User login
Lamotrigine-Induced Cutaneous Pseudolymphoma
To the Editor:
An 8-year-old girl presented with new lesions on the scalp that were mildly painful to palpation and had been increasing in size and number over the last 2 months. Her medical history was remarkable for seizures, keratosis pilaris, and seborrheic dermatitis. The seizures had been well controlled on oxcarbazepine; however, she was switched to lamotrigine 6 months prior to presentation under the care of her neurologist. The patient was not taking other oral medications, and she denied any trauma/insect bites to the affected area or systemic symptoms such as fever, fatigue, weight loss, nausea, swollen lymph nodes, or night sweats. Physical examination revealed 3 well-circumscribed, pink, slightly scaly, indurated nodules on the frontal and vertex scalp (Figure 1). She reported pain on palpation of the lesions. Treatment with ketoconazole shampoo and high-potency topical corticosteroids was ineffective.
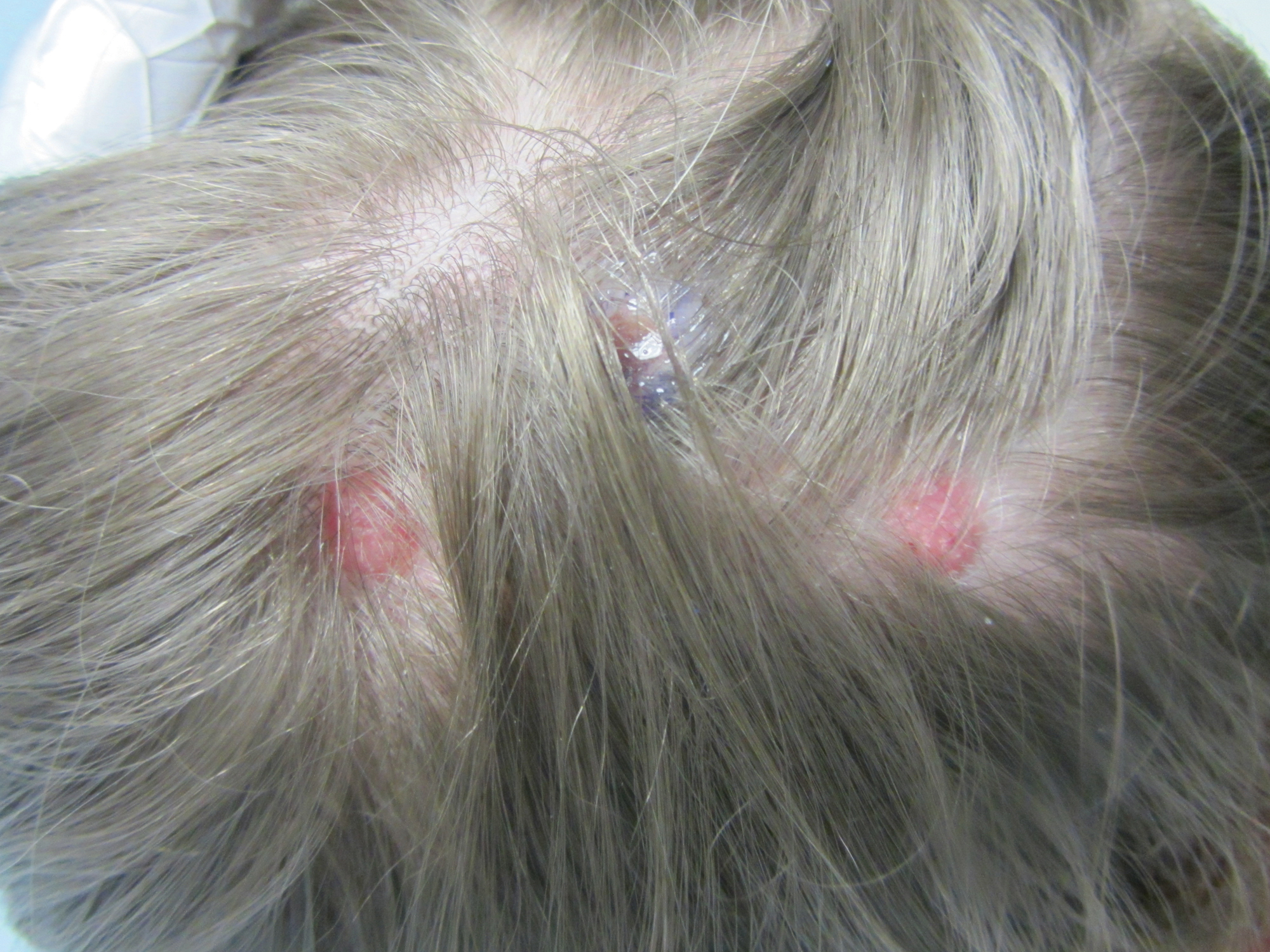
Over a period of 2 months after the initial presentation, the patient developed a total of 9 scalp lesions. Testing was performed 4 months after presentation of lesions. Bacterial and fungal cultures of the lesional skin of the scalp were negative. Two biopsies of lesions on the scalp were performed, the first of which showed a nonspecific lymphohistiocytic infiltrate. The second biopsy revealed a dense, nodular, atypical dermal lymphoid infiltrate composed primarily of round regular lymphocytes intermixed with some larger, more irregular lymphocytes and few scattered mitoses (Figure 2).
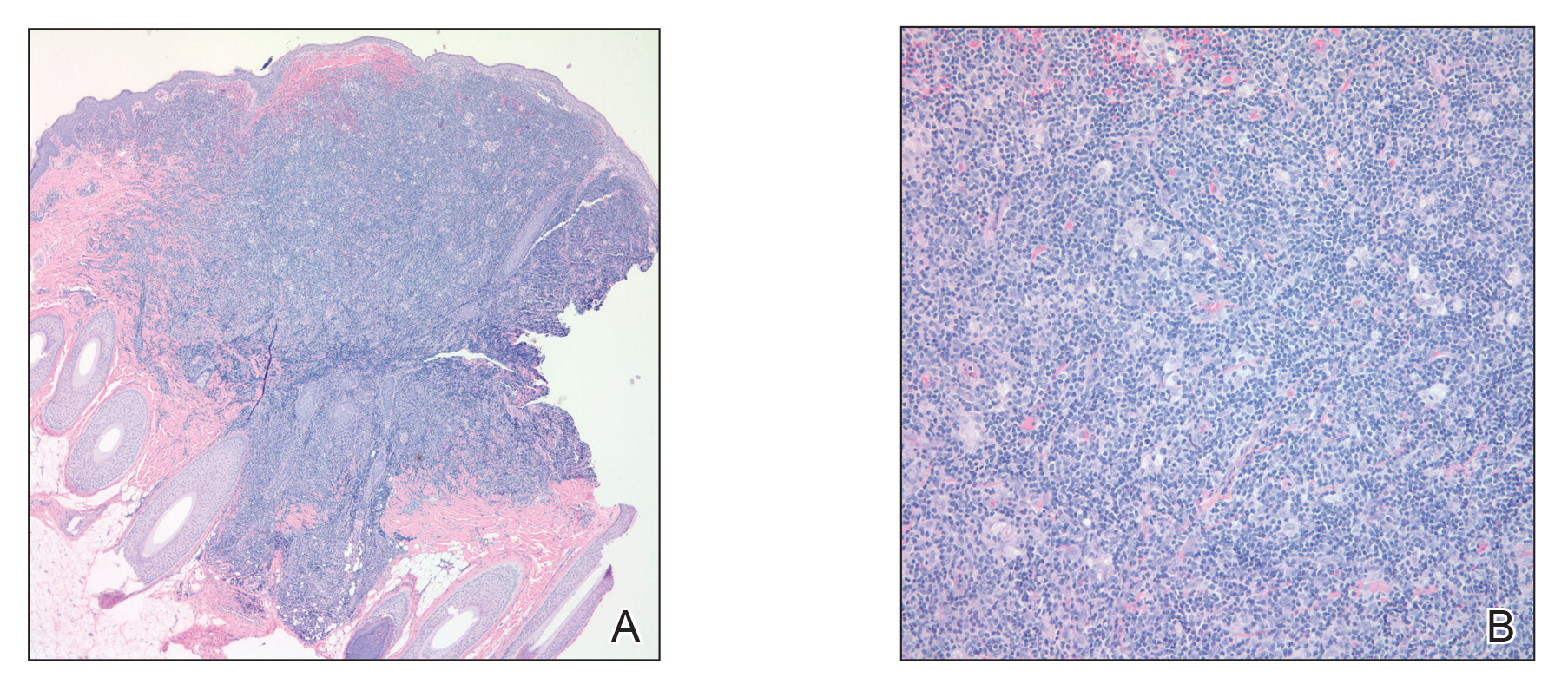
Immunohistochemical studies revealed small B-cell lymphoma 2–positive lymphocytes with a 2:1 mixture of CD3+ T cells and CD20+CD79a+ B cells. The T cells expressed CD2, CD5, and CD43, and a subset showed a loss of CD7. The CD4:CD8 ratio was 10 to 1. No follicular dendritic networks were noted with CD21 and CD23. Rare, scattered, medium-sized CD30 cells were noted. Staining for CD10, B-cell lymphoma 6, anaplastic lymphoma kinase, Epstein-Barr virus–encoded RNA 1, IgD, and IgM were negative. The plasma cells had a κ/λ free light chain ratio of 2 to 1. Ki-67 was positive in 15% of lymphoid cells. Polymerase chain reaction analysis of T-cell receptor gene rearrangement revealed a peak at 228 bp in a predominantly polyclonal background. A thorough systemic workup including complete blood cell count, immunoglobulin assay, bone marrow transplant panel, comprehensive metabolic panel, lactate dehydrogenase test, inflammatory markers, and viral testing failed to reveal any evidence of underlying malignancy.
After conferring with the patient’s neurologist, lamotrigine was discontinued. Within a few weeks of cessation, the scalp lesions resolved without recurrence at 9-month follow-up. In addition to the lack of clinical, histological, or immunohistochemical evidence of underlying malignancy, the temporal association of the development of lesions after starting lamotrigine and rapid resolution upon its discontinuation suggested a diagnosis of lamotrigine-induced cutaneous pseudolymphoma.
Cutaneous pseudolymphoma is a term used to describe a heterogenous group of benign reactive T-cell, B-cell, or mixed-cell lymphoproliferative processes that resemble cutaneous lymphomas clinically and/or histopathologically.1 Historically, these types of proliferations have been classified under many alternative names that originally served to describe only B-cell–type proliferations. With advances in immunohistochemistry allowing for more specific cell marker identification, cutaneous pseudolymphomas often are found to contain a mixture of T-cell and B-cell populations, which also led to identifying and describing T-cell–type pseudolymphomas.2
The clinical appearance of cutaneous pseudolymphoma is variable, ranging from discrete nodules or papules to even confluent erythroderma in certain cases.2 The high clinical variability further complicates diagnosis. Although our patient presented with 9 individual nodular lesions, this finding alone is not sufficient to have high suspicion for cutaneous pseudolymphoma without including a much broader differential diagnosis. In our case, the differential diagnosis also included cutaneous lymphoma, arthropod bite reaction, lymphomatoid papulosis, tumid lupus, follicular mucinosis, lymphocytic infiltrate of Jessner, and leukemia cutis.
The primary concern regarding diagnosis of cutaneous pseudolymphoma is the clinician’s ability to effectively differentiate this entity from a true malignant lymphoma. Immunostaining has some value by identification of heterogeneous cell–type populations with a mixed T-cell and B-cell infiltrate that is more characteristic of a benign reactive process. Subsequent polymerase chain reaction analysis can detect the presence or absence of monoclonal T-cell receptor gene rearrangement or immunoglobulin heavy chain rearrangement.3 If these monoclonal rearrangements are absent, a benign diagnosis is favored; however, these rearrangements also have been shown to exist in a case of cutaneous pseudolymphoma that earned the final diagnosis when removal of the offending agent led to spontaneous lesion regression, similar to our case.4
Many different entities have been described as causative factors for the development of cutaneous pseudolymphoma. Of those that have been considered causative, simple categories have emerged, including endogenous, exogenous, and iatrogenic causes. One potential endogenous etiology of cutaneous pseudolymphoma is IgG4-related disease.5 A multitude of exogenous causes have been reported, including several cases of cutaneous pseudolymphoma developing in a prior tattoo site.6 Viruses, specifically molluscum contagiosum, also have been implicated as exogenous causes, and a report of cutaneous pseudolymphoma development at a prior site of herpes zoster lesions has been described.7 Development of cutaneous pseudolymphoma in vaccination sites also has been reported,8 as well as more obscure inciting events such as Leishmania donovani infection and medicinal leech therapy.9
A considerable number of reported cases of cutaneous pseudolymphoma have been attributed to drugs, including monoclonal antibodies,10 herbal supplements,11 and a multitude of other medications.1 As a class, anticonvulsants are considered more likely to cause lymph node pseudolymphomas than strictly cutaneous pseudolymphomas12; however, many drugs in this class of medications have been described in the development of cutaneous pseudolymphoma.3 A review of the literature by Ploysangam et al1 revealed reports of the development of cutaneous pseudolymphomas after administration of phenytoin, carbamazepine, mephenytoin, trimethadione, phenobarbital, primidone, butabarbital, methsuximide, phensuximide, and valproic acid.
Our patient represents a rare case of strictly cutaneous pseudolymphoma caused by administration of lamotrigine. Our case demonstrated a clear temporal relation between the cessation of lamotrigine and rapid and spontaneous disappearance of cutaneous lesions. We found another case of pseudolymphoma in which lamotrigine was deemed causative, but only lymph node involvement was observed.12
Proper diagnosis of cutaneous pseudolymphoma is important not only with regard to the initial differentiation from true malignant lymphoma but in allowing for appropriate follow-up and vigilant surveillance. Cases of progression from cutaneous pseudolymphoma to true lymphoma have been reported.1,2 It is recommended that watchful follow-up for these patients be carried out until at least 5 years after the diagnosis of cutaneous pseudolymphoma is made to rule out the possibility of malignant transformation, particularly in idiopathic cases.13
- Ploysangam T, Breneman D, Mutasim D. Cutaneous pseudolymphomas. J Am Acad Dermatol. 1998;38:877-898.
- Bergman R. Pseudolymphoma and cutaneous lymphoma: facts and controversies. Clin Dermatol. 2010;28:568-574.
- Braddock S, Harrington D, Vose J. Generalized nodular cutaneous pseudolymphoma associated with phenytoin therapy. J Am Acad Dermatol. 1992;27:337-340.
- Cogrel O, Beylot-Barry M, Vergier B, et al. Sodium valproate-induced cutaneous pseudolymphoma followed by recurrence with carbamazepine. Br J Dermatol. 2001;144:1235-1238.
- Cheuk W, Lee K, Chong L, et al. IgG4-related sclerosing disease: a potential new etiology of cutaneous pseudolymphoma. Am J Surg Pathol. 2009;33:1713-1719.
- Marchesi A, Parodi P, Brioschi M, et al. Tattoo ink-related cutaneous pseudolymphomas: a rare but significant complication. case report and review of the literature. Aesthetic Plast Surg. 2014;38:471-478.
- Gonzalez J, Sanz A, Martin T, et al. Cutaneous pseudolymphoma associated with molluscum contagiosum: a case report. Int J Dermatol. 2008;47:502-504.
- Maubec E, Pinquier L, Viguier M, et al. Vaccination-induced cutaneous pseudolymphoma. J Am Acad Dermatol. 2005;52:623-629.
- Altamura D, Calonje E, Liau J, et al. Diffuse cutaneous pseudolymphoma due to therapy with medicinal leeches. JAMA Dermatol. 2014;150:783-784.
- Imafuku S, Ito K, Nakayama J. Cutaneous pseudolymphoma induced by adalimumab and reproduced by infliximab in a patient with arthopathic psoriasis. Br J Dermatol. 2011;166:675-678.
- Meyer S, Vogt T, Obermann EC, et al. Cutaneous pseudolymphoma induced by Cimicifuga racemosa. Dermatology. 2007;214:94-96.
- Pathak P, McLachlan R. Drug-induced pseudolymphoma secondary to lamotrigine. Neurology. 1998;50:1509-1510.
- Prabu V, Shivani A, Pawar V. Idiopathic cutaneous pseudolymphoma: an enigma. Indian Dermatol Online J. 2014;5:224-226.
To the Editor:
An 8-year-old girl presented with new lesions on the scalp that were mildly painful to palpation and had been increasing in size and number over the last 2 months. Her medical history was remarkable for seizures, keratosis pilaris, and seborrheic dermatitis. The seizures had been well controlled on oxcarbazepine; however, she was switched to lamotrigine 6 months prior to presentation under the care of her neurologist. The patient was not taking other oral medications, and she denied any trauma/insect bites to the affected area or systemic symptoms such as fever, fatigue, weight loss, nausea, swollen lymph nodes, or night sweats. Physical examination revealed 3 well-circumscribed, pink, slightly scaly, indurated nodules on the frontal and vertex scalp (Figure 1). She reported pain on palpation of the lesions. Treatment with ketoconazole shampoo and high-potency topical corticosteroids was ineffective.
Over a period of 2 months after the initial presentation, the patient developed a total of 9 scalp lesions. Testing was performed 4 months after presentation of lesions. Bacterial and fungal cultures of the lesional skin of the scalp were negative. Two biopsies of lesions on the scalp were performed, the first of which showed a nonspecific lymphohistiocytic infiltrate. The second biopsy revealed a dense, nodular, atypical dermal lymphoid infiltrate composed primarily of round regular lymphocytes intermixed with some larger, more irregular lymphocytes and few scattered mitoses (Figure 2).
Immunohistochemical studies revealed small B-cell lymphoma 2–positive lymphocytes with a 2:1 mixture of CD3+ T cells and CD20+CD79a+ B cells. The T cells expressed CD2, CD5, and CD43, and a subset showed a loss of CD7. The CD4:CD8 ratio was 10 to 1. No follicular dendritic networks were noted with CD21 and CD23. Rare, scattered, medium-sized CD30 cells were noted. Staining for CD10, B-cell lymphoma 6, anaplastic lymphoma kinase, Epstein-Barr virus–encoded RNA 1, IgD, and IgM were negative. The plasma cells had a κ/λ free light chain ratio of 2 to 1. Ki-67 was positive in 15% of lymphoid cells. Polymerase chain reaction analysis of T-cell receptor gene rearrangement revealed a peak at 228 bp in a predominantly polyclonal background. A thorough systemic workup including complete blood cell count, immunoglobulin assay, bone marrow transplant panel, comprehensive metabolic panel, lactate dehydrogenase test, inflammatory markers, and viral testing failed to reveal any evidence of underlying malignancy.
After conferring with the patient’s neurologist, lamotrigine was discontinued. Within a few weeks of cessation, the scalp lesions resolved without recurrence at 9-month follow-up. In addition to the lack of clinical, histological, or immunohistochemical evidence of underlying malignancy, the temporal association of the development of lesions after starting lamotrigine and rapid resolution upon its discontinuation suggested a diagnosis of lamotrigine-induced cutaneous pseudolymphoma.
Cutaneous pseudolymphoma is a term used to describe a heterogenous group of benign reactive T-cell, B-cell, or mixed-cell lymphoproliferative processes that resemble cutaneous lymphomas clinically and/or histopathologically.1 Historically, these types of proliferations have been classified under many alternative names that originally served to describe only B-cell–type proliferations. With advances in immunohistochemistry allowing for more specific cell marker identification, cutaneous pseudolymphomas often are found to contain a mixture of T-cell and B-cell populations, which also led to identifying and describing T-cell–type pseudolymphomas.2
The clinical appearance of cutaneous pseudolymphoma is variable, ranging from discrete nodules or papules to even confluent erythroderma in certain cases.2 The high clinical variability further complicates diagnosis. Although our patient presented with 9 individual nodular lesions, this finding alone is not sufficient to have high suspicion for cutaneous pseudolymphoma without including a much broader differential diagnosis. In our case, the differential diagnosis also included cutaneous lymphoma, arthropod bite reaction, lymphomatoid papulosis, tumid lupus, follicular mucinosis, lymphocytic infiltrate of Jessner, and leukemia cutis.
The primary concern regarding diagnosis of cutaneous pseudolymphoma is the clinician’s ability to effectively differentiate this entity from a true malignant lymphoma. Immunostaining has some value by identification of heterogeneous cell–type populations with a mixed T-cell and B-cell infiltrate that is more characteristic of a benign reactive process. Subsequent polymerase chain reaction analysis can detect the presence or absence of monoclonal T-cell receptor gene rearrangement or immunoglobulin heavy chain rearrangement.3 If these monoclonal rearrangements are absent, a benign diagnosis is favored; however, these rearrangements also have been shown to exist in a case of cutaneous pseudolymphoma that earned the final diagnosis when removal of the offending agent led to spontaneous lesion regression, similar to our case.4
Many different entities have been described as causative factors for the development of cutaneous pseudolymphoma. Of those that have been considered causative, simple categories have emerged, including endogenous, exogenous, and iatrogenic causes. One potential endogenous etiology of cutaneous pseudolymphoma is IgG4-related disease.5 A multitude of exogenous causes have been reported, including several cases of cutaneous pseudolymphoma developing in a prior tattoo site.6 Viruses, specifically molluscum contagiosum, also have been implicated as exogenous causes, and a report of cutaneous pseudolymphoma development at a prior site of herpes zoster lesions has been described.7 Development of cutaneous pseudolymphoma in vaccination sites also has been reported,8 as well as more obscure inciting events such as Leishmania donovani infection and medicinal leech therapy.9
A considerable number of reported cases of cutaneous pseudolymphoma have been attributed to drugs, including monoclonal antibodies,10 herbal supplements,11 and a multitude of other medications.1 As a class, anticonvulsants are considered more likely to cause lymph node pseudolymphomas than strictly cutaneous pseudolymphomas12; however, many drugs in this class of medications have been described in the development of cutaneous pseudolymphoma.3 A review of the literature by Ploysangam et al1 revealed reports of the development of cutaneous pseudolymphomas after administration of phenytoin, carbamazepine, mephenytoin, trimethadione, phenobarbital, primidone, butabarbital, methsuximide, phensuximide, and valproic acid.
Our patient represents a rare case of strictly cutaneous pseudolymphoma caused by administration of lamotrigine. Our case demonstrated a clear temporal relation between the cessation of lamotrigine and rapid and spontaneous disappearance of cutaneous lesions. We found another case of pseudolymphoma in which lamotrigine was deemed causative, but only lymph node involvement was observed.12
Proper diagnosis of cutaneous pseudolymphoma is important not only with regard to the initial differentiation from true malignant lymphoma but in allowing for appropriate follow-up and vigilant surveillance. Cases of progression from cutaneous pseudolymphoma to true lymphoma have been reported.1,2 It is recommended that watchful follow-up for these patients be carried out until at least 5 years after the diagnosis of cutaneous pseudolymphoma is made to rule out the possibility of malignant transformation, particularly in idiopathic cases.13
To the Editor:
An 8-year-old girl presented with new lesions on the scalp that were mildly painful to palpation and had been increasing in size and number over the last 2 months. Her medical history was remarkable for seizures, keratosis pilaris, and seborrheic dermatitis. The seizures had been well controlled on oxcarbazepine; however, she was switched to lamotrigine 6 months prior to presentation under the care of her neurologist. The patient was not taking other oral medications, and she denied any trauma/insect bites to the affected area or systemic symptoms such as fever, fatigue, weight loss, nausea, swollen lymph nodes, or night sweats. Physical examination revealed 3 well-circumscribed, pink, slightly scaly, indurated nodules on the frontal and vertex scalp (Figure 1). She reported pain on palpation of the lesions. Treatment with ketoconazole shampoo and high-potency topical corticosteroids was ineffective.
Over a period of 2 months after the initial presentation, the patient developed a total of 9 scalp lesions. Testing was performed 4 months after presentation of lesions. Bacterial and fungal cultures of the lesional skin of the scalp were negative. Two biopsies of lesions on the scalp were performed, the first of which showed a nonspecific lymphohistiocytic infiltrate. The second biopsy revealed a dense, nodular, atypical dermal lymphoid infiltrate composed primarily of round regular lymphocytes intermixed with some larger, more irregular lymphocytes and few scattered mitoses (Figure 2).
Immunohistochemical studies revealed small B-cell lymphoma 2–positive lymphocytes with a 2:1 mixture of CD3+ T cells and CD20+CD79a+ B cells. The T cells expressed CD2, CD5, and CD43, and a subset showed a loss of CD7. The CD4:CD8 ratio was 10 to 1. No follicular dendritic networks were noted with CD21 and CD23. Rare, scattered, medium-sized CD30 cells were noted. Staining for CD10, B-cell lymphoma 6, anaplastic lymphoma kinase, Epstein-Barr virus–encoded RNA 1, IgD, and IgM were negative. The plasma cells had a κ/λ free light chain ratio of 2 to 1. Ki-67 was positive in 15% of lymphoid cells. Polymerase chain reaction analysis of T-cell receptor gene rearrangement revealed a peak at 228 bp in a predominantly polyclonal background. A thorough systemic workup including complete blood cell count, immunoglobulin assay, bone marrow transplant panel, comprehensive metabolic panel, lactate dehydrogenase test, inflammatory markers, and viral testing failed to reveal any evidence of underlying malignancy.
After conferring with the patient’s neurologist, lamotrigine was discontinued. Within a few weeks of cessation, the scalp lesions resolved without recurrence at 9-month follow-up. In addition to the lack of clinical, histological, or immunohistochemical evidence of underlying malignancy, the temporal association of the development of lesions after starting lamotrigine and rapid resolution upon its discontinuation suggested a diagnosis of lamotrigine-induced cutaneous pseudolymphoma.
Cutaneous pseudolymphoma is a term used to describe a heterogenous group of benign reactive T-cell, B-cell, or mixed-cell lymphoproliferative processes that resemble cutaneous lymphomas clinically and/or histopathologically.1 Historically, these types of proliferations have been classified under many alternative names that originally served to describe only B-cell–type proliferations. With advances in immunohistochemistry allowing for more specific cell marker identification, cutaneous pseudolymphomas often are found to contain a mixture of T-cell and B-cell populations, which also led to identifying and describing T-cell–type pseudolymphomas.2
The clinical appearance of cutaneous pseudolymphoma is variable, ranging from discrete nodules or papules to even confluent erythroderma in certain cases.2 The high clinical variability further complicates diagnosis. Although our patient presented with 9 individual nodular lesions, this finding alone is not sufficient to have high suspicion for cutaneous pseudolymphoma without including a much broader differential diagnosis. In our case, the differential diagnosis also included cutaneous lymphoma, arthropod bite reaction, lymphomatoid papulosis, tumid lupus, follicular mucinosis, lymphocytic infiltrate of Jessner, and leukemia cutis.
The primary concern regarding diagnosis of cutaneous pseudolymphoma is the clinician’s ability to effectively differentiate this entity from a true malignant lymphoma. Immunostaining has some value by identification of heterogeneous cell–type populations with a mixed T-cell and B-cell infiltrate that is more characteristic of a benign reactive process. Subsequent polymerase chain reaction analysis can detect the presence or absence of monoclonal T-cell receptor gene rearrangement or immunoglobulin heavy chain rearrangement.3 If these monoclonal rearrangements are absent, a benign diagnosis is favored; however, these rearrangements also have been shown to exist in a case of cutaneous pseudolymphoma that earned the final diagnosis when removal of the offending agent led to spontaneous lesion regression, similar to our case.4
Many different entities have been described as causative factors for the development of cutaneous pseudolymphoma. Of those that have been considered causative, simple categories have emerged, including endogenous, exogenous, and iatrogenic causes. One potential endogenous etiology of cutaneous pseudolymphoma is IgG4-related disease.5 A multitude of exogenous causes have been reported, including several cases of cutaneous pseudolymphoma developing in a prior tattoo site.6 Viruses, specifically molluscum contagiosum, also have been implicated as exogenous causes, and a report of cutaneous pseudolymphoma development at a prior site of herpes zoster lesions has been described.7 Development of cutaneous pseudolymphoma in vaccination sites also has been reported,8 as well as more obscure inciting events such as Leishmania donovani infection and medicinal leech therapy.9
A considerable number of reported cases of cutaneous pseudolymphoma have been attributed to drugs, including monoclonal antibodies,10 herbal supplements,11 and a multitude of other medications.1 As a class, anticonvulsants are considered more likely to cause lymph node pseudolymphomas than strictly cutaneous pseudolymphomas12; however, many drugs in this class of medications have been described in the development of cutaneous pseudolymphoma.3 A review of the literature by Ploysangam et al1 revealed reports of the development of cutaneous pseudolymphomas after administration of phenytoin, carbamazepine, mephenytoin, trimethadione, phenobarbital, primidone, butabarbital, methsuximide, phensuximide, and valproic acid.
