User login
Von Willebrand Disease: Approach to Diagnosis and Management
Introduction
von Willebrand disease (VWD) is an inherited bleeding disorder caused by deficient or defective plasma von Willebrand factor (VWF). VWF is an adhesive multimeric plasma glycoprotein that performs 2 major functions in hemostasis: it mediates platelet adhesion to injured subendothelium via glycoprotein 1bα (GPIbα), and it binds and stabilizes factor VIII (FVIII) in circulation, protecting it from proteolytic degradation by enzymes. The current VWD classification recognizes 3 types (Table 1).1 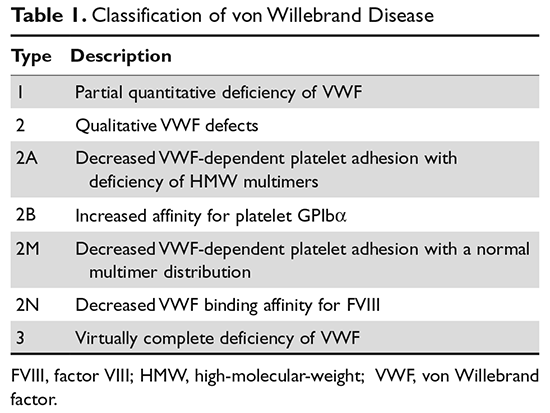
Prevalence
VWD is the most common inherited bleeding disorder. However, because VWF levels are highly variable and disease severity ranges from mild bleeding symptoms to severe or life-threatening bleeds, the reported prevalence of VWD depends on the diagnostic definition used. Two large epidemiologic studies have reported prevalence rates of approximately 1%.2,3 In these studies, healthy school-aged children were screened and diagnosed with VWD based on low VWF activity, measured as ristocetin cofactor, and a personal and family history of bleeding symptoms. At the other extreme, when considering patients whose bleeding symptoms are sufficiently severe to warrant referral to specialized centers, the reported prevalence of VWD ranges from 20 to 113 per million.4 These studies likely over- and underestimate clinically significant VWD. More recent studies suggest that the prevalence of VWD in individuals whose bleeding symptoms are significant enough to present to a primary care physician is approximately 0.1%.5 This figure is likely a more accurate estimate of the true prevalence of symptomatic VWD.
Although VWD is autosomally inherited, females are more likely to present with bleeding symptoms and be diagnosed because of increased exposure to bleeding challenges, such as menorrhagia and childbirth. VWD does not show any geographic or ethnic predilection, but there is an increased prevalence of the recessive forms, such as type 2N and type 3 VWD, in areas with high rates of consanguinity.
VWF Protein Structure and Function
The VWF gene is located on chromosome 12 at p13.3 and spans 178 kb comprising 52 exons.6 The expression of the VWF gene is tightly restricted to endothelial cells, platelets, and megakaryocytes, where VWF is stored in Weibel-Palade bodies and α-granules. VWF is a large multimeric glycoprotein with several important functional domains (Figure). 
Classification, Pathophysiology, and Genetics
The International Society of Thrombosis and Hemostasis (ISTH) classification of VWD was updated in 2006 (Table 1).1 It incorporates important aspects of clinical phenotype, pathophysiological mechanisms, and treatment considerations. The 3 categories are: type 1, which is a partial quantitative deficiency; type 2 with 4 subtypes (2A, 2B, 2M, and 2N), which is a qualitative defect; and type 3, which is a virtual absence of VWF. Although the diagnosis and categorization of VWD can be achieved with widely available laboratory testing, further subcategorization among type 2 VWD subtypes may require referral to a specialized laboratory. The current ISTH classification intentionally does not incorporate genotypic data. In type 2 or type 3 VWD disease, VWF mutations are identified in more than 90% of cases and are completely penetrant, whereas mutations are identified in only approximately 65% of type 1 VWD cases and have been associated with incomplete penetrance and variable expressivity.10 These studies suggest that type 1 VWD is an oligogenic disease with mutations in genes regulating secretion or clearance contributing to a VWD phenotype.
VWD Types
Type 1
Type 1 VWD is caused by a partial quantitative deficiency of VWF and represents approximately 75% of VWD cases. It is the most clinically heterogeneous type, with patients having a mild to moderate bleeding phenotype.11 Bleeding in type 1 VWD results from a decrease in the concentration of VWF. The VWF function is normal without a significant abnormality in the platelet, collagen, or FVIII binding sites or a significant decrease in HMW multimers. Functional assays of VWF, such as VWF ristocetin cofactor (VWF:RCo) or VWF activity (VWF:Act) (see section on Laboratory Testing for further details), are proportionally decreased relative to the VWF antigen level (VWF:Ag), and the ratio of functional activity as compared with the VWF level is normal (ie, VWF:RCo/VWF:Ag ratio is > 0.6). As noted, VWF mutations are identified in only 65% of type 1 VWD cases and have incomplete penetrance and variable expressivity.10 Approximately 70% of mutations identified are missense mutations. Missense mutations may affect VWF levels by affecting any part of the biosynthetic pathway, including trafficking, storage, secretion, and/or clearance of VWF.
Increased VWF clearance is a well-described mechanism for type 1 VWD, known as type 1C. These patients will typically have very low VWF levels, an increased VWF propeptide to antigen ratio (VWFpp/VWF:Ag), and a marked but short-lived response to DDAVP, limiting DDAVP’s clinical applicability.12 On the other hand, the half-life of VWF/FVIII concentrates is normal in these individuals. Type 1C VWD is caused by missense mutations which occur mainly in the D3 domain and reduce the half-life of VWF up to 15-fold. R1205H, known as the “Vicenza” variant, is the most common and severe as well as the best characterized of these mutations.13
Type 2
Accounting for approximately 25% of VWD cases, type 2 VWD is characterized by a qualitative deficiency of VWF activity and is further subcategorized based on the mechanism of VWF dysfunction. Type 2A, 2B, and 2M affect VWF–platelet interactions by way of loss of HMW multimers, a gain of function of the GPIbα binding site, or a loss of function of the same site, respectively. On the other hand, type 2N is caused by defective VWF binding to FVIII. Type 2 VWD is often suspected when investigations demonstrate a function-antigen discordance: the VWF:RCo or VWF:Act is decreased disproportionately to the decrease in VWF:Ag, and the VWF:RCo/VWF:Ag ratio is less than 0.6.
Type 2A VWD is the most common type 2 variant. It is characterized by disproportionately low functional activity compared to antigen level (ie, VWF:RCo/VWF:ag ratio is < 0.6) and a loss of HMW and sometimes intermediate molecular weight (IMW) multimers. Ristocetin-induced platelet agglutination (RIPA) will be decreased with standard doses of ristocetin and absent with low doses. Type 2A VWD is usually inherited as an autosomal dominant trait. This subtype encompasses missense mutations that impair dimerization or multimerization of VWF subunits (CK, D1, and D2 domains); disrupt intersubunit disulphide bonds (D3 and D2 domains); enhance susceptibility to ADAMTS13-mediated proteolysis (A2 and A1 domains); or result in intracellular retention of the HMW multimers (D3, A1, and A2 domains).10 The result is VWF that lacks HMW multimers, thereby possessing fewer GPIbα binding sites, and that is less effective in binding platelets.
Type 2B VWD is the result of gain-of-function mutations within the GPIbα binding site of VWF. Generally, the platelet-binding site of VWF within the A1 domain is only exposed once VWF is immobilized on injured collagen and subjected to shear forces, resulting in a conformational change.7 In type 2B VWD, the gain-of-function mutation results in spontaneous binding of VWF to platelets without the need for a VWF-collagen interaction and unfolding of VWF by shear forces. The VWF–platelet interaction selectively depletes the HMW multimers by the unfolding of the A2 domain and increasing ADAMTS13 proteolysis. The increased binding of mutant VWF to platelets also triggers the formation of platelet aggregates, which are removed from circulation resulting in thrombocytopenia. Increases in endogenous VWF seen with acute stressors or pregnancy can worsen thrombocytopenia and increase the risk of bleeding.14 Certain mutations, such as V1316M, alter megakaryocytopoiesis and are characterized by giant platelets with abnormal ultrastructure and further exacerbate the thrombocytopenia.15 The laboratory profile reveals a VWF:RCo/VWF:Ag ratio of < 0.6 and absence of HMW multimers. In contrast to type 2A, platelets will agglutinate with low-dose ristocetin. Missense mutations are highly penetrant dominant and occur in or close to the A1 domain.16
Type 2M VWD is characterized by loss-of-function mutations within the GPIbα binding site of VWF. Phenotypic characteristics include a reduced ratio of VWF:RCo/VWF:Ag of < 0.6 but a normal multimer pattern.17 Missense mutations are reported in the A1 domain affecting the GPIbα-binding site. In very rare instances, mutations in the A3 domain that impair the VWF/collagen interaction have been described.18 These collagen-binding mutations are not included in the last iteration of the ISTH classification in 2006,1 but fit best in the type 2M category. In these cases, VWF:RCo or VWF:Act, which reflect activity at the GPIbα-binding site, may be normal and the diagnosis requires VWF/collagen binding assays (VWF:CB).
Type 2N VWD results from mutations of the FVIII binding site or conformational changes that impair the VWF–FVIII interaction. Most (~80%) missense mutations are located in domains D’ and D3.19 These mutations are autosomal recessive, and affected individuals are either homozygous or compound heterozygous for type 2N/2N or type 1/2N mutations, or compound heterozygous for a missense mutation and a mutation resulting in a null allele (type 2N/3 mutations). The laboratory phenotype is a disproportionate reduction in the FVIII level relative to the VWF level, which may be low or normal. Most cases of type 2N VWD have a normal multimeric profile, but rare cases will demonstrate loss of HMW multimers. Definitive diagnosis requires evidence of reduced FVIII binding to VWF (VWF:FVIIIB) or the identification of causative mutations in the FVIII binding region of the VWF gene.20
Type 3
Type 3 VWD is defined by a virtual absence of VWF. The inheritance of type 3 VWD has often been reported as autosomal recessive. However, there is emerging evidence that it can also be inherited in a co-dominant pattern: obligate carriers of type 3 VWD mutations have more mucocutaneous bleeding symptoms than normal individuals, and in approximately 50% of cases may carry a diagnosis of type 1 VWD.21 This condition is characterized by prolongation of the activated partial thromboplastin time (aPTT), undetectable levels of VWF:Ag, and VWF:RCo and FVIII levels less than 10 IU/dL (10%). The majority (~80%) of type 3 VWD patients have 2 null alleles as a result of a variety of mutations, with nonsense mutations accounting for about one-third.10 The remainder of the mutational spectrum is made up of missense mutations predominantly located in the D1-D2 (exons 3–11) and D4-CK (exons 37–52) domains that result in intracellular VWF retention, or large deletions, resulting in frameshift mutations affecting 1 or more exons. Because there is little or no circulating VWF, patients with type 3 VWD may develop alloantibodies to VWF, which can complicate treatment.22
Diagnosis
Clinical Manifestations
VWD is a congenital bleeding disorder. The increased risk of bleeding is present from birth, but symptoms may only manifest when there is a hemostatic challenge. Bleeding symptoms become more apparent with increasing age and exposure to hemostatic challenges. As a result, the diagnosis is often delayed into adulthood in mild to moderate forms of VWD. On the other hand, with more severe bleeding phenotypes such as type 3 VWD, the diagnosis is often made in childhood. Individuals with VWD primarily complain of excessive mucocutaneous bleeding, which includes spontaneous bruising, recurrent epistaxis, and bleeding from the gums after brushing, dental cleaning, and extractions. In addition, prolonged or excessive bleeding after surgery or trauma is often reported. Females frequently experience menorrhagia, usually beginning at menarche, and can have prolonged or excessive bleeding after childbirth.23 Musculoskeletal bleeding is unusual, except in type 2N or type 3 VWD when the FVIII:C level may be less than 10 IU/dL.
Mucocutaneous bleeding symptoms such as epistaxis, gum bleeding, ecchymosis, and menorrhagia overlap with those experienced by a normal population, and therefore can be easily overlooked by both patients and physicians.11 The use of bleeding assessment tools (BATs) to standardize the bleeding history and interpretation of the severity of the bleeding phenotype is becoming part of routine clinical practice. Three different BATs, each an adaptation of its predecessor, have been created and validated.24 Each of the scores performs well in an undiagnosed population presenting with bleeding symptoms. The negative predictive value is typically greater than 0.99, meaning that a negative bleeding score nearly excludes a clinically significant bleeding disorder. Thus, the main utility of the current BATs is at the time of new patient assessments: a negative bleeding score will help avoid unnecessary laboratory testing and prevent false-positive diagnoses of VWD (borderline low VWF:Ag without a significant bleeding history). However, the currently available BATs have some limitations. When scoring severe bleeding disorders, BATs become saturated as they take into account the worst episode of bleeding within each category but not the frequency of bleeding. BATs need to be administered by an expert and are time consuming to complete. Finally, they are not useful for monitoring bleeding symptoms or response to therapy because of the cumulative nature of the scores. In an attempt to standardize the BAT and bleeding score, the ISTH/Scientific and Standardization Committee (SSC) Joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group has established a revised BAT, known as the ISTH-BAT, specifically designed to extend the utility of the earlier BATS by incorporating information on both symptom frequency and severity.25,26 The ISTH-BAT has been further modified to a patient- or self-administered BAT (SELF-BAT). The SELF-BAT has been shown to be a reliable and effective tool in the assessment of patients who are being evaluated for VWD.27
Laboratory Testing
Screening tests include a complete blood count (CBC), prothrombin time, aPTT, thrombin time, and fibrinogen concentration to exclude the presence of other hemostatic disorders. The CBC may show thrombocytopenia in type 2B VWD. The aPTT is often normal, but will be prolonged if the FVIII level is below 30 IU/dL, as can be seen in severe type 1, type 2N, or type 3 VWD. The platelet function analyzer (PFA-100) is a system for analyzing primary hemostasis under high shear rates, but its role in the diagnosis of VWD is controversial.11
The evaluation of VWD involves quantitative (VWF:Ag) and qualitative measurements of VWF (VWF:RCo, or one of the novel assays: VWF:Act or VWF:GPIbM) and FVIII activity (FVIII:C). Type 2 VWD is suspected when the VWF activity to VWF:Ag ratio is < 0.6, the FVIII:C is more significantly decreased as compared to VWF:Ag, or with the presence of thrombocytopenia. In these cases, further testing (multimer gel electrophoresis, VWF:CB, RIPA, VWF:FVIIIB, and genotyping) is required to discriminate the type 2 VWD subtype, but such testing may be available only in specialized laboratories. If type 1C VWD is suspected, the VWFpp/VWF:ag ratio may confirm the diagnosis. Table 2 summarizes the results seen with each subtype. These assays are described in detail below.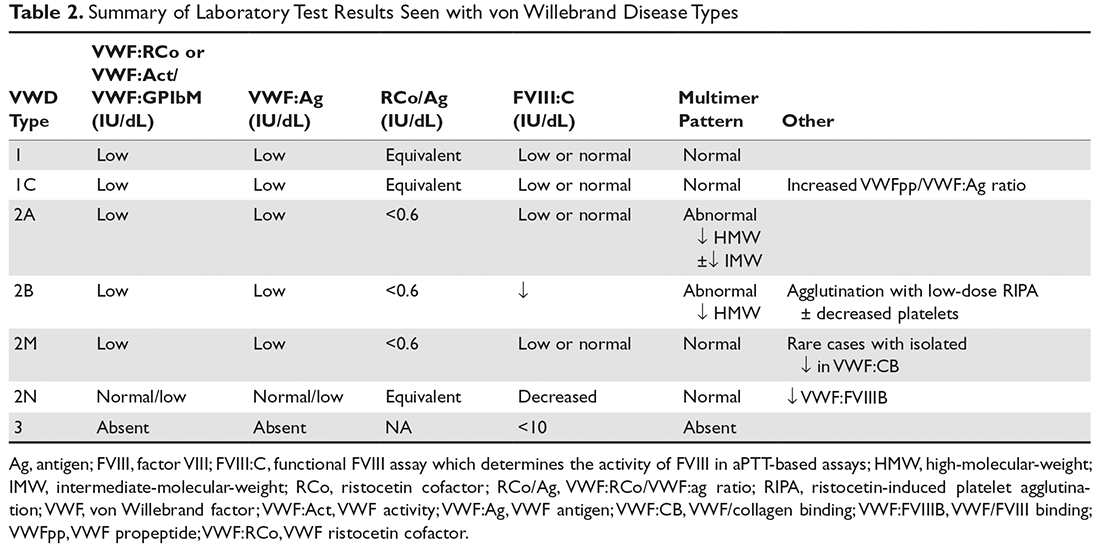
VWD Assays
VWF:Ag represents the quantity of VWF protein (antigen) in the plasma measured using an enzyme-linked immunosorbent assay (ELISA) or latex immunoassay. The normal range is approximately 50 to 200 IU/dL.
VWF:RCo is a functional assay that determines the capacity of VWF to agglutinate platelets via the platelet receptor GPIbα in the presence of ristocetin. The normal range is approximately 50 to 200 IU/dL. Novel methods of measuring VWF’s platelet-binding activity are increasingly being adopted by clinical laboratories and are associated with greater precision and improved lower limits of detection and coefficients of variation.28,29 The first is the VWF:Act, a rapid automated assay that measures VWF activity using an antibody directed to the GPIbα binding site of VWF.28 The second novel assay is VWF:GPIbM, which involves a gain-of-function GPIB construct that binds VWF without ristocetin.30,31 For simplicity, VWF:RCo will be used to refer to VWF platelet-binding activity in the ensuing text. Factor VIII:C is a functional FVIII assay that determines the activity of FVIII in aPTT-based assays. The normal range is approximately 50 to 150 IU/dL.
VWF multimer analysis by SDS-agarose electrophoresis assesses VWF oligomers in plasma.32 Normal plasma contains multimers composed of over 40 VWF dimers, and these multimers are classified as high (HMW), intermediate (IMW), or low molecular weight (LMW). HMW multimers are decreased or missing in types 2A and 2B VWD, and IMW multimers may also be absent in type 2A VWD.
Low-dose RIPA tests the capacity of the patient’s platelets to agglutinate at low concentrations of ristocetin (~0.5 mg/mL). This is in contrast to the VWF:RCo, in which formalin-fixed control platelets are used. With type 2B, the platelet membrane is “overloaded” with high-affinity mutant VWF, resulting in abnormal platelet agglutination at low ristocetin concentrations. In some cases of type 2B VWD, all variables except RIPA may be normal.29
VWF:FVIIIB is an ELISA-based assay that determines the ability of VWF to bind FVIII and is used to make the diagnosis of type 2N VWD.19
VWF:CB is an ELISA-based assay that measures the ability of VWF to bind to collagen, a function of VWF that is dependent on the collagen-binding domain (A3) and on the presence of HMW multimers. VWF:CB helps to distinguish between types 1 and 2 VWD by reflecting the loss of HMW multimer forms (type 2A VWD) or can reflect a specific collagen-binding deficiency (type 2M VWD).33 The normal range is approximately 50 to 200 IU/dL. This assay is not available in most clinical laboratories.
VWFpp/VWF:Ag takes advantage of 2 facts: the VWF propeptide is secreted in a one-to-one ratio to VWF subunits and has a stable half-life in plasma. Thus, an increased ratio identifies patients with mutations that increase VWF clearance, such as type 1C VWD.34 The mean ratio in normal individuals is 1.3, with a normal range of 0.54 to 1.98.
Genotyping should be considered when specialized testing with the VWF:FVIIIB, RIPA, or VWF:CB assays is unavailable and a diagnosis of type 2 VWD is suspected. A guideline on VWD genetic testing has been published by the UK Haemophilia Centre Doctors Organisation.35
Interpretation of Clinical History and Laboratory Investigations
Normal plasma levels of VWF are approximately 100 IU/dL (100%, corresponds to ~10 μg/mL) with a population range of 50 to 200 IU/dL (50%–200%). There are a number of preanalytical variables (patient specific or laboratory specific) that affect the results of VWF laboratory testing. Patient-specific variables that are associated with increased VWF levels include increasing age, African ethnicity, exercise, inflammatory disease states, blood group A or B, increased levels of epinephrine, cocaine use, and neuroendocrine hormone levels. Decreased VWF levels are associated with medications such as valproic acid, hypothyroidism, autoantibodies, and blood group O. Individuals with blood group O have VWF levels that are 25% lower than levels in other blood groups.36 Several analytical variables also can complicate the diagnosis of VWD: methods for established reference ranges, limitations to the sensitivity of assays, and sample handling issues.11 These factors (summarized in Table 3) must be considered when interpreting VWF laboratory results, and at least 2 sets of tests using fresh samples are needed to confirm the diagnosis of VWD. Testing should be avoided in stressed, ill, or pregnant patients.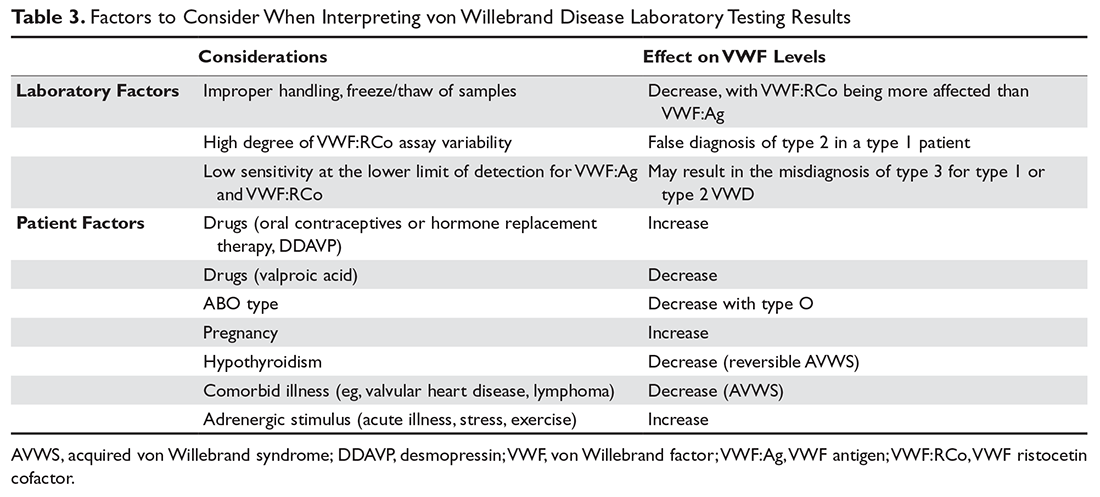
Mild type 1 VWD can be a difficult diagnosis to make because of the overlap of bleeding symptoms among normal individuals and those with mild type 1 VWD, as well as the variability of VWF levels. There is no consensus on the exact VWF levels required to confirm the diagnosis: the NHLBI Expert Panel recommends VWF:Ag and VWF:RCo levels less than 0.30 IU/mL to diagnose type 1 VWD,11 whereas the ISTH-SSC Subcommittee on von Willebrand factor recommends using VWF:RCo and VWF:Ag levels greater than 2 standard deviations below the population mean.37 In the absence of a bleeding history, slightly reduced VWF levels do not predict future significant bleeding events.38 Therefore, regardless of the laboratory cut-off used, the cornerstone of a VWD diagnosis should be a history of excessive mucocutaneous bleeding.
Differential Diagnosis
When considering a diagnosis of VWD, the differential diagnosis must be considered and includes acquired von Willebrand syndrome (AVWS), platelet-type VWD (PT-VWD), and hemophilia A. AVWS is the result of an acquired deficiency or defect of VWF and manifests with a mild to moderate bleeding disorder without a lifelong personal and family history of bleeding. AVWS has diverse pathology. The most common mechanism is proteolytic cleavage of VWF after shear stress–induced unfolding, as seen with aortic stenosis and ventricular assist devices, where as many as 79% of persons with aortic stenosis39 and up to 100% with left ventricular assist devices are affected.40 Other disease mechanisms include autoantibody formation that impairs VWF function or increases its clearance (autoimmune disease or lymphoproliferative disease), adsorption of HMW VWF multimers to malignant cells or platelets (myeloproliferative neoplasms and Wilm’s tumor), or decreased synthesis (hypothyroidism, valproic acid). The median age of diagnosis is 62 years, but the disorder may occur in any age-group (range 2–96 years).41 The approach to management of AVWS should focus on treatment of bleeding and induction of long-term remission. Treatment of bleeding will depend on the underlying mechanism of AVWS and may include a combination of DDAVP or VWF/FVIII concentrates, recombinant factor VIIa, antifibrinolytic agents, intravenous immunoglobulin, or plasmapheresis for AVWS associated with autoantibodies. Treatment of the underlying disorder (eg, aortic valve repair or treatment of a lymphoproliferative disorder) may result in remission of the AVWS.
Mild hemophilia A (caused by mutations in the F8 gene) and type 2N VWD can be difficult to differentiate clinically. Both present with reduced FVIII:C, and type 2N VWD may have normal or borderline low levels of VWF. Although the VWF:FVIIIB assay will distinguish between the 2 disorders, the test is not available in many centers. The pattern of inheritance may be helpful: hemophilia A is an X-linked disorder, whereas type 2N is autosomal recessive. Often, the diagnosis of type 2N VWD is suspected when genotyping of F8 does not identify a mutation in mild hemophilia A, when infused FVIII concentrates have a decreased half-life, or when DDAVP is associated with a brisk but short-lived response. In the absence of VWF:FVIIIB assay availability, genotyping of VWF will confirm the diagnosis, with missense mutations being located in exons 17–20 or 24–27.19
PT-VWD represents the phenocopy of type 2B VWD. The mutation is in the platelet receptor gene GPIBA and causes enhanced VWF-platelet binding. The disorders can be differentiated by RIPA plasma/platelet mixing studies or flow cytometry.42,43 However, these assays are technically challenging. In the absence of mutations in exon 28 of VWF, mutations in exon 2 of GPIBA may be identified in approximately 10% of persons misdiagnosed with type 2B VWD.
Management
Patients with VWD present to medical attention in a number of ways: excessive post-trauma or surgical bleeding, recurrent mucocutaneous bleeding such as epistaxis, menorrhagia, gastrointestinal bleeding, or, in severe cases, recurrent hemarthroses and muscle hematomas. Irrespective of the presentation, the goal is to minimize and control bleeding. Therapeutic options can be divided into 3 main categories: (1) localized measures to stop bleeding; (2) pharmacologic agents with indirect hemostatic benefit; and (3) treatments that directly increase plasma VWF and FVIII levels. A combination of all 3 of these modalities can be used depending on the bleeding location and severity.
Localized Measures
Localized measures to control bleeding in VWD will depend on the site of bleeding. Epistaxis can be particularly problematic for affected children, and patients should be armed with a step-wise action plan that escalates from pressure to packing and includes guidelines regarding how long to wait before seeking medical attention. In selected cases, nasal cautery may be required for prolonged or excessive epistaxis. Topical hemostatic agents such as gelatin foam/matrix, topical thrombin, and fibrin sealants are predominately used to achieve surgical hemostasis and may have a limited role in the treatment of VWD-associated bleeding. In the case of menorrhagia, hormonal treatments (ie, the combined oral contraceptive pill, OCP), levonorgestrel-releasing intrauterine systems, or endometrial ablation all effectively reduce menstrual blood loss through their local effects on the endometrial lining.44 In addition, older generations of OCP are associated with increases in VWF levels. This effect is mediated by the estrogen component and is evident with ethynylestradiol doses of 0.5 μg or higher. Lower estrogen doses, seen in currently used OCP, have little or no effect on VWF levels.11,45
Pharmacologic Therapy
Indirect therapies include the antifibrinolytic agents (eg, tranexamic acid and aminocaproic acid). These agents are used either as the sole therapy at the time of minor surgical and dental procedures, or as an adjunct in combination with DDAVP or VWF/FVIII concentrates. Antifibrinolytics are thought to be particularly useful for controlling mucosal bleeding in areas of high fibrinolytic activity: the oral cavity, gastrointestinal tract, or uterus. Tranexamic acid inhibits the conversion of plasminogen to plasmin, and is the more commonly used antifibrinolytic.11 Tranexamic acid can be administered either intravenously or orally at doses of 10 to 25 mg/kg, respectively. It is usually continued until bleeding is controlled or up to 7 to 10 days postoperatively. The most common adverse events associated with tranexamic acid are headache, back pain, and gastrointestinal side effects.46 Tranexamic acid is contraindicated in disseminated intravascular coagulation and bleeding from the upper urinary tract, where it can lead to urinary tract obstruction by clots.
DDAVP, a synthetic derivative of vasopressin, promotes release of stored VWF from endothelial cells. Most individuals with type 1 VWD and some with type 2A VWD respond to treatment with DDAVP: a therapeutic trial to confirm adequate DDAVP response should be performed prior to its clinical use. Assessment of VWF:Ag, VWF:RCo, and FVIII levels should be performed before and at several time points after the DDAVP administration up to and including 4 hours. Peak VWF levels are achieved 30 and 90 minutes after intravenous and intranasal delivery, respectively. An increase in VWF:Ag/VWF:RCo and FVIII levels to at least 30 IU/dL is adequate for most dental procedures, minor surgery, or the treatment of epistaxis or menorrhagia. DDAVP may be adequate to treat major bleeds or for major surgery when VWF levels increase well above 50 IU/dL. Decisions surrounding the use of DDAVP versus a VWF/FVIII concentrate will depend on the expected DDAVP response, the type of surgery, and the anticipated duration of therapy required to achieve hemostasis. If treatment is required for more than 3 days, concerns regarding tachyphylaxis and side effects may limit its use. Significantly decreased VWF:Ag/VWF:RCo or FVIII at the 4-hour time point of a DDAVP trial may indicate type 1C or type 2N VWD, which are associated with increased clearance of endogenous VWF or FVIII, respectively. Despite the transient response in these patients, DDAVP remains a therapeutic option and its use should be assessed on a case-by-case basis.47
The parenteral dose of DDAVP is 0.3 μg/kg infused in 30 to 50 mL of normal saline over approximately 30 minutes every 12 to 24 hours. The dose of the highly concentrated intranasal preparation is 150 μg for children under 50 kg and 300 μg for larger children and adults (1 spray per naris). It is important to note that the products used to treat VWD (eg, Stimate) deliver 150 μg per spray, a much higher concentration than that used to treat enuresis. Repeated DDAVP dosing is associated with the development of tachyphylaxis: with subsequent dosing, the magnitude of the VWF and FVIII increments can fall to approximately 70% of that obtained with the initial dose.48 DDAVP is safe and generally well tolerated. Side effects include facial flushing, headache, tachycardia, light-headedness, and mild hypotension. The most serious side effects, severe hyponatremia and seizures,49 can be avoided by fluid restriction for 24 hours after DDAVP administration. Serum sodium levels should be monitored with repeated doses. DDAVP is generally avoided in those younger than 2 years of age because of a higher risk of hyponatremia. Patients who are intolerant of DDAVP or have a poor VWF response need to be treated with a VWF/FVIII concentrate.
VWF/FVIII Concentrate
VWF/FVIII concentrates are required for patients who do not have an adequate response to DDAVP, who have side effects from or contraindications to DDAVP, or who require a long duration of treatment, rendering the use of DDAVP impractical. Purified, viral-inactivated, plasma-derived VWF/FVIII concentrates are the products most frequently used (eg, Humate-P, Wilate, Alphanate SD/HT). The quantity of VWF:RCo activity relative to FVIII:C varies by product; Humate-P contains 2.4 VWF:RCo units for each unit of FVIII:C; Wilate contains a 1:1 ratio; and Alphanate contains a 0.5:1 ratio. Both Humate-P and Wilate are reported to contain a full spectrum of VWF multimers, including HMW multimers, and closely resemble normal plasma, but Alphanate SD/HT lacks HMW mutimers.11,50 Thus, the available VWF/FVIII vary in terms of VWF:RCo to FVIII concentrate, HMW multimer composition, reported VWF:RCo, and FVIII half-lives and even approved indications. They should not be considered interchangeable, and further information should be sought from the respective product inserts.
Dosing recommendations are provided either in VWF:RCo (North America) or FVIII:C units (Europe) and are weight-based (Table 4); repeat infusions can be given every 8 to 24 hours depending on the type of surgery/injury and the product used. 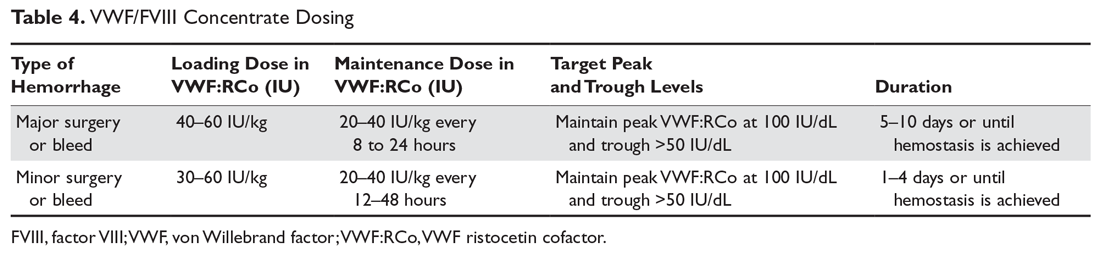
Long-term continuous use of concentrates to prevent bleeds, known as prophylaxis, is the standard of care in severe hemophilia A and B and is now being adopted in severe VWD. Patients with type 3 VWD or severe type 1 or type 2 VWD may experience recurrent bleeds into joints, nasal/oropharynx, or gastrointestinal tract or excessive menstrual bleeding. Retrospective cohort and case series suggest that prophylaxis improves quality of life; reduces the frequency of bleeding, need for transfusions, and hospitalizations; and prevents chronic joint disease.54,55 More recently, a prospective study confirmed that prophylaxis with VWF concentrates at doses ranging from 50 IU VWF RCo/kg 1 to 3 times per week was highly effective at reducing bleeding rates, with annualized bleeding rates decreasing from 25 to 6.1 in 11 participants with either type 2A or type 3 VWD.56
VWF/FVIII concentrates are effective in more than 97% of events.57 Rarely, when infusion of a VWF/FVIII concentrate is ineffective at stopping bleeding, transfusion of platelet concentrates may be beneficial, presumably because they facilitate the delivery of small amounts of platelet VWF to the site of vascular injury. Highly purified FVIII concentrates (monoclonal antibody purified and recombinant) should not be used to treat VWD because they lack VWF.
A recombinant VWF concentrate (Vonvendi) combined initially with recombinant FVIII concentrate in a 1.3:1 ratio of VWF:RCo to FVIII:C has been shown to be safe and efficacious for the on-demand treatment of bleeds.58,59 After the initial FVIII dose, the patients’ endogenous FVIII levels are stabilized within 6 hours and further FVIII administration may not required. A prospective phase 3 trial investigating the efficacy of recombinant VWF in the prophylaxis of severe VWD is ongoing. Vonvendi has been licensed for on-demand treatment in the United States since 2015. For further dosing information, please refer to the product insert.
Conclusion
VWF is a complex protein with several important and distinct functional domains: binding sites to collagen, FVIII, and platelet GPIbα; an ADAMTS13 cleavage site; and domains important for multimer formation. Mutations in any of these sites can result in a dysfunctional protein and as a result, VWD is a heterogeneous disorder with many specific assays available to determine the subtype. Despite this, the treatment of VWD is straightforward with only a small number of therapeutic options: indirect therapies such as antifibrinolytic agents, or direct therapies that increase VWF levels, DDAVP, or VWF/FVIII concentrates. Management focuses on preventing bleeding complications associated with invasive procedures or promptly treating bleeding episodes.
1. Sadler JE, Budde U, Eikenboom JCJ, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemost 2006;4:2103–14.
2. Rodeghiero F, Castaman G. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood 1987;69:454–9.
3. Werner EJ, Broxson EH, Tucker EL, et al. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr 1993;123:893–8.
4. Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost 2000;84:160–74.
5. Bowman M, Hopman WM, Rapson D, et al. The prevalence of symptomatic von Willebrand disease in primary care practice. J Thromb Haemost 2010;8:213–6.
6. Mancuso DJ, Tuley EA, Westfield LA, et al. Structure of the gene for human von Willebrand factor. J Biol Chem 1989;264:19514–27.
7. Kang I, Raghavachari M, Hofmann CM, Marchant RE. Surface-dependent expression in the platelet GPIb binding domain within human von Willebrand factor studied by atomic force microscopy. Thromb Res 2007;119:731–40.
8. Savage B, Saldívar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 1996;84:289–97.
9. Dong J, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002;100:4033–9.
10. Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev 2010;24:123–34.
11. Nichols WL, Hultin MB, James AH, et al. Von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) expert panel report (USA). Haemophilia 2008;14:171–232.
12. Haberichter SL, Castaman G, Budde U, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 vwd (MCMDM-1VWD). Blood 2008;111:4979–85.
13. Goodeve A. Vicenza deciphered: modeling the von Willebrand disease enigma: commentary on accelerated clearance alone explains ultralarge multimers in VWD Vicenza. J Thromb Haemost 2010;8:1271–2.
14. Federici AB, Mannucci PM, Castaman G, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood 2009;113:526–34.
15. Nurden AT, Federici AB, Nurden P. Altered megakaryocytopoiesis in von Willebrand type 2B disease. J Thromb Haemost 2009;7 Suppl 1:277–81.
16. Ruggeri ZM, Pareti FI, Mannucci PM, et al. Heightened interaction between platelets and factor VIII/von Willebrand factor in a new subtype of von Willebrand’s disease. New Engl J Med 1980;302:1047–51.
17. James PD, Notley C, Hegadorn C, et al. Challenges in defining type 2M von Willebrand disease: results from a Canadian cohort study. J Thromb Haemost 2007;5:1914–22.
18. Flood VH, Lederman CA, Wren JS, et al. Absent collagen binding in a VWF A3 domain mutant: utility of the VWF:CB in diagnosis of VWD. J Thromb Haemost 2010;8:1431–3.
19. Mazurier C, Hilbert L. Type 2N von Willebrand disease. Curr Hematol Rep 2005;4:350–8.
20. Nesbitt IM, Goodeve AC, Guilliatt AM, et al. Characterisation of type 2N von Willebrand disease using phenotypic and molecular techniques. Thromb Haemost 1996;75:959–64.
21. Bowman M, Tuttle A, Notley C, et al. The genetics of Canadian type 3 von Willebrand disease: further evidence for co-dominant inheritance of mutant alleles. J Thromb Haemost 2013;11:512–20.
22. James PD, Lillicrap D, Mannucci PM. Alloantibodies in von Willebrand disease. Blood 2013;122:636–40.
23. James AH, Jamison MG. Bleeding events and other complications during pregnancy and childbirth in women with von Willebrand disease. J Thromb Haemost 2007;5:1165–9.
24. Rydz N, James PD. The evolution and value of bleeding assessment tools. J Thromb Haemost 2012;2223–9.
25. Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost 2010;8:2063–5.
26. Elbatarny M, Mollah S, Grabell J, et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia 2014;20:831–5.
27. Deforest M, Grabell J, Alberta S et al. Generation and optimization of the self-administered bleeding assessment tool and its validation as a screening test for von Willebrand disease. Haemophilia 2015;21:e384-8.
28. Castaman G, Hillarp A, Goodeve A. Laboratory aspects of von Willebrand disease: test repertoire and options for activity assays and genetic analysis. Haemophilia 2014;20(Suppl. 4):65–70.
29. Favaloro EJ. Von Willebrand disease, type 2B: a diagnosis more elusive than previously thought. Thromb Haemost 2008;99:630–1.
30. Budde U. Diagnosis of von Willebrand disease subtypes: implications for treatment. Haemophilia 2008;14 Suppl 5:27–38.
31. Favaloro EJ. Von Willebrand factor collagen-binding (activity) assay in the diagnosis of von Willebrand disease: a 15-year journey. Sem Thromb Hemost 2002;28:191–202.
32. Patzke J, Budde U, Huber A, et al. Performance evaluation and multicenter study of a von Willebrand factor activity assay based on GPIb binding in the absence of ristocetin. Blood Coagul Fibrinolysis 2014;25:860-70.
33. Graf L, Moffat KA, Carlino SA, et al. Evaluation of an automated method for measuring von Willebrand factor activity in clinical samples without ristocetin. Int J Lab Hematol 2014;36:341–51.
34. Haberichter SL, Balistreri M, Christopherson P, et al. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood 2006;108:3344–51.
35. Keeney S, Bowen D, Cumming A, et al. The molecular analysis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors’ Organisation Haemophilia genetics laboratory network. Haemophilia 2008;14:1099–111.
36. Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ, Montgomery RR. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood 1987;69:1691–5.
37. Sadler JE, Rodeghiero F. Provisional criteria for the diagnosis of VWD type 1. J Thromb Haemost 2005;3:775–7.
38. Tosetto A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM- 1VWD). J Thromb Haemost 2006;4:766–73.
39. Vincentelli A, Susen S, Le Tourneau T, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med 2003;349:343–9.
40. Uriel N, Pak S-W, Jorde UP, et al. Acquired von Willebrand syndrome after continuous-flow mechanical device support contributes to a high prevalence of bleeding during long-term support and at the time of transplantation. J Am Coll Cardiol 2010;56:1207–13.
41. Federici AB, Rand JH, Bucciarelli P, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost 2000;84:345–9.
42. Favaloro EJ, Patterson D, Denholm A, et al. Differential identification of a rare form of platelet-type (pseudo-) von Willebrand disease (VWD) from type 2B VWD using a simplified ristocetin-induced-platelet-agglutination mixing assay and confirmed by genetic analysis. Brit J Haematol 2007;139:621–8.
43. Giannini S, Cecchetti L, Mezzasoma AM, Gresele P. Diagnosis of platelet-type von Willebrand disease by flow cytometry. Haematologica 2010;95:1021–4.
44. Farquhar C, Brown J. Oral contraceptive pill for heavy menstrual bleeding. Cochrane Database Syst Rev 2009 Oct 7;(4):CD000154.
45. Kadir R, Economides DL, Sabin C, et al. Variations in coagulation factors in women: effects of age, ethnicity, menstrual cycle and combined oral contraceptive. Thromb Haemost 1999;82:1456–61.
46. Muse K, Lukes AS, Gersten J, et al. Long-term evaluation of safety and health-related quality of life in women with heavy menstrual bleeding treated with oral tranexamic acid. Womens Health 2011;7:699–707.
47. Castaman G, Tosetto A, Federici AB, Rodeghiero F. Bleeding tendency and efficacy of anti-haemorrhagic treatments in patients with type 1 von Willebrand disease and increased von Willebrand factor clearance. Thromb Haemost 2011;105:647–54.
48. Mannucci PM, Bettega D, Cattaneo M. Patterns of development of tachyphylaxis in patients with haemophilia and von Willebrand disease after repeated doses of desmopressin (DDAVP). Brit J Haematol 1992;82:87–93.
49. Greaves M, Watson HG. Approach to the diagnosis and management of mild bleeding disorders. J Thromb Haemost 2007;5 Suppl 1:167–74.
50. Kessler CM, Friedman K, Schwartz BA, Gill JC, Powell JS. The pharmacokinetic diversity of two von Willebrand factor (VWF) / factor VIII (FVIII) concentrates in subjects with congenital von Willebrand disease. results from a prospective, randomised crossover study. Thromb Haemost 2011;106:279–88.
51. Weiss HJ, Sussman II, Hoyer LW. Stabilization of factor VIII in plasma by the von Willebrand factor. Studies on posttransfusion and dissociated factor VIII and in patients with von Willebrand’s disease. J Clin Invest 1977;60:390–404.
52. Berntorp E. Haemate P/Humate-P: a systematic review. Thromb Res 2009;124:S11–14.
53. Lillicrap D, Poon MC, Walker I, et al. Efficacy and safety of the factor VIII/von Willebrand factor concentrate, Haemate-P/Humate-P: ristocetin cofactor unit dosing in patients with von Willebrand disease. Thromb Haemost 2002;87:224–30.
54. Halimeh S, Krümpel A, Rott H, et al. Long-term secondary prophylaxis in children, adolescents and young adults with von Willebrand disease. results of a cohort study. Thromb Haemost 2011;105:597–604.
55. Abshire TC, Federici AB, Alvárez MT, et al. Prophylaxis in severe forms of von Willebrand’s disease: results from the von Willebrand disease prophylaxis network (VWD PN). Haemophilia 2013;19:76–81.
56. Abshire T, Cox-Gill J, Kempton CL, et al. Prophylaxis escalation in severe von Willebrand disease: a prospective study from the von Willebrand Disease Prophylaxis Network. J Thromb Haemost 2015;13:1585– 9.
57. Auerswald G, Kreuz W. Haemate P/Humate-P for the treatment of von Willebrand disease: considerations for use and clinical experience. Sem Thromb Hemost 2008;14 (Suppl 5):39–46.
58. Mannucci PM, Kempton C, Millar C, et al. Pharmacokinetics and safety of a novel recombinant human von Willebrand factor manufactured with a plasma-free method: a prospective clinical trial. Blood 2013;122:648–57.
59. Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood 2015;126:2038–46.
Introduction
von Willebrand disease (VWD) is an inherited bleeding disorder caused by deficient or defective plasma von Willebrand factor (VWF). VWF is an adhesive multimeric plasma glycoprotein that performs 2 major functions in hemostasis: it mediates platelet adhesion to injured subendothelium via glycoprotein 1bα (GPIbα), and it binds and stabilizes factor VIII (FVIII) in circulation, protecting it from proteolytic degradation by enzymes. The current VWD classification recognizes 3 types (Table 1).1
Prevalence
VWD is the most common inherited bleeding disorder. However, because VWF levels are highly variable and disease severity ranges from mild bleeding symptoms to severe or life-threatening bleeds, the reported prevalence of VWD depends on the diagnostic definition used. Two large epidemiologic studies have reported prevalence rates of approximately 1%.2,3 In these studies, healthy school-aged children were screened and diagnosed with VWD based on low VWF activity, measured as ristocetin cofactor, and a personal and family history of bleeding symptoms. At the other extreme, when considering patients whose bleeding symptoms are sufficiently severe to warrant referral to specialized centers, the reported prevalence of VWD ranges from 20 to 113 per million.4 These studies likely over- and underestimate clinically significant VWD. More recent studies suggest that the prevalence of VWD in individuals whose bleeding symptoms are significant enough to present to a primary care physician is approximately 0.1%.5 This figure is likely a more accurate estimate of the true prevalence of symptomatic VWD.
Although VWD is autosomally inherited, females are more likely to present with bleeding symptoms and be diagnosed because of increased exposure to bleeding challenges, such as menorrhagia and childbirth. VWD does not show any geographic or ethnic predilection, but there is an increased prevalence of the recessive forms, such as type 2N and type 3 VWD, in areas with high rates of consanguinity.
VWF Protein Structure and Function
The VWF gene is located on chromosome 12 at p13.3 and spans 178 kb comprising 52 exons.6 The expression of the VWF gene is tightly restricted to endothelial cells, platelets, and megakaryocytes, where VWF is stored in Weibel-Palade bodies and α-granules. VWF is a large multimeric glycoprotein with several important functional domains (Figure).
Classification, Pathophysiology, and Genetics
The International Society of Thrombosis and Hemostasis (ISTH) classification of VWD was updated in 2006 (Table 1).1 It incorporates important aspects of clinical phenotype, pathophysiological mechanisms, and treatment considerations. The 3 categories are: type 1, which is a partial quantitative deficiency; type 2 with 4 subtypes (2A, 2B, 2M, and 2N), which is a qualitative defect; and type 3, which is a virtual absence of VWF. Although the diagnosis and categorization of VWD can be achieved with widely available laboratory testing, further subcategorization among type 2 VWD subtypes may require referral to a specialized laboratory. The current ISTH classification intentionally does not incorporate genotypic data. In type 2 or type 3 VWD disease, VWF mutations are identified in more than 90% of cases and are completely penetrant, whereas mutations are identified in only approximately 65% of type 1 VWD cases and have been associated with incomplete penetrance and variable expressivity.10 These studies suggest that type 1 VWD is an oligogenic disease with mutations in genes regulating secretion or clearance contributing to a VWD phenotype.
VWD Types
Type 1
Type 1 VWD is caused by a partial quantitative deficiency of VWF and represents approximately 75% of VWD cases. It is the most clinically heterogeneous type, with patients having a mild to moderate bleeding phenotype.11 Bleeding in type 1 VWD results from a decrease in the concentration of VWF. The VWF function is normal without a significant abnormality in the platelet, collagen, or FVIII binding sites or a significant decrease in HMW multimers. Functional assays of VWF, such as VWF ristocetin cofactor (VWF:RCo) or VWF activity (VWF:Act) (see section on Laboratory Testing for further details), are proportionally decreased relative to the VWF antigen level (VWF:Ag), and the ratio of functional activity as compared with the VWF level is normal (ie, VWF:RCo/VWF:Ag ratio is > 0.6). As noted, VWF mutations are identified in only 65% of type 1 VWD cases and have incomplete penetrance and variable expressivity.10 Approximately 70% of mutations identified are missense mutations. Missense mutations may affect VWF levels by affecting any part of the biosynthetic pathway, including trafficking, storage, secretion, and/or clearance of VWF.
Increased VWF clearance is a well-described mechanism for type 1 VWD, known as type 1C. These patients will typically have very low VWF levels, an increased VWF propeptide to antigen ratio (VWFpp/VWF:Ag), and a marked but short-lived response to DDAVP, limiting DDAVP’s clinical applicability.12 On the other hand, the half-life of VWF/FVIII concentrates is normal in these individuals. Type 1C VWD is caused by missense mutations which occur mainly in the D3 domain and reduce the half-life of VWF up to 15-fold. R1205H, known as the “Vicenza” variant, is the most common and severe as well as the best characterized of these mutations.13
Type 2
Accounting for approximately 25% of VWD cases, type 2 VWD is characterized by a qualitative deficiency of VWF activity and is further subcategorized based on the mechanism of VWF dysfunction. Type 2A, 2B, and 2M affect VWF–platelet interactions by way of loss of HMW multimers, a gain of function of the GPIbα binding site, or a loss of function of the same site, respectively. On the other hand, type 2N is caused by defective VWF binding to FVIII. Type 2 VWD is often suspected when investigations demonstrate a function-antigen discordance: the VWF:RCo or VWF:Act is decreased disproportionately to the decrease in VWF:Ag, and the VWF:RCo/VWF:Ag ratio is less than 0.6.
Type 2A VWD is the most common type 2 variant. It is characterized by disproportionately low functional activity compared to antigen level (ie, VWF:RCo/VWF:ag ratio is < 0.6) and a loss of HMW and sometimes intermediate molecular weight (IMW) multimers. Ristocetin-induced platelet agglutination (RIPA) will be decreased with standard doses of ristocetin and absent with low doses. Type 2A VWD is usually inherited as an autosomal dominant trait. This subtype encompasses missense mutations that impair dimerization or multimerization of VWF subunits (CK, D1, and D2 domains); disrupt intersubunit disulphide bonds (D3 and D2 domains); enhance susceptibility to ADAMTS13-mediated proteolysis (A2 and A1 domains); or result in intracellular retention of the HMW multimers (D3, A1, and A2 domains).10 The result is VWF that lacks HMW multimers, thereby possessing fewer GPIbα binding sites, and that is less effective in binding platelets.
Type 2B VWD is the result of gain-of-function mutations within the GPIbα binding site of VWF. Generally, the platelet-binding site of VWF within the A1 domain is only exposed once VWF is immobilized on injured collagen and subjected to shear forces, resulting in a conformational change.7 In type 2B VWD, the gain-of-function mutation results in spontaneous binding of VWF to platelets without the need for a VWF-collagen interaction and unfolding of VWF by shear forces. The VWF–platelet interaction selectively depletes the HMW multimers by the unfolding of the A2 domain and increasing ADAMTS13 proteolysis. The increased binding of mutant VWF to platelets also triggers the formation of platelet aggregates, which are removed from circulation resulting in thrombocytopenia. Increases in endogenous VWF seen with acute stressors or pregnancy can worsen thrombocytopenia and increase the risk of bleeding.14 Certain mutations, such as V1316M, alter megakaryocytopoiesis and are characterized by giant platelets with abnormal ultrastructure and further exacerbate the thrombocytopenia.15 The laboratory profile reveals a VWF:RCo/VWF:Ag ratio of < 0.6 and absence of HMW multimers. In contrast to type 2A, platelets will agglutinate with low-dose ristocetin. Missense mutations are highly penetrant dominant and occur in or close to the A1 domain.16
Type 2M VWD is characterized by loss-of-function mutations within the GPIbα binding site of VWF. Phenotypic characteristics include a reduced ratio of VWF:RCo/VWF:Ag of < 0.6 but a normal multimer pattern.17 Missense mutations are reported in the A1 domain affecting the GPIbα-binding site. In very rare instances, mutations in the A3 domain that impair the VWF/collagen interaction have been described.18 These collagen-binding mutations are not included in the last iteration of the ISTH classification in 2006,1 but fit best in the type 2M category. In these cases, VWF:RCo or VWF:Act, which reflect activity at the GPIbα-binding site, may be normal and the diagnosis requires VWF/collagen binding assays (VWF:CB).
Type 2N VWD results from mutations of the FVIII binding site or conformational changes that impair the VWF–FVIII interaction. Most (~80%) missense mutations are located in domains D’ and D3.19 These mutations are autosomal recessive, and affected individuals are either homozygous or compound heterozygous for type 2N/2N or type 1/2N mutations, or compound heterozygous for a missense mutation and a mutation resulting in a null allele (type 2N/3 mutations). The laboratory phenotype is a disproportionate reduction in the FVIII level relative to the VWF level, which may be low or normal. Most cases of type 2N VWD have a normal multimeric profile, but rare cases will demonstrate loss of HMW multimers. Definitive diagnosis requires evidence of reduced FVIII binding to VWF (VWF:FVIIIB) or the identification of causative mutations in the FVIII binding region of the VWF gene.20
Type 3
Type 3 VWD is defined by a virtual absence of VWF. The inheritance of type 3 VWD has often been reported as autosomal recessive. However, there is emerging evidence that it can also be inherited in a co-dominant pattern: obligate carriers of type 3 VWD mutations have more mucocutaneous bleeding symptoms than normal individuals, and in approximately 50% of cases may carry a diagnosis of type 1 VWD.21 This condition is characterized by prolongation of the activated partial thromboplastin time (aPTT), undetectable levels of VWF:Ag, and VWF:RCo and FVIII levels less than 10 IU/dL (10%). The majority (~80%) of type 3 VWD patients have 2 null alleles as a result of a variety of mutations, with nonsense mutations accounting for about one-third.10 The remainder of the mutational spectrum is made up of missense mutations predominantly located in the D1-D2 (exons 3–11) and D4-CK (exons 37–52) domains that result in intracellular VWF retention, or large deletions, resulting in frameshift mutations affecting 1 or more exons. Because there is little or no circulating VWF, patients with type 3 VWD may develop alloantibodies to VWF, which can complicate treatment.22
Diagnosis
Clinical Manifestations
VWD is a congenital bleeding disorder. The increased risk of bleeding is present from birth, but symptoms may only manifest when there is a hemostatic challenge. Bleeding symptoms become more apparent with increasing age and exposure to hemostatic challenges. As a result, the diagnosis is often delayed into adulthood in mild to moderate forms of VWD. On the other hand, with more severe bleeding phenotypes such as type 3 VWD, the diagnosis is often made in childhood. Individuals with VWD primarily complain of excessive mucocutaneous bleeding, which includes spontaneous bruising, recurrent epistaxis, and bleeding from the gums after brushing, dental cleaning, and extractions. In addition, prolonged or excessive bleeding after surgery or trauma is often reported. Females frequently experience menorrhagia, usually beginning at menarche, and can have prolonged or excessive bleeding after childbirth.23 Musculoskeletal bleeding is unusual, except in type 2N or type 3 VWD when the FVIII:C level may be less than 10 IU/dL.
Mucocutaneous bleeding symptoms such as epistaxis, gum bleeding, ecchymosis, and menorrhagia overlap with those experienced by a normal population, and therefore can be easily overlooked by both patients and physicians.11 The use of bleeding assessment tools (BATs) to standardize the bleeding history and interpretation of the severity of the bleeding phenotype is becoming part of routine clinical practice. Three different BATs, each an adaptation of its predecessor, have been created and validated.24 Each of the scores performs well in an undiagnosed population presenting with bleeding symptoms. The negative predictive value is typically greater than 0.99, meaning that a negative bleeding score nearly excludes a clinically significant bleeding disorder. Thus, the main utility of the current BATs is at the time of new patient assessments: a negative bleeding score will help avoid unnecessary laboratory testing and prevent false-positive diagnoses of VWD (borderline low VWF:Ag without a significant bleeding history). However, the currently available BATs have some limitations. When scoring severe bleeding disorders, BATs become saturated as they take into account the worst episode of bleeding within each category but not the frequency of bleeding. BATs need to be administered by an expert and are time consuming to complete. Finally, they are not useful for monitoring bleeding symptoms or response to therapy because of the cumulative nature of the scores. In an attempt to standardize the BAT and bleeding score, the ISTH/Scientific and Standardization Committee (SSC) Joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group has established a revised BAT, known as the ISTH-BAT, specifically designed to extend the utility of the earlier BATS by incorporating information on both symptom frequency and severity.25,26 The ISTH-BAT has been further modified to a patient- or self-administered BAT (SELF-BAT). The SELF-BAT has been shown to be a reliable and effective tool in the assessment of patients who are being evaluated for VWD.27
Laboratory Testing
Screening tests include a complete blood count (CBC), prothrombin time, aPTT, thrombin time, and fibrinogen concentration to exclude the presence of other hemostatic disorders. The CBC may show thrombocytopenia in type 2B VWD. The aPTT is often normal, but will be prolonged if the FVIII level is below 30 IU/dL, as can be seen in severe type 1, type 2N, or type 3 VWD. The platelet function analyzer (PFA-100) is a system for analyzing primary hemostasis under high shear rates, but its role in the diagnosis of VWD is controversial.11
The evaluation of VWD involves quantitative (VWF:Ag) and qualitative measurements of VWF (VWF:RCo, or one of the novel assays: VWF:Act or VWF:GPIbM) and FVIII activity (FVIII:C). Type 2 VWD is suspected when the VWF activity to VWF:Ag ratio is < 0.6, the FVIII:C is more significantly decreased as compared to VWF:Ag, or with the presence of thrombocytopenia. In these cases, further testing (multimer gel electrophoresis, VWF:CB, RIPA, VWF:FVIIIB, and genotyping) is required to discriminate the type 2 VWD subtype, but such testing may be available only in specialized laboratories. If type 1C VWD is suspected, the VWFpp/VWF:ag ratio may confirm the diagnosis. Table 2 summarizes the results seen with each subtype. These assays are described in detail below.
VWD Assays
VWF:Ag represents the quantity of VWF protein (antigen) in the plasma measured using an enzyme-linked immunosorbent assay (ELISA) or latex immunoassay. The normal range is approximately 50 to 200 IU/dL.
VWF:RCo is a functional assay that determines the capacity of VWF to agglutinate platelets via the platelet receptor GPIbα in the presence of ristocetin. The normal range is approximately 50 to 200 IU/dL. Novel methods of measuring VWF’s platelet-binding activity are increasingly being adopted by clinical laboratories and are associated with greater precision and improved lower limits of detection and coefficients of variation.28,29 The first is the VWF:Act, a rapid automated assay that measures VWF activity using an antibody directed to the GPIbα binding site of VWF.28 The second novel assay is VWF:GPIbM, which involves a gain-of-function GPIB construct that binds VWF without ristocetin.30,31 For simplicity, VWF:RCo will be used to refer to VWF platelet-binding activity in the ensuing text. Factor VIII:C is a functional FVIII assay that determines the activity of FVIII in aPTT-based assays. The normal range is approximately 50 to 150 IU/dL.
VWF multimer analysis by SDS-agarose electrophoresis assesses VWF oligomers in plasma.32 Normal plasma contains multimers composed of over 40 VWF dimers, and these multimers are classified as high (HMW), intermediate (IMW), or low molecular weight (LMW). HMW multimers are decreased or missing in types 2A and 2B VWD, and IMW multimers may also be absent in type 2A VWD.
Low-dose RIPA tests the capacity of the patient’s platelets to agglutinate at low concentrations of ristocetin (~0.5 mg/mL). This is in contrast to the VWF:RCo, in which formalin-fixed control platelets are used. With type 2B, the platelet membrane is “overloaded” with high-affinity mutant VWF, resulting in abnormal platelet agglutination at low ristocetin concentrations. In some cases of type 2B VWD, all variables except RIPA may be normal.29
VWF:FVIIIB is an ELISA-based assay that determines the ability of VWF to bind FVIII and is used to make the diagnosis of type 2N VWD.19
VWF:CB is an ELISA-based assay that measures the ability of VWF to bind to collagen, a function of VWF that is dependent on the collagen-binding domain (A3) and on the presence of HMW multimers. VWF:CB helps to distinguish between types 1 and 2 VWD by reflecting the loss of HMW multimer forms (type 2A VWD) or can reflect a specific collagen-binding deficiency (type 2M VWD).33 The normal range is approximately 50 to 200 IU/dL. This assay is not available in most clinical laboratories.
VWFpp/VWF:Ag takes advantage of 2 facts: the VWF propeptide is secreted in a one-to-one ratio to VWF subunits and has a stable half-life in plasma. Thus, an increased ratio identifies patients with mutations that increase VWF clearance, such as type 1C VWD.34 The mean ratio in normal individuals is 1.3, with a normal range of 0.54 to 1.98.
Genotyping should be considered when specialized testing with the VWF:FVIIIB, RIPA, or VWF:CB assays is unavailable and a diagnosis of type 2 VWD is suspected. A guideline on VWD genetic testing has been published by the UK Haemophilia Centre Doctors Organisation.35
Interpretation of Clinical History and Laboratory Investigations
Normal plasma levels of VWF are approximately 100 IU/dL (100%, corresponds to ~10 μg/mL) with a population range of 50 to 200 IU/dL (50%–200%). There are a number of preanalytical variables (patient specific or laboratory specific) that affect the results of VWF laboratory testing. Patient-specific variables that are associated with increased VWF levels include increasing age, African ethnicity, exercise, inflammatory disease states, blood group A or B, increased levels of epinephrine, cocaine use, and neuroendocrine hormone levels. Decreased VWF levels are associated with medications such as valproic acid, hypothyroidism, autoantibodies, and blood group O. Individuals with blood group O have VWF levels that are 25% lower than levels in other blood groups.36 Several analytical variables also can complicate the diagnosis of VWD: methods for established reference ranges, limitations to the sensitivity of assays, and sample handling issues.11 These factors (summarized in Table 3) must be considered when interpreting VWF laboratory results, and at least 2 sets of tests using fresh samples are needed to confirm the diagnosis of VWD. Testing should be avoided in stressed, ill, or pregnant patients.
Mild type 1 VWD can be a difficult diagnosis to make because of the overlap of bleeding symptoms among normal individuals and those with mild type 1 VWD, as well as the variability of VWF levels. There is no consensus on the exact VWF levels required to confirm the diagnosis: the NHLBI Expert Panel recommends VWF:Ag and VWF:RCo levels less than 0.30 IU/mL to diagnose type 1 VWD,11 whereas the ISTH-SSC Subcommittee on von Willebrand factor recommends using VWF:RCo and VWF:Ag levels greater than 2 standard deviations below the population mean.37 In the absence of a bleeding history, slightly reduced VWF levels do not predict future significant bleeding events.38 Therefore, regardless of the laboratory cut-off used, the cornerstone of a VWD diagnosis should be a history of excessive mucocutaneous bleeding.
Differential Diagnosis
When considering a diagnosis of VWD, the differential diagnosis must be considered and includes acquired von Willebrand syndrome (AVWS), platelet-type VWD (PT-VWD), and hemophilia A. AVWS is the result of an acquired deficiency or defect of VWF and manifests with a mild to moderate bleeding disorder without a lifelong personal and family history of bleeding. AVWS has diverse pathology. The most common mechanism is proteolytic cleavage of VWF after shear stress–induced unfolding, as seen with aortic stenosis and ventricular assist devices, where as many as 79% of persons with aortic stenosis39 and up to 100% with left ventricular assist devices are affected.40 Other disease mechanisms include autoantibody formation that impairs VWF function or increases its clearance (autoimmune disease or lymphoproliferative disease), adsorption of HMW VWF multimers to malignant cells or platelets (myeloproliferative neoplasms and Wilm’s tumor), or decreased synthesis (hypothyroidism, valproic acid). The median age of diagnosis is 62 years, but the disorder may occur in any age-group (range 2–96 years).41 The approach to management of AVWS should focus on treatment of bleeding and induction of long-term remission. Treatment of bleeding will depend on the underlying mechanism of AVWS and may include a combination of DDAVP or VWF/FVIII concentrates, recombinant factor VIIa, antifibrinolytic agents, intravenous immunoglobulin, or plasmapheresis for AVWS associated with autoantibodies. Treatment of the underlying disorder (eg, aortic valve repair or treatment of a lymphoproliferative disorder) may result in remission of the AVWS.
Mild hemophilia A (caused by mutations in the F8 gene) and type 2N VWD can be difficult to differentiate clinically. Both present with reduced FVIII:C, and type 2N VWD may have normal or borderline low levels of VWF. Although the VWF:FVIIIB assay will distinguish between the 2 disorders, the test is not available in many centers. The pattern of inheritance may be helpful: hemophilia A is an X-linked disorder, whereas type 2N is autosomal recessive. Often, the diagnosis of type 2N VWD is suspected when genotyping of F8 does not identify a mutation in mild hemophilia A, when infused FVIII concentrates have a decreased half-life, or when DDAVP is associated with a brisk but short-lived response. In the absence of VWF:FVIIIB assay availability, genotyping of VWF will confirm the diagnosis, with missense mutations being located in exons 17–20 or 24–27.19
PT-VWD represents the phenocopy of type 2B VWD. The mutation is in the platelet receptor gene GPIBA and causes enhanced VWF-platelet binding. The disorders can be differentiated by RIPA plasma/platelet mixing studies or flow cytometry.42,43 However, these assays are technically challenging. In the absence of mutations in exon 28 of VWF, mutations in exon 2 of GPIBA may be identified in approximately 10% of persons misdiagnosed with type 2B VWD.
Management
Patients with VWD present to medical attention in a number of ways: excessive post-trauma or surgical bleeding, recurrent mucocutaneous bleeding such as epistaxis, menorrhagia, gastrointestinal bleeding, or, in severe cases, recurrent hemarthroses and muscle hematomas. Irrespective of the presentation, the goal is to minimize and control bleeding. Therapeutic options can be divided into 3 main categories: (1) localized measures to stop bleeding; (2) pharmacologic agents with indirect hemostatic benefit; and (3) treatments that directly increase plasma VWF and FVIII levels. A combination of all 3 of these modalities can be used depending on the bleeding location and severity.
Localized Measures
Localized measures to control bleeding in VWD will depend on the site of bleeding. Epistaxis can be particularly problematic for affected children, and patients should be armed with a step-wise action plan that escalates from pressure to packing and includes guidelines regarding how long to wait before seeking medical attention. In selected cases, nasal cautery may be required for prolonged or excessive epistaxis. Topical hemostatic agents such as gelatin foam/matrix, topical thrombin, and fibrin sealants are predominately used to achieve surgical hemostasis and may have a limited role in the treatment of VWD-associated bleeding. In the case of menorrhagia, hormonal treatments (ie, the combined oral contraceptive pill, OCP), levonorgestrel-releasing intrauterine systems, or endometrial ablation all effectively reduce menstrual blood loss through their local effects on the endometrial lining.44 In addition, older generations of OCP are associated with increases in VWF levels. This effect is mediated by the estrogen component and is evident with ethynylestradiol doses of 0.5 μg or higher. Lower estrogen doses, seen in currently used OCP, have little or no effect on VWF levels.11,45
Pharmacologic Therapy
Indirect therapies include the antifibrinolytic agents (eg, tranexamic acid and aminocaproic acid). These agents are used either as the sole therapy at the time of minor surgical and dental procedures, or as an adjunct in combination with DDAVP or VWF/FVIII concentrates. Antifibrinolytics are thought to be particularly useful for controlling mucosal bleeding in areas of high fibrinolytic activity: the oral cavity, gastrointestinal tract, or uterus. Tranexamic acid inhibits the conversion of plasminogen to plasmin, and is the more commonly used antifibrinolytic.11 Tranexamic acid can be administered either intravenously or orally at doses of 10 to 25 mg/kg, respectively. It is usually continued until bleeding is controlled or up to 7 to 10 days postoperatively. The most common adverse events associated with tranexamic acid are headache, back pain, and gastrointestinal side effects.46 Tranexamic acid is contraindicated in disseminated intravascular coagulation and bleeding from the upper urinary tract, where it can lead to urinary tract obstruction by clots.
DDAVP, a synthetic derivative of vasopressin, promotes release of stored VWF from endothelial cells. Most individuals with type 1 VWD and some with type 2A VWD respond to treatment with DDAVP: a therapeutic trial to confirm adequate DDAVP response should be performed prior to its clinical use. Assessment of VWF:Ag, VWF:RCo, and FVIII levels should be performed before and at several time points after the DDAVP administration up to and including 4 hours. Peak VWF levels are achieved 30 and 90 minutes after intravenous and intranasal delivery, respectively. An increase in VWF:Ag/VWF:RCo and FVIII levels to at least 30 IU/dL is adequate for most dental procedures, minor surgery, or the treatment of epistaxis or menorrhagia. DDAVP may be adequate to treat major bleeds or for major surgery when VWF levels increase well above 50 IU/dL. Decisions surrounding the use of DDAVP versus a VWF/FVIII concentrate will depend on the expected DDAVP response, the type of surgery, and the anticipated duration of therapy required to achieve hemostasis. If treatment is required for more than 3 days, concerns regarding tachyphylaxis and side effects may limit its use. Significantly decreased VWF:Ag/VWF:RCo or FVIII at the 4-hour time point of a DDAVP trial may indicate type 1C or type 2N VWD, which are associated with increased clearance of endogenous VWF or FVIII, respectively. Despite the transient response in these patients, DDAVP remains a therapeutic option and its use should be assessed on a case-by-case basis.47
The parenteral dose of DDAVP is 0.3 μg/kg infused in 30 to 50 mL of normal saline over approximately 30 minutes every 12 to 24 hours. The dose of the highly concentrated intranasal preparation is 150 μg for children under 50 kg and 300 μg for larger children and adults (1 spray per naris). It is important to note that the products used to treat VWD (eg, Stimate) deliver 150 μg per spray, a much higher concentration than that used to treat enuresis. Repeated DDAVP dosing is associated with the development of tachyphylaxis: with subsequent dosing, the magnitude of the VWF and FVIII increments can fall to approximately 70% of that obtained with the initial dose.48 DDAVP is safe and generally well tolerated. Side effects include facial flushing, headache, tachycardia, light-headedness, and mild hypotension. The most serious side effects, severe hyponatremia and seizures,49 can be avoided by fluid restriction for 24 hours after DDAVP administration. Serum sodium levels should be monitored with repeated doses. DDAVP is generally avoided in those younger than 2 years of age because of a higher risk of hyponatremia. Patients who are intolerant of DDAVP or have a poor VWF response need to be treated with a VWF/FVIII concentrate.
VWF/FVIII Concentrate
VWF/FVIII concentrates are required for patients who do not have an adequate response to DDAVP, who have side effects from or contraindications to DDAVP, or who require a long duration of treatment, rendering the use of DDAVP impractical. Purified, viral-inactivated, plasma-derived VWF/FVIII concentrates are the products most frequently used (eg, Humate-P, Wilate, Alphanate SD/HT). The quantity of VWF:RCo activity relative to FVIII:C varies by product; Humate-P contains 2.4 VWF:RCo units for each unit of FVIII:C; Wilate contains a 1:1 ratio; and Alphanate contains a 0.5:1 ratio. Both Humate-P and Wilate are reported to contain a full spectrum of VWF multimers, including HMW multimers, and closely resemble normal plasma, but Alphanate SD/HT lacks HMW mutimers.11,50 Thus, the available VWF/FVIII vary in terms of VWF:RCo to FVIII concentrate, HMW multimer composition, reported VWF:RCo, and FVIII half-lives and even approved indications. They should not be considered interchangeable, and further information should be sought from the respective product inserts.
Dosing recommendations are provided either in VWF:RCo (North America) or FVIII:C units (Europe) and are weight-based (Table 4); repeat infusions can be given every 8 to 24 hours depending on the type of surgery/injury and the product used.
Long-term continuous use of concentrates to prevent bleeds, known as prophylaxis, is the standard of care in severe hemophilia A and B and is now being adopted in severe VWD. Patients with type 3 VWD or severe type 1 or type 2 VWD may experience recurrent bleeds into joints, nasal/oropharynx, or gastrointestinal tract or excessive menstrual bleeding. Retrospective cohort and case series suggest that prophylaxis improves quality of life; reduces the frequency of bleeding, need for transfusions, and hospitalizations; and prevents chronic joint disease.54,55 More recently, a prospective study confirmed that prophylaxis with VWF concentrates at doses ranging from 50 IU VWF RCo/kg 1 to 3 times per week was highly effective at reducing bleeding rates, with annualized bleeding rates decreasing from 25 to 6.1 in 11 participants with either type 2A or type 3 VWD.56
VWF/FVIII concentrates are effective in more than 97% of events.57 Rarely, when infusion of a VWF/FVIII concentrate is ineffective at stopping bleeding, transfusion of platelet concentrates may be beneficial, presumably because they facilitate the delivery of small amounts of platelet VWF to the site of vascular injury. Highly purified FVIII concentrates (monoclonal antibody purified and recombinant) should not be used to treat VWD because they lack VWF.
A recombinant VWF concentrate (Vonvendi) combined initially with recombinant FVIII concentrate in a 1.3:1 ratio of VWF:RCo to FVIII:C has been shown to be safe and efficacious for the on-demand treatment of bleeds.58,59 After the initial FVIII dose, the patients’ endogenous FVIII levels are stabilized within 6 hours and further FVIII administration may not required. A prospective phase 3 trial investigating the efficacy of recombinant VWF in the prophylaxis of severe VWD is ongoing. Vonvendi has been licensed for on-demand treatment in the United States since 2015. For further dosing information, please refer to the product insert.
Conclusion
VWF is a complex protein with several important and distinct functional domains: binding sites to collagen, FVIII, and platelet GPIbα; an ADAMTS13 cleavage site; and domains important for multimer formation. Mutations in any of these sites can result in a dysfunctional protein and as a result, VWD is a heterogeneous disorder with many specific assays available to determine the subtype. Despite this, the treatment of VWD is straightforward with only a small number of therapeutic options: indirect therapies such as antifibrinolytic agents, or direct therapies that increase VWF levels, DDAVP, or VWF/FVIII concentrates. Management focuses on preventing bleeding complications associated with invasive procedures or promptly treating bleeding episodes.
Introduction
von Willebrand disease (VWD) is an inherited bleeding disorder caused by deficient or defective plasma von Willebrand factor (VWF). VWF is an adhesive multimeric plasma glycoprotein that performs 2 major functions in hemostasis: it mediates platelet adhesion to injured subendothelium via glycoprotein 1bα (GPIbα), and it binds and stabilizes factor VIII (FVIII) in circulation, protecting it from proteolytic degradation by enzymes. The current VWD classification recognizes 3 types (Table 1).1
Prevalence
VWD is the most common inherited bleeding disorder. However, because VWF levels are highly variable and disease severity ranges from mild bleeding symptoms to severe or life-threatening bleeds, the reported prevalence of VWD depends on the diagnostic definition used. Two large epidemiologic studies have reported prevalence rates of approximately 1%.2,3 In these studies, healthy school-aged children were screened and diagnosed with VWD based on low VWF activity, measured as ristocetin cofactor, and a personal and family history of bleeding symptoms. At the other extreme, when considering patients whose bleeding symptoms are sufficiently severe to warrant referral to specialized centers, the reported prevalence of VWD ranges from 20 to 113 per million.4 These studies likely over- and underestimate clinically significant VWD. More recent studies suggest that the prevalence of VWD in individuals whose bleeding symptoms are significant enough to present to a primary care physician is approximately 0.1%.5 This figure is likely a more accurate estimate of the true prevalence of symptomatic VWD.
Although VWD is autosomally inherited, females are more likely to present with bleeding symptoms and be diagnosed because of increased exposure to bleeding challenges, such as menorrhagia and childbirth. VWD does not show any geographic or ethnic predilection, but there is an increased prevalence of the recessive forms, such as type 2N and type 3 VWD, in areas with high rates of consanguinity.
VWF Protein Structure and Function
The VWF gene is located on chromosome 12 at p13.3 and spans 178 kb comprising 52 exons.6 The expression of the VWF gene is tightly restricted to endothelial cells, platelets, and megakaryocytes, where VWF is stored in Weibel-Palade bodies and α-granules. VWF is a large multimeric glycoprotein with several important functional domains (Figure).
Classification, Pathophysiology, and Genetics
The International Society of Thrombosis and Hemostasis (ISTH) classification of VWD was updated in 2006 (Table 1).1 It incorporates important aspects of clinical phenotype, pathophysiological mechanisms, and treatment considerations. The 3 categories are: type 1, which is a partial quantitative deficiency; type 2 with 4 subtypes (2A, 2B, 2M, and 2N), which is a qualitative defect; and type 3, which is a virtual absence of VWF. Although the diagnosis and categorization of VWD can be achieved with widely available laboratory testing, further subcategorization among type 2 VWD subtypes may require referral to a specialized laboratory. The current ISTH classification intentionally does not incorporate genotypic data. In type 2 or type 3 VWD disease, VWF mutations are identified in more than 90% of cases and are completely penetrant, whereas mutations are identified in only approximately 65% of type 1 VWD cases and have been associated with incomplete penetrance and variable expressivity.10 These studies suggest that type 1 VWD is an oligogenic disease with mutations in genes regulating secretion or clearance contributing to a VWD phenotype.
VWD Types
Type 1
Type 1 VWD is caused by a partial quantitative deficiency of VWF and represents approximately 75% of VWD cases. It is the most clinically heterogeneous type, with patients having a mild to moderate bleeding phenotype.11 Bleeding in type 1 VWD results from a decrease in the concentration of VWF. The VWF function is normal without a significant abnormality in the platelet, collagen, or FVIII binding sites or a significant decrease in HMW multimers. Functional assays of VWF, such as VWF ristocetin cofactor (VWF:RCo) or VWF activity (VWF:Act) (see section on Laboratory Testing for further details), are proportionally decreased relative to the VWF antigen level (VWF:Ag), and the ratio of functional activity as compared with the VWF level is normal (ie, VWF:RCo/VWF:Ag ratio is > 0.6). As noted, VWF mutations are identified in only 65% of type 1 VWD cases and have incomplete penetrance and variable expressivity.10 Approximately 70% of mutations identified are missense mutations. Missense mutations may affect VWF levels by affecting any part of the biosynthetic pathway, including trafficking, storage, secretion, and/or clearance of VWF.
Increased VWF clearance is a well-described mechanism for type 1 VWD, known as type 1C. These patients will typically have very low VWF levels, an increased VWF propeptide to antigen ratio (VWFpp/VWF:Ag), and a marked but short-lived response to DDAVP, limiting DDAVP’s clinical applicability.12 On the other hand, the half-life of VWF/FVIII concentrates is normal in these individuals. Type 1C VWD is caused by missense mutations which occur mainly in the D3 domain and reduce the half-life of VWF up to 15-fold. R1205H, known as the “Vicenza” variant, is the most common and severe as well as the best characterized of these mutations.13
Type 2
Accounting for approximately 25% of VWD cases, type 2 VWD is characterized by a qualitative deficiency of VWF activity and is further subcategorized based on the mechanism of VWF dysfunction. Type 2A, 2B, and 2M affect VWF–platelet interactions by way of loss of HMW multimers, a gain of function of the GPIbα binding site, or a loss of function of the same site, respectively. On the other hand, type 2N is caused by defective VWF binding to FVIII. Type 2 VWD is often suspected when investigations demonstrate a function-antigen discordance: the VWF:RCo or VWF:Act is decreased disproportionately to the decrease in VWF:Ag, and the VWF:RCo/VWF:Ag ratio is less than 0.6.
Type 2A VWD is the most common type 2 variant. It is characterized by disproportionately low functional activity compared to antigen level (ie, VWF:RCo/VWF:ag ratio is < 0.6) and a loss of HMW and sometimes intermediate molecular weight (IMW) multimers. Ristocetin-induced platelet agglutination (RIPA) will be decreased with standard doses of ristocetin and absent with low doses. Type 2A VWD is usually inherited as an autosomal dominant trait. This subtype encompasses missense mutations that impair dimerization or multimerization of VWF subunits (CK, D1, and D2 domains); disrupt intersubunit disulphide bonds (D3 and D2 domains); enhance susceptibility to ADAMTS13-mediated proteolysis (A2 and A1 domains); or result in intracellular retention of the HMW multimers (D3, A1, and A2 domains).10 The result is VWF that lacks HMW multimers, thereby possessing fewer GPIbα binding sites, and that is less effective in binding platelets.
Type 2B VWD is the result of gain-of-function mutations within the GPIbα binding site of VWF. Generally, the platelet-binding site of VWF within the A1 domain is only exposed once VWF is immobilized on injured collagen and subjected to shear forces, resulting in a conformational change.7 In type 2B VWD, the gain-of-function mutation results in spontaneous binding of VWF to platelets without the need for a VWF-collagen interaction and unfolding of VWF by shear forces. The VWF–platelet interaction selectively depletes the HMW multimers by the unfolding of the A2 domain and increasing ADAMTS13 proteolysis. The increased binding of mutant VWF to platelets also triggers the formation of platelet aggregates, which are removed from circulation resulting in thrombocytopenia. Increases in endogenous VWF seen with acute stressors or pregnancy can worsen thrombocytopenia and increase the risk of bleeding.14 Certain mutations, such as V1316M, alter megakaryocytopoiesis and are characterized by giant platelets with abnormal ultrastructure and further exacerbate the thrombocytopenia.15 The laboratory profile reveals a VWF:RCo/VWF:Ag ratio of < 0.6 and absence of HMW multimers. In contrast to type 2A, platelets will agglutinate with low-dose ristocetin. Missense mutations are highly penetrant dominant and occur in or close to the A1 domain.16
Type 2M VWD is characterized by loss-of-function mutations within the GPIbα binding site of VWF. Phenotypic characteristics include a reduced ratio of VWF:RCo/VWF:Ag of < 0.6 but a normal multimer pattern.17 Missense mutations are reported in the A1 domain affecting the GPIbα-binding site. In very rare instances, mutations in the A3 domain that impair the VWF/collagen interaction have been described.18 These collagen-binding mutations are not included in the last iteration of the ISTH classification in 2006,1 but fit best in the type 2M category. In these cases, VWF:RCo or VWF:Act, which reflect activity at the GPIbα-binding site, may be normal and the diagnosis requires VWF/collagen binding assays (VWF:CB).
Type 2N VWD results from mutations of the FVIII binding site or conformational changes that impair the VWF–FVIII interaction. Most (~80%) missense mutations are located in domains D’ and D3.19 These mutations are autosomal recessive, and affected individuals are either homozygous or compound heterozygous for type 2N/2N or type 1/2N mutations, or compound heterozygous for a missense mutation and a mutation resulting in a null allele (type 2N/3 mutations). The laboratory phenotype is a disproportionate reduction in the FVIII level relative to the VWF level, which may be low or normal. Most cases of type 2N VWD have a normal multimeric profile, but rare cases will demonstrate loss of HMW multimers. Definitive diagnosis requires evidence of reduced FVIII binding to VWF (VWF:FVIIIB) or the identification of causative mutations in the FVIII binding region of the VWF gene.20
Type 3
Type 3 VWD is defined by a virtual absence of VWF. The inheritance of type 3 VWD has often been reported as autosomal recessive. However, there is emerging evidence that it can also be inherited in a co-dominant pattern: obligate carriers of type 3 VWD mutations have more mucocutaneous bleeding symptoms than normal individuals, and in approximately 50% of cases may carry a diagnosis of type 1 VWD.21 This condition is characterized by prolongation of the activated partial thromboplastin time (aPTT), undetectable levels of VWF:Ag, and VWF:RCo and FVIII levels less than 10 IU/dL (10%). The majority (~80%) of type 3 VWD patients have 2 null alleles as a result of a variety of mutations, with nonsense mutations accounting for about one-third.10 The remainder of the mutational spectrum is made up of missense mutations predominantly located in the D1-D2 (exons 3–11) and D4-CK (exons 37–52) domains that result in intracellular VWF retention, or large deletions, resulting in frameshift mutations affecting 1 or more exons. Because there is little or no circulating VWF, patients with type 3 VWD may develop alloantibodies to VWF, which can complicate treatment.22
Diagnosis
Clinical Manifestations
VWD is a congenital bleeding disorder. The increased risk of bleeding is present from birth, but symptoms may only manifest when there is a hemostatic challenge. Bleeding symptoms become more apparent with increasing age and exposure to hemostatic challenges. As a result, the diagnosis is often delayed into adulthood in mild to moderate forms of VWD. On the other hand, with more severe bleeding phenotypes such as type 3 VWD, the diagnosis is often made in childhood. Individuals with VWD primarily complain of excessive mucocutaneous bleeding, which includes spontaneous bruising, recurrent epistaxis, and bleeding from the gums after brushing, dental cleaning, and extractions. In addition, prolonged or excessive bleeding after surgery or trauma is often reported. Females frequently experience menorrhagia, usually beginning at menarche, and can have prolonged or excessive bleeding after childbirth.23 Musculoskeletal bleeding is unusual, except in type 2N or type 3 VWD when the FVIII:C level may be less than 10 IU/dL.
