User login
Clinical Conundrum
A 49‐year‐old man presented with 2 days of chills, fever, anorexia, and increased cough and dyspnea. The patient had a history of chronic obstructive pulmonary disease (COPD) and noted that his cough and dyspnea had increased above normal for several days. He was now dyspneic with minimal activity and had slept at a 45‐degree incline the night prior to evaluation due to dyspnea. He noted less improvement than usual with the use of his metered dose inhaler. His cough was occasionally productive of small amounts of white phlegm. He had vomited once. During a coughing episode the patient experienced a sudden onset of sharp right upper quadrant abdominal pain that worsened with coughing and sudden position changes. The patient denied a prior history of abdominal pain or surgery. The patient's last bowel movement was 2 days prior to admission. He denied melena or bright red blood per rectum.
My initial differential diagnosis for this patient's dyspnea and cough is pneumonia, acute exacerbation of COPD, or congestive heart failure. The presence of fever and anorexia increases the likelihood of infectious etiologies, whereas the presence of orthopnea points toward congestive heart failure. Noncardiac processessuch as a large pleural effusion or apical lung diseasecould also cause orthopnea. His abdominal pain could be a result of pneumonia alone (perhaps in the right lower lobe with diaphragmatic irritation), but I am also considering complications of pneumonia such as empyema. Although his abdominal pain, dyspnea, and cough could also be a result of hepatobiliary disease, a perforated viscus, or pancreatitis, we currently have little reason to suspect a direct abdominal etiology. My top diagnosis is community‐acquired pneumonia, perhaps accompanied by pleural effusion.
His medical history was significant for dilated cardiomyopathy and heavy alcohol use. His medications included various meter‐dosed inhalers, bupropion, digoxin, spironolactone, lisinopril, and metoprolol. He had never received corticosteroid therapy and had not previously been hospitalized for COPD‐related problems. He had smoked one pack of cigarettes daily for 40 years.
Heavy alcohol use is associated with an increased risk of several pulmonary infections such as gram‐negative necrotizing pneumonia (classically, Klebsiella pneumoniae), pneumococcal pneumonia, aspiration pneumonia, anaerobic lung abscesses, and tuberculosis. Given his right upper quadrant pain, acute alcoholic hepatitis and alcohol‐related pancreatitis enter the differential. His history of cardiomyopathy makes me consider congestive heart failure as more likely than before, and perhaps his abdominal pain is a result of hepatic congestion from right heart failure. His fever, however, cannot be attributed to cardiac failure. Less likely diagnoses include ischemic conditions related to his cardiomyopathy such as mesenteric ischemia from low perfusion or embolism from a cardiac thrombus. A pulmonary infection remains the most likely diagnosis.
He was an ill‐appearing man in moderate respiratory distress, looking older than his stated age. His temperature was 38.4C, heart rate 129 beats/minute, blood pressure 85/56 mm Hg, respiratory rate 24 breaths/minute, and oxygen saturation 92% on room air. A cardiovascular exam revealed no murmur, gallop, or rub. The jugular venous pulse was not elevated. His lungs were clear to auscultation. Abdominal exam revealed right‐sided abdominal tenderness that appeared to localize to the rectus sheath. Otherwise, the abdomen was soft, with normal bowel sounds and no organomegaly. Rectal examination revealed guaiac negative stool and no focal tenderness. His extremities were normal.
His vital signs are worrisome for impending cardiovascular collapse and shock, possibly due to sepsis. The relatively nonfocal cardiopulmonary exam is surprising given his initial symptoms and makes me wonder if his dyspnea is primarily related to an abdominal process leading to diaphragmatic irritation rather than to a thoracic process. Congestive heart failure seems unlikely given the lack of supportive physical examination findings. His abdominal exam findings are puzzling. Although his abdominal wall tenderness could be benignperhaps from muscular strain or a tear from coughingit could represent a more worrisome process such as infection or a hematoma in the abdominal wall muscles. Mesenteric ischemia is still possible, as the exam is often unimpressive. A hepatic abscess or subphrenic abscess should be considered, as physical exam findings in these conditions can be subtle.
My differential remains relatively unchanged, but I have now put consideration of a hepatic or subphrenic abscess higher on my list. Early empiric broad‐spectrum antibiotics seem necessary.
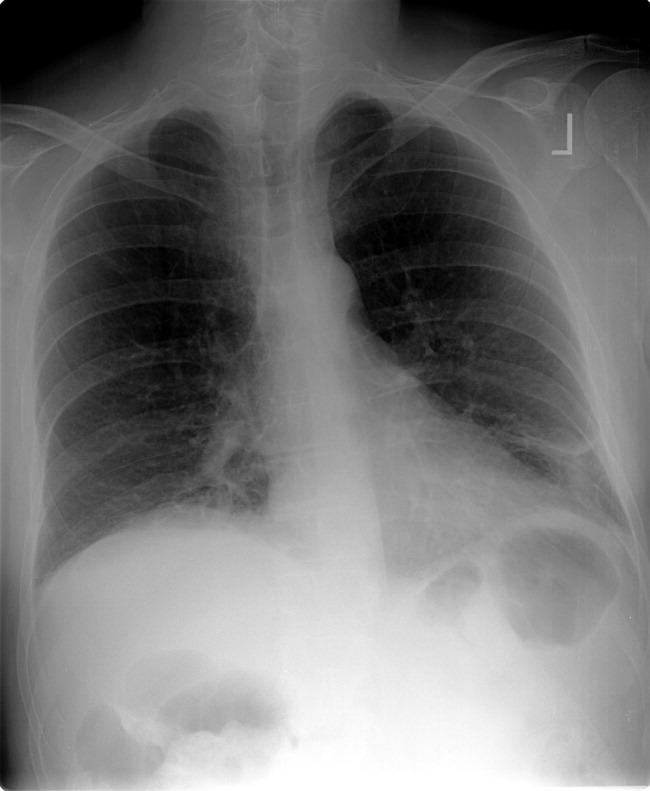
He had a white blood cell count of 26,700/mL with 92% neutrophils, a hemoglobin of 14.6 g/dL, and a platelet count of 312,000/mL. Sodium was 134 mmol/L, potassium was 4.3 mmol/L, chloride was 94 mmol/L, bicarbonate was 23 mmol/L, blood urea nitrogen was 23 mg/dL, and creatinine was 2.1 mg/dL. The results of the calcium, protein, albumin, and liver function tests were normal. Urinalysis was negative for protein and red blood cells. An electrocardiogram revealed sinus tachycardia. A chest radiograph at admission revealed mild opacities in both lower lobes and the right middle lobe consistent with either atelectasis or pneumonia (Fig. 1). A very small left effusion was also identified.
The additional data reinforce my clinical impression that this process is likely to be infectious. The chest radiograph is consistent with community‐acquired pneumonia, possibly from an atypical pathogen. Given his elevated creatinine, I am also considering a pulmonary‐renal syndrome such as vasculitis, though hematuria was not present. A subphrenic abscess, mesenteric ischemia, or an abdominal wall process (because his abdominal tenderness on exam still needs an explanation) remain possibilities; my suspicion would increase if he does not respond appropriately to therapy for community‐acquired pneumonia.
The clinical team's working diagnosis also was community‐acquired pneumonia. Blood and sputum cultures were obtained, and the patient was treated with intravenous ceftriaxone, azithromycin, and intravenous fluid. By the second day, his creatinine had normalized; however, his hypoxemia had worsened, and he now required supplemental oxygen. His temperature was 39.3C, and his heart rate was 150 beats/minute. The findings of an abdominal ultrasound of the kidneys, spleen, and right upper quadrant were normal.
It is too early to say the patient has failed therapy because a patient can get worse before getting better during the course of antibiotic therapy for community‐acquired pneumonia. Fever, for example, may take up to 7 days to resolve, depending on host factors and the pathogen. Though I typically wait about 72 hours before assuming a patient is not appropriately responding to therapy, the additional information has made me concerned. The degree of tachycardia is significant and warrants an EKG to exclude an arrthymia. I would also repeat the chest radiograph to evaluate for worsening infiltrates or increased pleural effusion.
On the third hospital day, the patient's abdominal pain had decreased with analgesia, but his fever, cough, and dyspnea remained largely unchanged. Antibiotics were changed to intravenous levofloxacin. A repeat chest radiograph revealed elevation of the right hemidiaphragm and bilateral effusions (Fig. 2). An electrocardiogram revealed sinus tachycardia. Blood cultures revealed no growth, and sputum cultures grew oral flora.
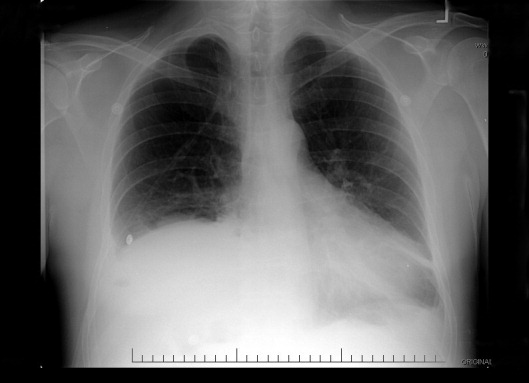
A significantly elevated right hemidiaphragm makes me reconsider the diagnosis of simple community‐acquired pneumonia. The differential diagnosis for an elevated hemidiaphragm is best considered by location in relation to the diaphragm. Causes above the diaphragm include rib fracture, atelectasis, pleural thickening, and volume loss of the lung for another reason (e.g., surgery, bronchial obstruction due to tumor or mucus plugging), as well as mimics such as a densely consolidated pneumonia, pulmonary infarction, or a subpulmonary effusion. Diaphragmatic causes include eventration, rupture, phrenic nerve weakness, and intrinsic weakness because of neuromuscular disease (usually bilateral). Causes below the diaphragm that must be considered are subphrenic or liver abscess, liver (and other abdominal) malignancy, pancreatic pseudocyst, and distended bowel. Given the clinical picture, I am focusing below the diaphragmespecially on a possible hepatic or subphrenic abscess (which could be missed on ultrasound) and mimics of it such as dense consolidation or a subpulmonary effusion. Given the lack of response to antibiotics, I need to consider an infection that is not being treated, either because of location (abscess, effusion) or microbiology (tuberculosis, a parasite, a fungus, resistant bacteria). After confirming that the patient has a substantive pleural effusion, he needs a thoracentesis.
On the fourth hospital day, his temperature was 38.8C, and his white blood cell count was 21,000/mL. A right‐sided thoracentesis was performed; approximately 250 cc of fluid was obtained. Pleural fluid analysis revealed bloody fluid, with a white blood cell count of 16,750/mL with 94% neutrophils, 40,000 red blood cells/mL, lactate dehydrogenase of 278 U/L (normal serum value 80200 U/L), protein of 3.7 g/dL, and glucose of 81 mg/dL. A pleural fluid pH was not obtained. A gram stain revealed many white blood cells with no organisms noted. Serum protein was 7.4 g/dL. These results were thought to represent an exudative parapneumonic effusion; levofloxacin and supplemental oxygen were continued.
The pleural fluid appears exudative, but I am not sure this man has a parapneumonic effusion because, despite clinical deterioration, an obvious infiltrate is not seen on interval chest radiography. We must look closely at the fluid because this is a bloody effusion and somewhat atypical for a parapneumonic effusion. Also, the effusion does not appear large enough to explain why he has not improved on the current antibiotics. We should thus reconsider our diagnosis and management. I would obtain additional imaging (such as an abdominal and chest computed tomography [CT]) and perhaps obtain a consultation from the pulmonary team regarding the postulated initial diagnosis of pneumonia with effusion.
On the fifth day of hospitalization, the patient's dyspnea and cough persisted but were improved. His abdominal pain was minimal and felt improved with flatus. Fever continued to 38.8C, and the white blood cell count was 20,000/mL. On examination the patient had decreased breath sounds at the right base and bibasilar crackles. His abdomen was soft, with tenderness in his right upper quadrant only with deep palpation; bowel sounds remained. An ultrasound of the chest was performed to look for a loculated effusion; however, no fluid was identified. The pulmonary consultant thought it likely that the patient had a subpulmonic effusion and recommended CT of the abdomen and chest.
His right upper quadrant tenderness is still unexplained. I would agree with the CT, primarily to evaluate other causes of his elevated diaphragm such as subphrenic or hepatic abscess. For now, I would make no change in antibiotic therapy.
On the sixth hospital day, the patient had an episode of bilious emesis. Chest and abdominal CT revealed collapse of the right middle and lower lobes with a small adjacent effusion, and a 6 6 16 cm abscess intimately opposed to the right lobe of the liver. Extending from the inferior extent of the abscess was a tubular thick‐walled structure connecting to the cecum that was suspicious of a thickened inflamed appendix. There was periappendiceal stranding suggesting inflammation. The small bowel was diffusely dilated up to 4.5 cm, suggesting a small bowel obstruction.
I suspect that his abscess is related to a perforated appendix and that the dilated small bowel is most likely a result of localized irritation of the bowel by the abscess and appendicitis. The collapsed lung is most likely due to local inflammation from the subdiaphragmatic abscess. Treatment should now be changed substantially. I would ask a surgeon to evaluate the patient because the most likely diagnosis is perforated appendicitis with abscess formation.
When the periappendiceal abscess was drained percutaneously, 190 mL of purulent fluid was removed. The cultures were positive for Klebsiella pneumonia, Enterococcus faecalis,and Streptococcus milleri. The patient was given 6 weeks of intravenous antibiotics with improvement in his clinical symptoms. During the interval the findings on his chest radiograph resolved completely. A laproscopic appendectomy 3 months later revealed significant right lower quadrant adhesions. The pathology specimen identified a distorted appendix with regeneration consistent with prior appendicitis. The patient was contacted 4 months after his surgery, and he reported that he was doing well, with no cardiopulmonary or gastrointestinal symptoms.
COMMENTARY
Community‐acquired pneumonia (CAP) is a common cause of acute illness and accounts for nearly 1 million admissions per year in the United States.1 The diagnosis of CAP is made when symptoms including dyspnea, fever, cough, or leukocytosis are present, with confirmation provided by a chest radiograph. Often the diagnosis is clear; however, there is no pathognomonic constellation of signs or symptoms that establish the diagnosis with certainty.2 Many physicians learn that pneumoniaespecially lower‐lobe pneumoniacan lead to abdominal findings such as upper quadrant pain, vomiting, and tenderness to palpation. Conversely, the patient discussed above illustrates that a primary abdominal process can also result in a symptom complex that mimics pneumonia.
The prevalence of CAP coupled with the inherent uncertainty of a clinical diagnosis of CAP leads to an important question: How long is too long before questioning the diagnosis? An analysis of the pneumonia Patient Outcomes Research Trial (PORT) limited to inpatients with CAP examined time to clinical stability. For the majority of patients, abnormal vital signs resolved within 23 days.3 In this study, 29% of patients had severe disease, and not surprisingly, these patients took longer to improve. Using the pneumonia severity index score, which accounts for age, comorbidity, abnormal vital signs, and laboratory data, the patient described in this article would be considered at high risk for death and complication with an estimated mortality of 9%.4 Using a combination of defervescence, resolution of tachycardia, tachypnea, and hypoxemia as markers of clinical stability, a patient like ours should respond within 4 days (with a range of 27 days). On the basis of these dataand the discrepancy between the patient's severe illness and relatively minor pulmonary infiltratesit seems reasonable to have considered this patient as failing CAP therapy as early as the fourth day of hospitalization.
In approximately 10% of hospitalized patients with CAP, the clinical course is protracted.5 When patients do not improve as quickly as expected, the reasons that could explain this should be investigated. In a cohort of 49 patients with CAP who failed therapy the most common reasons for failure to improve were severity of the pneumonia and drug resistance.6 A multicenter study found that the incidence of resistance to penicillin by Streptococcus pneumoniae, the most common bacterial pathogen in CAP, was 30%, with a 4% in vitro resistance rate to ceftriaxone.7 How well in vitro resistance predicts clinical response, however, is unclear. Risk factors for antibiotic resistance include close exposure to children, recent antibiotic use, and recent hospitalization. Immunosuppressive conditions should also be considered in patients who fail to improve. Suppurative complications of pneumoniasuch as empyema, parapneumonic effusion, and lung abscessalso delay recovery.
Another consideration in a patient with what appears to be a nonresolving pneumonia with pleural effusion is that the initial diagnosis is incorrect and the cause is extrathoracic. Pulmonary and cardiac diseases account for more than 90% of effusions, whereas less than 5% of pleural effusions result from intraabdominal causes.8 When should intraabdominal diseases be sought in patients with an effusion, fever, dyspnea, and cough? Light suggests that intraabdominal pathology should be investigated in patients who have pleural effusions without significant parenchymal disease.8 This point is underscored by the experience of our patient, whose chest radiographs showed, despite clinical decline, minimal airspace disease.
Several abdominal entities cause pleural effusion. Pancreatitis, either acute or chronic, with pseudocyst formation is the most common abdominal cause of exudative pleural effusions. Approximately 10% of patients with pancreatic disease will develop effusions, usually left‐sided.9 These left‐sided effusions are also seen in splenic abscesses, usually as a result of endocarditis. Intrahepatic abscess is associated with effusions in 20% of patients.10 A subphrenic abscess, as seen in our patient, is an uncommon cause of exudative pleural effusions. Historically, subphrenic abscesses resulted from a perforated viscus, with ruptured appendicitis the most common cause,11 followed by perforated peptic ulcers and biliary tract disease. With the advent of antibiotics, the causes of subphrenic abscess changed considerably, with the majority of current cases resulting from postsurgical complications.12 The findings of a chest radiograph are abnormal in 80% of patients with subphrenic abscess;1214 an elevated hemidiaphragm and pleural effusion are found in the majority of cases. The symptoms of a subphrenic abscess are nonspecific, and patient's complaints are equally split between predominantly thoracic and predomninantly abdominal complaints.15
Appendicitis, a common disease predominantly of the young, may lead to atypical presentations in older individuals. In a retrospective analysis of 113 patients older than 60 years with appendicitis, 70% presented in an atypical fashion.16 Typical symptoms include right lower quadrant pain, fever, anorexia and a white blood cell count greater than 10,000/mL. Fever was the most frequently absent symptom, seen in only 37% of older patients. In this cohort, approximately one third of older patients waited more than 48 hours prior to presentation. The time between symptom onset and clinical presentation is a strong predictor of perforation risk.17 As in this case, roughly 2% of patients with acute appendicitis will present with perforation and abscess formation.18 In such patients the management is initially conservative. Percutaneous drainage and broad spectrum antibiotics are the treatment of choice, followed by an interval appendectomy in 612 weeks.19 The rationale for delayed surgery is that earlier surgery may disseminate a localized inflammatory process.20
Community‐acquired pneumonia is a more frequent cause of hospital admission than is intraabdominal abscess. Physicians often face the dilemma of when to pursue alternative diagnoses after a patient who is thought to have an atypical presentation of a common disease (ie, CAP) fails to respond to conventional therapy. Although clinicians learn that right upper quadrant pain may be a symptom of pneumonia, our patient revealed that abdominal causes may mimic pneumonia and produce a pleural effusion. Determining whether the primary disease originates above or below the diaphragm is critical to guiding therapy. When patients fail to respond adequately to therapy, clinicians should set a low threshold for deciding to image the abdomen in a patient with modest pulmonary infiltrates, pleural effusion, and abdominal pain.
- ,,, et al.The cost of treating community‐acquired pneumonia.Clin Ther.1998;20:820–827.
- ,,.Does this patient have community‐acquired pneumonia? Diagnosing pneumonia by history and physical examination.JAMA.1997;278:1440–1445.
- ,,, et al.Time to clinical stability in patients hospitalized with community acquired pneumonia. Implications for practice guidelines.JAMA.1998;279:1452–1457.
- ,,, et al.A prediction rule to identify low‐risk patients with community‐acquired pneumonia.N Engl J Med.1997;336:243–250.
- ,,, et al.Utility of fiberoptic bronchoscopy in non resolving pneumonia.Chest.1990;98:1322–1326.
- ,,, et al.Antimicrobial treatment failures in patients with community acquired pneumonia. Causes and prognostic implications.Am J Respir Crit Care Med.2000;162:154–160.
- ,,, et al.Antimicrobial resistance with Streptococcus pneumoniae in the United States, 1997–98.Emerg Infect Dis.1999;5:757–765.
- ,.Pleural effusion. In:Murray JF,Nadel JA, eds.Textbook of respiratory medicine. 3rd ed.Philadelphia:WB Saunders,2000:2013–2041.
- ,,.Significance of pleural effusion in patients with acute pancreatitis.Am J Gastroenterol.1992;87:871–874.
- .Exudative pleural effusions secondary to gastrointestinal diseases.Clin Chest Med.1985;6(1):103–111.
- .Subphrenic abscess.Ann Surg.1963;158:240–248.
- ,,,.Upper abdominal abscess: a continuing and deadly problem.Am J Roentgenol.1980;134:759–765.
- .Subphrenic abscess. A clinical study of 101 cases.Acta Chir Scand.1959;117:388–408.
- ,,.Subphrenic abscess a continuing hazard.Am J Surg.1969:117–122.
- ,.Subphrenic abscess: a thoracoabdominal clinical complex. The changing picture with antibiotics.Am J Surg.1964;108:165–172.
- ,.What have we learned over the past 20 years about appendicitis in the elderly.Am J Surg.2003;185:198–201.
- ,,, et al.Appendicitis: why so complicated? Analysis of 5755 consecutive appendectomies.Am Surg.2000;66:548–554.
- ,,.Appendicitis with a palpable mass.Ann Surg.1981;193:227–229.
- ,,, et al.Nonoperative management of perforated appendicitis without periappendiceal mass.Am J Surg.2000;179:177–181.
- ,,.Appendix. In:Townsend CM, ed.Sabiston textbook of surgery. The biologic basis of modern surgical practice. 16th ed.Philadelphia:W. B. Saunders,2001:917–928.
A 49‐year‐old man presented with 2 days of chills, fever, anorexia, and increased cough and dyspnea. The patient had a history of chronic obstructive pulmonary disease (COPD) and noted that his cough and dyspnea had increased above normal for several days. He was now dyspneic with minimal activity and had slept at a 45‐degree incline the night prior to evaluation due to dyspnea. He noted less improvement than usual with the use of his metered dose inhaler. His cough was occasionally productive of small amounts of white phlegm. He had vomited once. During a coughing episode the patient experienced a sudden onset of sharp right upper quadrant abdominal pain that worsened with coughing and sudden position changes. The patient denied a prior history of abdominal pain or surgery. The patient's last bowel movement was 2 days prior to admission. He denied melena or bright red blood per rectum.
My initial differential diagnosis for this patient's dyspnea and cough is pneumonia, acute exacerbation of COPD, or congestive heart failure. The presence of fever and anorexia increases the likelihood of infectious etiologies, whereas the presence of orthopnea points toward congestive heart failure. Noncardiac processessuch as a large pleural effusion or apical lung diseasecould also cause orthopnea. His abdominal pain could be a result of pneumonia alone (perhaps in the right lower lobe with diaphragmatic irritation), but I am also considering complications of pneumonia such as empyema. Although his abdominal pain, dyspnea, and cough could also be a result of hepatobiliary disease, a perforated viscus, or pancreatitis, we currently have little reason to suspect a direct abdominal etiology. My top diagnosis is community‐acquired pneumonia, perhaps accompanied by pleural effusion.
His medical history was significant for dilated cardiomyopathy and heavy alcohol use. His medications included various meter‐dosed inhalers, bupropion, digoxin, spironolactone, lisinopril, and metoprolol. He had never received corticosteroid therapy and had not previously been hospitalized for COPD‐related problems. He had smoked one pack of cigarettes daily for 40 years.
Heavy alcohol use is associated with an increased risk of several pulmonary infections such as gram‐negative necrotizing pneumonia (classically, Klebsiella pneumoniae), pneumococcal pneumonia, aspiration pneumonia, anaerobic lung abscesses, and tuberculosis. Given his right upper quadrant pain, acute alcoholic hepatitis and alcohol‐related pancreatitis enter the differential. His history of cardiomyopathy makes me consider congestive heart failure as more likely than before, and perhaps his abdominal pain is a result of hepatic congestion from right heart failure. His fever, however, cannot be attributed to cardiac failure. Less likely diagnoses include ischemic conditions related to his cardiomyopathy such as mesenteric ischemia from low perfusion or embolism from a cardiac thrombus. A pulmonary infection remains the most likely diagnosis.
He was an ill‐appearing man in moderate respiratory distress, looking older than his stated age. His temperature was 38.4C, heart rate 129 beats/minute, blood pressure 85/56 mm Hg, respiratory rate 24 breaths/minute, and oxygen saturation 92% on room air. A cardiovascular exam revealed no murmur, gallop, or rub. The jugular venous pulse was not elevated. His lungs were clear to auscultation. Abdominal exam revealed right‐sided abdominal tenderness that appeared to localize to the rectus sheath. Otherwise, the abdomen was soft, with normal bowel sounds and no organomegaly. Rectal examination revealed guaiac negative stool and no focal tenderness. His extremities were normal.
His vital signs are worrisome for impending cardiovascular collapse and shock, possibly due to sepsis. The relatively nonfocal cardiopulmonary exam is surprising given his initial symptoms and makes me wonder if his dyspnea is primarily related to an abdominal process leading to diaphragmatic irritation rather than to a thoracic process. Congestive heart failure seems unlikely given the lack of supportive physical examination findings. His abdominal exam findings are puzzling. Although his abdominal wall tenderness could be benignperhaps from muscular strain or a tear from coughingit could represent a more worrisome process such as infection or a hematoma in the abdominal wall muscles. Mesenteric ischemia is still possible, as the exam is often unimpressive. A hepatic abscess or subphrenic abscess should be considered, as physical exam findings in these conditions can be subtle.
My differential remains relatively unchanged, but I have now put consideration of a hepatic or subphrenic abscess higher on my list. Early empiric broad‐spectrum antibiotics seem necessary.
He had a white blood cell count of 26,700/mL with 92% neutrophils, a hemoglobin of 14.6 g/dL, and a platelet count of 312,000/mL. Sodium was 134 mmol/L, potassium was 4.3 mmol/L, chloride was 94 mmol/L, bicarbonate was 23 mmol/L, blood urea nitrogen was 23 mg/dL, and creatinine was 2.1 mg/dL. The results of the calcium, protein, albumin, and liver function tests were normal. Urinalysis was negative for protein and red blood cells. An electrocardiogram revealed sinus tachycardia. A chest radiograph at admission revealed mild opacities in both lower lobes and the right middle lobe consistent with either atelectasis or pneumonia (Fig. 1). A very small left effusion was also identified.
The additional data reinforce my clinical impression that this process is likely to be infectious. The chest radiograph is consistent with community‐acquired pneumonia, possibly from an atypical pathogen. Given his elevated creatinine, I am also considering a pulmonary‐renal syndrome such as vasculitis, though hematuria was not present. A subphrenic abscess, mesenteric ischemia, or an abdominal wall process (because his abdominal tenderness on exam still needs an explanation) remain possibilities; my suspicion would increase if he does not respond appropriately to therapy for community‐acquired pneumonia.
The clinical team's working diagnosis also was community‐acquired pneumonia. Blood and sputum cultures were obtained, and the patient was treated with intravenous ceftriaxone, azithromycin, and intravenous fluid. By the second day, his creatinine had normalized; however, his hypoxemia had worsened, and he now required supplemental oxygen. His temperature was 39.3C, and his heart rate was 150 beats/minute. The findings of an abdominal ultrasound of the kidneys, spleen, and right upper quadrant were normal.
It is too early to say the patient has failed therapy because a patient can get worse before getting better during the course of antibiotic therapy for community‐acquired pneumonia. Fever, for example, may take up to 7 days to resolve, depending on host factors and the pathogen. Though I typically wait about 72 hours before assuming a patient is not appropriately responding to therapy, the additional information has made me concerned. The degree of tachycardia is significant and warrants an EKG to exclude an arrthymia. I would also repeat the chest radiograph to evaluate for worsening infiltrates or increased pleural effusion.
On the third hospital day, the patient's abdominal pain had decreased with analgesia, but his fever, cough, and dyspnea remained largely unchanged. Antibiotics were changed to intravenous levofloxacin. A repeat chest radiograph revealed elevation of the right hemidiaphragm and bilateral effusions (Fig. 2). An electrocardiogram revealed sinus tachycardia. Blood cultures revealed no growth, and sputum cultures grew oral flora.
A significantly elevated right hemidiaphragm makes me reconsider the diagnosis of simple community‐acquired pneumonia. The differential diagnosis for an elevated hemidiaphragm is best considered by location in relation to the diaphragm. Causes above the diaphragm include rib fracture, atelectasis, pleural thickening, and volume loss of the lung for another reason (e.g., surgery, bronchial obstruction due to tumor or mucus plugging), as well as mimics such as a densely consolidated pneumonia, pulmonary infarction, or a subpulmonary effusion. Diaphragmatic causes include eventration, rupture, phrenic nerve weakness, and intrinsic weakness because of neuromuscular disease (usually bilateral). Causes below the diaphragm that must be considered are subphrenic or liver abscess, liver (and other abdominal) malignancy, pancreatic pseudocyst, and distended bowel. Given the clinical picture, I am focusing below the diaphragmespecially on a possible hepatic or subphrenic abscess (which could be missed on ultrasound) and mimics of it such as dense consolidation or a subpulmonary effusion. Given the lack of response to antibiotics, I need to consider an infection that is not being treated, either because of location (abscess, effusion) or microbiology (tuberculosis, a parasite, a fungus, resistant bacteria). After confirming that the patient has a substantive pleural effusion, he needs a thoracentesis.
On the fourth hospital day, his temperature was 38.8C, and his white blood cell count was 21,000/mL. A right‐sided thoracentesis was performed; approximately 250 cc of fluid was obtained. Pleural fluid analysis revealed bloody fluid, with a white blood cell count of 16,750/mL with 94% neutrophils, 40,000 red blood cells/mL, lactate dehydrogenase of 278 U/L (normal serum value 80200 U/L), protein of 3.7 g/dL, and glucose of 81 mg/dL. A pleural fluid pH was not obtained. A gram stain revealed many white blood cells with no organisms noted. Serum protein was 7.4 g/dL. These results were thought to represent an exudative parapneumonic effusion; levofloxacin and supplemental oxygen were continued.
The pleural fluid appears exudative, but I am not sure this man has a parapneumonic effusion because, despite clinical deterioration, an obvious infiltrate is not seen on interval chest radiography. We must look closely at the fluid because this is a bloody effusion and somewhat atypical for a parapneumonic effusion. Also, the effusion does not appear large enough to explain why he has not improved on the current antibiotics. We should thus reconsider our diagnosis and management. I would obtain additional imaging (such as an abdominal and chest computed tomography [CT]) and perhaps obtain a consultation from the pulmonary team regarding the postulated initial diagnosis of pneumonia with effusion.
On the fifth day of hospitalization, the patient's dyspnea and cough persisted but were improved. His abdominal pain was minimal and felt improved with flatus. Fever continued to 38.8C, and the white blood cell count was 20,000/mL. On examination the patient had decreased breath sounds at the right base and bibasilar crackles. His abdomen was soft, with tenderness in his right upper quadrant only with deep palpation; bowel sounds remained. An ultrasound of the chest was performed to look for a loculated effusion; however, no fluid was identified. The pulmonary consultant thought it likely that the patient had a subpulmonic effusion and recommended CT of the abdomen and chest.
His right upper quadrant tenderness is still unexplained. I would agree with the CT, primarily to evaluate other causes of his elevated diaphragm such as subphrenic or hepatic abscess. For now, I would make no change in antibiotic therapy.
On the sixth hospital day, the patient had an episode of bilious emesis. Chest and abdominal CT revealed collapse of the right middle and lower lobes with a small adjacent effusion, and a 6 6 16 cm abscess intimately opposed to the right lobe of the liver. Extending from the inferior extent of the abscess was a tubular thick‐walled structure connecting to the cecum that was suspicious of a thickened inflamed appendix. There was periappendiceal stranding suggesting inflammation. The small bowel was diffusely dilated up to 4.5 cm, suggesting a small bowel obstruction.
I suspect that his abscess is related to a perforated appendix and that the dilated small bowel is most likely a result of localized irritation of the bowel by the abscess and appendicitis. The collapsed lung is most likely due to local inflammation from the subdiaphragmatic abscess. Treatment should now be changed substantially. I would ask a surgeon to evaluate the patient because the most likely diagnosis is perforated appendicitis with abscess formation.
When the periappendiceal abscess was drained percutaneously, 190 mL of purulent fluid was removed. The cultures were positive for Klebsiella pneumonia, Enterococcus faecalis,and Streptococcus milleri. The patient was given 6 weeks of intravenous antibiotics with improvement in his clinical symptoms. During the interval the findings on his chest radiograph resolved completely. A laproscopic appendectomy 3 months later revealed significant right lower quadrant adhesions. The pathology specimen identified a distorted appendix with regeneration consistent with prior appendicitis. The patient was contacted 4 months after his surgery, and he reported that he was doing well, with no cardiopulmonary or gastrointestinal symptoms.
COMMENTARY
Community‐acquired pneumonia (CAP) is a common cause of acute illness and accounts for nearly 1 million admissions per year in the United States.1 The diagnosis of CAP is made when symptoms including dyspnea, fever, cough, or leukocytosis are present, with confirmation provided by a chest radiograph. Often the diagnosis is clear; however, there is no pathognomonic constellation of signs or symptoms that establish the diagnosis with certainty.2 Many physicians learn that pneumoniaespecially lower‐lobe pneumoniacan lead to abdominal findings such as upper quadrant pain, vomiting, and tenderness to palpation. Conversely, the patient discussed above illustrates that a primary abdominal process can also result in a symptom complex that mimics pneumonia.
The prevalence of CAP coupled with the inherent uncertainty of a clinical diagnosis of CAP leads to an important question: How long is too long before questioning the diagnosis? An analysis of the pneumonia Patient Outcomes Research Trial (PORT) limited to inpatients with CAP examined time to clinical stability. For the majority of patients, abnormal vital signs resolved within 23 days.3 In this study, 29% of patients had severe disease, and not surprisingly, these patients took longer to improve. Using the pneumonia severity index score, which accounts for age, comorbidity, abnormal vital signs, and laboratory data, the patient described in this article would be considered at high risk for death and complication with an estimated mortality of 9%.4 Using a combination of defervescence, resolution of tachycardia, tachypnea, and hypoxemia as markers of clinical stability, a patient like ours should respond within 4 days (with a range of 27 days). On the basis of these dataand the discrepancy between the patient's severe illness and relatively minor pulmonary infiltratesit seems reasonable to have considered this patient as failing CAP therapy as early as the fourth day of hospitalization.
In approximately 10% of hospitalized patients with CAP, the clinical course is protracted.5 When patients do not improve as quickly as expected, the reasons that could explain this should be investigated. In a cohort of 49 patients with CAP who failed therapy the most common reasons for failure to improve were severity of the pneumonia and drug resistance.6 A multicenter study found that the incidence of resistance to penicillin by Streptococcus pneumoniae, the most common bacterial pathogen in CAP, was 30%, with a 4% in vitro resistance rate to ceftriaxone.7 How well in vitro resistance predicts clinical response, however, is unclear. Risk factors for antibiotic resistance include close exposure to children, recent antibiotic use, and recent hospitalization. Immunosuppressive conditions should also be considered in patients who fail to improve. Suppurative complications of pneumoniasuch as empyema, parapneumonic effusion, and lung abscessalso delay recovery.
Another consideration in a patient with what appears to be a nonresolving pneumonia with pleural effusion is that the initial diagnosis is incorrect and the cause is extrathoracic. Pulmonary and cardiac diseases account for more than 90% of effusions, whereas less than 5% of pleural effusions result from intraabdominal causes.8 When should intraabdominal diseases be sought in patients with an effusion, fever, dyspnea, and cough? Light suggests that intraabdominal pathology should be investigated in patients who have pleural effusions without significant parenchymal disease.8 This point is underscored by the experience of our patient, whose chest radiographs showed, despite clinical decline, minimal airspace disease.
Several abdominal entities cause pleural effusion. Pancreatitis, either acute or chronic, with pseudocyst formation is the most common abdominal cause of exudative pleural effusions. Approximately 10% of patients with pancreatic disease will develop effusions, usually left‐sided.9 These left‐sided effusions are also seen in splenic abscesses, usually as a result of endocarditis. Intrahepatic abscess is associated with effusions in 20% of patients.10 A subphrenic abscess, as seen in our patient, is an uncommon cause of exudative pleural effusions. Historically, subphrenic abscesses resulted from a perforated viscus, with ruptured appendicitis the most common cause,11 followed by perforated peptic ulcers and biliary tract disease. With the advent of antibiotics, the causes of subphrenic abscess changed considerably, with the majority of current cases resulting from postsurgical complications.12 The findings of a chest radiograph are abnormal in 80% of patients with subphrenic abscess;1214 an elevated hemidiaphragm and pleural effusion are found in the majority of cases. The symptoms of a subphrenic abscess are nonspecific, and patient's complaints are equally split between predominantly thoracic and predomninantly abdominal complaints.15
Appendicitis, a common disease predominantly of the young, may lead to atypical presentations in older individuals. In a retrospective analysis of 113 patients older than 60 years with appendicitis, 70% presented in an atypical fashion.16 Typical symptoms include right lower quadrant pain, fever, anorexia and a white blood cell count greater than 10,000/mL. Fever was the most frequently absent symptom, seen in only 37% of older patients. In this cohort, approximately one third of older patients waited more than 48 hours prior to presentation. The time between symptom onset and clinical presentation is a strong predictor of perforation risk.17 As in this case, roughly 2% of patients with acute appendicitis will present with perforation and abscess formation.18 In such patients the management is initially conservative. Percutaneous drainage and broad spectrum antibiotics are the treatment of choice, followed by an interval appendectomy in 612 weeks.19 The rationale for delayed surgery is that earlier surgery may disseminate a localized inflammatory process.20
Community‐acquired pneumonia is a more frequent cause of hospital admission than is intraabdominal abscess. Physicians often face the dilemma of when to pursue alternative diagnoses after a patient who is thought to have an atypical presentation of a common disease (ie, CAP) fails to respond to conventional therapy. Although clinicians learn that right upper quadrant pain may be a symptom of pneumonia, our patient revealed that abdominal causes may mimic pneumonia and produce a pleural effusion. Determining whether the primary disease originates above or below the diaphragm is critical to guiding therapy. When patients fail to respond adequately to therapy, clinicians should set a low threshold for deciding to image the abdomen in a patient with modest pulmonary infiltrates, pleural effusion, and abdominal pain.
A 49‐year‐old man presented with 2 days of chills, fever, anorexia, and increased cough and dyspnea. The patient had a history of chronic obstructive pulmonary disease (COPD) and noted that his cough and dyspnea had increased above normal for several days. He was now dyspneic with minimal activity and had slept at a 45‐degree incline the night prior to evaluation due to dyspnea. He noted less improvement than usual with the use of his metered dose inhaler. His cough was occasionally productive of small amounts of white phlegm. He had vomited once. During a coughing episode the patient experienced a sudden onset of sharp right upper quadrant abdominal pain that worsened with coughing and sudden position changes. The patient denied a prior history of abdominal pain or surgery. The patient's last bowel movement was 2 days prior to admission. He denied melena or bright red blood per rectum.
My initial differential diagnosis for this patient's dyspnea and cough is pneumonia, acute exacerbation of COPD, or congestive heart failure. The presence of fever and anorexia increases the likelihood of infectious etiologies, whereas the presence of orthopnea points toward congestive heart failure. Noncardiac processessuch as a large pleural effusion or apical lung diseasecould also cause orthopnea. His abdominal pain could be a result of pneumonia alone (perhaps in the right lower lobe with diaphragmatic irritation), but I am also considering complications of pneumonia such as empyema. Although his abdominal pain, dyspnea, and cough could also be a result of hepatobiliary disease, a perforated viscus, or pancreatitis, we currently have little reason to suspect a direct abdominal etiology. My top diagnosis is community‐acquired pneumonia, perhaps accompanied by pleural effusion.
His medical history was significant for dilated cardiomyopathy and heavy alcohol use. His medications included various meter‐dosed inhalers, bupropion, digoxin, spironolactone, lisinopril, and metoprolol. He had never received corticosteroid therapy and had not previously been hospitalized for COPD‐related problems. He had smoked one pack of cigarettes daily for 40 years.
Heavy alcohol use is associated with an increased risk of several pulmonary infections such as gram‐negative necrotizing pneumonia (classically, Klebsiella pneumoniae), pneumococcal pneumonia, aspiration pneumonia, anaerobic lung abscesses, and tuberculosis. Given his right upper quadrant pain, acute alcoholic hepatitis and alcohol‐related pancreatitis enter the differential. His history of cardiomyopathy makes me consider congestive heart failure as more likely than before, and perhaps his abdominal pain is a result of hepatic congestion from right heart failure. His fever, however, cannot be attributed to cardiac failure. Less likely diagnoses include ischemic conditions related to his cardiomyopathy such as mesenteric ischemia from low perfusion or embolism from a cardiac thrombus. A pulmonary infection remains the most likely diagnosis.
He was an ill‐appearing man in moderate respiratory distress, looking older than his stated age. His temperature was 38.4C, heart rate 129 beats/minute, blood pressure 85/56 mm Hg, respiratory rate 24 breaths/minute, and oxygen saturation 92% on room air. A cardiovascular exam revealed no murmur, gallop, or rub. The jugular venous pulse was not elevated. His lungs were clear to auscultation. Abdominal exam revealed right‐sided abdominal tenderness that appeared to localize to the rectus sheath. Otherwise, the abdomen was soft, with normal bowel sounds and no organomegaly. Rectal examination revealed guaiac negative stool and no focal tenderness. His extremities were normal.
His vital signs are worrisome for impending cardiovascular collapse and shock, possibly due to sepsis. The relatively nonfocal cardiopulmonary exam is surprising given his initial symptoms and makes me wonder if his dyspnea is primarily related to an abdominal process leading to diaphragmatic irritation rather than to a thoracic process. Congestive heart failure seems unlikely given the lack of supportive physical examination findings. His abdominal exam findings are puzzling. Although his abdominal wall tenderness could be benignperhaps from muscular strain or a tear from coughingit could represent a more worrisome process such as infection or a hematoma in the abdominal wall muscles. Mesenteric ischemia is still possible, as the exam is often unimpressive. A hepatic abscess or subphrenic abscess should be considered, as physical exam findings in these conditions can be subtle.
My differential remains relatively unchanged, but I have now put consideration of a hepatic or subphrenic abscess higher on my list. Early empiric broad‐spectrum antibiotics seem necessary.
He had a white blood cell count of 26,700/mL with 92% neutrophils, a hemoglobin of 14.6 g/dL, and a platelet count of 312,000/mL. Sodium was 134 mmol/L, potassium was 4.3 mmol/L, chloride was 94 mmol/L, bicarbonate was 23 mmol/L, blood urea nitrogen was 23 mg/dL, and creatinine was 2.1 mg/dL. The results of the calcium, protein, albumin, and liver function tests were normal. Urinalysis was negative for protein and red blood cells. An electrocardiogram revealed sinus tachycardia. A chest radiograph at admission revealed mild opacities in both lower lobes and the right middle lobe consistent with either atelectasis or pneumonia (Fig. 1). A very small left effusion was also identified.
The additional data reinforce my clinical impression that this process is likely to be infectious. The chest radiograph is consistent with community‐acquired pneumonia, possibly from an atypical pathogen. Given his elevated creatinine, I am also considering a pulmonary‐renal syndrome such as vasculitis, though hematuria was not present. A subphrenic abscess, mesenteric ischemia, or an abdominal wall process (because his abdominal tenderness on exam still needs an explanation) remain possibilities; my suspicion would increase if he does not respond appropriately to therapy for community‐acquired pneumonia.
The clinical team's working diagnosis also was community‐acquired pneumonia. Blood and sputum cultures were obtained, and the patient was treated with intravenous ceftriaxone, azithromycin, and intravenous fluid. By the second day, his creatinine had normalized; however, his hypoxemia had worsened, and he now required supplemental oxygen. His temperature was 39.3C, and his heart rate was 150 beats/minute. The findings of an abdominal ultrasound of the kidneys, spleen, and right upper quadrant were normal.
It is too early to say the patient has failed therapy because a patient can get worse before getting better during the course of antibiotic therapy for community‐acquired pneumonia. Fever, for example, may take up to 7 days to resolve, depending on host factors and the pathogen. Though I typically wait about 72 hours before assuming a patient is not appropriately responding to therapy, the additional information has made me concerned. The degree of tachycardia is significant and warrants an EKG to exclude an arrthymia. I would also repeat the chest radiograph to evaluate for worsening infiltrates or increased pleural effusion.
On the third hospital day, the patient's abdominal pain had decreased with analgesia, but his fever, cough, and dyspnea remained largely unchanged. Antibiotics were changed to intravenous levofloxacin. A repeat chest radiograph revealed elevation of the right hemidiaphragm and bilateral effusions (Fig. 2). An electrocardiogram revealed sinus tachycardia. Blood cultures revealed no growth, and sputum cultures grew oral flora.
A significantly elevated right hemidiaphragm makes me reconsider the diagnosis of simple community‐acquired pneumonia. The differential diagnosis for an elevated hemidiaphragm is best considered by location in relation to the diaphragm. Causes above the diaphragm include rib fracture, atelectasis, pleural thickening, and volume loss of the lung for another reason (e.g., surgery, bronchial obstruction due to tumor or mucus plugging), as well as mimics such as a densely consolidated pneumonia, pulmonary infarction, or a subpulmonary effusion. Diaphragmatic causes include eventration, rupture, phrenic nerve weakness, and intrinsic weakness because of neuromuscular disease (usually bilateral). Causes below the diaphragm that must be considered are subphrenic or liver abscess, liver (and other abdominal) malignancy, pancreatic pseudocyst, and distended bowel. Given the clinical picture, I am focusing below the diaphragmespecially on a possible hepatic or subphrenic abscess (which could be missed on ultrasound) and mimics of it such as dense consolidation or a subpulmonary effusion. Given the lack of response to antibiotics, I need to consider an infection that is not being treated, either because of location (abscess, effusion) or microbiology (tuberculosis, a parasite, a fungus, resistant bacteria). After confirming that the patient has a substantive pleural effusion, he needs a thoracentesis.
On the fourth hospital day, his temperature was 38.8C, and his white blood cell count was 21,000/mL. A right‐sided thoracentesis was performed; approximately 250 cc of fluid was obtained. Pleural fluid analysis revealed bloody fluid, with a white blood cell count of 16,750/mL with 94% neutrophils, 40,000 red blood cells/mL, lactate dehydrogenase of 278 U/L (normal serum value 80200 U/L), protein of 3.7 g/dL, and glucose of 81 mg/dL. A pleural fluid pH was not obtained. A gram stain revealed many white blood cells with no organisms noted. Serum protein was 7.4 g/dL. These results were thought to represent an exudative parapneumonic effusion; levofloxacin and supplemental oxygen were continued.
The pleural fluid appears exudative, but I am not sure this man has a parapneumonic effusion because, despite clinical deterioration, an obvious infiltrate is not seen on interval chest radiography. We must look closely at the fluid because this is a bloody effusion and somewhat atypical for a parapneumonic effusion. Also, the effusion does not appear large enough to explain why he has not improved on the current antibiotics. We should thus reconsider our diagnosis and management. I would obtain additional imaging (such as an abdominal and chest computed tomography [CT]) and perhaps obtain a consultation from the pulmonary team regarding the postulated initial diagnosis of pneumonia with effusion.
On the fifth day of hospitalization, the patient's dyspnea and cough persisted but were improved. His abdominal pain was minimal and felt improved with flatus. Fever continued to 38.8C, and the white blood cell count was 20,000/mL. On examination the patient had decreased breath sounds at the right base and bibasilar crackles. His abdomen was soft, with tenderness in his right upper quadrant only with deep palpation; bowel sounds remained. An ultrasound of the chest was performed to look for a loculated effusion; however, no fluid was identified. The pulmonary consultant thought it likely that the patient had a subpulmonic effusion and recommended CT of the abdomen and chest.
His right upper quadrant tenderness is still unexplained. I would agree with the CT, primarily to evaluate other causes of his elevated diaphragm such as subphrenic or hepatic abscess. For now, I would make no change in antibiotic therapy.
On the sixth hospital day, the patient had an episode of bilious emesis. Chest and abdominal CT revealed collapse of the right middle and lower lobes with a small adjacent effusion, and a 6 6 16 cm abscess intimately opposed to the right lobe of the liver. Extending from the inferior extent of the abscess was a tubular thick‐walled structure connecting to the cecum that was suspicious of a thickened inflamed appendix. There was periappendiceal stranding suggesting inflammation. The small bowel was diffusely dilated up to 4.5 cm, suggesting a small bowel obstruction.
I suspect that his abscess is related to a perforated appendix and that the dilated small bowel is most likely a result of localized irritation of the bowel by the abscess and appendicitis. The collapsed lung is most likely due to local inflammation from the subdiaphragmatic abscess. Treatment should now be changed substantially. I would ask a surgeon to evaluate the patient because the most likely diagnosis is perforated appendicitis with abscess formation.
When the periappendiceal abscess was drained percutaneously, 190 mL of purulent fluid was removed. The cultures were positive for Klebsiella pneumonia, Enterococcus faecalis,and Streptococcus milleri. The patient was given 6 weeks of intravenous antibiotics with improvement in his clinical symptoms. During the interval the findings on his chest radiograph resolved completely. A laproscopic appendectomy 3 months later revealed significant right lower quadrant adhesions. The pathology specimen identified a distorted appendix with regeneration consistent with prior appendicitis. The patient was contacted 4 months after his surgery, and he reported that he was doing well, with no cardiopulmonary or gastrointestinal symptoms.
COMMENTARY
Community‐acquired pneumonia (CAP) is a common cause of acute illness and accounts for nearly 1 million admissions per year in the United States.1 The diagnosis of CAP is made when symptoms including dyspnea, fever, cough, or leukocytosis are present, with confirmation provided by a chest radiograph. Often the diagnosis is clear; however, there is no pathognomonic constellation of signs or symptoms that establish the diagnosis with certainty.2 Many physicians learn that pneumoniaespecially lower‐lobe pneumoniacan lead to abdominal findings such as upper quadrant pain, vomiting, and tenderness to palpation. Conversely, the patient discussed above illustrates that a primary abdominal process can also result in a symptom complex that mimics pneumonia.
The prevalence of CAP coupled with the inherent uncertainty of a clinical diagnosis of CAP leads to an important question: How long is too long before questioning the diagnosis? An analysis of the pneumonia Patient Outcomes Research Trial (PORT) limited to inpatients with CAP examined time to clinical stability. For the majority of patients, abnormal vital signs resolved within 23 days.3 In this study, 29% of patients had severe disease, and not surprisingly, these patients took longer to improve. Using the pneumonia severity index score, which accounts for age, comorbidity, abnormal vital signs, and laboratory data, the patient described in this article would be considered at high risk for death and complication with an estimated mortality of 9%.4 Using a combination of defervescence, resolution of tachycardia, tachypnea, and hypoxemia as markers of clinical stability, a patient like ours should respond within 4 days (with a range of 27 days). On the basis of these dataand the discrepancy between the patient's severe illness and relatively minor pulmonary infiltratesit seems reasonable to have considered this patient as failing CAP therapy as early as the fourth day of hospitalization.
