User login
Answers to your questions about SSRIs
• Avoid prescribing a highly activating SSRI, such as fluoxetine, for patients for whom agitation is a presenting symptom. B
• Consider citalopram and escitalopram, which have less potential for drug interactions and less complex dose titration compared with other SSRIs, as first-line agents for depressed patients with complex medication profiles. C
• For patients with sexual side effects caused by SSRIs, consider augmentation therapy with bupropion or mirtazapine; for male sexual dysfunction, a trial of a phosphodiesterase inhibitor is another alternative. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Shortly after the US Food and Drug Administration (FDA) approved fluoxetine for the treatment of major depression nearly a quarter of a century ago, Prozac became a household name. And, as a handful of additional selective serotonin reuptake inhibitors (SSRIs) were approved, the popularity of this category of antidepressant continued to grow.1 In 2008, 5 of the 6 SSRIs on the US market (fluvoxamine was the exception) were among the 200 most frequently dispensed prescription drugs.2
While much has been written about the properties of SSRIs, uncertainty about many features of particular agents and what should be considered in selecting an antidepressant for a particular patient remains. To help clear up the confusion, we’ve addressed 7 questions about SSRIs that we are often asked. Although there are no simple answers, the evidence presented, both in the answers and in the tables that follow, will help primary care physicians make more informed choices for patients who require antidepressant therapy.
1. Which is the best SSRI to start a patient on?
A recent meta-analysis of 117 head-to-head studies assessed the efficacy and acceptability of 12 newer antidepressants, including all 6 SSRIs (citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, and sertraline).3 The researchers found 2 SSRIs, sertraline and escitalopram, to be superior to the other medications studied on both counts. A choice between sertraline or escitalopram may be a reasonable starting point in many cases, but it is impossible to recommend 1 or 2 SSRIs that are effective for all patients. There are several reasons for this.
The first is addressed by the Cipriani meta-analysis. The researchers assessed the efficacy of initial antidepressant therapy at 8 weeks, so the results cannot be extrapolated to long-term response rates or acceptability. (For a detailed review of the meta-analysis, see “Try these 2 drugs first for depression,” J Fam Pract. 2009;58:365-369.)
A second reason is the substantial publication bias associated with studies of antidepressants. Turner et al assessed 74 studies registered with the FDA, determined whether the results were positive or negative, and categorized the studies based on publication status.4 Their conclusions: 97% of studies with positive findings (n=38) were published.
The majority of the remaining studies, all of which were determined to have negative findings, were either not published or published in a manner that suggested a positive outcome. When these additional studies are taken into account, the percentage of published studies with a positive response drops to 51%.4 In addition, the effect size of each agent is reduced when all the studies are included.
There are many confounding variables associated with publication, so Turner and his colleagues were unable to definitively determine the reason for the disparity. Nonetheless, their evaluation raises questions about the reported effectiveness, not only of SSRIs, but of antidepressants in general.
A recent Cochrane review of SSRIs for treatment of depression in children and adolescents (a topic covered in greater detail in the answer to Question 6) raised similar concerns. The reviewers cited study methodology and recruitment methods as potential sources of the conflicting results they found.5
Finally, individual differences, such as age, comorbidities, and medication history, require an individualized approach to SSRI treatment.
2. Which side effects are common to all SSRIs, and which can be resolved by switching agents?
As a class, SSRIs are well tolerated, but they do have some common adverse effects, most notably gastrointestinal (GI) problems, sexual dysfunction, and sleep disturbances. There are also considerable differences in SSRI profiles and a few adverse effects that a switch to another SSRI may alleviate or resolve.
Compared with other SSRIs, for example, sertraline has a higher risk of diarrhea, but this adverse event does not usually lead to medication discontinuation. If it is bothersome to the patient or does not resolve with continued therapy, a change to another agent might eliminate this adverse effect.
Weight gain has been found to be more significant with paroxetine than with fluoxetine or sertraline, which may be due to the anticholinergic action of the drug.6 For someone who is particularly concerned about extra weight, avoiding paroxetine in the first place, or changing a patient who’s already taking it to fluoxetine or sertraline, might be an effective treatment strategy.
Overall, paroxetine has more anticholinergic side effects than other SSRIs, the likely result of its higher affinity for muscarinic receptors ( TABLE 1 ). Its potential sedating effect,7 which can be extremely disturbing to some patients, may be a desired feature for others. A patient with insomnia, for example, might benefit from taking paroxetine at bedtime or switching to a less activating SSRI, such as citalopram or escitalopram. Excessive activation, such as significant insomnia, may also be a warning sign of undiagnosed bipolar disorder. Thus, careful screening is needed prior to switching medications or adding a hypnotic agent. (See the box below “Treating depression in primary care: Tips from a psychiatrist”.)
A switch to another SSRI is not the only way to alleviate a troublesome side effect, of course. Insomnia can also be managed by adding a short course of trazodone to the drug regimen or by switching the patient to mirtazapine or a tricyclic antidepressant (TCA).8
Augmentation with either bupropion or mirtazapine may alleviate sexual side effects and should be tried prior to switching the patient to a different antidepressant; however, the positive effects of augmentation should be balanced with any additional adverse events either agent may cause. For male sexual dysfunction, a trial of a phosphodiesterase inhibitor such as sildenafil is another alternative.
TABLE 1
SSRIs: Neurotransmitter affinity and side effects9
| Neurotransmitter/enzyme | SSRIs with most potent affinity* | Likely side effects |
|---|---|---|
| Serotonin | Paroxetine Sertraline Fluoxetine Citalopram Fluvoxamine | Sexual dysfunction, including anorgasmy; GI disturbance; activation |
| Dopamine | Sertraline Paroxetine Fluoxetine Fluvoxamine Citalopram | Extrapyramidal |
| Norepinephrine | Paroxetine Fluoxetine Sertraline Fluvoxamine Citalopram | Tremor; tachycardia; elevated BP |
| Muscarinic receptors | Paroxetine Sertraline Fluoxetine Citalopram Fluvoxamine | Anticholinergic (blurred vision, constipation, dry mouth) |
| *SSRIs listed in order of potency; escitalopram would be expected to have effects comparable to citalopram. | ||
| BP, blood pressure; GI, gastrointestinal; SSRI, selective serotonin reuptake inhibitors. | ||
- Use a validated instrument for depression screening and monitoring treatment success—preferably the PHQ-9 (http://www.lphi.org/LPHIadmin/uploads/.PHQ-9-Review-Kroenke-63754.PDF), which is validated for primary care practice. Use it at subsequent visits to track treatment response. The goal is not just response to treatment but remission of depressive symptoms, both of which are correlated with a decrease in the PHQ-9 total score.
- Take a careful history of bipolar symptoms and family history before starting a new patient on an SSRI. The DIGFAST mnemonic (http://www.usmle-forums.com/usmle-step-1-mnemonics/793-dig-fast-mnemonic-mania-symptoms.html) can help. Failure to respond to an SSRI or exacerbation of symptoms such as restlessness or insomnia once treatment has started should prompt you to consider a missed diagnosis.
- Consider augmentation strategies for patients who show partial improvement on an SSRI, rather than throwing out the gain with a wholesale switch to another drug. If a patient is only 30% better on the first agent you prescribe, try adding a second agent, which may significantly boost the response rate. Suggestions for SSRI augmentation include lithium, thyroid hormone, bupropion, and aripiprazole.
- Make sure the patient has been on a therapeutic dose for a sufficient period of time—at least 4 weeks—before chalking up a lack of response to an SSRI failure. Very often a patient is started on, say, 50 mg of sertraline and never titrated up. Many patients will not respond to the starting dose; those who don’t should be tried on higher doses of a single agent until tolerability factors predominate, maximum approved dose is reached, or remission of symptoms is obtained.—Christopher White, MD, JD
3. What drug interactions with SSRIs should I be most concerned about?
Like other psychotropic medications, SSRIs interact with drugs in a number of ways. There are interactions that occur at the cytochrome (CYP) 450 level and can result in toxicity or loss of effect, interactions that increase the likelihood of bleeding, and interactions that can lead to serotonin syndrome.
CYP 450 interactions. Depending on the CYP substrates that the SSRI and the other medication act upon, the result could be an increased concentration of the other agent and increased or decreased concentrations of the SSRI. The additive toxicity that could result has the potential to result in rare SSRI-associated adverse events, such as seizures and syndrome of inappropriate antidiuretic hormone (SIADH). Three exceptions to the increased concentration interaction are codeine, tamoxifen, and clopidogrel. Codeine, which relies on CYP metabolism to morphine, may have less analgesic effect if given with a CYP inhibitor. Tamoxifen may not be converted to endoxifen if given with a CYP inhibitor, resulting in a potentially lower therapeutic effect. Theoretically, a similar interaction could occur with clopidogrel when a CYP inhibitor is administered concurrently.
Fluoxetine, fluvoxamine, and paroxetine are the SSRIs with the greatest likelihood of having a significant CYP 450 interaction by inhibiting the metabolism of medications mediated by CYP 2D6, CYP 1A2, and CYP 2C19.9,10 (A partial list of medications and drug classes mediated by these substrates appears in TABLE 2 .)
Risk of bleeding. Combining any SSRI with a nonsteroidal anti-inflammatory drug (NSAID) without the addition of an acid-suppressing agent would cause 1 in 250 patients to experience an upper GI bleed, a recent study found.11 One in 500 patients treated with an SSRI and an antiplatelet agent developed an upper GI bleed. If a patient taking an SSRI requires antiplatelet or anticoagulant therapy, it is crucial to alert him or her to the risk and to carefully review signs and symptoms of bleeding. Hepatitis C, cirrhosis, hepatic failure, and portal hypertension are independent causes of coagulopathy, so patients with any of these conditions face an elevated risk of bleeding and would need to be monitored more closely.11 Avoid prescribing fluoxetine for patients with severe liver disease; an SSRI with a shorter half-life would be a more appropriate choice.
Serotonin syndrome. Combining an SSRI with a drug that affects serotonin (venlafaxine, mirtazapine, and serotonin receptor agonists such as sumatriptan, TCAs, St. John’s wort, meperidine, and tryptophan) or a drug that exhibits monoamine oxidase inhibition properties (isocarboxazid, linezolid, phenelzine, phentermine, selegiline, and tranylcypromine) may lead to serotonin syndrome. This toxidrome is identified by autonomic instability, neuromuscular changes, and altered mental status in a patient who has ingested a substance that could elevate serotonin levels, but typically resolves within 24 hours after the serotonergic agent is discontinued.12
Because of the high risk of serotonin syndrome when a monoamine oxidase inhibitor (MAOI) is combined with an SSRI, do not prescribe a drug in this class until the patient has been off the SSRI for at least 2 weeks. Fluoxetine has a longer half-life, so a patient should be off of it for 5 weeks before taking an MAOI.13
TABLE 2
CYP 450 interactions: Beware of these drug pairings9,10,13-17
| SSRI* | Other medications |
|---|---|
| Fluoxetine | Aripiprazole Clopidogrel Codeine Dextromethorphan Diazepam Duloxetine Haloperidol Metoprolol Phenobarbital Phenytoin PPIs Risperidone Tamoxifen TCAs Tramadol Venlafaxine |
| Fluvoxamine | Amitriptyline Clopidogrel Clozapine Cyclobenzaprine Diazepam Imipramine Naproxen Phenobarbital Phenytoin PPIs Theophylline |
| Paroxetine | Aripiprazole Codeine Dextromethorphan Duloxetine Haloperidol Metoprolol Risperidone TCAs Tramadol Venlafaxine |
| *Sertraline is a modest CYP 2D6 inhibitor. | |
| CYP, cytochrome; PPIs, proton pump inhibitors; SSRI, selective serotonin reuptake inhibitor; TCAs, tricyclic antidepressants. | |
4. What precautions are necessary when starting a patient on an SSRI or modifying therapy?
Dosing is the initial concern, with adjustments made based on specific patient factors. Elderly patients should be started on a low dose and titrated up more slowly than younger patients, for example. Low starting doses are also recommended for patients with hepatic dysfunction.14-17
“Start low and go slow” is a good rule to follow when prescribing an SSRI to anyone whom you suspect may be intolerant to common side effects—a patient with GI symptoms associated with depression, for example.
Patient comorbidities affect choice of agent as well as dose. For a patient with a creatinine clearance <20 mL/min, citalopram and escitalopram should be used with caution.14,15 Paroxetine should be initiated at lower doses for patients with a creatinine clearance <30 mL/min. While citalopram and escitalopram may not be ideal SSRIs for patients with renal impairment because of the potential for accumulation, they lack the substantial drug interactions and marked discontinuation syndrome seen with SSRIs such as paroxetine.
Two key concerns when changing from an SSRI to another class of antidepressant, or vice versa, are the increased risk of adverse events and a reduction in symptom control. A cross-titration strategy is appropriate for most such changes, provided the other drug is not an MAOI.
Discontinuation syndrome, which can be remembered by the mnemonic FInISH (Flu-like symptoms, Insomnia, Imbalance, Sensory disturbances, and Hyperarousal),18 is also a concern when antidepressant therapy is modified. The likelihood that a patient will develop discontinuation syndrome appears to be related to dose and agent, but not to the duration of treatment.19
While discontinuation syndrome is self-limiting, it is prudent to taper SSRI therapy whenever possible to minimize the risk of this adverse event, especially with paroxetine. A sample taper would be to reduce paroxetine by 10 mg per day every 5 to 7 days until the dose is down to 5 to 10 mg daily, then to discontinue the drug completely.20 Cross-titration to a different medication will also prevent withdrawal symptoms and minimize the risk that a patient who was taking the maximum dose of an SSRI will develop serotonin syndrome.21 Because of the long half-life of fluoxetine and its metabolite, norfluoxetine, fluoxetine is less likely than other SSRIs to cause discontinuation syndrome. Basically, it self-tapers.
5. What should I tell pregnant patients about the risks of SSRIs?
Be upfront with them that depression in pregnancy presents a dilemma.
Tell them that on the one hand, untreated depression has been found to increase the risk of preterm labor, low birth weight, decreased fetal growth, preeclampsia, and a worsening psychiatric condition after childbirth.22,23 In a 2006 study of 201 pregnant women with a previous diagnosis of depression, 43% relapsed during pregnancy. Those who were not taking antidepressants were 2.6 times more likely to relapse than women being treated for depression.24
Patients also need to be informed that antidepressant therapy during pregnancy carries its own set of risks. Five SSRIs are pregnancy category C,25 indicating either animal studies have found the drug to be harmful to fetuses and there are no well-done studies in pregnant women or that no animal studies and no human studies have tested its safety during pregnancy (the data were gathered after pregnancy). The sole exception is paroxetine, which has a D rating.25 Studies have linked paroxetine to an increased risk of cardiovascular malformations in babies who were exposed to it during the first trimester.26 These ratings may change shortly, however, as they are under FDA review. In May 2008, a new classification system for medication use in pregnancy was proposed.27 While this system would have great clinical utility, no target date for its release has been identified.
Use of SSRIs during the second and third trimester increases the risk of neonatal pulmonary hypertension. One study found that exposed newborns were 6 times more likely to experience persistent pulmonary hypertension, compared with newborns who were not exposed to SSRIs in the second and third trimesters.28
In addition, a derivative of the discontinuation syndrome is associated with neonatal withdrawal after in utero exposure, especially during the third trimester. Up to 30% of infants exposed to an SSRI may experience withdrawal symptoms, including increased or decreased muscle tone, jitteriness, feeding problems, irritability, sleep disturbance, and respiratory distress.29
Under the circumstances, the best you can do is to provide the patient with as much information as possible about the benefits and risks of each strategy. In any case, pregnant women suffering from depression should receive frequent follow-up and a referral to a mental health professional. Emphasize the importance of discussing their current medications and symptoms of depression with their obstetrician and psychiatrist or psychotherapist.
6. What can I tell adolescents and their parents about SSRI safety?
Explain that 4 SSRIs—escitalopram, fluoxetine, fluvoxamine, and sertraline—are approved for use in this age group, for specific indications. Fluoxetine and escitalopram are approved for the treatment of depression in children ≥8 and ≥12 years of age, respectively. Fluvoxamine, fluoxetine, and sertraline are approved for obsessive-compulsive disorder in children ≥8, ≥7, and ≥6 years, respectively.
You can also tell patients and parents that adolescents typically fare better when they receive a combination of medication and psychotherapy, compared with medications or therapy alone.
The FDA issued an initial warning about antidepressant use in pediatric and adolescent patients in 2003, based on data from 23 randomized controlled trials submitted by 8 different drug manufacturers.30 Most of the studies reported roughly twice the risk for suicidal ideation in adolescents taking SSRIs, compared with placebo. It is noteworthy, however, that there were no reports of completed suicides in the submitted trials.30 In fact, data suggest that despite some increased suicidal ideation when SSRIs are initiated, these antidepressants result in improved symptom control. In 2007, after a data review, the FDA issued an advisory warning physicians about increased suicidality in young patients.31
The FDA has recommended increased monitoring of adolescents taking SSRIs, with office visits once a week for the first month of treatment and every 2 weeks for the second month, followed by 1 visit every 3 months. This stringent schedule has proven difficult to adhere to. One study showed that only 5% of adolescent patients received this level of attention.32 The American Academy of Child and Adolescent Psychiatry and the American Psychiatric Association advocate an individualized treatment plan instead.33
If you prescribe SSRIs for depressed adolescents, educate patients and parents about the atypical presentation of depression that is common in patients of this age group. Advise them to watch closely for, and promptly report, increases in agitation, anxiety, impulsiveness, and restlessness, and symptoms of mania or hypomania.33
7. When should I refer a patient to a mental health professional?
Refer patients to a mental health specialist when the optimal treatment calls for a combination of psychotherapy and medication, as is the case with depressed adolescents. Referral is recommended, too, for any complex patients. Examples include elderly individuals who are taking multiple medications or have comorbidities that can interfere with optimal treatment, and pregnant women who need additional help weighing the benefits and risks of antidepressant therapy vs nonpharmacologic treatments.
Finally, referral is critical for any patient who does not respond to treatment, even after dose adjustments, for patients who need cross-tapering that may be better handled by a specialist, and certainly for any patient who you suspect may have suicidal ideation.
CORRESPONDENCE
Patricia R. Wigle, PharmD, BCPS, University of Cincinnati, The James L. Winkle College of Pharmacy, 3225 Eden Ave., Cincinnati, OH 45267; [email protected]
1. Thase ME. Are SNRIs more effective than SSRIs? Medscape. Available at: http://www.medscape.com/viewarticle/578077. Accessed July 28, 2008.
2. Lamb E. Top 200 prescription drugs of 2008. Pharmacy Times. May 2009. Available at: http://www.pharmacytimes.com/issue/pharmacy/2009/2009-05/RxFocusTop200Drugs-0509. Accessed August 17, 2009.
3. Cipriani A, Funkawa TA, Salanti G, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373:746-758.
4. Turner EH, Matthews AM, Linardatos E, et al. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med. 2008;358:252-260.
5. Hetrick SE, Merry SN, McKenzie J, et al. Selective serotonin reuptake inhibitors (SSRIs) for depressive disorders in children and adolescents. Cochrane Database Sys Rev. 2007(3);CD004851.-
6. Agency for Healthcare Research and Quality. Newer class of antidepressants similar in effectiveness, but side effects differ. January 24, 2007. Available at: http://www.ahrq.gov/news/press/pr2007/antideppr.htm. Accessed August 20, 2009.
7. Sanchez C, Hyttel J. Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cell Mol Neurobiol. 1999;19:467-489.
8. Dording CM, Mischoulon D, Peterson TJ, et al. The pharmacologic management of selective serotonin reuptake inhibitor-induced side effects: a survey of psychiatrists. Ann Clin Psychiatr. 2002;14:143-147.
9. Richelson E. Pharmacology of antidepressants. Mayo Clin Proc. 2001;76:511-527.
10. Goodnick PJ, Goldstein BJ. Selective serotonin reuptake inhibitors in affective disorders–I. Basic pharmacology. J Psychopharmacol. 1998;12(suppl B):S5-S20.
11. de Abajo FJ, Garcia-Rodriguez LA. Risk of upper gastrointestinal tract bleeding associated with selective serotonin reuptake inhibitors and venlafaxine therapy. Arch Gen Psychiatry. 2008;65:795-803.
12. Boyer E, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352:1112-1120.
13. Prozac [package insert]. Indianapolis: Eli Lilly and Company; 2008.
14. Celexa [package insert]. St. Louis: Forest Pharmaceuticals, Inc.; 2008.
15. Lexapro [package insert]. St. Louis: Forest Pharmaceuticals, Inc.; 2008.
16. Paxil CR [package insert]. Research Triangle Park, NC: Glaxo SmithKline; 2008.
17. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmith-Kline; 2008.
18. Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46:464-494.
19. Baldwin D, Montgomery SA, Nil R, et al. Discontinuation symptoms in depression and anxiety disorders. Int J Neuropsychopharmacol. 2007;10:73-84.
20. Shelton RC. Steps following attainment of remission: discontinuation of antidepressant therapy. Prim Care Companion J Clin Psychiatry. 2001;3:168-174.
21. van Geffen EC, Hugtenburg JG, Heerdink ER, et al. Discontinuation symptoms in users of selective serotonin reuptake inhibitors in clinical practice: tapering versus abrupt discontinuation. Eur J Clin Pharmacol. 2005;61:303-307.
22. Alder J, Fink N, Bitzer J, et al. Depression and anxiety during pregnancy: a risk factor for obstetric, fetal and neonatal outcome? A critical review of the literature. J Matern Fetal Neonatal Med. 2007;20:189-209.
23. ACOG Committee on Practice Bulletins—Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists No. 92, April 2008. Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol. 2008;111:1001-1020.
24. Cohen LS, Altshuler LL, Harlow BL, et al. Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA. 2006;295:499-507.
25. US Health and Human Services. Pregnancy and medicines: frequently asked questions. Available at: http://womenshealth.gov/FAQ/pregnancy-medicines.cfm. Last updated May 1, 2007. Accessed December 17, 2009.
26. Food and Drug Administration. Public Health Advisory Paroxetine. Available at: www.drugs.com/news/fda-public-health-advisory-paroxetine-1646.html. Accessed December 17, 2009.
27. FDA proposes new rule to provide updated information on the use of prescription drugs and biological products during pregnancy and breastfeeding. May 28, 2008. Available at: www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2008/ucm116902.htm. Accessed December 17, 2009.
28. Chambers CD, Hernandez-Diaz S, Van Marter LS, et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med. 2006;354:579-587.
29. Levinson-Castiel R, Merlob P, Linder N, et al. Neonatal abstinence syndrome after in utero exposure to selective serotonin reuptake inhibitors in term infants. Arch Pediatr Adolesc Med. 2006;160:173-176.
30. Food and Drug Administration. Relationship between psychotropic drugs and pediatric suicidality. August 16, 2004. Available at: http://www.fda.gov/ohrms/dockets/ac/04/briefing/2004-4065b1-10-TAB08-Hammads-Review.pdf. Accessed August 20, 2009.
31. Food and Drug Administration. FDA proposes new warnings about suicidal thinking, behavior in young adults who take antidepressant medications. May 2, 2007. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2007/ucm108905.htm. Accessed August 20, 2009.
32. Morrato EH, Libby AM, Orton HD, et al. Frequency of provider contact after FDA advisory on risk of pediatric suicidality with SSRIs. Am J Psychiatry. 2008;165:42-50.
33. Hammerness PG, Vivas FM, Geller DA. Selective serotonin reuptake inhibitors in pediatric psychopharmacology: a review of the evidence. J Pediatr. 2006;148:158-165.
• Avoid prescribing a highly activating SSRI, such as fluoxetine, for patients for whom agitation is a presenting symptom. B
• Consider citalopram and escitalopram, which have less potential for drug interactions and less complex dose titration compared with other SSRIs, as first-line agents for depressed patients with complex medication profiles. C
• For patients with sexual side effects caused by SSRIs, consider augmentation therapy with bupropion or mirtazapine; for male sexual dysfunction, a trial of a phosphodiesterase inhibitor is another alternative. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Shortly after the US Food and Drug Administration (FDA) approved fluoxetine for the treatment of major depression nearly a quarter of a century ago, Prozac became a household name. And, as a handful of additional selective serotonin reuptake inhibitors (SSRIs) were approved, the popularity of this category of antidepressant continued to grow.1 In 2008, 5 of the 6 SSRIs on the US market (fluvoxamine was the exception) were among the 200 most frequently dispensed prescription drugs.2
While much has been written about the properties of SSRIs, uncertainty about many features of particular agents and what should be considered in selecting an antidepressant for a particular patient remains. To help clear up the confusion, we’ve addressed 7 questions about SSRIs that we are often asked. Although there are no simple answers, the evidence presented, both in the answers and in the tables that follow, will help primary care physicians make more informed choices for patients who require antidepressant therapy.
1. Which is the best SSRI to start a patient on?
A recent meta-analysis of 117 head-to-head studies assessed the efficacy and acceptability of 12 newer antidepressants, including all 6 SSRIs (citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, and sertraline).3 The researchers found 2 SSRIs, sertraline and escitalopram, to be superior to the other medications studied on both counts. A choice between sertraline or escitalopram may be a reasonable starting point in many cases, but it is impossible to recommend 1 or 2 SSRIs that are effective for all patients. There are several reasons for this.
The first is addressed by the Cipriani meta-analysis. The researchers assessed the efficacy of initial antidepressant therapy at 8 weeks, so the results cannot be extrapolated to long-term response rates or acceptability. (For a detailed review of the meta-analysis, see “Try these 2 drugs first for depression,” J Fam Pract. 2009;58:365-369.)
A second reason is the substantial publication bias associated with studies of antidepressants. Turner et al assessed 74 studies registered with the FDA, determined whether the results were positive or negative, and categorized the studies based on publication status.4 Their conclusions: 97% of studies with positive findings (n=38) were published.
The majority of the remaining studies, all of which were determined to have negative findings, were either not published or published in a manner that suggested a positive outcome. When these additional studies are taken into account, the percentage of published studies with a positive response drops to 51%.4 In addition, the effect size of each agent is reduced when all the studies are included.
There are many confounding variables associated with publication, so Turner and his colleagues were unable to definitively determine the reason for the disparity. Nonetheless, their evaluation raises questions about the reported effectiveness, not only of SSRIs, but of antidepressants in general.
A recent Cochrane review of SSRIs for treatment of depression in children and adolescents (a topic covered in greater detail in the answer to Question 6) raised similar concerns. The reviewers cited study methodology and recruitment methods as potential sources of the conflicting results they found.5
Finally, individual differences, such as age, comorbidities, and medication history, require an individualized approach to SSRI treatment.
2. Which side effects are common to all SSRIs, and which can be resolved by switching agents?
As a class, SSRIs are well tolerated, but they do have some common adverse effects, most notably gastrointestinal (GI) problems, sexual dysfunction, and sleep disturbances. There are also considerable differences in SSRI profiles and a few adverse effects that a switch to another SSRI may alleviate or resolve.
Compared with other SSRIs, for example, sertraline has a higher risk of diarrhea, but this adverse event does not usually lead to medication discontinuation. If it is bothersome to the patient or does not resolve with continued therapy, a change to another agent might eliminate this adverse effect.
Weight gain has been found to be more significant with paroxetine than with fluoxetine or sertraline, which may be due to the anticholinergic action of the drug.6 For someone who is particularly concerned about extra weight, avoiding paroxetine in the first place, or changing a patient who’s already taking it to fluoxetine or sertraline, might be an effective treatment strategy.
Overall, paroxetine has more anticholinergic side effects than other SSRIs, the likely result of its higher affinity for muscarinic receptors ( TABLE 1 ). Its potential sedating effect,7 which can be extremely disturbing to some patients, may be a desired feature for others. A patient with insomnia, for example, might benefit from taking paroxetine at bedtime or switching to a less activating SSRI, such as citalopram or escitalopram. Excessive activation, such as significant insomnia, may also be a warning sign of undiagnosed bipolar disorder. Thus, careful screening is needed prior to switching medications or adding a hypnotic agent. (See the box below “Treating depression in primary care: Tips from a psychiatrist”.)
A switch to another SSRI is not the only way to alleviate a troublesome side effect, of course. Insomnia can also be managed by adding a short course of trazodone to the drug regimen or by switching the patient to mirtazapine or a tricyclic antidepressant (TCA).8
Augmentation with either bupropion or mirtazapine may alleviate sexual side effects and should be tried prior to switching the patient to a different antidepressant; however, the positive effects of augmentation should be balanced with any additional adverse events either agent may cause. For male sexual dysfunction, a trial of a phosphodiesterase inhibitor such as sildenafil is another alternative.
TABLE 1
SSRIs: Neurotransmitter affinity and side effects9
| Neurotransmitter/enzyme | SSRIs with most potent affinity* | Likely side effects |
|---|---|---|
| Serotonin | Paroxetine Sertraline Fluoxetine Citalopram Fluvoxamine | Sexual dysfunction, including anorgasmy; GI disturbance; activation |
| Dopamine | Sertraline Paroxetine Fluoxetine Fluvoxamine Citalopram | Extrapyramidal |
| Norepinephrine | Paroxetine Fluoxetine Sertraline Fluvoxamine Citalopram | Tremor; tachycardia; elevated BP |
| Muscarinic receptors | Paroxetine Sertraline Fluoxetine Citalopram Fluvoxamine | Anticholinergic (blurred vision, constipation, dry mouth) |
| *SSRIs listed in order of potency; escitalopram would be expected to have effects comparable to citalopram. | ||
| BP, blood pressure; GI, gastrointestinal; SSRI, selective serotonin reuptake inhibitors. | ||
- Use a validated instrument for depression screening and monitoring treatment success—preferably the PHQ-9 (http://www.lphi.org/LPHIadmin/uploads/.PHQ-9-Review-Kroenke-63754.PDF), which is validated for primary care practice. Use it at subsequent visits to track treatment response. The goal is not just response to treatment but remission of depressive symptoms, both of which are correlated with a decrease in the PHQ-9 total score.
- Take a careful history of bipolar symptoms and family history before starting a new patient on an SSRI. The DIGFAST mnemonic (http://www.usmle-forums.com/usmle-step-1-mnemonics/793-dig-fast-mnemonic-mania-symptoms.html) can help. Failure to respond to an SSRI or exacerbation of symptoms such as restlessness or insomnia once treatment has started should prompt you to consider a missed diagnosis.
- Consider augmentation strategies for patients who show partial improvement on an SSRI, rather than throwing out the gain with a wholesale switch to another drug. If a patient is only 30% better on the first agent you prescribe, try adding a second agent, which may significantly boost the response rate. Suggestions for SSRI augmentation include lithium, thyroid hormone, bupropion, and aripiprazole.
- Make sure the patient has been on a therapeutic dose for a sufficient period of time—at least 4 weeks—before chalking up a lack of response to an SSRI failure. Very often a patient is started on, say, 50 mg of sertraline and never titrated up. Many patients will not respond to the starting dose; those who don’t should be tried on higher doses of a single agent until tolerability factors predominate, maximum approved dose is reached, or remission of symptoms is obtained.—Christopher White, MD, JD
3. What drug interactions with SSRIs should I be most concerned about?
Like other psychotropic medications, SSRIs interact with drugs in a number of ways. There are interactions that occur at the cytochrome (CYP) 450 level and can result in toxicity or loss of effect, interactions that increase the likelihood of bleeding, and interactions that can lead to serotonin syndrome.
CYP 450 interactions. Depending on the CYP substrates that the SSRI and the other medication act upon, the result could be an increased concentration of the other agent and increased or decreased concentrations of the SSRI. The additive toxicity that could result has the potential to result in rare SSRI-associated adverse events, such as seizures and syndrome of inappropriate antidiuretic hormone (SIADH). Three exceptions to the increased concentration interaction are codeine, tamoxifen, and clopidogrel. Codeine, which relies on CYP metabolism to morphine, may have less analgesic effect if given with a CYP inhibitor. Tamoxifen may not be converted to endoxifen if given with a CYP inhibitor, resulting in a potentially lower therapeutic effect. Theoretically, a similar interaction could occur with clopidogrel when a CYP inhibitor is administered concurrently.
Fluoxetine, fluvoxamine, and paroxetine are the SSRIs with the greatest likelihood of having a significant CYP 450 interaction by inhibiting the metabolism of medications mediated by CYP 2D6, CYP 1A2, and CYP 2C19.9,10 (A partial list of medications and drug classes mediated by these substrates appears in TABLE 2 .)
Risk of bleeding. Combining any SSRI with a nonsteroidal anti-inflammatory drug (NSAID) without the addition of an acid-suppressing agent would cause 1 in 250 patients to experience an upper GI bleed, a recent study found.11 One in 500 patients treated with an SSRI and an antiplatelet agent developed an upper GI bleed. If a patient taking an SSRI requires antiplatelet or anticoagulant therapy, it is crucial to alert him or her to the risk and to carefully review signs and symptoms of bleeding. Hepatitis C, cirrhosis, hepatic failure, and portal hypertension are independent causes of coagulopathy, so patients with any of these conditions face an elevated risk of bleeding and would need to be monitored more closely.11 Avoid prescribing fluoxetine for patients with severe liver disease; an SSRI with a shorter half-life would be a more appropriate choice.
Serotonin syndrome. Combining an SSRI with a drug that affects serotonin (venlafaxine, mirtazapine, and serotonin receptor agonists such as sumatriptan, TCAs, St. John’s wort, meperidine, and tryptophan) or a drug that exhibits monoamine oxidase inhibition properties (isocarboxazid, linezolid, phenelzine, phentermine, selegiline, and tranylcypromine) may lead to serotonin syndrome. This toxidrome is identified by autonomic instability, neuromuscular changes, and altered mental status in a patient who has ingested a substance that could elevate serotonin levels, but typically resolves within 24 hours after the serotonergic agent is discontinued.12
Because of the high risk of serotonin syndrome when a monoamine oxidase inhibitor (MAOI) is combined with an SSRI, do not prescribe a drug in this class until the patient has been off the SSRI for at least 2 weeks. Fluoxetine has a longer half-life, so a patient should be off of it for 5 weeks before taking an MAOI.13
TABLE 2
CYP 450 interactions: Beware of these drug pairings9,10,13-17
| SSRI* | Other medications |
|---|---|
| Fluoxetine | Aripiprazole Clopidogrel Codeine Dextromethorphan Diazepam Duloxetine Haloperidol Metoprolol Phenobarbital Phenytoin PPIs Risperidone Tamoxifen TCAs Tramadol Venlafaxine |
| Fluvoxamine | Amitriptyline Clopidogrel Clozapine Cyclobenzaprine Diazepam Imipramine Naproxen Phenobarbital Phenytoin PPIs Theophylline |
| Paroxetine | Aripiprazole Codeine Dextromethorphan Duloxetine Haloperidol Metoprolol Risperidone TCAs Tramadol Venlafaxine |
| *Sertraline is a modest CYP 2D6 inhibitor. | |
| CYP, cytochrome; PPIs, proton pump inhibitors; SSRI, selective serotonin reuptake inhibitor; TCAs, tricyclic antidepressants. | |
4. What precautions are necessary when starting a patient on an SSRI or modifying therapy?
Dosing is the initial concern, with adjustments made based on specific patient factors. Elderly patients should be started on a low dose and titrated up more slowly than younger patients, for example. Low starting doses are also recommended for patients with hepatic dysfunction.14-17
“Start low and go slow” is a good rule to follow when prescribing an SSRI to anyone whom you suspect may be intolerant to common side effects—a patient with GI symptoms associated with depression, for example.
Patient comorbidities affect choice of agent as well as dose. For a patient with a creatinine clearance <20 mL/min, citalopram and escitalopram should be used with caution.14,15 Paroxetine should be initiated at lower doses for patients with a creatinine clearance <30 mL/min. While citalopram and escitalopram may not be ideal SSRIs for patients with renal impairment because of the potential for accumulation, they lack the substantial drug interactions and marked discontinuation syndrome seen with SSRIs such as paroxetine.
Two key concerns when changing from an SSRI to another class of antidepressant, or vice versa, are the increased risk of adverse events and a reduction in symptom control. A cross-titration strategy is appropriate for most such changes, provided the other drug is not an MAOI.
Discontinuation syndrome, which can be remembered by the mnemonic FInISH (Flu-like symptoms, Insomnia, Imbalance, Sensory disturbances, and Hyperarousal),18 is also a concern when antidepressant therapy is modified. The likelihood that a patient will develop discontinuation syndrome appears to be related to dose and agent, but not to the duration of treatment.19
While discontinuation syndrome is self-limiting, it is prudent to taper SSRI therapy whenever possible to minimize the risk of this adverse event, especially with paroxetine. A sample taper would be to reduce paroxetine by 10 mg per day every 5 to 7 days until the dose is down to 5 to 10 mg daily, then to discontinue the drug completely.20 Cross-titration to a different medication will also prevent withdrawal symptoms and minimize the risk that a patient who was taking the maximum dose of an SSRI will develop serotonin syndrome.21 Because of the long half-life of fluoxetine and its metabolite, norfluoxetine, fluoxetine is less likely than other SSRIs to cause discontinuation syndrome. Basically, it self-tapers.
5. What should I tell pregnant patients about the risks of SSRIs?
Be upfront with them that depression in pregnancy presents a dilemma.
Tell them that on the one hand, untreated depression has been found to increase the risk of preterm labor, low birth weight, decreased fetal growth, preeclampsia, and a worsening psychiatric condition after childbirth.22,23 In a 2006 study of 201 pregnant women with a previous diagnosis of depression, 43% relapsed during pregnancy. Those who were not taking antidepressants were 2.6 times more likely to relapse than women being treated for depression.24
Patients also need to be informed that antidepressant therapy during pregnancy carries its own set of risks. Five SSRIs are pregnancy category C,25 indicating either animal studies have found the drug to be harmful to fetuses and there are no well-done studies in pregnant women or that no animal studies and no human studies have tested its safety during pregnancy (the data were gathered after pregnancy). The sole exception is paroxetine, which has a D rating.25 Studies have linked paroxetine to an increased risk of cardiovascular malformations in babies who were exposed to it during the first trimester.26 These ratings may change shortly, however, as they are under FDA review. In May 2008, a new classification system for medication use in pregnancy was proposed.27 While this system would have great clinical utility, no target date for its release has been identified.
Use of SSRIs during the second and third trimester increases the risk of neonatal pulmonary hypertension. One study found that exposed newborns were 6 times more likely to experience persistent pulmonary hypertension, compared with newborns who were not exposed to SSRIs in the second and third trimesters.28
In addition, a derivative of the discontinuation syndrome is associated with neonatal withdrawal after in utero exposure, especially during the third trimester. Up to 30% of infants exposed to an SSRI may experience withdrawal symptoms, including increased or decreased muscle tone, jitteriness, feeding problems, irritability, sleep disturbance, and respiratory distress.29
Under the circumstances, the best you can do is to provide the patient with as much information as possible about the benefits and risks of each strategy. In any case, pregnant women suffering from depression should receive frequent follow-up and a referral to a mental health professional. Emphasize the importance of discussing their current medications and symptoms of depression with their obstetrician and psychiatrist or psychotherapist.
6. What can I tell adolescents and their parents about SSRI safety?
Explain that 4 SSRIs—escitalopram, fluoxetine, fluvoxamine, and sertraline—are approved for use in this age group, for specific indications. Fluoxetine and escitalopram are approved for the treatment of depression in children ≥8 and ≥12 years of age, respectively. Fluvoxamine, fluoxetine, and sertraline are approved for obsessive-compulsive disorder in children ≥8, ≥7, and ≥6 years, respectively.
You can also tell patients and parents that adolescents typically fare better when they receive a combination of medication and psychotherapy, compared with medications or therapy alone.
The FDA issued an initial warning about antidepressant use in pediatric and adolescent patients in 2003, based on data from 23 randomized controlled trials submitted by 8 different drug manufacturers.30 Most of the studies reported roughly twice the risk for suicidal ideation in adolescents taking SSRIs, compared with placebo. It is noteworthy, however, that there were no reports of completed suicides in the submitted trials.30 In fact, data suggest that despite some increased suicidal ideation when SSRIs are initiated, these antidepressants result in improved symptom control. In 2007, after a data review, the FDA issued an advisory warning physicians about increased suicidality in young patients.31
The FDA has recommended increased monitoring of adolescents taking SSRIs, with office visits once a week for the first month of treatment and every 2 weeks for the second month, followed by 1 visit every 3 months. This stringent schedule has proven difficult to adhere to. One study showed that only 5% of adolescent patients received this level of attention.32 The American Academy of Child and Adolescent Psychiatry and the American Psychiatric Association advocate an individualized treatment plan instead.33
If you prescribe SSRIs for depressed adolescents, educate patients and parents about the atypical presentation of depression that is common in patients of this age group. Advise them to watch closely for, and promptly report, increases in agitation, anxiety, impulsiveness, and restlessness, and symptoms of mania or hypomania.33
7. When should I refer a patient to a mental health professional?
Refer patients to a mental health specialist when the optimal treatment calls for a combination of psychotherapy and medication, as is the case with depressed adolescents. Referral is recommended, too, for any complex patients. Examples include elderly individuals who are taking multiple medications or have comorbidities that can interfere with optimal treatment, and pregnant women who need additional help weighing the benefits and risks of antidepressant therapy vs nonpharmacologic treatments.
Finally, referral is critical for any patient who does not respond to treatment, even after dose adjustments, for patients who need cross-tapering that may be better handled by a specialist, and certainly for any patient who you suspect may have suicidal ideation.
CORRESPONDENCE
Patricia R. Wigle, PharmD, BCPS, University of Cincinnati, The James L. Winkle College of Pharmacy, 3225 Eden Ave., Cincinnati, OH 45267; [email protected]
• Avoid prescribing a highly activating SSRI, such as fluoxetine, for patients for whom agitation is a presenting symptom. B
• Consider citalopram and escitalopram, which have less potential for drug interactions and less complex dose titration compared with other SSRIs, as first-line agents for depressed patients with complex medication profiles. C
• For patients with sexual side effects caused by SSRIs, consider augmentation therapy with bupropion or mirtazapine; for male sexual dysfunction, a trial of a phosphodiesterase inhibitor is another alternative. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Shortly after the US Food and Drug Administration (FDA) approved fluoxetine for the treatment of major depression nearly a quarter of a century ago, Prozac became a household name. And, as a handful of additional selective serotonin reuptake inhibitors (SSRIs) were approved, the popularity of this category of antidepressant continued to grow.1 In 2008, 5 of the 6 SSRIs on the US market (fluvoxamine was the exception) were among the 200 most frequently dispensed prescription drugs.2
While much has been written about the properties of SSRIs, uncertainty about many features of particular agents and what should be considered in selecting an antidepressant for a particular patient remains. To help clear up the confusion, we’ve addressed 7 questions about SSRIs that we are often asked. Although there are no simple answers, the evidence presented, both in the answers and in the tables that follow, will help primary care physicians make more informed choices for patients who require antidepressant therapy.
1. Which is the best SSRI to start a patient on?
A recent meta-analysis of 117 head-to-head studies assessed the efficacy and acceptability of 12 newer antidepressants, including all 6 SSRIs (citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, and sertraline).3 The researchers found 2 SSRIs, sertraline and escitalopram, to be superior to the other medications studied on both counts. A choice between sertraline or escitalopram may be a reasonable starting point in many cases, but it is impossible to recommend 1 or 2 SSRIs that are effective for all patients. There are several reasons for this.
The first is addressed by the Cipriani meta-analysis. The researchers assessed the efficacy of initial antidepressant therapy at 8 weeks, so the results cannot be extrapolated to long-term response rates or acceptability. (For a detailed review of the meta-analysis, see “Try these 2 drugs first for depression,” J Fam Pract. 2009;58:365-369.)
A second reason is the substantial publication bias associated with studies of antidepressants. Turner et al assessed 74 studies registered with the FDA, determined whether the results were positive or negative, and categorized the studies based on publication status.4 Their conclusions: 97% of studies with positive findings (n=38) were published.
The majority of the remaining studies, all of which were determined to have negative findings, were either not published or published in a manner that suggested a positive outcome. When these additional studies are taken into account, the percentage of published studies with a positive response drops to 51%.4 In addition, the effect size of each agent is reduced when all the studies are included.
There are many confounding variables associated with publication, so Turner and his colleagues were unable to definitively determine the reason for the disparity. Nonetheless, their evaluation raises questions about the reported effectiveness, not only of SSRIs, but of antidepressants in general.
A recent Cochrane review of SSRIs for treatment of depression in children and adolescents (a topic covered in greater detail in the answer to Question 6) raised similar concerns. The reviewers cited study methodology and recruitment methods as potential sources of the conflicting results they found.5
Finally, individual differences, such as age, comorbidities, and medication history, require an individualized approach to SSRI treatment.
2. Which side effects are common to all SSRIs, and which can be resolved by switching agents?
As a class, SSRIs are well tolerated, but they do have some common adverse effects, most notably gastrointestinal (GI) problems, sexual dysfunction, and sleep disturbances. There are also considerable differences in SSRI profiles and a few adverse effects that a switch to another SSRI may alleviate or resolve.
Compared with other SSRIs, for example, sertraline has a higher risk of diarrhea, but this adverse event does not usually lead to medication discontinuation. If it is bothersome to the patient or does not resolve with continued therapy, a change to another agent might eliminate this adverse effect.
Weight gain has been found to be more significant with paroxetine than with fluoxetine or sertraline, which may be due to the anticholinergic action of the drug.6 For someone who is particularly concerned about extra weight, avoiding paroxetine in the first place, or changing a patient who’s already taking it to fluoxetine or sertraline, might be an effective treatment strategy.
Overall, paroxetine has more anticholinergic side effects than other SSRIs, the likely result of its higher affinity for muscarinic receptors ( TABLE 1 ). Its potential sedating effect,7 which can be extremely disturbing to some patients, may be a desired feature for others. A patient with insomnia, for example, might benefit from taking paroxetine at bedtime or switching to a less activating SSRI, such as citalopram or escitalopram. Excessive activation, such as significant insomnia, may also be a warning sign of undiagnosed bipolar disorder. Thus, careful screening is needed prior to switching medications or adding a hypnotic agent. (See the box below “Treating depression in primary care: Tips from a psychiatrist”.)
A switch to another SSRI is not the only way to alleviate a troublesome side effect, of course. Insomnia can also be managed by adding a short course of trazodone to the drug regimen or by switching the patient to mirtazapine or a tricyclic antidepressant (TCA).8
Augmentation with either bupropion or mirtazapine may alleviate sexual side effects and should be tried prior to switching the patient to a different antidepressant; however, the positive effects of augmentation should be balanced with any additional adverse events either agent may cause. For male sexual dysfunction, a trial of a phosphodiesterase inhibitor such as sildenafil is another alternative.
TABLE 1
SSRIs: Neurotransmitter affinity and side effects9
| Neurotransmitter/enzyme | SSRIs with most potent affinity* | Likely side effects |
|---|---|---|
| Serotonin | Paroxetine Sertraline Fluoxetine Citalopram Fluvoxamine | Sexual dysfunction, including anorgasmy; GI disturbance; activation |
| Dopamine | Sertraline Paroxetine Fluoxetine Fluvoxamine Citalopram | Extrapyramidal |
| Norepinephrine | Paroxetine Fluoxetine Sertraline Fluvoxamine Citalopram | Tremor; tachycardia; elevated BP |
| Muscarinic receptors | Paroxetine Sertraline Fluoxetine Citalopram Fluvoxamine | Anticholinergic (blurred vision, constipation, dry mouth) |
| *SSRIs listed in order of potency; escitalopram would be expected to have effects comparable to citalopram. | ||
| BP, blood pressure; GI, gastrointestinal; SSRI, selective serotonin reuptake inhibitors. | ||
- Use a validated instrument for depression screening and monitoring treatment success—preferably the PHQ-9 (http://www.lphi.org/LPHIadmin/uploads/.PHQ-9-Review-Kroenke-63754.PDF), which is validated for primary care practice. Use it at subsequent visits to track treatment response. The goal is not just response to treatment but remission of depressive symptoms, both of which are correlated with a decrease in the PHQ-9 total score.
- Take a careful history of bipolar symptoms and family history before starting a new patient on an SSRI. The DIGFAST mnemonic (http://www.usmle-forums.com/usmle-step-1-mnemonics/793-dig-fast-mnemonic-mania-symptoms.html) can help. Failure to respond to an SSRI or exacerbation of symptoms such as restlessness or insomnia once treatment has started should prompt you to consider a missed diagnosis.
- Consider augmentation strategies for patients who show partial improvement on an SSRI, rather than throwing out the gain with a wholesale switch to another drug. If a patient is only 30% better on the first agent you prescribe, try adding a second agent, which may significantly boost the response rate. Suggestions for SSRI augmentation include lithium, thyroid hormone, bupropion, and aripiprazole.
- Make sure the patient has been on a therapeutic dose for a sufficient period of time—at least 4 weeks—before chalking up a lack of response to an SSRI failure. Very often a patient is started on, say, 50 mg of sertraline and never titrated up. Many patients will not respond to the starting dose; those who don’t should be tried on higher doses of a single agent until tolerability factors predominate, maximum approved dose is reached, or remission of symptoms is obtained.—Christopher White, MD, JD
3. What drug interactions with SSRIs should I be most concerned about?
Like other psychotropic medications, SSRIs interact with drugs in a number of ways. There are interactions that occur at the cytochrome (CYP) 450 level and can result in toxicity or loss of effect, interactions that increase the likelihood of bleeding, and interactions that can lead to serotonin syndrome.
CYP 450 interactions. Depending on the CYP substrates that the SSRI and the other medication act upon, the result could be an increased concentration of the other agent and increased or decreased concentrations of the SSRI. The additive toxicity that could result has the potential to result in rare SSRI-associated adverse events, such as seizures and syndrome of inappropriate antidiuretic hormone (SIADH). Three exceptions to the increased concentration interaction are codeine, tamoxifen, and clopidogrel. Codeine, which relies on CYP metabolism to morphine, may have less analgesic effect if given with a CYP inhibitor. Tamoxifen may not be converted to endoxifen if given with a CYP inhibitor, resulting in a potentially lower therapeutic effect. Theoretically, a similar interaction could occur with clopidogrel when a CYP inhibitor is administered concurrently.
Fluoxetine, fluvoxamine, and paroxetine are the SSRIs with the greatest likelihood of having a significant CYP 450 interaction by inhibiting the metabolism of medications mediated by CYP 2D6, CYP 1A2, and CYP 2C19.9,10 (A partial list of medications and drug classes mediated by these substrates appears in TABLE 2 .)
Risk of bleeding. Combining any SSRI with a nonsteroidal anti-inflammatory drug (NSAID) without the addition of an acid-suppressing agent would cause 1 in 250 patients to experience an upper GI bleed, a recent study found.11 One in 500 patients treated with an SSRI and an antiplatelet agent developed an upper GI bleed. If a patient taking an SSRI requires antiplatelet or anticoagulant therapy, it is crucial to alert him or her to the risk and to carefully review signs and symptoms of bleeding. Hepatitis C, cirrhosis, hepatic failure, and portal hypertension are independent causes of coagulopathy, so patients with any of these conditions face an elevated risk of bleeding and would need to be monitored more closely.11 Avoid prescribing fluoxetine for patients with severe liver disease; an SSRI with a shorter half-life would be a more appropriate choice.
Serotonin syndrome. Combining an SSRI with a drug that affects serotonin (venlafaxine, mirtazapine, and serotonin receptor agonists such as sumatriptan, TCAs, St. John’s wort, meperidine, and tryptophan) or a drug that exhibits monoamine oxidase inhibition properties (isocarboxazid, linezolid, phenelzine, phentermine, selegiline, and tranylcypromine) may lead to serotonin syndrome. This toxidrome is identified by autonomic instability, neuromuscular changes, and altered mental status in a patient who has ingested a substance that could elevate serotonin levels, but typically resolves within 24 hours after the serotonergic agent is discontinued.12
Because of the high risk of serotonin syndrome when a monoamine oxidase inhibitor (MAOI) is combined with an SSRI, do not prescribe a drug in this class until the patient has been off the SSRI for at least 2 weeks. Fluoxetine has a longer half-life, so a patient should be off of it for 5 weeks before taking an MAOI.13
TABLE 2
CYP 450 interactions: Beware of these drug pairings9,10,13-17
| SSRI* | Other medications |
|---|---|
| Fluoxetine | Aripiprazole Clopidogrel Codeine Dextromethorphan Diazepam Duloxetine Haloperidol Metoprolol Phenobarbital Phenytoin PPIs Risperidone Tamoxifen TCAs Tramadol Venlafaxine |
| Fluvoxamine | Amitriptyline Clopidogrel Clozapine Cyclobenzaprine Diazepam Imipramine Naproxen Phenobarbital Phenytoin PPIs Theophylline |
| Paroxetine | Aripiprazole Codeine Dextromethorphan Duloxetine Haloperidol Metoprolol Risperidone TCAs Tramadol Venlafaxine |
| *Sertraline is a modest CYP 2D6 inhibitor. | |
| CYP, cytochrome; PPIs, proton pump inhibitors; SSRI, selective serotonin reuptake inhibitor; TCAs, tricyclic antidepressants. | |
4. What precautions are necessary when starting a patient on an SSRI or modifying therapy?
Dosing is the initial concern, with adjustments made based on specific patient factors. Elderly patients should be started on a low dose and titrated up more slowly than younger patients, for example. Low starting doses are also recommended for patients with hepatic dysfunction.14-17
“Start low and go slow” is a good rule to follow when prescribing an SSRI to anyone whom you suspect may be intolerant to common side effects—a patient with GI symptoms associated with depression, for example.
Patient comorbidities affect choice of agent as well as dose. For a patient with a creatinine clearance <20 mL/min, citalopram and escitalopram should be used with caution.14,15 Paroxetine should be initiated at lower doses for patients with a creatinine clearance <30 mL/min. While citalopram and escitalopram may not be ideal SSRIs for patients with renal impairment because of the potential for accumulation, they lack the substantial drug interactions and marked discontinuation syndrome seen with SSRIs such as paroxetine.
Two key concerns when changing from an SSRI to another class of antidepressant, or vice versa, are the increased risk of adverse events and a reduction in symptom control. A cross-titration strategy is appropriate for most such changes, provided the other drug is not an MAOI.
Discontinuation syndrome, which can be remembered by the mnemonic FInISH (Flu-like symptoms, Insomnia, Imbalance, Sensory disturbances, and Hyperarousal),18 is also a concern when antidepressant therapy is modified. The likelihood that a patient will develop discontinuation syndrome appears to be related to dose and agent, but not to the duration of treatment.19
While discontinuation syndrome is self-limiting, it is prudent to taper SSRI therapy whenever possible to minimize the risk of this adverse event, especially with paroxetine. A sample taper would be to reduce paroxetine by 10 mg per day every 5 to 7 days until the dose is down to 5 to 10 mg daily, then to discontinue the drug completely.20 Cross-titration to a different medication will also prevent withdrawal symptoms and minimize the risk that a patient who was taking the maximum dose of an SSRI will develop serotonin syndrome.21 Because of the long half-life of fluoxetine and its metabolite, norfluoxetine, fluoxetine is less likely than other SSRIs to cause discontinuation syndrome. Basically, it self-tapers.
5. What should I tell pregnant patients about the risks of SSRIs?
Be upfront with them that depression in pregnancy presents a dilemma.
Tell them that on the one hand, untreated depression has been found to increase the risk of preterm labor, low birth weight, decreased fetal growth, preeclampsia, and a worsening psychiatric condition after childbirth.22,23 In a 2006 study of 201 pregnant women with a previous diagnosis of depression, 43% relapsed during pregnancy. Those who were not taking antidepressants were 2.6 times more likely to relapse than women being treated for depression.24
Patients also need to be informed that antidepressant therapy during pregnancy carries its own set of risks. Five SSRIs are pregnancy category C,25 indicating either animal studies have found the drug to be harmful to fetuses and there are no well-done studies in pregnant women or that no animal studies and no human studies have tested its safety during pregnancy (the data were gathered after pregnancy). The sole exception is paroxetine, which has a D rating.25 Studies have linked paroxetine to an increased risk of cardiovascular malformations in babies who were exposed to it during the first trimester.26 These ratings may change shortly, however, as they are under FDA review. In May 2008, a new classification system for medication use in pregnancy was proposed.27 While this system would have great clinical utility, no target date for its release has been identified.
Use of SSRIs during the second and third trimester increases the risk of neonatal pulmonary hypertension. One study found that exposed newborns were 6 times more likely to experience persistent pulmonary hypertension, compared with newborns who were not exposed to SSRIs in the second and third trimesters.28
In addition, a derivative of the discontinuation syndrome is associated with neonatal withdrawal after in utero exposure, especially during the third trimester. Up to 30% of infants exposed to an SSRI may experience withdrawal symptoms, including increased or decreased muscle tone, jitteriness, feeding problems, irritability, sleep disturbance, and respiratory distress.29
Under the circumstances, the best you can do is to provide the patient with as much information as possible about the benefits and risks of each strategy. In any case, pregnant women suffering from depression should receive frequent follow-up and a referral to a mental health professional. Emphasize the importance of discussing their current medications and symptoms of depression with their obstetrician and psychiatrist or psychotherapist.
6. What can I tell adolescents and their parents about SSRI safety?
Explain that 4 SSRIs—escitalopram, fluoxetine, fluvoxamine, and sertraline—are approved for use in this age group, for specific indications. Fluoxetine and escitalopram are approved for the treatment of depression in children ≥8 and ≥12 years of age, respectively. Fluvoxamine, fluoxetine, and sertraline are approved for obsessive-compulsive disorder in children ≥8, ≥7, and ≥6 years, respectively.
You can also tell patients and parents that adolescents typically fare better when they receive a combination of medication and psychotherapy, compared with medications or therapy alone.
The FDA issued an initial warning about antidepressant use in pediatric and adolescent patients in 2003, based on data from 23 randomized controlled trials submitted by 8 different drug manufacturers.30 Most of the studies reported roughly twice the risk for suicidal ideation in adolescents taking SSRIs, compared with placebo. It is noteworthy, however, that there were no reports of completed suicides in the submitted trials.30 In fact, data suggest that despite some increased suicidal ideation when SSRIs are initiated, these antidepressants result in improved symptom control. In 2007, after a data review, the FDA issued an advisory warning physicians about increased suicidality in young patients.31
The FDA has recommended increased monitoring of adolescents taking SSRIs, with office visits once a week for the first month of treatment and every 2 weeks for the second month, followed by 1 visit every 3 months. This stringent schedule has proven difficult to adhere to. One study showed that only 5% of adolescent patients received this level of attention.32 The American Academy of Child and Adolescent Psychiatry and the American Psychiatric Association advocate an individualized treatment plan instead.33
If you prescribe SSRIs for depressed adolescents, educate patients and parents about the atypical presentation of depression that is common in patients of this age group. Advise them to watch closely for, and promptly report, increases in agitation, anxiety, impulsiveness, and restlessness, and symptoms of mania or hypomania.33
7. When should I refer a patient to a mental health professional?
Refer patients to a mental health specialist when the optimal treatment calls for a combination of psychotherapy and medication, as is the case with depressed adolescents. Referral is recommended, too, for any complex patients. Examples include elderly individuals who are taking multiple medications or have comorbidities that can interfere with optimal treatment, and pregnant women who need additional help weighing the benefits and risks of antidepressant therapy vs nonpharmacologic treatments.
Finally, referral is critical for any patient who does not respond to treatment, even after dose adjustments, for patients who need cross-tapering that may be better handled by a specialist, and certainly for any patient who you suspect may have suicidal ideation.
CORRESPONDENCE
Patricia R. Wigle, PharmD, BCPS, University of Cincinnati, The James L. Winkle College of Pharmacy, 3225 Eden Ave., Cincinnati, OH 45267; [email protected]
1. Thase ME. Are SNRIs more effective than SSRIs? Medscape. Available at: http://www.medscape.com/viewarticle/578077. Accessed July 28, 2008.
2. Lamb E. Top 200 prescription drugs of 2008. Pharmacy Times. May 2009. Available at: http://www.pharmacytimes.com/issue/pharmacy/2009/2009-05/RxFocusTop200Drugs-0509. Accessed August 17, 2009.
3. Cipriani A, Funkawa TA, Salanti G, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373:746-758.
4. Turner EH, Matthews AM, Linardatos E, et al. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med. 2008;358:252-260.
5. Hetrick SE, Merry SN, McKenzie J, et al. Selective serotonin reuptake inhibitors (SSRIs) for depressive disorders in children and adolescents. Cochrane Database Sys Rev. 2007(3);CD004851.-
6. Agency for Healthcare Research and Quality. Newer class of antidepressants similar in effectiveness, but side effects differ. January 24, 2007. Available at: http://www.ahrq.gov/news/press/pr2007/antideppr.htm. Accessed August 20, 2009.
7. Sanchez C, Hyttel J. Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cell Mol Neurobiol. 1999;19:467-489.
8. Dording CM, Mischoulon D, Peterson TJ, et al. The pharmacologic management of selective serotonin reuptake inhibitor-induced side effects: a survey of psychiatrists. Ann Clin Psychiatr. 2002;14:143-147.
9. Richelson E. Pharmacology of antidepressants. Mayo Clin Proc. 2001;76:511-527.
10. Goodnick PJ, Goldstein BJ. Selective serotonin reuptake inhibitors in affective disorders–I. Basic pharmacology. J Psychopharmacol. 1998;12(suppl B):S5-S20.
11. de Abajo FJ, Garcia-Rodriguez LA. Risk of upper gastrointestinal tract bleeding associated with selective serotonin reuptake inhibitors and venlafaxine therapy. Arch Gen Psychiatry. 2008;65:795-803.
12. Boyer E, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352:1112-1120.
13. Prozac [package insert]. Indianapolis: Eli Lilly and Company; 2008.
14. Celexa [package insert]. St. Louis: Forest Pharmaceuticals, Inc.; 2008.
15. Lexapro [package insert]. St. Louis: Forest Pharmaceuticals, Inc.; 2008.
16. Paxil CR [package insert]. Research Triangle Park, NC: Glaxo SmithKline; 2008.
17. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmith-Kline; 2008.
18. Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46:464-494.
19. Baldwin D, Montgomery SA, Nil R, et al. Discontinuation symptoms in depression and anxiety disorders. Int J Neuropsychopharmacol. 2007;10:73-84.
20. Shelton RC. Steps following attainment of remission: discontinuation of antidepressant therapy. Prim Care Companion J Clin Psychiatry. 2001;3:168-174.
21. van Geffen EC, Hugtenburg JG, Heerdink ER, et al. Discontinuation symptoms in users of selective serotonin reuptake inhibitors in clinical practice: tapering versus abrupt discontinuation. Eur J Clin Pharmacol. 2005;61:303-307.
22. Alder J, Fink N, Bitzer J, et al. Depression and anxiety during pregnancy: a risk factor for obstetric, fetal and neonatal outcome? A critical review of the literature. J Matern Fetal Neonatal Med. 2007;20:189-209.
23. ACOG Committee on Practice Bulletins—Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists No. 92, April 2008. Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol. 2008;111:1001-1020.
24. Cohen LS, Altshuler LL, Harlow BL, et al. Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA. 2006;295:499-507.
25. US Health and Human Services. Pregnancy and medicines: frequently asked questions. Available at: http://womenshealth.gov/FAQ/pregnancy-medicines.cfm. Last updated May 1, 2007. Accessed December 17, 2009.
26. Food and Drug Administration. Public Health Advisory Paroxetine. Available at: www.drugs.com/news/fda-public-health-advisory-paroxetine-1646.html. Accessed December 17, 2009.
27. FDA proposes new rule to provide updated information on the use of prescription drugs and biological products during pregnancy and breastfeeding. May 28, 2008. Available at: www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2008/ucm116902.htm. Accessed December 17, 2009.
28. Chambers CD, Hernandez-Diaz S, Van Marter LS, et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med. 2006;354:579-587.
29. Levinson-Castiel R, Merlob P, Linder N, et al. Neonatal abstinence syndrome after in utero exposure to selective serotonin reuptake inhibitors in term infants. Arch Pediatr Adolesc Med. 2006;160:173-176.
30. Food and Drug Administration. Relationship between psychotropic drugs and pediatric suicidality. August 16, 2004. Available at: http://www.fda.gov/ohrms/dockets/ac/04/briefing/2004-4065b1-10-TAB08-Hammads-Review.pdf. Accessed August 20, 2009.
31. Food and Drug Administration. FDA proposes new warnings about suicidal thinking, behavior in young adults who take antidepressant medications. May 2, 2007. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2007/ucm108905.htm. Accessed August 20, 2009.
32. Morrato EH, Libby AM, Orton HD, et al. Frequency of provider contact after FDA advisory on risk of pediatric suicidality with SSRIs. Am J Psychiatry. 2008;165:42-50.
33. Hammerness PG, Vivas FM, Geller DA. Selective serotonin reuptake inhibitors in pediatric psychopharmacology: a review of the evidence. J Pediatr. 2006;148:158-165.
1. Thase ME. Are SNRIs more effective than SSRIs? Medscape. Available at: http://www.medscape.com/viewarticle/578077. Accessed July 28, 2008.
2. Lamb E. Top 200 prescription drugs of 2008. Pharmacy Times. May 2009. Available at: http://www.pharmacytimes.com/issue/pharmacy/2009/2009-05/RxFocusTop200Drugs-0509. Accessed August 17, 2009.
3. Cipriani A, Funkawa TA, Salanti G, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373:746-758.
4. Turner EH, Matthews AM, Linardatos E, et al. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med. 2008;358:252-260.
5. Hetrick SE, Merry SN, McKenzie J, et al. Selective serotonin reuptake inhibitors (SSRIs) for depressive disorders in children and adolescents. Cochrane Database Sys Rev. 2007(3);CD004851.-
6. Agency for Healthcare Research and Quality. Newer class of antidepressants similar in effectiveness, but side effects differ. January 24, 2007. Available at: http://www.ahrq.gov/news/press/pr2007/antideppr.htm. Accessed August 20, 2009.
7. Sanchez C, Hyttel J. Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cell Mol Neurobiol. 1999;19:467-489.
8. Dording CM, Mischoulon D, Peterson TJ, et al. The pharmacologic management of selective serotonin reuptake inhibitor-induced side effects: a survey of psychiatrists. Ann Clin Psychiatr. 2002;14:143-147.
9. Richelson E. Pharmacology of antidepressants. Mayo Clin Proc. 2001;76:511-527.
10. Goodnick PJ, Goldstein BJ. Selective serotonin reuptake inhibitors in affective disorders–I. Basic pharmacology. J Psychopharmacol. 1998;12(suppl B):S5-S20.
11. de Abajo FJ, Garcia-Rodriguez LA. Risk of upper gastrointestinal tract bleeding associated with selective serotonin reuptake inhibitors and venlafaxine therapy. Arch Gen Psychiatry. 2008;65:795-803.
12. Boyer E, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352:1112-1120.
13. Prozac [package insert]. Indianapolis: Eli Lilly and Company; 2008.
14. Celexa [package insert]. St. Louis: Forest Pharmaceuticals, Inc.; 2008.
15. Lexapro [package insert]. St. Louis: Forest Pharmaceuticals, Inc.; 2008.
16. Paxil CR [package insert]. Research Triangle Park, NC: Glaxo SmithKline; 2008.
17. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmith-Kline; 2008.
18. Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46:464-494.
19. Baldwin D, Montgomery SA, Nil R, et al. Discontinuation symptoms in depression and anxiety disorders. Int J Neuropsychopharmacol. 2007;10:73-84.
20. Shelton RC. Steps following attainment of remission: discontinuation of antidepressant therapy. Prim Care Companion J Clin Psychiatry. 2001;3:168-174.
21. van Geffen EC, Hugtenburg JG, Heerdink ER, et al. Discontinuation symptoms in users of selective serotonin reuptake inhibitors in clinical practice: tapering versus abrupt discontinuation. Eur J Clin Pharmacol. 2005;61:303-307.
22. Alder J, Fink N, Bitzer J, et al. Depression and anxiety during pregnancy: a risk factor for obstetric, fetal and neonatal outcome? A critical review of the literature. J Matern Fetal Neonatal Med. 2007;20:189-209.
23. ACOG Committee on Practice Bulletins—Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists No. 92, April 2008. Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol. 2008;111:1001-1020.
24. Cohen LS, Altshuler LL, Harlow BL, et al. Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA. 2006;295:499-507.
25. US Health and Human Services. Pregnancy and medicines: frequently asked questions. Available at: http://womenshealth.gov/FAQ/pregnancy-medicines.cfm. Last updated May 1, 2007. Accessed December 17, 2009.
