User login
An 11-year-old boy presents with small itchy bumps on the wrists, face, arms, and legs
The patient was diagnosed with lichen nitidus, given the characteristic clinical presentation.
Lichen nitidus is a rare chronic inflammatory condition of the skin that most commonly presents in children and young adults and does not seem to be restricted to any sex or race. The classic lesions are described as asymptomatic to slightly pruritic, small (1 mm), skin-colored to hypopigmented flat-topped papules.
Koebner phenomenon is usually seen in which the skin lesions appear in areas of traumatized healthy skin. The extremities, abdomen, chest, and penis are common locations for the lesions to occur. Rarely, the oral mucosa or nails can be involved. It has been described in patients with a diagnosis of Crohn’s disease, Niemann-Pick disease, Down syndrome, and HIV. The rare, generalized purpuric variant has been reported in a few cases associated with interferon and ribavirin treatment for hepatitis C infection and nivolumab treatment for cancer. The pathophysiology of lichen nitidus is unknown.
Lichen nitidus can occur in the presence of other skin conditions like lichen planus, atopic dermatitis, vitiligo, erythema nodosum, and lichen spinulosus. Histopathologic characteristics of lichen nitidus are described as a “ball and claw” of epidermal rete around a lymphohistiocytic infiltrate. Parakeratosis overlying epidermal atrophy and focal basal liquefaction degeneration is also seen.
The differential diagnosis of lichen nitidus includes flat warts, which can present as clusters of small flat-topped papules that can show a pseudo-Koebner phenomenon (where the virus is seeded in traumatized skin). The morphological difference between the condition is that lichen nitidus lesions are usually monomorphic, compared with flat warts, which usually present with different sizes and shapes.

Patients with a history of allergic contact dermatitis may present with a generalized monomorphic eruption of skin-colored papules (known as ID reaction) that can sometimes be very similar to lichen nitidus. Allergic contact dermatitis tends to respond fairly quickly to topical or systemic corticosteroids, unlike lichen nitidus. There are a few reports that consider lichen nitidus to be a variant of lichen planus, although they have different histopathologic findings. Lichen planus lesions are described as polygonal, pruritic, purple to pink papules most commonly seen on the wrists, lower back, and ankles. Lichen planus can be seen in patients with hepatitis C and may also occur secondary to medication.
Milia are small keratin cysts on the skin that are commonly seen in babies as primary milia and can be seen in older children secondary to trauma (commonly on the eyelids) or medications. Given their size and monomorphic appearance, they can sometimes be confused with lichen nitidus.
Lichen nitidus is often asymptomatic and the lesions resolve within a few months to years. Topical corticosteroids can be helpful to alleviate the symptoms in patients who present with pruritus. In more persistent and generalized cases, phototherapy, systemic corticosteroids, acitretin, isotretinoin, or cyclosporine can be considered.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Chu J and Lam JM. CMAJ. 2014 Dec 9;186(18):E688.
Lestringant G et al. Dermatology 1996;192:171-3.
Peterson JA et al. Proc (Bayl Univ Med Cent). 2021 Aug 25;35(1):70-2.
Schwartz C and Goodman MB. “Lichen nitidus,” in StatPearls. Treasure Island, Fla.: StatPearls Publishing, 2022.
The patient was diagnosed with lichen nitidus, given the characteristic clinical presentation.
Lichen nitidus is a rare chronic inflammatory condition of the skin that most commonly presents in children and young adults and does not seem to be restricted to any sex or race. The classic lesions are described as asymptomatic to slightly pruritic, small (1 mm), skin-colored to hypopigmented flat-topped papules.
Koebner phenomenon is usually seen in which the skin lesions appear in areas of traumatized healthy skin. The extremities, abdomen, chest, and penis are common locations for the lesions to occur. Rarely, the oral mucosa or nails can be involved. It has been described in patients with a diagnosis of Crohn’s disease, Niemann-Pick disease, Down syndrome, and HIV. The rare, generalized purpuric variant has been reported in a few cases associated with interferon and ribavirin treatment for hepatitis C infection and nivolumab treatment for cancer. The pathophysiology of lichen nitidus is unknown.
Lichen nitidus can occur in the presence of other skin conditions like lichen planus, atopic dermatitis, vitiligo, erythema nodosum, and lichen spinulosus. Histopathologic characteristics of lichen nitidus are described as a “ball and claw” of epidermal rete around a lymphohistiocytic infiltrate. Parakeratosis overlying epidermal atrophy and focal basal liquefaction degeneration is also seen.
The differential diagnosis of lichen nitidus includes flat warts, which can present as clusters of small flat-topped papules that can show a pseudo-Koebner phenomenon (where the virus is seeded in traumatized skin). The morphological difference between the condition is that lichen nitidus lesions are usually monomorphic, compared with flat warts, which usually present with different sizes and shapes.
Patients with a history of allergic contact dermatitis may present with a generalized monomorphic eruption of skin-colored papules (known as ID reaction) that can sometimes be very similar to lichen nitidus. Allergic contact dermatitis tends to respond fairly quickly to topical or systemic corticosteroids, unlike lichen nitidus. There are a few reports that consider lichen nitidus to be a variant of lichen planus, although they have different histopathologic findings. Lichen planus lesions are described as polygonal, pruritic, purple to pink papules most commonly seen on the wrists, lower back, and ankles. Lichen planus can be seen in patients with hepatitis C and may also occur secondary to medication.
Milia are small keratin cysts on the skin that are commonly seen in babies as primary milia and can be seen in older children secondary to trauma (commonly on the eyelids) or medications. Given their size and monomorphic appearance, they can sometimes be confused with lichen nitidus.
Lichen nitidus is often asymptomatic and the lesions resolve within a few months to years. Topical corticosteroids can be helpful to alleviate the symptoms in patients who present with pruritus. In more persistent and generalized cases, phototherapy, systemic corticosteroids, acitretin, isotretinoin, or cyclosporine can be considered.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Chu J and Lam JM. CMAJ. 2014 Dec 9;186(18):E688.
Lestringant G et al. Dermatology 1996;192:171-3.
Peterson JA et al. Proc (Bayl Univ Med Cent). 2021 Aug 25;35(1):70-2.
Schwartz C and Goodman MB. “Lichen nitidus,” in StatPearls. Treasure Island, Fla.: StatPearls Publishing, 2022.
The patient was diagnosed with lichen nitidus, given the characteristic clinical presentation.
Lichen nitidus is a rare chronic inflammatory condition of the skin that most commonly presents in children and young adults and does not seem to be restricted to any sex or race. The classic lesions are described as asymptomatic to slightly pruritic, small (1 mm), skin-colored to hypopigmented flat-topped papules.
Koebner phenomenon is usually seen in which the skin lesions appear in areas of traumatized healthy skin. The extremities, abdomen, chest, and penis are common locations for the lesions to occur. Rarely, the oral mucosa or nails can be involved. It has been described in patients with a diagnosis of Crohn’s disease, Niemann-Pick disease, Down syndrome, and HIV. The rare, generalized purpuric variant has been reported in a few cases associated with interferon and ribavirin treatment for hepatitis C infection and nivolumab treatment for cancer. The pathophysiology of lichen nitidus is unknown.
Lichen nitidus can occur in the presence of other skin conditions like lichen planus, atopic dermatitis, vitiligo, erythema nodosum, and lichen spinulosus. Histopathologic characteristics of lichen nitidus are described as a “ball and claw” of epidermal rete around a lymphohistiocytic infiltrate. Parakeratosis overlying epidermal atrophy and focal basal liquefaction degeneration is also seen.
The differential diagnosis of lichen nitidus includes flat warts, which can present as clusters of small flat-topped papules that can show a pseudo-Koebner phenomenon (where the virus is seeded in traumatized skin). The morphological difference between the condition is that lichen nitidus lesions are usually monomorphic, compared with flat warts, which usually present with different sizes and shapes.
Patients with a history of allergic contact dermatitis may present with a generalized monomorphic eruption of skin-colored papules (known as ID reaction) that can sometimes be very similar to lichen nitidus. Allergic contact dermatitis tends to respond fairly quickly to topical or systemic corticosteroids, unlike lichen nitidus. There are a few reports that consider lichen nitidus to be a variant of lichen planus, although they have different histopathologic findings. Lichen planus lesions are described as polygonal, pruritic, purple to pink papules most commonly seen on the wrists, lower back, and ankles. Lichen planus can be seen in patients with hepatitis C and may also occur secondary to medication.
Milia are small keratin cysts on the skin that are commonly seen in babies as primary milia and can be seen in older children secondary to trauma (commonly on the eyelids) or medications. Given their size and monomorphic appearance, they can sometimes be confused with lichen nitidus.
Lichen nitidus is often asymptomatic and the lesions resolve within a few months to years. Topical corticosteroids can be helpful to alleviate the symptoms in patients who present with pruritus. In more persistent and generalized cases, phototherapy, systemic corticosteroids, acitretin, isotretinoin, or cyclosporine can be considered.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Chu J and Lam JM. CMAJ. 2014 Dec 9;186(18):E688.
Lestringant G et al. Dermatology 1996;192:171-3.
Peterson JA et al. Proc (Bayl Univ Med Cent). 2021 Aug 25;35(1):70-2.
Schwartz C and Goodman MB. “Lichen nitidus,” in StatPearls. Treasure Island, Fla.: StatPearls Publishing, 2022.
An 11-year-old male with a prior history of atopic dermatitis as a young child, presents with 6 months of slightly itchy, small bumps on the wrists, face, arms, and legs. Has been treated with fluocinolone oil and hydrocortisone 2.5% for a month with no change in the lesions. Besides the use of topical corticosteroids, he has not been taking any other medications.
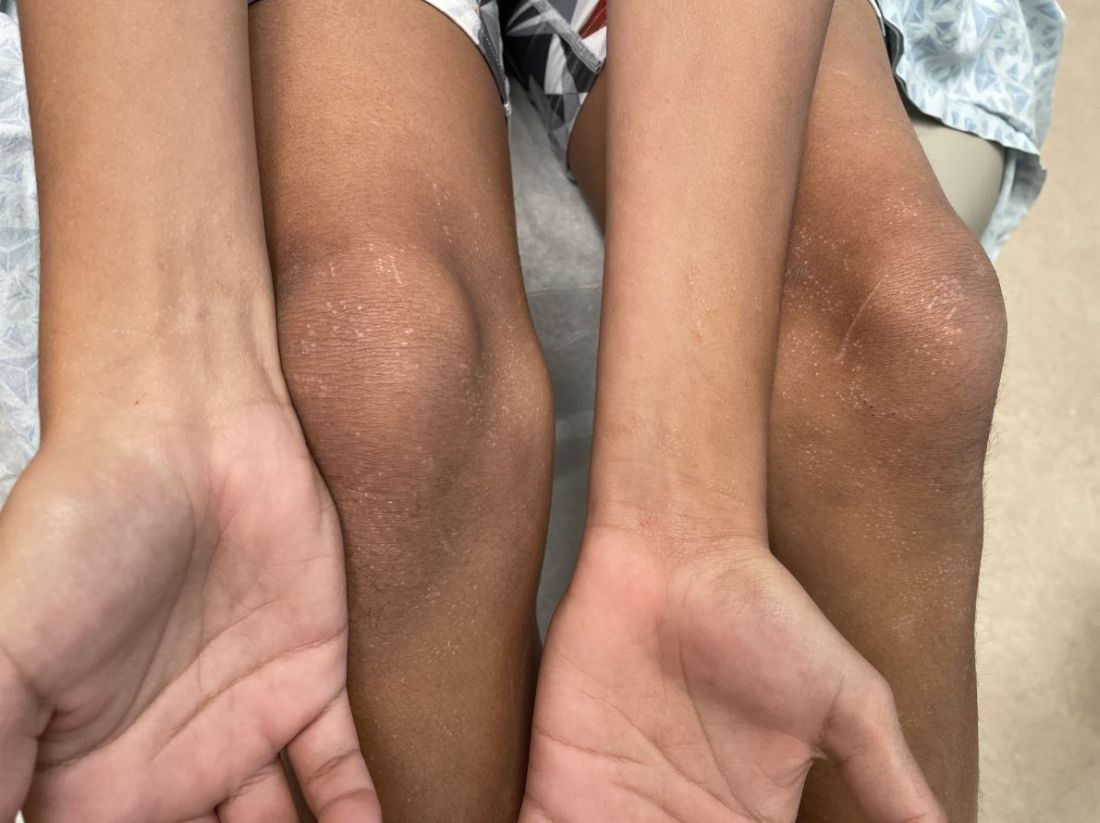
On physical examination he has multiple skin-colored, flat-topped papules that coalesce into plaques on the arms, legs, chest, and back (Photo 1). Koebner phenomenon was also seen on the knees and arms. There were no lesions in the mouth or on the nails.
A toddler presents with a dark line on a fingernail
Given the over 1-year history of an unchanging longitudinal band of pigment without extension to the proximal or lateral nailfolds or any other nail findings, the most likely diagnosis is benign longitudinal melanonychia.
Longitudinal melanonychia, also known as melanonychia striata, describes a brown to black streak of pigment extending from the nail matrix to the free edge of the nail.1,2
This disorder can occur secondary to a wide variety of benign and pathologic causes including lentigines, nevi, melanoma, chronic trauma, inflammatory skin diseases, systemic diseases, iatrogenic causes, and genetic syndromes.3 In melanocytic causes of longitudinal melanonychia, either melanocytic activation or hyperplasia drive pigmentary development leading to the brown to black band seen in the nail.4 Benign causes of longitudinal melanonychia include benign melanocyte activation, lentigo, and benign nevus.1
What’s the differential diagnosis?
The differential diagnosis for longitudinal melanonychia can include a wide variety of local and systemic causes. For our discussion, we will limit our differential to other locally involved disorders of the nail including subungual melanoma, subungual hematoma, onychomycosis, and glomus tumor.

Subungual melanoma is a rare subtype of acral lentiginous melanoma that most often presents as longitudinal melanonychia. Subungual melanoma is more common in those aged 50-70 years, individuals with personal or family history of melanoma or dysplastic nevus syndrome, and persons with African American, Native American, and Asian descent. Longitudinal melanonychia features that can be concerning for subungual melanoma include the presence of multiple colors, width greater than or equal to 3 mm, blurry borders, rapid increase in size, and extension to the proximal or lateral nailfolds (Hutchinson’s sign). Biopsy is required to make the diagnosis of subungual melanoma but is not necessary for melanonychia without atypical features.

Treatment of subungual melanoma depends on disease stage and can range from wide local excision of the nail apparatus to amputation of the affected digit and management with a medical oncologist. Given the absence of concerning neoplastic findings or personal or family history of melanoma, subungual melanoma is unlikely in this patient.

Subungual hematoma is an accumulation of blood underneath the nail plate that is typically the result of acute or chronic trauma to the distal phalanx. It can present as purple, red, pink, brown, or black discoloration under the nail plate and is most commonly found on the first toe. With acute trauma, pain is usually present upon initial injury. Subungual hematomas typically resolve on their own with normal nail growth. The absence of a history of trauma or pain, and the linear appearance of the lesion in our patient are inconsistent with a subungual hematoma.
Onychomycosis is a fungal infection of the nail caused by dermatophytes, nondermatophytes, or yeasts. It may present with longitudinal melanonychia; however, it more often presents with other nail abnormalities such as nail thickening, yellow discoloration, onycholysis, splitting, subungual hyperkeratosis, and nail plate destruction, which are not present in this patient. Furthermore, onychomycosis is more common in adults than children. Diagnosis is usually made with potassium hydroxide (KOH) preparations, histopathologic examination of nail clippings with a periodic acid-Schiff stain, fungal culture, or PCR.
Glomus tumor is a rare, benign neoplasm originating from cells of the glomus body. It is often found in the subungual region, in addition to other areas rich in glomus bodies such as the fingertips, palms, wrists, and forearms. Subungual glomus tumors present as a red, purple, or blueish lesions under the nail plate. Distal notching or an overlying longitudinal fissure may be present. Subungual glomus tumors are typically associated with pinpoint tenderness, paroxysmal pain, and cold sensitivity, features that are not present in our patient. The history and examination of our patient are much more consistent with benign longitudinal melanonychia.
It appears that melanoma associated with longitudinal melanonychia is very rare in children. According to one review published in 2020, only 12 cases of pediatric subungual melanoma have been reported.5 Recent series have observed longitudinal melanonychia in large sets of children, with findings that demonstrate that the vast majority of longitudinal melanonychia either stops progressing or regresses. These investigations therefore recommend serial observation of longitudinal melanonychia except in rare circumstances.6,7
Given the lack of troubling findings or concerning history, our patient was managed with observation. On follow-up 6 months later, he was found to have no change in his nail pigmentation.
Dr. Haft is an inflammatory skin disease fellow in the division of pediatric and adolescent dermatology; Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology; and Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics, all at the University of California and Rady Children’s Hospital, San Diego. They have no relevant disclosures.
References
1. Mannava KA et al. Hand Surg. 2013;18(1):133-9.
2. Leung AKC et al. Int J Dermatol. 2019;58(11):1239-45.
3. Andre J and Lateur N. Dermatol Clin. 2006;24(3):329-39.
4. Lee DK and Lipner SR. Ann Med. 2022;54(1):694-712.
5. Smith RJ and Rubin AI. Curr Opin Pediatr. 2020;32(4):506-15. .
6. Matsui Y et al. J Am Acad Dermatol. 2022;86(4):946-8.
7. Lee JS et al. J Am Acad Dermatol. 2022;87(2):366-72.
Given the over 1-year history of an unchanging longitudinal band of pigment without extension to the proximal or lateral nailfolds or any other nail findings, the most likely diagnosis is benign longitudinal melanonychia.
Longitudinal melanonychia, also known as melanonychia striata, describes a brown to black streak of pigment extending from the nail matrix to the free edge of the nail.1,2
This disorder can occur secondary to a wide variety of benign and pathologic causes including lentigines, nevi, melanoma, chronic trauma, inflammatory skin diseases, systemic diseases, iatrogenic causes, and genetic syndromes.3 In melanocytic causes of longitudinal melanonychia, either melanocytic activation or hyperplasia drive pigmentary development leading to the brown to black band seen in the nail.4 Benign causes of longitudinal melanonychia include benign melanocyte activation, lentigo, and benign nevus.1
What’s the differential diagnosis?
The differential diagnosis for longitudinal melanonychia can include a wide variety of local and systemic causes. For our discussion, we will limit our differential to other locally involved disorders of the nail including subungual melanoma, subungual hematoma, onychomycosis, and glomus tumor.
Subungual melanoma is a rare subtype of acral lentiginous melanoma that most often presents as longitudinal melanonychia. Subungual melanoma is more common in those aged 50-70 years, individuals with personal or family history of melanoma or dysplastic nevus syndrome, and persons with African American, Native American, and Asian descent. Longitudinal melanonychia features that can be concerning for subungual melanoma include the presence of multiple colors, width greater than or equal to 3 mm, blurry borders, rapid increase in size, and extension to the proximal or lateral nailfolds (Hutchinson’s sign). Biopsy is required to make the diagnosis of subungual melanoma but is not necessary for melanonychia without atypical features.