In approximately 10% of hospitalized patients with CAP, the clinical course is protracted.5 When patients do not improve as quickly as expected, the reasons that could explain this should be investigated. In a cohort of 49 patients with CAP who failed therapy the most common reasons for failure to improve were severity of the pneumonia and drug resistance.6 A multicenter study found that the incidence of resistance to penicillin by Streptococcus pneumoniae, the most common bacterial pathogen in CAP, was 30%, with a 4% in vitro resistance rate to ceftriaxone.7 How well in vitro resistance predicts clinical response, however, is unclear. Risk factors for antibiotic resistance include close exposure to children, recent antibiotic use, and recent hospitalization. Immunosuppressive conditions should also be considered in patients who fail to improve. Suppurative complications of pneumoniasuch as empyema, parapneumonic effusion, and lung abscessalso delay recovery.
Another consideration in a patient with what appears to be a nonresolving pneumonia with pleural effusion is that the initial diagnosis is incorrect and the cause is extrathoracic. Pulmonary and cardiac diseases account for more than 90% of effusions, whereas less than 5% of pleural effusions result from intraabdominal causes.8 When should intraabdominal diseases be sought in patients with an effusion, fever, dyspnea, and cough? Light suggests that intraabdominal pathology should be investigated in patients who have pleural effusions without significant parenchymal disease.8 This point is underscored by the experience of our patient, whose chest radiographs showed, despite clinical decline, minimal airspace disease.
Several abdominal entities cause pleural effusion. Pancreatitis, either acute or chronic, with pseudocyst formation is the most common abdominal cause of exudative pleural effusions. Approximately 10% of patients with pancreatic disease will develop effusions, usually left‐sided.9 These left‐sided effusions are also seen in splenic abscesses, usually as a result of endocarditis. Intrahepatic abscess is associated with effusions in 20% of patients.10 A subphrenic abscess, as seen in our patient, is an uncommon cause of exudative pleural effusions. Historically, subphrenic abscesses resulted from a perforated viscus, with ruptured appendicitis the most common cause,11 followed by perforated peptic ulcers and biliary tract disease. With the advent of antibiotics, the causes of subphrenic abscess changed considerably, with the majority of current cases resulting from postsurgical complications.12 The findings of a chest radiograph are abnormal in 80% of patients with subphrenic abscess;1214 an elevated hemidiaphragm and pleural effusion are found in the majority of cases. The symptoms of a subphrenic abscess are nonspecific, and patient's complaints are equally split between predominantly thoracic and predomninantly abdominal complaints.15
Appendicitis, a common disease predominantly of the young, may lead to atypical presentations in older individuals. In a retrospective analysis of 113 patients older than 60 years with appendicitis, 70% presented in an atypical fashion.16 Typical symptoms include right lower quadrant pain, fever, anorexia and a white blood cell count greater than 10,000/mL. Fever was the most frequently absent symptom, seen in only 37% of older patients. In this cohort, approximately one third of older patients waited more than 48 hours prior to presentation. The time between symptom onset and clinical presentation is a strong predictor of perforation risk.17 As in this case, roughly 2% of patients with acute appendicitis will present with perforation and abscess formation.18 In such patients the management is initially conservative. Percutaneous drainage and broad spectrum antibiotics are the treatment of choice, followed by an interval appendectomy in 612 weeks.19 The rationale for delayed surgery is that earlier surgery may disseminate a localized inflammatory process.20
Community‐acquired pneumonia is a more frequent cause of hospital admission than is intraabdominal abscess. Physicians often face the dilemma of when to pursue alternative diagnoses after a patient who is thought to have an atypical presentation of a common disease (ie, CAP) fails to respond to conventional therapy. Although clinicians learn that right upper quadrant pain may be a symptom of pneumonia, our patient revealed that abdominal causes may mimic pneumonia and produce a pleural effusion. Determining whether the primary disease originates above or below the diaphragm is critical to guiding therapy. When patients fail to respond adequately to therapy, clinicians should set a low threshold for deciding to image the abdomen in a patient with modest pulmonary infiltrates, pleural effusion, and abdominal pain.
- ,,, et al.The cost of treating community‐acquired pneumonia.Clin Ther.1998;20:820–827.
- ,,.Does this patient have community‐acquired pneumonia? Diagnosing pneumonia by history and physical examination.JAMA.1997;278:1440–1445.
- ,,, et al.Time to clinical stability in patients hospitalized with community acquired pneumonia. Implications for practice guidelines.JAMA.1998;279:1452–1457.
- ,,, et al.A prediction rule to identify low‐risk patients with community‐acquired pneumonia.N Engl J Med.1997;336:243–250.
- ,,, et al.Utility of fiberoptic bronchoscopy in non resolving pneumonia.Chest.1990;98:1322–1326.
- ,,, et al.Antimicrobial treatment failures in patients with community acquired pneumonia. Causes and prognostic implications.Am J Respir Crit Care Med.2000;162:154–160.
- ,,, et al.Antimicrobial resistance with Streptococcus pneumoniae in the United States, 1997–98.Emerg Infect Dis.1999;5:757–765.
- ,.Pleural effusion. In:Murray JF,Nadel JA, eds.Textbook of respiratory medicine. 3rd ed.Philadelphia:WB Saunders,2000:2013–2041.
- ,,.Significance of pleural effusion in patients with acute pancreatitis.Am J Gastroenterol.1992;87:871–874.
- .Exudative pleural effusions secondary to gastrointestinal diseases.Clin Chest Med.1985;6(1):103–111.
- .Subphrenic abscess.Ann Surg.1963;158:240–248.
- ,,,.Upper abdominal abscess: a continuing and deadly problem.Am J Roentgenol.1980;134:759–765.
- .Subphrenic abscess. A clinical study of 101 cases.Acta Chir Scand.1959;117:388–408.
- ,,.Subphrenic abscess a continuing hazard.Am J Surg.1969:117–122.
- ,.Subphrenic abscess: a thoracoabdominal clinical complex. The changing picture with antibiotics.Am J Surg.1964;108:165–172.
- ,.What have we learned over the past 20 years about appendicitis in the elderly.Am J Surg.2003;185:198–201.
- ,,, et al.Appendicitis: why so complicated? Analysis of 5755 consecutive appendectomies.Am Surg.2000;66:548–554.
- ,,.Appendicitis with a palpable mass.Ann Surg.1981;193:227–229.
- ,,, et al.Nonoperative management of perforated appendicitis without periappendiceal mass.Am J Surg.2000;179:177–181.
- ,,.Appendix. In:Townsend CM, ed.Sabiston textbook of surgery. The biologic basis of modern surgical practice. 16th ed.Philadelphia:W. B. Saunders,2001:917–928.
- ,,, et al.The cost of treating community‐acquired pneumonia.Clin Ther.1998;20:820–827.
- ,,.Does this patient have community‐acquired pneumonia? Diagnosing pneumonia by history and physical examination.JAMA.1997;278:1440–1445.
- ,,, et al.Time to clinical stability in patients hospitalized with community acquired pneumonia. Implications for practice guidelines.JAMA.1998;279:1452–1457.
- ,,, et al.A prediction rule to identify low‐risk patients with community‐acquired pneumonia.N Engl J Med.1997;336:243–250.
- ,,, et al.Utility of fiberoptic bronchoscopy in non resolving pneumonia.Chest.1990;98:1322–1326.
- ,,, et al.Antimicrobial treatment failures in patients with community acquired pneumonia. Causes and prognostic implications.Am J Respir Crit Care Med.2000;162:154–160.
- ,,, et al.Antimicrobial resistance with Streptococcus pneumoniae in the United States, 1997–98.Emerg Infect Dis.1999;5:757–765.
- ,.Pleural effusion. In:Murray JF,Nadel JA, eds.Textbook of respiratory medicine. 3rd ed.Philadelphia:WB Saunders,2000:2013–2041.
- ,,.Significance of pleural effusion in patients with acute pancreatitis.Am J Gastroenterol.1992;87:871–874.
- .Exudative pleural effusions secondary to gastrointestinal diseases.Clin Chest Med.1985;6(1):103–111.
- .Subphrenic abscess.Ann Surg.1963;158:240–248.
- ,,,.Upper abdominal abscess: a continuing and deadly problem.Am J Roentgenol.1980;134:759–765.
- .Subphrenic abscess. A clinical study of 101 cases.Acta Chir Scand.1959;117:388–408.
- ,,.Subphrenic abscess a continuing hazard.Am J Surg.1969:117–122.
- ,.Subphrenic abscess: a thoracoabdominal clinical complex. The changing picture with antibiotics.Am J Surg.1964;108:165–172.
- ,.What have we learned over the past 20 years about appendicitis in the elderly.Am J Surg.2003;185:198–201.
- ,,, et al.Appendicitis: why so complicated? Analysis of 5755 consecutive appendectomies.Am Surg.2000;66:548–554.
- ,,.Appendicitis with a palpable mass.Ann Surg.1981;193:227–229.
- ,,, et al.Nonoperative management of perforated appendicitis without periappendiceal mass.Am J Surg.2000;179:177–181.
- ,,.Appendix. In:Townsend CM, ed.Sabiston textbook of surgery. The biologic basis of modern surgical practice. 16th ed.Philadelphia:W. B. Saunders,2001:917–928.
Hospital‐Acquired Gastrointestinal Bleeding
Gastrointestinal bleeding occurring in hospitalized patients admitted for nongastrointestinal disorders has been extensively studied in intensive care unit patients. However, a systematic study in noncritically ill medical patients has not yet been done. In critically ill patients the incidence of hospital‐acquired gastrointestinal bleeding (GIB) varies from 0.17% to 5%, depending on its definition.16 These bleeding events significantly increase the morbidity and duration of hospitalization.1, 5, 79
Risk factors for bleeding in the intensive care unit include mechanical ventilation, coagulopathy, burns, chronic renal failure, and neurological insults.15 Several studies have found that stress ulcer prophylaxis with histamine‐2 (H2) receptor antagonists, sucralfate, or proton pump inhibitors (PPIs) decreases bleeding in this group of patients, with a relative risk reduction of 29%61%.10, 11 However, use of these drugs outside this high‐risk group has been questioned because of the low overall risk of bleeding.1, 11, 12 Despite their being an unproven benefit in the noncritically ill population, prophylactic H2 antagonists or PPIs are prescribed in an indiscriminant fashion to up to 30%50% of patients admitted to the hospital,13, 14 suggesting that physician preference dictates this practice. To shed light on this issue in noncritically ill patients, we conducted a retrospective casecontrol study in order to identify risk factors that predict hospital‐acquired gastrointestinal bleeding in this group of patients and to assess whether treatment with prophylactic acid suppression was associated with fewer bleeding events. We also sought to characterize the endoscopic lesions in these patients.
MATERIALS AND METHODS
Study Patients
The institutional review board of the Cleveland Clinic Foundation (Cleveland, OH) approved this study. All patients admitted to the General Internal Medicine service between January 1, 1999, and December 31, 2002, were eligible for inclusion. Two types of cases were included: 1) patients admitted for nongastrointestinal illnesses who developed bleeding at least 24 hours after admission and required esophagogastroduodenoscopy (EGD) during hospitalization (designated in‐hospital bleeding), and 2) patients admitted with gastrointestinal bleeding (requiring EGD) who had been hospitalized on the General Medical service during the preceding 4 weeks for a nongastrointestinal illness (designated out‐of‐hospital bleeding). This second group was included to identify risk factors for delayed bleeding that might not be obvious during hospitalization.
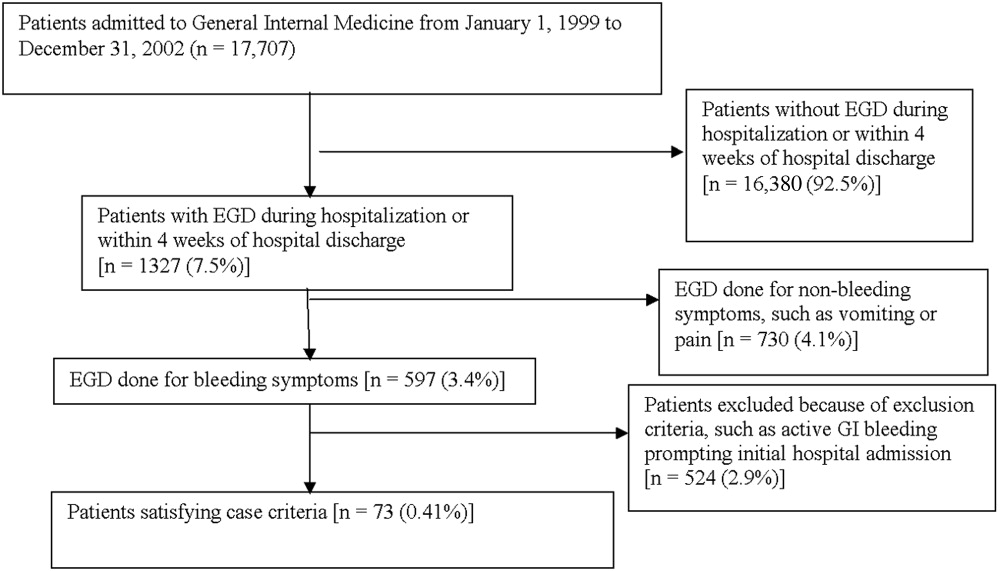
Medical records of all General Medicine patients who underwent EGD were reviewed in a standardized fashion (Fig. 1). We excluded patients with documented gastrointestinal complaints (including bleeding) at the time of the index admission or within 24 hours of admission, bleeding in the intensive care unit (ICU) or in another hospital prior to transfer to the General Medicine service, or a history of gastrointestinal bleeding during the month prior to admission. ICU stay prior to General Medicine admission, if not associated with GI bleeding, was not an exclusion criterion for our study.
Controls, also without any acute gastrointestinal symptoms at admission, were randomly matched to cases in a 1:1 ratio by date of admission. We used this liberal matching strategy because any factors matched for would no longer be eligible to be risk factors for bleeding. If more than one control was admitted on the same day as a case, then a random number was used to select the control.
Definition of Prophylactic Acid Suppression
We defined prophylactic acid suppression as in‐hospital de novo treatment with histamine‐2 receptor antagonists and/or proton pump inhibitors received prior to the onset of any symptoms that would suggest GI bleeding (for cases) or any time during hospitalization (for controls). Patients taking these drugs prior to admission were deemed ineligible for in‐hospital prophylactic acid blockade and were excluded from the related analyses.
Data Collection
We extracted demographic information, medical history, medication usage, and laboratory data by chart review. For those patients readmitted for gastrointestinal bleeding following discharge, data from the initial (nongastrointestinal illnessassociated) hospitalization were recorded. Bleeding symptoms triggering endoscopy were grouped into four categories: 1) melena or hematochezia; 2) hematemesis (frank blood in vomitus or coffee‐grounds emesis); 3) melena or hematochezia plus hematemesis (both 1 and 2); 4) stool positivity for occult blood or unexplained drop in hemoglobin in the absence of overt bleeding. Endoscopic findings were categorized by the nature of the visualized lesions, and if multiple lesions were noted, the endoscopist's impression of the most likely bleeding site was used to define the source of bleeding. We recorded colonoscopy findings for patients undergoing this evaluation.
Statistical Analysis
We analyzed data utilizing JMP 5.1 (SAS Institute, Cary, NC). Random controls were chosen using computer‐generated random numbers. The proportions of patients with various categorical characteristics were compared using the chi‐square test or Fisher's exact test as appropriate. We used the Student t test or Wilcoxon's test to compare continuous variables. Odds ratios and adjusted odds ratios were calculated by logistic regression. Two‐tailed P values less than .05 were considered statistically significant.
RESULTS: Identification of Cases and Controls
Of 17,707 patients admitted to the General Medicine service, 1327 (7.5%) underwent EGD during hospitalization or within 1 month of discharge. Only 73 (0.41%) of the total number of patients met the case definition (Fig. 1). Of these cases, 62 (84.9%) had developed gastrointestinal bleeding during the index hospitalization, whereas 11 (15.1%) were readmitted for bleeding within 4 weeks of hospital discharge. The remaining 1254 patients who underwent EGD were excluded based on exclusion criteria, including an absence of documented bleeding prompting the EGD.
Clinical Risk Factors for Bleeding
In univariate analysis, as shown in Table 1, predictors of GIB included: 1) age (P = .02); 2) admission diagnosis (P = .01); 3) preexisting coronary artery disease (P = .004); 4) treatment with blood‐thinning medications, including warfarin (P = .0004), intravenous heparin (P = .0003), and clopidogrel (P = .02); and 5) treatment with PPIs (P = .02). After adjusting for the use of full‐dose anticoagulation and/or clopidogrel, the only of these risk factors that remained significantly associated with GIB was treatment with PPIs prior to hospitalization (adjusted OR = 2.1; 95% CI 1.17.0; P = .04), suggesting that PPI treatment in the outpatient setting may be a marker for GI vulnerability.
| Characteristic | Cases n = 73 | Controls n = 73 | Unadjusted | Adjusted for treatment with full‐dose anticoagulants or clopidogrel | ||
|---|---|---|---|---|---|---|
| Odds ratio (95% CI) | P value (2‐tailed) | Odds ratio (95% CI) | P value (2‐tailed) | |||
| ||||||
| Demographics | ||||||
| Women | 36 (49.3%) | 29 (39.7%) | 1.5 (0.82.9) | .24 | 1.6 (0.83.3) | .19 |
| Age (years), mean (SD) | 71.6 (13.7) | 65.7 (17.2) | 1.5 (1.12.1)c | .02 | 1.3 (0.91.8) | .19 |
| Caucasian | 42 (58.3%) | 32 (44.4%) | 1.7 (0.93.4) | .09 | 1.3 (0.62.6) | .50 |
| Nursing home residents | 5 (6.9%) | 5 (6.9%) | 1.0 (0.33.7) | >.99 | 0.5 (0.12.2) | .35 |
| Admission diagnosisa | .01d | .30d | ||||
| Cardiovascular (non‐thrombotic) | 15 (20.5%) | 6 (8.2%) | 2.9 (1.18.5) | .04 | 2.1 (0.76.5) | .19 |
| Arterial or venous thrombosis | 13 (17.8%) | 2 (2.7%) | 7.9 (2.050.4) | .009 | 3.3 (0.822.1) | .15 |
| Infection | 21 (28.8%) | 24 (32.9%) | 0.8 (0.41.7) | .59 | 1.1 (0.52.3) | .86 |
| Pulmonary (noninfectious) | 4 (5.5%) | 10 (13.7%) | 0.4 (0.11.2) | .10 | 0.5 (0.11.7) | .31 |
| Altered level of consciousness | 7 (9.6%) | 10 (13.7%) | 0.7 (0.21.8) | .44 | 0.7 (0.22.2) | .59 |
| Other | 13 (17.8%) | 21 (28.8%) | 0.5 (0.21.2) | .12 | 0.6 (0.31.5) | .29 |
| Baseline medical conditions | ||||||
| Diabetes mellitus | 28 (38.4%) | 25 (34.3%) | 1.2 (0.62.4) | .61 | 1.3 (0.62.7) | .48 |
| Hypertension | 50 (68.5%) | 48 (65.8%) | 1.1 (0.62.3) | .72 | 1.2 (0.52.5) | .71 |
| Coronary artery disease | 36 (49.3%) | 19 (26.0%) | 2.8 (1.45.6) | .004 | 2.0 (1.04.3) | .06 |
| Atrial fibrillation | 18 (24.7%) | 10 (13.7%) | 2.1 (0.95.0) | .09 | 1.4 (0.53.6) | .49 |
| Congestive heart failure | 25 (34.3%) | 16 (21.9%) | 1.9 (0.93.9) | .10 | 1.5 (0.73.3) | .35 |
| Renal insufficiency (creatinine > 2) | 18 (24.7%) | 11 (15.1%) | 1.8 (0.84.4) | .14 | 1.9 (0.84.7) | .33 |
| Chronic obstructive pulmonary disease | 21 (28.8%) | 20 (27.4%) | 1.1 (0.52.2) | .85 | 1.5 (0.73.4) | .29 |
| Stroke | 13 (17.8%) | 16 (21.9%) | 0.8 (0.31.7) | .53 | 0.7 (0.31.6) | .39 |
| Active malignancy | 6 (8.2%) | 8 (11.0%) | 0.7 (0.32.2) | .57 | 1.0 (0.33.5) | .80 |
| Gastroesophageal reflux (GERD) | 10 (13.7%) | 10 (13.7%) | 1.0 (0.42.6) | >.99 | 1.0 (0.32.7) | .92 |
| Liver disease | 7 (9.6%) | 6 (8.2%) | 1.2 (0.43.9) | .77 | 1.4 (0.44.9) | .59 |
| Peptic ulcer disease | 13 (17.8%) | 5 (6.9%) | 2.9 (1.09.6) | .04 | 2.7 (0.99.4) | .09 |
| Colonic disease (diverticulosis, polyp, or AVM) | 7 (9.6%) | 4 (5.5%) | 1.8 (0.57.3) | .34 | 1.2 (0.35.2) | .79 |
| Prior gastrointestinal hemorrhage | 15 (20.1%) | 7 (9.6%) | 2.4 (1.06.8) | .06 | 2.0 (0.75.8) | .20 |
| Tobacco abuse (current smoking) | 9 (12.3%) | 18 (24.7%) | 0.4 (0.21.0) | .05 | 0.6 (0.21.5) | .26 |
| Heavy drinking (>8 drinks/day) | 2 (2.7%) | 2 (2.7%) | 1.0 (0.18.5) | >.99 | 1.3 (0.111.7) | .83 |
| Medication exposure prior to bleeding (excluding acid blockade)b | ||||||
| Aspirin (with or without NSAID) | 34 (46.6%) | 32 (43.8%) | 1.1 (0.62.1) | .74 | 0.7 (0.31.5) | .42 |
| Nonselective NSAID (without aspirin) | 3 (4.1%) | 5 (6.9%) | 0.6 (0.12.5) | .72 | 0.6 (0.12.6) | .44 |
| COX‐2 inhibitors | 3 (4.1%) | 7 (9.6%) | 0.4 (0.11.5) | .18 | 0.3 (0.11.4) | .15 |
| Glucocorticoids | 17 (23.3%) | 20 (27.4%) | 0.8 (0.41.7) | .57 | 0.9 (0.42.1) | .89 |
| Warfarin | 24 (32.9%) | 7 (9.6%) | 4.6 (1.912.4) | .004 | N/A | N/A |
| Unfractionated heparin, UFH (full‐dose intravenous | 23 (31.5%) | 6 (20.7%) | 5.1 (2.114.8) | .0003 | N/A | N/A |
| Full‐dose low‐molecular‐weight heparin (LMWH) | 2 (2.7%) | 0 (0%) | infinity | .50 | N/A | N/A |
| Clopidogrel | 9 (12.3%) | 2 (2.7%) | 5.0 (1.233.5) | .02 | N/A | N/A |
| Prophylactic LMWH or UFH (among 103 patients not on full‐dose anticoagulants) | 19 (47.5%) | 32 (50.8%) | 0.9 (0.41.9) | .74 | N/A | N/A |
| Any treatment with warfarin, full‐dose UFH, full‐ dose LMWH, and/or clopidogrel | 41 (56.2%) | 14 (19.2%) | 5.4 (2.611.7) | <.0001 | N/A | N/A |
| Gastric acid suppression (prior to any gastrointestinal hemorrhage) | ||||||
| H2‐receptor antagonists (H2RA) (total) | 11 (15.1%) | 19 (26.0%) | 0.5 (0.21.1) | .10 | 0.6 (0.31.5) | .31 |
| Taken prior to admission | 6 (8.2%) | 9 (12.3%) | 0.6 (0.21.9) | .41 | 0.6 (0.22.1) | .47 |
| Started de novo at admission | 5 (6.9%) | 10 (13.7%) | 0.5 (0.11.4) | .17 | 0.7 (0.22.2) | .53 |
| Proton‐pump inhibitor (PPI) (total) | 28 (38.6%) | 16 (21.9%) | 2.2 (1.14.7) | .03 | 2.1 (1.04.6) | .07 |
| Taken prior to admission | 20 (27.4%) | 9 (12.3%) | 2.2 (1.14.7) | .02 | 2.7 (1.17.0) | .04 |
| Started de novo at admission | 8 (11.0%) | 7 (9.6%) | 1.2 (0.43.5) | .79 | 1.0 (0.33.2) | .99 |
| Any treatment with PPI or H2RA prior to hemorrhage (total) | 39 (53.4%) | 33 (45.2%) | 1.4 (0.72.7) | .32 | 1.5 (0.73.0) | .28 |
| Taken prior to admission | 26 (35.6%) | 18 (24.7%) | 1.7 (0.83.5) | .15 | 1.7 (0.83.7) | .18 |
| Started de novo at admission (among the 102 patients not taking prior to admission) | 13 (27.7%) | 15 (27.3%) | 1.0 (0.42.4) | .97 | 1.1 (0.42.9) | .80 |
Among patients on warfarin, the peak international normalized ratio (median [IQR]) was 3.0 (1.25.0) for cases and 1.9 (1.64.8) for controls (P = .52). For those on heparin (23 cases and 6 controls), the median peak activated partial thromboplastin time (aPTT) was 67 (5082) and 128 (67180) seconds for cases and controls, respectively (P = .03), a surprising finding that was likely a result of type III error and small sample size.
Outcomes
We found no evidence of major complications from bleeding, as shown in Table 2. As expected, cases were more likely to receive blood transfusions than were controls, but clinically serious outcomes were uncommon in both groups.
| Characteristic | Cases n = 73 | Controls n = 73 | P value |
|---|---|---|---|
| |||
| Pulmonary complicationsa | 4 ( 5.5%) | 2 (2.7%) | .68 |
| Cardiac complicationsb | 4 ( 5.5%) | 3 (4.1%) | >.99 |
| Acute renal failure requiring dialysis | 0 ( 0.0%) | 1 (1.4%) | >.99 |
| Stroke or transient cerebral ischemia | 1 ( 1.4%) | 1 (1.4%) | >.99 |
| Transfer to intensive care unit | 9 (12.3%) | 4 (5.5%) | .14 |
| Blood transfusion required | 46 (63.0%) | 3 (4.1%) | <.0001 |
| All‐cause mortality | 3 ( 4.1%) | 2 (2.7%) | >.99 |
Gastrointestinal Symptoms and Endoscopic Findings
Bleeding symptoms prompting EGD and associated endoscopic findings are shown in Table 3. Findings on colonoscopy (performed in 34 patients) are included. Overall, 54 (74%) patients had a detected abnormality on EGD and/or colonoscopy that was believed to be a likely source of bleeding by the endoscopist, and 19 (26%) had no apparent culprit lesions. Melena and stool positivity for occult blood were the most common manifestations of gastrointestinal bleeding (77%) and also accounted for all the normal endoscopic evaluations. Of the 21 ulcers, 18 (85.7%) had a clean base, 1 (4.8%) had a red spot, and 2 (9.5%) had an adherent clot. None had a bleeding vessel. Endoscopic treatment was performed in one patient and angiography in one patient. A possible gastric stromal tumor (not the source of bleeding) was seen in one patient, but no mucosal malignant lesions were identified. Of the 73 cases, 41 (56.2% of cases and 0.2% of the total cohort of 17,707 patients) had culprit lesions that might have been preventable with gastric acid suppression (including peptic ulcers, esophagitis, and duodenitis).
| Most likely primary source of bleeding based on EGD with or without colonoscopya | Hematemesis only n = 10 (13.7% of cases) | Melena or hematochezia n = 33 (45.2% of cases) | Hematemesis plus either melena or hematochezia n = 4 (5.5% of cases) | Occult blood (+) and/or drop in hemoglobin (without overt bleeding) n = 26 (35.6% of cases) |
|---|---|---|---|---|
| ||||
| Normal (no lesions identified n = 19 (26.0% of cases) | 0 | 12 | 0 | 7 |
| Peptic ulcer n = 21 (28.8% of cases) | 4 | 10 | 0 | 7 |
| Esophagitis n = 8 (11.0% of cases) | 2 | 2 | 2 | 2 |
| Gastritis or duodenitis n = 12 (16.4% of cases) | 1 | 6 | 1 | 4 |
| Lower GI source only n = 1 (1.4% of cases) | 0 | 0 | 0 | 1 |
| Miscellaneous upper GI sourceb n = 12 (16.4% of cases) | 3 | 3 | 1 | 5 |
Prophylactic Gastric Acid Suppression
One hundred and two patients were not taking any acid‐suppressive prophylaxis on admission to the hospital. Of these patients, on admission 28 (27.5%) were prescribed either histamine‐2 receptor antagonists or proton pump inhibitors. We identified no clinical features associated with the prescriptions for these medications (Table 4), suggesting that physician preference, rather than perceived risk factors for bleeding, determined which patients received prophylactic acid blockade. There was no association between this prophylaxis and GI bleeding, but because of the small size of our sample, the confidence interval was wide (OR = 1.0; 95% CI 0.42.4; P = .97). In the analysis of the subgroup of patients receiving anticoagulation or clopidogrel, prophylaxis showed a nonsignificant trend toward benefit (OR = 0.71; 95% CI 0.23.9; P = .67). There was no significant interaction between the presence of anticoagulation or clopidogrel and prophylaxis (P = .61). Similarly, when we excluded those without prior GI bleeding from analysis, there was still no apparent protective effect of acid‐suppressive prophylaxis (OR = 1.0; 95% CI 0.42.5; P = .97). Finally, there was no significant association between the use of prophylaxis and lesions (theoretically) preventable by acid blockade (OR = 0.9; 95% CI 0.32.3; P = .84).
| Characteristic | Prophylaxis | |||
|---|---|---|---|---|
| Initiated n = 28 (27.5%) | Withheld n = 74 (72.5%) | Odds ratio (95% CI) | P value | |
| ||||
| Cases | ||||
| All lesions | 13 (46.4%) | 34 (46.0%) | 1.0 (0.42.4) | .97 |
| Lesions preventable with acid blockadeb | 8 (28.6%) | 20 (27.0%) | 1.1 (0.42.8) | .88 |
| Demographics | ||||
| Age, in years (SD) | 70.3 (18.6) | 66.9 (15.7) | 1.2 (0.81.9) | .40 |
| Female | 10 (35.7%) | 36 (48.7%) | 0.6 (0.21.4) | .24 |
| Medical history | ||||
| Prior gastrointestinal bleeding | 4 (14.3%) | 7 ( 9.5%) | 1.6 (0.45.8) | .49 |
| History of GERD | 1 ( 3.6%) | 4 ( 5.4%) | 0.6 (0.04.6) | >.99 |
| History of peptic ulcer disease | 3 (10.7%) | 5 ( 6.8%) | 1.7 (0.37.3) | .68 |
| Hospitalization variables | ||||
| Transferred from ICU | 2 ( 7.1%) | 4 ( 5.4%) | 1.3 (0.27.3) | .67 |
| Cardiovascular admission diagnosis | 7 (25.0%) | 21 (28.4%) | 0.8 (0.32.2) | .73 |
| Medication exposure | ||||
| Aspirin (with or without NSAID) | 13 (46.4%) | 30 (40.5%) | 1.3 (0.53.1) | .59 |
| NSAID alone (nonselective) | 2 ( 7.1%) | 4 ( 5.4%) | 1.3 (0.27.3) | .67 |
| Glucocorticoids | 6 (21.4%) | 19 (25.7%) | 0.8 (0.32.2) | .65 |
| Warfarin, clopidogrel, or IV heparin | 9 (32.1%) | 28 (37.8%) | 0.8 (0.31.9) | .59 |
DISCUSSION
Our data suggest that the incidence of hospital‐acquired gastrointestinal bleeding in noncritically ill medical patients is low (approximately 0.4%) and that treatment with anticoagulants or clopidogrel predisposes to this complication. Anticoagulation is a well‐known risk factor for gastrointestinal bleeding, with an estimated odds ratio of 2416; our study confirmed this risk.
Although some studies have questioned the utility of prophylactic acid blockade in the intensive care unit,15 the weight of current evidence supports prophylaxis in selected critically ill patients. In a randomized double‐blind study of 1200 mechanically ventilated patients, the relative risk of gastrointestinal bleeding in patients treated with ranitidine was 0.44 (95% CI 0.210.92 P = .02).11 Many experts discourage indiscriminant use of prophylaxes, even by patients in intensive care units, recommending that it be used only in patients with established risk factors for bleeding.1, 12
Despite the absence of evidence of any benefit of the use of prophylactic acid blockade outside the intensive care unit, this practice is common. In our study, 27.5% of patients who were not on outpatient acid suppression medications (PPIs or H2 antagonists) were started on them on admission to the hospital, presumably as prophylaxes, as we excluded patients admitted for acute gastrointestinal complaints. Other studies have reported prophylaxis rates of 30%50%.13, 14 Many patients started on this prophylaxis during hospitalization go on to take these drugs following discharge, creating an unnecessary economic burden.13, 14 In our study, GI prophylaxis did not appear to prevent hospital‐acquired gastrointestinal bleeding. However, the odds ratio associating the use of prophylactic acid suppression with gastrointestinal bleeding (1.0) was associated with a wide 95% confidence interval (0.42.4), so we cannot exclude the possibility that these medications might provide a relative risk reduction that we were unable to detect. Finally, although gastrointestinal bleeding in the intensive care unit is associated with significant morbidity and mortality,8, 9 we found no evidence to suggest that gastrointestinal bleeding in our patients was associated with poor outcomes.
In interpreting the data from this study, it is important to note that the definition of hospital‐acquired gastrointestinal bleeding in the literature has been inconsistent. Some studies have required that bleeding be hemodynamically significant1, 2, 5, 11a stringent criterion that may be present in only 10%15% of patients with bleeding16whereas other studies defined gastrointestinal bleeding on the basis of occult‐blood‐positive nasogastric aspirates or positive endoscopic findings.7, 15 Because the definition used in the present study required a hard clinical event (EGD), it excluded bleeding events that were considered clinically insignificant by treating physicians. We justified this definition on our belief that any bleeding that warrants invasive evaluation is clinically relevant because it is expensive and puts the patient at some physical risk. Even though some of our patients were diagnosed with GIB without obvious melena or hematemesis (ie, based on stool positivity for occult blood), many of these patients had significant drops in hemoglobin during hospitalization, which, accompanied by occult blood positivity, justified inpatient EGD. We do not believe our definition of GI bleeding was too restrictive, at least for our institution, as physicians at the Cleveland Clinic generally pursue inpatient EGD with clinically apparent gastrointestinal bleeding; we maintain that bleeding that is minor enough not to change management is of limited clinical relevance. Nevertheless, the threshold for EGD at a given institution could affect the rate of EGD for soft indications and the overall prevalence of nosocomial GI bleeding based on our definition.
It also is worth noting that our definition of nosocomial bleeding encompassed some patients with recent hospitalization on the medical service who bled following discharge (15% of cases in this study). This inclusion criterion was chosen because of our concern that the stress of hospitalization might lead to complications even after discharge. We chose an arbitrary postdischarge cutoff of 4 weeks. When we excluded these patients from analysis, the results were similar (data not shown). Although it is possible that we missed some patient who presented to other institutions with GI bleeding following discharge from the Cleveland Clinic, we suspect that the number of such patients was very small based on current referral patterns.
We do not have complete information to determine exactly why patients were on acid‐suppressive therapy prior to admission, but the available data suggest that many had gastroesophageal reflux disease (GERD), PUD, or prior GI bleeding. For this reason, we focused the investigation of the potential efficacy of prophylactic initiation of acid blockade among patients who at presentation were not taking these medications, as prior GERD (or undocumented GIB) leading to chronic use of acid blockade may predispose to subsequent GIB. Although we analyzed only those patients who had newly started taking acid‐suppressive medications, we acknowledge that a few of them may have been started on these medications for other reasons, like chest pain or GERD. However, the evidence suggests that an overwhelming number are started on these medications for the sole purpose of GI prophylaxis.13, 14
Our study was limited by its retrospective casecontrol design. However, because of the low prevalence of hospital‐acquired gastrointestinal bleeding outside the critical care unit, a prospective study would have to enroll thousands of patients in order to generate statistically meaningful results.
In summary, hospital‐acquired gastrointestinal bleeding outside the intensive care unit is uncommon, with an incidence of about 0.4% according to our definition of bleeding. We found no evidence that these bleeding episodes are associated with increased mortality or with occult malignancy. Furthermore, we found no evidence that prophylactic gastric acid suppression prevents these events, and only 41 patients (0.2% of the total cohort) had lesions that might be preventable with gastric acid blockade. We discourage the indiscriminant use of prophylactic acid suppressants in general medical patients.
Acknowledgements
The authors thank Donna M. Richey and Betty Lou Harrison for clerical support.
- ,,, et al.Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group.N Engl J Med.1994;330:377–381.
- ,,, et al.Risk factors for clinically important upper gastrointestinal bleeding in patients requiring mechanical ventilation. Canadian Critical Care Trials Group.Crit Care Med.1999;27:2812–2817.
- ,,,,.Prospective evaluation of the risk of upper gastrointestinal bleeding after admission to a medical intensive care unit.Am J Med.1984;76:623–630.
- ,,, et al.Risk factors for hospitalized gastrointestinal bleeding among older persons. Cardiovascular Health Study Investigators.J Am Geriatr Soc.2001;49:126–133.
- ,.Gastrointestinal bleeding in the hospitalized patient: a case–control study to assess risk factors, causes, and outcome.Am J Med.1998;104:349–354.
- ,,.Characterization of gastrointestinal bleeding in severely ill hospitalized patients.Crit Care Med.2000;28:46–50.
- ,,,,.Clinically significant gastrointestinal bleeding in critically ill patients in an era of prophylaxis.Am J Gastroenterol.2000;95:2801–2806.
- ,,, et al.The attributable mortality and length of intensive care unit stay of clinically important gastrointestinal bleeding in critically ill patients.Crit Care.2001;5:368–375.
- ,,,.Risks for developing critical illness with GI hemorrhage.Chest.2000;118:473–478.
- .Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Best evidence synthesis.Scand J GastroenterolSuppl.1995;210:48–52.
- ,,, et al.A comparison of sucralfate and ranitidine for the prevention of upper gastrointestinal bleeding in patients requiring mechanical ventilation. Canadian Critical Care Trials Group.N Engl J Med.1998;338:791–797.
- ,.Stress ulcer: is routine prophylaxis necessary?Am J Gastroenterol.1995;90:708–712.
- ,,, et al.Hospital use of acid‐suppressive medications and its fall‐out on prescribing in general practice: a 1‐month survey.Aliment Pharmacol Ther.2003;17:1503–1506.
- ,,.Overuse of acid‐suppressive therapy in hospitalized patients.Am J Gastroenterol.2000;95:3118–122.
- ,,, et al.Prophylaxis for stress‐related gastric hemorrhage in the medical intensive care unit. A randomized, controlled, single‐blind study.Ann Intern Med.1994;121:568–575.
- .Low incidence of hemodynamic instability in patients with gastrointestinal hemorrhage.South Med J.1996;89:386–390.
Gastrointestinal bleeding occurring in hospitalized patients admitted for nongastrointestinal disorders has been extensively studied in intensive care unit patients. However, a systematic study in noncritically ill medical patients has not yet been done. In critically ill patients the incidence of hospital‐acquired gastrointestinal bleeding (GIB) varies from 0.17% to 5%, depending on its definition.16 These bleeding events significantly increase the morbidity and duration of hospitalization.1, 5, 79
Risk factors for bleeding in the intensive care unit include mechanical ventilation, coagulopathy, burns, chronic renal failure, and neurological insults.15 Several studies have found that stress ulcer prophylaxis with histamine‐2 (H2) receptor antagonists, sucralfate, or proton pump inhibitors (PPIs) decreases bleeding in this group of patients, with a relative risk reduction of 29%61%.10, 11 However, use of these drugs outside this high‐risk group has been questioned because of the low overall risk of bleeding.1, 11, 12 Despite their being an unproven benefit in the noncritically ill population, prophylactic H2 antagonists or PPIs are prescribed in an indiscriminant fashion to up to 30%50% of patients admitted to the hospital,13, 14 suggesting that physician preference dictates this practice. To shed light on this issue in noncritically ill patients, we conducted a retrospective casecontrol study in order to identify risk factors that predict hospital‐acquired gastrointestinal bleeding in this group of patients and to assess whether treatment with prophylactic acid suppression was associated with fewer bleeding events. We also sought to characterize the endoscopic lesions in these patients.
MATERIALS AND METHODS
Study Patients
The institutional review board of the Cleveland Clinic Foundation (Cleveland, OH) approved this study. All patients admitted to the General Internal Medicine service between January 1, 1999, and December 31, 2002, were eligible for inclusion. Two types of cases were included: 1) patients admitted for nongastrointestinal illnesses who developed bleeding at least 24 hours after admission and required esophagogastroduodenoscopy (EGD) during hospitalization (designated in‐hospital bleeding), and 2) patients admitted with gastrointestinal bleeding (requiring EGD) who had been hospitalized on the General Medical service during the preceding 4 weeks for a nongastrointestinal illness (designated out‐of‐hospital bleeding). This second group was included to identify risk factors for delayed bleeding that might not be obvious during hospitalization.
Medical records of all General Medicine patients who underwent EGD were reviewed in a standardized fashion (Fig. 1). We excluded patients with documented gastrointestinal complaints (including bleeding) at the time of the index admission or within 24 hours of admission, bleeding in the intensive care unit (ICU) or in another hospital prior to transfer to the General Medicine service, or a history of gastrointestinal bleeding during the month prior to admission. ICU stay prior to General Medicine admission, if not associated with GI bleeding, was not an exclusion criterion for our study.
Controls, also without any acute gastrointestinal symptoms at admission, were randomly matched to cases in a 1:1 ratio by date of admission. We used this liberal matching strategy because any factors matched for would no longer be eligible to be risk factors for bleeding. If more than one control was admitted on the same day as a case, then a random number was used to select the control.
Definition of Prophylactic Acid Suppression
We defined prophylactic acid suppression as in‐hospital de novo treatment with histamine‐2 receptor antagonists and/or proton pump inhibitors received prior to the onset of any symptoms that would suggest GI bleeding (for cases) or any time during hospitalization (for controls). Patients taking these drugs prior to admission were deemed ineligible for in‐hospital prophylactic acid blockade and were excluded from the related analyses.
Data Collection
We extracted demographic information, medical history, medication usage, and laboratory data by chart review. For those patients readmitted for gastrointestinal bleeding following discharge, data from the initial (nongastrointestinal illnessassociated) hospitalization were recorded. Bleeding symptoms triggering endoscopy were grouped into four categories: 1) melena or hematochezia; 2) hematemesis (frank blood in vomitus or coffee‐grounds emesis); 3) melena or hematochezia plus hematemesis (both 1 and 2); 4) stool positivity for occult blood or unexplained drop in hemoglobin in the absence of overt bleeding. Endoscopic findings were categorized by the nature of the visualized lesions, and if multiple lesions were noted, the endoscopist's impression of the most likely bleeding site was used to define the source of bleeding. We recorded colonoscopy findings for patients undergoing this evaluation.
Statistical Analysis
We analyzed data utilizing JMP 5.1 (SAS Institute, Cary, NC). Random controls were chosen using computer‐generated random numbers. The proportions of patients with various categorical characteristics were compared using the chi‐square test or Fisher's exact test as appropriate. We used the Student t test or Wilcoxon's test to compare continuous variables. Odds ratios and adjusted odds ratios were calculated by logistic regression. Two‐tailed P values less than .05 were considered statistically significant.
RESULTS: Identification of Cases and Controls
Of 17,707 patients admitted to the General Medicine service, 1327 (7.5%) underwent EGD during hospitalization or within 1 month of discharge. Only 73 (0.41%) of the total number of patients met the case definition (Fig. 1). Of these cases, 62 (84.9%) had developed gastrointestinal bleeding during the index hospitalization, whereas 11 (15.1%) were readmitted for bleeding within 4 weeks of hospital discharge. The remaining 1254 patients who underwent EGD were excluded based on exclusion criteria, including an absence of documented bleeding prompting the EGD.
Clinical Risk Factors for Bleeding
In univariate analysis, as shown in Table 1, predictors of GIB included: 1) age (P = .02); 2) admission diagnosis (P = .01); 3) preexisting coronary artery disease (P = .004); 4) treatment with blood‐thinning medications, including warfarin (P = .0004), intravenous heparin (P = .0003), and clopidogrel (P = .02); and 5) treatment with PPIs (P = .02). After adjusting for the use of full‐dose anticoagulation and/or clopidogrel, the only of these risk factors that remained significantly associated with GIB was treatment with PPIs prior to hospitalization (adjusted OR = 2.1; 95% CI 1.17.0; P = .04), suggesting that PPI treatment in the outpatient setting may be a marker for GI vulnerability.
| Characteristic | Cases n = 73 | Controls n = 73 | Unadjusted | Adjusted for treatment with full‐dose anticoagulants or clopidogrel | ||
|---|---|---|---|---|---|---|
| Odds ratio (95% CI) | P value (2‐tailed) | Odds ratio (95% CI) | P value (2‐tailed) | |||
| ||||||
| Demographics | ||||||
| Women | 36 (49.3%) | 29 (39.7%) | 1.5 (0.82.9) | .24 | 1.6 (0.83.3) | .19 |
| Age (years), mean (SD) | 71.6 (13.7) | 65.7 (17.2) | 1.5 (1.12.1)c | .02 | 1.3 (0.91.8) | .19 |
| Caucasian | 42 (58.3%) | 32 (44.4%) | 1.7 (0.93.4) | .09 | 1.3 (0.62.6) | .50 |
| Nursing home residents | 5 (6.9%) | 5 (6.9%) | 1.0 (0.33.7) | >.99 | 0.5 (0.12.2) | .35 |
| Admission diagnosisa | .01d | .30d | ||||
| Cardiovascular (non‐thrombotic) | 15 (20.5%) | 6 (8.2%) | 2.9 (1.18.5) | .04 | 2.1 (0.76.5) | .19 |
| Arterial or venous thrombosis | 13 (17.8%) | 2 (2.7%) | 7.9 (2.050.4) | .009 | 3.3 (0.822.1) | .15 |
| Infection | 21 (28.8%) | 24 (32.9%) | 0.8 (0.41.7) | .59 | 1.1 (0.52.3) | .86 |
| Pulmonary (noninfectious) | 4 (5.5%) | 10 (13.7%) | 0.4 (0.11.2) | .10 | 0.5 (0.11.7) | .31 |
| Altered level of consciousness | 7 (9.6%) | 10 (13.7%) | 0.7 (0.21.8) | .44 | 0.7 (0.22.2) | .59 |
| Other | 13 (17.8%) | 21 (28.8%) | 0.5 (0.21.2) | .12 | 0.6 (0.31.5) | .29 |
| Baseline medical conditions | ||||||
| Diabetes mellitus | 28 (38.4%) | 25 (34.3%) | 1.2 (0.62.4) | .61 | 1.3 (0.62.7) | .48 |
| Hypertension | 50 (68.5%) | 48 (65.8%) | 1.1 (0.62.3) | .72 | 1.2 (0.52.5) | .71 |
| Coronary artery disease | 36 (49.3%) | 19 (26.0%) | 2.8 (1.45.6) | .004 | 2.0 (1.04.3) | .06 |
| Atrial fibrillation | 18 (24.7%) | 10 (13.7%) | 2.1 (0.95.0) | .09 | 1.4 (0.53.6) | .49 |
| Congestive heart failure | 25 (34.3%) | 16 (21.9%) | 1.9 (0.93.9) | .10 | 1.5 (0.73.3) | .35 |
| Renal insufficiency (creatinine > 2) | 18 (24.7%) | 11 (15.1%) | 1.8 (0.84.4) | .14 | 1.9 (0.84.7) | .33 |
| Chronic obstructive pulmonary disease | 21 (28.8%) | 20 (27.4%) | 1.1 (0.52.2) | .85 | 1.5 (0.73.4) | .29 |
| Stroke | 13 (17.8%) | 16 (21.9%) | 0.8 (0.31.7) | .53 | 0.7 (0.31.6) | .39 |
| Active malignancy | 6 (8.2%) | 8 (11.0%) | 0.7 (0.32.2) | .57 | 1.0 (0.33.5) | .80 |
| Gastroesophageal reflux (GERD) | 10 (13.7%) | 10 (13.7%) | 1.0 (0.42.6) | >.99 | 1.0 (0.32.7) | .92 |
| Liver disease | 7 (9.6%) | 6 (8.2%) | 1.2 (0.43.9) | .77 | 1.4 (0.44.9) | .59 |
| Peptic ulcer disease | 13 (17.8%) | 5 (6.9%) | 2.9 (1.09.6) | .04 | 2.7 (0.99.4) | .09 |
| Colonic disease (diverticulosis, polyp, or AVM) | 7 (9.6%) | 4 (5.5%) | 1.8 (0.57.3) | .34 | 1.2 (0.35.2) | .79 |
| Prior gastrointestinal hemorrhage | 15 (20.1%) | 7 (9.6%) | 2.4 (1.06.8) | .06 | 2.0 (0.75.8) | .20 |
| Tobacco abuse (current smoking) | 9 (12.3%) | 18 (24.7%) | 0.4 (0.21.0) | .05 | 0.6 (0.21.5) | .26 |
| Heavy drinking (>8 drinks/day) | 2 (2.7%) | 2 (2.7%) | 1.0 (0.18.5) | >.99 | 1.3 (0.111.7) | .83 |
| Medication exposure prior to bleeding (excluding acid blockade)b | ||||||
| Aspirin (with or without NSAID) | 34 (46.6%) | 32 (43.8%) | 1.1 (0.62.1) | .74 | 0.7 (0.31.5) | .42 |
| Nonselective NSAID (without aspirin) | 3 (4.1%) | 5 (6.9%) | 0.6 (0.12.5) | .72 | 0.6 (0.12.6) | .44 |
| COX‐2 inhibitors | 3 (4.1%) | 7 (9.6%) | 0.4 (0.11.5) | .18 | 0.3 (0.11.4) | .15 |
| Glucocorticoids | 17 (23.3%) | 20 (27.4%) | 0.8 (0.41.7) | .57 | 0.9 (0.42.1) | .89 |
| Warfarin | 24 (32.9%) | 7 (9.6%) | 4.6 (1.912.4) | .004 | N/A | N/A |
| Unfractionated heparin, UFH (full‐dose intravenous | 23 (31.5%) | 6 (20.7%) | 5.1 (2.114.8) | .0003 | N/A | N/A |
| Full‐dose low‐molecular‐weight heparin (LMWH) | 2 (2.7%) | 0 (0%) | infinity | .50 | N/A | N/A |
| Clopidogrel | 9 (12.3%) | 2 (2.7%) | 5.0 (1.233.5) | .02 | N/A | N/A |
| Prophylactic LMWH or UFH (among 103 patients not on full‐dose anticoagulants) | 19 (47.5%) | 32 (50.8%) | 0.9 (0.41.9) | .74 | N/A | N/A |
| Any treatment with warfarin, full‐dose UFH, full‐ dose LMWH, and/or clopidogrel | 41 (56.2%) | 14 (19.2%) | 5.4 (2.611.7) | <.0001 | N/A | N/A |
| Gastric acid suppression (prior to any gastrointestinal hemorrhage) | ||||||
| H2‐receptor antagonists (H2RA) (total) | 11 (15.1%) | 19 (26.0%) | 0.5 (0.21.1) | .10 | 0.6 (0.31.5) | .31 |
| Taken prior to admission | 6 (8.2%) | 9 (12.3%) | 0.6 (0.21.9) | .41 | 0.6 (0.22.1) | .47 |
| Started de novo at admission | 5 (6.9%) | 10 (13.7%) | 0.5 (0.11.4) | .17 | 0.7 (0.22.2) | .53 |
| Proton‐pump inhibitor (PPI) (total) | 28 (38.6%) | 16 (21.9%) | 2.2 (1.14.7) | .03 | 2.1 (1.04.6) | .07 |
| Taken prior to admission | 20 (27.4%) | 9 (12.3%) | 2.2 (1.14.7) | .02 | 2.7 (1.17.0) | .04 |
| Started de novo at admission | 8 (11.0%) | 7 (9.6%) | 1.2 (0.43.5) | .79 | 1.0 (0.33.2) | .99 |
| Any treatment with PPI or H2RA prior to hemorrhage (total) | 39 (53.4%) | 33 (45.2%) | 1.4 (0.72.7) | .32 | 1.5 (0.73.0) | .28 |
| Taken prior to admission | 26 (35.6%) | 18 (24.7%) | 1.7 (0.83.5) | .15 | 1.7 (0.83.7) | .18 |
| Started de novo at admission (among the 102 patients not taking prior to admission) | 13 (27.7%) | 15 (27.3%) | 1.0 (0.42.4) | .97 | 1.1 (0.42.9) | .80 |
Among patients on warfarin, the peak international normalized ratio (median [IQR]) was 3.0 (1.25.0) for cases and 1.9 (1.64.8) for controls (P = .52). For those on heparin (23 cases and 6 controls), the median peak activated partial thromboplastin time (aPTT) was 67 (5082) and 128 (67180) seconds for cases and controls, respectively (P = .03), a surprising finding that was likely a result of type III error and small sample size.
