User login
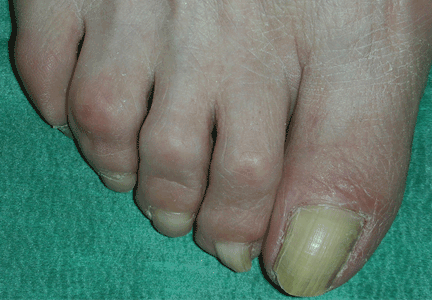
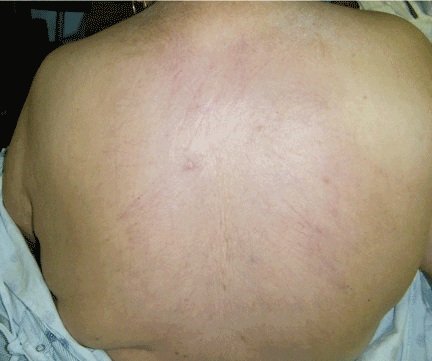
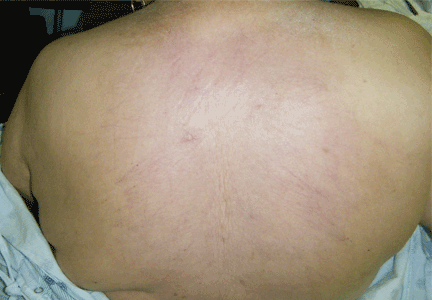
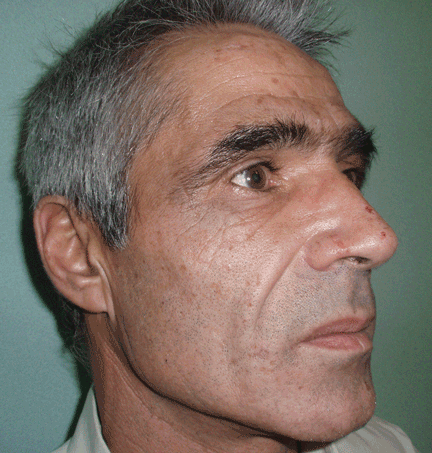
Thick skin on the back
Q: Which is the correct diagnosis?
- Scleroderma (systemic sclerosis)
- Scleredema diabeticorum
- Amyloidosis
- Cutaneous sarcoidosis
- Porphyria cutanea tarda
A: The correct answer is scleredema diabeticorum, a common, underdiagnosed skin manifestation of uncontrolled diabetes mellitus seen in 2.5% to 14% of diabetic patients.1,2 It most often presents with the insidious onset of painless induration and nonpitting thickening of the skin, predominantly on the upper back and neck. Biopsy of the skin usually reveals thickening of the dermis with deposition of collagen and hyaluronic acid without an inflammatory infiltrate.3
Of note, patients may present with similar skin changes acutely in conditions such as postinfectious scleredema (scleredema of Buschke) and paraproteinemias.
Treatment of scleredema is usually difficult, but options include radiotherapy, ultraviolet light therapy, low-dose methotrexate, psoralen, and extracorporeal photopheresis.4–7
- Cole GW, Headley J, Skowsky R. Scleredema diabeticorum: a common and distinct cutaneous manifestation of diabetes mellitus. Diabetes Care 1983; 6:189–192.
- Sattar MA, Diab S, Sugathan TN, Sivanandasingham P, Fenech FF. Scleroedema diabeticorum: a minor but often unrecognized complication of diabetes mellitus. Diabet Med 1988; 5:465–468.
- Varga J, Gotta S, Li L, Sollberg S, Di Leonardo M. Scleredema adultorum: case report and demonstration of abnormal expression of extracellular matrix genes in skin fibroblasts in vivo and in vitro. Br J Dermatol 1995; 132:992–999.
- Seyger MM, van den Hoogen FH, de Mare S, van Haelst U, de Jong EM. A patient with a severe scleroedema diabeticorum, partially responding to low-dose methotrexate. Dermatology 1999; 198:177–179.
- Lee MW, Choi JH, Sung KJ, Moon KC, Koh JK. Electron beam therapy in patients with scleredema. Acta Derm Venereol 2000; 80:307–308.
- Bowen AR, Smith L, Zone JJ. Scleredema adultorum of Buschke treated with radiation. Arch Dermatol 2003; 139:780–784.
- Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum 2006; 35:355–359.
Q: Which is the correct diagnosis?
- Scleroderma (systemic sclerosis)
- Scleredema diabeticorum
- Amyloidosis
- Cutaneous sarcoidosis
- Porphyria cutanea tarda
A: The correct answer is scleredema diabeticorum, a common, underdiagnosed skin manifestation of uncontrolled diabetes mellitus seen in 2.5% to 14% of diabetic patients.1,2 It most often presents with the insidious onset of painless induration and nonpitting thickening of the skin, predominantly on the upper back and neck. Biopsy of the skin usually reveals thickening of the dermis with deposition of collagen and hyaluronic acid without an inflammatory infiltrate.3
Of note, patients may present with similar skin changes acutely in conditions such as postinfectious scleredema (scleredema of Buschke) and paraproteinemias.
Treatment of scleredema is usually difficult, but options include radiotherapy, ultraviolet light therapy, low-dose methotrexate, psoralen, and extracorporeal photopheresis.4–7
Q: Which is the correct diagnosis?
- Scleroderma (systemic sclerosis)
- Scleredema diabeticorum
- Amyloidosis
- Cutaneous sarcoidosis
- Porphyria cutanea tarda
A: The correct answer is scleredema diabeticorum, a common, underdiagnosed skin manifestation of uncontrolled diabetes mellitus seen in 2.5% to 14% of diabetic patients.1,2 It most often presents with the insidious onset of painless induration and nonpitting thickening of the skin, predominantly on the upper back and neck. Biopsy of the skin usually reveals thickening of the dermis with deposition of collagen and hyaluronic acid without an inflammatory infiltrate.3
Of note, patients may present with similar skin changes acutely in conditions such as postinfectious scleredema (scleredema of Buschke) and paraproteinemias.
Treatment of scleredema is usually difficult, but options include radiotherapy, ultraviolet light therapy, low-dose methotrexate, psoralen, and extracorporeal photopheresis.4–7
- Cole GW, Headley J, Skowsky R. Scleredema diabeticorum: a common and distinct cutaneous manifestation of diabetes mellitus. Diabetes Care 1983; 6:189–192.
- Sattar MA, Diab S, Sugathan TN, Sivanandasingham P, Fenech FF. Scleroedema diabeticorum: a minor but often unrecognized complication of diabetes mellitus. Diabet Med 1988; 5:465–468.
- Varga J, Gotta S, Li L, Sollberg S, Di Leonardo M. Scleredema adultorum: case report and demonstration of abnormal expression of extracellular matrix genes in skin fibroblasts in vivo and in vitro. Br J Dermatol 1995; 132:992–999.
- Seyger MM, van den Hoogen FH, de Mare S, van Haelst U, de Jong EM. A patient with a severe scleroedema diabeticorum, partially responding to low-dose methotrexate. Dermatology 1999; 198:177–179.
- Lee MW, Choi JH, Sung KJ, Moon KC, Koh JK. Electron beam therapy in patients with scleredema. Acta Derm Venereol 2000; 80:307–308.
- Bowen AR, Smith L, Zone JJ. Scleredema adultorum of Buschke treated with radiation. Arch Dermatol 2003; 139:780–784.
- Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum 2006; 35:355–359.
- Cole GW, Headley J, Skowsky R. Scleredema diabeticorum: a common and distinct cutaneous manifestation of diabetes mellitus. Diabetes Care 1983; 6:189–192.
- Sattar MA, Diab S, Sugathan TN, Sivanandasingham P, Fenech FF. Scleroedema diabeticorum: a minor but often unrecognized complication of diabetes mellitus. Diabet Med 1988; 5:465–468.
- Varga J, Gotta S, Li L, Sollberg S, Di Leonardo M. Scleredema adultorum: case report and demonstration of abnormal expression of extracellular matrix genes in skin fibroblasts in vivo and in vitro. Br J Dermatol 1995; 132:992–999.
- Seyger MM, van den Hoogen FH, de Mare S, van Haelst U, de Jong EM. A patient with a severe scleroedema diabeticorum, partially responding to low-dose methotrexate. Dermatology 1999; 198:177–179.
- Lee MW, Choi JH, Sung KJ, Moon KC, Koh JK. Electron beam therapy in patients with scleredema. Acta Derm Venereol 2000; 80:307–308.
- Bowen AR, Smith L, Zone JJ. Scleredema adultorum of Buschke treated with radiation. Arch Dermatol 2003; 139:780–784.
- Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum 2006; 35:355–359.
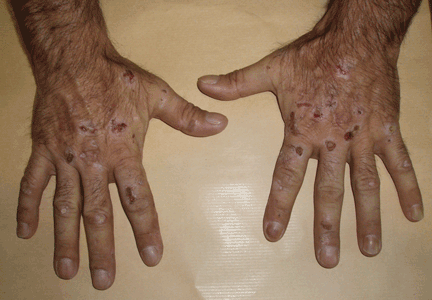
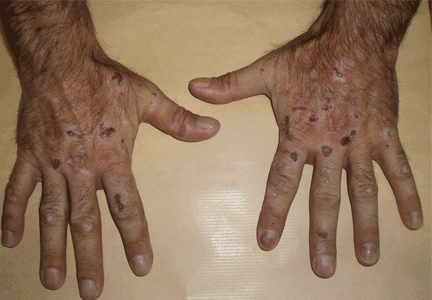
Lesions on the hands, high aminotransferase levels
Q: Which is the most likely diagnosis?
- Addison disease
- Lupus erythematosus
- Polymorphous light eruption
- Porphyria cutanea tarda
- Bullous pemphigoid
A: Urine testing, including examination under ultraviolet light with a Wood lamp, indicates porphyria cutanea tarda. This is the most common porphyria, occurring mainly in men. Its true prevalence is not known but is estimated to be from 1:5,000 to 1:25,000.1
There are three types of porphyria cutanea tarda. About 80% of cases are type I, also referred to as “sporadic.” In type I, levels of uroporphyrinogen decarboxylase (UROD) in red blood cells are normal, but are low in the liver during episodes of the disease. In type II, UROD levels are about 50% below normal in all tissues. Type III is similar to type I, except that it occurs in more than one family member.
The genetic mutation that produces a deficiency of UROD leads to an excess of uroporphyrins and porphyrins that are partially decarboxylated and that irreversibly oxidize. When they are deposited in the skin and the skin is exposed to the sun, they cause the classic cutaneous manifestations.1
Risk factors2 for porphyria cutanea tarda can be extrinsic (eg, high iron blood levels,2,3 excessive ethanol intake, hepatitis C,2,4 human immunodeficiency virus, estrogen use,5 dialysis for end-stage renal disease) or intrinsic (altered iron metabolism or cytochrome P450 function2).
CLINICAL PRESENTATION
The cardinal symptom of porphyria cutanea tarda is photosensitivity, with the development of chronic blistering lesions on sun-exposed areas such as the hands, face, and forearms. Fluid-filled vesicles develop and rupture easily, and the denuded areas become crusted and heal slowly.5 Secondary infections can occur. Previous areas of blisters may appear atrophic, brownish, or violaceous. Small white plaques (milia) are also common and may precede or follow vesicle formation. These cutaneous lesions, however, are not specific to porphyria cutanea tarda and can appear in variegated porphyria and coproporphyria. Hypertrichosis5 and hyperpigmentation are usually present, mainly over the cheekbones and around the eyes. Patches of alopecia and hypopigmented sclerodermiform lesions may also be observed.
Porphyria cutanea tarda is usually accompanied by alterations in liver metabolism, affecting mainly aminotransferases and gammaglutamyltransferase. The absence of hepatitis C infection does not rule out porphyria cutanea tarda. About 50% of patients have pathologic structural changes in the liver such as lobular necrosis or fibrotic tracts, and 15% of patients have cirrhosis at presentation.6 The risk of hepatocellular carcinoma is clearly increased.6 Hepatitis C virus infection, iron overload, and excessive ethanol intake lead to a more severe liver disease.
DIAGNOSIS
The diagnosis of porphyria cutanea tarda is strongly suggested by the characteristic skin lesions in sun-exposed areas, but confirmation requires demonstration of high levels of uroporphyrins or coproporphyrins, or both.
Porphyrins accumulate in the liver, plasma, urine, and feces. Plasma porphyrin levels in porphyria cutanea tarda are usually above 10 μg/dL (normal < 1.4 μg/dL), and plasma fluorescence scanning usually shows a maximum fluorescence emission at an excitation wavelength of 619 nm. In this patient, however, the definitive diagnosis was made by chromatographic separation and the quantification of porphyrins in the urine and feces, which showed a predominance of uroporphyrins and heptacarboxyporphyrins in the urine and an excess of isocoproporphyrins in the feces.1
Analysis of UROD activity in erythrocytes can help determine the type of porphyria cutanea tarda. Type I and type III and porphyria cutanea tarda secondary to hepatotoxin exposure have normal levels, whereas type II and the hepatoerythropoietic form have abnormally low levels. Examination of the urine with a Wood lamp reveals coral pink fluorescence due to elimination of porphyrins, and this is another diagnostic clue.
Conditions that need to be ruled out include viral infection with hepatitis B or C or human immunodeficiency virus, iron overload, and hereditary hemochromatosis. Serum alpha fetoprotein level assessment, liver ultrasonography, or even biopsy may be indicated to exclude hepatocellular carcinoma.
TREATMENT
Once secondary causes of porphyria are excluded or treated (eg, advising the patient to avoid alcohol, discontinuing estrogens or iron intake), the next step in management is to reduce the patient’s porphyrin and iron loads. Phlebotomy is the standard way to reduce stores of iron throughout the body and particularly in the liver. It works by interrupting iron-mediated oxidative inhibition of hepatic UROD and the oxidation of hepatic porphyrinogens to porphyrinogens.
This adjustment must be gradual, with about 450 mL of blood removed at intervals of 1 to 2 weeks.7 This improves the cutaneous symptoms progressively, with resolution of vesicles in 2 to 3 months, improvement of skin fragility in 6 to 9 months, and normalization of porphyrin levels in 13 months. The scleroderma, atrophy, hyperpigmentation, and hypertrichosis respond more slowly and may take years to resolve.
Porphyria cutanea tarda can recur, usually with new exposure to risk factors. Treatment by phlebotomy may be stopped when the serum ferritin level has reached low-normal levels; the porphyrin levels may not yet be normal at that point but may continue to decline without additional phlebotomy sessions.
If phlebotomy is contraindicated, alternatives include iron chelation with deferoxamine (Desferal),7 or a low dose of chloroquine (Aralen) (125–250 mg orally twice a week) or hydroxychloroquine (Plaquenil) (100 mg orally twice a week) to avoid acute hepatic damage that may be caused by the release of large amounts of porphyrins that accompany standard dosing levels.
- Elder GH. Porphyria cutanea tarda. Semin Liver Dis 1998; 18:67–75.
- Mendez M, Rossetti MV, Del C, Batlle AM, Parera VE. The role of inherited and acquired factors in the development of porphyria cutanea tarda in the Argentinean population. J Am Acad Dermatol 2005; 52:417–424.
- Bonkovsky HL, Poh-Fitzpatrick M, Pimstone N, et al. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America. Hepatology 1998; 27:1661–1669.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005–1016.
- Grossman ME, Poh-Fitzpatrick MB. Porphyria cutanea tarda. diagnosis and management. Med Clin North Am 1980; 64:807–827.
- Cortés JM, Oliva H, Paradinas FJ, Hernandez-Guío C. The pathology of the liver in porphyria cutanea tarda. Histopathology 1980; 4:471–485.
- Rocchi E, Gibertini P, Cassanelli M, et al. Iron removal therapy in porphyria cutanea tarda: phlebotomy versus slow subcutaneous desferrioxamine infusion. Br J Dermatol 1986; 114:621–629.
Q: Which is the most likely diagnosis?
- Addison disease
- Lupus erythematosus
- Polymorphous light eruption
- Porphyria cutanea tarda
- Bullous pemphigoid
A: Urine testing, including examination under ultraviolet light with a Wood lamp, indicates porphyria cutanea tarda. This is the most common porphyria, occurring mainly in men. Its true prevalence is not known but is estimated to be from 1:5,000 to 1:25,000.1
There are three types of porphyria cutanea tarda. About 80% of cases are type I, also referred to as “sporadic.” In type I, levels of uroporphyrinogen decarboxylase (UROD) in red blood cells are normal, but are low in the liver during episodes of the disease. In type II, UROD levels are about 50% below normal in all tissues. Type III is similar to type I, except that it occurs in more than one family member.
The genetic mutation that produces a deficiency of UROD leads to an excess of uroporphyrins and porphyrins that are partially decarboxylated and that irreversibly oxidize. When they are deposited in the skin and the skin is exposed to the sun, they cause the classic cutaneous manifestations.1
Risk factors2 for porphyria cutanea tarda can be extrinsic (eg, high iron blood levels,2,3 excessive ethanol intake, hepatitis C,2,4 human immunodeficiency virus, estrogen use,5 dialysis for end-stage renal disease) or intrinsic (altered iron metabolism or cytochrome P450 function2).
CLINICAL PRESENTATION
The cardinal symptom of porphyria cutanea tarda is photosensitivity, with the development of chronic blistering lesions on sun-exposed areas such as the hands, face, and forearms. Fluid-filled vesicles develop and rupture easily, and the denuded areas become crusted and heal slowly.5 Secondary infections can occur. Previous areas of blisters may appear atrophic, brownish, or violaceous. Small white plaques (milia) are also common and may precede or follow vesicle formation. These cutaneous lesions, however, are not specific to porphyria cutanea tarda and can appear in variegated porphyria and coproporphyria. Hypertrichosis5 and hyperpigmentation are usually present, mainly over the cheekbones and around the eyes. Patches of alopecia and hypopigmented sclerodermiform lesions may also be observed.
