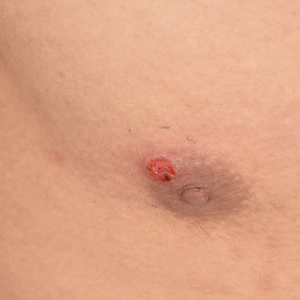
User login
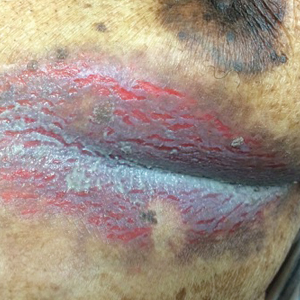
Inframammary Macerated Erosion
The Diagnosis: Hailey-Hailey Disease (Benign Familial Chronic Pemphigus)
Our patient had a long-standing history of Hailey-Hailey disease, as confirmed by multiple prior skin biopsies at outside institutions as well as our affiliated site. He began treatment with oral doxycycline 50 mg twice daily for 2 weeks, triamcinolone cream 0.1% twice daily to the affected region, and aluminum acetate solution soaks and chlorhexidine wash daily along with petroleum jelly, which resulted in good control of the disease. The differential diagnosis of eroded plaques, particularly in the axillary, crural, and inframammary folds, is broad and includes candidiasis, inverse psoriasis, contact dermatitis, dermatophyte infection, pemphigus vegetans or foliaceus, and granular parakeratosis.
Hailey-Hailey disease is a genetic disorder with a prevalence of 1 in 50,000 individuals. Most patients develop symptoms during the second or third decades of life.1 Hailey-Hailey disease exhibits an autosomal-dominant pattern of inheritance secondary to mutation in the human ATP2C1 gene, which codes for the ATPase secretory pathway of the Ca2+ transporting pump type 1 (SPCA1) localized in the Golgi apparatus.2 Altered SPCA1 protein reduces concentration of Ca2+ within the Golgi lumen, which in turn impairs the processing of junctional proteins needed for normal cell-to-cell adhesion.1
Clinically, Hailey-Hailey disease is characterized by vesicular or erosive plaques that have a predilection for intertriginous areas of the body.1 The primary lesions often are flaccid vesicles that easily rupture, leaving behind crusted erosions that spread peripherally. The lesions also can appear as macerated plaques resembling torn tissue paper, as in our case. Friction, heat, and sweat exacerbate the disease. Complications occur from secondary bacterial, fungal, and viral colonization. Malodor and vegetations can indicate bacterial or fungal infections and can lead to persistence of skin lesions. Herpes simplex virus infections can exacerbate preexisting lesions.3 Hailey-Hailey disease of the anogenital region also can be complicated by infection with oncogenic strains of human papillomavirus and lead to cutaneous squamous cell carcinoma.4
Hailey-Hailey disease histologically appears as suprabasal and intraepidermal keratinocyte acantholysis,5 which typically is widespread in the epidermis, with large areas of dyscohesion with a dilapidated brick wall-like appearance.1 In more chronic lesions, epidermal hyperplasia, parakeratosis, and focal crusts may be observed. A moderate perivascular lymphocytic infiltrate can be observed in the superficial dermis. Direct immunofluorescence typically is negative.
Topical corticosteroids and antimicrobials are first-line therapies that often only provide temporary suppression. When the disease is refractory to topical therapies, intralesional corticosteroids may be attempted. There is no strong evidence to support the use of systemic therapy, aside from antimicrobial agents (eg, doxycycline) for the use of superinfections. In severe cases, immunomodulating therapies such as prednisone, cyclosporine, methotrexate, dapsone, alefacept, and oral retinoids may be effective.6-8 Surgical therapy also can be considered for recalcitrant disease, including wide excision and grafting, though these techniques can be associated with morbidity.9
Superficial ablative techniques including dermabrasion, laser therapy with CO2 and erbium-doped YAG, photodynamic therapy, and electron beam radiation have been shown to be effective modalities in severe cases.5,9-11 It has been hypothesized that keratinocytes expressing the molecular defect are ablated, while the surrounding normal adnexal epithelium can regenerate normal epithelium. It also is thought that dermal fibrosis leads to better support of the diseased epidermis and decreases the risk for ulceration and fissuring.9
- Hohl D. Darier disease and Hailey-Hailey disease. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:887-896.
- Micaroni M, Giacchetti G, Plebani R, et al. ATP2C1 gene mutations in Hailey-Hailey disease and possible roles of SPCA1 isoforms in membrane trafficking. Cell Death Dis. 2016;7:E2259.
- Peppiatt T, Keefe M,White JE. Hailey-Hailey disease--exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 1992;17:201-202.
- Chen MY, Chiu HC, Su LH, et al. Presence of human papillomavirus type 6 DNA in the perineal verrucoid lesions of Hailey-Hailey disease. J Eur Acad Dermatol Venereol. 2006;20:1356-1357.
- Graham PM, Melkonian A, Fivenson D. Familial benign chronic pemphigus (Hailey-Hailey disease) treated with electron beam radiation. JAAD Case Rep. 2016;2:159-161.
- Berth-Jones J, Smith SG, Graham-Brown RA, et al. Benign familial chronic pemphigus (Hailey-Hailey disease) responds to cyclosporin. Clin Exp Dermatol. 1995;20:70-72.
- Sire DJ, Johnson BL. Benign familial chronic pemphigus treated with dapsone. Arch Dermatol. 1971;103:262-265.
- Hunt MJ, Salisbury EL, Painter DM. Vesiculobullous Hailey-Hailey disease: successful treatment with oral retinoids. Australas J Dermatol. 1996;37:196-198.
- Ortiz AE, Zachary CB. Laser therapy for Hailey-Hailey disease: review of the literature and a case report. Dermatol Reports. 2011;3:E28.
- Don PC, Carney PS, Lynch WS, et al. Carbon dioxide laserabrasion: a new approach to management of familial benign chronic pemphigus (Hailey-Hailey disease). J Dermatol Surg Oncol. 1987;13:1187-1194.
- Beier C, Kaufmann R. Efficacy of erbium:YAG laser ablation in Darier disease and Hailey-Hailey disease. Arch Dermatol. 1999;135:423-427.
The Diagnosis: Hailey-Hailey Disease (Benign Familial Chronic Pemphigus)
Our patient had a long-standing history of Hailey-Hailey disease, as confirmed by multiple prior skin biopsies at outside institutions as well as our affiliated site. He began treatment with oral doxycycline 50 mg twice daily for 2 weeks, triamcinolone cream 0.1% twice daily to the affected region, and aluminum acetate solution soaks and chlorhexidine wash daily along with petroleum jelly, which resulted in good control of the disease. The differential diagnosis of eroded plaques, particularly in the axillary, crural, and inframammary folds, is broad and includes candidiasis, inverse psoriasis, contact dermatitis, dermatophyte infection, pemphigus vegetans or foliaceus, and granular parakeratosis.
Hailey-Hailey disease is a genetic disorder with a prevalence of 1 in 50,000 individuals. Most patients develop symptoms during the second or third decades of life.1 Hailey-Hailey disease exhibits an autosomal-dominant pattern of inheritance secondary to mutation in the human ATP2C1 gene, which codes for the ATPase secretory pathway of the Ca2+ transporting pump type 1 (SPCA1) localized in the Golgi apparatus.2 Altered SPCA1 protein reduces concentration of Ca2+ within the Golgi lumen, which in turn impairs the processing of junctional proteins needed for normal cell-to-cell adhesion.1
Clinically, Hailey-Hailey disease is characterized by vesicular or erosive plaques that have a predilection for intertriginous areas of the body.1 The primary lesions often are flaccid vesicles that easily rupture, leaving behind crusted erosions that spread peripherally. The lesions also can appear as macerated plaques resembling torn tissue paper, as in our case. Friction, heat, and sweat exacerbate the disease. Complications occur from secondary bacterial, fungal, and viral colonization. Malodor and vegetations can indicate bacterial or fungal infections and can lead to persistence of skin lesions. Herpes simplex virus infections can exacerbate preexisting lesions.3 Hailey-Hailey disease of the anogenital region also can be complicated by infection with oncogenic strains of human papillomavirus and lead to cutaneous squamous cell carcinoma.4
Hailey-Hailey disease histologically appears as suprabasal and intraepidermal keratinocyte acantholysis,5 which typically is widespread in the epidermis, with large areas of dyscohesion with a dilapidated brick wall-like appearance.1 In more chronic lesions, epidermal hyperplasia, parakeratosis, and focal crusts may be observed. A moderate perivascular lymphocytic infiltrate can be observed in the superficial dermis. Direct immunofluorescence typically is negative.
Topical corticosteroids and antimicrobials are first-line therapies that often only provide temporary suppression. When the disease is refractory to topical therapies, intralesional corticosteroids may be attempted. There is no strong evidence to support the use of systemic therapy, aside from antimicrobial agents (eg, doxycycline) for the use of superinfections. In severe cases, immunomodulating therapies such as prednisone, cyclosporine, methotrexate, dapsone, alefacept, and oral retinoids may be effective.6-8 Surgical therapy also can be considered for recalcitrant disease, including wide excision and grafting, though these techniques can be associated with morbidity.9
Superficial ablative techniques including dermabrasion, laser therapy with CO2 and erbium-doped YAG, photodynamic therapy, and electron beam radiation have been shown to be effective modalities in severe cases.5,9-11 It has been hypothesized that keratinocytes expressing the molecular defect are ablated, while the surrounding normal adnexal epithelium can regenerate normal epithelium. It also is thought that dermal fibrosis leads to better support of the diseased epidermis and decreases the risk for ulceration and fissuring.9
The Diagnosis: Hailey-Hailey Disease (Benign Familial Chronic Pemphigus)
Our patient had a long-standing history of Hailey-Hailey disease, as confirmed by multiple prior skin biopsies at outside institutions as well as our affiliated site. He began treatment with oral doxycycline 50 mg twice daily for 2 weeks, triamcinolone cream 0.1% twice daily to the affected region, and aluminum acetate solution soaks and chlorhexidine wash daily along with petroleum jelly, which resulted in good control of the disease. The differential diagnosis of eroded plaques, particularly in the axillary, crural, and inframammary folds, is broad and includes candidiasis, inverse psoriasis, contact dermatitis, dermatophyte infection, pemphigus vegetans or foliaceus, and granular parakeratosis.
Hailey-Hailey disease is a genetic disorder with a prevalence of 1 in 50,000 individuals. Most patients develop symptoms during the second or third decades of life.1 Hailey-Hailey disease exhibits an autosomal-dominant pattern of inheritance secondary to mutation in the human ATP2C1 gene, which codes for the ATPase secretory pathway of the Ca2+ transporting pump type 1 (SPCA1) localized in the Golgi apparatus.2 Altered SPCA1 protein reduces concentration of Ca2+ within the Golgi lumen, which in turn impairs the processing of junctional proteins needed for normal cell-to-cell adhesion.1
Clinically, Hailey-Hailey disease is characterized by vesicular or erosive plaques that have a predilection for intertriginous areas of the body.1 The primary lesions often are flaccid vesicles that easily rupture, leaving behind crusted erosions that spread peripherally. The lesions also can appear as macerated plaques resembling torn tissue paper, as in our case. Friction, heat, and sweat exacerbate the disease. Complications occur from secondary bacterial, fungal, and viral colonization. Malodor and vegetations can indicate bacterial or fungal infections and can lead to persistence of skin lesions. Herpes simplex virus infections can exacerbate preexisting lesions.3 Hailey-Hailey disease of the anogenital region also can be complicated by infection with oncogenic strains of human papillomavirus and lead to cutaneous squamous cell carcinoma.4
Hailey-Hailey disease histologically appears as suprabasal and intraepidermal keratinocyte acantholysis,5 which typically is widespread in the epidermis, with large areas of dyscohesion with a dilapidated brick wall-like appearance.1 In more chronic lesions, epidermal hyperplasia, parakeratosis, and focal crusts may be observed. A moderate perivascular lymphocytic infiltrate can be observed in the superficial dermis. Direct immunofluorescence typically is negative.
Topical corticosteroids and antimicrobials are first-line therapies that often only provide temporary suppression. When the disease is refractory to topical therapies, intralesional corticosteroids may be attempted. There is no strong evidence to support the use of systemic therapy, aside from antimicrobial agents (eg, doxycycline) for the use of superinfections. In severe cases, immunomodulating therapies such as prednisone, cyclosporine, methotrexate, dapsone, alefacept, and oral retinoids may be effective.6-8 Surgical therapy also can be considered for recalcitrant disease, including wide excision and grafting, though these techniques can be associated with morbidity.9
Superficial ablative techniques including dermabrasion, laser therapy with CO2 and erbium-doped YAG, photodynamic therapy, and electron beam radiation have been shown to be effective modalities in severe cases.5,9-11 It has been hypothesized that keratinocytes expressing the molecular defect are ablated, while the surrounding normal adnexal epithelium can regenerate normal epithelium. It also is thought that dermal fibrosis leads to better support of the diseased epidermis and decreases the risk for ulceration and fissuring.9
- Hohl D. Darier disease and Hailey-Hailey disease. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:887-896.
- Micaroni M, Giacchetti G, Plebani R, et al. ATP2C1 gene mutations in Hailey-Hailey disease and possible roles of SPCA1 isoforms in membrane trafficking. Cell Death Dis. 2016;7:E2259.
- Peppiatt T, Keefe M,White JE. Hailey-Hailey disease--exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 1992;17:201-202.
- Chen MY, Chiu HC, Su LH, et al. Presence of human papillomavirus type 6 DNA in the perineal verrucoid lesions of Hailey-Hailey disease. J Eur Acad Dermatol Venereol. 2006;20:1356-1357.
- Graham PM, Melkonian A, Fivenson D. Familial benign chronic pemphigus (Hailey-Hailey disease) treated with electron beam radiation. JAAD Case Rep. 2016;2:159-161.
- Berth-Jones J, Smith SG, Graham-Brown RA, et al. Benign familial chronic pemphigus (Hailey-Hailey disease) responds to cyclosporin. Clin Exp Dermatol. 1995;20:70-72.
- Sire DJ, Johnson BL. Benign familial chronic pemphigus treated with dapsone. Arch Dermatol. 1971;103:262-265.
- Hunt MJ, Salisbury EL, Painter DM. Vesiculobullous Hailey-Hailey disease: successful treatment with oral retinoids. Australas J Dermatol. 1996;37:196-198.
- Ortiz AE, Zachary CB. Laser therapy for Hailey-Hailey disease: review of the literature and a case report. Dermatol Reports. 2011;3:E28.
- Don PC, Carney PS, Lynch WS, et al. Carbon dioxide laserabrasion: a new approach to management of familial benign chronic pemphigus (Hailey-Hailey disease). J Dermatol Surg Oncol. 1987;13:1187-1194.
- Beier C, Kaufmann R. Efficacy of erbium:YAG laser ablation in Darier disease and Hailey-Hailey disease. Arch Dermatol. 1999;135:423-427.
- Hohl D. Darier disease and Hailey-Hailey disease. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:887-896.
- Micaroni M, Giacchetti G, Plebani R, et al. ATP2C1 gene mutations in Hailey-Hailey disease and possible roles of SPCA1 isoforms in membrane trafficking. Cell Death Dis. 2016;7:E2259.
- Peppiatt T, Keefe M,White JE. Hailey-Hailey disease--exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 1992;17:201-202.
- Chen MY, Chiu HC, Su LH, et al. Presence of human papillomavirus type 6 DNA in the perineal verrucoid lesions of Hailey-Hailey disease. J Eur Acad Dermatol Venereol. 2006;20:1356-1357.
- Graham PM, Melkonian A, Fivenson D. Familial benign chronic pemphigus (Hailey-Hailey disease) treated with electron beam radiation. JAAD Case Rep. 2016;2:159-161.
- Berth-Jones J, Smith SG, Graham-Brown RA, et al. Benign familial chronic pemphigus (Hailey-Hailey disease) responds to cyclosporin. Clin Exp Dermatol. 1995;20:70-72.
- Sire DJ, Johnson BL. Benign familial chronic pemphigus treated with dapsone. Arch Dermatol. 1971;103:262-265.
- Hunt MJ, Salisbury EL, Painter DM. Vesiculobullous Hailey-Hailey disease: successful treatment with oral retinoids. Australas J Dermatol. 1996;37:196-198.
- Ortiz AE, Zachary CB. Laser therapy for Hailey-Hailey disease: review of the literature and a case report. Dermatol Reports. 2011;3:E28.
- Don PC, Carney PS, Lynch WS, et al. Carbon dioxide laserabrasion: a new approach to management of familial benign chronic pemphigus (Hailey-Hailey disease). J Dermatol Surg Oncol. 1987;13:1187-1194.
- Beier C, Kaufmann R. Efficacy of erbium:YAG laser ablation in Darier disease and Hailey-Hailey disease. Arch Dermatol. 1999;135:423-427.
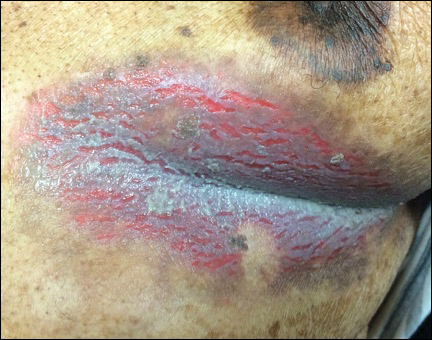
An 81-year-old man presented with a painful erosion in the left inframammary region of 2 weeks' duration. He described the lesion as pruritic and burning. He reported having prior similar episodes in the bilateral groin, axilla, and lower abdomen that often were malodorous. Use of triamcinolone cream 0.1% up to 4 times daily resulted in little relief of the erosion. Of note, the patient reported therapies for prior sites had included oral doxycycline 50 mg twice daily, clobetasol cream, and clindamycin solution, which provided limited relief but eventual resolution. Application of cold aluminum acetate solution compresses for 5 minutes daily irritated the skin even further and led to bleeding at the affected sites. The patient's father had a history of similar skin lesions.
Agminated Papules on the Neck
The Diagnosis: Pseudoxanthoma Elasticum
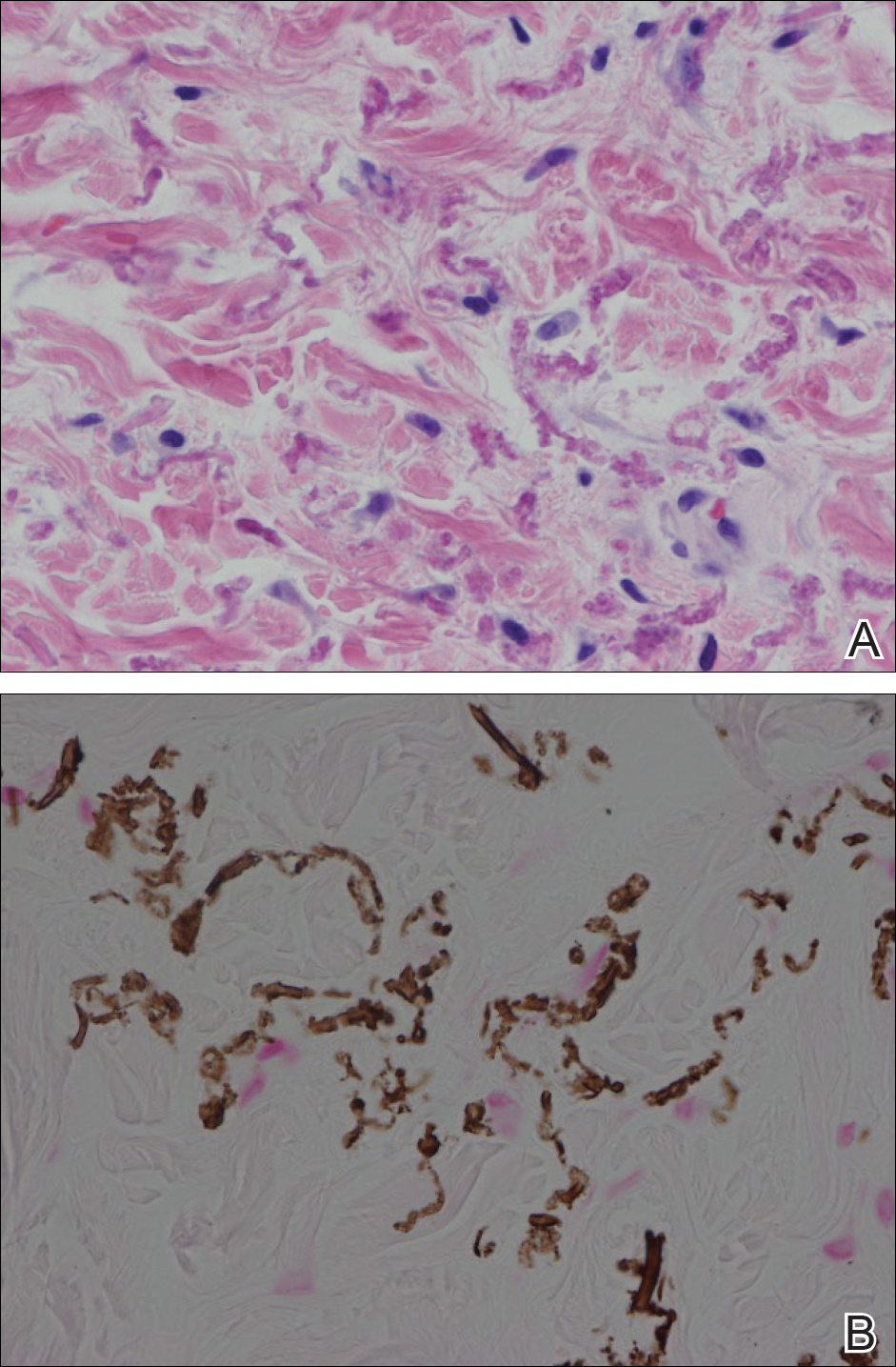
Histopathology showed abnormal curled frayed elastic fibers in the mid dermis (Figure, A); von Kossa stain was positive for calcified and fragmented elastic fibers (Figure, B). Based on clinical and histological findings, a diagnosis of pseudoxanthoma elasticum (PXE) was made.
Pseudoxanthoma elasticum is a rare multisystem heterogeneous genetic disorder that causes abnormal mineralization and fragmentation of tissue elastin fibers. Clinically, accumulation of mineralized elastin fibers leads to soft tissue calcification and late-onset pathology in the dermis, retinal Bruch membrane, and medial layers of large- and medium-sized arterial walls.
Pseudoxanthoma elasticum is an autosomal-recessive disease associated with more than 300 loss mutations in the ATP-binding cassette subfamily C member 6 gene, ABCC6.1,2 However, PXE clinically is characterized by wide variability in clinical progression and outcome as well as phenotypic overlap with other disorders such as generalized arterial calcification of infancy. Pseudoxanthoma elasticum affects an estimated 1 in 25,000 to 100,000 individuals with a female preponderance (2:1 ratio).1-3 Age of onset typically is in the second to third decades of life, with 80% of cases demonstrating skin manifestations before 20 years of age.2,3
The first and most benign finding often is the appearance of small soft asymptomatic yellow papules with a plucked chicken skin-like appearance that occur on the flexural areas such as the neck, axilla, antecubital, popliteal, inguinal, and periumbilical areas. These papules may progress to irregularly shaped, yellowish plaques with a leathery appearance; mucous membranes, often occurring on the inner aspect of the lower lips, also may be involved. More severe abdominal striae also may affect some but not all women with PXE. Histologic examination demonstrates swollen, clumped, and fragmented elastin fibers with calcium deposits in the mid dermis. Elastin-specific stains such as orcein and calcium-specific stains such as the von Kossa stain aid in the diagnosis.
