User login
MSBase study validates therapy for relapse in secondary progressive MS
An observational cohort study of a prospective international database of patients with multiple sclerosis has reported that medical therapy can reduce disability progression in patients with active secondary progressive MS (SPMS) who are prone to inflammatory relapses.

The international researchers, led by Nathanial Lizak, MMBS, of the University of Melbourne, conducted the cohort study of 1,621 patients with SPMS from the MSBase international registry, which prospectively collected the information from 1995 to 2018. Their findings were published in JAMA Neurology.
“ wrote Dr. Lizak and colleagues of the MSBase Study Group.
Therapy’s impact on disease progression
To ensure that they had timely data on the early disease course of all study patients, they researchers only included patients whose diagnosis and first documented Expanded Disability Status Scale (EDSS) score were no more than 24 months apart. At SPMS conversion, 1,494 patients had an EDSS score of less than 7 (on a scale of 0-10); during the follow-up period, 267 of them (17.9%) crossed over the threshold of 7.
Dr. Lizak and colleagues noted that early treatment during relapsing remitting MS didn’t impact outcomes after SPMS conversion.
For evaluating the MS Severity Score (MSSS), the study split the cohort into three groups depending on the efficacy of therapy: low-efficacy treatments, mostly consisting of interferon-beta and glatiramer acetate; medium-efficacy treatments, mostly fingolimod and dimethyl fumarate; and high-efficacy treatments, predominately natalizumab and mitoxantrone.
The MSSS progression slope in the cohort had an average reduction of 0.02 points per year. “For patients who experienced superimposed relapses during SPMS, a reduced MSSS progression slope during SPMS was observed among those who received disease-modifying therapies for a greater proportion of time during SPMS,” Dr. Lizak and colleagues wrote.
MSSS progression slope reduction was more pronounced in the medium- and high-efficacy groups, with a reduction of beta 0.22 (P = .06) and 0.034 (P = .002), respectively.
“Based on our models, the level of disability in patients with active SPMS who are continuously treated with high-efficacy immunotherapies would progress more slowly in comparison with the general population with MS by a mean (standard deviation) of 1.56 (4.60) deciles over 10 years,” Dr. Lizak and colleagues wrote.
While the researchers cited a number of studies that didn’t support immunotherapy for SPMS, they also did cite the EXPAND trial to support treatment with siponimod in SPMS patients (Lancet. 2018;391:1263-73). The ASCEND trial of natalizumab enrolled a largely relapse-free cohort and found no link between treatment and disability progression in SPMS (Lancet Neurol. 2018;17:405-15), and a previous report by the MSBase Study Group found no benefit of therapy when adjusted for SPMS relapse rates (Neurology. 2017;89:1050-9).
“Together with the present study, the existing data converge on the suggestion that relapses during SPMS provide a therapeutic target and a marker of future response to immunotherapy during SPMS,” Dr. Lizak and colleagues wrote.
Challenging dogma
Commenting on the research, Mark Freedman, HBSc, MSc, MD, said that the MSBase study makes an important contribution to the literature on management of SPMS. “Up until this point we’ve been basing our assumptions on secondary progressive MS on natural history studies, which are actually quite old, dating back 20-30 years.” Dr. Freedman is senior scientist in the Neuroscience Program at the Ottawa Hospital Research Institute and professor of medicine in neurology at the University of Ottawa.

He said “the most damaging” of those studies was by the late Christian Confavreaux, MD, and colleagues in Lyon, France (N Engl J Med. 2000;343:1430-8), that reported relapses didn’t alter the progression of disability. “In other words, once you’re in EDSS of 4, it’s a runaway train; it doesn’t matter what you do,” Dr. Freedman said.
“That was kind of dogma for years,” he said. “The reason this publication is important is because it’s suggesting that’s not the case.” In other words, the MSBase cohort study is validating what neurologists have been doing in the real world for years: treating patients with SPMS who have relapses.
Dr Lizak reported receiving travel compensation from Merck outside the scope of the study. His coauthors reported numerous financial relationships.
SOURCE: Lizak N et al. JAMA Neurol. 2020 Jul 27. doi: 10.1001/jamaneurol.2020.2453
An observational cohort study of a prospective international database of patients with multiple sclerosis has reported that medical therapy can reduce disability progression in patients with active secondary progressive MS (SPMS) who are prone to inflammatory relapses.
The international researchers, led by Nathanial Lizak, MMBS, of the University of Melbourne, conducted the cohort study of 1,621 patients with SPMS from the MSBase international registry, which prospectively collected the information from 1995 to 2018. Their findings were published in JAMA Neurology.
“ wrote Dr. Lizak and colleagues of the MSBase Study Group.
Therapy’s impact on disease progression
To ensure that they had timely data on the early disease course of all study patients, they researchers only included patients whose diagnosis and first documented Expanded Disability Status Scale (EDSS) score were no more than 24 months apart. At SPMS conversion, 1,494 patients had an EDSS score of less than 7 (on a scale of 0-10); during the follow-up period, 267 of them (17.9%) crossed over the threshold of 7.
Dr. Lizak and colleagues noted that early treatment during relapsing remitting MS didn’t impact outcomes after SPMS conversion.
For evaluating the MS Severity Score (MSSS), the study split the cohort into three groups depending on the efficacy of therapy: low-efficacy treatments, mostly consisting of interferon-beta and glatiramer acetate; medium-efficacy treatments, mostly fingolimod and dimethyl fumarate; and high-efficacy treatments, predominately natalizumab and mitoxantrone.
The MSSS progression slope in the cohort had an average reduction of 0.02 points per year. “For patients who experienced superimposed relapses during SPMS, a reduced MSSS progression slope during SPMS was observed among those who received disease-modifying therapies for a greater proportion of time during SPMS,” Dr. Lizak and colleagues wrote.
MSSS progression slope reduction was more pronounced in the medium- and high-efficacy groups, with a reduction of beta 0.22 (P = .06) and 0.034 (P = .002), respectively.
“Based on our models, the level of disability in patients with active SPMS who are continuously treated with high-efficacy immunotherapies would progress more slowly in comparison with the general population with MS by a mean (standard deviation) of 1.56 (4.60) deciles over 10 years,” Dr. Lizak and colleagues wrote.
While the researchers cited a number of studies that didn’t support immunotherapy for SPMS, they also did cite the EXPAND trial to support treatment with siponimod in SPMS patients (Lancet. 2018;391:1263-73). The ASCEND trial of natalizumab enrolled a largely relapse-free cohort and found no link between treatment and disability progression in SPMS (Lancet Neurol. 2018;17:405-15), and a previous report by the MSBase Study Group found no benefit of therapy when adjusted for SPMS relapse rates (Neurology. 2017;89:1050-9).
“Together with the present study, the existing data converge on the suggestion that relapses during SPMS provide a therapeutic target and a marker of future response to immunotherapy during SPMS,” Dr. Lizak and colleagues wrote.
Challenging dogma
Commenting on the research, Mark Freedman, HBSc, MSc, MD, said that the MSBase study makes an important contribution to the literature on management of SPMS. “Up until this point we’ve been basing our assumptions on secondary progressive MS on natural history studies, which are actually quite old, dating back 20-30 years.” Dr. Freedman is senior scientist in the Neuroscience Program at the Ottawa Hospital Research Institute and professor of medicine in neurology at the University of Ottawa.
He said “the most damaging” of those studies was by the late Christian Confavreaux, MD, and colleagues in Lyon, France (N Engl J Med. 2000;343:1430-8), that reported relapses didn’t alter the progression of disability. “In other words, once you’re in EDSS of 4, it’s a runaway train; it doesn’t matter what you do,” Dr. Freedman said.
“That was kind of dogma for years,” he said. “The reason this publication is important is because it’s suggesting that’s not the case.” In other words, the MSBase cohort study is validating what neurologists have been doing in the real world for years: treating patients with SPMS who have relapses.
Dr Lizak reported receiving travel compensation from Merck outside the scope of the study. His coauthors reported numerous financial relationships.
SOURCE: Lizak N et al. JAMA Neurol. 2020 Jul 27. doi: 10.1001/jamaneurol.2020.2453
An observational cohort study of a prospective international database of patients with multiple sclerosis has reported that medical therapy can reduce disability progression in patients with active secondary progressive MS (SPMS) who are prone to inflammatory relapses.
The international researchers, led by Nathanial Lizak, MMBS, of the University of Melbourne, conducted the cohort study of 1,621 patients with SPMS from the MSBase international registry, which prospectively collected the information from 1995 to 2018. Their findings were published in JAMA Neurology.
“ wrote Dr. Lizak and colleagues of the MSBase Study Group.
Therapy’s impact on disease progression
To ensure that they had timely data on the early disease course of all study patients, they researchers only included patients whose diagnosis and first documented Expanded Disability Status Scale (EDSS) score were no more than 24 months apart. At SPMS conversion, 1,494 patients had an EDSS score of less than 7 (on a scale of 0-10); during the follow-up period, 267 of them (17.9%) crossed over the threshold of 7.
Dr. Lizak and colleagues noted that early treatment during relapsing remitting MS didn’t impact outcomes after SPMS conversion.
For evaluating the MS Severity Score (MSSS), the study split the cohort into three groups depending on the efficacy of therapy: low-efficacy treatments, mostly consisting of interferon-beta and glatiramer acetate; medium-efficacy treatments, mostly fingolimod and dimethyl fumarate; and high-efficacy treatments, predominately natalizumab and mitoxantrone.
The MSSS progression slope in the cohort had an average reduction of 0.02 points per year. “For patients who experienced superimposed relapses during SPMS, a reduced MSSS progression slope during SPMS was observed among those who received disease-modifying therapies for a greater proportion of time during SPMS,” Dr. Lizak and colleagues wrote.
MSSS progression slope reduction was more pronounced in the medium- and high-efficacy groups, with a reduction of beta 0.22 (P = .06) and 0.034 (P = .002), respectively.
“Based on our models, the level of disability in patients with active SPMS who are continuously treated with high-efficacy immunotherapies would progress more slowly in comparison with the general population with MS by a mean (standard deviation) of 1.56 (4.60) deciles over 10 years,” Dr. Lizak and colleagues wrote.
While the researchers cited a number of studies that didn’t support immunotherapy for SPMS, they also did cite the EXPAND trial to support treatment with siponimod in SPMS patients (Lancet. 2018;391:1263-73). The ASCEND trial of natalizumab enrolled a largely relapse-free cohort and found no link between treatment and disability progression in SPMS (Lancet Neurol. 2018;17:405-15), and a previous report by the MSBase Study Group found no benefit of therapy when adjusted for SPMS relapse rates (Neurology. 2017;89:1050-9).
“Together with the present study, the existing data converge on the suggestion that relapses during SPMS provide a therapeutic target and a marker of future response to immunotherapy during SPMS,” Dr. Lizak and colleagues wrote.
Challenging dogma
Commenting on the research, Mark Freedman, HBSc, MSc, MD, said that the MSBase study makes an important contribution to the literature on management of SPMS. “Up until this point we’ve been basing our assumptions on secondary progressive MS on natural history studies, which are actually quite old, dating back 20-30 years.” Dr. Freedman is senior scientist in the Neuroscience Program at the Ottawa Hospital Research Institute and professor of medicine in neurology at the University of Ottawa.
He said “the most damaging” of those studies was by the late Christian Confavreaux, MD, and colleagues in Lyon, France (N Engl J Med. 2000;343:1430-8), that reported relapses didn’t alter the progression of disability. “In other words, once you’re in EDSS of 4, it’s a runaway train; it doesn’t matter what you do,” Dr. Freedman said.
“That was kind of dogma for years,” he said. “The reason this publication is important is because it’s suggesting that’s not the case.” In other words, the MSBase cohort study is validating what neurologists have been doing in the real world for years: treating patients with SPMS who have relapses.
Dr Lizak reported receiving travel compensation from Merck outside the scope of the study. His coauthors reported numerous financial relationships.
SOURCE: Lizak N et al. JAMA Neurol. 2020 Jul 27. doi: 10.1001/jamaneurol.2020.2453
FROM JAMA NEUROLOGY
FDA approves cannabidiol for tuberous sclerosis complex
The cannabidiol (CBD) oral solution Epidiolex has been approved by the Food and Drug Administration for the new indication of treatment of seizures associated with tuberous sclerosis complex in patients 1 year of age and older.
The drug was approved by the FDA in 2018 for the treatment of seizures associated with two rare and severe forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome, as reported by Medscape Medical News.
This is the only FDA-approved drug that contains a purified drug substance derived from cannabis. It is also the second FDA approval of a drug for the treatment of seizures associated with tuberous sclerosis complex.
CBD is a chemical component of the cannabis sativa plant, but it does not cause intoxication or euphoria (the “high”) that comes from tetrahydrocannabinol (THC), which is the primary psychoactive component of cannabis.
“The FDA continues to believe the drug approval process represents the best way to make new medicines, including any drugs derived from cannabis, available to patients in need of appropriate medical therapy such as the treatment of seizures associated with these rare conditions,” Douglas Throckmorton, MD, deputy center director for regulatory programs in the FDA’s Center for Drug Evaluation and Research, said in an agency press release.
“This paradigm ensures new therapies are safe, effective, and manufactured to a high quality that provides uniform and reliable dosing for patients,” Dr. Throckmorton said.
He added that the FDA is committed to supporting research on the potential medical uses of cannabis-derived products.
Rare genetic disease
Tuberous sclerosis complex is a rare genetic disease that causes benign tumors to grow in the brain and other parts of the body, such as the eyes, heart, kidneys, lungs, and skin.
It usually affects the central nervous system and can result in a combination of symptoms, including seizures, developmental delay, and behavioral problems. The signs and symptoms of the condition, as well as the severity of symptoms, vary widely. The disease affects about 1 in 6,000 individuals.
The effectiveness of Epidiolex in the treatment of seizures associated with tuberous sclerosis complex was established in a randomized, double-blind, placebo-controlled trial in which 148 patients of a total of 224 in the study received the active drug, the FDA noted.
Results showed that for patients treated with CBD, there was a significantly greater reduction in seizure frequency during the treatment period than for patients who received placebo.
This effect was seen within 8 weeks and remained consistent throughout the 16-week treatment period.
The most common side effects that occurred in CBD-treated participants were diarrhea, elevated liver enzyme levels, decreased appetite, sleepiness, fever, and vomiting. Additional side effects that have been reported with the product include liver injury, decreased weight, anemia, and increased creatinine level.
