User login
Clozapine interrupted: APA, others seek FDA forum on REMS
In a Feb. 14 letter, the groups asked the FDA to reconsider its new risk evaluation and mitigation strategy (REMS) for clozapine because of concerns it had the potential to cause abrupt discontinuation of the medication.
The groups cite an Institute for Safe Medication Practices (ISMP) report of a 40-year-old woman who was a long-time clozapine user, had a cardiac arrest, and died after she stopped taking the drug because her psychiatrist was unable to register for the updated version of the REMS.
“It is unacceptable for a REMS with unproven effectiveness at meeting its goal to carry risks of interruptions that can result in rehospitalization, acute exacerbation of psychosis, increased risk of suicide, and potentially fatal orthostatic hypotension/bradycardic syndromes associated with incorrect restarts,” the groups said in the letter.
“We feel certain that this case reported in the literature is not the only serious adverse outcome from the REMS and the transition,” they added.
The letter was signed by the American Psychiatric Association, the American Association for Community Psychiatry, the American Psychiatric Nurses Association, the College of Psychiatric and Neurologic Pharmacists, the National Alliance on Mental Illness, the National Association of State Mental Health Program Directors, and the National Council for Mental Wellbeing.
Clozapine can decrease the neutrophil count, which can lead to severe neutropenia, serious infection, and death. Consequently, the FDA put additional safety measures in place governing clozapine prescribing.
In 2015, a centralized clozapine REMS replaced separate prescribing registries that the drug manufacturers maintained. There were technical issues with the 2015 start-up of that website, including data migration problems and long call wait times, the FDA said.
Subsequently, the drug’s manufacturers then decided to change the REMS platform, which created new issues that led to high call volume and long wait times for clinicians and pharmacists who were trying to enroll.
Maintaining access
In November 2021, the FDA announced it would put some aspects of a planned switch on hold. A month later, the agency made further modifications to its plan.
The FDA said it would exercise “enforcement discretion” to try to maintain access to clozapine amid hitches with the REMS transition efforts. The agency also said at the time that it would not object if pharmacists dispensed clozapine without the usual authorization. In addition, wholesalers could ship the drug to pharmacies and health care settings without confirming REMS enrollment.
The FDA also held two December meetings to allow various stakeholders to air concerns.
In their letter, the APA and other groups asked if the FDA intends to continue with accommodations, such as allowing pharmacies to order clozapine from wholesalers without restriction.
“We do not feel the issues are resolved,” the groups said.
A version of this article first appeared on Medscape.com.
In a Feb. 14 letter, the groups asked the FDA to reconsider its new risk evaluation and mitigation strategy (REMS) for clozapine because of concerns it had the potential to cause abrupt discontinuation of the medication.
The groups cite an Institute for Safe Medication Practices (ISMP) report of a 40-year-old woman who was a long-time clozapine user, had a cardiac arrest, and died after she stopped taking the drug because her psychiatrist was unable to register for the updated version of the REMS.
“It is unacceptable for a REMS with unproven effectiveness at meeting its goal to carry risks of interruptions that can result in rehospitalization, acute exacerbation of psychosis, increased risk of suicide, and potentially fatal orthostatic hypotension/bradycardic syndromes associated with incorrect restarts,” the groups said in the letter.
“We feel certain that this case reported in the literature is not the only serious adverse outcome from the REMS and the transition,” they added.
The letter was signed by the American Psychiatric Association, the American Association for Community Psychiatry, the American Psychiatric Nurses Association, the College of Psychiatric and Neurologic Pharmacists, the National Alliance on Mental Illness, the National Association of State Mental Health Program Directors, and the National Council for Mental Wellbeing.
Clozapine can decrease the neutrophil count, which can lead to severe neutropenia, serious infection, and death. Consequently, the FDA put additional safety measures in place governing clozapine prescribing.
In 2015, a centralized clozapine REMS replaced separate prescribing registries that the drug manufacturers maintained. There were technical issues with the 2015 start-up of that website, including data migration problems and long call wait times, the FDA said.
Subsequently, the drug’s manufacturers then decided to change the REMS platform, which created new issues that led to high call volume and long wait times for clinicians and pharmacists who were trying to enroll.
Maintaining access
In November 2021, the FDA announced it would put some aspects of a planned switch on hold. A month later, the agency made further modifications to its plan.
The FDA said it would exercise “enforcement discretion” to try to maintain access to clozapine amid hitches with the REMS transition efforts. The agency also said at the time that it would not object if pharmacists dispensed clozapine without the usual authorization. In addition, wholesalers could ship the drug to pharmacies and health care settings without confirming REMS enrollment.
The FDA also held two December meetings to allow various stakeholders to air concerns.
In their letter, the APA and other groups asked if the FDA intends to continue with accommodations, such as allowing pharmacies to order clozapine from wholesalers without restriction.
“We do not feel the issues are resolved,” the groups said.
A version of this article first appeared on Medscape.com.
In a Feb. 14 letter, the groups asked the FDA to reconsider its new risk evaluation and mitigation strategy (REMS) for clozapine because of concerns it had the potential to cause abrupt discontinuation of the medication.
The groups cite an Institute for Safe Medication Practices (ISMP) report of a 40-year-old woman who was a long-time clozapine user, had a cardiac arrest, and died after she stopped taking the drug because her psychiatrist was unable to register for the updated version of the REMS.
“It is unacceptable for a REMS with unproven effectiveness at meeting its goal to carry risks of interruptions that can result in rehospitalization, acute exacerbation of psychosis, increased risk of suicide, and potentially fatal orthostatic hypotension/bradycardic syndromes associated with incorrect restarts,” the groups said in the letter.
“We feel certain that this case reported in the literature is not the only serious adverse outcome from the REMS and the transition,” they added.
The letter was signed by the American Psychiatric Association, the American Association for Community Psychiatry, the American Psychiatric Nurses Association, the College of Psychiatric and Neurologic Pharmacists, the National Alliance on Mental Illness, the National Association of State Mental Health Program Directors, and the National Council for Mental Wellbeing.
Clozapine can decrease the neutrophil count, which can lead to severe neutropenia, serious infection, and death. Consequently, the FDA put additional safety measures in place governing clozapine prescribing.
In 2015, a centralized clozapine REMS replaced separate prescribing registries that the drug manufacturers maintained. There were technical issues with the 2015 start-up of that website, including data migration problems and long call wait times, the FDA said.
Subsequently, the drug’s manufacturers then decided to change the REMS platform, which created new issues that led to high call volume and long wait times for clinicians and pharmacists who were trying to enroll.
Maintaining access
In November 2021, the FDA announced it would put some aspects of a planned switch on hold. A month later, the agency made further modifications to its plan.
The FDA said it would exercise “enforcement discretion” to try to maintain access to clozapine amid hitches with the REMS transition efforts. The agency also said at the time that it would not object if pharmacists dispensed clozapine without the usual authorization. In addition, wholesalers could ship the drug to pharmacies and health care settings without confirming REMS enrollment.
The FDA also held two December meetings to allow various stakeholders to air concerns.
In their letter, the APA and other groups asked if the FDA intends to continue with accommodations, such as allowing pharmacies to order clozapine from wholesalers without restriction.
“We do not feel the issues are resolved,” the groups said.
A version of this article first appeared on Medscape.com.
Mental Health Pharmacists: Increasing Necessary Mental Health Service Delivery
The COVID-19 pandemic has significantly impacted mental health. Adolescents, adults, and health care professionals (HCPs) report worsening mental health outcomes since the pandemic.1-3 Anxiety rates have tripled, depression quadrupled, and substance and alcohol use also have increased.3 The World Health Organization (WHO) reported that during the COVID-19 pandemic, 93% of countries worldwide documented disruptions to mental health services.4 HCP shortages, worsened by the pandemic, have resulted in a mental health crisis. What can we do?
Over the past 20 years, pharmacists have assumed a more significant role in managing patients’ mental health conditions through multidisciplinary team engagement. Pharmacists’ training includes optimizing pharmacotherapy, identifying and managing adverse effects (AEs), improving medication adherence, and reducing unnecessary health care costs.5 Pharmacists have assumed pivotal roles in mental health management, including but not limited to screening, drug selection, medication management, and decision-making support for patients and HCPs. Pharmacist-provided services have led to improved medication therapy outcomes and patient satisfaction.6
According to the 2012 National Alliance on Mental Illness national survey, > 50% of patients treated for a mental health condition report having a strong relationship with their pharmacist.7 The US Department of Veterans Affairs (VA) has led the charge, engaging pharmacists in patient-oriented mental health care,including those specific to accessing mental health care (eg, fear of stigmatization).8 After obtaining a 4-year PharmD degree, psychiatric pharmacists receive additional postgraduate residency training (2 years) focused on direct patient care and then are eligible for board certification. There are about 2000 board-certified psychiatric pharmacists in the United States. Qualified psychiatric pharmacists, especially those in underresourced states, have increased the number of available patient-oriented mental health services.7 However, to continue expanding and improving access to care, we need more HCPs and pharmacists.
Mental health clinical pharmacy specialists (CPSs) within the VA work in a variety of settings, including but not limited to, the inpatient psychiatric unit; residential programs for posttraumatic stress disorder (PTSD) and substance misuse; as part of the Mental Health Intensive Case Management (MHICM) team; and in pain, telehealth, and other outpatient clinics. The VA’s mental health CPSs operate under an independent scope of practice (SOP) and manage a variety of mental health disorders. The SOP also allows pharmacists to independently manage medications for psychiatric conditions, request laboratory tests, and change therapy as needed based on patient response. The Table describes pharmacist-reported roles in a single VA facility in various mental health practice sites (eg, inpatient, outpatient, substance misuse). Pharmacist involvement in medication management with the interdisciplinary team improved symptoms, medication adherence, and reduced AEs for conditions such as depression.9
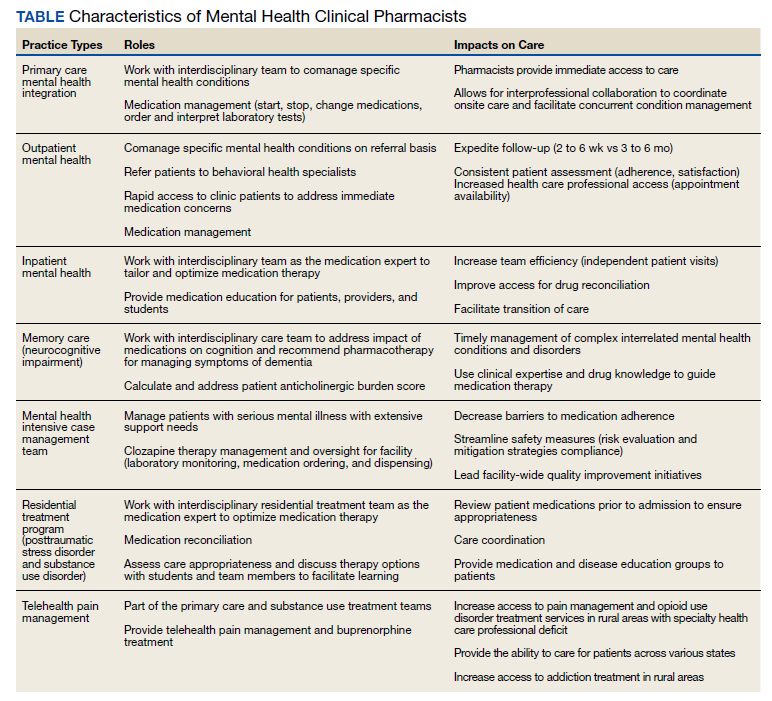
Within the VA, the outpatient mental health pharmacist works collaboratively with psychiatrists and HCPs to manage common psychiatric conditions on the phone and in person. VA pharmacists also are involved in the monitoring of patients on second-generation antipsychotics. Pharmacists assist with metabolic monitoring and assessing patients for movements disorders, using standardized rating scales. Pharmacists can manage complex psychiatric patients in collaboration with psychiatrists by providing medication management, laboratory test monitoring, medication counseling, and HCP referrals.
Pharmacists’ expertise is used in diverse ways in the VHA. At one facility, pharmacists functioned as interim prescribers when the facility experienced a turnover in behavioral health professionals. Pharmacists’ involvement decreased inappropriate use of psychiatric emergency services.10 VA pharmacists who manage patients’ mental health needs in primary care help achieve symptom improvement and medication adherence as well as lower referral rates for specialty mental health services.9 Pharmacist-managed electronic consult service provided a costs savings of about $40,000 a year.11
Pharmacists have shown that they can expand their roles. Pharmacists are versatile HCPs, currently working and collaborating with other HCPs in various settings to provide mental health services. Health care systems need to continue to use and expand the number of pharmacists. Including pharmacists in the primary and specialty care teams can increase access to care and improve health outcomes during the pandemic and beyond. The American Association of Colleges of Pharmacy in partnership with the American Medical Association established a resource to support and guide institutions interested in embedding pharmacists into different clinical sites.12 Opportunities for increased services by pharmacists can lead to improved outcomes, timely patient care, appropriate use of psychiatric medications and services, and cost savings.
Acknowledgments
We acknowledge the following Boise Veterans Affairs pharmacists: Paul Black, PharmD; Josh Gerving, PharmD; Kristin Helmboldt, PharmD; Samantha Patton, PharmD; Heather Walser, PharmD; and Andrea Winterswyk, PharmD, for contributing information about their practice roles and impact on patient care.
1. Panchal N, Kamal R. The implications of COVID-19 for mental health and substance use. Published February 10, 2021. Accessed February 8, 2022. https://www.kff.org/coronavirus-covid-19/issue-brief/the-implications-of-covid-19-for-mental-health-and-substance-use
2. How the pandemic has impacted teen mental health. National poll on children’s health. Published December 21, 2020. Accessed February 8, 2022. https://mottpoll.org/reports/how-pandemic-has-impacted-teen-mental-health
3. Substance Abuse and Mental Health Services Administration. A preliminary look at the mental health and substance use-related effects of the COVID-19 pandemic. Published May 2021. Accessed February 8, 2022. https://www.samhsa.gov/sites/default/files/dtac/mental-health-substance-use-effects-covid-pandemic-srb.pdf
4. World Health Organization. News release. COVID-19 disrupting mental health services in most countries, WHO survey. Published October 5, 2020. Accessed February 9, 2022. https://www.who.int/news/item/05-10-2020-covid-19-disrupting-mental-health-services-in-most-countries-who-survey
5. Avalere Health LLC. Exploring pharmacists’ role in a changing healthcare environment. Published May 2014. Accessed February 9, 2022. https://www.nacds.org/pdfs/comm/2014/pharmacist-role.pdf.