Vision impairment subsequently develops in 50% to 70% of patients, with severe vision loss in 3% to 8% of patients.4,5 Ophthalmologic examination identifies characteristic angioid streaks (ie, gray lines radiating from the optic disk) and subretinal hemorrhages caused by brittle new vessel formation.
Bleeding complications, especially from the gastrointestinal tract, caused by arterial wall fragility may affect 10% of PXE patients.5 Although bleeding complications also may affect the genitourinary system, the risk for fetal loss or adverse reproductive outcomes is considered low.6 More insidiously, progressive arterial calcification and peripheral arterial disease contribute to accelerated atherosclerosis, causing earlier presentations of claudication, angina pectoris, myocardial infarction, and hypertension by the third and fourth decades of life.
Management of PXE is limited. Primary care providers should be attentive to cardiovascular screening for coronary and peripheral arterial disease. Patients should receive regular eye examinations, and choroidal neovascularization should be aggressively treated with photocoagulation, photodynamic therapy, and vascular endothelial growth factor inhibitors.1,3
Collagenous fibromas are slow-growing tumors but are histologically distinct, showing fibrous or myxoid connective tissue arising within adipose tissue. Cutaneous leiomyomas may be solitary or grouped, often painful papules composed histologically of bundles of smooth muscle. Cutaneous sclerosis in sclerosing mesenteritis is a rare cutaneous manifestation of an internal disorder and presents as asymptomatic indurated subcutaneous nodules but histologically is distinctive, demonstrating sclerosis with fat necrosis. Xanthoma disseminatum is a rare form of histiocytosis that commonly presents as hundreds of small yellowish brown or reddish brown papules symmetrically distributed on the face, trunk, and intertriginous areas.
On follow-up within a year after initial presentation, our patient was found to have early subtle angioid streaks on ophthalmologic examination with no vision loss. A transthoracic echocardiogram was performed and showed no cardiac abnormalities. Her pregnancy was complicated by intrauterine growth retardation in the third trimester; however, the patient delivered a healthy-appearing 2835 g neonate (10th percentile for gestational age) at 39 weeks of gestations via an uncomplicated cesarean delivery.
- Uitto J, Bercovitch L, Terry SF, et al. Pseudoxanthoma elasticum: progress in diagnostics and research towards treatment: summary of the 2010 PXE International Research Meeting. Am J Med Genet A. 2011;155A:1517-1526.
- Li Q, Jiang Q, Pfendner E, et al. Pseudoxanthoma elasticum: clinical phenotypes, molecular genetics and putative pathomechanisms. Exp Dermatol. 2009;18:1-11.
- Finger RP, Charbel Issa P, Ladewig MS, et al. Pseudoxanthoma elasticum: genetics, clinical manifestations and therapeutic approaches. Surv Ophthalmol. 2009;54:272-285.
- Li Y, Cui Y, Zhao H, et al. Pseudoxanthoma elasticum: a review of 86 cases in China. Intractable Rare Dis Res. 2014;3:75-78.
- Laube S, Moss C. Pseudoxanthoma elasticum. Arch Dis Child. 2005;90:754-756.
- Bercovitch L, Leroux T, Terry S, et al. Pregnancy and obstetrical outcomes in pseudoxanthoma elasticum. Br J Dermatol. 2004;151:1011-1018.
The Diagnosis: Pseudoxanthoma Elasticum
Histopathology showed abnormal curled frayed elastic fibers in the mid dermis (Figure, A); von Kossa stain was positive for calcified and fragmented elastic fibers (Figure, B). Based on clinical and histological findings, a diagnosis of pseudoxanthoma elasticum (PXE) was made.
Pseudoxanthoma elasticum is a rare multisystem heterogeneous genetic disorder that causes abnormal mineralization and fragmentation of tissue elastin fibers. Clinically, accumulation of mineralized elastin fibers leads to soft tissue calcification and late-onset pathology in the dermis, retinal Bruch membrane, and medial layers of large- and medium-sized arterial walls.
Pseudoxanthoma elasticum is an autosomal-recessive disease associated with more than 300 loss mutations in the ATP-binding cassette subfamily C member 6 gene, ABCC6.1,2 However, PXE clinically is characterized by wide variability in clinical progression and outcome as well as phenotypic overlap with other disorders such as generalized arterial calcification of infancy. Pseudoxanthoma elasticum affects an estimated 1 in 25,000 to 100,000 individuals with a female preponderance (2:1 ratio).1-3 Age of onset typically is in the second to third decades of life, with 80% of cases demonstrating skin manifestations before 20 years of age.2,3
The first and most benign finding often is the appearance of small soft asymptomatic yellow papules with a plucked chicken skin-like appearance that occur on the flexural areas such as the neck, axilla, antecubital, popliteal, inguinal, and periumbilical areas. These papules may progress to irregularly shaped, yellowish plaques with a leathery appearance; mucous membranes, often occurring on the inner aspect of the lower lips, also may be involved. More severe abdominal striae also may affect some but not all women with PXE. Histologic examination demonstrates swollen, clumped, and fragmented elastin fibers with calcium deposits in the mid dermis. Elastin-specific stains such as orcein and calcium-specific stains such as the von Kossa stain aid in the diagnosis.
Vision impairment subsequently develops in 50% to 70% of patients, with severe vision loss in 3% to 8% of patients.4,5 Ophthalmologic examination identifies characteristic angioid streaks (ie, gray lines radiating from the optic disk) and subretinal hemorrhages caused by brittle new vessel formation.
Bleeding complications, especially from the gastrointestinal tract, caused by arterial wall fragility may affect 10% of PXE patients.5 Although bleeding complications also may affect the genitourinary system, the risk for fetal loss or adverse reproductive outcomes is considered low.6 More insidiously, progressive arterial calcification and peripheral arterial disease contribute to accelerated atherosclerosis, causing earlier presentations of claudication, angina pectoris, myocardial infarction, and hypertension by the third and fourth decades of life.
Management of PXE is limited. Primary care providers should be attentive to cardiovascular screening for coronary and peripheral arterial disease. Patients should receive regular eye examinations, and choroidal neovascularization should be aggressively treated with photocoagulation, photodynamic therapy, and vascular endothelial growth factor inhibitors.1,3
Collagenous fibromas are slow-growing tumors but are histologically distinct, showing fibrous or myxoid connective tissue arising within adipose tissue. Cutaneous leiomyomas may be solitary or grouped, often painful papules composed histologically of bundles of smooth muscle. Cutaneous sclerosis in sclerosing mesenteritis is a rare cutaneous manifestation of an internal disorder and presents as asymptomatic indurated subcutaneous nodules but histologically is distinctive, demonstrating sclerosis with fat necrosis. Xanthoma disseminatum is a rare form of histiocytosis that commonly presents as hundreds of small yellowish brown or reddish brown papules symmetrically distributed on the face, trunk, and intertriginous areas.
On follow-up within a year after initial presentation, our patient was found to have early subtle angioid streaks on ophthalmologic examination with no vision loss. A transthoracic echocardiogram was performed and showed no cardiac abnormalities. Her pregnancy was complicated by intrauterine growth retardation in the third trimester; however, the patient delivered a healthy-appearing 2835 g neonate (10th percentile for gestational age) at 39 weeks of gestations via an uncomplicated cesarean delivery.
The Diagnosis: Pseudoxanthoma Elasticum
Histopathology showed abnormal curled frayed elastic fibers in the mid dermis (Figure, A); von Kossa stain was positive for calcified and fragmented elastic fibers (Figure, B). Based on clinical and histological findings, a diagnosis of pseudoxanthoma elasticum (PXE) was made.
Pseudoxanthoma elasticum is a rare multisystem heterogeneous genetic disorder that causes abnormal mineralization and fragmentation of tissue elastin fibers. Clinically, accumulation of mineralized elastin fibers leads to soft tissue calcification and late-onset pathology in the dermis, retinal Bruch membrane, and medial layers of large- and medium-sized arterial walls.
Pseudoxanthoma elasticum is an autosomal-recessive disease associated with more than 300 loss mutations in the ATP-binding cassette subfamily C member 6 gene, ABCC6.1,2 However, PXE clinically is characterized by wide variability in clinical progression and outcome as well as phenotypic overlap with other disorders such as generalized arterial calcification of infancy. Pseudoxanthoma elasticum affects an estimated 1 in 25,000 to 100,000 individuals with a female preponderance (2:1 ratio).1-3 Age of onset typically is in the second to third decades of life, with 80% of cases demonstrating skin manifestations before 20 years of age.2,3
The first and most benign finding often is the appearance of small soft asymptomatic yellow papules with a plucked chicken skin-like appearance that occur on the flexural areas such as the neck, axilla, antecubital, popliteal, inguinal, and periumbilical areas. These papules may progress to irregularly shaped, yellowish plaques with a leathery appearance; mucous membranes, often occurring on the inner aspect of the lower lips, also may be involved. More severe abdominal striae also may affect some but not all women with PXE. Histologic examination demonstrates swollen, clumped, and fragmented elastin fibers with calcium deposits in the mid dermis. Elastin-specific stains such as orcein and calcium-specific stains such as the von Kossa stain aid in the diagnosis.
Vision impairment subsequently develops in 50% to 70% of patients, with severe vision loss in 3% to 8% of patients.4,5 Ophthalmologic examination identifies characteristic angioid streaks (ie, gray lines radiating from the optic disk) and subretinal hemorrhages caused by brittle new vessel formation.
Bleeding complications, especially from the gastrointestinal tract, caused by arterial wall fragility may affect 10% of PXE patients.5 Although bleeding complications also may affect the genitourinary system, the risk for fetal loss or adverse reproductive outcomes is considered low.6 More insidiously, progressive arterial calcification and peripheral arterial disease contribute to accelerated atherosclerosis, causing earlier presentations of claudication, angina pectoris, myocardial infarction, and hypertension by the third and fourth decades of life.
Management of PXE is limited. Primary care providers should be attentive to cardiovascular screening for coronary and peripheral arterial disease. Patients should receive regular eye examinations, and choroidal neovascularization should be aggressively treated with photocoagulation, photodynamic therapy, and vascular endothelial growth factor inhibitors.1,3
Collagenous fibromas are slow-growing tumors but are histologically distinct, showing fibrous or myxoid connective tissue arising within adipose tissue. Cutaneous leiomyomas may be solitary or grouped, often painful papules composed histologically of bundles of smooth muscle. Cutaneous sclerosis in sclerosing mesenteritis is a rare cutaneous manifestation of an internal disorder and presents as asymptomatic indurated subcutaneous nodules but histologically is distinctive, demonstrating sclerosis with fat necrosis. Xanthoma disseminatum is a rare form of histiocytosis that commonly presents as hundreds of small yellowish brown or reddish brown papules symmetrically distributed on the face, trunk, and intertriginous areas.
On follow-up within a year after initial presentation, our patient was found to have early subtle angioid streaks on ophthalmologic examination with no vision loss. A transthoracic echocardiogram was performed and showed no cardiac abnormalities. Her pregnancy was complicated by intrauterine growth retardation in the third trimester; however, the patient delivered a healthy-appearing 2835 g neonate (10th percentile for gestational age) at 39 weeks of gestations via an uncomplicated cesarean delivery.
- Uitto J, Bercovitch L, Terry SF, et al. Pseudoxanthoma elasticum: progress in diagnostics and research towards treatment: summary of the 2010 PXE International Research Meeting. Am J Med Genet A. 2011;155A:1517-1526.
- Li Q, Jiang Q, Pfendner E, et al. Pseudoxanthoma elasticum: clinical phenotypes, molecular genetics and putative pathomechanisms. Exp Dermatol. 2009;18:1-11.
- Finger RP, Charbel Issa P, Ladewig MS, et al. Pseudoxanthoma elasticum: genetics, clinical manifestations and therapeutic approaches. Surv Ophthalmol. 2009;54:272-285.
- Li Y, Cui Y, Zhao H, et al. Pseudoxanthoma elasticum: a review of 86 cases in China. Intractable Rare Dis Res. 2014;3:75-78.
- Laube S, Moss C. Pseudoxanthoma elasticum. Arch Dis Child. 2005;90:754-756.
- Bercovitch L, Leroux T, Terry S, et al. Pregnancy and obstetrical outcomes in pseudoxanthoma elasticum. Br J Dermatol. 2004;151:1011-1018.
- Uitto J, Bercovitch L, Terry SF, et al. Pseudoxanthoma elasticum: progress in diagnostics and research towards treatment: summary of the 2010 PXE International Research Meeting. Am J Med Genet A. 2011;155A:1517-1526.
- Li Q, Jiang Q, Pfendner E, et al. Pseudoxanthoma elasticum: clinical phenotypes, molecular genetics and putative pathomechanisms. Exp Dermatol. 2009;18:1-11.
- Finger RP, Charbel Issa P, Ladewig MS, et al. Pseudoxanthoma elasticum: genetics, clinical manifestations and therapeutic approaches. Surv Ophthalmol. 2009;54:272-285.
- Li Y, Cui Y, Zhao H, et al. Pseudoxanthoma elasticum: a review of 86 cases in China. Intractable Rare Dis Res. 2014;3:75-78.
- Laube S, Moss C. Pseudoxanthoma elasticum. Arch Dis Child. 2005;90:754-756.
- Bercovitch L, Leroux T, Terry S, et al. Pregnancy and obstetrical outcomes in pseudoxanthoma elasticum. Br J Dermatol. 2004;151:1011-1018.
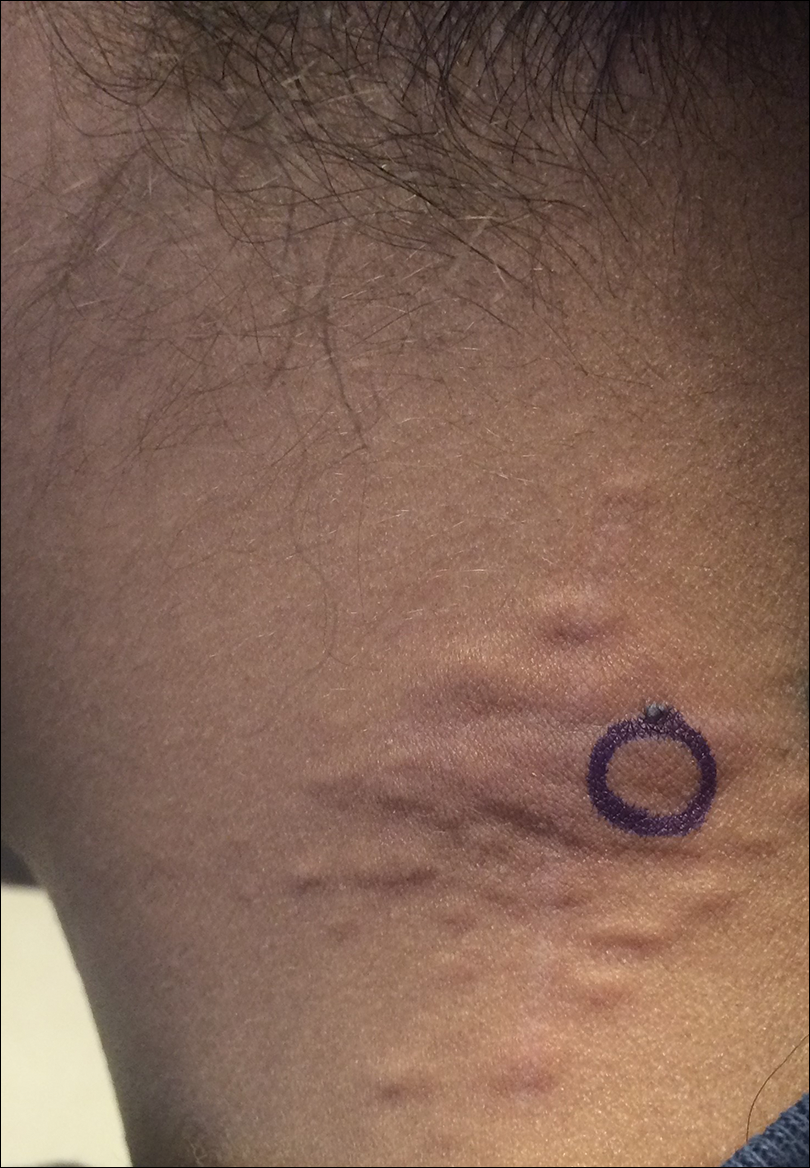
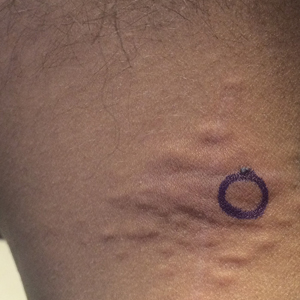
A 24-year-old woman presented with a lesion on the neck of 3 months' duration. She noted occasional mild pruritus at the site but no other symptoms or similar lesions elsewhere. At the time of presentation, she was at 17 weeks of gestation without any complications. Her medical history was notable for hypertension, unspecified chest pain with a normal electrocardiogram, and 2 spontaneous abortions. She denied a personal or family history of notable cardiovascular or gastrointestinal tract diseases. Examination of the skin showed indurated 3- to 5-mm papules coalescing into a 3- to 4-cm plaque on the left posterolateral neck.