As is true for all drugs that currently treat epilepsy, including Epidiolex, the most serious risks may include an increase in suicidal thoughts and behavior or thoughts of self-harm, the FDA reports.
Patients, their caregivers, and their families should be advised to monitor for any unusual changes in mood or behavior, such as worsening depression or suicidal thoughts or behavior. They should report behaviors of concern immediately to health care providers, the agency notes.
It also points out that Epidiolex can cause liver injury, of which most cases are generally mild. However, there is a risk for rare but more severe liver injury. More severe liver injury can cause nausea, vomiting, abdominal pain, fatigue, anorexia, jaundice, and/or dark urine.
A version of this story originally appeared on Medscape.com.
The cannabidiol (CBD) oral solution Epidiolex has been approved by the Food and Drug Administration for the new indication of treatment of seizures associated with tuberous sclerosis complex in patients 1 year of age and older.
The drug was approved by the FDA in 2018 for the treatment of seizures associated with two rare and severe forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome, as reported by Medscape Medical News.
This is the only FDA-approved drug that contains a purified drug substance derived from cannabis. It is also the second FDA approval of a drug for the treatment of seizures associated with tuberous sclerosis complex.
CBD is a chemical component of the cannabis sativa plant, but it does not cause intoxication or euphoria (the “high”) that comes from tetrahydrocannabinol (THC), which is the primary psychoactive component of cannabis.
“The FDA continues to believe the drug approval process represents the best way to make new medicines, including any drugs derived from cannabis, available to patients in need of appropriate medical therapy such as the treatment of seizures associated with these rare conditions,” Douglas Throckmorton, MD, deputy center director for regulatory programs in the FDA’s Center for Drug Evaluation and Research, said in an agency press release.
“This paradigm ensures new therapies are safe, effective, and manufactured to a high quality that provides uniform and reliable dosing for patients,” Dr. Throckmorton said.
He added that the FDA is committed to supporting research on the potential medical uses of cannabis-derived products.
Rare genetic disease
Tuberous sclerosis complex is a rare genetic disease that causes benign tumors to grow in the brain and other parts of the body, such as the eyes, heart, kidneys, lungs, and skin.
It usually affects the central nervous system and can result in a combination of symptoms, including seizures, developmental delay, and behavioral problems. The signs and symptoms of the condition, as well as the severity of symptoms, vary widely. The disease affects about 1 in 6,000 individuals.
The effectiveness of Epidiolex in the treatment of seizures associated with tuberous sclerosis complex was established in a randomized, double-blind, placebo-controlled trial in which 148 patients of a total of 224 in the study received the active drug, the FDA noted.
Results showed that for patients treated with CBD, there was a significantly greater reduction in seizure frequency during the treatment period than for patients who received placebo.
This effect was seen within 8 weeks and remained consistent throughout the 16-week treatment period.
The most common side effects that occurred in CBD-treated participants were diarrhea, elevated liver enzyme levels, decreased appetite, sleepiness, fever, and vomiting. Additional side effects that have been reported with the product include liver injury, decreased weight, anemia, and increased creatinine level.
As is true for all drugs that currently treat epilepsy, including Epidiolex, the most serious risks may include an increase in suicidal thoughts and behavior or thoughts of self-harm, the FDA reports.
Patients, their caregivers, and their families should be advised to monitor for any unusual changes in mood or behavior, such as worsening depression or suicidal thoughts or behavior. They should report behaviors of concern immediately to health care providers, the agency notes.
It also points out that Epidiolex can cause liver injury, of which most cases are generally mild. However, there is a risk for rare but more severe liver injury. More severe liver injury can cause nausea, vomiting, abdominal pain, fatigue, anorexia, jaundice, and/or dark urine.
A version of this story originally appeared on Medscape.com.
The cannabidiol (CBD) oral solution Epidiolex has been approved by the Food and Drug Administration for the new indication of treatment of seizures associated with tuberous sclerosis complex in patients 1 year of age and older.
The drug was approved by the FDA in 2018 for the treatment of seizures associated with two rare and severe forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome, as reported by Medscape Medical News.
This is the only FDA-approved drug that contains a purified drug substance derived from cannabis. It is also the second FDA approval of a drug for the treatment of seizures associated with tuberous sclerosis complex.
CBD is a chemical component of the cannabis sativa plant, but it does not cause intoxication or euphoria (the “high”) that comes from tetrahydrocannabinol (THC), which is the primary psychoactive component of cannabis.
“The FDA continues to believe the drug approval process represents the best way to make new medicines, including any drugs derived from cannabis, available to patients in need of appropriate medical therapy such as the treatment of seizures associated with these rare conditions,” Douglas Throckmorton, MD, deputy center director for regulatory programs in the FDA’s Center for Drug Evaluation and Research, said in an agency press release.
“This paradigm ensures new therapies are safe, effective, and manufactured to a high quality that provides uniform and reliable dosing for patients,” Dr. Throckmorton said.
He added that the FDA is committed to supporting research on the potential medical uses of cannabis-derived products.
Rare genetic disease
Tuberous sclerosis complex is a rare genetic disease that causes benign tumors to grow in the brain and other parts of the body, such as the eyes, heart, kidneys, lungs, and skin.
It usually affects the central nervous system and can result in a combination of symptoms, including seizures, developmental delay, and behavioral problems. The signs and symptoms of the condition, as well as the severity of symptoms, vary widely. The disease affects about 1 in 6,000 individuals.
The effectiveness of Epidiolex in the treatment of seizures associated with tuberous sclerosis complex was established in a randomized, double-blind, placebo-controlled trial in which 148 patients of a total of 224 in the study received the active drug, the FDA noted.
Results showed that for patients treated with CBD, there was a significantly greater reduction in seizure frequency during the treatment period than for patients who received placebo.
This effect was seen within 8 weeks and remained consistent throughout the 16-week treatment period.
The most common side effects that occurred in CBD-treated participants were diarrhea, elevated liver enzyme levels, decreased appetite, sleepiness, fever, and vomiting. Additional side effects that have been reported with the product include liver injury, decreased weight, anemia, and increased creatinine level.
As is true for all drugs that currently treat epilepsy, including Epidiolex, the most serious risks may include an increase in suicidal thoughts and behavior or thoughts of self-harm, the FDA reports.
Patients, their caregivers, and their families should be advised to monitor for any unusual changes in mood or behavior, such as worsening depression or suicidal thoughts or behavior. They should report behaviors of concern immediately to health care providers, the agency notes.
It also points out that Epidiolex can cause liver injury, of which most cases are generally mild. However, there is a risk for rare but more severe liver injury. More severe liver injury can cause nausea, vomiting, abdominal pain, fatigue, anorexia, jaundice, and/or dark urine.
A version of this story originally appeared on Medscape.com.
Positive phase 3 top-line results for migraine prevention drug
AbbVie, the company developing the drug, has announced.
Top-line results from the ADVANCE trial, which evaluated atogepant 10, 30, and 60 mg, showed all three doses were associated with a significant reduction from baseline in mean monthly migraine days, compared with placebo.
There were also significant improvements in all six secondary endpoints with the two higher doses.
Data from the ADVANCE trial and a previous phase 2/3 trial will be the basis for regulatory submissions in the United States and other countries, AbbVie reported.
Decreased migraine days
The phase 3, multicenter, randomized, double-blind, placebo-controlled, parallel-group trial was designed to evaluate the efficacy, safety, and tolerability of oral atogepant for the prevention of migraine in those who experienced 4-14 migraine days per month.
A total of 910 patients were randomized to one of four treatment groups: 10 mg, 30 mg, or 60 mg of atogepant once daily or placebo. Efficacy analyses were based on the modified intent-to-treat population of 873 patients.
The primary endpoint was change from baseline in mean monthly migraine days during the 12-week treatment period. All atogepant dose groups met the primary endpoint and demonstrated significantly greater decreases in mean monthly migraine days, compared with placebo.
Mean monthly migraine days were reduced by 3.69 days with the 10-mg dose, 3.86 days with the 30-mg dose, and 4.2 days with the 60-mg dose of atogepant, compared with a reduction of 2.48 migraine days in the placebo group (P < .0001, all dose groups vs. placebo).
A key secondary endpoint measured the proportion of patients who achieved at least a 50% reduction in mean monthly migraine days over 12 weeks. This outcome occurred in 55.6% of the 10-mg atogepant group, 58.7% of the 30-mg group, and 60.8% of the 60-mg group, compared with 29% of the placebo group (P < .0001, all dose groups vs. placebo).
Significant improvements
Additional secondary endpoints measured during the 12-week treatment period included change from baseline in mean monthly headache days, mean monthly acute–medication use days, mean monthly performance of daily activities and physical impairment domain scores on the Activity Impairment in Migraine-Diary, and change from baseline in the Migraine-Specific Quality of Life Questionnaire Role Function-Restrictive domain score at week 12. Treatment with the 30-mg and 60-mg doses resulted in significant improvements in all secondary endpoints, and treatment with the 10-mg dose resulted in significant improvements in four of the six secondary endpoints.
No new safety risks were observed when compared with the safety profile of atogepant observed in previous trials, AbbVie said. Serious adverse events occurred in 0.9% of patients in the atogepant 10-mg group versus 0.9% of patients in the placebo group. No patients in the atogepant 30-mg or 60-mg groups experienced a serious adverse event. The most common adverse events (reported in at least 5% of patients and at least one atogepant group and at a rate greater than placebo), across all doses versus placebo, were constipation (6.9%-7.7% vs. 0.5%), nausea (4.4%-6.1% vs. 1.8%), and upper respiratory tract infection (3.9%-5.7% vs. 4.5%).
Most cases of constipation, nausea, and upper respiratory tract infection were mild or moderate in severity and did not lead to discontinuation. There were no hepatic safety issues identified in the trial.
Full data results will be presented at an upcoming medical congress and/or published in a peer-reviewed journal, the company said.
A version of this article originally appeared on Medscape.com.
AbbVie, the company developing the drug, has announced.
Top-line results from the ADVANCE trial, which evaluated atogepant 10, 30, and 60 mg, showed all three doses were associated with a significant reduction from baseline in mean monthly migraine days, compared with placebo.
There were also significant improvements in all six secondary endpoints with the two higher doses.
Data from the ADVANCE trial and a previous phase 2/3 trial will be the basis for regulatory submissions in the United States and other countries, AbbVie reported.
Decreased migraine days
The phase 3, multicenter, randomized, double-blind, placebo-controlled, parallel-group trial was designed to evaluate the efficacy, safety, and tolerability of oral atogepant for the prevention of migraine in those who experienced 4-14 migraine days per month.
A total of 910 patients were randomized to one of four treatment groups: 10 mg, 30 mg, or 60 mg of atogepant once daily or placebo. Efficacy analyses were based on the modified intent-to-treat population of 873 patients.
The primary endpoint was change from baseline in mean monthly migraine days during the 12-week treatment period. All atogepant dose groups met the primary endpoint and demonstrated significantly greater decreases in mean monthly migraine days, compared with placebo.
Mean monthly migraine days were reduced by 3.69 days with the 10-mg dose, 3.86 days with the 30-mg dose, and 4.2 days with the 60-mg dose of atogepant, compared with a reduction of 2.48 migraine days in the placebo group (P < .0001, all dose groups vs. placebo).
A key secondary endpoint measured the proportion of patients who achieved at least a 50% reduction in mean monthly migraine days over 12 weeks. This outcome occurred in 55.6% of the 10-mg atogepant group, 58.7% of the 30-mg group, and 60.8% of the 60-mg group, compared with 29% of the placebo group (P < .0001, all dose groups vs. placebo).
Significant improvements
Additional secondary endpoints measured during the 12-week treatment period included change from baseline in mean monthly headache days, mean monthly acute–medication use days, mean monthly performance of daily activities and physical impairment domain scores on the Activity Impairment in Migraine-Diary, and change from baseline in the Migraine-Specific Quality of Life Questionnaire Role Function-Restrictive domain score at week 12. Treatment with the 30-mg and 60-mg doses resulted in significant improvements in all secondary endpoints, and treatment with the 10-mg dose resulted in significant improvements in four of the six secondary endpoints.
No new safety risks were observed when compared with the safety profile of atogepant observed in previous trials, AbbVie said. Serious adverse events occurred in 0.9% of patients in the atogepant 10-mg group versus 0.9% of patients in the placebo group. No patients in the atogepant 30-mg or 60-mg groups experienced a serious adverse event. The most common adverse events (reported in at least 5% of patients and at least one atogepant group and at a rate greater than placebo), across all doses versus placebo, were constipation (6.9%-7.7% vs. 0.5%), nausea (4.4%-6.1% vs. 1.8%), and upper respiratory tract infection (3.9%-5.7% vs. 4.5%).
Most cases of constipation, nausea, and upper respiratory tract infection were mild or moderate in severity and did not lead to discontinuation. There were no hepatic safety issues identified in the trial.
Full data results will be presented at an upcoming medical congress and/or published in a peer-reviewed journal, the company said.
A version of this article originally appeared on Medscape.com.
AbbVie, the company developing the drug, has announced.
Top-line results from the ADVANCE trial, which evaluated atogepant 10, 30, and 60 mg, showed all three doses were associated with a significant reduction from baseline in mean monthly migraine days, compared with placebo.
There were also significant improvements in all six secondary endpoints with the two higher doses.
Data from the ADVANCE trial and a previous phase 2/3 trial will be the basis for regulatory submissions in the United States and other countries, AbbVie reported.
Decreased migraine days
The phase 3, multicenter, randomized, double-blind, placebo-controlled, parallel-group trial was designed to evaluate the efficacy, safety, and tolerability of oral atogepant for the prevention of migraine in those who experienced 4-14 migraine days per month.
A total of 910 patients were randomized to one of four treatment groups: 10 mg, 30 mg, or 60 mg of atogepant once daily or placebo. Efficacy analyses were based on the modified intent-to-treat population of 873 patients.
The primary endpoint was change from baseline in mean monthly migraine days during the 12-week treatment period. All atogepant dose groups met the primary endpoint and demonstrated significantly greater decreases in mean monthly migraine days, compared with placebo.
Mean monthly migraine days were reduced by 3.69 days with the 10-mg dose, 3.86 days with the 30-mg dose, and 4.2 days with the 60-mg dose of atogepant, compared with a reduction of 2.48 migraine days in the placebo group (P < .0001, all dose groups vs. placebo).
A key secondary endpoint measured the proportion of patients who achieved at least a 50% reduction in mean monthly migraine days over 12 weeks. This outcome occurred in 55.6% of the 10-mg atogepant group, 58.7% of the 30-mg group, and 60.8% of the 60-mg group, compared with 29% of the placebo group (P < .0001, all dose groups vs. placebo).
