User login
Case Studies in Toxicology: The Perils of Playing Catch-up
Case
A 16-year-old girl, who recently emigrated from Haiti, was brought to the pediatric ED by her mother for evaluation of a 2-hour history of gastric discomfort. Upon arrival at the ED waiting area, the patient experienced a sudden onset of generalized tonic-clonic movement with altered sensorium, though she did not fall to the ground and was not injured. Vital signs from triage were: blood pressure, 110/76 mm Hg; heart rate, 112 beats/min; respiratory rate, 22 breaths/min; and temperature, 97°F. Oxygen saturation was 98% on room air.
The patient was immediately attached to a cardiac monitor, given oxygen via a face mask, and received airway suctioning. Despite receiving a total of 4 mg of lorazepam, the seizure continued. Physical examination revealed no signs of external injury, but the ongoing generalized status epilepticus made the examination difficult.
What are the causes of refractory seizures in an adolescent patient?
The differential diagnosis for pediatric patients presenting with refractory seizure is the same as that for adult patients and should include treatment noncompliance, infection, vascular event (eg, stroke, hemorrhage), trauma (eg, cerebral contusions), metabolic and electrolyte disturbances, anticonvulsant toxicity, and exposure to a convulsant toxin.
While certain drugs (eg, cocaine) may cause status epilepticus through a secondary effect such as ischemia or a bleed, some drugs can directly cause refractory seizures. A few drugs and toxins are responsible for the majority of such seizures: bupropion; carbon monoxide; diphenhydramine; ethanol (withdrawal); hypoglycemics; lead; theophylline; tramadol; and certain antibiotics, including cephalosporins, penicillins, quinolones, and, in particular, isoniazid (INH).1
Case Continuation
Upon further history-taking, the patient’s mother informed the ED staff that during a recent visit to a local clinic, her daughter tested positive on routine screening for tuberculosis and was given “some medications.” The patient’s mother further noted that her daughter was scheduled for a follow-up appointment at the same clinic later this morning. She believed the patient had taken “a few” of the prescribed pills at once to “catch-up” on missed doses prior to that appointment, and provided the ED staff with an empty bottle of INH that she had found in her daughter’s purse.
What are the signs and symptoms of acute isoniazid toxicity?
Isoniazid toxicity should be suspected in any patient who has access to INH—even if the drug was prescribed for someone other than the patient. Acute toxicity develops rapidly after the ingestion of supratherapeutic doses of INH and includes nausea, abdominal discomfort, vomiting, dizziness, and excessive fatigue or lethargy. Patients can present with tachycardia, stupor, agitation, mydriasis, increased anion gap metabolic acidosis, and encephalopathy.
Seizures occur due to an INH-induced functional pyridoxine deficiency. Isoniazid inhibits pyridoxine phosphokinase, the enzyme that converts pyridoxine (vitamin B6) to its physiologically active form, pyridoxal 5’-phosphate (PLP). Because the conversion of glutamate (an excitatory neurotransmitter) to gamma-aminobutyric acid (GABA; the body’s main inhibitory neurotransmitter) is dependent on PLP, an excess of glutamate and a deficiency of GABA occurs following INH overdose. The result is neuroexcitation, which manifests as generalized seizures in affected patients.
The most consequential effect of INH overdose, however, is the development of seizure refractory to conventional therapy, such as benzodiazepines. This occurs because benzodiazepines are indirect-acting GABA agonists, and require the presence of GABA to elicit their effect. Therefore, due to the impairment of GABA synthesis, benzodiazepines are limited or ineffective as anticonvulsants. Although INH doses in excess of 20 mg/kg may result in neuroexcitation, refractory seizures are uncommon with doses <70 mg/kg.
Complications of chronic INH use include hepatotoxicity, and patients will present with jaundice, hepatomegaly, and right upper quadrant pain and tenderness. Isoniazid must be discontinued rapidly in
How is acute isoniazid-induced seizure managed?
Management of patients with refractory seizure should initially include an assessment and management of the patient’s airway, breathing, and circulation. Although seizures induced by INH toxicity are often resistant to benzodiazepines, these agents remain the first-line therapy. For patients who fail to respond to a reasonable trial of benzodiazepines (eg, lorazepam 6 mg intravenously [IV]), pyridoxine should be administered.3 The recommended dose is 1 g pyridoxine per every 1 g of INH ingested—if the initial dose ingested is known—with a maximum dose of 5 g pyridoxine. If the initial dose of INH is not known, 70 mg/kg of pyridoxine, up to 5 g, is recommended. Repeated doses of pyridoxine can be administered if the seizure continues, up to a total dose of 10 g in an adult. At extremely high doses, pyridoxine itself can be neurotoxic, limiting the maximal antidotal dose.
Rapid initiation of pyridoxine is a challenge since typical stocks in most EDs are not in an adequate supply required for treatment. Additionally, a typical vial of pyridoxine contains 100 mg, highlighting the rare need to open dozens of vials for a single patient. Drawing up adequate doses of the IV formulation can be a challenge and time-consuming.
Regardless, the most reliable and rapid route of administration for pyridoxine is IV, at a rate of 0.5 to 1 g/min. Even if the seizure resolves prior to completion of the initial dose, the remaining doses should still be administered over a 4- to 6-hour period. Oral or (more likely) nasogastric administration of pyridoxine can be administered if the IV formulation is not available, but neither are optimal routes of delivery. Every effort should be made to stock pyridoxine in the antidote supply in the ED to avoid time delays involving finding, preparing, and administering the drug in these scenarios. Previous studies have found that most EDs are not prepared to handle pyridoxine replacement.4,5
Since benzodiazepines and barbiturates are GABA agonists with complementary mechanisms of actions to pyridoxine, they should be administered to potentiate the antiseizure effect of pyridoxine. If the seizure does not terminate, the use of propofol or general anesthesia may be required. Once the seizure is terminated, oral activated charcoal can be administered if the ingestion occurred within several hours of presentation. Given the rapid onset of effect of a large dose of INH, most patients will develop seizure shortly after exposure, limiting the benefits of both aggressive gastrointestinal decontamination and delayed activated charcoal. Charcoal also can be used for patients who overdose on INH but do not develop seizures.
Although the utility of a head computed tomography (CT) scan or laboratory studies is limited given the context of the exposure, these are generally obtained for patients with new-onset seizure. Since many patients with INH toxicity do not seize, such a patient may have a lower seizure threshold due to the existence of a subclinical cerebral lesion or metabolic abnormality.
Case Conclusion
The patient’s INH-induced refractory seizure was treated with pyridoxine. Her history suggested that she had ingested an unknown number of INH tablets within an hour. On this initial basis, an IV dose of 5,000 mg of pyridoxine was administered. The patient’s seizures terminated within 2 minutes of the infusion, and no additional doses of pyridoxine were required. Given the lack of concern for self-harm, an acetaminophen concentration was not obtained. A urine toxicology screen was negative for cocaine and amphetamines, and a CT scan of the head was negative for any abnormality. The patient was admitted to the pediatric intensive care unit for status epileptics and was discharged home on hospital day 2 after an uneventful stay.
1. Cock HR. Drug-induced status epilepticus. Epilepsy Behav. 2015;49:76-82. doi:10.1016/j.yebeh.2015.04.034.
2. Latent tuberculosis infection: a guide for primary health care providers. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/tb/publications/LTBI/treatment.htm. Updated August 5, 2016. Accessed December 13, 2016.
3. Howland MA. Antidotes in depth: pyridoxine. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:797-799.
4. Shah BR, Santucci K, Sinert R, Steiner P. Acute isoniazid neurotoxicity in an urban hospital. Pediatrics. 1995;95(5):700-704.
5. Santucci KA, Shah BR, Linakis JG. Acute isoniazid exposures and antidote availability. Pediatr Emerg Care. 1999;15(2):99-101.
Case
A 16-year-old girl, who recently emigrated from Haiti, was brought to the pediatric ED by her mother for evaluation of a 2-hour history of gastric discomfort. Upon arrival at the ED waiting area, the patient experienced a sudden onset of generalized tonic-clonic movement with altered sensorium, though she did not fall to the ground and was not injured. Vital signs from triage were: blood pressure, 110/76 mm Hg; heart rate, 112 beats/min; respiratory rate, 22 breaths/min; and temperature, 97°F. Oxygen saturation was 98% on room air.
The patient was immediately attached to a cardiac monitor, given oxygen via a face mask, and received airway suctioning. Despite receiving a total of 4 mg of lorazepam, the seizure continued. Physical examination revealed no signs of external injury, but the ongoing generalized status epilepticus made the examination difficult.
What are the causes of refractory seizures in an adolescent patient?
The differential diagnosis for pediatric patients presenting with refractory seizure is the same as that for adult patients and should include treatment noncompliance, infection, vascular event (eg, stroke, hemorrhage), trauma (eg, cerebral contusions), metabolic and electrolyte disturbances, anticonvulsant toxicity, and exposure to a convulsant toxin.
While certain drugs (eg, cocaine) may cause status epilepticus through a secondary effect such as ischemia or a bleed, some drugs can directly cause refractory seizures. A few drugs and toxins are responsible for the majority of such seizures: bupropion; carbon monoxide; diphenhydramine; ethanol (withdrawal); hypoglycemics; lead; theophylline; tramadol; and certain antibiotics, including cephalosporins, penicillins, quinolones, and, in particular, isoniazid (INH).1
Case Continuation
Upon further history-taking, the patient’s mother informed the ED staff that during a recent visit to a local clinic, her daughter tested positive on routine screening for tuberculosis and was given “some medications.” The patient’s mother further noted that her daughter was scheduled for a follow-up appointment at the same clinic later this morning. She believed the patient had taken “a few” of the prescribed pills at once to “catch-up” on missed doses prior to that appointment, and provided the ED staff with an empty bottle of INH that she had found in her daughter’s purse.
What are the signs and symptoms of acute isoniazid toxicity?
Isoniazid toxicity should be suspected in any patient who has access to INH—even if the drug was prescribed for someone other than the patient. Acute toxicity develops rapidly after the ingestion of supratherapeutic doses of INH and includes nausea, abdominal discomfort, vomiting, dizziness, and excessive fatigue or lethargy. Patients can present with tachycardia, stupor, agitation, mydriasis, increased anion gap metabolic acidosis, and encephalopathy.
Seizures occur due to an INH-induced functional pyridoxine deficiency. Isoniazid inhibits pyridoxine phosphokinase, the enzyme that converts pyridoxine (vitamin B6) to its physiologically active form, pyridoxal 5’-phosphate (PLP). Because the conversion of glutamate (an excitatory neurotransmitter) to gamma-aminobutyric acid (GABA; the body’s main inhibitory neurotransmitter) is dependent on PLP, an excess of glutamate and a deficiency of GABA occurs following INH overdose. The result is neuroexcitation, which manifests as generalized seizures in affected patients.
The most consequential effect of INH overdose, however, is the development of seizure refractory to conventional therapy, such as benzodiazepines. This occurs because benzodiazepines are indirect-acting GABA agonists, and require the presence of GABA to elicit their effect. Therefore, due to the impairment of GABA synthesis, benzodiazepines are limited or ineffective as anticonvulsants. Although INH doses in excess of 20 mg/kg may result in neuroexcitation, refractory seizures are uncommon with doses <70 mg/kg.
Complications of chronic INH use include hepatotoxicity, and patients will present with jaundice, hepatomegaly, and right upper quadrant pain and tenderness. Isoniazid must be discontinued rapidly in
How is acute isoniazid-induced seizure managed?
Management of patients with refractory seizure should initially include an assessment and management of the patient’s airway, breathing, and circulation. Although seizures induced by INH toxicity are often resistant to benzodiazepines, these agents remain the first-line therapy. For patients who fail to respond to a reasonable trial of benzodiazepines (eg, lorazepam 6 mg intravenously [IV]), pyridoxine should be administered.3 The recommended dose is 1 g pyridoxine per every 1 g of INH ingested—if the initial dose ingested is known—with a maximum dose of 5 g pyridoxine. If the initial dose of INH is not known, 70 mg/kg of pyridoxine, up to 5 g, is recommended. Repeated doses of pyridoxine can be administered if the seizure continues, up to a total dose of 10 g in an adult. At extremely high doses, pyridoxine itself can be neurotoxic, limiting the maximal antidotal dose.
Rapid initiation of pyridoxine is a challenge since typical stocks in most EDs are not in an adequate supply required for treatment. Additionally, a typical vial of pyridoxine contains 100 mg, highlighting the rare need to open dozens of vials for a single patient. Drawing up adequate doses of the IV formulation can be a challenge and time-consuming.
Regardless, the most reliable and rapid route of administration for pyridoxine is IV, at a rate of 0.5 to 1 g/min. Even if the seizure resolves prior to completion of the initial dose, the remaining doses should still be administered over a 4- to 6-hour period. Oral or (more likely) nasogastric administration of pyridoxine can be administered if the IV formulation is not available, but neither are optimal routes of delivery. Every effort should be made to stock pyridoxine in the antidote supply in the ED to avoid time delays involving finding, preparing, and administering the drug in these scenarios. Previous studies have found that most EDs are not prepared to handle pyridoxine replacement.4,5
Since benzodiazepines and barbiturates are GABA agonists with complementary mechanisms of actions to pyridoxine, they should be administered to potentiate the antiseizure effect of pyridoxine. If the seizure does not terminate, the use of propofol or general anesthesia may be required. Once the seizure is terminated, oral activated charcoal can be administered if the ingestion occurred within several hours of presentation. Given the rapid onset of effect of a large dose of INH, most patients will develop seizure shortly after exposure, limiting the benefits of both aggressive gastrointestinal decontamination and delayed activated charcoal. Charcoal also can be used for patients who overdose on INH but do not develop seizures.
Although the utility of a head computed tomography (CT) scan or laboratory studies is limited given the context of the exposure, these are generally obtained for patients with new-onset seizure. Since many patients with INH toxicity do not seize, such a patient may have a lower seizure threshold due to the existence of a subclinical cerebral lesion or metabolic abnormality.
Case Conclusion
The patient’s INH-induced refractory seizure was treated with pyridoxine. Her history suggested that she had ingested an unknown number of INH tablets within an hour. On this initial basis, an IV dose of 5,000 mg of pyridoxine was administered. The patient’s seizures terminated within 2 minutes of the infusion, and no additional doses of pyridoxine were required. Given the lack of concern for self-harm, an acetaminophen concentration was not obtained. A urine toxicology screen was negative for cocaine and amphetamines, and a CT scan of the head was negative for any abnormality. The patient was admitted to the pediatric intensive care unit for status epileptics and was discharged home on hospital day 2 after an uneventful stay.
Case
A 16-year-old girl, who recently emigrated from Haiti, was brought to the pediatric ED by her mother for evaluation of a 2-hour history of gastric discomfort. Upon arrival at the ED waiting area, the patient experienced a sudden onset of generalized tonic-clonic movement with altered sensorium, though she did not fall to the ground and was not injured. Vital signs from triage were: blood pressure, 110/76 mm Hg; heart rate, 112 beats/min; respiratory rate, 22 breaths/min; and temperature, 97°F. Oxygen saturation was 98% on room air.
The patient was immediately attached to a cardiac monitor, given oxygen via a face mask, and received airway suctioning. Despite receiving a total of 4 mg of lorazepam, the seizure continued. Physical examination revealed no signs of external injury, but the ongoing generalized status epilepticus made the examination difficult.
What are the causes of refractory seizures in an adolescent patient?
The differential diagnosis for pediatric patients presenting with refractory seizure is the same as that for adult patients and should include treatment noncompliance, infection, vascular event (eg, stroke, hemorrhage), trauma (eg, cerebral contusions), metabolic and electrolyte disturbances, anticonvulsant toxicity, and exposure to a convulsant toxin.
While certain drugs (eg, cocaine) may cause status epilepticus through a secondary effect such as ischemia or a bleed, some drugs can directly cause refractory seizures. A few drugs and toxins are responsible for the majority of such seizures: bupropion; carbon monoxide; diphenhydramine; ethanol (withdrawal); hypoglycemics; lead; theophylline; tramadol; and certain antibiotics, including cephalosporins, penicillins, quinolones, and, in particular, isoniazid (INH).1
Case Continuation
Upon further history-taking, the patient’s mother informed the ED staff that during a recent visit to a local clinic, her daughter tested positive on routine screening for tuberculosis and was given “some medications.” The patient’s mother further noted that her daughter was scheduled for a follow-up appointment at the same clinic later this morning. She believed the patient had taken “a few” of the prescribed pills at once to “catch-up” on missed doses prior to that appointment, and provided the ED staff with an empty bottle of INH that she had found in her daughter’s purse.
What are the signs and symptoms of acute isoniazid toxicity?
Isoniazid toxicity should be suspected in any patient who has access to INH—even if the drug was prescribed for someone other than the patient. Acute toxicity develops rapidly after the ingestion of supratherapeutic doses of INH and includes nausea, abdominal discomfort, vomiting, dizziness, and excessive fatigue or lethargy. Patients can present with tachycardia, stupor, agitation, mydriasis, increased anion gap metabolic acidosis, and encephalopathy.
Seizures occur due to an INH-induced functional pyridoxine deficiency. Isoniazid inhibits pyridoxine phosphokinase, the enzyme that converts pyridoxine (vitamin B6) to its physiologically active form, pyridoxal 5’-phosphate (PLP). Because the conversion of glutamate (an excitatory neurotransmitter) to gamma-aminobutyric acid (GABA; the body’s main inhibitory neurotransmitter) is dependent on PLP, an excess of glutamate and a deficiency of GABA occurs following INH overdose. The result is neuroexcitation, which manifests as generalized seizures in affected patients.
The most consequential effect of INH overdose, however, is the development of seizure refractory to conventional therapy, such as benzodiazepines. This occurs because benzodiazepines are indirect-acting GABA agonists, and require the presence of GABA to elicit their effect. Therefore, due to the impairment of GABA synthesis, benzodiazepines are limited or ineffective as anticonvulsants. Although INH doses in excess of 20 mg/kg may result in neuroexcitation, refractory seizures are uncommon with doses <70 mg/kg.
Complications of chronic INH use include hepatotoxicity, and patients will present with jaundice, hepatomegaly, and right upper quadrant pain and tenderness. Isoniazid must be discontinued rapidly in
How is acute isoniazid-induced seizure managed?
Management of patients with refractory seizure should initially include an assessment and management of the patient’s airway, breathing, and circulation. Although seizures induced by INH toxicity are often resistant to benzodiazepines, these agents remain the first-line therapy. For patients who fail to respond to a reasonable trial of benzodiazepines (eg, lorazepam 6 mg intravenously [IV]), pyridoxine should be administered.3 The recommended dose is 1 g pyridoxine per every 1 g of INH ingested—if the initial dose ingested is known—with a maximum dose of 5 g pyridoxine. If the initial dose of INH is not known, 70 mg/kg of pyridoxine, up to 5 g, is recommended. Repeated doses of pyridoxine can be administered if the seizure continues, up to a total dose of 10 g in an adult. At extremely high doses, pyridoxine itself can be neurotoxic, limiting the maximal antidotal dose.
Rapid initiation of pyridoxine is a challenge since typical stocks in most EDs are not in an adequate supply required for treatment. Additionally, a typical vial of pyridoxine contains 100 mg, highlighting the rare need to open dozens of vials for a single patient. Drawing up adequate doses of the IV formulation can be a challenge and time-consuming.
Regardless, the most reliable and rapid route of administration for pyridoxine is IV, at a rate of 0.5 to 1 g/min. Even if the seizure resolves prior to completion of the initial dose, the remaining doses should still be administered over a 4- to 6-hour period. Oral or (more likely) nasogastric administration of pyridoxine can be administered if the IV formulation is not available, but neither are optimal routes of delivery. Every effort should be made to stock pyridoxine in the antidote supply in the ED to avoid time delays involving finding, preparing, and administering the drug in these scenarios. Previous studies have found that most EDs are not prepared to handle pyridoxine replacement.4,5
Since benzodiazepines and barbiturates are GABA agonists with complementary mechanisms of actions to pyridoxine, they should be administered to potentiate the antiseizure effect of pyridoxine. If the seizure does not terminate, the use of propofol or general anesthesia may be required. Once the seizure is terminated, oral activated charcoal can be administered if the ingestion occurred within several hours of presentation. Given the rapid onset of effect of a large dose of INH, most patients will develop seizure shortly after exposure, limiting the benefits of both aggressive gastrointestinal decontamination and delayed activated charcoal. Charcoal also can be used for patients who overdose on INH but do not develop seizures.
Although the utility of a head computed tomography (CT) scan or laboratory studies is limited given the context of the exposure, these are generally obtained for patients with new-onset seizure. Since many patients with INH toxicity do not seize, such a patient may have a lower seizure threshold due to the existence of a subclinical cerebral lesion or metabolic abnormality.
Case Conclusion
The patient’s INH-induced refractory seizure was treated with pyridoxine. Her history suggested that she had ingested an unknown number of INH tablets within an hour. On this initial basis, an IV dose of 5,000 mg of pyridoxine was administered. The patient’s seizures terminated within 2 minutes of the infusion, and no additional doses of pyridoxine were required. Given the lack of concern for self-harm, an acetaminophen concentration was not obtained. A urine toxicology screen was negative for cocaine and amphetamines, and a CT scan of the head was negative for any abnormality. The patient was admitted to the pediatric intensive care unit for status epileptics and was discharged home on hospital day 2 after an uneventful stay.
1. Cock HR. Drug-induced status epilepticus. Epilepsy Behav. 2015;49:76-82. doi:10.1016/j.yebeh.2015.04.034.
2. Latent tuberculosis infection: a guide for primary health care providers. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/tb/publications/LTBI/treatment.htm. Updated August 5, 2016. Accessed December 13, 2016.
3. Howland MA. Antidotes in depth: pyridoxine. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:797-799.
4. Shah BR, Santucci K, Sinert R, Steiner P. Acute isoniazid neurotoxicity in an urban hospital. Pediatrics. 1995;95(5):700-704.
5. Santucci KA, Shah BR, Linakis JG. Acute isoniazid exposures and antidote availability. Pediatr Emerg Care. 1999;15(2):99-101.
1. Cock HR. Drug-induced status epilepticus. Epilepsy Behav. 2015;49:76-82. doi:10.1016/j.yebeh.2015.04.034.
2. Latent tuberculosis infection: a guide for primary health care providers. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/tb/publications/LTBI/treatment.htm. Updated August 5, 2016. Accessed December 13, 2016.
3. Howland MA. Antidotes in depth: pyridoxine. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:797-799.
4. Shah BR, Santucci K, Sinert R, Steiner P. Acute isoniazid neurotoxicity in an urban hospital. Pediatrics. 1995;95(5):700-704.
5. Santucci KA, Shah BR, Linakis JG. Acute isoniazid exposures and antidote availability. Pediatr Emerg Care. 1999;15(2):99-101.

Dissection of the Celiac Artery
Case
A 41-year-old man presented to our ED with a 4-day history of epigastric pain radiating to the bilateral flanks and back. His medical history was significant for hypertension, for which he was prescribed isosorbide dinitrite 30 mg four times per day; however, he reported that he did not regularly take this medication.
The patient had visited our ED 3 days earlier with the same complaint. Since his blood pressure (BP) reading at the first ED presentation was 213/141 mm Hg, he had been admitted for hypertensive urgency. The patient’s BP was controlled with antihypertensive agents during his stay, but he continued to experience epigastric pain. A basic work-up for abdominal pain was ordered, the results of which were normal. Based on these findings, the patient’s pain was attributed to gastritis, and he was discharged home with instructions to return to the ED if his pain became worse or persisted.
At both ED presentations, the patient denied experiencing any nausea, vomiting, diarrhea, or chest pain. At the second presentation, his triage BP was 158/106 mm Hg. A chest X-ray, complete blood count (CBC), basic metabolic profile (BMP), hepatic panel, and lipase evaluation were all unremarkable, with the exception of a mild increase in creatinine to 1.38 mg/dL. A point-of-care (POC) ultrasound study of the aorta was normal. 
Based on the CTA findings, a nicardipine infusion was immediately started, and the patient was admitted to the medical intensive care unit (MICU). Because his heart rate was in the range of 60 beats/min, an esmolol infusion was not required. Prior to transferring the patient to MICU, a second ultrasound study of the aorta was performed by our fellowship-trained director of emergency medicine ultrasound. 
In the MICU, the patient’s BP was stabilized on hospital day 2, and he was transitioned to oral antihypertensive medications. He was also started on a heparin infusion at the recommendation of vascular surgery services.
A repeat CTA of the abdomen taken on hospital day 3 showed an unchanged dissection in the celiac axis extending into the hepatic artery. The vascular surgeon recommended strict BP control, anticoagulation therapy, and a vascular surgery follow-up with a repeat CTA of the abdomen in 6 months.
On hospital day 6, repeat serial CBC, BMP, and hepatic panels revealed only slight increases in aspartate transaminase to 88 U/L and alanine aminotransferase to 117 U/L. The patient was transitioned to enoxaparin and discharged home on hospital day 6, and instructed to follow-up with his primary care physician for transition to warfarin. Unfortunately, this patient was lost to follow-up.
Discussion
Isolated DCA is a rare cause of abdominal pain. The first documented case of isolated DCA is often incorrectly attributed to Bauersfeld’s1 1947 case series on dissections,but that report described superior mesenteric artery dissection rather than a celiac artery dissection. Watson’s2 1956 dissection series is also incorrectly cited as the first DCA, but that series described a dissection of the splenic artery, which is a branch of the celiac artery. In a 1959 series, Foord and Lewis3 described what is most likely the first report of DCA as an incidental finding at autopsy. More frequent descriptions in recent years are thought to be due to the routine use of abdominal CTA.4
Dissection of the celiac artery is a rare occurrence, with less than 100 cases reported, and little evidence exists to guide its management.5 These dissections represent 36.8% of all visceral artery dissections,6 which themselves are less common than renal, carotid, and vertebral artery dissections.7 Dissection of visceral arteries occurs predominantly in men and more often in middle-aged patients.8 Risk factors for DCA are thought to mirror risk factors for dissection of other arteries, including atherosclerotic disease, hypertension, connective tissue disorders, trauma, vasculitis, and pregnancy.9-11
Signs and Symptoms
Patients with DCA typically present with sudden onset of epigastric, flank, and/or chest pain, though 50% of patients may be asymptomatic.12 This pain is easily overlooked because the physical examination and laboratory studies are typically unremarkable.13 Fortunately, DCA is rarely accompanied by fatal organ dysfunction due to collateral flow from other vessels.14
Diagnosis and Management
While CTA with contrast is considered the mainstay of diagnosis of DCA,15 optimal treatment for DCA has not been well established. Management options include medical management, operative repair, and endovascular embolization. Medical management is reserved for stable patients without signs of end organ dysfunction. Typical management involves anticoagulation with warfarin for 3 to 6 months and strict BP control accompanied by close surveillance for progression.10,13 Some clinicians have argued that anticoagulation therapy may be unnecessary and that risk factor modification and BP control alone may be sufficient.5,6 Others have advocated that surgical management should be favored in cases of persistent pain, development of aneurysm, or threatened or compromised flow to end organs.7
Point-of-Care Ultrasound
The American College of Emergency Physicians considers ultrasound of the abdominal aorta a core application of emergency ultrasound.16 While sensitivity and specificity of emergency ultrasound for abdominal aortic aneurysm are well established, data supporting its use for screening for dissections are less definitive. With a sensitivity of 67% to 80% and a specificity of 99% to 100% with visualization of an intimal flap, aortic dissection screening using ultrasound is less reliable than most emergency physicians (EPs) would prefer.17,18 There are no published data reporting the sensitivity or specificity of emergency ultrasound for DCA. However, the vascular surgery literature encourages color Doppler ultrasound as part of the initial diagnostic work-up for this rare entity.19 While this may seem like an area ripe for emergency ultrasound, it is important to note—as seen in our case—that the site of the dissection is not often seen. Instead, the use of Doppler allows a screening for an abnormal flow pattern suggestive of dissection.20
Conclusion
In our case, both resident EPs and an expert fellowship-trained emergency ultrasound attending physician were unable to visualize a dissection—even after knowledge of the lesion was established by CTA. This points out a limitation of emergency ultrasound. While a POC ultrasound may be able to effectively rule in dissections of the aorta and its branches, we cannot reliably rule out these lesions. As EPs continue to expand the use of ultrasound, it is important to balance the desire for efficiency and cost-effectiveness with a high index of suspicion, experience, and clinical acumen.
1. Bauersfeld SR. Dissecting aneurysm of the aorta; a presentation of 15 cases and a review of the recent literature. Ann Intern Med. 1947;26(6):873-889.