26. Food and Drug Administration. Public Health Advisory Paroxetine. Available at: www.drugs.com/news/fda-public-health-advisory-paroxetine-1646.html. Accessed December 17, 2009.
27. FDA proposes new rule to provide updated information on the use of prescription drugs and biological products during pregnancy and breastfeeding. May 28, 2008. Available at: www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2008/ucm116902.htm. Accessed December 17, 2009.
28. Chambers CD, Hernandez-Diaz S, Van Marter LS, et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med. 2006;354:579-587.
29. Levinson-Castiel R, Merlob P, Linder N, et al. Neonatal abstinence syndrome after in utero exposure to selective serotonin reuptake inhibitors in term infants. Arch Pediatr Adolesc Med. 2006;160:173-176.
30. Food and Drug Administration. Relationship between psychotropic drugs and pediatric suicidality. August 16, 2004. Available at: http://www.fda.gov/ohrms/dockets/ac/04/briefing/2004-4065b1-10-TAB08-Hammads-Review.pdf. Accessed August 20, 2009.
31. Food and Drug Administration. FDA proposes new warnings about suicidal thinking, behavior in young adults who take antidepressant medications. May 2, 2007. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2007/ucm108905.htm. Accessed August 20, 2009.
32. Morrato EH, Libby AM, Orton HD, et al. Frequency of provider contact after FDA advisory on risk of pediatric suicidality with SSRIs. Am J Psychiatry. 2008;165:42-50.
33. Hammerness PG, Vivas FM, Geller DA. Selective serotonin reuptake inhibitors in pediatric psychopharmacology: a review of the evidence. J Pediatr. 2006;148:158-165.
Is it stroke, or something else?
• Arrange for urgent transport to the hospital when a patient presents with stroke-like symptoms of acute onset, especially within the 3- to 6-hour therapeutic window. B
• Use a validated prehospital stroke identification algorithm such as the Face Arms Speech Time (FAST) test to identify possible acute stroke patients requiring urgent transport to the hospital. B
• Obtain a CBC and basic metabolic panel for all patients with signs and symptoms suggestive of stroke—and a blood alcohol, hepatic function, and toxicology screen, in select patients—to help rule out stroke mimics. C
• Ensure that patients undergo brain imaging to rule out stroke mimics before treatment for acute ischemic stroke is initiated. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Stroke is the third leading cause of death (claiming the life of 1 person every 3 to 4 minutes) and the No. 1 cause of adult disability in the United States.1 Advances in thrombolysis and clot removal can improve outcomes, but are dependent on swift and certain diagnosis. Amid the rush to ensure that treatment is initiated within the therapeutic window for cerebral reperfusion, “stroke mimics”—so called because of their ability to cause signs and symptoms similar to stroke—are sometimes mistaken for the real thing.
The prevalence of misdiagnosis ranges from about 4% of patients who receive tissue plasminogen activator (tPA) for reperfusion2 to 25% of patients who are rushed to the hospital because they are thought to be having a stroke.3 Seizures, migraine, sepsis, and peripheral vestibular disorders are among the many conditions that can masquerade as stroke.
Misdiagnosis can subject patients to unnecessary, and potentially harmful, invasive stroke therapies, and significantly delay the treatment they need. To prevent such outcomes, it is essential for primary care physicians, as well as emergency responders and emergency department (ED) physicians, to be aware of—and on the lookout for—the clues that can distinguish stroke from stroke mimics.
Suspect stroke? Establish a baseline
Despite a nationwide effort to increase public awareness of stroke and the importance of getting to the hospital within the therapeutic window for treatment,4 too few patients arrive within the time frame for cerebral reperfusion therapy. Primary care physicians can help by educating patients about stroke signs and symptoms.
When a patient presents with possible stroke, determine whether symptoms began gradually or abruptly. An acute ischemic stroke is heralded by the sudden onset of a focal neurologic deficit in a vascular pattern. Duration of symptoms can help distinguish stroke from a transient ischemic attack (TIA). Although TIAs were defined by the National Institutes of Health in 1975 as neurologic deficits that resolve within 24 hours of their onset, we now know that they typically last only 2 to 15 minutes, with the vast majority resolving within an hour.5
If the onset was sudden, find out when the patient was last seen at his or her neurologic baseline—information a family member, friend, or caregiver can often provide. This information is crucial because the neurologic baseline, rather than the time at which the symptoms were first noticed, is the basis for the therapeutic window for thrombolysis (3 hours for intravenous tPA and 6 hours for intra-arterial tPA). (Clot extraction with a mechanical embolus retrieval device [MERCI, Concentric Medical, Mountain View, Calif] has a 9-hour window.6,7)
Use a rapid stroke screening tool. To rapidly evaluate a patient with stroke-like signs and symptoms in a clinic or other outpatient setting, use a stroke screening tool with a high sensitivity,8 such as the Cincinnati Prehospital Stroke Scale (CPSS), the Face Arms Speech Time (FAST) test, or the Los Angeles Prehospital Stroke Screen (LAPSS) (TABLE 1). All 3 have a high positive predictive value (CPSS: 88%, FAST: 89%, LAPSS: 87%), but there is greater variation in the negative predictive value: 75%, 73%, and 55%, respectively.9
Patients with positive results typically require rapid transport to the ED—even if you notice red flags that may signal that you’re dealing with a stroke mimic.
TABLE 1
Stroke screening tools for outpatient use*30-32
| Cincinnati Prehospital Stroke Scale (CPSS) (www.strokecenter.org/trials/scales/cincinnati.html), which assesses the unilateral presence of any (or all) of the 3 key indicators—facial droop, arm drift, or slurred speech |
| Face Arms Speech Time (FAST) (www.stroke.org/site/PageServer?pagename=symp), a modification of CPSS based on the same criteria, has been validated in primary care clinics as well as emergency departments |
| Los Angeles Prehospital Stroke Screen (LAPSS) (www.strokecenter.org/trials/scales/lapss.html), a 1-page instrument that uses 5 criteria—age (>45 years), seizure history (none), onset of neurologic symptoms (within 24 hours), ambulatory status (ambulatory prior to event), and blood glucose level (60-400 mg/dL)—and 3 physical characteristics (facial smile/grimace, grip, and arm weakness) to screen for possible stroke |
| * A positive test is based on the presence of 1 or more key features for CPSS or FAST, and on a Yes (or Unknown) response to all the screening criteria in LAPSS. |
Be alert to the signs of conditions masquerading as stroke
Seizure at the onset of the episode; isolated mild neurological deficits, such as ataxia, sensory loss, or dysarthria alone; and/or minimal weakness are contraindications to thrombolytic therapy, according to the American Academy of Neurology (AAN).10
Rapidly improving neurological status is a probable indicator of a TIA or nonstroke etiology. Decreased level of consciousness with normal eye movements increases the likelihood that the patient has a condition that mimics stroke.11 Additional symptoms that strongly suggest a disorder other than stroke are convulsions (odds ratio [OR]: 0.1), loss of consciousness (OR: 0.1), confusion (OR: 0.2), headache (OR: 0.8), nausea (OR: 0.5), vomiting (OR: 0.6), and dizziness (OR: 0.3).9
Age is another consideration. The vast majority of patients with conditions that turn out to be stroke mimics are younger than 50 years of age. In patients older than 50, the prevalence of stroke misdiagnosis is just 3%.12
Watch for these stroke mimics
Seizures, either unwitnessed or unrecognized, and complex migraine are the most common stroke masqueraders. Other conditions frequently misdiagnosed as stroke include: systemic infections and early sepsis, central nervous system (CNS) tumors, and toxic-metabolic syndromes (including intoxication, hypoglycemia, hypercalcemia, and hyperosmolar nonketotic coma) (TABLE 2). Patients with cranial or peripheral neuropathy; dementia; labyrinthitis/benign positional vertigo; psychiatric disorders, in particular, conversion reaction; syncope; and transient global amnesia may also present with neurological symptoms suggestive of stroke. (For more on transient global amnesia, see this month’s Hospitalist Rounds at http://www.jfponline.com/CollectionContent.asp?CollectionID=286.) Characteristics of some of the more common mimics are detailed below.
Seizures. Neurologic deficits associated with seizures are reversible, with no structural CNS abnormalities. Postictal hemiparesis, also known as Todd’s paralysis—a focal weakness after a seizure, typically localized to 1 side of the body—occurs in approximately 13% of all seizures.13 Todd’s paralysis, which can be seen after either partial complex or generalized tonic-clonic seizures, may also affect speech and vision, producing a range of signs and symptoms easily mistaken for stroke. Duration ranges from minutes to 48 hours,14 but generally lasts only 3 to 22 minutes.13
Differentiating Todd’s paralysis from stroke is complicated by the fact that some strokes trigger focal seizures during the acute phase. However, a history of seizures or witnessed seizure activity points to Todd’s paralysis rather than stroke.
Complex migraine. Like Todd’s paralysis, complex migraine may result in hemiparesis. The presentation may also include vision loss, aphasia, or vertigo and other basilar symptoms—neurologic changes that can outlast the headache. Complex migraine is a diagnosis of exclusion, arrived at after a full neurologic assessment, including stroke work-up. Indeed, you can be certain of a diagnosis of complex migraine only after the patient has had recurrent complex migraine attacks.
Some basilar TIAs can also present with headache, but the onset is typically sudden, as opposed to the more gradual onset of migraine aura with posterior circulation symptoms.15 Age is a factor as well: Complex migraines usually develop well before the age of 40, while the mean age for ischemic stroke is 70. Although complex migraine is a risk factor for ischemic stroke, in most patients migraine is a benign condition.15,16
Systemic infections. Sepsis from almost any infectious agent can cause delirium, altered speech, weakness, and less specific stroke-like symptoms. Microbial seeding of the CNS can result in focal lesions (eg, the lesions shown in FIGURE 1B are associated with cryptococcal meningoencephalitis) or abscess formation with focal neurologic deficits.
Mass lesions. Primary CNS tumors, metastatic tumors, and cerebral abscesses are among the lesions that can cause symptoms that mimic stroke. In most cases, symptoms develop gradually as the lesion enlarges, but a small subset of patients have symptoms lasting less than 1 day. This is thought to be due to hemorrhage into the tumor or the acute development of obstructive hydrocephalus.17
Metabolic disorders. Diabetic hypoglycemia, among other metabolic disorders, is a classic stroke mimic, as well as a cause of seizures, so early evaluation of blood glucose is a crucial step in evaluating a patient with neurologic signs and symptoms. Patients with diabetic hypoglycemia may present with hemiplegia and aphasia; similar symptoms may occur in patients with hypoglycemia secondary to alcoholism, among other causes. Those with hyperglycemic nonketotic hyperosmolar states, severe hyponatremia, and hepatic encephalopathy may also present with focal stroke-like symptoms. Neurologic changes associated with metabolic disorders generally resolve rapidly with the administration of IV glucose, but on rare occasions may take several hours to resolve.14
Psychiatric illness. Patients with certain psychiatric disorders—including conversion reaction, a psychological condition that presents as an alteration in, or loss of, physical function—may present with dramatic focal problems and apparent deficits that mimic neurologic disease. Subtle disparities in the physical exam, such as Hoover’s sign, give-away weakness,18 and “la belle indifference,” as well as negative neuroimaging, will establish this difficult-to-treat stroke mimic.19 Grand mal pseudo-seizures can be differentiated from actual grand mal seizures by the failure of a prolactin level (drawn 10 to 20 minutes post-event) to rise at least 2-fold.20
Transient global amnesia. The rare, sudden development of dense anterograde amnesia occurs without alteration in level of consciousness, focal neurologic deficits, or seizure activity. It is self-limiting and mainly affects those older than 50. Transient global amnesia has an uncertain etiology, although atypical migraine, seizure discharge, and venous congestion with hippocampal ischemia are viewed as possible causes. Reported triggers include severe physical or emotional stress, strenuous physical activity, and orgasmic sexual intercourse.21
TABLE 2
Common stroke mimics9,11,12,14,22
| Condition | Misdiagnosed as stroke (%) |
|---|---|
| Brain tumor | 7-15 |
| Labyrinthitis | 5-6 |
| Metabolic disorder | 3-13 |
| Migraine | 11-47 |
| Psychiatric disorder | 1-40 |
| Seizures | 11-40 |
| Sepsis | 14-17 |
| Syncope | 5-22 |
| Transient global amnesia | 3-10 |
| Other | 11-37 |
In the ED: Evaluation is guided by a timeline
Current guidelines from the American Heart Association and American Stroke Association recommend that a possible stroke patient be evaluated by the physician in the ED within 10 minutes of his or her arrival—and that a decision on how to proceed be reached within 60 minutes of arrival. The guidelines call for the initial computed tomography (CT) to be completed within 25 minutes of the patient’s arrival and interpreted by a physician with expertise in reading CT studies within 45 minutes of arrival.6,24
In the ED, the National Institutes of Health Stroke Scale (NIHSS)25 (TABLE 3) is an ideal way to focus and record the neurological exam.6 The scale assesses 6 separate neurologic functions (level of consciousness, vision, motor function, sensory function, language, and cerebellar function) and can be performed within 5 to 8 minutes. It yields a score from 0 to 42, with the higher numbers indicating worse neurologic function.26 Although a score ≤10 is generally considered to be predictive of a stroke mimic, a recent study found that 19% of patients with an NIHSS score >10 also had conditions masquerading as stroke.27
Imaging leads to accurate diagnosis. The rate at which stroke mimics are mistaken for actual strokes varies with the population studied and the diagnostic tests performed. While stroke is largely a clinical diagnosis and a history and physical exam focused on onset, duration, and symptoms are key elements in differentiating stroke from a stroke mimic, studies have found that the incidence of misdiagnosis (19% with history, physical, and lab work alone) drops to 5% when noncontrast CT is added. When diffusion-weighted magnetic resonance imaging (MRI) is used instead, misdiagnosis drops to just 2%.11,12,14,22,23
Basic lab tests—a complete blood count and basic metabolic panel, with blood alcohol, hepatic function tests, and toxicology screens in select cases—help rule out stroke mimics. Radiographic imaging of the brain provides further clarification (FIGURE 1A AND 1B), serving 2 main purposes: to (1) evaluate diagnoses other than stroke and (2) identify the presence of any acute intracranial bleeding. Noncontrast CT scans detect acute hemorrhage with a sensitivity of 89% and specificity of 100%.27 CT angiography (which can identify the location of a clot) and CT perfusion (which allows an assessment of any existing penumbra) can also be obtained in a timely fashion with newer multislice scanners.
Some institutions, however, evaluate acute stroke patients with MRI. Depending on the sequences used, MRI has the advantage of being able to detect early ischemic changes, diffusion and perfusion mismatches, and abnormalities of the posterior fossa.29 In acute ischemic stroke, diffusion-weighted MRI has a sensitivity of 83% and specificity of 96%, compared with a sensitivity of 16% and specificity of 98% for noncontrast CT.28
TABLE 3
National Institutes of Health Stroke Scale25
| Item | Response score* |
|---|---|
| 1a. Level of consciousness | 0 = alert 1 = not alert 2 = obtunded 3 = unresponsive |
| 1b. Level of consciousness Questions | 0 = answers both correctly 1 = answers one correctly 2 = answers neither correctly |
| 1c. Level of consciousness Commands | 0 = performs both tasks correctly 1 = performs one task correctly 2 = performs neither task correctly |
| 2. Gaze | 0 = normal 1 = partial gaze palsy 2 = total gaze palsy |
| 3. Visual fields | 0 = no visual loss 1 = partial hemianopsia 2 = complete hemianopsia 3 = bilateral hemianopsia |
| 4. Facial palsy | 0 = normal 1 = minor paralysis 2 = partial paralysis 3 = complete paralysis |
| 5. Motor arm a. Left b. Right | 0 = no drift 1 = drifts before 5 sec 2 = falls before 10 sec 3 = no effort against gravity 4 = no movement |
| 6. Motor leg a. Left b. Right | 0 = no drift 1 = drifts before 5 sec 2 = falls before 10 sec 3 = no effort against gravity 4 = no movement |
| 7. Ataxia | 0 = absent 1 = 1 limb 2 = 2 limbs |
| 8. Sensory | 0 = normal 1 = mild loss 2 = severe loss |
| 9. Language | 0 = normal 1 = mild aphasia 2 = severe aphasia 3 = mute or global aphasia |
| 10. Dysarthria | 0 = normal 1 = mild 2 = severe |
| 11. Extinction/inattention | 0 = normal 1 = mild 2 = severe |
| * Yields a score from 0 to 42 (higher numbers indicate worse neurologic function). | |
FIGURE 1
2 patients with common symptoms, vastly different diagnoses
The markedly abnormal perfusion (arrows) this CT image reveals corresponds to an acute occlusion of the left vertebral artery and a subsequent infarct.
An axial postcontrast MRI reveals multiple lesions in the left temporal lobe (arrows) in a patient with rapid-onset mental changes. The diagnosis: cryptococcal meningoencephalitis.
CORRESPONDENCE
Konrad C. Nau, MD, West Virginia University Department of Family Medicine-Eastern Division, 171 Taylor Street, Harpers Ferry, WV 25425; [email protected]
1. Lloyd-Jones D, Adams R, Carnethon M, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480-486.
2. Scott PA, Silbergeit R. Misdiagnosis of stroke in tissue plasminogen activator-treated patients: characteristics and outcomes. Ann Emerg Med. 2003;42:611-618.
3. Morgenstern LB, Lisabeth LD, Mecozzi AC, et al. A population-based study of acute stroke and TIA diagnosis. Neurology. 2004;62:895-900.
4. Nicol MB, Thrift AG. Knowledge of risk factors and warning signs of stroke. Vasc Health Risk Manag. 2005;1:137-147.
5. Albers GW. Transient ischemic attack—proposal for a new definition. N Engl J Med. 2002;347:1713-1716.
6. Adams HP, del Zoppo G, Alberts MJ, et al. Guidelines for early management of adults with ischemic stroke. Circulation. 2007;115:e478-e534.
7. Rosamond WD, Reeves MJ, Johnson A, et al. Paul Coverdell National Acute Stroke Registry Prototype Investigators. Documentation of stroke onset time: challenges and recommendations. Am J Prev Med. 2006;6(suppl 2):S230-S234.
8. Crocco TJ. Streamlining stroke care: from symptom onset to emergency department. J Emerg Med. 2007;33:255-260.
9. Nor AM, Davis J, Sen B, et al. The recognition of stroke in the emergency room scale: development and validation of a stroke recognition scale. Lancet Neurol. 2005;4:727-734.
10. Practice Advisory: Thrombolytic therapy for acute ischemic stroke-summary statement. Report of the Quality standards subcommittee of the American Academy of Neurology. Neurology. 1996;47:835-839.
11. Libman RB, Wirkowski E, Alvir J, et al. Conditions that mimic stroke in the emergency department. Implications for acute stroke trials. Arch Neurol. 1995;52:1119-1122.
12. Vroomen P, Buddingh MK, Kuijckx G, et al. The incidence of stroke mimics among stroke department admissions in relation to age group. J Stroke Cerebrovasc Dis. 2008;17:418-422.
13. Gallmetzer P, Leutmezer F, Serles W, et al. Postictal paresis in focal epilepsies: incidence, duration, and causes. Neurology. 2004;12:2160-2164.
14. Huff JS. Stroke mimics and chameleons. Emerg Med Clin N Am. 2002;20:583-595.
15. Bousser MG, Welch KM. Relation between migraine and stroke. Lancet Neurol. 2005;4:533-542.
16. Bigal ME, Kurth T, Hu H, et al. Migraine and cardiovascular disease: possible mechanisms of interaction. Neurology. 2009;72:1864-1871.
17. Snyder H, Robinson K, Shah D, et al. Signs and symptoms of patients with brain tumors presenting to the emergency department. J Emerg Med. 1993;11:253-258.
18. Stone J, Zeman A, Sharpe M. Functional weakness and sensory disturbance. J Neurol Neurosurg Psychiatr. 2002;73:241-245.
19. Phoebe SC, Tobiano PS, Wang HE, et al. Case of conversion disorder presenting as a severe acute stroke. J Emerg Med. 2006;30:283-286.
20. Chen DK, So YT, Fischer RS. Use of serum prolactin in diagnosing epileptic seizures. Report of the therapeutics and technology subcommittee of the American Academy of Neurology. Neurology. 2005;65:668-675.
21. Quinette P, Guillery-Girard B, Dayan J, et al. What does transient global amnesia really mean? Review of the literature and thorough study of 142 cases. Brain. 2006;129:1640-1658.
22. Kothari RU, Brott T, Broderick JP, et al. Emergency physicians: accuracy in diagnosis of stroke. Stroke. 1995;26:2238-2241.
23. Ay H, Buonanno FS, Rordorf G, et al. Normal diffusion-weighted MRI during stroke-like deficits. Neurology. 1999;52:1784-1792.
24. Bock BF. Response system for patients presenting with acute stroke. In: Marler JR, Jones PM, Emr M, ed. Proceeding of a National Symposium on Rapid Identification and Treatment of Acute Stroke: 1997. Bethesda, MD: National Institute of Neurological Disorders and Stroke, National Institutes of Health; 1997.
25. National Institutes of Health. Know stroke. Available at: http://www.ninds.nih.gov/doctors/NIH_Stroke_Scale_Booklet.pdf. Accessed December 10, 2009.
26. Kasner SE. Clinical interpretation and use of stroke scales. Lancet Neurol. 2006;5:603-612.
27. Hand PJ, Kwan J, Lindley RI, et al. Distinguishing between stroke and mimic at the bedside: the Brain Attack Study. Stroke. 2006;36:769-775.
28. Chalela JA, Kidwell CS, Nentwich LM, et al. Magnetic resonance imaging and computerized tomography in emergency assessment of patients with suspected acute stroke—a prospective comparison. Lancet. 2007;369:293-298.
29. Kohrmann M, Jüttler E, Huttner HB, et al. Acute stroke imaging for thrombolytic therapy—an update. Cerebrovasc Dis. 2007;24:161-169.
30. Kothari RU, Panciolo A, Liu T, et al. Cincinnati prehospital stroke scale: reproducibility and validity. Ann Emerg Med. 1999;33:373-378.
31. Harbison J, Hossain O, Jenkinson D, et al. Diagnostic accuracy of stroke referrals from primary care, emergency room physicians, and ambulance staff using the face arm speech test. Stroke. 2003;34:71-76.
32. Kidwell CS, Starkman S, Eckstein M, et al. Identifying stroke in the field: prospective validation of the Los Angeles prehospital stroke screen (LAPPS). Stroke. 2000;31:71-76.
• Arrange for urgent transport to the hospital when a patient presents with stroke-like symptoms of acute onset, especially within the 3- to 6-hour therapeutic window. B
• Use a validated prehospital stroke identification algorithm such as the Face Arms Speech Time (FAST) test to identify possible acute stroke patients requiring urgent transport to the hospital. B
• Obtain a CBC and basic metabolic panel for all patients with signs and symptoms suggestive of stroke—and a blood alcohol, hepatic function, and toxicology screen, in select patients—to help rule out stroke mimics. C
• Ensure that patients undergo brain imaging to rule out stroke mimics before treatment for acute ischemic stroke is initiated. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Stroke is the third leading cause of death (claiming the life of 1 person every 3 to 4 minutes) and the No. 1 cause of adult disability in the United States.1 Advances in thrombolysis and clot removal can improve outcomes, but are dependent on swift and certain diagnosis. Amid the rush to ensure that treatment is initiated within the therapeutic window for cerebral reperfusion, “stroke mimics”—so called because of their ability to cause signs and symptoms similar to stroke—are sometimes mistaken for the real thing.
The prevalence of misdiagnosis ranges from about 4% of patients who receive tissue plasminogen activator (tPA) for reperfusion2 to 25% of patients who are rushed to the hospital because they are thought to be having a stroke.3 Seizures, migraine, sepsis, and peripheral vestibular disorders are among the many conditions that can masquerade as stroke.
Misdiagnosis can subject patients to unnecessary, and potentially harmful, invasive stroke therapies, and significantly delay the treatment they need. To prevent such outcomes, it is essential for primary care physicians, as well as emergency responders and emergency department (ED) physicians, to be aware of—and on the lookout for—the clues that can distinguish stroke from stroke mimics.
Suspect stroke? Establish a baseline
Despite a nationwide effort to increase public awareness of stroke and the importance of getting to the hospital within the therapeutic window for treatment,4 too few patients arrive within the time frame for cerebral reperfusion therapy. Primary care physicians can help by educating patients about stroke signs and symptoms.
When a patient presents with possible stroke, determine whether symptoms began gradually or abruptly. An acute ischemic stroke is heralded by the sudden onset of a focal neurologic deficit in a vascular pattern. Duration of symptoms can help distinguish stroke from a transient ischemic attack (TIA). Although TIAs were defined by the National Institutes of Health in 1975 as neurologic deficits that resolve within 24 hours of their onset, we now know that they typically last only 2 to 15 minutes, with the vast majority resolving within an hour.5
If the onset was sudden, find out when the patient was last seen at his or her neurologic baseline—information a family member, friend, or caregiver can often provide. This information is crucial because the neurologic baseline, rather than the time at which the symptoms were first noticed, is the basis for the therapeutic window for thrombolysis (3 hours for intravenous tPA and 6 hours for intra-arterial tPA). (Clot extraction with a mechanical embolus retrieval device [MERCI, Concentric Medical, Mountain View, Calif] has a 9-hour window.6,7)
Use a rapid stroke screening tool. To rapidly evaluate a patient with stroke-like signs and symptoms in a clinic or other outpatient setting, use a stroke screening tool with a high sensitivity,8 such as the Cincinnati Prehospital Stroke Scale (CPSS), the Face Arms Speech Time (FAST) test, or the Los Angeles Prehospital Stroke Screen (LAPSS) (TABLE 1). All 3 have a high positive predictive value (CPSS: 88%, FAST: 89%, LAPSS: 87%), but there is greater variation in the negative predictive value: 75%, 73%, and 55%, respectively.9
Patients with positive results typically require rapid transport to the ED—even if you notice red flags that may signal that you’re dealing with a stroke mimic.
TABLE 1
Stroke screening tools for outpatient use*30-32
| Cincinnati Prehospital Stroke Scale (CPSS) (www.strokecenter.org/trials/scales/cincinnati.html), which assesses the unilateral presence of any (or all) of the 3 key indicators—facial droop, arm drift, or slurred speech |
| Face Arms Speech Time (FAST) (www.stroke.org/site/PageServer?pagename=symp), a modification of CPSS based on the same criteria, has been validated in primary care clinics as well as emergency departments |
| Los Angeles Prehospital Stroke Screen (LAPSS) (www.strokecenter.org/trials/scales/lapss.html), a 1-page instrument that uses 5 criteria—age (>45 years), seizure history (none), onset of neurologic symptoms (within 24 hours), ambulatory status (ambulatory prior to event), and blood glucose level (60-400 mg/dL)—and 3 physical characteristics (facial smile/grimace, grip, and arm weakness) to screen for possible stroke |
| * A positive test is based on the presence of 1 or more key features for CPSS or FAST, and on a Yes (or Unknown) response to all the screening criteria in LAPSS. |
Be alert to the signs of conditions masquerading as stroke
Seizure at the onset of the episode; isolated mild neurological deficits, such as ataxia, sensory loss, or dysarthria alone; and/or minimal weakness are contraindications to thrombolytic therapy, according to the American Academy of Neurology (AAN).10
Rapidly improving neurological status is a probable indicator of a TIA or nonstroke etiology. Decreased level of consciousness with normal eye movements increases the likelihood that the patient has a condition that mimics stroke.11 Additional symptoms that strongly suggest a disorder other than stroke are convulsions (odds ratio [OR]: 0.1), loss of consciousness (OR: 0.1), confusion (OR: 0.2), headache (OR: 0.8), nausea (OR: 0.5), vomiting (OR: 0.6), and dizziness (OR: 0.3).9
Age is another consideration. The vast majority of patients with conditions that turn out to be stroke mimics are younger than 50 years of age. In patients older than 50, the prevalence of stroke misdiagnosis is just 3%.12
Watch for these stroke mimics
Seizures, either unwitnessed or unrecognized, and complex migraine are the most common stroke masqueraders. Other conditions frequently misdiagnosed as stroke include: systemic infections and early sepsis, central nervous system (CNS) tumors, and toxic-metabolic syndromes (including intoxication, hypoglycemia, hypercalcemia, and hyperosmolar nonketotic coma) (TABLE 2). Patients with cranial or peripheral neuropathy; dementia; labyrinthitis/benign positional vertigo; psychiatric disorders, in particular, conversion reaction; syncope; and transient global amnesia may also present with neurological symptoms suggestive of stroke. (For more on transient global amnesia, see this month’s Hospitalist Rounds at http://www.jfponline.com/CollectionContent.asp?CollectionID=286.) Characteristics of some of the more common mimics are detailed below.
Seizures. Neurologic deficits associated with seizures are reversible, with no structural CNS abnormalities. Postictal hemiparesis, also known as Todd’s paralysis—a focal weakness after a seizure, typically localized to 1 side of the body—occurs in approximately 13% of all seizures.13 Todd’s paralysis, which can be seen after either partial complex or generalized tonic-clonic seizures, may also affect speech and vision, producing a range of signs and symptoms easily mistaken for stroke. Duration ranges from minutes to 48 hours,14 but generally lasts only 3 to 22 minutes.13
Differentiating Todd’s paralysis from stroke is complicated by the fact that some strokes trigger focal seizures during the acute phase. However, a history of seizures or witnessed seizure activity points to Todd’s paralysis rather than stroke.
Complex migraine. Like Todd’s paralysis, complex migraine may result in hemiparesis. The presentation may also include vision loss, aphasia, or vertigo and other basilar symptoms—neurologic changes that can outlast the headache. Complex migraine is a diagnosis of exclusion, arrived at after a full neurologic assessment, including stroke work-up. Indeed, you can be certain of a diagnosis of complex migraine only after the patient has had recurrent complex migraine attacks.
Some basilar TIAs can also present with headache, but the onset is typically sudden, as opposed to the more gradual onset of migraine aura with posterior circulation symptoms.15 Age is a factor as well: Complex migraines usually develop well before the age of 40, while the mean age for ischemic stroke is 70. Although complex migraine is a risk factor for ischemic stroke, in most patients migraine is a benign condition.15,16
Systemic infections. Sepsis from almost any infectious agent can cause delirium, altered speech, weakness, and less specific stroke-like symptoms. Microbial seeding of the CNS can result in focal lesions (eg, the lesions shown in FIGURE 1B are associated with cryptococcal meningoencephalitis) or abscess formation with focal neurologic deficits.
Mass lesions. Primary CNS tumors, metastatic tumors, and cerebral abscesses are among the lesions that can cause symptoms that mimic stroke. In most cases, symptoms develop gradually as the lesion enlarges, but a small subset of patients have symptoms lasting less than 1 day. This is thought to be due to hemorrhage into the tumor or the acute development of obstructive hydrocephalus.17
Metabolic disorders. Diabetic hypoglycemia, among other metabolic disorders, is a classic stroke mimic, as well as a cause of seizures, so early evaluation of blood glucose is a crucial step in evaluating a patient with neurologic signs and symptoms. Patients with diabetic hypoglycemia may present with hemiplegia and aphasia; similar symptoms may occur in patients with hypoglycemia secondary to alcoholism, among other causes. Those with hyperglycemic nonketotic hyperosmolar states, severe hyponatremia, and hepatic encephalopathy may also present with focal stroke-like symptoms. Neurologic changes associated with metabolic disorders generally resolve rapidly with the administration of IV glucose, but on rare occasions may take several hours to resolve.14
Psychiatric illness. Patients with certain psychiatric disorders—including conversion reaction, a psychological condition that presents as an alteration in, or loss of, physical function—may present with dramatic focal problems and apparent deficits that mimic neurologic disease. Subtle disparities in the physical exam, such as Hoover’s sign, give-away weakness,18 and “la belle indifference,” as well as negative neuroimaging, will establish this difficult-to-treat stroke mimic.19 Grand mal pseudo-seizures can be differentiated from actual grand mal seizures by the failure of a prolactin level (drawn 10 to 20 minutes post-event) to rise at least 2-fold.20
Transient global amnesia. The rare, sudden development of dense anterograde amnesia occurs without alteration in level of consciousness, focal neurologic deficits, or seizure activity. It is self-limiting and mainly affects those older than 50. Transient global amnesia has an uncertain etiology, although atypical migraine, seizure discharge, and venous congestion with hippocampal ischemia are viewed as possible causes. Reported triggers include severe physical or emotional stress, strenuous physical activity, and orgasmic sexual intercourse.21
TABLE 2
Common stroke mimics9,11,12,14,22
| Condition | Misdiagnosed as stroke (%) |
|---|---|
| Brain tumor | 7-15 |
| Labyrinthitis | 5-6 |
| Metabolic disorder | 3-13 |
| Migraine | 11-47 |
| Psychiatric disorder | 1-40 |
| Seizures | 11-40 |
| Sepsis | 14-17 |
| Syncope | 5-22 |
| Transient global amnesia | 3-10 |
| Other | 11-37 |
In the ED: Evaluation is guided by a timeline
Current guidelines from the American Heart Association and American Stroke Association recommend that a possible stroke patient be evaluated by the physician in the ED within 10 minutes of his or her arrival—and that a decision on how to proceed be reached within 60 minutes of arrival. The guidelines call for the initial computed tomography (CT) to be completed within 25 minutes of the patient’s arrival and interpreted by a physician with expertise in reading CT studies within 45 minutes of arrival.6,24
In the ED, the National Institutes of Health Stroke Scale (NIHSS)25 (TABLE 3) is an ideal way to focus and record the neurological exam.6 The scale assesses 6 separate neurologic functions (level of consciousness, vision, motor function, sensory function, language, and cerebellar function) and can be performed within 5 to 8 minutes. It yields a score from 0 to 42, with the higher numbers indicating worse neurologic function.26 Although a score ≤10 is generally considered to be predictive of a stroke mimic, a recent study found that 19% of patients with an NIHSS score >10 also had conditions masquerading as stroke.27
Imaging leads to accurate diagnosis. The rate at which stroke mimics are mistaken for actual strokes varies with the population studied and the diagnostic tests performed. While stroke is largely a clinical diagnosis and a history and physical exam focused on onset, duration, and symptoms are key elements in differentiating stroke from a stroke mimic, studies have found that the incidence of misdiagnosis (19% with history, physical, and lab work alone) drops to 5% when noncontrast CT is added. When diffusion-weighted magnetic resonance imaging (MRI) is used instead, misdiagnosis drops to just 2%.11,12,14,22,23
Basic lab tests—a complete blood count and basic metabolic panel, with blood alcohol, hepatic function tests, and toxicology screens in select cases—help rule out stroke mimics. Radiographic imaging of the brain provides further clarification (FIGURE 1A AND 1B), serving 2 main purposes: to (1) evaluate diagnoses other than stroke and (2) identify the presence of any acute intracranial bleeding. Noncontrast CT scans detect acute hemorrhage with a sensitivity of 89% and specificity of 100%.27 CT angiography (which can identify the location of a clot) and CT perfusion (which allows an assessment of any existing penumbra) can also be obtained in a timely fashion with newer multislice scanners.
Some institutions, however, evaluate acute stroke patients with MRI. Depending on the sequences used, MRI has the advantage of being able to detect early ischemic changes, diffusion and perfusion mismatches, and abnormalities of the posterior fossa.29 In acute ischemic stroke, diffusion-weighted MRI has a sensitivity of 83% and specificity of 96%, compared with a sensitivity of 16% and specificity of 98% for noncontrast CT.28
TABLE 3
National Institutes of Health Stroke Scale25
| Item | Response score* |
|---|---|
| 1a. Level of consciousness | 0 = alert 1 = not alert 2 = obtunded 3 = unresponsive |
| 1b. Level of consciousness Questions | 0 = answers both correctly 1 = answers one correctly 2 = answers neither correctly |
| 1c. Level of consciousness Commands | 0 = performs both tasks correctly 1 = performs one task correctly 2 = performs neither task correctly |
| 2. Gaze | 0 = normal 1 = partial gaze palsy 2 = total gaze palsy |
| 3. Visual fields | 0 = no visual loss 1 = partial hemianopsia 2 = complete hemianopsia 3 = bilateral hemianopsia |
| 4. Facial palsy | 0 = normal 1 = minor paralysis 2 = partial paralysis 3 = complete paralysis |
| 5. Motor arm a. Left b. Right | 0 = no drift 1 = drifts before 5 sec 2 = falls before 10 sec 3 = no effort against gravity 4 = no movement |
| 6. Motor leg a. Left b. Right | 0 = no drift 1 = drifts before 5 sec 2 = falls before 10 sec 3 = no effort against gravity 4 = no movement |
| 7. Ataxia | 0 = absent 1 = 1 limb 2 = 2 limbs |
| 8. Sensory | 0 = normal 1 = mild loss 2 = severe loss |
| 9. Language | 0 = normal 1 = mild aphasia 2 = severe aphasia 3 = mute or global aphasia |
| 10. Dysarthria | 0 = normal 1 = mild 2 = severe |
| 11. Extinction/inattention | 0 = normal 1 = mild 2 = severe |
| * Yields a score from 0 to 42 (higher numbers indicate worse neurologic function). | |
FIGURE 1
2 patients with common symptoms, vastly different diagnoses
The markedly abnormal perfusion (arrows) this CT image reveals corresponds to an acute occlusion of the left vertebral artery and a subsequent infarct.
An axial postcontrast MRI reveals multiple lesions in the left temporal lobe (arrows) in a patient with rapid-onset mental changes. The diagnosis: cryptococcal meningoencephalitis.
CORRESPONDENCE
Konrad C. Nau, MD, West Virginia University Department of Family Medicine-Eastern Division, 171 Taylor Street, Harpers Ferry, WV 25425; [email protected]
• Arrange for urgent transport to the hospital when a patient presents with stroke-like symptoms of acute onset, especially within the 3- to 6-hour therapeutic window. B
• Use a validated prehospital stroke identification algorithm such as the Face Arms Speech Time (FAST) test to identify possible acute stroke patients requiring urgent transport to the hospital. B
• Obtain a CBC and basic metabolic panel for all patients with signs and symptoms suggestive of stroke—and a blood alcohol, hepatic function, and toxicology screen, in select patients—to help rule out stroke mimics. C
• Ensure that patients undergo brain imaging to rule out stroke mimics before treatment for acute ischemic stroke is initiated. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Stroke is the third leading cause of death (claiming the life of 1 person every 3 to 4 minutes) and the No. 1 cause of adult disability in the United States.1 Advances in thrombolysis and clot removal can improve outcomes, but are dependent on swift and certain diagnosis. Amid the rush to ensure that treatment is initiated within the therapeutic window for cerebral reperfusion, “stroke mimics”—so called because of their ability to cause signs and symptoms similar to stroke—are sometimes mistaken for the real thing.
The prevalence of misdiagnosis ranges from about 4% of patients who receive tissue plasminogen activator (tPA) for reperfusion2 to 25% of patients who are rushed to the hospital because they are thought to be having a stroke.3 Seizures, migraine, sepsis, and peripheral vestibular disorders are among the many conditions that can masquerade as stroke.
Misdiagnosis can subject patients to unnecessary, and potentially harmful, invasive stroke therapies, and significantly delay the treatment they need. To prevent such outcomes, it is essential for primary care physicians, as well as emergency responders and emergency department (ED) physicians, to be aware of—and on the lookout for—the clues that can distinguish stroke from stroke mimics.
Suspect stroke? Establish a baseline
Despite a nationwide effort to increase public awareness of stroke and the importance of getting to the hospital within the therapeutic window for treatment,4 too few patients arrive within the time frame for cerebral reperfusion therapy. Primary care physicians can help by educating patients about stroke signs and symptoms.
When a patient presents with possible stroke, determine whether symptoms began gradually or abruptly. An acute ischemic stroke is heralded by the sudden onset of a focal neurologic deficit in a vascular pattern. Duration of symptoms can help distinguish stroke from a transient ischemic attack (TIA). Although TIAs were defined by the National Institutes of Health in 1975 as neurologic deficits that resolve within 24 hours of their onset, we now know that they typically last only 2 to 15 minutes, with the vast majority resolving within an hour.5
If the onset was sudden, find out when the patient was last seen at his or her neurologic baseline—information a family member, friend, or caregiver can often provide. This information is crucial because the neurologic baseline, rather than the time at which the symptoms were first noticed, is the basis for the therapeutic window for thrombolysis (3 hours for intravenous tPA and 6 hours for intra-arterial tPA). (Clot extraction with a mechanical embolus retrieval device [MERCI, Concentric Medical, Mountain View, Calif] has a 9-hour window.6,7)
Use a rapid stroke screening tool. To rapidly evaluate a patient with stroke-like signs and symptoms in a clinic or other outpatient setting, use a stroke screening tool with a high sensitivity,8 such as the Cincinnati Prehospital Stroke Scale (CPSS), the Face Arms Speech Time (FAST) test, or the Los Angeles Prehospital Stroke Screen (LAPSS) (TABLE 1). All 3 have a high positive predictive value (CPSS: 88%, FAST: 89%, LAPSS: 87%), but there is greater variation in the negative predictive value: 75%, 73%, and 55%, respectively.9
Patients with positive results typically require rapid transport to the ED—even if you notice red flags that may signal that you’re dealing with a stroke mimic.
TABLE 1
Stroke screening tools for outpatient use*30-32
| Cincinnati Prehospital Stroke Scale (CPSS) (www.strokecenter.org/trials/scales/cincinnati.html), which assesses the unilateral presence of any (or all) of the 3 key indicators—facial droop, arm drift, or slurred speech |
| Face Arms Speech Time (FAST) (www.stroke.org/site/PageServer?pagename=symp), a modification of CPSS based on the same criteria, has been validated in primary care clinics as well as emergency departments |
| Los Angeles Prehospital Stroke Screen (LAPSS) (www.strokecenter.org/trials/scales/lapss.html), a 1-page instrument that uses 5 criteria—age (>45 years), seizure history (none), onset of neurologic symptoms (within 24 hours), ambulatory status (ambulatory prior to event), and blood glucose level (60-400 mg/dL)—and 3 physical characteristics (facial smile/grimace, grip, and arm weakness) to screen for possible stroke |
| * A positive test is based on the presence of 1 or more key features for CPSS or FAST, and on a Yes (or Unknown) response to all the screening criteria in LAPSS. |
Be alert to the signs of conditions masquerading as stroke
Seizure at the onset of the episode; isolated mild neurological deficits, such as ataxia, sensory loss, or dysarthria alone; and/or minimal weakness are contraindications to thrombolytic therapy, according to the American Academy of Neurology (AAN).10
Rapidly improving neurological status is a probable indicator of a TIA or nonstroke etiology. Decreased level of consciousness with normal eye movements increases the likelihood that the patient has a condition that mimics stroke.11 Additional symptoms that strongly suggest a disorder other than stroke are convulsions (odds ratio [OR]: 0.1), loss of consciousness (OR: 0.1), confusion (OR: 0.2), headache (OR: 0.8), nausea (OR: 0.5), vomiting (OR: 0.6), and dizziness (OR: 0.3).9
Age is another consideration. The vast majority of patients with conditions that turn out to be stroke mimics are younger than 50 years of age. In patients older than 50, the prevalence of stroke misdiagnosis is just 3%.12
Watch for these stroke mimics
Seizures, either unwitnessed or unrecognized, and complex migraine are the most common stroke masqueraders. Other conditions frequently misdiagnosed as stroke include: systemic infections and early sepsis, central nervous system (CNS) tumors, and toxic-metabolic syndromes (including intoxication, hypoglycemia, hypercalcemia, and hyperosmolar nonketotic coma) (TABLE 2). Patients with cranial or peripheral neuropathy; dementia; labyrinthitis/benign positional vertigo; psychiatric disorders, in particular, conversion reaction; syncope; and transient global amnesia may also present with neurological symptoms suggestive of stroke. (For more on transient global amnesia, see this month’s Hospitalist Rounds at http://www.jfponline.com/CollectionContent.asp?CollectionID=286.) Characteristics of some of the more common mimics are detailed below.
Seizures. Neurologic deficits associated with seizures are reversible, with no structural CNS abnormalities. Postictal hemiparesis, also known as Todd’s paralysis—a focal weakness after a seizure, typically localized to 1 side of the body—occurs in approximately 13% of all seizures.13 Todd’s paralysis, which can be seen after either partial complex or generalized tonic-clonic seizures, may also affect speech and vision, producing a range of signs and symptoms easily mistaken for stroke. Duration ranges from minutes to 48 hours,14 but generally lasts only 3 to 22 minutes.13
Differentiating Todd’s paralysis from stroke is complicated by the fact that some strokes trigger focal seizures during the acute phase. However, a history of seizures or witnessed seizure activity points to Todd’s paralysis rather than stroke.
Complex migraine. Like Todd’s paralysis, complex migraine may result in hemiparesis. The presentation may also include vision loss, aphasia, or vertigo and other basilar symptoms—neurologic changes that can outlast the headache. Complex migraine is a diagnosis of exclusion, arrived at after a full neurologic assessment, including stroke work-up. Indeed, you can be certain of a diagnosis of complex migraine only after the patient has had recurrent complex migraine attacks.
Some basilar TIAs can also present with headache, but the onset is typically sudden, as opposed to the more gradual onset of migraine aura with posterior circulation symptoms.15 Age is a factor as well: Complex migraines usually develop well before the age of 40, while the mean age for ischemic stroke is 70. Although complex migraine is a risk factor for ischemic stroke, in most patients migraine is a benign condition.15,16
Systemic infections. Sepsis from almost any infectious agent can cause delirium, altered speech, weakness, and less specific stroke-like symptoms. Microbial seeding of the CNS can result in focal lesions (eg, the lesions shown in FIGURE 1B are associated with cryptococcal meningoencephalitis) or abscess formation with focal neurologic deficits.
Mass lesions. Primary CNS tumors, metastatic tumors, and cerebral abscesses are among the lesions that can cause symptoms that mimic stroke. In most cases, symptoms develop gradually as the lesion enlarges, but a small subset of patients have symptoms lasting less than 1 day. This is thought to be due to hemorrhage into the tumor or the acute development of obstructive hydrocephalus.17
Metabolic disorders. Diabetic hypoglycemia, among other metabolic disorders, is a classic stroke mimic, as well as a cause of seizures, so early evaluation of blood glucose is a crucial step in evaluating a patient with neurologic signs and symptoms. Patients with diabetic hypoglycemia may present with hemiplegia and aphasia; similar symptoms may occur in patients with hypoglycemia secondary to alcoholism, among other causes. Those with hyperglycemic nonketotic hyperosmolar states, severe hyponatremia, and hepatic encephalopathy may also present with focal stroke-like symptoms. Neurologic changes associated with metabolic disorders generally resolve rapidly with the administration of IV glucose, but on rare occasions may take several hours to resolve.14
Psychiatric illness. Patients with certain psychiatric disorders—including conversion reaction, a psychological condition that presents as an alteration in, or loss of, physical function—may present with dramatic focal problems and apparent deficits that mimic neurologic disease. Subtle disparities in the physical exam, such as Hoover’s sign, give-away weakness,18 and “la belle indifference,” as well as negative neuroimaging, will establish this difficult-to-treat stroke mimic.19 Grand mal pseudo-seizures can be differentiated from actual grand mal seizures by the failure of a prolactin level (drawn 10 to 20 minutes post-event) to rise at least 2-fold.20
Transient global amnesia. The rare, sudden development of dense anterograde amnesia occurs without alteration in level of consciousness, focal neurologic deficits, or seizure activity. It is self-limiting and mainly affects those older than 50. Transient global amnesia has an uncertain etiology, although atypical migraine, seizure discharge, and venous congestion with hippocampal ischemia are viewed as possible causes. Reported triggers include severe physical or emotional stress, strenuous physical activity, and orgasmic sexual intercourse.21
TABLE 2
Common stroke mimics9,11,12,14,22
| Condition | Misdiagnosed as stroke (%) |
|---|---|
| Brain tumor | 7-15 |
| Labyrinthitis | 5-6 |
| Metabolic disorder | 3-13 |
| Migraine | 11-47 |
| Psychiatric disorder | 1-40 |
| Seizures | 11-40 |
| Sepsis | 14-17 |
| Syncope | 5-22 |
| Transient global amnesia | 3-10 |
| Other | 11-37 |
In the ED: Evaluation is guided by a timeline
Current guidelines from the American Heart Association and American Stroke Association recommend that a possible stroke patient be evaluated by the physician in the ED within 10 minutes of his or her arrival—and that a decision on how to proceed be reached within 60 minutes of arrival. The guidelines call for the initial computed tomography (CT) to be completed within 25 minutes of the patient’s arrival and interpreted by a physician with expertise in reading CT studies within 45 minutes of arrival.6,24
In the ED, the National Institutes of Health Stroke Scale (NIHSS)25 (TABLE 3) is an ideal way to focus and record the neurological exam.6 The scale assesses 6 separate neurologic functions (level of consciousness, vision, motor function, sensory function, language, and cerebellar function) and can be performed within 5 to 8 minutes. It yields a score from 0 to 42, with the higher numbers indicating worse neurologic function.26 Although a score ≤10 is generally considered to be predictive of a stroke mimic, a recent study found that 19% of patients with an NIHSS score >10 also had conditions masquerading as stroke.27
Imaging leads to accurate diagnosis. The rate at which stroke mimics are mistaken for actual strokes varies with the population studied and the diagnostic tests performed. While stroke is largely a clinical diagnosis and a history and physical exam focused on onset, duration, and symptoms are key elements in differentiating stroke from a stroke mimic, studies have found that the incidence of misdiagnosis (19% with history, physical, and lab work alone) drops to 5% when noncontrast CT is added. When diffusion-weighted magnetic resonance imaging (MRI) is used instead, misdiagnosis drops to just 2%.11,12,14,22,23
Basic lab tests—a complete blood count and basic metabolic panel, with blood alcohol, hepatic function tests, and toxicology screens in select cases—help rule out stroke mimics. Radiographic imaging of the brain provides further clarification (FIGURE 1A AND 1B), serving 2 main purposes: to (1) evaluate diagnoses other than stroke and (2) identify the presence of any acute intracranial bleeding. Noncontrast CT scans detect acute hemorrhage with a sensitivity of 89% and specificity of 100%.27 CT angiography (which can identify the location of a clot) and CT perfusion (which allows an assessment of any existing penumbra) can also be obtained in a timely fashion with newer multislice scanners.
Some institutions, however, evaluate acute stroke patients with MRI. Depending on the sequences used, MRI has the advantage of being able to detect early ischemic changes, diffusion and perfusion mismatches, and abnormalities of the posterior fossa.29 In acute ischemic stroke, diffusion-weighted MRI has a sensitivity of 83% and specificity of 96%, compared with a sensitivity of 16% and specificity of 98% for noncontrast CT.28
TABLE 3
National Institutes of Health Stroke Scale25
| Item | Response score* |
|---|---|
| 1a. Level of consciousness | 0 = alert 1 = not alert 2 = obtunded 3 = unresponsive |
| 1b. Level of consciousness Questions | 0 = answers both correctly 1 = answers one correctly 2 = answers neither correctly |
| 1c. Level of consciousness Commands | 0 = performs both tasks correctly 1 = performs one task correctly 2 = performs neither task correctly |
| 2. Gaze | 0 = normal 1 = partial gaze palsy 2 = total gaze palsy |
| 3. Visual fields | 0 = no visual loss 1 = partial hemianopsia 2 = complete hemianopsia 3 = bilateral hemianopsia |
| 4. Facial palsy | 0 = normal 1 = minor paralysis 2 = partial paralysis 3 = complete paralysis |
| 5. Motor arm a. Left b. Right | 0 = no drift 1 = drifts before 5 sec 2 = falls before 10 sec 3 = no effort against gravity 4 = no movement |
| 6. Motor leg a. Left b. Right | 0 = no drift 1 = drifts before 5 sec 2 = falls before 10 sec 3 = no effort against gravity 4 = no movement |
| 7. Ataxia | 0 = absent 1 = 1 limb 2 = 2 limbs |
| 8. Sensory | 0 = normal 1 = mild loss 2 = severe loss |
| 9. Language | 0 = normal 1 = mild aphasia 2 = severe aphasia 3 = mute or global aphasia |
| 10. Dysarthria | 0 = normal 1 = mild 2 = severe |
| 11. Extinction/inattention | 0 = normal 1 = mild 2 = severe |
| * Yields a score from 0 to 42 (higher numbers indicate worse neurologic function). | |
FIGURE 1
2 patients with common symptoms, vastly different diagnoses
The markedly abnormal perfusion (arrows) this CT image reveals corresponds to an acute occlusion of the left vertebral artery and a subsequent infarct.
An axial postcontrast MRI reveals multiple lesions in the left temporal lobe (arrows) in a patient with rapid-onset mental changes. The diagnosis: cryptococcal meningoencephalitis.
CORRESPONDENCE
Konrad C. Nau, MD, West Virginia University Department of Family Medicine-Eastern Division, 171 Taylor Street, Harpers Ferry, WV 25425; [email protected]
1. Lloyd-Jones D, Adams R, Carnethon M, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480-486.
2. Scott PA, Silbergeit R. Misdiagnosis of stroke in tissue plasminogen activator-treated patients: characteristics and outcomes. Ann Emerg Med. 2003;42:611-618.
3. Morgenstern LB, Lisabeth LD, Mecozzi AC, et al. A population-based study of acute stroke and TIA diagnosis. Neurology. 2004;62:895-900.
4. Nicol MB, Thrift AG. Knowledge of risk factors and warning signs of stroke. Vasc Health Risk Manag. 2005;1:137-147.
5. Albers GW. Transient ischemic attack—proposal for a new definition. N Engl J Med. 2002;347:1713-1716.
6. Adams HP, del Zoppo G, Alberts MJ, et al. Guidelines for early management of adults with ischemic stroke. Circulation. 2007;115:e478-e534.
7. Rosamond WD, Reeves MJ, Johnson A, et al. Paul Coverdell National Acute Stroke Registry Prototype Investigators. Documentation of stroke onset time: challenges and recommendations. Am J Prev Med. 2006;6(suppl 2):S230-S234.
8. Crocco TJ. Streamlining stroke care: from symptom onset to emergency department. J Emerg Med. 2007;33:255-260.
9. Nor AM, Davis J, Sen B, et al. The recognition of stroke in the emergency room scale: development and validation of a stroke recognition scale. Lancet Neurol. 2005;4:727-734.
10. Practice Advisory: Thrombolytic therapy for acute ischemic stroke-summary statement. Report of the Quality standards subcommittee of the American Academy of Neurology. Neurology. 1996;47:835-839.
11. Libman RB, Wirkowski E, Alvir J, et al. Conditions that mimic stroke in the emergency department. Implications for acute stroke trials. Arch Neurol. 1995;52:1119-1122.
12. Vroomen P, Buddingh MK, Kuijckx G, et al. The incidence of stroke mimics among stroke department admissions in relation to age group. J Stroke Cerebrovasc Dis. 2008;17:418-422.
13. Gallmetzer P, Leutmezer F, Serles W, et al. Postictal paresis in focal epilepsies: incidence, duration, and causes. Neurology. 2004;12:2160-2164.
14. Huff JS. Stroke mimics and chameleons. Emerg Med Clin N Am. 2002;20:583-595.
15. Bousser MG, Welch KM. Relation between migraine and stroke. Lancet Neurol. 2005;4:533-542.
16. Bigal ME, Kurth T, Hu H, et al. Migraine and cardiovascular disease: possible mechanisms of interaction. Neurology. 2009;72:1864-1871.
17. Snyder H, Robinson K, Shah D, et al. Signs and symptoms of patients with brain tumors presenting to the emergency department. J Emerg Med. 1993;11:253-258.
18. Stone J, Zeman A, Sharpe M. Functional weakness and sensory disturbance. J Neurol Neurosurg Psychiatr. 2002;73:241-245.
19. Phoebe SC, Tobiano PS, Wang HE, et al. Case of conversion disorder presenting as a severe acute stroke. J Emerg Med. 2006;30:283-286.
20. Chen DK, So YT, Fischer RS. Use of serum prolactin in diagnosing epileptic seizures. Report of the therapeutics and technology subcommittee of the American Academy of Neurology. Neurology. 2005;65:668-675.
21. Quinette P, Guillery-Girard B, Dayan J, et al. What does transient global amnesia really mean? Review of the literature and thorough study of 142 cases. Brain. 2006;129:1640-1658.
22. Kothari RU, Brott T, Broderick JP, et al. Emergency physicians: accuracy in diagnosis of stroke. Stroke. 1995;26:2238-2241.
23. Ay H, Buonanno FS, Rordorf G, et al. Normal diffusion-weighted MRI during stroke-like deficits. Neurology. 1999;52:1784-1792.
24. Bock BF. Response system for patients presenting with acute stroke. In: Marler JR, Jones PM, Emr M, ed. Proceeding of a National Symposium on Rapid Identification and Treatment of Acute Stroke: 1997. Bethesda, MD: National Institute of Neurological Disorders and Stroke, National Institutes of Health; 1997.
25. National Institutes of Health. Know stroke. Available at: http://www.ninds.nih.gov/doctors/NIH_Stroke_Scale_Booklet.pdf. Accessed December 10, 2009.
26. Kasner SE. Clinical interpretation and use of stroke scales. Lancet Neurol. 2006;5:603-612.
27. Hand PJ, Kwan J, Lindley RI, et al. Distinguishing between stroke and mimic at the bedside: the Brain Attack Study. Stroke. 2006;36:769-775.
28. Chalela JA, Kidwell CS, Nentwich LM, et al. Magnetic resonance imaging and computerized tomography in emergency assessment of patients with suspected acute stroke—a prospective comparison. Lancet. 2007;369:293-298.
29. Kohrmann M, Jüttler E, Huttner HB, et al. Acute stroke imaging for thrombolytic therapy—an update. Cerebrovasc Dis. 2007;24:161-169.
30. Kothari RU, Panciolo A, Liu T, et al. Cincinnati prehospital stroke scale: reproducibility and validity. Ann Emerg Med. 1999;33:373-378.
31. Harbison J, Hossain O, Jenkinson D, et al. Diagnostic accuracy of stroke referrals from primary care, emergency room physicians, and ambulance staff using the face arm speech test. Stroke. 2003;34:71-76.
32. Kidwell CS, Starkman S, Eckstein M, et al. Identifying stroke in the field: prospective validation of the Los Angeles prehospital stroke screen (LAPPS). Stroke. 2000;31:71-76.
1. Lloyd-Jones D, Adams R, Carnethon M, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480-486.
2. Scott PA, Silbergeit R. Misdiagnosis of stroke in tissue plasminogen activator-treated patients: characteristics and outcomes. Ann Emerg Med. 2003;42:611-618.
3. Morgenstern LB, Lisabeth LD, Mecozzi AC, et al. A population-based study of acute stroke and TIA diagnosis. Neurology. 2004;62:895-900.
4. Nicol MB, Thrift AG. Knowledge of risk factors and warning signs of stroke. Vasc Health Risk Manag. 2005;1:137-147.
5. Albers GW. Transient ischemic attack—proposal for a new definition. N Engl J Med. 2002;347:1713-1716.
6. Adams HP, del Zoppo G, Alberts MJ, et al. Guidelines for early management of adults with ischemic stroke. Circulation. 2007;115:e478-e534.
7. Rosamond WD, Reeves MJ, Johnson A, et al. Paul Coverdell National Acute Stroke Registry Prototype Investigators. Documentation of stroke onset time: challenges and recommendations. Am J Prev Med. 2006;6(suppl 2):S230-S234.
8. Crocco TJ. Streamlining stroke care: from symptom onset to emergency department. J Emerg Med. 2007;33:255-260.
9. Nor AM, Davis J, Sen B, et al. The recognition of stroke in the emergency room scale: development and validation of a stroke recognition scale. Lancet Neurol. 2005;4:727-734.
10. Practice Advisory: Thrombolytic therapy for acute ischemic stroke-summary statement. Report of the Quality standards subcommittee of the American Academy of Neurology. Neurology. 1996;47:835-839.
11. Libman RB, Wirkowski E, Alvir J, et al. Conditions that mimic stroke in the emergency department. Implications for acute stroke trials. Arch Neurol. 1995;52:1119-1122.
12. Vroomen P, Buddingh MK, Kuijckx G, et al. The incidence of stroke mimics among stroke department admissions in relation to age group. J Stroke Cerebrovasc Dis. 2008;17:418-422.
13. Gallmetzer P, Leutmezer F, Serles W, et al. Postictal paresis in focal epilepsies: incidence, duration, and causes. Neurology. 2004;12:2160-2164.
14. Huff JS. Stroke mimics and chameleons. Emerg Med Clin N Am. 2002;20:583-595.
15. Bousser MG, Welch KM. Relation between migraine and stroke. Lancet Neurol. 2005;4:533-542.
16. Bigal ME, Kurth T, Hu H, et al. Migraine and cardiovascular disease: possible mechanisms of interaction. Neurology. 2009;72:1864-1871.
17. Snyder H, Robinson K, Shah D, et al. Signs and symptoms of patients with brain tumors presenting to the emergency department. J Emerg Med. 1993;11:253-258.
18. Stone J, Zeman A, Sharpe M. Functional weakness and sensory disturbance. J Neurol Neurosurg Psychiatr. 2002;73:241-245.
19. Phoebe SC, Tobiano PS, Wang HE, et al. Case of conversion disorder presenting as a severe acute stroke. J Emerg Med. 2006;30:283-286.
20. Chen DK, So YT, Fischer RS. Use of serum prolactin in diagnosing epileptic seizures. Report of the therapeutics and technology subcommittee of the American Academy of Neurology. Neurology. 2005;65:668-675.
21. Quinette P, Guillery-Girard B, Dayan J, et al. What does transient global amnesia really mean? Review of the literature and thorough study of 142 cases. Brain. 2006;129:1640-1658.
22. Kothari RU, Brott T, Broderick JP, et al. Emergency physicians: accuracy in diagnosis of stroke. Stroke. 1995;26:2238-2241.
23. Ay H, Buonanno FS, Rordorf G, et al. Normal diffusion-weighted MRI during stroke-like deficits. Neurology. 1999;52:1784-1792.
24. Bock BF. Response system for patients presenting with acute stroke. In: Marler JR, Jones PM, Emr M, ed. Proceeding of a National Symposium on Rapid Identification and Treatment of Acute Stroke: 1997. Bethesda, MD: National Institute of Neurological Disorders and Stroke, National Institutes of Health; 1997.
25. National Institutes of Health. Know stroke. Available at: http://www.ninds.nih.gov/doctors/NIH_Stroke_Scale_Booklet.pdf. Accessed December 10, 2009.
26. Kasner SE. Clinical interpretation and use of stroke scales. Lancet Neurol. 2006;5:603-612.
27. Hand PJ, Kwan J, Lindley RI, et al. Distinguishing between stroke and mimic at the bedside: the Brain Attack Study. Stroke. 2006;36:769-775.
28. Chalela JA, Kidwell CS, Nentwich LM, et al. Magnetic resonance imaging and computerized tomography in emergency assessment of patients with suspected acute stroke—a prospective comparison. Lancet. 2007;369:293-298.
29. Kohrmann M, Jüttler E, Huttner HB, et al. Acute stroke imaging for thrombolytic therapy—an update. Cerebrovasc Dis. 2007;24:161-169.
30. Kothari RU, Panciolo A, Liu T, et al. Cincinnati prehospital stroke scale: reproducibility and validity. Ann Emerg Med. 1999;33:373-378.
31. Harbison J, Hossain O, Jenkinson D, et al. Diagnostic accuracy of stroke referrals from primary care, emergency room physicians, and ambulance staff using the face arm speech test. Stroke. 2003;34:71-76.
32. Kidwell CS, Starkman S, Eckstein M, et al. Identifying stroke in the field: prospective validation of the Los Angeles prehospital stroke screen (LAPPS). Stroke. 2000;31:71-76.
Anemia and chronic kidney disease: What’s the connection?
• Evaluate for chronic kidney disease (CKD) anemia when a patient has a serum creatinine ≥2 mg/dL and hemoglobin <12 g/dL (adult males and postmenopausal females) or <11 g/dL (premenopausal females). A
• Before you treat CKD anemia, correct any underlying iron deficiency. A
• Start anemia therapy with erythropoietin-stimulating agents when hemoglobin is ≤10 g/dL, and maintain target hemoglobin levels between 11 and 12 g/dL, in accordance with National Kidney Foundation guidelines. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Mary J, a 65-year-old woman with stage 3 chronic kidney disease (CKD), is in your office for a follow-up appointment. Over the past 6 months, she has noticed a decrease in her energy level. On her routine blood work, you see that her hemoglobin has been slowly declining over the past year. It is now 9 g/dL and her estimated glomerular filtration rate (GFR) is 40 mL/min.
How would you evaluate Mary’s anemia, and would you suspect that it was related to her CKD?
Most physicians are aware that CKD—which affects approximately 10% of the US population1—has a deleterious effect on cardiovascular disease, but many fail to recognize the impact it has on the hematopoietic system. Managing the anemia that accompanies CKD in patients like Mary requires a finely tuned diagnostic approach and treatment strategy. This article will help toward that end.
Anemia of CKD: A common problem
Anemia of CKD is one of the first signs of kidney dysfunction, yet it often goes undetected because of its insidious onset. Anemia develops gradually as kidney function declines and the GFR drops to 70 mL/min in male patients and 50 mL/min in females.2 Epidemiologic data indicate that two-thirds of patients in the early stages of kidney failure are also anemic, with a hemoglobin level of less than 11 g/dL, yet only one-third of these patients have ever received erythropoietin-stimulating agents (ESAs) to treat their anemia.1 The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines recommend that the evaluation of anemia of CKD begin in patients with a serum creatinine ≥2 mg/dL when the hemoglobin is <12 g/dL in adult males and postmenopausal females and <11 g/dL in premenopausal females.3
How kidney failure leads to anemia
Patients like Mary develop anemia of CKD because failing kidneys produce less erythropoietin (EPO) than the body requires for the production of red blood cells. EPO is an endogenous hormone produced by peritubular fibroblasts in the renal cortex.4 Most of this hormone (90%) is produced in the kidney, with the remainder manufactured by hepatocytes.
Erythropoiesis is stimulated by blood loss, decreased oxygen tension, and an increase in oxygen affinity, which leads to an increase in EPO production via upregulation of the EPO gene. In healthy individuals, detection of hypoxia by the kidney can result in a 1000-fold increase in EPO production.5 Patients with CKD don’t have that kind of robust response, and their EPO levels remain normal or below normal even when challenged by lack of oxygen. Anemia in CKD can also be caused by nutritional deficiencies, decreased red blood cell survival because of uremic toxins, oxidative stress, inflammation, and the use of angiotensin-converting enzyme (ACE) inhibitors.
Chronic anemia, CKD, and CV disease: A deadly triad
The leading cause of death in patients with CKD is cardiovascular disease. Patients with cardiorenal anemia syndrome develop a self-perpetuating triad that increases the risk of death when all 3 conditions are present. Anemic patients double their relative risk of death when CKD is present and triple their risk if they have anemia, CKD, and cardiovascular disease.6
Epidemiologic studies suggest an association among anemia, left ventricular hypertrophy (LVH), mortality, and cardiovascular outcomes. One study evaluated 2423 stage 3 and 4 CKD patients with anemia, defined as hemoglobin <13 g/dL in males and <12 g/dL in females. The results showed an increase in composite outcomes of myocardial infarction, stroke, and death.7 A prospective study evaluating 246 people with stages 2 to 4 CKD reported anemia to be an independent risk factor for the development of LVH.8 The stages of CKD are shown in the TABLE.
Suspected mechanisms of cardiovascular disease progression due to chronic anemia include tissue hypoxia, free radical formation, endothelial dysfunction, and vascular damage. Compensatory neurohumeral adaptations result in an increased sympathetic response and upregulation of the reninangiotensin-aldosterone system.9
TABLE
Stages of chronic kidney disease
| Stage | Description | GFR (mL/min/1.73 m2) |
|---|---|---|
| 1 | Kidney damage with normal or increased GFR | ≥90 |
| 2 | Kidney damage with mildly decreased GFR | 60-89 |
| 3 | Moderately decreased GFR | 30-59 |
| 4 | Severely decreased GFR | 15-29 |
| 5 | Kidney failure | <15 or dialysis |
| GFR, glomerular filtration rate. | ||
| Source: KDOQI Clinical Practice Guideline and Clinical Practice Recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis. 2007.3 | ||
Anemia of CKD: A diagnosis of exclusion
Because anemia can have many causes, other possibilities must be ruled out before a diagnosis of CKD anemia can be made. Testing should be tailored to each individual situation, determined by a thorough history and physical. Steps in the diagnosis are shown in the FLOW CHART. A basic work-up should include complete blood count with differential, iron studies (ferritin, serum Fe, and total iron binding capacity), reticulocyte count, and a guaiac test. Other blood tests, such as thyroid-stimulating hormone (TSH), B12, and folate levels, and a hemolysis panel (lactate dehydrogenase, haptoglobin), should be obtained if the history suggests these disorders. A peripheral blood smear showing normocytic red blood cells with a normochromic pattern would favor the diagnosis of anemia of CKD.