6. Silvia R. Collaborative treatment of depression by a psychiatric pharmacist integrated within a community health center primary care clinic. J Pharm Practice. 2016;29(3):270-341. doi:10.1177/0897190016645328
7. Caley C, Stimmel G. Characterizing the relationship between individuals with mental health conditions and community pharmacists. Published 2012. Accessed February 9, 2022. https://www.nami.org/About-NAMI/Publications-Reports/Survey-Reports/nami-cpnp-survey-report2012.pdf
8. Bovin MJ, Koenig CJ, Zamora KA, et al. Veterans’ experiences initiating VA-based mental health care. Psychol Serv. 2019;16(4):612-620. doi:10.1037/ser0000233
9. Herbert C, Winkler H. Impact of a clinical pharmacist–managed clinic in primary care mental health integration at a Veterans Affairs health system. Ment Health Clin. 2018;8(3):105-109. doi:10.9740/mhc.2018.05.105
10. Gibu M, Clark J, Gold J. Mental health pharmacists as interim prescribers. Ment Health Clin. 2018;7(3):111-115. doi:10.9740/mhc.2017.05.111
11. Herbert C, Winkler H, Moore TA. Outcomes of mental health pharmacist-managed electronic consults at a Veterans Affairs health care system. Ment Health Clin. 2018;7(3):131-136. doi:10.9740/mhc.2017.05.131
12. AACP. Embedding pharmacists into the practice. Accessed February 9, 2022. https://edhub.ama-assn.org/steps-forward/module/2702554
The COVID-19 pandemic has significantly impacted mental health. Adolescents, adults, and health care professionals (HCPs) report worsening mental health outcomes since the pandemic.1-3 Anxiety rates have tripled, depression quadrupled, and substance and alcohol use also have increased.3 The World Health Organization (WHO) reported that during the COVID-19 pandemic, 93% of countries worldwide documented disruptions to mental health services.4 HCP shortages, worsened by the pandemic, have resulted in a mental health crisis. What can we do?
Over the past 20 years, pharmacists have assumed a more significant role in managing patients’ mental health conditions through multidisciplinary team engagement. Pharmacists’ training includes optimizing pharmacotherapy, identifying and managing adverse effects (AEs), improving medication adherence, and reducing unnecessary health care costs.5 Pharmacists have assumed pivotal roles in mental health management, including but not limited to screening, drug selection, medication management, and decision-making support for patients and HCPs. Pharmacist-provided services have led to improved medication therapy outcomes and patient satisfaction.6
According to the 2012 National Alliance on Mental Illness national survey, > 50% of patients treated for a mental health condition report having a strong relationship with their pharmacist.7 The US Department of Veterans Affairs (VA) has led the charge, engaging pharmacists in patient-oriented mental health care,including those specific to accessing mental health care (eg, fear of stigmatization).8 After obtaining a 4-year PharmD degree, psychiatric pharmacists receive additional postgraduate residency training (2 years) focused on direct patient care and then are eligible for board certification. There are about 2000 board-certified psychiatric pharmacists in the United States. Qualified psychiatric pharmacists, especially those in underresourced states, have increased the number of available patient-oriented mental health services.7 However, to continue expanding and improving access to care, we need more HCPs and pharmacists.
Mental health clinical pharmacy specialists (CPSs) within the VA work in a variety of settings, including but not limited to, the inpatient psychiatric unit; residential programs for posttraumatic stress disorder (PTSD) and substance misuse; as part of the Mental Health Intensive Case Management (MHICM) team; and in pain, telehealth, and other outpatient clinics. The VA’s mental health CPSs operate under an independent scope of practice (SOP) and manage a variety of mental health disorders. The SOP also allows pharmacists to independently manage medications for psychiatric conditions, request laboratory tests, and change therapy as needed based on patient response. The Table describes pharmacist-reported roles in a single VA facility in various mental health practice sites (eg, inpatient, outpatient, substance misuse). Pharmacist involvement in medication management with the interdisciplinary team improved symptoms, medication adherence, and reduced AEs for conditions such as depression.9
Within the VA, the outpatient mental health pharmacist works collaboratively with psychiatrists and HCPs to manage common psychiatric conditions on the phone and in person. VA pharmacists also are involved in the monitoring of patients on second-generation antipsychotics. Pharmacists assist with metabolic monitoring and assessing patients for movements disorders, using standardized rating scales. Pharmacists can manage complex psychiatric patients in collaboration with psychiatrists by providing medication management, laboratory test monitoring, medication counseling, and HCP referrals.
Pharmacists’ expertise is used in diverse ways in the VHA. At one facility, pharmacists functioned as interim prescribers when the facility experienced a turnover in behavioral health professionals. Pharmacists’ involvement decreased inappropriate use of psychiatric emergency services.10 VA pharmacists who manage patients’ mental health needs in primary care help achieve symptom improvement and medication adherence as well as lower referral rates for specialty mental health services.9 Pharmacist-managed electronic consult service provided a costs savings of about $40,000 a year.11
Pharmacists have shown that they can expand their roles. Pharmacists are versatile HCPs, currently working and collaborating with other HCPs in various settings to provide mental health services. Health care systems need to continue to use and expand the number of pharmacists. Including pharmacists in the primary and specialty care teams can increase access to care and improve health outcomes during the pandemic and beyond. The American Association of Colleges of Pharmacy in partnership with the American Medical Association established a resource to support and guide institutions interested in embedding pharmacists into different clinical sites.12 Opportunities for increased services by pharmacists can lead to improved outcomes, timely patient care, appropriate use of psychiatric medications and services, and cost savings.
Acknowledgments
We acknowledge the following Boise Veterans Affairs pharmacists: Paul Black, PharmD; Josh Gerving, PharmD; Kristin Helmboldt, PharmD; Samantha Patton, PharmD; Heather Walser, PharmD; and Andrea Winterswyk, PharmD, for contributing information about their practice roles and impact on patient care.
The COVID-19 pandemic has significantly impacted mental health. Adolescents, adults, and health care professionals (HCPs) report worsening mental health outcomes since the pandemic.1-3 Anxiety rates have tripled, depression quadrupled, and substance and alcohol use also have increased.3 The World Health Organization (WHO) reported that during the COVID-19 pandemic, 93% of countries worldwide documented disruptions to mental health services.4 HCP shortages, worsened by the pandemic, have resulted in a mental health crisis. What can we do?
Over the past 20 years, pharmacists have assumed a more significant role in managing patients’ mental health conditions through multidisciplinary team engagement. Pharmacists’ training includes optimizing pharmacotherapy, identifying and managing adverse effects (AEs), improving medication adherence, and reducing unnecessary health care costs.5 Pharmacists have assumed pivotal roles in mental health management, including but not limited to screening, drug selection, medication management, and decision-making support for patients and HCPs. Pharmacist-provided services have led to improved medication therapy outcomes and patient satisfaction.6
According to the 2012 National Alliance on Mental Illness national survey, > 50% of patients treated for a mental health condition report having a strong relationship with their pharmacist.7 The US Department of Veterans Affairs (VA) has led the charge, engaging pharmacists in patient-oriented mental health care,including those specific to accessing mental health care (eg, fear of stigmatization).8 After obtaining a 4-year PharmD degree, psychiatric pharmacists receive additional postgraduate residency training (2 years) focused on direct patient care and then are eligible for board certification. There are about 2000 board-certified psychiatric pharmacists in the United States. Qualified psychiatric pharmacists, especially those in underresourced states, have increased the number of available patient-oriented mental health services.7 However, to continue expanding and improving access to care, we need more HCPs and pharmacists.
Mental health clinical pharmacy specialists (CPSs) within the VA work in a variety of settings, including but not limited to, the inpatient psychiatric unit; residential programs for posttraumatic stress disorder (PTSD) and substance misuse; as part of the Mental Health Intensive Case Management (MHICM) team; and in pain, telehealth, and other outpatient clinics. The VA’s mental health CPSs operate under an independent scope of practice (SOP) and manage a variety of mental health disorders. The SOP also allows pharmacists to independently manage medications for psychiatric conditions, request laboratory tests, and change therapy as needed based on patient response. The Table describes pharmacist-reported roles in a single VA facility in various mental health practice sites (eg, inpatient, outpatient, substance misuse). Pharmacist involvement in medication management with the interdisciplinary team improved symptoms, medication adherence, and reduced AEs for conditions such as depression.9
Within the VA, the outpatient mental health pharmacist works collaboratively with psychiatrists and HCPs to manage common psychiatric conditions on the phone and in person. VA pharmacists also are involved in the monitoring of patients on second-generation antipsychotics. Pharmacists assist with metabolic monitoring and assessing patients for movements disorders, using standardized rating scales. Pharmacists can manage complex psychiatric patients in collaboration with psychiatrists by providing medication management, laboratory test monitoring, medication counseling, and HCP referrals.
Pharmacists’ expertise is used in diverse ways in the VHA. At one facility, pharmacists functioned as interim prescribers when the facility experienced a turnover in behavioral health professionals. Pharmacists’ involvement decreased inappropriate use of psychiatric emergency services.10 VA pharmacists who manage patients’ mental health needs in primary care help achieve symptom improvement and medication adherence as well as lower referral rates for specialty mental health services.9 Pharmacist-managed electronic consult service provided a costs savings of about $40,000 a year.11
Pharmacists have shown that they can expand their roles. Pharmacists are versatile HCPs, currently working and collaborating with other HCPs in various settings to provide mental health services. Health care systems need to continue to use and expand the number of pharmacists. Including pharmacists in the primary and specialty care teams can increase access to care and improve health outcomes during the pandemic and beyond. The American Association of Colleges of Pharmacy in partnership with the American Medical Association established a resource to support and guide institutions interested in embedding pharmacists into different clinical sites.12 Opportunities for increased services by pharmacists can lead to improved outcomes, timely patient care, appropriate use of psychiatric medications and services, and cost savings.
Acknowledgments
We acknowledge the following Boise Veterans Affairs pharmacists: Paul Black, PharmD; Josh Gerving, PharmD; Kristin Helmboldt, PharmD; Samantha Patton, PharmD; Heather Walser, PharmD; and Andrea Winterswyk, PharmD, for contributing information about their practice roles and impact on patient care.
1. Panchal N, Kamal R. The implications of COVID-19 for mental health and substance use. Published February 10, 2021. Accessed February 8, 2022. https://www.kff.org/coronavirus-covid-19/issue-brief/the-implications-of-covid-19-for-mental-health-and-substance-use
2. How the pandemic has impacted teen mental health. National poll on children’s health. Published December 21, 2020. Accessed February 8, 2022. https://mottpoll.org/reports/how-pandemic-has-impacted-teen-mental-health
3. Substance Abuse and Mental Health Services Administration. A preliminary look at the mental health and substance use-related effects of the COVID-19 pandemic. Published May 2021. Accessed February 8, 2022. https://www.samhsa.gov/sites/default/files/dtac/mental-health-substance-use-effects-covid-pandemic-srb.pdf
4. World Health Organization. News release. COVID-19 disrupting mental health services in most countries, WHO survey. Published October 5, 2020. Accessed February 9, 2022. https://www.who.int/news/item/05-10-2020-covid-19-disrupting-mental-health-services-in-most-countries-who-survey
5. Avalere Health LLC. Exploring pharmacists’ role in a changing healthcare environment. Published May 2014. Accessed February 9, 2022. https://www.nacds.org/pdfs/comm/2014/pharmacist-role.pdf.
6. Silvia R. Collaborative treatment of depression by a psychiatric pharmacist integrated within a community health center primary care clinic. J Pharm Practice. 2016;29(3):270-341. doi:10.1177/0897190016645328
7. Caley C, Stimmel G. Characterizing the relationship between individuals with mental health conditions and community pharmacists. Published 2012. Accessed February 9, 2022. https://www.nami.org/About-NAMI/Publications-Reports/Survey-Reports/nami-cpnp-survey-report2012.pdf
8. Bovin MJ, Koenig CJ, Zamora KA, et al. Veterans’ experiences initiating VA-based mental health care. Psychol Serv. 2019;16(4):612-620. doi:10.1037/ser0000233
9. Herbert C, Winkler H. Impact of a clinical pharmacist–managed clinic in primary care mental health integration at a Veterans Affairs health system. Ment Health Clin. 2018;8(3):105-109. doi:10.9740/mhc.2018.05.105
10. Gibu M, Clark J, Gold J. Mental health pharmacists as interim prescribers. Ment Health Clin. 2018;7(3):111-115. doi:10.9740/mhc.2017.05.111
11. Herbert C, Winkler H, Moore TA. Outcomes of mental health pharmacist-managed electronic consults at a Veterans Affairs health care system. Ment Health Clin. 2018;7(3):131-136. doi:10.9740/mhc.2017.05.131
12. AACP. Embedding pharmacists into the practice. Accessed February 9, 2022. https://edhub.ama-assn.org/steps-forward/module/2702554
1. Panchal N, Kamal R. The implications of COVID-19 for mental health and substance use. Published February 10, 2021. Accessed February 8, 2022. https://www.kff.org/coronavirus-covid-19/issue-brief/the-implications-of-covid-19-for-mental-health-and-substance-use
2. How the pandemic has impacted teen mental health. National poll on children’s health. Published December 21, 2020. Accessed February 8, 2022. https://mottpoll.org/reports/how-pandemic-has-impacted-teen-mental-health
3. Substance Abuse and Mental Health Services Administration. A preliminary look at the mental health and substance use-related effects of the COVID-19 pandemic. Published May 2021. Accessed February 8, 2022. https://www.samhsa.gov/sites/default/files/dtac/mental-health-substance-use-effects-covid-pandemic-srb.pdf
4. World Health Organization. News release. COVID-19 disrupting mental health services in most countries, WHO survey. Published October 5, 2020. Accessed February 9, 2022. https://www.who.int/news/item/05-10-2020-covid-19-disrupting-mental-health-services-in-most-countries-who-survey
5. Avalere Health LLC. Exploring pharmacists’ role in a changing healthcare environment. Published May 2014. Accessed February 9, 2022. https://www.nacds.org/pdfs/comm/2014/pharmacist-role.pdf.