Porphyria cutanea tarda is usually accompanied by alterations in liver metabolism, affecting mainly aminotransferases and gammaglutamyltransferase. The absence of hepatitis C infection does not rule out porphyria cutanea tarda. About 50% of patients have pathologic structural changes in the liver such as lobular necrosis or fibrotic tracts, and 15% of patients have cirrhosis at presentation.6 The risk of hepatocellular carcinoma is clearly increased.6 Hepatitis C virus infection, iron overload, and excessive ethanol intake lead to a more severe liver disease.
DIAGNOSIS
The diagnosis of porphyria cutanea tarda is strongly suggested by the characteristic skin lesions in sun-exposed areas, but confirmation requires demonstration of high levels of uroporphyrins or coproporphyrins, or both.
Porphyrins accumulate in the liver, plasma, urine, and feces. Plasma porphyrin levels in porphyria cutanea tarda are usually above 10 μg/dL (normal < 1.4 μg/dL), and plasma fluorescence scanning usually shows a maximum fluorescence emission at an excitation wavelength of 619 nm. In this patient, however, the definitive diagnosis was made by chromatographic separation and the quantification of porphyrins in the urine and feces, which showed a predominance of uroporphyrins and heptacarboxyporphyrins in the urine and an excess of isocoproporphyrins in the feces.1
Analysis of UROD activity in erythrocytes can help determine the type of porphyria cutanea tarda. Type I and type III and porphyria cutanea tarda secondary to hepatotoxin exposure have normal levels, whereas type II and the hepatoerythropoietic form have abnormally low levels. Examination of the urine with a Wood lamp reveals coral pink fluorescence due to elimination of porphyrins, and this is another diagnostic clue.
Conditions that need to be ruled out include viral infection with hepatitis B or C or human immunodeficiency virus, iron overload, and hereditary hemochromatosis. Serum alpha fetoprotein level assessment, liver ultrasonography, or even biopsy may be indicated to exclude hepatocellular carcinoma.
TREATMENT
Once secondary causes of porphyria are excluded or treated (eg, advising the patient to avoid alcohol, discontinuing estrogens or iron intake), the next step in management is to reduce the patient’s porphyrin and iron loads. Phlebotomy is the standard way to reduce stores of iron throughout the body and particularly in the liver. It works by interrupting iron-mediated oxidative inhibition of hepatic UROD and the oxidation of hepatic porphyrinogens to porphyrinogens.
This adjustment must be gradual, with about 450 mL of blood removed at intervals of 1 to 2 weeks.7 This improves the cutaneous symptoms progressively, with resolution of vesicles in 2 to 3 months, improvement of skin fragility in 6 to 9 months, and normalization of porphyrin levels in 13 months. The scleroderma, atrophy, hyperpigmentation, and hypertrichosis respond more slowly and may take years to resolve.
Porphyria cutanea tarda can recur, usually with new exposure to risk factors. Treatment by phlebotomy may be stopped when the serum ferritin level has reached low-normal levels; the porphyrin levels may not yet be normal at that point but may continue to decline without additional phlebotomy sessions.
If phlebotomy is contraindicated, alternatives include iron chelation with deferoxamine (Desferal),7 or a low dose of chloroquine (Aralen) (125–250 mg orally twice a week) or hydroxychloroquine (Plaquenil) (100 mg orally twice a week) to avoid acute hepatic damage that may be caused by the release of large amounts of porphyrins that accompany standard dosing levels.
Q: Which is the most likely diagnosis?
- Addison disease
- Lupus erythematosus
- Polymorphous light eruption
- Porphyria cutanea tarda
- Bullous pemphigoid
A: Urine testing, including examination under ultraviolet light with a Wood lamp, indicates porphyria cutanea tarda. This is the most common porphyria, occurring mainly in men. Its true prevalence is not known but is estimated to be from 1:5,000 to 1:25,000.1
There are three types of porphyria cutanea tarda. About 80% of cases are type I, also referred to as “sporadic.” In type I, levels of uroporphyrinogen decarboxylase (UROD) in red blood cells are normal, but are low in the liver during episodes of the disease. In type II, UROD levels are about 50% below normal in all tissues. Type III is similar to type I, except that it occurs in more than one family member.
The genetic mutation that produces a deficiency of UROD leads to an excess of uroporphyrins and porphyrins that are partially decarboxylated and that irreversibly oxidize. When they are deposited in the skin and the skin is exposed to the sun, they cause the classic cutaneous manifestations.1
Risk factors2 for porphyria cutanea tarda can be extrinsic (eg, high iron blood levels,2,3 excessive ethanol intake, hepatitis C,2,4 human immunodeficiency virus, estrogen use,5 dialysis for end-stage renal disease) or intrinsic (altered iron metabolism or cytochrome P450 function2).
CLINICAL PRESENTATION
The cardinal symptom of porphyria cutanea tarda is photosensitivity, with the development of chronic blistering lesions on sun-exposed areas such as the hands, face, and forearms. Fluid-filled vesicles develop and rupture easily, and the denuded areas become crusted and heal slowly.5 Secondary infections can occur. Previous areas of blisters may appear atrophic, brownish, or violaceous. Small white plaques (milia) are also common and may precede or follow vesicle formation. These cutaneous lesions, however, are not specific to porphyria cutanea tarda and can appear in variegated porphyria and coproporphyria. Hypertrichosis5 and hyperpigmentation are usually present, mainly over the cheekbones and around the eyes. Patches of alopecia and hypopigmented sclerodermiform lesions may also be observed.
Porphyria cutanea tarda is usually accompanied by alterations in liver metabolism, affecting mainly aminotransferases and gammaglutamyltransferase. The absence of hepatitis C infection does not rule out porphyria cutanea tarda. About 50% of patients have pathologic structural changes in the liver such as lobular necrosis or fibrotic tracts, and 15% of patients have cirrhosis at presentation.6 The risk of hepatocellular carcinoma is clearly increased.6 Hepatitis C virus infection, iron overload, and excessive ethanol intake lead to a more severe liver disease.
DIAGNOSIS
The diagnosis of porphyria cutanea tarda is strongly suggested by the characteristic skin lesions in sun-exposed areas, but confirmation requires demonstration of high levels of uroporphyrins or coproporphyrins, or both.
Porphyrins accumulate in the liver, plasma, urine, and feces. Plasma porphyrin levels in porphyria cutanea tarda are usually above 10 μg/dL (normal < 1.4 μg/dL), and plasma fluorescence scanning usually shows a maximum fluorescence emission at an excitation wavelength of 619 nm. In this patient, however, the definitive diagnosis was made by chromatographic separation and the quantification of porphyrins in the urine and feces, which showed a predominance of uroporphyrins and heptacarboxyporphyrins in the urine and an excess of isocoproporphyrins in the feces.1
Analysis of UROD activity in erythrocytes can help determine the type of porphyria cutanea tarda. Type I and type III and porphyria cutanea tarda secondary to hepatotoxin exposure have normal levels, whereas type II and the hepatoerythropoietic form have abnormally low levels. Examination of the urine with a Wood lamp reveals coral pink fluorescence due to elimination of porphyrins, and this is another diagnostic clue.
Conditions that need to be ruled out include viral infection with hepatitis B or C or human immunodeficiency virus, iron overload, and hereditary hemochromatosis. Serum alpha fetoprotein level assessment, liver ultrasonography, or even biopsy may be indicated to exclude hepatocellular carcinoma.
TREATMENT
Once secondary causes of porphyria are excluded or treated (eg, advising the patient to avoid alcohol, discontinuing estrogens or iron intake), the next step in management is to reduce the patient’s porphyrin and iron loads. Phlebotomy is the standard way to reduce stores of iron throughout the body and particularly in the liver. It works by interrupting iron-mediated oxidative inhibition of hepatic UROD and the oxidation of hepatic porphyrinogens to porphyrinogens.
This adjustment must be gradual, with about 450 mL of blood removed at intervals of 1 to 2 weeks.7 This improves the cutaneous symptoms progressively, with resolution of vesicles in 2 to 3 months, improvement of skin fragility in 6 to 9 months, and normalization of porphyrin levels in 13 months. The scleroderma, atrophy, hyperpigmentation, and hypertrichosis respond more slowly and may take years to resolve.
Porphyria cutanea tarda can recur, usually with new exposure to risk factors. Treatment by phlebotomy may be stopped when the serum ferritin level has reached low-normal levels; the porphyrin levels may not yet be normal at that point but may continue to decline without additional phlebotomy sessions.
If phlebotomy is contraindicated, alternatives include iron chelation with deferoxamine (Desferal),7 or a low dose of chloroquine (Aralen) (125–250 mg orally twice a week) or hydroxychloroquine (Plaquenil) (100 mg orally twice a week) to avoid acute hepatic damage that may be caused by the release of large amounts of porphyrins that accompany standard dosing levels.
- Elder GH. Porphyria cutanea tarda. Semin Liver Dis 1998; 18:67–75.
- Mendez M, Rossetti MV, Del C, Batlle AM, Parera VE. The role of inherited and acquired factors in the development of porphyria cutanea tarda in the Argentinean population. J Am Acad Dermatol 2005; 52:417–424.
- Bonkovsky HL, Poh-Fitzpatrick M, Pimstone N, et al. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America. Hepatology 1998; 27:1661–1669.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005–1016.
- Grossman ME, Poh-Fitzpatrick MB. Porphyria cutanea tarda. diagnosis and management. Med Clin North Am 1980; 64:807–827.
- Cortés JM, Oliva H, Paradinas FJ, Hernandez-Guío C. The pathology of the liver in porphyria cutanea tarda. Histopathology 1980; 4:471–485.
- Rocchi E, Gibertini P, Cassanelli M, et al. Iron removal therapy in porphyria cutanea tarda: phlebotomy versus slow subcutaneous desferrioxamine infusion. Br J Dermatol 1986; 114:621–629.
- Elder GH. Porphyria cutanea tarda. Semin Liver Dis 1998; 18:67–75.
- Mendez M, Rossetti MV, Del C, Batlle AM, Parera VE. The role of inherited and acquired factors in the development of porphyria cutanea tarda in the Argentinean population. J Am Acad Dermatol 2005; 52:417–424.
- Bonkovsky HL, Poh-Fitzpatrick M, Pimstone N, et al. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America. Hepatology 1998; 27:1661–1669.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005–1016.
- Grossman ME, Poh-Fitzpatrick MB. Porphyria cutanea tarda. diagnosis and management. Med Clin North Am 1980; 64:807–827.
- Cortés JM, Oliva H, Paradinas FJ, Hernandez-Guío C. The pathology of the liver in porphyria cutanea tarda. Histopathology 1980; 4:471–485.
- Rocchi E, Gibertini P, Cassanelli M, et al. Iron removal therapy in porphyria cutanea tarda: phlebotomy versus slow subcutaneous desferrioxamine infusion. Br J Dermatol 1986; 114:621–629.
Pleural effusion from a candy wrapper
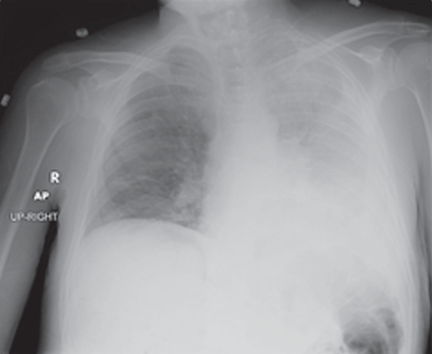
The patient is started on ceftriaxone (Rocephin) and levofloxacin (Levaquin). Left thoracentesis reveals blood-tinged exudative pleural fluid with 1.01 × 109/L nucleated cells, 43% neutrophils, 35% macrophages, and 15% lymphocytes; glucose and pH are normal. Pleural fluid culture is negative, and cytologic study does not reveal malignant cells. Computed tomography of the chest reveals consolidated pneumonia in the left upper lobe, with atelectasis of the left lower lobe and left pleural effusion. The combination of exudative pleural fluid with pH of 7.35, a glucose level of 87 mg/dL, a lactate dehydrogenase level of 406 U/L (normal range 100–200), negative microbiologic test results, and pneumonia supports the diagnosis of uncomplicated parapneumonic pleural effusion, which occurs in 40% of cases of bacterial pneumonia.
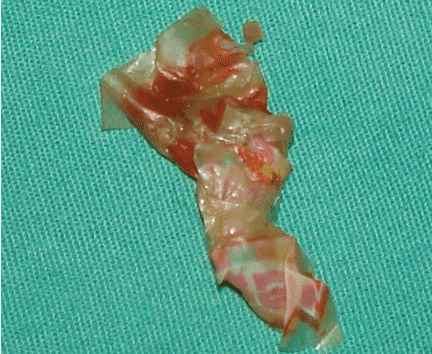
The patient could have been treated only for complicated left pneumonia with parapneumonic pleural effusion. But the high suspicion of foreign-body aspiration, with the history of mild mental retardation and previous recurrent right pneumonia, led to the further workup and appropriate treatment.
Risk factors for tracheobronchial foreign-body aspiration in adults are a depressed mental status or an impairment in the swallowing reflex.2
Aspiration commonly occurs into the right lung because of the almost straight axis between the trachea and the right mainstem bronchus.3 However, aspiration can occur into any part of the lung depending on the position of the patient. Common foreign bodies described in adults are teeth or dental appliances, pins, and semisolid food, especially in elderly people.4 The clinical presentation varies from dyspnea and wheezing to asphyxia and cardiac arrest. Unilateral wheezing—particularly in young children with asthma, cough, or cold symptoms—should raise suspicion of foreign body aspiration.
Delayed complications of bronchial obstruction manifest usually as pneumonia. Subsequently, pleural effusion may develop because of an increased capillary permeability secondary to endothelial injury,5 and may progress to empyema.
Fiberoptic bronchoscopy has become the cornerstone of the diagnostic evaluation in adults with suspected foreign-body aspiration. Treatment options include extraction of the foreign body via flexible bronchoscopy or rigid bronchoscopy. Rigid bronchoscopy has higher success rates but requires general anesthesia.6
A history of choking is not always obtained. Imaging studies do not localize the foreign body in all cases. Many other medical conditions mimic breathing abnormalities similar to those associated with tracheobronchial foreign body in adults. Only a high index of suspicion can ensure proper diagnosis and treatment.
- Light RW, Girard WM, Jenkinson SG, George RB. Parapneumonic effusions. Am J Med 1980; 69:507–512.
- Boyd M, Chatterjee A, Chiles C, Chin R. Tracheobronchial foreign body aspiration in adults. South Med J 2009; 102:171–174.
- Debeljak A, Sorli J, Music E, Kecelj P. Bronchoscopic removal of foreign bodies in adults: experience with 62 patients from 1974–1998. Eur Respir J 1999; 14:792–795.
- Paintal HS, Kuschner WG. Aspiration syndromes: 10 clinical pearls every physician should know. Int J Clin Pract 2007; 61:846–852.
- Sahn SA. Diagnosis and management of parapneumonic effusions and empyema. Clin Infect Dis 2007; 45:1480–1486.
- Swanson KL, Edell ES. Tracheobronchial foreign bodies. Chest Surg Clin North Am 2001; 11:861–872.
The patient is started on ceftriaxone (Rocephin) and levofloxacin (Levaquin). Left thoracentesis reveals blood-tinged exudative pleural fluid with 1.01 × 109/L nucleated cells, 43% neutrophils, 35% macrophages, and 15% lymphocytes; glucose and pH are normal. Pleural fluid culture is negative, and cytologic study does not reveal malignant cells. Computed tomography of the chest reveals consolidated pneumonia in the left upper lobe, with atelectasis of the left lower lobe and left pleural effusion. The combination of exudative pleural fluid with pH of 7.35, a glucose level of 87 mg/dL, a lactate dehydrogenase level of 406 U/L (normal range 100–200), negative microbiologic test results, and pneumonia supports the diagnosis of uncomplicated parapneumonic pleural effusion, which occurs in 40% of cases of bacterial pneumonia.
The patient could have been treated only for complicated left pneumonia with parapneumonic pleural effusion. But the high suspicion of foreign-body aspiration, with the history of mild mental retardation and previous recurrent right pneumonia, led to the further workup and appropriate treatment.
Risk factors for tracheobronchial foreign-body aspiration in adults are a depressed mental status or an impairment in the swallowing reflex.2
Aspiration commonly occurs into the right lung because of the almost straight axis between the trachea and the right mainstem bronchus.3 However, aspiration can occur into any part of the lung depending on the position of the patient. Common foreign bodies described in adults are teeth or dental appliances, pins, and semisolid food, especially in elderly people.4 The clinical presentation varies from dyspnea and wheezing to asphyxia and cardiac arrest. Unilateral wheezing—particularly in young children with asthma, cough, or cold symptoms—should raise suspicion of foreign body aspiration.
Delayed complications of bronchial obstruction manifest usually as pneumonia. Subsequently, pleural effusion may develop because of an increased capillary permeability secondary to endothelial injury,5 and may progress to empyema.
Fiberoptic bronchoscopy has become the cornerstone of the diagnostic evaluation in adults with suspected foreign-body aspiration. Treatment options include extraction of the foreign body via flexible bronchoscopy or rigid bronchoscopy. Rigid bronchoscopy has higher success rates but requires general anesthesia.6
A history of choking is not always obtained. Imaging studies do not localize the foreign body in all cases. Many other medical conditions mimic breathing abnormalities similar to those associated with tracheobronchial foreign body in adults. Only a high index of suspicion can ensure proper diagnosis and treatment.
The patient is started on ceftriaxone (Rocephin) and levofloxacin (Levaquin). Left thoracentesis reveals blood-tinged exudative pleural fluid with 1.01 × 109/L nucleated cells, 43% neutrophils, 35% macrophages, and 15% lymphocytes; glucose and pH are normal. Pleural fluid culture is negative, and cytologic study does not reveal malignant cells. Computed tomography of the chest reveals consolidated pneumonia in the left upper lobe, with atelectasis of the left lower lobe and left pleural effusion. The combination of exudative pleural fluid with pH of 7.35, a glucose level of 87 mg/dL, a lactate dehydrogenase level of 406 U/L (normal range 100–200), negative microbiologic test results, and pneumonia supports the diagnosis of uncomplicated parapneumonic pleural effusion, which occurs in 40% of cases of bacterial pneumonia.