Significant improvements
Additional secondary endpoints measured during the 12-week treatment period included change from baseline in mean monthly headache days, mean monthly acute–medication use days, mean monthly performance of daily activities and physical impairment domain scores on the Activity Impairment in Migraine-Diary, and change from baseline in the Migraine-Specific Quality of Life Questionnaire Role Function-Restrictive domain score at week 12. Treatment with the 30-mg and 60-mg doses resulted in significant improvements in all secondary endpoints, and treatment with the 10-mg dose resulted in significant improvements in four of the six secondary endpoints.
No new safety risks were observed when compared with the safety profile of atogepant observed in previous trials, AbbVie said. Serious adverse events occurred in 0.9% of patients in the atogepant 10-mg group versus 0.9% of patients in the placebo group. No patients in the atogepant 30-mg or 60-mg groups experienced a serious adverse event. The most common adverse events (reported in at least 5% of patients and at least one atogepant group and at a rate greater than placebo), across all doses versus placebo, were constipation (6.9%-7.7% vs. 0.5%), nausea (4.4%-6.1% vs. 1.8%), and upper respiratory tract infection (3.9%-5.7% vs. 4.5%).
Most cases of constipation, nausea, and upper respiratory tract infection were mild or moderate in severity and did not lead to discontinuation. There were no hepatic safety issues identified in the trial.
Full data results will be presented at an upcoming medical congress and/or published in a peer-reviewed journal, the company said.
A version of this article originally appeared on Medscape.com.
CCC19, other registries help define COVID/cancer landscape
Initial results from the CCC19 registry were reported as part of the American Society of Clinical Oncology (ASCO) virtual scientific program and published in The Lancet (Lancet. 2020 Jun 20;395[10241]:1907-18).
The latest data were presented at the AACR virtual meeting: COVID-19 and Cancer by Brian I. Rini, MD, of Vanderbilt University, Nashville, Tenn. They were simultaneously published in Cancer Discovery (Cancer Discov. 2020 Jul 22;CD-20-0941).
The CCC19 registry was launched in March by a few institutions as part of “a grassroots idea ... to collect granular data regarding cancer patients and their outcomes with COVID,” Dr. Rini said.
Within a few months of its inception, the registry had partnered with more than 100 institutions worldwide and accrued data from more than 2,000 patients.
The reports in The Lancet and at ASCO included outcomes for the first 928 patients and showed a 13% mortality rate as well as a fivefold increase in the risk of 30-day mortality among patients with COVID-19 and progressing cancer.
The data also showed an increased mortality risk among older patients, men, former smokers, those with poor performance status, those with multiple comorbidities, and those treated with hydroxychloroquine and azithromycin.
The latest data
The CCC19 registry has grown to include 114 sites worldwide, including major comprehensive cancer centers and community sites. As of June 26, there were 2,749 patients enrolled.
Since the last data were reported, the mortality rate increased from 13% to 16% (versus 5% globally). In addition, the increased mortality risk among non-Hispanic black patients and patients with hematologic malignancies reached statistical significance, Dr. Rini said. He noted that the increase in mortality rate was largely attributable to improved follow-up.
Mechanical ventilation was required in 12% of patients, ICU admission was required in 16%, oxygen was required in 45%, and hospitalization was required in 60%. The composite outcome of death, severe illness requiring hospitalization, ICU admission, or mechanical ventilation was reached in 29% of patients, Dr. Rini said.
Mortality rates across cancer types ranged from 3% to 26%, with thyroid and breast cancer patients having the lowest rates (3% and 8%, respectively), and with lymphoma and lung cancer patients having the highest (22% and 26%, respectively), Dr. Rini said.
He noted that the TERAVOLT registry, a COVID-19 registry for patients with thoracic cancers, also showed a very high mortality rate in this subgroup of patients.
Results from TERAVOLT were reported at the AACR virtual meeting I, presented at ASCO, and published in The Lancet (Lancet Oncol. 2020 Jul;21[7]:914-22). The most recent results showed a mortality rate of nearly 36% and reinforce the high mortality rate seen in lung cancer patients in CCC19, Dr. Rini said.
Increased mortality risk
After adjustment for several demographic and disease characteristics, the updated CCC19 data showed a significantly increased risk of mortality among:
- Older patients (adjusted odds ratio [aOR] per decade of age, 1.52).
- Men (aOR, 1.43).
- Current or former smokers vs. never smokers (aOR, 1.28).
- Patients with Eastern Cooperative Oncology Group performance scores of 1 vs. 0 (aOR of 1.80) or 2 vs. 0 (aOR, 4.22).
- Stable cancer vs. remission (aOR, 1.47).
- Progressive cancer vs. remission (aOR, 2.96).
- Non-Hispanic Black vs. White patients (aOR, 1.56).
- Hematologic malignancies vs. solid tumors (aOR, 1.80).
“Importantly, there were some factors that did not reach statistical significance,” Dr. Rini said. These include obesity (aOR, 1.23), recent surgery (aOR, 1.05), receipt of cytotoxic chemotherapy vs. no chemotherapy (aOR, 1.14), and receipt of noncytotoxic chemotherapy vs. no chemotherapy (aOR, 0.75).
“I think this provides some reassurance that cancer care can and should continue for these patients,” Dr. Rini said.
He noted, however, that in TERAVOLT, chemotherapy with or without other treatment was a risk factor for mortality in lung cancer patients when compared with no chemotherapy (OR, 1.71) and when compared with immunotherapy or targeted therapy (OR, 1.64).
NCCAPS and other registries
Dr. Rini discussed a number of registries looking at outcomes in COVID-19 patients with cancer, and he said the findings to date appear to confirm a higher mortality rate among cancer patients, particularly those with lung cancer.
Several factors are emerging that appear to be related to risk, including both cancer-related and non–cancer-related factors, he added.
The ongoing prospective National Cancer Institute COVID-19 in Cancer Patients Study (NCCAPS) “will provide much needed longitudinal data and, importantly, biospecimen collection in a large cohort of patients who have active cancer and are receiving treatment, said Dr. Rini, who is the study’s protocol chair. NCCAPS is a natural history study in that population, he said.
The planned accrual is about 2,000 patients who will be followed for up to 2 years for data collection, imaging scans, and research specimens.
The use of specimens is “a unique and special part of this study,” Dr. Rini said, explaining that the specimens will be used to look for development of antibodies over time, to describe the trajectory of cytokine abnormalities – especially in patients with more acute inpatient courses – to perform DNA-based genome-wide association studies, and to assess coagulation parameters.
NCCAPS is activated at 546 sties, 10 patients were enrolled as of June 21, and rapid accrual is expected over the next several months, he said.
Gypsyamber D’Souza, PhD, session moderator and an infectious disease epidemiologist at Johns Hopkins University in Baltimore, acknowledged the challenge that registry administrators face when trying to balance the need to get data out against the desire to ask the right questions and to have the right comparison groups, stratification, and analyses, especially amid a crisis like the COVID-19 pandemic.
Dr. Rini said it has indeed been a bit of a struggle with CCC19 to determine what information should be published and when, and what constitutes an important update.
“It’s been a learning experience, and frankly, I think we’re still learning,” he said. “This has been such a unique time in terms of a rush to get data out, balanced against making sure that there’s quality data and that you’re actually answering important questions.”
In fact, a number of ongoing registries “should start to produce great data [that will be presented] at upcoming big conferences,” Dr. Rini said. He added that those data “will help piece together different important aspects of this and different hypotheses, and hopefully complement the clinical data that’s starting to come out.”
The CCC19 registry is sponsored by Vanderbilt-Ingram Cancer Center. Dr. Rini disclosed relationships with Pfizer, Merck, Genentech/Roche, Aveo, AstraZeneca, Bristol Myers Squibb, Exelixis, Synthorx, Peloton, Compugen, Corvus, Surface Oncology, 3DMedicines, Aravive, Alkermes, Arrowhead, and PTC Therapeutics. Dr. D’Souza did not disclose any conflicts.
SOURCE: Rini BI. AACR: COVID-19 and Cancer. Abstract IA26.
Initial results from the CCC19 registry were reported as part of the American Society of Clinical Oncology (ASCO) virtual scientific program and published in The Lancet (Lancet. 2020 Jun 20;395[10241]:1907-18).
The latest data were presented at the AACR virtual meeting: COVID-19 and Cancer by Brian I. Rini, MD, of Vanderbilt University, Nashville, Tenn. They were simultaneously published in Cancer Discovery (Cancer Discov. 2020 Jul 22;CD-20-0941).
The CCC19 registry was launched in March by a few institutions as part of “a grassroots idea ... to collect granular data regarding cancer patients and their outcomes with COVID,” Dr. Rini said.
Within a few months of its inception, the registry had partnered with more than 100 institutions worldwide and accrued data from more than 2,000 patients.
The reports in The Lancet and at ASCO included outcomes for the first 928 patients and showed a 13% mortality rate as well as a fivefold increase in the risk of 30-day mortality among patients with COVID-19 and progressing cancer.
The data also showed an increased mortality risk among older patients, men, former smokers, those with poor performance status, those with multiple comorbidities, and those treated with hydroxychloroquine and azithromycin.
The latest data
The CCC19 registry has grown to include 114 sites worldwide, including major comprehensive cancer centers and community sites. As of June 26, there were 2,749 patients enrolled.
Since the last data were reported, the mortality rate increased from 13% to 16% (versus 5% globally). In addition, the increased mortality risk among non-Hispanic black patients and patients with hematologic malignancies reached statistical significance, Dr. Rini said. He noted that the increase in mortality rate was largely attributable to improved follow-up.
Mechanical ventilation was required in 12% of patients, ICU admission was required in 16%, oxygen was required in 45%, and hospitalization was required in 60%. The composite outcome of death, severe illness requiring hospitalization, ICU admission, or mechanical ventilation was reached in 29% of patients, Dr. Rini said.
Mortality rates across cancer types ranged from 3% to 26%, with thyroid and breast cancer patients having the lowest rates (3% and 8%, respectively), and with lymphoma and lung cancer patients having the highest (22% and 26%, respectively), Dr. Rini said.
He noted that the TERAVOLT registry, a COVID-19 registry for patients with thoracic cancers, also showed a very high mortality rate in this subgroup of patients.
Results from TERAVOLT were reported at the AACR virtual meeting I, presented at ASCO, and published in The Lancet (Lancet Oncol. 2020 Jul;21[7]:914-22). The most recent results showed a mortality rate of nearly 36% and reinforce the high mortality rate seen in lung cancer patients in CCC19, Dr. Rini said.
Increased mortality risk
After adjustment for several demographic and disease characteristics, the updated CCC19 data showed a significantly increased risk of mortality among:
- Older patients (adjusted odds ratio [aOR] per decade of age, 1.52).
- Men (aOR, 1.43).
- Current or former smokers vs. never smokers (aOR, 1.28).
- Patients with Eastern Cooperative Oncology Group performance scores of 1 vs. 0 (aOR of 1.80) or 2 vs. 0 (aOR, 4.22).
- Stable cancer vs. remission (aOR, 1.47).
- Progressive cancer vs. remission (aOR, 2.96).
- Non-Hispanic Black vs. White patients (aOR, 1.56).
- Hematologic malignancies vs. solid tumors (aOR, 1.80).
“Importantly, there were some factors that did not reach statistical significance,” Dr. Rini said. These include obesity (aOR, 1.23), recent surgery (aOR, 1.05), receipt of cytotoxic chemotherapy vs. no chemotherapy (aOR, 1.14), and receipt of noncytotoxic chemotherapy vs. no chemotherapy (aOR, 0.75).
“I think this provides some reassurance that cancer care can and should continue for these patients,” Dr. Rini said.
He noted, however, that in TERAVOLT, chemotherapy with or without other treatment was a risk factor for mortality in lung cancer patients when compared with no chemotherapy (OR, 1.71) and when compared with immunotherapy or targeted therapy (OR, 1.64).
NCCAPS and other registries
Dr. Rini discussed a number of registries looking at outcomes in COVID-19 patients with cancer, and he said the findings to date appear to confirm a higher mortality rate among cancer patients, particularly those with lung cancer.
Several factors are emerging that appear to be related to risk, including both cancer-related and non–cancer-related factors, he added.
The ongoing prospective National Cancer Institute COVID-19 in Cancer Patients Study (NCCAPS) “will provide much needed longitudinal data and, importantly, biospecimen collection in a large cohort of patients who have active cancer and are receiving treatment, said Dr. Rini, who is the study’s protocol chair. NCCAPS is a natural history study in that population, he said.
The planned accrual is about 2,000 patients who will be followed for up to 2 years for data collection, imaging scans, and research specimens.
The use of specimens is “a unique and special part of this study,” Dr. Rini said, explaining that the specimens will be used to look for development of antibodies over time, to describe the trajectory of cytokine abnormalities – especially in patients with more acute inpatient courses – to perform DNA-based genome-wide association studies, and to assess coagulation parameters.
NCCAPS is activated at 546 sties, 10 patients were enrolled as of June 21, and rapid accrual is expected over the next several months, he said.
Gypsyamber D’Souza, PhD, session moderator and an infectious disease epidemiologist at Johns Hopkins University in Baltimore, acknowledged the challenge that registry administrators face when trying to balance the need to get data out against the desire to ask the right questions and to have the right comparison groups, stratification, and analyses, especially amid a crisis like the COVID-19 pandemic.
Dr. Rini said it has indeed been a bit of a struggle with CCC19 to determine what information should be published and when, and what constitutes an important update.
“It’s been a learning experience, and frankly, I think we’re still learning,” he said. “This has been such a unique time in terms of a rush to get data out, balanced against making sure that there’s quality data and that you’re actually answering important questions.”
In fact, a number of ongoing registries “should start to produce great data [that will be presented] at upcoming big conferences,” Dr. Rini said. He added that those data “will help piece together different important aspects of this and different hypotheses, and hopefully complement the clinical data that’s starting to come out.”
The CCC19 registry is sponsored by Vanderbilt-Ingram Cancer Center. Dr. Rini disclosed relationships with Pfizer, Merck, Genentech/Roche, Aveo, AstraZeneca, Bristol Myers Squibb, Exelixis, Synthorx, Peloton, Compugen, Corvus, Surface Oncology, 3DMedicines, Aravive, Alkermes, Arrowhead, and PTC Therapeutics. Dr. D’Souza did not disclose any conflicts.