2. Watson AJ. Dissecting aneurysm of arteries other than the aorta. J Pathol. 1956;72(2):439-449. doi:10.1002/path.1700720209.
3. Foord AG, Lewis RD. Primary dissecting aneurysms of peripheral and pulmonary arteries: dissecting hemorrhage of media. Arch Pathol. 1959;68:553-577.
4. Neychev V, Krol E, Dietzek A. Unusual presentation and treatment of spontaneous celiac artery dissection. J Vasc Surg. 2013;58(2):491-495. doi:10.1016/j.jvs.2012.10.136.
5. DiMusto PD, Oberdoerster MM, Criado E. Isolated celiac artery dissection. J Vasc Surg. 2015;61(4):972-976. doi: 10.1016/j.jvs.2014.10.108.
6. Takayama T, Miyata T, Shirakawa M, Nagawa H. J Vasc Surg. 2008;48(2):329-333. doi:10.1016/j.jvs.2008.03.002.
7. Glehen O, Feugier P, Aleksic Y, Delannoy P, Chevalier JM. Spontaneous dissection of the celiac artery. Ann Vasc Surg. 2001;15(6):687-692.
8. Patel KS, Benshar O, Vrabie R, Patel A, Adler M, Hines G. A major pain in the … back and epigastrium: an unusual case of spontaneous celiac artery dissection. J Community Hosp Intern Med Perspect. 2014;4(5):23840. doi:10.3402/jchimp.v4.23840.
9. Kang TL, Teich DL, McGillicuddy DC. Isolated, spontaneous superior mesenteric and celiac artery dissection: case report and review of literature. J Emerg Med. 2011;40(2):e21-e25. doi:10.1016/j.jemermed.2007.12.038.
10. Galastri FL, Cavalcante RN, Motta-Leal-Filho JM, et al. Evaluation and management of symptomatic isolated spontaneous celiac trunk dissection. Vasc Med. 2015;20(4):358-363. doi:10.1177/1358863X15581447.
11. Wang HC, Chen JH, Hsiao CC, Jeng CM, Chen WL. Spontaneous dissection of the celiac artery: a case report and literature review. Am J Emerg Med. 2013;31(6):1000.e3-e5. doi:10.1016/j.ajem.2013.02.007.
12. Oh S, Cho YP, Kim JH, Shin S, Kwon TW, Ko GY. Symptomatic spontaneous celiac artery dissection treated by conservative management: serial imaging findings. Abdom Imaging. 2011;36(1):79-82. doi:10.1007/s00261-010-9657-x.
13. Wang JL, Hsieh MJ, Lee CH, Chen CC, Hsieh IC. Celiac artery dissection presenting with abdominal and chest pain. Am J Emerg Med. 2010;28(1):111.e3-e5. doi:10.1016/j.ajem.2009.02.023.
14. Takayama Y, Takao M, Inoue T, Yoshimi F, Koyama K, Nagai H. Isolated spontaneous dissection of the celiac artery: report of two cases. Ann Vasc Dis. 2014;7(1):64-67. doi:10.3400/avd.cr.13-00102.
15. Rehman AU, Almanfi A, Nadella S, Sohail U. Isolated spontaneous celiac artery dissection in a 47-year-old man with von Willebrand disease. Tex Heart Inst J. 2014;41(3):344-345. doi:10.14503/THIJ-13-3404.
16. American College of Emergency Physicians. Policy statement. Ultrasound Guidelines: Emergency, Point-of-Care, and Clinical Ultrasound Guidelines in Medicine, June 2016. https://www.acep.org/Clinical---Practice-Management/Ultrasound/. Accessed November 15, 2016.
17. Williams J, Heiner JD, Perreault MD, McArthur TJ. Aortic dissection diagnosed by ultrasound. West J Emerg Med. 2010;11(1):98-99.
18. Fojtik JP, Costantino TG, Dean AJ. The diagnosis of aortic dissection by emergency medicine ultrasound. J Emerg Med. 2007;32(2):191-196.
19. Woolard JD, Ammar AD. Spontaneous dissection of the celiac artery: a case report. J Vasc Surg. 2007;45(6):1256-1258.
20. Fenoglio L, Allione A, Scalabrino E, et al. Spontaneous dissection of the celiac artery: a pitfall in the diagnosis of acute abdominal pain. Presentation of two cases. Dig Dis Sci. 2004;49(7-8):1223-1227.
Case
A 41-year-old man presented to our ED with a 4-day history of epigastric pain radiating to the bilateral flanks and back. His medical history was significant for hypertension, for which he was prescribed isosorbide dinitrite 30 mg four times per day; however, he reported that he did not regularly take this medication.
The patient had visited our ED 3 days earlier with the same complaint. Since his blood pressure (BP) reading at the first ED presentation was 213/141 mm Hg, he had been admitted for hypertensive urgency. The patient’s BP was controlled with antihypertensive agents during his stay, but he continued to experience epigastric pain. A basic work-up for abdominal pain was ordered, the results of which were normal. Based on these findings, the patient’s pain was attributed to gastritis, and he was discharged home with instructions to return to the ED if his pain became worse or persisted.
At both ED presentations, the patient denied experiencing any nausea, vomiting, diarrhea, or chest pain. At the second presentation, his triage BP was 158/106 mm Hg. A chest X-ray, complete blood count (CBC), basic metabolic profile (BMP), hepatic panel, and lipase evaluation were all unremarkable, with the exception of a mild increase in creatinine to 1.38 mg/dL. A point-of-care (POC) ultrasound study of the aorta was normal.
Based on the CTA findings, a nicardipine infusion was immediately started, and the patient was admitted to the medical intensive care unit (MICU). Because his heart rate was in the range of 60 beats/min, an esmolol infusion was not required. Prior to transferring the patient to MICU, a second ultrasound study of the aorta was performed by our fellowship-trained director of emergency medicine ultrasound.
In the MICU, the patient’s BP was stabilized on hospital day 2, and he was transitioned to oral antihypertensive medications. He was also started on a heparin infusion at the recommendation of vascular surgery services.
A repeat CTA of the abdomen taken on hospital day 3 showed an unchanged dissection in the celiac axis extending into the hepatic artery. The vascular surgeon recommended strict BP control, anticoagulation therapy, and a vascular surgery follow-up with a repeat CTA of the abdomen in 6 months.
On hospital day 6, repeat serial CBC, BMP, and hepatic panels revealed only slight increases in aspartate transaminase to 88 U/L and alanine aminotransferase to 117 U/L. The patient was transitioned to enoxaparin and discharged home on hospital day 6, and instructed to follow-up with his primary care physician for transition to warfarin. Unfortunately, this patient was lost to follow-up.
Discussion
Isolated DCA is a rare cause of abdominal pain. The first documented case of isolated DCA is often incorrectly attributed to Bauersfeld’s1 1947 case series on dissections,but that report described superior mesenteric artery dissection rather than a celiac artery dissection. Watson’s2 1956 dissection series is also incorrectly cited as the first DCA, but that series described a dissection of the splenic artery, which is a branch of the celiac artery. In a 1959 series, Foord and Lewis3 described what is most likely the first report of DCA as an incidental finding at autopsy. More frequent descriptions in recent years are thought to be due to the routine use of abdominal CTA.4
Dissection of the celiac artery is a rare occurrence, with less than 100 cases reported, and little evidence exists to guide its management.5 These dissections represent 36.8% of all visceral artery dissections,6 which themselves are less common than renal, carotid, and vertebral artery dissections.7 Dissection of visceral arteries occurs predominantly in men and more often in middle-aged patients.8 Risk factors for DCA are thought to mirror risk factors for dissection of other arteries, including atherosclerotic disease, hypertension, connective tissue disorders, trauma, vasculitis, and pregnancy.9-11
Signs and Symptoms
Patients with DCA typically present with sudden onset of epigastric, flank, and/or chest pain, though 50% of patients may be asymptomatic.12 This pain is easily overlooked because the physical examination and laboratory studies are typically unremarkable.13 Fortunately, DCA is rarely accompanied by fatal organ dysfunction due to collateral flow from other vessels.14
Diagnosis and Management
While CTA with contrast is considered the mainstay of diagnosis of DCA,15 optimal treatment for DCA has not been well established. Management options include medical management, operative repair, and endovascular embolization. Medical management is reserved for stable patients without signs of end organ dysfunction. Typical management involves anticoagulation with warfarin for 3 to 6 months and strict BP control accompanied by close surveillance for progression.10,13 Some clinicians have argued that anticoagulation therapy may be unnecessary and that risk factor modification and BP control alone may be sufficient.5,6 Others have advocated that surgical management should be favored in cases of persistent pain, development of aneurysm, or threatened or compromised flow to end organs.7
Point-of-Care Ultrasound
The American College of Emergency Physicians considers ultrasound of the abdominal aorta a core application of emergency ultrasound.16 While sensitivity and specificity of emergency ultrasound for abdominal aortic aneurysm are well established, data supporting its use for screening for dissections are less definitive. With a sensitivity of 67% to 80% and a specificity of 99% to 100% with visualization of an intimal flap, aortic dissection screening using ultrasound is less reliable than most emergency physicians (EPs) would prefer.17,18 There are no published data reporting the sensitivity or specificity of emergency ultrasound for DCA. However, the vascular surgery literature encourages color Doppler ultrasound as part of the initial diagnostic work-up for this rare entity.19 While this may seem like an area ripe for emergency ultrasound, it is important to note—as seen in our case—that the site of the dissection is not often seen. Instead, the use of Doppler allows a screening for an abnormal flow pattern suggestive of dissection.20
Conclusion
In our case, both resident EPs and an expert fellowship-trained emergency ultrasound attending physician were unable to visualize a dissection—even after knowledge of the lesion was established by CTA. This points out a limitation of emergency ultrasound. While a POC ultrasound may be able to effectively rule in dissections of the aorta and its branches, we cannot reliably rule out these lesions. As EPs continue to expand the use of ultrasound, it is important to balance the desire for efficiency and cost-effectiveness with a high index of suspicion, experience, and clinical acumen.
Case
A 41-year-old man presented to our ED with a 4-day history of epigastric pain radiating to the bilateral flanks and back. His medical history was significant for hypertension, for which he was prescribed isosorbide dinitrite 30 mg four times per day; however, he reported that he did not regularly take this medication.
The patient had visited our ED 3 days earlier with the same complaint. Since his blood pressure (BP) reading at the first ED presentation was 213/141 mm Hg, he had been admitted for hypertensive urgency. The patient’s BP was controlled with antihypertensive agents during his stay, but he continued to experience epigastric pain. A basic work-up for abdominal pain was ordered, the results of which were normal. Based on these findings, the patient’s pain was attributed to gastritis, and he was discharged home with instructions to return to the ED if his pain became worse or persisted.
At both ED presentations, the patient denied experiencing any nausea, vomiting, diarrhea, or chest pain. At the second presentation, his triage BP was 158/106 mm Hg. A chest X-ray, complete blood count (CBC), basic metabolic profile (BMP), hepatic panel, and lipase evaluation were all unremarkable, with the exception of a mild increase in creatinine to 1.38 mg/dL. A point-of-care (POC) ultrasound study of the aorta was normal.
Based on the CTA findings, a nicardipine infusion was immediately started, and the patient was admitted to the medical intensive care unit (MICU). Because his heart rate was in the range of 60 beats/min, an esmolol infusion was not required. Prior to transferring the patient to MICU, a second ultrasound study of the aorta was performed by our fellowship-trained director of emergency medicine ultrasound.
In the MICU, the patient’s BP was stabilized on hospital day 2, and he was transitioned to oral antihypertensive medications. He was also started on a heparin infusion at the recommendation of vascular surgery services.
A repeat CTA of the abdomen taken on hospital day 3 showed an unchanged dissection in the celiac axis extending into the hepatic artery. The vascular surgeon recommended strict BP control, anticoagulation therapy, and a vascular surgery follow-up with a repeat CTA of the abdomen in 6 months.
On hospital day 6, repeat serial CBC, BMP, and hepatic panels revealed only slight increases in aspartate transaminase to 88 U/L and alanine aminotransferase to 117 U/L. The patient was transitioned to enoxaparin and discharged home on hospital day 6, and instructed to follow-up with his primary care physician for transition to warfarin. Unfortunately, this patient was lost to follow-up.
Discussion
Isolated DCA is a rare cause of abdominal pain. The first documented case of isolated DCA is often incorrectly attributed to Bauersfeld’s1 1947 case series on dissections,but that report described superior mesenteric artery dissection rather than a celiac artery dissection. Watson’s2 1956 dissection series is also incorrectly cited as the first DCA, but that series described a dissection of the splenic artery, which is a branch of the celiac artery. In a 1959 series, Foord and Lewis3 described what is most likely the first report of DCA as an incidental finding at autopsy. More frequent descriptions in recent years are thought to be due to the routine use of abdominal CTA.4
Dissection of the celiac artery is a rare occurrence, with less than 100 cases reported, and little evidence exists to guide its management.5 These dissections represent 36.8% of all visceral artery dissections,6 which themselves are less common than renal, carotid, and vertebral artery dissections.7 Dissection of visceral arteries occurs predominantly in men and more often in middle-aged patients.8 Risk factors for DCA are thought to mirror risk factors for dissection of other arteries, including atherosclerotic disease, hypertension, connective tissue disorders, trauma, vasculitis, and pregnancy.9-11
Signs and Symptoms
Patients with DCA typically present with sudden onset of epigastric, flank, and/or chest pain, though 50% of patients may be asymptomatic.12 This pain is easily overlooked because the physical examination and laboratory studies are typically unremarkable.13 Fortunately, DCA is rarely accompanied by fatal organ dysfunction due to collateral flow from other vessels.14
Diagnosis and Management
While CTA with contrast is considered the mainstay of diagnosis of DCA,15 optimal treatment for DCA has not been well established. Management options include medical management, operative repair, and endovascular embolization. Medical management is reserved for stable patients without signs of end organ dysfunction. Typical management involves anticoagulation with warfarin for 3 to 6 months and strict BP control accompanied by close surveillance for progression.10,13 Some clinicians have argued that anticoagulation therapy may be unnecessary and that risk factor modification and BP control alone may be sufficient.5,6 Others have advocated that surgical management should be favored in cases of persistent pain, development of aneurysm, or threatened or compromised flow to end organs.7
Point-of-Care Ultrasound
The American College of Emergency Physicians considers ultrasound of the abdominal aorta a core application of emergency ultrasound.16 While sensitivity and specificity of emergency ultrasound for abdominal aortic aneurysm are well established, data supporting its use for screening for dissections are less definitive. With a sensitivity of 67% to 80% and a specificity of 99% to 100% with visualization of an intimal flap, aortic dissection screening using ultrasound is less reliable than most emergency physicians (EPs) would prefer.17,18 There are no published data reporting the sensitivity or specificity of emergency ultrasound for DCA. However, the vascular surgery literature encourages color Doppler ultrasound as part of the initial diagnostic work-up for this rare entity.19 While this may seem like an area ripe for emergency ultrasound, it is important to note—as seen in our case—that the site of the dissection is not often seen. Instead, the use of Doppler allows a screening for an abnormal flow pattern suggestive of dissection.20
Conclusion
In our case, both resident EPs and an expert fellowship-trained emergency ultrasound attending physician were unable to visualize a dissection—even after knowledge of the lesion was established by CTA. This points out a limitation of emergency ultrasound. While a POC ultrasound may be able to effectively rule in dissections of the aorta and its branches, we cannot reliably rule out these lesions. As EPs continue to expand the use of ultrasound, it is important to balance the desire for efficiency and cost-effectiveness with a high index of suspicion, experience, and clinical acumen.
1. Bauersfeld SR. Dissecting aneurysm of the aorta; a presentation of 15 cases and a review of the recent literature. Ann Intern Med. 1947;26(6):873-889.
2. Watson AJ. Dissecting aneurysm of arteries other than the aorta. J Pathol. 1956;72(2):439-449. doi:10.1002/path.1700720209.
3. Foord AG, Lewis RD. Primary dissecting aneurysms of peripheral and pulmonary arteries: dissecting hemorrhage of media. Arch Pathol. 1959;68:553-577.
4. Neychev V, Krol E, Dietzek A. Unusual presentation and treatment of spontaneous celiac artery dissection. J Vasc Surg. 2013;58(2):491-495. doi:10.1016/j.jvs.2012.10.136.
5. DiMusto PD, Oberdoerster MM, Criado E. Isolated celiac artery dissection. J Vasc Surg. 2015;61(4):972-976. doi: 10.1016/j.jvs.2014.10.108.
6. Takayama T, Miyata T, Shirakawa M, Nagawa H. J Vasc Surg. 2008;48(2):329-333. doi:10.1016/j.jvs.2008.03.002.
7. Glehen O, Feugier P, Aleksic Y, Delannoy P, Chevalier JM. Spontaneous dissection of the celiac artery. Ann Vasc Surg. 2001;15(6):687-692.
8. Patel KS, Benshar O, Vrabie R, Patel A, Adler M, Hines G. A major pain in the … back and epigastrium: an unusual case of spontaneous celiac artery dissection. J Community Hosp Intern Med Perspect. 2014;4(5):23840. doi:10.3402/jchimp.v4.23840.
9. Kang TL, Teich DL, McGillicuddy DC. Isolated, spontaneous superior mesenteric and celiac artery dissection: case report and review of literature. J Emerg Med. 2011;40(2):e21-e25. doi:10.1016/j.jemermed.2007.12.038.
10. Galastri FL, Cavalcante RN, Motta-Leal-Filho JM, et al. Evaluation and management of symptomatic isolated spontaneous celiac trunk dissection. Vasc Med. 2015;20(4):358-363. doi:10.1177/1358863X15581447.
11. Wang HC, Chen JH, Hsiao CC, Jeng CM, Chen WL. Spontaneous dissection of the celiac artery: a case report and literature review. Am J Emerg Med. 2013;31(6):1000.e3-e5. doi:10.1016/j.ajem.2013.02.007.
12. Oh S, Cho YP, Kim JH, Shin S, Kwon TW, Ko GY. Symptomatic spontaneous celiac artery dissection treated by conservative management: serial imaging findings. Abdom Imaging. 2011;36(1):79-82. doi:10.1007/s00261-010-9657-x.
13. Wang JL, Hsieh MJ, Lee CH, Chen CC, Hsieh IC. Celiac artery dissection presenting with abdominal and chest pain. Am J Emerg Med. 2010;28(1):111.e3-e5. doi:10.1016/j.ajem.2009.02.023.
14. Takayama Y, Takao M, Inoue T, Yoshimi F, Koyama K, Nagai H. Isolated spontaneous dissection of the celiac artery: report of two cases. Ann Vasc Dis. 2014;7(1):64-67. doi:10.3400/avd.cr.13-00102.
15. Rehman AU, Almanfi A, Nadella S, Sohail U. Isolated spontaneous celiac artery dissection in a 47-year-old man with von Willebrand disease. Tex Heart Inst J. 2014;41(3):344-345. doi:10.14503/THIJ-13-3404.
16. American College of Emergency Physicians. Policy statement. Ultrasound Guidelines: Emergency, Point-of-Care, and Clinical Ultrasound Guidelines in Medicine, June 2016. https://www.acep.org/Clinical---Practice-Management/Ultrasound/. Accessed November 15, 2016.
17. Williams J, Heiner JD, Perreault MD, McArthur TJ. Aortic dissection diagnosed by ultrasound. West J Emerg Med. 2010;11(1):98-99.
18. Fojtik JP, Costantino TG, Dean AJ. The diagnosis of aortic dissection by emergency medicine ultrasound. J Emerg Med. 2007;32(2):191-196.
19. Woolard JD, Ammar AD. Spontaneous dissection of the celiac artery: a case report. J Vasc Surg. 2007;45(6):1256-1258.
20. Fenoglio L, Allione A, Scalabrino E, et al. Spontaneous dissection of the celiac artery: a pitfall in the diagnosis of acute abdominal pain. Presentation of two cases. Dig Dis Sci. 2004;49(7-8):1223-1227.
1. Bauersfeld SR. Dissecting aneurysm of the aorta; a presentation of 15 cases and a review of the recent literature. Ann Intern Med. 1947;26(6):873-889.
2. Watson AJ. Dissecting aneurysm of arteries other than the aorta. J Pathol. 1956;72(2):439-449. doi:10.1002/path.1700720209.
3. Foord AG, Lewis RD. Primary dissecting aneurysms of peripheral and pulmonary arteries: dissecting hemorrhage of media. Arch Pathol. 1959;68:553-577.
4. Neychev V, Krol E, Dietzek A. Unusual presentation and treatment of spontaneous celiac artery dissection. J Vasc Surg. 2013;58(2):491-495. doi:10.1016/j.jvs.2012.10.136.
5. DiMusto PD, Oberdoerster MM, Criado E. Isolated celiac artery dissection. J Vasc Surg. 2015;61(4):972-976. doi: 10.1016/j.jvs.2014.10.108.
6. Takayama T, Miyata T, Shirakawa M, Nagawa H. J Vasc Surg. 2008;48(2):329-333. doi:10.1016/j.jvs.2008.03.002.
7. Glehen O, Feugier P, Aleksic Y, Delannoy P, Chevalier JM. Spontaneous dissection of the celiac artery. Ann Vasc Surg. 2001;15(6):687-692.
8. Patel KS, Benshar O, Vrabie R, Patel A, Adler M, Hines G. A major pain in the … back and epigastrium: an unusual case of spontaneous celiac artery dissection. J Community Hosp Intern Med Perspect. 2014;4(5):23840. doi:10.3402/jchimp.v4.23840.
9. Kang TL, Teich DL, McGillicuddy DC. Isolated, spontaneous superior mesenteric and celiac artery dissection: case report and review of literature. J Emerg Med. 2011;40(2):e21-e25. doi:10.1016/j.jemermed.2007.12.038.
10. Galastri FL, Cavalcante RN, Motta-Leal-Filho JM, et al. Evaluation and management of symptomatic isolated spontaneous celiac trunk dissection. Vasc Med. 2015;20(4):358-363. doi:10.1177/1358863X15581447.
11. Wang HC, Chen JH, Hsiao CC, Jeng CM, Chen WL. Spontaneous dissection of the celiac artery: a case report and literature review. Am J Emerg Med. 2013;31(6):1000.e3-e5. doi:10.1016/j.ajem.2013.02.007.
12. Oh S, Cho YP, Kim JH, Shin S, Kwon TW, Ko GY. Symptomatic spontaneous celiac artery dissection treated by conservative management: serial imaging findings. Abdom Imaging. 2011;36(1):79-82. doi:10.1007/s00261-010-9657-x.
13. Wang JL, Hsieh MJ, Lee CH, Chen CC, Hsieh IC. Celiac artery dissection presenting with abdominal and chest pain. Am J Emerg Med. 2010;28(1):111.e3-e5. doi:10.1016/j.ajem.2009.02.023.
14. Takayama Y, Takao M, Inoue T, Yoshimi F, Koyama K, Nagai H. Isolated spontaneous dissection of the celiac artery: report of two cases. Ann Vasc Dis. 2014;7(1):64-67. doi:10.3400/avd.cr.13-00102.
15. Rehman AU, Almanfi A, Nadella S, Sohail U. Isolated spontaneous celiac artery dissection in a 47-year-old man with von Willebrand disease. Tex Heart Inst J. 2014;41(3):344-345. doi:10.14503/THIJ-13-3404.
16. American College of Emergency Physicians. Policy statement. Ultrasound Guidelines: Emergency, Point-of-Care, and Clinical Ultrasound Guidelines in Medicine, June 2016. https://www.acep.org/Clinical---Practice-Management/Ultrasound/. Accessed November 15, 2016.
17. Williams J, Heiner JD, Perreault MD, McArthur TJ. Aortic dissection diagnosed by ultrasound. West J Emerg Med. 2010;11(1):98-99.
18. Fojtik JP, Costantino TG, Dean AJ. The diagnosis of aortic dissection by emergency medicine ultrasound. J Emerg Med. 2007;32(2):191-196.
19. Woolard JD, Ammar AD. Spontaneous dissection of the celiac artery: a case report. J Vasc Surg. 2007;45(6):1256-1258.
20. Fenoglio L, Allione A, Scalabrino E, et al. Spontaneous dissection of the celiac artery: a pitfall in the diagnosis of acute abdominal pain. Presentation of two cases. Dig Dis Sci. 2004;49(7-8):1223-1227.
Relapsing Polychondritis With Meningoencephalitis
Relapsing polychondritis (RP) is an autoimmune disease affecting cartilaginous structures such as the ears, respiratory passages, joints, and cardiovascular system.1,2 In rare cases, the systemic effects of this autoimmune process can cause central nervous system (CNS) involvement such as meningoencephalitis (ME).3 In 2011, Wang et al4 described 4 cases of RP with ME and reviewed 24 cases from the literature. We present a case of a man with RP-associated ME that was responsive to steroid treatment. We also provide an updated review of the literature.
Case Report
A 44-year-old man developed gradually worsening bilateral ear pain, headaches, and seizures. He was briefly hospitalized and discharged with levetiracetam and quetiapine. However, his mental status continued to deteriorate and he was subsequently hospitalized 3 months later with confusion, hallucinations, and seizures.
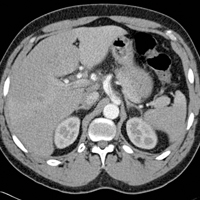
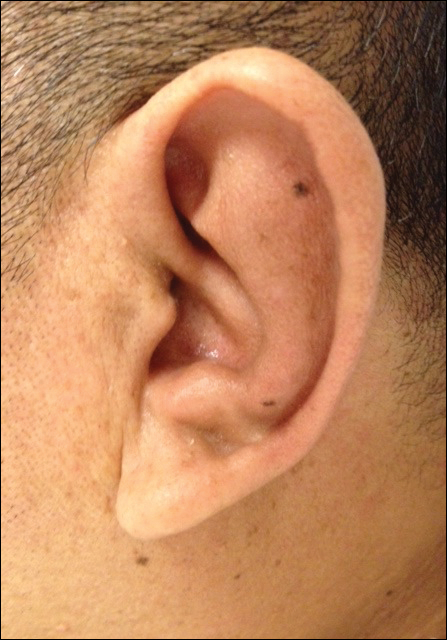
On physical examination the patient was disoriented and unable to form cohesive sentences. He had bilateral tenderness, erythema, and edema of the auricles, which notably spared the lobules (Figure 1). The conjunctivae were injected bilaterally, and joint involvement included bilateral knee tenderness and swelling. Neurologic examination revealed questionable meningeal signs but no motor or sensory deficits. An extensive laboratory workup for the etiology of his altered mental status was unremarkable, except for a mildly elevated white blood cell count in the cerebrospinal fluid with predominantly lymphocytes. No infectious etiologies were identified on laboratory testing, and rheumatologic markers were negative including antinuclear antibody, rheumatoid factor, and anti–Sjögren syndrome antigen A/Sjögren syndrome antigen B. Magnetic resonance imaging revealed nonspecific findings of bilateral T2 hyperdensities in the subcortical white matter; however, cerebral angiography revealed no evidence of vasculitis. A biopsy of the right antihelix revealed prominent perichondritis and a neutrophilic inflammatory infiltrate with several lymphocytes and histiocytes (Figure 2). There was degeneration of the cartilaginous tissue with evidence of pyknotic nuclei, eosinophilia, and vacuolization of the chondrocytes. He was diagnosed with RP on the basis of clinical and histologic inflammation of the auricular cartilage, polyarthritis, and ocular inflammation.
The patient was treated with high-dose immunosuppression with methylprednisolone (1000 mg intravenous once daily for 5 days) and cyclophosphamide (one dose at 500 mg/m2), which resulted in remarkable improvement in his mental status, auricular inflammation, and knee pain. After 31 days of hospitalization the patient was discharged with a course of oral prednisone (starting at 60 mg/d, then tapered over the following 2 months) and monthly cyclophosphamide infusions (5 months total; starting at 500 mg/m2, then uptitrated to goal of 1000 mg/m2). Maintenance suppression was achieved with azathioprine (starting at 50 mg daily, then uptitrated to 100 mg daily), which was continued without any evidence of relapsed disease through his last outpatient visit 1 year after the diagnosis.
