User login
Gestational, umbilical cord vitamin D levels don’t predict atopic disease in offspring
according to study results published in the journal Allergy.

Áine Hennessy, PhD, from the School of Food and Nutritional Sciences at the University College Cork (Ireland), and her colleagues performed a prospective cohort study of 1,537 women in the Cork BASELINE Birth Cohort Study who underwent measurement of serum 25-hydroxyvitamin D (25[OH]D) from maternal sera followed by measurement of 25(OH)D in umbilical cord blood (1,050 cases). They then measured the prevalence of eczema, food allergy, allergic rhinitis, and asthma in infants at aged 2 and 5 years.
The researchers found at 2 years old, 5% of infants had persistent eczema, 4% of infants had a food allergy and 8% of infants had aeroallergen sensitization. At age 5 years, 15% of infants had asthma, while 5% had allergic rhinitis. Mothers whose children went on to have atopy did not differ in their 25(OH)D levels at 15 weeks’ gestation (mean 58.4 nmol/L vs. 58.5 nmol/L) or in the levels in umbilical cord blood (mean 35.2 nmol/L and 35.4 nmol/L).
Of the women in the cohort, 74% ranged in age from 25 to 34 years; 49% reported a personal history of allergy and 37% reported a paternal allergy. The mean birth weight of the infants was 3,458 g; infants were breastfed for mean 11.9 weeks, 73% of infants were breastfeeding by the time they left the hospital and 45% of infants were breastfeeding by age 2 months.
Limitations of the study included that parental atopy status was self-reported and that the researchers noted they did not examine genetic variants of immunoglobulin E synthesis or vitamin D receptor polymorphisms.
“To fully characterize relationships between intrauterine vitamin D exposure and allergic disease, analysis of well‐constructed, large‐scale prospective cohorts of maternal‐infant dyads, which take due consideration of an individual’s inherited risk, early‐life exposures and environmental confounders, is still needed,” Dr. Hennessy and her colleagues wrote.
The study was funded by grants from the European Commission, Ireland Health Research Board, National Children’s Research Centre, Food Standards Agency and Science Foundation Ireland. The authors report no relevant conflicts of interest.
SOURCE: Hennessy A et al. Allergy. 2018 Aug 7. doi: 10.1111/all.13590.
according to study results published in the journal Allergy.
Áine Hennessy, PhD, from the School of Food and Nutritional Sciences at the University College Cork (Ireland), and her colleagues performed a prospective cohort study of 1,537 women in the Cork BASELINE Birth Cohort Study who underwent measurement of serum 25-hydroxyvitamin D (25[OH]D) from maternal sera followed by measurement of 25(OH)D in umbilical cord blood (1,050 cases). They then measured the prevalence of eczema, food allergy, allergic rhinitis, and asthma in infants at aged 2 and 5 years.
The researchers found at 2 years old, 5% of infants had persistent eczema, 4% of infants had a food allergy and 8% of infants had aeroallergen sensitization. At age 5 years, 15% of infants had asthma, while 5% had allergic rhinitis. Mothers whose children went on to have atopy did not differ in their 25(OH)D levels at 15 weeks’ gestation (mean 58.4 nmol/L vs. 58.5 nmol/L) or in the levels in umbilical cord blood (mean 35.2 nmol/L and 35.4 nmol/L).
Of the women in the cohort, 74% ranged in age from 25 to 34 years; 49% reported a personal history of allergy and 37% reported a paternal allergy. The mean birth weight of the infants was 3,458 g; infants were breastfed for mean 11.9 weeks, 73% of infants were breastfeeding by the time they left the hospital and 45% of infants were breastfeeding by age 2 months.
Limitations of the study included that parental atopy status was self-reported and that the researchers noted they did not examine genetic variants of immunoglobulin E synthesis or vitamin D receptor polymorphisms.
“To fully characterize relationships between intrauterine vitamin D exposure and allergic disease, analysis of well‐constructed, large‐scale prospective cohorts of maternal‐infant dyads, which take due consideration of an individual’s inherited risk, early‐life exposures and environmental confounders, is still needed,” Dr. Hennessy and her colleagues wrote.
The study was funded by grants from the European Commission, Ireland Health Research Board, National Children’s Research Centre, Food Standards Agency and Science Foundation Ireland. The authors report no relevant conflicts of interest.
SOURCE: Hennessy A et al. Allergy. 2018 Aug 7. doi: 10.1111/all.13590.
according to study results published in the journal Allergy.
Áine Hennessy, PhD, from the School of Food and Nutritional Sciences at the University College Cork (Ireland), and her colleagues performed a prospective cohort study of 1,537 women in the Cork BASELINE Birth Cohort Study who underwent measurement of serum 25-hydroxyvitamin D (25[OH]D) from maternal sera followed by measurement of 25(OH)D in umbilical cord blood (1,050 cases). They then measured the prevalence of eczema, food allergy, allergic rhinitis, and asthma in infants at aged 2 and 5 years.
The researchers found at 2 years old, 5% of infants had persistent eczema, 4% of infants had a food allergy and 8% of infants had aeroallergen sensitization. At age 5 years, 15% of infants had asthma, while 5% had allergic rhinitis. Mothers whose children went on to have atopy did not differ in their 25(OH)D levels at 15 weeks’ gestation (mean 58.4 nmol/L vs. 58.5 nmol/L) or in the levels in umbilical cord blood (mean 35.2 nmol/L and 35.4 nmol/L).
Of the women in the cohort, 74% ranged in age from 25 to 34 years; 49% reported a personal history of allergy and 37% reported a paternal allergy. The mean birth weight of the infants was 3,458 g; infants were breastfed for mean 11.9 weeks, 73% of infants were breastfeeding by the time they left the hospital and 45% of infants were breastfeeding by age 2 months.
Limitations of the study included that parental atopy status was self-reported and that the researchers noted they did not examine genetic variants of immunoglobulin E synthesis or vitamin D receptor polymorphisms.
“To fully characterize relationships between intrauterine vitamin D exposure and allergic disease, analysis of well‐constructed, large‐scale prospective cohorts of maternal‐infant dyads, which take due consideration of an individual’s inherited risk, early‐life exposures and environmental confounders, is still needed,” Dr. Hennessy and her colleagues wrote.
The study was funded by grants from the European Commission, Ireland Health Research Board, National Children’s Research Centre, Food Standards Agency and Science Foundation Ireland. The authors report no relevant conflicts of interest.
SOURCE: Hennessy A et al. Allergy. 2018 Aug 7. doi: 10.1111/all.13590.
FROM ALLERGY
Key clinical point: There was no association between prevalence of atopic disease and vitamin D levels measured in maternal sera during pregnancy or in umbilical cord blood.
Major finding: Maternal vitamin D levels at 15 weeks of gestation (mean 58.4 nmol/L vs. 58.5 nmol/L) and concentrations in umbilical cord blood (mean 35.2 nmol/L and 35.4 nmol/L) were not associated with such atopic diseases as eczema, food allergy, asthma, and allergic rhinitis in children.
Study details: A prospective group of 1,537 women and infant pairs from the Cork BASELINE Birth Cohort Study.
Disclosures: This study was funded by grants from the European Commission, Ireland Health Research Board, National Children’s Research Centre, Food Standards Agency and Science Foundation Ireland. The authors report no relevant conflicts of interest.
Source: Hennessy A et al. Allergy 2018 Aug 7. doi:10.1111/all.13590.

Diffuse facial rash in a former collegiate wrestler
A 22-year-old Caucasian man with a history of atopic dermatitis (AD) was referred to our dermatology clinic for evaluation of a diffuse facial rash that had been present for the previous 7 days. The rash initially presented as erythema on the right malar cheek that rapidly spread to the entire face. Initially diagnosed as impetigo, empiric treatment with sulfamethoxazole/trimethoprim (800 mg/160 mg PO BID for 7 days), dicloxacillin (500 mg PO BID for 6 days), cephalexin (500 mg TID for 5 days), and mupirocin (2% topical cream applied TID for 6 days) failed to improve the patient’s symptoms. He reported mild pain associated with facial movements.
The patient had a history of similar (but more limited) rashes, which he described as “recurrent impetigo,” that began during his career as a high school and collegiate wrestler. These rashes were different from the rashes he described as his history of AD, which consisted of pruritic and erythematous skin in his antecubital and popliteal fossae. He denied any history of herpes simplex virus (HSV) infection.
A physical examination revealed numerous monomorphic, 1- to 3-mm, punched-out erosions and ulcers with overlying yellow-brown crust encompassing the patient’s entire face and portions of his anterior neck. Several clustered vesicles on erythematous bases also were noted (FIGUREs 1A and 1B). We used a Dermablade to unroof some of the vesicles and sent the scrapings to the lab for Tzanck, direct fluorescent antibody assay (DFA), and HSV polymerase chain reaction (PCR) testing.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Eczema herpeticum secondary to herpes gladiatorum
The patient’s laboratory results came back and the Tzanck preparation was positive for multinucleated giant cells, and both the DFA and HSV PCR were positive for HSV infection. This, paired with the widely disseminated rash observed on examination and the patient’s history of AD, was consistent with a diagnosis of eczema herpeticum (EH).
Rather than primary impetigo, the patient’s self-described history of recurrent rashes was felt to represent a history of HSV outbreaks. Given his denial of prior oral or genital HSV infection, as well as the coincident onset of these outbreaks during his career as a competitive wrestler, the most likely primary infection source was direct contact with another HSV-infected wrestler.
Herpes gladiatorum refers to a primary cutaneous HSV infection contracted by an athlete through direct skin-to-skin contact with another athlete.1 It is common in contact sports, such as rugby and wrestling, and particularly common at organized wrestling camps, where mass outbreaks are a frequent occurrence.2 Herpes gladiatorum is so common at these camps that many recommend prophylactic valacyclovir treatment for all participants to mitigate the risk of contracting HSV. In a 2016 review, Anderson et al concluded that prophylactic valacyclovir treatment at a 28-day high school wrestling camp effectively reduced outbreak incidence by 89.5%.2
The lesions of herpes gladiatorum are classically limited in distribution and reflective of the areas of direct contact with infected skin, most commonly the face, neck, and arms. Our patient’s history of more limited outbreaks on his face was consistent with this typical presentation. His current outbreak, however, had become much more widely disseminated, which led to the diagnosis of EH secondary to herpes gladiatorum.
Eczema herpeticum: Pathogenesis and diagnosis
Also known as Kaposi’s varicelliform eruption, EH is a rapid, widespread cutaneous dissemination of HSV infection in areas of dermatitis or skin barrier disruption, most commonly caused by HSV-1 infection.3 It is classically associated with AD, but also can occur in patients with impaired epidermal barrier function due to other conditions, such as burns, pemphigus vulgaris, mycosis fungoides, and Darier disease.4 It occurs in <3% of patients with AD and is more commonly observed in infants and children with AD than adults.5
Continue to: Clinically, the most common manifestations are discrete..
Clinically, the most common manifestations are discrete, monomorphic, 2- to 3-mm, punched-out erosions with hemorrhagic crusts; intact vesicles are less commonly observed.4 Involved skin is typically painful and may be pruritic. Clinical diagnosis should be confirmed by laboratory evaluation, typically Tzanck preparation, DFA, and/or HSV PCR.
Complications and the importance of rapid treatment
The most common complication of EH is bacterial superinfection (impetigo), usually by Staphylococcus aureus or group A streptococci. Signs of bacterial superinfection include weeping lesions, pustules, honey-colored/golden crusting, worsening of existing dermatitis, and failure to respond to antiviral treatment. Topical mupirocin 2% cream is generally effective for controlling limited infection. However, systemic antibiotics (cephalosporins or penicillinase-resistant penicillins) may be necessary to control widespread disease.4 Clinical improvement should be observed within a single course of an appropriate antibiotic.
In contrast to impetigo, less common but more serious complications of EH can be life threatening. Systemic dissemination of disease is of particular importance in vulnerable populations such as pediatric and immunocompromised patients. Meningoencephalitis, secondary bacteremia, and herpes keratitis can all develop secondary to EH and incur significant morbidity and mortality.1
Fever, malaise, lymphadenopathy, or eye pain should prompt immediate consideration of inpatient evaluation and treatment for these potentially deadly or debilitating complications. All patients with EH distributed near the eyes should be referred to ophthalmology to rule out ocular involvement.
Immediately treat with antivirals
Due to the potential complications discussed above, a diagnosis of EH necessitates immediate treatment with oral or intravenous antiviral medication. Acyclovir, valacyclovir, or famciclovir may be used, with typical treatment courses ranging from 10 to 14 days or until all mucocutaneous lesions are healed.4 Although typically reserved for patients with recurrent genital herpes resulting in 6 or more outbreaks annually, chronic suppressive therapy also may be considered for patients with EH who suffer from frequent or severe recurrent outbreaks.
Continue to: Our patient
Our patient. Given his otherwise excellent health and the absence of symptoms of potentially serious complications, our patient was treated as an outpatient with a 10-day course of valacyclovir 1000 mg PO BID. He was additionally prescribed a 7-day course of cephalexin 500 mg PO TID for coverage of bacterial superinfection. He responded well to treatment.
Ten days after his initial presentation to our clinic, his erosions and vesicles had completely cleared, and the associated erythema had significantly improved (FIGURE 2). Given the severity of his presentation and his history of 2 to 3 outbreaks annually, he opted to continue prophylactic valacyclovir (500 mg/d) for long-term suppression.

CORRESPONDENCE
Jonathan Madden, MD, 221 3rd Street West, JBSA-Randolph, TX 78150, [email protected]
1. Shenoy R, Mostow E, Cain G. Eczema herpeticum in a wrestler. Clin J Sport Med. 2015;25:e18-e19.
2. Anderson BJ, McGuire DP, Reed M, et al. Prophylactic valacyclovir to prevent outbreaks of primary herpes gladiatorum at a 28-day wrestling camp: a 10-year review. Clin J Sport Med. 2016;26:272-278.
3. Olson J, Robles DT, Kirby P, et al. Kaposi varicelliform eruption (eczema herpeticum). Dermatol Online J. 2008;14:18.
4. Downing C, Mendoza N, Tyring S. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier; 2018:1400-1424.
5. Leung DY. Why is eczema herpeticum unexpectedly rare? Antiviral Res. 2013;98:153-157.
A 22-year-old Caucasian man with a history of atopic dermatitis (AD) was referred to our dermatology clinic for evaluation of a diffuse facial rash that had been present for the previous 7 days. The rash initially presented as erythema on the right malar cheek that rapidly spread to the entire face. Initially diagnosed as impetigo, empiric treatment with sulfamethoxazole/trimethoprim (800 mg/160 mg PO BID for 7 days), dicloxacillin (500 mg PO BID for 6 days), cephalexin (500 mg TID for 5 days), and mupirocin (2% topical cream applied TID for 6 days) failed to improve the patient’s symptoms. He reported mild pain associated with facial movements.
The patient had a history of similar (but more limited) rashes, which he described as “recurrent impetigo,” that began during his career as a high school and collegiate wrestler. These rashes were different from the rashes he described as his history of AD, which consisted of pruritic and erythematous skin in his antecubital and popliteal fossae. He denied any history of herpes simplex virus (HSV) infection.