Treatment of subungual melanoma depends on disease stage and can range from wide local excision of the nail apparatus to amputation of the affected digit and management with a medical oncologist. Given the absence of concerning neoplastic findings or personal or family history of melanoma, subungual melanoma is unlikely in this patient.
Subungual hematoma is an accumulation of blood underneath the nail plate that is typically the result of acute or chronic trauma to the distal phalanx. It can present as purple, red, pink, brown, or black discoloration under the nail plate and is most commonly found on the first toe. With acute trauma, pain is usually present upon initial injury. Subungual hematomas typically resolve on their own with normal nail growth. The absence of a history of trauma or pain, and the linear appearance of the lesion in our patient are inconsistent with a subungual hematoma.
Onychomycosis is a fungal infection of the nail caused by dermatophytes, nondermatophytes, or yeasts. It may present with longitudinal melanonychia; however, it more often presents with other nail abnormalities such as nail thickening, yellow discoloration, onycholysis, splitting, subungual hyperkeratosis, and nail plate destruction, which are not present in this patient. Furthermore, onychomycosis is more common in adults than children. Diagnosis is usually made with potassium hydroxide (KOH) preparations, histopathologic examination of nail clippings with a periodic acid-Schiff stain, fungal culture, or PCR.
Glomus tumor is a rare, benign neoplasm originating from cells of the glomus body. It is often found in the subungual region, in addition to other areas rich in glomus bodies such as the fingertips, palms, wrists, and forearms. Subungual glomus tumors present as a red, purple, or blueish lesions under the nail plate. Distal notching or an overlying longitudinal fissure may be present. Subungual glomus tumors are typically associated with pinpoint tenderness, paroxysmal pain, and cold sensitivity, features that are not present in our patient. The history and examination of our patient are much more consistent with benign longitudinal melanonychia.
It appears that melanoma associated with longitudinal melanonychia is very rare in children. According to one review published in 2020, only 12 cases of pediatric subungual melanoma have been reported.5 Recent series have observed longitudinal melanonychia in large sets of children, with findings that demonstrate that the vast majority of longitudinal melanonychia either stops progressing or regresses. These investigations therefore recommend serial observation of longitudinal melanonychia except in rare circumstances.6,7
Given the lack of troubling findings or concerning history, our patient was managed with observation. On follow-up 6 months later, he was found to have no change in his nail pigmentation.
Dr. Haft is an inflammatory skin disease fellow in the division of pediatric and adolescent dermatology; Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology; and Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics, all at the University of California and Rady Children’s Hospital, San Diego. They have no relevant disclosures.
References
1. Mannava KA et al. Hand Surg. 2013;18(1):133-9.
2. Leung AKC et al. Int J Dermatol. 2019;58(11):1239-45.
3. Andre J and Lateur N. Dermatol Clin. 2006;24(3):329-39.
4. Lee DK and Lipner SR. Ann Med. 2022;54(1):694-712.
5. Smith RJ and Rubin AI. Curr Opin Pediatr. 2020;32(4):506-15. .
6. Matsui Y et al. J Am Acad Dermatol. 2022;86(4):946-8.
7. Lee JS et al. J Am Acad Dermatol. 2022;87(2):366-72.
Given the over 1-year history of an unchanging longitudinal band of pigment without extension to the proximal or lateral nailfolds or any other nail findings, the most likely diagnosis is benign longitudinal melanonychia.
Longitudinal melanonychia, also known as melanonychia striata, describes a brown to black streak of pigment extending from the nail matrix to the free edge of the nail.1,2
This disorder can occur secondary to a wide variety of benign and pathologic causes including lentigines, nevi, melanoma, chronic trauma, inflammatory skin diseases, systemic diseases, iatrogenic causes, and genetic syndromes.3 In melanocytic causes of longitudinal melanonychia, either melanocytic activation or hyperplasia drive pigmentary development leading to the brown to black band seen in the nail.4 Benign causes of longitudinal melanonychia include benign melanocyte activation, lentigo, and benign nevus.1
What’s the differential diagnosis?
The differential diagnosis for longitudinal melanonychia can include a wide variety of local and systemic causes. For our discussion, we will limit our differential to other locally involved disorders of the nail including subungual melanoma, subungual hematoma, onychomycosis, and glomus tumor.
Subungual melanoma is a rare subtype of acral lentiginous melanoma that most often presents as longitudinal melanonychia. Subungual melanoma is more common in those aged 50-70 years, individuals with personal or family history of melanoma or dysplastic nevus syndrome, and persons with African American, Native American, and Asian descent. Longitudinal melanonychia features that can be concerning for subungual melanoma include the presence of multiple colors, width greater than or equal to 3 mm, blurry borders, rapid increase in size, and extension to the proximal or lateral nailfolds (Hutchinson’s sign). Biopsy is required to make the diagnosis of subungual melanoma but is not necessary for melanonychia without atypical features.
Treatment of subungual melanoma depends on disease stage and can range from wide local excision of the nail apparatus to amputation of the affected digit and management with a medical oncologist. Given the absence of concerning neoplastic findings or personal or family history of melanoma, subungual melanoma is unlikely in this patient.
Subungual hematoma is an accumulation of blood underneath the nail plate that is typically the result of acute or chronic trauma to the distal phalanx. It can present as purple, red, pink, brown, or black discoloration under the nail plate and is most commonly found on the first toe. With acute trauma, pain is usually present upon initial injury. Subungual hematomas typically resolve on their own with normal nail growth. The absence of a history of trauma or pain, and the linear appearance of the lesion in our patient are inconsistent with a subungual hematoma.
Onychomycosis is a fungal infection of the nail caused by dermatophytes, nondermatophytes, or yeasts. It may present with longitudinal melanonychia; however, it more often presents with other nail abnormalities such as nail thickening, yellow discoloration, onycholysis, splitting, subungual hyperkeratosis, and nail plate destruction, which are not present in this patient. Furthermore, onychomycosis is more common in adults than children. Diagnosis is usually made with potassium hydroxide (KOH) preparations, histopathologic examination of nail clippings with a periodic acid-Schiff stain, fungal culture, or PCR.
Glomus tumor is a rare, benign neoplasm originating from cells of the glomus body. It is often found in the subungual region, in addition to other areas rich in glomus bodies such as the fingertips, palms, wrists, and forearms. Subungual glomus tumors present as a red, purple, or blueish lesions under the nail plate. Distal notching or an overlying longitudinal fissure may be present. Subungual glomus tumors are typically associated with pinpoint tenderness, paroxysmal pain, and cold sensitivity, features that are not present in our patient. The history and examination of our patient are much more consistent with benign longitudinal melanonychia.
It appears that melanoma associated with longitudinal melanonychia is very rare in children. According to one review published in 2020, only 12 cases of pediatric subungual melanoma have been reported.5 Recent series have observed longitudinal melanonychia in large sets of children, with findings that demonstrate that the vast majority of longitudinal melanonychia either stops progressing or regresses. These investigations therefore recommend serial observation of longitudinal melanonychia except in rare circumstances.6,7
Given the lack of troubling findings or concerning history, our patient was managed with observation. On follow-up 6 months later, he was found to have no change in his nail pigmentation.
Dr. Haft is an inflammatory skin disease fellow in the division of pediatric and adolescent dermatology; Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology; and Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics, all at the University of California and Rady Children’s Hospital, San Diego. They have no relevant disclosures.
References
1. Mannava KA et al. Hand Surg. 2013;18(1):133-9.
2. Leung AKC et al. Int J Dermatol. 2019;58(11):1239-45.
3. Andre J and Lateur N. Dermatol Clin. 2006;24(3):329-39.
4. Lee DK and Lipner SR. Ann Med. 2022;54(1):694-712.
5. Smith RJ and Rubin AI. Curr Opin Pediatr. 2020;32(4):506-15. .
6. Matsui Y et al. J Am Acad Dermatol. 2022;86(4):946-8.
7. Lee JS et al. J Am Acad Dermatol. 2022;87(2):366-72.
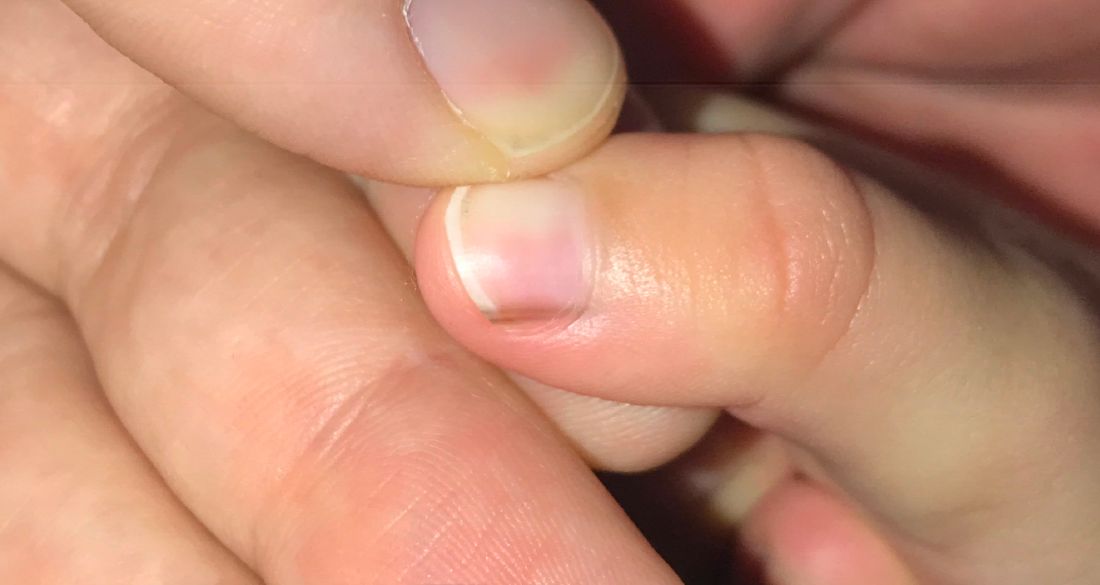
Examination findings reveal a 2-mm brown longitudinal band on the radial aspect of the right thumbnail that does not extend into the proximal or lateral nailfolds. The rest of the skin and nail exam is unremarkable.
Hyperpigmented Papules on the Tongue of a Child
The Diagnosis: Pigmented Fungiform Papillae of the Tongue
Our patient’s hyperpigmentation was confined to the fungiform papillae, leading to a diagnosis of pigmented fungiform papillae of the tongue (PFPT). A biopsy was not performed, and reassurance was provided regarding the benign nature of this finding, which did not require treatment.
Pigmented fungiform papillae of the tongue is a benign, nonprogressive, asymptomatic pigmentary condition that is most common among patients with skin of color and typically develops within the second or third decade of life.1,2 The pathogenesis is unclear, but activation of subepithelial melanophages without evidence of inflammation has been implicated.2 Although no standard treatment exists, cosmetic improvement with the use of the Q-switched ruby laser has been reported.3,4 Clinically, PFPT presents as asymptomatic hyperpigmentation confined to the fungiform papillae along the anterior and lateral portions of the tongue.1,2
Pigmented fungiform papillae of the tongue typically is an isolated finding but rarely can be associated with hyperpigmentation of the nails (as in our patient) or gingiva.2 Three different clinical patterns of presentation have been described: (1) a single well-circumscribed collection of pigmented fungiform papillae, (2) few scattered pigmented fungiform papillae admixed with many nonpigmented fungiform papillae, or (3) pigmentation of all fungiform papillae on the dorsal aspect of the tongue.2,5,6 Pigmented fungiform papillae of the tongue is a clinical diagnosis based on visual recognition. Dermoscopic examination revealing a cobblestonelike or rose petal–like pattern may be helpful in diagnosing PFPT.2,5-7 Although not typically recommended in the evaluation of PFPT, a biopsy will reveal papillary structures with hyperpigmentation of basilar keratinocytes as well as melanophages in the lamina propria.8 The latter finding suggests a transient inflammatory process despite the hallmark absence of inflammation.5 Melanocytic neoplasia and exogenous granules of pigment typically are not seen.8
Other conditions that may present with dark-colored macules or papules on the tongue should be considered in the evaluation of a patient with these clinical findings. Black hairy tongue (BHT), or lingua villosa nigra, is a benign finding due to filiform papillae hypertrophy on the dorsum of the tongue.9 Food particle debris caught in BHT can lead to porphyrin production by chromogenic bacteria and fungi. These porphyrins result in discoloration ranging from brown-black to yellow and green occurring anteriorly to the circumvallate papillae while usually sparing the tip and lateral sides of the tongue. Dermoscopy can show thin discolored fibers with a hairy appearance. Although normal filiform papillae are less than 1-mm long, 3-mm long papillae are considered diagnostic of BHT.9 Treatment includes effective oral hygiene and desquamation measures, which can lead to complete resolution.10
Peutz-Jeghers syndrome is a rare genodermatosis that is characterized by focal hyperpigmentation and multiple gastrointestinal mucosal hamartomatous polyps. Peutz-Jeghers syndrome should be suspected in a patient with discrete, 1- to 5-mm, brown to black macules on the perioral or periocular skin, tongue, genitals, palms, soles, and buccal mucosa with a history of abdominal symptoms.11,12
Addison disease, or primary adrenal insufficiency, may present with brown hyperpigmentation on chronically sun-exposed areas; regions of friction or pressure; surrounding scar tissue; and mucosal surfaces such as the tongue, inner surface of the lip, and buccal and gingival mucosa.13 Addison disease is differentiated from PFPT by a more generalized hyperpigmentation due to increased melanin production as well as the presence of systemic symptoms related to hypocortisolism. The pigmentation seen on the buccal mucosa in Addison disease is patchy and diffuse, and histology reveals basal melanin hyperpigmentation with superficial dermal melanophages.13
Hereditary hemorrhagic telangiectasia is an inherited disorder featuring telangiectasia and generally appears in the third decade of life.14 Telangiectases classically are 1 to 3 mm in diameter with or without slight elevation. Dermoscopic findings include small red clots, lacunae, and serpentine or linear vessels arranged in a radial conformation surrounding a homogenous pink center.15 These telangiectases typically occur on the skin or mucosa, particularly the face, lips, tongue, nail beds, and nasal mucosa; however, any organ can be affected with arteriovenous malformations. Recurrent epistaxis occurs in more than half of patients with hereditary hemorrhagic telangiectasia.14 Histopathology reveals dilated vessels and lacunae near the dermoepidermal junction displacing the epidermis and papillary dermis.15 It is distinguished from PFPT by the vascular nature of the lesions and by the presence of other characteristic symptoms such as recurrent epistaxis and visceral arteriovenous malformations.
- Romiti R, Molina De Medeiros L. Pigmented fungiform papillae of the tongue. Pediatr Dermatol. 2010;27:398-399. doi:10.1111/j .1525-1470.2010.01183.x
- Chessa MA, Patrizi A, Sechi A, et al. Pigmented fungiform lingual papillae: dermoscopic and clinical features. J Eur Acad Dermatol Venereol. 2018;32:935-939. doi:10.1111/jdv.14809
- Rice SM, Lal K. Successful treatment of pigmented fungiform papillae of the tongue with Q-switched ruby laser. Dermatol Surg. 2022;48:368-369. doi:10.1097/DSS.0000000000003371
- Mizawa M, Makino T, Furukawa F, et al. Efficacy of Q-switched ruby laser treatment for pigmented fungiform papillae of the tongue. J Dermatol. 2022;49:E133-E134. doi:10.1111/1346-8138.16270
- Holzwanger JM, Rudolph RI, Heaton CL. Pigmented fungiform papillae of the tongue: a common variant of oral pigmentation. Int J Dermatol. 1974;13:403-408. doi:10.1111/j.1365-4362.1974. tb05073.x
- Mukamal LV, Ormiga P, Ramos-E-Silva M. Dermoscopy of the pigmented fungiform papillae of the tongue. J Dermatol. 2012;39:397-399. doi:10.1111/j.1346-8138.2011.01328.x
- Surboyo MDC, Santosh ABR, Hariyani N, et al. Clinical utility of dermoscopy on diagnosing pigmented papillary fungiform papillae of the tongue: a systematic review. J Oral Biol Craniofac Res. 2021;11:618-623. doi:10.1016/j.jobcr.2021.09.008
- Chamseddin B, Vandergriff T. Pigmented fungiform papillae of the tongue: a clinical and histologic description [published online September 15, 2019]. Dermatol Online J. 2019;25:13030/qt8674c519.
- Jayasree P, Kaliyadan F, Ashique KT. Black hairy tongue. JAMA Dermatol. 2022;158:573. doi:10.1001/jamadermatol.2021.5314
- Schlager E, St Claire C, Ashack K, et al. Black hairy tongue: predisposing factors, diagnosis, and treatment. Am J Clin Dermatol. 2017;18:563-569. doi:10.1007/s40257-017-0268-y
- Sandru F, Petca A, Dumitrascu MC, et al. Peutz-Jeghers syndrome: skin manifestations and endocrine anomalies (review). Exp Ther Med. 2021;22:1387. doi:10.3892/etm.2021.10823
- Shah KR, Boland CR, Patel M, et al. Cutaneous manifestations of gastrointestinal disease: part I. J Am Acad Dermatol. 2013;68:189.e1-210. doi:10.1016/j.jaad.2012.10.037
- Lee K, Lian C, Vaidya A, et al. Oral mucosal hyperpigmentation. JAAD Case Rep. 2020;6:993-995. doi:10.1016/j.jdcr.2020.08.013
- Haitjema T, Westermann CJ, Overtoom TT, et al. Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease): new insights in pathogenesis, complications, and treatment. Arch Intern Med. 1996;156:714-719.
- Tokoro S, Namiki T, Ugajin T, et al. Hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber’s disease): detailed assessment of skin lesions by dermoscopy and ultrasound. Int J Dermatol. 2019;58:E224-E226. doi:10.1111/ijd.14578
The Diagnosis: Pigmented Fungiform Papillae of the Tongue
Our patient’s hyperpigmentation was confined to the fungiform papillae, leading to a diagnosis of pigmented fungiform papillae of the tongue (PFPT). A biopsy was not performed, and reassurance was provided regarding the benign nature of this finding, which did not require treatment.