6. Silvia R. Collaborative treatment of depression by a psychiatric pharmacist integrated within a community health center primary care clinic. J Pharm Practice. 2016;29(3):270-341. doi:10.1177/0897190016645328
7. Caley C, Stimmel G. Characterizing the relationship between individuals with mental health conditions and community pharmacists. Published 2012. Accessed February 9, 2022. https://www.nami.org/About-NAMI/Publications-Reports/Survey-Reports/nami-cpnp-survey-report2012.pdf
8. Bovin MJ, Koenig CJ, Zamora KA, et al. Veterans’ experiences initiating VA-based mental health care. Psychol Serv. 2019;16(4):612-620. doi:10.1037/ser0000233
9. Herbert C, Winkler H. Impact of a clinical pharmacist–managed clinic in primary care mental health integration at a Veterans Affairs health system. Ment Health Clin. 2018;8(3):105-109. doi:10.9740/mhc.2018.05.105
10. Gibu M, Clark J, Gold J. Mental health pharmacists as interim prescribers. Ment Health Clin. 2018;7(3):111-115. doi:10.9740/mhc.2017.05.111
11. Herbert C, Winkler H, Moore TA. Outcomes of mental health pharmacist-managed electronic consults at a Veterans Affairs health care system. Ment Health Clin. 2018;7(3):131-136. doi:10.9740/mhc.2017.05.131
12. AACP. Embedding pharmacists into the practice. Accessed February 9, 2022. https://edhub.ama-assn.org/steps-forward/module/2702554
Analysis questions tocilizumab in ventilated COVID patients
A new statistical analysis of an existing meta-analysis reaffirms a finding that hospitalized patients with COVID-19 who are on simple oxygen or noninvasive ventilation can benefit from treatment with the arthritis drug tocilizumab (Actemra) in conjunction with corticosteroids. But the report also casts doubt on the effectiveness of tocilizumab in patients who are on ventilators.
“Clinicians should prescribe steroids and tocilizumab for hospitalized patients needing simple oxygen or noninvasive ventilation,” epidemiologist and study coauthor James (Jay) Brophy, MD, PhD, of McGill University, Montreal, said in an interview. “Further research is required to answer the question of whether tocilizumab is beneficial in patients requiring invasive ventilation, and consideration of participation in further tocilizumab studies seems reasonable.”
The new analysis was published Feb. 28, 2022, in JAMA Network Open.
The initial meta-analysis, published in 2021 in JAMA, was conducted by the WHO Rapid Evidence Appraisal for COVID-19 Therapies Working Group. It analyzed the results of 27 randomized trials that explored the use of interleukin-6 antagonists, including tocilizumab, and found that “28-day all-cause mortality was lower among patients who received IL-6 antagonists, compared with those who received usual care or placebo (summary odds ratio, 0.86). The summary ORs for the association of IL-6 antagonist treatment with 28-day all-cause mortality were 0.78 with concomitant administration of corticosteroids versus 1.09 without administration of corticosteroids.”
For the new report, researchers conducted a Bayesian statistical analysis of 15 studies within the meta-analysis that specifically examined the use of the rheumatoid arthritis drug tocilizumab. “Bayesian analysis allows one to make direct probability statements regarding the exact magnitude and the certainty of any benefit,” Dr. Brophy said. “This provides clinicians with the information they require to make well-informed decisions.”
The analysis estimated that the probability of a “clinically meaningful association” (absolute mortality risk difference, >1%) because of use of tocilizumab was higher than 95% in patients receiving simple oxygen and higher than 90% in those receiving noninvasive ventilation. But the probability was only about 67% higher in those receiving invasive mechanical ventilation.
Also, the researchers estimated that about 72% of future tocilizumab studies in patients on invasive mechanical ventilation would show a benefit.
The new analysis findings don’t add much to existing knowledge, said nephrologist David E. Leaf, MD, MMSc, of Harvard Medical School, Boston, who’s studied tocilizumab in COVID-19.
“The signal seems to be consistent that there is a greater benefit of tocilizumab in less ill patients than those who are more ill – e.g., those who are receiving invasive mechanical ventilation,” Dr. Leaf said in an interview. “This is interesting because in clinical practice the opposite approach is often undertaken, with tocilizumab use only being used in the sickest patients, even though the patients most likely to benefit seem to be those who are less ill.”
Clinically, he said, “hospitalized patients with COVID-19 should receive tocilizumab unless they have a clear contraindication and assuming it can be administered relatively early in their disease course. Earlier administration, before the onset of irreversible organ injury, is likely to have greater benefit.”
Dr. Leaf also noted it’s unknown whether the drug is helpful in several groups – patients presenting later in the course of COVID-19 illness, patients with additional infections, and immunocompromised patients.
It’s also not clear if tocilizumab benefits patients with lower levels of C-reactive protein, Shruti Gupta, MD, MPH, a nephrologist at Brigham and Women’s Hospital in Boston, said in an interview. The RECOVERY trial, for example, limited subjects to those with C-reactive protein of at least 75 mg/L.
Dr. Leaf and Dr. Gupta coauthored a 2021 cohort study analyzing mortality rates in patients with COVID-19 who were treated with tocilizumab versus those who were not.
No study funding was reported. Dr. Brophy, Dr. Leaf, and Dr. Gupta disclosed no relevant financial relationships. One study author reported participating in one of the randomized clinical trials included in the analysis.
A version of this article first appeared on Medscape.com.
A new statistical analysis of an existing meta-analysis reaffirms a finding that hospitalized patients with COVID-19 who are on simple oxygen or noninvasive ventilation can benefit from treatment with the arthritis drug tocilizumab (Actemra) in conjunction with corticosteroids. But the report also casts doubt on the effectiveness of tocilizumab in patients who are on ventilators.
“Clinicians should prescribe steroids and tocilizumab for hospitalized patients needing simple oxygen or noninvasive ventilation,” epidemiologist and study coauthor James (Jay) Brophy, MD, PhD, of McGill University, Montreal, said in an interview. “Further research is required to answer the question of whether tocilizumab is beneficial in patients requiring invasive ventilation, and consideration of participation in further tocilizumab studies seems reasonable.”
The new analysis was published Feb. 28, 2022, in JAMA Network Open.
The initial meta-analysis, published in 2021 in JAMA, was conducted by the WHO Rapid Evidence Appraisal for COVID-19 Therapies Working Group. It analyzed the results of 27 randomized trials that explored the use of interleukin-6 antagonists, including tocilizumab, and found that “28-day all-cause mortality was lower among patients who received IL-6 antagonists, compared with those who received usual care or placebo (summary odds ratio, 0.86). The summary ORs for the association of IL-6 antagonist treatment with 28-day all-cause mortality were 0.78 with concomitant administration of corticosteroids versus 1.09 without administration of corticosteroids.”
For the new report, researchers conducted a Bayesian statistical analysis of 15 studies within the meta-analysis that specifically examined the use of the rheumatoid arthritis drug tocilizumab. “Bayesian analysis allows one to make direct probability statements regarding the exact magnitude and the certainty of any benefit,” Dr. Brophy said. “This provides clinicians with the information they require to make well-informed decisions.”
The analysis estimated that the probability of a “clinically meaningful association” (absolute mortality risk difference, >1%) because of use of tocilizumab was higher than 95% in patients receiving simple oxygen and higher than 90% in those receiving noninvasive ventilation. But the probability was only about 67% higher in those receiving invasive mechanical ventilation.
Also, the researchers estimated that about 72% of future tocilizumab studies in patients on invasive mechanical ventilation would show a benefit.
The new analysis findings don’t add much to existing knowledge, said nephrologist David E. Leaf, MD, MMSc, of Harvard Medical School, Boston, who’s studied tocilizumab in COVID-19.
“The signal seems to be consistent that there is a greater benefit of tocilizumab in less ill patients than those who are more ill – e.g., those who are receiving invasive mechanical ventilation,” Dr. Leaf said in an interview. “This is interesting because in clinical practice the opposite approach is often undertaken, with tocilizumab use only being used in the sickest patients, even though the patients most likely to benefit seem to be those who are less ill.”
Clinically, he said, “hospitalized patients with COVID-19 should receive tocilizumab unless they have a clear contraindication and assuming it can be administered relatively early in their disease course. Earlier administration, before the onset of irreversible organ injury, is likely to have greater benefit.”
Dr. Leaf also noted it’s unknown whether the drug is helpful in several groups – patients presenting later in the course of COVID-19 illness, patients with additional infections, and immunocompromised patients.
It’s also not clear if tocilizumab benefits patients with lower levels of C-reactive protein, Shruti Gupta, MD, MPH, a nephrologist at Brigham and Women’s Hospital in Boston, said in an interview. The RECOVERY trial, for example, limited subjects to those with C-reactive protein of at least 75 mg/L.
Dr. Leaf and Dr. Gupta coauthored a 2021 cohort study analyzing mortality rates in patients with COVID-19 who were treated with tocilizumab versus those who were not.
No study funding was reported. Dr. Brophy, Dr. Leaf, and Dr. Gupta disclosed no relevant financial relationships. One study author reported participating in one of the randomized clinical trials included in the analysis.
A version of this article first appeared on Medscape.com.
A new statistical analysis of an existing meta-analysis reaffirms a finding that hospitalized patients with COVID-19 who are on simple oxygen or noninvasive ventilation can benefit from treatment with the arthritis drug tocilizumab (Actemra) in conjunction with corticosteroids. But the report also casts doubt on the effectiveness of tocilizumab in patients who are on ventilators.
“Clinicians should prescribe steroids and tocilizumab for hospitalized patients needing simple oxygen or noninvasive ventilation,” epidemiologist and study coauthor James (Jay) Brophy, MD, PhD, of McGill University, Montreal, said in an interview. “Further research is required to answer the question of whether tocilizumab is beneficial in patients requiring invasive ventilation, and consideration of participation in further tocilizumab studies seems reasonable.”
The new analysis was published Feb. 28, 2022, in JAMA Network Open.
The initial meta-analysis, published in 2021 in JAMA, was conducted by the WHO Rapid Evidence Appraisal for COVID-19 Therapies Working Group. It analyzed the results of 27 randomized trials that explored the use of interleukin-6 antagonists, including tocilizumab, and found that “28-day all-cause mortality was lower among patients who received IL-6 antagonists, compared with those who received usual care or placebo (summary odds ratio, 0.86). The summary ORs for the association of IL-6 antagonist treatment with 28-day all-cause mortality were 0.78 with concomitant administration of corticosteroids versus 1.09 without administration of corticosteroids.”
For the new report, researchers conducted a Bayesian statistical analysis of 15 studies within the meta-analysis that specifically examined the use of the rheumatoid arthritis drug tocilizumab. “Bayesian analysis allows one to make direct probability statements regarding the exact magnitude and the certainty of any benefit,” Dr. Brophy said. “This provides clinicians with the information they require to make well-informed decisions.”
The analysis estimated that the probability of a “clinically meaningful association” (absolute mortality risk difference, >1%) because of use of tocilizumab was higher than 95% in patients receiving simple oxygen and higher than 90% in those receiving noninvasive ventilation. But the probability was only about 67% higher in those receiving invasive mechanical ventilation.
Also, the researchers estimated that about 72% of future tocilizumab studies in patients on invasive mechanical ventilation would show a benefit.
The new analysis findings don’t add much to existing knowledge, said nephrologist David E. Leaf, MD, MMSc, of Harvard Medical School, Boston, who’s studied tocilizumab in COVID-19.
“The signal seems to be consistent that there is a greater benefit of tocilizumab in less ill patients than those who are more ill – e.g., those who are receiving invasive mechanical ventilation,” Dr. Leaf said in an interview. “This is interesting because in clinical practice the opposite approach is often undertaken, with tocilizumab use only being used in the sickest patients, even though the patients most likely to benefit seem to be those who are less ill.”
Clinically, he said, “hospitalized patients with COVID-19 should receive tocilizumab unless they have a clear contraindication and assuming it can be administered relatively early in their disease course. Earlier administration, before the onset of irreversible organ injury, is likely to have greater benefit.”
Dr. Leaf also noted it’s unknown whether the drug is helpful in several groups – patients presenting later in the course of COVID-19 illness, patients with additional infections, and immunocompromised patients.
It’s also not clear if tocilizumab benefits patients with lower levels of C-reactive protein, Shruti Gupta, MD, MPH, a nephrologist at Brigham and Women’s Hospital in Boston, said in an interview. The RECOVERY trial, for example, limited subjects to those with C-reactive protein of at least 75 mg/L.
Dr. Leaf and Dr. Gupta coauthored a 2021 cohort study analyzing mortality rates in patients with COVID-19 who were treated with tocilizumab versus those who were not.
No study funding was reported. Dr. Brophy, Dr. Leaf, and Dr. Gupta disclosed no relevant financial relationships. One study author reported participating in one of the randomized clinical trials included in the analysis.
A version of this article first appeared on Medscape.com.
FROM JAMA NETWORK OPEN
Needle-free epinephrine products could be available in 2023
Longstanding anxiety around use of epinephrine autoinjectors has prompted research into alternative delivery routes for this life-saving medication. Several companies presented posters on their needle-free epinephrine products at the American Academy of Allergy, Asthma & Immunology (AAAAI) Annual Meeting.
Intranasal formulations are under development at ARS Pharmaceuticals (San Diego) and Bryn Pharma (Raleigh, N.C.). And Aquestive Therapeutics (Warren, N.J.) is working on a sublingual film that delivers epinephrine prodrug when applied under the tongue.
Epinephrine is essential for stopping life-threatening allergic reactions, yet patients often don’t carry their autoinjectors and many hesitate to use them. “It’s needle phobia,” said ARS Pharmaceuticals CEO Richard Lowenthal in an interview with this news organization. “They’re afraid to use it. They don’t like to inject their children, so they hesitate.”
Both nasal sprays reached maximal plasma concentration in 20-30 minutes. ARS Pharmaceuticals compared its intranasal product (Neffy 1 mg) against manual intramuscular injection (0.3 mg) and two autoinjectors (EpiPen 0.3 mg and Symjepi 0.3 mg) by analyzing data from multiple randomized crossover Phase 1 studies examining pharmacokinetics and pharmacodynamics in 175 healthy adults. In this integrated analysis, EpiPen was fastest (20 minutes) at reaching maximal concentration (Tmax), followed by Symjepi and Neffy (both 30 minutes) and epinephrine 0.3 mg IM (45 minutes). In a human factors analysis, ARS Pharmaceuticals reported that untrained participants were able to administer the Neffy spray to themselves or another participant safely and effectively during a simulated emergency scenario.
Bryn Pharma compared pharmacokinetics of its nasal spray product (BRYN-NDS1C 6.6 mg) when self-administered or administered by trained professionals and found comparable profiles for each. Tmax values were also similar: 21.63 minutes (trained professional) and 19.82 minutes (self-administered).
Aquestive Therapeutics is developing a postage stamp-sized product (AQST-109) that delivers epinephrine and begins dissolving when placed under the tongue. No water or swallowing is required for administration, and its packaging is thinner and smaller than a credit card, according to CEO Keith Kendall.
Its analysis showed that the epinephrine reaches maximum plasma concentration in about 15 minutes, with a Tmax range narrower than that of the EpiPen. “The results showed dosing with AQST-109 resulted in PK concentration and Tmax values comparable to published data from autoinjectors,” said John Oppenheimer, MD, of Rutgers University School of Medicine, in a prerecorded poster summary.
Aquestive aims to move forward to the manufacture of registration batches and a pivotal pharmacokinetic study in the second half of 2022. Mr. Lowenthal said ARS Pharmaceuticals is hoping for approval and launch of its nasal spray by summer 2023.
