotic regimen to reduce late ischemic and death rates has not been determined.
Study design: Prospective, open-label randomized control trial.
Setting: 450 academic and community-based institutions in 17 countries.
Synopsis: A total of 13,819 patients were enrolled between August 2003 and December 2005. They were assigned to heparin plus glycoprotein (GP) IIb/IIIa inhibitors (n=4,603), bivalirudin (Angiomax) plus IIb/IIIa inhibitors (n=4,604), or bivalirudin monotherapy (n=4,612).
For patients receiving GP IIb/IIIa inhibitors, a 2x2 factorial design assigned half the heparin and bivalirudin groups to routine upstream GP inhibitor administration (4,605 patients). The other half received selective GP IIb/IIIa inhibitors administration if PCI was indicated (4,602 patients).
At one year, there was no statistically significant difference in ischemia or mortality rate among the three therapy groups. No difference in ischemia rate was detected between the two GP IIb/IIIa inhibitor utilization strategies.
Since the hypotheses and the power for the one-year analysis in this trial were not prospectively determined, the results are considered to be exploratory and hypothesis generating.
Bottom line: At one year, there is no statistically significant difference in ischemia or mortality rate for the three antithrombotic regiments and the two glycoprotein utilization strategies.
Citation: Stone GW, Ware JH, Bertrand ME, et. al. Antithrombotic strategies in patients with acute coronary syndromes undergoing early invasive management. One-year results from the ACUITY trial. JAMA 2007;298:2497-2505.
What Is the PE Risk after Discontinuing Anticoagulation in Patients with Symptomatic VTE?
Background: The natural history of patients with symptomatic VTE who have completed anticoagulation is not well understood.
Study design: Inception cohort using pooled data from a prospective cohort study and one arm of an open-label randomized trial.
Setting: Academic medical centers in Canada, Sweden, and Italy.
Synopsis: Using pooled data from two previous studies, 2,052 patients with a first diagnosis of symptomatic VTE (lower-extremity deep-vein thrombosis [DVT], PE, or both) were evaluated for fatal PE after a standard course of therapy (mean of six months) with a vitamin K antagonist.
Patients were followed for up to 120 months. The investigators found an annual event risk of 0.19-0.49 per 100 person-years for fatal PE. Patients with prolonged immobility, active cancer, and thrombophilia were excluded, as were those with recurrent acute DVT.
Secondary analysis revealed an incidence of any fatal, definite or probable PE within the first year of discontinuing therapy of 0.35%-0.81%.
After the first year, the annual event risk ranged from 0.15-0.40 events per 100 person-years. Patients with advanced age, idiopathic VTE as well as those presenting with PE had higher rates of fatal PE.
Bottom line: There is a real though small (less than 1%) risk of fatal PE in the first year following discontinuation of anticoagulation for the first VTE episode. The optimal course of treatment for patients with idiopathic VTE is yet to be determined.
Citation: Douketis JD, Gu CS, Schulman S, et al. The risk for fatal pulmonary embolism after discontinuing anticoagulant therapy for venous thromboembolism. Ann Intern Med. 2007;147(11):766-774.
Do the Beers Criteria Predict ED Visits Associated with Adverse Drug Events?
Background: Adverse drug events are common in the elderly. The Beers criteria are a consensus-based list of 41 medications that are considered inappropriate for use in older adults and often lead to poor outcomes.
Study design: Retrospective medical record review and data analysis.
Setting: Three nationally representative, U.S. public health surveillance systems: the National Electronic Injury Surveillance System-Cooperative Adverse Drug Event Surveillance System (NEISS-CADES), 2004-2005; the National Ambulatory Medical Care Survey (NAMCS), 2004; and National Hospital Ambulatory Medical Care Survey (NHAMCS), 2004.
Synopsis: Using data collected from ED visits at 58 hospitals in the NEISS-CADES system, this study estimated that 177,504 visits for adverse drug events occur annually in the United States. Only 8.8% of such visits were attributable to the 41 medications included in the Beers criteria. Three drug classes (anticoagulant and antiplatelet agents, antidiabetic agents, and narrow therapeutic index agents) accounted for nearly half of all such ED visits. Warfarin (17.3%), insulin (13%), and digoxin (3.2%) were the most commonly implicated medications, collectively accounting for 33% of visits (CI, 27.8% to 38.7%).
This study suggests that because of the common use and high risk of adverse events associated with these three drugs, interventions targeting their use may prevent ED visits for adverse drug events in the elderly, compared with interventions aimed at reducing the use of medications identified in the Beers criteria.
This study only included adverse drug events identified in the ED and relied on the diagnosis and documentation of such events by the ED physician.
Bottom line: Beers criteria medications, although considered inappropriate for use in the elderly, were associated with significantly fewer ED visits for adverse events compared with warfarin, digoxin, and insulin.
Citation: Budnitz DS, Shehab N, Kegler SR, et. al. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med. 2007;147:755-765. TH