Pigmented fungiform papillae of the tongue is a benign, nonprogressive, asymptomatic pigmentary condition that is most common among patients with skin of color and typically develops within the second or third decade of life.1,2 The pathogenesis is unclear, but activation of subepithelial melanophages without evidence of inflammation has been implicated.2 Although no standard treatment exists, cosmetic improvement with the use of the Q-switched ruby laser has been reported.3,4 Clinically, PFPT presents as asymptomatic hyperpigmentation confined to the fungiform papillae along the anterior and lateral portions of the tongue.1,2
Pigmented fungiform papillae of the tongue typically is an isolated finding but rarely can be associated with hyperpigmentation of the nails (as in our patient) or gingiva.2 Three different clinical patterns of presentation have been described: (1) a single well-circumscribed collection of pigmented fungiform papillae, (2) few scattered pigmented fungiform papillae admixed with many nonpigmented fungiform papillae, or (3) pigmentation of all fungiform papillae on the dorsal aspect of the tongue.2,5,6 Pigmented fungiform papillae of the tongue is a clinical diagnosis based on visual recognition. Dermoscopic examination revealing a cobblestonelike or rose petal–like pattern may be helpful in diagnosing PFPT.2,5-7 Although not typically recommended in the evaluation of PFPT, a biopsy will reveal papillary structures with hyperpigmentation of basilar keratinocytes as well as melanophages in the lamina propria.8 The latter finding suggests a transient inflammatory process despite the hallmark absence of inflammation.5 Melanocytic neoplasia and exogenous granules of pigment typically are not seen.8
Other conditions that may present with dark-colored macules or papules on the tongue should be considered in the evaluation of a patient with these clinical findings. Black hairy tongue (BHT), or lingua villosa nigra, is a benign finding due to filiform papillae hypertrophy on the dorsum of the tongue.9 Food particle debris caught in BHT can lead to porphyrin production by chromogenic bacteria and fungi. These porphyrins result in discoloration ranging from brown-black to yellow and green occurring anteriorly to the circumvallate papillae while usually sparing the tip and lateral sides of the tongue. Dermoscopy can show thin discolored fibers with a hairy appearance. Although normal filiform papillae are less than 1-mm long, 3-mm long papillae are considered diagnostic of BHT.9 Treatment includes effective oral hygiene and desquamation measures, which can lead to complete resolution.10
Peutz-Jeghers syndrome is a rare genodermatosis that is characterized by focal hyperpigmentation and multiple gastrointestinal mucosal hamartomatous polyps. Peutz-Jeghers syndrome should be suspected in a patient with discrete, 1- to 5-mm, brown to black macules on the perioral or periocular skin, tongue, genitals, palms, soles, and buccal mucosa with a history of abdominal symptoms.11,12
Addison disease, or primary adrenal insufficiency, may present with brown hyperpigmentation on chronically sun-exposed areas; regions of friction or pressure; surrounding scar tissue; and mucosal surfaces such as the tongue, inner surface of the lip, and buccal and gingival mucosa.13 Addison disease is differentiated from PFPT by a more generalized hyperpigmentation due to increased melanin production as well as the presence of systemic symptoms related to hypocortisolism. The pigmentation seen on the buccal mucosa in Addison disease is patchy and diffuse, and histology reveals basal melanin hyperpigmentation with superficial dermal melanophages.13
Hereditary hemorrhagic telangiectasia is an inherited disorder featuring telangiectasia and generally appears in the third decade of life.14 Telangiectases classically are 1 to 3 mm in diameter with or without slight elevation. Dermoscopic findings include small red clots, lacunae, and serpentine or linear vessels arranged in a radial conformation surrounding a homogenous pink center.15 These telangiectases typically occur on the skin or mucosa, particularly the face, lips, tongue, nail beds, and nasal mucosa; however, any organ can be affected with arteriovenous malformations. Recurrent epistaxis occurs in more than half of patients with hereditary hemorrhagic telangiectasia.14 Histopathology reveals dilated vessels and lacunae near the dermoepidermal junction displacing the epidermis and papillary dermis.15 It is distinguished from PFPT by the vascular nature of the lesions and by the presence of other characteristic symptoms such as recurrent epistaxis and visceral arteriovenous malformations.
The Diagnosis: Pigmented Fungiform Papillae of the Tongue
Our patient’s hyperpigmentation was confined to the fungiform papillae, leading to a diagnosis of pigmented fungiform papillae of the tongue (PFPT). A biopsy was not performed, and reassurance was provided regarding the benign nature of this finding, which did not require treatment.
Pigmented fungiform papillae of the tongue is a benign, nonprogressive, asymptomatic pigmentary condition that is most common among patients with skin of color and typically develops within the second or third decade of life.1,2 The pathogenesis is unclear, but activation of subepithelial melanophages without evidence of inflammation has been implicated.2 Although no standard treatment exists, cosmetic improvement with the use of the Q-switched ruby laser has been reported.3,4 Clinically, PFPT presents as asymptomatic hyperpigmentation confined to the fungiform papillae along the anterior and lateral portions of the tongue.1,2
Pigmented fungiform papillae of the tongue typically is an isolated finding but rarely can be associated with hyperpigmentation of the nails (as in our patient) or gingiva.2 Three different clinical patterns of presentation have been described: (1) a single well-circumscribed collection of pigmented fungiform papillae, (2) few scattered pigmented fungiform papillae admixed with many nonpigmented fungiform papillae, or (3) pigmentation of all fungiform papillae on the dorsal aspect of the tongue.2,5,6 Pigmented fungiform papillae of the tongue is a clinical diagnosis based on visual recognition. Dermoscopic examination revealing a cobblestonelike or rose petal–like pattern may be helpful in diagnosing PFPT.2,5-7 Although not typically recommended in the evaluation of PFPT, a biopsy will reveal papillary structures with hyperpigmentation of basilar keratinocytes as well as melanophages in the lamina propria.8 The latter finding suggests a transient inflammatory process despite the hallmark absence of inflammation.5 Melanocytic neoplasia and exogenous granules of pigment typically are not seen.8
Other conditions that may present with dark-colored macules or papules on the tongue should be considered in the evaluation of a patient with these clinical findings. Black hairy tongue (BHT), or lingua villosa nigra, is a benign finding due to filiform papillae hypertrophy on the dorsum of the tongue.9 Food particle debris caught in BHT can lead to porphyrin production by chromogenic bacteria and fungi. These porphyrins result in discoloration ranging from brown-black to yellow and green occurring anteriorly to the circumvallate papillae while usually sparing the tip and lateral sides of the tongue. Dermoscopy can show thin discolored fibers with a hairy appearance. Although normal filiform papillae are less than 1-mm long, 3-mm long papillae are considered diagnostic of BHT.9 Treatment includes effective oral hygiene and desquamation measures, which can lead to complete resolution.10
Peutz-Jeghers syndrome is a rare genodermatosis that is characterized by focal hyperpigmentation and multiple gastrointestinal mucosal hamartomatous polyps. Peutz-Jeghers syndrome should be suspected in a patient with discrete, 1- to 5-mm, brown to black macules on the perioral or periocular skin, tongue, genitals, palms, soles, and buccal mucosa with a history of abdominal symptoms.11,12
Addison disease, or primary adrenal insufficiency, may present with brown hyperpigmentation on chronically sun-exposed areas; regions of friction or pressure; surrounding scar tissue; and mucosal surfaces such as the tongue, inner surface of the lip, and buccal and gingival mucosa.13 Addison disease is differentiated from PFPT by a more generalized hyperpigmentation due to increased melanin production as well as the presence of systemic symptoms related to hypocortisolism. The pigmentation seen on the buccal mucosa in Addison disease is patchy and diffuse, and histology reveals basal melanin hyperpigmentation with superficial dermal melanophages.13
Hereditary hemorrhagic telangiectasia is an inherited disorder featuring telangiectasia and generally appears in the third decade of life.14 Telangiectases classically are 1 to 3 mm in diameter with or without slight elevation. Dermoscopic findings include small red clots, lacunae, and serpentine or linear vessels arranged in a radial conformation surrounding a homogenous pink center.15 These telangiectases typically occur on the skin or mucosa, particularly the face, lips, tongue, nail beds, and nasal mucosa; however, any organ can be affected with arteriovenous malformations. Recurrent epistaxis occurs in more than half of patients with hereditary hemorrhagic telangiectasia.14 Histopathology reveals dilated vessels and lacunae near the dermoepidermal junction displacing the epidermis and papillary dermis.15 It is distinguished from PFPT by the vascular nature of the lesions and by the presence of other characteristic symptoms such as recurrent epistaxis and visceral arteriovenous malformations.
- Romiti R, Molina De Medeiros L. Pigmented fungiform papillae of the tongue. Pediatr Dermatol. 2010;27:398-399. doi:10.1111/j .1525-1470.2010.01183.x
- Chessa MA, Patrizi A, Sechi A, et al. Pigmented fungiform lingual papillae: dermoscopic and clinical features. J Eur Acad Dermatol Venereol. 2018;32:935-939. doi:10.1111/jdv.14809
- Rice SM, Lal K. Successful treatment of pigmented fungiform papillae of the tongue with Q-switched ruby laser. Dermatol Surg. 2022;48:368-369. doi:10.1097/DSS.0000000000003371
- Mizawa M, Makino T, Furukawa F, et al. Efficacy of Q-switched ruby laser treatment for pigmented fungiform papillae of the tongue. J Dermatol. 2022;49:E133-E134. doi:10.1111/1346-8138.16270
- Holzwanger JM, Rudolph RI, Heaton CL. Pigmented fungiform papillae of the tongue: a common variant of oral pigmentation. Int J Dermatol. 1974;13:403-408. doi:10.1111/j.1365-4362.1974. tb05073.x
- Mukamal LV, Ormiga P, Ramos-E-Silva M. Dermoscopy of the pigmented fungiform papillae of the tongue. J Dermatol. 2012;39:397-399. doi:10.1111/j.1346-8138.2011.01328.x
- Surboyo MDC, Santosh ABR, Hariyani N, et al. Clinical utility of dermoscopy on diagnosing pigmented papillary fungiform papillae of the tongue: a systematic review. J Oral Biol Craniofac Res. 2021;11:618-623. doi:10.1016/j.jobcr.2021.09.008
- Chamseddin B, Vandergriff T. Pigmented fungiform papillae of the tongue: a clinical and histologic description [published online September 15, 2019]. Dermatol Online J. 2019;25:13030/qt8674c519.
- Jayasree P, Kaliyadan F, Ashique KT. Black hairy tongue. JAMA Dermatol. 2022;158:573. doi:10.1001/jamadermatol.2021.5314
- Schlager E, St Claire C, Ashack K, et al. Black hairy tongue: predisposing factors, diagnosis, and treatment. Am J Clin Dermatol. 2017;18:563-569. doi:10.1007/s40257-017-0268-y
- Sandru F, Petca A, Dumitrascu MC, et al. Peutz-Jeghers syndrome: skin manifestations and endocrine anomalies (review). Exp Ther Med. 2021;22:1387. doi:10.3892/etm.2021.10823
- Shah KR, Boland CR, Patel M, et al. Cutaneous manifestations of gastrointestinal disease: part I. J Am Acad Dermatol. 2013;68:189.e1-210. doi:10.1016/j.jaad.2012.10.037
- Lee K, Lian C, Vaidya A, et al. Oral mucosal hyperpigmentation. JAAD Case Rep. 2020;6:993-995. doi:10.1016/j.jdcr.2020.08.013
- Haitjema T, Westermann CJ, Overtoom TT, et al. Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease): new insights in pathogenesis, complications, and treatment. Arch Intern Med. 1996;156:714-719.
- Tokoro S, Namiki T, Ugajin T, et al. Hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber’s disease): detailed assessment of skin lesions by dermoscopy and ultrasound. Int J Dermatol. 2019;58:E224-E226. doi:10.1111/ijd.14578
- Romiti R, Molina De Medeiros L. Pigmented fungiform papillae of the tongue. Pediatr Dermatol. 2010;27:398-399. doi:10.1111/j .1525-1470.2010.01183.x
- Chessa MA, Patrizi A, Sechi A, et al. Pigmented fungiform lingual papillae: dermoscopic and clinical features. J Eur Acad Dermatol Venereol. 2018;32:935-939. doi:10.1111/jdv.14809
- Rice SM, Lal K. Successful treatment of pigmented fungiform papillae of the tongue with Q-switched ruby laser. Dermatol Surg. 2022;48:368-369. doi:10.1097/DSS.0000000000003371
- Mizawa M, Makino T, Furukawa F, et al. Efficacy of Q-switched ruby laser treatment for pigmented fungiform papillae of the tongue. J Dermatol. 2022;49:E133-E134. doi:10.1111/1346-8138.16270
- Holzwanger JM, Rudolph RI, Heaton CL. Pigmented fungiform papillae of the tongue: a common variant of oral pigmentation. Int J Dermatol. 1974;13:403-408. doi:10.1111/j.1365-4362.1974. tb05073.x
- Mukamal LV, Ormiga P, Ramos-E-Silva M. Dermoscopy of the pigmented fungiform papillae of the tongue. J Dermatol. 2012;39:397-399. doi:10.1111/j.1346-8138.2011.01328.x
- Surboyo MDC, Santosh ABR, Hariyani N, et al. Clinical utility of dermoscopy on diagnosing pigmented papillary fungiform papillae of the tongue: a systematic review. J Oral Biol Craniofac Res. 2021;11:618-623. doi:10.1016/j.jobcr.2021.09.008
- Chamseddin B, Vandergriff T. Pigmented fungiform papillae of the tongue: a clinical and histologic description [published online September 15, 2019]. Dermatol Online J. 2019;25:13030/qt8674c519.
- Jayasree P, Kaliyadan F, Ashique KT. Black hairy tongue. JAMA Dermatol. 2022;158:573. doi:10.1001/jamadermatol.2021.5314
- Schlager E, St Claire C, Ashack K, et al. Black hairy tongue: predisposing factors, diagnosis, and treatment. Am J Clin Dermatol. 2017;18:563-569. doi:10.1007/s40257-017-0268-y
- Sandru F, Petca A, Dumitrascu MC, et al. Peutz-Jeghers syndrome: skin manifestations and endocrine anomalies (review). Exp Ther Med. 2021;22:1387. doi:10.3892/etm.2021.10823
- Shah KR, Boland CR, Patel M, et al. Cutaneous manifestations of gastrointestinal disease: part I. J Am Acad Dermatol. 2013;68:189.e1-210. doi:10.1016/j.jaad.2012.10.037
- Lee K, Lian C, Vaidya A, et al. Oral mucosal hyperpigmentation. JAAD Case Rep. 2020;6:993-995. doi:10.1016/j.jdcr.2020.08.013
- Haitjema T, Westermann CJ, Overtoom TT, et al. Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease): new insights in pathogenesis, complications, and treatment. Arch Intern Med. 1996;156:714-719.
- Tokoro S, Namiki T, Ugajin T, et al. Hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber’s disease): detailed assessment of skin lesions by dermoscopy and ultrasound. Int J Dermatol. 2019;58:E224-E226. doi:10.1111/ijd.14578
A 9-year-old Black boy presented to the dermatology clinic for evaluation of dark spots on the tongue. The family first noted these spots 5 months prior and reported that they remained stable during that time. The patient’s medical history was notable for autism spectrum disorder and multiple food allergies. His family history was negative for similar oral pigmentation or other pigmentary anomalies. A review of systems was positive only for selective eating and rare nosebleeds. Physical examination revealed numerous dark brown, pinpoint papules across the dorsal aspect of the tongue. No hyperpigmentation of the buccal mucosae, lips, palms, or soles was identified. Several light brown streaks were present on the fingernails and toenails, consistent with longitudinal melanonychia. A prior complete blood cell count was within reference range.


A 17-year-old male was referred by his pediatrician for evaluation of a year-long rash
A biopsy of the edge of one of lesions on the torso was performed. Histopathology demonstrated hyperkeratosis of the stratum corneum with focal thickening of the granular cell layer, basal layer degeneration of the epidermis, and a band-like subepidermal lymphocytic infiltrate with Civatte bodies consistent with lichen planus. There was some reduction in the elastic fibers on the papillary dermis.

Given the morphology of the lesions and the histopathologic presentation, he was diagnosed with annular atrophic lichen planus (AALP). Lichen planus is a chronic inflammatory condition that can affect the skin, nails, hair, and mucosa. Lichen planus is seen in less than 1% of the population, occurring mainly in middle-aged adults and rarely seen in children. Though, there appears to be no clear racial predilection, a small study in the United States showed a higher incidence of lichen planus in Black children. Lesions with classic characteristics are pruritic, polygonal, violaceous, flat-topped papules and plaques.
There are different subtypes of lichen planus, which include papular or classic form, hypertrophic, vesiculobullous, actinic, annular, atrophic, annular atrophic, linear, follicular, lichen planus pigmentosus, lichen pigmentosa pigmentosus-inversus, lichen planus–lupus erythematosus overlap syndrome, and lichen planus pemphigoides. The annular atrophic form is the least common of all, and there are few reports in the pediatric population. AALP presents as annular papules and plaques with atrophic centers that resolve within a few months leaving postinflammatory hypo- or hyperpigmentation and, in some patients, permanent atrophic scarring.
In histopathology, the lesions show the classic characteristics of lichen planus including vacuolar interface changes and necrotic keratinocytes, hypergranulosis, band-like infiltrate in the dermis, melanin incontinence, and Civatte bodies. In AALP, the center of the lesion shows an atrophic epidermis, and there is also a characteristic partial reduction to complete destruction of elastic fibers in the papillary dermis in the center of the lesion and sometimes in the periphery as well, which helps differentiate AALP from other forms of lichen planus.
The differential diagnosis for AALP includes tinea corporis, which can present with annular lesions, but they are usually scaly and rarely resolve on their own. Pityriasis rosea lesions can also look very similar to AALP lesions, but the difference is the presence of an inner collaret of scale and a lack of atrophy in pityriasis rosea. Pityriasis rosea is a rash that can be triggered by viral infections, medications, and vaccines and self-resolves within 10-12 weeks. Secondary syphilis can also be annular and resemble lesions of AALP. Syphilis patients are usually sexually active and may have lesions present on the palms and soles, which were not seen in our patient.
Granuloma annulare should also be included in the differential diagnosis of AALP. Granuloma annulare lesions present as annular papules or plaques with raised borders and a slightly hyperpigmented center that may appear more depressed compared to the edges of the lesion, though not atrophic as seen in AALP. Pityriasis lichenoides chronica is an inflammatory condition of the skin in which patients present with erythematous to brown papules in different stages which may have a mica-like scale, usually not seen on AALP. Sometimes a skin biopsy will be needed to differentiate between these conditions.
It is very important to make a timely diagnosis of AALP and treat the lesions early as it may leave long-lasting dyspigmentation and scarring. Though AAPL lesions can be resistant to treatment with topical medications, there are reports of improvement with superpotent topical corticosteroids and calcineurin inhibitors. In recalcitrant cases, systemic therapy with isotretinoin, acitretin, methotrexate, systemic corticosteroids, dapsone, and hydroxychloroquine can be considered. Our patient was treated with clobetasol propionate ointment 0.05% with good response.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Bowers S and Warshaw EM. J Am Acad Dermatol. 2006 Oct;55(4):557-72; quiz 573-6.
Gorouhi F et al. Scientific World Journal. 2014 Jan 30;2014:742826.
Santhosh P and George M. Int J Dermatol. 2022.61:1213-7.
Sears S et al. Pediatr Dermatol. 2021;38:1283-7.
Weston G and Payette M. Int J Womens Dermatol. 2015 Sep 16;1(3):140-9.
A biopsy of the edge of one of lesions on the torso was performed. Histopathology demonstrated hyperkeratosis of the stratum corneum with focal thickening of the granular cell layer, basal layer degeneration of the epidermis, and a band-like subepidermal lymphocytic infiltrate with Civatte bodies consistent with lichen planus. There was some reduction in the elastic fibers on the papillary dermis.
Given the morphology of the lesions and the histopathologic presentation, he was diagnosed with annular atrophic lichen planus (AALP). Lichen planus is a chronic inflammatory condition that can affect the skin, nails, hair, and mucosa. Lichen planus is seen in less than 1% of the population, occurring mainly in middle-aged adults and rarely seen in children. Though, there appears to be no clear racial predilection, a small study in the United States showed a higher incidence of lichen planus in Black children. Lesions with classic characteristics are pruritic, polygonal, violaceous, flat-topped papules and plaques.