Outcomes
We found no evidence of major complications from bleeding, as shown in Table 2. As expected, cases were more likely to receive blood transfusions than were controls, but clinically serious outcomes were uncommon in both groups.
| Characteristic | Cases n = 73 | Controls n = 73 | P value |
|---|---|---|---|
| |||
| Pulmonary complicationsa | 4 ( 5.5%) | 2 (2.7%) | .68 |
| Cardiac complicationsb | 4 ( 5.5%) | 3 (4.1%) | >.99 |
| Acute renal failure requiring dialysis | 0 ( 0.0%) | 1 (1.4%) | >.99 |
| Stroke or transient cerebral ischemia | 1 ( 1.4%) | 1 (1.4%) | >.99 |
| Transfer to intensive care unit | 9 (12.3%) | 4 (5.5%) | .14 |
| Blood transfusion required | 46 (63.0%) | 3 (4.1%) | <.0001 |
| All‐cause mortality | 3 ( 4.1%) | 2 (2.7%) | >.99 |
Gastrointestinal Symptoms and Endoscopic Findings
Bleeding symptoms prompting EGD and associated endoscopic findings are shown in Table 3. Findings on colonoscopy (performed in 34 patients) are included. Overall, 54 (74%) patients had a detected abnormality on EGD and/or colonoscopy that was believed to be a likely source of bleeding by the endoscopist, and 19 (26%) had no apparent culprit lesions. Melena and stool positivity for occult blood were the most common manifestations of gastrointestinal bleeding (77%) and also accounted for all the normal endoscopic evaluations. Of the 21 ulcers, 18 (85.7%) had a clean base, 1 (4.8%) had a red spot, and 2 (9.5%) had an adherent clot. None had a bleeding vessel. Endoscopic treatment was performed in one patient and angiography in one patient. A possible gastric stromal tumor (not the source of bleeding) was seen in one patient, but no mucosal malignant lesions were identified. Of the 73 cases, 41 (56.2% of cases and 0.2% of the total cohort of 17,707 patients) had culprit lesions that might have been preventable with gastric acid suppression (including peptic ulcers, esophagitis, and duodenitis).
| Most likely primary source of bleeding based on EGD with or without colonoscopya | Hematemesis only n = 10 (13.7% of cases) | Melena or hematochezia n = 33 (45.2% of cases) | Hematemesis plus either melena or hematochezia n = 4 (5.5% of cases) | Occult blood (+) and/or drop in hemoglobin (without overt bleeding) n = 26 (35.6% of cases) |
|---|---|---|---|---|
| ||||
| Normal (no lesions identified n = 19 (26.0% of cases) | 0 | 12 | 0 | 7 |
| Peptic ulcer n = 21 (28.8% of cases) | 4 | 10 | 0 | 7 |
| Esophagitis n = 8 (11.0% of cases) | 2 | 2 | 2 | 2 |
| Gastritis or duodenitis n = 12 (16.4% of cases) | 1 | 6 | 1 | 4 |
| Lower GI source only n = 1 (1.4% of cases) | 0 | 0 | 0 | 1 |
| Miscellaneous upper GI sourceb n = 12 (16.4% of cases) | 3 | 3 | 1 | 5 |
Prophylactic Gastric Acid Suppression
One hundred and two patients were not taking any acid‐suppressive prophylaxis on admission to the hospital. Of these patients, on admission 28 (27.5%) were prescribed either histamine‐2 receptor antagonists or proton pump inhibitors. We identified no clinical features associated with the prescriptions for these medications (Table 4), suggesting that physician preference, rather than perceived risk factors for bleeding, determined which patients received prophylactic acid blockade. There was no association between this prophylaxis and GI bleeding, but because of the small size of our sample, the confidence interval was wide (OR = 1.0; 95% CI 0.42.4; P = .97). In the analysis of the subgroup of patients receiving anticoagulation or clopidogrel, prophylaxis showed a nonsignificant trend toward benefit (OR = 0.71; 95% CI 0.23.9; P = .67). There was no significant interaction between the presence of anticoagulation or clopidogrel and prophylaxis (P = .61). Similarly, when we excluded those without prior GI bleeding from analysis, there was still no apparent protective effect of acid‐suppressive prophylaxis (OR = 1.0; 95% CI 0.42.5; P = .97). Finally, there was no significant association between the use of prophylaxis and lesions (theoretically) preventable by acid blockade (OR = 0.9; 95% CI 0.32.3; P = .84).
| Characteristic | Prophylaxis | |||
|---|---|---|---|---|
| Initiated n = 28 (27.5%) | Withheld n = 74 (72.5%) | Odds ratio (95% CI) | P value | |
| ||||
| Cases | ||||
| All lesions | 13 (46.4%) | 34 (46.0%) | 1.0 (0.42.4) | .97 |
| Lesions preventable with acid blockadeb | 8 (28.6%) | 20 (27.0%) | 1.1 (0.42.8) | .88 |
| Demographics | ||||
| Age, in years (SD) | 70.3 (18.6) | 66.9 (15.7) | 1.2 (0.81.9) | .40 |
| Female | 10 (35.7%) | 36 (48.7%) | 0.6 (0.21.4) | .24 |
| Medical history | ||||
| Prior gastrointestinal bleeding | 4 (14.3%) | 7 ( 9.5%) | 1.6 (0.45.8) | .49 |
| History of GERD | 1 ( 3.6%) | 4 ( 5.4%) | 0.6 (0.04.6) | >.99 |
| History of peptic ulcer disease | 3 (10.7%) | 5 ( 6.8%) | 1.7 (0.37.3) | .68 |
| Hospitalization variables | ||||
| Transferred from ICU | 2 ( 7.1%) | 4 ( 5.4%) | 1.3 (0.27.3) | .67 |
| Cardiovascular admission diagnosis | 7 (25.0%) | 21 (28.4%) | 0.8 (0.32.2) | .73 |
| Medication exposure | ||||
| Aspirin (with or without NSAID) | 13 (46.4%) | 30 (40.5%) | 1.3 (0.53.1) | .59 |
| NSAID alone (nonselective) | 2 ( 7.1%) | 4 ( 5.4%) | 1.3 (0.27.3) | .67 |
| Glucocorticoids | 6 (21.4%) | 19 (25.7%) | 0.8 (0.32.2) | .65 |
| Warfarin, clopidogrel, or IV heparin | 9 (32.1%) | 28 (37.8%) | 0.8 (0.31.9) | .59 |
DISCUSSION
Our data suggest that the incidence of hospital‐acquired gastrointestinal bleeding in noncritically ill medical patients is low (approximately 0.4%) and that treatment with anticoagulants or clopidogrel predisposes to this complication. Anticoagulation is a well‐known risk factor for gastrointestinal bleeding, with an estimated odds ratio of 2416; our study confirmed this risk.
Although some studies have questioned the utility of prophylactic acid blockade in the intensive care unit,15 the weight of current evidence supports prophylaxis in selected critically ill patients. In a randomized double‐blind study of 1200 mechanically ventilated patients, the relative risk of gastrointestinal bleeding in patients treated with ranitidine was 0.44 (95% CI 0.210.92 P = .02).11 Many experts discourage indiscriminant use of prophylaxes, even by patients in intensive care units, recommending that it be used only in patients with established risk factors for bleeding.1, 12
Despite the absence of evidence of any benefit of the use of prophylactic acid blockade outside the intensive care unit, this practice is common. In our study, 27.5% of patients who were not on outpatient acid suppression medications (PPIs or H2 antagonists) were started on them on admission to the hospital, presumably as prophylaxes, as we excluded patients admitted for acute gastrointestinal complaints. Other studies have reported prophylaxis rates of 30%50%.13, 14 Many patients started on this prophylaxis during hospitalization go on to take these drugs following discharge, creating an unnecessary economic burden.13, 14 In our study, GI prophylaxis did not appear to prevent hospital‐acquired gastrointestinal bleeding. However, the odds ratio associating the use of prophylactic acid suppression with gastrointestinal bleeding (1.0) was associated with a wide 95% confidence interval (0.42.4), so we cannot exclude the possibility that these medications might provide a relative risk reduction that we were unable to detect. Finally, although gastrointestinal bleeding in the intensive care unit is associated with significant morbidity and mortality,8, 9 we found no evidence to suggest that gastrointestinal bleeding in our patients was associated with poor outcomes.
In interpreting the data from this study, it is important to note that the definition of hospital‐acquired gastrointestinal bleeding in the literature has been inconsistent. Some studies have required that bleeding be hemodynamically significant1, 2, 5, 11a stringent criterion that may be present in only 10%15% of patients with bleeding16whereas other studies defined gastrointestinal bleeding on the basis of occult‐blood‐positive nasogastric aspirates or positive endoscopic findings.7, 15 Because the definition used in the present study required a hard clinical event (EGD), it excluded bleeding events that were considered clinically insignificant by treating physicians. We justified this definition on our belief that any bleeding that warrants invasive evaluation is clinically relevant because it is expensive and puts the patient at some physical risk. Even though some of our patients were diagnosed with GIB without obvious melena or hematemesis (ie, based on stool positivity for occult blood), many of these patients had significant drops in hemoglobin during hospitalization, which, accompanied by occult blood positivity, justified inpatient EGD. We do not believe our definition of GI bleeding was too restrictive, at least for our institution, as physicians at the Cleveland Clinic generally pursue inpatient EGD with clinically apparent gastrointestinal bleeding; we maintain that bleeding that is minor enough not to change management is of limited clinical relevance. Nevertheless, the threshold for EGD at a given institution could affect the rate of EGD for soft indications and the overall prevalence of nosocomial GI bleeding based on our definition.
It also is worth noting that our definition of nosocomial bleeding encompassed some patients with recent hospitalization on the medical service who bled following discharge (15% of cases in this study). This inclusion criterion was chosen because of our concern that the stress of hospitalization might lead to complications even after discharge. We chose an arbitrary postdischarge cutoff of 4 weeks. When we excluded these patients from analysis, the results were similar (data not shown). Although it is possible that we missed some patient who presented to other institutions with GI bleeding following discharge from the Cleveland Clinic, we suspect that the number of such patients was very small based on current referral patterns.
We do not have complete information to determine exactly why patients were on acid‐suppressive therapy prior to admission, but the available data suggest that many had gastroesophageal reflux disease (GERD), PUD, or prior GI bleeding. For this reason, we focused the investigation of the potential efficacy of prophylactic initiation of acid blockade among patients who at presentation were not taking these medications, as prior GERD (or undocumented GIB) leading to chronic use of acid blockade may predispose to subsequent GIB. Although we analyzed only those patients who had newly started taking acid‐suppressive medications, we acknowledge that a few of them may have been started on these medications for other reasons, like chest pain or GERD. However, the evidence suggests that an overwhelming number are started on these medications for the sole purpose of GI prophylaxis.13, 14
Our study was limited by its retrospective casecontrol design. However, because of the low prevalence of hospital‐acquired gastrointestinal bleeding outside the critical care unit, a prospective study would have to enroll thousands of patients in order to generate statistically meaningful results.
In summary, hospital‐acquired gastrointestinal bleeding outside the intensive care unit is uncommon, with an incidence of about 0.4% according to our definition of bleeding. We found no evidence that these bleeding episodes are associated with increased mortality or with occult malignancy. Furthermore, we found no evidence that prophylactic gastric acid suppression prevents these events, and only 41 patients (0.2% of the total cohort) had lesions that might be preventable with gastric acid blockade. We discourage the indiscriminant use of prophylactic acid suppressants in general medical patients.
Acknowledgements
The authors thank Donna M. Richey and Betty Lou Harrison for clerical support.
Gastrointestinal bleeding occurring in hospitalized patients admitted for nongastrointestinal disorders has been extensively studied in intensive care unit patients. However, a systematic study in noncritically ill medical patients has not yet been done. In critically ill patients the incidence of hospital‐acquired gastrointestinal bleeding (GIB) varies from 0.17% to 5%, depending on its definition.16 These bleeding events significantly increase the morbidity and duration of hospitalization.1, 5, 79
Risk factors for bleeding in the intensive care unit include mechanical ventilation, coagulopathy, burns, chronic renal failure, and neurological insults.15 Several studies have found that stress ulcer prophylaxis with histamine‐2 (H2) receptor antagonists, sucralfate, or proton pump inhibitors (PPIs) decreases bleeding in this group of patients, with a relative risk reduction of 29%61%.10, 11 However, use of these drugs outside this high‐risk group has been questioned because of the low overall risk of bleeding.1, 11, 12 Despite their being an unproven benefit in the noncritically ill population, prophylactic H2 antagonists or PPIs are prescribed in an indiscriminant fashion to up to 30%50% of patients admitted to the hospital,13, 14 suggesting that physician preference dictates this practice. To shed light on this issue in noncritically ill patients, we conducted a retrospective casecontrol study in order to identify risk factors that predict hospital‐acquired gastrointestinal bleeding in this group of patients and to assess whether treatment with prophylactic acid suppression was associated with fewer bleeding events. We also sought to characterize the endoscopic lesions in these patients.
MATERIALS AND METHODS
Study Patients
The institutional review board of the Cleveland Clinic Foundation (Cleveland, OH) approved this study. All patients admitted to the General Internal Medicine service between January 1, 1999, and December 31, 2002, were eligible for inclusion. Two types of cases were included: 1) patients admitted for nongastrointestinal illnesses who developed bleeding at least 24 hours after admission and required esophagogastroduodenoscopy (EGD) during hospitalization (designated in‐hospital bleeding), and 2) patients admitted with gastrointestinal bleeding (requiring EGD) who had been hospitalized on the General Medical service during the preceding 4 weeks for a nongastrointestinal illness (designated out‐of‐hospital bleeding). This second group was included to identify risk factors for delayed bleeding that might not be obvious during hospitalization.
Medical records of all General Medicine patients who underwent EGD were reviewed in a standardized fashion (Fig. 1). We excluded patients with documented gastrointestinal complaints (including bleeding) at the time of the index admission or within 24 hours of admission, bleeding in the intensive care unit (ICU) or in another hospital prior to transfer to the General Medicine service, or a history of gastrointestinal bleeding during the month prior to admission. ICU stay prior to General Medicine admission, if not associated with GI bleeding, was not an exclusion criterion for our study.
Controls, also without any acute gastrointestinal symptoms at admission, were randomly matched to cases in a 1:1 ratio by date of admission. We used this liberal matching strategy because any factors matched for would no longer be eligible to be risk factors for bleeding. If more than one control was admitted on the same day as a case, then a random number was used to select the control.
Definition of Prophylactic Acid Suppression
We defined prophylactic acid suppression as in‐hospital de novo treatment with histamine‐2 receptor antagonists and/or proton pump inhibitors received prior to the onset of any symptoms that would suggest GI bleeding (for cases) or any time during hospitalization (for controls). Patients taking these drugs prior to admission were deemed ineligible for in‐hospital prophylactic acid blockade and were excluded from the related analyses.
Data Collection
We extracted demographic information, medical history, medication usage, and laboratory data by chart review. For those patients readmitted for gastrointestinal bleeding following discharge, data from the initial (nongastrointestinal illnessassociated) hospitalization were recorded. Bleeding symptoms triggering endoscopy were grouped into four categories: 1) melena or hematochezia; 2) hematemesis (frank blood in vomitus or coffee‐grounds emesis); 3) melena or hematochezia plus hematemesis (both 1 and 2); 4) stool positivity for occult blood or unexplained drop in hemoglobin in the absence of overt bleeding. Endoscopic findings were categorized by the nature of the visualized lesions, and if multiple lesions were noted, the endoscopist's impression of the most likely bleeding site was used to define the source of bleeding. We recorded colonoscopy findings for patients undergoing this evaluation.
Statistical Analysis
We analyzed data utilizing JMP 5.1 (SAS Institute, Cary, NC). Random controls were chosen using computer‐generated random numbers. The proportions of patients with various categorical characteristics were compared using the chi‐square test or Fisher's exact test as appropriate. We used the Student t test or Wilcoxon's test to compare continuous variables. Odds ratios and adjusted odds ratios were calculated by logistic regression. Two‐tailed P values less than .05 were considered statistically significant.
RESULTS: Identification of Cases and Controls
Of 17,707 patients admitted to the General Medicine service, 1327 (7.5%) underwent EGD during hospitalization or within 1 month of discharge. Only 73 (0.41%) of the total number of patients met the case definition (Fig. 1). Of these cases, 62 (84.9%) had developed gastrointestinal bleeding during the index hospitalization, whereas 11 (15.1%) were readmitted for bleeding within 4 weeks of hospital discharge. The remaining 1254 patients who underwent EGD were excluded based on exclusion criteria, including an absence of documented bleeding prompting the EGD.
Clinical Risk Factors for Bleeding
In univariate analysis, as shown in Table 1, predictors of GIB included: 1) age (P = .02); 2) admission diagnosis (P = .01); 3) preexisting coronary artery disease (P = .004); 4) treatment with blood‐thinning medications, including warfarin (P = .0004), intravenous heparin (P = .0003), and clopidogrel (P = .02); and 5) treatment with PPIs (P = .02). After adjusting for the use of full‐dose anticoagulation and/or clopidogrel, the only of these risk factors that remained significantly associated with GIB was treatment with PPIs prior to hospitalization (adjusted OR = 2.1; 95% CI 1.17.0; P = .04), suggesting that PPI treatment in the outpatient setting may be a marker for GI vulnerability.
| Characteristic | Cases n = 73 | Controls n = 73 | Unadjusted | Adjusted for treatment with full‐dose anticoagulants or clopidogrel | ||
|---|---|---|---|---|---|---|
| Odds ratio (95% CI) | P value (2‐tailed) | Odds ratio (95% CI) | P value (2‐tailed) | |||
| ||||||
| Demographics | ||||||
| Women | 36 (49.3%) | 29 (39.7%) | 1.5 (0.82.9) | .24 | 1.6 (0.83.3) | .19 |
| Age (years), mean (SD) | 71.6 (13.7) | 65.7 (17.2) | 1.5 (1.12.1)c | .02 | 1.3 (0.91.8) | .19 |
| Caucasian | 42 (58.3%) | 32 (44.4%) | 1.7 (0.93.4) | .09 | 1.3 (0.62.6) | .50 |
| Nursing home residents | 5 (6.9%) | 5 (6.9%) | 1.0 (0.33.7) | >.99 | 0.5 (0.12.2) | .35 |
| Admission diagnosisa | .01d | .30d | ||||
| Cardiovascular (non‐thrombotic) | 15 (20.5%) | 6 (8.2%) | 2.9 (1.18.5) | .04 | 2.1 (0.76.5) | .19 |
| Arterial or venous thrombosis | 13 (17.8%) | 2 (2.7%) | 7.9 (2.050.4) | .009 | 3.3 (0.822.1) | .15 |
| Infection | 21 (28.8%) | 24 (32.9%) | 0.8 (0.41.7) | .59 | 1.1 (0.52.3) | .86 |
| Pulmonary (noninfectious) | 4 (5.5%) | 10 (13.7%) | 0.4 (0.11.2) | .10 | 0.5 (0.11.7) | .31 |
| Altered level of consciousness | 7 (9.6%) | 10 (13.7%) | 0.7 (0.21.8) | .44 | 0.7 (0.22.2) | .59 |
| Other | 13 (17.8%) | 21 (28.8%) | 0.5 (0.21.2) | .12 | 0.6 (0.31.5) | .29 |
| Baseline medical conditions | ||||||
| Diabetes mellitus | 28 (38.4%) | 25 (34.3%) | 1.2 (0.62.4) | .61 | 1.3 (0.62.7) | .48 |
| Hypertension | 50 (68.5%) | 48 (65.8%) | 1.1 (0.62.3) | .72 | 1.2 (0.52.5) | .71 |
| Coronary artery disease | 36 (49.3%) | 19 (26.0%) | 2.8 (1.45.6) | .004 | 2.0 (1.04.3) | .06 |
| Atrial fibrillation | 18 (24.7%) | 10 (13.7%) | 2.1 (0.95.0) | .09 | 1.4 (0.53.6) | .49 |
| Congestive heart failure | 25 (34.3%) | 16 (21.9%) | 1.9 (0.93.9) | .10 | 1.5 (0.73.3) | .35 |
| Renal insufficiency (creatinine > 2) | 18 (24.7%) | 11 (15.1%) | 1.8 (0.84.4) | .14 | 1.9 (0.84.7) | .33 |
| Chronic obstructive pulmonary disease | 21 (28.8%) | 20 (27.4%) | 1.1 (0.52.2) | .85 | 1.5 (0.73.4) | .29 |
| Stroke | 13 (17.8%) | 16 (21.9%) | 0.8 (0.31.7) | .53 | 0.7 (0.31.6) | .39 |
| Active malignancy | 6 (8.2%) | 8 (11.0%) | 0.7 (0.32.2) | .57 | 1.0 (0.33.5) | .80 |
| Gastroesophageal reflux (GERD) | 10 (13.7%) | 10 (13.7%) | 1.0 (0.42.6) | >.99 | 1.0 (0.32.7) | .92 |
| Liver disease | 7 (9.6%) | 6 (8.2%) | 1.2 (0.43.9) | .77 | 1.4 (0.44.9) | .59 |
| Peptic ulcer disease | 13 (17.8%) | 5 (6.9%) | 2.9 (1.09.6) | .04 | 2.7 (0.99.4) | .09 |
| Colonic disease (diverticulosis, polyp, or AVM) | 7 (9.6%) | 4 (5.5%) | 1.8 (0.57.3) | .34 | 1.2 (0.35.2) | .79 |
| Prior gastrointestinal hemorrhage | 15 (20.1%) | 7 (9.6%) | 2.4 (1.06.8) | .06 | 2.0 (0.75.8) | .20 |
| Tobacco abuse (current smoking) | 9 (12.3%) | 18 (24.7%) | 0.4 (0.21.0) | .05 | 0.6 (0.21.5) | .26 |
| Heavy drinking (>8 drinks/day) | 2 (2.7%) | 2 (2.7%) | 1.0 (0.18.5) | >.99 | 1.3 (0.111.7) | .83 |
| Medication exposure prior to bleeding (excluding acid blockade)b | ||||||
| Aspirin (with or without NSAID) | 34 (46.6%) | 32 (43.8%) | 1.1 (0.62.1) | .74 | 0.7 (0.31.5) | .42 |
| Nonselective NSAID (without aspirin) | 3 (4.1%) | 5 (6.9%) | 0.6 (0.12.5) | .72 | 0.6 (0.12.6) | .44 |
| COX‐2 inhibitors | 3 (4.1%) | 7 (9.6%) | 0.4 (0.11.5) | .18 | 0.3 (0.11.4) | .15 |
| Glucocorticoids | 17 (23.3%) | 20 (27.4%) | 0.8 (0.41.7) | .57 | 0.9 (0.42.1) | .89 |
| Warfarin | 24 (32.9%) | 7 (9.6%) | 4.6 (1.912.4) | .004 | N/A | N/A |
| Unfractionated heparin, UFH (full‐dose intravenous | 23 (31.5%) | 6 (20.7%) | 5.1 (2.114.8) | .0003 | N/A | N/A |
| Full‐dose low‐molecular‐weight heparin (LMWH) | 2 (2.7%) | 0 (0%) | infinity | .50 | N/A | N/A |
| Clopidogrel | 9 (12.3%) | 2 (2.7%) | 5.0 (1.233.5) | .02 | N/A | N/A |
| Prophylactic LMWH or UFH (among 103 patients not on full‐dose anticoagulants) | 19 (47.5%) | 32 (50.8%) | 0.9 (0.41.9) | .74 | N/A | N/A |
| Any treatment with warfarin, full‐dose UFH, full‐ dose LMWH, and/or clopidogrel | 41 (56.2%) | 14 (19.2%) | 5.4 (2.611.7) | <.0001 | N/A | N/A |
| Gastric acid suppression (prior to any gastrointestinal hemorrhage) | ||||||
| H2‐receptor antagonists (H2RA) (total) | 11 (15.1%) | 19 (26.0%) | 0.5 (0.21.1) | .10 | 0.6 (0.31.5) | .31 |
| Taken prior to admission | 6 (8.2%) | 9 (12.3%) | 0.6 (0.21.9) | .41 | 0.6 (0.22.1) | .47 |
| Started de novo at admission | 5 (6.9%) | 10 (13.7%) | 0.5 (0.11.4) | .17 | 0.7 (0.22.2) | .53 |
| Proton‐pump inhibitor (PPI) (total) | 28 (38.6%) | 16 (21.9%) | 2.2 (1.14.7) | .03 | 2.1 (1.04.6) | .07 |
| Taken prior to admission | 20 (27.4%) | 9 (12.3%) | 2.2 (1.14.7) | .02 | 2.7 (1.17.0) | .04 |
| Started de novo at admission | 8 (11.0%) | 7 (9.6%) | 1.2 (0.43.5) | .79 | 1.0 (0.33.2) | .99 |
| Any treatment with PPI or H2RA prior to hemorrhage (total) | 39 (53.4%) | 33 (45.2%) | 1.4 (0.72.7) | .32 | 1.5 (0.73.0) | .28 |
| Taken prior to admission | 26 (35.6%) | 18 (24.7%) | 1.7 (0.83.5) | .15 | 1.7 (0.83.7) | .18 |
| Started de novo at admission (among the 102 patients not taking prior to admission) | 13 (27.7%) | 15 (27.3%) | 1.0 (0.42.4) | .97 | 1.1 (0.42.9) | .80 |
Among patients on warfarin, the peak international normalized ratio (median [IQR]) was 3.0 (1.25.0) for cases and 1.9 (1.64.8) for controls (P = .52). For those on heparin (23 cases and 6 controls), the median peak activated partial thromboplastin time (aPTT) was 67 (5082) and 128 (67180) seconds for cases and controls, respectively (P = .03), a surprising finding that was likely a result of type III error and small sample size.
Outcomes
We found no evidence of major complications from bleeding, as shown in Table 2. As expected, cases were more likely to receive blood transfusions than were controls, but clinically serious outcomes were uncommon in both groups.
| Characteristic | Cases n = 73 | Controls n = 73 | P value |
|---|---|---|---|
| |||
| Pulmonary complicationsa | 4 ( 5.5%) | 2 (2.7%) | .68 |
| Cardiac complicationsb | 4 ( 5.5%) | 3 (4.1%) | >.99 |
| Acute renal failure requiring dialysis | 0 ( 0.0%) | 1 (1.4%) | >.99 |
| Stroke or transient cerebral ischemia | 1 ( 1.4%) | 1 (1.4%) | >.99 |
| Transfer to intensive care unit | 9 (12.3%) | 4 (5.5%) | .14 |
| Blood transfusion required | 46 (63.0%) | 3 (4.1%) | <.0001 |
| All‐cause mortality | 3 ( 4.1%) | 2 (2.7%) | >.99 |
Gastrointestinal Symptoms and Endoscopic Findings
Bleeding symptoms prompting EGD and associated endoscopic findings are shown in Table 3. Findings on colonoscopy (performed in 34 patients) are included. Overall, 54 (74%) patients had a detected abnormality on EGD and/or colonoscopy that was believed to be a likely source of bleeding by the endoscopist, and 19 (26%) had no apparent culprit lesions. Melena and stool positivity for occult blood were the most common manifestations of gastrointestinal bleeding (77%) and also accounted for all the normal endoscopic evaluations. Of the 21 ulcers, 18 (85.7%) had a clean base, 1 (4.8%) had a red spot, and 2 (9.5%) had an adherent clot. None had a bleeding vessel. Endoscopic treatment was performed in one patient and angiography in one patient. A possible gastric stromal tumor (not the source of bleeding) was seen in one patient, but no mucosal malignant lesions were identified. Of the 73 cases, 41 (56.2% of cases and 0.2% of the total cohort of 17,707 patients) had culprit lesions that might have been preventable with gastric acid suppression (including peptic ulcers, esophagitis, and duodenitis).
| Most likely primary source of bleeding based on EGD with or without colonoscopya | Hematemesis only n = 10 (13.7% of cases) | Melena or hematochezia n = 33 (45.2% of cases) | Hematemesis plus either melena or hematochezia n = 4 (5.5% of cases) | Occult blood (+) and/or drop in hemoglobin (without overt bleeding) n = 26 (35.6% of cases) |
|---|---|---|---|---|
| ||||
| Normal (no lesions identified n = 19 (26.0% of cases) | 0 | 12 | 0 | 7 |
| Peptic ulcer n = 21 (28.8% of cases) | 4 | 10 | 0 | 7 |
| Esophagitis n = 8 (11.0% of cases) | 2 | 2 | 2 | 2 |
| Gastritis or duodenitis n = 12 (16.4% of cases) | 1 | 6 | 1 | 4 |
| Lower GI source only n = 1 (1.4% of cases) | 0 | 0 | 0 | 1 |
| Miscellaneous upper GI sourceb n = 12 (16.4% of cases) | 3 | 3 | 1 | 5 |
Prophylactic Gastric Acid Suppression
One hundred and two patients were not taking any acid‐suppressive prophylaxis on admission to the hospital. Of these patients, on admission 28 (27.5%) were prescribed either histamine‐2 receptor antagonists or proton pump inhibitors. We identified no clinical features associated with the prescriptions for these medications (Table 4), suggesting that physician preference, rather than perceived risk factors for bleeding, determined which patients received prophylactic acid blockade. There was no association between this prophylaxis and GI bleeding, but because of the small size of our sample, the confidence interval was wide (OR = 1.0; 95% CI 0.42.4; P = .97). In the analysis of the subgroup of patients receiving anticoagulation or clopidogrel, prophylaxis showed a nonsignificant trend toward benefit (OR = 0.71; 95% CI 0.23.9; P = .67). There was no significant interaction between the presence of anticoagulation or clopidogrel and prophylaxis (P = .61). Similarly, when we excluded those without prior GI bleeding from analysis, there was still no apparent protective effect of acid‐suppressive prophylaxis (OR = 1.0; 95% CI 0.42.5; P = .97). Finally, there was no significant association between the use of prophylaxis and lesions (theoretically) preventable by acid blockade (OR = 0.9; 95% CI 0.32.3; P = .84).
| Characteristic | Prophylaxis | |||
|---|---|---|---|---|
| Initiated n = 28 (27.5%) | Withheld n = 74 (72.5%) | Odds ratio (95% CI) | P value | |
| ||||
| Cases | ||||
| All lesions | 13 (46.4%) | 34 (46.0%) | 1.0 (0.42.4) | .97 |
| Lesions preventable with acid blockadeb | 8 (28.6%) | 20 (27.0%) | 1.1 (0.42.8) | .88 |
| Demographics | ||||
| Age, in years (SD) | 70.3 (18.6) | 66.9 (15.7) | 1.2 (0.81.9) | .40 |
| Female | 10 (35.7%) | 36 (48.7%) | 0.6 (0.21.4) | .24 |
| Medical history | ||||
| Prior gastrointestinal bleeding | 4 (14.3%) | 7 ( 9.5%) | 1.6 (0.45.8) | .49 |
| History of GERD | 1 ( 3.6%) | 4 ( 5.4%) | 0.6 (0.04.6) | >.99 |
| History of peptic ulcer disease | 3 (10.7%) | 5 ( 6.8%) | 1.7 (0.37.3) | .68 |
| Hospitalization variables | ||||
| Transferred from ICU | 2 ( 7.1%) | 4 ( 5.4%) | 1.3 (0.27.3) | .67 |
| Cardiovascular admission diagnosis | 7 (25.0%) | 21 (28.4%) | 0.8 (0.32.2) | .73 |
| Medication exposure | ||||
| Aspirin (with or without NSAID) | 13 (46.4%) | 30 (40.5%) | 1.3 (0.53.1) | .59 |
| NSAID alone (nonselective) | 2 ( 7.1%) | 4 ( 5.4%) | 1.3 (0.27.3) | .67 |
| Glucocorticoids | 6 (21.4%) | 19 (25.7%) | 0.8 (0.32.2) | .65 |
| Warfarin, clopidogrel, or IV heparin | 9 (32.1%) | 28 (37.8%) | 0.8 (0.31.9) | .59 |
DISCUSSION
Our data suggest that the incidence of hospital‐acquired gastrointestinal bleeding in noncritically ill medical patients is low (approximately 0.4%) and that treatment with anticoagulants or clopidogrel predisposes to this complication. Anticoagulation is a well‐known risk factor for gastrointestinal bleeding, with an estimated odds ratio of 2416; our study confirmed this risk.
Although some studies have questioned the utility of prophylactic acid blockade in the intensive care unit,15 the weight of current evidence supports prophylaxis in selected critically ill patients. In a randomized double‐blind study of 1200 mechanically ventilated patients, the relative risk of gastrointestinal bleeding in patients treated with ranitidine was 0.44 (95% CI 0.210.92 P = .02).11 Many experts discourage indiscriminant use of prophylaxes, even by patients in intensive care units, recommending that it be used only in patients with established risk factors for bleeding.1, 12
Despite the absence of evidence of any benefit of the use of prophylactic acid blockade outside the intensive care unit, this practice is common. In our study, 27.5% of patients who were not on outpatient acid suppression medications (PPIs or H2 antagonists) were started on them on admission to the hospital, presumably as prophylaxes, as we excluded patients admitted for acute gastrointestinal complaints. Other studies have reported prophylaxis rates of 30%50%.13, 14 Many patients started on this prophylaxis during hospitalization go on to take these drugs following discharge, creating an unnecessary economic burden.13, 14 In our study, GI prophylaxis did not appear to prevent hospital‐acquired gastrointestinal bleeding. However, the odds ratio associating the use of prophylactic acid suppression with gastrointestinal bleeding (1.0) was associated with a wide 95% confidence interval (0.42.4), so we cannot exclude the possibility that these medications might provide a relative risk reduction that we were unable to detect. Finally, although gastrointestinal bleeding in the intensive care unit is associated with significant morbidity and mortality,8, 9 we found no evidence to suggest that gastrointestinal bleeding in our patients was associated with poor outcomes.
In interpreting the data from this study, it is important to note that the definition of hospital‐acquired gastrointestinal bleeding in the literature has been inconsistent. Some studies have required that bleeding be hemodynamically significant1, 2, 5, 11a stringent criterion that may be present in only 10%15% of patients with bleeding16whereas other studies defined gastrointestinal bleeding on the basis of occult‐blood‐positive nasogastric aspirates or positive endoscopic findings.7, 15 Because the definition used in the present study required a hard clinical event (EGD), it excluded bleeding events that were considered clinically insignificant by treating physicians. We justified this definition on our belief that any bleeding that warrants invasive evaluation is clinically relevant because it is expensive and puts the patient at some physical risk. Even though some of our patients were diagnosed with GIB without obvious melena or hematemesis (ie, based on stool positivity for occult blood), many of these patients had significant drops in hemoglobin during hospitalization, which, accompanied by occult blood positivity, justified inpatient EGD. We do not believe our definition of GI bleeding was too restrictive, at least for our institution, as physicians at the Cleveland Clinic generally pursue inpatient EGD with clinically apparent gastrointestinal bleeding; we maintain that bleeding that is minor enough not to change management is of limited clinical relevance. Nevertheless, the threshold for EGD at a given institution could affect the rate of EGD for soft indications and the overall prevalence of nosocomial GI bleeding based on our definition.
It also is worth noting that our definition of nosocomial bleeding encompassed some patients with recent hospitalization on the medical service who bled following discharge (15% of cases in this study). This inclusion criterion was chosen because of our concern that the stress of hospitalization might lead to complications even after discharge. We chose an arbitrary postdischarge cutoff of 4 weeks. When we excluded these patients from analysis, the results were similar (data not shown). Although it is possible that we missed some patient who presented to other institutions with GI bleeding following discharge from the Cleveland Clinic, we suspect that the number of such patients was very small based on current referral patterns.
We do not have complete information to determine exactly why patients were on acid‐suppressive therapy prior to admission, but the available data suggest that many had gastroesophageal reflux disease (GERD), PUD, or prior GI bleeding. For this reason, we focused the investigation of the potential efficacy of prophylactic initiation of acid blockade among patients who at presentation were not taking these medications, as prior GERD (or undocumented GIB) leading to chronic use of acid blockade may predispose to subsequent GIB. Although we analyzed only those patients who had newly started taking acid‐suppressive medications, we acknowledge that a few of them may have been started on these medications for other reasons, like chest pain or GERD. However, the evidence suggests that an overwhelming number are started on these medications for the sole purpose of GI prophylaxis.13, 14
Our study was limited by its retrospective casecontrol design. However, because of the low prevalence of hospital‐acquired gastrointestinal bleeding outside the critical care unit, a prospective study would have to enroll thousands of patients in order to generate statistically meaningful results.
In summary, hospital‐acquired gastrointestinal bleeding outside the intensive care unit is uncommon, with an incidence of about 0.4% according to our definition of bleeding. We found no evidence that these bleeding episodes are associated with increased mortality or with occult malignancy. Furthermore, we found no evidence that prophylactic gastric acid suppression prevents these events, and only 41 patients (0.2% of the total cohort) had lesions that might be preventable with gastric acid blockade. We discourage the indiscriminant use of prophylactic acid suppressants in general medical patients.
Acknowledgements
The authors thank Donna M. Richey and Betty Lou Harrison for clerical support.
- ,,, et al.Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group.N Engl J Med.1994;330:377–381.
- ,,, et al.Risk factors for clinically important upper gastrointestinal bleeding in patients requiring mechanical ventilation. Canadian Critical Care Trials Group.Crit Care Med.1999;27:2812–2817.
- ,,,,.Prospective evaluation of the risk of upper gastrointestinal bleeding after admission to a medical intensive care unit.Am J Med.1984;76:623–630.
- ,,, et al.Risk factors for hospitalized gastrointestinal bleeding among older persons. Cardiovascular Health Study Investigators.J Am Geriatr Soc.2001;49:126–133.
- ,.Gastrointestinal bleeding in the hospitalized patient: a case–control study to assess risk factors, causes, and outcome.Am J Med.1998;104:349–354.
- ,,.Characterization of gastrointestinal bleeding in severely ill hospitalized patients.Crit Care Med.2000;28:46–50.
- ,,,,.Clinically significant gastrointestinal bleeding in critically ill patients in an era of prophylaxis.Am J Gastroenterol.2000;95:2801–2806.
- ,,, et al.The attributable mortality and length of intensive care unit stay of clinically important gastrointestinal bleeding in critically ill patients.Crit Care.2001;5:368–375.
- ,,,.Risks for developing critical illness with GI hemorrhage.Chest.2000;118:473–478.
- .Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Best evidence synthesis.Scand J GastroenterolSuppl.1995;210:48–52.
- ,,, et al.A comparison of sucralfate and ranitidine for the prevention of upper gastrointestinal bleeding in patients requiring mechanical ventilation. Canadian Critical Care Trials Group.N Engl J Med.1998;338:791–797.
- ,.Stress ulcer: is routine prophylaxis necessary?Am J Gastroenterol.1995;90:708–712.
- ,,, et al.Hospital use of acid‐suppressive medications and its fall‐out on prescribing in general practice: a 1‐month survey.Aliment Pharmacol Ther.2003;17:1503–1506.
- ,,.Overuse of acid‐suppressive therapy in hospitalized patients.Am J Gastroenterol.2000;95:3118–122.
- ,,, et al.Prophylaxis for stress‐related gastric hemorrhage in the medical intensive care unit. A randomized, controlled, single‐blind study.Ann Intern Med.1994;121:568–575.
- .Low incidence of hemodynamic instability in patients with gastrointestinal hemorrhage.South Med J.1996;89:386–390.
- ,,, et al.Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group.N Engl J Med.1994;330:377–381.
- ,,, et al.Risk factors for clinically important upper gastrointestinal bleeding in patients requiring mechanical ventilation. Canadian Critical Care Trials Group.Crit Care Med.1999;27:2812–2817.
- ,,,,.Prospective evaluation of the risk of upper gastrointestinal bleeding after admission to a medical intensive care unit.Am J Med.1984;76:623–630.
- ,,, et al.Risk factors for hospitalized gastrointestinal bleeding among older persons. Cardiovascular Health Study Investigators.J Am Geriatr Soc.2001;49:126–133.
- ,.Gastrointestinal bleeding in the hospitalized patient: a case–control study to assess risk factors, causes, and outcome.Am J Med.1998;104:349–354.
- ,,.Characterization of gastrointestinal bleeding in severely ill hospitalized patients.Crit Care Med.2000;28:46–50.
- ,,,,.Clinically significant gastrointestinal bleeding in critically ill patients in an era of prophylaxis.Am J Gastroenterol.2000;95:2801–2806.
- ,,, et al.The attributable mortality and length of intensive care unit stay of clinically important gastrointestinal bleeding in critically ill patients.Crit Care.2001;5:368–375.
- ,,,.Risks for developing critical illness with GI hemorrhage.Chest.2000;118:473–478.
- .Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Best evidence synthesis.Scand J GastroenterolSuppl.1995;210:48–52.
- ,,, et al.A comparison of sucralfate and ranitidine for the prevention of upper gastrointestinal bleeding in patients requiring mechanical ventilation. Canadian Critical Care Trials Group.N Engl J Med.1998;338:791–797.
- ,.Stress ulcer: is routine prophylaxis necessary?Am J Gastroenterol.1995;90:708–712.
- ,,, et al.Hospital use of acid‐suppressive medications and its fall‐out on prescribing in general practice: a 1‐month survey.Aliment Pharmacol Ther.2003;17:1503–1506.
- ,,.Overuse of acid‐suppressive therapy in hospitalized patients.Am J Gastroenterol.2000;95:3118–122.
- ,,, et al.Prophylaxis for stress‐related gastric hemorrhage in the medical intensive care unit. A randomized, controlled, single‐blind study.Ann Intern Med.1994;121:568–575.
- .Low incidence of hemodynamic instability in patients with gastrointestinal hemorrhage.South Med J.1996;89:386–390.
Copyright © 2006 Society of Hospital Medicine
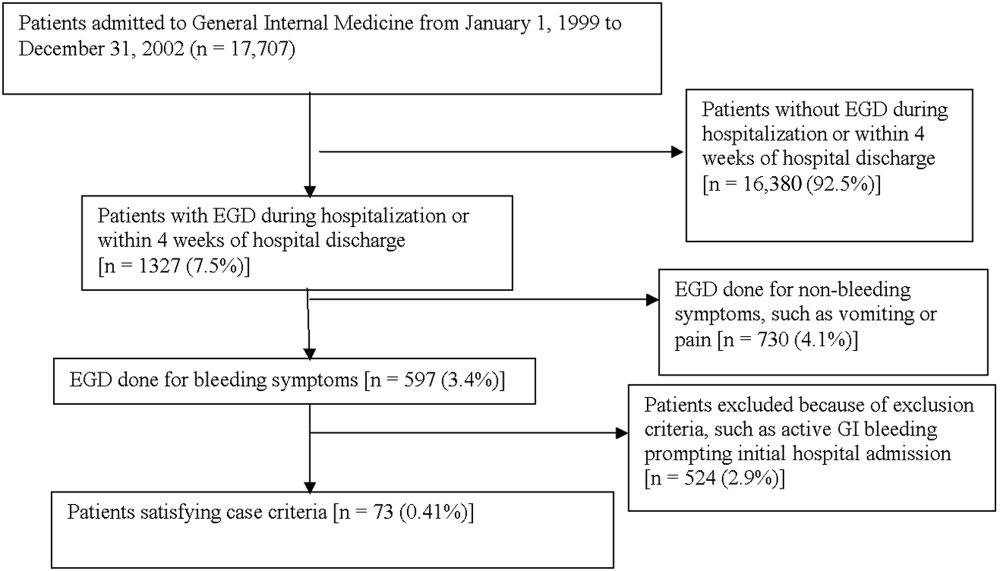
Editorial
It is right and fitting that an article focused on palliative care appears in the inaugural issue of the Journal of Hospital Medicine (JHM).1 Both hospital medicine and palliative care are rapidly growing fields expanding in response to quality and economic imperatives. Both fields recognize the need to develop systems to care for seriously ill patients and to work within interdisciplinary teams. In fact, a natural and mutually beneficial relationship should exist between these two fields. For palliative care, hospital medicine and hospitalists offer the physicians and systems approach to care that could guarantee access to high‐quality palliative care for all hospitalized patients. In addition, hospitalists offer the promise of increasing the number of hospital‐based palliative care programs as the presence of a hospitalist program is strongly associated with having or starting such a program.2, 3 For hospital medicine and hospitalists, palliative care offers a compassionate and high‐quality response to the challenge of caring for seriously and terminally ill patients and their families. By each embracing the other, both fields could find willing and eager partners in the quest to provide the highest possible quality of care for hospitalized patients.
In this first issue of JHM, Dr. Meier offers hospitalists an intriguing and attractive picture of palliative care. She describes how the growth of palliative care is driven by the needs of an ever‐larger group of patients living with chronic and life‐threatening illness and evidence of high quality and satisfaction for these patients who have many physical, emotional, psychological, and spiritual concerns. Dr. Meier also demonstrates how hospital‐based palliative care can coordinate with hospices to provide the continuity of care for terminally ill patients that is often elusive at hospital discharge. Finally, Dr. Meier provides a practical list of resources for clinicians seeking further training in the field. No doubt hospitalists will appreciate this list as the core competencies in hospital medicine, published as a supplement to this issue of JHM, include palliative care, pain management, communication, and discharge planning.
As Dr. Meier states in her article Palliative Care in Hospitals, many types of clinicians can provide palliative care in hospitals, including general internists, nurses, geriatricians, oncologists, hospitalists, and others, yet hospitalists are likely to emerge as the predominant providers of palliative care to hospitalized patients.4 That 75% of Americans die in institutionalized settings, where hospitalists are becoming the dominant providers of care, will drive this prediction.5 In addition, hospitalists are increasingly leading efforts in quality improvement, patient satisfaction, and patient safety.6 Of necessity these initiatives will involve the sickest hospitalized patients and will look to palliative care as a proven response for improving quality and increasing satisfaction.
Hospital medicine and palliative care have other aspects in common that make a melding of the two fields beneficial. Both fields recognize and emphasize the need for interdisciplinary care; good communication between members of the health care team and between health care providers and patients; and timely, effective, and responsible discharge planning. Finally, both fields often rely on multiple sources of funding including professional fee billing and support from the hospital for the added value that programs provide. Sharing so many issues in common should help hospital medicine and palliative care form strong links.
For these links to take hold and for the benefits of this partnership to bear fruit, members of both fields, and especially those with a foot in each, need to reach out. For hospitalists this means getting educated in palliative care, an area for which hospitalists recognize they are underprepared.7 Each hospitalist must be able to provide primary, basic palliative care to each patient.8 Some hospitalists will discover the rewards of palliative care and seek further training and even board certification. These hospitalists can start or join palliative care teams in their institutions. Finally, some hospitalists will become experts in palliative care and join or lead palliative care programs at tertiary care centers. In turn, palliative care providers must reach out to hospitalists. Palliative care clinicians should seek out hospitalists at their institutions and hospices should contact hospitalists at their local hospitals. These programs need to invite hospitalists to participate in the palliative care team and suggest how their services can help the patients of hospitalists. This natural alliance can come about only if both sides reach out.
A partnership between palliative care and hospital medicine will be good for patients and their families as well as for each field, as hospitalists enable realization of the goal of providing palliative care to every patient in the United States. In addition, this partnership will be good for hospitalists who embrace this work. Palliative care can connect us to the humanism and compassion that brought so many of us to medicine and can serve as an antidote to burnout. Furthermore, by caring for patients with life‐threatening illnesses we remember that our time is limited and that each day is a gift. We recognize the importance of making the most of our time regardless of how long we have and of choosing carefully how and with whom we spend our time.
In this first issue of JHM, Dr. Meier makes a strong argument for the need and continued growth of palliative care in hospitals, lays out a strategy for achieving this growth through education and program development, and in doing so, opens the door to hope for the future. Through palliative care we can offer patients hope for healing when cure is not possible, for comfort in the face of suffering, and for what can still be despite all that cannot. The possibility that hospitalists could provide all patients access to palliative care is cause enough for hope. The knowledge that hospitalists will play a major role in making this possibility a reality and may become the predominant providers of palliative care can make that hope a reality.
- .Palliative care in hospitals.J Hosp Med.2006;1:21–28.
- ,.Prevalence and structure of palliative care services in California hospitals.Arch Intern Med.2003;163:1084–1088.
- ,,,,,.Evaluation of the California Hospital Initiative in Palliative Services (CHIPS).Arch Intern Med. In press.
- ,.Palliative care and the hospitalist: an opportunity for cross‐fertilization.Am J Med.2001;111:10S–14S.
- Field MJ,Cassell CK, Eds.Approaching death: improving care at the end of life.Washington, DC:National Academy Press,1997.
- ,.The hospitalist movement 5 years later.JAMA.2002;287:487–494.
- ,,,.Hospitalists' perceptions of their residency training needs: results of a national survey.Am J Med.2001;111:247–254.
- .Secondary and tertiary palliative care in US hospitals.JAMA.2002;287:875–881.
It is right and fitting that an article focused on palliative care appears in the inaugural issue of the Journal of Hospital Medicine (JHM).1 Both hospital medicine and palliative care are rapidly growing fields expanding in response to quality and economic imperatives. Both fields recognize the need to develop systems to care for seriously ill patients and to work within interdisciplinary teams. In fact, a natural and mutually beneficial relationship should exist between these two fields. For palliative care, hospital medicine and hospitalists offer the physicians and systems approach to care that could guarantee access to high‐quality palliative care for all hospitalized patients. In addition, hospitalists offer the promise of increasing the number of hospital‐based palliative care programs as the presence of a hospitalist program is strongly associated with having or starting such a program.2, 3 For hospital medicine and hospitalists, palliative care offers a compassionate and high‐quality response to the challenge of caring for seriously and terminally ill patients and their families. By each embracing the other, both fields could find willing and eager partners in the quest to provide the highest possible quality of care for hospitalized patients.
In this first issue of JHM, Dr. Meier offers hospitalists an intriguing and attractive picture of palliative care. She describes how the growth of palliative care is driven by the needs of an ever‐larger group of patients living with chronic and life‐threatening illness and evidence of high quality and satisfaction for these patients who have many physical, emotional, psychological, and spiritual concerns. Dr. Meier also demonstrates how hospital‐based palliative care can coordinate with hospices to provide the continuity of care for terminally ill patients that is often elusive at hospital discharge. Finally, Dr. Meier provides a practical list of resources for clinicians seeking further training in the field. No doubt hospitalists will appreciate this list as the core competencies in hospital medicine, published as a supplement to this issue of JHM, include palliative care, pain management, communication, and discharge planning.