Comment
Auricular inflammation is a hallmark of RP and is present in 83% to 95% of patients.1,3 The affected ears can appear erythematous to violaceous with tender edema of the auricle that spares the lobules where no cartilage is present. The inflammation can extend into the ear canal and cause hearing loss, tinnitus, and vertigo. Histologically, RP can present with a nonspecific leukocytoclastic vasculitis and inflammatory destruction of the cartilage. Therefore, diagnosis of RP is reliant mainly on clinical characteristics rather than pathologic findings. In 1976, McAdam et al5 established diagnostic criteria for RP based on the presence of common clinical manifestations (eg, auricular chondritis, seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation). Michet et al6 later proposed major and minor criteria to classify and diagnose RP based on clinical manifestations. Diagnosis of our patient was confirmed by the presence of auricular chondritis, polyarthritis, and ocular inflammation. Diagnosing RP can be difficult because it has many systemic manifestations that can evoke a broad differential diagnosis. The time to diagnosis in our patient was 3 months, but the mean delay in diagnosis for patients with RP and ME is 2.9 years.4
The etiology of RP remains unclear, but current evidence supports an immune-mediated process directed toward proteins found in cartilage. Animal studies have suggested that RP may be driven by antibodies to matrillin 1 and type II collagen. There also may be a familial association with HLA-DR4 and genetic predisposition to autoimmune diseases in individuals affected by RP.1,3 The pathogenesis of CNS involvement in RP is thought to be due to a localized small vessel vasculitis.7,8 In our patient, however, cerebral angiography was negative for vasculitis, and thus our case may represent another mechanism for CNS involvement. There have been cases of encephalitis in RP caused by pathways other than CNS vasculitis. Kashihara et al9 reported a case of RP with encephalitis associated with antiglutamate receptor antibodies found in the cerebrospinal fluid and blood.
Treatment of RP has been based on pathophysiological considerations rather than empiric data due to its rarity. Relapsing polychondritis has been responsive to steroid treatment in reported cases as well as in our patient; however, in cases in which RP did not respond to steroids, infliximab may be effective for RP with ME.10 Further research regarding the treatment outcomes of RP with ME may be warranted.
Although rare, additional cases of RP with ME have been reported (Table). Wang et al4 described a series of 28 patients with RP and ME from 1960 to 2010. A PubMed search of articles indexed for MEDLINE that were published in the English-language literature from 2010 to 2016 was performed using the search terms relapsing polychondritis and nervous system. Including our patient, RP with ME was reported in 17 additional cases since Wang et al4 published their findings. These cases involved adults ranging in age from 44 to 73 years who were mainly men (14/17 [82%]). All of the patients presented with bilateral auricular chondritis, except for a case of unilateral ear involvement reported by Storey et al.11 Common neurologic manifestations included fever, headache, and altered mental status. Motor symptoms ranged from dysarthria and agraphia12 to hemiparesis.13 The mechanism of CNS involvement in RP was not identified in most cases; however, Mattiassich et al14 documented cerebral vasculitis in their patient, and Niwa et al16 found diffuse cerebral vasculitis on autopsy. Eleven of 17 (65%) cases responded to steroid treatment. Of the 6 cases in which RP did not respond to steroids, 2 patients died despite high-dose steroid treatment,11,20 2 responded to infliximab,10,15 1 responded to tocilizumab,21 and 1 was lost to follow-up after initial treatment failure.20
Conclusion
Although rare, RP should not be overlooked in the inpatient setting due to its potential for life-threatening systemic effects. Early diagnosis of this condition may be of benefit to this select population of patients, and further research regarding the prognosis, mechanisms, and treatment of RP may be necessary in the future.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis: a 2013 update. Autoimmun Rev. 2014;13:90-95.
- Ostrowski RA, Takagishi T, Robinson J. Rheumatoid arthritis, spondyloarthropathies, and relapsing polychondritis. Handb Clin Neurol. 2014;119:449-461.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Wang ZJ, Pu CQ, Wang ZJ, et al. Meningoencephalitis or meningitis in relapsing polychondritis: four case reports and a literature review. J Clin Neurosci. 2011;18:1608-1615.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet C, McKenna C, Luthra H, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Sampaio L, Silva L, Mariz E, et al. CNS involvement in relapsing polychondritis. Joint Bone Spine. 2010;77:619-620.
- Prinz S, Dafotakis M, Schneider RK, et al. The red puffy ear sign—a clinical sign to diagnose a rare cause of meningoencephalitis. Fortschr Neurol Psychiatr. 2012;80:463-467.
- Kashihara K, Kawada S, Takahashi Y. Autoantibodies to glutamate receptor GluR2 in a patient with limic encephalitis associated with relapsing polychondritis. J Neurol Sci. 2009;287:275-277.
- Garcia-Egido A, Gutierrez C, de la Fuente C, et al. Relapsing polychondritis-associated meningitis and encephalitis: response to infliximab. Rheumatology (Oxford). 2011;50:1721-1723.
- Storey K, Matej R, Rusina R. Unusual association of seronegative, nonparaneoplastic limbic encephalitis and relapsing polychondritis in a patient with history of thymectomy for myasthemia: a case study. J Neurol. 2010;258:159-161.
- Choi HJ, Lee HJ. Relapsing polychondritis with encephalitis. J Clin Rheum. 2011;6:329-331.
- Fujiwara S, Zenke K, Iwata S, et al. Relapsing polychondritis presenting as encephalitis. No Shinkei Geka. 2012;40:247-253.
- Mattiassich G, Egger M, Semlitsch G, et al. Occurrence of relapsing polychondritis with a rising cANCA titre in a cANCA-positive systemic and cerebral vasculitis patient [published online February 5, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-008717.
- Kondo T, Fukuta M, Takemoto A, et al. Limbic encephalitis associated with relapsing polychondritis responded to infliximab and maintained its condition without recurrence after discontinuation: a case report and review of the literature. Nagoya J Med Sci. 2014;76:361-368.
- Niwa A, Okamoto Y, Kondo T, et al. Perivasculitic pancencephalitis with relapsing polychondritis: an autopsy case report and review of previous cases. Intern Med. 2014;53:1191-1195.
- Coban EK, Xanmemmedoy E, Colak M, et al. A rare complication of a rare disease; stroke due to relapsing polychondritis. Ideggyogy Sz. 2015;68:429-432.
- Ducci R, Germiniani F, Czecko L, et al. Relapsing polychondritis and lymphocytic meningitis with varied neurological symptoms [published online February 5, 2016]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.09.005.
- Baba T, Kanno S, Shijo T, et al. Callosal disconnection syndrome associated with relapsing polychondritis. Intern Med. 2016;55:1191-1193.
- Jeon C. Relapsing polychondritis with central nervous system involvement: experience of three different cases in a single center. J Korean Med. 2016;31:1846-1850.
- Liu L, Liu S, Guan W, et al. Efficacy of tocilizumab for psychiatric symptoms associated with relapsing polychondritis: the first case report and review of the literature. Rheumatol Int. 2016;36:1185-1189.
Relapsing polychondritis (RP) is an autoimmune disease affecting cartilaginous structures such as the ears, respiratory passages, joints, and cardiovascular system.1,2 In rare cases, the systemic effects of this autoimmune process can cause central nervous system (CNS) involvement such as meningoencephalitis (ME).3 In 2011, Wang et al4 described 4 cases of RP with ME and reviewed 24 cases from the literature. We present a case of a man with RP-associated ME that was responsive to steroid treatment. We also provide an updated review of the literature.
Case Report
A 44-year-old man developed gradually worsening bilateral ear pain, headaches, and seizures. He was briefly hospitalized and discharged with levetiracetam and quetiapine. However, his mental status continued to deteriorate and he was subsequently hospitalized 3 months later with confusion, hallucinations, and seizures.
On physical examination the patient was disoriented and unable to form cohesive sentences. He had bilateral tenderness, erythema, and edema of the auricles, which notably spared the lobules (Figure 1). The conjunctivae were injected bilaterally, and joint involvement included bilateral knee tenderness and swelling. Neurologic examination revealed questionable meningeal signs but no motor or sensory deficits. An extensive laboratory workup for the etiology of his altered mental status was unremarkable, except for a mildly elevated white blood cell count in the cerebrospinal fluid with predominantly lymphocytes. No infectious etiologies were identified on laboratory testing, and rheumatologic markers were negative including antinuclear antibody, rheumatoid factor, and anti–Sjögren syndrome antigen A/Sjögren syndrome antigen B. Magnetic resonance imaging revealed nonspecific findings of bilateral T2 hyperdensities in the subcortical white matter; however, cerebral angiography revealed no evidence of vasculitis. A biopsy of the right antihelix revealed prominent perichondritis and a neutrophilic inflammatory infiltrate with several lymphocytes and histiocytes (Figure 2). There was degeneration of the cartilaginous tissue with evidence of pyknotic nuclei, eosinophilia, and vacuolization of the chondrocytes. He was diagnosed with RP on the basis of clinical and histologic inflammation of the auricular cartilage, polyarthritis, and ocular inflammation.
The patient was treated with high-dose immunosuppression with methylprednisolone (1000 mg intravenous once daily for 5 days) and cyclophosphamide (one dose at 500 mg/m2), which resulted in remarkable improvement in his mental status, auricular inflammation, and knee pain. After 31 days of hospitalization the patient was discharged with a course of oral prednisone (starting at 60 mg/d, then tapered over the following 2 months) and monthly cyclophosphamide infusions (5 months total; starting at 500 mg/m2, then uptitrated to goal of 1000 mg/m2). Maintenance suppression was achieved with azathioprine (starting at 50 mg daily, then uptitrated to 100 mg daily), which was continued without any evidence of relapsed disease through his last outpatient visit 1 year after the diagnosis.
Comment
Auricular inflammation is a hallmark of RP and is present in 83% to 95% of patients.1,3 The affected ears can appear erythematous to violaceous with tender edema of the auricle that spares the lobules where no cartilage is present. The inflammation can extend into the ear canal and cause hearing loss, tinnitus, and vertigo. Histologically, RP can present with a nonspecific leukocytoclastic vasculitis and inflammatory destruction of the cartilage. Therefore, diagnosis of RP is reliant mainly on clinical characteristics rather than pathologic findings. In 1976, McAdam et al5 established diagnostic criteria for RP based on the presence of common clinical manifestations (eg, auricular chondritis, seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation). Michet et al6 later proposed major and minor criteria to classify and diagnose RP based on clinical manifestations. Diagnosis of our patient was confirmed by the presence of auricular chondritis, polyarthritis, and ocular inflammation. Diagnosing RP can be difficult because it has many systemic manifestations that can evoke a broad differential diagnosis. The time to diagnosis in our patient was 3 months, but the mean delay in diagnosis for patients with RP and ME is 2.9 years.4
The etiology of RP remains unclear, but current evidence supports an immune-mediated process directed toward proteins found in cartilage. Animal studies have suggested that RP may be driven by antibodies to matrillin 1 and type II collagen. There also may be a familial association with HLA-DR4 and genetic predisposition to autoimmune diseases in individuals affected by RP.1,3 The pathogenesis of CNS involvement in RP is thought to be due to a localized small vessel vasculitis.7,8 In our patient, however, cerebral angiography was negative for vasculitis, and thus our case may represent another mechanism for CNS involvement. There have been cases of encephalitis in RP caused by pathways other than CNS vasculitis. Kashihara et al9 reported a case of RP with encephalitis associated with antiglutamate receptor antibodies found in the cerebrospinal fluid and blood.
Treatment of RP has been based on pathophysiological considerations rather than empiric data due to its rarity. Relapsing polychondritis has been responsive to steroid treatment in reported cases as well as in our patient; however, in cases in which RP did not respond to steroids, infliximab may be effective for RP with ME.10 Further research regarding the treatment outcomes of RP with ME may be warranted.
Although rare, additional cases of RP with ME have been reported (Table). Wang et al4 described a series of 28 patients with RP and ME from 1960 to 2010. A PubMed search of articles indexed for MEDLINE that were published in the English-language literature from 2010 to 2016 was performed using the search terms relapsing polychondritis and nervous system. Including our patient, RP with ME was reported in 17 additional cases since Wang et al4 published their findings. These cases involved adults ranging in age from 44 to 73 years who were mainly men (14/17 [82%]). All of the patients presented with bilateral auricular chondritis, except for a case of unilateral ear involvement reported by Storey et al.11 Common neurologic manifestations included fever, headache, and altered mental status. Motor symptoms ranged from dysarthria and agraphia12 to hemiparesis.13 The mechanism of CNS involvement in RP was not identified in most cases; however, Mattiassich et al14 documented cerebral vasculitis in their patient, and Niwa et al16 found diffuse cerebral vasculitis on autopsy. Eleven of 17 (65%) cases responded to steroid treatment. Of the 6 cases in which RP did not respond to steroids, 2 patients died despite high-dose steroid treatment,11,20 2 responded to infliximab,10,15 1 responded to tocilizumab,21 and 1 was lost to follow-up after initial treatment failure.20
Conclusion
Although rare, RP should not be overlooked in the inpatient setting due to its potential for life-threatening systemic effects. Early diagnosis of this condition may be of benefit to this select population of patients, and further research regarding the prognosis, mechanisms, and treatment of RP may be necessary in the future.
Relapsing polychondritis (RP) is an autoimmune disease affecting cartilaginous structures such as the ears, respiratory passages, joints, and cardiovascular system.1,2 In rare cases, the systemic effects of this autoimmune process can cause central nervous system (CNS) involvement such as meningoencephalitis (ME).3 In 2011, Wang et al4 described 4 cases of RP with ME and reviewed 24 cases from the literature. We present a case of a man with RP-associated ME that was responsive to steroid treatment. We also provide an updated review of the literature.
Case Report
A 44-year-old man developed gradually worsening bilateral ear pain, headaches, and seizures. He was briefly hospitalized and discharged with levetiracetam and quetiapine. However, his mental status continued to deteriorate and he was subsequently hospitalized 3 months later with confusion, hallucinations, and seizures.
On physical examination the patient was disoriented and unable to form cohesive sentences. He had bilateral tenderness, erythema, and edema of the auricles, which notably spared the lobules (Figure 1). The conjunctivae were injected bilaterally, and joint involvement included bilateral knee tenderness and swelling. Neurologic examination revealed questionable meningeal signs but no motor or sensory deficits. An extensive laboratory workup for the etiology of his altered mental status was unremarkable, except for a mildly elevated white blood cell count in the cerebrospinal fluid with predominantly lymphocytes. No infectious etiologies were identified on laboratory testing, and rheumatologic markers were negative including antinuclear antibody, rheumatoid factor, and anti–Sjögren syndrome antigen A/Sjögren syndrome antigen B. Magnetic resonance imaging revealed nonspecific findings of bilateral T2 hyperdensities in the subcortical white matter; however, cerebral angiography revealed no evidence of vasculitis. A biopsy of the right antihelix revealed prominent perichondritis and a neutrophilic inflammatory infiltrate with several lymphocytes and histiocytes (Figure 2). There was degeneration of the cartilaginous tissue with evidence of pyknotic nuclei, eosinophilia, and vacuolization of the chondrocytes. He was diagnosed with RP on the basis of clinical and histologic inflammation of the auricular cartilage, polyarthritis, and ocular inflammation.
The patient was treated with high-dose immunosuppression with methylprednisolone (1000 mg intravenous once daily for 5 days) and cyclophosphamide (one dose at 500 mg/m2), which resulted in remarkable improvement in his mental status, auricular inflammation, and knee pain. After 31 days of hospitalization the patient was discharged with a course of oral prednisone (starting at 60 mg/d, then tapered over the following 2 months) and monthly cyclophosphamide infusions (5 months total; starting at 500 mg/m2, then uptitrated to goal of 1000 mg/m2). Maintenance suppression was achieved with azathioprine (starting at 50 mg daily, then uptitrated to 100 mg daily), which was continued without any evidence of relapsed disease through his last outpatient visit 1 year after the diagnosis.
Comment
Auricular inflammation is a hallmark of RP and is present in 83% to 95% of patients.1,3 The affected ears can appear erythematous to violaceous with tender edema of the auricle that spares the lobules where no cartilage is present. The inflammation can extend into the ear canal and cause hearing loss, tinnitus, and vertigo. Histologically, RP can present with a nonspecific leukocytoclastic vasculitis and inflammatory destruction of the cartilage. Therefore, diagnosis of RP is reliant mainly on clinical characteristics rather than pathologic findings. In 1976, McAdam et al5 established diagnostic criteria for RP based on the presence of common clinical manifestations (eg, auricular chondritis, seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation). Michet et al6 later proposed major and minor criteria to classify and diagnose RP based on clinical manifestations. Diagnosis of our patient was confirmed by the presence of auricular chondritis, polyarthritis, and ocular inflammation. Diagnosing RP can be difficult because it has many systemic manifestations that can evoke a broad differential diagnosis. The time to diagnosis in our patient was 3 months, but the mean delay in diagnosis for patients with RP and ME is 2.9 years.4
The etiology of RP remains unclear, but current evidence supports an immune-mediated process directed toward proteins found in cartilage. Animal studies have suggested that RP may be driven by antibodies to matrillin 1 and type II collagen. There also may be a familial association with HLA-DR4 and genetic predisposition to autoimmune diseases in individuals affected by RP.1,3 The pathogenesis of CNS involvement in RP is thought to be due to a localized small vessel vasculitis.7,8 In our patient, however, cerebral angiography was negative for vasculitis, and thus our case may represent another mechanism for CNS involvement. There have been cases of encephalitis in RP caused by pathways other than CNS vasculitis. Kashihara et al9 reported a case of RP with encephalitis associated with antiglutamate receptor antibodies found in the cerebrospinal fluid and blood.
Treatment of RP has been based on pathophysiological considerations rather than empiric data due to its rarity. Relapsing polychondritis has been responsive to steroid treatment in reported cases as well as in our patient; however, in cases in which RP did not respond to steroids, infliximab may be effective for RP with ME.10 Further research regarding the treatment outcomes of RP with ME may be warranted.
Although rare, additional cases of RP with ME have been reported (Table). Wang et al4 described a series of 28 patients with RP and ME from 1960 to 2010. A PubMed search of articles indexed for MEDLINE that were published in the English-language literature from 2010 to 2016 was performed using the search terms relapsing polychondritis and nervous system. Including our patient, RP with ME was reported in 17 additional cases since Wang et al4 published their findings. These cases involved adults ranging in age from 44 to 73 years who were mainly men (14/17 [82%]). All of the patients presented with bilateral auricular chondritis, except for a case of unilateral ear involvement reported by Storey et al.11 Common neurologic manifestations included fever, headache, and altered mental status. Motor symptoms ranged from dysarthria and agraphia12 to hemiparesis.13 The mechanism of CNS involvement in RP was not identified in most cases; however, Mattiassich et al14 documented cerebral vasculitis in their patient, and Niwa et al16 found diffuse cerebral vasculitis on autopsy. Eleven of 17 (65%) cases responded to steroid treatment. Of the 6 cases in which RP did not respond to steroids, 2 patients died despite high-dose steroid treatment,11,20 2 responded to infliximab,10,15 1 responded to tocilizumab,21 and 1 was lost to follow-up after initial treatment failure.20
Conclusion
Although rare, RP should not be overlooked in the inpatient setting due to its potential for life-threatening systemic effects. Early diagnosis of this condition may be of benefit to this select population of patients, and further research regarding the prognosis, mechanisms, and treatment of RP may be necessary in the future.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis: a 2013 update. Autoimmun Rev. 2014;13:90-95.
- Ostrowski RA, Takagishi T, Robinson J. Rheumatoid arthritis, spondyloarthropathies, and relapsing polychondritis. Handb Clin Neurol. 2014;119:449-461.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Wang ZJ, Pu CQ, Wang ZJ, et al. Meningoencephalitis or meningitis in relapsing polychondritis: four case reports and a literature review. J Clin Neurosci. 2011;18:1608-1615.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet C, McKenna C, Luthra H, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Sampaio L, Silva L, Mariz E, et al. CNS involvement in relapsing polychondritis. Joint Bone Spine. 2010;77:619-620.
- Prinz S, Dafotakis M, Schneider RK, et al. The red puffy ear sign—a clinical sign to diagnose a rare cause of meningoencephalitis. Fortschr Neurol Psychiatr. 2012;80:463-467.
- Kashihara K, Kawada S, Takahashi Y. Autoantibodies to glutamate receptor GluR2 in a patient with limic encephalitis associated with relapsing polychondritis. J Neurol Sci. 2009;287:275-277.
- Garcia-Egido A, Gutierrez C, de la Fuente C, et al. Relapsing polychondritis-associated meningitis and encephalitis: response to infliximab. Rheumatology (Oxford). 2011;50:1721-1723.
- Storey K, Matej R, Rusina R. Unusual association of seronegative, nonparaneoplastic limbic encephalitis and relapsing polychondritis in a patient with history of thymectomy for myasthemia: a case study. J Neurol. 2010;258:159-161.
- Choi HJ, Lee HJ. Relapsing polychondritis with encephalitis. J Clin Rheum. 2011;6:329-331.
- Fujiwara S, Zenke K, Iwata S, et al. Relapsing polychondritis presenting as encephalitis. No Shinkei Geka. 2012;40:247-253.
- Mattiassich G, Egger M, Semlitsch G, et al. Occurrence of relapsing polychondritis with a rising cANCA titre in a cANCA-positive systemic and cerebral vasculitis patient [published online February 5, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-008717.
- Kondo T, Fukuta M, Takemoto A, et al. Limbic encephalitis associated with relapsing polychondritis responded to infliximab and maintained its condition without recurrence after discontinuation: a case report and review of the literature. Nagoya J Med Sci. 2014;76:361-368.
- Niwa A, Okamoto Y, Kondo T, et al. Perivasculitic pancencephalitis with relapsing polychondritis: an autopsy case report and review of previous cases. Intern Med. 2014;53:1191-1195.
- Coban EK, Xanmemmedoy E, Colak M, et al. A rare complication of a rare disease; stroke due to relapsing polychondritis. Ideggyogy Sz. 2015;68:429-432.
- Ducci R, Germiniani F, Czecko L, et al. Relapsing polychondritis and lymphocytic meningitis with varied neurological symptoms [published online February 5, 2016]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.09.005.
- Baba T, Kanno S, Shijo T, et al. Callosal disconnection syndrome associated with relapsing polychondritis. Intern Med. 2016;55:1191-1193.
- Jeon C. Relapsing polychondritis with central nervous system involvement: experience of three different cases in a single center. J Korean Med. 2016;31:1846-1850.
- Liu L, Liu S, Guan W, et al. Efficacy of tocilizumab for psychiatric symptoms associated with relapsing polychondritis: the first case report and review of the literature. Rheumatol Int. 2016;36:1185-1189.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis: a 2013 update. Autoimmun Rev. 2014;13:90-95.
- Ostrowski RA, Takagishi T, Robinson J. Rheumatoid arthritis, spondyloarthropathies, and relapsing polychondritis. Handb Clin Neurol. 2014;119:449-461.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Wang ZJ, Pu CQ, Wang ZJ, et al. Meningoencephalitis or meningitis in relapsing polychondritis: four case reports and a literature review. J Clin Neurosci. 2011;18:1608-1615.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet C, McKenna C, Luthra H, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Sampaio L, Silva L, Mariz E, et al. CNS involvement in relapsing polychondritis. Joint Bone Spine. 2010;77:619-620.
- Prinz S, Dafotakis M, Schneider RK, et al. The red puffy ear sign—a clinical sign to diagnose a rare cause of meningoencephalitis. Fortschr Neurol Psychiatr. 2012;80:463-467.
- Kashihara K, Kawada S, Takahashi Y. Autoantibodies to glutamate receptor GluR2 in a patient with limic encephalitis associated with relapsing polychondritis. J Neurol Sci. 2009;287:275-277.
- Garcia-Egido A, Gutierrez C, de la Fuente C, et al. Relapsing polychondritis-associated meningitis and encephalitis: response to infliximab. Rheumatology (Oxford). 2011;50:1721-1723.
- Storey K, Matej R, Rusina R. Unusual association of seronegative, nonparaneoplastic limbic encephalitis and relapsing polychondritis in a patient with history of thymectomy for myasthemia: a case study. J Neurol. 2010;258:159-161.
- Choi HJ, Lee HJ. Relapsing polychondritis with encephalitis. J Clin Rheum. 2011;6:329-331.
- Fujiwara S, Zenke K, Iwata S, et al. Relapsing polychondritis presenting as encephalitis. No Shinkei Geka. 2012;40:247-253.
- Mattiassich G, Egger M, Semlitsch G, et al. Occurrence of relapsing polychondritis with a rising cANCA titre in a cANCA-positive systemic and cerebral vasculitis patient [published online February 5, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-008717.
- Kondo T, Fukuta M, Takemoto A, et al. Limbic encephalitis associated with relapsing polychondritis responded to infliximab and maintained its condition without recurrence after discontinuation: a case report and review of the literature. Nagoya J Med Sci. 2014;76:361-368.
- Niwa A, Okamoto Y, Kondo T, et al. Perivasculitic pancencephalitis with relapsing polychondritis: an autopsy case report and review of previous cases. Intern Med. 2014;53:1191-1195.
- Coban EK, Xanmemmedoy E, Colak M, et al. A rare complication of a rare disease; stroke due to relapsing polychondritis. Ideggyogy Sz. 2015;68:429-432.
- Ducci R, Germiniani F, Czecko L, et al. Relapsing polychondritis and lymphocytic meningitis with varied neurological symptoms [published online February 5, 2016]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.09.005.
- Baba T, Kanno S, Shijo T, et al. Callosal disconnection syndrome associated with relapsing polychondritis. Intern Med. 2016;55:1191-1193.
- Jeon C. Relapsing polychondritis with central nervous system involvement: experience of three different cases in a single center. J Korean Med. 2016;31:1846-1850.
- Liu L, Liu S, Guan W, et al. Efficacy of tocilizumab for psychiatric symptoms associated with relapsing polychondritis: the first case report and review of the literature. Rheumatol Int. 2016;36:1185-1189.
Practice Points
- Meningoencephalitis (ME) is a potentially rare complication of relapsing polychondritis (RP).
- Treatment of ME due to RP can include high-dose steroids and biologics.
Papillary Transitional Cell Bladder Carcinoma and Systematized Epidermal Nevus Syndrome
Epidermal nevi can occur in isolation or in association with internal abnormalities. Epidermal nevus syndrome (ENS) is a heterogeneous group of neurocutaneous disorders characterized by mosaicism and epidermal nevi found in association with various systemic abnormalities.1-4 There are many possible associated systemic findings, including abnormalities of the central nervous, musculoskeletal, renal, and hematologic systems. Epidermal nevi have been associated with internal malignancies. We present the case of a patient with epidermal nevi associated with papillary transitional cell bladder carcinoma. According to a PubMed search of articles indexed for MEDLINE using the search terms transitional cell bladder carcinoma and epidermal nevus, there have only been 4 other cases of transitional cell bladder carcinoma and ENS reported in the literature,5-8 2 of which were reports of papillary transitional cell bladder carcinoma.5,6
Case Report
A 29-year-old woman presented to our clinic with a rash that had been present since 3 years of age. The emergency department consulted dermatology for evaluation of what was believed to be contact dermatitis; however, upon questioning the patient, it was revealed that the rash was chronic and persistent.
The rash was nonpruritic and was located on the face, hands (Figure 1), chest, buttocks, thighs, legs, and back (Figure 2). Although asymptomatic, the appearance of the skin caused the patient some emotional distress. As a child she had been evaluated by a dermatologist and a biopsy was performed, but she did not recall the results or have any records. She had been prescribed an oral medication by the dermatologist, but treatment was terminated early due to nausea. The skin lesions did not improve with the short course of treatment.
Eighteen months prior to presentation to our clinic, the patient was discovered to have hematuria on routine examination by her primary care physician. At that time, the patient underwent a workup for hematuria and a mass was discovered in the bladder via cystoscopy. A diagnosis of low-grade papillary transitional cell bladder carcinoma was made, and she underwent a partial cystectomy. No radiation or chemotherapy was required. The remainder of her medical history was only remarkable for asthma, which was well controlled with albuterol. On examination, generalized, hyperpigmented, reticulated patches, macules, and hyperpigmented verrucous plaques were distributed along the Blaschko lines, sparing the face. No limb abnormalities or dental or nail abnormalities were noted. Examination of the axillary and cervical lymph nodes was unremarkable, and no neurological abnormalities were noted. A 3-mm punch biopsy of the mid upper back was performed. Histopathology revealed papillomatous, nonorganoid, nonepidermolytic hyperplasia of the epidermis with elongated rete ridges (Figure 3), which was diagnosed as a nonorganoid nonepidermolytic epidermal nevus.