A physical examination revealed numerous monomorphic, 1- to 3-mm, punched-out erosions and ulcers with overlying yellow-brown crust encompassing the patient’s entire face and portions of his anterior neck. Several clustered vesicles on erythematous bases also were noted (FIGUREs 1A and 1B). We used a Dermablade to unroof some of the vesicles and sent the scrapings to the lab for Tzanck, direct fluorescent antibody assay (DFA), and HSV polymerase chain reaction (PCR) testing.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Eczema herpeticum secondary to herpes gladiatorum
The patient’s laboratory results came back and the Tzanck preparation was positive for multinucleated giant cells, and both the DFA and HSV PCR were positive for HSV infection. This, paired with the widely disseminated rash observed on examination and the patient’s history of AD, was consistent with a diagnosis of eczema herpeticum (EH).
Rather than primary impetigo, the patient’s self-described history of recurrent rashes was felt to represent a history of HSV outbreaks. Given his denial of prior oral or genital HSV infection, as well as the coincident onset of these outbreaks during his career as a competitive wrestler, the most likely primary infection source was direct contact with another HSV-infected wrestler.
Herpes gladiatorum refers to a primary cutaneous HSV infection contracted by an athlete through direct skin-to-skin contact with another athlete.1 It is common in contact sports, such as rugby and wrestling, and particularly common at organized wrestling camps, where mass outbreaks are a frequent occurrence.2 Herpes gladiatorum is so common at these camps that many recommend prophylactic valacyclovir treatment for all participants to mitigate the risk of contracting HSV. In a 2016 review, Anderson et al concluded that prophylactic valacyclovir treatment at a 28-day high school wrestling camp effectively reduced outbreak incidence by 89.5%.2
The lesions of herpes gladiatorum are classically limited in distribution and reflective of the areas of direct contact with infected skin, most commonly the face, neck, and arms. Our patient’s history of more limited outbreaks on his face was consistent with this typical presentation. His current outbreak, however, had become much more widely disseminated, which led to the diagnosis of EH secondary to herpes gladiatorum.
Eczema herpeticum: Pathogenesis and diagnosis
Also known as Kaposi’s varicelliform eruption, EH is a rapid, widespread cutaneous dissemination of HSV infection in areas of dermatitis or skin barrier disruption, most commonly caused by HSV-1 infection.3 It is classically associated with AD, but also can occur in patients with impaired epidermal barrier function due to other conditions, such as burns, pemphigus vulgaris, mycosis fungoides, and Darier disease.4 It occurs in <3% of patients with AD and is more commonly observed in infants and children with AD than adults.5
Continue to: Clinically, the most common manifestations are discrete..
Clinically, the most common manifestations are discrete, monomorphic, 2- to 3-mm, punched-out erosions with hemorrhagic crusts; intact vesicles are less commonly observed.4 Involved skin is typically painful and may be pruritic. Clinical diagnosis should be confirmed by laboratory evaluation, typically Tzanck preparation, DFA, and/or HSV PCR.
Complications and the importance of rapid treatment
The most common complication of EH is bacterial superinfection (impetigo), usually by Staphylococcus aureus or group A streptococci. Signs of bacterial superinfection include weeping lesions, pustules, honey-colored/golden crusting, worsening of existing dermatitis, and failure to respond to antiviral treatment. Topical mupirocin 2% cream is generally effective for controlling limited infection. However, systemic antibiotics (cephalosporins or penicillinase-resistant penicillins) may be necessary to control widespread disease.4 Clinical improvement should be observed within a single course of an appropriate antibiotic.
In contrast to impetigo, less common but more serious complications of EH can be life threatening. Systemic dissemination of disease is of particular importance in vulnerable populations such as pediatric and immunocompromised patients. Meningoencephalitis, secondary bacteremia, and herpes keratitis can all develop secondary to EH and incur significant morbidity and mortality.1
Fever, malaise, lymphadenopathy, or eye pain should prompt immediate consideration of inpatient evaluation and treatment for these potentially deadly or debilitating complications. All patients with EH distributed near the eyes should be referred to ophthalmology to rule out ocular involvement.
Immediately treat with antivirals
Due to the potential complications discussed above, a diagnosis of EH necessitates immediate treatment with oral or intravenous antiviral medication. Acyclovir, valacyclovir, or famciclovir may be used, with typical treatment courses ranging from 10 to 14 days or until all mucocutaneous lesions are healed.4 Although typically reserved for patients with recurrent genital herpes resulting in 6 or more outbreaks annually, chronic suppressive therapy also may be considered for patients with EH who suffer from frequent or severe recurrent outbreaks.
Continue to: Our patient
Our patient. Given his otherwise excellent health and the absence of symptoms of potentially serious complications, our patient was treated as an outpatient with a 10-day course of valacyclovir 1000 mg PO BID. He was additionally prescribed a 7-day course of cephalexin 500 mg PO TID for coverage of bacterial superinfection. He responded well to treatment.
Ten days after his initial presentation to our clinic, his erosions and vesicles had completely cleared, and the associated erythema had significantly improved (FIGURE 2). Given the severity of his presentation and his history of 2 to 3 outbreaks annually, he opted to continue prophylactic valacyclovir (500 mg/d) for long-term suppression.
CORRESPONDENCE
Jonathan Madden, MD, 221 3rd Street West, JBSA-Randolph, TX 78150, [email protected]
A 22-year-old Caucasian man with a history of atopic dermatitis (AD) was referred to our dermatology clinic for evaluation of a diffuse facial rash that had been present for the previous 7 days. The rash initially presented as erythema on the right malar cheek that rapidly spread to the entire face. Initially diagnosed as impetigo, empiric treatment with sulfamethoxazole/trimethoprim (800 mg/160 mg PO BID for 7 days), dicloxacillin (500 mg PO BID for 6 days), cephalexin (500 mg TID for 5 days), and mupirocin (2% topical cream applied TID for 6 days) failed to improve the patient’s symptoms. He reported mild pain associated with facial movements.
The patient had a history of similar (but more limited) rashes, which he described as “recurrent impetigo,” that began during his career as a high school and collegiate wrestler. These rashes were different from the rashes he described as his history of AD, which consisted of pruritic and erythematous skin in his antecubital and popliteal fossae. He denied any history of herpes simplex virus (HSV) infection.
A physical examination revealed numerous monomorphic, 1- to 3-mm, punched-out erosions and ulcers with overlying yellow-brown crust encompassing the patient’s entire face and portions of his anterior neck. Several clustered vesicles on erythematous bases also were noted (FIGUREs 1A and 1B). We used a Dermablade to unroof some of the vesicles and sent the scrapings to the lab for Tzanck, direct fluorescent antibody assay (DFA), and HSV polymerase chain reaction (PCR) testing.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Eczema herpeticum secondary to herpes gladiatorum
The patient’s laboratory results came back and the Tzanck preparation was positive for multinucleated giant cells, and both the DFA and HSV PCR were positive for HSV infection. This, paired with the widely disseminated rash observed on examination and the patient’s history of AD, was consistent with a diagnosis of eczema herpeticum (EH).
Rather than primary impetigo, the patient’s self-described history of recurrent rashes was felt to represent a history of HSV outbreaks. Given his denial of prior oral or genital HSV infection, as well as the coincident onset of these outbreaks during his career as a competitive wrestler, the most likely primary infection source was direct contact with another HSV-infected wrestler.
Herpes gladiatorum refers to a primary cutaneous HSV infection contracted by an athlete through direct skin-to-skin contact with another athlete.1 It is common in contact sports, such as rugby and wrestling, and particularly common at organized wrestling camps, where mass outbreaks are a frequent occurrence.2 Herpes gladiatorum is so common at these camps that many recommend prophylactic valacyclovir treatment for all participants to mitigate the risk of contracting HSV. In a 2016 review, Anderson et al concluded that prophylactic valacyclovir treatment at a 28-day high school wrestling camp effectively reduced outbreak incidence by 89.5%.2
The lesions of herpes gladiatorum are classically limited in distribution and reflective of the areas of direct contact with infected skin, most commonly the face, neck, and arms. Our patient’s history of more limited outbreaks on his face was consistent with this typical presentation. His current outbreak, however, had become much more widely disseminated, which led to the diagnosis of EH secondary to herpes gladiatorum.
Eczema herpeticum: Pathogenesis and diagnosis
Also known as Kaposi’s varicelliform eruption, EH is a rapid, widespread cutaneous dissemination of HSV infection in areas of dermatitis or skin barrier disruption, most commonly caused by HSV-1 infection.3 It is classically associated with AD, but also can occur in patients with impaired epidermal barrier function due to other conditions, such as burns, pemphigus vulgaris, mycosis fungoides, and Darier disease.4 It occurs in <3% of patients with AD and is more commonly observed in infants and children with AD than adults.5
Continue to: Clinically, the most common manifestations are discrete..
Clinically, the most common manifestations are discrete, monomorphic, 2- to 3-mm, punched-out erosions with hemorrhagic crusts; intact vesicles are less commonly observed.4 Involved skin is typically painful and may be pruritic. Clinical diagnosis should be confirmed by laboratory evaluation, typically Tzanck preparation, DFA, and/or HSV PCR.
Complications and the importance of rapid treatment
The most common complication of EH is bacterial superinfection (impetigo), usually by Staphylococcus aureus or group A streptococci. Signs of bacterial superinfection include weeping lesions, pustules, honey-colored/golden crusting, worsening of existing dermatitis, and failure to respond to antiviral treatment. Topical mupirocin 2% cream is generally effective for controlling limited infection. However, systemic antibiotics (cephalosporins or penicillinase-resistant penicillins) may be necessary to control widespread disease.4 Clinical improvement should be observed within a single course of an appropriate antibiotic.
In contrast to impetigo, less common but more serious complications of EH can be life threatening. Systemic dissemination of disease is of particular importance in vulnerable populations such as pediatric and immunocompromised patients. Meningoencephalitis, secondary bacteremia, and herpes keratitis can all develop secondary to EH and incur significant morbidity and mortality.1
Fever, malaise, lymphadenopathy, or eye pain should prompt immediate consideration of inpatient evaluation and treatment for these potentially deadly or debilitating complications. All patients with EH distributed near the eyes should be referred to ophthalmology to rule out ocular involvement.
Immediately treat with antivirals
Due to the potential complications discussed above, a diagnosis of EH necessitates immediate treatment with oral or intravenous antiviral medication. Acyclovir, valacyclovir, or famciclovir may be used, with typical treatment courses ranging from 10 to 14 days or until all mucocutaneous lesions are healed.4 Although typically reserved for patients with recurrent genital herpes resulting in 6 or more outbreaks annually, chronic suppressive therapy also may be considered for patients with EH who suffer from frequent or severe recurrent outbreaks.
Continue to: Our patient
Our patient. Given his otherwise excellent health and the absence of symptoms of potentially serious complications, our patient was treated as an outpatient with a 10-day course of valacyclovir 1000 mg PO BID. He was additionally prescribed a 7-day course of cephalexin 500 mg PO TID for coverage of bacterial superinfection. He responded well to treatment.
Ten days after his initial presentation to our clinic, his erosions and vesicles had completely cleared, and the associated erythema had significantly improved (FIGURE 2). Given the severity of his presentation and his history of 2 to 3 outbreaks annually, he opted to continue prophylactic valacyclovir (500 mg/d) for long-term suppression.
CORRESPONDENCE
Jonathan Madden, MD, 221 3rd Street West, JBSA-Randolph, TX 78150, [email protected]
1. Shenoy R, Mostow E, Cain G. Eczema herpeticum in a wrestler. Clin J Sport Med. 2015;25:e18-e19.
2. Anderson BJ, McGuire DP, Reed M, et al. Prophylactic valacyclovir to prevent outbreaks of primary herpes gladiatorum at a 28-day wrestling camp: a 10-year review. Clin J Sport Med. 2016;26:272-278.
3. Olson J, Robles DT, Kirby P, et al. Kaposi varicelliform eruption (eczema herpeticum). Dermatol Online J. 2008;14:18.
4. Downing C, Mendoza N, Tyring S. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier; 2018:1400-1424.
5. Leung DY. Why is eczema herpeticum unexpectedly rare? Antiviral Res. 2013;98:153-157.
1. Shenoy R, Mostow E, Cain G. Eczema herpeticum in a wrestler. Clin J Sport Med. 2015;25:e18-e19.
2. Anderson BJ, McGuire DP, Reed M, et al. Prophylactic valacyclovir to prevent outbreaks of primary herpes gladiatorum at a 28-day wrestling camp: a 10-year review. Clin J Sport Med. 2016;26:272-278.
3. Olson J, Robles DT, Kirby P, et al. Kaposi varicelliform eruption (eczema herpeticum). Dermatol Online J. 2008;14:18.
4. Downing C, Mendoza N, Tyring S. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier; 2018:1400-1424.
5. Leung DY. Why is eczema herpeticum unexpectedly rare? Antiviral Res. 2013;98:153-157.

Tanning use disorder should be added to the DSM-5
Raghav Tripathi and his associates maintained in a letter to the editor of the Journal of the European Academy of Dermatology and Venereology.

“Strong evidence suggests that tanning use disorder should be included in the DSM-5,” they wrote, noting that individuals who show signs of tanning use disorder have problems quitting, and also are more likely to smoke cigarettes, drink alcohol excessively, and engage in other high-risk behaviors.
In the letter, Mr. Tripathi, a medical student at Case Western Reserve University, Cleveland, and his associates also noted that frequent tanners have been found to prefer tanning beds that used UV radiation (UVR) over beds that did not, even though they were blinded to which ones did and did not. Reports of pain relief and improved mood following UVR exposure, withdrawal symptoms upon discontinuation of UVR, and the successful use of opioid antagonists to reduce UVR dependence “underscore the importance of viewing tanning as a use disorder.”
The same “brain circuitry and neurotransmitters involved in the reward pathways of other use disorders” are associated with the addictive characteristics of UVR, they wrote.
In one study, people who were compulsive tanners were found to have an increase in “cerebral blood flow in the mesostriatal reward pathway when exposed to UVR.” In another study, opioid antagonism using naltrexone was found to reduce “UVR preference in frequent tanners.”
Understanding the biologic connections is crucial to advocating for formalization of the condition as a recognized disorder in the DSM-5; classification would not only increase awareness of the condition but also standardize approaches to diagnosis and treatment that are key to improving patient care, the authors wrote. Moreover, inclusion of the disorder in the DSM-5 could help to pave the way for inclusion in the ICD-10, which would have broader implications for limiting the overall harmful effects that tanning poses.
The authors had no relevant financial disclosures to report.
SOURCE: Tripathi R et al. J Eur Acad Dermatol Venereol. 2018 Oct 13. doi: 10.1111/jdv.15286.
Raghav Tripathi and his associates maintained in a letter to the editor of the Journal of the European Academy of Dermatology and Venereology.
“Strong evidence suggests that tanning use disorder should be included in the DSM-5,” they wrote, noting that individuals who show signs of tanning use disorder have problems quitting, and also are more likely to smoke cigarettes, drink alcohol excessively, and engage in other high-risk behaviors.
In the letter, Mr. Tripathi, a medical student at Case Western Reserve University, Cleveland, and his associates also noted that frequent tanners have been found to prefer tanning beds that used UV radiation (UVR) over beds that did not, even though they were blinded to which ones did and did not. Reports of pain relief and improved mood following UVR exposure, withdrawal symptoms upon discontinuation of UVR, and the successful use of opioid antagonists to reduce UVR dependence “underscore the importance of viewing tanning as a use disorder.”
The same “brain circuitry and neurotransmitters involved in the reward pathways of other use disorders” are associated with the addictive characteristics of UVR, they wrote.
In one study, people who were compulsive tanners were found to have an increase in “cerebral blood flow in the mesostriatal reward pathway when exposed to UVR.” In another study, opioid antagonism using naltrexone was found to reduce “UVR preference in frequent tanners.”