Mucocutaneous bleeding symptoms such as epistaxis, gum bleeding, ecchymosis, and menorrhagia overlap with those experienced by a normal population, and therefore can be easily overlooked by both patients and physicians.11 The use of bleeding assessment tools (BATs) to standardize the bleeding history and interpretation of the severity of the bleeding phenotype is becoming part of routine clinical practice. Three different BATs, each an adaptation of its predecessor, have been created and validated.24 Each of the scores performs well in an undiagnosed population presenting with bleeding symptoms. The negative predictive value is typically greater than 0.99, meaning that a negative bleeding score nearly excludes a clinically significant bleeding disorder. Thus, the main utility of the current BATs is at the time of new patient assessments: a negative bleeding score will help avoid unnecessary laboratory testing and prevent false-positive diagnoses of VWD (borderline low VWF:Ag without a significant bleeding history). However, the currently available BATs have some limitations. When scoring severe bleeding disorders, BATs become saturated as they take into account the worst episode of bleeding within each category but not the frequency of bleeding. BATs need to be administered by an expert and are time consuming to complete. Finally, they are not useful for monitoring bleeding symptoms or response to therapy because of the cumulative nature of the scores. In an attempt to standardize the BAT and bleeding score, the ISTH/Scientific and Standardization Committee (SSC) Joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group has established a revised BAT, known as the ISTH-BAT, specifically designed to extend the utility of the earlier BATS by incorporating information on both symptom frequency and severity.25,26 The ISTH-BAT has been further modified to a patient- or self-administered BAT (SELF-BAT). The SELF-BAT has been shown to be a reliable and effective tool in the assessment of patients who are being evaluated for VWD.27
Laboratory Testing
Screening tests include a complete blood count (CBC), prothrombin time, aPTT, thrombin time, and fibrinogen concentration to exclude the presence of other hemostatic disorders. The CBC may show thrombocytopenia in type 2B VWD. The aPTT is often normal, but will be prolonged if the FVIII level is below 30 IU/dL, as can be seen in severe type 1, type 2N, or type 3 VWD. The platelet function analyzer (PFA-100) is a system for analyzing primary hemostasis under high shear rates, but its role in the diagnosis of VWD is controversial.11
The evaluation of VWD involves quantitative (VWF:Ag) and qualitative measurements of VWF (VWF:RCo, or one of the novel assays: VWF:Act or VWF:GPIbM) and FVIII activity (FVIII:C). Type 2 VWD is suspected when the VWF activity to VWF:Ag ratio is < 0.6, the FVIII:C is more significantly decreased as compared to VWF:Ag, or with the presence of thrombocytopenia. In these cases, further testing (multimer gel electrophoresis, VWF:CB, RIPA, VWF:FVIIIB, and genotyping) is required to discriminate the type 2 VWD subtype, but such testing may be available only in specialized laboratories. If type 1C VWD is suspected, the VWFpp/VWF:ag ratio may confirm the diagnosis. Table 2 summarizes the results seen with each subtype. These assays are described in detail below.
VWD Assays
VWF:Ag represents the quantity of VWF protein (antigen) in the plasma measured using an enzyme-linked immunosorbent assay (ELISA) or latex immunoassay. The normal range is approximately 50 to 200 IU/dL.
VWF:RCo is a functional assay that determines the capacity of VWF to agglutinate platelets via the platelet receptor GPIbα in the presence of ristocetin. The normal range is approximately 50 to 200 IU/dL. Novel methods of measuring VWF’s platelet-binding activity are increasingly being adopted by clinical laboratories and are associated with greater precision and improved lower limits of detection and coefficients of variation.28,29 The first is the VWF:Act, a rapid automated assay that measures VWF activity using an antibody directed to the GPIbα binding site of VWF.28 The second novel assay is VWF:GPIbM, which involves a gain-of-function GPIB construct that binds VWF without ristocetin.30,31 For simplicity, VWF:RCo will be used to refer to VWF platelet-binding activity in the ensuing text. Factor VIII:C is a functional FVIII assay that determines the activity of FVIII in aPTT-based assays. The normal range is approximately 50 to 150 IU/dL.
VWF multimer analysis by SDS-agarose electrophoresis assesses VWF oligomers in plasma.32 Normal plasma contains multimers composed of over 40 VWF dimers, and these multimers are classified as high (HMW), intermediate (IMW), or low molecular weight (LMW). HMW multimers are decreased or missing in types 2A and 2B VWD, and IMW multimers may also be absent in type 2A VWD.
Low-dose RIPA tests the capacity of the patient’s platelets to agglutinate at low concentrations of ristocetin (~0.5 mg/mL). This is in contrast to the VWF:RCo, in which formalin-fixed control platelets are used. With type 2B, the platelet membrane is “overloaded” with high-affinity mutant VWF, resulting in abnormal platelet agglutination at low ristocetin concentrations. In some cases of type 2B VWD, all variables except RIPA may be normal.29
VWF:FVIIIB is an ELISA-based assay that determines the ability of VWF to bind FVIII and is used to make the diagnosis of type 2N VWD.19
VWF:CB is an ELISA-based assay that measures the ability of VWF to bind to collagen, a function of VWF that is dependent on the collagen-binding domain (A3) and on the presence of HMW multimers. VWF:CB helps to distinguish between types 1 and 2 VWD by reflecting the loss of HMW multimer forms (type 2A VWD) or can reflect a specific collagen-binding deficiency (type 2M VWD).33 The normal range is approximately 50 to 200 IU/dL. This assay is not available in most clinical laboratories.
VWFpp/VWF:Ag takes advantage of 2 facts: the VWF propeptide is secreted in a one-to-one ratio to VWF subunits and has a stable half-life in plasma. Thus, an increased ratio identifies patients with mutations that increase VWF clearance, such as type 1C VWD.34 The mean ratio in normal individuals is 1.3, with a normal range of 0.54 to 1.98.
Genotyping should be considered when specialized testing with the VWF:FVIIIB, RIPA, or VWF:CB assays is unavailable and a diagnosis of type 2 VWD is suspected. A guideline on VWD genetic testing has been published by the UK Haemophilia Centre Doctors Organisation.35
Interpretation of Clinical History and Laboratory Investigations
Normal plasma levels of VWF are approximately 100 IU/dL (100%, corresponds to ~10 μg/mL) with a population range of 50 to 200 IU/dL (50%–200%). There are a number of preanalytical variables (patient specific or laboratory specific) that affect the results of VWF laboratory testing. Patient-specific variables that are associated with increased VWF levels include increasing age, African ethnicity, exercise, inflammatory disease states, blood group A or B, increased levels of epinephrine, cocaine use, and neuroendocrine hormone levels. Decreased VWF levels are associated with medications such as valproic acid, hypothyroidism, autoantibodies, and blood group O. Individuals with blood group O have VWF levels that are 25% lower than levels in other blood groups.36 Several analytical variables also can complicate the diagnosis of VWD: methods for established reference ranges, limitations to the sensitivity of assays, and sample handling issues.11 These factors (summarized in Table 3) must be considered when interpreting VWF laboratory results, and at least 2 sets of tests using fresh samples are needed to confirm the diagnosis of VWD. Testing should be avoided in stressed, ill, or pregnant patients.
Mild type 1 VWD can be a difficult diagnosis to make because of the overlap of bleeding symptoms among normal individuals and those with mild type 1 VWD, as well as the variability of VWF levels. There is no consensus on the exact VWF levels required to confirm the diagnosis: the NHLBI Expert Panel recommends VWF:Ag and VWF:RCo levels less than 0.30 IU/mL to diagnose type 1 VWD,11 whereas the ISTH-SSC Subcommittee on von Willebrand factor recommends using VWF:RCo and VWF:Ag levels greater than 2 standard deviations below the population mean.37 In the absence of a bleeding history, slightly reduced VWF levels do not predict future significant bleeding events.38 Therefore, regardless of the laboratory cut-off used, the cornerstone of a VWD diagnosis should be a history of excessive mucocutaneous bleeding.
Differential Diagnosis
When considering a diagnosis of VWD, the differential diagnosis must be considered and includes acquired von Willebrand syndrome (AVWS), platelet-type VWD (PT-VWD), and hemophilia A. AVWS is the result of an acquired deficiency or defect of VWF and manifests with a mild to moderate bleeding disorder without a lifelong personal and family history of bleeding. AVWS has diverse pathology. The most common mechanism is proteolytic cleavage of VWF after shear stress–induced unfolding, as seen with aortic stenosis and ventricular assist devices, where as many as 79% of persons with aortic stenosis39 and up to 100% with left ventricular assist devices are affected.40 Other disease mechanisms include autoantibody formation that impairs VWF function or increases its clearance (autoimmune disease or lymphoproliferative disease), adsorption of HMW VWF multimers to malignant cells or platelets (myeloproliferative neoplasms and Wilm’s tumor), or decreased synthesis (hypothyroidism, valproic acid). The median age of diagnosis is 62 years, but the disorder may occur in any age-group (range 2–96 years).41 The approach to management of AVWS should focus on treatment of bleeding and induction of long-term remission. Treatment of bleeding will depend on the underlying mechanism of AVWS and may include a combination of DDAVP or VWF/FVIII concentrates, recombinant factor VIIa, antifibrinolytic agents, intravenous immunoglobulin, or plasmapheresis for AVWS associated with autoantibodies. Treatment of the underlying disorder (eg, aortic valve repair or treatment of a lymphoproliferative disorder) may result in remission of the AVWS.
Mild hemophilia A (caused by mutations in the F8 gene) and type 2N VWD can be difficult to differentiate clinically. Both present with reduced FVIII:C, and type 2N VWD may have normal or borderline low levels of VWF. Although the VWF:FVIIIB assay will distinguish between the 2 disorders, the test is not available in many centers. The pattern of inheritance may be helpful: hemophilia A is an X-linked disorder, whereas type 2N is autosomal recessive. Often, the diagnosis of type 2N VWD is suspected when genotyping of F8 does not identify a mutation in mild hemophilia A, when infused FVIII concentrates have a decreased half-life, or when DDAVP is associated with a brisk but short-lived response. In the absence of VWF:FVIIIB assay availability, genotyping of VWF will confirm the diagnosis, with missense mutations being located in exons 17–20 or 24–27.19
PT-VWD represents the phenocopy of type 2B VWD. The mutation is in the platelet receptor gene GPIBA and causes enhanced VWF-platelet binding. The disorders can be differentiated by RIPA plasma/platelet mixing studies or flow cytometry.42,43 However, these assays are technically challenging. In the absence of mutations in exon 28 of VWF, mutations in exon 2 of GPIBA may be identified in approximately 10% of persons misdiagnosed with type 2B VWD.
Management
Patients with VWD present to medical attention in a number of ways: excessive post-trauma or surgical bleeding, recurrent mucocutaneous bleeding such as epistaxis, menorrhagia, gastrointestinal bleeding, or, in severe cases, recurrent hemarthroses and muscle hematomas. Irrespective of the presentation, the goal is to minimize and control bleeding. Therapeutic options can be divided into 3 main categories: (1) localized measures to stop bleeding; (2) pharmacologic agents with indirect hemostatic benefit; and (3) treatments that directly increase plasma VWF and FVIII levels. A combination of all 3 of these modalities can be used depending on the bleeding location and severity.
Localized Measures
Localized measures to control bleeding in VWD will depend on the site of bleeding. Epistaxis can be particularly problematic for affected children, and patients should be armed with a step-wise action plan that escalates from pressure to packing and includes guidelines regarding how long to wait before seeking medical attention. In selected cases, nasal cautery may be required for prolonged or excessive epistaxis. Topical hemostatic agents such as gelatin foam/matrix, topical thrombin, and fibrin sealants are predominately used to achieve surgical hemostasis and may have a limited role in the treatment of VWD-associated bleeding. In the case of menorrhagia, hormonal treatments (ie, the combined oral contraceptive pill, OCP), levonorgestrel-releasing intrauterine systems, or endometrial ablation all effectively reduce menstrual blood loss through their local effects on the endometrial lining.44 In addition, older generations of OCP are associated with increases in VWF levels. This effect is mediated by the estrogen component and is evident with ethynylestradiol doses of 0.5 μg or higher. Lower estrogen doses, seen in currently used OCP, have little or no effect on VWF levels.11,45
Pharmacologic Therapy
Indirect therapies include the antifibrinolytic agents (eg, tranexamic acid and aminocaproic acid). These agents are used either as the sole therapy at the time of minor surgical and dental procedures, or as an adjunct in combination with DDAVP or VWF/FVIII concentrates. Antifibrinolytics are thought to be particularly useful for controlling mucosal bleeding in areas of high fibrinolytic activity: the oral cavity, gastrointestinal tract, or uterus. Tranexamic acid inhibits the conversion of plasminogen to plasmin, and is the more commonly used antifibrinolytic.11 Tranexamic acid can be administered either intravenously or orally at doses of 10 to 25 mg/kg, respectively. It is usually continued until bleeding is controlled or up to 7 to 10 days postoperatively. The most common adverse events associated with tranexamic acid are headache, back pain, and gastrointestinal side effects.46 Tranexamic acid is contraindicated in disseminated intravascular coagulation and bleeding from the upper urinary tract, where it can lead to urinary tract obstruction by clots.
DDAVP, a synthetic derivative of vasopressin, promotes release of stored VWF from endothelial cells. Most individuals with type 1 VWD and some with type 2A VWD respond to treatment with DDAVP: a therapeutic trial to confirm adequate DDAVP response should be performed prior to its clinical use. Assessment of VWF:Ag, VWF:RCo, and FVIII levels should be performed before and at several time points after the DDAVP administration up to and including 4 hours. Peak VWF levels are achieved 30 and 90 minutes after intravenous and intranasal delivery, respectively. An increase in VWF:Ag/VWF:RCo and FVIII levels to at least 30 IU/dL is adequate for most dental procedures, minor surgery, or the treatment of epistaxis or menorrhagia. DDAVP may be adequate to treat major bleeds or for major surgery when VWF levels increase well above 50 IU/dL. Decisions surrounding the use of DDAVP versus a VWF/FVIII concentrate will depend on the expected DDAVP response, the type of surgery, and the anticipated duration of therapy required to achieve hemostasis. If treatment is required for more than 3 days, concerns regarding tachyphylaxis and side effects may limit its use. Significantly decreased VWF:Ag/VWF:RCo or FVIII at the 4-hour time point of a DDAVP trial may indicate type 1C or type 2N VWD, which are associated with increased clearance of endogenous VWF or FVIII, respectively. Despite the transient response in these patients, DDAVP remains a therapeutic option and its use should be assessed on a case-by-case basis.47
The parenteral dose of DDAVP is 0.3 μg/kg infused in 30 to 50 mL of normal saline over approximately 30 minutes every 12 to 24 hours. The dose of the highly concentrated intranasal preparation is 150 μg for children under 50 kg and 300 μg for larger children and adults (1 spray per naris). It is important to note that the products used to treat VWD (eg, Stimate) deliver 150 μg per spray, a much higher concentration than that used to treat enuresis. Repeated DDAVP dosing is associated with the development of tachyphylaxis: with subsequent dosing, the magnitude of the VWF and FVIII increments can fall to approximately 70% of that obtained with the initial dose.48 DDAVP is safe and generally well tolerated. Side effects include facial flushing, headache, tachycardia, light-headedness, and mild hypotension. The most serious side effects, severe hyponatremia and seizures,49 can be avoided by fluid restriction for 24 hours after DDAVP administration. Serum sodium levels should be monitored with repeated doses. DDAVP is generally avoided in those younger than 2 years of age because of a higher risk of hyponatremia. Patients who are intolerant of DDAVP or have a poor VWF response need to be treated with a VWF/FVIII concentrate.
VWF/FVIII Concentrate
VWF/FVIII concentrates are required for patients who do not have an adequate response to DDAVP, who have side effects from or contraindications to DDAVP, or who require a long duration of treatment, rendering the use of DDAVP impractical. Purified, viral-inactivated, plasma-derived VWF/FVIII concentrates are the products most frequently used (eg, Humate-P, Wilate, Alphanate SD/HT). The quantity of VWF:RCo activity relative to FVIII:C varies by product; Humate-P contains 2.4 VWF:RCo units for each unit of FVIII:C; Wilate contains a 1:1 ratio; and Alphanate contains a 0.5:1 ratio. Both Humate-P and Wilate are reported to contain a full spectrum of VWF multimers, including HMW multimers, and closely resemble normal plasma, but Alphanate SD/HT lacks HMW mutimers.11,50 Thus, the available VWF/FVIII vary in terms of VWF:RCo to FVIII concentrate, HMW multimer composition, reported VWF:RCo, and FVIII half-lives and even approved indications. They should not be considered interchangeable, and further information should be sought from the respective product inserts.
Dosing recommendations are provided either in VWF:RCo (North America) or FVIII:C units (Europe) and are weight-based (Table 4); repeat infusions can be given every 8 to 24 hours depending on the type of surgery/injury and the product used.
Long-term continuous use of concentrates to prevent bleeds, known as prophylaxis, is the standard of care in severe hemophilia A and B and is now being adopted in severe VWD. Patients with type 3 VWD or severe type 1 or type 2 VWD may experience recurrent bleeds into joints, nasal/oropharynx, or gastrointestinal tract or excessive menstrual bleeding. Retrospective cohort and case series suggest that prophylaxis improves quality of life; reduces the frequency of bleeding, need for transfusions, and hospitalizations; and prevents chronic joint disease.54,55 More recently, a prospective study confirmed that prophylaxis with VWF concentrates at doses ranging from 50 IU VWF RCo/kg 1 to 3 times per week was highly effective at reducing bleeding rates, with annualized bleeding rates decreasing from 25 to 6.1 in 11 participants with either type 2A or type 3 VWD.56
VWF/FVIII concentrates are effective in more than 97% of events.57 Rarely, when infusion of a VWF/FVIII concentrate is ineffective at stopping bleeding, transfusion of platelet concentrates may be beneficial, presumably because they facilitate the delivery of small amounts of platelet VWF to the site of vascular injury. Highly purified FVIII concentrates (monoclonal antibody purified and recombinant) should not be used to treat VWD because they lack VWF.
A recombinant VWF concentrate (Vonvendi) combined initially with recombinant FVIII concentrate in a 1.3:1 ratio of VWF:RCo to FVIII:C has been shown to be safe and efficacious for the on-demand treatment of bleeds.58,59 After the initial FVIII dose, the patients’ endogenous FVIII levels are stabilized within 6 hours and further FVIII administration may not required. A prospective phase 3 trial investigating the efficacy of recombinant VWF in the prophylaxis of severe VWD is ongoing. Vonvendi has been licensed for on-demand treatment in the United States since 2015. For further dosing information, please refer to the product insert.
Conclusion
VWF is a complex protein with several important and distinct functional domains: binding sites to collagen, FVIII, and platelet GPIbα; an ADAMTS13 cleavage site; and domains important for multimer formation. Mutations in any of these sites can result in a dysfunctional protein and as a result, VWD is a heterogeneous disorder with many specific assays available to determine the subtype. Despite this, the treatment of VWD is straightforward with only a small number of therapeutic options: indirect therapies such as antifibrinolytic agents, or direct therapies that increase VWF levels, DDAVP, or VWF/FVIII concentrates. Management focuses on preventing bleeding complications associated with invasive procedures or promptly treating bleeding episodes.
1. Sadler JE, Budde U, Eikenboom JCJ, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemost 2006;4:2103–14.
2. Rodeghiero F, Castaman G. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood 1987;69:454–9.
3. Werner EJ, Broxson EH, Tucker EL, et al. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr 1993;123:893–8.
4. Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost 2000;84:160–74.
5. Bowman M, Hopman WM, Rapson D, et al. The prevalence of symptomatic von Willebrand disease in primary care practice. J Thromb Haemost 2010;8:213–6.
6. Mancuso DJ, Tuley EA, Westfield LA, et al. Structure of the gene for human von Willebrand factor. J Biol Chem 1989;264:19514–27.
7. Kang I, Raghavachari M, Hofmann CM, Marchant RE. Surface-dependent expression in the platelet GPIb binding domain within human von Willebrand factor studied by atomic force microscopy. Thromb Res 2007;119:731–40.
8. Savage B, Saldívar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 1996;84:289–97.
9. Dong J, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002;100:4033–9.
10. Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev 2010;24:123–34.
11. Nichols WL, Hultin MB, James AH, et al. Von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) expert panel report (USA). Haemophilia 2008;14:171–232.
12. Haberichter SL, Castaman G, Budde U, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 vwd (MCMDM-1VWD). Blood 2008;111:4979–85.
13. Goodeve A. Vicenza deciphered: modeling the von Willebrand disease enigma: commentary on accelerated clearance alone explains ultralarge multimers in VWD Vicenza. J Thromb Haemost 2010;8:1271–2.
14. Federici AB, Mannucci PM, Castaman G, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood 2009;113:526–34.
15. Nurden AT, Federici AB, Nurden P. Altered megakaryocytopoiesis in von Willebrand type 2B disease. J Thromb Haemost 2009;7 Suppl 1:277–81.
16. Ruggeri ZM, Pareti FI, Mannucci PM, et al. Heightened interaction between platelets and factor VIII/von Willebrand factor in a new subtype of von Willebrand’s disease. New Engl J Med 1980;302:1047–51.
17. James PD, Notley C, Hegadorn C, et al. Challenges in defining type 2M von Willebrand disease: results from a Canadian cohort study. J Thromb Haemost 2007;5:1914–22.
18. Flood VH, Lederman CA, Wren JS, et al. Absent collagen binding in a VWF A3 domain mutant: utility of the VWF:CB in diagnosis of VWD. J Thromb Haemost 2010;8:1431–3.
19. Mazurier C, Hilbert L. Type 2N von Willebrand disease. Curr Hematol Rep 2005;4:350–8.
20. Nesbitt IM, Goodeve AC, Guilliatt AM, et al. Characterisation of type 2N von Willebrand disease using phenotypic and molecular techniques. Thromb Haemost 1996;75:959–64.
21. Bowman M, Tuttle A, Notley C, et al. The genetics of Canadian type 3 von Willebrand disease: further evidence for co-dominant inheritance of mutant alleles. J Thromb Haemost 2013;11:512–20.
22. James PD, Lillicrap D, Mannucci PM. Alloantibodies in von Willebrand disease. Blood 2013;122:636–40.
23. James AH, Jamison MG. Bleeding events and other complications during pregnancy and childbirth in women with von Willebrand disease. J Thromb Haemost 2007;5:1165–9.
24. Rydz N, James PD. The evolution and value of bleeding assessment tools. J Thromb Haemost 2012;2223–9.
25. Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost 2010;8:2063–5.
26. Elbatarny M, Mollah S, Grabell J, et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia 2014;20:831–5.
27. Deforest M, Grabell J, Alberta S et al. Generation and optimization of the self-administered bleeding assessment tool and its validation as a screening test for von Willebrand disease. Haemophilia 2015;21:e384-8.
28. Castaman G, Hillarp A, Goodeve A. Laboratory aspects of von Willebrand disease: test repertoire and options for activity assays and genetic analysis. Haemophilia 2014;20(Suppl. 4):65–70.
29. Favaloro EJ. Von Willebrand disease, type 2B: a diagnosis more elusive than previously thought. Thromb Haemost 2008;99:630–1.
30. Budde U. Diagnosis of von Willebrand disease subtypes: implications for treatment. Haemophilia 2008;14 Suppl 5:27–38.
31. Favaloro EJ. Von Willebrand factor collagen-binding (activity) assay in the diagnosis of von Willebrand disease: a 15-year journey. Sem Thromb Hemost 2002;28:191–202.
32. Patzke J, Budde U, Huber A, et al. Performance evaluation and multicenter study of a von Willebrand factor activity assay based on GPIb binding in the absence of ristocetin. Blood Coagul Fibrinolysis 2014;25:860-70.
33. Graf L, Moffat KA, Carlino SA, et al. Evaluation of an automated method for measuring von Willebrand factor activity in clinical samples without ristocetin. Int J Lab Hematol 2014;36:341–51.
34. Haberichter SL, Balistreri M, Christopherson P, et al. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood 2006;108:3344–51.
35. Keeney S, Bowen D, Cumming A, et al. The molecular analysis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors’ Organisation Haemophilia genetics laboratory network. Haemophilia 2008;14:1099–111.
36. Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ, Montgomery RR. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood 1987;69:1691–5.
37. Sadler JE, Rodeghiero F. Provisional criteria for the diagnosis of VWD type 1. J Thromb Haemost 2005;3:775–7.
38. Tosetto A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM- 1VWD). J Thromb Haemost 2006;4:766–73.
39. Vincentelli A, Susen S, Le Tourneau T, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med 2003;349:343–9.
40. Uriel N, Pak S-W, Jorde UP, et al. Acquired von Willebrand syndrome after continuous-flow mechanical device support contributes to a high prevalence of bleeding during long-term support and at the time of transplantation. J Am Coll Cardiol 2010;56:1207–13.
41. Federici AB, Rand JH, Bucciarelli P, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost 2000;84:345–9.
42. Favaloro EJ, Patterson D, Denholm A, et al. Differential identification of a rare form of platelet-type (pseudo-) von Willebrand disease (VWD) from type 2B VWD using a simplified ristocetin-induced-platelet-agglutination mixing assay and confirmed by genetic analysis. Brit J Haematol 2007;139:621–8.
43. Giannini S, Cecchetti L, Mezzasoma AM, Gresele P. Diagnosis of platelet-type von Willebrand disease by flow cytometry. Haematologica 2010;95:1021–4.
44. Farquhar C, Brown J. Oral contraceptive pill for heavy menstrual bleeding. Cochrane Database Syst Rev 2009 Oct 7;(4):CD000154.
45. Kadir R, Economides DL, Sabin C, et al. Variations in coagulation factors in women: effects of age, ethnicity, menstrual cycle and combined oral contraceptive. Thromb Haemost 1999;82:1456–61.
46. Muse K, Lukes AS, Gersten J, et al. Long-term evaluation of safety and health-related quality of life in women with heavy menstrual bleeding treated with oral tranexamic acid. Womens Health 2011;7:699–707.
47. Castaman G, Tosetto A, Federici AB, Rodeghiero F. Bleeding tendency and efficacy of anti-haemorrhagic treatments in patients with type 1 von Willebrand disease and increased von Willebrand factor clearance. Thromb Haemost 2011;105:647–54.
48. Mannucci PM, Bettega D, Cattaneo M. Patterns of development of tachyphylaxis in patients with haemophilia and von Willebrand disease after repeated doses of desmopressin (DDAVP). Brit J Haematol 1992;82:87–93.
49. Greaves M, Watson HG. Approach to the diagnosis and management of mild bleeding disorders. J Thromb Haemost 2007;5 Suppl 1:167–74.
50. Kessler CM, Friedman K, Schwartz BA, Gill JC, Powell JS. The pharmacokinetic diversity of two von Willebrand factor (VWF) / factor VIII (FVIII) concentrates in subjects with congenital von Willebrand disease. results from a prospective, randomised crossover study. Thromb Haemost 2011;106:279–88.
51. Weiss HJ, Sussman II, Hoyer LW. Stabilization of factor VIII in plasma by the von Willebrand factor. Studies on posttransfusion and dissociated factor VIII and in patients with von Willebrand’s disease. J Clin Invest 1977;60:390–404.
52. Berntorp E. Haemate P/Humate-P: a systematic review. Thromb Res 2009;124:S11–14.
53. Lillicrap D, Poon MC, Walker I, et al. Efficacy and safety of the factor VIII/von Willebrand factor concentrate, Haemate-P/Humate-P: ristocetin cofactor unit dosing in patients with von Willebrand disease. Thromb Haemost 2002;87:224–30.
54. Halimeh S, Krümpel A, Rott H, et al. Long-term secondary prophylaxis in children, adolescents and young adults with von Willebrand disease. results of a cohort study. Thromb Haemost 2011;105:597–604.
55. Abshire TC, Federici AB, Alvárez MT, et al. Prophylaxis in severe forms of von Willebrand’s disease: results from the von Willebrand disease prophylaxis network (VWD PN). Haemophilia 2013;19:76–81.
56. Abshire T, Cox-Gill J, Kempton CL, et al. Prophylaxis escalation in severe von Willebrand disease: a prospective study from the von Willebrand Disease Prophylaxis Network. J Thromb Haemost 2015;13:1585– 9.
57. Auerswald G, Kreuz W. Haemate P/Humate-P for the treatment of von Willebrand disease: considerations for use and clinical experience. Sem Thromb Hemost 2008;14 (Suppl 5):39–46.
58. Mannucci PM, Kempton C, Millar C, et al. Pharmacokinetics and safety of a novel recombinant human von Willebrand factor manufactured with a plasma-free method: a prospective clinical trial. Blood 2013;122:648–57.
59. Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood 2015;126:2038–46.
1. Sadler JE, Budde U, Eikenboom JCJ, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemost 2006;4:2103–14.
2. Rodeghiero F, Castaman G. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood 1987;69:454–9.
3. Werner EJ, Broxson EH, Tucker EL, et al. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr 1993;123:893–8.
4. Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost 2000;84:160–74.
5. Bowman M, Hopman WM, Rapson D, et al. The prevalence of symptomatic von Willebrand disease in primary care practice. J Thromb Haemost 2010;8:213–6.
6. Mancuso DJ, Tuley EA, Westfield LA, et al. Structure of the gene for human von Willebrand factor. J Biol Chem 1989;264:19514–27.
7. Kang I, Raghavachari M, Hofmann CM, Marchant RE. Surface-dependent expression in the platelet GPIb binding domain within human von Willebrand factor studied by atomic force microscopy. Thromb Res 2007;119:731–40.
8. Savage B, Saldívar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 1996;84:289–97.
9. Dong J, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002;100:4033–9.
10. Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev 2010;24:123–34.
11. Nichols WL, Hultin MB, James AH, et al. Von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) expert panel report (USA). Haemophilia 2008;14:171–232.
12. Haberichter SL, Castaman G, Budde U, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 vwd (MCMDM-1VWD). Blood 2008;111:4979–85.
13. Goodeve A. Vicenza deciphered: modeling the von Willebrand disease enigma: commentary on accelerated clearance alone explains ultralarge multimers in VWD Vicenza. J Thromb Haemost 2010;8:1271–2.
14. Federici AB, Mannucci PM, Castaman G, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood 2009;113:526–34.
15. Nurden AT, Federici AB, Nurden P. Altered megakaryocytopoiesis in von Willebrand type 2B disease. J Thromb Haemost 2009;7 Suppl 1:277–81.
16. Ruggeri ZM, Pareti FI, Mannucci PM, et al. Heightened interaction between platelets and factor VIII/von Willebrand factor in a new subtype of von Willebrand’s disease. New Engl J Med 1980;302:1047–51.
17. James PD, Notley C, Hegadorn C, et al. Challenges in defining type 2M von Willebrand disease: results from a Canadian cohort study. J Thromb Haemost 2007;5:1914–22.
18. Flood VH, Lederman CA, Wren JS, et al. Absent collagen binding in a VWF A3 domain mutant: utility of the VWF:CB in diagnosis of VWD. J Thromb Haemost 2010;8:1431–3.
19. Mazurier C, Hilbert L. Type 2N von Willebrand disease. Curr Hematol Rep 2005;4:350–8.
20. Nesbitt IM, Goodeve AC, Guilliatt AM, et al. Characterisation of type 2N von Willebrand disease using phenotypic and molecular techniques. Thromb Haemost 1996;75:959–64.
21. Bowman M, Tuttle A, Notley C, et al. The genetics of Canadian type 3 von Willebrand disease: further evidence for co-dominant inheritance of mutant alleles. J Thromb Haemost 2013;11:512–20.
22. James PD, Lillicrap D, Mannucci PM. Alloantibodies in von Willebrand disease. Blood 2013;122:636–40.
23. James AH, Jamison MG. Bleeding events and other complications during pregnancy and childbirth in women with von Willebrand disease. J Thromb Haemost 2007;5:1165–9.
24. Rydz N, James PD. The evolution and value of bleeding assessment tools. J Thromb Haemost 2012;2223–9.
25. Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost 2010;8:2063–5.
26. Elbatarny M, Mollah S, Grabell J, et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia 2014;20:831–5.
27. Deforest M, Grabell J, Alberta S et al. Generation and optimization of the self-administered bleeding assessment tool and its validation as a screening test for von Willebrand disease. Haemophilia 2015;21:e384-8.
28. Castaman G, Hillarp A, Goodeve A. Laboratory aspects of von Willebrand disease: test repertoire and options for activity assays and genetic analysis. Haemophilia 2014;20(Suppl. 4):65–70.
29. Favaloro EJ. Von Willebrand disease, type 2B: a diagnosis more elusive than previously thought. Thromb Haemost 2008;99:630–1.
30. Budde U. Diagnosis of von Willebrand disease subtypes: implications for treatment. Haemophilia 2008;14 Suppl 5:27–38.
31. Favaloro EJ. Von Willebrand factor collagen-binding (activity) assay in the diagnosis of von Willebrand disease: a 15-year journey. Sem Thromb Hemost 2002;28:191–202.
32. Patzke J, Budde U, Huber A, et al. Performance evaluation and multicenter study of a von Willebrand factor activity assay based on GPIb binding in the absence of ristocetin. Blood Coagul Fibrinolysis 2014;25:860-70.
33. Graf L, Moffat KA, Carlino SA, et al. Evaluation of an automated method for measuring von Willebrand factor activity in clinical samples without ristocetin. Int J Lab Hematol 2014;36:341–51.
34. Haberichter SL, Balistreri M, Christopherson P, et al. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood 2006;108:3344–51.
35. Keeney S, Bowen D, Cumming A, et al. The molecular analysis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors’ Organisation Haemophilia genetics laboratory network. Haemophilia 2008;14:1099–111.
36. Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ, Montgomery RR. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood 1987;69:1691–5.
37. Sadler JE, Rodeghiero F. Provisional criteria for the diagnosis of VWD type 1. J Thromb Haemost 2005;3:775–7.
38. Tosetto A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM- 1VWD). J Thromb Haemost 2006;4:766–73.
39. Vincentelli A, Susen S, Le Tourneau T, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med 2003;349:343–9.
40. Uriel N, Pak S-W, Jorde UP, et al. Acquired von Willebrand syndrome after continuous-flow mechanical device support contributes to a high prevalence of bleeding during long-term support and at the time of transplantation. J Am Coll Cardiol 2010;56:1207–13.
41. Federici AB, Rand JH, Bucciarelli P, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost 2000;84:345–9.
42. Favaloro EJ, Patterson D, Denholm A, et al. Differential identification of a rare form of platelet-type (pseudo-) von Willebrand disease (VWD) from type 2B VWD using a simplified ristocetin-induced-platelet-agglutination mixing assay and confirmed by genetic analysis. Brit J Haematol 2007;139:621–8.
43. Giannini S, Cecchetti L, Mezzasoma AM, Gresele P. Diagnosis of platelet-type von Willebrand disease by flow cytometry. Haematologica 2010;95:1021–4.
44. Farquhar C, Brown J. Oral contraceptive pill for heavy menstrual bleeding. Cochrane Database Syst Rev 2009 Oct 7;(4):CD000154.
45. Kadir R, Economides DL, Sabin C, et al. Variations in coagulation factors in women: effects of age, ethnicity, menstrual cycle and combined oral contraceptive. Thromb Haemost 1999;82:1456–61.
46. Muse K, Lukes AS, Gersten J, et al. Long-term evaluation of safety and health-related quality of life in women with heavy menstrual bleeding treated with oral tranexamic acid. Womens Health 2011;7:699–707.
47. Castaman G, Tosetto A, Federici AB, Rodeghiero F. Bleeding tendency and efficacy of anti-haemorrhagic treatments in patients with type 1 von Willebrand disease and increased von Willebrand factor clearance. Thromb Haemost 2011;105:647–54.
48. Mannucci PM, Bettega D, Cattaneo M. Patterns of development of tachyphylaxis in patients with haemophilia and von Willebrand disease after repeated doses of desmopressin (DDAVP). Brit J Haematol 1992;82:87–93.
49. Greaves M, Watson HG. Approach to the diagnosis and management of mild bleeding disorders. J Thromb Haemost 2007;5 Suppl 1:167–74.
50. Kessler CM, Friedman K, Schwartz BA, Gill JC, Powell JS. The pharmacokinetic diversity of two von Willebrand factor (VWF) / factor VIII (FVIII) concentrates in subjects with congenital von Willebrand disease. results from a prospective, randomised crossover study. Thromb Haemost 2011;106:279–88.
51. Weiss HJ, Sussman II, Hoyer LW. Stabilization of factor VIII in plasma by the von Willebrand factor. Studies on posttransfusion and dissociated factor VIII and in patients with von Willebrand’s disease. J Clin Invest 1977;60:390–404.
52. Berntorp E. Haemate P/Humate-P: a systematic review. Thromb Res 2009;124:S11–14.
53. Lillicrap D, Poon MC, Walker I, et al. Efficacy and safety of the factor VIII/von Willebrand factor concentrate, Haemate-P/Humate-P: ristocetin cofactor unit dosing in patients with von Willebrand disease. Thromb Haemost 2002;87:224–30.
54. Halimeh S, Krümpel A, Rott H, et al. Long-term secondary prophylaxis in children, adolescents and young adults with von Willebrand disease. results of a cohort study. Thromb Haemost 2011;105:597–604.
55. Abshire TC, Federici AB, Alvárez MT, et al. Prophylaxis in severe forms of von Willebrand’s disease: results from the von Willebrand disease prophylaxis network (VWD PN). Haemophilia 2013;19:76–81.
56. Abshire T, Cox-Gill J, Kempton CL, et al. Prophylaxis escalation in severe von Willebrand disease: a prospective study from the von Willebrand Disease Prophylaxis Network. J Thromb Haemost 2015;13:1585– 9.
57. Auerswald G, Kreuz W. Haemate P/Humate-P for the treatment of von Willebrand disease: considerations for use and clinical experience. Sem Thromb Hemost 2008;14 (Suppl 5):39–46.
58. Mannucci PM, Kempton C, Millar C, et al. Pharmacokinetics and safety of a novel recombinant human von Willebrand factor manufactured with a plasma-free method: a prospective clinical trial. Blood 2013;122:648–57.
59. Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood 2015;126:2038–46.
Pancreatic Adenocarcinoma: Update on Neoadjuvant and Adjuvant Treatment
Introduction
Exocrine pancreatic cancer refers to pancreatic adenocarcinomas that arise from ductal epithelial cells. Pancreatic ductal adenocarcinoma is a highly lethal malignancy, ranking as the fourth most common cause of cancer-related death in the United States1 and the eighth most common worldwide.2 In the United States, the pancreas is the second most common site of gastrointestinal malignancy after the colon.1 The only potentially curative modality for pancreatic adenocarcinomas is complete resection, followed by adjuvant therapy; unfortunately, only around 20% of patients are surgical candidates at the time of presentation due to delayed development of symptoms and consequently diagnosis.3 Most symptomatic patients with pancreatic cancer have locally advanced disease at diagnosis, and only a select group of patients with good performance status and borderline resectable disease can be offered neoadjuvant therapy. Adjuvant chemotherapy is typically recommended for patients who undergo potentially curative resection for pancreatic cancer.
Epidemiology
In the United States, pancreatic cancer has an annual estimated incidence of 55,440 new cases.1 It causes an estimated 44,330 deaths per year, with a 5-year overall survival (OS) rate of 8.2%.1 Worldwide an estimated 138,100 men and 127,900 women die of pancreatic cancer each year.2 In general, pancreatic cancers occur more commonly in persons living in Western/industrialized countries, older persons (age > 60 years), males (ratio 1.3:1 female), and African-Americans and native Hawaiians.4
Etiology
The major preventable environmental risk factor for pancreatic cancer is cigarette smoking, which accounts for 25% of all cases.5 A prospective study that estimated the excess incidence of pancreatic cancer among cigarette smokers and assessed the influence of smoking cessation on the risk for pancreatic cancer showed that persons who quit smoking reduced their risk of pancreatic cancer by 48% after 2 years of cessation, compared with smokers who did not quit, and reduced their risk to near the level of a never smoker after 10 years of cessation.5 Risk is higher for heavy smokers and those with homozygous deletions of the glutathione S-transferase theta 1 gene (GSTT1), which results in the absence of the carcinogen-metabolizing function of the glutathione S-transferase enzyme. High body mass index and sedentary lifestyle have been linked to pancreatic cancer.6 Data regarding aspirin, diet, coffee, and excess alcohol consumption are insufficient, inconclusive, and even conflicting, and thus the effect of these factors on risk for pancreatic cancer remains unclear. Infectious risk factors such as Helicobacter pylori and hepatitis B and C virus have weak associations with pancreatic cancer. Chronic pancreatitis and pancreatic cysts (eg, intraductal papillary mucinous neoplasm [IPMN] of the pancreas) carry a risk for malignant transformation, and hence may require surveillance. Multiple epidemiologic studies have shown a strong association between pancreatic cancer and recently diagnosed diabetes mellitus (relative risk [RR] 1.97 [95% confidence interval {CI} 1.78 to 2.18]); the presence of diabetes also may be a long-term predisposing factor for pancreatic cancer, and cancer screening needs to be considered for selected patients.7
A predisposing genetic anomaly accounts for 15% of all cases of pancreatic cancer.8 Hereditary risk factors are divided into 2 broad categories: defined genetic syndromes and familial pancreatic cancer. Familial predispositions that do not meet genetic syndrome criteria account for approximately 5% to 10% of all cases associated with hereditary factors; in one study, 29% of tested kindreds with an incident pancreatic cancer had a germline BRCA2 mutation.9 Other predisposing genetic syndromes that have been linked to pancreatic cancer include:
- Peutz-Jeghers syndrome with germline STK11 mutations (RR 132);
- Hereditary pancreatitis with germline PRSS1, SPINK1, and CFTR mutations (RR 26–87);
- Familial atypical multiple mole melanoma syndrome with CDKN2A mutations (RR 20–40);
- Familial breast and ovarian cancer with BRCA2 (RR 10) and BRCA1 (RR 2.8) mutations;
- Hereditary nonpolyposis colorectal cancer (HNPCC, Lynch II syndrome) with MLH1, MSH2, MSH6, and PMS2 mutations (RR 9–11); and
- Familial adenomatous polyposis with APC mutations (RR 5).10
Other gene mutations with unknown relative risk for pancreatic cancer include mutations affecting PALB2, ATM, and TP53.
The International Cancer of the Pancreas Screening consortium consensus on screening for pancreatic cancer in patients with increased risk for familial pancreatic cancer recommends screening those at high risk: first-degree relatives (FDRs) of patients with pancreatic cancer from a familial pancreatic kindred with at least 2 affected FDRs; patients with Peutz-Jeghers syndrome; and p16, BRCA2, and HNPCC mutation carriers with 1 or more affected FDRs and hereditary pancreatitis. The guidelines emphasize that screening should be done only in those who are surgical candidates and are evaluated at an experienced multidisciplinary center.8
Deleterious germline mutations in pancreatic cancer can account for 33% of patients with apparent sporadic cancers and no hereditary risk. These include germline mutations affecting BRCA1/2, PALB2, ATM, MLH1, CHK-2, CDKN2A, and TP53.11
Pathogenesis
Pancreatic neoplasms can be benign or malignant and thus a tissue histologic diagnosis is paramount. Pancreatic adenocarcinomas with exocrine features represent more than 95% of all pancreatic neoplasms, with only 5% arising from the endocrine pancreas (ie, neuroendocrine tumors). Pancreatic neuroendocrine tumors and pancreatic adenocarcinoma must be distinguished histologically because treatment of the 2 neoplasms is completely different. Other malignant pancreatic tumors are signet ring cell carcinoma, adenosquamous carcinoma, undifferentiated (anaplastic) carcinoma, and mucinous noncystic (colloid) carcinoma; the latter tumor has a better prognosis.12 It is essential to characterize and distinguish among benign cystic neoplasms, as some require surgical resection due to the risk of malignant transformation. IPMN, pancreatic intraepithelial neoplasia, and mucinous cystic neoplasms are thought to be premalignant lesions of invasive ductal adenocarcinomas, and the pathological report should highlight the degree of dysplasia for adequate risk stratification.13 This information could be the deciding factor in whether a pancreatectomy is recommended by a multidisciplinary team.
Most pancreatic cancers harbor activating or silencing genetic mutations, and multiple combinations of altered genes can be detected by next-generation sequencing (average of 63 genetic alterations per cancer).14 Mutational activated KRAS is the most frequent (> 90%) genetic alteration in pancreatic cancer, even in early neoplastic precursors (IPMN > 75%). KRAS is a highly complex, dynamic proto-oncogene involved in signaling of various receptor kinases such as the epidermal growth factor receptor and the insulin-like growth factor receptor-I. It also engages in canonical downstream effector pathways, mainly Raf/MEK/ERK, PI3K/PDK1/Akt, and the Ral guanine nucleotide exchange factor pathway, which drive much of the pathogenesis of malignancy. These pathways lead to sustained proliferation, metabolic reprogramming, anti-apoptosis, remodeling of the tumor microenvironment, evasion of the immune response, cell migration, and metastasis. An activating point mutation in codon G12 is the most common (98%) locus of KRAS mutation in pancreatic adenocarcinoma, but all drugs targeting this mutation have failed in clinical practice.15 Additionally, inactivation of tumor suppressor genes such as p53, DPC4 (SMAD4/MADH4), CDKN2A (p16/MTS1), and BRCA2 can be found in 75%, 30%, 35%, and 4% of pancreatic adenocarcinoma cases, respectively.14 Another pancreatic cancer hallmark is inactivation of DNA damage repair genes, which include MLH1 and MSH2.16
Diagnosis and Staging
Case Presentation
A 71-year-old male veteran with no significant past medical history other than hypertension and hyperlipidemia and an excellent performance status presents to the emergency department after noticing a yellowish skin and sclera color. He denies weight loss, abdominal pain, or any other pertinent symptom or sign. Physical examination reveals a healthy developed man with yellowish discoloration of the skin and sclera and a soft, nontender benign abdomen; physical examination is otherwise unremarkable. Laboratory evaluation reveals a direct bilirubin level of 4.5 mg/dL and normal values for complete blood count and renal, liver, and coagulation panels. Abdominal and pelvis computed tomography (CT) with intravenous contrast shows a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein, which remains patent. Follow-up endoscopic ultrasound (EUS) confirms an irregular mass at the head of the pancreas measuring 3.2 × 2.6 cm with sonographic evidence suggesting invasion into the portal vein. During the procedure, the bile duct is successfully stented, the mass is biopsied, and bile duct brushing is performed. Pathology report is consistent with pancreatic adenocarcinoma.
- What is the typical presentation of pancreatic cancer?
The most common symptoms of pancreatic cancer at the time of presentation include weight loss (85%), asthenia/anorexia (86%), and/or abdominal pain (79%).17 The most frequent signs are jaundice (55%), hepatomegaly (39%), and cachexia (13%). Courvoisier sign, a nontender but palpable distended gallbladder at the right costal margin, is neither sensitive nor specific for pancreatic cancer (13% of cases). Trousseau syndrome, a superficial thrombophlebitis, is another classic sign that reflects the hypercoagulable nature of pancreatic cancer (3% of cases).17 The pathophysiology of this syndrome is not completely understood, but it may occur secondary to the release of cancer microparticles in the blood stream which in turn stimulate the coagulation cascade. Other nonspecific symptoms are dark urine, nausea, vomiting, diarrhea, steatorrhea, and epigastric and back pain. Because symptoms early in the course of the disease are nonspecific, pancreatic cancer is typically diagnosed late, after the cancer has invaded local structures or metastasized. The initial presentation varies depending on tumor location, with 70% of pancreatic head malignancies presenting with jaundice and pain correlating to an advanced stage.18 Although data supporting an association between new-onset diabetes mellitus and pancreatic cancer are inconclusive, pancreatic cancer should still be a consideration in patients with new-onset diabetes mellitus and other symptoms such as pain and weight loss. Early signs of incurable disease include a palpable mass, ascites, lymphadenopathy (classic Virchow node), and an umbilical mass (Sister Mary Joseph node). Incidentally discovered pancreatic masses on imaging are rare, but the incidence is increasing due to frequent imaging for other reasons and improved diagnostic techniques.
- What is the approach to diagnosis and staging?
History and physical examination findings are not sufficiently sensitive or specific to diagnose pancreatic cancer. High clinical suspicion in a patient with risk factors can lead to a comprehensive evaluation and potential early diagnosis. In general, an initial diagnostic work-up for suspected pancreatic cancer will include serologic evaluation (liver function test, lipase, tumor markers) and abdominal imaging (ultrasound, CT scans, or magnetic resonance imaging [MRI]). Ultrasound is a first-line diagnostic tool with a sensitivity of 90% and specificity of 98.8% for pancreatic cancer, but it is investigator-dependent and is less accurate in detecting tumors smaller than 3 cm in diameter.19 Multiphasic helical CT of the abdomen has better sensitivity (100%) and specificity (100%) for detecting tumors larger than 2 cm, but this modality is less accurate in detecting pancreatic masses smaller than 2 cm (77%).20 Percutaneous fine-needle aspiration (FNA) performed by ultrasound or CT guidance is avoided due to theoretical concerns about intraperitoneal seeding and bleeding.
If a pancreatic mass is detected by ultrasound or CT, additional interventions may be indicated depending on the clinical scenario. EUS-guided biopsy can provide histological confirmation and is currently utilized frequently for diagnosis and early resectability staging. Endoscopic retrograde cholangiopancreatography (ERCP) is indicated for patients with biliary obstruction requiring stent placement, and this procedure may provide tissue confirmation by forceps biopsy or brush cytology (lower accuracy than EUS). In a meta-analysis that evaluated the diagnostic value of tests for pancreatic cancer, ERCP had the highest sensitivity (92%) and specificity (96%) compared to ultrasound and CT,21 but this modality carries a risk for pancreatitis, bleeding, and cholangitis. Magnetic resonance cholangiopancreatography has not replaced ERCP, but it but may be an alternative for patients who cannot undergo ERCP (eg, gastric outlet obstruction, duodenal stenosis, anatomical surgical disruption, unsuccessful ERCP). ERCP is used frequently because many patients present with obstructive jaundice due to pancreatic mass compression, specifically if the mass is located in the head, and must undergo ERCP and stenting of the common bile duct.
The carbohydrate antigen (CA) 19-9 level has variable sensitivity and specificity in pancreatic cancer, as levels can be elevated in many benign pancreaticobiliary disorders. Elevated CA 19-9, in the appropriate clinical scenario (ie, a suspicious pancreatic mass and a value greater than 37 U/mL) demonstrated a sensitivity of 77% and specificity of 87% when differentiating pancreaticobiliary cancer from benign clinical conditions such as acute cholangitis or cholestasis.22 CA 19-9 level has prognostic value, as it may predict occult disease and correlates with survival rates, but no specific cutoff value has been established to guide perioperative therapy for high-risk resectable tumors.23
The American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) tumor, node, metastasis (TNM) system is the preferred method for staging pancreatic cancer (Table 1). 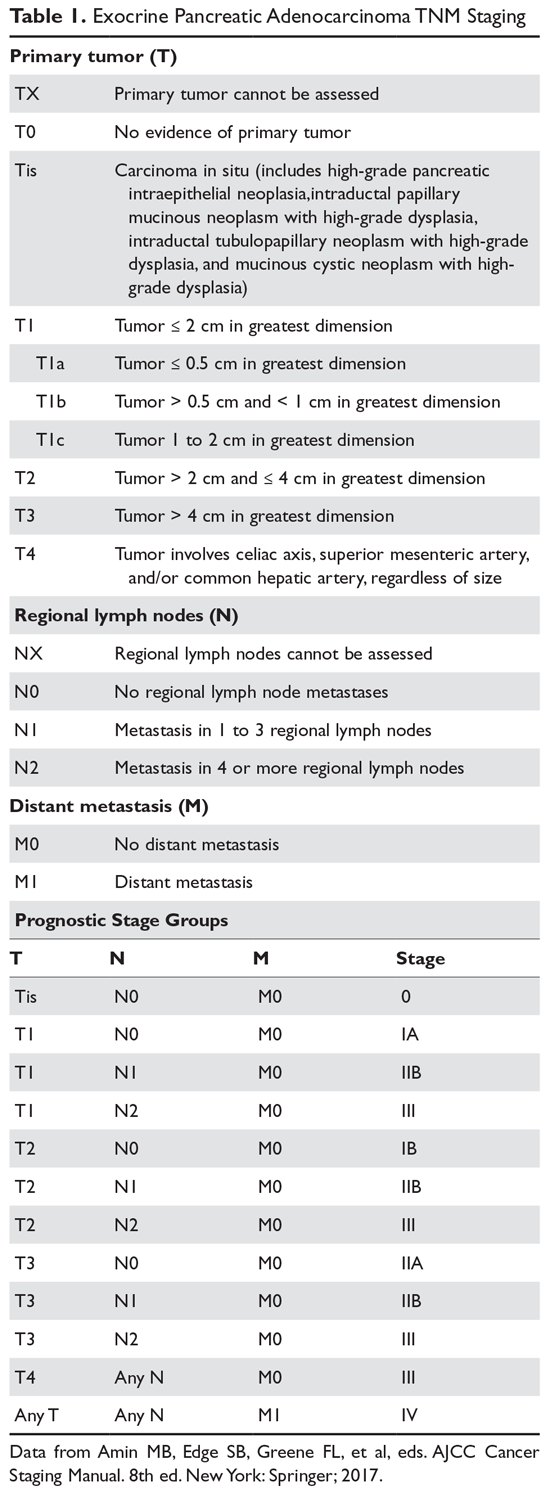

Positron emission tomography with CT scan is occasionally utilized in practice to assess tumor burden by evaluating anatomical structures and assessing physiologic uptake, which aids in establishing the extent of disease in equivocal cases. Staging laparoscopy with or without peritoneal biopsy is sometimes used to establish appropriate staging in cases that are questionable for occult metastatic disease. This procedure helps avoid unnecessary morbid surgeries.
Neoadjuvant Therapy
Case Continued
The patient is referred to oncology. Blood work reveals a CA 19-9 level of 100 U/mL (reference range < 35 U/mL) and a staging CT scan of the chest reveals a benign-appearing 3-mm nodule (no prior imaging for comparison). CT scan of the abdomen and pelvis does not define venous vasculature involvement appropriately and hence MRI of the abdomen and pelvis is performed. MRI reveals a pancreatic head mass measuring 3.0 × 2.7 cm, without arterial or venous vasculature invasion. However, the mass is abutting the portal vein and superior mesenteric vein and there is a new nonspecific 8-mm aortocaval lymph node.
- What are the current approaches to treating patients with resectable, unresectable, and metastatic disease?
Accurate staging and assessment of surgical resectability in pancreatic cancer are paramount as these steps prevent a futile morbid Whipple procedure in patients with advanced disease and a high risk of recurrence. Conversely, it allows patients with low-volume disease to undergo a potentially curative surgery. Approximately 20% of patients present with resectable disease, 40% present with locally advanced unresectable tumors (eg, involvement of critical vascular structures), and 40% present with metastatic disease.3 Treatment for resectable pancreatic cancer continues to be upfront surgery, although neoadjuvant therapy with either chemoradiation, radiation alone, or chemotherapy is an option per guidelines from the American Society of Clinical Oncology (ASCO),28 the NCCN,26 and the European Society for Medical Oncology (ESMO),29 particularly for patients with borderline resectable tumors (Table 3). 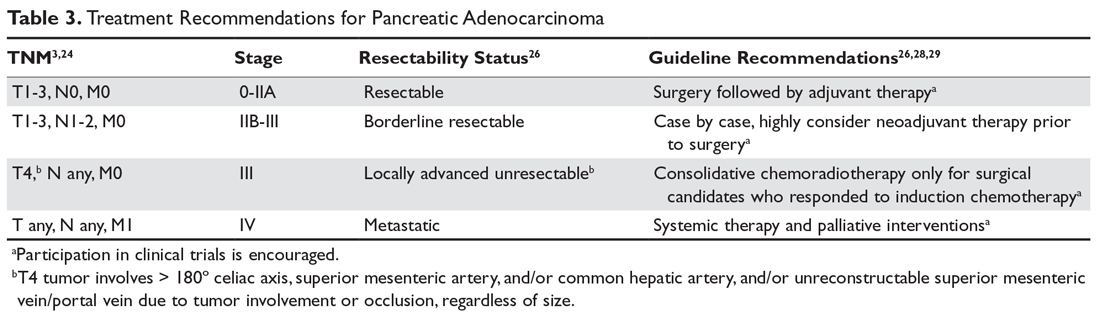
Systemic chemotherapy is recommended for fit candidates with locally advanced unresectable or metastatic disease, with an emphasis on supportive palliative measures. Palliative interventions include biliary stenting, duodenal stent for relieving gastric-outlet obstruction, and celiac axis nerve blocks, when indicated. Routine preoperative biliary stent placement/drainage in patients undergoing subsequent surgery for pancreatic cancer located in the head is associated with an increased risk of surgical complications when compared with up-front surgery without prior biliary drainage, and thus stent placement/drainage is not recommended.26 Aggressive supportive management of symptoms, such as cancer-associated pain, anorexia-cachexia syndromes, and anxiety-depression disorders, should remain a primary palliative focus.
Case Continued
A multidisciplinary tumor board discusses the patient’s case and deems the cancer borderline resectable; neoadjuvant therapy is recommended. The patient is started on treatment with gemcitabine and nab-paclitaxel as first-line neoadjuvant therapy. After 4 cycles, the CA 19-9 level drops to 14 U/mL, and MRI reveals a smaller head mass of 1.3 × 1.4 cm with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remains stable. At tumor board, it is evident that the patient has responded to therapy and the recommendation is to treat with gemcitabine chemoradiotherapy before surgery.
- What neoadjuvant therapy strategies are used in the treatment of pancreatic adenocarcinoma?
There are no established evidence-based recommendations for neoadjuvant therapy in patients with borderline resectable pancreatic cancer or patients with unresectable locally advanced pancreatic cancer. However, there are ongoing trials to investigate this treatment approach, and it is offered off-label in specific clinical scenarios, such as in the case patient described here. In patients with borderline resectable disease, preoperative chemotherapy followed by chemoradiation is a routine practice in most cancer centers,32 and ongoing clinical trials are an option for this cohort of patients (eg, Southwest Oncology Group Trial 1505, NCT02562716). The definitions of borderline resectable and unresectable pancreatic cancer have been described by the NCCN,26 although most surgeons consider involvement of the major upper abdominal blood vessels the main unresectability criterion; oncologists also consider other parameters such as suspicious lesions on scans, worsening performance status, and a significantly elevated CA 19-9 level suggestive of disseminated disease.28 The goal of a conversion approach by chemotherapy with or without radiation for borderline and unresectable cancers is to deliver a tolerable regimen leading to tumor downstaging, allowing for surgical resection. No randomized clinical trial has shown a survival advantage of this approach. Enrollment in clinical trials is preferred for patients with borderline and unresectable cancer, and there are trials that are currently enrolling patients.
The main treatment strategies for patients with locally advanced borderline and unresectable pancreatic cancer outside of a clinical trial are primary radiotherapy, systemic chemotherapy, and chemoradiation therapy. Guidelines from ASCO, NCCN, and ESMO recommend induction chemotherapy followed by restaging and consolidation chemoradiotherapy in the absence of progression.26,28,29 There is no standard chemoradiation regimen and the role of chemotherapy sensitizers, including fluorouracil, gemcitabine, and capecitabine (an oral fluoropyrimidine substitute), and targeted agents in combination with different radiation modalities is now being investigated.
Fluorouracil is a radio-sensitizer that has been used in locally advanced pancreatic cancer based on experience in other gastrointestinal malignancies; data shows conflicting results with this drug. Capecitabine and tegafur/gimeracil/oteracil (S-1) are oral prodrugs that can safely replace infusional fluorouracil. Gemcitabine, a more potent radiation sensitizer, is very toxic, even at low-doses twice weekly, and does not provide a survival benefit, as demonstrated in the Cancer and Leukemia Group B (CALGB) 89805 trial, a phase 2 study of patients with surgically staged locally advanced pancreatic cancer.33 Gemcitabine-based chemoradiotherapy was also evaluated in the Eastern Cooperative Group (ECOG) E4201 trial, which randomly assigned patients to receive gemcitabine alone (at 1000 mg/m2/wk for weeks 1 through 6, followed by 1 week rest, then weekly for 3 out of 4 weeks) or gemcitabine (600 mg/m2/wk for weeks 1 to 5, then 4 weeks later 1000 mg/m2 for 3 out of 4 weeks) plus radiotherapy (starting on day 1, 1.8 Gy/fraction for total of 50.4 Gy).34 Patients with locally advanced unresectable pancreatic cancer had a better OS outcome with gemcitabine in combination with radiation therapy (11.1 months) as compared with patients who received gemcitabine alone (9.2 months). Although there was a greater incidence of grade 4 and 5 treatment-related toxicities in the combination arm, no statistical differences in quality-of-life measurements were reported. Gemcitabine-based and capecitabine-based chemoradiotherapy were compared in the open-label phase 2 multicenter randomized SCALOP trial.35 Patients with locally advanced pancreatic cancer were assigned to receive 3 cycles of induction with gemcitabine 1000 mg/m2 days 1, 8, and 15 and capecitabine 830 mg/m2 days 1 to 21 every 28 days; patients who had stable or responding disease were randomly assigned to receive a fourth cycle followed by capecitabine (830 mg/m2 twice daily on weekdays only) or gemcitabine (300 mg/m2 weekly) with radiation (50.4 Gy over 28 fractions). Patients treated with capecitabine-based chemoradiotherapy had higher nonsignificant median OS (17.6 months) and median progression-free survival (12 months) compared to those treated with gemcitabine (14.6 months and 10.4 months, respectively).
The benefit of radiation therapy in the treatment of locally advanced pancreatic cancer was further explored by the Fédération Francophone de Cancérologie Digestive 2000-01 phase 3 trial. This study compared induction chemoradiotherapy (60 Gy, 2 Gy/fraction; concomitant fluorouracil infusion, 300 mg/m2/day, days 1–5 for 6 weeks; cisplatin, 20 mg/m2/day, days 1–5 during weeks 1 and 5) to gemcitabine alone (1000 mg/m2 weekly for 7 weeks) followed by maintenance gemcitabine in both arms.36 Unexpectedly, the median OS was significantly shorter in the chemoradiotherapy arm than in the chemotherapy alone arm (8.6 months versus 13 months, respectively, P = 0.03) and the combination arm had more toxicities. The phase 3 open-label LAP07 study explored the role of radiation therapy in patients with locally advanced pancreatic cancer who had controlled disease after 4 months of induction therapy.37 LAP07 had 2 randomizations: first, patients with locally advanced pancreatic cancer were assigned to receive weekly gemcitabine alone (1000 mg/m2) or this same dose of gemcitabine plus erlotinib 100 mg/day; second, patients with progression-free disease (61% of initial cohort) after 4 months of therapy were assigned to receive 2 months of the same chemotherapy or chemoradiotherapy (54 Gy plus capecitabine). This study showed that the addition of erlotinib to gemcitabine did not improve survival and in fact affected survival adversely. Of note, no survival benefit was observed after the first randomization from chemotherapy to consolidating chemoradiotherapy. Chemoradiotherapy achieved better locoregional tumor control with significantly less local tumor progression (32% versus 46%, P < 0.03) and no increase in toxicity. Based on prior moderate-quality evidence, guidelines recommend consolidative chemoradiotherapy only for surgical resection candidates following induction chemotherapy; for those who are not surgical candidates, guidelines recommend continuing systemic therapy.26,28,29
Gemcitabine and fluorouracil-based chemotherapies were the standard induction regimens until evidence from studies of metastatic systemic treatment protocols with FOLFIRINOX (ACCORD trial38) and nanoparticle albumin-bound paclitaxel (nab-paclitaxel) plus gemcitabine (MPACT trial39) was extrapolated to clinical practice. These regimens were shown to achieve higher objective response rates when compared to single-agent gemcitabine in patients with metastatic pancreatic cancer. Due to the broad heterogeneity of results in small retrospective series with neoadjuvant trials in borderline resectable pancreatic cancer, the quality of the evidence is low and any recommendation is limited. Many individual series have demonstrated improved complete resection rates and promising survival rates. In the largest single-institution retrospective review of patients with borderline resectable pancreatic adenocarcinoma who completed neoadjuvant gemcitabine-based chemoradiotherapy (50 Gy in 28 fractions or 30 Gy in 10 fractions), 94% achieved a margin-negative pancreatectomy; the median OS in those who completed preoperative therapy and had surgery was 40 months, with a 5-year OS of 36%.40 A meta-analysis by Andriulli and colleagues included 20 prospective studies of patients with initially resectable (366 lesions) or unresectable (341 lesions) disease who were treated with neoadjuvant/preoperative gemcitabine with or without radiotherapy.41 In the group with initially unresectable disease, 39% underwent surgery after restaging and 68% of explored patients were resected; the R0 resection rate was 60%. After restaging, 91% of patients with resectable disease underwent surgery, with 82% of explored patients undergoing surgical resection and 89% of these achieving R0 resection. The estimated 1- and 2-year survival probabilities after resection among patients with initially unresectable disease were 86.3% and 54.2%.41
The largest single-institution retrospective review of FOLFIRINOX (fluorouracil, oxaliplatin, irinotecan, and leucovorin), an alternative to gemcitabine, for neoadjuvant induction therapy for patients with locally advanced unresectable disease was conducted at Memorial Sloan Kettering Cancer Center. In this study (n = 101), 31% of patients initially deemed unresectable who completed FOLFIRINOX induction therapy with or without chemoradiation underwent resection. The R0 resection rate in these patients was 55%, and patients who did not progress during induction FOLFIRINOX therapy had a median OS of 26 months.42 A systematic review and meta-analysis of FOLFIRINOX chemotherapy with or without radiotherapy in patients with locally advanced unresectable pancreatic cancer reported that 25.9% of patients underwent resection after FOLFIRINOX therapy, and the R0 resection rate in these patients was 78.4%.43 The median OS in this study was 24.2 months, which was longer than the previously reported median OS rates for gemcitabine.
There is no strong evidence published for the use of combination nab-paclitaxel plus gemcitabine in the neoadjuvant setting, but it is used in clinical practice based on evidence from the MPACT trial, which showed the combination improved OS and progression-free survival in patients with metastatic pancreatic cancer.39 An early-phase 1-arm clinical trial of neoadjuvant gemcitabine, docetaxel, and capecitabine (GTX) followed by radiotherapy showed an increased response rate and survival for locally advanced disease; however, the NCCN expert panel has reached a consensus but not a uniform recommendation regarding this regimen due to significant toxicities and low patient accrual.26 Selected patients with pancreatic cancer with BRCA1/2 mutations are more sensitive to platinum-based chemotherapy. Although studies of neoadjuvant platinum-based chemotherapy in this population have not been reported, the NCCN guidelines list it as an alternative option based on extrapolated data.26 A clinical trial of gemcitabine, nab-paclitaxel, and cisplatin in the neoadjuvant setting in patients with resectable pancreatic cancer is currently enrolling patients (NGC triple regimen NCT0339257).
Summary
Chemotherapy alone or followed by chemoradiotherapy may be used as initial treatment for patients with borderline and unresectable pancreatic adenocarcinoma without distant metastases who are potential surgical candidates. Chemoradiotherapy remains a preferred treatment option for patients with poorly controlled pain from local tumor invasion, in view of the well-documented analgesic palliative effect of radiation therapy. FOLFIRINOX with or without radiation therapy may offer the highest documented response rates, but it also results in higher rates of treatment-related toxicities. FOLFIRINOX can be offered to selected fit patients (< 65 years old, no comorbidity contraindication, good functional status [ECOG 0–1]) who can tolerate triple therapy with a more toxic adverse-effect profile. A clinical trial evaluating neoadjuvant FOLFIRINOX with or without preoperative chemoradiotherapy in patients with borderline resectable pancreatic cancer is ongoing (PANDAS-PRODIGE 44, NCT02676349). Gemcitabine with or without radiation therapy is a tolerable combination, although it is potentially more toxic when combined with radiation. The addition of nab-paclitaxel to gemcitabine without radiation may emerge as a preferred neoadjuvant treatment for selected patients; a clinical trial investigating this modality in patients with resectable and borderline resectable disease is ongoing (NCT02723331).
Adjuvant Therapy
Case Continued
Prior to the planned surgical resection and after undergoing chemoradiation therapy, the patient has an excellent performance status and repeat MRI shows a 1.3 × 1.4 cm head mass with no further vasculature involvement, no evidence of lymphadenopathy, and no distant metastasis. The CA 19-9 level is stable at 18 U/mL. The patient undergoes an uncomplicated partial pancreaticoduodenectomy, and analysis of a surgical pathology specimen reveals T3N0 disease with closest margin of 0.1 cm.
- Would the patient benefit from adjuvant therapy?
Adjuvant chemotherapy for 6 months after pancreatic cancer resection should be offered to all patients based on mature data. Gemcitabine and capecitabine are the current standard of care in adjuvant therapy; alternatively, single-agent gemcitabine can be offered to patients with poor performance status or patients who cannot tolerate the toxicities associated with this combination.28 Adjuvant treatment should be initiated within approximately 8 weeks of surgical resection. The value of radiation therapy remains controversial, but it can be offered within the context of a clinical trial or to patients with positive margins after surgical resection and/or lymph node–positive disease. Based on low-quality supportive evidence, it is strongly recommended that patients who receive neoadjuvant therapy complete a total of 6 months of chemotherapy, factoring in the duration of the preoperative regimen.28 Different adjuvant strategies have been investigated, including chemotherapy alone with a fluoropyrimidine and/or gemcitabine with or without combined chemoradiation therapy.
The European Study Group for Pancreatic Cancer 1 (ESPAC)-1 trial was a randomized clinical trial that evaluated several adjuvant strategies in pancreatic cancer treatment. This trial assigned patients who underwent pancreatic adenocarcinoma resection to adjuvant chemotherapy alone (intravenous fluorouracil 425 mg/m2 and leucovorin 20 mg/m2 daily for 5 days, monthly for 6 months), chemoradiotherapy (20 Gy in 10 daily fractions over 2 weeks with 500 mg/m2 intravenous fluorouracil on days 1–3, repeated after 2 weeks), both chemotherapy and chemoradiation, and observation.44 The results showed no added benefit for adjuvant chemoradiotherapy, with a median OS of 15.5 months in the chemoradiotherapy cohort, as compared to a median OS of 16.1 months in the chemotherapy-alone cohort (hazard ratio [HR] 1.18 [95% CI 0.90 to 1.55], P = 0.24). In addition, there was evidence of a survival benefit for the chemotherapy-alone arm when compared to the combined modality arm, with a median OS of 19.7 versus 14.0 months, respectively (HR 0.66 [95% CI 0.52 to 0.83], P = 0.0005). Although ESPAC-1 has been criticized for being underpowered to perform statistical comparison, it is still considered a landmark trial demonstrating benefit with single-agent chemotherapy alone. A follow-up analysis of ESPAC-1 showed that adjuvant chemotherapy alone conferred a significant 5-year survival benefit while the combined modality had a deleterious effect on survival. 45 Hence, adjuvant chemotherapy alone became the standard of care in the United States following resection.
The results of the multicenter randomized controlled phase 3 CONKO-001 (CharitéOnkologie 001) trial, which were reported in 2007, supported the use of adjuvant gemcitabine for 6 months in patients with resected pancreatic adenocarcinoma. In this study, patients treated with adjuvant gemcitabine (1000 mg/m2 days 1, 8, and 15 every 4 weeks for 6 months) had superior disease-free survival compared with those who received surgery alone.30 A long-term outcome update of this study demonstrated a significant improvement in 5-year OS for patients treated with adjuvant gemcitabine (20.7% [95% CI 14.7% to 26.6%]) compared to those who received surgical resection alone (10.4% [95% CI 5.9% to 15.0%]). This benefit persisted at 10-year follow-up, with an OS of 12.2% (95% CI 7.3% to 17.2%) in the adjuvant gemcitabine group, as compared to 7.7% (95% CI 3.6% to 11.8%) in the resection alone group.31
Fluorouracil and gemcitabine remained equivalent adjuvant treatment options until the results of the ESPAC-3 trial were reported in 2010.32 This large phase 3 trial, conducted mainly in the United Kingdom, compared weekly gemcitabine (1000 mg/m2 weekly for 3 of every 4 weeks) to leucovorin-modulated fluorouracil (Mayo Clinic regimen: leucovorin 20 mg/m2 followed by fluorouracil 425 mg/m2 intravenous bolus days 1 through 5 every 28 days) as adjuvant therapy in resected pancreatic adenocarcinoma. After a median follow-up of 34.2 months, the median OS was similar in the 2 groups (fluorouracil/leucovorin 23.0 months versus gemcitabine 23.6 months; P = 0.39). However, the fluorouracil/leucovorin group experienced more grade 3/4 treatment-related toxicities (mucositis, stomatitis, diarrhea, and hosptializations; 14% versus 7.5%; P < 0.001).46 Following this trial, gemcitabine became the standard of care for adjuvant chemotherapy for resected pancreatic cancer.
The U.S. Radiation Therapy Oncology Group (RTOG) 9704 trial was conducted to investigate the potential benefit of adding radiation therapy to gemcitabine. This trial demonstrated an improved trend among patients with pancreatic head tumors (but not with cancers of the pancreatic body or tail) who received adjuvant gemcitabine followed by chemoradiotherapy (50.4 Gy in 1.8 Gy daily fractions for 5.5 weeks with concurrent infusional fluorouracil 250 mg/m2 daily) and subsequent gemcitabine monotherapy compared to postoperative fluorouracil-based chemoradiotherapy. Results showed a 5-year OS of 22% versus 18%, respectively, although this improvement was not statistically significant (P = 0.08). This trial failed to show a benefit of adding radiotherapy to gemcitabine.47
The ESPAC-4 trial, reported in 2017, evaluated the combination of gemcitabine and capecitabine compared to gemcitabine alone as adjuvant therapy for resected pancreatic adenocarcinoma.48 Patients were randomly assigned after surgical resection, regardless of margin or node status, to 6 months of gemcitabine alone (1000 mg/m2/day on days 1, 8, and 15 of each 28-day cycle) or gemcitabine plus capecitabine (1660 mg/m2/day on days 1 through 21 of each 28-day cycle). Combination therapy had a significant survival benefit compared to single therapy, with median OS durations of 28 months and 25.5 months, respectively (HR for death 0.82 [95% CI 0.68 to 0.98]). The 5-year OS for patients who received combination treatment was 29 months (95% CI 22.9 to 35.2) versus 16 months (95% CI 10.2 to 23.7) for those in the monotherapy group. As expected, grade 3 or 4 treatment-related toxicities (diarrhea, hand-foot syndrome, and neutropenia) were significantly more common with combined therapy, although there were no significant differences in the rates of serious adverse events. The adjuvant combination of gemcitabine and capecitabine became the current and preferred new standard of care following resection of pancreatic ductal adenocarcinoma,28 but single-agent gemcitabine and fluorouracil/leucovorin continue to be viable options,26,28,29 particularly for elderly patients, patients with borderline performance status, or patients with multiple comorbidities.
Evidence showing that a more intensive regimen can improve outcome in the adjuvant setting remains elusive. The phase 3 APACT study (Adjuvant Therapy for Patients with Resected Pancreatic Cancer, NCT01964430) comparing gemcitabine alone to gemcitabine plus nab-paclitaxel in patients with surgically resected pancreatic adenocarcinoma has concluded, with the results projected to be released in 2018. Another phase 3 trial investigating the efficacy of FOLFIRINOX versus gemcitabine alone as adjuvant therapy is underway in France and Canada (PRODIGE24/ACCORD24, NCT01526135). Other strategies with newer targeted therapies and immunotherapy are in the development phase.
Follow-Up and Surveillance
Case Conclusion
After recovery from surgery, the patient is offered and completes 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine. He is started on surveillance at 3 and 6 months, and he maintains an excellent performance status. He develops clinical evidence of pancreatic enzyme insufficiency and is placed on oral replacement therapy. He has no other complaints, and there is no evidence of recurrence on MRI and CA 19-9 levels.
- What is the recommended duration of surveillance following curative resection?
Surveillance after curative resection of pancreatic adenocarcinoma is recommended by NCCN guidelines.26 However, pancreatic adenocarcinoma has a poor prognosis, and surveillance after curative surgical resection with or without perioperative therapy has not been shown to impact survival. Most recurrences will occur within 2 years after treatment. Surveillance recommendations differ among expert groups.26,28,29 NCCN guidelines recommend evaluating patients by history and physical examination every 3 to 6 months for the first 2 years, then every 6 to 12 months for 3 years. CA 19-9 level and CT scan should be obtained every 3 to 6 months for 2 years and then every 6 to 12 months for 3 years. Follow-up with CA 19-9 levels and CT scans after 5 years is not routinely performed unless guided by signs, symptoms, or laboratory findings that raise suspicion for recurrence. Follow-up visits should also include evaluation of treatment-related toxicities, symptom management, nutrition support of pancreatic insufficiency, and psychosocial support.
Conclusion
Pancreatic cancer is a leading cause of cancer-related death that frequently presents with locally advanced or metastatic disease due to nonspecific symptoms and lack of a screening modality. Histological tissue biopsy confirmation and accurate resectability staging guide treatment planning and prognosis. The only potentially curative therapy is surgical resection plus adjuvant therapy for those with resectable disease. Surgical candidates with borderline resectable and unresectable disease can be offered induction preoperative chemotherapy followed by consolidation chemoradiation, based on clinical consensus practice. Enrollment in clinical trials should be encouraged for all patients, as evidence from clinical trials is essential to making progress in pancreatic cancer treatment.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
2. Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69.
3. Kamarajah SK, Burns WR, Frankel TL, et al. Validation of the American Joint Commission on Cancer (AJCC) 8th edition staging system for patients with pancreatic adenocarcinoma: a Surveillance, Epidemiology and End Results (SEER) analysis. Ann Surg Oncol 2017;24:2023–30.
4. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer.gov/statfacts/html/pancreas.html. Accessed 17 February 2018.
5. Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med 1996;156:2255–60.
6. Michaud DS, Giovannucci E, Willett WC, et al. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001;286:921–9.
7. Batabyal P, Vander Hoorn S, Christophi C, Nikfarjam M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann Surg Oncol 2014;21:2453–62. Epub 2014 Mar 9.
8. Canto MI, Harinck F, Hruban RH, et al, on behalf of the International Cancer of the Pancreas Screening (CAPS) Consortium. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013;62:339–47. Epub 2012 Nov 7.
9. Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res 2004;64:2634–8.
10. McKay SH,Humphris JL, Johns AL, et al. Inherited pancreatic cancer. Cancer Forum 2016;40(1).
11. Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol 2017;35:3382–90.
12. Hruban RH, Pitman MB, Klimstra DS. Tumors of the pancreas. AFIP Atlas of Tumor Pathology. 4th series, fascicle 6. Washington, DC: Armed Forces Institute of Pathology; 2007.
13. Vege SS, Ziring B, Jain R, Moayyedi P, Clinical Guidelines Committee, American Gastroenterology Association. American gastroenterological association institute guideline on the diagnosis and management of asymptomatic neoplastic pancreatic cysts. Gastroenterology 2015;148:819–22.
14. Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501.
15. Choi M, Bien H, Mofunanya A, Powers S. Challenges in Ras therapeutics in pancreatic cancer. Semin Cancer Biol 2017 Nov 21. pii: S1044-579X(17)30235-3.
16. Humphris JL, Patch AM, Nones K, et al. Hypermutation in pancreatic cancer. Gastroenterology 2017;152:68. Epub 2016 Nov 15.
17. Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol 2005;7:189–97.
18. Modolell I, Guarner L, Malagelada JR. Vagaries of clinical presentation of pancreatic and biliary tract cancer. Ann Oncol 1999;10 Suppl 4:82–4.
19. Karlson BM, Ekbom A, Lindgren PG, et al. Abdominal US for diagnosis of pancreatic tumor: prospective cohort analysis. Radiology 1999;213:107–11.
20. Bronstein YL, Loyer EM, Kaur H, et al. Detection of small pancreatic tumors with multiphasic helical CT. AJR Am J Roentgenol 2004;182:619–23.
21. Niederau C, Grendell JH. Diagnosis of pancreatic carcinoma. Imaging techniques and tumor markers. Pancreas 1992;7:66–86.
22. Kim HJ, Kim MH, Myung SJ, et al. A new strategy for the application of CA19-9 in the differentiation of pancreaticobiliary cancer: analysis using a receiver operating characteristic curve. Am J Gastroenterol 1999;94:1941–6.
23. Khorana AA, Mangu PB, Berlin J, et al. Potentially curable pancreatic cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2016;34:2541–56.
24. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
25. Soriano A, Castells A, Ayuso C, et al. Preoperative staging and tumor resectability assessment of pancreatic cancer: prospective study comparing endoscopic ultrasonography, helical computed tomography, magnetic resonance imaging, and angiography. Am J Gastroenterol 2004;99:492–501.
26. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
27. Al-Hawary MM, Francis IR, Chari ST, et al. Pancreatic ductal adenocarcinoma radiology reporting template: consensus statement of the Society of Abdominal Radiology and the American Pancreatic Association. Radiology 2014;270:248–60.
28. Khorana AA, Mangu PB, Berlin J, et al. Potentially curable pancreatic cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol 2017;35:2324–8.
28. Ducreux M, Cuhna AS, Caramella C, et al; ESMO Guidelines Committee. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2015;26 Suppl 5:v56–68.
30. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
31. Oettle H, Neuhaus P, Hochhaus A, et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. JAMA 2013;310:1473–81.
32. Huguet F, Girard N, Guerche CS, et al. Chemoradiotherapy in the management of locally advanced pancreatic carcinoma: a qualitative systematic review. J Clin Oncol 2009;27:2269–77.
33. Blackstock AW, Tepper JE, Niedwiecki D, et al. Cancer and leukemia group B (CALGB) 89805: phase II chemoradiation trial using gemcitabine in patients with locoregional adenocarcinoma of the pancreas. Int J Gastrointest Cancer 2003;34(2-3):107–16.
34. Loehrer PJ Sr, Feng Y, Cardenes H, et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an Eastern Cooperative Oncology Group trial. J Clin Oncol 2011;29:4105–12.
35. Hurt CN, Falk S, Crosby T, et al. Long-term results and recurrence patterns from SCALOP: a phase II randomised trial of gemcitabine- or capecitabine-based chemoradiation for locally advanced pancreatic cancer. Br J Cancer 2017;116:1264–70.
36. Chauffert B, Mornex F, Bonnetain F, et al. Phase III trial comparing intensive induction chemoradiotherapy (60 Gy, infusional 5-FU and intermittent cisplatin) followed by maintenance gemcitabine with gemcitabine alone for locally advanced unresectable pancreatic cancer. Definitive results of the 2000-01 FFCD/SFRO study. Ann Oncol 2008;19:1592–9.
37. Hammel P, Huguet F, van Laethem JL, et al, LAP07 Trial Group. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA 2016;315:1844–53.
38. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
39. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–703.
40. Katz MH, Pisters PW, Evans DB, et al. Borderline resectable pancreatic cancer: the importance of this emerging stage of disease. J Am Coll Surg 2008;206:833–46.
41. Andriulli A, Festa V, Botteri E, et al. Neoadjuvant/preoperative gemcitabine for patients with localized pancreatic cancer: a meta-analysis of prospective studies. Ann Surg Oncol 2012;19:1644–62.
42. Sadot E, Doussot A, O’Reilly EM, et al. FOLFIRINOX induction therapy for stage 3 pancreatic adenocarcinoma. Ann Surg Oncol 2015;22:3512–21.
43. Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol 2016;17:801–10.
44. Neoptolemos JP, Dunn JA, Stocken DD, et al, European Study Group for Pancreatic Cancer. Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: a randomised controlled trial. Lancet 2001;358:1576–85.
45. Neoptolemos JP, Stocken DD, Friess H, et al, European Study Group for Pancreatic Cancer. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 2004;350:1200–10.
46. Neoptolemos JP, Stocken DD, Bassi C, et al, European Study Group for Pancreatic Cancer. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA 2010;304:1073–81.
47. Regine WF, Winter KA, Abrams RA, et al. Fluorouracil vs gemcitabine chemotherapy before and after fluorouracil-based chemoradiation following resection of pancreatic adenocarcinoma: a randomized controlled trial. JAMA 2008;299:1019–26.
48. Neoptolemos JP, Palmer DH, Ghaneh P, et al, European Study Group for Pancreatic Cancer. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389:1011–24. Epub 2017 Jan 25.
Introduction
Exocrine pancreatic cancer refers to pancreatic adenocarcinomas that arise from ductal epithelial cells. Pancreatic ductal adenocarcinoma is a highly lethal malignancy, ranking as the fourth most common cause of cancer-related death in the United States1 and the eighth most common worldwide.2 In the United States, the pancreas is the second most common site of gastrointestinal malignancy after the colon.1 The only potentially curative modality for pancreatic adenocarcinomas is complete resection, followed by adjuvant therapy; unfortunately, only around 20% of patients are surgical candidates at the time of presentation due to delayed development of symptoms and consequently diagnosis.3 Most symptomatic patients with pancreatic cancer have locally advanced disease at diagnosis, and only a select group of patients with good performance status and borderline resectable disease can be offered neoadjuvant therapy. Adjuvant chemotherapy is typically recommended for patients who undergo potentially curative resection for pancreatic cancer.