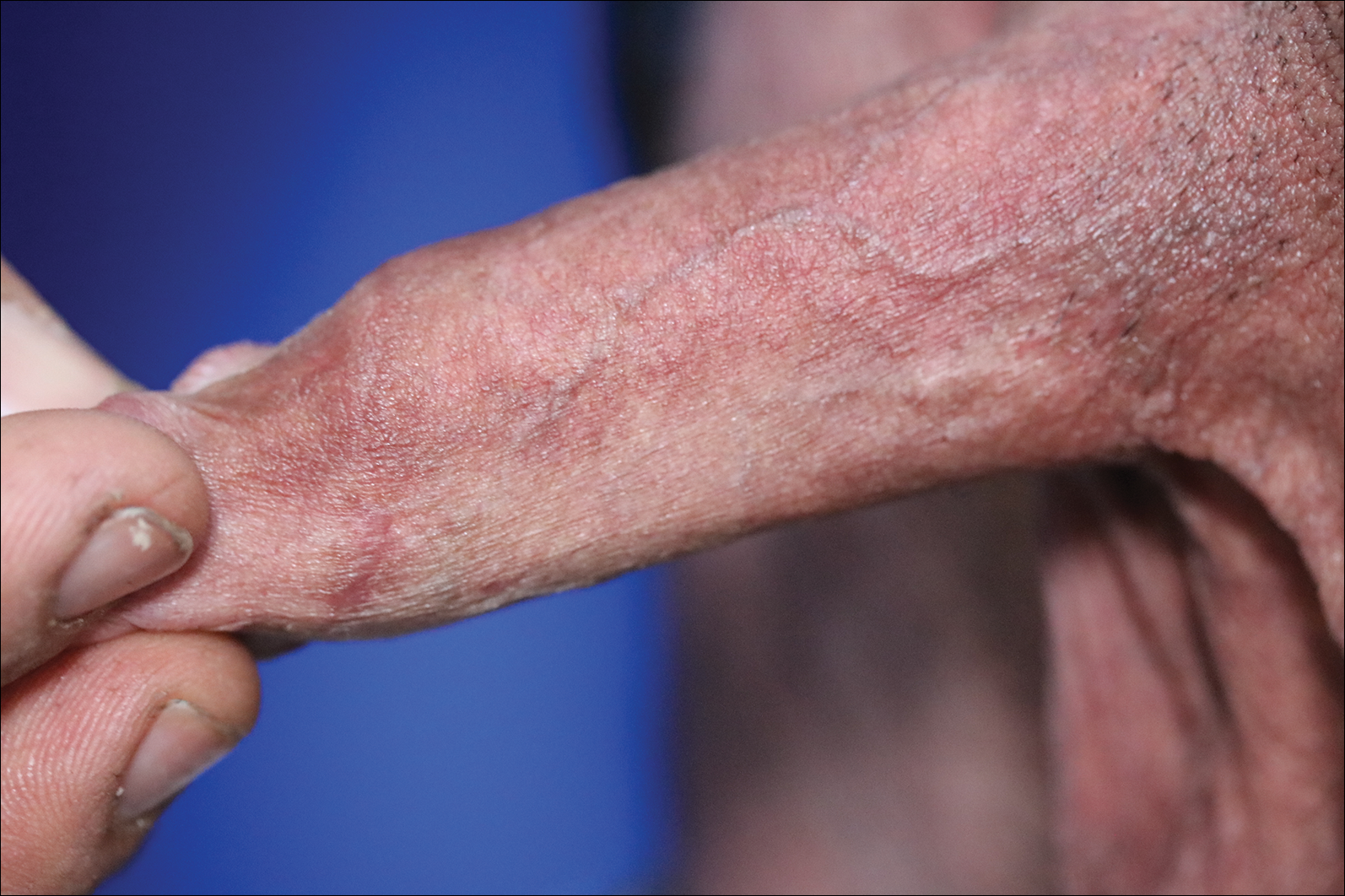
Brown Papules on the Penis
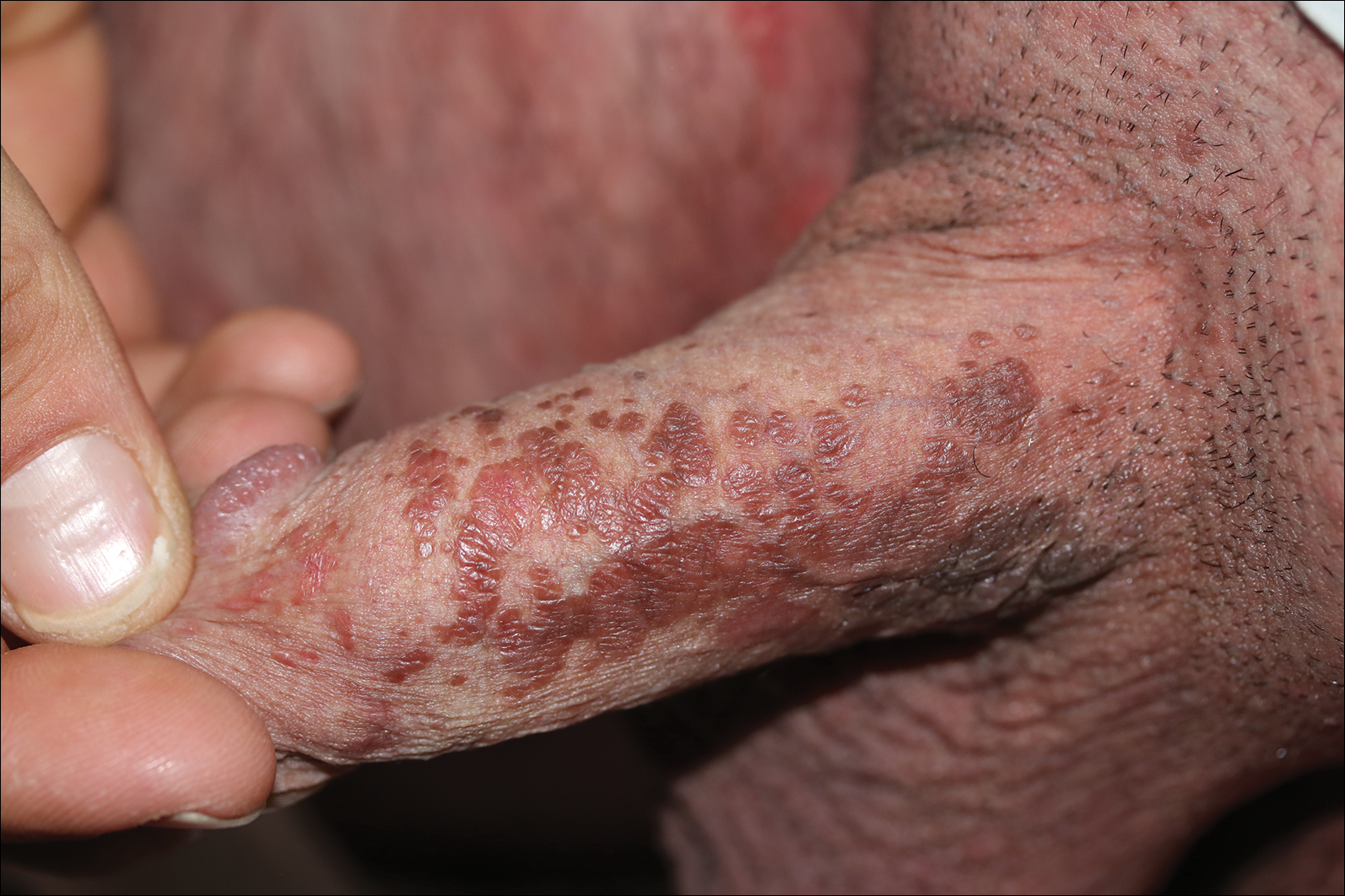
The Diagnosis: Bowenoid Papulosis
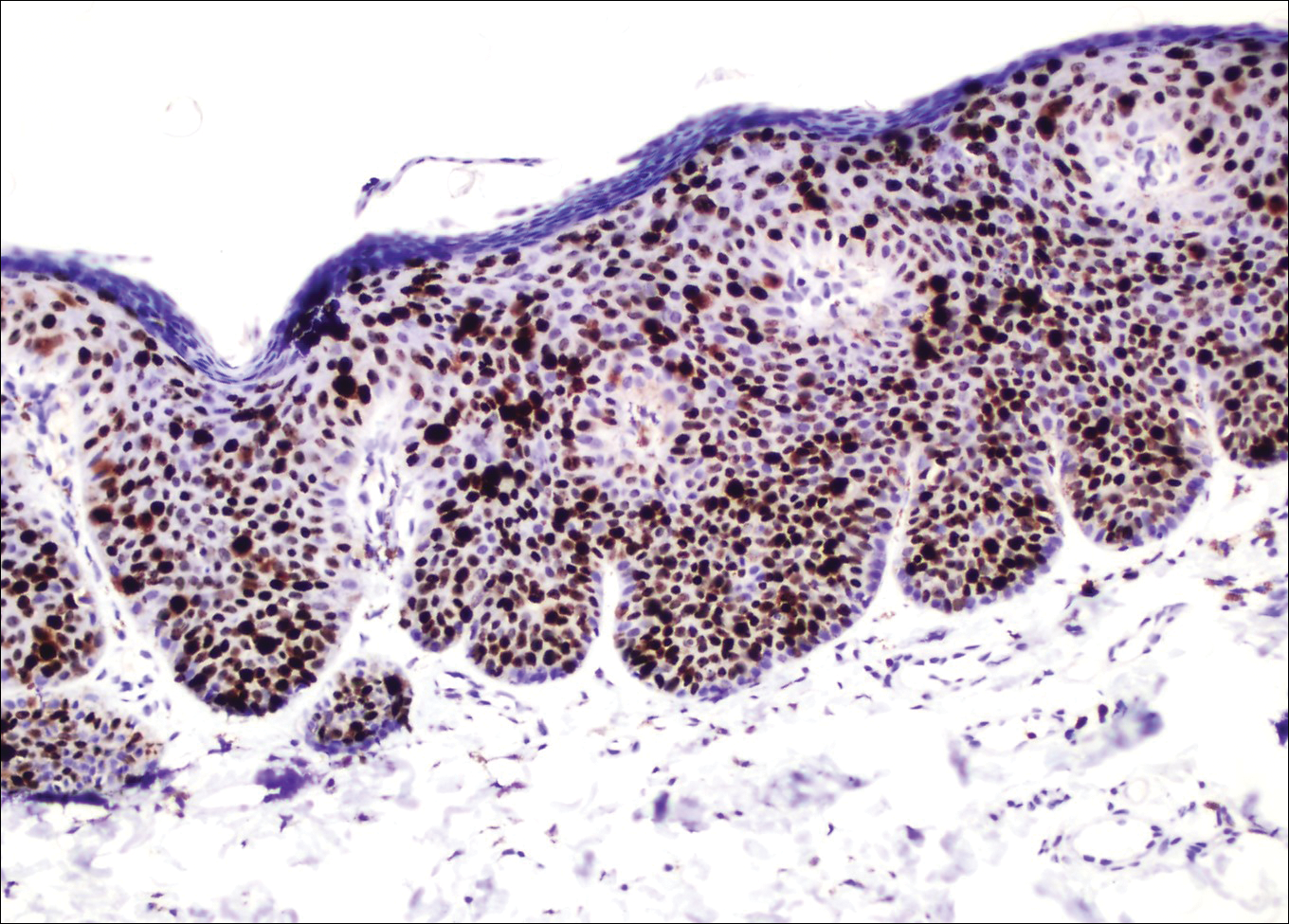
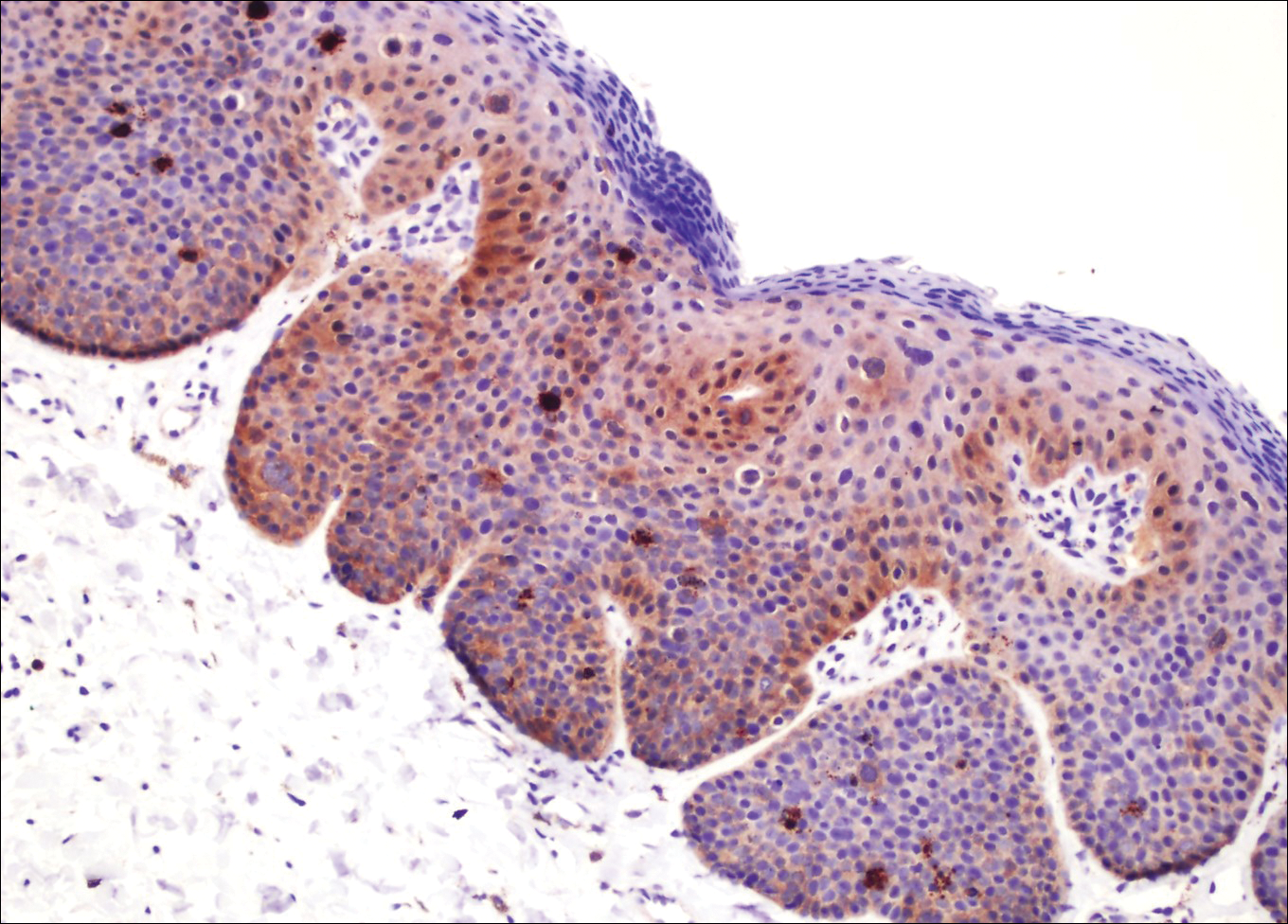
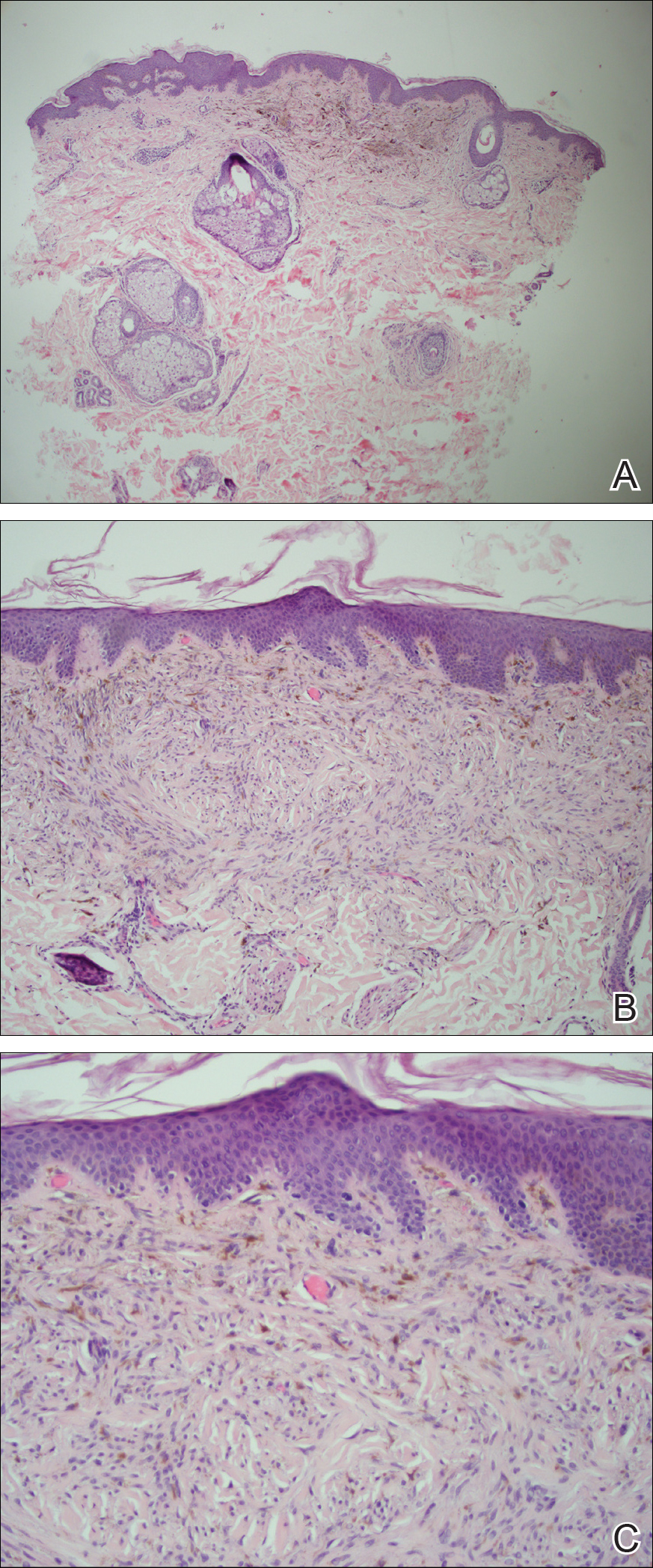
A 4-mm punch biopsy was performed from the active border of brown plaques on the dorsal penis. Histopathology revealed parakeratotic hyperkeratosis, acanthosis, loss of maturation in epithelium, and full-size atypia (Figure 1). Ki-67 index was 90% positive in the epidermis (Figure 2). Staining for p16 and human papillomavirus (HPV) screening was positive for HPV type 16 (Figure 3). Serologic tests for other sexually transmitted infections were negative. A diagnosis of penile bowenoid papulosis (BP) with grade 3 penile intraepithelial neoplasia was made, and treatment with topical 5-fluorouracil (5-FU) was initiated. Almost total regression was appreciated at 1-month follow-up (Figure 4), and he also was recurrence free at 1-year follow-up.

Penile intraepithelial neoplasia (PIN), or penile squamous cell carcinoma in situ, is a rare disease with high morbidity and mortality rates. Clinically, PIN is comprised of a clinical spectrum including 3 different entities: erythroplasia of Queyrat, Bowen disease, and BP.1 Histologically, PIN also is classified into 3 subtypes according to histological depth of epidermal atypia.1
Bowenoid papulosis usually is characterized by multiple red-brown or flesh-colored papules that most commonly appear on the shaft or glans of the penis. Bowenoid papulosis frequently is associated with high-risk types of HPV, such as HPV type 16, and is sometimes difficult to differentiate clinically from pigmented condyloma acuminatum. The clinical lesions of BP usually are less papillomatous, smoother topped, more polymorphic, and more coalescent compared to common genital viral condyloma acuminatum.2 Bowenoid papulosis usually is seen in young (<30 years of age) sexually active men, unlike the patches or plaques of erythroplasia of Queyrat or Bowen disease, which are seen in older men aged 45 to 75 years. Bowenoid papulosis also has a lower malignancy potential than erythroplasia of Queyrat and Bowen disease.2
Penile melanosis, penile lentigo, and seborrheic keratosis comprise the differential diagnosis of dark spots on the penis and also should be kept in mind. Penile melanosis is the most common cause of dark spots on the penis. When the dark spots have irregular borders and change in color, they may be misdiagnosed as malignant lesions such as melanoma.3 In most cases, biopsy is indicated. Histologically, penile melanosis is characterized by hyperpigmentation of the basal cell layer with no melanocytic hyperplasia. Treatment is unnecessary in most cases.
Penile lentigo presents as small flat pigmented spots on the penile skin with clearly defined margins surrounded by normal-appearing skin. Histologically, it is characterized by hyperplasia of melanocytes above the basement membrane of the epidermis.3
Penile pigmented seborrheic keratosis is a rare clinical entity that can be easily misinterpreted as condyloma acuminatum. Histologically, it is characterized by basal cell hyperplasia with cystic formation in the thickened epidermis. Excisional biopsy may be the only way to rule out malignant disease.
Treatment options for PIN include cryotherapy, CO2 or Nd:YAG lasers, photodynamic therapy, topical 5-FU or imiquimod therapy, and surgical excision such as Mohs micrographic surgery.4-9 Although these therapeutic modalities usually are effective, recurrence is common.6 The patients' discomfort and poor cosmetic and functional outcomes from the surgical removal of lesions also present a challenge in treatment planning.
In our patient, we quickly achieved a good result with topical 5-FU, though the disease was in local advanced stage. It is important for clinicians to consider 5-FU as an effective treatment option for PIN before planning surgery.
- Deen K, Burdon-Jones D. Imiquimod in the treatment of penile intraepithelial neoplasia: an update. Australas J Dermatol. 2017;58:86-92.
- Porter WM, Francis N, Hawkins D, et al. Penile intraepithelial neoplasia: clinical spectrum and treatment of 35 cases. Br J Dermatol. 2002;147:1159-1165.
- Fahmy M. Dermatological disease of the penis. In: Fahmy M. Congenital Anomalies of the Penis. Cham, Switzerland: Springer; 2017:257-264.
- Shimizu A, Kato M, Ishikawa O. Bowenoid papulosis successfully treated with imiquimod 5% cream. J Dermatol. 2014;41:545-546.
- Lucky M, Murthy KV, Rogers B, et al. The treatment of penile carcinoma in situ (CIS) within a UK supra-regional network [published online December 15, 2014]. BJU Int. 2015;115:595-598.
- Alnajjar HM, Lam W, Bolgeri M, et al. Treatment of carcinoma in situ of the glans penis with topical chemotherapy agents. Eur Urol. 2012;62:923-928.
- Wang XL, Wang HW, Guo MX, et al. Combination of immunotherapy and photodynamic therapy in the treatment of bowenoid papulosis. Photodiagnosis Photodyn Ther. 2007;4:88-93.
- Zreik A, Rewhorn M, Vint R, et al. Carbon dioxide laser treatment of penile intraepithelial neoplasia [published online December 7, 2016]. Surgeon. 2017;15:321-324.
- Machan M, Brodland D, Zitelli J. Penile squamous cell carcinoma: penis-preserving treatment with Mohs micrographic surgery. Dermatol Surg. 2016;42:936-944.
The Diagnosis: Bowenoid Papulosis
A 4-mm punch biopsy was performed from the active border of brown plaques on the dorsal penis. Histopathology revealed parakeratotic hyperkeratosis, acanthosis, loss of maturation in epithelium, and full-size atypia (Figure 1). Ki-67 index was 90% positive in the epidermis (Figure 2). Staining for p16 and human papillomavirus (HPV) screening was positive for HPV type 16 (Figure 3). Serologic tests for other sexually transmitted infections were negative. A diagnosis of penile bowenoid papulosis (BP) with grade 3 penile intraepithelial neoplasia was made, and treatment with topical 5-fluorouracil (5-FU) was initiated. Almost total regression was appreciated at 1-month follow-up (Figure 4), and he also was recurrence free at 1-year follow-up.
Penile intraepithelial neoplasia (PIN), or penile squamous cell carcinoma in situ, is a rare disease with high morbidity and mortality rates. Clinically, PIN is comprised of a clinical spectrum including 3 different entities: erythroplasia of Queyrat, Bowen disease, and BP.1 Histologically, PIN also is classified into 3 subtypes according to histological depth of epidermal atypia.1
Bowenoid papulosis usually is characterized by multiple red-brown or flesh-colored papules that most commonly appear on the shaft or glans of the penis. Bowenoid papulosis frequently is associated with high-risk types of HPV, such as HPV type 16, and is sometimes difficult to differentiate clinically from pigmented condyloma acuminatum. The clinical lesions of BP usually are less papillomatous, smoother topped, more polymorphic, and more coalescent compared to common genital viral condyloma acuminatum.2 Bowenoid papulosis usually is seen in young (<30 years of age) sexually active men, unlike the patches or plaques of erythroplasia of Queyrat or Bowen disease, which are seen in older men aged 45 to 75 years. Bowenoid papulosis also has a lower malignancy potential than erythroplasia of Queyrat and Bowen disease.2
Penile melanosis, penile lentigo, and seborrheic keratosis comprise the differential diagnosis of dark spots on the penis and also should be kept in mind. Penile melanosis is the most common cause of dark spots on the penis. When the dark spots have irregular borders and change in color, they may be misdiagnosed as malignant lesions such as melanoma.3 In most cases, biopsy is indicated. Histologically, penile melanosis is characterized by hyperpigmentation of the basal cell layer with no melanocytic hyperplasia. Treatment is unnecessary in most cases.
Penile lentigo presents as small flat pigmented spots on the penile skin with clearly defined margins surrounded by normal-appearing skin. Histologically, it is characterized by hyperplasia of melanocytes above the basement membrane of the epidermis.3
Penile pigmented seborrheic keratosis is a rare clinical entity that can be easily misinterpreted as condyloma acuminatum. Histologically, it is characterized by basal cell hyperplasia with cystic formation in the thickened epidermis. Excisional biopsy may be the only way to rule out malignant disease.
Treatment options for PIN include cryotherapy, CO2 or Nd:YAG lasers, photodynamic therapy, topical 5-FU or imiquimod therapy, and surgical excision such as Mohs micrographic surgery.4-9 Although these therapeutic modalities usually are effective, recurrence is common.6 The patients' discomfort and poor cosmetic and functional outcomes from the surgical removal of lesions also present a challenge in treatment planning.
In our patient, we quickly achieved a good result with topical 5-FU, though the disease was in local advanced stage. It is important for clinicians to consider 5-FU as an effective treatment option for PIN before planning surgery.
The Diagnosis: Bowenoid Papulosis
A 4-mm punch biopsy was performed from the active border of brown plaques on the dorsal penis. Histopathology revealed parakeratotic hyperkeratosis, acanthosis, loss of maturation in epithelium, and full-size atypia (Figure 1). Ki-67 index was 90% positive in the epidermis (Figure 2). Staining for p16 and human papillomavirus (HPV) screening was positive for HPV type 16 (Figure 3). Serologic tests for other sexually transmitted infections were negative. A diagnosis of penile bowenoid papulosis (BP) with grade 3 penile intraepithelial neoplasia was made, and treatment with topical 5-fluorouracil (5-FU) was initiated. Almost total regression was appreciated at 1-month follow-up (Figure 4), and he also was recurrence free at 1-year follow-up.
Penile intraepithelial neoplasia (PIN), or penile squamous cell carcinoma in situ, is a rare disease with high morbidity and mortality rates. Clinically, PIN is comprised of a clinical spectrum including 3 different entities: erythroplasia of Queyrat, Bowen disease, and BP.1 Histologically, PIN also is classified into 3 subtypes according to histological depth of epidermal atypia.1
Bowenoid papulosis usually is characterized by multiple red-brown or flesh-colored papules that most commonly appear on the shaft or glans of the penis. Bowenoid papulosis frequently is associated with high-risk types of HPV, such as HPV type 16, and is sometimes difficult to differentiate clinically from pigmented condyloma acuminatum. The clinical lesions of BP usually are less papillomatous, smoother topped, more polymorphic, and more coalescent compared to common genital viral condyloma acuminatum.2 Bowenoid papulosis usually is seen in young (<30 years of age) sexually active men, unlike the patches or plaques of erythroplasia of Queyrat or Bowen disease, which are seen in older men aged 45 to 75 years. Bowenoid papulosis also has a lower malignancy potential than erythroplasia of Queyrat and Bowen disease.2
Penile melanosis, penile lentigo, and seborrheic keratosis comprise the differential diagnosis of dark spots on the penis and also should be kept in mind. Penile melanosis is the most common cause of dark spots on the penis. When the dark spots have irregular borders and change in color, they may be misdiagnosed as malignant lesions such as melanoma.3 In most cases, biopsy is indicated. Histologically, penile melanosis is characterized by hyperpigmentation of the basal cell layer with no melanocytic hyperplasia. Treatment is unnecessary in most cases.
Penile lentigo presents as small flat pigmented spots on the penile skin with clearly defined margins surrounded by normal-appearing skin. Histologically, it is characterized by hyperplasia of melanocytes above the basement membrane of the epidermis.3
Penile pigmented seborrheic keratosis is a rare clinical entity that can be easily misinterpreted as condyloma acuminatum. Histologically, it is characterized by basal cell hyperplasia with cystic formation in the thickened epidermis. Excisional biopsy may be the only way to rule out malignant disease.
Treatment options for PIN include cryotherapy, CO2 or Nd:YAG lasers, photodynamic therapy, topical 5-FU or imiquimod therapy, and surgical excision such as Mohs micrographic surgery.4-9 Although these therapeutic modalities usually are effective, recurrence is common.6 The patients' discomfort and poor cosmetic and functional outcomes from the surgical removal of lesions also present a challenge in treatment planning.
In our patient, we quickly achieved a good result with topical 5-FU, though the disease was in local advanced stage. It is important for clinicians to consider 5-FU as an effective treatment option for PIN before planning surgery.
- Deen K, Burdon-Jones D. Imiquimod in the treatment of penile intraepithelial neoplasia: an update. Australas J Dermatol. 2017;58:86-92.