Understanding the biologic connections is crucial to advocating for formalization of the condition as a recognized disorder in the DSM-5; classification would not only increase awareness of the condition but also standardize approaches to diagnosis and treatment that are key to improving patient care, the authors wrote. Moreover, inclusion of the disorder in the DSM-5 could help to pave the way for inclusion in the ICD-10, which would have broader implications for limiting the overall harmful effects that tanning poses.
The authors had no relevant financial disclosures to report.
SOURCE: Tripathi R et al. J Eur Acad Dermatol Venereol. 2018 Oct 13. doi: 10.1111/jdv.15286.
Raghav Tripathi and his associates maintained in a letter to the editor of the Journal of the European Academy of Dermatology and Venereology.
“Strong evidence suggests that tanning use disorder should be included in the DSM-5,” they wrote, noting that individuals who show signs of tanning use disorder have problems quitting, and also are more likely to smoke cigarettes, drink alcohol excessively, and engage in other high-risk behaviors.
In the letter, Mr. Tripathi, a medical student at Case Western Reserve University, Cleveland, and his associates also noted that frequent tanners have been found to prefer tanning beds that used UV radiation (UVR) over beds that did not, even though they were blinded to which ones did and did not. Reports of pain relief and improved mood following UVR exposure, withdrawal symptoms upon discontinuation of UVR, and the successful use of opioid antagonists to reduce UVR dependence “underscore the importance of viewing tanning as a use disorder.”
The same “brain circuitry and neurotransmitters involved in the reward pathways of other use disorders” are associated with the addictive characteristics of UVR, they wrote.
In one study, people who were compulsive tanners were found to have an increase in “cerebral blood flow in the mesostriatal reward pathway when exposed to UVR.” In another study, opioid antagonism using naltrexone was found to reduce “UVR preference in frequent tanners.”
Understanding the biologic connections is crucial to advocating for formalization of the condition as a recognized disorder in the DSM-5; classification would not only increase awareness of the condition but also standardize approaches to diagnosis and treatment that are key to improving patient care, the authors wrote. Moreover, inclusion of the disorder in the DSM-5 could help to pave the way for inclusion in the ICD-10, which would have broader implications for limiting the overall harmful effects that tanning poses.
The authors had no relevant financial disclosures to report.
SOURCE: Tripathi R et al. J Eur Acad Dermatol Venereol. 2018 Oct 13. doi: 10.1111/jdv.15286.
FROM THE JOURNAL OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
RELIEF: In Behçet’s, apremilast improves oral ulcers for up to 28 weeks
CHICAGO – Apremilast was effective and well tolerated for up to 28 weeks for the treatment of oral ulcers in patients with active Behçet’s disease, based on findings from the randomized, placebo-controlled, phase 3 RELIEF trial.
At baseline, mean oral ulcer counts were 4.2 in 104 patients randomized to receive the oral phosphodiesterase-4 inhibitor and 3.9 in 103 patients in the placebo group. Mean visual analog scale (VAS) pain scores were 61.2 and 60.8 in the two groups, respectively.
The primary study endpoint of area under the curve for total number of oral ulcers over a 12-week period (AUCWk0-12) – a measure that reflects the number of oral ulcers that occur over time and also accounts for the recurring-remitting course of oral ulcers – was achieved. AUCWk0-12 was significantly lower in the apremilast group than in the placebo group (129.54 vs. 222.14, respectively; P less than .0001), Gulen Hatemi, MD, reported at the annual meeting of the American College of Rheumatology.
From baseline to week 12, apremilast treatment also resulted in a significantly lower number of oral ulcers (mean of 1.1 vs. 2.0 for placebo at 12 weeks) and significantly reduced pain from oral ulcers at every visit from week 1 through week 12 of the study, compared with placebo (mean VAS score change from baseline, –40.7 vs. –15.9), said Dr. Hatemi, a professor of medicine at Istanbul University.
“The [12-week] complete response rate ... was 53% in the apremilast group and 22.3% in the placebo group. The [12-week] partial response rate ...was 76% in the apremilast group and 48% in the placebo group,” she said, adding that the efficacy of apremilast was sustained with continued treatment through 28 weeks.
Study participants were adults (mean age, 40 years) with active Behçet’s disease and three or more oral ulcers at randomization or two or more at screening and at randomization. All had been previously treated with at least one nonbiologic medication for oral ulcers and were allowed to have received previous biologic therapies for other disease manifestations. Those with active major organ involvement were excluded.
Treatment included a 30-mg dose of apremilast twice daily for 12 weeks or placebo. After 12 weeks, all patients received apremilast through at least 28 weeks of the 64-week study.
At the 28-week analysis, patients who were initially randomized to placebo and who switched to apremilast after week 12 had benefits comparable with those seen in those randomized to apremilast at the start of the study. A complete response was seen in 59% and 62% of patients in the groups, respectively, and a partial response was seen in 90% and 85%, respectively. Additionally, the mean change in the VAS score for oral ulcer pain in the groups at that time was –40.6 and –41.9, Dr. Hatemi said.
Apremilast was well tolerated in this study; the incidence of adverse events was comparable in the treatment and placebo groups during the 12-week placebo-controlled phase of the study – 78.8% and 71.8%, respectively. The most common events were diarrhea, nausea, headache, and upper respiratory tract infection, she said.
“These were generally mild to moderate, and only two patients had to discontinue the study due to gastrointestinal adverse events,” she said, noting that no new safety signals were observed.
Behçet’s disease is a chronic, relapsing, multisystem inflammatory disorder characterized by recurrent oral ulcers that can be disabling and have a substantial effect on quality of life. These findings, which include efficacy data up to 28 weeks and safety data for at least 100 patients exposed to apremilast for at least 1 year, demonstrate the efficacy of apremilast for the treatment oral ulcers in patients with Behçet’s disease, she said, noting that “the safety findings were consistent with the known safety profile of apremilast.”
The RELIEF study was supported by Celgene. Dr. Hatemi reported receiving grant/research support from Celgene and serving as a speaker for AbbVie, Mustafa Nevzet Pharmaceuticals, and UCB.
SOURCE: Hatemi G et al. Arthritis Rheumatol. 2018;70(Suppl 10), Abstract 2789.
CHICAGO – Apremilast was effective and well tolerated for up to 28 weeks for the treatment of oral ulcers in patients with active Behçet’s disease, based on findings from the randomized, placebo-controlled, phase 3 RELIEF trial.
At baseline, mean oral ulcer counts were 4.2 in 104 patients randomized to receive the oral phosphodiesterase-4 inhibitor and 3.9 in 103 patients in the placebo group. Mean visual analog scale (VAS) pain scores were 61.2 and 60.8 in the two groups, respectively.
The primary study endpoint of area under the curve for total number of oral ulcers over a 12-week period (AUCWk0-12) – a measure that reflects the number of oral ulcers that occur over time and also accounts for the recurring-remitting course of oral ulcers – was achieved. AUCWk0-12 was significantly lower in the apremilast group than in the placebo group (129.54 vs. 222.14, respectively; P less than .0001), Gulen Hatemi, MD, reported at the annual meeting of the American College of Rheumatology.
From baseline to week 12, apremilast treatment also resulted in a significantly lower number of oral ulcers (mean of 1.1 vs. 2.0 for placebo at 12 weeks) and significantly reduced pain from oral ulcers at every visit from week 1 through week 12 of the study, compared with placebo (mean VAS score change from baseline, –40.7 vs. –15.9), said Dr. Hatemi, a professor of medicine at Istanbul University.
“The [12-week] complete response rate ... was 53% in the apremilast group and 22.3% in the placebo group. The [12-week] partial response rate ...was 76% in the apremilast group and 48% in the placebo group,” she said, adding that the efficacy of apremilast was sustained with continued treatment through 28 weeks.
Study participants were adults (mean age, 40 years) with active Behçet’s disease and three or more oral ulcers at randomization or two or more at screening and at randomization. All had been previously treated with at least one nonbiologic medication for oral ulcers and were allowed to have received previous biologic therapies for other disease manifestations. Those with active major organ involvement were excluded.
Treatment included a 30-mg dose of apremilast twice daily for 12 weeks or placebo. After 12 weeks, all patients received apremilast through at least 28 weeks of the 64-week study.
At the 28-week analysis, patients who were initially randomized to placebo and who switched to apremilast after week 12 had benefits comparable with those seen in those randomized to apremilast at the start of the study. A complete response was seen in 59% and 62% of patients in the groups, respectively, and a partial response was seen in 90% and 85%, respectively. Additionally, the mean change in the VAS score for oral ulcer pain in the groups at that time was –40.6 and –41.9, Dr. Hatemi said.
Apremilast was well tolerated in this study; the incidence of adverse events was comparable in the treatment and placebo groups during the 12-week placebo-controlled phase of the study – 78.8% and 71.8%, respectively. The most common events were diarrhea, nausea, headache, and upper respiratory tract infection, she said.
“These were generally mild to moderate, and only two patients had to discontinue the study due to gastrointestinal adverse events,” she said, noting that no new safety signals were observed.
Behçet’s disease is a chronic, relapsing, multisystem inflammatory disorder characterized by recurrent oral ulcers that can be disabling and have a substantial effect on quality of life. These findings, which include efficacy data up to 28 weeks and safety data for at least 100 patients exposed to apremilast for at least 1 year, demonstrate the efficacy of apremilast for the treatment oral ulcers in patients with Behçet’s disease, she said, noting that “the safety findings were consistent with the known safety profile of apremilast.”
The RELIEF study was supported by Celgene. Dr. Hatemi reported receiving grant/research support from Celgene and serving as a speaker for AbbVie, Mustafa Nevzet Pharmaceuticals, and UCB.
SOURCE: Hatemi G et al. Arthritis Rheumatol. 2018;70(Suppl 10), Abstract 2789.
CHICAGO – Apremilast was effective and well tolerated for up to 28 weeks for the treatment of oral ulcers in patients with active Behçet’s disease, based on findings from the randomized, placebo-controlled, phase 3 RELIEF trial.
At baseline, mean oral ulcer counts were 4.2 in 104 patients randomized to receive the oral phosphodiesterase-4 inhibitor and 3.9 in 103 patients in the placebo group. Mean visual analog scale (VAS) pain scores were 61.2 and 60.8 in the two groups, respectively.
The primary study endpoint of area under the curve for total number of oral ulcers over a 12-week period (AUCWk0-12) – a measure that reflects the number of oral ulcers that occur over time and also accounts for the recurring-remitting course of oral ulcers – was achieved. AUCWk0-12 was significantly lower in the apremilast group than in the placebo group (129.54 vs. 222.14, respectively; P less than .0001), Gulen Hatemi, MD, reported at the annual meeting of the American College of Rheumatology.
From baseline to week 12, apremilast treatment also resulted in a significantly lower number of oral ulcers (mean of 1.1 vs. 2.0 for placebo at 12 weeks) and significantly reduced pain from oral ulcers at every visit from week 1 through week 12 of the study, compared with placebo (mean VAS score change from baseline, –40.7 vs. –15.9), said Dr. Hatemi, a professor of medicine at Istanbul University.
“The [12-week] complete response rate ... was 53% in the apremilast group and 22.3% in the placebo group. The [12-week] partial response rate ...was 76% in the apremilast group and 48% in the placebo group,” she said, adding that the efficacy of apremilast was sustained with continued treatment through 28 weeks.
Study participants were adults (mean age, 40 years) with active Behçet’s disease and three or more oral ulcers at randomization or two or more at screening and at randomization. All had been previously treated with at least one nonbiologic medication for oral ulcers and were allowed to have received previous biologic therapies for other disease manifestations. Those with active major organ involvement were excluded.
Treatment included a 30-mg dose of apremilast twice daily for 12 weeks or placebo. After 12 weeks, all patients received apremilast through at least 28 weeks of the 64-week study.
At the 28-week analysis, patients who were initially randomized to placebo and who switched to apremilast after week 12 had benefits comparable with those seen in those randomized to apremilast at the start of the study. A complete response was seen in 59% and 62% of patients in the groups, respectively, and a partial response was seen in 90% and 85%, respectively. Additionally, the mean change in the VAS score for oral ulcer pain in the groups at that time was –40.6 and –41.9, Dr. Hatemi said.
Apremilast was well tolerated in this study; the incidence of adverse events was comparable in the treatment and placebo groups during the 12-week placebo-controlled phase of the study – 78.8% and 71.8%, respectively. The most common events were diarrhea, nausea, headache, and upper respiratory tract infection, she said.
“These were generally mild to moderate, and only two patients had to discontinue the study due to gastrointestinal adverse events,” she said, noting that no new safety signals were observed.
Behçet’s disease is a chronic, relapsing, multisystem inflammatory disorder characterized by recurrent oral ulcers that can be disabling and have a substantial effect on quality of life. These findings, which include efficacy data up to 28 weeks and safety data for at least 100 patients exposed to apremilast for at least 1 year, demonstrate the efficacy of apremilast for the treatment oral ulcers in patients with Behçet’s disease, she said, noting that “the safety findings were consistent with the known safety profile of apremilast.”
The RELIEF study was supported by Celgene. Dr. Hatemi reported receiving grant/research support from Celgene and serving as a speaker for AbbVie, Mustafa Nevzet Pharmaceuticals, and UCB.
SOURCE: Hatemi G et al. Arthritis Rheumatol. 2018;70(Suppl 10), Abstract 2789.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point: Apremilast is safe and effective for treating oral ulcers in patients with Behçet’s disease.
Major finding: The AUCWk0-12 was significantly lower with apremilast (129.54) versus placebo (222.14).
Study details: A randomized, placebo-controlled, phase 3 study of 207 patients.
Disclosures: The RELIEF study was supported by Celgene. Dr. Hatemi reported receiving grant/research support from Celgene and serving as a speaker for AbbVie, Mustafa Nevzet Pharmaceuticals, and UCB.
Source: Hatemi G et al. Arthritis Rheumatol. 2018;70(Suppl 10), Abstract 2789
Weight loss cuts risk of psoriatic arthritis
CHICAGO – Overweight and obese psoriasis patients have it within their power to reduce their risk of developing psoriatic arthritis through weight loss, according to a large British longitudinal study.

Of the three modifiable lifestyle factors evaluated in the study as potential risk factors for the development of psoriatic arthritis in psoriasis patients – body mass index, smoking, and alcohol intake – reduction in BMI over time was clearly the winning strategy, Neil McHugh, MD, said at the annual meeting of the American College of Rheumatology.
The message from this study of 90,189 incident cases of psoriasis identified in the U.K. Clinical Practice Research Datalink was unequivocal: “If you’re overweight and have psoriasis and you lose weight, you reduce your chance of developing a nasty form of arthritis,” said Dr. McHugh, professor of pharmacoepidemiology and a rheumatologist at the University of Bath, England.
“As psoriatic arthritis affects around 20% of people with psoriasis, weight reduction amongst those who are obese may have the potential to greatly reduce their risk of psoriatic arthritis in addition to providing additional health benefits,” he added.
Among the more than 90,000 patients diagnosed with psoriasis, 1,409 subsequently developed psoriatic arthritis, with an overall incidence rate of 2.72 cases per 1,000 person-years. Baseline BMI was strongly associated in stepwise fashion with subsequent psoriatic arthritis. Psoriasis patients with a baseline BMI of 25-29.9 kg/m2 were at an adjusted 1.76-fold increased risk of later developing psoriatic arthritis, compared with psoriasis patients having a BMI of less than 25. For those with a BMI of 30-34.9 kg/m2, the risk of subsequent psoriatic arthritis was increased 2.04-fold. And for those with a baseline BMI of 35 kg/m2 or more, the risk was increased 2.42-fold in analyses adjusted for age, sex, psoriasis duration and severity, history of trauma, and diabetes.