Epidemiology
In the United States, pancreatic cancer has an annual estimated incidence of 55,440 new cases.1 It causes an estimated 44,330 deaths per year, with a 5-year overall survival (OS) rate of 8.2%.1 Worldwide an estimated 138,100 men and 127,900 women die of pancreatic cancer each year.2 In general, pancreatic cancers occur more commonly in persons living in Western/industrialized countries, older persons (age > 60 years), males (ratio 1.3:1 female), and African-Americans and native Hawaiians.4
Etiology
The major preventable environmental risk factor for pancreatic cancer is cigarette smoking, which accounts for 25% of all cases.5 A prospective study that estimated the excess incidence of pancreatic cancer among cigarette smokers and assessed the influence of smoking cessation on the risk for pancreatic cancer showed that persons who quit smoking reduced their risk of pancreatic cancer by 48% after 2 years of cessation, compared with smokers who did not quit, and reduced their risk to near the level of a never smoker after 10 years of cessation.5 Risk is higher for heavy smokers and those with homozygous deletions of the glutathione S-transferase theta 1 gene (GSTT1), which results in the absence of the carcinogen-metabolizing function of the glutathione S-transferase enzyme. High body mass index and sedentary lifestyle have been linked to pancreatic cancer.6 Data regarding aspirin, diet, coffee, and excess alcohol consumption are insufficient, inconclusive, and even conflicting, and thus the effect of these factors on risk for pancreatic cancer remains unclear. Infectious risk factors such as Helicobacter pylori and hepatitis B and C virus have weak associations with pancreatic cancer. Chronic pancreatitis and pancreatic cysts (eg, intraductal papillary mucinous neoplasm [IPMN] of the pancreas) carry a risk for malignant transformation, and hence may require surveillance. Multiple epidemiologic studies have shown a strong association between pancreatic cancer and recently diagnosed diabetes mellitus (relative risk [RR] 1.97 [95% confidence interval {CI} 1.78 to 2.18]); the presence of diabetes also may be a long-term predisposing factor for pancreatic cancer, and cancer screening needs to be considered for selected patients.7
A predisposing genetic anomaly accounts for 15% of all cases of pancreatic cancer.8 Hereditary risk factors are divided into 2 broad categories: defined genetic syndromes and familial pancreatic cancer. Familial predispositions that do not meet genetic syndrome criteria account for approximately 5% to 10% of all cases associated with hereditary factors; in one study, 29% of tested kindreds with an incident pancreatic cancer had a germline BRCA2 mutation.9 Other predisposing genetic syndromes that have been linked to pancreatic cancer include:
- Peutz-Jeghers syndrome with germline STK11 mutations (RR 132);
- Hereditary pancreatitis with germline PRSS1, SPINK1, and CFTR mutations (RR 26–87);
- Familial atypical multiple mole melanoma syndrome with CDKN2A mutations (RR 20–40);
- Familial breast and ovarian cancer with BRCA2 (RR 10) and BRCA1 (RR 2.8) mutations;
- Hereditary nonpolyposis colorectal cancer (HNPCC, Lynch II syndrome) with MLH1, MSH2, MSH6, and PMS2 mutations (RR 9–11); and
- Familial adenomatous polyposis with APC mutations (RR 5).10
Other gene mutations with unknown relative risk for pancreatic cancer include mutations affecting PALB2, ATM, and TP53.
The International Cancer of the Pancreas Screening consortium consensus on screening for pancreatic cancer in patients with increased risk for familial pancreatic cancer recommends screening those at high risk: first-degree relatives (FDRs) of patients with pancreatic cancer from a familial pancreatic kindred with at least 2 affected FDRs; patients with Peutz-Jeghers syndrome; and p16, BRCA2, and HNPCC mutation carriers with 1 or more affected FDRs and hereditary pancreatitis. The guidelines emphasize that screening should be done only in those who are surgical candidates and are evaluated at an experienced multidisciplinary center.8
Deleterious germline mutations in pancreatic cancer can account for 33% of patients with apparent sporadic cancers and no hereditary risk. These include germline mutations affecting BRCA1/2, PALB2, ATM, MLH1, CHK-2, CDKN2A, and TP53.11
Pathogenesis
Pancreatic neoplasms can be benign or malignant and thus a tissue histologic diagnosis is paramount. Pancreatic adenocarcinomas with exocrine features represent more than 95% of all pancreatic neoplasms, with only 5% arising from the endocrine pancreas (ie, neuroendocrine tumors). Pancreatic neuroendocrine tumors and pancreatic adenocarcinoma must be distinguished histologically because treatment of the 2 neoplasms is completely different. Other malignant pancreatic tumors are signet ring cell carcinoma, adenosquamous carcinoma, undifferentiated (anaplastic) carcinoma, and mucinous noncystic (colloid) carcinoma; the latter tumor has a better prognosis.12 It is essential to characterize and distinguish among benign cystic neoplasms, as some require surgical resection due to the risk of malignant transformation. IPMN, pancreatic intraepithelial neoplasia, and mucinous cystic neoplasms are thought to be premalignant lesions of invasive ductal adenocarcinomas, and the pathological report should highlight the degree of dysplasia for adequate risk stratification.13 This information could be the deciding factor in whether a pancreatectomy is recommended by a multidisciplinary team.
Most pancreatic cancers harbor activating or silencing genetic mutations, and multiple combinations of altered genes can be detected by next-generation sequencing (average of 63 genetic alterations per cancer).14 Mutational activated KRAS is the most frequent (> 90%) genetic alteration in pancreatic cancer, even in early neoplastic precursors (IPMN > 75%). KRAS is a highly complex, dynamic proto-oncogene involved in signaling of various receptor kinases such as the epidermal growth factor receptor and the insulin-like growth factor receptor-I. It also engages in canonical downstream effector pathways, mainly Raf/MEK/ERK, PI3K/PDK1/Akt, and the Ral guanine nucleotide exchange factor pathway, which drive much of the pathogenesis of malignancy. These pathways lead to sustained proliferation, metabolic reprogramming, anti-apoptosis, remodeling of the tumor microenvironment, evasion of the immune response, cell migration, and metastasis. An activating point mutation in codon G12 is the most common (98%) locus of KRAS mutation in pancreatic adenocarcinoma, but all drugs targeting this mutation have failed in clinical practice.15 Additionally, inactivation of tumor suppressor genes such as p53, DPC4 (SMAD4/MADH4), CDKN2A (p16/MTS1), and BRCA2 can be found in 75%, 30%, 35%, and 4% of pancreatic adenocarcinoma cases, respectively.14 Another pancreatic cancer hallmark is inactivation of DNA damage repair genes, which include MLH1 and MSH2.16
Diagnosis and Staging
Case Presentation
A 71-year-old male veteran with no significant past medical history other than hypertension and hyperlipidemia and an excellent performance status presents to the emergency department after noticing a yellowish skin and sclera color. He denies weight loss, abdominal pain, or any other pertinent symptom or sign. Physical examination reveals a healthy developed man with yellowish discoloration of the skin and sclera and a soft, nontender benign abdomen; physical examination is otherwise unremarkable. Laboratory evaluation reveals a direct bilirubin level of 4.5 mg/dL and normal values for complete blood count and renal, liver, and coagulation panels. Abdominal and pelvis computed tomography (CT) with intravenous contrast shows a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein, which remains patent. Follow-up endoscopic ultrasound (EUS) confirms an irregular mass at the head of the pancreas measuring 3.2 × 2.6 cm with sonographic evidence suggesting invasion into the portal vein. During the procedure, the bile duct is successfully stented, the mass is biopsied, and bile duct brushing is performed. Pathology report is consistent with pancreatic adenocarcinoma.
- What is the typical presentation of pancreatic cancer?
The most common symptoms of pancreatic cancer at the time of presentation include weight loss (85%), asthenia/anorexia (86%), and/or abdominal pain (79%).17 The most frequent signs are jaundice (55%), hepatomegaly (39%), and cachexia (13%). Courvoisier sign, a nontender but palpable distended gallbladder at the right costal margin, is neither sensitive nor specific for pancreatic cancer (13% of cases). Trousseau syndrome, a superficial thrombophlebitis, is another classic sign that reflects the hypercoagulable nature of pancreatic cancer (3% of cases).17 The pathophysiology of this syndrome is not completely understood, but it may occur secondary to the release of cancer microparticles in the blood stream which in turn stimulate the coagulation cascade. Other nonspecific symptoms are dark urine, nausea, vomiting, diarrhea, steatorrhea, and epigastric and back pain. Because symptoms early in the course of the disease are nonspecific, pancreatic cancer is typically diagnosed late, after the cancer has invaded local structures or metastasized. The initial presentation varies depending on tumor location, with 70% of pancreatic head malignancies presenting with jaundice and pain correlating to an advanced stage.18 Although data supporting an association between new-onset diabetes mellitus and pancreatic cancer are inconclusive, pancreatic cancer should still be a consideration in patients with new-onset diabetes mellitus and other symptoms such as pain and weight loss. Early signs of incurable disease include a palpable mass, ascites, lymphadenopathy (classic Virchow node), and an umbilical mass (Sister Mary Joseph node). Incidentally discovered pancreatic masses on imaging are rare, but the incidence is increasing due to frequent imaging for other reasons and improved diagnostic techniques.
- What is the approach to diagnosis and staging?
History and physical examination findings are not sufficiently sensitive or specific to diagnose pancreatic cancer. High clinical suspicion in a patient with risk factors can lead to a comprehensive evaluation and potential early diagnosis. In general, an initial diagnostic work-up for suspected pancreatic cancer will include serologic evaluation (liver function test, lipase, tumor markers) and abdominal imaging (ultrasound, CT scans, or magnetic resonance imaging [MRI]). Ultrasound is a first-line diagnostic tool with a sensitivity of 90% and specificity of 98.8% for pancreatic cancer, but it is investigator-dependent and is less accurate in detecting tumors smaller than 3 cm in diameter.19 Multiphasic helical CT of the abdomen has better sensitivity (100%) and specificity (100%) for detecting tumors larger than 2 cm, but this modality is less accurate in detecting pancreatic masses smaller than 2 cm (77%).20 Percutaneous fine-needle aspiration (FNA) performed by ultrasound or CT guidance is avoided due to theoretical concerns about intraperitoneal seeding and bleeding.
If a pancreatic mass is detected by ultrasound or CT, additional interventions may be indicated depending on the clinical scenario. EUS-guided biopsy can provide histological confirmation and is currently utilized frequently for diagnosis and early resectability staging. Endoscopic retrograde cholangiopancreatography (ERCP) is indicated for patients with biliary obstruction requiring stent placement, and this procedure may provide tissue confirmation by forceps biopsy or brush cytology (lower accuracy than EUS). In a meta-analysis that evaluated the diagnostic value of tests for pancreatic cancer, ERCP had the highest sensitivity (92%) and specificity (96%) compared to ultrasound and CT,21 but this modality carries a risk for pancreatitis, bleeding, and cholangitis. Magnetic resonance cholangiopancreatography has not replaced ERCP, but it but may be an alternative for patients who cannot undergo ERCP (eg, gastric outlet obstruction, duodenal stenosis, anatomical surgical disruption, unsuccessful ERCP). ERCP is used frequently because many patients present with obstructive jaundice due to pancreatic mass compression, specifically if the mass is located in the head, and must undergo ERCP and stenting of the common bile duct.
The carbohydrate antigen (CA) 19-9 level has variable sensitivity and specificity in pancreatic cancer, as levels can be elevated in many benign pancreaticobiliary disorders. Elevated CA 19-9, in the appropriate clinical scenario (ie, a suspicious pancreatic mass and a value greater than 37 U/mL) demonstrated a sensitivity of 77% and specificity of 87% when differentiating pancreaticobiliary cancer from benign clinical conditions such as acute cholangitis or cholestasis.22 CA 19-9 level has prognostic value, as it may predict occult disease and correlates with survival rates, but no specific cutoff value has been established to guide perioperative therapy for high-risk resectable tumors.23
The American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) tumor, node, metastasis (TNM) system is the preferred method for staging pancreatic cancer (Table 1).
Positron emission tomography with CT scan is occasionally utilized in practice to assess tumor burden by evaluating anatomical structures and assessing physiologic uptake, which aids in establishing the extent of disease in equivocal cases. Staging laparoscopy with or without peritoneal biopsy is sometimes used to establish appropriate staging in cases that are questionable for occult metastatic disease. This procedure helps avoid unnecessary morbid surgeries.
Neoadjuvant Therapy
Case Continued
The patient is referred to oncology. Blood work reveals a CA 19-9 level of 100 U/mL (reference range < 35 U/mL) and a staging CT scan of the chest reveals a benign-appearing 3-mm nodule (no prior imaging for comparison). CT scan of the abdomen and pelvis does not define venous vasculature involvement appropriately and hence MRI of the abdomen and pelvis is performed. MRI reveals a pancreatic head mass measuring 3.0 × 2.7 cm, without arterial or venous vasculature invasion. However, the mass is abutting the portal vein and superior mesenteric vein and there is a new nonspecific 8-mm aortocaval lymph node.
- What are the current approaches to treating patients with resectable, unresectable, and metastatic disease?
Accurate staging and assessment of surgical resectability in pancreatic cancer are paramount as these steps prevent a futile morbid Whipple procedure in patients with advanced disease and a high risk of recurrence. Conversely, it allows patients with low-volume disease to undergo a potentially curative surgery. Approximately 20% of patients present with resectable disease, 40% present with locally advanced unresectable tumors (eg, involvement of critical vascular structures), and 40% present with metastatic disease.3 Treatment for resectable pancreatic cancer continues to be upfront surgery, although neoadjuvant therapy with either chemoradiation, radiation alone, or chemotherapy is an option per guidelines from the American Society of Clinical Oncology (ASCO),28 the NCCN,26 and the European Society for Medical Oncology (ESMO),29 particularly for patients with borderline resectable tumors (Table 3).
Systemic chemotherapy is recommended for fit candidates with locally advanced unresectable or metastatic disease, with an emphasis on supportive palliative measures. Palliative interventions include biliary stenting, duodenal stent for relieving gastric-outlet obstruction, and celiac axis nerve blocks, when indicated. Routine preoperative biliary stent placement/drainage in patients undergoing subsequent surgery for pancreatic cancer located in the head is associated with an increased risk of surgical complications when compared with up-front surgery without prior biliary drainage, and thus stent placement/drainage is not recommended.26 Aggressive supportive management of symptoms, such as cancer-associated pain, anorexia-cachexia syndromes, and anxiety-depression disorders, should remain a primary palliative focus.
Case Continued
A multidisciplinary tumor board discusses the patient’s case and deems the cancer borderline resectable; neoadjuvant therapy is recommended. The patient is started on treatment with gemcitabine and nab-paclitaxel as first-line neoadjuvant therapy. After 4 cycles, the CA 19-9 level drops to 14 U/mL, and MRI reveals a smaller head mass of 1.3 × 1.4 cm with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remains stable. At tumor board, it is evident that the patient has responded to therapy and the recommendation is to treat with gemcitabine chemoradiotherapy before surgery.
- What neoadjuvant therapy strategies are used in the treatment of pancreatic adenocarcinoma?
There are no established evidence-based recommendations for neoadjuvant therapy in patients with borderline resectable pancreatic cancer or patients with unresectable locally advanced pancreatic cancer. However, there are ongoing trials to investigate this treatment approach, and it is offered off-label in specific clinical scenarios, such as in the case patient described here. In patients with borderline resectable disease, preoperative chemotherapy followed by chemoradiation is a routine practice in most cancer centers,32 and ongoing clinical trials are an option for this cohort of patients (eg, Southwest Oncology Group Trial 1505, NCT02562716). The definitions of borderline resectable and unresectable pancreatic cancer have been described by the NCCN,26 although most surgeons consider involvement of the major upper abdominal blood vessels the main unresectability criterion; oncologists also consider other parameters such as suspicious lesions on scans, worsening performance status, and a significantly elevated CA 19-9 level suggestive of disseminated disease.28 The goal of a conversion approach by chemotherapy with or without radiation for borderline and unresectable cancers is to deliver a tolerable regimen leading to tumor downstaging, allowing for surgical resection. No randomized clinical trial has shown a survival advantage of this approach. Enrollment in clinical trials is preferred for patients with borderline and unresectable cancer, and there are trials that are currently enrolling patients.
The main treatment strategies for patients with locally advanced borderline and unresectable pancreatic cancer outside of a clinical trial are primary radiotherapy, systemic chemotherapy, and chemoradiation therapy. Guidelines from ASCO, NCCN, and ESMO recommend induction chemotherapy followed by restaging and consolidation chemoradiotherapy in the absence of progression.26,28,29 There is no standard chemoradiation regimen and the role of chemotherapy sensitizers, including fluorouracil, gemcitabine, and capecitabine (an oral fluoropyrimidine substitute), and targeted agents in combination with different radiation modalities is now being investigated.
Fluorouracil is a radio-sensitizer that has been used in locally advanced pancreatic cancer based on experience in other gastrointestinal malignancies; data shows conflicting results with this drug. Capecitabine and tegafur/gimeracil/oteracil (S-1) are oral prodrugs that can safely replace infusional fluorouracil. Gemcitabine, a more potent radiation sensitizer, is very toxic, even at low-doses twice weekly, and does not provide a survival benefit, as demonstrated in the Cancer and Leukemia Group B (CALGB) 89805 trial, a phase 2 study of patients with surgically staged locally advanced pancreatic cancer.33 Gemcitabine-based chemoradiotherapy was also evaluated in the Eastern Cooperative Group (ECOG) E4201 trial, which randomly assigned patients to receive gemcitabine alone (at 1000 mg/m2/wk for weeks 1 through 6, followed by 1 week rest, then weekly for 3 out of 4 weeks) or gemcitabine (600 mg/m2/wk for weeks 1 to 5, then 4 weeks later 1000 mg/m2 for 3 out of 4 weeks) plus radiotherapy (starting on day 1, 1.8 Gy/fraction for total of 50.4 Gy).34 Patients with locally advanced unresectable pancreatic cancer had a better OS outcome with gemcitabine in combination with radiation therapy (11.1 months) as compared with patients who received gemcitabine alone (9.2 months). Although there was a greater incidence of grade 4 and 5 treatment-related toxicities in the combination arm, no statistical differences in quality-of-life measurements were reported. Gemcitabine-based and capecitabine-based chemoradiotherapy were compared in the open-label phase 2 multicenter randomized SCALOP trial.35 Patients with locally advanced pancreatic cancer were assigned to receive 3 cycles of induction with gemcitabine 1000 mg/m2 days 1, 8, and 15 and capecitabine 830 mg/m2 days 1 to 21 every 28 days; patients who had stable or responding disease were randomly assigned to receive a fourth cycle followed by capecitabine (830 mg/m2 twice daily on weekdays only) or gemcitabine (300 mg/m2 weekly) with radiation (50.4 Gy over 28 fractions). Patients treated with capecitabine-based chemoradiotherapy had higher nonsignificant median OS (17.6 months) and median progression-free survival (12 months) compared to those treated with gemcitabine (14.6 months and 10.4 months, respectively).
The benefit of radiation therapy in the treatment of locally advanced pancreatic cancer was further explored by the Fédération Francophone de Cancérologie Digestive 2000-01 phase 3 trial. This study compared induction chemoradiotherapy (60 Gy, 2 Gy/fraction; concomitant fluorouracil infusion, 300 mg/m2/day, days 1–5 for 6 weeks; cisplatin, 20 mg/m2/day, days 1–5 during weeks 1 and 5) to gemcitabine alone (1000 mg/m2 weekly for 7 weeks) followed by maintenance gemcitabine in both arms.36 Unexpectedly, the median OS was significantly shorter in the chemoradiotherapy arm than in the chemotherapy alone arm (8.6 months versus 13 months, respectively, P = 0.03) and the combination arm had more toxicities. The phase 3 open-label LAP07 study explored the role of radiation therapy in patients with locally advanced pancreatic cancer who had controlled disease after 4 months of induction therapy.37 LAP07 had 2 randomizations: first, patients with locally advanced pancreatic cancer were assigned to receive weekly gemcitabine alone (1000 mg/m2) or this same dose of gemcitabine plus erlotinib 100 mg/day; second, patients with progression-free disease (61% of initial cohort) after 4 months of therapy were assigned to receive 2 months of the same chemotherapy or chemoradiotherapy (54 Gy plus capecitabine). This study showed that the addition of erlotinib to gemcitabine did not improve survival and in fact affected survival adversely. Of note, no survival benefit was observed after the first randomization from chemotherapy to consolidating chemoradiotherapy. Chemoradiotherapy achieved better locoregional tumor control with significantly less local tumor progression (32% versus 46%, P < 0.03) and no increase in toxicity. Based on prior moderate-quality evidence, guidelines recommend consolidative chemoradiotherapy only for surgical resection candidates following induction chemotherapy; for those who are not surgical candidates, guidelines recommend continuing systemic therapy.26,28,29
Gemcitabine and fluorouracil-based chemotherapies were the standard induction regimens until evidence from studies of metastatic systemic treatment protocols with FOLFIRINOX (ACCORD trial38) and nanoparticle albumin-bound paclitaxel (nab-paclitaxel) plus gemcitabine (MPACT trial39) was extrapolated to clinical practice. These regimens were shown to achieve higher objective response rates when compared to single-agent gemcitabine in patients with metastatic pancreatic cancer. Due to the broad heterogeneity of results in small retrospective series with neoadjuvant trials in borderline resectable pancreatic cancer, the quality of the evidence is low and any recommendation is limited. Many individual series have demonstrated improved complete resection rates and promising survival rates. In the largest single-institution retrospective review of patients with borderline resectable pancreatic adenocarcinoma who completed neoadjuvant gemcitabine-based chemoradiotherapy (50 Gy in 28 fractions or 30 Gy in 10 fractions), 94% achieved a margin-negative pancreatectomy; the median OS in those who completed preoperative therapy and had surgery was 40 months, with a 5-year OS of 36%.40 A meta-analysis by Andriulli and colleagues included 20 prospective studies of patients with initially resectable (366 lesions) or unresectable (341 lesions) disease who were treated with neoadjuvant/preoperative gemcitabine with or without radiotherapy.41 In the group with initially unresectable disease, 39% underwent surgery after restaging and 68% of explored patients were resected; the R0 resection rate was 60%. After restaging, 91% of patients with resectable disease underwent surgery, with 82% of explored patients undergoing surgical resection and 89% of these achieving R0 resection. The estimated 1- and 2-year survival probabilities after resection among patients with initially unresectable disease were 86.3% and 54.2%.41
The largest single-institution retrospective review of FOLFIRINOX (fluorouracil, oxaliplatin, irinotecan, and leucovorin), an alternative to gemcitabine, for neoadjuvant induction therapy for patients with locally advanced unresectable disease was conducted at Memorial Sloan Kettering Cancer Center. In this study (n = 101), 31% of patients initially deemed unresectable who completed FOLFIRINOX induction therapy with or without chemoradiation underwent resection. The R0 resection rate in these patients was 55%, and patients who did not progress during induction FOLFIRINOX therapy had a median OS of 26 months.42 A systematic review and meta-analysis of FOLFIRINOX chemotherapy with or without radiotherapy in patients with locally advanced unresectable pancreatic cancer reported that 25.9% of patients underwent resection after FOLFIRINOX therapy, and the R0 resection rate in these patients was 78.4%.43 The median OS in this study was 24.2 months, which was longer than the previously reported median OS rates for gemcitabine.
There is no strong evidence published for the use of combination nab-paclitaxel plus gemcitabine in the neoadjuvant setting, but it is used in clinical practice based on evidence from the MPACT trial, which showed the combination improved OS and progression-free survival in patients with metastatic pancreatic cancer.39 An early-phase 1-arm clinical trial of neoadjuvant gemcitabine, docetaxel, and capecitabine (GTX) followed by radiotherapy showed an increased response rate and survival for locally advanced disease; however, the NCCN expert panel has reached a consensus but not a uniform recommendation regarding this regimen due to significant toxicities and low patient accrual.26 Selected patients with pancreatic cancer with BRCA1/2 mutations are more sensitive to platinum-based chemotherapy. Although studies of neoadjuvant platinum-based chemotherapy in this population have not been reported, the NCCN guidelines list it as an alternative option based on extrapolated data.26 A clinical trial of gemcitabine, nab-paclitaxel, and cisplatin in the neoadjuvant setting in patients with resectable pancreatic cancer is currently enrolling patients (NGC triple regimen NCT0339257).
Summary
Chemotherapy alone or followed by chemoradiotherapy may be used as initial treatment for patients with borderline and unresectable pancreatic adenocarcinoma without distant metastases who are potential surgical candidates. Chemoradiotherapy remains a preferred treatment option for patients with poorly controlled pain from local tumor invasion, in view of the well-documented analgesic palliative effect of radiation therapy. FOLFIRINOX with or without radiation therapy may offer the highest documented response rates, but it also results in higher rates of treatment-related toxicities. FOLFIRINOX can be offered to selected fit patients (< 65 years old, no comorbidity contraindication, good functional status [ECOG 0–1]) who can tolerate triple therapy with a more toxic adverse-effect profile. A clinical trial evaluating neoadjuvant FOLFIRINOX with or without preoperative chemoradiotherapy in patients with borderline resectable pancreatic cancer is ongoing (PANDAS-PRODIGE 44, NCT02676349). Gemcitabine with or without radiation therapy is a tolerable combination, although it is potentially more toxic when combined with radiation. The addition of nab-paclitaxel to gemcitabine without radiation may emerge as a preferred neoadjuvant treatment for selected patients; a clinical trial investigating this modality in patients with resectable and borderline resectable disease is ongoing (NCT02723331).
Adjuvant Therapy
Case Continued
Prior to the planned surgical resection and after undergoing chemoradiation therapy, the patient has an excellent performance status and repeat MRI shows a 1.3 × 1.4 cm head mass with no further vasculature involvement, no evidence of lymphadenopathy, and no distant metastasis. The CA 19-9 level is stable at 18 U/mL. The patient undergoes an uncomplicated partial pancreaticoduodenectomy, and analysis of a surgical pathology specimen reveals T3N0 disease with closest margin of 0.1 cm.
- Would the patient benefit from adjuvant therapy?
Adjuvant chemotherapy for 6 months after pancreatic cancer resection should be offered to all patients based on mature data. Gemcitabine and capecitabine are the current standard of care in adjuvant therapy; alternatively, single-agent gemcitabine can be offered to patients with poor performance status or patients who cannot tolerate the toxicities associated with this combination.28 Adjuvant treatment should be initiated within approximately 8 weeks of surgical resection. The value of radiation therapy remains controversial, but it can be offered within the context of a clinical trial or to patients with positive margins after surgical resection and/or lymph node–positive disease. Based on low-quality supportive evidence, it is strongly recommended that patients who receive neoadjuvant therapy complete a total of 6 months of chemotherapy, factoring in the duration of the preoperative regimen.28 Different adjuvant strategies have been investigated, including chemotherapy alone with a fluoropyrimidine and/or gemcitabine with or without combined chemoradiation therapy.
The European Study Group for Pancreatic Cancer 1 (ESPAC)-1 trial was a randomized clinical trial that evaluated several adjuvant strategies in pancreatic cancer treatment. This trial assigned patients who underwent pancreatic adenocarcinoma resection to adjuvant chemotherapy alone (intravenous fluorouracil 425 mg/m2 and leucovorin 20 mg/m2 daily for 5 days, monthly for 6 months), chemoradiotherapy (20 Gy in 10 daily fractions over 2 weeks with 500 mg/m2 intravenous fluorouracil on days 1–3, repeated after 2 weeks), both chemotherapy and chemoradiation, and observation.44 The results showed no added benefit for adjuvant chemoradiotherapy, with a median OS of 15.5 months in the chemoradiotherapy cohort, as compared to a median OS of 16.1 months in the chemotherapy-alone cohort (hazard ratio [HR] 1.18 [95% CI 0.90 to 1.55], P = 0.24). In addition, there was evidence of a survival benefit for the chemotherapy-alone arm when compared to the combined modality arm, with a median OS of 19.7 versus 14.0 months, respectively (HR 0.66 [95% CI 0.52 to 0.83], P = 0.0005). Although ESPAC-1 has been criticized for being underpowered to perform statistical comparison, it is still considered a landmark trial demonstrating benefit with single-agent chemotherapy alone. A follow-up analysis of ESPAC-1 showed that adjuvant chemotherapy alone conferred a significant 5-year survival benefit while the combined modality had a deleterious effect on survival. 45 Hence, adjuvant chemotherapy alone became the standard of care in the United States following resection.
The results of the multicenter randomized controlled phase 3 CONKO-001 (CharitéOnkologie 001) trial, which were reported in 2007, supported the use of adjuvant gemcitabine for 6 months in patients with resected pancreatic adenocarcinoma. In this study, patients treated with adjuvant gemcitabine (1000 mg/m2 days 1, 8, and 15 every 4 weeks for 6 months) had superior disease-free survival compared with those who received surgery alone.30 A long-term outcome update of this study demonstrated a significant improvement in 5-year OS for patients treated with adjuvant gemcitabine (20.7% [95% CI 14.7% to 26.6%]) compared to those who received surgical resection alone (10.4% [95% CI 5.9% to 15.0%]). This benefit persisted at 10-year follow-up, with an OS of 12.2% (95% CI 7.3% to 17.2%) in the adjuvant gemcitabine group, as compared to 7.7% (95% CI 3.6% to 11.8%) in the resection alone group.31
Fluorouracil and gemcitabine remained equivalent adjuvant treatment options until the results of the ESPAC-3 trial were reported in 2010.32 This large phase 3 trial, conducted mainly in the United Kingdom, compared weekly gemcitabine (1000 mg/m2 weekly for 3 of every 4 weeks) to leucovorin-modulated fluorouracil (Mayo Clinic regimen: leucovorin 20 mg/m2 followed by fluorouracil 425 mg/m2 intravenous bolus days 1 through 5 every 28 days) as adjuvant therapy in resected pancreatic adenocarcinoma. After a median follow-up of 34.2 months, the median OS was similar in the 2 groups (fluorouracil/leucovorin 23.0 months versus gemcitabine 23.6 months; P = 0.39). However, the fluorouracil/leucovorin group experienced more grade 3/4 treatment-related toxicities (mucositis, stomatitis, diarrhea, and hosptializations; 14% versus 7.5%; P < 0.001).46 Following this trial, gemcitabine became the standard of care for adjuvant chemotherapy for resected pancreatic cancer.
The U.S. Radiation Therapy Oncology Group (RTOG) 9704 trial was conducted to investigate the potential benefit of adding radiation therapy to gemcitabine. This trial demonstrated an improved trend among patients with pancreatic head tumors (but not with cancers of the pancreatic body or tail) who received adjuvant gemcitabine followed by chemoradiotherapy (50.4 Gy in 1.8 Gy daily fractions for 5.5 weeks with concurrent infusional fluorouracil 250 mg/m2 daily) and subsequent gemcitabine monotherapy compared to postoperative fluorouracil-based chemoradiotherapy. Results showed a 5-year OS of 22% versus 18%, respectively, although this improvement was not statistically significant (P = 0.08). This trial failed to show a benefit of adding radiotherapy to gemcitabine.47
The ESPAC-4 trial, reported in 2017, evaluated the combination of gemcitabine and capecitabine compared to gemcitabine alone as adjuvant therapy for resected pancreatic adenocarcinoma.48 Patients were randomly assigned after surgical resection, regardless of margin or node status, to 6 months of gemcitabine alone (1000 mg/m2/day on days 1, 8, and 15 of each 28-day cycle) or gemcitabine plus capecitabine (1660 mg/m2/day on days 1 through 21 of each 28-day cycle). Combination therapy had a significant survival benefit compared to single therapy, with median OS durations of 28 months and 25.5 months, respectively (HR for death 0.82 [95% CI 0.68 to 0.98]). The 5-year OS for patients who received combination treatment was 29 months (95% CI 22.9 to 35.2) versus 16 months (95% CI 10.2 to 23.7) for those in the monotherapy group. As expected, grade 3 or 4 treatment-related toxicities (diarrhea, hand-foot syndrome, and neutropenia) were significantly more common with combined therapy, although there were no significant differences in the rates of serious adverse events. The adjuvant combination of gemcitabine and capecitabine became the current and preferred new standard of care following resection of pancreatic ductal adenocarcinoma,28 but single-agent gemcitabine and fluorouracil/leucovorin continue to be viable options,26,28,29 particularly for elderly patients, patients with borderline performance status, or patients with multiple comorbidities.
Evidence showing that a more intensive regimen can improve outcome in the adjuvant setting remains elusive. The phase 3 APACT study (Adjuvant Therapy for Patients with Resected Pancreatic Cancer, NCT01964430) comparing gemcitabine alone to gemcitabine plus nab-paclitaxel in patients with surgically resected pancreatic adenocarcinoma has concluded, with the results projected to be released in 2018. Another phase 3 trial investigating the efficacy of FOLFIRINOX versus gemcitabine alone as adjuvant therapy is underway in France and Canada (PRODIGE24/ACCORD24, NCT01526135). Other strategies with newer targeted therapies and immunotherapy are in the development phase.
Follow-Up and Surveillance
Case Conclusion
After recovery from surgery, the patient is offered and completes 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine. He is started on surveillance at 3 and 6 months, and he maintains an excellent performance status. He develops clinical evidence of pancreatic enzyme insufficiency and is placed on oral replacement therapy. He has no other complaints, and there is no evidence of recurrence on MRI and CA 19-9 levels.
- What is the recommended duration of surveillance following curative resection?
Surveillance after curative resection of pancreatic adenocarcinoma is recommended by NCCN guidelines.26 However, pancreatic adenocarcinoma has a poor prognosis, and surveillance after curative surgical resection with or without perioperative therapy has not been shown to impact survival. Most recurrences will occur within 2 years after treatment. Surveillance recommendations differ among expert groups.26,28,29 NCCN guidelines recommend evaluating patients by history and physical examination every 3 to 6 months for the first 2 years, then every 6 to 12 months for 3 years. CA 19-9 level and CT scan should be obtained every 3 to 6 months for 2 years and then every 6 to 12 months for 3 years. Follow-up with CA 19-9 levels and CT scans after 5 years is not routinely performed unless guided by signs, symptoms, or laboratory findings that raise suspicion for recurrence. Follow-up visits should also include evaluation of treatment-related toxicities, symptom management, nutrition support of pancreatic insufficiency, and psychosocial support.
Conclusion
Pancreatic cancer is a leading cause of cancer-related death that frequently presents with locally advanced or metastatic disease due to nonspecific symptoms and lack of a screening modality. Histological tissue biopsy confirmation and accurate resectability staging guide treatment planning and prognosis. The only potentially curative therapy is surgical resection plus adjuvant therapy for those with resectable disease. Surgical candidates with borderline resectable and unresectable disease can be offered induction preoperative chemotherapy followed by consolidation chemoradiation, based on clinical consensus practice. Enrollment in clinical trials should be encouraged for all patients, as evidence from clinical trials is essential to making progress in pancreatic cancer treatment.
Introduction
Exocrine pancreatic cancer refers to pancreatic adenocarcinomas that arise from ductal epithelial cells. Pancreatic ductal adenocarcinoma is a highly lethal malignancy, ranking as the fourth most common cause of cancer-related death in the United States1 and the eighth most common worldwide.2 In the United States, the pancreas is the second most common site of gastrointestinal malignancy after the colon.1 The only potentially curative modality for pancreatic adenocarcinomas is complete resection, followed by adjuvant therapy; unfortunately, only around 20% of patients are surgical candidates at the time of presentation due to delayed development of symptoms and consequently diagnosis.3 Most symptomatic patients with pancreatic cancer have locally advanced disease at diagnosis, and only a select group of patients with good performance status and borderline resectable disease can be offered neoadjuvant therapy. Adjuvant chemotherapy is typically recommended for patients who undergo potentially curative resection for pancreatic cancer.
Epidemiology
In the United States, pancreatic cancer has an annual estimated incidence of 55,440 new cases.1 It causes an estimated 44,330 deaths per year, with a 5-year overall survival (OS) rate of 8.2%.1 Worldwide an estimated 138,100 men and 127,900 women die of pancreatic cancer each year.2 In general, pancreatic cancers occur more commonly in persons living in Western/industrialized countries, older persons (age > 60 years), males (ratio 1.3:1 female), and African-Americans and native Hawaiians.4
Etiology
The major preventable environmental risk factor for pancreatic cancer is cigarette smoking, which accounts for 25% of all cases.5 A prospective study that estimated the excess incidence of pancreatic cancer among cigarette smokers and assessed the influence of smoking cessation on the risk for pancreatic cancer showed that persons who quit smoking reduced their risk of pancreatic cancer by 48% after 2 years of cessation, compared with smokers who did not quit, and reduced their risk to near the level of a never smoker after 10 years of cessation.5 Risk is higher for heavy smokers and those with homozygous deletions of the glutathione S-transferase theta 1 gene (GSTT1), which results in the absence of the carcinogen-metabolizing function of the glutathione S-transferase enzyme. High body mass index and sedentary lifestyle have been linked to pancreatic cancer.6 Data regarding aspirin, diet, coffee, and excess alcohol consumption are insufficient, inconclusive, and even conflicting, and thus the effect of these factors on risk for pancreatic cancer remains unclear. Infectious risk factors such as Helicobacter pylori and hepatitis B and C virus have weak associations with pancreatic cancer. Chronic pancreatitis and pancreatic cysts (eg, intraductal papillary mucinous neoplasm [IPMN] of the pancreas) carry a risk for malignant transformation, and hence may require surveillance. Multiple epidemiologic studies have shown a strong association between pancreatic cancer and recently diagnosed diabetes mellitus (relative risk [RR] 1.97 [95% confidence interval {CI} 1.78 to 2.18]); the presence of diabetes also may be a long-term predisposing factor for pancreatic cancer, and cancer screening needs to be considered for selected patients.7
A predisposing genetic anomaly accounts for 15% of all cases of pancreatic cancer.8 Hereditary risk factors are divided into 2 broad categories: defined genetic syndromes and familial pancreatic cancer. Familial predispositions that do not meet genetic syndrome criteria account for approximately 5% to 10% of all cases associated with hereditary factors; in one study, 29% of tested kindreds with an incident pancreatic cancer had a germline BRCA2 mutation.9 Other predisposing genetic syndromes that have been linked to pancreatic cancer include:
- Peutz-Jeghers syndrome with germline STK11 mutations (RR 132);
- Hereditary pancreatitis with germline PRSS1, SPINK1, and CFTR mutations (RR 26–87);
- Familial atypical multiple mole melanoma syndrome with CDKN2A mutations (RR 20–40);
- Familial breast and ovarian cancer with BRCA2 (RR 10) and BRCA1 (RR 2.8) mutations;
- Hereditary nonpolyposis colorectal cancer (HNPCC, Lynch II syndrome) with MLH1, MSH2, MSH6, and PMS2 mutations (RR 9–11); and
- Familial adenomatous polyposis with APC mutations (RR 5).10
Other gene mutations with unknown relative risk for pancreatic cancer include mutations affecting PALB2, ATM, and TP53.
The International Cancer of the Pancreas Screening consortium consensus on screening for pancreatic cancer in patients with increased risk for familial pancreatic cancer recommends screening those at high risk: first-degree relatives (FDRs) of patients with pancreatic cancer from a familial pancreatic kindred with at least 2 affected FDRs; patients with Peutz-Jeghers syndrome; and p16, BRCA2, and HNPCC mutation carriers with 1 or more affected FDRs and hereditary pancreatitis. The guidelines emphasize that screening should be done only in those who are surgical candidates and are evaluated at an experienced multidisciplinary center.8
Deleterious germline mutations in pancreatic cancer can account for 33% of patients with apparent sporadic cancers and no hereditary risk. These include germline mutations affecting BRCA1/2, PALB2, ATM, MLH1, CHK-2, CDKN2A, and TP53.11
Pathogenesis
Pancreatic neoplasms can be benign or malignant and thus a tissue histologic diagnosis is paramount. Pancreatic adenocarcinomas with exocrine features represent more than 95% of all pancreatic neoplasms, with only 5% arising from the endocrine pancreas (ie, neuroendocrine tumors). Pancreatic neuroendocrine tumors and pancreatic adenocarcinoma must be distinguished histologically because treatment of the 2 neoplasms is completely different. Other malignant pancreatic tumors are signet ring cell carcinoma, adenosquamous carcinoma, undifferentiated (anaplastic) carcinoma, and mucinous noncystic (colloid) carcinoma; the latter tumor has a better prognosis.12 It is essential to characterize and distinguish among benign cystic neoplasms, as some require surgical resection due to the risk of malignant transformation. IPMN, pancreatic intraepithelial neoplasia, and mucinous cystic neoplasms are thought to be premalignant lesions of invasive ductal adenocarcinomas, and the pathological report should highlight the degree of dysplasia for adequate risk stratification.13 This information could be the deciding factor in whether a pancreatectomy is recommended by a multidisciplinary team.
Most pancreatic cancers harbor activating or silencing genetic mutations, and multiple combinations of altered genes can be detected by next-generation sequencing (average of 63 genetic alterations per cancer).14 Mutational activated KRAS is the most frequent (> 90%) genetic alteration in pancreatic cancer, even in early neoplastic precursors (IPMN > 75%). KRAS is a highly complex, dynamic proto-oncogene involved in signaling of various receptor kinases such as the epidermal growth factor receptor and the insulin-like growth factor receptor-I. It also engages in canonical downstream effector pathways, mainly Raf/MEK/ERK, PI3K/PDK1/Akt, and the Ral guanine nucleotide exchange factor pathway, which drive much of the pathogenesis of malignancy. These pathways lead to sustained proliferation, metabolic reprogramming, anti-apoptosis, remodeling of the tumor microenvironment, evasion of the immune response, cell migration, and metastasis. An activating point mutation in codon G12 is the most common (98%) locus of KRAS mutation in pancreatic adenocarcinoma, but all drugs targeting this mutation have failed in clinical practice.15 Additionally, inactivation of tumor suppressor genes such as p53, DPC4 (SMAD4/MADH4), CDKN2A (p16/MTS1), and BRCA2 can be found in 75%, 30%, 35%, and 4% of pancreatic adenocarcinoma cases, respectively.14 Another pancreatic cancer hallmark is inactivation of DNA damage repair genes, which include MLH1 and MSH2.16
Diagnosis and Staging
Case Presentation
A 71-year-old male veteran with no significant past medical history other than hypertension and hyperlipidemia and an excellent performance status presents to the emergency department after noticing a yellowish skin and sclera color. He denies weight loss, abdominal pain, or any other pertinent symptom or sign. Physical examination reveals a healthy developed man with yellowish discoloration of the skin and sclera and a soft, nontender benign abdomen; physical examination is otherwise unremarkable. Laboratory evaluation reveals a direct bilirubin level of 4.5 mg/dL and normal values for complete blood count and renal, liver, and coagulation panels. Abdominal and pelvis computed tomography (CT) with intravenous contrast shows a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein, which remains patent. Follow-up endoscopic ultrasound (EUS) confirms an irregular mass at the head of the pancreas measuring 3.2 × 2.6 cm with sonographic evidence suggesting invasion into the portal vein. During the procedure, the bile duct is successfully stented, the mass is biopsied, and bile duct brushing is performed. Pathology report is consistent with pancreatic adenocarcinoma.
- What is the typical presentation of pancreatic cancer?
The most common symptoms of pancreatic cancer at the time of presentation include weight loss (85%), asthenia/anorexia (86%), and/or abdominal pain (79%).17 The most frequent signs are jaundice (55%), hepatomegaly (39%), and cachexia (13%). Courvoisier sign, a nontender but palpable distended gallbladder at the right costal margin, is neither sensitive nor specific for pancreatic cancer (13% of cases). Trousseau syndrome, a superficial thrombophlebitis, is another classic sign that reflects the hypercoagulable nature of pancreatic cancer (3% of cases).17 The pathophysiology of this syndrome is not completely understood, but it may occur secondary to the release of cancer microparticles in the blood stream which in turn stimulate the coagulation cascade. Other nonspecific symptoms are dark urine, nausea, vomiting, diarrhea, steatorrhea, and epigastric and back pain. Because symptoms early in the course of the disease are nonspecific, pancreatic cancer is typically diagnosed late, after the cancer has invaded local structures or metastasized. The initial presentation varies depending on tumor location, with 70% of pancreatic head malignancies presenting with jaundice and pain correlating to an advanced stage.18 Although data supporting an association between new-onset diabetes mellitus and pancreatic cancer are inconclusive, pancreatic cancer should still be a consideration in patients with new-onset diabetes mellitus and other symptoms such as pain and weight loss. Early signs of incurable disease include a palpable mass, ascites, lymphadenopathy (classic Virchow node), and an umbilical mass (Sister Mary Joseph node). Incidentally discovered pancreatic masses on imaging are rare, but the incidence is increasing due to frequent imaging for other reasons and improved diagnostic techniques.
- What is the approach to diagnosis and staging?
History and physical examination findings are not sufficiently sensitive or specific to diagnose pancreatic cancer. High clinical suspicion in a patient with risk factors can lead to a comprehensive evaluation and potential early diagnosis. In general, an initial diagnostic work-up for suspected pancreatic cancer will include serologic evaluation (liver function test, lipase, tumor markers) and abdominal imaging (ultrasound, CT scans, or magnetic resonance imaging [MRI]). Ultrasound is a first-line diagnostic tool with a sensitivity of 90% and specificity of 98.8% for pancreatic cancer, but it is investigator-dependent and is less accurate in detecting tumors smaller than 3 cm in diameter.19 Multiphasic helical CT of the abdomen has better sensitivity (100%) and specificity (100%) for detecting tumors larger than 2 cm, but this modality is less accurate in detecting pancreatic masses smaller than 2 cm (77%).20 Percutaneous fine-needle aspiration (FNA) performed by ultrasound or CT guidance is avoided due to theoretical concerns about intraperitoneal seeding and bleeding.
If a pancreatic mass is detected by ultrasound or CT, additional interventions may be indicated depending on the clinical scenario. EUS-guided biopsy can provide histological confirmation and is currently utilized frequently for diagnosis and early resectability staging. Endoscopic retrograde cholangiopancreatography (ERCP) is indicated for patients with biliary obstruction requiring stent placement, and this procedure may provide tissue confirmation by forceps biopsy or brush cytology (lower accuracy than EUS). In a meta-analysis that evaluated the diagnostic value of tests for pancreatic cancer, ERCP had the highest sensitivity (92%) and specificity (96%) compared to ultrasound and CT,21 but this modality carries a risk for pancreatitis, bleeding, and cholangitis. Magnetic resonance cholangiopancreatography has not replaced ERCP, but it but may be an alternative for patients who cannot undergo ERCP (eg, gastric outlet obstruction, duodenal stenosis, anatomical surgical disruption, unsuccessful ERCP). ERCP is used frequently because many patients present with obstructive jaundice due to pancreatic mass compression, specifically if the mass is located in the head, and must undergo ERCP and stenting of the common bile duct.
The carbohydrate antigen (CA) 19-9 level has variable sensitivity and specificity in pancreatic cancer, as levels can be elevated in many benign pancreaticobiliary disorders. Elevated CA 19-9, in the appropriate clinical scenario (ie, a suspicious pancreatic mass and a value greater than 37 U/mL) demonstrated a sensitivity of 77% and specificity of 87% when differentiating pancreaticobiliary cancer from benign clinical conditions such as acute cholangitis or cholestasis.22 CA 19-9 level has prognostic value, as it may predict occult disease and correlates with survival rates, but no specific cutoff value has been established to guide perioperative therapy for high-risk resectable tumors.23
The American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) tumor, node, metastasis (TNM) system is the preferred method for staging pancreatic cancer (Table 1).
Positron emission tomography with CT scan is occasionally utilized in practice to assess tumor burden by evaluating anatomical structures and assessing physiologic uptake, which aids in establishing the extent of disease in equivocal cases. Staging laparoscopy with or without peritoneal biopsy is sometimes used to establish appropriate staging in cases that are questionable for occult metastatic disease. This procedure helps avoid unnecessary morbid surgeries.
Neoadjuvant Therapy
Case Continued
The patient is referred to oncology. Blood work reveals a CA 19-9 level of 100 U/mL (reference range < 35 U/mL) and a staging CT scan of the chest reveals a benign-appearing 3-mm nodule (no prior imaging for comparison). CT scan of the abdomen and pelvis does not define venous vasculature involvement appropriately and hence MRI of the abdomen and pelvis is performed. MRI reveals a pancreatic head mass measuring 3.0 × 2.7 cm, without arterial or venous vasculature invasion. However, the mass is abutting the portal vein and superior mesenteric vein and there is a new nonspecific 8-mm aortocaval lymph node.
- What are the current approaches to treating patients with resectable, unresectable, and metastatic disease?
Accurate staging and assessment of surgical resectability in pancreatic cancer are paramount as these steps prevent a futile morbid Whipple procedure in patients with advanced disease and a high risk of recurrence. Conversely, it allows patients with low-volume disease to undergo a potentially curative surgery. Approximately 20% of patients present with resectable disease, 40% present with locally advanced unresectable tumors (eg, involvement of critical vascular structures), and 40% present with metastatic disease.3 Treatment for resectable pancreatic cancer continues to be upfront surgery, although neoadjuvant therapy with either chemoradiation, radiation alone, or chemotherapy is an option per guidelines from the American Society of Clinical Oncology (ASCO),28 the NCCN,26 and the European Society for Medical Oncology (ESMO),29 particularly for patients with borderline resectable tumors (Table 3).
Systemic chemotherapy is recommended for fit candidates with locally advanced unresectable or metastatic disease, with an emphasis on supportive palliative measures. Palliative interventions include biliary stenting, duodenal stent for relieving gastric-outlet obstruction, and celiac axis nerve blocks, when indicated. Routine preoperative biliary stent placement/drainage in patients undergoing subsequent surgery for pancreatic cancer located in the head is associated with an increased risk of surgical complications when compared with up-front surgery without prior biliary drainage, and thus stent placement/drainage is not recommended.26 Aggressive supportive management of symptoms, such as cancer-associated pain, anorexia-cachexia syndromes, and anxiety-depression disorders, should remain a primary palliative focus.
Case Continued
A multidisciplinary tumor board discusses the patient’s case and deems the cancer borderline resectable; neoadjuvant therapy is recommended. The patient is started on treatment with gemcitabine and nab-paclitaxel as first-line neoadjuvant therapy. After 4 cycles, the CA 19-9 level drops to 14 U/mL, and MRI reveals a smaller head mass of 1.3 × 1.4 cm with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remains stable. At tumor board, it is evident that the patient has responded to therapy and the recommendation is to treat with gemcitabine chemoradiotherapy before surgery.
- What neoadjuvant therapy strategies are used in the treatment of pancreatic adenocarcinoma?
There are no established evidence-based recommendations for neoadjuvant therapy in patients with borderline resectable pancreatic cancer or patients with unresectable locally advanced pancreatic cancer. However, there are ongoing trials to investigate this treatment approach, and it is offered off-label in specific clinical scenarios, such as in the case patient described here. In patients with borderline resectable disease, preoperative chemotherapy followed by chemoradiation is a routine practice in most cancer centers,32 and ongoing clinical trials are an option for this cohort of patients (eg, Southwest Oncology Group Trial 1505, NCT02562716). The definitions of borderline resectable and unresectable pancreatic cancer have been described by the NCCN,26 although most surgeons consider involvement of the major upper abdominal blood vessels the main unresectability criterion; oncologists also consider other parameters such as suspicious lesions on scans, worsening performance status, and a significantly elevated CA 19-9 level suggestive of disseminated disease.28 The goal of a conversion approach by chemotherapy with or without radiation for borderline and unresectable cancers is to deliver a tolerable regimen leading to tumor downstaging, allowing for surgical resection. No randomized clinical trial has shown a survival advantage of this approach. Enrollment in clinical trials is preferred for patients with borderline and unresectable cancer, and there are trials that are currently enrolling patients.
The main treatment strategies for patients with locally advanced borderline and unresectable pancreatic cancer outside of a clinical trial are primary radiotherapy, systemic chemotherapy, and chemoradiation therapy. Guidelines from ASCO, NCCN, and ESMO recommend induction chemotherapy followed by restaging and consolidation chemoradiotherapy in the absence of progression.26,28,29 There is no standard chemoradiation regimen and the role of chemotherapy sensitizers, including fluorouracil, gemcitabine, and capecitabine (an oral fluoropyrimidine substitute), and targeted agents in combination with different radiation modalities is now being investigated.
Fluorouracil is a radio-sensitizer that has been used in locally advanced pancreatic cancer based on experience in other gastrointestinal malignancies; data shows conflicting results with this drug. Capecitabine and tegafur/gimeracil/oteracil (S-1) are oral prodrugs that can safely replace infusional fluorouracil. Gemcitabine, a more potent radiation sensitizer, is very toxic, even at low-doses twice weekly, and does not provide a survival benefit, as demonstrated in the Cancer and Leukemia Group B (CALGB) 89805 trial, a phase 2 study of patients with surgically staged locally advanced pancreatic cancer.33 Gemcitabine-based chemoradiotherapy was also evaluated in the Eastern Cooperative Group (ECOG) E4201 trial, which randomly assigned patients to receive gemcitabine alone (at 1000 mg/m2/wk for weeks 1 through 6, followed by 1 week rest, then weekly for 3 out of 4 weeks) or gemcitabine (600 mg/m2/wk for weeks 1 to 5, then 4 weeks later 1000 mg/m2 for 3 out of 4 weeks) plus radiotherapy (starting on day 1, 1.8 Gy/fraction for total of 50.4 Gy).34 Patients with locally advanced unresectable pancreatic cancer had a better OS outcome with gemcitabine in combination with radiation therapy (11.1 months) as compared with patients who received gemcitabine alone (9.2 months). Although there was a greater incidence of grade 4 and 5 treatment-related toxicities in the combination arm, no statistical differences in quality-of-life measurements were reported. Gemcitabine-based and capecitabine-based chemoradiotherapy were compared in the open-label phase 2 multicenter randomized SCALOP trial.35 Patients with locally advanced pancreatic cancer were assigned to receive 3 cycles of induction with gemcitabine 1000 mg/m2 days 1, 8, and 15 and capecitabine 830 mg/m2 days 1 to 21 every 28 days; patients who had stable or responding disease were randomly assigned to receive a fourth cycle followed by capecitabine (830 mg/m2 twice daily on weekdays only) or gemcitabine (300 mg/m2 weekly) with radiation (50.4 Gy over 28 fractions). Patients treated with capecitabine-based chemoradiotherapy had higher nonsignificant median OS (17.6 months) and median progression-free survival (12 months) compared to those treated with gemcitabine (14.6 months and 10.4 months, respectively).
The benefit of radiation therapy in the treatment of locally advanced pancreatic cancer was further explored by the Fédération Francophone de Cancérologie Digestive 2000-01 phase 3 trial. This study compared induction chemoradiotherapy (60 Gy, 2 Gy/fraction; concomitant fluorouracil infusion, 300 mg/m2/day, days 1–5 for 6 weeks; cisplatin, 20 mg/m2/day, days 1–5 during weeks 1 and 5) to gemcitabine alone (1000 mg/m2 weekly for 7 weeks) followed by maintenance gemcitabine in both arms.36 Unexpectedly, the median OS was significantly shorter in the chemoradiotherapy arm than in the chemotherapy alone arm (8.6 months versus 13 months, respectively, P = 0.03) and the combination arm had more toxicities. The phase 3 open-label LAP07 study explored the role of radiation therapy in patients with locally advanced pancreatic cancer who had controlled disease after 4 months of induction therapy.37 LAP07 had 2 randomizations: first, patients with locally advanced pancreatic cancer were assigned to receive weekly gemcitabine alone (1000 mg/m2) or this same dose of gemcitabine plus erlotinib 100 mg/day; second, patients with progression-free disease (61% of initial cohort) after 4 months of therapy were assigned to receive 2 months of the same chemotherapy or chemoradiotherapy (54 Gy plus capecitabine). This study showed that the addition of erlotinib to gemcitabine did not improve survival and in fact affected survival adversely. Of note, no survival benefit was observed after the first randomization from chemotherapy to consolidating chemoradiotherapy. Chemoradiotherapy achieved better locoregional tumor control with significantly less local tumor progression (32% versus 46%, P < 0.03) and no increase in toxicity. Based on prior moderate-quality evidence, guidelines recommend consolidative chemoradiotherapy only for surgical resection candidates following induction chemotherapy; for those who are not surgical candidates, guidelines recommend continuing systemic therapy.26,28,29
Gemcitabine and fluorouracil-based chemotherapies were the standard induction regimens until evidence from studies of metastatic systemic treatment protocols with FOLFIRINOX (ACCORD trial38) and nanoparticle albumin-bound paclitaxel (nab-paclitaxel) plus gemcitabine (MPACT trial39) was extrapolated to clinical practice. These regimens were shown to achieve higher objective response rates when compared to single-agent gemcitabine in patients with metastatic pancreatic cancer. Due to the broad heterogeneity of results in small retrospective series with neoadjuvant trials in borderline resectable pancreatic cancer, the quality of the evidence is low and any recommendation is limited. Many individual series have demonstrated improved complete resection rates and promising survival rates. In the largest single-institution retrospective review of patients with borderline resectable pancreatic adenocarcinoma who completed neoadjuvant gemcitabine-based chemoradiotherapy (50 Gy in 28 fractions or 30 Gy in 10 fractions), 94% achieved a margin-negative pancreatectomy; the median OS in those who completed preoperative therapy and had surgery was 40 months, with a 5-year OS of 36%.40 A meta-analysis by Andriulli and colleagues included 20 prospective studies of patients with initially resectable (366 lesions) or unresectable (341 lesions) disease who were treated with neoadjuvant/preoperative gemcitabine with or without radiotherapy.41 In the group with initially unresectable disease, 39% underwent surgery after restaging and 68% of explored patients were resected; the R0 resection rate was 60%. After restaging, 91% of patients with resectable disease underwent surgery, with 82% of explored patients undergoing surgical resection and 89% of these achieving R0 resection. The estimated 1- and 2-year survival probabilities after resection among patients with initially unresectable disease were 86.3% and 54.2%.41
The largest single-institution retrospective review of FOLFIRINOX (fluorouracil, oxaliplatin, irinotecan, and leucovorin), an alternative to gemcitabine, for neoadjuvant induction therapy for patients with locally advanced unresectable disease was conducted at Memorial Sloan Kettering Cancer Center. In this study (n = 101), 31% of patients initially deemed unresectable who completed FOLFIRINOX induction therapy with or without chemoradiation underwent resection. The R0 resection rate in these patients was 55%, and patients who did not progress during induction FOLFIRINOX therapy had a median OS of 26 months.42 A systematic review and meta-analysis of FOLFIRINOX chemotherapy with or without radiotherapy in patients with locally advanced unresectable pancreatic cancer reported that 25.9% of patients underwent resection after FOLFIRINOX therapy, and the R0 resection rate in these patients was 78.4%.43 The median OS in this study was 24.2 months, which was longer than the previously reported median OS rates for gemcitabine.
There is no strong evidence published for the use of combination nab-paclitaxel plus gemcitabine in the neoadjuvant setting, but it is used in clinical practice based on evidence from the MPACT trial, which showed the combination improved OS and progression-free survival in patients with metastatic pancreatic cancer.39 An early-phase 1-arm clinical trial of neoadjuvant gemcitabine, docetaxel, and capecitabine (GTX) followed by radiotherapy showed an increased response rate and survival for locally advanced disease; however, the NCCN expert panel has reached a consensus but not a uniform recommendation regarding this regimen due to significant toxicities and low patient accrual.26 Selected patients with pancreatic cancer with BRCA1/2 mutations are more sensitive to platinum-based chemotherapy. Although studies of neoadjuvant platinum-based chemotherapy in this population have not been reported, the NCCN guidelines list it as an alternative option based on extrapolated data.26 A clinical trial of gemcitabine, nab-paclitaxel, and cisplatin in the neoadjuvant setting in patients with resectable pancreatic cancer is currently enrolling patients (NGC triple regimen NCT0339257).
Summary
Chemotherapy alone or followed by chemoradiotherapy may be used as initial treatment for patients with borderline and unresectable pancreatic adenocarcinoma without distant metastases who are potential surgical candidates. Chemoradiotherapy remains a preferred treatment option for patients with poorly controlled pain from local tumor invasion, in view of the well-documented analgesic palliative effect of radiation therapy. FOLFIRINOX with or without radiation therapy may offer the highest documented response rates, but it also results in higher rates of treatment-related toxicities. FOLFIRINOX can be offered to selected fit patients (< 65 years old, no comorbidity contraindication, good functional status [ECOG 0–1]) who can tolerate triple therapy with a more toxic adverse-effect profile. A clinical trial evaluating neoadjuvant FOLFIRINOX with or without preoperative chemoradiotherapy in patients with borderline resectable pancreatic cancer is ongoing (PANDAS-PRODIGE 44, NCT02676349). Gemcitabine with or without radiation therapy is a tolerable combination, although it is potentially more toxic when combined with radiation. The addition of nab-paclitaxel to gemcitabine without radiation may emerge as a preferred neoadjuvant treatment for selected patients; a clinical trial investigating this modality in patients with resectable and borderline resectable disease is ongoing (NCT02723331).
Adjuvant Therapy
Case Continued
Prior to the planned surgical resection and after undergoing chemoradiation therapy, the patient has an excellent performance status and repeat MRI shows a 1.3 × 1.4 cm head mass with no further vasculature involvement, no evidence of lymphadenopathy, and no distant metastasis. The CA 19-9 level is stable at 18 U/mL. The patient undergoes an uncomplicated partial pancreaticoduodenectomy, and analysis of a surgical pathology specimen reveals T3N0 disease with closest margin of 0.1 cm.
- Would the patient benefit from adjuvant therapy?
Adjuvant chemotherapy for 6 months after pancreatic cancer resection should be offered to all patients based on mature data. Gemcitabine and capecitabine are the current standard of care in adjuvant therapy; alternatively, single-agent gemcitabine can be offered to patients with poor performance status or patients who cannot tolerate the toxicities associated with this combination.28 Adjuvant treatment should be initiated within approximately 8 weeks of surgical resection. The value of radiation therapy remains controversial, but it can be offered within the context of a clinical trial or to patients with positive margins after surgical resection and/or lymph node–positive disease. Based on low-quality supportive evidence, it is strongly recommended that patients who receive neoadjuvant therapy complete a total of 6 months of chemotherapy, factoring in the duration of the preoperative regimen.28 Different adjuvant strategies have been investigated, including chemotherapy alone with a fluoropyrimidine and/or gemcitabine with or without combined chemoradiation therapy.
The European Study Group for Pancreatic Cancer 1 (ESPAC)-1 trial was a randomized clinical trial that evaluated several adjuvant strategies in pancreatic cancer treatment. This trial assigned patients who underwent pancreatic adenocarcinoma resection to adjuvant chemotherapy alone (intravenous fluorouracil 425 mg/m2 and leucovorin 20 mg/m2 daily for 5 days, monthly for 6 months), chemoradiotherapy (20 Gy in 10 daily fractions over 2 weeks with 500 mg/m2 intravenous fluorouracil on days 1–3, repeated after 2 weeks), both chemotherapy and chemoradiation, and observation.44 The results showed no added benefit for adjuvant chemoradiotherapy, with a median OS of 15.5 months in the chemoradiotherapy cohort, as compared to a median OS of 16.1 months in the chemotherapy-alone cohort (hazard ratio [HR] 1.18 [95% CI 0.90 to 1.55], P = 0.24). In addition, there was evidence of a survival benefit for the chemotherapy-alone arm when compared to the combined modality arm, with a median OS of 19.7 versus 14.0 months, respectively (HR 0.66 [95% CI 0.52 to 0.83], P = 0.0005). Although ESPAC-1 has been criticized for being underpowered to perform statistical comparison, it is still considered a landmark trial demonstrating benefit with single-agent chemotherapy alone. A follow-up analysis of ESPAC-1 showed that adjuvant chemotherapy alone conferred a significant 5-year survival benefit while the combined modality had a deleterious effect on survival. 45 Hence, adjuvant chemotherapy alone became the standard of care in the United States following resection.
The results of the multicenter randomized controlled phase 3 CONKO-001 (CharitéOnkologie 001) trial, which were reported in 2007, supported the use of adjuvant gemcitabine for 6 months in patients with resected pancreatic adenocarcinoma. In this study, patients treated with adjuvant gemcitabine (1000 mg/m2 days 1, 8, and 15 every 4 weeks for 6 months) had superior disease-free survival compared with those who received surgery alone.30 A long-term outcome update of this study demonstrated a significant improvement in 5-year OS for patients treated with adjuvant gemcitabine (20.7% [95% CI 14.7% to 26.6%]) compared to those who received surgical resection alone (10.4% [95% CI 5.9% to 15.0%]). This benefit persisted at 10-year follow-up, with an OS of 12.2% (95% CI 7.3% to 17.2%) in the adjuvant gemcitabine group, as compared to 7.7% (95% CI 3.6% to 11.8%) in the resection alone group.31
Fluorouracil and gemcitabine remained equivalent adjuvant treatment options until the results of the ESPAC-3 trial were reported in 2010.32 This large phase 3 trial, conducted mainly in the United Kingdom, compared weekly gemcitabine (1000 mg/m2 weekly for 3 of every 4 weeks) to leucovorin-modulated fluorouracil (Mayo Clinic regimen: leucovorin 20 mg/m2 followed by fluorouracil 425 mg/m2 intravenous bolus days 1 through 5 every 28 days) as adjuvant therapy in resected pancreatic adenocarcinoma. After a median follow-up of 34.2 months, the median OS was similar in the 2 groups (fluorouracil/leucovorin 23.0 months versus gemcitabine 23.6 months; P = 0.39). However, the fluorouracil/leucovorin group experienced more grade 3/4 treatment-related toxicities (mucositis, stomatitis, diarrhea, and hosptializations; 14% versus 7.5%; P < 0.001).46 Following this trial, gemcitabine became the standard of care for adjuvant chemotherapy for resected pancreatic cancer.
The U.S. Radiation Therapy Oncology Group (RTOG) 9704 trial was conducted to investigate the potential benefit of adding radiation therapy to gemcitabine. This trial demonstrated an improved trend among patients with pancreatic head tumors (but not with cancers of the pancreatic body or tail) who received adjuvant gemcitabine followed by chemoradiotherapy (50.4 Gy in 1.8 Gy daily fractions for 5.5 weeks with concurrent infusional fluorouracil 250 mg/m2 daily) and subsequent gemcitabine monotherapy compared to postoperative fluorouracil-based chemoradiotherapy. Results showed a 5-year OS of 22% versus 18%, respectively, although this improvement was not statistically significant (P = 0.08). This trial failed to show a benefit of adding radiotherapy to gemcitabine.47
The ESPAC-4 trial, reported in 2017, evaluated the combination of gemcitabine and capecitabine compared to gemcitabine alone as adjuvant therapy for resected pancreatic adenocarcinoma.48 Patients were randomly assigned after surgical resection, regardless of margin or node status, to 6 months of gemcitabine alone (1000 mg/m2/day on days 1, 8, and 15 of each 28-day cycle) or gemcitabine plus capecitabine (1660 mg/m2/day on days 1 through 21 of each 28-day cycle). Combination therapy had a significant survival benefit compared to single therapy, with median OS durations of 28 months and 25.5 months, respectively (HR for death 0.82 [95% CI 0.68 to 0.98]). The 5-year OS for patients who received combination treatment was 29 months (95% CI 22.9 to 35.2) versus 16 months (95% CI 10.2 to 23.7) for those in the monotherapy group. As expected, grade 3 or 4 treatment-related toxicities (diarrhea, hand-foot syndrome, and neutropenia) were significantly more common with combined therapy, although there were no significant differences in the rates of serious adverse events. The adjuvant combination of gemcitabine and capecitabine became the current and preferred new standard of care following resection of pancreatic ductal adenocarcinoma,28 but single-agent gemcitabine and fluorouracil/leucovorin continue to be viable options,26,28,29 particularly for elderly patients, patients with borderline performance status, or patients with multiple comorbidities.
Evidence showing that a more intensive regimen can improve outcome in the adjuvant setting remains elusive. The phase 3 APACT study (Adjuvant Therapy for Patients with Resected Pancreatic Cancer, NCT01964430) comparing gemcitabine alone to gemcitabine plus nab-paclitaxel in patients with surgically resected pancreatic adenocarcinoma has concluded, with the results projected to be released in 2018. Another phase 3 trial investigating the efficacy of FOLFIRINOX versus gemcitabine alone as adjuvant therapy is underway in France and Canada (PRODIGE24/ACCORD24, NCT01526135). Other strategies with newer targeted therapies and immunotherapy are in the development phase.
Follow-Up and Surveillance
Case Conclusion
After recovery from surgery, the patient is offered and completes 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine. He is started on surveillance at 3 and 6 months, and he maintains an excellent performance status. He develops clinical evidence of pancreatic enzyme insufficiency and is placed on oral replacement therapy. He has no other complaints, and there is no evidence of recurrence on MRI and CA 19-9 levels.
- What is the recommended duration of surveillance following curative resection?
Surveillance after curative resection of pancreatic adenocarcinoma is recommended by NCCN guidelines.26 However, pancreatic adenocarcinoma has a poor prognosis, and surveillance after curative surgical resection with or without perioperative therapy has not been shown to impact survival. Most recurrences will occur within 2 years after treatment. Surveillance recommendations differ among expert groups.26,28,29 NCCN guidelines recommend evaluating patients by history and physical examination every 3 to 6 months for the first 2 years, then every 6 to 12 months for 3 years. CA 19-9 level and CT scan should be obtained every 3 to 6 months for 2 years and then every 6 to 12 months for 3 years. Follow-up with CA 19-9 levels and CT scans after 5 years is not routinely performed unless guided by signs, symptoms, or laboratory findings that raise suspicion for recurrence. Follow-up visits should also include evaluation of treatment-related toxicities, symptom management, nutrition support of pancreatic insufficiency, and psychosocial support.
Conclusion
Pancreatic cancer is a leading cause of cancer-related death that frequently presents with locally advanced or metastatic disease due to nonspecific symptoms and lack of a screening modality. Histological tissue biopsy confirmation and accurate resectability staging guide treatment planning and prognosis. The only potentially curative therapy is surgical resection plus adjuvant therapy for those with resectable disease. Surgical candidates with borderline resectable and unresectable disease can be offered induction preoperative chemotherapy followed by consolidation chemoradiation, based on clinical consensus practice. Enrollment in clinical trials should be encouraged for all patients, as evidence from clinical trials is essential to making progress in pancreatic cancer treatment.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
2. Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69.
3. Kamarajah SK, Burns WR, Frankel TL, et al. Validation of the American Joint Commission on Cancer (AJCC) 8th edition staging system for patients with pancreatic adenocarcinoma: a Surveillance, Epidemiology and End Results (SEER) analysis. Ann Surg Oncol 2017;24:2023–30.
4. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer.gov/statfacts/html/pancreas.html. Accessed 17 February 2018.
5. Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med 1996;156:2255–60.
6. Michaud DS, Giovannucci E, Willett WC, et al. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001;286:921–9.
7. Batabyal P, Vander Hoorn S, Christophi C, Nikfarjam M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann Surg Oncol 2014;21:2453–62. Epub 2014 Mar 9.
8. Canto MI, Harinck F, Hruban RH, et al, on behalf of the International Cancer of the Pancreas Screening (CAPS) Consortium. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013;62:339–47. Epub 2012 Nov 7.
9. Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res 2004;64:2634–8.
10. McKay SH,Humphris JL, Johns AL, et al. Inherited pancreatic cancer. Cancer Forum 2016;40(1).
11. Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol 2017;35:3382–90.
12. Hruban RH, Pitman MB, Klimstra DS. Tumors of the pancreas. AFIP Atlas of Tumor Pathology. 4th series, fascicle 6. Washington, DC: Armed Forces Institute of Pathology; 2007.
13. Vege SS, Ziring B, Jain R, Moayyedi P, Clinical Guidelines Committee, American Gastroenterology Association. American gastroenterological association institute guideline on the diagnosis and management of asymptomatic neoplastic pancreatic cysts. Gastroenterology 2015;148:819–22.
14. Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501.
15. Choi M, Bien H, Mofunanya A, Powers S. Challenges in Ras therapeutics in pancreatic cancer. Semin Cancer Biol 2017 Nov 21. pii: S1044-579X(17)30235-3.
16. Humphris JL, Patch AM, Nones K, et al. Hypermutation in pancreatic cancer. Gastroenterology 2017;152:68. Epub 2016 Nov 15.
17. Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol 2005;7:189–97.
18. Modolell I, Guarner L, Malagelada JR. Vagaries of clinical presentation of pancreatic and biliary tract cancer. Ann Oncol 1999;10 Suppl 4:82–4.
19. Karlson BM, Ekbom A, Lindgren PG, et al. Abdominal US for diagnosis of pancreatic tumor: prospective cohort analysis. Radiology 1999;213:107–11.
20. Bronstein YL, Loyer EM, Kaur H, et al. Detection of small pancreatic tumors with multiphasic helical CT. AJR Am J Roentgenol 2004;182:619–23.
21. Niederau C, Grendell JH. Diagnosis of pancreatic carcinoma. Imaging techniques and tumor markers. Pancreas 1992;7:66–86.
22. Kim HJ, Kim MH, Myung SJ, et al. A new strategy for the application of CA19-9 in the differentiation of pancreaticobiliary cancer: analysis using a receiver operating characteristic curve. Am J Gastroenterol 1999;94:1941–6.
23. Khorana AA, Mangu PB, Berlin J, et al. Potentially curable pancreatic cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2016;34:2541–56.
24. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
25. Soriano A, Castells A, Ayuso C, et al. Preoperative staging and tumor resectability assessment of pancreatic cancer: prospective study comparing endoscopic ultrasonography, helical computed tomography, magnetic resonance imaging, and angiography. Am J Gastroenterol 2004;99:492–501.
26. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
27. Al-Hawary MM, Francis IR, Chari ST, et al. Pancreatic ductal adenocarcinoma radiology reporting template: consensus statement of the Society of Abdominal Radiology and the American Pancreatic Association. Radiology 2014;270:248–60.
28. Khorana AA, Mangu PB, Berlin J, et al. Potentially curable pancreatic cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol 2017;35:2324–8.
28. Ducreux M, Cuhna AS, Caramella C, et al; ESMO Guidelines Committee. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2015;26 Suppl 5:v56–68.
30. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
31. Oettle H, Neuhaus P, Hochhaus A, et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. JAMA 2013;310:1473–81.
32. Huguet F, Girard N, Guerche CS, et al. Chemoradiotherapy in the management of locally advanced pancreatic carcinoma: a qualitative systematic review. J Clin Oncol 2009;27:2269–77.
33. Blackstock AW, Tepper JE, Niedwiecki D, et al. Cancer and leukemia group B (CALGB) 89805: phase II chemoradiation trial using gemcitabine in patients with locoregional adenocarcinoma of the pancreas. Int J Gastrointest Cancer 2003;34(2-3):107–16.
34. Loehrer PJ Sr, Feng Y, Cardenes H, et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an Eastern Cooperative Oncology Group trial. J Clin Oncol 2011;29:4105–12.
35. Hurt CN, Falk S, Crosby T, et al. Long-term results and recurrence patterns from SCALOP: a phase II randomised trial of gemcitabine- or capecitabine-based chemoradiation for locally advanced pancreatic cancer. Br J Cancer 2017;116:1264–70.
36. Chauffert B, Mornex F, Bonnetain F, et al. Phase III trial comparing intensive induction chemoradiotherapy (60 Gy, infusional 5-FU and intermittent cisplatin) followed by maintenance gemcitabine with gemcitabine alone for locally advanced unresectable pancreatic cancer. Definitive results of the 2000-01 FFCD/SFRO study. Ann Oncol 2008;19:1592–9.
37. Hammel P, Huguet F, van Laethem JL, et al, LAP07 Trial Group. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA 2016;315:1844–53.
38. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
39. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–703.
40. Katz MH, Pisters PW, Evans DB, et al. Borderline resectable pancreatic cancer: the importance of this emerging stage of disease. J Am Coll Surg 2008;206:833–46.
41. Andriulli A, Festa V, Botteri E, et al. Neoadjuvant/preoperative gemcitabine for patients with localized pancreatic cancer: a meta-analysis of prospective studies. Ann Surg Oncol 2012;19:1644–62.
42. Sadot E, Doussot A, O’Reilly EM, et al. FOLFIRINOX induction therapy for stage 3 pancreatic adenocarcinoma. Ann Surg Oncol 2015;22:3512–21.
43. Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol 2016;17:801–10.
44. Neoptolemos JP, Dunn JA, Stocken DD, et al, European Study Group for Pancreatic Cancer. Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: a randomised controlled trial. Lancet 2001;358:1576–85.
45. Neoptolemos JP, Stocken DD, Friess H, et al, European Study Group for Pancreatic Cancer. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 2004;350:1200–10.
46. Neoptolemos JP, Stocken DD, Bassi C, et al, European Study Group for Pancreatic Cancer. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA 2010;304:1073–81.
47. Regine WF, Winter KA, Abrams RA, et al. Fluorouracil vs gemcitabine chemotherapy before and after fluorouracil-based chemoradiation following resection of pancreatic adenocarcinoma: a randomized controlled trial. JAMA 2008;299:1019–26.
48. Neoptolemos JP, Palmer DH, Ghaneh P, et al, European Study Group for Pancreatic Cancer. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389:1011–24. Epub 2017 Jan 25.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
2. Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69.
3. Kamarajah SK, Burns WR, Frankel TL, et al. Validation of the American Joint Commission on Cancer (AJCC) 8th edition staging system for patients with pancreatic adenocarcinoma: a Surveillance, Epidemiology and End Results (SEER) analysis. Ann Surg Oncol 2017;24:2023–30.
4. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer.gov/statfacts/html/pancreas.html. Accessed 17 February 2018.
5. Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med 1996;156:2255–60.
6. Michaud DS, Giovannucci E, Willett WC, et al. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001;286:921–9.
7. Batabyal P, Vander Hoorn S, Christophi C, Nikfarjam M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann Surg Oncol 2014;21:2453–62. Epub 2014 Mar 9.
8. Canto MI, Harinck F, Hruban RH, et al, on behalf of the International Cancer of the Pancreas Screening (CAPS) Consortium. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013;62:339–47. Epub 2012 Nov 7.
9. Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res 2004;64:2634–8.
10. McKay SH,Humphris JL, Johns AL, et al. Inherited pancreatic cancer. Cancer Forum 2016;40(1).
11. Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol 2017;35:3382–90.
12. Hruban RH, Pitman MB, Klimstra DS. Tumors of the pancreas. AFIP Atlas of Tumor Pathology. 4th series, fascicle 6. Washington, DC: Armed Forces Institute of Pathology; 2007.
13. Vege SS, Ziring B, Jain R, Moayyedi P, Clinical Guidelines Committee, American Gastroenterology Association. American gastroenterological association institute guideline on the diagnosis and management of asymptomatic neoplastic pancreatic cysts. Gastroenterology 2015;148:819–22.
14. Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501.
15. Choi M, Bien H, Mofunanya A, Powers S. Challenges in Ras therapeutics in pancreatic cancer. Semin Cancer Biol 2017 Nov 21. pii: S1044-579X(17)30235-3.
16. Humphris JL, Patch AM, Nones K, et al. Hypermutation in pancreatic cancer. Gastroenterology 2017;152:68. Epub 2016 Nov 15.
17. Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol 2005;7:189–97.
18. Modolell I, Guarner L, Malagelada JR. Vagaries of clinical presentation of pancreatic and biliary tract cancer. Ann Oncol 1999;10 Suppl 4:82–4.
19. Karlson BM, Ekbom A, Lindgren PG, et al. Abdominal US for diagnosis of pancreatic tumor: prospective cohort analysis. Radiology 1999;213:107–11.
20. Bronstein YL, Loyer EM, Kaur H, et al. Detection of small pancreatic tumors with multiphasic helical CT. AJR Am J Roentgenol 2004;182:619–23.
21. Niederau C, Grendell JH. Diagnosis of pancreatic carcinoma. Imaging techniques and tumor markers. Pancreas 1992;7:66–86.
22. Kim HJ, Kim MH, Myung SJ, et al. A new strategy for the application of CA19-9 in the differentiation of pancreaticobiliary cancer: analysis using a receiver operating characteristic curve. Am J Gastroenterol 1999;94:1941–6.
23. Khorana AA, Mangu PB, Berlin J, et al. Potentially curable pancreatic cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2016;34:2541–56.
24. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
25. Soriano A, Castells A, Ayuso C, et al. Preoperative staging and tumor resectability assessment of pancreatic cancer: prospective study comparing endoscopic ultrasonography, helical computed tomography, magnetic resonance imaging, and angiography. Am J Gastroenterol 2004;99:492–501.
26. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
27. Al-Hawary MM, Francis IR, Chari ST, et al. Pancreatic ductal adenocarcinoma radiology reporting template: consensus statement of the Society of Abdominal Radiology and the American Pancreatic Association. Radiology 2014;270:248–60.
28. Khorana AA, Mangu PB, Berlin J, et al. Potentially curable pancreatic cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol 2017;35:2324–8.
28. Ducreux M, Cuhna AS, Caramella C, et al; ESMO Guidelines Committee. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2015;26 Suppl 5:v56–68.
30. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
31. Oettle H, Neuhaus P, Hochhaus A, et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. JAMA 2013;310:1473–81.
32. Huguet F, Girard N, Guerche CS, et al. Chemoradiotherapy in the management of locally advanced pancreatic carcinoma: a qualitative systematic review. J Clin Oncol 2009;27:2269–77.
33. Blackstock AW, Tepper JE, Niedwiecki D, et al. Cancer and leukemia group B (CALGB) 89805: phase II chemoradiation trial using gemcitabine in patients with locoregional adenocarcinoma of the pancreas. Int J Gastrointest Cancer 2003;34(2-3):107–16.
34. Loehrer PJ Sr, Feng Y, Cardenes H, et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an Eastern Cooperative Oncology Group trial. J Clin Oncol 2011;29:4105–12.
35. Hurt CN, Falk S, Crosby T, et al. Long-term results and recurrence patterns from SCALOP: a phase II randomised trial of gemcitabine- or capecitabine-based chemoradiation for locally advanced pancreatic cancer. Br J Cancer 2017;116:1264–70.
36. Chauffert B, Mornex F, Bonnetain F, et al. Phase III trial comparing intensive induction chemoradiotherapy (60 Gy, infusional 5-FU and intermittent cisplatin) followed by maintenance gemcitabine with gemcitabine alone for locally advanced unresectable pancreatic cancer. Definitive results of the 2000-01 FFCD/SFRO study. Ann Oncol 2008;19:1592–9.
37. Hammel P, Huguet F, van Laethem JL, et al, LAP07 Trial Group. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA 2016;315:1844–53.
38. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
39. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–703.
40. Katz MH, Pisters PW, Evans DB, et al. Borderline resectable pancreatic cancer: the importance of this emerging stage of disease. J Am Coll Surg 2008;206:833–46.
41. Andriulli A, Festa V, Botteri E, et al. Neoadjuvant/preoperative gemcitabine for patients with localized pancreatic cancer: a meta-analysis of prospective studies. Ann Surg Oncol 2012;19:1644–62.
42. Sadot E, Doussot A, O’Reilly EM, et al. FOLFIRINOX induction therapy for stage 3 pancreatic adenocarcinoma. Ann Surg Oncol 2015;22:3512–21.
43. Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol 2016;17:801–10.
44. Neoptolemos JP, Dunn JA, Stocken DD, et al, European Study Group for Pancreatic Cancer. Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: a randomised controlled trial. Lancet 2001;358:1576–85.
45. Neoptolemos JP, Stocken DD, Friess H, et al, European Study Group for Pancreatic Cancer. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 2004;350:1200–10.
46. Neoptolemos JP, Stocken DD, Bassi C, et al, European Study Group for Pancreatic Cancer. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA 2010;304:1073–81.
47. Regine WF, Winter KA, Abrams RA, et al. Fluorouracil vs gemcitabine chemotherapy before and after fluorouracil-based chemoradiation following resection of pancreatic adenocarcinoma: a randomized controlled trial. JAMA 2008;299:1019–26.
48. Neoptolemos JP, Palmer DH, Ghaneh P, et al, European Study Group for Pancreatic Cancer. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389:1011–24. Epub 2017 Jan 25.
A Mission for Graduate Medical Education at VA
More than 65% of all physicians who train in the U.S. rotate through a VA hospital at some point during their training. In 2015 alone, more than 43,000 residents received some or all of their clinical training through VA.1 Of the approximately 120 VAMCs that hold academic affiliations
with medical schools and residency training programs, several hold affiliations with multiple institutions, including VA Boston Healthcare System (VABHS) in Massachusetts. The West Roxbury campus is the home of VA Boston’s acute care hospital, where residents and fellows from Boston Medical Center (BMC), Beth Israel Deaconess Medical Center (BIDMC), and Brigham and Women’s Hospital (BWH) train together. These are 3 of the largest medical training programs in Boston, though each provides a unique training experience for residents due to differences in patient population, faculty expertise, and hospital network affiliations (Table 1). 
This diversity brings differences in cultural norms, institutional preferences, and educational expectations. Furthermore, residents from different programs who work together at VA Boston are often meeting one another for the first time, as opportunities for interinstitutional collaboration among these 3 training programs do not exist outside of VA. This training environment presents both an opportunity
and a challenge for medical educators: offering the best possible learning experience for physiciansin-training from multiple programs while providing the best possible care for U.S. veterans.
To guide educators charged with meeting this challenge, the VA Office of Academic Affiliations put forth a mission statement describing its overarching teaching mission (Table 2).2 
To address this gap, chief medical residents from the 3 affiliate residency training programs came together to develop a shared mission statement for what we envision as the educational experience for all medical trainees rotating through VABHS (Table 2). In this article, we describe the development of a mission statement for graduate medical education in internal medicine at VABHS and provides examples of how our mission statement guided educational programming.
Methods
Whereas the affiliated institutions assign generic competency-based learning objectives to rotations at VABHS, no specific overarching educational objectives for residents have been defined previously. The directors of the internal medicine residency programs at each of the VABHS affiliate institutions grant their respective VA-based chief medical residents the autonomy to deliver graduate medical education at VA as they see fit, in collaboration with their colleagues from the other affiliated institutions and the VA director of medical resident education. This autonomy and flexibility allowed each of the chief medical residents to articulate an individual vision for VA graduate medical education based on their affiliate program’s goals, values, and mission.
At the beginning of the 2016/2017 academic year, in partnership with the director of medical resident education at VABHS, the chief medical residents met to reconcile these into a single shared mission statement. Special attention was paid to educational gaps at each affiliate institution that could be filled while residents were rotating at VABHS. Once all educational goals and priorities of the shared mission statement were identified, the chief medical residents and director of medical resident education adopted the mission statement as the blueprint for all educational programming for the academic year. Progress toward enacting the various components of the mission statement was reviewed monthly and changes in educational programming to ensure adequate emphasis of all components were made accordingly.
Results
Our first goal was to promote the personal and professional development of residents who rotate through VABHS, particularly interns, in a setting that fosters cross-institutional collaboration, respect, and friendship. The West Roxbury campus of VABHS is the only hospital in the city where internal medicine residents from 3 large training programs work together on teams that have been intentionally built to place residents from different institutions with one another. In educational conferences, we encouraged residents from different training programs to share their experiences with patient populations that others may not see at their home institutions, based on the specialized care that each institution provides. The conferences also give residents the opportunity to provide and receive near-peer teaching in a collegial environment.