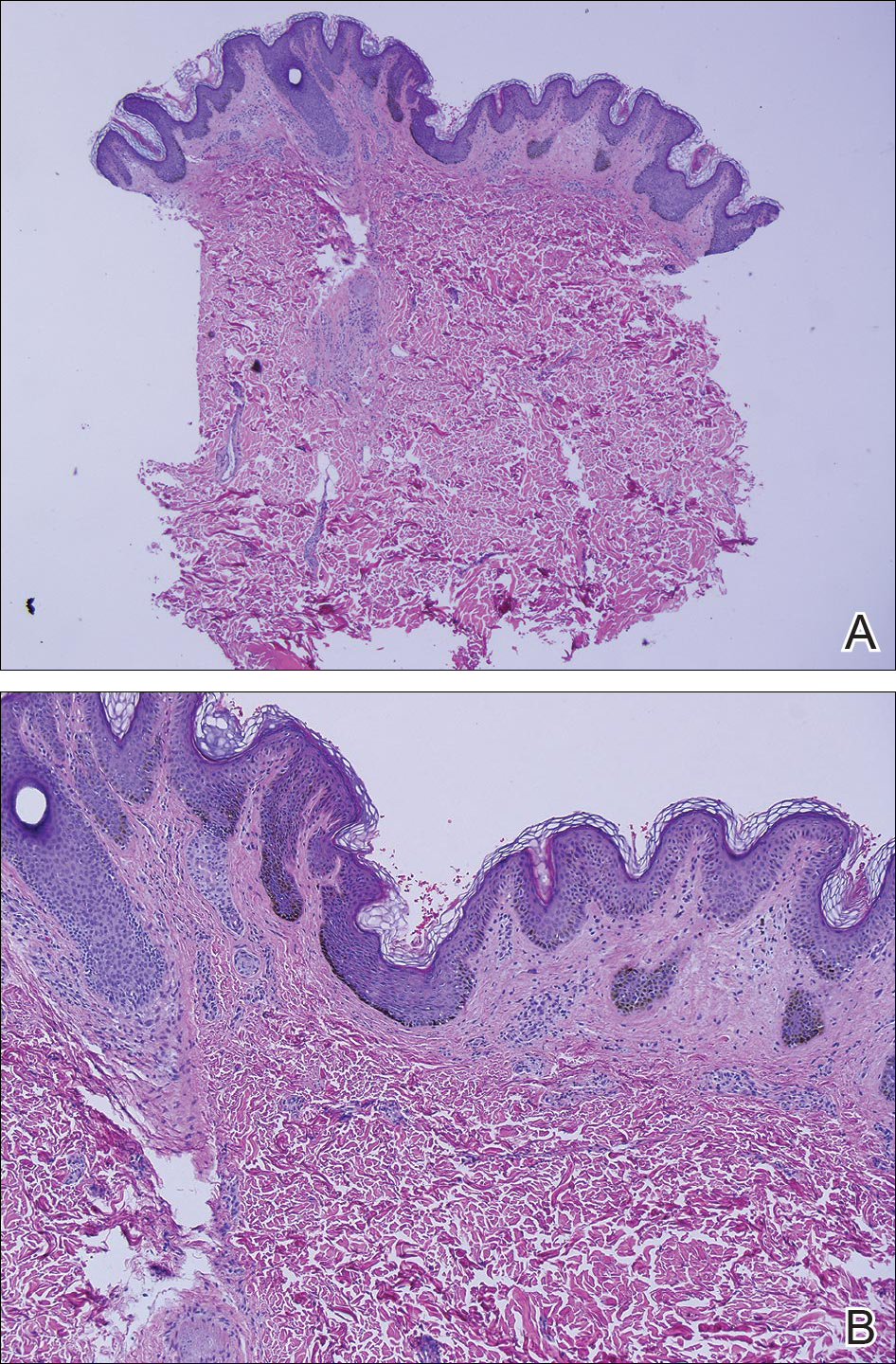
Comment
Epidermal nevus syndrome is a group of disorders characterized by both local or systematized epidermal nevi and systemic findings. Solomon et al4 first coined the term epidermal nevus syndrome more than 40 years ago; however, since then there has been confusion about how to define ENS. Epidermal nevus syndrome has been considered an umbrella term that includes more specific syndromes involving epidermal nevi, such as Proteus syndrome and Schimmelpenning syndrome; conversely, it also has been considered a term for those who do not meet the criteria for more specific syndromes.1,9 Happle1 discussed that the genetic variations found in ENS warrant recognition. Simply put, ENS is a heterogeneous group of syndromes that are similar in that they involve epidermal nevi and internal abnormalities but are genetically distinct. The list of definitive ENSs, as suggested by Happle1 and others, will likely continue to grow.3,5
The exact pathomechanism of ENS is unknown, but the clinical presentation most likely represents a lethal disorder mitigated by mosaicism.2,9 Gene defects vary depending on the specific ENS. For instance, the phosphatase and tensin homolog gene, PTEN, mutations have been associated with type 2 segmental Cowden disease. Fibroblast growth factor receptor 3, FGFR3, mutations have been linked to Garcia-Hafner-Happle syndrome.3FGFR3 mutations have been found in nonepidermolytic epidermal nevi, and some suggest that the majority of epidermal nevi exhibit mutations in FGFR3.5,10,11 On the other hand, other gene defects have not been elucidated, such as in Schimmelpenning syndrome.3
Clinically, ENS may involve nonepidermolytic verrucous nevi, sebaceous nevi, organoid nevi, linear Cowden nevi, and woolly hair nevi. Lesions may be flesh-colored, pink, yellow, or hyperpigmented plaques in a blaschkoid distribution and may be localized or systematized. Nevi typically are present at birth or develop within the first year of life.9,12,13 Other cutaneous findings may be noted apart from epidermal nevi, including melanocytic nevi, aplasia cutis congenita, and hemangiomas.13,14
Extracutaneous findings include central nervous system, skeletal, ocular, cardiac, and genitourinary defects, which are often observed in these patients.3,9,13,14 Central nervous system findings are seen in 50% to 70% of cases, with seizures and mental retardation among the most common.13-15 Genitourinary abnormalities associated with epidermal nevi, including horseshoe kidney, cystic kidney, duplicated collecting system, testicular and paratesticular tumors, and hypospadias have been documented in the literature.16 Our patient had a history of papillary transitional cell bladder carcinoma, which is rare for a patient younger than 30 years. The overall median age of diagnosis of bladder cancer is 65 years, and it is more common in men than in women.17 Transitional cell carcinomas account for approximately 90% of all bladder cancers in the United States. Other common types of bladder cancer include squamous cell carcinoma, adenocarcinoma, and rhabdomyosarcoma.16 Typically, transitional cell carcinoma is associated with smoking, exposure to aniline dyes, cyclophosphamide, and living in industrialized areas.16,17 Individuals who work with textiles, dyes, leather, tires, rubber, and/or petroleum; painters; truck drivers; drill press operators; and hairdressers are at an increased risk for development of bladder cancer.16
Interestingly, it has been shown in some studies that papillary transitional cell bladder carcinoma frequently is associated with FGFR3 mutations, which may be the missing link in the rare finding of papillary transitional cell bladder carcinoma and epidermal nevi.5,18,19 In addition, PTEN mutations also have been identified in low-grade papillary transitional cell carcinomas of the bladder, another gene linked to an ENS with type 2 segmental Cowden disease.3,20
Histopathologically, epidermal nevi have 10 different descriptions. Our patient had a nonorganoid nonepidermolytic epidermal nevus characterized by hyperkeratosis, acanthosis, papillomatosis, and elongated rete ridges. Focal acantholysis and epidermolytic hyperkeratosis also is seen in some epidermal nevi but was not seen in this case.9,21
Simple epidermal nevi occur in approximately 1 in 1000 newborns; however, when a child presents with multiple or systematized epidermal nevi, investigation should be undertaken for other possible associations.13,14 Of note, there have been several cases of squamous cell, verrucous, basal cell, and adnexal carcinomas arising in linear epidermal nevi.22-24
Epidermal nevi can be difficult to treat. Some patients are troubled by the appearance of these nevi, especially those with systematized disease. Unfortunately, for patients with multiple nevi or systematized disease, there are no consistently effective treatment options; however, there are case reports25,26 in the literature citing improvement or cure of epidermal nevi with full-thickness excision, continuous and pulsed CO2 laser, pulsed dye laser, and erbium-doped YAG laser.25 Other therapies that have been purported to help improve epidermal nevi are topical and oral retinoids, corticosteroids, topical 5-fluorouracil, anthralin, and podophyllin.26
Conclusion
Transitional cell bladder carcinoma is rare in patients in the third decade of life and younger. Given the age of our patient and her concomitant lack of risk factors, such as older age, history of smoking, and exposure to certain chemicals (eg, aniline dyes) and medications (eg, cyclophosphamide), it is more likely that the finding of papillary transitional cell bladder carcinoma and ENS are related. A clear genetic link between ENS and transitional cell papillary bladder carcinoma has yet to be elucidated, but the FGFR3 gene is promising.
- Happle R. What is a nevus? a proposed definition of a common medical term. Dermatology. 1995;191:1-5.
- Gonzalez ME, Jabbari A, Tlougan BE, et al. Epidermal nevus. Dermatol Online J. 2010;16:12.
- Happle R. The group of epidermal nevus syndromes. part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22.
- Solomon LM, Fretzin DF, Dewald RL. The epidermal nevus syndrome. Arch Dermatol. 1968;97:273-285.
- Flosadottir E, Bjarnason B. A non-epidermolytic epidermal nevus of a soft, papillomatous type with transitional cell cancer of the bladder: a case report and review of non-cutaneous cancers associated with epidermal naevi. Acta Derm Venerol. 2008;88:173-175.
- Rosenthal D, Fretzin DF. Epidermal nevus syndrome: report of association with transitional cell carcinoma of the bladder. Pediatr Dermatol. 1986;3:455-458.
- Garcia de Jalon A, Azua-Romea J, Trivez MA, et al. Epidermal naevus syndrome (Solomon’s syndrome) associated with bladder cancer in a 20-year-old female. Scand J Urol Nephrol. 2004;38:85-87.
- Rongioletti F, Rebora A. Epidermal nevus with transitional cell carcinomas of the urinary tract. J Am Acad Dermatol. 1991;25:856-858.
- Moss C. Mosacism and linear lesions. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:943-962.
- Hafner C, van Oers JM, Vogt T, et al. Mosaicisim of activating FGFR3 mutations in human skin causes epidermal nevi. J Clin Invest. 2006;116:2201-2207.
- Bygum A, Fagerberg CR, Clemmensen OJ, et al. Systemic epidermal nevus with involvement of the oral mucosa due to FGFR3 mutation. BMC Med Genet. 2011;12:79.
- Happle R. Linear Cowden nevus: a new distinct epidermal nevus. Eur J Dermatol. 2007;17:133-136.
- Vujevich JJ, Mancini AJ. The epidermal nevus syndromes: multisystem disorders. J Am Acad Dermatol. 2004;50:957-961.
- Solomon L, Esterly N. Epidermal and other congenital organoid nevi. Curr Probl Pediatr. 1975;6:1-56.
- Grebe TA, Rimsa ME, Richter SF, et al. Further delineation of the epidermal nevus syndrome: two cases with new findings and literature review. Am J Med Genet. 1993;47:24-30.
- Lamm DL, Torti FM. Bladder cancer, 1996. Ca Cancer J Clin. 1996;46:93-112.
- Metts MC, Metts JC, Milito SJ, et al. Bladder cancer: a review of diagnosis and management. J Natl Med Assoc. 2000;92:285-294.
- Kimura T, Suzuki H, Ohashi T, et al. The incidence of thanatophoric dysplasia mutations in FGFR3 gene is higher in low-grade or superficial bladder carcinomas. Cancer. 2001;92:2555-2561.
- Cappellen D, DeOliveira C, Ricol D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18-20.
- Knowles MA, Platt FM, Ross RL, et al. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305-316.
- Luzar B, Calonje E, Bastian B. Tumors of the surface epithelium. In: Calonje JE, Breen T, McKee PH, eds. McKee’s Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012:1076-1149.
- Masood Q, Narayan D. Squamous cell carcinoma in a linear epidermal nevus. J Plast Reconstr Aesthet Surg. 2009;62:693-694.
- Cramer SF, Mandel MA, Hauler R, et al. Squamous cell carcinoma arising in a linear epidermal nevus. Arch Dermatol. 1981;117:222-224.
- Affleck AG, Leach IJ, Varma S. Two squamous cell carcinomas arising in a linear epidermal nevus in a 28-year-old female. Clin Exp Dermatol. 2005;30:382-384.
- Alam M, Arndt KA. A method for pulsed carbon dioxide laser treatment of epidermal nevi. J Am Acad Dermatol. 2002;46:554-556.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:1809-1810.
Epidermal nevi can occur in isolation or in association with internal abnormalities. Epidermal nevus syndrome (ENS) is a heterogeneous group of neurocutaneous disorders characterized by mosaicism and epidermal nevi found in association with various systemic abnormalities.1-4 There are many possible associated systemic findings, including abnormalities of the central nervous, musculoskeletal, renal, and hematologic systems. Epidermal nevi have been associated with internal malignancies. We present the case of a patient with epidermal nevi associated with papillary transitional cell bladder carcinoma. According to a PubMed search of articles indexed for MEDLINE using the search terms transitional cell bladder carcinoma and epidermal nevus, there have only been 4 other cases of transitional cell bladder carcinoma and ENS reported in the literature,5-8 2 of which were reports of papillary transitional cell bladder carcinoma.5,6
Case Report
A 29-year-old woman presented to our clinic with a rash that had been present since 3 years of age. The emergency department consulted dermatology for evaluation of what was believed to be contact dermatitis; however, upon questioning the patient, it was revealed that the rash was chronic and persistent.
The rash was nonpruritic and was located on the face, hands (Figure 1), chest, buttocks, thighs, legs, and back (Figure 2). Although asymptomatic, the appearance of the skin caused the patient some emotional distress. As a child she had been evaluated by a dermatologist and a biopsy was performed, but she did not recall the results or have any records. She had been prescribed an oral medication by the dermatologist, but treatment was terminated early due to nausea. The skin lesions did not improve with the short course of treatment.
Eighteen months prior to presentation to our clinic, the patient was discovered to have hematuria on routine examination by her primary care physician. At that time, the patient underwent a workup for hematuria and a mass was discovered in the bladder via cystoscopy. A diagnosis of low-grade papillary transitional cell bladder carcinoma was made, and she underwent a partial cystectomy. No radiation or chemotherapy was required. The remainder of her medical history was only remarkable for asthma, which was well controlled with albuterol. On examination, generalized, hyperpigmented, reticulated patches, macules, and hyperpigmented verrucous plaques were distributed along the Blaschko lines, sparing the face. No limb abnormalities or dental or nail abnormalities were noted. Examination of the axillary and cervical lymph nodes was unremarkable, and no neurological abnormalities were noted. A 3-mm punch biopsy of the mid upper back was performed. Histopathology revealed papillomatous, nonorganoid, nonepidermolytic hyperplasia of the epidermis with elongated rete ridges (Figure 3), which was diagnosed as a nonorganoid nonepidermolytic epidermal nevus.
Comment
Epidermal nevus syndrome is a group of disorders characterized by both local or systematized epidermal nevi and systemic findings. Solomon et al4 first coined the term epidermal nevus syndrome more than 40 years ago; however, since then there has been confusion about how to define ENS. Epidermal nevus syndrome has been considered an umbrella term that includes more specific syndromes involving epidermal nevi, such as Proteus syndrome and Schimmelpenning syndrome; conversely, it also has been considered a term for those who do not meet the criteria for more specific syndromes.1,9 Happle1 discussed that the genetic variations found in ENS warrant recognition. Simply put, ENS is a heterogeneous group of syndromes that are similar in that they involve epidermal nevi and internal abnormalities but are genetically distinct. The list of definitive ENSs, as suggested by Happle1 and others, will likely continue to grow.3,5
The exact pathomechanism of ENS is unknown, but the clinical presentation most likely represents a lethal disorder mitigated by mosaicism.2,9 Gene defects vary depending on the specific ENS. For instance, the phosphatase and tensin homolog gene, PTEN, mutations have been associated with type 2 segmental Cowden disease. Fibroblast growth factor receptor 3, FGFR3, mutations have been linked to Garcia-Hafner-Happle syndrome.3FGFR3 mutations have been found in nonepidermolytic epidermal nevi, and some suggest that the majority of epidermal nevi exhibit mutations in FGFR3.5,10,11 On the other hand, other gene defects have not been elucidated, such as in Schimmelpenning syndrome.3
Clinically, ENS may involve nonepidermolytic verrucous nevi, sebaceous nevi, organoid nevi, linear Cowden nevi, and woolly hair nevi. Lesions may be flesh-colored, pink, yellow, or hyperpigmented plaques in a blaschkoid distribution and may be localized or systematized. Nevi typically are present at birth or develop within the first year of life.9,12,13 Other cutaneous findings may be noted apart from epidermal nevi, including melanocytic nevi, aplasia cutis congenita, and hemangiomas.13,14
Extracutaneous findings include central nervous system, skeletal, ocular, cardiac, and genitourinary defects, which are often observed in these patients.3,9,13,14 Central nervous system findings are seen in 50% to 70% of cases, with seizures and mental retardation among the most common.13-15 Genitourinary abnormalities associated with epidermal nevi, including horseshoe kidney, cystic kidney, duplicated collecting system, testicular and paratesticular tumors, and hypospadias have been documented in the literature.16 Our patient had a history of papillary transitional cell bladder carcinoma, which is rare for a patient younger than 30 years. The overall median age of diagnosis of bladder cancer is 65 years, and it is more common in men than in women.17 Transitional cell carcinomas account for approximately 90% of all bladder cancers in the United States. Other common types of bladder cancer include squamous cell carcinoma, adenocarcinoma, and rhabdomyosarcoma.16 Typically, transitional cell carcinoma is associated with smoking, exposure to aniline dyes, cyclophosphamide, and living in industrialized areas.16,17 Individuals who work with textiles, dyes, leather, tires, rubber, and/or petroleum; painters; truck drivers; drill press operators; and hairdressers are at an increased risk for development of bladder cancer.16
Interestingly, it has been shown in some studies that papillary transitional cell bladder carcinoma frequently is associated with FGFR3 mutations, which may be the missing link in the rare finding of papillary transitional cell bladder carcinoma and epidermal nevi.5,18,19 In addition, PTEN mutations also have been identified in low-grade papillary transitional cell carcinomas of the bladder, another gene linked to an ENS with type 2 segmental Cowden disease.3,20
Histopathologically, epidermal nevi have 10 different descriptions. Our patient had a nonorganoid nonepidermolytic epidermal nevus characterized by hyperkeratosis, acanthosis, papillomatosis, and elongated rete ridges. Focal acantholysis and epidermolytic hyperkeratosis also is seen in some epidermal nevi but was not seen in this case.9,21
Simple epidermal nevi occur in approximately 1 in 1000 newborns; however, when a child presents with multiple or systematized epidermal nevi, investigation should be undertaken for other possible associations.13,14 Of note, there have been several cases of squamous cell, verrucous, basal cell, and adnexal carcinomas arising in linear epidermal nevi.22-24
Epidermal nevi can be difficult to treat. Some patients are troubled by the appearance of these nevi, especially those with systematized disease. Unfortunately, for patients with multiple nevi or systematized disease, there are no consistently effective treatment options; however, there are case reports25,26 in the literature citing improvement or cure of epidermal nevi with full-thickness excision, continuous and pulsed CO2 laser, pulsed dye laser, and erbium-doped YAG laser.25 Other therapies that have been purported to help improve epidermal nevi are topical and oral retinoids, corticosteroids, topical 5-fluorouracil, anthralin, and podophyllin.26
Conclusion
Transitional cell bladder carcinoma is rare in patients in the third decade of life and younger. Given the age of our patient and her concomitant lack of risk factors, such as older age, history of smoking, and exposure to certain chemicals (eg, aniline dyes) and medications (eg, cyclophosphamide), it is more likely that the finding of papillary transitional cell bladder carcinoma and ENS are related. A clear genetic link between ENS and transitional cell papillary bladder carcinoma has yet to be elucidated, but the FGFR3 gene is promising.
Epidermal nevi can occur in isolation or in association with internal abnormalities. Epidermal nevus syndrome (ENS) is a heterogeneous group of neurocutaneous disorders characterized by mosaicism and epidermal nevi found in association with various systemic abnormalities.1-4 There are many possible associated systemic findings, including abnormalities of the central nervous, musculoskeletal, renal, and hematologic systems. Epidermal nevi have been associated with internal malignancies. We present the case of a patient with epidermal nevi associated with papillary transitional cell bladder carcinoma. According to a PubMed search of articles indexed for MEDLINE using the search terms transitional cell bladder carcinoma and epidermal nevus, there have only been 4 other cases of transitional cell bladder carcinoma and ENS reported in the literature,5-8 2 of which were reports of papillary transitional cell bladder carcinoma.5,6
Case Report
A 29-year-old woman presented to our clinic with a rash that had been present since 3 years of age. The emergency department consulted dermatology for evaluation of what was believed to be contact dermatitis; however, upon questioning the patient, it was revealed that the rash was chronic and persistent.
The rash was nonpruritic and was located on the face, hands (Figure 1), chest, buttocks, thighs, legs, and back (Figure 2). Although asymptomatic, the appearance of the skin caused the patient some emotional distress. As a child she had been evaluated by a dermatologist and a biopsy was performed, but she did not recall the results or have any records. She had been prescribed an oral medication by the dermatologist, but treatment was terminated early due to nausea. The skin lesions did not improve with the short course of treatment.
Eighteen months prior to presentation to our clinic, the patient was discovered to have hematuria on routine examination by her primary care physician. At that time, the patient underwent a workup for hematuria and a mass was discovered in the bladder via cystoscopy. A diagnosis of low-grade papillary transitional cell bladder carcinoma was made, and she underwent a partial cystectomy. No radiation or chemotherapy was required. The remainder of her medical history was only remarkable for asthma, which was well controlled with albuterol. On examination, generalized, hyperpigmented, reticulated patches, macules, and hyperpigmented verrucous plaques were distributed along the Blaschko lines, sparing the face. No limb abnormalities or dental or nail abnormalities were noted. Examination of the axillary and cervical lymph nodes was unremarkable, and no neurological abnormalities were noted. A 3-mm punch biopsy of the mid upper back was performed. Histopathology revealed papillomatous, nonorganoid, nonepidermolytic hyperplasia of the epidermis with elongated rete ridges (Figure 3), which was diagnosed as a nonorganoid nonepidermolytic epidermal nevus.
Comment
Epidermal nevus syndrome is a group of disorders characterized by both local or systematized epidermal nevi and systemic findings. Solomon et al4 first coined the term epidermal nevus syndrome more than 40 years ago; however, since then there has been confusion about how to define ENS. Epidermal nevus syndrome has been considered an umbrella term that includes more specific syndromes involving epidermal nevi, such as Proteus syndrome and Schimmelpenning syndrome; conversely, it also has been considered a term for those who do not meet the criteria for more specific syndromes.1,9 Happle1 discussed that the genetic variations found in ENS warrant recognition. Simply put, ENS is a heterogeneous group of syndromes that are similar in that they involve epidermal nevi and internal abnormalities but are genetically distinct. The list of definitive ENSs, as suggested by Happle1 and others, will likely continue to grow.3,5
The exact pathomechanism of ENS is unknown, but the clinical presentation most likely represents a lethal disorder mitigated by mosaicism.2,9 Gene defects vary depending on the specific ENS. For instance, the phosphatase and tensin homolog gene, PTEN, mutations have been associated with type 2 segmental Cowden disease. Fibroblast growth factor receptor 3, FGFR3, mutations have been linked to Garcia-Hafner-Happle syndrome.3FGFR3 mutations have been found in nonepidermolytic epidermal nevi, and some suggest that the majority of epidermal nevi exhibit mutations in FGFR3.5,10,11 On the other hand, other gene defects have not been elucidated, such as in Schimmelpenning syndrome.3
Clinically, ENS may involve nonepidermolytic verrucous nevi, sebaceous nevi, organoid nevi, linear Cowden nevi, and woolly hair nevi. Lesions may be flesh-colored, pink, yellow, or hyperpigmented plaques in a blaschkoid distribution and may be localized or systematized. Nevi typically are present at birth or develop within the first year of life.9,12,13 Other cutaneous findings may be noted apart from epidermal nevi, including melanocytic nevi, aplasia cutis congenita, and hemangiomas.13,14
Extracutaneous findings include central nervous system, skeletal, ocular, cardiac, and genitourinary defects, which are often observed in these patients.3,9,13,14 Central nervous system findings are seen in 50% to 70% of cases, with seizures and mental retardation among the most common.13-15 Genitourinary abnormalities associated with epidermal nevi, including horseshoe kidney, cystic kidney, duplicated collecting system, testicular and paratesticular tumors, and hypospadias have been documented in the literature.16 Our patient had a history of papillary transitional cell bladder carcinoma, which is rare for a patient younger than 30 years. The overall median age of diagnosis of bladder cancer is 65 years, and it is more common in men than in women.17 Transitional cell carcinomas account for approximately 90% of all bladder cancers in the United States. Other common types of bladder cancer include squamous cell carcinoma, adenocarcinoma, and rhabdomyosarcoma.16 Typically, transitional cell carcinoma is associated with smoking, exposure to aniline dyes, cyclophosphamide, and living in industrialized areas.16,17 Individuals who work with textiles, dyes, leather, tires, rubber, and/or petroleum; painters; truck drivers; drill press operators; and hairdressers are at an increased risk for development of bladder cancer.16
Interestingly, it has been shown in some studies that papillary transitional cell bladder carcinoma frequently is associated with FGFR3 mutations, which may be the missing link in the rare finding of papillary transitional cell bladder carcinoma and epidermal nevi.5,18,19 In addition, PTEN mutations also have been identified in low-grade papillary transitional cell carcinomas of the bladder, another gene linked to an ENS with type 2 segmental Cowden disease.3,20
Histopathologically, epidermal nevi have 10 different descriptions. Our patient had a nonorganoid nonepidermolytic epidermal nevus characterized by hyperkeratosis, acanthosis, papillomatosis, and elongated rete ridges. Focal acantholysis and epidermolytic hyperkeratosis also is seen in some epidermal nevi but was not seen in this case.9,21
Simple epidermal nevi occur in approximately 1 in 1000 newborns; however, when a child presents with multiple or systematized epidermal nevi, investigation should be undertaken for other possible associations.13,14 Of note, there have been several cases of squamous cell, verrucous, basal cell, and adnexal carcinomas arising in linear epidermal nevi.22-24
Epidermal nevi can be difficult to treat. Some patients are troubled by the appearance of these nevi, especially those with systematized disease. Unfortunately, for patients with multiple nevi or systematized disease, there are no consistently effective treatment options; however, there are case reports25,26 in the literature citing improvement or cure of epidermal nevi with full-thickness excision, continuous and pulsed CO2 laser, pulsed dye laser, and erbium-doped YAG laser.25 Other therapies that have been purported to help improve epidermal nevi are topical and oral retinoids, corticosteroids, topical 5-fluorouracil, anthralin, and podophyllin.26
Conclusion
Transitional cell bladder carcinoma is rare in patients in the third decade of life and younger. Given the age of our patient and her concomitant lack of risk factors, such as older age, history of smoking, and exposure to certain chemicals (eg, aniline dyes) and medications (eg, cyclophosphamide), it is more likely that the finding of papillary transitional cell bladder carcinoma and ENS are related. A clear genetic link between ENS and transitional cell papillary bladder carcinoma has yet to be elucidated, but the FGFR3 gene is promising.
- Happle R. What is a nevus? a proposed definition of a common medical term. Dermatology. 1995;191:1-5.
- Gonzalez ME, Jabbari A, Tlougan BE, et al. Epidermal nevus. Dermatol Online J. 2010;16:12.
- Happle R. The group of epidermal nevus syndromes. part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22.
- Solomon LM, Fretzin DF, Dewald RL. The epidermal nevus syndrome. Arch Dermatol. 1968;97:273-285.
- Flosadottir E, Bjarnason B. A non-epidermolytic epidermal nevus of a soft, papillomatous type with transitional cell cancer of the bladder: a case report and review of non-cutaneous cancers associated with epidermal naevi. Acta Derm Venerol. 2008;88:173-175.
- Rosenthal D, Fretzin DF. Epidermal nevus syndrome: report of association with transitional cell carcinoma of the bladder. Pediatr Dermatol. 1986;3:455-458.
- Garcia de Jalon A, Azua-Romea J, Trivez MA, et al. Epidermal naevus syndrome (Solomon’s syndrome) associated with bladder cancer in a 20-year-old female. Scand J Urol Nephrol. 2004;38:85-87.