As Dr. Meier states in her article Palliative Care in Hospitals, many types of clinicians can provide palliative care in hospitals, including general internists, nurses, geriatricians, oncologists, hospitalists, and others, yet hospitalists are likely to emerge as the predominant providers of palliative care to hospitalized patients.4 That 75% of Americans die in institutionalized settings, where hospitalists are becoming the dominant providers of care, will drive this prediction.5 In addition, hospitalists are increasingly leading efforts in quality improvement, patient satisfaction, and patient safety.6 Of necessity these initiatives will involve the sickest hospitalized patients and will look to palliative care as a proven response for improving quality and increasing satisfaction.
Hospital medicine and palliative care have other aspects in common that make a melding of the two fields beneficial. Both fields recognize and emphasize the need for interdisciplinary care; good communication between members of the health care team and between health care providers and patients; and timely, effective, and responsible discharge planning. Finally, both fields often rely on multiple sources of funding including professional fee billing and support from the hospital for the added value that programs provide. Sharing so many issues in common should help hospital medicine and palliative care form strong links.
For these links to take hold and for the benefits of this partnership to bear fruit, members of both fields, and especially those with a foot in each, need to reach out. For hospitalists this means getting educated in palliative care, an area for which hospitalists recognize they are underprepared.7 Each hospitalist must be able to provide primary, basic palliative care to each patient.8 Some hospitalists will discover the rewards of palliative care and seek further training and even board certification. These hospitalists can start or join palliative care teams in their institutions. Finally, some hospitalists will become experts in palliative care and join or lead palliative care programs at tertiary care centers. In turn, palliative care providers must reach out to hospitalists. Palliative care clinicians should seek out hospitalists at their institutions and hospices should contact hospitalists at their local hospitals. These programs need to invite hospitalists to participate in the palliative care team and suggest how their services can help the patients of hospitalists. This natural alliance can come about only if both sides reach out.
A partnership between palliative care and hospital medicine will be good for patients and their families as well as for each field, as hospitalists enable realization of the goal of providing palliative care to every patient in the United States. In addition, this partnership will be good for hospitalists who embrace this work. Palliative care can connect us to the humanism and compassion that brought so many of us to medicine and can serve as an antidote to burnout. Furthermore, by caring for patients with life‐threatening illnesses we remember that our time is limited and that each day is a gift. We recognize the importance of making the most of our time regardless of how long we have and of choosing carefully how and with whom we spend our time.
In this first issue of JHM, Dr. Meier makes a strong argument for the need and continued growth of palliative care in hospitals, lays out a strategy for achieving this growth through education and program development, and in doing so, opens the door to hope for the future. Through palliative care we can offer patients hope for healing when cure is not possible, for comfort in the face of suffering, and for what can still be despite all that cannot. The possibility that hospitalists could provide all patients access to palliative care is cause enough for hope. The knowledge that hospitalists will play a major role in making this possibility a reality and may become the predominant providers of palliative care can make that hope a reality.
It is right and fitting that an article focused on palliative care appears in the inaugural issue of the Journal of Hospital Medicine (JHM).1 Both hospital medicine and palliative care are rapidly growing fields expanding in response to quality and economic imperatives. Both fields recognize the need to develop systems to care for seriously ill patients and to work within interdisciplinary teams. In fact, a natural and mutually beneficial relationship should exist between these two fields. For palliative care, hospital medicine and hospitalists offer the physicians and systems approach to care that could guarantee access to high‐quality palliative care for all hospitalized patients. In addition, hospitalists offer the promise of increasing the number of hospital‐based palliative care programs as the presence of a hospitalist program is strongly associated with having or starting such a program.2, 3 For hospital medicine and hospitalists, palliative care offers a compassionate and high‐quality response to the challenge of caring for seriously and terminally ill patients and their families. By each embracing the other, both fields could find willing and eager partners in the quest to provide the highest possible quality of care for hospitalized patients.
In this first issue of JHM, Dr. Meier offers hospitalists an intriguing and attractive picture of palliative care. She describes how the growth of palliative care is driven by the needs of an ever‐larger group of patients living with chronic and life‐threatening illness and evidence of high quality and satisfaction for these patients who have many physical, emotional, psychological, and spiritual concerns. Dr. Meier also demonstrates how hospital‐based palliative care can coordinate with hospices to provide the continuity of care for terminally ill patients that is often elusive at hospital discharge. Finally, Dr. Meier provides a practical list of resources for clinicians seeking further training in the field. No doubt hospitalists will appreciate this list as the core competencies in hospital medicine, published as a supplement to this issue of JHM, include palliative care, pain management, communication, and discharge planning.
As Dr. Meier states in her article Palliative Care in Hospitals, many types of clinicians can provide palliative care in hospitals, including general internists, nurses, geriatricians, oncologists, hospitalists, and others, yet hospitalists are likely to emerge as the predominant providers of palliative care to hospitalized patients.4 That 75% of Americans die in institutionalized settings, where hospitalists are becoming the dominant providers of care, will drive this prediction.5 In addition, hospitalists are increasingly leading efforts in quality improvement, patient satisfaction, and patient safety.6 Of necessity these initiatives will involve the sickest hospitalized patients and will look to palliative care as a proven response for improving quality and increasing satisfaction.
Hospital medicine and palliative care have other aspects in common that make a melding of the two fields beneficial. Both fields recognize and emphasize the need for interdisciplinary care; good communication between members of the health care team and between health care providers and patients; and timely, effective, and responsible discharge planning. Finally, both fields often rely on multiple sources of funding including professional fee billing and support from the hospital for the added value that programs provide. Sharing so many issues in common should help hospital medicine and palliative care form strong links.
For these links to take hold and for the benefits of this partnership to bear fruit, members of both fields, and especially those with a foot in each, need to reach out. For hospitalists this means getting educated in palliative care, an area for which hospitalists recognize they are underprepared.7 Each hospitalist must be able to provide primary, basic palliative care to each patient.8 Some hospitalists will discover the rewards of palliative care and seek further training and even board certification. These hospitalists can start or join palliative care teams in their institutions. Finally, some hospitalists will become experts in palliative care and join or lead palliative care programs at tertiary care centers. In turn, palliative care providers must reach out to hospitalists. Palliative care clinicians should seek out hospitalists at their institutions and hospices should contact hospitalists at their local hospitals. These programs need to invite hospitalists to participate in the palliative care team and suggest how their services can help the patients of hospitalists. This natural alliance can come about only if both sides reach out.
A partnership between palliative care and hospital medicine will be good for patients and their families as well as for each field, as hospitalists enable realization of the goal of providing palliative care to every patient in the United States. In addition, this partnership will be good for hospitalists who embrace this work. Palliative care can connect us to the humanism and compassion that brought so many of us to medicine and can serve as an antidote to burnout. Furthermore, by caring for patients with life‐threatening illnesses we remember that our time is limited and that each day is a gift. We recognize the importance of making the most of our time regardless of how long we have and of choosing carefully how and with whom we spend our time.
In this first issue of JHM, Dr. Meier makes a strong argument for the need and continued growth of palliative care in hospitals, lays out a strategy for achieving this growth through education and program development, and in doing so, opens the door to hope for the future. Through palliative care we can offer patients hope for healing when cure is not possible, for comfort in the face of suffering, and for what can still be despite all that cannot. The possibility that hospitalists could provide all patients access to palliative care is cause enough for hope. The knowledge that hospitalists will play a major role in making this possibility a reality and may become the predominant providers of palliative care can make that hope a reality.
- .Palliative care in hospitals.J Hosp Med.2006;1:21–28.
- ,.Prevalence and structure of palliative care services in California hospitals.Arch Intern Med.2003;163:1084–1088.
- ,,,,,.Evaluation of the California Hospital Initiative in Palliative Services (CHIPS).Arch Intern Med. In press.
- ,.Palliative care and the hospitalist: an opportunity for cross‐fertilization.Am J Med.2001;111:10S–14S.
- Field MJ,Cassell CK, Eds.Approaching death: improving care at the end of life.Washington, DC:National Academy Press,1997.
- ,.The hospitalist movement 5 years later.JAMA.2002;287:487–494.
- ,,,.Hospitalists' perceptions of their residency training needs: results of a national survey.Am J Med.2001;111:247–254.
- .Secondary and tertiary palliative care in US hospitals.JAMA.2002;287:875–881.
- .Palliative care in hospitals.J Hosp Med.2006;1:21–28.
- ,.Prevalence and structure of palliative care services in California hospitals.Arch Intern Med.2003;163:1084–1088.
- ,,,,,.Evaluation of the California Hospital Initiative in Palliative Services (CHIPS).Arch Intern Med. In press.
- ,.Palliative care and the hospitalist: an opportunity for cross‐fertilization.Am J Med.2001;111:10S–14S.
- Field MJ,Cassell CK, Eds.Approaching death: improving care at the end of life.Washington, DC:National Academy Press,1997.
- ,.The hospitalist movement 5 years later.JAMA.2002;287:487–494.
- ,,,.Hospitalists' perceptions of their residency training needs: results of a national survey.Am J Med.2001;111:247–254.
- .Secondary and tertiary palliative care in US hospitals.JAMA.2002;287:875–881.
How to Use The Core Competencies
The seminal article that coined the term hospitalist, in 1996, attributed the role of the hospitalist to enhancing throughput and cost reduction, primarily through reduction in length of stay, accomplished by having a dedicated clinician on site in the hospital.1 Since that time the role of the hospitalist has evolved to address the needs of multiple stakeholders at a time when traditional residency programs in inpatient adult medicine do not adequately train physicians to become effective agents of change in complex and potentially unsafe hospital systems. Continuing the trend of pediatrics, obstetrics, gynecology, and geriatrics, hospitalists have emerged as a distinct group of physicians who fill a needed clinical niche and are demonstrating the benefits of bringing a unique role and skill sets to the general hospital ward.2
The eligibility requirements for certification by the American Board of Internal Medicine specify that the discipline must 1) have a distinct and unique body of knowledge, 2) have clinical applicability sufficient to support a distinct clinical practice, 3) generate new information and research, 4) require a minimum training period of 12 months, and 5) have a substantial number of trainees and training programs nationwide.3 The Society of Hospital Medicine (SHM), the national professional organization of hospitalists, commissioned a task force to develop The Core Competencies in Hospital Medicine: A Framework for Curriculum Development (referred to from here on as the Core Competencies) to standardize the expectations of practicing hospitalists, serve as a foundation for curricula and other professional development experiences, prioritize educational scholarship and research strategies, and assess the adequacy and improvement opportunities for current training and accreditation of hospital medicine physicians.4 The preceding companion article The Core Competencies in Hospital Medicine: Development and Methodology, describes in detail the rationale for the development of the Core Competencies and the methods by which the document was created.5
PURPOSE
The purpose of this article is to illustrate how curriculum developers can apply the Core Competencies in Hospital Medicine to educate trainees and faculty, to prioritize educational scholarship and research strategies, and thus to improve the care of our patients.
TARGET AUDIENCE
The Core Competencies specifically targets directors of continuing medical education (CME), hospitalist programs and fellowships, residency programs, and medical school internal medicine clerkships. It is also intended for health educators, hospital administrators, potential employers, policy makers, and agencies funding quality‐improvement initiatives in the hospital setting. For residency program directors and clerkship directors, the chapters can guide in the development of curricula for inpatient medicine rotations or in meeting the Accreditation Council on Graduate Medical Education's Outcomes Project. For directors developing medical education curricula, The Core Competencies in Hospital Medicine can serve as a template for CME. For hospitalists, hospital administrators, and potential employers, the Core Competencies can be used to as the starting point in local program development and as a resource for refining the skills of all hospitalists, even very experienced practicing clinicians.
DEFINITION OF CORE COMPETENCIES IN HOSPITAL MEDICINE
The Core Competencies in Hospital Medicine provides a framework for curricular development based on a shared understanding of the essential knowledge, skills, and attitudes expected of physicians working as hospitalists. The development process will be ongoing, with revisions reflecting the evolving specialty of hospital medicine, the needs of practicing hospitalists, and feedback from users of the Core Competencies.
PROBLEM IDENTIFICATION AND GENERAL NEEDS ASSESSMENT
Delivery of health care has large gaps compared to ideal performance. Since the publication by the Institute of Medicine of To Err Is Human, in 1999, multiple agencies including the American Hospital Association, the National Quality Forum, and the U.S. Agency for Health Care Research and Quality (AHRQ) have reported on the incidence of medical errors in U.S. hospitals.6, 7 Recognizing that medical errors represent a major health concern in the United States, the Joint Commission on the Accreditation of Health Care Organizations (JCAHO) now requires patient safety initiatives for hospital accreditation.8 Problem‐based learning and improvement and systems based practice are now required competencies in medical residency curricula by the Accreditation Council for Graduate Medical Education (ACGME) and these requirements have led to the development of continuous quality techniques for preventing errors and a variety of patient safety initiatives.9
In 2002 the SHM recognized the need for identifying a distinct set of competencies in hospital medicine. The published competencies highlight the current gap in training of hospitalists and the imperative for revising curricula relating to inpatient care, hospital systems, and teaching.4 With adequate training and preparation, hospitalists can take the lead in implementing systems for best practices from admission through discharge and care transition, and they can direct the development of a safer, more patient‐centered, and cost‐efficient culture.
By defining the role of the hospitalist, the Core Competencies reflects the view of the SHM about what is possible but does not suggest how a training program might be modified to achieve desired outcomes or provide any content, resources, or teaching strategies. It will be up to curriculum developers to determine the scope of cognitive, psychomotor, and affective objectives that targeted learnershospitalists, residents, and other members of the multidisciplinary teamshould be required to acquire through lectures, discussions, syllabus material, clinical experience, and other venues. We agree with a broader definition of the term curriculum for graduate medical education, one that goes beyond curriculum as a plan and takes into account the learners' experiences, both planned and unplanned in the hospital setting.10 In contrast to the technologic theory of curriculum, in which lists of knowledge and skills represent final destinations, in the experiential model of curriculum, the lists provide only points of departure.11 The goal of the Core Competencies is to facilitate curriculum development using complex teaching environments as building blocks through which learning can occur.
CORE COMPETENCIES FOR HOSPITALISTS: OVERVIEW
The Core Competencies in Hospital Medicine is the first published competency‐based framework for professional development of hospitalists and provides the basis for accreditation in hospital medicine.12 The Core Competencies is organized into three sectionsClinical Conditions, Procedures, and Healthcare Systems. The supplement intentionally does not focus on content; rather, specific competencies describe unambiguous, measurable learning objectives. Each chapter can be used as a stand‐alone chapter to develop training and curricula for a particular topic area. Each chapter divides competencies into three domains of educational outcomes: cognitive (knowledge), affective (attitudes), and psychomotor (skills). Each domain has defined levels of proficiency going from knowledge, the lowest level, to evaluation, the highest.12, 13 A specific level of proficiency is articulated in the competencies through careful selection of corresponding action verbs, which clearly indicate how mastery could be assessed (see Table 1).
| GI Bleed ExampleLevels of Proficiency in the Cognitive Domain (Knowledge) | |
|---|---|
| UNDERSTAND the advantages and disadvantages of medical, endoscopic, and surgical treatments for patients with upper and lower GI bleeding | The first option, use of the verb understand gives little insight into level of proficiency. A patient could read a list on a pamphlet and truthfully claim to have achieved understanding of the advantages of each approach. An experienced gastroenterologist could make the same claim. Yet the two obviously differ in their level of comprehension. |
| LIST the advantages and disadvantages of medical, endoscopic, and surgical treatments for patients with upper and lower GI bleeding | In the second option, use of the verb list indicates that the expectation for a learner is to be able to literally make a quick list of advantages, perhaps merely regurgitating what was read in a text, indicating the lowest level of learning outcome, or knowledge. |
| COMPARE the advantages and disadvantages of medical, endoscopic, and surgical treatments for patients with upper and lower GI bleeding | In this option, use of the verb compare indicates that a clinician must be able to grasp the meaning of material and consider all options, indicating a higher level of learning outcome, or comprehension. |
| Although the differences in these statements may seem subtle, they are essential to discerning a level of proficiency. Verbs that convey higher levels of proficiency in the cognitive domain include: | |
| Apply, or the ability to use learned material in new and concrete situations, | |
| Analyze, which requires an understanding of both content and its organizational structure, | |
| Synthesize, or the ability to create new patterns of structures, and | |
| Evaluate, or the ability to judge the value of material (statement, research) for a given purpose, the highest level. | |
| Learning outcomes in the evaluation category are the highest because they contain elements of all other categories plus conscious value judgments based on clearly defined criteria.13 | |
| Each competency in the Core Competencies was crafted to indicate the relevant concept, its level of proficiency, and how mastery could be evaluated. The teaching processes and learning experiences that must take place to achieve competency is left to the design of the curriculum developers and instructors. | |
In addition to specific competencies in these commonly accepted learning domains, the Clinical Conditions and Procedure sections of the Core Competencies articulate the proficiencies that hospitalists should possess in systems organization and improvement. The clinical topics were selected to set expectations of leading or participating in system improvements specific to a clinical area and to prevent predictable complications of acute illness. Competencies in the Systems Organization and Improvement section indicate mastery of multiple competencies across categories. The Core Competencies describes how the hospitalist approach facilitates coordination among all participants within the hospital system (clinical and nonclinical) and effects system changes that improve patient care processes. At the same time, the statements indicate a range of involvement from participation to leadership. For example, lead, coordination or participate in acknowledges the unique needs of different practice settings and suggests a potential professional evolution. The Systems Organization and Improvement competencies of each clinical and procedure chapter strive to capture the essence of hospitalists whose goals are to improve patient outcomes for a specific population of patients. Hospitalists do not solely focus on the care of the patient with x disease, but rather develop systems to provide the best and most efficient care for all patients with x disease, successfully transitioning these patients to outpatient care and avoiding readmission.
The third section of chapters in the Core Competencies, Healthcare Systems, distinguishes a hospitalist from others working in the inpatient setting whether practicing at academic medical centers, community hospitals, teaching hospitals, managed‐care settings, or for‐profit settings. The Healthcare Systems section identifies the integral components of the successful practice of hospital medicine and mastery of multiple competencies. This section highlights how hospitalists can facilitate coordination among all care providers within the hospital and with outpatient care providers. Hospitalists can effect system changes that improve complex care processes. It is likely that additional work experience and training beyond residency are required to attain global proficiency in the care of hospital medicine patients.
HOW TO USE THE CORE COMPETENCIES TO DEVELOP A CURRICULUM
The whole document, three sections and 51 chapters, develops expectations about the role of the hospitalist. Proficiency can be acquired through multiple means and should match the needs of the targeted learners in order to develop and maintain the necessary level of performance within the discipline of hospital medicine. Specific cases that hospitalists may encounter in their daily practice are used to illustrate how the Core Competencies can be applied to curriculum development.
The cases will employ the following six‐step approach described in Curriculum Development in Medical Education14:
A problem and a need for improvement (the actual case and quality gap)
Needs assessment of targeted learners (hospitalists, clinicians‐in‐training)
Goals and specific measurable objectives (with competencies bridging the gap between traditional roles and setting expectations about the hospitalist role)
Educational strategies (with competencies providing structure and guidance to educational efforts)
Implementation (applying competencies to a variety of training opportunities and curricula)
Evaluation and feedback (ongoing nationally, regionally, locally).
Like any quality‐improvement educational initiative, subsequent steps in curriculum development for hospitalists should include, after evaluation and feedback, dissemination of core competencies and promotion of rigorous ongoing evaluation and adaptation as needs and expectations evolve.
The first case example, failure to prevent and diagnose pulmonary embolism (see Table 2), illustrates quality issues relating to prevention of predictable complications of illness, clinical problem solving in complex conditions of uncertainty, repetitive and nondiagnostic testing, and triage of a critically ill patient between services. The Core Competencies sets expectations about the ideal role of the hospitalist that might lead to improved outcomes.
| A Common Problem That Seemed to Defy the Right Approach to Solving It |
|---|
| A 52‐year‐old female, status posthysterectomy for endometrial cancer, presents with shortness of breath. |
| High pretest probability of pulmonary embolism (PE): suggestive symptoms, major risk factors, and omission of appropriate perioperative venous thromboembolism (VTE) prophylaxis. |
| Her presentation complicated by emesis, hypotension, hypoxia after presumed aspiration, and likely PE. |
| Chest computed tomography (CT), PE protocol, reportedly negative for PE but positive for multilobar pneumonia. |
| Small bowel obstruction, 51% bandemia, and acute renal failure. |
| Subsequent emergency incarcerated hernia repair without VTE prophylaxis. |
| She is transferred to general medicine for hemodynamic monitoring and evaluation of hemoptysis and elevated troponin, presumably caused by a PE. |
| Transthoracic echocardiogram notable for right ventricular (RV) dilation and pulmonary hypertension. |
| Review of two chest CT scans, one PE protocol significant for an enlarged right ventricle and multilobar pneumonia but no PE. |
| Absence of confirmatory evidence of suspected PE by subsequent extensive testing, including beta‐natriuretic peptide (BNP) level, repeat PE protocol CT, repeat transthoracic echocardiogram, bilateral lower extremity ultrasound, persantine positron emission tomography (PET) scan, cardiac magnetic resonance imaging (MRI), and right heart catheterization. |
| Discharge plan: home on warfarin. |
| Repetitive testing did not alter management. |
| Retrospective review: Using the enlarged right atrium and ventricle as the radiographic clue to look more closely for PE, an experienced chest radiologist was able to diagnose the presence of acute PE on the first chest CT. |
Using this case example, the Evidence‐Based Medicine (EBM) chapter establishes explicit expectations for hospitalists in clinical problem solving, including 1) explaining how the tests help to verify a suspected diagnosis, 2) describing the human factor in test interpretation (e.g., technical limitations of the most recent multi‐detector‐row spiral CT), and 3) explaining how timing relative to the onset of symptoms affects test results. Rather than an overreliance on technology, leading to repeating the chest CT with PE protocol and subsequent excessive nondiagnostic testing, the hospitalist would use knowledge of pretest probability and test characteristics to determine the best diagnostic strategy. The hospitalist approach to patient care, articulated in the affective (attitudes) domains of each chapter, integrates the application of EBM principles to clinical problem solving with deliberation of cost effectiveness and efficiency.
Continuing with this case example, the Team Approach and Communication chapters establish explicit expectations for practicing hospitalists who would take the extra steps to communicate with multiple members of the care team. Knowledgeable about the hospital, the hospitalist would review the chest CT with a radiologist skilled in chest interpretation and specifically query about the significance of an enlarged right atrium and right ventricle in the setting of a high pretest clinical probability of PE. Together the radiologist and hospitalist would consider a different imaging modality if the patient flunked the chest CT when the pretest probability was high. Rather than simply deferring to the medical specialist who is consulting, the hospitalist would be expected to improve the efficiency of care and reduce cost by only ordering tests that would change clinical management, perhaps with improved outcomes.
The Hospitalist as Teacher chapter provides a frameworkcore competencies for impromptu learningbased on the patient encounter. Members of the multidisciplinary care team can be exposed to explicit clinical decision making, an approach made possible by hospitalists on site, who can provide teaching moments in real time when decisions have to be made and educational feedback is needed. Teaching expectations for hospitalists include unambiguous clinical problem solving at the bedside and possibly directing the education of residents, physician assistants, and nurses on how to initiate a quality improvement (QI) project in a hospital setting.
The Quality Improvement and Venous Thromboembolism chapters clarify the role of the hospitalist, who should direct therapy against predictable complications of serious illness, critically review prophylaxis, provide hospital‐specific data to clinicians, identify and lower barriers to prevention, devise strategies to bridge the gap between knowledge and practice, develop automated reminder systems, and participate in clinical research.
The SHM has used the Core Competencies to develop educational resources to better meet the needs of the healthcare system. Although patient safety initiatives are mandated by JCAHO for hospital accreditation and AHRQ has identified areas for safety improvement that lists venous thromboembolism (VTE) prevention as the number one priority, VTE prophylaxis is still underutilized in the United States. Although some mechanisms are in place to educate residents and hospitalists about how to manage a specific disease, traditional medical education does not focus on teaching students and residents how to manage complex patients with multiple comorbidities, to prevent predictable complications of illness, and to examine and improve care processes.15, 16 When it comes to leading quality improvement (QI), individual feedback and traditional curricula, which may include didactic lectures on the pathophysiology of VTE and morbidity and mortality conferences, have not demonstrated improved outcomes.17
The SHM QI Web‐based resource rooms offer support to any QI effort and raise collective awareness of a performance gap.18 Each resource room will describe the evidence‐based practices that should be put into effect and will leverage experience with the disease as well as with the improvement process. The underlying goal of the resource rooms is to enhance the ability of hospitalists to actually improve inpatient outcomes through self‐directed learning (see Fig. 1).
Hospitalists, residency directors, and directors of hospitalist fellowships and continuing education can use The Core Competencies in Hospital Medicine to develop curricula for their local hospitalist service and request that invited speakers develop learning objectives and content based on core competencies rather than giving a prepared lecture on a specific clinical condition. This case of PE illustrates that risk assessment, prophylaxis, EBM clinical problem solving, and QI are core topics that should be emphasized in the training of hospitalists and physicians in training.0
| STEP 1 The current problem and the need for improvement | Quality Issues |
| Prevention of predictable complications of illness: VTE still underutilized. | |
| Clinical problem‐solving in complex systems, cost‐effective, diagnostic testing. | |
| Triage of patients between services. | |
| STEP 2 Needs assessment of hospitalists and other | The Current Approach: The focus of traditional medical education. |
| members of the inpatient team | How to manage a specific disease rather than how to manage complex patients with multiple co‐morbidities. |
| Didactic lectures on the pathophysiology of VTE. rather than prevention, QI. | |
| Individual feedback, morbidity and mortality conferences | |
| STEP 3 Goals and specific measurable objectives | The Ideal Approach: Competencies as a framework for setting expectations about the role of the hospitalist |
| Direct therapy against predictable complications of serious illness. | |
| Critically review prophylaxis. | |
| Devise strategies to bridge the gap between knowledge and practice. | |
| STEP 4 Educational strategies | The first in a new online series: The VTE Resource Room, by SHM |
| Key knowledge, approaches, methods, and tools can be applied to improve performance despite variances due to particular systems and advances in medicine. | |
| Enhance the ability of hospitalists as self‐directed learners to improve inpatient outcomes. | |
| STEP 5 Implementation | The VTE Resource Room |
| A downloadable workbook and companion project outline for the improvement process. | |
| A slide set to disseminate valuable information about a safer system for VTE prevention. | |
| A moderated forum of VTE and QI experts to pose questions. | |
| STEP 6 Evaluation and feedback | Ongoing Evaluation and Feedback |
Continuous with other steps (see Fig. 1). | |
| STEP 7 Remaining questionsthe need for additional | Research Questions |
| research | Identifying barriers to VTE prophylaxis in the hospital setting. |
| Root cause analysis to determine prevention, process improvements, and training practices to encourage the safety of hospitalized patients. |
The second case example, the hand‐off (see Table 3), illustrates quality issues related to transfer of care from one physician to another. In this example, if the patient with moderate pleural effusion had been signed out, an earlier thoracentesis to drain a presumptive parapneumonic infection might have relieved this patient's shortness of breath and saved her from undergoing a subsequent VATS procedure. This case also demonstrates the importance of correlating imaging abnormalities with a patient's clinical presentation rather than using the traditional approach of just ruling out potential diagnoses to determine the cause of a problem. This case highlights elements of the process and system of care that can be modified to improve patient outcomes. Being proficient in transferring care of patients can save the hospitalist from error and prevent adverse events.
| The Hand‐Off: Avoiding Pitfalls in the Hospitalist System |
|---|
| A 30‐year‐old female, status postruptured uterus and caesarian section for pregnancy, presents with hypotension. |
| Shortness of breath postexploratory laparoscopy during fluid resuscitation. |
| Spiral CT performed to rule out pulmonary embolism, signed out as negative based on verbal report. |
| Estimated pulmonary arterial systolic pressure of 70 mmHg by transthoracic echocardiogram. |
| Extensive testing for underlying causes of pulmonary hypertension, hypercoagulable states. |
| Outpatient right heart catheterization scheduled by cardiology. |
| Sleep study advised to complete the workup of pulmonary hypertension. |
| After diuresis with a corresponding reduction in pulmonary capillary wedge pressure, her pulmonary hypertension resolves and her outpatient right heart catheterization is cancelled. |
| Final reading of chest CT (not signed out to receiving attending) reportedly notable for moderate right‐sided pleural effusion, small left‐sided effusion, and an apparent filling defect of right subclavian vein |
| Six days after the original spiral CT, unsuccessful thoracentesis attempted, with removal of 1 cc of fluid consistent with exudate. |
| Video‐assisted thorascopic surgery (VATS) procedure required to avoid chronic disability from trapped lung. |
| Retrospective review: Early drainage of a parapneumonic infection in the setting of sepsis might have avoided this complication. |
The Team Approach chapter establishes the need to acquire proficiencies not ordinarily obtained during residency in order to lead a multidisciplinary care team. This role requires a level of functioning beyond that of simply being the attending of record. The hospitalist must be able to synthesize information rather than simply defer to the consultant. Competencies specified in the Diagnostic Decision‐Making chapter can be used to identify the educational needs of hospitalists, who are expected to minimize diagnostic errors by knowing when to ask for help and where to get it, recognizing common diseases with uncommon presentations, and generating a broad differential diagnosis where there is uncertainty. The Patient Handoff chapter defines the proficiencies hospitalists need to facilitate the safe transfer of patients to other physicians on their service.0
| STEP 1 | |
| The current problem and the need for | Quality issues in the transfer of care. |
| improvement | Failure to review radiographic study. |
| Signing out pending test results. | |
| Failure to correlate imaging abnormalities with the patient's clinical presentation. | |
| STEP 2 | |
| Seeds assessment of hospitalists and other | The Current Approach: Inherent discontinuities of inpatient care. |
| embers of the inpatient team | ACGME legislated work hours: resident shifts. |
| Transfer of care to and from primary care physicians to hospitalists and between hospitalists. | |
| STEP 3 | |
| Goals and specific measurable objectives | The Ideal Approach: Development of a standardized method of communication between hospitalists and between residents. |
| A hand‐off checklist would include pending tests, including final readings of radiographic studies. | |
| Systematic review of all films with a radiologist. | |
| STEP 4 | |
| Educational strategies | Critical examination of local practice for variability in sign‐outs. |
| Development of curricula with an agreed‐upon standard using the Core Competencies as a templatethe Patient Hand‐Offs chapter. | |
| Measure quality of hand‐off and provide feedback. | |
| STEP 5 | |
| Implementation | Dissemination of the expectations of the hand‐off. |
| Series of didactic talks for residents, physician assistants, and medical students by hospitalists based on specific cases.19 | |
| Using the core competencies as a framework; didactic lectures on hospital medicine topics can be revised to better reflect the continuing educational needs of hospitalists and their roles and responsibilities. | |
| STEP 6 | |
| Evaluation and feedback | A Framework for Educational Scholarship: the process of evaluation. |
| Innovative educational pilots, designed for members of the multidisciplinary care team | |
| Clear goals, adequate preparation, appropriate methods, significant results, effective presentation, and reflective critique. | |
| New curricular designs and materials development in topics not traditionally taught during medical school and residency such as patient hand‐offs20, 21 | |
| Not limited to publication; educational scholarship can be funded through risk management and hospital‐funded seed grants. | |
| STEP 7 | |
| Remaining questionsthe need for | Research Questions |
| additional research | What are the key components of the sign‐out process? |
| How can an electronic medical record or other system be utilized to standardize and improve the process? |
The third case example, which expands the responsibilities of hospitalist to include meeting important needs in the hospital (see Table 4), illustrates that hospitalist services cannot succeed by offering all things to all people, a distraction that that keeps the members of these services from concentrating on their goals. Always saying yes to whoever asks for help is a band‐aid, a short‐term fix that impedes the effort and creativity required for durable long‐term solutions to problems.
| No Problem |
|---|
| A proposal has been made that a new academic hospitalist service care for neurosurgical patients in order to meet the goals of the neurosurgical residency program to maximize the operating room exposure of surgeons in training. |
| Patients would be admitted to the hospitalist service, with subsequent neurosurgical consultation. |
| Another proposal has been made that the hospitalist service care for uncovered patients without residents in order to meet the goals of the medical residency program. |
| Hospital leaders assume the hospitalist service would have no problem with this proposal. |
| The hospitalists, who are not in‐house at night, are asked to handle off‐hours triage issues when there is disagreement between two services; their proposed role would be to support the medical residents who do not feel empowered to say no to the surgical team seeing patients in the emergency department. |
| The hospitalist service has the following concerns: |
| Assuming responsibility for a nonteaching service undermines the vision of this new hospitalist service in an academic tertiary care facility. |
| Assuming responsibility for a surgical specialty service increases medical legal risk and concerns about timely backup. |
| Setting a bad precedent sends the wrong message. |
| Hospitalists functioning as superresidents damages the reputation of the service. |
| The proposal comes with a price, namely, accelerating physician burnout, declining job satisfaction, and inevitable turnover. |
| The proposal would adversely affect future physician recruitment and promotion through the medical school clinician educator track. |
| Existing problems with the work environment of this new hospitalist service include: |
| The service already does not have time to meet the responsibilities of inpatient care expected of hospitalists because of rapid growth and the need for further recruitment. |
| Lack of advocacy by hospital administrators who may not understand the role of the hospitalist and entertain other solutions is an ongoing concern. |
| Lack of support for other missions of teaching and quality improvement research, coupled with a changing job description and the daily unpredictability of the work, promotes the view that hospital medicine may not be sustainable as a career. |
| The challenge and opportunity: Expertise in strategic planning and operations management is needed in order to effectively respond to conflicting pressures and focus on goals that will sustain the ability to change, grow, and continuously improve. |
The Core Competencies sets expectations about the roles of hospitalists, who serve as well‐informed clinicians and clinical opinion leaders; effective educators, mentors, and role models; empathetic and timely communicators; efficient caregivers; and creative problem solvers arriving at durable, longer‐term solutions. The competencies demonstrate the knowledge, skills, and attitudes required to be effective agents of change. Changing business as usual almost always requires significant improvements in the underlying system, however uncomfortable. The Leadership chapter articulates competencies that hospitalists need in order to define their roles within the hospital, promote group cohesiveness, expand their practices intelligently, and anticipate and respond to change. This chapter details the proficiencies that hospitalists need in order to develop personal, team, and program goals and to identify and resolve conflicts using specific negotiation techniques. The Business Practices chapter articulates the fundamental skills needed to enhance program development and growth. Hospitalists can use the Core Competencies to identify educational needs and develop curricula to enhance their leadership and business skill sets.0
| STEP 1 | |
| The current problem and the need for improvement | Hospitalist Services cannot succeed by attempting to offer all things to all people. |
| Distracting members from their work and from concentrating on their goals. | |
| Always saying yes to whoever asks for help as a Band‐Aid, a short‐term fix that impedes the effort and creativity required for durable long‐term solutions to problems. | |
| STEP 2 | |
| Needs assessment of hospitalists and other members | The Current Approach: Problems with the work environment |
| of the inpatient team | Hospital medicine, a new specialty, does not yet have a similar supportive infrastructure analogous to other well‐established specialties with most hospitalist programs within divisions of general medicine. |
| Multiple stakeholdersadministrators, primary care providers, residency and clerkship directors, specialty services. | |
| Leadership and administrative skills are not consistently acquired proficiencies during residency training. | |
| STEP 3 | |
| Goals and specific measurable objectives | The Ideal Approach: Hospitalists can proactively improve their work life by developing skills and knowledge in hospital systems. |
| Develop personal, team, and program goals. | |
| Identify and resolve conflicts using specific negotiation techniques | |
| Enhance program development and growth. | |
| Identify senior physician leaders as mentors and advocates. | |
| STEP 4 | |
| Educational strategies | Annual retreats to generate enthusiasm, establish a strategic plan, continue a trajectory of success. |
| Invite an outside expert in QI or professional development to facilitate discussion. | |
| Recruit hospitalists and colleagues with expertise in healthcare systems to mentor and educate other members of the hospitalist service how to lead QI and other initiatives. | |
| STEP 5 | |
| Implementation | Use the core competencies to advocate for resources to support professional goals. |
| Funding for leadership courses and further training in business. | |
| Directors of CME sponsored by SHM have begun the process of using the core competencies as the framework for the development of hospital medicine curricula in leadership and QI. | |
| STEP 6 | |
| Evaluation and feedback | Consider using the Core Competencies to develop an internal report card on performance. |
| A self‐assessment tool based on the core values and goals of the hospitalist program. | |
| A means to help identify areas for improvement, modifiable risk factors for turnover, and opportunities to provide incentives to measure interventions, reward success, and ultimately deliver on the mandate to improve inpatient care. | |
| STEP 7 | |
| Remaining questionsthe need for additional | Challenges facing hospitalists practicing in multiple settings. |
| research | How to make processes of care efficient by examining specific tasks that hospitalists do and determining what tools, technologies, organizational structure, and supporting staff need to be available to make the performance of these tasks efficient. |
| How to make hospital medicine a sustainable and satisfying career. |
Medical educators should examine the outcomes of current training practices and assess what modifications of objectives, content, and instructional strategies should be made to better prepare the current and next generations of physicians to practice hospital medicine and to improve the hospital setting. Given the scope of the field of hospital medicine, the Core Competencies should guide: 1) what to teach and how much to teach; 2) how to teach and assess trainees, and how to assess and compare faculty development programs; 3) how to design systems for improving quality of care and assuring patient safety; and 4) how to establish priorities for hospital medicine research.
TRANSLATING A SET OF COMPETENCIES INTO CURRICULA: POTENTIAL BENEFITS
The Core Competencies in Hospital Medicine transcends hospital type, size, and setting and standardizes what the expectations for and proficiencies of a practicing hospitalist should be. By defining the role of the hospitalist, the Core Competencies serves as a resource for refining inpatient skills and assists in program development at the local, regional, and national levels. In addition, by using the Core Competencies as the standard and framework for the development of preparatory curricula, hospital administrators and other employers can rely on hospitalists having had a common preparation.
The medical profession is constantly evolving. Internal medicine curricula address the challenges hospital medicine physicians faced yesterday but could improve the training and preparation of physicians to serve in their new and emerging roles as leaders of multidisciplinary healthcare teams working to improve patient outcomes and the system of inpatient care. Hospital medicine no longer represents a group of physicians merely supporting other specialists and primary care physicians; it is itself a specialty, composed of physicians leading, directing, and improving inpatient care. The competencies presented in The Core Competencies in Hospital Medicine: A Framework for Curriculum Development, by the Society of Hospital Medicine, should spark debate about the adequacy and appropriateness of current training and certification expectations and serve as a foundation for the development of curricula to improve hospital medicine education.
- ,.The emerging role of “hospitalists” in the American health care system.N Engl J Med.1996;335:514–517.
- ,.The hospitalist movement 5 years later.JAMA.2002;287:487–494.
- .The hospitalist: a new medical specialty?Ann Intern Med.1999;130:373–375.
- ,,,,, eds.The Core Competencies in Hospital Medicine: A Framework for Curriculum Development.J Hosp Med.2006;1 (supplement 1).
- ,,,,.Core competencies in hospital medicine: development and methodology.J Hosp Med.2006;1:48–56.
- Koh LT,Corrigan JM,Donaldson MS, eds.To err is human.Washington, DC:National Academy Press,2000.
- ,,,,.Making healthcare safer: a critical analysis of patient safety practices. AHRQ publication 01‐E058,2001.
- Joint Commission on the Accreditation of Health Care Organizations. Available at URL: http://www.jcaho.org[accessed November 2005].
- Accreditation Council for Graduate Medical Education. Available at URL: http://www.acgme.org[accessed November 2005].
- ,.What is a curriculum?Ann Intern Med.1992;116:1055–1056.
- ,.Conceptualizing curriculum for graduate medical education.Acad Med.1992;67:528–534.
- American Association for Health Education,National Commission for Health Education Credentialing, Inc.,Society for Public Health Education.A competency‐based framework for graduate‐level health educators.Allentown, PA:NCHEC,1999.
- .How to write and use instructional objectives.6th ed.Upper Saddle River, NJ:Prentice Hall,2000.
- ,,, et al.Curriculum development for medical education: a six‐step approach.Baltimore:Johns Hopkins University Press,1998.
- ,.Needs assessment in postgraduate medical education: a review.Med Educ Online [serial online].2002;7. Available at URL:http://www.med‐ed‐online.org/pdf/f0000040.pdf[accessed December 7, 2005].
- .Identifying, appraising, and implementing medical education curricula: a guide for medical educators.Ann Intern Med.2001;135:889–896.
- ,,, et al.A quality improvement initiative at Brigham and Women's Hospital.N Engl J Med.2005;352:969.
- The Society of Hospital Medicine. Available from URL: http://www.hospitalmedicine.org[accessed November 2005].
- ,,.Teaching and the case method.3rd ed.Cambridge, MA:Harvard Business School,1994.
- .Scholarship reconsidered: priorities of the professoriate.Princeton, NJ:Carnegie Foundation for the Advance of Teaching,1990.
- ,Scholarly activities of faculty promoted in a teacher–clinician ladder.Acad Med.2000;75:649–52.
The seminal article that coined the term hospitalist, in 1996, attributed the role of the hospitalist to enhancing throughput and cost reduction, primarily through reduction in length of stay, accomplished by having a dedicated clinician on site in the hospital.1 Since that time the role of the hospitalist has evolved to address the needs of multiple stakeholders at a time when traditional residency programs in inpatient adult medicine do not adequately train physicians to become effective agents of change in complex and potentially unsafe hospital systems. Continuing the trend of pediatrics, obstetrics, gynecology, and geriatrics, hospitalists have emerged as a distinct group of physicians who fill a needed clinical niche and are demonstrating the benefits of bringing a unique role and skill sets to the general hospital ward.2
The eligibility requirements for certification by the American Board of Internal Medicine specify that the discipline must 1) have a distinct and unique body of knowledge, 2) have clinical applicability sufficient to support a distinct clinical practice, 3) generate new information and research, 4) require a minimum training period of 12 months, and 5) have a substantial number of trainees and training programs nationwide.3 The Society of Hospital Medicine (SHM), the national professional organization of hospitalists, commissioned a task force to develop The Core Competencies in Hospital Medicine: A Framework for Curriculum Development (referred to from here on as the Core Competencies) to standardize the expectations of practicing hospitalists, serve as a foundation for curricula and other professional development experiences, prioritize educational scholarship and research strategies, and assess the adequacy and improvement opportunities for current training and accreditation of hospital medicine physicians.4 The preceding companion article The Core Competencies in Hospital Medicine: Development and Methodology, describes in detail the rationale for the development of the Core Competencies and the methods by which the document was created.5
PURPOSE
The purpose of this article is to illustrate how curriculum developers can apply the Core Competencies in Hospital Medicine to educate trainees and faculty, to prioritize educational scholarship and research strategies, and thus to improve the care of our patients.
TARGET AUDIENCE
The Core Competencies specifically targets directors of continuing medical education (CME), hospitalist programs and fellowships, residency programs, and medical school internal medicine clerkships. It is also intended for health educators, hospital administrators, potential employers, policy makers, and agencies funding quality‐improvement initiatives in the hospital setting. For residency program directors and clerkship directors, the chapters can guide in the development of curricula for inpatient medicine rotations or in meeting the Accreditation Council on Graduate Medical Education's Outcomes Project. For directors developing medical education curricula, The Core Competencies in Hospital Medicine can serve as a template for CME. For hospitalists, hospital administrators, and potential employers, the Core Competencies can be used to as the starting point in local program development and as a resource for refining the skills of all hospitalists, even very experienced practicing clinicians.
DEFINITION OF CORE COMPETENCIES IN HOSPITAL MEDICINE
The Core Competencies in Hospital Medicine provides a framework for curricular development based on a shared understanding of the essential knowledge, skills, and attitudes expected of physicians working as hospitalists. The development process will be ongoing, with revisions reflecting the evolving specialty of hospital medicine, the needs of practicing hospitalists, and feedback from users of the Core Competencies.
PROBLEM IDENTIFICATION AND GENERAL NEEDS ASSESSMENT
Delivery of health care has large gaps compared to ideal performance. Since the publication by the Institute of Medicine of To Err Is Human, in 1999, multiple agencies including the American Hospital Association, the National Quality Forum, and the U.S. Agency for Health Care Research and Quality (AHRQ) have reported on the incidence of medical errors in U.S. hospitals.6, 7 Recognizing that medical errors represent a major health concern in the United States, the Joint Commission on the Accreditation of Health Care Organizations (JCAHO) now requires patient safety initiatives for hospital accreditation.8 Problem‐based learning and improvement and systems based practice are now required competencies in medical residency curricula by the Accreditation Council for Graduate Medical Education (ACGME) and these requirements have led to the development of continuous quality techniques for preventing errors and a variety of patient safety initiatives.9
In 2002 the SHM recognized the need for identifying a distinct set of competencies in hospital medicine. The published competencies highlight the current gap in training of hospitalists and the imperative for revising curricula relating to inpatient care, hospital systems, and teaching.4 With adequate training and preparation, hospitalists can take the lead in implementing systems for best practices from admission through discharge and care transition, and they can direct the development of a safer, more patient‐centered, and cost‐efficient culture.
By defining the role of the hospitalist, the Core Competencies reflects the view of the SHM about what is possible but does not suggest how a training program might be modified to achieve desired outcomes or provide any content, resources, or teaching strategies. It will be up to curriculum developers to determine the scope of cognitive, psychomotor, and affective objectives that targeted learnershospitalists, residents, and other members of the multidisciplinary teamshould be required to acquire through lectures, discussions, syllabus material, clinical experience, and other venues. We agree with a broader definition of the term curriculum for graduate medical education, one that goes beyond curriculum as a plan and takes into account the learners' experiences, both planned and unplanned in the hospital setting.10 In contrast to the technologic theory of curriculum, in which lists of knowledge and skills represent final destinations, in the experiential model of curriculum, the lists provide only points of departure.11 The goal of the Core Competencies is to facilitate curriculum development using complex teaching environments as building blocks through which learning can occur.
CORE COMPETENCIES FOR HOSPITALISTS: OVERVIEW
The Core Competencies in Hospital Medicine is the first published competency‐based framework for professional development of hospitalists and provides the basis for accreditation in hospital medicine.12 The Core Competencies is organized into three sectionsClinical Conditions, Procedures, and Healthcare Systems. The supplement intentionally does not focus on content; rather, specific competencies describe unambiguous, measurable learning objectives. Each chapter can be used as a stand‐alone chapter to develop training and curricula for a particular topic area. Each chapter divides competencies into three domains of educational outcomes: cognitive (knowledge), affective (attitudes), and psychomotor (skills). Each domain has defined levels of proficiency going from knowledge, the lowest level, to evaluation, the highest.12, 13 A specific level of proficiency is articulated in the competencies through careful selection of corresponding action verbs, which clearly indicate how mastery could be assessed (see Table 1).
| GI Bleed ExampleLevels of Proficiency in the Cognitive Domain (Knowledge) | |
|---|---|
| UNDERSTAND the advantages and disadvantages of medical, endoscopic, and surgical treatments for patients with upper and lower GI bleeding | The first option, use of the verb understand gives little insight into level of proficiency. A patient could read a list on a pamphlet and truthfully claim to have achieved understanding of the advantages of each approach. An experienced gastroenterologist could make the same claim. Yet the two obviously differ in their level of comprehension. |
| LIST the advantages and disadvantages of medical, endoscopic, and surgical treatments for patients with upper and lower GI bleeding | In the second option, use of the verb list indicates that the expectation for a learner is to be able to literally make a quick list of advantages, perhaps merely regurgitating what was read in a text, indicating the lowest level of learning outcome, or knowledge. |
| COMPARE the advantages and disadvantages of medical, endoscopic, and surgical treatments for patients with upper and lower GI bleeding | In this option, use of the verb compare indicates that a clinician must be able to grasp the meaning of material and consider all options, indicating a higher level of learning outcome, or comprehension. |
| Although the differences in these statements may seem subtle, they are essential to discerning a level of proficiency. Verbs that convey higher levels of proficiency in the cognitive domain include: | |
| Apply, or the ability to use learned material in new and concrete situations, | |
| Analyze, which requires an understanding of both content and its organizational structure, | |
| Synthesize, or the ability to create new patterns of structures, and | |
| Evaluate, or the ability to judge the value of material (statement, research) for a given purpose, the highest level. | |
| Learning outcomes in the evaluation category are the highest because they contain elements of all other categories plus conscious value judgments based on clearly defined criteria.13 | |
| Each competency in the Core Competencies was crafted to indicate the relevant concept, its level of proficiency, and how mastery could be evaluated. The teaching processes and learning experiences that must take place to achieve competency is left to the design of the curriculum developers and instructors. | |
In addition to specific competencies in these commonly accepted learning domains, the Clinical Conditions and Procedure sections of the Core Competencies articulate the proficiencies that hospitalists should possess in systems organization and improvement. The clinical topics were selected to set expectations of leading or participating in system improvements specific to a clinical area and to prevent predictable complications of acute illness. Competencies in the Systems Organization and Improvement section indicate mastery of multiple competencies across categories. The Core Competencies describes how the hospitalist approach facilitates coordination among all participants within the hospital system (clinical and nonclinical) and effects system changes that improve patient care processes. At the same time, the statements indicate a range of involvement from participation to leadership. For example, lead, coordination or participate in acknowledges the unique needs of different practice settings and suggests a potential professional evolution. The Systems Organization and Improvement competencies of each clinical and procedure chapter strive to capture the essence of hospitalists whose goals are to improve patient outcomes for a specific population of patients. Hospitalists do not solely focus on the care of the patient with x disease, but rather develop systems to provide the best and most efficient care for all patients with x disease, successfully transitioning these patients to outpatient care and avoiding readmission.
The third section of chapters in the Core Competencies, Healthcare Systems, distinguishes a hospitalist from others working in the inpatient setting whether practicing at academic medical centers, community hospitals, teaching hospitals, managed‐care settings, or for‐profit settings. The Healthcare Systems section identifies the integral components of the successful practice of hospital medicine and mastery of multiple competencies. This section highlights how hospitalists can facilitate coordination among all care providers within the hospital and with outpatient care providers. Hospitalists can effect system changes that improve complex care processes. It is likely that additional work experience and training beyond residency are required to attain global proficiency in the care of hospital medicine patients.
HOW TO USE THE CORE COMPETENCIES TO DEVELOP A CURRICULUM
The whole document, three sections and 51 chapters, develops expectations about the role of the hospitalist. Proficiency can be acquired through multiple means and should match the needs of the targeted learners in order to develop and maintain the necessary level of performance within the discipline of hospital medicine. Specific cases that hospitalists may encounter in their daily practice are used to illustrate how the Core Competencies can be applied to curriculum development.
The cases will employ the following six‐step approach described in Curriculum Development in Medical Education14:
A problem and a need for improvement (the actual case and quality gap)
Needs assessment of targeted learners (hospitalists, clinicians‐in‐training)
Goals and specific measurable objectives (with competencies bridging the gap between traditional roles and setting expectations about the hospitalist role)
Educational strategies (with competencies providing structure and guidance to educational efforts)
Implementation (applying competencies to a variety of training opportunities and curricula)
Evaluation and feedback (ongoing nationally, regionally, locally).
Like any quality‐improvement educational initiative, subsequent steps in curriculum development for hospitalists should include, after evaluation and feedback, dissemination of core competencies and promotion of rigorous ongoing evaluation and adaptation as needs and expectations evolve.