The patient could have been treated only for complicated left pneumonia with parapneumonic pleural effusion. But the high suspicion of foreign-body aspiration, with the history of mild mental retardation and previous recurrent right pneumonia, led to the further workup and appropriate treatment.
Risk factors for tracheobronchial foreign-body aspiration in adults are a depressed mental status or an impairment in the swallowing reflex.2
Aspiration commonly occurs into the right lung because of the almost straight axis between the trachea and the right mainstem bronchus.3 However, aspiration can occur into any part of the lung depending on the position of the patient. Common foreign bodies described in adults are teeth or dental appliances, pins, and semisolid food, especially in elderly people.4 The clinical presentation varies from dyspnea and wheezing to asphyxia and cardiac arrest. Unilateral wheezing—particularly in young children with asthma, cough, or cold symptoms—should raise suspicion of foreign body aspiration.
Delayed complications of bronchial obstruction manifest usually as pneumonia. Subsequently, pleural effusion may develop because of an increased capillary permeability secondary to endothelial injury,5 and may progress to empyema.
Fiberoptic bronchoscopy has become the cornerstone of the diagnostic evaluation in adults with suspected foreign-body aspiration. Treatment options include extraction of the foreign body via flexible bronchoscopy or rigid bronchoscopy. Rigid bronchoscopy has higher success rates but requires general anesthesia.6
A history of choking is not always obtained. Imaging studies do not localize the foreign body in all cases. Many other medical conditions mimic breathing abnormalities similar to those associated with tracheobronchial foreign body in adults. Only a high index of suspicion can ensure proper diagnosis and treatment.
- Light RW, Girard WM, Jenkinson SG, George RB. Parapneumonic effusions. Am J Med 1980; 69:507–512.
- Boyd M, Chatterjee A, Chiles C, Chin R. Tracheobronchial foreign body aspiration in adults. South Med J 2009; 102:171–174.
- Debeljak A, Sorli J, Music E, Kecelj P. Bronchoscopic removal of foreign bodies in adults: experience with 62 patients from 1974–1998. Eur Respir J 1999; 14:792–795.
- Paintal HS, Kuschner WG. Aspiration syndromes: 10 clinical pearls every physician should know. Int J Clin Pract 2007; 61:846–852.
- Sahn SA. Diagnosis and management of parapneumonic effusions and empyema. Clin Infect Dis 2007; 45:1480–1486.
- Swanson KL, Edell ES. Tracheobronchial foreign bodies. Chest Surg Clin North Am 2001; 11:861–872.
- Light RW, Girard WM, Jenkinson SG, George RB. Parapneumonic effusions. Am J Med 1980; 69:507–512.
- Boyd M, Chatterjee A, Chiles C, Chin R. Tracheobronchial foreign body aspiration in adults. South Med J 2009; 102:171–174.
- Debeljak A, Sorli J, Music E, Kecelj P. Bronchoscopic removal of foreign bodies in adults: experience with 62 patients from 1974–1998. Eur Respir J 1999; 14:792–795.
- Paintal HS, Kuschner WG. Aspiration syndromes: 10 clinical pearls every physician should know. Int J Clin Pract 2007; 61:846–852.
- Sahn SA. Diagnosis and management of parapneumonic effusions and empyema. Clin Infect Dis 2007; 45:1480–1486.
- Swanson KL, Edell ES. Tracheobronchial foreign bodies. Chest Surg Clin North Am 2001; 11:861–872.
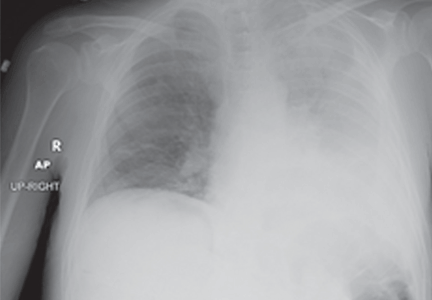
A 19-year-old man with progressive lung infiltrates
A 19-year-old man received induction chemotherapy with idarubicin and cytarabine for secondary acute myeloid leukemia. Subsequently, he developed fever, progressive lung infiltrates, and severe neutropenia. His white blood cell count was 1.1 × 109/L (reference range 4.0–11.0) with 100% lymphocytes; his blood glucose level remained normal.
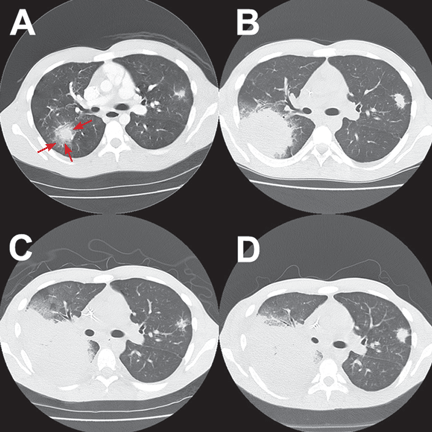
The patient was treated with voriconazole (Vfend) and broad-spectrum antibiotics.
Q: Which is the most likely cause?
- A bacterium
- A virus
- Cryptococcus
- Aspergillus
- Zygomycetes
A: The correct answer is Zygomycetes, the second most common cause of fungal respiratory disease in patients with hematologic malignancies.
UNCOMMON BUT OFTEN FATAL
Zygomycetes is a class of fungi that contains two orders, Mucorales and Entomophthorales. Human disease, which is uncommon but frequently fatal, is predominantly associated with Mucorales and is commonly called mucormycosis.1,2
The major mode of transmission is through inhalation of spores from diverse decaying environmental sources. As Zygomycetes are aerogenous pathogens, they predominantly affect the paranasal sinuses and the lungs. The main risk factors for Zygomycetes infection include diabetes mellitus, hematologic malignancies (predominantly acute leukemias treated with aggressive chemotherapeutic regimens), pharmacologic immunosuppression, solid organ or bone marrow transplantation, and therapy with deferoxamine (Desferal), an iron-chelating agent.1,3
Overall, rhinocerebral disease is the most common manifestation, especially in the setting of diabetic ketoacidosis.1 In hematologic malignancies, the most common presentation is pulmonary zygomycosis with associated profound neutropenia, as neutrophils are the central defense against filamentous fungal hyphae.1,4
The incidence of zygomycosis is increasing, likely owing to the greater number of patients receiving stem cell or solid organ transplants, the use of more aggressive immunosuppressive regimens, prolonged survival, and the frequent prophylactic use of antifungal agents without activity against Zygomycetes.2,5
INFECTION PROGRESSES RAPIDLY
Most patients present with fever, cough, thoracic pain, and dyspnea in association with hypoxemia and pulmonary infiltrates refractory to broad-spectrum antibiotics. Zygomycosis can also present radiographically as pulmonary nodules or consolidations with or without the halo sign or cavitations.6,7
The disease usually progresses rapidly, invading vessels and causing infarction, bleeding, and dissemination to extrapulmonary sites.1,5
In reported cases in patients with acute leukemia who received aggressive chemotherapy, the fungal infection occurred several days after profound neutropenia developed.3,4
SPUTUM, LAVAGE, BIOPSY
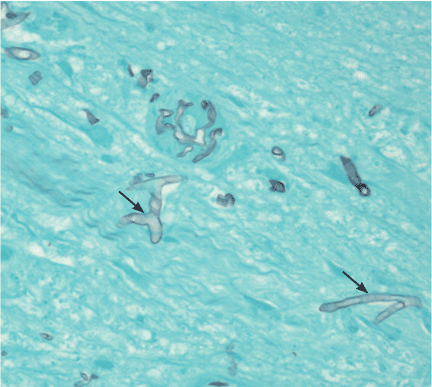
The diagnosis is based on directly identifying the fungus morphologically and on culturing it. However, cultures of sputum, BAL fluid, and blood are usually negative.
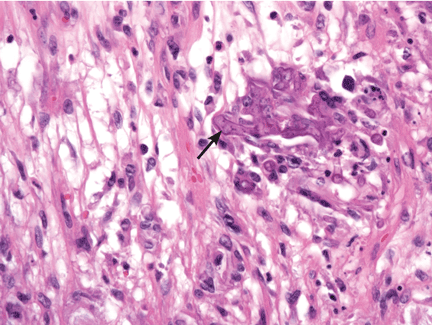
Morphologically, the fungus is broad with irregular walls; it is also nearly aseptate and frequently has right-angle branching. In contrast, Aspergillus is narrow with parallel walls, distinctive septae, and acute branching.2
Of note: physicians need to alert the microbiology laboratory about their clinical suspicion of Zygomycetes infection, because the recovery rate of Zygomycetes in culture is increased by slicing the biopsy specimen in small pieces but not dicing it (to avoid breaking the septae).1
The diagnostic tests include microscopic examination and culture of sputum, BAL fluid, and transbronchial biopsy specimens. If the initial tests are negative but the suspicion of zygomycosis is strong on clinical grounds, then fine-needle aspiration or open lung biopsy should be considered.1
Useful predictors that favor the diagnosis of pulmonary zygomycosis instead of the main alternative, invasive pulmonary aspergillosis, include concomitant sinusitis, voriconazole prophylaxis (due to antifungal pressure), a negative galactomannan test in serum, multiple pulmonary nodules, and pleural effusion.2,8
TREATMENT WITH AMPHOTERICIN
The treatment includes giving effective antifungal agents promptly, correcting hyperglycemia and metabolic acidosis, reversing immunosuppression (if possible), and considering surgical debridement.1,2
Antifungal therapy is with conventional amphotericin B (Amphocin) or its lipid formulation (Abelcet). The lipid formulation is at least as effective as conventional amphotericin B and less nephrotoxic, thus allowing higher doses.1,9 The optimal duration of therapy has not been evaluated, but experts in general treat until the pulmonary and sinus lesions have resolved.2
Posaconazole (Noxafil), a broad-spectrum oral azole, has activity in vitro and is a valuable alternative for patients who have refractory zygomycosis or who cannot tolerate amphotericin B.5,10
The role of echinocandins is unclear, as they do not have in vitro activity against Zygomycetes. However, tests in animals have shown a synergistic effect between the echinocandin caspofungin (Cancidas) and amphotericin B lipid complex.11 Other antifungal agents such as azoles lack activity against Zygomycetes.5
The return of neutrophils plays a substantial role in resolving the infection in neutropenic patients, a proposition supported by reports of the failure of antifungal therapy in patients with persistent neutropenia.1 The addition of granulocyte colony-stimulating factor may accelerate neutrophil recovery and enhance neutrophil activity against opportunistic fungal pathogens.12
Even though progress has been made in the treatment of this disease, the prognosis continues to be poor in patients with hematologic malignancies and pulmonary or disseminated zygomycosis.9
ENDOBRONCHIAL ZYGOMYCOSIS
Aspergillosis is the most common endobronchial fungal disease. Zygomycosis is the third most common, after coccidioidomycosis. In zygomycosis, endobronchial lesions can be found in a third of patients who have pulmonary involvement.6,13,14
The most common predisposing conditions for the development of endobronchial zygomycosis are diabetes and hematologic malignancies associated with neutropenia.14
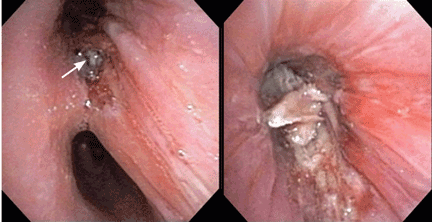
Endobronchial zygomycosis is characterized by a locally invasive gray-white mucoid lesion that blocks a major airway.13 The involved airway is usually edematous and necrotic. The diagnosis can be made by visualizing the organism in bronchial washings, brushings, or endobronchial biopsies.14
If the disease is not promptly diagnosed, the risk of death is very high. The management includes high-dose conventional or lipid amphotericin B and surgical or endobronchial resection.13,15
OUR CASE CONTINUED
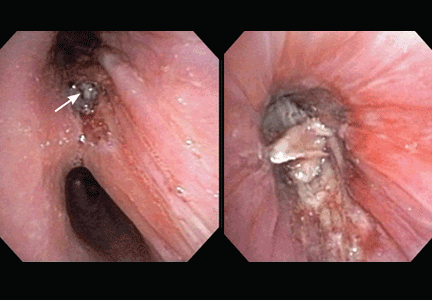
After Zygomycetes was seen in the tissue from his bronchial biopsy, our patient received amphotericin B lipid complex at 5 mg/kg/day (started between images C and D in Figure 1). He had a good initial clinical response, but the infection progressed (image D in Figure 1).
The patient died as a result of massive hemoptysis attributable to the angioinvasive nature of the fungus, which most likely caused an erosion of a major pulmonary vessel.
TAKE-HOME POINTS
- Pulmonary disease is the most common manifestation of zygomycosis in patients with underlying hematologic malignancy. In this setting, zygomycosis has a high rate of morbidity and death.
- Endobronchial lesions can be seen in up to a third of patients with pulmonary zygomycosis.
- Prompt and effective therapy is essential for treatment to be successful.
- Gonzalez CE, Rinaldi MG, Sugar AM. Zygomycosis. Infect Dis Clin North Am 2002; 16:895–914.
- Pyrgos V, Shoham S, Walsh TJ. Pulmonary zygomycosis. Semin Respir Crit Care Med 2008; 29:111–120.
- Kara IO, Tasova Y, Uguz A, Sahin B. Mucormycosis-associated fungal infections in patients with haematologic malignancies. Int J Clin Pract 2007; 63:134–139.
- Pagano L, Offidani M, Fianchi L, et al. Mucormycosis in hematologic patients. Haematologica 2004; 8:207–214.
- van Burik JA, Hare RS, Solomon HF, Corrado ML, Kontoyiannis DP. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis 2006; 42:e61–e65.
- Jamadar DA, Kazerooni EA, Daly BD, White CS, Gross BH. Pulmonary zygomycosis: CT appearance. J Comput Assist Tomogr 1995; 19:733–738.
- Lee YR, Choi YW, Lee KJ, Jeon SC, Park CK, Heo JN. CT halo sign: the spectrum of pulmonary diseases. Br J Radiol 2005; 78:862–865.
- Chamilos G, Marom EM, Lewis RE, Lionakis MS, Kontoyiannis DP. Predictors of pulmonary zygomycosis versus invasive pulmonary aspergillosis in patients with cancer. Clin Infect Dis 2005; 41:60–66.
- Gleissner B, Schilling A, Anagnostopolous I, Siehl I, Thiel E. Improved outcome of zygomycosis in patients with hematological diseases? Leuk Lymphoma 2004; 45:1351–1360.
- Greenberg RN, Mullane K, van Burik JA, et al. Posaconazole as salvage therapy for zygomycosis. Antimicrob Agents Chemother 2006; 50:126–133.
- Spellberg B, Fu Y, Edwards JE, Ibrahim AS. Combination therapy with amphotericin B lipid complex and caspofungin acetate of disseminated zygomycosis in diabetic ketoacidotic mice. Antimicrob Agents Chemother 2005; 49:830–832.
- Liles WC, Huang JE, van Burik JA, Bowden RA, Dale DC. Granulocyte colony-stimulating factor administered in vivo augments neutrophilmediated activity against opportunistic fungal pathogens. J Infect Dis 1997; 175:1012–1015.
- Husari AW, Jensen WA, Kirsch CM, et al. Pulmonary mucormycosis presenting as an endobronchial lesion. Chest 1994; 106:1889–1891.
- Karnak D, Avery RK, Gildea TR, Sahoo D, Mehta AC. Endobronchial fungal disease: an under-recognized entity. Respiration 2007; 74:88–104.
- al-Majed S, al-Kassimi F, Ashour M, Mekki MO, Sadiq S. Removal of endobronchial mucormycosis lesion through a rigid bronchoscope. Thorax 1992; 47:203–204.
A 19-year-old man received induction chemotherapy with idarubicin and cytarabine for secondary acute myeloid leukemia. Subsequently, he developed fever, progressive lung infiltrates, and severe neutropenia. His white blood cell count was 1.1 × 109/L (reference range 4.0–11.0) with 100% lymphocytes; his blood glucose level remained normal.
The patient was treated with voriconazole (Vfend) and broad-spectrum antibiotics.
Q: Which is the most likely cause?
- A bacterium
- A virus
- Cryptococcus
- Aspergillus
- Zygomycetes
A: The correct answer is Zygomycetes, the second most common cause of fungal respiratory disease in patients with hematologic malignancies.
UNCOMMON BUT OFTEN FATAL
Zygomycetes is a class of fungi that contains two orders, Mucorales and Entomophthorales. Human disease, which is uncommon but frequently fatal, is predominantly associated with Mucorales and is commonly called mucormycosis.1,2
The major mode of transmission is through inhalation of spores from diverse decaying environmental sources. As Zygomycetes are aerogenous pathogens, they predominantly affect the paranasal sinuses and the lungs. The main risk factors for Zygomycetes infection include diabetes mellitus, hematologic malignancies (predominantly acute leukemias treated with aggressive chemotherapeutic regimens), pharmacologic immunosuppression, solid organ or bone marrow transplantation, and therapy with deferoxamine (Desferal), an iron-chelating agent.1,3
Overall, rhinocerebral disease is the most common manifestation, especially in the setting of diabetic ketoacidosis.1 In hematologic malignancies, the most common presentation is pulmonary zygomycosis with associated profound neutropenia, as neutrophils are the central defense against filamentous fungal hyphae.1,4
The incidence of zygomycosis is increasing, likely owing to the greater number of patients receiving stem cell or solid organ transplants, the use of more aggressive immunosuppressive regimens, prolonged survival, and the frequent prophylactic use of antifungal agents without activity against Zygomycetes.2,5
INFECTION PROGRESSES RAPIDLY
Most patients present with fever, cough, thoracic pain, and dyspnea in association with hypoxemia and pulmonary infiltrates refractory to broad-spectrum antibiotics. Zygomycosis can also present radiographically as pulmonary nodules or consolidations with or without the halo sign or cavitations.6,7
The disease usually progresses rapidly, invading vessels and causing infarction, bleeding, and dissemination to extrapulmonary sites.1,5
In reported cases in patients with acute leukemia who received aggressive chemotherapy, the fungal infection occurred several days after profound neutropenia developed.3,4
SPUTUM, LAVAGE, BIOPSY
The diagnosis is based on directly identifying the fungus morphologically and on culturing it. However, cultures of sputum, BAL fluid, and blood are usually negative.