- Rongioletti F, Rebora A. Epidermal nevus with transitional cell carcinomas of the urinary tract. J Am Acad Dermatol. 1991;25:856-858.
- Moss C. Mosacism and linear lesions. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:943-962.
- Hafner C, van Oers JM, Vogt T, et al. Mosaicisim of activating FGFR3 mutations in human skin causes epidermal nevi. J Clin Invest. 2006;116:2201-2207.
- Bygum A, Fagerberg CR, Clemmensen OJ, et al. Systemic epidermal nevus with involvement of the oral mucosa due to FGFR3 mutation. BMC Med Genet. 2011;12:79.
- Happle R. Linear Cowden nevus: a new distinct epidermal nevus. Eur J Dermatol. 2007;17:133-136.
- Vujevich JJ, Mancini AJ. The epidermal nevus syndromes: multisystem disorders. J Am Acad Dermatol. 2004;50:957-961.
- Solomon L, Esterly N. Epidermal and other congenital organoid nevi. Curr Probl Pediatr. 1975;6:1-56.
- Grebe TA, Rimsa ME, Richter SF, et al. Further delineation of the epidermal nevus syndrome: two cases with new findings and literature review. Am J Med Genet. 1993;47:24-30.
- Lamm DL, Torti FM. Bladder cancer, 1996. Ca Cancer J Clin. 1996;46:93-112.
- Metts MC, Metts JC, Milito SJ, et al. Bladder cancer: a review of diagnosis and management. J Natl Med Assoc. 2000;92:285-294.
- Kimura T, Suzuki H, Ohashi T, et al. The incidence of thanatophoric dysplasia mutations in FGFR3 gene is higher in low-grade or superficial bladder carcinomas. Cancer. 2001;92:2555-2561.
- Cappellen D, DeOliveira C, Ricol D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18-20.
- Knowles MA, Platt FM, Ross RL, et al. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305-316.
- Luzar B, Calonje E, Bastian B. Tumors of the surface epithelium. In: Calonje JE, Breen T, McKee PH, eds. McKee’s Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012:1076-1149.
- Masood Q, Narayan D. Squamous cell carcinoma in a linear epidermal nevus. J Plast Reconstr Aesthet Surg. 2009;62:693-694.
- Cramer SF, Mandel MA, Hauler R, et al. Squamous cell carcinoma arising in a linear epidermal nevus. Arch Dermatol. 1981;117:222-224.
- Affleck AG, Leach IJ, Varma S. Two squamous cell carcinomas arising in a linear epidermal nevus in a 28-year-old female. Clin Exp Dermatol. 2005;30:382-384.
- Alam M, Arndt KA. A method for pulsed carbon dioxide laser treatment of epidermal nevi. J Am Acad Dermatol. 2002;46:554-556.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:1809-1810.
- Happle R. What is a nevus? a proposed definition of a common medical term. Dermatology. 1995;191:1-5.
- Gonzalez ME, Jabbari A, Tlougan BE, et al. Epidermal nevus. Dermatol Online J. 2010;16:12.
- Happle R. The group of epidermal nevus syndromes. part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22.
- Solomon LM, Fretzin DF, Dewald RL. The epidermal nevus syndrome. Arch Dermatol. 1968;97:273-285.
- Flosadottir E, Bjarnason B. A non-epidermolytic epidermal nevus of a soft, papillomatous type with transitional cell cancer of the bladder: a case report and review of non-cutaneous cancers associated with epidermal naevi. Acta Derm Venerol. 2008;88:173-175.
- Rosenthal D, Fretzin DF. Epidermal nevus syndrome: report of association with transitional cell carcinoma of the bladder. Pediatr Dermatol. 1986;3:455-458.
- Garcia de Jalon A, Azua-Romea J, Trivez MA, et al. Epidermal naevus syndrome (Solomon’s syndrome) associated with bladder cancer in a 20-year-old female. Scand J Urol Nephrol. 2004;38:85-87.
- Rongioletti F, Rebora A. Epidermal nevus with transitional cell carcinomas of the urinary tract. J Am Acad Dermatol. 1991;25:856-858.
- Moss C. Mosacism and linear lesions. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:943-962.
- Hafner C, van Oers JM, Vogt T, et al. Mosaicisim of activating FGFR3 mutations in human skin causes epidermal nevi. J Clin Invest. 2006;116:2201-2207.
- Bygum A, Fagerberg CR, Clemmensen OJ, et al. Systemic epidermal nevus with involvement of the oral mucosa due to FGFR3 mutation. BMC Med Genet. 2011;12:79.
- Happle R. Linear Cowden nevus: a new distinct epidermal nevus. Eur J Dermatol. 2007;17:133-136.
- Vujevich JJ, Mancini AJ. The epidermal nevus syndromes: multisystem disorders. J Am Acad Dermatol. 2004;50:957-961.
- Solomon L, Esterly N. Epidermal and other congenital organoid nevi. Curr Probl Pediatr. 1975;6:1-56.
- Grebe TA, Rimsa ME, Richter SF, et al. Further delineation of the epidermal nevus syndrome: two cases with new findings and literature review. Am J Med Genet. 1993;47:24-30.
- Lamm DL, Torti FM. Bladder cancer, 1996. Ca Cancer J Clin. 1996;46:93-112.
- Metts MC, Metts JC, Milito SJ, et al. Bladder cancer: a review of diagnosis and management. J Natl Med Assoc. 2000;92:285-294.
- Kimura T, Suzuki H, Ohashi T, et al. The incidence of thanatophoric dysplasia mutations in FGFR3 gene is higher in low-grade or superficial bladder carcinomas. Cancer. 2001;92:2555-2561.
- Cappellen D, DeOliveira C, Ricol D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18-20.
- Knowles MA, Platt FM, Ross RL, et al. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305-316.
- Luzar B, Calonje E, Bastian B. Tumors of the surface epithelium. In: Calonje JE, Breen T, McKee PH, eds. McKee’s Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012:1076-1149.
- Masood Q, Narayan D. Squamous cell carcinoma in a linear epidermal nevus. J Plast Reconstr Aesthet Surg. 2009;62:693-694.
- Cramer SF, Mandel MA, Hauler R, et al. Squamous cell carcinoma arising in a linear epidermal nevus. Arch Dermatol. 1981;117:222-224.
- Affleck AG, Leach IJ, Varma S. Two squamous cell carcinomas arising in a linear epidermal nevus in a 28-year-old female. Clin Exp Dermatol. 2005;30:382-384.
- Alam M, Arndt KA. A method for pulsed carbon dioxide laser treatment of epidermal nevi. J Am Acad Dermatol. 2002;46:554-556.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:1809-1810.
Practice Points
- Epidermal nevi are common benign cutaneous neoplasms.
- Extensive systematized epidermal nevi can be a sign of internal disease.
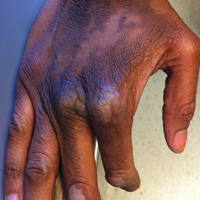
Firm, non-tender mass in right breast • worsening, nonproductive cough • pleuritic pain • Dx?
THE CASE
A 44-year-old woman with a 15-year history of type 2 diabetes sought care for a firm, non-tender mass in the medial lower quadrant of her right breast. She hadn’t experienced any skin changes or axillary lymphadenopathy. The patient had immigrated to California from Afghanistan 22 years earlier, at which time she was briefly married to an Afghan man suffering from a chronic cough.
Mammography revealed a 3.5 x 4 x 4 cm lesion at the chest wall, which was highly suspicious for carcinoma (FIGURES 1A AND 1B). Sonography showed a heterogenous hypoechoic and isoechoic mass with posterior acoustic enhancement (FIGURE 1C). An excisional biopsy was performed.
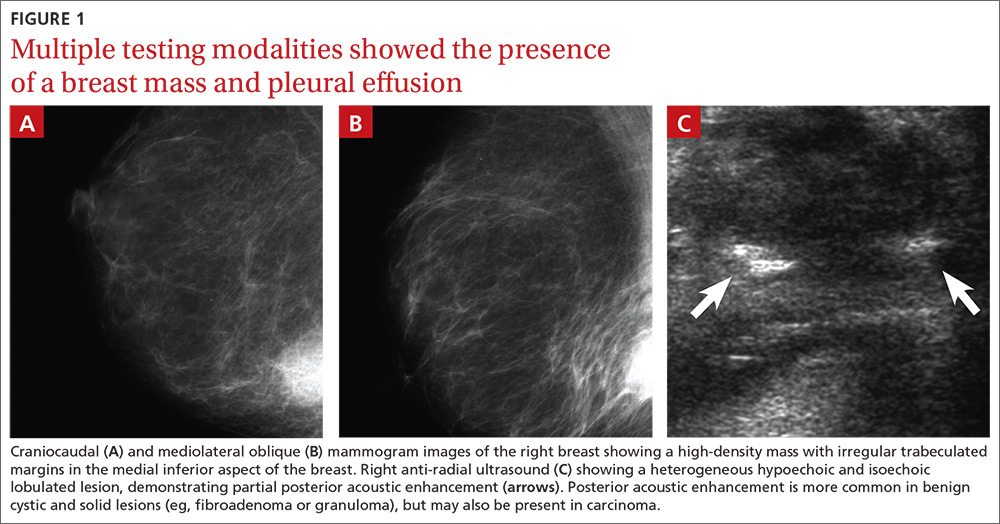
One week postoperatively, the patient presented to the emergency department for a worsening nonproductive cough that intensified when supine, and was associated with subscapular pleuritic pain. She denied fever or weight loss. Biopsy results were pending.
THE DIAGNOSIS
Chest x-rays revealed a large right pleural effusion that was presumed to be malignant (FIGURES 1D AND 1E). Thoracentesis yielded 1.5 liters of tea-colored exudate containing 2800 nucleated cells/mL—63% lymphocytes and 37% neutrophils—and a pleural fluid to serum protein ratio >0.5. Adenosine deaminase was <1 U/L. Fluid Gram stain, acid-fast bacillus (AFB) fluorescent antibody testing, AFB cultures, and cytology were negative. Computed tomography (CT) subsequently demonstrated recurrent effusion without hilar or mediastinal lymphadenopathy or pleural enhancement (FIGURE 1F).
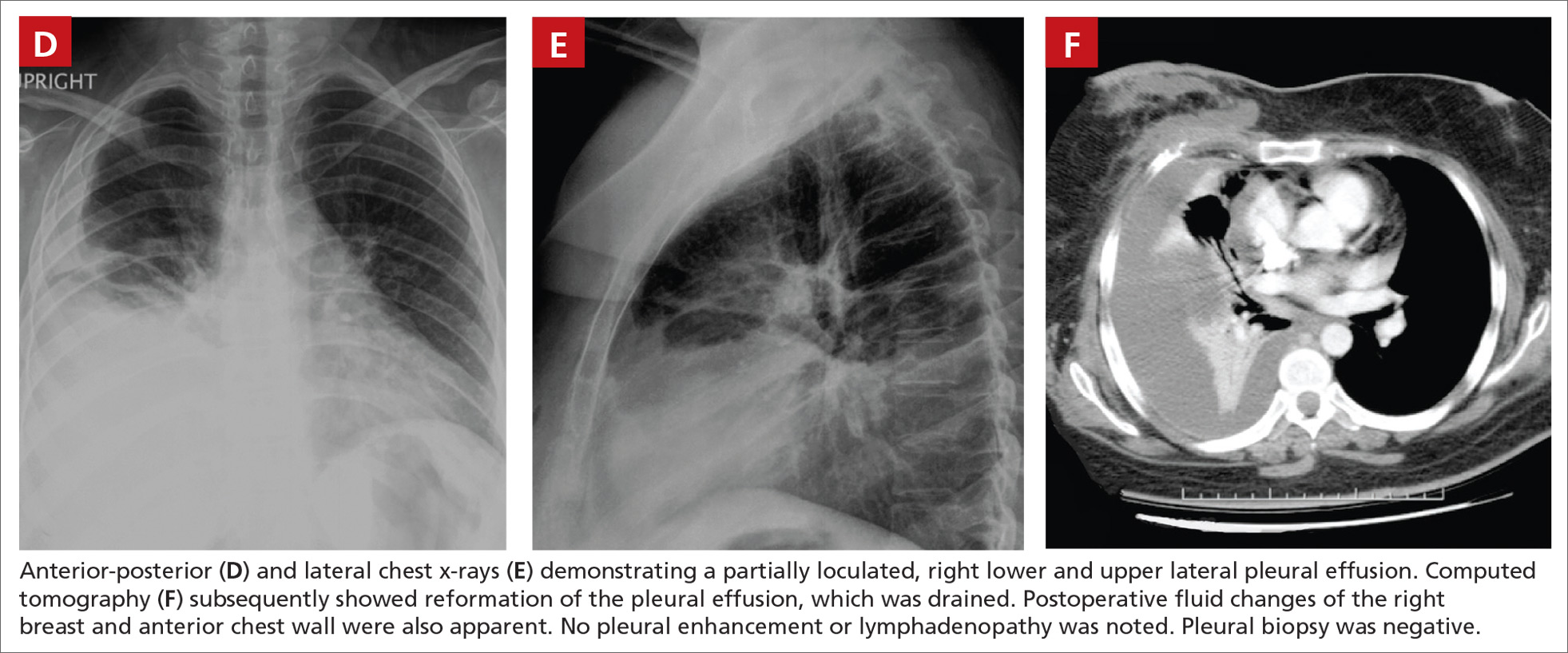
Histologically, the breast mass showed caseating granulomatous inflammation (FIGURES 1G AND 1H). An AFB stain was negative. Polymerase chain reaction (PCR) performed on DNA extracted from the formalin-fixed, paraffin-embedded biopsy material was positive for Mycobacterium tuberculosis.1 A CT-guided pleural biopsy showed only normal tissue. A follow-up tuberculin skin test (purified protein derivative [PPD]) yielded a 10-mm indurated reaction.
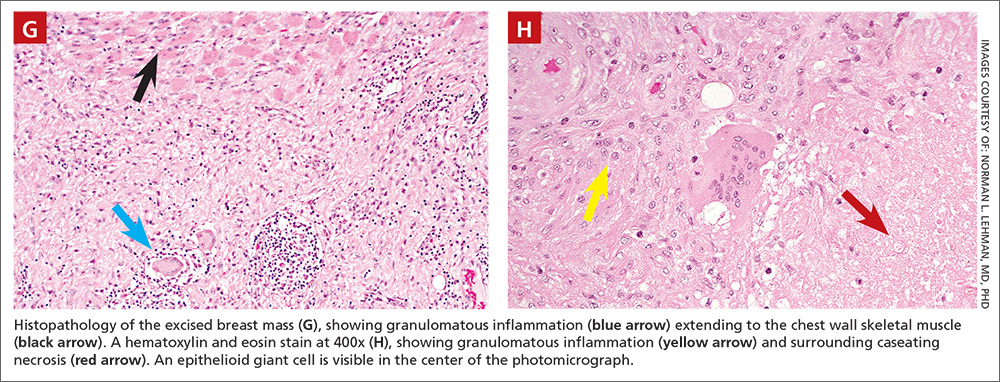
DISCUSSION
Granulomatous lesions, such as foreign body granuloma, idiopathic granulomatous mastitis (IGM), and sarcoidosis can mimic breast carcinoma.2,3 IGM is associated with elevated prolactin (eg, pregnancy or oral contraceptive use) and is usually subareolar.2 Infection, however, is also commonly subareolar. Sarcoidosis rarely exhibits unilateral pleural effusion and usually manifests with bilateral interstitial lung disease, hilar lymphadenopathy, and non-necrotizing granulomas.3,4
M tuberculosis and other granulomatous infections may also feign breast cancer.5-13 Breast TB, which is highly uncommon in the developed world, often demonstrates imaging similar to that which was seen in this case. Breast TB may appear nodular with ill-defined contours. Masses are sometimes attached to the chest wall and usually lack microcalcifications on mammography; they are also typically hypoechoic and heterogenous on ultrasound, often showing posterior enhancement.5,7,8 Like other breast infections, tuberculosis may show cutaneous sinus tract formation, which is seen in about one-third of patients.6,7 Alternatively, it may manifest as a diffuse mastitis with skin thickening and axillary lymphadenopathy.8
Primary breast TB without chest disease comprises up to 86% of mammary tuberculosis.6,7 Infection may occur via contamination of the skin or nipple.5-7 Lactation, pregnancy, and other causes of immunosuppression (especially human immunodeficiency virus) have been associated with an increased risk of breast infection.6-8 This patient was at risk for immunosuppression from longstanding diabetes.14
Many patients from TB-endemic areas have received the bacille Calmette-Guerin (BCG) vaccine and may exhibit equivocal or false-positive PPD results. Because interferon-gamma release assay TB blood tests (eg, QuantiFERON-TB Gold or T-SPOT.TB) are not affected by BCG, they are not associated with false-positive repeat testing results.15
Biopsy is necessary to rule out malignancy and diagnose breast TB
A pleural fluid to serum protein ratio >0.5 is consistent with infection, but also with sarcoidosis or malignancy.3,16 Elevated pleural fluid adenosine deaminase (>40 U/L) is sensitive, albeit nonspecific, for the presence of TB microorganisms. If a lymphocyte-dominant exudate is also present, however, its reliability greatly increases.16,17 Increased pleural fluid interferon-gamma is also sensitive and specific for TB pleurisy.18 Culture, along with drug sensitivity testing, should be performed on all unexplained pleural effusions.
A biopsy is often required to diagnose breast TB and should be performed on all suspicious lesions to exclude malignancy.5-7,9 AFB stains and cultures of aspirate fluids or tissue are often negative.7,9 PCR or other nucleic acid amplification tests of sputum, body fluids, or biopsy material may be positive in culture-negative cases and can rapidly confirm M tuberculosis infection.17,19 No testing modality offers 100% sensitivity or specificity; therefore, an additional confirmatory test is desirable.
Possible routes of transmission include activation of latent pulmonary tuberculosis and direct, lymphatic, or hematologic extension to the chest wall and breast.5-7 In this patient, we believe that activation of a latent breast granuloma may have resulted in a secondary or “sympathetic” pleural effusion, possibly triggered by surgical manipulation. This is compatible with her negative pleural adenosine deaminase result, negative culture, absence of pulmonary parenchymal disease, and negative pleural biopsy. Although we conducted a PubMed search, reviewing material as far back as 1966, we were unable to find a previous case of apparent sympathetic effusion associated with breast TB.
Our patient was treated with daily oral isoniazid, rifabutin, pyrazinamide, and ethambutol for 2 months, followed by isoniazid and rifabutin for 4 months. She has been disease-free for over 10 years.
THE TAKEAWAY
We describe a rare case of breast TB mimicking carcinoma that was associated with unilateral pleural effusion in a woman who had emigrated from Afghanistan. Patients at particular risk for breast TB include immigrants from endemic regions—especially parous females,6,7 those with a history of TB contacts, and those who are immunosuppressed.8 This case emphasizes the need for increased awareness of extrapulmonary TB by physicians in developed countries.
ACKNOWLEDGEMENTS
The authors thank Drs. Margie Scott, Harpreet Dhillon, Samir Vora, Todd Williams, Jeffrey Hawley, and Mr. Sergio Landeros. This report is dedicated to the memory of our friend and colleague in medicine, Dr. Jeanie Care Gillinta.
1. Bayer-Garner IB, Cox MD, Scott MA, et al. Mycobacteria other than Mycobacterium tuberculosis are not present in erythema induratum/nodular vasculitis: a case series and literature review of the clinical and histologic findings. J Cutan Pathol. 2005;32:220-226.
2. Verfaillie G, Breucq C, Sacre R, et al. Granulomatous lobular mastitis: a rare chronic inflammatory disease of the breast which can mimic breast carcinoma. Acta Chir Belg. 2006;106:222-224.
3. Fiorucci F, Conti V, Lucantoni G, et al. Sarcoidosis of the breast: a rare case report and a review. Eur Rev Med Pharmacol Sci. 2006;10:47-50.
4. Huggins JT, Doelken P, Sahn SA, et al. Pleural effusions in a series of 181 outpatients with sarcoidosis. Chest. 2006;129:1599-1604.
5. Zandrino F, Monetti F, Gandolfo N. Primary tuberculosis of the breast. A case report. Acta Radiol. 2000;41:61-63.
6. Khanna R, Prasanna GV, Gupta P, et al. Mammary tuberculosis: report on 52 cases. Postgrad Med J. 2002;78:422-424.
7. Harris SH, Khan MA, Khan R, et al. Mammary tuberculosis: analysis of thirty-eight patients. ANZ J Surg. 2006;76:234-237.
8. Meerkotter D, Spiegel K, Page-Shipp LS. Imaging of tuberculosis of the breast: 21 cases and a review of the literature. J Med Imaging Radiat Oncol. 2011;55:453-460.
9. Khodabakhshi B, Mehravar F. Breast tuberculosis in northeast Iran: review of 22 cases. BMC Womens Health. 2014;14:72.
10. Osborne BM. Granulomatous mastitis caused by histoplasma and mimicking inflammatory breast carcinoma. Hum Pathol. 1989;20:47-52.
11. Bocian JJ, Fahmy RN, Michas CA. A rare case of ‘coccidioidoma’ of the breast. Arch Pathol Lab Med. 1991;115:1064-1067.
12. Haddow LJ, Sahid F, Moosa MY. Cryptococcal breast abscess in an HIV-positive patient: arguments for reviewing the definition of immune reconstitution inflammatory syndrome. J Infect. 2008;57:82-84.
13. Lefkowitz M, Wear DJ. Cat-scratch disease masquerading as a solitary tumor of the breast. Arch Pathol Lab Med. 1989;113:473-475.
14. Ponce-De-Leon A, Garcia-Garcia Md Mde L, Garcia-Sancho MC, et al. Tuberculosis and diabetes in southern Mexico. Diabetes Care. 2004;27:1584-1590.
15. Mazurek GH, LoBue PA, Daley CL, et al. Comparison of a whole-blood interferon gamma assay with tuberculin skin testing for detecting latent Mycobacterium tuberculosis infection. JAMA. 2001;286:1740-1747.
16. Porcel JM, Light RW. Diagnostic approach to pleural effusion in adults. Am Fam Physician. 2006;73:1211-1220.
17. Burgess LJ, Maritz FJ, Le Roux I, et al. Combined use of pleural adenosine deaminase with lymphocyte/neutrophil ratio. Increased specificity for the diagnosis of tuberculous pleuritis. Chest. 1996;109:414-419.
18. Klimiuk J, Krenke R, Safianowska A, et al. Diagnostic performance of different pleural fluid biomarkers in tuberculous pleurisy. Adv Exp Med Biol. 2015;852:21-30.
19. Gopi A, Madhavan SM, Sharma SK, et al. Diagnosis and treatment of tuberculous pleural effusion in 2006. Chest. 2007;131:880-889.
THE CASE
A 44-year-old woman with a 15-year history of type 2 diabetes sought care for a firm, non-tender mass in the medial lower quadrant of her right breast. She hadn’t experienced any skin changes or axillary lymphadenopathy. The patient had immigrated to California from Afghanistan 22 years earlier, at which time she was briefly married to an Afghan man suffering from a chronic cough.
Mammography revealed a 3.5 x 4 x 4 cm lesion at the chest wall, which was highly suspicious for carcinoma (FIGURES 1A AND 1B). Sonography showed a heterogenous hypoechoic and isoechoic mass with posterior acoustic enhancement (FIGURE 1C). An excisional biopsy was performed.
One week postoperatively, the patient presented to the emergency department for a worsening nonproductive cough that intensified when supine, and was associated with subscapular pleuritic pain. She denied fever or weight loss. Biopsy results were pending.
THE DIAGNOSIS
Chest x-rays revealed a large right pleural effusion that was presumed to be malignant (FIGURES 1D AND 1E). Thoracentesis yielded 1.5 liters of tea-colored exudate containing 2800 nucleated cells/mL—63% lymphocytes and 37% neutrophils—and a pleural fluid to serum protein ratio >0.5. Adenosine deaminase was <1 U/L. Fluid Gram stain, acid-fast bacillus (AFB) fluorescent antibody testing, AFB cultures, and cytology were negative. Computed tomography (CT) subsequently demonstrated recurrent effusion without hilar or mediastinal lymphadenopathy or pleural enhancement (FIGURE 1F).
Histologically, the breast mass showed caseating granulomatous inflammation (FIGURES 1G AND 1H). An AFB stain was negative. Polymerase chain reaction (PCR) performed on DNA extracted from the formalin-fixed, paraffin-embedded biopsy material was positive for Mycobacterium tuberculosis.1 A CT-guided pleural biopsy showed only normal tissue. A follow-up tuberculin skin test (purified protein derivative [PPD]) yielded a 10-mm indurated reaction.
DISCUSSION
Granulomatous lesions, such as foreign body granuloma, idiopathic granulomatous mastitis (IGM), and sarcoidosis can mimic breast carcinoma.2,3 IGM is associated with elevated prolactin (eg, pregnancy or oral contraceptive use) and is usually subareolar.2 Infection, however, is also commonly subareolar. Sarcoidosis rarely exhibits unilateral pleural effusion and usually manifests with bilateral interstitial lung disease, hilar lymphadenopathy, and non-necrotizing granulomas.3,4
M tuberculosis and other granulomatous infections may also feign breast cancer.5-13 Breast TB, which is highly uncommon in the developed world, often demonstrates imaging similar to that which was seen in this case. Breast TB may appear nodular with ill-defined contours. Masses are sometimes attached to the chest wall and usually lack microcalcifications on mammography; they are also typically hypoechoic and heterogenous on ultrasound, often showing posterior enhancement.5,7,8 Like other breast infections, tuberculosis may show cutaneous sinus tract formation, which is seen in about one-third of patients.6,7 Alternatively, it may manifest as a diffuse mastitis with skin thickening and axillary lymphadenopathy.8
Primary breast TB without chest disease comprises up to 86% of mammary tuberculosis.6,7 Infection may occur via contamination of the skin or nipple.5-7 Lactation, pregnancy, and other causes of immunosuppression (especially human immunodeficiency virus) have been associated with an increased risk of breast infection.6-8 This patient was at risk for immunosuppression from longstanding diabetes.14
Many patients from TB-endemic areas have received the bacille Calmette-Guerin (BCG) vaccine and may exhibit equivocal or false-positive PPD results. Because interferon-gamma release assay TB blood tests (eg, QuantiFERON-TB Gold or T-SPOT.TB) are not affected by BCG, they are not associated with false-positive repeat testing results.15
Biopsy is necessary to rule out malignancy and diagnose breast TB
A pleural fluid to serum protein ratio >0.5 is consistent with infection, but also with sarcoidosis or malignancy.3,16 Elevated pleural fluid adenosine deaminase (>40 U/L) is sensitive, albeit nonspecific, for the presence of TB microorganisms. If a lymphocyte-dominant exudate is also present, however, its reliability greatly increases.16,17 Increased pleural fluid interferon-gamma is also sensitive and specific for TB pleurisy.18 Culture, along with drug sensitivity testing, should be performed on all unexplained pleural effusions.
A biopsy is often required to diagnose breast TB and should be performed on all suspicious lesions to exclude malignancy.5-7,9 AFB stains and cultures of aspirate fluids or tissue are often negative.7,9 PCR or other nucleic acid amplification tests of sputum, body fluids, or biopsy material may be positive in culture-negative cases and can rapidly confirm M tuberculosis infection.17,19 No testing modality offers 100% sensitivity or specificity; therefore, an additional confirmatory test is desirable.
Possible routes of transmission include activation of latent pulmonary tuberculosis and direct, lymphatic, or hematologic extension to the chest wall and breast.5-7 In this patient, we believe that activation of a latent breast granuloma may have resulted in a secondary or “sympathetic” pleural effusion, possibly triggered by surgical manipulation. This is compatible with her negative pleural adenosine deaminase result, negative culture, absence of pulmonary parenchymal disease, and negative pleural biopsy. Although we conducted a PubMed search, reviewing material as far back as 1966, we were unable to find a previous case of apparent sympathetic effusion associated with breast TB.
Our patient was treated with daily oral isoniazid, rifabutin, pyrazinamide, and ethambutol for 2 months, followed by isoniazid and rifabutin for 4 months. She has been disease-free for over 10 years.
THE TAKEAWAY
We describe a rare case of breast TB mimicking carcinoma that was associated with unilateral pleural effusion in a woman who had emigrated from Afghanistan. Patients at particular risk for breast TB include immigrants from endemic regions—especially parous females,6,7 those with a history of TB contacts, and those who are immunosuppressed.8 This case emphasizes the need for increased awareness of extrapulmonary TB by physicians in developed countries.