FLOW CHART
A step-by-step guide to CKD anemia diagnosis and treatment
CBC, complete blood count; CKD, chronic kidney disease; ESA, erythropoietin-stimulating agents; R/O, rule out; TIBC/TSAT, total iron-binding capacity/transferrin saturation.
A look at the iron connection
Many patients with CKD anemia have iron deficiency and are unable to produce adequate numbers of red blood cells. Iron deficiency can have many causes: not enough iron-rich food in the diet, chronic bleeding, malabsorption, or an occult gastrointestinal malignancy. Once iron deficiency anemia is diagnosed, a colonoscopy is warranted to rule out occult malignancy. Ferritin, a protein found mostly in macrophages and hepatocytes, stores iron and serves as a marker for total iron stores. Using stored iron requires transferrin, a transporting protein, to shuttle iron from the reticuloendothelial system and gut to the bone marrow. CKD is a pro-inflammatory state that results in a limited ability to use iron stores. For this reason, patients with CKD require higher levels of iron.
Absolute iron deficiency. Iron deficiency in CKD patients with serum ferritin <100 ng/mL and transferrin saturation (TSAT) <20% is characterized as absolute iron deficiency. The TSAT represents the percent of iron bound to transferrin and is a good indicator of the body’s functional capacity to use stored iron.
Relative iron deficiency and iron block. Patients who do not respond to ESA therapy even though they have adequate iron stores are said to have a functional or relative iron deficiency. Iron block is a condition that results in anemia from a chronic inflammatory state such as infection, autoimmune disorders, or malignancies. It resolves once the inflammatory process abates. Both conditions have similar anemia profiles, with a serum ferritin >100 ng/mL and a TSAT <20%. Differentiating between these conditions requires dynamic testing using serial iron studies and observing responses to ESAs and iron supplementation.
Options for correcting iron deficiency
After a thorough history and physical with appropriate screening, you find that Mary has an iron deficiency that must be corrected before her anemia can be treated effectively. Treatment for iron deficiency is usually initiated with oral therapy, at the recommended dose of 200 mg oral elemental iron a day in 3 divided doses.
If the oral therapy does not correct iron deficiency within 3 months, or a patient cannot tolerate the constipation that is often a side effect of this therapy, IV iron administration can be considered. Because CKD patients do not have the ongoing iron losses seen in patients with end-stage renal disease (ESRD), a conservative approach using a single IV dose followed by repeat testing is warranted. The goal is to achieve ferritin levels >100 ng/dL and TSAT >20%. A number of products for IV iron administration are available. The most widely used are iron dextran (INFeD), ferric gluconate (Ferrlecit), and iron sucrose (Venofer).
Iron stores are replenished? Time to treat the anemia
When ferritin levels and TSAT show that iron deficiency has been corrected, ESA treatment for anemia can begin. Two major brands of ESAs currently in use in the United States are a recombinant human erythropoietin (rHuEPO) known as epoetin alfa (Procrit, Epogen), and darbepoetin alpha (Aranesp). Both medications are effective and can be given intravenously or subcutaneously. Subcutaneous darbepoetin alpha has a longer half-life compared with epoetin alpha (70 vs 24 hours), so dosing intervals can be longer.10,11 ESAs should not be started in patients with uncontrolled hypertension until the blood pressure is controlled, or in patients with an active malignancy unless the treatment is directly supervised by an oncologist.
Aim for complete anemia resolution? That’s controversial
Treatment of CKD anemia with ESAs is widely practiced, but controversy over whether it is beneficial to aim for complete resolution of anemia is ongoing. The CREATE (Cardiovascular Risk Reduction by Early Anemia Treatment) and CHOIR (Correction of Hemoglobin and Outcomes in Renal Insufficiency) trials published in 2006 failed to resolve the issue.12,13
In the CREATE trial, patients targeted to achieve normal hemoglobin levels did no better in avoiding cardiovascular events than patients targeted for lower levels. The CHOIR trial was stopped early because of an increased trend toward death and hospitalization for congestive heart failure in the group with therapy targeted to achieve normal hemoglobin levels.
The recently published TREAT (Trial to Reduce Cardiovascular Events with Aranesp Therapy) study of patients with type 2 diabetes and CKD showed no reduction in all-cause mortality, cardiovascular morbidity, or ESRD in patients receiving Aranesp targeted to achieve a hemoglobin level of approximately 13 g/dL, compared with placebo.14 The study did demonstrate, however, that patients receiving Aranesp were about twice as likely to have a stroke than the placebo subjects (101 vs 53)—which might lead clinicians to ponder whether the gains, if any, were worth the risk.
Revised labeling. Late last year, the US Food and Drug Administration approved a label change for Procrit and Aranesp, warning that patients with renal failure “experienced greater risks for death and serious cardiovascular events when administered ESAs to target higher vs lower hemoglobin levels” and advising physicians to “individualize dosing to achieve and maintain hemoglobin levels within the range of 10 to 12 g/dL.”10,11 The 2007 NKF KDOQI guidelines suggest maintaining a hemoglobin level between 11 and 12 g/dL and have not incorporated the results of the TREAT trial.
Some patients don’t respond to ESAs
Inadequate response to ESAs is most commonly caused by underdosing or inadequate iron stores. NKF KDOQI guidelines recommend checking TSAT and ferritin prior to initiating therapy and monitoring these levels every 3 months.3 True nonresponders are individuals with good iron stores who are unable to achieve target hemoglobin within 4 to 6 months despite receiving subcutaneous epoetin 300 IU/kg per week. Inadequate response to ESAs can be caused by ongoing occult blood loss, infection, inflammation, nutritional deficiencies, hemolysis, hemoglobinemias, aluminum toxicity, anti-EPO antibody, hyperparathyroidism, multiple myeloma, and bone marrow dysfunction.10,11 If patients do not respond to ESA therapy, the NKF KDOQI guidelines recommend referral to a nephrologist or hematologist.3
How did Mary fare?
Mary did well taking oral iron supplementation. Once her iron deficiency was corrected, you were able to begin treating her anemia. After appropriate titration of her ESA, she was able to maintain a hemoglobin level between 11 and 12 g/dL 4 months into therapy. On a follow-up visit, she had no side effects from the medication and reported an increase in her energy level.
CORRESPONDENCE
Jonathan Taliercio, DO, Cleveland Clinic, Department of Nephrology and Hypertension, 9500 Euclid Avenue, Cleveland, OH 44195; [email protected]
1. United States Renal Data System, USRDS. 2009 Annual Data Report. Atlas of Chronic Kidney Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2009.
2. Hsu CJ, McCulloch CE, Curhan GC. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol. 2002;13:504-510.
3. KDOQI Clinical Practice Guideline and Clinical Practice Recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis. 2007;50:471-530.
4. Donnelly S. Why is erythropoietin made in the kidney? The kidney functions as a critmeter. Am J Kidney Dis. 2001;38:415-425.
5. Ebert B, Franklin H. Regulation of the erythropoietin gene. Blood. 1999;94:1864-1877.
6. Silverberg D, Wexler D, Blum M, et al. The cardio-renal anaemia syndrome: does it exist? Nephrol Dial Transplant. 2003;18(suppl 8):viii 7-viii 12.
7. Weiner D, Tighiouart H, Vlagopoulos P, et al. Effects of anemia and left ventricular hypertrophy on cardiovascular disease in patients with chronic kidney disease. J Am Soc Nephrol. 2005;16:1803-1810.
8. Levin A, Thompson C, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am J Kidney Dis. 1999;34:125-134.
9. Rao M, Pereira B. Optimal anemia management reduces cardiovascular morbidity, mortality, and costs in chronic kidney disease. Kidney Int. 2005;68:1432-1438.
10. Amgen. Aranesp (Darbepoetin Alpha) package insert. Available at www.aranesp.com/professional/crf/full_prescribing_info/pi.jsp. Accessed November 16, 2009.
11. Amgen. Procrit (Epoetin Alpha) package insert. Available at www.procrit.com/sites/default/files/shared/OBI/PI/ProcritBooklet.pdf#page=1. Accessed November 16, 2009.
12. Drueke T, Locatelli F, Clyne N, et al. Normalization of hemoglobin level in patients with CKD and anemia. N Engl J Med. 2006;355:2071-2084.
13. Singh A, Szczech L, Tang K. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355:2085-2098.
14. Pfeffer MA, Burdmann EA, Chen CY, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361:2019-2032.
• Evaluate for chronic kidney disease (CKD) anemia when a patient has a serum creatinine ≥2 mg/dL and hemoglobin <12 g/dL (adult males and postmenopausal females) or <11 g/dL (premenopausal females). A
• Before you treat CKD anemia, correct any underlying iron deficiency. A
• Start anemia therapy with erythropoietin-stimulating agents when hemoglobin is ≤10 g/dL, and maintain target hemoglobin levels between 11 and 12 g/dL, in accordance with National Kidney Foundation guidelines. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Mary J, a 65-year-old woman with stage 3 chronic kidney disease (CKD), is in your office for a follow-up appointment. Over the past 6 months, she has noticed a decrease in her energy level. On her routine blood work, you see that her hemoglobin has been slowly declining over the past year. It is now 9 g/dL and her estimated glomerular filtration rate (GFR) is 40 mL/min.
How would you evaluate Mary’s anemia, and would you suspect that it was related to her CKD?
Most physicians are aware that CKD—which affects approximately 10% of the US population1—has a deleterious effect on cardiovascular disease, but many fail to recognize the impact it has on the hematopoietic system. Managing the anemia that accompanies CKD in patients like Mary requires a finely tuned diagnostic approach and treatment strategy. This article will help toward that end.
Anemia of CKD: A common problem
Anemia of CKD is one of the first signs of kidney dysfunction, yet it often goes undetected because of its insidious onset. Anemia develops gradually as kidney function declines and the GFR drops to 70 mL/min in male patients and 50 mL/min in females.2 Epidemiologic data indicate that two-thirds of patients in the early stages of kidney failure are also anemic, with a hemoglobin level of less than 11 g/dL, yet only one-third of these patients have ever received erythropoietin-stimulating agents (ESAs) to treat their anemia.1 The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines recommend that the evaluation of anemia of CKD begin in patients with a serum creatinine ≥2 mg/dL when the hemoglobin is <12 g/dL in adult males and postmenopausal females and <11 g/dL in premenopausal females.3
How kidney failure leads to anemia
Patients like Mary develop anemia of CKD because failing kidneys produce less erythropoietin (EPO) than the body requires for the production of red blood cells. EPO is an endogenous hormone produced by peritubular fibroblasts in the renal cortex.4 Most of this hormone (90%) is produced in the kidney, with the remainder manufactured by hepatocytes.
Erythropoiesis is stimulated by blood loss, decreased oxygen tension, and an increase in oxygen affinity, which leads to an increase in EPO production via upregulation of the EPO gene. In healthy individuals, detection of hypoxia by the kidney can result in a 1000-fold increase in EPO production.5 Patients with CKD don’t have that kind of robust response, and their EPO levels remain normal or below normal even when challenged by lack of oxygen. Anemia in CKD can also be caused by nutritional deficiencies, decreased red blood cell survival because of uremic toxins, oxidative stress, inflammation, and the use of angiotensin-converting enzyme (ACE) inhibitors.
Chronic anemia, CKD, and CV disease: A deadly triad
The leading cause of death in patients with CKD is cardiovascular disease. Patients with cardiorenal anemia syndrome develop a self-perpetuating triad that increases the risk of death when all 3 conditions are present. Anemic patients double their relative risk of death when CKD is present and triple their risk if they have anemia, CKD, and cardiovascular disease.6
Epidemiologic studies suggest an association among anemia, left ventricular hypertrophy (LVH), mortality, and cardiovascular outcomes. One study evaluated 2423 stage 3 and 4 CKD patients with anemia, defined as hemoglobin <13 g/dL in males and <12 g/dL in females. The results showed an increase in composite outcomes of myocardial infarction, stroke, and death.7 A prospective study evaluating 246 people with stages 2 to 4 CKD reported anemia to be an independent risk factor for the development of LVH.8 The stages of CKD are shown in the TABLE.
Suspected mechanisms of cardiovascular disease progression due to chronic anemia include tissue hypoxia, free radical formation, endothelial dysfunction, and vascular damage. Compensatory neurohumeral adaptations result in an increased sympathetic response and upregulation of the reninangiotensin-aldosterone system.9
TABLE
Stages of chronic kidney disease
| Stage | Description | GFR (mL/min/1.73 m2) |
|---|---|---|
| 1 | Kidney damage with normal or increased GFR | ≥90 |
| 2 | Kidney damage with mildly decreased GFR | 60-89 |
| 3 | Moderately decreased GFR | 30-59 |
| 4 | Severely decreased GFR | 15-29 |
| 5 | Kidney failure | <15 or dialysis |
| GFR, glomerular filtration rate. | ||
| Source: KDOQI Clinical Practice Guideline and Clinical Practice Recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis. 2007.3 | ||
Anemia of CKD: A diagnosis of exclusion
Because anemia can have many causes, other possibilities must be ruled out before a diagnosis of CKD anemia can be made. Testing should be tailored to each individual situation, determined by a thorough history and physical. Steps in the diagnosis are shown in the FLOW CHART. A basic work-up should include complete blood count with differential, iron studies (ferritin, serum Fe, and total iron binding capacity), reticulocyte count, and a guaiac test. Other blood tests, such as thyroid-stimulating hormone (TSH), B12, and folate levels, and a hemolysis panel (lactate dehydrogenase, haptoglobin), should be obtained if the history suggests these disorders. A peripheral blood smear showing normocytic red blood cells with a normochromic pattern would favor the diagnosis of anemia of CKD.
FLOW CHART
A step-by-step guide to CKD anemia diagnosis and treatment
CBC, complete blood count; CKD, chronic kidney disease; ESA, erythropoietin-stimulating agents; R/O, rule out; TIBC/TSAT, total iron-binding capacity/transferrin saturation.
A look at the iron connection
Many patients with CKD anemia have iron deficiency and are unable to produce adequate numbers of red blood cells. Iron deficiency can have many causes: not enough iron-rich food in the diet, chronic bleeding, malabsorption, or an occult gastrointestinal malignancy. Once iron deficiency anemia is diagnosed, a colonoscopy is warranted to rule out occult malignancy. Ferritin, a protein found mostly in macrophages and hepatocytes, stores iron and serves as a marker for total iron stores. Using stored iron requires transferrin, a transporting protein, to shuttle iron from the reticuloendothelial system and gut to the bone marrow. CKD is a pro-inflammatory state that results in a limited ability to use iron stores. For this reason, patients with CKD require higher levels of iron.
Absolute iron deficiency. Iron deficiency in CKD patients with serum ferritin <100 ng/mL and transferrin saturation (TSAT) <20% is characterized as absolute iron deficiency. The TSAT represents the percent of iron bound to transferrin and is a good indicator of the body’s functional capacity to use stored iron.
Relative iron deficiency and iron block. Patients who do not respond to ESA therapy even though they have adequate iron stores are said to have a functional or relative iron deficiency. Iron block is a condition that results in anemia from a chronic inflammatory state such as infection, autoimmune disorders, or malignancies. It resolves once the inflammatory process abates. Both conditions have similar anemia profiles, with a serum ferritin >100 ng/mL and a TSAT <20%. Differentiating between these conditions requires dynamic testing using serial iron studies and observing responses to ESAs and iron supplementation.
Options for correcting iron deficiency
After a thorough history and physical with appropriate screening, you find that Mary has an iron deficiency that must be corrected before her anemia can be treated effectively. Treatment for iron deficiency is usually initiated with oral therapy, at the recommended dose of 200 mg oral elemental iron a day in 3 divided doses.
If the oral therapy does not correct iron deficiency within 3 months, or a patient cannot tolerate the constipation that is often a side effect of this therapy, IV iron administration can be considered. Because CKD patients do not have the ongoing iron losses seen in patients with end-stage renal disease (ESRD), a conservative approach using a single IV dose followed by repeat testing is warranted. The goal is to achieve ferritin levels >100 ng/dL and TSAT >20%. A number of products for IV iron administration are available. The most widely used are iron dextran (INFeD), ferric gluconate (Ferrlecit), and iron sucrose (Venofer).
Iron stores are replenished? Time to treat the anemia
When ferritin levels and TSAT show that iron deficiency has been corrected, ESA treatment for anemia can begin. Two major brands of ESAs currently in use in the United States are a recombinant human erythropoietin (rHuEPO) known as epoetin alfa (Procrit, Epogen), and darbepoetin alpha (Aranesp). Both medications are effective and can be given intravenously or subcutaneously. Subcutaneous darbepoetin alpha has a longer half-life compared with epoetin alpha (70 vs 24 hours), so dosing intervals can be longer.10,11 ESAs should not be started in patients with uncontrolled hypertension until the blood pressure is controlled, or in patients with an active malignancy unless the treatment is directly supervised by an oncologist.
Aim for complete anemia resolution? That’s controversial
Treatment of CKD anemia with ESAs is widely practiced, but controversy over whether it is beneficial to aim for complete resolution of anemia is ongoing. The CREATE (Cardiovascular Risk Reduction by Early Anemia Treatment) and CHOIR (Correction of Hemoglobin and Outcomes in Renal Insufficiency) trials published in 2006 failed to resolve the issue.12,13
In the CREATE trial, patients targeted to achieve normal hemoglobin levels did no better in avoiding cardiovascular events than patients targeted for lower levels. The CHOIR trial was stopped early because of an increased trend toward death and hospitalization for congestive heart failure in the group with therapy targeted to achieve normal hemoglobin levels.
The recently published TREAT (Trial to Reduce Cardiovascular Events with Aranesp Therapy) study of patients with type 2 diabetes and CKD showed no reduction in all-cause mortality, cardiovascular morbidity, or ESRD in patients receiving Aranesp targeted to achieve a hemoglobin level of approximately 13 g/dL, compared with placebo.14 The study did demonstrate, however, that patients receiving Aranesp were about twice as likely to have a stroke than the placebo subjects (101 vs 53)—which might lead clinicians to ponder whether the gains, if any, were worth the risk.
Revised labeling. Late last year, the US Food and Drug Administration approved a label change for Procrit and Aranesp, warning that patients with renal failure “experienced greater risks for death and serious cardiovascular events when administered ESAs to target higher vs lower hemoglobin levels” and advising physicians to “individualize dosing to achieve and maintain hemoglobin levels within the range of 10 to 12 g/dL.”10,11 The 2007 NKF KDOQI guidelines suggest maintaining a hemoglobin level between 11 and 12 g/dL and have not incorporated the results of the TREAT trial.
Some patients don’t respond to ESAs
Inadequate response to ESAs is most commonly caused by underdosing or inadequate iron stores. NKF KDOQI guidelines recommend checking TSAT and ferritin prior to initiating therapy and monitoring these levels every 3 months.3 True nonresponders are individuals with good iron stores who are unable to achieve target hemoglobin within 4 to 6 months despite receiving subcutaneous epoetin 300 IU/kg per week. Inadequate response to ESAs can be caused by ongoing occult blood loss, infection, inflammation, nutritional deficiencies, hemolysis, hemoglobinemias, aluminum toxicity, anti-EPO antibody, hyperparathyroidism, multiple myeloma, and bone marrow dysfunction.10,11 If patients do not respond to ESA therapy, the NKF KDOQI guidelines recommend referral to a nephrologist or hematologist.3
How did Mary fare?
Mary did well taking oral iron supplementation. Once her iron deficiency was corrected, you were able to begin treating her anemia. After appropriate titration of her ESA, she was able to maintain a hemoglobin level between 11 and 12 g/dL 4 months into therapy. On a follow-up visit, she had no side effects from the medication and reported an increase in her energy level.
CORRESPONDENCE
Jonathan Taliercio, DO, Cleveland Clinic, Department of Nephrology and Hypertension, 9500 Euclid Avenue, Cleveland, OH 44195; [email protected]
• Evaluate for chronic kidney disease (CKD) anemia when a patient has a serum creatinine ≥2 mg/dL and hemoglobin <12 g/dL (adult males and postmenopausal females) or <11 g/dL (premenopausal females). A
• Before you treat CKD anemia, correct any underlying iron deficiency. A
• Start anemia therapy with erythropoietin-stimulating agents when hemoglobin is ≤10 g/dL, and maintain target hemoglobin levels between 11 and 12 g/dL, in accordance with National Kidney Foundation guidelines. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Mary J, a 65-year-old woman with stage 3 chronic kidney disease (CKD), is in your office for a follow-up appointment. Over the past 6 months, she has noticed a decrease in her energy level. On her routine blood work, you see that her hemoglobin has been slowly declining over the past year. It is now 9 g/dL and her estimated glomerular filtration rate (GFR) is 40 mL/min.
How would you evaluate Mary’s anemia, and would you suspect that it was related to her CKD?
Most physicians are aware that CKD—which affects approximately 10% of the US population1—has a deleterious effect on cardiovascular disease, but many fail to recognize the impact it has on the hematopoietic system. Managing the anemia that accompanies CKD in patients like Mary requires a finely tuned diagnostic approach and treatment strategy. This article will help toward that end.
Anemia of CKD: A common problem
Anemia of CKD is one of the first signs of kidney dysfunction, yet it often goes undetected because of its insidious onset. Anemia develops gradually as kidney function declines and the GFR drops to 70 mL/min in male patients and 50 mL/min in females.2 Epidemiologic data indicate that two-thirds of patients in the early stages of kidney failure are also anemic, with a hemoglobin level of less than 11 g/dL, yet only one-third of these patients have ever received erythropoietin-stimulating agents (ESAs) to treat their anemia.1 The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines recommend that the evaluation of anemia of CKD begin in patients with a serum creatinine ≥2 mg/dL when the hemoglobin is <12 g/dL in adult males and postmenopausal females and <11 g/dL in premenopausal females.3
How kidney failure leads to anemia
Patients like Mary develop anemia of CKD because failing kidneys produce less erythropoietin (EPO) than the body requires for the production of red blood cells. EPO is an endogenous hormone produced by peritubular fibroblasts in the renal cortex.4 Most of this hormone (90%) is produced in the kidney, with the remainder manufactured by hepatocytes.
Erythropoiesis is stimulated by blood loss, decreased oxygen tension, and an increase in oxygen affinity, which leads to an increase in EPO production via upregulation of the EPO gene. In healthy individuals, detection of hypoxia by the kidney can result in a 1000-fold increase in EPO production.5 Patients with CKD don’t have that kind of robust response, and their EPO levels remain normal or below normal even when challenged by lack of oxygen. Anemia in CKD can also be caused by nutritional deficiencies, decreased red blood cell survival because of uremic toxins, oxidative stress, inflammation, and the use of angiotensin-converting enzyme (ACE) inhibitors.
Chronic anemia, CKD, and CV disease: A deadly triad
The leading cause of death in patients with CKD is cardiovascular disease. Patients with cardiorenal anemia syndrome develop a self-perpetuating triad that increases the risk of death when all 3 conditions are present. Anemic patients double their relative risk of death when CKD is present and triple their risk if they have anemia, CKD, and cardiovascular disease.6
Epidemiologic studies suggest an association among anemia, left ventricular hypertrophy (LVH), mortality, and cardiovascular outcomes. One study evaluated 2423 stage 3 and 4 CKD patients with anemia, defined as hemoglobin <13 g/dL in males and <12 g/dL in females. The results showed an increase in composite outcomes of myocardial infarction, stroke, and death.7 A prospective study evaluating 246 people with stages 2 to 4 CKD reported anemia to be an independent risk factor for the development of LVH.8 The stages of CKD are shown in the TABLE.
Suspected mechanisms of cardiovascular disease progression due to chronic anemia include tissue hypoxia, free radical formation, endothelial dysfunction, and vascular damage. Compensatory neurohumeral adaptations result in an increased sympathetic response and upregulation of the reninangiotensin-aldosterone system.9
TABLE
Stages of chronic kidney disease
| Stage | Description | GFR (mL/min/1.73 m2) |
|---|---|---|
| 1 | Kidney damage with normal or increased GFR | ≥90 |
| 2 | Kidney damage with mildly decreased GFR | 60-89 |
| 3 | Moderately decreased GFR | 30-59 |
| 4 | Severely decreased GFR | 15-29 |
| 5 | Kidney failure | <15 or dialysis |
| GFR, glomerular filtration rate. | ||
| Source: KDOQI Clinical Practice Guideline and Clinical Practice Recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis. 2007.3 | ||
Anemia of CKD: A diagnosis of exclusion
Because anemia can have many causes, other possibilities must be ruled out before a diagnosis of CKD anemia can be made. Testing should be tailored to each individual situation, determined by a thorough history and physical. Steps in the diagnosis are shown in the FLOW CHART. A basic work-up should include complete blood count with differential, iron studies (ferritin, serum Fe, and total iron binding capacity), reticulocyte count, and a guaiac test. Other blood tests, such as thyroid-stimulating hormone (TSH), B12, and folate levels, and a hemolysis panel (lactate dehydrogenase, haptoglobin), should be obtained if the history suggests these disorders. A peripheral blood smear showing normocytic red blood cells with a normochromic pattern would favor the diagnosis of anemia of CKD.
FLOW CHART
A step-by-step guide to CKD anemia diagnosis and treatment
CBC, complete blood count; CKD, chronic kidney disease; ESA, erythropoietin-stimulating agents; R/O, rule out; TIBC/TSAT, total iron-binding capacity/transferrin saturation.
A look at the iron connection
Many patients with CKD anemia have iron deficiency and are unable to produce adequate numbers of red blood cells. Iron deficiency can have many causes: not enough iron-rich food in the diet, chronic bleeding, malabsorption, or an occult gastrointestinal malignancy. Once iron deficiency anemia is diagnosed, a colonoscopy is warranted to rule out occult malignancy. Ferritin, a protein found mostly in macrophages and hepatocytes, stores iron and serves as a marker for total iron stores. Using stored iron requires transferrin, a transporting protein, to shuttle iron from the reticuloendothelial system and gut to the bone marrow. CKD is a pro-inflammatory state that results in a limited ability to use iron stores. For this reason, patients with CKD require higher levels of iron.
Absolute iron deficiency. Iron deficiency in CKD patients with serum ferritin <100 ng/mL and transferrin saturation (TSAT) <20% is characterized as absolute iron deficiency. The TSAT represents the percent of iron bound to transferrin and is a good indicator of the body’s functional capacity to use stored iron.
Relative iron deficiency and iron block. Patients who do not respond to ESA therapy even though they have adequate iron stores are said to have a functional or relative iron deficiency. Iron block is a condition that results in anemia from a chronic inflammatory state such as infection, autoimmune disorders, or malignancies. It resolves once the inflammatory process abates. Both conditions have similar anemia profiles, with a serum ferritin >100 ng/mL and a TSAT <20%. Differentiating between these conditions requires dynamic testing using serial iron studies and observing responses to ESAs and iron supplementation.
Options for correcting iron deficiency
After a thorough history and physical with appropriate screening, you find that Mary has an iron deficiency that must be corrected before her anemia can be treated effectively. Treatment for iron deficiency is usually initiated with oral therapy, at the recommended dose of 200 mg oral elemental iron a day in 3 divided doses.
If the oral therapy does not correct iron deficiency within 3 months, or a patient cannot tolerate the constipation that is often a side effect of this therapy, IV iron administration can be considered. Because CKD patients do not have the ongoing iron losses seen in patients with end-stage renal disease (ESRD), a conservative approach using a single IV dose followed by repeat testing is warranted. The goal is to achieve ferritin levels >100 ng/dL and TSAT >20%. A number of products for IV iron administration are available. The most widely used are iron dextran (INFeD), ferric gluconate (Ferrlecit), and iron sucrose (Venofer).
Iron stores are replenished? Time to treat the anemia
When ferritin levels and TSAT show that iron deficiency has been corrected, ESA treatment for anemia can begin. Two major brands of ESAs currently in use in the United States are a recombinant human erythropoietin (rHuEPO) known as epoetin alfa (Procrit, Epogen), and darbepoetin alpha (Aranesp). Both medications are effective and can be given intravenously or subcutaneously. Subcutaneous darbepoetin alpha has a longer half-life compared with epoetin alpha (70 vs 24 hours), so dosing intervals can be longer.10,11 ESAs should not be started in patients with uncontrolled hypertension until the blood pressure is controlled, or in patients with an active malignancy unless the treatment is directly supervised by an oncologist.
Aim for complete anemia resolution? That’s controversial
Treatment of CKD anemia with ESAs is widely practiced, but controversy over whether it is beneficial to aim for complete resolution of anemia is ongoing. The CREATE (Cardiovascular Risk Reduction by Early Anemia Treatment) and CHOIR (Correction of Hemoglobin and Outcomes in Renal Insufficiency) trials published in 2006 failed to resolve the issue.12,13
In the CREATE trial, patients targeted to achieve normal hemoglobin levels did no better in avoiding cardiovascular events than patients targeted for lower levels. The CHOIR trial was stopped early because of an increased trend toward death and hospitalization for congestive heart failure in the group with therapy targeted to achieve normal hemoglobin levels.
The recently published TREAT (Trial to Reduce Cardiovascular Events with Aranesp Therapy) study of patients with type 2 diabetes and CKD showed no reduction in all-cause mortality, cardiovascular morbidity, or ESRD in patients receiving Aranesp targeted to achieve a hemoglobin level of approximately 13 g/dL, compared with placebo.14 The study did demonstrate, however, that patients receiving Aranesp were about twice as likely to have a stroke than the placebo subjects (101 vs 53)—which might lead clinicians to ponder whether the gains, if any, were worth the risk.
Revised labeling. Late last year, the US Food and Drug Administration approved a label change for Procrit and Aranesp, warning that patients with renal failure “experienced greater risks for death and serious cardiovascular events when administered ESAs to target higher vs lower hemoglobin levels” and advising physicians to “individualize dosing to achieve and maintain hemoglobin levels within the range of 10 to 12 g/dL.”10,11 The 2007 NKF KDOQI guidelines suggest maintaining a hemoglobin level between 11 and 12 g/dL and have not incorporated the results of the TREAT trial.
Some patients don’t respond to ESAs
Inadequate response to ESAs is most commonly caused by underdosing or inadequate iron stores. NKF KDOQI guidelines recommend checking TSAT and ferritin prior to initiating therapy and monitoring these levels every 3 months.3 True nonresponders are individuals with good iron stores who are unable to achieve target hemoglobin within 4 to 6 months despite receiving subcutaneous epoetin 300 IU/kg per week. Inadequate response to ESAs can be caused by ongoing occult blood loss, infection, inflammation, nutritional deficiencies, hemolysis, hemoglobinemias, aluminum toxicity, anti-EPO antibody, hyperparathyroidism, multiple myeloma, and bone marrow dysfunction.10,11 If patients do not respond to ESA therapy, the NKF KDOQI guidelines recommend referral to a nephrologist or hematologist.3
How did Mary fare?
Mary did well taking oral iron supplementation. Once her iron deficiency was corrected, you were able to begin treating her anemia. After appropriate titration of her ESA, she was able to maintain a hemoglobin level between 11 and 12 g/dL 4 months into therapy. On a follow-up visit, she had no side effects from the medication and reported an increase in her energy level.
CORRESPONDENCE
Jonathan Taliercio, DO, Cleveland Clinic, Department of Nephrology and Hypertension, 9500 Euclid Avenue, Cleveland, OH 44195; [email protected]
1. United States Renal Data System, USRDS. 2009 Annual Data Report. Atlas of Chronic Kidney Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2009.
2. Hsu CJ, McCulloch CE, Curhan GC. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol. 2002;13:504-510.
3. KDOQI Clinical Practice Guideline and Clinical Practice Recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis. 2007;50:471-530.
4. Donnelly S. Why is erythropoietin made in the kidney? The kidney functions as a critmeter. Am J Kidney Dis. 2001;38:415-425.
5. Ebert B, Franklin H. Regulation of the erythropoietin gene. Blood. 1999;94:1864-1877.
6. Silverberg D, Wexler D, Blum M, et al. The cardio-renal anaemia syndrome: does it exist? Nephrol Dial Transplant. 2003;18(suppl 8):viii 7-viii 12.
7. Weiner D, Tighiouart H, Vlagopoulos P, et al. Effects of anemia and left ventricular hypertrophy on cardiovascular disease in patients with chronic kidney disease. J Am Soc Nephrol. 2005;16:1803-1810.
8. Levin A, Thompson C, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am J Kidney Dis. 1999;34:125-134.
9. Rao M, Pereira B. Optimal anemia management reduces cardiovascular morbidity, mortality, and costs in chronic kidney disease. Kidney Int. 2005;68:1432-1438.
10. Amgen. Aranesp (Darbepoetin Alpha) package insert. Available at www.aranesp.com/professional/crf/full_prescribing_info/pi.jsp. Accessed November 16, 2009.
11. Amgen. Procrit (Epoetin Alpha) package insert. Available at www.procrit.com/sites/default/files/shared/OBI/PI/ProcritBooklet.pdf#page=1. Accessed November 16, 2009.
12. Drueke T, Locatelli F, Clyne N, et al. Normalization of hemoglobin level in patients with CKD and anemia. N Engl J Med. 2006;355:2071-2084.
13. Singh A, Szczech L, Tang K. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355:2085-2098.
14. Pfeffer MA, Burdmann EA, Chen CY, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361:2019-2032.
1. United States Renal Data System, USRDS. 2009 Annual Data Report. Atlas of Chronic Kidney Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2009.
2. Hsu CJ, McCulloch CE, Curhan GC. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol. 2002;13:504-510.
3. KDOQI Clinical Practice Guideline and Clinical Practice Recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis. 2007;50:471-530.
4. Donnelly S. Why is erythropoietin made in the kidney? The kidney functions as a critmeter. Am J Kidney Dis. 2001;38:415-425.
5. Ebert B, Franklin H. Regulation of the erythropoietin gene. Blood. 1999;94:1864-1877.
6. Silverberg D, Wexler D, Blum M, et al. The cardio-renal anaemia syndrome: does it exist? Nephrol Dial Transplant. 2003;18(suppl 8):viii 7-viii 12.
7. Weiner D, Tighiouart H, Vlagopoulos P, et al. Effects of anemia and left ventricular hypertrophy on cardiovascular disease in patients with chronic kidney disease. J Am Soc Nephrol. 2005;16:1803-1810.
8. Levin A, Thompson C, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am J Kidney Dis. 1999;34:125-134.
9. Rao M, Pereira B. Optimal anemia management reduces cardiovascular morbidity, mortality, and costs in chronic kidney disease. Kidney Int. 2005;68:1432-1438.
10. Amgen. Aranesp (Darbepoetin Alpha) package insert. Available at www.aranesp.com/professional/crf/full_prescribing_info/pi.jsp. Accessed November 16, 2009.
11. Amgen. Procrit (Epoetin Alpha) package insert. Available at www.procrit.com/sites/default/files/shared/OBI/PI/ProcritBooklet.pdf#page=1. Accessed November 16, 2009.
12. Drueke T, Locatelli F, Clyne N, et al. Normalization of hemoglobin level in patients with CKD and anemia. N Engl J Med. 2006;355:2071-2084.
13. Singh A, Szczech L, Tang K. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355:2085-2098.
14. Pfeffer MA, Burdmann EA, Chen CY, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361:2019-2032.
Diabetic foot care: Tips and tools to streamline your approach
• Treat burning foot pain in patients with diabetes with tricyclic antidepressants or anticonvulsants. A
• Prescribe custom-fitted extra-depth shoes for patients with diabetes and neuropathy or foot deformity. B
• Consider hyperbaric oxygen therapy for ulcers that fail to respond to standard therapy. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE: AN OBESE PATIENT WITH BURNING FOOT PAIN
Mr. F., an obese 47-year-old with hypertension and type 2 diabetes, is trying to lose weight, and comes in to discuss a new exercise program. He recently had a negative cardiac workup for chest pain, which was ultimately diagnosed as gastroesophageal reflux disease.
The patient, whose most recent glycosylated hemoglobin (HbA1c) is 7.5, reports painful burning in his feet at night. A foot exam reveals no ulcers, lesions, or calluses; 2+ dorsalis pedis pulses bilaterally; and loss of sensation to 10-g monofilament testing at 3 sites on the bottom of each foot.
Based on Mr. F.’s presentation, it seems safe from a cardiac standpoint for him to embark on an exercise program, but what about his risk for a foot ulcer? An accurate assessment would enable you to tailor your recommendations and take the appropriate steps to modify the patient’s risk now—and to minimize complications down the road.
The incidence of diabetic foot ulcers—like that of diabetes itself—has risen in recent years.1 More than 15% of the approximately 23.5 million US adults with diabetes are expected to develop a foot ulcer at some point in their lives.2 Improvements in wound care and increased use of revascularization techniques have led to a decline in the number of ulcer-related amputations.3 But for 7% of those who develop diabetic foot ulcers, amputation is still the end result.4
As a family physician, you can play a key role in guarding against that outcome. This review—and the risk classification, ulcer grading, and treatment mnemonic tools that are detailed in the pages that follow—will help you optimize the foot care you provide to patients with diabetes.
Simple classification system accurately predicts risk
The longer an individual lives with diabetes and the poorer the level of blood glucose control, the greater the risk of ulceration.1,5 Other major risk factors include neuropathy, peripheral arterial disease, and a previous foot ulcer or amputation. All patients with diabetes should undergo an annual risk assessment for foot ulcers, which can be easily incorporated into their yearly physical. Although the causes of ulceration are numerous and complex, classification of risk based on simple measures has been found to accurately predict risk.6
Risk stratification tool. Medical history and in-office assessment of pulses, sensation, and foot deformities form the basis for a handy assessment tool (TABLE 1). In a study of 3256 patients, these simple criteria successfully predicted ulcer risk. During the nearly 2 years of the study, fewer than 1% of those categorized as low risk (n=2253) developed ulcers, while more than 29% of the high-risk group (n=477) did.6
To assess a patient’s risk, visually examine both feet, inspecting the skin, nails, and structure. Autonomic neuropathy may cause decreased sweating, leading to dry, breakable skin, while motor neuropathy can cause deformities such as hammer or claw toes, which also increase ulcer risk. Palpate for dorsalis pedis and posterior tibialis pulses. Although posterior tibialis pulses may be congenitally absent, the absence of a dorsalis pedis pulse is associated with a 6-fold increased risk of foot ulcer.1
Compared with patients with diabetes who have never had a foot ulcer, patients with a history of foot ulcer have 13 times the risk.7 As already noted, glucose control is also a key risk factor. For every 2-point increase in HbA1c, the odds ratio for ulcer development increases by 1.6,5 which is likely the result of hyperglycemia’s contribution to microvascular disease and peripheral neuropathy.
TABLE 1
Diabetic foot ulcer: Risk stratification
| Risk level/criteria | Incidence of ulcer* |
|---|---|
| Low risk All of the following: No history of ulcer At least 1 pulse per foot ≤1 of 10 sites insensate to monofilament testing No foot deformity or physical or visual impairment | 0.36% |
| Moderate risk 1 or more of the following: Missing both pulses in either foot ≥2 insensate sites to monofilament testing Foot deformity Unable to see or reach foot | 2.3% |
| High risk 1 or more of the following: Prior ulcer or amputation Neuropathy and absent pulses Neuropathy or absent pulses and calluses or foot deformity | 29.4% |
| * Percentage of patients in each risk category who developed foot ulcers over the nearly 2-year study period. | |
| Source: Leese GP, et al. Int J Clin Pract. 2006.6 | |
Screening for neuropathy, ulceration’s most consistent risk factor
Forty percent of patients with diabetes develop distal peripheral neuropathy.8 Damage to sensory nerves often results in the burning foot pain described by Mr. F., which can significantly affect quality of life. It may also present simply as decreased sensation or vibration sense. While decreased sensation is not painful and may not be troubling to the patient, it substantially heightens the risk of foot injury.
To screen for neuropathy, place a 10-g monofilament on noncallused plantar surfaces of the distal hallux and metatarsal heads, with enough pressure to slightly bend the filament. Instructing patients to close their eyes and report any sensation is more effective than prompting them for a response.
Recent guidelines from the American Diabetes Association and American Association of Clinical Endocrinologists recommend 1 additional screening test for neuropathy,9 such as pinprick or vibration sense.
Patients with neuropathy or other risk factors require more frequent follow-up to check for early signs of ulceration. Take the opportunity to provide education about the importance of self-examination of the feet, among other preventive measures (See “Preventing ulcers in high-risk feet”). Be alert to evidence of Charcot neuroarthropathy (CN). This progressive and irreversible condition of bone and joint slippage, dislocation, or fracture can affect any part of the foot, although it is typically found in the midfoot.
Acute CN is a clinical emergency characterized by pain, warmth, and erythema—making it clinically indistinguishable from cellulitis, osteomyelitis, or gout. Blood tests, including a white blood cell count, sedimentary rate, and uric acid level, may be necessary for diagnosis, as well as radiographs or magnetic resonance imaging. Treatment is long-term (4-6 months) immobilization of the joint to prevent further destruction,10 and bisphosphonates for pain during the acute phase.11 Untreated CN typically results in a rocker-bottom foot deformity (FIGURE) that puts patients at greater risk for plantar ulceration.
Although it is possible to successfully treat the majority of diabetic foot ulcers, the wounds result in considerable morbidity, lower quality of life, and increased health care costs. A far better approach is to focus on prevention, with appropriate interventions and frequent follow-up for those at high risk.
ENSURE THAT THE PATIENT HAS THE RIGHT FOOTWEAR.
Properly fitting shoes with ample room for the toes is a priority for all patients with diabetes, but “high-risk feet” need therapeutic footwear, which Medicare covers as a yearly benefit. For those with a history of foot ulcer or amputation, custom insoles, rigid rocker shoes, and orthotics can help prevent re-ulceration.34 For socks, synthetic blends are preferable to cotton, which can chafe when wet with sweat.
PROVIDE EDUCATIONAL MATERIALS; EMPHASIZE FOOT INSPECTION.
All patients with diabetes should receive general education regarding foot care, as there is evidence that it improves behavior and may prevent injuries.7,35 Educational materials emphasizing the importance of nightly foot inspection, overall foot care, and physician inspection are crucial for patients at high risk for developing foot ulcers. Excellent patient education materials are available from the National Diabetes Education Program (See “Take Care of Your Feet for a Lifetime”).
STRESS FREQUENT FOLLOW-UP.
Patients should be informed of their risk level for diabetic foot ulcers after screening. Advise those at high risk to have their feet inspected by a podiatrist or other knowledgeable clinician every 1 to 2 months.9
ENCOURAGE EXERCISE.
Non-weight-bearing exercise programs, including swimming, and a consistent level of daily weight-bearing activity should be encouraged. Caution patients to increase weight-bearing exercise gradually, however, ideally in a closely supervised setting, and to do everything possible to avoid even minimal foot trauma.
REVIEW MEDICATIONS AND ADJUST THERAPY, AS NEEDED.
Tight glycemic control and the use of angiotensin-converting enzyme inhibitors may help prevent the development of neuropathy.36,37 For those who already have neuropathy, tricyclic antidepressants38 or anticonvulsants may bring pain relief.
GUARD AGAINST CHARCOT NEUROARTHROPATHY.
Be alert to this diagnosis in patients who present with a warm, red, painful midfoot. Patients with long-standing neuropathy may benefit from preventive bracing to limit joint movement and lower the risk of Charcot neuroarthropathy.
FIGURE
Rocker-bottom foot deformity
This radiograph reveals extensive collapse of the inner arch and a “rocker-bottom” foot deformity, the result of untreated Charcot neuroarthropathy.
CASE: MR. F.’S RISK BECOMES A REALITY
Mr. F. has a moderate risk of foot ulceration, based on evidence of neuropathy with 6 insensate sites (TABLE 1). You emphasize the importance of foot care, including appropriate footwear, and refer him to a podiatrist. You strongly support his decision to begin an exercise program to improve his glycemic control, decrease his cardiovascular mortality risk, and possibly help him lose weight.
Non-weight-bearing activity can be safely recommended to patients with diabetic neuropathy. So can daily weight-bearing activity, which actually decreases the risk of foot ulcer by maintaining leg muscle tone and plantar tissue tolerance to stress.12,13 Recent studies suggest that increasing weight-bearing activity slowly–walking daily and adding a total of 100 additional steps every 2 weeks, for example—in a carefully monitored program is not associated with increased risk of foot ulceration.14 You report these findings to Mr. F.
When he returns to your office in 2 months, Mr. F. has lost 7 pounds and his HbA1c has fallen to 6.5. Despite these positive developments, an examination of his feet reveals a full thickness ulcer on the left metatarsal head, which the patient had not noticed.
You recognize the clinical urgency of effectively treating Mr. F.’s diabetic foot ulcer, as size, duration, and grade are the greatest predictors of healing. Ulcers that are larger than 2 cm, have been present for more than 6 months,15 or have a higher grade on a scale such as the Wagner Foot Classification System16 (TABLE 2) are far less likely to heal than smaller, low-grade ulcers of shorter duration.
AIM DOC mnemonic guides ulcer care
Comprehensive, coordinated care improves outcomes for diabetic ulcers and has repeatedly been shown to reduce amputation rates.17-19 Large clinical trials have not evaluated each aspect of diabetic ulcer care, however, so the recommendations that follow are based on expert opinion and available evidence. These include assessing the limb’s arterial supply and ensuring that the patient undergoes revascularization, as needed; promptly treating infection; and providing optimal wound care, including debridement of callused and necrotic tissue, off-loading pressure, and applying moist wound dressings.20
AIM DOC, developed by 1 of the authors (JE), is a handy treatment tool. The mnemonic represents both the elements of treatment and the order in which they should be carried out. The letters stand for:
- Arterial disease
- Infection
- Measure
- Debride
- Off-load
- Cover
Here’s how to use AIM DOC, step by step:
Step 1: Assess for arterial disease
Start by assessing the vascular supply to the affected limb, which can be presumed to be adequate if pulses are palpable. If pulses cannot be palpated, the patient should undergo an ankle-brachial index (ABI) test and, if necessary, referred to a specialist to be evaluated for angioplasty or vascular bypass surgery. An ABI <0.9 is abnormal; 0.5 is considered the threshold for healing without such intervention.3 Keep in mind, however, that the ABI is falsely elevated in approximately 15% of patients with diabetes. If classic signs and symptoms of arterial disease are present, further evaluation is needed even if the ABI is normal.
Classic signs and symptoms of arterial disease include cool, hairless feet with shiny skin, and claudication. Location may provide another clue to etiology: Ulcers located on the heel, the outside of the foot, or between the toes tend to be associated with vascular disease, while ulcers with surrounding callus, such as the classic mal perforans ulcer on the metatarsal head, are neuropathic.
Step 2: Treat—or rule out—infection
While patients with diabetes have a 5-fold increase in risk of infection21 compared with individuals without the disease, there is no value in treating an uninfected ulcer with antibiotics. Diagnosing infection can be challenging because patients with diabetes may be less likely to demonstrate evidence of infection.21
An elevated white blood cell count, purulent drainage, foul odor, and/or erythema >2 cm around the wound clearly indicates a need for systemic antibiotics.22,23 Tissue necrosis, often assumed to represent ischemia, may result from neutrophilic vasculitis from soft tissue infection.24 Superficial cultures reflect colonization and should not be used to diagnose infection. In inspecting the wound for signs of infection, probe the ulcer and evaluate for osteomyelitis if it reaches bone.
Gram-positive cocci, especially Staphylococcus aureus, are the predominant pathogens in diabetic foot infections, and antibiotics effective against them may be sufficient for mild-to-moderate infections in patients who have not recently received antibiotic therapy. Gram-negative rods may be found in chronic wounds, however, and anaerobic pathogens may be present in patients with foot ischemia or gangrene; in both cases, broader-spectrum antimicrobials are required.22 Highly bioavailable oral antibiotics are indicated for infection, including some cases of osteomyelitis. Silver dressings may be helpful as a topical antimicrobial; there have been reports of successful treatment with honey, as well.25,26
Step 3: Measure (and grade) the wound
Accurate measurement of ulcer size is critical, both when you initially detect it and at each subsequent visit. To get an accurate measure, simply multiply the greatest length by the greatest width. The percent change in wound size after 4 weeks of treatment is a significant predictor of healing.25
Some clinicians also use photographs to track wound size, but these can be misleading. A better approach is to trace the wound on a sheet of acetate to document progress over time. If you see no improvement within 2 weeks, treatment should be modified.
Use a grading tool. There are a number of systems used for grading ulcers, none of which is universally accepted. One well-known tool is the Wagner Classification System16 (TABLE 2) referred to earlier. The higher the grade, the lower the likelihood that the ulcer will heal.
TABLE 2
Grading the ulcer: The Wagner system
| Grade | Description |
|---|---|
| 0 | No open lesions; may have deformity or cellulitis |
| 1 | Superficial diabetic ulcer (partial or full thickness) |
| 2 | Ulcer extension to ligament, tendon, joint capsule, or deep fascia (without abscess or osteomyelitis) |
| 3 | Deep ulcer with abscess, osteomyelitis, or joint sepsis |
| 4 | Gangrene localized to portion of forefoot or heel |
| 5 | Extensive gangrenous involvement of the entire foot |
| Adapted from: Wagner FW Jr. Orthopedics. 1987.16 | |
Step 4: Debride the ulcer
Frequent sharp debridement—to remove necrotic, callused, infected, and hypergranulation tissue—has long been considered essential in the treatment of neuropathic ulcers,17,21 especially for chronic or infected wounds. Debridement is thought to aid in healing by reducing pressure on the ulcer, decreasing bacterial contamination, enhancing platelet activation, releasing growth factors, and stimulating granulation tissue.
While some physicians are hesitant to perform debridement in the office, the process can actually be carried out without difficulty in an outpatient setting. Because of the neuropathy associated with most diabetic ulcers, no anesthesia is required. While the procedure is not typically painful, you will need to provide patient education to prepare the patient for the possibility of bleeding. Debride the wound to the outer edge of the hyperkeratotic tissue. If bleeding occurs, simply apply pressure until it stops.
When not to debride. Debridement is contraindicated under certain circumstances—if the limb has poor circulation, for example. Similarly, avoid debriding heel ulcers covered by eschar if there is no fluctuance in the underlying tissue, as the eschar provides a protective barrier.24 When sharp debridement is not possible, consider topical hydrogel or maggot therapy, an adjuvant treatment we’ll discuss in a bit.
Step 5: Off-load the wound
Mechanical load relief is vital for treating neuropathic ulcers, both to redistribute plantar pressures and protect granulation tissue. Total contact casting (TCC) is the gold standard, healing 90% of ulcers within 6 to 8 weeks.28 TCC is costly when applied weekly, however, and should only be done by a specialist, as an incorrectly applied cast can lead to the creation of new ulcers.
Because of the heaviness and inconvenience of the casts, many patients prefer removable devices, but these devices are much less effective. One study found that the average removable off-loading device is worn no more than 30% of the time that the patient is walking.29 Removable devices can be temporarily secured with plaster of Paris (a process that is sometimes referred to as instant contact casting) to ensure compliance.
Acceptable removable devices include a heel pressure relief shoe for heel ulcers and a CAM (controlled ankle motion) walker for metatarsal ulcers. Be sure off-loading devices are applied securely so no slippage can occur.
Step 6: Cover with moist dressings
The purpose of any topical dressing is to keep wounds moist, absorb exudate, and prevent contamination. A variety of moist dressings have been successfully used to treat ulcers, although evidence to recommend any particular dressing is insufficient.19 While wound vacuum-assisted closure (VAC) devices are widely used, there is little support for their use. A review of 7 trials comparing VAC devices with moistened gauze dressings or other topical agents found no evidence that topical negative pressure increases chronic wound healing.30
When foot ulcers do not heal
AIM DOC highlights the steps of diabetic ulcer care that are most likely to result in healing. When ulcers are slow to heal, review the 6 steps of treatment, paying particular attention to off-loading. If you establish that these have been appropriately applied and the wound is still not responding, consider alternative diagnoses such as venous insufficiency, vasculitis, or malignancy. Venous insufficiency ulcers may be difficult to differentiate from neuropathic ulcers, but they won’t heal without compression dressings. Diagnosis of vasculitis and malignancy can be made by biopsying the ulcer edge.
2 adjunctive therapies to consider
If there is no evidence to support an alternative diagnosis, consider adjunctive treatments, with hyperbaric oxygen therapy (HBOT) foremost among them.
A Cochrane review of HBOT found that it reduced the number of major amputations in diabetes patients with chronic foot ulcers, and improved healing at 1 year.31 Both Medicare and Medicaid cover HBOT for patients with diabetic ulcers classified as Wagner Grade 3 or higher that have not responded to 30 days of standard treatment.
Maggots, scientifically known as Lucilia sericata (Greenbottle) fly larvae, secrete proteolytic enzymes that debride necrotic tissue but are inactivated by living tissue. One meta-analysis found that neither surgical debridement nor larval therapy showed significant benefit over hydrogel.32 A subsequent small study did show statistically improved healing in ulcers debrided with maggots, compared with surgical debridement.33
CASE: MR. F.’S ULCER HEALS
Because Mr. F.’s ulcer is small and shallow and has been present for a short time, it has an excellent chance of healing if you follow the AIM DOC steps. You determine that he has adequate arterial supply and that the wound is uninfected (there is a strong dorsalis pedis pulse and no warmth, exudate, or erythema around the wound). Using a #15 blade, you pare away the callus surrounding the ulcer and document the length and width of the wound. You cover the ulcer with a moist dressing and instruct Mr. F to replace it twice a day, cautioning him against using alcohol or hydrogen peroxide, which could harm the healing skin. You discuss the importance of avoiding all weight bearing on the ulcerated foot, prescribe a CAM walker to wear at all times except while he’s sleeping, and schedule weekly follow-up visits to track progress.
In 2 weeks, the wound has resolved. You educate Mr. F. about his risk of ulcer recurrence and outline appropriate preventive steps. You also refer him for fitted extra-depth diabetic shoes and ongoing podiatry follow-up.
ACKNOWLEDGMENT
The authors wish to thank Gregory Mack, DPM, University of Wisconsin School of Medicine and Public Health, for his teaching and collaboration.
CORRESPONDENCE
Jennifer Eddy, MD, University of Wisconsin School of Medicine and Public Health, 617 W. Clairemont Avenue, Eau Claire, WI 54701; [email protected]
1. LeMaster JW, Reiber GE. Epidemiology and economic impact of foot ulcers. In: Boulton AJM, Cavanaugh PR, Rayman G, eds. The Foot in Diabetes. 4th ed. London, England: John Wiley & Sons Ltd; 2006:1–16.
2. Department of Health and Human Services Centers for Disease Control and Health Prevention. National Diabetes Fact Sheet 2007. Available at: http://www.cdc.gov/diabetes/pubs/pdf/ndfs_2007.pdf. Accessed May 15, 2009.
3. Pinzur M. Amputations in the diabetic foot. In: Boulton AJM, Cavanaugh PR, Rayman G, eds. The Foot in Diabetes. 4th ed. London, England: John Wiley & Sons Ltd; 2006:308–322.
4. Margolis DJ, Allen-Taylor L, Hoffstad O, et al. Diabetic neuropathic foot ulcers and amputation. Wound Repair Regen. 2005;13:230-236.
5. Moss SE, Klein R, Kelin BE. The prevalence and incidence of lower extremity amputation in a diabetic population. Arch Intern Med. 1992;152:610-616.
6. Leese GP, Reid F, Green V, et al. Stratification of foot ulcer risk in patients with diabetes: a population based study. Int J Clin Pract. 2006;60:541-545.
7. Litzelman DR, Slemenda CW, Langefeld CD. Reduction of lower extremity clinical abnormalities in patient with non-insulin-dependent diabetes mellitus: a randomized controlled trial. Ann Intern Med. 1993;119:36-41.
8. Palumbo PJ, Melton LJ, III. Peripheral vascular disease and diabetes. In: National Diabetes Data Group, National Institute of Diabetes and Digestive and Kidney Disease, eds. Diabetes in America. 2nd ed. Bethesda, Md: National Institutes of Health, NIDDKD; 1995:401–408.
9. Boulton AJM, Armstrong DG, Albert SF, et al. Comprehensive foot examination and risk assessment: a report of the task force of the foot care interest group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care. 2008;31:1679-1685.
10. Jude EB. Charcot foot: what’s new in pathogensis and medical management? In: Boulton AJM, Cavanaugh PR, Rayman G, eds. The Foot in Diabetes. 4th ed. London, England: John Wiley & Sons Ltd; 2006: 265–273.
11. Pitocco D, Ruotolo V, Caputo S, et al. Six month treatment with alendronate in acute Charcot neuroarthropathy: a randomized controlled trial. Diabetes Care. 2005;28:1214-1215.
12. LeMaster JW, Reiber GE, Smith DG, et al. Daily weight-bearing activity does not increase the risk of diabetic foot ulcers. Med Sci Sports Exerc. 2003;35:1093-1099.
13. Lott DJ, Malug KS, Sinacore DR, et al. Relationship between changes in activity and plantar ulcer recurrence in a patient with diabetes mellitus. Phys Ther. 2005;85:579-588.
14. LeMaster JW, Mueller MJ, Reiber GE, et al. Effect of weight-bearing activity on foot ulcer incidence in people with diabetic peripheral neuropathy: Feet First randomized controlled trial. Phys Ther. 2008;88:1-13.
15. Margolis DJ, Kantor J, Santanna J, et al. Risk factors for delayed healing of neuropathic foot ulcers: a pooled analysis. Arch Dermatol. 2000;136:1531-1535.
16. Wagner FW, Jr. The diabetic foot. Orthopedics. 1987;10:163-172.
17. Rith-Najarian S, Branchaid C, Beaulieu O, et al. Reducing lower-extremity amputations due to diabetes: application of the staged diabetes management approach in a primary care setting. J Fam Pract. 1998;47:127-132.
18. Canavan RJ, Unwin NC, Connolly VM, et al. Diabetes and non-diabetes related lower extremity amputation incidence before and after the introduction of better organized diabetic foot care. Diabetes Care. 2008;31.-
19. Krishnan St, Nash F, Baker NR, et al. Reduction in diabetes amputations over 11 years in a defined UK population. Diabetes Care. 2008;31:99-101.
20. Clayton W, Elasy TA. A review of the pathophysiology, classification, and treatment of foot ulcers in diabetic patients. Clin Diabetes. 2009;27:52-58.
21. Majno G, Joris I. Cells, Tissues, and Disease: Principles of General Pathology, 2nd ed. Oxford, England: Oxford University Press; 2004.
22. Lipsky BA, Berendt AR, Deery HG, et al. Diagnosis and treatment of diabetic foot infections. Clin Infect Dis. 2004;39:885-910.
23. Frykberg RG, Zgonis T, Armstrong DG, et al. Diabetic foot disorders: a clinical practice guideline (2006 revision). J Foot Ankle Surg. 2006;45(5 suppl):S2-S66.
24. Edmonds M, Foster A. The use of antibiotics in the diabetic foot. Am J Surg. 2004;187:S25-S28.
25. Eddy JJ, Gideonsen MD. Topical honey therapy for diabetic foot ulcers. J Fam Pract. 2005;54:533-535.
26. Remmen R, Coenen S, Seuntjens R, et al. Honey for refractory diabetic foot ulcers. J Fam Pract. 2005;54:863.-
27. Sheehan P, Jones P, Caselli A, et al. Percent change in wound area in diabetic foot ulcers over a 4-week period is a robust predictor of complete healing in a 12-week prospective trial. Diabetes Care. 2003;26:1879-1882.
28. Lavery LA, Murdoch DP. Conventional offloading and activity monitoring. In: The Foot in Diabetes. 4th ed. Boulton AJM, Cavanaugh PR, Rayman G, eds. London, England: John Wiley & Sons Ltd; 2006: PG NUM.
29. Armstrong DG, Lavery LA, Kimbrel HR, et al. Activity patterns of patients with diabetic foot ulceration: patients with active ulceration may not adhere to a standard pressure off-loading regimen. Diabetes Care. 2003;26:2595-2597.
30. Ubbink DT, Westerbos SJ, Evans D, et al. H. Topical negative pressure for treating chronic wounds. Cochrane Database Syst Rev. 2008(3);CD001898.-
31. Kranke P, Bennett M, Roeckl-Wiedmann I, et al. Hyperbaric oxygen therapy for chronic wounds. Cochrane Database Syst Rev. 2004(2);CD004123.-
32. Edwards J. Debridement of diabetic foot ulcers. Cochrane Database Syst Rev. 2002(4);CD003556.-
33. Sherman RA. Maggot therapy for treating diabetic foot ulcers unresponsive to conventional therapy. Diabetes Care. 2003;26:446-451.
34. Spencer SA. Pressure relieving interventions for preventing and treating diabetic foot ulcers. Cochrane Database Syst Rev. 2000(3);CD002302.-
35. Valk GD, Kriegsman DM, Assendelft WJ. Patient education for preventing diabetic foot ulceration. Cochrane Database Syst Rev. 2001(4);CD001488.-(AU: 2005 update..?)
36. The Diabetes Control and Complications Trial Research Group. The effect of intensive diabetes therapy on the development and progression of neuropathy. Ann Intern Med. 1995;122:561-568.
37. Boulton AJM, Viniv AL, Arezzo JC, et al. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care. 2005;28:956-962.
38. Wong MC, Chung JW, Wong TK. Effects of treatments for symptoms of painful diabetic neuropathy: systematic review. BMJ. 2007;335:87.-
• Treat burning foot pain in patients with diabetes with tricyclic antidepressants or anticonvulsants. A
• Prescribe custom-fitted extra-depth shoes for patients with diabetes and neuropathy or foot deformity. B
• Consider hyperbaric oxygen therapy for ulcers that fail to respond to standard therapy. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE: AN OBESE PATIENT WITH BURNING FOOT PAIN
Mr. F., an obese 47-year-old with hypertension and type 2 diabetes, is trying to lose weight, and comes in to discuss a new exercise program. He recently had a negative cardiac workup for chest pain, which was ultimately diagnosed as gastroesophageal reflux disease.
The patient, whose most recent glycosylated hemoglobin (HbA1c) is 7.5, reports painful burning in his feet at night. A foot exam reveals no ulcers, lesions, or calluses; 2+ dorsalis pedis pulses bilaterally; and loss of sensation to 10-g monofilament testing at 3 sites on the bottom of each foot.
Based on Mr. F.’s presentation, it seems safe from a cardiac standpoint for him to embark on an exercise program, but what about his risk for a foot ulcer? An accurate assessment would enable you to tailor your recommendations and take the appropriate steps to modify the patient’s risk now—and to minimize complications down the road.
The incidence of diabetic foot ulcers—like that of diabetes itself—has risen in recent years.1 More than 15% of the approximately 23.5 million US adults with diabetes are expected to develop a foot ulcer at some point in their lives.2 Improvements in wound care and increased use of revascularization techniques have led to a decline in the number of ulcer-related amputations.3 But for 7% of those who develop diabetic foot ulcers, amputation is still the end result.4
As a family physician, you can play a key role in guarding against that outcome. This review—and the risk classification, ulcer grading, and treatment mnemonic tools that are detailed in the pages that follow—will help you optimize the foot care you provide to patients with diabetes.
Simple classification system accurately predicts risk
The longer an individual lives with diabetes and the poorer the level of blood glucose control, the greater the risk of ulceration.1,5 Other major risk factors include neuropathy, peripheral arterial disease, and a previous foot ulcer or amputation. All patients with diabetes should undergo an annual risk assessment for foot ulcers, which can be easily incorporated into their yearly physical. Although the causes of ulceration are numerous and complex, classification of risk based on simple measures has been found to accurately predict risk.6
Risk stratification tool. Medical history and in-office assessment of pulses, sensation, and foot deformities form the basis for a handy assessment tool (TABLE 1). In a study of 3256 patients, these simple criteria successfully predicted ulcer risk. During the nearly 2 years of the study, fewer than 1% of those categorized as low risk (n=2253) developed ulcers, while more than 29% of the high-risk group (n=477) did.6
To assess a patient’s risk, visually examine both feet, inspecting the skin, nails, and structure. Autonomic neuropathy may cause decreased sweating, leading to dry, breakable skin, while motor neuropathy can cause deformities such as hammer or claw toes, which also increase ulcer risk. Palpate for dorsalis pedis and posterior tibialis pulses. Although posterior tibialis pulses may be congenitally absent, the absence of a dorsalis pedis pulse is associated with a 6-fold increased risk of foot ulcer.1
Compared with patients with diabetes who have never had a foot ulcer, patients with a history of foot ulcer have 13 times the risk.7 As already noted, glucose control is also a key risk factor. For every 2-point increase in HbA1c, the odds ratio for ulcer development increases by 1.6,5 which is likely the result of hyperglycemia’s contribution to microvascular disease and peripheral neuropathy.
TABLE 1
Diabetic foot ulcer: Risk stratification
| Risk level/criteria | Incidence of ulcer* |
|---|---|
| Low risk All of the following: No history of ulcer At least 1 pulse per foot ≤1 of 10 sites insensate to monofilament testing No foot deformity or physical or visual impairment | 0.36% |
| Moderate risk 1 or more of the following: Missing both pulses in either foot ≥2 insensate sites to monofilament testing Foot deformity Unable to see or reach foot | 2.3% |
| High risk 1 or more of the following: Prior ulcer or amputation Neuropathy and absent pulses Neuropathy or absent pulses and calluses or foot deformity | 29.4% |
| * Percentage of patients in each risk category who developed foot ulcers over the nearly 2-year study period. | |
| Source: Leese GP, et al. Int J Clin Pract. 2006.6 | |
Screening for neuropathy, ulceration’s most consistent risk factor
Forty percent of patients with diabetes develop distal peripheral neuropathy.8 Damage to sensory nerves often results in the burning foot pain described by Mr. F., which can significantly affect quality of life. It may also present simply as decreased sensation or vibration sense. While decreased sensation is not painful and may not be troubling to the patient, it substantially heightens the risk of foot injury.
To screen for neuropathy, place a 10-g monofilament on noncallused plantar surfaces of the distal hallux and metatarsal heads, with enough pressure to slightly bend the filament. Instructing patients to close their eyes and report any sensation is more effective than prompting them for a response.
Recent guidelines from the American Diabetes Association and American Association of Clinical Endocrinologists recommend 1 additional screening test for neuropathy,9 such as pinprick or vibration sense.
Patients with neuropathy or other risk factors require more frequent follow-up to check for early signs of ulceration. Take the opportunity to provide education about the importance of self-examination of the feet, among other preventive measures (See “Preventing ulcers in high-risk feet”). Be alert to evidence of Charcot neuroarthropathy (CN). This progressive and irreversible condition of bone and joint slippage, dislocation, or fracture can affect any part of the foot, although it is typically found in the midfoot.
Acute CN is a clinical emergency characterized by pain, warmth, and erythema—making it clinically indistinguishable from cellulitis, osteomyelitis, or gout. Blood tests, including a white blood cell count, sedimentary rate, and uric acid level, may be necessary for diagnosis, as well as radiographs or magnetic resonance imaging. Treatment is long-term (4-6 months) immobilization of the joint to prevent further destruction,10 and bisphosphonates for pain during the acute phase.11 Untreated CN typically results in a rocker-bottom foot deformity (FIGURE) that puts patients at greater risk for plantar ulceration.
Although it is possible to successfully treat the majority of diabetic foot ulcers, the wounds result in considerable morbidity, lower quality of life, and increased health care costs. A far better approach is to focus on prevention, with appropriate interventions and frequent follow-up for those at high risk.
ENSURE THAT THE PATIENT HAS THE RIGHT FOOTWEAR.
Properly fitting shoes with ample room for the toes is a priority for all patients with diabetes, but “high-risk feet” need therapeutic footwear, which Medicare covers as a yearly benefit. For those with a history of foot ulcer or amputation, custom insoles, rigid rocker shoes, and orthotics can help prevent re-ulceration.34 For socks, synthetic blends are preferable to cotton, which can chafe when wet with sweat.
PROVIDE EDUCATIONAL MATERIALS; EMPHASIZE FOOT INSPECTION.
All patients with diabetes should receive general education regarding foot care, as there is evidence that it improves behavior and may prevent injuries.7,35 Educational materials emphasizing the importance of nightly foot inspection, overall foot care, and physician inspection are crucial for patients at high risk for developing foot ulcers. Excellent patient education materials are available from the National Diabetes Education Program (See “Take Care of Your Feet for a Lifetime”).