Morphologically, the fungus is broad with irregular walls; it is also nearly aseptate and frequently has right-angle branching. In contrast, Aspergillus is narrow with parallel walls, distinctive septae, and acute branching.2
Of note: physicians need to alert the microbiology laboratory about their clinical suspicion of Zygomycetes infection, because the recovery rate of Zygomycetes in culture is increased by slicing the biopsy specimen in small pieces but not dicing it (to avoid breaking the septae).1
The diagnostic tests include microscopic examination and culture of sputum, BAL fluid, and transbronchial biopsy specimens. If the initial tests are negative but the suspicion of zygomycosis is strong on clinical grounds, then fine-needle aspiration or open lung biopsy should be considered.1
Useful predictors that favor the diagnosis of pulmonary zygomycosis instead of the main alternative, invasive pulmonary aspergillosis, include concomitant sinusitis, voriconazole prophylaxis (due to antifungal pressure), a negative galactomannan test in serum, multiple pulmonary nodules, and pleural effusion.2,8
TREATMENT WITH AMPHOTERICIN
The treatment includes giving effective antifungal agents promptly, correcting hyperglycemia and metabolic acidosis, reversing immunosuppression (if possible), and considering surgical debridement.1,2
Antifungal therapy is with conventional amphotericin B (Amphocin) or its lipid formulation (Abelcet). The lipid formulation is at least as effective as conventional amphotericin B and less nephrotoxic, thus allowing higher doses.1,9 The optimal duration of therapy has not been evaluated, but experts in general treat until the pulmonary and sinus lesions have resolved.2
Posaconazole (Noxafil), a broad-spectrum oral azole, has activity in vitro and is a valuable alternative for patients who have refractory zygomycosis or who cannot tolerate amphotericin B.5,10
The role of echinocandins is unclear, as they do not have in vitro activity against Zygomycetes. However, tests in animals have shown a synergistic effect between the echinocandin caspofungin (Cancidas) and amphotericin B lipid complex.11 Other antifungal agents such as azoles lack activity against Zygomycetes.5
The return of neutrophils plays a substantial role in resolving the infection in neutropenic patients, a proposition supported by reports of the failure of antifungal therapy in patients with persistent neutropenia.1 The addition of granulocyte colony-stimulating factor may accelerate neutrophil recovery and enhance neutrophil activity against opportunistic fungal pathogens.12
Even though progress has been made in the treatment of this disease, the prognosis continues to be poor in patients with hematologic malignancies and pulmonary or disseminated zygomycosis.9
ENDOBRONCHIAL ZYGOMYCOSIS
Aspergillosis is the most common endobronchial fungal disease. Zygomycosis is the third most common, after coccidioidomycosis. In zygomycosis, endobronchial lesions can be found in a third of patients who have pulmonary involvement.6,13,14
The most common predisposing conditions for the development of endobronchial zygomycosis are diabetes and hematologic malignancies associated with neutropenia.14
Endobronchial zygomycosis is characterized by a locally invasive gray-white mucoid lesion that blocks a major airway.13 The involved airway is usually edematous and necrotic. The diagnosis can be made by visualizing the organism in bronchial washings, brushings, or endobronchial biopsies.14
If the disease is not promptly diagnosed, the risk of death is very high. The management includes high-dose conventional or lipid amphotericin B and surgical or endobronchial resection.13,15
OUR CASE CONTINUED
After Zygomycetes was seen in the tissue from his bronchial biopsy, our patient received amphotericin B lipid complex at 5 mg/kg/day (started between images C and D in Figure 1). He had a good initial clinical response, but the infection progressed (image D in Figure 1).
The patient died as a result of massive hemoptysis attributable to the angioinvasive nature of the fungus, which most likely caused an erosion of a major pulmonary vessel.
TAKE-HOME POINTS
- Pulmonary disease is the most common manifestation of zygomycosis in patients with underlying hematologic malignancy. In this setting, zygomycosis has a high rate of morbidity and death.
- Endobronchial lesions can be seen in up to a third of patients with pulmonary zygomycosis.
- Prompt and effective therapy is essential for treatment to be successful.
A 19-year-old man received induction chemotherapy with idarubicin and cytarabine for secondary acute myeloid leukemia. Subsequently, he developed fever, progressive lung infiltrates, and severe neutropenia. His white blood cell count was 1.1 × 109/L (reference range 4.0–11.0) with 100% lymphocytes; his blood glucose level remained normal.
The patient was treated with voriconazole (Vfend) and broad-spectrum antibiotics.
Q: Which is the most likely cause?
- A bacterium
- A virus
- Cryptococcus
- Aspergillus
- Zygomycetes
A: The correct answer is Zygomycetes, the second most common cause of fungal respiratory disease in patients with hematologic malignancies.
UNCOMMON BUT OFTEN FATAL
Zygomycetes is a class of fungi that contains two orders, Mucorales and Entomophthorales. Human disease, which is uncommon but frequently fatal, is predominantly associated with Mucorales and is commonly called mucormycosis.1,2
The major mode of transmission is through inhalation of spores from diverse decaying environmental sources. As Zygomycetes are aerogenous pathogens, they predominantly affect the paranasal sinuses and the lungs. The main risk factors for Zygomycetes infection include diabetes mellitus, hematologic malignancies (predominantly acute leukemias treated with aggressive chemotherapeutic regimens), pharmacologic immunosuppression, solid organ or bone marrow transplantation, and therapy with deferoxamine (Desferal), an iron-chelating agent.1,3
Overall, rhinocerebral disease is the most common manifestation, especially in the setting of diabetic ketoacidosis.1 In hematologic malignancies, the most common presentation is pulmonary zygomycosis with associated profound neutropenia, as neutrophils are the central defense against filamentous fungal hyphae.1,4
The incidence of zygomycosis is increasing, likely owing to the greater number of patients receiving stem cell or solid organ transplants, the use of more aggressive immunosuppressive regimens, prolonged survival, and the frequent prophylactic use of antifungal agents without activity against Zygomycetes.2,5
INFECTION PROGRESSES RAPIDLY
Most patients present with fever, cough, thoracic pain, and dyspnea in association with hypoxemia and pulmonary infiltrates refractory to broad-spectrum antibiotics. Zygomycosis can also present radiographically as pulmonary nodules or consolidations with or without the halo sign or cavitations.6,7
The disease usually progresses rapidly, invading vessels and causing infarction, bleeding, and dissemination to extrapulmonary sites.1,5
In reported cases in patients with acute leukemia who received aggressive chemotherapy, the fungal infection occurred several days after profound neutropenia developed.3,4
SPUTUM, LAVAGE, BIOPSY
The diagnosis is based on directly identifying the fungus morphologically and on culturing it. However, cultures of sputum, BAL fluid, and blood are usually negative.
Morphologically, the fungus is broad with irregular walls; it is also nearly aseptate and frequently has right-angle branching. In contrast, Aspergillus is narrow with parallel walls, distinctive septae, and acute branching.2
Of note: physicians need to alert the microbiology laboratory about their clinical suspicion of Zygomycetes infection, because the recovery rate of Zygomycetes in culture is increased by slicing the biopsy specimen in small pieces but not dicing it (to avoid breaking the septae).1
The diagnostic tests include microscopic examination and culture of sputum, BAL fluid, and transbronchial biopsy specimens. If the initial tests are negative but the suspicion of zygomycosis is strong on clinical grounds, then fine-needle aspiration or open lung biopsy should be considered.1
Useful predictors that favor the diagnosis of pulmonary zygomycosis instead of the main alternative, invasive pulmonary aspergillosis, include concomitant sinusitis, voriconazole prophylaxis (due to antifungal pressure), a negative galactomannan test in serum, multiple pulmonary nodules, and pleural effusion.2,8
TREATMENT WITH AMPHOTERICIN
The treatment includes giving effective antifungal agents promptly, correcting hyperglycemia and metabolic acidosis, reversing immunosuppression (if possible), and considering surgical debridement.1,2
Antifungal therapy is with conventional amphotericin B (Amphocin) or its lipid formulation (Abelcet). The lipid formulation is at least as effective as conventional amphotericin B and less nephrotoxic, thus allowing higher doses.1,9 The optimal duration of therapy has not been evaluated, but experts in general treat until the pulmonary and sinus lesions have resolved.2
Posaconazole (Noxafil), a broad-spectrum oral azole, has activity in vitro and is a valuable alternative for patients who have refractory zygomycosis or who cannot tolerate amphotericin B.5,10
The role of echinocandins is unclear, as they do not have in vitro activity against Zygomycetes. However, tests in animals have shown a synergistic effect between the echinocandin caspofungin (Cancidas) and amphotericin B lipid complex.11 Other antifungal agents such as azoles lack activity against Zygomycetes.5
The return of neutrophils plays a substantial role in resolving the infection in neutropenic patients, a proposition supported by reports of the failure of antifungal therapy in patients with persistent neutropenia.1 The addition of granulocyte colony-stimulating factor may accelerate neutrophil recovery and enhance neutrophil activity against opportunistic fungal pathogens.12
Even though progress has been made in the treatment of this disease, the prognosis continues to be poor in patients with hematologic malignancies and pulmonary or disseminated zygomycosis.9
ENDOBRONCHIAL ZYGOMYCOSIS
Aspergillosis is the most common endobronchial fungal disease. Zygomycosis is the third most common, after coccidioidomycosis. In zygomycosis, endobronchial lesions can be found in a third of patients who have pulmonary involvement.6,13,14
The most common predisposing conditions for the development of endobronchial zygomycosis are diabetes and hematologic malignancies associated with neutropenia.14
Endobronchial zygomycosis is characterized by a locally invasive gray-white mucoid lesion that blocks a major airway.13 The involved airway is usually edematous and necrotic. The diagnosis can be made by visualizing the organism in bronchial washings, brushings, or endobronchial biopsies.14
If the disease is not promptly diagnosed, the risk of death is very high. The management includes high-dose conventional or lipid amphotericin B and surgical or endobronchial resection.13,15
OUR CASE CONTINUED
After Zygomycetes was seen in the tissue from his bronchial biopsy, our patient received amphotericin B lipid complex at 5 mg/kg/day (started between images C and D in Figure 1). He had a good initial clinical response, but the infection progressed (image D in Figure 1).
The patient died as a result of massive hemoptysis attributable to the angioinvasive nature of the fungus, which most likely caused an erosion of a major pulmonary vessel.
TAKE-HOME POINTS
- Pulmonary disease is the most common manifestation of zygomycosis in patients with underlying hematologic malignancy. In this setting, zygomycosis has a high rate of morbidity and death.
- Endobronchial lesions can be seen in up to a third of patients with pulmonary zygomycosis.
- Prompt and effective therapy is essential for treatment to be successful.
- Gonzalez CE, Rinaldi MG, Sugar AM. Zygomycosis. Infect Dis Clin North Am 2002; 16:895–914.
- Pyrgos V, Shoham S, Walsh TJ. Pulmonary zygomycosis. Semin Respir Crit Care Med 2008; 29:111–120.
- Kara IO, Tasova Y, Uguz A, Sahin B. Mucormycosis-associated fungal infections in patients with haematologic malignancies. Int J Clin Pract 2007; 63:134–139.
- Pagano L, Offidani M, Fianchi L, et al. Mucormycosis in hematologic patients. Haematologica 2004; 8:207–214.
- van Burik JA, Hare RS, Solomon HF, Corrado ML, Kontoyiannis DP. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis 2006; 42:e61–e65.
- Jamadar DA, Kazerooni EA, Daly BD, White CS, Gross BH. Pulmonary zygomycosis: CT appearance. J Comput Assist Tomogr 1995; 19:733–738.
- Lee YR, Choi YW, Lee KJ, Jeon SC, Park CK, Heo JN. CT halo sign: the spectrum of pulmonary diseases. Br J Radiol 2005; 78:862–865.
- Chamilos G, Marom EM, Lewis RE, Lionakis MS, Kontoyiannis DP. Predictors of pulmonary zygomycosis versus invasive pulmonary aspergillosis in patients with cancer. Clin Infect Dis 2005; 41:60–66.
- Gleissner B, Schilling A, Anagnostopolous I, Siehl I, Thiel E. Improved outcome of zygomycosis in patients with hematological diseases? Leuk Lymphoma 2004; 45:1351–1360.
- Greenberg RN, Mullane K, van Burik JA, et al. Posaconazole as salvage therapy for zygomycosis. Antimicrob Agents Chemother 2006; 50:126–133.
- Spellberg B, Fu Y, Edwards JE, Ibrahim AS. Combination therapy with amphotericin B lipid complex and caspofungin acetate of disseminated zygomycosis in diabetic ketoacidotic mice. Antimicrob Agents Chemother 2005; 49:830–832.
- Liles WC, Huang JE, van Burik JA, Bowden RA, Dale DC. Granulocyte colony-stimulating factor administered in vivo augments neutrophilmediated activity against opportunistic fungal pathogens. J Infect Dis 1997; 175:1012–1015.
- Husari AW, Jensen WA, Kirsch CM, et al. Pulmonary mucormycosis presenting as an endobronchial lesion. Chest 1994; 106:1889–1891.
- Karnak D, Avery RK, Gildea TR, Sahoo D, Mehta AC. Endobronchial fungal disease: an under-recognized entity. Respiration 2007; 74:88–104.
- al-Majed S, al-Kassimi F, Ashour M, Mekki MO, Sadiq S. Removal of endobronchial mucormycosis lesion through a rigid bronchoscope. Thorax 1992; 47:203–204.
- Gonzalez CE, Rinaldi MG, Sugar AM. Zygomycosis. Infect Dis Clin North Am 2002; 16:895–914.
- Pyrgos V, Shoham S, Walsh TJ. Pulmonary zygomycosis. Semin Respir Crit Care Med 2008; 29:111–120.
- Kara IO, Tasova Y, Uguz A, Sahin B. Mucormycosis-associated fungal infections in patients with haematologic malignancies. Int J Clin Pract 2007; 63:134–139.
- Pagano L, Offidani M, Fianchi L, et al. Mucormycosis in hematologic patients. Haematologica 2004; 8:207–214.
- van Burik JA, Hare RS, Solomon HF, Corrado ML, Kontoyiannis DP. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis 2006; 42:e61–e65.
- Jamadar DA, Kazerooni EA, Daly BD, White CS, Gross BH. Pulmonary zygomycosis: CT appearance. J Comput Assist Tomogr 1995; 19:733–738.
- Lee YR, Choi YW, Lee KJ, Jeon SC, Park CK, Heo JN. CT halo sign: the spectrum of pulmonary diseases. Br J Radiol 2005; 78:862–865.
- Chamilos G, Marom EM, Lewis RE, Lionakis MS, Kontoyiannis DP. Predictors of pulmonary zygomycosis versus invasive pulmonary aspergillosis in patients with cancer. Clin Infect Dis 2005; 41:60–66.
- Gleissner B, Schilling A, Anagnostopolous I, Siehl I, Thiel E. Improved outcome of zygomycosis in patients with hematological diseases? Leuk Lymphoma 2004; 45:1351–1360.
- Greenberg RN, Mullane K, van Burik JA, et al. Posaconazole as salvage therapy for zygomycosis. Antimicrob Agents Chemother 2006; 50:126–133.
- Spellberg B, Fu Y, Edwards JE, Ibrahim AS. Combination therapy with amphotericin B lipid complex and caspofungin acetate of disseminated zygomycosis in diabetic ketoacidotic mice. Antimicrob Agents Chemother 2005; 49:830–832.
- Liles WC, Huang JE, van Burik JA, Bowden RA, Dale DC. Granulocyte colony-stimulating factor administered in vivo augments neutrophilmediated activity against opportunistic fungal pathogens. J Infect Dis 1997; 175:1012–1015.
- Husari AW, Jensen WA, Kirsch CM, et al. Pulmonary mucormycosis presenting as an endobronchial lesion. Chest 1994; 106:1889–1891.
- Karnak D, Avery RK, Gildea TR, Sahoo D, Mehta AC. Endobronchial fungal disease: an under-recognized entity. Respiration 2007; 74:88–104.
- al-Majed S, al-Kassimi F, Ashour M, Mekki MO, Sadiq S. Removal of endobronchial mucormycosis lesion through a rigid bronchoscope. Thorax 1992; 47:203–204.
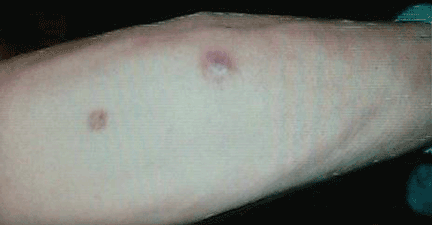
Ulcerative leg nodules in a transplant recipient
Q: What is the probable diagnosis?
- Kaposi sarcoma
- Majocchi (trichophytic) granuloma
- Staphylococcus aureus furunculosis
- Primary cutaneous mucormycosis
- Disseminated zoster
CAUSES AND RISK FACTORS
Mucormycosis is an invasive infection caused by fungi of a variety of genera (Rhizopus, Rhizomucor, Mucor, Absidia, Apophysomyces, Cunninghamella, Saksenaea, Conidiobolus, and Basidiobolus species) belonging to the class Zygomycetes in the order Mucorales. These ubiquitous environmental fungi have been found widely distributed in hospital sources, having been cultured from plaster casts,1 tongue depressors,2 cloth tapes for securing endotracheal tubes,3 peritoneal dialysis catheters,4 surgical wounds,5 contaminated dressings, needles, and intravenous catheters.6
These fungi rarely cause disease in healthy people, but patients with certain risk factors may develop disseminated disease, in which the death rate is high.6 Risk factors include diabetes mellitus, renal failure, trauma, burns, malnutrition, solid organ transplantation, hematologic malignancies, use of immunosuppressive drugs (eg, chemotherapeutic agents, corticosteroids, cyclosporine, methotrexate, infliximab [Remicade]7), and iron overload and iron-chelating therapy with deferoxamine. 8 Patients such as ours—an organ transplant recipient with concomitant diabetes mellitus—are highly susceptible to this infection.