SOURCE: Rini BI. AACR: COVID-19 and Cancer. Abstract IA26.
Initial results from the CCC19 registry were reported as part of the American Society of Clinical Oncology (ASCO) virtual scientific program and published in The Lancet (Lancet. 2020 Jun 20;395[10241]:1907-18).
The latest data were presented at the AACR virtual meeting: COVID-19 and Cancer by Brian I. Rini, MD, of Vanderbilt University, Nashville, Tenn. They were simultaneously published in Cancer Discovery (Cancer Discov. 2020 Jul 22;CD-20-0941).
The CCC19 registry was launched in March by a few institutions as part of “a grassroots idea ... to collect granular data regarding cancer patients and their outcomes with COVID,” Dr. Rini said.
Within a few months of its inception, the registry had partnered with more than 100 institutions worldwide and accrued data from more than 2,000 patients.
The reports in The Lancet and at ASCO included outcomes for the first 928 patients and showed a 13% mortality rate as well as a fivefold increase in the risk of 30-day mortality among patients with COVID-19 and progressing cancer.
The data also showed an increased mortality risk among older patients, men, former smokers, those with poor performance status, those with multiple comorbidities, and those treated with hydroxychloroquine and azithromycin.
The latest data
The CCC19 registry has grown to include 114 sites worldwide, including major comprehensive cancer centers and community sites. As of June 26, there were 2,749 patients enrolled.
Since the last data were reported, the mortality rate increased from 13% to 16% (versus 5% globally). In addition, the increased mortality risk among non-Hispanic black patients and patients with hematologic malignancies reached statistical significance, Dr. Rini said. He noted that the increase in mortality rate was largely attributable to improved follow-up.
Mechanical ventilation was required in 12% of patients, ICU admission was required in 16%, oxygen was required in 45%, and hospitalization was required in 60%. The composite outcome of death, severe illness requiring hospitalization, ICU admission, or mechanical ventilation was reached in 29% of patients, Dr. Rini said.
Mortality rates across cancer types ranged from 3% to 26%, with thyroid and breast cancer patients having the lowest rates (3% and 8%, respectively), and with lymphoma and lung cancer patients having the highest (22% and 26%, respectively), Dr. Rini said.
He noted that the TERAVOLT registry, a COVID-19 registry for patients with thoracic cancers, also showed a very high mortality rate in this subgroup of patients.
Results from TERAVOLT were reported at the AACR virtual meeting I, presented at ASCO, and published in The Lancet (Lancet Oncol. 2020 Jul;21[7]:914-22). The most recent results showed a mortality rate of nearly 36% and reinforce the high mortality rate seen in lung cancer patients in CCC19, Dr. Rini said.
Increased mortality risk
After adjustment for several demographic and disease characteristics, the updated CCC19 data showed a significantly increased risk of mortality among:
- Older patients (adjusted odds ratio [aOR] per decade of age, 1.52).
- Men (aOR, 1.43).
- Current or former smokers vs. never smokers (aOR, 1.28).
- Patients with Eastern Cooperative Oncology Group performance scores of 1 vs. 0 (aOR of 1.80) or 2 vs. 0 (aOR, 4.22).
- Stable cancer vs. remission (aOR, 1.47).
- Progressive cancer vs. remission (aOR, 2.96).
- Non-Hispanic Black vs. White patients (aOR, 1.56).
- Hematologic malignancies vs. solid tumors (aOR, 1.80).
“Importantly, there were some factors that did not reach statistical significance,” Dr. Rini said. These include obesity (aOR, 1.23), recent surgery (aOR, 1.05), receipt of cytotoxic chemotherapy vs. no chemotherapy (aOR, 1.14), and receipt of noncytotoxic chemotherapy vs. no chemotherapy (aOR, 0.75).
“I think this provides some reassurance that cancer care can and should continue for these patients,” Dr. Rini said.
He noted, however, that in TERAVOLT, chemotherapy with or without other treatment was a risk factor for mortality in lung cancer patients when compared with no chemotherapy (OR, 1.71) and when compared with immunotherapy or targeted therapy (OR, 1.64).
NCCAPS and other registries
Dr. Rini discussed a number of registries looking at outcomes in COVID-19 patients with cancer, and he said the findings to date appear to confirm a higher mortality rate among cancer patients, particularly those with lung cancer.
Several factors are emerging that appear to be related to risk, including both cancer-related and non–cancer-related factors, he added.
The ongoing prospective National Cancer Institute COVID-19 in Cancer Patients Study (NCCAPS) “will provide much needed longitudinal data and, importantly, biospecimen collection in a large cohort of patients who have active cancer and are receiving treatment, said Dr. Rini, who is the study’s protocol chair. NCCAPS is a natural history study in that population, he said.
The planned accrual is about 2,000 patients who will be followed for up to 2 years for data collection, imaging scans, and research specimens.
The use of specimens is “a unique and special part of this study,” Dr. Rini said, explaining that the specimens will be used to look for development of antibodies over time, to describe the trajectory of cytokine abnormalities – especially in patients with more acute inpatient courses – to perform DNA-based genome-wide association studies, and to assess coagulation parameters.
NCCAPS is activated at 546 sties, 10 patients were enrolled as of June 21, and rapid accrual is expected over the next several months, he said.
Gypsyamber D’Souza, PhD, session moderator and an infectious disease epidemiologist at Johns Hopkins University in Baltimore, acknowledged the challenge that registry administrators face when trying to balance the need to get data out against the desire to ask the right questions and to have the right comparison groups, stratification, and analyses, especially amid a crisis like the COVID-19 pandemic.
Dr. Rini said it has indeed been a bit of a struggle with CCC19 to determine what information should be published and when, and what constitutes an important update.
“It’s been a learning experience, and frankly, I think we’re still learning,” he said. “This has been such a unique time in terms of a rush to get data out, balanced against making sure that there’s quality data and that you’re actually answering important questions.”
In fact, a number of ongoing registries “should start to produce great data [that will be presented] at upcoming big conferences,” Dr. Rini said. He added that those data “will help piece together different important aspects of this and different hypotheses, and hopefully complement the clinical data that’s starting to come out.”
The CCC19 registry is sponsored by Vanderbilt-Ingram Cancer Center. Dr. Rini disclosed relationships with Pfizer, Merck, Genentech/Roche, Aveo, AstraZeneca, Bristol Myers Squibb, Exelixis, Synthorx, Peloton, Compugen, Corvus, Surface Oncology, 3DMedicines, Aravive, Alkermes, Arrowhead, and PTC Therapeutics. Dr. D’Souza did not disclose any conflicts.
SOURCE: Rini BI. AACR: COVID-19 and Cancer. Abstract IA26.
FROM AACR: COVID-19 and CANCER
Genetic differences by ancestry shouldn’t impact efficacy of prostate cancer therapies
, according to an analysis published in Clinical Cancer Research.
“[N]o significant differences were seen in clinically actionable DNA repair genes, MSI-high [microsatellite instability–high] status, and tumor mutation burden, suggesting that current therapeutic strategies may be equally beneficial in both populations,” wrote study author Yusuke Koga, of the Boston University, and colleagues.
“Since these findings suggest that the frequency of targetable genetic alterations is similar in patients of predominantly African versus European ancestry, offering comprehensive genomic profiling and biomarker-based therapies to all patients, including African American patients, is a critical component of promoting equity in the management of metastatic prostate cancer,” said Atish D. Choudhury, MD, PhD, of the Dana-Farber Cancer Institute in Boston, who was not involved in this study.
Mr. Koga and colleagues noted that, when compared with European-American men, African American men have a higher incidence of prostate cancer, present with more advanced disease at an earlier age, and have increased mortality. These differences persist even after adjustment for socioeconomic covariates. That raises the question of the role of genetics.
“There is emerging evidence that, across some clinical trials and equal-access health systems, outcomes between AFR [African-American] men and European-American men with prostate cancer are similar,” the investigators wrote. “Although these data suggest that disparities can be ameliorated, there is limited knowledge of the genomic alterations that differ between groups and that could impact clinical outcomes.”
Study details and results
To get a handle on the issue, the investigators performed a meta-analysis of tumors from 250 African American men and 611 European-American men to compare the frequencies of somatic alterations across datasets from the Cancer Genome Atlas, the African Ancestry prostate cancer cohort, and the Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets panel.
The team also compared prostate cancer sequencing data from a commercial platform, the Foundation Medicine assay, from 436 African-American men and 3,018 European-American men.
In the meta-analysis, mutations in ZFHX3 and focal deletions in ETV3 were more common in tumors from African American men than in tumors from European-American men. Both genes are putative prostate cancer tumor suppressors, the investigators noted.
TP53 mutations, meanwhile, were associated with increasing Gleason scores in both groups, suggesting “that if TP53 mutations are found in low-grade disease, they may potentially indicate a more aggressive clinical trajectory,” the investigators wrote.
In the analysis with the commercial assay, MYC amplifications were more frequent in African American men with metastatic disease, raising “the possibility that MYC amplifications may also contribute to high-risk disease in this population,” the team wrote.
Deletions in PTEN and rearrangements in TMPRSS2-ERG were less frequent in tumors from African American men, but KMT2D truncations and CCND1 amplifications were more frequent.
“Higher expression of CCND1 has been implicated with perineural invasion in prostate cancer, an aggressive histological feature in prostate cancer. Truncating mutations in KMT2D have been reported in both localized and metastatic prostate cancer patients with unclear clinical significance,” the investigators noted.
“The genomic differences seen in genes such as MYC, ZFHX3, PTEN, and TMPRSS2-ERG suggest that different pathways of carcinogenesis may be active in AFR [African American] men, which could lead to further disparities if targeted therapies for some of these alterations become available,” the team wrote.
They noted that the meta-analysis was limited by the fact that some cohorts lacked matched tumors from European-American men, which limited the investigators’ ability to control for differences in region, clinical setting, or sequencing assay. Furthermore, age, tumor stage, and Gleason grade were unavailable in the cohort analyzed with the commercial assay.
This research was funded by the Department of Defense, the National Cancer Institute, and the Prostate Cancer Foundation. Two authors are employees of Foundation Medicine.
SOURCE: Koga Y et al. Clin Cancer Res. 2020 Jul 10. doi: 10.1158/1078-0432.CCR-19-4112.
, according to an analysis published in Clinical Cancer Research.
“[N]o significant differences were seen in clinically actionable DNA repair genes, MSI-high [microsatellite instability–high] status, and tumor mutation burden, suggesting that current therapeutic strategies may be equally beneficial in both populations,” wrote study author Yusuke Koga, of the Boston University, and colleagues.
“Since these findings suggest that the frequency of targetable genetic alterations is similar in patients of predominantly African versus European ancestry, offering comprehensive genomic profiling and biomarker-based therapies to all patients, including African American patients, is a critical component of promoting equity in the management of metastatic prostate cancer,” said Atish D. Choudhury, MD, PhD, of the Dana-Farber Cancer Institute in Boston, who was not involved in this study.
Mr. Koga and colleagues noted that, when compared with European-American men, African American men have a higher incidence of prostate cancer, present with more advanced disease at an earlier age, and have increased mortality. These differences persist even after adjustment for socioeconomic covariates. That raises the question of the role of genetics.
“There is emerging evidence that, across some clinical trials and equal-access health systems, outcomes between AFR [African-American] men and European-American men with prostate cancer are similar,” the investigators wrote. “Although these data suggest that disparities can be ameliorated, there is limited knowledge of the genomic alterations that differ between groups and that could impact clinical outcomes.”
Study details and results
To get a handle on the issue, the investigators performed a meta-analysis of tumors from 250 African American men and 611 European-American men to compare the frequencies of somatic alterations across datasets from the Cancer Genome Atlas, the African Ancestry prostate cancer cohort, and the Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets panel.
The team also compared prostate cancer sequencing data from a commercial platform, the Foundation Medicine assay, from 436 African-American men and 3,018 European-American men.
In the meta-analysis, mutations in ZFHX3 and focal deletions in ETV3 were more common in tumors from African American men than in tumors from European-American men. Both genes are putative prostate cancer tumor suppressors, the investigators noted.
TP53 mutations, meanwhile, were associated with increasing Gleason scores in both groups, suggesting “that if TP53 mutations are found in low-grade disease, they may potentially indicate a more aggressive clinical trajectory,” the investigators wrote.
In the analysis with the commercial assay, MYC amplifications were more frequent in African American men with metastatic disease, raising “the possibility that MYC amplifications may also contribute to high-risk disease in this population,” the team wrote.
Deletions in PTEN and rearrangements in TMPRSS2-ERG were less frequent in tumors from African American men, but KMT2D truncations and CCND1 amplifications were more frequent.
“Higher expression of CCND1 has been implicated with perineural invasion in prostate cancer, an aggressive histological feature in prostate cancer. Truncating mutations in KMT2D have been reported in both localized and metastatic prostate cancer patients with unclear clinical significance,” the investigators noted.
“The genomic differences seen in genes such as MYC, ZFHX3, PTEN, and TMPRSS2-ERG suggest that different pathways of carcinogenesis may be active in AFR [African American] men, which could lead to further disparities if targeted therapies for some of these alterations become available,” the team wrote.
They noted that the meta-analysis was limited by the fact that some cohorts lacked matched tumors from European-American men, which limited the investigators’ ability to control for differences in region, clinical setting, or sequencing assay. Furthermore, age, tumor stage, and Gleason grade were unavailable in the cohort analyzed with the commercial assay.
This research was funded by the Department of Defense, the National Cancer Institute, and the Prostate Cancer Foundation. Two authors are employees of Foundation Medicine.
SOURCE: Koga Y et al. Clin Cancer Res. 2020 Jul 10. doi: 10.1158/1078-0432.CCR-19-4112.
, according to an analysis published in Clinical Cancer Research.
“[N]o significant differences were seen in clinically actionable DNA repair genes, MSI-high [microsatellite instability–high] status, and tumor mutation burden, suggesting that current therapeutic strategies may be equally beneficial in both populations,” wrote study author Yusuke Koga, of the Boston University, and colleagues.
“Since these findings suggest that the frequency of targetable genetic alterations is similar in patients of predominantly African versus European ancestry, offering comprehensive genomic profiling and biomarker-based therapies to all patients, including African American patients, is a critical component of promoting equity in the management of metastatic prostate cancer,” said Atish D. Choudhury, MD, PhD, of the Dana-Farber Cancer Institute in Boston, who was not involved in this study.
Mr. Koga and colleagues noted that, when compared with European-American men, African American men have a higher incidence of prostate cancer, present with more advanced disease at an earlier age, and have increased mortality. These differences persist even after adjustment for socioeconomic covariates. That raises the question of the role of genetics.