STRESS FREQUENT FOLLOW-UP.
Patients should be informed of their risk level for diabetic foot ulcers after screening. Advise those at high risk to have their feet inspected by a podiatrist or other knowledgeable clinician every 1 to 2 months.9
ENCOURAGE EXERCISE.
Non-weight-bearing exercise programs, including swimming, and a consistent level of daily weight-bearing activity should be encouraged. Caution patients to increase weight-bearing exercise gradually, however, ideally in a closely supervised setting, and to do everything possible to avoid even minimal foot trauma.
REVIEW MEDICATIONS AND ADJUST THERAPY, AS NEEDED.
Tight glycemic control and the use of angiotensin-converting enzyme inhibitors may help prevent the development of neuropathy.36,37 For those who already have neuropathy, tricyclic antidepressants38 or anticonvulsants may bring pain relief.
GUARD AGAINST CHARCOT NEUROARTHROPATHY.
Be alert to this diagnosis in patients who present with a warm, red, painful midfoot. Patients with long-standing neuropathy may benefit from preventive bracing to limit joint movement and lower the risk of Charcot neuroarthropathy.
FIGURE
Rocker-bottom foot deformity
This radiograph reveals extensive collapse of the inner arch and a “rocker-bottom” foot deformity, the result of untreated Charcot neuroarthropathy.
CASE: MR. F.’S RISK BECOMES A REALITY
Mr. F. has a moderate risk of foot ulceration, based on evidence of neuropathy with 6 insensate sites (TABLE 1). You emphasize the importance of foot care, including appropriate footwear, and refer him to a podiatrist. You strongly support his decision to begin an exercise program to improve his glycemic control, decrease his cardiovascular mortality risk, and possibly help him lose weight.
Non-weight-bearing activity can be safely recommended to patients with diabetic neuropathy. So can daily weight-bearing activity, which actually decreases the risk of foot ulcer by maintaining leg muscle tone and plantar tissue tolerance to stress.12,13 Recent studies suggest that increasing weight-bearing activity slowly–walking daily and adding a total of 100 additional steps every 2 weeks, for example—in a carefully monitored program is not associated with increased risk of foot ulceration.14 You report these findings to Mr. F.
When he returns to your office in 2 months, Mr. F. has lost 7 pounds and his HbA1c has fallen to 6.5. Despite these positive developments, an examination of his feet reveals a full thickness ulcer on the left metatarsal head, which the patient had not noticed.
You recognize the clinical urgency of effectively treating Mr. F.’s diabetic foot ulcer, as size, duration, and grade are the greatest predictors of healing. Ulcers that are larger than 2 cm, have been present for more than 6 months,15 or have a higher grade on a scale such as the Wagner Foot Classification System16 (TABLE 2) are far less likely to heal than smaller, low-grade ulcers of shorter duration.
AIM DOC mnemonic guides ulcer care
Comprehensive, coordinated care improves outcomes for diabetic ulcers and has repeatedly been shown to reduce amputation rates.17-19 Large clinical trials have not evaluated each aspect of diabetic ulcer care, however, so the recommendations that follow are based on expert opinion and available evidence. These include assessing the limb’s arterial supply and ensuring that the patient undergoes revascularization, as needed; promptly treating infection; and providing optimal wound care, including debridement of callused and necrotic tissue, off-loading pressure, and applying moist wound dressings.20
AIM DOC, developed by 1 of the authors (JE), is a handy treatment tool. The mnemonic represents both the elements of treatment and the order in which they should be carried out. The letters stand for:
- Arterial disease
- Infection
- Measure
- Debride
- Off-load
- Cover
Here’s how to use AIM DOC, step by step:
Step 1: Assess for arterial disease
Start by assessing the vascular supply to the affected limb, which can be presumed to be adequate if pulses are palpable. If pulses cannot be palpated, the patient should undergo an ankle-brachial index (ABI) test and, if necessary, referred to a specialist to be evaluated for angioplasty or vascular bypass surgery. An ABI <0.9 is abnormal; 0.5 is considered the threshold for healing without such intervention.3 Keep in mind, however, that the ABI is falsely elevated in approximately 15% of patients with diabetes. If classic signs and symptoms of arterial disease are present, further evaluation is needed even if the ABI is normal.
Classic signs and symptoms of arterial disease include cool, hairless feet with shiny skin, and claudication. Location may provide another clue to etiology: Ulcers located on the heel, the outside of the foot, or between the toes tend to be associated with vascular disease, while ulcers with surrounding callus, such as the classic mal perforans ulcer on the metatarsal head, are neuropathic.
Step 2: Treat—or rule out—infection
While patients with diabetes have a 5-fold increase in risk of infection21 compared with individuals without the disease, there is no value in treating an uninfected ulcer with antibiotics. Diagnosing infection can be challenging because patients with diabetes may be less likely to demonstrate evidence of infection.21
An elevated white blood cell count, purulent drainage, foul odor, and/or erythema >2 cm around the wound clearly indicates a need for systemic antibiotics.22,23 Tissue necrosis, often assumed to represent ischemia, may result from neutrophilic vasculitis from soft tissue infection.24 Superficial cultures reflect colonization and should not be used to diagnose infection. In inspecting the wound for signs of infection, probe the ulcer and evaluate for osteomyelitis if it reaches bone.
Gram-positive cocci, especially Staphylococcus aureus, are the predominant pathogens in diabetic foot infections, and antibiotics effective against them may be sufficient for mild-to-moderate infections in patients who have not recently received antibiotic therapy. Gram-negative rods may be found in chronic wounds, however, and anaerobic pathogens may be present in patients with foot ischemia or gangrene; in both cases, broader-spectrum antimicrobials are required.22 Highly bioavailable oral antibiotics are indicated for infection, including some cases of osteomyelitis. Silver dressings may be helpful as a topical antimicrobial; there have been reports of successful treatment with honey, as well.25,26
Step 3: Measure (and grade) the wound
Accurate measurement of ulcer size is critical, both when you initially detect it and at each subsequent visit. To get an accurate measure, simply multiply the greatest length by the greatest width. The percent change in wound size after 4 weeks of treatment is a significant predictor of healing.25
Some clinicians also use photographs to track wound size, but these can be misleading. A better approach is to trace the wound on a sheet of acetate to document progress over time. If you see no improvement within 2 weeks, treatment should be modified.
Use a grading tool. There are a number of systems used for grading ulcers, none of which is universally accepted. One well-known tool is the Wagner Classification System16 (TABLE 2) referred to earlier. The higher the grade, the lower the likelihood that the ulcer will heal.
TABLE 2
Grading the ulcer: The Wagner system
| Grade | Description |
|---|---|
| 0 | No open lesions; may have deformity or cellulitis |
| 1 | Superficial diabetic ulcer (partial or full thickness) |
| 2 | Ulcer extension to ligament, tendon, joint capsule, or deep fascia (without abscess or osteomyelitis) |
| 3 | Deep ulcer with abscess, osteomyelitis, or joint sepsis |
| 4 | Gangrene localized to portion of forefoot or heel |
| 5 | Extensive gangrenous involvement of the entire foot |
| Adapted from: Wagner FW Jr. Orthopedics. 1987.16 | |
Step 4: Debride the ulcer
Frequent sharp debridement—to remove necrotic, callused, infected, and hypergranulation tissue—has long been considered essential in the treatment of neuropathic ulcers,17,21 especially for chronic or infected wounds. Debridement is thought to aid in healing by reducing pressure on the ulcer, decreasing bacterial contamination, enhancing platelet activation, releasing growth factors, and stimulating granulation tissue.
While some physicians are hesitant to perform debridement in the office, the process can actually be carried out without difficulty in an outpatient setting. Because of the neuropathy associated with most diabetic ulcers, no anesthesia is required. While the procedure is not typically painful, you will need to provide patient education to prepare the patient for the possibility of bleeding. Debride the wound to the outer edge of the hyperkeratotic tissue. If bleeding occurs, simply apply pressure until it stops.
When not to debride. Debridement is contraindicated under certain circumstances—if the limb has poor circulation, for example. Similarly, avoid debriding heel ulcers covered by eschar if there is no fluctuance in the underlying tissue, as the eschar provides a protective barrier.24 When sharp debridement is not possible, consider topical hydrogel or maggot therapy, an adjuvant treatment we’ll discuss in a bit.
Step 5: Off-load the wound
Mechanical load relief is vital for treating neuropathic ulcers, both to redistribute plantar pressures and protect granulation tissue. Total contact casting (TCC) is the gold standard, healing 90% of ulcers within 6 to 8 weeks.28 TCC is costly when applied weekly, however, and should only be done by a specialist, as an incorrectly applied cast can lead to the creation of new ulcers.
Because of the heaviness and inconvenience of the casts, many patients prefer removable devices, but these devices are much less effective. One study found that the average removable off-loading device is worn no more than 30% of the time that the patient is walking.29 Removable devices can be temporarily secured with plaster of Paris (a process that is sometimes referred to as instant contact casting) to ensure compliance.
Acceptable removable devices include a heel pressure relief shoe for heel ulcers and a CAM (controlled ankle motion) walker for metatarsal ulcers. Be sure off-loading devices are applied securely so no slippage can occur.
Step 6: Cover with moist dressings
The purpose of any topical dressing is to keep wounds moist, absorb exudate, and prevent contamination. A variety of moist dressings have been successfully used to treat ulcers, although evidence to recommend any particular dressing is insufficient.19 While wound vacuum-assisted closure (VAC) devices are widely used, there is little support for their use. A review of 7 trials comparing VAC devices with moistened gauze dressings or other topical agents found no evidence that topical negative pressure increases chronic wound healing.30
When foot ulcers do not heal
AIM DOC highlights the steps of diabetic ulcer care that are most likely to result in healing. When ulcers are slow to heal, review the 6 steps of treatment, paying particular attention to off-loading. If you establish that these have been appropriately applied and the wound is still not responding, consider alternative diagnoses such as venous insufficiency, vasculitis, or malignancy. Venous insufficiency ulcers may be difficult to differentiate from neuropathic ulcers, but they won’t heal without compression dressings. Diagnosis of vasculitis and malignancy can be made by biopsying the ulcer edge.
2 adjunctive therapies to consider
If there is no evidence to support an alternative diagnosis, consider adjunctive treatments, with hyperbaric oxygen therapy (HBOT) foremost among them.
A Cochrane review of HBOT found that it reduced the number of major amputations in diabetes patients with chronic foot ulcers, and improved healing at 1 year.31 Both Medicare and Medicaid cover HBOT for patients with diabetic ulcers classified as Wagner Grade 3 or higher that have not responded to 30 days of standard treatment.
Maggots, scientifically known as Lucilia sericata (Greenbottle) fly larvae, secrete proteolytic enzymes that debride necrotic tissue but are inactivated by living tissue. One meta-analysis found that neither surgical debridement nor larval therapy showed significant benefit over hydrogel.32 A subsequent small study did show statistically improved healing in ulcers debrided with maggots, compared with surgical debridement.33
CASE: MR. F.’S ULCER HEALS
Because Mr. F.’s ulcer is small and shallow and has been present for a short time, it has an excellent chance of healing if you follow the AIM DOC steps. You determine that he has adequate arterial supply and that the wound is uninfected (there is a strong dorsalis pedis pulse and no warmth, exudate, or erythema around the wound). Using a #15 blade, you pare away the callus surrounding the ulcer and document the length and width of the wound. You cover the ulcer with a moist dressing and instruct Mr. F to replace it twice a day, cautioning him against using alcohol or hydrogen peroxide, which could harm the healing skin. You discuss the importance of avoiding all weight bearing on the ulcerated foot, prescribe a CAM walker to wear at all times except while he’s sleeping, and schedule weekly follow-up visits to track progress.
In 2 weeks, the wound has resolved. You educate Mr. F. about his risk of ulcer recurrence and outline appropriate preventive steps. You also refer him for fitted extra-depth diabetic shoes and ongoing podiatry follow-up.
ACKNOWLEDGMENT
The authors wish to thank Gregory Mack, DPM, University of Wisconsin School of Medicine and Public Health, for his teaching and collaboration.
CORRESPONDENCE
Jennifer Eddy, MD, University of Wisconsin School of Medicine and Public Health, 617 W. Clairemont Avenue, Eau Claire, WI 54701; [email protected]
• Treat burning foot pain in patients with diabetes with tricyclic antidepressants or anticonvulsants. A
• Prescribe custom-fitted extra-depth shoes for patients with diabetes and neuropathy or foot deformity. B
• Consider hyperbaric oxygen therapy for ulcers that fail to respond to standard therapy. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE: AN OBESE PATIENT WITH BURNING FOOT PAIN
Mr. F., an obese 47-year-old with hypertension and type 2 diabetes, is trying to lose weight, and comes in to discuss a new exercise program. He recently had a negative cardiac workup for chest pain, which was ultimately diagnosed as gastroesophageal reflux disease.
The patient, whose most recent glycosylated hemoglobin (HbA1c) is 7.5, reports painful burning in his feet at night. A foot exam reveals no ulcers, lesions, or calluses; 2+ dorsalis pedis pulses bilaterally; and loss of sensation to 10-g monofilament testing at 3 sites on the bottom of each foot.
Based on Mr. F.’s presentation, it seems safe from a cardiac standpoint for him to embark on an exercise program, but what about his risk for a foot ulcer? An accurate assessment would enable you to tailor your recommendations and take the appropriate steps to modify the patient’s risk now—and to minimize complications down the road.
The incidence of diabetic foot ulcers—like that of diabetes itself—has risen in recent years.1 More than 15% of the approximately 23.5 million US adults with diabetes are expected to develop a foot ulcer at some point in their lives.2 Improvements in wound care and increased use of revascularization techniques have led to a decline in the number of ulcer-related amputations.3 But for 7% of those who develop diabetic foot ulcers, amputation is still the end result.4
As a family physician, you can play a key role in guarding against that outcome. This review—and the risk classification, ulcer grading, and treatment mnemonic tools that are detailed in the pages that follow—will help you optimize the foot care you provide to patients with diabetes.
Simple classification system accurately predicts risk
The longer an individual lives with diabetes and the poorer the level of blood glucose control, the greater the risk of ulceration.1,5 Other major risk factors include neuropathy, peripheral arterial disease, and a previous foot ulcer or amputation. All patients with diabetes should undergo an annual risk assessment for foot ulcers, which can be easily incorporated into their yearly physical. Although the causes of ulceration are numerous and complex, classification of risk based on simple measures has been found to accurately predict risk.6
Risk stratification tool. Medical history and in-office assessment of pulses, sensation, and foot deformities form the basis for a handy assessment tool (TABLE 1). In a study of 3256 patients, these simple criteria successfully predicted ulcer risk. During the nearly 2 years of the study, fewer than 1% of those categorized as low risk (n=2253) developed ulcers, while more than 29% of the high-risk group (n=477) did.6
To assess a patient’s risk, visually examine both feet, inspecting the skin, nails, and structure. Autonomic neuropathy may cause decreased sweating, leading to dry, breakable skin, while motor neuropathy can cause deformities such as hammer or claw toes, which also increase ulcer risk. Palpate for dorsalis pedis and posterior tibialis pulses. Although posterior tibialis pulses may be congenitally absent, the absence of a dorsalis pedis pulse is associated with a 6-fold increased risk of foot ulcer.1
Compared with patients with diabetes who have never had a foot ulcer, patients with a history of foot ulcer have 13 times the risk.7 As already noted, glucose control is also a key risk factor. For every 2-point increase in HbA1c, the odds ratio for ulcer development increases by 1.6,5 which is likely the result of hyperglycemia’s contribution to microvascular disease and peripheral neuropathy.
TABLE 1
Diabetic foot ulcer: Risk stratification
| Risk level/criteria | Incidence of ulcer* |
|---|---|
| Low risk All of the following: No history of ulcer At least 1 pulse per foot ≤1 of 10 sites insensate to monofilament testing No foot deformity or physical or visual impairment | 0.36% |
| Moderate risk 1 or more of the following: Missing both pulses in either foot ≥2 insensate sites to monofilament testing Foot deformity Unable to see or reach foot | 2.3% |
| High risk 1 or more of the following: Prior ulcer or amputation Neuropathy and absent pulses Neuropathy or absent pulses and calluses or foot deformity | 29.4% |
| * Percentage of patients in each risk category who developed foot ulcers over the nearly 2-year study period. | |
| Source: Leese GP, et al. Int J Clin Pract. 2006.6 | |
Screening for neuropathy, ulceration’s most consistent risk factor
Forty percent of patients with diabetes develop distal peripheral neuropathy.8 Damage to sensory nerves often results in the burning foot pain described by Mr. F., which can significantly affect quality of life. It may also present simply as decreased sensation or vibration sense. While decreased sensation is not painful and may not be troubling to the patient, it substantially heightens the risk of foot injury.
To screen for neuropathy, place a 10-g monofilament on noncallused plantar surfaces of the distal hallux and metatarsal heads, with enough pressure to slightly bend the filament. Instructing patients to close their eyes and report any sensation is more effective than prompting them for a response.
Recent guidelines from the American Diabetes Association and American Association of Clinical Endocrinologists recommend 1 additional screening test for neuropathy,9 such as pinprick or vibration sense.
Patients with neuropathy or other risk factors require more frequent follow-up to check for early signs of ulceration. Take the opportunity to provide education about the importance of self-examination of the feet, among other preventive measures (See “Preventing ulcers in high-risk feet”). Be alert to evidence of Charcot neuroarthropathy (CN). This progressive and irreversible condition of bone and joint slippage, dislocation, or fracture can affect any part of the foot, although it is typically found in the midfoot.
Acute CN is a clinical emergency characterized by pain, warmth, and erythema—making it clinically indistinguishable from cellulitis, osteomyelitis, or gout. Blood tests, including a white blood cell count, sedimentary rate, and uric acid level, may be necessary for diagnosis, as well as radiographs or magnetic resonance imaging. Treatment is long-term (4-6 months) immobilization of the joint to prevent further destruction,10 and bisphosphonates for pain during the acute phase.11 Untreated CN typically results in a rocker-bottom foot deformity (FIGURE) that puts patients at greater risk for plantar ulceration.
Although it is possible to successfully treat the majority of diabetic foot ulcers, the wounds result in considerable morbidity, lower quality of life, and increased health care costs. A far better approach is to focus on prevention, with appropriate interventions and frequent follow-up for those at high risk.
ENSURE THAT THE PATIENT HAS THE RIGHT FOOTWEAR.
Properly fitting shoes with ample room for the toes is a priority for all patients with diabetes, but “high-risk feet” need therapeutic footwear, which Medicare covers as a yearly benefit. For those with a history of foot ulcer or amputation, custom insoles, rigid rocker shoes, and orthotics can help prevent re-ulceration.34 For socks, synthetic blends are preferable to cotton, which can chafe when wet with sweat.
PROVIDE EDUCATIONAL MATERIALS; EMPHASIZE FOOT INSPECTION.
All patients with diabetes should receive general education regarding foot care, as there is evidence that it improves behavior and may prevent injuries.7,35 Educational materials emphasizing the importance of nightly foot inspection, overall foot care, and physician inspection are crucial for patients at high risk for developing foot ulcers. Excellent patient education materials are available from the National Diabetes Education Program (See “Take Care of Your Feet for a Lifetime”).
STRESS FREQUENT FOLLOW-UP.
Patients should be informed of their risk level for diabetic foot ulcers after screening. Advise those at high risk to have their feet inspected by a podiatrist or other knowledgeable clinician every 1 to 2 months.9
ENCOURAGE EXERCISE.
Non-weight-bearing exercise programs, including swimming, and a consistent level of daily weight-bearing activity should be encouraged. Caution patients to increase weight-bearing exercise gradually, however, ideally in a closely supervised setting, and to do everything possible to avoid even minimal foot trauma.
REVIEW MEDICATIONS AND ADJUST THERAPY, AS NEEDED.
Tight glycemic control and the use of angiotensin-converting enzyme inhibitors may help prevent the development of neuropathy.36,37 For those who already have neuropathy, tricyclic antidepressants38 or anticonvulsants may bring pain relief.
GUARD AGAINST CHARCOT NEUROARTHROPATHY.
Be alert to this diagnosis in patients who present with a warm, red, painful midfoot. Patients with long-standing neuropathy may benefit from preventive bracing to limit joint movement and lower the risk of Charcot neuroarthropathy.
FIGURE
Rocker-bottom foot deformity
This radiograph reveals extensive collapse of the inner arch and a “rocker-bottom” foot deformity, the result of untreated Charcot neuroarthropathy.
CASE: MR. F.’S RISK BECOMES A REALITY
Mr. F. has a moderate risk of foot ulceration, based on evidence of neuropathy with 6 insensate sites (TABLE 1). You emphasize the importance of foot care, including appropriate footwear, and refer him to a podiatrist. You strongly support his decision to begin an exercise program to improve his glycemic control, decrease his cardiovascular mortality risk, and possibly help him lose weight.
Non-weight-bearing activity can be safely recommended to patients with diabetic neuropathy. So can daily weight-bearing activity, which actually decreases the risk of foot ulcer by maintaining leg muscle tone and plantar tissue tolerance to stress.12,13 Recent studies suggest that increasing weight-bearing activity slowly–walking daily and adding a total of 100 additional steps every 2 weeks, for example—in a carefully monitored program is not associated with increased risk of foot ulceration.14 You report these findings to Mr. F.
When he returns to your office in 2 months, Mr. F. has lost 7 pounds and his HbA1c has fallen to 6.5. Despite these positive developments, an examination of his feet reveals a full thickness ulcer on the left metatarsal head, which the patient had not noticed.
You recognize the clinical urgency of effectively treating Mr. F.’s diabetic foot ulcer, as size, duration, and grade are the greatest predictors of healing. Ulcers that are larger than 2 cm, have been present for more than 6 months,15 or have a higher grade on a scale such as the Wagner Foot Classification System16 (TABLE 2) are far less likely to heal than smaller, low-grade ulcers of shorter duration.
AIM DOC mnemonic guides ulcer care
Comprehensive, coordinated care improves outcomes for diabetic ulcers and has repeatedly been shown to reduce amputation rates.17-19 Large clinical trials have not evaluated each aspect of diabetic ulcer care, however, so the recommendations that follow are based on expert opinion and available evidence. These include assessing the limb’s arterial supply and ensuring that the patient undergoes revascularization, as needed; promptly treating infection; and providing optimal wound care, including debridement of callused and necrotic tissue, off-loading pressure, and applying moist wound dressings.20
AIM DOC, developed by 1 of the authors (JE), is a handy treatment tool. The mnemonic represents both the elements of treatment and the order in which they should be carried out. The letters stand for:
- Arterial disease
- Infection
- Measure
- Debride
- Off-load
- Cover
Here’s how to use AIM DOC, step by step:
Step 1: Assess for arterial disease
Start by assessing the vascular supply to the affected limb, which can be presumed to be adequate if pulses are palpable. If pulses cannot be palpated, the patient should undergo an ankle-brachial index (ABI) test and, if necessary, referred to a specialist to be evaluated for angioplasty or vascular bypass surgery. An ABI <0.9 is abnormal; 0.5 is considered the threshold for healing without such intervention.3 Keep in mind, however, that the ABI is falsely elevated in approximately 15% of patients with diabetes. If classic signs and symptoms of arterial disease are present, further evaluation is needed even if the ABI is normal.
Classic signs and symptoms of arterial disease include cool, hairless feet with shiny skin, and claudication. Location may provide another clue to etiology: Ulcers located on the heel, the outside of the foot, or between the toes tend to be associated with vascular disease, while ulcers with surrounding callus, such as the classic mal perforans ulcer on the metatarsal head, are neuropathic.
Step 2: Treat—or rule out—infection
While patients with diabetes have a 5-fold increase in risk of infection21 compared with individuals without the disease, there is no value in treating an uninfected ulcer with antibiotics. Diagnosing infection can be challenging because patients with diabetes may be less likely to demonstrate evidence of infection.21
An elevated white blood cell count, purulent drainage, foul odor, and/or erythema >2 cm around the wound clearly indicates a need for systemic antibiotics.22,23 Tissue necrosis, often assumed to represent ischemia, may result from neutrophilic vasculitis from soft tissue infection.24 Superficial cultures reflect colonization and should not be used to diagnose infection. In inspecting the wound for signs of infection, probe the ulcer and evaluate for osteomyelitis if it reaches bone.
Gram-positive cocci, especially Staphylococcus aureus, are the predominant pathogens in diabetic foot infections, and antibiotics effective against them may be sufficient for mild-to-moderate infections in patients who have not recently received antibiotic therapy. Gram-negative rods may be found in chronic wounds, however, and anaerobic pathogens may be present in patients with foot ischemia or gangrene; in both cases, broader-spectrum antimicrobials are required.22 Highly bioavailable oral antibiotics are indicated for infection, including some cases of osteomyelitis. Silver dressings may be helpful as a topical antimicrobial; there have been reports of successful treatment with honey, as well.25,26
Step 3: Measure (and grade) the wound
Accurate measurement of ulcer size is critical, both when you initially detect it and at each subsequent visit. To get an accurate measure, simply multiply the greatest length by the greatest width. The percent change in wound size after 4 weeks of treatment is a significant predictor of healing.25
Some clinicians also use photographs to track wound size, but these can be misleading. A better approach is to trace the wound on a sheet of acetate to document progress over time. If you see no improvement within 2 weeks, treatment should be modified.
Use a grading tool. There are a number of systems used for grading ulcers, none of which is universally accepted. One well-known tool is the Wagner Classification System16 (TABLE 2) referred to earlier. The higher the grade, the lower the likelihood that the ulcer will heal.
TABLE 2
Grading the ulcer: The Wagner system
| Grade | Description |
|---|---|
| 0 | No open lesions; may have deformity or cellulitis |
| 1 | Superficial diabetic ulcer (partial or full thickness) |
| 2 | Ulcer extension to ligament, tendon, joint capsule, or deep fascia (without abscess or osteomyelitis) |
| 3 | Deep ulcer with abscess, osteomyelitis, or joint sepsis |
| 4 | Gangrene localized to portion of forefoot or heel |
| 5 | Extensive gangrenous involvement of the entire foot |
| Adapted from: Wagner FW Jr. Orthopedics. 1987.16 | |
Step 4: Debride the ulcer
Frequent sharp debridement—to remove necrotic, callused, infected, and hypergranulation tissue—has long been considered essential in the treatment of neuropathic ulcers,17,21 especially for chronic or infected wounds. Debridement is thought to aid in healing by reducing pressure on the ulcer, decreasing bacterial contamination, enhancing platelet activation, releasing growth factors, and stimulating granulation tissue.
While some physicians are hesitant to perform debridement in the office, the process can actually be carried out without difficulty in an outpatient setting. Because of the neuropathy associated with most diabetic ulcers, no anesthesia is required. While the procedure is not typically painful, you will need to provide patient education to prepare the patient for the possibility of bleeding. Debride the wound to the outer edge of the hyperkeratotic tissue. If bleeding occurs, simply apply pressure until it stops.
When not to debride. Debridement is contraindicated under certain circumstances—if the limb has poor circulation, for example. Similarly, avoid debriding heel ulcers covered by eschar if there is no fluctuance in the underlying tissue, as the eschar provides a protective barrier.24 When sharp debridement is not possible, consider topical hydrogel or maggot therapy, an adjuvant treatment we’ll discuss in a bit.
Step 5: Off-load the wound
Mechanical load relief is vital for treating neuropathic ulcers, both to redistribute plantar pressures and protect granulation tissue. Total contact casting (TCC) is the gold standard, healing 90% of ulcers within 6 to 8 weeks.28 TCC is costly when applied weekly, however, and should only be done by a specialist, as an incorrectly applied cast can lead to the creation of new ulcers.
Because of the heaviness and inconvenience of the casts, many patients prefer removable devices, but these devices are much less effective. One study found that the average removable off-loading device is worn no more than 30% of the time that the patient is walking.29 Removable devices can be temporarily secured with plaster of Paris (a process that is sometimes referred to as instant contact casting) to ensure compliance.
Acceptable removable devices include a heel pressure relief shoe for heel ulcers and a CAM (controlled ankle motion) walker for metatarsal ulcers. Be sure off-loading devices are applied securely so no slippage can occur.
Step 6: Cover with moist dressings
The purpose of any topical dressing is to keep wounds moist, absorb exudate, and prevent contamination. A variety of moist dressings have been successfully used to treat ulcers, although evidence to recommend any particular dressing is insufficient.19 While wound vacuum-assisted closure (VAC) devices are widely used, there is little support for their use. A review of 7 trials comparing VAC devices with moistened gauze dressings or other topical agents found no evidence that topical negative pressure increases chronic wound healing.30
When foot ulcers do not heal
AIM DOC highlights the steps of diabetic ulcer care that are most likely to result in healing. When ulcers are slow to heal, review the 6 steps of treatment, paying particular attention to off-loading. If you establish that these have been appropriately applied and the wound is still not responding, consider alternative diagnoses such as venous insufficiency, vasculitis, or malignancy. Venous insufficiency ulcers may be difficult to differentiate from neuropathic ulcers, but they won’t heal without compression dressings. Diagnosis of vasculitis and malignancy can be made by biopsying the ulcer edge.
2 adjunctive therapies to consider
If there is no evidence to support an alternative diagnosis, consider adjunctive treatments, with hyperbaric oxygen therapy (HBOT) foremost among them.
A Cochrane review of HBOT found that it reduced the number of major amputations in diabetes patients with chronic foot ulcers, and improved healing at 1 year.31 Both Medicare and Medicaid cover HBOT for patients with diabetic ulcers classified as Wagner Grade 3 or higher that have not responded to 30 days of standard treatment.
Maggots, scientifically known as Lucilia sericata (Greenbottle) fly larvae, secrete proteolytic enzymes that debride necrotic tissue but are inactivated by living tissue. One meta-analysis found that neither surgical debridement nor larval therapy showed significant benefit over hydrogel.32 A subsequent small study did show statistically improved healing in ulcers debrided with maggots, compared with surgical debridement.33
CASE: MR. F.’S ULCER HEALS
Because Mr. F.’s ulcer is small and shallow and has been present for a short time, it has an excellent chance of healing if you follow the AIM DOC steps. You determine that he has adequate arterial supply and that the wound is uninfected (there is a strong dorsalis pedis pulse and no warmth, exudate, or erythema around the wound). Using a #15 blade, you pare away the callus surrounding the ulcer and document the length and width of the wound. You cover the ulcer with a moist dressing and instruct Mr. F to replace it twice a day, cautioning him against using alcohol or hydrogen peroxide, which could harm the healing skin. You discuss the importance of avoiding all weight bearing on the ulcerated foot, prescribe a CAM walker to wear at all times except while he’s sleeping, and schedule weekly follow-up visits to track progress.
In 2 weeks, the wound has resolved. You educate Mr. F. about his risk of ulcer recurrence and outline appropriate preventive steps. You also refer him for fitted extra-depth diabetic shoes and ongoing podiatry follow-up.
ACKNOWLEDGMENT
The authors wish to thank Gregory Mack, DPM, University of Wisconsin School of Medicine and Public Health, for his teaching and collaboration.
CORRESPONDENCE
Jennifer Eddy, MD, University of Wisconsin School of Medicine and Public Health, 617 W. Clairemont Avenue, Eau Claire, WI 54701; [email protected]
1. LeMaster JW, Reiber GE. Epidemiology and economic impact of foot ulcers. In: Boulton AJM, Cavanaugh PR, Rayman G, eds. The Foot in Diabetes. 4th ed. London, England: John Wiley & Sons Ltd; 2006:1–16.
2. Department of Health and Human Services Centers for Disease Control and Health Prevention. National Diabetes Fact Sheet 2007. Available at: http://www.cdc.gov/diabetes/pubs/pdf/ndfs_2007.pdf. Accessed May 15, 2009.
3. Pinzur M. Amputations in the diabetic foot. In: Boulton AJM, Cavanaugh PR, Rayman G, eds. The Foot in Diabetes. 4th ed. London, England: John Wiley & Sons Ltd; 2006:308–322.
4. Margolis DJ, Allen-Taylor L, Hoffstad O, et al. Diabetic neuropathic foot ulcers and amputation. Wound Repair Regen. 2005;13:230-236.
5. Moss SE, Klein R, Kelin BE. The prevalence and incidence of lower extremity amputation in a diabetic population. Arch Intern Med. 1992;152:610-616.
6. Leese GP, Reid F, Green V, et al. Stratification of foot ulcer risk in patients with diabetes: a population based study. Int J Clin Pract. 2006;60:541-545.
7. Litzelman DR, Slemenda CW, Langefeld CD. Reduction of lower extremity clinical abnormalities in patient with non-insulin-dependent diabetes mellitus: a randomized controlled trial. Ann Intern Med. 1993;119:36-41.
8. Palumbo PJ, Melton LJ, III. Peripheral vascular disease and diabetes. In: National Diabetes Data Group, National Institute of Diabetes and Digestive and Kidney Disease, eds. Diabetes in America. 2nd ed. Bethesda, Md: National Institutes of Health, NIDDKD; 1995:401–408.
9. Boulton AJM, Armstrong DG, Albert SF, et al. Comprehensive foot examination and risk assessment: a report of the task force of the foot care interest group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care. 2008;31:1679-1685.
10. Jude EB. Charcot foot: what’s new in pathogensis and medical management? In: Boulton AJM, Cavanaugh PR, Rayman G, eds. The Foot in Diabetes. 4th ed. London, England: John Wiley & Sons Ltd; 2006: 265–273.
11. Pitocco D, Ruotolo V, Caputo S, et al. Six month treatment with alendronate in acute Charcot neuroarthropathy: a randomized controlled trial. Diabetes Care. 2005;28:1214-1215.
12. LeMaster JW, Reiber GE, Smith DG, et al. Daily weight-bearing activity does not increase the risk of diabetic foot ulcers. Med Sci Sports Exerc. 2003;35:1093-1099.
13. Lott DJ, Malug KS, Sinacore DR, et al. Relationship between changes in activity and plantar ulcer recurrence in a patient with diabetes mellitus. Phys Ther. 2005;85:579-588.
14. LeMaster JW, Mueller MJ, Reiber GE, et al. Effect of weight-bearing activity on foot ulcer incidence in people with diabetic peripheral neuropathy: Feet First randomized controlled trial. Phys Ther. 2008;88:1-13.
15. Margolis DJ, Kantor J, Santanna J, et al. Risk factors for delayed healing of neuropathic foot ulcers: a pooled analysis. Arch Dermatol. 2000;136:1531-1535.
16. Wagner FW, Jr. The diabetic foot. Orthopedics. 1987;10:163-172.
17. Rith-Najarian S, Branchaid C, Beaulieu O, et al. Reducing lower-extremity amputations due to diabetes: application of the staged diabetes management approach in a primary care setting. J Fam Pract. 1998;47:127-132.
18. Canavan RJ, Unwin NC, Connolly VM, et al. Diabetes and non-diabetes related lower extremity amputation incidence before and after the introduction of better organized diabetic foot care. Diabetes Care. 2008;31.-
19. Krishnan St, Nash F, Baker NR, et al. Reduction in diabetes amputations over 11 years in a defined UK population. Diabetes Care. 2008;31:99-101.
20. Clayton W, Elasy TA. A review of the pathophysiology, classification, and treatment of foot ulcers in diabetic patients. Clin Diabetes. 2009;27:52-58.
21. Majno G, Joris I. Cells, Tissues, and Disease: Principles of General Pathology, 2nd ed. Oxford, England: Oxford University Press; 2004.
22. Lipsky BA, Berendt AR, Deery HG, et al. Diagnosis and treatment of diabetic foot infections. Clin Infect Dis. 2004;39:885-910.
23. Frykberg RG, Zgonis T, Armstrong DG, et al. Diabetic foot disorders: a clinical practice guideline (2006 revision). J Foot Ankle Surg. 2006;45(5 suppl):S2-S66.
24. Edmonds M, Foster A. The use of antibiotics in the diabetic foot. Am J Surg. 2004;187:S25-S28.
25. Eddy JJ, Gideonsen MD. Topical honey therapy for diabetic foot ulcers. J Fam Pract. 2005;54:533-535.
26. Remmen R, Coenen S, Seuntjens R, et al. Honey for refractory diabetic foot ulcers. J Fam Pract. 2005;54:863.-
27. Sheehan P, Jones P, Caselli A, et al. Percent change in wound area in diabetic foot ulcers over a 4-week period is a robust predictor of complete healing in a 12-week prospective trial. Diabetes Care. 2003;26:1879-1882.
28. Lavery LA, Murdoch DP. Conventional offloading and activity monitoring. In: The Foot in Diabetes. 4th ed. Boulton AJM, Cavanaugh PR, Rayman G, eds. London, England: John Wiley & Sons Ltd; 2006: PG NUM.
29. Armstrong DG, Lavery LA, Kimbrel HR, et al. Activity patterns of patients with diabetic foot ulceration: patients with active ulceration may not adhere to a standard pressure off-loading regimen. Diabetes Care. 2003;26:2595-2597.
30. Ubbink DT, Westerbos SJ, Evans D, et al. H. Topical negative pressure for treating chronic wounds. Cochrane Database Syst Rev. 2008(3);CD001898.-
31. Kranke P, Bennett M, Roeckl-Wiedmann I, et al. Hyperbaric oxygen therapy for chronic wounds. Cochrane Database Syst Rev. 2004(2);CD004123.-
32. Edwards J. Debridement of diabetic foot ulcers. Cochrane Database Syst Rev. 2002(4);CD003556.-
33. Sherman RA. Maggot therapy for treating diabetic foot ulcers unresponsive to conventional therapy. Diabetes Care. 2003;26:446-451.
34. Spencer SA. Pressure relieving interventions for preventing and treating diabetic foot ulcers. Cochrane Database Syst Rev. 2000(3);CD002302.-
35. Valk GD, Kriegsman DM, Assendelft WJ. Patient education for preventing diabetic foot ulceration. Cochrane Database Syst Rev. 2001(4);CD001488.-(AU: 2005 update..?)
36. The Diabetes Control and Complications Trial Research Group. The effect of intensive diabetes therapy on the development and progression of neuropathy. Ann Intern Med. 1995;122:561-568.
37. Boulton AJM, Viniv AL, Arezzo JC, et al. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care. 2005;28:956-962.
38. Wong MC, Chung JW, Wong TK. Effects of treatments for symptoms of painful diabetic neuropathy: systematic review. BMJ. 2007;335:87.-
1. LeMaster JW, Reiber GE. Epidemiology and economic impact of foot ulcers. In: Boulton AJM, Cavanaugh PR, Rayman G, eds. The Foot in Diabetes. 4th ed. London, England: John Wiley & Sons Ltd; 2006:1–16.
2. Department of Health and Human Services Centers for Disease Control and Health Prevention. National Diabetes Fact Sheet 2007. Available at: http://www.cdc.gov/diabetes/pubs/pdf/ndfs_2007.pdf. Accessed May 15, 2009.
3. Pinzur M. Amputations in the diabetic foot. In: Boulton AJM, Cavanaugh PR, Rayman G, eds. The Foot in Diabetes. 4th ed. London, England: John Wiley & Sons Ltd; 2006:308–322.
4. Margolis DJ, Allen-Taylor L, Hoffstad O, et al. Diabetic neuropathic foot ulcers and amputation. Wound Repair Regen. 2005;13:230-236.
5. Moss SE, Klein R, Kelin BE. The prevalence and incidence of lower extremity amputation in a diabetic population. Arch Intern Med. 1992;152:610-616.
6. Leese GP, Reid F, Green V, et al. Stratification of foot ulcer risk in patients with diabetes: a population based study. Int J Clin Pract. 2006;60:541-545.
7. Litzelman DR, Slemenda CW, Langefeld CD. Reduction of lower extremity clinical abnormalities in patient with non-insulin-dependent diabetes mellitus: a randomized controlled trial. Ann Intern Med. 1993;119:36-41.
8. Palumbo PJ, Melton LJ, III. Peripheral vascular disease and diabetes. In: National Diabetes Data Group, National Institute of Diabetes and Digestive and Kidney Disease, eds. Diabetes in America. 2nd ed. Bethesda, Md: National Institutes of Health, NIDDKD; 1995:401–408.
9. Boulton AJM, Armstrong DG, Albert SF, et al. Comprehensive foot examination and risk assessment: a report of the task force of the foot care interest group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care. 2008;31:1679-1685.
10. Jude EB. Charcot foot: what’s new in pathogensis and medical management? In: Boulton AJM, Cavanaugh PR, Rayman G, eds. The Foot in Diabetes. 4th ed. London, England: John Wiley & Sons Ltd; 2006: 265–273.
11. Pitocco D, Ruotolo V, Caputo S, et al. Six month treatment with alendronate in acute Charcot neuroarthropathy: a randomized controlled trial. Diabetes Care. 2005;28:1214-1215.
12. LeMaster JW, Reiber GE, Smith DG, et al. Daily weight-bearing activity does not increase the risk of diabetic foot ulcers. Med Sci Sports Exerc. 2003;35:1093-1099.
13. Lott DJ, Malug KS, Sinacore DR, et al. Relationship between changes in activity and plantar ulcer recurrence in a patient with diabetes mellitus. Phys Ther. 2005;85:579-588.
14. LeMaster JW, Mueller MJ, Reiber GE, et al. Effect of weight-bearing activity on foot ulcer incidence in people with diabetic peripheral neuropathy: Feet First randomized controlled trial. Phys Ther. 2008;88:1-13.
15. Margolis DJ, Kantor J, Santanna J, et al. Risk factors for delayed healing of neuropathic foot ulcers: a pooled analysis. Arch Dermatol. 2000;136:1531-1535.
16. Wagner FW, Jr. The diabetic foot. Orthopedics. 1987;10:163-172.
17. Rith-Najarian S, Branchaid C, Beaulieu O, et al. Reducing lower-extremity amputations due to diabetes: application of the staged diabetes management approach in a primary care setting. J Fam Pract. 1998;47:127-132.
18. Canavan RJ, Unwin NC, Connolly VM, et al. Diabetes and non-diabetes related lower extremity amputation incidence before and after the introduction of better organized diabetic foot care. Diabetes Care. 2008;31.-
19. Krishnan St, Nash F, Baker NR, et al. Reduction in diabetes amputations over 11 years in a defined UK population. Diabetes Care. 2008;31:99-101.
20. Clayton W, Elasy TA. A review of the pathophysiology, classification, and treatment of foot ulcers in diabetic patients. Clin Diabetes. 2009;27:52-58.
21. Majno G, Joris I. Cells, Tissues, and Disease: Principles of General Pathology, 2nd ed. Oxford, England: Oxford University Press; 2004.
22. Lipsky BA, Berendt AR, Deery HG, et al. Diagnosis and treatment of diabetic foot infections. Clin Infect Dis. 2004;39:885-910.
23. Frykberg RG, Zgonis T, Armstrong DG, et al. Diabetic foot disorders: a clinical practice guideline (2006 revision). J Foot Ankle Surg. 2006;45(5 suppl):S2-S66.
24. Edmonds M, Foster A. The use of antibiotics in the diabetic foot. Am J Surg. 2004;187:S25-S28.
25. Eddy JJ, Gideonsen MD. Topical honey therapy for diabetic foot ulcers. J Fam Pract. 2005;54:533-535.
26. Remmen R, Coenen S, Seuntjens R, et al. Honey for refractory diabetic foot ulcers. J Fam Pract. 2005;54:863.-
27. Sheehan P, Jones P, Caselli A, et al. Percent change in wound area in diabetic foot ulcers over a 4-week period is a robust predictor of complete healing in a 12-week prospective trial. Diabetes Care. 2003;26:1879-1882.
28. Lavery LA, Murdoch DP. Conventional offloading and activity monitoring. In: The Foot in Diabetes. 4th ed. Boulton AJM, Cavanaugh PR, Rayman G, eds. London, England: John Wiley & Sons Ltd; 2006: PG NUM.
29. Armstrong DG, Lavery LA, Kimbrel HR, et al. Activity patterns of patients with diabetic foot ulceration: patients with active ulceration may not adhere to a standard pressure off-loading regimen. Diabetes Care. 2003;26:2595-2597.
30. Ubbink DT, Westerbos SJ, Evans D, et al. H. Topical negative pressure for treating chronic wounds. Cochrane Database Syst Rev. 2008(3);CD001898.-
31. Kranke P, Bennett M, Roeckl-Wiedmann I, et al. Hyperbaric oxygen therapy for chronic wounds. Cochrane Database Syst Rev. 2004(2);CD004123.-
32. Edwards J. Debridement of diabetic foot ulcers. Cochrane Database Syst Rev. 2002(4);CD003556.-
33. Sherman RA. Maggot therapy for treating diabetic foot ulcers unresponsive to conventional therapy. Diabetes Care. 2003;26:446-451.
34. Spencer SA. Pressure relieving interventions for preventing and treating diabetic foot ulcers. Cochrane Database Syst Rev. 2000(3);CD002302.-
35. Valk GD, Kriegsman DM, Assendelft WJ. Patient education for preventing diabetic foot ulceration. Cochrane Database Syst Rev. 2001(4);CD001488.-(AU: 2005 update..?)
36. The Diabetes Control and Complications Trial Research Group. The effect of intensive diabetes therapy on the development and progression of neuropathy. Ann Intern Med. 1995;122:561-568.
37. Boulton AJM, Viniv AL, Arezzo JC, et al. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care. 2005;28:956-962.
38. Wong MC, Chung JW, Wong TK. Effects of treatments for symptoms of painful diabetic neuropathy: systematic review. BMJ. 2007;335:87.-
Primary care access: The view from the trenches
When The Journal of Family Practice asked family physicians across the country whether access to primary care was a serious problem in their communities, the answer was a unanimous Yes. Each of the physicians we interviewed is taking steps to increase access. But all agree that the problems contributing to the growing crisis—including a shrinking pool of primary care physicians (PCPs)—are too big to be solved without a national policy shift.
“Physicians owe so much by the time they finish medical school that unless reimbursement is substantially increased, deciding on primary care will be a very difficult decision to make,” says KAROL DAVIS, MD, a family physician (FP) at Heartland Primary Care in Kansas City, Kan.

Karol Davis, MD, Heartland Primary Care, Kansas City, Kan
STANLEY KOZAKOWSKI, MD, director of the Hunterdon Medical Center Family Medicine Residency in Flemington, NJ, echoes Davis’s concern. “We need to address the inequities between the earnings of primary care and non-primary care physicians,” he says, adding that by some accounts, what’s needed is an increase in PCP salaries in the order of 60%.

Stanley Kozakowski, MD, Hunterdon Medical Center Family Medicine Residency, Flemington, NJ
But money is neither the sole problem, nor the sole solution. An advocate of the patient-centered medical home, Kozakowski would end the “hamster wheel model” of practice—in which PCPs squeeze more and more patients into shorter and shorter time slots, but can never catch up. To enable FPs to partner with and coordinate care for their patients, he maintains, “We need to redesign our system to support an advanced primary care model.” In the model he envisions, a PCP’s panel might be closer to 1000 than 2300.
That’s in sharp contrast to the 3300-patient panel of DAVID SWITZER, MD. An FP with Page Healthcare Associates in rural Luray, Va, Switzer has spent the last 12 years in this community, a designated primary care shortage area. “As the population ages and fewer doctors go into primary care,” he says, “the problem is only going to get worse.”

David Switzer, MD, Page Healthcare Associates, Luray, Va
Improving access, 1 practice at a time
While Switzer agrees that “as a society, we need to figure out ways to make it more attractive for physicians to enter primary care disciplines,” he also believes—as do the other FPs we spoke to—that increasing access is the responsibility of individual physicians, too.
With that in mind, his rural practice added 1 full-time and 1 half-time physician a year ago. That increased access in terms of “warm bodies,” Switzer notes, “but we were hampered by space constraints because we had no additional exam rooms.” The solution? The practice instituted staggered schedules, which resulted in opening up earlier in the morning and remaining open a little later in the day.
To further improve access, Switzer and his colleagues implemented an open-access scheduling system. “That has certainly made us more accessible than the conventional scheduling system we previously used,” he acknowledges. “But when you’re limited in terms of capacity, as we are, open access can only go so far.”
If Switzer is not counting on health care reform to fix the PCP access problem, he is hopeful that the recent affliation of his employer, Page Memorial Hospital, with a larger regional health system will make the group more attractive to recruits over the next year or 2. “We now have access to capital to expand the office space, which will help increase our capacity,” he says. Equally helpful is the hospital’s new “convenient care” clinic, adjacent to the emergency department (ED), where patients can go for after-hours care at primary care—not ED—prices.
Heartland Primary Care in Kansas City runs its own urgent care clinic, which is open until 8 pm during the week and until 2 pm on Saturdays and Sundays. Although Davis rotates with several other physicians and nurse practitioners (NPs) in the practice (she works 1 evening a week and 1 weekend a month), after-hours care outside of the hospital is not the norm in their area.
“We routinely see patients at the clinic who have their own PCP, but their doctors close their doors at the end of the day and tell them if they’re sick to go to the ED or to our urgent care clinic, “Davis says. Those who choose the clinic, according to Davis, fare considerably better, in terms of convenience as well as costs: “We’ve gotten the wait down to a point where it’s usually less than 20 minutes,” she says. “At the ED down the street, I’m told, patients can wait as long as 3 hours.”
Davis has taken additional steps to improve patient access. After having a closed panel for 2 years, she hired an NP to help her work down the backlog and see more patients. Now she’s able to accommodate 2 new patients every workday. “I love meeting new people and would like to accept more new patients,” says Davis. But her panel size—now at 2700—is approaching the point where she may have to close it again.
While some FPs are frustrated by the focus of reform efforts, Davis sees the proposed legislation as an important first step. Another step in the right direction would be to institute reimbursement for electronic patient interactions, she adds, such as those supported by the patient portal Heartland is about to launch. Another key step: Require public and private health plans alike to pay higher fees for after-hours care.
“Plumbers get paid extra for working on Sundays,” Davis points out. “Why shouldn’t we?”
KATE ROWLAND, MD, an FP at Advocate Illinois Masonic Family Medicine Residency in Chicago, says that the residency clinic where she works has taken another step toward increasing access: The group has instituted formal walk-in hours. Between 8 and 10 am, established patients with acute care needs can come in without an appointment and be guaranteed that they’ll be seen by noon.

Kate Rowland, MD, Advocate Illinois Masonic Family Medicine Residency, Chicago
The new approach—a modified version of same-day scheduling—has been tremendously successful, she says, and relieved a good deal of pressure. Still, “We see some 3000 patients per month, and have just gotten busier and busier.”
What else can be done?
Rowland favors taking small steps, 1 at a time: extending clinic hours (it is now open until 7 pm 2 evenings a week and from 8 am until noon on Saturdays); reallocating physician duties, as needed; and establishing classes for patients with similar issues, such as asthma and diabetes, among other innovations.
She maintains, however, that the US health system is devoted to curing disease and prolonging life rather than on keeping the population healthy. A major refocus is needed for lasting change to occur, Rowland says. But will governmental policy changes and physician culture shift in the right direction?
“Medicine is incredibly stagnant,” the 31-year-old Rowland says. “I wonder whether we’ll still have the same chaotic system when I’m ready to retire.”
When The Journal of Family Practice asked family physicians across the country whether access to primary care was a serious problem in their communities, the answer was a unanimous Yes. Each of the physicians we interviewed is taking steps to increase access. But all agree that the problems contributing to the growing crisis—including a shrinking pool of primary care physicians (PCPs)—are too big to be solved without a national policy shift.
“Physicians owe so much by the time they finish medical school that unless reimbursement is substantially increased, deciding on primary care will be a very difficult decision to make,” says KAROL DAVIS, MD, a family physician (FP) at Heartland Primary Care in Kansas City, Kan.
Karol Davis, MD, Heartland Primary Care, Kansas City, Kan
STANLEY KOZAKOWSKI, MD, director of the Hunterdon Medical Center Family Medicine Residency in Flemington, NJ, echoes Davis’s concern. “We need to address the inequities between the earnings of primary care and non-primary care physicians,” he says, adding that by some accounts, what’s needed is an increase in PCP salaries in the order of 60%.
Stanley Kozakowski, MD, Hunterdon Medical Center Family Medicine Residency, Flemington, NJ
But money is neither the sole problem, nor the sole solution. An advocate of the patient-centered medical home, Kozakowski would end the “hamster wheel model” of practice—in which PCPs squeeze more and more patients into shorter and shorter time slots, but can never catch up. To enable FPs to partner with and coordinate care for their patients, he maintains, “We need to redesign our system to support an advanced primary care model.” In the model he envisions, a PCP’s panel might be closer to 1000 than 2300.
That’s in sharp contrast to the 3300-patient panel of DAVID SWITZER, MD. An FP with Page Healthcare Associates in rural Luray, Va, Switzer has spent the last 12 years in this community, a designated primary care shortage area. “As the population ages and fewer doctors go into primary care,” he says, “the problem is only going to get worse.”
David Switzer, MD, Page Healthcare Associates, Luray, Va
Improving access, 1 practice at a time
While Switzer agrees that “as a society, we need to figure out ways to make it more attractive for physicians to enter primary care disciplines,” he also believes—as do the other FPs we spoke to—that increasing access is the responsibility of individual physicians, too.
With that in mind, his rural practice added 1 full-time and 1 half-time physician a year ago. That increased access in terms of “warm bodies,” Switzer notes, “but we were hampered by space constraints because we had no additional exam rooms.” The solution? The practice instituted staggered schedules, which resulted in opening up earlier in the morning and remaining open a little later in the day.
To further improve access, Switzer and his colleagues implemented an open-access scheduling system. “That has certainly made us more accessible than the conventional scheduling system we previously used,” he acknowledges. “But when you’re limited in terms of capacity, as we are, open access can only go so far.”
If Switzer is not counting on health care reform to fix the PCP access problem, he is hopeful that the recent affliation of his employer, Page Memorial Hospital, with a larger regional health system will make the group more attractive to recruits over the next year or 2. “We now have access to capital to expand the office space, which will help increase our capacity,” he says. Equally helpful is the hospital’s new “convenient care” clinic, adjacent to the emergency department (ED), where patients can go for after-hours care at primary care—not ED—prices.
Heartland Primary Care in Kansas City runs its own urgent care clinic, which is open until 8 pm during the week and until 2 pm on Saturdays and Sundays. Although Davis rotates with several other physicians and nurse practitioners (NPs) in the practice (she works 1 evening a week and 1 weekend a month), after-hours care outside of the hospital is not the norm in their area.
“We routinely see patients at the clinic who have their own PCP, but their doctors close their doors at the end of the day and tell them if they’re sick to go to the ED or to our urgent care clinic, “Davis says. Those who choose the clinic, according to Davis, fare considerably better, in terms of convenience as well as costs: “We’ve gotten the wait down to a point where it’s usually less than 20 minutes,” she says. “At the ED down the street, I’m told, patients can wait as long as 3 hours.”
Davis has taken additional steps to improve patient access. After having a closed panel for 2 years, she hired an NP to help her work down the backlog and see more patients. Now she’s able to accommodate 2 new patients every workday. “I love meeting new people and would like to accept more new patients,” says Davis. But her panel size—now at 2700—is approaching the point where she may have to close it again.
While some FPs are frustrated by the focus of reform efforts, Davis sees the proposed legislation as an important first step. Another step in the right direction would be to institute reimbursement for electronic patient interactions, she adds, such as those supported by the patient portal Heartland is about to launch. Another key step: Require public and private health plans alike to pay higher fees for after-hours care.
“Plumbers get paid extra for working on Sundays,” Davis points out. “Why shouldn’t we?”
KATE ROWLAND, MD, an FP at Advocate Illinois Masonic Family Medicine Residency in Chicago, says that the residency clinic where she works has taken another step toward increasing access: The group has instituted formal walk-in hours. Between 8 and 10 am, established patients with acute care needs can come in without an appointment and be guaranteed that they’ll be seen by noon.
Kate Rowland, MD, Advocate Illinois Masonic Family Medicine Residency, Chicago
The new approach—a modified version of same-day scheduling—has been tremendously successful, she says, and relieved a good deal of pressure. Still, “We see some 3000 patients per month, and have just gotten busier and busier.”
What else can be done?
Rowland favors taking small steps, 1 at a time: extending clinic hours (it is now open until 7 pm 2 evenings a week and from 8 am until noon on Saturdays); reallocating physician duties, as needed; and establishing classes for patients with similar issues, such as asthma and diabetes, among other innovations.
She maintains, however, that the US health system is devoted to curing disease and prolonging life rather than on keeping the population healthy. A major refocus is needed for lasting change to occur, Rowland says. But will governmental policy changes and physician culture shift in the right direction?
“Medicine is incredibly stagnant,” the 31-year-old Rowland says. “I wonder whether we’ll still have the same chaotic system when I’m ready to retire.”
When The Journal of Family Practice asked family physicians across the country whether access to primary care was a serious problem in their communities, the answer was a unanimous Yes. Each of the physicians we interviewed is taking steps to increase access. But all agree that the problems contributing to the growing crisis—including a shrinking pool of primary care physicians (PCPs)—are too big to be solved without a national policy shift.
“Physicians owe so much by the time they finish medical school that unless reimbursement is substantially increased, deciding on primary care will be a very difficult decision to make,” says KAROL DAVIS, MD, a family physician (FP) at Heartland Primary Care in Kansas City, Kan.
Karol Davis, MD, Heartland Primary Care, Kansas City, Kan
STANLEY KOZAKOWSKI, MD, director of the Hunterdon Medical Center Family Medicine Residency in Flemington, NJ, echoes Davis’s concern. “We need to address the inequities between the earnings of primary care and non-primary care physicians,” he says, adding that by some accounts, what’s needed is an increase in PCP salaries in the order of 60%.
Stanley Kozakowski, MD, Hunterdon Medical Center Family Medicine Residency, Flemington, NJ
But money is neither the sole problem, nor the sole solution. An advocate of the patient-centered medical home, Kozakowski would end the “hamster wheel model” of practice—in which PCPs squeeze more and more patients into shorter and shorter time slots, but can never catch up. To enable FPs to partner with and coordinate care for their patients, he maintains, “We need to redesign our system to support an advanced primary care model.” In the model he envisions, a PCP’s panel might be closer to 1000 than 2300.
That’s in sharp contrast to the 3300-patient panel of DAVID SWITZER, MD. An FP with Page Healthcare Associates in rural Luray, Va, Switzer has spent the last 12 years in this community, a designated primary care shortage area. “As the population ages and fewer doctors go into primary care,” he says, “the problem is only going to get worse.”
David Switzer, MD, Page Healthcare Associates, Luray, Va
Improving access, 1 practice at a time
While Switzer agrees that “as a society, we need to figure out ways to make it more attractive for physicians to enter primary care disciplines,” he also believes—as do the other FPs we spoke to—that increasing access is the responsibility of individual physicians, too.
With that in mind, his rural practice added 1 full-time and 1 half-time physician a year ago. That increased access in terms of “warm bodies,” Switzer notes, “but we were hampered by space constraints because we had no additional exam rooms.” The solution? The practice instituted staggered schedules, which resulted in opening up earlier in the morning and remaining open a little later in the day.
To further improve access, Switzer and his colleagues implemented an open-access scheduling system. “That has certainly made us more accessible than the conventional scheduling system we previously used,” he acknowledges. “But when you’re limited in terms of capacity, as we are, open access can only go so far.”
If Switzer is not counting on health care reform to fix the PCP access problem, he is hopeful that the recent affliation of his employer, Page Memorial Hospital, with a larger regional health system will make the group more attractive to recruits over the next year or 2. “We now have access to capital to expand the office space, which will help increase our capacity,” he says. Equally helpful is the hospital’s new “convenient care” clinic, adjacent to the emergency department (ED), where patients can go for after-hours care at primary care—not ED—prices.
Heartland Primary Care in Kansas City runs its own urgent care clinic, which is open until 8 pm during the week and until 2 pm on Saturdays and Sundays. Although Davis rotates with several other physicians and nurse practitioners (NPs) in the practice (she works 1 evening a week and 1 weekend a month), after-hours care outside of the hospital is not the norm in their area.
“We routinely see patients at the clinic who have their own PCP, but their doctors close their doors at the end of the day and tell them if they’re sick to go to the ED or to our urgent care clinic, “Davis says. Those who choose the clinic, according to Davis, fare considerably better, in terms of convenience as well as costs: “We’ve gotten the wait down to a point where it’s usually less than 20 minutes,” she says. “At the ED down the street, I’m told, patients can wait as long as 3 hours.”
Davis has taken additional steps to improve patient access. After having a closed panel for 2 years, she hired an NP to help her work down the backlog and see more patients. Now she’s able to accommodate 2 new patients every workday. “I love meeting new people and would like to accept more new patients,” says Davis. But her panel size—now at 2700—is approaching the point where she may have to close it again.
While some FPs are frustrated by the focus of reform efforts, Davis sees the proposed legislation as an important first step. Another step in the right direction would be to institute reimbursement for electronic patient interactions, she adds, such as those supported by the patient portal Heartland is about to launch. Another key step: Require public and private health plans alike to pay higher fees for after-hours care.
“Plumbers get paid extra for working on Sundays,” Davis points out. “Why shouldn’t we?”
KATE ROWLAND, MD, an FP at Advocate Illinois Masonic Family Medicine Residency in Chicago, says that the residency clinic where she works has taken another step toward increasing access: The group has instituted formal walk-in hours. Between 8 and 10 am, established patients with acute care needs can come in without an appointment and be guaranteed that they’ll be seen by noon.
Kate Rowland, MD, Advocate Illinois Masonic Family Medicine Residency, Chicago
The new approach—a modified version of same-day scheduling—has been tremendously successful, she says, and relieved a good deal of pressure. Still, “We see some 3000 patients per month, and have just gotten busier and busier.”
What else can be done?
Rowland favors taking small steps, 1 at a time: extending clinic hours (it is now open until 7 pm 2 evenings a week and from 8 am until noon on Saturdays); reallocating physician duties, as needed; and establishing classes for patients with similar issues, such as asthma and diabetes, among other innovations.
She maintains, however, that the US health system is devoted to curing disease and prolonging life rather than on keeping the population healthy. A major refocus is needed for lasting change to occur, Rowland says. But will governmental policy changes and physician culture shift in the right direction?
“Medicine is incredibly stagnant,” the 31-year-old Rowland says. “I wonder whether we’ll still have the same chaotic system when I’m ready to retire.”
The health care problem no one’s talking about
As the nation moves closer to a health reform bill designed to extend coverage to the vast majority of the uninsured, a key concern is being overlooked: Simply having health insurance—crucial as that is—is not enough. People also need access to care, particularly to primary care. Yet a growing body of evidence suggests that even among insured Americans, access to primary care is on the decline.
Consider the following:
- In 2005, more than 56 million Americans (nearly half of whom had some form of coverage) were “medically disenfranchised”—lacking sufficient access to primary care services. Two years later, that number had grown to 60 million.1
- In a 2006 national survey, little more than 1 in 4 adults (ages 18 to 64) said they could easily reach their doctors by phone, get after-hours care, or schedule timely office visits.2
- The number of medical school graduates in the United States choosing careers in family medicine fell from 2340 in 1997 to 1132 in 2006; during that same period, the percentage of internal medicine residents entering primary care dropped from 54% to 20%.3
- In 2008, nearly 30% of Medicare beneficiaries seeking a new primary care physician (PCP) reported difficulty finding a doctor—a 17% increase since 2006.4
A shrinking pool of primary care physicians
Compared with other Western nations, the United States has a smaller proportion of its physician workforce engaged in primary care.5 Shortages of PCPs already exist in numerous states, with Alabama, Alaska, Florida, Kansas, Mississippi, Missouri, Oregon, South Carolina, and Utah among them.1 In the decade ahead, the Council on Graduate Medical Education, among other professional groups, expects the shortages to become more widespread and more severe.
Recruiting PCPs is increasingly difficult—something that comes as no surprise to physician recruiters. Merritt Hawkins, a large physician recruitment firm, reports that in 2005 the number of searches for open positions for PCPs exceeded searches for specialists for the first time.6 And in 2006, nearly half of all primary care residents were contacted by recruiters more than 50 times. In a survey of physician groups that same year, 94% of respondents ranked either internists or family physicians as the most difficult to recruit.7
Nurse practitioners (NPs) and physician assistants (PAs) have been important contributors to the primary care workforce, but they, too, may soon be in shorter supply. That’s especially true of PAs, given that less than one third (31%) of them are choosing careers in adult primary care.8
Quality of care pays the price. Ironically, the shrinking pool of PCPs coincides with the growing recognition of the importance of the patient-centered medical home. There is increasing evidence, too, of the link between lack of access to primary care and higher mortality rates9 and poorer outcomes.10 Lack of access appears to have an adverse effect on health care spending, as well. While the administration searches for ways to lower the cost of care in order to pay for the expansion in coverage, directors of health plans and medical groups expect medical spending to rise as the looming PCP shortage leads to greater use of specialists and more emergency visits.11
A broader look at inadequate access
The PCP shortage alone, however, is not the whole story. A number of other potent factors related to, but not the direct result of, the shortage contribute to the growing inability of insured Americans to have timely access to primary care. Chief among them are a mismatch between demand for appointments and physicians’ capacity to provide them, limited after-hours care, and organizational problems in primary care practices. We’ve identified the following barriers—and the policy changes and shifts in physician culture that are needed to overcome them.
BARRIER #1: Panel size
The average full-time primary care practitioner has an estimated panel of 2300 patients12—too many for a single physician to provide adequate patient care for, according to a recent study.13,14 Some practices have excessively large panels because they’re located in areas with a shortage of primary care providers. (In an area with 25 PCPs per 100,000 population, for instance, the average panel size would be 4000.) Other practices accept too many patients in order to stay afloat financially. In either case, a situation in which the demand for appointments exceeds the available time slots impedes a patient’s ability to get timely care.
BARRIER #2: Capacity
The number of hours per week that a PCP sees patients and the number of patients scheduled per hour determine that physician’s visit capacity. Quality of care is also at stake. Although physicians who schedule 1 patient every 10 or 15 minutes can, of course, accommodate more patients than doctors who spend 20 or 30 minutes per visit, shorter appointments have been found to adversely affect quality.15
Further complicating things is the increasing number of physicians who are opting for part-time work.16 Added to the hospital and nursing home responsibilities many PCPs share, working fewer hours imposes further limits on the number of patients they can care for.
BARRIER #3: Distance
The uneven geographic distribution of PCPs makes access difficult for patients living at a distance from the nearest primary care practice, a particular problem for the homebound and those without transportation. Telemedicine could help mitigate this problem, but few primary care practices are equipped to practice “distance” medicine.
BARRIER #4: Medicaid/Medicare issues
Some primary care practices make decisions about which new patients to accept based on the kind of coverage held by the prospective patients. Medicaid patients have an especially difficult time finding a PCP—far harder than privately insured individuals. Also, in areas in which Medicare fees are below the market rates paid by private insurers, many practices limit the number of Medicare patients they accept.
The bottom line: Already stressed by the economy and low fees, some PCPs say they have little choice but to restrict the number of patients whose care costs them more than they’re paid to provide it.
BARRIER #5: After-hours care
Many patients try, unsuccessfully, to reach their PCP in the evening or on the weekend. The dearth of after-hours access has led to an explosion of “convenience clinics” in pharmacies and shopping malls—and to overuse of the emergency department (ED). In a 2007 national survey, 67% of adults said they had difficulty getting care at night or on weekends unless they went to the ED.17 In another survey, conducted in California in 2006, nearly half of those who sought care in the ED believed their condition could have been handled in a primary care setting, had it been available.18
BARRIER #6: Scheduling
Many patients call their PCP’s office for an appointment, only to find that the next available opening is 3 weeks away. While some groups have introduced open-access scheduling—also called same-day scheduling or advanced access—such a system can only be sustained if the demand for appointments is in balance with the practice’s capacity to see patients.
Part of the problem appears to be organizational. Some physicians routinely make monthly follow-up appointments for patients with chronic conditions, such as hypertension, diabetes, or arthritis. However, the return visit interval is often based on the habits of the individual physician or provider group, rather than on the medical needs of the patients. Indeed, 1 study found that prolonging the visit interval resulted in an improvement rather than a decline in quality of care.19
BARRIER #7: Virtual visits
Many chronic care and preventive care issues could be handled in brief patient encounters via telephone or e-mail. In addition to being convenient for many patients, such virtual visits would increase the practice’s capacity for patients who require in-person visits.20 Here, too, the problem is financial: Insurers generally do not provide reimbursement for virtual visits.