In contrast, the risk of developing psoriatic arthritis wasn’t significantly different between psoriasis patients who were nonsmokers, ex-smokers, or current smokers. And while there was a significantly increased risk of developing psoriatic arthritis in psoriasis patients who were current drinkers, compared with nondrinkers, the risk in ex-drinkers and heavy drinkers was similar to that in nondrinkers, a counterintuitive finding Dr. McHugh suspects was a distortion due to small numbers.
While the observed relationship between baseline BMI and subsequent risk of psoriatic arthritis was informative, it only tells part of the story, since body weight so often changes over time. Dr. McHugh and his coinvestigators had data on change in BMI over the course of 10 years of follow-up in 15,627 psoriasis patients free of psoriatic arthritis at the time their psoriasis was diagnosed. The researchers developed a BMI risk calculator that expressed the effect of change in BMI over time on the cumulative risk of developing psoriatic arthritis.
“We were able to show that if, for instance, you started with a BMI of 25 at baseline and ended up with a BMI of 30, your risk of psoriatic arthritis goes up by 13%, whereas if you start at 30 and come down to 25, your risk decreases by 13%. And the more weight you lose, the greater you reduce your risk of developing psoriatic arthritis,” the rheumatologist explained in an interview.
Indeed, with more extreme changes in BMI over the course of a decade following diagnosis of psoriasis – for example, dropping from a baseline BMI of 36 kg/m2 to 23 kg/m2 – the risk of developing psoriatic arthritis fell by close to 30%.
Dr. McHugh reported having no financial conflicts regarding this study, funded by the U.K. National Institute for Health Research.
SOURCE: Green A et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2134.
CHICAGO – Overweight and obese psoriasis patients have it within their power to reduce their risk of developing psoriatic arthritis through weight loss, according to a large British longitudinal study.
Of the three modifiable lifestyle factors evaluated in the study as potential risk factors for the development of psoriatic arthritis in psoriasis patients – body mass index, smoking, and alcohol intake – reduction in BMI over time was clearly the winning strategy, Neil McHugh, MD, said at the annual meeting of the American College of Rheumatology.
The message from this study of 90,189 incident cases of psoriasis identified in the U.K. Clinical Practice Research Datalink was unequivocal: “If you’re overweight and have psoriasis and you lose weight, you reduce your chance of developing a nasty form of arthritis,” said Dr. McHugh, professor of pharmacoepidemiology and a rheumatologist at the University of Bath, England.
“As psoriatic arthritis affects around 20% of people with psoriasis, weight reduction amongst those who are obese may have the potential to greatly reduce their risk of psoriatic arthritis in addition to providing additional health benefits,” he added.
Among the more than 90,000 patients diagnosed with psoriasis, 1,409 subsequently developed psoriatic arthritis, with an overall incidence rate of 2.72 cases per 1,000 person-years. Baseline BMI was strongly associated in stepwise fashion with subsequent psoriatic arthritis. Psoriasis patients with a baseline BMI of 25-29.9 kg/m2 were at an adjusted 1.76-fold increased risk of later developing psoriatic arthritis, compared with psoriasis patients having a BMI of less than 25. For those with a BMI of 30-34.9 kg/m2, the risk of subsequent psoriatic arthritis was increased 2.04-fold. And for those with a baseline BMI of 35 kg/m2 or more, the risk was increased 2.42-fold in analyses adjusted for age, sex, psoriasis duration and severity, history of trauma, and diabetes.
In contrast, the risk of developing psoriatic arthritis wasn’t significantly different between psoriasis patients who were nonsmokers, ex-smokers, or current smokers. And while there was a significantly increased risk of developing psoriatic arthritis in psoriasis patients who were current drinkers, compared with nondrinkers, the risk in ex-drinkers and heavy drinkers was similar to that in nondrinkers, a counterintuitive finding Dr. McHugh suspects was a distortion due to small numbers.
While the observed relationship between baseline BMI and subsequent risk of psoriatic arthritis was informative, it only tells part of the story, since body weight so often changes over time. Dr. McHugh and his coinvestigators had data on change in BMI over the course of 10 years of follow-up in 15,627 psoriasis patients free of psoriatic arthritis at the time their psoriasis was diagnosed. The researchers developed a BMI risk calculator that expressed the effect of change in BMI over time on the cumulative risk of developing psoriatic arthritis.
“We were able to show that if, for instance, you started with a BMI of 25 at baseline and ended up with a BMI of 30, your risk of psoriatic arthritis goes up by 13%, whereas if you start at 30 and come down to 25, your risk decreases by 13%. And the more weight you lose, the greater you reduce your risk of developing psoriatic arthritis,” the rheumatologist explained in an interview.
Indeed, with more extreme changes in BMI over the course of a decade following diagnosis of psoriasis – for example, dropping from a baseline BMI of 36 kg/m2 to 23 kg/m2 – the risk of developing psoriatic arthritis fell by close to 30%.
Dr. McHugh reported having no financial conflicts regarding this study, funded by the U.K. National Institute for Health Research.
SOURCE: Green A et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2134.
CHICAGO – Overweight and obese psoriasis patients have it within their power to reduce their risk of developing psoriatic arthritis through weight loss, according to a large British longitudinal study.
Of the three modifiable lifestyle factors evaluated in the study as potential risk factors for the development of psoriatic arthritis in psoriasis patients – body mass index, smoking, and alcohol intake – reduction in BMI over time was clearly the winning strategy, Neil McHugh, MD, said at the annual meeting of the American College of Rheumatology.
The message from this study of 90,189 incident cases of psoriasis identified in the U.K. Clinical Practice Research Datalink was unequivocal: “If you’re overweight and have psoriasis and you lose weight, you reduce your chance of developing a nasty form of arthritis,” said Dr. McHugh, professor of pharmacoepidemiology and a rheumatologist at the University of Bath, England.
“As psoriatic arthritis affects around 20% of people with psoriasis, weight reduction amongst those who are obese may have the potential to greatly reduce their risk of psoriatic arthritis in addition to providing additional health benefits,” he added.
Among the more than 90,000 patients diagnosed with psoriasis, 1,409 subsequently developed psoriatic arthritis, with an overall incidence rate of 2.72 cases per 1,000 person-years. Baseline BMI was strongly associated in stepwise fashion with subsequent psoriatic arthritis. Psoriasis patients with a baseline BMI of 25-29.9 kg/m2 were at an adjusted 1.76-fold increased risk of later developing psoriatic arthritis, compared with psoriasis patients having a BMI of less than 25. For those with a BMI of 30-34.9 kg/m2, the risk of subsequent psoriatic arthritis was increased 2.04-fold. And for those with a baseline BMI of 35 kg/m2 or more, the risk was increased 2.42-fold in analyses adjusted for age, sex, psoriasis duration and severity, history of trauma, and diabetes.
In contrast, the risk of developing psoriatic arthritis wasn’t significantly different between psoriasis patients who were nonsmokers, ex-smokers, or current smokers. And while there was a significantly increased risk of developing psoriatic arthritis in psoriasis patients who were current drinkers, compared with nondrinkers, the risk in ex-drinkers and heavy drinkers was similar to that in nondrinkers, a counterintuitive finding Dr. McHugh suspects was a distortion due to small numbers.
While the observed relationship between baseline BMI and subsequent risk of psoriatic arthritis was informative, it only tells part of the story, since body weight so often changes over time. Dr. McHugh and his coinvestigators had data on change in BMI over the course of 10 years of follow-up in 15,627 psoriasis patients free of psoriatic arthritis at the time their psoriasis was diagnosed. The researchers developed a BMI risk calculator that expressed the effect of change in BMI over time on the cumulative risk of developing psoriatic arthritis.
“We were able to show that if, for instance, you started with a BMI of 25 at baseline and ended up with a BMI of 30, your risk of psoriatic arthritis goes up by 13%, whereas if you start at 30 and come down to 25, your risk decreases by 13%. And the more weight you lose, the greater you reduce your risk of developing psoriatic arthritis,” the rheumatologist explained in an interview.
Indeed, with more extreme changes in BMI over the course of a decade following diagnosis of psoriasis – for example, dropping from a baseline BMI of 36 kg/m2 to 23 kg/m2 – the risk of developing psoriatic arthritis fell by close to 30%.
Dr. McHugh reported having no financial conflicts regarding this study, funded by the U.K. National Institute for Health Research.
SOURCE: Green A et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2134.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point:
Major finding: A psoriasis patient’s risk of developing psoriatic arthritis increases stepwise with greater body mass index, and the converse is true as well.
Study details: This study included more than 90,000 patients with a diagnosis of psoriasis in the U.K. Clinical Practice Research Datalink.
Disclosures: The presenter reported having no financial conflicts regarding this study, funded by the U.K. National Institute for Health Research.
Source: Green A et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2134.
Anthralin shows promise as second-line agent for pediatric alopecia areata
Given its limited systemic toxicity, , according to Sean Z. Wu, MD, of the department of dermatology, University of Cincinnati, and his associates.
In a retrospective study of 37 pediatric patients with AA, published in Pediatric Dermatology, Dr. Wu and his colleagues found that almost two-thirds experienced at least 50% regrowth of hair with topical anthralin treatment, but they described severe dermatitis and relapses as “potential drawbacks” of treatment.
The 37 patients were in the Cleveland Clinic AA areata database and began treatment with anthralin between 2004 and 2015, at aged 2-17 years (mean age 9). Over half (22) were females and most (31) were white. About 65% had patchy AA; the remainder had either alopecia totalis or alopecia universalis. Prior treatments included topical corticosteroids, minoxidil, and intralesional corticosteroids; four patients had not been treated previously. Patients were followed up from 51 days to more than 10 years, with a mean duration of 2.5 years. Treatment regimens, titrated up to achieve a mild to moderate dermatitis, included application of 0.5% cream for 5 minutes twice a week up to 1.0% cream for 30 minutes a day.
With topical anthralin, 12 (32%) of patients had complete scalp regrowth, 25 (68%) experienced at least 50% regrowth, and 5 (14%) had no response; in five patients, no follow-up information was available. Among those with at least 50% regrowth, the initial response was first noted at a mean of 3.4 months, and the mean time to maximal response was 15 months. This timeline suggests that treatment with anthralin should be continued beyond 1 year to ensure maximum beneficial results with hair regrowth, the authors wrote.
Factors associated with a positive response to treatment included less than 50% of scalp involvement. The two patients who used anthralin as monotherapy did not achieve a 50% scalp response, but the four treatment-naive patients were among those with at least 50% scalp regrowth, versus three of the five patients (60%) who had been treated previously with systemic steroids.
Two potential clinical limitations were noted during the study. Four patients had to stop treatment because of dermatitis, which suggests that patients and parents should be counseled about the potential for severe skin irritation with this treatment, the authors said. And among those who achieved at least 50% scalp regrowth, 16 of the 25 (64%) relapsed. The authors speculated that the effects of the drug could be temporary “or that the disease process may be able to overcome the anthralin effect over time.”
Dr. Wu and his coauthors cited the retrospective design and the small population size as major limitations of the study. Because some patients continued other treatments, it is “difficult to attribute scalp regrowth entirely to anthralin,” and variations in formulation and in treatment regimens are also factors to be considered, they cautioned. That AA has been found to spontaneously resolve, depending upon the severity of the disease, presents additional challenges for clinicians attempting to determine the extent to which anthralin offers therapeutic benefit, they pointed out.
In spite of the drug’s limitations, the authors concluded that the treatment was a safe and useful option as an “adjunct for those who fail first-line therapy with topical or intralesional corticosteroids.” They added that more work is needed to tailor treatment formulation, frequency, and duration to the specific needs of pediatric patients.
The authors had no relevant financial disclosures to report.
SOURCE: Wu SZ et al. Pediatr Dermatol. 2018;35:817-20.
Given its limited systemic toxicity, , according to Sean Z. Wu, MD, of the department of dermatology, University of Cincinnati, and his associates.
In a retrospective study of 37 pediatric patients with AA, published in Pediatric Dermatology, Dr. Wu and his colleagues found that almost two-thirds experienced at least 50% regrowth of hair with topical anthralin treatment, but they described severe dermatitis and relapses as “potential drawbacks” of treatment.
The 37 patients were in the Cleveland Clinic AA areata database and began treatment with anthralin between 2004 and 2015, at aged 2-17 years (mean age 9). Over half (22) were females and most (31) were white. About 65% had patchy AA; the remainder had either alopecia totalis or alopecia universalis. Prior treatments included topical corticosteroids, minoxidil, and intralesional corticosteroids; four patients had not been treated previously. Patients were followed up from 51 days to more than 10 years, with a mean duration of 2.5 years. Treatment regimens, titrated up to achieve a mild to moderate dermatitis, included application of 0.5% cream for 5 minutes twice a week up to 1.0% cream for 30 minutes a day.
With topical anthralin, 12 (32%) of patients had complete scalp regrowth, 25 (68%) experienced at least 50% regrowth, and 5 (14%) had no response; in five patients, no follow-up information was available. Among those with at least 50% regrowth, the initial response was first noted at a mean of 3.4 months, and the mean time to maximal response was 15 months. This timeline suggests that treatment with anthralin should be continued beyond 1 year to ensure maximum beneficial results with hair regrowth, the authors wrote.
Factors associated with a positive response to treatment included less than 50% of scalp involvement. The two patients who used anthralin as monotherapy did not achieve a 50% scalp response, but the four treatment-naive patients were among those with at least 50% scalp regrowth, versus three of the five patients (60%) who had been treated previously with systemic steroids.
Two potential clinical limitations were noted during the study. Four patients had to stop treatment because of dermatitis, which suggests that patients and parents should be counseled about the potential for severe skin irritation with this treatment, the authors said. And among those who achieved at least 50% scalp regrowth, 16 of the 25 (64%) relapsed. The authors speculated that the effects of the drug could be temporary “or that the disease process may be able to overcome the anthralin effect over time.”
Dr. Wu and his coauthors cited the retrospective design and the small population size as major limitations of the study. Because some patients continued other treatments, it is “difficult to attribute scalp regrowth entirely to anthralin,” and variations in formulation and in treatment regimens are also factors to be considered, they cautioned. That AA has been found to spontaneously resolve, depending upon the severity of the disease, presents additional challenges for clinicians attempting to determine the extent to which anthralin offers therapeutic benefit, they pointed out.
In spite of the drug’s limitations, the authors concluded that the treatment was a safe and useful option as an “adjunct for those who fail first-line therapy with topical or intralesional corticosteroids.” They added that more work is needed to tailor treatment formulation, frequency, and duration to the specific needs of pediatric patients.
The authors had no relevant financial disclosures to report.
SOURCE: Wu SZ et al. Pediatr Dermatol. 2018;35:817-20.
Given its limited systemic toxicity, , according to Sean Z. Wu, MD, of the department of dermatology, University of Cincinnati, and his associates.
In a retrospective study of 37 pediatric patients with AA, published in Pediatric Dermatology, Dr. Wu and his colleagues found that almost two-thirds experienced at least 50% regrowth of hair with topical anthralin treatment, but they described severe dermatitis and relapses as “potential drawbacks” of treatment.