There are different subtypes of lichen planus, which include papular or classic form, hypertrophic, vesiculobullous, actinic, annular, atrophic, annular atrophic, linear, follicular, lichen planus pigmentosus, lichen pigmentosa pigmentosus-inversus, lichen planus–lupus erythematosus overlap syndrome, and lichen planus pemphigoides. The annular atrophic form is the least common of all, and there are few reports in the pediatric population. AALP presents as annular papules and plaques with atrophic centers that resolve within a few months leaving postinflammatory hypo- or hyperpigmentation and, in some patients, permanent atrophic scarring.
In histopathology, the lesions show the classic characteristics of lichen planus including vacuolar interface changes and necrotic keratinocytes, hypergranulosis, band-like infiltrate in the dermis, melanin incontinence, and Civatte bodies. In AALP, the center of the lesion shows an atrophic epidermis, and there is also a characteristic partial reduction to complete destruction of elastic fibers in the papillary dermis in the center of the lesion and sometimes in the periphery as well, which helps differentiate AALP from other forms of lichen planus.
The differential diagnosis for AALP includes tinea corporis, which can present with annular lesions, but they are usually scaly and rarely resolve on their own. Pityriasis rosea lesions can also look very similar to AALP lesions, but the difference is the presence of an inner collaret of scale and a lack of atrophy in pityriasis rosea. Pityriasis rosea is a rash that can be triggered by viral infections, medications, and vaccines and self-resolves within 10-12 weeks. Secondary syphilis can also be annular and resemble lesions of AALP. Syphilis patients are usually sexually active and may have lesions present on the palms and soles, which were not seen in our patient.
Granuloma annulare should also be included in the differential diagnosis of AALP. Granuloma annulare lesions present as annular papules or plaques with raised borders and a slightly hyperpigmented center that may appear more depressed compared to the edges of the lesion, though not atrophic as seen in AALP. Pityriasis lichenoides chronica is an inflammatory condition of the skin in which patients present with erythematous to brown papules in different stages which may have a mica-like scale, usually not seen on AALP. Sometimes a skin biopsy will be needed to differentiate between these conditions.
It is very important to make a timely diagnosis of AALP and treat the lesions early as it may leave long-lasting dyspigmentation and scarring. Though AAPL lesions can be resistant to treatment with topical medications, there are reports of improvement with superpotent topical corticosteroids and calcineurin inhibitors. In recalcitrant cases, systemic therapy with isotretinoin, acitretin, methotrexate, systemic corticosteroids, dapsone, and hydroxychloroquine can be considered. Our patient was treated with clobetasol propionate ointment 0.05% with good response.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Bowers S and Warshaw EM. J Am Acad Dermatol. 2006 Oct;55(4):557-72; quiz 573-6.
Gorouhi F et al. Scientific World Journal. 2014 Jan 30;2014:742826.
Santhosh P and George M. Int J Dermatol. 2022.61:1213-7.
Sears S et al. Pediatr Dermatol. 2021;38:1283-7.
Weston G and Payette M. Int J Womens Dermatol. 2015 Sep 16;1(3):140-9.
A biopsy of the edge of one of lesions on the torso was performed. Histopathology demonstrated hyperkeratosis of the stratum corneum with focal thickening of the granular cell layer, basal layer degeneration of the epidermis, and a band-like subepidermal lymphocytic infiltrate with Civatte bodies consistent with lichen planus. There was some reduction in the elastic fibers on the papillary dermis.
Given the morphology of the lesions and the histopathologic presentation, he was diagnosed with annular atrophic lichen planus (AALP). Lichen planus is a chronic inflammatory condition that can affect the skin, nails, hair, and mucosa. Lichen planus is seen in less than 1% of the population, occurring mainly in middle-aged adults and rarely seen in children. Though, there appears to be no clear racial predilection, a small study in the United States showed a higher incidence of lichen planus in Black children. Lesions with classic characteristics are pruritic, polygonal, violaceous, flat-topped papules and plaques.
There are different subtypes of lichen planus, which include papular or classic form, hypertrophic, vesiculobullous, actinic, annular, atrophic, annular atrophic, linear, follicular, lichen planus pigmentosus, lichen pigmentosa pigmentosus-inversus, lichen planus–lupus erythematosus overlap syndrome, and lichen planus pemphigoides. The annular atrophic form is the least common of all, and there are few reports in the pediatric population. AALP presents as annular papules and plaques with atrophic centers that resolve within a few months leaving postinflammatory hypo- or hyperpigmentation and, in some patients, permanent atrophic scarring.
In histopathology, the lesions show the classic characteristics of lichen planus including vacuolar interface changes and necrotic keratinocytes, hypergranulosis, band-like infiltrate in the dermis, melanin incontinence, and Civatte bodies. In AALP, the center of the lesion shows an atrophic epidermis, and there is also a characteristic partial reduction to complete destruction of elastic fibers in the papillary dermis in the center of the lesion and sometimes in the periphery as well, which helps differentiate AALP from other forms of lichen planus.
The differential diagnosis for AALP includes tinea corporis, which can present with annular lesions, but they are usually scaly and rarely resolve on their own. Pityriasis rosea lesions can also look very similar to AALP lesions, but the difference is the presence of an inner collaret of scale and a lack of atrophy in pityriasis rosea. Pityriasis rosea is a rash that can be triggered by viral infections, medications, and vaccines and self-resolves within 10-12 weeks. Secondary syphilis can also be annular and resemble lesions of AALP. Syphilis patients are usually sexually active and may have lesions present on the palms and soles, which were not seen in our patient.
Granuloma annulare should also be included in the differential diagnosis of AALP. Granuloma annulare lesions present as annular papules or plaques with raised borders and a slightly hyperpigmented center that may appear more depressed compared to the edges of the lesion, though not atrophic as seen in AALP. Pityriasis lichenoides chronica is an inflammatory condition of the skin in which patients present with erythematous to brown papules in different stages which may have a mica-like scale, usually not seen on AALP. Sometimes a skin biopsy will be needed to differentiate between these conditions.
It is very important to make a timely diagnosis of AALP and treat the lesions early as it may leave long-lasting dyspigmentation and scarring. Though AAPL lesions can be resistant to treatment with topical medications, there are reports of improvement with superpotent topical corticosteroids and calcineurin inhibitors. In recalcitrant cases, systemic therapy with isotretinoin, acitretin, methotrexate, systemic corticosteroids, dapsone, and hydroxychloroquine can be considered. Our patient was treated with clobetasol propionate ointment 0.05% with good response.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Bowers S and Warshaw EM. J Am Acad Dermatol. 2006 Oct;55(4):557-72; quiz 573-6.
Gorouhi F et al. Scientific World Journal. 2014 Jan 30;2014:742826.
Santhosh P and George M. Int J Dermatol. 2022.61:1213-7.
Sears S et al. Pediatr Dermatol. 2021;38:1283-7.
Weston G and Payette M. Int J Womens Dermatol. 2015 Sep 16;1(3):140-9.
A 17-year-old healthy male was referred by his pediatrician for evaluation of a rash on the skin which has been present on and off for a year. During the initial presentation, the lesions were clustered on the back, were slightly itchy, and resolved after 3 months. Several new lesions have developed on the neck, torso, and extremities, leaving hypopigmented marks on the skin. He has previously been treated with topical antifungal creams, oral fluconazole, and triamcinolone ointment without resolution of the lesions.
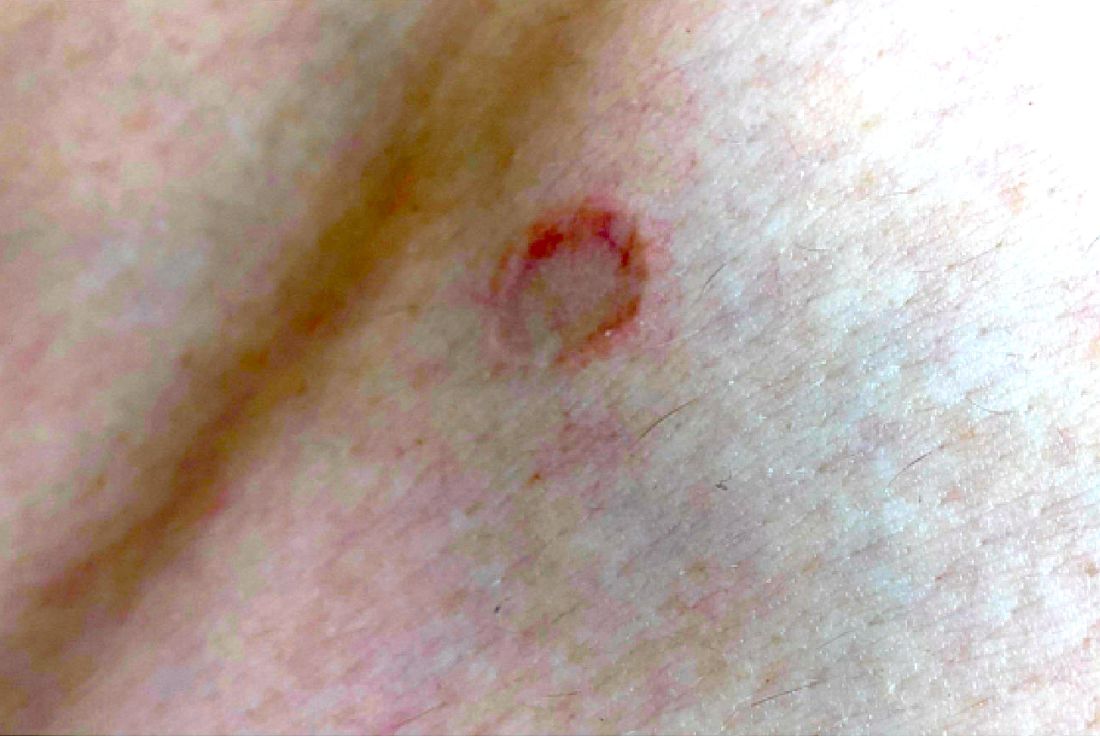
He is not involved in any contact sports, he has not traveled outside the country, and is not taking any other medications. He is not sexually active. He also has a diagnosis of mild acne that he is currently treating with over-the-counter medications.
On physical exam he had several annular plaques with central atrophic centers and no scale. He also had some hypo- and hyperpigmented macules at the sites of prior skin lesions
An adolescent male presents with an eroded bump on the temple
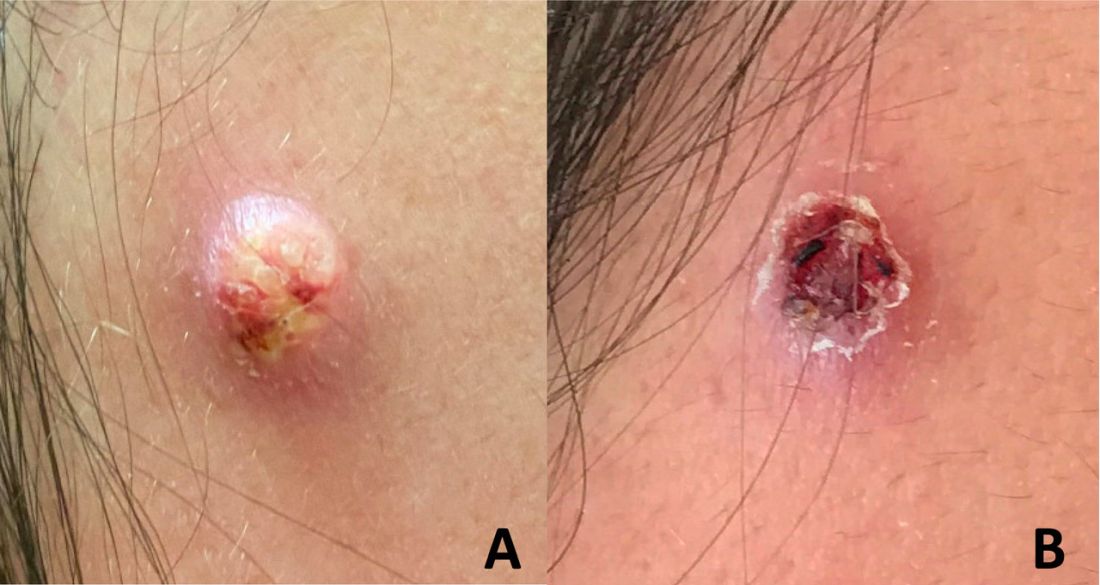
The correct answer is (D), molluscum contagiosum. Upon surgical excision, the pathology indicated the lesion was consistent with molluscum contagiosum.
Molluscum contagiosum is a benign skin disorder caused by a pox virus and is frequently seen in children. This disease is transmitted primarily through direct skin contact with an infected individual.1 Contaminated fomites have been suggested as another source of infection.2 The typical lesion appears dome-shaped, round, and pinkish-purple in color.1 The incubation period ranges from 2 weeks to 6 months and is typically self-limited in immunocompetent hosts; however, in immunocompromised persons, molluscum contagiosum lesions may present atypically such that they are larger in size and/or resemble malignancies, such as basal cell carcinoma or keratoacanthoma (for single lesions), or other infectious diseases, such as cryptococcosis and histoplasmosis (for more numerous lesions).3,4 A giant atypical molluscum contagiosum is rarely seen in healthy individuals.
What’s on the differential?
The recent episode of bleeding raises concern for other neoplastic processes of the skin including squamous cell carcinoma or basal cell carcinoma as well as cutaneous metastatic rhabdoid tumor, given the patient’s history.

Eruptive keratoacanthomas are also reported in patients taking nivolumab, an anti-PD-1 immunotherapy, which the patient has received for treatment of his recurrent metastatic rhabdoid tumor.5 More common entities such as a pyogenic granuloma or verruca are also included on the differential. The initial presentation of the lesion, however, is more consistent with the pearly umbilicated papules associated with molluscum contagiosum.
Comments from Dr. Eichenfield
This is a very hard diagnosis to make with the clinical findings and history.
Molluscum contagiosum infections are common, but with this patient’s medical history, biopsy and excision with pathologic examination was an appropriate approach to make a certain diagnosis.
Ms. Moyal is a research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego.
References
1. Brown J et al. Int J Dermatol. 2006 Feb;45(2):93-9.
2. Hanson D and Diven DG. Dermatol Online J. 2003 Mar;9(2).
3. Badri T and Gandhi GR. Molluscum contagiosum. 2022. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing.
4. Schwartz JJ and Myskowski PL. J Am Acad Dermatol. 1992 Oct 1;27(4):583-8.
5. Antonov NK et al. JAAD Case Rep. 2019 Apr 5;5(4):342-5.
The correct answer is (D), molluscum contagiosum. Upon surgical excision, the pathology indicated the lesion was consistent with molluscum contagiosum.
Molluscum contagiosum is a benign skin disorder caused by a pox virus and is frequently seen in children. This disease is transmitted primarily through direct skin contact with an infected individual.1 Contaminated fomites have been suggested as another source of infection.2 The typical lesion appears dome-shaped, round, and pinkish-purple in color.1 The incubation period ranges from 2 weeks to 6 months and is typically self-limited in immunocompetent hosts; however, in immunocompromised persons, molluscum contagiosum lesions may present atypically such that they are larger in size and/or resemble malignancies, such as basal cell carcinoma or keratoacanthoma (for single lesions), or other infectious diseases, such as cryptococcosis and histoplasmosis (for more numerous lesions).3,4 A giant atypical molluscum contagiosum is rarely seen in healthy individuals.
What’s on the differential?
The recent episode of bleeding raises concern for other neoplastic processes of the skin including squamous cell carcinoma or basal cell carcinoma as well as cutaneous metastatic rhabdoid tumor, given the patient’s history.
Eruptive keratoacanthomas are also reported in patients taking nivolumab, an anti-PD-1 immunotherapy, which the patient has received for treatment of his recurrent metastatic rhabdoid tumor.5 More common entities such as a pyogenic granuloma or verruca are also included on the differential. The initial presentation of the lesion, however, is more consistent with the pearly umbilicated papules associated with molluscum contagiosum.
Comments from Dr. Eichenfield
This is a very hard diagnosis to make with the clinical findings and history.
Molluscum contagiosum infections are common, but with this patient’s medical history, biopsy and excision with pathologic examination was an appropriate approach to make a certain diagnosis.
Ms. Moyal is a research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego.
References
1. Brown J et al. Int J Dermatol. 2006 Feb;45(2):93-9.
2. Hanson D and Diven DG. Dermatol Online J. 2003 Mar;9(2).
3. Badri T and Gandhi GR. Molluscum contagiosum. 2022. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing.
4. Schwartz JJ and Myskowski PL. J Am Acad Dermatol. 1992 Oct 1;27(4):583-8.
5. Antonov NK et al. JAAD Case Rep. 2019 Apr 5;5(4):342-5.
The correct answer is (D), molluscum contagiosum. Upon surgical excision, the pathology indicated the lesion was consistent with molluscum contagiosum.
Molluscum contagiosum is a benign skin disorder caused by a pox virus and is frequently seen in children. This disease is transmitted primarily through direct skin contact with an infected individual.1 Contaminated fomites have been suggested as another source of infection.2 The typical lesion appears dome-shaped, round, and pinkish-purple in color.1 The incubation period ranges from 2 weeks to 6 months and is typically self-limited in immunocompetent hosts; however, in immunocompromised persons, molluscum contagiosum lesions may present atypically such that they are larger in size and/or resemble malignancies, such as basal cell carcinoma or keratoacanthoma (for single lesions), or other infectious diseases, such as cryptococcosis and histoplasmosis (for more numerous lesions).3,4 A giant atypical molluscum contagiosum is rarely seen in healthy individuals.
What’s on the differential?
The recent episode of bleeding raises concern for other neoplastic processes of the skin including squamous cell carcinoma or basal cell carcinoma as well as cutaneous metastatic rhabdoid tumor, given the patient’s history.
Eruptive keratoacanthomas are also reported in patients taking nivolumab, an anti-PD-1 immunotherapy, which the patient has received for treatment of his recurrent metastatic rhabdoid tumor.5 More common entities such as a pyogenic granuloma or verruca are also included on the differential. The initial presentation of the lesion, however, is more consistent with the pearly umbilicated papules associated with molluscum contagiosum.
Comments from Dr. Eichenfield
This is a very hard diagnosis to make with the clinical findings and history.
Molluscum contagiosum infections are common, but with this patient’s medical history, biopsy and excision with pathologic examination was an appropriate approach to make a certain diagnosis.
Ms. Moyal is a research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego.
References
1. Brown J et al. Int J Dermatol. 2006 Feb;45(2):93-9.
2. Hanson D and Diven DG. Dermatol Online J. 2003 Mar;9(2).
3. Badri T and Gandhi GR. Molluscum contagiosum. 2022. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing.
4. Schwartz JJ and Myskowski PL. J Am Acad Dermatol. 1992 Oct 1;27(4):583-8.
5. Antonov NK et al. JAAD Case Rep. 2019 Apr 5;5(4):342-5.
Itchy Red-Brown Spots on a Child
The Diagnosis: Maculopapular Cutaneous Mastocytosis (Urticaria Pigmentosa)
A stroke test revealed urtication at the exact traumatized site (Figure). A skin biopsy performed 2 years prior by another physician in the same hospital had revealed mast cell infiltration of virtually the entire dermis. The diagnosis was then firmly established as maculopapular cutaneous mastocytosis (CM)(also known as urticaria pigmentosa) with both the pathology results and a confirmative stroke test, and no additional biopsy was attempted. Serum IgE and tryptase levels were within the reference range. General recommendations about the avoidance of trigger factors were given to the family, and a new-generation H1 blocker antihistaminic syrup was prescribed for flushing, itching, and urtication.