“Having a non-needle delivery device would help many people overcome that fear and hopefully increase use in anaphylaxis,” said David Stukus, MD, an allergist-immunologist and professor of clinical pediatrics at Nationwide Children’s Hospital, Columbus, who was not involved with any of the studies on EpiPen alternatives. And “it’s not just food allergy – anaphylaxis can occur from venom stings, medications, or idiopathic causes.”
Mr. Lowenthal is the CEO of ARS Pharmaceuticals. Mr. Kendall is CEO of Aquestive Therapeutics. Dr. Oppenheimer is a consultant for Aquestive, GSK, Amgen, Sanofi, and Aimmune and sits on Aquestive’s advisory board. Dr. Stukus is a consultant for Novartis.
A version of this article first appeared on Medscape.com.
Longstanding anxiety around use of epinephrine autoinjectors has prompted research into alternative delivery routes for this life-saving medication. Several companies presented posters on their needle-free epinephrine products at the American Academy of Allergy, Asthma & Immunology (AAAAI) Annual Meeting.
Intranasal formulations are under development at ARS Pharmaceuticals (San Diego) and Bryn Pharma (Raleigh, N.C.). And Aquestive Therapeutics (Warren, N.J.) is working on a sublingual film that delivers epinephrine prodrug when applied under the tongue.
Epinephrine is essential for stopping life-threatening allergic reactions, yet patients often don’t carry their autoinjectors and many hesitate to use them. “It’s needle phobia,” said ARS Pharmaceuticals CEO Richard Lowenthal in an interview with this news organization. “They’re afraid to use it. They don’t like to inject their children, so they hesitate.”
Both nasal sprays reached maximal plasma concentration in 20-30 minutes. ARS Pharmaceuticals compared its intranasal product (Neffy 1 mg) against manual intramuscular injection (0.3 mg) and two autoinjectors (EpiPen 0.3 mg and Symjepi 0.3 mg) by analyzing data from multiple randomized crossover Phase 1 studies examining pharmacokinetics and pharmacodynamics in 175 healthy adults. In this integrated analysis, EpiPen was fastest (20 minutes) at reaching maximal concentration (Tmax), followed by Symjepi and Neffy (both 30 minutes) and epinephrine 0.3 mg IM (45 minutes). In a human factors analysis, ARS Pharmaceuticals reported that untrained participants were able to administer the Neffy spray to themselves or another participant safely and effectively during a simulated emergency scenario.
Bryn Pharma compared pharmacokinetics of its nasal spray product (BRYN-NDS1C 6.6 mg) when self-administered or administered by trained professionals and found comparable profiles for each. Tmax values were also similar: 21.63 minutes (trained professional) and 19.82 minutes (self-administered).
Aquestive Therapeutics is developing a postage stamp-sized product (AQST-109) that delivers epinephrine and begins dissolving when placed under the tongue. No water or swallowing is required for administration, and its packaging is thinner and smaller than a credit card, according to CEO Keith Kendall.
Its analysis showed that the epinephrine reaches maximum plasma concentration in about 15 minutes, with a Tmax range narrower than that of the EpiPen. “The results showed dosing with AQST-109 resulted in PK concentration and Tmax values comparable to published data from autoinjectors,” said John Oppenheimer, MD, of Rutgers University School of Medicine, in a prerecorded poster summary.
Aquestive aims to move forward to the manufacture of registration batches and a pivotal pharmacokinetic study in the second half of 2022. Mr. Lowenthal said ARS Pharmaceuticals is hoping for approval and launch of its nasal spray by summer 2023.
“Having a non-needle delivery device would help many people overcome that fear and hopefully increase use in anaphylaxis,” said David Stukus, MD, an allergist-immunologist and professor of clinical pediatrics at Nationwide Children’s Hospital, Columbus, who was not involved with any of the studies on EpiPen alternatives. And “it’s not just food allergy – anaphylaxis can occur from venom stings, medications, or idiopathic causes.”
Mr. Lowenthal is the CEO of ARS Pharmaceuticals. Mr. Kendall is CEO of Aquestive Therapeutics. Dr. Oppenheimer is a consultant for Aquestive, GSK, Amgen, Sanofi, and Aimmune and sits on Aquestive’s advisory board. Dr. Stukus is a consultant for Novartis.
A version of this article first appeared on Medscape.com.
Longstanding anxiety around use of epinephrine autoinjectors has prompted research into alternative delivery routes for this life-saving medication. Several companies presented posters on their needle-free epinephrine products at the American Academy of Allergy, Asthma & Immunology (AAAAI) Annual Meeting.
Intranasal formulations are under development at ARS Pharmaceuticals (San Diego) and Bryn Pharma (Raleigh, N.C.). And Aquestive Therapeutics (Warren, N.J.) is working on a sublingual film that delivers epinephrine prodrug when applied under the tongue.
Epinephrine is essential for stopping life-threatening allergic reactions, yet patients often don’t carry their autoinjectors and many hesitate to use them. “It’s needle phobia,” said ARS Pharmaceuticals CEO Richard Lowenthal in an interview with this news organization. “They’re afraid to use it. They don’t like to inject their children, so they hesitate.”
Both nasal sprays reached maximal plasma concentration in 20-30 minutes. ARS Pharmaceuticals compared its intranasal product (Neffy 1 mg) against manual intramuscular injection (0.3 mg) and two autoinjectors (EpiPen 0.3 mg and Symjepi 0.3 mg) by analyzing data from multiple randomized crossover Phase 1 studies examining pharmacokinetics and pharmacodynamics in 175 healthy adults. In this integrated analysis, EpiPen was fastest (20 minutes) at reaching maximal concentration (Tmax), followed by Symjepi and Neffy (both 30 minutes) and epinephrine 0.3 mg IM (45 minutes). In a human factors analysis, ARS Pharmaceuticals reported that untrained participants were able to administer the Neffy spray to themselves or another participant safely and effectively during a simulated emergency scenario.
Bryn Pharma compared pharmacokinetics of its nasal spray product (BRYN-NDS1C 6.6 mg) when self-administered or administered by trained professionals and found comparable profiles for each. Tmax values were also similar: 21.63 minutes (trained professional) and 19.82 minutes (self-administered).
Aquestive Therapeutics is developing a postage stamp-sized product (AQST-109) that delivers epinephrine and begins dissolving when placed under the tongue. No water or swallowing is required for administration, and its packaging is thinner and smaller than a credit card, according to CEO Keith Kendall.
Its analysis showed that the epinephrine reaches maximum plasma concentration in about 15 minutes, with a Tmax range narrower than that of the EpiPen. “The results showed dosing with AQST-109 resulted in PK concentration and Tmax values comparable to published data from autoinjectors,” said John Oppenheimer, MD, of Rutgers University School of Medicine, in a prerecorded poster summary.
Aquestive aims to move forward to the manufacture of registration batches and a pivotal pharmacokinetic study in the second half of 2022. Mr. Lowenthal said ARS Pharmaceuticals is hoping for approval and launch of its nasal spray by summer 2023.
“Having a non-needle delivery device would help many people overcome that fear and hopefully increase use in anaphylaxis,” said David Stukus, MD, an allergist-immunologist and professor of clinical pediatrics at Nationwide Children’s Hospital, Columbus, who was not involved with any of the studies on EpiPen alternatives. And “it’s not just food allergy – anaphylaxis can occur from venom stings, medications, or idiopathic causes.”
Mr. Lowenthal is the CEO of ARS Pharmaceuticals. Mr. Kendall is CEO of Aquestive Therapeutics. Dr. Oppenheimer is a consultant for Aquestive, GSK, Amgen, Sanofi, and Aimmune and sits on Aquestive’s advisory board. Dr. Stukus is a consultant for Novartis.
A version of this article first appeared on Medscape.com.
FROM AAAAI
FDA approves new CAR T-cell treatment for multiple myeloma
A new treatment option for patients with refractory/relapsed multiple myeloma who have already tried four or more therapies has been approved by the U.S. Food and Drug Administration.
There are already two other therapies on the market that target BCMA – another CAR T cell, idecabtagene vicleucel (Abecma), which was approved by the FDA in March 2021, and a drug conjugate, belantamab mafodotin (Blenrep), which was approved in August 2020.
The approval of cilta-cel was based on clinical data from the CARTITUDE-1 study, which were initially presented in December 2020 at the annual meeting of the American Society of Hematology, as reported at the time by this news organization.
The trial involved 97 patients with relapsed/refractory multiple myeloma who had already received a median of six previous treatments (range, three to 18), including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.
“The treatment journey for the majority of patients living with multiple myeloma is a relentless cycle of remission and relapse, with fewer patients achieving a deep response as they progress through later lines of therapy,” commented Sundar Jagannath, MBBS, professor of medicine, hematology, and medical oncology at Mount Sinai, who was a principal investigator on the pivotal study.
“That is why I have been really excited about the results from the CARTITUDE-1 study, which has demonstrated that cilta-cel can provide deep and durable responses and long-term treatment-free intervals, even in this heavily pretreated multiple myeloma patient population,” he said.
“Today’s approval of Carvykti helps address a great unmet need for these patients,” he commented in a press release from the manufacturer.
Like other CAR T-cell therapies, ciltacabtagene autoleucel is a one-time treatment. It involves collecting blood from the patient, extracting T cells, genetically engineering them, then transfusing them back to the patient, who in the meantime has undergone conditioning.
The results from CARTITUDE-1 show that this one-time treatment resulted in deep and durable responses.
The overall response rate was 98%, and the majority of patients (78%) achieved a stringent complete response, in which physicians are unable to observe any signs or symptoms of disease via imaging or other tests after treatment.
At a median of 18 months’ follow-up, the median duration of response was 21.8 months.
“The responses in the CARTITUDE-1 study showed durability over time and resulted in the majority of heavily pretreated patients achieving deep responses after 18-month follow-up,” commented Mr. Jagannath.
“The approval of cilta-cel provides physicians an immunotherapy treatment option that offers patients an opportunity to be free from anti-myeloma therapies for a period of time,” he added.
As with other CAR T-cell therapies, there were serious side effects, and these products are available only through restricted programs under a risk evaluation and mitigation strategy.
The product information for Cartykti includes a boxed warning that mentions cytokine release syndrome (CRS), immune effector cell–associated neurotoxicity syndrome, parkinsonism, Guillain-Barré syndrome, hemophagocytic lymphohistiocytosis/macrophage activation syndrome, and prolonged and/or recurrent cytopenias.
The most common adverse reactions (reported in greater than or equal to 20% of patients) are pyrexia, CRS, hypogammaglobulinemia, hypotension, musculoskeletal pain, fatigue, infections–pathogens unspecified, cough, chills, diarrhea, nausea, encephalopathy, decreased appetite, upper respiratory tract infection, headache, tachycardia, dizziness, dyspnea, edema, viral infections, coagulopathy, constipation, and vomiting.
A version of this article first appeared on Medscape.com.
A new treatment option for patients with refractory/relapsed multiple myeloma who have already tried four or more therapies has been approved by the U.S. Food and Drug Administration.
There are already two other therapies on the market that target BCMA – another CAR T cell, idecabtagene vicleucel (Abecma), which was approved by the FDA in March 2021, and a drug conjugate, belantamab mafodotin (Blenrep), which was approved in August 2020.
The approval of cilta-cel was based on clinical data from the CARTITUDE-1 study, which were initially presented in December 2020 at the annual meeting of the American Society of Hematology, as reported at the time by this news organization.
The trial involved 97 patients with relapsed/refractory multiple myeloma who had already received a median of six previous treatments (range, three to 18), including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.
“The treatment journey for the majority of patients living with multiple myeloma is a relentless cycle of remission and relapse, with fewer patients achieving a deep response as they progress through later lines of therapy,” commented Sundar Jagannath, MBBS, professor of medicine, hematology, and medical oncology at Mount Sinai, who was a principal investigator on the pivotal study.
“That is why I have been really excited about the results from the CARTITUDE-1 study, which has demonstrated that cilta-cel can provide deep and durable responses and long-term treatment-free intervals, even in this heavily pretreated multiple myeloma patient population,” he said.
“Today’s approval of Carvykti helps address a great unmet need for these patients,” he commented in a press release from the manufacturer.
Like other CAR T-cell therapies, ciltacabtagene autoleucel is a one-time treatment. It involves collecting blood from the patient, extracting T cells, genetically engineering them, then transfusing them back to the patient, who in the meantime has undergone conditioning.
The results from CARTITUDE-1 show that this one-time treatment resulted in deep and durable responses.
The overall response rate was 98%, and the majority of patients (78%) achieved a stringent complete response, in which physicians are unable to observe any signs or symptoms of disease via imaging or other tests after treatment.
At a median of 18 months’ follow-up, the median duration of response was 21.8 months.
“The responses in the CARTITUDE-1 study showed durability over time and resulted in the majority of heavily pretreated patients achieving deep responses after 18-month follow-up,” commented Mr. Jagannath.
“The approval of cilta-cel provides physicians an immunotherapy treatment option that offers patients an opportunity to be free from anti-myeloma therapies for a period of time,” he added.
As with other CAR T-cell therapies, there were serious side effects, and these products are available only through restricted programs under a risk evaluation and mitigation strategy.
The product information for Cartykti includes a boxed warning that mentions cytokine release syndrome (CRS), immune effector cell–associated neurotoxicity syndrome, parkinsonism, Guillain-Barré syndrome, hemophagocytic lymphohistiocytosis/macrophage activation syndrome, and prolonged and/or recurrent cytopenias.
The most common adverse reactions (reported in greater than or equal to 20% of patients) are pyrexia, CRS, hypogammaglobulinemia, hypotension, musculoskeletal pain, fatigue, infections–pathogens unspecified, cough, chills, diarrhea, nausea, encephalopathy, decreased appetite, upper respiratory tract infection, headache, tachycardia, dizziness, dyspnea, edema, viral infections, coagulopathy, constipation, and vomiting.
A version of this article first appeared on Medscape.com.
A new treatment option for patients with refractory/relapsed multiple myeloma who have already tried four or more therapies has been approved by the U.S. Food and Drug Administration.
There are already two other therapies on the market that target BCMA – another CAR T cell, idecabtagene vicleucel (Abecma), which was approved by the FDA in March 2021, and a drug conjugate, belantamab mafodotin (Blenrep), which was approved in August 2020.
The approval of cilta-cel was based on clinical data from the CARTITUDE-1 study, which were initially presented in December 2020 at the annual meeting of the American Society of Hematology, as reported at the time by this news organization.
The trial involved 97 patients with relapsed/refractory multiple myeloma who had already received a median of six previous treatments (range, three to 18), including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.
“The treatment journey for the majority of patients living with multiple myeloma is a relentless cycle of remission and relapse, with fewer patients achieving a deep response as they progress through later lines of therapy,” commented Sundar Jagannath, MBBS, professor of medicine, hematology, and medical oncology at Mount Sinai, who was a principal investigator on the pivotal study.