Our patient represents a rare case of strictly cutaneous pseudolymphoma caused by administration of lamotrigine. Our case demonstrated a clear temporal relation between the cessation of lamotrigine and rapid and spontaneous disappearance of cutaneous lesions. We found another case of pseudolymphoma in which lamotrigine was deemed causative, but only lymph node involvement was observed.12
Proper diagnosis of cutaneous pseudolymphoma is important not only with regard to the initial differentiation from true malignant lymphoma but in allowing for appropriate follow-up and vigilant surveillance. Cases of progression from cutaneous pseudolymphoma to true lymphoma have been reported.1,2 It is recommended that watchful follow-up for these patients be carried out until at least 5 years after the diagnosis of cutaneous pseudolymphoma is made to rule out the possibility of malignant transformation, particularly in idiopathic cases.13
- Ploysangam T, Breneman D, Mutasim D. Cutaneous pseudolymphomas. J Am Acad Dermatol. 1998;38:877-898.
- Bergman R. Pseudolymphoma and cutaneous lymphoma: facts and controversies. Clin Dermatol. 2010;28:568-574.
- Braddock S, Harrington D, Vose J. Generalized nodular cutaneous pseudolymphoma associated with phenytoin therapy. J Am Acad Dermatol. 1992;27:337-340.
- Cogrel O, Beylot-Barry M, Vergier B, et al. Sodium valproate-induced cutaneous pseudolymphoma followed by recurrence with carbamazepine. Br J Dermatol. 2001;144:1235-1238.
- Cheuk W, Lee K, Chong L, et al. IgG4-related sclerosing disease: a potential new etiology of cutaneous pseudolymphoma. Am J Surg Pathol. 2009;33:1713-1719.
- Marchesi A, Parodi P, Brioschi M, et al. Tattoo ink-related cutaneous pseudolymphomas: a rare but significant complication. case report and review of the literature. Aesthetic Plast Surg. 2014;38:471-478.
- Gonzalez J, Sanz A, Martin T, et al. Cutaneous pseudolymphoma associated with molluscum contagiosum: a case report. Int J Dermatol. 2008;47:502-504.
- Maubec E, Pinquier L, Viguier M, et al. Vaccination-induced cutaneous pseudolymphoma. J Am Acad Dermatol. 2005;52:623-629.
- Altamura D, Calonje E, Liau J, et al. Diffuse cutaneous pseudolymphoma due to therapy with medicinal leeches. JAMA Dermatol. 2014;150:783-784.
- Imafuku S, Ito K, Nakayama J. Cutaneous pseudolymphoma induced by adalimumab and reproduced by infliximab in a patient with arthopathic psoriasis. Br J Dermatol. 2011;166:675-678.
- Meyer S, Vogt T, Obermann EC, et al. Cutaneous pseudolymphoma induced by Cimicifuga racemosa. Dermatology. 2007;214:94-96.
- Pathak P, McLachlan R. Drug-induced pseudolymphoma secondary to lamotrigine. Neurology. 1998;50:1509-1510.
- Prabu V, Shivani A, Pawar V. Idiopathic cutaneous pseudolymphoma: an enigma. Indian Dermatol Online J. 2014;5:224-226.
- Ploysangam T, Breneman D, Mutasim D. Cutaneous pseudolymphomas. J Am Acad Dermatol. 1998;38:877-898.
- Bergman R. Pseudolymphoma and cutaneous lymphoma: facts and controversies. Clin Dermatol. 2010;28:568-574.
- Braddock S, Harrington D, Vose J. Generalized nodular cutaneous pseudolymphoma associated with phenytoin therapy. J Am Acad Dermatol. 1992;27:337-340.
- Cogrel O, Beylot-Barry M, Vergier B, et al. Sodium valproate-induced cutaneous pseudolymphoma followed by recurrence with carbamazepine. Br J Dermatol. 2001;144:1235-1238.
- Cheuk W, Lee K, Chong L, et al. IgG4-related sclerosing disease: a potential new etiology of cutaneous pseudolymphoma. Am J Surg Pathol. 2009;33:1713-1719.
- Marchesi A, Parodi P, Brioschi M, et al. Tattoo ink-related cutaneous pseudolymphomas: a rare but significant complication. case report and review of the literature. Aesthetic Plast Surg. 2014;38:471-478.
- Gonzalez J, Sanz A, Martin T, et al. Cutaneous pseudolymphoma associated with molluscum contagiosum: a case report. Int J Dermatol. 2008;47:502-504.
- Maubec E, Pinquier L, Viguier M, et al. Vaccination-induced cutaneous pseudolymphoma. J Am Acad Dermatol. 2005;52:623-629.
- Altamura D, Calonje E, Liau J, et al. Diffuse cutaneous pseudolymphoma due to therapy with medicinal leeches. JAMA Dermatol. 2014;150:783-784.
- Imafuku S, Ito K, Nakayama J. Cutaneous pseudolymphoma induced by adalimumab and reproduced by infliximab in a patient with arthopathic psoriasis. Br J Dermatol. 2011;166:675-678.
- Meyer S, Vogt T, Obermann EC, et al. Cutaneous pseudolymphoma induced by Cimicifuga racemosa. Dermatology. 2007;214:94-96.
- Pathak P, McLachlan R. Drug-induced pseudolymphoma secondary to lamotrigine. Neurology. 1998;50:1509-1510.
- Prabu V, Shivani A, Pawar V. Idiopathic cutaneous pseudolymphoma: an enigma. Indian Dermatol Online J. 2014;5:224-226.
Practice Points
- Cutaneous pseudolymphomas are a heterogenous group of benign T-cell, B-cell, or mixed-cell lymphoproliferative processes that resemble cutaneous lymphomas clinically and/or histopathologically.
- Cutaneous pseudolymphomas have many causative factors, including medications, infections, tattoo ink, vaccinations, and insect bites.
- Lamotrigine is a potential inciting factor of cutaneous pseudolymphoma.
Darkening and Eruptive Nevi During Treatment With Erlotinib
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
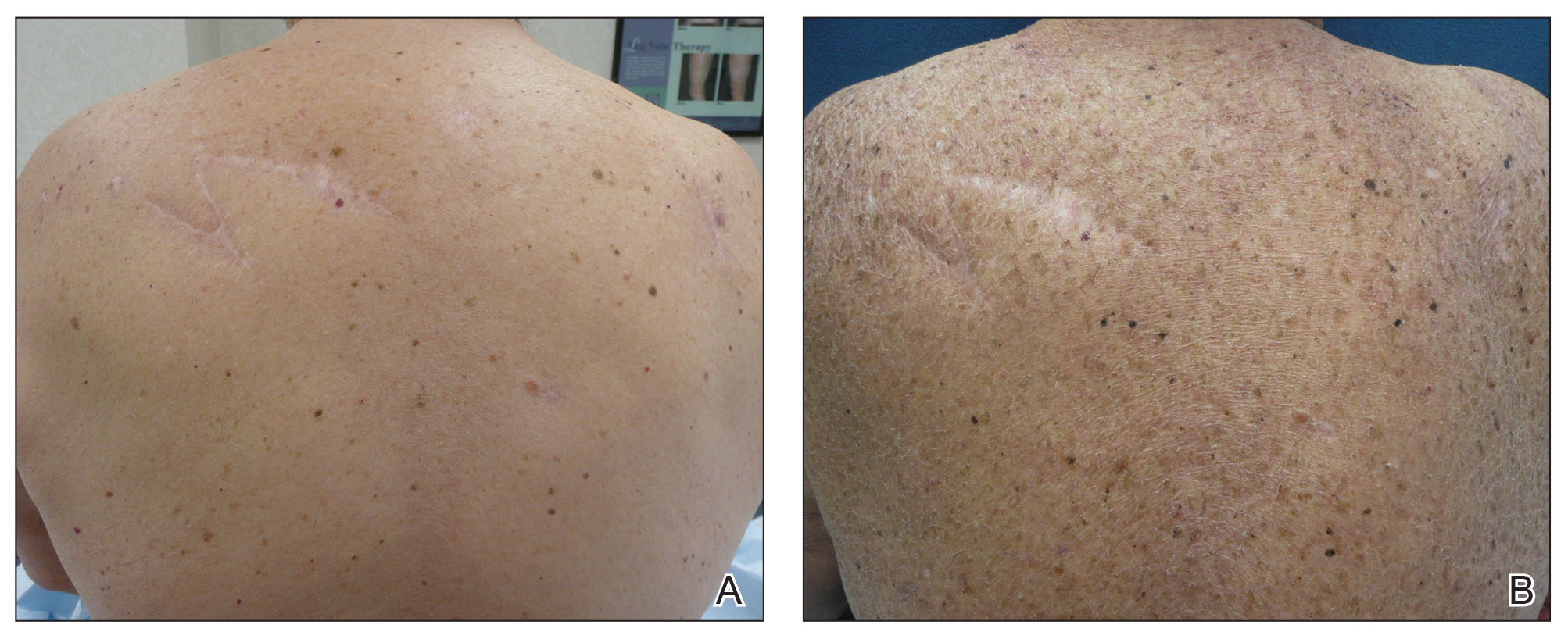
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
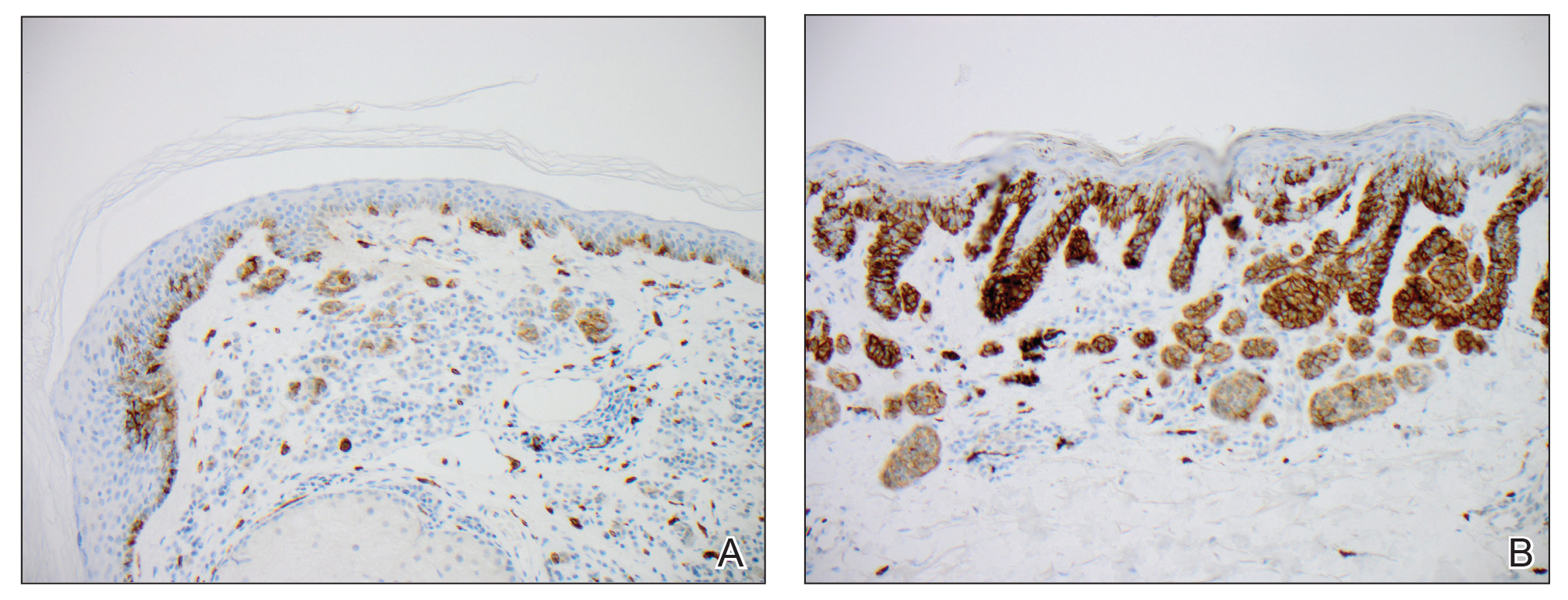
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
Practice Points
- Cutaneous side effects of erlotinib include acneform eruption, xerosis, paronychia, and pruritus.
- Clinicians should monitor patients for darkening and/or eruptive nevi as well as melanoma during treatment with erlotinib.
Metastatic Adamantinoma Presenting as a Cutaneous Papule
To the Editor:
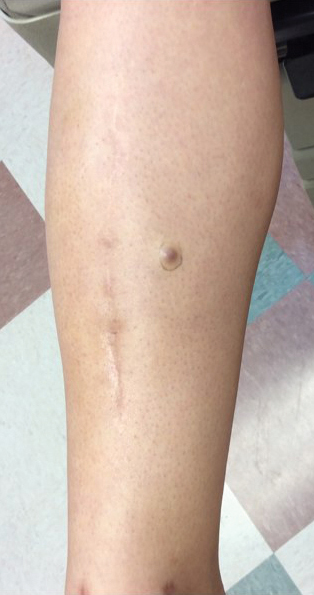
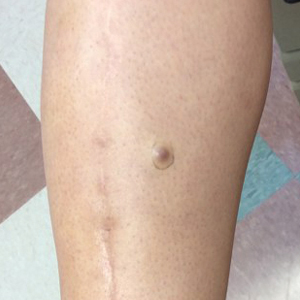
A 34-year-old woman with a history of adamantinoma of the right tibia that had been surgically resected with tibial reconstruction 5 years prior presented with a mildly tender, enlarging lesion on the right distal shin of 6 months’ duration that had started to change color. Review of systems was otherwise negative. Physical examination revealed an 8-mm, slightly tender, rubbery, pink papule adjacent to the surgical scar over the right tibia (Figure 1). Given the rapid growth of the lesion and its proximity to the surgical site, a punch biopsy was performed.
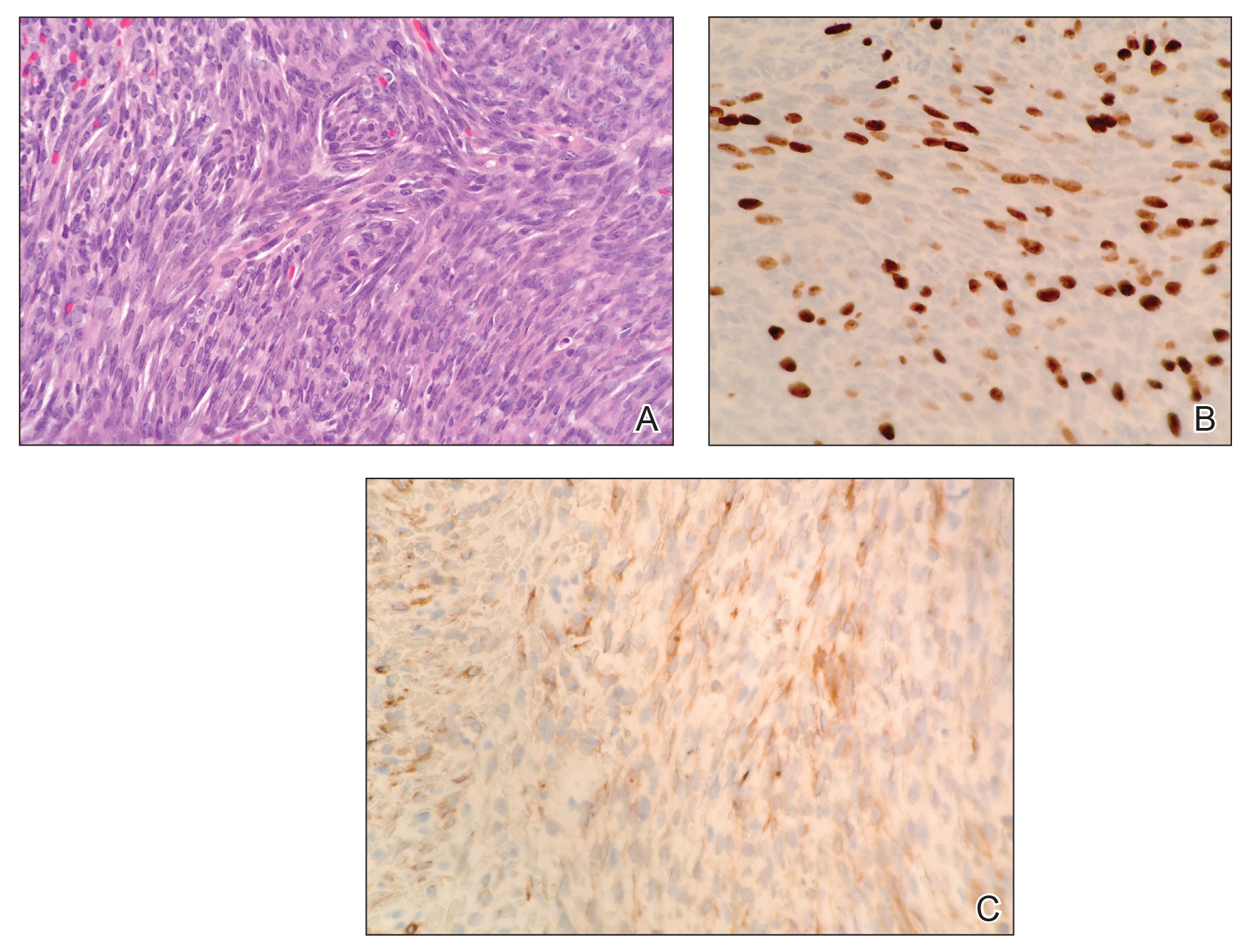
Histopathologic examination demonstrated a densely cellular dermal tumor composed of spindle cells with large hyperchromatic nuclei, numerous mitotic figures, and minimal eosinophilic cytoplasm (Figure 2A). Immunohistochemical studies revealed that approximately 40% of the tumor nuclei were immunoreactive to Ki-67 (Figure 2B), and total cytokeratin was focally positive (Figure 2C). A diagnosis of metastatic adamantinoma was made. Positron emission tomography and magnetic resonance imaging revealed new lytic lesions involving the T10 and L2 vertebrae (without frank spinal cord compression) and the right superior sacrum. Additionally, a small pulmonary nodule on the left upper lobe was noted on positron emission tomography, but it was below the size threshold for reliable detection. A computed tomography–guided biopsy of the T10 lesion demonstrated metastatic adamantinoma. The patient underwent a spinal stabilization procedure and discussed options regarding further oncologic and palliative management.
Adamantinoma is an extremely rare primary malignant bone tumor that typically involves the anterior portion of the tibial metaphysis or diaphysis in approximately 90% of cases. Young adults most commonly are affected in the third or fourth decades of life.1 Although the histogenesis is not clearly understood, experts have theorized that fetal implantation during embryogenesis or traumatic implantation of epithelial cells may be causes of this tumor and may explain the close pathologic similarity to basal cell carcinoma.2
Adamantinomas are slow growing, and as a result, patients often present with gradual onset of pain and swelling that persists for years.3,4 Metastasis occurs in 10% to 30% of patients, typically located in regional lymph nodes, the lungs, and distant bone.1,4 Our case represents a rare instance of adamantinoma metastasis to the skin. Although primary adamantinomas consist of both epithelial and stromal components, the typical metastatic lesions of adamantinomas are solely epithelial (often in a spindle-cell pattern),1 as was seen in our patient.
Operative removal via amputation or en bloc resection with limb salvage is the current treatment of choice. Adamantinomas are highly radioresistant, and chemotherapy has shown minimal efficacy.3,5
In conclusion, the presence of cutaneous metastasis from an adamantinoma is rare. Our case emphasizes this tumor’s potential for late metastasis as well as late recurrence.3,6 Most importantly, dermatologists should be made aware of this rare bone tumor and its unusual presentation, as early detection can aid in prognosis.
- Schowinsky JT, Ormond DR, Kleinschmidt-DeMasters BK. Tibial adamantinoma: late metastasis to the brain. J Neuropathol Exp Neurol. 2015;74:95-97.
- Jain D, Jain VK, Vasishta RK, et al. Adamantinoma: a clinicopathological review and update. Diagn Pathol. 2008;3:8.
- Qureshi AA, Shott S, Mallin BA, et al. Current trends in the management of adamantinoma of long bones. an international study. J Bone Joint Surg Am. 2000;82-A:1122-1131.
- Desai SS, Jambhekar N, Agarwal M, et al. Adamantinoma of tibia: a study of 12 cases. J Surg Oncol. 2006;93:429-433.
- Weiss SW, Dorfman HD. Adamantinoma of long bone. an analysis of nine new cases with emphasis on metastasizing lesions and fibrous dysplasia-like changes. Hum Pathol. 1977;8:141-153.
- Szendroi M, Antal I, Arató G. Adamantinoma of long bones: a long-term follow-up study of 11 cases. Pathol Oncol Res. 2009;15:209-216.
To the Editor:
A 34-year-old woman with a history of adamantinoma of the right tibia that had been surgically resected with tibial reconstruction 5 years prior presented with a mildly tender, enlarging lesion on the right distal shin of 6 months’ duration that had started to change color. Review of systems was otherwise negative. Physical examination revealed an 8-mm, slightly tender, rubbery, pink papule adjacent to the surgical scar over the right tibia (Figure 1). Given the rapid growth of the lesion and its proximity to the surgical site, a punch biopsy was performed.
Histopathologic examination demonstrated a densely cellular dermal tumor composed of spindle cells with large hyperchromatic nuclei, numerous mitotic figures, and minimal eosinophilic cytoplasm (Figure 2A). Immunohistochemical studies revealed that approximately 40% of the tumor nuclei were immunoreactive to Ki-67 (Figure 2B), and total cytokeratin was focally positive (Figure 2C). A diagnosis of metastatic adamantinoma was made. Positron emission tomography and magnetic resonance imaging revealed new lytic lesions involving the T10 and L2 vertebrae (without frank spinal cord compression) and the right superior sacrum. Additionally, a small pulmonary nodule on the left upper lobe was noted on positron emission tomography, but it was below the size threshold for reliable detection. A computed tomography–guided biopsy of the T10 lesion demonstrated metastatic adamantinoma. The patient underwent a spinal stabilization procedure and discussed options regarding further oncologic and palliative management.
Adamantinoma is an extremely rare primary malignant bone tumor that typically involves the anterior portion of the tibial metaphysis or diaphysis in approximately 90% of cases. Young adults most commonly are affected in the third or fourth decades of life.1 Although the histogenesis is not clearly understood, experts have theorized that fetal implantation during embryogenesis or traumatic implantation of epithelial cells may be causes of this tumor and may explain the close pathologic similarity to basal cell carcinoma.2
Adamantinomas are slow growing, and as a result, patients often present with gradual onset of pain and swelling that persists for years.3,4 Metastasis occurs in 10% to 30% of patients, typically located in regional lymph nodes, the lungs, and distant bone.1,4 Our case represents a rare instance of adamantinoma metastasis to the skin. Although primary adamantinomas consist of both epithelial and stromal components, the typical metastatic lesions of adamantinomas are solely epithelial (often in a spindle-cell pattern),1 as was seen in our patient.
Operative removal via amputation or en bloc resection with limb salvage is the current treatment of choice. Adamantinomas are highly radioresistant, and chemotherapy has shown minimal efficacy.3,5
In conclusion, the presence of cutaneous metastasis from an adamantinoma is rare. Our case emphasizes this tumor’s potential for late metastasis as well as late recurrence.3,6 Most importantly, dermatologists should be made aware of this rare bone tumor and its unusual presentation, as early detection can aid in prognosis.
To the Editor:
A 34-year-old woman with a history of adamantinoma of the right tibia that had been surgically resected with tibial reconstruction 5 years prior presented with a mildly tender, enlarging lesion on the right distal shin of 6 months’ duration that had started to change color. Review of systems was otherwise negative. Physical examination revealed an 8-mm, slightly tender, rubbery, pink papule adjacent to the surgical scar over the right tibia (Figure 1). Given the rapid growth of the lesion and its proximity to the surgical site, a punch biopsy was performed.
Histopathologic examination demonstrated a densely cellular dermal tumor composed of spindle cells with large hyperchromatic nuclei, numerous mitotic figures, and minimal eosinophilic cytoplasm (Figure 2A). Immunohistochemical studies revealed that approximately 40% of the tumor nuclei were immunoreactive to Ki-67 (Figure 2B), and total cytokeratin was focally positive (Figure 2C). A diagnosis of metastatic adamantinoma was made. Positron emission tomography and magnetic resonance imaging revealed new lytic lesions involving the T10 and L2 vertebrae (without frank spinal cord compression) and the right superior sacrum. Additionally, a small pulmonary nodule on the left upper lobe was noted on positron emission tomography, but it was below the size threshold for reliable detection. A computed tomography–guided biopsy of the T10 lesion demonstrated metastatic adamantinoma. The patient underwent a spinal stabilization procedure and discussed options regarding further oncologic and palliative management.
Adamantinoma is an extremely rare primary malignant bone tumor that typically involves the anterior portion of the tibial metaphysis or diaphysis in approximately 90% of cases. Young adults most commonly are affected in the third or fourth decades of life.1 Although the histogenesis is not clearly understood, experts have theorized that fetal implantation during embryogenesis or traumatic implantation of epithelial cells may be causes of this tumor and may explain the close pathologic similarity to basal cell carcinoma.2
Adamantinomas are slow growing, and as a result, patients often present with gradual onset of pain and swelling that persists for years.3,4 Metastasis occurs in 10% to 30% of patients, typically located in regional lymph nodes, the lungs, and distant bone.1,4 Our case represents a rare instance of adamantinoma metastasis to the skin. Although primary adamantinomas consist of both epithelial and stromal components, the typical metastatic lesions of adamantinomas are solely epithelial (often in a spindle-cell pattern),1 as was seen in our patient.
Operative removal via amputation or en bloc resection with limb salvage is the current treatment of choice. Adamantinomas are highly radioresistant, and chemotherapy has shown minimal efficacy.3,5
In conclusion, the presence of cutaneous metastasis from an adamantinoma is rare. Our case emphasizes this tumor’s potential for late metastasis as well as late recurrence.3,6 Most importantly, dermatologists should be made aware of this rare bone tumor and its unusual presentation, as early detection can aid in prognosis.
- Schowinsky JT, Ormond DR, Kleinschmidt-DeMasters BK. Tibial adamantinoma: late metastasis to the brain. J Neuropathol Exp Neurol. 2015;74:95-97.
- Jain D, Jain VK, Vasishta RK, et al. Adamantinoma: a clinicopathological review and update. Diagn Pathol. 2008;3:8.
- Qureshi AA, Shott S, Mallin BA, et al. Current trends in the management of adamantinoma of long bones. an international study. J Bone Joint Surg Am. 2000;82-A:1122-1131.
- Desai SS, Jambhekar N, Agarwal M, et al. Adamantinoma of tibia: a study of 12 cases. J Surg Oncol. 2006;93:429-433.