- Porter WM, Francis N, Hawkins D, et al. Penile intraepithelial neoplasia: clinical spectrum and treatment of 35 cases. Br J Dermatol. 2002;147:1159-1165.
- Fahmy M. Dermatological disease of the penis. In: Fahmy M. Congenital Anomalies of the Penis. Cham, Switzerland: Springer; 2017:257-264.
- Shimizu A, Kato M, Ishikawa O. Bowenoid papulosis successfully treated with imiquimod 5% cream. J Dermatol. 2014;41:545-546.
- Lucky M, Murthy KV, Rogers B, et al. The treatment of penile carcinoma in situ (CIS) within a UK supra-regional network [published online December 15, 2014]. BJU Int. 2015;115:595-598.
- Alnajjar HM, Lam W, Bolgeri M, et al. Treatment of carcinoma in situ of the glans penis with topical chemotherapy agents. Eur Urol. 2012;62:923-928.
- Wang XL, Wang HW, Guo MX, et al. Combination of immunotherapy and photodynamic therapy in the treatment of bowenoid papulosis. Photodiagnosis Photodyn Ther. 2007;4:88-93.
- Zreik A, Rewhorn M, Vint R, et al. Carbon dioxide laser treatment of penile intraepithelial neoplasia [published online December 7, 2016]. Surgeon. 2017;15:321-324.
- Machan M, Brodland D, Zitelli J. Penile squamous cell carcinoma: penis-preserving treatment with Mohs micrographic surgery. Dermatol Surg. 2016;42:936-944.
- Deen K, Burdon-Jones D. Imiquimod in the treatment of penile intraepithelial neoplasia: an update. Australas J Dermatol. 2017;58:86-92.
- Porter WM, Francis N, Hawkins D, et al. Penile intraepithelial neoplasia: clinical spectrum and treatment of 35 cases. Br J Dermatol. 2002;147:1159-1165.
- Fahmy M. Dermatological disease of the penis. In: Fahmy M. Congenital Anomalies of the Penis. Cham, Switzerland: Springer; 2017:257-264.
- Shimizu A, Kato M, Ishikawa O. Bowenoid papulosis successfully treated with imiquimod 5% cream. J Dermatol. 2014;41:545-546.
- Lucky M, Murthy KV, Rogers B, et al. The treatment of penile carcinoma in situ (CIS) within a UK supra-regional network [published online December 15, 2014]. BJU Int. 2015;115:595-598.
- Alnajjar HM, Lam W, Bolgeri M, et al. Treatment of carcinoma in situ of the glans penis with topical chemotherapy agents. Eur Urol. 2012;62:923-928.
- Wang XL, Wang HW, Guo MX, et al. Combination of immunotherapy and photodynamic therapy in the treatment of bowenoid papulosis. Photodiagnosis Photodyn Ther. 2007;4:88-93.
- Zreik A, Rewhorn M, Vint R, et al. Carbon dioxide laser treatment of penile intraepithelial neoplasia [published online December 7, 2016]. Surgeon. 2017;15:321-324.
- Machan M, Brodland D, Zitelli J. Penile squamous cell carcinoma: penis-preserving treatment with Mohs micrographic surgery. Dermatol Surg. 2016;42:936-944.
A 32-year-old man presented to the outpatient clinic with reddish brown lesions on the penis of 5 months' duration. Dermatologic examination revealed multiple mildly infiltrated, bright reddish brown papules and plaques on the dorsal penis.
Agminated Heterogeneous Papules on the Neck
The Diagnosis: Eruptive Blue Nevus
All biopsies demonstrated similar histologic features, including an intradermal proliferation of heavily pigmented, spindle-shaped dendritic melanocytes (Figure). The dermal pigment was most pronounced in the grossly darker papules, and there was not a substantial amount of background pigmentation at the stratum basale. Cytologic atypia, foci of necrosis, and mitotic activity were absent from all sections. There was no definitive junctional component identified, no multinucleated giant cells, and there was no overlying epidermal aberration. With some background pigmentation seen histologically, nevus spilus was considered, but because this acute eruption occurred in a young adult without appreciable gross background hyperpigmentation, the clinical context led to a diagnosis of eruptive blue nevus. After communicating the findings to the patient, he declined further treatment.
Eruptive blue nevus is an exceptionally rare subtype of blue nevus with few cases reported since the 1940s.1-9 Generally, each case report found a triggering event that could possibly have precipitated the acute proliferation and evolution of nevi. Triggering events can include bullous processes such as erythema multiforme2 and Stevens-Johnson syndrome,3 severe sunburn,4 trauma,5 immunosuppression,6 and a variety of endocrinopathies. No such history could be identified in our patient, except the biopsy.
Common blue nevi are benign, usually congenital, well-circumscribed, solitary, blue-gray macules or papules. Half of blue nevi cases are found on the dorsal aspects of the hands and feet but can present anywhere (eg, face, scalp, wrists, sacrum, buttocks). The blue-gray color appreciated clinically is attributed to the Tyndall effect, which occurs when long-wavelength light--red, orange, and yellow--is absorbed by the melanin deep in the dermis, while short-wavelength visible light--blue, violet, and indigo--is reflected with backscattering. On polarized dermoscopy, a homogeneous blue-gray hue is appreciated, but lighter segments may be present when collagen deposition is robust. Histopathologic findings confirm spindle-shaped dendritic melanocytes in the dermis without epidermal involvement. It generally is accepted that the etiology of these benign nevi is a failed migration of neural crest cells to the epidermis.10,11 Although the common blue nevus may be simple to diagnose, several subtypes have been described in the literature, including combined blue nevus, desmoplastic blue nevus, hypomelanotic/amelanotic blue nevus, and epithelioid blue nevus of Carney complex, and excluding a malignant process is of monumental importance.7,12
Biopsy is recommended for common blue nevi in the evaluation of newly acquired lesions, expansion of previously stable nevi, or for nevi larger than 10 mm in diameter. The nature of eruptive blue nevi warrants a biopsy to exclude melanoma or another malignant process. While the Becker nevus may manifest in adolescent males, it is clinically distinct from an eruptive blue nevus due to the size, relative homogeneity, and presence of hair within the lesion. Cutaneous amyloidosis may appear clinically similar to an eruptive blue nevus, but globular or amorphous material was not present in the papillary dermis of biopsied lesions in our patient. Since there was no cellular atypia or mitotic activity, melanoma and other malignancies were ruled out. Lastly, NAME syndrome by definition must include atrial myxomas, myxoid neurofibromas, and ephelides in addition to the nevi; however, our patient had only nevi and few ephelides. Once the diagnosis is established and benign nature confirmed, treatment is not necessarily required. If the patient elects to remove the lesion for aesthetic reasons, an excision into the subcutaneous fat is required to ensure complete removal of deep dermal melanocytes. Prior excisions of eruptive blue nevi have had no recurrence after more than 10 months.8,9
- Krause M, Bonnekoh B, Weisshaar E, et al. Coincidence of multiple, disseminated, tardive-eruptive blue nevi with cutis marmorata teleangiectatica congenita. Dermatology. 2000;200:134-138.
- Soltani K, Bernstein J, Lorincz A. Eruptive nevocytic nevi after erythema multiforme. J Am Acad Dermatol. 1979;1:503-505.
- Shoji T, Cockerell C, Koff A, et al. Eruptive melanocytic nevi after Stevens-Johnson syndrome. J Am Acad Dermatol. 1997;37:337-339.
- Hendricks W. Eruptive blue nevi. J Am Acad Dermatol. 1981;4:50-53.
- Kesty K, Zargari O. Eruptive blue nevi. Indian J Dermatol Venereol Leprol. 2015;81:198-201.
- Chen T, Kurwa H, Trotter M, et al. Agminated blue nevi in a patient with dermatomyositis. J Am Acad Dermatol. 2013;68:52-53.
- Walsh M. Correspondence: eruptive disseminated blue naevi of the scalp. Br J Dermatol. 1999;141:581-582.
- Nardini P, De Giorgi V, Massi D, et al. Eruptive disseminated blue naevi of the scalp. Br J Dermatol. 1999;140:178-180.
- de Giorgi V, Massi D, Brunasso G, et al. Eruptive multiple blue nevi of the penis: a clinical dermoscopic pathologic case study. J Cutan Pathol. 2004;31:185-188.
- Zimmermann AH, Becker SA. Precursors of epidermal melanocytes in the negro fetus. In: Gordon M, ed. Pigment Cell Biology. New York, NY: Academic Press Inc; 1959:159-170.
- Leopold JG, Richards DB. The interrelationship of blue and common naevi. J Pathol Bacteriol. 1968;95:37-46.
- Zembowicz A, Phadke P. Blue nevi and variants: an update. Arch Pathol Lab Med. 2011;135:327-336.
The Diagnosis: Eruptive Blue Nevus
All biopsies demonstrated similar histologic features, including an intradermal proliferation of heavily pigmented, spindle-shaped dendritic melanocytes (Figure). The dermal pigment was most pronounced in the grossly darker papules, and there was not a substantial amount of background pigmentation at the stratum basale. Cytologic atypia, foci of necrosis, and mitotic activity were absent from all sections. There was no definitive junctional component identified, no multinucleated giant cells, and there was no overlying epidermal aberration. With some background pigmentation seen histologically, nevus spilus was considered, but because this acute eruption occurred in a young adult without appreciable gross background hyperpigmentation, the clinical context led to a diagnosis of eruptive blue nevus. After communicating the findings to the patient, he declined further treatment.
Eruptive blue nevus is an exceptionally rare subtype of blue nevus with few cases reported since the 1940s.1-9 Generally, each case report found a triggering event that could possibly have precipitated the acute proliferation and evolution of nevi. Triggering events can include bullous processes such as erythema multiforme2 and Stevens-Johnson syndrome,3 severe sunburn,4 trauma,5 immunosuppression,6 and a variety of endocrinopathies. No such history could be identified in our patient, except the biopsy.
Common blue nevi are benign, usually congenital, well-circumscribed, solitary, blue-gray macules or papules. Half of blue nevi cases are found on the dorsal aspects of the hands and feet but can present anywhere (eg, face, scalp, wrists, sacrum, buttocks). The blue-gray color appreciated clinically is attributed to the Tyndall effect, which occurs when long-wavelength light--red, orange, and yellow--is absorbed by the melanin deep in the dermis, while short-wavelength visible light--blue, violet, and indigo--is reflected with backscattering. On polarized dermoscopy, a homogeneous blue-gray hue is appreciated, but lighter segments may be present when collagen deposition is robust. Histopathologic findings confirm spindle-shaped dendritic melanocytes in the dermis without epidermal involvement. It generally is accepted that the etiology of these benign nevi is a failed migration of neural crest cells to the epidermis.10,11 Although the common blue nevus may be simple to diagnose, several subtypes have been described in the literature, including combined blue nevus, desmoplastic blue nevus, hypomelanotic/amelanotic blue nevus, and epithelioid blue nevus of Carney complex, and excluding a malignant process is of monumental importance.7,12
Biopsy is recommended for common blue nevi in the evaluation of newly acquired lesions, expansion of previously stable nevi, or for nevi larger than 10 mm in diameter. The nature of eruptive blue nevi warrants a biopsy to exclude melanoma or another malignant process. While the Becker nevus may manifest in adolescent males, it is clinically distinct from an eruptive blue nevus due to the size, relative homogeneity, and presence of hair within the lesion. Cutaneous amyloidosis may appear clinically similar to an eruptive blue nevus, but globular or amorphous material was not present in the papillary dermis of biopsied lesions in our patient. Since there was no cellular atypia or mitotic activity, melanoma and other malignancies were ruled out. Lastly, NAME syndrome by definition must include atrial myxomas, myxoid neurofibromas, and ephelides in addition to the nevi; however, our patient had only nevi and few ephelides. Once the diagnosis is established and benign nature confirmed, treatment is not necessarily required. If the patient elects to remove the lesion for aesthetic reasons, an excision into the subcutaneous fat is required to ensure complete removal of deep dermal melanocytes. Prior excisions of eruptive blue nevi have had no recurrence after more than 10 months.8,9
The Diagnosis: Eruptive Blue Nevus
All biopsies demonstrated similar histologic features, including an intradermal proliferation of heavily pigmented, spindle-shaped dendritic melanocytes (Figure). The dermal pigment was most pronounced in the grossly darker papules, and there was not a substantial amount of background pigmentation at the stratum basale. Cytologic atypia, foci of necrosis, and mitotic activity were absent from all sections. There was no definitive junctional component identified, no multinucleated giant cells, and there was no overlying epidermal aberration. With some background pigmentation seen histologically, nevus spilus was considered, but because this acute eruption occurred in a young adult without appreciable gross background hyperpigmentation, the clinical context led to a diagnosis of eruptive blue nevus. After communicating the findings to the patient, he declined further treatment.
Eruptive blue nevus is an exceptionally rare subtype of blue nevus with few cases reported since the 1940s.1-9 Generally, each case report found a triggering event that could possibly have precipitated the acute proliferation and evolution of nevi. Triggering events can include bullous processes such as erythema multiforme2 and Stevens-Johnson syndrome,3 severe sunburn,4 trauma,5 immunosuppression,6 and a variety of endocrinopathies. No such history could be identified in our patient, except the biopsy.
Common blue nevi are benign, usually congenital, well-circumscribed, solitary, blue-gray macules or papules. Half of blue nevi cases are found on the dorsal aspects of the hands and feet but can present anywhere (eg, face, scalp, wrists, sacrum, buttocks). The blue-gray color appreciated clinically is attributed to the Tyndall effect, which occurs when long-wavelength light--red, orange, and yellow--is absorbed by the melanin deep in the dermis, while short-wavelength visible light--blue, violet, and indigo--is reflected with backscattering. On polarized dermoscopy, a homogeneous blue-gray hue is appreciated, but lighter segments may be present when collagen deposition is robust. Histopathologic findings confirm spindle-shaped dendritic melanocytes in the dermis without epidermal involvement. It generally is accepted that the etiology of these benign nevi is a failed migration of neural crest cells to the epidermis.10,11 Although the common blue nevus may be simple to diagnose, several subtypes have been described in the literature, including combined blue nevus, desmoplastic blue nevus, hypomelanotic/amelanotic blue nevus, and epithelioid blue nevus of Carney complex, and excluding a malignant process is of monumental importance.7,12
Biopsy is recommended for common blue nevi in the evaluation of newly acquired lesions, expansion of previously stable nevi, or for nevi larger than 10 mm in diameter. The nature of eruptive blue nevi warrants a biopsy to exclude melanoma or another malignant process. While the Becker nevus may manifest in adolescent males, it is clinically distinct from an eruptive blue nevus due to the size, relative homogeneity, and presence of hair within the lesion. Cutaneous amyloidosis may appear clinically similar to an eruptive blue nevus, but globular or amorphous material was not present in the papillary dermis of biopsied lesions in our patient. Since there was no cellular atypia or mitotic activity, melanoma and other malignancies were ruled out. Lastly, NAME syndrome by definition must include atrial myxomas, myxoid neurofibromas, and ephelides in addition to the nevi; however, our patient had only nevi and few ephelides. Once the diagnosis is established and benign nature confirmed, treatment is not necessarily required. If the patient elects to remove the lesion for aesthetic reasons, an excision into the subcutaneous fat is required to ensure complete removal of deep dermal melanocytes. Prior excisions of eruptive blue nevi have had no recurrence after more than 10 months.8,9
- Krause M, Bonnekoh B, Weisshaar E, et al. Coincidence of multiple, disseminated, tardive-eruptive blue nevi with cutis marmorata teleangiectatica congenita. Dermatology. 2000;200:134-138.
- Soltani K, Bernstein J, Lorincz A. Eruptive nevocytic nevi after erythema multiforme. J Am Acad Dermatol. 1979;1:503-505.
- Shoji T, Cockerell C, Koff A, et al. Eruptive melanocytic nevi after Stevens-Johnson syndrome. J Am Acad Dermatol. 1997;37:337-339.
- Hendricks W. Eruptive blue nevi. J Am Acad Dermatol. 1981;4:50-53.
- Kesty K, Zargari O. Eruptive blue nevi. Indian J Dermatol Venereol Leprol. 2015;81:198-201.
- Chen T, Kurwa H, Trotter M, et al. Agminated blue nevi in a patient with dermatomyositis. J Am Acad Dermatol. 2013;68:52-53.
- Walsh M. Correspondence: eruptive disseminated blue naevi of the scalp. Br J Dermatol. 1999;141:581-582.
- Nardini P, De Giorgi V, Massi D, et al. Eruptive disseminated blue naevi of the scalp. Br J Dermatol. 1999;140:178-180.
- de Giorgi V, Massi D, Brunasso G, et al. Eruptive multiple blue nevi of the penis: a clinical dermoscopic pathologic case study. J Cutan Pathol. 2004;31:185-188.
- Zimmermann AH, Becker SA. Precursors of epidermal melanocytes in the negro fetus. In: Gordon M, ed. Pigment Cell Biology. New York, NY: Academic Press Inc; 1959:159-170.
- Leopold JG, Richards DB. The interrelationship of blue and common naevi. J Pathol Bacteriol. 1968;95:37-46.
- Zembowicz A, Phadke P. Blue nevi and variants: an update. Arch Pathol Lab Med. 2011;135:327-336.
- Krause M, Bonnekoh B, Weisshaar E, et al. Coincidence of multiple, disseminated, tardive-eruptive blue nevi with cutis marmorata teleangiectatica congenita. Dermatology. 2000;200:134-138.
- Soltani K, Bernstein J, Lorincz A. Eruptive nevocytic nevi after erythema multiforme. J Am Acad Dermatol. 1979;1:503-505.
- Shoji T, Cockerell C, Koff A, et al. Eruptive melanocytic nevi after Stevens-Johnson syndrome. J Am Acad Dermatol. 1997;37:337-339.
- Hendricks W. Eruptive blue nevi. J Am Acad Dermatol. 1981;4:50-53.
- Kesty K, Zargari O. Eruptive blue nevi. Indian J Dermatol Venereol Leprol. 2015;81:198-201.
- Chen T, Kurwa H, Trotter M, et al. Agminated blue nevi in a patient with dermatomyositis. J Am Acad Dermatol. 2013;68:52-53.
- Walsh M. Correspondence: eruptive disseminated blue naevi of the scalp. Br J Dermatol. 1999;141:581-582.
- Nardini P, De Giorgi V, Massi D, et al. Eruptive disseminated blue naevi of the scalp. Br J Dermatol. 1999;140:178-180.
- de Giorgi V, Massi D, Brunasso G, et al. Eruptive multiple blue nevi of the penis: a clinical dermoscopic pathologic case study. J Cutan Pathol. 2004;31:185-188.
- Zimmermann AH, Becker SA. Precursors of epidermal melanocytes in the negro fetus. In: Gordon M, ed. Pigment Cell Biology. New York, NY: Academic Press Inc; 1959:159-170.
- Leopold JG, Richards DB. The interrelationship of blue and common naevi. J Pathol Bacteriol. 1968;95:37-46.
- Zembowicz A, Phadke P. Blue nevi and variants: an update. Arch Pathol Lab Med. 2011;135:327-336.
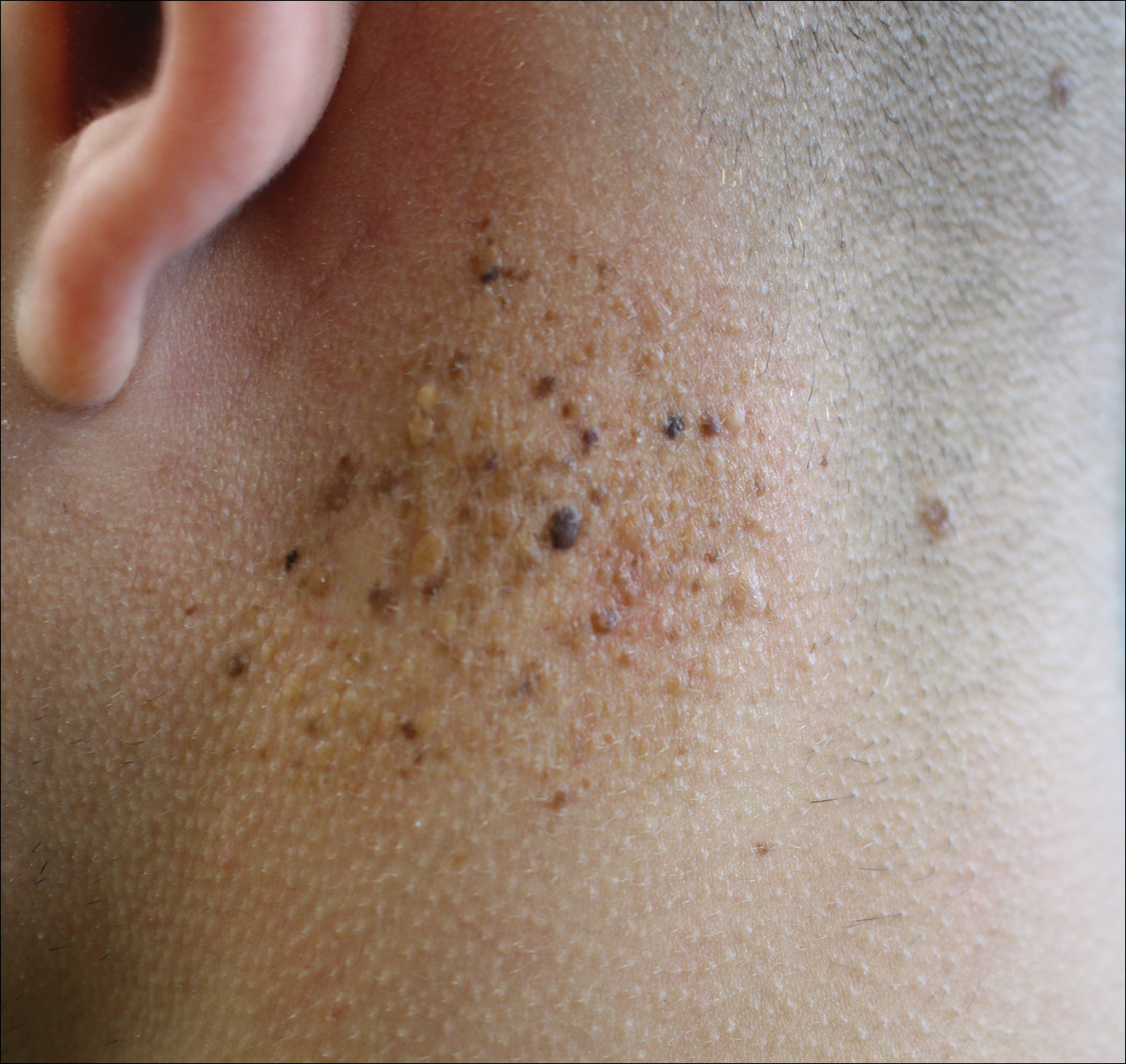
A 19-year-old man presented to the dermatology clinic for evaluation of several new dark papules on the neck of 1 year's duration. He denied any personal or family history of skin cancer, cardiac abnormalities, or endocrine dysfunction. He also denied any recent changes in health or use of medication. A biopsy was performed at the site 2 years prior for a single blue nevus, but the patient denied history of other trauma or cutaneous eruptions localized to the area. Physical examination revealed numerous dark brown, blue, white, and flesh-colored papules and macules agminated into a well-circumscribed plaque on the left posterolateral neck without background hyperpigmentation. The total area of the plaque was roughly 3×4 cm. There was no associated edema or erythema. Cardiac murmur, thyromegaly, exophthalmos, neurologic deficits, regional lymphadenopathy, and similar skin findings on other areas of the body were not appreciated. Three scouting punch biopsies were taken of the various morphologies present.