“That is why I have been really excited about the results from the CARTITUDE-1 study, which has demonstrated that cilta-cel can provide deep and durable responses and long-term treatment-free intervals, even in this heavily pretreated multiple myeloma patient population,” he said.
“Today’s approval of Carvykti helps address a great unmet need for these patients,” he commented in a press release from the manufacturer.
Like other CAR T-cell therapies, ciltacabtagene autoleucel is a one-time treatment. It involves collecting blood from the patient, extracting T cells, genetically engineering them, then transfusing them back to the patient, who in the meantime has undergone conditioning.
The results from CARTITUDE-1 show that this one-time treatment resulted in deep and durable responses.
The overall response rate was 98%, and the majority of patients (78%) achieved a stringent complete response, in which physicians are unable to observe any signs or symptoms of disease via imaging or other tests after treatment.
At a median of 18 months’ follow-up, the median duration of response was 21.8 months.
“The responses in the CARTITUDE-1 study showed durability over time and resulted in the majority of heavily pretreated patients achieving deep responses after 18-month follow-up,” commented Mr. Jagannath.
“The approval of cilta-cel provides physicians an immunotherapy treatment option that offers patients an opportunity to be free from anti-myeloma therapies for a period of time,” he added.
As with other CAR T-cell therapies, there were serious side effects, and these products are available only through restricted programs under a risk evaluation and mitigation strategy.
The product information for Cartykti includes a boxed warning that mentions cytokine release syndrome (CRS), immune effector cell–associated neurotoxicity syndrome, parkinsonism, Guillain-Barré syndrome, hemophagocytic lymphohistiocytosis/macrophage activation syndrome, and prolonged and/or recurrent cytopenias.
The most common adverse reactions (reported in greater than or equal to 20% of patients) are pyrexia, CRS, hypogammaglobulinemia, hypotension, musculoskeletal pain, fatigue, infections–pathogens unspecified, cough, chills, diarrhea, nausea, encephalopathy, decreased appetite, upper respiratory tract infection, headache, tachycardia, dizziness, dyspnea, edema, viral infections, coagulopathy, constipation, and vomiting.
A version of this article first appeared on Medscape.com.
Excess sodium in soluble acetaminophen tied to CVD risk, death
a large observational study of more than 300,000 adults suggests.
“Numerous studies have reported that high sodium intake is associated with increased risks of cardiovascular disease,” Yuqing Zhang, DSc, with Massachusetts General Hospital and Harvard Medical School, Boston, told this news organization. “Given that the pain relief effect of non–sodium-containing acetaminophen is similar to that of sodium-containing acetaminophen, clinicians may prescribe non–sodium-containing acetaminophen to their patients to minimize the risk of CVD and mortality,” Dr. Zhang said.
The study was published online Feb. 24 in the European Heart Journal.
‘Compelling results’
Dr. Zhang and colleagues note that the effervescent and soluble formulations of 0.5 g acetaminophen contain 0.44 and 0.39 g of sodium, respectively.
Therefore, the intake of maximum daily dose (4 g/day) of sodium-containing acetaminophen corresponds to the ingestion of more than 3 g of sodium, a dose that alone exceeds the recommended total daily sodium intake allowance of the World Health Organization (2 g/day).
“This hidden extra sodium intake is often overlooked,” Dr. Zhang told this news organization.
Using data from the Health Improvement Network, a U.K. primary care database, the researchers examined 4,532 patients with hypertension taking sodium-containing acetaminophen and compared them with 146,866 patients with hypertension taking non–sodium-containing acetaminophen (tablet, capsule, or oral suspension formulations).
After 1 year, the risk of incident CVD (myocardial infarction, stroke, and heart failure) was 5.6% in those taking sodium-containing acetaminophen, compared with 4.6% in those taking non–sodium-containing acetaminophen (average weighted hazard ratio, 1.59; 95% confidence interval, 1.32-1.92).
A separate analysis of normotensive patients taking sodium-containing acetaminophen (n = 5,351) or non–sodium-containing acetaminophen (n = 141,948) gave similar results.
The 1-year risk of incident CVD was 4.4% in those taking sodium-containing acetaminophen vs. 3.7% among those taking non–sodium-containing acetaminophen (average weighted HR, 1.45; 95% CI, 1.18-1.79).
There was also evidence of a dose-response relationship.
In those with hypertension, CVD risk increased by roughly one-quarter (odds ratio, 1.26) for those with one prescription of sodium-containing acetaminophen and by nearly one half (OR, 1.45) for those with five or more prescriptions of sodium-containing acetaminophen. Similar findings were observed among adults without hypertension.
Mortality at 1 year was also higher in those taking sodium-containing acetaminophen than non–sodium-containing acetaminophen, in patients with hypertension (7.6% vs. 6.1%) and without hypertension (7.3% vs. 5.9%).
“The results are compelling,” write the authors of an editorial published with the study.
“The direct message from this study is clear – there are likely to be millions of people worldwide taking paracetamol on a daily basis in a ‘fast-acting’ effervescent or soluble formulation who are increasing their risks of cardiovascular disease and premature death,” say Aletta Schutte, PhD, and Bruce Neal, MBChB, PhD, of the George Institute for Global Health, Sydney.
“The weight of the evidence makes ongoing inaction on sodium-containing medications untenable. The widespread use of effervescent medication in the general population, and the enormous doses of sodium that can be consumed inadvertently by unsuspecting consumers requires urgent action,” Dr. Schutte and Dr. Neal say.
The study was supported by the National Natural Science Foundation of China, the National Key Research and Development Project, the Project Program of National Clinical Research Center for Geriatric Disorders, the Key Research and Development Program of Hunan Province, and the Science and Technology Program of Hunan Province. Dr. Zhang, Dr. Schutte, and Dr. Neal have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
a large observational study of more than 300,000 adults suggests.
“Numerous studies have reported that high sodium intake is associated with increased risks of cardiovascular disease,” Yuqing Zhang, DSc, with Massachusetts General Hospital and Harvard Medical School, Boston, told this news organization. “Given that the pain relief effect of non–sodium-containing acetaminophen is similar to that of sodium-containing acetaminophen, clinicians may prescribe non–sodium-containing acetaminophen to their patients to minimize the risk of CVD and mortality,” Dr. Zhang said.
The study was published online Feb. 24 in the European Heart Journal.
‘Compelling results’
Dr. Zhang and colleagues note that the effervescent and soluble formulations of 0.5 g acetaminophen contain 0.44 and 0.39 g of sodium, respectively.
Therefore, the intake of maximum daily dose (4 g/day) of sodium-containing acetaminophen corresponds to the ingestion of more than 3 g of sodium, a dose that alone exceeds the recommended total daily sodium intake allowance of the World Health Organization (2 g/day).
“This hidden extra sodium intake is often overlooked,” Dr. Zhang told this news organization.
Using data from the Health Improvement Network, a U.K. primary care database, the researchers examined 4,532 patients with hypertension taking sodium-containing acetaminophen and compared them with 146,866 patients with hypertension taking non–sodium-containing acetaminophen (tablet, capsule, or oral suspension formulations).
After 1 year, the risk of incident CVD (myocardial infarction, stroke, and heart failure) was 5.6% in those taking sodium-containing acetaminophen, compared with 4.6% in those taking non–sodium-containing acetaminophen (average weighted hazard ratio, 1.59; 95% confidence interval, 1.32-1.92).
A separate analysis of normotensive patients taking sodium-containing acetaminophen (n = 5,351) or non–sodium-containing acetaminophen (n = 141,948) gave similar results.
The 1-year risk of incident CVD was 4.4% in those taking sodium-containing acetaminophen vs. 3.7% among those taking non–sodium-containing acetaminophen (average weighted HR, 1.45; 95% CI, 1.18-1.79).
There was also evidence of a dose-response relationship.
In those with hypertension, CVD risk increased by roughly one-quarter (odds ratio, 1.26) for those with one prescription of sodium-containing acetaminophen and by nearly one half (OR, 1.45) for those with five or more prescriptions of sodium-containing acetaminophen. Similar findings were observed among adults without hypertension.
Mortality at 1 year was also higher in those taking sodium-containing acetaminophen than non–sodium-containing acetaminophen, in patients with hypertension (7.6% vs. 6.1%) and without hypertension (7.3% vs. 5.9%).
“The results are compelling,” write the authors of an editorial published with the study.
“The direct message from this study is clear – there are likely to be millions of people worldwide taking paracetamol on a daily basis in a ‘fast-acting’ effervescent or soluble formulation who are increasing their risks of cardiovascular disease and premature death,” say Aletta Schutte, PhD, and Bruce Neal, MBChB, PhD, of the George Institute for Global Health, Sydney.
“The weight of the evidence makes ongoing inaction on sodium-containing medications untenable. The widespread use of effervescent medication in the general population, and the enormous doses of sodium that can be consumed inadvertently by unsuspecting consumers requires urgent action,” Dr. Schutte and Dr. Neal say.
The study was supported by the National Natural Science Foundation of China, the National Key Research and Development Project, the Project Program of National Clinical Research Center for Geriatric Disorders, the Key Research and Development Program of Hunan Province, and the Science and Technology Program of Hunan Province. Dr. Zhang, Dr. Schutte, and Dr. Neal have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
a large observational study of more than 300,000 adults suggests.
“Numerous studies have reported that high sodium intake is associated with increased risks of cardiovascular disease,” Yuqing Zhang, DSc, with Massachusetts General Hospital and Harvard Medical School, Boston, told this news organization. “Given that the pain relief effect of non–sodium-containing acetaminophen is similar to that of sodium-containing acetaminophen, clinicians may prescribe non–sodium-containing acetaminophen to their patients to minimize the risk of CVD and mortality,” Dr. Zhang said.
The study was published online Feb. 24 in the European Heart Journal.
‘Compelling results’
Dr. Zhang and colleagues note that the effervescent and soluble formulations of 0.5 g acetaminophen contain 0.44 and 0.39 g of sodium, respectively.
Therefore, the intake of maximum daily dose (4 g/day) of sodium-containing acetaminophen corresponds to the ingestion of more than 3 g of sodium, a dose that alone exceeds the recommended total daily sodium intake allowance of the World Health Organization (2 g/day).
“This hidden extra sodium intake is often overlooked,” Dr. Zhang told this news organization.
Using data from the Health Improvement Network, a U.K. primary care database, the researchers examined 4,532 patients with hypertension taking sodium-containing acetaminophen and compared them with 146,866 patients with hypertension taking non–sodium-containing acetaminophen (tablet, capsule, or oral suspension formulations).
After 1 year, the risk of incident CVD (myocardial infarction, stroke, and heart failure) was 5.6% in those taking sodium-containing acetaminophen, compared with 4.6% in those taking non–sodium-containing acetaminophen (average weighted hazard ratio, 1.59; 95% confidence interval, 1.32-1.92).
A separate analysis of normotensive patients taking sodium-containing acetaminophen (n = 5,351) or non–sodium-containing acetaminophen (n = 141,948) gave similar results.
The 1-year risk of incident CVD was 4.4% in those taking sodium-containing acetaminophen vs. 3.7% among those taking non–sodium-containing acetaminophen (average weighted HR, 1.45; 95% CI, 1.18-1.79).
There was also evidence of a dose-response relationship.
In those with hypertension, CVD risk increased by roughly one-quarter (odds ratio, 1.26) for those with one prescription of sodium-containing acetaminophen and by nearly one half (OR, 1.45) for those with five or more prescriptions of sodium-containing acetaminophen. Similar findings were observed among adults without hypertension.
Mortality at 1 year was also higher in those taking sodium-containing acetaminophen than non–sodium-containing acetaminophen, in patients with hypertension (7.6% vs. 6.1%) and without hypertension (7.3% vs. 5.9%).
“The results are compelling,” write the authors of an editorial published with the study.
“The direct message from this study is clear – there are likely to be millions of people worldwide taking paracetamol on a daily basis in a ‘fast-acting’ effervescent or soluble formulation who are increasing their risks of cardiovascular disease and premature death,” say Aletta Schutte, PhD, and Bruce Neal, MBChB, PhD, of the George Institute for Global Health, Sydney.
“The weight of the evidence makes ongoing inaction on sodium-containing medications untenable. The widespread use of effervescent medication in the general population, and the enormous doses of sodium that can be consumed inadvertently by unsuspecting consumers requires urgent action,” Dr. Schutte and Dr. Neal say.
The study was supported by the National Natural Science Foundation of China, the National Key Research and Development Project, the Project Program of National Clinical Research Center for Geriatric Disorders, the Key Research and Development Program of Hunan Province, and the Science and Technology Program of Hunan Province. Dr. Zhang, Dr. Schutte, and Dr. Neal have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE EUROPEAN HEART JOURNAL
In-hospital detox or not, anti-CGRPs show efficacy for medication overuse headache
, according to investigators.
Abruptly discontinuing overused analgesics with health care provider oversight – a frequently resource-intensive and challenging process – is no more effective for controlling medication overuse headache than simply advising patients to stop, reported lead author Umberto Pensato, MD, of the University of Bologna, Italy, and colleagues.
“[C]urrently, the abrupt discontinuation of the overused painkiller(s), accompanied by the start of a pharmacological preventive therapy, is the most recommended strategy [for medication overuse headache],” the investigators wrote in Cephalalgia. “While painkiller(s) withdrawal could be accomplished on an outpatient basis in most cases, an in-hospital setting may be required to achieve successful discontinuation in a subgroup of patients with medication overuse headache, further weighing on individual and hospital costs. Additionally hampering this approach, the abrupt discontinuation of the overused painkiller(s) invariably results in disabling withdrawal symptoms for up to 2 weeks, including a transitory worsening of headache, the so-called ‘rebound headache.’ ”
Inpatient or outpatient: Does it matter?
According to Dr. Pensato and colleagues, early evidence suggests that previous painkiller withdrawal does not impact the efficacy of anti-CGRPs for medication overuse headache, yet relevant data remain scarce. To address this knowledge gap, they conducted a prospective, real-world study exploring the relationship between detoxification and outcomes after starting anti-CGRP therapy.
Out of 401 patients enrolled based on initiation of erenumab or galcanezumab, 111 satisfied inclusion criteria, including diagnosis of chronic migraine and medication overuse headache, at least 28 days of analgesic usage and headache days per month in the preceding 3 months, and other factors. Of these 111 patients, 83 underwent in-hospital detox, while the remaining 28 patients, who declined detox based on personal reasons or COVID-19–related bed shortage, were advised to discontinue overused medication on an outpatient basis (without oversight).
The primary endpoint was medication overuse headache responder rate after 3 months, as defined by ICHD-3 diagnostic criteria. Secondary endpoints included 6-item headache impact test (HIT-6), monthly headache days (MHD), migraine disability assessment score (MIDAS), mean pain intensity (MPI), monthly pain medication intake (MPMI), baseline predictors of response/refractoriness, and safety.