BARRIER #8: Troubles with team care
At some medical groups, nonphysician providers, including registered nurses and pharmacists, use doctor-created protocols and standing orders to address routine chronic care issues and preventive measures for individuals with certain conditions—identified via patient registries. Similarly, medical assistants and community health workers may be trained as health coaches to work with patients on behavior change and adherence to medication regimens, thus freeing up physician time.21 Despite the benefits of team care, most insurers only reimburse the services of MDs, NPs and PAs, meaning that no incentives exist for primary care practices to hire other team members.
The solutions: Policy shifts and culture change
What will it take to improve access to primary care and tear down these barriers? First and foremost, we believe the following policy changes are needed:
- Increase reimbursement for primary care.
- Increase loan repayment programs for medical students who establish primary care practices in areas with established shortages.
- Standardize fees paid by private insurers, Medicare, and Medicaid plans.
- Provide financial incentives for PCPs to deliver after-hours care.
- Invest in a national program aimed at helping primary care practices implement same-day scheduling, team care, and other access improvements.22
- Provide reimbursement for e-mail and telephone encounters and team care, including fees for all allied health professionals who assist PCPs in managing chronic disease and preventive care.
These reforms, if they were to truly come to pass, would ease much of the pressure on PCPs. No matter what policy changes are implemented to increase access to primary care, however, it is clear that a substantial culture change is required on the part of PCPs, as well. Physicians can begin to make changes on their own to increase patient access—expanding the interval between follow-up visits for stable patients, for instance, and reorganizing work schedules so that the practice can remain open for more hours.
It’s clear that providing health insurance to the uninsured without guaranteeing access to primary care can turn a potentially positive development into widespread patient frustration. Unless Americans have greater access to primary care, we fear, the US health care system will undergo significant change without substantial improvement.
CORRESPONDENCE
Thomas Bodenheimer MD, MPH, Department of Family and Community Medicine, University of California at San Francisco, Bldg 80-83, SF General Hospital, 995 Potrero Avenue, San Francisco, CA 94110; [email protected]
1. Primary Care Access: An Essential Building Block of Health Care Reform. Bethesda, Md: National Association of Community Health Centers; March 2009. Available at: http://www.nachc.com/client/documents/pressreleases/PrimaryCareAccessRPT.pdf. Accessed November 11, 2009.
2. Beal A, Doty M, Hernandez S, et al. Closing the Divide: How Medical Homes Promote Equity in Health Care. New York: The Commonwealth Fund; 2007.
3. Bodenheimer T. Primary care–will it survive? N Engl J Med. 2006;355:861-864.
4. Medicare Payment Advisory Commission (MedPAC). Report to the Congress. Medicare Payment Policy. Washington, DC: MedPAC; March 2009, p. 88.
5. Starfield B. Primary Care. New York: Oxford University Press; 1998.
6. Merritt Hawkins & Associates. 2007 Review of Physician and CRNA Recruiting Incentives. 2007. Available at: http://www.merritthawkins.com/pdf/2007_Review_of_Physician_and_CRNA_Recruiting_Incentives.pdf. Accessed June 20, 2009.
7. American Medical Group Association, Cejka Search. 2006 Physician Retention Survey. March 2007. Available at: http://www.cejkasearch.com/surveys/physician-retention-surveys/2007/default.htm. Accessed June 20, 2009.
8. American Academy of Physician Assistants. 2008 AAPA Physician Assistant Census Report. September 5, 2008. Available at: http://www.aapa.org/about-pas/data-and-statistics/1116. Accessed June 20, 2009.
9. Shi L, Macinko J, Starfield B, et al. Primary care, infant mortality, and low birth weight in the states of the USA. J Epidiol Community Health. 2004;58:374-380.
10. Shi L, Stevens GD, Wulu JT, Jr, et al. America’s health centers: reducing racial and ethnic disparities in perinatal care and birth outcomes. Health Serv Res. 2004;39:1881-1901.
11. Cross MA. What the primary care physician shortage means for health plans. Managed Care. 2007. Available at: http://www.managedcaremag.com/archives/0706/0706.shortage.html. Accessed June 20, 2009.
12. Alexander GC, Kurlander J, Wynia MK. Physicians in retainer (concierge) practice. J Gen Intern Med. 2005;20:1069-1083.
13. Yarnall KS, Pollak KI, Ostbye T, et al. Primary care: is there enough time for prevention? Am J Public Health. 2003;93:635-641.
14. Ostbye T, Yarnall KS, Krause KM, et al. Is there time for management of patients with chronic diseases in primary care? Ann Fam Med. 2005;3:209-214.
15. Zyzanski SF, Stange KC, Langa D, et al. Trade-offs in high-volume primary care practice. J Fam Pract. 1998;46:397-402.
16. 46% jump in number of physicians working part-time. Managed Care. 2008;17(5):19.-
17. Schoen C, Osborn R, Doty MM, et al. Toward higher-performance health systems: adults’ health care experiences in seven countries, 2007. Health Aff. 2007;26(6):w717-w734.
18. California HealthCare Foundation. Overuse of Emergency Departments Among Insured Californians. October 2006. Available at: http://www.chcf.org/topics/hospitals/index.cfm?itemID=126089. Accessed June 20, 2009.
19. Schectman G, Barnas G, Laud PG, et al. Prolonging the return visit interval in primary care. Am J Med. 2005;118:393-399.
20. Bergmo TS, Kummervold PE, Gammon D, et al. Electronic patient-provider communication: will it offset office visits and telephone consultations in primary care? Int J Med Inform. 2005;74:705-710.
21. Bodenheimer T. Building teams in primary care: lessons from 15 case studies. California HealthCare Foundation; July 2007. Available at: http://www.fiercehealthcare.com/pages/building-teams-primary-care-lessons-15-case-studies. Accessed June 20, 2009.
22. Grumbach K, Mold JW. A health care cooperative extension service: transforming primary care and community health. JAMA. 2009;301:2589-2591.
As the nation moves closer to a health reform bill designed to extend coverage to the vast majority of the uninsured, a key concern is being overlooked: Simply having health insurance—crucial as that is—is not enough. People also need access to care, particularly to primary care. Yet a growing body of evidence suggests that even among insured Americans, access to primary care is on the decline.
Consider the following:
- In 2005, more than 56 million Americans (nearly half of whom had some form of coverage) were “medically disenfranchised”—lacking sufficient access to primary care services. Two years later, that number had grown to 60 million.1
- In a 2006 national survey, little more than 1 in 4 adults (ages 18 to 64) said they could easily reach their doctors by phone, get after-hours care, or schedule timely office visits.2
- The number of medical school graduates in the United States choosing careers in family medicine fell from 2340 in 1997 to 1132 in 2006; during that same period, the percentage of internal medicine residents entering primary care dropped from 54% to 20%.3
- In 2008, nearly 30% of Medicare beneficiaries seeking a new primary care physician (PCP) reported difficulty finding a doctor—a 17% increase since 2006.4
A shrinking pool of primary care physicians
Compared with other Western nations, the United States has a smaller proportion of its physician workforce engaged in primary care.5 Shortages of PCPs already exist in numerous states, with Alabama, Alaska, Florida, Kansas, Mississippi, Missouri, Oregon, South Carolina, and Utah among them.1 In the decade ahead, the Council on Graduate Medical Education, among other professional groups, expects the shortages to become more widespread and more severe.
Recruiting PCPs is increasingly difficult—something that comes as no surprise to physician recruiters. Merritt Hawkins, a large physician recruitment firm, reports that in 2005 the number of searches for open positions for PCPs exceeded searches for specialists for the first time.6 And in 2006, nearly half of all primary care residents were contacted by recruiters more than 50 times. In a survey of physician groups that same year, 94% of respondents ranked either internists or family physicians as the most difficult to recruit.7
Nurse practitioners (NPs) and physician assistants (PAs) have been important contributors to the primary care workforce, but they, too, may soon be in shorter supply. That’s especially true of PAs, given that less than one third (31%) of them are choosing careers in adult primary care.8
Quality of care pays the price. Ironically, the shrinking pool of PCPs coincides with the growing recognition of the importance of the patient-centered medical home. There is increasing evidence, too, of the link between lack of access to primary care and higher mortality rates9 and poorer outcomes.10 Lack of access appears to have an adverse effect on health care spending, as well. While the administration searches for ways to lower the cost of care in order to pay for the expansion in coverage, directors of health plans and medical groups expect medical spending to rise as the looming PCP shortage leads to greater use of specialists and more emergency visits.11
A broader look at inadequate access
The PCP shortage alone, however, is not the whole story. A number of other potent factors related to, but not the direct result of, the shortage contribute to the growing inability of insured Americans to have timely access to primary care. Chief among them are a mismatch between demand for appointments and physicians’ capacity to provide them, limited after-hours care, and organizational problems in primary care practices. We’ve identified the following barriers—and the policy changes and shifts in physician culture that are needed to overcome them.
BARRIER #1: Panel size
The average full-time primary care practitioner has an estimated panel of 2300 patients12—too many for a single physician to provide adequate patient care for, according to a recent study.13,14 Some practices have excessively large panels because they’re located in areas with a shortage of primary care providers. (In an area with 25 PCPs per 100,000 population, for instance, the average panel size would be 4000.) Other practices accept too many patients in order to stay afloat financially. In either case, a situation in which the demand for appointments exceeds the available time slots impedes a patient’s ability to get timely care.
BARRIER #2: Capacity
The number of hours per week that a PCP sees patients and the number of patients scheduled per hour determine that physician’s visit capacity. Quality of care is also at stake. Although physicians who schedule 1 patient every 10 or 15 minutes can, of course, accommodate more patients than doctors who spend 20 or 30 minutes per visit, shorter appointments have been found to adversely affect quality.15
Further complicating things is the increasing number of physicians who are opting for part-time work.16 Added to the hospital and nursing home responsibilities many PCPs share, working fewer hours imposes further limits on the number of patients they can care for.
BARRIER #3: Distance
The uneven geographic distribution of PCPs makes access difficult for patients living at a distance from the nearest primary care practice, a particular problem for the homebound and those without transportation. Telemedicine could help mitigate this problem, but few primary care practices are equipped to practice “distance” medicine.
BARRIER #4: Medicaid/Medicare issues
Some primary care practices make decisions about which new patients to accept based on the kind of coverage held by the prospective patients. Medicaid patients have an especially difficult time finding a PCP—far harder than privately insured individuals. Also, in areas in which Medicare fees are below the market rates paid by private insurers, many practices limit the number of Medicare patients they accept.
The bottom line: Already stressed by the economy and low fees, some PCPs say they have little choice but to restrict the number of patients whose care costs them more than they’re paid to provide it.
BARRIER #5: After-hours care
Many patients try, unsuccessfully, to reach their PCP in the evening or on the weekend. The dearth of after-hours access has led to an explosion of “convenience clinics” in pharmacies and shopping malls—and to overuse of the emergency department (ED). In a 2007 national survey, 67% of adults said they had difficulty getting care at night or on weekends unless they went to the ED.17 In another survey, conducted in California in 2006, nearly half of those who sought care in the ED believed their condition could have been handled in a primary care setting, had it been available.18
BARRIER #6: Scheduling
Many patients call their PCP’s office for an appointment, only to find that the next available opening is 3 weeks away. While some groups have introduced open-access scheduling—also called same-day scheduling or advanced access—such a system can only be sustained if the demand for appointments is in balance with the practice’s capacity to see patients.
Part of the problem appears to be organizational. Some physicians routinely make monthly follow-up appointments for patients with chronic conditions, such as hypertension, diabetes, or arthritis. However, the return visit interval is often based on the habits of the individual physician or provider group, rather than on the medical needs of the patients. Indeed, 1 study found that prolonging the visit interval resulted in an improvement rather than a decline in quality of care.19
BARRIER #7: Virtual visits
Many chronic care and preventive care issues could be handled in brief patient encounters via telephone or e-mail. In addition to being convenient for many patients, such virtual visits would increase the practice’s capacity for patients who require in-person visits.20 Here, too, the problem is financial: Insurers generally do not provide reimbursement for virtual visits.
BARRIER #8: Troubles with team care
At some medical groups, nonphysician providers, including registered nurses and pharmacists, use doctor-created protocols and standing orders to address routine chronic care issues and preventive measures for individuals with certain conditions—identified via patient registries. Similarly, medical assistants and community health workers may be trained as health coaches to work with patients on behavior change and adherence to medication regimens, thus freeing up physician time.21 Despite the benefits of team care, most insurers only reimburse the services of MDs, NPs and PAs, meaning that no incentives exist for primary care practices to hire other team members.
The solutions: Policy shifts and culture change
What will it take to improve access to primary care and tear down these barriers? First and foremost, we believe the following policy changes are needed:
- Increase reimbursement for primary care.
- Increase loan repayment programs for medical students who establish primary care practices in areas with established shortages.
- Standardize fees paid by private insurers, Medicare, and Medicaid plans.
- Provide financial incentives for PCPs to deliver after-hours care.
- Invest in a national program aimed at helping primary care practices implement same-day scheduling, team care, and other access improvements.22
- Provide reimbursement for e-mail and telephone encounters and team care, including fees for all allied health professionals who assist PCPs in managing chronic disease and preventive care.
These reforms, if they were to truly come to pass, would ease much of the pressure on PCPs. No matter what policy changes are implemented to increase access to primary care, however, it is clear that a substantial culture change is required on the part of PCPs, as well. Physicians can begin to make changes on their own to increase patient access—expanding the interval between follow-up visits for stable patients, for instance, and reorganizing work schedules so that the practice can remain open for more hours.
It’s clear that providing health insurance to the uninsured without guaranteeing access to primary care can turn a potentially positive development into widespread patient frustration. Unless Americans have greater access to primary care, we fear, the US health care system will undergo significant change without substantial improvement.
CORRESPONDENCE
Thomas Bodenheimer MD, MPH, Department of Family and Community Medicine, University of California at San Francisco, Bldg 80-83, SF General Hospital, 995 Potrero Avenue, San Francisco, CA 94110; [email protected]
As the nation moves closer to a health reform bill designed to extend coverage to the vast majority of the uninsured, a key concern is being overlooked: Simply having health insurance—crucial as that is—is not enough. People also need access to care, particularly to primary care. Yet a growing body of evidence suggests that even among insured Americans, access to primary care is on the decline.
Consider the following:
- In 2005, more than 56 million Americans (nearly half of whom had some form of coverage) were “medically disenfranchised”—lacking sufficient access to primary care services. Two years later, that number had grown to 60 million.1
- In a 2006 national survey, little more than 1 in 4 adults (ages 18 to 64) said they could easily reach their doctors by phone, get after-hours care, or schedule timely office visits.2
- The number of medical school graduates in the United States choosing careers in family medicine fell from 2340 in 1997 to 1132 in 2006; during that same period, the percentage of internal medicine residents entering primary care dropped from 54% to 20%.3
- In 2008, nearly 30% of Medicare beneficiaries seeking a new primary care physician (PCP) reported difficulty finding a doctor—a 17% increase since 2006.4
A shrinking pool of primary care physicians
Compared with other Western nations, the United States has a smaller proportion of its physician workforce engaged in primary care.5 Shortages of PCPs already exist in numerous states, with Alabama, Alaska, Florida, Kansas, Mississippi, Missouri, Oregon, South Carolina, and Utah among them.1 In the decade ahead, the Council on Graduate Medical Education, among other professional groups, expects the shortages to become more widespread and more severe.
Recruiting PCPs is increasingly difficult—something that comes as no surprise to physician recruiters. Merritt Hawkins, a large physician recruitment firm, reports that in 2005 the number of searches for open positions for PCPs exceeded searches for specialists for the first time.6 And in 2006, nearly half of all primary care residents were contacted by recruiters more than 50 times. In a survey of physician groups that same year, 94% of respondents ranked either internists or family physicians as the most difficult to recruit.7
Nurse practitioners (NPs) and physician assistants (PAs) have been important contributors to the primary care workforce, but they, too, may soon be in shorter supply. That’s especially true of PAs, given that less than one third (31%) of them are choosing careers in adult primary care.8
Quality of care pays the price. Ironically, the shrinking pool of PCPs coincides with the growing recognition of the importance of the patient-centered medical home. There is increasing evidence, too, of the link between lack of access to primary care and higher mortality rates9 and poorer outcomes.10 Lack of access appears to have an adverse effect on health care spending, as well. While the administration searches for ways to lower the cost of care in order to pay for the expansion in coverage, directors of health plans and medical groups expect medical spending to rise as the looming PCP shortage leads to greater use of specialists and more emergency visits.11
A broader look at inadequate access
The PCP shortage alone, however, is not the whole story. A number of other potent factors related to, but not the direct result of, the shortage contribute to the growing inability of insured Americans to have timely access to primary care. Chief among them are a mismatch between demand for appointments and physicians’ capacity to provide them, limited after-hours care, and organizational problems in primary care practices. We’ve identified the following barriers—and the policy changes and shifts in physician culture that are needed to overcome them.
BARRIER #1: Panel size
The average full-time primary care practitioner has an estimated panel of 2300 patients12—too many for a single physician to provide adequate patient care for, according to a recent study.13,14 Some practices have excessively large panels because they’re located in areas with a shortage of primary care providers. (In an area with 25 PCPs per 100,000 population, for instance, the average panel size would be 4000.) Other practices accept too many patients in order to stay afloat financially. In either case, a situation in which the demand for appointments exceeds the available time slots impedes a patient’s ability to get timely care.
BARRIER #2: Capacity
The number of hours per week that a PCP sees patients and the number of patients scheduled per hour determine that physician’s visit capacity. Quality of care is also at stake. Although physicians who schedule 1 patient every 10 or 15 minutes can, of course, accommodate more patients than doctors who spend 20 or 30 minutes per visit, shorter appointments have been found to adversely affect quality.15
Further complicating things is the increasing number of physicians who are opting for part-time work.16 Added to the hospital and nursing home responsibilities many PCPs share, working fewer hours imposes further limits on the number of patients they can care for.
BARRIER #3: Distance
The uneven geographic distribution of PCPs makes access difficult for patients living at a distance from the nearest primary care practice, a particular problem for the homebound and those without transportation. Telemedicine could help mitigate this problem, but few primary care practices are equipped to practice “distance” medicine.
BARRIER #4: Medicaid/Medicare issues
Some primary care practices make decisions about which new patients to accept based on the kind of coverage held by the prospective patients. Medicaid patients have an especially difficult time finding a PCP—far harder than privately insured individuals. Also, in areas in which Medicare fees are below the market rates paid by private insurers, many practices limit the number of Medicare patients they accept.
The bottom line: Already stressed by the economy and low fees, some PCPs say they have little choice but to restrict the number of patients whose care costs them more than they’re paid to provide it.
BARRIER #5: After-hours care
Many patients try, unsuccessfully, to reach their PCP in the evening or on the weekend. The dearth of after-hours access has led to an explosion of “convenience clinics” in pharmacies and shopping malls—and to overuse of the emergency department (ED). In a 2007 national survey, 67% of adults said they had difficulty getting care at night or on weekends unless they went to the ED.17 In another survey, conducted in California in 2006, nearly half of those who sought care in the ED believed their condition could have been handled in a primary care setting, had it been available.18
BARRIER #6: Scheduling
Many patients call their PCP’s office for an appointment, only to find that the next available opening is 3 weeks away. While some groups have introduced open-access scheduling—also called same-day scheduling or advanced access—such a system can only be sustained if the demand for appointments is in balance with the practice’s capacity to see patients.
Part of the problem appears to be organizational. Some physicians routinely make monthly follow-up appointments for patients with chronic conditions, such as hypertension, diabetes, or arthritis. However, the return visit interval is often based on the habits of the individual physician or provider group, rather than on the medical needs of the patients. Indeed, 1 study found that prolonging the visit interval resulted in an improvement rather than a decline in quality of care.19
BARRIER #7: Virtual visits
Many chronic care and preventive care issues could be handled in brief patient encounters via telephone or e-mail. In addition to being convenient for many patients, such virtual visits would increase the practice’s capacity for patients who require in-person visits.20 Here, too, the problem is financial: Insurers generally do not provide reimbursement for virtual visits.
BARRIER #8: Troubles with team care
At some medical groups, nonphysician providers, including registered nurses and pharmacists, use doctor-created protocols and standing orders to address routine chronic care issues and preventive measures for individuals with certain conditions—identified via patient registries. Similarly, medical assistants and community health workers may be trained as health coaches to work with patients on behavior change and adherence to medication regimens, thus freeing up physician time.21 Despite the benefits of team care, most insurers only reimburse the services of MDs, NPs and PAs, meaning that no incentives exist for primary care practices to hire other team members.
The solutions: Policy shifts and culture change
What will it take to improve access to primary care and tear down these barriers? First and foremost, we believe the following policy changes are needed:
- Increase reimbursement for primary care.
- Increase loan repayment programs for medical students who establish primary care practices in areas with established shortages.
- Standardize fees paid by private insurers, Medicare, and Medicaid plans.
- Provide financial incentives for PCPs to deliver after-hours care.
- Invest in a national program aimed at helping primary care practices implement same-day scheduling, team care, and other access improvements.22
- Provide reimbursement for e-mail and telephone encounters and team care, including fees for all allied health professionals who assist PCPs in managing chronic disease and preventive care.
These reforms, if they were to truly come to pass, would ease much of the pressure on PCPs. No matter what policy changes are implemented to increase access to primary care, however, it is clear that a substantial culture change is required on the part of PCPs, as well. Physicians can begin to make changes on their own to increase patient access—expanding the interval between follow-up visits for stable patients, for instance, and reorganizing work schedules so that the practice can remain open for more hours.
It’s clear that providing health insurance to the uninsured without guaranteeing access to primary care can turn a potentially positive development into widespread patient frustration. Unless Americans have greater access to primary care, we fear, the US health care system will undergo significant change without substantial improvement.
CORRESPONDENCE
Thomas Bodenheimer MD, MPH, Department of Family and Community Medicine, University of California at San Francisco, Bldg 80-83, SF General Hospital, 995 Potrero Avenue, San Francisco, CA 94110; [email protected]
1. Primary Care Access: An Essential Building Block of Health Care Reform. Bethesda, Md: National Association of Community Health Centers; March 2009. Available at: http://www.nachc.com/client/documents/pressreleases/PrimaryCareAccessRPT.pdf. Accessed November 11, 2009.
2. Beal A, Doty M, Hernandez S, et al. Closing the Divide: How Medical Homes Promote Equity in Health Care. New York: The Commonwealth Fund; 2007.
3. Bodenheimer T. Primary care–will it survive? N Engl J Med. 2006;355:861-864.
4. Medicare Payment Advisory Commission (MedPAC). Report to the Congress. Medicare Payment Policy. Washington, DC: MedPAC; March 2009, p. 88.
5. Starfield B. Primary Care. New York: Oxford University Press; 1998.
6. Merritt Hawkins & Associates. 2007 Review of Physician and CRNA Recruiting Incentives. 2007. Available at: http://www.merritthawkins.com/pdf/2007_Review_of_Physician_and_CRNA_Recruiting_Incentives.pdf. Accessed June 20, 2009.
7. American Medical Group Association, Cejka Search. 2006 Physician Retention Survey. March 2007. Available at: http://www.cejkasearch.com/surveys/physician-retention-surveys/2007/default.htm. Accessed June 20, 2009.
8. American Academy of Physician Assistants. 2008 AAPA Physician Assistant Census Report. September 5, 2008. Available at: http://www.aapa.org/about-pas/data-and-statistics/1116. Accessed June 20, 2009.
9. Shi L, Macinko J, Starfield B, et al. Primary care, infant mortality, and low birth weight in the states of the USA. J Epidiol Community Health. 2004;58:374-380.
10. Shi L, Stevens GD, Wulu JT, Jr, et al. America’s health centers: reducing racial and ethnic disparities in perinatal care and birth outcomes. Health Serv Res. 2004;39:1881-1901.
11. Cross MA. What the primary care physician shortage means for health plans. Managed Care. 2007. Available at: http://www.managedcaremag.com/archives/0706/0706.shortage.html. Accessed June 20, 2009.
12. Alexander GC, Kurlander J, Wynia MK. Physicians in retainer (concierge) practice. J Gen Intern Med. 2005;20:1069-1083.
13. Yarnall KS, Pollak KI, Ostbye T, et al. Primary care: is there enough time for prevention? Am J Public Health. 2003;93:635-641.
14. Ostbye T, Yarnall KS, Krause KM, et al. Is there time for management of patients with chronic diseases in primary care? Ann Fam Med. 2005;3:209-214.
15. Zyzanski SF, Stange KC, Langa D, et al. Trade-offs in high-volume primary care practice. J Fam Pract. 1998;46:397-402.
16. 46% jump in number of physicians working part-time. Managed Care. 2008;17(5):19.-
17. Schoen C, Osborn R, Doty MM, et al. Toward higher-performance health systems: adults’ health care experiences in seven countries, 2007. Health Aff. 2007;26(6):w717-w734.
18. California HealthCare Foundation. Overuse of Emergency Departments Among Insured Californians. October 2006. Available at: http://www.chcf.org/topics/hospitals/index.cfm?itemID=126089. Accessed June 20, 2009.
19. Schectman G, Barnas G, Laud PG, et al. Prolonging the return visit interval in primary care. Am J Med. 2005;118:393-399.
20. Bergmo TS, Kummervold PE, Gammon D, et al. Electronic patient-provider communication: will it offset office visits and telephone consultations in primary care? Int J Med Inform. 2005;74:705-710.
21. Bodenheimer T. Building teams in primary care: lessons from 15 case studies. California HealthCare Foundation; July 2007. Available at: http://www.fiercehealthcare.com/pages/building-teams-primary-care-lessons-15-case-studies. Accessed June 20, 2009.
22. Grumbach K, Mold JW. A health care cooperative extension service: transforming primary care and community health. JAMA. 2009;301:2589-2591.
1. Primary Care Access: An Essential Building Block of Health Care Reform. Bethesda, Md: National Association of Community Health Centers; March 2009. Available at: http://www.nachc.com/client/documents/pressreleases/PrimaryCareAccessRPT.pdf. Accessed November 11, 2009.
2. Beal A, Doty M, Hernandez S, et al. Closing the Divide: How Medical Homes Promote Equity in Health Care. New York: The Commonwealth Fund; 2007.
3. Bodenheimer T. Primary care–will it survive? N Engl J Med. 2006;355:861-864.
4. Medicare Payment Advisory Commission (MedPAC). Report to the Congress. Medicare Payment Policy. Washington, DC: MedPAC; March 2009, p. 88.
5. Starfield B. Primary Care. New York: Oxford University Press; 1998.
6. Merritt Hawkins & Associates. 2007 Review of Physician and CRNA Recruiting Incentives. 2007. Available at: http://www.merritthawkins.com/pdf/2007_Review_of_Physician_and_CRNA_Recruiting_Incentives.pdf. Accessed June 20, 2009.
7. American Medical Group Association, Cejka Search. 2006 Physician Retention Survey. March 2007. Available at: http://www.cejkasearch.com/surveys/physician-retention-surveys/2007/default.htm. Accessed June 20, 2009.
8. American Academy of Physician Assistants. 2008 AAPA Physician Assistant Census Report. September 5, 2008. Available at: http://www.aapa.org/about-pas/data-and-statistics/1116. Accessed June 20, 2009.
9. Shi L, Macinko J, Starfield B, et al. Primary care, infant mortality, and low birth weight in the states of the USA. J Epidiol Community Health. 2004;58:374-380.
10. Shi L, Stevens GD, Wulu JT, Jr, et al. America’s health centers: reducing racial and ethnic disparities in perinatal care and birth outcomes. Health Serv Res. 2004;39:1881-1901.
11. Cross MA. What the primary care physician shortage means for health plans. Managed Care. 2007. Available at: http://www.managedcaremag.com/archives/0706/0706.shortage.html. Accessed June 20, 2009.
12. Alexander GC, Kurlander J, Wynia MK. Physicians in retainer (concierge) practice. J Gen Intern Med. 2005;20:1069-1083.
13. Yarnall KS, Pollak KI, Ostbye T, et al. Primary care: is there enough time for prevention? Am J Public Health. 2003;93:635-641.
14. Ostbye T, Yarnall KS, Krause KM, et al. Is there time for management of patients with chronic diseases in primary care? Ann Fam Med. 2005;3:209-214.
15. Zyzanski SF, Stange KC, Langa D, et al. Trade-offs in high-volume primary care practice. J Fam Pract. 1998;46:397-402.
16. 46% jump in number of physicians working part-time. Managed Care. 2008;17(5):19.-
17. Schoen C, Osborn R, Doty MM, et al. Toward higher-performance health systems: adults’ health care experiences in seven countries, 2007. Health Aff. 2007;26(6):w717-w734.
18. California HealthCare Foundation. Overuse of Emergency Departments Among Insured Californians. October 2006. Available at: http://www.chcf.org/topics/hospitals/index.cfm?itemID=126089. Accessed June 20, 2009.
19. Schectman G, Barnas G, Laud PG, et al. Prolonging the return visit interval in primary care. Am J Med. 2005;118:393-399.
20. Bergmo TS, Kummervold PE, Gammon D, et al. Electronic patient-provider communication: will it offset office visits and telephone consultations in primary care? Int J Med Inform. 2005;74:705-710.
21. Bodenheimer T. Building teams in primary care: lessons from 15 case studies. California HealthCare Foundation; July 2007. Available at: http://www.fiercehealthcare.com/pages/building-teams-primary-care-lessons-15-case-studies. Accessed June 20, 2009.
22. Grumbach K, Mold JW. A health care cooperative extension service: transforming primary care and community health. JAMA. 2009;301:2589-2591.
When your patient’s blood pressure won’t come down
• Encourage home BP monitoring. Home readings are often lower than those taken in the office and closer to the average BP recorded by 24-hour ambulatory monitors. C
• Tell patients that reducing sodium intake not only reduces mortality, but it has positive cardiovascular effects separate from BP reduction, such as improved endothelium-dependent vasodilation. A
• Search for secondary causes of resistant hypertension, such as renal artery stenosis, pheochromocytoma, obstructive sleep apnea, and hyperaldosteronism. A
• Consider pseudohypertension in elderly patients who exhibit postural hypotension and fail to respond to increased doses of medication. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 Mr. Brown is a 60-year-old African American man who works as a mid-level executive at a local bank. About a year and a half ago, he was diagnosed with hypertension, joining a number of other family members who also have the condition. Reviewing his chart, you note that at many of his visits—but not all of them—his systolic blood pressure runs close to 150 mm Hg, with diastolic pressure <90 mm Hg. Today his blood pressure is 148/88 mm Hg, numbers that exceed the parameters of the currently accepted definition of hypertension: blood pressure ≥140 mm Hg systolic or ≥90 mm Hg diastolic, taken on 2 separate occasions with the patient sitting down.1
Mr. Brown is one of the more than 65 million American adults suffering from high blood pressure—the No. 1 diagnosis reported in outpatient medical offices.2,3 Despite such prevalence, blood pressure control in the United States is suboptimal, with only one-third of hypertensive patients under adequate control.1 In 2007, the total estimated cost of treating high blood pressure in the United States exceeded $66 billion.4
When you tell Mr. Brown that his blood pressure doesn’t meet the therapeutic goal of <140/90 mm Hg, he gives you his reasons: He was late, he rushed, the traffic was bad, and the nurse rushed him into the exam room before he even had time to catch his breath. He insists his blood pressure is “normal” at home, and blames the elevated numbers on anxiety. He also tells you he was up working most of the night before, drinking coffee to stay awake to finish an urgent project.
Mr. Brown’s current medication regimen includes daily doses of lisinopril-hydrochlorothiazide (HCTZ) 20/25 mg daily and simvastatin 20 mg. He tells you he has no chest pain, shortness of breath, cough, edema, claudication, paroxysmal nocturnal dyspnea, or orthopnea. When you ask if he takes his lisinopril-HCTZ every day, he says Yes, but you have your suspicions.
Consider pseudo-resistance
Suboptimal blood pressure control can be classified as either pseudo-resistant or resistant hypertension. According to the definition used in the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC7), resistance is the “failure to achieve goal BP in patients who are adhering to full doses of an appropriate 3-drug regimen that includes a diuretic.”1
Hypertension is described as “pseudo-resistant” when persistent elevations in blood pressure are the result of a failure to comply with the medication regimen, “white-coat” syndrome, poor blood pressure technique, or a combination of these.5,6 Physician failure to prescribe adequate doses of medication, so-called clinical inertia, may also be a factor.5,7 Causes and remedies for pseudo-resistance are summarized in the algorithm.
In Mr. Brown’s case, he’s told you that he’s taking his lisinopril-HCTZ every day, but when you check your records, you see that the intervals between his refill requests range between 34 and 36 days. So you ask him, again, whether he is taking his lisinopril-HCTZ daily, and this time he says he takes the medication “at least 5 or 6 days a week.”
Encourage compliance. To motivate Mr. Brown, you tell him his blood pressure does not meet the goal of <140/90 mm Hg and that pressure higher than goal is a significant risk factor for cardiovascular disease. You emphasize the importance of taking his medication every day—a mantra you’ve repeated to countless patients over the years. In fact, it’s estimated that up to 40% of patients will discontinue their medication at some point during treatment.8 Patients’ reasons vary and may include medication side effects, the cost of treatment, or a patient’s personal philosophy. Cultural differences may also play a role in noncompliance.9
You talk with Mr. Brown a bit more and confirm that his poor compliance is due to simple forgetfulness and not another underlying reason that would need to be addressed. You hand him a pill calendar to help him keep track of his medication. You review the dosage and feel confident that you’ve prescribed a regimen simple enough for Mr. Brown to stick to and adequate to bring his pressure down.
When your white coat is the problem. Though not the case with Mr. Brown, a patient’s elevated readings could be a case of white coat hypertension—a conditioned response in some patients that is probably the result of anxiety in the medical setting.10 Patients with white coat hypertension have significantly less risk of death and reduced target organ damage than patients with truly resistant hypertension.11,12
If you suspect that a patient of yours has white coat hypertension, you can test your hypothesis by encouraging him or her to buy a blood pressure monitor to use at home, keep a log of the readings, and bring the log in to the next appointment. Improved technology has made home blood pressure monitoring an important tool in the treatment of high blood pressure.13-15
According to a 2008 statement issued jointly by the American Heart Association, the American Society of Hypertension, and the Preventive Cardiovascular Nurses Association, “There is a rapidly growing literature showing that measurements taken by patients at home are often lower than readings taken in the office and closer to the average blood pressure recorded by 24-hour ambulatory monitors, which is the blood pressure that best predicts cardiovascular risk.”14
Arm monitors are the most accurate because they measure brachial artery blood pressure.14 Most wrist monitors have not been validated in studies, but obese patients who cannot find a properly sized cuff may need to use them.14 Finger devices should never be used.14
Sometimes technique is at fault. To ensure that you get proper readings when your patient is in the office, advise the nursing staff not to take a patient’s blood pressure for at least 5 minutes after bringing him or her to an exam room. Specifically, the nurse can either make blood pressure measurement the last thing she (or he) does when “rooming” the patient, or she may prefer to go back to the exam room a few minutes after the patient is there to take the reading. The nurse should take care that the cuff is the right size and that the pressure is taken with the patient’s arm at heart level. In hypertensive patients, placing the arm below the horizontal in the dependent position can raise blood pressure 23/10 mm Hg.1,16,17
Newer blood pressure monitoring devices for the office are available that obviate artificially high readings caused by errors in technique and by white coat syndrome. Researchers have found that readings taken with the BpTRU automatic device, which obtains and records 5 blood pressure readings over a 5-minute period, are lower than those taken by office personnel, and that the white coat effect is eliminated.18,19
One pseudo-resistant case solved. At Mr. Brown’s next visit, you review his pill calendar with him and note that his blood pressure is now running 138/88 mm Hg and he is within his target goal of less than 140/90 mm Hg.
If Mr. Brown’s blood pressure control had continued to be suboptimal even after you’d taken steps to address cause(s) of pseudo-resistance, you would have had to shift gears and consider his case one of resistant hypertension.1
Consider resistant hypertension
Causes of resistant hypertension include lifestyle factors, side effects of medications, and secondary causes.5,6 Tobacco use, obesity, lack of exercise, a high sodium diet, and alcohol consumption can all contribute to hypertension.1,5,6,20,21 Dietary sodium in particular has significant adverse effects. Reducing dietary sodium not only reduces mortality, but has positive cardiovascular effects separate from blood pressure reduction, such as improved endothelium-dependent vasodilation.22-24
Medications including nonsteroidal anti-inflammatory drugs (NSAIDs), oral contraceptives, sympathomimetics, glucocorticoids, and black licorice (found in some herbal supplements) may all raise blood pressure.5 Medications and other substances associated with increased blood pressure are listed in the TABLE. A search for such products, with changes or discontinuations that are clinically appropriate, should be part of any evaluation for resistant hypertension.
CASE 2 Ms. Stevens is a 30-year-old woman who was diagnosed with high blood pressure 7 months ago. She is currently taking triamterene-HCTZ 37.5/25 mg daily, amlodipine 10 mg daily, and metoprolol succinate 100 mg daily. She is compliant with medication, sticks to a low-sodium diet, and exercises 5 days a week. She does not drink alcohol. A review of her chart tells you her baseline serum chemistries, chest x-ray, and urinalysis are all normal. Today’s blood pressure taken in the sitting position after a 5-minute rest is 160/92 mm Hg. How should you proceed?
TABLE
Medications and other agents that may cause hypertension
| • Alcohol | • Cyclosporine | • Nicotine |
| • Antidepressants | • Erythropoietin | • NSAIDs |
| • Buspirone (Buspar) | • Estrogen preparations | • St. John’s wort |
| • Cocaine | • Licorice | • Tyramine-containing foods |
| • Corticosteroids | • Metoclopramide (Reglan) | |
| NSAIDs, nonsteroidal anti-infiammatory drugs. | ||
| Source: Chobanian A, et al. Hypertension. 2003.1 | ||
Is it resistant hypertension? Look for clues
You go over Ms. Stevens’ history carefully. Her hypertension medications should be adequate, and she is compliant with the prescribed regimen. Lifestyle factors are clearly not to blame: She’s not obese, she follows a low-sodium diet, doesn’t drink or smoke, and gets plenty of exercise. You ask her about other medications or over-the-counter supplements she takes, and nothing on her list raises a red flag. This doesn’t seem to be pseudo-resistance, and though you’re aware that only about 5% of cases of hypertension can be attributed to secondary causes, you think Ms. Stevens may be one of those patients.25
Numerous causes of secondary hypertension exist, and evaluations for such causes should be considered in all patients with resistant hypertension. Renal artery stenosis and pheochromocytoma are the most well-known causes, but less well known but significant contributors are obstructive sleep apnea and hyperaldosteronism.1,6,26,27
The relationship between obstructive sleep apnea and hypertension is not clear, and most of the studies linking the 2 conditions are population based.28 Nevertheless, because studies show that continuous positive airway pressure (CPAP) does improve blood pressure control in patients with obstructive sleep apnea, it is appropriate to investigate patients with resistant hypertension for this condition.29
Aldosterone’s role in resistant hypertension appears to be greatest in those with insulin resistance. The mechanism appears to be amplified sodium retention resulting in increased volume expansion, negative effects on oxidative stress, and elevated inflammation.27 Many patients with hypertension also have insulin resistance, so testing for hyperaldosteronism should be part of your investigation.
Test, and as necessary, test some more
Taking into consideration that 2 of the more common causes of secondary hypertension are obstructive sleep apnea and renal artery stenosis, you schedule an overnight sleep study for Ms. Stevens and a magnetic resonance arteriogram of the kidneys.26,30 (For more on the diagnostic tests and treatments for secondary causes of resistant hypertension, see the ALGORITHM.)
Reconsider medications. Ms. Stevens is already receiving the maximum dose of amlodipine, so you increase her metoprolol succinate to 200 mg daily and ask her to return in 1 month after the results of her tests come in.
Change medications and keep testing. At her next visit, Ms. Stevens’ blood pressure is 150/88 mm Hg, and her pulse rate is 64. Results of her overnight sleep study and magnetic resonance arteriogram are normal. At this juncture, with her work-up for common secondary causes of hypertension negative, you consider changes in her medication regimen. Because her pulse rate of 64 rules out any increase in the metoprolol, you decide to add lisinopril-HCTZ 10/12.5 mg to her regimen and discontinue her triamterene-HCTZ. Because her blood pressure is still elevated at this visit, you schedule additional testing to look for other secondary causes.
More tests, higher doses. One month later, Ms. Stevens returns with a blood pressure of 146/88 mm Hg. Appropriate testing for primary aldosteronism, pheochromocytoma, Cushing’s disease, and coarctation are all normal. Her lisinopril-HCTZ dose is increased to 20/12.5 mg, and when she returns 1 month later her blood pressure reading is 138/88 mm Hg.
You conclude that she is a patient with hard-to-control hypertension with no underlying cause who requires a significant amount of medication. That is not an uncommon situation. Although her blood pressure is now controlled to an appropriate level, you make a chart notation to consider referral to a hypertension specialist if her blood pressure become elevated again and treatment changes are not effective.6
ALGORITHM
Hard-to-control hypertension? This treatment algorithm can help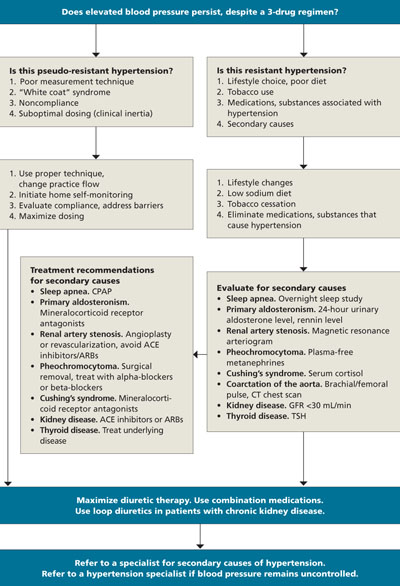
ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker; CPAP, continuous positive airway pressure; CT, computed tomography; GFR, glomerular filtration rate; TSH, thyroid-stimulating hormone.
Adapted from: Calhoun DA, et al. Hypertension. 2008.6
Another consideration: Pseudohypertension
This uncommon condition sometimes occurs in elderly patients who suffer from a stiffened or thickened brachial artery.1 Because the blood pressure cuff cannot compress the artery, blood pressure readings overestimate true arterial pressure. The condition should be considered in patients when increased doses of medication fail to achieve the desired response, particularly in patients who exhibit postural hypotension.1
This was not a likely explanation for Ms. Stevens, however, given the clinical scenario and her young age.
CORRESPONDENCE Randy Wexler, MD, MPH, B0902B Cramblett Hall, 456 W 10th Avenue, Columbus, OH 43210; [email protected]
1. Chobanian A, Bakris GL, Black HR, et al. Seventh Report of The Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC-7). Hypertension. 2003;42:1206-1252.
2. Fields L, Burt V, Cutler J, et al. The burden of adult hypertension in the United States 1999-2000: A rising tide. Hypertension. 2004;44:1-7.
3. Fang J, Alderman MH, Keenan NL, et al. Hypertension control at physicians’ offices in the United States. Am J Hypertens. 2008;21:136-142.
4. American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics–2007 update. Circulation. 2007;115:e69-e171.
5. Sarafidis PA, Bakris GL. Resistant hypertension: an overview of evaluation and treatment. J Am Coll Cardiol. 2008;52:1749-1757.
6. Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension. 2008;51:1403-1419.
7. Phillips LS, Branch WT, Book CB, et al. Clinical inertia. Ann Intern Med. 2001;135:825-834.
8. Van Wijk BL, Klungel OH, Heerdink ER, et al. Rate and determinants of 10-year persistence with antihypertensive drugs. J Hypertens. 2005;23:2101-2107.
9. Wexler R, Pleister A, Feldman D. Sociology meets genetics: sociogenetic implications for future management of hypertension and heart failure. Curr Treat Options Cardiovasc Med. 2009;11:305-315.
10. Ogedegbe G, Pickering TG, Clemow L, et al. The misdiagnosis of hypertension: the role of patient anxiety. Arch Intern Med. 2008;168:2459-2465.
11. Dawes MG, Bartlett G, Coats AJ, et al. Comparing the effects of white coat hypertension and sustained hypertension on mortality in a UK primary care setting. Ann Fam Med. 2008;6:390-396.
12. Pierdomenico SD, Lapenna D, Bucci A, et al. Cardiovascular outcome in treated hypertensive patients with responder, masked, false resistant, and true resistant hypertension. Am J Hypertens. 2005;18:1422-1428.
13. Verberk WJ, Kroon AA, Kessels AGH, et al. Home blood pressure monitoring: a systematic review. J Am Coll Cardiol. 2005;46:743-751.
14. Pickering TG, Miller NH, Ogedegbe G, et al. AHA/ASH/PCNA scientific statement. Call to action on use and reimbursement for home blood pressure monitoring. Hypertension. 2008;52:10-29.
15. O’Brien E. Ambulatory blood pressure measurement: the case for implementation in primary care. Hypertension. 2008;51:1435-1441.
16. O’Brien E, Asmar R, Beilin L, et al. On behalf of the European Society of Hypertension Working Group on Blood Pressure Monitoring. European Society of Hypertension recommendations for conventional ambulatory and home blood pressure measurements. J Hypertens. 2003;21:821-848.
17. Mourad A, Carney S, Gillies A, et al. Arm position and blood pressure: a risk factor for hypertension. J Hum Hypertens. 2003;17:389-395.
18. Myers MG. Automated blood pressure measurement in routine clinical practice. Blood Press Monit. 2006;11:59-62.
19. Myers MG, Valdivieso M, Kiss A. Use of automated office blood pressure measurement to reduce white coat hypertension. J Hypertens. 2009;27:280-286.
20. He J, Paul J, Whelton P, Appel L, et al. Long-term effects of weight loss and dietary sodium reduction on incidence of hypertension. Hypertension. 2000;35:544-549.
21. PREMIER Collaborative Research Writing Group. Effects of comprehensive lifestyle modification on blood pressure control. JAMA. 2003;289:2083-2093.
22. Cook NR, Cutler JA, Obarzanek E, et al. Long term effects of dietary sodium reduction on cardiovascular disease outcomes: observational follow-up of the trials of hypertension prevention (TOHP). BMJ. 2007;334:885-888.
23. Dickinson BD, Havas S. Reducing the population burden of cardiovascular disease by reducing sodium intake. Arch Intern Med. 2007;167:1460-1468.
24. Li J, White J, Guo L, et al. Salt inactivates endothelial nitric oxide synthase in endothelial cells. J Nutr. 2009;139:1-5.
25. Beevers G, Lip G, O’Brien E. ABCs of hypertension: the pathophysiology of hypertension. BMJ. 2001;322:912-916.
26. Peppard PE, Young T, Palta M, et al. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342:1378-1384.
27. Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150:776-783.
28. Young T, Peppard P, Palta M, et al. Population-based study of sleep-disordered breathing as a risk factor for hypertension. Arch Intern Med. 1997;157:1746-1752.
29. Haentjens P, Van Meerhaeghe A, Moscariello A. The impact of continuous positive airway pressure on blood pressure in patients with obstructive sleep apnea syndrome: evidence from a meta-analysis of placebo-controlled randomized trials. Arch Intern Med. 2007;167:757-764.
30. Kawashima A, Francis IR, Baumgarten DA, et al. For the Expert Panel on Urologic Imaging. Renovascular hypertension. Reston, Va: American College of Radiology; 2007. Available at: www.guideline.gov/summary/summary.aspx?ss=15&doc_id=11590&nbr=6003. Accessed April 4, 2009.
• Encourage home BP monitoring. Home readings are often lower than those taken in the office and closer to the average BP recorded by 24-hour ambulatory monitors. C
• Tell patients that reducing sodium intake not only reduces mortality, but it has positive cardiovascular effects separate from BP reduction, such as improved endothelium-dependent vasodilation. A
• Search for secondary causes of resistant hypertension, such as renal artery stenosis, pheochromocytoma, obstructive sleep apnea, and hyperaldosteronism. A
• Consider pseudohypertension in elderly patients who exhibit postural hypotension and fail to respond to increased doses of medication. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 Mr. Brown is a 60-year-old African American man who works as a mid-level executive at a local bank. About a year and a half ago, he was diagnosed with hypertension, joining a number of other family members who also have the condition. Reviewing his chart, you note that at many of his visits—but not all of them—his systolic blood pressure runs close to 150 mm Hg, with diastolic pressure <90 mm Hg. Today his blood pressure is 148/88 mm Hg, numbers that exceed the parameters of the currently accepted definition of hypertension: blood pressure ≥140 mm Hg systolic or ≥90 mm Hg diastolic, taken on 2 separate occasions with the patient sitting down.1
Mr. Brown is one of the more than 65 million American adults suffering from high blood pressure—the No. 1 diagnosis reported in outpatient medical offices.2,3 Despite such prevalence, blood pressure control in the United States is suboptimal, with only one-third of hypertensive patients under adequate control.1 In 2007, the total estimated cost of treating high blood pressure in the United States exceeded $66 billion.4
When you tell Mr. Brown that his blood pressure doesn’t meet the therapeutic goal of <140/90 mm Hg, he gives you his reasons: He was late, he rushed, the traffic was bad, and the nurse rushed him into the exam room before he even had time to catch his breath. He insists his blood pressure is “normal” at home, and blames the elevated numbers on anxiety. He also tells you he was up working most of the night before, drinking coffee to stay awake to finish an urgent project.
Mr. Brown’s current medication regimen includes daily doses of lisinopril-hydrochlorothiazide (HCTZ) 20/25 mg daily and simvastatin 20 mg. He tells you he has no chest pain, shortness of breath, cough, edema, claudication, paroxysmal nocturnal dyspnea, or orthopnea. When you ask if he takes his lisinopril-HCTZ every day, he says Yes, but you have your suspicions.
Consider pseudo-resistance
Suboptimal blood pressure control can be classified as either pseudo-resistant or resistant hypertension. According to the definition used in the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC7), resistance is the “failure to achieve goal BP in patients who are adhering to full doses of an appropriate 3-drug regimen that includes a diuretic.”1
Hypertension is described as “pseudo-resistant” when persistent elevations in blood pressure are the result of a failure to comply with the medication regimen, “white-coat” syndrome, poor blood pressure technique, or a combination of these.5,6 Physician failure to prescribe adequate doses of medication, so-called clinical inertia, may also be a factor.5,7 Causes and remedies for pseudo-resistance are summarized in the algorithm.
In Mr. Brown’s case, he’s told you that he’s taking his lisinopril-HCTZ every day, but when you check your records, you see that the intervals between his refill requests range between 34 and 36 days. So you ask him, again, whether he is taking his lisinopril-HCTZ daily, and this time he says he takes the medication “at least 5 or 6 days a week.”
Encourage compliance. To motivate Mr. Brown, you tell him his blood pressure does not meet the goal of <140/90 mm Hg and that pressure higher than goal is a significant risk factor for cardiovascular disease. You emphasize the importance of taking his medication every day—a mantra you’ve repeated to countless patients over the years. In fact, it’s estimated that up to 40% of patients will discontinue their medication at some point during treatment.8 Patients’ reasons vary and may include medication side effects, the cost of treatment, or a patient’s personal philosophy. Cultural differences may also play a role in noncompliance.9
You talk with Mr. Brown a bit more and confirm that his poor compliance is due to simple forgetfulness and not another underlying reason that would need to be addressed. You hand him a pill calendar to help him keep track of his medication. You review the dosage and feel confident that you’ve prescribed a regimen simple enough for Mr. Brown to stick to and adequate to bring his pressure down.
When your white coat is the problem. Though not the case with Mr. Brown, a patient’s elevated readings could be a case of white coat hypertension—a conditioned response in some patients that is probably the result of anxiety in the medical setting.10 Patients with white coat hypertension have significantly less risk of death and reduced target organ damage than patients with truly resistant hypertension.11,12
If you suspect that a patient of yours has white coat hypertension, you can test your hypothesis by encouraging him or her to buy a blood pressure monitor to use at home, keep a log of the readings, and bring the log in to the next appointment. Improved technology has made home blood pressure monitoring an important tool in the treatment of high blood pressure.13-15
According to a 2008 statement issued jointly by the American Heart Association, the American Society of Hypertension, and the Preventive Cardiovascular Nurses Association, “There is a rapidly growing literature showing that measurements taken by patients at home are often lower than readings taken in the office and closer to the average blood pressure recorded by 24-hour ambulatory monitors, which is the blood pressure that best predicts cardiovascular risk.”14
Arm monitors are the most accurate because they measure brachial artery blood pressure.14 Most wrist monitors have not been validated in studies, but obese patients who cannot find a properly sized cuff may need to use them.14 Finger devices should never be used.14
Sometimes technique is at fault. To ensure that you get proper readings when your patient is in the office, advise the nursing staff not to take a patient’s blood pressure for at least 5 minutes after bringing him or her to an exam room. Specifically, the nurse can either make blood pressure measurement the last thing she (or he) does when “rooming” the patient, or she may prefer to go back to the exam room a few minutes after the patient is there to take the reading. The nurse should take care that the cuff is the right size and that the pressure is taken with the patient’s arm at heart level. In hypertensive patients, placing the arm below the horizontal in the dependent position can raise blood pressure 23/10 mm Hg.1,16,17
Newer blood pressure monitoring devices for the office are available that obviate artificially high readings caused by errors in technique and by white coat syndrome. Researchers have found that readings taken with the BpTRU automatic device, which obtains and records 5 blood pressure readings over a 5-minute period, are lower than those taken by office personnel, and that the white coat effect is eliminated.18,19
One pseudo-resistant case solved. At Mr. Brown’s next visit, you review his pill calendar with him and note that his blood pressure is now running 138/88 mm Hg and he is within his target goal of less than 140/90 mm Hg.
If Mr. Brown’s blood pressure control had continued to be suboptimal even after you’d taken steps to address cause(s) of pseudo-resistance, you would have had to shift gears and consider his case one of resistant hypertension.1
Consider resistant hypertension
Causes of resistant hypertension include lifestyle factors, side effects of medications, and secondary causes.5,6 Tobacco use, obesity, lack of exercise, a high sodium diet, and alcohol consumption can all contribute to hypertension.1,5,6,20,21 Dietary sodium in particular has significant adverse effects. Reducing dietary sodium not only reduces mortality, but has positive cardiovascular effects separate from blood pressure reduction, such as improved endothelium-dependent vasodilation.22-24
Medications including nonsteroidal anti-inflammatory drugs (NSAIDs), oral contraceptives, sympathomimetics, glucocorticoids, and black licorice (found in some herbal supplements) may all raise blood pressure.5 Medications and other substances associated with increased blood pressure are listed in the TABLE. A search for such products, with changes or discontinuations that are clinically appropriate, should be part of any evaluation for resistant hypertension.
CASE 2 Ms. Stevens is a 30-year-old woman who was diagnosed with high blood pressure 7 months ago. She is currently taking triamterene-HCTZ 37.5/25 mg daily, amlodipine 10 mg daily, and metoprolol succinate 100 mg daily. She is compliant with medication, sticks to a low-sodium diet, and exercises 5 days a week. She does not drink alcohol. A review of her chart tells you her baseline serum chemistries, chest x-ray, and urinalysis are all normal. Today’s blood pressure taken in the sitting position after a 5-minute rest is 160/92 mm Hg. How should you proceed?
TABLE
Medications and other agents that may cause hypertension
| • Alcohol | • Cyclosporine | • Nicotine |
| • Antidepressants | • Erythropoietin | • NSAIDs |
| • Buspirone (Buspar) | • Estrogen preparations | • St. John’s wort |
| • Cocaine | • Licorice | • Tyramine-containing foods |
| • Corticosteroids | • Metoclopramide (Reglan) | |
| NSAIDs, nonsteroidal anti-infiammatory drugs. | ||
| Source: Chobanian A, et al. Hypertension. 2003.1 | ||
Is it resistant hypertension? Look for clues
You go over Ms. Stevens’ history carefully. Her hypertension medications should be adequate, and she is compliant with the prescribed regimen. Lifestyle factors are clearly not to blame: She’s not obese, she follows a low-sodium diet, doesn’t drink or smoke, and gets plenty of exercise. You ask her about other medications or over-the-counter supplements she takes, and nothing on her list raises a red flag. This doesn’t seem to be pseudo-resistance, and though you’re aware that only about 5% of cases of hypertension can be attributed to secondary causes, you think Ms. Stevens may be one of those patients.25
Numerous causes of secondary hypertension exist, and evaluations for such causes should be considered in all patients with resistant hypertension. Renal artery stenosis and pheochromocytoma are the most well-known causes, but less well known but significant contributors are obstructive sleep apnea and hyperaldosteronism.1,6,26,27
The relationship between obstructive sleep apnea and hypertension is not clear, and most of the studies linking the 2 conditions are population based.28 Nevertheless, because studies show that continuous positive airway pressure (CPAP) does improve blood pressure control in patients with obstructive sleep apnea, it is appropriate to investigate patients with resistant hypertension for this condition.29
Aldosterone’s role in resistant hypertension appears to be greatest in those with insulin resistance. The mechanism appears to be amplified sodium retention resulting in increased volume expansion, negative effects on oxidative stress, and elevated inflammation.27 Many patients with hypertension also have insulin resistance, so testing for hyperaldosteronism should be part of your investigation.
Test, and as necessary, test some more
Taking into consideration that 2 of the more common causes of secondary hypertension are obstructive sleep apnea and renal artery stenosis, you schedule an overnight sleep study for Ms. Stevens and a magnetic resonance arteriogram of the kidneys.26,30 (For more on the diagnostic tests and treatments for secondary causes of resistant hypertension, see the ALGORITHM.)
Reconsider medications. Ms. Stevens is already receiving the maximum dose of amlodipine, so you increase her metoprolol succinate to 200 mg daily and ask her to return in 1 month after the results of her tests come in.
Change medications and keep testing. At her next visit, Ms. Stevens’ blood pressure is 150/88 mm Hg, and her pulse rate is 64. Results of her overnight sleep study and magnetic resonance arteriogram are normal. At this juncture, with her work-up for common secondary causes of hypertension negative, you consider changes in her medication regimen. Because her pulse rate of 64 rules out any increase in the metoprolol, you decide to add lisinopril-HCTZ 10/12.5 mg to her regimen and discontinue her triamterene-HCTZ. Because her blood pressure is still elevated at this visit, you schedule additional testing to look for other secondary causes.
More tests, higher doses. One month later, Ms. Stevens returns with a blood pressure of 146/88 mm Hg. Appropriate testing for primary aldosteronism, pheochromocytoma, Cushing’s disease, and coarctation are all normal. Her lisinopril-HCTZ dose is increased to 20/12.5 mg, and when she returns 1 month later her blood pressure reading is 138/88 mm Hg.
You conclude that she is a patient with hard-to-control hypertension with no underlying cause who requires a significant amount of medication. That is not an uncommon situation. Although her blood pressure is now controlled to an appropriate level, you make a chart notation to consider referral to a hypertension specialist if her blood pressure become elevated again and treatment changes are not effective.6
ALGORITHM
Hard-to-control hypertension? This treatment algorithm can help
ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker; CPAP, continuous positive airway pressure; CT, computed tomography; GFR, glomerular filtration rate; TSH, thyroid-stimulating hormone.
Adapted from: Calhoun DA, et al. Hypertension. 2008.6
Another consideration: Pseudohypertension
This uncommon condition sometimes occurs in elderly patients who suffer from a stiffened or thickened brachial artery.1 Because the blood pressure cuff cannot compress the artery, blood pressure readings overestimate true arterial pressure. The condition should be considered in patients when increased doses of medication fail to achieve the desired response, particularly in patients who exhibit postural hypotension.1
This was not a likely explanation for Ms. Stevens, however, given the clinical scenario and her young age.
CORRESPONDENCE Randy Wexler, MD, MPH, B0902B Cramblett Hall, 456 W 10th Avenue, Columbus, OH 43210; [email protected]
• Encourage home BP monitoring. Home readings are often lower than those taken in the office and closer to the average BP recorded by 24-hour ambulatory monitors. C
• Tell patients that reducing sodium intake not only reduces mortality, but it has positive cardiovascular effects separate from BP reduction, such as improved endothelium-dependent vasodilation. A
• Search for secondary causes of resistant hypertension, such as renal artery stenosis, pheochromocytoma, obstructive sleep apnea, and hyperaldosteronism. A
• Consider pseudohypertension in elderly patients who exhibit postural hypotension and fail to respond to increased doses of medication. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 Mr. Brown is a 60-year-old African American man who works as a mid-level executive at a local bank. About a year and a half ago, he was diagnosed with hypertension, joining a number of other family members who also have the condition. Reviewing his chart, you note that at many of his visits—but not all of them—his systolic blood pressure runs close to 150 mm Hg, with diastolic pressure <90 mm Hg. Today his blood pressure is 148/88 mm Hg, numbers that exceed the parameters of the currently accepted definition of hypertension: blood pressure ≥140 mm Hg systolic or ≥90 mm Hg diastolic, taken on 2 separate occasions with the patient sitting down.1
Mr. Brown is one of the more than 65 million American adults suffering from high blood pressure—the No. 1 diagnosis reported in outpatient medical offices.2,3 Despite such prevalence, blood pressure control in the United States is suboptimal, with only one-third of hypertensive patients under adequate control.1 In 2007, the total estimated cost of treating high blood pressure in the United States exceeded $66 billion.4
When you tell Mr. Brown that his blood pressure doesn’t meet the therapeutic goal of <140/90 mm Hg, he gives you his reasons: He was late, he rushed, the traffic was bad, and the nurse rushed him into the exam room before he even had time to catch his breath. He insists his blood pressure is “normal” at home, and blames the elevated numbers on anxiety. He also tells you he was up working most of the night before, drinking coffee to stay awake to finish an urgent project.
Mr. Brown’s current medication regimen includes daily doses of lisinopril-hydrochlorothiazide (HCTZ) 20/25 mg daily and simvastatin 20 mg. He tells you he has no chest pain, shortness of breath, cough, edema, claudication, paroxysmal nocturnal dyspnea, or orthopnea. When you ask if he takes his lisinopril-HCTZ every day, he says Yes, but you have your suspicions.
Consider pseudo-resistance
Suboptimal blood pressure control can be classified as either pseudo-resistant or resistant hypertension. According to the definition used in the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC7), resistance is the “failure to achieve goal BP in patients who are adhering to full doses of an appropriate 3-drug regimen that includes a diuretic.”1
Hypertension is described as “pseudo-resistant” when persistent elevations in blood pressure are the result of a failure to comply with the medication regimen, “white-coat” syndrome, poor blood pressure technique, or a combination of these.5,6 Physician failure to prescribe adequate doses of medication, so-called clinical inertia, may also be a factor.5,7 Causes and remedies for pseudo-resistance are summarized in the algorithm.
In Mr. Brown’s case, he’s told you that he’s taking his lisinopril-HCTZ every day, but when you check your records, you see that the intervals between his refill requests range between 34 and 36 days. So you ask him, again, whether he is taking his lisinopril-HCTZ daily, and this time he says he takes the medication “at least 5 or 6 days a week.”
Encourage compliance. To motivate Mr. Brown, you tell him his blood pressure does not meet the goal of <140/90 mm Hg and that pressure higher than goal is a significant risk factor for cardiovascular disease. You emphasize the importance of taking his medication every day—a mantra you’ve repeated to countless patients over the years. In fact, it’s estimated that up to 40% of patients will discontinue their medication at some point during treatment.8 Patients’ reasons vary and may include medication side effects, the cost of treatment, or a patient’s personal philosophy. Cultural differences may also play a role in noncompliance.9
You talk with Mr. Brown a bit more and confirm that his poor compliance is due to simple forgetfulness and not another underlying reason that would need to be addressed. You hand him a pill calendar to help him keep track of his medication. You review the dosage and feel confident that you’ve prescribed a regimen simple enough for Mr. Brown to stick to and adequate to bring his pressure down.
When your white coat is the problem. Though not the case with Mr. Brown, a patient’s elevated readings could be a case of white coat hypertension—a conditioned response in some patients that is probably the result of anxiety in the medical setting.10 Patients with white coat hypertension have significantly less risk of death and reduced target organ damage than patients with truly resistant hypertension.11,12
If you suspect that a patient of yours has white coat hypertension, you can test your hypothesis by encouraging him or her to buy a blood pressure monitor to use at home, keep a log of the readings, and bring the log in to the next appointment. Improved technology has made home blood pressure monitoring an important tool in the treatment of high blood pressure.13-15
According to a 2008 statement issued jointly by the American Heart Association, the American Society of Hypertension, and the Preventive Cardiovascular Nurses Association, “There is a rapidly growing literature showing that measurements taken by patients at home are often lower than readings taken in the office and closer to the average blood pressure recorded by 24-hour ambulatory monitors, which is the blood pressure that best predicts cardiovascular risk.”14
Arm monitors are the most accurate because they measure brachial artery blood pressure.14 Most wrist monitors have not been validated in studies, but obese patients who cannot find a properly sized cuff may need to use them.14 Finger devices should never be used.14
Sometimes technique is at fault. To ensure that you get proper readings when your patient is in the office, advise the nursing staff not to take a patient’s blood pressure for at least 5 minutes after bringing him or her to an exam room. Specifically, the nurse can either make blood pressure measurement the last thing she (or he) does when “rooming” the patient, or she may prefer to go back to the exam room a few minutes after the patient is there to take the reading. The nurse should take care that the cuff is the right size and that the pressure is taken with the patient’s arm at heart level. In hypertensive patients, placing the arm below the horizontal in the dependent position can raise blood pressure 23/10 mm Hg.1,16,17
Newer blood pressure monitoring devices for the office are available that obviate artificially high readings caused by errors in technique and by white coat syndrome. Researchers have found that readings taken with the BpTRU automatic device, which obtains and records 5 blood pressure readings over a 5-minute period, are lower than those taken by office personnel, and that the white coat effect is eliminated.18,19
One pseudo-resistant case solved. At Mr. Brown’s next visit, you review his pill calendar with him and note that his blood pressure is now running 138/88 mm Hg and he is within his target goal of less than 140/90 mm Hg.
If Mr. Brown’s blood pressure control had continued to be suboptimal even after you’d taken steps to address cause(s) of pseudo-resistance, you would have had to shift gears and consider his case one of resistant hypertension.1
Consider resistant hypertension
Causes of resistant hypertension include lifestyle factors, side effects of medications, and secondary causes.5,6 Tobacco use, obesity, lack of exercise, a high sodium diet, and alcohol consumption can all contribute to hypertension.1,5,6,20,21 Dietary sodium in particular has significant adverse effects. Reducing dietary sodium not only reduces mortality, but has positive cardiovascular effects separate from blood pressure reduction, such as improved endothelium-dependent vasodilation.22-24
Medications including nonsteroidal anti-inflammatory drugs (NSAIDs), oral contraceptives, sympathomimetics, glucocorticoids, and black licorice (found in some herbal supplements) may all raise blood pressure.5 Medications and other substances associated with increased blood pressure are listed in the TABLE. A search for such products, with changes or discontinuations that are clinically appropriate, should be part of any evaluation for resistant hypertension.
CASE 2 Ms. Stevens is a 30-year-old woman who was diagnosed with high blood pressure 7 months ago. She is currently taking triamterene-HCTZ 37.5/25 mg daily, amlodipine 10 mg daily, and metoprolol succinate 100 mg daily. She is compliant with medication, sticks to a low-sodium diet, and exercises 5 days a week. She does not drink alcohol. A review of her chart tells you her baseline serum chemistries, chest x-ray, and urinalysis are all normal. Today’s blood pressure taken in the sitting position after a 5-minute rest is 160/92 mm Hg. How should you proceed?
TABLE
Medications and other agents that may cause hypertension
| • Alcohol | • Cyclosporine | • Nicotine |
| • Antidepressants | • Erythropoietin | • NSAIDs |
| • Buspirone (Buspar) | • Estrogen preparations | • St. John’s wort |
| • Cocaine | • Licorice | • Tyramine-containing foods |
| • Corticosteroids | • Metoclopramide (Reglan) | |
| NSAIDs, nonsteroidal anti-infiammatory drugs. | ||
| Source: Chobanian A, et al. Hypertension. 2003.1 | ||
Is it resistant hypertension? Look for clues
You go over Ms. Stevens’ history carefully. Her hypertension medications should be adequate, and she is compliant with the prescribed regimen. Lifestyle factors are clearly not to blame: She’s not obese, she follows a low-sodium diet, doesn’t drink or smoke, and gets plenty of exercise. You ask her about other medications or over-the-counter supplements she takes, and nothing on her list raises a red flag. This doesn’t seem to be pseudo-resistance, and though you’re aware that only about 5% of cases of hypertension can be attributed to secondary causes, you think Ms. Stevens may be one of those patients.25
Numerous causes of secondary hypertension exist, and evaluations for such causes should be considered in all patients with resistant hypertension. Renal artery stenosis and pheochromocytoma are the most well-known causes, but less well known but significant contributors are obstructive sleep apnea and hyperaldosteronism.1,6,26,27
The relationship between obstructive sleep apnea and hypertension is not clear, and most of the studies linking the 2 conditions are population based.28 Nevertheless, because studies show that continuous positive airway pressure (CPAP) does improve blood pressure control in patients with obstructive sleep apnea, it is appropriate to investigate patients with resistant hypertension for this condition.29
Aldosterone’s role in resistant hypertension appears to be greatest in those with insulin resistance. The mechanism appears to be amplified sodium retention resulting in increased volume expansion, negative effects on oxidative stress, and elevated inflammation.27 Many patients with hypertension also have insulin resistance, so testing for hyperaldosteronism should be part of your investigation.