Telangiectatic Patch on the Neck
The Diagnosis: Unilateral Nevoid Telangiectasia
Unilateral nevoid telangiectasia (UNT) is an uncommon, or perhaps underreported, cutaneous condition involving telangiectatic patches in a unilateral dermatomal or blaschkoid pattern.1 The condition has been described as either congenital or acquired. Congenital UNT is thought to be a result of somatic mosaicism, whereby a mutation during embryogenesis leads to a distinct population of cells expressing the vascular malformation.1 Congenital UNT has been associated with Becker nevus, which also is thought to be a result of somatic mosaicism, further providing evidence for this theory, though it is unclear whether this finding is incidental.2 The acquired form often is associated with fluctuation of hormones, such as in pregnancy or with oral contraceptive initiation, as well as with hepatic disease as seen in our patient. However, there are many cases of acquired UNT with no implicated underlying disease, alcohol abuse, or hormonal changes, which calls into question if UNT is definitively an estrogen-related condition.3 One study demonstrated an increased level of estrogen and progesterone receptors in affected skin, which may have led to expression of the cutaneous changes at that site.4 More research is needed to elucidate this point, as other studies have not reproduced similar findings.
Congenital UNT occurs more commonly in males, whereas the acquired variant is seen more frequently in females. The third and fourth cervical dermatomes most often are involved.5 Most lesions persist without spontaneous resolution. Treatment options are limited and include pulsed dye laser treatment and makeup application to cover the telangiectatic patches. The main side effect seen with pulsed dye laser treatment is reversible pigmentary changes, with 1 report of textural skin change.6
A biopsy was deemed unnecessary for the clinical diagnosis in our patient because there was a clear explanation for the physical examination findings due to long-standing underlying liver disease. When biopsied, UNT characteristically demonstrates dilated dermal capillaries.5 Our patient elected not to pursue laser therapy but expressed interest in using makeup to camouflage the lesion.
The differential diagnosis includes acquired nevus flammeus, which typically is present on the face and often appears following mechanical or thermal trauma. Angioma serpiginosum most often occurs on the buttocks and legs as small red papules or puncta coalescing into a serpiginous linear arrangement. It often appears in childhood. Angiosarcoma is an aggressive malignancy that often occurs on the head and neck in elderly patients. It is associated with areas of long-standing lymphedema and often appears as a bruiselike lesion. Rosacea typically is not fixed in its clinical appearance and presents as transitory flushing of the head and neck with or without a history of acneform eruptions on the face. It typically is not unilateral.
- Wilkin JK. Unilateral dermatomal superficial telangiectasia. Arch Dermatol. 1984;120:579-580.
- Karakaş M, Durdu M, Sönmezoğlu S, et al. Unilateral nevoid telangiectasia. J Dermatol. 2004;31:109-112.
- Taskapan O, Harmanyeri Y, Sener O, et al. Acquired unilateral nevoid telangiectasia syndrome. Acta Derm Venereol. 1997;77:62-63.
- Uhlin SR, McCarty KS Jr. Unilateral nevoid telangiectatic syndrome: the role of estrogen and progesterone receptors. Arch Dermatol. 1983;119:226-228.
- Derrow AE, Adams BB, Timani S, et al. Acquired unilateral nevoid telangiectasia in a 51-year-old female. Int J Dermatol. 2008;47:1331-1333.
- Sharma VK, Khandpur S. Unilateral nevoid telangiectasia--response to pulsed dye laser. Int J Dermatol. 2006;45:960-964.
The Diagnosis: Unilateral Nevoid Telangiectasia
Unilateral nevoid telangiectasia (UNT) is an uncommon, or perhaps underreported, cutaneous condition involving telangiectatic patches in a unilateral dermatomal or blaschkoid pattern.1 The condition has been described as either congenital or acquired. Congenital UNT is thought to be a result of somatic mosaicism, whereby a mutation during embryogenesis leads to a distinct population of cells expressing the vascular malformation.1 Congenital UNT has been associated with Becker nevus, which also is thought to be a result of somatic mosaicism, further providing evidence for this theory, though it is unclear whether this finding is incidental.2 The acquired form often is associated with fluctuation of hormones, such as in pregnancy or with oral contraceptive initiation, as well as with hepatic disease as seen in our patient. However, there are many cases of acquired UNT with no implicated underlying disease, alcohol abuse, or hormonal changes, which calls into question if UNT is definitively an estrogen-related condition.3 One study demonstrated an increased level of estrogen and progesterone receptors in affected skin, which may have led to expression of the cutaneous changes at that site.4 More research is needed to elucidate this point, as other studies have not reproduced similar findings.
Congenital UNT occurs more commonly in males, whereas the acquired variant is seen more frequently in females. The third and fourth cervical dermatomes most often are involved.5 Most lesions persist without spontaneous resolution. Treatment options are limited and include pulsed dye laser treatment and makeup application to cover the telangiectatic patches. The main side effect seen with pulsed dye laser treatment is reversible pigmentary changes, with 1 report of textural skin change.6
A biopsy was deemed unnecessary for the clinical diagnosis in our patient because there was a clear explanation for the physical examination findings due to long-standing underlying liver disease. When biopsied, UNT characteristically demonstrates dilated dermal capillaries.5 Our patient elected not to pursue laser therapy but expressed interest in using makeup to camouflage the lesion.
The differential diagnosis includes acquired nevus flammeus, which typically is present on the face and often appears following mechanical or thermal trauma. Angioma serpiginosum most often occurs on the buttocks and legs as small red papules or puncta coalescing into a serpiginous linear arrangement. It often appears in childhood. Angiosarcoma is an aggressive malignancy that often occurs on the head and neck in elderly patients. It is associated with areas of long-standing lymphedema and often appears as a bruiselike lesion. Rosacea typically is not fixed in its clinical appearance and presents as transitory flushing of the head and neck with or without a history of acneform eruptions on the face. It typically is not unilateral.
The Diagnosis: Unilateral Nevoid Telangiectasia
Unilateral nevoid telangiectasia (UNT) is an uncommon, or perhaps underreported, cutaneous condition involving telangiectatic patches in a unilateral dermatomal or blaschkoid pattern.1 The condition has been described as either congenital or acquired. Congenital UNT is thought to be a result of somatic mosaicism, whereby a mutation during embryogenesis leads to a distinct population of cells expressing the vascular malformation.1 Congenital UNT has been associated with Becker nevus, which also is thought to be a result of somatic mosaicism, further providing evidence for this theory, though it is unclear whether this finding is incidental.2 The acquired form often is associated with fluctuation of hormones, such as in pregnancy or with oral contraceptive initiation, as well as with hepatic disease as seen in our patient. However, there are many cases of acquired UNT with no implicated underlying disease, alcohol abuse, or hormonal changes, which calls into question if UNT is definitively an estrogen-related condition.3 One study demonstrated an increased level of estrogen and progesterone receptors in affected skin, which may have led to expression of the cutaneous changes at that site.4 More research is needed to elucidate this point, as other studies have not reproduced similar findings.
Congenital UNT occurs more commonly in males, whereas the acquired variant is seen more frequently in females. The third and fourth cervical dermatomes most often are involved.5 Most lesions persist without spontaneous resolution. Treatment options are limited and include pulsed dye laser treatment and makeup application to cover the telangiectatic patches. The main side effect seen with pulsed dye laser treatment is reversible pigmentary changes, with 1 report of textural skin change.6
A biopsy was deemed unnecessary for the clinical diagnosis in our patient because there was a clear explanation for the physical examination findings due to long-standing underlying liver disease. When biopsied, UNT characteristically demonstrates dilated dermal capillaries.5 Our patient elected not to pursue laser therapy but expressed interest in using makeup to camouflage the lesion.
The differential diagnosis includes acquired nevus flammeus, which typically is present on the face and often appears following mechanical or thermal trauma. Angioma serpiginosum most often occurs on the buttocks and legs as small red papules or puncta coalescing into a serpiginous linear arrangement. It often appears in childhood. Angiosarcoma is an aggressive malignancy that often occurs on the head and neck in elderly patients. It is associated with areas of long-standing lymphedema and often appears as a bruiselike lesion. Rosacea typically is not fixed in its clinical appearance and presents as transitory flushing of the head and neck with or without a history of acneform eruptions on the face. It typically is not unilateral.
- Wilkin JK. Unilateral dermatomal superficial telangiectasia. Arch Dermatol. 1984;120:579-580.
- Karakaş M, Durdu M, Sönmezoğlu S, et al. Unilateral nevoid telangiectasia. J Dermatol. 2004;31:109-112.
- Taskapan O, Harmanyeri Y, Sener O, et al. Acquired unilateral nevoid telangiectasia syndrome. Acta Derm Venereol. 1997;77:62-63.
- Uhlin SR, McCarty KS Jr. Unilateral nevoid telangiectatic syndrome: the role of estrogen and progesterone receptors. Arch Dermatol. 1983;119:226-228.
- Derrow AE, Adams BB, Timani S, et al. Acquired unilateral nevoid telangiectasia in a 51-year-old female. Int J Dermatol. 2008;47:1331-1333.
- Sharma VK, Khandpur S. Unilateral nevoid telangiectasia--response to pulsed dye laser. Int J Dermatol. 2006;45:960-964.
- Wilkin JK. Unilateral dermatomal superficial telangiectasia. Arch Dermatol. 1984;120:579-580.
- Karakaş M, Durdu M, Sönmezoğlu S, et al. Unilateral nevoid telangiectasia. J Dermatol. 2004;31:109-112.
- Taskapan O, Harmanyeri Y, Sener O, et al. Acquired unilateral nevoid telangiectasia syndrome. Acta Derm Venereol. 1997;77:62-63.
- Uhlin SR, McCarty KS Jr. Unilateral nevoid telangiectatic syndrome: the role of estrogen and progesterone receptors. Arch Dermatol. 1983;119:226-228.
- Derrow AE, Adams BB, Timani S, et al. Acquired unilateral nevoid telangiectasia in a 51-year-old female. Int J Dermatol. 2008;47:1331-1333.
- Sharma VK, Khandpur S. Unilateral nevoid telangiectasia--response to pulsed dye laser. Int J Dermatol. 2006;45:960-964.
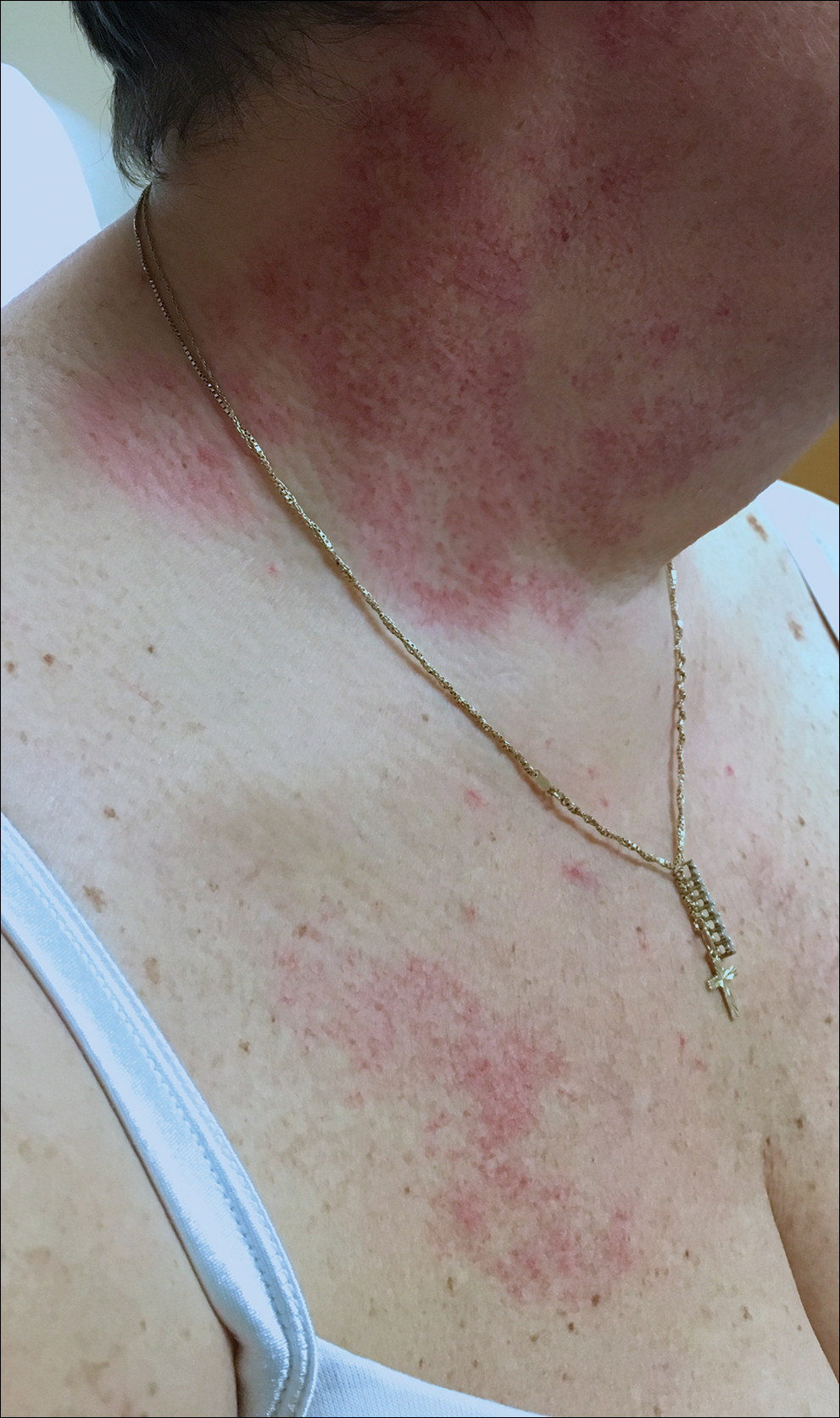
A 55-year-old woman presented to our clinic for a total-body skin examination and was noted to have a completely blanchable telangiectatic patch on the right side of the neck extending down onto the chest and breast. The patient reported that it had been present for 15 years and had slowly expanded in size. The lesion was asymptomatic. Pertinent medical history included cryptogenic cirrhosis of the liver, and she was undergoing a workup for a liver transplant.
Friable Erythema and Erosions on the Mouth
The Diagnosis: Radiation Mucositis
The patient was undergoing active radiation therapy for squamous cell carcinoma of the tongue, and according to the oncology team, the findings were in the precise location of radiation exposure. Radiation mucositis is a major and limiting side effect of radiation therapy for head and neck mucosal cancers, and symptom management is critical to ensure completion of the full radiation dose. Although infectious etiologies must be considered, the patient was already on prophylactic antiviral and antibacterial therapies. Moreover, the focal involvement with sparing of more mucosal tissue is atypical for most infections. Fixed drug reactions can present with localized mucosal and nonmucosal inflammation leading to erosion or ulceration. In this case, the only potential culprit was levofloxacin; however, it was initiated 2 days prior, and the patient never had reactions to this medication in the past.
Acute radiation mucositis is a transient but major limiting side effect of radiation therapy. The associated odynophagia, secondary infection, and reduced oral intake often can lead to diminished disease control secondary to treatment interruption and subsequent development of resistant tumor burden. Concurrent chemotherapy and alternated fractionation radiation therapy increase the incidence of mucositis. Trotti et al1 (n=6181) reported that severe mucositis (grades 3 to 4) was found in 56% of patients receiving altered fractionation radiation therapy compared to 34% of patients who received conventional radiation therapy. Other risk factors related to the development of acute radiation mucositis include associated chemotherapy, age (>65 years), poor oral hygiene, diabetes mellitus, and prior periodontal disease.2
Radiation causes direct cellular damage to keratinocytes, leading to ulceration and erythema, as well as keratinocyte stem cells, which interferes with the healing process. Typical symptoms of mucosal radiation injury may include erythema (asymptomatic or causing intolerance of warm foods) that develops at the end of the second week of radiation therapy, focal areas of desquamation that develops in week 3, and confluent mucositis that can further progress to ulceration and necrosis in weeks 4 to 5.2 The development of dysgeusia, which is estimated to occur in 67% of patients receiving radiotherapy and 76% of patients receiving combination therapy, also can contribute to nutritional difficulties and weight loss.3
Avoiding overtreatment by constraining radiation volume and limiting concurrent chemotherapy are important preventative measures. The mainstay for managing mucositis includes symptomatic relief with oral hygiene, topical agents, topical plus systemic analgesia, dietary changes, and treatment of associated infections. Benzydamine, a nonsteroidal anti-inflammatory drug, is not available in the United States but has been shown to effectively improve symptoms.4 Various formulations of topical anesthetics consisting of diphenhydramine with or without corticosteroids, antibiotics, and antifungals help alleviate symptoms of mucositis; however, no single formulation has been studied. Low-level laser therapy also has shown efficacy in managing symptoms of mucositis.5,6 For persistent odynophagia, systemic opioid therapy should be attempted to achieve uninterrupted radiation therapy. Severe mucositis requires balancing risks and benefits of interrupting treatment, as additional damage may cause permanent mucosal injury.
Our patient had adequate symptom control with benzocaine lozenges and a combination mouthwash containing diphenhydramine, nystatin, lidocaine, hydrocortisone, and tetracycline. He required only occasional doses of systemic oxycodone. After a 1-week hospital admission for treatment of the pneumonia, he resumed radiation therapy and completed a full 8-week radiation course.
- Trotti A, Bellm LA, Epstein JB, et al. Mucositis incidence, severity and associated outcomes in patients with head and neck cancer receiving radiotherapy with or without chemotherapy: a systematic literature review. Radiother Oncol. 2003;66:253-262.
- Mallick S, Benson R, Rath GK. Radiation induced oral mucositis: a review of current literature on prevention and management. Eur Arch Otorhinolaryngol. 2016;273:2285-2293.
- Hovan AJ, Williams PM, Stevenson-Moore P, et al; Dysgeusia Section, Oral Care Study Group, Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO). A systematic review of dysgeusia induced by cancer therapies. Support Care Cancer. 2010;18:1081-1087.
- Epstein JB, Silverman S, Paggiarino DA, et al. Benzydamine HCl for prophylaxis of radiation‐induced oral mucositis. Cancer. 2001;92:875-885.
- Henke M, Alfonsi M, Foa P, et al. Palifermin decreases severe oral mucositis of patients undergoing postoperative radiochemotherapy for head and neck cancer: a randomized, placebo-controlled trial. J Clin Oncol. 2011;29:2815-2820.
- Bensadoun RJ, Nair RG. Low-level laser therapy in the prevention and treatment of cancer therapy-induced mucositis: 2012 state of the art based on literature review and meta-analysis. Curr Opin Oncol. 2012;24:363-370.
The Diagnosis: Radiation Mucositis
The patient was undergoing active radiation therapy for squamous cell carcinoma of the tongue, and according to the oncology team, the findings were in the precise location of radiation exposure. Radiation mucositis is a major and limiting side effect of radiation therapy for head and neck mucosal cancers, and symptom management is critical to ensure completion of the full radiation dose. Although infectious etiologies must be considered, the patient was already on prophylactic antiviral and antibacterial therapies. Moreover, the focal involvement with sparing of more mucosal tissue is atypical for most infections. Fixed drug reactions can present with localized mucosal and nonmucosal inflammation leading to erosion or ulceration. In this case, the only potential culprit was levofloxacin; however, it was initiated 2 days prior, and the patient never had reactions to this medication in the past.
Acute radiation mucositis is a transient but major limiting side effect of radiation therapy. The associated odynophagia, secondary infection, and reduced oral intake often can lead to diminished disease control secondary to treatment interruption and subsequent development of resistant tumor burden. Concurrent chemotherapy and alternated fractionation radiation therapy increase the incidence of mucositis. Trotti et al1 (n=6181) reported that severe mucositis (grades 3 to 4) was found in 56% of patients receiving altered fractionation radiation therapy compared to 34% of patients who received conventional radiation therapy. Other risk factors related to the development of acute radiation mucositis include associated chemotherapy, age (>65 years), poor oral hygiene, diabetes mellitus, and prior periodontal disease.2
Radiation causes direct cellular damage to keratinocytes, leading to ulceration and erythema, as well as keratinocyte stem cells, which interferes with the healing process. Typical symptoms of mucosal radiation injury may include erythema (asymptomatic or causing intolerance of warm foods) that develops at the end of the second week of radiation therapy, focal areas of desquamation that develops in week 3, and confluent mucositis that can further progress to ulceration and necrosis in weeks 4 to 5.2 The development of dysgeusia, which is estimated to occur in 67% of patients receiving radiotherapy and 76% of patients receiving combination therapy, also can contribute to nutritional difficulties and weight loss.3
Avoiding overtreatment by constraining radiation volume and limiting concurrent chemotherapy are important preventative measures. The mainstay for managing mucositis includes symptomatic relief with oral hygiene, topical agents, topical plus systemic analgesia, dietary changes, and treatment of associated infections. Benzydamine, a nonsteroidal anti-inflammatory drug, is not available in the United States but has been shown to effectively improve symptoms.4 Various formulations of topical anesthetics consisting of diphenhydramine with or without corticosteroids, antibiotics, and antifungals help alleviate symptoms of mucositis; however, no single formulation has been studied. Low-level laser therapy also has shown efficacy in managing symptoms of mucositis.5,6 For persistent odynophagia, systemic opioid therapy should be attempted to achieve uninterrupted radiation therapy. Severe mucositis requires balancing risks and benefits of interrupting treatment, as additional damage may cause permanent mucosal injury.
Our patient had adequate symptom control with benzocaine lozenges and a combination mouthwash containing diphenhydramine, nystatin, lidocaine, hydrocortisone, and tetracycline. He required only occasional doses of systemic oxycodone. After a 1-week hospital admission for treatment of the pneumonia, he resumed radiation therapy and completed a full 8-week radiation course.
The Diagnosis: Radiation Mucositis
The patient was undergoing active radiation therapy for squamous cell carcinoma of the tongue, and according to the oncology team, the findings were in the precise location of radiation exposure. Radiation mucositis is a major and limiting side effect of radiation therapy for head and neck mucosal cancers, and symptom management is critical to ensure completion of the full radiation dose. Although infectious etiologies must be considered, the patient was already on prophylactic antiviral and antibacterial therapies. Moreover, the focal involvement with sparing of more mucosal tissue is atypical for most infections. Fixed drug reactions can present with localized mucosal and nonmucosal inflammation leading to erosion or ulceration. In this case, the only potential culprit was levofloxacin; however, it was initiated 2 days prior, and the patient never had reactions to this medication in the past.
Acute radiation mucositis is a transient but major limiting side effect of radiation therapy. The associated odynophagia, secondary infection, and reduced oral intake often can lead to diminished disease control secondary to treatment interruption and subsequent development of resistant tumor burden. Concurrent chemotherapy and alternated fractionation radiation therapy increase the incidence of mucositis. Trotti et al1 (n=6181) reported that severe mucositis (grades 3 to 4) was found in 56% of patients receiving altered fractionation radiation therapy compared to 34% of patients who received conventional radiation therapy. Other risk factors related to the development of acute radiation mucositis include associated chemotherapy, age (>65 years), poor oral hygiene, diabetes mellitus, and prior periodontal disease.2
Radiation causes direct cellular damage to keratinocytes, leading to ulceration and erythema, as well as keratinocyte stem cells, which interferes with the healing process. Typical symptoms of mucosal radiation injury may include erythema (asymptomatic or causing intolerance of warm foods) that develops at the end of the second week of radiation therapy, focal areas of desquamation that develops in week 3, and confluent mucositis that can further progress to ulceration and necrosis in weeks 4 to 5.2 The development of dysgeusia, which is estimated to occur in 67% of patients receiving radiotherapy and 76% of patients receiving combination therapy, also can contribute to nutritional difficulties and weight loss.3
Avoiding overtreatment by constraining radiation volume and limiting concurrent chemotherapy are important preventative measures. The mainstay for managing mucositis includes symptomatic relief with oral hygiene, topical agents, topical plus systemic analgesia, dietary changes, and treatment of associated infections. Benzydamine, a nonsteroidal anti-inflammatory drug, is not available in the United States but has been shown to effectively improve symptoms.4 Various formulations of topical anesthetics consisting of diphenhydramine with or without corticosteroids, antibiotics, and antifungals help alleviate symptoms of mucositis; however, no single formulation has been studied. Low-level laser therapy also has shown efficacy in managing symptoms of mucositis.5,6 For persistent odynophagia, systemic opioid therapy should be attempted to achieve uninterrupted radiation therapy. Severe mucositis requires balancing risks and benefits of interrupting treatment, as additional damage may cause permanent mucosal injury.