Three months after starting anti-CGRP therapy, 59% of patients had resolution of medication overuse headache, including 57% in the inpatient detox group and 64% in the outpatient group, a difference that was not statistically significant (P = .4788). Approximately half of the patients (51%) had at least 50% reduction in monthly headache days; although the rate was numerically lower in the inpatient group compared with the outpatient group, the difference was again not significant (51% vs. 54%; P = .8393).
“Our results support the emerging evidence that anti-CGRP drugs may be effective in these patients irrespective of the detoxification program,” the investigators concluded. “Further studies are needed to definitively confirm these results, potentially leading to a paradigm shift in the management of medication overuse headache.”
Abrupt or gradual detox?
According to Alan M. Rapoport, MD, clinical professor of neurology at the University of California, Los Angeles, and editor-in-chief of Neurology Reviews, the study was hampered by two major design limitations.
“The biggest problem I see is that the two groups were treated very differently for their detoxification,” Dr. Rapoport said. “One group was detoxified abruptly in the hospital, so the authors were sure that the patients were off acute-care medication before they started their preventives. The other group was advised to stop their medication on an outpatient basis. The issue is that we have no follow-up as to whether the outpatients did or did not abruptly detoxify. A bigger issue was that the two groups were not randomized so there are many other variables that may have come into consideration.”
Still, Dr. Rapoport, a past president of the International Headache Society (IHS), noted that the findings strengthen a growing body of evidence supporting the efficacy of monoclonal antibodies for medication overuse headache regardless of detoxification strategy. He cited a 2020 study by Carlsen and colleagues conducted at the Danish Headache Center in Copenhagen, which reported similar medication overuse headache outcomes across three randomized cohorts whether they received preventive therapy with detoxification, preventive therapy without detoxification, or detoxification followed 2 months later by preventive therapy.
“What I have noticed since we have had monoclonal antibodies in our armamentarium is that these drugs work very well even when the patient has not fully detoxified,” Dr. Rapoport said. “What I do with my patients is not teach them how to detoxify now, but simply educate them to take fewer acute care medications as their headaches get better from the monoclonal antibodies; they should try to take fewer acute care medications for milder, shorter headaches, and just let them go away on their own. Previous research suggests that even when a patient is not educated at all about medication overuse headache and the reason for detoxification, monoclonal antibodies still work in the presence of medication overuse headache, and improve it.”
The investigators disclosed relationships with Allergan, Novartis, Teva, and others. Dr. Rapoport is on the speakers bureau for AbbVie.
, according to investigators.
Abruptly discontinuing overused analgesics with health care provider oversight – a frequently resource-intensive and challenging process – is no more effective for controlling medication overuse headache than simply advising patients to stop, reported lead author Umberto Pensato, MD, of the University of Bologna, Italy, and colleagues.
“[C]urrently, the abrupt discontinuation of the overused painkiller(s), accompanied by the start of a pharmacological preventive therapy, is the most recommended strategy [for medication overuse headache],” the investigators wrote in Cephalalgia. “While painkiller(s) withdrawal could be accomplished on an outpatient basis in most cases, an in-hospital setting may be required to achieve successful discontinuation in a subgroup of patients with medication overuse headache, further weighing on individual and hospital costs. Additionally hampering this approach, the abrupt discontinuation of the overused painkiller(s) invariably results in disabling withdrawal symptoms for up to 2 weeks, including a transitory worsening of headache, the so-called ‘rebound headache.’ ”
Inpatient or outpatient: Does it matter?
According to Dr. Pensato and colleagues, early evidence suggests that previous painkiller withdrawal does not impact the efficacy of anti-CGRPs for medication overuse headache, yet relevant data remain scarce. To address this knowledge gap, they conducted a prospective, real-world study exploring the relationship between detoxification and outcomes after starting anti-CGRP therapy.
Out of 401 patients enrolled based on initiation of erenumab or galcanezumab, 111 satisfied inclusion criteria, including diagnosis of chronic migraine and medication overuse headache, at least 28 days of analgesic usage and headache days per month in the preceding 3 months, and other factors. Of these 111 patients, 83 underwent in-hospital detox, while the remaining 28 patients, who declined detox based on personal reasons or COVID-19–related bed shortage, were advised to discontinue overused medication on an outpatient basis (without oversight).
The primary endpoint was medication overuse headache responder rate after 3 months, as defined by ICHD-3 diagnostic criteria. Secondary endpoints included 6-item headache impact test (HIT-6), monthly headache days (MHD), migraine disability assessment score (MIDAS), mean pain intensity (MPI), monthly pain medication intake (MPMI), baseline predictors of response/refractoriness, and safety.
Three months after starting anti-CGRP therapy, 59% of patients had resolution of medication overuse headache, including 57% in the inpatient detox group and 64% in the outpatient group, a difference that was not statistically significant (P = .4788). Approximately half of the patients (51%) had at least 50% reduction in monthly headache days; although the rate was numerically lower in the inpatient group compared with the outpatient group, the difference was again not significant (51% vs. 54%; P = .8393).
“Our results support the emerging evidence that anti-CGRP drugs may be effective in these patients irrespective of the detoxification program,” the investigators concluded. “Further studies are needed to definitively confirm these results, potentially leading to a paradigm shift in the management of medication overuse headache.”
Abrupt or gradual detox?
According to Alan M. Rapoport, MD, clinical professor of neurology at the University of California, Los Angeles, and editor-in-chief of Neurology Reviews, the study was hampered by two major design limitations.
“The biggest problem I see is that the two groups were treated very differently for their detoxification,” Dr. Rapoport said. “One group was detoxified abruptly in the hospital, so the authors were sure that the patients were off acute-care medication before they started their preventives. The other group was advised to stop their medication on an outpatient basis. The issue is that we have no follow-up as to whether the outpatients did or did not abruptly detoxify. A bigger issue was that the two groups were not randomized so there are many other variables that may have come into consideration.”
Still, Dr. Rapoport, a past president of the International Headache Society (IHS), noted that the findings strengthen a growing body of evidence supporting the efficacy of monoclonal antibodies for medication overuse headache regardless of detoxification strategy. He cited a 2020 study by Carlsen and colleagues conducted at the Danish Headache Center in Copenhagen, which reported similar medication overuse headache outcomes across three randomized cohorts whether they received preventive therapy with detoxification, preventive therapy without detoxification, or detoxification followed 2 months later by preventive therapy.
“What I have noticed since we have had monoclonal antibodies in our armamentarium is that these drugs work very well even when the patient has not fully detoxified,” Dr. Rapoport said. “What I do with my patients is not teach them how to detoxify now, but simply educate them to take fewer acute care medications as their headaches get better from the monoclonal antibodies; they should try to take fewer acute care medications for milder, shorter headaches, and just let them go away on their own. Previous research suggests that even when a patient is not educated at all about medication overuse headache and the reason for detoxification, monoclonal antibodies still work in the presence of medication overuse headache, and improve it.”
The investigators disclosed relationships with Allergan, Novartis, Teva, and others. Dr. Rapoport is on the speakers bureau for AbbVie.
, according to investigators.
Abruptly discontinuing overused analgesics with health care provider oversight – a frequently resource-intensive and challenging process – is no more effective for controlling medication overuse headache than simply advising patients to stop, reported lead author Umberto Pensato, MD, of the University of Bologna, Italy, and colleagues.
“[C]urrently, the abrupt discontinuation of the overused painkiller(s), accompanied by the start of a pharmacological preventive therapy, is the most recommended strategy [for medication overuse headache],” the investigators wrote in Cephalalgia. “While painkiller(s) withdrawal could be accomplished on an outpatient basis in most cases, an in-hospital setting may be required to achieve successful discontinuation in a subgroup of patients with medication overuse headache, further weighing on individual and hospital costs. Additionally hampering this approach, the abrupt discontinuation of the overused painkiller(s) invariably results in disabling withdrawal symptoms for up to 2 weeks, including a transitory worsening of headache, the so-called ‘rebound headache.’ ”
Inpatient or outpatient: Does it matter?
According to Dr. Pensato and colleagues, early evidence suggests that previous painkiller withdrawal does not impact the efficacy of anti-CGRPs for medication overuse headache, yet relevant data remain scarce. To address this knowledge gap, they conducted a prospective, real-world study exploring the relationship between detoxification and outcomes after starting anti-CGRP therapy.
Out of 401 patients enrolled based on initiation of erenumab or galcanezumab, 111 satisfied inclusion criteria, including diagnosis of chronic migraine and medication overuse headache, at least 28 days of analgesic usage and headache days per month in the preceding 3 months, and other factors. Of these 111 patients, 83 underwent in-hospital detox, while the remaining 28 patients, who declined detox based on personal reasons or COVID-19–related bed shortage, were advised to discontinue overused medication on an outpatient basis (without oversight).
The primary endpoint was medication overuse headache responder rate after 3 months, as defined by ICHD-3 diagnostic criteria. Secondary endpoints included 6-item headache impact test (HIT-6), monthly headache days (MHD), migraine disability assessment score (MIDAS), mean pain intensity (MPI), monthly pain medication intake (MPMI), baseline predictors of response/refractoriness, and safety.
Three months after starting anti-CGRP therapy, 59% of patients had resolution of medication overuse headache, including 57% in the inpatient detox group and 64% in the outpatient group, a difference that was not statistically significant (P = .4788). Approximately half of the patients (51%) had at least 50% reduction in monthly headache days; although the rate was numerically lower in the inpatient group compared with the outpatient group, the difference was again not significant (51% vs. 54%; P = .8393).
“Our results support the emerging evidence that anti-CGRP drugs may be effective in these patients irrespective of the detoxification program,” the investigators concluded. “Further studies are needed to definitively confirm these results, potentially leading to a paradigm shift in the management of medication overuse headache.”
Abrupt or gradual detox?
According to Alan M. Rapoport, MD, clinical professor of neurology at the University of California, Los Angeles, and editor-in-chief of Neurology Reviews, the study was hampered by two major design limitations.
“The biggest problem I see is that the two groups were treated very differently for their detoxification,” Dr. Rapoport said. “One group was detoxified abruptly in the hospital, so the authors were sure that the patients were off acute-care medication before they started their preventives. The other group was advised to stop their medication on an outpatient basis. The issue is that we have no follow-up as to whether the outpatients did or did not abruptly detoxify. A bigger issue was that the two groups were not randomized so there are many other variables that may have come into consideration.”
Still, Dr. Rapoport, a past president of the International Headache Society (IHS), noted that the findings strengthen a growing body of evidence supporting the efficacy of monoclonal antibodies for medication overuse headache regardless of detoxification strategy. He cited a 2020 study by Carlsen and colleagues conducted at the Danish Headache Center in Copenhagen, which reported similar medication overuse headache outcomes across three randomized cohorts whether they received preventive therapy with detoxification, preventive therapy without detoxification, or detoxification followed 2 months later by preventive therapy.
“What I have noticed since we have had monoclonal antibodies in our armamentarium is that these drugs work very well even when the patient has not fully detoxified,” Dr. Rapoport said. “What I do with my patients is not teach them how to detoxify now, but simply educate them to take fewer acute care medications as their headaches get better from the monoclonal antibodies; they should try to take fewer acute care medications for milder, shorter headaches, and just let them go away on their own. Previous research suggests that even when a patient is not educated at all about medication overuse headache and the reason for detoxification, monoclonal antibodies still work in the presence of medication overuse headache, and improve it.”
The investigators disclosed relationships with Allergan, Novartis, Teva, and others. Dr. Rapoport is on the speakers bureau for AbbVie.
FROM CEPHALALGIA
FDA okays empagliflozin for HF regardless of ejection fraction
The Food and Drug Administration has approved an expanded heart failure indication for the sodium-glucose transporter 2 inhibitor empagliflozin (Jardiance) that now includes HF with mid-range or preserved left ventricular ejection fraction (LVEF), the agency announced on Feb. 24.
That means the SGLT2 inhibitor, once considered primarily an antidiabetic agent, is approved for use in patients with HF per se without regard to ventricular function. The drug received approval for HF with reduced LVEF in August 2021.
The expanded indication, specifically for reducing the risk of cardiovascular death and HF hospitalization in adults, was widely anticipated based on the landmark results from the EMPEROR-Preserved trial. The study saw a significant 21% relative reduction in that composite endpoint over about 2 years in patients with New York Heart Association class II-IV heart failure and an LVEF greater than 40% who received empagliflozin along with other standard care.
Interestingly, the drug’s expanded indication in HF resembles that approved for sacubitril/valsartan (Entresto) in February 2021 based mostly on the PARAGON-HF trial, which entered patients with HF and an LVEF at least 45%. The trial was “negative” in that it saw no significant advantage to the drug for its primary clinical outcome but did suggest benefit for some secondary endpoints.
The FDA had used more cautionary language in its expanded indication for sacubitril/valsartan, “to reduce the risk of cardiovascular death and hospitalization for heart failure in adult patients with chronic heart failure. Benefits are most clearly evident in patients with left ventricular ejection fraction below normal.”
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved an expanded heart failure indication for the sodium-glucose transporter 2 inhibitor empagliflozin (Jardiance) that now includes HF with mid-range or preserved left ventricular ejection fraction (LVEF), the agency announced on Feb. 24.
That means the SGLT2 inhibitor, once considered primarily an antidiabetic agent, is approved for use in patients with HF per se without regard to ventricular function. The drug received approval for HF with reduced LVEF in August 2021.
The expanded indication, specifically for reducing the risk of cardiovascular death and HF hospitalization in adults, was widely anticipated based on the landmark results from the EMPEROR-Preserved trial. The study saw a significant 21% relative reduction in that composite endpoint over about 2 years in patients with New York Heart Association class II-IV heart failure and an LVEF greater than 40% who received empagliflozin along with other standard care.
Interestingly, the drug’s expanded indication in HF resembles that approved for sacubitril/valsartan (Entresto) in February 2021 based mostly on the PARAGON-HF trial, which entered patients with HF and an LVEF at least 45%. The trial was “negative” in that it saw no significant advantage to the drug for its primary clinical outcome but did suggest benefit for some secondary endpoints.
The FDA had used more cautionary language in its expanded indication for sacubitril/valsartan, “to reduce the risk of cardiovascular death and hospitalization for heart failure in adult patients with chronic heart failure. Benefits are most clearly evident in patients with left ventricular ejection fraction below normal.”
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved an expanded heart failure indication for the sodium-glucose transporter 2 inhibitor empagliflozin (Jardiance) that now includes HF with mid-range or preserved left ventricular ejection fraction (LVEF), the agency announced on Feb. 24.
That means the SGLT2 inhibitor, once considered primarily an antidiabetic agent, is approved for use in patients with HF per se without regard to ventricular function. The drug received approval for HF with reduced LVEF in August 2021.