Mastocytosis is a canopy term for a heterogeneous group of disorders caused by clonal proliferation and accumulation of abnormal mast cells within the skin and visceral organs (ie, bone marrow, liver, spleen, lymph nodes, gastrointestinal tract). Cutaneous mastocytosis, the skin-restricted variant, is by far the most common form of childhood mastocytosis (90% of mastocytosis cases in children)1 and generally appears within the first 2 years of life.1-7 Pediatric CM usually is a benign and transient disease with an excellent prognosis and a negligible risk for systemic involvement.2,3,5
The pathogenesis of CM in children is obscure1; however, somatic or germline gain-of-function mutations of the c-KIT proto-oncogene, which encodes KIT (ie, a tyrosine kinase membrane receptor for stem cell factor), may account for most pediatric CM phenotypes.1,3,6 Activating c-KIT mutations leads to constitutive activation of the KIT receptor (expressed on the surface membrane of mast cells) and instigates autonomous (stem cell factor– independent) clonal proliferation, enhanced survival, and accumulation of mast cells.2
Maculopapular CM is the most common clinical form of CM.2,4,5 In children, maculopapular CM usually presents with polymorphous red-brown lesions of varying sizes and types—macule, papule, plaque, or nodule—on the torso and extremities.1-5 The distribution may be widespread and rarely is almost universal, as in our patient.2 Darier sign typically is positive, with a wheal and flare developing upon stroking or rubbing 1 or several lesions.1-6 The lesions gradually involute and often spontaneously regress at the time of puberty.1-3,5-7
The clinical signs and symptoms of mastocytosis are not only related to mast cell infiltration but also to mast cell activation within the tissues. The release of intracellular mediators from activated mast cells may have local and/or systemic consequences.4,7 Erythema, edema, flushing, pruritus, urticaria, blistering, and dermatographism are among the local cutaneous symptoms of mast cell activation.2-4,7 Systemic symptoms are rare in childhood CM and consist of wheezing, shortness of breath, nausea, vomiting, reflux, abdominal cramping, diarrhea, tachycardia, hypotension, syncope, anaphylaxis, and cyanotic spells.1-7 An elevated serum tryptase level is an indicator of both mast cell burden and risk for mast cell activation in the skin.4,7
Treatment of pediatric CM is conservative and symptomatic.3 Prevention of mediator release may be accomplished through avoidance of trigger factors.1 Alleviation of mediator-related symptoms might be attained using H1 and H2 histamine receptor blockers, oral cromolyn sodium, leukotriene antagonists, and epinephrine autoinjectors.1-3,5 Short-term topical or oral corticosteroids; calcineurin inhibitors (eg, pimecrolimus, tacrolimus); phototherapy; psoralen plus UVA; omalizumab; and innovative agents such as topical miltefosine, nemolizumab (an IL-31 antagonist), kinase inhibitors such as midostaurin, and tyrosine kinase inhibitors such as imatinib and masitinib may be tried in refractory or extensive pediatric CM.1,2,5,6
Although several disorders in childhood may present with red-brown macules and papules, Darier sign is unique to cutaneous mastocytosis. A biopsy also will be helpful in establishing the definitive diagnosis.
Histiocytosis X (also referred to as Langerhans cell histiocytosis) is the most common proliferative histiocytic disorder. Cutaneous lesions are polymorphic and consist of seborrheic involvement of the scalp with yellow, scaly or crusted papules; eroded patches; pustules; vesicles; petechiae; purpura; or red to purplish papules on the groin, abdomen, back, or chest.8
LEOPARD syndrome (also known as Noonan syndrome with multiple lentigines) is an acronym denoting lentigines (multiple), electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of the genitalia, retarded growth, and deafness (sensorineural). The disorder is caused by a genetic mutation involving the PTPN11 gene and currently is categorized under the canopy of RASopathies. Cutaneous findings consist of lentiginous and café-au-lait macules and patches.9
Neurofibromatosis is a genetic disorder with a plethora of cutaneous and systemic manifestations. The type 1 variant that constitutes more than 95% of cases is caused by mutations in the neurofibromin gene. The main cutaneous findings include café-au-lait macules, freckling in axillary and inguinal locations (Crowe sign), and neurofibromas. These lesions may present as macules, patches, papules, or nodules.10
Xanthoma disseminatum is a rare sporadic proliferative histiocyte disorder involving the skin and mucosa. The disorder may be a harbinger of diabetes insipidus. Cutaneous lesions consist of asymptomatic, symmetrical, discrete, erythematous to yellow-brown papules and nodules.11
- Sandru F, Petca RC, Costescu M, et al. Cutaneous mastocytosis in childhood: update from the literature. J Clin Med. 2021;10:1474. doi:10.3390/jcm10071474
- Lange M, Hartmann K, Carter MC, et al. Molecular background, clinical features and management of pediatric mastocytosis: status 2021. Int J Mol Sci. 2021;22:2586. doi:10.3390/ijms22052586
- Castells M, Metcalfe DD, Escribano L. Diagnosis and treatment of cutaneous mastocytosis in children: practical recommendations. Am J Clin Dermatol. 2011;12:259-270. doi:10.2165/11588890-000000000-00000
- Nedoszytko B, Arock M, Lyons JJ, et al. Clinical impact of inherited and acquired genetic variants in mastocytosis. Int J Mol Sci. 2021;22:411. doi:10.3390/ijms22010411
- Nemat K, Abraham S. Cutaneous mastocytosis in childhood. Allergol Select. 2022;6:1-10. doi:10.5414/ALX02304E
- Giona F. Pediatric mastocytosis: an update. Mediterr J Hematol Infect Dis. 2021;13:E2021069. doi:10.4084/MJHID.2021.069
- Brockow K, Plata-Nazar K, Lange M, et al. Mediator-related symptoms and anaphylaxis in children with mastocytosis. Int J Mol Sci. 2021;22:2684. doi:10.3390/ijms22052684
- Grana N. Langerhans cell histiocytosis. Cancer Control. 2014;21: 328-334.
- García-Gil MF, Álvarez-Salafranca M, Valero-Torres A, et al. Melanoma in Noonan syndrome with multiple lentigines (LEOPARD syndrome): a new case. Actas Dermosifiliogr (Engl Ed). 2020;111:619-621.
- Ozarslan B, Russo T, Argenziano G, et al. Cutaneous findings in neurofibromatosis type 1. Cancers (Basel). 2021;13:463.
- Behra A, Sa DK, Naik R, et al. A rare case of persistent xanthoma disseminatum without any systemic involvement. Indian J Dermatol. 2020;65:239-241.
The Diagnosis: Maculopapular Cutaneous Mastocytosis (Urticaria Pigmentosa)
A stroke test revealed urtication at the exact traumatized site (Figure). A skin biopsy performed 2 years prior by another physician in the same hospital had revealed mast cell infiltration of virtually the entire dermis. The diagnosis was then firmly established as maculopapular cutaneous mastocytosis (CM)(also known as urticaria pigmentosa) with both the pathology results and a confirmative stroke test, and no additional biopsy was attempted. Serum IgE and tryptase levels were within the reference range. General recommendations about the avoidance of trigger factors were given to the family, and a new-generation H1 blocker antihistaminic syrup was prescribed for flushing, itching, and urtication.
Mastocytosis is a canopy term for a heterogeneous group of disorders caused by clonal proliferation and accumulation of abnormal mast cells within the skin and visceral organs (ie, bone marrow, liver, spleen, lymph nodes, gastrointestinal tract). Cutaneous mastocytosis, the skin-restricted variant, is by far the most common form of childhood mastocytosis (90% of mastocytosis cases in children)1 and generally appears within the first 2 years of life.1-7 Pediatric CM usually is a benign and transient disease with an excellent prognosis and a negligible risk for systemic involvement.2,3,5
The pathogenesis of CM in children is obscure1; however, somatic or germline gain-of-function mutations of the c-KIT proto-oncogene, which encodes KIT (ie, a tyrosine kinase membrane receptor for stem cell factor), may account for most pediatric CM phenotypes.1,3,6 Activating c-KIT mutations leads to constitutive activation of the KIT receptor (expressed on the surface membrane of mast cells) and instigates autonomous (stem cell factor– independent) clonal proliferation, enhanced survival, and accumulation of mast cells.2
Maculopapular CM is the most common clinical form of CM.2,4,5 In children, maculopapular CM usually presents with polymorphous red-brown lesions of varying sizes and types—macule, papule, plaque, or nodule—on the torso and extremities.1-5 The distribution may be widespread and rarely is almost universal, as in our patient.2 Darier sign typically is positive, with a wheal and flare developing upon stroking or rubbing 1 or several lesions.1-6 The lesions gradually involute and often spontaneously regress at the time of puberty.1-3,5-7
The clinical signs and symptoms of mastocytosis are not only related to mast cell infiltration but also to mast cell activation within the tissues. The release of intracellular mediators from activated mast cells may have local and/or systemic consequences.4,7 Erythema, edema, flushing, pruritus, urticaria, blistering, and dermatographism are among the local cutaneous symptoms of mast cell activation.2-4,7 Systemic symptoms are rare in childhood CM and consist of wheezing, shortness of breath, nausea, vomiting, reflux, abdominal cramping, diarrhea, tachycardia, hypotension, syncope, anaphylaxis, and cyanotic spells.1-7 An elevated serum tryptase level is an indicator of both mast cell burden and risk for mast cell activation in the skin.4,7
Treatment of pediatric CM is conservative and symptomatic.3 Prevention of mediator release may be accomplished through avoidance of trigger factors.1 Alleviation of mediator-related symptoms might be attained using H1 and H2 histamine receptor blockers, oral cromolyn sodium, leukotriene antagonists, and epinephrine autoinjectors.1-3,5 Short-term topical or oral corticosteroids; calcineurin inhibitors (eg, pimecrolimus, tacrolimus); phototherapy; psoralen plus UVA; omalizumab; and innovative agents such as topical miltefosine, nemolizumab (an IL-31 antagonist), kinase inhibitors such as midostaurin, and tyrosine kinase inhibitors such as imatinib and masitinib may be tried in refractory or extensive pediatric CM.1,2,5,6
Although several disorders in childhood may present with red-brown macules and papules, Darier sign is unique to cutaneous mastocytosis. A biopsy also will be helpful in establishing the definitive diagnosis.
Histiocytosis X (also referred to as Langerhans cell histiocytosis) is the most common proliferative histiocytic disorder. Cutaneous lesions are polymorphic and consist of seborrheic involvement of the scalp with yellow, scaly or crusted papules; eroded patches; pustules; vesicles; petechiae; purpura; or red to purplish papules on the groin, abdomen, back, or chest.8
LEOPARD syndrome (also known as Noonan syndrome with multiple lentigines) is an acronym denoting lentigines (multiple), electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of the genitalia, retarded growth, and deafness (sensorineural). The disorder is caused by a genetic mutation involving the PTPN11 gene and currently is categorized under the canopy of RASopathies. Cutaneous findings consist of lentiginous and café-au-lait macules and patches.9
Neurofibromatosis is a genetic disorder with a plethora of cutaneous and systemic manifestations. The type 1 variant that constitutes more than 95% of cases is caused by mutations in the neurofibromin gene. The main cutaneous findings include café-au-lait macules, freckling in axillary and inguinal locations (Crowe sign), and neurofibromas. These lesions may present as macules, patches, papules, or nodules.10
Xanthoma disseminatum is a rare sporadic proliferative histiocyte disorder involving the skin and mucosa. The disorder may be a harbinger of diabetes insipidus. Cutaneous lesions consist of asymptomatic, symmetrical, discrete, erythematous to yellow-brown papules and nodules.11
The Diagnosis: Maculopapular Cutaneous Mastocytosis (Urticaria Pigmentosa)
A stroke test revealed urtication at the exact traumatized site (Figure). A skin biopsy performed 2 years prior by another physician in the same hospital had revealed mast cell infiltration of virtually the entire dermis. The diagnosis was then firmly established as maculopapular cutaneous mastocytosis (CM)(also known as urticaria pigmentosa) with both the pathology results and a confirmative stroke test, and no additional biopsy was attempted. Serum IgE and tryptase levels were within the reference range. General recommendations about the avoidance of trigger factors were given to the family, and a new-generation H1 blocker antihistaminic syrup was prescribed for flushing, itching, and urtication.
Mastocytosis is a canopy term for a heterogeneous group of disorders caused by clonal proliferation and accumulation of abnormal mast cells within the skin and visceral organs (ie, bone marrow, liver, spleen, lymph nodes, gastrointestinal tract). Cutaneous mastocytosis, the skin-restricted variant, is by far the most common form of childhood mastocytosis (90% of mastocytosis cases in children)1 and generally appears within the first 2 years of life.1-7 Pediatric CM usually is a benign and transient disease with an excellent prognosis and a negligible risk for systemic involvement.2,3,5
The pathogenesis of CM in children is obscure1; however, somatic or germline gain-of-function mutations of the c-KIT proto-oncogene, which encodes KIT (ie, a tyrosine kinase membrane receptor for stem cell factor), may account for most pediatric CM phenotypes.1,3,6 Activating c-KIT mutations leads to constitutive activation of the KIT receptor (expressed on the surface membrane of mast cells) and instigates autonomous (stem cell factor– independent) clonal proliferation, enhanced survival, and accumulation of mast cells.2
Maculopapular CM is the most common clinical form of CM.2,4,5 In children, maculopapular CM usually presents with polymorphous red-brown lesions of varying sizes and types—macule, papule, plaque, or nodule—on the torso and extremities.1-5 The distribution may be widespread and rarely is almost universal, as in our patient.2 Darier sign typically is positive, with a wheal and flare developing upon stroking or rubbing 1 or several lesions.1-6 The lesions gradually involute and often spontaneously regress at the time of puberty.1-3,5-7
The clinical signs and symptoms of mastocytosis are not only related to mast cell infiltration but also to mast cell activation within the tissues. The release of intracellular mediators from activated mast cells may have local and/or systemic consequences.4,7 Erythema, edema, flushing, pruritus, urticaria, blistering, and dermatographism are among the local cutaneous symptoms of mast cell activation.2-4,7 Systemic symptoms are rare in childhood CM and consist of wheezing, shortness of breath, nausea, vomiting, reflux, abdominal cramping, diarrhea, tachycardia, hypotension, syncope, anaphylaxis, and cyanotic spells.1-7 An elevated serum tryptase level is an indicator of both mast cell burden and risk for mast cell activation in the skin.4,7
Treatment of pediatric CM is conservative and symptomatic.3 Prevention of mediator release may be accomplished through avoidance of trigger factors.1 Alleviation of mediator-related symptoms might be attained using H1 and H2 histamine receptor blockers, oral cromolyn sodium, leukotriene antagonists, and epinephrine autoinjectors.1-3,5 Short-term topical or oral corticosteroids; calcineurin inhibitors (eg, pimecrolimus, tacrolimus); phototherapy; psoralen plus UVA; omalizumab; and innovative agents such as topical miltefosine, nemolizumab (an IL-31 antagonist), kinase inhibitors such as midostaurin, and tyrosine kinase inhibitors such as imatinib and masitinib may be tried in refractory or extensive pediatric CM.1,2,5,6
Although several disorders in childhood may present with red-brown macules and papules, Darier sign is unique to cutaneous mastocytosis. A biopsy also will be helpful in establishing the definitive diagnosis.
Histiocytosis X (also referred to as Langerhans cell histiocytosis) is the most common proliferative histiocytic disorder. Cutaneous lesions are polymorphic and consist of seborrheic involvement of the scalp with yellow, scaly or crusted papules; eroded patches; pustules; vesicles; petechiae; purpura; or red to purplish papules on the groin, abdomen, back, or chest.8
LEOPARD syndrome (also known as Noonan syndrome with multiple lentigines) is an acronym denoting lentigines (multiple), electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of the genitalia, retarded growth, and deafness (sensorineural). The disorder is caused by a genetic mutation involving the PTPN11 gene and currently is categorized under the canopy of RASopathies. Cutaneous findings consist of lentiginous and café-au-lait macules and patches.9
Neurofibromatosis is a genetic disorder with a plethora of cutaneous and systemic manifestations. The type 1 variant that constitutes more than 95% of cases is caused by mutations in the neurofibromin gene. The main cutaneous findings include café-au-lait macules, freckling in axillary and inguinal locations (Crowe sign), and neurofibromas. These lesions may present as macules, patches, papules, or nodules.10
Xanthoma disseminatum is a rare sporadic proliferative histiocyte disorder involving the skin and mucosa. The disorder may be a harbinger of diabetes insipidus. Cutaneous lesions consist of asymptomatic, symmetrical, discrete, erythematous to yellow-brown papules and nodules.11
- Sandru F, Petca RC, Costescu M, et al. Cutaneous mastocytosis in childhood: update from the literature. J Clin Med. 2021;10:1474. doi:10.3390/jcm10071474
- Lange M, Hartmann K, Carter MC, et al. Molecular background, clinical features and management of pediatric mastocytosis: status 2021. Int J Mol Sci. 2021;22:2586. doi:10.3390/ijms22052586
- Castells M, Metcalfe DD, Escribano L. Diagnosis and treatment of cutaneous mastocytosis in children: practical recommendations. Am J Clin Dermatol. 2011;12:259-270. doi:10.2165/11588890-000000000-00000
- Nedoszytko B, Arock M, Lyons JJ, et al. Clinical impact of inherited and acquired genetic variants in mastocytosis. Int J Mol Sci. 2021;22:411. doi:10.3390/ijms22010411
- Nemat K, Abraham S. Cutaneous mastocytosis in childhood. Allergol Select. 2022;6:1-10. doi:10.5414/ALX02304E
- Giona F. Pediatric mastocytosis: an update. Mediterr J Hematol Infect Dis. 2021;13:E2021069. doi:10.4084/MJHID.2021.069
- Brockow K, Plata-Nazar K, Lange M, et al. Mediator-related symptoms and anaphylaxis in children with mastocytosis. Int J Mol Sci. 2021;22:2684. doi:10.3390/ijms22052684
- Grana N. Langerhans cell histiocytosis. Cancer Control. 2014;21: 328-334.
- García-Gil MF, Álvarez-Salafranca M, Valero-Torres A, et al. Melanoma in Noonan syndrome with multiple lentigines (LEOPARD syndrome): a new case. Actas Dermosifiliogr (Engl Ed). 2020;111:619-621.
- Ozarslan B, Russo T, Argenziano G, et al. Cutaneous findings in neurofibromatosis type 1. Cancers (Basel). 2021;13:463.
- Behra A, Sa DK, Naik R, et al. A rare case of persistent xanthoma disseminatum without any systemic involvement. Indian J Dermatol. 2020;65:239-241.