The first case example, failure to prevent and diagnose pulmonary embolism (see Table 2), illustrates quality issues relating to prevention of predictable complications of illness, clinical problem solving in complex conditions of uncertainty, repetitive and nondiagnostic testing, and triage of a critically ill patient between services. The Core Competencies sets expectations about the ideal role of the hospitalist that might lead to improved outcomes.
| A Common Problem That Seemed to Defy the Right Approach to Solving It |
|---|
| A 52‐year‐old female, status posthysterectomy for endometrial cancer, presents with shortness of breath. |
| High pretest probability of pulmonary embolism (PE): suggestive symptoms, major risk factors, and omission of appropriate perioperative venous thromboembolism (VTE) prophylaxis. |
| Her presentation complicated by emesis, hypotension, hypoxia after presumed aspiration, and likely PE. |
| Chest computed tomography (CT), PE protocol, reportedly negative for PE but positive for multilobar pneumonia. |
| Small bowel obstruction, 51% bandemia, and acute renal failure. |
| Subsequent emergency incarcerated hernia repair without VTE prophylaxis. |
| She is transferred to general medicine for hemodynamic monitoring and evaluation of hemoptysis and elevated troponin, presumably caused by a PE. |
| Transthoracic echocardiogram notable for right ventricular (RV) dilation and pulmonary hypertension. |
| Review of two chest CT scans, one PE protocol significant for an enlarged right ventricle and multilobar pneumonia but no PE. |
| Absence of confirmatory evidence of suspected PE by subsequent extensive testing, including beta‐natriuretic peptide (BNP) level, repeat PE protocol CT, repeat transthoracic echocardiogram, bilateral lower extremity ultrasound, persantine positron emission tomography (PET) scan, cardiac magnetic resonance imaging (MRI), and right heart catheterization. |
| Discharge plan: home on warfarin. |
| Repetitive testing did not alter management. |
| Retrospective review: Using the enlarged right atrium and ventricle as the radiographic clue to look more closely for PE, an experienced chest radiologist was able to diagnose the presence of acute PE on the first chest CT. |
Using this case example, the Evidence‐Based Medicine (EBM) chapter establishes explicit expectations for hospitalists in clinical problem solving, including 1) explaining how the tests help to verify a suspected diagnosis, 2) describing the human factor in test interpretation (e.g., technical limitations of the most recent multi‐detector‐row spiral CT), and 3) explaining how timing relative to the onset of symptoms affects test results. Rather than an overreliance on technology, leading to repeating the chest CT with PE protocol and subsequent excessive nondiagnostic testing, the hospitalist would use knowledge of pretest probability and test characteristics to determine the best diagnostic strategy. The hospitalist approach to patient care, articulated in the affective (attitudes) domains of each chapter, integrates the application of EBM principles to clinical problem solving with deliberation of cost effectiveness and efficiency.
Continuing with this case example, the Team Approach and Communication chapters establish explicit expectations for practicing hospitalists who would take the extra steps to communicate with multiple members of the care team. Knowledgeable about the hospital, the hospitalist would review the chest CT with a radiologist skilled in chest interpretation and specifically query about the significance of an enlarged right atrium and right ventricle in the setting of a high pretest clinical probability of PE. Together the radiologist and hospitalist would consider a different imaging modality if the patient flunked the chest CT when the pretest probability was high. Rather than simply deferring to the medical specialist who is consulting, the hospitalist would be expected to improve the efficiency of care and reduce cost by only ordering tests that would change clinical management, perhaps with improved outcomes.
The Hospitalist as Teacher chapter provides a frameworkcore competencies for impromptu learningbased on the patient encounter. Members of the multidisciplinary care team can be exposed to explicit clinical decision making, an approach made possible by hospitalists on site, who can provide teaching moments in real time when decisions have to be made and educational feedback is needed. Teaching expectations for hospitalists include unambiguous clinical problem solving at the bedside and possibly directing the education of residents, physician assistants, and nurses on how to initiate a quality improvement (QI) project in a hospital setting.
The Quality Improvement and Venous Thromboembolism chapters clarify the role of the hospitalist, who should direct therapy against predictable complications of serious illness, critically review prophylaxis, provide hospital‐specific data to clinicians, identify and lower barriers to prevention, devise strategies to bridge the gap between knowledge and practice, develop automated reminder systems, and participate in clinical research.
The SHM has used the Core Competencies to develop educational resources to better meet the needs of the healthcare system. Although patient safety initiatives are mandated by JCAHO for hospital accreditation and AHRQ has identified areas for safety improvement that lists venous thromboembolism (VTE) prevention as the number one priority, VTE prophylaxis is still underutilized in the United States. Although some mechanisms are in place to educate residents and hospitalists about how to manage a specific disease, traditional medical education does not focus on teaching students and residents how to manage complex patients with multiple comorbidities, to prevent predictable complications of illness, and to examine and improve care processes.15, 16 When it comes to leading quality improvement (QI), individual feedback and traditional curricula, which may include didactic lectures on the pathophysiology of VTE and morbidity and mortality conferences, have not demonstrated improved outcomes.17
The SHM QI Web‐based resource rooms offer support to any QI effort and raise collective awareness of a performance gap.18 Each resource room will describe the evidence‐based practices that should be put into effect and will leverage experience with the disease as well as with the improvement process. The underlying goal of the resource rooms is to enhance the ability of hospitalists to actually improve inpatient outcomes through self‐directed learning (see Fig. 1).
Hospitalists, residency directors, and directors of hospitalist fellowships and continuing education can use The Core Competencies in Hospital Medicine to develop curricula for their local hospitalist service and request that invited speakers develop learning objectives and content based on core competencies rather than giving a prepared lecture on a specific clinical condition. This case of PE illustrates that risk assessment, prophylaxis, EBM clinical problem solving, and QI are core topics that should be emphasized in the training of hospitalists and physicians in training.0
| STEP 1 The current problem and the need for improvement | Quality Issues |
| Prevention of predictable complications of illness: VTE still underutilized. | |
| Clinical problem‐solving in complex systems, cost‐effective, diagnostic testing. | |
| Triage of patients between services. | |
| STEP 2 Needs assessment of hospitalists and other | The Current Approach: The focus of traditional medical education. |
| members of the inpatient team | How to manage a specific disease rather than how to manage complex patients with multiple co‐morbidities. |
| Didactic lectures on the pathophysiology of VTE. rather than prevention, QI. | |
| Individual feedback, morbidity and mortality conferences | |
| STEP 3 Goals and specific measurable objectives | The Ideal Approach: Competencies as a framework for setting expectations about the role of the hospitalist |
| Direct therapy against predictable complications of serious illness. | |
| Critically review prophylaxis. | |
| Devise strategies to bridge the gap between knowledge and practice. | |
| STEP 4 Educational strategies | The first in a new online series: The VTE Resource Room, by SHM |
| Key knowledge, approaches, methods, and tools can be applied to improve performance despite variances due to particular systems and advances in medicine. | |
| Enhance the ability of hospitalists as self‐directed learners to improve inpatient outcomes. | |
| STEP 5 Implementation | The VTE Resource Room |
| A downloadable workbook and companion project outline for the improvement process. | |
| A slide set to disseminate valuable information about a safer system for VTE prevention. | |
| A moderated forum of VTE and QI experts to pose questions. | |
| STEP 6 Evaluation and feedback | Ongoing Evaluation and Feedback |
Continuous with other steps (see Fig. 1). | |
| STEP 7 Remaining questionsthe need for additional | Research Questions |
| research | Identifying barriers to VTE prophylaxis in the hospital setting. |
| Root cause analysis to determine prevention, process improvements, and training practices to encourage the safety of hospitalized patients. |
The second case example, the hand‐off (see Table 3), illustrates quality issues related to transfer of care from one physician to another. In this example, if the patient with moderate pleural effusion had been signed out, an earlier thoracentesis to drain a presumptive parapneumonic infection might have relieved this patient's shortness of breath and saved her from undergoing a subsequent VATS procedure. This case also demonstrates the importance of correlating imaging abnormalities with a patient's clinical presentation rather than using the traditional approach of just ruling out potential diagnoses to determine the cause of a problem. This case highlights elements of the process and system of care that can be modified to improve patient outcomes. Being proficient in transferring care of patients can save the hospitalist from error and prevent adverse events.
| The Hand‐Off: Avoiding Pitfalls in the Hospitalist System |
|---|
| A 30‐year‐old female, status postruptured uterus and caesarian section for pregnancy, presents with hypotension. |
| Shortness of breath postexploratory laparoscopy during fluid resuscitation. |
| Spiral CT performed to rule out pulmonary embolism, signed out as negative based on verbal report. |
| Estimated pulmonary arterial systolic pressure of 70 mmHg by transthoracic echocardiogram. |
| Extensive testing for underlying causes of pulmonary hypertension, hypercoagulable states. |
| Outpatient right heart catheterization scheduled by cardiology. |
| Sleep study advised to complete the workup of pulmonary hypertension. |
| After diuresis with a corresponding reduction in pulmonary capillary wedge pressure, her pulmonary hypertension resolves and her outpatient right heart catheterization is cancelled. |
| Final reading of chest CT (not signed out to receiving attending) reportedly notable for moderate right‐sided pleural effusion, small left‐sided effusion, and an apparent filling defect of right subclavian vein |
| Six days after the original spiral CT, unsuccessful thoracentesis attempted, with removal of 1 cc of fluid consistent with exudate. |
| Video‐assisted thorascopic surgery (VATS) procedure required to avoid chronic disability from trapped lung. |
| Retrospective review: Early drainage of a parapneumonic infection in the setting of sepsis might have avoided this complication. |
The Team Approach chapter establishes the need to acquire proficiencies not ordinarily obtained during residency in order to lead a multidisciplinary care team. This role requires a level of functioning beyond that of simply being the attending of record. The hospitalist must be able to synthesize information rather than simply defer to the consultant. Competencies specified in the Diagnostic Decision‐Making chapter can be used to identify the educational needs of hospitalists, who are expected to minimize diagnostic errors by knowing when to ask for help and where to get it, recognizing common diseases with uncommon presentations, and generating a broad differential diagnosis where there is uncertainty. The Patient Handoff chapter defines the proficiencies hospitalists need to facilitate the safe transfer of patients to other physicians on their service.0
| STEP 1 | |
| The current problem and the need for | Quality issues in the transfer of care. |
| improvement | Failure to review radiographic study. |
| Signing out pending test results. | |
| Failure to correlate imaging abnormalities with the patient's clinical presentation. | |
| STEP 2 | |
| Seeds assessment of hospitalists and other | The Current Approach: Inherent discontinuities of inpatient care. |
| embers of the inpatient team | ACGME legislated work hours: resident shifts. |
| Transfer of care to and from primary care physicians to hospitalists and between hospitalists. | |
| STEP 3 | |
| Goals and specific measurable objectives | The Ideal Approach: Development of a standardized method of communication between hospitalists and between residents. |
| A hand‐off checklist would include pending tests, including final readings of radiographic studies. | |
| Systematic review of all films with a radiologist. | |
| STEP 4 | |
| Educational strategies | Critical examination of local practice for variability in sign‐outs. |
| Development of curricula with an agreed‐upon standard using the Core Competencies as a templatethe Patient Hand‐Offs chapter. | |
| Measure quality of hand‐off and provide feedback. | |
| STEP 5 | |
| Implementation | Dissemination of the expectations of the hand‐off. |
| Series of didactic talks for residents, physician assistants, and medical students by hospitalists based on specific cases.19 | |
| Using the core competencies as a framework; didactic lectures on hospital medicine topics can be revised to better reflect the continuing educational needs of hospitalists and their roles and responsibilities. | |
| STEP 6 | |
| Evaluation and feedback | A Framework for Educational Scholarship: the process of evaluation. |
| Innovative educational pilots, designed for members of the multidisciplinary care team | |
| Clear goals, adequate preparation, appropriate methods, significant results, effective presentation, and reflective critique. | |
| New curricular designs and materials development in topics not traditionally taught during medical school and residency such as patient hand‐offs20, 21 | |
| Not limited to publication; educational scholarship can be funded through risk management and hospital‐funded seed grants. | |
| STEP 7 | |
| Remaining questionsthe need for | Research Questions |
| additional research | What are the key components of the sign‐out process? |
| How can an electronic medical record or other system be utilized to standardize and improve the process? |
The third case example, which expands the responsibilities of hospitalist to include meeting important needs in the hospital (see Table 4), illustrates that hospitalist services cannot succeed by offering all things to all people, a distraction that that keeps the members of these services from concentrating on their goals. Always saying yes to whoever asks for help is a band‐aid, a short‐term fix that impedes the effort and creativity required for durable long‐term solutions to problems.
| No Problem |
|---|
| A proposal has been made that a new academic hospitalist service care for neurosurgical patients in order to meet the goals of the neurosurgical residency program to maximize the operating room exposure of surgeons in training. |
| Patients would be admitted to the hospitalist service, with subsequent neurosurgical consultation. |
| Another proposal has been made that the hospitalist service care for uncovered patients without residents in order to meet the goals of the medical residency program. |
| Hospital leaders assume the hospitalist service would have no problem with this proposal. |
| The hospitalists, who are not in‐house at night, are asked to handle off‐hours triage issues when there is disagreement between two services; their proposed role would be to support the medical residents who do not feel empowered to say no to the surgical team seeing patients in the emergency department. |
| The hospitalist service has the following concerns: |
| Assuming responsibility for a nonteaching service undermines the vision of this new hospitalist service in an academic tertiary care facility. |
| Assuming responsibility for a surgical specialty service increases medical legal risk and concerns about timely backup. |
| Setting a bad precedent sends the wrong message. |
| Hospitalists functioning as superresidents damages the reputation of the service. |
| The proposal comes with a price, namely, accelerating physician burnout, declining job satisfaction, and inevitable turnover. |
| The proposal would adversely affect future physician recruitment and promotion through the medical school clinician educator track. |
| Existing problems with the work environment of this new hospitalist service include: |
| The service already does not have time to meet the responsibilities of inpatient care expected of hospitalists because of rapid growth and the need for further recruitment. |
| Lack of advocacy by hospital administrators who may not understand the role of the hospitalist and entertain other solutions is an ongoing concern. |
| Lack of support for other missions of teaching and quality improvement research, coupled with a changing job description and the daily unpredictability of the work, promotes the view that hospital medicine may not be sustainable as a career. |
| The challenge and opportunity: Expertise in strategic planning and operations management is needed in order to effectively respond to conflicting pressures and focus on goals that will sustain the ability to change, grow, and continuously improve. |
The Core Competencies sets expectations about the roles of hospitalists, who serve as well‐informed clinicians and clinical opinion leaders; effective educators, mentors, and role models; empathetic and timely communicators; efficient caregivers; and creative problem solvers arriving at durable, longer‐term solutions. The competencies demonstrate the knowledge, skills, and attitudes required to be effective agents of change. Changing business as usual almost always requires significant improvements in the underlying system, however uncomfortable. The Leadership chapter articulates competencies that hospitalists need in order to define their roles within the hospital, promote group cohesiveness, expand their practices intelligently, and anticipate and respond to change. This chapter details the proficiencies that hospitalists need in order to develop personal, team, and program goals and to identify and resolve conflicts using specific negotiation techniques. The Business Practices chapter articulates the fundamental skills needed to enhance program development and growth. Hospitalists can use the Core Competencies to identify educational needs and develop curricula to enhance their leadership and business skill sets.0
| STEP 1 | |
| The current problem and the need for improvement | Hospitalist Services cannot succeed by attempting to offer all things to all people. |
| Distracting members from their work and from concentrating on their goals. | |
| Always saying yes to whoever asks for help as a Band‐Aid, a short‐term fix that impedes the effort and creativity required for durable long‐term solutions to problems. | |
| STEP 2 | |
| Needs assessment of hospitalists and other members | The Current Approach: Problems with the work environment |
| of the inpatient team | Hospital medicine, a new specialty, does not yet have a similar supportive infrastructure analogous to other well‐established specialties with most hospitalist programs within divisions of general medicine. |
| Multiple stakeholdersadministrators, primary care providers, residency and clerkship directors, specialty services. | |
| Leadership and administrative skills are not consistently acquired proficiencies during residency training. | |
| STEP 3 | |
| Goals and specific measurable objectives | The Ideal Approach: Hospitalists can proactively improve their work life by developing skills and knowledge in hospital systems. |
| Develop personal, team, and program goals. | |
| Identify and resolve conflicts using specific negotiation techniques | |
| Enhance program development and growth. | |
| Identify senior physician leaders as mentors and advocates. | |
| STEP 4 | |
| Educational strategies | Annual retreats to generate enthusiasm, establish a strategic plan, continue a trajectory of success. |
| Invite an outside expert in QI or professional development to facilitate discussion. | |
| Recruit hospitalists and colleagues with expertise in healthcare systems to mentor and educate other members of the hospitalist service how to lead QI and other initiatives. | |
| STEP 5 | |
| Implementation | Use the core competencies to advocate for resources to support professional goals. |
| Funding for leadership courses and further training in business. | |
| Directors of CME sponsored by SHM have begun the process of using the core competencies as the framework for the development of hospital medicine curricula in leadership and QI. | |
| STEP 6 | |
| Evaluation and feedback | Consider using the Core Competencies to develop an internal report card on performance. |
| A self‐assessment tool based on the core values and goals of the hospitalist program. | |
| A means to help identify areas for improvement, modifiable risk factors for turnover, and opportunities to provide incentives to measure interventions, reward success, and ultimately deliver on the mandate to improve inpatient care. | |
| STEP 7 | |
| Remaining questionsthe need for additional | Challenges facing hospitalists practicing in multiple settings. |
| research | How to make processes of care efficient by examining specific tasks that hospitalists do and determining what tools, technologies, organizational structure, and supporting staff need to be available to make the performance of these tasks efficient. |
| How to make hospital medicine a sustainable and satisfying career. |
Medical educators should examine the outcomes of current training practices and assess what modifications of objectives, content, and instructional strategies should be made to better prepare the current and next generations of physicians to practice hospital medicine and to improve the hospital setting. Given the scope of the field of hospital medicine, the Core Competencies should guide: 1) what to teach and how much to teach; 2) how to teach and assess trainees, and how to assess and compare faculty development programs; 3) how to design systems for improving quality of care and assuring patient safety; and 4) how to establish priorities for hospital medicine research.
TRANSLATING A SET OF COMPETENCIES INTO CURRICULA: POTENTIAL BENEFITS
The Core Competencies in Hospital Medicine transcends hospital type, size, and setting and standardizes what the expectations for and proficiencies of a practicing hospitalist should be. By defining the role of the hospitalist, the Core Competencies serves as a resource for refining inpatient skills and assists in program development at the local, regional, and national levels. In addition, by using the Core Competencies as the standard and framework for the development of preparatory curricula, hospital administrators and other employers can rely on hospitalists having had a common preparation.
The medical profession is constantly evolving. Internal medicine curricula address the challenges hospital medicine physicians faced yesterday but could improve the training and preparation of physicians to serve in their new and emerging roles as leaders of multidisciplinary healthcare teams working to improve patient outcomes and the system of inpatient care. Hospital medicine no longer represents a group of physicians merely supporting other specialists and primary care physicians; it is itself a specialty, composed of physicians leading, directing, and improving inpatient care. The competencies presented in The Core Competencies in Hospital Medicine: A Framework for Curriculum Development, by the Society of Hospital Medicine, should spark debate about the adequacy and appropriateness of current training and certification expectations and serve as a foundation for the development of curricula to improve hospital medicine education.
The seminal article that coined the term hospitalist, in 1996, attributed the role of the hospitalist to enhancing throughput and cost reduction, primarily through reduction in length of stay, accomplished by having a dedicated clinician on site in the hospital.1 Since that time the role of the hospitalist has evolved to address the needs of multiple stakeholders at a time when traditional residency programs in inpatient adult medicine do not adequately train physicians to become effective agents of change in complex and potentially unsafe hospital systems. Continuing the trend of pediatrics, obstetrics, gynecology, and geriatrics, hospitalists have emerged as a distinct group of physicians who fill a needed clinical niche and are demonstrating the benefits of bringing a unique role and skill sets to the general hospital ward.2
The eligibility requirements for certification by the American Board of Internal Medicine specify that the discipline must 1) have a distinct and unique body of knowledge, 2) have clinical applicability sufficient to support a distinct clinical practice, 3) generate new information and research, 4) require a minimum training period of 12 months, and 5) have a substantial number of trainees and training programs nationwide.3 The Society of Hospital Medicine (SHM), the national professional organization of hospitalists, commissioned a task force to develop The Core Competencies in Hospital Medicine: A Framework for Curriculum Development (referred to from here on as the Core Competencies) to standardize the expectations of practicing hospitalists, serve as a foundation for curricula and other professional development experiences, prioritize educational scholarship and research strategies, and assess the adequacy and improvement opportunities for current training and accreditation of hospital medicine physicians.4 The preceding companion article The Core Competencies in Hospital Medicine: Development and Methodology, describes in detail the rationale for the development of the Core Competencies and the methods by which the document was created.5
PURPOSE
The purpose of this article is to illustrate how curriculum developers can apply the Core Competencies in Hospital Medicine to educate trainees and faculty, to prioritize educational scholarship and research strategies, and thus to improve the care of our patients.
TARGET AUDIENCE
The Core Competencies specifically targets directors of continuing medical education (CME), hospitalist programs and fellowships, residency programs, and medical school internal medicine clerkships. It is also intended for health educators, hospital administrators, potential employers, policy makers, and agencies funding quality‐improvement initiatives in the hospital setting. For residency program directors and clerkship directors, the chapters can guide in the development of curricula for inpatient medicine rotations or in meeting the Accreditation Council on Graduate Medical Education's Outcomes Project. For directors developing medical education curricula, The Core Competencies in Hospital Medicine can serve as a template for CME. For hospitalists, hospital administrators, and potential employers, the Core Competencies can be used to as the starting point in local program development and as a resource for refining the skills of all hospitalists, even very experienced practicing clinicians.
DEFINITION OF CORE COMPETENCIES IN HOSPITAL MEDICINE
The Core Competencies in Hospital Medicine provides a framework for curricular development based on a shared understanding of the essential knowledge, skills, and attitudes expected of physicians working as hospitalists. The development process will be ongoing, with revisions reflecting the evolving specialty of hospital medicine, the needs of practicing hospitalists, and feedback from users of the Core Competencies.
PROBLEM IDENTIFICATION AND GENERAL NEEDS ASSESSMENT
Delivery of health care has large gaps compared to ideal performance. Since the publication by the Institute of Medicine of To Err Is Human, in 1999, multiple agencies including the American Hospital Association, the National Quality Forum, and the U.S. Agency for Health Care Research and Quality (AHRQ) have reported on the incidence of medical errors in U.S. hospitals.6, 7 Recognizing that medical errors represent a major health concern in the United States, the Joint Commission on the Accreditation of Health Care Organizations (JCAHO) now requires patient safety initiatives for hospital accreditation.8 Problem‐based learning and improvement and systems based practice are now required competencies in medical residency curricula by the Accreditation Council for Graduate Medical Education (ACGME) and these requirements have led to the development of continuous quality techniques for preventing errors and a variety of patient safety initiatives.9
In 2002 the SHM recognized the need for identifying a distinct set of competencies in hospital medicine. The published competencies highlight the current gap in training of hospitalists and the imperative for revising curricula relating to inpatient care, hospital systems, and teaching.4 With adequate training and preparation, hospitalists can take the lead in implementing systems for best practices from admission through discharge and care transition, and they can direct the development of a safer, more patient‐centered, and cost‐efficient culture.
By defining the role of the hospitalist, the Core Competencies reflects the view of the SHM about what is possible but does not suggest how a training program might be modified to achieve desired outcomes or provide any content, resources, or teaching strategies. It will be up to curriculum developers to determine the scope of cognitive, psychomotor, and affective objectives that targeted learnershospitalists, residents, and other members of the multidisciplinary teamshould be required to acquire through lectures, discussions, syllabus material, clinical experience, and other venues. We agree with a broader definition of the term curriculum for graduate medical education, one that goes beyond curriculum as a plan and takes into account the learners' experiences, both planned and unplanned in the hospital setting.10 In contrast to the technologic theory of curriculum, in which lists of knowledge and skills represent final destinations, in the experiential model of curriculum, the lists provide only points of departure.11 The goal of the Core Competencies is to facilitate curriculum development using complex teaching environments as building blocks through which learning can occur.
CORE COMPETENCIES FOR HOSPITALISTS: OVERVIEW
The Core Competencies in Hospital Medicine is the first published competency‐based framework for professional development of hospitalists and provides the basis for accreditation in hospital medicine.12 The Core Competencies is organized into three sectionsClinical Conditions, Procedures, and Healthcare Systems. The supplement intentionally does not focus on content; rather, specific competencies describe unambiguous, measurable learning objectives. Each chapter can be used as a stand‐alone chapter to develop training and curricula for a particular topic area. Each chapter divides competencies into three domains of educational outcomes: cognitive (knowledge), affective (attitudes), and psychomotor (skills). Each domain has defined levels of proficiency going from knowledge, the lowest level, to evaluation, the highest.12, 13 A specific level of proficiency is articulated in the competencies through careful selection of corresponding action verbs, which clearly indicate how mastery could be assessed (see Table 1).
| GI Bleed ExampleLevels of Proficiency in the Cognitive Domain (Knowledge) | |
|---|---|
| UNDERSTAND the advantages and disadvantages of medical, endoscopic, and surgical treatments for patients with upper and lower GI bleeding | The first option, use of the verb understand gives little insight into level of proficiency. A patient could read a list on a pamphlet and truthfully claim to have achieved understanding of the advantages of each approach. An experienced gastroenterologist could make the same claim. Yet the two obviously differ in their level of comprehension. |
| LIST the advantages and disadvantages of medical, endoscopic, and surgical treatments for patients with upper and lower GI bleeding | In the second option, use of the verb list indicates that the expectation for a learner is to be able to literally make a quick list of advantages, perhaps merely regurgitating what was read in a text, indicating the lowest level of learning outcome, or knowledge. |
| COMPARE the advantages and disadvantages of medical, endoscopic, and surgical treatments for patients with upper and lower GI bleeding | In this option, use of the verb compare indicates that a clinician must be able to grasp the meaning of material and consider all options, indicating a higher level of learning outcome, or comprehension. |
| Although the differences in these statements may seem subtle, they are essential to discerning a level of proficiency. Verbs that convey higher levels of proficiency in the cognitive domain include: | |
| Apply, or the ability to use learned material in new and concrete situations, | |
| Analyze, which requires an understanding of both content and its organizational structure, | |
| Synthesize, or the ability to create new patterns of structures, and | |
| Evaluate, or the ability to judge the value of material (statement, research) for a given purpose, the highest level. | |
| Learning outcomes in the evaluation category are the highest because they contain elements of all other categories plus conscious value judgments based on clearly defined criteria.13 | |
| Each competency in the Core Competencies was crafted to indicate the relevant concept, its level of proficiency, and how mastery could be evaluated. The teaching processes and learning experiences that must take place to achieve competency is left to the design of the curriculum developers and instructors. | |
In addition to specific competencies in these commonly accepted learning domains, the Clinical Conditions and Procedure sections of the Core Competencies articulate the proficiencies that hospitalists should possess in systems organization and improvement. The clinical topics were selected to set expectations of leading or participating in system improvements specific to a clinical area and to prevent predictable complications of acute illness. Competencies in the Systems Organization and Improvement section indicate mastery of multiple competencies across categories. The Core Competencies describes how the hospitalist approach facilitates coordination among all participants within the hospital system (clinical and nonclinical) and effects system changes that improve patient care processes. At the same time, the statements indicate a range of involvement from participation to leadership. For example, lead, coordination or participate in acknowledges the unique needs of different practice settings and suggests a potential professional evolution. The Systems Organization and Improvement competencies of each clinical and procedure chapter strive to capture the essence of hospitalists whose goals are to improve patient outcomes for a specific population of patients. Hospitalists do not solely focus on the care of the patient with x disease, but rather develop systems to provide the best and most efficient care for all patients with x disease, successfully transitioning these patients to outpatient care and avoiding readmission.
The third section of chapters in the Core Competencies, Healthcare Systems, distinguishes a hospitalist from others working in the inpatient setting whether practicing at academic medical centers, community hospitals, teaching hospitals, managed‐care settings, or for‐profit settings. The Healthcare Systems section identifies the integral components of the successful practice of hospital medicine and mastery of multiple competencies. This section highlights how hospitalists can facilitate coordination among all care providers within the hospital and with outpatient care providers. Hospitalists can effect system changes that improve complex care processes. It is likely that additional work experience and training beyond residency are required to attain global proficiency in the care of hospital medicine patients.
HOW TO USE THE CORE COMPETENCIES TO DEVELOP A CURRICULUM
The whole document, three sections and 51 chapters, develops expectations about the role of the hospitalist. Proficiency can be acquired through multiple means and should match the needs of the targeted learners in order to develop and maintain the necessary level of performance within the discipline of hospital medicine. Specific cases that hospitalists may encounter in their daily practice are used to illustrate how the Core Competencies can be applied to curriculum development.
The cases will employ the following six‐step approach described in Curriculum Development in Medical Education14:
A problem and a need for improvement (the actual case and quality gap)
Needs assessment of targeted learners (hospitalists, clinicians‐in‐training)
Goals and specific measurable objectives (with competencies bridging the gap between traditional roles and setting expectations about the hospitalist role)
Educational strategies (with competencies providing structure and guidance to educational efforts)
Implementation (applying competencies to a variety of training opportunities and curricula)
Evaluation and feedback (ongoing nationally, regionally, locally).
Like any quality‐improvement educational initiative, subsequent steps in curriculum development for hospitalists should include, after evaluation and feedback, dissemination of core competencies and promotion of rigorous ongoing evaluation and adaptation as needs and expectations evolve.
The first case example, failure to prevent and diagnose pulmonary embolism (see Table 2), illustrates quality issues relating to prevention of predictable complications of illness, clinical problem solving in complex conditions of uncertainty, repetitive and nondiagnostic testing, and triage of a critically ill patient between services. The Core Competencies sets expectations about the ideal role of the hospitalist that might lead to improved outcomes.
| A Common Problem That Seemed to Defy the Right Approach to Solving It |
|---|
| A 52‐year‐old female, status posthysterectomy for endometrial cancer, presents with shortness of breath. |
| High pretest probability of pulmonary embolism (PE): suggestive symptoms, major risk factors, and omission of appropriate perioperative venous thromboembolism (VTE) prophylaxis. |
| Her presentation complicated by emesis, hypotension, hypoxia after presumed aspiration, and likely PE. |
| Chest computed tomography (CT), PE protocol, reportedly negative for PE but positive for multilobar pneumonia. |
| Small bowel obstruction, 51% bandemia, and acute renal failure. |
| Subsequent emergency incarcerated hernia repair without VTE prophylaxis. |
| She is transferred to general medicine for hemodynamic monitoring and evaluation of hemoptysis and elevated troponin, presumably caused by a PE. |
| Transthoracic echocardiogram notable for right ventricular (RV) dilation and pulmonary hypertension. |
| Review of two chest CT scans, one PE protocol significant for an enlarged right ventricle and multilobar pneumonia but no PE. |
| Absence of confirmatory evidence of suspected PE by subsequent extensive testing, including beta‐natriuretic peptide (BNP) level, repeat PE protocol CT, repeat transthoracic echocardiogram, bilateral lower extremity ultrasound, persantine positron emission tomography (PET) scan, cardiac magnetic resonance imaging (MRI), and right heart catheterization. |
| Discharge plan: home on warfarin. |
| Repetitive testing did not alter management. |
| Retrospective review: Using the enlarged right atrium and ventricle as the radiographic clue to look more closely for PE, an experienced chest radiologist was able to diagnose the presence of acute PE on the first chest CT. |
Using this case example, the Evidence‐Based Medicine (EBM) chapter establishes explicit expectations for hospitalists in clinical problem solving, including 1) explaining how the tests help to verify a suspected diagnosis, 2) describing the human factor in test interpretation (e.g., technical limitations of the most recent multi‐detector‐row spiral CT), and 3) explaining how timing relative to the onset of symptoms affects test results. Rather than an overreliance on technology, leading to repeating the chest CT with PE protocol and subsequent excessive nondiagnostic testing, the hospitalist would use knowledge of pretest probability and test characteristics to determine the best diagnostic strategy. The hospitalist approach to patient care, articulated in the affective (attitudes) domains of each chapter, integrates the application of EBM principles to clinical problem solving with deliberation of cost effectiveness and efficiency.
Continuing with this case example, the Team Approach and Communication chapters establish explicit expectations for practicing hospitalists who would take the extra steps to communicate with multiple members of the care team. Knowledgeable about the hospital, the hospitalist would review the chest CT with a radiologist skilled in chest interpretation and specifically query about the significance of an enlarged right atrium and right ventricle in the setting of a high pretest clinical probability of PE. Together the radiologist and hospitalist would consider a different imaging modality if the patient flunked the chest CT when the pretest probability was high. Rather than simply deferring to the medical specialist who is consulting, the hospitalist would be expected to improve the efficiency of care and reduce cost by only ordering tests that would change clinical management, perhaps with improved outcomes.
The Hospitalist as Teacher chapter provides a frameworkcore competencies for impromptu learningbased on the patient encounter. Members of the multidisciplinary care team can be exposed to explicit clinical decision making, an approach made possible by hospitalists on site, who can provide teaching moments in real time when decisions have to be made and educational feedback is needed. Teaching expectations for hospitalists include unambiguous clinical problem solving at the bedside and possibly directing the education of residents, physician assistants, and nurses on how to initiate a quality improvement (QI) project in a hospital setting.
The Quality Improvement and Venous Thromboembolism chapters clarify the role of the hospitalist, who should direct therapy against predictable complications of serious illness, critically review prophylaxis, provide hospital‐specific data to clinicians, identify and lower barriers to prevention, devise strategies to bridge the gap between knowledge and practice, develop automated reminder systems, and participate in clinical research.
The SHM has used the Core Competencies to develop educational resources to better meet the needs of the healthcare system. Although patient safety initiatives are mandated by JCAHO for hospital accreditation and AHRQ has identified areas for safety improvement that lists venous thromboembolism (VTE) prevention as the number one priority, VTE prophylaxis is still underutilized in the United States. Although some mechanisms are in place to educate residents and hospitalists about how to manage a specific disease, traditional medical education does not focus on teaching students and residents how to manage complex patients with multiple comorbidities, to prevent predictable complications of illness, and to examine and improve care processes.15, 16 When it comes to leading quality improvement (QI), individual feedback and traditional curricula, which may include didactic lectures on the pathophysiology of VTE and morbidity and mortality conferences, have not demonstrated improved outcomes.17
The SHM QI Web‐based resource rooms offer support to any QI effort and raise collective awareness of a performance gap.18 Each resource room will describe the evidence‐based practices that should be put into effect and will leverage experience with the disease as well as with the improvement process. The underlying goal of the resource rooms is to enhance the ability of hospitalists to actually improve inpatient outcomes through self‐directed learning (see Fig. 1).
Hospitalists, residency directors, and directors of hospitalist fellowships and continuing education can use The Core Competencies in Hospital Medicine to develop curricula for their local hospitalist service and request that invited speakers develop learning objectives and content based on core competencies rather than giving a prepared lecture on a specific clinical condition. This case of PE illustrates that risk assessment, prophylaxis, EBM clinical problem solving, and QI are core topics that should be emphasized in the training of hospitalists and physicians in training.0
| STEP 1 The current problem and the need for improvement | Quality Issues |
| Prevention of predictable complications of illness: VTE still underutilized. | |
| Clinical problem‐solving in complex systems, cost‐effective, diagnostic testing. | |
| Triage of patients between services. | |
| STEP 2 Needs assessment of hospitalists and other | The Current Approach: The focus of traditional medical education. |
| members of the inpatient team | How to manage a specific disease rather than how to manage complex patients with multiple co‐morbidities. |
| Didactic lectures on the pathophysiology of VTE. rather than prevention, QI. | |
| Individual feedback, morbidity and mortality conferences | |
| STEP 3 Goals and specific measurable objectives | The Ideal Approach: Competencies as a framework for setting expectations about the role of the hospitalist |
| Direct therapy against predictable complications of serious illness. | |
| Critically review prophylaxis. | |
| Devise strategies to bridge the gap between knowledge and practice. | |
| STEP 4 Educational strategies | The first in a new online series: The VTE Resource Room, by SHM |
| Key knowledge, approaches, methods, and tools can be applied to improve performance despite variances due to particular systems and advances in medicine. | |
| Enhance the ability of hospitalists as self‐directed learners to improve inpatient outcomes. | |
| STEP 5 Implementation | The VTE Resource Room |
| A downloadable workbook and companion project outline for the improvement process. | |
| A slide set to disseminate valuable information about a safer system for VTE prevention. | |
| A moderated forum of VTE and QI experts to pose questions. | |
| STEP 6 Evaluation and feedback | Ongoing Evaluation and Feedback |
Continuous with other steps (see Fig. 1). | |
| STEP 7 Remaining questionsthe need for additional | Research Questions |
| research | Identifying barriers to VTE prophylaxis in the hospital setting. |
| Root cause analysis to determine prevention, process improvements, and training practices to encourage the safety of hospitalized patients. |
The second case example, the hand‐off (see Table 3), illustrates quality issues related to transfer of care from one physician to another. In this example, if the patient with moderate pleural effusion had been signed out, an earlier thoracentesis to drain a presumptive parapneumonic infection might have relieved this patient's shortness of breath and saved her from undergoing a subsequent VATS procedure. This case also demonstrates the importance of correlating imaging abnormalities with a patient's clinical presentation rather than using the traditional approach of just ruling out potential diagnoses to determine the cause of a problem. This case highlights elements of the process and system of care that can be modified to improve patient outcomes. Being proficient in transferring care of patients can save the hospitalist from error and prevent adverse events.
| The Hand‐Off: Avoiding Pitfalls in the Hospitalist System |
|---|
| A 30‐year‐old female, status postruptured uterus and caesarian section for pregnancy, presents with hypotension. |
| Shortness of breath postexploratory laparoscopy during fluid resuscitation. |
| Spiral CT performed to rule out pulmonary embolism, signed out as negative based on verbal report. |
| Estimated pulmonary arterial systolic pressure of 70 mmHg by transthoracic echocardiogram. |
| Extensive testing for underlying causes of pulmonary hypertension, hypercoagulable states. |
| Outpatient right heart catheterization scheduled by cardiology. |
| Sleep study advised to complete the workup of pulmonary hypertension. |
| After diuresis with a corresponding reduction in pulmonary capillary wedge pressure, her pulmonary hypertension resolves and her outpatient right heart catheterization is cancelled. |
| Final reading of chest CT (not signed out to receiving attending) reportedly notable for moderate right‐sided pleural effusion, small left‐sided effusion, and an apparent filling defect of right subclavian vein |
| Six days after the original spiral CT, unsuccessful thoracentesis attempted, with removal of 1 cc of fluid consistent with exudate. |
| Video‐assisted thorascopic surgery (VATS) procedure required to avoid chronic disability from trapped lung. |
| Retrospective review: Early drainage of a parapneumonic infection in the setting of sepsis might have avoided this complication. |
The Team Approach chapter establishes the need to acquire proficiencies not ordinarily obtained during residency in order to lead a multidisciplinary care team. This role requires a level of functioning beyond that of simply being the attending of record. The hospitalist must be able to synthesize information rather than simply defer to the consultant. Competencies specified in the Diagnostic Decision‐Making chapter can be used to identify the educational needs of hospitalists, who are expected to minimize diagnostic errors by knowing when to ask for help and where to get it, recognizing common diseases with uncommon presentations, and generating a broad differential diagnosis where there is uncertainty. The Patient Handoff chapter defines the proficiencies hospitalists need to facilitate the safe transfer of patients to other physicians on their service.0
| STEP 1 | |
| The current problem and the need for | Quality issues in the transfer of care. |
| improvement | Failure to review radiographic study. |
| Signing out pending test results. | |
| Failure to correlate imaging abnormalities with the patient's clinical presentation. | |
| STEP 2 | |
| Seeds assessment of hospitalists and other | The Current Approach: Inherent discontinuities of inpatient care. |
| embers of the inpatient team | ACGME legislated work hours: resident shifts. |
| Transfer of care to and from primary care physicians to hospitalists and between hospitalists. | |
| STEP 3 | |
| Goals and specific measurable objectives | The Ideal Approach: Development of a standardized method of communication between hospitalists and between residents. |
| A hand‐off checklist would include pending tests, including final readings of radiographic studies. | |
| Systematic review of all films with a radiologist. | |
| STEP 4 | |
| Educational strategies | Critical examination of local practice for variability in sign‐outs. |
| Development of curricula with an agreed‐upon standard using the Core Competencies as a templatethe Patient Hand‐Offs chapter. | |
| Measure quality of hand‐off and provide feedback. | |
| STEP 5 | |
| Implementation | Dissemination of the expectations of the hand‐off. |
| Series of didactic talks for residents, physician assistants, and medical students by hospitalists based on specific cases.19 | |
| Using the core competencies as a framework; didactic lectures on hospital medicine topics can be revised to better reflect the continuing educational needs of hospitalists and their roles and responsibilities. | |
| STEP 6 | |
| Evaluation and feedback | A Framework for Educational Scholarship: the process of evaluation. |
| Innovative educational pilots, designed for members of the multidisciplinary care team | |
| Clear goals, adequate preparation, appropriate methods, significant results, effective presentation, and reflective critique. | |
| New curricular designs and materials development in topics not traditionally taught during medical school and residency such as patient hand‐offs20, 21 | |
| Not limited to publication; educational scholarship can be funded through risk management and hospital‐funded seed grants. | |
| STEP 7 | |
| Remaining questionsthe need for | Research Questions |
| additional research | What are the key components of the sign‐out process? |
| How can an electronic medical record or other system be utilized to standardize and improve the process? |
The third case example, which expands the responsibilities of hospitalist to include meeting important needs in the hospital (see Table 4), illustrates that hospitalist services cannot succeed by offering all things to all people, a distraction that that keeps the members of these services from concentrating on their goals. Always saying yes to whoever asks for help is a band‐aid, a short‐term fix that impedes the effort and creativity required for durable long‐term solutions to problems.
| No Problem |
|---|
| A proposal has been made that a new academic hospitalist service care for neurosurgical patients in order to meet the goals of the neurosurgical residency program to maximize the operating room exposure of surgeons in training. |
| Patients would be admitted to the hospitalist service, with subsequent neurosurgical consultation. |
| Another proposal has been made that the hospitalist service care for uncovered patients without residents in order to meet the goals of the medical residency program. |
| Hospital leaders assume the hospitalist service would have no problem with this proposal. |
| The hospitalists, who are not in‐house at night, are asked to handle off‐hours triage issues when there is disagreement between two services; their proposed role would be to support the medical residents who do not feel empowered to say no to the surgical team seeing patients in the emergency department. |
| The hospitalist service has the following concerns: |
| Assuming responsibility for a nonteaching service undermines the vision of this new hospitalist service in an academic tertiary care facility. |
| Assuming responsibility for a surgical specialty service increases medical legal risk and concerns about timely backup. |
| Setting a bad precedent sends the wrong message. |
| Hospitalists functioning as superresidents damages the reputation of the service. |
| The proposal comes with a price, namely, accelerating physician burnout, declining job satisfaction, and inevitable turnover. |
| The proposal would adversely affect future physician recruitment and promotion through the medical school clinician educator track. |
| Existing problems with the work environment of this new hospitalist service include: |
| The service already does not have time to meet the responsibilities of inpatient care expected of hospitalists because of rapid growth and the need for further recruitment. |
| Lack of advocacy by hospital administrators who may not understand the role of the hospitalist and entertain other solutions is an ongoing concern. |
| Lack of support for other missions of teaching and quality improvement research, coupled with a changing job description and the daily unpredictability of the work, promotes the view that hospital medicine may not be sustainable as a career. |
| The challenge and opportunity: Expertise in strategic planning and operations management is needed in order to effectively respond to conflicting pressures and focus on goals that will sustain the ability to change, grow, and continuously improve. |
The Core Competencies sets expectations about the roles of hospitalists, who serve as well‐informed clinicians and clinical opinion leaders; effective educators, mentors, and role models; empathetic and timely communicators; efficient caregivers; and creative problem solvers arriving at durable, longer‐term solutions. The competencies demonstrate the knowledge, skills, and attitudes required to be effective agents of change. Changing business as usual almost always requires significant improvements in the underlying system, however uncomfortable. The Leadership chapter articulates competencies that hospitalists need in order to define their roles within the hospital, promote group cohesiveness, expand their practices intelligently, and anticipate and respond to change. This chapter details the proficiencies that hospitalists need in order to develop personal, team, and program goals and to identify and resolve conflicts using specific negotiation techniques. The Business Practices chapter articulates the fundamental skills needed to enhance program development and growth. Hospitalists can use the Core Competencies to identify educational needs and develop curricula to enhance their leadership and business skill sets.0
| STEP 1 | |
| The current problem and the need for improvement | Hospitalist Services cannot succeed by attempting to offer all things to all people. |
| Distracting members from their work and from concentrating on their goals. | |
| Always saying yes to whoever asks for help as a Band‐Aid, a short‐term fix that impedes the effort and creativity required for durable long‐term solutions to problems. | |
| STEP 2 | |
| Needs assessment of hospitalists and other members | The Current Approach: Problems with the work environment |
| of the inpatient team | Hospital medicine, a new specialty, does not yet have a similar supportive infrastructure analogous to other well‐established specialties with most hospitalist programs within divisions of general medicine. |
| Multiple stakeholdersadministrators, primary care providers, residency and clerkship directors, specialty services. | |
| Leadership and administrative skills are not consistently acquired proficiencies during residency training. | |
| STEP 3 | |
| Goals and specific measurable objectives | The Ideal Approach: Hospitalists can proactively improve their work life by developing skills and knowledge in hospital systems. |
| Develop personal, team, and program goals. | |
| Identify and resolve conflicts using specific negotiation techniques | |
| Enhance program development and growth. | |
| Identify senior physician leaders as mentors and advocates. | |
| STEP 4 | |
| Educational strategies | Annual retreats to generate enthusiasm, establish a strategic plan, continue a trajectory of success. |
| Invite an outside expert in QI or professional development to facilitate discussion. | |
| Recruit hospitalists and colleagues with expertise in healthcare systems to mentor and educate other members of the hospitalist service how to lead QI and other initiatives. | |
| STEP 5 | |
| Implementation | Use the core competencies to advocate for resources to support professional goals. |
| Funding for leadership courses and further training in business. | |
| Directors of CME sponsored by SHM have begun the process of using the core competencies as the framework for the development of hospital medicine curricula in leadership and QI. | |
| STEP 6 | |
| Evaluation and feedback | Consider using the Core Competencies to develop an internal report card on performance. |
| A self‐assessment tool based on the core values and goals of the hospitalist program. | |
| A means to help identify areas for improvement, modifiable risk factors for turnover, and opportunities to provide incentives to measure interventions, reward success, and ultimately deliver on the mandate to improve inpatient care. | |
| STEP 7 | |
| Remaining questionsthe need for additional | Challenges facing hospitalists practicing in multiple settings. |
| research | How to make processes of care efficient by examining specific tasks that hospitalists do and determining what tools, technologies, organizational structure, and supporting staff need to be available to make the performance of these tasks efficient. |
| How to make hospital medicine a sustainable and satisfying career. |
Medical educators should examine the outcomes of current training practices and assess what modifications of objectives, content, and instructional strategies should be made to better prepare the current and next generations of physicians to practice hospital medicine and to improve the hospital setting. Given the scope of the field of hospital medicine, the Core Competencies should guide: 1) what to teach and how much to teach; 2) how to teach and assess trainees, and how to assess and compare faculty development programs; 3) how to design systems for improving quality of care and assuring patient safety; and 4) how to establish priorities for hospital medicine research.
TRANSLATING A SET OF COMPETENCIES INTO CURRICULA: POTENTIAL BENEFITS
The Core Competencies in Hospital Medicine transcends hospital type, size, and setting and standardizes what the expectations for and proficiencies of a practicing hospitalist should be. By defining the role of the hospitalist, the Core Competencies serves as a resource for refining inpatient skills and assists in program development at the local, regional, and national levels. In addition, by using the Core Competencies as the standard and framework for the development of preparatory curricula, hospital administrators and other employers can rely on hospitalists having had a common preparation.
The medical profession is constantly evolving. Internal medicine curricula address the challenges hospital medicine physicians faced yesterday but could improve the training and preparation of physicians to serve in their new and emerging roles as leaders of multidisciplinary healthcare teams working to improve patient outcomes and the system of inpatient care. Hospital medicine no longer represents a group of physicians merely supporting other specialists and primary care physicians; it is itself a specialty, composed of physicians leading, directing, and improving inpatient care. The competencies presented in The Core Competencies in Hospital Medicine: A Framework for Curriculum Development, by the Society of Hospital Medicine, should spark debate about the adequacy and appropriateness of current training and certification expectations and serve as a foundation for the development of curricula to improve hospital medicine education.
- ,.The emerging role of “hospitalists” in the American health care system.N Engl J Med.1996;335:514–517.
- ,.The hospitalist movement 5 years later.JAMA.2002;287:487–494.
- .The hospitalist: a new medical specialty?Ann Intern Med.1999;130:373–375.
- ,,,,, eds.The Core Competencies in Hospital Medicine: A Framework for Curriculum Development.J Hosp Med.2006;1 (supplement 1).
- ,,,,.Core competencies in hospital medicine: development and methodology.J Hosp Med.2006;1:48–56.
- Koh LT,Corrigan JM,Donaldson MS, eds.To err is human.Washington, DC:National Academy Press,2000.
- ,,,,.Making healthcare safer: a critical analysis of patient safety practices. AHRQ publication 01‐E058,2001.
- Joint Commission on the Accreditation of Health Care Organizations. Available at URL: http://www.jcaho.org[accessed November 2005].
- Accreditation Council for Graduate Medical Education. Available at URL: http://www.acgme.org[accessed November 2005].
- ,.What is a curriculum?Ann Intern Med.1992;116:1055–1056.
- ,.Conceptualizing curriculum for graduate medical education.Acad Med.1992;67:528–534.
- American Association for Health Education,National Commission for Health Education Credentialing, Inc.,Society for Public Health Education.A competency‐based framework for graduate‐level health educators.Allentown, PA:NCHEC,1999.
- .How to write and use instructional objectives.6th ed.Upper Saddle River, NJ:Prentice Hall,2000.
- ,,, et al.Curriculum development for medical education: a six‐step approach.Baltimore:Johns Hopkins University Press,1998.
- ,.Needs assessment in postgraduate medical education: a review.Med Educ Online [serial online].2002;7. Available at URL:http://www.med‐ed‐online.org/pdf/f0000040.pdf[accessed December 7, 2005].
- .Identifying, appraising, and implementing medical education curricula: a guide for medical educators.Ann Intern Med.2001;135:889–896.
- ,,, et al.A quality improvement initiative at Brigham and Women's Hospital.N Engl J Med.2005;352:969.
- The Society of Hospital Medicine. Available from URL: http://www.hospitalmedicine.org[accessed November 2005].
- ,,.Teaching and the case method.3rd ed.Cambridge, MA:Harvard Business School,1994.
- .Scholarship reconsidered: priorities of the professoriate.Princeton, NJ:Carnegie Foundation for the Advance of Teaching,1990.
- ,Scholarly activities of faculty promoted in a teacher–clinician ladder.Acad Med.2000;75:649–52.
- ,.The emerging role of “hospitalists” in the American health care system.N Engl J Med.1996;335:514–517.
- ,.The hospitalist movement 5 years later.JAMA.2002;287:487–494.
- .The hospitalist: a new medical specialty?Ann Intern Med.1999;130:373–375.
- ,,,,, eds.The Core Competencies in Hospital Medicine: A Framework for Curriculum Development.J Hosp Med.2006;1 (supplement 1).
- ,,,,.Core competencies in hospital medicine: development and methodology.J Hosp Med.2006;1:48–56.