Test, and as necessary, test some more
Taking into consideration that 2 of the more common causes of secondary hypertension are obstructive sleep apnea and renal artery stenosis, you schedule an overnight sleep study for Ms. Stevens and a magnetic resonance arteriogram of the kidneys.26,30 (For more on the diagnostic tests and treatments for secondary causes of resistant hypertension, see the ALGORITHM.)
Reconsider medications. Ms. Stevens is already receiving the maximum dose of amlodipine, so you increase her metoprolol succinate to 200 mg daily and ask her to return in 1 month after the results of her tests come in.
Change medications and keep testing. At her next visit, Ms. Stevens’ blood pressure is 150/88 mm Hg, and her pulse rate is 64. Results of her overnight sleep study and magnetic resonance arteriogram are normal. At this juncture, with her work-up for common secondary causes of hypertension negative, you consider changes in her medication regimen. Because her pulse rate of 64 rules out any increase in the metoprolol, you decide to add lisinopril-HCTZ 10/12.5 mg to her regimen and discontinue her triamterene-HCTZ. Because her blood pressure is still elevated at this visit, you schedule additional testing to look for other secondary causes.
More tests, higher doses. One month later, Ms. Stevens returns with a blood pressure of 146/88 mm Hg. Appropriate testing for primary aldosteronism, pheochromocytoma, Cushing’s disease, and coarctation are all normal. Her lisinopril-HCTZ dose is increased to 20/12.5 mg, and when she returns 1 month later her blood pressure reading is 138/88 mm Hg.
You conclude that she is a patient with hard-to-control hypertension with no underlying cause who requires a significant amount of medication. That is not an uncommon situation. Although her blood pressure is now controlled to an appropriate level, you make a chart notation to consider referral to a hypertension specialist if her blood pressure become elevated again and treatment changes are not effective.6
ALGORITHM
Hard-to-control hypertension? This treatment algorithm can help
ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker; CPAP, continuous positive airway pressure; CT, computed tomography; GFR, glomerular filtration rate; TSH, thyroid-stimulating hormone.
Adapted from: Calhoun DA, et al. Hypertension. 2008.6
Another consideration: Pseudohypertension
This uncommon condition sometimes occurs in elderly patients who suffer from a stiffened or thickened brachial artery.1 Because the blood pressure cuff cannot compress the artery, blood pressure readings overestimate true arterial pressure. The condition should be considered in patients when increased doses of medication fail to achieve the desired response, particularly in patients who exhibit postural hypotension.1
This was not a likely explanation for Ms. Stevens, however, given the clinical scenario and her young age.
CORRESPONDENCE Randy Wexler, MD, MPH, B0902B Cramblett Hall, 456 W 10th Avenue, Columbus, OH 43210; [email protected]
1. Chobanian A, Bakris GL, Black HR, et al. Seventh Report of The Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC-7). Hypertension. 2003;42:1206-1252.
2. Fields L, Burt V, Cutler J, et al. The burden of adult hypertension in the United States 1999-2000: A rising tide. Hypertension. 2004;44:1-7.
3. Fang J, Alderman MH, Keenan NL, et al. Hypertension control at physicians’ offices in the United States. Am J Hypertens. 2008;21:136-142.
4. American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics–2007 update. Circulation. 2007;115:e69-e171.
5. Sarafidis PA, Bakris GL. Resistant hypertension: an overview of evaluation and treatment. J Am Coll Cardiol. 2008;52:1749-1757.
6. Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension. 2008;51:1403-1419.
7. Phillips LS, Branch WT, Book CB, et al. Clinical inertia. Ann Intern Med. 2001;135:825-834.
8. Van Wijk BL, Klungel OH, Heerdink ER, et al. Rate and determinants of 10-year persistence with antihypertensive drugs. J Hypertens. 2005;23:2101-2107.
9. Wexler R, Pleister A, Feldman D. Sociology meets genetics: sociogenetic implications for future management of hypertension and heart failure. Curr Treat Options Cardiovasc Med. 2009;11:305-315.
10. Ogedegbe G, Pickering TG, Clemow L, et al. The misdiagnosis of hypertension: the role of patient anxiety. Arch Intern Med. 2008;168:2459-2465.
11. Dawes MG, Bartlett G, Coats AJ, et al. Comparing the effects of white coat hypertension and sustained hypertension on mortality in a UK primary care setting. Ann Fam Med. 2008;6:390-396.
12. Pierdomenico SD, Lapenna D, Bucci A, et al. Cardiovascular outcome in treated hypertensive patients with responder, masked, false resistant, and true resistant hypertension. Am J Hypertens. 2005;18:1422-1428.
13. Verberk WJ, Kroon AA, Kessels AGH, et al. Home blood pressure monitoring: a systematic review. J Am Coll Cardiol. 2005;46:743-751.
14. Pickering TG, Miller NH, Ogedegbe G, et al. AHA/ASH/PCNA scientific statement. Call to action on use and reimbursement for home blood pressure monitoring. Hypertension. 2008;52:10-29.
15. O’Brien E. Ambulatory blood pressure measurement: the case for implementation in primary care. Hypertension. 2008;51:1435-1441.
16. O’Brien E, Asmar R, Beilin L, et al. On behalf of the European Society of Hypertension Working Group on Blood Pressure Monitoring. European Society of Hypertension recommendations for conventional ambulatory and home blood pressure measurements. J Hypertens. 2003;21:821-848.
17. Mourad A, Carney S, Gillies A, et al. Arm position and blood pressure: a risk factor for hypertension. J Hum Hypertens. 2003;17:389-395.
18. Myers MG. Automated blood pressure measurement in routine clinical practice. Blood Press Monit. 2006;11:59-62.
19. Myers MG, Valdivieso M, Kiss A. Use of automated office blood pressure measurement to reduce white coat hypertension. J Hypertens. 2009;27:280-286.
20. He J, Paul J, Whelton P, Appel L, et al. Long-term effects of weight loss and dietary sodium reduction on incidence of hypertension. Hypertension. 2000;35:544-549.
21. PREMIER Collaborative Research Writing Group. Effects of comprehensive lifestyle modification on blood pressure control. JAMA. 2003;289:2083-2093.
22. Cook NR, Cutler JA, Obarzanek E, et al. Long term effects of dietary sodium reduction on cardiovascular disease outcomes: observational follow-up of the trials of hypertension prevention (TOHP). BMJ. 2007;334:885-888.
23. Dickinson BD, Havas S. Reducing the population burden of cardiovascular disease by reducing sodium intake. Arch Intern Med. 2007;167:1460-1468.
24. Li J, White J, Guo L, et al. Salt inactivates endothelial nitric oxide synthase in endothelial cells. J Nutr. 2009;139:1-5.
25. Beevers G, Lip G, O’Brien E. ABCs of hypertension: the pathophysiology of hypertension. BMJ. 2001;322:912-916.
26. Peppard PE, Young T, Palta M, et al. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342:1378-1384.
27. Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150:776-783.
28. Young T, Peppard P, Palta M, et al. Population-based study of sleep-disordered breathing as a risk factor for hypertension. Arch Intern Med. 1997;157:1746-1752.
29. Haentjens P, Van Meerhaeghe A, Moscariello A. The impact of continuous positive airway pressure on blood pressure in patients with obstructive sleep apnea syndrome: evidence from a meta-analysis of placebo-controlled randomized trials. Arch Intern Med. 2007;167:757-764.
30. Kawashima A, Francis IR, Baumgarten DA, et al. For the Expert Panel on Urologic Imaging. Renovascular hypertension. Reston, Va: American College of Radiology; 2007. Available at: www.guideline.gov/summary/summary.aspx?ss=15&doc_id=11590&nbr=6003. Accessed April 4, 2009.
1. Chobanian A, Bakris GL, Black HR, et al. Seventh Report of The Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC-7). Hypertension. 2003;42:1206-1252.
2. Fields L, Burt V, Cutler J, et al. The burden of adult hypertension in the United States 1999-2000: A rising tide. Hypertension. 2004;44:1-7.
3. Fang J, Alderman MH, Keenan NL, et al. Hypertension control at physicians’ offices in the United States. Am J Hypertens. 2008;21:136-142.
4. American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics–2007 update. Circulation. 2007;115:e69-e171.
5. Sarafidis PA, Bakris GL. Resistant hypertension: an overview of evaluation and treatment. J Am Coll Cardiol. 2008;52:1749-1757.
6. Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension. 2008;51:1403-1419.
7. Phillips LS, Branch WT, Book CB, et al. Clinical inertia. Ann Intern Med. 2001;135:825-834.
8. Van Wijk BL, Klungel OH, Heerdink ER, et al. Rate and determinants of 10-year persistence with antihypertensive drugs. J Hypertens. 2005;23:2101-2107.
9. Wexler R, Pleister A, Feldman D. Sociology meets genetics: sociogenetic implications for future management of hypertension and heart failure. Curr Treat Options Cardiovasc Med. 2009;11:305-315.
10. Ogedegbe G, Pickering TG, Clemow L, et al. The misdiagnosis of hypertension: the role of patient anxiety. Arch Intern Med. 2008;168:2459-2465.
11. Dawes MG, Bartlett G, Coats AJ, et al. Comparing the effects of white coat hypertension and sustained hypertension on mortality in a UK primary care setting. Ann Fam Med. 2008;6:390-396.
12. Pierdomenico SD, Lapenna D, Bucci A, et al. Cardiovascular outcome in treated hypertensive patients with responder, masked, false resistant, and true resistant hypertension. Am J Hypertens. 2005;18:1422-1428.
13. Verberk WJ, Kroon AA, Kessels AGH, et al. Home blood pressure monitoring: a systematic review. J Am Coll Cardiol. 2005;46:743-751.
14. Pickering TG, Miller NH, Ogedegbe G, et al. AHA/ASH/PCNA scientific statement. Call to action on use and reimbursement for home blood pressure monitoring. Hypertension. 2008;52:10-29.
15. O’Brien E. Ambulatory blood pressure measurement: the case for implementation in primary care. Hypertension. 2008;51:1435-1441.
16. O’Brien E, Asmar R, Beilin L, et al. On behalf of the European Society of Hypertension Working Group on Blood Pressure Monitoring. European Society of Hypertension recommendations for conventional ambulatory and home blood pressure measurements. J Hypertens. 2003;21:821-848.
17. Mourad A, Carney S, Gillies A, et al. Arm position and blood pressure: a risk factor for hypertension. J Hum Hypertens. 2003;17:389-395.
18. Myers MG. Automated blood pressure measurement in routine clinical practice. Blood Press Monit. 2006;11:59-62.
19. Myers MG, Valdivieso M, Kiss A. Use of automated office blood pressure measurement to reduce white coat hypertension. J Hypertens. 2009;27:280-286.
20. He J, Paul J, Whelton P, Appel L, et al. Long-term effects of weight loss and dietary sodium reduction on incidence of hypertension. Hypertension. 2000;35:544-549.
21. PREMIER Collaborative Research Writing Group. Effects of comprehensive lifestyle modification on blood pressure control. JAMA. 2003;289:2083-2093.
22. Cook NR, Cutler JA, Obarzanek E, et al. Long term effects of dietary sodium reduction on cardiovascular disease outcomes: observational follow-up of the trials of hypertension prevention (TOHP). BMJ. 2007;334:885-888.
23. Dickinson BD, Havas S. Reducing the population burden of cardiovascular disease by reducing sodium intake. Arch Intern Med. 2007;167:1460-1468.
24. Li J, White J, Guo L, et al. Salt inactivates endothelial nitric oxide synthase in endothelial cells. J Nutr. 2009;139:1-5.
25. Beevers G, Lip G, O’Brien E. ABCs of hypertension: the pathophysiology of hypertension. BMJ. 2001;322:912-916.
26. Peppard PE, Young T, Palta M, et al. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342:1378-1384.
27. Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150:776-783.
28. Young T, Peppard P, Palta M, et al. Population-based study of sleep-disordered breathing as a risk factor for hypertension. Arch Intern Med. 1997;157:1746-1752.
29. Haentjens P, Van Meerhaeghe A, Moscariello A. The impact of continuous positive airway pressure on blood pressure in patients with obstructive sleep apnea syndrome: evidence from a meta-analysis of placebo-controlled randomized trials. Arch Intern Med. 2007;167:757-764.
30. Kawashima A, Francis IR, Baumgarten DA, et al. For the Expert Panel on Urologic Imaging. Renovascular hypertension. Reston, Va: American College of Radiology; 2007. Available at: www.guideline.gov/summary/summary.aspx?ss=15&doc_id=11590&nbr=6003. Accessed April 4, 2009.
Professional obligations when patients pay out of pocket
During a long-overdue routine check-up with a 65-year-old Caucasian patient we’ll call Dan, you discover his blood pressure is 150/90—this, despite the prescription you wrote for him nearly a year ago. When you recommend that he increase the daily dose and suggest he may want to try a newer drug, he tells you he never filled the original prescription. Soon it’s apparent that he and his wife, who has diabetes, hypercholesterolemia, and severe arthritis, have been deciding which medicines to take and how often to schedule visits based on their monthly budget.
This is one of countless common scenarios playing out as a result of consumer-directed health care (CDHC), the latest movement in the constant struggle to control the costs and improve the quality of health care. CDHC uses mechanisms like steeply tiered co-payments, high deductibles coupled with health savings accounts, and reduced coverage (such as the infamous “donut hole” in Medicare Part D) to compel patients to spend their own money, not insurance money. This approach is intended to give patients more “skin in the game” so that they look harder for thriftier options and accept only treatments they really think are worth the money.1,2
CDHC may be wise; it may be foolish. But many patients must use it, and they urgently need their doctors’ help. This asks a lot of you, but the medical profession has a long and honorable tradition of pursuing what is best for patients, not just what is most cost-effective.3 Patients’ modest understanding of health insurance, providers, and medicine rarely equips them to make medically and financially prudent decisions. For sound information and sage guidance, your patients must rely on you, their physician. In other words, under CDHC, patients present not just with medical symptoms and a social history, but also with a financial condition.
So what kinds of practical and ethical problems does this create? Official oracles of medical ethics are virtually silent, yet patients have paid their own costs from time immemorial, and much can be learned from the collective wisdom of doctors’ accumulated experience.4
With this in mind, we interviewed a convenience sample of 7 primary care physicians in North Carolina who treat lower-income patients (3 in family practice, 2 in geriatrics, 1 in internal medicine, and 1 in pediatrics), and we reviewed the relevant professional ethics literature.5-10 From this body of practical and professional knowledge, we synthesized the following principles and strategies.
Talking about money is fraught with difficulty
If you are to help patients in the new consumerist health care world, you need to know to what extent money is an issue for a patient. But both doctors and patients often dislike discussing money.11,12 In 1 study, women were more uncomfortable talking about their income than their abortion.13 Many patients hesitate to broach the topic for fear of offending their doctor, who recommends services and may be selling them.11,14 And many doctors fear that mentioning costs during examination and treatment will alienate patients who take offense at, or misunderstand the motives behind, discussions of money.11,15
Medical anthropologist Howard Stein even suggests there is a “taboo in official American health culture: namely, a prohibition upon allowing the physician to appear concerned with financial matters” because introducing money violates “the sacred by the profane.”16 Nevertheless, the purpose of CDHC is precisely to place cost in the front of patients’ minds. So patients may be grateful for help in acknowledging the elephant in the room.
Approach finances as forthrightly as you would a potentially embarrassing clinical problem. You can work to help patients feel comfortable discussing costs by treating financial issues in the same matter-of-fact way you address sexual concerns.17 One doctor we interviewed at a low-income clinic said that his patients may be ashamed or embarrassed to acknowledge their financial problems. So he normalizes cost concerns by routinely asking patients if insurance coverage will be an issue—gracefully putting the discussion more in terms of third party rules than the patient’s ability to pay.
Other doctors we interviewed recommend watching for clues patients give when they are concerned about costs, just as doctors attend to patients’ clues about clinical problems.18 Patients may, for example, delay scheduling visits or neglect to fill prescriptions. As a fine doctor said a century ago, “Just remember that people generally care little how you collect your facts. They want to help you to help them, and are ready to accept your methods, especially if tactfully applied.”19
Patients often feel relieved to address cost problems, but finding out exactly what financial obligations a patient faces can be challenging. Different health plans allocate costs between patient and insurer in dismayingly different ways. Furthermore, those allocations fluctuate depending on each policy’s annual cycle of deductibles and out-of-pocket limits, which in turn depend on each patient’s renewal date. Patients are better situated than doctors to know the particulars of their own plans, but most people find their insurance baffling. Advances in information systems someday will prune this thicket, but today insurance coverage must often be added to the list of concerns about which doctors need better information.
Share your knowledge of treatment costs. If patients who pay out of pocket really are to make wise economic and medical choices, they need to know what tests and treatments cost.20 This information, too, may be elusive. For instance, physicians often are unfamiliar with, or mistaken about, the plethora of drug prices. One literature review reports, “With…the median [physician’s] estimate 243% away from the true cost, many of the estimates appear to be wild guesses.”21 And hospital charge masters are impossible to master. They can list more than 40,000 items whose prices are negotiated by insurance companies in a tumultuous market that regards prices as trade secrets.22
Precision may be unachievable, but there is room at least for a better understanding of large-magnitude cost differences. For instance, physicians we interviewed said that their computers or handheld devices provide basic information about the costs of prescription drugs, and some states and leading insurers are starting to post comparative provider and procedure prices online.23 Without these aids, doctors still appear very able and practiced in discussing the costs of different options in general qualitative terms, even if they lack exact price information.
How to factor cost into your discussions of treatment
The law of malpractice enforces the medical profession’s minimum standards for treatments, and the culture of medicine expects doctors to provide the best care available—to apply the gold standard of treatment. Patients (and perhaps juries) share that preference. But CDHC gives patients reasons to seek something less than the gold standard.
So once approximate costs are known, how should you factor them into discussions about treatments? When care is needed, do you merely inform the patient of less expensive options but always recommend the optimal one? When might you press a more effective option on a reluctant patient? Once again, these questions raise dilemmas doctors know all too well.24,25 You face them every day when patients assert other reasons to refuse treatment, like discomfort or inconvenience, or when their reticence amounts to little more than caprice. Here are 3 situations to consider:
When some treatment is better than none at all. The easiest situation arises when a more expensive option would be superior in an ideal world, but not in the real world. Sometimes, the best can be the enemy of the good. For example, if a patient who is offered only the medically optimal treatment leans toward forgoing treatment altogether, doctors often recommend a suboptimal but still useful alternative.26 Based on examples we heard, a physician might order a generic medication to control blood pressure when much costlier options are only moderately more effective, or an x-ray rather than a computed tomography scan, or 1 return visit rather than 2.
But what about malpractice liability for suboptimal care? Within reasonable ranges of professional judgment, the liability threat is not serious, since there are 2 legal defenses:27
- If a less expensive treatment, or no treatment at all, is within the broad prevailing standard of care or a recognized alternative school of thought, then doctors may recommend this, even if it is not the course they normally counsel.
- Even substandard options are defensible if reasonably well-informed patients understand their options and reject the doctor’s first recommendation.
When medical consequences of refusing a treatment are not dire. A second situation is also comparatively easy, at least in theory. Where the long-term medical outcomes are not dire and patients experience the health consequences directly, patients can reasonably be left to make suboptimal choices. Examples we were given include physical therapy or pain control. When a patient is considering direct-impact, lower-stakes treatments, a doctor should not feel great ethical or liability qualms in acceding to the patient’s wish to sacrifice health for wealth.
When a patient’s decision and your opinion are at odds. In the third category, physicians’ role as healers conflicts with their role as patients’ agents.28,29 If you suspect that a cost-reluctant patient can afford the gold standard and the patient chooses the pyrite standard, what should—or may—you do? This, too, is a variant of a familiar problem: Even well-informed patients may make bad decisions. To cope, doctors have developed an array of techniques (from soft to firm) that can be applied when decisions seem “penny wise and pound foolish.”30
First, and most coercively, doctors can simply refuse to treat a noncompliant patient. Except in emergencies, this is professionally and legally permissible; however, it is hardly ideal. When patients flatly cannot afford decent care, doctors often help by discounting fees or by arranging financial assistance.31
When patients are simply penurious rather than penniless, doctors can try arguing a patient into a wise choice. This tactic is not necessarily impermissible paternalism; it can be an act of respect and friendship. In our interviews, for instance, 1 doctor told a woman who balked at a mammogram that he was scheduling it anyway. Another called a taxi to drive a patient directly to the hospital out of concern that she might just go home. Yet another doctor enlisted family members in convincing recalcitrant patients. In sum, doctors dance a delicate dance to accommodate patients’ ambivalent wants and ambiguous needs.
Finding a new balance
You can accommodate the theory and policy of CDHC by acceding to a patient’s desire to pay less and get less.32 Professional obligations can be met by recommending the same care to each patient with a given condition, but informing patients of the costs and consequences of alternatives. Properly documented, these economically impartial conversations should protect physicians from malpractice liability. However, you need not go as far as having patients sign “Against Medical Advice” forms in order to continue seeing those who refuse optimal care. Doctors we interviewed thought it would be excessive to do this routinely and would threaten good relationships with their patients.
Unavoidably, solving consumerism’s problems will require conversations between you and your patients that take time—time that is already maddeningly limited. “Current practice guidelines for only 10 chronic illnesses require more time than primary care physicians have available for patient care overall.”33 For preventive care alone, providing all recommended services “to a panel of 2500 patients could require up to 7½ hours a day of physician time.”34
Furthermore, some doctors may feel that expecting patients to pay more out of pocket is an unwise policy. That may be right; even well-intentioned social reforms sometimes make ill-conceived demands of professionals. But rightly or wrongly, our political economy, having resisted managed care (at the urging of doctors and patients), has accepted consumerism as another means to restrain unsustainable spending. In public policy forums, doctors may argue against government or market initiatives, but in clinical forums, there is a professional obligation to cooperate with prevailing social policy—especially when the policy forges the interests that patients bring to the examination room.
Acknowledgments
This work was supported by a Robert Wood Johnson Foundation Investigator Award in Health Policy Research. The views expressed imply no endorsement by that foundation.
CORRESPONDENCE Mark A. Hall, JD, Wake Forest University, Division of Public Health Sciences, Medical Center Blvd., Winston-Salem, NC 27157-1063; [email protected]
1. Robinson JC. Reinvention of health insurance in the consumer era. JAMA. 2004;291:1880-1886.
2. Bloche MG. Consumer-directed health care. N Engl J Med. 2006;355:1756-1759.
3. Shrank WH, Asch SM, Joseph GJ, et al. Physicians’ perceived knowledge of and responsibility for managing patients’ out-of-pocket costs for prescription drugs. Ann Pharmacother. 2006;40:1534-1540.
4. Hall MA, Schneider CE. Learning from the legal history of billing for medical fees. J Gen Intern Med. 2008;23:257-260.
5. Cassel CK. Doctors and allocation decisions: a new role in the new Medicare. J Health Polit Policy Law. 1985;10:549-564.
6. Weiner S. “I can’t afford that!” Dilemmas in the care of the uninsured and underinsured. J Gen Intern Med. 2001;16:412-418.
7. Hurst SA, Hull SC, DuVal G, et al. Physicians’ responses to resource constraints. Arch Intern Med. 2005;165:639-644.
8. Hurst SA, Slowther AM, Forde R, et al. Prevalence and determinants of physician bedside rationing: data from Europe. J Gen Intern Med. 2006;21:1138-1143.
9. Whitney SN, McCullough LB. Physicians’ silent decisions: because patient autonomy does not always come first. Am J Bioeth. 2007;7:33-38.
10. Mort E, Edwards JN, Emmons D, et al. Physician response to patient insurance status in ambulatory care clinical decision-making. Med Care. 1996;34:783-797.
11. Schneider CE, Hall MA. The patient life: can consumers direct health care? Am J Law Med. 2009;35:7-65.
12. Alexander GC, Casalino LP, Meltzer DO. Patient-physician communication about out-of-pocket costs. JAMA. 2003;290:953-958.
13. Becker-Blease KA, Freyd JJ. Research participants telling the truth about their lives: the ethics of asking and not asking about abuse. Am Psychol. 2006;61:218-226.
14. Alexander GC, Casalino LP, Tseng CW, et al. Barriers to patient-physician communication about out-of-pocket costs. J Gen Intern Med. 2004;19:856-860.
15. Federman AD. Don’t ask, don’t tell: the status of doctor-patient communication about healthcare costs. Arch Intern Med. 2004;164:1723-1724.
16. Stein HF. The money taboo in American medicine. Med Anthropol. 1983;7:1-11.
17. Hardee JT, Platt FW, Kasper IK. Discussing health care costs with patients: an opportunity for empathic communication. J Gen Intern Med. 2005;20:666-669.
18. Alexander GC, Tseng CW. Six strategies to identify and assist patients burdened by out-of-pocket prescription costs. Cleve Clin J Med. 2004;71:433-438.
19. Shattuck FC. The science and art of medicine in some of their aspects. Boston Med Surg J. 1907;157:63-67.
20. Wilkes MS, Schriger DL. Caution: the meter is running informing patients about health care costs. West J Med. 1996;165:74-79.
21. Allan GM, Lexchin J, Wiebe N. Physician awareness of drug cost: a systematic review. PLoS Med. 2007;4:1486-1496.
22. Hall MA, Schneider CE. Patients as consumers: courts, contracts, and the new medical marketplace. Mich Law Rev. 2008;106:643-689.
23. Austin DA, Gravelle JG. Does price transparency improve market efficiency? Implications of empirical evidence in other markets for the health sector. Congressional Res Service, April 29, 2008. Available at: www.fas.org/sgp/crs/misc/RL34101.pdf. Accessed October 9, 2009.
24. Meyers DS, Mishori R, McCann J, et al. Primary care physicians’ perceptions of the effect of insurance status on clinical decision making. Ann Fam Med. 2006;4:399-402.
25. Pham HH, Alexander GC, O’Malley AS. Physician consideration of patients’ out-of-pocket costs in making common clinical decisions. Arch Intern Med. 2007;167:663-668.
26. Reichert S, Simon T, Halm EA. Physicians’ attitudes about prescribing and knowledge of the costs of common medications. Arch Intern Med. 2000;160:2799-2803.
27. Hall MA, Schneider CE. Physician liability when patients refuse to pay for recommended care. Conn Law Rev. 2009;4:743-780.
28. Alexander GC, Hall MA, Lantos JD. Rethinking professional ethics in the cost-sharing era. Am J Bioeth. 2006;6:W17-W22.
29. Gafni A, Charles C, Whelan T. The physician-patient encounter: the physician as a perfect agent for the patient versus the informed decision-making model. Soc Sci Med. 1998;47:347-354.
30. Carrese JA. Refusal of care: patients’ well-being and physicians’ ethical obligations: “But doctor, I want to go home.” JAMA. 2006;296:691-695.
31. Hall MA, Schneider CE. The professional ethics of billing and collections. JAMA. 2008;300:1806-1808.
32. Hall MA. Paying for what you get, and getting what you pay for. Law Contemp Probl. 2006;69:159-180.
33. Ostbye T, Yarnall KS, Krause KM, et al. Is there time for management of patients with chronic diseases in primary care? Ann Fam Med. 2005;3:209-214.
34. Goodson JD. Unintended consequences of resource-based relative value scale reimbursement. JAMA. 2007;298:2308-2310.
During a long-overdue routine check-up with a 65-year-old Caucasian patient we’ll call Dan, you discover his blood pressure is 150/90—this, despite the prescription you wrote for him nearly a year ago. When you recommend that he increase the daily dose and suggest he may want to try a newer drug, he tells you he never filled the original prescription. Soon it’s apparent that he and his wife, who has diabetes, hypercholesterolemia, and severe arthritis, have been deciding which medicines to take and how often to schedule visits based on their monthly budget.
This is one of countless common scenarios playing out as a result of consumer-directed health care (CDHC), the latest movement in the constant struggle to control the costs and improve the quality of health care. CDHC uses mechanisms like steeply tiered co-payments, high deductibles coupled with health savings accounts, and reduced coverage (such as the infamous “donut hole” in Medicare Part D) to compel patients to spend their own money, not insurance money. This approach is intended to give patients more “skin in the game” so that they look harder for thriftier options and accept only treatments they really think are worth the money.1,2
CDHC may be wise; it may be foolish. But many patients must use it, and they urgently need their doctors’ help. This asks a lot of you, but the medical profession has a long and honorable tradition of pursuing what is best for patients, not just what is most cost-effective.3 Patients’ modest understanding of health insurance, providers, and medicine rarely equips them to make medically and financially prudent decisions. For sound information and sage guidance, your patients must rely on you, their physician. In other words, under CDHC, patients present not just with medical symptoms and a social history, but also with a financial condition.
So what kinds of practical and ethical problems does this create? Official oracles of medical ethics are virtually silent, yet patients have paid their own costs from time immemorial, and much can be learned from the collective wisdom of doctors’ accumulated experience.4
With this in mind, we interviewed a convenience sample of 7 primary care physicians in North Carolina who treat lower-income patients (3 in family practice, 2 in geriatrics, 1 in internal medicine, and 1 in pediatrics), and we reviewed the relevant professional ethics literature.5-10 From this body of practical and professional knowledge, we synthesized the following principles and strategies.
Talking about money is fraught with difficulty
If you are to help patients in the new consumerist health care world, you need to know to what extent money is an issue for a patient. But both doctors and patients often dislike discussing money.11,12 In 1 study, women were more uncomfortable talking about their income than their abortion.13 Many patients hesitate to broach the topic for fear of offending their doctor, who recommends services and may be selling them.11,14 And many doctors fear that mentioning costs during examination and treatment will alienate patients who take offense at, or misunderstand the motives behind, discussions of money.11,15
Medical anthropologist Howard Stein even suggests there is a “taboo in official American health culture: namely, a prohibition upon allowing the physician to appear concerned with financial matters” because introducing money violates “the sacred by the profane.”16 Nevertheless, the purpose of CDHC is precisely to place cost in the front of patients’ minds. So patients may be grateful for help in acknowledging the elephant in the room.
Approach finances as forthrightly as you would a potentially embarrassing clinical problem. You can work to help patients feel comfortable discussing costs by treating financial issues in the same matter-of-fact way you address sexual concerns.17 One doctor we interviewed at a low-income clinic said that his patients may be ashamed or embarrassed to acknowledge their financial problems. So he normalizes cost concerns by routinely asking patients if insurance coverage will be an issue—gracefully putting the discussion more in terms of third party rules than the patient’s ability to pay.
Other doctors we interviewed recommend watching for clues patients give when they are concerned about costs, just as doctors attend to patients’ clues about clinical problems.18 Patients may, for example, delay scheduling visits or neglect to fill prescriptions. As a fine doctor said a century ago, “Just remember that people generally care little how you collect your facts. They want to help you to help them, and are ready to accept your methods, especially if tactfully applied.”19
Patients often feel relieved to address cost problems, but finding out exactly what financial obligations a patient faces can be challenging. Different health plans allocate costs between patient and insurer in dismayingly different ways. Furthermore, those allocations fluctuate depending on each policy’s annual cycle of deductibles and out-of-pocket limits, which in turn depend on each patient’s renewal date. Patients are better situated than doctors to know the particulars of their own plans, but most people find their insurance baffling. Advances in information systems someday will prune this thicket, but today insurance coverage must often be added to the list of concerns about which doctors need better information.
Share your knowledge of treatment costs. If patients who pay out of pocket really are to make wise economic and medical choices, they need to know what tests and treatments cost.20 This information, too, may be elusive. For instance, physicians often are unfamiliar with, or mistaken about, the plethora of drug prices. One literature review reports, “With…the median [physician’s] estimate 243% away from the true cost, many of the estimates appear to be wild guesses.”21 And hospital charge masters are impossible to master. They can list more than 40,000 items whose prices are negotiated by insurance companies in a tumultuous market that regards prices as trade secrets.22
Precision may be unachievable, but there is room at least for a better understanding of large-magnitude cost differences. For instance, physicians we interviewed said that their computers or handheld devices provide basic information about the costs of prescription drugs, and some states and leading insurers are starting to post comparative provider and procedure prices online.23 Without these aids, doctors still appear very able and practiced in discussing the costs of different options in general qualitative terms, even if they lack exact price information.
How to factor cost into your discussions of treatment
The law of malpractice enforces the medical profession’s minimum standards for treatments, and the culture of medicine expects doctors to provide the best care available—to apply the gold standard of treatment. Patients (and perhaps juries) share that preference. But CDHC gives patients reasons to seek something less than the gold standard.
So once approximate costs are known, how should you factor them into discussions about treatments? When care is needed, do you merely inform the patient of less expensive options but always recommend the optimal one? When might you press a more effective option on a reluctant patient? Once again, these questions raise dilemmas doctors know all too well.24,25 You face them every day when patients assert other reasons to refuse treatment, like discomfort or inconvenience, or when their reticence amounts to little more than caprice. Here are 3 situations to consider:
When some treatment is better than none at all. The easiest situation arises when a more expensive option would be superior in an ideal world, but not in the real world. Sometimes, the best can be the enemy of the good. For example, if a patient who is offered only the medically optimal treatment leans toward forgoing treatment altogether, doctors often recommend a suboptimal but still useful alternative.26 Based on examples we heard, a physician might order a generic medication to control blood pressure when much costlier options are only moderately more effective, or an x-ray rather than a computed tomography scan, or 1 return visit rather than 2.
But what about malpractice liability for suboptimal care? Within reasonable ranges of professional judgment, the liability threat is not serious, since there are 2 legal defenses:27
- If a less expensive treatment, or no treatment at all, is within the broad prevailing standard of care or a recognized alternative school of thought, then doctors may recommend this, even if it is not the course they normally counsel.
- Even substandard options are defensible if reasonably well-informed patients understand their options and reject the doctor’s first recommendation.
When medical consequences of refusing a treatment are not dire. A second situation is also comparatively easy, at least in theory. Where the long-term medical outcomes are not dire and patients experience the health consequences directly, patients can reasonably be left to make suboptimal choices. Examples we were given include physical therapy or pain control. When a patient is considering direct-impact, lower-stakes treatments, a doctor should not feel great ethical or liability qualms in acceding to the patient’s wish to sacrifice health for wealth.
When a patient’s decision and your opinion are at odds. In the third category, physicians’ role as healers conflicts with their role as patients’ agents.28,29 If you suspect that a cost-reluctant patient can afford the gold standard and the patient chooses the pyrite standard, what should—or may—you do? This, too, is a variant of a familiar problem: Even well-informed patients may make bad decisions. To cope, doctors have developed an array of techniques (from soft to firm) that can be applied when decisions seem “penny wise and pound foolish.”30
First, and most coercively, doctors can simply refuse to treat a noncompliant patient. Except in emergencies, this is professionally and legally permissible; however, it is hardly ideal. When patients flatly cannot afford decent care, doctors often help by discounting fees or by arranging financial assistance.31
When patients are simply penurious rather than penniless, doctors can try arguing a patient into a wise choice. This tactic is not necessarily impermissible paternalism; it can be an act of respect and friendship. In our interviews, for instance, 1 doctor told a woman who balked at a mammogram that he was scheduling it anyway. Another called a taxi to drive a patient directly to the hospital out of concern that she might just go home. Yet another doctor enlisted family members in convincing recalcitrant patients. In sum, doctors dance a delicate dance to accommodate patients’ ambivalent wants and ambiguous needs.
Finding a new balance
You can accommodate the theory and policy of CDHC by acceding to a patient’s desire to pay less and get less.32 Professional obligations can be met by recommending the same care to each patient with a given condition, but informing patients of the costs and consequences of alternatives. Properly documented, these economically impartial conversations should protect physicians from malpractice liability. However, you need not go as far as having patients sign “Against Medical Advice” forms in order to continue seeing those who refuse optimal care. Doctors we interviewed thought it would be excessive to do this routinely and would threaten good relationships with their patients.
Unavoidably, solving consumerism’s problems will require conversations between you and your patients that take time—time that is already maddeningly limited. “Current practice guidelines for only 10 chronic illnesses require more time than primary care physicians have available for patient care overall.”33 For preventive care alone, providing all recommended services “to a panel of 2500 patients could require up to 7½ hours a day of physician time.”34
Furthermore, some doctors may feel that expecting patients to pay more out of pocket is an unwise policy. That may be right; even well-intentioned social reforms sometimes make ill-conceived demands of professionals. But rightly or wrongly, our political economy, having resisted managed care (at the urging of doctors and patients), has accepted consumerism as another means to restrain unsustainable spending. In public policy forums, doctors may argue against government or market initiatives, but in clinical forums, there is a professional obligation to cooperate with prevailing social policy—especially when the policy forges the interests that patients bring to the examination room.
Acknowledgments
This work was supported by a Robert Wood Johnson Foundation Investigator Award in Health Policy Research. The views expressed imply no endorsement by that foundation.
CORRESPONDENCE Mark A. Hall, JD, Wake Forest University, Division of Public Health Sciences, Medical Center Blvd., Winston-Salem, NC 27157-1063; [email protected]
During a long-overdue routine check-up with a 65-year-old Caucasian patient we’ll call Dan, you discover his blood pressure is 150/90—this, despite the prescription you wrote for him nearly a year ago. When you recommend that he increase the daily dose and suggest he may want to try a newer drug, he tells you he never filled the original prescription. Soon it’s apparent that he and his wife, who has diabetes, hypercholesterolemia, and severe arthritis, have been deciding which medicines to take and how often to schedule visits based on their monthly budget.
This is one of countless common scenarios playing out as a result of consumer-directed health care (CDHC), the latest movement in the constant struggle to control the costs and improve the quality of health care. CDHC uses mechanisms like steeply tiered co-payments, high deductibles coupled with health savings accounts, and reduced coverage (such as the infamous “donut hole” in Medicare Part D) to compel patients to spend their own money, not insurance money. This approach is intended to give patients more “skin in the game” so that they look harder for thriftier options and accept only treatments they really think are worth the money.1,2
CDHC may be wise; it may be foolish. But many patients must use it, and they urgently need their doctors’ help. This asks a lot of you, but the medical profession has a long and honorable tradition of pursuing what is best for patients, not just what is most cost-effective.3 Patients’ modest understanding of health insurance, providers, and medicine rarely equips them to make medically and financially prudent decisions. For sound information and sage guidance, your patients must rely on you, their physician. In other words, under CDHC, patients present not just with medical symptoms and a social history, but also with a financial condition.
So what kinds of practical and ethical problems does this create? Official oracles of medical ethics are virtually silent, yet patients have paid their own costs from time immemorial, and much can be learned from the collective wisdom of doctors’ accumulated experience.4
With this in mind, we interviewed a convenience sample of 7 primary care physicians in North Carolina who treat lower-income patients (3 in family practice, 2 in geriatrics, 1 in internal medicine, and 1 in pediatrics), and we reviewed the relevant professional ethics literature.5-10 From this body of practical and professional knowledge, we synthesized the following principles and strategies.
Talking about money is fraught with difficulty
If you are to help patients in the new consumerist health care world, you need to know to what extent money is an issue for a patient. But both doctors and patients often dislike discussing money.11,12 In 1 study, women were more uncomfortable talking about their income than their abortion.13 Many patients hesitate to broach the topic for fear of offending their doctor, who recommends services and may be selling them.11,14 And many doctors fear that mentioning costs during examination and treatment will alienate patients who take offense at, or misunderstand the motives behind, discussions of money.11,15
Medical anthropologist Howard Stein even suggests there is a “taboo in official American health culture: namely, a prohibition upon allowing the physician to appear concerned with financial matters” because introducing money violates “the sacred by the profane.”16 Nevertheless, the purpose of CDHC is precisely to place cost in the front of patients’ minds. So patients may be grateful for help in acknowledging the elephant in the room.
Approach finances as forthrightly as you would a potentially embarrassing clinical problem. You can work to help patients feel comfortable discussing costs by treating financial issues in the same matter-of-fact way you address sexual concerns.17 One doctor we interviewed at a low-income clinic said that his patients may be ashamed or embarrassed to acknowledge their financial problems. So he normalizes cost concerns by routinely asking patients if insurance coverage will be an issue—gracefully putting the discussion more in terms of third party rules than the patient’s ability to pay.
Other doctors we interviewed recommend watching for clues patients give when they are concerned about costs, just as doctors attend to patients’ clues about clinical problems.18 Patients may, for example, delay scheduling visits or neglect to fill prescriptions. As a fine doctor said a century ago, “Just remember that people generally care little how you collect your facts. They want to help you to help them, and are ready to accept your methods, especially if tactfully applied.”19
Patients often feel relieved to address cost problems, but finding out exactly what financial obligations a patient faces can be challenging. Different health plans allocate costs between patient and insurer in dismayingly different ways. Furthermore, those allocations fluctuate depending on each policy’s annual cycle of deductibles and out-of-pocket limits, which in turn depend on each patient’s renewal date. Patients are better situated than doctors to know the particulars of their own plans, but most people find their insurance baffling. Advances in information systems someday will prune this thicket, but today insurance coverage must often be added to the list of concerns about which doctors need better information.
Share your knowledge of treatment costs. If patients who pay out of pocket really are to make wise economic and medical choices, they need to know what tests and treatments cost.20 This information, too, may be elusive. For instance, physicians often are unfamiliar with, or mistaken about, the plethora of drug prices. One literature review reports, “With…the median [physician’s] estimate 243% away from the true cost, many of the estimates appear to be wild guesses.”21 And hospital charge masters are impossible to master. They can list more than 40,000 items whose prices are negotiated by insurance companies in a tumultuous market that regards prices as trade secrets.22
Precision may be unachievable, but there is room at least for a better understanding of large-magnitude cost differences. For instance, physicians we interviewed said that their computers or handheld devices provide basic information about the costs of prescription drugs, and some states and leading insurers are starting to post comparative provider and procedure prices online.23 Without these aids, doctors still appear very able and practiced in discussing the costs of different options in general qualitative terms, even if they lack exact price information.
How to factor cost into your discussions of treatment
The law of malpractice enforces the medical profession’s minimum standards for treatments, and the culture of medicine expects doctors to provide the best care available—to apply the gold standard of treatment. Patients (and perhaps juries) share that preference. But CDHC gives patients reasons to seek something less than the gold standard.
So once approximate costs are known, how should you factor them into discussions about treatments? When care is needed, do you merely inform the patient of less expensive options but always recommend the optimal one? When might you press a more effective option on a reluctant patient? Once again, these questions raise dilemmas doctors know all too well.24,25 You face them every day when patients assert other reasons to refuse treatment, like discomfort or inconvenience, or when their reticence amounts to little more than caprice. Here are 3 situations to consider:
When some treatment is better than none at all. The easiest situation arises when a more expensive option would be superior in an ideal world, but not in the real world. Sometimes, the best can be the enemy of the good. For example, if a patient who is offered only the medically optimal treatment leans toward forgoing treatment altogether, doctors often recommend a suboptimal but still useful alternative.26 Based on examples we heard, a physician might order a generic medication to control blood pressure when much costlier options are only moderately more effective, or an x-ray rather than a computed tomography scan, or 1 return visit rather than 2.
But what about malpractice liability for suboptimal care? Within reasonable ranges of professional judgment, the liability threat is not serious, since there are 2 legal defenses:27
- If a less expensive treatment, or no treatment at all, is within the broad prevailing standard of care or a recognized alternative school of thought, then doctors may recommend this, even if it is not the course they normally counsel.
- Even substandard options are defensible if reasonably well-informed patients understand their options and reject the doctor’s first recommendation.
When medical consequences of refusing a treatment are not dire. A second situation is also comparatively easy, at least in theory. Where the long-term medical outcomes are not dire and patients experience the health consequences directly, patients can reasonably be left to make suboptimal choices. Examples we were given include physical therapy or pain control. When a patient is considering direct-impact, lower-stakes treatments, a doctor should not feel great ethical or liability qualms in acceding to the patient’s wish to sacrifice health for wealth.
When a patient’s decision and your opinion are at odds. In the third category, physicians’ role as healers conflicts with their role as patients’ agents.28,29 If you suspect that a cost-reluctant patient can afford the gold standard and the patient chooses the pyrite standard, what should—or may—you do? This, too, is a variant of a familiar problem: Even well-informed patients may make bad decisions. To cope, doctors have developed an array of techniques (from soft to firm) that can be applied when decisions seem “penny wise and pound foolish.”30
First, and most coercively, doctors can simply refuse to treat a noncompliant patient. Except in emergencies, this is professionally and legally permissible; however, it is hardly ideal. When patients flatly cannot afford decent care, doctors often help by discounting fees or by arranging financial assistance.31
When patients are simply penurious rather than penniless, doctors can try arguing a patient into a wise choice. This tactic is not necessarily impermissible paternalism; it can be an act of respect and friendship. In our interviews, for instance, 1 doctor told a woman who balked at a mammogram that he was scheduling it anyway. Another called a taxi to drive a patient directly to the hospital out of concern that she might just go home. Yet another doctor enlisted family members in convincing recalcitrant patients. In sum, doctors dance a delicate dance to accommodate patients’ ambivalent wants and ambiguous needs.
Finding a new balance
You can accommodate the theory and policy of CDHC by acceding to a patient’s desire to pay less and get less.32 Professional obligations can be met by recommending the same care to each patient with a given condition, but informing patients of the costs and consequences of alternatives. Properly documented, these economically impartial conversations should protect physicians from malpractice liability. However, you need not go as far as having patients sign “Against Medical Advice” forms in order to continue seeing those who refuse optimal care. Doctors we interviewed thought it would be excessive to do this routinely and would threaten good relationships with their patients.
Unavoidably, solving consumerism’s problems will require conversations between you and your patients that take time—time that is already maddeningly limited. “Current practice guidelines for only 10 chronic illnesses require more time than primary care physicians have available for patient care overall.”33 For preventive care alone, providing all recommended services “to a panel of 2500 patients could require up to 7½ hours a day of physician time.”34
Furthermore, some doctors may feel that expecting patients to pay more out of pocket is an unwise policy. That may be right; even well-intentioned social reforms sometimes make ill-conceived demands of professionals. But rightly or wrongly, our political economy, having resisted managed care (at the urging of doctors and patients), has accepted consumerism as another means to restrain unsustainable spending. In public policy forums, doctors may argue against government or market initiatives, but in clinical forums, there is a professional obligation to cooperate with prevailing social policy—especially when the policy forges the interests that patients bring to the examination room.
Acknowledgments
This work was supported by a Robert Wood Johnson Foundation Investigator Award in Health Policy Research. The views expressed imply no endorsement by that foundation.
CORRESPONDENCE Mark A. Hall, JD, Wake Forest University, Division of Public Health Sciences, Medical Center Blvd., Winston-Salem, NC 27157-1063; [email protected]
1. Robinson JC. Reinvention of health insurance in the consumer era. JAMA. 2004;291:1880-1886.
2. Bloche MG. Consumer-directed health care. N Engl J Med. 2006;355:1756-1759.
3. Shrank WH, Asch SM, Joseph GJ, et al. Physicians’ perceived knowledge of and responsibility for managing patients’ out-of-pocket costs for prescription drugs. Ann Pharmacother. 2006;40:1534-1540.
4. Hall MA, Schneider CE. Learning from the legal history of billing for medical fees. J Gen Intern Med. 2008;23:257-260.
5. Cassel CK. Doctors and allocation decisions: a new role in the new Medicare. J Health Polit Policy Law. 1985;10:549-564.
6. Weiner S. “I can’t afford that!” Dilemmas in the care of the uninsured and underinsured. J Gen Intern Med. 2001;16:412-418.
7. Hurst SA, Hull SC, DuVal G, et al. Physicians’ responses to resource constraints. Arch Intern Med. 2005;165:639-644.
8. Hurst SA, Slowther AM, Forde R, et al. Prevalence and determinants of physician bedside rationing: data from Europe. J Gen Intern Med. 2006;21:1138-1143.
9. Whitney SN, McCullough LB. Physicians’ silent decisions: because patient autonomy does not always come first. Am J Bioeth. 2007;7:33-38.
10. Mort E, Edwards JN, Emmons D, et al. Physician response to patient insurance status in ambulatory care clinical decision-making. Med Care. 1996;34:783-797.
11. Schneider CE, Hall MA. The patient life: can consumers direct health care? Am J Law Med. 2009;35:7-65.
12. Alexander GC, Casalino LP, Meltzer DO. Patient-physician communication about out-of-pocket costs. JAMA. 2003;290:953-958.
13. Becker-Blease KA, Freyd JJ. Research participants telling the truth about their lives: the ethics of asking and not asking about abuse. Am Psychol. 2006;61:218-226.
14. Alexander GC, Casalino LP, Tseng CW, et al. Barriers to patient-physician communication about out-of-pocket costs. J Gen Intern Med. 2004;19:856-860.
15. Federman AD. Don’t ask, don’t tell: the status of doctor-patient communication about healthcare costs. Arch Intern Med. 2004;164:1723-1724.
16. Stein HF. The money taboo in American medicine. Med Anthropol. 1983;7:1-11.
17. Hardee JT, Platt FW, Kasper IK. Discussing health care costs with patients: an opportunity for empathic communication. J Gen Intern Med. 2005;20:666-669.
18. Alexander GC, Tseng CW. Six strategies to identify and assist patients burdened by out-of-pocket prescription costs. Cleve Clin J Med. 2004;71:433-438.
19. Shattuck FC. The science and art of medicine in some of their aspects. Boston Med Surg J. 1907;157:63-67.
20. Wilkes MS, Schriger DL. Caution: the meter is running informing patients about health care costs. West J Med. 1996;165:74-79.
21. Allan GM, Lexchin J, Wiebe N. Physician awareness of drug cost: a systematic review. PLoS Med. 2007;4:1486-1496.
22. Hall MA, Schneider CE. Patients as consumers: courts, contracts, and the new medical marketplace. Mich Law Rev. 2008;106:643-689.
23. Austin DA, Gravelle JG. Does price transparency improve market efficiency? Implications of empirical evidence in other markets for the health sector. Congressional Res Service, April 29, 2008. Available at: www.fas.org/sgp/crs/misc/RL34101.pdf. Accessed October 9, 2009.
24. Meyers DS, Mishori R, McCann J, et al. Primary care physicians’ perceptions of the effect of insurance status on clinical decision making. Ann Fam Med. 2006;4:399-402.
25. Pham HH, Alexander GC, O’Malley AS. Physician consideration of patients’ out-of-pocket costs in making common clinical decisions. Arch Intern Med. 2007;167:663-668.
26. Reichert S, Simon T, Halm EA. Physicians’ attitudes about prescribing and knowledge of the costs of common medications. Arch Intern Med. 2000;160:2799-2803.
27. Hall MA, Schneider CE. Physician liability when patients refuse to pay for recommended care. Conn Law Rev. 2009;4:743-780.
28. Alexander GC, Hall MA, Lantos JD. Rethinking professional ethics in the cost-sharing era. Am J Bioeth. 2006;6:W17-W22.
29. Gafni A, Charles C, Whelan T. The physician-patient encounter: the physician as a perfect agent for the patient versus the informed decision-making model. Soc Sci Med. 1998;47:347-354.
30. Carrese JA. Refusal of care: patients’ well-being and physicians’ ethical obligations: “But doctor, I want to go home.” JAMA. 2006;296:691-695.
31. Hall MA, Schneider CE. The professional ethics of billing and collections. JAMA. 2008;300:1806-1808.
32. Hall MA. Paying for what you get, and getting what you pay for. Law Contemp Probl. 2006;69:159-180.
33. Ostbye T, Yarnall KS, Krause KM, et al. Is there time for management of patients with chronic diseases in primary care? Ann Fam Med. 2005;3:209-214.
34. Goodson JD. Unintended consequences of resource-based relative value scale reimbursement. JAMA. 2007;298:2308-2310.
1. Robinson JC. Reinvention of health insurance in the consumer era. JAMA. 2004;291:1880-1886.
2. Bloche MG. Consumer-directed health care. N Engl J Med. 2006;355:1756-1759.
3. Shrank WH, Asch SM, Joseph GJ, et al. Physicians’ perceived knowledge of and responsibility for managing patients’ out-of-pocket costs for prescription drugs. Ann Pharmacother. 2006;40:1534-1540.
4. Hall MA, Schneider CE. Learning from the legal history of billing for medical fees. J Gen Intern Med. 2008;23:257-260.
5. Cassel CK. Doctors and allocation decisions: a new role in the new Medicare. J Health Polit Policy Law. 1985;10:549-564.
6. Weiner S. “I can’t afford that!” Dilemmas in the care of the uninsured and underinsured. J Gen Intern Med. 2001;16:412-418.
7. Hurst SA, Hull SC, DuVal G, et al. Physicians’ responses to resource constraints. Arch Intern Med. 2005;165:639-644.
8. Hurst SA, Slowther AM, Forde R, et al. Prevalence and determinants of physician bedside rationing: data from Europe. J Gen Intern Med. 2006;21:1138-1143.
9. Whitney SN, McCullough LB. Physicians’ silent decisions: because patient autonomy does not always come first. Am J Bioeth. 2007;7:33-38.
10. Mort E, Edwards JN, Emmons D, et al. Physician response to patient insurance status in ambulatory care clinical decision-making. Med Care. 1996;34:783-797.
11. Schneider CE, Hall MA. The patient life: can consumers direct health care? Am J Law Med. 2009;35:7-65.
12. Alexander GC, Casalino LP, Meltzer DO. Patient-physician communication about out-of-pocket costs. JAMA. 2003;290:953-958.
13. Becker-Blease KA, Freyd JJ. Research participants telling the truth about their lives: the ethics of asking and not asking about abuse. Am Psychol. 2006;61:218-226.
14. Alexander GC, Casalino LP, Tseng CW, et al. Barriers to patient-physician communication about out-of-pocket costs. J Gen Intern Med. 2004;19:856-860.
15. Federman AD. Don’t ask, don’t tell: the status of doctor-patient communication about healthcare costs. Arch Intern Med. 2004;164:1723-1724.
16. Stein HF. The money taboo in American medicine. Med Anthropol. 1983;7:1-11.
17. Hardee JT, Platt FW, Kasper IK. Discussing health care costs with patients: an opportunity for empathic communication. J Gen Intern Med. 2005;20:666-669.
18. Alexander GC, Tseng CW. Six strategies to identify and assist patients burdened by out-of-pocket prescription costs. Cleve Clin J Med. 2004;71:433-438.
19. Shattuck FC. The science and art of medicine in some of their aspects. Boston Med Surg J. 1907;157:63-67.
20. Wilkes MS, Schriger DL. Caution: the meter is running informing patients about health care costs. West J Med. 1996;165:74-79.
21. Allan GM, Lexchin J, Wiebe N. Physician awareness of drug cost: a systematic review. PLoS Med. 2007;4:1486-1496.
22. Hall MA, Schneider CE. Patients as consumers: courts, contracts, and the new medical marketplace. Mich Law Rev. 2008;106:643-689.
23. Austin DA, Gravelle JG. Does price transparency improve market efficiency? Implications of empirical evidence in other markets for the health sector. Congressional Res Service, April 29, 2008. Available at: www.fas.org/sgp/crs/misc/RL34101.pdf. Accessed October 9, 2009.
24. Meyers DS, Mishori R, McCann J, et al. Primary care physicians’ perceptions of the effect of insurance status on clinical decision making. Ann Fam Med. 2006;4:399-402.
25. Pham HH, Alexander GC, O’Malley AS. Physician consideration of patients’ out-of-pocket costs in making common clinical decisions. Arch Intern Med. 2007;167:663-668.
26. Reichert S, Simon T, Halm EA. Physicians’ attitudes about prescribing and knowledge of the costs of common medications. Arch Intern Med. 2000;160:2799-2803.
27. Hall MA, Schneider CE. Physician liability when patients refuse to pay for recommended care. Conn Law Rev. 2009;4:743-780.
28. Alexander GC, Hall MA, Lantos JD. Rethinking professional ethics in the cost-sharing era. Am J Bioeth. 2006;6:W17-W22.
29. Gafni A, Charles C, Whelan T. The physician-patient encounter: the physician as a perfect agent for the patient versus the informed decision-making model. Soc Sci Med. 1998;47:347-354.
30. Carrese JA. Refusal of care: patients’ well-being and physicians’ ethical obligations: “But doctor, I want to go home.” JAMA. 2006;296:691-695.
31. Hall MA, Schneider CE. The professional ethics of billing and collections. JAMA. 2008;300:1806-1808.
32. Hall MA. Paying for what you get, and getting what you pay for. Law Contemp Probl. 2006;69:159-180.
33. Ostbye T, Yarnall KS, Krause KM, et al. Is there time for management of patients with chronic diseases in primary care? Ann Fam Med. 2005;3:209-214.
34. Goodson JD. Unintended consequences of resource-based relative value scale reimbursement. JAMA. 2007;298:2308-2310.
Early pregnancy loss needn’t require a trip to the hospital
• Family physicians can provide in-office treatment for patients with early pregnancy loss, as long as they are hemodynamically stable. A
• Manual vacuum aspiration in the office is as effective as surgical emptying of the uterus (dilatation and curettage) in the operating room. A
• No single in-office option for managing early pregnancy loss—wait and watch, misoprostol, or vacuum aspiration—is clearly more beneficial than another. Patients should be free to choose the method they prefer. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1: Janet C is 22 years old, excited about her first pregnancy and eager to do all the right things to have a healthy baby. But now, in her third month, she has started to bleed and has pelvic pain. She calls in a panic. You tell her to come in immediately. In the office, an ultrasound shows a residual gestational sac.
CASE 2: Lizbeth G, 40, is a successful professional, recently married, with a down-to-earth, decisive personality. She and her husband are eager to start a family. But now, in her second month, she calls to say she’s been having severe cramps and heavy bleeding. She knows she is having a miscarriage. When you see her in the examining room, she’s saddened but calm, eager to know what went wrong and what she needs to do now.
CASE 3: Lola M, 36, mother of 2, is in your office for a routine prenatal visit. She’s in her third month, expecting this pregnancy to be as uneventful as her previous ones. But your ultrasound exam reveals that her fetus has no heartbeat.
What would you tell each of these patients? What options would you offer them?
Chances are good that you’ve cared for any number of patients like Janet, Lizbeth, and Lola. Approximately 15% of clinically recognized pregnancies end in early pregnancy loss (EPL), defined as a miscarriage that occurs earlier than the 12th week of pregnancy. When clinically unrecognized miscarriages are included, the EPL rate may be as high as 30%.1 Most pregnancy losses (80%) occur during the first trimester.2
In the past, EPL was routinely considered an indication for uterine dilatation and curettage (D&C) performed in the operating room.3 This approach was effective, but had serious drawbacks: Costs were high and women had to undergo a surgical procedure that many would prefer to avoid.4
More recently, professional organizations such as the American Academy of Family Physicians and the United Kingdom’s Royal College of Obstetricians and Gynecologists have encouraged a wider range of treatment options that can be provided in an outpatient setting.5,6 These choices, which are available to women with confirmed intrauterine—not ectopic—pregnancy, include “watch and wait” (expectant management), medical management with misoprostol, and outpatient manual vacuum aspiration (MVA) of the uterus.
But before you can even discuss these options, it’s important to find out how your patient has been feeling about her pregnancy: Was it planned or unplanned? Is she happy or unhappy about being pregnant? Does she have a supportive partner, or is her relationship in turmoil? Having a clear sense of where she is emotionally will better enable you to counsel her on her options.
Know, too, that managing EPL patients in their family “medical home” has many advantages. Patients can remain with a caregiver they know and trust. Because they can choose the treatment option they prefer, they are more likely to be satisfied with their care.7 Their quality of life after treatment is better, and the emotional support they can receive in these familiar surroundings has been shown to decrease the psychological sequelae of a miscarriage.8-10
How best to define, and describe, what’s happened
Providing your patient with an accurate description of her situation is essential to adequately counseling her on treatment options. Types of EPL include:
Missed abortion, which occurs when a nonviable pregnancy is detected on ultrasound. The patient is usually without bleeding. A missed abortion is further distinguished sonographically as either an “anembryonic pregnancy”—a mean sac diameter of >10 mm and no yolk sac or a mean sac diameter of 20 mm and no embryo on transvaginal ultrasound—or as an “embryonic demise”—a crown rump length of ≥6 mm without cardiac activity on transvaginal ultrasound.5
Incomplete abortion occurs when a residual gestational sac is detected on ultrasound, and vaginal bleeding and pelvic pain are present.
Inevitable abortion occurs when the internal os is open, but the pregnancy has not yet passed.
Complete abortion occurs when no gestational sac is detected on ultrasound, the cervical os is generally closed, and significant cramping and bleeding have resolved.
Women experiencing a complete abortion require no treatment; they have already successfully passed the pregnancy. Women with a missed, incomplete, or inevitable abortion can be offered the choice of expectant management, medication, or uterine aspiration.
Does your patient want to wait it out?
The success rate for expectant management depends on the time-frame studied and the type of EPL.11 (Success in EPL is defined as complete uterine evacuation.) Patients who choose this approach are usually seen every 1 to 2 weeks so that you can evaluate symptoms and do a physical examination. In some cases, assessment also includes serial serum human chorionic gonadotropin (hCG) testing or ultrasonography.
Expectant management is usually more efficacious for women with an incomplete abortion than for women with anembryonic gestation or embryonic demise.12-16 TABLE W1, available at jfponline.com, provides a comparison of the efficacy of expectant management and misoprostol. In 1 observational study of 1096 women who chose expectant management, 91% of those with incomplete abortion were successful and 84% completed within 14 days of diagnosis. By comparison, only 59% of those with a missed abortion completed within 14 days.17
According to a study performed by Wieringa-de Waard and colleagues, increased bleeding appears to be the greatest predictor of completion. They showed that the median blood flow and pain were heaviest on the third day of vaginal bleeding, which then decreased steeply after 8 days to slight bleeding and spotting. Of the patients they followed, 50% completed during the first 8 days of bleeding.18
A Cochrane review of 5 studies comparing expectant management with vacuum aspiration found expectant management carried a higher risk of incomplete miscarriage, need for vacuum aspiration, and bleeding. In contrast, vacuum aspiration was associated with a significantly higher risk of infection.19
A low-cost option that can speed things up
EPL can be treated with prostaglandins to hasten the time to completion.20 Misoprostol is a synthetic prostaglandin E1 analog that causes contractions of the uterus and gastrointestinal tract. This medication is approved by the US Food and Drug Administration (FDA) only for the treatment of gastric ulcers, but it is commonly used off-label for labor induction, postpartum hemorrhage, and cervical ripening prior to gynecological procedures—as well as for the management of miscarriage.21 Misoprostol’s low cost and stability at room temperature make it easy to use.22
Route of administration. Although misoprostol is manufactured and approved for oral use only, administration by vaginal, buccal, or sublingual routes can increase the desired effect on the uterus, with the added benefit of decreased gastrointestinal side effects.23
The dosage and dosing intervals for misoprostol for treatment of EPL have not been well established. A comprehensive review article recommends a single dose of 800 mcg vaginal misoprostol or, alternatively, 600 mcg sublingual misoprostol for anembryonic pregnancy or embryonic/fetal demise.24 A single dose of 600 mcg oral or 400 mcg sublingual misoprostol is recommended for incomplete abortion.25 The vaginal route may not be feasible when bleeding is heavy.
Safety and efficacy. Multiple studies have found that misoprostol is a safe and acceptable alternative to vacuum aspiration or expectant management.11,26-29
A study comparing 652 women randomized to misoprostol vaginally or vacuum aspiration found that 84% of the misoprostol group had complete expulsion within 8 days of treatment initiation.30
Infection rates. The Miscarriage Treatment (MIST) trial randomized 1200 women with a diagnosis of embryonic demise or incomplete abortion at <13 weeks to medical (n=398), expectant (n=399), or vacuum aspiration management (n=403).31 Overall, the researchers found a low incidence of gynecologic infection (2.3%), and no evidence of difference in the infection rate attributable to the type of management selected.
Antibiotic use to reduce infection rates after misoprostol for EPL has not been studied. Nonetheless, a recent retrospective study examined infection rates after medical abortion with mifepristone and misoprostol.32 The study demonstrated a reduction in severe infection rates from 0.25 per 1000 abortions to 0.06 per 1000 (absolute reduction, 0.19 per 1000; 95% confidence interval [CI], 0.02-0.34; P=.03) with the routine use of doxycycline 100 mg PO twice daily for 7 days. The risk reduction is in comparison to the prior practice of either testing for sexually transmitted infection (STI) or using prophylactic doxycycline. The authors also reported a decrease in infection rate with a change from vaginal to buccal administration of misoprostol. The benefit of this change is unknown, because the practice of routine screening for STI or routine antibiotic provision was introduced at the same time.32
Follow-up. After misoprostol administration, follow-up includes confirmation of passage of the embryo or gestational sac by a combination of history, clinical examination, and serial hCG measurement or ultrasound. A completed abortion can be demonstrated by quantitative serum hCG showing a 50% drop between first and repeat test 48 to 72 hours after the passage of tissue.33 Follow-up 1 to 2 weeks after treatment is common practice, but can be scheduled sooner if the patient has not had bleeding and cramping. In that situation, you can give her the option of proceeding to uterine aspiration or trying a second dose of misoprostol (see doses given earlier), as long as she remains hemodynamically stable.24
Women who experience successful treatment with misoprostol like the method. In a multicenter, randomized clinical trial, 154 women with EPL confirmed by ultrasonography who had not passed the pregnancy after a week were randomly assigned to treatment with misoprostol (n=79) or curettage (n=75). In cases where misoprostol had caused complete evacuation, 76% of the women would opt for the same treatment, whereas only 38% of women who needed vacuum aspiration after unsuccessful misoprostol would do so (P<.01).34
A sample protocol for medical management of EPL is provided in the box.
Candidates
Women with ultrasound diagnosis of a nonviable pregnancy up to 10 weeks’ gestation. Nonviable pregnancy is diagnosed by ultrasound and subnormal, rising quantitative human chorionic gonadotropin (hCG) levels. Misoprostol treatment is not suitable in ectopic pregnancy, which must be excluded before treatment is begun.
Laboratory workup
Rh screen, hemoglobin, and quantitative serum hCG.
Procedure
Insert 800 mcg misoprostol in the vagina. (This can also be done by the patient at home.) If passage of tissue does not occur, the physician can give the patient a second dose of 800 mcg misoprostol. Anembryonic pregnancy or fetal demise can also be treated with 600 mcg given sublingually. Incomplete abortion is treated with a single dose of 600 mcg orally or 400 mcg sublingually.
Pain management
Provide a prescription for ibuprofen 800 mg and Tylenol #3 to the patient. Instruct her to take a tablet of ibuprofen at the time of misoprostol insertion and then every 6 hours as needed for pain. If pain is severe, she may take 1 to 2 tablets of Tylenol #3 every 3 to 4 hours as needed.
Instructions to patient
Tell the patient to call the office for “heavy bleeding,” defined as soaking 2 pads an hour for more than 2 hours. Tell the patient that there is no need to bring the expelled material for your inspection. Make sure she has your phone and pager numbers. If she needs to go to an emergency department or a hospital, tell her to request that you be called.
Follow-up
Schedule a follow-up visit 1 to 2 weeks after misoprostol insertion. A completed abortion can be demonstrated by quantitative serum hCG showing a 50% drop between first and repeat test 48 to 72 hours after the passage of tissue. Alternatively, a transvaginal ultrasound should show absence of a sac.
Of note: If one of these criteria is met, no further follow-up of serum hCG is warranted. Patients may elect manual vacuum aspiration at any time if the gestational sac and/or embryo have not passed.