DIAGNOSIS
The clinical presentation of mucormycosis can be rhino-orbitocerebral (most common),9 pulmonary, primary cutaneous, gastrointestinal, cardiac, or disseminated infection.6 Primary cutaneous mucormycosis occurs from fungal inoculation of the dermis. Small areas of trauma may be all that is needed for this inoculation to occur.
The initial lesion may be an erythematous patch, plaque, or nodule that may subsequently ulcerate and become gangrenous or necrotic.10
The differential diagnosis of new cutaneous nodules in an immunocompromised patient includes a wide variety of infections, such as ecthyma gangrenosum caused by Pseudomonas or Candida species, herpes simplex virus infection, cryptococcal infection, phaeohyphomycosis, and cutaneous aspergillosis.
Because the morphology of cutaneous mucormycosis is not distinctive, and because we need to cast our net wide for the numerous, potentially lethal diagnostic possibilities in an immunosuppressed patient, it is crucial that we have a low threshold for prompt biopsy to establish the diagnosis and initiate definitive treatment. While skin biopsy for microscopy can suggest certain fungi, the exact diagnosis is confirmed by a fungal culture of a biopsy specimen.
TREATMENT
Localized soft tissue infection is more amenable to therapy and therefore carries a better prognosis than visceral or disseminated disease.
The best treatment outcomes of primary cutaneous mucormycosis are achieved with both complete excision and debridement of necrotic tissue and systemic antifungal therapy. Amphotericin B in conventional form (Fungizone) and liposomal form (AmBisome) and posaconazole (Noxafil)11–12 are effective.
Paradoxically, in contrast to deferoxamine, other iron chelators such as deferiprone (Ferriprox, not available in the United States) and deferasirox (Exjade), which do not supply iron to the fungus, had been shown to be effective against Zygomycetes in in vitro and animal models. The role of iron chelators as adjunctive therapy for mucormycosis needs further investigation.8
Primary cutaneous mucormycosis may become a disseminated infection. Therefore, one should have a high index of suspicion. When a transplant recipient develops a cutaneous plaque or nodule, biopsy should be performed promptly. Failure to do so can increase the risk of morbidity and death.
- Johnson AS, Ranson M, Scarffe JH, Morgenstern GR, Shaw AJ, Oppenheim BA. Cutaneous infection with Rhizopus oryzae and Aspergillus niger following bone marrow transplantation. J Hosp Infect 1993; 25:293–296.
- Paydas S, Yavuz S, Disel U, et al. Mucormycosis of the tongue in a patient with acute lymphoblastic leukemia: a possible relation with use of a tongue depressor. Am J Med 2003; 114:618–620.
- Dickinson M, Kalayanamit T, Yang CA, Pomper GJ, Franco-Webb C, Rodman D. Cutaneous zygomycosis (mucormycosis) complicating endotracheal intubation: diagnosis and successful treatment. Chest 1998; 114:340–342.
- Nayak S, Satish R, Gokulnath , Savio J, Rajalakshmi T. Peritoneal mucormycosis in a patient on CAPD. Perit Dial Int 2007; 27:216–217.
- Chew HH, Abuzeid A, Singh D, Tai CC. Surgical wound mucormycosis necessitating hand amputation: a case report. J Orthop Surg (Hong Kong) 2008; 16:267–269.
- Benbow EW, Stoddart RW. Systemic zygomycosis. Postgrad Med J 1986; 62:985–996.
- Gadadhar H, Hawkins S, Huffstutter JE, Panda M. Cutaneous mucormycosis complicating methotrexate, prednisone, and infliximab therapy. J Clin Rheumatol 2007; 13:361–362.
- Ibrahim AS, Spellberg B, Edwards J. Iron acquisition: a novel perspective on mucormycosis pathogenesis and treatment. Curr Opin Infect Dis 2008; 21:620–625.
- Chakrabarti A, Das A, Sharma A, et al. Ten years’ experience in zygomycosis at a tertiary care centre in India. J Infect 2001; 42:261–266.
- Nouri-Majalan N, Moghimi M. Skin mucormycosis presenting as an erythema-nodosum-like rash in a renal transplant recipient: a case report. J Med Case Reports 2008, 2:112.
- Sun QN, Fothergill AW, McCarthy DI, Rinaldi MG, Graybill JR. In vitro activities of posaconazole, itraconazole, voriconazole, amphotericin B, and fluconazole against 37 clinical isolates of zygomycetes. Antimicrob Agents Chemother 2002; 46:1581–1582.
- Spellberg B, Edwards J, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation and management. Clin Microbial Rev 2005; 18:556–569.
Q: What is the probable diagnosis?
- Kaposi sarcoma
- Majocchi (trichophytic) granuloma
- Staphylococcus aureus furunculosis
- Primary cutaneous mucormycosis
- Disseminated zoster
CAUSES AND RISK FACTORS
Mucormycosis is an invasive infection caused by fungi of a variety of genera (Rhizopus, Rhizomucor, Mucor, Absidia, Apophysomyces, Cunninghamella, Saksenaea, Conidiobolus, and Basidiobolus species) belonging to the class Zygomycetes in the order Mucorales. These ubiquitous environmental fungi have been found widely distributed in hospital sources, having been cultured from plaster casts,1 tongue depressors,2 cloth tapes for securing endotracheal tubes,3 peritoneal dialysis catheters,4 surgical wounds,5 contaminated dressings, needles, and intravenous catheters.6
These fungi rarely cause disease in healthy people, but patients with certain risk factors may develop disseminated disease, in which the death rate is high.6 Risk factors include diabetes mellitus, renal failure, trauma, burns, malnutrition, solid organ transplantation, hematologic malignancies, use of immunosuppressive drugs (eg, chemotherapeutic agents, corticosteroids, cyclosporine, methotrexate, infliximab [Remicade]7), and iron overload and iron-chelating therapy with deferoxamine. 8 Patients such as ours—an organ transplant recipient with concomitant diabetes mellitus—are highly susceptible to this infection.
DIAGNOSIS
The clinical presentation of mucormycosis can be rhino-orbitocerebral (most common),9 pulmonary, primary cutaneous, gastrointestinal, cardiac, or disseminated infection.6 Primary cutaneous mucormycosis occurs from fungal inoculation of the dermis. Small areas of trauma may be all that is needed for this inoculation to occur.
The initial lesion may be an erythematous patch, plaque, or nodule that may subsequently ulcerate and become gangrenous or necrotic.10
The differential diagnosis of new cutaneous nodules in an immunocompromised patient includes a wide variety of infections, such as ecthyma gangrenosum caused by Pseudomonas or Candida species, herpes simplex virus infection, cryptococcal infection, phaeohyphomycosis, and cutaneous aspergillosis.
Because the morphology of cutaneous mucormycosis is not distinctive, and because we need to cast our net wide for the numerous, potentially lethal diagnostic possibilities in an immunosuppressed patient, it is crucial that we have a low threshold for prompt biopsy to establish the diagnosis and initiate definitive treatment. While skin biopsy for microscopy can suggest certain fungi, the exact diagnosis is confirmed by a fungal culture of a biopsy specimen.
TREATMENT
Localized soft tissue infection is more amenable to therapy and therefore carries a better prognosis than visceral or disseminated disease.
The best treatment outcomes of primary cutaneous mucormycosis are achieved with both complete excision and debridement of necrotic tissue and systemic antifungal therapy. Amphotericin B in conventional form (Fungizone) and liposomal form (AmBisome) and posaconazole (Noxafil)11–12 are effective.
Paradoxically, in contrast to deferoxamine, other iron chelators such as deferiprone (Ferriprox, not available in the United States) and deferasirox (Exjade), which do not supply iron to the fungus, had been shown to be effective against Zygomycetes in in vitro and animal models. The role of iron chelators as adjunctive therapy for mucormycosis needs further investigation.8
Primary cutaneous mucormycosis may become a disseminated infection. Therefore, one should have a high index of suspicion. When a transplant recipient develops a cutaneous plaque or nodule, biopsy should be performed promptly. Failure to do so can increase the risk of morbidity and death.
Q: What is the probable diagnosis?
- Kaposi sarcoma
- Majocchi (trichophytic) granuloma
- Staphylococcus aureus furunculosis
- Primary cutaneous mucormycosis
- Disseminated zoster
CAUSES AND RISK FACTORS
Mucormycosis is an invasive infection caused by fungi of a variety of genera (Rhizopus, Rhizomucor, Mucor, Absidia, Apophysomyces, Cunninghamella, Saksenaea, Conidiobolus, and Basidiobolus species) belonging to the class Zygomycetes in the order Mucorales. These ubiquitous environmental fungi have been found widely distributed in hospital sources, having been cultured from plaster casts,1 tongue depressors,2 cloth tapes for securing endotracheal tubes,3 peritoneal dialysis catheters,4 surgical wounds,5 contaminated dressings, needles, and intravenous catheters.6
These fungi rarely cause disease in healthy people, but patients with certain risk factors may develop disseminated disease, in which the death rate is high.6 Risk factors include diabetes mellitus, renal failure, trauma, burns, malnutrition, solid organ transplantation, hematologic malignancies, use of immunosuppressive drugs (eg, chemotherapeutic agents, corticosteroids, cyclosporine, methotrexate, infliximab [Remicade]7), and iron overload and iron-chelating therapy with deferoxamine. 8 Patients such as ours—an organ transplant recipient with concomitant diabetes mellitus—are highly susceptible to this infection.
DIAGNOSIS
The clinical presentation of mucormycosis can be rhino-orbitocerebral (most common),9 pulmonary, primary cutaneous, gastrointestinal, cardiac, or disseminated infection.6 Primary cutaneous mucormycosis occurs from fungal inoculation of the dermis. Small areas of trauma may be all that is needed for this inoculation to occur.
The initial lesion may be an erythematous patch, plaque, or nodule that may subsequently ulcerate and become gangrenous or necrotic.10
The differential diagnosis of new cutaneous nodules in an immunocompromised patient includes a wide variety of infections, such as ecthyma gangrenosum caused by Pseudomonas or Candida species, herpes simplex virus infection, cryptococcal infection, phaeohyphomycosis, and cutaneous aspergillosis.
Because the morphology of cutaneous mucormycosis is not distinctive, and because we need to cast our net wide for the numerous, potentially lethal diagnostic possibilities in an immunosuppressed patient, it is crucial that we have a low threshold for prompt biopsy to establish the diagnosis and initiate definitive treatment. While skin biopsy for microscopy can suggest certain fungi, the exact diagnosis is confirmed by a fungal culture of a biopsy specimen.
TREATMENT
Localized soft tissue infection is more amenable to therapy and therefore carries a better prognosis than visceral or disseminated disease.
The best treatment outcomes of primary cutaneous mucormycosis are achieved with both complete excision and debridement of necrotic tissue and systemic antifungal therapy. Amphotericin B in conventional form (Fungizone) and liposomal form (AmBisome) and posaconazole (Noxafil)11–12 are effective.
Paradoxically, in contrast to deferoxamine, other iron chelators such as deferiprone (Ferriprox, not available in the United States) and deferasirox (Exjade), which do not supply iron to the fungus, had been shown to be effective against Zygomycetes in in vitro and animal models. The role of iron chelators as adjunctive therapy for mucormycosis needs further investigation.8
Primary cutaneous mucormycosis may become a disseminated infection. Therefore, one should have a high index of suspicion. When a transplant recipient develops a cutaneous plaque or nodule, biopsy should be performed promptly. Failure to do so can increase the risk of morbidity and death.
- Johnson AS, Ranson M, Scarffe JH, Morgenstern GR, Shaw AJ, Oppenheim BA. Cutaneous infection with Rhizopus oryzae and Aspergillus niger following bone marrow transplantation. J Hosp Infect 1993; 25:293–296.
- Paydas S, Yavuz S, Disel U, et al. Mucormycosis of the tongue in a patient with acute lymphoblastic leukemia: a possible relation with use of a tongue depressor. Am J Med 2003; 114:618–620.
- Dickinson M, Kalayanamit T, Yang CA, Pomper GJ, Franco-Webb C, Rodman D. Cutaneous zygomycosis (mucormycosis) complicating endotracheal intubation: diagnosis and successful treatment. Chest 1998; 114:340–342.
- Nayak S, Satish R, Gokulnath , Savio J, Rajalakshmi T. Peritoneal mucormycosis in a patient on CAPD. Perit Dial Int 2007; 27:216–217.
- Chew HH, Abuzeid A, Singh D, Tai CC. Surgical wound mucormycosis necessitating hand amputation: a case report. J Orthop Surg (Hong Kong) 2008; 16:267–269.
- Benbow EW, Stoddart RW. Systemic zygomycosis. Postgrad Med J 1986; 62:985–996.
- Gadadhar H, Hawkins S, Huffstutter JE, Panda M. Cutaneous mucormycosis complicating methotrexate, prednisone, and infliximab therapy. J Clin Rheumatol 2007; 13:361–362.
- Ibrahim AS, Spellberg B, Edwards J. Iron acquisition: a novel perspective on mucormycosis pathogenesis and treatment. Curr Opin Infect Dis 2008; 21:620–625.
- Chakrabarti A, Das A, Sharma A, et al. Ten years’ experience in zygomycosis at a tertiary care centre in India. J Infect 2001; 42:261–266.
- Nouri-Majalan N, Moghimi M. Skin mucormycosis presenting as an erythema-nodosum-like rash in a renal transplant recipient: a case report. J Med Case Reports 2008, 2:112.
- Sun QN, Fothergill AW, McCarthy DI, Rinaldi MG, Graybill JR. In vitro activities of posaconazole, itraconazole, voriconazole, amphotericin B, and fluconazole against 37 clinical isolates of zygomycetes. Antimicrob Agents Chemother 2002; 46:1581–1582.
- Spellberg B, Edwards J, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation and management. Clin Microbial Rev 2005; 18:556–569.
- Johnson AS, Ranson M, Scarffe JH, Morgenstern GR, Shaw AJ, Oppenheim BA. Cutaneous infection with Rhizopus oryzae and Aspergillus niger following bone marrow transplantation. J Hosp Infect 1993; 25:293–296.
- Paydas S, Yavuz S, Disel U, et al. Mucormycosis of the tongue in a patient with acute lymphoblastic leukemia: a possible relation with use of a tongue depressor. Am J Med 2003; 114:618–620.
- Dickinson M, Kalayanamit T, Yang CA, Pomper GJ, Franco-Webb C, Rodman D. Cutaneous zygomycosis (mucormycosis) complicating endotracheal intubation: diagnosis and successful treatment. Chest 1998; 114:340–342.
- Nayak S, Satish R, Gokulnath , Savio J, Rajalakshmi T. Peritoneal mucormycosis in a patient on CAPD. Perit Dial Int 2007; 27:216–217.
- Chew HH, Abuzeid A, Singh D, Tai CC. Surgical wound mucormycosis necessitating hand amputation: a case report. J Orthop Surg (Hong Kong) 2008; 16:267–269.
- Benbow EW, Stoddart RW. Systemic zygomycosis. Postgrad Med J 1986; 62:985–996.
- Gadadhar H, Hawkins S, Huffstutter JE, Panda M. Cutaneous mucormycosis complicating methotrexate, prednisone, and infliximab therapy. J Clin Rheumatol 2007; 13:361–362.
- Ibrahim AS, Spellberg B, Edwards J. Iron acquisition: a novel perspective on mucormycosis pathogenesis and treatment. Curr Opin Infect Dis 2008; 21:620–625.
- Chakrabarti A, Das A, Sharma A, et al. Ten years’ experience in zygomycosis at a tertiary care centre in India. J Infect 2001; 42:261–266.
- Nouri-Majalan N, Moghimi M. Skin mucormycosis presenting as an erythema-nodosum-like rash in a renal transplant recipient: a case report. J Med Case Reports 2008, 2:112.
- Sun QN, Fothergill AW, McCarthy DI, Rinaldi MG, Graybill JR. In vitro activities of posaconazole, itraconazole, voriconazole, amphotericin B, and fluconazole against 37 clinical isolates of zygomycetes. Antimicrob Agents Chemother 2002; 46:1581–1582.
- Spellberg B, Edwards J, Ibrahim A. Novel perspectives on mucormycosis: pathophysiology, presentation and management. Clin Microbial Rev 2005; 18:556–569.
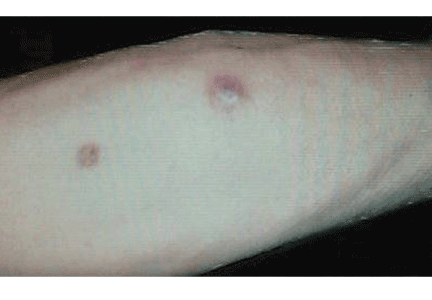
Recurrent pyelonephritis as a sign of ‘sponge kidney’
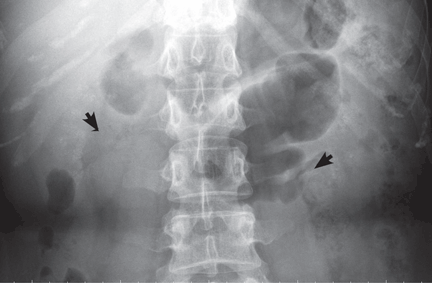
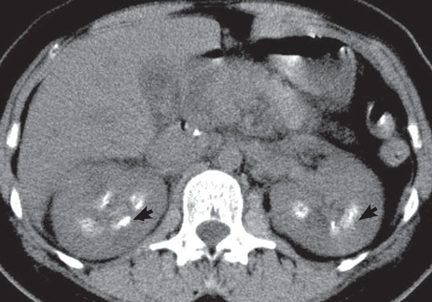
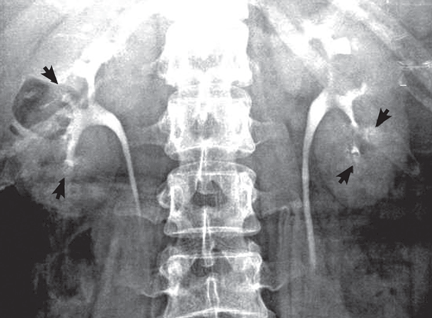
KEY FEATURES
Medullary sponge kidney causes extensive cystic dilation of medullary collecting tubules.1 It is usually an incidental finding in patients undergoing intravenous urography as part of the evaluation for infection, hematuria, or kidney stones.