The expanded indication, specifically for reducing the risk of cardiovascular death and HF hospitalization in adults, was widely anticipated based on the landmark results from the EMPEROR-Preserved trial. The study saw a significant 21% relative reduction in that composite endpoint over about 2 years in patients with New York Heart Association class II-IV heart failure and an LVEF greater than 40% who received empagliflozin along with other standard care.
Interestingly, the drug’s expanded indication in HF resembles that approved for sacubitril/valsartan (Entresto) in February 2021 based mostly on the PARAGON-HF trial, which entered patients with HF and an LVEF at least 45%. The trial was “negative” in that it saw no significant advantage to the drug for its primary clinical outcome but did suggest benefit for some secondary endpoints.
The FDA had used more cautionary language in its expanded indication for sacubitril/valsartan, “to reduce the risk of cardiovascular death and hospitalization for heart failure in adult patients with chronic heart failure. Benefits are most clearly evident in patients with left ventricular ejection fraction below normal.”
A version of this article first appeared on Medscape.com.
Ivermectin does not stop progression to severe COVID: randomized trial
Ivermectin treatment given to high-risk patients with mild-to-moderate COVID-19 during the first week of illness did not prevent progression to severe disease, according to results from a randomized clinical trial.
“The study findings do not support the use of ivermectin for patients with COVID-19,” researchers conclude in the paper published online in JAMA Internal Medicine.
The open-label trial was conducted at 20 public hospitals and a COVID-19 quarantine center in Malaysia between May 31 and Oct. 25, 2021. It was led by Steven Chee Loon Lim, MRCP, department of medicine, Raja Permaisuri Bainun Hospital, Perak, Malaysia.
Among 490 patients in the primary analysis, 52 of 241 patients (21.6%) in the ivermectin group and 43 of 249 patients (17.3%) in the control group progressed to severe disease (relative risk, 1.25; 95% confidence interval, 0.87-1.80; P = .25). All major ethnic groups in Malaysia were well represented, the researchers write.
Participants (average age 62.5 and 54.5% women) were randomly assigned 1:1 to receive either a 5-day course of oral ivermectin (0.4 mg/kg body weight daily for 5 days) plus standard of care (n = 241) or standard of care alone (n = 249). Standard of care included symptomatic therapy and monitoring for early deterioration based on clinical findings, laboratory tests, and chest imaging.
Secondary outcomes
Secondary outcomes included rates of mechanical ventilation, intensive care unit (ICU) admission, 28-day in-hospital mortality, and side effects.
In all the secondary outcomes, there were no significant differences between groups.
Mechanical ventilation occurred in four patients on the ivermectin protocol (1.7%) versus 10 patients in the control group (4.0%) (RR, 0.41; 95% CI, 0.13-1.30; P = .17); ICU admission occurred in six (2.4%) versus eight (3.2%) (RR, 0.78; 95% CI, 0.27-2.20; P = .79); and 28-day in-hospital death occurred in three (1.2%) versus 10 (4.0%) (RR, 0.31; 95% CI, 0.09-1.11; P = .09).
The most common adverse event was diarrhea, reported by 5.8% in the ivermectin group and 1.6% in the control group.
No difference by vaccine status
The researchers conducted a subgroup analysis to evaluate any differences in whether participants were vaccinated. They said that analysis was “unremarkable.”
Just more than half of participants (51.8%) were fully vaccinated, with two doses of COVID-19 vaccines. Among the fully vaccinated patients, 17.7% in the ivermectin group and 9.2% in the control group developed severe disease (RR, 1.92; 95% CI, 0.99-3.71; P = .06).
Ivermectin, an inexpensive and widely available antiparasitic drug, is prescribed to treat COVID-19 but has not been approved by the U.S. Food and Drug Administration for that purpose. Evidence-based data for or against use has been sparse.
The authors write that “although some early clinical studies suggested the potential efficacy of ivermectin in the treatment and prevention of COVID-19, these studies had methodologic weaknesses.”
Dr. Lim and colleagues point out that their findings are consistent with those of the IVERCOR-COVID19 trial, which found ivermectin ineffective in reducing hospitalization risk.
Previous randomized trials of ivermectin for COVID-19 patients that have included at least 400 patients have focused on outpatients.
In the current study, the authors note, patients were hospitalized, which allowed investigators to observe administration of ivermectin with a high adherence rate. Additionally, the researchers used clearly defined criteria for determining progression to severe disease.
Limitations of the current study include that the open-label design might lead to under-reporting of adverse events in the control group while overestimating the drug effects of ivermectin. The study was also not designed to assess the effects of ivermectin on mortality from COVID-19.
A version of this article first appeared on Medscape.com.
Ivermectin treatment given to high-risk patients with mild-to-moderate COVID-19 during the first week of illness did not prevent progression to severe disease, according to results from a randomized clinical trial.
“The study findings do not support the use of ivermectin for patients with COVID-19,” researchers conclude in the paper published online in JAMA Internal Medicine.
The open-label trial was conducted at 20 public hospitals and a COVID-19 quarantine center in Malaysia between May 31 and Oct. 25, 2021. It was led by Steven Chee Loon Lim, MRCP, department of medicine, Raja Permaisuri Bainun Hospital, Perak, Malaysia.
Among 490 patients in the primary analysis, 52 of 241 patients (21.6%) in the ivermectin group and 43 of 249 patients (17.3%) in the control group progressed to severe disease (relative risk, 1.25; 95% confidence interval, 0.87-1.80; P = .25). All major ethnic groups in Malaysia were well represented, the researchers write.
Participants (average age 62.5 and 54.5% women) were randomly assigned 1:1 to receive either a 5-day course of oral ivermectin (0.4 mg/kg body weight daily for 5 days) plus standard of care (n = 241) or standard of care alone (n = 249). Standard of care included symptomatic therapy and monitoring for early deterioration based on clinical findings, laboratory tests, and chest imaging.
Secondary outcomes
Secondary outcomes included rates of mechanical ventilation, intensive care unit (ICU) admission, 28-day in-hospital mortality, and side effects.
In all the secondary outcomes, there were no significant differences between groups.
Mechanical ventilation occurred in four patients on the ivermectin protocol (1.7%) versus 10 patients in the control group (4.0%) (RR, 0.41; 95% CI, 0.13-1.30; P = .17); ICU admission occurred in six (2.4%) versus eight (3.2%) (RR, 0.78; 95% CI, 0.27-2.20; P = .79); and 28-day in-hospital death occurred in three (1.2%) versus 10 (4.0%) (RR, 0.31; 95% CI, 0.09-1.11; P = .09).
The most common adverse event was diarrhea, reported by 5.8% in the ivermectin group and 1.6% in the control group.
No difference by vaccine status
The researchers conducted a subgroup analysis to evaluate any differences in whether participants were vaccinated. They said that analysis was “unremarkable.”
Just more than half of participants (51.8%) were fully vaccinated, with two doses of COVID-19 vaccines. Among the fully vaccinated patients, 17.7% in the ivermectin group and 9.2% in the control group developed severe disease (RR, 1.92; 95% CI, 0.99-3.71; P = .06).
Ivermectin, an inexpensive and widely available antiparasitic drug, is prescribed to treat COVID-19 but has not been approved by the U.S. Food and Drug Administration for that purpose. Evidence-based data for or against use has been sparse.
The authors write that “although some early clinical studies suggested the potential efficacy of ivermectin in the treatment and prevention of COVID-19, these studies had methodologic weaknesses.”
Dr. Lim and colleagues point out that their findings are consistent with those of the IVERCOR-COVID19 trial, which found ivermectin ineffective in reducing hospitalization risk.
Previous randomized trials of ivermectin for COVID-19 patients that have included at least 400 patients have focused on outpatients.
In the current study, the authors note, patients were hospitalized, which allowed investigators to observe administration of ivermectin with a high adherence rate. Additionally, the researchers used clearly defined criteria for determining progression to severe disease.
Limitations of the current study include that the open-label design might lead to under-reporting of adverse events in the control group while overestimating the drug effects of ivermectin. The study was also not designed to assess the effects of ivermectin on mortality from COVID-19.
A version of this article first appeared on Medscape.com.
Ivermectin treatment given to high-risk patients with mild-to-moderate COVID-19 during the first week of illness did not prevent progression to severe disease, according to results from a randomized clinical trial.
“The study findings do not support the use of ivermectin for patients with COVID-19,” researchers conclude in the paper published online in JAMA Internal Medicine.
The open-label trial was conducted at 20 public hospitals and a COVID-19 quarantine center in Malaysia between May 31 and Oct. 25, 2021. It was led by Steven Chee Loon Lim, MRCP, department of medicine, Raja Permaisuri Bainun Hospital, Perak, Malaysia.
Among 490 patients in the primary analysis, 52 of 241 patients (21.6%) in the ivermectin group and 43 of 249 patients (17.3%) in the control group progressed to severe disease (relative risk, 1.25; 95% confidence interval, 0.87-1.80; P = .25). All major ethnic groups in Malaysia were well represented, the researchers write.
Participants (average age 62.5 and 54.5% women) were randomly assigned 1:1 to receive either a 5-day course of oral ivermectin (0.4 mg/kg body weight daily for 5 days) plus standard of care (n = 241) or standard of care alone (n = 249). Standard of care included symptomatic therapy and monitoring for early deterioration based on clinical findings, laboratory tests, and chest imaging.
Secondary outcomes
Secondary outcomes included rates of mechanical ventilation, intensive care unit (ICU) admission, 28-day in-hospital mortality, and side effects.
In all the secondary outcomes, there were no significant differences between groups.
Mechanical ventilation occurred in four patients on the ivermectin protocol (1.7%) versus 10 patients in the control group (4.0%) (RR, 0.41; 95% CI, 0.13-1.30; P = .17); ICU admission occurred in six (2.4%) versus eight (3.2%) (RR, 0.78; 95% CI, 0.27-2.20; P = .79); and 28-day in-hospital death occurred in three (1.2%) versus 10 (4.0%) (RR, 0.31; 95% CI, 0.09-1.11; P = .09).
The most common adverse event was diarrhea, reported by 5.8% in the ivermectin group and 1.6% in the control group.
No difference by vaccine status
The researchers conducted a subgroup analysis to evaluate any differences in whether participants were vaccinated. They said that analysis was “unremarkable.”
Just more than half of participants (51.8%) were fully vaccinated, with two doses of COVID-19 vaccines. Among the fully vaccinated patients, 17.7% in the ivermectin group and 9.2% in the control group developed severe disease (RR, 1.92; 95% CI, 0.99-3.71; P = .06).
Ivermectin, an inexpensive and widely available antiparasitic drug, is prescribed to treat COVID-19 but has not been approved by the U.S. Food and Drug Administration for that purpose. Evidence-based data for or against use has been sparse.
The authors write that “although some early clinical studies suggested the potential efficacy of ivermectin in the treatment and prevention of COVID-19, these studies had methodologic weaknesses.”
Dr. Lim and colleagues point out that their findings are consistent with those of the IVERCOR-COVID19 trial, which found ivermectin ineffective in reducing hospitalization risk.
Previous randomized trials of ivermectin for COVID-19 patients that have included at least 400 patients have focused on outpatients.
In the current study, the authors note, patients were hospitalized, which allowed investigators to observe administration of ivermectin with a high adherence rate. Additionally, the researchers used clearly defined criteria for determining progression to severe disease.
Limitations of the current study include that the open-label design might lead to under-reporting of adverse events in the control group while overestimating the drug effects of ivermectin. The study was also not designed to assess the effects of ivermectin on mortality from COVID-19.
A version of this article first appeared on Medscape.com.
FROM JAMA INTERNAL MEDICNE
DOACs comparable to warfarin in CVT
and are less likely to result in major bleeding, a retrospective study suggests.
The ACTION CVT study was presented at the International Stroke Conference (ISC) 2022 by Ekaterina Bakradze, MD, assistant professor of neurology at the University of Alabama at Birmingham.
It was also simultaneously published online in Stroke.
“This real-world data supports use of direct oral anticoagulant drugs as a reasonable alternative to warfarin in patients with cerebral venous thrombosis,” Dr. Bakradze concluded.
But she added that because this study was based on retrospective observational data, the findings should be interpreted with caution and require confirmation by larger prospective studies.
Two such studies are now underway: the Direct Oral Anticoagulants in the Treatment of Cerebral Venous Thrombosis (DOAC-CVT) study and the randomized Study of Rivaroxaban for Cerebral Venous Thrombosis (SECRET) trial.
Dr. Bakradze explained that cerebral venous thrombosis is a less common cause of stroke and occurs more often in women and younger patients, with a median age of 37 years. Current recommended treatment consists of heparin followed by oral anticoagulation.
She noted that although randomized trials and current guidelines indicate that DOACs are a preferred alternative to warfarin for the treatment of patients with venous thromboembolism, there are limited data on their use in patients with CVT.
A small, randomized trial (RESPECT-CVT) showed no significant difference in efficacy and safety outcomes between dabigatran and warfarin in patients with cerebral venous thrombosis, but with only 120 patients, this trial was too small for definite answers to this question.
A better understanding of this issue is important, because the mechanisms underlying cerebral venous thrombosis and other thromboembolism and their subsequent risks may differ, Dr. Bakradze said.
As randomized trials in patients with cerebral venous thrombosis are difficult to perform because the condition has a low incidence and low event rates, the researchers decided to look at this question with a large retrospective multicenter study.
The ACTION-CVT study involved 845 consecutive patients with cerebral venous thrombosis over 6 years (from January 2015 and December 2020) from 27 centers in Italy, New Zealand, Switzerland, and the United States. Patients were identified from medical records with diagnostic codes and confirmed with imaging.
The primary predictor in the study was oral anticoagulant type (DOAC vs. warfarin). Study outcomes were abstracted by individual sites through review of all available medical records.
The primary outcome was recurrent venous thrombosis (venous thromboembolism or cerebral venous thrombosis) during follow-up. Imaging outcomes based on recanalization status on last venous imaging study abstracted from radiology reports were also reported.
The safety outcome was major hemorrhage, defined as new or worsening intracranial hemorrhage (ICH), or major extracranial hemorrhage. Results were adjusted for age, sex, and relevant medical conditions.
The mean age of the patients included was 44.8 years, 64.7% were women, 33% received DOAC only, 51.8% received warfarin only, and 15.1% received both treatments at different times.
Results showed that during a median follow-up of 345 days, there were 5.68 recurrent venous thrombosis events, 3.77 major hemorrhages, and 1.84 deaths per 100 patient-years.
Among 525 patients who met recanalization analysis inclusion criteria, 36.6% had complete, 48.2% had partial, and 15.2% had no recanalization.