- Sandru F, Petca RC, Costescu M, et al. Cutaneous mastocytosis in childhood: update from the literature. J Clin Med. 2021;10:1474. doi:10.3390/jcm10071474
- Lange M, Hartmann K, Carter MC, et al. Molecular background, clinical features and management of pediatric mastocytosis: status 2021. Int J Mol Sci. 2021;22:2586. doi:10.3390/ijms22052586
- Castells M, Metcalfe DD, Escribano L. Diagnosis and treatment of cutaneous mastocytosis in children: practical recommendations. Am J Clin Dermatol. 2011;12:259-270. doi:10.2165/11588890-000000000-00000
- Nedoszytko B, Arock M, Lyons JJ, et al. Clinical impact of inherited and acquired genetic variants in mastocytosis. Int J Mol Sci. 2021;22:411. doi:10.3390/ijms22010411
- Nemat K, Abraham S. Cutaneous mastocytosis in childhood. Allergol Select. 2022;6:1-10. doi:10.5414/ALX02304E
- Giona F. Pediatric mastocytosis: an update. Mediterr J Hematol Infect Dis. 2021;13:E2021069. doi:10.4084/MJHID.2021.069
- Brockow K, Plata-Nazar K, Lange M, et al. Mediator-related symptoms and anaphylaxis in children with mastocytosis. Int J Mol Sci. 2021;22:2684. doi:10.3390/ijms22052684
- Grana N. Langerhans cell histiocytosis. Cancer Control. 2014;21: 328-334.
- García-Gil MF, Álvarez-Salafranca M, Valero-Torres A, et al. Melanoma in Noonan syndrome with multiple lentigines (LEOPARD syndrome): a new case. Actas Dermosifiliogr (Engl Ed). 2020;111:619-621.
- Ozarslan B, Russo T, Argenziano G, et al. Cutaneous findings in neurofibromatosis type 1. Cancers (Basel). 2021;13:463.
- Behra A, Sa DK, Naik R, et al. A rare case of persistent xanthoma disseminatum without any systemic involvement. Indian J Dermatol. 2020;65:239-241.
A 5-year-old boy presented with red-brown spots diffusely spread over the body that were present since birth. There were no subjective symptoms, except for rare instances of flushing, itching, and urtication following hot baths and abrasive scrubs. Dermatologic examination revealed widespread brown polymorphic macules and papules of varying sizes on the forehead, neck, torso, and extremities. Physical examination was otherwise normal.


An infant with a tender bump on her ear
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.
Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.
Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.
Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A 4-month-old female was referred to our clinic for evaluation of a bump on the right ear. The lesion was first noted at 2 months of age as a little pimple. She was evaluated by her pediatrician and was treated with topical and oral antibiotics without resolution of the lesion. The bump continued to grow and seemed tender to palpation, so she was referred to dermatology for evaluation.
She was born via normal vaginal delivery at 40 weeks. Her mother has no medical conditions and the pregnancy was uneventful. She has been growing and developing well. She takes vitamin D and is currently breast fed.
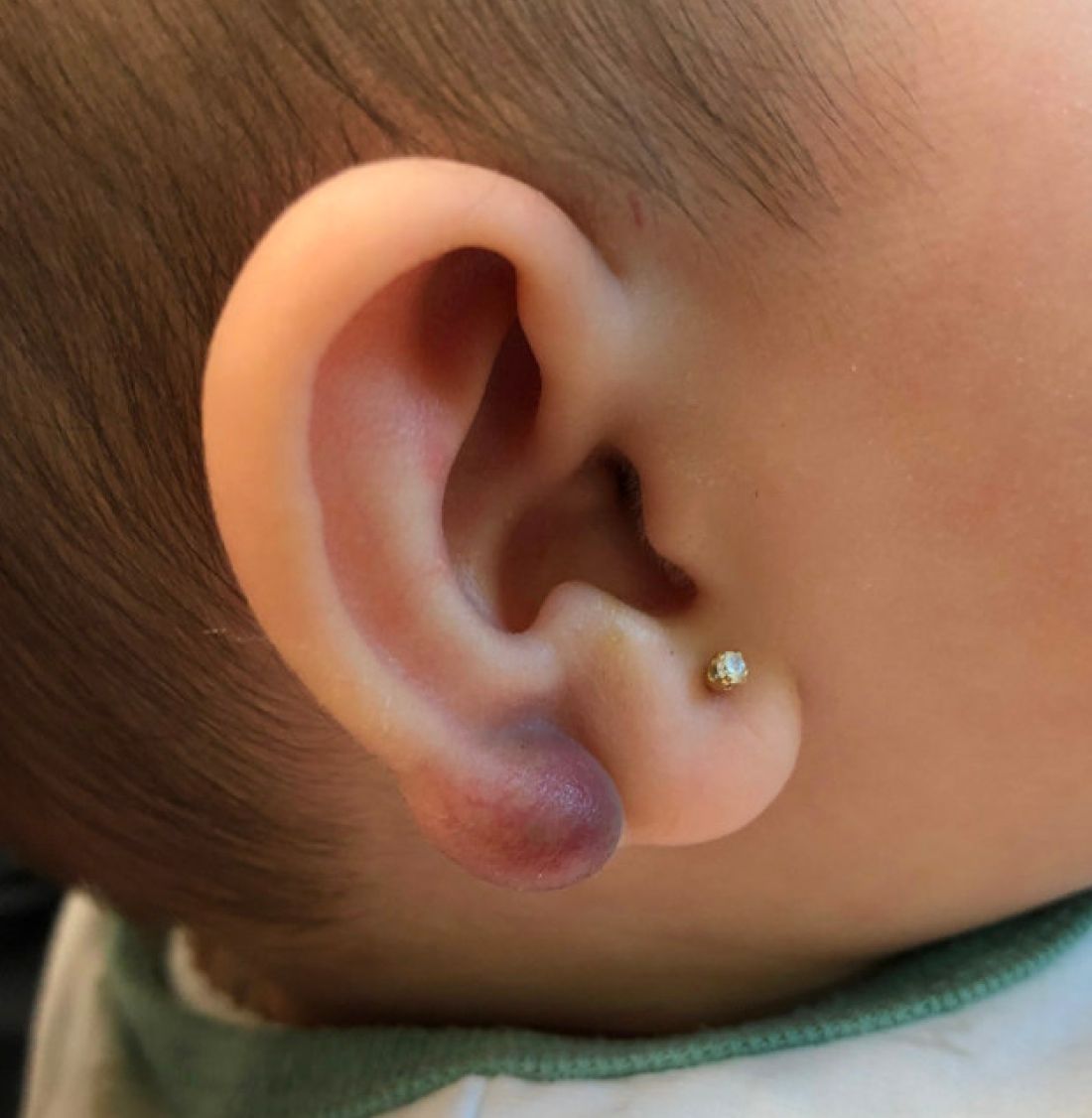
There have been no other family members with similar lesions. She had her ears pierced at a month of age without any complications.
On skin examination she has a firm red nodule on the right ear that appears slightly tender to touch. She has no other skin lesions of concern. She has normal muscle tone and there are no other abnormalities noted on the physical exam. She has no hepatomegaly, splenomegaly, or lymphadenopathy.
A 10-year-old with a red bump on her lower lip
The patient’s history and examination are consistent with a diagnosis of pyogenic granuloma. Specifically, the history of rapid growth, friable nature, associated bleeding, and hemorrhagic crusting point to pyogenic granuloma as the most likely diagnosis.
Pyogenic granuloma is an acquired benign vascular growth of the skin or mucous membranes.1 It most frequently occurs in children and young adults and most commonly affects the skin of the head, trunk, and extremities.2 Common mucosal sites include the gingiva, lips, and tongue.2 The etiology of pyogenic granuloma is unknown, though it is thought to be a process akin to the overgrowth of granulation tissue.3,4 Expression of angiogenic factors and subsequent vascular hyperplasia are also implicated as key players in the pathogenesis of pyogenic granuloma.1,4 In addition, several associated factors and inciting triggers have been proposed including trauma, infections, and hormonal fluctuations.3-5 However, the majority of patients do not report predisposing factors or a history of prior trauma at the site.3,6
Clinically, pyogenic granuloma usually presents as a painless, erythematous, dome-shaped friable papule or nodule that easily bleeds and may ulcerate. It typically undergoes a period of growth over weeks to months followed by stabilization. Occasionally, pyogenic granulomas will spontaneously involute, though most do not.7 Pyogenic granuloma may occur within an existing capillary malformation, such as a port wine stain, spontaneously or as a sequela of laser treatment.8,9 Diagnosis of pyogenic granuloma can typically be made clinically on the basis of history and exam. Dermoscopic evaluation of pyogenic granuloma will reveal a homogeneous papule with a surrounding white-brown collarette, and potentially white intersecting lines.10 Histopathologic evaluation may be necessary to differentiate lesions from conditions that may mimic pyogenic granuloma.
What’s on the differential?
The differential diagnosis for pyogenic granuloma consists of Spitz nevus, cherry hemangioma, amelanotic melanoma, and glomus tumor.
Spitz nevus
Spitz nevus (spindle and epithelial cell nevus) is a benign melanocytic lesion that classically appears as a sharply circumscribed, smooth, dome-shaped, pink-red, or brown papule or plaque. There is typically a history of rapid growth over several months followed by stabilization. It usually presents in childhood or adolescence and is most commonly located on the face and extremities. While there are similarities in the appearance of Spitz nevi and pyogenic granuloma, Spitz nevi are not usually friable nor associated with bleeding as in our patient. Furthermore, on dermoscopy, Spitz nevus typically exhibits a starburst pattern with regularly distributed dotted vessels, or a peripheral globular pattern with reticular depigmentation. The definitive diagnosis of Spitz nevi relies on histopathologic evaluation, which is critical for discriminating Spitz nevi from melanoma.
Cherry hemangioma
Cherry angiomas are the most common type of acquired benign vascular proliferation. They present as small, bright red or violaceous macules or papules. However, they typically appear in early to midadulthood and increase in number with age. The age of our patient and solitary presentation of the lesion make this diagnosis unlikely. In addition, cherry angiomas are not usually associated with bleeding. It is important to note that, depending on the age of the patient, pyogenic granuloma may also be confused with infantile hemangioma. Infantile hemangiomas may become bright red papules, nodules, or plaques that appear in early infancy. They characteristically involute, which does not typically happen with pyogenic granuloma.
Amelanotic melanoma
Amelanotic melanoma is an uncommon variant of melanoma with little to no pigmentation. It may appear as a skin-colored to light-brown, pink, or red macule, papule, or nodule. The lesion may be asymmetric with irregular and well-defined borders. The variable and uncharacteristic appearance of this melanoma variant makes it diagnostically challenging and it is often confused with benign lesions including pyogenic granuloma. Dermoscopy can help distinguish amelanotic melanoma from other benign conditions, and will reveal areas of pink to white, polymorphous vessels and crystalline structures. However, ultimately biopsy and histopathological evaluation is necessary for accurate diagnosis.
Glomus tumor
Glomus tumors are rare, benign neoplasms originating from cells of the glomus body that presents as a red-purple, vascular papule or nodule. They are usually found in areas rich in glomus bodies, such as the subungual regions, fingertips, palms, wrists, and forearms. Glomus tumors are typically associated with tenderness, paroxysmal pain, and cold sensitivity. They do not bleed or ulcerate. While pyogenic granuloma may be confused for glomus tumor when present on the fingers or extremities, the location of the lesion in our patient is not consistent with a diagnosis of glomus tumor.
Management and disease course
Management with procedural or topical interventions is usually pursued for pyogenic granuloma because of frequent bleeding and ulceration of lesions. The most common approach is simple excision by a scoop or shave technique, with or without curettage and most commonly with electrocautery of the base. Other options include full-thickness excision, destruction with laser therapy, cryotherapy, or topical treatments (for example, timolol).11 Lesion recurrence can occur with both surgical and nonsurgical management.11 Regardless of management technique, it is useful to obtain histopathologic evaluation of tissue for accurate diagnosis.
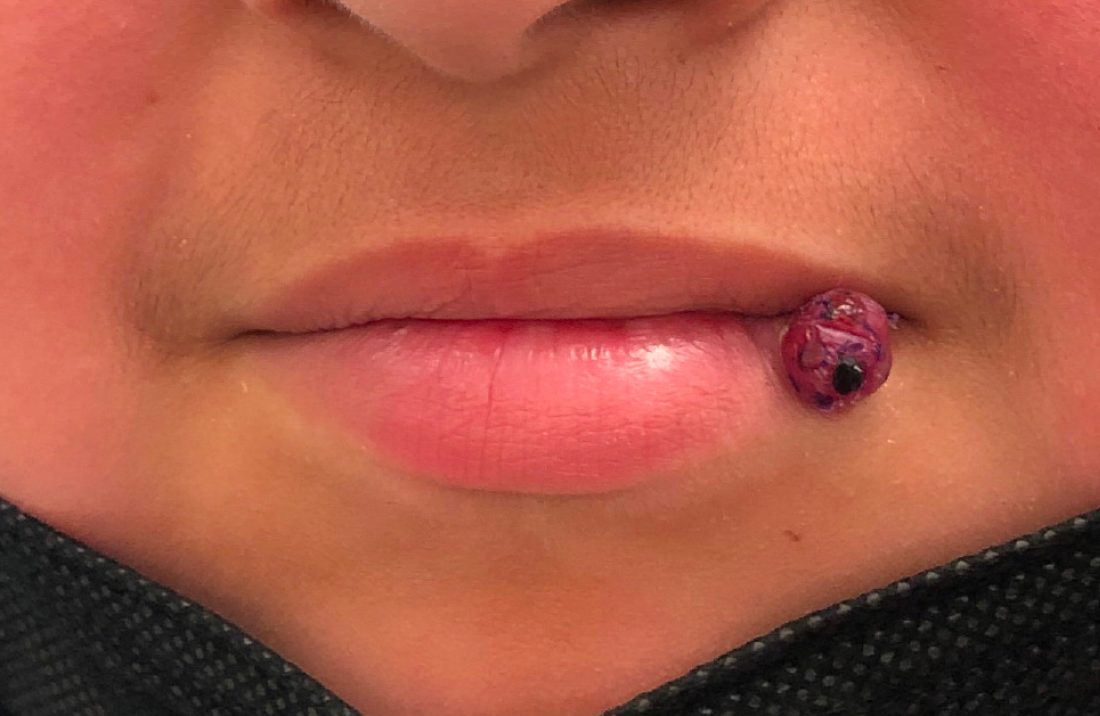
Our patient underwent surgical destruction of her lower-lip lesion with shave excision followed by electrocautery. The surgical specimen was sent for pathology, which confirmed the diagnosis of pyogenic granuloma. The patient experienced no complications from the procedure and did not have recurrence of the lesion.
Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Ms. Sui nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Lin RL and Janniger CK. Cutis. 2004 Oct;74(4):229-33.
2. Harris MN et al. J Am Acad Dermatol. 2000 Jun;42(6):1012-6.
3. Pagliai KA and Cohen BA. Pediatr Dermatol. 2004 Jan-Feb;21(1):10-3.
4. Kamal R et al. J Oral Maxillofac Pathol. 2012 Jan;16(1):79-82.
5. Requena L and Sangueza OP. J Am Acad Dermatol. 1997 Dec;37(6):887-919.
6. Patrice SJ et al. Pediatr Dermatol. 1991 Dec;8(4):267-76.
7. Luba MC et al. Am Fam Physician. 2003 Feb 15;67(4):729-38.
8. Swerlick RA and Cooper PH. J Am Acad Dermatol. 1983 May;8(5):627-30.
9. Sheehan DJ and Lesher JL Jr. Cutis. 2004 Mar;73(3):175-80.
10. Zaballos P et al. Br J Dermatol. 2006 Jun;154(6):1108-11.
11. Lee J et al. J Plast Reconstr Aesthet Surg. 2011 Sep;64(9):1216-20. .
The patient’s history and examination are consistent with a diagnosis of pyogenic granuloma. Specifically, the history of rapid growth, friable nature, associated bleeding, and hemorrhagic crusting point to pyogenic granuloma as the most likely diagnosis.
Pyogenic granuloma is an acquired benign vascular growth of the skin or mucous membranes.1 It most frequently occurs in children and young adults and most commonly affects the skin of the head, trunk, and extremities.2 Common mucosal sites include the gingiva, lips, and tongue.2 The etiology of pyogenic granuloma is unknown, though it is thought to be a process akin to the overgrowth of granulation tissue.3,4 Expression of angiogenic factors and subsequent vascular hyperplasia are also implicated as key players in the pathogenesis of pyogenic granuloma.1,4 In addition, several associated factors and inciting triggers have been proposed including trauma, infections, and hormonal fluctuations.3-5 However, the majority of patients do not report predisposing factors or a history of prior trauma at the site.3,6
Clinically, pyogenic granuloma usually presents as a painless, erythematous, dome-shaped friable papule or nodule that easily bleeds and may ulcerate. It typically undergoes a period of growth over weeks to months followed by stabilization. Occasionally, pyogenic granulomas will spontaneously involute, though most do not.7 Pyogenic granuloma may occur within an existing capillary malformation, such as a port wine stain, spontaneously or as a sequela of laser treatment.8,9 Diagnosis of pyogenic granuloma can typically be made clinically on the basis of history and exam. Dermoscopic evaluation of pyogenic granuloma will reveal a homogeneous papule with a surrounding white-brown collarette, and potentially white intersecting lines.10 Histopathologic evaluation may be necessary to differentiate lesions from conditions that may mimic pyogenic granuloma.
What’s on the differential?
The differential diagnosis for pyogenic granuloma consists of Spitz nevus, cherry hemangioma, amelanotic melanoma, and glomus tumor.
Spitz nevus
Spitz nevus (spindle and epithelial cell nevus) is a benign melanocytic lesion that classically appears as a sharply circumscribed, smooth, dome-shaped, pink-red, or brown papule or plaque. There is typically a history of rapid growth over several months followed by stabilization. It usually presents in childhood or adolescence and is most commonly located on the face and extremities. While there are similarities in the appearance of Spitz nevi and pyogenic granuloma, Spitz nevi are not usually friable nor associated with bleeding as in our patient. Furthermore, on dermoscopy, Spitz nevus typically exhibits a starburst pattern with regularly distributed dotted vessels, or a peripheral globular pattern with reticular depigmentation. The definitive diagnosis of Spitz nevi relies on histopathologic evaluation, which is critical for discriminating Spitz nevi from melanoma.
Cherry hemangioma
Cherry angiomas are the most common type of acquired benign vascular proliferation. They present as small, bright red or violaceous macules or papules. However, they typically appear in early to midadulthood and increase in number with age. The age of our patient and solitary presentation of the lesion make this diagnosis unlikely. In addition, cherry angiomas are not usually associated with bleeding. It is important to note that, depending on the age of the patient, pyogenic granuloma may also be confused with infantile hemangioma. Infantile hemangiomas may become bright red papules, nodules, or plaques that appear in early infancy. They characteristically involute, which does not typically happen with pyogenic granuloma.
Amelanotic melanoma
Amelanotic melanoma is an uncommon variant of melanoma with little to no pigmentation. It may appear as a skin-colored to light-brown, pink, or red macule, papule, or nodule. The lesion may be asymmetric with irregular and well-defined borders. The variable and uncharacteristic appearance of this melanoma variant makes it diagnostically challenging and it is often confused with benign lesions including pyogenic granuloma. Dermoscopy can help distinguish amelanotic melanoma from other benign conditions, and will reveal areas of pink to white, polymorphous vessels and crystalline structures. However, ultimately biopsy and histopathological evaluation is necessary for accurate diagnosis.