Manual vacuum aspiration means less blood loss
A Cochrane review that compared vacuum aspiration with surgical D&C found that vacuum aspiration was associated with significantly less blood loss, pain, and time needed for the procedure.35 Traditionally, vacuum aspiration for EPL has occurred in the OR, using electrical suction and general anesthesia. Recently, a manual vacuum aspirator that allows women to have the procedure done in the outpatient setting has become available. It is used with analgesia given PO and a paracervical block.4,36
The manual vacuum aspirator (MVA) is a handheld syringe that works well in the ambulatory setting because it is small, quiet, portable, and inexpensive. The MVA is safe, provides the same degree of suction as an electric pump, and is as effective as electrical vacuum aspiration for the management of both spontaneous and induced abortion.37
As safe, as effective. A study by Goldberg and colleagues compared complication rates with MVA and electric suction in EPL of up to 10 weeks’ gestation.38 The researchers found no significant difference in perforation or need for re-aspiration. A comparison of the 2 methods in gestations of less than 6 weeks found a similar, small risk of failed abortion (<3%).39 A study of 1677 women treated with MVA as outpatients in a primary care practice had a complication rate of only 1.25%.40
Faster, cheaper. Blumenthal and Remsburg demonstrated that MVA in an outpatient setting decreases anesthesia requirements, hospital stay times, patient waiting times, and procedure times when compared with aspiration done in the OR. They showed a substantial saving, with the cost of uterine evacuation in the OR estimated at $1404 vs $827 per case when the aspiration was done as an outpatient procedure in the labor and delivery suite.41 The MVA syringe costs about $30 and is reusable after appropriate cleaning through sterilization or high-level disinfectant. The disposable plastic suction cannulas cost less than $3 each.
Pain management. A combination of an oral nonsteroidal anti-inflammatory (NSAID) medication and a paracervical block is a practical approach to managing the pain of this procedure. No published reports demonstrate that 1 type of local anesthetic is better than another, and many different techniques and combinations of medicines used for the paracervical block have been described.42
To minimize the effects of accidental blood vessel injection, the lowest anesthetic dose should be used, usually 10 to 20 mL of a 0.5% to 1% lidocaine or 0.25% bupivacaine solution. A common technique is to inject 8 to 10 cc of 1% lidocaine with epinephrine or vasopressin at 4 and 8 o’clock at the cervicovaginal reflection after careful aspiration to ensure the needle is not in a blood vessel.
Oral narcotics. Clinicians can also choose to manage pain with oral narcotics, benzodiazepines, or intravenous conscious sedation. Moderate cramping during and immediately after the procedure is common and can often be alleviated with verbal support.
For patients whose anxiety level is high, conscious sedation or general anesthesia may be the most appropriate choice. Your patient’s preference and your evaluation of her medical risk and emotional state together determine the most appropriate course.43 The technique for MVA is described inTABLE W2, available at jfponline.com.
Which approach is best for your patient?
Because all 3 approaches to managing EPL are effective and safe, family physicians can empower patients to make the choice themselves. Counseling about treatment options should include consideration of the patient’s support at home, availability of transportation in case of emergency, her desire to avoid surgery, and her need for a definitive resolution.
Counseling should also include information on the likely efficacy of each option, given the type of EPL the patient has experienced. For example, women who have had a missed abortion (embryonic demise or anembryonic gestation) are less likely to complete with expectant management than women with an incomplete abortion. Efficacy rates for different types of EPL are shown in TABLE W1, available at jfponline.com.
There’s time for your patient to change her mind
A woman may opt for 1 approach to start with, but choose a different option later. She may chose expectant management for a week, and then if the pregnancy has not passed on its own, decide that she wants to try misoprostol. If that fails, too, she may want a uterine aspiration procedure.
How did our 3 patients fare?
CASE 1: At first, Janet was content to wait and see whether her miscarriage would pass without further intervention. But when a week went by and nothing happened, she wanted to get it over with. She asked to try MVA, under conscious sedation. The procedure was successful. Now, a year later, she’s very happy to be pregnant again and confidently awaits a happy outcome.
CASE 2: By the time Lizbeth called, you suspected her abortion was complete. Your examination confirmed that diagnosis. She required no treatment, and a year later was ready to try again.
CASE 3: Lola was shocked when she learned her fetus had died in utero. But once she and her husband had taken in the sad news, they wanted to know what options were available. They talked it over and chose treatment with misoprostol. The miscarriage was completed 8 days later. They are content with their current family size and have decided not to try for another pregnancy.
CORRESPONDENCE Emily M. Godfrey, MD, MPH, UIC Department of Family Medicine, 1919 W. Taylor Ave., Room 145, M/C 663, Chicago, IL 60612; [email protected]
1. Wilcox AJ, Weinberg CR, O’Connor JF, et al. Incidence of early loss of pregnancy. N Engl J Med. 1988;319:189-194.
2. Tang OS, Ho PC. The use of misoprostol for early pregnancy failure. Curr Opin Obstet Gynecol. 2006;18:581-586.
3. Jurkovic D. Modern management of miscarriage: is there a place for non-surgical treatment? Ultrasound Obstet Gynecol. 1998;11:161-163.
4. Dalton VK, Harris L, Weisman CS, et al. Patient p, satisfaction, and resource use in office evacuation of early pregnancy failure. Obstet Gynecol. 2006;108:103-110.
5. Royal College of Obstetricians and Gynaecologists. The Management of Early Pregnancy Loss. London: Royal College of Obstetricians and Gynaecologists; 2006. Guideline No. 25.
6. Deutchman M. Advanced Life Support in Obstetrics Syllabus Update: First Trimester Pregnancy Complications. Leawood, Kan: American Academy of Family Physicians; 2007:1-2.
7. Wieringa-de Waard M, Bindels PJ, Vos J, et al. Patient p for expectant management vs. surgical evacuation in first-trimester uncomplicated miscarriage. J Clin Epidemiol. 2004;57:167-173.
8. Edwards S, Tureck R, Fredrick M, et al. Patient acceptability of manual versus electric vacuum aspiration for early pregnancy loss. J Womens Health (Larchmt). 2007;16:1429-1436.
9. Smith LF, Frost J, Levitas R, Bradley H, et al. Women’s experiences of three early miscarriage management options: a qualitative study. Br J Gen Pract. 2006;56:198-205.
10. Nikcevic AV, Kuczmierczyk AR, Nicolaides KH. The influence of medical and psychological interventions on women’s distress after miscarriage. J Psychosom Res. 2007;63:283-290.
11. Sotiriadis A, Makrydimas G, Papatheodorou S, et al. Expectant, medical, or surgical management of first-trimester miscarriage: a meta-analysis. Obstet Gynecol. 2005;105(5 Pt 1):1104-1113.
12. Bagratee JS, Khullar V, Regan L, et al. A randomized controlled trial comparing medical and expectant management of first trimester miscarriage. Hum Reprod. 2004;19:266-271.
13. Blohm F, Friden B, Platz-Christensen JJ, et al. Expectant management of first-trimester miscarriage in clinical practice. Acta Obstet Gynecol Scand. 2003;82:654-658.
14. Gronlund L, Gronlund AL, Clevin L, et al. Spontaneous abortion: expectant management, medical treatment or surgical evacuation. Acta Obstet Gynecol Scand. 2002;81:781-782.
15. Graziosi GC, Mol BW, Ankum WM, et al. Management of early pregnancy loss. Int J Gynaecol Obstet. 2004;86:337-346.
16. Wood SL, Brain PH. Medical management of missed abortion: a randomized clinical trial. Obstet Gynecol. 2002;99:563-566.
17. Luise C, Jermy K, May C, et al. Outcome of expectant management of spontaneous first trimester miscarriage: observational study [see comment]. BMJ. 2002;324:873-875.
18. Wieringa-de Waard M, Ankum WM, Bonsel GJ, et al. The natural course of spontaneous miscarriage: analysis of signs and symptoms in 188 expectantly managed women. Br J Gen Pract. 2003;53:704-708.
19. Nanda K, Peloggia A, Grimes D, et al. Expectant care versus surgical treatment for miscarriage. Cochrane Database Syst Rev. 2006(2):CD003518.-
20. Neilson JP, Hickey M, Vazquez J. Medical treatment for early fetal death (less than 24 weeks). Cochrane Database Syst Rev. 2006;(3):CD002253.-
21. Weeks A, Faundes A. Misoprostol in obstetrics and gynecology. Int J Gynaecol Obstet. 2007;99(suppl 2):S156-S159.
22. World Health Organization. Medical Methods for Termination of Pregnancy. Geneva: WHO; 1997.
23. Tang OS, Gemzell-Danielsson K, Ho PC. Misoprostol: pharmacokinetic profiles, effects on the uterus and side-effects. Int J Gynaecol Obstet. 2007;99(suppl 2):S160-S167.
24. Gemzell-Danielsson K, Ho PC, Gomez Ponce de Leon R, et al. Misoprostol to treat missed abortion in the first trimester. Int J Gynaecol Obstet. 2007;99(suppl 2):S182-S185.
25. Blum J, Winikoff B, Gemzell-Danielsson K, et al. Treatment of incomplete abortion and miscarriage with misoprostol. Int J Gynaecol Obstet. 2007;99(suppl 2):S186-S189.
26. Moodliar S, Bagratee JS, Moodley J. Medical vs. surgical evacuation of first-trimester spontaneous abortion. Int J Gynaecol Obstet. 2005;91:21-26.
27. Graziosi GC, Bruinse HW, Reuwer PJ, et al. Women’s p for misoprostol in case of early pregnancy failure. Eur J Obstet Gynecol Reprod Biol. 2006;124:184-186.
28. Weeks A, Alia G, Blum J, et al. A randomized trial of misoprostol compared with manual vacuum aspiration for incomplete abortion. Obstet Gynecol. 2005;106:540-547.
29. Demetroulis C, Saridogan E, Kunde D, et al. A prospective randomized control trial comparing medical and surgical treatment for early pregnancy failure. Hum Reprod. 2001;16:365-369.
30. Zhang J, Gilles JM, Barnhart K, et al. A comparison of medical management with misoprostol and surgical management for early pregnancy failure [see comment]. N Engl J Med. 2005;353:761-769.
31. Trinder J, Brocklehurst P, Porter R, et al. Management of miscarriage: expectant, medical, or surgical? Results of randomized controlled trial (miscarriage treatment (MIST) trial) [see comment]. BMJ. 2006;332:1235-1240.
32. Fjerstad M, Trussell J, Sivin I, et al. Rates of serious infection after changes in regimens for medical abortion. N Engl J Med. 2009;61:145-151.
33. Creinin MD. Change in serum beta-human chorionic gonadotropin after abortion with methotrexate and misoprostol. Am J Ob Gyn. 1996;174:776-778.
34. Graziosi GC, Bruinse HW, Reuwer PJ, et al. Misoprostol versus curettage in women with early pregnancy failure: impact on women’s health-related quality of life. A randomized controlled trial. Hum Reprod. 2005;20:2340-2347.
35. Forna F, Gulmezoglu AM. Surgical procedures to evacuate incomplete miscarriage. Cochrane Database Syst Rev. 2001;(1):CD001993.-
36. Castleman L, Mann C. Manual Vacuum Aspiration (MVA) for Uterine Evacuation: Pain Management. Chapel Hill, NC: Ipas; 2002.
37. Baird T, Flinn SK. Manual Vacuum Aspiration: Expanding Women’s Access to Safe Abortion Services. Chapel Hill, NC: Ipas; 2001.
38. Goldberg AB, Dean G, Kang MS, et al. Manual versus electric vacuum aspiration for early first-trimester abortion: a controlled study of complication rates. Obstet Gynecol. 2004;103:101-107.
39. Paul ME, Mitchell CM, Rogers AJ, et al. Early surgical abortion: efficacy and safety. Am J Obstet Gynecol. 2002;187:407-411.
40. Westfall JM, Sophocles A, Burggraf H, et al. Manual vacuum aspiration for first-trimester abortion. Arch Fam Med. 1998;7:559-562.
41. Blumenthal PD, Remsburg RE. A time and cost analysis of the management of incomplete abortion with manual vacuum aspiration. Int J Gynaecol Obstet. 1994;45:261-267.
42. Maltzer DS, Maltzer MC, Wiebe ER, et al. Pain management. In: Paul M, Lichtenberg ES, Borgatta L, Grimes DA, Stubblefield PG, eds. A Clinician’s Guide to Medical and Surgical Abortion. Philadelphia: Churchill Livingstone; 1999.
43. Hansen GR, Streltzer J. The psychology of pain. Emerg Med Clin North Am. 2005;23:339-348.
• Family physicians can provide in-office treatment for patients with early pregnancy loss, as long as they are hemodynamically stable. A
• Manual vacuum aspiration in the office is as effective as surgical emptying of the uterus (dilatation and curettage) in the operating room. A
• No single in-office option for managing early pregnancy loss—wait and watch, misoprostol, or vacuum aspiration—is clearly more beneficial than another. Patients should be free to choose the method they prefer. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1: Janet C is 22 years old, excited about her first pregnancy and eager to do all the right things to have a healthy baby. But now, in her third month, she has started to bleed and has pelvic pain. She calls in a panic. You tell her to come in immediately. In the office, an ultrasound shows a residual gestational sac.
CASE 2: Lizbeth G, 40, is a successful professional, recently married, with a down-to-earth, decisive personality. She and her husband are eager to start a family. But now, in her second month, she calls to say she’s been having severe cramps and heavy bleeding. She knows she is having a miscarriage. When you see her in the examining room, she’s saddened but calm, eager to know what went wrong and what she needs to do now.
CASE 3: Lola M, 36, mother of 2, is in your office for a routine prenatal visit. She’s in her third month, expecting this pregnancy to be as uneventful as her previous ones. But your ultrasound exam reveals that her fetus has no heartbeat.
What would you tell each of these patients? What options would you offer them?
Chances are good that you’ve cared for any number of patients like Janet, Lizbeth, and Lola. Approximately 15% of clinically recognized pregnancies end in early pregnancy loss (EPL), defined as a miscarriage that occurs earlier than the 12th week of pregnancy. When clinically unrecognized miscarriages are included, the EPL rate may be as high as 30%.1 Most pregnancy losses (80%) occur during the first trimester.2
In the past, EPL was routinely considered an indication for uterine dilatation and curettage (D&C) performed in the operating room.3 This approach was effective, but had serious drawbacks: Costs were high and women had to undergo a surgical procedure that many would prefer to avoid.4
More recently, professional organizations such as the American Academy of Family Physicians and the United Kingdom’s Royal College of Obstetricians and Gynecologists have encouraged a wider range of treatment options that can be provided in an outpatient setting.5,6 These choices, which are available to women with confirmed intrauterine—not ectopic—pregnancy, include “watch and wait” (expectant management), medical management with misoprostol, and outpatient manual vacuum aspiration (MVA) of the uterus.
But before you can even discuss these options, it’s important to find out how your patient has been feeling about her pregnancy: Was it planned or unplanned? Is she happy or unhappy about being pregnant? Does she have a supportive partner, or is her relationship in turmoil? Having a clear sense of where she is emotionally will better enable you to counsel her on her options.
Know, too, that managing EPL patients in their family “medical home” has many advantages. Patients can remain with a caregiver they know and trust. Because they can choose the treatment option they prefer, they are more likely to be satisfied with their care.7 Their quality of life after treatment is better, and the emotional support they can receive in these familiar surroundings has been shown to decrease the psychological sequelae of a miscarriage.8-10
How best to define, and describe, what’s happened
Providing your patient with an accurate description of her situation is essential to adequately counseling her on treatment options. Types of EPL include:
Missed abortion, which occurs when a nonviable pregnancy is detected on ultrasound. The patient is usually without bleeding. A missed abortion is further distinguished sonographically as either an “anembryonic pregnancy”—a mean sac diameter of >10 mm and no yolk sac or a mean sac diameter of 20 mm and no embryo on transvaginal ultrasound—or as an “embryonic demise”—a crown rump length of ≥6 mm without cardiac activity on transvaginal ultrasound.5
Incomplete abortion occurs when a residual gestational sac is detected on ultrasound, and vaginal bleeding and pelvic pain are present.
Inevitable abortion occurs when the internal os is open, but the pregnancy has not yet passed.
Complete abortion occurs when no gestational sac is detected on ultrasound, the cervical os is generally closed, and significant cramping and bleeding have resolved.
Women experiencing a complete abortion require no treatment; they have already successfully passed the pregnancy. Women with a missed, incomplete, or inevitable abortion can be offered the choice of expectant management, medication, or uterine aspiration.
Does your patient want to wait it out?
The success rate for expectant management depends on the time-frame studied and the type of EPL.11 (Success in EPL is defined as complete uterine evacuation.) Patients who choose this approach are usually seen every 1 to 2 weeks so that you can evaluate symptoms and do a physical examination. In some cases, assessment also includes serial serum human chorionic gonadotropin (hCG) testing or ultrasonography.
Expectant management is usually more efficacious for women with an incomplete abortion than for women with anembryonic gestation or embryonic demise.12-16 TABLE W1, available at jfponline.com, provides a comparison of the efficacy of expectant management and misoprostol. In 1 observational study of 1096 women who chose expectant management, 91% of those with incomplete abortion were successful and 84% completed within 14 days of diagnosis. By comparison, only 59% of those with a missed abortion completed within 14 days.17
According to a study performed by Wieringa-de Waard and colleagues, increased bleeding appears to be the greatest predictor of completion. They showed that the median blood flow and pain were heaviest on the third day of vaginal bleeding, which then decreased steeply after 8 days to slight bleeding and spotting. Of the patients they followed, 50% completed during the first 8 days of bleeding.18
A Cochrane review of 5 studies comparing expectant management with vacuum aspiration found expectant management carried a higher risk of incomplete miscarriage, need for vacuum aspiration, and bleeding. In contrast, vacuum aspiration was associated with a significantly higher risk of infection.19
A low-cost option that can speed things up
EPL can be treated with prostaglandins to hasten the time to completion.20 Misoprostol is a synthetic prostaglandin E1 analog that causes contractions of the uterus and gastrointestinal tract. This medication is approved by the US Food and Drug Administration (FDA) only for the treatment of gastric ulcers, but it is commonly used off-label for labor induction, postpartum hemorrhage, and cervical ripening prior to gynecological procedures—as well as for the management of miscarriage.21 Misoprostol’s low cost and stability at room temperature make it easy to use.22
Route of administration. Although misoprostol is manufactured and approved for oral use only, administration by vaginal, buccal, or sublingual routes can increase the desired effect on the uterus, with the added benefit of decreased gastrointestinal side effects.23
The dosage and dosing intervals for misoprostol for treatment of EPL have not been well established. A comprehensive review article recommends a single dose of 800 mcg vaginal misoprostol or, alternatively, 600 mcg sublingual misoprostol for anembryonic pregnancy or embryonic/fetal demise.24 A single dose of 600 mcg oral or 400 mcg sublingual misoprostol is recommended for incomplete abortion.25 The vaginal route may not be feasible when bleeding is heavy.
Safety and efficacy. Multiple studies have found that misoprostol is a safe and acceptable alternative to vacuum aspiration or expectant management.11,26-29
A study comparing 652 women randomized to misoprostol vaginally or vacuum aspiration found that 84% of the misoprostol group had complete expulsion within 8 days of treatment initiation.30
Infection rates. The Miscarriage Treatment (MIST) trial randomized 1200 women with a diagnosis of embryonic demise or incomplete abortion at <13 weeks to medical (n=398), expectant (n=399), or vacuum aspiration management (n=403).31 Overall, the researchers found a low incidence of gynecologic infection (2.3%), and no evidence of difference in the infection rate attributable to the type of management selected.
Antibiotic use to reduce infection rates after misoprostol for EPL has not been studied. Nonetheless, a recent retrospective study examined infection rates after medical abortion with mifepristone and misoprostol.32 The study demonstrated a reduction in severe infection rates from 0.25 per 1000 abortions to 0.06 per 1000 (absolute reduction, 0.19 per 1000; 95% confidence interval [CI], 0.02-0.34; P=.03) with the routine use of doxycycline 100 mg PO twice daily for 7 days. The risk reduction is in comparison to the prior practice of either testing for sexually transmitted infection (STI) or using prophylactic doxycycline. The authors also reported a decrease in infection rate with a change from vaginal to buccal administration of misoprostol. The benefit of this change is unknown, because the practice of routine screening for STI or routine antibiotic provision was introduced at the same time.32
Follow-up. After misoprostol administration, follow-up includes confirmation of passage of the embryo or gestational sac by a combination of history, clinical examination, and serial hCG measurement or ultrasound. A completed abortion can be demonstrated by quantitative serum hCG showing a 50% drop between first and repeat test 48 to 72 hours after the passage of tissue.33 Follow-up 1 to 2 weeks after treatment is common practice, but can be scheduled sooner if the patient has not had bleeding and cramping. In that situation, you can give her the option of proceeding to uterine aspiration or trying a second dose of misoprostol (see doses given earlier), as long as she remains hemodynamically stable.24
Women who experience successful treatment with misoprostol like the method. In a multicenter, randomized clinical trial, 154 women with EPL confirmed by ultrasonography who had not passed the pregnancy after a week were randomly assigned to treatment with misoprostol (n=79) or curettage (n=75). In cases where misoprostol had caused complete evacuation, 76% of the women would opt for the same treatment, whereas only 38% of women who needed vacuum aspiration after unsuccessful misoprostol would do so (P<.01).34
A sample protocol for medical management of EPL is provided in the box.
Candidates
Women with ultrasound diagnosis of a nonviable pregnancy up to 10 weeks’ gestation. Nonviable pregnancy is diagnosed by ultrasound and subnormal, rising quantitative human chorionic gonadotropin (hCG) levels. Misoprostol treatment is not suitable in ectopic pregnancy, which must be excluded before treatment is begun.
Laboratory workup
Rh screen, hemoglobin, and quantitative serum hCG.
Procedure
Insert 800 mcg misoprostol in the vagina. (This can also be done by the patient at home.) If passage of tissue does not occur, the physician can give the patient a second dose of 800 mcg misoprostol. Anembryonic pregnancy or fetal demise can also be treated with 600 mcg given sublingually. Incomplete abortion is treated with a single dose of 600 mcg orally or 400 mcg sublingually.
Pain management
Provide a prescription for ibuprofen 800 mg and Tylenol #3 to the patient. Instruct her to take a tablet of ibuprofen at the time of misoprostol insertion and then every 6 hours as needed for pain. If pain is severe, she may take 1 to 2 tablets of Tylenol #3 every 3 to 4 hours as needed.
Instructions to patient
Tell the patient to call the office for “heavy bleeding,” defined as soaking 2 pads an hour for more than 2 hours. Tell the patient that there is no need to bring the expelled material for your inspection. Make sure she has your phone and pager numbers. If she needs to go to an emergency department or a hospital, tell her to request that you be called.
Follow-up
Schedule a follow-up visit 1 to 2 weeks after misoprostol insertion. A completed abortion can be demonstrated by quantitative serum hCG showing a 50% drop between first and repeat test 48 to 72 hours after the passage of tissue. Alternatively, a transvaginal ultrasound should show absence of a sac.
Of note: If one of these criteria is met, no further follow-up of serum hCG is warranted. Patients may elect manual vacuum aspiration at any time if the gestational sac and/or embryo have not passed.
Manual vacuum aspiration means less blood loss
A Cochrane review that compared vacuum aspiration with surgical D&C found that vacuum aspiration was associated with significantly less blood loss, pain, and time needed for the procedure.35 Traditionally, vacuum aspiration for EPL has occurred in the OR, using electrical suction and general anesthesia. Recently, a manual vacuum aspirator that allows women to have the procedure done in the outpatient setting has become available. It is used with analgesia given PO and a paracervical block.4,36
The manual vacuum aspirator (MVA) is a handheld syringe that works well in the ambulatory setting because it is small, quiet, portable, and inexpensive. The MVA is safe, provides the same degree of suction as an electric pump, and is as effective as electrical vacuum aspiration for the management of both spontaneous and induced abortion.37
As safe, as effective. A study by Goldberg and colleagues compared complication rates with MVA and electric suction in EPL of up to 10 weeks’ gestation.38 The researchers found no significant difference in perforation or need for re-aspiration. A comparison of the 2 methods in gestations of less than 6 weeks found a similar, small risk of failed abortion (<3%).39 A study of 1677 women treated with MVA as outpatients in a primary care practice had a complication rate of only 1.25%.40
Faster, cheaper. Blumenthal and Remsburg demonstrated that MVA in an outpatient setting decreases anesthesia requirements, hospital stay times, patient waiting times, and procedure times when compared with aspiration done in the OR. They showed a substantial saving, with the cost of uterine evacuation in the OR estimated at $1404 vs $827 per case when the aspiration was done as an outpatient procedure in the labor and delivery suite.41 The MVA syringe costs about $30 and is reusable after appropriate cleaning through sterilization or high-level disinfectant. The disposable plastic suction cannulas cost less than $3 each.
Pain management. A combination of an oral nonsteroidal anti-inflammatory (NSAID) medication and a paracervical block is a practical approach to managing the pain of this procedure. No published reports demonstrate that 1 type of local anesthetic is better than another, and many different techniques and combinations of medicines used for the paracervical block have been described.42
To minimize the effects of accidental blood vessel injection, the lowest anesthetic dose should be used, usually 10 to 20 mL of a 0.5% to 1% lidocaine or 0.25% bupivacaine solution. A common technique is to inject 8 to 10 cc of 1% lidocaine with epinephrine or vasopressin at 4 and 8 o’clock at the cervicovaginal reflection after careful aspiration to ensure the needle is not in a blood vessel.
Oral narcotics. Clinicians can also choose to manage pain with oral narcotics, benzodiazepines, or intravenous conscious sedation. Moderate cramping during and immediately after the procedure is common and can often be alleviated with verbal support.
For patients whose anxiety level is high, conscious sedation or general anesthesia may be the most appropriate choice. Your patient’s preference and your evaluation of her medical risk and emotional state together determine the most appropriate course.43 The technique for MVA is described inTABLE W2, available at jfponline.com.
Which approach is best for your patient?
Because all 3 approaches to managing EPL are effective and safe, family physicians can empower patients to make the choice themselves. Counseling about treatment options should include consideration of the patient’s support at home, availability of transportation in case of emergency, her desire to avoid surgery, and her need for a definitive resolution.
Counseling should also include information on the likely efficacy of each option, given the type of EPL the patient has experienced. For example, women who have had a missed abortion (embryonic demise or anembryonic gestation) are less likely to complete with expectant management than women with an incomplete abortion. Efficacy rates for different types of EPL are shown in TABLE W1, available at jfponline.com.
There’s time for your patient to change her mind
A woman may opt for 1 approach to start with, but choose a different option later. She may chose expectant management for a week, and then if the pregnancy has not passed on its own, decide that she wants to try misoprostol. If that fails, too, she may want a uterine aspiration procedure.
How did our 3 patients fare?
CASE 1: At first, Janet was content to wait and see whether her miscarriage would pass without further intervention. But when a week went by and nothing happened, she wanted to get it over with. She asked to try MVA, under conscious sedation. The procedure was successful. Now, a year later, she’s very happy to be pregnant again and confidently awaits a happy outcome.
CASE 2: By the time Lizbeth called, you suspected her abortion was complete. Your examination confirmed that diagnosis. She required no treatment, and a year later was ready to try again.
CASE 3: Lola was shocked when she learned her fetus had died in utero. But once she and her husband had taken in the sad news, they wanted to know what options were available. They talked it over and chose treatment with misoprostol. The miscarriage was completed 8 days later. They are content with their current family size and have decided not to try for another pregnancy.
CORRESPONDENCE Emily M. Godfrey, MD, MPH, UIC Department of Family Medicine, 1919 W. Taylor Ave., Room 145, M/C 663, Chicago, IL 60612; [email protected]
• Family physicians can provide in-office treatment for patients with early pregnancy loss, as long as they are hemodynamically stable. A
• Manual vacuum aspiration in the office is as effective as surgical emptying of the uterus (dilatation and curettage) in the operating room. A
• No single in-office option for managing early pregnancy loss—wait and watch, misoprostol, or vacuum aspiration—is clearly more beneficial than another. Patients should be free to choose the method they prefer. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1: Janet C is 22 years old, excited about her first pregnancy and eager to do all the right things to have a healthy baby. But now, in her third month, she has started to bleed and has pelvic pain. She calls in a panic. You tell her to come in immediately. In the office, an ultrasound shows a residual gestational sac.
CASE 2: Lizbeth G, 40, is a successful professional, recently married, with a down-to-earth, decisive personality. She and her husband are eager to start a family. But now, in her second month, she calls to say she’s been having severe cramps and heavy bleeding. She knows she is having a miscarriage. When you see her in the examining room, she’s saddened but calm, eager to know what went wrong and what she needs to do now.
CASE 3: Lola M, 36, mother of 2, is in your office for a routine prenatal visit. She’s in her third month, expecting this pregnancy to be as uneventful as her previous ones. But your ultrasound exam reveals that her fetus has no heartbeat.
What would you tell each of these patients? What options would you offer them?
Chances are good that you’ve cared for any number of patients like Janet, Lizbeth, and Lola. Approximately 15% of clinically recognized pregnancies end in early pregnancy loss (EPL), defined as a miscarriage that occurs earlier than the 12th week of pregnancy. When clinically unrecognized miscarriages are included, the EPL rate may be as high as 30%.1 Most pregnancy losses (80%) occur during the first trimester.2
In the past, EPL was routinely considered an indication for uterine dilatation and curettage (D&C) performed in the operating room.3 This approach was effective, but had serious drawbacks: Costs were high and women had to undergo a surgical procedure that many would prefer to avoid.4
More recently, professional organizations such as the American Academy of Family Physicians and the United Kingdom’s Royal College of Obstetricians and Gynecologists have encouraged a wider range of treatment options that can be provided in an outpatient setting.5,6 These choices, which are available to women with confirmed intrauterine—not ectopic—pregnancy, include “watch and wait” (expectant management), medical management with misoprostol, and outpatient manual vacuum aspiration (MVA) of the uterus.
But before you can even discuss these options, it’s important to find out how your patient has been feeling about her pregnancy: Was it planned or unplanned? Is she happy or unhappy about being pregnant? Does she have a supportive partner, or is her relationship in turmoil? Having a clear sense of where she is emotionally will better enable you to counsel her on her options.
Know, too, that managing EPL patients in their family “medical home” has many advantages. Patients can remain with a caregiver they know and trust. Because they can choose the treatment option they prefer, they are more likely to be satisfied with their care.7 Their quality of life after treatment is better, and the emotional support they can receive in these familiar surroundings has been shown to decrease the psychological sequelae of a miscarriage.8-10
How best to define, and describe, what’s happened
Providing your patient with an accurate description of her situation is essential to adequately counseling her on treatment options. Types of EPL include:
Missed abortion, which occurs when a nonviable pregnancy is detected on ultrasound. The patient is usually without bleeding. A missed abortion is further distinguished sonographically as either an “anembryonic pregnancy”—a mean sac diameter of >10 mm and no yolk sac or a mean sac diameter of 20 mm and no embryo on transvaginal ultrasound—or as an “embryonic demise”—a crown rump length of ≥6 mm without cardiac activity on transvaginal ultrasound.5
Incomplete abortion occurs when a residual gestational sac is detected on ultrasound, and vaginal bleeding and pelvic pain are present.
Inevitable abortion occurs when the internal os is open, but the pregnancy has not yet passed.
Complete abortion occurs when no gestational sac is detected on ultrasound, the cervical os is generally closed, and significant cramping and bleeding have resolved.
Women experiencing a complete abortion require no treatment; they have already successfully passed the pregnancy. Women with a missed, incomplete, or inevitable abortion can be offered the choice of expectant management, medication, or uterine aspiration.
Does your patient want to wait it out?
The success rate for expectant management depends on the time-frame studied and the type of EPL.11 (Success in EPL is defined as complete uterine evacuation.) Patients who choose this approach are usually seen every 1 to 2 weeks so that you can evaluate symptoms and do a physical examination. In some cases, assessment also includes serial serum human chorionic gonadotropin (hCG) testing or ultrasonography.
Expectant management is usually more efficacious for women with an incomplete abortion than for women with anembryonic gestation or embryonic demise.12-16 TABLE W1, available at jfponline.com, provides a comparison of the efficacy of expectant management and misoprostol. In 1 observational study of 1096 women who chose expectant management, 91% of those with incomplete abortion were successful and 84% completed within 14 days of diagnosis. By comparison, only 59% of those with a missed abortion completed within 14 days.17
According to a study performed by Wieringa-de Waard and colleagues, increased bleeding appears to be the greatest predictor of completion. They showed that the median blood flow and pain were heaviest on the third day of vaginal bleeding, which then decreased steeply after 8 days to slight bleeding and spotting. Of the patients they followed, 50% completed during the first 8 days of bleeding.18
A Cochrane review of 5 studies comparing expectant management with vacuum aspiration found expectant management carried a higher risk of incomplete miscarriage, need for vacuum aspiration, and bleeding. In contrast, vacuum aspiration was associated with a significantly higher risk of infection.19
A low-cost option that can speed things up
EPL can be treated with prostaglandins to hasten the time to completion.20 Misoprostol is a synthetic prostaglandin E1 analog that causes contractions of the uterus and gastrointestinal tract. This medication is approved by the US Food and Drug Administration (FDA) only for the treatment of gastric ulcers, but it is commonly used off-label for labor induction, postpartum hemorrhage, and cervical ripening prior to gynecological procedures—as well as for the management of miscarriage.21 Misoprostol’s low cost and stability at room temperature make it easy to use.22
Route of administration. Although misoprostol is manufactured and approved for oral use only, administration by vaginal, buccal, or sublingual routes can increase the desired effect on the uterus, with the added benefit of decreased gastrointestinal side effects.23
The dosage and dosing intervals for misoprostol for treatment of EPL have not been well established. A comprehensive review article recommends a single dose of 800 mcg vaginal misoprostol or, alternatively, 600 mcg sublingual misoprostol for anembryonic pregnancy or embryonic/fetal demise.24 A single dose of 600 mcg oral or 400 mcg sublingual misoprostol is recommended for incomplete abortion.25 The vaginal route may not be feasible when bleeding is heavy.
Safety and efficacy. Multiple studies have found that misoprostol is a safe and acceptable alternative to vacuum aspiration or expectant management.11,26-29
A study comparing 652 women randomized to misoprostol vaginally or vacuum aspiration found that 84% of the misoprostol group had complete expulsion within 8 days of treatment initiation.30
Infection rates. The Miscarriage Treatment (MIST) trial randomized 1200 women with a diagnosis of embryonic demise or incomplete abortion at <13 weeks to medical (n=398), expectant (n=399), or vacuum aspiration management (n=403).31 Overall, the researchers found a low incidence of gynecologic infection (2.3%), and no evidence of difference in the infection rate attributable to the type of management selected.
Antibiotic use to reduce infection rates after misoprostol for EPL has not been studied. Nonetheless, a recent retrospective study examined infection rates after medical abortion with mifepristone and misoprostol.32 The study demonstrated a reduction in severe infection rates from 0.25 per 1000 abortions to 0.06 per 1000 (absolute reduction, 0.19 per 1000; 95% confidence interval [CI], 0.02-0.34; P=.03) with the routine use of doxycycline 100 mg PO twice daily for 7 days. The risk reduction is in comparison to the prior practice of either testing for sexually transmitted infection (STI) or using prophylactic doxycycline. The authors also reported a decrease in infection rate with a change from vaginal to buccal administration of misoprostol. The benefit of this change is unknown, because the practice of routine screening for STI or routine antibiotic provision was introduced at the same time.32
Follow-up. After misoprostol administration, follow-up includes confirmation of passage of the embryo or gestational sac by a combination of history, clinical examination, and serial hCG measurement or ultrasound. A completed abortion can be demonstrated by quantitative serum hCG showing a 50% drop between first and repeat test 48 to 72 hours after the passage of tissue.33 Follow-up 1 to 2 weeks after treatment is common practice, but can be scheduled sooner if the patient has not had bleeding and cramping. In that situation, you can give her the option of proceeding to uterine aspiration or trying a second dose of misoprostol (see doses given earlier), as long as she remains hemodynamically stable.24
Women who experience successful treatment with misoprostol like the method. In a multicenter, randomized clinical trial, 154 women with EPL confirmed by ultrasonography who had not passed the pregnancy after a week were randomly assigned to treatment with misoprostol (n=79) or curettage (n=75). In cases where misoprostol had caused complete evacuation, 76% of the women would opt for the same treatment, whereas only 38% of women who needed vacuum aspiration after unsuccessful misoprostol would do so (P<.01).34
A sample protocol for medical management of EPL is provided in the box.
Candidates
Women with ultrasound diagnosis of a nonviable pregnancy up to 10 weeks’ gestation. Nonviable pregnancy is diagnosed by ultrasound and subnormal, rising quantitative human chorionic gonadotropin (hCG) levels. Misoprostol treatment is not suitable in ectopic pregnancy, which must be excluded before treatment is begun.
Laboratory workup
Rh screen, hemoglobin, and quantitative serum hCG.
Procedure
Insert 800 mcg misoprostol in the vagina. (This can also be done by the patient at home.) If passage of tissue does not occur, the physician can give the patient a second dose of 800 mcg misoprostol. Anembryonic pregnancy or fetal demise can also be treated with 600 mcg given sublingually. Incomplete abortion is treated with a single dose of 600 mcg orally or 400 mcg sublingually.
Pain management
Provide a prescription for ibuprofen 800 mg and Tylenol #3 to the patient. Instruct her to take a tablet of ibuprofen at the time of misoprostol insertion and then every 6 hours as needed for pain. If pain is severe, she may take 1 to 2 tablets of Tylenol #3 every 3 to 4 hours as needed.
Instructions to patient
Tell the patient to call the office for “heavy bleeding,” defined as soaking 2 pads an hour for more than 2 hours. Tell the patient that there is no need to bring the expelled material for your inspection. Make sure she has your phone and pager numbers. If she needs to go to an emergency department or a hospital, tell her to request that you be called.
Follow-up
Schedule a follow-up visit 1 to 2 weeks after misoprostol insertion. A completed abortion can be demonstrated by quantitative serum hCG showing a 50% drop between first and repeat test 48 to 72 hours after the passage of tissue. Alternatively, a transvaginal ultrasound should show absence of a sac.
Of note: If one of these criteria is met, no further follow-up of serum hCG is warranted. Patients may elect manual vacuum aspiration at any time if the gestational sac and/or embryo have not passed.
Manual vacuum aspiration means less blood loss
A Cochrane review that compared vacuum aspiration with surgical D&C found that vacuum aspiration was associated with significantly less blood loss, pain, and time needed for the procedure.35 Traditionally, vacuum aspiration for EPL has occurred in the OR, using electrical suction and general anesthesia. Recently, a manual vacuum aspirator that allows women to have the procedure done in the outpatient setting has become available. It is used with analgesia given PO and a paracervical block.4,36
The manual vacuum aspirator (MVA) is a handheld syringe that works well in the ambulatory setting because it is small, quiet, portable, and inexpensive. The MVA is safe, provides the same degree of suction as an electric pump, and is as effective as electrical vacuum aspiration for the management of both spontaneous and induced abortion.37
As safe, as effective. A study by Goldberg and colleagues compared complication rates with MVA and electric suction in EPL of up to 10 weeks’ gestation.38 The researchers found no significant difference in perforation or need for re-aspiration. A comparison of the 2 methods in gestations of less than 6 weeks found a similar, small risk of failed abortion (<3%).39 A study of 1677 women treated with MVA as outpatients in a primary care practice had a complication rate of only 1.25%.40
Faster, cheaper. Blumenthal and Remsburg demonstrated that MVA in an outpatient setting decreases anesthesia requirements, hospital stay times, patient waiting times, and procedure times when compared with aspiration done in the OR. They showed a substantial saving, with the cost of uterine evacuation in the OR estimated at $1404 vs $827 per case when the aspiration was done as an outpatient procedure in the labor and delivery suite.41 The MVA syringe costs about $30 and is reusable after appropriate cleaning through sterilization or high-level disinfectant. The disposable plastic suction cannulas cost less than $3 each.
Pain management. A combination of an oral nonsteroidal anti-inflammatory (NSAID) medication and a paracervical block is a practical approach to managing the pain of this procedure. No published reports demonstrate that 1 type of local anesthetic is better than another, and many different techniques and combinations of medicines used for the paracervical block have been described.42
To minimize the effects of accidental blood vessel injection, the lowest anesthetic dose should be used, usually 10 to 20 mL of a 0.5% to 1% lidocaine or 0.25% bupivacaine solution. A common technique is to inject 8 to 10 cc of 1% lidocaine with epinephrine or vasopressin at 4 and 8 o’clock at the cervicovaginal reflection after careful aspiration to ensure the needle is not in a blood vessel.
Oral narcotics. Clinicians can also choose to manage pain with oral narcotics, benzodiazepines, or intravenous conscious sedation. Moderate cramping during and immediately after the procedure is common and can often be alleviated with verbal support.
For patients whose anxiety level is high, conscious sedation or general anesthesia may be the most appropriate choice. Your patient’s preference and your evaluation of her medical risk and emotional state together determine the most appropriate course.43 The technique for MVA is described inTABLE W2, available at jfponline.com.
Which approach is best for your patient?
Because all 3 approaches to managing EPL are effective and safe, family physicians can empower patients to make the choice themselves. Counseling about treatment options should include consideration of the patient’s support at home, availability of transportation in case of emergency, her desire to avoid surgery, and her need for a definitive resolution.
Counseling should also include information on the likely efficacy of each option, given the type of EPL the patient has experienced. For example, women who have had a missed abortion (embryonic demise or anembryonic gestation) are less likely to complete with expectant management than women with an incomplete abortion. Efficacy rates for different types of EPL are shown in TABLE W1, available at jfponline.com.
There’s time for your patient to change her mind
A woman may opt for 1 approach to start with, but choose a different option later. She may chose expectant management for a week, and then if the pregnancy has not passed on its own, decide that she wants to try misoprostol. If that fails, too, she may want a uterine aspiration procedure.
How did our 3 patients fare?
CASE 1: At first, Janet was content to wait and see whether her miscarriage would pass without further intervention. But when a week went by and nothing happened, she wanted to get it over with. She asked to try MVA, under conscious sedation. The procedure was successful. Now, a year later, she’s very happy to be pregnant again and confidently awaits a happy outcome.
CASE 2: By the time Lizbeth called, you suspected her abortion was complete. Your examination confirmed that diagnosis. She required no treatment, and a year later was ready to try again.
CASE 3: Lola was shocked when she learned her fetus had died in utero. But once she and her husband had taken in the sad news, they wanted to know what options were available. They talked it over and chose treatment with misoprostol. The miscarriage was completed 8 days later. They are content with their current family size and have decided not to try for another pregnancy.
CORRESPONDENCE Emily M. Godfrey, MD, MPH, UIC Department of Family Medicine, 1919 W. Taylor Ave., Room 145, M/C 663, Chicago, IL 60612; [email protected]
1. Wilcox AJ, Weinberg CR, O’Connor JF, et al. Incidence of early loss of pregnancy. N Engl J Med. 1988;319:189-194.
2. Tang OS, Ho PC. The use of misoprostol for early pregnancy failure. Curr Opin Obstet Gynecol. 2006;18:581-586.
3. Jurkovic D. Modern management of miscarriage: is there a place for non-surgical treatment? Ultrasound Obstet Gynecol. 1998;11:161-163.
4. Dalton VK, Harris L, Weisman CS, et al. Patient p, satisfaction, and resource use in office evacuation of early pregnancy failure. Obstet Gynecol. 2006;108:103-110.
5. Royal College of Obstetricians and Gynaecologists. The Management of Early Pregnancy Loss. London: Royal College of Obstetricians and Gynaecologists; 2006. Guideline No. 25.
6. Deutchman M. Advanced Life Support in Obstetrics Syllabus Update: First Trimester Pregnancy Complications. Leawood, Kan: American Academy of Family Physicians; 2007:1-2.
7. Wieringa-de Waard M, Bindels PJ, Vos J, et al. Patient p for expectant management vs. surgical evacuation in first-trimester uncomplicated miscarriage. J Clin Epidemiol. 2004;57:167-173.
8. Edwards S, Tureck R, Fredrick M, et al. Patient acceptability of manual versus electric vacuum aspiration for early pregnancy loss. J Womens Health (Larchmt). 2007;16:1429-1436.
9. Smith LF, Frost J, Levitas R, Bradley H, et al. Women’s experiences of three early miscarriage management options: a qualitative study. Br J Gen Pract. 2006;56:198-205.
10. Nikcevic AV, Kuczmierczyk AR, Nicolaides KH. The influence of medical and psychological interventions on women’s distress after miscarriage. J Psychosom Res. 2007;63:283-290.
11. Sotiriadis A, Makrydimas G, Papatheodorou S, et al. Expectant, medical, or surgical management of first-trimester miscarriage: a meta-analysis. Obstet Gynecol. 2005;105(5 Pt 1):1104-1113.
12. Bagratee JS, Khullar V, Regan L, et al. A randomized controlled trial comparing medical and expectant management of first trimester miscarriage. Hum Reprod. 2004;19:266-271.
13. Blohm F, Friden B, Platz-Christensen JJ, et al. Expectant management of first-trimester miscarriage in clinical practice. Acta Obstet Gynecol Scand. 2003;82:654-658.
14. Gronlund L, Gronlund AL, Clevin L, et al. Spontaneous abortion: expectant management, medical treatment or surgical evacuation. Acta Obstet Gynecol Scand. 2002;81:781-782.
15. Graziosi GC, Mol BW, Ankum WM, et al. Management of early pregnancy loss. Int J Gynaecol Obstet. 2004;86:337-346.
16. Wood SL, Brain PH. Medical management of missed abortion: a randomized clinical trial. Obstet Gynecol. 2002;99:563-566.
17. Luise C, Jermy K, May C, et al. Outcome of expectant management of spontaneous first trimester miscarriage: observational study [see comment]. BMJ. 2002;324:873-875.
18. Wieringa-de Waard M, Ankum WM, Bonsel GJ, et al. The natural course of spontaneous miscarriage: analysis of signs and symptoms in 188 expectantly managed women. Br J Gen Pract. 2003;53:704-708.
19. Nanda K, Peloggia A, Grimes D, et al. Expectant care versus surgical treatment for miscarriage. Cochrane Database Syst Rev. 2006(2):CD003518.-
20. Neilson JP, Hickey M, Vazquez J. Medical treatment for early fetal death (less than 24 weeks). Cochrane Database Syst Rev. 2006;(3):CD002253.-
21. Weeks A, Faundes A. Misoprostol in obstetrics and gynecology. Int J Gynaecol Obstet. 2007;99(suppl 2):S156-S159.
22. World Health Organization. Medical Methods for Termination of Pregnancy. Geneva: WHO; 1997.
23. Tang OS, Gemzell-Danielsson K, Ho PC. Misoprostol: pharmacokinetic profiles, effects on the uterus and side-effects. Int J Gynaecol Obstet. 2007;99(suppl 2):S160-S167.
24. Gemzell-Danielsson K, Ho PC, Gomez Ponce de Leon R, et al. Misoprostol to treat missed abortion in the first trimester. Int J Gynaecol Obstet. 2007;99(suppl 2):S182-S185.
25. Blum J, Winikoff B, Gemzell-Danielsson K, et al. Treatment of incomplete abortion and miscarriage with misoprostol. Int J Gynaecol Obstet. 2007;99(suppl 2):S186-S189.
26. Moodliar S, Bagratee JS, Moodley J. Medical vs. surgical evacuation of first-trimester spontaneous abortion. Int J Gynaecol Obstet. 2005;91:21-26.
27. Graziosi GC, Bruinse HW, Reuwer PJ, et al. Women’s p for misoprostol in case of early pregnancy failure. Eur J Obstet Gynecol Reprod Biol. 2006;124:184-186.
28. Weeks A, Alia G, Blum J, et al. A randomized trial of misoprostol compared with manual vacuum aspiration for incomplete abortion. Obstet Gynecol. 2005;106:540-547.
29. Demetroulis C, Saridogan E, Kunde D, et al. A prospective randomized control trial comparing medical and surgical treatment for early pregnancy failure. Hum Reprod. 2001;16:365-369.
30. Zhang J, Gilles JM, Barnhart K, et al. A comparison of medical management with misoprostol and surgical management for early pregnancy failure [see comment]. N Engl J Med. 2005;353:761-769.
31. Trinder J, Brocklehurst P, Porter R, et al. Management of miscarriage: expectant, medical, or surgical? Results of randomized controlled trial (miscarriage treatment (MIST) trial) [see comment]. BMJ. 2006;332:1235-1240.
32. Fjerstad M, Trussell J, Sivin I, et al. Rates of serious infection after changes in regimens for medical abortion. N Engl J Med. 2009;61:145-151.
33. Creinin MD. Change in serum beta-human chorionic gonadotropin after abortion with methotrexate and misoprostol. Am J Ob Gyn. 1996;174:776-778.
34. Graziosi GC, Bruinse HW, Reuwer PJ, et al. Misoprostol versus curettage in women with early pregnancy failure: impact on women’s health-related quality of life. A randomized controlled trial. Hum Reprod. 2005;20:2340-2347.
35. Forna F, Gulmezoglu AM. Surgical procedures to evacuate incomplete miscarriage. Cochrane Database Syst Rev. 2001;(1):CD001993.-
36. Castleman L, Mann C. Manual Vacuum Aspiration (MVA) for Uterine Evacuation: Pain Management. Chapel Hill, NC: Ipas; 2002.
37. Baird T, Flinn SK. Manual Vacuum Aspiration: Expanding Women’s Access to Safe Abortion Services. Chapel Hill, NC: Ipas; 2001.
38. Goldberg AB, Dean G, Kang MS, et al. Manual versus electric vacuum aspiration for early first-trimester abortion: a controlled study of complication rates. Obstet Gynecol. 2004;103:101-107.
39. Paul ME, Mitchell CM, Rogers AJ, et al. Early surgical abortion: efficacy and safety. Am J Obstet Gynecol. 2002;187:407-411.
40. Westfall JM, Sophocles A, Burggraf H, et al. Manual vacuum aspiration for first-trimester abortion. Arch Fam Med. 1998;7:559-562.
41. Blumenthal PD, Remsburg RE. A time and cost analysis of the management of incomplete abortion with manual vacuum aspiration. Int J Gynaecol Obstet. 1994;45:261-267.
42. Maltzer DS, Maltzer MC, Wiebe ER, et al. Pain management. In: Paul M, Lichtenberg ES, Borgatta L, Grimes DA, Stubblefield PG, eds. A Clinician’s Guide to Medical and Surgical Abortion. Philadelphia: Churchill Livingstone; 1999.
43. Hansen GR, Streltzer J. The psychology of pain. Emerg Med Clin North Am. 2005;23:339-348.
1. Wilcox AJ, Weinberg CR, O’Connor JF, et al. Incidence of early loss of pregnancy. N Engl J Med. 1988;319:189-194.
2. Tang OS, Ho PC. The use of misoprostol for early pregnancy failure. Curr Opin Obstet Gynecol. 2006;18:581-586.
3. Jurkovic D. Modern management of miscarriage: is there a place for non-surgical treatment? Ultrasound Obstet Gynecol. 1998;11:161-163.
4. Dalton VK, Harris L, Weisman CS, et al. Patient p, satisfaction, and resource use in office evacuation of early pregnancy failure. Obstet Gynecol. 2006;108:103-110.
5. Royal College of Obstetricians and Gynaecologists. The Management of Early Pregnancy Loss. London: Royal College of Obstetricians and Gynaecologists; 2006. Guideline No. 25.
6. Deutchman M. Advanced Life Support in Obstetrics Syllabus Update: First Trimester Pregnancy Complications. Leawood, Kan: American Academy of Family Physicians; 2007:1-2.
7. Wieringa-de Waard M, Bindels PJ, Vos J, et al. Patient p for expectant management vs. surgical evacuation in first-trimester uncomplicated miscarriage. J Clin Epidemiol. 2004;57:167-173.
8. Edwards S, Tureck R, Fredrick M, et al. Patient acceptability of manual versus electric vacuum aspiration for early pregnancy loss. J Womens Health (Larchmt). 2007;16:1429-1436.
9. Smith LF, Frost J, Levitas R, Bradley H, et al. Women’s experiences of three early miscarriage management options: a qualitative study. Br J Gen Pract. 2006;56:198-205.
10. Nikcevic AV, Kuczmierczyk AR, Nicolaides KH. The influence of medical and psychological interventions on women’s distress after miscarriage. J Psychosom Res. 2007;63:283-290.
11. Sotiriadis A, Makrydimas G, Papatheodorou S, et al. Expectant, medical, or surgical management of first-trimester miscarriage: a meta-analysis. Obstet Gynecol. 2005;105(5 Pt 1):1104-1113.
12. Bagratee JS, Khullar V, Regan L, et al. A randomized controlled trial comparing medical and expectant management of first trimester miscarriage. Hum Reprod. 2004;19:266-271.
13. Blohm F, Friden B, Platz-Christensen JJ, et al. Expectant management of first-trimester miscarriage in clinical practice. Acta Obstet Gynecol Scand. 2003;82:654-658.
14. Gronlund L, Gronlund AL, Clevin L, et al. Spontaneous abortion: expectant management, medical treatment or surgical evacuation. Acta Obstet Gynecol Scand. 2002;81:781-782.
15. Graziosi GC, Mol BW, Ankum WM, et al. Management of early pregnancy loss. Int J Gynaecol Obstet. 2004;86:337-346.
16. Wood SL, Brain PH. Medical management of missed abortion: a randomized clinical trial. Obstet Gynecol. 2002;99:563-566.
17. Luise C, Jermy K, May C, et al. Outcome of expectant management of spontaneous first trimester miscarriage: observational study [see comment]. BMJ. 2002;324:873-875.
18. Wieringa-de Waard M, Ankum WM, Bonsel GJ, et al. The natural course of spontaneous miscarriage: analysis of signs and symptoms in 188 expectantly managed women. Br J Gen Pract. 2003;53:704-708.
19. Nanda K, Peloggia A, Grimes D, et al. Expectant care versus surgical treatment for miscarriage. Cochrane Database Syst Rev. 2006(2):CD003518.-
20. Neilson JP, Hickey M, Vazquez J. Medical treatment for early fetal death (less than 24 weeks). Cochrane Database Syst Rev. 2006;(3):CD002253.-
21. Weeks A, Faundes A. Misoprostol in obstetrics and gynecology. Int J Gynaecol Obstet. 2007;99(suppl 2):S156-S159.
22. World Health Organization. Medical Methods for Termination of Pregnancy. Geneva: WHO; 1997.
23. Tang OS, Gemzell-Danielsson K, Ho PC. Misoprostol: pharmacokinetic profiles, effects on the uterus and side-effects. Int J Gynaecol Obstet. 2007;99(suppl 2):S160-S167.
24. Gemzell-Danielsson K, Ho PC, Gomez Ponce de Leon R, et al. Misoprostol to treat missed abortion in the first trimester. Int J Gynaecol Obstet. 2007;99(suppl 2):S182-S185.
25. Blum J, Winikoff B, Gemzell-Danielsson K, et al. Treatment of incomplete abortion and miscarriage with misoprostol. Int J Gynaecol Obstet. 2007;99(suppl 2):S186-S189.
26. Moodliar S, Bagratee JS, Moodley J. Medical vs. surgical evacuation of first-trimester spontaneous abortion. Int J Gynaecol Obstet. 2005;91:21-26.
27. Graziosi GC, Bruinse HW, Reuwer PJ, et al. Women’s p for misoprostol in case of early pregnancy failure. Eur J Obstet Gynecol Reprod Biol. 2006;124:184-186.
28. Weeks A, Alia G, Blum J, et al. A randomized trial of misoprostol compared with manual vacuum aspiration for incomplete abortion. Obstet Gynecol. 2005;106:540-547.
29. Demetroulis C, Saridogan E, Kunde D, et al. A prospective randomized control trial comparing medical and surgical treatment for early pregnancy failure. Hum Reprod. 2001;16:365-369.
30. Zhang J, Gilles JM, Barnhart K, et al. A comparison of medical management with misoprostol and surgical management for early pregnancy failure [see comment]. N Engl J Med. 2005;353:761-769.
31. Trinder J, Brocklehurst P, Porter R, et al. Management of miscarriage: expectant, medical, or surgical? Results of randomized controlled trial (miscarriage treatment (MIST) trial) [see comment]. BMJ. 2006;332:1235-1240.
32. Fjerstad M, Trussell J, Sivin I, et al. Rates of serious infection after changes in regimens for medical abortion. N Engl J Med. 2009;61:145-151.
33. Creinin MD. Change in serum beta-human chorionic gonadotropin after abortion with methotrexate and misoprostol. Am J Ob Gyn. 1996;174:776-778.
34. Graziosi GC, Bruinse HW, Reuwer PJ, et al. Misoprostol versus curettage in women with early pregnancy failure: impact on women’s health-related quality of life. A randomized controlled trial. Hum Reprod. 2005;20:2340-2347.
35. Forna F, Gulmezoglu AM. Surgical procedures to evacuate incomplete miscarriage. Cochrane Database Syst Rev. 2001;(1):CD001993.-
36. Castleman L, Mann C. Manual Vacuum Aspiration (MVA) for Uterine Evacuation: Pain Management. Chapel Hill, NC: Ipas; 2002.
37. Baird T, Flinn SK. Manual Vacuum Aspiration: Expanding Women’s Access to Safe Abortion Services. Chapel Hill, NC: Ipas; 2001.
38. Goldberg AB, Dean G, Kang MS, et al. Manual versus electric vacuum aspiration for early first-trimester abortion: a controlled study of complication rates. Obstet Gynecol. 2004;103:101-107.
39. Paul ME, Mitchell CM, Rogers AJ, et al. Early surgical abortion: efficacy and safety. Am J Obstet Gynecol. 2002;187:407-411.
40. Westfall JM, Sophocles A, Burggraf H, et al. Manual vacuum aspiration for first-trimester abortion. Arch Fam Med. 1998;7:559-562.
41. Blumenthal PD, Remsburg RE. A time and cost analysis of the management of incomplete abortion with manual vacuum aspiration. Int J Gynaecol Obstet. 1994;45:261-267.
42. Maltzer DS, Maltzer MC, Wiebe ER, et al. Pain management. In: Paul M, Lichtenberg ES, Borgatta L, Grimes DA, Stubblefield PG, eds. A Clinician’s Guide to Medical and Surgical Abortion. Philadelphia: Churchill Livingstone; 1999.
43. Hansen GR, Streltzer J. The psychology of pain. Emerg Med Clin North Am. 2005;23:339-348.
Hypertrophic cardiomyopathy: Ask athletes these 9 questions
• Screen for hypertrophic cardiomyopathy (HCM) during preparticipation sports physicals. C
• Patients who have symptoms of, or are diagnosed with, HCM should be cleared by a cardiologist before being allowed to participate in organized sports or engaging in physical exercise. B
• Patients with HCM and a history of cardiac arrest should be given the opportunity to receive an automatic implantable cardioverter defibrillator (AICD). A
• Beta-blockers are first-line therapy for patients with HCM who have symptoms of heart failure. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Hypertrophic cardiomyopathy (HCM) is the most common cardiac genetic disorder.1 But it is best known for its tragic outcomes—the sudden cardiac death (SCD) of young athletes, many of whom are in high school or college.
In fact, HCM can present at any time from infancy through old age, and has a broad range of manifestations. Some individuals remain asymptomatic throughout their lives and enjoy a normal life span; others experience a range of symptoms, from chest pain and dyspnea on exertion to dizziness, light-headedness, palpitations, and syncope. Diagnosis is difficult, and predicting which patients will develop severe complications and face the greatest risk of premature SCD remains an inexact science.
What is clear is that in the United States, HCM causes SCD far more than any other condition.2 HCM has been positively identified in well over a third of cases (36%) of SCD in athletes under the age of 30, and cited as a possible cause in another 8%. Coronary artery anomalies is a distant second, responsible for 17% of sudden deaths in this patient population; myocarditis follows, at just 6%.2
We also know that HCM itself occurs much more frequently than previously believed. In the early 1980s, its prevalence was estimated at <1 in 100,000;3 today, HCM is believed to affect approximately 1 in 500 adults.2,4 Because this potentially fatal condition often remains undetected until an athlete collapses on the playing field, it is crucial that family physicians—who conduct many of the sports physicals typically required before students are permitted to play competitive sports—maintain a high degree of suspicion.
Knowing what signs and symptoms to look for and, perhaps more importantly, the key questions to ask, will help you identify young athletes at risk. This update, which begins with a review of the genetic basis and pathophysiology of HCM, will prepare you to take steps to protect your young patients.
Genetics and pathophysiology
HCM is associated with an autosomal-dominant family of disorders affecting any of 11 genes encoding for proteins in the myocardial sarcomere.5 Hundreds of mutations have been identified that can cause HCM disease. (You’ll find an updated list at http://genetics.med.harvard.edu/~seidman/cg3/index.html.)
The disease occurs equally in men and women,6 although far more males than females die on the playing field.3 Postulated reasons for the lower death rate in female athletes include differences in training regimen; lack of participation in high-risk sports such as football; and, until recently, fewer opportunities for young women to participate in high school and college athletic programs.3 HCM has no known racial predilection, although it may be underdiagnosed in women, minorities, and underserved populations.6
Myocardial tissue is disordered
The pathophysiology of HCM directly relates to the disordered myocardial tissue arising from the various gene mutations. Left ventricular myocytes appear hypertrophied and are chaotically arranged in bizarre structural patterns.1,7 This results in asymmetrically thickened and stiff ventricular walls, which can lead to diastolic dysfunction from poor left ventricle filling.
The abnormally thickened walls and septum also push the mitral valve anteriorly during systole. In some HCM patients, the anterior motion of the mitral valve, in conjunction with septal hypertrophy, result in obstruction of the subaortic left ventricular outflow tract. This obstruction—known as left ventricular outflow tract obstruction, or LVOTO—changes with physical position as well as physiological factors such as hydration level, heart rate, and vascular preload, making the murmur and abnormal pulses associated with it hard to detect during a physical exam.6
Ischemia and scarring result
The coronary arteries of patients with HCM are often tortuous, with thickened walls and narrow lumens. This leads to uneven perfusion and areas of relative myocardial ischemia, which result in patchy scarring. The ischemia, disorganized cellular architecture, and myocardial scarring create an environment that promotes the ventricular and atrial arrhythmias that commonly complicate HCM.1
While patients with HCM may develop heart failure at any age, only a minority ever experience severe heart failure—and more than 25% of patients with HCM live beyond the age of 75.8 Overall, adults with HCM have a 1% annual mortality rate, which is similar to that of the general adult population in the United States. For adults with HCM who have had an episode of cardiac arrest or have more than 1 major risk factor for SCD (TABLE 1), the annual mortality rate climbs to 6%.1
TABLE 1
HCM: Assessing risk of sudden cardiac death6,21,33
| Major risk factors Prior cardiac arrest* Unexplained syncope Family history of SCD Left ventricular wall thickness ≥30 mm Abnormal BP response to exercise Nonsustained spontaneous ventricular tachycardia |
| Possible risk factors LVOTO Late gadolinium enhancement on MRI Myocardial ischemia Specific troponin T and I mutations Intense physical exertion Atrial fibrillation |
| *Prior cardiac arrest is the most predictive of any major factor. |
| BP, blood pressure; HCM, hypertrophic cardiomyopathy; LVOTO, left ventricular outflow tract obstruction; MRI, magnetic resonance imaging; SCD, sudden cardiac death. |
The sports physical: Frustratingly “normal”
The mainstay of screening for HCM and other cardiac abnormalities associated with exertional SCD is the preparticipation physical examination, coupled with the medical history. The physical exam itself, however, is an insensitive screening tool for this condition. That’s because most patients with HCM have nonobstructive disease, meaning there is no murmur to be heard.1 Even among HCM patients with LVOTO, the murmur may be difficult to detect. Typically, it can be heard only when the patient stands or performs the Valsalva maneuver, movements that decrease preload. Clinicians who manage to detect the murmur generally describe it as a late-systolic ejection murmur best heard at the left sternal border radiating to the aortic and mitral areas, but not into the neck.9
Resting pulses, too, are typically normal in a patient with HCM/LVOTO, although “water hammer” and double-peak pulses may be present.10 At high levels of exertion, such patients may exhibit decreased peripheral pulses and an ominous decrease in systolic blood pressure.
It’s important to realize, however, that such findings are the exception. Because most patients with HCM have normal physical exams, the medical history plays a particularly important role in pinpointing patients at risk.
It’s time to tweak your preparticipation questionnaire
In recent years, efforts have been made to improve preparticipation questionnaires.11 Despite these efforts, only 17% of the preparticipation evaluation forms currently used by US high schools contain all the recommended screening elements.12 Validated screening questions are recommended by the American College of Cardiology (ACC), American Academy of Family Physicians, and other major organizations (TABLE 2). These 9 questions address symptoms triggered by exertion, such as chest pain, palpitations, syncope, or near syncope; history of heart murmur or need for an electrocardiogram (EKG); and family history of unexplained sudden death or premature heart disease.13 Further evaluation is critical if the answers indicate a suggestive patient or family history. (See “The sports physical: Should EKG be mandatory?”.)
TABLE 2
Screening for HCM: 9 questions you need to ask13
| 1. Have you ever passed out or nearly passed out during exercise? |
| 2. Have you ever passed out or nearly passed out after exercise? |
| 3. Have you ever had discomfort, pain, or pressure in your chest during exercise? |
| 4. Does your heart race or skip beats during exercise? |
| 5. Has a doctor ever told you that you have a heart murmur? |
| 6. Has a doctor ever ordered a test for your heart (for example, EKG, echocardiogram)? |
| 7. Has anyone in your family died for no apparent reason? |
| 8. Does anyone in your family have a heart problem? |
| 9. Has any family member or relative died of heart problems or of sudden death before age 50? |
| EKG, electrocardiogram; HCM, hypertrophic cardiomyopathy. |
Because both the physical exam and medical history are imperfect screening modalities, some clinicians have proposed the 12-lead EKG as an additional HCM screening tool. In the United States, the proposal is controversial, but the debate has intensified as a result of Italy’s experience: a 90% reduction in SCD following the implementation of a national EKG screening program for young athletes.37
Advocates in the United States cite the success of the Italian model and the lack of sensitivity in the standard history-physical HCM screening. Indeed, a retrospective study of US athletes who died suddenly showed that only 3% had been identified as having HCM during the traditional preparticipation screening, and none had been disqualified.38 Opponents of universal EKG screening point to the large number of potential candidates—approximately 12 million young people, ranging in age from adolescence through college, would need to be screened. Opponents also cite differences in the Italian and American populations; cost-benefit considerations; the large number of false-positive EKGs expected (10% to 15%); and most importantly, the lack of medical personnel to perform and interpret the EKGs.39 While advances in EKG technique may minimize false-positive readings and changes in the health care system may eventually create an environment more favorable to uniform screening procedures, current recommendations for preparticipation screening call for history and physical alone.
From suspicion to diagnosis
Patients with any abnormal findings or features suggestive of HCM should be referred to a cardiologist for further work-up.13 Accompany the referral with an order for complete exercise restriction until a more detailed analysis has been completed or HCM has been ruled out.
2-dimensional echocardiogram shows LV wall hypertrophy
Diagnosis is primarily made on the basis of 2-dimensional echocardiography showing asymmetric LV wall hypertrophy without chamber dilation (in the absence of another condition that might cause hypertrophy, such as hypertension, valvular disease, or amyloidosis). The anterior septum is commonly thickened; abnormal systolic anterior motion of the mitral valve may be evident, as well.1
Although increased LV wall thickness (≥13 mm) is the most common finding in HCM—and thicknesses up to 60 mm have been recorded—this is not a universal sign. Many people with genetic evidence of HCM have normal LV wall thickness. Conversely, some patients have increased LV wall thickness but do not have HCM.
HCM or “athlete’s heart”? Mild concentric LV hypertrophy (13-14 mm)—a level of thickening often referred to as the “athlete’s heart”—may be present in healthy individuals who exercise strenuously. In borderline cases, calculation of the LV mass distribution index by 3-dimensional echocardiography has been shown to have 100% specificity in distinguishing HCM from both athlete’s heart and hypertensive cardiomyopathy.14
Cardiac magnetic resonance imaging (MRI). With gadolinium as the contrast agent, cardiac MRI is another diagnostic tool. Imaging may reveal certain delayed enhancement patterns that are highly suggestive of HCM.15 Cardiac MRI can accurately quantify LV wall thickness and LV mass distribution index and identify subtle areas of patchy LV wall thickness that echocardiography may miss.16
Ensure that family members undergo screening
When physical exam, medical history, and follow-up tests are highly suggestive of HCM, clinical screening of asymptomatic first-degree relatives is recommended. In screening family members, it is important to remember that a normal physical exam, echocardiogram, and EKG do not definitively rule out HCM, as many people who have genetic mutations for this condition do not develop physical abnormalities until they reach adulthood.1 In such cases, genetic screening—the most definitive means of HCM diagnosis—may be considered, in consultation with a specialist.