- Weiss SW, Dorfman HD. Adamantinoma of long bone. an analysis of nine new cases with emphasis on metastasizing lesions and fibrous dysplasia-like changes. Hum Pathol. 1977;8:141-153.
- Szendroi M, Antal I, Arató G. Adamantinoma of long bones: a long-term follow-up study of 11 cases. Pathol Oncol Res. 2009;15:209-216.
- Schowinsky JT, Ormond DR, Kleinschmidt-DeMasters BK. Tibial adamantinoma: late metastasis to the brain. J Neuropathol Exp Neurol. 2015;74:95-97.
- Jain D, Jain VK, Vasishta RK, et al. Adamantinoma: a clinicopathological review and update. Diagn Pathol. 2008;3:8.
- Qureshi AA, Shott S, Mallin BA, et al. Current trends in the management of adamantinoma of long bones. an international study. J Bone Joint Surg Am. 2000;82-A:1122-1131.
- Desai SS, Jambhekar N, Agarwal M, et al. Adamantinoma of tibia: a study of 12 cases. J Surg Oncol. 2006;93:429-433.
- Weiss SW, Dorfman HD. Adamantinoma of long bone. an analysis of nine new cases with emphasis on metastasizing lesions and fibrous dysplasia-like changes. Hum Pathol. 1977;8:141-153.
- Szendroi M, Antal I, Arató G. Adamantinoma of long bones: a long-term follow-up study of 11 cases. Pathol Oncol Res. 2009;15:209-216.
Practice Points
- Metastatic adamantinoma of the skin is a rare clinical scenario.
- Dermatologists should be made aware of this rare bone tumor and its unusual presentation, as early detection can aid in prognosis.
A Unique Presentation of Lupus Erythematosus Tumidus in an Adolescent Boy
To the Editor:
Lupus erythematosus tumidus (LET) is a rarely diagnosed condition that was first described in 1909 by Hoffmann.1 Limited cases have been reported in the literature, with few documenting the disease in children.2 We report a unique clinical case of LET in a 14-year-old adolescent boy that was distributed solely on the hands. With slight heterogeneity in regards to clinical presentation and histopathology, there is a need for further exploration with regard to LET.
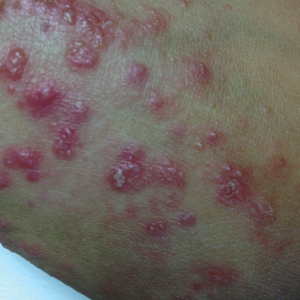
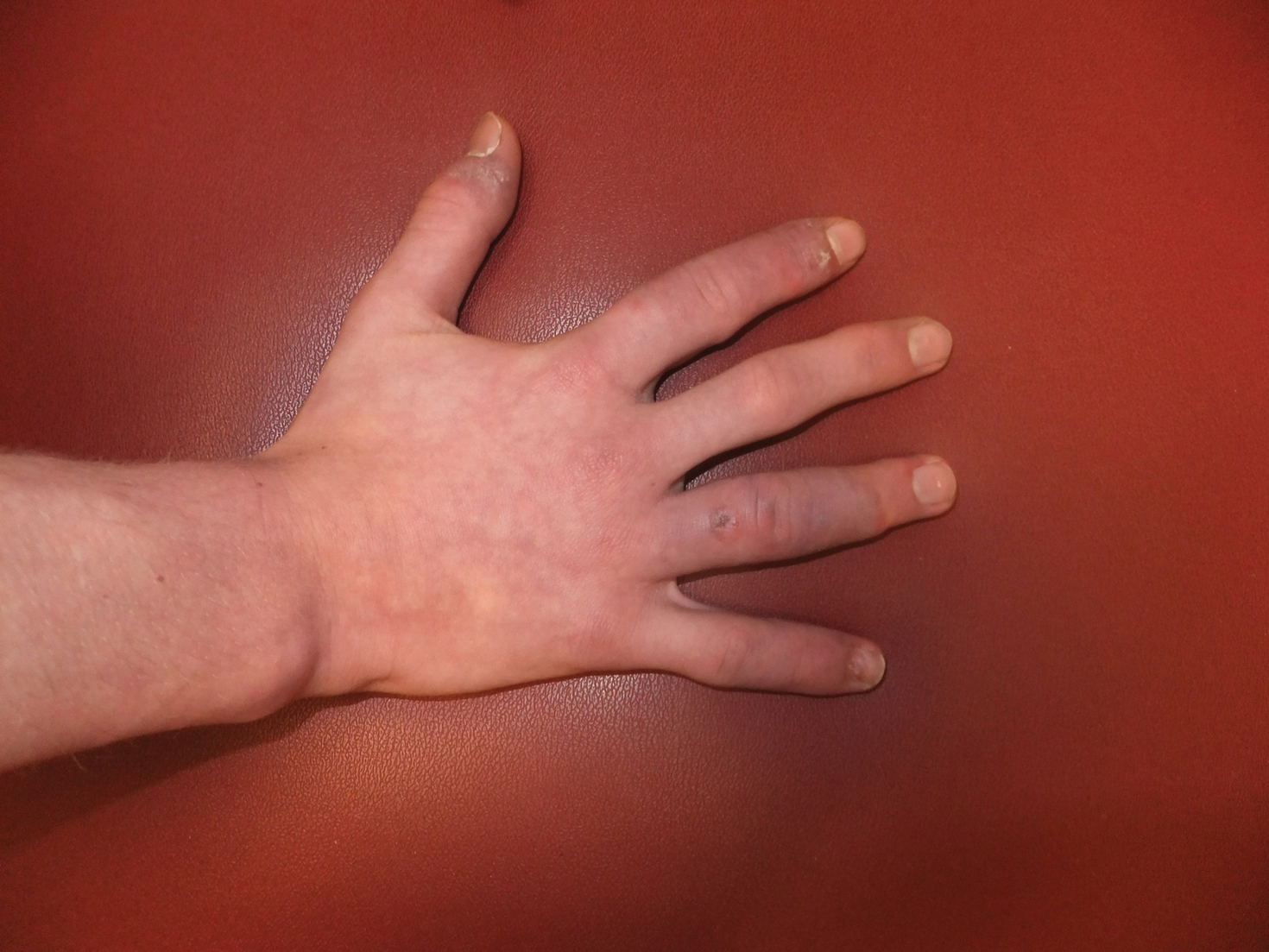
A 14-year-old adolescent boy presented to the dermatology clinic with progressive bilateral edema of 1 year’s duration with plaques and some scaling on the dorsal aspects of the digits and the nail bases predominantly on the right hand (Figure 1) and to a lesser extent on the left hand. The edema, erythema, and tenderness started in the right fifth digit; soon after the edema appeared, plaques began to form at the base of each nail bed, and the edema and erythema progressively spread to the other digits. He denied worsening of symptoms when exposed to cold temperatures. A complete review of systems was negative. The differential diagnoses included chilblain lupus erythematosus, perniosis, dermatomyositis, and polymorphous light eruption. A punch biopsy from the right fourth digit was performed.
The biopsy showed superficial and deep perivascular and periadnexal mononuclear inflammation with large amounts of interstitial mucin deposition (Figure 2). The epidermis exhibited a loose orthokeratotic scale with no signs of interface damage. A diagnosis of perniosis was entertained but was ruled out due to the lack of papillary dermal edema and large amounts of mucin. With the lack of interface change and large amounts of mucin, a diagnosis of LET was favored over chilblain lupus erythematosus, as the latter diagnosis typically demonstrates interface change. The patient was started on hydroxychloroquine 200 mg twice daily and a short course of prednisone, and improvement of the lesions/plaques was noted at follow-up 6 weeks later. Continued improvement was noted 2 years after the initial presentation. His condition recurred when the hydroxychloroquine dosage was reduced to 200 mg once daily after 1 year. The patient did not report any adverse sequelae to treatment.
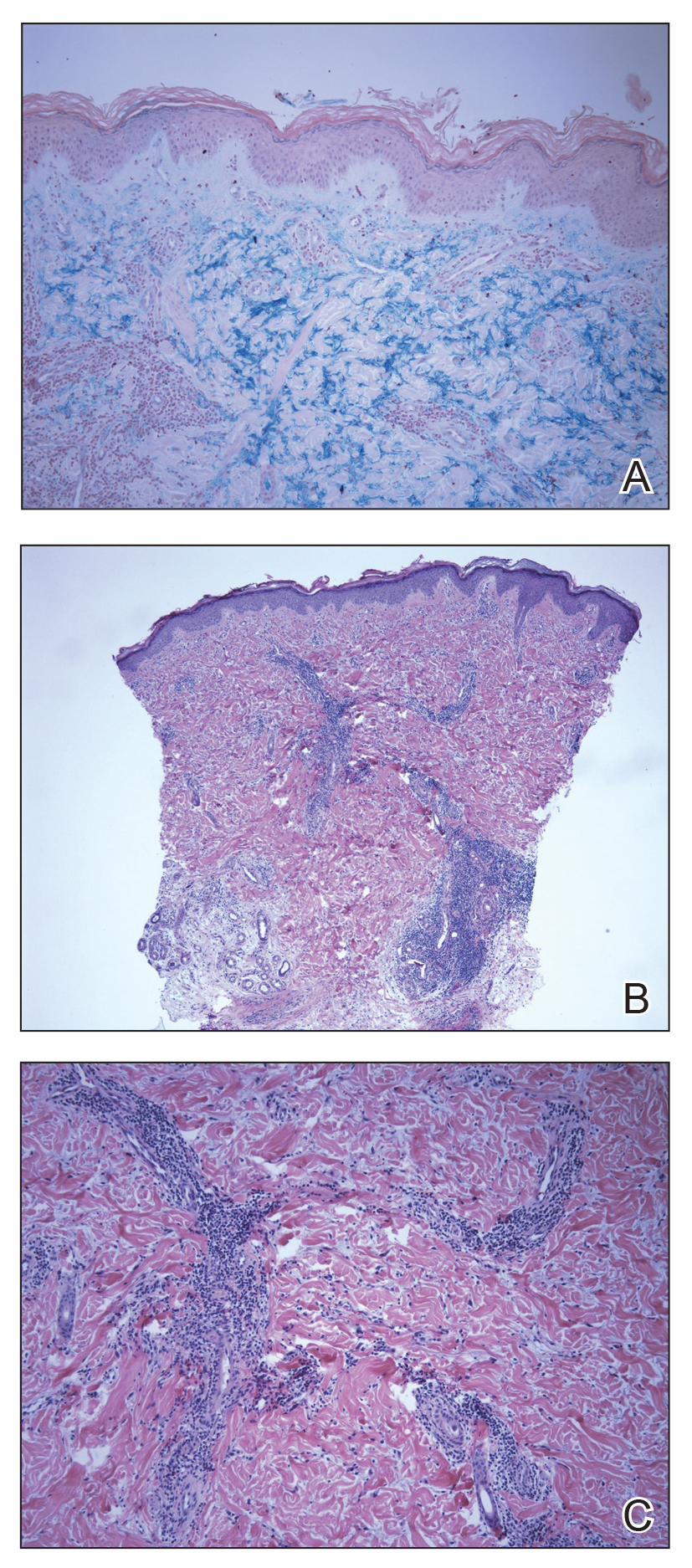
Histopathologic findings of superficial and deep perivascular and periadnexal lymphocytic infiltrates and interstitial dermal deposition of mucin in LET have remained consistent in the literature. Direct immunofluorescence has not revealed any complement or immunoglobulin deposition on the basement membrane.3,4 The epidermal characteristics are not as uniform, with the majority of cases in one review showing no epidermal changes and a minority showing minimal epidermal changes (eg, epidermal atrophy, hyperkeratosis, parakeratosis, acanthosis, spongiosis).5 When working up patients for LET, blood work usually is unremarkable, as LET rarely is associated with antinuclear antibodies or anti-Ro, anti-La, and anti-DNA antibodies.3,4 Lupus erythematosus tumidus generally is an independent process, but it has been reported to coexist with discoid lupus erythematosus and systemic lupus erythematosus in rare cases.6
The lesions of LET have been consistently described in the literature as photosensitive, erythematous, non-scarring, annular plaques and papules commonly occurring on the head/neck and other sun-exposed areas that do not cause hypopigmentation.3 Treatment of LET consists of systemic treatment with antimalarial drugs, sunscreens, and topical steroids for flares.
Lupus erythematosus tumidus is rare in children, with few case reports noted in the literature. Sonntag et al2 documented the disease in 3 children ranging from 3 to 8 years of age. Furthermore, Ruiz and Sanchez7 reported a case of LET in a 16-year-old adolescent girl. Our case is unique in that the lesions only occurred on the hands, whereas most case reports document distribution of the lesions on the head, neck, face, arms, back, and chest. Our patient’s age and the location of the lesions make it a unique clinical presentation of LET.
Reports in the literature show evidence of heterogeneity in the presentation, classification, and some of the histopathologic features of LET; however, there are minimal data on childhood LET. Further research and investigations are needed to more precisely define this condition.
Acknowledgment
The authors acknowledge Richard Schwartz, MD (Akron, Ohio), for reading the biopsy reports and assisting with photomicrographs.
- Hoffmann E. Demonstrationen: lupus erythematosus tumidus. Derm Zeitschr. 1909;16:159-160.
- Sonntag M, Lehmann P, Megahed M, et al. Lupus erythematosus tumidus in childhood. Dermatology. 2003;207:188-192.
- Schmitt V, Meuth AM, Amler S, et al. Lupus erythematosus tumidus is a separate subtype of cutaneous lupus erythematosus. Br J Dermatol. 2010;162:64-73.
- Vieira V, Del Pozo J, Yebra-Pimentel MT, et al. Lupus erythematosus tumidus: a series of 26 cases. Int J Dermatol. 2006;45:512-517.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Chen X, Wang S, Li L. A case report of lupus erythematosus tumidus converted from discoid lupus erythematosus. Medicine (Baltimore). 2018;97:e0375.
- Ruiz H, Sanchez J. Tumid lupus erythematosus. Am J Dermatopathol. 1999;21:356-360.
To the Editor:
Lupus erythematosus tumidus (LET) is a rarely diagnosed condition that was first described in 1909 by Hoffmann.1 Limited cases have been reported in the literature, with few documenting the disease in children.2 We report a unique clinical case of LET in a 14-year-old adolescent boy that was distributed solely on the hands. With slight heterogeneity in regards to clinical presentation and histopathology, there is a need for further exploration with regard to LET.
A 14-year-old adolescent boy presented to the dermatology clinic with progressive bilateral edema of 1 year’s duration with plaques and some scaling on the dorsal aspects of the digits and the nail bases predominantly on the right hand (Figure 1) and to a lesser extent on the left hand. The edema, erythema, and tenderness started in the right fifth digit; soon after the edema appeared, plaques began to form at the base of each nail bed, and the edema and erythema progressively spread to the other digits. He denied worsening of symptoms when exposed to cold temperatures. A complete review of systems was negative. The differential diagnoses included chilblain lupus erythematosus, perniosis, dermatomyositis, and polymorphous light eruption. A punch biopsy from the right fourth digit was performed.
The biopsy showed superficial and deep perivascular and periadnexal mononuclear inflammation with large amounts of interstitial mucin deposition (Figure 2). The epidermis exhibited a loose orthokeratotic scale with no signs of interface damage. A diagnosis of perniosis was entertained but was ruled out due to the lack of papillary dermal edema and large amounts of mucin. With the lack of interface change and large amounts of mucin, a diagnosis of LET was favored over chilblain lupus erythematosus, as the latter diagnosis typically demonstrates interface change. The patient was started on hydroxychloroquine 200 mg twice daily and a short course of prednisone, and improvement of the lesions/plaques was noted at follow-up 6 weeks later. Continued improvement was noted 2 years after the initial presentation. His condition recurred when the hydroxychloroquine dosage was reduced to 200 mg once daily after 1 year. The patient did not report any adverse sequelae to treatment.
Histopathologic findings of superficial and deep perivascular and periadnexal lymphocytic infiltrates and interstitial dermal deposition of mucin in LET have remained consistent in the literature. Direct immunofluorescence has not revealed any complement or immunoglobulin deposition on the basement membrane.3,4 The epidermal characteristics are not as uniform, with the majority of cases in one review showing no epidermal changes and a minority showing minimal epidermal changes (eg, epidermal atrophy, hyperkeratosis, parakeratosis, acanthosis, spongiosis).5 When working up patients for LET, blood work usually is unremarkable, as LET rarely is associated with antinuclear antibodies or anti-Ro, anti-La, and anti-DNA antibodies.3,4 Lupus erythematosus tumidus generally is an independent process, but it has been reported to coexist with discoid lupus erythematosus and systemic lupus erythematosus in rare cases.6
The lesions of LET have been consistently described in the literature as photosensitive, erythematous, non-scarring, annular plaques and papules commonly occurring on the head/neck and other sun-exposed areas that do not cause hypopigmentation.3 Treatment of LET consists of systemic treatment with antimalarial drugs, sunscreens, and topical steroids for flares.
Lupus erythematosus tumidus is rare in children, with few case reports noted in the literature. Sonntag et al2 documented the disease in 3 children ranging from 3 to 8 years of age. Furthermore, Ruiz and Sanchez7 reported a case of LET in a 16-year-old adolescent girl. Our case is unique in that the lesions only occurred on the hands, whereas most case reports document distribution of the lesions on the head, neck, face, arms, back, and chest. Our patient’s age and the location of the lesions make it a unique clinical presentation of LET.
Reports in the literature show evidence of heterogeneity in the presentation, classification, and some of the histopathologic features of LET; however, there are minimal data on childhood LET. Further research and investigations are needed to more precisely define this condition.
Acknowledgment
The authors acknowledge Richard Schwartz, MD (Akron, Ohio), for reading the biopsy reports and assisting with photomicrographs.
To the Editor:
Lupus erythematosus tumidus (LET) is a rarely diagnosed condition that was first described in 1909 by Hoffmann.1 Limited cases have been reported in the literature, with few documenting the disease in children.2 We report a unique clinical case of LET in a 14-year-old adolescent boy that was distributed solely on the hands. With slight heterogeneity in regards to clinical presentation and histopathology, there is a need for further exploration with regard to LET.
A 14-year-old adolescent boy presented to the dermatology clinic with progressive bilateral edema of 1 year’s duration with plaques and some scaling on the dorsal aspects of the digits and the nail bases predominantly on the right hand (Figure 1) and to a lesser extent on the left hand. The edema, erythema, and tenderness started in the right fifth digit; soon after the edema appeared, plaques began to form at the base of each nail bed, and the edema and erythema progressively spread to the other digits. He denied worsening of symptoms when exposed to cold temperatures. A complete review of systems was negative. The differential diagnoses included chilblain lupus erythematosus, perniosis, dermatomyositis, and polymorphous light eruption. A punch biopsy from the right fourth digit was performed.
The biopsy showed superficial and deep perivascular and periadnexal mononuclear inflammation with large amounts of interstitial mucin deposition (Figure 2). The epidermis exhibited a loose orthokeratotic scale with no signs of interface damage. A diagnosis of perniosis was entertained but was ruled out due to the lack of papillary dermal edema and large amounts of mucin. With the lack of interface change and large amounts of mucin, a diagnosis of LET was favored over chilblain lupus erythematosus, as the latter diagnosis typically demonstrates interface change. The patient was started on hydroxychloroquine 200 mg twice daily and a short course of prednisone, and improvement of the lesions/plaques was noted at follow-up 6 weeks later. Continued improvement was noted 2 years after the initial presentation. His condition recurred when the hydroxychloroquine dosage was reduced to 200 mg once daily after 1 year. The patient did not report any adverse sequelae to treatment.
Histopathologic findings of superficial and deep perivascular and periadnexal lymphocytic infiltrates and interstitial dermal deposition of mucin in LET have remained consistent in the literature. Direct immunofluorescence has not revealed any complement or immunoglobulin deposition on the basement membrane.3,4 The epidermal characteristics are not as uniform, with the majority of cases in one review showing no epidermal changes and a minority showing minimal epidermal changes (eg, epidermal atrophy, hyperkeratosis, parakeratosis, acanthosis, spongiosis).5 When working up patients for LET, blood work usually is unremarkable, as LET rarely is associated with antinuclear antibodies or anti-Ro, anti-La, and anti-DNA antibodies.3,4 Lupus erythematosus tumidus generally is an independent process, but it has been reported to coexist with discoid lupus erythematosus and systemic lupus erythematosus in rare cases.6
The lesions of LET have been consistently described in the literature as photosensitive, erythematous, non-scarring, annular plaques and papules commonly occurring on the head/neck and other sun-exposed areas that do not cause hypopigmentation.3 Treatment of LET consists of systemic treatment with antimalarial drugs, sunscreens, and topical steroids for flares.
Lupus erythematosus tumidus is rare in children, with few case reports noted in the literature. Sonntag et al2 documented the disease in 3 children ranging from 3 to 8 years of age. Furthermore, Ruiz and Sanchez7 reported a case of LET in a 16-year-old adolescent girl. Our case is unique in that the lesions only occurred on the hands, whereas most case reports document distribution of the lesions on the head, neck, face, arms, back, and chest. Our patient’s age and the location of the lesions make it a unique clinical presentation of LET.
Reports in the literature show evidence of heterogeneity in the presentation, classification, and some of the histopathologic features of LET; however, there are minimal data on childhood LET. Further research and investigations are needed to more precisely define this condition.
Acknowledgment
The authors acknowledge Richard Schwartz, MD (Akron, Ohio), for reading the biopsy reports and assisting with photomicrographs.
- Hoffmann E. Demonstrationen: lupus erythematosus tumidus. Derm Zeitschr. 1909;16:159-160.
- Sonntag M, Lehmann P, Megahed M, et al. Lupus erythematosus tumidus in childhood. Dermatology. 2003;207:188-192.
- Schmitt V, Meuth AM, Amler S, et al. Lupus erythematosus tumidus is a separate subtype of cutaneous lupus erythematosus. Br J Dermatol. 2010;162:64-73.
- Vieira V, Del Pozo J, Yebra-Pimentel MT, et al. Lupus erythematosus tumidus: a series of 26 cases. Int J Dermatol. 2006;45:512-517.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Chen X, Wang S, Li L. A case report of lupus erythematosus tumidus converted from discoid lupus erythematosus. Medicine (Baltimore). 2018;97:e0375.
- Ruiz H, Sanchez J. Tumid lupus erythematosus. Am J Dermatopathol. 1999;21:356-360.
- Hoffmann E. Demonstrationen: lupus erythematosus tumidus. Derm Zeitschr. 1909;16:159-160.
- Sonntag M, Lehmann P, Megahed M, et al. Lupus erythematosus tumidus in childhood. Dermatology. 2003;207:188-192.
- Schmitt V, Meuth AM, Amler S, et al. Lupus erythematosus tumidus is a separate subtype of cutaneous lupus erythematosus. Br J Dermatol. 2010;162:64-73.
- Vieira V, Del Pozo J, Yebra-Pimentel MT, et al. Lupus erythematosus tumidus: a series of 26 cases. Int J Dermatol. 2006;45:512-517.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Chen X, Wang S, Li L. A case report of lupus erythematosus tumidus converted from discoid lupus erythematosus. Medicine (Baltimore). 2018;97:e0375.
- Ruiz H, Sanchez J. Tumid lupus erythematosus. Am J Dermatopathol. 1999;21:356-360.
Practice Points
- Lupus erythematosus tumidus rarely occurs in the pediatric population.
- Lupus erythematosus tumidus is a unique subset of lupus associated with lack of interface change on histology and large amounts of mucin.
- Lesions typically present on the face and trunk but can very rarely present on the extremities and hands.
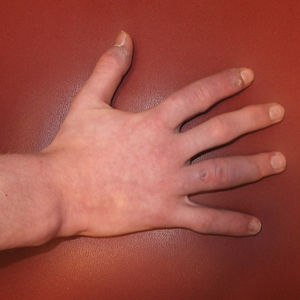
Solar Urticaria Treated With Omalizumab
To the Editor:
First documented in 1904,1 solar urticaria is an IgE-induced condition that predominantly occurs in women aged 20 to 50 years. Worldwide prevalence and incidence information is lacking, but it is known to occur in up to 0.4% of urticaria cases.2 Solar urticaria is characterized by pruritus of the skin with erythematous wheals and flares in reaction to sunlight exposure, even despite partial protection by barriers such as glass or clothing.2,3 It can have an acute or chronic presentation caused by visible or UV light wavelengths. Solar urticaria can lead to debilitating symptoms and psychological stressors that can severely impact a patient’s well-being and also may be accompanied by conditions such as polymorphous light eruption, angioedema, or vasculitis.4 Standard treatments include first- and second-generation antihistamines, which are efficacious approximately 50% of the time, as well as phototherapy, which can be time consuming and a burden on patients who work or go to school full time.2 Other possible treatment modalities include plasmapheresis, intravenous immunoglobulins, steroids, cyclosporine, and anti-IgE recombinant monoclonal antibody injections.5,6 We present the case of a patient who was successfully treated with subcutaneous injections of omalizumab every 3 weeks to add to the growing number of case reports of treatment of solar urticaria.
A 30-year-old woman with Fitzpatrick skin type III and a 9-year history of solar urticaria was referred to the Department of Allergy and Immunology by her primary care physician. The patient reported that redness, swelling, and itching would occur on sun-exposed areas of the skin after approximately 10 minutes of exposure despite daily sunscreen application. She had been successfully treated with hydroxychloroquine 400 mg once daily after her first formal evaluation by dermatology 4 years prior to the current presentation. She subsequently self-discontinued treatment after 8 months of treatment due to resolution of symptoms. She noted the symptoms had returned upon relocating to Hawaii after living in the continental United States and Italy. Initially she was restarted on hydroxychloroquine 200 mg once daily and 4-times the recommended daily dose of second-generation antihistamines without relief. The hydroxychloroquine dosage subsequently was increased to 400 mg once daily, but her symptoms did not resolve.
On physical examination, sun-exposed areas of the skin showed marked macular erythema with discrete erythematous lines of demarcation observed between exposed and unexposed skin. The patient also reported concomitant pruritus, which antihistamines did not alleviate. A maximum 1-year course of cyclosporine 300 mg once daily initially was planned but was discontinued due to immediate onset of severe nausea and emesis after the first dose as well as continued outbreaks of urticaria for 1 month after incrementally increasing by 100 mg from a starting dose of 100 mg.
After discussion with the dermatology department, a trial of omalizumab was started because the daily impact of a UV light sensitization course was not feasible with her work schedule, and serum IgE blood level was 560.4 µg/L (reference range, 0–1500 µg/L). The patient was started on a regimen of omalizumab 300 mg (subcutaneous injections) every 2 weeks with noted improvement after the third dose, with no urticarial symptoms after sun exposure. After 2 months, the dosage interval was increased to every 4 weeks given her level of improvement, but her symptoms recurred. The treatment regimen was then changed to every 3 weeks. The patient was symptom free for a period of 10 months on this regimen, followed by only 1 outbreak of erythema and urticaria, which occurred 1 day prior to a scheduled omalizumab injection. Symptoms have otherwise been well controlled to date on omalizumab.