The 37 patients were in the Cleveland Clinic AA areata database and began treatment with anthralin between 2004 and 2015, at aged 2-17 years (mean age 9). Over half (22) were females and most (31) were white. About 65% had patchy AA; the remainder had either alopecia totalis or alopecia universalis. Prior treatments included topical corticosteroids, minoxidil, and intralesional corticosteroids; four patients had not been treated previously. Patients were followed up from 51 days to more than 10 years, with a mean duration of 2.5 years. Treatment regimens, titrated up to achieve a mild to moderate dermatitis, included application of 0.5% cream for 5 minutes twice a week up to 1.0% cream for 30 minutes a day.
With topical anthralin, 12 (32%) of patients had complete scalp regrowth, 25 (68%) experienced at least 50% regrowth, and 5 (14%) had no response; in five patients, no follow-up information was available. Among those with at least 50% regrowth, the initial response was first noted at a mean of 3.4 months, and the mean time to maximal response was 15 months. This timeline suggests that treatment with anthralin should be continued beyond 1 year to ensure maximum beneficial results with hair regrowth, the authors wrote.
Factors associated with a positive response to treatment included less than 50% of scalp involvement. The two patients who used anthralin as monotherapy did not achieve a 50% scalp response, but the four treatment-naive patients were among those with at least 50% scalp regrowth, versus three of the five patients (60%) who had been treated previously with systemic steroids.
Two potential clinical limitations were noted during the study. Four patients had to stop treatment because of dermatitis, which suggests that patients and parents should be counseled about the potential for severe skin irritation with this treatment, the authors said. And among those who achieved at least 50% scalp regrowth, 16 of the 25 (64%) relapsed. The authors speculated that the effects of the drug could be temporary “or that the disease process may be able to overcome the anthralin effect over time.”
Dr. Wu and his coauthors cited the retrospective design and the small population size as major limitations of the study. Because some patients continued other treatments, it is “difficult to attribute scalp regrowth entirely to anthralin,” and variations in formulation and in treatment regimens are also factors to be considered, they cautioned. That AA has been found to spontaneously resolve, depending upon the severity of the disease, presents additional challenges for clinicians attempting to determine the extent to which anthralin offers therapeutic benefit, they pointed out.
In spite of the drug’s limitations, the authors concluded that the treatment was a safe and useful option as an “adjunct for those who fail first-line therapy with topical or intralesional corticosteroids.” They added that more work is needed to tailor treatment formulation, frequency, and duration to the specific needs of pediatric patients.
The authors had no relevant financial disclosures to report.
SOURCE: Wu SZ et al. Pediatr Dermatol. 2018;35:817-20.
FROM PEDIATRIC DERMATOLOGY
Key clinical point: Additional research is needed to pinpoint protocols for optimal formulation, dosage, and duration of anthralin in pediatric patients.
Major finding: At least 50% regrowth of scalp hair was achieved in 68% of patients treated with topical anthralin.
Study details: A retrospective chart review of 37 patients with alopecia areata who started treatment at a mean age of 9 years.
Disclosures: The authors reported no relevant financial disclosures.
Source: Wu SZ et al. Pediatr Dermatol. 2018;35:817-20.
What is your diagnosis? - December 2018
A KOH (potassium hydroxide) test done at the visit was negative as well as a fungal culture of each toenail.

The patient was diagnosed with congenital malalignment of the great toenails (CMGTN) based on history and morphologic appearance.
Congenital malalignment of the great toenails is an underrecognized and underreported nail disorder characterized by lateral deviation of the nail plate, which is not parallel to the longitudinal axis of the distal phalanx.1 The cause is unknown. Some reports suggest a genetic cause being transmitted in an autosomal dominant fashion with variable expression.2 There have been reports of CMGTN in monozygotic and dizygotic twins making this theory likely.3 Other authors consider an external cause such as amniotic bands, neonatal asphyxia, vascular malformations, and uterine pressure. This condition also has been reported in patients with Rubinstein-Taybi syndrome.4
The nail changes can occur at birth but in some cases, such as our patient, the nails become dystrophic months to years after birth. Characteristic nail changes include shorter, discolored, hyperkeratotic nails with transverse groove or ridges. In some cases, the dystrophic nails may cause inflammation and tenderness and is the most common cause of ingrown toenails in children.
The differential diagnosis includes onychomycosis, traumatic nails, nail psoriasis, pachyonychia congenital (PC), and onychomadesis. Onychomycosis can present with white or yellow discoloration of the nail that in some cases can be associated with nail breakage, hyperkeratosis, onycholysis, and subungual debris. Either fungal culture or periodic acid shift stain of nail clippings can help confirm or exclude this diagnosis. Psoriatic nails present with nail pits, oils spots, and onycholysis. Traumatic nail changes may occur from using small shoes and trauma from running or playing soccer, and presents with subungual hemorrhage and nail dystrophy of the first or second toenail. PC is a genetic disorder caused by a mutation in certain keratin proteins of the skin (k6a, k6b, K16 and K17). These patients usually have other skin findings including palmoplantar keratoderma, white plaques on the mouth, and skin cysts (steatocystoma multiplex and vellus hair cysts). Nail changes characteristic of PC includes subungual hyperkeratosis that causes a wedge shape thickening of the nail bed (pincer nails).5 Onychomadesis can be seen after viral infections such as hand-foot-mouth disease or in patients taking chemotherapy drugs that affect nail growth.
CMGTN usually resolves with time, but some patients with severe deviation and paronychia may need surgical correction.6
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at [email protected].
References
1. Dermatol Online J. 2014 Jan 15;20(1):21251.
2. J Dtsch Dermatol Ges. 2012 May;10(5):326-30.
3. J Am Acad Dermatol. 2007 Oct;57(4):711-5.
4. Pediatr Dermatol. 2004 Jan-Feb;21(1):44-7.
5. Curr Opin Pediatr. 2014 Aug;26(4):440-5.
6. Skin Appendage Disord. 2018 Oct;4(4):230-5.
A KOH (potassium hydroxide) test done at the visit was negative as well as a fungal culture of each toenail.
The patient was diagnosed with congenital malalignment of the great toenails (CMGTN) based on history and morphologic appearance.
Congenital malalignment of the great toenails is an underrecognized and underreported nail disorder characterized by lateral deviation of the nail plate, which is not parallel to the longitudinal axis of the distal phalanx.1 The cause is unknown. Some reports suggest a genetic cause being transmitted in an autosomal dominant fashion with variable expression.2 There have been reports of CMGTN in monozygotic and dizygotic twins making this theory likely.3 Other authors consider an external cause such as amniotic bands, neonatal asphyxia, vascular malformations, and uterine pressure. This condition also has been reported in patients with Rubinstein-Taybi syndrome.4
The nail changes can occur at birth but in some cases, such as our patient, the nails become dystrophic months to years after birth. Characteristic nail changes include shorter, discolored, hyperkeratotic nails with transverse groove or ridges. In some cases, the dystrophic nails may cause inflammation and tenderness and is the most common cause of ingrown toenails in children.
The differential diagnosis includes onychomycosis, traumatic nails, nail psoriasis, pachyonychia congenital (PC), and onychomadesis. Onychomycosis can present with white or yellow discoloration of the nail that in some cases can be associated with nail breakage, hyperkeratosis, onycholysis, and subungual debris. Either fungal culture or periodic acid shift stain of nail clippings can help confirm or exclude this diagnosis. Psoriatic nails present with nail pits, oils spots, and onycholysis. Traumatic nail changes may occur from using small shoes and trauma from running or playing soccer, and presents with subungual hemorrhage and nail dystrophy of the first or second toenail. PC is a genetic disorder caused by a mutation in certain keratin proteins of the skin (k6a, k6b, K16 and K17). These patients usually have other skin findings including palmoplantar keratoderma, white plaques on the mouth, and skin cysts (steatocystoma multiplex and vellus hair cysts). Nail changes characteristic of PC includes subungual hyperkeratosis that causes a wedge shape thickening of the nail bed (pincer nails).5 Onychomadesis can be seen after viral infections such as hand-foot-mouth disease or in patients taking chemotherapy drugs that affect nail growth.
CMGTN usually resolves with time, but some patients with severe deviation and paronychia may need surgical correction.6
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at [email protected].
References
1. Dermatol Online J. 2014 Jan 15;20(1):21251.
2. J Dtsch Dermatol Ges. 2012 May;10(5):326-30.
3. J Am Acad Dermatol. 2007 Oct;57(4):711-5.
4. Pediatr Dermatol. 2004 Jan-Feb;21(1):44-7.
5. Curr Opin Pediatr. 2014 Aug;26(4):440-5.
6. Skin Appendage Disord. 2018 Oct;4(4):230-5.
A KOH (potassium hydroxide) test done at the visit was negative as well as a fungal culture of each toenail.
The patient was diagnosed with congenital malalignment of the great toenails (CMGTN) based on history and morphologic appearance.
Congenital malalignment of the great toenails is an underrecognized and underreported nail disorder characterized by lateral deviation of the nail plate, which is not parallel to the longitudinal axis of the distal phalanx.1 The cause is unknown. Some reports suggest a genetic cause being transmitted in an autosomal dominant fashion with variable expression.2 There have been reports of CMGTN in monozygotic and dizygotic twins making this theory likely.3 Other authors consider an external cause such as amniotic bands, neonatal asphyxia, vascular malformations, and uterine pressure. This condition also has been reported in patients with Rubinstein-Taybi syndrome.4
The nail changes can occur at birth but in some cases, such as our patient, the nails become dystrophic months to years after birth. Characteristic nail changes include shorter, discolored, hyperkeratotic nails with transverse groove or ridges. In some cases, the dystrophic nails may cause inflammation and tenderness and is the most common cause of ingrown toenails in children.
The differential diagnosis includes onychomycosis, traumatic nails, nail psoriasis, pachyonychia congenital (PC), and onychomadesis. Onychomycosis can present with white or yellow discoloration of the nail that in some cases can be associated with nail breakage, hyperkeratosis, onycholysis, and subungual debris. Either fungal culture or periodic acid shift stain of nail clippings can help confirm or exclude this diagnosis. Psoriatic nails present with nail pits, oils spots, and onycholysis. Traumatic nail changes may occur from using small shoes and trauma from running or playing soccer, and presents with subungual hemorrhage and nail dystrophy of the first or second toenail. PC is a genetic disorder caused by a mutation in certain keratin proteins of the skin (k6a, k6b, K16 and K17). These patients usually have other skin findings including palmoplantar keratoderma, white plaques on the mouth, and skin cysts (steatocystoma multiplex and vellus hair cysts). Nail changes characteristic of PC includes subungual hyperkeratosis that causes a wedge shape thickening of the nail bed (pincer nails).5 Onychomadesis can be seen after viral infections such as hand-foot-mouth disease or in patients taking chemotherapy drugs that affect nail growth.
CMGTN usually resolves with time, but some patients with severe deviation and paronychia may need surgical correction.6
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at [email protected].
References
1. Dermatol Online J. 2014 Jan 15;20(1):21251.
2. J Dtsch Dermatol Ges. 2012 May;10(5):326-30.
3. J Am Acad Dermatol. 2007 Oct;57(4):711-5.
4. Pediatr Dermatol. 2004 Jan-Feb;21(1):44-7.
5. Curr Opin Pediatr. 2014 Aug;26(4):440-5.
6. Skin Appendage Disord. 2018 Oct;4(4):230-5.
A 4-year-old boy is brought to our pediatric dermatology clinic by his mother with the concern of difficult to treat toenail fungus.
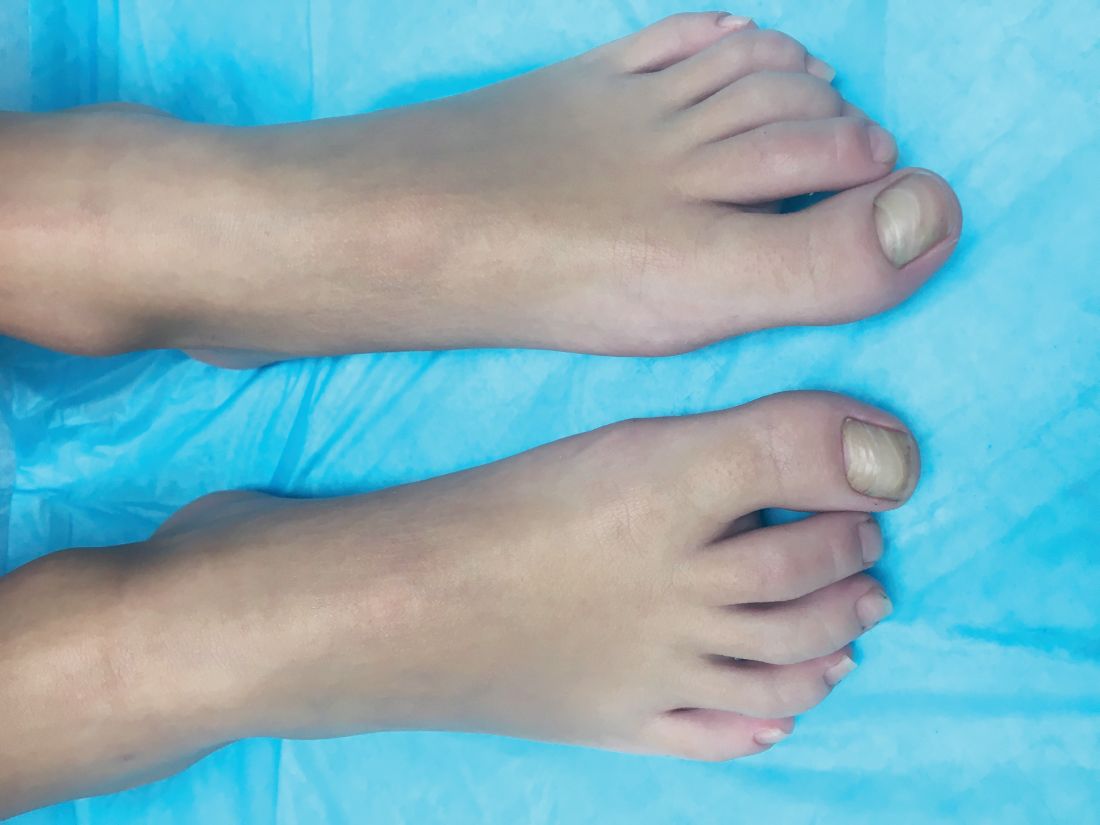
The mother reported that she started noticing the toenail changes at around 8 months of age, and it has been progressively getting worse.
He has been treated with several courses of topical antifungals and 3 months of oral terbinafine without success.
A fungal culture done 1 year prior showed slight growth of Cladosporium Sp., but the nails failed to improve after systemic therapy. He denied any associated pain or inflammation. He likes playing softball and plays soccer sometimes. The mother is very worried because the father also has a history of onychomycosis that he has not been able to clear for years.
On physical exam, he is a very pleasant young boy. His cutaneous exam is normal including hair and teeth except for thickening of the bilateral first toenails associated with transverse ridging and yellow discoloration.
Topical retinoid found effective as microneedling for acne scars
according to a new study.
In a prospective, randomized, split-face study of adults with postacne scarring, both treatments resulted in similar efficacy after 6 months, reported T.P. Afra, MD, and associates from the department of dermatology, venereology, and leprology at the Postgraduate Institute of Medical Education and Research in Chandigarh, India. While the clinical usefulness of microneedling as a procedure for postacne scarring is well established, research exploring the effectiveness of topical therapies for acne scarring that could be used at home is lacking. “A home-based topical treatment with a comparable efficacy to microneedling and that is well tolerated would be a useful addition in the armamentarium of acne scar management,” they wrote in the study, published in JAMA Facial Plastic Surgery.