“There is emerging evidence that, across some clinical trials and equal-access health systems, outcomes between AFR [African-American] men and European-American men with prostate cancer are similar,” the investigators wrote. “Although these data suggest that disparities can be ameliorated, there is limited knowledge of the genomic alterations that differ between groups and that could impact clinical outcomes.”
Study details and results
To get a handle on the issue, the investigators performed a meta-analysis of tumors from 250 African American men and 611 European-American men to compare the frequencies of somatic alterations across datasets from the Cancer Genome Atlas, the African Ancestry prostate cancer cohort, and the Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets panel.
The team also compared prostate cancer sequencing data from a commercial platform, the Foundation Medicine assay, from 436 African-American men and 3,018 European-American men.
In the meta-analysis, mutations in ZFHX3 and focal deletions in ETV3 were more common in tumors from African American men than in tumors from European-American men. Both genes are putative prostate cancer tumor suppressors, the investigators noted.
TP53 mutations, meanwhile, were associated with increasing Gleason scores in both groups, suggesting “that if TP53 mutations are found in low-grade disease, they may potentially indicate a more aggressive clinical trajectory,” the investigators wrote.
In the analysis with the commercial assay, MYC amplifications were more frequent in African American men with metastatic disease, raising “the possibility that MYC amplifications may also contribute to high-risk disease in this population,” the team wrote.
Deletions in PTEN and rearrangements in TMPRSS2-ERG were less frequent in tumors from African American men, but KMT2D truncations and CCND1 amplifications were more frequent.
“Higher expression of CCND1 has been implicated with perineural invasion in prostate cancer, an aggressive histological feature in prostate cancer. Truncating mutations in KMT2D have been reported in both localized and metastatic prostate cancer patients with unclear clinical significance,” the investigators noted.
“The genomic differences seen in genes such as MYC, ZFHX3, PTEN, and TMPRSS2-ERG suggest that different pathways of carcinogenesis may be active in AFR [African American] men, which could lead to further disparities if targeted therapies for some of these alterations become available,” the team wrote.
They noted that the meta-analysis was limited by the fact that some cohorts lacked matched tumors from European-American men, which limited the investigators’ ability to control for differences in region, clinical setting, or sequencing assay. Furthermore, age, tumor stage, and Gleason grade were unavailable in the cohort analyzed with the commercial assay.
This research was funded by the Department of Defense, the National Cancer Institute, and the Prostate Cancer Foundation. Two authors are employees of Foundation Medicine.
SOURCE: Koga Y et al. Clin Cancer Res. 2020 Jul 10. doi: 10.1158/1078-0432.CCR-19-4112.
FROM CLINICAL CANCER RESEARCH
Early data support further study of ivosidenib in mIDH1 glioma
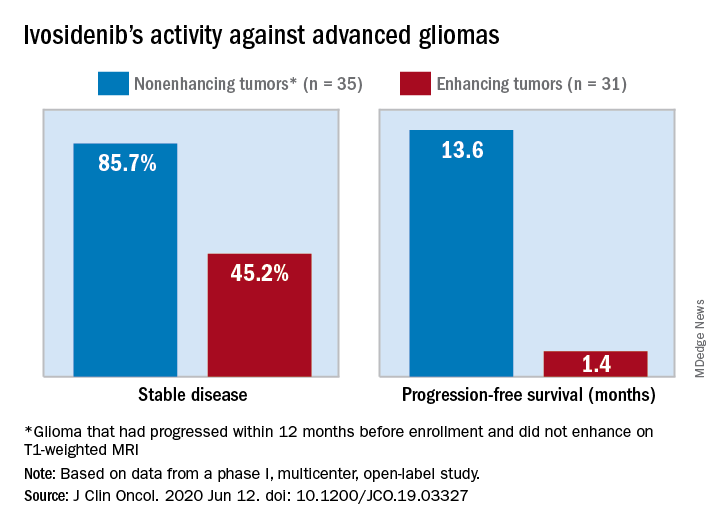
The median progression-free survival was 13.6 months for patients with nonenhancing tumors and 1.4 months for patients with enhancing tumors in a study of 66 adults with mIDH1 advanced glioma.
“On the basis of these data, additional clinical development of mIDH inhibitors for mIDH low-grade gliomas is warranted,” Ingo Mellinghoff, MD, of Memorial Sloan Kettering Cancer Center in New York, and colleagues wrote in the Journal of Clinical Oncology.
“This is not a home run but is of interest to the community,” said Lawrence Recht, MD, of Stanford (Calif.) University, who was not involved in this study. “Other companies are also developing agents like this.”
Considering that the ivosidenib study “is uncontrolled, one cannot say for sure that this wasn’t just the natural history of the disease,” Dr. Recht continued. “This type of tumor can behave very indolently, and patients can survive years without treatment, so this is rather a short interval to make a long-time statement. I think the authors are a bit overenthusiastic.”
The authors tested ivosidenib in 66 adults with mIDH1 glioma – 35 with nonenhancing glioma and 31 with enhancing glioma. Tumors had recurred after, or did not respond to, initial surgery, radiation, or chemotherapy.
The patients’ median age was 41 years (range, 21-71 years), and 25 patients (37.9%) were women. The most common tumor type at screening was oligodendroglioma in 23 patients (34.8%).
Patients received ivosidenib at doses ranging from 100 mg twice a day to 900 mg once a day. A total of 50 patients received the phase 2 recommended dose – 500 mg once a day. There were no dose-limiting toxicities, and there was no maximum-tolerated dose.
Adverse events of grade 3 or higher occurred in 19.7% of patients and included headache, seizure, hyperglycemia, neutropenia, and hypophosphatemia. Grade 3 or higher treatment-related adverse events occurred in two patients.
A total of 30 patients with nonenhancing tumors (85.7%) and 14 with enhancing tumors (45.2%) had a best response of stable disease. There was one partial response in a nonenhancing patient on 500 mg/day. The rest of the subjects had a best response of progressive disease.
The median treatment duration was 18.4 months among patients with nonenhancing tumors and 1.9 months among those with enhancing tumors. Discontinuation was caused byo progression in all but one case.
Among patients with measurable disease, tumor measurements decreased from baseline in 22 nonenhancing tumors (66.7%) and in 9 enhancing tumors (33.3%).
“Despite the heterogeneous patient population in our trial, the nonrandomized design, and the lack of central pathology review, the data from our trial suggest that ivosidenib has greater activity against nonenhancing gliomas than against enhancing gliomas,” the investigators wrote. “This finding may seem surprising because the absence of contrast enhancement is typically associated with impaired drug delivery.
“We hypothesize that ivosidenib may be more effective in nonenhancing gliomas because these tumors represent an earlier disease stage with fewer genetic alterations, reminiscent of the greater antitumor activity of the BCR-ABL inhibitor imatinib in earlier stages of chronic myeloid leukemia,” the investigators wrote.
The team also noted that the median progression-free survival for patients with nonenhancing gliomas in the current study “compares favorably to that reported for temozolomide” in advanced mIDH1 low-grade glioma, which was approximately 7 months.
This research was funded by Agios Pharmaceuticals, the company developing ivosidenib. Dr. Mellinghoff receives travel compensation from and is an adviser to the company. Several other investigators are employees. Dr. Recht disclosed no conflicts of interest.
SOURCE: Mellinghoff I et al. J Clin Oncol. 2020 Jun 12. doi: 10.1200/JCO.19.03327
The median progression-free survival was 13.6 months for patients with nonenhancing tumors and 1.4 months for patients with enhancing tumors in a study of 66 adults with mIDH1 advanced glioma.
“On the basis of these data, additional clinical development of mIDH inhibitors for mIDH low-grade gliomas is warranted,” Ingo Mellinghoff, MD, of Memorial Sloan Kettering Cancer Center in New York, and colleagues wrote in the Journal of Clinical Oncology.
“This is not a home run but is of interest to the community,” said Lawrence Recht, MD, of Stanford (Calif.) University, who was not involved in this study. “Other companies are also developing agents like this.”
Considering that the ivosidenib study “is uncontrolled, one cannot say for sure that this wasn’t just the natural history of the disease,” Dr. Recht continued. “This type of tumor can behave very indolently, and patients can survive years without treatment, so this is rather a short interval to make a long-time statement. I think the authors are a bit overenthusiastic.”
The authors tested ivosidenib in 66 adults with mIDH1 glioma – 35 with nonenhancing glioma and 31 with enhancing glioma. Tumors had recurred after, or did not respond to, initial surgery, radiation, or chemotherapy.
The patients’ median age was 41 years (range, 21-71 years), and 25 patients (37.9%) were women. The most common tumor type at screening was oligodendroglioma in 23 patients (34.8%).
Patients received ivosidenib at doses ranging from 100 mg twice a day to 900 mg once a day. A total of 50 patients received the phase 2 recommended dose – 500 mg once a day. There were no dose-limiting toxicities, and there was no maximum-tolerated dose.
Adverse events of grade 3 or higher occurred in 19.7% of patients and included headache, seizure, hyperglycemia, neutropenia, and hypophosphatemia. Grade 3 or higher treatment-related adverse events occurred in two patients.
A total of 30 patients with nonenhancing tumors (85.7%) and 14 with enhancing tumors (45.2%) had a best response of stable disease. There was one partial response in a nonenhancing patient on 500 mg/day. The rest of the subjects had a best response of progressive disease.
The median treatment duration was 18.4 months among patients with nonenhancing tumors and 1.9 months among those with enhancing tumors. Discontinuation was caused byo progression in all but one case.
Among patients with measurable disease, tumor measurements decreased from baseline in 22 nonenhancing tumors (66.7%) and in 9 enhancing tumors (33.3%).
“Despite the heterogeneous patient population in our trial, the nonrandomized design, and the lack of central pathology review, the data from our trial suggest that ivosidenib has greater activity against nonenhancing gliomas than against enhancing gliomas,” the investigators wrote. “This finding may seem surprising because the absence of contrast enhancement is typically associated with impaired drug delivery.
“We hypothesize that ivosidenib may be more effective in nonenhancing gliomas because these tumors represent an earlier disease stage with fewer genetic alterations, reminiscent of the greater antitumor activity of the BCR-ABL inhibitor imatinib in earlier stages of chronic myeloid leukemia,” the investigators wrote.
The team also noted that the median progression-free survival for patients with nonenhancing gliomas in the current study “compares favorably to that reported for temozolomide” in advanced mIDH1 low-grade glioma, which was approximately 7 months.
This research was funded by Agios Pharmaceuticals, the company developing ivosidenib. Dr. Mellinghoff receives travel compensation from and is an adviser to the company. Several other investigators are employees. Dr. Recht disclosed no conflicts of interest.
SOURCE: Mellinghoff I et al. J Clin Oncol. 2020 Jun 12. doi: 10.1200/JCO.19.03327
The median progression-free survival was 13.6 months for patients with nonenhancing tumors and 1.4 months for patients with enhancing tumors in a study of 66 adults with mIDH1 advanced glioma.
“On the basis of these data, additional clinical development of mIDH inhibitors for mIDH low-grade gliomas is warranted,” Ingo Mellinghoff, MD, of Memorial Sloan Kettering Cancer Center in New York, and colleagues wrote in the Journal of Clinical Oncology.
“This is not a home run but is of interest to the community,” said Lawrence Recht, MD, of Stanford (Calif.) University, who was not involved in this study. “Other companies are also developing agents like this.”
Considering that the ivosidenib study “is uncontrolled, one cannot say for sure that this wasn’t just the natural history of the disease,” Dr. Recht continued. “This type of tumor can behave very indolently, and patients can survive years without treatment, so this is rather a short interval to make a long-time statement. I think the authors are a bit overenthusiastic.”
The authors tested ivosidenib in 66 adults with mIDH1 glioma – 35 with nonenhancing glioma and 31 with enhancing glioma. Tumors had recurred after, or did not respond to, initial surgery, radiation, or chemotherapy.
The patients’ median age was 41 years (range, 21-71 years), and 25 patients (37.9%) were women. The most common tumor type at screening was oligodendroglioma in 23 patients (34.8%).
Patients received ivosidenib at doses ranging from 100 mg twice a day to 900 mg once a day. A total of 50 patients received the phase 2 recommended dose – 500 mg once a day. There were no dose-limiting toxicities, and there was no maximum-tolerated dose.
Adverse events of grade 3 or higher occurred in 19.7% of patients and included headache, seizure, hyperglycemia, neutropenia, and hypophosphatemia. Grade 3 or higher treatment-related adverse events occurred in two patients.
A total of 30 patients with nonenhancing tumors (85.7%) and 14 with enhancing tumors (45.2%) had a best response of stable disease. There was one partial response in a nonenhancing patient on 500 mg/day. The rest of the subjects had a best response of progressive disease.
The median treatment duration was 18.4 months among patients with nonenhancing tumors and 1.9 months among those with enhancing tumors. Discontinuation was caused byo progression in all but one case.
Among patients with measurable disease, tumor measurements decreased from baseline in 22 nonenhancing tumors (66.7%) and in 9 enhancing tumors (33.3%).
“Despite the heterogeneous patient population in our trial, the nonrandomized design, and the lack of central pathology review, the data from our trial suggest that ivosidenib has greater activity against nonenhancing gliomas than against enhancing gliomas,” the investigators wrote. “This finding may seem surprising because the absence of contrast enhancement is typically associated with impaired drug delivery.
“We hypothesize that ivosidenib may be more effective in nonenhancing gliomas because these tumors represent an earlier disease stage with fewer genetic alterations, reminiscent of the greater antitumor activity of the BCR-ABL inhibitor imatinib in earlier stages of chronic myeloid leukemia,” the investigators wrote.
The team also noted that the median progression-free survival for patients with nonenhancing gliomas in the current study “compares favorably to that reported for temozolomide” in advanced mIDH1 low-grade glioma, which was approximately 7 months.
This research was funded by Agios Pharmaceuticals, the company developing ivosidenib. Dr. Mellinghoff receives travel compensation from and is an adviser to the company. Several other investigators are employees. Dr. Recht disclosed no conflicts of interest.
SOURCE: Mellinghoff I et al. J Clin Oncol. 2020 Jun 12. doi: 10.1200/JCO.19.03327
FROM THE JOURNAL OF CLINICAL ONCOLOGY
FDA approves new treatment for Dravet syndrome
Dravet syndrome is a rare childhood-onset epilepsy characterized by frequent, drug-resistant convulsive seizures that may contribute to intellectual disability and impairments in motor control, behavior, and cognition, as well as an increased risk of sudden unexpected death in epilepsy (SUDEP).