Solar urticaria is a poorly understood phenomenon that has no clear prognostic indicators; therefore, diagnosis often is made based on the patient’s history and physical examination. Further testing to confirm the diagnosis can be performed using specific wavelengths of UV light to determine which band of light affects patients most; however, the wavelength can change over time, leading to less clinical significance, and may decrease efficacy of phototherapy.2 Solar urticaria has no clear predisposing factors, and treatments to date have been moderately successful. Exposure to sunlight is thought to initiate an alteration in a skin or serum chromophore or photoallergen, which then causes subsequent cross-linking and IgE-dependent release of histamine as well as other mediators such as cytokines, eicosanoids, and proteases with mast cell degranulation.7
Omalizumab is a recombinant humanized monoclonal IgG1 antibody targeting the methylated IgE Cε3 domain that initially was marketed toward controlling IgE-mediated moderate to severe asthma recalcitrant to standard treatments. It has since received approval from the US Food and Drug Administration for treatment of chronic idiopathic urticaria after first being noticed to serendipitously treat a patient with cold urticaria and asthma in 2006.4,7,8 It was then first documented to successfully treat solar urticaria in 2008.6 The safety profile of omalizumab makes it a more favorable choice when compared to other immunomodulating treatments, with the most serious adverse reaction being anaphylaxis, occurring in 0.2% of patients in a postmarketing study.9 It functions through binding to free IgE at a region necessary for IgE to bind at low- and high-affinity receptors but not to immunoglobulins already bound to cells, thus theoretically preventing activation of mast cells or basophils.10 It also has been suggested that low steady-state values are needed to see continued benefit from the drug,10 which may have been seen in our patient after having an outbreak just prior to receiving an injection; however, prior reports have shown benefit unrelated to total IgE levels, with improvement after days to 4 months.4,10,11 One case report showed no response after 4 doses; it is unknown if this patient was tested for clinical improvement to omalizumab through further immunoglobulin analysis, but treatment response is important to consider when deciding on whether to use this drug in future patients.12 It is unknown why some patients will respond to omalizumab, others will partially respond, and others will not respond, which can be ascertained either through quality-of-life improvement or lack thereof.
In our experience, omalizumab is a viable option to consider in patients with solar urticaria that is recalcitrant to standard treatments and elevated IgE levels for whom other treatments are either too time consuming or have side-effect profiles that are not tolerable to the patient. If the patient has concomitant asthma, there may be additional therapeutic benefit. Further research is needed with regard to a cost-benefit analysis of omalizumab and whether using such a costly drug outweighs the cost associated with time and resources utilized with repeat clinic visits if other standard treatments are not effective.13
- Merkin P. Pratique Dermatologique. Paris, France: Masso; 1904.
- Beattie PE, Dawe RS, Ibbotson SH, et al. Characteristics and prognosis of idiopathic solar urticaria: a cohort of 87 cases. Arch Dermatol. 2003;139:1149-1154.
- Kaplan AP. Therapy of chronic urticaria: a simple, modern approach. Ann Allergy Asthma Immunol. 2014;112:419-425.
- Metz M, Maurer M. Omalizumab in chronic urticaria. Curr Opin Allergy Clin Immunol. 2012;12:406-410.
- Aubin F, Porcher R, Jeanmougin M, et al. Severe and refractory solar urticaria treated with intravenous immunoglobulins: a phase II multicenter study. J Am Acad Dermatol. 2014;71:948-953.e1.
- Güzelbey O, Ardelean E, Magerl M, et al. Successful treatment of solar urticaria with anti-immunoglobulin E therapy. Allergy. 2008;63:1563-1565.
- Wu K, Jabbar-Lopez Z. Omalizumab, an anti-IgE mAb, receives approval for the treatment of chronic idiopathic/spontaneous urticaria. J Invest Dermatol. 2015;135:13-15.
- Boyce JA. Successful treatment of cold-induced urticaria/anaphylaxis with anti-IgE. J Allergy Clin Immunol. 2006;117:1415-1418.
- Corren J, Casale TB, Lanier B, et al. Safety and tolerability of omalizumab. Clin Exp Allergy. 2009;39:788-797.
- Wu K, Long H. Omalizumab for chronic urticaria. N Engl J Med. 2013;368:2527-2528.
- Morgado-Carrasco D, Giacaman-Von der Weth M, Fusta-Novell X, et al. Clinical response and long-term follow-up of 20 patients with refractory solar urticarial under treatment with omalizumab [published online May 28, 2019]. J Am Acad Dermatol. doi:10.1016/j.jaad.2019.05.070.
- Duchini G, Bäumler W, Bircher AJ, et al. Failure of omalizumab (Xolair®) in the treatment of a case of solar urticaria caused by ultraviolet A and visible light. Photodermatol Photoimmunol Photomed. 2011;27:336-337.
- Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133:1270-1277.
To the Editor:
First documented in 1904,1 solar urticaria is an IgE-induced condition that predominantly occurs in women aged 20 to 50 years. Worldwide prevalence and incidence information is lacking, but it is known to occur in up to 0.4% of urticaria cases.2 Solar urticaria is characterized by pruritus of the skin with erythematous wheals and flares in reaction to sunlight exposure, even despite partial protection by barriers such as glass or clothing.2,3 It can have an acute or chronic presentation caused by visible or UV light wavelengths. Solar urticaria can lead to debilitating symptoms and psychological stressors that can severely impact a patient’s well-being and also may be accompanied by conditions such as polymorphous light eruption, angioedema, or vasculitis.4 Standard treatments include first- and second-generation antihistamines, which are efficacious approximately 50% of the time, as well as phototherapy, which can be time consuming and a burden on patients who work or go to school full time.2 Other possible treatment modalities include plasmapheresis, intravenous immunoglobulins, steroids, cyclosporine, and anti-IgE recombinant monoclonal antibody injections.5,6 We present the case of a patient who was successfully treated with subcutaneous injections of omalizumab every 3 weeks to add to the growing number of case reports of treatment of solar urticaria.
A 30-year-old woman with Fitzpatrick skin type III and a 9-year history of solar urticaria was referred to the Department of Allergy and Immunology by her primary care physician. The patient reported that redness, swelling, and itching would occur on sun-exposed areas of the skin after approximately 10 minutes of exposure despite daily sunscreen application. She had been successfully treated with hydroxychloroquine 400 mg once daily after her first formal evaluation by dermatology 4 years prior to the current presentation. She subsequently self-discontinued treatment after 8 months of treatment due to resolution of symptoms. She noted the symptoms had returned upon relocating to Hawaii after living in the continental United States and Italy. Initially she was restarted on hydroxychloroquine 200 mg once daily and 4-times the recommended daily dose of second-generation antihistamines without relief. The hydroxychloroquine dosage subsequently was increased to 400 mg once daily, but her symptoms did not resolve.
On physical examination, sun-exposed areas of the skin showed marked macular erythema with discrete erythematous lines of demarcation observed between exposed and unexposed skin. The patient also reported concomitant pruritus, which antihistamines did not alleviate. A maximum 1-year course of cyclosporine 300 mg once daily initially was planned but was discontinued due to immediate onset of severe nausea and emesis after the first dose as well as continued outbreaks of urticaria for 1 month after incrementally increasing by 100 mg from a starting dose of 100 mg.
After discussion with the dermatology department, a trial of omalizumab was started because the daily impact of a UV light sensitization course was not feasible with her work schedule, and serum IgE blood level was 560.4 µg/L (reference range, 0–1500 µg/L). The patient was started on a regimen of omalizumab 300 mg (subcutaneous injections) every 2 weeks with noted improvement after the third dose, with no urticarial symptoms after sun exposure. After 2 months, the dosage interval was increased to every 4 weeks given her level of improvement, but her symptoms recurred. The treatment regimen was then changed to every 3 weeks. The patient was symptom free for a period of 10 months on this regimen, followed by only 1 outbreak of erythema and urticaria, which occurred 1 day prior to a scheduled omalizumab injection. Symptoms have otherwise been well controlled to date on omalizumab.
Solar urticaria is a poorly understood phenomenon that has no clear prognostic indicators; therefore, diagnosis often is made based on the patient’s history and physical examination. Further testing to confirm the diagnosis can be performed using specific wavelengths of UV light to determine which band of light affects patients most; however, the wavelength can change over time, leading to less clinical significance, and may decrease efficacy of phototherapy.2 Solar urticaria has no clear predisposing factors, and treatments to date have been moderately successful. Exposure to sunlight is thought to initiate an alteration in a skin or serum chromophore or photoallergen, which then causes subsequent cross-linking and IgE-dependent release of histamine as well as other mediators such as cytokines, eicosanoids, and proteases with mast cell degranulation.7
Omalizumab is a recombinant humanized monoclonal IgG1 antibody targeting the methylated IgE Cε3 domain that initially was marketed toward controlling IgE-mediated moderate to severe asthma recalcitrant to standard treatments. It has since received approval from the US Food and Drug Administration for treatment of chronic idiopathic urticaria after first being noticed to serendipitously treat a patient with cold urticaria and asthma in 2006.4,7,8 It was then first documented to successfully treat solar urticaria in 2008.6 The safety profile of omalizumab makes it a more favorable choice when compared to other immunomodulating treatments, with the most serious adverse reaction being anaphylaxis, occurring in 0.2% of patients in a postmarketing study.9 It functions through binding to free IgE at a region necessary for IgE to bind at low- and high-affinity receptors but not to immunoglobulins already bound to cells, thus theoretically preventing activation of mast cells or basophils.10 It also has been suggested that low steady-state values are needed to see continued benefit from the drug,10 which may have been seen in our patient after having an outbreak just prior to receiving an injection; however, prior reports have shown benefit unrelated to total IgE levels, with improvement after days to 4 months.4,10,11 One case report showed no response after 4 doses; it is unknown if this patient was tested for clinical improvement to omalizumab through further immunoglobulin analysis, but treatment response is important to consider when deciding on whether to use this drug in future patients.12 It is unknown why some patients will respond to omalizumab, others will partially respond, and others will not respond, which can be ascertained either through quality-of-life improvement or lack thereof.
In our experience, omalizumab is a viable option to consider in patients with solar urticaria that is recalcitrant to standard treatments and elevated IgE levels for whom other treatments are either too time consuming or have side-effect profiles that are not tolerable to the patient. If the patient has concomitant asthma, there may be additional therapeutic benefit. Further research is needed with regard to a cost-benefit analysis of omalizumab and whether using such a costly drug outweighs the cost associated with time and resources utilized with repeat clinic visits if other standard treatments are not effective.13
To the Editor:
First documented in 1904,1 solar urticaria is an IgE-induced condition that predominantly occurs in women aged 20 to 50 years. Worldwide prevalence and incidence information is lacking, but it is known to occur in up to 0.4% of urticaria cases.2 Solar urticaria is characterized by pruritus of the skin with erythematous wheals and flares in reaction to sunlight exposure, even despite partial protection by barriers such as glass or clothing.2,3 It can have an acute or chronic presentation caused by visible or UV light wavelengths. Solar urticaria can lead to debilitating symptoms and psychological stressors that can severely impact a patient’s well-being and also may be accompanied by conditions such as polymorphous light eruption, angioedema, or vasculitis.4 Standard treatments include first- and second-generation antihistamines, which are efficacious approximately 50% of the time, as well as phototherapy, which can be time consuming and a burden on patients who work or go to school full time.2 Other possible treatment modalities include plasmapheresis, intravenous immunoglobulins, steroids, cyclosporine, and anti-IgE recombinant monoclonal antibody injections.5,6 We present the case of a patient who was successfully treated with subcutaneous injections of omalizumab every 3 weeks to add to the growing number of case reports of treatment of solar urticaria.
A 30-year-old woman with Fitzpatrick skin type III and a 9-year history of solar urticaria was referred to the Department of Allergy and Immunology by her primary care physician. The patient reported that redness, swelling, and itching would occur on sun-exposed areas of the skin after approximately 10 minutes of exposure despite daily sunscreen application. She had been successfully treated with hydroxychloroquine 400 mg once daily after her first formal evaluation by dermatology 4 years prior to the current presentation. She subsequently self-discontinued treatment after 8 months of treatment due to resolution of symptoms. She noted the symptoms had returned upon relocating to Hawaii after living in the continental United States and Italy. Initially she was restarted on hydroxychloroquine 200 mg once daily and 4-times the recommended daily dose of second-generation antihistamines without relief. The hydroxychloroquine dosage subsequently was increased to 400 mg once daily, but her symptoms did not resolve.
On physical examination, sun-exposed areas of the skin showed marked macular erythema with discrete erythematous lines of demarcation observed between exposed and unexposed skin. The patient also reported concomitant pruritus, which antihistamines did not alleviate. A maximum 1-year course of cyclosporine 300 mg once daily initially was planned but was discontinued due to immediate onset of severe nausea and emesis after the first dose as well as continued outbreaks of urticaria for 1 month after incrementally increasing by 100 mg from a starting dose of 100 mg.
After discussion with the dermatology department, a trial of omalizumab was started because the daily impact of a UV light sensitization course was not feasible with her work schedule, and serum IgE blood level was 560.4 µg/L (reference range, 0–1500 µg/L). The patient was started on a regimen of omalizumab 300 mg (subcutaneous injections) every 2 weeks with noted improvement after the third dose, with no urticarial symptoms after sun exposure. After 2 months, the dosage interval was increased to every 4 weeks given her level of improvement, but her symptoms recurred. The treatment regimen was then changed to every 3 weeks. The patient was symptom free for a period of 10 months on this regimen, followed by only 1 outbreak of erythema and urticaria, which occurred 1 day prior to a scheduled omalizumab injection. Symptoms have otherwise been well controlled to date on omalizumab.
Solar urticaria is a poorly understood phenomenon that has no clear prognostic indicators; therefore, diagnosis often is made based on the patient’s history and physical examination. Further testing to confirm the diagnosis can be performed using specific wavelengths of UV light to determine which band of light affects patients most; however, the wavelength can change over time, leading to less clinical significance, and may decrease efficacy of phototherapy.2 Solar urticaria has no clear predisposing factors, and treatments to date have been moderately successful. Exposure to sunlight is thought to initiate an alteration in a skin or serum chromophore or photoallergen, which then causes subsequent cross-linking and IgE-dependent release of histamine as well as other mediators such as cytokines, eicosanoids, and proteases with mast cell degranulation.7
Omalizumab is a recombinant humanized monoclonal IgG1 antibody targeting the methylated IgE Cε3 domain that initially was marketed toward controlling IgE-mediated moderate to severe asthma recalcitrant to standard treatments. It has since received approval from the US Food and Drug Administration for treatment of chronic idiopathic urticaria after first being noticed to serendipitously treat a patient with cold urticaria and asthma in 2006.4,7,8 It was then first documented to successfully treat solar urticaria in 2008.6 The safety profile of omalizumab makes it a more favorable choice when compared to other immunomodulating treatments, with the most serious adverse reaction being anaphylaxis, occurring in 0.2% of patients in a postmarketing study.9 It functions through binding to free IgE at a region necessary for IgE to bind at low- and high-affinity receptors but not to immunoglobulins already bound to cells, thus theoretically preventing activation of mast cells or basophils.10 It also has been suggested that low steady-state values are needed to see continued benefit from the drug,10 which may have been seen in our patient after having an outbreak just prior to receiving an injection; however, prior reports have shown benefit unrelated to total IgE levels, with improvement after days to 4 months.4,10,11 One case report showed no response after 4 doses; it is unknown if this patient was tested for clinical improvement to omalizumab through further immunoglobulin analysis, but treatment response is important to consider when deciding on whether to use this drug in future patients.12 It is unknown why some patients will respond to omalizumab, others will partially respond, and others will not respond, which can be ascertained either through quality-of-life improvement or lack thereof.
In our experience, omalizumab is a viable option to consider in patients with solar urticaria that is recalcitrant to standard treatments and elevated IgE levels for whom other treatments are either too time consuming or have side-effect profiles that are not tolerable to the patient. If the patient has concomitant asthma, there may be additional therapeutic benefit. Further research is needed with regard to a cost-benefit analysis of omalizumab and whether using such a costly drug outweighs the cost associated with time and resources utilized with repeat clinic visits if other standard treatments are not effective.13
- Merkin P. Pratique Dermatologique. Paris, France: Masso; 1904.
- Beattie PE, Dawe RS, Ibbotson SH, et al. Characteristics and prognosis of idiopathic solar urticaria: a cohort of 87 cases. Arch Dermatol. 2003;139:1149-1154.
- Kaplan AP. Therapy of chronic urticaria: a simple, modern approach. Ann Allergy Asthma Immunol. 2014;112:419-425.
- Metz M, Maurer M. Omalizumab in chronic urticaria. Curr Opin Allergy Clin Immunol. 2012;12:406-410.
- Aubin F, Porcher R, Jeanmougin M, et al. Severe and refractory solar urticaria treated with intravenous immunoglobulins: a phase II multicenter study. J Am Acad Dermatol. 2014;71:948-953.e1.
- Güzelbey O, Ardelean E, Magerl M, et al. Successful treatment of solar urticaria with anti-immunoglobulin E therapy. Allergy. 2008;63:1563-1565.
- Wu K, Jabbar-Lopez Z. Omalizumab, an anti-IgE mAb, receives approval for the treatment of chronic idiopathic/spontaneous urticaria. J Invest Dermatol. 2015;135:13-15.
- Boyce JA. Successful treatment of cold-induced urticaria/anaphylaxis with anti-IgE. J Allergy Clin Immunol. 2006;117:1415-1418.
- Corren J, Casale TB, Lanier B, et al. Safety and tolerability of omalizumab. Clin Exp Allergy. 2009;39:788-797.
- Wu K, Long H. Omalizumab for chronic urticaria. N Engl J Med. 2013;368:2527-2528.
- Morgado-Carrasco D, Giacaman-Von der Weth M, Fusta-Novell X, et al. Clinical response and long-term follow-up of 20 patients with refractory solar urticarial under treatment with omalizumab [published online May 28, 2019]. J Am Acad Dermatol. doi:10.1016/j.jaad.2019.05.070.
- Duchini G, Bäumler W, Bircher AJ, et al. Failure of omalizumab (Xolair®) in the treatment of a case of solar urticaria caused by ultraviolet A and visible light. Photodermatol Photoimmunol Photomed. 2011;27:336-337.
- Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133:1270-1277.
- Merkin P. Pratique Dermatologique. Paris, France: Masso; 1904.
- Beattie PE, Dawe RS, Ibbotson SH, et al. Characteristics and prognosis of idiopathic solar urticaria: a cohort of 87 cases. Arch Dermatol. 2003;139:1149-1154.
- Kaplan AP. Therapy of chronic urticaria: a simple, modern approach. Ann Allergy Asthma Immunol. 2014;112:419-425.
- Metz M, Maurer M. Omalizumab in chronic urticaria. Curr Opin Allergy Clin Immunol. 2012;12:406-410.
- Aubin F, Porcher R, Jeanmougin M, et al. Severe and refractory solar urticaria treated with intravenous immunoglobulins: a phase II multicenter study. J Am Acad Dermatol. 2014;71:948-953.e1.
- Güzelbey O, Ardelean E, Magerl M, et al. Successful treatment of solar urticaria with anti-immunoglobulin E therapy. Allergy. 2008;63:1563-1565.
- Wu K, Jabbar-Lopez Z. Omalizumab, an anti-IgE mAb, receives approval for the treatment of chronic idiopathic/spontaneous urticaria. J Invest Dermatol. 2015;135:13-15.
- Boyce JA. Successful treatment of cold-induced urticaria/anaphylaxis with anti-IgE. J Allergy Clin Immunol. 2006;117:1415-1418.
- Corren J, Casale TB, Lanier B, et al. Safety and tolerability of omalizumab. Clin Exp Allergy. 2009;39:788-797.
- Wu K, Long H. Omalizumab for chronic urticaria. N Engl J Med. 2013;368:2527-2528.
- Morgado-Carrasco D, Giacaman-Von der Weth M, Fusta-Novell X, et al. Clinical response and long-term follow-up of 20 patients with refractory solar urticarial under treatment with omalizumab [published online May 28, 2019]. J Am Acad Dermatol. doi:10.1016/j.jaad.2019.05.070.
- Duchini G, Bäumler W, Bircher AJ, et al. Failure of omalizumab (Xolair®) in the treatment of a case of solar urticaria caused by ultraviolet A and visible light. Photodermatol Photoimmunol Photomed. 2011;27:336-337.
- Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133:1270-1277.
Practice Points
- Recurrent solar urticaria can be recalcitrant to treatment.
- Omalizumab may be an effective treatment option for solar urticaria, especially in patients with a concomitant asthma diagnosis.
Scrotal Ulceration: A Complication of Hyperthermic Intraperitoneal Chemotherapy and Subsequent Treatment With Dimethyl Sulfoxide
To the Editor:
A 54-year-old man with a history of stage IV appendiceal carcinoid adenocarcinoma treated approximately 3 months prior with intraoperative hyperthermic intraperitoneal chemotherapy (HIPEC) presented to our clinic with scrotal pain of 5 days’ duration. He had no history of genital herpes, topical contactants, other cutaneous lesions on the body, fever, or chills. On physical examination the patient had an erythematous, purpuric, indurated, tender plaque on the left anterolateral and anterior midline of the scrotum (Figure 1). No other areas of acral purpura or livedoid cutaneous changes were identified. There was no inguinal lymphadenopathy. Biopsy was performed for histologic examination as well as tissue culture. Histology demonstrated epidermal necrosis without evidence of vasculitis. Tissue culture was unremarkable.

Two days after clinic evaluation, the patient presented to the emergency department with progression of the lesions, and he was admitted to the hospital for pain control. Computed tomography of the pelvis showed bilateral hydroceles without evidence of abscess. Ultrasonography showed scrotal thickening without abscess or fluid collection. On day 5 in the hospital, a regimen of topical 60% dimethyl sulfoxide (DMSO) was applied every 8 hours to the affected area. The patient experienced notable pain relief and a decrease in erythema within 7 hours of application (Figure 2). This regimen was continued for 7 days with improvement in surrounding erythema and pain; however, the patient’s pain persisted in the areas of necrosis. Fourteen days following completion of therapy (27 days following presentation), the patient underwent debridement and partial scrotal resection for eschar removal. Histologic examination of the debrided scrotal tissue showed necrosis extending into the dermis and no evidence of vasculitis.

Our case demonstrates a unique presentation of scrotal necrosis secondary to mitomycin C (MitC) extravasation subsequently managed with DMSO. Imaging and biopsy findings effectively ruled out infection or vasculitis and led us to consider extravasation reactions that typically occur at peripheral intravenous (IV) infusion sites. Suspected cases of scrotal necrosis following HIPEC with MitC have been reported in the literature, along with hypothesized pathophysiology.1-3
In consideration of the proposed pathophysiology, individuals with hydroceles may be more likely to experience this complication due to an abnormal but not uncommon communication between the intraperitoneal cavity and the scrotum via a patent processus vaginalis. The location of necrosis on the anterior scrotum remains unexplained. It may be a consequence of the anatomic location of the hydrocele, a collection of fluid within the tunica vaginalis. The tunica vaginalis is composed of an inner visceral and outer parietal layer, enveloping the testis at the anterior border but not the superior or posterior border. Thus, sequestration of MitC in a hydrocele would correlate anatomically to necrosis of the anterior wall of the scrotum.
Akhavan et al1 proposed the testes are unaffected because of the presence of the tough fibrous coat of the tunica albuginea that directly adheres to the testes, in addition to the adjacent visceral layer of the tunica vaginalis. These 2 layers separating the testes and the hydrocele may provide a double barrier of protection for the testes.1
According to a PubMed search of articles indexed for MEDLINE using the terms scrotal or cutaneous, pain or ulceration, and HIPEC or hyperthermic in
Hyperthermic intraperitoneal chemotherapy involves installation of high-concentration chemotherapeutics into the peritoneal cavity at the conclusion of surgical cytoreductive therapy. Cell cycle–nonspecific agents such as MitC commonly are used for this procedure.4 It is classified as a vesicant, which is the designation given to drugs known to produce the most severe extravasation reactions of skin ulceration and necrosis.5,6 Symptoms typically include an early area of localized edema, erythema, and severe pain that progresses to superficial soft tissue and skin necrosis.7 Unfortunately, no well-studied antidote exists for MitC, though empirical guidelines suggest therapeutic management with DMSO and ice packs.6,8
Dimethyl sulfoxide is thought to work as a free radical scavenger as well as a solvent that facilitates diffusion of chemotherapeutics through tissues and thus down a concentration gradient, ideal in the circumstance of an extravasation reaction.8 Topical DMSO has been studied as a nonsurgical treatment in a small number of patients to prevent progression to necrosis following MitC extravasation.5,7 However, these cases only report extravasation reactions from IV infiltration.5,7,9 Dimethyl sulfoxide is rapidly absorbed and acts as a theoretical carrier for MitC as well as other topical substances.5,10,11 Caution is advised when using topical lidocaine or steroids in combination with DMSO, as they will be rapidly absorbed systemically. Patients also should be informed about a mild local burning sensation after DMSO application and a garliclike odor of the breath, which have occurred in 5.5% and 27.5% of patients, respectively (N=144).5 Dimethyl sulfoxide has no known toxic side effects but can cause erythema, pruritus, and very rarely allergic contact dermatitis.5,12 Abdul Aziz et al2 postulated that DMSO might be used as a method to prevent the progression of necrosis in symptomatic patients following HIPEC with MitC. Reports of its use on the scrotum are absent in the current available literature.
Treatment with DMSO was attempted in our patient with limited success secondary to delayed recognition and lack of supporting literature for DMSO treatment of scrotal necrosis. Treatment was delayed by 11 days after the onset of symptoms, which is far beyond the recommendation of starting within 10 minutes.8 Irreversible tissue necrosis had already occurred as evidenced by the presence of eschar. However, it seems apparent that DMSO provided some benefit given the clear improvement in erythema and pain 7 hours after application (Figure 2). It is unknown to what extent the necrosis would have progressed if not treated with DMSO.
Scrotal necrosis following HIPEC with MitC is a rare and incompletely understood but important chemotherapy reaction. The presentation is fairly specific with the presence of intractable and constant scrotal pain along with erythema and induration progressing to eschar. Although DMSO has been found to be effective for certain vesicant extravasation reactions at IV sites, it is not well studied for MitC, and no reports exist regarding its use on the scrotum. The presented characterization and explanation of the pathophysiology of this entity will aid in early recognition and timely institution of topical mitigating agents such as DMSO, which may prevent progression to scrotal necrosis and need for surgical debridement. More effective strategies may be geared toward prevention with thorough washout following HIPEC, preprocedural radiologic imaging or intraoperative visualization of the patent processus vaginalis, internal inguinal canal plugs, and patient education with anticipatory guidance should a reaction occur.2
- Akhavan A, Yin M, Benoit R. Scrotal ulcer after intraperitoneal hyperthermic chemotherapy. Urology. 2007;69:778.E9-E10.
- Abdul Aziz NH, Wang W, Teo MC. Scrotal pain and ulceration post HIPEC: a case report. J Gastrointest Cancer. 2015;46:60-63.
- Silva F, Avancini J, Criado P, et al. Scrotum ulcer developed after intraperitoneal hyperthermic chemotherapy with mitomycin-C [published October 21, 2012]. Bjui International. doi:10.1002/BJUIw-2012-019-web.
- González-Moreno S, González-Bayón LA, Ortega-Pérez G.Hyperthermic intraperitoneal chemotherapy: rationale and technique. World J Gastrointest Oncol. 2010;15:68-75.
- Bertelli G, Gozza A, Forno GB, et al. Topical dimethyl sulfoxide for the prevention of soft tissue injury after extravasation of vesicant cytotoxic drugs: a prospective clinical study. J Clin Oncol. 1995;13:2851-2855.
- Bertelli G. Prevention and management of extravasation of cytotoxic drugs. Drug Saf. 1995;12:245-255.
- Alberts DS, Dorr RT. Case report: topical DMSO for mitomycin-C-induced skin ulceration. Oncol Nurs Forum. 1991;18:693-695.
- Pérez Fidalgo JA, García Fabregat L, Cervantes A, et al; ESMO Guidelines Working Group. Management of chemotherapy extravasation: ESMO-EONS Clinical Practice Guidelines. Ann Oncol. 2012;23(suppl 5):167-173.
- Ludwig CU, Stoll HR, Obrist R, et al. Prevention of cytotoxic drug induced skin ulcers with dimethyl sulfoxide (DMSO) and alpha-tocopherole. Eur J Cancer Clin Oncol. 1987;23:327-329.