When compared with warfarin, DOAC treatment was associated with similar risk for recurrent venous thrombosis (adjusted hazard ratio, 0.94; 95% confidence interval, 0.51-1.73; P = .84), death (aHR, 0.71, 95% CI, 0.24-2.08; P = .53), and rate of partial/complete recanalization (aHR, 0.92, 95% CI, 0.48-1.73; P = .79).
But patients who received a DOAC had a significantly lower rate of major hemorrhage (aHR, 0.35; 95% CI, 0.15-0.81; P = .02).
When examined separately, the occurrence of ICH per 100 patient-years was much lower among the patients prescribed DOACs than those who were prescribed warfarin (1.52 vs. 3.51), whereas the occurrence of major bleeding outside the brain was similar (0.91 vs. 1.15).
Similar efficacy, better safety
Commenting on the study at an ISC press conference, Mitchell Elkind, MD, immediate past president of the American Heart Association/American Stroke Association and professor of neurology at Columbia University, New York, said: “The community has been concerned about extending the use of these new direct-acting oral anticoagulant drugs to cerebral venous thrombosis, but this study suggests that these patients may benefit from these new agents too.”
Tudor Jovin, MD, chair of neurology at Cooper University Hospital, Cherry Hill, New Jersey, also commented: “This study confirms what we already know from other indications about these DOAC drugs: that they have similar efficacy to warfarin but a better safety profile. These results are really spot on with that. These drugs are also much easier and more convenient to use than warfarin.”
“This is a great step forward,” he added. “Only 30% of patients in this study received DOACs, reflecting the fact that clinicians may be a little reluctant to use them in this condition. But this study now has the potential to change practice.”
In an editorial accompanying the publication in Stroke, Johnathon Gorman, MD, and Thalia Field, MD, from the Vancouver Stroke Program at the University of British Columbia, say that despite its methodological limitations, the ACTION-CVT study “provides added value to the current state of knowledge by virtue of its size and ‘real world’ setting that is reflective of how DOACs are being used to manage CVT in current clinical practice.”
They point out that although baseline characteristics between the DOAC and warfarin groups were similar, the possibility of confounding cannot be excluded, and “other characteristics not easily captured in a retrospective study may sway anticoagulation strategy.”
They acknowledge, however, that an additional propensity score analysis “provides reassurance that the groups are reasonably balanced, adjusting for variables associated with recurrent cerebral venous thrombosis, recanalization, and hemorrhage.”
The editorialists conclude that ACTION-CVT gives additional reassurance for DOACs as an alternative approach to warfarin as a treatment for cerebral venous thrombosis and for the shifts in clinical practice that are already occurring at many centers.
The study was partially supported by the Italian Ministry of Health Ricerca Corrente–IRCCS MultiMedica. Dr. Bakradze reports no disclosures. Dr. Field is the principal investigator of the SECRET trial, which received in-kind study medication from Bayer Canada. She reports honoraria from HLS Therapeutics outside the submitted work and is on the board of Destine Health. The other editorialist reports no conflicts.
A version of this article first appeared on Medscape.com.
and are less likely to result in major bleeding, a retrospective study suggests.
The ACTION CVT study was presented at the International Stroke Conference (ISC) 2022 by Ekaterina Bakradze, MD, assistant professor of neurology at the University of Alabama at Birmingham.
It was also simultaneously published online in Stroke.
“This real-world data supports use of direct oral anticoagulant drugs as a reasonable alternative to warfarin in patients with cerebral venous thrombosis,” Dr. Bakradze concluded.
But she added that because this study was based on retrospective observational data, the findings should be interpreted with caution and require confirmation by larger prospective studies.
Two such studies are now underway: the Direct Oral Anticoagulants in the Treatment of Cerebral Venous Thrombosis (DOAC-CVT) study and the randomized Study of Rivaroxaban for Cerebral Venous Thrombosis (SECRET) trial.
Dr. Bakradze explained that cerebral venous thrombosis is a less common cause of stroke and occurs more often in women and younger patients, with a median age of 37 years. Current recommended treatment consists of heparin followed by oral anticoagulation.
She noted that although randomized trials and current guidelines indicate that DOACs are a preferred alternative to warfarin for the treatment of patients with venous thromboembolism, there are limited data on their use in patients with CVT.
A small, randomized trial (RESPECT-CVT) showed no significant difference in efficacy and safety outcomes between dabigatran and warfarin in patients with cerebral venous thrombosis, but with only 120 patients, this trial was too small for definite answers to this question.
A better understanding of this issue is important, because the mechanisms underlying cerebral venous thrombosis and other thromboembolism and their subsequent risks may differ, Dr. Bakradze said.
As randomized trials in patients with cerebral venous thrombosis are difficult to perform because the condition has a low incidence and low event rates, the researchers decided to look at this question with a large retrospective multicenter study.
The ACTION-CVT study involved 845 consecutive patients with cerebral venous thrombosis over 6 years (from January 2015 and December 2020) from 27 centers in Italy, New Zealand, Switzerland, and the United States. Patients were identified from medical records with diagnostic codes and confirmed with imaging.
The primary predictor in the study was oral anticoagulant type (DOAC vs. warfarin). Study outcomes were abstracted by individual sites through review of all available medical records.
The primary outcome was recurrent venous thrombosis (venous thromboembolism or cerebral venous thrombosis) during follow-up. Imaging outcomes based on recanalization status on last venous imaging study abstracted from radiology reports were also reported.
The safety outcome was major hemorrhage, defined as new or worsening intracranial hemorrhage (ICH), or major extracranial hemorrhage. Results were adjusted for age, sex, and relevant medical conditions.
The mean age of the patients included was 44.8 years, 64.7% were women, 33% received DOAC only, 51.8% received warfarin only, and 15.1% received both treatments at different times.
Results showed that during a median follow-up of 345 days, there were 5.68 recurrent venous thrombosis events, 3.77 major hemorrhages, and 1.84 deaths per 100 patient-years.
Among 525 patients who met recanalization analysis inclusion criteria, 36.6% had complete, 48.2% had partial, and 15.2% had no recanalization.
When compared with warfarin, DOAC treatment was associated with similar risk for recurrent venous thrombosis (adjusted hazard ratio, 0.94; 95% confidence interval, 0.51-1.73; P = .84), death (aHR, 0.71, 95% CI, 0.24-2.08; P = .53), and rate of partial/complete recanalization (aHR, 0.92, 95% CI, 0.48-1.73; P = .79).
But patients who received a DOAC had a significantly lower rate of major hemorrhage (aHR, 0.35; 95% CI, 0.15-0.81; P = .02).
When examined separately, the occurrence of ICH per 100 patient-years was much lower among the patients prescribed DOACs than those who were prescribed warfarin (1.52 vs. 3.51), whereas the occurrence of major bleeding outside the brain was similar (0.91 vs. 1.15).
Similar efficacy, better safety
Commenting on the study at an ISC press conference, Mitchell Elkind, MD, immediate past president of the American Heart Association/American Stroke Association and professor of neurology at Columbia University, New York, said: “The community has been concerned about extending the use of these new direct-acting oral anticoagulant drugs to cerebral venous thrombosis, but this study suggests that these patients may benefit from these new agents too.”
Tudor Jovin, MD, chair of neurology at Cooper University Hospital, Cherry Hill, New Jersey, also commented: “This study confirms what we already know from other indications about these DOAC drugs: that they have similar efficacy to warfarin but a better safety profile. These results are really spot on with that. These drugs are also much easier and more convenient to use than warfarin.”
“This is a great step forward,” he added. “Only 30% of patients in this study received DOACs, reflecting the fact that clinicians may be a little reluctant to use them in this condition. But this study now has the potential to change practice.”
In an editorial accompanying the publication in Stroke, Johnathon Gorman, MD, and Thalia Field, MD, from the Vancouver Stroke Program at the University of British Columbia, say that despite its methodological limitations, the ACTION-CVT study “provides added value to the current state of knowledge by virtue of its size and ‘real world’ setting that is reflective of how DOACs are being used to manage CVT in current clinical practice.”
They point out that although baseline characteristics between the DOAC and warfarin groups were similar, the possibility of confounding cannot be excluded, and “other characteristics not easily captured in a retrospective study may sway anticoagulation strategy.”
They acknowledge, however, that an additional propensity score analysis “provides reassurance that the groups are reasonably balanced, adjusting for variables associated with recurrent cerebral venous thrombosis, recanalization, and hemorrhage.”
The editorialists conclude that ACTION-CVT gives additional reassurance for DOACs as an alternative approach to warfarin as a treatment for cerebral venous thrombosis and for the shifts in clinical practice that are already occurring at many centers.
The study was partially supported by the Italian Ministry of Health Ricerca Corrente–IRCCS MultiMedica. Dr. Bakradze reports no disclosures. Dr. Field is the principal investigator of the SECRET trial, which received in-kind study medication from Bayer Canada. She reports honoraria from HLS Therapeutics outside the submitted work and is on the board of Destine Health. The other editorialist reports no conflicts.
A version of this article first appeared on Medscape.com.
and are less likely to result in major bleeding, a retrospective study suggests.
The ACTION CVT study was presented at the International Stroke Conference (ISC) 2022 by Ekaterina Bakradze, MD, assistant professor of neurology at the University of Alabama at Birmingham.
It was also simultaneously published online in Stroke.
“This real-world data supports use of direct oral anticoagulant drugs as a reasonable alternative to warfarin in patients with cerebral venous thrombosis,” Dr. Bakradze concluded.
But she added that because this study was based on retrospective observational data, the findings should be interpreted with caution and require confirmation by larger prospective studies.
Two such studies are now underway: the Direct Oral Anticoagulants in the Treatment of Cerebral Venous Thrombosis (DOAC-CVT) study and the randomized Study of Rivaroxaban for Cerebral Venous Thrombosis (SECRET) trial.
Dr. Bakradze explained that cerebral venous thrombosis is a less common cause of stroke and occurs more often in women and younger patients, with a median age of 37 years. Current recommended treatment consists of heparin followed by oral anticoagulation.
She noted that although randomized trials and current guidelines indicate that DOACs are a preferred alternative to warfarin for the treatment of patients with venous thromboembolism, there are limited data on their use in patients with CVT.
A small, randomized trial (RESPECT-CVT) showed no significant difference in efficacy and safety outcomes between dabigatran and warfarin in patients with cerebral venous thrombosis, but with only 120 patients, this trial was too small for definite answers to this question.
A better understanding of this issue is important, because the mechanisms underlying cerebral venous thrombosis and other thromboembolism and their subsequent risks may differ, Dr. Bakradze said.
As randomized trials in patients with cerebral venous thrombosis are difficult to perform because the condition has a low incidence and low event rates, the researchers decided to look at this question with a large retrospective multicenter study.
The ACTION-CVT study involved 845 consecutive patients with cerebral venous thrombosis over 6 years (from January 2015 and December 2020) from 27 centers in Italy, New Zealand, Switzerland, and the United States. Patients were identified from medical records with diagnostic codes and confirmed with imaging.
The primary predictor in the study was oral anticoagulant type (DOAC vs. warfarin). Study outcomes were abstracted by individual sites through review of all available medical records.
The primary outcome was recurrent venous thrombosis (venous thromboembolism or cerebral venous thrombosis) during follow-up. Imaging outcomes based on recanalization status on last venous imaging study abstracted from radiology reports were also reported.
The safety outcome was major hemorrhage, defined as new or worsening intracranial hemorrhage (ICH), or major extracranial hemorrhage. Results were adjusted for age, sex, and relevant medical conditions.
The mean age of the patients included was 44.8 years, 64.7% were women, 33% received DOAC only, 51.8% received warfarin only, and 15.1% received both treatments at different times.
Results showed that during a median follow-up of 345 days, there were 5.68 recurrent venous thrombosis events, 3.77 major hemorrhages, and 1.84 deaths per 100 patient-years.
Among 525 patients who met recanalization analysis inclusion criteria, 36.6% had complete, 48.2% had partial, and 15.2% had no recanalization.
When compared with warfarin, DOAC treatment was associated with similar risk for recurrent venous thrombosis (adjusted hazard ratio, 0.94; 95% confidence interval, 0.51-1.73; P = .84), death (aHR, 0.71, 95% CI, 0.24-2.08; P = .53), and rate of partial/complete recanalization (aHR, 0.92, 95% CI, 0.48-1.73; P = .79).
But patients who received a DOAC had a significantly lower rate of major hemorrhage (aHR, 0.35; 95% CI, 0.15-0.81; P = .02).
When examined separately, the occurrence of ICH per 100 patient-years was much lower among the patients prescribed DOACs than those who were prescribed warfarin (1.52 vs. 3.51), whereas the occurrence of major bleeding outside the brain was similar (0.91 vs. 1.15).
Similar efficacy, better safety
Commenting on the study at an ISC press conference, Mitchell Elkind, MD, immediate past president of the American Heart Association/American Stroke Association and professor of neurology at Columbia University, New York, said: “The community has been concerned about extending the use of these new direct-acting oral anticoagulant drugs to cerebral venous thrombosis, but this study suggests that these patients may benefit from these new agents too.”
Tudor Jovin, MD, chair of neurology at Cooper University Hospital, Cherry Hill, New Jersey, also commented: “This study confirms what we already know from other indications about these DOAC drugs: that they have similar efficacy to warfarin but a better safety profile. These results are really spot on with that. These drugs are also much easier and more convenient to use than warfarin.”
“This is a great step forward,” he added. “Only 30% of patients in this study received DOACs, reflecting the fact that clinicians may be a little reluctant to use them in this condition. But this study now has the potential to change practice.”
In an editorial accompanying the publication in Stroke, Johnathon Gorman, MD, and Thalia Field, MD, from the Vancouver Stroke Program at the University of British Columbia, say that despite its methodological limitations, the ACTION-CVT study “provides added value to the current state of knowledge by virtue of its size and ‘real world’ setting that is reflective of how DOACs are being used to manage CVT in current clinical practice.”
They point out that although baseline characteristics between the DOAC and warfarin groups were similar, the possibility of confounding cannot be excluded, and “other characteristics not easily captured in a retrospective study may sway anticoagulation strategy.”
They acknowledge, however, that an additional propensity score analysis “provides reassurance that the groups are reasonably balanced, adjusting for variables associated with recurrent cerebral venous thrombosis, recanalization, and hemorrhage.”
The editorialists conclude that ACTION-CVT gives additional reassurance for DOACs as an alternative approach to warfarin as a treatment for cerebral venous thrombosis and for the shifts in clinical practice that are already occurring at many centers.
The study was partially supported by the Italian Ministry of Health Ricerca Corrente–IRCCS MultiMedica. Dr. Bakradze reports no disclosures. Dr. Field is the principal investigator of the SECRET trial, which received in-kind study medication from Bayer Canada. She reports honoraria from HLS Therapeutics outside the submitted work and is on the board of Destine Health. The other editorialist reports no conflicts.
A version of this article first appeared on Medscape.com.
From ISC 2022