Our patient had adequate symptom control with benzocaine lozenges and a combination mouthwash containing diphenhydramine, nystatin, lidocaine, hydrocortisone, and tetracycline. He required only occasional doses of systemic oxycodone. After a 1-week hospital admission for treatment of the pneumonia, he resumed radiation therapy and completed a full 8-week radiation course.
- Trotti A, Bellm LA, Epstein JB, et al. Mucositis incidence, severity and associated outcomes in patients with head and neck cancer receiving radiotherapy with or without chemotherapy: a systematic literature review. Radiother Oncol. 2003;66:253-262.
- Mallick S, Benson R, Rath GK. Radiation induced oral mucositis: a review of current literature on prevention and management. Eur Arch Otorhinolaryngol. 2016;273:2285-2293.
- Hovan AJ, Williams PM, Stevenson-Moore P, et al; Dysgeusia Section, Oral Care Study Group, Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO). A systematic review of dysgeusia induced by cancer therapies. Support Care Cancer. 2010;18:1081-1087.
- Epstein JB, Silverman S, Paggiarino DA, et al. Benzydamine HCl for prophylaxis of radiation‐induced oral mucositis. Cancer. 2001;92:875-885.
- Henke M, Alfonsi M, Foa P, et al. Palifermin decreases severe oral mucositis of patients undergoing postoperative radiochemotherapy for head and neck cancer: a randomized, placebo-controlled trial. J Clin Oncol. 2011;29:2815-2820.
- Bensadoun RJ, Nair RG. Low-level laser therapy in the prevention and treatment of cancer therapy-induced mucositis: 2012 state of the art based on literature review and meta-analysis. Curr Opin Oncol. 2012;24:363-370.
- Trotti A, Bellm LA, Epstein JB, et al. Mucositis incidence, severity and associated outcomes in patients with head and neck cancer receiving radiotherapy with or without chemotherapy: a systematic literature review. Radiother Oncol. 2003;66:253-262.
- Mallick S, Benson R, Rath GK. Radiation induced oral mucositis: a review of current literature on prevention and management. Eur Arch Otorhinolaryngol. 2016;273:2285-2293.
- Hovan AJ, Williams PM, Stevenson-Moore P, et al; Dysgeusia Section, Oral Care Study Group, Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO). A systematic review of dysgeusia induced by cancer therapies. Support Care Cancer. 2010;18:1081-1087.
- Epstein JB, Silverman S, Paggiarino DA, et al. Benzydamine HCl for prophylaxis of radiation‐induced oral mucositis. Cancer. 2001;92:875-885.
- Henke M, Alfonsi M, Foa P, et al. Palifermin decreases severe oral mucositis of patients undergoing postoperative radiochemotherapy for head and neck cancer: a randomized, placebo-controlled trial. J Clin Oncol. 2011;29:2815-2820.
- Bensadoun RJ, Nair RG. Low-level laser therapy in the prevention and treatment of cancer therapy-induced mucositis: 2012 state of the art based on literature review and meta-analysis. Curr Opin Oncol. 2012;24:363-370.
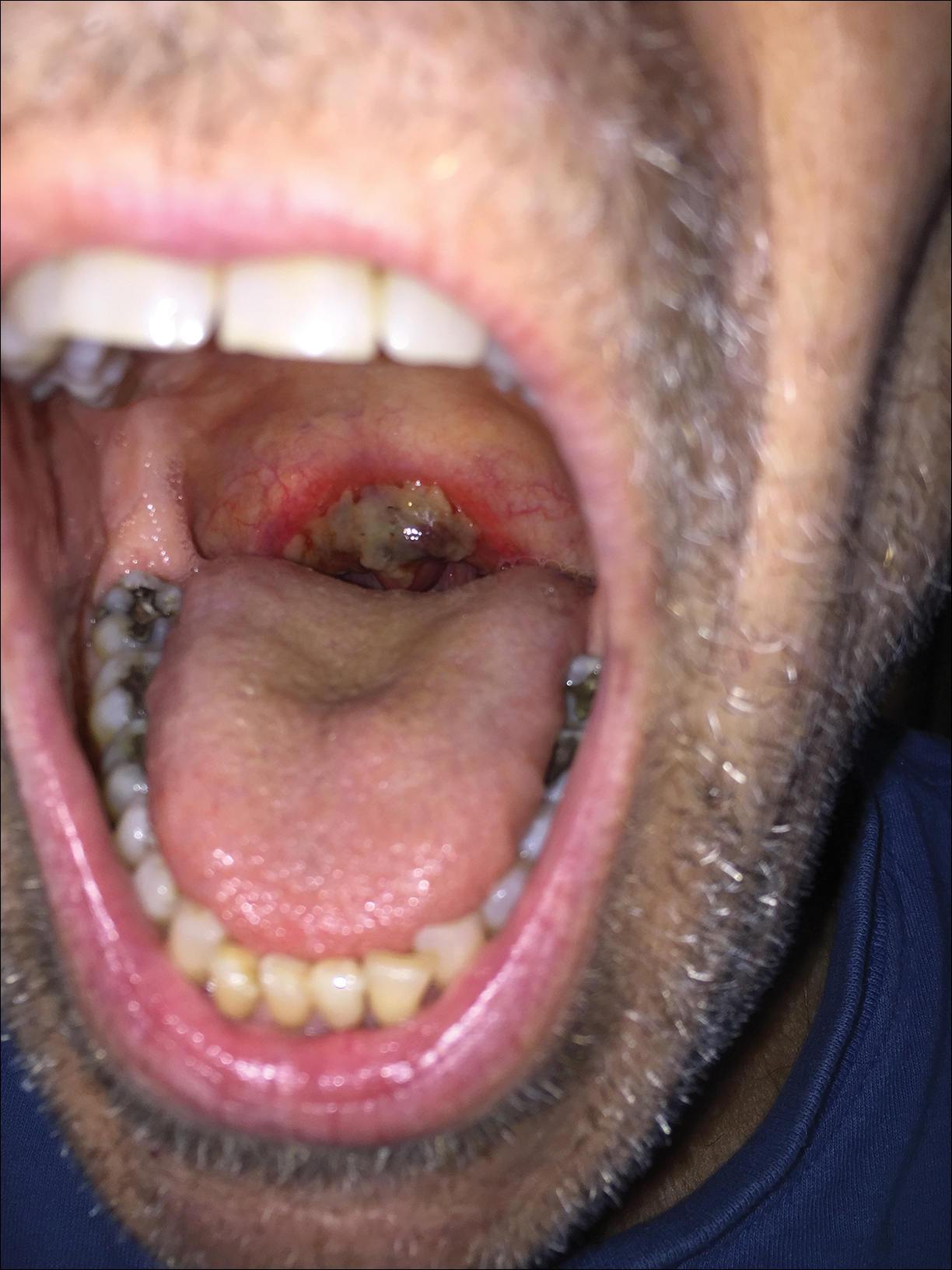
A 68-year-old man with squamous cell carcinoma of the tongue presented with a sore throat and odynophagia of 4 days' duration. At the time he was undergoing radiation therapy for the squamous cell carcinoma, and multiple myeloma was being actively treated with carfilzomib and pomalidomide. At the time of symptom onset he also was undergoing treatment with levofloxacin for community-acquired pneumonia. On day 2 of antibiotic therapy he noted pain with swallowing and an intolerance to warm foods. He was unaware of any new rash or lesions of the lips or mouth. He denied dysgeusia, changes in speech, bleeding, trauma, or recent smoking. He was taking prophylactic acyclovir and trimethoprim-sulfamethoxazole due to chemotherapy. Physical examination revealed a posterior oropharynx and uvula with well-defined friable erythema and erosions covered by white patches. There was no mucosal ulceration and no notable skin findings. The remainder of the physical examination was unremarkable.
Acute Painful Rash on the Cheek
The Diagnosis: Acute Contact Dermatitis
Dermoscopy demonstrated caterpillar hairs (Figure) and established the diagnosis of acute contact dermatitis due to a caterpillar. Upon further questioning, the patient recalled that something dustlike fell on the left cheek as she walked under some trees. The clinical and dermoscopic findings suggested a diagnosis of caterpillar dermatitis (family Limacodidae or Lymantriinae, order Lepidoptera). During the life cycle from young larvae to mature larvae, the quantities of the toxic thorns and hairs increase from 60,000 to 80,000 to 2,000,000 to 3,000,000. The toxic hairs measure 0.5 to 2.0 mm in length. They drop from the mature larvae's skin during desquamation as well as from the cocoon curing maturation to a moth. The hairs appear tubular and arrowlike with terminal spines.1 The larvae are called "the fiery hot" in Chinese, which vividly describes the swelling and sensation of burning with immediate contact.

Eruption severity and distribution depend on exposure modality and intensity. Exposed body parts, including the face, neck, forearms, interdigital spaces, and dorsal aspects of the hands, most commonly are involved. The eruption can be immediate or delayed, occurring hours or even days after the first contact.2 Itching is intense and continuous, with intermittent worsening. Clinically, the eruption manifests with rose to bright red, round macules and papules. Although rare, skin manifestations can be accompanied by systemic symptoms, such as malaise, fever, and anaphylaxis syndrome.3 The cutaneous lesions may last for 3 to 4 days and subside, leaving a brownish macule.1,4
The differential diagnosis includes acute herpes simplex, which presents as grouped vesicles on an erythematous base with itching or burning, and recurrences in the same location are common. Acute Sweet syndrome may appear as erythematous or edematous painful plaques with fever and neutrophilia. Acute urticaria appears as wheals with severe pruritus, and individual lesions can resolve within several hours. Insect bites often appear as itching or painful erythema or papules.
We sterilized the lesion with alcohol, removed the thorns as much as possible with ophthalmic forceps under the guidance of dermoscopy, and prescribed chloramphenicol ointment 1% twice daily. Our patient was completely cured within 24 hours with no systemic symptoms or pigmentation.
This case directly showed a novel usage of dermoscopy in diagnosis and therapy, especially in acute contact dermatitis. Small irritants such as caterpillar thorns and hairs easily can be observed and removed by dermoscopy devices with higher magnification.
- Fangan H, Yun H, Yuhua G, et al. Observations on the pathogenicity of Lepidoptera, Euileidae caterpillar and the clinical pathological pictures of patients with dermatitis. Chinese J Zoonoses. 2005,21:414-416.
- Bonamonte D, Foti C, Vestita M, et al. Skin reactions to pine processionary caterpillar Thaumetopoea pityocampa Schiff. ScientificWorldJournal. 2013;2013:867431.
- Burns T, Breathnach S, Cox N, et al, eds. Rook's Textbook of Dermatology. 8th ed. Vol 2. Oxford, United Kingdom: Blackwell; 2010.
- Henwood BP, MacDonald DM. Caterpillar dermatitis. Clin Exp Dermatol. 1983;8:77-93.
The Diagnosis: Acute Contact Dermatitis
Dermoscopy demonstrated caterpillar hairs (Figure) and established the diagnosis of acute contact dermatitis due to a caterpillar. Upon further questioning, the patient recalled that something dustlike fell on the left cheek as she walked under some trees. The clinical and dermoscopic findings suggested a diagnosis of caterpillar dermatitis (family Limacodidae or Lymantriinae, order Lepidoptera). During the life cycle from young larvae to mature larvae, the quantities of the toxic thorns and hairs increase from 60,000 to 80,000 to 2,000,000 to 3,000,000. The toxic hairs measure 0.5 to 2.0 mm in length. They drop from the mature larvae's skin during desquamation as well as from the cocoon curing maturation to a moth. The hairs appear tubular and arrowlike with terminal spines.1 The larvae are called "the fiery hot" in Chinese, which vividly describes the swelling and sensation of burning with immediate contact.
Eruption severity and distribution depend on exposure modality and intensity. Exposed body parts, including the face, neck, forearms, interdigital spaces, and dorsal aspects of the hands, most commonly are involved. The eruption can be immediate or delayed, occurring hours or even days after the first contact.2 Itching is intense and continuous, with intermittent worsening. Clinically, the eruption manifests with rose to bright red, round macules and papules. Although rare, skin manifestations can be accompanied by systemic symptoms, such as malaise, fever, and anaphylaxis syndrome.3 The cutaneous lesions may last for 3 to 4 days and subside, leaving a brownish macule.1,4
The differential diagnosis includes acute herpes simplex, which presents as grouped vesicles on an erythematous base with itching or burning, and recurrences in the same location are common. Acute Sweet syndrome may appear as erythematous or edematous painful plaques with fever and neutrophilia. Acute urticaria appears as wheals with severe pruritus, and individual lesions can resolve within several hours. Insect bites often appear as itching or painful erythema or papules.
We sterilized the lesion with alcohol, removed the thorns as much as possible with ophthalmic forceps under the guidance of dermoscopy, and prescribed chloramphenicol ointment 1% twice daily. Our patient was completely cured within 24 hours with no systemic symptoms or pigmentation.
This case directly showed a novel usage of dermoscopy in diagnosis and therapy, especially in acute contact dermatitis. Small irritants such as caterpillar thorns and hairs easily can be observed and removed by dermoscopy devices with higher magnification.
The Diagnosis: Acute Contact Dermatitis
Dermoscopy demonstrated caterpillar hairs (Figure) and established the diagnosis of acute contact dermatitis due to a caterpillar. Upon further questioning, the patient recalled that something dustlike fell on the left cheek as she walked under some trees. The clinical and dermoscopic findings suggested a diagnosis of caterpillar dermatitis (family Limacodidae or Lymantriinae, order Lepidoptera). During the life cycle from young larvae to mature larvae, the quantities of the toxic thorns and hairs increase from 60,000 to 80,000 to 2,000,000 to 3,000,000. The toxic hairs measure 0.5 to 2.0 mm in length. They drop from the mature larvae's skin during desquamation as well as from the cocoon curing maturation to a moth. The hairs appear tubular and arrowlike with terminal spines.1 The larvae are called "the fiery hot" in Chinese, which vividly describes the swelling and sensation of burning with immediate contact.
Eruption severity and distribution depend on exposure modality and intensity. Exposed body parts, including the face, neck, forearms, interdigital spaces, and dorsal aspects of the hands, most commonly are involved. The eruption can be immediate or delayed, occurring hours or even days after the first contact.2 Itching is intense and continuous, with intermittent worsening. Clinically, the eruption manifests with rose to bright red, round macules and papules. Although rare, skin manifestations can be accompanied by systemic symptoms, such as malaise, fever, and anaphylaxis syndrome.3 The cutaneous lesions may last for 3 to 4 days and subside, leaving a brownish macule.1,4
The differential diagnosis includes acute herpes simplex, which presents as grouped vesicles on an erythematous base with itching or burning, and recurrences in the same location are common. Acute Sweet syndrome may appear as erythematous or edematous painful plaques with fever and neutrophilia. Acute urticaria appears as wheals with severe pruritus, and individual lesions can resolve within several hours. Insect bites often appear as itching or painful erythema or papules.
We sterilized the lesion with alcohol, removed the thorns as much as possible with ophthalmic forceps under the guidance of dermoscopy, and prescribed chloramphenicol ointment 1% twice daily. Our patient was completely cured within 24 hours with no systemic symptoms or pigmentation.
This case directly showed a novel usage of dermoscopy in diagnosis and therapy, especially in acute contact dermatitis. Small irritants such as caterpillar thorns and hairs easily can be observed and removed by dermoscopy devices with higher magnification.
- Fangan H, Yun H, Yuhua G, et al. Observations on the pathogenicity of Lepidoptera, Euileidae caterpillar and the clinical pathological pictures of patients with dermatitis. Chinese J Zoonoses. 2005,21:414-416.
- Bonamonte D, Foti C, Vestita M, et al. Skin reactions to pine processionary caterpillar Thaumetopoea pityocampa Schiff. ScientificWorldJournal. 2013;2013:867431.
- Burns T, Breathnach S, Cox N, et al, eds. Rook's Textbook of Dermatology. 8th ed. Vol 2. Oxford, United Kingdom: Blackwell; 2010.
- Henwood BP, MacDonald DM. Caterpillar dermatitis. Clin Exp Dermatol. 1983;8:77-93.
- Fangan H, Yun H, Yuhua G, et al. Observations on the pathogenicity of Lepidoptera, Euileidae caterpillar and the clinical pathological pictures of patients with dermatitis. Chinese J Zoonoses. 2005,21:414-416.
- Bonamonte D, Foti C, Vestita M, et al. Skin reactions to pine processionary caterpillar Thaumetopoea pityocampa Schiff. ScientificWorldJournal. 2013;2013:867431.
- Burns T, Breathnach S, Cox N, et al, eds. Rook's Textbook of Dermatology. 8th ed. Vol 2. Oxford, United Kingdom: Blackwell; 2010.
- Henwood BP, MacDonald DM. Caterpillar dermatitis. Clin Exp Dermatol. 1983;8:77-93.
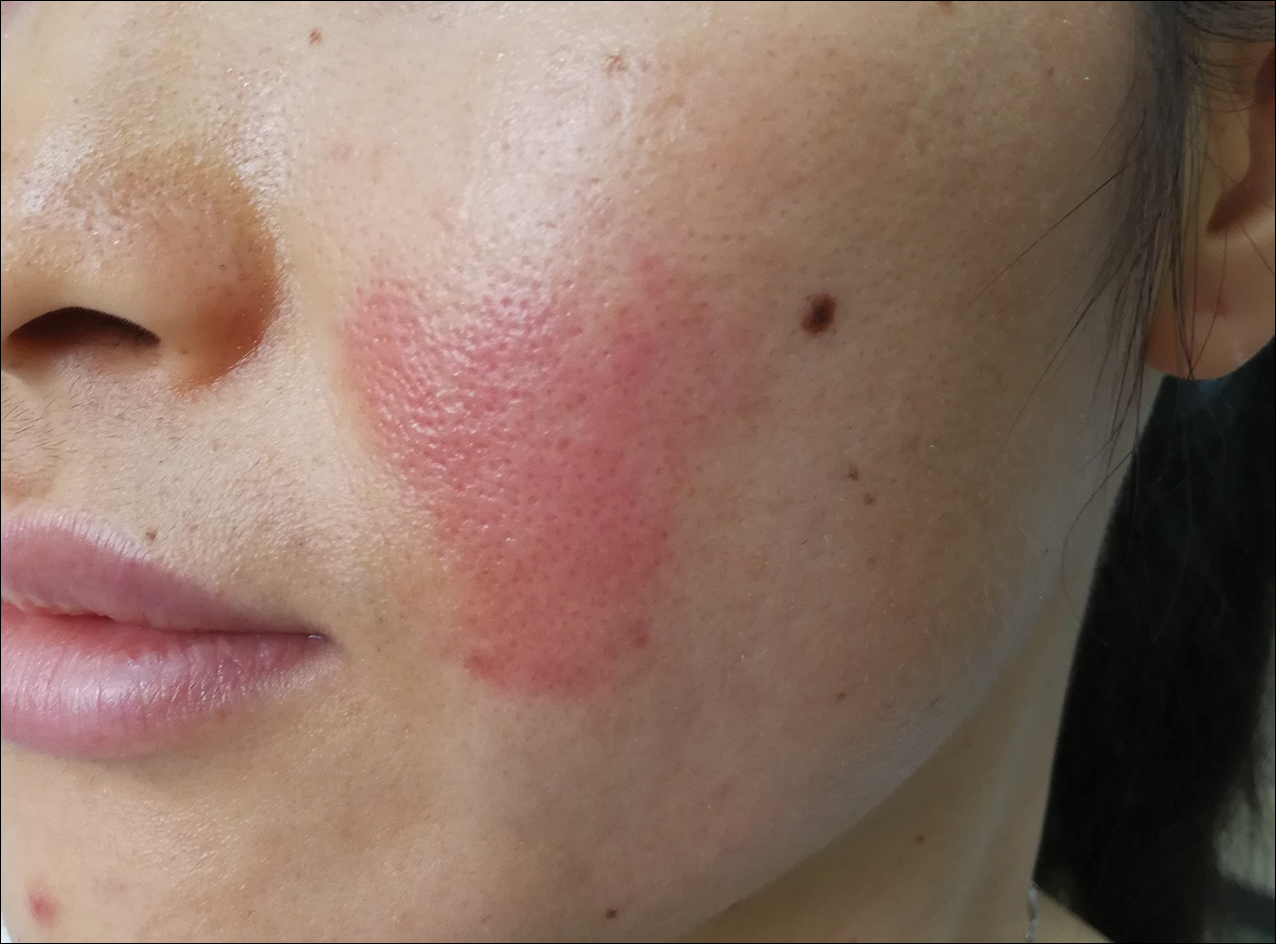
A 31-year-old woman presented to an outpatient dermatology department with acute pruritus, burning, and moderate swelling of the left cheek of 10 minutes' duration that occurred while waiting to see a hematologist in the same building. The patient was diagnosed with aplastic anemia 11 years prior and was awaiting bone marrow transplantation. Physical examination showed an edematous erythematous wheal with a relatively distinct border measuring 3 cm in diameter. No foreign material could be identified on the surface with the naked eye. Dermoscopy was performed.
Solitary Nodule on the Proximal Nail Fold
The Diagnosis: Superficial Acral Fibromyxoma
A shave biopsy revealed an uninvolved grenz zone and mildly cellular spindle cell dermal proliferation in a collagenous and myxoid background (Figure 1). Spindle cells were seen in a myxoid background among dense coarse collagen (Figure 2A). Spindle cells also were seen in a myxoid background with mast cells and capillary network (Figure 2B). Histopathologic examination of the biopsy specimen revealed spindle cells that were diffusely positive for CD34 (Figure 3); focally positive for epithelial membrane antigen; and negative for melanocytic markers, smooth muscle markers, and cytokeratin. A diagnosis of superficial acral fibromyxoma (SAFM) was made based on clinical, histopathologic, and immunohistochemical findings.
.")
 as well as with mast cells and capillary network (B)(H&E, original magnifications ×200 and ×400).")
.")
Superficial acral fibromyxomas, also known as digital fibromyxomas, are soft, slow-growing tumors that have a predilection for subungual or periungual regions of the hands and feet. Superficial acral fibromyxomas most frequently occur on the hallux and rarely occur on the ankle or leg. They can present as nodular, dome-shaped, polyploid, or verrucous masses. They can be soft to firm, gelatinous or solid, off-white to gray-white and can have fasciculate cut surfaces. Superficial acral fibromyxomas can be either painful or painless and present with a deformed nail in 9% of cases. Superficial acral fibromyxoma is a superficial lesion with frequent infiltration of the dermal collagen and subcutaneous tissue and may even erode or infiltrate into the underlying bone in rare cases.1-4 Although SAFMs are rare tumors, documented cases of SAFM have been reported at an increasing rate since the first published report by Fetsch et al2 in 2001.