- Groel JT. Dimethyl sulfoxide as a vehicle for corticosteroids. a comparison with the occlusive dressing technique. Arch Dermatol. 1968;97:110-114.
- Simon LS, Grierson LM, Naseer Z. Efficacy and safety of topical diclofenac containing dimethyl sulfoxide (DMSO) compared with those of topical placebo, DMSO vehicle and oral diclofenac for knee osteoarthritis [published online April 19, 2009]. Pain. 2009;143:238-245.
- Nishimura M, Takano Y, Toshitani S. Systemic contact dermatitis medicamentosa occurring after intravesical dimethyl sulfoxide treatment for interstitial cystitis. Arch Dermatol. 1988;124:182-183.
To the Editor:
A 54-year-old man with a history of stage IV appendiceal carcinoid adenocarcinoma treated approximately 3 months prior with intraoperative hyperthermic intraperitoneal chemotherapy (HIPEC) presented to our clinic with scrotal pain of 5 days’ duration. He had no history of genital herpes, topical contactants, other cutaneous lesions on the body, fever, or chills. On physical examination the patient had an erythematous, purpuric, indurated, tender plaque on the left anterolateral and anterior midline of the scrotum (Figure 1). No other areas of acral purpura or livedoid cutaneous changes were identified. There was no inguinal lymphadenopathy. Biopsy was performed for histologic examination as well as tissue culture. Histology demonstrated epidermal necrosis without evidence of vasculitis. Tissue culture was unremarkable.
Two days after clinic evaluation, the patient presented to the emergency department with progression of the lesions, and he was admitted to the hospital for pain control. Computed tomography of the pelvis showed bilateral hydroceles without evidence of abscess. Ultrasonography showed scrotal thickening without abscess or fluid collection. On day 5 in the hospital, a regimen of topical 60% dimethyl sulfoxide (DMSO) was applied every 8 hours to the affected area. The patient experienced notable pain relief and a decrease in erythema within 7 hours of application (Figure 2). This regimen was continued for 7 days with improvement in surrounding erythema and pain; however, the patient’s pain persisted in the areas of necrosis. Fourteen days following completion of therapy (27 days following presentation), the patient underwent debridement and partial scrotal resection for eschar removal. Histologic examination of the debrided scrotal tissue showed necrosis extending into the dermis and no evidence of vasculitis.
Our case demonstrates a unique presentation of scrotal necrosis secondary to mitomycin C (MitC) extravasation subsequently managed with DMSO. Imaging and biopsy findings effectively ruled out infection or vasculitis and led us to consider extravasation reactions that typically occur at peripheral intravenous (IV) infusion sites. Suspected cases of scrotal necrosis following HIPEC with MitC have been reported in the literature, along with hypothesized pathophysiology.1-3
In consideration of the proposed pathophysiology, individuals with hydroceles may be more likely to experience this complication due to an abnormal but not uncommon communication between the intraperitoneal cavity and the scrotum via a patent processus vaginalis. The location of necrosis on the anterior scrotum remains unexplained. It may be a consequence of the anatomic location of the hydrocele, a collection of fluid within the tunica vaginalis. The tunica vaginalis is composed of an inner visceral and outer parietal layer, enveloping the testis at the anterior border but not the superior or posterior border. Thus, sequestration of MitC in a hydrocele would correlate anatomically to necrosis of the anterior wall of the scrotum.
Akhavan et al1 proposed the testes are unaffected because of the presence of the tough fibrous coat of the tunica albuginea that directly adheres to the testes, in addition to the adjacent visceral layer of the tunica vaginalis. These 2 layers separating the testes and the hydrocele may provide a double barrier of protection for the testes.1
According to a PubMed search of articles indexed for MEDLINE using the terms scrotal or cutaneous, pain or ulceration, and HIPEC or hyperthermic in
Hyperthermic intraperitoneal chemotherapy involves installation of high-concentration chemotherapeutics into the peritoneal cavity at the conclusion of surgical cytoreductive therapy. Cell cycle–nonspecific agents such as MitC commonly are used for this procedure.4 It is classified as a vesicant, which is the designation given to drugs known to produce the most severe extravasation reactions of skin ulceration and necrosis.5,6 Symptoms typically include an early area of localized edema, erythema, and severe pain that progresses to superficial soft tissue and skin necrosis.7 Unfortunately, no well-studied antidote exists for MitC, though empirical guidelines suggest therapeutic management with DMSO and ice packs.6,8
Dimethyl sulfoxide is thought to work as a free radical scavenger as well as a solvent that facilitates diffusion of chemotherapeutics through tissues and thus down a concentration gradient, ideal in the circumstance of an extravasation reaction.8 Topical DMSO has been studied as a nonsurgical treatment in a small number of patients to prevent progression to necrosis following MitC extravasation.5,7 However, these cases only report extravasation reactions from IV infiltration.5,7,9 Dimethyl sulfoxide is rapidly absorbed and acts as a theoretical carrier for MitC as well as other topical substances.5,10,11 Caution is advised when using topical lidocaine or steroids in combination with DMSO, as they will be rapidly absorbed systemically. Patients also should be informed about a mild local burning sensation after DMSO application and a garliclike odor of the breath, which have occurred in 5.5% and 27.5% of patients, respectively (N=144).5 Dimethyl sulfoxide has no known toxic side effects but can cause erythema, pruritus, and very rarely allergic contact dermatitis.5,12 Abdul Aziz et al2 postulated that DMSO might be used as a method to prevent the progression of necrosis in symptomatic patients following HIPEC with MitC. Reports of its use on the scrotum are absent in the current available literature.
Treatment with DMSO was attempted in our patient with limited success secondary to delayed recognition and lack of supporting literature for DMSO treatment of scrotal necrosis. Treatment was delayed by 11 days after the onset of symptoms, which is far beyond the recommendation of starting within 10 minutes.8 Irreversible tissue necrosis had already occurred as evidenced by the presence of eschar. However, it seems apparent that DMSO provided some benefit given the clear improvement in erythema and pain 7 hours after application (Figure 2). It is unknown to what extent the necrosis would have progressed if not treated with DMSO.
Scrotal necrosis following HIPEC with MitC is a rare and incompletely understood but important chemotherapy reaction. The presentation is fairly specific with the presence of intractable and constant scrotal pain along with erythema and induration progressing to eschar. Although DMSO has been found to be effective for certain vesicant extravasation reactions at IV sites, it is not well studied for MitC, and no reports exist regarding its use on the scrotum. The presented characterization and explanation of the pathophysiology of this entity will aid in early recognition and timely institution of topical mitigating agents such as DMSO, which may prevent progression to scrotal necrosis and need for surgical debridement. More effective strategies may be geared toward prevention with thorough washout following HIPEC, preprocedural radiologic imaging or intraoperative visualization of the patent processus vaginalis, internal inguinal canal plugs, and patient education with anticipatory guidance should a reaction occur.2
To the Editor:
A 54-year-old man with a history of stage IV appendiceal carcinoid adenocarcinoma treated approximately 3 months prior with intraoperative hyperthermic intraperitoneal chemotherapy (HIPEC) presented to our clinic with scrotal pain of 5 days’ duration. He had no history of genital herpes, topical contactants, other cutaneous lesions on the body, fever, or chills. On physical examination the patient had an erythematous, purpuric, indurated, tender plaque on the left anterolateral and anterior midline of the scrotum (Figure 1). No other areas of acral purpura or livedoid cutaneous changes were identified. There was no inguinal lymphadenopathy. Biopsy was performed for histologic examination as well as tissue culture. Histology demonstrated epidermal necrosis without evidence of vasculitis. Tissue culture was unremarkable.
Two days after clinic evaluation, the patient presented to the emergency department with progression of the lesions, and he was admitted to the hospital for pain control. Computed tomography of the pelvis showed bilateral hydroceles without evidence of abscess. Ultrasonography showed scrotal thickening without abscess or fluid collection. On day 5 in the hospital, a regimen of topical 60% dimethyl sulfoxide (DMSO) was applied every 8 hours to the affected area. The patient experienced notable pain relief and a decrease in erythema within 7 hours of application (Figure 2). This regimen was continued for 7 days with improvement in surrounding erythema and pain; however, the patient’s pain persisted in the areas of necrosis. Fourteen days following completion of therapy (27 days following presentation), the patient underwent debridement and partial scrotal resection for eschar removal. Histologic examination of the debrided scrotal tissue showed necrosis extending into the dermis and no evidence of vasculitis.
Our case demonstrates a unique presentation of scrotal necrosis secondary to mitomycin C (MitC) extravasation subsequently managed with DMSO. Imaging and biopsy findings effectively ruled out infection or vasculitis and led us to consider extravasation reactions that typically occur at peripheral intravenous (IV) infusion sites. Suspected cases of scrotal necrosis following HIPEC with MitC have been reported in the literature, along with hypothesized pathophysiology.1-3
In consideration of the proposed pathophysiology, individuals with hydroceles may be more likely to experience this complication due to an abnormal but not uncommon communication between the intraperitoneal cavity and the scrotum via a patent processus vaginalis. The location of necrosis on the anterior scrotum remains unexplained. It may be a consequence of the anatomic location of the hydrocele, a collection of fluid within the tunica vaginalis. The tunica vaginalis is composed of an inner visceral and outer parietal layer, enveloping the testis at the anterior border but not the superior or posterior border. Thus, sequestration of MitC in a hydrocele would correlate anatomically to necrosis of the anterior wall of the scrotum.
Akhavan et al1 proposed the testes are unaffected because of the presence of the tough fibrous coat of the tunica albuginea that directly adheres to the testes, in addition to the adjacent visceral layer of the tunica vaginalis. These 2 layers separating the testes and the hydrocele may provide a double barrier of protection for the testes.1
According to a PubMed search of articles indexed for MEDLINE using the terms scrotal or cutaneous, pain or ulceration, and HIPEC or hyperthermic in
Hyperthermic intraperitoneal chemotherapy involves installation of high-concentration chemotherapeutics into the peritoneal cavity at the conclusion of surgical cytoreductive therapy. Cell cycle–nonspecific agents such as MitC commonly are used for this procedure.4 It is classified as a vesicant, which is the designation given to drugs known to produce the most severe extravasation reactions of skin ulceration and necrosis.5,6 Symptoms typically include an early area of localized edema, erythema, and severe pain that progresses to superficial soft tissue and skin necrosis.7 Unfortunately, no well-studied antidote exists for MitC, though empirical guidelines suggest therapeutic management with DMSO and ice packs.6,8
Dimethyl sulfoxide is thought to work as a free radical scavenger as well as a solvent that facilitates diffusion of chemotherapeutics through tissues and thus down a concentration gradient, ideal in the circumstance of an extravasation reaction.8 Topical DMSO has been studied as a nonsurgical treatment in a small number of patients to prevent progression to necrosis following MitC extravasation.5,7 However, these cases only report extravasation reactions from IV infiltration.5,7,9 Dimethyl sulfoxide is rapidly absorbed and acts as a theoretical carrier for MitC as well as other topical substances.5,10,11 Caution is advised when using topical lidocaine or steroids in combination with DMSO, as they will be rapidly absorbed systemically. Patients also should be informed about a mild local burning sensation after DMSO application and a garliclike odor of the breath, which have occurred in 5.5% and 27.5% of patients, respectively (N=144).5 Dimethyl sulfoxide has no known toxic side effects but can cause erythema, pruritus, and very rarely allergic contact dermatitis.5,12 Abdul Aziz et al2 postulated that DMSO might be used as a method to prevent the progression of necrosis in symptomatic patients following HIPEC with MitC. Reports of its use on the scrotum are absent in the current available literature.
Treatment with DMSO was attempted in our patient with limited success secondary to delayed recognition and lack of supporting literature for DMSO treatment of scrotal necrosis. Treatment was delayed by 11 days after the onset of symptoms, which is far beyond the recommendation of starting within 10 minutes.8 Irreversible tissue necrosis had already occurred as evidenced by the presence of eschar. However, it seems apparent that DMSO provided some benefit given the clear improvement in erythema and pain 7 hours after application (Figure 2). It is unknown to what extent the necrosis would have progressed if not treated with DMSO.
Scrotal necrosis following HIPEC with MitC is a rare and incompletely understood but important chemotherapy reaction. The presentation is fairly specific with the presence of intractable and constant scrotal pain along with erythema and induration progressing to eschar. Although DMSO has been found to be effective for certain vesicant extravasation reactions at IV sites, it is not well studied for MitC, and no reports exist regarding its use on the scrotum. The presented characterization and explanation of the pathophysiology of this entity will aid in early recognition and timely institution of topical mitigating agents such as DMSO, which may prevent progression to scrotal necrosis and need for surgical debridement. More effective strategies may be geared toward prevention with thorough washout following HIPEC, preprocedural radiologic imaging or intraoperative visualization of the patent processus vaginalis, internal inguinal canal plugs, and patient education with anticipatory guidance should a reaction occur.2
- Akhavan A, Yin M, Benoit R. Scrotal ulcer after intraperitoneal hyperthermic chemotherapy. Urology. 2007;69:778.E9-E10.
- Abdul Aziz NH, Wang W, Teo MC. Scrotal pain and ulceration post HIPEC: a case report. J Gastrointest Cancer. 2015;46:60-63.
- Silva F, Avancini J, Criado P, et al. Scrotum ulcer developed after intraperitoneal hyperthermic chemotherapy with mitomycin-C [published October 21, 2012]. Bjui International. doi:10.1002/BJUIw-2012-019-web.
- González-Moreno S, González-Bayón LA, Ortega-Pérez G.Hyperthermic intraperitoneal chemotherapy: rationale and technique. World J Gastrointest Oncol. 2010;15:68-75.
- Bertelli G, Gozza A, Forno GB, et al. Topical dimethyl sulfoxide for the prevention of soft tissue injury after extravasation of vesicant cytotoxic drugs: a prospective clinical study. J Clin Oncol. 1995;13:2851-2855.
- Bertelli G. Prevention and management of extravasation of cytotoxic drugs. Drug Saf. 1995;12:245-255.
- Alberts DS, Dorr RT. Case report: topical DMSO for mitomycin-C-induced skin ulceration. Oncol Nurs Forum. 1991;18:693-695.
- Pérez Fidalgo JA, García Fabregat L, Cervantes A, et al; ESMO Guidelines Working Group. Management of chemotherapy extravasation: ESMO-EONS Clinical Practice Guidelines. Ann Oncol. 2012;23(suppl 5):167-173.
- Ludwig CU, Stoll HR, Obrist R, et al. Prevention of cytotoxic drug induced skin ulcers with dimethyl sulfoxide (DMSO) and alpha-tocopherole. Eur J Cancer Clin Oncol. 1987;23:327-329.
- Groel JT. Dimethyl sulfoxide as a vehicle for corticosteroids. a comparison with the occlusive dressing technique. Arch Dermatol. 1968;97:110-114.
- Simon LS, Grierson LM, Naseer Z. Efficacy and safety of topical diclofenac containing dimethyl sulfoxide (DMSO) compared with those of topical placebo, DMSO vehicle and oral diclofenac for knee osteoarthritis [published online April 19, 2009]. Pain. 2009;143:238-245.
- Nishimura M, Takano Y, Toshitani S. Systemic contact dermatitis medicamentosa occurring after intravesical dimethyl sulfoxide treatment for interstitial cystitis. Arch Dermatol. 1988;124:182-183.
- Akhavan A, Yin M, Benoit R. Scrotal ulcer after intraperitoneal hyperthermic chemotherapy. Urology. 2007;69:778.E9-E10.
- Abdul Aziz NH, Wang W, Teo MC. Scrotal pain and ulceration post HIPEC: a case report. J Gastrointest Cancer. 2015;46:60-63.
- Silva F, Avancini J, Criado P, et al. Scrotum ulcer developed after intraperitoneal hyperthermic chemotherapy with mitomycin-C [published October 21, 2012]. Bjui International. doi:10.1002/BJUIw-2012-019-web.
- González-Moreno S, González-Bayón LA, Ortega-Pérez G.Hyperthermic intraperitoneal chemotherapy: rationale and technique. World J Gastrointest Oncol. 2010;15:68-75.
- Bertelli G, Gozza A, Forno GB, et al. Topical dimethyl sulfoxide for the prevention of soft tissue injury after extravasation of vesicant cytotoxic drugs: a prospective clinical study. J Clin Oncol. 1995;13:2851-2855.
- Bertelli G. Prevention and management of extravasation of cytotoxic drugs. Drug Saf. 1995;12:245-255.
- Alberts DS, Dorr RT. Case report: topical DMSO for mitomycin-C-induced skin ulceration. Oncol Nurs Forum. 1991;18:693-695.
- Pérez Fidalgo JA, García Fabregat L, Cervantes A, et al; ESMO Guidelines Working Group. Management of chemotherapy extravasation: ESMO-EONS Clinical Practice Guidelines. Ann Oncol. 2012;23(suppl 5):167-173.
- Ludwig CU, Stoll HR, Obrist R, et al. Prevention of cytotoxic drug induced skin ulcers with dimethyl sulfoxide (DMSO) and alpha-tocopherole. Eur J Cancer Clin Oncol. 1987;23:327-329.
- Groel JT. Dimethyl sulfoxide as a vehicle for corticosteroids. a comparison with the occlusive dressing technique. Arch Dermatol. 1968;97:110-114.
- Simon LS, Grierson LM, Naseer Z. Efficacy and safety of topical diclofenac containing dimethyl sulfoxide (DMSO) compared with those of topical placebo, DMSO vehicle and oral diclofenac for knee osteoarthritis [published online April 19, 2009]. Pain. 2009;143:238-245.
- Nishimura M, Takano Y, Toshitani S. Systemic contact dermatitis medicamentosa occurring after intravesical dimethyl sulfoxide treatment for interstitial cystitis. Arch Dermatol. 1988;124:182-183.
Practice Points
- Scrotal ulceration following hyperthermic intraperitoneal chemotherapy has been reported only a few times in the literature and is likely underreported. The presentation in all reported cases was similar, with a delay in symptom onset of weeks to months, involvement of the anterior scrotum, and pain.
- Dimethyl sulfoxide, used in other vesicant reactions, may have a role in mitigating tissue damage. Alternatively, methods to prevent sequestration of vesicants in the potential space of the tunica vaginalis layers can be employed.
Severe Acne Fulminans Following Low-Dose Isotretinoin and Testosterone Use
To the Editor:
Acne fulminans (AF), the most severe form of acne, is a rare condition with an incidence of less than 1% of total acne cases.1 Adolescent boys are the most susceptible group of patients.2 Painful inflammatory pustules that transform into deep ulcerations covered by abundant hemorrhagic crust are typical of AF. Commonly affected areas include the face, back, neck, and chest. Additionally, fever and polyarthralgia may be present, and there often is myopathy due to rapid weight loss.3,4 Less often, erythema nodosum and splenomegaly may be observed.5 Laboratory testing also may reveal markers of systemic inflammation such as leukocytosis with neutrophilia, elevated C-reactive protein levels, increased erythrocyte sedimentation rate, and thrombocytosis. Anemia and elevated hepatic enzyme levels also may be present in AF.2 It is suspected that AF may be induced by low doses of isotretinoin therapy with concomitant inherited susceptibility.6
We report the case of a 21-year-old man who was referred to the Department of Dermatology by his primary care physician for evaluation of severe hemorrhagic lesions on the trunk following use of oral isotretinoin (Figure 1). Prior to development of the lesions, the patient had started weekly intramuscular injections of testosterone 500 mg, which he purchased online without consulting a physician, to address muscle mass reduction associated with sudden weight loss from intense physical training. After 8 months of testosterone supplementation along with continued physical training, the patient presented to his primary care physician for treatment of acne vulgaris on the back and trunk of 2 months’ duration. Oral isotretinoin 20 mg once daily was initiated; however, the patient reported that the acne lesions showed progression after 1 month of treatment. Isotretinoin was increased to a more weight-appropriate dosage of 60 mg once daily 2 weeks before admission to our dermatology clinic.
At the current presentation, dermatologic examination revealed numerous inflamed ulcerations covered by a hemorrhagic crust on the back and trunk. The patient also reported knee, elbow, and inguinal pain, especially at night. No fever or loss of appetite was reported. The patient was otherwise healthy and had no remarkable family history of acne or other dermatologic diseases.
Laboratory testing showed leukocytosis (11,000/µL [reference range, 4500–11,000/µL]), an elevated C-reactive protein level (66 mg/L [reference range, 0.08–3.1 mg/L]), and an elevated erythrocyte sedimentation rate (46 mm/h [reference range, 0–20 mm/h]). There were laboratory and clinical signs of a secondary bacterial infection in the affected areas, and a culture of secretions collected from lesions on the back grew Staphylococcus aureus with sensitivity to erythromycin, clindamycin, doxycycline, and trimethoprim-sulfamethoxazole and resistance to penicillin. A diagnosis of AF was made based on the clinical presentation and systemic symptoms, and anabolic-androgenic steroids and low-dose isotretinoin were identified as etiologic factors.
Treatment initially included cessation of isotretinoin and administration of prednisone, omeprazole, clindamycin, and doxycycline. Prednisone was given at a dosage of 40 mg once daily for 1 week, then decreased by 5 mg every 7 days. Omeprazole was given concurrently as prophylaxis for the gastrointestinal tract side effects of long-term prednisone use. Clindamycin was given at a dosage of 300 mg 3 times daily. Doxycycline was given for 6 weeks at a dosage of 100 mg twice daily. Topical octenidine dihydrochloride also was given.
Marked improvement was noted after 24 hours (Figure 2) as well as on the third day of treatment (Figure 3A). After 6 weeks, only disfiguring scars were visible (Figure 3B). Oral isotretinoin was reincorporated after 8 weeks and was subsequently discontinued after 5 months of therapy with a cumulative dose of 150 mg/kg.

It is important to differentiate AF from exacerbation of acne vulgaris because patients typically have mild or moderate acne vulgaris before the onset of acute symptoms.1 Acne fulminans is characterized by systemic symptoms such as myalgia, polyarthralgia, fatigue, and osteolytic bone lesions.1,7 Additionally, hematologic symptoms such as fever, leukocytosis, anemia, splenomegaly, and hepatomegaly may be present.1,5,7 Our patient demonstrated the polysymptomatic form of AF. The patient had severe acne with a tendency to scar. There also were some systemic manifestations such as polyarthralgia, weight loss, leukocytosis, an elevated erythrocyte sedimentation rate, and an elevated C-reactive protein level.
The clinical diagnosis in our patient also was supported by the hypothesis that heredity, overactive immune reactions, bacterial infections, and use of some drugs (eg, isotretinoin, tetracycline, testosterone) can trigger AF.8 The most well-known theory is that low doses of isotretinoin induce AF.6 The majority of cases are caused by doses of less than 20 mg/kg once daily, but there have been reports of patients using full doses and developing this condition.9 The fact that the use of low- and high-dose isotretinoin can provoke AF suggests an idiosyncratic reaction that is not clearly dose related. The most dangerous triggering factor of AF is concomitant usage of testosterone and isotretinoin.10 Our patient used testosterone injections to increase muscle mass and underwent treatment with isotretinoin for acne.
Treatment of AF is controversial, as there is no standard therapy. Currently, steroids and isotretinoin are the treatments of choice. Antibiotic use is controversial because of a lack of randomized trials.11
In the first stage of treatment, prednisone 0.5 to 1 mg/kg once daily is recommended as an initial anti-inflammatory therapy, with gradual dose reduction. According to evidence-based recommendations, a low dose of isotretinoin can be introduced after crusted lesions have healed. The overlapping therapy with steroids and isotretinoin should be provided for at least 4 weeks. High-potency topical corticosteroids can be used on granulation tissue, which can shorten the systemic treatment with prednisone or can be an alternative treatment for patients with contraindications to systemic corticosteroids.11
Additionally, local care of the lesions including compresses and topical emollients is crucial. There are some case reports in which there is introduction of high doses of isotretinoin, subsequently with systemic steroids.7,8,12 Seukeran and Cunliffe5 proved that it is beneficial to give acne prophylaxis to prevent further episodes. Our patient was similarly treated with systemic steroids and isotretinoin. Treatment guidelines for AF do not recommend oral antibiotics,11 but data are limited in the case of isotretinoin-induced AF. Our patient was given doxycycline concomitant with systemic steroids, which was necessary due to signs of secondary infection from a lesion culture. Doxycycline was stopped when isotretinoin treatment was initiated to prevent pseudotumor cerebri. The patient achieved good clinical improvement with no relapse.
Using isotretinoin to treat acne vulgaris has many benefits, despite the possibility of developing AF as an extremely rare complication. Clinicians should be aware of the risk of this complication to make the diagnosis and provide appropriate care, especially in young men. It is particularly important to consider the possibility of concomitant testosterone and isotretinoin when documenting the patient’s medical history.
- Romiti R, Jansen T, Plewig G. Acne fulminans. An Bras Dermatol. 2000;75:611-617.
- Karvonen SL. Acne fulminans: report of clinical findings and treatment of twenty-four patients. J Am Acad Dermatol. 1993;28:572-579.
- Kelly AP, Burns RE. Acute febrile ulcerative conglobate acne with polyarthralgia. Arch Dermatol. 1971;104:182-187.
- Plewig G, Kligman AM. Vitamin A acid in acneiform dermatoses. Acta Derm Venereol Suppl. 1975;74:119-127.
- Seukeran DC, Cunliffe WJ. The treatment of acne fulminans: a review of 25 cases. Br J Dermatol. 1999;141:307-309.
- Kraus SL, Emmert S, Schön MP, et al. The dark side of beauty: acne fulminans induced by anabolic steroids in a male bodybuilder. Arch Dermatol. 2012;148:1210-1212.
- Jansen T, Plewig G. Acne fulminans. Int J Dermatol. 1998;37:254-257.
- Zanelato TP, Gontijo GM, Alves CA, et al. Disabling acne fulminans. An Bras Dermatol. 2011;86:9-12.
- Azulay DR, Abulafia LA, Costa JAN, et al. Tecido de granulação exuberante. efeito colateral da terapêutica com isotretinoína. An Bras Dermatol. 1985;60:179-182.
- Traupe H, von Mühlendahl KE, Brämswig J, et al. Acne of the fulminans type following testosterone therapy in three excessively tall boys. Arch Dermatol. 1988;124:414-417.
- Greywal T, Zaenglein AL, Baldwin HE, et al. Evidence-based recommendations for the management of acne fulminans and its variants. J Am Acad Dermatol. 2017;77:109-117.
- Honma M, Murakami M, Iinuma S, et al. Acne fulminans following measles infection. J Dermatol. 2009;36:471-473.
To the Editor:
Acne fulminans (AF), the most severe form of acne, is a rare condition with an incidence of less than 1% of total acne cases.1 Adolescent boys are the most susceptible group of patients.2 Painful inflammatory pustules that transform into deep ulcerations covered by abundant hemorrhagic crust are typical of AF. Commonly affected areas include the face, back, neck, and chest. Additionally, fever and polyarthralgia may be present, and there often is myopathy due to rapid weight loss.3,4 Less often, erythema nodosum and splenomegaly may be observed.5 Laboratory testing also may reveal markers of systemic inflammation such as leukocytosis with neutrophilia, elevated C-reactive protein levels, increased erythrocyte sedimentation rate, and thrombocytosis. Anemia and elevated hepatic enzyme levels also may be present in AF.2 It is suspected that AF may be induced by low doses of isotretinoin therapy with concomitant inherited susceptibility.6
We report the case of a 21-year-old man who was referred to the Department of Dermatology by his primary care physician for evaluation of severe hemorrhagic lesions on the trunk following use of oral isotretinoin (Figure 1). Prior to development of the lesions, the patient had started weekly intramuscular injections of testosterone 500 mg, which he purchased online without consulting a physician, to address muscle mass reduction associated with sudden weight loss from intense physical training. After 8 months of testosterone supplementation along with continued physical training, the patient presented to his primary care physician for treatment of acne vulgaris on the back and trunk of 2 months’ duration. Oral isotretinoin 20 mg once daily was initiated; however, the patient reported that the acne lesions showed progression after 1 month of treatment. Isotretinoin was increased to a more weight-appropriate dosage of 60 mg once daily 2 weeks before admission to our dermatology clinic.