Recognize the limits of genetic testing. Genetic testing is not universally recommended, however, for a number of reasons. Cost (about $3000) is a key factor. In addition, the test for HCM is not widely available. Nor is it highly sensitive, identifying only 50% to 60% of those with genetic mutations associated with HCM.4 What’s more, the presence of a genetic mutation does not guarantee that cardiac abnormalities will ever develop. Lifestyle, coexisting hypertension, and modifier genes may all play a role in determining whether an individual is affected.1,4
Provide follow-up. When genetic screening is not available, is declined, or is negative, stress the need for periodic clinical follow-up of family members. If the first-degree relative is an adolescent, he or she will need a history and physical examination, 12-lead EKG, and 2-dimensional echocardiography annually from the age of 12 to 18 years. If the relative is older than 18, he or she should be evaluated every 5 years.6
For patients themselves, SCD risk assessment is the next step
While family physicians may be involved in the care of a patient with HCM, an assessment of the individual’s risk for SCD is best done by a specialist. Risk stratification is typically based on the presence (or absence) of LVOTO, atrial fibrillation (AF), and heart failure.
LVOTO. Defined as a subaortic gradient of 30 mm Hg or more, LVOTO is present at rest in 25% of HCM patients.17 Because the obstruction is typically dynamic, treadmill or bicycle exercise testing in conjunction with Doppler echocardiography may be needed to identify it.6 LVOTO is a strong risk factor for death due to heart failure or stroke (relative risk [RR], 4.4, compared with HCM patients who do not have LVOTO) and death from any HCM-related cause (RR=2.0). Patients with LVOTO and left atrial enlargement are also at increased risk for bacterial infective endocarditis.18
AF, which occurs in approximately 25% of those with HCM and is more common in patients with LVOTO,19 often presents clinically as acute heart failure because of the reduced diastolic filling. Although AF is not as ominous as ventricular arrhythmia, it increases the risk for embolic stroke, the likelihood of severe functional disability, and the risk of death from HCM.19
Heart failure. This is the most common complication of HCM. In some cases, patients progress to a dilated cardiomyopathy that resembles classic systolic heart failure—and responds well to conventional treatments for systolic failure. More often, the condition more closely resembles diastolic failure and responds best to negative inotropic agents and the avoidance of volume depletion, both of which increase cardiac filling.20
Consensus guidelines weigh in on SCD risk
Age is another consideration in risk stratification for SCD, which primarily strikes those with HCM in adolescence or early adulthood. Consensus guidelines from the American College of Cardiology (ACC), American Heart Association (AHA), and European Society of Cardiology (ESC)21 (TABLE 1) offer additional considerations in assessing SCD risk.
Risk factors identified as “major” in the consensus guidelines include unexplained syncope, family history of premature cardiac death, left ventricular wall thickness ≥30 mm, abnormal blood pressure response to exercise, and nonsustained ventricular tachycardia, as well as a prior episode of cardiac arrest—the single most predictive risk factor.22 Because of the high risk of sudden death, exercise is absolutely contraindicated for many patients with certain HCM phenotypes and major risks.
The organizations also cite “possible” risk factors, and indicate in consensus statements that patients with more than 1 major or other possible risk factors are at higher risk for SCD.6,21 In cohort studies, however, other than a prior episode of cardiac arrest, no other risk factor has been found to predict SCD.
Cardiac MRI, discussed earlier for diagnostic purposes, may also have a role in stratifying risk. In small studies conducted recently, the presence of myocardial fibrosis (as demonstrated by delayed gadolinium enhancement) correlates with increased risk of nonsustained ventricular tachycardia (RR, 1.6, compared with HCM patients without myocardial fibrosis).23
HCM management: Pharmacologic and surgical options
There are no large-scale studies of medical treatments for HCM, and therapy is largely empiric and individualized based on complications, symptoms, and risk (FIGURE).24
FIGURE
HCM: A guide to screening, diagnosis, and treatment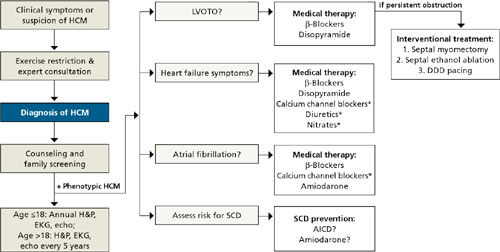
*Relatively contraindicated in LVOTO patients.
AICD, automatic implantable cardioverter defibrillator; DDD, dual-chamber; echo, echocardiography; EKG, electrocardiography; HCM, hypertrophic cardiomyopathy; H&P, history and physical; LVOTO, left ventricular outflow tract obstruction; SCD, sudden cardiac death.
Adapted from: Soor GS, et al. J Clin Pathol.4
Which drugs for which patients?
For those with symptoms of heart failure, beta-blockers are first-line therapy. Additional therapeutic options for patients without LVOTO include calcium channel blockers, nitrates, and diuretics. But these additional therapies are relatively contraindicated in patients with LVOTO. For LVOTO patients, disopyramide can be added to the beta-blocker regimen, if needed for symptom control.4,24
Patients with LVOTO or abnormal mitral motion are at moderate risk of spontaneous bacterial endocarditis (SBE).18 But evolving evidence shows low baseline rates of SBE and increased complications in patients routinely receiving antimicrobial prophylaxis, so the most recent guidelines do not recommend such treatment for any HCM patient.25
Amiodarone is effective for atrial fibrillation in HCM when beta- or calcium channel blockers fail to provide sufficient rate control.4 Amiodarone can also be used to prevent SCD.26 Recent data show that an automatic implantable cardioverter defibrillator (AICD)—which we’ll discuss later—is superior to amiodarone in preventing SCD,27 but the drug may be used in addition to an AICD or given to patients who are not candidates for, or not interested in, an implantable device.22,24
Invasive treatments may be considered for patients with LVOTO who do not respond to medical management.
Septal myomectomy or ethanol ablation: Which is better?
Septal myomectomy is the gold standard for refractory LVOTO.28 This open-heart procedure, which involves the resection of a portion of the septum to remove the obstructing cardiac tissue, has an operative mortality rate <1%.28 Surgical complication rates are also low, but include aortic regurgitation, left bundle or complete heart block, and iatrogenic ventricular septal defect.
Septal ethanol ablation is a percutaneous alternative to surgical myomectomy. In this minimally invasive procedure, ethanol is injected into the first septal perforating branch of the left anterior descending (LAD) artery, resulting in myocardial necrosis and septal wall thinning, which relieves the obstruction. Complications include ablation of inappropriate myocardium, heart block, pericardial effusion, and LAD dissection.29 Maximum effects of the ablation are delayed, typically occurring 6 to 12 months after the procedure.
Although limited by a lack of randomized controlled trials, a recent meta-analysis found surgical myomectomy and ethanol ablation to be equally effective in decreasing the LV outflow gradient to <20 mm Hg. Notably, though, surgical myomectomy reduced the gradient to nearly 10 mm Hg, compared with an average of 18 mm Hg for ethanol ablation.28 What’s more, several studies have found a higher incidence of complete heart block in patients who had ethanol ablation compared with those who underwent myomectomy.30 For patients who cannot tolerate or are not interested in invasive surgery, ablation offers an effective option.
Dual-chamber (DDD) pacing can also be used to treat LVOTO, but studies comparing pacing with myomectomy and ablation have found mixed results.6 Despite recent data showing the benefits of pacing in HCM,31 DDD pacing is typically reserved for patients who are not candidates for either surgical myomectomy or ablation.4
AICDs for which patients? It’s not always clear
AICDs effectively prevent SCD in patients with HCM,1,6,22 but the substantial cost and high rate of complications (>36%) make the devices impractical and inadvisable for universal use. Adverse events include pneumothorax, pericardial effusion, device infection or malfunction, and physical and psychological sequelae from inappropriate shocks.32 In fact, several studies of AICDs in patients with HCM have found the yearly rate of inappropriate shocks to be higher than the rate of appropriate discharges.22,31 And, because HCM patients are typically decades younger than coronary disease patients when they undergo implantation, they have a significantly higher burden of complication.32
Consensus statements vs actual practice. The central question of HCM risk stratification is how to identify patients at risk of SCD, thereby making it possible for them to reap the benefits of an AICD and drug treatment while sparing low-risk candidates the morbidity and the expense. So far, that question has not been definitively answered. As noted earlier, consensus statements agree that patients with more than 1 major risk factor have a higher risk of SCD6,28 than those with only 1; yet many patients with a single risk factor (and no prior cardiac arrest) have received AICDs.33
Studies highlight limitations of consensus guidelines’ assessment of risk. In recent case studies of HCM patients with AICDs based on registry data, roughly 25% of those studied22 received AICDs for secondary prevention—that is, after surviving cardiac arrest; the rest received them for primary prevention based on clinical risk factors. Rates of appropriate AICD discharge were 3-fold higher in patients who had survived previous cardiac arrest than in those who had not,22,32 a finding that supports aggressive AICD implantation among these high-risk patients.
Among patients who had received AICDs for primary prevention, however, appropriate discharges occurred at statistically identical rates whether they had 1, 2, or 3 major risk factors. Further, there was no association between the number of risk factors and the likelihood of appropriate discharge. Given these results, the decision to use an AICD in an HCM patient for primary prevention should be made after careful consultation with the patient and an HCM disease specialist.
What to tell patients about sports activities
Just as there is no definitive means of deciding when, or whether, a patient who has never experienced cardiac arrest should receive an AICD, there is no clear, evidence-based consensus on exercise restriction. Recommendations, which are based on expert opinion, leave room for individualized decision-making.
For those with genetic mutations consistent with HCM but no associated cardiac abnormalities and no family history of sudden death, no objective data support exercise limitations.34 For such patients, education regarding warning signs and symptoms and annual follow-up should be sufficient.
For athletes with a probable or unequivocal diagnosis of HCM, the ACC recommends restriction from competitive sports, with the possible exception of low-intensity activities such as billiards, bowling, and golf.35 This recommendation is not dependent on the presence of LVOTO or on patient symptoms, medical or surgical therapy, or the placement of an AICD.
A consensus statement from the ESC also lists recreational doubles tennis and biking, lap swimming, and weight lifting as permissible activities for patients with probable or unequivocal HCM, with a cautionary note about avoiding the Valsalva maneuver.36 The society discourages those with HCM from engaging in any activity that provokes dyspnea. Primary care physicians, too, must be sure that young patients with this condition understand the importance of avoiding intense exertion—and immediately stopping any physical activity if they notice any signs or symptoms associated with HCM.
Disclosure
The views expressed here are those of the authors and do not represent the policy of the United States Air Force, United States Army, or Department of Defense.
CORRESPONDENCE Anthony Beutler, MD, FAAFP, Department of Family Medicine A-1038, Uniformed Services University, 4301 Jones Bridge Road, Bethesda, MD 20814; [email protected]
1. Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308-1320.
2. Maron BJ. Hypertrophic cardiomyopathy and other causes of sudden cardiac death in young competitive athletes, with considerations for preparticipation screening and criteria for disqualification. Cardiol Clin. 2007;25:399-414.
3. McKenna W, Deanfield J, Faruqui A, et al. Prognosis in hypertrophic cardiomyopathy: role of age and clinical, electrocardiographic and hemodynamic features. Am J Cardiol. 1981;47:532-538.
4. Soor GS, Luk A, Ahn E, et al. Hypertrophic cardiomyopathy: current understanding and treatment objectives. J Clin Pathol. 2009;62:226-235.
5. Watkins H, Ashrafian H, McKenna WJ. The genetics of hypertrophic cardiomyopathy: Teare redux. Heart. 2008;94:1264-1268.
6. Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687-1713.
7. Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;2:1-8.
8. Maron BJ, Casey SA, Poliac LC, et al. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA. 1999;281:650-655.
9. Makenna W. Diseases of the Myocardium and Endocardium. Goldman L, Ausiello D, ed. Philadelphia: Saunders; 2008.
10. Giese EA, O’Connor FG, Brennan FH, et al. The athletic preparticipation evaluation: cardiovascular assessment. Am Fam Physician. 2007;75:1008-1114.
11. Glover DW, Maron BJ. Evolution in the process of screening United States high school student-athletes for cardiovascular disease. Am J Cardiol. 2007;100:1709-1712.
12. Rauch CM, Phillips GC. Adherence to guidelines for cardiovascular screening in current high school preparticipation evaluation forms. J Pediatr. 2009;155:584-586.
13. American Academy of Family Physicians, American Academy of Pediatrics, American Medical Society for Sports Medicine, American Osteopathic Society for Sports Medicine. The Preparticipation Physical Evaluation. 3rd ed. Minneapolis, Minn: McGrawHill; 2005:19–23.
14. Caselli S, Pelliccia A, Maron M, et al. Differentiation of hypertrophic cardiomyopathy from other forms of left ventricular hypertrophy by means of three-dimensional echocardiography. Am J Cardiol. 2008;102:616-620.
15. Olivotto I, Maron MS, Autore C, et al. Assessment and significance of left ventricular mass by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;52:559-566.
16. Maron MS, Maron BJ, Harrigan C, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. 2009;54:220-228.
17. Maron MS, Olivotto I, Betocchi S, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003;348:295-303.
18. Spirito P, Rapezzi C, Bellone P, et al. Infective endocarditis in hypertrophic cardiomyopathy: prevalence, incidence, and indications for antibiotic prophylaxis. Circulation. 1999;99:2132-2137.
19. Olivotto I, Cecchi F, Casey SA, et al. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation. 2001;104:2517-2524.
20. Spirito P, Seidman CE, McKenna WJ, et al. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775-785.
21. Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death—executive summary: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Eur Heart J. 2006;27:2099-2140.
22. Maron BJ, Spirito P, Shen WK, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405-412.
23. Nazarian S, Lima JA. Cardiovascular magnetic resonance for risk stratification of arrhythmia in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;51:1375-1376.
24. Fifer MA, Vlahakes GJ. Management of symptoms in hypertrophic cardiomyopathy. Circulation. 2008;117:429-439.
25. Nishimura RA, Carabello BA, Faxon DP, et al. ACC/AHA 2008 Guideline update on valvular heart disease: focused update on infective endocarditis: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;52:676-685.
26. McKenna WJ, Oakley CM, Krikler DM, et al. Improved survival with amiodarone in patients with hypertrophic cardiomyopathy and ventricular tachycardia. Br Heart J. 1985;53:412-416.
27. Maron BJ, Shen WK, Link MS, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000;343:365-373.
28. Alam M, Dokainish H, Lakkis NM. Hypertrophic obstructive cardiomyopathy-alcohol septal ablation vs. myectomy: a meta-analysis. Eur Heart J. 2009;30:1080-1087.
29. Alam M, Dokainish H, Lakkis N. Alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a systematic review of published studies. J Interv Cardiol. 2006;19:319-327.
30. Fernandes VL, Nielsen C, Nagueh SF, et al. Follow-up of alcohol septal ablation for symptomatic hypertrophic obstructive cardiomyopathy the Baylor and Medical University of South Carolina experience 1996 to 2007. JACC Cardiovasc Interv. 2008;1:561-570.
31. Galve E, Sambola A, Saldana G, et al. Late benefits of dual-chamber pacing in obstructive hypertrophic cardiomyopathy. A 10-year follow-up study. Heart. 2009; May 28 [E-pub ahead of print.]
32. Lin G, Nishimura RA, Gersh BJ, et al. Device complications and inappropriate implantable cardioverter defibrillator shocks in patients with hypertrophic cardiomyopathy. Heart. 2009;95:709-714.
33. Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212-2218.
34. Elliott P, Spirito P. Prevention of hypertrophic cardiomyopathy-related deaths: theory and practice. Heart. 2008;94:1269-1275.
35. Maron BJ, Ackerman MJ, Nishimura RA, et al. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol. 2005;45:1340-1345.
36. Pelliccia A, Fagard R, Bjornstad HH, et al. Recommendations for competitive sports participation in athletes with cardiovascular disease: a consensus document from the Study Group of Sports Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur Heart J. 2005;26:1422-1445.
37. Corrado D, Basso C, Pavei A, et al. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006;296:1593-1601.
38. Maron BJ, Shirani J, Poliac LC, et al. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. JAMA. 1996;276:199-204.
39. Maron BJ, Thompson PD, Ackerman MJ, et al. Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: a scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: endorsed by the American College of Cardiology Foundation. Circulation. 2007;115:1643-1655.
• Screen for hypertrophic cardiomyopathy (HCM) during preparticipation sports physicals. C
• Patients who have symptoms of, or are diagnosed with, HCM should be cleared by a cardiologist before being allowed to participate in organized sports or engaging in physical exercise. B
• Patients with HCM and a history of cardiac arrest should be given the opportunity to receive an automatic implantable cardioverter defibrillator (AICD). A
• Beta-blockers are first-line therapy for patients with HCM who have symptoms of heart failure. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Hypertrophic cardiomyopathy (HCM) is the most common cardiac genetic disorder.1 But it is best known for its tragic outcomes—the sudden cardiac death (SCD) of young athletes, many of whom are in high school or college.
In fact, HCM can present at any time from infancy through old age, and has a broad range of manifestations. Some individuals remain asymptomatic throughout their lives and enjoy a normal life span; others experience a range of symptoms, from chest pain and dyspnea on exertion to dizziness, light-headedness, palpitations, and syncope. Diagnosis is difficult, and predicting which patients will develop severe complications and face the greatest risk of premature SCD remains an inexact science.
What is clear is that in the United States, HCM causes SCD far more than any other condition.2 HCM has been positively identified in well over a third of cases (36%) of SCD in athletes under the age of 30, and cited as a possible cause in another 8%. Coronary artery anomalies is a distant second, responsible for 17% of sudden deaths in this patient population; myocarditis follows, at just 6%.2
We also know that HCM itself occurs much more frequently than previously believed. In the early 1980s, its prevalence was estimated at <1 in 100,000;3 today, HCM is believed to affect approximately 1 in 500 adults.2,4 Because this potentially fatal condition often remains undetected until an athlete collapses on the playing field, it is crucial that family physicians—who conduct many of the sports physicals typically required before students are permitted to play competitive sports—maintain a high degree of suspicion.
Knowing what signs and symptoms to look for and, perhaps more importantly, the key questions to ask, will help you identify young athletes at risk. This update, which begins with a review of the genetic basis and pathophysiology of HCM, will prepare you to take steps to protect your young patients.
Genetics and pathophysiology
HCM is associated with an autosomal-dominant family of disorders affecting any of 11 genes encoding for proteins in the myocardial sarcomere.5 Hundreds of mutations have been identified that can cause HCM disease. (You’ll find an updated list at http://genetics.med.harvard.edu/~seidman/cg3/index.html.)
The disease occurs equally in men and women,6 although far more males than females die on the playing field.3 Postulated reasons for the lower death rate in female athletes include differences in training regimen; lack of participation in high-risk sports such as football; and, until recently, fewer opportunities for young women to participate in high school and college athletic programs.3 HCM has no known racial predilection, although it may be underdiagnosed in women, minorities, and underserved populations.6
Myocardial tissue is disordered
The pathophysiology of HCM directly relates to the disordered myocardial tissue arising from the various gene mutations. Left ventricular myocytes appear hypertrophied and are chaotically arranged in bizarre structural patterns.1,7 This results in asymmetrically thickened and stiff ventricular walls, which can lead to diastolic dysfunction from poor left ventricle filling.
The abnormally thickened walls and septum also push the mitral valve anteriorly during systole. In some HCM patients, the anterior motion of the mitral valve, in conjunction with septal hypertrophy, result in obstruction of the subaortic left ventricular outflow tract. This obstruction—known as left ventricular outflow tract obstruction, or LVOTO—changes with physical position as well as physiological factors such as hydration level, heart rate, and vascular preload, making the murmur and abnormal pulses associated with it hard to detect during a physical exam.6
Ischemia and scarring result
The coronary arteries of patients with HCM are often tortuous, with thickened walls and narrow lumens. This leads to uneven perfusion and areas of relative myocardial ischemia, which result in patchy scarring. The ischemia, disorganized cellular architecture, and myocardial scarring create an environment that promotes the ventricular and atrial arrhythmias that commonly complicate HCM.1
While patients with HCM may develop heart failure at any age, only a minority ever experience severe heart failure—and more than 25% of patients with HCM live beyond the age of 75.8 Overall, adults with HCM have a 1% annual mortality rate, which is similar to that of the general adult population in the United States. For adults with HCM who have had an episode of cardiac arrest or have more than 1 major risk factor for SCD (TABLE 1), the annual mortality rate climbs to 6%.1
TABLE 1
HCM: Assessing risk of sudden cardiac death6,21,33
| Major risk factors Prior cardiac arrest* Unexplained syncope Family history of SCD Left ventricular wall thickness ≥30 mm Abnormal BP response to exercise Nonsustained spontaneous ventricular tachycardia |
| Possible risk factors LVOTO Late gadolinium enhancement on MRI Myocardial ischemia Specific troponin T and I mutations Intense physical exertion Atrial fibrillation |
| *Prior cardiac arrest is the most predictive of any major factor. |
| BP, blood pressure; HCM, hypertrophic cardiomyopathy; LVOTO, left ventricular outflow tract obstruction; MRI, magnetic resonance imaging; SCD, sudden cardiac death. |
The sports physical: Frustratingly “normal”
The mainstay of screening for HCM and other cardiac abnormalities associated with exertional SCD is the preparticipation physical examination, coupled with the medical history. The physical exam itself, however, is an insensitive screening tool for this condition. That’s because most patients with HCM have nonobstructive disease, meaning there is no murmur to be heard.1 Even among HCM patients with LVOTO, the murmur may be difficult to detect. Typically, it can be heard only when the patient stands or performs the Valsalva maneuver, movements that decrease preload. Clinicians who manage to detect the murmur generally describe it as a late-systolic ejection murmur best heard at the left sternal border radiating to the aortic and mitral areas, but not into the neck.9
Resting pulses, too, are typically normal in a patient with HCM/LVOTO, although “water hammer” and double-peak pulses may be present.10 At high levels of exertion, such patients may exhibit decreased peripheral pulses and an ominous decrease in systolic blood pressure.
It’s important to realize, however, that such findings are the exception. Because most patients with HCM have normal physical exams, the medical history plays a particularly important role in pinpointing patients at risk.
It’s time to tweak your preparticipation questionnaire
In recent years, efforts have been made to improve preparticipation questionnaires.11 Despite these efforts, only 17% of the preparticipation evaluation forms currently used by US high schools contain all the recommended screening elements.12 Validated screening questions are recommended by the American College of Cardiology (ACC), American Academy of Family Physicians, and other major organizations (TABLE 2). These 9 questions address symptoms triggered by exertion, such as chest pain, palpitations, syncope, or near syncope; history of heart murmur or need for an electrocardiogram (EKG); and family history of unexplained sudden death or premature heart disease.13 Further evaluation is critical if the answers indicate a suggestive patient or family history. (See “The sports physical: Should EKG be mandatory?”.)
TABLE 2
Screening for HCM: 9 questions you need to ask13
| 1. Have you ever passed out or nearly passed out during exercise? |
| 2. Have you ever passed out or nearly passed out after exercise? |
| 3. Have you ever had discomfort, pain, or pressure in your chest during exercise? |
| 4. Does your heart race or skip beats during exercise? |
| 5. Has a doctor ever told you that you have a heart murmur? |
| 6. Has a doctor ever ordered a test for your heart (for example, EKG, echocardiogram)? |
| 7. Has anyone in your family died for no apparent reason? |
| 8. Does anyone in your family have a heart problem? |
| 9. Has any family member or relative died of heart problems or of sudden death before age 50? |
| EKG, electrocardiogram; HCM, hypertrophic cardiomyopathy. |
Because both the physical exam and medical history are imperfect screening modalities, some clinicians have proposed the 12-lead EKG as an additional HCM screening tool. In the United States, the proposal is controversial, but the debate has intensified as a result of Italy’s experience: a 90% reduction in SCD following the implementation of a national EKG screening program for young athletes.37
Advocates in the United States cite the success of the Italian model and the lack of sensitivity in the standard history-physical HCM screening. Indeed, a retrospective study of US athletes who died suddenly showed that only 3% had been identified as having HCM during the traditional preparticipation screening, and none had been disqualified.38 Opponents of universal EKG screening point to the large number of potential candidates—approximately 12 million young people, ranging in age from adolescence through college, would need to be screened. Opponents also cite differences in the Italian and American populations; cost-benefit considerations; the large number of false-positive EKGs expected (10% to 15%); and most importantly, the lack of medical personnel to perform and interpret the EKGs.39 While advances in EKG technique may minimize false-positive readings and changes in the health care system may eventually create an environment more favorable to uniform screening procedures, current recommendations for preparticipation screening call for history and physical alone.
From suspicion to diagnosis
Patients with any abnormal findings or features suggestive of HCM should be referred to a cardiologist for further work-up.13 Accompany the referral with an order for complete exercise restriction until a more detailed analysis has been completed or HCM has been ruled out.
2-dimensional echocardiogram shows LV wall hypertrophy
Diagnosis is primarily made on the basis of 2-dimensional echocardiography showing asymmetric LV wall hypertrophy without chamber dilation (in the absence of another condition that might cause hypertrophy, such as hypertension, valvular disease, or amyloidosis). The anterior septum is commonly thickened; abnormal systolic anterior motion of the mitral valve may be evident, as well.1
Although increased LV wall thickness (≥13 mm) is the most common finding in HCM—and thicknesses up to 60 mm have been recorded—this is not a universal sign. Many people with genetic evidence of HCM have normal LV wall thickness. Conversely, some patients have increased LV wall thickness but do not have HCM.
HCM or “athlete’s heart”? Mild concentric LV hypertrophy (13-14 mm)—a level of thickening often referred to as the “athlete’s heart”—may be present in healthy individuals who exercise strenuously. In borderline cases, calculation of the LV mass distribution index by 3-dimensional echocardiography has been shown to have 100% specificity in distinguishing HCM from both athlete’s heart and hypertensive cardiomyopathy.14
Cardiac magnetic resonance imaging (MRI). With gadolinium as the contrast agent, cardiac MRI is another diagnostic tool. Imaging may reveal certain delayed enhancement patterns that are highly suggestive of HCM.15 Cardiac MRI can accurately quantify LV wall thickness and LV mass distribution index and identify subtle areas of patchy LV wall thickness that echocardiography may miss.16
Ensure that family members undergo screening
When physical exam, medical history, and follow-up tests are highly suggestive of HCM, clinical screening of asymptomatic first-degree relatives is recommended. In screening family members, it is important to remember that a normal physical exam, echocardiogram, and EKG do not definitively rule out HCM, as many people who have genetic mutations for this condition do not develop physical abnormalities until they reach adulthood.1 In such cases, genetic screening—the most definitive means of HCM diagnosis—may be considered, in consultation with a specialist.
Recognize the limits of genetic testing. Genetic testing is not universally recommended, however, for a number of reasons. Cost (about $3000) is a key factor. In addition, the test for HCM is not widely available. Nor is it highly sensitive, identifying only 50% to 60% of those with genetic mutations associated with HCM.4 What’s more, the presence of a genetic mutation does not guarantee that cardiac abnormalities will ever develop. Lifestyle, coexisting hypertension, and modifier genes may all play a role in determining whether an individual is affected.1,4
Provide follow-up. When genetic screening is not available, is declined, or is negative, stress the need for periodic clinical follow-up of family members. If the first-degree relative is an adolescent, he or she will need a history and physical examination, 12-lead EKG, and 2-dimensional echocardiography annually from the age of 12 to 18 years. If the relative is older than 18, he or she should be evaluated every 5 years.6
For patients themselves, SCD risk assessment is the next step
While family physicians may be involved in the care of a patient with HCM, an assessment of the individual’s risk for SCD is best done by a specialist. Risk stratification is typically based on the presence (or absence) of LVOTO, atrial fibrillation (AF), and heart failure.
LVOTO. Defined as a subaortic gradient of 30 mm Hg or more, LVOTO is present at rest in 25% of HCM patients.17 Because the obstruction is typically dynamic, treadmill or bicycle exercise testing in conjunction with Doppler echocardiography may be needed to identify it.6 LVOTO is a strong risk factor for death due to heart failure or stroke (relative risk [RR], 4.4, compared with HCM patients who do not have LVOTO) and death from any HCM-related cause (RR=2.0). Patients with LVOTO and left atrial enlargement are also at increased risk for bacterial infective endocarditis.18
AF, which occurs in approximately 25% of those with HCM and is more common in patients with LVOTO,19 often presents clinically as acute heart failure because of the reduced diastolic filling. Although AF is not as ominous as ventricular arrhythmia, it increases the risk for embolic stroke, the likelihood of severe functional disability, and the risk of death from HCM.19
Heart failure. This is the most common complication of HCM. In some cases, patients progress to a dilated cardiomyopathy that resembles classic systolic heart failure—and responds well to conventional treatments for systolic failure. More often, the condition more closely resembles diastolic failure and responds best to negative inotropic agents and the avoidance of volume depletion, both of which increase cardiac filling.20
Consensus guidelines weigh in on SCD risk
Age is another consideration in risk stratification for SCD, which primarily strikes those with HCM in adolescence or early adulthood. Consensus guidelines from the American College of Cardiology (ACC), American Heart Association (AHA), and European Society of Cardiology (ESC)21 (TABLE 1) offer additional considerations in assessing SCD risk.
Risk factors identified as “major” in the consensus guidelines include unexplained syncope, family history of premature cardiac death, left ventricular wall thickness ≥30 mm, abnormal blood pressure response to exercise, and nonsustained ventricular tachycardia, as well as a prior episode of cardiac arrest—the single most predictive risk factor.22 Because of the high risk of sudden death, exercise is absolutely contraindicated for many patients with certain HCM phenotypes and major risks.
The organizations also cite “possible” risk factors, and indicate in consensus statements that patients with more than 1 major or other possible risk factors are at higher risk for SCD.6,21 In cohort studies, however, other than a prior episode of cardiac arrest, no other risk factor has been found to predict SCD.
Cardiac MRI, discussed earlier for diagnostic purposes, may also have a role in stratifying risk. In small studies conducted recently, the presence of myocardial fibrosis (as demonstrated by delayed gadolinium enhancement) correlates with increased risk of nonsustained ventricular tachycardia (RR, 1.6, compared with HCM patients without myocardial fibrosis).23
HCM management: Pharmacologic and surgical options
There are no large-scale studies of medical treatments for HCM, and therapy is largely empiric and individualized based on complications, symptoms, and risk (FIGURE).24
FIGURE
HCM: A guide to screening, diagnosis, and treatment
*Relatively contraindicated in LVOTO patients.
AICD, automatic implantable cardioverter defibrillator; DDD, dual-chamber; echo, echocardiography; EKG, electrocardiography; HCM, hypertrophic cardiomyopathy; H&P, history and physical; LVOTO, left ventricular outflow tract obstruction; SCD, sudden cardiac death.
Adapted from: Soor GS, et al. J Clin Pathol.4
Which drugs for which patients?
For those with symptoms of heart failure, beta-blockers are first-line therapy. Additional therapeutic options for patients without LVOTO include calcium channel blockers, nitrates, and diuretics. But these additional therapies are relatively contraindicated in patients with LVOTO. For LVOTO patients, disopyramide can be added to the beta-blocker regimen, if needed for symptom control.4,24
Patients with LVOTO or abnormal mitral motion are at moderate risk of spontaneous bacterial endocarditis (SBE).18 But evolving evidence shows low baseline rates of SBE and increased complications in patients routinely receiving antimicrobial prophylaxis, so the most recent guidelines do not recommend such treatment for any HCM patient.25
Amiodarone is effective for atrial fibrillation in HCM when beta- or calcium channel blockers fail to provide sufficient rate control.4 Amiodarone can also be used to prevent SCD.26 Recent data show that an automatic implantable cardioverter defibrillator (AICD)—which we’ll discuss later—is superior to amiodarone in preventing SCD,27 but the drug may be used in addition to an AICD or given to patients who are not candidates for, or not interested in, an implantable device.22,24
Invasive treatments may be considered for patients with LVOTO who do not respond to medical management.
Septal myomectomy or ethanol ablation: Which is better?
Septal myomectomy is the gold standard for refractory LVOTO.28 This open-heart procedure, which involves the resection of a portion of the septum to remove the obstructing cardiac tissue, has an operative mortality rate <1%.28 Surgical complication rates are also low, but include aortic regurgitation, left bundle or complete heart block, and iatrogenic ventricular septal defect.
Septal ethanol ablation is a percutaneous alternative to surgical myomectomy. In this minimally invasive procedure, ethanol is injected into the first septal perforating branch of the left anterior descending (LAD) artery, resulting in myocardial necrosis and septal wall thinning, which relieves the obstruction. Complications include ablation of inappropriate myocardium, heart block, pericardial effusion, and LAD dissection.29 Maximum effects of the ablation are delayed, typically occurring 6 to 12 months after the procedure.
Although limited by a lack of randomized controlled trials, a recent meta-analysis found surgical myomectomy and ethanol ablation to be equally effective in decreasing the LV outflow gradient to <20 mm Hg. Notably, though, surgical myomectomy reduced the gradient to nearly 10 mm Hg, compared with an average of 18 mm Hg for ethanol ablation.28 What’s more, several studies have found a higher incidence of complete heart block in patients who had ethanol ablation compared with those who underwent myomectomy.30 For patients who cannot tolerate or are not interested in invasive surgery, ablation offers an effective option.
Dual-chamber (DDD) pacing can also be used to treat LVOTO, but studies comparing pacing with myomectomy and ablation have found mixed results.6 Despite recent data showing the benefits of pacing in HCM,31 DDD pacing is typically reserved for patients who are not candidates for either surgical myomectomy or ablation.4
AICDs for which patients? It’s not always clear
AICDs effectively prevent SCD in patients with HCM,1,6,22 but the substantial cost and high rate of complications (>36%) make the devices impractical and inadvisable for universal use. Adverse events include pneumothorax, pericardial effusion, device infection or malfunction, and physical and psychological sequelae from inappropriate shocks.32 In fact, several studies of AICDs in patients with HCM have found the yearly rate of inappropriate shocks to be higher than the rate of appropriate discharges.22,31 And, because HCM patients are typically decades younger than coronary disease patients when they undergo implantation, they have a significantly higher burden of complication.32
Consensus statements vs actual practice. The central question of HCM risk stratification is how to identify patients at risk of SCD, thereby making it possible for them to reap the benefits of an AICD and drug treatment while sparing low-risk candidates the morbidity and the expense. So far, that question has not been definitively answered. As noted earlier, consensus statements agree that patients with more than 1 major risk factor have a higher risk of SCD6,28 than those with only 1; yet many patients with a single risk factor (and no prior cardiac arrest) have received AICDs.33
Studies highlight limitations of consensus guidelines’ assessment of risk. In recent case studies of HCM patients with AICDs based on registry data, roughly 25% of those studied22 received AICDs for secondary prevention—that is, after surviving cardiac arrest; the rest received them for primary prevention based on clinical risk factors. Rates of appropriate AICD discharge were 3-fold higher in patients who had survived previous cardiac arrest than in those who had not,22,32 a finding that supports aggressive AICD implantation among these high-risk patients.
Among patients who had received AICDs for primary prevention, however, appropriate discharges occurred at statistically identical rates whether they had 1, 2, or 3 major risk factors. Further, there was no association between the number of risk factors and the likelihood of appropriate discharge. Given these results, the decision to use an AICD in an HCM patient for primary prevention should be made after careful consultation with the patient and an HCM disease specialist.
What to tell patients about sports activities
Just as there is no definitive means of deciding when, or whether, a patient who has never experienced cardiac arrest should receive an AICD, there is no clear, evidence-based consensus on exercise restriction. Recommendations, which are based on expert opinion, leave room for individualized decision-making.
For those with genetic mutations consistent with HCM but no associated cardiac abnormalities and no family history of sudden death, no objective data support exercise limitations.34 For such patients, education regarding warning signs and symptoms and annual follow-up should be sufficient.
For athletes with a probable or unequivocal diagnosis of HCM, the ACC recommends restriction from competitive sports, with the possible exception of low-intensity activities such as billiards, bowling, and golf.35 This recommendation is not dependent on the presence of LVOTO or on patient symptoms, medical or surgical therapy, or the placement of an AICD.
A consensus statement from the ESC also lists recreational doubles tennis and biking, lap swimming, and weight lifting as permissible activities for patients with probable or unequivocal HCM, with a cautionary note about avoiding the Valsalva maneuver.36 The society discourages those with HCM from engaging in any activity that provokes dyspnea. Primary care physicians, too, must be sure that young patients with this condition understand the importance of avoiding intense exertion—and immediately stopping any physical activity if they notice any signs or symptoms associated with HCM.
Disclosure
The views expressed here are those of the authors and do not represent the policy of the United States Air Force, United States Army, or Department of Defense.
CORRESPONDENCE Anthony Beutler, MD, FAAFP, Department of Family Medicine A-1038, Uniformed Services University, 4301 Jones Bridge Road, Bethesda, MD 20814; [email protected]
• Screen for hypertrophic cardiomyopathy (HCM) during preparticipation sports physicals. C
• Patients who have symptoms of, or are diagnosed with, HCM should be cleared by a cardiologist before being allowed to participate in organized sports or engaging in physical exercise. B
• Patients with HCM and a history of cardiac arrest should be given the opportunity to receive an automatic implantable cardioverter defibrillator (AICD). A
• Beta-blockers are first-line therapy for patients with HCM who have symptoms of heart failure. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Hypertrophic cardiomyopathy (HCM) is the most common cardiac genetic disorder.1 But it is best known for its tragic outcomes—the sudden cardiac death (SCD) of young athletes, many of whom are in high school or college.
In fact, HCM can present at any time from infancy through old age, and has a broad range of manifestations. Some individuals remain asymptomatic throughout their lives and enjoy a normal life span; others experience a range of symptoms, from chest pain and dyspnea on exertion to dizziness, light-headedness, palpitations, and syncope. Diagnosis is difficult, and predicting which patients will develop severe complications and face the greatest risk of premature SCD remains an inexact science.
What is clear is that in the United States, HCM causes SCD far more than any other condition.2 HCM has been positively identified in well over a third of cases (36%) of SCD in athletes under the age of 30, and cited as a possible cause in another 8%. Coronary artery anomalies is a distant second, responsible for 17% of sudden deaths in this patient population; myocarditis follows, at just 6%.2
We also know that HCM itself occurs much more frequently than previously believed. In the early 1980s, its prevalence was estimated at <1 in 100,000;3 today, HCM is believed to affect approximately 1 in 500 adults.2,4 Because this potentially fatal condition often remains undetected until an athlete collapses on the playing field, it is crucial that family physicians—who conduct many of the sports physicals typically required before students are permitted to play competitive sports—maintain a high degree of suspicion.
Knowing what signs and symptoms to look for and, perhaps more importantly, the key questions to ask, will help you identify young athletes at risk. This update, which begins with a review of the genetic basis and pathophysiology of HCM, will prepare you to take steps to protect your young patients.
Genetics and pathophysiology
HCM is associated with an autosomal-dominant family of disorders affecting any of 11 genes encoding for proteins in the myocardial sarcomere.5 Hundreds of mutations have been identified that can cause HCM disease. (You’ll find an updated list at http://genetics.med.harvard.edu/~seidman/cg3/index.html.)
The disease occurs equally in men and women,6 although far more males than females die on the playing field.3 Postulated reasons for the lower death rate in female athletes include differences in training regimen; lack of participation in high-risk sports such as football; and, until recently, fewer opportunities for young women to participate in high school and college athletic programs.3 HCM has no known racial predilection, although it may be underdiagnosed in women, minorities, and underserved populations.6
Myocardial tissue is disordered
The pathophysiology of HCM directly relates to the disordered myocardial tissue arising from the various gene mutations. Left ventricular myocytes appear hypertrophied and are chaotically arranged in bizarre structural patterns.1,7 This results in asymmetrically thickened and stiff ventricular walls, which can lead to diastolic dysfunction from poor left ventricle filling.
The abnormally thickened walls and septum also push the mitral valve anteriorly during systole. In some HCM patients, the anterior motion of the mitral valve, in conjunction with septal hypertrophy, result in obstruction of the subaortic left ventricular outflow tract. This obstruction—known as left ventricular outflow tract obstruction, or LVOTO—changes with physical position as well as physiological factors such as hydration level, heart rate, and vascular preload, making the murmur and abnormal pulses associated with it hard to detect during a physical exam.6
Ischemia and scarring result
The coronary arteries of patients with HCM are often tortuous, with thickened walls and narrow lumens. This leads to uneven perfusion and areas of relative myocardial ischemia, which result in patchy scarring. The ischemia, disorganized cellular architecture, and myocardial scarring create an environment that promotes the ventricular and atrial arrhythmias that commonly complicate HCM.1
While patients with HCM may develop heart failure at any age, only a minority ever experience severe heart failure—and more than 25% of patients with HCM live beyond the age of 75.8 Overall, adults with HCM have a 1% annual mortality rate, which is similar to that of the general adult population in the United States. For adults with HCM who have had an episode of cardiac arrest or have more than 1 major risk factor for SCD (TABLE 1), the annual mortality rate climbs to 6%.1
TABLE 1
HCM: Assessing risk of sudden cardiac death6,21,33
| Major risk factors Prior cardiac arrest* Unexplained syncope Family history of SCD Left ventricular wall thickness ≥30 mm Abnormal BP response to exercise Nonsustained spontaneous ventricular tachycardia |
| Possible risk factors LVOTO Late gadolinium enhancement on MRI Myocardial ischemia Specific troponin T and I mutations Intense physical exertion Atrial fibrillation |
| *Prior cardiac arrest is the most predictive of any major factor. |
| BP, blood pressure; HCM, hypertrophic cardiomyopathy; LVOTO, left ventricular outflow tract obstruction; MRI, magnetic resonance imaging; SCD, sudden cardiac death. |
The sports physical: Frustratingly “normal”
The mainstay of screening for HCM and other cardiac abnormalities associated with exertional SCD is the preparticipation physical examination, coupled with the medical history. The physical exam itself, however, is an insensitive screening tool for this condition. That’s because most patients with HCM have nonobstructive disease, meaning there is no murmur to be heard.1 Even among HCM patients with LVOTO, the murmur may be difficult to detect. Typically, it can be heard only when the patient stands or performs the Valsalva maneuver, movements that decrease preload. Clinicians who manage to detect the murmur generally describe it as a late-systolic ejection murmur best heard at the left sternal border radiating to the aortic and mitral areas, but not into the neck.9
Resting pulses, too, are typically normal in a patient with HCM/LVOTO, although “water hammer” and double-peak pulses may be present.10 At high levels of exertion, such patients may exhibit decreased peripheral pulses and an ominous decrease in systolic blood pressure.
It’s important to realize, however, that such findings are the exception. Because most patients with HCM have normal physical exams, the medical history plays a particularly important role in pinpointing patients at risk.
It’s time to tweak your preparticipation questionnaire
In recent years, efforts have been made to improve preparticipation questionnaires.11 Despite these efforts, only 17% of the preparticipation evaluation forms currently used by US high schools contain all the recommended screening elements.12 Validated screening questions are recommended by the American College of Cardiology (ACC), American Academy of Family Physicians, and other major organizations (TABLE 2). These 9 questions address symptoms triggered by exertion, such as chest pain, palpitations, syncope, or near syncope; history of heart murmur or need for an electrocardiogram (EKG); and family history of unexplained sudden death or premature heart disease.13 Further evaluation is critical if the answers indicate a suggestive patient or family history. (See “The sports physical: Should EKG be mandatory?”.)
TABLE 2
Screening for HCM: 9 questions you need to ask13
| 1. Have you ever passed out or nearly passed out during exercise? |
| 2. Have you ever passed out or nearly passed out after exercise? |
| 3. Have you ever had discomfort, pain, or pressure in your chest during exercise? |
| 4. Does your heart race or skip beats during exercise? |
| 5. Has a doctor ever told you that you have a heart murmur? |
| 6. Has a doctor ever ordered a test for your heart (for example, EKG, echocardiogram)? |
| 7. Has anyone in your family died for no apparent reason? |
| 8. Does anyone in your family have a heart problem? |
| 9. Has any family member or relative died of heart problems or of sudden death before age 50? |
| EKG, electrocardiogram; HCM, hypertrophic cardiomyopathy. |
Because both the physical exam and medical history are imperfect screening modalities, some clinicians have proposed the 12-lead EKG as an additional HCM screening tool. In the United States, the proposal is controversial, but the debate has intensified as a result of Italy’s experience: a 90% reduction in SCD following the implementation of a national EKG screening program for young athletes.37
Advocates in the United States cite the success of the Italian model and the lack of sensitivity in the standard history-physical HCM screening. Indeed, a retrospective study of US athletes who died suddenly showed that only 3% had been identified as having HCM during the traditional preparticipation screening, and none had been disqualified.38 Opponents of universal EKG screening point to the large number of potential candidates—approximately 12 million young people, ranging in age from adolescence through college, would need to be screened. Opponents also cite differences in the Italian and American populations; cost-benefit considerations; the large number of false-positive EKGs expected (10% to 15%); and most importantly, the lack of medical personnel to perform and interpret the EKGs.39 While advances in EKG technique may minimize false-positive readings and changes in the health care system may eventually create an environment more favorable to uniform screening procedures, current recommendations for preparticipation screening call for history and physical alone.
From suspicion to diagnosis
Patients with any abnormal findings or features suggestive of HCM should be referred to a cardiologist for further work-up.13 Accompany the referral with an order for complete exercise restriction until a more detailed analysis has been completed or HCM has been ruled out.
2-dimensional echocardiogram shows LV wall hypertrophy
Diagnosis is primarily made on the basis of 2-dimensional echocardiography showing asymmetric LV wall hypertrophy without chamber dilation (in the absence of another condition that might cause hypertrophy, such as hypertension, valvular disease, or amyloidosis). The anterior septum is commonly thickened; abnormal systolic anterior motion of the mitral valve may be evident, as well.1
Although increased LV wall thickness (≥13 mm) is the most common finding in HCM—and thicknesses up to 60 mm have been recorded—this is not a universal sign. Many people with genetic evidence of HCM have normal LV wall thickness. Conversely, some patients have increased LV wall thickness but do not have HCM.
HCM or “athlete’s heart”? Mild concentric LV hypertrophy (13-14 mm)—a level of thickening often referred to as the “athlete’s heart”—may be present in healthy individuals who exercise strenuously. In borderline cases, calculation of the LV mass distribution index by 3-dimensional echocardiography has been shown to have 100% specificity in distinguishing HCM from both athlete’s heart and hypertensive cardiomyopathy.14
Cardiac magnetic resonance imaging (MRI). With gadolinium as the contrast agent, cardiac MRI is another diagnostic tool. Imaging may reveal certain delayed enhancement patterns that are highly suggestive of HCM.15 Cardiac MRI can accurately quantify LV wall thickness and LV mass distribution index and identify subtle areas of patchy LV wall thickness that echocardiography may miss.16
Ensure that family members undergo screening
When physical exam, medical history, and follow-up tests are highly suggestive of HCM, clinical screening of asymptomatic first-degree relatives is recommended. In screening family members, it is important to remember that a normal physical exam, echocardiogram, and EKG do not definitively rule out HCM, as many people who have genetic mutations for this condition do not develop physical abnormalities until they reach adulthood.1 In such cases, genetic screening—the most definitive means of HCM diagnosis—may be considered, in consultation with a specialist.
Recognize the limits of genetic testing. Genetic testing is not universally recommended, however, for a number of reasons. Cost (about $3000) is a key factor. In addition, the test for HCM is not widely available. Nor is it highly sensitive, identifying only 50% to 60% of those with genetic mutations associated with HCM.4 What’s more, the presence of a genetic mutation does not guarantee that cardiac abnormalities will ever develop. Lifestyle, coexisting hypertension, and modifier genes may all play a role in determining whether an individual is affected.1,4
Provide follow-up. When genetic screening is not available, is declined, or is negative, stress the need for periodic clinical follow-up of family members. If the first-degree relative is an adolescent, he or she will need a history and physical examination, 12-lead EKG, and 2-dimensional echocardiography annually from the age of 12 to 18 years. If the relative is older than 18, he or she should be evaluated every 5 years.6
For patients themselves, SCD risk assessment is the next step
While family physicians may be involved in the care of a patient with HCM, an assessment of the individual’s risk for SCD is best done by a specialist. Risk stratification is typically based on the presence (or absence) of LVOTO, atrial fibrillation (AF), and heart failure.
LVOTO. Defined as a subaortic gradient of 30 mm Hg or more, LVOTO is present at rest in 25% of HCM patients.17 Because the obstruction is typically dynamic, treadmill or bicycle exercise testing in conjunction with Doppler echocardiography may be needed to identify it.6 LVOTO is a strong risk factor for death due to heart failure or stroke (relative risk [RR], 4.4, compared with HCM patients who do not have LVOTO) and death from any HCM-related cause (RR=2.0). Patients with LVOTO and left atrial enlargement are also at increased risk for bacterial infective endocarditis.18
AF, which occurs in approximately 25% of those with HCM and is more common in patients with LVOTO,19 often presents clinically as acute heart failure because of the reduced diastolic filling. Although AF is not as ominous as ventricular arrhythmia, it increases the risk for embolic stroke, the likelihood of severe functional disability, and the risk of death from HCM.19
Heart failure. This is the most common complication of HCM. In some cases, patients progress to a dilated cardiomyopathy that resembles classic systolic heart failure—and responds well to conventional treatments for systolic failure. More often, the condition more closely resembles diastolic failure and responds best to negative inotropic agents and the avoidance of volume depletion, both of which increase cardiac filling.20
Consensus guidelines weigh in on SCD risk
Age is another consideration in risk stratification for SCD, which primarily strikes those with HCM in adolescence or early adulthood. Consensus guidelines from the American College of Cardiology (ACC), American Heart Association (AHA), and European Society of Cardiology (ESC)21 (TABLE 1) offer additional considerations in assessing SCD risk.
Risk factors identified as “major” in the consensus guidelines include unexplained syncope, family history of premature cardiac death, left ventricular wall thickness ≥30 mm, abnormal blood pressure response to exercise, and nonsustained ventricular tachycardia, as well as a prior episode of cardiac arrest—the single most predictive risk factor.22 Because of the high risk of sudden death, exercise is absolutely contraindicated for many patients with certain HCM phenotypes and major risks.
The organizations also cite “possible” risk factors, and indicate in consensus statements that patients with more than 1 major or other possible risk factors are at higher risk for SCD.6,21 In cohort studies, however, other than a prior episode of cardiac arrest, no other risk factor has been found to predict SCD.
Cardiac MRI, discussed earlier for diagnostic purposes, may also have a role in stratifying risk. In small studies conducted recently, the presence of myocardial fibrosis (as demonstrated by delayed gadolinium enhancement) correlates with increased risk of nonsustained ventricular tachycardia (RR, 1.6, compared with HCM patients without myocardial fibrosis).23
HCM management: Pharmacologic and surgical options
There are no large-scale studies of medical treatments for HCM, and therapy is largely empiric and individualized based on complications, symptoms, and risk (FIGURE).24
FIGURE
HCM: A guide to screening, diagnosis, and treatment
*Relatively contraindicated in LVOTO patients.
AICD, automatic implantable cardioverter defibrillator; DDD, dual-chamber; echo, echocardiography; EKG, electrocardiography; HCM, hypertrophic cardiomyopathy; H&P, history and physical; LVOTO, left ventricular outflow tract obstruction; SCD, sudden cardiac death.
Adapted from: Soor GS, et al. J Clin Pathol.4
Which drugs for which patients?
For those with symptoms of heart failure, beta-blockers are first-line therapy. Additional therapeutic options for patients without LVOTO include calcium channel blockers, nitrates, and diuretics. But these additional therapies are relatively contraindicated in patients with LVOTO. For LVOTO patients, disopyramide can be added to the beta-blocker regimen, if needed for symptom control.4,24
Patients with LVOTO or abnormal mitral motion are at moderate risk of spontaneous bacterial endocarditis (SBE).18 But evolving evidence shows low baseline rates of SBE and increased complications in patients routinely receiving antimicrobial prophylaxis, so the most recent guidelines do not recommend such treatment for any HCM patient.25
Amiodarone is effective for atrial fibrillation in HCM when beta- or calcium channel blockers fail to provide sufficient rate control.4 Amiodarone can also be used to prevent SCD.26 Recent data show that an automatic implantable cardioverter defibrillator (AICD)—which we’ll discuss later—is superior to amiodarone in preventing SCD,27 but the drug may be used in addition to an AICD or given to patients who are not candidates for, or not interested in, an implantable device.22,24
Invasive treatments may be considered for patients with LVOTO who do not respond to medical management.
Septal myomectomy or ethanol ablation: Which is better?
Septal myomectomy is the gold standard for refractory LVOTO.28 This open-heart procedure, which involves the resection of a portion of the septum to remove the obstructing cardiac tissue, has an operative mortality rate <1%.28 Surgical complication rates are also low, but include aortic regurgitation, left bundle or complete heart block, and iatrogenic ventricular septal defect.
Septal ethanol ablation is a percutaneous alternative to surgical myomectomy. In this minimally invasive procedure, ethanol is injected into the first septal perforating branch of the left anterior descending (LAD) artery, resulting in myocardial necrosis and septal wall thinning, which relieves the obstruction. Complications include ablation of inappropriate myocardium, heart block, pericardial effusion, and LAD dissection.29 Maximum effects of the ablation are delayed, typically occurring 6 to 12 months after the procedure.
Although limited by a lack of randomized controlled trials, a recent meta-analysis found surgical myomectomy and ethanol ablation to be equally effective in decreasing the LV outflow gradient to <20 mm Hg. Notably, though, surgical myomectomy reduced the gradient to nearly 10 mm Hg, compared with an average of 18 mm Hg for ethanol ablation.28 What’s more, several studies have found a higher incidence of complete heart block in patients who had ethanol ablation compared with those who underwent myomectomy.30 For patients who cannot tolerate or are not interested in invasive surgery, ablation offers an effective option.
Dual-chamber (DDD) pacing can also be used to treat LVOTO, but studies comparing pacing with myomectomy and ablation have found mixed results.6 Despite recent data showing the benefits of pacing in HCM,31 DDD pacing is typically reserved for patients who are not candidates for either surgical myomectomy or ablation.4
AICDs for which patients? It’s not always clear
AICDs effectively prevent SCD in patients with HCM,1,6,22 but the substantial cost and high rate of complications (>36%) make the devices impractical and inadvisable for universal use. Adverse events include pneumothorax, pericardial effusion, device infection or malfunction, and physical and psychological sequelae from inappropriate shocks.32 In fact, several studies of AICDs in patients with HCM have found the yearly rate of inappropriate shocks to be higher than the rate of appropriate discharges.22,31 And, because HCM patients are typically decades younger than coronary disease patients when they undergo implantation, they have a significantly higher burden of complication.32
Consensus statements vs actual practice. The central question of HCM risk stratification is how to identify patients at risk of SCD, thereby making it possible for them to reap the benefits of an AICD and drug treatment while sparing low-risk candidates the morbidity and the expense. So far, that question has not been definitively answered. As noted earlier, consensus statements agree that patients with more than 1 major risk factor have a higher risk of SCD6,28 than those with only 1; yet many patients with a single risk factor (and no prior cardiac arrest) have received AICDs.33
Studies highlight limitations of consensus guidelines’ assessment of risk. In recent case studies of HCM patients with AICDs based on registry data, roughly 25% of those studied22 received AICDs for secondary prevention—that is, after surviving cardiac arrest; the rest received them for primary prevention based on clinical risk factors. Rates of appropriate AICD discharge were 3-fold higher in patients who had survived previous cardiac arrest than in those who had not,22,32 a finding that supports aggressive AICD implantation among these high-risk patients.
Among patients who had received AICDs for primary prevention, however, appropriate discharges occurred at statistically identical rates whether they had 1, 2, or 3 major risk factors. Further, there was no association between the number of risk factors and the likelihood of appropriate discharge. Given these results, the decision to use an AICD in an HCM patient for primary prevention should be made after careful consultation with the patient and an HCM disease specialist.
What to tell patients about sports activities
Just as there is no definitive means of deciding when, or whether, a patient who has never experienced cardiac arrest should receive an AICD, there is no clear, evidence-based consensus on exercise restriction. Recommendations, which are based on expert opinion, leave room for individualized decision-making.
For those with genetic mutations consistent with HCM but no associated cardiac abnormalities and no family history of sudden death, no objective data support exercise limitations.34 For such patients, education regarding warning signs and symptoms and annual follow-up should be sufficient.
For athletes with a probable or unequivocal diagnosis of HCM, the ACC recommends restriction from competitive sports, with the possible exception of low-intensity activities such as billiards, bowling, and golf.35 This recommendation is not dependent on the presence of LVOTO or on patient symptoms, medical or surgical therapy, or the placement of an AICD.
A consensus statement from the ESC also lists recreational doubles tennis and biking, lap swimming, and weight lifting as permissible activities for patients with probable or unequivocal HCM, with a cautionary note about avoiding the Valsalva maneuver.36 The society discourages those with HCM from engaging in any activity that provokes dyspnea. Primary care physicians, too, must be sure that young patients with this condition understand the importance of avoiding intense exertion—and immediately stopping any physical activity if they notice any signs or symptoms associated with HCM.
Disclosure
The views expressed here are those of the authors and do not represent the policy of the United States Air Force, United States Army, or Department of Defense.
CORRESPONDENCE Anthony Beutler, MD, FAAFP, Department of Family Medicine A-1038, Uniformed Services University, 4301 Jones Bridge Road, Bethesda, MD 20814; [email protected]
1. Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308-1320.
2. Maron BJ. Hypertrophic cardiomyopathy and other causes of sudden cardiac death in young competitive athletes, with considerations for preparticipation screening and criteria for disqualification. Cardiol Clin. 2007;25:399-414.
3. McKenna W, Deanfield J, Faruqui A, et al. Prognosis in hypertrophic cardiomyopathy: role of age and clinical, electrocardiographic and hemodynamic features. Am J Cardiol. 1981;47:532-538.
4. Soor GS, Luk A, Ahn E, et al. Hypertrophic cardiomyopathy: current understanding and treatment objectives. J Clin Pathol. 2009;62:226-235.
5. Watkins H, Ashrafian H, McKenna WJ. The genetics of hypertrophic cardiomyopathy: Teare redux. Heart. 2008;94:1264-1268.
6. Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687-1713.
7. Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;2:1-8.
8. Maron BJ, Casey SA, Poliac LC, et al. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA. 1999;281:650-655.
9. Makenna W. Diseases of the Myocardium and Endocardium. Goldman L, Ausiello D, ed. Philadelphia: Saunders; 2008.
10. Giese EA, O’Connor FG, Brennan FH, et al. The athletic preparticipation evaluation: cardiovascular assessment. Am Fam Physician. 2007;75:1008-1114.
11. Glover DW, Maron BJ. Evolution in the process of screening United States high school student-athletes for cardiovascular disease. Am J Cardiol. 2007;100:1709-1712.
12. Rauch CM, Phillips GC. Adherence to guidelines for cardiovascular screening in current high school preparticipation evaluation forms. J Pediatr. 2009;155:584-586.
13. American Academy of Family Physicians, American Academy of Pediatrics, American Medical Society for Sports Medicine, American Osteopathic Society for Sports Medicine. The Preparticipation Physical Evaluation. 3rd ed. Minneapolis, Minn: McGrawHill; 2005:19–23.
14. Caselli S, Pelliccia A, Maron M, et al. Differentiation of hypertrophic cardiomyopathy from other forms of left ventricular hypertrophy by means of three-dimensional echocardiography. Am J Cardiol. 2008;102:616-620.
15. Olivotto I, Maron MS, Autore C, et al. Assessment and significance of left ventricular mass by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;52:559-566.
16. Maron MS, Maron BJ, Harrigan C, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. 2009;54:220-228.
17. Maron MS, Olivotto I, Betocchi S, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003;348:295-303.
18. Spirito P, Rapezzi C, Bellone P, et al. Infective endocarditis in hypertrophic cardiomyopathy: prevalence, incidence, and indications for antibiotic prophylaxis. Circulation. 1999;99:2132-2137.
19. Olivotto I, Cecchi F, Casey SA, et al. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation. 2001;104:2517-2524.
20. Spirito P, Seidman CE, McKenna WJ, et al. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775-785.
21. Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death—executive summary: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Eur Heart J. 2006;27:2099-2140.
22. Maron BJ, Spirito P, Shen WK, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405-412.
23. Nazarian S, Lima JA. Cardiovascular magnetic resonance for risk stratification of arrhythmia in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;51:1375-1376.
24. Fifer MA, Vlahakes GJ. Management of symptoms in hypertrophic cardiomyopathy. Circulation. 2008;117:429-439.
25. Nishimura RA, Carabello BA, Faxon DP, et al. ACC/AHA 2008 Guideline update on valvular heart disease: focused update on infective endocarditis: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;52:676-685.
26. McKenna WJ, Oakley CM, Krikler DM, et al. Improved survival with amiodarone in patients with hypertrophic cardiomyopathy and ventricular tachycardia. Br Heart J. 1985;53:412-416.
27. Maron BJ, Shen WK, Link MS, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000;343:365-373.
28. Alam M, Dokainish H, Lakkis NM. Hypertrophic obstructive cardiomyopathy-alcohol septal ablation vs. myectomy: a meta-analysis. Eur Heart J. 2009;30:1080-1087.
29. Alam M, Dokainish H, Lakkis N. Alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a systematic review of published studies. J Interv Cardiol. 2006;19:319-327.
30. Fernandes VL, Nielsen C, Nagueh SF, et al. Follow-up of alcohol septal ablation for symptomatic hypertrophic obstructive cardiomyopathy the Baylor and Medical University of South Carolina experience 1996 to 2007. JACC Cardiovasc Interv. 2008;1:561-570.
31. Galve E, Sambola A, Saldana G, et al. Late benefits of dual-chamber pacing in obstructive hypertrophic cardiomyopathy. A 10-year follow-up study. Heart. 2009; May 28 [E-pub ahead of print.]
32. Lin G, Nishimura RA, Gersh BJ, et al. Device complications and inappropriate implantable cardioverter defibrillator shocks in patients with hypertrophic cardiomyopathy. Heart. 2009;95:709-714.
33. Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212-2218.
34. Elliott P, Spirito P. Prevention of hypertrophic cardiomyopathy-related deaths: theory and practice. Heart. 2008;94:1269-1275.
35. Maron BJ, Ackerman MJ, Nishimura RA, et al. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol. 2005;45:1340-1345.
36. Pelliccia A, Fagard R, Bjornstad HH, et al. Recommendations for competitive sports participation in athletes with cardiovascular disease: a consensus document from the Study Group of Sports Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur Heart J. 2005;26:1422-1445.
37. Corrado D, Basso C, Pavei A, et al. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006;296:1593-1601.
38. Maron BJ, Shirani J, Poliac LC, et al. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. JAMA. 1996;276:199-204.
39. Maron BJ, Thompson PD, Ackerman MJ, et al. Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: a scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: endorsed by the American College of Cardiology Foundation. Circulation. 2007;115:1643-1655.
1. Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308-1320.
2. Maron BJ. Hypertrophic cardiomyopathy and other causes of sudden cardiac death in young competitive athletes, with considerations for preparticipation screening and criteria for disqualification. Cardiol Clin. 2007;25:399-414.
3. McKenna W, Deanfield J, Faruqui A, et al. Prognosis in hypertrophic cardiomyopathy: role of age and clinical, electrocardiographic and hemodynamic features. Am J Cardiol. 1981;47:532-538.
4. Soor GS, Luk A, Ahn E, et al. Hypertrophic cardiomyopathy: current understanding and treatment objectives. J Clin Pathol. 2009;62:226-235.
5. Watkins H, Ashrafian H, McKenna WJ. The genetics of hypertrophic cardiomyopathy: Teare redux. Heart. 2008;94:1264-1268.
6. Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687-1713.
7. Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;2:1-8.
8. Maron BJ, Casey SA, Poliac LC, et al. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA. 1999;281:650-655.
9. Makenna W. Diseases of the Myocardium and Endocardium. Goldman L, Ausiello D, ed. Philadelphia: Saunders; 2008.
10. Giese EA, O’Connor FG, Brennan FH, et al. The athletic preparticipation evaluation: cardiovascular assessment. Am Fam Physician. 2007;75:1008-1114.
11. Glover DW, Maron BJ. Evolution in the process of screening United States high school student-athletes for cardiovascular disease. Am J Cardiol. 2007;100:1709-1712.
12. Rauch CM, Phillips GC. Adherence to guidelines for cardiovascular screening in current high school preparticipation evaluation forms. J Pediatr. 2009;155:584-586.
13. American Academy of Family Physicians, American Academy of Pediatrics, American Medical Society for Sports Medicine, American Osteopathic Society for Sports Medicine. The Preparticipation Physical Evaluation. 3rd ed. Minneapolis, Minn: McGrawHill; 2005:19–23.
14. Caselli S, Pelliccia A, Maron M, et al. Differentiation of hypertrophic cardiomyopathy from other forms of left ventricular hypertrophy by means of three-dimensional echocardiography. Am J Cardiol. 2008;102:616-620.
15. Olivotto I, Maron MS, Autore C, et al. Assessment and significance of left ventricular mass by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;52:559-566.
16. Maron MS, Maron BJ, Harrigan C, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. 2009;54:220-228.
17. Maron MS, Olivotto I, Betocchi S, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003;348:295-303.
18. Spirito P, Rapezzi C, Bellone P, et al. Infective endocarditis in hypertrophic cardiomyopathy: prevalence, incidence, and indications for antibiotic prophylaxis. Circulation. 1999;99:2132-2137.
19. Olivotto I, Cecchi F, Casey SA, et al. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation. 2001;104:2517-2524.
20. Spirito P, Seidman CE, McKenna WJ, et al. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775-785.
21. Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death—executive summary: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Eur Heart J. 2006;27:2099-2140.
22. Maron BJ, Spirito P, Shen WK, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405-412.
23. Nazarian S, Lima JA. Cardiovascular magnetic resonance for risk stratification of arrhythmia in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;51:1375-1376.
24. Fifer MA, Vlahakes GJ. Management of symptoms in hypertrophic cardiomyopathy. Circulation. 2008;117:429-439.
25. Nishimura RA, Carabello BA, Faxon DP, et al. ACC/AHA 2008 Guideline update on valvular heart disease: focused update on infective endocarditis: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;52:676-685.
26. McKenna WJ, Oakley CM, Krikler DM, et al. Improved survival with amiodarone in patients with hypertrophic cardiomyopathy and ventricular tachycardia. Br Heart J. 1985;53:412-416.
27. Maron BJ, Shen WK, Link MS, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000;343:365-373.
28. Alam M, Dokainish H, Lakkis NM. Hypertrophic obstructive cardiomyopathy-alcohol septal ablation vs. myectomy: a meta-analysis. Eur Heart J. 2009;30:1080-1087.
29. Alam M, Dokainish H, Lakkis N. Alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a systematic review of published studies. J Interv Cardiol. 2006;19:319-327.
30. Fernandes VL, Nielsen C, Nagueh SF, et al. Follow-up of alcohol septal ablation for symptomatic hypertrophic obstructive cardiomyopathy the Baylor and Medical University of South Carolina experience 1996 to 2007. JACC Cardiovasc Interv. 2008;1:561-570.
31. Galve E, Sambola A, Saldana G, et al. Late benefits of dual-chamber pacing in obstructive hypertrophic cardiomyopathy. A 10-year follow-up study. Heart. 2009; May 28 [E-pub ahead of print.]
32. Lin G, Nishimura RA, Gersh BJ, et al. Device complications and inappropriate implantable cardioverter defibrillator shocks in patients with hypertrophic cardiomyopathy. Heart. 2009;95:709-714.
33. Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212-2218.
34. Elliott P, Spirito P. Prevention of hypertrophic cardiomyopathy-related deaths: theory and practice. Heart. 2008;94:1269-1275.
35. Maron BJ, Ackerman MJ, Nishimura RA, et al. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol. 2005;45:1340-1345.
36. Pelliccia A, Fagard R, Bjornstad HH, et al. Recommendations for competitive sports participation in athletes with cardiovascular disease: a consensus document from the Study Group of Sports Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur Heart J. 2005;26:1422-1445.
37. Corrado D, Basso C, Pavei A, et al. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006;296:1593-1601.
38. Maron BJ, Shirani J, Poliac LC, et al. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. JAMA. 1996;276:199-204.
39. Maron BJ, Thompson PD, Ackerman MJ, et al. Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: a scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: endorsed by the American College of Cardiology Foundation. Circulation. 2007;115:1643-1655.