The study included 34 patients, aged 18-30 years, with grade 2-4 facial atrophic acne scars at their initial visit to the research team’s skin clinic. One side of each participants face was randomized to receive microneedling treatment for four sessions over 3 months (using a dermaroller with 1.5-mm needles). Topical tazarotene gel 0.1%, a retinoid approved by the Food and Drug Administration as a treatment for mild to moderate facial acne, was applied to the other side of their face once a night during the same time. Almost 81% were skin phototypes IV, the rest were type III or V. Patients followed up every month for 3 months, then at 6 months.
Changes in acne scar severity from baseline, the primary outcome, were assessed using Goodman and Baron quantitative and qualitative scores and a subjective dermatologist score. Patient satisfaction measured with a Patient Global Assessment (PGA) score and adverse events were secondary outcomes.
In 31 patients (91.2%), overall improvements from baseline to the 6-month visit in quantitative acne scar severity scores for both treatments were seen, with significant improvements from baseline to 6 months: A median improvement of 3 on the sides of the face treated with microneedling and a median improvement of 2.5 on the sides of the face treated with tazarotene (between-group comparison, P = .42). The qualitative acne scar severity score did not significantly improve with either treatment, the investigators noted.
The median improvement in the independent dermatologist score was also comparable for both methods at 3 and 6 months.
At 6 months, improvement in the mean PGA score was “slightly but significantly superior” for the microneedling treatment, compared with that for tazarotene (mean of 5.86 vs. 5.76, respectively; P less than .001), with both falling into the “satisfactory” range for the PGA, the investigators wrote. They also noted a positive correlation between previous exposure to oral isotretinoin and patient satisfaction.
“Although collagen accumulation has been considered a drawback of isotretinoin therapy owing to the development of hypertrophic scars, the better atrophic acne scar outcomes observed for both the present treatment groups in patients with a history of isotretinoin treatment indicates that the collagen accumulation in this case may actually be beneficial,” they wrote.
The topical retinoid was well tolerated by participants, with less than a third reporting dryness and scaling, and adverse effects associated with microneedling were described as “minimal.”
“The use of a modality such as tazarotene that prevents acne flares while addressing acne scarring is a practical addition to clinical practice,” the investigators concluded. “Tazarotene gel 0.1% would be a useful alternative to microneedling in the management of atrophic acne scars. Such a home-based medical management option for acne scarring may decrease physician dependence and health care expenditures for patients with postacne scarring.”
The study authors noted that, as collagen remodeling is a continuous process lasting more than 1 year, a limitation of their study was its short-follow-up of 6 months. However, a strength of the study was its use of validated acne scar severity scoring tools as well as patient and physician assessment of scar improvement in the outcome assessments.
The authors had no disclosures to report.
SOURCE: Afra TP et al. JAMA Facial Plast Surg. 2018 Nov 15. doi: 10.1001/jamafacial.2018.1404.
according to a new study.
In a prospective, randomized, split-face study of adults with postacne scarring, both treatments resulted in similar efficacy after 6 months, reported T.P. Afra, MD, and associates from the department of dermatology, venereology, and leprology at the Postgraduate Institute of Medical Education and Research in Chandigarh, India. While the clinical usefulness of microneedling as a procedure for postacne scarring is well established, research exploring the effectiveness of topical therapies for acne scarring that could be used at home is lacking. “A home-based topical treatment with a comparable efficacy to microneedling and that is well tolerated would be a useful addition in the armamentarium of acne scar management,” they wrote in the study, published in JAMA Facial Plastic Surgery.
The study included 34 patients, aged 18-30 years, with grade 2-4 facial atrophic acne scars at their initial visit to the research team’s skin clinic. One side of each participants face was randomized to receive microneedling treatment for four sessions over 3 months (using a dermaroller with 1.5-mm needles). Topical tazarotene gel 0.1%, a retinoid approved by the Food and Drug Administration as a treatment for mild to moderate facial acne, was applied to the other side of their face once a night during the same time. Almost 81% were skin phototypes IV, the rest were type III or V. Patients followed up every month for 3 months, then at 6 months.
Changes in acne scar severity from baseline, the primary outcome, were assessed using Goodman and Baron quantitative and qualitative scores and a subjective dermatologist score. Patient satisfaction measured with a Patient Global Assessment (PGA) score and adverse events were secondary outcomes.
In 31 patients (91.2%), overall improvements from baseline to the 6-month visit in quantitative acne scar severity scores for both treatments were seen, with significant improvements from baseline to 6 months: A median improvement of 3 on the sides of the face treated with microneedling and a median improvement of 2.5 on the sides of the face treated with tazarotene (between-group comparison, P = .42). The qualitative acne scar severity score did not significantly improve with either treatment, the investigators noted.
The median improvement in the independent dermatologist score was also comparable for both methods at 3 and 6 months.
At 6 months, improvement in the mean PGA score was “slightly but significantly superior” for the microneedling treatment, compared with that for tazarotene (mean of 5.86 vs. 5.76, respectively; P less than .001), with both falling into the “satisfactory” range for the PGA, the investigators wrote. They also noted a positive correlation between previous exposure to oral isotretinoin and patient satisfaction.
“Although collagen accumulation has been considered a drawback of isotretinoin therapy owing to the development of hypertrophic scars, the better atrophic acne scar outcomes observed for both the present treatment groups in patients with a history of isotretinoin treatment indicates that the collagen accumulation in this case may actually be beneficial,” they wrote.
The topical retinoid was well tolerated by participants, with less than a third reporting dryness and scaling, and adverse effects associated with microneedling were described as “minimal.”
“The use of a modality such as tazarotene that prevents acne flares while addressing acne scarring is a practical addition to clinical practice,” the investigators concluded. “Tazarotene gel 0.1% would be a useful alternative to microneedling in the management of atrophic acne scars. Such a home-based medical management option for acne scarring may decrease physician dependence and health care expenditures for patients with postacne scarring.”
The study authors noted that, as collagen remodeling is a continuous process lasting more than 1 year, a limitation of their study was its short-follow-up of 6 months. However, a strength of the study was its use of validated acne scar severity scoring tools as well as patient and physician assessment of scar improvement in the outcome assessments.
The authors had no disclosures to report.
SOURCE: Afra TP et al. JAMA Facial Plast Surg. 2018 Nov 15. doi: 10.1001/jamafacial.2018.1404.
according to a new study.
In a prospective, randomized, split-face study of adults with postacne scarring, both treatments resulted in similar efficacy after 6 months, reported T.P. Afra, MD, and associates from the department of dermatology, venereology, and leprology at the Postgraduate Institute of Medical Education and Research in Chandigarh, India. While the clinical usefulness of microneedling as a procedure for postacne scarring is well established, research exploring the effectiveness of topical therapies for acne scarring that could be used at home is lacking. “A home-based topical treatment with a comparable efficacy to microneedling and that is well tolerated would be a useful addition in the armamentarium of acne scar management,” they wrote in the study, published in JAMA Facial Plastic Surgery.
The study included 34 patients, aged 18-30 years, with grade 2-4 facial atrophic acne scars at their initial visit to the research team’s skin clinic. One side of each participants face was randomized to receive microneedling treatment for four sessions over 3 months (using a dermaroller with 1.5-mm needles). Topical tazarotene gel 0.1%, a retinoid approved by the Food and Drug Administration as a treatment for mild to moderate facial acne, was applied to the other side of their face once a night during the same time. Almost 81% were skin phototypes IV, the rest were type III or V. Patients followed up every month for 3 months, then at 6 months.
Changes in acne scar severity from baseline, the primary outcome, were assessed using Goodman and Baron quantitative and qualitative scores and a subjective dermatologist score. Patient satisfaction measured with a Patient Global Assessment (PGA) score and adverse events were secondary outcomes.
In 31 patients (91.2%), overall improvements from baseline to the 6-month visit in quantitative acne scar severity scores for both treatments were seen, with significant improvements from baseline to 6 months: A median improvement of 3 on the sides of the face treated with microneedling and a median improvement of 2.5 on the sides of the face treated with tazarotene (between-group comparison, P = .42). The qualitative acne scar severity score did not significantly improve with either treatment, the investigators noted.
The median improvement in the independent dermatologist score was also comparable for both methods at 3 and 6 months.
At 6 months, improvement in the mean PGA score was “slightly but significantly superior” for the microneedling treatment, compared with that for tazarotene (mean of 5.86 vs. 5.76, respectively; P less than .001), with both falling into the “satisfactory” range for the PGA, the investigators wrote. They also noted a positive correlation between previous exposure to oral isotretinoin and patient satisfaction.
“Although collagen accumulation has been considered a drawback of isotretinoin therapy owing to the development of hypertrophic scars, the better atrophic acne scar outcomes observed for both the present treatment groups in patients with a history of isotretinoin treatment indicates that the collagen accumulation in this case may actually be beneficial,” they wrote.
The topical retinoid was well tolerated by participants, with less than a third reporting dryness and scaling, and adverse effects associated with microneedling were described as “minimal.”
“The use of a modality such as tazarotene that prevents acne flares while addressing acne scarring is a practical addition to clinical practice,” the investigators concluded. “Tazarotene gel 0.1% would be a useful alternative to microneedling in the management of atrophic acne scars. Such a home-based medical management option for acne scarring may decrease physician dependence and health care expenditures for patients with postacne scarring.”
The study authors noted that, as collagen remodeling is a continuous process lasting more than 1 year, a limitation of their study was its short-follow-up of 6 months. However, a strength of the study was its use of validated acne scar severity scoring tools as well as patient and physician assessment of scar improvement in the outcome assessments.
The authors had no disclosures to report.
SOURCE: Afra TP et al. JAMA Facial Plast Surg. 2018 Nov 15. doi: 10.1001/jamafacial.2018.1404.
FROM JAMA FACIAL PLASTIC SURGERY
Key clinical point: The topical retinoid tazarotene could be a home-based option for treating atrophic acne scarring.
Major finding: Improvements in acne scarring were similar with microneedling and nightly applications of tazarotene gel 0.1% after 6 months.
Study details: A prospective, observer-blinded, split-face, randomized, clinical trial involving 34 patients with grade 2-4 facial atrophic postacne scars.
Disclosures: The authors had no disclosures to report.
Source: Afra TP et al. JAMA Facial Plast Surg. 2018 Nov 15. doi: 10.1001/jamafacial.2018.1404.
Skin rashes often accompany drug-induced liver injury
SAN FRANCISCO – More than a quarter of drug-induced liver injury (DILI) cases also involve skin reactions, most often drug rash with eosinophilia and system symptoms (DRESS) syndrome. These dual cases of DILI and drug-induced skin injury (DISI) underscore the need for hepatologists to pay attention to dermatologic conditions and emphasize the need for the two specialties to work together.
The findings suggest that DISI/DILI comorbidity is not uncommon, and may hint at underlying mechanisms that could be used to tailor treatment, according to Harshad Devarbhavi, MD, who presented the study at the annual meeting of the American Association for the Study of Liver Diseases. “My message was that people should work more and see if there’s any type of genotype or HLA [human leukocyte antigen] that produces this reaction. It’s a multisystem disease. It doesn’t belong to dermatologists, it’s a domain that also belongs to hepatologists,” said Dr. Devarbhavi, who is a hepatology fellow at St. John’s Medical College in Bangalore, India.
DISI is more common than DILI, and may or may not be caused by an immune response. The two conditions were previously known to co-occur, but it is rarely reported because dermatologists and hepatologists report findings in different journals.
The researchers defined DILI as a fivefold or greater increase in aspartate aminotransferase (AST) or alanine aminotransferase (ALT); a threefold or greater increase with symptoms, including cutaneous reactions; any elevation of AST, ALT, or alkaline phosphatase (ALP) accompanying a bilirubin increase of 2 mg/dL or more; or a twofold or higher increase in ALP combined with a cutaneous reaction.
They analyzed 921 DILI patients from a single registry in India, who were seen between 1997 and April 2018. All patients with skin reactions were seen by dermatologists and competing causes were excluded. A total of 28% of patients with DILI also had DISI, 13% of whom were also HIV positive; 56% developed jaundice. The mean age of patients with DILI/DISI was 35 years, compared with 42 years in DILI only patients (P = .001) and the mean duration of drug therapy was 42 days, compared with 89 days (P = .002). Twelve percent of DILI/DISI patients died, which was lower than the 17% mortality in those with DILI alone.
Of the DILI/DISI patients, 59% experienced DRESS, and 19% had Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN). Six percent of patients with DRESS died, as did 22% of those with SJS/TEN. Mortality was 16% among those with other skin manifestations. Eighteen percent of those with jaundice died, compared with 3% of those without jaundice.
Thirty patients with DILI/DISI died; 37% (11) of them had SJS/TEN, compared with 17% of survivors (P = .01). DRESS was more common in survivors (62% vs. 33%; P = .02).
Of DILI/DISI and SJS/TEN cases, 75% were associated with four drug classes: antiepileptic drugs, dapsone, antiretroviral therapies, and leflunomide.
“The liver is the biggest internal organ in the body, and skin is the largest external organ, so there is some correlation between the two, but people haven’t looked at it. People should come together and see why some drugs produce both these injuries. I think there is some mechanistic information in these drugs,” said Dr. Devarbhavi.
Source: Hepatology 2018 Oct 1;68[S1], Abstract 37.
SAN FRANCISCO – More than a quarter of drug-induced liver injury (DILI) cases also involve skin reactions, most often drug rash with eosinophilia and system symptoms (DRESS) syndrome. These dual cases of DILI and drug-induced skin injury (DISI) underscore the need for hepatologists to pay attention to dermatologic conditions and emphasize the need for the two specialties to work together.
The findings suggest that DISI/DILI comorbidity is not uncommon, and may hint at underlying mechanisms that could be used to tailor treatment, according to Harshad Devarbhavi, MD, who presented the study at the annual meeting of the American Association for the Study of Liver Diseases. “My message was that people should work more and see if there’s any type of genotype or HLA [human leukocyte antigen] that produces this reaction. It’s a multisystem disease. It doesn’t belong to dermatologists, it’s a domain that also belongs to hepatologists,” said Dr. Devarbhavi, who is a hepatology fellow at St. John’s Medical College in Bangalore, India.
DISI is more common than DILI, and may or may not be caused by an immune response. The two conditions were previously known to co-occur, but it is rarely reported because dermatologists and hepatologists report findings in different journals.
The researchers defined DILI as a fivefold or greater increase in aspartate aminotransferase (AST) or alanine aminotransferase (ALT); a threefold or greater increase with symptoms, including cutaneous reactions; any elevation of AST, ALT, or alkaline phosphatase (ALP) accompanying a bilirubin increase of 2 mg/dL or more; or a twofold or higher increase in ALP combined with a cutaneous reaction.
They analyzed 921 DILI patients from a single registry in India, who were seen between 1997 and April 2018. All patients with skin reactions were seen by dermatologists and competing causes were excluded. A total of 28% of patients with DILI also had DISI, 13% of whom were also HIV positive; 56% developed jaundice. The mean age of patients with DILI/DISI was 35 years, compared with 42 years in DILI only patients (P = .001) and the mean duration of drug therapy was 42 days, compared with 89 days (P = .002). Twelve percent of DILI/DISI patients died, which was lower than the 17% mortality in those with DILI alone.
Of the DILI/DISI patients, 59% experienced DRESS, and 19% had Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN). Six percent of patients with DRESS died, as did 22% of those with SJS/TEN. Mortality was 16% among those with other skin manifestations. Eighteen percent of those with jaundice died, compared with 3% of those without jaundice.