Patients often delay seeking medical treatment and present with a solitary mass that has been slowly growing for months to years. In a study of 124 patients, Hollmann et al1 found that symptoms exist for a mean of 35 months and present with a small mass with a mean tumor size of 1.7 cm before biopsy or excision. Although the age range is broad, SAFM mostly affects middle-aged adults (median age, 49 years).1 Hollmann et al1 also reported a male predominance (1.3:1 ratio), and preexisting local trauma is reported in 25% of cases.2-4
The differential for SAFM should include dermatofibroma, keloid, dermatofibrosarcoma protuberans, acquired digital fibrokeratoma, infantile digital fibromatosis, neurolemmoma, sclerosing perineurioma, superficial angiomyxoma, low-grade fibromyxoid sarcoma, and acral myxoinflammatory fibroblastic sarcoma.1-4
Superficial acral fibromyxomas are composed of CD34+ spindle or stellate-shaped cells that are embedded in a myxoid and/or dense hyalinized collagenous stroma in a random or loosely fascicular growth pattern. The spindle or stellate-shaped cells in SAFMs also have been found to be focally positive for epithelial membrane antigen and CD99. Lesions have accentuated microvasculature and increased mast cells.5-8
Conservative management is reasonable, but patients presenting with persistent pain and/or local deformity should be definitively treated with complete excision and follow-up. Hollmann et al1 found that 24% of tumors recurred locally upon incomplete excision after a mean interval of 27 months. All recurrent tumors had positive margins at excision or initial biopsy.1 To date, no reports of tumors metastasizing have been documented.1-4
- Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
- Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
- Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
- Ashby-Richardson H, Rogers GS, Stadecker MJ. Superficial acral fibromyxoma: an overview. Arch Pathol Lab Med. 2011;135:1064-1066.
- Quaba O, Evans A, Al-Nafussi AA, et al. Superficial acral fibromyxoma. Br J Plast Surg. 2005;58:561-564.
- Oteo-Alvaro A, Meizoso T, Scarpellini A, et al. Superficial acral fibromyxoma of the toe, with erosion of the distal phalanx: a clinical report. Arch Orthop Trauma Surg. 2008;128:271-274.
- Meyerle J, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
- Kazakov DV, Mentzel T, Buro G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
The Diagnosis: Superficial Acral Fibromyxoma
A shave biopsy revealed an uninvolved grenz zone and mildly cellular spindle cell dermal proliferation in a collagenous and myxoid background (Figure 1). Spindle cells were seen in a myxoid background among dense coarse collagen (Figure 2A). Spindle cells also were seen in a myxoid background with mast cells and capillary network (Figure 2B). Histopathologic examination of the biopsy specimen revealed spindle cells that were diffusely positive for CD34 (Figure 3); focally positive for epithelial membrane antigen; and negative for melanocytic markers, smooth muscle markers, and cytokeratin. A diagnosis of superficial acral fibromyxoma (SAFM) was made based on clinical, histopathologic, and immunohistochemical findings.
Superficial acral fibromyxomas, also known as digital fibromyxomas, are soft, slow-growing tumors that have a predilection for subungual or periungual regions of the hands and feet. Superficial acral fibromyxomas most frequently occur on the hallux and rarely occur on the ankle or leg. They can present as nodular, dome-shaped, polyploid, or verrucous masses. They can be soft to firm, gelatinous or solid, off-white to gray-white and can have fasciculate cut surfaces. Superficial acral fibromyxomas can be either painful or painless and present with a deformed nail in 9% of cases. Superficial acral fibromyxoma is a superficial lesion with frequent infiltration of the dermal collagen and subcutaneous tissue and may even erode or infiltrate into the underlying bone in rare cases.1-4 Although SAFMs are rare tumors, documented cases of SAFM have been reported at an increasing rate since the first published report by Fetsch et al2 in 2001.
Patients often delay seeking medical treatment and present with a solitary mass that has been slowly growing for months to years. In a study of 124 patients, Hollmann et al1 found that symptoms exist for a mean of 35 months and present with a small mass with a mean tumor size of 1.7 cm before biopsy or excision. Although the age range is broad, SAFM mostly affects middle-aged adults (median age, 49 years).1 Hollmann et al1 also reported a male predominance (1.3:1 ratio), and preexisting local trauma is reported in 25% of cases.2-4
The differential for SAFM should include dermatofibroma, keloid, dermatofibrosarcoma protuberans, acquired digital fibrokeratoma, infantile digital fibromatosis, neurolemmoma, sclerosing perineurioma, superficial angiomyxoma, low-grade fibromyxoid sarcoma, and acral myxoinflammatory fibroblastic sarcoma.1-4
Superficial acral fibromyxomas are composed of CD34+ spindle or stellate-shaped cells that are embedded in a myxoid and/or dense hyalinized collagenous stroma in a random or loosely fascicular growth pattern. The spindle or stellate-shaped cells in SAFMs also have been found to be focally positive for epithelial membrane antigen and CD99. Lesions have accentuated microvasculature and increased mast cells.5-8
Conservative management is reasonable, but patients presenting with persistent pain and/or local deformity should be definitively treated with complete excision and follow-up. Hollmann et al1 found that 24% of tumors recurred locally upon incomplete excision after a mean interval of 27 months. All recurrent tumors had positive margins at excision or initial biopsy.1 To date, no reports of tumors metastasizing have been documented.1-4
The Diagnosis: Superficial Acral Fibromyxoma
A shave biopsy revealed an uninvolved grenz zone and mildly cellular spindle cell dermal proliferation in a collagenous and myxoid background (Figure 1). Spindle cells were seen in a myxoid background among dense coarse collagen (Figure 2A). Spindle cells also were seen in a myxoid background with mast cells and capillary network (Figure 2B). Histopathologic examination of the biopsy specimen revealed spindle cells that were diffusely positive for CD34 (Figure 3); focally positive for epithelial membrane antigen; and negative for melanocytic markers, smooth muscle markers, and cytokeratin. A diagnosis of superficial acral fibromyxoma (SAFM) was made based on clinical, histopathologic, and immunohistochemical findings.
Superficial acral fibromyxomas, also known as digital fibromyxomas, are soft, slow-growing tumors that have a predilection for subungual or periungual regions of the hands and feet. Superficial acral fibromyxomas most frequently occur on the hallux and rarely occur on the ankle or leg. They can present as nodular, dome-shaped, polyploid, or verrucous masses. They can be soft to firm, gelatinous or solid, off-white to gray-white and can have fasciculate cut surfaces. Superficial acral fibromyxomas can be either painful or painless and present with a deformed nail in 9% of cases. Superficial acral fibromyxoma is a superficial lesion with frequent infiltration of the dermal collagen and subcutaneous tissue and may even erode or infiltrate into the underlying bone in rare cases.1-4 Although SAFMs are rare tumors, documented cases of SAFM have been reported at an increasing rate since the first published report by Fetsch et al2 in 2001.
Patients often delay seeking medical treatment and present with a solitary mass that has been slowly growing for months to years. In a study of 124 patients, Hollmann et al1 found that symptoms exist for a mean of 35 months and present with a small mass with a mean tumor size of 1.7 cm before biopsy or excision. Although the age range is broad, SAFM mostly affects middle-aged adults (median age, 49 years).1 Hollmann et al1 also reported a male predominance (1.3:1 ratio), and preexisting local trauma is reported in 25% of cases.2-4
The differential for SAFM should include dermatofibroma, keloid, dermatofibrosarcoma protuberans, acquired digital fibrokeratoma, infantile digital fibromatosis, neurolemmoma, sclerosing perineurioma, superficial angiomyxoma, low-grade fibromyxoid sarcoma, and acral myxoinflammatory fibroblastic sarcoma.1-4
Superficial acral fibromyxomas are composed of CD34+ spindle or stellate-shaped cells that are embedded in a myxoid and/or dense hyalinized collagenous stroma in a random or loosely fascicular growth pattern. The spindle or stellate-shaped cells in SAFMs also have been found to be focally positive for epithelial membrane antigen and CD99. Lesions have accentuated microvasculature and increased mast cells.5-8
Conservative management is reasonable, but patients presenting with persistent pain and/or local deformity should be definitively treated with complete excision and follow-up. Hollmann et al1 found that 24% of tumors recurred locally upon incomplete excision after a mean interval of 27 months. All recurrent tumors had positive margins at excision or initial biopsy.1 To date, no reports of tumors metastasizing have been documented.1-4
- Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
- Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
- Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
- Ashby-Richardson H, Rogers GS, Stadecker MJ. Superficial acral fibromyxoma: an overview. Arch Pathol Lab Med. 2011;135:1064-1066.
- Quaba O, Evans A, Al-Nafussi AA, et al. Superficial acral fibromyxoma. Br J Plast Surg. 2005;58:561-564.
- Oteo-Alvaro A, Meizoso T, Scarpellini A, et al. Superficial acral fibromyxoma of the toe, with erosion of the distal phalanx: a clinical report. Arch Orthop Trauma Surg. 2008;128:271-274.
- Meyerle J, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
- Kazakov DV, Mentzel T, Buro G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
- Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
- Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
- Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
- Ashby-Richardson H, Rogers GS, Stadecker MJ. Superficial acral fibromyxoma: an overview. Arch Pathol Lab Med. 2011;135:1064-1066.
- Quaba O, Evans A, Al-Nafussi AA, et al. Superficial acral fibromyxoma. Br J Plast Surg. 2005;58:561-564.
- Oteo-Alvaro A, Meizoso T, Scarpellini A, et al. Superficial acral fibromyxoma of the toe, with erosion of the distal phalanx: a clinical report. Arch Orthop Trauma Surg. 2008;128:271-274.
- Meyerle J, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
- Kazakov DV, Mentzel T, Buro G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
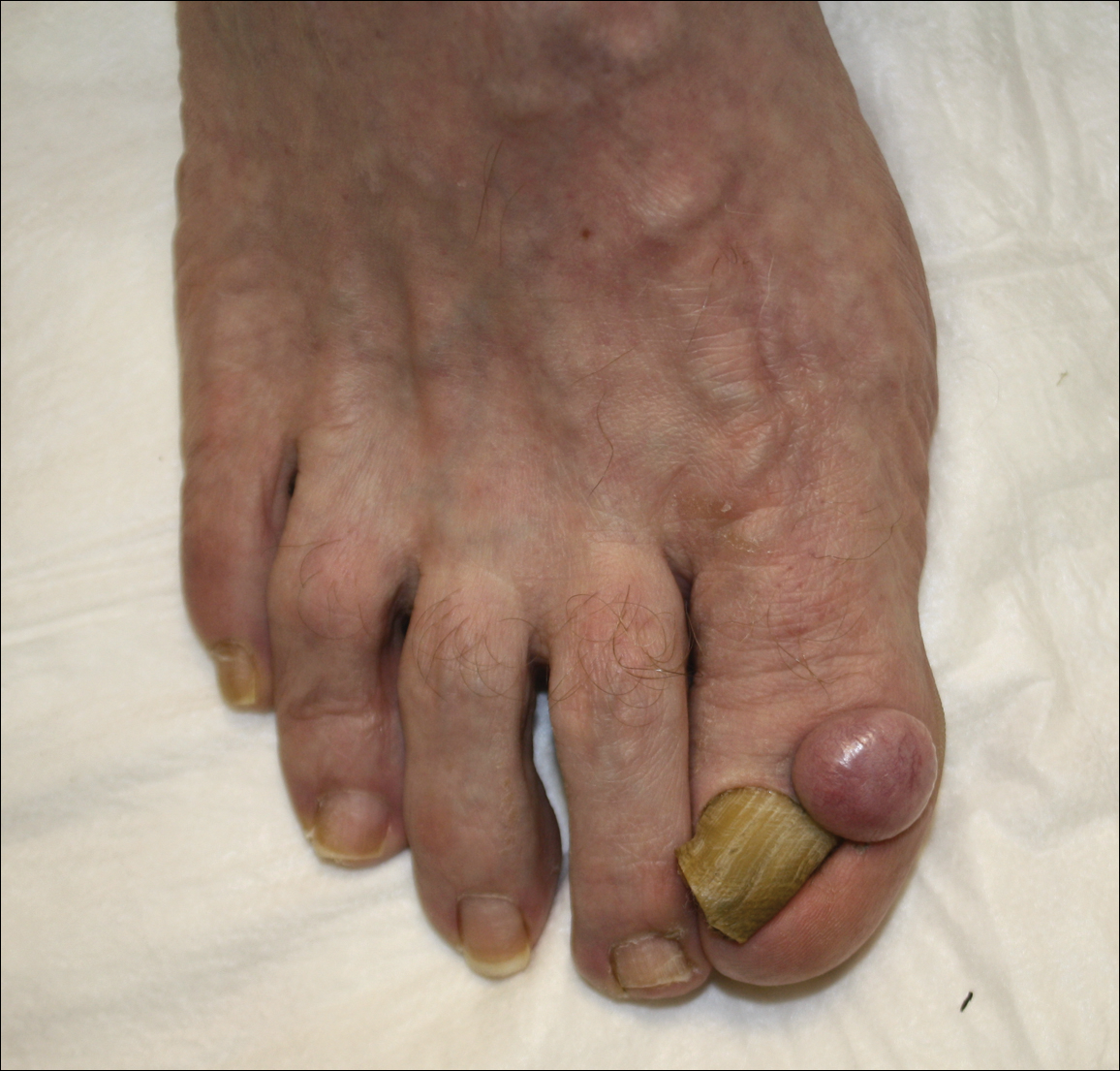
A 62-year-old man presented for evaluation of a slowly growing, nonpainful nodule on the first proximal toenail fold of the right foot of 6 years' duration. He reported that the nail plate of the affected toe was thickened and malaligned. He denied a history of trauma. Physical examination revealed a 2.0×1.6-cm, flesh-colored, nontender, well-defined, rubbery nodule with prominent overlying tortuous telangiectases on the medial aspect of the first proximal toenail fold of the right foot. The associated nail plate was yellow, thickened, and angled laterally into the second toe. Radiograph of the right hallux identified a soft tissue density contiguous with the dorsal aspect of the distal portion of the phalanx. There was no evidence of bony involvement. A shave saucerization biopsy specimen was obtained and sent for hematoxylin and eosin and immunohistochemical staining. The spindle cells were diffusely positive for CD34.
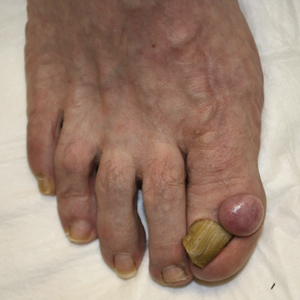
Hypopigmentation on the Ear
The Diagnosis: Corticosteroid-Induced Hypopigmentation
This patient received several intralesional injections of triamcinolone acetonide once monthly for treatment of the keloid scar on the left ear at an outside institution. There was improvement in the size of the keloid over time. On physical examination during the most recent visit there was a prominent streak of hypopigmentation and atrophy near the corticosteroid injection site with extension to the postauricular region. There also was telangiectasia noted within the area of hypopigmentation. Intralesional triamcinolone injections were discontinued and the patient was advised to return for monitoring.
Intra-articular and intralesional corticosteroid injections frequently are used by clinicians. Cutaneous complications associated with these injections include atrophy, pigmentary changes, hypersensitivity reactions, flushing, cellulitis, and necrotizing fasciitis. Tendon rupture also has been reported.1
There are several case reports in the literature describing hypopigmentation and/or subcutaneous atrophy after intralesional or intra-articular corticosteroid injections. A variety of underlying conditions were treated including alopecia areata, keloids, rheumatoid arthritis, de Quervain tendonitis, and psoriasis.2-6 The lesions typically are described as linear rays of atrophy and hypopigmentation at or near the injection site, with some cases noting extension along lymph channels and proximal veins.4,6 There usually is no associated pruritus or pain.3 This phenomenon can be seen after single or multiple injections.4,6
Extension of hypopigmentation from the site of injection has been postulated to be due to venous or lymphatic uptake.2,4-6 The mechanism of hypopigmentation is not known. Biopsy of a previously described case showed intact melanocytes along the dermoepidermal junction.2 Biopsy from another case revealed a decrease in melanin staining, which suggests a decrease in number or activity of melanocytes.4 It was proposed that hypopigmentation was secondary to loss of melanocyte function instead of loss of melanocytes.2 Spontaneous improvement or resolution of the hypopigmentation were noted in some cases ranging from 1 month to 1 year after initial presentation, but the hypopigmentation also can be persistent.3-6
Hypopigmented sarcoidosis and hypopigmented mycosis fungoides, both often present on dark-skinned individuals, are included in the differential diagnosis. Hypopigmented sarcoidosis presents with hypopigmented macules or patches, some with central papules, and hypopigmented mycosis fungoides presents with hypopigmented patches or plaques with fine scale and onset often in childhood or adolescence.7,8 Morphea can present with an initial inflammatory stage that develops into a sclerotic firm plaque or nodule with hyperpigmentation or hypopigmentation.9 Vitiligo usually presents with depigmented macules or patches and depigmented hair within the lesion.10
- Brinks A, Koes BW, Volkers AC, et al. Adverse effects of extra-articular corticosteroid injections: a systematic review. BMC Musculoskelet Disord. 2010;11:206.
- Venkatesan P, Fangman WL. Linear hypopigmentation and cutaneous atrophy following intra-articular steroid injections for de Quervain's tendonitis. J Drugs Dermatol. 2009;8:492-493.
- Evans AV, McGibbon DH. Symmetrical hypopigmentation following triamcinolone injection for de Quervain's tenosynovitis. Clin Exp Dermatol. 2002;27:247-251.
- Friedman SJ, Butler DF, Pittelkow MR. Perilesional linear atrophy and hypopigmentation after intralesional corticosteroid therapy. report of two cases and review of the literature. J Am Acad Dermatol. 1988;19:537-541.
- van Vendeloo SN, Ettema HB. Skin depigmentation along lymph vessels of the lower leg following local corticosteroid injection for interdigital neuroma. Foot Ankle Surg. 2016;22:139-141.
- Kumar P, Adolph S. Hypopigmentation along subcutaneous veins following intrakeloid triamcinolone injection: a case report and review of literature. Burns. 1998;24:487-488.
- Elgart ML. Cutaneous sarcoidosis: definitions and types of lesions. Clin Dermatol. 1986;4:35-45.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
- Yaghoobi R, Omidian M, Bagherani N. Vitiligo: a review of the published work. J Dermatol. 2011;38:419-431.
The Diagnosis: Corticosteroid-Induced Hypopigmentation
This patient received several intralesional injections of triamcinolone acetonide once monthly for treatment of the keloid scar on the left ear at an outside institution. There was improvement in the size of the keloid over time. On physical examination during the most recent visit there was a prominent streak of hypopigmentation and atrophy near the corticosteroid injection site with extension to the postauricular region. There also was telangiectasia noted within the area of hypopigmentation. Intralesional triamcinolone injections were discontinued and the patient was advised to return for monitoring.
Intra-articular and intralesional corticosteroid injections frequently are used by clinicians. Cutaneous complications associated with these injections include atrophy, pigmentary changes, hypersensitivity reactions, flushing, cellulitis, and necrotizing fasciitis. Tendon rupture also has been reported.1
There are several case reports in the literature describing hypopigmentation and/or subcutaneous atrophy after intralesional or intra-articular corticosteroid injections. A variety of underlying conditions were treated including alopecia areata, keloids, rheumatoid arthritis, de Quervain tendonitis, and psoriasis.2-6 The lesions typically are described as linear rays of atrophy and hypopigmentation at or near the injection site, with some cases noting extension along lymph channels and proximal veins.4,6 There usually is no associated pruritus or pain.3 This phenomenon can be seen after single or multiple injections.4,6
Extension of hypopigmentation from the site of injection has been postulated to be due to venous or lymphatic uptake.2,4-6 The mechanism of hypopigmentation is not known. Biopsy of a previously described case showed intact melanocytes along the dermoepidermal junction.2 Biopsy from another case revealed a decrease in melanin staining, which suggests a decrease in number or activity of melanocytes.4 It was proposed that hypopigmentation was secondary to loss of melanocyte function instead of loss of melanocytes.2 Spontaneous improvement or resolution of the hypopigmentation were noted in some cases ranging from 1 month to 1 year after initial presentation, but the hypopigmentation also can be persistent.3-6
Hypopigmented sarcoidosis and hypopigmented mycosis fungoides, both often present on dark-skinned individuals, are included in the differential diagnosis. Hypopigmented sarcoidosis presents with hypopigmented macules or patches, some with central papules, and hypopigmented mycosis fungoides presents with hypopigmented patches or plaques with fine scale and onset often in childhood or adolescence.7,8 Morphea can present with an initial inflammatory stage that develops into a sclerotic firm plaque or nodule with hyperpigmentation or hypopigmentation.9 Vitiligo usually presents with depigmented macules or patches and depigmented hair within the lesion.10
The Diagnosis: Corticosteroid-Induced Hypopigmentation
This patient received several intralesional injections of triamcinolone acetonide once monthly for treatment of the keloid scar on the left ear at an outside institution. There was improvement in the size of the keloid over time. On physical examination during the most recent visit there was a prominent streak of hypopigmentation and atrophy near the corticosteroid injection site with extension to the postauricular region. There also was telangiectasia noted within the area of hypopigmentation. Intralesional triamcinolone injections were discontinued and the patient was advised to return for monitoring.
Intra-articular and intralesional corticosteroid injections frequently are used by clinicians. Cutaneous complications associated with these injections include atrophy, pigmentary changes, hypersensitivity reactions, flushing, cellulitis, and necrotizing fasciitis. Tendon rupture also has been reported.1
There are several case reports in the literature describing hypopigmentation and/or subcutaneous atrophy after intralesional or intra-articular corticosteroid injections. A variety of underlying conditions were treated including alopecia areata, keloids, rheumatoid arthritis, de Quervain tendonitis, and psoriasis.2-6 The lesions typically are described as linear rays of atrophy and hypopigmentation at or near the injection site, with some cases noting extension along lymph channels and proximal veins.4,6 There usually is no associated pruritus or pain.3 This phenomenon can be seen after single or multiple injections.4,6
Extension of hypopigmentation from the site of injection has been postulated to be due to venous or lymphatic uptake.2,4-6 The mechanism of hypopigmentation is not known. Biopsy of a previously described case showed intact melanocytes along the dermoepidermal junction.2 Biopsy from another case revealed a decrease in melanin staining, which suggests a decrease in number or activity of melanocytes.4 It was proposed that hypopigmentation was secondary to loss of melanocyte function instead of loss of melanocytes.2 Spontaneous improvement or resolution of the hypopigmentation were noted in some cases ranging from 1 month to 1 year after initial presentation, but the hypopigmentation also can be persistent.3-6
Hypopigmented sarcoidosis and hypopigmented mycosis fungoides, both often present on dark-skinned individuals, are included in the differential diagnosis. Hypopigmented sarcoidosis presents with hypopigmented macules or patches, some with central papules, and hypopigmented mycosis fungoides presents with hypopigmented patches or plaques with fine scale and onset often in childhood or adolescence.7,8 Morphea can present with an initial inflammatory stage that develops into a sclerotic firm plaque or nodule with hyperpigmentation or hypopigmentation.9 Vitiligo usually presents with depigmented macules or patches and depigmented hair within the lesion.10
- Brinks A, Koes BW, Volkers AC, et al. Adverse effects of extra-articular corticosteroid injections: a systematic review. BMC Musculoskelet Disord. 2010;11:206.
- Venkatesan P, Fangman WL. Linear hypopigmentation and cutaneous atrophy following intra-articular steroid injections for de Quervain's tendonitis. J Drugs Dermatol. 2009;8:492-493.
- Evans AV, McGibbon DH. Symmetrical hypopigmentation following triamcinolone injection for de Quervain's tenosynovitis. Clin Exp Dermatol. 2002;27:247-251.