The classic urographic appearance is linear striations with small brushes or “bouquets of flowers,” which represent the collection of contrast material in small papillary cysts.
Medullary sponge kidney has long been considered a congenital disorder, but the genetic defect has not yet been identified, and the pathogenesis is not yet known. It has only rarely been reported in children.2
It is usually asymptomatic, but complications may occur, including nephrocalcinosis or lithiasis, urinary tract infection, renal tubular acidosis, and impaired urine concentrating ability.
RISK OF LITHIASIS
Gambaro et al3 estimated that medullary sponge kidney is found in up to 20% of patients with urolithiasis, and that more than 70% of patients with medullary sponge kidney develop stones.
Cystic dilation of medullary collecting tubules (an anatomic abnormality) inevitably causes urinary stasis. This, combined with hypercalciuria and reduced excretion of urinary citrate and magnesium (metabolic abnormalities), contributes to lithiasis.4 Lithiasis in medullary sponge kidney is a well-known cause of urinary tract infection, and it tends to facilitate infective stone formation after episodes of urinary tract infection.5
The unique anatomic derangement of medullary sponge kidney contributes to the recurrence of pyelonephritis, in addition to the conventional risk factors—frequent sexual intercourse, avoidance of voiding because it is inconvenient, incomplete bladder emptying, ureteropelvic junction obstruction, ectopic ureter, and impaired immunity.6
DIAGNOSIS AND TREATMENT
Intravenous urography is the gold standard for the diagnosis of medullary sponge kidney; computed tomography and ultrasonography are generally limited in their ability to clearly show the tubular ectasia.7
Treatment includes antibiotics for acute pyelonephritis and thiazide diuretics and potassium citrate to prevent stone formation and renal tubular acidosis. Due to its silent course, medullary sponge kidney should be considered not only as a cause of nephrocalcinosis and nephrolithiasis, but also as a distinct entity complicating recurrent pyelonephritis.
- Gambaro G, Feltrin GP, Lupo A, Bonfante L, D'Angelo A, Antonello A. Medullary sponge kidney (Lenarduzzi-Cacchi-Ricci disease): a Padua Medical School discovery in the 1930s. Kidney Int 2006; 69:663–670.
- Kasap B, Soylu A, Oren O, Turkmen M, Kavukcu S. Medullary sponge kidney associated with distal renal tubular acidosis in a 5-year-old girl. Eur J Pediatr 2006; 165:648–651.
- Gambaro G, Fabris A, Puliatta D, Lupo A. Lithiasis in cystic kidney disease and malformations of the urinary tract. Urol Res 2006; 34:102–107.
- O'Neill M, Breslau NA, Pak CY. Metabolic evaluation of nephrolithiasis in patients with medullary sponge kidney. JAMA 1981; 245:1233–1236.
- Miano R, Germani S, Vespasiani G. Stones and urinary tract infections. Urol Int 2007; 79(suppl 1):32–36.
- Scholes D, Hooton TM, Roberts PL, Gupta K, Stapleton AE, Stamm WE. Risk factors associated with acute pyelonephritis in healthy women. Ann Intern Med 2005; 142:20–27.
- Maw AM, Megibow AJ, Grasso M, Goldfarb DS. Diagnosis of medullary sponge kidney by computed tomographic urography. Am J Kidney Dis 2007; 50:146–150.
KEY FEATURES
Medullary sponge kidney causes extensive cystic dilation of medullary collecting tubules.1 It is usually an incidental finding in patients undergoing intravenous urography as part of the evaluation for infection, hematuria, or kidney stones.
The classic urographic appearance is linear striations with small brushes or “bouquets of flowers,” which represent the collection of contrast material in small papillary cysts.
Medullary sponge kidney has long been considered a congenital disorder, but the genetic defect has not yet been identified, and the pathogenesis is not yet known. It has only rarely been reported in children.2
It is usually asymptomatic, but complications may occur, including nephrocalcinosis or lithiasis, urinary tract infection, renal tubular acidosis, and impaired urine concentrating ability.
RISK OF LITHIASIS
Gambaro et al3 estimated that medullary sponge kidney is found in up to 20% of patients with urolithiasis, and that more than 70% of patients with medullary sponge kidney develop stones.
Cystic dilation of medullary collecting tubules (an anatomic abnormality) inevitably causes urinary stasis. This, combined with hypercalciuria and reduced excretion of urinary citrate and magnesium (metabolic abnormalities), contributes to lithiasis.4 Lithiasis in medullary sponge kidney is a well-known cause of urinary tract infection, and it tends to facilitate infective stone formation after episodes of urinary tract infection.5
The unique anatomic derangement of medullary sponge kidney contributes to the recurrence of pyelonephritis, in addition to the conventional risk factors—frequent sexual intercourse, avoidance of voiding because it is inconvenient, incomplete bladder emptying, ureteropelvic junction obstruction, ectopic ureter, and impaired immunity.6
DIAGNOSIS AND TREATMENT
Intravenous urography is the gold standard for the diagnosis of medullary sponge kidney; computed tomography and ultrasonography are generally limited in their ability to clearly show the tubular ectasia.7
Treatment includes antibiotics for acute pyelonephritis and thiazide diuretics and potassium citrate to prevent stone formation and renal tubular acidosis. Due to its silent course, medullary sponge kidney should be considered not only as a cause of nephrocalcinosis and nephrolithiasis, but also as a distinct entity complicating recurrent pyelonephritis.
KEY FEATURES
Medullary sponge kidney causes extensive cystic dilation of medullary collecting tubules.1 It is usually an incidental finding in patients undergoing intravenous urography as part of the evaluation for infection, hematuria, or kidney stones.
The classic urographic appearance is linear striations with small brushes or “bouquets of flowers,” which represent the collection of contrast material in small papillary cysts.
Medullary sponge kidney has long been considered a congenital disorder, but the genetic defect has not yet been identified, and the pathogenesis is not yet known. It has only rarely been reported in children.2
It is usually asymptomatic, but complications may occur, including nephrocalcinosis or lithiasis, urinary tract infection, renal tubular acidosis, and impaired urine concentrating ability.
RISK OF LITHIASIS
Gambaro et al3 estimated that medullary sponge kidney is found in up to 20% of patients with urolithiasis, and that more than 70% of patients with medullary sponge kidney develop stones.
Cystic dilation of medullary collecting tubules (an anatomic abnormality) inevitably causes urinary stasis. This, combined with hypercalciuria and reduced excretion of urinary citrate and magnesium (metabolic abnormalities), contributes to lithiasis.4 Lithiasis in medullary sponge kidney is a well-known cause of urinary tract infection, and it tends to facilitate infective stone formation after episodes of urinary tract infection.5
The unique anatomic derangement of medullary sponge kidney contributes to the recurrence of pyelonephritis, in addition to the conventional risk factors—frequent sexual intercourse, avoidance of voiding because it is inconvenient, incomplete bladder emptying, ureteropelvic junction obstruction, ectopic ureter, and impaired immunity.6
DIAGNOSIS AND TREATMENT
Intravenous urography is the gold standard for the diagnosis of medullary sponge kidney; computed tomography and ultrasonography are generally limited in their ability to clearly show the tubular ectasia.7
Treatment includes antibiotics for acute pyelonephritis and thiazide diuretics and potassium citrate to prevent stone formation and renal tubular acidosis. Due to its silent course, medullary sponge kidney should be considered not only as a cause of nephrocalcinosis and nephrolithiasis, but also as a distinct entity complicating recurrent pyelonephritis.
- Gambaro G, Feltrin GP, Lupo A, Bonfante L, D'Angelo A, Antonello A. Medullary sponge kidney (Lenarduzzi-Cacchi-Ricci disease): a Padua Medical School discovery in the 1930s. Kidney Int 2006; 69:663–670.
- Kasap B, Soylu A, Oren O, Turkmen M, Kavukcu S. Medullary sponge kidney associated with distal renal tubular acidosis in a 5-year-old girl. Eur J Pediatr 2006; 165:648–651.
- Gambaro G, Fabris A, Puliatta D, Lupo A. Lithiasis in cystic kidney disease and malformations of the urinary tract. Urol Res 2006; 34:102–107.
- O'Neill M, Breslau NA, Pak CY. Metabolic evaluation of nephrolithiasis in patients with medullary sponge kidney. JAMA 1981; 245:1233–1236.
- Miano R, Germani S, Vespasiani G. Stones and urinary tract infections. Urol Int 2007; 79(suppl 1):32–36.
- Scholes D, Hooton TM, Roberts PL, Gupta K, Stapleton AE, Stamm WE. Risk factors associated with acute pyelonephritis in healthy women. Ann Intern Med 2005; 142:20–27.
- Maw AM, Megibow AJ, Grasso M, Goldfarb DS. Diagnosis of medullary sponge kidney by computed tomographic urography. Am J Kidney Dis 2007; 50:146–150.
- Gambaro G, Feltrin GP, Lupo A, Bonfante L, D'Angelo A, Antonello A. Medullary sponge kidney (Lenarduzzi-Cacchi-Ricci disease): a Padua Medical School discovery in the 1930s. Kidney Int 2006; 69:663–670.
- Kasap B, Soylu A, Oren O, Turkmen M, Kavukcu S. Medullary sponge kidney associated with distal renal tubular acidosis in a 5-year-old girl. Eur J Pediatr 2006; 165:648–651.
- Gambaro G, Fabris A, Puliatta D, Lupo A. Lithiasis in cystic kidney disease and malformations of the urinary tract. Urol Res 2006; 34:102–107.
- O'Neill M, Breslau NA, Pak CY. Metabolic evaluation of nephrolithiasis in patients with medullary sponge kidney. JAMA 1981; 245:1233–1236.
- Miano R, Germani S, Vespasiani G. Stones and urinary tract infections. Urol Int 2007; 79(suppl 1):32–36.
- Scholes D, Hooton TM, Roberts PL, Gupta K, Stapleton AE, Stamm WE. Risk factors associated with acute pyelonephritis in healthy women. Ann Intern Med 2005; 142:20–27.
- Maw AM, Megibow AJ, Grasso M, Goldfarb DS. Diagnosis of medullary sponge kidney by computed tomographic urography. Am J Kidney Dis 2007; 50:146–150.
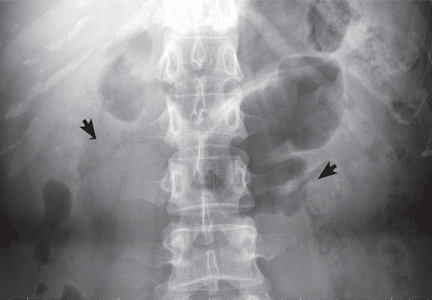
Painful eye with a facial rash
A 75-year-old man presents 4 days after painful cutaneous lesions appeared on the left side of his face, associated with severe ocular pain. Two days before the eruption, he had had an intense headache, which was diagnosed as a tension headache and was treated with oral acetaminophen (Tylenol), but with no improvement.
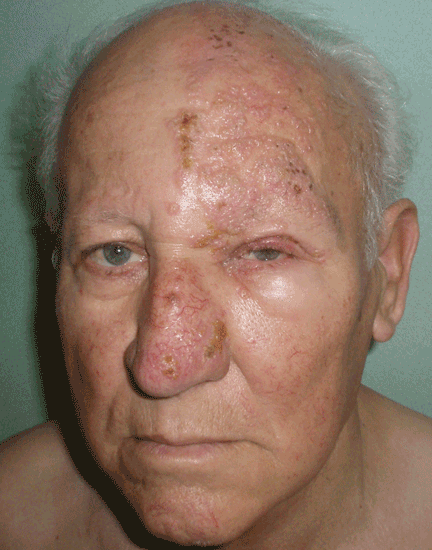
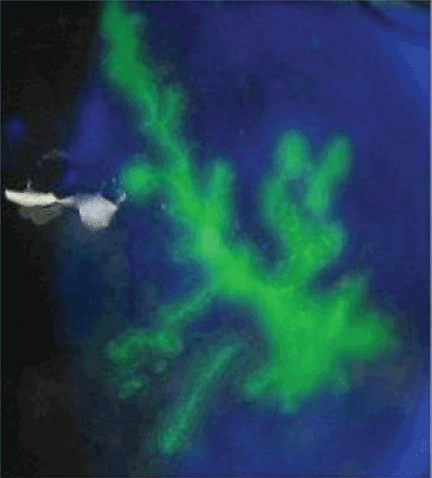
The remainder of his physical examination is normal. Laboratory tests, including red and white blood cell counts, hemoglobin, and basic metabolic and coagulation tests reveal no abnormalities.
Q: What is your diagnosis?
- Allergic contact dermatitis
- Herpes simplex
- Varicella
- Ramsay-Hunt syndrome
- Herpes zoster ophthalmicus and herpetic keratitis
A: Herpes zoster ophthalmicus is the correct diagnosis. It represents a reactivation of the varicella zoster virus.1
Varicella zoster virus, like others of the herpes family, has developed a complex control of virus-host interactions to ensure its survival in humans. It lies dormant in the sensory ganglia and, when reactivated, moves down the neurons and satellite cells along the sensory axons to the skin.1 The reactivation is related to diminished cell-mediated immunity, which occurs as a physiologic part of aging, which is why the elderly tend to be the most often affected. 2 The incidence of herpes zoster varies from 2.2 to 3.4 per 1,000 people per year.3 Its incidence in people over age 80 is about 10 per 1,000 people per year.3
CLINICAL PRESENTATION
Herpes zoster typically presents as a dermatome-grouped vesicular eruption over an erythematous base, accompanied or preceded by local pain. It has two main complications, postherpetic neuralgia and ocular involvement. Postherpetic neuralgia is neuropathic pain that persists or develops after the dermatomal rash has healed.4 Independent predictors of postherpetic neuralgia are older age, severe acute pain, severe rash, a shorter duration of rash before consultation, and ocular involvement.5 It occurs in 36.6% of patients over age 60, and in 47.5% over 70.6 Persistent postherpetic neuralgia has been linked to suicide in patients over 70.7
Ocular infection occurs with involvement of the ophthalmic division of the fifth cranial nerve. Before the antiviral era, it was seen in as many as 50% of patients.8 Hutchinson’s sign is skin lesions on the tip, side, or root of the nose and is an important predictor of ocular involvement.1 Lesions may include folliculopapillar conjunctivitis, episcleritis, scleritis, keratitis (dendritic, pseudodendritic, and interstitial), uveitis, and necrotizing retinitis.
DIAGNOSIS
The diagnosis of herpes zoster is usually based on clinical observation of the characteristic rash, although viral culture and molecular techniques are available when definitive diagnosis is required. When ophthalmic division is affected and Hutchinson’s sign, unexplained ocular redness with pain, or complaints of visual problems are present, the patient should be referred promptly to an ophthalmologist, because serious visual impairment can occur. The fluorescein dye may show no staining or the typical dendritic keratitis (Figure 2).
TREATMENT
Oral antiviral drugs have made the treatment of zoster possible when, effectively, no treatment existed before. Ideally, an antiviral should be given within 72 hours of symptom onset. Starting treatment as early as possible—especially within 72 hours of onset—has been shown to be effective in alleviating acute pain and in preventing or limiting the duration and severity of postherpetic neuralgia.3
Acyclovir (Zovirax) 800 mg five times a day for 7 days or one of its derivatives—eg, famciclovir (Famvir), penciclovir (Denavir), or valacyclovir (Valtrex)—has been shown to be safe and effective in the treatment of active disease, as well as in preventing or shortening the duration of postherpetic neuralgia.3 It has also been shown to reduce the rate of eye involvement from 50% to 20% or 30%.9 This is why all patients with this dermatomal involvement must be treated.
Second-generation antivirals
Valacyclovir 1,000 mg three times a day and famciclovir 500 mg three times a day seem to be as effective as acyclovir in reducing zoster-associated pain, but their efficacy in reducing eye involvement has not been studied. In clinical practice, however, these second-generation antivirals may be more effective than acyclovir because patients are more likely to comply with the treatment regimen of three rather than five daily doses.
Other considerations
In patients with kidney failure, the non-nephrotoxic antiviral brivudine is preferred, but it is not available in the United States. Therefore, one must use acyclovir or one of the other drugs, carefully adjusting the dose according to the creatinine clearance and making sure the patient is well hydrated.
The efficacy of antiviral treatment that is started more than 72 hours after the onset of skin rash has never been confirmed.
Although the additional effectiveness of acyclovir eye ointment has never been established, topical acyclovir can be considered in cases of dendritic or pseudodendritic keratitis.
- Liesegang TJ. Herpes zoster ophthalmicus: natural history, risk factors, clinical presentation, and morbidity. Ophthalmology 2008; 115( suppl 2):S3–S12.
- Opstelten W, Eekhof J, Neven AK, Verheij T. Treatment of herpes zoster. Can Fam Physician 2008; 54:373–377.
- Opstelten W, Zaal MJ. Managing ophthalmic herpes zoster in primary care. BMJ 2005; 331:147–151.
- Donahue JG, Choo PW, Manson JE, Platt R. The incidence of herpes zoster. Arch Intern Med 1995; 155:1605–1609.
- Opstelten W, Zuithoff NP, van Essen GA, et al. Predicting postherpetic neuralgia in elderly primary care patients with herpes zoster: prospective prognostic study. Pain 2007; 132( suppl 1):S52–S59.
- De Morgas JM, Kierland RR. The outcome of patients with herpes zoster. AMA Arch Derm 1957; 75:193–196.
- Hess TM, Lutz LJ, Nauss LA, Lamer TJ. Treatment of acute herpetic neuralgia. A case report and review of the literature. Minn Med 1990; 73:37–40.