- Koh LT,Corrigan JM,Donaldson MS, eds.To err is human.Washington, DC:National Academy Press,2000.
- ,,,,.Making healthcare safer: a critical analysis of patient safety practices. AHRQ publication 01‐E058,2001.
- Joint Commission on the Accreditation of Health Care Organizations. Available at URL: http://www.jcaho.org[accessed November 2005].
- Accreditation Council for Graduate Medical Education. Available at URL: http://www.acgme.org[accessed November 2005].
- ,.What is a curriculum?Ann Intern Med.1992;116:1055–1056.
- ,.Conceptualizing curriculum for graduate medical education.Acad Med.1992;67:528–534.
- American Association for Health Education,National Commission for Health Education Credentialing, Inc.,Society for Public Health Education.A competency‐based framework for graduate‐level health educators.Allentown, PA:NCHEC,1999.
- .How to write and use instructional objectives.6th ed.Upper Saddle River, NJ:Prentice Hall,2000.
- ,,, et al.Curriculum development for medical education: a six‐step approach.Baltimore:Johns Hopkins University Press,1998.
- ,.Needs assessment in postgraduate medical education: a review.Med Educ Online [serial online].2002;7. Available at URL:http://www.med‐ed‐online.org/pdf/f0000040.pdf[accessed December 7, 2005].
- .Identifying, appraising, and implementing medical education curricula: a guide for medical educators.Ann Intern Med.2001;135:889–896.
- ,,, et al.A quality improvement initiative at Brigham and Women's Hospital.N Engl J Med.2005;352:969.
- The Society of Hospital Medicine. Available from URL: http://www.hospitalmedicine.org[accessed November 2005].
- ,,.Teaching and the case method.3rd ed.Cambridge, MA:Harvard Business School,1994.
- .Scholarship reconsidered: priorities of the professoriate.Princeton, NJ:Carnegie Foundation for the Advance of Teaching,1990.
- ,Scholarly activities of faculty promoted in a teacher–clinician ladder.Acad Med.2000;75:649–52.
Copyright © 2006 Society of Hospital Medicine
Core Competencies: Development and Methodology
Identification of the core competencies of a medical specialty provides the necessary framework for that specialty to develop, refine itself, and evolve. It also provides a structure from which training, testing, and curricula can be developed and effectively utilized. For nearly a decade, since the coining of the term hospitalist,1 the field of hospital medicine has been emerging as the next generation of site‐defined specialties, after emergency medicine and critical care medicine. The Core Competencies in Hospital Medicine: A Framework for Curriculum Development (referred to as the Core Competencies from this point on) introduces the expectations of hospitalists, helps to define their role, and suggests how knowledge, skill, and attitude acquisition might be evaluated. Furthermore, this document provides an initial structural framework from which curricula in adult hospital medicine may be developed.
The Core Competencies document, produced by the Society of Hospital Medicine (SHM) and published as a supplement to the first issue of the Journal of Hospital Medicine,2 is meant to serve as a framework for educators at all levels of medical education to develop curricula, training, and evaluations for students, clinicians‐in‐training, and practicing hospitalists. The Core Competencies document is not meant to contain a complete compilation of inpatient clinical topics or to re‐create what many residency training programs in adult inpatient care already provide. It should not limit and does not define every aspect of hospitalist practice. It includes the most common and fundamental elements of inpatient care without exhaustively listing every clinical entity that may be encountered by a hospitalist. Some of the more common clinical topics encountered by inpatient physicians are included, with an emphasis on subject areas that stress a systems‐based approach to health care, which is central to the practice of hospital medicine. This initial version of the Core Competencies document also focuses on potential areas of deficiency in the training of physicians to become hospitalists. It provides developers of curricula and content with a standardized set of measurable learning objectives, while allowing them the flexibility needed to address specific contexts and incorporate advances in medicine.
The SHM, the sole professional organization representing inpatient physicians, defines hospitalists as physicians whose primary professional focus is the general medical care of hospitalized patients. Their activities include patient care, teaching, research, and leadership related to Hospital Medicine.3 An estimated 12,000 hospitalists are currently practicing in the United States, with a projected workforce need of an estimated 20,00030,000 practicing hospitalists in the United States in the next 510 years.4 Various factors have contributed to the rapid growth and expansion of hospital medicine, including factors related to care efficiency, care quality, and inpatient teaching.512 The pressures that have contributed to the development of and evolution toward the hospitalist model of care over the past decade are facilitating the transformation from a traditional model of inpatient care to the care of inpatients by hospitalist physicians dedicated primarily to the inpatient setting. As a result of this growth in hospital medicine, the SHM realized that core competencies were needed to help define the field.
The purpose of this article is to describe the developmental process and content structure of the Core Competencies document. It delineates the process from initial needs assessment to topic list development to chapter production to internal and external review and revisions of individual chapters and the complete document. The supplement to this first issue of the Journal of Hospital Medicine contains 1) the Core Competencies,2 2) a reprint of this article, and 3) a reprint of the article by McKean et al. in this issue detailing how to use the Core Competencies,13 with examples and suggestions related to curriculum development. The authors propose that this combined compilation may spur curriculum development in hospital medicine that will help to define the field and set expectations for practice.
PROCESS AND TIMELINE
Education Summit
Early in the growth of hospital medicine, the Society of Hospital Medicine identified a need to better define a common educational and practice framework for hospitalist physicians. Such a framework could help to define hospitalists as a distinct group of practicing physicians with common goals and a common set of competencies. The importance of identifying and delineating the common knowledge, skills, and attitudes of hospitalists was paramount. Figure 1 shows the details of the 4‐year process of developing the Core Competencies.
In 2002, the SHM drew together educational leaders in hospital medicine in its first educational summit. One of the primary charges that the SHM received from this summit was to develop the needed core curriculum in hospital medicine. After the summit, the SHM's Education Committee formed the Core Curriculum Task Force (CCTF), composed of approximately 15 member hospitalists, with representation from university and community hospitals, teaching and nonteaching programs, and for‐profit and not‐for‐profit settings from various geographic regions of the country. The selection process ensured that the task force was representative of practicing hospitalists and SHM membership throughout the United States.
The CCTF
The task force met through frequent conference‐call meetings and at least one in‐person meeting annually. The primary goal set forth by the task force was the initial development of a distinct set of core competencies in hospital medicine that could then guide curriculum development within the field.
Topic List
The task force determined that the topics (or chapters) should be divided into three sectionsClinical Conditions, Procedures, and Healthcare Systems (Table 1, Chapter List)all integral components of the practice of hospital medicine. For Clinical Conditions chapters, the task force decided that an exhaustive listing of all potential clinical entities that hospitalists might encounter during their clinical practice was not the goal of the Core Competencies. Rather, clinical topics were selected to reflect conditions in the hospital setting that are encountered with significant frequency, that might be significantly life‐threatening, or that are likely to have the significant involvement and impact of hospitalists in altering or refining care processes, leading to improvement in care quality and efficiency. The list of Clinical Condition chapters should not limit or rigidly define the scope of practice of hospitalist physicians. Instead, it should help those entering the field of hospital medicine better understand some of the core clinical topics on which hospitalists focus in the design of institutional or global quality initiatives.
| Clinical Conditions* | Procedures | Healthcare Systems |
|---|---|---|
| ||
|
|
|
Clinical Conditions Section
In an effort to both narrow and delineate the core Clinical Condition areas necessary for practicing hospital medicine, the task force elected first to draw from national data the most common diagnosis‐related groups (DRGs) discharged from U.S. hospitals. Utilizing the Medicare database, the top 15 nonsurgical discharge diagnoses were initially selected. Certain clinical conditions that the task force believed to be highly relevant to the practice of hospital medicine but that did not neatly fall into a specific DRG, such as pain management and perioperative medicine, were proposed for and then added to the list of Clinical Conditions chapters by the task force. Other chapters, such as that on venous thromboembolism, were added because a particular disease, although not necessarily a high‐ranked discharge DRG, showed high inpatient morbidity and mortality and reflected the role of the hospitalist in the prevention of predictable complications during hospitalization. When possible, some diagnoses were consolidated to better incorporate crosscutting competencies or to highlight opportunities for leadership in systems‐based improvements. For example, upper and lower gastrointestinal bleeding were consolidated into the chapter on gastrointestinal bleeding. Similarly, all relevant arrhythmias that a hospitalist might encounter were consolidated into a single chapter. For at least one clinical topic, pneumonia, the task force believed it necessary to have two distinct chapters, one on community‐acquired pneumonia and the other on hospital‐acquired pneumonia, because these two entities are significantly different and have distinct competencies. The final listing of Clinical Conditions chapters reflects 19 clinical areas that hospitalists encounter on a frequent basis and for which they can have an effect on systems and processes of care. These clinical chapters form a foundation of topics for which hospitalists have already begun quality and efficiency initiatives.
The task force further decided that symptom evaluation and management could be consolidated into a systems chapter dedicated to diagnostic decision making. A reasonably large constellation of symptoms, including but not limited to chest pain, shortness of breath, syncope, and altered mental status, are encountered by hospitalists daily. Although evaluation and management of these symptoms are extremely important parts of triage, subsequent testing, and hospital care, the ability to develop a differential diagnosis and proceed with the indicated testing and its interpretation is common to all symptom evaluation. Such evaluation and diagnostic decision making are therefore summarized in a single chapter in the Healthcare Systems section, and no symptom chapters are found in the Clinical section.
Procedures Section
The initial topic lists for the Procedures and Systems sections were developed through input from the broad representation of the Core Curriculum Task Force. The chapters in the Procedures section contain competencies expected for the inpatient procedures that hospitalists are most likely to perform or supervise in their day‐to‐day care of hospitalized patients. The presence of a procedural skill in the Core Competencies does not necessarily indicate that every hospitalist will perform or be proficient in that procedure. Similarly, the absence of a procedure from the Core Competencies should not exclude trained and experienced hospitalists from performing that procedure. The task force recognizes that the individual hospital setting, including local and regional variations, determines who might perform certain procedures depending on many factors, which may include whether there are trainees, specialty support including radiology, and procedure teams. The Procedures section outlines those procedures frequently performed in the everyday practice of hospital medicine and incorporates relevant competencies to afford proper performance, patient education and involvement, prevention of complications, and quality improvement for these procedures.
Healthcare Systems Section
Although many competencies delineated in the Clinical Conditions and Procedures sections of the supplement may be taught well during medical school and residency training, that is not true of the chapters and competencies in the Healthcare Systems section, many of which are not extensively taught in most undergraduate or graduate medical education programs. Therefore, many hospitalists must gain or supplant their knowledge, skills, and attitudes in system areas posttraining.
The Healthcare Systems section delineates themes integral to the successful practice of hospital medicine in diverse hospital settings. Many chapters in this section focus on processes and systems of care that typically span multiple disease entities and frequently require multidisciplinary input to create a coordinated effort for care quality and efficiency. The chapters and core competencies in the Healthcare Systems section direct hospitalists to lead and innovate in their own hospital practices and to convey the principles of evidence‐based inpatient medical care and systems‐based practice to medical students, physicians‐in‐training, other medical staff, colleagues, and patients. The task force expects that many new hospitalists will still be learning many of the competencies in the Healthcare Systems section during the early stages of their posttraining practice. However, as training of hospitalists during undergraduate and graduate medical education further evolves, we expect that more hospitalists will enter the workforce with more of the skills necessary to prepare them for their careers.
Some Healthcare Systems chapters have clinical themes but were included in this section because it is believed that the clinical approach always spans multiple clinical entities and always requires an organizational approach crossing several disciplines in medicine in order to optimize the hospital care. Such chapters include Care of the Elderly Patient, Prevention of Healthcare Associated Infections and Antimicrobial Resistance, Nutrition and the Hospitalized Patient, and Palliative Care. Other chapters in the Healthcare Systems section focus on educational themes that drive the practice of hospital medicine and the lifelong learning and teaching required of hospitalists. Some of these chapters include Evidence‐Based Medicine, Hospitalist as Teacher, Patient Education, and Practice‐Based Learning and Improvement. Still other chapters in the Healthcare Systems section identify much of the organizational approachboth from clinical practice and practice management standpointsthat must be adopted by hospitalists in order to provide high‐quality care while maintaining functional and sound practice. Examples of chapters focusing on clinical practice organization include Patient Safety, Quality Improvement, Team Approach and Multidisciplinary Care, Transitions of Care, and Patient Handoffs. Although the Transitions of Care chapter focuses on the processes and communication required for the safe transition of patients from one clinical setting to another; the Patient Handoffs (or sign‐out) chapter focuses on the hospitalist‐to‐hospitalist communication essential when one hospitalist assumes care of a patient from another (either from dayshift to nightshift on the same service or assuming care of service from a different service). Examples of chapters focusing on practice management organization include Business Practices, Equitable Allocation of Resources, Leadership, and Risk Management. Overall, the Healthcare Systems chapters help to characterize and delineate the practice and scope of hospital medicine, especially with topics not taught in detail during most residency training programs.
Editorial Board, Content Survey, and Topic List Refinement
Once the initial topic list was created, a five‐member editorial board was chosen from the CCTF membership, including the SHM CCTF chair, the Education Committee chair, two member hospitalists, and a health education specialist. The purpose of this board was to interpret survey feedback, solicit contributors to write competency chapters, review and revise the chapters submitted, and prepare the larger document for review and final publication. The Core Curriculum Task Force developed a survey to obtain feedback on the initial topic list. Face validity was established through a survey sent electronically in 2003 to the SHM Board of Directors and Education Committee, as well as to 10 representatives of each SHM regional council and local chapter. In all, more than 250 hospitalists representing diverse geographic and practice backgrounds were surveyed. Feedback from the survey was reviewed by the CCTF. The topic list was then revised with additions and modifications incorporated from survey feedback. The scope of individual topics also was modified in multiple iterations congruent with the internal and external review processes.
Contributors
Contributors were solicited by the task force, utilizing SHM databasesbelieved to be the most comprehensive registry of hospitalist physiciansand an electronic call for nominations to practicing hospitalists from around the United States. Other recognized content experts were solicited independently on the basis of chapter or content needs. Efforts were taken to identify hospitalists with expertise in specific topic areas, particularly those with a history of presentations or publications on individual chapter subject matter. Potential contributors submitted credentials, including curricula vitae and other supporting documents or information, when requesting to write a specific chapter for the Core Competencies compendium. Contributors were competitively selected on the basis of their submitted information compared to those of others requesting to write the same chapter. In some cases practicing hospitalists were paired with nonhospitalist expert contributors to create a chapter. Contributors were provided with guidelines with which to prepare their chapter.
Review and Revision
The editorial board reviewed all the chapters, rigorously evaluating each chapter through at least five stages of review and revision. First, chapters were reviewed by the editorial boardinitially by at least two physician members and then by the entire editorial board. Chapters were reviewed for the scope and completeness of concepts, adherence to educational theory, and consistency in chapter format. Changes in content and for consistency were extensive in some chapters, whereas others required only small or moderate changes. Significant editing was required to create chapters as a compilation of specific, measurable competencies as opposed to topic‐related content. All chapters required some level of modification to assist with consistency in style, language, and overall goals. Where appropriate, individual chapters were also reviewed by relevant SHM committees, task forces, or content experts, and initial feedback was provided. For example, the Leadership chapter was reviewed by the SHM Leadership Task Force. Other SHM committees and task forces involved in chapter reviews included the Education, Healthcare Quality and Patient Safety, and Ethics committees as well as the Geriatric Task Force. Changes recommended changes on the basis of committee and task force feedback were incorporated into the relevant chapters.
Second, revisions of individual chapters from the editorial board were sent back to contributors for final comment, revision, and approval. Third, the compilation of all chapters and sections was reviewed (as a whole) and underwent further revision by the editorial board based on feedback from the contributors and the relevant SHM committees. Fourth, the entire revised supplement was sent for an internal review by the SHM board and relevant SHM committees or committee representatives.
Fifth, final reviews were solicited from external reviewers of medical professional organizations and academic organizations. Feedback from the internal and external reviews were compiled and systematically evaluated by the CCTF editorial board. Recommended changes were incorporated into individual chapters or throughout the Core Competencies compendium on the basis of the evaluation and consensus approval of the editorial board. For example, one reviewer believed that quality improvement initiatives were necessary for all procedures that hospitalists perform in order to help reduce the risk of complications. Therefore, each procedure chapter was revised to reflect this competency. Similarly, another reviewer thought that in many chapters the involvement of nursing and other medical staff in the implementation of multidisciplinary teams was underemphasized. Therefore, efforts were taken to improve the emphasis of these key participants in multidisciplinary hospital care.
The efforts of many individuals and professional organizations have helped the CCTF to refine the expectations of a professional trained in the discipline of hospital medicine. Table 2 has a complete listing of those solicited to be internal and external reviewers. Although aggressive efforts were undertaken to encourage feedback from all solicited reviewers of the Core Competencies document, time or other constraints prevented some reviewers from responding to the review request (overall response or review rate: 52%). Nevertheless, the multiple review and revision process brought what was initially disparate content and organization together in a much more cohesive and consistent approach and structure to competencies in hospital medicine.
|
| Accreditation Council of Graduate Medical Education (ACGME) |
| Agency for Healthcare Research & Quality (AHRQ) |
| American Academy of Family Practice (AAFP) |
| American Association of Critical Care Nurses (AACCN) |
| American Association of Subspecialty Professors |
| American Board of Family Practice |
| American Board of Internal Medicine (ABIM) |
| American College of Chest Physicians (ACCP) |
| American College of Emergency Physicians (ACEP) |
| American College of Physicians (ACP) |
| American Geriatrics Society |
| American Hospital Association (AHA) |
| Association of American Medical Colleges (AAMC) |
| Institute for Healthcare Improvement (IHI) |
| John A. Hartford Foundation |
| Joint Commission on Accreditation of Healthcare Organizations (JCAHO) |
| Residency Review Committee Internal Medicine (RRC‐IM) |
| Reynolds Foundation |
| Robert Wood Johnson Foundation (RWJF) |
| Society of Critical Care Medicine (SCCM) |
| Society of General Internal Medicine (SGIM) |
| Society of Hospital Medicine |
| ○ Board of Directors (9 members solicited) |
| ○ CCTF Members (3 members solicited exclusive of editorial board) |
CHAPTER CONTENT DESCRIPTION
As previously delineated, the Core Competencies document has three sections: Clinical Conditions, Procedures, and Healthcare Systems. The chapters in the entire compendium and within each section have been designed to stand alone and to be used either individually or collectively to assist with curriculum development in hospital medicine. However, each chapter should be used in the context of the entire document because a particular issue may only be touched on in one chapter but may be more elaborately detailed in another. For example, all clinical conditions chapters include a competency on the issue of care transitions, but the specific competencies for care transitions are presented in a separate Transitions of Care chapter.
All chapters in each section begin with an introduction that provides brief background information and establishes the relevance of the topic to practicing hospitalists. Each chapter then utilizes the educational theory of learning domains. The learning domains include the cognitive domain (knowledge), the psychomotor domain (skills), and the affective domain (attitudes). The companion article How to Use The Core Competencies in Hospital Medicine: A Framework for Curriculum Development13 describes in detail the educational theory guiding the development of the Core Competencies document and suggested methods for applying it to the development and revision of curricula and other training activities.
The task force further decided that each chapter in the Clinical Conditions and Procedures sections should include a subsection dedicated to system organization and improvement, an added domain that requires integration of knowledge, skills, and attitudes and the involvement of other medical services and disciplines for optimal patient care. The editorial board believed that system organization and improvement was already an intrinsic feature embedded in the chapters of the Healthcare Systems section. Therefore, this subsection was not included in those chapters.
Hospitalists subscribe to a systems organizational approach to clinical management and processes of care within the hospital. This systems approach, more than any level of knowledge or skill, is required to effectively and efficiently practice in the hospital setting. Practicing with a systems approach, with the interest of improving processes of care, is embedded throughout the Core Competencies document and is a practice method that all hospitalists may strive to achieve as they develop and improve their inpatient care. The competencies within the Systems Organization and Improvement section may contain a range of competency expectation (eg, lead, coordinate, or participate in) to acknowledge their uniqueness and variation according to practice settings and locally instituted responsibilities.
Each competency within a chapter details a level of proficiency, providing guidance on learning activities and potential evaluation strategies. Several overarching themes are followed in the chapters that help to define hospitalists as physicians who specialize in the care of hospitalized patients. First, hospitalists strive to support and adhere to a multidisciplinary approach for the patients under their care. Such an approach involves active interaction with and integration of other hospital medical staff (eg, nursing, rehabilitation therapies, social services) and of specialty medical or surgical services when indicated. Recognizing that hospitalists vary in experience and mastery of their field, the task force and editorial board believed that, at minimum, hospitalists would participate in multidisciplinary teams for improvement of the care and process related to the clinical conditions within their organization. However, they might also lead and/or coordinate teams in such efforts. Therefore, most chapters contain competencies that expect hospitalists to lead, coordinate, or participate in multidisciplinary teams or initiatives that will facilitate optimal care within their organization.
Second, because hospital medicine centers around the quality of inpatient care, participation in quality improvement (QI) initiatives, focusing on improving processes or systems of care in a local institution or organization, may be common in hospitalist practices. The level of involvement and role in QI initiatives may vary according to the particular system, the resources available, and a hospitalist's experience. Finally, because hospitalist care intrinsically involves an increase in the number of care transitions and handoffs, hospitalists need to remain sensitive to and focused on the care transitions that occur with their patients. Such transitions may occur as patients enter the hospital, move from one location to another within the hospital, or leave the hospital. This vulnerable time for patients requires hospitalists to be vigilant in their communication effortswith patients, with medical staff, and with outpatient clinicians.
Each competency was crafted to indicate the relevant concept, the level of proficiency expected, and a way to evaluate mastery. The teaching processes and learning experiences that must take place to achieve competency are left for curriculum developers and instructors to design. These core competencies represent an initial step in curriculum development, creating an identity and core set of expectations for hospitalists that we believe will lead to progress and maturity within the field.
SUMMARY AND FUTURE DIRECTIONS
The practice of hospital medicine requires proficiency of interrelated aspects of practiceclinical, procedural, and system‐based competencies. For practicing hospitalists, the Core Competencies document may serve as a resource to refine skills and assist in program development at individual institutions, both regionally and nationally. For residency program directors and clerkship directors, the Core Competencies document can function as a guide for developing the curriculum of inpatient medicine rotations or for meeting the requirements of the Outcomes Project of the Accreditation Council on Graduate Medical Education's. Last, for those developing continuing medical education programs, the Core Competencies document or individual chapters or topics within it may serve as an outline around which specific or broad‐based programs can be developed. Although the development of such curricula and the recipients of them should be evaluated, the actual evaluation is left to the curriculum developers.
Hospitalists are invested in making hospitals run better. They are positioned to take leadership roles in addressing quality, efficiency, and cost interests in both community and academic hospital settings. Their goals include improving care processes, hospital work life, and the setting in which they practice. The key core competencies described in this compendium define hospitalists as agents of change 1) to develop and implement systems to enable best practices to occur from admission through discharge, and 2) to promote the development of a safer culture within the hospital.
Hospital medicine remains an evolving specialty. Although great care was taken to construct these competencies so they would retain their relevance over time, SHM, the Core Curriculum Task Force, and the editorial board recognize the need for their continual reevaluation and modification in the context of advances and changes in the practice of hospital medicine. Our intent is that these competencies be a common reference and foundation for the creation of hospital medicine curricula and serve to standardize and improve training practices.
- ,.The emerging role of “hospitalists” in the American health care system.N Engl J Med.1996;335:514–517.
- ,,,,, eds.The Core Competencies in Hospital Medicine: A Framework for Curriculum Development.J Hosp Med.2006;1(supplement 1).
- Society of Hospital Medicine. About SHM: What is a hospitalist? Available from URL: http://www.hospitalmedicine.org[accessed July 22, 2005].
- .The future of hospital medicine: evolution or revolution?Am J Med.2004;117:446–450.
- ,.The hospitalist movement 5 years later.JAMA.2002;287:487–494.
- ,,, et al.Implementation of a voluntary hospitalist service at a community teaching hospital: improved clinical efficiency and patient outcomes.Ann Intern Med.2002;137:859–865.
- ,,, et al.Effects of physician experience on costs and outcomes on an academic general medicine service: results of a trial of hospitalists.Ann Intern Med.2002;137:866–874.
- ,,, et al.Making Healthcare aafer: a critical analysis of patient safety practices.Rockville, MD:U.S. Dept. of Health and Human Services, Agency for Healthcare Research and Quality;2001. AHRQ publication 01‐E058. Available from URL: http://www.ahrq.gov.
- ,,, et al.Medical student evaluation of the quality of hospitalist and nonhospitalist teaching faculty on inpatient medicine rotations.Acad Med.2004;79:78–82.
- ,,, et al.Hospitalists as teachers.J Gen Intern Med.2004;19(1):8–15.
- ,,, et al.The positive impact of initiation of hospitalist clinician educators.J Gen Intern Med.2004;19:293–301.
- ,,, et al.Effects of hospitalist attending physicians on trainee satisfaction with teaching and with internal medicine rotations.Arch Intern Med.2004;164:1866–1887.
- ,,,,.How to use The Core Competencies in Hospital Medicine: A Framework for Curriculum Development.J Hosp Med.2006;1:57–67.
Identification of the core competencies of a medical specialty provides the necessary framework for that specialty to develop, refine itself, and evolve. It also provides a structure from which training, testing, and curricula can be developed and effectively utilized. For nearly a decade, since the coining of the term hospitalist,1 the field of hospital medicine has been emerging as the next generation of site‐defined specialties, after emergency medicine and critical care medicine. The Core Competencies in Hospital Medicine: A Framework for Curriculum Development (referred to as the Core Competencies from this point on) introduces the expectations of hospitalists, helps to define their role, and suggests how knowledge, skill, and attitude acquisition might be evaluated. Furthermore, this document provides an initial structural framework from which curricula in adult hospital medicine may be developed.
The Core Competencies document, produced by the Society of Hospital Medicine (SHM) and published as a supplement to the first issue of the Journal of Hospital Medicine,2 is meant to serve as a framework for educators at all levels of medical education to develop curricula, training, and evaluations for students, clinicians‐in‐training, and practicing hospitalists. The Core Competencies document is not meant to contain a complete compilation of inpatient clinical topics or to re‐create what many residency training programs in adult inpatient care already provide. It should not limit and does not define every aspect of hospitalist practice. It includes the most common and fundamental elements of inpatient care without exhaustively listing every clinical entity that may be encountered by a hospitalist. Some of the more common clinical topics encountered by inpatient physicians are included, with an emphasis on subject areas that stress a systems‐based approach to health care, which is central to the practice of hospital medicine. This initial version of the Core Competencies document also focuses on potential areas of deficiency in the training of physicians to become hospitalists. It provides developers of curricula and content with a standardized set of measurable learning objectives, while allowing them the flexibility needed to address specific contexts and incorporate advances in medicine.
The SHM, the sole professional organization representing inpatient physicians, defines hospitalists as physicians whose primary professional focus is the general medical care of hospitalized patients. Their activities include patient care, teaching, research, and leadership related to Hospital Medicine.3 An estimated 12,000 hospitalists are currently practicing in the United States, with a projected workforce need of an estimated 20,00030,000 practicing hospitalists in the United States in the next 510 years.4 Various factors have contributed to the rapid growth and expansion of hospital medicine, including factors related to care efficiency, care quality, and inpatient teaching.512 The pressures that have contributed to the development of and evolution toward the hospitalist model of care over the past decade are facilitating the transformation from a traditional model of inpatient care to the care of inpatients by hospitalist physicians dedicated primarily to the inpatient setting. As a result of this growth in hospital medicine, the SHM realized that core competencies were needed to help define the field.
The purpose of this article is to describe the developmental process and content structure of the Core Competencies document. It delineates the process from initial needs assessment to topic list development to chapter production to internal and external review and revisions of individual chapters and the complete document. The supplement to this first issue of the Journal of Hospital Medicine contains 1) the Core Competencies,2 2) a reprint of this article, and 3) a reprint of the article by McKean et al. in this issue detailing how to use the Core Competencies,13 with examples and suggestions related to curriculum development. The authors propose that this combined compilation may spur curriculum development in hospital medicine that will help to define the field and set expectations for practice.
PROCESS AND TIMELINE
Education Summit
Early in the growth of hospital medicine, the Society of Hospital Medicine identified a need to better define a common educational and practice framework for hospitalist physicians. Such a framework could help to define hospitalists as a distinct group of practicing physicians with common goals and a common set of competencies. The importance of identifying and delineating the common knowledge, skills, and attitudes of hospitalists was paramount. Figure 1 shows the details of the 4‐year process of developing the Core Competencies.
In 2002, the SHM drew together educational leaders in hospital medicine in its first educational summit. One of the primary charges that the SHM received from this summit was to develop the needed core curriculum in hospital medicine. After the summit, the SHM's Education Committee formed the Core Curriculum Task Force (CCTF), composed of approximately 15 member hospitalists, with representation from university and community hospitals, teaching and nonteaching programs, and for‐profit and not‐for‐profit settings from various geographic regions of the country. The selection process ensured that the task force was representative of practicing hospitalists and SHM membership throughout the United States.
The CCTF
The task force met through frequent conference‐call meetings and at least one in‐person meeting annually. The primary goal set forth by the task force was the initial development of a distinct set of core competencies in hospital medicine that could then guide curriculum development within the field.
Topic List
The task force determined that the topics (or chapters) should be divided into three sectionsClinical Conditions, Procedures, and Healthcare Systems (Table 1, Chapter List)all integral components of the practice of hospital medicine. For Clinical Conditions chapters, the task force decided that an exhaustive listing of all potential clinical entities that hospitalists might encounter during their clinical practice was not the goal of the Core Competencies. Rather, clinical topics were selected to reflect conditions in the hospital setting that are encountered with significant frequency, that might be significantly life‐threatening, or that are likely to have the significant involvement and impact of hospitalists in altering or refining care processes, leading to improvement in care quality and efficiency. The list of Clinical Condition chapters should not limit or rigidly define the scope of practice of hospitalist physicians. Instead, it should help those entering the field of hospital medicine better understand some of the core clinical topics on which hospitalists focus in the design of institutional or global quality initiatives.
| Clinical Conditions* | Procedures | Healthcare Systems |
|---|---|---|
| ||
|
|
|
Clinical Conditions Section
In an effort to both narrow and delineate the core Clinical Condition areas necessary for practicing hospital medicine, the task force elected first to draw from national data the most common diagnosis‐related groups (DRGs) discharged from U.S. hospitals. Utilizing the Medicare database, the top 15 nonsurgical discharge diagnoses were initially selected. Certain clinical conditions that the task force believed to be highly relevant to the practice of hospital medicine but that did not neatly fall into a specific DRG, such as pain management and perioperative medicine, were proposed for and then added to the list of Clinical Conditions chapters by the task force. Other chapters, such as that on venous thromboembolism, were added because a particular disease, although not necessarily a high‐ranked discharge DRG, showed high inpatient morbidity and mortality and reflected the role of the hospitalist in the prevention of predictable complications during hospitalization. When possible, some diagnoses were consolidated to better incorporate crosscutting competencies or to highlight opportunities for leadership in systems‐based improvements. For example, upper and lower gastrointestinal bleeding were consolidated into the chapter on gastrointestinal bleeding. Similarly, all relevant arrhythmias that a hospitalist might encounter were consolidated into a single chapter. For at least one clinical topic, pneumonia, the task force believed it necessary to have two distinct chapters, one on community‐acquired pneumonia and the other on hospital‐acquired pneumonia, because these two entities are significantly different and have distinct competencies. The final listing of Clinical Conditions chapters reflects 19 clinical areas that hospitalists encounter on a frequent basis and for which they can have an effect on systems and processes of care. These clinical chapters form a foundation of topics for which hospitalists have already begun quality and efficiency initiatives.
The task force further decided that symptom evaluation and management could be consolidated into a systems chapter dedicated to diagnostic decision making. A reasonably large constellation of symptoms, including but not limited to chest pain, shortness of breath, syncope, and altered mental status, are encountered by hospitalists daily. Although evaluation and management of these symptoms are extremely important parts of triage, subsequent testing, and hospital care, the ability to develop a differential diagnosis and proceed with the indicated testing and its interpretation is common to all symptom evaluation. Such evaluation and diagnostic decision making are therefore summarized in a single chapter in the Healthcare Systems section, and no symptom chapters are found in the Clinical section.
Procedures Section
The initial topic lists for the Procedures and Systems sections were developed through input from the broad representation of the Core Curriculum Task Force. The chapters in the Procedures section contain competencies expected for the inpatient procedures that hospitalists are most likely to perform or supervise in their day‐to‐day care of hospitalized patients. The presence of a procedural skill in the Core Competencies does not necessarily indicate that every hospitalist will perform or be proficient in that procedure. Similarly, the absence of a procedure from the Core Competencies should not exclude trained and experienced hospitalists from performing that procedure. The task force recognizes that the individual hospital setting, including local and regional variations, determines who might perform certain procedures depending on many factors, which may include whether there are trainees, specialty support including radiology, and procedure teams. The Procedures section outlines those procedures frequently performed in the everyday practice of hospital medicine and incorporates relevant competencies to afford proper performance, patient education and involvement, prevention of complications, and quality improvement for these procedures.
Healthcare Systems Section
Although many competencies delineated in the Clinical Conditions and Procedures sections of the supplement may be taught well during medical school and residency training, that is not true of the chapters and competencies in the Healthcare Systems section, many of which are not extensively taught in most undergraduate or graduate medical education programs. Therefore, many hospitalists must gain or supplant their knowledge, skills, and attitudes in system areas posttraining.
The Healthcare Systems section delineates themes integral to the successful practice of hospital medicine in diverse hospital settings. Many chapters in this section focus on processes and systems of care that typically span multiple disease entities and frequently require multidisciplinary input to create a coordinated effort for care quality and efficiency. The chapters and core competencies in the Healthcare Systems section direct hospitalists to lead and innovate in their own hospital practices and to convey the principles of evidence‐based inpatient medical care and systems‐based practice to medical students, physicians‐in‐training, other medical staff, colleagues, and patients. The task force expects that many new hospitalists will still be learning many of the competencies in the Healthcare Systems section during the early stages of their posttraining practice. However, as training of hospitalists during undergraduate and graduate medical education further evolves, we expect that more hospitalists will enter the workforce with more of the skills necessary to prepare them for their careers.
Some Healthcare Systems chapters have clinical themes but were included in this section because it is believed that the clinical approach always spans multiple clinical entities and always requires an organizational approach crossing several disciplines in medicine in order to optimize the hospital care. Such chapters include Care of the Elderly Patient, Prevention of Healthcare Associated Infections and Antimicrobial Resistance, Nutrition and the Hospitalized Patient, and Palliative Care. Other chapters in the Healthcare Systems section focus on educational themes that drive the practice of hospital medicine and the lifelong learning and teaching required of hospitalists. Some of these chapters include Evidence‐Based Medicine, Hospitalist as Teacher, Patient Education, and Practice‐Based Learning and Improvement. Still other chapters in the Healthcare Systems section identify much of the organizational approachboth from clinical practice and practice management standpointsthat must be adopted by hospitalists in order to provide high‐quality care while maintaining functional and sound practice. Examples of chapters focusing on clinical practice organization include Patient Safety, Quality Improvement, Team Approach and Multidisciplinary Care, Transitions of Care, and Patient Handoffs. Although the Transitions of Care chapter focuses on the processes and communication required for the safe transition of patients from one clinical setting to another; the Patient Handoffs (or sign‐out) chapter focuses on the hospitalist‐to‐hospitalist communication essential when one hospitalist assumes care of a patient from another (either from dayshift to nightshift on the same service or assuming care of service from a different service). Examples of chapters focusing on practice management organization include Business Practices, Equitable Allocation of Resources, Leadership, and Risk Management. Overall, the Healthcare Systems chapters help to characterize and delineate the practice and scope of hospital medicine, especially with topics not taught in detail during most residency training programs.
Editorial Board, Content Survey, and Topic List Refinement
Once the initial topic list was created, a five‐member editorial board was chosen from the CCTF membership, including the SHM CCTF chair, the Education Committee chair, two member hospitalists, and a health education specialist. The purpose of this board was to interpret survey feedback, solicit contributors to write competency chapters, review and revise the chapters submitted, and prepare the larger document for review and final publication. The Core Curriculum Task Force developed a survey to obtain feedback on the initial topic list. Face validity was established through a survey sent electronically in 2003 to the SHM Board of Directors and Education Committee, as well as to 10 representatives of each SHM regional council and local chapter. In all, more than 250 hospitalists representing diverse geographic and practice backgrounds were surveyed. Feedback from the survey was reviewed by the CCTF. The topic list was then revised with additions and modifications incorporated from survey feedback. The scope of individual topics also was modified in multiple iterations congruent with the internal and external review processes.
Contributors
Contributors were solicited by the task force, utilizing SHM databasesbelieved to be the most comprehensive registry of hospitalist physiciansand an electronic call for nominations to practicing hospitalists from around the United States. Other recognized content experts were solicited independently on the basis of chapter or content needs. Efforts were taken to identify hospitalists with expertise in specific topic areas, particularly those with a history of presentations or publications on individual chapter subject matter. Potential contributors submitted credentials, including curricula vitae and other supporting documents or information, when requesting to write a specific chapter for the Core Competencies compendium. Contributors were competitively selected on the basis of their submitted information compared to those of others requesting to write the same chapter. In some cases practicing hospitalists were paired with nonhospitalist expert contributors to create a chapter. Contributors were provided with guidelines with which to prepare their chapter.
Review and Revision
The editorial board reviewed all the chapters, rigorously evaluating each chapter through at least five stages of review and revision. First, chapters were reviewed by the editorial boardinitially by at least two physician members and then by the entire editorial board. Chapters were reviewed for the scope and completeness of concepts, adherence to educational theory, and consistency in chapter format. Changes in content and for consistency were extensive in some chapters, whereas others required only small or moderate changes. Significant editing was required to create chapters as a compilation of specific, measurable competencies as opposed to topic‐related content. All chapters required some level of modification to assist with consistency in style, language, and overall goals. Where appropriate, individual chapters were also reviewed by relevant SHM committees, task forces, or content experts, and initial feedback was provided. For example, the Leadership chapter was reviewed by the SHM Leadership Task Force. Other SHM committees and task forces involved in chapter reviews included the Education, Healthcare Quality and Patient Safety, and Ethics committees as well as the Geriatric Task Force. Changes recommended changes on the basis of committee and task force feedback were incorporated into the relevant chapters.
Second, revisions of individual chapters from the editorial board were sent back to contributors for final comment, revision, and approval. Third, the compilation of all chapters and sections was reviewed (as a whole) and underwent further revision by the editorial board based on feedback from the contributors and the relevant SHM committees. Fourth, the entire revised supplement was sent for an internal review by the SHM board and relevant SHM committees or committee representatives.
Fifth, final reviews were solicited from external reviewers of medical professional organizations and academic organizations. Feedback from the internal and external reviews were compiled and systematically evaluated by the CCTF editorial board. Recommended changes were incorporated into individual chapters or throughout the Core Competencies compendium on the basis of the evaluation and consensus approval of the editorial board. For example, one reviewer believed that quality improvement initiatives were necessary for all procedures that hospitalists perform in order to help reduce the risk of complications. Therefore, each procedure chapter was revised to reflect this competency. Similarly, another reviewer thought that in many chapters the involvement of nursing and other medical staff in the implementation of multidisciplinary teams was underemphasized. Therefore, efforts were taken to improve the emphasis of these key participants in multidisciplinary hospital care.
The efforts of many individuals and professional organizations have helped the CCTF to refine the expectations of a professional trained in the discipline of hospital medicine. Table 2 has a complete listing of those solicited to be internal and external reviewers. Although aggressive efforts were undertaken to encourage feedback from all solicited reviewers of the Core Competencies document, time or other constraints prevented some reviewers from responding to the review request (overall response or review rate: 52%). Nevertheless, the multiple review and revision process brought what was initially disparate content and organization together in a much more cohesive and consistent approach and structure to competencies in hospital medicine.
|
| Accreditation Council of Graduate Medical Education (ACGME) |
| Agency for Healthcare Research & Quality (AHRQ) |
| American Academy of Family Practice (AAFP) |
| American Association of Critical Care Nurses (AACCN) |
| American Association of Subspecialty Professors |
| American Board of Family Practice |
| American Board of Internal Medicine (ABIM) |
| American College of Chest Physicians (ACCP) |
| American College of Emergency Physicians (ACEP) |
| American College of Physicians (ACP) |
| American Geriatrics Society |
| American Hospital Association (AHA) |
| Association of American Medical Colleges (AAMC) |
| Institute for Healthcare Improvement (IHI) |
| John A. Hartford Foundation |
| Joint Commission on Accreditation of Healthcare Organizations (JCAHO) |
| Residency Review Committee Internal Medicine (RRC‐IM) |
| Reynolds Foundation |
| Robert Wood Johnson Foundation (RWJF) |
| Society of Critical Care Medicine (SCCM) |
| Society of General Internal Medicine (SGIM) |
| Society of Hospital Medicine |
| ○ Board of Directors (9 members solicited) |
| ○ CCTF Members (3 members solicited exclusive of editorial board) |
CHAPTER CONTENT DESCRIPTION
As previously delineated, the Core Competencies document has three sections: Clinical Conditions, Procedures, and Healthcare Systems. The chapters in the entire compendium and within each section have been designed to stand alone and to be used either individually or collectively to assist with curriculum development in hospital medicine. However, each chapter should be used in the context of the entire document because a particular issue may only be touched on in one chapter but may be more elaborately detailed in another. For example, all clinical conditions chapters include a competency on the issue of care transitions, but the specific competencies for care transitions are presented in a separate Transitions of Care chapter.
All chapters in each section begin with an introduction that provides brief background information and establishes the relevance of the topic to practicing hospitalists. Each chapter then utilizes the educational theory of learning domains. The learning domains include the cognitive domain (knowledge), the psychomotor domain (skills), and the affective domain (attitudes). The companion article How to Use The Core Competencies in Hospital Medicine: A Framework for Curriculum Development13 describes in detail the educational theory guiding the development of the Core Competencies document and suggested methods for applying it to the development and revision of curricula and other training activities.
The task force further decided that each chapter in the Clinical Conditions and Procedures sections should include a subsection dedicated to system organization and improvement, an added domain that requires integration of knowledge, skills, and attitudes and the involvement of other medical services and disciplines for optimal patient care. The editorial board believed that system organization and improvement was already an intrinsic feature embedded in the chapters of the Healthcare Systems section. Therefore, this subsection was not included in those chapters.
Hospitalists subscribe to a systems organizational approach to clinical management and processes of care within the hospital. This systems approach, more than any level of knowledge or skill, is required to effectively and efficiently practice in the hospital setting. Practicing with a systems approach, with the interest of improving processes of care, is embedded throughout the Core Competencies document and is a practice method that all hospitalists may strive to achieve as they develop and improve their inpatient care. The competencies within the Systems Organization and Improvement section may contain a range of competency expectation (eg, lead, coordinate, or participate in) to acknowledge their uniqueness and variation according to practice settings and locally instituted responsibilities.
Each competency within a chapter details a level of proficiency, providing guidance on learning activities and potential evaluation strategies. Several overarching themes are followed in the chapters that help to define hospitalists as physicians who specialize in the care of hospitalized patients. First, hospitalists strive to support and adhere to a multidisciplinary approach for the patients under their care. Such an approach involves active interaction with and integration of other hospital medical staff (eg, nursing, rehabilitation therapies, social services) and of specialty medical or surgical services when indicated. Recognizing that hospitalists vary in experience and mastery of their field, the task force and editorial board believed that, at minimum, hospitalists would participate in multidisciplinary teams for improvement of the care and process related to the clinical conditions within their organization. However, they might also lead and/or coordinate teams in such efforts. Therefore, most chapters contain competencies that expect hospitalists to lead, coordinate, or participate in multidisciplinary teams or initiatives that will facilitate optimal care within their organization.
Second, because hospital medicine centers around the quality of inpatient care, participation in quality improvement (QI) initiatives, focusing on improving processes or systems of care in a local institution or organization, may be common in hospitalist practices. The level of involvement and role in QI initiatives may vary according to the particular system, the resources available, and a hospitalist's experience. Finally, because hospitalist care intrinsically involves an increase in the number of care transitions and handoffs, hospitalists need to remain sensitive to and focused on the care transitions that occur with their patients. Such transitions may occur as patients enter the hospital, move from one location to another within the hospital, or leave the hospital. This vulnerable time for patients requires hospitalists to be vigilant in their communication effortswith patients, with medical staff, and with outpatient clinicians.
Each competency was crafted to indicate the relevant concept, the level of proficiency expected, and a way to evaluate mastery. The teaching processes and learning experiences that must take place to achieve competency are left for curriculum developers and instructors to design. These core competencies represent an initial step in curriculum development, creating an identity and core set of expectations for hospitalists that we believe will lead to progress and maturity within the field.
SUMMARY AND FUTURE DIRECTIONS
The practice of hospital medicine requires proficiency of interrelated aspects of practiceclinical, procedural, and system‐based competencies. For practicing hospitalists, the Core Competencies document may serve as a resource to refine skills and assist in program development at individual institutions, both regionally and nationally. For residency program directors and clerkship directors, the Core Competencies document can function as a guide for developing the curriculum of inpatient medicine rotations or for meeting the requirements of the Outcomes Project of the Accreditation Council on Graduate Medical Education's. Last, for those developing continuing medical education programs, the Core Competencies document or individual chapters or topics within it may serve as an outline around which specific or broad‐based programs can be developed. Although the development of such curricula and the recipients of them should be evaluated, the actual evaluation is left to the curriculum developers.
Hospitalists are invested in making hospitals run better. They are positioned to take leadership roles in addressing quality, efficiency, and cost interests in both community and academic hospital settings. Their goals include improving care processes, hospital work life, and the setting in which they practice. The key core competencies described in this compendium define hospitalists as agents of change 1) to develop and implement systems to enable best practices to occur from admission through discharge, and 2) to promote the development of a safer culture within the hospital.
Hospital medicine remains an evolving specialty. Although great care was taken to construct these competencies so they would retain their relevance over time, SHM, the Core Curriculum Task Force, and the editorial board recognize the need for their continual reevaluation and modification in the context of advances and changes in the practice of hospital medicine. Our intent is that these competencies be a common reference and foundation for the creation of hospital medicine curricula and serve to standardize and improve training practices.
Identification of the core competencies of a medical specialty provides the necessary framework for that specialty to develop, refine itself, and evolve. It also provides a structure from which training, testing, and curricula can be developed and effectively utilized. For nearly a decade, since the coining of the term hospitalist,1 the field of hospital medicine has been emerging as the next generation of site‐defined specialties, after emergency medicine and critical care medicine. The Core Competencies in Hospital Medicine: A Framework for Curriculum Development (referred to as the Core Competencies from this point on) introduces the expectations of hospitalists, helps to define their role, and suggests how knowledge, skill, and attitude acquisition might be evaluated. Furthermore, this document provides an initial structural framework from which curricula in adult hospital medicine may be developed.
The Core Competencies document, produced by the Society of Hospital Medicine (SHM) and published as a supplement to the first issue of the Journal of Hospital Medicine,2 is meant to serve as a framework for educators at all levels of medical education to develop curricula, training, and evaluations for students, clinicians‐in‐training, and practicing hospitalists. The Core Competencies document is not meant to contain a complete compilation of inpatient clinical topics or to re‐create what many residency training programs in adult inpatient care already provide. It should not limit and does not define every aspect of hospitalist practice. It includes the most common and fundamental elements of inpatient care without exhaustively listing every clinical entity that may be encountered by a hospitalist. Some of the more common clinical topics encountered by inpatient physicians are included, with an emphasis on subject areas that stress a systems‐based approach to health care, which is central to the practice of hospital medicine. This initial version of the Core Competencies document also focuses on potential areas of deficiency in the training of physicians to become hospitalists. It provides developers of curricula and content with a standardized set of measurable learning objectives, while allowing them the flexibility needed to address specific contexts and incorporate advances in medicine.
The SHM, the sole professional organization representing inpatient physicians, defines hospitalists as physicians whose primary professional focus is the general medical care of hospitalized patients. Their activities include patient care, teaching, research, and leadership related to Hospital Medicine.3 An estimated 12,000 hospitalists are currently practicing in the United States, with a projected workforce need of an estimated 20,00030,000 practicing hospitalists in the United States in the next 510 years.4 Various factors have contributed to the rapid growth and expansion of hospital medicine, including factors related to care efficiency, care quality, and inpatient teaching.512 The pressures that have contributed to the development of and evolution toward the hospitalist model of care over the past decade are facilitating the transformation from a traditional model of inpatient care to the care of inpatients by hospitalist physicians dedicated primarily to the inpatient setting. As a result of this growth in hospital medicine, the SHM realized that core competencies were needed to help define the field.
The purpose of this article is to describe the developmental process and content structure of the Core Competencies document. It delineates the process from initial needs assessment to topic list development to chapter production to internal and external review and revisions of individual chapters and the complete document. The supplement to this first issue of the Journal of Hospital Medicine contains 1) the Core Competencies,2 2) a reprint of this article, and 3) a reprint of the article by McKean et al. in this issue detailing how to use the Core Competencies,13 with examples and suggestions related to curriculum development. The authors propose that this combined compilation may spur curriculum development in hospital medicine that will help to define the field and set expectations for practice.
PROCESS AND TIMELINE
Education Summit
Early in the growth of hospital medicine, the Society of Hospital Medicine identified a need to better define a common educational and practice framework for hospitalist physicians. Such a framework could help to define hospitalists as a distinct group of practicing physicians with common goals and a common set of competencies. The importance of identifying and delineating the common knowledge, skills, and attitudes of hospitalists was paramount. Figure 1 shows the details of the 4‐year process of developing the Core Competencies.
In 2002, the SHM drew together educational leaders in hospital medicine in its first educational summit. One of the primary charges that the SHM received from this summit was to develop the needed core curriculum in hospital medicine. After the summit, the SHM's Education Committee formed the Core Curriculum Task Force (CCTF), composed of approximately 15 member hospitalists, with representation from university and community hospitals, teaching and nonteaching programs, and for‐profit and not‐for‐profit settings from various geographic regions of the country. The selection process ensured that the task force was representative of practicing hospitalists and SHM membership throughout the United States.
The CCTF
The task force met through frequent conference‐call meetings and at least one in‐person meeting annually. The primary goal set forth by the task force was the initial development of a distinct set of core competencies in hospital medicine that could then guide curriculum development within the field.