- Friedman SJ, Butler DF, Pittelkow MR. Perilesional linear atrophy and hypopigmentation after intralesional corticosteroid therapy. report of two cases and review of the literature. J Am Acad Dermatol. 1988;19:537-541.
- van Vendeloo SN, Ettema HB. Skin depigmentation along lymph vessels of the lower leg following local corticosteroid injection for interdigital neuroma. Foot Ankle Surg. 2016;22:139-141.
- Kumar P, Adolph S. Hypopigmentation along subcutaneous veins following intrakeloid triamcinolone injection: a case report and review of literature. Burns. 1998;24:487-488.
- Elgart ML. Cutaneous sarcoidosis: definitions and types of lesions. Clin Dermatol. 1986;4:35-45.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
- Yaghoobi R, Omidian M, Bagherani N. Vitiligo: a review of the published work. J Dermatol. 2011;38:419-431.
- Brinks A, Koes BW, Volkers AC, et al. Adverse effects of extra-articular corticosteroid injections: a systematic review. BMC Musculoskelet Disord. 2010;11:206.
- Venkatesan P, Fangman WL. Linear hypopigmentation and cutaneous atrophy following intra-articular steroid injections for de Quervain's tendonitis. J Drugs Dermatol. 2009;8:492-493.
- Evans AV, McGibbon DH. Symmetrical hypopigmentation following triamcinolone injection for de Quervain's tenosynovitis. Clin Exp Dermatol. 2002;27:247-251.
- Friedman SJ, Butler DF, Pittelkow MR. Perilesional linear atrophy and hypopigmentation after intralesional corticosteroid therapy. report of two cases and review of the literature. J Am Acad Dermatol. 1988;19:537-541.
- van Vendeloo SN, Ettema HB. Skin depigmentation along lymph vessels of the lower leg following local corticosteroid injection for interdigital neuroma. Foot Ankle Surg. 2016;22:139-141.
- Kumar P, Adolph S. Hypopigmentation along subcutaneous veins following intrakeloid triamcinolone injection: a case report and review of literature. Burns. 1998;24:487-488.
- Elgart ML. Cutaneous sarcoidosis: definitions and types of lesions. Clin Dermatol. 1986;4:35-45.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
- Yaghoobi R, Omidian M, Bagherani N. Vitiligo: a review of the published work. J Dermatol. 2011;38:419-431.
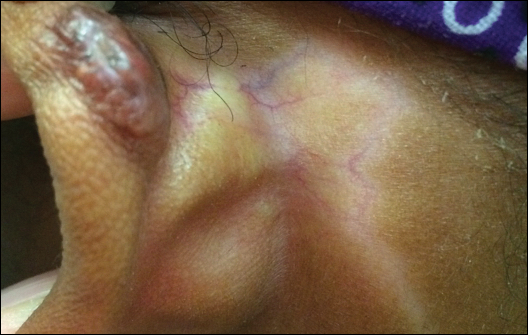
A 20-year-old black woman underwent multiple intralesional corticosteroid injections for treatment of a keloid on the superior aspect of the left helix and subsequently presented with a streak of atrophy and hypopigmentation in the postauricular region of unknown duration due to the lesion location.
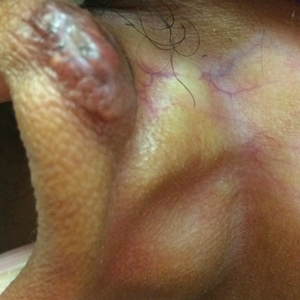
Painless Ulcer on the Areola
The Diagnosis: Primary Syphilitic Chancre of the Nipple
Because laboratory investigation was negative, a primary syphilitic chancre was suspected based on clinical findings, which was confirmed by a positive rapid plasma reagin with a titer of 1:32 and a positive Treponema pallidum particle agglutination assay. Results were negative for human immunodeficiency virus. On further inquiry, the patient acknowledged that the right areola had been traumatized during sexual activity with his regular male partner 1 month prior. In the last year he reported having had 5 different male partners. He was treated with a single dose of 2.4 million IU of intramuscular benzathine penicillin. Screening for other sexually transmitted infections revealed concomitant gonococcal infection of the pharynx and chlamydia proctitis, both of which were subsequently treated. On follow-up 2 weeks after presentation the ulcer had resolved, and he currently is undergoing serial rapid plasma reagin titer monitoring.
Primary syphilitic chancres can occur at any mucocutaneous site of inoculation, most frequently on the genitalia.1 Classically, after an incubation period of 9 to 90 days, a painless indurated ulcer forms2 and heals spontaneously after 3 to 6 weeks if left untreated.3 Chancres at extragenital sites are uncommon, occurring in approximately 2% of patients with primary syphilis.1 Of them, common sites include the lips and mouth (40%-70%),4 with areolar involvement rarely being reported. A PubMed search of articles indexed for MEDLINE using the terms nipple and chancre revealed 9 case reports in the English-language literature, with the first 2 cases being reported by Lee et al5 in 2006. The characteristics of these cases and our patient are summarized in the Table.5-12
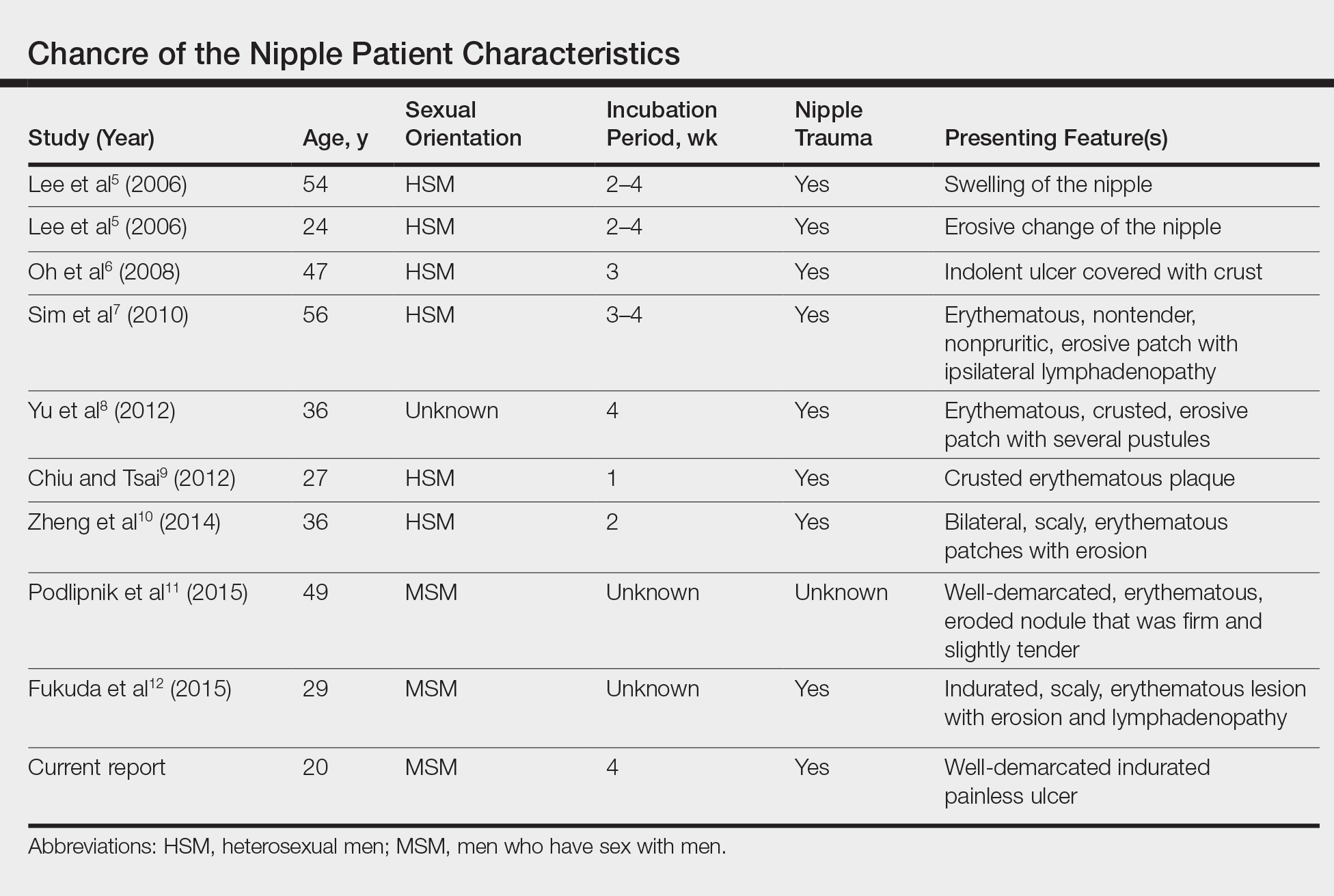
Oral contact or traumatization of the nipple by the patient's sexual partner was reported in all but one of these cases5-10,12; trauma was unknown in one case.11 Our patient reported a similar history of trauma to the nipple. It is known that transmission of syphilis can take place via kissing or oral contact, and it has been asserted that oral syphilitic lesions are highly infectious.13 Syphilis also can be transmitted by an already infected sexual partner sustaining minor trauma at the oral mucosa, allowing Treponema pallidum from the bloodstream to be inoculated onto the nipple. Another explanation for transmission could be the Koebner phenomenon, whereby trauma at the nipple of an already infected patient could lead to the formation of a chancre.6,8
The differential diagnosis includes erosive adenomatosis of the nipple, nipple eczema, Paget disease of the breast, and ulcerated basal cell carcinoma. Erosive adenomatosis of the nipple is a benign tumor of unilateral involvement that presents as an asymptomatic eroded/ulcerated papule. Clinically, it is similar to Paget disease of the breast. Eczema of the nipple usually is associated with pruritus and epidermal changes such as scaling.7,8 Paget disease of the breast arises from the extension of breast ductal carcinoma in situ onto the skin overlying the nipple. It can present as a unilateral nipple plaque with ulceration and bloody discharge. The diagnoses of erosive adenomatosis and Paget disease are confirmed with histologic examination. Basal cell carcinoma is the most common nonmelanoma skin cancer and can present as an ulcerated plaque, often with rolled borders, pearly edges, and overlying telangiectasia. It is known to be locally invasive. A punch biopsy and histopathologic examination would confirm the diagnosis of basal cell carcinoma.14
Extragenital chancres, especially those occurring at unusual sites, are uncommon. Therefore, a high index of suspicion is required to diagnose and initiate appropriate treatment for these patients.
- Mindel A, Tovey SJ, Timmins DJ, et al. Primary and secondary syphilis, 20 years' experience. 2. clinical features. Genitourin Med. 1989;65:1-3.
- Goh B. Syphilis in adults. Sex Transm Infect. 2005;81:448-452.
- Katz KA. Syphilis. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:2471-2492.
- Singh AE, Romanowski B. Syphilis: review with emphasis on clinical, epidemiologic, and some biologic features. Clin Microbiol Rev. 1999;12:187-209.
- Lee JY, Lin MH, Jung YC. Extragenital syphilitic chancre manifesting as a solitary nodule of the nipple. J Eur Acad Dermatol Venereol. 2006;20:886-887.
- Oh Y, Ahn S, Hong SP, et al. A case of extragenital chancre on a nipple from a human bite during sexual intercourse. Int J Dermatol. 2008;47:978-980.
- Sim JH, Lee MG, In SI, et al. Erythematous erosive patch on the left nipple--quiz case. diagnosis: extragenital syphilitic chancres. Arch Dermatol. 2010;146:81-86.
- Yu M, Lee HR, Han TY, et al. A solitary erosive patch on the left nipple. extragenital syphilitic chancres. Int J Dermatol. 2012;51:27-28.
- Chiu HY, Tsai TF. A crusted plaque on the right nipple. JAMA. 2012;308:403-404.
- Zheng S, Liu J, Xu XG, et al. Primary syphilis presenting as bilateral nipple-areola eczematoid lesions. Acta Derm Venereol. 2014;94:617-618.
- Podlipnik S, Giavedoni P, Alsina M, et al. An erythematous nodule on the nipple: an unusual presentation of primary syphilis. J Cutan Pathol. 2015;42:239-243.
- Fukuda H, Takahashi M, Kato K, et al. Multiple primary syphilis on the lip, nipple-areola and penis: an immunohistochemical examination of Treponema pallidum localization using an anti-T. pallidum antibody. J Dermatol. 2015;42:515-517.
- Yu X, Zheng H. Syphilitic chancre of the lips transmitted by kissing. Medicine (Baltimore). 2016;95:E3303.
- Carucci JA, Leffell DJ, Pettersen JS. Basal cell carcinoma. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:1294-1303.
The Diagnosis: Primary Syphilitic Chancre of the Nipple
Because laboratory investigation was negative, a primary syphilitic chancre was suspected based on clinical findings, which was confirmed by a positive rapid plasma reagin with a titer of 1:32 and a positive Treponema pallidum particle agglutination assay. Results were negative for human immunodeficiency virus. On further inquiry, the patient acknowledged that the right areola had been traumatized during sexual activity with his regular male partner 1 month prior. In the last year he reported having had 5 different male partners. He was treated with a single dose of 2.4 million IU of intramuscular benzathine penicillin. Screening for other sexually transmitted infections revealed concomitant gonococcal infection of the pharynx and chlamydia proctitis, both of which were subsequently treated. On follow-up 2 weeks after presentation the ulcer had resolved, and he currently is undergoing serial rapid plasma reagin titer monitoring.
Primary syphilitic chancres can occur at any mucocutaneous site of inoculation, most frequently on the genitalia.1 Classically, after an incubation period of 9 to 90 days, a painless indurated ulcer forms2 and heals spontaneously after 3 to 6 weeks if left untreated.3 Chancres at extragenital sites are uncommon, occurring in approximately 2% of patients with primary syphilis.1 Of them, common sites include the lips and mouth (40%-70%),4 with areolar involvement rarely being reported. A PubMed search of articles indexed for MEDLINE using the terms nipple and chancre revealed 9 case reports in the English-language literature, with the first 2 cases being reported by Lee et al5 in 2006. The characteristics of these cases and our patient are summarized in the Table.5-12
Oral contact or traumatization of the nipple by the patient's sexual partner was reported in all but one of these cases5-10,12; trauma was unknown in one case.11 Our patient reported a similar history of trauma to the nipple. It is known that transmission of syphilis can take place via kissing or oral contact, and it has been asserted that oral syphilitic lesions are highly infectious.13 Syphilis also can be transmitted by an already infected sexual partner sustaining minor trauma at the oral mucosa, allowing Treponema pallidum from the bloodstream to be inoculated onto the nipple. Another explanation for transmission could be the Koebner phenomenon, whereby trauma at the nipple of an already infected patient could lead to the formation of a chancre.6,8
The differential diagnosis includes erosive adenomatosis of the nipple, nipple eczema, Paget disease of the breast, and ulcerated basal cell carcinoma. Erosive adenomatosis of the nipple is a benign tumor of unilateral involvement that presents as an asymptomatic eroded/ulcerated papule. Clinically, it is similar to Paget disease of the breast. Eczema of the nipple usually is associated with pruritus and epidermal changes such as scaling.7,8 Paget disease of the breast arises from the extension of breast ductal carcinoma in situ onto the skin overlying the nipple. It can present as a unilateral nipple plaque with ulceration and bloody discharge. The diagnoses of erosive adenomatosis and Paget disease are confirmed with histologic examination. Basal cell carcinoma is the most common nonmelanoma skin cancer and can present as an ulcerated plaque, often with rolled borders, pearly edges, and overlying telangiectasia. It is known to be locally invasive. A punch biopsy and histopathologic examination would confirm the diagnosis of basal cell carcinoma.14
Extragenital chancres, especially those occurring at unusual sites, are uncommon. Therefore, a high index of suspicion is required to diagnose and initiate appropriate treatment for these patients.
The Diagnosis: Primary Syphilitic Chancre of the Nipple
Because laboratory investigation was negative, a primary syphilitic chancre was suspected based on clinical findings, which was confirmed by a positive rapid plasma reagin with a titer of 1:32 and a positive Treponema pallidum particle agglutination assay. Results were negative for human immunodeficiency virus. On further inquiry, the patient acknowledged that the right areola had been traumatized during sexual activity with his regular male partner 1 month prior. In the last year he reported having had 5 different male partners. He was treated with a single dose of 2.4 million IU of intramuscular benzathine penicillin. Screening for other sexually transmitted infections revealed concomitant gonococcal infection of the pharynx and chlamydia proctitis, both of which were subsequently treated. On follow-up 2 weeks after presentation the ulcer had resolved, and he currently is undergoing serial rapid plasma reagin titer monitoring.
Primary syphilitic chancres can occur at any mucocutaneous site of inoculation, most frequently on the genitalia.1 Classically, after an incubation period of 9 to 90 days, a painless indurated ulcer forms2 and heals spontaneously after 3 to 6 weeks if left untreated.3 Chancres at extragenital sites are uncommon, occurring in approximately 2% of patients with primary syphilis.1 Of them, common sites include the lips and mouth (40%-70%),4 with areolar involvement rarely being reported. A PubMed search of articles indexed for MEDLINE using the terms nipple and chancre revealed 9 case reports in the English-language literature, with the first 2 cases being reported by Lee et al5 in 2006. The characteristics of these cases and our patient are summarized in the Table.5-12
Oral contact or traumatization of the nipple by the patient's sexual partner was reported in all but one of these cases5-10,12; trauma was unknown in one case.11 Our patient reported a similar history of trauma to the nipple. It is known that transmission of syphilis can take place via kissing or oral contact, and it has been asserted that oral syphilitic lesions are highly infectious.13 Syphilis also can be transmitted by an already infected sexual partner sustaining minor trauma at the oral mucosa, allowing Treponema pallidum from the bloodstream to be inoculated onto the nipple. Another explanation for transmission could be the Koebner phenomenon, whereby trauma at the nipple of an already infected patient could lead to the formation of a chancre.6,8
The differential diagnosis includes erosive adenomatosis of the nipple, nipple eczema, Paget disease of the breast, and ulcerated basal cell carcinoma. Erosive adenomatosis of the nipple is a benign tumor of unilateral involvement that presents as an asymptomatic eroded/ulcerated papule. Clinically, it is similar to Paget disease of the breast. Eczema of the nipple usually is associated with pruritus and epidermal changes such as scaling.7,8 Paget disease of the breast arises from the extension of breast ductal carcinoma in situ onto the skin overlying the nipple. It can present as a unilateral nipple plaque with ulceration and bloody discharge. The diagnoses of erosive adenomatosis and Paget disease are confirmed with histologic examination. Basal cell carcinoma is the most common nonmelanoma skin cancer and can present as an ulcerated plaque, often with rolled borders, pearly edges, and overlying telangiectasia. It is known to be locally invasive. A punch biopsy and histopathologic examination would confirm the diagnosis of basal cell carcinoma.14
Extragenital chancres, especially those occurring at unusual sites, are uncommon. Therefore, a high index of suspicion is required to diagnose and initiate appropriate treatment for these patients.
- Mindel A, Tovey SJ, Timmins DJ, et al. Primary and secondary syphilis, 20 years' experience. 2. clinical features. Genitourin Med. 1989;65:1-3.
- Goh B. Syphilis in adults. Sex Transm Infect. 2005;81:448-452.
- Katz KA. Syphilis. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:2471-2492.
- Singh AE, Romanowski B. Syphilis: review with emphasis on clinical, epidemiologic, and some biologic features. Clin Microbiol Rev. 1999;12:187-209.
- Lee JY, Lin MH, Jung YC. Extragenital syphilitic chancre manifesting as a solitary nodule of the nipple. J Eur Acad Dermatol Venereol. 2006;20:886-887.
- Oh Y, Ahn S, Hong SP, et al. A case of extragenital chancre on a nipple from a human bite during sexual intercourse. Int J Dermatol. 2008;47:978-980.
- Sim JH, Lee MG, In SI, et al. Erythematous erosive patch on the left nipple--quiz case. diagnosis: extragenital syphilitic chancres. Arch Dermatol. 2010;146:81-86.
- Yu M, Lee HR, Han TY, et al. A solitary erosive patch on the left nipple. extragenital syphilitic chancres. Int J Dermatol. 2012;51:27-28.
- Chiu HY, Tsai TF. A crusted plaque on the right nipple. JAMA. 2012;308:403-404.
- Zheng S, Liu J, Xu XG, et al. Primary syphilis presenting as bilateral nipple-areola eczematoid lesions. Acta Derm Venereol. 2014;94:617-618.
- Podlipnik S, Giavedoni P, Alsina M, et al. An erythematous nodule on the nipple: an unusual presentation of primary syphilis. J Cutan Pathol. 2015;42:239-243.
- Fukuda H, Takahashi M, Kato K, et al. Multiple primary syphilis on the lip, nipple-areola and penis: an immunohistochemical examination of Treponema pallidum localization using an anti-T. pallidum antibody. J Dermatol. 2015;42:515-517.
- Yu X, Zheng H. Syphilitic chancre of the lips transmitted by kissing. Medicine (Baltimore). 2016;95:E3303.
- Carucci JA, Leffell DJ, Pettersen JS. Basal cell carcinoma. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:1294-1303.
- Mindel A, Tovey SJ, Timmins DJ, et al. Primary and secondary syphilis, 20 years' experience. 2. clinical features. Genitourin Med. 1989;65:1-3.
- Goh B. Syphilis in adults. Sex Transm Infect. 2005;81:448-452.
- Katz KA. Syphilis. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:2471-2492.
- Singh AE, Romanowski B. Syphilis: review with emphasis on clinical, epidemiologic, and some biologic features. Clin Microbiol Rev. 1999;12:187-209.
- Lee JY, Lin MH, Jung YC. Extragenital syphilitic chancre manifesting as a solitary nodule of the nipple. J Eur Acad Dermatol Venereol. 2006;20:886-887.
- Oh Y, Ahn S, Hong SP, et al. A case of extragenital chancre on a nipple from a human bite during sexual intercourse. Int J Dermatol. 2008;47:978-980.
- Sim JH, Lee MG, In SI, et al. Erythematous erosive patch on the left nipple--quiz case. diagnosis: extragenital syphilitic chancres. Arch Dermatol. 2010;146:81-86.
- Yu M, Lee HR, Han TY, et al. A solitary erosive patch on the left nipple. extragenital syphilitic chancres. Int J Dermatol. 2012;51:27-28.
- Chiu HY, Tsai TF. A crusted plaque on the right nipple. JAMA. 2012;308:403-404.
- Zheng S, Liu J, Xu XG, et al. Primary syphilis presenting as bilateral nipple-areola eczematoid lesions. Acta Derm Venereol. 2014;94:617-618.
- Podlipnik S, Giavedoni P, Alsina M, et al. An erythematous nodule on the nipple: an unusual presentation of primary syphilis. J Cutan Pathol. 2015;42:239-243.
- Fukuda H, Takahashi M, Kato K, et al. Multiple primary syphilis on the lip, nipple-areola and penis: an immunohistochemical examination of Treponema pallidum localization using an anti-T. pallidum antibody. J Dermatol. 2015;42:515-517.
- Yu X, Zheng H. Syphilitic chancre of the lips transmitted by kissing. Medicine (Baltimore). 2016;95:E3303.
- Carucci JA, Leffell DJ, Pettersen JS. Basal cell carcinoma. In: Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Medical; 2012:1294-1303.
A previously healthy 20-year-old Chinese man presented to our dermatology outpatient clinic with a solitary painless ulcer on the right areola of 1 week's duration. Examination showed a small, slightly indurated ulcer with well-defined borders. No lesions were noted elsewhere. Swabs for pyogenic culture and herpes simplex virus polymerase chain reaction tests were sent, and he was treated empirically with oral cephalexin and tetracycline ointment 3%. At 1-week follow-up the ulcer had dried up and begun to heal, and results from the laboratory investigations were negative.