ACKNOWLEDGEMENTS
The authors thank Drs. Margie Scott, Harpreet Dhillon, Samir Vora, Todd Williams, Jeffrey Hawley, and Mr. Sergio Landeros. This report is dedicated to the memory of our friend and colleague in medicine, Dr. Jeanie Care Gillinta.
THE CASE
A 44-year-old woman with a 15-year history of type 2 diabetes sought care for a firm, non-tender mass in the medial lower quadrant of her right breast. She hadn’t experienced any skin changes or axillary lymphadenopathy. The patient had immigrated to California from Afghanistan 22 years earlier, at which time she was briefly married to an Afghan man suffering from a chronic cough.
Mammography revealed a 3.5 x 4 x 4 cm lesion at the chest wall, which was highly suspicious for carcinoma (FIGURES 1A AND 1B). Sonography showed a heterogenous hypoechoic and isoechoic mass with posterior acoustic enhancement (FIGURE 1C). An excisional biopsy was performed.
One week postoperatively, the patient presented to the emergency department for a worsening nonproductive cough that intensified when supine, and was associated with subscapular pleuritic pain. She denied fever or weight loss. Biopsy results were pending.
THE DIAGNOSIS
Chest x-rays revealed a large right pleural effusion that was presumed to be malignant (FIGURES 1D AND 1E). Thoracentesis yielded 1.5 liters of tea-colored exudate containing 2800 nucleated cells/mL—63% lymphocytes and 37% neutrophils—and a pleural fluid to serum protein ratio >0.5. Adenosine deaminase was <1 U/L. Fluid Gram stain, acid-fast bacillus (AFB) fluorescent antibody testing, AFB cultures, and cytology were negative. Computed tomography (CT) subsequently demonstrated recurrent effusion without hilar or mediastinal lymphadenopathy or pleural enhancement (FIGURE 1F).
Histologically, the breast mass showed caseating granulomatous inflammation (FIGURES 1G AND 1H). An AFB stain was negative. Polymerase chain reaction (PCR) performed on DNA extracted from the formalin-fixed, paraffin-embedded biopsy material was positive for Mycobacterium tuberculosis.1 A CT-guided pleural biopsy showed only normal tissue. A follow-up tuberculin skin test (purified protein derivative [PPD]) yielded a 10-mm indurated reaction.
DISCUSSION
Granulomatous lesions, such as foreign body granuloma, idiopathic granulomatous mastitis (IGM), and sarcoidosis can mimic breast carcinoma.2,3 IGM is associated with elevated prolactin (eg, pregnancy or oral contraceptive use) and is usually subareolar.2 Infection, however, is also commonly subareolar. Sarcoidosis rarely exhibits unilateral pleural effusion and usually manifests with bilateral interstitial lung disease, hilar lymphadenopathy, and non-necrotizing granulomas.3,4
M tuberculosis and other granulomatous infections may also feign breast cancer.5-13 Breast TB, which is highly uncommon in the developed world, often demonstrates imaging similar to that which was seen in this case. Breast TB may appear nodular with ill-defined contours. Masses are sometimes attached to the chest wall and usually lack microcalcifications on mammography; they are also typically hypoechoic and heterogenous on ultrasound, often showing posterior enhancement.5,7,8 Like other breast infections, tuberculosis may show cutaneous sinus tract formation, which is seen in about one-third of patients.6,7 Alternatively, it may manifest as a diffuse mastitis with skin thickening and axillary lymphadenopathy.8
Primary breast TB without chest disease comprises up to 86% of mammary tuberculosis.6,7 Infection may occur via contamination of the skin or nipple.5-7 Lactation, pregnancy, and other causes of immunosuppression (especially human immunodeficiency virus) have been associated with an increased risk of breast infection.6-8 This patient was at risk for immunosuppression from longstanding diabetes.14
Many patients from TB-endemic areas have received the bacille Calmette-Guerin (BCG) vaccine and may exhibit equivocal or false-positive PPD results. Because interferon-gamma release assay TB blood tests (eg, QuantiFERON-TB Gold or T-SPOT.TB) are not affected by BCG, they are not associated with false-positive repeat testing results.15
Biopsy is necessary to rule out malignancy and diagnose breast TB
A pleural fluid to serum protein ratio >0.5 is consistent with infection, but also with sarcoidosis or malignancy.3,16 Elevated pleural fluid adenosine deaminase (>40 U/L) is sensitive, albeit nonspecific, for the presence of TB microorganisms. If a lymphocyte-dominant exudate is also present, however, its reliability greatly increases.16,17 Increased pleural fluid interferon-gamma is also sensitive and specific for TB pleurisy.18 Culture, along with drug sensitivity testing, should be performed on all unexplained pleural effusions.
A biopsy is often required to diagnose breast TB and should be performed on all suspicious lesions to exclude malignancy.5-7,9 AFB stains and cultures of aspirate fluids or tissue are often negative.7,9 PCR or other nucleic acid amplification tests of sputum, body fluids, or biopsy material may be positive in culture-negative cases and can rapidly confirm M tuberculosis infection.17,19 No testing modality offers 100% sensitivity or specificity; therefore, an additional confirmatory test is desirable.
Possible routes of transmission include activation of latent pulmonary tuberculosis and direct, lymphatic, or hematologic extension to the chest wall and breast.5-7 In this patient, we believe that activation of a latent breast granuloma may have resulted in a secondary or “sympathetic” pleural effusion, possibly triggered by surgical manipulation. This is compatible with her negative pleural adenosine deaminase result, negative culture, absence of pulmonary parenchymal disease, and negative pleural biopsy. Although we conducted a PubMed search, reviewing material as far back as 1966, we were unable to find a previous case of apparent sympathetic effusion associated with breast TB.
Our patient was treated with daily oral isoniazid, rifabutin, pyrazinamide, and ethambutol for 2 months, followed by isoniazid and rifabutin for 4 months. She has been disease-free for over 10 years.
THE TAKEAWAY
We describe a rare case of breast TB mimicking carcinoma that was associated with unilateral pleural effusion in a woman who had emigrated from Afghanistan. Patients at particular risk for breast TB include immigrants from endemic regions—especially parous females,6,7 those with a history of TB contacts, and those who are immunosuppressed.8 This case emphasizes the need for increased awareness of extrapulmonary TB by physicians in developed countries.
ACKNOWLEDGEMENTS
The authors thank Drs. Margie Scott, Harpreet Dhillon, Samir Vora, Todd Williams, Jeffrey Hawley, and Mr. Sergio Landeros. This report is dedicated to the memory of our friend and colleague in medicine, Dr. Jeanie Care Gillinta.
1. Bayer-Garner IB, Cox MD, Scott MA, et al. Mycobacteria other than Mycobacterium tuberculosis are not present in erythema induratum/nodular vasculitis: a case series and literature review of the clinical and histologic findings. J Cutan Pathol. 2005;32:220-226.
2. Verfaillie G, Breucq C, Sacre R, et al. Granulomatous lobular mastitis: a rare chronic inflammatory disease of the breast which can mimic breast carcinoma. Acta Chir Belg. 2006;106:222-224.
3. Fiorucci F, Conti V, Lucantoni G, et al. Sarcoidosis of the breast: a rare case report and a review. Eur Rev Med Pharmacol Sci. 2006;10:47-50.
4. Huggins JT, Doelken P, Sahn SA, et al. Pleural effusions in a series of 181 outpatients with sarcoidosis. Chest. 2006;129:1599-1604.
5. Zandrino F, Monetti F, Gandolfo N. Primary tuberculosis of the breast. A case report. Acta Radiol. 2000;41:61-63.
6. Khanna R, Prasanna GV, Gupta P, et al. Mammary tuberculosis: report on 52 cases. Postgrad Med J. 2002;78:422-424.
7. Harris SH, Khan MA, Khan R, et al. Mammary tuberculosis: analysis of thirty-eight patients. ANZ J Surg. 2006;76:234-237.
8. Meerkotter D, Spiegel K, Page-Shipp LS. Imaging of tuberculosis of the breast: 21 cases and a review of the literature. J Med Imaging Radiat Oncol. 2011;55:453-460.
9. Khodabakhshi B, Mehravar F. Breast tuberculosis in northeast Iran: review of 22 cases. BMC Womens Health. 2014;14:72.
10. Osborne BM. Granulomatous mastitis caused by histoplasma and mimicking inflammatory breast carcinoma. Hum Pathol. 1989;20:47-52.
11. Bocian JJ, Fahmy RN, Michas CA. A rare case of ‘coccidioidoma’ of the breast. Arch Pathol Lab Med. 1991;115:1064-1067.
12. Haddow LJ, Sahid F, Moosa MY. Cryptococcal breast abscess in an HIV-positive patient: arguments for reviewing the definition of immune reconstitution inflammatory syndrome. J Infect. 2008;57:82-84.
13. Lefkowitz M, Wear DJ. Cat-scratch disease masquerading as a solitary tumor of the breast. Arch Pathol Lab Med. 1989;113:473-475.
14. Ponce-De-Leon A, Garcia-Garcia Md Mde L, Garcia-Sancho MC, et al. Tuberculosis and diabetes in southern Mexico. Diabetes Care. 2004;27:1584-1590.
15. Mazurek GH, LoBue PA, Daley CL, et al. Comparison of a whole-blood interferon gamma assay with tuberculin skin testing for detecting latent Mycobacterium tuberculosis infection. JAMA. 2001;286:1740-1747.
16. Porcel JM, Light RW. Diagnostic approach to pleural effusion in adults. Am Fam Physician. 2006;73:1211-1220.
17. Burgess LJ, Maritz FJ, Le Roux I, et al. Combined use of pleural adenosine deaminase with lymphocyte/neutrophil ratio. Increased specificity for the diagnosis of tuberculous pleuritis. Chest. 1996;109:414-419.
18. Klimiuk J, Krenke R, Safianowska A, et al. Diagnostic performance of different pleural fluid biomarkers in tuberculous pleurisy. Adv Exp Med Biol. 2015;852:21-30.
19. Gopi A, Madhavan SM, Sharma SK, et al. Diagnosis and treatment of tuberculous pleural effusion in 2006. Chest. 2007;131:880-889.
1. Bayer-Garner IB, Cox MD, Scott MA, et al. Mycobacteria other than Mycobacterium tuberculosis are not present in erythema induratum/nodular vasculitis: a case series and literature review of the clinical and histologic findings. J Cutan Pathol. 2005;32:220-226.
2. Verfaillie G, Breucq C, Sacre R, et al. Granulomatous lobular mastitis: a rare chronic inflammatory disease of the breast which can mimic breast carcinoma. Acta Chir Belg. 2006;106:222-224.
3. Fiorucci F, Conti V, Lucantoni G, et al. Sarcoidosis of the breast: a rare case report and a review. Eur Rev Med Pharmacol Sci. 2006;10:47-50.
4. Huggins JT, Doelken P, Sahn SA, et al. Pleural effusions in a series of 181 outpatients with sarcoidosis. Chest. 2006;129:1599-1604.
5. Zandrino F, Monetti F, Gandolfo N. Primary tuberculosis of the breast. A case report. Acta Radiol. 2000;41:61-63.
6. Khanna R, Prasanna GV, Gupta P, et al. Mammary tuberculosis: report on 52 cases. Postgrad Med J. 2002;78:422-424.
7. Harris SH, Khan MA, Khan R, et al. Mammary tuberculosis: analysis of thirty-eight patients. ANZ J Surg. 2006;76:234-237.
8. Meerkotter D, Spiegel K, Page-Shipp LS. Imaging of tuberculosis of the breast: 21 cases and a review of the literature. J Med Imaging Radiat Oncol. 2011;55:453-460.
9. Khodabakhshi B, Mehravar F. Breast tuberculosis in northeast Iran: review of 22 cases. BMC Womens Health. 2014;14:72.
10. Osborne BM. Granulomatous mastitis caused by histoplasma and mimicking inflammatory breast carcinoma. Hum Pathol. 1989;20:47-52.
11. Bocian JJ, Fahmy RN, Michas CA. A rare case of ‘coccidioidoma’ of the breast. Arch Pathol Lab Med. 1991;115:1064-1067.
12. Haddow LJ, Sahid F, Moosa MY. Cryptococcal breast abscess in an HIV-positive patient: arguments for reviewing the definition of immune reconstitution inflammatory syndrome. J Infect. 2008;57:82-84.
13. Lefkowitz M, Wear DJ. Cat-scratch disease masquerading as a solitary tumor of the breast. Arch Pathol Lab Med. 1989;113:473-475.
14. Ponce-De-Leon A, Garcia-Garcia Md Mde L, Garcia-Sancho MC, et al. Tuberculosis and diabetes in southern Mexico. Diabetes Care. 2004;27:1584-1590.
15. Mazurek GH, LoBue PA, Daley CL, et al. Comparison of a whole-blood interferon gamma assay with tuberculin skin testing for detecting latent Mycobacterium tuberculosis infection. JAMA. 2001;286:1740-1747.
16. Porcel JM, Light RW. Diagnostic approach to pleural effusion in adults. Am Fam Physician. 2006;73:1211-1220.
17. Burgess LJ, Maritz FJ, Le Roux I, et al. Combined use of pleural adenosine deaminase with lymphocyte/neutrophil ratio. Increased specificity for the diagnosis of tuberculous pleuritis. Chest. 1996;109:414-419.
18. Klimiuk J, Krenke R, Safianowska A, et al. Diagnostic performance of different pleural fluid biomarkers in tuberculous pleurisy. Adv Exp Med Biol. 2015;852:21-30.
19. Gopi A, Madhavan SM, Sharma SK, et al. Diagnosis and treatment of tuberculous pleural effusion in 2006. Chest. 2007;131:880-889.

Unicentric Castleman disease disguised as a pancreatic neoplasm
Castleman disease or angiofollicular lymph node hyperplasia is an uncommon cause of an incidental abdominal mass found on imaging. The etiology of Castleman disease is relatively unknown, however, it is thought to be primarily associated with an oversecretion of interleukin-6. The oversecretion of this pro-inflammatory cytokine leads to lymph node hyperplasia. Castleman disease can be classified into 2 categories: unicentric or multicentric. Most cases of unicentric Castleman disease are asymptomatic and are found on routine imaging. It is found predominately in middle-aged persons of equal sex and is managed primarily by surgical resection. We present here a case of a peripancreatic mass diagnosed by surgical excision as Castleman disease, hyaline vascular type.
Click on the PDF icon at the top of this introduction to read the full article.
Castleman disease or angiofollicular lymph node hyperplasia is an uncommon cause of an incidental abdominal mass found on imaging. The etiology of Castleman disease is relatively unknown, however, it is thought to be primarily associated with an oversecretion of interleukin-6. The oversecretion of this pro-inflammatory cytokine leads to lymph node hyperplasia. Castleman disease can be classified into 2 categories: unicentric or multicentric. Most cases of unicentric Castleman disease are asymptomatic and are found on routine imaging. It is found predominately in middle-aged persons of equal sex and is managed primarily by surgical resection. We present here a case of a peripancreatic mass diagnosed by surgical excision as Castleman disease, hyaline vascular type.
Click on the PDF icon at the top of this introduction to read the full article.
Castleman disease or angiofollicular lymph node hyperplasia is an uncommon cause of an incidental abdominal mass found on imaging. The etiology of Castleman disease is relatively unknown, however, it is thought to be primarily associated with an oversecretion of interleukin-6. The oversecretion of this pro-inflammatory cytokine leads to lymph node hyperplasia. Castleman disease can be classified into 2 categories: unicentric or multicentric. Most cases of unicentric Castleman disease are asymptomatic and are found on routine imaging. It is found predominately in middle-aged persons of equal sex and is managed primarily by surgical resection. We present here a case of a peripancreatic mass diagnosed by surgical excision as Castleman disease, hyaline vascular type.
Click on the PDF icon at the top of this introduction to read the full article.
Paraneoplastic Isaacs syndrome leading to diagnosis of small-cell lung cancer
Paraneoplastic Isaacs syndrome is a rare disorder with distinct clinical and electromyographic characteristics. It is a consequence of neoplastic process that is not directly caused by the tumor itself, but usually mediated by immune response primarily against the tumor and neural tissues are damaged owing to bystander effect. Paraneoplastic neurologic disorders may precede cancer diagnosis. Here we report the case of 75-year-old woman who presented with numbness, tingling sensation, and weakness of lower extremities, and was diagnosed with Isaacs syndrome and subsequently small-cell lung cancer. Plasmapheresis and treatment of small-cell lung cancer produced signficant symptoms improvement. We also conduct a complete review of the published case reports and case series of Isaacs syndrome of paraneoplastic etiology, which usually has good response to carbamazepine and to specfic treatment of underlying neoplasm.
Click on the PDF icon at the top of this introduction to read the full article.
Paraneoplastic Isaacs syndrome is a rare disorder with distinct clinical and electromyographic characteristics. It is a consequence of neoplastic process that is not directly caused by the tumor itself, but usually mediated by immune response primarily against the tumor and neural tissues are damaged owing to bystander effect. Paraneoplastic neurologic disorders may precede cancer diagnosis. Here we report the case of 75-year-old woman who presented with numbness, tingling sensation, and weakness of lower extremities, and was diagnosed with Isaacs syndrome and subsequently small-cell lung cancer. Plasmapheresis and treatment of small-cell lung cancer produced signficant symptoms improvement. We also conduct a complete review of the published case reports and case series of Isaacs syndrome of paraneoplastic etiology, which usually has good response to carbamazepine and to specfic treatment of underlying neoplasm.
Click on the PDF icon at the top of this introduction to read the full article.
Paraneoplastic Isaacs syndrome is a rare disorder with distinct clinical and electromyographic characteristics. It is a consequence of neoplastic process that is not directly caused by the tumor itself, but usually mediated by immune response primarily against the tumor and neural tissues are damaged owing to bystander effect. Paraneoplastic neurologic disorders may precede cancer diagnosis. Here we report the case of 75-year-old woman who presented with numbness, tingling sensation, and weakness of lower extremities, and was diagnosed with Isaacs syndrome and subsequently small-cell lung cancer. Plasmapheresis and treatment of small-cell lung cancer produced signficant symptoms improvement. We also conduct a complete review of the published case reports and case series of Isaacs syndrome of paraneoplastic etiology, which usually has good response to carbamazepine and to specfic treatment of underlying neoplasm.
Click on the PDF icon at the top of this introduction to read the full article.
Cutaneous Adnexal Carcinoma With Apocrine Differentiation
Differentiation between a primary adnexal carcinoma and a metastatic carcinoma to the skin is a challenging yet critical task for dermatologists and pathologists. Carcinomas that have metastasized to the skin are a sign of widespread systemic involvement and poor prognosis, while primary adnexal carcinomas tend to progress with an indolent clinical course. Although many patients with cutaneous metastases from an internal primary neoplasm can expect a median survival of no more than 12 months,1 patients with primary adnexal carcinomas are reported to have a 5-year survival rate of 95.5% for localized disease and 85% with spread to regional lymph nodes.2 We report a case of multiple cutaneous neoplasms of unknown primary origin in a 71-year-old man and describe our approach to identification of the possible primary site as well as management of the disease.
Case Report
A 71-year-old man initially presented to his primary physician for evaluation of a mass on the left side of the neck of 3 months' duration. On physical examination, a firm 2.5×3.0-cm nodule was noted at the anterior border of the trapezius muscle. Palpation of the thyroid revealed an additional right-sided nodule. The submandibular and parotid glands were unremarkable to palpation. The patient was referred to general surgery for biopsy, which revealed an infiltrating, moderately differentiated adenocarcinoma with extensive lymphatic permeation. Immunohistochemical staining for cytokeratin (CK) 7 was positive, while CK20 and thyroid transcription factor 1 were negative. A positron emission tomography/computed tomography (CT) fusion scan demonstrated 3 areas of enhanced uptake: one in the right side of the thyroid, a second corresponding to the mass on the left side of the neck at the level of the trapezius muscle, and a third in the left masseter muscle. Surgical excision with negative margins with possible chemotherapy was recommended; however, the patient declined treatment and was lost to follow-up until 2 years later when he presented to his primary physician with an additional lesion on his scalp.
Four years after the biopsy, the patient presented to the dermatology department with additional tumor nodules including a 4-cm, annular, indurated, focally eroded plaque on the left side of the lateral neck (Figure 1); 3 separate 1-cm nodules on the right side of the lateral neck; and an ulcerated, crusted, 10×8-cm plaque on the posterior aspect of the scalp. Despite the extensive lesions, the patient remained in good health and reported no recent weight loss or signs or symptoms of systemic involvement. The posterior scalp lesion, which developed 2 years after the initial appearance of the mass on the neck and was thought to represent a possible metastasis of the tumor, was biopsied and showed diffuse infiltration of the dermis by poorly differentiated tumor cells with vacuolated cytoplasm arranged in nests and cords and sometimes in a single-file arrangement (Figure 2). A CT scan demonstrated pretracheal lymphadenopathy as well as small intraparenchymal and subpleural pulmonary nodules throughout both lung fields.
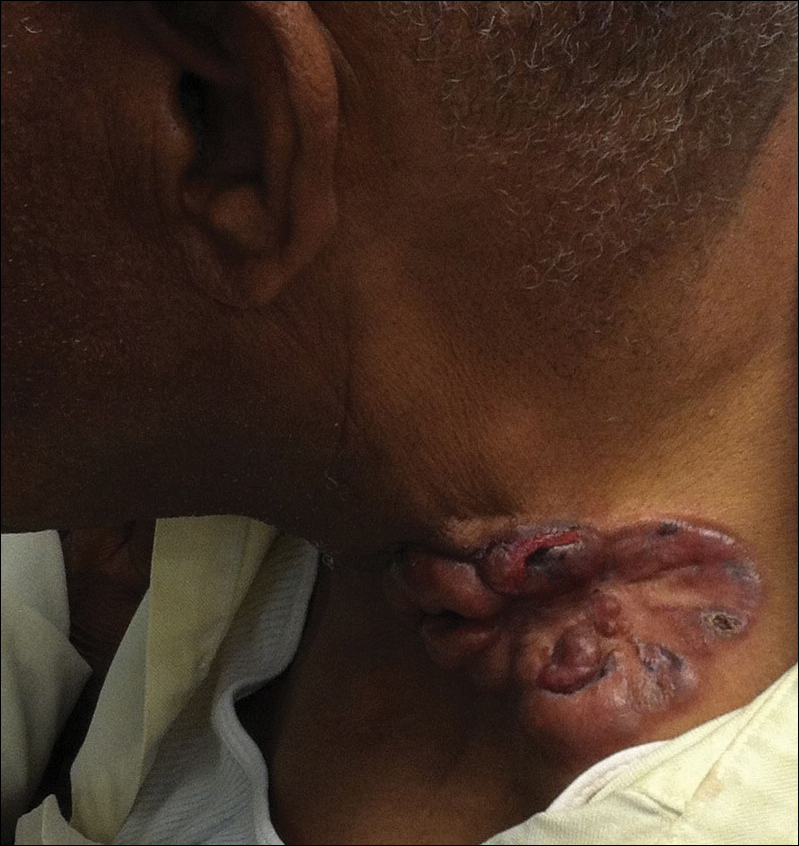
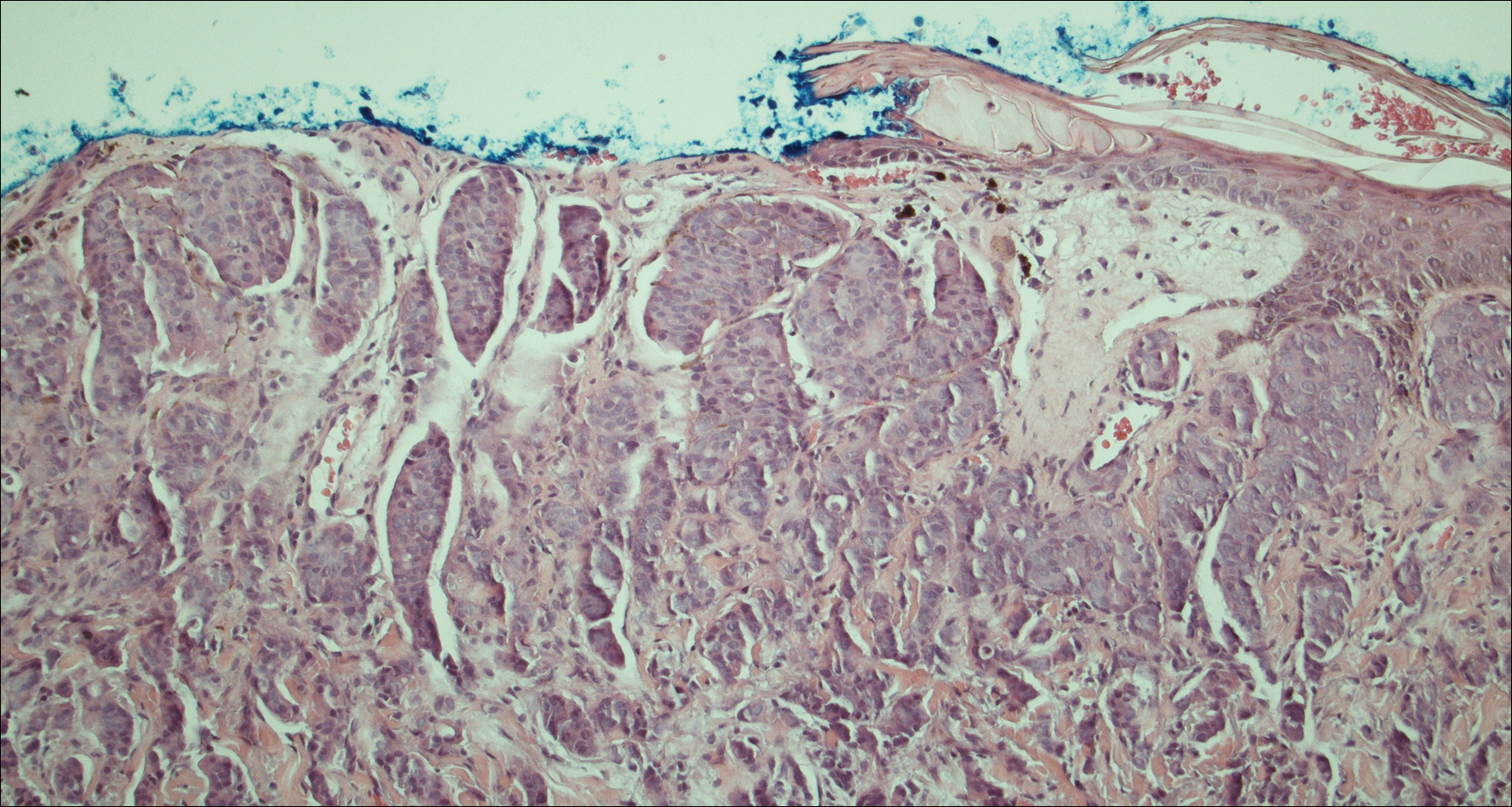
Another scalp biopsy was taken. Tumor cells were negative on mucicarmine staining. Additional immunohistochemical staining, including a periodic acid-Schiff stain with diastase digestion for epithelial mucin revealed minimal luminal positivity. Immunostaining was positive for CK7, carcinoembryonic antigen, CD15, estrogen receptor, progesterone receptor, gross cystic disease fluid protein 15 (GCDFP-15), and mammaglobin, and negative for CK20, podoplanin, thyroid transcription factor 1, S-100 protein, p63, and prostate specific antigen. ERBB2 (formerly HER2/neu) staining was negative according to fluorescence in situ hybridization analysis. Tumor cells showed a Ki-67 nuclear proliferation index of greater than 50%, indicating progression to aggressive carcinoma.
Based on the histological and immunochemical studies, the differential diagnosis included primary cutaneous apocrine carcinoma versus breast carcinoma; however, the prolonged clinical progression of these lesions favored a primary cutaneous adnexal tumor over a metastatic adenocarcinoma. Nevertheless, despite the initially indolent growth of the lesions over the first 5 years, the Ki-67 proliferation index and presence of widespread metastases on the posterior scalp indicated progression to an aggressive carcinoma. Chemotherapy was recommended as the treatment of choice. At his most recent follow-up visit 4 months later, the patient chose to begin treatment with tamoxifen and refused other treatment options.