Glomus tumor
Glomus tumors are rare, benign neoplasms originating from cells of the glomus body that presents as a red-purple, vascular papule or nodule. They are usually found in areas rich in glomus bodies, such as the subungual regions, fingertips, palms, wrists, and forearms. Glomus tumors are typically associated with tenderness, paroxysmal pain, and cold sensitivity. They do not bleed or ulcerate. While pyogenic granuloma may be confused for glomus tumor when present on the fingers or extremities, the location of the lesion in our patient is not consistent with a diagnosis of glomus tumor.
Management and disease course
Management with procedural or topical interventions is usually pursued for pyogenic granuloma because of frequent bleeding and ulceration of lesions. The most common approach is simple excision by a scoop or shave technique, with or without curettage and most commonly with electrocautery of the base. Other options include full-thickness excision, destruction with laser therapy, cryotherapy, or topical treatments (for example, timolol).11 Lesion recurrence can occur with both surgical and nonsurgical management.11 Regardless of management technique, it is useful to obtain histopathologic evaluation of tissue for accurate diagnosis.
Our patient underwent surgical destruction of her lower-lip lesion with shave excision followed by electrocautery. The surgical specimen was sent for pathology, which confirmed the diagnosis of pyogenic granuloma. The patient experienced no complications from the procedure and did not have recurrence of the lesion.
Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Ms. Sui nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Lin RL and Janniger CK. Cutis. 2004 Oct;74(4):229-33.
2. Harris MN et al. J Am Acad Dermatol. 2000 Jun;42(6):1012-6.
3. Pagliai KA and Cohen BA. Pediatr Dermatol. 2004 Jan-Feb;21(1):10-3.
4. Kamal R et al. J Oral Maxillofac Pathol. 2012 Jan;16(1):79-82.
5. Requena L and Sangueza OP. J Am Acad Dermatol. 1997 Dec;37(6):887-919.
6. Patrice SJ et al. Pediatr Dermatol. 1991 Dec;8(4):267-76.
7. Luba MC et al. Am Fam Physician. 2003 Feb 15;67(4):729-38.
8. Swerlick RA and Cooper PH. J Am Acad Dermatol. 1983 May;8(5):627-30.
9. Sheehan DJ and Lesher JL Jr. Cutis. 2004 Mar;73(3):175-80.
10. Zaballos P et al. Br J Dermatol. 2006 Jun;154(6):1108-11.
11. Lee J et al. J Plast Reconstr Aesthet Surg. 2011 Sep;64(9):1216-20. .
The patient’s history and examination are consistent with a diagnosis of pyogenic granuloma. Specifically, the history of rapid growth, friable nature, associated bleeding, and hemorrhagic crusting point to pyogenic granuloma as the most likely diagnosis.
Pyogenic granuloma is an acquired benign vascular growth of the skin or mucous membranes.1 It most frequently occurs in children and young adults and most commonly affects the skin of the head, trunk, and extremities.2 Common mucosal sites include the gingiva, lips, and tongue.2 The etiology of pyogenic granuloma is unknown, though it is thought to be a process akin to the overgrowth of granulation tissue.3,4 Expression of angiogenic factors and subsequent vascular hyperplasia are also implicated as key players in the pathogenesis of pyogenic granuloma.1,4 In addition, several associated factors and inciting triggers have been proposed including trauma, infections, and hormonal fluctuations.3-5 However, the majority of patients do not report predisposing factors or a history of prior trauma at the site.3,6
Clinically, pyogenic granuloma usually presents as a painless, erythematous, dome-shaped friable papule or nodule that easily bleeds and may ulcerate. It typically undergoes a period of growth over weeks to months followed by stabilization. Occasionally, pyogenic granulomas will spontaneously involute, though most do not.7 Pyogenic granuloma may occur within an existing capillary malformation, such as a port wine stain, spontaneously or as a sequela of laser treatment.8,9 Diagnosis of pyogenic granuloma can typically be made clinically on the basis of history and exam. Dermoscopic evaluation of pyogenic granuloma will reveal a homogeneous papule with a surrounding white-brown collarette, and potentially white intersecting lines.10 Histopathologic evaluation may be necessary to differentiate lesions from conditions that may mimic pyogenic granuloma.
What’s on the differential?
The differential diagnosis for pyogenic granuloma consists of Spitz nevus, cherry hemangioma, amelanotic melanoma, and glomus tumor.
Spitz nevus
Spitz nevus (spindle and epithelial cell nevus) is a benign melanocytic lesion that classically appears as a sharply circumscribed, smooth, dome-shaped, pink-red, or brown papule or plaque. There is typically a history of rapid growth over several months followed by stabilization. It usually presents in childhood or adolescence and is most commonly located on the face and extremities. While there are similarities in the appearance of Spitz nevi and pyogenic granuloma, Spitz nevi are not usually friable nor associated with bleeding as in our patient. Furthermore, on dermoscopy, Spitz nevus typically exhibits a starburst pattern with regularly distributed dotted vessels, or a peripheral globular pattern with reticular depigmentation. The definitive diagnosis of Spitz nevi relies on histopathologic evaluation, which is critical for discriminating Spitz nevi from melanoma.
Cherry hemangioma
Cherry angiomas are the most common type of acquired benign vascular proliferation. They present as small, bright red or violaceous macules or papules. However, they typically appear in early to midadulthood and increase in number with age. The age of our patient and solitary presentation of the lesion make this diagnosis unlikely. In addition, cherry angiomas are not usually associated with bleeding. It is important to note that, depending on the age of the patient, pyogenic granuloma may also be confused with infantile hemangioma. Infantile hemangiomas may become bright red papules, nodules, or plaques that appear in early infancy. They characteristically involute, which does not typically happen with pyogenic granuloma.
Amelanotic melanoma
Amelanotic melanoma is an uncommon variant of melanoma with little to no pigmentation. It may appear as a skin-colored to light-brown, pink, or red macule, papule, or nodule. The lesion may be asymmetric with irregular and well-defined borders. The variable and uncharacteristic appearance of this melanoma variant makes it diagnostically challenging and it is often confused with benign lesions including pyogenic granuloma. Dermoscopy can help distinguish amelanotic melanoma from other benign conditions, and will reveal areas of pink to white, polymorphous vessels and crystalline structures. However, ultimately biopsy and histopathological evaluation is necessary for accurate diagnosis.
Glomus tumor
Glomus tumors are rare, benign neoplasms originating from cells of the glomus body that presents as a red-purple, vascular papule or nodule. They are usually found in areas rich in glomus bodies, such as the subungual regions, fingertips, palms, wrists, and forearms. Glomus tumors are typically associated with tenderness, paroxysmal pain, and cold sensitivity. They do not bleed or ulcerate. While pyogenic granuloma may be confused for glomus tumor when present on the fingers or extremities, the location of the lesion in our patient is not consistent with a diagnosis of glomus tumor.
Management and disease course
Management with procedural or topical interventions is usually pursued for pyogenic granuloma because of frequent bleeding and ulceration of lesions. The most common approach is simple excision by a scoop or shave technique, with or without curettage and most commonly with electrocautery of the base. Other options include full-thickness excision, destruction with laser therapy, cryotherapy, or topical treatments (for example, timolol).11 Lesion recurrence can occur with both surgical and nonsurgical management.11 Regardless of management technique, it is useful to obtain histopathologic evaluation of tissue for accurate diagnosis.
Our patient underwent surgical destruction of her lower-lip lesion with shave excision followed by electrocautery. The surgical specimen was sent for pathology, which confirmed the diagnosis of pyogenic granuloma. The patient experienced no complications from the procedure and did not have recurrence of the lesion.
Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Ms. Sui nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Lin RL and Janniger CK. Cutis. 2004 Oct;74(4):229-33.
2. Harris MN et al. J Am Acad Dermatol. 2000 Jun;42(6):1012-6.
3. Pagliai KA and Cohen BA. Pediatr Dermatol. 2004 Jan-Feb;21(1):10-3.
4. Kamal R et al. J Oral Maxillofac Pathol. 2012 Jan;16(1):79-82.
5. Requena L and Sangueza OP. J Am Acad Dermatol. 1997 Dec;37(6):887-919.
6. Patrice SJ et al. Pediatr Dermatol. 1991 Dec;8(4):267-76.
7. Luba MC et al. Am Fam Physician. 2003 Feb 15;67(4):729-38.
8. Swerlick RA and Cooper PH. J Am Acad Dermatol. 1983 May;8(5):627-30.
9. Sheehan DJ and Lesher JL Jr. Cutis. 2004 Mar;73(3):175-80.
10. Zaballos P et al. Br J Dermatol. 2006 Jun;154(6):1108-11.
11. Lee J et al. J Plast Reconstr Aesthet Surg. 2011 Sep;64(9):1216-20. .
Polycyclic Scaly Eruption
The Diagnosis: Netherton Syndrome
A punch biopsy from the right lower back supported the clinical diagnosis of ichthyosis linearis circumflexa. The patient underwent genetic testing and was found to have a heterozygous mutation in the serine protease inhibitor Kazal type 5 gene, SPINK5, that was consistent with a diagnosis of Netherton syndrome.
Netherton syndrome is an autosomal-recessive genodermatosis characterized by a triad of congenital ichthyosis, hair shaft abnormalities, and atopic diatheses.1,2 It affects approximately 1 in 200,000 live births2,3; however, it is considered by many to be underdiagnosed due to the variability in the clinical appearance. Therefore, the incidence of Netherton syndrome may actually be closer 1 in 50,000 live births.1 The manifestations of the disease are caused by a germline mutation in the SPINK5 gene, which encodes the serine protease inhibitor LEKTI.1,2 Dysfunctional LEKTI results in increased proteolytic activity of the lipid-processing enzymes in the stratum corneum, resulting in a disruption in the lipid bilayer.1 Dysfunctional LEKTI also results in a loss of the antiinflammatory and antimicrobial function of the stratum corneum. Clinical features of Netherton syndrome usually present at birth or shortly thereafter.1 Congenital ichthyosiform erythroderma, or the continuous peeling of the skin, is a common presentation seen at birth and in the neonatal period.2 As the patient ages, the dermatologic manifestations evolve into serpiginous and circinate, erythematous plaques with a characteristic peripheral, double-edged scaling.1,2 This distinctive finding is termed ichthyosis linearis circumflexa and is pathognomonic for the syndrome.2 Lesions often affect the trunk and extremities and demonstrate an undulating course.1 Because eczematous and lichenified plaques in flexural areas as well as pruritus are common clinical features, this disease often is misdiagnosed as atopic dermatitis,1,3 as was the case in our patient.
Patients with Netherton syndrome can present with various hair abnormalities. Trichorrhexis invaginata, known as bamboo hair, is the intussusception of the hair shaft and is characteristic of the disease.3 It develops from a reduced number of disulfide bonds, which results in cortical softening.1 Trichorrhexis invaginata may not be present at birth and often improves with age.1,3 Other hair shaft abnormalities such as pili torti, trichorrhexis nodosa, and helical hair also may be observed in Netherton syndrome.1 Extracutaneous manifestations also are typical. There is immune dysregulation of memory B cells and natural killer cells, which manifests as frequent respiratory and skin infections as well as sepsis.1,2 Patients also may have increased levels of serum IgE and eosinophilia resulting in atopy and allergic reactions to various triggers such as foods.1 The neonatal period also may be complicated by dehydration, electrolyte imbalances, inability to regulate body temperature, and failure to thrive.1,3
When there is an extensive disruption of the skin barrier during the neonatal period, there may be severe electrolyte imbalances and thermoregulatory challenges necessitating treatment in the neonatal intensive care unit. Cutaneous disease can be treated with topical therapies with variable success.1 Topical therapies for symptom management include emollients, corticosteroids, calcineurin inhibitors, calcipotriene, and retinoids; however, utmost caution must be employed with these therapies due to the increased risk for systemic absorption resulting from the disturbance of the skin barrier. When therapy with topical tacrolimus is implemented, monitoring of serum drug levels is required.1 Pruritus may be treated symptomatically with oral antihistamines. Intravenous immunoglobulin has been shown to decrease the frequency of infections and improve skin inflammation. Systemic retinoids have unpredictable effects and result in improvement of disease in some patients but exacerbation in others. Phototherapy with narrowband UVB, psoralen plus UVA, UVA1, and balneophototherapy also are effective treatments for cutaneous disease.1 Dupilumab has been shown to decrease pruritus, improve hair abnormalities, and improve skin disease, thereby demonstrating its effectiveness in treating the atopy and ichthyosis in Netherton syndrome.4
The differential diagnosis includes other figurate erythemas including erythema marginatum and erythrokeratodermia variabilis. Erythema marginatum is a cutaneous manifestation of acute rheumatic fever and is characterized by migratory polycyclic erythematous plaques without overlying scale, usually on the trunk and proximal extremities.5 Erythrokeratodermia variabilis is caused by heterozygous mutations in gap junction protein beta 3, GJB3, and gap junction protein beta 4, GJB4, and is characterized by transient geographic and erythematous patches and stable scaly plaques; however, double-edged scaling is not a feature.1 Acrodermatitis enteropathica is an autosomal-recessive disorder caused by mutations in the zinc transporter SLC39A4. Cutaneous manifestations occur after weaning from breast milk and are characterized by erythematous plaques with erosions, vesicles, and scaling, which characteristically occur in the perioral and perianal locations.6 Neonatal lupus is a form of subacute cutaneous lupus erythematosus. Typical skin lesions are erythematous annular plaques with overlying scaling, which may be present at birth and have a predilection for the face and other sun-exposed areas. Lesions generally resolve after clearance of the pathogenic maternal antibodies.7
- Richard G, Ringpfeil F. Ichthyoses, erythrokeratodermas, and related disorders. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:888-923.
- Garza JI, Herz-Ruelas ME, Guerrero-González GA, et al. Netherton syndrome: a diagnostic and therapeutic challenge. J Am Acad Dermatol. 2016;74(suppl 1):AB129.
- Heymann W. Appending the appendages: new perspectives on Netherton syndrome and green nail syndrome. J Am Acad Dermatol. 2020;83:735-736.
- Murase C, Takeichi T, Taki T, et al. Successful dupilumab treatment for ichthyotic and atopic features of Netherton syndrome. J Dermatol Sci. 2021;102:126-129.
- España A. Figurate erythemas. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:320-331.
- Noguera-Morel L, McLeish Schaefer S, Hivnor C. Nutritional diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:793-809.
- Lee L, Werth V. Lupus erythematosus. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:662-680.
The Diagnosis: Netherton Syndrome
A punch biopsy from the right lower back supported the clinical diagnosis of ichthyosis linearis circumflexa. The patient underwent genetic testing and was found to have a heterozygous mutation in the serine protease inhibitor Kazal type 5 gene, SPINK5, that was consistent with a diagnosis of Netherton syndrome.
Netherton syndrome is an autosomal-recessive genodermatosis characterized by a triad of congenital ichthyosis, hair shaft abnormalities, and atopic diatheses.1,2 It affects approximately 1 in 200,000 live births2,3; however, it is considered by many to be underdiagnosed due to the variability in the clinical appearance. Therefore, the incidence of Netherton syndrome may actually be closer 1 in 50,000 live births.1 The manifestations of the disease are caused by a germline mutation in the SPINK5 gene, which encodes the serine protease inhibitor LEKTI.1,2 Dysfunctional LEKTI results in increased proteolytic activity of the lipid-processing enzymes in the stratum corneum, resulting in a disruption in the lipid bilayer.1 Dysfunctional LEKTI also results in a loss of the antiinflammatory and antimicrobial function of the stratum corneum. Clinical features of Netherton syndrome usually present at birth or shortly thereafter.1 Congenital ichthyosiform erythroderma, or the continuous peeling of the skin, is a common presentation seen at birth and in the neonatal period.2 As the patient ages, the dermatologic manifestations evolve into serpiginous and circinate, erythematous plaques with a characteristic peripheral, double-edged scaling.1,2 This distinctive finding is termed ichthyosis linearis circumflexa and is pathognomonic for the syndrome.2 Lesions often affect the trunk and extremities and demonstrate an undulating course.1 Because eczematous and lichenified plaques in flexural areas as well as pruritus are common clinical features, this disease often is misdiagnosed as atopic dermatitis,1,3 as was the case in our patient.
Patients with Netherton syndrome can present with various hair abnormalities. Trichorrhexis invaginata, known as bamboo hair, is the intussusception of the hair shaft and is characteristic of the disease.3 It develops from a reduced number of disulfide bonds, which results in cortical softening.1 Trichorrhexis invaginata may not be present at birth and often improves with age.1,3 Other hair shaft abnormalities such as pili torti, trichorrhexis nodosa, and helical hair also may be observed in Netherton syndrome.1 Extracutaneous manifestations also are typical. There is immune dysregulation of memory B cells and natural killer cells, which manifests as frequent respiratory and skin infections as well as sepsis.1,2 Patients also may have increased levels of serum IgE and eosinophilia resulting in atopy and allergic reactions to various triggers such as foods.1 The neonatal period also may be complicated by dehydration, electrolyte imbalances, inability to regulate body temperature, and failure to thrive.1,3
When there is an extensive disruption of the skin barrier during the neonatal period, there may be severe electrolyte imbalances and thermoregulatory challenges necessitating treatment in the neonatal intensive care unit. Cutaneous disease can be treated with topical therapies with variable success.1 Topical therapies for symptom management include emollients, corticosteroids, calcineurin inhibitors, calcipotriene, and retinoids; however, utmost caution must be employed with these therapies due to the increased risk for systemic absorption resulting from the disturbance of the skin barrier. When therapy with topical tacrolimus is implemented, monitoring of serum drug levels is required.1 Pruritus may be treated symptomatically with oral antihistamines. Intravenous immunoglobulin has been shown to decrease the frequency of infections and improve skin inflammation. Systemic retinoids have unpredictable effects and result in improvement of disease in some patients but exacerbation in others. Phototherapy with narrowband UVB, psoralen plus UVA, UVA1, and balneophototherapy also are effective treatments for cutaneous disease.1 Dupilumab has been shown to decrease pruritus, improve hair abnormalities, and improve skin disease, thereby demonstrating its effectiveness in treating the atopy and ichthyosis in Netherton syndrome.4
The differential diagnosis includes other figurate erythemas including erythema marginatum and erythrokeratodermia variabilis. Erythema marginatum is a cutaneous manifestation of acute rheumatic fever and is characterized by migratory polycyclic erythematous plaques without overlying scale, usually on the trunk and proximal extremities.5 Erythrokeratodermia variabilis is caused by heterozygous mutations in gap junction protein beta 3, GJB3, and gap junction protein beta 4, GJB4, and is characterized by transient geographic and erythematous patches and stable scaly plaques; however, double-edged scaling is not a feature.1 Acrodermatitis enteropathica is an autosomal-recessive disorder caused by mutations in the zinc transporter SLC39A4. Cutaneous manifestations occur after weaning from breast milk and are characterized by erythematous plaques with erosions, vesicles, and scaling, which characteristically occur in the perioral and perianal locations.6 Neonatal lupus is a form of subacute cutaneous lupus erythematosus. Typical skin lesions are erythematous annular plaques with overlying scaling, which may be present at birth and have a predilection for the face and other sun-exposed areas. Lesions generally resolve after clearance of the pathogenic maternal antibodies.7
The Diagnosis: Netherton Syndrome
A punch biopsy from the right lower back supported the clinical diagnosis of ichthyosis linearis circumflexa. The patient underwent genetic testing and was found to have a heterozygous mutation in the serine protease inhibitor Kazal type 5 gene, SPINK5, that was consistent with a diagnosis of Netherton syndrome.