Topic List
The task force determined that the topics (or chapters) should be divided into three sectionsClinical Conditions, Procedures, and Healthcare Systems (Table 1, Chapter List)all integral components of the practice of hospital medicine. For Clinical Conditions chapters, the task force decided that an exhaustive listing of all potential clinical entities that hospitalists might encounter during their clinical practice was not the goal of the Core Competencies. Rather, clinical topics were selected to reflect conditions in the hospital setting that are encountered with significant frequency, that might be significantly life‐threatening, or that are likely to have the significant involvement and impact of hospitalists in altering or refining care processes, leading to improvement in care quality and efficiency. The list of Clinical Condition chapters should not limit or rigidly define the scope of practice of hospitalist physicians. Instead, it should help those entering the field of hospital medicine better understand some of the core clinical topics on which hospitalists focus in the design of institutional or global quality initiatives.
| Clinical Conditions* | Procedures | Healthcare Systems |
|---|---|---|
| ||
|
|
|
Clinical Conditions Section
In an effort to both narrow and delineate the core Clinical Condition areas necessary for practicing hospital medicine, the task force elected first to draw from national data the most common diagnosis‐related groups (DRGs) discharged from U.S. hospitals. Utilizing the Medicare database, the top 15 nonsurgical discharge diagnoses were initially selected. Certain clinical conditions that the task force believed to be highly relevant to the practice of hospital medicine but that did not neatly fall into a specific DRG, such as pain management and perioperative medicine, were proposed for and then added to the list of Clinical Conditions chapters by the task force. Other chapters, such as that on venous thromboembolism, were added because a particular disease, although not necessarily a high‐ranked discharge DRG, showed high inpatient morbidity and mortality and reflected the role of the hospitalist in the prevention of predictable complications during hospitalization. When possible, some diagnoses were consolidated to better incorporate crosscutting competencies or to highlight opportunities for leadership in systems‐based improvements. For example, upper and lower gastrointestinal bleeding were consolidated into the chapter on gastrointestinal bleeding. Similarly, all relevant arrhythmias that a hospitalist might encounter were consolidated into a single chapter. For at least one clinical topic, pneumonia, the task force believed it necessary to have two distinct chapters, one on community‐acquired pneumonia and the other on hospital‐acquired pneumonia, because these two entities are significantly different and have distinct competencies. The final listing of Clinical Conditions chapters reflects 19 clinical areas that hospitalists encounter on a frequent basis and for which they can have an effect on systems and processes of care. These clinical chapters form a foundation of topics for which hospitalists have already begun quality and efficiency initiatives.
The task force further decided that symptom evaluation and management could be consolidated into a systems chapter dedicated to diagnostic decision making. A reasonably large constellation of symptoms, including but not limited to chest pain, shortness of breath, syncope, and altered mental status, are encountered by hospitalists daily. Although evaluation and management of these symptoms are extremely important parts of triage, subsequent testing, and hospital care, the ability to develop a differential diagnosis and proceed with the indicated testing and its interpretation is common to all symptom evaluation. Such evaluation and diagnostic decision making are therefore summarized in a single chapter in the Healthcare Systems section, and no symptom chapters are found in the Clinical section.
Procedures Section
The initial topic lists for the Procedures and Systems sections were developed through input from the broad representation of the Core Curriculum Task Force. The chapters in the Procedures section contain competencies expected for the inpatient procedures that hospitalists are most likely to perform or supervise in their day‐to‐day care of hospitalized patients. The presence of a procedural skill in the Core Competencies does not necessarily indicate that every hospitalist will perform or be proficient in that procedure. Similarly, the absence of a procedure from the Core Competencies should not exclude trained and experienced hospitalists from performing that procedure. The task force recognizes that the individual hospital setting, including local and regional variations, determines who might perform certain procedures depending on many factors, which may include whether there are trainees, specialty support including radiology, and procedure teams. The Procedures section outlines those procedures frequently performed in the everyday practice of hospital medicine and incorporates relevant competencies to afford proper performance, patient education and involvement, prevention of complications, and quality improvement for these procedures.
Healthcare Systems Section
Although many competencies delineated in the Clinical Conditions and Procedures sections of the supplement may be taught well during medical school and residency training, that is not true of the chapters and competencies in the Healthcare Systems section, many of which are not extensively taught in most undergraduate or graduate medical education programs. Therefore, many hospitalists must gain or supplant their knowledge, skills, and attitudes in system areas posttraining.
The Healthcare Systems section delineates themes integral to the successful practice of hospital medicine in diverse hospital settings. Many chapters in this section focus on processes and systems of care that typically span multiple disease entities and frequently require multidisciplinary input to create a coordinated effort for care quality and efficiency. The chapters and core competencies in the Healthcare Systems section direct hospitalists to lead and innovate in their own hospital practices and to convey the principles of evidence‐based inpatient medical care and systems‐based practice to medical students, physicians‐in‐training, other medical staff, colleagues, and patients. The task force expects that many new hospitalists will still be learning many of the competencies in the Healthcare Systems section during the early stages of their posttraining practice. However, as training of hospitalists during undergraduate and graduate medical education further evolves, we expect that more hospitalists will enter the workforce with more of the skills necessary to prepare them for their careers.
Some Healthcare Systems chapters have clinical themes but were included in this section because it is believed that the clinical approach always spans multiple clinical entities and always requires an organizational approach crossing several disciplines in medicine in order to optimize the hospital care. Such chapters include Care of the Elderly Patient, Prevention of Healthcare Associated Infections and Antimicrobial Resistance, Nutrition and the Hospitalized Patient, and Palliative Care. Other chapters in the Healthcare Systems section focus on educational themes that drive the practice of hospital medicine and the lifelong learning and teaching required of hospitalists. Some of these chapters include Evidence‐Based Medicine, Hospitalist as Teacher, Patient Education, and Practice‐Based Learning and Improvement. Still other chapters in the Healthcare Systems section identify much of the organizational approachboth from clinical practice and practice management standpointsthat must be adopted by hospitalists in order to provide high‐quality care while maintaining functional and sound practice. Examples of chapters focusing on clinical practice organization include Patient Safety, Quality Improvement, Team Approach and Multidisciplinary Care, Transitions of Care, and Patient Handoffs. Although the Transitions of Care chapter focuses on the processes and communication required for the safe transition of patients from one clinical setting to another; the Patient Handoffs (or sign‐out) chapter focuses on the hospitalist‐to‐hospitalist communication essential when one hospitalist assumes care of a patient from another (either from dayshift to nightshift on the same service or assuming care of service from a different service). Examples of chapters focusing on practice management organization include Business Practices, Equitable Allocation of Resources, Leadership, and Risk Management. Overall, the Healthcare Systems chapters help to characterize and delineate the practice and scope of hospital medicine, especially with topics not taught in detail during most residency training programs.
Editorial Board, Content Survey, and Topic List Refinement
Once the initial topic list was created, a five‐member editorial board was chosen from the CCTF membership, including the SHM CCTF chair, the Education Committee chair, two member hospitalists, and a health education specialist. The purpose of this board was to interpret survey feedback, solicit contributors to write competency chapters, review and revise the chapters submitted, and prepare the larger document for review and final publication. The Core Curriculum Task Force developed a survey to obtain feedback on the initial topic list. Face validity was established through a survey sent electronically in 2003 to the SHM Board of Directors and Education Committee, as well as to 10 representatives of each SHM regional council and local chapter. In all, more than 250 hospitalists representing diverse geographic and practice backgrounds were surveyed. Feedback from the survey was reviewed by the CCTF. The topic list was then revised with additions and modifications incorporated from survey feedback. The scope of individual topics also was modified in multiple iterations congruent with the internal and external review processes.
Contributors
Contributors were solicited by the task force, utilizing SHM databasesbelieved to be the most comprehensive registry of hospitalist physiciansand an electronic call for nominations to practicing hospitalists from around the United States. Other recognized content experts were solicited independently on the basis of chapter or content needs. Efforts were taken to identify hospitalists with expertise in specific topic areas, particularly those with a history of presentations or publications on individual chapter subject matter. Potential contributors submitted credentials, including curricula vitae and other supporting documents or information, when requesting to write a specific chapter for the Core Competencies compendium. Contributors were competitively selected on the basis of their submitted information compared to those of others requesting to write the same chapter. In some cases practicing hospitalists were paired with nonhospitalist expert contributors to create a chapter. Contributors were provided with guidelines with which to prepare their chapter.
Review and Revision
The editorial board reviewed all the chapters, rigorously evaluating each chapter through at least five stages of review and revision. First, chapters were reviewed by the editorial boardinitially by at least two physician members and then by the entire editorial board. Chapters were reviewed for the scope and completeness of concepts, adherence to educational theory, and consistency in chapter format. Changes in content and for consistency were extensive in some chapters, whereas others required only small or moderate changes. Significant editing was required to create chapters as a compilation of specific, measurable competencies as opposed to topic‐related content. All chapters required some level of modification to assist with consistency in style, language, and overall goals. Where appropriate, individual chapters were also reviewed by relevant SHM committees, task forces, or content experts, and initial feedback was provided. For example, the Leadership chapter was reviewed by the SHM Leadership Task Force. Other SHM committees and task forces involved in chapter reviews included the Education, Healthcare Quality and Patient Safety, and Ethics committees as well as the Geriatric Task Force. Changes recommended changes on the basis of committee and task force feedback were incorporated into the relevant chapters.
Second, revisions of individual chapters from the editorial board were sent back to contributors for final comment, revision, and approval. Third, the compilation of all chapters and sections was reviewed (as a whole) and underwent further revision by the editorial board based on feedback from the contributors and the relevant SHM committees. Fourth, the entire revised supplement was sent for an internal review by the SHM board and relevant SHM committees or committee representatives.
Fifth, final reviews were solicited from external reviewers of medical professional organizations and academic organizations. Feedback from the internal and external reviews were compiled and systematically evaluated by the CCTF editorial board. Recommended changes were incorporated into individual chapters or throughout the Core Competencies compendium on the basis of the evaluation and consensus approval of the editorial board. For example, one reviewer believed that quality improvement initiatives were necessary for all procedures that hospitalists perform in order to help reduce the risk of complications. Therefore, each procedure chapter was revised to reflect this competency. Similarly, another reviewer thought that in many chapters the involvement of nursing and other medical staff in the implementation of multidisciplinary teams was underemphasized. Therefore, efforts were taken to improve the emphasis of these key participants in multidisciplinary hospital care.
The efforts of many individuals and professional organizations have helped the CCTF to refine the expectations of a professional trained in the discipline of hospital medicine. Table 2 has a complete listing of those solicited to be internal and external reviewers. Although aggressive efforts were undertaken to encourage feedback from all solicited reviewers of the Core Competencies document, time or other constraints prevented some reviewers from responding to the review request (overall response or review rate: 52%). Nevertheless, the multiple review and revision process brought what was initially disparate content and organization together in a much more cohesive and consistent approach and structure to competencies in hospital medicine.
|
| Accreditation Council of Graduate Medical Education (ACGME) |
| Agency for Healthcare Research & Quality (AHRQ) |
| American Academy of Family Practice (AAFP) |
| American Association of Critical Care Nurses (AACCN) |
| American Association of Subspecialty Professors |
| American Board of Family Practice |
| American Board of Internal Medicine (ABIM) |
| American College of Chest Physicians (ACCP) |
| American College of Emergency Physicians (ACEP) |
| American College of Physicians (ACP) |
| American Geriatrics Society |
| American Hospital Association (AHA) |
| Association of American Medical Colleges (AAMC) |
| Institute for Healthcare Improvement (IHI) |
| John A. Hartford Foundation |
| Joint Commission on Accreditation of Healthcare Organizations (JCAHO) |
| Residency Review Committee Internal Medicine (RRC‐IM) |
| Reynolds Foundation |
| Robert Wood Johnson Foundation (RWJF) |
| Society of Critical Care Medicine (SCCM) |
| Society of General Internal Medicine (SGIM) |
| Society of Hospital Medicine |
| ○ Board of Directors (9 members solicited) |
| ○ CCTF Members (3 members solicited exclusive of editorial board) |
CHAPTER CONTENT DESCRIPTION
As previously delineated, the Core Competencies document has three sections: Clinical Conditions, Procedures, and Healthcare Systems. The chapters in the entire compendium and within each section have been designed to stand alone and to be used either individually or collectively to assist with curriculum development in hospital medicine. However, each chapter should be used in the context of the entire document because a particular issue may only be touched on in one chapter but may be more elaborately detailed in another. For example, all clinical conditions chapters include a competency on the issue of care transitions, but the specific competencies for care transitions are presented in a separate Transitions of Care chapter.
All chapters in each section begin with an introduction that provides brief background information and establishes the relevance of the topic to practicing hospitalists. Each chapter then utilizes the educational theory of learning domains. The learning domains include the cognitive domain (knowledge), the psychomotor domain (skills), and the affective domain (attitudes). The companion article How to Use The Core Competencies in Hospital Medicine: A Framework for Curriculum Development13 describes in detail the educational theory guiding the development of the Core Competencies document and suggested methods for applying it to the development and revision of curricula and other training activities.
The task force further decided that each chapter in the Clinical Conditions and Procedures sections should include a subsection dedicated to system organization and improvement, an added domain that requires integration of knowledge, skills, and attitudes and the involvement of other medical services and disciplines for optimal patient care. The editorial board believed that system organization and improvement was already an intrinsic feature embedded in the chapters of the Healthcare Systems section. Therefore, this subsection was not included in those chapters.
Hospitalists subscribe to a systems organizational approach to clinical management and processes of care within the hospital. This systems approach, more than any level of knowledge or skill, is required to effectively and efficiently practice in the hospital setting. Practicing with a systems approach, with the interest of improving processes of care, is embedded throughout the Core Competencies document and is a practice method that all hospitalists may strive to achieve as they develop and improve their inpatient care. The competencies within the Systems Organization and Improvement section may contain a range of competency expectation (eg, lead, coordinate, or participate in) to acknowledge their uniqueness and variation according to practice settings and locally instituted responsibilities.
Each competency within a chapter details a level of proficiency, providing guidance on learning activities and potential evaluation strategies. Several overarching themes are followed in the chapters that help to define hospitalists as physicians who specialize in the care of hospitalized patients. First, hospitalists strive to support and adhere to a multidisciplinary approach for the patients under their care. Such an approach involves active interaction with and integration of other hospital medical staff (eg, nursing, rehabilitation therapies, social services) and of specialty medical or surgical services when indicated. Recognizing that hospitalists vary in experience and mastery of their field, the task force and editorial board believed that, at minimum, hospitalists would participate in multidisciplinary teams for improvement of the care and process related to the clinical conditions within their organization. However, they might also lead and/or coordinate teams in such efforts. Therefore, most chapters contain competencies that expect hospitalists to lead, coordinate, or participate in multidisciplinary teams or initiatives that will facilitate optimal care within their organization.
Second, because hospital medicine centers around the quality of inpatient care, participation in quality improvement (QI) initiatives, focusing on improving processes or systems of care in a local institution or organization, may be common in hospitalist practices. The level of involvement and role in QI initiatives may vary according to the particular system, the resources available, and a hospitalist's experience. Finally, because hospitalist care intrinsically involves an increase in the number of care transitions and handoffs, hospitalists need to remain sensitive to and focused on the care transitions that occur with their patients. Such transitions may occur as patients enter the hospital, move from one location to another within the hospital, or leave the hospital. This vulnerable time for patients requires hospitalists to be vigilant in their communication effortswith patients, with medical staff, and with outpatient clinicians.
Each competency was crafted to indicate the relevant concept, the level of proficiency expected, and a way to evaluate mastery. The teaching processes and learning experiences that must take place to achieve competency are left for curriculum developers and instructors to design. These core competencies represent an initial step in curriculum development, creating an identity and core set of expectations for hospitalists that we believe will lead to progress and maturity within the field.
SUMMARY AND FUTURE DIRECTIONS
The practice of hospital medicine requires proficiency of interrelated aspects of practiceclinical, procedural, and system‐based competencies. For practicing hospitalists, the Core Competencies document may serve as a resource to refine skills and assist in program development at individual institutions, both regionally and nationally. For residency program directors and clerkship directors, the Core Competencies document can function as a guide for developing the curriculum of inpatient medicine rotations or for meeting the requirements of the Outcomes Project of the Accreditation Council on Graduate Medical Education's. Last, for those developing continuing medical education programs, the Core Competencies document or individual chapters or topics within it may serve as an outline around which specific or broad‐based programs can be developed. Although the development of such curricula and the recipients of them should be evaluated, the actual evaluation is left to the curriculum developers.
Hospitalists are invested in making hospitals run better. They are positioned to take leadership roles in addressing quality, efficiency, and cost interests in both community and academic hospital settings. Their goals include improving care processes, hospital work life, and the setting in which they practice. The key core competencies described in this compendium define hospitalists as agents of change 1) to develop and implement systems to enable best practices to occur from admission through discharge, and 2) to promote the development of a safer culture within the hospital.
Hospital medicine remains an evolving specialty. Although great care was taken to construct these competencies so they would retain their relevance over time, SHM, the Core Curriculum Task Force, and the editorial board recognize the need for their continual reevaluation and modification in the context of advances and changes in the practice of hospital medicine. Our intent is that these competencies be a common reference and foundation for the creation of hospital medicine curricula and serve to standardize and improve training practices.
- ,.The emerging role of “hospitalists” in the American health care system.N Engl J Med.1996;335:514–517.
- ,,,,, eds.The Core Competencies in Hospital Medicine: A Framework for Curriculum Development.J Hosp Med.2006;1(supplement 1).
- Society of Hospital Medicine. About SHM: What is a hospitalist? Available from URL: http://www.hospitalmedicine.org[accessed July 22, 2005].
- .The future of hospital medicine: evolution or revolution?Am J Med.2004;117:446–450.
- ,.The hospitalist movement 5 years later.JAMA.2002;287:487–494.
- ,,, et al.Implementation of a voluntary hospitalist service at a community teaching hospital: improved clinical efficiency and patient outcomes.Ann Intern Med.2002;137:859–865.
- ,,, et al.Effects of physician experience on costs and outcomes on an academic general medicine service: results of a trial of hospitalists.Ann Intern Med.2002;137:866–874.
- ,,, et al.Making Healthcare aafer: a critical analysis of patient safety practices.Rockville, MD:U.S. Dept. of Health and Human Services, Agency for Healthcare Research and Quality;2001. AHRQ publication 01‐E058. Available from URL: http://www.ahrq.gov.
- ,,, et al.Medical student evaluation of the quality of hospitalist and nonhospitalist teaching faculty on inpatient medicine rotations.Acad Med.2004;79:78–82.
- ,,, et al.Hospitalists as teachers.J Gen Intern Med.2004;19(1):8–15.
- ,,, et al.The positive impact of initiation of hospitalist clinician educators.J Gen Intern Med.2004;19:293–301.
- ,,, et al.Effects of hospitalist attending physicians on trainee satisfaction with teaching and with internal medicine rotations.Arch Intern Med.2004;164:1866–1887.
- ,,,,.How to use The Core Competencies in Hospital Medicine: A Framework for Curriculum Development.J Hosp Med.2006;1:57–67.
- ,.The emerging role of “hospitalists” in the American health care system.N Engl J Med.1996;335:514–517.
- ,,,,, eds.The Core Competencies in Hospital Medicine: A Framework for Curriculum Development.J Hosp Med.2006;1(supplement 1).
- Society of Hospital Medicine. About SHM: What is a hospitalist? Available from URL: http://www.hospitalmedicine.org[accessed July 22, 2005].
- .The future of hospital medicine: evolution or revolution?Am J Med.2004;117:446–450.
- ,.The hospitalist movement 5 years later.JAMA.2002;287:487–494.
- ,,, et al.Implementation of a voluntary hospitalist service at a community teaching hospital: improved clinical efficiency and patient outcomes.Ann Intern Med.2002;137:859–865.
- ,,, et al.Effects of physician experience on costs and outcomes on an academic general medicine service: results of a trial of hospitalists.Ann Intern Med.2002;137:866–874.
- ,,, et al.Making Healthcare aafer: a critical analysis of patient safety practices.Rockville, MD:U.S. Dept. of Health and Human Services, Agency for Healthcare Research and Quality;2001. AHRQ publication 01‐E058. Available from URL: http://www.ahrq.gov.
- ,,, et al.Medical student evaluation of the quality of hospitalist and nonhospitalist teaching faculty on inpatient medicine rotations.Acad Med.2004;79:78–82.
- ,,, et al.Hospitalists as teachers.J Gen Intern Med.2004;19(1):8–15.
- ,,, et al.The positive impact of initiation of hospitalist clinician educators.J Gen Intern Med.2004;19:293–301.
- ,,, et al.Effects of hospitalist attending physicians on trainee satisfaction with teaching and with internal medicine rotations.Arch Intern Med.2004;164:1866–1887.
- ,,,,.How to use The Core Competencies in Hospital Medicine: A Framework for Curriculum Development.J Hosp Med.2006;1:57–67.
Copyright © 2006 Society of Hospital Medicine
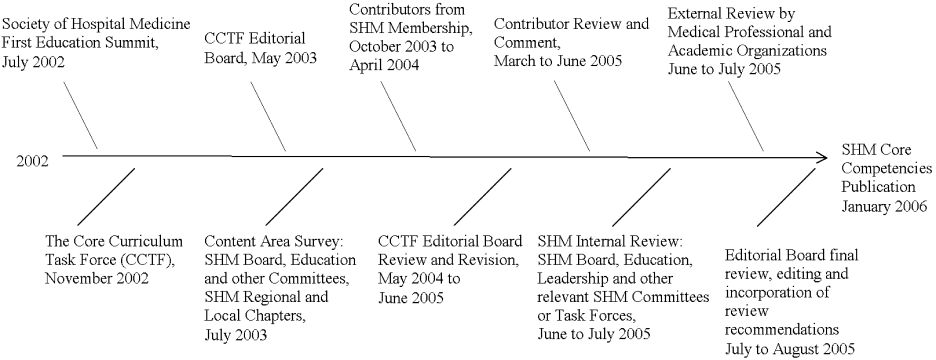
Editorial
Congratulations to the Society of Hospital Medicine (SHM) for launching this important new journal. Congratulations as well to the SHM members, who have identified an important patient care need and moved to meet that need by defining the special competencies of the hospitalist. The Core Competencies in Hospital Medicine: A Framework for Curriculum Development (the Core Competencies), by the Society of Hospital Medicine, accompanies this inaugural issue of the Journal of Hospital Medicine.
As a geriatrician, I can personally attest to the need to have skilled physicians on‐site in the hospital to care for elderly patients. Older people with complex illnesses are susceptible to multiple hospital complications, which often present subtly but can quickly turn into life‐threateningbut potentially reversibleillnesses. Given the demography of hospitalized patients in the 21st century in the United States, every good hospitalist also has to be a good geriatrician.
As evidenced in the Core Competencies, the hospitalist community recognizes as well the importance of developing expertise in caring for both the medical and surgical conditions of patients. Providing attentive diagnostic and management skills to pre‐ and postoperative patients, especially those with preexisting chronic conditions, will surely improve outcomes.
Continuity and coordination within a single hospital episode and across multiple hospitalizations are major challenges for our fragmented and often chaotic health care system. The Core Competencies recognizes the centrality of systems‐based practice to the foundation of hospitalist skills. We at the American Board of Internal Medicine (ABIM) share the belief that every physician must understand the principles of quality improvement; accordingly, this competency is now demanded of every resident and is assessed in the maintenance of certification (MOC) of every internal medicine specialist. That hospitalists have grabbed the quality‐improvement mantle is a welcome development and shows that hospitalists are likely to become key teachers of systems‐based care and quality‐improvement competencies in teaching hospitals.
The growth of hospital medicine in the United States has raised many important issues concerning quality of care that cannot be totally solved by the creation of a hospital‐based practice discipline. The vexing issues of continuity of care, continuing relationships, and efficient management of resources over the entire trajectory of a patient's illness (not just during a hospitalization) are not fundamentally addressed by the existence of hospital medicine as a discipline. However, hospitalists can partner with others in the health care system to create a clinically meaningful continuum that truly would serve patients, especially those with the greatest need such as the elderly and the chronically ill. The ABIM has been in discussions with the Society of Hospital Medicine, the Society of General Internal Medicine, the American College of Physicians, and the Alliance of Academic Internal Medicine to develop a response to the important and evolving arenas of specific expertise in hospital and outpatient medicine. The Core Competencies in Hospital Medicine will significantly help to further these discussions.
Let me raise two concerns whose resolution will need the input of hospitalists as the discipline of hospital medicine becomes more mature. First, hospitalist models are quite variable. Many academic physicians who call themselves hospitalists attend on an inpatient service 2, 3, or 5 months a year and still see outpatients. Many physicians who consider themselves general internists (and not hospitalists) have a weekly half‐day clinic and attend on the wards 3 months a year. Which is a hospitalist? Does it matter? Will the definition of a hospitalist be based on achievement of the competencies described here, or will it be based primarily on the amount of time in hospital‐based practice? This will be an important question to resolve, especially as we embark on a path toward offering a hospitalist credential.
Second, general internal medicine is becoming an increasingly vital part of the continuum of care for patients with multiple complex chronic illnesses, at the same time that poor reimbursement has undermined its vitality and threatens its existence. (Family medicine is also suffering from reduced interest among medical students.) Because most institutions function on an each tub on its own bottom model, it is unrealistic to expect the practice of ambulatory general internal medicine to support itself. Generalist practices thrive in integrated group models. These practices recognize the importance of the physician who provides a coordinating function for all the specialists who care for a complex patient. Such an outpatient generalist thus reduces excess and unnecessary care while identifying gaps where relevant specialties could improve function or quality of life. Ambulatory practice also requires skill in systems and improvement, but few of the 80% of generalists who practice in small groups have sufficient infrastructure and resources to support practice redesign. Indeed, a new report from Mercer consultants coined the phrase ambulatory intensivists to identify practices with Medicare patients and recognizes that these practices are every bit as intense and complex and in need of systems management as an inpatient practice. What the complex patient needs is a seamless interface between the two.
The authors of the Core Competencies in Hospital Medicine hope that this document will stand the test of time as it evolves. I would urge that the document remain flexiblea living documentbecause the one thing about which we can be sure is that hospital practice will change. More and more critical care will be delivered throughout the hospital, more and more of all kinds of care will be performed outside the hospital, and the nature of hospitals will surely change with shifts in reimbursement that we cannot yet imagine but that might be right around the corner. If able to provide hands‐on care less expensively, physician assistants and nurse‐practitioners functioning according to protocols developed by systems thinkers, only some of whom will be physicians, may replace the physician in some settings. What will become of hospitalists as these systems change? I hope that hospitalists, together with other general internists, will be at the forefront of ensuring that the changes in practice that result from the combination of new technologies and financing structures will ultimately also serve the needs of patients. The patient is at the center of our discipline and, as articulated so clearly in the Core Competencies, should always be the focus of our future thinking.
- .Health care system chaos should spur innovation: summary of a report of the Society of General Internal Medicine Task Force on the Domain of General Internal Medicine.Ann Intern Med.2004;140:639–643.
Congratulations to the Society of Hospital Medicine (SHM) for launching this important new journal. Congratulations as well to the SHM members, who have identified an important patient care need and moved to meet that need by defining the special competencies of the hospitalist. The Core Competencies in Hospital Medicine: A Framework for Curriculum Development (the Core Competencies), by the Society of Hospital Medicine, accompanies this inaugural issue of the Journal of Hospital Medicine.
As a geriatrician, I can personally attest to the need to have skilled physicians on‐site in the hospital to care for elderly patients. Older people with complex illnesses are susceptible to multiple hospital complications, which often present subtly but can quickly turn into life‐threateningbut potentially reversibleillnesses. Given the demography of hospitalized patients in the 21st century in the United States, every good hospitalist also has to be a good geriatrician.
As evidenced in the Core Competencies, the hospitalist community recognizes as well the importance of developing expertise in caring for both the medical and surgical conditions of patients. Providing attentive diagnostic and management skills to pre‐ and postoperative patients, especially those with preexisting chronic conditions, will surely improve outcomes.
Continuity and coordination within a single hospital episode and across multiple hospitalizations are major challenges for our fragmented and often chaotic health care system. The Core Competencies recognizes the centrality of systems‐based practice to the foundation of hospitalist skills. We at the American Board of Internal Medicine (ABIM) share the belief that every physician must understand the principles of quality improvement; accordingly, this competency is now demanded of every resident and is assessed in the maintenance of certification (MOC) of every internal medicine specialist. That hospitalists have grabbed the quality‐improvement mantle is a welcome development and shows that hospitalists are likely to become key teachers of systems‐based care and quality‐improvement competencies in teaching hospitals.
The growth of hospital medicine in the United States has raised many important issues concerning quality of care that cannot be totally solved by the creation of a hospital‐based practice discipline. The vexing issues of continuity of care, continuing relationships, and efficient management of resources over the entire trajectory of a patient's illness (not just during a hospitalization) are not fundamentally addressed by the existence of hospital medicine as a discipline. However, hospitalists can partner with others in the health care system to create a clinically meaningful continuum that truly would serve patients, especially those with the greatest need such as the elderly and the chronically ill. The ABIM has been in discussions with the Society of Hospital Medicine, the Society of General Internal Medicine, the American College of Physicians, and the Alliance of Academic Internal Medicine to develop a response to the important and evolving arenas of specific expertise in hospital and outpatient medicine. The Core Competencies in Hospital Medicine will significantly help to further these discussions.
Let me raise two concerns whose resolution will need the input of hospitalists as the discipline of hospital medicine becomes more mature. First, hospitalist models are quite variable. Many academic physicians who call themselves hospitalists attend on an inpatient service 2, 3, or 5 months a year and still see outpatients. Many physicians who consider themselves general internists (and not hospitalists) have a weekly half‐day clinic and attend on the wards 3 months a year. Which is a hospitalist? Does it matter? Will the definition of a hospitalist be based on achievement of the competencies described here, or will it be based primarily on the amount of time in hospital‐based practice? This will be an important question to resolve, especially as we embark on a path toward offering a hospitalist credential.
Second, general internal medicine is becoming an increasingly vital part of the continuum of care for patients with multiple complex chronic illnesses, at the same time that poor reimbursement has undermined its vitality and threatens its existence. (Family medicine is also suffering from reduced interest among medical students.) Because most institutions function on an each tub on its own bottom model, it is unrealistic to expect the practice of ambulatory general internal medicine to support itself. Generalist practices thrive in integrated group models. These practices recognize the importance of the physician who provides a coordinating function for all the specialists who care for a complex patient. Such an outpatient generalist thus reduces excess and unnecessary care while identifying gaps where relevant specialties could improve function or quality of life. Ambulatory practice also requires skill in systems and improvement, but few of the 80% of generalists who practice in small groups have sufficient infrastructure and resources to support practice redesign. Indeed, a new report from Mercer consultants coined the phrase ambulatory intensivists to identify practices with Medicare patients and recognizes that these practices are every bit as intense and complex and in need of systems management as an inpatient practice. What the complex patient needs is a seamless interface between the two.
The authors of the Core Competencies in Hospital Medicine hope that this document will stand the test of time as it evolves. I would urge that the document remain flexiblea living documentbecause the one thing about which we can be sure is that hospital practice will change. More and more critical care will be delivered throughout the hospital, more and more of all kinds of care will be performed outside the hospital, and the nature of hospitals will surely change with shifts in reimbursement that we cannot yet imagine but that might be right around the corner. If able to provide hands‐on care less expensively, physician assistants and nurse‐practitioners functioning according to protocols developed by systems thinkers, only some of whom will be physicians, may replace the physician in some settings. What will become of hospitalists as these systems change? I hope that hospitalists, together with other general internists, will be at the forefront of ensuring that the changes in practice that result from the combination of new technologies and financing structures will ultimately also serve the needs of patients. The patient is at the center of our discipline and, as articulated so clearly in the Core Competencies, should always be the focus of our future thinking.
Congratulations to the Society of Hospital Medicine (SHM) for launching this important new journal. Congratulations as well to the SHM members, who have identified an important patient care need and moved to meet that need by defining the special competencies of the hospitalist. The Core Competencies in Hospital Medicine: A Framework for Curriculum Development (the Core Competencies), by the Society of Hospital Medicine, accompanies this inaugural issue of the Journal of Hospital Medicine.
As a geriatrician, I can personally attest to the need to have skilled physicians on‐site in the hospital to care for elderly patients. Older people with complex illnesses are susceptible to multiple hospital complications, which often present subtly but can quickly turn into life‐threateningbut potentially reversibleillnesses. Given the demography of hospitalized patients in the 21st century in the United States, every good hospitalist also has to be a good geriatrician.
As evidenced in the Core Competencies, the hospitalist community recognizes as well the importance of developing expertise in caring for both the medical and surgical conditions of patients. Providing attentive diagnostic and management skills to pre‐ and postoperative patients, especially those with preexisting chronic conditions, will surely improve outcomes.
Continuity and coordination within a single hospital episode and across multiple hospitalizations are major challenges for our fragmented and often chaotic health care system. The Core Competencies recognizes the centrality of systems‐based practice to the foundation of hospitalist skills. We at the American Board of Internal Medicine (ABIM) share the belief that every physician must understand the principles of quality improvement; accordingly, this competency is now demanded of every resident and is assessed in the maintenance of certification (MOC) of every internal medicine specialist. That hospitalists have grabbed the quality‐improvement mantle is a welcome development and shows that hospitalists are likely to become key teachers of systems‐based care and quality‐improvement competencies in teaching hospitals.
The growth of hospital medicine in the United States has raised many important issues concerning quality of care that cannot be totally solved by the creation of a hospital‐based practice discipline. The vexing issues of continuity of care, continuing relationships, and efficient management of resources over the entire trajectory of a patient's illness (not just during a hospitalization) are not fundamentally addressed by the existence of hospital medicine as a discipline. However, hospitalists can partner with others in the health care system to create a clinically meaningful continuum that truly would serve patients, especially those with the greatest need such as the elderly and the chronically ill. The ABIM has been in discussions with the Society of Hospital Medicine, the Society of General Internal Medicine, the American College of Physicians, and the Alliance of Academic Internal Medicine to develop a response to the important and evolving arenas of specific expertise in hospital and outpatient medicine. The Core Competencies in Hospital Medicine will significantly help to further these discussions.
Let me raise two concerns whose resolution will need the input of hospitalists as the discipline of hospital medicine becomes more mature. First, hospitalist models are quite variable. Many academic physicians who call themselves hospitalists attend on an inpatient service 2, 3, or 5 months a year and still see outpatients. Many physicians who consider themselves general internists (and not hospitalists) have a weekly half‐day clinic and attend on the wards 3 months a year. Which is a hospitalist? Does it matter? Will the definition of a hospitalist be based on achievement of the competencies described here, or will it be based primarily on the amount of time in hospital‐based practice? This will be an important question to resolve, especially as we embark on a path toward offering a hospitalist credential.
Second, general internal medicine is becoming an increasingly vital part of the continuum of care for patients with multiple complex chronic illnesses, at the same time that poor reimbursement has undermined its vitality and threatens its existence. (Family medicine is also suffering from reduced interest among medical students.) Because most institutions function on an each tub on its own bottom model, it is unrealistic to expect the practice of ambulatory general internal medicine to support itself. Generalist practices thrive in integrated group models. These practices recognize the importance of the physician who provides a coordinating function for all the specialists who care for a complex patient. Such an outpatient generalist thus reduces excess and unnecessary care while identifying gaps where relevant specialties could improve function or quality of life. Ambulatory practice also requires skill in systems and improvement, but few of the 80% of generalists who practice in small groups have sufficient infrastructure and resources to support practice redesign. Indeed, a new report from Mercer consultants coined the phrase ambulatory intensivists to identify practices with Medicare patients and recognizes that these practices are every bit as intense and complex and in need of systems management as an inpatient practice. What the complex patient needs is a seamless interface between the two.
The authors of the Core Competencies in Hospital Medicine hope that this document will stand the test of time as it evolves. I would urge that the document remain flexiblea living documentbecause the one thing about which we can be sure is that hospital practice will change. More and more critical care will be delivered throughout the hospital, more and more of all kinds of care will be performed outside the hospital, and the nature of hospitals will surely change with shifts in reimbursement that we cannot yet imagine but that might be right around the corner. If able to provide hands‐on care less expensively, physician assistants and nurse‐practitioners functioning according to protocols developed by systems thinkers, only some of whom will be physicians, may replace the physician in some settings. What will become of hospitalists as these systems change? I hope that hospitalists, together with other general internists, will be at the forefront of ensuring that the changes in practice that result from the combination of new technologies and financing structures will ultimately also serve the needs of patients. The patient is at the center of our discipline and, as articulated so clearly in the Core Competencies, should always be the focus of our future thinking.
- .Health care system chaos should spur innovation: summary of a report of the Society of General Internal Medicine Task Force on the Domain of General Internal Medicine.Ann Intern Med.2004;140:639–643.
- .Health care system chaos should spur innovation: summary of a report of the Society of General Internal Medicine Task Force on the Domain of General Internal Medicine.Ann Intern Med.2004;140:639–643.
The Colonial Formulary
Pharmacies have plotted a dynamic course through history, arriving late to North America in relation to other nations. Before the origin of the first true hospital pharmacy, medicinal therapies were often distributed by public officials, heads of households, and religious leaders or sold in drug stores and other free-standing apothecaries. Colonists followed books on self-treatment and methods of cultivating herbs. With the introduction of hospitals came the hospital pharmacy.
Great Britain had long been a glowing example of how pharmacists could prepare, compound, and administer prescriptions ordered by physicians in an organized manner. In contrast, the role of physicians, surgeons, and apothecaries in the British colonies were blurred, each with overlapping responsibilities of caring for patients and treating ailments.
It was not until 1751 after Benjamin Franklin and Dr. Thomas Bond founded the first hospital in the British Colonies—Pennsylvania Hospital in Philadelphia—that the idea for a hospital pharmacy was cultivated in North America. Because the first hospital’s mission was to provide charity for the poor, there initially was resistance to Dr. Bond’s idea of creating an apothecary in the same institution. Colonists feared that it would become costly to those in Philadelphia being served by the hospital. However, with Franklin’s persuading, enough funds were eventually solicited to purchase more than 112 pounds worth of drugs from London. In 1752, these drugs filled shelves in the hospital president’s office in the Pine Building of Pennsylvania Hospital instituting the first “Apothecary’s Shop in the Hospital” in the British colonies.
The first salaried hospital pharmacist, making 15 pounds per year, was Jonathon Roberts who worked until 1755 fulfilling the role of preparing medications requested by physicians. Medical and surgical students were often hired for short tenures in the apothecary to obtain experience in pharmacy or to simply cover their room and board expenses. John Morgan replaced Roberts in May 1755, and he worked for only one year before using that experience as a springboard for stirring up great influence in the future direction of American pharmacy. Morgan went on to become a physician and a vocal advocate for a more distinct separation of professions among physicians, surgeons, and pharmacists in America.
Most of the drugs available in the first American hospital pharmacy could be found in the London Pharmacopoeia of 1650, whereas very few drugs were of North American origin. Contributions from the colonies came primarily from the American Indian traditions that involved the extraction of botanical drugs such as cascara, bloodroot, and jalap. Nearly 170 of these particular preparations used by Indians north of the Rio Grande or their derivatives are still used today.
Other drugs used at the time of the first hospital pharmacy included emetic ipecac, an expectorant made of benzoin known as “Jesuit’s Drops,” antimony in “Plummer’s Pills,” and tincture of lavender (originally referred to as “Palsy Drops” and used to treat muscle spasms and headaches).
The advent of the American Revolution made importing drugs nearly impossible, requiring an increase in the number of patented drugs from North America. The first colonial hospital pharmacy, thanks to the ingenuity and persistence of Benjamin Franklin and Dr. Bond, set the stage for the development and transformation of pharmacies as we know them to today. TH
Nordman is a senior medical student at Penn State University.
Resources
- Bender GA. The First Hospital Pharmacy in Colonial America. In: Great Moments in Pharmacy. Detroit: Northwood Institute Press; 1966:84-87.
- Franklin B. Some Account of the Pennsylvania Hospital. Baltimore: The Johns Hopkins Press; 1954.
- Harris MR, Paracandola J. Images of Hospital Pharmacy in America. Am J Hosp Pharm. Reprint. June 1992.
- Lawall CH. Four Thousand Years of Pharmacy: An Outline History of Pharmacy and the Allied Sciences. Philadelphia: Lippincott; 1927.
- Massengill SE. American Pharmacy. In: A Sketch of Medicine and Pharmacy. Bristol, Tenn.: The S.E. Massengill Company. Chapter XV.
- Osborne GE. Pharmacy in British Colonial America. In: Bender GA, Parascandolam J, eds. American Pharmacy in the Colonial and Revolutionary Periods: A Bicentential Symposium held April 5, 1976. Madison, Wis.: American Institute of Pharmacy; 1977.
- Williams WH. Pharmacists at America’s First Hospital, 1752–1841 [abstract]. Am J Health Sys Pharm. 1976;33:804-804.
Pharmacies have plotted a dynamic course through history, arriving late to North America in relation to other nations. Before the origin of the first true hospital pharmacy, medicinal therapies were often distributed by public officials, heads of households, and religious leaders or sold in drug stores and other free-standing apothecaries. Colonists followed books on self-treatment and methods of cultivating herbs. With the introduction of hospitals came the hospital pharmacy.
Great Britain had long been a glowing example of how pharmacists could prepare, compound, and administer prescriptions ordered by physicians in an organized manner. In contrast, the role of physicians, surgeons, and apothecaries in the British colonies were blurred, each with overlapping responsibilities of caring for patients and treating ailments.
It was not until 1751 after Benjamin Franklin and Dr. Thomas Bond founded the first hospital in the British Colonies—Pennsylvania Hospital in Philadelphia—that the idea for a hospital pharmacy was cultivated in North America. Because the first hospital’s mission was to provide charity for the poor, there initially was resistance to Dr. Bond’s idea of creating an apothecary in the same institution. Colonists feared that it would become costly to those in Philadelphia being served by the hospital. However, with Franklin’s persuading, enough funds were eventually solicited to purchase more than 112 pounds worth of drugs from London. In 1752, these drugs filled shelves in the hospital president’s office in the Pine Building of Pennsylvania Hospital instituting the first “Apothecary’s Shop in the Hospital” in the British colonies.
The first salaried hospital pharmacist, making 15 pounds per year, was Jonathon Roberts who worked until 1755 fulfilling the role of preparing medications requested by physicians. Medical and surgical students were often hired for short tenures in the apothecary to obtain experience in pharmacy or to simply cover their room and board expenses. John Morgan replaced Roberts in May 1755, and he worked for only one year before using that experience as a springboard for stirring up great influence in the future direction of American pharmacy. Morgan went on to become a physician and a vocal advocate for a more distinct separation of professions among physicians, surgeons, and pharmacists in America.
Most of the drugs available in the first American hospital pharmacy could be found in the London Pharmacopoeia of 1650, whereas very few drugs were of North American origin. Contributions from the colonies came primarily from the American Indian traditions that involved the extraction of botanical drugs such as cascara, bloodroot, and jalap. Nearly 170 of these particular preparations used by Indians north of the Rio Grande or their derivatives are still used today.
Other drugs used at the time of the first hospital pharmacy included emetic ipecac, an expectorant made of benzoin known as “Jesuit’s Drops,” antimony in “Plummer’s Pills,” and tincture of lavender (originally referred to as “Palsy Drops” and used to treat muscle spasms and headaches).
The advent of the American Revolution made importing drugs nearly impossible, requiring an increase in the number of patented drugs from North America. The first colonial hospital pharmacy, thanks to the ingenuity and persistence of Benjamin Franklin and Dr. Bond, set the stage for the development and transformation of pharmacies as we know them to today. TH
Nordman is a senior medical student at Penn State University.
Resources
- Bender GA. The First Hospital Pharmacy in Colonial America. In: Great Moments in Pharmacy. Detroit: Northwood Institute Press; 1966:84-87.
- Franklin B. Some Account of the Pennsylvania Hospital. Baltimore: The Johns Hopkins Press; 1954.
- Harris MR, Paracandola J. Images of Hospital Pharmacy in America. Am J Hosp Pharm. Reprint. June 1992.
- Lawall CH. Four Thousand Years of Pharmacy: An Outline History of Pharmacy and the Allied Sciences. Philadelphia: Lippincott; 1927.
- Massengill SE. American Pharmacy. In: A Sketch of Medicine and Pharmacy. Bristol, Tenn.: The S.E. Massengill Company. Chapter XV.
- Osborne GE. Pharmacy in British Colonial America. In: Bender GA, Parascandolam J, eds. American Pharmacy in the Colonial and Revolutionary Periods: A Bicentential Symposium held April 5, 1976. Madison, Wis.: American Institute of Pharmacy; 1977.
- Williams WH. Pharmacists at America’s First Hospital, 1752–1841 [abstract]. Am J Health Sys Pharm. 1976;33:804-804.
Pharmacies have plotted a dynamic course through history, arriving late to North America in relation to other nations. Before the origin of the first true hospital pharmacy, medicinal therapies were often distributed by public officials, heads of households, and religious leaders or sold in drug stores and other free-standing apothecaries. Colonists followed books on self-treatment and methods of cultivating herbs. With the introduction of hospitals came the hospital pharmacy.
Great Britain had long been a glowing example of how pharmacists could prepare, compound, and administer prescriptions ordered by physicians in an organized manner. In contrast, the role of physicians, surgeons, and apothecaries in the British colonies were blurred, each with overlapping responsibilities of caring for patients and treating ailments.
It was not until 1751 after Benjamin Franklin and Dr. Thomas Bond founded the first hospital in the British Colonies—Pennsylvania Hospital in Philadelphia—that the idea for a hospital pharmacy was cultivated in North America. Because the first hospital’s mission was to provide charity for the poor, there initially was resistance to Dr. Bond’s idea of creating an apothecary in the same institution. Colonists feared that it would become costly to those in Philadelphia being served by the hospital. However, with Franklin’s persuading, enough funds were eventually solicited to purchase more than 112 pounds worth of drugs from London. In 1752, these drugs filled shelves in the hospital president’s office in the Pine Building of Pennsylvania Hospital instituting the first “Apothecary’s Shop in the Hospital” in the British colonies.
The first salaried hospital pharmacist, making 15 pounds per year, was Jonathon Roberts who worked until 1755 fulfilling the role of preparing medications requested by physicians. Medical and surgical students were often hired for short tenures in the apothecary to obtain experience in pharmacy or to simply cover their room and board expenses. John Morgan replaced Roberts in May 1755, and he worked for only one year before using that experience as a springboard for stirring up great influence in the future direction of American pharmacy. Morgan went on to become a physician and a vocal advocate for a more distinct separation of professions among physicians, surgeons, and pharmacists in America.
Most of the drugs available in the first American hospital pharmacy could be found in the London Pharmacopoeia of 1650, whereas very few drugs were of North American origin. Contributions from the colonies came primarily from the American Indian traditions that involved the extraction of botanical drugs such as cascara, bloodroot, and jalap. Nearly 170 of these particular preparations used by Indians north of the Rio Grande or their derivatives are still used today.
Other drugs used at the time of the first hospital pharmacy included emetic ipecac, an expectorant made of benzoin known as “Jesuit’s Drops,” antimony in “Plummer’s Pills,” and tincture of lavender (originally referred to as “Palsy Drops” and used to treat muscle spasms and headaches).
The advent of the American Revolution made importing drugs nearly impossible, requiring an increase in the number of patented drugs from North America. The first colonial hospital pharmacy, thanks to the ingenuity and persistence of Benjamin Franklin and Dr. Bond, set the stage for the development and transformation of pharmacies as we know them to today. TH
Nordman is a senior medical student at Penn State University.
Resources
- Bender GA. The First Hospital Pharmacy in Colonial America. In: Great Moments in Pharmacy. Detroit: Northwood Institute Press; 1966:84-87.
- Franklin B. Some Account of the Pennsylvania Hospital. Baltimore: The Johns Hopkins Press; 1954.
- Harris MR, Paracandola J. Images of Hospital Pharmacy in America. Am J Hosp Pharm. Reprint. June 1992.
- Lawall CH. Four Thousand Years of Pharmacy: An Outline History of Pharmacy and the Allied Sciences. Philadelphia: Lippincott; 1927.
- Massengill SE. American Pharmacy. In: A Sketch of Medicine and Pharmacy. Bristol, Tenn.: The S.E. Massengill Company. Chapter XV.
- Osborne GE. Pharmacy in British Colonial America. In: Bender GA, Parascandolam J, eds. American Pharmacy in the Colonial and Revolutionary Periods: A Bicentential Symposium held April 5, 1976. Madison, Wis.: American Institute of Pharmacy; 1977.
- Williams WH. Pharmacists at America’s First Hospital, 1752–1841 [abstract]. Am J Health Sys Pharm. 1976;33:804-804.
The Case of the Perfect Performer

Junior Moleray rubbed his large hand against the jet-black stubble on his square jaw. His feet were on his desk, and a dead pint of Old Croup Whisky was in the dumpster. It was quiet in the Moleray detective agency. Too quiet.
Moleray specialized in medical insurance fraud. It had been a week since he had solved the last case. A cagey bird had been collecting disability payments on five different accounts. Moleray caught up with him on a double black diamond run at Jackson Hole. Case solved. It had been too easy.
Junior thought about how he’d gotten into the detective racket. It was an improbably sad story. He’d been a detective on the Philadelphia police force and quit to become a medical student—the second doctor in the family.
His older brother, Maurie, had just finished an internal medicine residency and had signed with a clinic in Punxatawney—an outpatient internal medicine and pain clinic. Things were great for Maurie those the first two weeks. He was still getting his feet wet when his boss, Dr. Rock, went on vacation and disappeared while climbing in Malta. Shortly thereafter Rock’s widow showed up at the clinic with legal papers in hand. She offered to sell the entire practice for $10,000 cash just to be done with it. It was a beautiful office and a busy practice. Dr. Rock’s misfortune was Maurie’s stroke of luck. The deal was closed in 24 hours.
A week later, moving men came for the furniture (it was all rented and payment was overdue). Then the building manager evicted Maurie because the rent was also unpaid. Within two weeks he was accused of Medicare and Medicaid fraud and prescription peddling and named as a codefendant on six separate malpractice cases. The malpractice premiums hadn’t been paid in months.
Mrs. Rock was nowhere to be found. Maurie had been framed. His license was suspended. His debt was magnificent. His career in tatters.
Junior got to his brother’s house just in time. He found Maurie knotting ties together into a noose.
Junior quit medical school to track down the wife. After months, he found Mrs. Rock living in a shotgun shack in St. Bernard Parish, La. It didn’t look like the home of a rich widow. When he saw Mrs. Rock, his draw dropped. She was a tall drink of water, and he wanted to be the straw. Later he couldn’t remember what her face looked like. She invited him in.
“Come on in, honey” was the last thing he heard as he stepped into the room. He woke up hours later in an empty room with an occipital goose egg. There was nothing left except some half filled out forms in a precise handwriting. It was a cold trail that he swore he’d pick up again one day.
The ringing of the phone was a welcome relief.
“Moleray, it’s your dime,” Junior barked into the receiver.
“Hey Mole,” came an annoyingly familiar voice on the phone, “I got a hot one for you.”
It was Benny “the Weasel” Rabinowitz from the Mutual International Reinsurance Corporation.
“We’ve got a hospital that just submitted some numbers that are hard to believe, perfect numbers,” said Rabinowitz. “Unless we figure out their scam, we are going to have to fork over a cool million in pay-for-performance bonus fees. I smell a rat.”
“What’s the matter, Weasel, don’t your handlers at the Mutt want to pay the piper?” Junior retorted. He never trusted these big companies, even if the clients were defrauding them. The companies were none too innocent themselves. He took the case. It beat boredom.
The hospital was a rickety old building on South Main. It didn’t seem like the type of facility that would have perfect utilization numbers, but looks can be deceiving.
The medical records office was in the basement. A grumpy woman directed him to a pile of charts and disappeared. Junior spent the next six hours sitting in the cramped cubicle, sipping lukewarm, weak coffee and marveling at the perfect records. They were all written by the same physician with the perfect handwriting. There was something familiar about the writing, but Junior couldn’t make the connection.
The records were meticulous. Every patient with an MI had been given an ace inhibitor, a beta-blocker, aspirin, and a statin. But every patient was identical; the EKGs identical; the lab values identical—even the vitals were the same. Something was very wrong with these charts. By day’s end Junior was ready to go back to his cheesy motel room and make some phone calls. He had lifted some phone numbers from the charts. (Junior didn’t let a little thing like confidentiality stand in his way.) He tried to call several patients, but every single one of them had a disconnected phone.
The next morning Junior returned to the record room and knocked on the door. There was a different clerk today. A lilting voice said, “Come on in, honey.”