Comment
The distinction between primary adnexal and metastatic adenocarcinomas of the skin is challenging both clinically and histologically. Some pathologists have argued that metastatic breast carcinomas and primary cutaneous apocrine carcinomas are essentially indistinguishable.3 Patients with cutaneous metastases, which occur in approximately 5.3% of all malignancies,4 typically can expect survival of no more than 12 months from the time of detection.1 In contrast, primary apocrine carcinomas of the skin, though much less common, carry a remarkably better prognosis, with 5-year relative survival rates of 95.5% and 85.5% reported for patients with localized disease and spread to regional lymph nodes, respectively.2
Fewer than 100 cases of primary cutaneous adnexal (apocrine) carcinomas have been reported overall, with the earliest known report dating back to 1944.5 According to the literature, primary apocrine carcinomas were diagnosed at a median age of 66 years and were slightly more common in females than males.2,6 Apocrine carcinomas were seen most frequently on the head, neck, and trunk,2 generally presenting in the form of asymptomatic nodules or plaques of 2 to 3 cm in size, with gradual progression occurring over months to years.6 Approximately 40% of patients have been reported with positive regional lymph nodes at diagnosis. Treatment of apocrine carcinoma typically has involved local excision with clear margins with or without lymph node dissection. Chemotherapy and radiation therapy have shown no proven benefit.7
Currently, there is no standardized approach to evaluating patients with possible cutaneous metastasis versus primary cutaneous adnexal carcinomas. Imaging studies such as mammography and abdominal CT typically reveal an internal primary cancer in one-third of patients. However, additional studies such as gastrointestinal radiography, chest and pelvic CT, barium enema, and intravenous pyelogram have shown to be of limited value.8 Although specificity and sensitivity of immunohistochemistry is limited, a number of immunomarkers, including CK7 and CK20, are routinely studied to narrow the differential diagnosis of a cutaneous neoplasm of unclear origin. Urothelial, gastric, colorectal, and pancreatic carcinomas generally are positive for CK20; CK7-positive adenocarcinomas include salivary, non-small cell lung, breast, ovarian, pancreatic, endometrial, and transitional cell adenocarcinomas. Carcinomas negative for both CK7 and CK20 include colorectal, hepatocellular, renal cell, prostate, and squamous cell carcinoma of the lung.
The presence of positive staining for estrogen and progesterone receptors as well as GCDFP-15 and mammaglobin raised the possibility of primary breast adenocarcinoma in our patient, but given that these markers can be positive in primary cutaneous adnexal tumors, immunohistochemistry results were not able to provide a definitive primary site. The overall staining pattern was nearly identical to 26 cases of primary cutaneous cribriform apocrine carcinoma, which was found to be positive for CK7 and carcinoembryonic antigen, and negative for CK20 and S-100. The only difference was in GCDFP-15 staining, which was positive in our case and negative in the cases of cribriform apocrine carcinoma.9 Histologic features favoring a primary apocrine origin include normal apocrine glands in the vicinity, glandular structures with decapitation secretion high in the dermis, and intracytoplasmic iron granules.10 Additionally, positive estrogen receptor staining appears to be much more common in apocrine carcinomas (5/10) than in eccrine carcinomas (1/7).11
A number of other markers have been investigated for possible diagnostic utility for distinction between primary adnexal carcinomas and metastatic adenocarcinomas. The nuclear transcription factor p63, which plays a role in keratinocyte differentiation, is preferentially expressed in a number of primary adnexal carcinomas and is purported to be the most sensitive marker overall, with a sensitivity of 78% to 91%.12-14 However, p63 has shown incomplete specificity for primary adnexal neoplasms, having been reported as positive in 11% to 22% of adenocarcinomas metastatic to skin.15-18 Nestin and CK15, which are expressed in hair follicle progenitor cells, also are potential specific markers for some primary adnexal lesions, specifically eccrine carcinoma, porocarcinoma, hidradenocarcinoma, and microcystic adnexal carcinoma; however, in one report, none of the apocrine carcinomas were positive for p63, cytokeratin 15, or D2-40.19 Thus, while markers for some primary adnexal neoplasms are emerging, specific tests at the immunohistochemical level for the apocrine carcinoma subgroup are still lacking.
Conclusion
In summary, a conclusive distinction between primary cutaneous apocrine carcinoma and metastatic adenocarcinoma to the skin remains challenging. Although new markers provide more specificity and sensitivity for neoplasms of eccrine origin, these markers do not appear to differentiate between primary apocrine carcinoma and metastatic breast carcinoma. In this case, as in other recent reports, diagnosis remained dependent on the clinical course of the patient. Although considerable progress has been made regarding immunohistochemical analysis of these cases, additional markers, especially ones more specific for primary skin cancers with apocrine differentiation, are still needed.
- Nashan D, Müller ML, Braun-Falco M, et al. Cutaneous metastases of visceral tumours: a review. J Cancer Res Clin Oncol. 2009;135:1-14.
- Blake PW, Bradford PT, Devesa SS, et al. Cutaneous appendageal carcinoma incidence and survival patterns in the United States: a population-based study. Arch Dermatol. 2010;146:625-632.
- Fernandez-Flores A. The elusive differential diagnosis of cutaneous apocrine adenocarcinoma vs. metastasis: the current role of clinical correlation. Acta Dermatovenerol Alp Pannonica Adriat. 2009;18:141-142.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma. A retrospective study of 7316 cancer patients. J Am Acad Dermatol. 1990;22:19-26.
- Horn RC. Malignant papillary cystadenoma of sweat glands with metastases to the regional lymph nodes. Surgery. 1944;16:348-355.
- Pucevich B, Catinchi-Jaime S, Ho J, et al. Invasive primary ductal apocrine adenocarcinoma of axilla: a case report with immunohistochemical profiling and a review of literature. Dermatol Online J. 2008;14:5.
- Vasilakaki T, Skafida E, Moustou E, et al. Primary cutaneous apocrine carcinoma of sweat glands: a rare case report [published online December 17, 2011]. Case Rep Oncol. 2011;4:597-601.
- Hainsworth JD, Greco FA. Treatment of patients with cancer of an unknown primary site. N Engl J Med. 1993;329:257-263.
- Rutten A, Kutzner H, Mentzel T, et al. Primary cutaneous cribriform apocrine carcinoma: a clinicopathologic and immunohistochemical study of 26 cases of an under-recognized cutaneous adnexal neoplasm. J Am Acad Dermatol. 2009;61:644-651.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever's Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott, Williams, and Wilkins; 2009.
- Le LP, Dias-Santagata D, Pawlak AC, et al. Apocrine-eccrine carcinomas: molecular and immunohistochemical analyses. PLoS One. 2012;7:e47290.
- Levrero M, De Laurenzi V, Costanzo A, et al. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci. 2000;113:1661-1670.
- Pellegrini G, Dellambra E, Golisano O, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A. 2001;98:3156-3161.
- Reis-Filho JS, Torio B, Albergaria A, et al. p63 expression in normal skin and usual cutaneous carcinomas. J Cutan Pathol. 2002;29:517-523.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Mahalingam M, Nguyen LP, Richards JE, et al. The diagnostic utility of immunohistochemistry in distinguishing primary skin adnexal carcinomas from metastatic adenocarcinoma to skin: an immunohistochemical reappraisal using cytokeratin 15, nestin, p63, D2-40, and calretinin. Mod Pathol. 2010;23:713-719.
Differentiation between a primary adnexal carcinoma and a metastatic carcinoma to the skin is a challenging yet critical task for dermatologists and pathologists. Carcinomas that have metastasized to the skin are a sign of widespread systemic involvement and poor prognosis, while primary adnexal carcinomas tend to progress with an indolent clinical course. Although many patients with cutaneous metastases from an internal primary neoplasm can expect a median survival of no more than 12 months,1 patients with primary adnexal carcinomas are reported to have a 5-year survival rate of 95.5% for localized disease and 85% with spread to regional lymph nodes.2 We report a case of multiple cutaneous neoplasms of unknown primary origin in a 71-year-old man and describe our approach to identification of the possible primary site as well as management of the disease.
Case Report
A 71-year-old man initially presented to his primary physician for evaluation of a mass on the left side of the neck of 3 months' duration. On physical examination, a firm 2.5×3.0-cm nodule was noted at the anterior border of the trapezius muscle. Palpation of the thyroid revealed an additional right-sided nodule. The submandibular and parotid glands were unremarkable to palpation. The patient was referred to general surgery for biopsy, which revealed an infiltrating, moderately differentiated adenocarcinoma with extensive lymphatic permeation. Immunohistochemical staining for cytokeratin (CK) 7 was positive, while CK20 and thyroid transcription factor 1 were negative. A positron emission tomography/computed tomography (CT) fusion scan demonstrated 3 areas of enhanced uptake: one in the right side of the thyroid, a second corresponding to the mass on the left side of the neck at the level of the trapezius muscle, and a third in the left masseter muscle. Surgical excision with negative margins with possible chemotherapy was recommended; however, the patient declined treatment and was lost to follow-up until 2 years later when he presented to his primary physician with an additional lesion on his scalp.
Four years after the biopsy, the patient presented to the dermatology department with additional tumor nodules including a 4-cm, annular, indurated, focally eroded plaque on the left side of the lateral neck (Figure 1); 3 separate 1-cm nodules on the right side of the lateral neck; and an ulcerated, crusted, 10×8-cm plaque on the posterior aspect of the scalp. Despite the extensive lesions, the patient remained in good health and reported no recent weight loss or signs or symptoms of systemic involvement. The posterior scalp lesion, which developed 2 years after the initial appearance of the mass on the neck and was thought to represent a possible metastasis of the tumor, was biopsied and showed diffuse infiltration of the dermis by poorly differentiated tumor cells with vacuolated cytoplasm arranged in nests and cords and sometimes in a single-file arrangement (Figure 2). A CT scan demonstrated pretracheal lymphadenopathy as well as small intraparenchymal and subpleural pulmonary nodules throughout both lung fields.
Another scalp biopsy was taken. Tumor cells were negative on mucicarmine staining. Additional immunohistochemical staining, including a periodic acid-Schiff stain with diastase digestion for epithelial mucin revealed minimal luminal positivity. Immunostaining was positive for CK7, carcinoembryonic antigen, CD15, estrogen receptor, progesterone receptor, gross cystic disease fluid protein 15 (GCDFP-15), and mammaglobin, and negative for CK20, podoplanin, thyroid transcription factor 1, S-100 protein, p63, and prostate specific antigen. ERBB2 (formerly HER2/neu) staining was negative according to fluorescence in situ hybridization analysis. Tumor cells showed a Ki-67 nuclear proliferation index of greater than 50%, indicating progression to aggressive carcinoma.
Based on the histological and immunochemical studies, the differential diagnosis included primary cutaneous apocrine carcinoma versus breast carcinoma; however, the prolonged clinical progression of these lesions favored a primary cutaneous adnexal tumor over a metastatic adenocarcinoma. Nevertheless, despite the initially indolent growth of the lesions over the first 5 years, the Ki-67 proliferation index and presence of widespread metastases on the posterior scalp indicated progression to an aggressive carcinoma. Chemotherapy was recommended as the treatment of choice. At his most recent follow-up visit 4 months later, the patient chose to begin treatment with tamoxifen and refused other treatment options.
Comment
The distinction between primary adnexal and metastatic adenocarcinomas of the skin is challenging both clinically and histologically. Some pathologists have argued that metastatic breast carcinomas and primary cutaneous apocrine carcinomas are essentially indistinguishable.3 Patients with cutaneous metastases, which occur in approximately 5.3% of all malignancies,4 typically can expect survival of no more than 12 months from the time of detection.1 In contrast, primary apocrine carcinomas of the skin, though much less common, carry a remarkably better prognosis, with 5-year relative survival rates of 95.5% and 85.5% reported for patients with localized disease and spread to regional lymph nodes, respectively.2
Fewer than 100 cases of primary cutaneous adnexal (apocrine) carcinomas have been reported overall, with the earliest known report dating back to 1944.5 According to the literature, primary apocrine carcinomas were diagnosed at a median age of 66 years and were slightly more common in females than males.2,6 Apocrine carcinomas were seen most frequently on the head, neck, and trunk,2 generally presenting in the form of asymptomatic nodules or plaques of 2 to 3 cm in size, with gradual progression occurring over months to years.6 Approximately 40% of patients have been reported with positive regional lymph nodes at diagnosis. Treatment of apocrine carcinoma typically has involved local excision with clear margins with or without lymph node dissection. Chemotherapy and radiation therapy have shown no proven benefit.7
Currently, there is no standardized approach to evaluating patients with possible cutaneous metastasis versus primary cutaneous adnexal carcinomas. Imaging studies such as mammography and abdominal CT typically reveal an internal primary cancer in one-third of patients. However, additional studies such as gastrointestinal radiography, chest and pelvic CT, barium enema, and intravenous pyelogram have shown to be of limited value.8 Although specificity and sensitivity of immunohistochemistry is limited, a number of immunomarkers, including CK7 and CK20, are routinely studied to narrow the differential diagnosis of a cutaneous neoplasm of unclear origin. Urothelial, gastric, colorectal, and pancreatic carcinomas generally are positive for CK20; CK7-positive adenocarcinomas include salivary, non-small cell lung, breast, ovarian, pancreatic, endometrial, and transitional cell adenocarcinomas. Carcinomas negative for both CK7 and CK20 include colorectal, hepatocellular, renal cell, prostate, and squamous cell carcinoma of the lung.
The presence of positive staining for estrogen and progesterone receptors as well as GCDFP-15 and mammaglobin raised the possibility of primary breast adenocarcinoma in our patient, but given that these markers can be positive in primary cutaneous adnexal tumors, immunohistochemistry results were not able to provide a definitive primary site. The overall staining pattern was nearly identical to 26 cases of primary cutaneous cribriform apocrine carcinoma, which was found to be positive for CK7 and carcinoembryonic antigen, and negative for CK20 and S-100. The only difference was in GCDFP-15 staining, which was positive in our case and negative in the cases of cribriform apocrine carcinoma.9 Histologic features favoring a primary apocrine origin include normal apocrine glands in the vicinity, glandular structures with decapitation secretion high in the dermis, and intracytoplasmic iron granules.10 Additionally, positive estrogen receptor staining appears to be much more common in apocrine carcinomas (5/10) than in eccrine carcinomas (1/7).11
A number of other markers have been investigated for possible diagnostic utility for distinction between primary adnexal carcinomas and metastatic adenocarcinomas. The nuclear transcription factor p63, which plays a role in keratinocyte differentiation, is preferentially expressed in a number of primary adnexal carcinomas and is purported to be the most sensitive marker overall, with a sensitivity of 78% to 91%.12-14 However, p63 has shown incomplete specificity for primary adnexal neoplasms, having been reported as positive in 11% to 22% of adenocarcinomas metastatic to skin.15-18 Nestin and CK15, which are expressed in hair follicle progenitor cells, also are potential specific markers for some primary adnexal lesions, specifically eccrine carcinoma, porocarcinoma, hidradenocarcinoma, and microcystic adnexal carcinoma; however, in one report, none of the apocrine carcinomas were positive for p63, cytokeratin 15, or D2-40.19 Thus, while markers for some primary adnexal neoplasms are emerging, specific tests at the immunohistochemical level for the apocrine carcinoma subgroup are still lacking.
Conclusion
In summary, a conclusive distinction between primary cutaneous apocrine carcinoma and metastatic adenocarcinoma to the skin remains challenging. Although new markers provide more specificity and sensitivity for neoplasms of eccrine origin, these markers do not appear to differentiate between primary apocrine carcinoma and metastatic breast carcinoma. In this case, as in other recent reports, diagnosis remained dependent on the clinical course of the patient. Although considerable progress has been made regarding immunohistochemical analysis of these cases, additional markers, especially ones more specific for primary skin cancers with apocrine differentiation, are still needed.
Differentiation between a primary adnexal carcinoma and a metastatic carcinoma to the skin is a challenging yet critical task for dermatologists and pathologists. Carcinomas that have metastasized to the skin are a sign of widespread systemic involvement and poor prognosis, while primary adnexal carcinomas tend to progress with an indolent clinical course. Although many patients with cutaneous metastases from an internal primary neoplasm can expect a median survival of no more than 12 months,1 patients with primary adnexal carcinomas are reported to have a 5-year survival rate of 95.5% for localized disease and 85% with spread to regional lymph nodes.2 We report a case of multiple cutaneous neoplasms of unknown primary origin in a 71-year-old man and describe our approach to identification of the possible primary site as well as management of the disease.
Case Report
A 71-year-old man initially presented to his primary physician for evaluation of a mass on the left side of the neck of 3 months' duration. On physical examination, a firm 2.5×3.0-cm nodule was noted at the anterior border of the trapezius muscle. Palpation of the thyroid revealed an additional right-sided nodule. The submandibular and parotid glands were unremarkable to palpation. The patient was referred to general surgery for biopsy, which revealed an infiltrating, moderately differentiated adenocarcinoma with extensive lymphatic permeation. Immunohistochemical staining for cytokeratin (CK) 7 was positive, while CK20 and thyroid transcription factor 1 were negative. A positron emission tomography/computed tomography (CT) fusion scan demonstrated 3 areas of enhanced uptake: one in the right side of the thyroid, a second corresponding to the mass on the left side of the neck at the level of the trapezius muscle, and a third in the left masseter muscle. Surgical excision with negative margins with possible chemotherapy was recommended; however, the patient declined treatment and was lost to follow-up until 2 years later when he presented to his primary physician with an additional lesion on his scalp.
Four years after the biopsy, the patient presented to the dermatology department with additional tumor nodules including a 4-cm, annular, indurated, focally eroded plaque on the left side of the lateral neck (Figure 1); 3 separate 1-cm nodules on the right side of the lateral neck; and an ulcerated, crusted, 10×8-cm plaque on the posterior aspect of the scalp. Despite the extensive lesions, the patient remained in good health and reported no recent weight loss or signs or symptoms of systemic involvement. The posterior scalp lesion, which developed 2 years after the initial appearance of the mass on the neck and was thought to represent a possible metastasis of the tumor, was biopsied and showed diffuse infiltration of the dermis by poorly differentiated tumor cells with vacuolated cytoplasm arranged in nests and cords and sometimes in a single-file arrangement (Figure 2). A CT scan demonstrated pretracheal lymphadenopathy as well as small intraparenchymal and subpleural pulmonary nodules throughout both lung fields.
Another scalp biopsy was taken. Tumor cells were negative on mucicarmine staining. Additional immunohistochemical staining, including a periodic acid-Schiff stain with diastase digestion for epithelial mucin revealed minimal luminal positivity. Immunostaining was positive for CK7, carcinoembryonic antigen, CD15, estrogen receptor, progesterone receptor, gross cystic disease fluid protein 15 (GCDFP-15), and mammaglobin, and negative for CK20, podoplanin, thyroid transcription factor 1, S-100 protein, p63, and prostate specific antigen. ERBB2 (formerly HER2/neu) staining was negative according to fluorescence in situ hybridization analysis. Tumor cells showed a Ki-67 nuclear proliferation index of greater than 50%, indicating progression to aggressive carcinoma.
Based on the histological and immunochemical studies, the differential diagnosis included primary cutaneous apocrine carcinoma versus breast carcinoma; however, the prolonged clinical progression of these lesions favored a primary cutaneous adnexal tumor over a metastatic adenocarcinoma. Nevertheless, despite the initially indolent growth of the lesions over the first 5 years, the Ki-67 proliferation index and presence of widespread metastases on the posterior scalp indicated progression to an aggressive carcinoma. Chemotherapy was recommended as the treatment of choice. At his most recent follow-up visit 4 months later, the patient chose to begin treatment with tamoxifen and refused other treatment options.
Comment
The distinction between primary adnexal and metastatic adenocarcinomas of the skin is challenging both clinically and histologically. Some pathologists have argued that metastatic breast carcinomas and primary cutaneous apocrine carcinomas are essentially indistinguishable.3 Patients with cutaneous metastases, which occur in approximately 5.3% of all malignancies,4 typically can expect survival of no more than 12 months from the time of detection.1 In contrast, primary apocrine carcinomas of the skin, though much less common, carry a remarkably better prognosis, with 5-year relative survival rates of 95.5% and 85.5% reported for patients with localized disease and spread to regional lymph nodes, respectively.2
Fewer than 100 cases of primary cutaneous adnexal (apocrine) carcinomas have been reported overall, with the earliest known report dating back to 1944.5 According to the literature, primary apocrine carcinomas were diagnosed at a median age of 66 years and were slightly more common in females than males.2,6 Apocrine carcinomas were seen most frequently on the head, neck, and trunk,2 generally presenting in the form of asymptomatic nodules or plaques of 2 to 3 cm in size, with gradual progression occurring over months to years.6 Approximately 40% of patients have been reported with positive regional lymph nodes at diagnosis. Treatment of apocrine carcinoma typically has involved local excision with clear margins with or without lymph node dissection. Chemotherapy and radiation therapy have shown no proven benefit.7
Currently, there is no standardized approach to evaluating patients with possible cutaneous metastasis versus primary cutaneous adnexal carcinomas. Imaging studies such as mammography and abdominal CT typically reveal an internal primary cancer in one-third of patients. However, additional studies such as gastrointestinal radiography, chest and pelvic CT, barium enema, and intravenous pyelogram have shown to be of limited value.8 Although specificity and sensitivity of immunohistochemistry is limited, a number of immunomarkers, including CK7 and CK20, are routinely studied to narrow the differential diagnosis of a cutaneous neoplasm of unclear origin. Urothelial, gastric, colorectal, and pancreatic carcinomas generally are positive for CK20; CK7-positive adenocarcinomas include salivary, non-small cell lung, breast, ovarian, pancreatic, endometrial, and transitional cell adenocarcinomas. Carcinomas negative for both CK7 and CK20 include colorectal, hepatocellular, renal cell, prostate, and squamous cell carcinoma of the lung.
The presence of positive staining for estrogen and progesterone receptors as well as GCDFP-15 and mammaglobin raised the possibility of primary breast adenocarcinoma in our patient, but given that these markers can be positive in primary cutaneous adnexal tumors, immunohistochemistry results were not able to provide a definitive primary site. The overall staining pattern was nearly identical to 26 cases of primary cutaneous cribriform apocrine carcinoma, which was found to be positive for CK7 and carcinoembryonic antigen, and negative for CK20 and S-100. The only difference was in GCDFP-15 staining, which was positive in our case and negative in the cases of cribriform apocrine carcinoma.9 Histologic features favoring a primary apocrine origin include normal apocrine glands in the vicinity, glandular structures with decapitation secretion high in the dermis, and intracytoplasmic iron granules.10 Additionally, positive estrogen receptor staining appears to be much more common in apocrine carcinomas (5/10) than in eccrine carcinomas (1/7).11
A number of other markers have been investigated for possible diagnostic utility for distinction between primary adnexal carcinomas and metastatic adenocarcinomas. The nuclear transcription factor p63, which plays a role in keratinocyte differentiation, is preferentially expressed in a number of primary adnexal carcinomas and is purported to be the most sensitive marker overall, with a sensitivity of 78% to 91%.12-14 However, p63 has shown incomplete specificity for primary adnexal neoplasms, having been reported as positive in 11% to 22% of adenocarcinomas metastatic to skin.15-18 Nestin and CK15, which are expressed in hair follicle progenitor cells, also are potential specific markers for some primary adnexal lesions, specifically eccrine carcinoma, porocarcinoma, hidradenocarcinoma, and microcystic adnexal carcinoma; however, in one report, none of the apocrine carcinomas were positive for p63, cytokeratin 15, or D2-40.19 Thus, while markers for some primary adnexal neoplasms are emerging, specific tests at the immunohistochemical level for the apocrine carcinoma subgroup are still lacking.
Conclusion
In summary, a conclusive distinction between primary cutaneous apocrine carcinoma and metastatic adenocarcinoma to the skin remains challenging. Although new markers provide more specificity and sensitivity for neoplasms of eccrine origin, these markers do not appear to differentiate between primary apocrine carcinoma and metastatic breast carcinoma. In this case, as in other recent reports, diagnosis remained dependent on the clinical course of the patient. Although considerable progress has been made regarding immunohistochemical analysis of these cases, additional markers, especially ones more specific for primary skin cancers with apocrine differentiation, are still needed.
- Nashan D, Müller ML, Braun-Falco M, et al. Cutaneous metastases of visceral tumours: a review. J Cancer Res Clin Oncol. 2009;135:1-14.
- Blake PW, Bradford PT, Devesa SS, et al. Cutaneous appendageal carcinoma incidence and survival patterns in the United States: a population-based study. Arch Dermatol. 2010;146:625-632.
- Fernandez-Flores A. The elusive differential diagnosis of cutaneous apocrine adenocarcinoma vs. metastasis: the current role of clinical correlation. Acta Dermatovenerol Alp Pannonica Adriat. 2009;18:141-142.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma. A retrospective study of 7316 cancer patients. J Am Acad Dermatol. 1990;22:19-26.
- Horn RC. Malignant papillary cystadenoma of sweat glands with metastases to the regional lymph nodes. Surgery. 1944;16:348-355.
- Pucevich B, Catinchi-Jaime S, Ho J, et al. Invasive primary ductal apocrine adenocarcinoma of axilla: a case report with immunohistochemical profiling and a review of literature. Dermatol Online J. 2008;14:5.
- Vasilakaki T, Skafida E, Moustou E, et al. Primary cutaneous apocrine carcinoma of sweat glands: a rare case report [published online December 17, 2011]. Case Rep Oncol. 2011;4:597-601.
- Hainsworth JD, Greco FA. Treatment of patients with cancer of an unknown primary site. N Engl J Med. 1993;329:257-263.
- Rutten A, Kutzner H, Mentzel T, et al. Primary cutaneous cribriform apocrine carcinoma: a clinicopathologic and immunohistochemical study of 26 cases of an under-recognized cutaneous adnexal neoplasm. J Am Acad Dermatol. 2009;61:644-651.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever's Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott, Williams, and Wilkins; 2009.
- Le LP, Dias-Santagata D, Pawlak AC, et al. Apocrine-eccrine carcinomas: molecular and immunohistochemical analyses. PLoS One. 2012;7:e47290.
- Levrero M, De Laurenzi V, Costanzo A, et al. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci. 2000;113:1661-1670.
- Pellegrini G, Dellambra E, Golisano O, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A. 2001;98:3156-3161.
- Reis-Filho JS, Torio B, Albergaria A, et al. p63 expression in normal skin and usual cutaneous carcinomas. J Cutan Pathol. 2002;29:517-523.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Mahalingam M, Nguyen LP, Richards JE, et al. The diagnostic utility of immunohistochemistry in distinguishing primary skin adnexal carcinomas from metastatic adenocarcinoma to skin: an immunohistochemical reappraisal using cytokeratin 15, nestin, p63, D2-40, and calretinin. Mod Pathol. 2010;23:713-719.
- Nashan D, Müller ML, Braun-Falco M, et al. Cutaneous metastases of visceral tumours: a review. J Cancer Res Clin Oncol. 2009;135:1-14.
- Blake PW, Bradford PT, Devesa SS, et al. Cutaneous appendageal carcinoma incidence and survival patterns in the United States: a population-based study. Arch Dermatol. 2010;146:625-632.
- Fernandez-Flores A. The elusive differential diagnosis of cutaneous apocrine adenocarcinoma vs. metastasis: the current role of clinical correlation. Acta Dermatovenerol Alp Pannonica Adriat. 2009;18:141-142.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma. A retrospective study of 7316 cancer patients. J Am Acad Dermatol. 1990;22:19-26.
- Horn RC. Malignant papillary cystadenoma of sweat glands with metastases to the regional lymph nodes. Surgery. 1944;16:348-355.
- Pucevich B, Catinchi-Jaime S, Ho J, et al. Invasive primary ductal apocrine adenocarcinoma of axilla: a case report with immunohistochemical profiling and a review of literature. Dermatol Online J. 2008;14:5.
- Vasilakaki T, Skafida E, Moustou E, et al. Primary cutaneous apocrine carcinoma of sweat glands: a rare case report [published online December 17, 2011]. Case Rep Oncol. 2011;4:597-601.
- Hainsworth JD, Greco FA. Treatment of patients with cancer of an unknown primary site. N Engl J Med. 1993;329:257-263.
- Rutten A, Kutzner H, Mentzel T, et al. Primary cutaneous cribriform apocrine carcinoma: a clinicopathologic and immunohistochemical study of 26 cases of an under-recognized cutaneous adnexal neoplasm. J Am Acad Dermatol. 2009;61:644-651.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever's Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott, Williams, and Wilkins; 2009.
- Le LP, Dias-Santagata D, Pawlak AC, et al. Apocrine-eccrine carcinomas: molecular and immunohistochemical analyses. PLoS One. 2012;7:e47290.
- Levrero M, De Laurenzi V, Costanzo A, et al. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci. 2000;113:1661-1670.
- Pellegrini G, Dellambra E, Golisano O, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A. 2001;98:3156-3161.