Netherton syndrome is an autosomal-recessive genodermatosis characterized by a triad of congenital ichthyosis, hair shaft abnormalities, and atopic diatheses.1,2 It affects approximately 1 in 200,000 live births2,3; however, it is considered by many to be underdiagnosed due to the variability in the clinical appearance. Therefore, the incidence of Netherton syndrome may actually be closer 1 in 50,000 live births.1 The manifestations of the disease are caused by a germline mutation in the SPINK5 gene, which encodes the serine protease inhibitor LEKTI.1,2 Dysfunctional LEKTI results in increased proteolytic activity of the lipid-processing enzymes in the stratum corneum, resulting in a disruption in the lipid bilayer.1 Dysfunctional LEKTI also results in a loss of the antiinflammatory and antimicrobial function of the stratum corneum. Clinical features of Netherton syndrome usually present at birth or shortly thereafter.1 Congenital ichthyosiform erythroderma, or the continuous peeling of the skin, is a common presentation seen at birth and in the neonatal period.2 As the patient ages, the dermatologic manifestations evolve into serpiginous and circinate, erythematous plaques with a characteristic peripheral, double-edged scaling.1,2 This distinctive finding is termed ichthyosis linearis circumflexa and is pathognomonic for the syndrome.2 Lesions often affect the trunk and extremities and demonstrate an undulating course.1 Because eczematous and lichenified plaques in flexural areas as well as pruritus are common clinical features, this disease often is misdiagnosed as atopic dermatitis,1,3 as was the case in our patient.
Patients with Netherton syndrome can present with various hair abnormalities. Trichorrhexis invaginata, known as bamboo hair, is the intussusception of the hair shaft and is characteristic of the disease.3 It develops from a reduced number of disulfide bonds, which results in cortical softening.1 Trichorrhexis invaginata may not be present at birth and often improves with age.1,3 Other hair shaft abnormalities such as pili torti, trichorrhexis nodosa, and helical hair also may be observed in Netherton syndrome.1 Extracutaneous manifestations also are typical. There is immune dysregulation of memory B cells and natural killer cells, which manifests as frequent respiratory and skin infections as well as sepsis.1,2 Patients also may have increased levels of serum IgE and eosinophilia resulting in atopy and allergic reactions to various triggers such as foods.1 The neonatal period also may be complicated by dehydration, electrolyte imbalances, inability to regulate body temperature, and failure to thrive.1,3
When there is an extensive disruption of the skin barrier during the neonatal period, there may be severe electrolyte imbalances and thermoregulatory challenges necessitating treatment in the neonatal intensive care unit. Cutaneous disease can be treated with topical therapies with variable success.1 Topical therapies for symptom management include emollients, corticosteroids, calcineurin inhibitors, calcipotriene, and retinoids; however, utmost caution must be employed with these therapies due to the increased risk for systemic absorption resulting from the disturbance of the skin barrier. When therapy with topical tacrolimus is implemented, monitoring of serum drug levels is required.1 Pruritus may be treated symptomatically with oral antihistamines. Intravenous immunoglobulin has been shown to decrease the frequency of infections and improve skin inflammation. Systemic retinoids have unpredictable effects and result in improvement of disease in some patients but exacerbation in others. Phototherapy with narrowband UVB, psoralen plus UVA, UVA1, and balneophototherapy also are effective treatments for cutaneous disease.1 Dupilumab has been shown to decrease pruritus, improve hair abnormalities, and improve skin disease, thereby demonstrating its effectiveness in treating the atopy and ichthyosis in Netherton syndrome.4
The differential diagnosis includes other figurate erythemas including erythema marginatum and erythrokeratodermia variabilis. Erythema marginatum is a cutaneous manifestation of acute rheumatic fever and is characterized by migratory polycyclic erythematous plaques without overlying scale, usually on the trunk and proximal extremities.5 Erythrokeratodermia variabilis is caused by heterozygous mutations in gap junction protein beta 3, GJB3, and gap junction protein beta 4, GJB4, and is characterized by transient geographic and erythematous patches and stable scaly plaques; however, double-edged scaling is not a feature.1 Acrodermatitis enteropathica is an autosomal-recessive disorder caused by mutations in the zinc transporter SLC39A4. Cutaneous manifestations occur after weaning from breast milk and are characterized by erythematous plaques with erosions, vesicles, and scaling, which characteristically occur in the perioral and perianal locations.6 Neonatal lupus is a form of subacute cutaneous lupus erythematosus. Typical skin lesions are erythematous annular plaques with overlying scaling, which may be present at birth and have a predilection for the face and other sun-exposed areas. Lesions generally resolve after clearance of the pathogenic maternal antibodies.7
- Richard G, Ringpfeil F. Ichthyoses, erythrokeratodermas, and related disorders. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:888-923.
- Garza JI, Herz-Ruelas ME, Guerrero-González GA, et al. Netherton syndrome: a diagnostic and therapeutic challenge. J Am Acad Dermatol. 2016;74(suppl 1):AB129.
- Heymann W. Appending the appendages: new perspectives on Netherton syndrome and green nail syndrome. J Am Acad Dermatol. 2020;83:735-736.
- Murase C, Takeichi T, Taki T, et al. Successful dupilumab treatment for ichthyotic and atopic features of Netherton syndrome. J Dermatol Sci. 2021;102:126-129.
- España A. Figurate erythemas. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:320-331.
- Noguera-Morel L, McLeish Schaefer S, Hivnor C. Nutritional diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:793-809.
- Lee L, Werth V. Lupus erythematosus. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:662-680.
- Richard G, Ringpfeil F. Ichthyoses, erythrokeratodermas, and related disorders. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:888-923.
- Garza JI, Herz-Ruelas ME, Guerrero-González GA, et al. Netherton syndrome: a diagnostic and therapeutic challenge. J Am Acad Dermatol. 2016;74(suppl 1):AB129.
- Heymann W. Appending the appendages: new perspectives on Netherton syndrome and green nail syndrome. J Am Acad Dermatol. 2020;83:735-736.
- Murase C, Takeichi T, Taki T, et al. Successful dupilumab treatment for ichthyotic and atopic features of Netherton syndrome. J Dermatol Sci. 2021;102:126-129.
- España A. Figurate erythemas. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:320-331.
- Noguera-Morel L, McLeish Schaefer S, Hivnor C. Nutritional diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:793-809.
- Lee L, Werth V. Lupus erythematosus. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:662-680.
A 9-year-old boy presented to the dermatology clinic with a scaly eruption distributed throughout the body that had been present since birth. He had been diagnosed with atopic dermatitis by multiple dermatologists prior to the current presentation and had been treated with various topical steroids with minimal improvement. He had no family history of similar eruptions and no personal history of asthma or allergies. Physical examination revealed erythematous, serpiginous, polycyclic plaques with peripheral, double-edged scaling. Decreased hair density of the lateral eyebrows also was observed.


A 9-year-old girl was evaluated for a week-long history of rash on the feet
A complete body examination failed to reveal any other lesions suggestive of a fungal infection. A blood count and urinalysis were within normal limits. She had no lymphadenopathy or hepatosplenomegaly. She was diagnosed with cutaneous larva migrans (CLM) given the clinical appearance of the lesions and the recent travel history.
CLM is a zoonotic infection caused by several hookworms such as Ancylostoma braziliense, Ancylostoma caninum, and Uncinaria stenocephala, as well as human hookworms such as Ancylostoma duodenale and Necator americanus. The hookworms can be present in contaminated soils and sandy beaches on the coastal regions of South America, the Caribbean, the Southeastern United States, Southeast Asia, and Africa.1-5
It is a common disease in the tourist population visiting tropical countries because of exposure to the hookworms in the soil without use of proper foot protection.
The clinical features are of an erythematous linear serpiginous plaque that is pruritic and can progress from millimeters to centimeters in size within a few days to weeks. Vesicles and multiple tracks can also be seen. The most common locations are the feet, buttocks, and thighs.
The larvae in the soil come from eggs excreted in the feces of infected cats and dogs. The infection is caused by direct contact of the larvae with the stratum corneum of the skin creating a burrow and an inflammatory response that will cause erythema, edema, track formation, and pruritus.
Diagnosis is made clinically. Rarely, a skin biopsy is warranted. The differential diagnosis includes tinea pedis, granuloma annulare, larva currens, contact dermatitis, and herpes zoster.

Tinea pedis is a fungal infection of the skin of the feet, commonly localized on the web spaces. The risk factors are a hot and humid environment, prolonged wear of occlusive footwear, excess sweating, and prolonged exposure to water.6 Diagnosis is confirmed by microscopic evaluation of skin scrapings with potassium hydroxide or a fungal culture. The infection is treated with topical antifungal creams and, in severe cases, systemic antifungals. Granuloma annulare is a benign chronic skin condition that presents with annular-shaped lesions. Its etiology is unknown. The lesions may be asymptomatic or mildly pruritic. Localized granuloma annulare typically presents as reddish-brown papules or plaques on the fingers, hands, elbows, dorsal feet, or ankles. The feature distinguishing granuloma annulare from other annular lesions is its absence of scale.
Allergic contact dermatitis is caused by skin exposure to an allergen and a secondary inflammatory response to this material on the skin causing inflammation, vesiculation, and pruritus. Lesions are treated with topical corticosteroids and avoidance of the allergen.
Herpes zoster is caused by a viral infection of the latent varicella-zoster virus. Its reactivation causes the presence of vesicles with an erythematous base that have a dermatomal distribution. The lesions are usually tender. Treatment is recommended to be started within 72 hours of the eruption with antivirals such as acyclovir or valacyclovir.
Cutaneous larva currens is caused by the cutaneous infection with Strongyloides stercoralis. In comparison with CLM, the lesions progress faster, at up to a centimeter within hours.
CLM is usually self-limited. If the patient has multiple lesions or more severe disease, oral albendazole or ivermectin can be prescribed. Other treatments, though not preferred, include freezing and topical thiabendazole solutions.
As our patient had several lesions, oral ivermectin was chosen as treatment and the lesions cleared within a week. Also, she was recommended to always wear shoes when walking on the beach.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Dr. Valderrama is a pediatric dermatologist at Fundación Cardioinfantil, Bogota, Colombia.
References
1. Feldmeier H and Schuster A. Eur J Clin Microbiol Infect Dis. 2012 Jun;31(6):915-8.
2. Jacobson CC and Abel EA. J Am Acad Dermatol. 2007 Jun;56(6):1026-43.
3. Kincaid L et al. Travel Med Infect Dis. 2015 Sep-Oct;13(5):382-7.
4. Gill N et al. Adv Skin Wound Care. 2020 Jul;33(7):356-9.
5. Rodenas-Herranz T et al. Dermatol Ther. 2020 May;33(3):e13316.
6. Pramod K et al. In: StatPearls [Internet]. Treasure Island (Fla): StatPearls Publishing; 2022 Jan.
A complete body examination failed to reveal any other lesions suggestive of a fungal infection. A blood count and urinalysis were within normal limits. She had no lymphadenopathy or hepatosplenomegaly. She was diagnosed with cutaneous larva migrans (CLM) given the clinical appearance of the lesions and the recent travel history.
CLM is a zoonotic infection caused by several hookworms such as Ancylostoma braziliense, Ancylostoma caninum, and Uncinaria stenocephala, as well as human hookworms such as Ancylostoma duodenale and Necator americanus. The hookworms can be present in contaminated soils and sandy beaches on the coastal regions of South America, the Caribbean, the Southeastern United States, Southeast Asia, and Africa.1-5
It is a common disease in the tourist population visiting tropical countries because of exposure to the hookworms in the soil without use of proper foot protection.
The clinical features are of an erythematous linear serpiginous plaque that is pruritic and can progress from millimeters to centimeters in size within a few days to weeks. Vesicles and multiple tracks can also be seen. The most common locations are the feet, buttocks, and thighs.
The larvae in the soil come from eggs excreted in the feces of infected cats and dogs. The infection is caused by direct contact of the larvae with the stratum corneum of the skin creating a burrow and an inflammatory response that will cause erythema, edema, track formation, and pruritus.
Diagnosis is made clinically. Rarely, a skin biopsy is warranted. The differential diagnosis includes tinea pedis, granuloma annulare, larva currens, contact dermatitis, and herpes zoster.
Tinea pedis is a fungal infection of the skin of the feet, commonly localized on the web spaces. The risk factors are a hot and humid environment, prolonged wear of occlusive footwear, excess sweating, and prolonged exposure to water.6 Diagnosis is confirmed by microscopic evaluation of skin scrapings with potassium hydroxide or a fungal culture. The infection is treated with topical antifungal creams and, in severe cases, systemic antifungals. Granuloma annulare is a benign chronic skin condition that presents with annular-shaped lesions. Its etiology is unknown. The lesions may be asymptomatic or mildly pruritic. Localized granuloma annulare typically presents as reddish-brown papules or plaques on the fingers, hands, elbows, dorsal feet, or ankles. The feature distinguishing granuloma annulare from other annular lesions is its absence of scale.
Allergic contact dermatitis is caused by skin exposure to an allergen and a secondary inflammatory response to this material on the skin causing inflammation, vesiculation, and pruritus. Lesions are treated with topical corticosteroids and avoidance of the allergen.
Herpes zoster is caused by a viral infection of the latent varicella-zoster virus. Its reactivation causes the presence of vesicles with an erythematous base that have a dermatomal distribution. The lesions are usually tender. Treatment is recommended to be started within 72 hours of the eruption with antivirals such as acyclovir or valacyclovir.
Cutaneous larva currens is caused by the cutaneous infection with Strongyloides stercoralis. In comparison with CLM, the lesions progress faster, at up to a centimeter within hours.
CLM is usually self-limited. If the patient has multiple lesions or more severe disease, oral albendazole or ivermectin can be prescribed. Other treatments, though not preferred, include freezing and topical thiabendazole solutions.
As our patient had several lesions, oral ivermectin was chosen as treatment and the lesions cleared within a week. Also, she was recommended to always wear shoes when walking on the beach.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Dr. Valderrama is a pediatric dermatologist at Fundación Cardioinfantil, Bogota, Colombia.
References
1. Feldmeier H and Schuster A. Eur J Clin Microbiol Infect Dis. 2012 Jun;31(6):915-8.
2. Jacobson CC and Abel EA. J Am Acad Dermatol. 2007 Jun;56(6):1026-43.
3. Kincaid L et al. Travel Med Infect Dis. 2015 Sep-Oct;13(5):382-7.
4. Gill N et al. Adv Skin Wound Care. 2020 Jul;33(7):356-9.
5. Rodenas-Herranz T et al. Dermatol Ther. 2020 May;33(3):e13316.
6. Pramod K et al. In: StatPearls [Internet]. Treasure Island (Fla): StatPearls Publishing; 2022 Jan.
A complete body examination failed to reveal any other lesions suggestive of a fungal infection. A blood count and urinalysis were within normal limits. She had no lymphadenopathy or hepatosplenomegaly. She was diagnosed with cutaneous larva migrans (CLM) given the clinical appearance of the lesions and the recent travel history.
CLM is a zoonotic infection caused by several hookworms such as Ancylostoma braziliense, Ancylostoma caninum, and Uncinaria stenocephala, as well as human hookworms such as Ancylostoma duodenale and Necator americanus. The hookworms can be present in contaminated soils and sandy beaches on the coastal regions of South America, the Caribbean, the Southeastern United States, Southeast Asia, and Africa.1-5
It is a common disease in the tourist population visiting tropical countries because of exposure to the hookworms in the soil without use of proper foot protection.
The clinical features are of an erythematous linear serpiginous plaque that is pruritic and can progress from millimeters to centimeters in size within a few days to weeks. Vesicles and multiple tracks can also be seen. The most common locations are the feet, buttocks, and thighs.
The larvae in the soil come from eggs excreted in the feces of infected cats and dogs. The infection is caused by direct contact of the larvae with the stratum corneum of the skin creating a burrow and an inflammatory response that will cause erythema, edema, track formation, and pruritus.
Diagnosis is made clinically. Rarely, a skin biopsy is warranted. The differential diagnosis includes tinea pedis, granuloma annulare, larva currens, contact dermatitis, and herpes zoster.
Tinea pedis is a fungal infection of the skin of the feet, commonly localized on the web spaces. The risk factors are a hot and humid environment, prolonged wear of occlusive footwear, excess sweating, and prolonged exposure to water.6 Diagnosis is confirmed by microscopic evaluation of skin scrapings with potassium hydroxide or a fungal culture. The infection is treated with topical antifungal creams and, in severe cases, systemic antifungals. Granuloma annulare is a benign chronic skin condition that presents with annular-shaped lesions. Its etiology is unknown. The lesions may be asymptomatic or mildly pruritic. Localized granuloma annulare typically presents as reddish-brown papules or plaques on the fingers, hands, elbows, dorsal feet, or ankles. The feature distinguishing granuloma annulare from other annular lesions is its absence of scale.
Allergic contact dermatitis is caused by skin exposure to an allergen and a secondary inflammatory response to this material on the skin causing inflammation, vesiculation, and pruritus. Lesions are treated with topical corticosteroids and avoidance of the allergen.
Herpes zoster is caused by a viral infection of the latent varicella-zoster virus. Its reactivation causes the presence of vesicles with an erythematous base that have a dermatomal distribution. The lesions are usually tender. Treatment is recommended to be started within 72 hours of the eruption with antivirals such as acyclovir or valacyclovir.
Cutaneous larva currens is caused by the cutaneous infection with Strongyloides stercoralis. In comparison with CLM, the lesions progress faster, at up to a centimeter within hours.
CLM is usually self-limited. If the patient has multiple lesions or more severe disease, oral albendazole or ivermectin can be prescribed. Other treatments, though not preferred, include freezing and topical thiabendazole solutions.
As our patient had several lesions, oral ivermectin was chosen as treatment and the lesions cleared within a week. Also, she was recommended to always wear shoes when walking on the beach.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Dr. Valderrama is a pediatric dermatologist at Fundación Cardioinfantil, Bogota, Colombia.
References
1. Feldmeier H and Schuster A. Eur J Clin Microbiol Infect Dis. 2012 Jun;31(6):915-8.
2. Jacobson CC and Abel EA. J Am Acad Dermatol. 2007 Jun;56(6):1026-43.
3. Kincaid L et al. Travel Med Infect Dis. 2015 Sep-Oct;13(5):382-7.
4. Gill N et al. Adv Skin Wound Care. 2020 Jul;33(7):356-9.
5. Rodenas-Herranz T et al. Dermatol Ther. 2020 May;33(3):e13316.
6. Pramod K et al. In: StatPearls [Internet]. Treasure Island (Fla): StatPearls Publishing; 2022 Jan.
Her mother reported recent travel to a beachside city in Colombia. A review of systems was negative. She was not taking any other medications or vitamin supplements. There were no pets at home and no other affected family members. Physical exam was notable for an erythematous curvilinear plaque on the feet and a small vesicle.