Junior started to get chills up and down his spine. He knew that voice … from somewhere. He felt her brush against his back. The smell of jasmine filled the air as she place a cup of coffee on the desk before him. As he finished off the foul morning brew it came to him. He knew her voice and her handwriting.
He stood quickly and turned toward her, but his head began to swim. Suddenly she was joined by a man whose photo he had seen before. It was Dr. Rock and his “wife.” Junior took a step toward them and then hit the ground, drugged into oblivion.
Fluorescent lights. Movement though a hallway. Junior found himself strapped on a gurney. He heard the orderly talking, maybe to a nurse.
“Is this Mr. Johnson?” asked the nurse. She checked his arm band; the ID was correct.
“He doesn’t say much does he?” the orderly commented.
“He’s going for resection of a huge brain mass,” the nurse replied.
Junior wondered who they were talking about as his stretcher was wheeled into the operating room. He felt the IV go into his hand. They checked his arm band again: “This is Hugo Johnson, birth date Oct. 5, 1957? OK.” They started to put the mask over his face, and Junior summoned all his strength and whispered, “Not me.”
The anesthesiologist shook his head, ”Sorry I know you’re scared but these things happen to everyone. You’ll be fine, Hugo.”
Junior tried again: “Not Hugo.”
The anesthesiologist nodded again, “I know we all feel immortal, but anyone—even you—can get sick.”
The mask came toward Junior again. He knew this was his last chance before his skull was cut open and his brain dissected in a hunt for a tumor that was not there.
Summoning his last reserve of energy, he yelled, “Sentinel event.” Everyone in the room stopped cold.
Junior sat in his office, his feet on his desk, and a newly deceased pint of Old Croup in the dumpster. He looked at his check from the Mutual for $25,000. He was ready for his next case. TH
Jamie Newman, MD, FACP, is the physician editor of The Hospitalist, senior associate consultant, Hospital Internal Medicine, and assistant professor of internal medicine and medical history, Mayo Clinic College of Medicine at the Mayo Clinic College of Medicine, Rochester, Minn.
Junior Moleray rubbed his large hand against the jet-black stubble on his square jaw. His feet were on his desk, and a dead pint of Old Croup Whisky was in the dumpster. It was quiet in the Moleray detective agency. Too quiet.
Moleray specialized in medical insurance fraud. It had been a week since he had solved the last case. A cagey bird had been collecting disability payments on five different accounts. Moleray caught up with him on a double black diamond run at Jackson Hole. Case solved. It had been too easy.
Junior thought about how he’d gotten into the detective racket. It was an improbably sad story. He’d been a detective on the Philadelphia police force and quit to become a medical student—the second doctor in the family.
His older brother, Maurie, had just finished an internal medicine residency and had signed with a clinic in Punxatawney—an outpatient internal medicine and pain clinic. Things were great for Maurie those the first two weeks. He was still getting his feet wet when his boss, Dr. Rock, went on vacation and disappeared while climbing in Malta. Shortly thereafter Rock’s widow showed up at the clinic with legal papers in hand. She offered to sell the entire practice for $10,000 cash just to be done with it. It was a beautiful office and a busy practice. Dr. Rock’s misfortune was Maurie’s stroke of luck. The deal was closed in 24 hours.
A week later, moving men came for the furniture (it was all rented and payment was overdue). Then the building manager evicted Maurie because the rent was also unpaid. Within two weeks he was accused of Medicare and Medicaid fraud and prescription peddling and named as a codefendant on six separate malpractice cases. The malpractice premiums hadn’t been paid in months.
Mrs. Rock was nowhere to be found. Maurie had been framed. His license was suspended. His debt was magnificent. His career in tatters.
Junior got to his brother’s house just in time. He found Maurie knotting ties together into a noose.
Junior quit medical school to track down the wife. After months, he found Mrs. Rock living in a shotgun shack in St. Bernard Parish, La. It didn’t look like the home of a rich widow. When he saw Mrs. Rock, his draw dropped. She was a tall drink of water, and he wanted to be the straw. Later he couldn’t remember what her face looked like. She invited him in.
“Come on in, honey” was the last thing he heard as he stepped into the room. He woke up hours later in an empty room with an occipital goose egg. There was nothing left except some half filled out forms in a precise handwriting. It was a cold trail that he swore he’d pick up again one day.
The ringing of the phone was a welcome relief.
“Moleray, it’s your dime,” Junior barked into the receiver.
“Hey Mole,” came an annoyingly familiar voice on the phone, “I got a hot one for you.”
It was Benny “the Weasel” Rabinowitz from the Mutual International Reinsurance Corporation.
“We’ve got a hospital that just submitted some numbers that are hard to believe, perfect numbers,” said Rabinowitz. “Unless we figure out their scam, we are going to have to fork over a cool million in pay-for-performance bonus fees. I smell a rat.”
“What’s the matter, Weasel, don’t your handlers at the Mutt want to pay the piper?” Junior retorted. He never trusted these big companies, even if the clients were defrauding them. The companies were none too innocent themselves. He took the case. It beat boredom.
The hospital was a rickety old building on South Main. It didn’t seem like the type of facility that would have perfect utilization numbers, but looks can be deceiving.
The medical records office was in the basement. A grumpy woman directed him to a pile of charts and disappeared. Junior spent the next six hours sitting in the cramped cubicle, sipping lukewarm, weak coffee and marveling at the perfect records. They were all written by the same physician with the perfect handwriting. There was something familiar about the writing, but Junior couldn’t make the connection.
The records were meticulous. Every patient with an MI had been given an ace inhibitor, a beta-blocker, aspirin, and a statin. But every patient was identical; the EKGs identical; the lab values identical—even the vitals were the same. Something was very wrong with these charts. By day’s end Junior was ready to go back to his cheesy motel room and make some phone calls. He had lifted some phone numbers from the charts. (Junior didn’t let a little thing like confidentiality stand in his way.) He tried to call several patients, but every single one of them had a disconnected phone.
The next morning Junior returned to the record room and knocked on the door. There was a different clerk today. A lilting voice said, “Come on in, honey.”
Junior started to get chills up and down his spine. He knew that voice … from somewhere. He felt her brush against his back. The smell of jasmine filled the air as she place a cup of coffee on the desk before him. As he finished off the foul morning brew it came to him. He knew her voice and her handwriting.
He stood quickly and turned toward her, but his head began to swim. Suddenly she was joined by a man whose photo he had seen before. It was Dr. Rock and his “wife.” Junior took a step toward them and then hit the ground, drugged into oblivion.
Fluorescent lights. Movement though a hallway. Junior found himself strapped on a gurney. He heard the orderly talking, maybe to a nurse.
“Is this Mr. Johnson?” asked the nurse. She checked his arm band; the ID was correct.
“He doesn’t say much does he?” the orderly commented.
“He’s going for resection of a huge brain mass,” the nurse replied.
Junior wondered who they were talking about as his stretcher was wheeled into the operating room. He felt the IV go into his hand. They checked his arm band again: “This is Hugo Johnson, birth date Oct. 5, 1957? OK.” They started to put the mask over his face, and Junior summoned all his strength and whispered, “Not me.”
The anesthesiologist shook his head, ”Sorry I know you’re scared but these things happen to everyone. You’ll be fine, Hugo.”
Junior tried again: “Not Hugo.”
The anesthesiologist nodded again, “I know we all feel immortal, but anyone—even you—can get sick.”
The mask came toward Junior again. He knew this was his last chance before his skull was cut open and his brain dissected in a hunt for a tumor that was not there.
Summoning his last reserve of energy, he yelled, “Sentinel event.” Everyone in the room stopped cold.
Junior sat in his office, his feet on his desk, and a newly deceased pint of Old Croup in the dumpster. He looked at his check from the Mutual for $25,000. He was ready for his next case. TH
Jamie Newman, MD, FACP, is the physician editor of The Hospitalist, senior associate consultant, Hospital Internal Medicine, and assistant professor of internal medicine and medical history, Mayo Clinic College of Medicine at the Mayo Clinic College of Medicine, Rochester, Minn.
Junior Moleray rubbed his large hand against the jet-black stubble on his square jaw. His feet were on his desk, and a dead pint of Old Croup Whisky was in the dumpster. It was quiet in the Moleray detective agency. Too quiet.
Moleray specialized in medical insurance fraud. It had been a week since he had solved the last case. A cagey bird had been collecting disability payments on five different accounts. Moleray caught up with him on a double black diamond run at Jackson Hole. Case solved. It had been too easy.
Junior thought about how he’d gotten into the detective racket. It was an improbably sad story. He’d been a detective on the Philadelphia police force and quit to become a medical student—the second doctor in the family.
His older brother, Maurie, had just finished an internal medicine residency and had signed with a clinic in Punxatawney—an outpatient internal medicine and pain clinic. Things were great for Maurie those the first two weeks. He was still getting his feet wet when his boss, Dr. Rock, went on vacation and disappeared while climbing in Malta. Shortly thereafter Rock’s widow showed up at the clinic with legal papers in hand. She offered to sell the entire practice for $10,000 cash just to be done with it. It was a beautiful office and a busy practice. Dr. Rock’s misfortune was Maurie’s stroke of luck. The deal was closed in 24 hours.
A week later, moving men came for the furniture (it was all rented and payment was overdue). Then the building manager evicted Maurie because the rent was also unpaid. Within two weeks he was accused of Medicare and Medicaid fraud and prescription peddling and named as a codefendant on six separate malpractice cases. The malpractice premiums hadn’t been paid in months.
Mrs. Rock was nowhere to be found. Maurie had been framed. His license was suspended. His debt was magnificent. His career in tatters.
Junior got to his brother’s house just in time. He found Maurie knotting ties together into a noose.
Junior quit medical school to track down the wife. After months, he found Mrs. Rock living in a shotgun shack in St. Bernard Parish, La. It didn’t look like the home of a rich widow. When he saw Mrs. Rock, his draw dropped. She was a tall drink of water, and he wanted to be the straw. Later he couldn’t remember what her face looked like. She invited him in.
“Come on in, honey” was the last thing he heard as he stepped into the room. He woke up hours later in an empty room with an occipital goose egg. There was nothing left except some half filled out forms in a precise handwriting. It was a cold trail that he swore he’d pick up again one day.
The ringing of the phone was a welcome relief.
“Moleray, it’s your dime,” Junior barked into the receiver.
“Hey Mole,” came an annoyingly familiar voice on the phone, “I got a hot one for you.”
It was Benny “the Weasel” Rabinowitz from the Mutual International Reinsurance Corporation.
“We’ve got a hospital that just submitted some numbers that are hard to believe, perfect numbers,” said Rabinowitz. “Unless we figure out their scam, we are going to have to fork over a cool million in pay-for-performance bonus fees. I smell a rat.”
“What’s the matter, Weasel, don’t your handlers at the Mutt want to pay the piper?” Junior retorted. He never trusted these big companies, even if the clients were defrauding them. The companies were none too innocent themselves. He took the case. It beat boredom.
The hospital was a rickety old building on South Main. It didn’t seem like the type of facility that would have perfect utilization numbers, but looks can be deceiving.
The medical records office was in the basement. A grumpy woman directed him to a pile of charts and disappeared. Junior spent the next six hours sitting in the cramped cubicle, sipping lukewarm, weak coffee and marveling at the perfect records. They were all written by the same physician with the perfect handwriting. There was something familiar about the writing, but Junior couldn’t make the connection.
The records were meticulous. Every patient with an MI had been given an ace inhibitor, a beta-blocker, aspirin, and a statin. But every patient was identical; the EKGs identical; the lab values identical—even the vitals were the same. Something was very wrong with these charts. By day’s end Junior was ready to go back to his cheesy motel room and make some phone calls. He had lifted some phone numbers from the charts. (Junior didn’t let a little thing like confidentiality stand in his way.) He tried to call several patients, but every single one of them had a disconnected phone.
The next morning Junior returned to the record room and knocked on the door. There was a different clerk today. A lilting voice said, “Come on in, honey.”
Junior started to get chills up and down his spine. He knew that voice … from somewhere. He felt her brush against his back. The smell of jasmine filled the air as she place a cup of coffee on the desk before him. As he finished off the foul morning brew it came to him. He knew her voice and her handwriting.
He stood quickly and turned toward her, but his head began to swim. Suddenly she was joined by a man whose photo he had seen before. It was Dr. Rock and his “wife.” Junior took a step toward them and then hit the ground, drugged into oblivion.
Fluorescent lights. Movement though a hallway. Junior found himself strapped on a gurney. He heard the orderly talking, maybe to a nurse.
“Is this Mr. Johnson?” asked the nurse. She checked his arm band; the ID was correct.
“He doesn’t say much does he?” the orderly commented.
“He’s going for resection of a huge brain mass,” the nurse replied.
Junior wondered who they were talking about as his stretcher was wheeled into the operating room. He felt the IV go into his hand. They checked his arm band again: “This is Hugo Johnson, birth date Oct. 5, 1957? OK.” They started to put the mask over his face, and Junior summoned all his strength and whispered, “Not me.”
The anesthesiologist shook his head, ”Sorry I know you’re scared but these things happen to everyone. You’ll be fine, Hugo.”
Junior tried again: “Not Hugo.”
The anesthesiologist nodded again, “I know we all feel immortal, but anyone—even you—can get sick.”
The mask came toward Junior again. He knew this was his last chance before his skull was cut open and his brain dissected in a hunt for a tumor that was not there.
Summoning his last reserve of energy, he yelled, “Sentinel event.” Everyone in the room stopped cold.
Junior sat in his office, his feet on his desk, and a newly deceased pint of Old Croup in the dumpster. He looked at his check from the Mutual for $25,000. He was ready for his next case. TH
Jamie Newman, MD, FACP, is the physician editor of The Hospitalist, senior associate consultant, Hospital Internal Medicine, and assistant professor of internal medicine and medical history, Mayo Clinic College of Medicine at the Mayo Clinic College of Medicine, Rochester, Minn.
Medication Compliance, the New C. Diff
The Tricky Nature of Medication Compliance
Review by Osterberg L, Blaschke T. Adherence to Medication. N Engl J Med. 2005;353:487-497.
Adherence to (or compliance with) a medication regimen is generally defined as the extent to which patients take medications as prescribed by their healthcare providers. Adherence rates are typically higher among patients with acute conditions, as compared with those with chronic conditions; persistence among patients with chronic conditions is disappointingly low, dropping most drastically after the first six months of therapy. Of all medication-related hospital admissions in the United States, 33% to 69% are because of poor medication adherence, with a resultant cost of approximately $100 billion a year.
Electronic medication-monitoring devices have provided very detailed information about the patterns of medication-taking behavior. Studies using these monitors have shown six general patterns of taking medication among patients treated for chronic illnesses who continue to take their medications. Approximately one-sixth come close to perfect adherence to a regimen; one-sixth take nearly all doses, but with some timing irregularity; one-sixth miss an occasional single day’s dose and have some timing inconsistency; one-sixth take drug holidays three to four times a year, with occasional omissions of doses; one-sixth have a drug holiday monthly or more often, with frequent omissions of doses; and one-sixth take few or no doses while giving the impression of good adherence.
Poor adherence to medication regimens is common, contributing to substantial worsening of disease, death, and increased healthcare costs. Practitioners should always look for poor adherence and can enhance adherence by emphasizing the value of a patient’s regimen, making the regimen simple, and customizing the regimen to the patient’s lifestyle. Asking patients nonjudgmentally about medication-taking behavior is a practical strategy for identifying poor adherence. A collaborative approach to care augments adherence. Patients who have difficulty maintaining adequate adherence need more intensive strategies than do patients who have less difficulty with adherence, a more forgiving medication regimen, or both. Innovative methods of managing chronic diseases have had some success in improving adherence when a regimen has been difficult to follow.
The New Clostridium Difficile—What Does It Mean?
McDonald LC, Killgore GE, Thompson A, et al. An epidemic, toxic gene-variant of Clostridium difficile. N Eng J Med. 2005;353;2433-2441.
Clostridium difficile is the only anaerobe that causes nosocomial infections. It colonizes the colon in 3% of the healthy population and about 20% to 40% of hospitalized patients.
This study was done in response to reports of increasing rate and severity of this infection. This study looked at healthcare facilities in Pennsylvania, Maine, Georgia, Oregon, Illinois, and New Jersey and did indeed find a new strain of Clostridium difficile isolate which showed 100% resistance to gatifloxacin and moxifloxacin, compared with no resistance in the historic strain.
Resistance to clindamycin was similar in both the groups, which was measured at 79%. This particular strain secretes 16 to 23 times more toxins A and B in vitro than other strains. And in this study the new strain accounted for 51% of the infections compared with 17% in the historic control isolates. Fluoroquinolones were implicated alone or in combination with other antibiotics in 52% of the cases. Those infected with the new strain were more likely to have higher rates of toxic megacolon, need for colectomy, leukemoid reaction, shock, and death. Like any disease, the interaction between host and pathogen is key to severity, thereby making patients who are chronically ill and elderly more susceptible.
For hospitalists the implications for this study are certainly important. We need to be aware of whether this strain is prevalent in our work environment. Close collaboration with our colleagues from infectious disease services along with monitoring clinical outcomes of patients with Clostridium difficile infection is the need of the hour. Also recommended is investigation of any increases in caseload of this infection. Simple measures such as judicious use of antibiotics, early diagnosis, and appropriate treatment of Clostridium difficile infection and strict isolation of the patients infected or colonized with Clostridium difficile would go a long way in controlling the spread of the new more virulent strain. It must be pointed out that alcohol-based waterless hand-sanitizing agents do not kill the Clostridium difficile spores; washing hands with soap and water is a prudent option after coming in contact with a patient with Clostridium difficile. TH
Nasal MRSA Carriage: A Study of Current Prevalence with Commentary
Creech CB, Kernodle DS, Alsectzer M, et al. Increasing rates of nasal carriage of methicillin-resistant Staphylococcus aureus in healthy children. Pediatr Infect Dis J. 2005;24:617-621.
Review by Laura Ortman, MD
The incidence of methicillin-resistant Staphylococcus aureus (MRSA) infections seen in outpatient clinics and emergency rooms appears to be on the rise. In 2001 a study done at Vanderbilt University Medical Center found the prevalence of MRSA in its pediatric community to be 0.8%1. Creech, et al., devised a study to describe the current prevalence of MRSA colonization in the same population.
The study population was children between the ages of two weeks and 21 years of age presenting for a health maintenance visit at two outpatient clinics. Nasal swabs were obtained and cultures preformed on plates with and without oxacillin containing media. Possible MRSA isolates were confirmed with PCR for the mecA gene, which codes for the protein responsible for beta-lactam resistance.
Of the 500 children enrolled 182 (36.4%) were found to be colonized with S. aureus. 46 (9.2%) isolates were positive for the mecA gene and considered MRSA. The only risk factor found to increase risk for MRSA colonization was having a family member who works in a hospital (odds ratio, 2.0; 95% confidence interval, 1.03-4.1). Fifty-four percent of MRSA isolates were resistant to erythromycin, and 32% of these had inducible clindamycin resistance.
Commentary: This study shows a greater than tenfold increase in MRSA colonization in a three-year time period in a healthy outpatient population. This finding is consistent with other studies that have shown increasing rates of colonization.2-3 This increase has led some institutions to attempt decolonization of MRSA, most often using nasal mupirocin. To determine if current evidence supports attempts to eradicate MRSA nasal colonization, the following literature search was performed: Cochrane DSR, ACP Journal Club, PubMed, and PubMed Clinical Queries were searched using the search terms “MRSA,” “colonization,” and “staphylococcus.”
One Cochrane review summarizes the evidence for use of antimicrobial agents on MRSA colonized patients4. Of six randomized controlled trials, only one compares rates of infection during follow-up between the study and control groups. The difference in infections was not statistically significant. Five other studies of inconsistent quality followed eradication rates of MRSA and varied widely in their results. The Cochrane review concluded that there was insufficient evidence to recommend nasal decolonization of MRSA.
One article reviewed the evidence for intranasal mupirocin for S. aureus.5 This review did not differentiate between MRSA and MSSA. The authors appraised clinical trials that evaluated the effect of mupirocin on MRSA colonization and infection. In a trial of patients undergoing dialysis there was no overall difference in the rate of infection between groups. In trials using mupirocin for preoperative prophylaxis there was no difference in number of surgical site infections. The authors concluded that mupirocin did not result in long-term clearance of S. aureus and that the available evidence does not support its use for prevention of infection. With the current evidence routine decolonization of patients colonized with MRSA cannot be recommended.
References
- Nakamura MM, Rohling KL, Shashaty M, et al. Prevalence of methicillin-resistant Staphylococcus aureus nasal carriage in the community pediatric population. Pediatr Infect Dis J. 2002;21:917-922.
- Herold BC, Immergluck LC, Maranan MC, et al. Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA. 1998;279:593-598.
- Fergie JE, Purcell K. Community-acquired methicillin-resistant Staphylococcus aureus infections in south Texas children. Pediatr Infect Dis J. 2001;20:860-863.
- Loeb M, Main C, Walker-Dilks C. Antimicrobial drugs for treating methicillin-resistant Staphylococcus aureus colonization. Cochrane Database Syst Rev. 2003;(4):CD003340.
- Laupland KB, Conly JM. Treatment of Staphylococcus aureus colonization and prophylaxis for infection with topical intranasal mupirocin: an evidence-based review. Clin Infect Dis. 2003;37:933-938.
Is Ultrasound Sufficient for Diagnosing Urolithiasis in the Pediatric Patient?
Palmer JS, Donaher ER, O’Riordan MA, et al. Diagnosis of Pediatric Urolithiasis: Role of ultrasound and computerized tomography. J Urol. 2005;174:1413-1416.
Review by Ann Mattison, RN, CPNP
Pediatric urolithiasis is uncommon and may present without the classic symptoms of renal colic, making diagnosis of pediatric urolithiasis problematic. Previously published data has revealed that unenhanced spiral CT is the gold standard in diagnosing urinary tract calculi in adults. However, CT carries the risk of exposure to ionizing radiation, which can be a significant issue in children.
Due to the low prevalence of urolithiasis in addition to concerns about radiation exposure, many primary care providers choose ultrasound as the initial radiographic study for children with symptoms that can be associated with urolithiasis, such as flank pain, abdominal pain, and gross hematuria. But the accuracy of ultrasound in detecting pediatric urolithiasis has not been well studied.
A retrospective chart review was performed in all patients 0-18 evaluated as outpatients and inpatients at the study institution. Subjects were identified by ICD-9 codes and billing records. The study showed the accuracy of ultrasounds performed was variable and dependent on the location of the calculi. In contrast, CT was highly accurate regardless of calculi location.
The study concluded that ultrasound may still be the appropriate initial study for the majority of children presenting with symptoms suggestive of urolithiasis; however, a negative ultrasound should not be considered sufficient to rule out the diagnosis of urolithiasis in pediatric patients. The authors recommended the patient with persistent symptoms and negative ultrasound undergo unenhanced CT. The retrospective design of this study limits application of these results; however, the study does highlight the need for a heightened index of suspicion for the diagnosis as well as the need for further prospective studies describing the most safe and efficient method for confirming the diagnosis. TH
The Tricky Nature of Medication Compliance
Review by Osterberg L, Blaschke T. Adherence to Medication. N Engl J Med. 2005;353:487-497.
Adherence to (or compliance with) a medication regimen is generally defined as the extent to which patients take medications as prescribed by their healthcare providers. Adherence rates are typically higher among patients with acute conditions, as compared with those with chronic conditions; persistence among patients with chronic conditions is disappointingly low, dropping most drastically after the first six months of therapy. Of all medication-related hospital admissions in the United States, 33% to 69% are because of poor medication adherence, with a resultant cost of approximately $100 billion a year.
Electronic medication-monitoring devices have provided very detailed information about the patterns of medication-taking behavior. Studies using these monitors have shown six general patterns of taking medication among patients treated for chronic illnesses who continue to take their medications. Approximately one-sixth come close to perfect adherence to a regimen; one-sixth take nearly all doses, but with some timing irregularity; one-sixth miss an occasional single day’s dose and have some timing inconsistency; one-sixth take drug holidays three to four times a year, with occasional omissions of doses; one-sixth have a drug holiday monthly or more often, with frequent omissions of doses; and one-sixth take few or no doses while giving the impression of good adherence.
Poor adherence to medication regimens is common, contributing to substantial worsening of disease, death, and increased healthcare costs. Practitioners should always look for poor adherence and can enhance adherence by emphasizing the value of a patient’s regimen, making the regimen simple, and customizing the regimen to the patient’s lifestyle. Asking patients nonjudgmentally about medication-taking behavior is a practical strategy for identifying poor adherence. A collaborative approach to care augments adherence. Patients who have difficulty maintaining adequate adherence need more intensive strategies than do patients who have less difficulty with adherence, a more forgiving medication regimen, or both. Innovative methods of managing chronic diseases have had some success in improving adherence when a regimen has been difficult to follow.
The New Clostridium Difficile—What Does It Mean?
McDonald LC, Killgore GE, Thompson A, et al. An epidemic, toxic gene-variant of Clostridium difficile. N Eng J Med. 2005;353;2433-2441.
Clostridium difficile is the only anaerobe that causes nosocomial infections. It colonizes the colon in 3% of the healthy population and about 20% to 40% of hospitalized patients.
This study was done in response to reports of increasing rate and severity of this infection. This study looked at healthcare facilities in Pennsylvania, Maine, Georgia, Oregon, Illinois, and New Jersey and did indeed find a new strain of Clostridium difficile isolate which showed 100% resistance to gatifloxacin and moxifloxacin, compared with no resistance in the historic strain.
Resistance to clindamycin was similar in both the groups, which was measured at 79%. This particular strain secretes 16 to 23 times more toxins A and B in vitro than other strains. And in this study the new strain accounted for 51% of the infections compared with 17% in the historic control isolates. Fluoroquinolones were implicated alone or in combination with other antibiotics in 52% of the cases. Those infected with the new strain were more likely to have higher rates of toxic megacolon, need for colectomy, leukemoid reaction, shock, and death. Like any disease, the interaction between host and pathogen is key to severity, thereby making patients who are chronically ill and elderly more susceptible.
For hospitalists the implications for this study are certainly important. We need to be aware of whether this strain is prevalent in our work environment. Close collaboration with our colleagues from infectious disease services along with monitoring clinical outcomes of patients with Clostridium difficile infection is the need of the hour. Also recommended is investigation of any increases in caseload of this infection. Simple measures such as judicious use of antibiotics, early diagnosis, and appropriate treatment of Clostridium difficile infection and strict isolation of the patients infected or colonized with Clostridium difficile would go a long way in controlling the spread of the new more virulent strain. It must be pointed out that alcohol-based waterless hand-sanitizing agents do not kill the Clostridium difficile spores; washing hands with soap and water is a prudent option after coming in contact with a patient with Clostridium difficile. TH
Nasal MRSA Carriage: A Study of Current Prevalence with Commentary
Creech CB, Kernodle DS, Alsectzer M, et al. Increasing rates of nasal carriage of methicillin-resistant Staphylococcus aureus in healthy children. Pediatr Infect Dis J. 2005;24:617-621.
Review by Laura Ortman, MD
The incidence of methicillin-resistant Staphylococcus aureus (MRSA) infections seen in outpatient clinics and emergency rooms appears to be on the rise. In 2001 a study done at Vanderbilt University Medical Center found the prevalence of MRSA in its pediatric community to be 0.8%1. Creech, et al., devised a study to describe the current prevalence of MRSA colonization in the same population.
The study population was children between the ages of two weeks and 21 years of age presenting for a health maintenance visit at two outpatient clinics. Nasal swabs were obtained and cultures preformed on plates with and without oxacillin containing media. Possible MRSA isolates were confirmed with PCR for the mecA gene, which codes for the protein responsible for beta-lactam resistance.
Of the 500 children enrolled 182 (36.4%) were found to be colonized with S. aureus. 46 (9.2%) isolates were positive for the mecA gene and considered MRSA. The only risk factor found to increase risk for MRSA colonization was having a family member who works in a hospital (odds ratio, 2.0; 95% confidence interval, 1.03-4.1). Fifty-four percent of MRSA isolates were resistant to erythromycin, and 32% of these had inducible clindamycin resistance.
Commentary: This study shows a greater than tenfold increase in MRSA colonization in a three-year time period in a healthy outpatient population. This finding is consistent with other studies that have shown increasing rates of colonization.2-3 This increase has led some institutions to attempt decolonization of MRSA, most often using nasal mupirocin. To determine if current evidence supports attempts to eradicate MRSA nasal colonization, the following literature search was performed: Cochrane DSR, ACP Journal Club, PubMed, and PubMed Clinical Queries were searched using the search terms “MRSA,” “colonization,” and “staphylococcus.”
One Cochrane review summarizes the evidence for use of antimicrobial agents on MRSA colonized patients4. Of six randomized controlled trials, only one compares rates of infection during follow-up between the study and control groups. The difference in infections was not statistically significant. Five other studies of inconsistent quality followed eradication rates of MRSA and varied widely in their results. The Cochrane review concluded that there was insufficient evidence to recommend nasal decolonization of MRSA.
One article reviewed the evidence for intranasal mupirocin for S. aureus.5 This review did not differentiate between MRSA and MSSA. The authors appraised clinical trials that evaluated the effect of mupirocin on MRSA colonization and infection. In a trial of patients undergoing dialysis there was no overall difference in the rate of infection between groups. In trials using mupirocin for preoperative prophylaxis there was no difference in number of surgical site infections. The authors concluded that mupirocin did not result in long-term clearance of S. aureus and that the available evidence does not support its use for prevention of infection. With the current evidence routine decolonization of patients colonized with MRSA cannot be recommended.
References
- Nakamura MM, Rohling KL, Shashaty M, et al. Prevalence of methicillin-resistant Staphylococcus aureus nasal carriage in the community pediatric population. Pediatr Infect Dis J. 2002;21:917-922.
- Herold BC, Immergluck LC, Maranan MC, et al. Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA. 1998;279:593-598.
- Fergie JE, Purcell K. Community-acquired methicillin-resistant Staphylococcus aureus infections in south Texas children. Pediatr Infect Dis J. 2001;20:860-863.
- Loeb M, Main C, Walker-Dilks C. Antimicrobial drugs for treating methicillin-resistant Staphylococcus aureus colonization. Cochrane Database Syst Rev. 2003;(4):CD003340.
- Laupland KB, Conly JM. Treatment of Staphylococcus aureus colonization and prophylaxis for infection with topical intranasal mupirocin: an evidence-based review. Clin Infect Dis. 2003;37:933-938.
Is Ultrasound Sufficient for Diagnosing Urolithiasis in the Pediatric Patient?
Palmer JS, Donaher ER, O’Riordan MA, et al. Diagnosis of Pediatric Urolithiasis: Role of ultrasound and computerized tomography. J Urol. 2005;174:1413-1416.
Review by Ann Mattison, RN, CPNP
Pediatric urolithiasis is uncommon and may present without the classic symptoms of renal colic, making diagnosis of pediatric urolithiasis problematic. Previously published data has revealed that unenhanced spiral CT is the gold standard in diagnosing urinary tract calculi in adults. However, CT carries the risk of exposure to ionizing radiation, which can be a significant issue in children.
Due to the low prevalence of urolithiasis in addition to concerns about radiation exposure, many primary care providers choose ultrasound as the initial radiographic study for children with symptoms that can be associated with urolithiasis, such as flank pain, abdominal pain, and gross hematuria. But the accuracy of ultrasound in detecting pediatric urolithiasis has not been well studied.
A retrospective chart review was performed in all patients 0-18 evaluated as outpatients and inpatients at the study institution. Subjects were identified by ICD-9 codes and billing records. The study showed the accuracy of ultrasounds performed was variable and dependent on the location of the calculi. In contrast, CT was highly accurate regardless of calculi location.
The study concluded that ultrasound may still be the appropriate initial study for the majority of children presenting with symptoms suggestive of urolithiasis; however, a negative ultrasound should not be considered sufficient to rule out the diagnosis of urolithiasis in pediatric patients. The authors recommended the patient with persistent symptoms and negative ultrasound undergo unenhanced CT. The retrospective design of this study limits application of these results; however, the study does highlight the need for a heightened index of suspicion for the diagnosis as well as the need for further prospective studies describing the most safe and efficient method for confirming the diagnosis. TH
The Tricky Nature of Medication Compliance
Review by Osterberg L, Blaschke T. Adherence to Medication. N Engl J Med. 2005;353:487-497.
Adherence to (or compliance with) a medication regimen is generally defined as the extent to which patients take medications as prescribed by their healthcare providers. Adherence rates are typically higher among patients with acute conditions, as compared with those with chronic conditions; persistence among patients with chronic conditions is disappointingly low, dropping most drastically after the first six months of therapy. Of all medication-related hospital admissions in the United States, 33% to 69% are because of poor medication adherence, with a resultant cost of approximately $100 billion a year.
Electronic medication-monitoring devices have provided very detailed information about the patterns of medication-taking behavior. Studies using these monitors have shown six general patterns of taking medication among patients treated for chronic illnesses who continue to take their medications. Approximately one-sixth come close to perfect adherence to a regimen; one-sixth take nearly all doses, but with some timing irregularity; one-sixth miss an occasional single day’s dose and have some timing inconsistency; one-sixth take drug holidays three to four times a year, with occasional omissions of doses; one-sixth have a drug holiday monthly or more often, with frequent omissions of doses; and one-sixth take few or no doses while giving the impression of good adherence.
Poor adherence to medication regimens is common, contributing to substantial worsening of disease, death, and increased healthcare costs. Practitioners should always look for poor adherence and can enhance adherence by emphasizing the value of a patient’s regimen, making the regimen simple, and customizing the regimen to the patient’s lifestyle. Asking patients nonjudgmentally about medication-taking behavior is a practical strategy for identifying poor adherence. A collaborative approach to care augments adherence. Patients who have difficulty maintaining adequate adherence need more intensive strategies than do patients who have less difficulty with adherence, a more forgiving medication regimen, or both. Innovative methods of managing chronic diseases have had some success in improving adherence when a regimen has been difficult to follow.
The New Clostridium Difficile—What Does It Mean?
McDonald LC, Killgore GE, Thompson A, et al. An epidemic, toxic gene-variant of Clostridium difficile. N Eng J Med. 2005;353;2433-2441.
Clostridium difficile is the only anaerobe that causes nosocomial infections. It colonizes the colon in 3% of the healthy population and about 20% to 40% of hospitalized patients.
This study was done in response to reports of increasing rate and severity of this infection. This study looked at healthcare facilities in Pennsylvania, Maine, Georgia, Oregon, Illinois, and New Jersey and did indeed find a new strain of Clostridium difficile isolate which showed 100% resistance to gatifloxacin and moxifloxacin, compared with no resistance in the historic strain.
Resistance to clindamycin was similar in both the groups, which was measured at 79%. This particular strain secretes 16 to 23 times more toxins A and B in vitro than other strains. And in this study the new strain accounted for 51% of the infections compared with 17% in the historic control isolates. Fluoroquinolones were implicated alone or in combination with other antibiotics in 52% of the cases. Those infected with the new strain were more likely to have higher rates of toxic megacolon, need for colectomy, leukemoid reaction, shock, and death. Like any disease, the interaction between host and pathogen is key to severity, thereby making patients who are chronically ill and elderly more susceptible.
For hospitalists the implications for this study are certainly important. We need to be aware of whether this strain is prevalent in our work environment. Close collaboration with our colleagues from infectious disease services along with monitoring clinical outcomes of patients with Clostridium difficile infection is the need of the hour. Also recommended is investigation of any increases in caseload of this infection. Simple measures such as judicious use of antibiotics, early diagnosis, and appropriate treatment of Clostridium difficile infection and strict isolation of the patients infected or colonized with Clostridium difficile would go a long way in controlling the spread of the new more virulent strain. It must be pointed out that alcohol-based waterless hand-sanitizing agents do not kill the Clostridium difficile spores; washing hands with soap and water is a prudent option after coming in contact with a patient with Clostridium difficile. TH
Nasal MRSA Carriage: A Study of Current Prevalence with Commentary
Creech CB, Kernodle DS, Alsectzer M, et al. Increasing rates of nasal carriage of methicillin-resistant Staphylococcus aureus in healthy children. Pediatr Infect Dis J. 2005;24:617-621.
Review by Laura Ortman, MD
The incidence of methicillin-resistant Staphylococcus aureus (MRSA) infections seen in outpatient clinics and emergency rooms appears to be on the rise. In 2001 a study done at Vanderbilt University Medical Center found the prevalence of MRSA in its pediatric community to be 0.8%1. Creech, et al., devised a study to describe the current prevalence of MRSA colonization in the same population.
The study population was children between the ages of two weeks and 21 years of age presenting for a health maintenance visit at two outpatient clinics. Nasal swabs were obtained and cultures preformed on plates with and without oxacillin containing media. Possible MRSA isolates were confirmed with PCR for the mecA gene, which codes for the protein responsible for beta-lactam resistance.
Of the 500 children enrolled 182 (36.4%) were found to be colonized with S. aureus. 46 (9.2%) isolates were positive for the mecA gene and considered MRSA. The only risk factor found to increase risk for MRSA colonization was having a family member who works in a hospital (odds ratio, 2.0; 95% confidence interval, 1.03-4.1). Fifty-four percent of MRSA isolates were resistant to erythromycin, and 32% of these had inducible clindamycin resistance.
Commentary: This study shows a greater than tenfold increase in MRSA colonization in a three-year time period in a healthy outpatient population. This finding is consistent with other studies that have shown increasing rates of colonization.2-3 This increase has led some institutions to attempt decolonization of MRSA, most often using nasal mupirocin. To determine if current evidence supports attempts to eradicate MRSA nasal colonization, the following literature search was performed: Cochrane DSR, ACP Journal Club, PubMed, and PubMed Clinical Queries were searched using the search terms “MRSA,” “colonization,” and “staphylococcus.”
One Cochrane review summarizes the evidence for use of antimicrobial agents on MRSA colonized patients4. Of six randomized controlled trials, only one compares rates of infection during follow-up between the study and control groups. The difference in infections was not statistically significant. Five other studies of inconsistent quality followed eradication rates of MRSA and varied widely in their results. The Cochrane review concluded that there was insufficient evidence to recommend nasal decolonization of MRSA.
One article reviewed the evidence for intranasal mupirocin for S. aureus.5 This review did not differentiate between MRSA and MSSA. The authors appraised clinical trials that evaluated the effect of mupirocin on MRSA colonization and infection. In a trial of patients undergoing dialysis there was no overall difference in the rate of infection between groups. In trials using mupirocin for preoperative prophylaxis there was no difference in number of surgical site infections. The authors concluded that mupirocin did not result in long-term clearance of S. aureus and that the available evidence does not support its use for prevention of infection. With the current evidence routine decolonization of patients colonized with MRSA cannot be recommended.
References
- Nakamura MM, Rohling KL, Shashaty M, et al. Prevalence of methicillin-resistant Staphylococcus aureus nasal carriage in the community pediatric population. Pediatr Infect Dis J. 2002;21:917-922.
- Herold BC, Immergluck LC, Maranan MC, et al. Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA. 1998;279:593-598.
- Fergie JE, Purcell K. Community-acquired methicillin-resistant Staphylococcus aureus infections in south Texas children. Pediatr Infect Dis J. 2001;20:860-863.
- Loeb M, Main C, Walker-Dilks C. Antimicrobial drugs for treating methicillin-resistant Staphylococcus aureus colonization. Cochrane Database Syst Rev. 2003;(4):CD003340.
- Laupland KB, Conly JM. Treatment of Staphylococcus aureus colonization and prophylaxis for infection with topical intranasal mupirocin: an evidence-based review. Clin Infect Dis. 2003;37:933-938.
Is Ultrasound Sufficient for Diagnosing Urolithiasis in the Pediatric Patient?
Palmer JS, Donaher ER, O’Riordan MA, et al. Diagnosis of Pediatric Urolithiasis: Role of ultrasound and computerized tomography. J Urol. 2005;174:1413-1416.
Review by Ann Mattison, RN, CPNP
Pediatric urolithiasis is uncommon and may present without the classic symptoms of renal colic, making diagnosis of pediatric urolithiasis problematic. Previously published data has revealed that unenhanced spiral CT is the gold standard in diagnosing urinary tract calculi in adults. However, CT carries the risk of exposure to ionizing radiation, which can be a significant issue in children.
Due to the low prevalence of urolithiasis in addition to concerns about radiation exposure, many primary care providers choose ultrasound as the initial radiographic study for children with symptoms that can be associated with urolithiasis, such as flank pain, abdominal pain, and gross hematuria. But the accuracy of ultrasound in detecting pediatric urolithiasis has not been well studied.
A retrospective chart review was performed in all patients 0-18 evaluated as outpatients and inpatients at the study institution. Subjects were identified by ICD-9 codes and billing records. The study showed the accuracy of ultrasounds performed was variable and dependent on the location of the calculi. In contrast, CT was highly accurate regardless of calculi location.
The study concluded that ultrasound may still be the appropriate initial study for the majority of children presenting with symptoms suggestive of urolithiasis; however, a negative ultrasound should not be considered sufficient to rule out the diagnosis of urolithiasis in pediatric patients. The authors recommended the patient with persistent symptoms and negative ultrasound undergo unenhanced CT. The retrospective design of this study limits application of these results; however, the study does highlight the need for a heightened index of suspicion for the diagnosis as well as the need for further prospective studies describing the most safe and efficient method for confirming the diagnosis. TH
A Return Visit to Mercy's Pay-For-Performance Program
Within a year of the implementation of a performance-based incentive program for its hospitalists, Mercy Hospital in Springfield, Mass., found itself leading the state in key composite compliance measures. Mercy was No. 2 on the list for two quarters in a row when MassPRO (the federally designated Quality Improvement Organization for Massachusetts) rated all 63 hospitals in the state on performance on quality indicators for heart failure, pneumonia, and MI.
Mercy Hospital ranked second in the state for both the fourth quarter of 2004 and the first quarter of 2005, whereas a different hospital ranked first for each quarter—and Massachusetts is the second-ranked state in the United States in these indicators.
How did Mercy rise so quickly to the top? Win Whitcomb, MD, who heads the hospitalist program at Mercy Hospital, credits the quality-based incentive program he helped initiate. “We had rapid improvement because we had a dedicated group of hospitalists and they were incentivized,” says Dr. Whitcomb.
Pay-for-Performance Program
A full 75% of the inpatients at Mercy Hospital are under the care of 10 hospitalists employed by the Mercy Inpatient Medicine Service (MIMS). As outlined in an article Dr. Whitcomb wrote for the July/August 2005 issue of The Hospitalist (“Physician Pay-for-Performance Comes to the Hospital”), MIMS implemented a unique incentive program for their hospitalists in January 2004.
The pay-for-performance, quality-based incentive program promised that each physician would receive a cash bonus every six months of more than 7.5% of his or her salary—but only if Mercy Medical Center reached the following targets for all hospital patients by the end of 2004:
- A 45% rate of pneumococcal vaccine screening and administration for all pneumonia patients;
- An 85% rate of documentation of ejection fraction for all heart failure patients; and
- Less than 40% rate of ejection fraction for heart failure patients and prescription of an ACE-inhibitor upon discharge (or documentation of a contraindication).
Dr. Whitcomb’s article shows how the MIMS group exceeded each of these quality improvement goals by the end of 2004. The MassPRO ranking shows how well they did in comparison with other hospitals in their state.
“The MassPRO recommendation for our performance is for the whole hospital—not just hospitalists,” says Dr. Whitcomb. “This is a good example of how hospitalists can carry the hospital. We also have traditional PCPs [primary care physicians] who are eager to measure up to our hospitalists; I feed back information to them, too.”
Update on the Incentive Program
MIMS is not resting on its laurels; they have continued to expand and update the incentive program. According to Bipinchandra Mistry, MD, MRCP, the current leader of the incentive program, 2005 has seen the addition of quality markers for reduction of decubitus ulcer rates, reduction of postoperative urinary tract infections, and discharge instructions for CHF. The annual bonus for physicians will be increased accordingly if these new markers are met.
“Of course, we must also maintain the previous quality markers at the same time,” explains Dr. Mistry.
Dr. Mistry attributes the success of the incentive program to its tie-in with a quality department. “The key is to have a person in your quality department involved to keep an eye on [markers in an incentive program] and see what barriers are coming up,” he says. “Otherwise, it’s harder for a group to forge ahead.”
Of the pay bonus that is tied to the markers, 30% relies on reaching the quality markers. “I think 30% to 40% is a reasonable target,” says Dr. Mistry.
Because these particular measures are difficult for the MIMS hospitalists to monitor alone, a quality improvement group headed by Dr. Whitcomb worked to include both a separate hospitalist group as well as PCPs. All were held accountable for quality through the addition of a “night-time coverage fee” that would be forgiven when the new quality goals were met.
Time will tell if the MIMS pay-for-performance program continues to pay off in increased quality of care for patients. TH
Contributing Writer Jane Jerrard is based in Chicago.
Within a year of the implementation of a performance-based incentive program for its hospitalists, Mercy Hospital in Springfield, Mass., found itself leading the state in key composite compliance measures. Mercy was No. 2 on the list for two quarters in a row when MassPRO (the federally designated Quality Improvement Organization for Massachusetts) rated all 63 hospitals in the state on performance on quality indicators for heart failure, pneumonia, and MI.
Mercy Hospital ranked second in the state for both the fourth quarter of 2004 and the first quarter of 2005, whereas a different hospital ranked first for each quarter—and Massachusetts is the second-ranked state in the United States in these indicators.
How did Mercy rise so quickly to the top? Win Whitcomb, MD, who heads the hospitalist program at Mercy Hospital, credits the quality-based incentive program he helped initiate. “We had rapid improvement because we had a dedicated group of hospitalists and they were incentivized,” says Dr. Whitcomb.
Pay-for-Performance Program
A full 75% of the inpatients at Mercy Hospital are under the care of 10 hospitalists employed by the Mercy Inpatient Medicine Service (MIMS). As outlined in an article Dr. Whitcomb wrote for the July/August 2005 issue of The Hospitalist (“Physician Pay-for-Performance Comes to the Hospital”), MIMS implemented a unique incentive program for their hospitalists in January 2004.
The pay-for-performance, quality-based incentive program promised that each physician would receive a cash bonus every six months of more than 7.5% of his or her salary—but only if Mercy Medical Center reached the following targets for all hospital patients by the end of 2004:
- A 45% rate of pneumococcal vaccine screening and administration for all pneumonia patients;
- An 85% rate of documentation of ejection fraction for all heart failure patients; and
- Less than 40% rate of ejection fraction for heart failure patients and prescription of an ACE-inhibitor upon discharge (or documentation of a contraindication).
Dr. Whitcomb’s article shows how the MIMS group exceeded each of these quality improvement goals by the end of 2004. The MassPRO ranking shows how well they did in comparison with other hospitals in their state.
“The MassPRO recommendation for our performance is for the whole hospital—not just hospitalists,” says Dr. Whitcomb. “This is a good example of how hospitalists can carry the hospital. We also have traditional PCPs [primary care physicians] who are eager to measure up to our hospitalists; I feed back information to them, too.”
Update on the Incentive Program
MIMS is not resting on its laurels; they have continued to expand and update the incentive program. According to Bipinchandra Mistry, MD, MRCP, the current leader of the incentive program, 2005 has seen the addition of quality markers for reduction of decubitus ulcer rates, reduction of postoperative urinary tract infections, and discharge instructions for CHF. The annual bonus for physicians will be increased accordingly if these new markers are met.
“Of course, we must also maintain the previous quality markers at the same time,” explains Dr. Mistry.
Dr. Mistry attributes the success of the incentive program to its tie-in with a quality department. “The key is to have a person in your quality department involved to keep an eye on [markers in an incentive program] and see what barriers are coming up,” he says. “Otherwise, it’s harder for a group to forge ahead.”
Of the pay bonus that is tied to the markers, 30% relies on reaching the quality markers. “I think 30% to 40% is a reasonable target,” says Dr. Mistry.
Because these particular measures are difficult for the MIMS hospitalists to monitor alone, a quality improvement group headed by Dr. Whitcomb worked to include both a separate hospitalist group as well as PCPs. All were held accountable for quality through the addition of a “night-time coverage fee” that would be forgiven when the new quality goals were met.
Time will tell if the MIMS pay-for-performance program continues to pay off in increased quality of care for patients. TH
Contributing Writer Jane Jerrard is based in Chicago.
Within a year of the implementation of a performance-based incentive program for its hospitalists, Mercy Hospital in Springfield, Mass., found itself leading the state in key composite compliance measures. Mercy was No. 2 on the list for two quarters in a row when MassPRO (the federally designated Quality Improvement Organization for Massachusetts) rated all 63 hospitals in the state on performance on quality indicators for heart failure, pneumonia, and MI.
Mercy Hospital ranked second in the state for both the fourth quarter of 2004 and the first quarter of 2005, whereas a different hospital ranked first for each quarter—and Massachusetts is the second-ranked state in the United States in these indicators.
How did Mercy rise so quickly to the top? Win Whitcomb, MD, who heads the hospitalist program at Mercy Hospital, credits the quality-based incentive program he helped initiate. “We had rapid improvement because we had a dedicated group of hospitalists and they were incentivized,” says Dr. Whitcomb.
Pay-for-Performance Program
A full 75% of the inpatients at Mercy Hospital are under the care of 10 hospitalists employed by the Mercy Inpatient Medicine Service (MIMS). As outlined in an article Dr. Whitcomb wrote for the July/August 2005 issue of The Hospitalist (“Physician Pay-for-Performance Comes to the Hospital”), MIMS implemented a unique incentive program for their hospitalists in January 2004.
The pay-for-performance, quality-based incentive program promised that each physician would receive a cash bonus every six months of more than 7.5% of his or her salary—but only if Mercy Medical Center reached the following targets for all hospital patients by the end of 2004:
- A 45% rate of pneumococcal vaccine screening and administration for all pneumonia patients;
- An 85% rate of documentation of ejection fraction for all heart failure patients; and
- Less than 40% rate of ejection fraction for heart failure patients and prescription of an ACE-inhibitor upon discharge (or documentation of a contraindication).
Dr. Whitcomb’s article shows how the MIMS group exceeded each of these quality improvement goals by the end of 2004. The MassPRO ranking shows how well they did in comparison with other hospitals in their state.
“The MassPRO recommendation for our performance is for the whole hospital—not just hospitalists,” says Dr. Whitcomb. “This is a good example of how hospitalists can carry the hospital. We also have traditional PCPs [primary care physicians] who are eager to measure up to our hospitalists; I feed back information to them, too.”
Update on the Incentive Program
MIMS is not resting on its laurels; they have continued to expand and update the incentive program. According to Bipinchandra Mistry, MD, MRCP, the current leader of the incentive program, 2005 has seen the addition of quality markers for reduction of decubitus ulcer rates, reduction of postoperative urinary tract infections, and discharge instructions for CHF. The annual bonus for physicians will be increased accordingly if these new markers are met.
“Of course, we must also maintain the previous quality markers at the same time,” explains Dr. Mistry.
Dr. Mistry attributes the success of the incentive program to its tie-in with a quality department. “The key is to have a person in your quality department involved to keep an eye on [markers in an incentive program] and see what barriers are coming up,” he says. “Otherwise, it’s harder for a group to forge ahead.”
Of the pay bonus that is tied to the markers, 30% relies on reaching the quality markers. “I think 30% to 40% is a reasonable target,” says Dr. Mistry.
Because these particular measures are difficult for the MIMS hospitalists to monitor alone, a quality improvement group headed by Dr. Whitcomb worked to include both a separate hospitalist group as well as PCPs. All were held accountable for quality through the addition of a “night-time coverage fee” that would be forgiven when the new quality goals were met.
Time will tell if the MIMS pay-for-performance program continues to pay off in increased quality of care for patients. TH
Contributing Writer Jane Jerrard is based in Chicago.