- Reis-Filho JS, Torio B, Albergaria A, et al. p63 expression in normal skin and usual cutaneous carcinomas. J Cutan Pathol. 2002;29:517-523.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Mahalingam M, Nguyen LP, Richards JE, et al. The diagnostic utility of immunohistochemistry in distinguishing primary skin adnexal carcinomas from metastatic adenocarcinoma to skin: an immunohistochemical reappraisal using cytokeratin 15, nestin, p63, D2-40, and calretinin. Mod Pathol. 2010;23:713-719.
Practice Points
- Despite advances in immunohistochemical analysis, differentiating between primary apocrine carcinoma and metastatic breast carcinoma remains largely dependent on the clinical course of the patient.
- Treatment of apocrine carcinoma typically involves local excision with clear margins with or without lymph node dissection.
Potential Operating Room Fire Hazard of Bone Cement
Approximately 600 cases of operating room (OR) fires are reported annually.1 The incidence of OR fires in the United States equals that of wrong-site surgeries, and 20% of cases have associated morbidity.1,2 The estimated mortality rate is 1 to 2 cases per year.3-5 The most commonly involved anatomical regions are the airway (33%) and the face (28%).4 Most surgical fires are reported in anesthetized patients with open oxygen delivery systems during head, neck, and upper chest surgeries; electrosurgical instruments are the ignition source in 90% of these cases.6 Despite extensive fire safety education and training, complete elimination of OR fires still has not been achieved.
Each fire requires an ignition source, a fuel source, and an oxidizer.7 In the OR, the 2 most common oxidizers are oxygen and nitrous oxide. Head and neck surgeries have a high concentration of these gases near the working field and therefore a higher risk and incidence of fires. Furthermore, surgical drapes and equipment (eg, closed or semi-closed breathing systems, masks) may potentiate this risk by reducing ventilation in areas where gases can accumulate and ignite. Ignition sources provide the energy that starts fires; common sources are electrocautery, lasers, fiber-optic light cords, drills/burrs, and defibrillator paddles. Fires are propagated by fuel sources, which encompass any flammable material, including tracheal tubes, sponges, alcohol-based solutions, hair, gastrointestinal tract gases, gloves, and packaging materials.8 Of note, alcohol-based skin-preparation agents emit flammable vapors that can ignite.9-14 Before draping or exposure to an ignition source, chlorhexidine gluconate-based preparations must be allowed to dry for at least 3 minutes after application to hairless skin and up to 1 hour after application to hair.15 Inadequate drying poses a risk of fire.10We present the case of an OR fire ignited by electrocautery near freshly applied bone cement. No patient information is disclosed in this report.
Case Report
Our patient was evaluated in clinic and scheduled for total knee arthroplasty (TKA). All preoperative safety checklists and time-out procedures were followed and documented at the start of surgery. The TKA was performed with a standard medial patellar arthrotomy. Tourniquet control was used after Esmarch exsanguination. The surgery proceeded uneventfully until just after the bone cement was applied to the tibial surface. The surgeon was using a Bovie to resect residual lateral meniscus tissue when a fire instantaneously erupted within the joint space. Fortunately, the surgeon quickly suffocated the fire with a dry towel. The ignited bone cement was removed, and the patient was examined. There was no injury to surrounding tissue or joint space. Surgery was resumed with application of new bone cement to the tibial surface. The artificial joint was then successfully implanted and the case completed without further incident. The patient was discharged from the hospital and followed up as an outpatient without any postoperative complications.
Discussion
Bone cement, which is commonly used in artificial joint anchoring, craniofacial reconstruction, and vertebroplasty, has liquid and powder components. The liquid monomer methyl methacrylate (MMA) is colorless and flammable and has a distinct odor.16 Exposure to heat or light can prematurely polymerize MMA, requiring the addition of hydroquinone to inhibit the reaction.16 The powder polymethylmethacrylate affords excellent structural support, radiopacity, and facility of use.17 Dibenzoyl peroxide and N,N-dimethyl-p-toluidine are added to the powder to facilitate the polymerization reaction at room temperature (ie, cold curing of cement). Premature application of unpolymerized cement increases the risk of fire from the volatile liquid component.
In the OR, bone cement is prepared by mixing together its powder and liquid components.18 The reaction is exothermic polymerization. The liquid is highly volatile and flammable in both liquid and vapor states.16,19 The vapors are denser than air and can concentrate in poorly ventilated areas. The OR and the application site must be adequately ventilated to eliminate any pockets of vapor accumulation.16 A vacuum mixer can be used to minimize fume exposure, enhance cement strength, and reduce fire risk while combining the 2 components.
MMA’s flash point, the temperature at which the fumes could ignite in the presence of an ignition source, is 10.5ºC. The auto-ignition point, the temperature at which MMA spontaneously combusts, is 421ºC.20 The OR is usually warmer than the flash point temperature, but the electrocautery tip can generate up to 1200ºC of heat.21 Therefore, bone cement is a potential fire hazard, and use of Bovies or other ignition sources in its vicinity must be avoided.
The Table lists the recommended times for preparing various bone cement products.22,23Mix time is the time needed to combine the liquid and powder into a homogenous putty. 
For OR fires, the standard guidelines for rapid containment and safety apply. These guidelines are detailed by the American Society of Anesthesiologists.8 Briefly, delivery of all airway gases to the patient is discontinued. Any burning material is removed and extinguished by the OR staff.1 Carbon dioxide fire extinguishers are used to put out any patient fires and minimize the risk of thermal injury. (Water-mist fire extinguishers can contaminate surgical wounds and present an electric shock hazard with surgical devices and should be avoided.24) If a fire occurs in a patient’s airway, the tracheal tube is removed, and airway patency is maintained with use of other invasive or noninvasive techniques. Often, noninvasive positive pressure ventilation without supplemental oxygen is used until the fire is controlled and the patient is safe. Once the patient fire is controlled, ventilation is restarted, and the patient is evacuated from the OR and away from any other hazards, as required. Last, the patient is physically examined for any injuries and treated.24 Specific to TKA, the procedure is resumed after removal of all bone cement, inspection of the operative site, and treatment of any fire-related injuries.
We have reported the case of an OR fire during TKA. Appropriate selection and use of bone cement products, proper assessment of set time, and avoidance of electrocautery near cement application sites may dramatically reduce associated fire risks.
Am J Orthop. 2016;45(7):E512-E514. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Hart SR, Yajnik A, Ashford J, Springer R, Harvey S. Operating room fire safety. Ochsner J. 2011;11(1):37-42.
2. American Society of Anesthesiologists Task Force on Operating Room Fires; Caplan RA, Barker SJ, Connis RT, et al. Practice advisory for the prevention and management of operating room fires. Anesthesiology. 2008;108(5):786-801.
3. Bruley M. Surgical fires: perioperative communication is essential to prevent this rare but devastating complication. Qual Saf HealthCare. 2004;13(6):467-471.
4. Daane SP, Toth BA. Fire in the operating room: principles and prevention. Plast Reconstr Surg. 2005;115(5):73e-75e.
5. Rinder CS. Fire safety in the operating room. Curr Opin Anaesthesiol. 2008;21(6):790-795.
6. Mathias JM. Fast action, team coordination critical when surgical fires occur. OR Manager. 2013;29(11):9-10.
7. Culp WC Jr, Kimbrough BA, Luna S. Flammability of surgical drapes and materials in varying concentrations of oxygen. Anesthesiology. 2013;119(4):770-776.
8. Apfelbaum JL, Caplan RA, Barker SJ, et al; American Society of Anesthesiologists Task Force on Operating Room Fires. Practice advisory for the prevention and management of operating room fires: an updated report by the American Society of Anesthesiologists Task Force on Operating Room Fires. Anesthesiology. 2013;118(2):271-290.
9. Barker SJ, Polson JS. Fire in the operating room: a case report and laboratory study. Anesth Analg. 2001;93(4):960-965.
10. Fire hazard created by the misuse of DuraPrep solution. Health Devices. 1998;27(11):400-402.
11. Hurt TL, Schweich PJ. Do not get burned: preventing iatrogenic fires and burns in the emergency department. Pediatr Emerg Care. 2003;19(4):255-259.
12. Prasad R, Quezado Z, St Andre A, O’Grady NP. Fires in the operating room and intensive care unit: awareness is the key to prevention. Anesth Analg. 2006;102(1):172-174.
13. Shah SC. Correspondence: operating room flash fire. Anesth Analg. 1974;53(2):288.
14. Tooher R, Maddern GJ, Simpson J. Surgical fires and alcohol-based skin preparations. ANZ J Surg. 2004;74(5):382-385.
15. Using ChloraPrep™ products and the skin prep portfolio. http://www.carefusion.com/medical-products/infection-prevention/skin-preparation/using-chloraprep.aspx. Accessed October 7, 2016.16. DePuy CMW. DePuy Orthopaedic Gentamicin Bone Cements. Blackpool, United Kingdom: DePuy International Ltd; 2008.
17. Dall’Oca C, Maluta T, Cavani F, et al. The biocompatibility of porous vs non-porous bone cements: a new methodological approach. Eur J Histochem. 2014;58(2):2255.
18. Zimmer Biomet. Bone Cement: Biomet Cement and Cementing Systems. http://www.biomet.com/wps/portal/internet/Biomet/Healthcare-Professionals/products/orthopedics. 2014. Accessed October 7, 2016.
19. Sigma-Aldrich. Methyl methacrylate. http://www.sigmaaldrich.com/catalog/product/aldrich/w400201?lang=en®ion=US. Accessed October 7, 2016.
20. DePuy Synthes. Unmedicated bone cements MSDS. Blackpool, United Kingdom: DePuy International Ltd. http://msdsdigital.com/unmedicated-bone-cements-msds. Accessed October 7, 2016.
21. Mir MR, Sun GS, Wang CM. Electrocautery. http://emedicine.medscape.com/article/2111163-overview#showall. Accessed October 7, 2016.
22. DePuy Synthes. Bone cement time setting.
23. Berry DJ, Lieberman JR, eds. Surgery of the Hip. New York, NY: Elsevier; 2011.
24. ECRI Institute. Surgical Fire Prevention. https://www.ecri.org/Accident_Investigation/Pages/Surgical-Fire-Prevention.aspx. 2014. Accessed October 7, 2016.
Approximately 600 cases of operating room (OR) fires are reported annually.1 The incidence of OR fires in the United States equals that of wrong-site surgeries, and 20% of cases have associated morbidity.1,2 The estimated mortality rate is 1 to 2 cases per year.3-5 The most commonly involved anatomical regions are the airway (33%) and the face (28%).4 Most surgical fires are reported in anesthetized patients with open oxygen delivery systems during head, neck, and upper chest surgeries; electrosurgical instruments are the ignition source in 90% of these cases.6 Despite extensive fire safety education and training, complete elimination of OR fires still has not been achieved.
Each fire requires an ignition source, a fuel source, and an oxidizer.7 In the OR, the 2 most common oxidizers are oxygen and nitrous oxide. Head and neck surgeries have a high concentration of these gases near the working field and therefore a higher risk and incidence of fires. Furthermore, surgical drapes and equipment (eg, closed or semi-closed breathing systems, masks) may potentiate this risk by reducing ventilation in areas where gases can accumulate and ignite. Ignition sources provide the energy that starts fires; common sources are electrocautery, lasers, fiber-optic light cords, drills/burrs, and defibrillator paddles. Fires are propagated by fuel sources, which encompass any flammable material, including tracheal tubes, sponges, alcohol-based solutions, hair, gastrointestinal tract gases, gloves, and packaging materials.8 Of note, alcohol-based skin-preparation agents emit flammable vapors that can ignite.9-14 Before draping or exposure to an ignition source, chlorhexidine gluconate-based preparations must be allowed to dry for at least 3 minutes after application to hairless skin and up to 1 hour after application to hair.15 Inadequate drying poses a risk of fire.10We present the case of an OR fire ignited by electrocautery near freshly applied bone cement. No patient information is disclosed in this report.
Case Report
Our patient was evaluated in clinic and scheduled for total knee arthroplasty (TKA). All preoperative safety checklists and time-out procedures were followed and documented at the start of surgery. The TKA was performed with a standard medial patellar arthrotomy. Tourniquet control was used after Esmarch exsanguination. The surgery proceeded uneventfully until just after the bone cement was applied to the tibial surface. The surgeon was using a Bovie to resect residual lateral meniscus tissue when a fire instantaneously erupted within the joint space. Fortunately, the surgeon quickly suffocated the fire with a dry towel. The ignited bone cement was removed, and the patient was examined. There was no injury to surrounding tissue or joint space. Surgery was resumed with application of new bone cement to the tibial surface. The artificial joint was then successfully implanted and the case completed without further incident. The patient was discharged from the hospital and followed up as an outpatient without any postoperative complications.
Discussion
Bone cement, which is commonly used in artificial joint anchoring, craniofacial reconstruction, and vertebroplasty, has liquid and powder components. The liquid monomer methyl methacrylate (MMA) is colorless and flammable and has a distinct odor.16 Exposure to heat or light can prematurely polymerize MMA, requiring the addition of hydroquinone to inhibit the reaction.16 The powder polymethylmethacrylate affords excellent structural support, radiopacity, and facility of use.17 Dibenzoyl peroxide and N,N-dimethyl-p-toluidine are added to the powder to facilitate the polymerization reaction at room temperature (ie, cold curing of cement). Premature application of unpolymerized cement increases the risk of fire from the volatile liquid component.
In the OR, bone cement is prepared by mixing together its powder and liquid components.18 The reaction is exothermic polymerization. The liquid is highly volatile and flammable in both liquid and vapor states.16,19 The vapors are denser than air and can concentrate in poorly ventilated areas. The OR and the application site must be adequately ventilated to eliminate any pockets of vapor accumulation.16 A vacuum mixer can be used to minimize fume exposure, enhance cement strength, and reduce fire risk while combining the 2 components.
MMA’s flash point, the temperature at which the fumes could ignite in the presence of an ignition source, is 10.5ºC. The auto-ignition point, the temperature at which MMA spontaneously combusts, is 421ºC.20 The OR is usually warmer than the flash point temperature, but the electrocautery tip can generate up to 1200ºC of heat.21 Therefore, bone cement is a potential fire hazard, and use of Bovies or other ignition sources in its vicinity must be avoided.
The Table lists the recommended times for preparing various bone cement products.22,23Mix time is the time needed to combine the liquid and powder into a homogenous putty.
For OR fires, the standard guidelines for rapid containment and safety apply. These guidelines are detailed by the American Society of Anesthesiologists.8 Briefly, delivery of all airway gases to the patient is discontinued. Any burning material is removed and extinguished by the OR staff.1 Carbon dioxide fire extinguishers are used to put out any patient fires and minimize the risk of thermal injury. (Water-mist fire extinguishers can contaminate surgical wounds and present an electric shock hazard with surgical devices and should be avoided.24) If a fire occurs in a patient’s airway, the tracheal tube is removed, and airway patency is maintained with use of other invasive or noninvasive techniques. Often, noninvasive positive pressure ventilation without supplemental oxygen is used until the fire is controlled and the patient is safe. Once the patient fire is controlled, ventilation is restarted, and the patient is evacuated from the OR and away from any other hazards, as required. Last, the patient is physically examined for any injuries and treated.24 Specific to TKA, the procedure is resumed after removal of all bone cement, inspection of the operative site, and treatment of any fire-related injuries.
We have reported the case of an OR fire during TKA. Appropriate selection and use of bone cement products, proper assessment of set time, and avoidance of electrocautery near cement application sites may dramatically reduce associated fire risks.
Am J Orthop. 2016;45(7):E512-E514. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
Approximately 600 cases of operating room (OR) fires are reported annually.1 The incidence of OR fires in the United States equals that of wrong-site surgeries, and 20% of cases have associated morbidity.1,2 The estimated mortality rate is 1 to 2 cases per year.3-5 The most commonly involved anatomical regions are the airway (33%) and the face (28%).4 Most surgical fires are reported in anesthetized patients with open oxygen delivery systems during head, neck, and upper chest surgeries; electrosurgical instruments are the ignition source in 90% of these cases.6 Despite extensive fire safety education and training, complete elimination of OR fires still has not been achieved.
Each fire requires an ignition source, a fuel source, and an oxidizer.7 In the OR, the 2 most common oxidizers are oxygen and nitrous oxide. Head and neck surgeries have a high concentration of these gases near the working field and therefore a higher risk and incidence of fires. Furthermore, surgical drapes and equipment (eg, closed or semi-closed breathing systems, masks) may potentiate this risk by reducing ventilation in areas where gases can accumulate and ignite. Ignition sources provide the energy that starts fires; common sources are electrocautery, lasers, fiber-optic light cords, drills/burrs, and defibrillator paddles. Fires are propagated by fuel sources, which encompass any flammable material, including tracheal tubes, sponges, alcohol-based solutions, hair, gastrointestinal tract gases, gloves, and packaging materials.8 Of note, alcohol-based skin-preparation agents emit flammable vapors that can ignite.9-14 Before draping or exposure to an ignition source, chlorhexidine gluconate-based preparations must be allowed to dry for at least 3 minutes after application to hairless skin and up to 1 hour after application to hair.15 Inadequate drying poses a risk of fire.10We present the case of an OR fire ignited by electrocautery near freshly applied bone cement. No patient information is disclosed in this report.
Case Report
Our patient was evaluated in clinic and scheduled for total knee arthroplasty (TKA). All preoperative safety checklists and time-out procedures were followed and documented at the start of surgery. The TKA was performed with a standard medial patellar arthrotomy. Tourniquet control was used after Esmarch exsanguination. The surgery proceeded uneventfully until just after the bone cement was applied to the tibial surface. The surgeon was using a Bovie to resect residual lateral meniscus tissue when a fire instantaneously erupted within the joint space. Fortunately, the surgeon quickly suffocated the fire with a dry towel. The ignited bone cement was removed, and the patient was examined. There was no injury to surrounding tissue or joint space. Surgery was resumed with application of new bone cement to the tibial surface. The artificial joint was then successfully implanted and the case completed without further incident. The patient was discharged from the hospital and followed up as an outpatient without any postoperative complications.
Discussion
Bone cement, which is commonly used in artificial joint anchoring, craniofacial reconstruction, and vertebroplasty, has liquid and powder components. The liquid monomer methyl methacrylate (MMA) is colorless and flammable and has a distinct odor.16 Exposure to heat or light can prematurely polymerize MMA, requiring the addition of hydroquinone to inhibit the reaction.16 The powder polymethylmethacrylate affords excellent structural support, radiopacity, and facility of use.17 Dibenzoyl peroxide and N,N-dimethyl-p-toluidine are added to the powder to facilitate the polymerization reaction at room temperature (ie, cold curing of cement). Premature application of unpolymerized cement increases the risk of fire from the volatile liquid component.
In the OR, bone cement is prepared by mixing together its powder and liquid components.18 The reaction is exothermic polymerization. The liquid is highly volatile and flammable in both liquid and vapor states.16,19 The vapors are denser than air and can concentrate in poorly ventilated areas. The OR and the application site must be adequately ventilated to eliminate any pockets of vapor accumulation.16 A vacuum mixer can be used to minimize fume exposure, enhance cement strength, and reduce fire risk while combining the 2 components.
MMA’s flash point, the temperature at which the fumes could ignite in the presence of an ignition source, is 10.5ºC. The auto-ignition point, the temperature at which MMA spontaneously combusts, is 421ºC.20 The OR is usually warmer than the flash point temperature, but the electrocautery tip can generate up to 1200ºC of heat.21 Therefore, bone cement is a potential fire hazard, and use of Bovies or other ignition sources in its vicinity must be avoided.
The Table lists the recommended times for preparing various bone cement products.22,23Mix time is the time needed to combine the liquid and powder into a homogenous putty.
For OR fires, the standard guidelines for rapid containment and safety apply. These guidelines are detailed by the American Society of Anesthesiologists.8 Briefly, delivery of all airway gases to the patient is discontinued. Any burning material is removed and extinguished by the OR staff.1 Carbon dioxide fire extinguishers are used to put out any patient fires and minimize the risk of thermal injury. (Water-mist fire extinguishers can contaminate surgical wounds and present an electric shock hazard with surgical devices and should be avoided.24) If a fire occurs in a patient’s airway, the tracheal tube is removed, and airway patency is maintained with use of other invasive or noninvasive techniques. Often, noninvasive positive pressure ventilation without supplemental oxygen is used until the fire is controlled and the patient is safe. Once the patient fire is controlled, ventilation is restarted, and the patient is evacuated from the OR and away from any other hazards, as required. Last, the patient is physically examined for any injuries and treated.24 Specific to TKA, the procedure is resumed after removal of all bone cement, inspection of the operative site, and treatment of any fire-related injuries.
We have reported the case of an OR fire during TKA. Appropriate selection and use of bone cement products, proper assessment of set time, and avoidance of electrocautery near cement application sites may dramatically reduce associated fire risks.
Am J Orthop. 2016;45(7):E512-E514. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Hart SR, Yajnik A, Ashford J, Springer R, Harvey S. Operating room fire safety. Ochsner J. 2011;11(1):37-42.
2. American Society of Anesthesiologists Task Force on Operating Room Fires; Caplan RA, Barker SJ, Connis RT, et al. Practice advisory for the prevention and management of operating room fires. Anesthesiology. 2008;108(5):786-801.
3. Bruley M. Surgical fires: perioperative communication is essential to prevent this rare but devastating complication. Qual Saf HealthCare. 2004;13(6):467-471.
4. Daane SP, Toth BA. Fire in the operating room: principles and prevention. Plast Reconstr Surg. 2005;115(5):73e-75e.
5. Rinder CS. Fire safety in the operating room. Curr Opin Anaesthesiol. 2008;21(6):790-795.
6. Mathias JM. Fast action, team coordination critical when surgical fires occur. OR Manager. 2013;29(11):9-10.
7. Culp WC Jr, Kimbrough BA, Luna S. Flammability of surgical drapes and materials in varying concentrations of oxygen. Anesthesiology. 2013;119(4):770-776.
8. Apfelbaum JL, Caplan RA, Barker SJ, et al; American Society of Anesthesiologists Task Force on Operating Room Fires. Practice advisory for the prevention and management of operating room fires: an updated report by the American Society of Anesthesiologists Task Force on Operating Room Fires. Anesthesiology. 2013;118(2):271-290.
9. Barker SJ, Polson JS. Fire in the operating room: a case report and laboratory study. Anesth Analg. 2001;93(4):960-965.
10. Fire hazard created by the misuse of DuraPrep solution. Health Devices. 1998;27(11):400-402.
11. Hurt TL, Schweich PJ. Do not get burned: preventing iatrogenic fires and burns in the emergency department. Pediatr Emerg Care. 2003;19(4):255-259.
12. Prasad R, Quezado Z, St Andre A, O’Grady NP. Fires in the operating room and intensive care unit: awareness is the key to prevention. Anesth Analg. 2006;102(1):172-174.
13. Shah SC. Correspondence: operating room flash fire. Anesth Analg. 1974;53(2):288.
14. Tooher R, Maddern GJ, Simpson J. Surgical fires and alcohol-based skin preparations. ANZ J Surg. 2004;74(5):382-385.
15. Using ChloraPrep™ products and the skin prep portfolio. http://www.carefusion.com/medical-products/infection-prevention/skin-preparation/using-chloraprep.aspx. Accessed October 7, 2016.16. DePuy CMW. DePuy Orthopaedic Gentamicin Bone Cements. Blackpool, United Kingdom: DePuy International Ltd; 2008.
17. Dall’Oca C, Maluta T, Cavani F, et al. The biocompatibility of porous vs non-porous bone cements: a new methodological approach. Eur J Histochem. 2014;58(2):2255.
18. Zimmer Biomet. Bone Cement: Biomet Cement and Cementing Systems. http://www.biomet.com/wps/portal/internet/Biomet/Healthcare-Professionals/products/orthopedics. 2014. Accessed October 7, 2016.
19. Sigma-Aldrich. Methyl methacrylate. http://www.sigmaaldrich.com/catalog/product/aldrich/w400201?lang=en®ion=US. Accessed October 7, 2016.
20. DePuy Synthes. Unmedicated bone cements MSDS. Blackpool, United Kingdom: DePuy International Ltd. http://msdsdigital.com/unmedicated-bone-cements-msds. Accessed October 7, 2016.
21. Mir MR, Sun GS, Wang CM. Electrocautery. http://emedicine.medscape.com/article/2111163-overview#showall. Accessed October 7, 2016.
22. DePuy Synthes. Bone cement time setting.
23. Berry DJ, Lieberman JR, eds. Surgery of the Hip. New York, NY: Elsevier; 2011.
24. ECRI Institute. Surgical Fire Prevention. https://www.ecri.org/Accident_Investigation/Pages/Surgical-Fire-Prevention.aspx. 2014. Accessed October 7, 2016.
1. Hart SR, Yajnik A, Ashford J, Springer R, Harvey S. Operating room fire safety. Ochsner J. 2011;11(1):37-42.
2. American Society of Anesthesiologists Task Force on Operating Room Fires; Caplan RA, Barker SJ, Connis RT, et al. Practice advisory for the prevention and management of operating room fires. Anesthesiology. 2008;108(5):786-801.
3. Bruley M. Surgical fires: perioperative communication is essential to prevent this rare but devastating complication. Qual Saf HealthCare. 2004;13(6):467-471.
4. Daane SP, Toth BA. Fire in the operating room: principles and prevention. Plast Reconstr Surg. 2005;115(5):73e-75e.
5. Rinder CS. Fire safety in the operating room. Curr Opin Anaesthesiol. 2008;21(6):790-795.
6. Mathias JM. Fast action, team coordination critical when surgical fires occur. OR Manager. 2013;29(11):9-10.
7. Culp WC Jr, Kimbrough BA, Luna S. Flammability of surgical drapes and materials in varying concentrations of oxygen. Anesthesiology. 2013;119(4):770-776.
8. Apfelbaum JL, Caplan RA, Barker SJ, et al; American Society of Anesthesiologists Task Force on Operating Room Fires. Practice advisory for the prevention and management of operating room fires: an updated report by the American Society of Anesthesiologists Task Force on Operating Room Fires. Anesthesiology. 2013;118(2):271-290.
9. Barker SJ, Polson JS. Fire in the operating room: a case report and laboratory study. Anesth Analg. 2001;93(4):960-965.
10. Fire hazard created by the misuse of DuraPrep solution. Health Devices. 1998;27(11):400-402.
11. Hurt TL, Schweich PJ. Do not get burned: preventing iatrogenic fires and burns in the emergency department. Pediatr Emerg Care. 2003;19(4):255-259.
12. Prasad R, Quezado Z, St Andre A, O’Grady NP. Fires in the operating room and intensive care unit: awareness is the key to prevention. Anesth Analg. 2006;102(1):172-174.
13. Shah SC. Correspondence: operating room flash fire. Anesth Analg. 1974;53(2):288.
14. Tooher R, Maddern GJ, Simpson J. Surgical fires and alcohol-based skin preparations. ANZ J Surg. 2004;74(5):382-385.
15. Using ChloraPrep™ products and the skin prep portfolio. http://www.carefusion.com/medical-products/infection-prevention/skin-preparation/using-chloraprep.aspx. Accessed October 7, 2016.16. DePuy CMW. DePuy Orthopaedic Gentamicin Bone Cements. Blackpool, United Kingdom: DePuy International Ltd; 2008.
17. Dall’Oca C, Maluta T, Cavani F, et al. The biocompatibility of porous vs non-porous bone cements: a new methodological approach. Eur J Histochem. 2014;58(2):2255.
18. Zimmer Biomet. Bone Cement: Biomet Cement and Cementing Systems. http://www.biomet.com/wps/portal/internet/Biomet/Healthcare-Professionals/products/orthopedics. 2014. Accessed October 7, 2016.
19. Sigma-Aldrich. Methyl methacrylate. http://www.sigmaaldrich.com/catalog/product/aldrich/w400201?lang=en®ion=US. Accessed October 7, 2016.
20. DePuy Synthes. Unmedicated bone cements MSDS. Blackpool, United Kingdom: DePuy International Ltd. http://msdsdigital.com/unmedicated-bone-cements-msds. Accessed October 7, 2016.
21. Mir MR, Sun GS, Wang CM. Electrocautery. http://emedicine.medscape.com/article/2111163-overview#showall. Accessed October 7, 2016.
22. DePuy Synthes. Bone cement time setting.
23. Berry DJ, Lieberman JR, eds. Surgery of the Hip. New York, NY: Elsevier; 2011.
24. ECRI Institute. Surgical Fire Prevention. https://www.ecri.org/Accident_Investigation/Pages/Surgical-Fire-Prevention.aspx. 2014. Accessed October 7, 2016.