- Harding SP, Lipton JR, Wells JC. Natural history of herpes zoster ophthalmicus: predictors of postherpetic neuralgia and ocular involvement. Br J Ophthalmol 1987; 71:353–358.
- Cobo LM, Foulks GN, Liesegang T, et al. Oral acyclovir in the treatment of acute herpes zoster ophthalmicus. Ophthalmology 1986; 93:763–770.
A 75-year-old man presents 4 days after painful cutaneous lesions appeared on the left side of his face, associated with severe ocular pain. Two days before the eruption, he had had an intense headache, which was diagnosed as a tension headache and was treated with oral acetaminophen (Tylenol), but with no improvement.
The remainder of his physical examination is normal. Laboratory tests, including red and white blood cell counts, hemoglobin, and basic metabolic and coagulation tests reveal no abnormalities.
Q: What is your diagnosis?
- Allergic contact dermatitis
- Herpes simplex
- Varicella
- Ramsay-Hunt syndrome
- Herpes zoster ophthalmicus and herpetic keratitis
A: Herpes zoster ophthalmicus is the correct diagnosis. It represents a reactivation of the varicella zoster virus.1
Varicella zoster virus, like others of the herpes family, has developed a complex control of virus-host interactions to ensure its survival in humans. It lies dormant in the sensory ganglia and, when reactivated, moves down the neurons and satellite cells along the sensory axons to the skin.1 The reactivation is related to diminished cell-mediated immunity, which occurs as a physiologic part of aging, which is why the elderly tend to be the most often affected. 2 The incidence of herpes zoster varies from 2.2 to 3.4 per 1,000 people per year.3 Its incidence in people over age 80 is about 10 per 1,000 people per year.3
CLINICAL PRESENTATION
Herpes zoster typically presents as a dermatome-grouped vesicular eruption over an erythematous base, accompanied or preceded by local pain. It has two main complications, postherpetic neuralgia and ocular involvement. Postherpetic neuralgia is neuropathic pain that persists or develops after the dermatomal rash has healed.4 Independent predictors of postherpetic neuralgia are older age, severe acute pain, severe rash, a shorter duration of rash before consultation, and ocular involvement.5 It occurs in 36.6% of patients over age 60, and in 47.5% over 70.6 Persistent postherpetic neuralgia has been linked to suicide in patients over 70.7
Ocular infection occurs with involvement of the ophthalmic division of the fifth cranial nerve. Before the antiviral era, it was seen in as many as 50% of patients.8 Hutchinson’s sign is skin lesions on the tip, side, or root of the nose and is an important predictor of ocular involvement.1 Lesions may include folliculopapillar conjunctivitis, episcleritis, scleritis, keratitis (dendritic, pseudodendritic, and interstitial), uveitis, and necrotizing retinitis.
DIAGNOSIS
The diagnosis of herpes zoster is usually based on clinical observation of the characteristic rash, although viral culture and molecular techniques are available when definitive diagnosis is required. When ophthalmic division is affected and Hutchinson’s sign, unexplained ocular redness with pain, or complaints of visual problems are present, the patient should be referred promptly to an ophthalmologist, because serious visual impairment can occur. The fluorescein dye may show no staining or the typical dendritic keratitis (Figure 2).
TREATMENT
Oral antiviral drugs have made the treatment of zoster possible when, effectively, no treatment existed before. Ideally, an antiviral should be given within 72 hours of symptom onset. Starting treatment as early as possible—especially within 72 hours of onset—has been shown to be effective in alleviating acute pain and in preventing or limiting the duration and severity of postherpetic neuralgia.3
Acyclovir (Zovirax) 800 mg five times a day for 7 days or one of its derivatives—eg, famciclovir (Famvir), penciclovir (Denavir), or valacyclovir (Valtrex)—has been shown to be safe and effective in the treatment of active disease, as well as in preventing or shortening the duration of postherpetic neuralgia.3 It has also been shown to reduce the rate of eye involvement from 50% to 20% or 30%.9 This is why all patients with this dermatomal involvement must be treated.
Second-generation antivirals
Valacyclovir 1,000 mg three times a day and famciclovir 500 mg three times a day seem to be as effective as acyclovir in reducing zoster-associated pain, but their efficacy in reducing eye involvement has not been studied. In clinical practice, however, these second-generation antivirals may be more effective than acyclovir because patients are more likely to comply with the treatment regimen of three rather than five daily doses.
Other considerations
In patients with kidney failure, the non-nephrotoxic antiviral brivudine is preferred, but it is not available in the United States. Therefore, one must use acyclovir or one of the other drugs, carefully adjusting the dose according to the creatinine clearance and making sure the patient is well hydrated.
The efficacy of antiviral treatment that is started more than 72 hours after the onset of skin rash has never been confirmed.
Although the additional effectiveness of acyclovir eye ointment has never been established, topical acyclovir can be considered in cases of dendritic or pseudodendritic keratitis.
A 75-year-old man presents 4 days after painful cutaneous lesions appeared on the left side of his face, associated with severe ocular pain. Two days before the eruption, he had had an intense headache, which was diagnosed as a tension headache and was treated with oral acetaminophen (Tylenol), but with no improvement.
The remainder of his physical examination is normal. Laboratory tests, including red and white blood cell counts, hemoglobin, and basic metabolic and coagulation tests reveal no abnormalities.
Q: What is your diagnosis?
- Allergic contact dermatitis
- Herpes simplex
- Varicella
- Ramsay-Hunt syndrome
- Herpes zoster ophthalmicus and herpetic keratitis
A: Herpes zoster ophthalmicus is the correct diagnosis. It represents a reactivation of the varicella zoster virus.1
Varicella zoster virus, like others of the herpes family, has developed a complex control of virus-host interactions to ensure its survival in humans. It lies dormant in the sensory ganglia and, when reactivated, moves down the neurons and satellite cells along the sensory axons to the skin.1 The reactivation is related to diminished cell-mediated immunity, which occurs as a physiologic part of aging, which is why the elderly tend to be the most often affected. 2 The incidence of herpes zoster varies from 2.2 to 3.4 per 1,000 people per year.3 Its incidence in people over age 80 is about 10 per 1,000 people per year.3
CLINICAL PRESENTATION
Herpes zoster typically presents as a dermatome-grouped vesicular eruption over an erythematous base, accompanied or preceded by local pain. It has two main complications, postherpetic neuralgia and ocular involvement. Postherpetic neuralgia is neuropathic pain that persists or develops after the dermatomal rash has healed.4 Independent predictors of postherpetic neuralgia are older age, severe acute pain, severe rash, a shorter duration of rash before consultation, and ocular involvement.5 It occurs in 36.6% of patients over age 60, and in 47.5% over 70.6 Persistent postherpetic neuralgia has been linked to suicide in patients over 70.7
Ocular infection occurs with involvement of the ophthalmic division of the fifth cranial nerve. Before the antiviral era, it was seen in as many as 50% of patients.8 Hutchinson’s sign is skin lesions on the tip, side, or root of the nose and is an important predictor of ocular involvement.1 Lesions may include folliculopapillar conjunctivitis, episcleritis, scleritis, keratitis (dendritic, pseudodendritic, and interstitial), uveitis, and necrotizing retinitis.
DIAGNOSIS
The diagnosis of herpes zoster is usually based on clinical observation of the characteristic rash, although viral culture and molecular techniques are available when definitive diagnosis is required. When ophthalmic division is affected and Hutchinson’s sign, unexplained ocular redness with pain, or complaints of visual problems are present, the patient should be referred promptly to an ophthalmologist, because serious visual impairment can occur. The fluorescein dye may show no staining or the typical dendritic keratitis (Figure 2).
TREATMENT
Oral antiviral drugs have made the treatment of zoster possible when, effectively, no treatment existed before. Ideally, an antiviral should be given within 72 hours of symptom onset. Starting treatment as early as possible—especially within 72 hours of onset—has been shown to be effective in alleviating acute pain and in preventing or limiting the duration and severity of postherpetic neuralgia.3
Acyclovir (Zovirax) 800 mg five times a day for 7 days or one of its derivatives—eg, famciclovir (Famvir), penciclovir (Denavir), or valacyclovir (Valtrex)—has been shown to be safe and effective in the treatment of active disease, as well as in preventing or shortening the duration of postherpetic neuralgia.3 It has also been shown to reduce the rate of eye involvement from 50% to 20% or 30%.9 This is why all patients with this dermatomal involvement must be treated.
Second-generation antivirals
Valacyclovir 1,000 mg three times a day and famciclovir 500 mg three times a day seem to be as effective as acyclovir in reducing zoster-associated pain, but their efficacy in reducing eye involvement has not been studied. In clinical practice, however, these second-generation antivirals may be more effective than acyclovir because patients are more likely to comply with the treatment regimen of three rather than five daily doses.
Other considerations
In patients with kidney failure, the non-nephrotoxic antiviral brivudine is preferred, but it is not available in the United States. Therefore, one must use acyclovir or one of the other drugs, carefully adjusting the dose according to the creatinine clearance and making sure the patient is well hydrated.
The efficacy of antiviral treatment that is started more than 72 hours after the onset of skin rash has never been confirmed.
Although the additional effectiveness of acyclovir eye ointment has never been established, topical acyclovir can be considered in cases of dendritic or pseudodendritic keratitis.
- Liesegang TJ. Herpes zoster ophthalmicus: natural history, risk factors, clinical presentation, and morbidity. Ophthalmology 2008; 115( suppl 2):S3–S12.
- Opstelten W, Eekhof J, Neven AK, Verheij T. Treatment of herpes zoster. Can Fam Physician 2008; 54:373–377.
- Opstelten W, Zaal MJ. Managing ophthalmic herpes zoster in primary care. BMJ 2005; 331:147–151.
- Donahue JG, Choo PW, Manson JE, Platt R. The incidence of herpes zoster. Arch Intern Med 1995; 155:1605–1609.
- Opstelten W, Zuithoff NP, van Essen GA, et al. Predicting postherpetic neuralgia in elderly primary care patients with herpes zoster: prospective prognostic study. Pain 2007; 132( suppl 1):S52–S59.
- De Morgas JM, Kierland RR. The outcome of patients with herpes zoster. AMA Arch Derm 1957; 75:193–196.
- Hess TM, Lutz LJ, Nauss LA, Lamer TJ. Treatment of acute herpetic neuralgia. A case report and review of the literature. Minn Med 1990; 73:37–40.
- Harding SP, Lipton JR, Wells JC. Natural history of herpes zoster ophthalmicus: predictors of postherpetic neuralgia and ocular involvement. Br J Ophthalmol 1987; 71:353–358.
- Cobo LM, Foulks GN, Liesegang T, et al. Oral acyclovir in the treatment of acute herpes zoster ophthalmicus. Ophthalmology 1986; 93:763–770.
- Liesegang TJ. Herpes zoster ophthalmicus: natural history, risk factors, clinical presentation, and morbidity. Ophthalmology 2008; 115( suppl 2):S3–S12.
- Opstelten W, Eekhof J, Neven AK, Verheij T. Treatment of herpes zoster. Can Fam Physician 2008; 54:373–377.
- Opstelten W, Zaal MJ. Managing ophthalmic herpes zoster in primary care. BMJ 2005; 331:147–151.
- Donahue JG, Choo PW, Manson JE, Platt R. The incidence of herpes zoster. Arch Intern Med 1995; 155:1605–1609.
- Opstelten W, Zuithoff NP, van Essen GA, et al. Predicting postherpetic neuralgia in elderly primary care patients with herpes zoster: prospective prognostic study. Pain 2007; 132( suppl 1):S52–S59.
- De Morgas JM, Kierland RR. The outcome of patients with herpes zoster. AMA Arch Derm 1957; 75:193–196.
- Hess TM, Lutz LJ, Nauss LA, Lamer TJ. Treatment of acute herpetic neuralgia. A case report and review of the literature. Minn Med 1990; 73:37–40.
- Harding SP, Lipton JR, Wells JC. Natural history of herpes zoster ophthalmicus: predictors of postherpetic neuralgia and ocular involvement. Br J Ophthalmol 1987; 71:353–358.
- Cobo LM, Foulks GN, Liesegang T, et al. Oral acyclovir in the treatment of acute herpes zoster ophthalmicus. Ophthalmology 1986; 93:763–770.
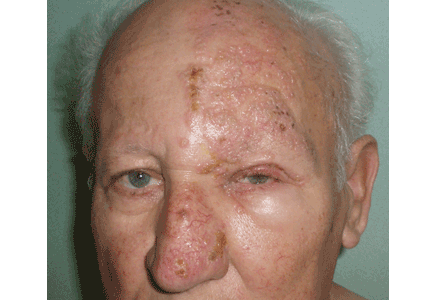
Black esophagus
A 60-year-old man with a history of alcoholic cirrhosis and gastrointestinal bleeding was admitted after being found unconscious. He had coffee-ground emesis and melena.
ASSOCIATED CONDITIONS
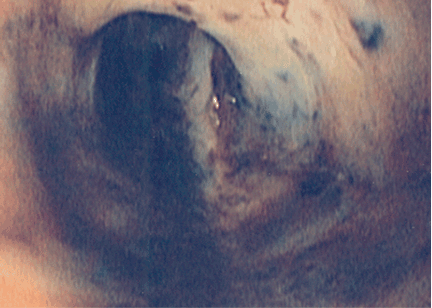
TREATMENT AND COURSE
The treatment is mainly supportive, with intravenous fluids, proton pump inhibitors, and nothing taken orally.1 Complications include stricture, perforation, and death.1,3 The death rate is about 30% in these patients.3
- Khan AM, Hundal R, Ramaswamy V, Korsten M, Dhuper S. Acute esophageal necrosis and liver pathology, a rare combination. World J Gastroenterol 2004; 10:2457–2458.
- Le K, Ahmed A. Acute necrotizing esophagitis: case report and review of the literature. J La State Med Soc 2007; 159:330,333–338.
- Katsinelos P, Pilpilidis I, Dimiropoulos S, et al. Black esophagus induced by severe vomiting in a healthy young man. Surg Endosc 2003; 17:521.
A 60-year-old man with a history of alcoholic cirrhosis and gastrointestinal bleeding was admitted after being found unconscious. He had coffee-ground emesis and melena.
ASSOCIATED CONDITIONS
TREATMENT AND COURSE
The treatment is mainly supportive, with intravenous fluids, proton pump inhibitors, and nothing taken orally.1 Complications include stricture, perforation, and death.1,3 The death rate is about 30% in these patients.3
A 60-year-old man with a history of alcoholic cirrhosis and gastrointestinal bleeding was admitted after being found unconscious. He had coffee-ground emesis and melena.
ASSOCIATED CONDITIONS
TREATMENT AND COURSE
The treatment is mainly supportive, with intravenous fluids, proton pump inhibitors, and nothing taken orally.1 Complications include stricture, perforation, and death.1,3 The death rate is about 30% in these patients.3
- Khan AM, Hundal R, Ramaswamy V, Korsten M, Dhuper S. Acute esophageal necrosis and liver pathology, a rare combination. World J Gastroenterol 2004; 10:2457–2458.
- Le K, Ahmed A. Acute necrotizing esophagitis: case report and review of the literature. J La State Med Soc 2007; 159:330,333–338.
- Katsinelos P, Pilpilidis I, Dimiropoulos S, et al. Black esophagus induced by severe vomiting in a healthy young man. Surg Endosc 2003; 17:521.
- Khan AM, Hundal R, Ramaswamy V, Korsten M, Dhuper S. Acute esophageal necrosis and liver pathology, a rare combination. World J Gastroenterol 2004; 10:2457–2458.
- Le K, Ahmed A. Acute necrotizing esophagitis: case report and review of the literature. J La State Med Soc 2007; 159:330,333–338.
- Katsinelos P, Pilpilidis I, Dimiropoulos S, et al. Black esophagus induced by severe vomiting in a healthy young man. Surg Endosc 2003; 17:521.
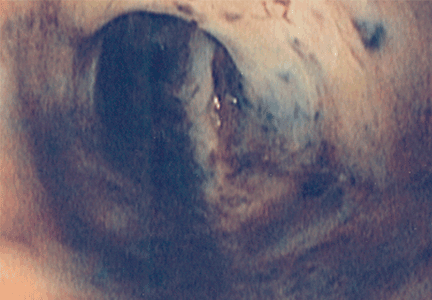
Soft tissue atrophy after corticosteroid injection
A 27-year-old woman presents with pain and tenderness over her right radial styloid. Examination reveals tenderness to palpation and a positive Finkelstein test, and her condition is diagnosed as de Quervain tenosynovitis. She is referred for occupational therapy and for a corticosteroid injection.
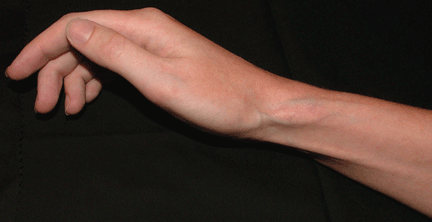
Q: On the basis of the skin findings, which corticosteroid injection was most likely used?
- Triamcinolone hexacetonide (Aristospan)
- Dexamethasone sodium phosphate (Decadron)
- Betamethasone sodium phosphate and betamethasone acetate (Celestone Soluspan)
- Triamcinolone acetonide (Kenalog-40)
A: Both triamcinolone hexacetonide and triamcinolone acetonide are correct, as they are the least soluble of the agents listed.
ADVERSE EFFECTS OF STEROID INJECTIONS
Soft tissue atrophy and local depigmentation are possible adverse effects of any steroid injection, particularly when given at a superficial site.1,2 Although these are rare, with an estimated risk of less than 1%, patients still need to be told about these potential side effects.3 In addition, these adverse effects of injection may be prevented by applying pressure with gauze over the injection site as the needle is withdrawn to prevent leakage of corticosteroid along the needle track.3
Soft tissue atrophy generally appears in 1 to 4 months and resolves 6 to 30 months later. 4 Patients with darker skin are at greater risk of depigmentation.