Dravet syndrome takes a “tremendous toll on both patients and their families. Fintepla offers an additional effective treatment option for the treatment of seizures associated with Dravet syndrome,” Billy Dunn, MD, director, Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research, said in a news release.
The FDA approved fenfluramine for Dravet syndrome based on the results of two randomized, double-blind, placebo-controlled phase 3 trials involving children ages 2 to 18 years with Dravet syndrome.
In both studies, children treated with fenfluramine experienced significantly greater reductions in the frequency of convulsive seizures than did their peers who received placebo. These reductions occurred within 3 to 4 weeks, and remained generally consistent over the 14- to 15-week treatment periods, the FDA said.
“There remains a huge unmet need for the many Dravet syndrome patients who continue to experience frequent severe seizures even while taking one or more of the currently available antiseizure medications,” Joseph Sullivan, MD, who worked on the fenfluramine for Dravet syndrome studies, said in a news release.
Given the “profound reductions” in convulsive seizure frequency seen in the clinical trials, combined with the “ongoing, robust safety monitoring,” fenfluramine offers “an extremely important treatment option for Dravet syndrome patients,” said Dr. Sullivan, director of the Pediatric Epilepsy Center of Excellence at the University of California San Francisco (UCSF) Benioff Children’s Hospital.
Fenfluramine is an anorectic agent that was used to treat obesity until it was removed from the market in 1997 over reports of increased risk of valvular heart disease when prescribed in higher doses and most often when prescribed with phentermine. The combination of the two drugs was known as fen-phen.
In the clinical trials of Dravet syndrome, the most common adverse reactions were decreased appetite; somnolence, sedation, lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue, malaise, asthenia; ataxia, balance disorder, gait disturbance; increased blood pressure; drooling, salivary hypersecretion; pyrexia; upper respiratory tract infection; vomiting; decreased weight; fall; and status epilepticus.
The Fintepla label has a boxed warning stating that the drug is associated with valvular heart disease (VHD) and pulmonary arterial hypertension (PAH). Due to these risks, patients must undergo echocardiography before treatment, every 6 months during treatment, and once 3 to 6 months after treatment is stopped.
If signs of VHD, PAH, or other cardiac abnormalities are present, clinicians should weigh the benefits and risks of continuing treatment with Fintepla, the FDA said.
Fintepla is available only through a risk evaluation and mitigation strategy (REMS) program, which requires physicians who prescribe the drug and pharmacies that dispense it to be certified in the Fintepla REMS and that patients be enrolled in the program.
As part of the REMS requirements, prescribers and patients must adhere to the required cardiac monitoring to receive the drug.
Fintepla will be available to certified prescribers in the United States in July. Zogenix is launching Zogenix Central, a comprehensive support service that will provide ongoing product assistance to patients, caregivers, and their medical teams. Further information is available online.
This article first appeared on Medscape.com.
Dravet syndrome is a rare childhood-onset epilepsy characterized by frequent, drug-resistant convulsive seizures that may contribute to intellectual disability and impairments in motor control, behavior, and cognition, as well as an increased risk of sudden unexpected death in epilepsy (SUDEP).
Dravet syndrome takes a “tremendous toll on both patients and their families. Fintepla offers an additional effective treatment option for the treatment of seizures associated with Dravet syndrome,” Billy Dunn, MD, director, Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research, said in a news release.
The FDA approved fenfluramine for Dravet syndrome based on the results of two randomized, double-blind, placebo-controlled phase 3 trials involving children ages 2 to 18 years with Dravet syndrome.
In both studies, children treated with fenfluramine experienced significantly greater reductions in the frequency of convulsive seizures than did their peers who received placebo. These reductions occurred within 3 to 4 weeks, and remained generally consistent over the 14- to 15-week treatment periods, the FDA said.
“There remains a huge unmet need for the many Dravet syndrome patients who continue to experience frequent severe seizures even while taking one or more of the currently available antiseizure medications,” Joseph Sullivan, MD, who worked on the fenfluramine for Dravet syndrome studies, said in a news release.
Given the “profound reductions” in convulsive seizure frequency seen in the clinical trials, combined with the “ongoing, robust safety monitoring,” fenfluramine offers “an extremely important treatment option for Dravet syndrome patients,” said Dr. Sullivan, director of the Pediatric Epilepsy Center of Excellence at the University of California San Francisco (UCSF) Benioff Children’s Hospital.
Fenfluramine is an anorectic agent that was used to treat obesity until it was removed from the market in 1997 over reports of increased risk of valvular heart disease when prescribed in higher doses and most often when prescribed with phentermine. The combination of the two drugs was known as fen-phen.
In the clinical trials of Dravet syndrome, the most common adverse reactions were decreased appetite; somnolence, sedation, lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue, malaise, asthenia; ataxia, balance disorder, gait disturbance; increased blood pressure; drooling, salivary hypersecretion; pyrexia; upper respiratory tract infection; vomiting; decreased weight; fall; and status epilepticus.
The Fintepla label has a boxed warning stating that the drug is associated with valvular heart disease (VHD) and pulmonary arterial hypertension (PAH). Due to these risks, patients must undergo echocardiography before treatment, every 6 months during treatment, and once 3 to 6 months after treatment is stopped.
If signs of VHD, PAH, or other cardiac abnormalities are present, clinicians should weigh the benefits and risks of continuing treatment with Fintepla, the FDA said.
Fintepla is available only through a risk evaluation and mitigation strategy (REMS) program, which requires physicians who prescribe the drug and pharmacies that dispense it to be certified in the Fintepla REMS and that patients be enrolled in the program.
As part of the REMS requirements, prescribers and patients must adhere to the required cardiac monitoring to receive the drug.
Fintepla will be available to certified prescribers in the United States in July. Zogenix is launching Zogenix Central, a comprehensive support service that will provide ongoing product assistance to patients, caregivers, and their medical teams. Further information is available online.
This article first appeared on Medscape.com.
Dravet syndrome is a rare childhood-onset epilepsy characterized by frequent, drug-resistant convulsive seizures that may contribute to intellectual disability and impairments in motor control, behavior, and cognition, as well as an increased risk of sudden unexpected death in epilepsy (SUDEP).
Dravet syndrome takes a “tremendous toll on both patients and their families. Fintepla offers an additional effective treatment option for the treatment of seizures associated with Dravet syndrome,” Billy Dunn, MD, director, Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research, said in a news release.
The FDA approved fenfluramine for Dravet syndrome based on the results of two randomized, double-blind, placebo-controlled phase 3 trials involving children ages 2 to 18 years with Dravet syndrome.
In both studies, children treated with fenfluramine experienced significantly greater reductions in the frequency of convulsive seizures than did their peers who received placebo. These reductions occurred within 3 to 4 weeks, and remained generally consistent over the 14- to 15-week treatment periods, the FDA said.
“There remains a huge unmet need for the many Dravet syndrome patients who continue to experience frequent severe seizures even while taking one or more of the currently available antiseizure medications,” Joseph Sullivan, MD, who worked on the fenfluramine for Dravet syndrome studies, said in a news release.
Given the “profound reductions” in convulsive seizure frequency seen in the clinical trials, combined with the “ongoing, robust safety monitoring,” fenfluramine offers “an extremely important treatment option for Dravet syndrome patients,” said Dr. Sullivan, director of the Pediatric Epilepsy Center of Excellence at the University of California San Francisco (UCSF) Benioff Children’s Hospital.
Fenfluramine is an anorectic agent that was used to treat obesity until it was removed from the market in 1997 over reports of increased risk of valvular heart disease when prescribed in higher doses and most often when prescribed with phentermine. The combination of the two drugs was known as fen-phen.
In the clinical trials of Dravet syndrome, the most common adverse reactions were decreased appetite; somnolence, sedation, lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue, malaise, asthenia; ataxia, balance disorder, gait disturbance; increased blood pressure; drooling, salivary hypersecretion; pyrexia; upper respiratory tract infection; vomiting; decreased weight; fall; and status epilepticus.
The Fintepla label has a boxed warning stating that the drug is associated with valvular heart disease (VHD) and pulmonary arterial hypertension (PAH). Due to these risks, patients must undergo echocardiography before treatment, every 6 months during treatment, and once 3 to 6 months after treatment is stopped.
If signs of VHD, PAH, or other cardiac abnormalities are present, clinicians should weigh the benefits and risks of continuing treatment with Fintepla, the FDA said.
Fintepla is available only through a risk evaluation and mitigation strategy (REMS) program, which requires physicians who prescribe the drug and pharmacies that dispense it to be certified in the Fintepla REMS and that patients be enrolled in the program.
As part of the REMS requirements, prescribers and patients must adhere to the required cardiac monitoring to receive the drug.
Fintepla will be available to certified prescribers in the United States in July. Zogenix is launching Zogenix Central, a comprehensive support service that will provide ongoing product assistance to patients, caregivers, and their medical teams. Further information is available online.
This article first appeared on Medscape.com.
Relapsing, progressive MS classifications should be abandoned
Most disability accumulation in relapsing multiple sclerosis (MS) is not associated with overt relapses, challenging the current clinical distinction of relapsing and progressive forms of the disease, a new analysis shows. “We have to abandon the distinction between relapsing and progressive MS being different populations,” said lead author Ludwig Kappos, MD, University of Basel (Switzerland). “The disease appears to be more of a continuum of disability progression, which is sometimes also accompanied by relapses.”

The analysis was published online June 8 in JAMA Neurology.
Assessing disability progression
Noting that there are mounting data to suggest patients with relapsing MS frequently experience worsening disability over time – even when relapse activity appears well controlled – the researchers aimed to investigate the relative contributions of progression independent of relapse activity and relapse-associated worsening to overall accumulating disability in patients with relapsing multiple sclerosis. To do this, they analyzed data from two identical randomized clinical trials (OPERA I and OPERA II) conducted between 2011 and 2015, which compared treatment with the new B-cell–depleting therapy ocrelizumab with interferon beta-1a in 1,656 patients with relapsing MS.
Confirmed disability accumulation was defined by an increase in 1 or more of 3 measures (Expanded Disability Status Scale, timed 25-ft walk, or 9-hole peg test), confirmed after 3 or 6 months, and was classified as being related to a clinical relapse or occurring in the absence of a relapse.
Results showed that after 96 weeks (1.8 years) of treatment, 12-week composite confirmed disability accumulation had occurred in 29.6% of patients receiving interferon beta-1a and 21.1% of those given ocrelizumab; 24-week composite confirmed disability accumulation occurred in 22.7% of interferon beta-1a patients and 16.2% of the ocrelizumab group.
In both treatment groups, the vast majority of events contributing to disability accumulation occurred independently of relapse activity. In the interferon group, 78% of events contributing to 12-week confirmed disability accumulation and 80.6% of events contributing to 24-week confirmed disability accumulation occurred in the absence of clinical relapses, with the corresponding figures in the ocrelizumab group being 88.0% (12 weeks) and 89.1% (24 weeks).
Only a minority of patients (about 17% in both groups) had confirmed disability accumulation accompanied by clinical relapses. Very few patients with confirmed disability accumulation (4% to 5%) experienced disability worsening both associated and independent of relapses. Ocrelizumab was associated with a reduced risk of both relapse-associated and relapse-independent confirmed disability accumulation, compared with interferon beta-1a.
“We found that there was progression of disability in both groups, and the really astonishing finding was that although all patients were classified as having relapsing remitting MS, actually most of the disability progression occurred without preceding relapses,” Dr. Kappos commented. He noted that there have been two previous observational studies that have shown a high rate of disability progressions without temporal association to relapses in relapsing remitting patients, but this is the first time that this progression of disability independent of relapses has been shown in the controlled setting of two prospective, randomized clinical trials over a 2-year period.
“While we expected to see some disability progression independent of relapses, we were surprised to see that the disability progression occurring in both studies was almost exclusively happening without temporal relation to relapses. That was certainly an unexpected finding,” Dr. Kappos said. “These observations make it difficult to keep the current definitions of ‘relapsing remitting’ and ‘secondary progressive’ MS, [ones] that suggest a clear-cut distinction marked by the presence or absence of relapses. This can no longer be justified,” he stressed.
“We are not saying that relapses do not contribute to disability progression. There are a lot of data to support the fact that they do. But I think what we might be seeing is that the drug therapy is quite effective in reducing disability due to relapses but only partially effective in reducing progression independent of relapses,” Dr. Kappos explained.
Although there have been many advances in reducing relapses with drug therapy, focus now needs to shift to the other more continuous process of disability progression independent of relapses, Dr. Kappos said. “There is still a lot of room for improvement here.”
“If continuous progression independent of relapses is already present in the early phases of MS, it is reasonable to study the effects of intervention on steady progression already in this early phase,” he noted. “This might help to capture patients at earlier stages who better respond to treatment aimed at halting progression.”
Dr. Kappos also called for more subtle measurements of disability than the EDSS alone, including measures such as the 9-hole peg test and the 25-ft walk as they did in this analysis. But other measures could also be added that would characterize continuous disease activity and progression, such as laboratory values (e.g., neurofilament light chain) and advanced, more tissue-specific quantitative MRI techniques and digital biomarkers to detect subtle changes in neurologic function.
An artificial distinction?
Commenting on the study, Jeffrey Cohen, MD, director of the experimental therapeutics program at the Mellen Center for Multiple Sclerosis Treatment and Research, Cleveland Clinic, said he too sees very little distinction between relapsing remitting and progressive forms of the disease.
“This study confirms what has been suspected for quite a few years –that if one looks sufficiently and carefully, there is gradual worsening of some aspects of the disease in many patients from the earliest stages,” Dr. Cohen said. “Conversely, some patients with progressive MS have superimposed relapses or MRI lesion activity.
“Thus, the distinction between relapsing-remitting and progressive MS subtypes appears artificial,” he concluded.
This study was sponsored by F. Hoffmann–La Roche. Dr. Kappos has received research support from the company.
This article first appeared on Medscape.com.
Most disability accumulation in relapsing multiple sclerosis (MS) is not associated with overt relapses, challenging the current clinical distinction of relapsing and progressive forms of the disease, a new analysis shows. “We have to abandon the distinction between relapsing and progressive MS being different populations,” said lead author Ludwig Kappos, MD, University of Basel (Switzerland). “The disease appears to be more of a continuum of disability progression, which is sometimes also accompanied by relapses.”
The analysis was published online June 8 in JAMA Neurology.