Our second goal was to maintain an environment of educational excellence. We produced thought-provoking conferences that prioritized inspiring curiosity and teaching systems of thought over the dissemination of facts. We regularly focused on the broader context of medicine in case conferences and journal club, including topics such as public health, health policy, advocacy, health economics, quality improvement (QI), and high-value care. Our morning reports were interactive and participatory, emphasizing both technical skill practice and sophisticated clinical reasoning.
We embraced the principles of cognitive learning theory by priming learners with preconference “teasers” that previewed conference topics to be discussed. Every Friday, we played a medical version of Jeopardy!, which used spaced learning to consolidate the week’s teaching points in a fun, collaborative, and collegial atmosphere. Our dedicated patient safety conference gave residents the chance to use QI tools to dissect and tackle real problems in the hospital, and our monthly Morbidity and Mortality conference served as inspiration for many of the resident-driven QI projects.
Our third goal was to challenge physicians to provide the best possible care to veterans, including learning about issues unique to this often-marginalized population. We emphasized that training at a VA hospital is a privilege and that the best way to honor our veterans is to take advantage of the unique learning opportunities available at VA. To that end, we exposed residents to veteran-specific educational content, ranging from the structure and payment model of VHA to service-related medical conditions, such as posttraumatic stress disorder, other mental health issues, traumatic brain injury, Agent Orange exposure, and Gulf War Syndrome.
Discussion
Findings from the recently published Accreditation Council for Graduate Medical Education’s (ACGME) 2016 Clinical Learning Environment Review (CLER) Report support the need for mission statements like ours to guide the delivery of graduate medical education.3 A major finding of this report was that the development and implementation of graduate medical education largely occurs separately from other areas of organizational and strategic focus within clinical learning environments. Our mission statement has served as a road map for aligning the delivery of graduate medical education at VABHS with the specific strengths of the clinical learning environment that VA affords.
Additionally, the 2016 CLER report identified a lack of specificity in training on health care disparities and cultural competency for the specific populations served by the surveyed residency programs. The emphasis we placed on learning about issues specific to the care of the veteran population highlights the potential for other mission statements like ours to bridge the gap between articulation and execution of educational priorities. Finally, through the academic partnerships it holds with more than 90% of medical schools in the U.S., VA already has an integral role in both undergraduate and graduate medical education that positions its hospitals as ideal training environments in which to address shortcomings in medical training like those identified by the ACGME.4
Conclusion
We propose this mission statement as a model for the delivery of graduate medical education throughout all VA hospitals with academic affiliations and especially those where trainees from multiple institutions work together. As embodied in our mission statement, our goal was to provide a clinical training experience at VA that complements that of our residents’ home institutions and fosters a respect for and interest in the special care provided at VA. The development of a shared mission statement provides an invaluable tool in accomplishing that goal. We encourage chief medical residents and other leaders in medical education in all specialties at VAMCs to develop their own mission statements that reflect and embody the values of each affiliated training program. For our residents, rotating at VA is an opportunity to learn the practice of medicine for veterans, rather than practicing medicine on veterans. It is our sincere hope that shaping our residents’ educational experience in this fashion will foster a greater appreciation for the care of our nation’s veterans.
1. VA Office of Academic Affiliations. 2015 statistics: health professions trainees. http://www.va.gov/oaa/docs/OAA_Statistics.pdf. Published 2016. Accessed September 18, 2017.
2. VA Office of Academic Affiliations. Mission of the Office of Academic Affiliations. http://www.va.gov/oaa/oaa_mission.asp. Updated June 23, 2017. Accessed September 18, 2017.
3. Accreditation Council for Graduate Medical Education. Clinical learning environment review – national report of findings 2016 – executive summary. https://www.acgme.org/Portals/0/PDFs/CLER/ACGME-CLER-ExecutiveSummary.pdf. Published 2016. Accessed September 18, 2017.
4. Association of American Medical Colleges. The VA and academic medicine: partners in health care, training, and research. https://www.aamc.org/download/385612/data/07182014.pdf. Accessed September 14, 2017.
More than 65% of all physicians who train in the U.S. rotate through a VA hospital at some point during their training. In 2015 alone, more than 43,000 residents received some or all of their clinical training through VA.1 Of the approximately 120 VAMCs that hold academic affiliations
with medical schools and residency training programs, several hold affiliations with multiple institutions, including VA Boston Healthcare System (VABHS) in Massachusetts. The West Roxbury campus is the home of VA Boston’s acute care hospital, where residents and fellows from Boston Medical Center (BMC), Beth Israel Deaconess Medical Center (BIDMC), and Brigham and Women’s Hospital (BWH) train together. These are 3 of the largest medical training programs in Boston, though each provides a unique training experience for residents due to differences in patient population, faculty expertise, and hospital network affiliations (Table 1).
This diversity brings differences in cultural norms, institutional preferences, and educational expectations. Furthermore, residents from different programs who work together at VA Boston are often meeting one another for the first time, as opportunities for interinstitutional collaboration among these 3 training programs do not exist outside of VA. This training environment presents both an opportunity
and a challenge for medical educators: offering the best possible learning experience for physiciansin-training from multiple programs while providing the best possible care for U.S. veterans.
To guide educators charged with meeting this challenge, the VA Office of Academic Affiliations put forth a mission statement describing its overarching teaching mission (Table 2).2
To address this gap, chief medical residents from the 3 affiliate residency training programs came together to develop a shared mission statement for what we envision as the educational experience for all medical trainees rotating through VABHS (Table 2). In this article, we describe the development of a mission statement for graduate medical education in internal medicine at VABHS and provides examples of how our mission statement guided educational programming.
Methods
Whereas the affiliated institutions assign generic competency-based learning objectives to rotations at VABHS, no specific overarching educational objectives for residents have been defined previously. The directors of the internal medicine residency programs at each of the VABHS affiliate institutions grant their respective VA-based chief medical residents the autonomy to deliver graduate medical education at VA as they see fit, in collaboration with their colleagues from the other affiliated institutions and the VA director of medical resident education. This autonomy and flexibility allowed each of the chief medical residents to articulate an individual vision for VA graduate medical education based on their affiliate program’s goals, values, and mission.
At the beginning of the 2016/2017 academic year, in partnership with the director of medical resident education at VABHS, the chief medical residents met to reconcile these into a single shared mission statement. Special attention was paid to educational gaps at each affiliate institution that could be filled while residents were rotating at VABHS. Once all educational goals and priorities of the shared mission statement were identified, the chief medical residents and director of medical resident education adopted the mission statement as the blueprint for all educational programming for the academic year. Progress toward enacting the various components of the mission statement was reviewed monthly and changes in educational programming to ensure adequate emphasis of all components were made accordingly.
Results
Our first goal was to promote the personal and professional development of residents who rotate through VABHS, particularly interns, in a setting that fosters cross-institutional collaboration, respect, and friendship. The West Roxbury campus of VABHS is the only hospital in the city where internal medicine residents from 3 large training programs work together on teams that have been intentionally built to place residents from different institutions with one another. In educational conferences, we encouraged residents from different training programs to share their experiences with patient populations that others may not see at their home institutions, based on the specialized care that each institution provides. The conferences also give residents the opportunity to provide and receive near-peer teaching in a collegial environment.
Our second goal was to maintain an environment of educational excellence. We produced thought-provoking conferences that prioritized inspiring curiosity and teaching systems of thought over the dissemination of facts. We regularly focused on the broader context of medicine in case conferences and journal club, including topics such as public health, health policy, advocacy, health economics, quality improvement (QI), and high-value care. Our morning reports were interactive and participatory, emphasizing both technical skill practice and sophisticated clinical reasoning.
We embraced the principles of cognitive learning theory by priming learners with preconference “teasers” that previewed conference topics to be discussed. Every Friday, we played a medical version of Jeopardy!, which used spaced learning to consolidate the week’s teaching points in a fun, collaborative, and collegial atmosphere. Our dedicated patient safety conference gave residents the chance to use QI tools to dissect and tackle real problems in the hospital, and our monthly Morbidity and Mortality conference served as inspiration for many of the resident-driven QI projects.
Our third goal was to challenge physicians to provide the best possible care to veterans, including learning about issues unique to this often-marginalized population. We emphasized that training at a VA hospital is a privilege and that the best way to honor our veterans is to take advantage of the unique learning opportunities available at VA. To that end, we exposed residents to veteran-specific educational content, ranging from the structure and payment model of VHA to service-related medical conditions, such as posttraumatic stress disorder, other mental health issues, traumatic brain injury, Agent Orange exposure, and Gulf War Syndrome.
Discussion
Findings from the recently published Accreditation Council for Graduate Medical Education’s (ACGME) 2016 Clinical Learning Environment Review (CLER) Report support the need for mission statements like ours to guide the delivery of graduate medical education.3 A major finding of this report was that the development and implementation of graduate medical education largely occurs separately from other areas of organizational and strategic focus within clinical learning environments. Our mission statement has served as a road map for aligning the delivery of graduate medical education at VABHS with the specific strengths of the clinical learning environment that VA affords.
Additionally, the 2016 CLER report identified a lack of specificity in training on health care disparities and cultural competency for the specific populations served by the surveyed residency programs. The emphasis we placed on learning about issues specific to the care of the veteran population highlights the potential for other mission statements like ours to bridge the gap between articulation and execution of educational priorities. Finally, through the academic partnerships it holds with more than 90% of medical schools in the U.S., VA already has an integral role in both undergraduate and graduate medical education that positions its hospitals as ideal training environments in which to address shortcomings in medical training like those identified by the ACGME.4
Conclusion
We propose this mission statement as a model for the delivery of graduate medical education throughout all VA hospitals with academic affiliations and especially those where trainees from multiple institutions work together. As embodied in our mission statement, our goal was to provide a clinical training experience at VA that complements that of our residents’ home institutions and fosters a respect for and interest in the special care provided at VA. The development of a shared mission statement provides an invaluable tool in accomplishing that goal. We encourage chief medical residents and other leaders in medical education in all specialties at VAMCs to develop their own mission statements that reflect and embody the values of each affiliated training program. For our residents, rotating at VA is an opportunity to learn the practice of medicine for veterans, rather than practicing medicine on veterans. It is our sincere hope that shaping our residents’ educational experience in this fashion will foster a greater appreciation for the care of our nation’s veterans.
More than 65% of all physicians who train in the U.S. rotate through a VA hospital at some point during their training. In 2015 alone, more than 43,000 residents received some or all of their clinical training through VA.1 Of the approximately 120 VAMCs that hold academic affiliations
with medical schools and residency training programs, several hold affiliations with multiple institutions, including VA Boston Healthcare System (VABHS) in Massachusetts. The West Roxbury campus is the home of VA Boston’s acute care hospital, where residents and fellows from Boston Medical Center (BMC), Beth Israel Deaconess Medical Center (BIDMC), and Brigham and Women’s Hospital (BWH) train together. These are 3 of the largest medical training programs in Boston, though each provides a unique training experience for residents due to differences in patient population, faculty expertise, and hospital network affiliations (Table 1).
This diversity brings differences in cultural norms, institutional preferences, and educational expectations. Furthermore, residents from different programs who work together at VA Boston are often meeting one another for the first time, as opportunities for interinstitutional collaboration among these 3 training programs do not exist outside of VA. This training environment presents both an opportunity
and a challenge for medical educators: offering the best possible learning experience for physiciansin-training from multiple programs while providing the best possible care for U.S. veterans.
To guide educators charged with meeting this challenge, the VA Office of Academic Affiliations put forth a mission statement describing its overarching teaching mission (Table 2).2
To address this gap, chief medical residents from the 3 affiliate residency training programs came together to develop a shared mission statement for what we envision as the educational experience for all medical trainees rotating through VABHS (Table 2). In this article, we describe the development of a mission statement for graduate medical education in internal medicine at VABHS and provides examples of how our mission statement guided educational programming.
Methods
Whereas the affiliated institutions assign generic competency-based learning objectives to rotations at VABHS, no specific overarching educational objectives for residents have been defined previously. The directors of the internal medicine residency programs at each of the VABHS affiliate institutions grant their respective VA-based chief medical residents the autonomy to deliver graduate medical education at VA as they see fit, in collaboration with their colleagues from the other affiliated institutions and the VA director of medical resident education. This autonomy and flexibility allowed each of the chief medical residents to articulate an individual vision for VA graduate medical education based on their affiliate program’s goals, values, and mission.
At the beginning of the 2016/2017 academic year, in partnership with the director of medical resident education at VABHS, the chief medical residents met to reconcile these into a single shared mission statement. Special attention was paid to educational gaps at each affiliate institution that could be filled while residents were rotating at VABHS. Once all educational goals and priorities of the shared mission statement were identified, the chief medical residents and director of medical resident education adopted the mission statement as the blueprint for all educational programming for the academic year. Progress toward enacting the various components of the mission statement was reviewed monthly and changes in educational programming to ensure adequate emphasis of all components were made accordingly.
Results
Our first goal was to promote the personal and professional development of residents who rotate through VABHS, particularly interns, in a setting that fosters cross-institutional collaboration, respect, and friendship. The West Roxbury campus of VABHS is the only hospital in the city where internal medicine residents from 3 large training programs work together on teams that have been intentionally built to place residents from different institutions with one another. In educational conferences, we encouraged residents from different training programs to share their experiences with patient populations that others may not see at their home institutions, based on the specialized care that each institution provides. The conferences also give residents the opportunity to provide and receive near-peer teaching in a collegial environment.
Our second goal was to maintain an environment of educational excellence. We produced thought-provoking conferences that prioritized inspiring curiosity and teaching systems of thought over the dissemination of facts. We regularly focused on the broader context of medicine in case conferences and journal club, including topics such as public health, health policy, advocacy, health economics, quality improvement (QI), and high-value care. Our morning reports were interactive and participatory, emphasizing both technical skill practice and sophisticated clinical reasoning.
We embraced the principles of cognitive learning theory by priming learners with preconference “teasers” that previewed conference topics to be discussed. Every Friday, we played a medical version of Jeopardy!, which used spaced learning to consolidate the week’s teaching points in a fun, collaborative, and collegial atmosphere. Our dedicated patient safety conference gave residents the chance to use QI tools to dissect and tackle real problems in the hospital, and our monthly Morbidity and Mortality conference served as inspiration for many of the resident-driven QI projects.
Our third goal was to challenge physicians to provide the best possible care to veterans, including learning about issues unique to this often-marginalized population. We emphasized that training at a VA hospital is a privilege and that the best way to honor our veterans is to take advantage of the unique learning opportunities available at VA. To that end, we exposed residents to veteran-specific educational content, ranging from the structure and payment model of VHA to service-related medical conditions, such as posttraumatic stress disorder, other mental health issues, traumatic brain injury, Agent Orange exposure, and Gulf War Syndrome.
Discussion
Findings from the recently published Accreditation Council for Graduate Medical Education’s (ACGME) 2016 Clinical Learning Environment Review (CLER) Report support the need for mission statements like ours to guide the delivery of graduate medical education.3 A major finding of this report was that the development and implementation of graduate medical education largely occurs separately from other areas of organizational and strategic focus within clinical learning environments. Our mission statement has served as a road map for aligning the delivery of graduate medical education at VABHS with the specific strengths of the clinical learning environment that VA affords.
Additionally, the 2016 CLER report identified a lack of specificity in training on health care disparities and cultural competency for the specific populations served by the surveyed residency programs. The emphasis we placed on learning about issues specific to the care of the veteran population highlights the potential for other mission statements like ours to bridge the gap between articulation and execution of educational priorities. Finally, through the academic partnerships it holds with more than 90% of medical schools in the U.S., VA already has an integral role in both undergraduate and graduate medical education that positions its hospitals as ideal training environments in which to address shortcomings in medical training like those identified by the ACGME.4
Conclusion
We propose this mission statement as a model for the delivery of graduate medical education throughout all VA hospitals with academic affiliations and especially those where trainees from multiple institutions work together. As embodied in our mission statement, our goal was to provide a clinical training experience at VA that complements that of our residents’ home institutions and fosters a respect for and interest in the special care provided at VA. The development of a shared mission statement provides an invaluable tool in accomplishing that goal. We encourage chief medical residents and other leaders in medical education in all specialties at VAMCs to develop their own mission statements that reflect and embody the values of each affiliated training program. For our residents, rotating at VA is an opportunity to learn the practice of medicine for veterans, rather than practicing medicine on veterans. It is our sincere hope that shaping our residents’ educational experience in this fashion will foster a greater appreciation for the care of our nation’s veterans.
1. VA Office of Academic Affiliations. 2015 statistics: health professions trainees. http://www.va.gov/oaa/docs/OAA_Statistics.pdf. Published 2016. Accessed September 18, 2017.
2. VA Office of Academic Affiliations. Mission of the Office of Academic Affiliations. http://www.va.gov/oaa/oaa_mission.asp. Updated June 23, 2017. Accessed September 18, 2017.
3. Accreditation Council for Graduate Medical Education. Clinical learning environment review – national report of findings 2016 – executive summary. https://www.acgme.org/Portals/0/PDFs/CLER/ACGME-CLER-ExecutiveSummary.pdf. Published 2016. Accessed September 18, 2017.
4. Association of American Medical Colleges. The VA and academic medicine: partners in health care, training, and research. https://www.aamc.org/download/385612/data/07182014.pdf. Accessed September 14, 2017.
1. VA Office of Academic Affiliations. 2015 statistics: health professions trainees. http://www.va.gov/oaa/docs/OAA_Statistics.pdf. Published 2016. Accessed September 18, 2017.
2. VA Office of Academic Affiliations. Mission of the Office of Academic Affiliations. http://www.va.gov/oaa/oaa_mission.asp. Updated June 23, 2017. Accessed September 18, 2017.
3. Accreditation Council for Graduate Medical Education. Clinical learning environment review – national report of findings 2016 – executive summary. https://www.acgme.org/Portals/0/PDFs/CLER/ACGME-CLER-ExecutiveSummary.pdf. Published 2016. Accessed September 18, 2017.
4. Association of American Medical Colleges. The VA and academic medicine: partners in health care, training, and research. https://www.aamc.org/download/385612/data/07182014.pdf. Accessed September 14, 2017.
Open Clinical Trials for Patients With Multiple Sclerosis (FULL)
Providing access to clinical trials for veteran and active-duty military patients can be a challenge, but a significant number of trials are now recruiting patients from those patient populations. More than 51,000 open trials are listed on the ClinicalTrials.gov website. Many explicitly recruit patients from the VA (212 studies), the military (132 studies), and IHS and native health organizations (4 studies). The VA Health Services Research and Development department alone sponsors > 250 research initiatives, and many more are sponsored by Walter Reed National Medical Center and other major defense and VA facilities. The clinical trials listed below are all open as of February 22, 2017, and are focused on the treatment of multiple sclerosis (MS) and other neurologic disorders. For additional information and full inclusion/exclusion criteria, please consult https://clinicaltrials.gov.
Integrating Caregiver Support Into MS Care (MS Caregiver)
With loss of mobility in MS comes an increase in amount and types of caregiver assistance, with a concomitant increase in burden for the caregiver. This feasibility study will test integration of a successful behavioral caregiving intervention into clinical practice to improve functioning of veterans with MS and their caregivers. Caregivers of veterans with MS will receive a behavioral caregiver intervention designed to address caregiver coping and management of patient concerns, with special focus on patient mobility and walking. A pre-post intervention design will compare outcomes for veterans and caregivers.
ID: NCT02835677
Sponsor: Memphis VA Medical Center
Contact: Linda O. Nichols, PhD, [email protected], Jennifer L. Martindale-Adams, [email protected]
Study and Treatment of Visual Dysfunction and Motor Fatigue in Multiple Sclerosis
Primary fatigue represents a major cause of disability in patients with MS, being reported in about 90% of cases. The investigators propose a characteristic eye movement abnormality (internuclear ophthalmoparesis), commonly encountered in MS, as a simple model for primary motor fatigue. The investigators propose a medical treatment to improve ocular performance/fatigue in internuclear ophthalmoparesis, which can reduce visual disability and improve quality of life in veterans with MS.
ID: NCT02391961
Sponsor: VA Office of Research and Development
Location (contact): Louis Stokes VA Medical Center, Cleveland, Ohio (Holly B. Henry)
Dysport Treatment of Urinary Incontinence in Adults Subjects With Neurogenic Detrusor Overactivity Due to Spinal Cord Injury or Multiple Sclerosis
The purpose of this study is to provide confirmatory evidence of the safety and efficacy of 2 Dysport (AbobotulinumtoxinA) doses (600 units [U] and 800 U), compared to placebo in reducing urinary incontinence in adult subjects treated for neurogenic detrusor overactivity due to spinal cord injury or MS.
ID: NCT02660138
Sponsor: Ipsen
Location (contact Ipsen Recruitment Enquiries, clinical.trials@ ipsen.com): VA Long Beach Healthcare System, California, VA Palo Alto Health Care System, California, Raymond G. Murphy VAMC Albuquerque, New Mexico, Louis Stokes Cleveland Veterans Affairs Medical Center, Ohio.

Click here to read the digital edition.
Providing access to clinical trials for veteran and active-duty military patients can be a challenge, but a significant number of trials are now recruiting patients from those patient populations. More than 51,000 open trials are listed on the ClinicalTrials.gov website. Many explicitly recruit patients from the VA (212 studies), the military (132 studies), and IHS and native health organizations (4 studies). The VA Health Services Research and Development department alone sponsors > 250 research initiatives, and many more are sponsored by Walter Reed National Medical Center and other major defense and VA facilities. The clinical trials listed below are all open as of February 22, 2017, and are focused on the treatment of multiple sclerosis (MS) and other neurologic disorders. For additional information and full inclusion/exclusion criteria, please consult https://clinicaltrials.gov.
Integrating Caregiver Support Into MS Care (MS Caregiver)
With loss of mobility in MS comes an increase in amount and types of caregiver assistance, with a concomitant increase in burden for the caregiver. This feasibility study will test integration of a successful behavioral caregiving intervention into clinical practice to improve functioning of veterans with MS and their caregivers. Caregivers of veterans with MS will receive a behavioral caregiver intervention designed to address caregiver coping and management of patient concerns, with special focus on patient mobility and walking. A pre-post intervention design will compare outcomes for veterans and caregivers.
ID: NCT02835677
Sponsor: Memphis VA Medical Center
Contact: Linda O. Nichols, PhD, [email protected], Jennifer L. Martindale-Adams, [email protected]
Study and Treatment of Visual Dysfunction and Motor Fatigue in Multiple Sclerosis
Primary fatigue represents a major cause of disability in patients with MS, being reported in about 90% of cases. The investigators propose a characteristic eye movement abnormality (internuclear ophthalmoparesis), commonly encountered in MS, as a simple model for primary motor fatigue. The investigators propose a medical treatment to improve ocular performance/fatigue in internuclear ophthalmoparesis, which can reduce visual disability and improve quality of life in veterans with MS.
ID: NCT02391961
Sponsor: VA Office of Research and Development
Location (contact): Louis Stokes VA Medical Center, Cleveland, Ohio (Holly B. Henry)
Dysport Treatment of Urinary Incontinence in Adults Subjects With Neurogenic Detrusor Overactivity Due to Spinal Cord Injury or Multiple Sclerosis
The purpose of this study is to provide confirmatory evidence of the safety and efficacy of 2 Dysport (AbobotulinumtoxinA) doses (600 units [U] and 800 U), compared to placebo in reducing urinary incontinence in adult subjects treated for neurogenic detrusor overactivity due to spinal cord injury or MS.
ID: NCT02660138
Sponsor: Ipsen
Location (contact Ipsen Recruitment Enquiries, clinical.trials@ ipsen.com): VA Long Beach Healthcare System, California, VA Palo Alto Health Care System, California, Raymond G. Murphy VAMC Albuquerque, New Mexico, Louis Stokes Cleveland Veterans Affairs Medical Center, Ohio.
Click here to read the digital edition.
Providing access to clinical trials for veteran and active-duty military patients can be a challenge, but a significant number of trials are now recruiting patients from those patient populations. More than 51,000 open trials are listed on the ClinicalTrials.gov website. Many explicitly recruit patients from the VA (212 studies), the military (132 studies), and IHS and native health organizations (4 studies). The VA Health Services Research and Development department alone sponsors > 250 research initiatives, and many more are sponsored by Walter Reed National Medical Center and other major defense and VA facilities. The clinical trials listed below are all open as of February 22, 2017, and are focused on the treatment of multiple sclerosis (MS) and other neurologic disorders. For additional information and full inclusion/exclusion criteria, please consult https://clinicaltrials.gov.
Integrating Caregiver Support Into MS Care (MS Caregiver)
With loss of mobility in MS comes an increase in amount and types of caregiver assistance, with a concomitant increase in burden for the caregiver. This feasibility study will test integration of a successful behavioral caregiving intervention into clinical practice to improve functioning of veterans with MS and their caregivers. Caregivers of veterans with MS will receive a behavioral caregiver intervention designed to address caregiver coping and management of patient concerns, with special focus on patient mobility and walking. A pre-post intervention design will compare outcomes for veterans and caregivers.
ID: NCT02835677
Sponsor: Memphis VA Medical Center
Contact: Linda O. Nichols, PhD, [email protected], Jennifer L. Martindale-Adams, [email protected]
Study and Treatment of Visual Dysfunction and Motor Fatigue in Multiple Sclerosis
Primary fatigue represents a major cause of disability in patients with MS, being reported in about 90% of cases. The investigators propose a characteristic eye movement abnormality (internuclear ophthalmoparesis), commonly encountered in MS, as a simple model for primary motor fatigue. The investigators propose a medical treatment to improve ocular performance/fatigue in internuclear ophthalmoparesis, which can reduce visual disability and improve quality of life in veterans with MS.
ID: NCT02391961
Sponsor: VA Office of Research and Development
Location (contact): Louis Stokes VA Medical Center, Cleveland, Ohio (Holly B. Henry)
Dysport Treatment of Urinary Incontinence in Adults Subjects With Neurogenic Detrusor Overactivity Due to Spinal Cord Injury or Multiple Sclerosis
The purpose of this study is to provide confirmatory evidence of the safety and efficacy of 2 Dysport (AbobotulinumtoxinA) doses (600 units [U] and 800 U), compared to placebo in reducing urinary incontinence in adult subjects treated for neurogenic detrusor overactivity due to spinal cord injury or MS.
ID: NCT02660138
Sponsor: Ipsen
Location (contact Ipsen Recruitment Enquiries, clinical.trials@ ipsen.com): VA Long Beach Healthcare System, California, VA Palo Alto Health Care System, California, Raymond G. Murphy VAMC Albuquerque, New Mexico, Louis Stokes Cleveland Veterans Affairs Medical Center, Ohio.
Click here to read the digital edition.
Panic Disorder: Ensuring Prompt Recognition and Treatment
Lacey, 37, is seen by her primary care provider (PCP) as follow-up to a visit she made to the emergency department (ED). She has gone to the ED four times in the past year. Each time, she presents with tachycardia, dyspnea, nausea, numbness in her extremities, and a fear that she is having a heart attack. Despite negative workups at each visit (ECG, cardiac enzymes, complete blood count, toxicology screen, Holter monitoring), Lacey is terrified that the ED doctors are missing something. She is still “rattled” by the chest pain and shortness of breath she experiences. Mild symptoms are persisting, and she is worried that she will have a heart attack and die without the treatment she believes she needs. How do you proceed?
Panic disorder (PD) is characterized by the spontaneous and unexpected occurrence of panic attacks and by at least one month of persistent worry about having another attack or significant maladaptive behaviors related to the attack. Frequency of such attacks can vary from several a day to only a few per year. In a panic attack, an intense fear develops abruptly and peaks within 10 minutes of onset. At least four of the following 13 symptoms must accompany the attack, according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth edition (DSM-5)
- Palpitations, pounding heart, or accelerated heart rate
- Sweating
- Trembling or shaking
- Sensations of shortness of breath or smothering
- Feeling of choking
- Chest pain or discomfort
- Feeling dizzy, unsteady, lightheaded, or faint
- Nausea or abdominal distress
- Derealization (feelings of unreality) or depersonalization (being detached from oneself)
- Fear of losing control or going crazy
- Fear of dying
- Paresthesia (numbness or tingling sensations)
- Chills or hot flushes.1
Lifetime incidence rates of PD are 1% to 3% for the general population.2 A closer look at patients presenting to the ED with chest pain reveals that 17% to 25% meet criteria for PD.3,4 And an estimated 6% of individuals experiencing a panic attack present to their primary care provider.5 Patients with PD tend to use health care resources at a disproportionately high rate.6
An international review of PD research suggests the average age of onset is 32 years.7 Triggers can vary widely, and no single stressor has been identified. The exact cause of PD is unknown, but a convergence of social and biological influences (including involvement of the amygdala) are implicated in its development.6 For individuals who have had a panic attack, 66.5% will have recurrent attacks.7 Lifetime prevalence of panic attacks is 13.2%.7
Differential goes far beyond myocardial infarction. Many medical conditions can mimic PD symptoms: cardiovascular, pulmonary, and neurologic diseases; endocrine diseases (eg, hyperthyroidism); drug intoxication (eg, stimulants [cocaine, amphetamines]); drug withdrawal (eg, benzodiazepines, alcohol, sedative-hypnotics); and ingestion of excessive quantities of caffeine. Common comorbid medical disorders include asthma, coronary artery disease, cancer, thyroid disease, hypertension, ulcer, and migraine headaches.8
When patients present with paniclike symptoms, suspect a possible medical condition when those symptoms include ataxia, altered mental status, or loss of bladder control, or when onset of panic symptoms occur later in life for a patient with no significant psychiatric history.
RULE OUT ORGANIC CAUSES
In addition to obtaining a complete history and doing a physical exam on patients with paniclike symptoms, you’ll also need to ensure that the following are done: a neurologic examination, standard laboratory testing (thyroid function, complete blood cell count, chemistry panel), and possible additional testing (eg, urine toxicology screen and
If organic causes are ruled out, focus on a psychiatric assessment, including
- History of the present illness (onset, symptoms, frequency, predisposing/precipitating factors)
- Psychiatric history
- History of substance use
- Family history of psychiatric disorders (especially anxiety disorders)
- Social history (life events, including those preceding the onset of panic; history of child abuse)
- Medications
- Mental status examination
- Safety (PD is associated with higher risk for suicidal ideation).9
TREATMENT INCLUDES CBT AND MEDICATION
PD is a chronic disease with a variable course, but the long-term prognosis is good. PD is usually treated in an outpatient setting. Consider hospitalization if the patient is suicidal, if the potential for life-threatening withdrawal symptoms is high (as with alcohol or benzodiazepines), or if the symptoms are severely debilitating or attempted outpatient treatment is unsuccessful. Pharmacologic and psychotherapeutic interventions are used for PD (see Figure), although there is not enough evidence to recommend one versus the other or combination therapy versus monotherapy.9
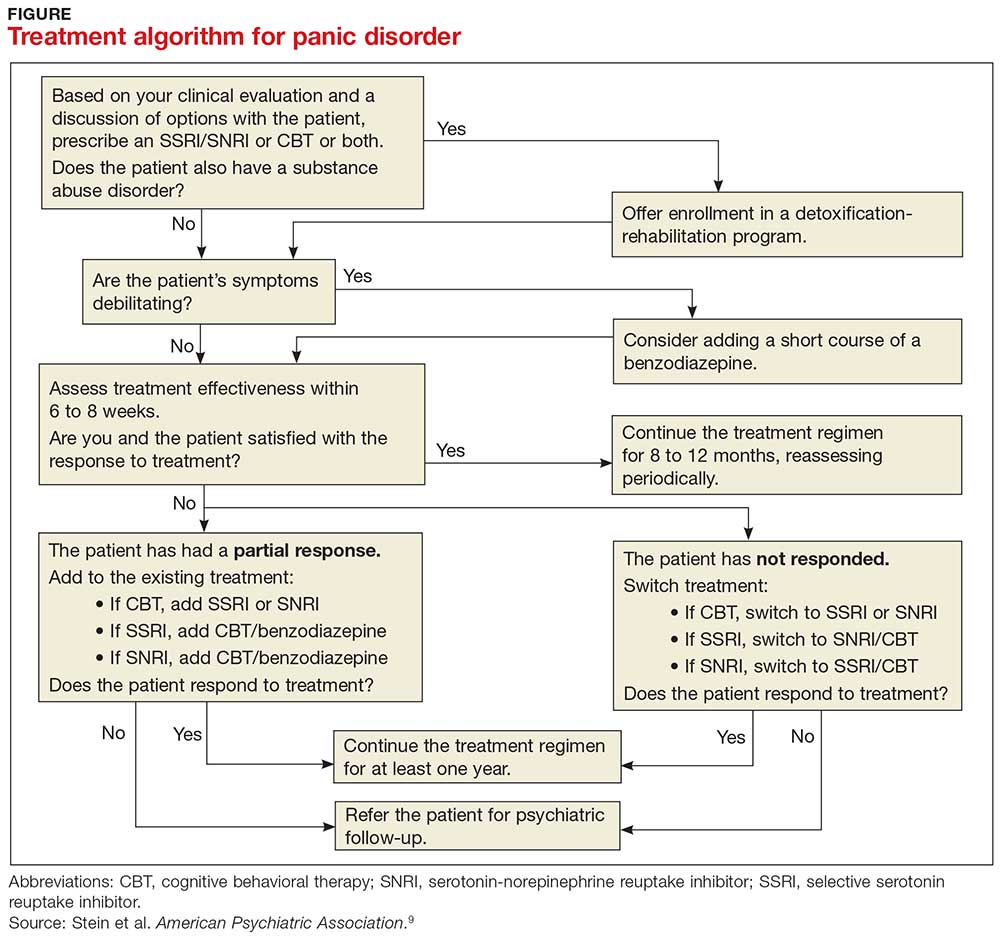
All Lacey’s test results come back negative, and the psychiatric assessment reveals that she meets the DSM-5 criteria for PD. Counting on the strength of their relationship, her PCP talks to her about PD and discusses treatment options, which include counseling, medication, or both. Lacey agrees to a referral for cognitive behavioral therapy (CBT) with a psychologist embedded at her primary care clinic and to begin taking medication. Her PCP starts her on sertraline 25 mg/d.
In CBT, Lacey’s psychologist teaches her about “fight or flight” and explains that it is a normal physiologic response that can lead to panic. Lacey learns to approach her physical symptoms in a different way, and how to breathe in a way that slows her panic reaction.
Consider SSRIs and SNRIs
Firstline medication is a selective serotonin reuptake inhibitor (SSRI) or a serotonin-norepinephrine reuptake inhibitor (SNRI), due to the better tolerability and lower adverse effect profile of these classes compared with the tricyclic antidepressants or monoamine oxidase inhibitors (MAOIs). MAOIs are usually reserved for patients in whom multiple medication trials have failed.
Special considerations. American Psychiatric Association guidelines advise starting with a very low dose of an SSRI or SNRI, such as paroxetine 10 mg/d (although
Keep in mind that the onset of therapeutic effect is between two and four weeks, but that clinical response can take eight to 12 weeks. Continue pharmacotherapy for at least one year. When discontinuing the medication, taper it slowly, and monitor the patient for withdrawal symptoms and recurrence of PD.9
Consider adding a benzodiazepine if symptoms are debilitating.9 Keep in mind, though, that the potential for addiction with these medications is high and they are intended to be used for only four to 12 weeks.8 Onset of action is within the first week, and a scheduled dosing regimen is preferred to giving the medication as needed. The starting dose (eg, clonazepam 0.25 mg bid) may be increased three to five days following initiation.9
Evidence supports the use of CBT for PD
CBT is an evidenced-based treatment for PD.10-13 Up to 75% of patients treated with CBT are panic free within four months.10 Other techniques proven effective are progressive muscle relaxation training, breathing retraining, psychoeducation, exposure, and imagery.14
Treatment with medications and CBT, either combined or used individually, is effective in 80% to 90% of cases.15 CBT has been shown to decrease the likelihood of relapse in the year following treatment.15 Good premorbid functioning and a brief duration of symptoms increase the likelihood of a good prognosis.15
WHEN TO REFER TO A PSYCHIATRIST
Consider referral to a psychiatrist when patients have a comorbid psychiatric condition that complicates the clinical picture (eg, substance abuse disorder), if the diagnosis is uncertain, or if the patient does not respond to one or two adequate trials of medication and psychotherapy. Although psychiatric follow-up is sometimes difficult due to a lack of psychiatrist availability locally, it is a best-practice recommendation.
Ten days after Lacey starts the sertraline 25 mg/d, she calls the PCP to report daily diarrhea. She stopped the sertraline on her own and is asking for another medication. She also expresses her frustration with the severity of the symptoms. She is having three to five panic attacks daily and has been missing many days of work.
On the day of her follow-up PCP appointment, Lacey also sees the psychologist. She reports that she’s been practicing relaxation breathing, tracking her panic attacks, limiting her caffeine intake, and exercising regularly. But the attacks are still occurring.
The PCP switches her to paroxetine 10 mg/d and, due to the severity of the symptoms, prescribes clonazepam 0.5 mg bid. Two weeks later, Lacey reports that she is feeling a little better, has returned to work, and is hopeful that she will be her “normal self again.” The PCP plans to encourage continuation of CBT, titrate the paroxetine to 20 to 40 mg/d based on symptoms, and slowly taper the clonazepam toward discontinuation in the near future.
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Publishing; 2013.
2. Kumar S, Oakley-Browne M. Panic disorder. Clin Evid. 2006;15:1438-1452.
3. Yingling KW, Wulsin LR, Arnold LM, et al. Estimated prevalences of panic disorder and depression among consecutive patients seen in an emergency department with acute chest pain. J Gen Intern Med. 1993;8:231-235.
4. Fleet RP, Dupuis G, Marchand A, et al. Panic disorder in emergency department chest pain patients: prevalence, comorbidity, suicidal ideation, and physician recognition. Am J Med. 1996;101:371-380.
5. Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA. 1994;272:1749-1756.
6. Taylor CB. Panic disorder. BMJ. 2006;332:951-955.
7. de Jonge P, Roest AM, Lim CC, et al. Cross-national epidemiology of panic disorder and panic attacks in the world mental health surveys. Depress Anxiety. 2016;33: 1155-1177.
8. Sadock BJ, Sadock VA, Ruiz P. Panic disorder. In: Kaplan & Sadock’s Synopsis of Psychiatry. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2015:392-397.
9. Stein MB, Goin MK, Pollack MH, et al. ractice Guideline for the Treatment of Patients with Panic Disorder. 2nd ed. Washington, DC: American Psychiatric Association; 2010. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/panicdisorder.pdf. Accessed February 14, 2018.
10. Westen D, Morrison K. A multidimensional meta-analysis of treatments for depression, panic, and generalized anxiety disorder: an empirical examination of the status of empirically supported therapies. J Consult Clin Psychol. 2001;69:875-899.
11. Gould RA, Otto MW, Pollack MH. A meta-analysis of treatment outcome for panic disorder. www.ncbi.nlm.nih.gov/books/NBK66380/. Accessed February 14, 2018.
12. Clum GA, Clum GA, Surls R. A meta-analysis of treatments for panic disorder. J Consult Clin Psychol. 1993; 61:317-326.
13. Shear MK, Houck P, Greeno C, et al. Emotion-focused psychotherapy for patients with panic disorder. Am J Psychiatry. 2001;158:1993-1998.
14. Stewart RE, Chambless DL. Cognitive-behavioral therapy for adult anxiety disorders in clinical practice: a meta-analysis of effectiveness studies. J Consult Clin Psychol. 2009;77:595-606.
15. Craske M. Psychotherapy for panic disorder in adults. Up to Date. 2017. www.uptodate.com/contents/psychotherapy-for-panic-disorder-with-or-without-agoraphobia-in-adults. Accessed February 14, 2018.
Lacey, 37, is seen by her primary care provider (PCP) as follow-up to a visit she made to the emergency department (ED). She has gone to the ED four times in the past year. Each time, she presents with tachycardia, dyspnea, nausea, numbness in her extremities, and a fear that she is having a heart attack. Despite negative workups at each visit (ECG, cardiac enzymes, complete blood count, toxicology screen, Holter monitoring), Lacey is terrified that the ED doctors are missing something. She is still “rattled” by the chest pain and shortness of breath she experiences. Mild symptoms are persisting, and she is worried that she will have a heart attack and die without the treatment she believes she needs. How do you proceed?
Panic disorder (PD) is characterized by the spontaneous and unexpected occurrence of panic attacks and by at least one month of persistent worry about having another attack or significant maladaptive behaviors related to the attack. Frequency of such attacks can vary from several a day to only a few per year. In a panic attack, an intense fear develops abruptly and peaks within 10 minutes of onset. At least four of the following 13 symptoms must accompany the attack, according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth edition (DSM-5)
- Palpitations, pounding heart, or accelerated heart rate
- Sweating
- Trembling or shaking
- Sensations of shortness of breath or smothering
- Feeling of choking
- Chest pain or discomfort
- Feeling dizzy, unsteady, lightheaded, or faint
- Nausea or abdominal distress
- Derealization (feelings of unreality) or depersonalization (being detached from oneself)
- Fear of losing control or going crazy
- Fear of dying
- Paresthesia (numbness or tingling sensations)
- Chills or hot flushes.1
Lifetime incidence rates of PD are 1% to 3% for the general population.2 A closer look at patients presenting to the ED with chest pain reveals that 17% to 25% meet criteria for PD.3,4 And an estimated 6% of individuals experiencing a panic attack present to their primary care provider.5 Patients with PD tend to use health care resources at a disproportionately high rate.6
An international review of PD research suggests the average age of onset is 32 years.7 Triggers can vary widely, and no single stressor has been identified. The exact cause of PD is unknown, but a convergence of social and biological influences (including involvement of the amygdala) are implicated in its development.6 For individuals who have had a panic attack, 66.5% will have recurrent attacks.7 Lifetime prevalence of panic attacks is 13.2%.7
Differential goes far beyond myocardial infarction. Many medical conditions can mimic PD symptoms: cardiovascular, pulmonary, and neurologic diseases; endocrine diseases (eg, hyperthyroidism); drug intoxication (eg, stimulants [cocaine, amphetamines]); drug withdrawal (eg, benzodiazepines, alcohol, sedative-hypnotics); and ingestion of excessive quantities of caffeine. Common comorbid medical disorders include asthma, coronary artery disease, cancer, thyroid disease, hypertension, ulcer, and migraine headaches.8
When patients present with paniclike symptoms, suspect a possible medical condition when those symptoms include ataxia, altered mental status, or loss of bladder control, or when onset of panic symptoms occur later in life for a patient with no significant psychiatric history.
RULE OUT ORGANIC CAUSES
In addition to obtaining a complete history and doing a physical exam on patients with paniclike symptoms, you’ll also need to ensure that the following are done: a neurologic examination, standard laboratory testing (thyroid function, complete blood cell count, chemistry panel), and possible additional testing (eg, urine toxicology screen and
If organic causes are ruled out, focus on a psychiatric assessment, including
- History of the present illness (onset, symptoms, frequency, predisposing/precipitating factors)
- Psychiatric history
- History of substance use
- Family history of psychiatric disorders (especially anxiety disorders)
- Social history (life events, including those preceding the onset of panic; history of child abuse)
- Medications
- Mental status examination
- Safety (PD is associated with higher risk for suicidal ideation).9
TREATMENT INCLUDES CBT AND MEDICATION
PD is a chronic disease with a variable course, but the long-term prognosis is good. PD is usually treated in an outpatient setting. Consider hospitalization if the patient is suicidal, if the potential for life-threatening withdrawal symptoms is high (as with alcohol or benzodiazepines), or if the symptoms are severely debilitating or attempted outpatient treatment is unsuccessful. Pharmacologic and psychotherapeutic interventions are used for PD (see Figure), although there is not enough evidence to recommend one versus the other or combination therapy versus monotherapy.9
All Lacey’s test results come back negative, and the psychiatric assessment reveals that she meets the DSM-5 criteria for PD. Counting on the strength of their relationship, her PCP talks to her about PD and discusses treatment options, which include counseling, medication, or both. Lacey agrees to a referral for cognitive behavioral therapy (CBT) with a psychologist embedded at her primary care clinic and to begin taking medication. Her PCP starts her on sertraline 25 mg/d.
In CBT, Lacey’s psychologist teaches her about “fight or flight” and explains that it is a normal physiologic response that can lead to panic. Lacey learns to approach her physical symptoms in a different way, and how to breathe in a way that slows her panic reaction.
Consider SSRIs and SNRIs
Firstline medication is a selective serotonin reuptake inhibitor (SSRI) or a serotonin-norepinephrine reuptake inhibitor (SNRI), due to the better tolerability and lower adverse effect profile of these classes compared with the tricyclic antidepressants or monoamine oxidase inhibitors (MAOIs). MAOIs are usually reserved for patients in whom multiple medication trials have failed.
Special considerations. American Psychiatric Association guidelines advise starting with a very low dose of an SSRI or SNRI, such as paroxetine 10 mg/d (although
Keep in mind that the onset of therapeutic effect is between two and four weeks, but that clinical response can take eight to 12 weeks. Continue pharmacotherapy for at least one year. When discontinuing the medication, taper it slowly, and monitor the patient for withdrawal symptoms and recurrence of PD.9
Consider adding a benzodiazepine if symptoms are debilitating.9 Keep in mind, though, that the potential for addiction with these medications is high and they are intended to be used for only four to 12 weeks.8 Onset of action is within the first week, and a scheduled dosing regimen is preferred to giving the medication as needed. The starting dose (eg, clonazepam 0.25 mg bid) may be increased three to five days following initiation.9
Evidence supports the use of CBT for PD
CBT is an evidenced-based treatment for PD.10-13 Up to 75% of patients treated with CBT are panic free within four months.10 Other techniques proven effective are progressive muscle relaxation training, breathing retraining, psychoeducation, exposure, and imagery.14
Treatment with medications and CBT, either combined or used individually, is effective in 80% to 90% of cases.15 CBT has been shown to decrease the likelihood of relapse in the year following treatment.15 Good premorbid functioning and a brief duration of symptoms increase the likelihood of a good prognosis.15
WHEN TO REFER TO A PSYCHIATRIST
Consider referral to a psychiatrist when patients have a comorbid psychiatric condition that complicates the clinical picture (eg, substance abuse disorder), if the diagnosis is uncertain, or if the patient does not respond to one or two adequate trials of medication and psychotherapy. Although psychiatric follow-up is sometimes difficult due to a lack of psychiatrist availability locally, it is a best-practice recommendation.
Ten days after Lacey starts the sertraline 25 mg/d, she calls the PCP to report daily diarrhea. She stopped the sertraline on her own and is asking for another medication. She also expresses her frustration with the severity of the symptoms. She is having three to five panic attacks daily and has been missing many days of work.
On the day of her follow-up PCP appointment, Lacey also sees the psychologist. She reports that she’s been practicing relaxation breathing, tracking her panic attacks, limiting her caffeine intake, and exercising regularly. But the attacks are still occurring.
The PCP switches her to paroxetine 10 mg/d and, due to the severity of the symptoms, prescribes clonazepam 0.5 mg bid. Two weeks later, Lacey reports that she is feeling a little better, has returned to work, and is hopeful that she will be her “normal self again.” The PCP plans to encourage continuation of CBT, titrate the paroxetine to 20 to 40 mg/d based on symptoms, and slowly taper the clonazepam toward discontinuation in the near future.
Lacey, 37, is seen by her primary care provider (PCP) as follow-up to a visit she made to the emergency department (ED). She has gone to the ED four times in the past year. Each time, she presents with tachycardia, dyspnea, nausea, numbness in her extremities, and a fear that she is having a heart attack. Despite negative workups at each visit (ECG, cardiac enzymes, complete blood count, toxicology screen, Holter monitoring), Lacey is terrified that the ED doctors are missing something. She is still “rattled” by the chest pain and shortness of breath she experiences. Mild symptoms are persisting, and she is worried that she will have a heart attack and die without the treatment she believes she needs. How do you proceed?
Panic disorder (PD) is characterized by the spontaneous and unexpected occurrence of panic attacks and by at least one month of persistent worry about having another attack or significant maladaptive behaviors related to the attack. Frequency of such attacks can vary from several a day to only a few per year. In a panic attack, an intense fear develops abruptly and peaks within 10 minutes of onset. At least four of the following 13 symptoms must accompany the attack, according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth edition (DSM-5)
- Palpitations, pounding heart, or accelerated heart rate
- Sweating
- Trembling or shaking
- Sensations of shortness of breath or smothering
- Feeling of choking
- Chest pain or discomfort
- Feeling dizzy, unsteady, lightheaded, or faint
- Nausea or abdominal distress
- Derealization (feelings of unreality) or depersonalization (being detached from oneself)
- Fear of losing control or going crazy
- Fear of dying
- Paresthesia (numbness or tingling sensations)
- Chills or hot flushes.1
Lifetime incidence rates of PD are 1% to 3% for the general population.2 A closer look at patients presenting to the ED with chest pain reveals that 17% to 25% meet criteria for PD.3,4 And an estimated 6% of individuals experiencing a panic attack present to their primary care provider.5 Patients with PD tend to use health care resources at a disproportionately high rate.6
An international review of PD research suggests the average age of onset is 32 years.7 Triggers can vary widely, and no single stressor has been identified. The exact cause of PD is unknown, but a convergence of social and biological influences (including involvement of the amygdala) are implicated in its development.6 For individuals who have had a panic attack, 66.5% will have recurrent attacks.7 Lifetime prevalence of panic attacks is 13.2%.7
Differential goes far beyond myocardial infarction. Many medical conditions can mimic PD symptoms: cardiovascular, pulmonary, and neurologic diseases; endocrine diseases (eg, hyperthyroidism); drug intoxication (eg, stimulants [cocaine, amphetamines]); drug withdrawal (eg, benzodiazepines, alcohol, sedative-hypnotics); and ingestion of excessive quantities of caffeine. Common comorbid medical disorders include asthma, coronary artery disease, cancer, thyroid disease, hypertension, ulcer, and migraine headaches.8
When patients present with paniclike symptoms, suspect a possible medical condition when those symptoms include ataxia, altered mental status, or loss of bladder control, or when onset of panic symptoms occur later in life for a patient with no significant psychiatric history.
RULE OUT ORGANIC CAUSES
In addition to obtaining a complete history and doing a physical exam on patients with paniclike symptoms, you’ll also need to ensure that the following are done: a neurologic examination, standard laboratory testing (thyroid function, complete blood cell count, chemistry panel), and possible additional testing (eg, urine toxicology screen and
If organic causes are ruled out, focus on a psychiatric assessment, including
- History of the present illness (onset, symptoms, frequency, predisposing/precipitating factors)
- Psychiatric history
- History of substance use
- Family history of psychiatric disorders (especially anxiety disorders)
- Social history (life events, including those preceding the onset of panic; history of child abuse)
- Medications
- Mental status examination
- Safety (PD is associated with higher risk for suicidal ideation).9
TREATMENT INCLUDES CBT AND MEDICATION
PD is a chronic disease with a variable course, but the long-term prognosis is good. PD is usually treated in an outpatient setting. Consider hospitalization if the patient is suicidal, if the potential for life-threatening withdrawal symptoms is high (as with alcohol or benzodiazepines), or if the symptoms are severely debilitating or attempted outpatient treatment is unsuccessful. Pharmacologic and psychotherapeutic interventions are used for PD (see Figure), although there is not enough evidence to recommend one versus the other or combination therapy versus monotherapy.9
All Lacey’s test results come back negative, and the psychiatric assessment reveals that she meets the DSM-5 criteria for PD. Counting on the strength of their relationship, her PCP talks to her about PD and discusses treatment options, which include counseling, medication, or both. Lacey agrees to a referral for cognitive behavioral therapy (CBT) with a psychologist embedded at her primary care clinic and to begin taking medication. Her PCP starts her on sertraline 25 mg/d.
In CBT, Lacey’s psychologist teaches her about “fight or flight” and explains that it is a normal physiologic response that can lead to panic. Lacey learns to approach her physical symptoms in a different way, and how to breathe in a way that slows her panic reaction.
Consider SSRIs and SNRIs
Firstline medication is a selective serotonin reuptake inhibitor (SSRI) or a serotonin-norepinephrine reuptake inhibitor (SNRI), due to the better tolerability and lower adverse effect profile of these classes compared with the tricyclic antidepressants or monoamine oxidase inhibitors (MAOIs). MAOIs are usually reserved for patients in whom multiple medication trials have failed.
Special considerations. American Psychiatric Association guidelines advise starting with a very low dose of an SSRI or SNRI, such as paroxetine 10 mg/d (although
Keep in mind that the onset of therapeutic effect is between two and four weeks, but that clinical response can take eight to 12 weeks. Continue pharmacotherapy for at least one year. When discontinuing the medication, taper it slowly, and monitor the patient for withdrawal symptoms and recurrence of PD.9
Consider adding a benzodiazepine if symptoms are debilitating.9 Keep in mind, though, that the potential for addiction with these medications is high and they are intended to be used for only four to 12 weeks.8 Onset of action is within the first week, and a scheduled dosing regimen is preferred to giving the medication as needed. The starting dose (eg, clonazepam 0.25 mg bid) may be increased three to five days following initiation.9
Evidence supports the use of CBT for PD
CBT is an evidenced-based treatment for PD.10-13 Up to 75% of patients treated with CBT are panic free within four months.10 Other techniques proven effective are progressive muscle relaxation training, breathing retraining, psychoeducation, exposure, and imagery.14
Treatment with medications and CBT, either combined or used individually, is effective in 80% to 90% of cases.15 CBT has been shown to decrease the likelihood of relapse in the year following treatment.15 Good premorbid functioning and a brief duration of symptoms increase the likelihood of a good prognosis.15
WHEN TO REFER TO A PSYCHIATRIST
Consider referral to a psychiatrist when patients have a comorbid psychiatric condition that complicates the clinical picture (eg, substance abuse disorder), if the diagnosis is uncertain, or if the patient does not respond to one or two adequate trials of medication and psychotherapy. Although psychiatric follow-up is sometimes difficult due to a lack of psychiatrist availability locally, it is a best-practice recommendation.
Ten days after Lacey starts the sertraline 25 mg/d, she calls the PCP to report daily diarrhea. She stopped the sertraline on her own and is asking for another medication. She also expresses her frustration with the severity of the symptoms. She is having three to five panic attacks daily and has been missing many days of work.
On the day of her follow-up PCP appointment, Lacey also sees the psychologist. She reports that she’s been practicing relaxation breathing, tracking her panic attacks, limiting her caffeine intake, and exercising regularly. But the attacks are still occurring.
The PCP switches her to paroxetine 10 mg/d and, due to the severity of the symptoms, prescribes clonazepam 0.5 mg bid. Two weeks later, Lacey reports that she is feeling a little better, has returned to work, and is hopeful that she will be her “normal self again.” The PCP plans to encourage continuation of CBT, titrate the paroxetine to 20 to 40 mg/d based on symptoms, and slowly taper the clonazepam toward discontinuation in the near future.
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Publishing; 2013.
2. Kumar S, Oakley-Browne M. Panic disorder. Clin Evid. 2006;15:1438-1452.
3. Yingling KW, Wulsin LR, Arnold LM, et al. Estimated prevalences of panic disorder and depression among consecutive patients seen in an emergency department with acute chest pain. J Gen Intern Med. 1993;8:231-235.
4. Fleet RP, Dupuis G, Marchand A, et al. Panic disorder in emergency department chest pain patients: prevalence, comorbidity, suicidal ideation, and physician recognition. Am J Med. 1996;101:371-380.
5. Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA. 1994;272:1749-1756.
6. Taylor CB. Panic disorder. BMJ. 2006;332:951-955.
7. de Jonge P, Roest AM, Lim CC, et al. Cross-national epidemiology of panic disorder and panic attacks in the world mental health surveys. Depress Anxiety. 2016;33: 1155-1177.
8. Sadock BJ, Sadock VA, Ruiz P. Panic disorder. In: Kaplan & Sadock’s Synopsis of Psychiatry. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2015:392-397.
9. Stein MB, Goin MK, Pollack MH, et al. ractice Guideline for the Treatment of Patients with Panic Disorder. 2nd ed. Washington, DC: American Psychiatric Association; 2010. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/panicdisorder.pdf. Accessed February 14, 2018.
10. Westen D, Morrison K. A multidimensional meta-analysis of treatments for depression, panic, and generalized anxiety disorder: an empirical examination of the status of empirically supported therapies. J Consult Clin Psychol. 2001;69:875-899.
11. Gould RA, Otto MW, Pollack MH. A meta-analysis of treatment outcome for panic disorder. www.ncbi.nlm.nih.gov/books/NBK66380/. Accessed February 14, 2018.
12. Clum GA, Clum GA, Surls R. A meta-analysis of treatments for panic disorder. J Consult Clin Psychol. 1993; 61:317-326.
13. Shear MK, Houck P, Greeno C, et al. Emotion-focused psychotherapy for patients with panic disorder. Am J Psychiatry. 2001;158:1993-1998.
14. Stewart RE, Chambless DL. Cognitive-behavioral therapy for adult anxiety disorders in clinical practice: a meta-analysis of effectiveness studies. J Consult Clin Psychol. 2009;77:595-606.
15. Craske M. Psychotherapy for panic disorder in adults. Up to Date. 2017. www.uptodate.com/contents/psychotherapy-for-panic-disorder-with-or-without-agoraphobia-in-adults. Accessed February 14, 2018.
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Publishing; 2013.
2. Kumar S, Oakley-Browne M. Panic disorder. Clin Evid. 2006;15:1438-1452.
3. Yingling KW, Wulsin LR, Arnold LM, et al. Estimated prevalences of panic disorder and depression among consecutive patients seen in an emergency department with acute chest pain. J Gen Intern Med. 1993;8:231-235.
4. Fleet RP, Dupuis G, Marchand A, et al. Panic disorder in emergency department chest pain patients: prevalence, comorbidity, suicidal ideation, and physician recognition. Am J Med. 1996;101:371-380.
5. Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA. 1994;272:1749-1756.
6. Taylor CB. Panic disorder. BMJ. 2006;332:951-955.
7. de Jonge P, Roest AM, Lim CC, et al. Cross-national epidemiology of panic disorder and panic attacks in the world mental health surveys. Depress Anxiety. 2016;33: 1155-1177.
8. Sadock BJ, Sadock VA, Ruiz P. Panic disorder. In: Kaplan & Sadock’s Synopsis of Psychiatry. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2015:392-397.
9. Stein MB, Goin MK, Pollack MH, et al. ractice Guideline for the Treatment of Patients with Panic Disorder. 2nd ed. Washington, DC: American Psychiatric Association; 2010. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/panicdisorder.pdf. Accessed February 14, 2018.
10. Westen D, Morrison K. A multidimensional meta-analysis of treatments for depression, panic, and generalized anxiety disorder: an empirical examination of the status of empirically supported therapies. J Consult Clin Psychol. 2001;69:875-899.
11. Gould RA, Otto MW, Pollack MH. A meta-analysis of treatment outcome for panic disorder. www.ncbi.nlm.nih.gov/books/NBK66380/. Accessed February 14, 2018.
12. Clum GA, Clum GA, Surls R. A meta-analysis of treatments for panic disorder. J Consult Clin Psychol. 1993; 61:317-326.
13. Shear MK, Houck P, Greeno C, et al. Emotion-focused psychotherapy for patients with panic disorder. Am J Psychiatry. 2001;158:1993-1998.
14. Stewart RE, Chambless DL. Cognitive-behavioral therapy for adult anxiety disorders in clinical practice: a meta-analysis of effectiveness studies. J Consult Clin Psychol. 2009;77:595-606.
15. Craske M. Psychotherapy for panic disorder in adults. Up to Date. 2017. www.uptodate.com/contents/psychotherapy-for-panic-disorder-with-or-without-agoraphobia-in-adults. Accessed February 14, 2018.
Update on Management of Barrett’s Esophagus for Primary Care Providers
From the Gastroenterology and Hepatology Section, Baylor College of Medicine, Houston, TX.
Abstract
- Objective: To provide an update on management of Barrett’s esophagus.
- Methods: Review of the literature.
- Results: Management of Barrett’s esophagus depends on the degree of dysplasia. Surveillance by endoscopy every 3–5 years is recommended in patients with Barrett’s esophagus without dysplasia. Patients with Barrett’s esophagus and low-grade dysplasia should undergo surveillance by endoscopy in 3 months for confirmation of the diagnosis; if the diagnosis is confirmed then surveillance by endoscopy or eradication of Barrett’s epithelium by ablation or endoscopic resection are recommended. There is a sufficient evidence to recommend radiofrequency ablation of high-grade dysplasia within Barrett’s esophagus or to perform endoscopic mucosal resection of nodular Barrett’s esophagus with any degree of dysplasia. Early esophageal cancers that are limited to the mucosa can be treated by endoscopic resection, while cancer invading into the deep submucosa or muscularis propria may need esophagectomy with or without chemoradiation.
- Conclusion: The management of Barrett’s esophagus depends on the degree of dysplasia. Radiofrequency ablation and endoscopic mucosal resection are the most commonly used treatment for Barrett’s esophagus with dysplasia.
Keywords: Barrett’s esophagus; radiofrequency ablation; endoscopic submucosal dissection; endoscopic mucosal resection; early esophageal cancer.
Barrett’s esophagus is a common complication of chronic reflux disease [1]. Metaplastic changes that occur at the distal esophageal epithelium are usually asymptomatic [2,3] and occur as reparative adaptations to the insult of the gastric acid [4]. The management of Barrett’s esophagus after diagnosis is currently debated amongst experts without a clear consensus [5,6]. This review is generally consistent with the 2016 guidelines from the American College of Gastroenterology [1], a 2012 guideline from the American Society of Gastrointestinal Endoscopy [7], a 2011 guideline, and a 2016 expert review from the American Gastroenterological Association [8,9].
D efinition
Barrett’s esophagus is a metaplasia of the stratified squamous epithelium to a specialized columnar intestinal epithelium of mucus cells and goblet cells at the distal esophagus secondary to gastroesophageal reflux disease (GERD) [7,10]. Barrett’s esophagus is unstable tissue which can progress to esophageal adenocarcinoma. When unmanaged, the risk of cancer in dysplastic mucosa is at least thirty-fold greater than that for the general population [11–14], with recent studies suggesting a 0.4–0.7 occurrence rate per year [11,15]. With no dysplasia, the risk is low [16].
Epidemiology
The prevalence of Barrett’s esophagus is ~10% in patients with GERD [11–13,17], an estimate tied to the prevalence of GERD. However, due to the lack of symptoms of Barrett’s esophagus, no solid data supports this assumption [18–20]. A European study estimated the prevalence of Barrett’s esophagus to be 1.6% among the general population [21,22]. Barrett’s esophagus is usually diagnosed during endoscopic examinations of middle-aged and older adults, with the mean age being 55 years of age. It is most commonly found in Caucasian males and associated with the use of smoking tobacco. The male-to-female ratio is approximately 2:1 [1] and it appears to be uncommon in African Americans [23,24]. Abdominal obesity as measured by an increased waist-to-hip ratio is associated with an increased risk of Barrett’s esophagus [25,26]. Germline mutations in the MSR1, ASCC1, and CTHRC1 genes have been associated with the presence of Barrett’s esophagus and esophageal adenocarcinoma [27]. Risk factors are listed in Table 1. 
Clinical Symptoms
Columnar metaplasia itself does not cause any symptoms but is merely the adaptation of the cells to the repeated effect of the acid. The main clinical symptoms of the disease would initially be symptoms associated with GERD, such as heartburn, water brash, and dysphagia [1]. Severe presentations of GERD, such as esophageal ulceration, stricture, and hemorrhage, usually occur with long-segment Barrett’s esophagus [29,30]. However, 40% of patients presenting with adenocarcinoma had no history of GERD or symptoms of heartburn [13]. Furthermore, as few as 5% of those presenting with adenocarcinoma were known to have Barrett’s esophagus [31].
D iagnosis
Barrett’s esophagus generally requires an endoscopic examination with biopsy confirmation from the distal esophagus showing specialized intestinal columnar epithelium [32]. The biopsy specimen is acquired from the cellular lining proximal to gastroesophageal junction [5,33]. Barrett’s esophagus is classified into long- and short-segment based on the length of salmon-colored mucosa in the distal esophagus. A distance longer than 3 cm is 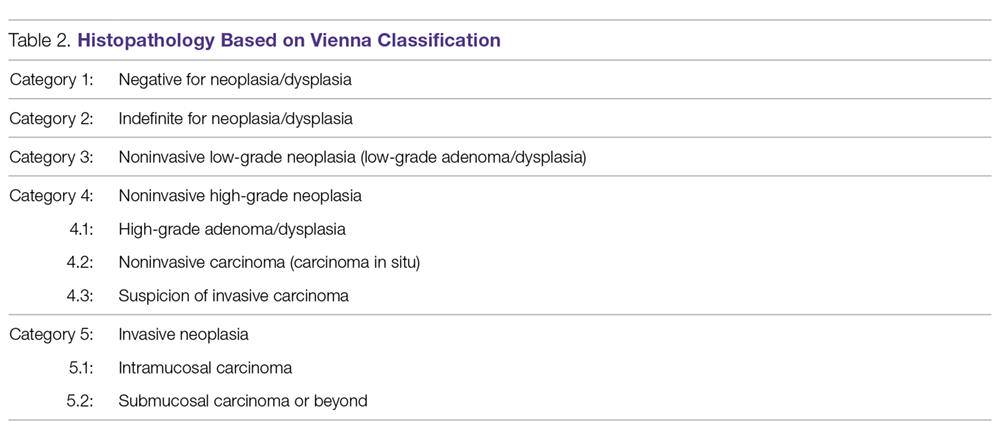
The Prague classification was presented by an international research group in 2006 and is regarded as the standard for measuring the length of Barrett’s esophagus. The lower measurement boundary is formed by the proximal cardial notch, and the 2 upper measurement boundaries are marked by the proximal limit of the circumferential Barrett’s segment and the longest tongue of Barrett’s [37].Confirmation of the diagnosis of dysplastic 
Once the initial diagnosis of Barrett’s esophagus is made, we recommend referring the patient to a Barrett’s esophagus specialized center in order to offer the patient a second opinion from a team of experts in Barrett’s esophagus. This would avoid possible false-positive results, which can be as high as 40% [1,8]. It would also offer the patient a comprehensive multidisciplinary approach and adequate long-term management. It is further preferred if an advanced intervention is offered to the patient such as endoscopic mucosal resection or endoscopic submucosal dissection. Expert endoscopists 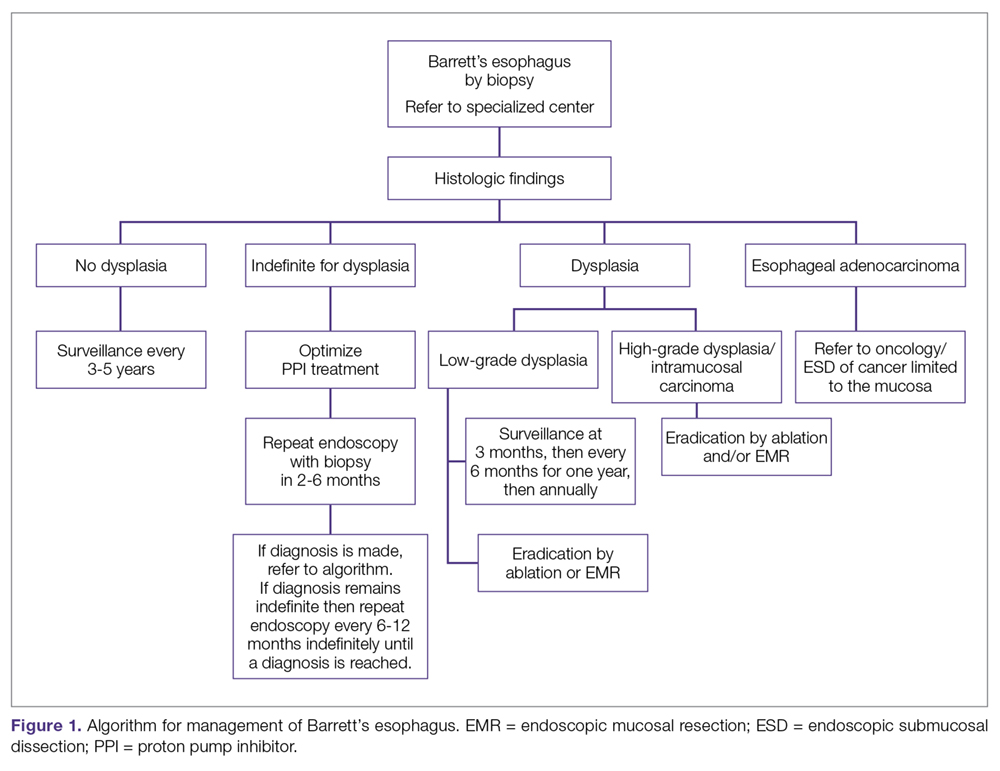
The availability of advanced endoscopic tools improves diagnostic yield. Adopting advanced techniques can reduce errors during biopsy sampling [40–42]. Some of the newer advanced imaging endoscopic tools include chromoendoscopy, optical coherence tomography, confocal microendoscopy, autofluorescence endoscopy, narrow band imaging (NBI), and Fujinon intelligent chromoendoscopy (FICE) [43,44]. In a meta-analysis examining whether advanced techniques improved diagnostic yield, it was found that advanced imaging increased the diagnostic yield by 34%. Advanced technology mentioned here is not mandated in current guidelines [5,45].
Histopathology categories and TNM staging system for Barrett’s esophagus are shown in Table 2 and Table 3. A management algorithm based on histologic findings is presented in Figure 1.
S creening
Logically, Barrett’s esophagus screening should be offered to every patient with GERD; however, this is against current recommendations because it is cost-prohibitive [46,47]. It is a given that the rationale behind the screening is to decrease morbidity and mortality from esophageal adenocarcinoma by offering early and definitive intervention [14,48,49]. The gold standard for screening is upper endoscopy, although nonendoscopic methods are being studied [1]. For example, a capsule attached to a string can be swallowed by the patient, the capsule is then deployed and pulled through the esophagus to obtains a brush sample of the cells. It is a promising technology due to high sensitivity and specificity [50]; however, it is not regularly utilized in practice.
Approaches to screening for Barrett’s esophagus has been addressed by multiple societies with several guidelines currently available [51]. None of these approaches have been proven to be superior in clinical studies. The American Gastroenterological Association (AGA) recommends screening patients with multiple risk factors associated with esophageal adenocarcinoma for Barrett’s esophagus. Risk factors listed by the AGA include white patients, male patients, patients above the age of 50 with a history of chronic GERD, hiatal hernia, elevated body mass index, and certain body fat distribution. The AGA recommends against screening the general population with GERD [9]. The American College of Gastroenterology (ACG) recommends upper endoscopy only in the presence of alarm symptoms (eg, dysphagia, weight loss, gastrointestinal bleeding) and for screening of patients at high risk for complications [52]. The American College of Physicians recommends upper endoscopy for screening for Barrett’s esophagus in men older than 50 years with GERD symptoms for more than 5 years with any risk factors like nocturnal reflux symptoms, hiatus hernia, elevated body mass index, tobacco use, and intra-abdominal distribution of fat [1].
Overall, the sensitivity of endoscopy to diagnose Barrett’s esophagus, as seen in a Veterans Affairs (VA) cohort study for the detection of Barrett’s, is about 80%. The concluded 80% sensitivity rate was based on the performance of 2 endoscopies, 6 weeks apart for each patient, with subsequent labeling of the diagnosis if intestinal metaplasia was found in either of the 2 biopsy samples taken from the 2 procedures [53].
Promising molecular biomarkers have been associated with Barrett’s esophagus including p53 and cyclin D1 expression. However, additional studies are needed before they are incorporated as part of screening practices [5].
General Management
Medical Management of GERD
All patients with Barrett’s esophagus should be treated with a proton pump inhibitor (PPI) indefinitely based on multiple studies [54–56]. Effective control of GERD was associated with a decreased risk of dysplasia and adenocarcinoma (adjusted odds ratio 0.29, 95% CI 0.12–0.79) [57].
Effective control of GERD decreases chronic esophageal inflammation, which could progress to Barrett’s esophagus and has a risk of possible progression to adenocarcinoma. In one study, patients with proven Barrett’s esophagus showed partial regression of the intestinal metaplasia with aggressive PPI therapy [57–59]. Despite this, it is not clear whether the regression decreased risk of malignant progression [58,60,61]. In a study of 68 patients, aggressive acid reduction with omeprazole 40 mg twice a day lead to partial regression of Barrett’s esophagus when compared to mild suppression with ranitidine 150 mg twice a day. However, there was no reduction in the risk of cancer [59].
Surveillance
The reasoning behind surveillance is to detect dysplasia or adenocarcinoma in patients known to have Barrett’s esophagus early enough to provide early and efficient treatment to improve the outcome. Surveillance is done by endoscopy with biopsies in addition to sampling any irregularity [1,62,63]; however, the evidence to back up the benefit of surveillance is not clear [5,64]. It should be noted that surveillance carries risks, and morbidity associated with repeated procedures may affect patients psychologically and financially. Patients who have Barrett’s esophagus are more likely to die from other more common diseases, such as coronary heart disease, prior to developing adenocarcinoma [65]. In a recent meta-analysis, the mortality rate due to esophageal adenocarcinoma was 3.0 per 1000 person-years, whereas the mortality rate due to other causes was 37.1 per 1000 person-years [65]. An ongoing randomized, multicenter trial which assesses scheduled endoscopy every 2 years will shed more light on the overall survival after applying surveillance recommendations for each grade of dysplasia [66].
The following are ACG recommendations for surveillance of Barrett’s esophagus based on the histopathology report. The histopathology report delineates 1 of 3 types of columnar epithelium [68]: cardiac epithelium of mucus-secreting cells, atrophic gastric fundic type epithelium, or specialized columnar cells with goblet cells. The latter type is the most common with high potential for cancer [32]. Degrees of dysplasia and possible adenocarcinoma is usually described in the report as well. The degree of dysplasia if found helps the endoscopist plan the next step in management. It is worth mentioning that sampling errors can lead to missing a diagnosis. In a meta-analysis, 13% of patients diagnosed with high-grade dysplasia who underwent resection were found to have invasive cancer [69].
Surveillance for Patients with No Dysplasia
We suggest surveillance every 3 to 5 years since the rate of neoplasia is low [14]. For management of select patients with no dysplasia and with additional risk factors, radiofrequency ablation (RFA) may be an option, although it remains a controversial approach. For example, in a patient under 50 years of age, family history would be an argument for proceeding with RFA instead of prolonged surveillance. In a prospective cohort study of 139 patients with 10-year follow-up after ablation, recurrent Barrett’s occurred in less than 5% of the patients. [70]
Surveillance for Patients with Biopsy Showing “Indefinite for Dysplasia”
Aggressive treatment with PPI twice daily is recommended to avoid the misinterpretation of reactive esophageal changes secondary to reflux as dysplasia on the following endoscopy with biopsy. These patients will require a repeat endoscopy with biopsies after 3 months of aggressive treatment with PPI. Biopsies should be taken every 1 centimeter within Barrett’s epithelium [1].
If it remains indefinite, biopsies should be examined by a second pathologist with expertise in Barrett’s esophagus. If the second pathologist agrees on the indefinite diagnosis for dysplasia, then endoscopy every 12 months is recommended [1]. Treatment versus surveillance after repeat endoscopy and biopsy should be tailored to the new histopathology results on the most recent exam.
Surveillance for Patients with Biopsy Results showing Low-Grade Dysplasia (LGD), High-Grade Dysplasia (HGD), or Intramucosal Carcinoma
Surveillance recommendations are discussed under Management of Dysplasia or Intramucosal Carcinoma, below.
Efficacy of Surveillance
Asymptomatic adenocarcinoma could be discovered during surveillance, and neoplasia detected during surveillance is usually less advanced than those found after development of symptoms such as dysphagia, bleeding or weight loss [2,3,71–75]. These studies obviously had lead–time bias and did not document terminal cancer in patients adherent to surveillance protocol.
Management of Dysplasia or Intramucosal Carcinoma
Overview
Historically, dysplasia was managed with esophagectomy, which was associated with high morbidity and mortality. With advancement in the field of endoscopy, dysplasia is managed quite differently today, with endoscopic eradication therapy, which includes the use of endoscopic ablation techniques and endoscopic resection. The advantage of endoscopic resection is preservation of resected tissue for further examination, thus providing valuable information regarding the stage of the tumor (depth). Histological examination is not possible with photo or thermal ablation techniques as destroyed mucosa cannot be submitted for tissue analysis.
Low-Grade Dysplasia
If low-grade dysplasia is found, it is followed by a repeat endoscopy 8 weeks after aggressive PPI therapy. The repeat endoscopy should be performed with high definition/high-resolution endoscopy. The rationale of a second endoscopy is to ensure that the metaplastic mucosa was adequately inspected and biopsied prior to further intervention [1,9]. If the diagnosis is confirmed as low-grade dysplasia, and the patient prefers to go with the path of intervention instead of conservative management (endoscopic surveillance every 6 months for 1 year then annually with biopsies), then multiple options are available for the patient, including RFA or cryotherapy [8] [76]. In a randomized clinical trial in patients with Barrett’s esophagus and LGD, RFA was shown to reduce risk of neoplastic progression over a 3-year follow-up. The study included 136 patients randomized to receive ablation and 68 patients who underwent endoscopic surveillance. In the ablation group, the risk of progression to HGD or esophageal adenocarcinoma was reduced by 25% and the risk of progression to adenocarcinoma was reduced by 7.4%. In the ablation group, complete eradication of dysplasia and intestinal metaplasia occurred in 92.6% and 88.2% of patients respectively. In the endoscopic surveillance group, complete eradication of dysplasia and intestinal metaplasia was seen in 27.9% and 0.0% of patients respectively. [76]. Treatment-related adverse events occurred in 19.1% of patients receiving ablation (P < 0.001). The most common adverse event was stricture, occurring in 8 patients receiving ablation (11.8%), all resolved by endoscopic dilation [76,78].
Surveillance after ablation of LGD is still an ongoing debate, and further evidence is needed to establish guidelines [8]. Due to lack of evidence, we would lean towards surveillance for those patients with an annual esophagogastroduodenoscopy with biopsy examination (author’s opinion, no associated level of evidence).
High-Grade Dysplasia or Intramucosal Carcinoma
For patients with HGD or intramucosal carcinoma without submucosal invasion, eradication is the treatment of choice. Current guidelines advocate for endoscopic eradication therapy for most if not all patients with HGD or intramucosal carcinoma with a goal of removing all metaplastic and dysplastic tissue [1,5,62,64]. It should be noted that the biopsy specimen should be extensive to decrease the error margin. If the diagnosis were made on endoscopy without procuring extensive biopsies, then repeat endoscopy with extensive biopsies is needed prior to deciding the treatment path. The rationale behind extensive biopsies is to confirm the diagnosis and to determine the extent of dysplasia. Other factors potentially influencing the treatment path include the patient’s age, comorbid conditions, quality of life, and available access to an advanced endoscopist or specialized surgeon. Patient’s preferences and adherence to follow-up visits should also be a consideration.
The long-term benefits of endoscopic intervention versus surgical intervention are not well established. Esophagectomy is no longer a preferred method of treatment due to high morbidity and mortality associated with the procedure when compared to endoscopic interventions. However, it is still a preferred choice amongst a select group of patients unwilling to follow-up. A cost-effective analysis found that endoscopic ablation provided the longest quality adjusted life expectancy for Barrett’s esophagus with HGD [79,80].
Endoscopic Therapy in Barrett’s Esophagus
Endoscopic Ablative Therapies
With the advancement of endoscopic intervention, we now have multiple tools to ablate abnormal epithelium in Barrett’s esophagus. Examples of ablation techniques include thermal, photochemical, and mechanical techniques [81,82]. RFA is the treatment of choice for ablation [83]. However, non-contact ablative therapy, such as cryoablation, may be prefered if topography of the esophagus doesn’t allow contact ablation.
Radiofrequency Ablation (Figure 2). RFA is a procedure in which heat is generated from medium frequency alternating current and leads to thermal injury [84]. In Barrett’s esophagus, RFA uses radiofrequency energy delivered by a balloon that has a series of closely spaced electrodes in a 
Most patients will require multiple sessions of RFA to achieve eradication. It is very rarely a one-time procedure. In a meta-analysis of 18 studies including 3082 patients, the most common adverse effects of RFA were stricture in 5% [76,98], bleeding in 1%, and pain in 3% of patients [99].
It is crucial for successful RFA to continue medical treatment for acid suppression, in order to allow healthy regeneration of the squamous cell lining. It is suggested to use PPI twice a day with sucralfate and ranitidine after the intervention [100,101]. Adhering to a liquid diet for 24 hours is needed, followed by a soft diet to allow faster regeneration of the epithelium.
The caveat with RFA is that new evidence shows a higher rate of recurrence than previously thought. In one study of 246 patients, recurrence of dysplasia occurred in 25% of patients at 48 months after eradication in 80% of the patients, and metaplasia occurred in 50% at 60 months [102]. The other risk is buried Barrett’s, a condition occurring after incomplete ablation, in which squamous cell epithelium covers patches of incompletely destroyed intestinal lining, leading to possible progression of the disease to adenocarcinoma under the surface [103].
It has been reported that patients who underwent RFA had remarkable improvement in quality of life even if RFA did not achieve eradication. Patients reported less depression, less stress and better quality of life [104].
Based on a survey of experts, follow-up at 3 months, 6 months, and then annually is recommended after ablation [1,105]. Biopsies should be taken distal to neosquamous epithelium and from suspicious areas [97,106].
Endoscopic Spray Cryotherapy. This technique involves application of liquid nitrogen or carbon dioxide gas by endoscope on the tissue to freeze it off. Although it has been shown to eliminate HGD in over 95% of the cases and all dysplasia in over 85% of the cases, it was effective in eradicating intestinal metaplasia in only 55% of patients [103,108,109]. Thus, RFA as ablation therapy is still superior to cryotherapy and is still the first-line treatment for dysplastic Barrett’s esophagus. In comparison to cryotherapy, RFA efficacy has been studied extensively with well documented outcomes. However, there is a role for cryotherapy over RFA in certain clinical situations (such as severe chest pain from RFA or lack of efficacy in eradicating intestinal metaplasia or dysplasia by RFA).
Similar to RFA, on occasions of partial ablation, the remaining metaplastic tissue may get buried beneath a layer of squamous epithelium and can possibly progress to adenocarcinoma [110].
Photodynamic Therapy (PDT). This technique works by producing cytotoxicity at the cellular level by exposure to light at a specific wavelength in the presence of a chemical agent known as photosensitize [107]. Although superior to omeprazole, PDT has a significant rate of complications, mainly stricture, and a high occurrence of esophageal cancer during follow-up. For this reason, it is less favorable compared to RFA [107] and mentioned here as a historical therapy.
Endoscopic Resection Techniques: Endoscopic Mucosal Resection (EMR) and Endoscopic Submucosal Dissection (ESD)
Unlike flat mucosa in Barrett’s esophagus, which respond to ablative techniques such as RFA or cryotherapy, nodular Barrett’s esophagus is hard to treat and requires endoscopic resection prior to ablation. Endoscopic mucosal resection (EMR) is the most widely used technique and it is available in most tertiary referral centers. Another technique named endoscopic submucosal dissection (ESD) allows the removal of large nodular areas of Barrett’s esophagus in one piece to ensure complete removal of nodular dysplasia. ESD is technically challenging and it is only available in a handful of centers in the US. Endoscopic resection techniques are the preferred interventions for nodular dysplasia due to their ability to provide valuable information for staging the lesion [111–113]. Endoscopic resection techniques are safer and more effective with similar or better results when compared with other approaches [114].
EMR is completed by the excision of esophageal mucosa down to the submucosa and submitting a large tissue specimen to the pathologist. It additionally serves as a therapeutic measure in cases of no submucosal extension. Another advantage of EMR is the ability to predict lymph node metastasis. The rationale is based on the fact that the most important predictor of lymph node metastasis is the depth of the tumor; hence, invasive tumors would likely be associated with lymph node metastasis [115,116].
In a systematic review of 11 studies, complete EMR was as equally effective in the short-term treatment of dysplastic Barrett’s esophagus when compared to RFA, but adverse event rates were greater with complete EMR (mainly strictures). Strictures are more likely to occur in patients undergoing extensive EMR. In another meta-analysis of 22 studies comparing the efficacy of EMR to RFA, both techniques were effective in eradicating dysplasia (95% in EMR group and 92% in RFA group). However, extensive EMR was associated with higher complication rates suggesting that a combined endoscopic approach of focal EMR followed by RFA is preferred over extensive EMR alone [86].
It should be noted that EMR and ESD information were derived from highly specialized center and these results may not be duplicated in community settings [113,117].
Efficacy of Endoscopic Resection. Endoscopic resection has a success rate comparable to surgical esophagectomy with fewer complications [113,114,118–121] in patients with HGD and early stages of esophageal cancer [122]. Complete remission can be as high as 89%. Recurrence occurred in 6% to 30% of patients [114,118,119], which was attributed to incomplete removal, large lesions, failure to use adjunct therapy, or lack of follow-up [123]. Even when recurrence occurred, it was successfully managed by endoscopic intervention [124].
In a large cohort study of 1000 patients with early mucosal adenocarcinoma who were treated with endoscopic resection, long-term complete remission occurred in 94% of patients. There was no mortality and less than 2% of patients had major complications. Infrequent complications include bleeding, perforations, and strictures [123,125,126]. The rate of complications is lower in highly specialized centers [127–129].
Surgery was necessary in 12 patients (3.7%) after endoscopic therapy failed [123]. Post-resection care and follow-up is similar to the post-RFA care discussed above.
Management of Invasive Esophageal Adenocarcinoma
Patients diagnosed with an invasive adenocarcinoma need to be referred to an oncologist for staging and to discuss treatment options. A select number of patients may be referred by oncology for endoscopic resection, yet the need for a multidisciplinary approach in these situations is absolutely necessary [1].
Esophagectomy
Esophagectomy offers the complete removal of the HGD along with any adenocarcinoma in the regional lymph nodes. However, mortality rates are as high as 12% immediately after the procedure [130]. The multitude of short- and long-term morbidity has significant effects on quality of life. Short-term morbidity is as high as 30%. Patients may develop serious postoperative complications such as myocardial infarction, hospital associated pneumonia, or anatomic leak [131].