At the current presentation, dermatologic examination revealed numerous inflamed ulcerations covered by a hemorrhagic crust on the back and trunk. The patient also reported knee, elbow, and inguinal pain, especially at night. No fever or loss of appetite was reported. The patient was otherwise healthy and had no remarkable family history of acne or other dermatologic diseases.
Laboratory testing showed leukocytosis (11,000/µL [reference range, 4500–11,000/µL]), an elevated C-reactive protein level (66 mg/L [reference range, 0.08–3.1 mg/L]), and an elevated erythrocyte sedimentation rate (46 mm/h [reference range, 0–20 mm/h]). There were laboratory and clinical signs of a secondary bacterial infection in the affected areas, and a culture of secretions collected from lesions on the back grew Staphylococcus aureus with sensitivity to erythromycin, clindamycin, doxycycline, and trimethoprim-sulfamethoxazole and resistance to penicillin. A diagnosis of AF was made based on the clinical presentation and systemic symptoms, and anabolic-androgenic steroids and low-dose isotretinoin were identified as etiologic factors.
Treatment initially included cessation of isotretinoin and administration of prednisone, omeprazole, clindamycin, and doxycycline. Prednisone was given at a dosage of 40 mg once daily for 1 week, then decreased by 5 mg every 7 days. Omeprazole was given concurrently as prophylaxis for the gastrointestinal tract side effects of long-term prednisone use. Clindamycin was given at a dosage of 300 mg 3 times daily. Doxycycline was given for 6 weeks at a dosage of 100 mg twice daily. Topical octenidine dihydrochloride also was given.
Marked improvement was noted after 24 hours (Figure 2) as well as on the third day of treatment (Figure 3A). After 6 weeks, only disfiguring scars were visible (Figure 3B). Oral isotretinoin was reincorporated after 8 weeks and was subsequently discontinued after 5 months of therapy with a cumulative dose of 150 mg/kg.
It is important to differentiate AF from exacerbation of acne vulgaris because patients typically have mild or moderate acne vulgaris before the onset of acute symptoms.1 Acne fulminans is characterized by systemic symptoms such as myalgia, polyarthralgia, fatigue, and osteolytic bone lesions.1,7 Additionally, hematologic symptoms such as fever, leukocytosis, anemia, splenomegaly, and hepatomegaly may be present.1,5,7 Our patient demonstrated the polysymptomatic form of AF. The patient had severe acne with a tendency to scar. There also were some systemic manifestations such as polyarthralgia, weight loss, leukocytosis, an elevated erythrocyte sedimentation rate, and an elevated C-reactive protein level.
The clinical diagnosis in our patient also was supported by the hypothesis that heredity, overactive immune reactions, bacterial infections, and use of some drugs (eg, isotretinoin, tetracycline, testosterone) can trigger AF.8 The most well-known theory is that low doses of isotretinoin induce AF.6 The majority of cases are caused by doses of less than 20 mg/kg once daily, but there have been reports of patients using full doses and developing this condition.9 The fact that the use of low- and high-dose isotretinoin can provoke AF suggests an idiosyncratic reaction that is not clearly dose related. The most dangerous triggering factor of AF is concomitant usage of testosterone and isotretinoin.10 Our patient used testosterone injections to increase muscle mass and underwent treatment with isotretinoin for acne.
Treatment of AF is controversial, as there is no standard therapy. Currently, steroids and isotretinoin are the treatments of choice. Antibiotic use is controversial because of a lack of randomized trials.11
In the first stage of treatment, prednisone 0.5 to 1 mg/kg once daily is recommended as an initial anti-inflammatory therapy, with gradual dose reduction. According to evidence-based recommendations, a low dose of isotretinoin can be introduced after crusted lesions have healed. The overlapping therapy with steroids and isotretinoin should be provided for at least 4 weeks. High-potency topical corticosteroids can be used on granulation tissue, which can shorten the systemic treatment with prednisone or can be an alternative treatment for patients with contraindications to systemic corticosteroids.11
Additionally, local care of the lesions including compresses and topical emollients is crucial. There are some case reports in which there is introduction of high doses of isotretinoin, subsequently with systemic steroids.7,8,12 Seukeran and Cunliffe5 proved that it is beneficial to give acne prophylaxis to prevent further episodes. Our patient was similarly treated with systemic steroids and isotretinoin. Treatment guidelines for AF do not recommend oral antibiotics,11 but data are limited in the case of isotretinoin-induced AF. Our patient was given doxycycline concomitant with systemic steroids, which was necessary due to signs of secondary infection from a lesion culture. Doxycycline was stopped when isotretinoin treatment was initiated to prevent pseudotumor cerebri. The patient achieved good clinical improvement with no relapse.
Using isotretinoin to treat acne vulgaris has many benefits, despite the possibility of developing AF as an extremely rare complication. Clinicians should be aware of the risk of this complication to make the diagnosis and provide appropriate care, especially in young men. It is particularly important to consider the possibility of concomitant testosterone and isotretinoin when documenting the patient’s medical history.
To the Editor:
Acne fulminans (AF), the most severe form of acne, is a rare condition with an incidence of less than 1% of total acne cases.1 Adolescent boys are the most susceptible group of patients.2 Painful inflammatory pustules that transform into deep ulcerations covered by abundant hemorrhagic crust are typical of AF. Commonly affected areas include the face, back, neck, and chest. Additionally, fever and polyarthralgia may be present, and there often is myopathy due to rapid weight loss.3,4 Less often, erythema nodosum and splenomegaly may be observed.5 Laboratory testing also may reveal markers of systemic inflammation such as leukocytosis with neutrophilia, elevated C-reactive protein levels, increased erythrocyte sedimentation rate, and thrombocytosis. Anemia and elevated hepatic enzyme levels also may be present in AF.2 It is suspected that AF may be induced by low doses of isotretinoin therapy with concomitant inherited susceptibility.6
We report the case of a 21-year-old man who was referred to the Department of Dermatology by his primary care physician for evaluation of severe hemorrhagic lesions on the trunk following use of oral isotretinoin (Figure 1). Prior to development of the lesions, the patient had started weekly intramuscular injections of testosterone 500 mg, which he purchased online without consulting a physician, to address muscle mass reduction associated with sudden weight loss from intense physical training. After 8 months of testosterone supplementation along with continued physical training, the patient presented to his primary care physician for treatment of acne vulgaris on the back and trunk of 2 months’ duration. Oral isotretinoin 20 mg once daily was initiated; however, the patient reported that the acne lesions showed progression after 1 month of treatment. Isotretinoin was increased to a more weight-appropriate dosage of 60 mg once daily 2 weeks before admission to our dermatology clinic.
At the current presentation, dermatologic examination revealed numerous inflamed ulcerations covered by a hemorrhagic crust on the back and trunk. The patient also reported knee, elbow, and inguinal pain, especially at night. No fever or loss of appetite was reported. The patient was otherwise healthy and had no remarkable family history of acne or other dermatologic diseases.
Laboratory testing showed leukocytosis (11,000/µL [reference range, 4500–11,000/µL]), an elevated C-reactive protein level (66 mg/L [reference range, 0.08–3.1 mg/L]), and an elevated erythrocyte sedimentation rate (46 mm/h [reference range, 0–20 mm/h]). There were laboratory and clinical signs of a secondary bacterial infection in the affected areas, and a culture of secretions collected from lesions on the back grew Staphylococcus aureus with sensitivity to erythromycin, clindamycin, doxycycline, and trimethoprim-sulfamethoxazole and resistance to penicillin. A diagnosis of AF was made based on the clinical presentation and systemic symptoms, and anabolic-androgenic steroids and low-dose isotretinoin were identified as etiologic factors.
Treatment initially included cessation of isotretinoin and administration of prednisone, omeprazole, clindamycin, and doxycycline. Prednisone was given at a dosage of 40 mg once daily for 1 week, then decreased by 5 mg every 7 days. Omeprazole was given concurrently as prophylaxis for the gastrointestinal tract side effects of long-term prednisone use. Clindamycin was given at a dosage of 300 mg 3 times daily. Doxycycline was given for 6 weeks at a dosage of 100 mg twice daily. Topical octenidine dihydrochloride also was given.
Marked improvement was noted after 24 hours (Figure 2) as well as on the third day of treatment (Figure 3A). After 6 weeks, only disfiguring scars were visible (Figure 3B). Oral isotretinoin was reincorporated after 8 weeks and was subsequently discontinued after 5 months of therapy with a cumulative dose of 150 mg/kg.
It is important to differentiate AF from exacerbation of acne vulgaris because patients typically have mild or moderate acne vulgaris before the onset of acute symptoms.1 Acne fulminans is characterized by systemic symptoms such as myalgia, polyarthralgia, fatigue, and osteolytic bone lesions.1,7 Additionally, hematologic symptoms such as fever, leukocytosis, anemia, splenomegaly, and hepatomegaly may be present.1,5,7 Our patient demonstrated the polysymptomatic form of AF. The patient had severe acne with a tendency to scar. There also were some systemic manifestations such as polyarthralgia, weight loss, leukocytosis, an elevated erythrocyte sedimentation rate, and an elevated C-reactive protein level.
The clinical diagnosis in our patient also was supported by the hypothesis that heredity, overactive immune reactions, bacterial infections, and use of some drugs (eg, isotretinoin, tetracycline, testosterone) can trigger AF.8 The most well-known theory is that low doses of isotretinoin induce AF.6 The majority of cases are caused by doses of less than 20 mg/kg once daily, but there have been reports of patients using full doses and developing this condition.9 The fact that the use of low- and high-dose isotretinoin can provoke AF suggests an idiosyncratic reaction that is not clearly dose related. The most dangerous triggering factor of AF is concomitant usage of testosterone and isotretinoin.10 Our patient used testosterone injections to increase muscle mass and underwent treatment with isotretinoin for acne.
Treatment of AF is controversial, as there is no standard therapy. Currently, steroids and isotretinoin are the treatments of choice. Antibiotic use is controversial because of a lack of randomized trials.11
In the first stage of treatment, prednisone 0.5 to 1 mg/kg once daily is recommended as an initial anti-inflammatory therapy, with gradual dose reduction. According to evidence-based recommendations, a low dose of isotretinoin can be introduced after crusted lesions have healed. The overlapping therapy with steroids and isotretinoin should be provided for at least 4 weeks. High-potency topical corticosteroids can be used on granulation tissue, which can shorten the systemic treatment with prednisone or can be an alternative treatment for patients with contraindications to systemic corticosteroids.11
Additionally, local care of the lesions including compresses and topical emollients is crucial. There are some case reports in which there is introduction of high doses of isotretinoin, subsequently with systemic steroids.7,8,12 Seukeran and Cunliffe5 proved that it is beneficial to give acne prophylaxis to prevent further episodes. Our patient was similarly treated with systemic steroids and isotretinoin. Treatment guidelines for AF do not recommend oral antibiotics,11 but data are limited in the case of isotretinoin-induced AF. Our patient was given doxycycline concomitant with systemic steroids, which was necessary due to signs of secondary infection from a lesion culture. Doxycycline was stopped when isotretinoin treatment was initiated to prevent pseudotumor cerebri. The patient achieved good clinical improvement with no relapse.
Using isotretinoin to treat acne vulgaris has many benefits, despite the possibility of developing AF as an extremely rare complication. Clinicians should be aware of the risk of this complication to make the diagnosis and provide appropriate care, especially in young men. It is particularly important to consider the possibility of concomitant testosterone and isotretinoin when documenting the patient’s medical history.
- Romiti R, Jansen T, Plewig G. Acne fulminans. An Bras Dermatol. 2000;75:611-617.
- Karvonen SL. Acne fulminans: report of clinical findings and treatment of twenty-four patients. J Am Acad Dermatol. 1993;28:572-579.
- Kelly AP, Burns RE. Acute febrile ulcerative conglobate acne with polyarthralgia. Arch Dermatol. 1971;104:182-187.
- Plewig G, Kligman AM. Vitamin A acid in acneiform dermatoses. Acta Derm Venereol Suppl. 1975;74:119-127.
- Seukeran DC, Cunliffe WJ. The treatment of acne fulminans: a review of 25 cases. Br J Dermatol. 1999;141:307-309.
- Kraus SL, Emmert S, Schön MP, et al. The dark side of beauty: acne fulminans induced by anabolic steroids in a male bodybuilder. Arch Dermatol. 2012;148:1210-1212.
- Jansen T, Plewig G. Acne fulminans. Int J Dermatol. 1998;37:254-257.
- Zanelato TP, Gontijo GM, Alves CA, et al. Disabling acne fulminans. An Bras Dermatol. 2011;86:9-12.
- Azulay DR, Abulafia LA, Costa JAN, et al. Tecido de granulação exuberante. efeito colateral da terapêutica com isotretinoína. An Bras Dermatol. 1985;60:179-182.
- Traupe H, von Mühlendahl KE, Brämswig J, et al. Acne of the fulminans type following testosterone therapy in three excessively tall boys. Arch Dermatol. 1988;124:414-417.
- Greywal T, Zaenglein AL, Baldwin HE, et al. Evidence-based recommendations for the management of acne fulminans and its variants. J Am Acad Dermatol. 2017;77:109-117.
- Honma M, Murakami M, Iinuma S, et al. Acne fulminans following measles infection. J Dermatol. 2009;36:471-473.
- Romiti R, Jansen T, Plewig G. Acne fulminans. An Bras Dermatol. 2000;75:611-617.
- Karvonen SL. Acne fulminans: report of clinical findings and treatment of twenty-four patients. J Am Acad Dermatol. 1993;28:572-579.
- Kelly AP, Burns RE. Acute febrile ulcerative conglobate acne with polyarthralgia. Arch Dermatol. 1971;104:182-187.
- Plewig G, Kligman AM. Vitamin A acid in acneiform dermatoses. Acta Derm Venereol Suppl. 1975;74:119-127.
- Seukeran DC, Cunliffe WJ. The treatment of acne fulminans: a review of 25 cases. Br J Dermatol. 1999;141:307-309.
- Kraus SL, Emmert S, Schön MP, et al. The dark side of beauty: acne fulminans induced by anabolic steroids in a male bodybuilder. Arch Dermatol. 2012;148:1210-1212.
- Jansen T, Plewig G. Acne fulminans. Int J Dermatol. 1998;37:254-257.
- Zanelato TP, Gontijo GM, Alves CA, et al. Disabling acne fulminans. An Bras Dermatol. 2011;86:9-12.
- Azulay DR, Abulafia LA, Costa JAN, et al. Tecido de granulação exuberante. efeito colateral da terapêutica com isotretinoína. An Bras Dermatol. 1985;60:179-182.
- Traupe H, von Mühlendahl KE, Brämswig J, et al. Acne of the fulminans type following testosterone therapy in three excessively tall boys. Arch Dermatol. 1988;124:414-417.
- Greywal T, Zaenglein AL, Baldwin HE, et al. Evidence-based recommendations for the management of acne fulminans and its variants. J Am Acad Dermatol. 2017;77:109-117.
- Honma M, Murakami M, Iinuma S, et al. Acne fulminans following measles infection. J Dermatol. 2009;36:471-473.
Practice Points
- Acne fulminans, the most severe form of acne, is characterized by deep ulcerations covered by a hemorrhagic crust. It is commonly associated with fever, polyarthralgia, and myopathy caused by rapid weight loss.
- This rare condition is recognized as a potential complication of oral isotretinoin therapy.
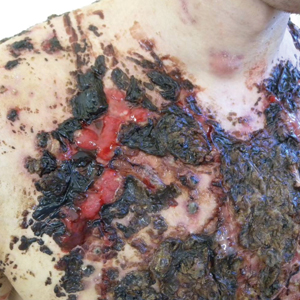
Allergic Contact Dermatitis With Sparing of Exposed Psoriasis Plaques
To the Editor:
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity reaction against antigens to which the skin’s immune system was previously sensitized. The initial sensitization requires penetration of the antigen through the stratum corneum. Thus, the ability of a particle to cause ACD is related to its molecular structure and size, lipophilicity, and protein-binding affinity, as well as the dose and duration of exposure.1 Psoriasis typically presents as well-demarcated areas of skin that may be erythematous, indurated, and scaly to variable degrees. Histologically, psoriasis plaques are characterized by epidermal hyperplasia in the presence of a T-cell infiltrate and neutrophilic microabscesses. We report a case of a patient with plaque-type psoriasis who experienced ACD with sparing of exposed psoriatic plaques.
A 45-year-old man with a 5-year history of generalized moderate to severe psoriasis undergoing therapy with ustekinumab 45 mg subcutaneously once every 12 weeks presented to the emergency department with intensely erythematous, pruritic, vesicular lesions on the trunk, arms, and legs within 24 hours of exposure to poison oak while hiking. The patient reported pruritus, pain, and swelling of the affected areas. On physical examination, he was afebrile. Widespread erythematous vesicular lesions were noted on the face, trunk, arms, and legs, sparing the well-demarcated scaly psoriatic plaques on the arms and legs (Figure). The patient was given intravenous fluids and intravenous diphenhydramine. After responding to initial treatment, the patient was discharged with ibuprofen and a tapering dose of oral prednisone from 60 mg 5 times daily, to 40 mg 5 times daily, to 20 mg 5 times daily over 15 days.
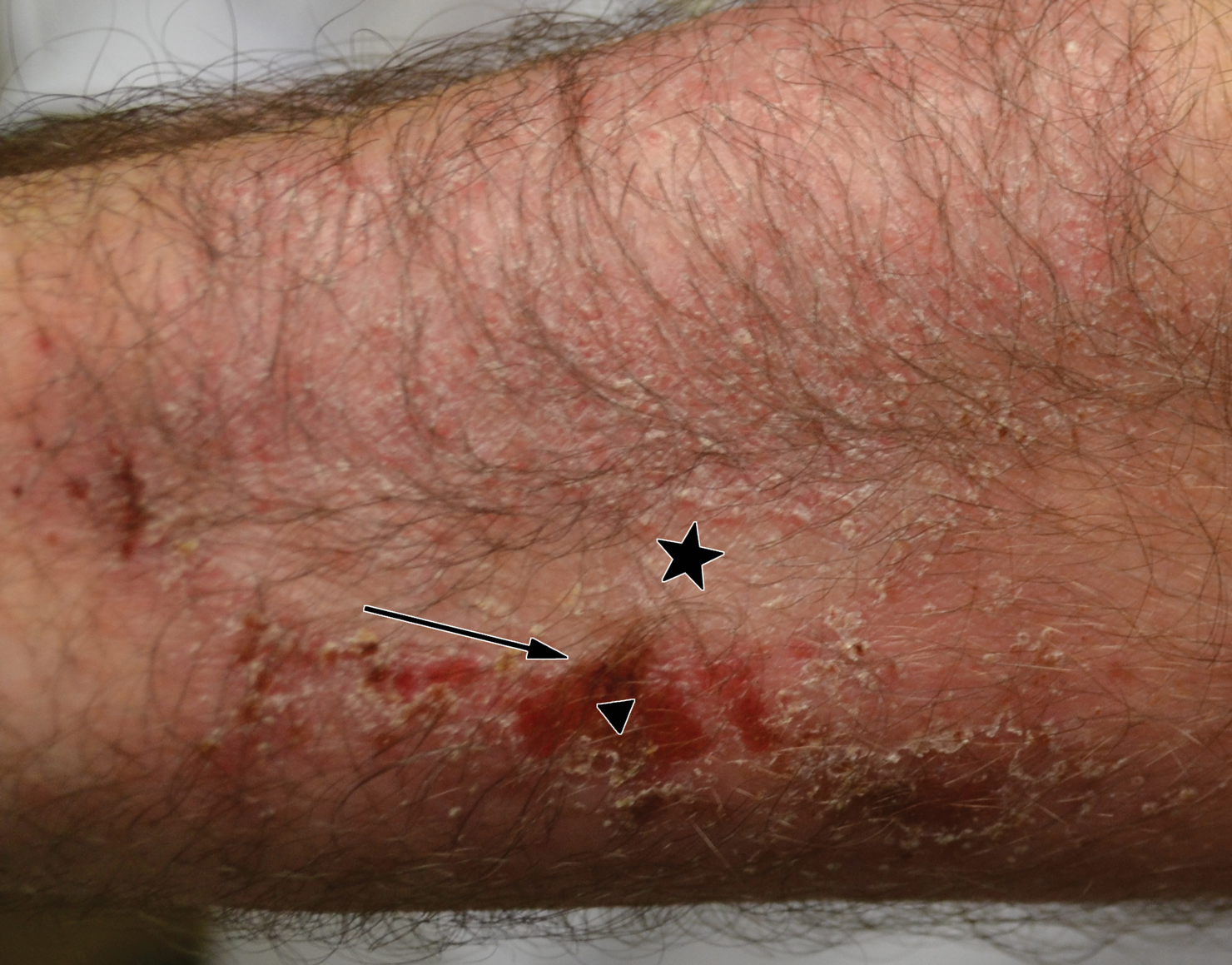
star), with a linear border demarcating the ACD lesion and the unaffected psoriatic plaque (black arrow).
Allergic contact dermatitis occurs after sensitization to environmental allergens or haptens. Clinically, ACD is characterized by pruritic, erythematous, vesicular papules and plaques. The predominant effector cells in ACD are CD8+ T cells, along with contributions from helper T cells (TH2). Together, these cell types produce an environment enriched in IFN-γ, IL-2, IL-4, IL-10, IL-17, and tumor necrosis factor α.2 Ultimately, the ACD response induces keratinocyte apoptosis via cytotoxic effects.3,4
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease that presents clinically as erythematous well-demarcated plaques with a micaceous scale. The immunologic environment of psoriasis plaques is characterized by infiltration of CD4+ TH17 cells and elevated levels of IL-17, IL-23, tumor necrosis factor α, and IL-1β, which induce keratinocyte hyperproliferation through a complex mechanism resulting in hyperkeratosis composed of orthokeratosis and parakeratosis, a neutrophilic infiltrate, and Munro microabscesses.5
The predominant effector cells and the final effects on keratinocyte survival are divergent in psoriasis and ACD. The possibly antagonistic relationship between these immunologic processes is further supported by epidemiologic studies demonstrating a decreased incidence of ACD in patients with psoriasis.6,7
Our patient demonstrated a typical ACD reaction in response to exposure to urushiol, the allergen present in poison oak, in areas unaffected by psoriasis plaques. Interestingly, the patient displayed this response even while undergoing therapy with ustekinumab, a fully humanized antibody that binds IL-12 and IL-23 and ultimately downregulates TH17 cell-mediated release of IL-17 in the treatment of psoriasis. Although IL-17 also has been implicated in ACD, the lack of inhibition of ACD with ustekinumab treatment was previously demonstrated in a small retrospective study, indicating a potentially different source of IL-17 in ACD.8
Our patient did not demonstrate a typical ACD response in areas of active psoriasis plaques. This phenomenon was of great interest to us. It is possible that the presence of hyperkeratosis, manifested clinically as scaling, served as a mechanical barrier preventing the diffusion and exposure of cutaneous immune cells to urushiol. On the other hand, it is possible that the immunologic environment of the active psoriasis plaque was altered in such a way that it did not demonstrate the typical response to allergen exposure.
We hypothesize that the lack of a typical ACD response at sites of psoriatic plaques in our patient may be attributed to the intensity and duration of exposure to the allergen. Quaranta et al9 reported a typical ACD clinical response and a mixed immunohistologic response to nickel patch testing at sites of active plaques in nickel-sensitized psoriasis patients. Patch testing involves 48 hours of direct contact with an allergen, while our patient experienced an estimated 8 to 10 hours of exposure to the allergen prior to removal via washing. Supporting this line of reasoning, a proportion of patients who are responsive to nickel patch testing do not exhibit clinical symptoms in response to casual nickel exposure.10 Although a physical barrier effect due to hyperkeratosis may have contributed to the lack of ACD response in sites of psoriasis plaques in our patient, it remains possible that a more limited duration of exposure to the allergen is not sufficient to overcome the native immunologic milieu of the psoriasis plaque and induce the immunologic cascade resulting in ACD. Further research into the potentially antagonistic relationship of psoriasis and ACD should be performed to elucidate the interaction between these two common conditions.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Vocanson M, Hennino A, Cluzel-Tailhardat M, et al. CD8+ T cells are effector cells of contact dermatitis to common skin allergens in mice. J Invest Dermatol. 2006;126:815-820.
- Akiba H, Kehren J, Ducluzeau MT, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol. 2002;168:3079-3087.
- Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25-35.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Bangsgaard N, Engkilde K, Thyssen JP, et al. Inverse relationship between contact allergy and psoriasis: results from a patient- and a population-based study. Br J Dermatol. 2009;161:1119-1123.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Bangsgaard N, Zachariae C, Menne T, et al. Lack of effect of ustekinumab in treatment of allergic contact dermatitis. Contact Dermatitis. 2011;65:227-230.
- Quaranta M, Eyerich S, Knapp B, et al. Allergic contact dermatitis in psoriasis patients: typical, delayed, and non-interacting. PLoS One. 2014;9:e101814.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
To the Editor:
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity reaction against antigens to which the skin’s immune system was previously sensitized. The initial sensitization requires penetration of the antigen through the stratum corneum. Thus, the ability of a particle to cause ACD is related to its molecular structure and size, lipophilicity, and protein-binding affinity, as well as the dose and duration of exposure.1 Psoriasis typically presents as well-demarcated areas of skin that may be erythematous, indurated, and scaly to variable degrees. Histologically, psoriasis plaques are characterized by epidermal hyperplasia in the presence of a T-cell infiltrate and neutrophilic microabscesses. We report a case of a patient with plaque-type psoriasis who experienced ACD with sparing of exposed psoriatic plaques.
A 45-year-old man with a 5-year history of generalized moderate to severe psoriasis undergoing therapy with ustekinumab 45 mg subcutaneously once every 12 weeks presented to the emergency department with intensely erythematous, pruritic, vesicular lesions on the trunk, arms, and legs within 24 hours of exposure to poison oak while hiking. The patient reported pruritus, pain, and swelling of the affected areas. On physical examination, he was afebrile. Widespread erythematous vesicular lesions were noted on the face, trunk, arms, and legs, sparing the well-demarcated scaly psoriatic plaques on the arms and legs (Figure). The patient was given intravenous fluids and intravenous diphenhydramine. After responding to initial treatment, the patient was discharged with ibuprofen and a tapering dose of oral prednisone from 60 mg 5 times daily, to 40 mg 5 times daily, to 20 mg 5 times daily over 15 days.
star), with a linear border demarcating the ACD lesion and the unaffected psoriatic plaque (black arrow).