Assessing disability progression
Noting that there are mounting data to suggest patients with relapsing MS frequently experience worsening disability over time – even when relapse activity appears well controlled – the researchers aimed to investigate the relative contributions of progression independent of relapse activity and relapse-associated worsening to overall accumulating disability in patients with relapsing multiple sclerosis. To do this, they analyzed data from two identical randomized clinical trials (OPERA I and OPERA II) conducted between 2011 and 2015, which compared treatment with the new B-cell–depleting therapy ocrelizumab with interferon beta-1a in 1,656 patients with relapsing MS.
Confirmed disability accumulation was defined by an increase in 1 or more of 3 measures (Expanded Disability Status Scale, timed 25-ft walk, or 9-hole peg test), confirmed after 3 or 6 months, and was classified as being related to a clinical relapse or occurring in the absence of a relapse.
Results showed that after 96 weeks (1.8 years) of treatment, 12-week composite confirmed disability accumulation had occurred in 29.6% of patients receiving interferon beta-1a and 21.1% of those given ocrelizumab; 24-week composite confirmed disability accumulation occurred in 22.7% of interferon beta-1a patients and 16.2% of the ocrelizumab group.
In both treatment groups, the vast majority of events contributing to disability accumulation occurred independently of relapse activity. In the interferon group, 78% of events contributing to 12-week confirmed disability accumulation and 80.6% of events contributing to 24-week confirmed disability accumulation occurred in the absence of clinical relapses, with the corresponding figures in the ocrelizumab group being 88.0% (12 weeks) and 89.1% (24 weeks).
Only a minority of patients (about 17% in both groups) had confirmed disability accumulation accompanied by clinical relapses. Very few patients with confirmed disability accumulation (4% to 5%) experienced disability worsening both associated and independent of relapses. Ocrelizumab was associated with a reduced risk of both relapse-associated and relapse-independent confirmed disability accumulation, compared with interferon beta-1a.
“We found that there was progression of disability in both groups, and the really astonishing finding was that although all patients were classified as having relapsing remitting MS, actually most of the disability progression occurred without preceding relapses,” Dr. Kappos commented. He noted that there have been two previous observational studies that have shown a high rate of disability progressions without temporal association to relapses in relapsing remitting patients, but this is the first time that this progression of disability independent of relapses has been shown in the controlled setting of two prospective, randomized clinical trials over a 2-year period.
“While we expected to see some disability progression independent of relapses, we were surprised to see that the disability progression occurring in both studies was almost exclusively happening without temporal relation to relapses. That was certainly an unexpected finding,” Dr. Kappos said. “These observations make it difficult to keep the current definitions of ‘relapsing remitting’ and ‘secondary progressive’ MS, [ones] that suggest a clear-cut distinction marked by the presence or absence of relapses. This can no longer be justified,” he stressed.
“We are not saying that relapses do not contribute to disability progression. There are a lot of data to support the fact that they do. But I think what we might be seeing is that the drug therapy is quite effective in reducing disability due to relapses but only partially effective in reducing progression independent of relapses,” Dr. Kappos explained.
Although there have been many advances in reducing relapses with drug therapy, focus now needs to shift to the other more continuous process of disability progression independent of relapses, Dr. Kappos said. “There is still a lot of room for improvement here.”
“If continuous progression independent of relapses is already present in the early phases of MS, it is reasonable to study the effects of intervention on steady progression already in this early phase,” he noted. “This might help to capture patients at earlier stages who better respond to treatment aimed at halting progression.”
Dr. Kappos also called for more subtle measurements of disability than the EDSS alone, including measures such as the 9-hole peg test and the 25-ft walk as they did in this analysis. But other measures could also be added that would characterize continuous disease activity and progression, such as laboratory values (e.g., neurofilament light chain) and advanced, more tissue-specific quantitative MRI techniques and digital biomarkers to detect subtle changes in neurologic function.
An artificial distinction?
Commenting on the study, Jeffrey Cohen, MD, director of the experimental therapeutics program at the Mellen Center for Multiple Sclerosis Treatment and Research, Cleveland Clinic, said he too sees very little distinction between relapsing remitting and progressive forms of the disease.
“This study confirms what has been suspected for quite a few years –that if one looks sufficiently and carefully, there is gradual worsening of some aspects of the disease in many patients from the earliest stages,” Dr. Cohen said. “Conversely, some patients with progressive MS have superimposed relapses or MRI lesion activity.
“Thus, the distinction between relapsing-remitting and progressive MS subtypes appears artificial,” he concluded.
This study was sponsored by F. Hoffmann–La Roche. Dr. Kappos has received research support from the company.
This article first appeared on Medscape.com.
Most disability accumulation in relapsing multiple sclerosis (MS) is not associated with overt relapses, challenging the current clinical distinction of relapsing and progressive forms of the disease, a new analysis shows. “We have to abandon the distinction between relapsing and progressive MS being different populations,” said lead author Ludwig Kappos, MD, University of Basel (Switzerland). “The disease appears to be more of a continuum of disability progression, which is sometimes also accompanied by relapses.”
The analysis was published online June 8 in JAMA Neurology.
Assessing disability progression
Noting that there are mounting data to suggest patients with relapsing MS frequently experience worsening disability over time – even when relapse activity appears well controlled – the researchers aimed to investigate the relative contributions of progression independent of relapse activity and relapse-associated worsening to overall accumulating disability in patients with relapsing multiple sclerosis. To do this, they analyzed data from two identical randomized clinical trials (OPERA I and OPERA II) conducted between 2011 and 2015, which compared treatment with the new B-cell–depleting therapy ocrelizumab with interferon beta-1a in 1,656 patients with relapsing MS.
Confirmed disability accumulation was defined by an increase in 1 or more of 3 measures (Expanded Disability Status Scale, timed 25-ft walk, or 9-hole peg test), confirmed after 3 or 6 months, and was classified as being related to a clinical relapse or occurring in the absence of a relapse.
Results showed that after 96 weeks (1.8 years) of treatment, 12-week composite confirmed disability accumulation had occurred in 29.6% of patients receiving interferon beta-1a and 21.1% of those given ocrelizumab; 24-week composite confirmed disability accumulation occurred in 22.7% of interferon beta-1a patients and 16.2% of the ocrelizumab group.
In both treatment groups, the vast majority of events contributing to disability accumulation occurred independently of relapse activity. In the interferon group, 78% of events contributing to 12-week confirmed disability accumulation and 80.6% of events contributing to 24-week confirmed disability accumulation occurred in the absence of clinical relapses, with the corresponding figures in the ocrelizumab group being 88.0% (12 weeks) and 89.1% (24 weeks).
Only a minority of patients (about 17% in both groups) had confirmed disability accumulation accompanied by clinical relapses. Very few patients with confirmed disability accumulation (4% to 5%) experienced disability worsening both associated and independent of relapses. Ocrelizumab was associated with a reduced risk of both relapse-associated and relapse-independent confirmed disability accumulation, compared with interferon beta-1a.
“We found that there was progression of disability in both groups, and the really astonishing finding was that although all patients were classified as having relapsing remitting MS, actually most of the disability progression occurred without preceding relapses,” Dr. Kappos commented. He noted that there have been two previous observational studies that have shown a high rate of disability progressions without temporal association to relapses in relapsing remitting patients, but this is the first time that this progression of disability independent of relapses has been shown in the controlled setting of two prospective, randomized clinical trials over a 2-year period.
“While we expected to see some disability progression independent of relapses, we were surprised to see that the disability progression occurring in both studies was almost exclusively happening without temporal relation to relapses. That was certainly an unexpected finding,” Dr. Kappos said. “These observations make it difficult to keep the current definitions of ‘relapsing remitting’ and ‘secondary progressive’ MS, [ones] that suggest a clear-cut distinction marked by the presence or absence of relapses. This can no longer be justified,” he stressed.
“We are not saying that relapses do not contribute to disability progression. There are a lot of data to support the fact that they do. But I think what we might be seeing is that the drug therapy is quite effective in reducing disability due to relapses but only partially effective in reducing progression independent of relapses,” Dr. Kappos explained.
Although there have been many advances in reducing relapses with drug therapy, focus now needs to shift to the other more continuous process of disability progression independent of relapses, Dr. Kappos said. “There is still a lot of room for improvement here.”
“If continuous progression independent of relapses is already present in the early phases of MS, it is reasonable to study the effects of intervention on steady progression already in this early phase,” he noted. “This might help to capture patients at earlier stages who better respond to treatment aimed at halting progression.”
Dr. Kappos also called for more subtle measurements of disability than the EDSS alone, including measures such as the 9-hole peg test and the 25-ft walk as they did in this analysis. But other measures could also be added that would characterize continuous disease activity and progression, such as laboratory values (e.g., neurofilament light chain) and advanced, more tissue-specific quantitative MRI techniques and digital biomarkers to detect subtle changes in neurologic function.
An artificial distinction?
Commenting on the study, Jeffrey Cohen, MD, director of the experimental therapeutics program at the Mellen Center for Multiple Sclerosis Treatment and Research, Cleveland Clinic, said he too sees very little distinction between relapsing remitting and progressive forms of the disease.
“This study confirms what has been suspected for quite a few years –that if one looks sufficiently and carefully, there is gradual worsening of some aspects of the disease in many patients from the earliest stages,” Dr. Cohen said. “Conversely, some patients with progressive MS have superimposed relapses or MRI lesion activity.
“Thus, the distinction between relapsing-remitting and progressive MS subtypes appears artificial,” he concluded.
This study was sponsored by F. Hoffmann–La Roche. Dr. Kappos has received research support from the company.
This article first appeared on Medscape.com.
FDA approves Uplizna for treatment of anti-AQP4 antibody–positive NMOSD
The Food and Drug Administration has approved Uplizna (inebilizumab-cdon) for the treatment of adult patients with neuromyelitis optica spectrum disorder (NMOSD) who are anti-AQP4 antibody positive. Uplizna is the second approved treatment for the disorder.
Approval was based on results from the global, placebo-controlled N-MOmentum trial, which included 213 anti-AQP4 antibody–positive patients and 17 anti-AQP4 antibody–negative patients who received inebilizumab-cdon or placebo. Just under 90% of patients in the positive group remained relapse free 6 months after the initial dosing, compared with 58% of patients taking placebo. People who took inebilizumab also saw a reduction in NMOSD-related hospitalizations. There was no evidence of a benefit in patients who were anti-AQP4 antibody negative.
Inebilizumab-cdon was safe and well tolerated during the trial, with the most common adverse events being urinary tract infection (20%), nasopharyngitis (13%), infusion reaction (12%), arthralgia (11%), and headache (10%). The drug is approved as twice-yearly maintenance after initial dosing. The prescribing information for Uplizna includes a warning for infusion reactions, potential depletion of certain proteins (hypogammaglobulinemia), and potential increased risk of infection—including progressive multifocal leukoencephalopathy—and potential reactivation of hepatitis B and tuberculosis.
“NMOSD is an extremely challenging disease to treat. Patients experience unpredictable attacks that can lead to permanent disability from blindness and paralysis. In addition, each subsequent attack may result in a cumulative worsening of disability,” Bruce Cree, MD, PhD, lead investigator for the N-MOmentum trial and professor of clinical neurology at the University of California, San Francisco, said in a press release. “Uplizna is an important new treatment option that provides prescribing physicians and patients living with NMOSD a therapy with proven efficacy, a favorable safety profile and a twice-a-year maintenance dosing schedule.”
The Food and Drug Administration has approved Uplizna (inebilizumab-cdon) for the treatment of adult patients with neuromyelitis optica spectrum disorder (NMOSD) who are anti-AQP4 antibody positive. Uplizna is the second approved treatment for the disorder.
Approval was based on results from the global, placebo-controlled N-MOmentum trial, which included 213 anti-AQP4 antibody–positive patients and 17 anti-AQP4 antibody–negative patients who received inebilizumab-cdon or placebo. Just under 90% of patients in the positive group remained relapse free 6 months after the initial dosing, compared with 58% of patients taking placebo. People who took inebilizumab also saw a reduction in NMOSD-related hospitalizations. There was no evidence of a benefit in patients who were anti-AQP4 antibody negative.
Inebilizumab-cdon was safe and well tolerated during the trial, with the most common adverse events being urinary tract infection (20%), nasopharyngitis (13%), infusion reaction (12%), arthralgia (11%), and headache (10%). The drug is approved as twice-yearly maintenance after initial dosing. The prescribing information for Uplizna includes a warning for infusion reactions, potential depletion of certain proteins (hypogammaglobulinemia), and potential increased risk of infection—including progressive multifocal leukoencephalopathy—and potential reactivation of hepatitis B and tuberculosis.
“NMOSD is an extremely challenging disease to treat. Patients experience unpredictable attacks that can lead to permanent disability from blindness and paralysis. In addition, each subsequent attack may result in a cumulative worsening of disability,” Bruce Cree, MD, PhD, lead investigator for the N-MOmentum trial and professor of clinical neurology at the University of California, San Francisco, said in a press release. “Uplizna is an important new treatment option that provides prescribing physicians and patients living with NMOSD a therapy with proven efficacy, a favorable safety profile and a twice-a-year maintenance dosing schedule.”
The Food and Drug Administration has approved Uplizna (inebilizumab-cdon) for the treatment of adult patients with neuromyelitis optica spectrum disorder (NMOSD) who are anti-AQP4 antibody positive. Uplizna is the second approved treatment for the disorder.
Approval was based on results from the global, placebo-controlled N-MOmentum trial, which included 213 anti-AQP4 antibody–positive patients and 17 anti-AQP4 antibody–negative patients who received inebilizumab-cdon or placebo. Just under 90% of patients in the positive group remained relapse free 6 months after the initial dosing, compared with 58% of patients taking placebo. People who took inebilizumab also saw a reduction in NMOSD-related hospitalizations. There was no evidence of a benefit in patients who were anti-AQP4 antibody negative.
Inebilizumab-cdon was safe and well tolerated during the trial, with the most common adverse events being urinary tract infection (20%), nasopharyngitis (13%), infusion reaction (12%), arthralgia (11%), and headache (10%). The drug is approved as twice-yearly maintenance after initial dosing. The prescribing information for Uplizna includes a warning for infusion reactions, potential depletion of certain proteins (hypogammaglobulinemia), and potential increased risk of infection—including progressive multifocal leukoencephalopathy—and potential reactivation of hepatitis B and tuberculosis.