Examples of long-term morbidity include dysphagia, transection of vagal nerve, and dumping syndrome. Recent development in minimally invasive surgeries for esophagectomy has not reduced postoperative morbidity rates [132].
Advocates of esophagectomy illustrate the advantage of eradication of occult lymph node metastasis. The counter argument has been established by a systemic review in which occult lymph node metastasis occurred in less than 2% of patients with HGD and intramucosal carcinoma; whereas the mortality rate after esophagectomy is substantially higher with no guarantee of curing metastatic disease [133].
Prevention of Barrett’s Esophagus
Since Barrett’s esophagus precedes most of the cases of EAC if not all [1,134], methods that aim at decreasing the incidence of Barrett’s esophagus could help in prevention. The modifiable risk factors listed by the AGA include BMI, GERD, and hiatal hernia management. Along with diet and exercise, the advent of new therapies to help patients manage their weight could in return help in avoiding a plethora of medical conditions including Barrett’s esophagus. Hiatal hernia management could lower the risk of Barrett’s by restoring normal anatomy. Lastly, proper management of GERD would lower the risk of developing Barrett’s esophagus as discussed in this article [1,9].
It is worth noting that a large trial on the efficacy and safety of aspirin for prevention of adenocarcinoma progression in Barrett’s esophagus is ongoing in the UK (AspECT trial). The AspECT trial examines the efficacy of low dose vs. high dose PPI with or without aspirin for the chemoprevention of esophageal adenocarcinoma. The theory behind the study is the inhibition of COX 2 receptors in Barrett’s cells can decrease tissue progression to cancer. This chempreventive effect of nonsteroidal anti-inflammatory drugs was shown to be augmented when combined with statin intake [56,135–138].
Conclusion
Barrett’s esophagus is usually diagnosed during routine endoscopic examination. The initial symptoms are those associated with GERD, like heartburn, dyspepsia, and regurgitation. Specialized columnar epithelium is the hallmark of histopathological diagnosis. Recommendations of the ACG and AGA suggest treatment based on biopsy results. The intervention would vary on a wide spectrum starting from acid suppression, radiofrequency ablation, endoscopic resection therapy, and rarely, esophagectomy.
Corresponding author: Mohamed O. Othman, MD, Gastroenterology and Hepatology Section, Baylor College of Medicine, 7200 Cambridge St., Suite 8C, Houston, TX 77030, [email protected].
Financial disclosures: Dr. Othman has received grant support from Abbvie and has served as a consultant for Olympus.
1. Shaheen NJ, Falk GW, Iyer PG, Gerson LB; American College of Gastroenterology. ACG Clinical Guideline: diagnosis and management of Barrett’s esophagus. Am J Gastroenterol 2016;111:30–50; quiz 51.
2. Peters JH, Clark GW, Ireland AP. Outcome of adenocarcinoma arising in Barrett’s esophagus in endoscopically surveyed and nonsurveyed patients. J Thorac Cardiovasc Surg 1994;108:813–21; discussion 821–2.
3. Streitz JM Jr, Andrews CW Jr, Ellis FH Jr. Endoscopic surveillance of Barrett’s esophagus. Does it help? J Thorac Cardiovasc Surg 1993;105:383–7; discussion 387–8.
4. Spechler SJ, Souza RF. Barrett’s esophagus. N Engl J Med 2014;371:836–45.
5. Spechler SJ, Sharma P, Souza RF, et al; American Gastroenterological Association. American Gastroenterological Association medical position statement on the management of Barrett’s esophagus. Gastroenterology 2011;140:1084–91.
6. Sharma P, McQuaid K, Dent J, et al; AGA Chicago Workshop. A critical review of the diagnosis and management of Barrett’s esophagus: the AGA Chicago Workshop. Gastroenterology 2004;127:310–30.
7. ASGE Standards of Practice Committee, Evans JA, Early DS; Standards of Practice Committee of the American Society for Gastrointestinal Endoscopy. The role of endoscopy in Barrett’s esophagus and other premalignant conditions of the esophagus. Gastrointest Endosc 2012;76(6):1087–94.
8. Wani S, Rubenstein JH, Vieth M, Bergman J. Diagnosis and management of low–grade dysplasia in Barrett’s esophagus: Expert review from the clinical practice updates Committee of the American Gastroenterological Association. Gastroenterology 2016;151:822–835.
9. American Gastroenterological, A., et al., American Gastroenterological Association medical position statement on the management of Barrett’s esophagus. Gastroenterology, 2011. 140(3): p. 1084–91. [Same as #5]
10. Van Eyken P. Definition of Barrett’s oesophagus. Acta Gastroenterol Belg 2000;63:10–2.
11. Hirota WK, Loughney TM, Lazas DJ, et al. Specialized intestinal metaplasia, dysplasia, and cancer of the esophagus and esophagogastric junction: prevalence and clinical data. Gastroenterology 1999;116(2):277–85.
12. Cameron AJ, Zinsmeister AR, Ballard DJ, Carney JA. Prevalence of columnar–lined (Barrett’s) esophagus. Comparison of population–based clinical and autopsy findings. Gastroenterology 1990;99:918–22.
13. Gerson LB, Shetler K, Triadafilopoulos G. Prevalence of Barrett’s esophagus in asymptomatic individuals. Gastroenterology 2002;123:461–7.
14. Van der Veen AH, Dees J, Blankensteijn JD, Van Blankenstein M. Adenocarcinoma in Barrett’s oesophagus: an overrated risk. Gut 1989;30:14–8.
15. Winters C Jr, Spurling TJ, Chobanian SJ, et al. Barrett’s esophagus. A prevalent, occult complication of gastroesophageal reflux disease. Gastroenterology 1987;92:118–24.
16. Wani S, Falk G, Hall M, et al. Patients with nondysplastic Barrett’s esophagus have low risks for developing dysplasia or esophageal adenocarcinoma. Clin Gastroenterol Hepatol 2011;9:220–7;quiz e26.
17. Ward EM, Wolfsen HC, Achem SR, et al. Barrett’s esophagus is common in older men and women undergoing screening colonoscopy regardless of reflux symptoms. Am J Gastroenterol 2006;101:12–7.
18. Rex DK, Cummings OW, Shaw M. Screening for Barrett’s esophagus in colonoscopy patients with and without heartburn. Gastroenterology 2003;125:1670–7.
19. Rubenstein JH, Taylor JB. Meta–analysis: the association of oesophageal adenocarcinoma with symptoms of gastro–oesophageal reflux. Aliment Pharmacol Ther 2010;32:1222–7.
20. Lagergren J, Bergström R, Lindgren A, Nyrén O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med 1999;340:825–31.
21. Ronkainen J, Aro P, Storskrubb T, et al. Prevalence of Barrett’s esophagus in the general population: an endoscopic study. Gastroenterology, 2005;129:1825–31.
22. Sampliner RE. A population prevalence of Barrett’s esophagus––finally. Gastroenterology 2005; 129:2101–3.
23. Abrams JA, Fields S, Lightdale CJ, Neugut AI. Racial and ethnic disparities in the prevalence of Barrett’s esophagus among patients who undergo upper endoscopy. Clin Gastroenterol Hepatol 2008;6:30–4.
24. Corley DA, Kubo A, Levin TR, et al. Race, ethnicity, sex and temporal differences in Barrett’s oesophagus diagnosis: a large community–based study, 1994–2006. Gut 2009;58:182–8.
25. Kamat P, Wen S, Morris J, Anandasabapathy S. Exploring the association between elevated body mass index and Barrett’s esophagus: a systematic review and meta–analysis. Ann Thorac Surg 2009;87:655–62.
26. Jacobson BC, Chan AT, Giovannucci EL, Fuchs CS. Body mass index and Barrett’s oesophagus in women. Gut 2009;58:1460–6.
27. Orloff M, Peterson C, He X, et al. Germline mutations in MSR1, ASCC1, and CTHRC1 in patients with Barrett esophagus and esophageal adenocarcinoma. JAMA 2011;306:410–9.
28. Sharma N, Ho KY. Risk Factors for Barrett’s oesophagus. Gastrointest Tumors 2016;3:103–8.
29. Spechler SJ. Barrett’s esophagus. Semin Gastrointest Dis 1996;7:51–60.
30. Ronkainen J, Talley NJ, Storskrubb T. Erosive esophagitis is a risk factor for Barrett’s esophagus: a community–based endoscopic follow–up study. Am J Gastroenterol 2011;106:1946–52.
31. Kim SL, Wo JM, Hunter JG. The prevalence of intestinal metaplasia in patients with and without peptic strictures. Am J Gastroenterol 1998;93:53–5.
32. Spechler SJ. Clinical practice. Barrett’s esophagus. N Engl J Med 2002;346:836–42.
33. Riddell RH, Odze RD. Definition of Barrett’s esophagus: time for a rethink––is intestinal metaplasia dead? Am J Gastroenterol 2009;104:2588–94.
34. Sharma P, Morales TG, Sampliner RE. Short segment Barrett’s esophagus––the need for standardization of the definition and of endoscopic criteria. Am J Gastroenterol 1998;93:1033–6.
35. Eloubeidi MA, Provenzale D. Does this patient have Barrett’s esophagus? The utility of predicting Barrett’s esophagus at the index endoscopy. Am J Gastroenterol 1999;94:937–43.
36. Rudolph RE, Vaughan TL, Storer BE, et al. Effect of segment length on risk for neoplastic progression in patients with Barrett esophagus. Ann Intern Med 2000;132:612–20.
37. Sharma P, Dent J, Armstrong D, et al. The development and validation of an endoscopic grading system for Barrett’s esophagus: the Prague C & M criteria. Gastroenterology 2006;131:1392–9.
38. Bhat S, Coleman HG, Yousef F, et al. Risk of malignant progression in Barrett’s esophagus patients: results from a large population–based study. J Natl Cancer Inst 2011;103:1049–57.
39. Shaheen NJ, Dulai GS, Ascher B, et al. Effect of a new diagnosis of Barrett’s esophagus on insurance status. Am J Gastroenterol 2005;100:577–80.
40. Canto MI, Setrakian S, Willis J, et al. Methylene blue–directed biopsies improve detection of intestinal metaplasia and dysplasia in Barrett’s esophagus. Gastrointest Endosc 2000;51:560–8.
41. Scotiniotis IA, Kochman ML, Lewis JD. Accuracy of EUS in the evaluation of Barrett’s esophagus and high–grade dysplasia or intramucosal carcinoma. Gastrointest Endosc 2001;54:689–96.
42. Kobayashi K, Izatt JA, Kulkarni MD, et al. High–resolution cross–sectional imaging of the gastrointestinal tract using optical coherence tomography: preliminary results. Gastrointest Endosc 1998;47:515–23.
43. Georgakoudi I, Jacobson BC, Van Dam J, et al. Fluorescence, reflectance, and light–scattering spectroscopy for evaluating dysplasia in patients with Barrett’s esophagus. Gastroenterology 2001. 120: 1620–9.
44. Wallace MB, Sharma P, Lightdale C, et al. Preliminary accuracy and interobserver agreement for the detection of intraepithelial neoplasia in Barrett’s esophagus with probe–based confocal laser endomicroscopy. Gastrointest Endosc 2010;72:19–24.
45. Qumseya BJ, Wang H, Badie N, et al. Advanced imaging technologies increase detection of dysplasia and neoplasia in patients with Barrett’s esophagus: a meta–analysis and systematic review. Clin Gastroenterol Hepatol 2013;11:1562–70.e1–2.–
46. DeVault KR, Castell DO. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. The Practice Parameters Committee of the American College of Gastroenterology. Am J Gastroenterol 1999;94:1434–42.
47. Inadomi JM, Sampliner R, Lagergren J. Screening and surveillance for Barrett esophagus in high–risk groups: a cost–utility analysis. Ann Intern Med 2003;138:176–86.
48. Conio M, Blanchi S, Lapertosa G, et al. Long–term endoscopic surveillance of patients with Barrett’s esophagus. Incidence of dysplasia and adenocarcinoma: a prospective study. Am J Gastroenterol 2003;98:1931–9.
49. Rastogi A, Puli S, El–Serag HB, et al. Incidence of esophageal adenocarcinoma in patients with Barrett’s esophagus and high–grade dysplasia: a meta–analysis. Gastrointest Endosc 2008;67:394–8.
50. Kadri SR, Lao–Sirieix P, O’Donovan M, et al. Acceptability and accuracy of a non–endoscopic screening test for Barrett’s oesophagus in primary care: cohort study. BMJ 2010;341:c4372.
51. Sharma, P., et al., A critical review of the diagnosis and management of Barrett’s esophagus: the AGA Chicago Workshop. Gastroenterology, 2004. 127(1): p. 310–30. [Same as #6]
52. Katz PO, Gerson LB, Vela MF. Guidelines for the diagnosis and management of gastroesophageal reflux disease. Am J Gastroenterol 2013;108:308–28; quiz 329.
53. Kim SL, Waring JP, Spechler SJ, et al. Diagnostic inconsistencies in Barrett’s esophagus. Department of Veterans Affairs Gastroesophageal Reflux Study Group. Gastroenterology 1994;107:945–9.
54. Kastelein F, Spaander MC, Steyerberg EW, et al; ProBar Study Group. Proton pump inhibitors reduce the risk of neoplastic progression in patients with Barrett’s esophagus. Clin Gastroenterol Hepatol 2013;11:382–8.
55. El–Serag HB, Aguirre TV, Davis S, et al. Proton pump inhibitors are associated with reduced incidence of dysplasia in Barrett’s esophagus. Am J Gastroenterol 2004;99:1877–83.
56. Nguyen DM, El–Serag HB, Henderson L, et al. Medication usage and the risk of neoplasia in patients with Barrett’s esophagus. Clin Gastroenterol Hepatol 2009;7:1299–304.
57. Singh S, Garg SK, Singh PP, et al. Acid–suppressive medications and risk of oesophageal adenocarcinoma in patients with Barrett’s oesophagus: a systematic review and meta–analysis. Gut 2014;63:1229–37.
58. Spechler SJ, Lee E, Ahnen D, et al. Long–term outcome of medical and surgical therapies for gastroesophageal reflux disease: follow–up of a randomized controlled trial. JAMA 2001;285:2331–8.
59. Peters FT, Ganesh S, Kuipers EJ, et al. Endoscopic regression of Barrett’s oesophagus during omeprazole treatment; a randomised double blind study. Gut 1999;45:489–94.
60. Ouatu–Lascar R, Triadafilopoulos G. –Complete elimination of reflux symptoms does not guarantee normalization of intraesophageal acid reflux in patients with Barrett’s esophagus. Am J Gastroenterol 1998;93:711–6.
61. Maret–Ouda J, Konings P, Lagergren J, Brusselaers N. Antireflux surgery and risk of esophageal adenocarcinoma: A systematic review and meta–analysis. Ann Surg 2016;263:251–7.
62. Evans, J.A., et al., The role of endoscopy in Barrett’s esophagus and other premalignant conditions of the esophagus. Gastrointest Endosc, 2012. 76(6): p. 1087–94. [Same as #7]
63. ASGE Standards of Practice Committee, Evans JA, Early DS,et al; American Society for Gastrointestinal Endoscopy. The role of endoscopy in the assessment and treatment of esophageal cancer. Gastrointest Endosc 2013;77:328–34.
64. Bennett C, Moayyedi P, Corley DA, et al; BOB CAT Consortium. BOB CAT: A large–scale review and delphi consensus for ,anagement of Barrett’s esophagus with no dysplasia, indefinite for, or low–grade dysplasia. Am J Gastroenterol 2015;110:662–82; quiz 683.
65. Sikkema M, de Jonge PJ, Steyerberg EW, Kuipers EJ. Risk of esophageal adenocarcinoma and mortality in patients with Barrett’s esophagus: a systematic review and meta–analysis. Clin Gastroenterol Hepatol 2010;8:235–44; quiz e32.
66. Old O, Moayyedi P, Love S, et al; BOSS Trial Team. Barrett’s Oesophagus Surveillance versus endoscopy at need Study (BOSS): protocol and analysis plan for a multicentre randomized controlled trial. J Med Screen 2015;22:158–64.
67. Weston AP, Sharma P, Topalovski M, et al. Long–term follow–up of Barrett’s high–grade dysplasia. Am J Gastroenterol 2000;95:1888–93.
68. Paull A, Trier JS, Dalton MD, et al. The histologic spectrum of Barrett’s esophagus. N Engl J Med 1976;295:476–80.
69. Konda VJ, Ross AS, Ferguson MK, et al. Is the risk of concomitant invasive esophageal cancer in high–grade dysplasia in Barrett’s esophagus overestimated? Clin Gastroenterol Hepatol 2008;6:159–64.
70. Allison H, Banchs MA, Bonis PA, Guelrud M. Long–term remission of nondysplastic Barrett’s esophagus after multipolar electrocoagulation ablation: report of 139 patients with 10 years of follow–up. Gastrointest Endosc 2011;73:651–8.
71. Corley DA, Levin TR, Habel LA, Weiss NS, et al. Surveillance and survival in Barrett’s adenocarcinomas: a population–based study. Gastroenterology 2002;122:633–40.
72. Wong T, Tian J, Nagar AB. Barrett’s surveillance identifies patients with early esophageal adenocarcinoma. Am J Med 2010;123:462–7.
73. Fountoulakis A, Zafirellis KD, Dolan K, et al. Effect of surveillance of Barrett’s oesophagus on the clinical outcome of oesophageal cancer. Br J Surg 2004;91:997–1003.
74. Verbeek RE, Leenders M, Ten Kate FJ, et al. Surveillance of Barrett’s esophagus and mortality from esophageal adenocarcinoma: a population–based cohort study. Am J Gastroenterol 2014;109:1215–22.
75. Kastelein F, van Olphen SH, Steyerberg EW, et al. Impact of surveillance for Barrett’s oesophagus on tumour stage and survival of patients with neoplastic progression. Gut 2016;65:548–54.
76. Phoa KN, van Vilsteren FG, Weusten BL, et al. Radiofrequency ablation vs endoscopic surveillance for patients with Barrett esophagus and low–grade dysplasia: a randomized clinical trial. JAMA 2014;311: 1209–17.
77. Wang WL, Chang IW, Chen CC, et al. Radiofrequency ablation versus endoscopic submucosal dissection in treating large early esophageal squamous cell neoplasia. Medicine (Baltimore) 2015;94:e2240.
78. Lim CH, Treanor D, Dixon MF, Axon AT. Low–grade dysplasia in Barrett’s esophagus has a high risk of progression. Endoscopy 2007;39:581–7.
79. Shaheen NJ, Inadomi JM, Overholt BF, Sharma P. What is the best management strategy for high grade dysplasia in Barrett’s oesophagus? A cost effectiveness analysis. Gut 2004;53:1736–44.
80. Vij R, Triadafilopoulos G, Owens DK, et al. Cost–effectiveness of photodynamic therapy for high–grade dysplasia in Barrett’s esophagus. Gastrointest Endosc 2004;60:739–56.
81. van den Boogert J, v an Hillegersberg R, Siersema PD, et al. Endoscopic ablation therapy for Barrett’s esophagus with high–grade dysplasia: a review. Am J Gastroenterol 1999;94:1153–60.
82. Sampliner RE. Endoscopic ablative therapy for Barrett’s esophagus: current status. Gastrointest Endosc 2004;59:66–9.
83. Sharma VK, Wang KK, Overholt BF, et al. Balloon–based, circumferential, endoscopic radiofrequency ablation of Barrett’s esophagus: 1–year follow–up of 100 patients. Gastrointest Endosc 2007;65:185–95.
84. Hanlon CR. Textbook of surgery: The biological basis of modern surgical practice, 14th edition. Ann Surg 1992;216:94.
85. Bright T, Watson DI, Tam W, et al. Randomized trial of argon plasma coagulation versus endoscopic surveillance for barrett esophagus after antireflux surgery: late results. Ann Surg 2007;246:1016–20.
86. Chadwick G, Groene O, Markar SR, et al. Systematic review comparing radiofrequency ablation and complete endoscopic resection in treating dysplastic Barrett’s esophagus: a critical assessment of histologic outcomes and adverse events. Gastrointest Endosc 2014;79:718–31.e3.
87. Gondrie JJ, Pouw RE, Sondermeijer CM, et al. Effective treatment of early Barrett’s neoplasia with stepwise circumferential and focal ablation using the HALO system. Endoscopy 2008;40:370–9.
88. Pouw RE, Wirths K, Eisendrath P, et al. Efficacy of radiofrequency ablation combined with endoscopic resection for barrett’s esophagus with early neoplasia. Clin Gastroenterol Hepatol 2010;8:23–9.
89. Kim HP, Bulsiewicz WJ, Cotton CC, et al. Focal endoscopic mucosal resection before radiofrequency ablation is equally effective and safe compared with radiofrequency ablation alone for the eradication of Barrett’s esophagus with advanced neoplasia. Gastrointest Endosc 2012;76:733–9.
90. Fleischer DE, Overholt BF, Sharma VK, et al. Endoscopic ablation of Barrett’s esophagus: a multicenter study with 2.5–year follow–up. Gastrointest Endosc 2008;68:867–76.
91. Sharma VK, Jae Kim H, Das A, et al. Circumferential and focal ablation of Barrett’s esophagus containing dysplasia. Am J Gastroenterol 2009;104:310–7.
92. Ganz RA, Overholt BF, Sharma VK, et al. Circumferential ablation of Barrett’s esophagus that contains high–grade dysplasia: a U.S. multicenter registry. Gastrointest Endosc 2008;68:35–40.
93. Gupta M, Iyer PG, Lutzke L, et al. Recurrence of esophageal intestinal metaplasia after endoscopic mucosal resection and radiofrequency ablation of Barrett’s esophagus: results from a US Multicenter Consortium. Gastroenterology 2013;145:79–86.e1.
94. Lyday WD, Corbett FS, Kuperman DA, et al. Radiofrequency ablation of Barrett’s esophagus: outcomes of 429 patients from a multicenter community practice registry. Endoscopy 2010;42:272–8.
95. Pasricha S, Bulsiewicz WJ, Hathorn KE, et al. Durability and predictors of successful radiofrequency ablation for Barrett’s esophagus. Clin Gastroenterol Hepatol 2014;12:1840–7.e1.
96. Fleischer DE, Overholt BF, Sharma VK, et al. Endoscopic radiofrequency ablation for Barrett’s esophagus: 5–year outcomes from a prospective multicenter trial. Endoscopy 2010;42:781–9.
97. Weston AP, Sharma P, Banerjee S, et al. Visible endoscopic and histologic changes in the cardia, before and after complete Barrett’s esophagus ablation. Gastrointest Endosc 2005;61:515–21.
98. Beaumont H, Gondrie JJ, McMahon BP, et al. Stepwise radiofrequency ablation of Barrett’s esophagus preserves esophageal inner diameter, compliance, and motility. Endoscopy 2009;41:2–8.
99. Orman ES, Li N, Shaheen NJ.Efficacy and durability of radiofrequency ablation for Barrett’s Esophagus: systematic review and meta–analysis. Clin Gastroenterol Hepatol 2013;11:1245–55.
100. Krishnan K, Pandolfino JE, Kahrilas PJ, et al. Increased risk for persistent intestinal metaplasia in patients with Barrett’s esophagus and uncontrolled reflux exposure before radiofrequency ablation. Gastroenterology 2012;143:576–81.
101. Akiyama J, Marcus SN, Triadafilopoulos G. Effective intra–esophageal acid control is associated with improved radiofrequency ablation outcomes in Barrett’s esophagus. Dig Dis Sci 2012;57:2625–32.
102. Small AJ, Sutherland SE, Hightower JS, et al. Comparative risk of recurrence of dysplasia and carcinoma after endoluminal eradication therapy of high–grade dysplasia versus intramucosal carcinoma in Barrett’s esophagus. Gastrointest Endosc 2015;81:1158–66.e1–4.
103. Shaheen NJ, Greenwald BD, Peery AF, et al. Safety and efficacy of endoscopic spray cryotherapy for Barrett’s esophagus with high–grade dysplasia. Gastrointest Endosc 2010;71:680–5.
104. Shaheen NJ, Peery AF, Hawes RH, et al. Quality of life following radiofrequency ablation of dysplastic Barrett’s esophagus. Endoscopy 2010;42:790–9.
105. Bedi AO, Kwon RS, Rubenstein JH, et al. A survey of expert follow–up practices after successful endoscopic eradication therapy for Barrett’s esophagus with high–grade dysplasia and intramucosal adenocarcinoma. Gastrointest Endosc 2013;78:696–701.
106. Sampliner RE, Camargo E, Prasad AR. Prasad, Association of ablation of Barrett’s esophagus with high grade dysplasia and adenocarcinoma of the gastric cardia. Dis Esophagus 2006;19:277–9.
107. Overholt BF, Panjehpour M, Halberg DL. Photodynamic therapy for Barrett’s esophagus with dysplasia and/or early stage carcinoma: long–term results. Gastrointest Endosc 2003;58:183–8.
108. Gosain S, Mercer K, Twaddell WS, et al. Liquid nitrogen spray cryotherapy in Barrett’s esophagus with high–grade dysplasia: long–term results. Gastrointest Endosc 2013;78:260–5.
109. Canto MI, Shin EJ, Khashab MA, et al. Safety and efficacy of carbon dioxide cryotherapy for treatment of neoplastic Barrett’s esophagus. Endoscopy 2015;47:582–91.
110. Van Laethem JL, Peny MO, Salmon I, et al. Intramucosal adenocarcinoma arising under squamous re–epithelialisation of Barrett’s oesophagus. Gut 2000;46:574–7.
111. Pech O, May A, Gossner L, et al. Management of pre–malignant and malignant lesions by endoscopic resection. Best Pract Res Clin Gastroenterol 2004;18:61–76.
112. Soetikno RM, Gotoda T, Nakanishi Y, Soehendra N. Endoscopic mucosal resection. Gastrointest Endosc 2003;57:567–79.
113. Ell C, May A, Gossner L, et al. Endoscopic mucosal resection of early cancer and high–grade dysplasia in Barrett’s esophagus. Gastroenterology 2000;118:670–7.
114. Pech O, Behrens A, May A, et al. Long–term results and risk factor analysis for recurrence after curative endoscopic therapy in 349 patients with high–grade intraepithelial neoplasia and mucosal adenocarcinoma in Barrett’s oesophagus. Gut 2008;57:1200–6.
115. Vieth M, Ell C, Gossner L, et al. Histological analysis of endoscopic resection specimens from 326 patients with Barrett’s esophagus and early neoplasia. Endoscopy 2004;36:776–81.
116. Buskens CJ, Westerterp M, Lagarde SM, et al. Prediction of appropriateness of local endoscopic treatment for high–grade dysplasia and early adenocarcinoma by EUS and histopathologic features. Gastrointest Endosc 2004;60:703–10.
117. Nijhawan PK, Wang KK. Endoscopic mucosal resection for lesions with endoscopic features suggestive of malignancy and high–grade dysplasia within Barrett’s esophagus. Gastrointest Endosc 2000;52:328–32.
118. Esaki M, Matsumoto T, Hirakawa K, et al. Risk factors for local recurrence of superficial esophageal cancer after treatment by endoscopic mucosal resection. Endoscopy 2007;39:41–5.
119. Ell C, May A, Pech O, et al. Curative endoscopic resection of early esophageal adenocarcinomas (Barrett’s cancer). Gastrointest Endosc 2007;65:3–10.
120. Chennat J, Konda VJ, Ross AS, et al. Complete Barrett’s eradication endoscopic mucosal resection: an effective treatment modality for high–grade dysplasia and intramucosal carcinoma––an American single–center experience. Am J Gastroenterol 2009;104:2684–92.
121. Pech O, Bollschweiler E, Manner H, et al. Comparison between endoscopic and surgical resection of mucosal esophageal adenocarcinoma in Barrett’s esophagus at two high–volume centers. Ann Surg 2011;254:67–72.
122. Wu J, Pan YM, Wang TT, et al. Endotherapy versus surgery for early neoplasia in Barrett’s esophagus: a meta–analysis. Gastrointest Endosc 2014;79:233–241.e2.
123. Pech O, May A, Manner H, et al. Long–term efficacy and safety of endoscopic resection for patients with mucosal adenocarcinoma of the esophagus. Gastroenterology 2014;146:652–660.e1.
124. Prasad GA, Wu TT, Wigle DA, et al. Endoscopic and surgical treatment of mucosal (T1a) esophageal adenocarcinoma in Barrett’s esophagus. Gastroenterology 2009;137:815–23.
125. May A, Gossner L, Pech O, et al. Local endoscopic therapy for intraepithelial high–grade neoplasia and early adenocarcinoma in Barrett’s oesophagus: acute–phase and intermediate results of a new treatment approach. Eur J Gastroenterol Hepatol 2002;14(10):1085–91.
126. Gerke H, Siddiqui J, Nasr I, et al. Efficacy and safety of EMR to completely remove Barrett’s esophagus: experience in 41 patients. Gastrointest Endosc 2011;74:761–71.
127. Lewis JJ, Rubenstein JH, Singal AG, et al. Factors associated with esophageal stricture formation after endoscopic mucosal resection for neoplastic Barrett’s esophagus. Gastrointest Endosc 2011;74:753–60.
128. Choi IJ, Kim CG, Chang HJ, et al. The learning curve for EMR with circumferential mucosal incision in treating intramucosal gastric neoplasm. Gastrointest Endosc 2005;62:860–5.
129. Deprez PH, Bergman JJ, Meisner S, et al. Current practice with endoscopic submucosal dissection in Europe: position statement from a panel of experts. Endoscopy 2010;42:853–8.
130. van Lanschot JJ, Hulscher JB, Buskens CJ, et al. Hospital volume and hospital mortality for esophagectomy. Cancer 2001;91:1574–8.
131. Karl RC, Schreiber R, Boulware D, et al. Factors affecting morbidity, mortality, and survival in patients undergoing Ivor Lewis esophagogastrectomy. Ann Surg 2000;231:635–43.
132. Young MM, Deschamps C, Trastek VF, et al. Esophageal reconstruction for benign disease: early morbidity, mortality, and functional results. Ann Thorac Surg 2000;70:1651–5.
133. Dunbar KB, Spechler SJ. The risk of lymph–node metastases in patients with high–grade dysplasia or intramucosal carcinoma in Barrett’s esophagus: a systematic review. Am J Gastroenterol 2012;107:850–62; quiz 863.
134. Morales CP, Souza RF, Spechler SJ. Hallmarks of cancer progression in Barrett’s oesophagus. Lancet 2002;360:1587–9.
135. Omer ZB, Ananthakrishnan AN, Nattinger KJ, et al. Aspirin protects against Barrett’s esophagus in a multivariate logistic regression analysis. Clin Gastroenterol Hepatol 2012;10:722–7.
136. Abnet CC, Freedman ND, Kamangar F, et al. Non–steroidal anti–inflammatory drugs and risk of gastric and oesophageal adenocarcinomas: results from a cohort study and a meta–analysis. Br J Cancer 2009;100:551–7.
137. Kastelein F, Spaander MC, Biermann K, et al. Nonsteroidal anti–inflammatory drugs and statins have chemopreventative effects in patients with Barrett’s esophagus. Gastroenterology 2011;141:2000–8; quiz e13–4.
138. Zhang S, Zhang XQ, Ding XW, et al. Cyclooxygenase inhibitors use is associated with reduced risk of esophageal adenocarcinoma in patients with Barrett’s esophagus: a meta–analysis. Br J Cancer 2014;110: 2378–88.
From the Gastroenterology and Hepatology Section, Baylor College of Medicine, Houston, TX.
Abstract
- Objective: To provide an update on management of Barrett’s esophagus.
- Methods: Review of the literature.
- Results: Management of Barrett’s esophagus depends on the degree of dysplasia. Surveillance by endoscopy every 3–5 years is recommended in patients with Barrett’s esophagus without dysplasia. Patients with Barrett’s esophagus and low-grade dysplasia should undergo surveillance by endoscopy in 3 months for confirmation of the diagnosis; if the diagnosis is confirmed then surveillance by endoscopy or eradication of Barrett’s epithelium by ablation or endoscopic resection are recommended. There is a sufficient evidence to recommend radiofrequency ablation of high-grade dysplasia within Barrett’s esophagus or to perform endoscopic mucosal resection of nodular Barrett’s esophagus with any degree of dysplasia. Early esophageal cancers that are limited to the mucosa can be treated by endoscopic resection, while cancer invading into the deep submucosa or muscularis propria may need esophagectomy with or without chemoradiation.
- Conclusion: The management of Barrett’s esophagus depends on the degree of dysplasia. Radiofrequency ablation and endoscopic mucosal resection are the most commonly used treatment for Barrett’s esophagus with dysplasia.
Keywords: Barrett’s esophagus; radiofrequency ablation; endoscopic submucosal dissection; endoscopic mucosal resection; early esophageal cancer.
Barrett’s esophagus is a common complication of chronic reflux disease [1]. Metaplastic changes that occur at the distal esophageal epithelium are usually asymptomatic [2,3] and occur as reparative adaptations to the insult of the gastric acid [4]. The management of Barrett’s esophagus after diagnosis is currently debated amongst experts without a clear consensus [5,6]. This review is generally consistent with the 2016 guidelines from the American College of Gastroenterology [1], a 2012 guideline from the American Society of Gastrointestinal Endoscopy [7], a 2011 guideline, and a 2016 expert review from the American Gastroenterological Association [8,9].
D efinition
Barrett’s esophagus is a metaplasia of the stratified squamous epithelium to a specialized columnar intestinal epithelium of mucus cells and goblet cells at the distal esophagus secondary to gastroesophageal reflux disease (GERD) [7,10]. Barrett’s esophagus is unstable tissue which can progress to esophageal adenocarcinoma. When unmanaged, the risk of cancer in dysplastic mucosa is at least thirty-fold greater than that for the general population [11–14], with recent studies suggesting a 0.4–0.7 occurrence rate per year [11,15]. With no dysplasia, the risk is low [16].
Epidemiology
The prevalence of Barrett’s esophagus is ~10% in patients with GERD [11–13,17], an estimate tied to the prevalence of GERD. However, due to the lack of symptoms of Barrett’s esophagus, no solid data supports this assumption [18–20]. A European study estimated the prevalence of Barrett’s esophagus to be 1.6% among the general population [21,22]. Barrett’s esophagus is usually diagnosed during endoscopic examinations of middle-aged and older adults, with the mean age being 55 years of age. It is most commonly found in Caucasian males and associated with the use of smoking tobacco. The male-to-female ratio is approximately 2:1 [1] and it appears to be uncommon in African Americans [23,24]. Abdominal obesity as measured by an increased waist-to-hip ratio is associated with an increased risk of Barrett’s esophagus [25,26]. Germline mutations in the MSR1, ASCC1, and CTHRC1 genes have been associated with the presence of Barrett’s esophagus and esophageal adenocarcinoma [27]. Risk factors are listed in Table 1.
Clinical Symptoms
Columnar metaplasia itself does not cause any symptoms but is merely the adaptation of the cells to the repeated effect of the acid. The main clinical symptoms of the disease would initially be symptoms associated with GERD, such as heartburn, water brash, and dysphagia [1]. Severe presentations of GERD, such as esophageal ulceration, stricture, and hemorrhage, usually occur with long-segment Barrett’s esophagus [29,30]. However, 40% of patients presenting with adenocarcinoma had no history of GERD or symptoms of heartburn [13]. Furthermore, as few as 5% of those presenting with adenocarcinoma were known to have Barrett’s esophagus [31].
D iagnosis
Barrett’s esophagus generally requires an endoscopic examination with biopsy confirmation from the distal esophagus showing specialized intestinal columnar epithelium [32]. The biopsy specimen is acquired from the cellular lining proximal to gastroesophageal junction [5,33]. Barrett’s esophagus is classified into long- and short-segment based on the length of salmon-colored mucosa in the distal esophagus. A distance longer than 3 cm is
The Prague classification was presented by an international research group in 2006 and is regarded as the standard for measuring the length of Barrett’s esophagus. The lower measurement boundary is formed by the proximal cardial notch, and the 2 upper measurement boundaries are marked by the proximal limit of the circumferential Barrett’s segment and the longest tongue of Barrett’s [37].Confirmation of the diagnosis of dysplastic
Once the initial diagnosis of Barrett’s esophagus is made, we recommend referring the patient to a Barrett’s esophagus specialized center in order to offer the patient a second opinion from a team of experts in Barrett’s esophagus. This would avoid possible false-positive results, which can be as high as 40% [1,8]. It would also offer the patient a comprehensive multidisciplinary approach and adequate long-term management. It is further preferred if an advanced intervention is offered to the patient such as endoscopic mucosal resection or endoscopic submucosal dissection. Expert endoscopists
The availability of advanced endoscopic tools improves diagnostic yield. Adopting advanced techniques can reduce errors during biopsy sampling [40–42]. Some of the newer advanced imaging endoscopic tools include chromoendoscopy, optical coherence tomography, confocal microendoscopy, autofluorescence endoscopy, narrow band imaging (NBI), and Fujinon intelligent chromoendoscopy (FICE) [43,44]. In a meta-analysis examining whether advanced techniques improved diagnostic yield, it was found that advanced imaging increased the diagnostic yield by 34%. Advanced technology mentioned here is not mandated in current guidelines [5,45].
Histopathology categories and TNM staging system for Barrett’s esophagus are shown in Table 2 and Table 3. A management algorithm based on histologic findings is presented in Figure 1.
S creening
Logically, Barrett’s esophagus screening should be offered to every patient with GERD; however, this is against current recommendations because it is cost-prohibitive [46,47]. It is a given that the rationale behind the screening is to decrease morbidity and mortality from esophageal adenocarcinoma by offering early and definitive intervention [14,48,49]. The gold standard for screening is upper endoscopy, although nonendoscopic methods are being studied [1]. For example, a capsule attached to a string can be swallowed by the patient, the capsule is then deployed and pulled through the esophagus to obtains a brush sample of the cells. It is a promising technology due to high sensitivity and specificity [50]; however, it is not regularly utilized in practice.
Approaches to screening for Barrett’s esophagus has been addressed by multiple societies with several guidelines currently available [51]. None of these approaches have been proven to be superior in clinical studies. The American Gastroenterological Association (AGA) recommends screening patients with multiple risk factors associated with esophageal adenocarcinoma for Barrett’s esophagus. Risk factors listed by the AGA include white patients, male patients, patients above the age of 50 with a history of chronic GERD, hiatal hernia, elevated body mass index, and certain body fat distribution. The AGA recommends against screening the general population with GERD [9]. The American College of Gastroenterology (ACG) recommends upper endoscopy only in the presence of alarm symptoms (eg, dysphagia, weight loss, gastrointestinal bleeding) and for screening of patients at high risk for complications [52]. The American College of Physicians recommends upper endoscopy for screening for Barrett’s esophagus in men older than 50 years with GERD symptoms for more than 5 years with any risk factors like nocturnal reflux symptoms, hiatus hernia, elevated body mass index, tobacco use, and intra-abdominal distribution of fat [1].
Overall, the sensitivity of endoscopy to diagnose Barrett’s esophagus, as seen in a Veterans Affairs (VA) cohort study for the detection of Barrett’s, is about 80%. The concluded 80% sensitivity rate was based on the performance of 2 endoscopies, 6 weeks apart for each patient, with subsequent labeling of the diagnosis if intestinal metaplasia was found in either of the 2 biopsy samples taken from the 2 procedures [53].
Promising molecular biomarkers have been associated with Barrett’s esophagus including p53 and cyclin D1 expression. However, additional studies are needed before they are incorporated as part of screening practices [5].
General Management
Medical Management of GERD
All patients with Barrett’s esophagus should be treated with a proton pump inhibitor (PPI) indefinitely based on multiple studies [54–56]. Effective control of GERD was associated with a decreased risk of dysplasia and adenocarcinoma (adjusted odds ratio 0.29, 95% CI 0.12–0.79) [57].
Effective control of GERD decreases chronic esophageal inflammation, which could progress to Barrett’s esophagus and has a risk of possible progression to adenocarcinoma. In one study, patients with proven Barrett’s esophagus showed partial regression of the intestinal metaplasia with aggressive PPI therapy [57–59]. Despite this, it is not clear whether the regression decreased risk of malignant progression [58,60,61]. In a study of 68 patients, aggressive acid reduction with omeprazole 40 mg twice a day lead to partial regression of Barrett’s esophagus when compared to mild suppression with ranitidine 150 mg twice a day. However, there was no reduction in the risk of cancer [59].
Surveillance
The reasoning behind surveillance is to detect dysplasia or adenocarcinoma in patients known to have Barrett’s esophagus early enough to provide early and efficient treatment to improve the outcome. Surveillance is done by endoscopy with biopsies in addition to sampling any irregularity [1,62,63]; however, the evidence to back up the benefit of surveillance is not clear [5,64]. It should be noted that surveillance carries risks, and morbidity associated with repeated procedures may affect patients psychologically and financially. Patients who have Barrett’s esophagus are more likely to die from other more common diseases, such as coronary heart disease, prior to developing adenocarcinoma [65]. In a recent meta-analysis, the mortality rate due to esophageal adenocarcinoma was 3.0 per 1000 person-years, whereas the mortality rate due to other causes was 37.1 per 1000 person-years [65]. An ongoing randomized, multicenter trial which assesses scheduled endoscopy every 2 years will shed more light on the overall survival after applying surveillance recommendations for each grade of dysplasia [66].
The following are ACG recommendations for surveillance of Barrett’s esophagus based on the histopathology report. The histopathology report delineates 1 of 3 types of columnar epithelium [68]: cardiac epithelium of mucus-secreting cells, atrophic gastric fundic type epithelium, or specialized columnar cells with goblet cells. The latter type is the most common with high potential for cancer [32]. Degrees of dysplasia and possible adenocarcinoma is usually described in the report as well. The degree of dysplasia if found helps the endoscopist plan the next step in management. It is worth mentioning that sampling errors can lead to missing a diagnosis. In a meta-analysis, 13% of patients diagnosed with high-grade dysplasia who underwent resection were found to have invasive cancer [69].
Surveillance for Patients with No Dysplasia
We suggest surveillance every 3 to 5 years since the rate of neoplasia is low [14]. For management of select patients with no dysplasia and with additional risk factors, radiofrequency ablation (RFA) may be an option, although it remains a controversial approach. For example, in a patient under 50 years of age, family history would be an argument for proceeding with RFA instead of prolonged surveillance. In a prospective cohort study of 139 patients with 10-year follow-up after ablation, recurrent Barrett’s occurred in less than 5% of the patients. [70]
Surveillance for Patients with Biopsy Showing “Indefinite for Dysplasia”
Aggressive treatment with PPI twice daily is recommended to avoid the misinterpretation of reactive esophageal changes secondary to reflux as dysplasia on the following endoscopy with biopsy. These patients will require a repeat endoscopy with biopsies after 3 months of aggressive treatment with PPI. Biopsies should be taken every 1 centimeter within Barrett’s epithelium [1].
If it remains indefinite, biopsies should be examined by a second pathologist with expertise in Barrett’s esophagus. If the second pathologist agrees on the indefinite diagnosis for dysplasia, then endoscopy every 12 months is recommended [1]. Treatment versus surveillance after repeat endoscopy and biopsy should be tailored to the new histopathology results on the most recent exam.
Surveillance for Patients with Biopsy Results showing Low-Grade Dysplasia (LGD), High-Grade Dysplasia (HGD), or Intramucosal Carcinoma
Surveillance recommendations are discussed under Management of Dysplasia or Intramucosal Carcinoma, below.
Efficacy of Surveillance
Asymptomatic adenocarcinoma could be discovered during surveillance, and neoplasia detected during surveillance is usually less advanced than those found after development of symptoms such as dysphagia, bleeding or weight loss [2,3,71–75]. These studies obviously had lead–time bias and did not document terminal cancer in patients adherent to surveillance protocol.
Management of Dysplasia or Intramucosal Carcinoma
Overview
Historically, dysplasia was managed with esophagectomy, which was associated with high morbidity and mortality. With advancement in the field of endoscopy, dysplasia is managed quite differently today, with endoscopic eradication therapy, which includes the use of endoscopic ablation techniques and endoscopic resection. The advantage of endoscopic resection is preservation of resected tissue for further examination, thus providing valuable information regarding the stage of the tumor (depth). Histological examination is not possible with photo or thermal ablation techniques as destroyed mucosa cannot be submitted for tissue analysis.
Low-Grade Dysplasia
If low-grade dysplasia is found, it is followed by a repeat endoscopy 8 weeks after aggressive PPI therapy. The repeat endoscopy should be performed with high definition/high-resolution endoscopy. The rationale of a second endoscopy is to ensure that the metaplastic mucosa was adequately inspected and biopsied prior to further intervention [1,9]. If the diagnosis is confirmed as low-grade dysplasia, and the patient prefers to go with the path of intervention instead of conservative management (endoscopic surveillance every 6 months for 1 year then annually with biopsies), then multiple options are available for the patient, including RFA or cryotherapy [8] [76]. In a randomized clinical trial in patients with Barrett’s esophagus and LGD, RFA was shown to reduce risk of neoplastic progression over a 3-year follow-up. The study included 136 patients randomized to receive ablation and 68 patients who underwent endoscopic surveillance. In the ablation group, the risk of progression to HGD or esophageal adenocarcinoma was reduced by 25% and the risk of progression to adenocarcinoma was reduced by 7.4%. In the ablation group, complete eradication of dysplasia and intestinal metaplasia occurred in 92.6% and 88.2% of patients respectively. In the endoscopic surveillance group, complete eradication of dysplasia and intestinal metaplasia was seen in 27.9% and 0.0% of patients respectively. [76]. Treatment-related adverse events occurred in 19.1% of patients receiving ablation (P < 0.001). The most common adverse event was stricture, occurring in 8 patients receiving ablation (11.8%), all resolved by endoscopic dilation [76,78].
Surveillance after ablation of LGD is still an ongoing debate, and further evidence is needed to establish guidelines [8]. Due to lack of evidence, we would lean towards surveillance for those patients with an annual esophagogastroduodenoscopy with biopsy examination (author’s opinion, no associated level of evidence).
High-Grade Dysplasia or Intramucosal Carcinoma
For patients with HGD or intramucosal carcinoma without submucosal invasion, eradication is the treatment of choice. Current guidelines advocate for endoscopic eradication therapy for most if not all patients with HGD or intramucosal carcinoma with a goal of removing all metaplastic and dysplastic tissue [1,5,62,64]. It should be noted that the biopsy specimen should be extensive to decrease the error margin. If the diagnosis were made on endoscopy without procuring extensive biopsies, then repeat endoscopy with extensive biopsies is needed prior to deciding the treatment path. The rationale behind extensive biopsies is to confirm the diagnosis and to determine the extent of dysplasia. Other factors potentially influencing the treatment path include the patient’s age, comorbid conditions, quality of life, and available access to an advanced endoscopist or specialized surgeon. Patient’s preferences and adherence to follow-up visits should also be a consideration.
The long-term benefits of endoscopic intervention versus surgical intervention are not well established. Esophagectomy is no longer a preferred method of treatment due to high morbidity and mortality associated with the procedure when compared to endoscopic interventions. However, it is still a preferred choice amongst a select group of patients unwilling to follow-up. A cost-effective analysis found that endoscopic ablation provided the longest quality adjusted life expectancy for Barrett’s esophagus with HGD [79,80].
Endoscopic Therapy in Barrett’s Esophagus
Endoscopic Ablative Therapies
With the advancement of endoscopic intervention, we now have multiple tools to ablate abnormal epithelium in Barrett’s esophagus. Examples of ablation techniques include thermal, photochemical, and mechanical techniques [81,82]. RFA is the treatment of choice for ablation [83]. However, non-contact ablative therapy, such as cryoablation, may be prefered if topography of the esophagus doesn’t allow contact ablation.
Radiofrequency Ablation (Figure 2). RFA is a procedure in which heat is generated from medium frequency alternating current and leads to thermal injury [84]. In Barrett’s esophagus, RFA uses radiofrequency energy delivered by a balloon that has a series of closely spaced electrodes in a
Most patients will require multiple sessions of RFA to achieve eradication. It is very rarely a one-time procedure. In a meta-analysis of 18 studies including 3082 patients, the most common adverse effects of RFA were stricture in 5% [76,98], bleeding in 1%, and pain in 3% of patients [99].
It is crucial for successful RFA to continue medical treatment for acid suppression, in order to allow healthy regeneration of the squamous cell lining. It is suggested to use PPI twice a day with sucralfate and ranitidine after the intervention [100,101]. Adhering to a liquid diet for 24 hours is needed, followed by a soft diet to allow faster regeneration of the epithelium.
The caveat with RFA is that new evidence shows a higher rate of recurrence than previously thought. In one study of 246 patients, recurrence of dysplasia occurred in 25% of patients at 48 months after eradication in 80% of the patients, and metaplasia occurred in 50% at 60 months [102]. The other risk is buried Barrett’s, a condition occurring after incomplete ablation, in which squamous cell epithelium covers patches of incompletely destroyed intestinal lining, leading to possible progression of the disease to adenocarcinoma under the surface [103].
It has been reported that patients who underwent RFA had remarkable improvement in quality of life even if RFA did not achieve eradication. Patients reported less depression, less stress and better quality of life [104].
Based on a survey of experts, follow-up at 3 months, 6 months, and then annually is recommended after ablation [1,105]. Biopsies should be taken distal to neosquamous epithelium and from suspicious areas [97,106].
Endoscopic Spray Cryotherapy. This technique involves application of liquid nitrogen or carbon dioxide gas by endoscope on the tissue to freeze it off. Although it has been shown to eliminate HGD in over 95% of the cases and all dysplasia in over 85% of the cases, it was effective in eradicating intestinal metaplasia in only 55% of patients [103,108,109]. Thus, RFA as ablation therapy is still superior to cryotherapy and is still the first-line treatment for dysplastic Barrett’s esophagus. In comparison to cryotherapy, RFA efficacy has been studied extensively with well documented outcomes. However, there is a role for cryotherapy over RFA in certain clinical situations (such as severe chest pain from RFA or lack of efficacy in eradicating intestinal metaplasia or dysplasia by RFA).
Similar to RFA, on occasions of partial ablation, the remaining metaplastic tissue may get buried beneath a layer of squamous epithelium and can possibly progress to adenocarcinoma [110].
Photodynamic Therapy (PDT). This technique works by producing cytotoxicity at the cellular level by exposure to light at a specific wavelength in the presence of a chemical agent known as photosensitize [107]. Although superior to omeprazole, PDT has a significant rate of complications, mainly stricture, and a high occurrence of esophageal cancer during follow-up. For this reason, it is less favorable compared to RFA [107] and mentioned here as a historical therapy.
Endoscopic Resection Techniques: Endoscopic Mucosal Resection (EMR) and Endoscopic Submucosal Dissection (ESD)
Unlike flat mucosa in Barrett’s esophagus, which respond to ablative techniques such as RFA or cryotherapy, nodular Barrett’s esophagus is hard to treat and requires endoscopic resection prior to ablation. Endoscopic mucosal resection (EMR) is the most widely used technique and it is available in most tertiary referral centers. Another technique named endoscopic submucosal dissection (ESD) allows the removal of large nodular areas of Barrett’s esophagus in one piece to ensure complete removal of nodular dysplasia. ESD is technically challenging and it is only available in a handful of centers in the US. Endoscopic resection techniques are the preferred interventions for nodular dysplasia due to their ability to provide valuable information for staging the lesion [111–113]. Endoscopic resection techniques are safer and more effective with similar or better results when compared with other approaches [114].
EMR is completed by the excision of esophageal mucosa down to the submucosa and submitting a large tissue specimen to the pathologist. It additionally serves as a therapeutic measure in cases of no submucosal extension. Another advantage of EMR is the ability to predict lymph node metastasis. The rationale is based on the fact that the most important predictor of lymph node metastasis is the depth of the tumor; hence, invasive tumors would likely be associated with lymph node metastasis [115,116].
In a systematic review of 11 studies, complete EMR was as equally effective in the short-term treatment of dysplastic Barrett’s esophagus when compared to RFA, but adverse event rates were greater with complete EMR (mainly strictures). Strictures are more likely to occur in patients undergoing extensive EMR. In another meta-analysis of 22 studies comparing the efficacy of EMR to RFA, both techniques were effective in eradicating dysplasia (95% in EMR group and 92% in RFA group). However, extensive EMR was associated with higher complication rates suggesting that a combined endoscopic approach of focal EMR followed by RFA is preferred over extensive EMR alone [86].
It should be noted that EMR and ESD information were derived from highly specialized center and these results may not be duplicated in community settings [113,117].
Efficacy of Endoscopic Resection. Endoscopic resection has a success rate comparable to surgical esophagectomy with fewer complications [113,114,118–121] in patients with HGD and early stages of esophageal cancer [122]. Complete remission can be as high as 89%. Recurrence occurred in 6% to 30% of patients [114,118,119], which was attributed to incomplete removal, large lesions, failure to use adjunct therapy, or lack of follow-up [123]. Even when recurrence occurred, it was successfully managed by endoscopic intervention [124].
In a large cohort study of 1000 patients with early mucosal adenocarcinoma who were treated with endoscopic resection, long-term complete remission occurred in 94% of patients. There was no mortality and less than 2% of patients had major complications. Infrequent complications include bleeding, perforations, and strictures [123,125,126]. The rate of complications is lower in highly specialized centers [127–129].
Surgery was necessary in 12 patients (3.7%) after endoscopic therapy failed [123]. Post-resection care and follow-up is similar to the post-RFA care discussed above.
Management of Invasive Esophageal Adenocarcinoma
Patients diagnosed with an invasive adenocarcinoma need to be referred to an oncologist for staging and to discuss treatment options. A select number of patients may be referred by oncology for endoscopic resection, yet the need for a multidisciplinary approach in these situations is absolutely necessary [1].
Esophagectomy
Esophagectomy offers the complete removal of the HGD along with any adenocarcinoma in the regional lymph nodes. However, mortality rates are as high as 12% immediately after the procedure [130]. The multitude of short- and long-term morbidity has significant effects on quality of life. Short-term morbidity is as high as 30%. Patients may develop serious postoperative complications such as myocardial infarction, hospital associated pneumonia, or anatomic leak [131].
Examples of long-term morbidity include dysphagia, transection of vagal nerve, and dumping syndrome. Recent development in minimally invasive surgeries for esophagectomy has not reduced postoperative morbidity rates [132].
Advocates of esophagectomy illustrate the advantage of eradication of occult lymph node metastasis. The counter argument has been established by a systemic review in which occult lymph node metastasis occurred in less than 2% of patients with HGD and intramucosal carcinoma; whereas the mortality rate after esophagectomy is substantially higher with no guarantee of curing metastatic disease [133].
Prevention of Barrett’s Esophagus
Since Barrett’s esophagus precedes most of the cases of EAC if not all [1,134], methods that aim at decreasing the incidence of Barrett’s esophagus could help in prevention. The modifiable risk factors listed by the AGA include BMI, GERD, and hiatal hernia management. Along with diet and exercise, the advent of new therapies to help patients manage their weight could in return help in avoiding a plethora of medical conditions including Barrett’s esophagus. Hiatal hernia management could lower the risk of Barrett’s by restoring normal anatomy. Lastly, proper management of GERD would lower the risk of developing Barrett’s esophagus as discussed in this article [1,9].
It is worth noting that a large trial on the efficacy and safety of aspirin for prevention of adenocarcinoma progression in Barrett’s esophagus is ongoing in the UK (AspECT trial). The AspECT trial examines the efficacy of low dose vs. high dose PPI with or without aspirin for the chemoprevention of esophageal adenocarcinoma. The theory behind the study is the inhibition of COX 2 receptors in Barrett’s cells can decrease tissue progression to cancer. This chempreventive effect of nonsteroidal anti-inflammatory drugs was shown to be augmented when combined with statin intake [56,135–138].
Conclusion
Barrett’s esophagus is usually diagnosed during routine endoscopic examination. The initial symptoms are those associated with GERD, like heartburn, dyspepsia, and regurgitation. Specialized columnar epithelium is the hallmark of histopathological diagnosis. Recommendations of the ACG and AGA suggest treatment based on biopsy results. The intervention would vary on a wide spectrum starting from acid suppression, radiofrequency ablation, endoscopic resection therapy, and rarely, esophagectomy.
Corresponding author: Mohamed O. Othman, MD, Gastroenterology and Hepatology Section, Baylor College of Medicine, 7200 Cambridge St., Suite 8C, Houston, TX 77030, [email protected].
Financial disclosures: Dr. Othman has received grant support from Abbvie and has served as a consultant for Olympus.
From the Gastroenterology and Hepatology Section, Baylor College of Medicine, Houston, TX.
Abstract
- Objective: To provide an update on management of Barrett’s esophagus.
- Methods: Review of the literature.
- Results: Management of Barrett’s esophagus depends on the degree of dysplasia. Surveillance by endoscopy every 3–5 years is recommended in patients with Barrett’s esophagus without dysplasia. Patients with Barrett’s esophagus and low-grade dysplasia should undergo surveillance by endoscopy in 3 months for confirmation of the diagnosis; if the diagnosis is confirmed then surveillance by endoscopy or eradication of Barrett’s epithelium by ablation or endoscopic resection are recommended. There is a sufficient evidence to recommend radiofrequency ablation of high-grade dysplasia within Barrett’s esophagus or to perform endoscopic mucosal resection of nodular Barrett’s esophagus with any degree of dysplasia. Early esophageal cancers that are limited to the mucosa can be treated by endoscopic resection, while cancer invading into the deep submucosa or muscularis propria may need esophagectomy with or without chemoradiation.
- Conclusion: The management of Barrett’s esophagus depends on the degree of dysplasia. Radiofrequency ablation and endoscopic mucosal resection are the most commonly used treatment for Barrett’s esophagus with dysplasia.
Keywords: Barrett’s esophagus; radiofrequency ablation; endoscopic submucosal dissection; endoscopic mucosal resection; early esophageal cancer.
Barrett’s esophagus is a common complication of chronic reflux disease [1]. Metaplastic changes that occur at the distal esophageal epithelium are usually asymptomatic [2,3] and occur as reparative adaptations to the insult of the gastric acid [4]. The management of Barrett’s esophagus after diagnosis is currently debated amongst experts without a clear consensus [5,6]. This review is generally consistent with the 2016 guidelines from the American College of Gastroenterology [1], a 2012 guideline from the American Society of Gastrointestinal Endoscopy [7], a 2011 guideline, and a 2016 expert review from the American Gastroenterological Association [8,9].
D efinition
Barrett’s esophagus is a metaplasia of the stratified squamous epithelium to a specialized columnar intestinal epithelium of mucus cells and goblet cells at the distal esophagus secondary to gastroesophageal reflux disease (GERD) [7,10]. Barrett’s esophagus is unstable tissue which can progress to esophageal adenocarcinoma. When unmanaged, the risk of cancer in dysplastic mucosa is at least thirty-fold greater than that for the general population [11–14], with recent studies suggesting a 0.4–0.7 occurrence rate per year [11,15]. With no dysplasia, the risk is low [16].
Epidemiology
The prevalence of Barrett’s esophagus is ~10% in patients with GERD [11–13,17], an estimate tied to the prevalence of GERD. However, due to the lack of symptoms of Barrett’s esophagus, no solid data supports this assumption [18–20]. A European study estimated the prevalence of Barrett’s esophagus to be 1.6% among the general population [21,22]. Barrett’s esophagus is usually diagnosed during endoscopic examinations of middle-aged and older adults, with the mean age being 55 years of age. It is most commonly found in Caucasian males and associated with the use of smoking tobacco. The male-to-female ratio is approximately 2:1 [1] and it appears to be uncommon in African Americans [23,24]. Abdominal obesity as measured by an increased waist-to-hip ratio is associated with an increased risk of Barrett’s esophagus [25,26]. Germline mutations in the MSR1, ASCC1, and CTHRC1 genes have been associated with the presence of Barrett’s esophagus and esophageal adenocarcinoma [27]. Risk factors are listed in Table 1.
Clinical Symptoms
Columnar metaplasia itself does not cause any symptoms but is merely the adaptation of the cells to the repeated effect of the acid. The main clinical symptoms of the disease would initially be symptoms associated with GERD, such as heartburn, water brash, and dysphagia [1]. Severe presentations of GERD, such as esophageal ulceration, stricture, and hemorrhage, usually occur with long-segment Barrett’s esophagus [29,30]. However, 40% of patients presenting with adenocarcinoma had no history of GERD or symptoms of heartburn [13]. Furthermore, as few as 5% of those presenting with adenocarcinoma were known to have Barrett’s esophagus [31].
D iagnosis
Barrett’s esophagus generally requires an endoscopic examination with biopsy confirmation from the distal esophagus showing specialized intestinal columnar epithelium [32]. The biopsy specimen is acquired from the cellular lining proximal to gastroesophageal junction [5,33]. Barrett’s esophagus is classified into long- and short-segment based on the length of salmon-colored mucosa in the distal esophagus. A distance longer than 3 cm is
The Prague classification was presented by an international research group in 2006 and is regarded as the standard for measuring the length of Barrett’s esophagus. The lower measurement boundary is formed by the proximal cardial notch, and the 2 upper measurement boundaries are marked by the proximal limit of the circumferential Barrett’s segment and the longest tongue of Barrett’s [37].Confirmation of the diagnosis of dysplastic
Once the initial diagnosis of Barrett’s esophagus is made, we recommend referring the patient to a Barrett’s esophagus specialized center in order to offer the patient a second opinion from a team of experts in Barrett’s esophagus. This would avoid possible false-positive results, which can be as high as 40% [1,8]. It would also offer the patient a comprehensive multidisciplinary approach and adequate long-term management. It is further preferred if an advanced intervention is offered to the patient such as endoscopic mucosal resection or endoscopic submucosal dissection. Expert endoscopists
The availability of advanced endoscopic tools improves diagnostic yield. Adopting advanced techniques can reduce errors during biopsy sampling [40–42]. Some of the newer advanced imaging endoscopic tools include chromoendoscopy, optical coherence tomography, confocal microendoscopy, autofluorescence endoscopy, narrow band imaging (NBI), and Fujinon intelligent chromoendoscopy (FICE) [43,44]. In a meta-analysis examining whether advanced techniques improved diagnostic yield, it was found that advanced imaging increased the diagnostic yield by 34%. Advanced technology mentioned here is not mandated in current guidelines [5,45].
Histopathology categories and TNM staging system for Barrett’s esophagus are shown in Table 2 and Table 3. A management algorithm based on histologic findings is presented in Figure 1.
S creening
Logically, Barrett’s esophagus screening should be offered to every patient with GERD; however, this is against current recommendations because it is cost-prohibitive [46,47]. It is a given that the rationale behind the screening is to decrease morbidity and mortality from esophageal adenocarcinoma by offering early and definitive intervention [14,48,49]. The gold standard for screening is upper endoscopy, although nonendoscopic methods are being studied [1]. For example, a capsule attached to a string can be swallowed by the patient, the capsule is then deployed and pulled through the esophagus to obtains a brush sample of the cells. It is a promising technology due to high sensitivity and specificity [50]; however, it is not regularly utilized in practice.
Approaches to screening for Barrett’s esophagus has been addressed by multiple societies with several guidelines currently available [51]. None of these approaches have been proven to be superior in clinical studies. The American Gastroenterological Association (AGA) recommends screening patients with multiple risk factors associated with esophageal adenocarcinoma for Barrett’s esophagus. Risk factors listed by the AGA include white patients, male patients, patients above the age of 50 with a history of chronic GERD, hiatal hernia, elevated body mass index, and certain body fat distribution. The AGA recommends against screening the general population with GERD [9]. The American College of Gastroenterology (ACG) recommends upper endoscopy only in the presence of alarm symptoms (eg, dysphagia, weight loss, gastrointestinal bleeding) and for screening of patients at high risk for complications [52]. The American College of Physicians recommends upper endoscopy for screening for Barrett’s esophagus in men older than 50 years with GERD symptoms for more than 5 years with any risk factors like nocturnal reflux symptoms, hiatus hernia, elevated body mass index, tobacco use, and intra-abdominal distribution of fat [1].
Overall, the sensitivity of endoscopy to diagnose Barrett’s esophagus, as seen in a Veterans Affairs (VA) cohort study for the detection of Barrett’s, is about 80%. The concluded 80% sensitivity rate was based on the performance of 2 endoscopies, 6 weeks apart for each patient, with subsequent labeling of the diagnosis if intestinal metaplasia was found in either of the 2 biopsy samples taken from the 2 procedures [53].
Promising molecular biomarkers have been associated with Barrett’s esophagus including p53 and cyclin D1 expression. However, additional studies are needed before they are incorporated as part of screening practices [5].
General Management
Medical Management of GERD
All patients with Barrett’s esophagus should be treated with a proton pump inhibitor (PPI) indefinitely based on multiple studies [54–56]. Effective control of GERD was associated with a decreased risk of dysplasia and adenocarcinoma (adjusted odds ratio 0.29, 95% CI 0.12–0.79) [57].
Effective control of GERD decreases chronic esophageal inflammation, which could progress to Barrett’s esophagus and has a risk of possible progression to adenocarcinoma. In one study, patients with proven Barrett’s esophagus showed partial regression of the intestinal metaplasia with aggressive PPI therapy [57–59]. Despite this, it is not clear whether the regression decreased risk of malignant progression [58,60,61]. In a study of 68 patients, aggressive acid reduction with omeprazole 40 mg twice a day lead to partial regression of Barrett’s esophagus when compared to mild suppression with ranitidine 150 mg twice a day. However, there was no reduction in the risk of cancer [59].
Surveillance
The reasoning behind surveillance is to detect dysplasia or adenocarcinoma in patients known to have Barrett’s esophagus early enough to provide early and efficient treatment to improve the outcome. Surveillance is done by endoscopy with biopsies in addition to sampling any irregularity [1,62,63]; however, the evidence to back up the benefit of surveillance is not clear [5,64]. It should be noted that surveillance carries risks, and morbidity associated with repeated procedures may affect patients psychologically and financially. Patients who have Barrett’s esophagus are more likely to die from other more common diseases, such as coronary heart disease, prior to developing adenocarcinoma [65]. In a recent meta-analysis, the mortality rate due to esophageal adenocarcinoma was 3.0 per 1000 person-years, whereas the mortality rate due to other causes was 37.1 per 1000 person-years [65]. An ongoing randomized, multicenter trial which assesses scheduled endoscopy every 2 years will shed more light on the overall survival after applying surveillance recommendations for each grade of dysplasia [66].
The following are ACG recommendations for surveillance of Barrett’s esophagus based on the histopathology report. The histopathology report delineates 1 of 3 types of columnar epithelium [68]: cardiac epithelium of mucus-secreting cells, atrophic gastric fundic type epithelium, or specialized columnar cells with goblet cells. The latter type is the most common with high potential for cancer [32]. Degrees of dysplasia and possible adenocarcinoma is usually described in the report as well. The degree of dysplasia if found helps the endoscopist plan the next step in management. It is worth mentioning that sampling errors can lead to missing a diagnosis. In a meta-analysis, 13% of patients diagnosed with high-grade dysplasia who underwent resection were found to have invasive cancer [69].
Surveillance for Patients with No Dysplasia
We suggest surveillance every 3 to 5 years since the rate of neoplasia is low [14]. For management of select patients with no dysplasia and with additional risk factors, radiofrequency ablation (RFA) may be an option, although it remains a controversial approach. For example, in a patient under 50 years of age, family history would be an argument for proceeding with RFA instead of prolonged surveillance. In a prospective cohort study of 139 patients with 10-year follow-up after ablation, recurrent Barrett’s occurred in less than 5% of the patients. [70]
Surveillance for Patients with Biopsy Showing “Indefinite for Dysplasia”
Aggressive treatment with PPI twice daily is recommended to avoid the misinterpretation of reactive esophageal changes secondary to reflux as dysplasia on the following endoscopy with biopsy. These patients will require a repeat endoscopy with biopsies after 3 months of aggressive treatment with PPI. Biopsies should be taken every 1 centimeter within Barrett’s epithelium [1].
If it remains indefinite, biopsies should be examined by a second pathologist with expertise in Barrett’s esophagus. If the second pathologist agrees on the indefinite diagnosis for dysplasia, then endoscopy every 12 months is recommended [1]. Treatment versus surveillance after repeat endoscopy and biopsy should be tailored to the new histopathology results on the most recent exam.
Surveillance for Patients with Biopsy Results showing Low-Grade Dysplasia (LGD), High-Grade Dysplasia (HGD), or Intramucosal Carcinoma
Surveillance recommendations are discussed under Management of Dysplasia or Intramucosal Carcinoma, below.
Efficacy of Surveillance
Asymptomatic adenocarcinoma could be discovered during surveillance, and neoplasia detected during surveillance is usually less advanced than those found after development of symptoms such as dysphagia, bleeding or weight loss [2,3,71–75]. These studies obviously had lead–time bias and did not document terminal cancer in patients adherent to surveillance protocol.
Management of Dysplasia or Intramucosal Carcinoma
Overview
Historically, dysplasia was managed with esophagectomy, which was associated with high morbidity and mortality. With advancement in the field of endoscopy, dysplasia is managed quite differently today, with endoscopic eradication therapy, which includes the use of endoscopic ablation techniques and endoscopic resection. The advantage of endoscopic resection is preservation of resected tissue for further examination, thus providing valuable information regarding the stage of the tumor (depth). Histological examination is not possible with photo or thermal ablation techniques as destroyed mucosa cannot be submitted for tissue analysis.
Low-Grade Dysplasia
If low-grade dysplasia is found, it is followed by a repeat endoscopy 8 weeks after aggressive PPI therapy. The repeat endoscopy should be performed with high definition/high-resolution endoscopy. The rationale of a second endoscopy is to ensure that the metaplastic mucosa was adequately inspected and biopsied prior to further intervention [1,9]. If the diagnosis is confirmed as low-grade dysplasia, and the patient prefers to go with the path of intervention instead of conservative management (endoscopic surveillance every 6 months for 1 year then annually with biopsies), then multiple options are available for the patient, including RFA or cryotherapy [8] [76]. In a randomized clinical trial in patients with Barrett’s esophagus and LGD, RFA was shown to reduce risk of neoplastic progression over a 3-year follow-up. The study included 136 patients randomized to receive ablation and 68 patients who underwent endoscopic surveillance. In the ablation group, the risk of progression to HGD or esophageal adenocarcinoma was reduced by 25% and the risk of progression to adenocarcinoma was reduced by 7.4%. In the ablation group, complete eradication of dysplasia and intestinal metaplasia occurred in 92.6% and 88.2% of patients respectively. In the endoscopic surveillance group, complete eradication of dysplasia and intestinal metaplasia was seen in 27.9% and 0.0% of patients respectively. [76]. Treatment-related adverse events occurred in 19.1% of patients receiving ablation (P < 0.001). The most common adverse event was stricture, occurring in 8 patients receiving ablation (11.8%), all resolved by endoscopic dilation [76,78].
Surveillance after ablation of LGD is still an ongoing debate, and further evidence is needed to establish guidelines [8]. Due to lack of evidence, we would lean towards surveillance for those patients with an annual esophagogastroduodenoscopy with biopsy examination (author’s opinion, no associated level of evidence).
High-Grade Dysplasia or Intramucosal Carcinoma
For patients with HGD or intramucosal carcinoma without submucosal invasion, eradication is the treatment of choice. Current guidelines advocate for endoscopic eradication therapy for most if not all patients with HGD or intramucosal carcinoma with a goal of removing all metaplastic and dysplastic tissue [1,5,62,64]. It should be noted that the biopsy specimen should be extensive to decrease the error margin. If the diagnosis were made on endoscopy without procuring extensive biopsies, then repeat endoscopy with extensive biopsies is needed prior to deciding the treatment path. The rationale behind extensive biopsies is to confirm the diagnosis and to determine the extent of dysplasia. Other factors potentially influencing the treatment path include the patient’s age, comorbid conditions, quality of life, and available access to an advanced endoscopist or specialized surgeon. Patient’s preferences and adherence to follow-up visits should also be a consideration.
The long-term benefits of endoscopic intervention versus surgical intervention are not well established. Esophagectomy is no longer a preferred method of treatment due to high morbidity and mortality associated with the procedure when compared to endoscopic interventions. However, it is still a preferred choice amongst a select group of patients unwilling to follow-up. A cost-effective analysis found that endoscopic ablation provided the longest quality adjusted life expectancy for Barrett’s esophagus with HGD [79,80].
Endoscopic Therapy in Barrett’s Esophagus
Endoscopic Ablative Therapies
With the advancement of endoscopic intervention, we now have multiple tools to ablate abnormal epithelium in Barrett’s esophagus. Examples of ablation techniques include thermal, photochemical, and mechanical techniques [81,82]. RFA is the treatment of choice for ablation [83]. However, non-contact ablative therapy, such as cryoablation, may be prefered if topography of the esophagus doesn’t allow contact ablation.
Radiofrequency Ablation (Figure 2). RFA is a procedure in which heat is generated from medium frequency alternating current and leads to thermal injury [84]. In Barrett’s esophagus, RFA uses radiofrequency energy delivered by a balloon that has a series of closely spaced electrodes in a
Most patients will require multiple sessions of RFA to achieve eradication. It is very rarely a one-time procedure. In a meta-analysis of 18 studies including 3082 patients, the most common adverse effects of RFA were stricture in 5% [76,98], bleeding in 1%, and pain in 3% of patients [99].
It is crucial for successful RFA to continue medical treatment for acid suppression, in order to allow healthy regeneration of the squamous cell lining. It is suggested to use PPI twice a day with sucralfate and ranitidine after the intervention [100,101]. Adhering to a liquid diet for 24 hours is needed, followed by a soft diet to allow faster regeneration of the epithelium.
The caveat with RFA is that new evidence shows a higher rate of recurrence than previously thought. In one study of 246 patients, recurrence of dysplasia occurred in 25% of patients at 48 months after eradication in 80% of the patients, and metaplasia occurred in 50% at 60 months [102]. The other risk is buried Barrett’s, a condition occurring after incomplete ablation, in which squamous cell epithelium covers patches of incompletely destroyed intestinal lining, leading to possible progression of the disease to adenocarcinoma under the surface [103].
It has been reported that patients who underwent RFA had remarkable improvement in quality of life even if RFA did not achieve eradication. Patients reported less depression, less stress and better quality of life [104].
Based on a survey of experts, follow-up at 3 months, 6 months, and then annually is recommended after ablation [1,105]. Biopsies should be taken distal to neosquamous epithelium and from suspicious areas [97,106].
Endoscopic Spray Cryotherapy. This technique involves application of liquid nitrogen or carbon dioxide gas by endoscope on the tissue to freeze it off. Although it has been shown to eliminate HGD in over 95% of the cases and all dysplasia in over 85% of the cases, it was effective in eradicating intestinal metaplasia in only 55% of patients [103,108,109]. Thus, RFA as ablation therapy is still superior to cryotherapy and is still the first-line treatment for dysplastic Barrett’s esophagus. In comparison to cryotherapy, RFA efficacy has been studied extensively with well documented outcomes. However, there is a role for cryotherapy over RFA in certain clinical situations (such as severe chest pain from RFA or lack of efficacy in eradicating intestinal metaplasia or dysplasia by RFA).
Similar to RFA, on occasions of partial ablation, the remaining metaplastic tissue may get buried beneath a layer of squamous epithelium and can possibly progress to adenocarcinoma [110].
Photodynamic Therapy (PDT). This technique works by producing cytotoxicity at the cellular level by exposure to light at a specific wavelength in the presence of a chemical agent known as photosensitize [107]. Although superior to omeprazole, PDT has a significant rate of complications, mainly stricture, and a high occurrence of esophageal cancer during follow-up. For this reason, it is less favorable compared to RFA [107] and mentioned here as a historical therapy.
Endoscopic Resection Techniques: Endoscopic Mucosal Resection (EMR) and Endoscopic Submucosal Dissection (ESD)
Unlike flat mucosa in Barrett’s esophagus, which respond to ablative techniques such as RFA or cryotherapy, nodular Barrett’s esophagus is hard to treat and requires endoscopic resection prior to ablation. Endoscopic mucosal resection (EMR) is the most widely used technique and it is available in most tertiary referral centers. Another technique named endoscopic submucosal dissection (ESD) allows the removal of large nodular areas of Barrett’s esophagus in one piece to ensure complete removal of nodular dysplasia. ESD is technically challenging and it is only available in a handful of centers in the US. Endoscopic resection techniques are the preferred interventions for nodular dysplasia due to their ability to provide valuable information for staging the lesion [111–113]. Endoscopic resection techniques are safer and more effective with similar or better results when compared with other approaches [114].
EMR is completed by the excision of esophageal mucosa down to the submucosa and submitting a large tissue specimen to the pathologist. It additionally serves as a therapeutic measure in cases of no submucosal extension. Another advantage of EMR is the ability to predict lymph node metastasis. The rationale is based on the fact that the most important predictor of lymph node metastasis is the depth of the tumor; hence, invasive tumors would likely be associated with lymph node metastasis [115,116].
In a systematic review of 11 studies, complete EMR was as equally effective in the short-term treatment of dysplastic Barrett’s esophagus when compared to RFA, but adverse event rates were greater with complete EMR (mainly strictures). Strictures are more likely to occur in patients undergoing extensive EMR. In another meta-analysis of 22 studies comparing the efficacy of EMR to RFA, both techniques were effective in eradicating dysplasia (95% in EMR group and 92% in RFA group). However, extensive EMR was associated with higher complication rates suggesting that a combined endoscopic approach of focal EMR followed by RFA is preferred over extensive EMR alone [86].
It should be noted that EMR and ESD information were derived from highly specialized center and these results may not be duplicated in community settings [113,117].
Efficacy of Endoscopic Resection. Endoscopic resection has a success rate comparable to surgical esophagectomy with fewer complications [113,114,118–121] in patients with HGD and early stages of esophageal cancer [122]. Complete remission can be as high as 89%. Recurrence occurred in 6% to 30% of patients [114,118,119], which was attributed to incomplete removal, large lesions, failure to use adjunct therapy, or lack of follow-up [123]. Even when recurrence occurred, it was successfully managed by endoscopic intervention [124].
In a large cohort study of 1000 patients with early mucosal adenocarcinoma who were treated with endoscopic resection, long-term complete remission occurred in 94% of patients. There was no mortality and less than 2% of patients had major complications. Infrequent complications include bleeding, perforations, and strictures [123,125,126]. The rate of complications is lower in highly specialized centers [127–129].
Surgery was necessary in 12 patients (3.7%) after endoscopic therapy failed [123]. Post-resection care and follow-up is similar to the post-RFA care discussed above.
Management of Invasive Esophageal Adenocarcinoma
Patients diagnosed with an invasive adenocarcinoma need to be referred to an oncologist for staging and to discuss treatment options. A select number of patients may be referred by oncology for endoscopic resection, yet the need for a multidisciplinary approach in these situations is absolutely necessary [1].
Esophagectomy
Esophagectomy offers the complete removal of the HGD along with any adenocarcinoma in the regional lymph nodes. However, mortality rates are as high as 12% immediately after the procedure [130]. The multitude of short- and long-term morbidity has significant effects on quality of life. Short-term morbidity is as high as 30%. Patients may develop serious postoperative complications such as myocardial infarction, hospital associated pneumonia, or anatomic leak [131].
Examples of long-term morbidity include dysphagia, transection of vagal nerve, and dumping syndrome. Recent development in minimally invasive surgeries for esophagectomy has not reduced postoperative morbidity rates [132].
Advocates of esophagectomy illustrate the advantage of eradication of occult lymph node metastasis. The counter argument has been established by a systemic review in which occult lymph node metastasis occurred in less than 2% of patients with HGD and intramucosal carcinoma; whereas the mortality rate after esophagectomy is substantially higher with no guarantee of curing metastatic disease [133].
Prevention of Barrett’s Esophagus
Since Barrett’s esophagus precedes most of the cases of EAC if not all [1,134], methods that aim at decreasing the incidence of Barrett’s esophagus could help in prevention. The modifiable risk factors listed by the AGA include BMI, GERD, and hiatal hernia management. Along with diet and exercise, the advent of new therapies to help patients manage their weight could in return help in avoiding a plethora of medical conditions including Barrett’s esophagus. Hiatal hernia management could lower the risk of Barrett’s by restoring normal anatomy. Lastly, proper management of GERD would lower the risk of developing Barrett’s esophagus as discussed in this article [1,9].
It is worth noting that a large trial on the efficacy and safety of aspirin for prevention of adenocarcinoma progression in Barrett’s esophagus is ongoing in the UK (AspECT trial). The AspECT trial examines the efficacy of low dose vs. high dose PPI with or without aspirin for the chemoprevention of esophageal adenocarcinoma. The theory behind the study is the inhibition of COX 2 receptors in Barrett’s cells can decrease tissue progression to cancer. This chempreventive effect of nonsteroidal anti-inflammatory drugs was shown to be augmented when combined with statin intake [56,135–138].
Conclusion
Barrett’s esophagus is usually diagnosed during routine endoscopic examination. The initial symptoms are those associated with GERD, like heartburn, dyspepsia, and regurgitation. Specialized columnar epithelium is the hallmark of histopathological diagnosis. Recommendations of the ACG and AGA suggest treatment based on biopsy results. The intervention would vary on a wide spectrum starting from acid suppression, radiofrequency ablation, endoscopic resection therapy, and rarely, esophagectomy.
Corresponding author: Mohamed O. Othman, MD, Gastroenterology and Hepatology Section, Baylor College of Medicine, 7200 Cambridge St., Suite 8C, Houston, TX 77030, [email protected].
Financial disclosures: Dr. Othman has received grant support from Abbvie and has served as a consultant for Olympus.
1. Shaheen NJ, Falk GW, Iyer PG, Gerson LB; American College of Gastroenterology. ACG Clinical Guideline: diagnosis and management of Barrett’s esophagus. Am J Gastroenterol 2016;111:30–50; quiz 51.
2. Peters JH, Clark GW, Ireland AP. Outcome of adenocarcinoma arising in Barrett’s esophagus in endoscopically surveyed and nonsurveyed patients. J Thorac Cardiovasc Surg 1994;108:813–21; discussion 821–2.
3. Streitz JM Jr, Andrews CW Jr, Ellis FH Jr. Endoscopic surveillance of Barrett’s esophagus. Does it help? J Thorac Cardiovasc Surg 1993;105:383–7; discussion 387–8.
4. Spechler SJ, Souza RF. Barrett’s esophagus. N Engl J Med 2014;371:836–45.
5. Spechler SJ, Sharma P, Souza RF, et al; American Gastroenterological Association. American Gastroenterological Association medical position statement on the management of Barrett’s esophagus. Gastroenterology 2011;140:1084–91.
6. Sharma P, McQuaid K, Dent J, et al; AGA Chicago Workshop. A critical review of the diagnosis and management of Barrett’s esophagus: the AGA Chicago Workshop. Gastroenterology 2004;127:310–30.
7. ASGE Standards of Practice Committee, Evans JA, Early DS; Standards of Practice Committee of the American Society for Gastrointestinal Endoscopy. The role of endoscopy in Barrett’s esophagus and other premalignant conditions of the esophagus. Gastrointest Endosc 2012;76(6):1087–94.
8. Wani S, Rubenstein JH, Vieth M, Bergman J. Diagnosis and management of low–grade dysplasia in Barrett’s esophagus: Expert review from the clinical practice updates Committee of the American Gastroenterological Association. Gastroenterology 2016;151:822–835.
9. American Gastroenterological, A., et al., American Gastroenterological Association medical position statement on the management of Barrett’s esophagus. Gastroenterology, 2011. 140(3): p. 1084–91. [Same as #5]
10. Van Eyken P. Definition of Barrett’s oesophagus. Acta Gastroenterol Belg 2000;63:10–2.
11. Hirota WK, Loughney TM, Lazas DJ, et al. Specialized intestinal metaplasia, dysplasia, and cancer of the esophagus and esophagogastric junction: prevalence and clinical data. Gastroenterology 1999;116(2):277–85.
12. Cameron AJ, Zinsmeister AR, Ballard DJ, Carney JA. Prevalence of columnar–lined (Barrett’s) esophagus. Comparison of population–based clinical and autopsy findings. Gastroenterology 1990;99:918–22.
13. Gerson LB, Shetler K, Triadafilopoulos G. Prevalence of Barrett’s esophagus in asymptomatic individuals. Gastroenterology 2002;123:461–7.
14. Van der Veen AH, Dees J, Blankensteijn JD, Van Blankenstein M. Adenocarcinoma in Barrett’s oesophagus: an overrated risk. Gut 1989;30:14–8.
15. Winters C Jr, Spurling TJ, Chobanian SJ, et al. Barrett’s esophagus. A prevalent, occult complication of gastroesophageal reflux disease. Gastroenterology 1987;92:118–24.
16. Wani S, Falk G, Hall M, et al. Patients with nondysplastic Barrett’s esophagus have low risks for developing dysplasia or esophageal adenocarcinoma. Clin Gastroenterol Hepatol 2011;9:220–7;quiz e26.
17. Ward EM, Wolfsen HC, Achem SR, et al. Barrett’s esophagus is common in older men and women undergoing screening colonoscopy regardless of reflux symptoms. Am J Gastroenterol 2006;101:12–7.
18. Rex DK, Cummings OW, Shaw M. Screening for Barrett’s esophagus in colonoscopy patients with and without heartburn. Gastroenterology 2003;125:1670–7.
19. Rubenstein JH, Taylor JB. Meta–analysis: the association of oesophageal adenocarcinoma with symptoms of gastro–oesophageal reflux. Aliment Pharmacol Ther 2010;32:1222–7.
20. Lagergren J, Bergström R, Lindgren A, Nyrén O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med 1999;340:825–31.
21. Ronkainen J, Aro P, Storskrubb T, et al. Prevalence of Barrett’s esophagus in the general population: an endoscopic study. Gastroenterology, 2005;129:1825–31.
22. Sampliner RE. A population prevalence of Barrett’s esophagus––finally. Gastroenterology 2005; 129:2101–3.
23. Abrams JA, Fields S, Lightdale CJ, Neugut AI. Racial and ethnic disparities in the prevalence of Barrett’s esophagus among patients who undergo upper endoscopy. Clin Gastroenterol Hepatol 2008;6:30–4.