Thirty patients with DILI/DISI died; 37% (11) of them had SJS/TEN, compared with 17% of survivors (P = .01). DRESS was more common in survivors (62% vs. 33%; P = .02).
Of DILI/DISI and SJS/TEN cases, 75% were associated with four drug classes: antiepileptic drugs, dapsone, antiretroviral therapies, and leflunomide.
“The liver is the biggest internal organ in the body, and skin is the largest external organ, so there is some correlation between the two, but people haven’t looked at it. People should come together and see why some drugs produce both these injuries. I think there is some mechanistic information in these drugs,” said Dr. Devarbhavi.
Source: Hepatology 2018 Oct 1;68[S1], Abstract 37.
SAN FRANCISCO – More than a quarter of drug-induced liver injury (DILI) cases also involve skin reactions, most often drug rash with eosinophilia and system symptoms (DRESS) syndrome. These dual cases of DILI and drug-induced skin injury (DISI) underscore the need for hepatologists to pay attention to dermatologic conditions and emphasize the need for the two specialties to work together.
The findings suggest that DISI/DILI comorbidity is not uncommon, and may hint at underlying mechanisms that could be used to tailor treatment, according to Harshad Devarbhavi, MD, who presented the study at the annual meeting of the American Association for the Study of Liver Diseases. “My message was that people should work more and see if there’s any type of genotype or HLA [human leukocyte antigen] that produces this reaction. It’s a multisystem disease. It doesn’t belong to dermatologists, it’s a domain that also belongs to hepatologists,” said Dr. Devarbhavi, who is a hepatology fellow at St. John’s Medical College in Bangalore, India.
DISI is more common than DILI, and may or may not be caused by an immune response. The two conditions were previously known to co-occur, but it is rarely reported because dermatologists and hepatologists report findings in different journals.
The researchers defined DILI as a fivefold or greater increase in aspartate aminotransferase (AST) or alanine aminotransferase (ALT); a threefold or greater increase with symptoms, including cutaneous reactions; any elevation of AST, ALT, or alkaline phosphatase (ALP) accompanying a bilirubin increase of 2 mg/dL or more; or a twofold or higher increase in ALP combined with a cutaneous reaction.
They analyzed 921 DILI patients from a single registry in India, who were seen between 1997 and April 2018. All patients with skin reactions were seen by dermatologists and competing causes were excluded. A total of 28% of patients with DILI also had DISI, 13% of whom were also HIV positive; 56% developed jaundice. The mean age of patients with DILI/DISI was 35 years, compared with 42 years in DILI only patients (P = .001) and the mean duration of drug therapy was 42 days, compared with 89 days (P = .002). Twelve percent of DILI/DISI patients died, which was lower than the 17% mortality in those with DILI alone.
Of the DILI/DISI patients, 59% experienced DRESS, and 19% had Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN). Six percent of patients with DRESS died, as did 22% of those with SJS/TEN. Mortality was 16% among those with other skin manifestations. Eighteen percent of those with jaundice died, compared with 3% of those without jaundice.
Thirty patients with DILI/DISI died; 37% (11) of them had SJS/TEN, compared with 17% of survivors (P = .01). DRESS was more common in survivors (62% vs. 33%; P = .02).
Of DILI/DISI and SJS/TEN cases, 75% were associated with four drug classes: antiepileptic drugs, dapsone, antiretroviral therapies, and leflunomide.
“The liver is the biggest internal organ in the body, and skin is the largest external organ, so there is some correlation between the two, but people haven’t looked at it. People should come together and see why some drugs produce both these injuries. I think there is some mechanistic information in these drugs,” said Dr. Devarbhavi.
Source: Hepatology 2018 Oct 1;68[S1], Abstract 37.
REPORTING FROM THE LIVER MEETING 2018
Key clinical point: Researchers hope the findings will shed light on the mechanism of injury.
Major finding: 28% of patients with DILI also had a skin rash.
Study details: Retrospective analysis of 921 DILI patients.
Disclosures: No source of funding was disclosed. Dr. Devarbhavi disclosed no relevant conflicts.
Source: Hepatology 2018 Oct 1;68[S1], Abstract 37.
Three drugs disappoint in SSc trials, but show some promise
CHICAGO – Recent randomized, placebo-controlled, phase 3 trials of tocilizumab, abatacept, and riociguat for the treatment of systemic sclerosis each failed to reach its primary endpoint of change from baseline in modified Rodnan Skin Score (mRSS).

Still, findings with respect to secondary endpoints and certain exploratory outcomes suggest each of the agents holds some promise in the systemic sclerosis (SSc) arena, according to the data presented at the annual meeting of the American College of Rheumatology.
Tocilizumab (Actemra)
In the double-blind portion of the phase 3 focuSSced trial of 212 patients with SSc, numerical improvement was observed for the primary endpoint of mean change in mRSS from baseline to week 48 with tocilizumab versus placebo (–6.14 vs. –4.41 points, respectively). The change in the treatment group was comparable with what was seen in the phase 2 faSScinate trial, but the decline in mRSS in the placebo group was much greater in phase 3 than in phase 2, and so the difference between the groups in the current study failed to reach statistical significance (P = .098), reported Dinesh Khanna, MBBS, a professor of medicine and director of the scleroderma program at the University of Michigan, Ann Arbor.
The interleukin-6 (IL-6) receptor–alpha antibody was previously shown in the faSScinate trial to lead to numeric improvements in skin thickening as measured by the mRSS, as well as to clinically meaningful lung function preservation as measured by percent predicted forced vital capacity (FVC).
In the current phase 3 study, key secondary end points also appeared to favor tocilizumab, but since the primary endpoint for mRSS was not met, all other P values cannot be considered statistically significant despite the strength of the evidence and were reported for informational purposes only, he noted.
The median cumulative distribution of change from baseline to week 48 in percent predicted FVC with tocilizumab versus placebo was –0.6 vs. –3.9, respectively (descriptive P = .0015), and the mean change from baseline in FVC at week 48 was –24 mL vs. –190 mL (difference of 167 mL in favor of tocilizumab; descriptive P = .0001).
Time to treatment failure also favored tocilizumab, he said (hazard ratio, 0.63; descriptive P = .082), he said.
Patients were randomly assigned to receive either weekly 162-mg injections of subcutaneous tocilizumab or placebo for 48 weeks. Escape therapy was allowed beginning at week 16 if patients experienced declines in FVC or beginning at week 24 if they experienced worsened mRSS or worsened SSc complications, Dr. Khanna said.
“The key part is that no immunotherapy was allowed. ... So it’s a true randomized, placebo-controlled trial,” he said.
Most (81%) of the patients were women, and they had a mean age of 48 years, mean SSc duration of 23 months, mean mRSS of 20.4 units on a 0-51 scale, and a normal mean percent predicted FVC of 82.1%.
“HAQ-DI showed moderate disability of 1.2,” he noted.
Safety in the study was consistent with that seen in prior tocilizumab studies; no new safety signals were identified. Serious adverse events occurred in 13% and 17% of tocilizumab and placebo group patients , respectively, and serious infections were reported by 7% and 2%.
Although clinically meaningful and consistent differences in FVC favoring tocilizumab were shown in this study, the primary endpoint was not met, Dr. Khanna said.
“There were no statistically significant differences, largely driven by unexpected improvement in the placebo group, which was different than what we found in [the faSScinate] trial,” he said, noting, however, that the FVC findings in the current study were clinically meaningful.
Also, in a separate presentation at the meeting, he explained that the differences favoring tocilizumab were statistically significant when patient-level data from the trial were analyzed based on the ACR Composite Response Index in Systemic Sclerosis (CRISS). Those findings provide validation of the novel outcomes measure, he said.
Abatacept (Orencia)
Dr. Khanna also reported results of the 12-month, double-blind, randomized, placebo-controlled phase 2 ASSET trial of abatacept, which showed no significant difference in mRSS in patients with early diffuse cutaneous SSc (dfSSc) who were treated with 125 mg of the recombinant fusion protein weekly and those who received placebo. However, certain secondary outcomes favored abatacept. No concomitant immunotherapy was allowed.
The adjusted mean decrease in the mRSS among patients who completed the 12-month treatment period was –6.24 vs. –4.49 in 34 patients in the abatacept group and 35 in the placebo group, respectively (P = .28).
The secondary outcome measures of mean change in Health Assessment Questionnaire Disability Index (HAQ-DI), patients global assessment, physician global assessment, and ACR CRISS scores were statistically significant or showed numerical results favoring abatacept over placebo: mean decrease in HAQ-DI, –0.17 vs. –0.11 (P = .05), respectively; mean change in physician global assessment scores, –1.30 vs. –0.35 (P = .03); median ACR CRISS index, 0.68 vs. 0.01 (P = .03), decline in percent predicted FVC of 4.13% and 1.34% (P = .11).
Escape therapy was allowed at 6 months for worsening SSc, but it did not change the outcomes trajectory, he said. A larger proportion of placebo vs. abatacept subjects required escape immunosuppressive therapy (36% vs. 16%; P = .03).
Patients were enrolled between 2014 and 2018 at 27 U.S., Canadian, and U.K. sites. At baseline, participants had a mean age of 49 years, 75% were women, and mean disease duration was very short at 1.59 years, with 60% having disease duration of 18 months or less. The mean baseline mRSS was 22.4, mean percent predicted FVC was 85.3%, and mean HAQ-DI was 1.0.
Compliance with both treatments was greater than 98%. Abatacept was well tolerated with comparable adverse events (AEs), serious AEs, and AEs of special interest such as infections and malignancies between treatments, Dr. Khanna said, noting that two deaths occurred in the abatacept group (caused by scleroderma renal crisis in both cases at days 11 and 46) and one occurred in a placebo group patient who experienced sudden cardiac arrest at day 310.
Of note, mRSS showed large variability, despite recruiting an early dcSSc population, Dr. Khanna said.
The finding with respect to the primary outcome is consistent with other recent trials because of improvement in mRSS that’s part of the natural history of the disease, including the tocilizumab findings that he reported at the meeting. The findings with respect to secondary endpoints and safety show promise.
“Stay tuned for robust ongoing work on the relationship between clinical changes and ongoing mechanistic work,” he said.
Riociguat (Adempas)

Similarly, in the randomized, placebo-controlled phase 2b RISE-SSc study comparing riociguat and placebo for early dcSSc, the primary efficacy endpoint of mean change in mRSS did not reach statistical significance, but exploratory data suggested that the soluble guanylate cyclase stimulator prevented disease progression in patients with early dcSSc, reported Oliver Distler, MD, head of the connective tissue diseases program at University Hospital Zurich (Switzerland).
The mean mRSS at baseline was comparable in 60 patients randomized to receive riociguat and 61 in the placebo group (16.8 and 16.71, respectively). These mean values at week 52 dropped to 14.63 vs. 15.73, respectively (P = .08).
“So it was close, but it didn’t reach significance,” he said.
The difference in the mRSS progression rate, however, suggested significant effects favoring riociguat (descriptive P = .02), he said.
Further, mean change from baseline to week 52 in percent predicted FVC was not different overall between the groups, but a large difference favoring riociguat was seen among patients with scleroderma interstitial lung disease at baseline (mean change of –2.7 vs. –8.9), he said.
No differences were seen between the groups in HAQ-DI or patient and physician global assessment. The proportion of patients with probability of improvement at 52 weeks as measured using ACR CRISS was also the same at 18% in both treatment arms, he noted, ”but the CRISS is designed more for assessing disease regression than for assessing prevention of progression.”
Treatment was, however, well tolerated. At week 52, fewer serious adverse events occurred with riociguat group than in the placebo group (15% vs. 25%, respectively), and no new safety signals were observed, he said.
Riociguat has previously shown antifibrotic effects in animal models and efficacy in patients with pulmonary arterial hypertension associated with connective tissue disease, so it was hypothesized that patients with dcSSc might benefit from riociguat therapy, Dr. Distler explained.
Study subjects had very early dcSSc (duration of 18 months or less; mean of 9 months), mRSS of 10-22 units, FVC of 45% predicted or greater, and diffusion capacity of the lung for carbon monoxide of at least 40% of predicted at screening.
Riociguat was given at an individually adjusted dose between 0.5 mg and 2.5 mg three times daily.
The findings demonstrate a numeric decrease in mRSS over time with riociguat versus placebo and a prevention of progression with riociguat; the failure to reach the primary endpoint may be related to the small study size and the higher than expected regression rate in the placebo group, Dr. Distler said.
Dr. Khanna is a consultant to Roche/Genentech and Bayer, which markets riociguat, and other companies. He has received research grants from Bayer, Bristol-Myers Squibb (which markets abatacept), and Pfizer. The ASSET trial he presented was sponsored by an National Institutes of Health/National Institute of Allergy and Infectious Diseases Clinical ACE grant and an investigator-initiated grant by Bristol-Myers Squibb. Dr. Distler has a consultancy relationship and/or has received research funding from Bayer, Roche/Genentech, and other companies. In addition, he has a patent on mir-29 for the treatment of systemic sclerosis.
SOURCES: Khanna D et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 898 and Abstract 900; Distler O et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 903.
CHICAGO – Recent randomized, placebo-controlled, phase 3 trials of tocilizumab, abatacept, and riociguat for the treatment of systemic sclerosis each failed to reach its primary endpoint of change from baseline in modified Rodnan Skin Score (mRSS).
Still, findings with respect to secondary endpoints and certain exploratory outcomes suggest each of the agents holds some promise in the systemic sclerosis (SSc) arena, according to the data presented at the annual meeting of the American College of Rheumatology.
Tocilizumab (Actemra)
In the double-blind portion of the phase 3 focuSSced trial of 212 patients with SSc, numerical improvement was observed for the primary endpoint of mean change in mRSS from baseline to week 48 with tocilizumab versus placebo (–6.14 vs. –4.41 points, respectively). The change in the treatment group was comparable with what was seen in the phase 2 faSScinate trial, but the decline in mRSS in the placebo group was much greater in phase 3 than in phase 2, and so the difference between the groups in the current study failed to reach statistical significance (P = .098), reported Dinesh Khanna, MBBS, a professor of medicine and director of the scleroderma program at the University of Michigan, Ann Arbor.
The interleukin-6 (IL-6) receptor–alpha antibody was previously shown in the faSScinate trial to lead to numeric improvements in skin thickening as measured by the mRSS, as well as to clinically meaningful lung function preservation as measured by percent predicted forced vital capacity (FVC).
In the current phase 3 study, key secondary end points also appeared to favor tocilizumab, but since the primary endpoint for mRSS was not met, all other P values cannot be considered statistically significant despite the strength of the evidence and were reported for informational purposes only, he noted.
The median cumulative distribution of change from baseline to week 48 in percent predicted FVC with tocilizumab versus placebo was –0.6 vs. –3.9, respectively (descriptive P = .0015), and the mean change from baseline in FVC at week 48 was –24 mL vs. –190 mL (difference of 167 mL in favor of tocilizumab; descriptive P = .0001).
Time to treatment failure also favored tocilizumab, he said (hazard ratio, 0.63; descriptive P = .082), he said.
Patients were randomly assigned to receive either weekly 162-mg injections of subcutaneous tocilizumab or placebo for 48 weeks. Escape therapy was allowed beginning at week 16 if patients experienced declines in FVC or beginning at week 24 if they experienced worsened mRSS or worsened SSc complications, Dr. Khanna said.
“The key part is that no immunotherapy was allowed. ... So it’s a true randomized, placebo-controlled trial,” he said.