Allergic contact dermatitis occurs after sensitization to environmental allergens or haptens. Clinically, ACD is characterized by pruritic, erythematous, vesicular papules and plaques. The predominant effector cells in ACD are CD8+ T cells, along with contributions from helper T cells (TH2). Together, these cell types produce an environment enriched in IFN-γ, IL-2, IL-4, IL-10, IL-17, and tumor necrosis factor α.2 Ultimately, the ACD response induces keratinocyte apoptosis via cytotoxic effects.3,4
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease that presents clinically as erythematous well-demarcated plaques with a micaceous scale. The immunologic environment of psoriasis plaques is characterized by infiltration of CD4+ TH17 cells and elevated levels of IL-17, IL-23, tumor necrosis factor α, and IL-1β, which induce keratinocyte hyperproliferation through a complex mechanism resulting in hyperkeratosis composed of orthokeratosis and parakeratosis, a neutrophilic infiltrate, and Munro microabscesses.5
The predominant effector cells and the final effects on keratinocyte survival are divergent in psoriasis and ACD. The possibly antagonistic relationship between these immunologic processes is further supported by epidemiologic studies demonstrating a decreased incidence of ACD in patients with psoriasis.6,7
Our patient demonstrated a typical ACD reaction in response to exposure to urushiol, the allergen present in poison oak, in areas unaffected by psoriasis plaques. Interestingly, the patient displayed this response even while undergoing therapy with ustekinumab, a fully humanized antibody that binds IL-12 and IL-23 and ultimately downregulates TH17 cell-mediated release of IL-17 in the treatment of psoriasis. Although IL-17 also has been implicated in ACD, the lack of inhibition of ACD with ustekinumab treatment was previously demonstrated in a small retrospective study, indicating a potentially different source of IL-17 in ACD.8
Our patient did not demonstrate a typical ACD response in areas of active psoriasis plaques. This phenomenon was of great interest to us. It is possible that the presence of hyperkeratosis, manifested clinically as scaling, served as a mechanical barrier preventing the diffusion and exposure of cutaneous immune cells to urushiol. On the other hand, it is possible that the immunologic environment of the active psoriasis plaque was altered in such a way that it did not demonstrate the typical response to allergen exposure.
We hypothesize that the lack of a typical ACD response at sites of psoriatic plaques in our patient may be attributed to the intensity and duration of exposure to the allergen. Quaranta et al9 reported a typical ACD clinical response and a mixed immunohistologic response to nickel patch testing at sites of active plaques in nickel-sensitized psoriasis patients. Patch testing involves 48 hours of direct contact with an allergen, while our patient experienced an estimated 8 to 10 hours of exposure to the allergen prior to removal via washing. Supporting this line of reasoning, a proportion of patients who are responsive to nickel patch testing do not exhibit clinical symptoms in response to casual nickel exposure.10 Although a physical barrier effect due to hyperkeratosis may have contributed to the lack of ACD response in sites of psoriasis plaques in our patient, it remains possible that a more limited duration of exposure to the allergen is not sufficient to overcome the native immunologic milieu of the psoriasis plaque and induce the immunologic cascade resulting in ACD. Further research into the potentially antagonistic relationship of psoriasis and ACD should be performed to elucidate the interaction between these two common conditions.
To the Editor:
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity reaction against antigens to which the skin’s immune system was previously sensitized. The initial sensitization requires penetration of the antigen through the stratum corneum. Thus, the ability of a particle to cause ACD is related to its molecular structure and size, lipophilicity, and protein-binding affinity, as well as the dose and duration of exposure.1 Psoriasis typically presents as well-demarcated areas of skin that may be erythematous, indurated, and scaly to variable degrees. Histologically, psoriasis plaques are characterized by epidermal hyperplasia in the presence of a T-cell infiltrate and neutrophilic microabscesses. We report a case of a patient with plaque-type psoriasis who experienced ACD with sparing of exposed psoriatic plaques.
A 45-year-old man with a 5-year history of generalized moderate to severe psoriasis undergoing therapy with ustekinumab 45 mg subcutaneously once every 12 weeks presented to the emergency department with intensely erythematous, pruritic, vesicular lesions on the trunk, arms, and legs within 24 hours of exposure to poison oak while hiking. The patient reported pruritus, pain, and swelling of the affected areas. On physical examination, he was afebrile. Widespread erythematous vesicular lesions were noted on the face, trunk, arms, and legs, sparing the well-demarcated scaly psoriatic plaques on the arms and legs (Figure). The patient was given intravenous fluids and intravenous diphenhydramine. After responding to initial treatment, the patient was discharged with ibuprofen and a tapering dose of oral prednisone from 60 mg 5 times daily, to 40 mg 5 times daily, to 20 mg 5 times daily over 15 days.
star), with a linear border demarcating the ACD lesion and the unaffected psoriatic plaque (black arrow).
Allergic contact dermatitis occurs after sensitization to environmental allergens or haptens. Clinically, ACD is characterized by pruritic, erythematous, vesicular papules and plaques. The predominant effector cells in ACD are CD8+ T cells, along with contributions from helper T cells (TH2). Together, these cell types produce an environment enriched in IFN-γ, IL-2, IL-4, IL-10, IL-17, and tumor necrosis factor α.2 Ultimately, the ACD response induces keratinocyte apoptosis via cytotoxic effects.3,4
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease that presents clinically as erythematous well-demarcated plaques with a micaceous scale. The immunologic environment of psoriasis plaques is characterized by infiltration of CD4+ TH17 cells and elevated levels of IL-17, IL-23, tumor necrosis factor α, and IL-1β, which induce keratinocyte hyperproliferation through a complex mechanism resulting in hyperkeratosis composed of orthokeratosis and parakeratosis, a neutrophilic infiltrate, and Munro microabscesses.5
The predominant effector cells and the final effects on keratinocyte survival are divergent in psoriasis and ACD. The possibly antagonistic relationship between these immunologic processes is further supported by epidemiologic studies demonstrating a decreased incidence of ACD in patients with psoriasis.6,7
Our patient demonstrated a typical ACD reaction in response to exposure to urushiol, the allergen present in poison oak, in areas unaffected by psoriasis plaques. Interestingly, the patient displayed this response even while undergoing therapy with ustekinumab, a fully humanized antibody that binds IL-12 and IL-23 and ultimately downregulates TH17 cell-mediated release of IL-17 in the treatment of psoriasis. Although IL-17 also has been implicated in ACD, the lack of inhibition of ACD with ustekinumab treatment was previously demonstrated in a small retrospective study, indicating a potentially different source of IL-17 in ACD.8
Our patient did not demonstrate a typical ACD response in areas of active psoriasis plaques. This phenomenon was of great interest to us. It is possible that the presence of hyperkeratosis, manifested clinically as scaling, served as a mechanical barrier preventing the diffusion and exposure of cutaneous immune cells to urushiol. On the other hand, it is possible that the immunologic environment of the active psoriasis plaque was altered in such a way that it did not demonstrate the typical response to allergen exposure.
We hypothesize that the lack of a typical ACD response at sites of psoriatic plaques in our patient may be attributed to the intensity and duration of exposure to the allergen. Quaranta et al9 reported a typical ACD clinical response and a mixed immunohistologic response to nickel patch testing at sites of active plaques in nickel-sensitized psoriasis patients. Patch testing involves 48 hours of direct contact with an allergen, while our patient experienced an estimated 8 to 10 hours of exposure to the allergen prior to removal via washing. Supporting this line of reasoning, a proportion of patients who are responsive to nickel patch testing do not exhibit clinical symptoms in response to casual nickel exposure.10 Although a physical barrier effect due to hyperkeratosis may have contributed to the lack of ACD response in sites of psoriasis plaques in our patient, it remains possible that a more limited duration of exposure to the allergen is not sufficient to overcome the native immunologic milieu of the psoriasis plaque and induce the immunologic cascade resulting in ACD. Further research into the potentially antagonistic relationship of psoriasis and ACD should be performed to elucidate the interaction between these two common conditions.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Vocanson M, Hennino A, Cluzel-Tailhardat M, et al. CD8+ T cells are effector cells of contact dermatitis to common skin allergens in mice. J Invest Dermatol. 2006;126:815-820.
- Akiba H, Kehren J, Ducluzeau MT, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol. 2002;168:3079-3087.
- Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25-35.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Bangsgaard N, Engkilde K, Thyssen JP, et al. Inverse relationship between contact allergy and psoriasis: results from a patient- and a population-based study. Br J Dermatol. 2009;161:1119-1123.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Bangsgaard N, Zachariae C, Menne T, et al. Lack of effect of ustekinumab in treatment of allergic contact dermatitis. Contact Dermatitis. 2011;65:227-230.
- Quaranta M, Eyerich S, Knapp B, et al. Allergic contact dermatitis in psoriasis patients: typical, delayed, and non-interacting. PLoS One. 2014;9:e101814.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Vocanson M, Hennino A, Cluzel-Tailhardat M, et al. CD8+ T cells are effector cells of contact dermatitis to common skin allergens in mice. J Invest Dermatol. 2006;126:815-820.
- Akiba H, Kehren J, Ducluzeau MT, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol. 2002;168:3079-3087.
- Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25-35.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Bangsgaard N, Engkilde K, Thyssen JP, et al. Inverse relationship between contact allergy and psoriasis: results from a patient- and a population-based study. Br J Dermatol. 2009;161:1119-1123.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Bangsgaard N, Zachariae C, Menne T, et al. Lack of effect of ustekinumab in treatment of allergic contact dermatitis. Contact Dermatitis. 2011;65:227-230.
- Quaranta M, Eyerich S, Knapp B, et al. Allergic contact dermatitis in psoriasis patients: typical, delayed, and non-interacting. PLoS One. 2014;9:e101814.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
Practice Points
- Patients with plaque-type psoriasis who experience allergic contact dermatitis (ACD) may present with sparing of exposed psoriatic plaques.
- The divergent immunologic milieus present in ACD and psoriasis likely underly the decreased incidence of ACD in patients with psoriasis.
Generalized Granuloma Annulare Responsive to Narrowband UVB
To the Editor:
Granuloma annulare (GA) is a common dermatosis that usually presents with dermal papules and annular plaques in a symmetric distribution.1 The etiology is unknown, but a delayed-type hypersensitivity reaction is the favored pathogenesis. Several systemic associations have been reported with generalized GA including diabetes mellitus, hyperlipidemia, autoimmune thyroiditis, rheumatoid arthritis, and lymphoproliferative malignancies, as well as other malignancies and viral infections such as human immunodeficiency virus and hepatitis C. Localized GA often is self-limiting, but generalized disease can be chronic and progressive. Although asymptomatic in most cases, the lesions can be cosmetically bothersome, and many patients desire treatment. There are few well-controlled studies of treatment, and most are limited to case reports and series. A review of GA treatment noted only 3 randomized studies: 2 relating to photodynamic therapy and 1 to cryosurgery. Well-accepted therapies, such as topical and intralesional corticosteroids, antimalarials, immunosuppressants, antibiotics, and phototherapy, are substantiated by lesser-quality evidence.1 Phototherapy has been studied for the treatment of GA and other disorders with altered dermal matrix deposition for which there are limited effective treatment options. UV irradiation promotes degradation of structural components of the dermis and inhibition of collagen production.2 Granuloma annulare generally is resistant to therapy. We report a case of generalized GA of long duration that responded well to phototherapy with narrowband UVB (NB-UVB).
A 60-year-old woman presented with generalized GA of 4 years’ duration that was confirmed on biopsy on 2 occasions (Figure 1). The lesions were asymptomatic but disfiguring and consisted of extensive pink, thin, annular plaques and papules on the torso, arms, and legs (Figure 2A). Apart from mild depression for which she was being treated with paroxetine and trazodone, she was otherwise healthy without evidence of thyroid disease, hyperlipidemia, or diabetes mellitus. Prior treatments for GA had included tapering courses of prednisone (up to 30 mg/d, tapered by 5 mg every 4 days) and betamethasone dipropionate cream 0.05%. She was started on NB-UVB therapy 5 times weekly in incremental doses with no adjuvant therapy. After 100 treatments, there was notable improvement with lesions becoming paler and flatter, with some involuting completely (Figure 2B). The frequency of treatment was reduced to 3 times weekly with continued improvement. An NB-UVB device was used containing 48 TL 100W/01-FS72 lamps with a mean irradiance of 2.9 mW/cm2. Her starting dose was 90 mJ/cm2. The cumulative dose after 100 treatments was 35,600 mJ/cm2. Apart from occasional mild erythema, there were no adverse effects.
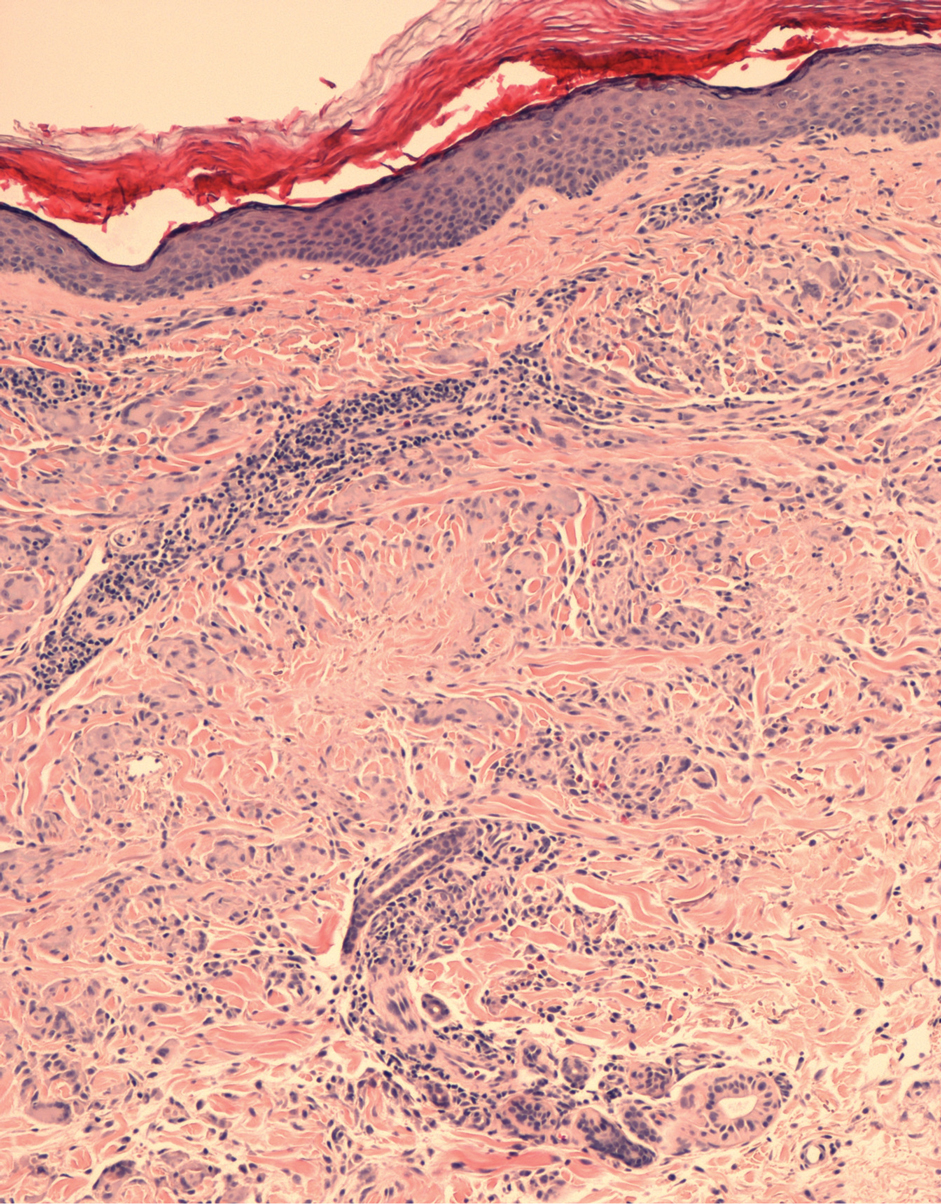

Inui et al3 described the successful treatment of generalized GA with NB-UVB. A retrospective review of NB-UVB for vitiligo, pruritus, and inflammatory dermatoses included 2 cases of generalized GA that were noted to have only a minimal to mild improvement.4 Most reports relating to phototherapy of GA have focused on psoralen plus UVA (PUVA). A retrospective study of 33 patients treated with systemic PUVA showed improvement in two-thirds of patients.5 Older studies showed systemic PUVA was effective in 1 patient after 53 treatments6 and in 4 patients using a high-dose protocol7; topical PUVA was effective in 4 patients after an average of 26 treatments.8 Psoralen plus UVA bath was reported as an effective treatment of generalized GA in a child.9 UVA1 phototherapy provided good or excellent results in half of patients (10/20) studied with generalized GA; however, discontinuation of treatment resulted in early recurrence of disease.10 In general, NB-UVB has been preferred over PUVA and UVA1 due to long-term safety, tolerability, and access. Although further clinical trials are needed, our report suggests that NB-UVB could be a useful modality in generalized GA.
- Thornsberry LA, English JC III. Etiology, diagnosis, and therapeutic management of granuloma annulare: an update. Am J Clin Dermatol. 2013;14:279-290.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Inui S, Nishida Y, Itami S, et al. Disseminated granuloma annulare responsive to narrowband ultraviolet B therapy. J Am Acad Dermatol. 2005;53:532-533.
- Samson Yashar S, Gielczyk R, Scherschun L, et al. Narrow-band ultraviolet B treatment for vitiligo, pruritus, and inflammatory dermatoses. Photodermatol Photoimmunol Photomed. 2003;19:164-168.
- Browne F, Turner D, Goulden V. Psoralen and ultraviolet A in the treatment of granuloma annulare. Photodermatol Photoimmunol Photomed. 2011;27:81-84.
- Setterfield J, Huilgol SC, Black MM. Generalised granuloma annulare successfully treated with PUVA. Clin Exp Dermatol. 1999;24:458-460.
- Munchenberger S, Schopf E, Simon JC. Phototherapy with UVA-1 for generalized granuloma annulare. Arch Dermatol. 1997;133:1605.
- Grundmann-Kollmann M, Ochsendorf FR, Zollner TM, et al. Cream psoralen plus ultraviolet A therapy for granuloma annulare. Br J Dermatol. 2001;144:996-999.
- Batchelor R, Clark S. Clearance of generalized popular umbilicated granuloma annulare in a child with bath PUVA therapy. Pediatr Dermatol. 2006;23:72-74.
- Schnopp C, Tzaneva S, Mempel M, et al. UVA1 phototherapy for disseminated granuloma annulare. Photodermatol Photoimmunol Photomed. 2005;21:68-71.
To the Editor:
Granuloma annulare (GA) is a common dermatosis that usually presents with dermal papules and annular plaques in a symmetric distribution.1 The etiology is unknown, but a delayed-type hypersensitivity reaction is the favored pathogenesis. Several systemic associations have been reported with generalized GA including diabetes mellitus, hyperlipidemia, autoimmune thyroiditis, rheumatoid arthritis, and lymphoproliferative malignancies, as well as other malignancies and viral infections such as human immunodeficiency virus and hepatitis C. Localized GA often is self-limiting, but generalized disease can be chronic and progressive. Although asymptomatic in most cases, the lesions can be cosmetically bothersome, and many patients desire treatment. There are few well-controlled studies of treatment, and most are limited to case reports and series. A review of GA treatment noted only 3 randomized studies: 2 relating to photodynamic therapy and 1 to cryosurgery. Well-accepted therapies, such as topical and intralesional corticosteroids, antimalarials, immunosuppressants, antibiotics, and phototherapy, are substantiated by lesser-quality evidence.1 Phototherapy has been studied for the treatment of GA and other disorders with altered dermal matrix deposition for which there are limited effective treatment options. UV irradiation promotes degradation of structural components of the dermis and inhibition of collagen production.2 Granuloma annulare generally is resistant to therapy. We report a case of generalized GA of long duration that responded well to phototherapy with narrowband UVB (NB-UVB).
A 60-year-old woman presented with generalized GA of 4 years’ duration that was confirmed on biopsy on 2 occasions (Figure 1). The lesions were asymptomatic but disfiguring and consisted of extensive pink, thin, annular plaques and papules on the torso, arms, and legs (Figure 2A). Apart from mild depression for which she was being treated with paroxetine and trazodone, she was otherwise healthy without evidence of thyroid disease, hyperlipidemia, or diabetes mellitus. Prior treatments for GA had included tapering courses of prednisone (up to 30 mg/d, tapered by 5 mg every 4 days) and betamethasone dipropionate cream 0.05%. She was started on NB-UVB therapy 5 times weekly in incremental doses with no adjuvant therapy. After 100 treatments, there was notable improvement with lesions becoming paler and flatter, with some involuting completely (Figure 2B). The frequency of treatment was reduced to 3 times weekly with continued improvement. An NB-UVB device was used containing 48 TL 100W/01-FS72 lamps with a mean irradiance of 2.9 mW/cm2. Her starting dose was 90 mJ/cm2. The cumulative dose after 100 treatments was 35,600 mJ/cm2. Apart from occasional mild erythema, there were no adverse effects.
Inui et al3 described the successful treatment of generalized GA with NB-UVB. A retrospective review of NB-UVB for vitiligo, pruritus, and inflammatory dermatoses included 2 cases of generalized GA that were noted to have only a minimal to mild improvement.4 Most reports relating to phototherapy of GA have focused on psoralen plus UVA (PUVA). A retrospective study of 33 patients treated with systemic PUVA showed improvement in two-thirds of patients.5 Older studies showed systemic PUVA was effective in 1 patient after 53 treatments6 and in 4 patients using a high-dose protocol7; topical PUVA was effective in 4 patients after an average of 26 treatments.8 Psoralen plus UVA bath was reported as an effective treatment of generalized GA in a child.9 UVA1 phototherapy provided good or excellent results in half of patients (10/20) studied with generalized GA; however, discontinuation of treatment resulted in early recurrence of disease.10 In general, NB-UVB has been preferred over PUVA and UVA1 due to long-term safety, tolerability, and access. Although further clinical trials are needed, our report suggests that NB-UVB could be a useful modality in generalized GA.
To the Editor:
Granuloma annulare (GA) is a common dermatosis that usually presents with dermal papules and annular plaques in a symmetric distribution.1 The etiology is unknown, but a delayed-type hypersensitivity reaction is the favored pathogenesis. Several systemic associations have been reported with generalized GA including diabetes mellitus, hyperlipidemia, autoimmune thyroiditis, rheumatoid arthritis, and lymphoproliferative malignancies, as well as other malignancies and viral infections such as human immunodeficiency virus and hepatitis C. Localized GA often is self-limiting, but generalized disease can be chronic and progressive. Although asymptomatic in most cases, the lesions can be cosmetically bothersome, and many patients desire treatment. There are few well-controlled studies of treatment, and most are limited to case reports and series. A review of GA treatment noted only 3 randomized studies: 2 relating to photodynamic therapy and 1 to cryosurgery. Well-accepted therapies, such as topical and intralesional corticosteroids, antimalarials, immunosuppressants, antibiotics, and phototherapy, are substantiated by lesser-quality evidence.1 Phototherapy has been studied for the treatment of GA and other disorders with altered dermal matrix deposition for which there are limited effective treatment options. UV irradiation promotes degradation of structural components of the dermis and inhibition of collagen production.2 Granuloma annulare generally is resistant to therapy. We report a case of generalized GA of long duration that responded well to phototherapy with narrowband UVB (NB-UVB).
A 60-year-old woman presented with generalized GA of 4 years’ duration that was confirmed on biopsy on 2 occasions (Figure 1). The lesions were asymptomatic but disfiguring and consisted of extensive pink, thin, annular plaques and papules on the torso, arms, and legs (Figure 2A). Apart from mild depression for which she was being treated with paroxetine and trazodone, she was otherwise healthy without evidence of thyroid disease, hyperlipidemia, or diabetes mellitus. Prior treatments for GA had included tapering courses of prednisone (up to 30 mg/d, tapered by 5 mg every 4 days) and betamethasone dipropionate cream 0.05%. She was started on NB-UVB therapy 5 times weekly in incremental doses with no adjuvant therapy. After 100 treatments, there was notable improvement with lesions becoming paler and flatter, with some involuting completely (Figure 2B). The frequency of treatment was reduced to 3 times weekly with continued improvement. An NB-UVB device was used containing 48 TL 100W/01-FS72 lamps with a mean irradiance of 2.9 mW/cm2. Her starting dose was 90 mJ/cm2. The cumulative dose after 100 treatments was 35,600 mJ/cm2. Apart from occasional mild erythema, there were no adverse effects.
Inui et al3 described the successful treatment of generalized GA with NB-UVB. A retrospective review of NB-UVB for vitiligo, pruritus, and inflammatory dermatoses included 2 cases of generalized GA that were noted to have only a minimal to mild improvement.4 Most reports relating to phototherapy of GA have focused on psoralen plus UVA (PUVA). A retrospective study of 33 patients treated with systemic PUVA showed improvement in two-thirds of patients.5 Older studies showed systemic PUVA was effective in 1 patient after 53 treatments6 and in 4 patients using a high-dose protocol7; topical PUVA was effective in 4 patients after an average of 26 treatments.8 Psoralen plus UVA bath was reported as an effective treatment of generalized GA in a child.9 UVA1 phototherapy provided good or excellent results in half of patients (10/20) studied with generalized GA; however, discontinuation of treatment resulted in early recurrence of disease.10 In general, NB-UVB has been preferred over PUVA and UVA1 due to long-term safety, tolerability, and access. Although further clinical trials are needed, our report suggests that NB-UVB could be a useful modality in generalized GA.
- Thornsberry LA, English JC III. Etiology, diagnosis, and therapeutic management of granuloma annulare: an update. Am J Clin Dermatol. 2013;14:279-290.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Inui S, Nishida Y, Itami S, et al. Disseminated granuloma annulare responsive to narrowband ultraviolet B therapy. J Am Acad Dermatol. 2005;53:532-533.
- Samson Yashar S, Gielczyk R, Scherschun L, et al. Narrow-band ultraviolet B treatment for vitiligo, pruritus, and inflammatory dermatoses. Photodermatol Photoimmunol Photomed. 2003;19:164-168.
- Browne F, Turner D, Goulden V. Psoralen and ultraviolet A in the treatment of granuloma annulare. Photodermatol Photoimmunol Photomed. 2011;27:81-84.
- Setterfield J, Huilgol SC, Black MM. Generalised granuloma annulare successfully treated with PUVA. Clin Exp Dermatol. 1999;24:458-460.
- Munchenberger S, Schopf E, Simon JC. Phototherapy with UVA-1 for generalized granuloma annulare. Arch Dermatol. 1997;133:1605.
- Grundmann-Kollmann M, Ochsendorf FR, Zollner TM, et al. Cream psoralen plus ultraviolet A therapy for granuloma annulare. Br J Dermatol. 2001;144:996-999.
- Batchelor R, Clark S. Clearance of generalized popular umbilicated granuloma annulare in a child with bath PUVA therapy. Pediatr Dermatol. 2006;23:72-74.
- Schnopp C, Tzaneva S, Mempel M, et al. UVA1 phototherapy for disseminated granuloma annulare. Photodermatol Photoimmunol Photomed. 2005;21:68-71.
- Thornsberry LA, English JC III. Etiology, diagnosis, and therapeutic management of granuloma annulare: an update. Am J Clin Dermatol. 2013;14:279-290.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Inui S, Nishida Y, Itami S, et al. Disseminated granuloma annulare responsive to narrowband ultraviolet B therapy. J Am Acad Dermatol. 2005;53:532-533.
- Samson Yashar S, Gielczyk R, Scherschun L, et al. Narrow-band ultraviolet B treatment for vitiligo, pruritus, and inflammatory dermatoses. Photodermatol Photoimmunol Photomed. 2003;19:164-168.
- Browne F, Turner D, Goulden V. Psoralen and ultraviolet A in the treatment of granuloma annulare. Photodermatol Photoimmunol Photomed. 2011;27:81-84.
- Setterfield J, Huilgol SC, Black MM. Generalised granuloma annulare successfully treated with PUVA. Clin Exp Dermatol. 1999;24:458-460.
- Munchenberger S, Schopf E, Simon JC. Phototherapy with UVA-1 for generalized granuloma annulare. Arch Dermatol. 1997;133:1605.
- Grundmann-Kollmann M, Ochsendorf FR, Zollner TM, et al. Cream psoralen plus ultraviolet A therapy for granuloma annulare. Br J Dermatol. 2001;144:996-999.
- Batchelor R, Clark S. Clearance of generalized popular umbilicated granuloma annulare in a child with bath PUVA therapy. Pediatr Dermatol. 2006;23:72-74.
- Schnopp C, Tzaneva S, Mempel M, et al. UVA1 phototherapy for disseminated granuloma annulare. Photodermatol Photoimmunol Photomed. 2005;21:68-71.