24. Corley DA, Kubo A, Levin TR, et al. Race, ethnicity, sex and temporal differences in Barrett’s oesophagus diagnosis: a large community–based study, 1994–2006. Gut 2009;58:182–8.
25. Kamat P, Wen S, Morris J, Anandasabapathy S. Exploring the association between elevated body mass index and Barrett’s esophagus: a systematic review and meta–analysis. Ann Thorac Surg 2009;87:655–62.
26. Jacobson BC, Chan AT, Giovannucci EL, Fuchs CS. Body mass index and Barrett’s oesophagus in women. Gut 2009;58:1460–6.
27. Orloff M, Peterson C, He X, et al. Germline mutations in MSR1, ASCC1, and CTHRC1 in patients with Barrett esophagus and esophageal adenocarcinoma. JAMA 2011;306:410–9.
28. Sharma N, Ho KY. Risk Factors for Barrett’s oesophagus. Gastrointest Tumors 2016;3:103–8.
29. Spechler SJ. Barrett’s esophagus. Semin Gastrointest Dis 1996;7:51–60.
30. Ronkainen J, Talley NJ, Storskrubb T. Erosive esophagitis is a risk factor for Barrett’s esophagus: a community–based endoscopic follow–up study. Am J Gastroenterol 2011;106:1946–52.
31. Kim SL, Wo JM, Hunter JG. The prevalence of intestinal metaplasia in patients with and without peptic strictures. Am J Gastroenterol 1998;93:53–5.
32. Spechler SJ. Clinical practice. Barrett’s esophagus. N Engl J Med 2002;346:836–42.
33. Riddell RH, Odze RD. Definition of Barrett’s esophagus: time for a rethink––is intestinal metaplasia dead? Am J Gastroenterol 2009;104:2588–94.
34. Sharma P, Morales TG, Sampliner RE. Short segment Barrett’s esophagus––the need for standardization of the definition and of endoscopic criteria. Am J Gastroenterol 1998;93:1033–6.
35. Eloubeidi MA, Provenzale D. Does this patient have Barrett’s esophagus? The utility of predicting Barrett’s esophagus at the index endoscopy. Am J Gastroenterol 1999;94:937–43.
36. Rudolph RE, Vaughan TL, Storer BE, et al. Effect of segment length on risk for neoplastic progression in patients with Barrett esophagus. Ann Intern Med 2000;132:612–20.
37. Sharma P, Dent J, Armstrong D, et al. The development and validation of an endoscopic grading system for Barrett’s esophagus: the Prague C & M criteria. Gastroenterology 2006;131:1392–9.
38. Bhat S, Coleman HG, Yousef F, et al. Risk of malignant progression in Barrett’s esophagus patients: results from a large population–based study. J Natl Cancer Inst 2011;103:1049–57.
39. Shaheen NJ, Dulai GS, Ascher B, et al. Effect of a new diagnosis of Barrett’s esophagus on insurance status. Am J Gastroenterol 2005;100:577–80.
40. Canto MI, Setrakian S, Willis J, et al. Methylene blue–directed biopsies improve detection of intestinal metaplasia and dysplasia in Barrett’s esophagus. Gastrointest Endosc 2000;51:560–8.
41. Scotiniotis IA, Kochman ML, Lewis JD. Accuracy of EUS in the evaluation of Barrett’s esophagus and high–grade dysplasia or intramucosal carcinoma. Gastrointest Endosc 2001;54:689–96.
42. Kobayashi K, Izatt JA, Kulkarni MD, et al. High–resolution cross–sectional imaging of the gastrointestinal tract using optical coherence tomography: preliminary results. Gastrointest Endosc 1998;47:515–23.
43. Georgakoudi I, Jacobson BC, Van Dam J, et al. Fluorescence, reflectance, and light–scattering spectroscopy for evaluating dysplasia in patients with Barrett’s esophagus. Gastroenterology 2001. 120: 1620–9.
44. Wallace MB, Sharma P, Lightdale C, et al. Preliminary accuracy and interobserver agreement for the detection of intraepithelial neoplasia in Barrett’s esophagus with probe–based confocal laser endomicroscopy. Gastrointest Endosc 2010;72:19–24.
45. Qumseya BJ, Wang H, Badie N, et al. Advanced imaging technologies increase detection of dysplasia and neoplasia in patients with Barrett’s esophagus: a meta–analysis and systematic review. Clin Gastroenterol Hepatol 2013;11:1562–70.e1–2.–
46. DeVault KR, Castell DO. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. The Practice Parameters Committee of the American College of Gastroenterology. Am J Gastroenterol 1999;94:1434–42.
47. Inadomi JM, Sampliner R, Lagergren J. Screening and surveillance for Barrett esophagus in high–risk groups: a cost–utility analysis. Ann Intern Med 2003;138:176–86.
48. Conio M, Blanchi S, Lapertosa G, et al. Long–term endoscopic surveillance of patients with Barrett’s esophagus. Incidence of dysplasia and adenocarcinoma: a prospective study. Am J Gastroenterol 2003;98:1931–9.
49. Rastogi A, Puli S, El–Serag HB, et al. Incidence of esophageal adenocarcinoma in patients with Barrett’s esophagus and high–grade dysplasia: a meta–analysis. Gastrointest Endosc 2008;67:394–8.
50. Kadri SR, Lao–Sirieix P, O’Donovan M, et al. Acceptability and accuracy of a non–endoscopic screening test for Barrett’s oesophagus in primary care: cohort study. BMJ 2010;341:c4372.
51. Sharma, P., et al., A critical review of the diagnosis and management of Barrett’s esophagus: the AGA Chicago Workshop. Gastroenterology, 2004. 127(1): p. 310–30. [Same as #6]
52. Katz PO, Gerson LB, Vela MF. Guidelines for the diagnosis and management of gastroesophageal reflux disease. Am J Gastroenterol 2013;108:308–28; quiz 329.
53. Kim SL, Waring JP, Spechler SJ, et al. Diagnostic inconsistencies in Barrett’s esophagus. Department of Veterans Affairs Gastroesophageal Reflux Study Group. Gastroenterology 1994;107:945–9.
54. Kastelein F, Spaander MC, Steyerberg EW, et al; ProBar Study Group. Proton pump inhibitors reduce the risk of neoplastic progression in patients with Barrett’s esophagus. Clin Gastroenterol Hepatol 2013;11:382–8.
55. El–Serag HB, Aguirre TV, Davis S, et al. Proton pump inhibitors are associated with reduced incidence of dysplasia in Barrett’s esophagus. Am J Gastroenterol 2004;99:1877–83.
56. Nguyen DM, El–Serag HB, Henderson L, et al. Medication usage and the risk of neoplasia in patients with Barrett’s esophagus. Clin Gastroenterol Hepatol 2009;7:1299–304.
57. Singh S, Garg SK, Singh PP, et al. Acid–suppressive medications and risk of oesophageal adenocarcinoma in patients with Barrett’s oesophagus: a systematic review and meta–analysis. Gut 2014;63:1229–37.
58. Spechler SJ, Lee E, Ahnen D, et al. Long–term outcome of medical and surgical therapies for gastroesophageal reflux disease: follow–up of a randomized controlled trial. JAMA 2001;285:2331–8.
59. Peters FT, Ganesh S, Kuipers EJ, et al. Endoscopic regression of Barrett’s oesophagus during omeprazole treatment; a randomised double blind study. Gut 1999;45:489–94.
60. Ouatu–Lascar R, Triadafilopoulos G. –Complete elimination of reflux symptoms does not guarantee normalization of intraesophageal acid reflux in patients with Barrett’s esophagus. Am J Gastroenterol 1998;93:711–6.
61. Maret–Ouda J, Konings P, Lagergren J, Brusselaers N. Antireflux surgery and risk of esophageal adenocarcinoma: A systematic review and meta–analysis. Ann Surg 2016;263:251–7.
62. Evans, J.A., et al., The role of endoscopy in Barrett’s esophagus and other premalignant conditions of the esophagus. Gastrointest Endosc, 2012. 76(6): p. 1087–94. [Same as #7]
63. ASGE Standards of Practice Committee, Evans JA, Early DS,et al; American Society for Gastrointestinal Endoscopy. The role of endoscopy in the assessment and treatment of esophageal cancer. Gastrointest Endosc 2013;77:328–34.
64. Bennett C, Moayyedi P, Corley DA, et al; BOB CAT Consortium. BOB CAT: A large–scale review and delphi consensus for ,anagement of Barrett’s esophagus with no dysplasia, indefinite for, or low–grade dysplasia. Am J Gastroenterol 2015;110:662–82; quiz 683.
65. Sikkema M, de Jonge PJ, Steyerberg EW, Kuipers EJ. Risk of esophageal adenocarcinoma and mortality in patients with Barrett’s esophagus: a systematic review and meta–analysis. Clin Gastroenterol Hepatol 2010;8:235–44; quiz e32.
66. Old O, Moayyedi P, Love S, et al; BOSS Trial Team. Barrett’s Oesophagus Surveillance versus endoscopy at need Study (BOSS): protocol and analysis plan for a multicentre randomized controlled trial. J Med Screen 2015;22:158–64.
67. Weston AP, Sharma P, Topalovski M, et al. Long–term follow–up of Barrett’s high–grade dysplasia. Am J Gastroenterol 2000;95:1888–93.
68. Paull A, Trier JS, Dalton MD, et al. The histologic spectrum of Barrett’s esophagus. N Engl J Med 1976;295:476–80.
69. Konda VJ, Ross AS, Ferguson MK, et al. Is the risk of concomitant invasive esophageal cancer in high–grade dysplasia in Barrett’s esophagus overestimated? Clin Gastroenterol Hepatol 2008;6:159–64.
70. Allison H, Banchs MA, Bonis PA, Guelrud M. Long–term remission of nondysplastic Barrett’s esophagus after multipolar electrocoagulation ablation: report of 139 patients with 10 years of follow–up. Gastrointest Endosc 2011;73:651–8.
71. Corley DA, Levin TR, Habel LA, Weiss NS, et al. Surveillance and survival in Barrett’s adenocarcinomas: a population–based study. Gastroenterology 2002;122:633–40.
72. Wong T, Tian J, Nagar AB. Barrett’s surveillance identifies patients with early esophageal adenocarcinoma. Am J Med 2010;123:462–7.
73. Fountoulakis A, Zafirellis KD, Dolan K, et al. Effect of surveillance of Barrett’s oesophagus on the clinical outcome of oesophageal cancer. Br J Surg 2004;91:997–1003.
74. Verbeek RE, Leenders M, Ten Kate FJ, et al. Surveillance of Barrett’s esophagus and mortality from esophageal adenocarcinoma: a population–based cohort study. Am J Gastroenterol 2014;109:1215–22.
75. Kastelein F, van Olphen SH, Steyerberg EW, et al. Impact of surveillance for Barrett’s oesophagus on tumour stage and survival of patients with neoplastic progression. Gut 2016;65:548–54.
76. Phoa KN, van Vilsteren FG, Weusten BL, et al. Radiofrequency ablation vs endoscopic surveillance for patients with Barrett esophagus and low–grade dysplasia: a randomized clinical trial. JAMA 2014;311: 1209–17.
77. Wang WL, Chang IW, Chen CC, et al. Radiofrequency ablation versus endoscopic submucosal dissection in treating large early esophageal squamous cell neoplasia. Medicine (Baltimore) 2015;94:e2240.
78. Lim CH, Treanor D, Dixon MF, Axon AT. Low–grade dysplasia in Barrett’s esophagus has a high risk of progression. Endoscopy 2007;39:581–7.
79. Shaheen NJ, Inadomi JM, Overholt BF, Sharma P. What is the best management strategy for high grade dysplasia in Barrett’s oesophagus? A cost effectiveness analysis. Gut 2004;53:1736–44.
80. Vij R, Triadafilopoulos G, Owens DK, et al. Cost–effectiveness of photodynamic therapy for high–grade dysplasia in Barrett’s esophagus. Gastrointest Endosc 2004;60:739–56.
81. van den Boogert J, v an Hillegersberg R, Siersema PD, et al. Endoscopic ablation therapy for Barrett’s esophagus with high–grade dysplasia: a review. Am J Gastroenterol 1999;94:1153–60.
82. Sampliner RE. Endoscopic ablative therapy for Barrett’s esophagus: current status. Gastrointest Endosc 2004;59:66–9.
83. Sharma VK, Wang KK, Overholt BF, et al. Balloon–based, circumferential, endoscopic radiofrequency ablation of Barrett’s esophagus: 1–year follow–up of 100 patients. Gastrointest Endosc 2007;65:185–95.
84. Hanlon CR. Textbook of surgery: The biological basis of modern surgical practice, 14th edition. Ann Surg 1992;216:94.
85. Bright T, Watson DI, Tam W, et al. Randomized trial of argon plasma coagulation versus endoscopic surveillance for barrett esophagus after antireflux surgery: late results. Ann Surg 2007;246:1016–20.
86. Chadwick G, Groene O, Markar SR, et al. Systematic review comparing radiofrequency ablation and complete endoscopic resection in treating dysplastic Barrett’s esophagus: a critical assessment of histologic outcomes and adverse events. Gastrointest Endosc 2014;79:718–31.e3.
87. Gondrie JJ, Pouw RE, Sondermeijer CM, et al. Effective treatment of early Barrett’s neoplasia with stepwise circumferential and focal ablation using the HALO system. Endoscopy 2008;40:370–9.
88. Pouw RE, Wirths K, Eisendrath P, et al. Efficacy of radiofrequency ablation combined with endoscopic resection for barrett’s esophagus with early neoplasia. Clin Gastroenterol Hepatol 2010;8:23–9.
89. Kim HP, Bulsiewicz WJ, Cotton CC, et al. Focal endoscopic mucosal resection before radiofrequency ablation is equally effective and safe compared with radiofrequency ablation alone for the eradication of Barrett’s esophagus with advanced neoplasia. Gastrointest Endosc 2012;76:733–9.
90. Fleischer DE, Overholt BF, Sharma VK, et al. Endoscopic ablation of Barrett’s esophagus: a multicenter study with 2.5–year follow–up. Gastrointest Endosc 2008;68:867–76.
91. Sharma VK, Jae Kim H, Das A, et al. Circumferential and focal ablation of Barrett’s esophagus containing dysplasia. Am J Gastroenterol 2009;104:310–7.
92. Ganz RA, Overholt BF, Sharma VK, et al. Circumferential ablation of Barrett’s esophagus that contains high–grade dysplasia: a U.S. multicenter registry. Gastrointest Endosc 2008;68:35–40.
93. Gupta M, Iyer PG, Lutzke L, et al. Recurrence of esophageal intestinal metaplasia after endoscopic mucosal resection and radiofrequency ablation of Barrett’s esophagus: results from a US Multicenter Consortium. Gastroenterology 2013;145:79–86.e1.
94. Lyday WD, Corbett FS, Kuperman DA, et al. Radiofrequency ablation of Barrett’s esophagus: outcomes of 429 patients from a multicenter community practice registry. Endoscopy 2010;42:272–8.
95. Pasricha S, Bulsiewicz WJ, Hathorn KE, et al. Durability and predictors of successful radiofrequency ablation for Barrett’s esophagus. Clin Gastroenterol Hepatol 2014;12:1840–7.e1.
96. Fleischer DE, Overholt BF, Sharma VK, et al. Endoscopic radiofrequency ablation for Barrett’s esophagus: 5–year outcomes from a prospective multicenter trial. Endoscopy 2010;42:781–9.
97. Weston AP, Sharma P, Banerjee S, et al. Visible endoscopic and histologic changes in the cardia, before and after complete Barrett’s esophagus ablation. Gastrointest Endosc 2005;61:515–21.
98. Beaumont H, Gondrie JJ, McMahon BP, et al. Stepwise radiofrequency ablation of Barrett’s esophagus preserves esophageal inner diameter, compliance, and motility. Endoscopy 2009;41:2–8.
99. Orman ES, Li N, Shaheen NJ.Efficacy and durability of radiofrequency ablation for Barrett’s Esophagus: systematic review and meta–analysis. Clin Gastroenterol Hepatol 2013;11:1245–55.
100. Krishnan K, Pandolfino JE, Kahrilas PJ, et al. Increased risk for persistent intestinal metaplasia in patients with Barrett’s esophagus and uncontrolled reflux exposure before radiofrequency ablation. Gastroenterology 2012;143:576–81.
101. Akiyama J, Marcus SN, Triadafilopoulos G. Effective intra–esophageal acid control is associated with improved radiofrequency ablation outcomes in Barrett’s esophagus. Dig Dis Sci 2012;57:2625–32.
102. Small AJ, Sutherland SE, Hightower JS, et al. Comparative risk of recurrence of dysplasia and carcinoma after endoluminal eradication therapy of high–grade dysplasia versus intramucosal carcinoma in Barrett’s esophagus. Gastrointest Endosc 2015;81:1158–66.e1–4.
103. Shaheen NJ, Greenwald BD, Peery AF, et al. Safety and efficacy of endoscopic spray cryotherapy for Barrett’s esophagus with high–grade dysplasia. Gastrointest Endosc 2010;71:680–5.
104. Shaheen NJ, Peery AF, Hawes RH, et al. Quality of life following radiofrequency ablation of dysplastic Barrett’s esophagus. Endoscopy 2010;42:790–9.
105. Bedi AO, Kwon RS, Rubenstein JH, et al. A survey of expert follow–up practices after successful endoscopic eradication therapy for Barrett’s esophagus with high–grade dysplasia and intramucosal adenocarcinoma. Gastrointest Endosc 2013;78:696–701.
106. Sampliner RE, Camargo E, Prasad AR. Prasad, Association of ablation of Barrett’s esophagus with high grade dysplasia and adenocarcinoma of the gastric cardia. Dis Esophagus 2006;19:277–9.
107. Overholt BF, Panjehpour M, Halberg DL. Photodynamic therapy for Barrett’s esophagus with dysplasia and/or early stage carcinoma: long–term results. Gastrointest Endosc 2003;58:183–8.
108. Gosain S, Mercer K, Twaddell WS, et al. Liquid nitrogen spray cryotherapy in Barrett’s esophagus with high–grade dysplasia: long–term results. Gastrointest Endosc 2013;78:260–5.
109. Canto MI, Shin EJ, Khashab MA, et al. Safety and efficacy of carbon dioxide cryotherapy for treatment of neoplastic Barrett’s esophagus. Endoscopy 2015;47:582–91.
110. Van Laethem JL, Peny MO, Salmon I, et al. Intramucosal adenocarcinoma arising under squamous re–epithelialisation of Barrett’s oesophagus. Gut 2000;46:574–7.
111. Pech O, May A, Gossner L, et al. Management of pre–malignant and malignant lesions by endoscopic resection. Best Pract Res Clin Gastroenterol 2004;18:61–76.
112. Soetikno RM, Gotoda T, Nakanishi Y, Soehendra N. Endoscopic mucosal resection. Gastrointest Endosc 2003;57:567–79.
113. Ell C, May A, Gossner L, et al. Endoscopic mucosal resection of early cancer and high–grade dysplasia in Barrett’s esophagus. Gastroenterology 2000;118:670–7.
114. Pech O, Behrens A, May A, et al. Long–term results and risk factor analysis for recurrence after curative endoscopic therapy in 349 patients with high–grade intraepithelial neoplasia and mucosal adenocarcinoma in Barrett’s oesophagus. Gut 2008;57:1200–6.
115. Vieth M, Ell C, Gossner L, et al. Histological analysis of endoscopic resection specimens from 326 patients with Barrett’s esophagus and early neoplasia. Endoscopy 2004;36:776–81.
116. Buskens CJ, Westerterp M, Lagarde SM, et al. Prediction of appropriateness of local endoscopic treatment for high–grade dysplasia and early adenocarcinoma by EUS and histopathologic features. Gastrointest Endosc 2004;60:703–10.
117. Nijhawan PK, Wang KK. Endoscopic mucosal resection for lesions with endoscopic features suggestive of malignancy and high–grade dysplasia within Barrett’s esophagus. Gastrointest Endosc 2000;52:328–32.
118. Esaki M, Matsumoto T, Hirakawa K, et al. Risk factors for local recurrence of superficial esophageal cancer after treatment by endoscopic mucosal resection. Endoscopy 2007;39:41–5.
119. Ell C, May A, Pech O, et al. Curative endoscopic resection of early esophageal adenocarcinomas (Barrett’s cancer). Gastrointest Endosc 2007;65:3–10.
120. Chennat J, Konda VJ, Ross AS, et al. Complete Barrett’s eradication endoscopic mucosal resection: an effective treatment modality for high–grade dysplasia and intramucosal carcinoma––an American single–center experience. Am J Gastroenterol 2009;104:2684–92.
121. Pech O, Bollschweiler E, Manner H, et al. Comparison between endoscopic and surgical resection of mucosal esophageal adenocarcinoma in Barrett’s esophagus at two high–volume centers. Ann Surg 2011;254:67–72.
122. Wu J, Pan YM, Wang TT, et al. Endotherapy versus surgery for early neoplasia in Barrett’s esophagus: a meta–analysis. Gastrointest Endosc 2014;79:233–241.e2.
123. Pech O, May A, Manner H, et al. Long–term efficacy and safety of endoscopic resection for patients with mucosal adenocarcinoma of the esophagus. Gastroenterology 2014;146:652–660.e1.
124. Prasad GA, Wu TT, Wigle DA, et al. Endoscopic and surgical treatment of mucosal (T1a) esophageal adenocarcinoma in Barrett’s esophagus. Gastroenterology 2009;137:815–23.
125. May A, Gossner L, Pech O, et al. Local endoscopic therapy for intraepithelial high–grade neoplasia and early adenocarcinoma in Barrett’s oesophagus: acute–phase and intermediate results of a new treatment approach. Eur J Gastroenterol Hepatol 2002;14(10):1085–91.
126. Gerke H, Siddiqui J, Nasr I, et al. Efficacy and safety of EMR to completely remove Barrett’s esophagus: experience in 41 patients. Gastrointest Endosc 2011;74:761–71.
127. Lewis JJ, Rubenstein JH, Singal AG, et al. Factors associated with esophageal stricture formation after endoscopic mucosal resection for neoplastic Barrett’s esophagus. Gastrointest Endosc 2011;74:753–60.
128. Choi IJ, Kim CG, Chang HJ, et al. The learning curve for EMR with circumferential mucosal incision in treating intramucosal gastric neoplasm. Gastrointest Endosc 2005;62:860–5.
129. Deprez PH, Bergman JJ, Meisner S, et al. Current practice with endoscopic submucosal dissection in Europe: position statement from a panel of experts. Endoscopy 2010;42:853–8.
130. van Lanschot JJ, Hulscher JB, Buskens CJ, et al. Hospital volume and hospital mortality for esophagectomy. Cancer 2001;91:1574–8.
131. Karl RC, Schreiber R, Boulware D, et al. Factors affecting morbidity, mortality, and survival in patients undergoing Ivor Lewis esophagogastrectomy. Ann Surg 2000;231:635–43.
132. Young MM, Deschamps C, Trastek VF, et al. Esophageal reconstruction for benign disease: early morbidity, mortality, and functional results. Ann Thorac Surg 2000;70:1651–5.
133. Dunbar KB, Spechler SJ. The risk of lymph–node metastases in patients with high–grade dysplasia or intramucosal carcinoma in Barrett’s esophagus: a systematic review. Am J Gastroenterol 2012;107:850–62; quiz 863.
134. Morales CP, Souza RF, Spechler SJ. Hallmarks of cancer progression in Barrett’s oesophagus. Lancet 2002;360:1587–9.
135. Omer ZB, Ananthakrishnan AN, Nattinger KJ, et al. Aspirin protects against Barrett’s esophagus in a multivariate logistic regression analysis. Clin Gastroenterol Hepatol 2012;10:722–7.
136. Abnet CC, Freedman ND, Kamangar F, et al. Non–steroidal anti–inflammatory drugs and risk of gastric and oesophageal adenocarcinomas: results from a cohort study and a meta–analysis. Br J Cancer 2009;100:551–7.
137. Kastelein F, Spaander MC, Biermann K, et al. Nonsteroidal anti–inflammatory drugs and statins have chemopreventative effects in patients with Barrett’s esophagus. Gastroenterology 2011;141:2000–8; quiz e13–4.
138. Zhang S, Zhang XQ, Ding XW, et al. Cyclooxygenase inhibitors use is associated with reduced risk of esophageal adenocarcinoma in patients with Barrett’s esophagus: a meta–analysis. Br J Cancer 2014;110: 2378–88.
1. Shaheen NJ, Falk GW, Iyer PG, Gerson LB; American College of Gastroenterology. ACG Clinical Guideline: diagnosis and management of Barrett’s esophagus. Am J Gastroenterol 2016;111:30–50; quiz 51.
2. Peters JH, Clark GW, Ireland AP. Outcome of adenocarcinoma arising in Barrett’s esophagus in endoscopically surveyed and nonsurveyed patients. J Thorac Cardiovasc Surg 1994;108:813–21; discussion 821–2.
3. Streitz JM Jr, Andrews CW Jr, Ellis FH Jr. Endoscopic surveillance of Barrett’s esophagus. Does it help? J Thorac Cardiovasc Surg 1993;105:383–7; discussion 387–8.
4. Spechler SJ, Souza RF. Barrett’s esophagus. N Engl J Med 2014;371:836–45.
5. Spechler SJ, Sharma P, Souza RF, et al; American Gastroenterological Association. American Gastroenterological Association medical position statement on the management of Barrett’s esophagus. Gastroenterology 2011;140:1084–91.
6. Sharma P, McQuaid K, Dent J, et al; AGA Chicago Workshop. A critical review of the diagnosis and management of Barrett’s esophagus: the AGA Chicago Workshop. Gastroenterology 2004;127:310–30.
7. ASGE Standards of Practice Committee, Evans JA, Early DS; Standards of Practice Committee of the American Society for Gastrointestinal Endoscopy. The role of endoscopy in Barrett’s esophagus and other premalignant conditions of the esophagus. Gastrointest Endosc 2012;76(6):1087–94.
8. Wani S, Rubenstein JH, Vieth M, Bergman J. Diagnosis and management of low–grade dysplasia in Barrett’s esophagus: Expert review from the clinical practice updates Committee of the American Gastroenterological Association. Gastroenterology 2016;151:822–835.
9. American Gastroenterological, A., et al., American Gastroenterological Association medical position statement on the management of Barrett’s esophagus. Gastroenterology, 2011. 140(3): p. 1084–91. [Same as #5]
10. Van Eyken P. Definition of Barrett’s oesophagus. Acta Gastroenterol Belg 2000;63:10–2.
11. Hirota WK, Loughney TM, Lazas DJ, et al. Specialized intestinal metaplasia, dysplasia, and cancer of the esophagus and esophagogastric junction: prevalence and clinical data. Gastroenterology 1999;116(2):277–85.
12. Cameron AJ, Zinsmeister AR, Ballard DJ, Carney JA. Prevalence of columnar–lined (Barrett’s) esophagus. Comparison of population–based clinical and autopsy findings. Gastroenterology 1990;99:918–22.
13. Gerson LB, Shetler K, Triadafilopoulos G. Prevalence of Barrett’s esophagus in asymptomatic individuals. Gastroenterology 2002;123:461–7.
14. Van der Veen AH, Dees J, Blankensteijn JD, Van Blankenstein M. Adenocarcinoma in Barrett’s oesophagus: an overrated risk. Gut 1989;30:14–8.
15. Winters C Jr, Spurling TJ, Chobanian SJ, et al. Barrett’s esophagus. A prevalent, occult complication of gastroesophageal reflux disease. Gastroenterology 1987;92:118–24.
16. Wani S, Falk G, Hall M, et al. Patients with nondysplastic Barrett’s esophagus have low risks for developing dysplasia or esophageal adenocarcinoma. Clin Gastroenterol Hepatol 2011;9:220–7;quiz e26.
17. Ward EM, Wolfsen HC, Achem SR, et al. Barrett’s esophagus is common in older men and women undergoing screening colonoscopy regardless of reflux symptoms. Am J Gastroenterol 2006;101:12–7.
18. Rex DK, Cummings OW, Shaw M. Screening for Barrett’s esophagus in colonoscopy patients with and without heartburn. Gastroenterology 2003;125:1670–7.
19. Rubenstein JH, Taylor JB. Meta–analysis: the association of oesophageal adenocarcinoma with symptoms of gastro–oesophageal reflux. Aliment Pharmacol Ther 2010;32:1222–7.
20. Lagergren J, Bergström R, Lindgren A, Nyrén O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med 1999;340:825–31.
21. Ronkainen J, Aro P, Storskrubb T, et al. Prevalence of Barrett’s esophagus in the general population: an endoscopic study. Gastroenterology, 2005;129:1825–31.
22. Sampliner RE. A population prevalence of Barrett’s esophagus––finally. Gastroenterology 2005; 129:2101–3.
23. Abrams JA, Fields S, Lightdale CJ, Neugut AI. Racial and ethnic disparities in the prevalence of Barrett’s esophagus among patients who undergo upper endoscopy. Clin Gastroenterol Hepatol 2008;6:30–4.
24. Corley DA, Kubo A, Levin TR, et al. Race, ethnicity, sex and temporal differences in Barrett’s oesophagus diagnosis: a large community–based study, 1994–2006. Gut 2009;58:182–8.
25. Kamat P, Wen S, Morris J, Anandasabapathy S. Exploring the association between elevated body mass index and Barrett’s esophagus: a systematic review and meta–analysis. Ann Thorac Surg 2009;87:655–62.
26. Jacobson BC, Chan AT, Giovannucci EL, Fuchs CS. Body mass index and Barrett’s oesophagus in women. Gut 2009;58:1460–6.
27. Orloff M, Peterson C, He X, et al. Germline mutations in MSR1, ASCC1, and CTHRC1 in patients with Barrett esophagus and esophageal adenocarcinoma. JAMA 2011;306:410–9.
28. Sharma N, Ho KY. Risk Factors for Barrett’s oesophagus. Gastrointest Tumors 2016;3:103–8.
29. Spechler SJ. Barrett’s esophagus. Semin Gastrointest Dis 1996;7:51–60.
30. Ronkainen J, Talley NJ, Storskrubb T. Erosive esophagitis is a risk factor for Barrett’s esophagus: a community–based endoscopic follow–up study. Am J Gastroenterol 2011;106:1946–52.
31. Kim SL, Wo JM, Hunter JG. The prevalence of intestinal metaplasia in patients with and without peptic strictures. Am J Gastroenterol 1998;93:53–5.
32. Spechler SJ. Clinical practice. Barrett’s esophagus. N Engl J Med 2002;346:836–42.
33. Riddell RH, Odze RD. Definition of Barrett’s esophagus: time for a rethink––is intestinal metaplasia dead? Am J Gastroenterol 2009;104:2588–94.
34. Sharma P, Morales TG, Sampliner RE. Short segment Barrett’s esophagus––the need for standardization of the definition and of endoscopic criteria. Am J Gastroenterol 1998;93:1033–6.
35. Eloubeidi MA, Provenzale D. Does this patient have Barrett’s esophagus? The utility of predicting Barrett’s esophagus at the index endoscopy. Am J Gastroenterol 1999;94:937–43.
36. Rudolph RE, Vaughan TL, Storer BE, et al. Effect of segment length on risk for neoplastic progression in patients with Barrett esophagus. Ann Intern Med 2000;132:612–20.
37. Sharma P, Dent J, Armstrong D, et al. The development and validation of an endoscopic grading system for Barrett’s esophagus: the Prague C & M criteria. Gastroenterology 2006;131:1392–9.
38. Bhat S, Coleman HG, Yousef F, et al. Risk of malignant progression in Barrett’s esophagus patients: results from a large population–based study. J Natl Cancer Inst 2011;103:1049–57.
39. Shaheen NJ, Dulai GS, Ascher B, et al. Effect of a new diagnosis of Barrett’s esophagus on insurance status. Am J Gastroenterol 2005;100:577–80.
40. Canto MI, Setrakian S, Willis J, et al. Methylene blue–directed biopsies improve detection of intestinal metaplasia and dysplasia in Barrett’s esophagus. Gastrointest Endosc 2000;51:560–8.
41. Scotiniotis IA, Kochman ML, Lewis JD. Accuracy of EUS in the evaluation of Barrett’s esophagus and high–grade dysplasia or intramucosal carcinoma. Gastrointest Endosc 2001;54:689–96.
42. Kobayashi K, Izatt JA, Kulkarni MD, et al. High–resolution cross–sectional imaging of the gastrointestinal tract using optical coherence tomography: preliminary results. Gastrointest Endosc 1998;47:515–23.
43. Georgakoudi I, Jacobson BC, Van Dam J, et al. Fluorescence, reflectance, and light–scattering spectroscopy for evaluating dysplasia in patients with Barrett’s esophagus. Gastroenterology 2001. 120: 1620–9.
44. Wallace MB, Sharma P, Lightdale C, et al. Preliminary accuracy and interobserver agreement for the detection of intraepithelial neoplasia in Barrett’s esophagus with probe–based confocal laser endomicroscopy. Gastrointest Endosc 2010;72:19–24.
45. Qumseya BJ, Wang H, Badie N, et al. Advanced imaging technologies increase detection of dysplasia and neoplasia in patients with Barrett’s esophagus: a meta–analysis and systematic review. Clin Gastroenterol Hepatol 2013;11:1562–70.e1–2.–
46. DeVault KR, Castell DO. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. The Practice Parameters Committee of the American College of Gastroenterology. Am J Gastroenterol 1999;94:1434–42.
47. Inadomi JM, Sampliner R, Lagergren J. Screening and surveillance for Barrett esophagus in high–risk groups: a cost–utility analysis. Ann Intern Med 2003;138:176–86.
48. Conio M, Blanchi S, Lapertosa G, et al. Long–term endoscopic surveillance of patients with Barrett’s esophagus. Incidence of dysplasia and adenocarcinoma: a prospective study. Am J Gastroenterol 2003;98:1931–9.
49. Rastogi A, Puli S, El–Serag HB, et al. Incidence of esophageal adenocarcinoma in patients with Barrett’s esophagus and high–grade dysplasia: a meta–analysis. Gastrointest Endosc 2008;67:394–8.
50. Kadri SR, Lao–Sirieix P, O’Donovan M, et al. Acceptability and accuracy of a non–endoscopic screening test for Barrett’s oesophagus in primary care: cohort study. BMJ 2010;341:c4372.
51. Sharma, P., et al., A critical review of the diagnosis and management of Barrett’s esophagus: the AGA Chicago Workshop. Gastroenterology, 2004. 127(1): p. 310–30. [Same as #6]
52. Katz PO, Gerson LB, Vela MF. Guidelines for the diagnosis and management of gastroesophageal reflux disease. Am J Gastroenterol 2013;108:308–28; quiz 329.
53. Kim SL, Waring JP, Spechler SJ, et al. Diagnostic inconsistencies in Barrett’s esophagus. Department of Veterans Affairs Gastroesophageal Reflux Study Group. Gastroenterology 1994;107:945–9.
54. Kastelein F, Spaander MC, Steyerberg EW, et al; ProBar Study Group. Proton pump inhibitors reduce the risk of neoplastic progression in patients with Barrett’s esophagus. Clin Gastroenterol Hepatol 2013;11:382–8.
55. El–Serag HB, Aguirre TV, Davis S, et al. Proton pump inhibitors are associated with reduced incidence of dysplasia in Barrett’s esophagus. Am J Gastroenterol 2004;99:1877–83.
56. Nguyen DM, El–Serag HB, Henderson L, et al. Medication usage and the risk of neoplasia in patients with Barrett’s esophagus. Clin Gastroenterol Hepatol 2009;7:1299–304.
57. Singh S, Garg SK, Singh PP, et al. Acid–suppressive medications and risk of oesophageal adenocarcinoma in patients with Barrett’s oesophagus: a systematic review and meta–analysis. Gut 2014;63:1229–37.
58. Spechler SJ, Lee E, Ahnen D, et al. Long–term outcome of medical and surgical therapies for gastroesophageal reflux disease: follow–up of a randomized controlled trial. JAMA 2001;285:2331–8.
59. Peters FT, Ganesh S, Kuipers EJ, et al. Endoscopic regression of Barrett’s oesophagus during omeprazole treatment; a randomised double blind study. Gut 1999;45:489–94.
60. Ouatu–Lascar R, Triadafilopoulos G. –Complete elimination of reflux symptoms does not guarantee normalization of intraesophageal acid reflux in patients with Barrett’s esophagus. Am J Gastroenterol 1998;93:711–6.
61. Maret–Ouda J, Konings P, Lagergren J, Brusselaers N. Antireflux surgery and risk of esophageal adenocarcinoma: A systematic review and meta–analysis. Ann Surg 2016;263:251–7.
62. Evans, J.A., et al., The role of endoscopy in Barrett’s esophagus and other premalignant conditions of the esophagus. Gastrointest Endosc, 2012. 76(6): p. 1087–94. [Same as #7]
63. ASGE Standards of Practice Committee, Evans JA, Early DS,et al; American Society for Gastrointestinal Endoscopy. The role of endoscopy in the assessment and treatment of esophageal cancer. Gastrointest Endosc 2013;77:328–34.
64. Bennett C, Moayyedi P, Corley DA, et al; BOB CAT Consortium. BOB CAT: A large–scale review and delphi consensus for ,anagement of Barrett’s esophagus with no dysplasia, indefinite for, or low–grade dysplasia. Am J Gastroenterol 2015;110:662–82; quiz 683.
65. Sikkema M, de Jonge PJ, Steyerberg EW, Kuipers EJ. Risk of esophageal adenocarcinoma and mortality in patients with Barrett’s esophagus: a systematic review and meta–analysis. Clin Gastroenterol Hepatol 2010;8:235–44; quiz e32.
66. Old O, Moayyedi P, Love S, et al; BOSS Trial Team. Barrett’s Oesophagus Surveillance versus endoscopy at need Study (BOSS): protocol and analysis plan for a multicentre randomized controlled trial. J Med Screen 2015;22:158–64.
67. Weston AP, Sharma P, Topalovski M, et al. Long–term follow–up of Barrett’s high–grade dysplasia. Am J Gastroenterol 2000;95:1888–93.
68. Paull A, Trier JS, Dalton MD, et al. The histologic spectrum of Barrett’s esophagus. N Engl J Med 1976;295:476–80.
69. Konda VJ, Ross AS, Ferguson MK, et al. Is the risk of concomitant invasive esophageal cancer in high–grade dysplasia in Barrett’s esophagus overestimated? Clin Gastroenterol Hepatol 2008;6:159–64.
70. Allison H, Banchs MA, Bonis PA, Guelrud M. Long–term remission of nondysplastic Barrett’s esophagus after multipolar electrocoagulation ablation: report of 139 patients with 10 years of follow–up. Gastrointest Endosc 2011;73:651–8.
71. Corley DA, Levin TR, Habel LA, Weiss NS, et al. Surveillance and survival in Barrett’s adenocarcinomas: a population–based study. Gastroenterology 2002;122:633–40.
72. Wong T, Tian J, Nagar AB. Barrett’s surveillance identifies patients with early esophageal adenocarcinoma. Am J Med 2010;123:462–7.
73. Fountoulakis A, Zafirellis KD, Dolan K, et al. Effect of surveillance of Barrett’s oesophagus on the clinical outcome of oesophageal cancer. Br J Surg 2004;91:997–1003.
74. Verbeek RE, Leenders M, Ten Kate FJ, et al. Surveillance of Barrett’s esophagus and mortality from esophageal adenocarcinoma: a population–based cohort study. Am J Gastroenterol 2014;109:1215–22.
75. Kastelein F, van Olphen SH, Steyerberg EW, et al. Impact of surveillance for Barrett’s oesophagus on tumour stage and survival of patients with neoplastic progression. Gut 2016;65:548–54.
76. Phoa KN, van Vilsteren FG, Weusten BL, et al. Radiofrequency ablation vs endoscopic surveillance for patients with Barrett esophagus and low–grade dysplasia: a randomized clinical trial. JAMA 2014;311: 1209–17.
77. Wang WL, Chang IW, Chen CC, et al. Radiofrequency ablation versus endoscopic submucosal dissection in treating large early esophageal squamous cell neoplasia. Medicine (Baltimore) 2015;94:e2240.
78. Lim CH, Treanor D, Dixon MF, Axon AT. Low–grade dysplasia in Barrett’s esophagus has a high risk of progression. Endoscopy 2007;39:581–7.
79. Shaheen NJ, Inadomi JM, Overholt BF, Sharma P. What is the best management strategy for high grade dysplasia in Barrett’s oesophagus? A cost effectiveness analysis. Gut 2004;53:1736–44.
80. Vij R, Triadafilopoulos G, Owens DK, et al. Cost–effectiveness of photodynamic therapy for high–grade dysplasia in Barrett’s esophagus. Gastrointest Endosc 2004;60:739–56.
81. van den Boogert J, v an Hillegersberg R, Siersema PD, et al. Endoscopic ablation therapy for Barrett’s esophagus with high–grade dysplasia: a review. Am J Gastroenterol 1999;94:1153–60.
82. Sampliner RE. Endoscopic ablative therapy for Barrett’s esophagus: current status. Gastrointest Endosc 2004;59:66–9.
83. Sharma VK, Wang KK, Overholt BF, et al. Balloon–based, circumferential, endoscopic radiofrequency ablation of Barrett’s esophagus: 1–year follow–up of 100 patients. Gastrointest Endosc 2007;65:185–95.
84. Hanlon CR. Textbook of surgery: The biological basis of modern surgical practice, 14th edition. Ann Surg 1992;216:94.
85. Bright T, Watson DI, Tam W, et al. Randomized trial of argon plasma coagulation versus endoscopic surveillance for barrett esophagus after antireflux surgery: late results. Ann Surg 2007;246:1016–20.
86. Chadwick G, Groene O, Markar SR, et al. Systematic review comparing radiofrequency ablation and complete endoscopic resection in treating dysplastic Barrett’s esophagus: a critical assessment of histologic outcomes and adverse events. Gastrointest Endosc 2014;79:718–31.e3.
87. Gondrie JJ, Pouw RE, Sondermeijer CM, et al. Effective treatment of early Barrett’s neoplasia with stepwise circumferential and focal ablation using the HALO system. Endoscopy 2008;40:370–9.
88. Pouw RE, Wirths K, Eisendrath P, et al. Efficacy of radiofrequency ablation combined with endoscopic resection for barrett’s esophagus with early neoplasia. Clin Gastroenterol Hepatol 2010;8:23–9.
89. Kim HP, Bulsiewicz WJ, Cotton CC, et al. Focal endoscopic mucosal resection before radiofrequency ablation is equally effective and safe compared with radiofrequency ablation alone for the eradication of Barrett’s esophagus with advanced neoplasia. Gastrointest Endosc 2012;76:733–9.
90. Fleischer DE, Overholt BF, Sharma VK, et al. Endoscopic ablation of Barrett’s esophagus: a multicenter study with 2.5–year follow–up. Gastrointest Endosc 2008;68:867–76.
91. Sharma VK, Jae Kim H, Das A, et al. Circumferential and focal ablation of Barrett’s esophagus containing dysplasia. Am J Gastroenterol 2009;104:310–7.
92. Ganz RA, Overholt BF, Sharma VK, et al. Circumferential ablation of Barrett’s esophagus that contains high–grade dysplasia: a U.S. multicenter registry. Gastrointest Endosc 2008;68:35–40.
93. Gupta M, Iyer PG, Lutzke L, et al. Recurrence of esophageal intestinal metaplasia after endoscopic mucosal resection and radiofrequency ablation of Barrett’s esophagus: results from a US Multicenter Consortium. Gastroenterology 2013;145:79–86.e1.
94. Lyday WD, Corbett FS, Kuperman DA, et al. Radiofrequency ablation of Barrett’s esophagus: outcomes of 429 patients from a multicenter community practice registry. Endoscopy 2010;42:272–8.
95. Pasricha S, Bulsiewicz WJ, Hathorn KE, et al. Durability and predictors of successful radiofrequency ablation for Barrett’s esophagus. Clin Gastroenterol Hepatol 2014;12:1840–7.e1.
96. Fleischer DE, Overholt BF, Sharma VK, et al. Endoscopic radiofrequency ablation for Barrett’s esophagus: 5–year outcomes from a prospective multicenter trial. Endoscopy 2010;42:781–9.
97. Weston AP, Sharma P, Banerjee S, et al. Visible endoscopic and histologic changes in the cardia, before and after complete Barrett’s esophagus ablation. Gastrointest Endosc 2005;61:515–21.
98. Beaumont H, Gondrie JJ, McMahon BP, et al. Stepwise radiofrequency ablation of Barrett’s esophagus preserves esophageal inner diameter, compliance, and motility. Endoscopy 2009;41:2–8.
99. Orman ES, Li N, Shaheen NJ.Efficacy and durability of radiofrequency ablation for Barrett’s Esophagus: systematic review and meta–analysis. Clin Gastroenterol Hepatol 2013;11:1245–55.
100. Krishnan K, Pandolfino JE, Kahrilas PJ, et al. Increased risk for persistent intestinal metaplasia in patients with Barrett’s esophagus and uncontrolled reflux exposure before radiofrequency ablation. Gastroenterology 2012;143:576–81.
101. Akiyama J, Marcus SN, Triadafilopoulos G. Effective intra–esophageal acid control is associated with improved radiofrequency ablation outcomes in Barrett’s esophagus. Dig Dis Sci 2012;57:2625–32.
102. Small AJ, Sutherland SE, Hightower JS, et al. Comparative risk of recurrence of dysplasia and carcinoma after endoluminal eradication therapy of high–grade dysplasia versus intramucosal carcinoma in Barrett’s esophagus. Gastrointest Endosc 2015;81:1158–66.e1–4.
103. Shaheen NJ, Greenwald BD, Peery AF, et al. Safety and efficacy of endoscopic spray cryotherapy for Barrett’s esophagus with high–grade dysplasia. Gastrointest Endosc 2010;71:680–5.
104. Shaheen NJ, Peery AF, Hawes RH, et al. Quality of life following radiofrequency ablation of dysplastic Barrett’s esophagus. Endoscopy 2010;42:790–9.
105. Bedi AO, Kwon RS, Rubenstein JH, et al. A survey of expert follow–up practices after successful endoscopic eradication therapy for Barrett’s esophagus with high–grade dysplasia and intramucosal adenocarcinoma. Gastrointest Endosc 2013;78:696–701.
106. Sampliner RE, Camargo E, Prasad AR. Prasad, Association of ablation of Barrett’s esophagus with high grade dysplasia and adenocarcinoma of the gastric cardia. Dis Esophagus 2006;19:277–9.
107. Overholt BF, Panjehpour M, Halberg DL. Photodynamic therapy for Barrett’s esophagus with dysplasia and/or early stage carcinoma: long–term results. Gastrointest Endosc 2003;58:183–8.
108. Gosain S, Mercer K, Twaddell WS, et al. Liquid nitrogen spray cryotherapy in Barrett’s esophagus with high–grade dysplasia: long–term results. Gastrointest Endosc 2013;78:260–5.
109. Canto MI, Shin EJ, Khashab MA, et al. Safety and efficacy of carbon dioxide cryotherapy for treatment of neoplastic Barrett’s esophagus. Endoscopy 2015;47:582–91.
110. Van Laethem JL, Peny MO, Salmon I, et al. Intramucosal adenocarcinoma arising under squamous re–epithelialisation of Barrett’s oesophagus. Gut 2000;46:574–7.
111. Pech O, May A, Gossner L, et al. Management of pre–malignant and malignant lesions by endoscopic resection. Best Pract Res Clin Gastroenterol 2004;18:61–76.
112. Soetikno RM, Gotoda T, Nakanishi Y, Soehendra N. Endoscopic mucosal resection. Gastrointest Endosc 2003;57:567–79.
113. Ell C, May A, Gossner L, et al. Endoscopic mucosal resection of early cancer and high–grade dysplasia in Barrett’s esophagus. Gastroenterology 2000;118:670–7.
114. Pech O, Behrens A, May A, et al. Long–term results and risk factor analysis for recurrence after curative endoscopic therapy in 349 patients with high–grade intraepithelial neoplasia and mucosal adenocarcinoma in Barrett’s oesophagus. Gut 2008;57:1200–6.
115. Vieth M, Ell C, Gossner L, et al. Histological analysis of endoscopic resection specimens from 326 patients with Barrett’s esophagus and early neoplasia. Endoscopy 2004;36:776–81.
116. Buskens CJ, Westerterp M, Lagarde SM, et al. Prediction of appropriateness of local endoscopic treatment for high–grade dysplasia and early adenocarcinoma by EUS and histopathologic features. Gastrointest Endosc 2004;60:703–10.
117. Nijhawan PK, Wang KK. Endoscopic mucosal resection for lesions with endoscopic features suggestive of malignancy and high–grade dysplasia within Barrett’s esophagus. Gastrointest Endosc 2000;52:328–32.
118. Esaki M, Matsumoto T, Hirakawa K, et al. Risk factors for local recurrence of superficial esophageal cancer after treatment by endoscopic mucosal resection. Endoscopy 2007;39:41–5.
119. Ell C, May A, Pech O, et al. Curative endoscopic resection of early esophageal adenocarcinomas (Barrett’s cancer). Gastrointest Endosc 2007;65:3–10.
120. Chennat J, Konda VJ, Ross AS, et al. Complete Barrett’s eradication endoscopic mucosal resection: an effective treatment modality for high–grade dysplasia and intramucosal carcinoma––an American single–center experience. Am J Gastroenterol 2009;104:2684–92.
121. Pech O, Bollschweiler E, Manner H, et al. Comparison between endoscopic and surgical resection of mucosal esophageal adenocarcinoma in Barrett’s esophagus at two high–volume centers. Ann Surg 2011;254:67–72.
122. Wu J, Pan YM, Wang TT, et al. Endotherapy versus surgery for early neoplasia in Barrett’s esophagus: a meta–analysis. Gastrointest Endosc 2014;79:233–241.e2.
123. Pech O, May A, Manner H, et al. Long–term efficacy and safety of endoscopic resection for patients with mucosal adenocarcinoma of the esophagus. Gastroenterology 2014;146:652–660.e1.
124. Prasad GA, Wu TT, Wigle DA, et al. Endoscopic and surgical treatment of mucosal (T1a) esophageal adenocarcinoma in Barrett’s esophagus. Gastroenterology 2009;137:815–23.
125. May A, Gossner L, Pech O, et al. Local endoscopic therapy for intraepithelial high–grade neoplasia and early adenocarcinoma in Barrett’s oesophagus: acute–phase and intermediate results of a new treatment approach. Eur J Gastroenterol Hepatol 2002;14(10):1085–91.
126. Gerke H, Siddiqui J, Nasr I, et al. Efficacy and safety of EMR to completely remove Barrett’s esophagus: experience in 41 patients. Gastrointest Endosc 2011;74:761–71.
127. Lewis JJ, Rubenstein JH, Singal AG, et al. Factors associated with esophageal stricture formation after endoscopic mucosal resection for neoplastic Barrett’s esophagus. Gastrointest Endosc 2011;74:753–60.
128. Choi IJ, Kim CG, Chang HJ, et al. The learning curve for EMR with circumferential mucosal incision in treating intramucosal gastric neoplasm. Gastrointest Endosc 2005;62:860–5.
129. Deprez PH, Bergman JJ, Meisner S, et al. Current practice with endoscopic submucosal dissection in Europe: position statement from a panel of experts. Endoscopy 2010;42:853–8.
130. van Lanschot JJ, Hulscher JB, Buskens CJ, et al. Hospital volume and hospital mortality for esophagectomy. Cancer 2001;91:1574–8.
131. Karl RC, Schreiber R, Boulware D, et al. Factors affecting morbidity, mortality, and survival in patients undergoing Ivor Lewis esophagogastrectomy. Ann Surg 2000;231:635–43.
132. Young MM, Deschamps C, Trastek VF, et al. Esophageal reconstruction for benign disease: early morbidity, mortality, and functional results. Ann Thorac Surg 2000;70:1651–5.
133. Dunbar KB, Spechler SJ. The risk of lymph–node metastases in patients with high–grade dysplasia or intramucosal carcinoma in Barrett’s esophagus: a systematic review. Am J Gastroenterol 2012;107:850–62; quiz 863.
134. Morales CP, Souza RF, Spechler SJ. Hallmarks of cancer progression in Barrett’s oesophagus. Lancet 2002;360:1587–9.
135. Omer ZB, Ananthakrishnan AN, Nattinger KJ, et al. Aspirin protects against Barrett’s esophagus in a multivariate logistic regression analysis. Clin Gastroenterol Hepatol 2012;10:722–7.
136. Abnet CC, Freedman ND, Kamangar F, et al. Non–steroidal anti–inflammatory drugs and risk of gastric and oesophageal adenocarcinomas: results from a cohort study and a meta–analysis. Br J Cancer 2009;100:551–7.
137. Kastelein F, Spaander MC, Biermann K, et al. Nonsteroidal anti–inflammatory drugs and statins have chemopreventative effects in patients with Barrett’s esophagus. Gastroenterology 2011;141:2000–8; quiz e13–4.
138. Zhang S, Zhang XQ, Ding XW, et al. Cyclooxygenase inhibitors use is associated with reduced risk of esophageal adenocarcinoma in patients with Barrett’s esophagus: a meta–analysis. Br J Cancer 2014;110: 2378–88.
Current Guidelines for Psoriasis Treatment: A Work in Progress
Psoriasis is a chronic autoinflammatory disorder affecting approximately 2% to 4% of the Western population.1 While there is no absolute cure for psoriasis, novel therapies allow for substantial reduction in symptoms and considerable improvement in quality of life (QoL). In the past few years, multiple treatment guidelines (recommendations based on evidence-based literature reviews) and consensus statements (a set of declarations determined and voted on by a panel of experts in the field) have been developed to guide physicians worldwide in treating psoriasis in the clinical setting (eTable).2-10
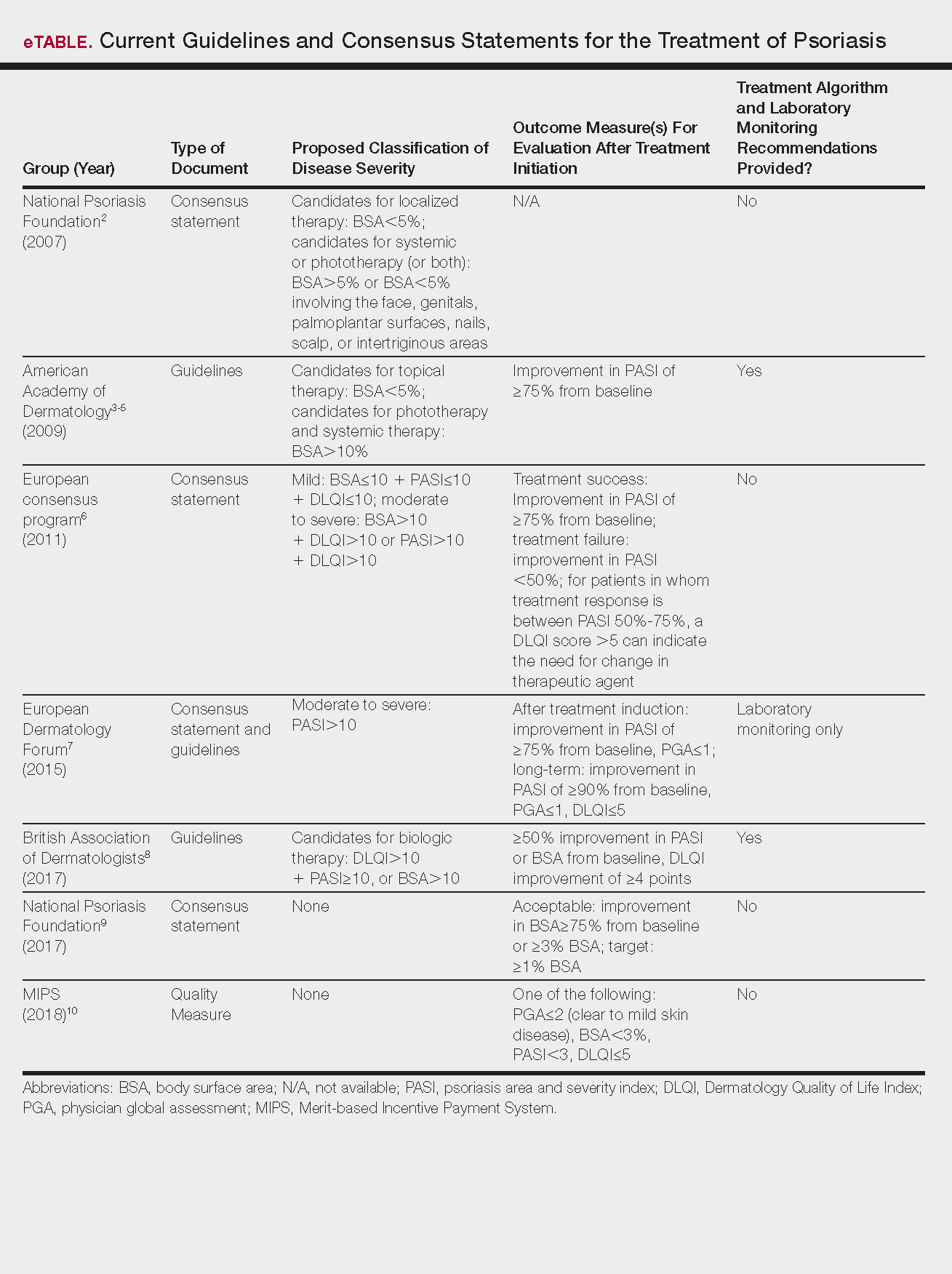
Because psoriasis is a complex disease with multiple comorbidities, applicability of these guidelines may be limited. Although some basic treatment algorithms exist, patient preference, disease severity, and other variables including comorbidities (eg, psoriatic arthritis [PsA], risk of major cardiac events, inflammatory bowel disease [IBD]), history of nonmelanoma skin cancer (NMSC), pregnancy and lactation, and specific contraindications to therapy (eg, renal failure, liver disease, active malignancy) should be considered. In this article, we summarize common themes across existing guidelines and consensus statements for the treatment of psoriasis and highlight areas where there is consistent agreement or lack of sufficient information.
Disease Severity and Treatment Outcomes
There currently are no consensus definitions for mild, moderate, and severe psoriasis, but several consensus statements have attempted to standardize grading systems based on objective values, such as body surface area (BSA) and psoriasis area and severity index (PASI)(a scoring system used to grade the degree of redness, thickness, and scaling of psoriasis plaques), as well as subjective QoL measures.2,6 Although classification of disease severity varies, mild psoriasis generally is characterized as disease that can be managed with local and topical therapy, and moderate to severe psoriasis typically warrants consideration for escalated treatment with phototherapy or systemic agents.
Most definitions of disease severity in psoriasis reference 5% to 10% BSA involvement as a cutoff that should trigger consideration of systemic treatment; however, these criteria could result in undertreatment of patients with substantial disease. For example, patients who have limited BSA involvement but whose disease has a considerable impact on QoL, as well as those who have debilitating disease in localized areas (eg, palms, soles, scalp, nails) or substantial joint involvement may also be appropriate candidates for systemic treatment.5,8
Once therapy is initiated, patients should be evaluated for appropriate treatment response at dedicated intervals. While the time to maximum therapeutic benefit depends on the agent of choice, European guidelines recommend that patients be evaluated after an induction phase (typically 16–24 weeks) and define treatment success as either (1) at least 75% improvement in PASI or (2) at least 50% improvement in PASI and a Dermatology Quality of Life Index (DLQI) score of 5 or lower.6
Alternatively, the National Psoriasis Foundation (NPF) recommended BSA as the preferred outcome measure in a recent consensus statement and concluded that an outcome of 3% or less BSA involvement or improvement in BSA of 75% or more is considered a desirable treatment response.9 Additionally, the Medicare Merit-based Incentive Payment System (MIPS) guidelines for successful systemic treatment response include at least 1 of the following: (1) physician global assessment score of 2 or lower, (2) BSA involvement of less than 3%, (3) PASI score lower than 3, or (4) DLQI score of 5 or lower.10
Although an array of outcome measures have been utilized in clinical trials and proposed in psoriasis guidelines and consensus statements, BSA is typically a manageable measure of treatment response in a clinical setting; however, DLQI should also be assessed if possible, particularly in patients with debilitating localized disease.9
Treatment Options
Because topical treatment regimens can be arduous and typically do not result in sustained clearance, patient expectations should be ascertained prior to initiation of therapy. Topical corticosteroids often can be used as monotherapy in patients with mild psoriasis.3 Topical vitamin D analogues and retinoids also can be effective; however, combined use of these agents with topical steroids should be considered to increase efficacy, and combination formulations can be prescribed to simplify application and improve adherence.
Treatment with UVB or psoralen plus UVA phototherapy is recommended for patients with moderate to severe psoriasis as well as in those who have had minimal response to topical therapy.4 Targeted phototherapy with an excimer laser can be used in patients with BSA involvement of less than 10%.
Methotrexate (MTX), cyclosporine, and acitretin are the most commonly prescribed systemic medications for severe psoriasis in the United States.5 Despite the risk for hepatotoxicity, MTX appears to have the best combined safety and efficacy profile in terms of serious adverse events compared to other systemic agents.11 Guidelines for MTX monitoring, especially with regard to when to do a liver biopsy, have been substantially liberalized over time, and the recommended interval for biopsy has been extended by years; biopsy was previously recommended after a cumulative MTX dose of 1 to 1.5 g, but guidelines now suggest biopsy after 3.5 to 4 g in low-risk patients.5 While abnormally elevated liver function tests during treatment with MTX may necessitate liver biopsy, the use of transient elastography and a panel of serum biomarkers for liver function also can be used to monitor noninvasively for hepatotoxicity before biopsy is considered; these recommendations are likely to be incorporated into newer guidelines in development.12 Methotrexate has demonstrated safety and increased efficacy when used in combination with biologic agents such as adalimumab, etanercept, infliximab, and secukinumab7 and has been studied in combination with many biologics indicated for PsA.13
Due to a considerable risk of glomerulosclerosis, cyclosporine is approved for a maximum of 1 year of continuous treatment of psoriasis in the United States and2 years in Europe.5,7 Cyclosporine is best used as induction therapy in psoriasis patients with severe disease who are seeking faster abatement of symptoms.
Acitretin is another systemic treatment option, although efficacy of this agent is dose dependent. Higher dosing often is limited due to lower tolerability.5
Given that many insurance formularies primarily cover traditional systemic therapies and that MTX and phototherapy are generally well tolerated and cost effective, patients may need to be treated with traditional agents before escalating to biologics. Prior to starting treatment with any biologic, patients should typically be screened for tuberculosis (TB), human immunodeficiency virus infection, and immunization for, exposure to, and/or infection with hepatitis B and C virus, and any other active infections. In patients who do not demonstrate hepatitis immunity, the hepatitis B vaccine should be administered prior to starting treatment with a biologic.14 In psoriasis patients with latent TB, 2 months of treatment should be completed before initiating biologic therapy8; once a biologic has been initiated, all patients should be screened annually for TB.
European guidelines for biologic treatment recommend that complete blood count and liver and renal function be evaluated at baseline, at months 1 and 3 of treatment, and then every 3 to 6 months thereafter while on the biologic agent.7 These recommendations are more stringent than those indicated in regulatory labeling and, based on the continual accumulation of data regarding the safety of these agents, some investigators have argued that laboratory testing might not be necessary at all.15
Treatment in Special Populations
Psoriasis patients often present with comorbidities or a complicated medical history, which can make it challenging to decide which therapy is most suitable. Patients with comorbid diseases (eg, PsA, risk of major cardiac event, IBD) or a history of NMSC and those who are pregnant or are lactating require special considerations to ensure treatment safety and efficacy.
Tumor necrosis factor α (TNF-α) and IL-17 inhibitors are used in the treatment of joint disorders and should be considered in patients with PsA. IL-23/IL-12 inhibition appears to have less benefit in patients with PsA, but studies on IL-23 inhibition (p19 antibodies) alone are ongoing.16 It has been reported that TNF-α inhibition may be beneficial in patients at risk for major cardiac events.8,17 In patients with IBD, IL-17 inhibitors should be avoided because they may exacerbate the condition; however, TNF-α and IL-23/IL-12 inhibition have shown to be safe in patients with IBD and many agents in these classes are approved by the US Food and Drug Administration for use in this population.18,19
Although biologics may increase the risk of developing NMSC20 and should generally be avoided in patients with any active malignancy, specific guidelines for screening and initiation of treatment in patients with a history of cancer are not clearly outlined. Prior to initiating systemic therapy in any patient, a careful medical history should be obtained. These agents often are not prescribed in patients with a history of cancer until remission has been established for at least 5 years, with the exception of patients with a history of treated NMSC.8 Annual skin monitoring for NMSC should be undertaken for psoriasis patients on most immunomodulating systemic therapies.
Recommendations for biologic treatment in psoriasis patients who are pregnant or lactating also are limited. European guidelines have noted pregnancy as an absolute contraindication to treatment with biologics,7but the regulatory guidance has recently changed for some agents, so this recommendation also may evolve.21 British8 and US5 guidelines do not consider pregnancy a contraindication for treatment with biologics.
Information on the safety of TNF-α antagonists during pregnancy comes primarily from use in patients with IBD and rheumatologic disease. To date, reports on the incidence of congenital malformations have been generally reassuring. Because IgG antibodies are actively transferred across the placenta in the late-second or the third trimesters, neonates born to mothers on biologic treatments may have high levels of some biologic drugs at birth. As a result, live vaccination should be avoided in neonates whose mothers were treated with IgG-based biologics.
Changing Treatment Agents
Patients may need to stop and change treatment agents due to ineffectiveness, personal preference, or worsening disease. When transitioning from any systemic or biologic agent to another (other than MTX), the British Association of Dermatologists recommends a washout period of at least 1 month before initiating a new therapy.8 Most guidelines do not define parameters for therapy escalation when patients fail multiple systemic agents, so physicians should use clinical judgment along with consideration of patient preference and comorbidity profile to ascertain which agent is most appropriate.
Conclusion
Keeping psoriasis treatment guidelines updated can be difficult, especially as new therapeutic options for psoriasis and treatment regimens rapidly evolve. Regulatory recommendations also vary worldwide, but most guidelines are reasonably consistent without being overly prescriptive, appropriately allowing for flexibility for application in clinical practice. Nonetheless, physicians should keep in mind new or changing guidelines while tailoring psoriasis treatment recommendations to best suit their individual patients.
- Parisi R, Symmons DP, Griffiths CE, et al; Identification and Management of Psoriasis and Associated ComorbidiTty (IMPACT) project team. Global epidemiology of psoriasis: a systematic review of incidence and prevalence [published online September 27, 2012]. J Invest Dermatol. 2013;133:377-385.
- Pariser DM, Bagel J, Gelfand JM, et al. National Psoriasis Foundation clinical consensus on disease severity. Arch Dermatol. 2007;143:239-242.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis. section 3. guidelines of care for the management and treatment of psoriasis with topical therapies. J Am Acad Dermatol. 2009;60:643-659.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 5. guidelines of care for the treatment of psoriasis with phototherapy and photochemotherapy. J Am Acad Dermatol. 2010;62:114-135.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol. 2009;61:451-485.
- Mrowietz U, Kragballe K, Reich K, et al. Definition of treatment goals for moderate to severe psoriasis: a European consensus. Arch Dermatol Res. 2011;303:1-10.
- Nast A, Gisondi P, Ormerod AD, et al. European S3-guidelines on the systemic treatment of psoriasis vulgaris—update 2015—short version—EDF in cooperation with EADV and IPC [published online October 9, 2015]. J Eur Acad Dermatol Venereol. 2015;29:2277-2294.
- Smith CH, Jabbar-Lopez ZK, Yiu ZZ, et al. British Association of Dermatologists guidelines for biologic therapy for psoriasis 2017. Br J Dermatol. 2017;177:628-636.
- Armstrong AW, Siegel MP, Bagel J, et al. From the medical board of the National Psoriasis Foundation: treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017;76:290-298.
- Quality ID #410: psoriasis: clinical response to oral systemic or biologic medications—national quality strategy domain: person and caregiver-centered experience and outcomes. Centers for Medicare and Medicaid Services website. https://www.cms.gov/Medicare/Quality-Payment-Program/Resource-Library/2018-Resources.html. Accessed February 27, 2018.
- Sbidian E, Chaimani A, Garcia-Doval I, et al. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis. Cochrane Database of Syst Rev. 2017;12:CD011535.
- Lynch M, Higgins E, McCormick PA, et al. The use of transient elastography and FibroTest for monitoring hepatotoxicity in patients receiving methotrexate for psoriasis. JAMA Dermatol. 2014;150:856-862.
- Behrens F, Canete J, Olivieri I, et al. Tumor necrosis factor inhibitor monotherapy versus combination with MTX in the treatment of PsA: a systemic review of the literature. Rheumatology. 2015;54:915-926.
- Karadağ Ö, Kaşifoğlu T, Özer B, et al. Viral hepatitis screening guideline before biological drug use in rheumatic patients. Eur J Rheumatol. 2016;3:25-28.
- Ahn CS, Dothard EH, Garner ML, et al. To test or not to test? an updated evidence-based assessment of the value of screening and monitoring tests when using systemic biologic agents to treat psoriasis and psoriatic arthritis. J Am Acad Dermatol. 2015;73:420-428.
- Reich K, Armstrong AW, Foley P, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double-blind, placebo- and active comparator–controlled VOYAGE 2 trial. J Am Acad Dermatol. 2017;76:418-431.
- Wu JJ, Guérin A, Sundaram M, et al. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-α inhibitors versus methotrexate. J Am Acad Dermatol. 2017;76:81-90.
- Humira [package insert]. North Chicago, IL: Abbott Laboratories; 2011.
- Stelara [package insert]. Bloomington, IN: Janssen Biotech, Inc; 2016.
- Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum. 2007;56:2886-2895.
- Cimzia [package insert]. UCB, Inc: Smyrna, GA; 2016.
Psoriasis is a chronic autoinflammatory disorder affecting approximately 2% to 4% of the Western population.1 While there is no absolute cure for psoriasis, novel therapies allow for substantial reduction in symptoms and considerable improvement in quality of life (QoL). In the past few years, multiple treatment guidelines (recommendations based on evidence-based literature reviews) and consensus statements (a set of declarations determined and voted on by a panel of experts in the field) have been developed to guide physicians worldwide in treating psoriasis in the clinical setting (eTable).2-10
Because psoriasis is a complex disease with multiple comorbidities, applicability of these guidelines may be limited. Although some basic treatment algorithms exist, patient preference, disease severity, and other variables including comorbidities (eg, psoriatic arthritis [PsA], risk of major cardiac events, inflammatory bowel disease [IBD]), history of nonmelanoma skin cancer (NMSC), pregnancy and lactation, and specific contraindications to therapy (eg, renal failure, liver disease, active malignancy) should be considered. In this article, we summarize common themes across existing guidelines and consensus statements for the treatment of psoriasis and highlight areas where there is consistent agreement or lack of sufficient information.
Disease Severity and Treatment Outcomes
There currently are no consensus definitions for mild, moderate, and severe psoriasis, but several consensus statements have attempted to standardize grading systems based on objective values, such as body surface area (BSA) and psoriasis area and severity index (PASI)(a scoring system used to grade the degree of redness, thickness, and scaling of psoriasis plaques), as well as subjective QoL measures.2,6 Although classification of disease severity varies, mild psoriasis generally is characterized as disease that can be managed with local and topical therapy, and moderate to severe psoriasis typically warrants consideration for escalated treatment with phototherapy or systemic agents.
Most definitions of disease severity in psoriasis reference 5% to 10% BSA involvement as a cutoff that should trigger consideration of systemic treatment; however, these criteria could result in undertreatment of patients with substantial disease. For example, patients who have limited BSA involvement but whose disease has a considerable impact on QoL, as well as those who have debilitating disease in localized areas (eg, palms, soles, scalp, nails) or substantial joint involvement may also be appropriate candidates for systemic treatment.5,8
Once therapy is initiated, patients should be evaluated for appropriate treatment response at dedicated intervals. While the time to maximum therapeutic benefit depends on the agent of choice, European guidelines recommend that patients be evaluated after an induction phase (typically 16–24 weeks) and define treatment success as either (1) at least 75% improvement in PASI or (2) at least 50% improvement in PASI and a Dermatology Quality of Life Index (DLQI) score of 5 or lower.6
Alternatively, the National Psoriasis Foundation (NPF) recommended BSA as the preferred outcome measure in a recent consensus statement and concluded that an outcome of 3% or less BSA involvement or improvement in BSA of 75% or more is considered a desirable treatment response.9 Additionally, the Medicare Merit-based Incentive Payment System (MIPS) guidelines for successful systemic treatment response include at least 1 of the following: (1) physician global assessment score of 2 or lower, (2) BSA involvement of less than 3%, (3) PASI score lower than 3, or (4) DLQI score of 5 or lower.10
Although an array of outcome measures have been utilized in clinical trials and proposed in psoriasis guidelines and consensus statements, BSA is typically a manageable measure of treatment response in a clinical setting; however, DLQI should also be assessed if possible, particularly in patients with debilitating localized disease.9
Treatment Options
Because topical treatment regimens can be arduous and typically do not result in sustained clearance, patient expectations should be ascertained prior to initiation of therapy. Topical corticosteroids often can be used as monotherapy in patients with mild psoriasis.3 Topical vitamin D analogues and retinoids also can be effective; however, combined use of these agents with topical steroids should be considered to increase efficacy, and combination formulations can be prescribed to simplify application and improve adherence.
Treatment with UVB or psoralen plus UVA phototherapy is recommended for patients with moderate to severe psoriasis as well as in those who have had minimal response to topical therapy.4 Targeted phototherapy with an excimer laser can be used in patients with BSA involvement of less than 10%.
Methotrexate (MTX), cyclosporine, and acitretin are the most commonly prescribed systemic medications for severe psoriasis in the United States.5 Despite the risk for hepatotoxicity, MTX appears to have the best combined safety and efficacy profile in terms of serious adverse events compared to other systemic agents.11 Guidelines for MTX monitoring, especially with regard to when to do a liver biopsy, have been substantially liberalized over time, and the recommended interval for biopsy has been extended by years; biopsy was previously recommended after a cumulative MTX dose of 1 to 1.5 g, but guidelines now suggest biopsy after 3.5 to 4 g in low-risk patients.5 While abnormally elevated liver function tests during treatment with MTX may necessitate liver biopsy, the use of transient elastography and a panel of serum biomarkers for liver function also can be used to monitor noninvasively for hepatotoxicity before biopsy is considered; these recommendations are likely to be incorporated into newer guidelines in development.12 Methotrexate has demonstrated safety and increased efficacy when used in combination with biologic agents such as adalimumab, etanercept, infliximab, and secukinumab7 and has been studied in combination with many biologics indicated for PsA.13
Due to a considerable risk of glomerulosclerosis, cyclosporine is approved for a maximum of 1 year of continuous treatment of psoriasis in the United States and2 years in Europe.5,7 Cyclosporine is best used as induction therapy in psoriasis patients with severe disease who are seeking faster abatement of symptoms.
Acitretin is another systemic treatment option, although efficacy of this agent is dose dependent. Higher dosing often is limited due to lower tolerability.5
Given that many insurance formularies primarily cover traditional systemic therapies and that MTX and phototherapy are generally well tolerated and cost effective, patients may need to be treated with traditional agents before escalating to biologics. Prior to starting treatment with any biologic, patients should typically be screened for tuberculosis (TB), human immunodeficiency virus infection, and immunization for, exposure to, and/or infection with hepatitis B and C virus, and any other active infections. In patients who do not demonstrate hepatitis immunity, the hepatitis B vaccine should be administered prior to starting treatment with a biologic.14 In psoriasis patients with latent TB, 2 months of treatment should be completed before initiating biologic therapy8; once a biologic has been initiated, all patients should be screened annually for TB.
European guidelines for biologic treatment recommend that complete blood count and liver and renal function be evaluated at baseline, at months 1 and 3 of treatment, and then every 3 to 6 months thereafter while on the biologic agent.7 These recommendations are more stringent than those indicated in regulatory labeling and, based on the continual accumulation of data regarding the safety of these agents, some investigators have argued that laboratory testing might not be necessary at all.15
Treatment in Special Populations
Psoriasis patients often present with comorbidities or a complicated medical history, which can make it challenging to decide which therapy is most suitable. Patients with comorbid diseases (eg, PsA, risk of major cardiac event, IBD) or a history of NMSC and those who are pregnant or are lactating require special considerations to ensure treatment safety and efficacy.
Tumor necrosis factor α (TNF-α) and IL-17 inhibitors are used in the treatment of joint disorders and should be considered in patients with PsA. IL-23/IL-12 inhibition appears to have less benefit in patients with PsA, but studies on IL-23 inhibition (p19 antibodies) alone are ongoing.16 It has been reported that TNF-α inhibition may be beneficial in patients at risk for major cardiac events.8,17 In patients with IBD, IL-17 inhibitors should be avoided because they may exacerbate the condition; however, TNF-α and IL-23/IL-12 inhibition have shown to be safe in patients with IBD and many agents in these classes are approved by the US Food and Drug Administration for use in this population.18,19
Although biologics may increase the risk of developing NMSC20 and should generally be avoided in patients with any active malignancy, specific guidelines for screening and initiation of treatment in patients with a history of cancer are not clearly outlined. Prior to initiating systemic therapy in any patient, a careful medical history should be obtained. These agents often are not prescribed in patients with a history of cancer until remission has been established for at least 5 years, with the exception of patients with a history of treated NMSC.8 Annual skin monitoring for NMSC should be undertaken for psoriasis patients on most immunomodulating systemic therapies.
Recommendations for biologic treatment in psoriasis patients who are pregnant or lactating also are limited. European guidelines have noted pregnancy as an absolute contraindication to treatment with biologics,7but the regulatory guidance has recently changed for some agents, so this recommendation also may evolve.21 British8 and US5 guidelines do not consider pregnancy a contraindication for treatment with biologics.
Information on the safety of TNF-α antagonists during pregnancy comes primarily from use in patients with IBD and rheumatologic disease. To date, reports on the incidence of congenital malformations have been generally reassuring. Because IgG antibodies are actively transferred across the placenta in the late-second or the third trimesters, neonates born to mothers on biologic treatments may have high levels of some biologic drugs at birth. As a result, live vaccination should be avoided in neonates whose mothers were treated with IgG-based biologics.
Changing Treatment Agents
Patients may need to stop and change treatment agents due to ineffectiveness, personal preference, or worsening disease. When transitioning from any systemic or biologic agent to another (other than MTX), the British Association of Dermatologists recommends a washout period of at least 1 month before initiating a new therapy.8 Most guidelines do not define parameters for therapy escalation when patients fail multiple systemic agents, so physicians should use clinical judgment along with consideration of patient preference and comorbidity profile to ascertain which agent is most appropriate.
Conclusion
Keeping psoriasis treatment guidelines updated can be difficult, especially as new therapeutic options for psoriasis and treatment regimens rapidly evolve. Regulatory recommendations also vary worldwide, but most guidelines are reasonably consistent without being overly prescriptive, appropriately allowing for flexibility for application in clinical practice. Nonetheless, physicians should keep in mind new or changing guidelines while tailoring psoriasis treatment recommendations to best suit their individual patients.
Psoriasis is a chronic autoinflammatory disorder affecting approximately 2% to 4% of the Western population.1 While there is no absolute cure for psoriasis, novel therapies allow for substantial reduction in symptoms and considerable improvement in quality of life (QoL). In the past few years, multiple treatment guidelines (recommendations based on evidence-based literature reviews) and consensus statements (a set of declarations determined and voted on by a panel of experts in the field) have been developed to guide physicians worldwide in treating psoriasis in the clinical setting (eTable).2-10
Because psoriasis is a complex disease with multiple comorbidities, applicability of these guidelines may be limited. Although some basic treatment algorithms exist, patient preference, disease severity, and other variables including comorbidities (eg, psoriatic arthritis [PsA], risk of major cardiac events, inflammatory bowel disease [IBD]), history of nonmelanoma skin cancer (NMSC), pregnancy and lactation, and specific contraindications to therapy (eg, renal failure, liver disease, active malignancy) should be considered. In this article, we summarize common themes across existing guidelines and consensus statements for the treatment of psoriasis and highlight areas where there is consistent agreement or lack of sufficient information.
Disease Severity and Treatment Outcomes
There currently are no consensus definitions for mild, moderate, and severe psoriasis, but several consensus statements have attempted to standardize grading systems based on objective values, such as body surface area (BSA) and psoriasis area and severity index (PASI)(a scoring system used to grade the degree of redness, thickness, and scaling of psoriasis plaques), as well as subjective QoL measures.2,6 Although classification of disease severity varies, mild psoriasis generally is characterized as disease that can be managed with local and topical therapy, and moderate to severe psoriasis typically warrants consideration for escalated treatment with phototherapy or systemic agents.
Most definitions of disease severity in psoriasis reference 5% to 10% BSA involvement as a cutoff that should trigger consideration of systemic treatment; however, these criteria could result in undertreatment of patients with substantial disease. For example, patients who have limited BSA involvement but whose disease has a considerable impact on QoL, as well as those who have debilitating disease in localized areas (eg, palms, soles, scalp, nails) or substantial joint involvement may also be appropriate candidates for systemic treatment.5,8
Once therapy is initiated, patients should be evaluated for appropriate treatment response at dedicated intervals. While the time to maximum therapeutic benefit depends on the agent of choice, European guidelines recommend that patients be evaluated after an induction phase (typically 16–24 weeks) and define treatment success as either (1) at least 75% improvement in PASI or (2) at least 50% improvement in PASI and a Dermatology Quality of Life Index (DLQI) score of 5 or lower.6
Alternatively, the National Psoriasis Foundation (NPF) recommended BSA as the preferred outcome measure in a recent consensus statement and concluded that an outcome of 3% or less BSA involvement or improvement in BSA of 75% or more is considered a desirable treatment response.9 Additionally, the Medicare Merit-based Incentive Payment System (MIPS) guidelines for successful systemic treatment response include at least 1 of the following: (1) physician global assessment score of 2 or lower, (2) BSA involvement of less than 3%, (3) PASI score lower than 3, or (4) DLQI score of 5 or lower.10
Although an array of outcome measures have been utilized in clinical trials and proposed in psoriasis guidelines and consensus statements, BSA is typically a manageable measure of treatment response in a clinical setting; however, DLQI should also be assessed if possible, particularly in patients with debilitating localized disease.9
Treatment Options
Because topical treatment regimens can be arduous and typically do not result in sustained clearance, patient expectations should be ascertained prior to initiation of therapy. Topical corticosteroids often can be used as monotherapy in patients with mild psoriasis.3 Topical vitamin D analogues and retinoids also can be effective; however, combined use of these agents with topical steroids should be considered to increase efficacy, and combination formulations can be prescribed to simplify application and improve adherence.
Treatment with UVB or psoralen plus UVA phototherapy is recommended for patients with moderate to severe psoriasis as well as in those who have had minimal response to topical therapy.4 Targeted phototherapy with an excimer laser can be used in patients with BSA involvement of less than 10%.
Methotrexate (MTX), cyclosporine, and acitretin are the most commonly prescribed systemic medications for severe psoriasis in the United States.5 Despite the risk for hepatotoxicity, MTX appears to have the best combined safety and efficacy profile in terms of serious adverse events compared to other systemic agents.11 Guidelines for MTX monitoring, especially with regard to when to do a liver biopsy, have been substantially liberalized over time, and the recommended interval for biopsy has been extended by years; biopsy was previously recommended after a cumulative MTX dose of 1 to 1.5 g, but guidelines now suggest biopsy after 3.5 to 4 g in low-risk patients.5 While abnormally elevated liver function tests during treatment with MTX may necessitate liver biopsy, the use of transient elastography and a panel of serum biomarkers for liver function also can be used to monitor noninvasively for hepatotoxicity before biopsy is considered; these recommendations are likely to be incorporated into newer guidelines in development.12 Methotrexate has demonstrated safety and increased efficacy when used in combination with biologic agents such as adalimumab, etanercept, infliximab, and secukinumab7 and has been studied in combination with many biologics indicated for PsA.13
Due to a considerable risk of glomerulosclerosis, cyclosporine is approved for a maximum of 1 year of continuous treatment of psoriasis in the United States and2 years in Europe.5,7 Cyclosporine is best used as induction therapy in psoriasis patients with severe disease who are seeking faster abatement of symptoms.
Acitretin is another systemic treatment option, although efficacy of this agent is dose dependent. Higher dosing often is limited due to lower tolerability.5
Given that many insurance formularies primarily cover traditional systemic therapies and that MTX and phototherapy are generally well tolerated and cost effective, patients may need to be treated with traditional agents before escalating to biologics. Prior to starting treatment with any biologic, patients should typically be screened for tuberculosis (TB), human immunodeficiency virus infection, and immunization for, exposure to, and/or infection with hepatitis B and C virus, and any other active infections. In patients who do not demonstrate hepatitis immunity, the hepatitis B vaccine should be administered prior to starting treatment with a biologic.14 In psoriasis patients with latent TB, 2 months of treatment should be completed before initiating biologic therapy8; once a biologic has been initiated, all patients should be screened annually for TB.
European guidelines for biologic treatment recommend that complete blood count and liver and renal function be evaluated at baseline, at months 1 and 3 of treatment, and then every 3 to 6 months thereafter while on the biologic agent.7 These recommendations are more stringent than those indicated in regulatory labeling and, based on the continual accumulation of data regarding the safety of these agents, some investigators have argued that laboratory testing might not be necessary at all.15
Treatment in Special Populations
Psoriasis patients often present with comorbidities or a complicated medical history, which can make it challenging to decide which therapy is most suitable. Patients with comorbid diseases (eg, PsA, risk of major cardiac event, IBD) or a history of NMSC and those who are pregnant or are lactating require special considerations to ensure treatment safety and efficacy.