“NMOSD is an extremely challenging disease to treat. Patients experience unpredictable attacks that can lead to permanent disability from blindness and paralysis. In addition, each subsequent attack may result in a cumulative worsening of disability,” Bruce Cree, MD, PhD, lead investigator for the N-MOmentum trial and professor of clinical neurology at the University of California, San Francisco, said in a press release. “Uplizna is an important new treatment option that provides prescribing physicians and patients living with NMOSD a therapy with proven efficacy, a favorable safety profile and a twice-a-year maintenance dosing schedule.”
Neurologists’ pay gets a boost, most happy with career choice
findings from the newly released Medscape Neurologist Compensation Report 2020 show.
Neurologists’ average annual income this year rose to $280,000, up from $267,000 last year. More than half of neurologists (53%) feel fairly compensated, similar to last year’s percentage.
Neurologists are below the middle earners of all physician specialties. At $280,000 in annual compensation for patient care, neurologists rank ninth from the bottom, just below allergists/immunologists ($301,000) but ahead of psychiatrists ($268,000), rheumatologists ($262,000), and internists ($251,000).
Orthopedists are the top earners ($511,000 annual pay), followed by plastic surgeons ($479,000), otolaryngologists ($455,000), and cardiologists ($438,000), according the overall Medscape Physician Compensation Report 2020, which covers U.S. physicians as a whole. The survey included more than 17,000 physicians in over 30 specialties.
COVID-19 impact
An important caveat is that the data for this year’s report were collected prior to Feb. 10, 2020, and therefore reflect physician salary and income prior to the COVID-19 crisis, which has had a huge impact on physicians.
For example, data show that since the start of the crisis, physician practices have seen a 55% dip in revenue and a 60% dip in patient volume on average. Hospitals and physician groups nationwide have implemented layoffs, furloughs, and pay cuts.
In March, 43,000 health care workers were laid off; 9% of independent medical practices reported that they had closed their practices, at least temporarily.
There continues to be a gender pay gap in neurology, with male neurologists earning about 26% more than their female peers ($299,000 vs. $237,000). Among all specialists, men earn 31% more than women, similar to last year’s figure of 33%. There continues to be a 25% gender pay gap among primary care physicians.
More than half of all physicians (56%) say they receive an incentive bonus. Neurologists report that they are eligible for an annual incentive bonus of $35,000. Average annual incentive bonuses are highest among orthopedists ($96,000) and lowest among family medicine physicians ($24,000).
Close to one third of physicians overall who receive incentive bonuses say the prospect of receiving the bonus has encouraged them to work longer hours. A higher percentage of neurologists (41%) say their potential bonus influenced them to increase their work hours.
Fifty-eight percent of neurologists achieve more than three quarters of their potential annual incentive bonus. On average, neurologists achieve about two thirds of their potential bonus, the same proportion as for physicians overall.
However, COVID-19 may change that. Experts who were interviewed recently by Medscape noted that productivity benchmarks for physicians are likely to be lowered in light of plunging patient numbers from COVID-19, and bonuses are expected to take a hit.
Happy at work
On average, male neurologists spend 37.7 hours per week seeing patients, somewhat more hours per week than female neurologists (36.1 hours); the average for all physicians is 37.9 hours per week.
Bureaucratic tasks continue to be a burden for physicians in all specialties. On average, neurologists spend 16.9 hours per week on paperwork and administration, about the same as physicians overall (15.6 hours).
Intensivists top the list regarding such tasks (19.1 hours), followed by internists (18.5), infectious disease physicians (18.5), and psychiatrists (18.3). Anesthesiologists and ophthalmologists spend the least amount of time on paperwork/administration (10.0 and 9.8 hours per week, respectively).
What is most rewarding about being a neurologist? Being good at what they do/finding answers, diagnoses tops the list (33%), followed by making the world a better place/helping others (26%), relationships with and gratitude from patients (18%), and making good money at a job they like (11%). A few cited teaching (5%) and pride in their profession (4%).
The most challenging part of practicing neurology is having to follow so many rules and regulations (26%). Other challenges include having to work long hours (18%), dealing with difficult patients (17%), trouble getting fair reimbursement (13%), and working with electronic health records (10%).
Despite the challenges, if they had to do it all over again, 73% of neurologists would still choose medicine as a career, and 86% would again choose neurology.
Other key findings in the latest report regarding neurologists include the following:
- At 18%, neurologists rank near the middle among physicians with regard to losing money on denied or resubmitted claims. Plastic surgery and emergency medicine have the highest percentage of claims denied or resubmitted (28% and 22%, respectively). One study found that, on average, 63% of denied claims are recoverable, but healthcare professionals spend about $118 per claim on appeals.
- 29% of neurologists say they use physician assistants (PAs) to treat patients in their practices, and 53% use nurse practitioners (NPs); 38% use neither for patient care. Of neurologists who work with PAs and NPs in their offices, 49% say these employees have helped boost profitability.
- Two-thirds of neurologists say they will continue taking new and current Medicare/Medicaid patients; none say they will not take new Medicare patients; and 26% are undecided.
- Neurologists participate in various payment methods; 78% are reimbursed via insurance, 35% have fee-for-service arrangements, and 28% are in accountable care organizations.
- Nearly 40% of neurologists expect to participate in the merit-based incentive payment system option, and 10% expect to participate in alternative payment models.
This article first appeared on Medscape.com.
findings from the newly released Medscape Neurologist Compensation Report 2020 show.
Neurologists’ average annual income this year rose to $280,000, up from $267,000 last year. More than half of neurologists (53%) feel fairly compensated, similar to last year’s percentage.
Neurologists are below the middle earners of all physician specialties. At $280,000 in annual compensation for patient care, neurologists rank ninth from the bottom, just below allergists/immunologists ($301,000) but ahead of psychiatrists ($268,000), rheumatologists ($262,000), and internists ($251,000).
Orthopedists are the top earners ($511,000 annual pay), followed by plastic surgeons ($479,000), otolaryngologists ($455,000), and cardiologists ($438,000), according the overall Medscape Physician Compensation Report 2020, which covers U.S. physicians as a whole. The survey included more than 17,000 physicians in over 30 specialties.
COVID-19 impact
An important caveat is that the data for this year’s report were collected prior to Feb. 10, 2020, and therefore reflect physician salary and income prior to the COVID-19 crisis, which has had a huge impact on physicians.
For example, data show that since the start of the crisis, physician practices have seen a 55% dip in revenue and a 60% dip in patient volume on average. Hospitals and physician groups nationwide have implemented layoffs, furloughs, and pay cuts.
In March, 43,000 health care workers were laid off; 9% of independent medical practices reported that they had closed their practices, at least temporarily.
There continues to be a gender pay gap in neurology, with male neurologists earning about 26% more than their female peers ($299,000 vs. $237,000). Among all specialists, men earn 31% more than women, similar to last year’s figure of 33%. There continues to be a 25% gender pay gap among primary care physicians.
More than half of all physicians (56%) say they receive an incentive bonus. Neurologists report that they are eligible for an annual incentive bonus of $35,000. Average annual incentive bonuses are highest among orthopedists ($96,000) and lowest among family medicine physicians ($24,000).
Close to one third of physicians overall who receive incentive bonuses say the prospect of receiving the bonus has encouraged them to work longer hours. A higher percentage of neurologists (41%) say their potential bonus influenced them to increase their work hours.
Fifty-eight percent of neurologists achieve more than three quarters of their potential annual incentive bonus. On average, neurologists achieve about two thirds of their potential bonus, the same proportion as for physicians overall.
However, COVID-19 may change that. Experts who were interviewed recently by Medscape noted that productivity benchmarks for physicians are likely to be lowered in light of plunging patient numbers from COVID-19, and bonuses are expected to take a hit.
Happy at work
On average, male neurologists spend 37.7 hours per week seeing patients, somewhat more hours per week than female neurologists (36.1 hours); the average for all physicians is 37.9 hours per week.
Bureaucratic tasks continue to be a burden for physicians in all specialties. On average, neurologists spend 16.9 hours per week on paperwork and administration, about the same as physicians overall (15.6 hours).
Intensivists top the list regarding such tasks (19.1 hours), followed by internists (18.5), infectious disease physicians (18.5), and psychiatrists (18.3). Anesthesiologists and ophthalmologists spend the least amount of time on paperwork/administration (10.0 and 9.8 hours per week, respectively).
What is most rewarding about being a neurologist? Being good at what they do/finding answers, diagnoses tops the list (33%), followed by making the world a better place/helping others (26%), relationships with and gratitude from patients (18%), and making good money at a job they like (11%). A few cited teaching (5%) and pride in their profession (4%).
The most challenging part of practicing neurology is having to follow so many rules and regulations (26%). Other challenges include having to work long hours (18%), dealing with difficult patients (17%), trouble getting fair reimbursement (13%), and working with electronic health records (10%).
Despite the challenges, if they had to do it all over again, 73% of neurologists would still choose medicine as a career, and 86% would again choose neurology.
Other key findings in the latest report regarding neurologists include the following:
- At 18%, neurologists rank near the middle among physicians with regard to losing money on denied or resubmitted claims. Plastic surgery and emergency medicine have the highest percentage of claims denied or resubmitted (28% and 22%, respectively). One study found that, on average, 63% of denied claims are recoverable, but healthcare professionals spend about $118 per claim on appeals.
- 29% of neurologists say they use physician assistants (PAs) to treat patients in their practices, and 53% use nurse practitioners (NPs); 38% use neither for patient care. Of neurologists who work with PAs and NPs in their offices, 49% say these employees have helped boost profitability.
- Two-thirds of neurologists say they will continue taking new and current Medicare/Medicaid patients; none say they will not take new Medicare patients; and 26% are undecided.
- Neurologists participate in various payment methods; 78% are reimbursed via insurance, 35% have fee-for-service arrangements, and 28% are in accountable care organizations.
- Nearly 40% of neurologists expect to participate in the merit-based incentive payment system option, and 10% expect to participate in alternative payment models.
This article first appeared on Medscape.com.
findings from the newly released Medscape Neurologist Compensation Report 2020 show.
Neurologists’ average annual income this year rose to $280,000, up from $267,000 last year. More than half of neurologists (53%) feel fairly compensated, similar to last year’s percentage.
Neurologists are below the middle earners of all physician specialties. At $280,000 in annual compensation for patient care, neurologists rank ninth from the bottom, just below allergists/immunologists ($301,000) but ahead of psychiatrists ($268,000), rheumatologists ($262,000), and internists ($251,000).
Orthopedists are the top earners ($511,000 annual pay), followed by plastic surgeons ($479,000), otolaryngologists ($455,000), and cardiologists ($438,000), according the overall Medscape Physician Compensation Report 2020, which covers U.S. physicians as a whole. The survey included more than 17,000 physicians in over 30 specialties.
COVID-19 impact
An important caveat is that the data for this year’s report were collected prior to Feb. 10, 2020, and therefore reflect physician salary and income prior to the COVID-19 crisis, which has had a huge impact on physicians.
For example, data show that since the start of the crisis, physician practices have seen a 55% dip in revenue and a 60% dip in patient volume on average. Hospitals and physician groups nationwide have implemented layoffs, furloughs, and pay cuts.
In March, 43,000 health care workers were laid off; 9% of independent medical practices reported that they had closed their practices, at least temporarily.
There continues to be a gender pay gap in neurology, with male neurologists earning about 26% more than their female peers ($299,000 vs. $237,000). Among all specialists, men earn 31% more than women, similar to last year’s figure of 33%. There continues to be a 25% gender pay gap among primary care physicians.
More than half of all physicians (56%) say they receive an incentive bonus. Neurologists report that they are eligible for an annual incentive bonus of $35,000. Average annual incentive bonuses are highest among orthopedists ($96,000) and lowest among family medicine physicians ($24,000).
Close to one third of physicians overall who receive incentive bonuses say the prospect of receiving the bonus has encouraged them to work longer hours. A higher percentage of neurologists (41%) say their potential bonus influenced them to increase their work hours.
Fifty-eight percent of neurologists achieve more than three quarters of their potential annual incentive bonus. On average, neurologists achieve about two thirds of their potential bonus, the same proportion as for physicians overall.
However, COVID-19 may change that. Experts who were interviewed recently by Medscape noted that productivity benchmarks for physicians are likely to be lowered in light of plunging patient numbers from COVID-19, and bonuses are expected to take a hit.
Happy at work
On average, male neurologists spend 37.7 hours per week seeing patients, somewhat more hours per week than female neurologists (36.1 hours); the average for all physicians is 37.9 hours per week.
Bureaucratic tasks continue to be a burden for physicians in all specialties. On average, neurologists spend 16.9 hours per week on paperwork and administration, about the same as physicians overall (15.6 hours).
Intensivists top the list regarding such tasks (19.1 hours), followed by internists (18.5), infectious disease physicians (18.5), and psychiatrists (18.3). Anesthesiologists and ophthalmologists spend the least amount of time on paperwork/administration (10.0 and 9.8 hours per week, respectively).
What is most rewarding about being a neurologist? Being good at what they do/finding answers, diagnoses tops the list (33%), followed by making the world a better place/helping others (26%), relationships with and gratitude from patients (18%), and making good money at a job they like (11%). A few cited teaching (5%) and pride in their profession (4%).
The most challenging part of practicing neurology is having to follow so many rules and regulations (26%). Other challenges include having to work long hours (18%), dealing with difficult patients (17%), trouble getting fair reimbursement (13%), and working with electronic health records (10%).
Despite the challenges, if they had to do it all over again, 73% of neurologists would still choose medicine as a career, and 86% would again choose neurology.
Other key findings in the latest report regarding neurologists include the following:
- At 18%, neurologists rank near the middle among physicians with regard to losing money on denied or resubmitted claims. Plastic surgery and emergency medicine have the highest percentage of claims denied or resubmitted (28% and 22%, respectively). One study found that, on average, 63% of denied claims are recoverable, but healthcare professionals spend about $118 per claim on appeals.
- 29% of neurologists say they use physician assistants (PAs) to treat patients in their practices, and 53% use nurse practitioners (NPs); 38% use neither for patient care. Of neurologists who work with PAs and NPs in their offices, 49% say these employees have helped boost profitability.
- Two-thirds of neurologists say they will continue taking new and current Medicare/Medicaid patients; none say they will not take new Medicare patients; and 26% are undecided.
- Neurologists participate in various payment methods; 78% are reimbursed via insurance, 35% have fee-for-service arrangements, and 28% are in accountable care organizations.
- Nearly 40% of neurologists expect to participate in the merit-based incentive payment system option, and 10% expect to participate in alternative payment models.
This article first appeared on Medscape.com.