Practice Points
- The generalized variant of granuloma annulare (GA) can be persistent, sometimes lasting years to decades; treatment is not always effective.
- The safety profile and tolerability of narrowband UVB phototherapy make it a suitable treatment option for generalized GA.
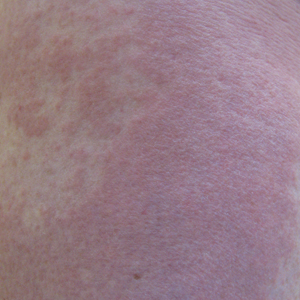
Pustular Tinea Id Reaction
To the Editor:
A 17-year-old adolescent girl presented to the dermatology clinic with a tender pruritic rash on the left wrist that was spreading to the bilateral arms and legs of several years’ duration. An area of a prior biopsy on the left wrist was healing well with use of petroleum jelly and halcinonide cream. The patient denied any constitutional symptoms.
Physical examination revealed numerous erythematous papules coalescing into plaques on the bilateral anterior and posterior arms and legs, including some erythematous macules and papules on the palms and soles. The original area of involvement on the left dorsal medial wrist demonstrated a background of erythema with overlying peripheral scaling and resolving violaceous to erythematous papules with signs of serosanguineous crusting (Figure 1). Scattered perifollicular erythema was present on the posterior aspects of the bilateral thighs and arms (Figure 2). Baseline complete blood cell count and complete metabolic panel were within reference range.
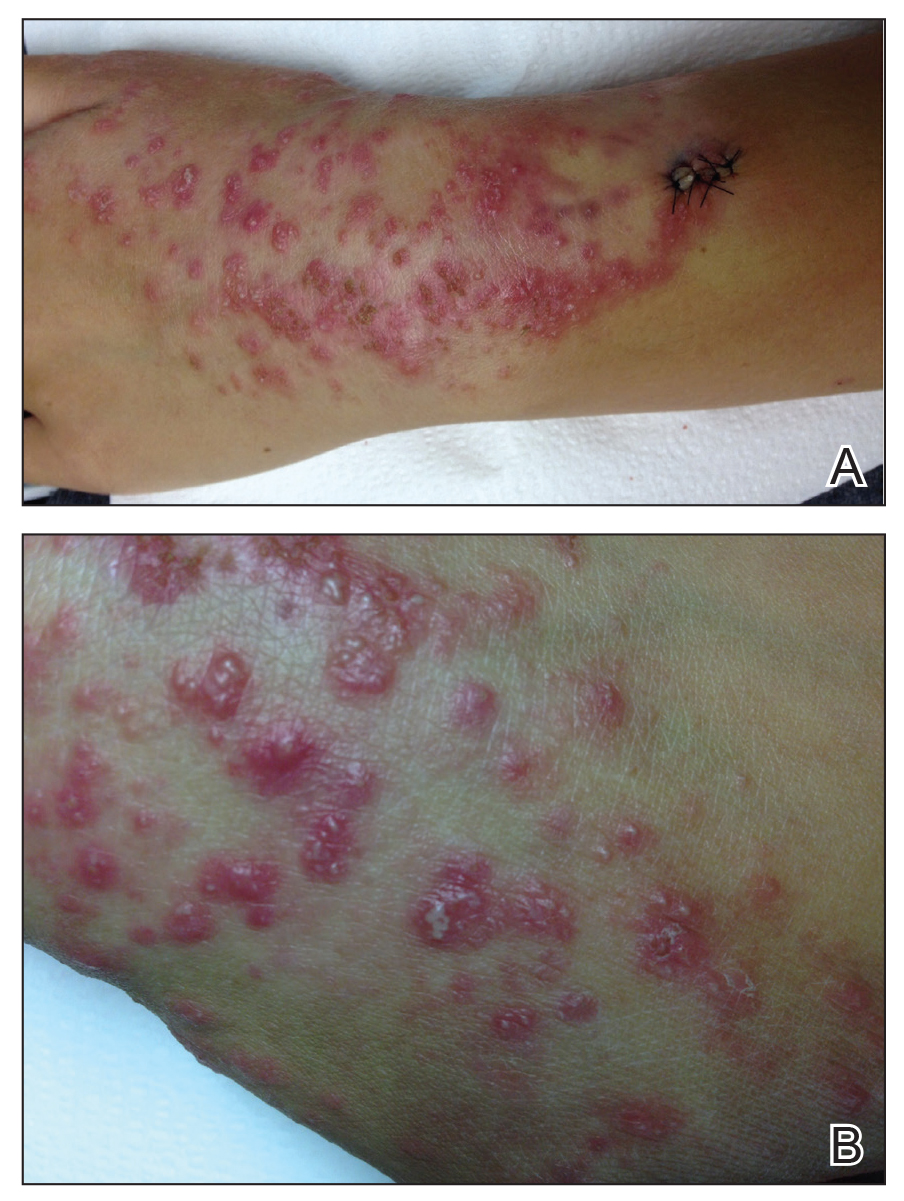
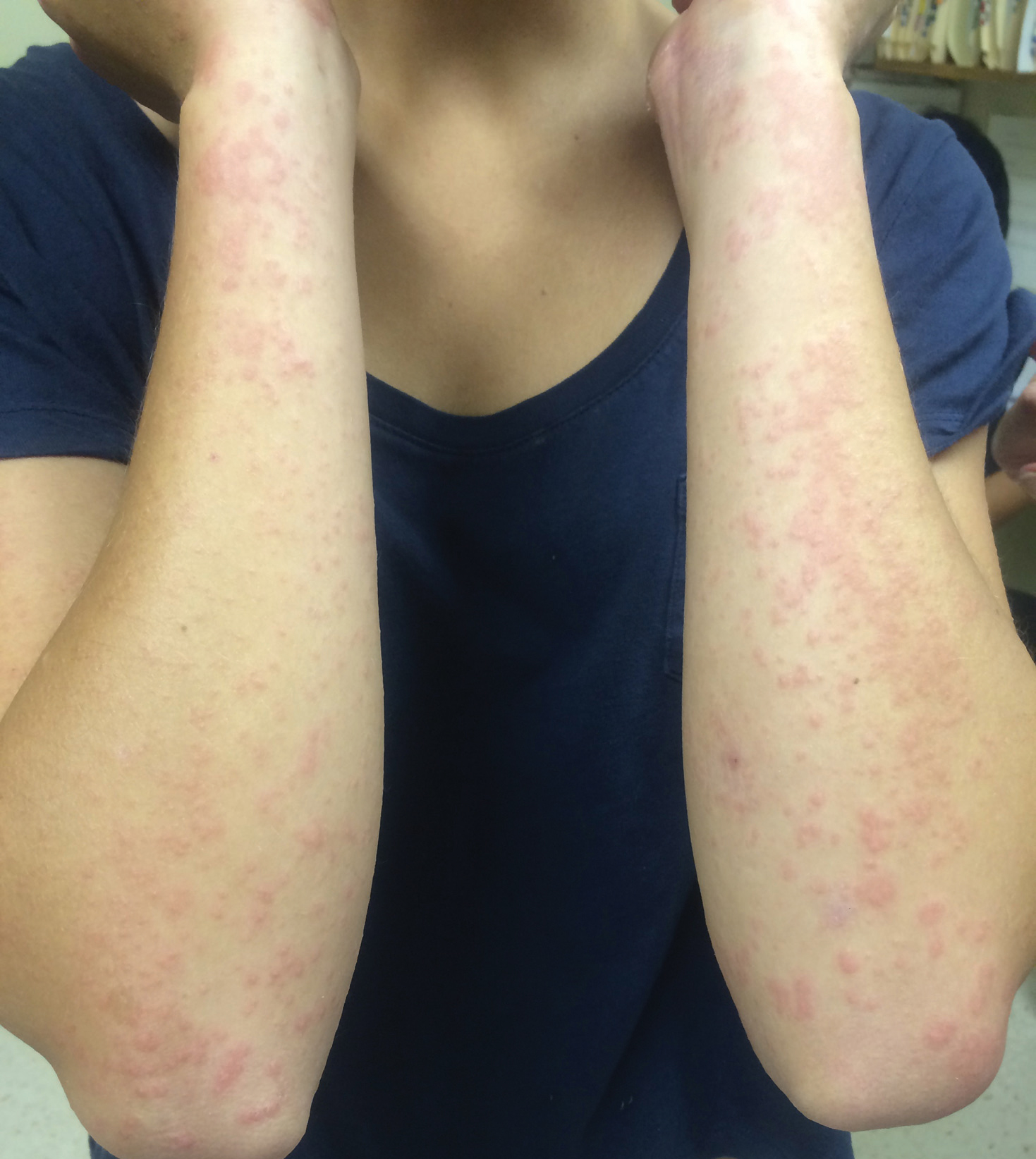
Clinical histopathology showed evidence of a pustular superficial dermatophyte infection, and Grocott-Gomori methenamine-silver stain demonstrated numerous fungal hyphae within subcorneal pustules, indicating pustular tinea. Based on the clinicopathologic correlation, the initial presentation was diagnosed as pustular tinea of the entire left wrist, followed by a generalized id reaction 1 week later.
The patient was prescribed oral terbinafine 250 mg once daily to treat the diffuse involvement of the pustular tinea as well as once-daily oral cetirizine, once-daily oral diphenhydramine, a topical emollient, and a topical nonsteroidal antipruritic gel.
Tinea is a superficial fungal infection commonly caused by the dermatophytes Epidermophyton, Trichophyton, and Microsporum. It has a variety of clinical presentations based on the anatomic location, including tinea capitis (hair/scalp), tinea pedis (feet), tinea corporis (face/trunk/extremities), tinea cruris (groin), and tinea unguium (nails).1 Tinea infections occur in the stratum corneum, hair, and nails, thriving on dead keratin in these areas.2 Tinea corporis usually appears as an erythematous ring-shaped lesion with a scaly border, but atypical cases presenting with vesicles, pustules, and bullae also have been reported.3 Additionally, secondary eruptions called id reactions, or autoeczematization, can present in the setting of dermatophyte infections. Such outbreaks may be due to a delayed hypersensitivity reaction to the fungal antigens. Id reactions can manifest in many forms of tinea with patients generally exhibiting pruritic papulovesicular lesions that can present far from the site of origin.4
Patients with id reactions can have atypical and varied presentations. In a case of id reaction due to tinea corporis, a patient presented with vesicles and pustules that grew in number and coalesced to form annular lesions.5 A case of an id reaction caused by tinea pedis also noted the presence of pustules, which are atypical in this form of tinea.6 In another case of tinea pedis, a generalized id reaction was noted, illustrating that such eruptions do not necessarily appear at the original site of infection.7 Additionally, in a rare presentation of tinea invading the nares, a patient developed an erythema multiforme id reaction.8 Id reactions also were noted in 14 patients with refractory otitis externa, illustrating the ability of this fungal infection to persist and infect distant locations.9
Because the differential diagnoses for tinea infection are extensive, pathology or laboratory confirmation is necessary for diagnosis, and potassium hydroxide preparation often is used to diagnose dermatophyte infections.1,2 Additionally, the possibility of a hypersensitivity drug rash should remain in the differential if the patient received allergy-inducing medications prior to the outbreak, which may in turn complicate the diagnosis.
Tinea infections typically can be treated with topical antifungals such as terbinafine, butenafine,1 and luliconazole10; however, more involved cases may require oral antifungal treatment.1 Systemic treatment of tinea corporis includes itraconazole, terbinafine, and fluconazole,11 all of which exhibit fewer side effects and greater efficacy when compared to griseofulvin.12-15
Treatment of id reactions centers on the proper clearance of the dermatophyte infection, and treatment with oral antifungals generally is sufficient. In the cases of id reaction in patients with refractory otitis, some success was achieved with treatment involving immunotherapy with dermatophyte and dust mite allergen extracts coupled with a yeast elimination diet.9 In acute id reactions, topical corticosteroids and antipruritic agents can be applied.4 Rarely, systemic glucocorticoids are required, such as in cases in which the id reaction persists despite proper treatment of the primary infection.16
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Hanover, NH: Elsevier, Inc; 2010.
- Ziemer M, Seyfarth F, Elsner P, et al. Atypical manifestations of tinea corporis. Mycoses. 2007;50(suppl 2):31-35.
- Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications [published online July 4, 2011]. Pediatrics. 2011;128:e453-e457.
- Ohno S, Tanabe H, Kawasaki M, et al. Tinea corporis with acute inflammation caused by Trichophyton tonsurans. J Dermatol. 2008;35:590-593.
- Hirschmann JV, Raugi GJ. Pustular tinea pedis. J Am Acad Dermatol. 2000;42:132-133.
- Iglesias ME, España A, Idoate MA, et al. Generalized skin reaction following tinea pedis (dermatophytids). J Dermatol. 1994;21:31-34.
- Atzori L, Pau M, Aste M. Erythema multiforme ID reaction in atypical dermatophytosis: a case report. J Eur Acad Dermatol Venereol. 2003;17:699-701.
- Derebery J, Berliner KI. Foot and ear disease—the dermatophytid reaction in otology. Laryngoscope. 1996;106(2 Pt 1):181-186.
- Khanna D, Bharti S. Luliconazole for the treatment of fungal infections: an evidence-based review. Core Evid. 2014;9:113-124.
- Korting HC, Schöllmann C. The significance of itraconazole for treatment of fungal infections of skin, nails and mucous membranes. J Dtsch Dermatol Ges. 2009;7:11-20.
- Goldstein AO, Goldstein BG. Dermatophyte (tinea) infections. UpToDate website. https://www.uptodate.com/contents/dermatophyte-tinea-infections. Updated December 28, 2018. Accessed April 24, 2019.
- Cole GW, Stricklin G. A comparison of a new oral antifungal, terbinafine, with griseofulvin as therapy for tinea corporis. Arch Dermatol. 1989;125:1537.
- Panagiotidou D, Kousidou T, Chaidemenos G, et al. A comparison of itraconazole and griseofulvin in the treatment of tinea corporis and tinea cruris: a double-blind study. J Int Med Res. 1992;20:392-400.
- Faergemann J, Mörk NJ, Haglund A, et al. A multicentre (double-blind) comparative study to assess the safety and efficacy of fluconazole and griseofulvin in the treatment of tinea corporis and tinea cruris. Br J Dermatol. 1997;136:575-577.
- Ilkit M, Durdu M, Karakas M. Cutaneous id reactions: a comprehensive review of clinical manifestations, epidemiology, etiology, and management. Crit Rev Microbiol. 2012;38:191-202.
To the Editor:
A 17-year-old adolescent girl presented to the dermatology clinic with a tender pruritic rash on the left wrist that was spreading to the bilateral arms and legs of several years’ duration. An area of a prior biopsy on the left wrist was healing well with use of petroleum jelly and halcinonide cream. The patient denied any constitutional symptoms.
Physical examination revealed numerous erythematous papules coalescing into plaques on the bilateral anterior and posterior arms and legs, including some erythematous macules and papules on the palms and soles. The original area of involvement on the left dorsal medial wrist demonstrated a background of erythema with overlying peripheral scaling and resolving violaceous to erythematous papules with signs of serosanguineous crusting (Figure 1). Scattered perifollicular erythema was present on the posterior aspects of the bilateral thighs and arms (Figure 2). Baseline complete blood cell count and complete metabolic panel were within reference range.
Clinical histopathology showed evidence of a pustular superficial dermatophyte infection, and Grocott-Gomori methenamine-silver stain demonstrated numerous fungal hyphae within subcorneal pustules, indicating pustular tinea. Based on the clinicopathologic correlation, the initial presentation was diagnosed as pustular tinea of the entire left wrist, followed by a generalized id reaction 1 week later.
The patient was prescribed oral terbinafine 250 mg once daily to treat the diffuse involvement of the pustular tinea as well as once-daily oral cetirizine, once-daily oral diphenhydramine, a topical emollient, and a topical nonsteroidal antipruritic gel.
Tinea is a superficial fungal infection commonly caused by the dermatophytes Epidermophyton, Trichophyton, and Microsporum. It has a variety of clinical presentations based on the anatomic location, including tinea capitis (hair/scalp), tinea pedis (feet), tinea corporis (face/trunk/extremities), tinea cruris (groin), and tinea unguium (nails).1 Tinea infections occur in the stratum corneum, hair, and nails, thriving on dead keratin in these areas.2 Tinea corporis usually appears as an erythematous ring-shaped lesion with a scaly border, but atypical cases presenting with vesicles, pustules, and bullae also have been reported.3 Additionally, secondary eruptions called id reactions, or autoeczematization, can present in the setting of dermatophyte infections. Such outbreaks may be due to a delayed hypersensitivity reaction to the fungal antigens. Id reactions can manifest in many forms of tinea with patients generally exhibiting pruritic papulovesicular lesions that can present far from the site of origin.4
Patients with id reactions can have atypical and varied presentations. In a case of id reaction due to tinea corporis, a patient presented with vesicles and pustules that grew in number and coalesced to form annular lesions.5 A case of an id reaction caused by tinea pedis also noted the presence of pustules, which are atypical in this form of tinea.6 In another case of tinea pedis, a generalized id reaction was noted, illustrating that such eruptions do not necessarily appear at the original site of infection.7 Additionally, in a rare presentation of tinea invading the nares, a patient developed an erythema multiforme id reaction.8 Id reactions also were noted in 14 patients with refractory otitis externa, illustrating the ability of this fungal infection to persist and infect distant locations.9
Because the differential diagnoses for tinea infection are extensive, pathology or laboratory confirmation is necessary for diagnosis, and potassium hydroxide preparation often is used to diagnose dermatophyte infections.1,2 Additionally, the possibility of a hypersensitivity drug rash should remain in the differential if the patient received allergy-inducing medications prior to the outbreak, which may in turn complicate the diagnosis.
Tinea infections typically can be treated with topical antifungals such as terbinafine, butenafine,1 and luliconazole10; however, more involved cases may require oral antifungal treatment.1 Systemic treatment of tinea corporis includes itraconazole, terbinafine, and fluconazole,11 all of which exhibit fewer side effects and greater efficacy when compared to griseofulvin.12-15
Treatment of id reactions centers on the proper clearance of the dermatophyte infection, and treatment with oral antifungals generally is sufficient. In the cases of id reaction in patients with refractory otitis, some success was achieved with treatment involving immunotherapy with dermatophyte and dust mite allergen extracts coupled with a yeast elimination diet.9 In acute id reactions, topical corticosteroids and antipruritic agents can be applied.4 Rarely, systemic glucocorticoids are required, such as in cases in which the id reaction persists despite proper treatment of the primary infection.16
To the Editor:
A 17-year-old adolescent girl presented to the dermatology clinic with a tender pruritic rash on the left wrist that was spreading to the bilateral arms and legs of several years’ duration. An area of a prior biopsy on the left wrist was healing well with use of petroleum jelly and halcinonide cream. The patient denied any constitutional symptoms.
Physical examination revealed numerous erythematous papules coalescing into plaques on the bilateral anterior and posterior arms and legs, including some erythematous macules and papules on the palms and soles. The original area of involvement on the left dorsal medial wrist demonstrated a background of erythema with overlying peripheral scaling and resolving violaceous to erythematous papules with signs of serosanguineous crusting (Figure 1). Scattered perifollicular erythema was present on the posterior aspects of the bilateral thighs and arms (Figure 2). Baseline complete blood cell count and complete metabolic panel were within reference range.
Clinical histopathology showed evidence of a pustular superficial dermatophyte infection, and Grocott-Gomori methenamine-silver stain demonstrated numerous fungal hyphae within subcorneal pustules, indicating pustular tinea. Based on the clinicopathologic correlation, the initial presentation was diagnosed as pustular tinea of the entire left wrist, followed by a generalized id reaction 1 week later.
The patient was prescribed oral terbinafine 250 mg once daily to treat the diffuse involvement of the pustular tinea as well as once-daily oral cetirizine, once-daily oral diphenhydramine, a topical emollient, and a topical nonsteroidal antipruritic gel.
Tinea is a superficial fungal infection commonly caused by the dermatophytes Epidermophyton, Trichophyton, and Microsporum. It has a variety of clinical presentations based on the anatomic location, including tinea capitis (hair/scalp), tinea pedis (feet), tinea corporis (face/trunk/extremities), tinea cruris (groin), and tinea unguium (nails).1 Tinea infections occur in the stratum corneum, hair, and nails, thriving on dead keratin in these areas.2 Tinea corporis usually appears as an erythematous ring-shaped lesion with a scaly border, but atypical cases presenting with vesicles, pustules, and bullae also have been reported.3 Additionally, secondary eruptions called id reactions, or autoeczematization, can present in the setting of dermatophyte infections. Such outbreaks may be due to a delayed hypersensitivity reaction to the fungal antigens. Id reactions can manifest in many forms of tinea with patients generally exhibiting pruritic papulovesicular lesions that can present far from the site of origin.4
Patients with id reactions can have atypical and varied presentations. In a case of id reaction due to tinea corporis, a patient presented with vesicles and pustules that grew in number and coalesced to form annular lesions.5 A case of an id reaction caused by tinea pedis also noted the presence of pustules, which are atypical in this form of tinea.6 In another case of tinea pedis, a generalized id reaction was noted, illustrating that such eruptions do not necessarily appear at the original site of infection.7 Additionally, in a rare presentation of tinea invading the nares, a patient developed an erythema multiforme id reaction.8 Id reactions also were noted in 14 patients with refractory otitis externa, illustrating the ability of this fungal infection to persist and infect distant locations.9
Because the differential diagnoses for tinea infection are extensive, pathology or laboratory confirmation is necessary for diagnosis, and potassium hydroxide preparation often is used to diagnose dermatophyte infections.1,2 Additionally, the possibility of a hypersensitivity drug rash should remain in the differential if the patient received allergy-inducing medications prior to the outbreak, which may in turn complicate the diagnosis.
Tinea infections typically can be treated with topical antifungals such as terbinafine, butenafine,1 and luliconazole10; however, more involved cases may require oral antifungal treatment.1 Systemic treatment of tinea corporis includes itraconazole, terbinafine, and fluconazole,11 all of which exhibit fewer side effects and greater efficacy when compared to griseofulvin.12-15
Treatment of id reactions centers on the proper clearance of the dermatophyte infection, and treatment with oral antifungals generally is sufficient. In the cases of id reaction in patients with refractory otitis, some success was achieved with treatment involving immunotherapy with dermatophyte and dust mite allergen extracts coupled with a yeast elimination diet.9 In acute id reactions, topical corticosteroids and antipruritic agents can be applied.4 Rarely, systemic glucocorticoids are required, such as in cases in which the id reaction persists despite proper treatment of the primary infection.16
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Hanover, NH: Elsevier, Inc; 2010.
- Ziemer M, Seyfarth F, Elsner P, et al. Atypical manifestations of tinea corporis. Mycoses. 2007;50(suppl 2):31-35.
- Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications [published online July 4, 2011]. Pediatrics. 2011;128:e453-e457.
- Ohno S, Tanabe H, Kawasaki M, et al. Tinea corporis with acute inflammation caused by Trichophyton tonsurans. J Dermatol. 2008;35:590-593.
- Hirschmann JV, Raugi GJ. Pustular tinea pedis. J Am Acad Dermatol. 2000;42:132-133.
- Iglesias ME, España A, Idoate MA, et al. Generalized skin reaction following tinea pedis (dermatophytids). J Dermatol. 1994;21:31-34.
- Atzori L, Pau M, Aste M. Erythema multiforme ID reaction in atypical dermatophytosis: a case report. J Eur Acad Dermatol Venereol. 2003;17:699-701.
- Derebery J, Berliner KI. Foot and ear disease—the dermatophytid reaction in otology. Laryngoscope. 1996;106(2 Pt 1):181-186.
- Khanna D, Bharti S. Luliconazole for the treatment of fungal infections: an evidence-based review. Core Evid. 2014;9:113-124.
- Korting HC, Schöllmann C. The significance of itraconazole for treatment of fungal infections of skin, nails and mucous membranes. J Dtsch Dermatol Ges. 2009;7:11-20.
- Goldstein AO, Goldstein BG. Dermatophyte (tinea) infections. UpToDate website. https://www.uptodate.com/contents/dermatophyte-tinea-infections. Updated December 28, 2018. Accessed April 24, 2019.
- Cole GW, Stricklin G. A comparison of a new oral antifungal, terbinafine, with griseofulvin as therapy for tinea corporis. Arch Dermatol. 1989;125:1537.
- Panagiotidou D, Kousidou T, Chaidemenos G, et al. A comparison of itraconazole and griseofulvin in the treatment of tinea corporis and tinea cruris: a double-blind study. J Int Med Res. 1992;20:392-400.
- Faergemann J, Mörk NJ, Haglund A, et al. A multicentre (double-blind) comparative study to assess the safety and efficacy of fluconazole and griseofulvin in the treatment of tinea corporis and tinea cruris. Br J Dermatol. 1997;136:575-577.
- Ilkit M, Durdu M, Karakas M. Cutaneous id reactions: a comprehensive review of clinical manifestations, epidemiology, etiology, and management. Crit Rev Microbiol. 2012;38:191-202.
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Hanover, NH: Elsevier, Inc; 2010.
- Ziemer M, Seyfarth F, Elsner P, et al. Atypical manifestations of tinea corporis. Mycoses. 2007;50(suppl 2):31-35.
- Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications [published online July 4, 2011]. Pediatrics. 2011;128:e453-e457.
- Ohno S, Tanabe H, Kawasaki M, et al. Tinea corporis with acute inflammation caused by Trichophyton tonsurans. J Dermatol. 2008;35:590-593.
- Hirschmann JV, Raugi GJ. Pustular tinea pedis. J Am Acad Dermatol. 2000;42:132-133.
- Iglesias ME, España A, Idoate MA, et al. Generalized skin reaction following tinea pedis (dermatophytids). J Dermatol. 1994;21:31-34.
- Atzori L, Pau M, Aste M. Erythema multiforme ID reaction in atypical dermatophytosis: a case report. J Eur Acad Dermatol Venereol. 2003;17:699-701.
- Derebery J, Berliner KI. Foot and ear disease—the dermatophytid reaction in otology. Laryngoscope. 1996;106(2 Pt 1):181-186.
- Khanna D, Bharti S. Luliconazole for the treatment of fungal infections: an evidence-based review. Core Evid. 2014;9:113-124.
- Korting HC, Schöllmann C. The significance of itraconazole for treatment of fungal infections of skin, nails and mucous membranes. J Dtsch Dermatol Ges. 2009;7:11-20.
- Goldstein AO, Goldstein BG. Dermatophyte (tinea) infections. UpToDate website. https://www.uptodate.com/contents/dermatophyte-tinea-infections. Updated December 28, 2018. Accessed April 24, 2019.
- Cole GW, Stricklin G. A comparison of a new oral antifungal, terbinafine, with griseofulvin as therapy for tinea corporis. Arch Dermatol. 1989;125:1537.
- Panagiotidou D, Kousidou T, Chaidemenos G, et al. A comparison of itraconazole and griseofulvin in the treatment of tinea corporis and tinea cruris: a double-blind study. J Int Med Res. 1992;20:392-400.
- Faergemann J, Mörk NJ, Haglund A, et al. A multicentre (double-blind) comparative study to assess the safety and efficacy of fluconazole and griseofulvin in the treatment of tinea corporis and tinea cruris. Br J Dermatol. 1997;136:575-577.
- Ilkit M, Durdu M, Karakas M. Cutaneous id reactions: a comprehensive review of clinical manifestations, epidemiology, etiology, and management. Crit Rev Microbiol. 2012;38:191-202.
Practice Points
• Id reactions, or autoeczematization, can occur secondary to dermatophyte infections, possibly due to a hypersensitivity reaction to the fungus. These eruptions can occur in many forms of tinea and in a variety of clinical presentations.
• Treatment is based on clearance of the original dermatophyte infection.