Tumor necrosis factor α (TNF-α) and IL-17 inhibitors are used in the treatment of joint disorders and should be considered in patients with PsA. IL-23/IL-12 inhibition appears to have less benefit in patients with PsA, but studies on IL-23 inhibition (p19 antibodies) alone are ongoing.16 It has been reported that TNF-α inhibition may be beneficial in patients at risk for major cardiac events.8,17 In patients with IBD, IL-17 inhibitors should be avoided because they may exacerbate the condition; however, TNF-α and IL-23/IL-12 inhibition have shown to be safe in patients with IBD and many agents in these classes are approved by the US Food and Drug Administration for use in this population.18,19
Although biologics may increase the risk of developing NMSC20 and should generally be avoided in patients with any active malignancy, specific guidelines for screening and initiation of treatment in patients with a history of cancer are not clearly outlined. Prior to initiating systemic therapy in any patient, a careful medical history should be obtained. These agents often are not prescribed in patients with a history of cancer until remission has been established for at least 5 years, with the exception of patients with a history of treated NMSC.8 Annual skin monitoring for NMSC should be undertaken for psoriasis patients on most immunomodulating systemic therapies.
Recommendations for biologic treatment in psoriasis patients who are pregnant or lactating also are limited. European guidelines have noted pregnancy as an absolute contraindication to treatment with biologics,7but the regulatory guidance has recently changed for some agents, so this recommendation also may evolve.21 British8 and US5 guidelines do not consider pregnancy a contraindication for treatment with biologics.
Information on the safety of TNF-α antagonists during pregnancy comes primarily from use in patients with IBD and rheumatologic disease. To date, reports on the incidence of congenital malformations have been generally reassuring. Because IgG antibodies are actively transferred across the placenta in the late-second or the third trimesters, neonates born to mothers on biologic treatments may have high levels of some biologic drugs at birth. As a result, live vaccination should be avoided in neonates whose mothers were treated with IgG-based biologics.
Changing Treatment Agents
Patients may need to stop and change treatment agents due to ineffectiveness, personal preference, or worsening disease. When transitioning from any systemic or biologic agent to another (other than MTX), the British Association of Dermatologists recommends a washout period of at least 1 month before initiating a new therapy.8 Most guidelines do not define parameters for therapy escalation when patients fail multiple systemic agents, so physicians should use clinical judgment along with consideration of patient preference and comorbidity profile to ascertain which agent is most appropriate.
Conclusion
Keeping psoriasis treatment guidelines updated can be difficult, especially as new therapeutic options for psoriasis and treatment regimens rapidly evolve. Regulatory recommendations also vary worldwide, but most guidelines are reasonably consistent without being overly prescriptive, appropriately allowing for flexibility for application in clinical practice. Nonetheless, physicians should keep in mind new or changing guidelines while tailoring psoriasis treatment recommendations to best suit their individual patients.
- Parisi R, Symmons DP, Griffiths CE, et al; Identification and Management of Psoriasis and Associated ComorbidiTty (IMPACT) project team. Global epidemiology of psoriasis: a systematic review of incidence and prevalence [published online September 27, 2012]. J Invest Dermatol. 2013;133:377-385.
- Pariser DM, Bagel J, Gelfand JM, et al. National Psoriasis Foundation clinical consensus on disease severity. Arch Dermatol. 2007;143:239-242.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis. section 3. guidelines of care for the management and treatment of psoriasis with topical therapies. J Am Acad Dermatol. 2009;60:643-659.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 5. guidelines of care for the treatment of psoriasis with phototherapy and photochemotherapy. J Am Acad Dermatol. 2010;62:114-135.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol. 2009;61:451-485.
- Mrowietz U, Kragballe K, Reich K, et al. Definition of treatment goals for moderate to severe psoriasis: a European consensus. Arch Dermatol Res. 2011;303:1-10.
- Nast A, Gisondi P, Ormerod AD, et al. European S3-guidelines on the systemic treatment of psoriasis vulgaris—update 2015—short version—EDF in cooperation with EADV and IPC [published online October 9, 2015]. J Eur Acad Dermatol Venereol. 2015;29:2277-2294.
- Smith CH, Jabbar-Lopez ZK, Yiu ZZ, et al. British Association of Dermatologists guidelines for biologic therapy for psoriasis 2017. Br J Dermatol. 2017;177:628-636.
- Armstrong AW, Siegel MP, Bagel J, et al. From the medical board of the National Psoriasis Foundation: treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017;76:290-298.
- Quality ID #410: psoriasis: clinical response to oral systemic or biologic medications—national quality strategy domain: person and caregiver-centered experience and outcomes. Centers for Medicare and Medicaid Services website. https://www.cms.gov/Medicare/Quality-Payment-Program/Resource-Library/2018-Resources.html. Accessed February 27, 2018.
- Sbidian E, Chaimani A, Garcia-Doval I, et al. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis. Cochrane Database of Syst Rev. 2017;12:CD011535.
- Lynch M, Higgins E, McCormick PA, et al. The use of transient elastography and FibroTest for monitoring hepatotoxicity in patients receiving methotrexate for psoriasis. JAMA Dermatol. 2014;150:856-862.
- Behrens F, Canete J, Olivieri I, et al. Tumor necrosis factor inhibitor monotherapy versus combination with MTX in the treatment of PsA: a systemic review of the literature. Rheumatology. 2015;54:915-926.
- Karadağ Ö, Kaşifoğlu T, Özer B, et al. Viral hepatitis screening guideline before biological drug use in rheumatic patients. Eur J Rheumatol. 2016;3:25-28.
- Ahn CS, Dothard EH, Garner ML, et al. To test or not to test? an updated evidence-based assessment of the value of screening and monitoring tests when using systemic biologic agents to treat psoriasis and psoriatic arthritis. J Am Acad Dermatol. 2015;73:420-428.
- Reich K, Armstrong AW, Foley P, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double-blind, placebo- and active comparator–controlled VOYAGE 2 trial. J Am Acad Dermatol. 2017;76:418-431.
- Wu JJ, Guérin A, Sundaram M, et al. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-α inhibitors versus methotrexate. J Am Acad Dermatol. 2017;76:81-90.
- Humira [package insert]. North Chicago, IL: Abbott Laboratories; 2011.
- Stelara [package insert]. Bloomington, IN: Janssen Biotech, Inc; 2016.
- Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum. 2007;56:2886-2895.
- Cimzia [package insert]. UCB, Inc: Smyrna, GA; 2016.
- Parisi R, Symmons DP, Griffiths CE, et al; Identification and Management of Psoriasis and Associated ComorbidiTty (IMPACT) project team. Global epidemiology of psoriasis: a systematic review of incidence and prevalence [published online September 27, 2012]. J Invest Dermatol. 2013;133:377-385.
- Pariser DM, Bagel J, Gelfand JM, et al. National Psoriasis Foundation clinical consensus on disease severity. Arch Dermatol. 2007;143:239-242.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis. section 3. guidelines of care for the management and treatment of psoriasis with topical therapies. J Am Acad Dermatol. 2009;60:643-659.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 5. guidelines of care for the treatment of psoriasis with phototherapy and photochemotherapy. J Am Acad Dermatol. 2010;62:114-135.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol. 2009;61:451-485.
- Mrowietz U, Kragballe K, Reich K, et al. Definition of treatment goals for moderate to severe psoriasis: a European consensus. Arch Dermatol Res. 2011;303:1-10.
- Nast A, Gisondi P, Ormerod AD, et al. European S3-guidelines on the systemic treatment of psoriasis vulgaris—update 2015—short version—EDF in cooperation with EADV and IPC [published online October 9, 2015]. J Eur Acad Dermatol Venereol. 2015;29:2277-2294.
- Smith CH, Jabbar-Lopez ZK, Yiu ZZ, et al. British Association of Dermatologists guidelines for biologic therapy for psoriasis 2017. Br J Dermatol. 2017;177:628-636.
- Armstrong AW, Siegel MP, Bagel J, et al. From the medical board of the National Psoriasis Foundation: treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017;76:290-298.
- Quality ID #410: psoriasis: clinical response to oral systemic or biologic medications—national quality strategy domain: person and caregiver-centered experience and outcomes. Centers for Medicare and Medicaid Services website. https://www.cms.gov/Medicare/Quality-Payment-Program/Resource-Library/2018-Resources.html. Accessed February 27, 2018.
- Sbidian E, Chaimani A, Garcia-Doval I, et al. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis. Cochrane Database of Syst Rev. 2017;12:CD011535.
- Lynch M, Higgins E, McCormick PA, et al. The use of transient elastography and FibroTest for monitoring hepatotoxicity in patients receiving methotrexate for psoriasis. JAMA Dermatol. 2014;150:856-862.
- Behrens F, Canete J, Olivieri I, et al. Tumor necrosis factor inhibitor monotherapy versus combination with MTX in the treatment of PsA: a systemic review of the literature. Rheumatology. 2015;54:915-926.
- Karadağ Ö, Kaşifoğlu T, Özer B, et al. Viral hepatitis screening guideline before biological drug use in rheumatic patients. Eur J Rheumatol. 2016;3:25-28.
- Ahn CS, Dothard EH, Garner ML, et al. To test or not to test? an updated evidence-based assessment of the value of screening and monitoring tests when using systemic biologic agents to treat psoriasis and psoriatic arthritis. J Am Acad Dermatol. 2015;73:420-428.
- Reich K, Armstrong AW, Foley P, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double-blind, placebo- and active comparator–controlled VOYAGE 2 trial. J Am Acad Dermatol. 2017;76:418-431.
- Wu JJ, Guérin A, Sundaram M, et al. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-α inhibitors versus methotrexate. J Am Acad Dermatol. 2017;76:81-90.
- Humira [package insert]. North Chicago, IL: Abbott Laboratories; 2011.
- Stelara [package insert]. Bloomington, IN: Janssen Biotech, Inc; 2016.
- Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum. 2007;56:2886-2895.
- Cimzia [package insert]. UCB, Inc: Smyrna, GA; 2016.
Practice Points
- Guidelines and consensus statements for psoriasis treatment are generally but not always consistent.
- As guidelines evolve, individual patient preferences, disease severity, and comorbid conditions remain important considerations when selecting treatment agents for psoriasis.
- More frequent updates to psoriasis treatment guidelines are becoming increasingly important given the rapid changes in the field.
Emerging Therapies In Psoriasis: A Systematic Review
Psoriasis is a chronic, autoimmune-mediated disease estimated to affect 2.8% of the US population.1 The pathogenesis of psoriasis is thought to involve a complex process triggered by a combination of genetic and environmental factors that induce tumor necrosis factor (TNF) α secretion by keratinocytes, which in turn activates dendritic cells. Activated dendritic cells produce IL-23, leading to helper T cell (TH17) differentiation.2,3 TH17 cells secrete IL-17A, which has been shown to promote psoriatic skin changes.4 Therefore, TNF-α, IL-23, and IL-17A have been recognized as key targets for psoriasis therapy.
The newest biologic agents targeting IL-17–mediated pathways include ixekizumab, brodalumab, and bimekizumab. Secukinumab, the first US Food and Drug Administration (FDA)–approved IL-17 inhibitor, has been available since 2015 and therefore is not included in this review. IL-23 inhibitors that are FDA approved or being evaluated in clinical trials include guselkumab, tildrakizumab, and risankizumab. In addition, certolizumab pegol, a TNF-α inhibitor, is being studied for use in psoriasis.
METHODS
We reviewed the published results of phase 3 clinical trials for ixekizumab, brodalumab, bimekizumab, guselkumab, tildrakizumab, risankizumab, and certolizumab pegol. We performed an English-language literature search (January 1, 2012 to October 15, 2017) of articles indexed for PubMed/MEDLINE using the following combinations of keywords: IL-23 and psoriasis; IL-17 and psoriasis; tumor necrosis factor and psoriasis; [drug name] and psoriasis. If data from phase 3 clinical trials were not yet available, data from phase 2 clinical trials were incorporated in our analysis. We also reviewed citations within articles to identify relevant sources.
RESULTS
Phase 3 clinical trial design, efficacy, and adverse events (AEs) for ixekizumab and brodalumab are reported in eTable 15-10 and for guselkumab and tildrakizumab in eTable 2.11-14 Phase 2 clinical trial design, efficacy, and AEs are presented for risankizumab in eTable 315-18 and for certolizumab pegol in eTable 4.17,19 No published clinical trial data were found for bimekizumab.
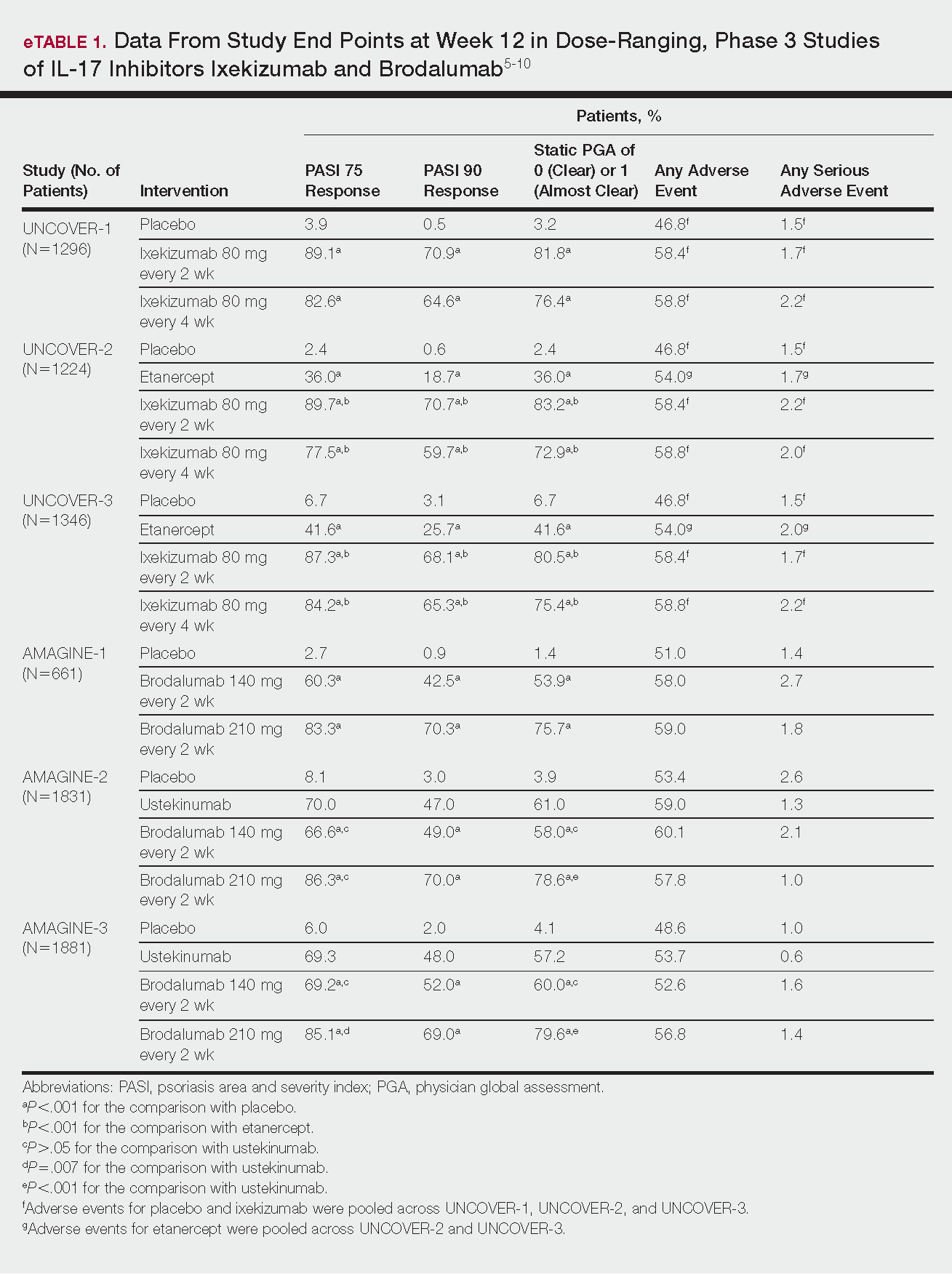
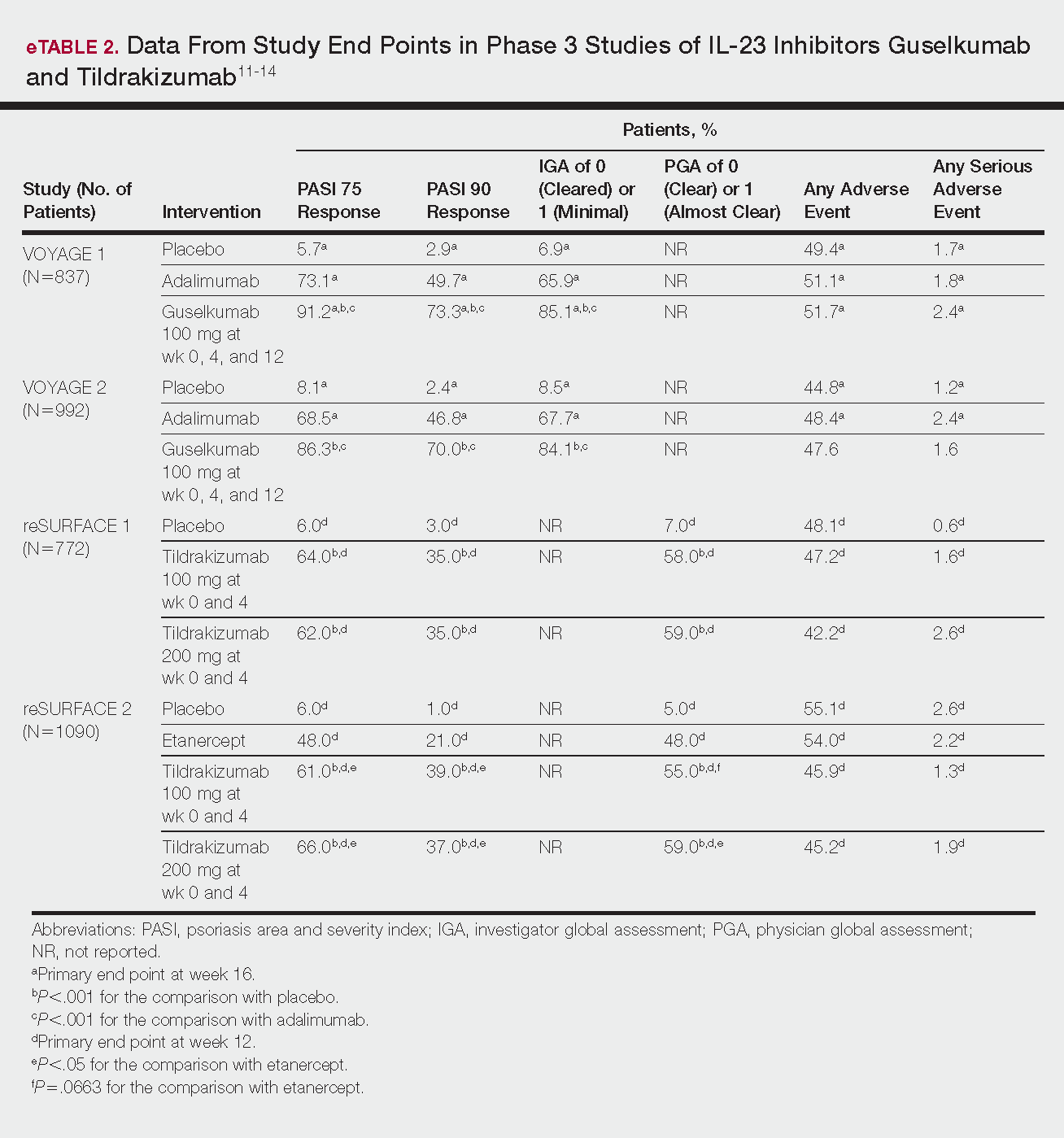
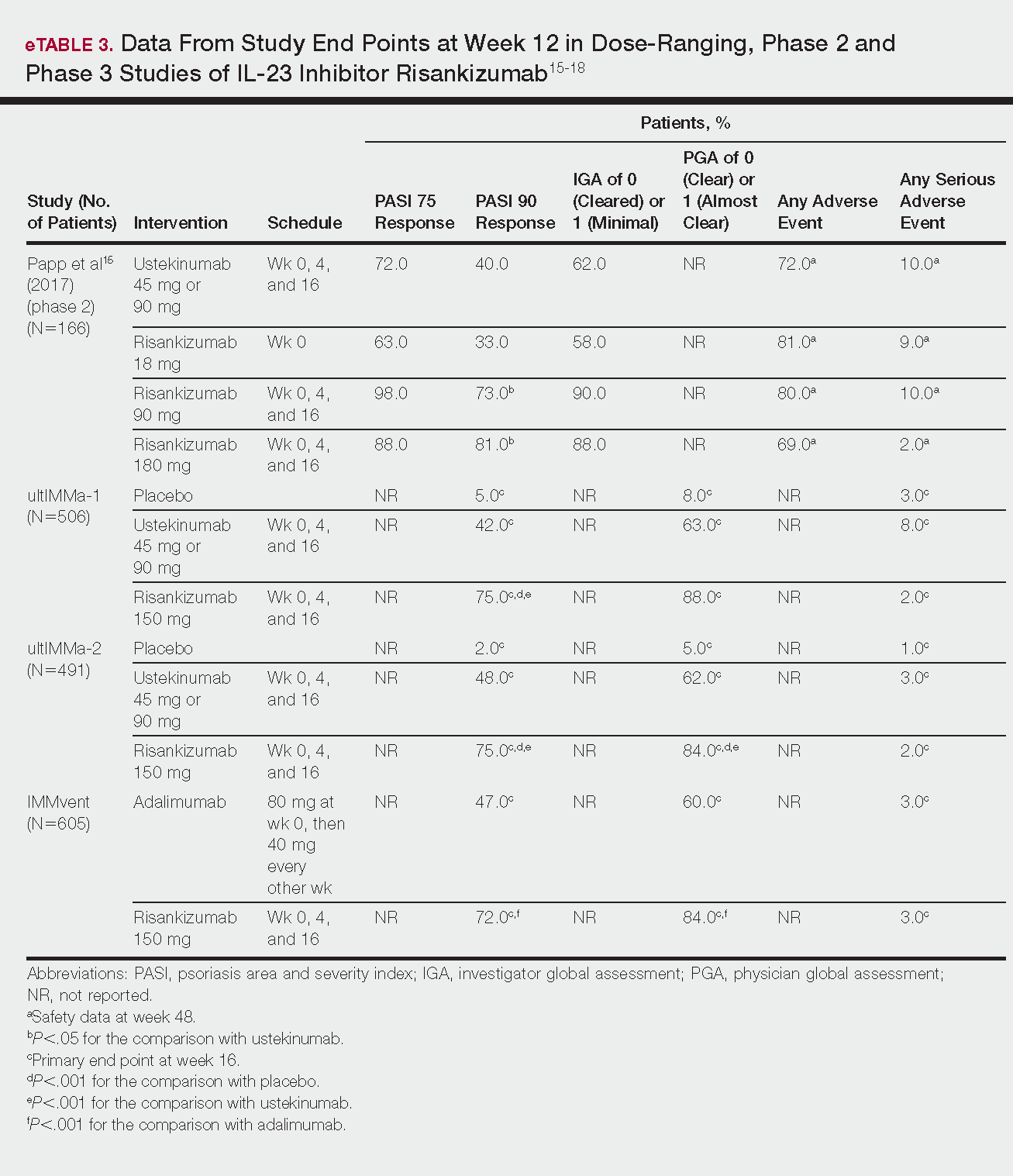
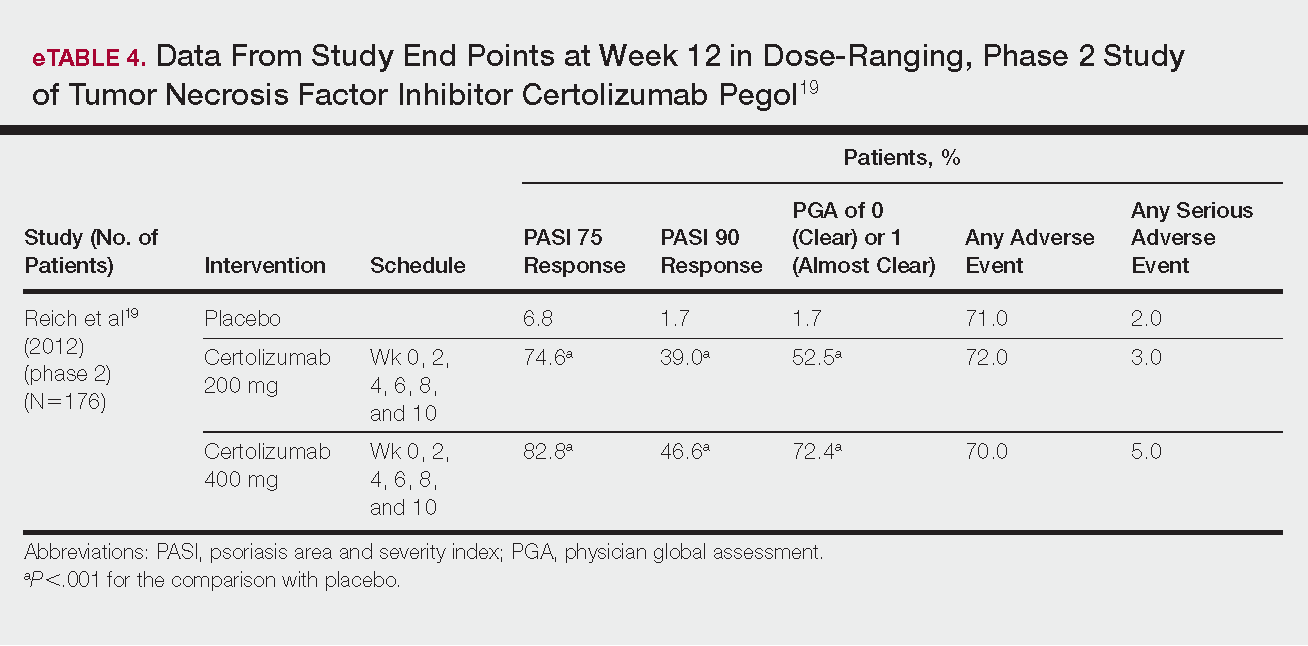
IL-17 Inhibitors
Ixekizumab
This recombinant, high-affinity IgG4κ antibody selectively binds and neutralizes IL-17A.5,6 Three phase 3 clinical trials—UNCOVER-1, UNCOVER-2, and UNCOVER-3—evaluated ixekizumab for moderate to severe plaque psoriasis.7
The 3 UNCOVER trials were randomized, double-blind, phase 3 trials of 1296, 1224, and 1346 patients, respectively, assigned to a placebo group; a group treated with ixekizumab 80 mg every 2 weeks; and a group treated with ixekizumab 80 mg every 4 weeks. Both ixekizumab groups received a loading dose of 160 mg at week 0.5,6 UNCOVER-2 and UNCOVER-3 also included a comparator group of patients on etanercept 50 mg.5 Co-primary end points included the percentage of patients reaching a psoriasis area and severity index (PASI) of 75 and with a static physician global assessment (PGA) score of clear (0) or almost clear (1) at week 12.5,6
Ixekizumab achieved greater efficacy than placebo: 89.1%, 89.7%, and 87.3% of patients achieved PASI 75 in the every 2-week dosing group, and 82.6%, 77.5% and 84.2% achieved PASI 75 in the every 4-week dosing group in UNCOVER-1, UNCOVER-2, and UNCOVER-3, respectively (P<.001 for both treatment arms compared to placebo in all trials). The percentage of patients achieving a static PGA score of 0 or 1 also was higher in the ixekizumab groups in the 2-week and 4-week dosing groups in all UNCOVER trials—81.8% and 76.4% in UNCOVER-1, 83.2% and 72.9% in UNCOVER-2, and 80.5% and 75.4% in UNCOVER-3—compared to 3.2%, 2.4%, and 6.7% in the placebo groups of the 3 trials (P<.001 for both ixekizumab groups compared to placebo in all trials).5,6 Ixekizumab also was found to be more effective than etanercept for both co-primary end points in both UNCOVER-2 and UNCOVER-3 (eTable 1).5
Safety data for all UNCOVER trials were pooled and reported.6 At week 12 the rate of at least 1 AE was 58.4% in patients on ixekizumab every 2 weeks and 58.8% in patients on ixekizumab every 4 weeks compared to 54.0% in the etanercept group in UNCOVER-2 and UNCOVER-3 and 46.8% in the placebo group. At week 12, 72 nonfatal serious AEs were reported: 12 in the placebo group, 14 in the etanercept group, 20 in the ixekizumab every 2 weeks group, and 26 in the ixekizumab every 4 weeks group.6
The most common AE across all groups was nasopharyngitis. Overall, infections were more frequent in patients treated with ixekizumab than in patients treated with placebo or etanercept. Specifically, oral candidiasis occurred more frequently in the ixekizumab groups, with a higher rate in the 2-week dosing group than in the 4-week dosing group.6 Two myocardial infarctions (MIs) occurred: 1 in the etanercept group and 1 in the placebo group.5
Brodalumab
This human monoclonal antibody binds to IL-17ra.8,9 Three double-blind, placebo-controlled, phase 3 trials—AMAGINE-1, AMAGINE-2, and AMAGINE-3—evaluated its use for plaque psoriasis.10
In AMAGINE-1 (N=661), patients were randomized to receive brodalumab 140 mg or 210 mg (every 2 weeks for 12 weeks), or placebo.8 In AMAGINE-2 (N=1831) and AMAGINE-3 (N=1881), patients were randomized to receive brodalumab 140 mg or 210 mg (every 2 weeks for 12 weeks), ustekinumab 45 mg or 90 mg by weight (at weeks 0 and 4, then every 12 weeks thereafter), or placebo. In all trials, patients on brodalumab received a dose at week 0 and week 1. Co-primary end points were PASI 75 and a static PGA score of 0 or 1 at 12 weeks compared to placebo and to ustekinumab (in AMAGINE-2 and AMAGINE-3 only).8
At week 12, 83.3%, 86.3%, and 85.1% of patients on brodalumab 210 mg, and 60.3%, 66.6%, and 69.2% of patients on brodalumab 140 mg, achieved PASI 75 in AMAGINE-1, AMAGINE-2, and AMAGINE-3, respectively, compared to 2.7%, 8.1%, and 6.0% in the placebo groups (P<.001 between both brodalumab groups and placebo in all trials).8 Both brodalumab groups were noninferior but not significantly superior to ustekinumab, which achieved a PASI 75 of 70.0% in AMAGINE-2 and 69.3% in AMAGINE-3. The PASI 90 rate was higher, however, in both brodalumab groups compared to ustekinumab but significance was not reported (eTable 1).9 For both brodalumab groups, significantly more patients achieved a static PGA value of 0 or 1 compared to placebo (P<.001 across all trials). However, only the brodalumab 210-mg group achieved a significantly higher rate of static PGA 0 or 1 compared to ustekinumab in AMAGINE-2 and AMAGINE-3 (P<.001).9
After 12 weeks, the percentage of patients reporting at least 1 AE was 59.0%, 57.8%, and 56.8% in the brodalumab 210-mg group in AMAGINE-1, AMAGINE-2, and AMAGINE-3, respectively; 58.0%, 60.1%, and 52.6% in the brodalumab 140-mg group; and 51.0%, 53.4%, and 48.6% in the placebo group. Patients taking ustekinumab had an AE rate of 59.0% in AMAGINE-2 and 53.7% in AMAGINE-3. The most common AE was nasopharyngitis, followed by upper respiratory infection (URI) and headache across all trials.8,9 Serious AEs were rare: 10 in AMAGINE-1, 31 in AMAGINE-2, and 24 in AMAGINE-3 across all groups. One death occurred from stroke in the brodalumab 210-mg group in AMAGINE-2.9
IL-23 Inhibitors
Guselkumab
This drug is a human IgG1κ antibody that binds to the p19 subunit of IL-23, thereby inhibiting IL-23 signaling.11,12 Guselkumab was approved by the FDA in July 2017 for moderate to severe plaque psoriasis.13
VOYAGE 1 and VOYAGE 2 were phase 3, double-blind, placebo- and active comparator–controlled trials of 837 and 992 patients, respectively, randomized to receive adalimumab (80 mg at week 0 and 40 mg at week 1, then at 40 mg every 2 weeks thereafter), guselkumab 100 mg at weeks 0, 4, and 12, or placebo.11 Co-primary end points for both trials were the percentage of patients reaching PASI 90 and an investigator global assessment (IGA) score of cleared (0) or minimal (1) at week 16.11
By week 16 of both trials, PASI 90 values were statistically superior for guselkumab (VOYAGE 1, 73.3%; VOYAGE 2, 70.0%) compared to adalimumab (VOYAGE 1, 49.7%; VOYAGE 2, 46.8%) and placebo (VOYAGE 1, 2.9%; VOYAGE 2, 2.4%)(P<.001). Moreover, patients on guselkumab achieved a higher rate of IGA values of 0 and 1 at week 12 (85.1% in VOYAGE 1 and 84.1% in VOYAGE 2) than patients on adalimumab (65.9% in VOYAGE 1 and 67.7% in VOYAGE 2) and placebo (6.9% in VOYAGE 1 and 8.5% in VOYAGE 2)(P<.001).11,12
The frequency of AEs was comparable across all groups in both trials.11,12 During the 16-week treatment period, 51.7% and 47.6% of the guselkumab groups in VOYAGE 1 and VOYAGE 2, respectively; 51.1% and 48.4% of the adalimumab groups; and 49.4% and 44.8% of the placebo groups reported at least 1 AE. The most common AEs in all groups were nasopharyngitis, headache, and URI.11,12
Serious AEs also occurred at similar rates: 2.4% and 1.6% in the guselkumab group in VOYAGE 1 and VOYAGE 2, respectively; 2.4% and 1.8% in the adalimumab group; and 1.7% and 1.2% in the placebo group.11,12 One case of malignancy occurred in the VOYAGE 1 trial: basal cell carcinoma in the guselkumab group.11 Three major cardiovascular events occurred across both trials: 1 MI in the guselkumab group in each trial and 1 MI in the adalimumab group in VOYAGE 1.11,12
Tildrakizumab
A high-affinity, humanized IgG1κ antibody, tildrakizumab targets the p19 subunit of IL-23. As of February 2018, 2 double-blind, randomized phase 3 trials have studied tildrakizumab with published results: reSURFACE 1 and reSURFACE 2.14
reSURFACE 1 (N=772) and reSURFACE 2 (N=1090) randomized patients to receive tildrakizumab 100 or 200 mg (at weeks 0 and 4), etanercept 50 mg (twice weekly) for 12 weeks (reSURFACE 2 only), or placebo. Co-primary end points were the percentage of patients achieving PASI 75 and the percentage of patients achieving a PGA score of 0 or 1 at week 12.14
In reSURFACE 1, significantly more patients receiving tildrakizumab attained PASI 75 at week 12 compared to placebo: 200 mg, 62.0%; 100 mg, 64.0%; and placebo, 6.0% (P<.001 for tildrakizumab groups compared to placebo). Moreover, significantly proportionally more patients received a PGA score of 0 or 1 compared to placebo: 100 mg, 59%; 200 mg, 58.0%; placebo, 7.0% (P<.001 for both tildrakizumab groups compared to placebo).14
In reSURFACE 2, significantly more patients receiving tildrakizumab achieved PASI 75 compared to etanercept and placebo at week 12: 200 mg, 66.0%; 100mg, 61.0%; etanercept, 48.0%; placebo, 6.0% (P<.001 for both tildrakizumab groups compared to placebo; P<.05 for both tildrakizumab groups compared to etanercept). Additionally, significantly more patients in the tildrakizumab groups experienced a PGA score of 0 or 1 at week 12 compared to placebo: 200 mg, 59%; 100 mg, 55.0%; placebo, 5% (P<.001 for both tildrakizumab groups compared to placebo).14
Adverse events were reported at a similar rate across all groups. For reSURFACE 1 and reSURFACE 2, at least 1 AE by week 12 was reported by 42.2% and 45.2% of patients in the 200-mg group; 47.2% and 45.9% in the 100-mg group; and 48.1% and 55.1% in the placebo groups.14The most common AEs were nasopharyngitis, URI (reSURFACE 1), and erythema at the injection site (reSURFACE 2). One case of serious infection was reported in each of the tildrakizumab groups: 1 case of drug-related hypersensitivity reaction in the 200-mg group, and 1 major cardiovascular event in the 100-mg group of reSURFACE 1. There was 1 serious AE in reSURFACE 2 that led to death in which the cause was undetermined.14
Risankizumab
This humanized IgG1 antibody binds the p19 unit of IL-23.15,16 The drug is undergoing 3 phase 3 trials—ultIMMa-1, ultIMMa-2, and IMMvent—for which only preliminary data have been published and are reported here.16,17 There is 1 phase 2 randomized, dose-ranging trial with published data.15
ultIMMa-1 and ultIMMa-2 comprised 506 and 491 patients, respectively, randomized to receive risankizumab (150 mg at weeks 0, 4, and 16), ustekinumab (45 mg or 90 mg, by weight, at weeks 0, 4, and 16), or placebo. Co-primary end points were PASI 90 and a PGA score of 0 or 1 at week 16.17
In ultIMMa-1 and ultIMMa-2, 75.0% and 75.0% of patients on risankizumab 150 mg achieved PASI 90 compared to 42.0% and 48.0% on ustekinumab and 5.0% and 2.0% on placebo at 16 weeks (P<.001 between both placebo and ustekinumab in both trials).17 In both trials, patients receiving risankizumab achieved higher rates of a static PGA score of 0 or 1 (88.0% and 84.0%) compared to ustekinumab (63.0% and 62.0%) and placebo (8.0% and 5.0%) at 16 weeks (P<.001 for both trials).18
At week 16, 2.0% of patients on risankizumab reported a serious AE in both trials, compared to 8.0% and 3.0% of patients on ustekinumab and 3.0% and 1.0% on placebo. No new safety concerns were noted.17
In the phase 3 IMMvent trial, 605 patients were randomized to receive risankizumab (150 mg at weeks 0, 4, and 16) or adalimumab (80 mg at week 0, 40 mg at week 1, then 40 mg every 2 weeks). Co-primary end points were PASI 90 and a static PGA score of 0 or 1 at week 16.17
In IMMvent, risankizumab was significantly more effective than adalimumab for PASI 75 (risankizumab, 72.0%; adalimumab, 47.0%) and a static PGA score of 0 or 1 (risankizumab 84.0%; adalimumab, 60.0%) (P<.001 risankizumab compared to adalimumab for both end points).17
At week 16, serious AEs were reported in 3.0% of patients on risankizumab and 3.0% of patients on adalimumab. One patient receiving risankizumab died of an acute MI during the treatment phase.17
TNF Inhibitor
Certolizumab Pegol
Certolizumab pegol is a human PEGylated anti-TNF agent. In vitro studies have shown that certolizumab binds to soluble and membrane-bound TNF.19 Unlike other TNF inhibitors, certolizumab pegol is a Fab‘ portion of anti-TNF conjugated to a molecule of polyethylene glycol.19 The drug is approved in the United States for treating psoriatic arthritis, Crohn disease, and rheumatoid arthritis; its potential for treating psoriasis has been confirmed. Results of 1 phase 2 trial have been published19; data from 3 phase 3 trials are forthcoming.
This randomized, placebo-controlled, double-blind phase 2 study comprised 176 patients who received certolizumab 200 mg, certolizumab 400 mg, or placebo. The dosing schedule was 400 mg at week 0, followed by either 200 or 400 mg every other week until week 10. Co-primary end points were PASI 75 and a PGA score of 0 or 1 at week 12.19
Certolizumab was significantly more effective than placebo at week 12: 74.6% of the 200-mg group and 82.8% of the 400-mg group achieved PASI 75 compared to 6.8% of the placebo group (P<.001). Certolizumab also performed better for the PGA score: 52.5% and 72.4% of patients attained a score of 0 or 1 in the 200-mg and 400-mg groups compared to 1.7% in the placebo group.19
Adverse events were reported equally across all groups: 72% of patients in the 200-mg group, 70% in the 400-mg group, and 71% in the placebo group reported at least 1 AE, most commonly nasopharyngitis, headache, and pruritis.19
COMMENT
With the development of new insights into the pathogenesis of psoriasis, therapies that are targeted toward key cytokines may contribute to improved management of the disease. The results of these clinical trials demonstrate numerous promising options for psoriatic patients.
IL-17 Inhibitors Ixekizumab and Brodalumab
When comparing these 2 biologics, it is important to consider that these studies were not performed head to head, thereby inhibiting direct comparisons. Moreover, dosage ranges of the investigative drugs were not identical, which also makes comparisons challenging. However, when looking at the highest dosages of ixekizumab and brodalumab, results indicate that ixekizumab may be slightly more effective than brodalumab based on the percentage of patients who achieved a PASI 75 and a static PGA score of 0 or 1 (eTable 1).
Phase 3 trials have shown ixekizumab to maintain efficacy over 60 weeks of treatment.6 Ixekizumab also has been shown to alleviate other symptoms of psoriasis, such as itching, pain, and nail involvement.20,21 Furthermore, ixekizumab appears to be equally effective in patients with or without prior exposure to biologics22; therefore, ixekizumab may benefit patients who have not experienced success with other biologics.
Across the UNCOVER trials, 11 cases of inflammatory bowel disease were reported in patients receiving ixekizumab (ulcerative colitis in 7; Crohn disease in 4)6; it appears that at least 3 of these cases were new diagnoses. In light of a study suggesting that IL-17A might have a protective function in the intestine,23 these findings may have important clinical implications and require follow-up studies.
Brodalumab also has been shown to maintain efficacy and acceptable safety for as long as 120 weeks.24 In the extension period of the AMAGINE-1 trial, patients who experienced a return of disease during a withdrawal period recaptured static PGA success with re-treatment for 12 weeks (re-treatment was successful in 97% of those given a dosage of 210 mg and in 84% of those given 140 mg).8
Furthermore, phase 2 trials also have shown that brodalumab is effective in patients with a history of biologic use.25 Across all AMAGINE trials, only 1 case of Crohn disease was reported in a patient taking brodalumab.9 There are concerns about depression, despite data from AMAGINE-1 stating patients on brodalumab actually had greater improvements in Hospital Anxiety and Depression Scale scores after 12 weeks of treatment (P<.001) for both brodalumab 140 mg and 210 mg compared to placebo.8 Regardless, brodalumab has a black-box warning for suicidal ideation and behavior, and availability is restricted through a Risk Evaluation and Mitigation Strategy (REMS) program.26
Bimekizumab
Although no phase 2 or phase 3 clinical trial data have been published for bimekizumab (phase 2 trials are underway), it has been shown in a phase 1 trial to be effective for psoriasis. Bimekizumab also is unique; it is the first dual inhibitor of IL-17A and IL-17F.18
IL-23 Inhibitors Guselkumab, Tildrakizumab, and Risankizumab
Making comparisons among the IL-23 inhibitors also is difficult; studies were not head-to-head comparison trials, and the VOYAGE and reSURFACE studies used different time points for primary end points. Furthermore, only phase 2 trial data are available for risankizumab. Despite these limitations, results of these trials suggest that guselkumab and risankizumab may be slightly more efficacious than tildrakizumab. However, future studies, including head-to-head studies, would ultimately provide further information on how these agents compare.
Guselkumab was shown to remain efficacious at 48 weeks, though patients on maintenance dosing had better results than those who were re-treated.12 Moreover, guselkumab was found to be effective in hard-to-treat areas, such as the scalp,11 and in patients who did not respond to adalimumab. Guselkumab may therefore benefit patients who have experienced limited clinical improvement on other biologics.12
Tildrakizumab was shown to improve PASI 75 and PGA scores through week 28 of treatment. Moreover, a higher percentage of patients taking tildrakizumab scored 0 or 1 on the dermatology life quality index, suggesting that the drug improves quality of life.14 No specific safety concerns arose in either reSURFACE trial; however, long-term studies are needed for further evaluation.
Risankizumab appears to be a promising new therapy based on phase 2 trial results. Improvements also were seen in dermatology life quality index scores, scalp and fingernail symptoms, and palmoplantar psoriasis.15 Of note, neutralizing antidrug antibodies were found in 3 patients during this study,15 which may present potential problems for long-term efficacy. However, preliminary data from 3 phase 3 trials—ultIMMa-1, ultIMMa-2, and IMMvent—are promising.17
CONCLUSION
Advances in the understanding of psoriasis have led to new targeted therapies. Ongoing clinical trials have shown encouraging results for treating physical and psychological symptoms of psoriasis. The findings of these trials support the idea that therapies targeting IL-23, specifically its p19 subunit, are effective against psoriasis while sparing IL-12. Long-term data from open-label extension studies would help guide clinical recommendations regarding the safety profiles of these agents and determine their long-term utility.
- Langley RG, Krueger GG, Griffiths CE. Psoriasis: epidemiology, clinical features, and quality of life. Ann Rheum Dis. 2005;64(suppl 2):ii18-ii23; discussion, ii24, ii25.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Amin M, Darji K, No DJ, et al. Review of phase III trial data on IL-23 inhibitors tildrakizumab and guselkumab for psoriasis. J Eur Acad Dermatol Venereol. 2017;31:1627-1632.
- Arican O, Aral M, Sasmaz S, et al. Levels of TNF-alpha, IFN-gamma, IL6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. 2005:273-279.
- Griffiths CE, Reich K, Lebwohl M, et al; UNCOVER-2 and UNCOVER-3 investigators. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386:541-551.
- Gordon KB, Blauvelt A, Papp KA, et al; UNCOVER-1 study group, UNCOVER-2 study group, UNCOVER-3 study group. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345-356.
- FDA approves new psoriasis drug Taltz [news release]. Silver Spring, MD: US Food and Drug Administration; March 22, 2016. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm491872.htm. Accessed January 29, 2018.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016;175:273-286.
- Lebwohl M, Strober B, Mentor A, et al. Phase 3 studies comparing brodalumab with ustekinumab for psoriasis. N Engl J Med. 2015;373:1318-1328.
- FDA approves new psoriasis drug [news release]. Silver Spring, MD: US Food and Drug Administration; February 15, 2017. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm541981.htm. Accessed January 29, 2018.
- Blauvelt A, Papp KA, Griffiths CE, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate-to-severe plaque psoriasis: results from the phase III, double-blinded placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76:405-417.
- Reich K, Armstrong AW, Foley P, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J Am Acad Dermatol. 2017;76:418-431.
- Janssen announces U.S. FDA approval of Tremfya™ (guselkumab) for the treatment of moderate to severe plaque psoriasis [news release]. Horsham, PA: Johnson & Johnson; July 13, 2017. https://www.jnj.com/media-center/press-releases/janssen-announces-us-fda-approval-of-tremfya-guselkumab-for-the-treatment-of-moderate-to-severe-plaque-psoriasis. Accessed January 29, 2018.
- Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE1 and reSURFACE 2): results from two randomized controlled, phase 3 trials. Lancet. 2017;390:276-288.
- Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376:1551-1560.
- Risankizumab. AbbVie Inc website. https://www.abbvie.com/our-science/pipeline/risankizumab.html. Accessed January 29, 2018.
- Risankizumab meets all co-primary and ranked secondary endpoints, achieving significantly greater efficacy versus standard biologic therapies in three pivotal phase 3 psoriasis studies [news release]. North Chicago, IL: AbbVie Inc; October 26, 2017. https://news.abbvie.com/news/risankizumab-meets-all-co-primary-and-ranked-secondary-endpoints-achieving-significantly-greater-efficacy-versus-standard-biologic-therapies-in-three-pivotal-phase-3-psoriasis-studies.htm. Accessed January 29, 2018.
- Glatt S, Helmer E, Haier B, et al. First-in-human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL-17A and IL-17F, in mild psoriasis. Br J Clin Pharmacol. 2017;83:991-1001.
- Reich K, Ortonne JP, Gottlieb AB, et al. Successful treatment of moderate to severe plaque psoriasis with the PEGylated Fab‘ certolizumab pegol: results of a phase II randomized, placebo-controlled trial with a re-treatment extension. Br J Dermatol. 2012;167:180-190.
- Kimball AB, Luger T, Gottlieb A, et al. Impact of ixekizumab on psoriasis itch severity and other psoriasis symptoms: results from 3 phase III psoriasis clinical trials. J Am Acad Dermatol. 2016;75:1156-1161.
- Dennehy EB, Zhang L, Amato D, et al. Ixekizumab is effective in subjects with moderate to severe plaque psoriasis with significant nail involvement: results from UNCOVER 3. J Drugs Dermatol. 2016;15:958-961.
- Gottlieb AB, Lacour JP, Korman N, et al. Treatment outcomes with ixekizumab in patients with moderate-to-severe psoriasis who have not received prior biological therapies: an integrated analysis of two phase III randomized studies. J Eur Acad Dermatol Venereol. 2017;31:679-685.
- Hueber W, Sands BE, Lewitsky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693-1700.
- Papp K, Leonardi C, Menter A, et al. Safety and efficacy of brodalumab for psoriasis after 120 weeks of treatment. J Am Acad Dermatol. 2014;71:1183-1190.
- Papp K, Menter A, Strober B, et al. Efficacy and safety of brodalumab in subpopulations of patients with difficult-to-treat moderate-to-severe plaque psoriasis. J Am Acad Dermatol. 2015;72:436-439.
- SILIQ [package insert]. Thousand Oaks, CA: Amgen, Inc; 2017.
Psoriasis is a chronic, autoimmune-mediated disease estimated to affect 2.8% of the US population.1 The pathogenesis of psoriasis is thought to involve a complex process triggered by a combination of genetic and environmental factors that induce tumor necrosis factor (TNF) α secretion by keratinocytes, which in turn activates dendritic cells. Activated dendritic cells produce IL-23, leading to helper T cell (TH17) differentiation.2,3 TH17 cells secrete IL-17A, which has been shown to promote psoriatic skin changes.4 Therefore, TNF-α, IL-23, and IL-17A have been recognized as key targets for psoriasis therapy.
The newest biologic agents targeting IL-17–mediated pathways include ixekizumab, brodalumab, and bimekizumab. Secukinumab, the first US Food and Drug Administration (FDA)–approved IL-17 inhibitor, has been available since 2015 and therefore is not included in this review. IL-23 inhibitors that are FDA approved or being evaluated in clinical trials include guselkumab, tildrakizumab, and risankizumab. In addition, certolizumab pegol, a TNF-α inhibitor, is being studied for use in psoriasis.
METHODS
We reviewed the published results of phase 3 clinical trials for ixekizumab, brodalumab, bimekizumab, guselkumab, tildrakizumab, risankizumab, and certolizumab pegol. We performed an English-language literature search (January 1, 2012 to October 15, 2017) of articles indexed for PubMed/MEDLINE using the following combinations of keywords: IL-23 and psoriasis; IL-17 and psoriasis; tumor necrosis factor and psoriasis; [drug name] and psoriasis. If data from phase 3 clinical trials were not yet available, data from phase 2 clinical trials were incorporated in our analysis. We also reviewed citations within articles to identify relevant sources.
RESULTS
Phase 3 clinical trial design, efficacy, and adverse events (AEs) for ixekizumab and brodalumab are reported in eTable 15-10 and for guselkumab and tildrakizumab in eTable 2.11-14 Phase 2 clinical trial design, efficacy, and AEs are presented for risankizumab in eTable 315-18 and for certolizumab pegol in eTable 4.17,19 No published clinical trial data were found for bimekizumab.
IL-17 Inhibitors
Ixekizumab
This recombinant, high-affinity IgG4κ antibody selectively binds and neutralizes IL-17A.5,6 Three phase 3 clinical trials—UNCOVER-1, UNCOVER-2, and UNCOVER-3—evaluated ixekizumab for moderate to severe plaque psoriasis.7
The 3 UNCOVER trials were randomized, double-blind, phase 3 trials of 1296, 1224, and 1346 patients, respectively, assigned to a placebo group; a group treated with ixekizumab 80 mg every 2 weeks; and a group treated with ixekizumab 80 mg every 4 weeks. Both ixekizumab groups received a loading dose of 160 mg at week 0.5,6 UNCOVER-2 and UNCOVER-3 also included a comparator group of patients on etanercept 50 mg.5 Co-primary end points included the percentage of patients reaching a psoriasis area and severity index (PASI) of 75 and with a static physician global assessment (PGA) score of clear (0) or almost clear (1) at week 12.5,6
Ixekizumab achieved greater efficacy than placebo: 89.1%, 89.7%, and 87.3% of patients achieved PASI 75 in the every 2-week dosing group, and 82.6%, 77.5% and 84.2% achieved PASI 75 in the every 4-week dosing group in UNCOVER-1, UNCOVER-2, and UNCOVER-3, respectively (P<.001 for both treatment arms compared to placebo in all trials). The percentage of patients achieving a static PGA score of 0 or 1 also was higher in the ixekizumab groups in the 2-week and 4-week dosing groups in all UNCOVER trials—81.8% and 76.4% in UNCOVER-1, 83.2% and 72.9% in UNCOVER-2, and 80.5% and 75.4% in UNCOVER-3—compared to 3.2%, 2.4%, and 6.7% in the placebo groups of the 3 trials (P<.001 for both ixekizumab groups compared to placebo in all trials).5,6 Ixekizumab also was found to be more effective than etanercept for both co-primary end points in both UNCOVER-2 and UNCOVER-3 (eTable 1).5
Safety data for all UNCOVER trials were pooled and reported.6 At week 12 the rate of at least 1 AE was 58.4% in patients on ixekizumab every 2 weeks and 58.8% in patients on ixekizumab every 4 weeks compared to 54.0% in the etanercept group in UNCOVER-2 and UNCOVER-3 and 46.8% in the placebo group. At week 12, 72 nonfatal serious AEs were reported: 12 in the placebo group, 14 in the etanercept group, 20 in the ixekizumab every 2 weeks group, and 26 in the ixekizumab every 4 weeks group.6
The most common AE across all groups was nasopharyngitis. Overall, infections were more frequent in patients treated with ixekizumab than in patients treated with placebo or etanercept. Specifically, oral candidiasis occurred more frequently in the ixekizumab groups, with a higher rate in the 2-week dosing group than in the 4-week dosing group.6 Two myocardial infarctions (MIs) occurred: 1 in the etanercept group and 1 in the placebo group.5
Brodalumab
This human monoclonal antibody binds to IL-17ra.8,9 Three double-blind, placebo-controlled, phase 3 trials—AMAGINE-1, AMAGINE-2, and AMAGINE-3—evaluated its use for plaque psoriasis.10
In AMAGINE-1 (N=661), patients were randomized to receive brodalumab 140 mg or 210 mg (every 2 weeks for 12 weeks), or placebo.8 In AMAGINE-2 (N=1831) and AMAGINE-3 (N=1881), patients were randomized to receive brodalumab 140 mg or 210 mg (every 2 weeks for 12 weeks), ustekinumab 45 mg or 90 mg by weight (at weeks 0 and 4, then every 12 weeks thereafter), or placebo. In all trials, patients on brodalumab received a dose at week 0 and week 1. Co-primary end points were PASI 75 and a static PGA score of 0 or 1 at 12 weeks compared to placebo and to ustekinumab (in AMAGINE-2 and AMAGINE-3 only).8
At week 12, 83.3%, 86.3%, and 85.1% of patients on brodalumab 210 mg, and 60.3%, 66.6%, and 69.2% of patients on brodalumab 140 mg, achieved PASI 75 in AMAGINE-1, AMAGINE-2, and AMAGINE-3, respectively, compared to 2.7%, 8.1%, and 6.0% in the placebo groups (P<.001 between both brodalumab groups and placebo in all trials).8 Both brodalumab groups were noninferior but not significantly superior to ustekinumab, which achieved a PASI 75 of 70.0% in AMAGINE-2 and 69.3% in AMAGINE-3. The PASI 90 rate was higher, however, in both brodalumab groups compared to ustekinumab but significance was not reported (eTable 1).9 For both brodalumab groups, significantly more patients achieved a static PGA value of 0 or 1 compared to placebo (P<.001 across all trials). However, only the brodalumab 210-mg group achieved a significantly higher rate of static PGA 0 or 1 compared to ustekinumab in AMAGINE-2 and AMAGINE-3 (P<.001).9
After 12 weeks, the percentage of patients reporting at least 1 AE was 59.0%, 57.8%, and 56.8% in the brodalumab 210-mg group in AMAGINE-1, AMAGINE-2, and AMAGINE-3, respectively; 58.0%, 60.1%, and 52.6% in the brodalumab 140-mg group; and 51.0%, 53.4%, and 48.6% in the placebo group. Patients taking ustekinumab had an AE rate of 59.0% in AMAGINE-2 and 53.7% in AMAGINE-3. The most common AE was nasopharyngitis, followed by upper respiratory infection (URI) and headache across all trials.8,9 Serious AEs were rare: 10 in AMAGINE-1, 31 in AMAGINE-2, and 24 in AMAGINE-3 across all groups. One death occurred from stroke in the brodalumab 210-mg group in AMAGINE-2.9
IL-23 Inhibitors
Guselkumab
This drug is a human IgG1κ antibody that binds to the p19 subunit of IL-23, thereby inhibiting IL-23 signaling.11,12 Guselkumab was approved by the FDA in July 2017 for moderate to severe plaque psoriasis.13
VOYAGE 1 and VOYAGE 2 were phase 3, double-blind, placebo- and active comparator–controlled trials of 837 and 992 patients, respectively, randomized to receive adalimumab (80 mg at week 0 and 40 mg at week 1, then at 40 mg every 2 weeks thereafter), guselkumab 100 mg at weeks 0, 4, and 12, or placebo.11 Co-primary end points for both trials were the percentage of patients reaching PASI 90 and an investigator global assessment (IGA) score of cleared (0) or minimal (1) at week 16.11
By week 16 of both trials, PASI 90 values were statistically superior for guselkumab (VOYAGE 1, 73.3%; VOYAGE 2, 70.0%) compared to adalimumab (VOYAGE 1, 49.7%; VOYAGE 2, 46.8%) and placebo (VOYAGE 1, 2.9%; VOYAGE 2, 2.4%)(P<.001). Moreover, patients on guselkumab achieved a higher rate of IGA values of 0 and 1 at week 12 (85.1% in VOYAGE 1 and 84.1% in VOYAGE 2) than patients on adalimumab (65.9% in VOYAGE 1 and 67.7% in VOYAGE 2) and placebo (6.9% in VOYAGE 1 and 8.5% in VOYAGE 2)(P<.001).11,12
The frequency of AEs was comparable across all groups in both trials.11,12 During the 16-week treatment period, 51.7% and 47.6% of the guselkumab groups in VOYAGE 1 and VOYAGE 2, respectively; 51.1% and 48.4% of the adalimumab groups; and 49.4% and 44.8% of the placebo groups reported at least 1 AE. The most common AEs in all groups were nasopharyngitis, headache, and URI.11,12
Serious AEs also occurred at similar rates: 2.4% and 1.6% in the guselkumab group in VOYAGE 1 and VOYAGE 2, respectively; 2.4% and 1.8% in the adalimumab group; and 1.7% and 1.2% in the placebo group.11,12 One case of malignancy occurred in the VOYAGE 1 trial: basal cell carcinoma in the guselkumab group.11 Three major cardiovascular events occurred across both trials: 1 MI in the guselkumab group in each trial and 1 MI in the adalimumab group in VOYAGE 1.11,12
Tildrakizumab
A high-affinity, humanized IgG1κ antibody, tildrakizumab targets the p19 subunit of IL-23. As of February 2018, 2 double-blind, randomized phase 3 trials have studied tildrakizumab with published results: reSURFACE 1 and reSURFACE 2.14
reSURFACE 1 (N=772) and reSURFACE 2 (N=1090) randomized patients to receive tildrakizumab 100 or 200 mg (at weeks 0 and 4), etanercept 50 mg (twice weekly) for 12 weeks (reSURFACE 2 only), or placebo. Co-primary end points were the percentage of patients achieving PASI 75 and the percentage of patients achieving a PGA score of 0 or 1 at week 12.14
In reSURFACE 1, significantly more patients receiving tildrakizumab attained PASI 75 at week 12 compared to placebo: 200 mg, 62.0%; 100 mg, 64.0%; and placebo, 6.0% (P<.001 for tildrakizumab groups compared to placebo). Moreover, significantly proportionally more patients received a PGA score of 0 or 1 compared to placebo: 100 mg, 59%; 200 mg, 58.0%; placebo, 7.0% (P<.001 for both tildrakizumab groups compared to placebo).14
In reSURFACE 2, significantly more patients receiving tildrakizumab achieved PASI 75 compared to etanercept and placebo at week 12: 200 mg, 66.0%; 100mg, 61.0%; etanercept, 48.0%; placebo, 6.0% (P<.001 for both tildrakizumab groups compared to placebo; P<.05 for both tildrakizumab groups compared to etanercept). Additionally, significantly more patients in the tildrakizumab groups experienced a PGA score of 0 or 1 at week 12 compared to placebo: 200 mg, 59%; 100 mg, 55.0%; placebo, 5% (P<.001 for both tildrakizumab groups compared to placebo).14
Adverse events were reported at a similar rate across all groups. For reSURFACE 1 and reSURFACE 2, at least 1 AE by week 12 was reported by 42.2% and 45.2% of patients in the 200-mg group; 47.2% and 45.9% in the 100-mg group; and 48.1% and 55.1% in the placebo groups.14The most common AEs were nasopharyngitis, URI (reSURFACE 1), and erythema at the injection site (reSURFACE 2). One case of serious infection was reported in each of the tildrakizumab groups: 1 case of drug-related hypersensitivity reaction in the 200-mg group, and 1 major cardiovascular event in the 100-mg group of reSURFACE 1. There was 1 serious AE in reSURFACE 2 that led to death in which the cause was undetermined.14
Risankizumab
This humanized IgG1 antibody binds the p19 unit of IL-23.15,16 The drug is undergoing 3 phase 3 trials—ultIMMa-1, ultIMMa-2, and IMMvent—for which only preliminary data have been published and are reported here.16,17 There is 1 phase 2 randomized, dose-ranging trial with published data.15
ultIMMa-1 and ultIMMa-2 comprised 506 and 491 patients, respectively, randomized to receive risankizumab (150 mg at weeks 0, 4, and 16), ustekinumab (45 mg or 90 mg, by weight, at weeks 0, 4, and 16), or placebo. Co-primary end points were PASI 90 and a PGA score of 0 or 1 at week 16.17
In ultIMMa-1 and ultIMMa-2, 75.0% and 75.0% of patients on risankizumab 150 mg achieved PASI 90 compared to 42.0% and 48.0% on ustekinumab and 5.0% and 2.0% on placebo at 16 weeks (P<.001 between both placebo and ustekinumab in both trials).17 In both trials, patients receiving risankizumab achieved higher rates of a static PGA score of 0 or 1 (88.0% and 84.0%) compared to ustekinumab (63.0% and 62.0%) and placebo (8.0% and 5.0%) at 16 weeks (P<.001 for both trials).18
At week 16, 2.0% of patients on risankizumab reported a serious AE in both trials, compared to 8.0% and 3.0% of patients on ustekinumab and 3.0% and 1.0% on placebo. No new safety concerns were noted.17
In the phase 3 IMMvent trial, 605 patients were randomized to receive risankizumab (150 mg at weeks 0, 4, and 16) or adalimumab (80 mg at week 0, 40 mg at week 1, then 40 mg every 2 weeks). Co-primary end points were PASI 90 and a static PGA score of 0 or 1 at week 16.17
In IMMvent, risankizumab was significantly more effective than adalimumab for PASI 75 (risankizumab, 72.0%; adalimumab, 47.0%) and a static PGA score of 0 or 1 (risankizumab 84.0%; adalimumab, 60.0%) (P<.001 risankizumab compared to adalimumab for both end points).17
At week 16, serious AEs were reported in 3.0% of patients on risankizumab and 3.0% of patients on adalimumab. One patient receiving risankizumab died of an acute MI during the treatment phase.17
TNF Inhibitor
Certolizumab Pegol
Certolizumab pegol is a human PEGylated anti-TNF agent. In vitro studies have shown that certolizumab binds to soluble and membrane-bound TNF.19 Unlike other TNF inhibitors, certolizumab pegol is a Fab‘ portion of anti-TNF conjugated to a molecule of polyethylene glycol.19 The drug is approved in the United States for treating psoriatic arthritis, Crohn disease, and rheumatoid arthritis; its potential for treating psoriasis has been confirmed. Results of 1 phase 2 trial have been published19; data from 3 phase 3 trials are forthcoming.
This randomized, placebo-controlled, double-blind phase 2 study comprised 176 patients who received certolizumab 200 mg, certolizumab 400 mg, or placebo. The dosing schedule was 400 mg at week 0, followed by either 200 or 400 mg every other week until week 10. Co-primary end points were PASI 75 and a PGA score of 0 or 1 at week 12.19
Certolizumab was significantly more effective than placebo at week 12: 74.6% of the 200-mg group and 82.8% of the 400-mg group achieved PASI 75 compared to 6.8% of the placebo group (P<.001). Certolizumab also performed better for the PGA score: 52.5% and 72.4% of patients attained a score of 0 or 1 in the 200-mg and 400-mg groups compared to 1.7% in the placebo group.19
Adverse events were reported equally across all groups: 72% of patients in the 200-mg group, 70% in the 400-mg group, and 71% in the placebo group reported at least 1 AE, most commonly nasopharyngitis, headache, and pruritis.19
COMMENT
With the development of new insights into the pathogenesis of psoriasis, therapies that are targeted toward key cytokines may contribute to improved management of the disease. The results of these clinical trials demonstrate numerous promising options for psoriatic patients.
IL-17 Inhibitors Ixekizumab and Brodalumab
When comparing these 2 biologics, it is important to consider that these studies were not performed head to head, thereby inhibiting direct comparisons. Moreover, dosage ranges of the investigative drugs were not identical, which also makes comparisons challenging. However, when looking at the highest dosages of ixekizumab and brodalumab, results indicate that ixekizumab may be slightly more effective than brodalumab based on the percentage of patients who achieved a PASI 75 and a static PGA score of 0 or 1 (eTable 1).
Phase 3 trials have shown ixekizumab to maintain efficacy over 60 weeks of treatment.6 Ixekizumab also has been shown to alleviate other symptoms of psoriasis, such as itching, pain, and nail involvement.20,21 Furthermore, ixekizumab appears to be equally effective in patients with or without prior exposure to biologics22; therefore, ixekizumab may benefit patients who have not experienced success with other biologics.
Across the UNCOVER trials, 11 cases of inflammatory bowel disease were reported in patients receiving ixekizumab (ulcerative colitis in 7; Crohn disease in 4)6; it appears that at least 3 of these cases were new diagnoses. In light of a study suggesting that IL-17A might have a protective function in the intestine,23 these findings may have important clinical implications and require follow-up studies.
Brodalumab also has been shown to maintain efficacy and acceptable safety for as long as 120 weeks.24 In the extension period of the AMAGINE-1 trial, patients who experienced a return of disease during a withdrawal period recaptured static PGA success with re-treatment for 12 weeks (re-treatment was successful in 97% of those given a dosage of 210 mg and in 84% of those given 140 mg).8
Furthermore, phase 2 trials also have shown that brodalumab is effective in patients with a history of biologic use.25 Across all AMAGINE trials, only 1 case of Crohn disease was reported in a patient taking brodalumab.9 There are concerns about depression, despite data from AMAGINE-1 stating patients on brodalumab actually had greater improvements in Hospital Anxiety and Depression Scale scores after 12 weeks of treatment (P<.001) for both brodalumab 140 mg and 210 mg compared to placebo.8 Regardless, brodalumab has a black-box warning for suicidal ideation and behavior, and availability is restricted through a Risk Evaluation and Mitigation Strategy (REMS) program.26
Bimekizumab
Although no phase 2 or phase 3 clinical trial data have been published for bimekizumab (phase 2 trials are underway), it has been shown in a phase 1 trial to be effective for psoriasis. Bimekizumab also is unique; it is the first dual inhibitor of IL-17A and IL-17F.18
IL-23 Inhibitors Guselkumab, Tildrakizumab, and Risankizumab
Making comparisons among the IL-23 inhibitors also is difficult; studies were not head-to-head comparison trials, and the VOYAGE and reSURFACE studies used different time points for primary end points. Furthermore, only phase 2 trial data are available for risankizumab. Despite these limitations, results of these trials suggest that guselkumab and risankizumab may be slightly more efficacious than tildrakizumab. However, future studies, including head-to-head studies, would ultimately provide further information on how these agents compare.
Guselkumab was shown to remain efficacious at 48 weeks, though patients on maintenance dosing had better results than those who were re-treated.12 Moreover, guselkumab was found to be effective in hard-to-treat areas, such as the scalp,11 and in patients who did not respond to adalimumab. Guselkumab may therefore benefit patients who have experienced limited clinical improvement on other biologics.12
Tildrakizumab was shown to improve PASI 75 and PGA scores through week 28 of treatment. Moreover, a higher percentage of patients taking tildrakizumab scored 0 or 1 on the dermatology life quality index, suggesting that the drug improves quality of life.14 No specific safety concerns arose in either reSURFACE trial; however, long-term studies are needed for further evaluation.
Risankizumab appears to be a promising new therapy based on phase 2 trial results. Improvements also were seen in dermatology life quality index scores, scalp and fingernail symptoms, and palmoplantar psoriasis.15 Of note, neutralizing antidrug antibodies were found in 3 patients during this study,15 which may present potential problems for long-term efficacy. However, preliminary data from 3 phase 3 trials—ultIMMa-1, ultIMMa-2, and IMMvent—are promising.17
CONCLUSION
Advances in the understanding of psoriasis have led to new targeted therapies. Ongoing clinical trials have shown encouraging results for treating physical and psychological symptoms of psoriasis. The findings of these trials support the idea that therapies targeting IL-23, specifically its p19 subunit, are effective against psoriasis while sparing IL-12. Long-term data from open-label extension studies would help guide clinical recommendations regarding the safety profiles of these agents and determine their long-term utility.
Psoriasis is a chronic, autoimmune-mediated disease estimated to affect 2.8% of the US population.1 The pathogenesis of psoriasis is thought to involve a complex process triggered by a combination of genetic and environmental factors that induce tumor necrosis factor (TNF) α secretion by keratinocytes, which in turn activates dendritic cells. Activated dendritic cells produce IL-23, leading to helper T cell (TH17) differentiation.2,3 TH17 cells secrete IL-17A, which has been shown to promote psoriatic skin changes.4 Therefore, TNF-α, IL-23, and IL-17A have been recognized as key targets for psoriasis therapy.
The newest biologic agents targeting IL-17–mediated pathways include ixekizumab, brodalumab, and bimekizumab. Secukinumab, the first US Food and Drug Administration (FDA)–approved IL-17 inhibitor, has been available since 2015 and therefore is not included in this review. IL-23 inhibitors that are FDA approved or being evaluated in clinical trials include guselkumab, tildrakizumab, and risankizumab. In addition, certolizumab pegol, a TNF-α inhibitor, is being studied for use in psoriasis.
METHODS
We reviewed the published results of phase 3 clinical trials for ixekizumab, brodalumab, bimekizumab, guselkumab, tildrakizumab, risankizumab, and certolizumab pegol. We performed an English-language literature search (January 1, 2012 to October 15, 2017) of articles indexed for PubMed/MEDLINE using the following combinations of keywords: IL-23 and psoriasis; IL-17 and psoriasis; tumor necrosis factor and psoriasis; [drug name] and psoriasis. If data from phase 3 clinical trials were not yet available, data from phase 2 clinical trials were incorporated in our analysis. We also reviewed citations within articles to identify relevant sources.
RESULTS
Phase 3 clinical trial design, efficacy, and adverse events (AEs) for ixekizumab and brodalumab are reported in eTable 15-10 and for guselkumab and tildrakizumab in eTable 2.11-14 Phase 2 clinical trial design, efficacy, and AEs are presented for risankizumab in eTable 315-18 and for certolizumab pegol in eTable 4.17,19 No published clinical trial data were found for bimekizumab.
IL-17 Inhibitors
Ixekizumab
This recombinant, high-affinity IgG4κ antibody selectively binds and neutralizes IL-17A.5,6 Three phase 3 clinical trials—UNCOVER-1, UNCOVER-2, and UNCOVER-3—evaluated ixekizumab for moderate to severe plaque psoriasis.7
The 3 UNCOVER trials were randomized, double-blind, phase 3 trials of 1296, 1224, and 1346 patients, respectively, assigned to a placebo group; a group treated with ixekizumab 80 mg every 2 weeks; and a group treated with ixekizumab 80 mg every 4 weeks. Both ixekizumab groups received a loading dose of 160 mg at week 0.5,6 UNCOVER-2 and UNCOVER-3 also included a comparator group of patients on etanercept 50 mg.5 Co-primary end points included the percentage of patients reaching a psoriasis area and severity index (PASI) of 75 and with a static physician global assessment (PGA) score of clear (0) or almost clear (1) at week 12.5,6
Ixekizumab achieved greater efficacy than placebo: 89.1%, 89.7%, and 87.3% of patients achieved PASI 75 in the every 2-week dosing group, and 82.6%, 77.5% and 84.2% achieved PASI 75 in the every 4-week dosing group in UNCOVER-1, UNCOVER-2, and UNCOVER-3, respectively (P<.001 for both treatment arms compared to placebo in all trials). The percentage of patients achieving a static PGA score of 0 or 1 also was higher in the ixekizumab groups in the 2-week and 4-week dosing groups in all UNCOVER trials—81.8% and 76.4% in UNCOVER-1, 83.2% and 72.9% in UNCOVER-2, and 80.5% and 75.4% in UNCOVER-3—compared to 3.2%, 2.4%, and 6.7% in the placebo groups of the 3 trials (P<.001 for both ixekizumab groups compared to placebo in all trials).5,6 Ixekizumab also was found to be more effective than etanercept for both co-primary end points in both UNCOVER-2 and UNCOVER-3 (eTable 1).5
Safety data for all UNCOVER trials were pooled and reported.6 At week 12 the rate of at least 1 AE was 58.4% in patients on ixekizumab every 2 weeks and 58.8% in patients on ixekizumab every 4 weeks compared to 54.0% in the etanercept group in UNCOVER-2 and UNCOVER-3 and 46.8% in the placebo group. At week 12, 72 nonfatal serious AEs were reported: 12 in the placebo group, 14 in the etanercept group, 20 in the ixekizumab every 2 weeks group, and 26 in the ixekizumab every 4 weeks group.6
The most common AE across all groups was nasopharyngitis. Overall, infections were more frequent in patients treated with ixekizumab than in patients treated with placebo or etanercept. Specifically, oral candidiasis occurred more frequently in the ixekizumab groups, with a higher rate in the 2-week dosing group than in the 4-week dosing group.6 Two myocardial infarctions (MIs) occurred: 1 in the etanercept group and 1 in the placebo group.5
Brodalumab
This human monoclonal antibody binds to IL-17ra.8,9 Three double-blind, placebo-controlled, phase 3 trials—AMAGINE-1, AMAGINE-2, and AMAGINE-3—evaluated its use for plaque psoriasis.10
In AMAGINE-1 (N=661), patients were randomized to receive brodalumab 140 mg or 210 mg (every 2 weeks for 12 weeks), or placebo.8 In AMAGINE-2 (N=1831) and AMAGINE-3 (N=1881), patients were randomized to receive brodalumab 140 mg or 210 mg (every 2 weeks for 12 weeks), ustekinumab 45 mg or 90 mg by weight (at weeks 0 and 4, then every 12 weeks thereafter), or placebo. In all trials, patients on brodalumab received a dose at week 0 and week 1. Co-primary end points were PASI 75 and a static PGA score of 0 or 1 at 12 weeks compared to placebo and to ustekinumab (in AMAGINE-2 and AMAGINE-3 only).8
At week 12, 83.3%, 86.3%, and 85.1% of patients on brodalumab 210 mg, and 60.3%, 66.6%, and 69.2% of patients on brodalumab 140 mg, achieved PASI 75 in AMAGINE-1, AMAGINE-2, and AMAGINE-3, respectively, compared to 2.7%, 8.1%, and 6.0% in the placebo groups (P<.001 between both brodalumab groups and placebo in all trials).8 Both brodalumab groups were noninferior but not significantly superior to ustekinumab, which achieved a PASI 75 of 70.0% in AMAGINE-2 and 69.3% in AMAGINE-3. The PASI 90 rate was higher, however, in both brodalumab groups compared to ustekinumab but significance was not reported (eTable 1).9 For both brodalumab groups, significantly more patients achieved a static PGA value of 0 or 1 compared to placebo (P<.001 across all trials). However, only the brodalumab 210-mg group achieved a significantly higher rate of static PGA 0 or 1 compared to ustekinumab in AMAGINE-2 and AMAGINE-3 (P<.001).9
After 12 weeks, the percentage of patients reporting at least 1 AE was 59.0%, 57.8%, and 56.8% in the brodalumab 210-mg group in AMAGINE-1, AMAGINE-2, and AMAGINE-3, respectively; 58.0%, 60.1%, and 52.6% in the brodalumab 140-mg group; and 51.0%, 53.4%, and 48.6% in the placebo group. Patients taking ustekinumab had an AE rate of 59.0% in AMAGINE-2 and 53.7% in AMAGINE-3. The most common AE was nasopharyngitis, followed by upper respiratory infection (URI) and headache across all trials.8,9 Serious AEs were rare: 10 in AMAGINE-1, 31 in AMAGINE-2, and 24 in AMAGINE-3 across all groups. One death occurred from stroke in the brodalumab 210-mg group in AMAGINE-2.9
IL-23 Inhibitors
Guselkumab
This drug is a human IgG1κ antibody that binds to the p19 subunit of IL-23, thereby inhibiting IL-23 signaling.11,12 Guselkumab was approved by the FDA in July 2017 for moderate to severe plaque psoriasis.13
VOYAGE 1 and VOYAGE 2 were phase 3, double-blind, placebo- and active comparator–controlled trials of 837 and 992 patients, respectively, randomized to receive adalimumab (80 mg at week 0 and 40 mg at week 1, then at 40 mg every 2 weeks thereafter), guselkumab 100 mg at weeks 0, 4, and 12, or placebo.11 Co-primary end points for both trials were the percentage of patients reaching PASI 90 and an investigator global assessment (IGA) score of cleared (0) or minimal (1) at week 16.11
By week 16 of both trials, PASI 90 values were statistically superior for guselkumab (VOYAGE 1, 73.3%; VOYAGE 2, 70.0%) compared to adalimumab (VOYAGE 1, 49.7%; VOYAGE 2, 46.8%) and placebo (VOYAGE 1, 2.9%; VOYAGE 2, 2.4%)(P<.001). Moreover, patients on guselkumab achieved a higher rate of IGA values of 0 and 1 at week 12 (85.1% in VOYAGE 1 and 84.1% in VOYAGE 2) than patients on adalimumab (65.9% in VOYAGE 1 and 67.7% in VOYAGE 2) and placebo (6.9% in VOYAGE 1 and 8.5% in VOYAGE 2)(P<.001).11,12
The frequency of AEs was comparable across all groups in both trials.11,12 During the 16-week treatment period, 51.7% and 47.6% of the guselkumab groups in VOYAGE 1 and VOYAGE 2, respectively; 51.1% and 48.4% of the adalimumab groups; and 49.4% and 44.8% of the placebo groups reported at least 1 AE. The most common AEs in all groups were nasopharyngitis, headache, and URI.11,12
Serious AEs also occurred at similar rates: 2.4% and 1.6% in the guselkumab group in VOYAGE 1 and VOYAGE 2, respectively; 2.4% and 1.8% in the adalimumab group; and 1.7% and 1.2% in the placebo group.11,12 One case of malignancy occurred in the VOYAGE 1 trial: basal cell carcinoma in the guselkumab group.11 Three major cardiovascular events occurred across both trials: 1 MI in the guselkumab group in each trial and 1 MI in the adalimumab group in VOYAGE 1.11,12
Tildrakizumab
A high-affinity, humanized IgG1κ antibody, tildrakizumab targets the p19 subunit of IL-23. As of February 2018, 2 double-blind, randomized phase 3 trials have studied tildrakizumab with published results: reSURFACE 1 and reSURFACE 2.14
reSURFACE 1 (N=772) and reSURFACE 2 (N=1090) randomized patients to receive tildrakizumab 100 or 200 mg (at weeks 0 and 4), etanercept 50 mg (twice weekly) for 12 weeks (reSURFACE 2 only), or placebo. Co-primary end points were the percentage of patients achieving PASI 75 and the percentage of patients achieving a PGA score of 0 or 1 at week 12.14
In reSURFACE 1, significantly more patients receiving tildrakizumab attained PASI 75 at week 12 compared to placebo: 200 mg, 62.0%; 100 mg, 64.0%; and placebo, 6.0% (P<.001 for tildrakizumab groups compared to placebo). Moreover, significantly proportionally more patients received a PGA score of 0 or 1 compared to placebo: 100 mg, 59%; 200 mg, 58.0%; placebo, 7.0% (P<.001 for both tildrakizumab groups compared to placebo).14
In reSURFACE 2, significantly more patients receiving tildrakizumab achieved PASI 75 compared to etanercept and placebo at week 12: 200 mg, 66.0%; 100mg, 61.0%; etanercept, 48.0%; placebo, 6.0% (P<.001 for both tildrakizumab groups compared to placebo; P<.05 for both tildrakizumab groups compared to etanercept). Additionally, significantly more patients in the tildrakizumab groups experienced a PGA score of 0 or 1 at week 12 compared to placebo: 200 mg, 59%; 100 mg, 55.0%; placebo, 5% (P<.001 for both tildrakizumab groups compared to placebo).14
Adverse events were reported at a similar rate across all groups. For reSURFACE 1 and reSURFACE 2, at least 1 AE by week 12 was reported by 42.2% and 45.2% of patients in the 200-mg group; 47.2% and 45.9% in the 100-mg group; and 48.1% and 55.1% in the placebo groups.14The most common AEs were nasopharyngitis, URI (reSURFACE 1), and erythema at the injection site (reSURFACE 2). One case of serious infection was reported in each of the tildrakizumab groups: 1 case of drug-related hypersensitivity reaction in the 200-mg group, and 1 major cardiovascular event in the 100-mg group of reSURFACE 1. There was 1 serious AE in reSURFACE 2 that led to death in which the cause was undetermined.14
Risankizumab
This humanized IgG1 antibody binds the p19 unit of IL-23.15,16 The drug is undergoing 3 phase 3 trials—ultIMMa-1, ultIMMa-2, and IMMvent—for which only preliminary data have been published and are reported here.16,17 There is 1 phase 2 randomized, dose-ranging trial with published data.15
ultIMMa-1 and ultIMMa-2 comprised 506 and 491 patients, respectively, randomized to receive risankizumab (150 mg at weeks 0, 4, and 16), ustekinumab (45 mg or 90 mg, by weight, at weeks 0, 4, and 16), or placebo. Co-primary end points were PASI 90 and a PGA score of 0 or 1 at week 16.17
In ultIMMa-1 and ultIMMa-2, 75.0% and 75.0% of patients on risankizumab 150 mg achieved PASI 90 compared to 42.0% and 48.0% on ustekinumab and 5.0% and 2.0% on placebo at 16 weeks (P<.001 between both placebo and ustekinumab in both trials).17 In both trials, patients receiving risankizumab achieved higher rates of a static PGA score of 0 or 1 (88.0% and 84.0%) compared to ustekinumab (63.0% and 62.0%) and placebo (8.0% and 5.0%) at 16 weeks (P<.001 for both trials).18
At week 16, 2.0% of patients on risankizumab reported a serious AE in both trials, compared to 8.0% and 3.0% of patients on ustekinumab and 3.0% and 1.0% on placebo. No new safety concerns were noted.17
In the phase 3 IMMvent trial, 605 patients were randomized to receive risankizumab (150 mg at weeks 0, 4, and 16) or adalimumab (80 mg at week 0, 40 mg at week 1, then 40 mg every 2 weeks). Co-primary end points were PASI 90 and a static PGA score of 0 or 1 at week 16.17
In IMMvent, risankizumab was significantly more effective than adalimumab for PASI 75 (risankizumab, 72.0%; adalimumab, 47.0%) and a static PGA score of 0 or 1 (risankizumab 84.0%; adalimumab, 60.0%) (P<.001 risankizumab compared to adalimumab for both end points).17
At week 16, serious AEs were reported in 3.0% of patients on risankizumab and 3.0% of patients on adalimumab. One patient receiving risankizumab died of an acute MI during the treatment phase.17
TNF Inhibitor
Certolizumab Pegol
Certolizumab pegol is a human PEGylated anti-TNF agent. In vitro studies have shown that certolizumab binds to soluble and membrane-bound TNF.19 Unlike other TNF inhibitors, certolizumab pegol is a Fab‘ portion of anti-TNF conjugated to a molecule of polyethylene glycol.19 The drug is approved in the United States for treating psoriatic arthritis, Crohn disease, and rheumatoid arthritis; its potential for treating psoriasis has been confirmed. Results of 1 phase 2 trial have been published19; data from 3 phase 3 trials are forthcoming.
This randomized, placebo-controlled, double-blind phase 2 study comprised 176 patients who received certolizumab 200 mg, certolizumab 400 mg, or placebo. The dosing schedule was 400 mg at week 0, followed by either 200 or 400 mg every other week until week 10. Co-primary end points were PASI 75 and a PGA score of 0 or 1 at week 12.19
Certolizumab was significantly more effective than placebo at week 12: 74.6% of the 200-mg group and 82.8% of the 400-mg group achieved PASI 75 compared to 6.8% of the placebo group (P<.001). Certolizumab also performed better for the PGA score: 52.5% and 72.4% of patients attained a score of 0 or 1 in the 200-mg and 400-mg groups compared to 1.7% in the placebo group.19
Adverse events were reported equally across all groups: 72% of patients in the 200-mg group, 70% in the 400-mg group, and 71% in the placebo group reported at least 1 AE, most commonly nasopharyngitis, headache, and pruritis.19
COMMENT
With the development of new insights into the pathogenesis of psoriasis, therapies that are targeted toward key cytokines may contribute to improved management of the disease. The results of these clinical trials demonstrate numerous promising options for psoriatic patients.
IL-17 Inhibitors Ixekizumab and Brodalumab
When comparing these 2 biologics, it is important to consider that these studies were not performed head to head, thereby inhibiting direct comparisons. Moreover, dosage ranges of the investigative drugs were not identical, which also makes comparisons challenging. However, when looking at the highest dosages of ixekizumab and brodalumab, results indicate that ixekizumab may be slightly more effective than brodalumab based on the percentage of patients who achieved a PASI 75 and a static PGA score of 0 or 1 (eTable 1).