The cause of the pigment changes is not fully understood but may be related either to the steroid or to the constituents of the vehicle in which the steroid is suspended.5
CHOOSING THE APPROPRIATE STEROID PREPARATION
Although soft tissue (fat) atrophy and local depigmentation are possible with any steroid preparation injected into soft tissue, the risk can be modulated by using a corticosteroid agent with appropriate solubility. A less soluble agent such as triamcinolone acetonide or hexacetonide is preferred for intra-articular injections of deep structures, such as the knee, elbow, or shoulder. A more soluble agent, such as betamethasone sodium phosphate and acetate or dexamethasone sodium phosphate, is preferred for soft tissue injections of bursae, tendon sheaths, metacarpophalangeal joints, proximal phalangeal joints, and the carpal tunnel.
OTHER POSSIBLE COMPLICATIONS
Other potential complications of corticosteroid injection include pain, bleeding, infection (risk 1 in 40,000), flushing, post-injection flare (< 1%), nerve damage, tendon weakening, and rarely, tendon rupture. In cases of tendonitis, it is very important to ensure that the drug is injected into the tendon sheath and not the tendon. A general rule for tendon sheath injection is to not inject if resistance is met.
PATIENT UNWILLING TO RECEIVE MORE INJECTIONS
This patient’s symptoms persist, with a painful right wrist, perhaps due to refractory de Quervain tenosynovitis, nerve damage, or tendinosis. In time, the tenosynovitis and atrophy may improve, but she is reluctant to receive any more injections, as she was not forewarned about the possibility of atrophy.
- Saunders S, Longworth S. Injection Techniques in Orthopaedics and Sports Medicine. 3rd ed. London: Elsevier; 2006.
- Cardone DA, Tallia AF. Joint and soft tissue injection. Am Fam Physician 2002; 66:283–288.
- Gray RG, Gottlieb NL. Intra-articular corticosteroids: an updated assessment. Clin Orthop Relat Res 1983; 177:235–263.
- Cassidy JT, Bole GG. Cutaneous atrophy secondary to intra-articular corticosteroid administration. Ann Intern Med 1966; 65:1008–1018.
- Newman RJ. Local skin depigmentation due to corticosteroid injection. Br Med J (Clin Res Ed) 1984; 288:1725–1726.
A 27-year-old woman presents with pain and tenderness over her right radial styloid. Examination reveals tenderness to palpation and a positive Finkelstein test, and her condition is diagnosed as de Quervain tenosynovitis. She is referred for occupational therapy and for a corticosteroid injection.
Q: On the basis of the skin findings, which corticosteroid injection was most likely used?
- Triamcinolone hexacetonide (Aristospan)
- Dexamethasone sodium phosphate (Decadron)
- Betamethasone sodium phosphate and betamethasone acetate (Celestone Soluspan)
- Triamcinolone acetonide (Kenalog-40)
A: Both triamcinolone hexacetonide and triamcinolone acetonide are correct, as they are the least soluble of the agents listed.
ADVERSE EFFECTS OF STEROID INJECTIONS
Soft tissue atrophy and local depigmentation are possible adverse effects of any steroid injection, particularly when given at a superficial site.1,2 Although these are rare, with an estimated risk of less than 1%, patients still need to be told about these potential side effects.3 In addition, these adverse effects of injection may be prevented by applying pressure with gauze over the injection site as the needle is withdrawn to prevent leakage of corticosteroid along the needle track.3
Soft tissue atrophy generally appears in 1 to 4 months and resolves 6 to 30 months later. 4 Patients with darker skin are at greater risk of depigmentation.
The cause of the pigment changes is not fully understood but may be related either to the steroid or to the constituents of the vehicle in which the steroid is suspended.5
CHOOSING THE APPROPRIATE STEROID PREPARATION
Although soft tissue (fat) atrophy and local depigmentation are possible with any steroid preparation injected into soft tissue, the risk can be modulated by using a corticosteroid agent with appropriate solubility. A less soluble agent such as triamcinolone acetonide or hexacetonide is preferred for intra-articular injections of deep structures, such as the knee, elbow, or shoulder. A more soluble agent, such as betamethasone sodium phosphate and acetate or dexamethasone sodium phosphate, is preferred for soft tissue injections of bursae, tendon sheaths, metacarpophalangeal joints, proximal phalangeal joints, and the carpal tunnel.
OTHER POSSIBLE COMPLICATIONS
Other potential complications of corticosteroid injection include pain, bleeding, infection (risk 1 in 40,000), flushing, post-injection flare (< 1%), nerve damage, tendon weakening, and rarely, tendon rupture. In cases of tendonitis, it is very important to ensure that the drug is injected into the tendon sheath and not the tendon. A general rule for tendon sheath injection is to not inject if resistance is met.
PATIENT UNWILLING TO RECEIVE MORE INJECTIONS
This patient’s symptoms persist, with a painful right wrist, perhaps due to refractory de Quervain tenosynovitis, nerve damage, or tendinosis. In time, the tenosynovitis and atrophy may improve, but she is reluctant to receive any more injections, as she was not forewarned about the possibility of atrophy.
A 27-year-old woman presents with pain and tenderness over her right radial styloid. Examination reveals tenderness to palpation and a positive Finkelstein test, and her condition is diagnosed as de Quervain tenosynovitis. She is referred for occupational therapy and for a corticosteroid injection.
Q: On the basis of the skin findings, which corticosteroid injection was most likely used?
- Triamcinolone hexacetonide (Aristospan)
- Dexamethasone sodium phosphate (Decadron)
- Betamethasone sodium phosphate and betamethasone acetate (Celestone Soluspan)
- Triamcinolone acetonide (Kenalog-40)
A: Both triamcinolone hexacetonide and triamcinolone acetonide are correct, as they are the least soluble of the agents listed.
ADVERSE EFFECTS OF STEROID INJECTIONS
Soft tissue atrophy and local depigmentation are possible adverse effects of any steroid injection, particularly when given at a superficial site.1,2 Although these are rare, with an estimated risk of less than 1%, patients still need to be told about these potential side effects.3 In addition, these adverse effects of injection may be prevented by applying pressure with gauze over the injection site as the needle is withdrawn to prevent leakage of corticosteroid along the needle track.3
Soft tissue atrophy generally appears in 1 to 4 months and resolves 6 to 30 months later. 4 Patients with darker skin are at greater risk of depigmentation.
The cause of the pigment changes is not fully understood but may be related either to the steroid or to the constituents of the vehicle in which the steroid is suspended.5
CHOOSING THE APPROPRIATE STEROID PREPARATION
Although soft tissue (fat) atrophy and local depigmentation are possible with any steroid preparation injected into soft tissue, the risk can be modulated by using a corticosteroid agent with appropriate solubility. A less soluble agent such as triamcinolone acetonide or hexacetonide is preferred for intra-articular injections of deep structures, such as the knee, elbow, or shoulder. A more soluble agent, such as betamethasone sodium phosphate and acetate or dexamethasone sodium phosphate, is preferred for soft tissue injections of bursae, tendon sheaths, metacarpophalangeal joints, proximal phalangeal joints, and the carpal tunnel.
OTHER POSSIBLE COMPLICATIONS
Other potential complications of corticosteroid injection include pain, bleeding, infection (risk 1 in 40,000), flushing, post-injection flare (< 1%), nerve damage, tendon weakening, and rarely, tendon rupture. In cases of tendonitis, it is very important to ensure that the drug is injected into the tendon sheath and not the tendon. A general rule for tendon sheath injection is to not inject if resistance is met.
PATIENT UNWILLING TO RECEIVE MORE INJECTIONS
This patient’s symptoms persist, with a painful right wrist, perhaps due to refractory de Quervain tenosynovitis, nerve damage, or tendinosis. In time, the tenosynovitis and atrophy may improve, but she is reluctant to receive any more injections, as she was not forewarned about the possibility of atrophy.
- Saunders S, Longworth S. Injection Techniques in Orthopaedics and Sports Medicine. 3rd ed. London: Elsevier; 2006.
- Cardone DA, Tallia AF. Joint and soft tissue injection. Am Fam Physician 2002; 66:283–288.
- Gray RG, Gottlieb NL. Intra-articular corticosteroids: an updated assessment. Clin Orthop Relat Res 1983; 177:235–263.
- Cassidy JT, Bole GG. Cutaneous atrophy secondary to intra-articular corticosteroid administration. Ann Intern Med 1966; 65:1008–1018.
- Newman RJ. Local skin depigmentation due to corticosteroid injection. Br Med J (Clin Res Ed) 1984; 288:1725–1726.
- Saunders S, Longworth S. Injection Techniques in Orthopaedics and Sports Medicine. 3rd ed. London: Elsevier; 2006.
- Cardone DA, Tallia AF. Joint and soft tissue injection. Am Fam Physician 2002; 66:283–288.
- Gray RG, Gottlieb NL. Intra-articular corticosteroids: an updated assessment. Clin Orthop Relat Res 1983; 177:235–263.
- Cassidy JT, Bole GG. Cutaneous atrophy secondary to intra-articular corticosteroid administration. Ann Intern Med 1966; 65:1008–1018.
- Newman RJ. Local skin depigmentation due to corticosteroid injection. Br Med J (Clin Res Ed) 1984; 288:1725–1726.
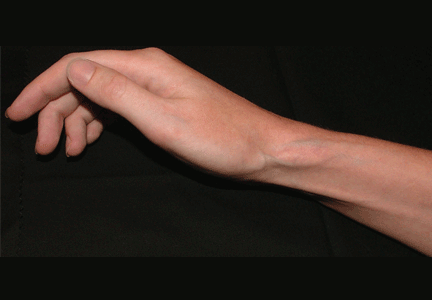
Yellow nails, ankle edema, and pleural effusion
A 57-year-old woman says that for the past 30 years she has been aware of thickening, reduction in the growth rate, and yellow discoloration of the nail beds of her fingers and toes. Nail clippings were cultured about 10 years ago, but no fungus was isolated. About 7 years after she first noticed the nail changes, she developed pitting edema of the ankles without evidence of cardiac failure, and she was put on a diuretic.
A chest radiograph taken 8 years ago during a health examination was normal. However, 1 month before admission to our department, she began experiencing exertional dyspnea, with productive cough but no fever.
She has never traveled abroad, has no history of hypertension or ischemic cardiomyopathy, has never undergone surgery, and has no history of tuberculosis.
EXAMINATION AND TESTING
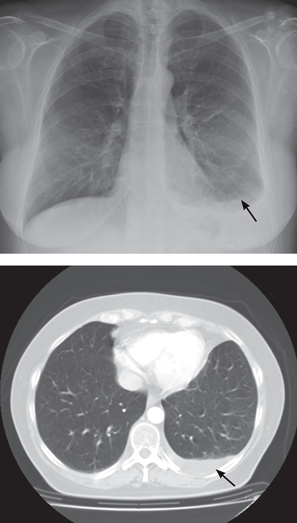
Q: Which of the following is the most likely diagnosis?
- Cardiac failure
- Yellow nail syndrome
- Infection
- Neoplastic disease
A: The correct diagnosis is yellow nail syndrome, a rare disorder characterized by yellow nails, lymphedema, and chronic respiratory manifestations. However, patients rarely present with all three parts of the triad, and fewer than half of patients present with pleural effusions.
The respiratory manifestations of yellow nail syndrome are diverse and include pleural effusion, bronchiectasis, rhinosinusitis, chronic cough, and recurrent lung infections. Furthermore, this syndrome is in the differential diagnosis of chronic pleural effusions lasting more than 1 year, and also of bronchiectasis and rhinosinusitis.
The pathophysiology of yellow nail syndrome remains unclear, but various anatomic or functional lymphatic drainage abnormalities have been proposed as the underlying cause.
Therapy includes bronchopulmonary hygiene (eg, postural drainage); inhaled steroids and antibiotics to control exacerbations of symptoms; serial thoracocentesis or pleurodesis to control pleural effusions; and lymphatic drainage and diuretic drugs for lymphedema. Some experts propose vitamin E for the treatment of yellow nails, but yellow nails often improve spontaneously.
CASE CONCLUDED
In this case, the diagnosis of yellow nail syndrome was based on the presence of its three major features. She had no history of hypertension or ischemic cardiomyopathy, and no echocardiographic signs of cardiac failure. She had no signs of infection or malignancy and had never taken antirheumatic drugs such as d-penicillamine (Cuprimine, Depen), which can cause pleural effusion, and nothing in her history or presentation suggested an immunosuppressed state.
Her treatment involved thoracocentesis to reduce the pleural effusion, correction of her nutritional status, a low-salt diet, vitamin E supplementation, and diuretics. At discharge, her pleural effusion and lymphedema were reduced, and her dyspnea had resolved.
SUGGESTED READING
Maldonado F, Tazelaar HD, Wang CW, Ryu JH. Yellow nail syndrome: analysis of 41 consecutive patients. Chest 2008; 134:375–381.
A 57-year-old woman says that for the past 30 years she has been aware of thickening, reduction in the growth rate, and yellow discoloration of the nail beds of her fingers and toes. Nail clippings were cultured about 10 years ago, but no fungus was isolated. About 7 years after she first noticed the nail changes, she developed pitting edema of the ankles without evidence of cardiac failure, and she was put on a diuretic.
A chest radiograph taken 8 years ago during a health examination was normal. However, 1 month before admission to our department, she began experiencing exertional dyspnea, with productive cough but no fever.
She has never traveled abroad, has no history of hypertension or ischemic cardiomyopathy, has never undergone surgery, and has no history of tuberculosis.
EXAMINATION AND TESTING
Q: Which of the following is the most likely diagnosis?
- Cardiac failure
- Yellow nail syndrome
- Infection
- Neoplastic disease
A: The correct diagnosis is yellow nail syndrome, a rare disorder characterized by yellow nails, lymphedema, and chronic respiratory manifestations. However, patients rarely present with all three parts of the triad, and fewer than half of patients present with pleural effusions.
The respiratory manifestations of yellow nail syndrome are diverse and include pleural effusion, bronchiectasis, rhinosinusitis, chronic cough, and recurrent lung infections. Furthermore, this syndrome is in the differential diagnosis of chronic pleural effusions lasting more than 1 year, and also of bronchiectasis and rhinosinusitis.
The pathophysiology of yellow nail syndrome remains unclear, but various anatomic or functional lymphatic drainage abnormalities have been proposed as the underlying cause.
Therapy includes bronchopulmonary hygiene (eg, postural drainage); inhaled steroids and antibiotics to control exacerbations of symptoms; serial thoracocentesis or pleurodesis to control pleural effusions; and lymphatic drainage and diuretic drugs for lymphedema. Some experts propose vitamin E for the treatment of yellow nails, but yellow nails often improve spontaneously.
CASE CONCLUDED
In this case, the diagnosis of yellow nail syndrome was based on the presence of its three major features. She had no history of hypertension or ischemic cardiomyopathy, and no echocardiographic signs of cardiac failure. She had no signs of infection or malignancy and had never taken antirheumatic drugs such as d-penicillamine (Cuprimine, Depen), which can cause pleural effusion, and nothing in her history or presentation suggested an immunosuppressed state.
Her treatment involved thoracocentesis to reduce the pleural effusion, correction of her nutritional status, a low-salt diet, vitamin E supplementation, and diuretics. At discharge, her pleural effusion and lymphedema were reduced, and her dyspnea had resolved.
A 57-year-old woman says that for the past 30 years she has been aware of thickening, reduction in the growth rate, and yellow discoloration of the nail beds of her fingers and toes. Nail clippings were cultured about 10 years ago, but no fungus was isolated. About 7 years after she first noticed the nail changes, she developed pitting edema of the ankles without evidence of cardiac failure, and she was put on a diuretic.
A chest radiograph taken 8 years ago during a health examination was normal. However, 1 month before admission to our department, she began experiencing exertional dyspnea, with productive cough but no fever.
She has never traveled abroad, has no history of hypertension or ischemic cardiomyopathy, has never undergone surgery, and has no history of tuberculosis.
EXAMINATION AND TESTING
Q: Which of the following is the most likely diagnosis?
- Cardiac failure
- Yellow nail syndrome
- Infection
- Neoplastic disease
A: The correct diagnosis is yellow nail syndrome, a rare disorder characterized by yellow nails, lymphedema, and chronic respiratory manifestations. However, patients rarely present with all three parts of the triad, and fewer than half of patients present with pleural effusions.
The respiratory manifestations of yellow nail syndrome are diverse and include pleural effusion, bronchiectasis, rhinosinusitis, chronic cough, and recurrent lung infections. Furthermore, this syndrome is in the differential diagnosis of chronic pleural effusions lasting more than 1 year, and also of bronchiectasis and rhinosinusitis.
The pathophysiology of yellow nail syndrome remains unclear, but various anatomic or functional lymphatic drainage abnormalities have been proposed as the underlying cause.
Therapy includes bronchopulmonary hygiene (eg, postural drainage); inhaled steroids and antibiotics to control exacerbations of symptoms; serial thoracocentesis or pleurodesis to control pleural effusions; and lymphatic drainage and diuretic drugs for lymphedema. Some experts propose vitamin E for the treatment of yellow nails, but yellow nails often improve spontaneously.
CASE CONCLUDED
In this case, the diagnosis of yellow nail syndrome was based on the presence of its three major features. She had no history of hypertension or ischemic cardiomyopathy, and no echocardiographic signs of cardiac failure. She had no signs of infection or malignancy and had never taken antirheumatic drugs such as d-penicillamine (Cuprimine, Depen), which can cause pleural effusion, and nothing in her history or presentation suggested an immunosuppressed state.
Her treatment involved thoracocentesis to reduce the pleural effusion, correction of her nutritional status, a low-salt diet, vitamin E supplementation, and diuretics. At discharge, her pleural effusion and lymphedema were reduced, and her dyspnea had resolved.
SUGGESTED READING
Maldonado F, Tazelaar HD, Wang CW, Ryu JH. Yellow nail syndrome: analysis of 41 consecutive patients. Chest 2008; 134:375–381.
SUGGESTED READING
Maldonado F, Tazelaar HD, Wang CW, Ryu JH. Yellow nail syndrome: analysis of 41 consecutive patients. Chest 2008; 134:375–381.