Most (81%) of the patients were women, and they had a mean age of 48 years, mean SSc duration of 23 months, mean mRSS of 20.4 units on a 0-51 scale, and a normal mean percent predicted FVC of 82.1%.
“HAQ-DI showed moderate disability of 1.2,” he noted.
Safety in the study was consistent with that seen in prior tocilizumab studies; no new safety signals were identified. Serious adverse events occurred in 13% and 17% of tocilizumab and placebo group patients , respectively, and serious infections were reported by 7% and 2%.
Although clinically meaningful and consistent differences in FVC favoring tocilizumab were shown in this study, the primary endpoint was not met, Dr. Khanna said.
“There were no statistically significant differences, largely driven by unexpected improvement in the placebo group, which was different than what we found in [the faSScinate] trial,” he said, noting, however, that the FVC findings in the current study were clinically meaningful.
Also, in a separate presentation at the meeting, he explained that the differences favoring tocilizumab were statistically significant when patient-level data from the trial were analyzed based on the ACR Composite Response Index in Systemic Sclerosis (CRISS). Those findings provide validation of the novel outcomes measure, he said.
Abatacept (Orencia)
Dr. Khanna also reported results of the 12-month, double-blind, randomized, placebo-controlled phase 2 ASSET trial of abatacept, which showed no significant difference in mRSS in patients with early diffuse cutaneous SSc (dfSSc) who were treated with 125 mg of the recombinant fusion protein weekly and those who received placebo. However, certain secondary outcomes favored abatacept. No concomitant immunotherapy was allowed.
The adjusted mean decrease in the mRSS among patients who completed the 12-month treatment period was –6.24 vs. –4.49 in 34 patients in the abatacept group and 35 in the placebo group, respectively (P = .28).
The secondary outcome measures of mean change in Health Assessment Questionnaire Disability Index (HAQ-DI), patients global assessment, physician global assessment, and ACR CRISS scores were statistically significant or showed numerical results favoring abatacept over placebo: mean decrease in HAQ-DI, –0.17 vs. –0.11 (P = .05), respectively; mean change in physician global assessment scores, –1.30 vs. –0.35 (P = .03); median ACR CRISS index, 0.68 vs. 0.01 (P = .03), decline in percent predicted FVC of 4.13% and 1.34% (P = .11).
Escape therapy was allowed at 6 months for worsening SSc, but it did not change the outcomes trajectory, he said. A larger proportion of placebo vs. abatacept subjects required escape immunosuppressive therapy (36% vs. 16%; P = .03).
Patients were enrolled between 2014 and 2018 at 27 U.S., Canadian, and U.K. sites. At baseline, participants had a mean age of 49 years, 75% were women, and mean disease duration was very short at 1.59 years, with 60% having disease duration of 18 months or less. The mean baseline mRSS was 22.4, mean percent predicted FVC was 85.3%, and mean HAQ-DI was 1.0.
Compliance with both treatments was greater than 98%. Abatacept was well tolerated with comparable adverse events (AEs), serious AEs, and AEs of special interest such as infections and malignancies between treatments, Dr. Khanna said, noting that two deaths occurred in the abatacept group (caused by scleroderma renal crisis in both cases at days 11 and 46) and one occurred in a placebo group patient who experienced sudden cardiac arrest at day 310.
Of note, mRSS showed large variability, despite recruiting an early dcSSc population, Dr. Khanna said.
The finding with respect to the primary outcome is consistent with other recent trials because of improvement in mRSS that’s part of the natural history of the disease, including the tocilizumab findings that he reported at the meeting. The findings with respect to secondary endpoints and safety show promise.
“Stay tuned for robust ongoing work on the relationship between clinical changes and ongoing mechanistic work,” he said.
Riociguat (Adempas)
Similarly, in the randomized, placebo-controlled phase 2b RISE-SSc study comparing riociguat and placebo for early dcSSc, the primary efficacy endpoint of mean change in mRSS did not reach statistical significance, but exploratory data suggested that the soluble guanylate cyclase stimulator prevented disease progression in patients with early dcSSc, reported Oliver Distler, MD, head of the connective tissue diseases program at University Hospital Zurich (Switzerland).
The mean mRSS at baseline was comparable in 60 patients randomized to receive riociguat and 61 in the placebo group (16.8 and 16.71, respectively). These mean values at week 52 dropped to 14.63 vs. 15.73, respectively (P = .08).
“So it was close, but it didn’t reach significance,” he said.
The difference in the mRSS progression rate, however, suggested significant effects favoring riociguat (descriptive P = .02), he said.
Further, mean change from baseline to week 52 in percent predicted FVC was not different overall between the groups, but a large difference favoring riociguat was seen among patients with scleroderma interstitial lung disease at baseline (mean change of –2.7 vs. –8.9), he said.
No differences were seen between the groups in HAQ-DI or patient and physician global assessment. The proportion of patients with probability of improvement at 52 weeks as measured using ACR CRISS was also the same at 18% in both treatment arms, he noted, ”but the CRISS is designed more for assessing disease regression than for assessing prevention of progression.”
Treatment was, however, well tolerated. At week 52, fewer serious adverse events occurred with riociguat group than in the placebo group (15% vs. 25%, respectively), and no new safety signals were observed, he said.
Riociguat has previously shown antifibrotic effects in animal models and efficacy in patients with pulmonary arterial hypertension associated with connective tissue disease, so it was hypothesized that patients with dcSSc might benefit from riociguat therapy, Dr. Distler explained.
Study subjects had very early dcSSc (duration of 18 months or less; mean of 9 months), mRSS of 10-22 units, FVC of 45% predicted or greater, and diffusion capacity of the lung for carbon monoxide of at least 40% of predicted at screening.
Riociguat was given at an individually adjusted dose between 0.5 mg and 2.5 mg three times daily.
The findings demonstrate a numeric decrease in mRSS over time with riociguat versus placebo and a prevention of progression with riociguat; the failure to reach the primary endpoint may be related to the small study size and the higher than expected regression rate in the placebo group, Dr. Distler said.
Dr. Khanna is a consultant to Roche/Genentech and Bayer, which markets riociguat, and other companies. He has received research grants from Bayer, Bristol-Myers Squibb (which markets abatacept), and Pfizer. The ASSET trial he presented was sponsored by an National Institutes of Health/National Institute of Allergy and Infectious Diseases Clinical ACE grant and an investigator-initiated grant by Bristol-Myers Squibb. Dr. Distler has a consultancy relationship and/or has received research funding from Bayer, Roche/Genentech, and other companies. In addition, he has a patent on mir-29 for the treatment of systemic sclerosis.
SOURCES: Khanna D et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 898 and Abstract 900; Distler O et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 903.
CHICAGO – Recent randomized, placebo-controlled, phase 3 trials of tocilizumab, abatacept, and riociguat for the treatment of systemic sclerosis each failed to reach its primary endpoint of change from baseline in modified Rodnan Skin Score (mRSS).
Still, findings with respect to secondary endpoints and certain exploratory outcomes suggest each of the agents holds some promise in the systemic sclerosis (SSc) arena, according to the data presented at the annual meeting of the American College of Rheumatology.
Tocilizumab (Actemra)
In the double-blind portion of the phase 3 focuSSced trial of 212 patients with SSc, numerical improvement was observed for the primary endpoint of mean change in mRSS from baseline to week 48 with tocilizumab versus placebo (–6.14 vs. –4.41 points, respectively). The change in the treatment group was comparable with what was seen in the phase 2 faSScinate trial, but the decline in mRSS in the placebo group was much greater in phase 3 than in phase 2, and so the difference between the groups in the current study failed to reach statistical significance (P = .098), reported Dinesh Khanna, MBBS, a professor of medicine and director of the scleroderma program at the University of Michigan, Ann Arbor.
The interleukin-6 (IL-6) receptor–alpha antibody was previously shown in the faSScinate trial to lead to numeric improvements in skin thickening as measured by the mRSS, as well as to clinically meaningful lung function preservation as measured by percent predicted forced vital capacity (FVC).
In the current phase 3 study, key secondary end points also appeared to favor tocilizumab, but since the primary endpoint for mRSS was not met, all other P values cannot be considered statistically significant despite the strength of the evidence and were reported for informational purposes only, he noted.
The median cumulative distribution of change from baseline to week 48 in percent predicted FVC with tocilizumab versus placebo was –0.6 vs. –3.9, respectively (descriptive P = .0015), and the mean change from baseline in FVC at week 48 was –24 mL vs. –190 mL (difference of 167 mL in favor of tocilizumab; descriptive P = .0001).
Time to treatment failure also favored tocilizumab, he said (hazard ratio, 0.63; descriptive P = .082), he said.
Patients were randomly assigned to receive either weekly 162-mg injections of subcutaneous tocilizumab or placebo for 48 weeks. Escape therapy was allowed beginning at week 16 if patients experienced declines in FVC or beginning at week 24 if they experienced worsened mRSS or worsened SSc complications, Dr. Khanna said.
“The key part is that no immunotherapy was allowed. ... So it’s a true randomized, placebo-controlled trial,” he said.
Most (81%) of the patients were women, and they had a mean age of 48 years, mean SSc duration of 23 months, mean mRSS of 20.4 units on a 0-51 scale, and a normal mean percent predicted FVC of 82.1%.
“HAQ-DI showed moderate disability of 1.2,” he noted.
Safety in the study was consistent with that seen in prior tocilizumab studies; no new safety signals were identified. Serious adverse events occurred in 13% and 17% of tocilizumab and placebo group patients , respectively, and serious infections were reported by 7% and 2%.
Although clinically meaningful and consistent differences in FVC favoring tocilizumab were shown in this study, the primary endpoint was not met, Dr. Khanna said.
“There were no statistically significant differences, largely driven by unexpected improvement in the placebo group, which was different than what we found in [the faSScinate] trial,” he said, noting, however, that the FVC findings in the current study were clinically meaningful.
Also, in a separate presentation at the meeting, he explained that the differences favoring tocilizumab were statistically significant when patient-level data from the trial were analyzed based on the ACR Composite Response Index in Systemic Sclerosis (CRISS). Those findings provide validation of the novel outcomes measure, he said.
Abatacept (Orencia)
Dr. Khanna also reported results of the 12-month, double-blind, randomized, placebo-controlled phase 2 ASSET trial of abatacept, which showed no significant difference in mRSS in patients with early diffuse cutaneous SSc (dfSSc) who were treated with 125 mg of the recombinant fusion protein weekly and those who received placebo. However, certain secondary outcomes favored abatacept. No concomitant immunotherapy was allowed.
The adjusted mean decrease in the mRSS among patients who completed the 12-month treatment period was –6.24 vs. –4.49 in 34 patients in the abatacept group and 35 in the placebo group, respectively (P = .28).
The secondary outcome measures of mean change in Health Assessment Questionnaire Disability Index (HAQ-DI), patients global assessment, physician global assessment, and ACR CRISS scores were statistically significant or showed numerical results favoring abatacept over placebo: mean decrease in HAQ-DI, –0.17 vs. –0.11 (P = .05), respectively; mean change in physician global assessment scores, –1.30 vs. –0.35 (P = .03); median ACR CRISS index, 0.68 vs. 0.01 (P = .03), decline in percent predicted FVC of 4.13% and 1.34% (P = .11).
Escape therapy was allowed at 6 months for worsening SSc, but it did not change the outcomes trajectory, he said. A larger proportion of placebo vs. abatacept subjects required escape immunosuppressive therapy (36% vs. 16%; P = .03).
Patients were enrolled between 2014 and 2018 at 27 U.S., Canadian, and U.K. sites. At baseline, participants had a mean age of 49 years, 75% were women, and mean disease duration was very short at 1.59 years, with 60% having disease duration of 18 months or less. The mean baseline mRSS was 22.4, mean percent predicted FVC was 85.3%, and mean HAQ-DI was 1.0.
Compliance with both treatments was greater than 98%. Abatacept was well tolerated with comparable adverse events (AEs), serious AEs, and AEs of special interest such as infections and malignancies between treatments, Dr. Khanna said, noting that two deaths occurred in the abatacept group (caused by scleroderma renal crisis in both cases at days 11 and 46) and one occurred in a placebo group patient who experienced sudden cardiac arrest at day 310.
Of note, mRSS showed large variability, despite recruiting an early dcSSc population, Dr. Khanna said.
The finding with respect to the primary outcome is consistent with other recent trials because of improvement in mRSS that’s part of the natural history of the disease, including the tocilizumab findings that he reported at the meeting. The findings with respect to secondary endpoints and safety show promise.
“Stay tuned for robust ongoing work on the relationship between clinical changes and ongoing mechanistic work,” he said.
Riociguat (Adempas)
Similarly, in the randomized, placebo-controlled phase 2b RISE-SSc study comparing riociguat and placebo for early dcSSc, the primary efficacy endpoint of mean change in mRSS did not reach statistical significance, but exploratory data suggested that the soluble guanylate cyclase stimulator prevented disease progression in patients with early dcSSc, reported Oliver Distler, MD, head of the connective tissue diseases program at University Hospital Zurich (Switzerland).
The mean mRSS at baseline was comparable in 60 patients randomized to receive riociguat and 61 in the placebo group (16.8 and 16.71, respectively). These mean values at week 52 dropped to 14.63 vs. 15.73, respectively (P = .08).
“So it was close, but it didn’t reach significance,” he said.
The difference in the mRSS progression rate, however, suggested significant effects favoring riociguat (descriptive P = .02), he said.
Further, mean change from baseline to week 52 in percent predicted FVC was not different overall between the groups, but a large difference favoring riociguat was seen among patients with scleroderma interstitial lung disease at baseline (mean change of –2.7 vs. –8.9), he said.
No differences were seen between the groups in HAQ-DI or patient and physician global assessment. The proportion of patients with probability of improvement at 52 weeks as measured using ACR CRISS was also the same at 18% in both treatment arms, he noted, ”but the CRISS is designed more for assessing disease regression than for assessing prevention of progression.”
Treatment was, however, well tolerated. At week 52, fewer serious adverse events occurred with riociguat group than in the placebo group (15% vs. 25%, respectively), and no new safety signals were observed, he said.
Riociguat has previously shown antifibrotic effects in animal models and efficacy in patients with pulmonary arterial hypertension associated with connective tissue disease, so it was hypothesized that patients with dcSSc might benefit from riociguat therapy, Dr. Distler explained.
Study subjects had very early dcSSc (duration of 18 months or less; mean of 9 months), mRSS of 10-22 units, FVC of 45% predicted or greater, and diffusion capacity of the lung for carbon monoxide of at least 40% of predicted at screening.
Riociguat was given at an individually adjusted dose between 0.5 mg and 2.5 mg three times daily.
The findings demonstrate a numeric decrease in mRSS over time with riociguat versus placebo and a prevention of progression with riociguat; the failure to reach the primary endpoint may be related to the small study size and the higher than expected regression rate in the placebo group, Dr. Distler said.
Dr. Khanna is a consultant to Roche/Genentech and Bayer, which markets riociguat, and other companies. He has received research grants from Bayer, Bristol-Myers Squibb (which markets abatacept), and Pfizer. The ASSET trial he presented was sponsored by an National Institutes of Health/National Institute of Allergy and Infectious Diseases Clinical ACE grant and an investigator-initiated grant by Bristol-Myers Squibb. Dr. Distler has a consultancy relationship and/or has received research funding from Bayer, Roche/Genentech, and other companies. In addition, he has a patent on mir-29 for the treatment of systemic sclerosis.
SOURCES: Khanna D et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 898 and Abstract 900; Distler O et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 903.
REPORTING FROM THE ACR ANNUAL MEETING