Phase 3 trials have shown ixekizumab to maintain efficacy over 60 weeks of treatment.6 Ixekizumab also has been shown to alleviate other symptoms of psoriasis, such as itching, pain, and nail involvement.20,21 Furthermore, ixekizumab appears to be equally effective in patients with or without prior exposure to biologics22; therefore, ixekizumab may benefit patients who have not experienced success with other biologics.
Across the UNCOVER trials, 11 cases of inflammatory bowel disease were reported in patients receiving ixekizumab (ulcerative colitis in 7; Crohn disease in 4)6; it appears that at least 3 of these cases were new diagnoses. In light of a study suggesting that IL-17A might have a protective function in the intestine,23 these findings may have important clinical implications and require follow-up studies.
Brodalumab also has been shown to maintain efficacy and acceptable safety for as long as 120 weeks.24 In the extension period of the AMAGINE-1 trial, patients who experienced a return of disease during a withdrawal period recaptured static PGA success with re-treatment for 12 weeks (re-treatment was successful in 97% of those given a dosage of 210 mg and in 84% of those given 140 mg).8
Furthermore, phase 2 trials also have shown that brodalumab is effective in patients with a history of biologic use.25 Across all AMAGINE trials, only 1 case of Crohn disease was reported in a patient taking brodalumab.9 There are concerns about depression, despite data from AMAGINE-1 stating patients on brodalumab actually had greater improvements in Hospital Anxiety and Depression Scale scores after 12 weeks of treatment (P<.001) for both brodalumab 140 mg and 210 mg compared to placebo.8 Regardless, brodalumab has a black-box warning for suicidal ideation and behavior, and availability is restricted through a Risk Evaluation and Mitigation Strategy (REMS) program.26
Bimekizumab
Although no phase 2 or phase 3 clinical trial data have been published for bimekizumab (phase 2 trials are underway), it has been shown in a phase 1 trial to be effective for psoriasis. Bimekizumab also is unique; it is the first dual inhibitor of IL-17A and IL-17F.18
IL-23 Inhibitors Guselkumab, Tildrakizumab, and Risankizumab
Making comparisons among the IL-23 inhibitors also is difficult; studies were not head-to-head comparison trials, and the VOYAGE and reSURFACE studies used different time points for primary end points. Furthermore, only phase 2 trial data are available for risankizumab. Despite these limitations, results of these trials suggest that guselkumab and risankizumab may be slightly more efficacious than tildrakizumab. However, future studies, including head-to-head studies, would ultimately provide further information on how these agents compare.
Guselkumab was shown to remain efficacious at 48 weeks, though patients on maintenance dosing had better results than those who were re-treated.12 Moreover, guselkumab was found to be effective in hard-to-treat areas, such as the scalp,11 and in patients who did not respond to adalimumab. Guselkumab may therefore benefit patients who have experienced limited clinical improvement on other biologics.12
Tildrakizumab was shown to improve PASI 75 and PGA scores through week 28 of treatment. Moreover, a higher percentage of patients taking tildrakizumab scored 0 or 1 on the dermatology life quality index, suggesting that the drug improves quality of life.14 No specific safety concerns arose in either reSURFACE trial; however, long-term studies are needed for further evaluation.
Risankizumab appears to be a promising new therapy based on phase 2 trial results. Improvements also were seen in dermatology life quality index scores, scalp and fingernail symptoms, and palmoplantar psoriasis.15 Of note, neutralizing antidrug antibodies were found in 3 patients during this study,15 which may present potential problems for long-term efficacy. However, preliminary data from 3 phase 3 trials—ultIMMa-1, ultIMMa-2, and IMMvent—are promising.17
CONCLUSION
Advances in the understanding of psoriasis have led to new targeted therapies. Ongoing clinical trials have shown encouraging results for treating physical and psychological symptoms of psoriasis. The findings of these trials support the idea that therapies targeting IL-23, specifically its p19 subunit, are effective against psoriasis while sparing IL-12. Long-term data from open-label extension studies would help guide clinical recommendations regarding the safety profiles of these agents and determine their long-term utility.
- Langley RG, Krueger GG, Griffiths CE. Psoriasis: epidemiology, clinical features, and quality of life. Ann Rheum Dis. 2005;64(suppl 2):ii18-ii23; discussion, ii24, ii25.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Amin M, Darji K, No DJ, et al. Review of phase III trial data on IL-23 inhibitors tildrakizumab and guselkumab for psoriasis. J Eur Acad Dermatol Venereol. 2017;31:1627-1632.
- Arican O, Aral M, Sasmaz S, et al. Levels of TNF-alpha, IFN-gamma, IL6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. 2005:273-279.
- Griffiths CE, Reich K, Lebwohl M, et al; UNCOVER-2 and UNCOVER-3 investigators. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386:541-551.
- Gordon KB, Blauvelt A, Papp KA, et al; UNCOVER-1 study group, UNCOVER-2 study group, UNCOVER-3 study group. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345-356.
- FDA approves new psoriasis drug Taltz [news release]. Silver Spring, MD: US Food and Drug Administration; March 22, 2016. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm491872.htm. Accessed January 29, 2018.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016;175:273-286.
- Lebwohl M, Strober B, Mentor A, et al. Phase 3 studies comparing brodalumab with ustekinumab for psoriasis. N Engl J Med. 2015;373:1318-1328.
- FDA approves new psoriasis drug [news release]. Silver Spring, MD: US Food and Drug Administration; February 15, 2017. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm541981.htm. Accessed January 29, 2018.
- Blauvelt A, Papp KA, Griffiths CE, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate-to-severe plaque psoriasis: results from the phase III, double-blinded placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76:405-417.
- Reich K, Armstrong AW, Foley P, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J Am Acad Dermatol. 2017;76:418-431.
- Janssen announces U.S. FDA approval of Tremfya™ (guselkumab) for the treatment of moderate to severe plaque psoriasis [news release]. Horsham, PA: Johnson & Johnson; July 13, 2017. https://www.jnj.com/media-center/press-releases/janssen-announces-us-fda-approval-of-tremfya-guselkumab-for-the-treatment-of-moderate-to-severe-plaque-psoriasis. Accessed January 29, 2018.
- Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE1 and reSURFACE 2): results from two randomized controlled, phase 3 trials. Lancet. 2017;390:276-288.
- Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376:1551-1560.
- Risankizumab. AbbVie Inc website. https://www.abbvie.com/our-science/pipeline/risankizumab.html. Accessed January 29, 2018.
- Risankizumab meets all co-primary and ranked secondary endpoints, achieving significantly greater efficacy versus standard biologic therapies in three pivotal phase 3 psoriasis studies [news release]. North Chicago, IL: AbbVie Inc; October 26, 2017. https://news.abbvie.com/news/risankizumab-meets-all-co-primary-and-ranked-secondary-endpoints-achieving-significantly-greater-efficacy-versus-standard-biologic-therapies-in-three-pivotal-phase-3-psoriasis-studies.htm. Accessed January 29, 2018.
- Glatt S, Helmer E, Haier B, et al. First-in-human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL-17A and IL-17F, in mild psoriasis. Br J Clin Pharmacol. 2017;83:991-1001.
- Reich K, Ortonne JP, Gottlieb AB, et al. Successful treatment of moderate to severe plaque psoriasis with the PEGylated Fab‘ certolizumab pegol: results of a phase II randomized, placebo-controlled trial with a re-treatment extension. Br J Dermatol. 2012;167:180-190.
- Kimball AB, Luger T, Gottlieb A, et al. Impact of ixekizumab on psoriasis itch severity and other psoriasis symptoms: results from 3 phase III psoriasis clinical trials. J Am Acad Dermatol. 2016;75:1156-1161.
- Dennehy EB, Zhang L, Amato D, et al. Ixekizumab is effective in subjects with moderate to severe plaque psoriasis with significant nail involvement: results from UNCOVER 3. J Drugs Dermatol. 2016;15:958-961.
- Gottlieb AB, Lacour JP, Korman N, et al. Treatment outcomes with ixekizumab in patients with moderate-to-severe psoriasis who have not received prior biological therapies: an integrated analysis of two phase III randomized studies. J Eur Acad Dermatol Venereol. 2017;31:679-685.
- Hueber W, Sands BE, Lewitsky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693-1700.
- Papp K, Leonardi C, Menter A, et al. Safety and efficacy of brodalumab for psoriasis after 120 weeks of treatment. J Am Acad Dermatol. 2014;71:1183-1190.
- Papp K, Menter A, Strober B, et al. Efficacy and safety of brodalumab in subpopulations of patients with difficult-to-treat moderate-to-severe plaque psoriasis. J Am Acad Dermatol. 2015;72:436-439.
- SILIQ [package insert]. Thousand Oaks, CA: Amgen, Inc; 2017.
- Langley RG, Krueger GG, Griffiths CE. Psoriasis: epidemiology, clinical features, and quality of life. Ann Rheum Dis. 2005;64(suppl 2):ii18-ii23; discussion, ii24, ii25.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Amin M, Darji K, No DJ, et al. Review of phase III trial data on IL-23 inhibitors tildrakizumab and guselkumab for psoriasis. J Eur Acad Dermatol Venereol. 2017;31:1627-1632.
- Arican O, Aral M, Sasmaz S, et al. Levels of TNF-alpha, IFN-gamma, IL6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. 2005:273-279.
- Griffiths CE, Reich K, Lebwohl M, et al; UNCOVER-2 and UNCOVER-3 investigators. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386:541-551.
- Gordon KB, Blauvelt A, Papp KA, et al; UNCOVER-1 study group, UNCOVER-2 study group, UNCOVER-3 study group. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345-356.
- FDA approves new psoriasis drug Taltz [news release]. Silver Spring, MD: US Food and Drug Administration; March 22, 2016. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm491872.htm. Accessed January 29, 2018.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016;175:273-286.
- Lebwohl M, Strober B, Mentor A, et al. Phase 3 studies comparing brodalumab with ustekinumab for psoriasis. N Engl J Med. 2015;373:1318-1328.
- FDA approves new psoriasis drug [news release]. Silver Spring, MD: US Food and Drug Administration; February 15, 2017. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm541981.htm. Accessed January 29, 2018.
- Blauvelt A, Papp KA, Griffiths CE, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate-to-severe plaque psoriasis: results from the phase III, double-blinded placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76:405-417.
- Reich K, Armstrong AW, Foley P, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J Am Acad Dermatol. 2017;76:418-431.
- Janssen announces U.S. FDA approval of Tremfya™ (guselkumab) for the treatment of moderate to severe plaque psoriasis [news release]. Horsham, PA: Johnson & Johnson; July 13, 2017. https://www.jnj.com/media-center/press-releases/janssen-announces-us-fda-approval-of-tremfya-guselkumab-for-the-treatment-of-moderate-to-severe-plaque-psoriasis. Accessed January 29, 2018.
- Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE1 and reSURFACE 2): results from two randomized controlled, phase 3 trials. Lancet. 2017;390:276-288.
- Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376:1551-1560.
- Risankizumab. AbbVie Inc website. https://www.abbvie.com/our-science/pipeline/risankizumab.html. Accessed January 29, 2018.
- Risankizumab meets all co-primary and ranked secondary endpoints, achieving significantly greater efficacy versus standard biologic therapies in three pivotal phase 3 psoriasis studies [news release]. North Chicago, IL: AbbVie Inc; October 26, 2017. https://news.abbvie.com/news/risankizumab-meets-all-co-primary-and-ranked-secondary-endpoints-achieving-significantly-greater-efficacy-versus-standard-biologic-therapies-in-three-pivotal-phase-3-psoriasis-studies.htm. Accessed January 29, 2018.
- Glatt S, Helmer E, Haier B, et al. First-in-human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL-17A and IL-17F, in mild psoriasis. Br J Clin Pharmacol. 2017;83:991-1001.
- Reich K, Ortonne JP, Gottlieb AB, et al. Successful treatment of moderate to severe plaque psoriasis with the PEGylated Fab‘ certolizumab pegol: results of a phase II randomized, placebo-controlled trial with a re-treatment extension. Br J Dermatol. 2012;167:180-190.
- Kimball AB, Luger T, Gottlieb A, et al. Impact of ixekizumab on psoriasis itch severity and other psoriasis symptoms: results from 3 phase III psoriasis clinical trials. J Am Acad Dermatol. 2016;75:1156-1161.
- Dennehy EB, Zhang L, Amato D, et al. Ixekizumab is effective in subjects with moderate to severe plaque psoriasis with significant nail involvement: results from UNCOVER 3. J Drugs Dermatol. 2016;15:958-961.
- Gottlieb AB, Lacour JP, Korman N, et al. Treatment outcomes with ixekizumab in patients with moderate-to-severe psoriasis who have not received prior biological therapies: an integrated analysis of two phase III randomized studies. J Eur Acad Dermatol Venereol. 2017;31:679-685.
- Hueber W, Sands BE, Lewitsky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693-1700.
- Papp K, Leonardi C, Menter A, et al. Safety and efficacy of brodalumab for psoriasis after 120 weeks of treatment. J Am Acad Dermatol. 2014;71:1183-1190.
- Papp K, Menter A, Strober B, et al. Efficacy and safety of brodalumab in subpopulations of patients with difficult-to-treat moderate-to-severe plaque psoriasis. J Am Acad Dermatol. 2015;72:436-439.
- SILIQ [package insert]. Thousand Oaks, CA: Amgen, Inc; 2017.
Practice Points
- Tumor necrosis factor α, IL-23, and IL-17A are key targets for psoriasis therapy based on an understanding of the key role that these cytokines play in the pathophysiology of disease.
- The biologic agents secukinumab and ixekizumab are approved for use in the management of psoriasis. Other biologics—brodalumab, bimekizumab, guselkumab, tildrakizumab, risankizumab, and certolizumab pegol—have been (and some continue to be) the focus of phase 2 and phase 3 clinical trials.
- Findings of several of those trials support the idea that therapies targeting IL-23, specifically its p19 subunit, but that spare IL-12 are effective against psoriasis.
- Longer-term studies are needed to determine whether the agents reviewed here, including those approved for clinical use, are suitable for prolonged administration.
Deepithelialized Flaps and Grafts: Applications in Dermatologic Surgery
Deepithelialized flaps and grafts have been widely used by reconstructive surgeons in a diverse range of medical specialties since the early 20th century. 1 These reconstructive modalities have more recently been applied to dermatologic surgery. Deepithelialized flaps and grafts involve removal of the epidermis from the dermis for a variety of surgical purposes. Although these techniques play an important role in dermatologic surgery, reports of application of deepithelialized flaps and grafts in the dermatology literature is limited. This article includes a presentation of the applications of deepithelialized flaps and grafts in procedural dermatology.
DEEPITHELIALIZATION TECHNIQUES
There are a variety of techniques for deepithelialization, although sharp deepithelialization generally is preferred by dermatologic surgeons. The scalpel technique can be accomplished by making an intradermal incision with a No. 15 blade. Traction is an essential component of the deepthelialization process and facilitates sharp removal of the epidermis and superficial dermis in an even plane. The peeling orange technique, which has been described in reduction mammoplasty, is a variant of the scalpel technique used for creating a large area of deepithelialized tissue.2 A No. 10 blade is used to make multiple partial-thickness intradermal incisions 1 to 2 cm apart along the pedicle. Traction facilitates rapid deepithelialization of the skin strips on the pedicle. A sharp curette is an alternative option for sharply removing the epithelium from a small area. Electric dermatome, laser, and electrocautery techniques for deepithelialization also can be considered.2,3
APPLICATION OF DEEPITHELIALIZED FLAPS
Deepithelialized flaps may be considered for single-stage reconstruction with tunneled interpolation flaps, reconstruction requiring contour preservation, and reconstruction involving free margins.4-17
Reconstruction With Single-Stage Tunneled Interpolated Flaps
Alar Base
A partially deepithelialized tunneled interpolated flap is an elegant reconstructive option for defects involving the upper cutaneous lip and alar base. The flap is elevated from the ipsilateral nasolabial fold, deepithelialized proximally, and tunneled under the intact portion of the cutaneous upper lip and ala. The flap is then deepithelialized superiorly to bolster the alar base and inset at the recipient site.4
Nasal Ala
The tunneled interpolated flap is useful for reconstruction of defects of the nasal ala. A flap with a superior deepithelialized pedicle and an anticipated inferior Burow triangle is designed along the axis of the nasolabial fold. The inferior Burow triangle and central flap are elevated at the level of the superficial subcutaneous fat and the pedicle is dissected. The donor and recipient sites are widely undermined, and the flap and pedicle pass through the tunnel. The donor site is closed primarily, the inferior Burow triangle is trimmed, and the flap is sutured into the defect.5 This flap allows for preservation of free margins and favorable placement of incision lines. Furthermore, pincushioning of the flap helps to recreate the rounded shape of the lateral ala.6
Nasal Tip
Nasal tip defects can be repaired with a retroangular flap, centered on the angular artery. The flap is elevated along the axis of the nasolabial fold, deepithelialized at its proximal base, and transferred through a subcutaneous tunnel to the nasal tip. The angular artery is ligated at the inferior aspect of the flap.7
Nasal Sidewall
A deepithelialized tunneled interpolated forehead flap, similar to the classic paramedian forehead flap, can be used to reconstruct nasal sidewall defects. A flap is elevated on the contralateral forehead and the proximal portion is deepithelialized. A tunnel is then bluntly dissected just above the periosteum, and the flap is introduced into the defect through the tunnel and inset. This flap has the advantages of being a single-stage procedure, restoring volume to the defect area, and maintaining excellent vascular supply.8
Eyelid
A tunneled interpolated forehead flap also can be used to repair medial canthal defects and for anterior lamellar repair of lower eyelid defects. In a study of 9 patients receiving a tunneled interpolated forehead flap in these anatomic locations, all flaps demonstrated viability, protection of the globe, and preservation of the concave architecture of the medial canthus.9
Earlobe
Earlobe defects may be repaired with a pull-through interpolated preauricular flap. A flap is elevated superiorly in the preauricular region and the proximal aspect of the flap is deepithelialized. The flap is pulled through a tunnel and inset at the anterior earlobe defect. The donor site is closed primarily.10,11
Concha
Reconstruction of anterior conchal defects with exposed cartilage can be accomplished with a pull-through interpolated postauricular flap based on the auriculomastoid fossa. The postauricular flap is elevated, the base is deepithelialized, an incision is made in the medial aspect of the defect, and the flap is moved through a tunnel between the posterior and anterior surfaces of the ear. The flap is secured to the anterior surface of the concha.12
Reconstruction Requiring Contour Preservation
Central Face
The hinge flap is optimal for reconstruction of deep central facial defects (Figure 1). The hinge flap is planned at a site contiguous with a margin of the defect and can include the dermis, subcutaneous tissue, muscle, or a combination of these. The desired tissue is folded over on the pedicle to fill the defect. Cutaneous coverage is accomplished through a primary closure, separate flap, or skin graft. In addition to restoring contour and therefore the cosmetic subunit, the hinge flap is performed in a single stage, resists wound contracture, and provides a well-vascularized wound bed resulting in a low incidence of graft failure.13,14 Muscular hinge flaps have been described for reconstruction of forehead defects with exposed bone based on the frontalis muscle.15
Lower Lip
A variant of a V-Y advancement flap has been described for reconstruction of defects greater than one-third the length of the lower lip. The top of the “V” is deepithelialized and the flap is advanced such that the top of the “V” abuts the inferior border of the defect. The “V” flap is inset at its advanced position, converting the “V”-shaped wound into a “Y.” An overlying buccal mucosal graft provides reconstruction of the lower red lip and labial mucosa.16
Helix of the Ear
Large defects of the scapha and helix of the ear can be reconstructed with the use of a staged interpolated postauricular flap. The postauricular flap is elevated into a subcutaneous plane. A full-thickness incision is made medial to the helical rim, and the flap is tunneled through and sutured into place. The pedicle is later divided, and the distal aspect of the flap is deepithelialized and inset into the helical rim for volume restoration.17
Reconstruction Involving Free Margins
Nasal Ala
For large defects involving the upper cutaneous lip with adjacent alar base involvement, a partially deepithelialized V-Y flap is a useful reconstructive option (Figure 2).
Infraorbital Region
A deepithelialized variant of a V-Y advancement flap can be used for closure of infraorbital defects. The limbs of the V-Y flap are deepithelialized and anchored to the medial and lateral canthal tendons or periosteum. Ectropion prevention is the primary advantage of this flap.18
APPLICATION OF DEEPITHELIALIZED GRAFTS
Deepithelialized grafts may be considered for volume replacement, reconstruction requiring contour preservation, and restoration of mechanical integrity in areas of high mechanical tension.3,19-21
Reconstruction Requiring Contour Preservation
Deepithelialized grafts are used to improve depressed nasal scars and restore volume in deep nasal wounds. One method involves deepithelialization of 2 postauricular punch biopsies. An 18-gauge needle is used to make a small hole in the depressed nasal scar, the dermal grafts are inserted, and the defect is closed primarily.19 Dermal grafts may be harvested from excess full-thickness skin grafts (FTSGs) or dog-ear tissue. When used under flaps, the dermal graft is trimmed to the size of the defect. When used under FTSGs, thin dermal graft strips are placed in a gridlike pattern to allow for revascularization. A study of 15 patients with contour deformities reconstructed with dermal graft insertions demonstrated that 14 (94%) patients had no significant complications and improvement of scar depression was achieved.20
Reconstruction in Areas of High Mechanical Tension
Plantar Foot
A combined dermal and full-thickness sandwich graft has been described for reconstruction of plantar foot defects.3 The graft is created by obtaining a FTSG twice the size of the wound defect and deepithelializing half of the graft. The graft is then defatted and the deepithelialized portion is folded beneath the other half, allowing the papillary dermis to make contact with the wound surface.
Scalp
Dermal graft reconstruction for scalp defects may be accomplished with a split-thickness skin flap. The flap is harvested using an electronic dermatome that ensures the proximal aspect is still attached to adjacent skin. The dermis is removed from the area underneath the back-folded split-thickness skin flap. The dermal graft is meshed and sutured into the recipient site. The split-thickness skin flap is replaced over the donor site. Meshed reversed dermal grafts have excellent survival rates, even with direct placement on bone without periosteum. Querings et al21 reported graft survival with no complications in 19 of 21 (90.4%) patients undergoing scalp or plantar sole reconstruction.
CONCLUSION
With the widespread adoption of the fresh-tissue technique for Mohs micrographic surgery and the establishment of the American Society for Dermatologic Surgery in 1970, the depth and scope of techniques used by dermatologic surgeons has dramatically expanded. Although the use of dermal flaps and grafts is not as widespread in dermatology as other reconstructive techniques, their unique advantages should be considered. Deepithelialized flaps and grafts should be considered when the following reconstructive goals are desired: (1) conversion of a 2-stage interpolation flap to a single-stage tunneled flap, (2) contour and cosmetic subunit preservation of deep defects through volume augmentation, (3) reconstruction in areas of high mechanical tension, and (4) free margin preservation. The multiple applications of deepithelialized flaps and grafts as described in this review demonstrate their continued applicability in dermatologic surgery.
- Straatsma CR. Use of the dermal graft in the repairs of small saddle defects of the nose. Arch Otolaryngol. 1932;16:506-509.
- Cydeli A, Hunter J. Peeling orange: rapid deepithelialization in reduction mammoplasty. J Aesthet Surg. 2004;24:580-581.
- Bechara F, Sand M, Radenhausen M, et al. Erbium:YAG laser-assisted preparation of a combined dermal/full thickness sandwich skin graft. Dermatol Surg. 2006;32:353-358.
- Cook JL. Tunneled and transposed island flaps in facial reconstructive surgery. Dermatol Surg. 2014;40(suppl 9):S16-S29.
- Krishnan RS, Clark DP. Tunneled transposition flap for reconstruction of defects of the nasal ala. Dermatol Surg. 2007;33:1496-1501.
- Mahlberg M. Tunneled melolabial pedicle flap for small but deep lateral alar rim defect. Dermatol Surg. 2013;39:1527-1529.
- Ascari-Raccagni A, Balderi U. The retroangular flap used in the surgery of nasal tip defects. Dermatol Surg. 2004;30:1131-1137.
- Hollmig ST, Leach BC, Cook J. Single-staged interpolation flaps in facial reconstruction. Dermatol Surg. 2014;40(suppl 9):S62-S70.
- Mombaerts I, Gillis A. The tunneled forehead flap in medial canthal and eyelid reconstruction. Dermatol Surg. 2010:36:1118-1125.
- Wang SQ, Goldberg LH, Kimyah-Asadi A. Tunneled island pedicle flap for an earlobe defect. Dermatol Surg. 2007;33:835-838.
- Hatoko M, Kuwahara M, Shiba A, et al. Earlobe reconstruction using a subcutaneous island pedicle flap after resection of “earlobe keloid.” Dermatol Surg. 1998;24:257-261.
- Alder N, Ad-El D, Azaria R. Reconstruction of nonhelical auricular defects with local flaps. Dermatol Surg. 2008;34:501-507.
- Fader DJ, Wang TS, Johnson TM. Nasal reconstruction utilizing a muscle hinge flap with overlying FTSG. J Am Acad Dermatol. 2000;43:837-840.
- Braun MA, Cook J. Hinge flaps in facial reconstruction. Dermatol Surg. 2007;33:213-221.
- Salmon PL, Mortimer NL, Hill SE. Muscular hinge flaps: utility and technique in facial reconstructive surgery. Dermatol Surg. 2010;36:227-234.
- Seo Y, Song S, Choi Y, et al. A lower lip reconstruction. Dermatol Surg. 2015;41:505-507.
- Malone CH, Wagner RF. Partially de-epithelialized postauricular flap for ear reconstruction. J Am Acad Dermatol. 2015;73:E219-E220.
- Yildrim S, Akoz T, Akan M, et al. Nasolabial V-Y advancement for closure of the midface defects. Dermatol Surg. 2001;27:656-662.
- Jensen DJ, Cohen JL. Nasal tip revision using a dermal graft. Dermatol Surg. 2014;40:1140-1142.
- Meyers S, Rohrer T. Use of dermal grafts in reconstructing deep nasal defects and shaping the ala nasi. Dermatol Surg. 2001;27:300-305.
- Querings K, Bachter D, Balda B. Meshed reversed dermal graft in patients with surgical defects of sole and scalp: technique and long-term results. Dermatol Surg. 2002;28:122-126.
Deepithelialized flaps and grafts have been widely used by reconstructive surgeons in a diverse range of medical specialties since the early 20th century. 1 These reconstructive modalities have more recently been applied to dermatologic surgery. Deepithelialized flaps and grafts involve removal of the epidermis from the dermis for a variety of surgical purposes. Although these techniques play an important role in dermatologic surgery, reports of application of deepithelialized flaps and grafts in the dermatology literature is limited. This article includes a presentation of the applications of deepithelialized flaps and grafts in procedural dermatology.
DEEPITHELIALIZATION TECHNIQUES
There are a variety of techniques for deepithelialization, although sharp deepithelialization generally is preferred by dermatologic surgeons. The scalpel technique can be accomplished by making an intradermal incision with a No. 15 blade. Traction is an essential component of the deepthelialization process and facilitates sharp removal of the epidermis and superficial dermis in an even plane. The peeling orange technique, which has been described in reduction mammoplasty, is a variant of the scalpel technique used for creating a large area of deepithelialized tissue.2 A No. 10 blade is used to make multiple partial-thickness intradermal incisions 1 to 2 cm apart along the pedicle. Traction facilitates rapid deepithelialization of the skin strips on the pedicle. A sharp curette is an alternative option for sharply removing the epithelium from a small area. Electric dermatome, laser, and electrocautery techniques for deepithelialization also can be considered.2,3
APPLICATION OF DEEPITHELIALIZED FLAPS
Deepithelialized flaps may be considered for single-stage reconstruction with tunneled interpolation flaps, reconstruction requiring contour preservation, and reconstruction involving free margins.4-17
Reconstruction With Single-Stage Tunneled Interpolated Flaps
Alar Base
A partially deepithelialized tunneled interpolated flap is an elegant reconstructive option for defects involving the upper cutaneous lip and alar base. The flap is elevated from the ipsilateral nasolabial fold, deepithelialized proximally, and tunneled under the intact portion of the cutaneous upper lip and ala. The flap is then deepithelialized superiorly to bolster the alar base and inset at the recipient site.4
Nasal Ala
The tunneled interpolated flap is useful for reconstruction of defects of the nasal ala. A flap with a superior deepithelialized pedicle and an anticipated inferior Burow triangle is designed along the axis of the nasolabial fold. The inferior Burow triangle and central flap are elevated at the level of the superficial subcutaneous fat and the pedicle is dissected. The donor and recipient sites are widely undermined, and the flap and pedicle pass through the tunnel. The donor site is closed primarily, the inferior Burow triangle is trimmed, and the flap is sutured into the defect.5 This flap allows for preservation of free margins and favorable placement of incision lines. Furthermore, pincushioning of the flap helps to recreate the rounded shape of the lateral ala.6
Nasal Tip
Nasal tip defects can be repaired with a retroangular flap, centered on the angular artery. The flap is elevated along the axis of the nasolabial fold, deepithelialized at its proximal base, and transferred through a subcutaneous tunnel to the nasal tip. The angular artery is ligated at the inferior aspect of the flap.7
Nasal Sidewall
A deepithelialized tunneled interpolated forehead flap, similar to the classic paramedian forehead flap, can be used to reconstruct nasal sidewall defects. A flap is elevated on the contralateral forehead and the proximal portion is deepithelialized. A tunnel is then bluntly dissected just above the periosteum, and the flap is introduced into the defect through the tunnel and inset. This flap has the advantages of being a single-stage procedure, restoring volume to the defect area, and maintaining excellent vascular supply.8
Eyelid
A tunneled interpolated forehead flap also can be used to repair medial canthal defects and for anterior lamellar repair of lower eyelid defects. In a study of 9 patients receiving a tunneled interpolated forehead flap in these anatomic locations, all flaps demonstrated viability, protection of the globe, and preservation of the concave architecture of the medial canthus.9
Earlobe
Earlobe defects may be repaired with a pull-through interpolated preauricular flap. A flap is elevated superiorly in the preauricular region and the proximal aspect of the flap is deepithelialized. The flap is pulled through a tunnel and inset at the anterior earlobe defect. The donor site is closed primarily.10,11
Concha
Reconstruction of anterior conchal defects with exposed cartilage can be accomplished with a pull-through interpolated postauricular flap based on the auriculomastoid fossa. The postauricular flap is elevated, the base is deepithelialized, an incision is made in the medial aspect of the defect, and the flap is moved through a tunnel between the posterior and anterior surfaces of the ear. The flap is secured to the anterior surface of the concha.12
Reconstruction Requiring Contour Preservation
Central Face
The hinge flap is optimal for reconstruction of deep central facial defects (Figure 1). The hinge flap is planned at a site contiguous with a margin of the defect and can include the dermis, subcutaneous tissue, muscle, or a combination of these. The desired tissue is folded over on the pedicle to fill the defect. Cutaneous coverage is accomplished through a primary closure, separate flap, or skin graft. In addition to restoring contour and therefore the cosmetic subunit, the hinge flap is performed in a single stage, resists wound contracture, and provides a well-vascularized wound bed resulting in a low incidence of graft failure.13,14 Muscular hinge flaps have been described for reconstruction of forehead defects with exposed bone based on the frontalis muscle.15
Lower Lip
A variant of a V-Y advancement flap has been described for reconstruction of defects greater than one-third the length of the lower lip. The top of the “V” is deepithelialized and the flap is advanced such that the top of the “V” abuts the inferior border of the defect. The “V” flap is inset at its advanced position, converting the “V”-shaped wound into a “Y.” An overlying buccal mucosal graft provides reconstruction of the lower red lip and labial mucosa.16
Helix of the Ear
Large defects of the scapha and helix of the ear can be reconstructed with the use of a staged interpolated postauricular flap. The postauricular flap is elevated into a subcutaneous plane. A full-thickness incision is made medial to the helical rim, and the flap is tunneled through and sutured into place. The pedicle is later divided, and the distal aspect of the flap is deepithelialized and inset into the helical rim for volume restoration.17
Reconstruction Involving Free Margins
Nasal Ala
For large defects involving the upper cutaneous lip with adjacent alar base involvement, a partially deepithelialized V-Y flap is a useful reconstructive option (Figure 2).
Infraorbital Region
A deepithelialized variant of a V-Y advancement flap can be used for closure of infraorbital defects. The limbs of the V-Y flap are deepithelialized and anchored to the medial and lateral canthal tendons or periosteum. Ectropion prevention is the primary advantage of this flap.18
APPLICATION OF DEEPITHELIALIZED GRAFTS
Deepithelialized grafts may be considered for volume replacement, reconstruction requiring contour preservation, and restoration of mechanical integrity in areas of high mechanical tension.3,19-21
Reconstruction Requiring Contour Preservation
Deepithelialized grafts are used to improve depressed nasal scars and restore volume in deep nasal wounds. One method involves deepithelialization of 2 postauricular punch biopsies. An 18-gauge needle is used to make a small hole in the depressed nasal scar, the dermal grafts are inserted, and the defect is closed primarily.19 Dermal grafts may be harvested from excess full-thickness skin grafts (FTSGs) or dog-ear tissue. When used under flaps, the dermal graft is trimmed to the size of the defect. When used under FTSGs, thin dermal graft strips are placed in a gridlike pattern to allow for revascularization. A study of 15 patients with contour deformities reconstructed with dermal graft insertions demonstrated that 14 (94%) patients had no significant complications and improvement of scar depression was achieved.20
Reconstruction in Areas of High Mechanical Tension
Plantar Foot
A combined dermal and full-thickness sandwich graft has been described for reconstruction of plantar foot defects.3 The graft is created by obtaining a FTSG twice the size of the wound defect and deepithelializing half of the graft. The graft is then defatted and the deepithelialized portion is folded beneath the other half, allowing the papillary dermis to make contact with the wound surface.
Scalp
Dermal graft reconstruction for scalp defects may be accomplished with a split-thickness skin flap. The flap is harvested using an electronic dermatome that ensures the proximal aspect is still attached to adjacent skin. The dermis is removed from the area underneath the back-folded split-thickness skin flap. The dermal graft is meshed and sutured into the recipient site. The split-thickness skin flap is replaced over the donor site. Meshed reversed dermal grafts have excellent survival rates, even with direct placement on bone without periosteum. Querings et al21 reported graft survival with no complications in 19 of 21 (90.4%) patients undergoing scalp or plantar sole reconstruction.
CONCLUSION
With the widespread adoption of the fresh-tissue technique for Mohs micrographic surgery and the establishment of the American Society for Dermatologic Surgery in 1970, the depth and scope of techniques used by dermatologic surgeons has dramatically expanded. Although the use of dermal flaps and grafts is not as widespread in dermatology as other reconstructive techniques, their unique advantages should be considered. Deepithelialized flaps and grafts should be considered when the following reconstructive goals are desired: (1) conversion of a 2-stage interpolation flap to a single-stage tunneled flap, (2) contour and cosmetic subunit preservation of deep defects through volume augmentation, (3) reconstruction in areas of high mechanical tension, and (4) free margin preservation. The multiple applications of deepithelialized flaps and grafts as described in this review demonstrate their continued applicability in dermatologic surgery.
Deepithelialized flaps and grafts have been widely used by reconstructive surgeons in a diverse range of medical specialties since the early 20th century. 1 These reconstructive modalities have more recently been applied to dermatologic surgery. Deepithelialized flaps and grafts involve removal of the epidermis from the dermis for a variety of surgical purposes. Although these techniques play an important role in dermatologic surgery, reports of application of deepithelialized flaps and grafts in the dermatology literature is limited. This article includes a presentation of the applications of deepithelialized flaps and grafts in procedural dermatology.
DEEPITHELIALIZATION TECHNIQUES
There are a variety of techniques for deepithelialization, although sharp deepithelialization generally is preferred by dermatologic surgeons. The scalpel technique can be accomplished by making an intradermal incision with a No. 15 blade. Traction is an essential component of the deepthelialization process and facilitates sharp removal of the epidermis and superficial dermis in an even plane. The peeling orange technique, which has been described in reduction mammoplasty, is a variant of the scalpel technique used for creating a large area of deepithelialized tissue.2 A No. 10 blade is used to make multiple partial-thickness intradermal incisions 1 to 2 cm apart along the pedicle. Traction facilitates rapid deepithelialization of the skin strips on the pedicle. A sharp curette is an alternative option for sharply removing the epithelium from a small area. Electric dermatome, laser, and electrocautery techniques for deepithelialization also can be considered.2,3
APPLICATION OF DEEPITHELIALIZED FLAPS
Deepithelialized flaps may be considered for single-stage reconstruction with tunneled interpolation flaps, reconstruction requiring contour preservation, and reconstruction involving free margins.4-17
Reconstruction With Single-Stage Tunneled Interpolated Flaps
Alar Base
A partially deepithelialized tunneled interpolated flap is an elegant reconstructive option for defects involving the upper cutaneous lip and alar base. The flap is elevated from the ipsilateral nasolabial fold, deepithelialized proximally, and tunneled under the intact portion of the cutaneous upper lip and ala. The flap is then deepithelialized superiorly to bolster the alar base and inset at the recipient site.4
Nasal Ala
The tunneled interpolated flap is useful for reconstruction of defects of the nasal ala. A flap with a superior deepithelialized pedicle and an anticipated inferior Burow triangle is designed along the axis of the nasolabial fold. The inferior Burow triangle and central flap are elevated at the level of the superficial subcutaneous fat and the pedicle is dissected. The donor and recipient sites are widely undermined, and the flap and pedicle pass through the tunnel. The donor site is closed primarily, the inferior Burow triangle is trimmed, and the flap is sutured into the defect.5 This flap allows for preservation of free margins and favorable placement of incision lines. Furthermore, pincushioning of the flap helps to recreate the rounded shape of the lateral ala.6
Nasal Tip
Nasal tip defects can be repaired with a retroangular flap, centered on the angular artery. The flap is elevated along the axis of the nasolabial fold, deepithelialized at its proximal base, and transferred through a subcutaneous tunnel to the nasal tip. The angular artery is ligated at the inferior aspect of the flap.7
Nasal Sidewall
A deepithelialized tunneled interpolated forehead flap, similar to the classic paramedian forehead flap, can be used to reconstruct nasal sidewall defects. A flap is elevated on the contralateral forehead and the proximal portion is deepithelialized. A tunnel is then bluntly dissected just above the periosteum, and the flap is introduced into the defect through the tunnel and inset. This flap has the advantages of being a single-stage procedure, restoring volume to the defect area, and maintaining excellent vascular supply.8
Eyelid
A tunneled interpolated forehead flap also can be used to repair medial canthal defects and for anterior lamellar repair of lower eyelid defects. In a study of 9 patients receiving a tunneled interpolated forehead flap in these anatomic locations, all flaps demonstrated viability, protection of the globe, and preservation of the concave architecture of the medial canthus.9
Earlobe
Earlobe defects may be repaired with a pull-through interpolated preauricular flap. A flap is elevated superiorly in the preauricular region and the proximal aspect of the flap is deepithelialized. The flap is pulled through a tunnel and inset at the anterior earlobe defect. The donor site is closed primarily.10,11
Concha
Reconstruction of anterior conchal defects with exposed cartilage can be accomplished with a pull-through interpolated postauricular flap based on the auriculomastoid fossa. The postauricular flap is elevated, the base is deepithelialized, an incision is made in the medial aspect of the defect, and the flap is moved through a tunnel between the posterior and anterior surfaces of the ear. The flap is secured to the anterior surface of the concha.12
Reconstruction Requiring Contour Preservation
Central Face
The hinge flap is optimal for reconstruction of deep central facial defects (Figure 1). The hinge flap is planned at a site contiguous with a margin of the defect and can include the dermis, subcutaneous tissue, muscle, or a combination of these. The desired tissue is folded over on the pedicle to fill the defect. Cutaneous coverage is accomplished through a primary closure, separate flap, or skin graft. In addition to restoring contour and therefore the cosmetic subunit, the hinge flap is performed in a single stage, resists wound contracture, and provides a well-vascularized wound bed resulting in a low incidence of graft failure.13,14 Muscular hinge flaps have been described for reconstruction of forehead defects with exposed bone based on the frontalis muscle.15
Lower Lip
A variant of a V-Y advancement flap has been described for reconstruction of defects greater than one-third the length of the lower lip. The top of the “V” is deepithelialized and the flap is advanced such that the top of the “V” abuts the inferior border of the defect. The “V” flap is inset at its advanced position, converting the “V”-shaped wound into a “Y.” An overlying buccal mucosal graft provides reconstruction of the lower red lip and labial mucosa.16
Helix of the Ear
Large defects of the scapha and helix of the ear can be reconstructed with the use of a staged interpolated postauricular flap. The postauricular flap is elevated into a subcutaneous plane. A full-thickness incision is made medial to the helical rim, and the flap is tunneled through and sutured into place. The pedicle is later divided, and the distal aspect of the flap is deepithelialized and inset into the helical rim for volume restoration.17
Reconstruction Involving Free Margins
Nasal Ala
For large defects involving the upper cutaneous lip with adjacent alar base involvement, a partially deepithelialized V-Y flap is a useful reconstructive option (Figure 2).
Infraorbital Region
A deepithelialized variant of a V-Y advancement flap can be used for closure of infraorbital defects. The limbs of the V-Y flap are deepithelialized and anchored to the medial and lateral canthal tendons or periosteum. Ectropion prevention is the primary advantage of this flap.18
APPLICATION OF DEEPITHELIALIZED GRAFTS
Deepithelialized grafts may be considered for volume replacement, reconstruction requiring contour preservation, and restoration of mechanical integrity in areas of high mechanical tension.3,19-21
Reconstruction Requiring Contour Preservation
Deepithelialized grafts are used to improve depressed nasal scars and restore volume in deep nasal wounds. One method involves deepithelialization of 2 postauricular punch biopsies. An 18-gauge needle is used to make a small hole in the depressed nasal scar, the dermal grafts are inserted, and the defect is closed primarily.19 Dermal grafts may be harvested from excess full-thickness skin grafts (FTSGs) or dog-ear tissue. When used under flaps, the dermal graft is trimmed to the size of the defect. When used under FTSGs, thin dermal graft strips are placed in a gridlike pattern to allow for revascularization. A study of 15 patients with contour deformities reconstructed with dermal graft insertions demonstrated that 14 (94%) patients had no significant complications and improvement of scar depression was achieved.20
Reconstruction in Areas of High Mechanical Tension
Plantar Foot
A combined dermal and full-thickness sandwich graft has been described for reconstruction of plantar foot defects.3 The graft is created by obtaining a FTSG twice the size of the wound defect and deepithelializing half of the graft. The graft is then defatted and the deepithelialized portion is folded beneath the other half, allowing the papillary dermis to make contact with the wound surface.
Scalp
Dermal graft reconstruction for scalp defects may be accomplished with a split-thickness skin flap. The flap is harvested using an electronic dermatome that ensures the proximal aspect is still attached to adjacent skin. The dermis is removed from the area underneath the back-folded split-thickness skin flap. The dermal graft is meshed and sutured into the recipient site. The split-thickness skin flap is replaced over the donor site. Meshed reversed dermal grafts have excellent survival rates, even with direct placement on bone without periosteum. Querings et al21 reported graft survival with no complications in 19 of 21 (90.4%) patients undergoing scalp or plantar sole reconstruction.
CONCLUSION
With the widespread adoption of the fresh-tissue technique for Mohs micrographic surgery and the establishment of the American Society for Dermatologic Surgery in 1970, the depth and scope of techniques used by dermatologic surgeons has dramatically expanded. Although the use of dermal flaps and grafts is not as widespread in dermatology as other reconstructive techniques, their unique advantages should be considered. Deepithelialized flaps and grafts should be considered when the following reconstructive goals are desired: (1) conversion of a 2-stage interpolation flap to a single-stage tunneled flap, (2) contour and cosmetic subunit preservation of deep defects through volume augmentation, (3) reconstruction in areas of high mechanical tension, and (4) free margin preservation. The multiple applications of deepithelialized flaps and grafts as described in this review demonstrate their continued applicability in dermatologic surgery.
- Straatsma CR. Use of the dermal graft in the repairs of small saddle defects of the nose. Arch Otolaryngol. 1932;16:506-509.
- Cydeli A, Hunter J. Peeling orange: rapid deepithelialization in reduction mammoplasty. J Aesthet Surg. 2004;24:580-581.
- Bechara F, Sand M, Radenhausen M, et al. Erbium:YAG laser-assisted preparation of a combined dermal/full thickness sandwich skin graft. Dermatol Surg. 2006;32:353-358.
- Cook JL. Tunneled and transposed island flaps in facial reconstructive surgery. Dermatol Surg. 2014;40(suppl 9):S16-S29.
- Krishnan RS, Clark DP. Tunneled transposition flap for reconstruction of defects of the nasal ala. Dermatol Surg. 2007;33:1496-1501.
- Mahlberg M. Tunneled melolabial pedicle flap for small but deep lateral alar rim defect. Dermatol Surg. 2013;39:1527-1529.
- Ascari-Raccagni A, Balderi U. The retroangular flap used in the surgery of nasal tip defects. Dermatol Surg. 2004;30:1131-1137.
- Hollmig ST, Leach BC, Cook J. Single-staged interpolation flaps in facial reconstruction. Dermatol Surg. 2014;40(suppl 9):S62-S70.
- Mombaerts I, Gillis A. The tunneled forehead flap in medial canthal and eyelid reconstruction. Dermatol Surg. 2010:36:1118-1125.
- Wang SQ, Goldberg LH, Kimyah-Asadi A. Tunneled island pedicle flap for an earlobe defect. Dermatol Surg. 2007;33:835-838.
- Hatoko M, Kuwahara M, Shiba A, et al. Earlobe reconstruction using a subcutaneous island pedicle flap after resection of “earlobe keloid.” Dermatol Surg. 1998;24:257-261.
- Alder N, Ad-El D, Azaria R. Reconstruction of nonhelical auricular defects with local flaps. Dermatol Surg. 2008;34:501-507.
- Fader DJ, Wang TS, Johnson TM. Nasal reconstruction utilizing a muscle hinge flap with overlying FTSG. J Am Acad Dermatol. 2000;43:837-840.
- Braun MA, Cook J. Hinge flaps in facial reconstruction. Dermatol Surg. 2007;33:213-221.
- Salmon PL, Mortimer NL, Hill SE. Muscular hinge flaps: utility and technique in facial reconstructive surgery. Dermatol Surg. 2010;36:227-234.
- Seo Y, Song S, Choi Y, et al. A lower lip reconstruction. Dermatol Surg. 2015;41:505-507.
- Malone CH, Wagner RF. Partially de-epithelialized postauricular flap for ear reconstruction. J Am Acad Dermatol. 2015;73:E219-E220.
- Yildrim S, Akoz T, Akan M, et al. Nasolabial V-Y advancement for closure of the midface defects. Dermatol Surg. 2001;27:656-662.
- Jensen DJ, Cohen JL. Nasal tip revision using a dermal graft. Dermatol Surg. 2014;40:1140-1142.
- Meyers S, Rohrer T. Use of dermal grafts in reconstructing deep nasal defects and shaping the ala nasi. Dermatol Surg. 2001;27:300-305.
- Querings K, Bachter D, Balda B. Meshed reversed dermal graft in patients with surgical defects of sole and scalp: technique and long-term results. Dermatol Surg. 2002;28:122-126.
- Straatsma CR. Use of the dermal graft in the repairs of small saddle defects of the nose. Arch Otolaryngol. 1932;16:506-509.
- Cydeli A, Hunter J. Peeling orange: rapid deepithelialization in reduction mammoplasty. J Aesthet Surg. 2004;24:580-581.
- Bechara F, Sand M, Radenhausen M, et al. Erbium:YAG laser-assisted preparation of a combined dermal/full thickness sandwich skin graft. Dermatol Surg. 2006;32:353-358.
- Cook JL. Tunneled and transposed island flaps in facial reconstructive surgery. Dermatol Surg. 2014;40(suppl 9):S16-S29.
- Krishnan RS, Clark DP. Tunneled transposition flap for reconstruction of defects of the nasal ala. Dermatol Surg. 2007;33:1496-1501.
- Mahlberg M. Tunneled melolabial pedicle flap for small but deep lateral alar rim defect. Dermatol Surg. 2013;39:1527-1529.
- Ascari-Raccagni A, Balderi U. The retroangular flap used in the surgery of nasal tip defects. Dermatol Surg. 2004;30:1131-1137.
- Hollmig ST, Leach BC, Cook J. Single-staged interpolation flaps in facial reconstruction. Dermatol Surg. 2014;40(suppl 9):S62-S70.
- Mombaerts I, Gillis A. The tunneled forehead flap in medial canthal and eyelid reconstruction. Dermatol Surg. 2010:36:1118-1125.
- Wang SQ, Goldberg LH, Kimyah-Asadi A. Tunneled island pedicle flap for an earlobe defect. Dermatol Surg. 2007;33:835-838.
- Hatoko M, Kuwahara M, Shiba A, et al. Earlobe reconstruction using a subcutaneous island pedicle flap after resection of “earlobe keloid.” Dermatol Surg. 1998;24:257-261.
- Alder N, Ad-El D, Azaria R. Reconstruction of nonhelical auricular defects with local flaps. Dermatol Surg. 2008;34:501-507.
- Fader DJ, Wang TS, Johnson TM. Nasal reconstruction utilizing a muscle hinge flap with overlying FTSG. J Am Acad Dermatol. 2000;43:837-840.
- Braun MA, Cook J. Hinge flaps in facial reconstruction. Dermatol Surg. 2007;33:213-221.
- Salmon PL, Mortimer NL, Hill SE. Muscular hinge flaps: utility and technique in facial reconstructive surgery. Dermatol Surg. 2010;36:227-234.
- Seo Y, Song S, Choi Y, et al. A lower lip reconstruction. Dermatol Surg. 2015;41:505-507.
- Malone CH, Wagner RF. Partially de-epithelialized postauricular flap for ear reconstruction. J Am Acad Dermatol. 2015;73:E219-E220.
- Yildrim S, Akoz T, Akan M, et al. Nasolabial V-Y advancement for closure of the midface defects. Dermatol Surg. 2001;27:656-662.
- Jensen DJ, Cohen JL. Nasal tip revision using a dermal graft. Dermatol Surg. 2014;40:1140-1142.
- Meyers S, Rohrer T. Use of dermal grafts in reconstructing deep nasal defects and shaping the ala nasi. Dermatol Surg. 2001;27:300-305.
- Querings K, Bachter D, Balda B. Meshed reversed dermal graft in patients with surgical defects of sole and scalp: technique and long-term results. Dermatol Surg. 2002;28:122-126.
Practice Points
- Deepithelialized flaps should be considered for single-stage reconstruction with tunneled interpolation flaps, reconstruction requiring contour preservation, and reconstruction involving free margins.
- Deepithelialized grafts may be considered for volume replacement, reconstruction requiring contour preservation, and reconstruction in areas of high mechanical tension.
Do Psoriasis Patients Engage In Vigorous Physical Activity?
Psoriasis is a chronic inflammatory disease that affects approximately 2% to 3% of the US population.1 Patients with psoriasis are more likely to have cardiovascular risk factors (eg, obesity, metabolic syndrome) than individuals without psoriasis.2 In fact, recent evidence has suggested that a diagnosis of psoriasis is an independent risk factor for cardiometabolic diseases including diabetes, major adverse cardiovascular events, and obesity.3 Given the well-recognized health benefits of physical activity and the associated reduction in coronary heart disease risk,4 patients with psoriasis specifically may benefit from regular participation in physical activity. Thus, an enhanced understanding of the relationship between psoriasis and vigorous physical activity would help determine the role of initiating and recommending interventions that implement physical activity for patients with psoriasis. A review was conducted to determine the relationship between psoriasis and vigorous physical activity.
Methods
An English-language literature search of PubMed articles indexed for MEDLINE (January 1, 1946–October 15, 2017) as well as articles in the Embase database (January 1, 1947–October 15, 2017) and Cochrane Library (January 1, 1992–October 15, 2017) using the terms psoriasis and physical activity was performed. The search strategy was established based on a prior review of vigorous physical activity in eczema.5 The article titles and/or abstracts were reviewed, and the studies were excluded if they did not evaluate physical activity in patients with psoriasis. Studies without a control group also were excluded. Articles on patients with psoriatic arthritis and studies that involved modification of dietary intake also were excluded.
Two reviewers (M.A. and E.B.L.) independently extracted data from the studies and compiled the results. The following factors were included in the data extracted: study year, location, and design; method of diagnosis of psoriasis; total number of patients included in the study; and age, gender, and level of physical activity of the study patients. Level of physical activity was the exposure, and diagnosis of psoriasis was the dependent variable. Physical activity was defined differently across the studies that were evaluated. To determine study quality, we implemented the Newcastle–Ottawa Scale (NOS), a 9-star scoring system that includes items such as selection criteria, comparability, and study outcome.6 Studies with an NOS score of 7 or higher were included in the meta-analysis.
Results
The literature search generated 353 nonduplicate articles. A thorough review of the articles yielded 4 studies that were incorporated in the final analysis.7-10 We aimed to perform a meta-analysis; however, only 1 of the studies included in the final analysis had an NOS score of 7 or higher along with adequate data to be incorporated into our study.10 As a result, the meta-analysis was converted to a regular review.
The cross-sectional study we reviewed, which had an NOS score of 7, included males and females in the United States aged 20 to 59 years.10 Data were collected using the population-based National Health and Nutrition Examination Survey from 2003 to 2006. The survey measured the likelihood of participation in leisure-time moderate to vigorous physical activity (MVPA) and metabolic equivalent task (MET) minutes of MVPA in the past 30 days. Of 6549 participants, 385 were excluded from the analysis due to missing values for 1 or more of the study variables. Of the remaining 6164 participants, 84 (1.4%) reported having a diagnosis of psoriasis with few or no psoriasis patches at the time of the survey, and 71 (1.2%) reported having a diagnosis of psoriasis with few to extensive patches at the time of the survey.10
Participants with psoriasis were less likely to participate in MVPA in the previous 30 days compared to participants without psoriasis, but the association was not statistically significant.10 The study demonstrated that, on average, participants with psoriasis spent 31% (95% confidence interval [CI], −0.57 to −0.05) fewer MET minutes on leisure-time MVPA versus participants without psoriasis; however, this association was not statistically significant. It is important to note that the diagnosis of psoriasis was self-reported, and measures of disease duration or areas of involvement were not incorporated.
Comment
Our review revealed that vigorous physical activity may be reduced in patients with psoriasis compared to those without psoriasis. Initially, we aimed to perform a systematic review of the literature; however, only 1 study met the criteria for the systematic review, highlighting the need for more robust studies evaluating this subject.
Do et al10 demonstrated that psoriasis patients were less likely to participate in MVPA, but the findings were not statistically significant. Of those who participated in MVPA, MET minutes were fewer among patients with few to extensive skin lesions compared to those without psoriasis. The investigators suggested that psoriasis patients with more severe disease tend to exercise less and ultimately would benefit from regular vigorous physical activity.
Frankel et al7 performed a prospective cohort study in US women to evaluate the role of physical activity in preventing psoriasis. The investigators reported that the most physically active quintile had a lower multivariate relative risk of psoriasis (0.72; 95% CI, 0.59–0.89; P<.001 for trend) compared to the least active quintile.7 Additionally, vigorous physical activity, which was defined as 6 or more MET minutes, was associated with a significantly lower risk of incident psoriasis (0.66; 95% CI, 0.54–0.81; P<.001 for trend), which maintained significance after adjusting for body mass index (BMI). The investigators suggested that, by decreasing chronic inflammation and lowering levels of proinflammatory cytokines, vigorous physical activity may reduce the risk of psoriasis development in women.7 It is plausible that vigorous physical activity modifies the state of chronic inflammation, which could subsequently reduce the risk of developing psoriasis; however, further long-term, randomized, prospective studies are needed to verify the relationship between physical activity and development of psoriasis.
Torres et al8 performed a cross-sectional questionnaire study to assess physical activity in patients with severe psoriasis (defined as >10% body surface area involvement and/or disease requiring systemic therapy or phototherapy) versus healthy controls. Physical activity level was measured using the International Physical Activity Questionnaire. The odds ratio of low-level physical activity compared to non–low-level physical activity among psoriasis patients versus controls was 3.42 (95% CI, 1.47–7.91; P=.002). Additionally, the average total MET minutes of psoriasis patients were significantly reduced compared to those of the healthy controls (P=.001). Thus, the investigators suggested that vigorous physical activity is less likely in psoriasis patients, which may contribute to the increased risk of cardiovascular disease in this population.8 Vigorous physical activity would benefit patients with psoriasis to help lower the chronic state of inflammation and cardiometabolic comorbidities.
Demirel et al9 performed a study to compare aerobic exercise capacity and daily physical activity level in psoriasis patients (n=30) compared to controls (n=30). Daily physical activity, measured with an accelerometer, was significantly higher in male patients with psoriasis compared to controls (P=.021). No significant difference was reported in maximal aerobic capacity in both male and female psoriasis patients versus controls. The investigators suggested that the level of daily physical activity is not limited in psoriasis patients, yet the small sample size may limit the generalizability of the study.
The ability to dissipate heat during exercise seems to be diminished in patients with psoriasis. Specifically, it has been suggested that psoriasis lesions interfere with normal perspiration.11 Moreover, joint involvement in patients with psoriatic arthritis may lead to physical functional disabilities that can interfere with the ability of these patients to participate in regular physical activity.12-14 For this reason, our review excluded articles that evaluated patients with psoriatic arthritis. Despite this exclusion, it is important to consider that comorbid psoriatic arthritis in clinical practice may impede patients with psoriasis from participating in physical activity. Additionally, various social aspects also may limit physical activity in psoriasis patients; for instance, psoriasis patients often avoid activities that involve increased exposure of the skin (eg, communal showers, wearing sports attire).15
Furthermore, obese psoriasis patients are less likely to exercise compared to obese individuals without psoriasis.16 In patients with higher BMI, the risk of psoriasis is increased.17 A systematic review suggested that weight loss may improve psoriasis severity.18 Bariatric surgery also may improve psoriasis.19 Moreover, obesity may interfere with response to biologic therapies for psoriasis. Specifically, higher BMI is linked with lower response to fixed-dose biologic therapies compared to weight-based biologic options (eg, infliximab).20,21
Conclusion
Given the increased risk of myocardial infarction in patients with psoriasis, it is important to recognize the barriers to physical activity that psoriasis patients face.22 Due to the considerable health benefits associated with regular physical activity, physicians should encourage patients with psoriasis to participate in physical activity as tolerated. Of note, the studies included in this review varied in their definitions of psoriasis disease severity and measures of physical activity level. Long-term, randomized, prospective studies are needed to clarify the relationship between psoriasis and physical activity. Evidence from these studies would help guide clinical recommendations regarding the role of physical activity for patients with psoriasis.
- Takeshita J, Gelfand JM, Li P, et al. Psoriasis in the US Medicare population: prevalence, treatment, and factors associated with biologic use. J Invest Dermatol. 2015;135:2955-2963.
- Prey S, Paul C, Bronsard V, et al. Cardiovascular risk factors in patients with plaque psoriasis: a systematic review of epidemiological studies. J Eur Acad Dermatol Venereol. 2010;24(suppl 2):23-30.
- Takeshita J, Grewal S, Langan SM, et al. Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol. 2017;76:377-390.
- Leon AS. Biological mechanisms for the cardioprotective effects of aerobic exercise. Am J Lifestyle Med. 2009;3:32S-34S.
- Kim A, Silverberg JI. A systematic review of vigorous physical activity in eczema. Br J Dermatol. 2016;174:660-662.
- Wells GA, Shea B, O’Connell D, et al. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomized studies in meta-analyses. The Ottawa Hospital Research Institute website. http://www.ohri.ca/programs/clinical_epidemiology/oxford.htm. Accessed February 23, 2018.
- Frankel HC, Han J, Li T, et al. The association between physical activity and the risk of incident psoriasis. Arch Dermatol. 2012;148:918-924.
- Torres T, Alexandre JM, Mendonça D, et al. Levels of physical activity in patients with severe psoriasis: a cross-sectional questionnaire study. Am J Clin Dermatol. 2014;15:129-135.
- Demirel R, Genc A, Ucok K, et al. Do patients with mild to moderate psoriasis really have a sedentary lifestyle? Int J Dermatol. 2013;52:1129-1134.
- Do YK, Lakhani N, Malhotra R, et al. Association between psoriasis and leisure‐time physical activity: findings from the National Health and Nutrition Examination Survey. J Dermatol. 2015;42:148-153.
- Leibowitz E, Seidman DS, Laor A, et al. Are psoriatic patients at risk of heat intolerance? Br J Dermatol. 1991;124:439-442.
- Husted JA, Tom BD, Farewell VT, et al. Description and prediction of physical functional disability in psoriatic arthritis: a longitudinal analysis using a Markov model approach. Arthritis Rheum. 2005;53:404-409.
- Wilson FC, Icen M, Crowson CS, et al. Incidence and clinical predictors of psoriatic arthritis in patients with psoriasis: a population‐based study. Arthritis Rheum. 2009;61:233-239.
- Shih M, Hootman JM, Kruger J, et al. Physical activity in men and women with arthritis: National Health Interview Survey, 2002. Am J Prev Med. 2006;30:385-393.
- Ramsay B, O’Reagan M. A survey of the social and psychological effects of psoriasis. Br J Dermatol. 1988;118:195-201.
- Herron MD, Hinckley M, Hoffman MS, et al. Impact of obesity and smoking on psoriasis presentation and management. Arch Dermatol. 2005;141:1527-1534.
- Kumar S, Han J, Li T, et al. Obesity, waist circumference, weight change and the risk of psoriasis in US women. J Eur Acad Dermatol Venereol. 2013;27:1293-1298.
- Upala S, Sanguankeo A. Effect of lifestyle weight loss intervention on disease severity in patients with psoriasis: a systematic review and meta-analysis. Int J Obes (Lond). 2015;39:1197-1202.
- Sako EY, Famenini S, Wu JJ. Bariatric surgery and psoriasis. J Am Acad Dermatol. 2014;70:774-779.
- Clark L, Lebwohl M. The effect of weight on the efficacy of biologic therapy in patients with psoriasis. J Am Acad Dermatol. 2008;58:443-446.
- Puig L. Obesity and psoriasis: body weight and body mass index influence the response to biological treatment. J Eur Acad Dermatol Venereol. 2011;25:1007-1011.
- Wu JJ, Choi YM, Bebchuk JD. Risk of myocardial infarction in psoriasis patients: a retrospective cohort study. J Dermatolog Treat. 2015;26:230-234.
Psoriasis is a chronic inflammatory disease that affects approximately 2% to 3% of the US population.1 Patients with psoriasis are more likely to have cardiovascular risk factors (eg, obesity, metabolic syndrome) than individuals without psoriasis.2 In fact, recent evidence has suggested that a diagnosis of psoriasis is an independent risk factor for cardiometabolic diseases including diabetes, major adverse cardiovascular events, and obesity.3 Given the well-recognized health benefits of physical activity and the associated reduction in coronary heart disease risk,4 patients with psoriasis specifically may benefit from regular participation in physical activity. Thus, an enhanced understanding of the relationship between psoriasis and vigorous physical activity would help determine the role of initiating and recommending interventions that implement physical activity for patients with psoriasis. A review was conducted to determine the relationship between psoriasis and vigorous physical activity.
Methods
An English-language literature search of PubMed articles indexed for MEDLINE (January 1, 1946–October 15, 2017) as well as articles in the Embase database (January 1, 1947–October 15, 2017) and Cochrane Library (January 1, 1992–October 15, 2017) using the terms psoriasis and physical activity was performed. The search strategy was established based on a prior review of vigorous physical activity in eczema.5 The article titles and/or abstracts were reviewed, and the studies were excluded if they did not evaluate physical activity in patients with psoriasis. Studies without a control group also were excluded. Articles on patients with psoriatic arthritis and studies that involved modification of dietary intake also were excluded.
Two reviewers (M.A. and E.B.L.) independently extracted data from the studies and compiled the results. The following factors were included in the data extracted: study year, location, and design; method of diagnosis of psoriasis; total number of patients included in the study; and age, gender, and level of physical activity of the study patients. Level of physical activity was the exposure, and diagnosis of psoriasis was the dependent variable. Physical activity was defined differently across the studies that were evaluated. To determine study quality, we implemented the Newcastle–Ottawa Scale (NOS), a 9-star scoring system that includes items such as selection criteria, comparability, and study outcome.6 Studies with an NOS score of 7 or higher were included in the meta-analysis.
Results
The literature search generated 353 nonduplicate articles. A thorough review of the articles yielded 4 studies that were incorporated in the final analysis.7-10 We aimed to perform a meta-analysis; however, only 1 of the studies included in the final analysis had an NOS score of 7 or higher along with adequate data to be incorporated into our study.10 As a result, the meta-analysis was converted to a regular review.
The cross-sectional study we reviewed, which had an NOS score of 7, included males and females in the United States aged 20 to 59 years.10 Data were collected using the population-based National Health and Nutrition Examination Survey from 2003 to 2006. The survey measured the likelihood of participation in leisure-time moderate to vigorous physical activity (MVPA) and metabolic equivalent task (MET) minutes of MVPA in the past 30 days. Of 6549 participants, 385 were excluded from the analysis due to missing values for 1 or more of the study variables. Of the remaining 6164 participants, 84 (1.4%) reported having a diagnosis of psoriasis with few or no psoriasis patches at the time of the survey, and 71 (1.2%) reported having a diagnosis of psoriasis with few to extensive patches at the time of the survey.10
Participants with psoriasis were less likely to participate in MVPA in the previous 30 days compared to participants without psoriasis, but the association was not statistically significant.10 The study demonstrated that, on average, participants with psoriasis spent 31% (95% confidence interval [CI], −0.57 to −0.05) fewer MET minutes on leisure-time MVPA versus participants without psoriasis; however, this association was not statistically significant. It is important to note that the diagnosis of psoriasis was self-reported, and measures of disease duration or areas of involvement were not incorporated.
Comment
Our review revealed that vigorous physical activity may be reduced in patients with psoriasis compared to those without psoriasis. Initially, we aimed to perform a systematic review of the literature; however, only 1 study met the criteria for the systematic review, highlighting the need for more robust studies evaluating this subject.
Do et al10 demonstrated that psoriasis patients were less likely to participate in MVPA, but the findings were not statistically significant. Of those who participated in MVPA, MET minutes were fewer among patients with few to extensive skin lesions compared to those without psoriasis. The investigators suggested that psoriasis patients with more severe disease tend to exercise less and ultimately would benefit from regular vigorous physical activity.
Frankel et al7 performed a prospective cohort study in US women to evaluate the role of physical activity in preventing psoriasis. The investigators reported that the most physically active quintile had a lower multivariate relative risk of psoriasis (0.72; 95% CI, 0.59–0.89; P<.001 for trend) compared to the least active quintile.7 Additionally, vigorous physical activity, which was defined as 6 or more MET minutes, was associated with a significantly lower risk of incident psoriasis (0.66; 95% CI, 0.54–0.81; P<.001 for trend), which maintained significance after adjusting for body mass index (BMI). The investigators suggested that, by decreasing chronic inflammation and lowering levels of proinflammatory cytokines, vigorous physical activity may reduce the risk of psoriasis development in women.7 It is plausible that vigorous physical activity modifies the state of chronic inflammation, which could subsequently reduce the risk of developing psoriasis; however, further long-term, randomized, prospective studies are needed to verify the relationship between physical activity and development of psoriasis.
Torres et al8 performed a cross-sectional questionnaire study to assess physical activity in patients with severe psoriasis (defined as >10% body surface area involvement and/or disease requiring systemic therapy or phototherapy) versus healthy controls. Physical activity level was measured using the International Physical Activity Questionnaire. The odds ratio of low-level physical activity compared to non–low-level physical activity among psoriasis patients versus controls was 3.42 (95% CI, 1.47–7.91; P=.002). Additionally, the average total MET minutes of psoriasis patients were significantly reduced compared to those of the healthy controls (P=.001). Thus, the investigators suggested that vigorous physical activity is less likely in psoriasis patients, which may contribute to the increased risk of cardiovascular disease in this population.8 Vigorous physical activity would benefit patients with psoriasis to help lower the chronic state of inflammation and cardiometabolic comorbidities.
Demirel et al9 performed a study to compare aerobic exercise capacity and daily physical activity level in psoriasis patients (n=30) compared to controls (n=30). Daily physical activity, measured with an accelerometer, was significantly higher in male patients with psoriasis compared to controls (P=.021). No significant difference was reported in maximal aerobic capacity in both male and female psoriasis patients versus controls. The investigators suggested that the level of daily physical activity is not limited in psoriasis patients, yet the small sample size may limit the generalizability of the study.
The ability to dissipate heat during exercise seems to be diminished in patients with psoriasis. Specifically, it has been suggested that psoriasis lesions interfere with normal perspiration.11 Moreover, joint involvement in patients with psoriatic arthritis may lead to physical functional disabilities that can interfere with the ability of these patients to participate in regular physical activity.12-14 For this reason, our review excluded articles that evaluated patients with psoriatic arthritis. Despite this exclusion, it is important to consider that comorbid psoriatic arthritis in clinical practice may impede patients with psoriasis from participating in physical activity. Additionally, various social aspects also may limit physical activity in psoriasis patients; for instance, psoriasis patients often avoid activities that involve increased exposure of the skin (eg, communal showers, wearing sports attire).15
Furthermore, obese psoriasis patients are less likely to exercise compared to obese individuals without psoriasis.16 In patients with higher BMI, the risk of psoriasis is increased.17 A systematic review suggested that weight loss may improve psoriasis severity.18 Bariatric surgery also may improve psoriasis.19 Moreover, obesity may interfere with response to biologic therapies for psoriasis. Specifically, higher BMI is linked with lower response to fixed-dose biologic therapies compared to weight-based biologic options (eg, infliximab).20,21
Conclusion
Given the increased risk of myocardial infarction in patients with psoriasis, it is important to recognize the barriers to physical activity that psoriasis patients face.22 Due to the considerable health benefits associated with regular physical activity, physicians should encourage patients with psoriasis to participate in physical activity as tolerated. Of note, the studies included in this review varied in their definitions of psoriasis disease severity and measures of physical activity level. Long-term, randomized, prospective studies are needed to clarify the relationship between psoriasis and physical activity. Evidence from these studies would help guide clinical recommendations regarding the role of physical activity for patients with psoriasis.
Psoriasis is a chronic inflammatory disease that affects approximately 2% to 3% of the US population.1 Patients with psoriasis are more likely to have cardiovascular risk factors (eg, obesity, metabolic syndrome) than individuals without psoriasis.2 In fact, recent evidence has suggested that a diagnosis of psoriasis is an independent risk factor for cardiometabolic diseases including diabetes, major adverse cardiovascular events, and obesity.3 Given the well-recognized health benefits of physical activity and the associated reduction in coronary heart disease risk,4 patients with psoriasis specifically may benefit from regular participation in physical activity. Thus, an enhanced understanding of the relationship between psoriasis and vigorous physical activity would help determine the role of initiating and recommending interventions that implement physical activity for patients with psoriasis. A review was conducted to determine the relationship between psoriasis and vigorous physical activity.
Methods
An English-language literature search of PubMed articles indexed for MEDLINE (January 1, 1946–October 15, 2017) as well as articles in the Embase database (January 1, 1947–October 15, 2017) and Cochrane Library (January 1, 1992–October 15, 2017) using the terms psoriasis and physical activity was performed. The search strategy was established based on a prior review of vigorous physical activity in eczema.5 The article titles and/or abstracts were reviewed, and the studies were excluded if they did not evaluate physical activity in patients with psoriasis. Studies without a control group also were excluded. Articles on patients with psoriatic arthritis and studies that involved modification of dietary intake also were excluded.
Two reviewers (M.A. and E.B.L.) independently extracted data from the studies and compiled the results. The following factors were included in the data extracted: study year, location, and design; method of diagnosis of psoriasis; total number of patients included in the study; and age, gender, and level of physical activity of the study patients. Level of physical activity was the exposure, and diagnosis of psoriasis was the dependent variable. Physical activity was defined differently across the studies that were evaluated. To determine study quality, we implemented the Newcastle–Ottawa Scale (NOS), a 9-star scoring system that includes items such as selection criteria, comparability, and study outcome.6 Studies with an NOS score of 7 or higher were included in the meta-analysis.
Results
The literature search generated 353 nonduplicate articles. A thorough review of the articles yielded 4 studies that were incorporated in the final analysis.7-10 We aimed to perform a meta-analysis; however, only 1 of the studies included in the final analysis had an NOS score of 7 or higher along with adequate data to be incorporated into our study.10 As a result, the meta-analysis was converted to a regular review.
The cross-sectional study we reviewed, which had an NOS score of 7, included males and females in the United States aged 20 to 59 years.10 Data were collected using the population-based National Health and Nutrition Examination Survey from 2003 to 2006. The survey measured the likelihood of participation in leisure-time moderate to vigorous physical activity (MVPA) and metabolic equivalent task (MET) minutes of MVPA in the past 30 days. Of 6549 participants, 385 were excluded from the analysis due to missing values for 1 or more of the study variables. Of the remaining 6164 participants, 84 (1.4%) reported having a diagnosis of psoriasis with few or no psoriasis patches at the time of the survey, and 71 (1.2%) reported having a diagnosis of psoriasis with few to extensive patches at the time of the survey.10
Participants with psoriasis were less likely to participate in MVPA in the previous 30 days compared to participants without psoriasis, but the association was not statistically significant.10 The study demonstrated that, on average, participants with psoriasis spent 31% (95% confidence interval [CI], −0.57 to −0.05) fewer MET minutes on leisure-time MVPA versus participants without psoriasis; however, this association was not statistically significant. It is important to note that the diagnosis of psoriasis was self-reported, and measures of disease duration or areas of involvement were not incorporated.
Comment
Our review revealed that vigorous physical activity may be reduced in patients with psoriasis compared to those without psoriasis. Initially, we aimed to perform a systematic review of the literature; however, only 1 study met the criteria for the systematic review, highlighting the need for more robust studies evaluating this subject.
Do et al10 demonstrated that psoriasis patients were less likely to participate in MVPA, but the findings were not statistically significant. Of those who participated in MVPA, MET minutes were fewer among patients with few to extensive skin lesions compared to those without psoriasis. The investigators suggested that psoriasis patients with more severe disease tend to exercise less and ultimately would benefit from regular vigorous physical activity.
Frankel et al7 performed a prospective cohort study in US women to evaluate the role of physical activity in preventing psoriasis. The investigators reported that the most physically active quintile had a lower multivariate relative risk of psoriasis (0.72; 95% CI, 0.59–0.89; P<.001 for trend) compared to the least active quintile.7 Additionally, vigorous physical activity, which was defined as 6 or more MET minutes, was associated with a significantly lower risk of incident psoriasis (0.66; 95% CI, 0.54–0.81; P<.001 for trend), which maintained significance after adjusting for body mass index (BMI). The investigators suggested that, by decreasing chronic inflammation and lowering levels of proinflammatory cytokines, vigorous physical activity may reduce the risk of psoriasis development in women.7 It is plausible that vigorous physical activity modifies the state of chronic inflammation, which could subsequently reduce the risk of developing psoriasis; however, further long-term, randomized, prospective studies are needed to verify the relationship between physical activity and development of psoriasis.
Torres et al8 performed a cross-sectional questionnaire study to assess physical activity in patients with severe psoriasis (defined as >10% body surface area involvement and/or disease requiring systemic therapy or phototherapy) versus healthy controls. Physical activity level was measured using the International Physical Activity Questionnaire. The odds ratio of low-level physical activity compared to non–low-level physical activity among psoriasis patients versus controls was 3.42 (95% CI, 1.47–7.91; P=.002). Additionally, the average total MET minutes of psoriasis patients were significantly reduced compared to those of the healthy controls (P=.001). Thus, the investigators suggested that vigorous physical activity is less likely in psoriasis patients, which may contribute to the increased risk of cardiovascular disease in this population.8 Vigorous physical activity would benefit patients with psoriasis to help lower the chronic state of inflammation and cardiometabolic comorbidities.
Demirel et al9 performed a study to compare aerobic exercise capacity and daily physical activity level in psoriasis patients (n=30) compared to controls (n=30). Daily physical activity, measured with an accelerometer, was significantly higher in male patients with psoriasis compared to controls (P=.021). No significant difference was reported in maximal aerobic capacity in both male and female psoriasis patients versus controls. The investigators suggested that the level of daily physical activity is not limited in psoriasis patients, yet the small sample size may limit the generalizability of the study.
The ability to dissipate heat during exercise seems to be diminished in patients with psoriasis. Specifically, it has been suggested that psoriasis lesions interfere with normal perspiration.11 Moreover, joint involvement in patients with psoriatic arthritis may lead to physical functional disabilities that can interfere with the ability of these patients to participate in regular physical activity.12-14 For this reason, our review excluded articles that evaluated patients with psoriatic arthritis. Despite this exclusion, it is important to consider that comorbid psoriatic arthritis in clinical practice may impede patients with psoriasis from participating in physical activity. Additionally, various social aspects also may limit physical activity in psoriasis patients; for instance, psoriasis patients often avoid activities that involve increased exposure of the skin (eg, communal showers, wearing sports attire).15
Furthermore, obese psoriasis patients are less likely to exercise compared to obese individuals without psoriasis.16 In patients with higher BMI, the risk of psoriasis is increased.17 A systematic review suggested that weight loss may improve psoriasis severity.18 Bariatric surgery also may improve psoriasis.19 Moreover, obesity may interfere with response to biologic therapies for psoriasis. Specifically, higher BMI is linked with lower response to fixed-dose biologic therapies compared to weight-based biologic options (eg, infliximab).20,21
Conclusion
Given the increased risk of myocardial infarction in patients with psoriasis, it is important to recognize the barriers to physical activity that psoriasis patients face.22 Due to the considerable health benefits associated with regular physical activity, physicians should encourage patients with psoriasis to participate in physical activity as tolerated. Of note, the studies included in this review varied in their definitions of psoriasis disease severity and measures of physical activity level. Long-term, randomized, prospective studies are needed to clarify the relationship between psoriasis and physical activity. Evidence from these studies would help guide clinical recommendations regarding the role of physical activity for patients with psoriasis.
- Takeshita J, Gelfand JM, Li P, et al. Psoriasis in the US Medicare population: prevalence, treatment, and factors associated with biologic use. J Invest Dermatol. 2015;135:2955-2963.
- Prey S, Paul C, Bronsard V, et al. Cardiovascular risk factors in patients with plaque psoriasis: a systematic review of epidemiological studies. J Eur Acad Dermatol Venereol. 2010;24(suppl 2):23-30.
- Takeshita J, Grewal S, Langan SM, et al. Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol. 2017;76:377-390.
- Leon AS. Biological mechanisms for the cardioprotective effects of aerobic exercise. Am J Lifestyle Med. 2009;3:32S-34S.
- Kim A, Silverberg JI. A systematic review of vigorous physical activity in eczema. Br J Dermatol. 2016;174:660-662.
- Wells GA, Shea B, O’Connell D, et al. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomized studies in meta-analyses. The Ottawa Hospital Research Institute website. http://www.ohri.ca/programs/clinical_epidemiology/oxford.htm. Accessed February 23, 2018.
- Frankel HC, Han J, Li T, et al. The association between physical activity and the risk of incident psoriasis. Arch Dermatol. 2012;148:918-924.
- Torres T, Alexandre JM, Mendonça D, et al. Levels of physical activity in patients with severe psoriasis: a cross-sectional questionnaire study. Am J Clin Dermatol. 2014;15:129-135.
- Demirel R, Genc A, Ucok K, et al. Do patients with mild to moderate psoriasis really have a sedentary lifestyle? Int J Dermatol. 2013;52:1129-1134.
- Do YK, Lakhani N, Malhotra R, et al. Association between psoriasis and leisure‐time physical activity: findings from the National Health and Nutrition Examination Survey. J Dermatol. 2015;42:148-153.
- Leibowitz E, Seidman DS, Laor A, et al. Are psoriatic patients at risk of heat intolerance? Br J Dermatol. 1991;124:439-442.
- Husted JA, Tom BD, Farewell VT, et al. Description and prediction of physical functional disability in psoriatic arthritis: a longitudinal analysis using a Markov model approach. Arthritis Rheum. 2005;53:404-409.
- Wilson FC, Icen M, Crowson CS, et al. Incidence and clinical predictors of psoriatic arthritis in patients with psoriasis: a population‐based study. Arthritis Rheum. 2009;61:233-239.
- Shih M, Hootman JM, Kruger J, et al. Physical activity in men and women with arthritis: National Health Interview Survey, 2002. Am J Prev Med. 2006;30:385-393.
- Ramsay B, O’Reagan M. A survey of the social and psychological effects of psoriasis. Br J Dermatol. 1988;118:195-201.
- Herron MD, Hinckley M, Hoffman MS, et al. Impact of obesity and smoking on psoriasis presentation and management. Arch Dermatol. 2005;141:1527-1534.
- Kumar S, Han J, Li T, et al. Obesity, waist circumference, weight change and the risk of psoriasis in US women. J Eur Acad Dermatol Venereol. 2013;27:1293-1298.
- Upala S, Sanguankeo A. Effect of lifestyle weight loss intervention on disease severity in patients with psoriasis: a systematic review and meta-analysis. Int J Obes (Lond). 2015;39:1197-1202.
- Sako EY, Famenini S, Wu JJ. Bariatric surgery and psoriasis. J Am Acad Dermatol. 2014;70:774-779.
- Clark L, Lebwohl M. The effect of weight on the efficacy of biologic therapy in patients with psoriasis. J Am Acad Dermatol. 2008;58:443-446.
- Puig L. Obesity and psoriasis: body weight and body mass index influence the response to biological treatment. J Eur Acad Dermatol Venereol. 2011;25:1007-1011.
- Wu JJ, Choi YM, Bebchuk JD. Risk of myocardial infarction in psoriasis patients: a retrospective cohort study. J Dermatolog Treat. 2015;26:230-234.
- Takeshita J, Gelfand JM, Li P, et al. Psoriasis in the US Medicare population: prevalence, treatment, and factors associated with biologic use. J Invest Dermatol. 2015;135:2955-2963.
- Prey S, Paul C, Bronsard V, et al. Cardiovascular risk factors in patients with plaque psoriasis: a systematic review of epidemiological studies. J Eur Acad Dermatol Venereol. 2010;24(suppl 2):23-30.
- Takeshita J, Grewal S, Langan SM, et al. Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol. 2017;76:377-390.
- Leon AS. Biological mechanisms for the cardioprotective effects of aerobic exercise. Am J Lifestyle Med. 2009;3:32S-34S.
- Kim A, Silverberg JI. A systematic review of vigorous physical activity in eczema. Br J Dermatol. 2016;174:660-662.
- Wells GA, Shea B, O’Connell D, et al. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomized studies in meta-analyses. The Ottawa Hospital Research Institute website. http://www.ohri.ca/programs/clinical_epidemiology/oxford.htm. Accessed February 23, 2018.
- Frankel HC, Han J, Li T, et al. The association between physical activity and the risk of incident psoriasis. Arch Dermatol. 2012;148:918-924.
- Torres T, Alexandre JM, Mendonça D, et al. Levels of physical activity in patients with severe psoriasis: a cross-sectional questionnaire study. Am J Clin Dermatol. 2014;15:129-135.
- Demirel R, Genc A, Ucok K, et al. Do patients with mild to moderate psoriasis really have a sedentary lifestyle? Int J Dermatol. 2013;52:1129-1134.
- Do YK, Lakhani N, Malhotra R, et al. Association between psoriasis and leisure‐time physical activity: findings from the National Health and Nutrition Examination Survey. J Dermatol. 2015;42:148-153.
- Leibowitz E, Seidman DS, Laor A, et al. Are psoriatic patients at risk of heat intolerance? Br J Dermatol. 1991;124:439-442.
- Husted JA, Tom BD, Farewell VT, et al. Description and prediction of physical functional disability in psoriatic arthritis: a longitudinal analysis using a Markov model approach. Arthritis Rheum. 2005;53:404-409.
- Wilson FC, Icen M, Crowson CS, et al. Incidence and clinical predictors of psoriatic arthritis in patients with psoriasis: a population‐based study. Arthritis Rheum. 2009;61:233-239.
- Shih M, Hootman JM, Kruger J, et al. Physical activity in men and women with arthritis: National Health Interview Survey, 2002. Am J Prev Med. 2006;30:385-393.
- Ramsay B, O’Reagan M. A survey of the social and psychological effects of psoriasis. Br J Dermatol. 1988;118:195-201.
- Herron MD, Hinckley M, Hoffman MS, et al. Impact of obesity and smoking on psoriasis presentation and management. Arch Dermatol. 2005;141:1527-1534.
- Kumar S, Han J, Li T, et al. Obesity, waist circumference, weight change and the risk of psoriasis in US women. J Eur Acad Dermatol Venereol. 2013;27:1293-1298.
- Upala S, Sanguankeo A. Effect of lifestyle weight loss intervention on disease severity in patients with psoriasis: a systematic review and meta-analysis. Int J Obes (Lond). 2015;39:1197-1202.
- Sako EY, Famenini S, Wu JJ. Bariatric surgery and psoriasis. J Am Acad Dermatol. 2014;70:774-779.
- Clark L, Lebwohl M. The effect of weight on the efficacy of biologic therapy in patients with psoriasis. J Am Acad Dermatol. 2008;58:443-446.
- Puig L. Obesity and psoriasis: body weight and body mass index influence the response to biological treatment. J Eur Acad Dermatol Venereol. 2011;25:1007-1011.
- Wu JJ, Choi YM, Bebchuk JD. Risk of myocardial infarction in psoriasis patients: a retrospective cohort study. J Dermatolog Treat. 2015;26:230-234.
Practice Points
- Psoriasis is associated with comorbid disease conditions, including cardiovascular disease.
- Regular physical activity is known to decrease the risk of developing cardiovascular disease.
- Patients with psoriasis would likely benefit from regular participation in vigorous physical activity to help reduce the risk of developing cardiovascular disease.