User login
Pretreatment ECG unwarranted for most infantile hemangioma patients starting propranolol
reported Emily B. Lund, MD, of the University of Chicago, and her associates.
This finding supports previously published studies that pretreatment ECG is not necessary, despite consensus guidelines published in 2013 that recommend ECG screening of high-risk infants presenting with below-normal heart rate, arrhythmia, or family history of either arrhythmia or congenital heart disease.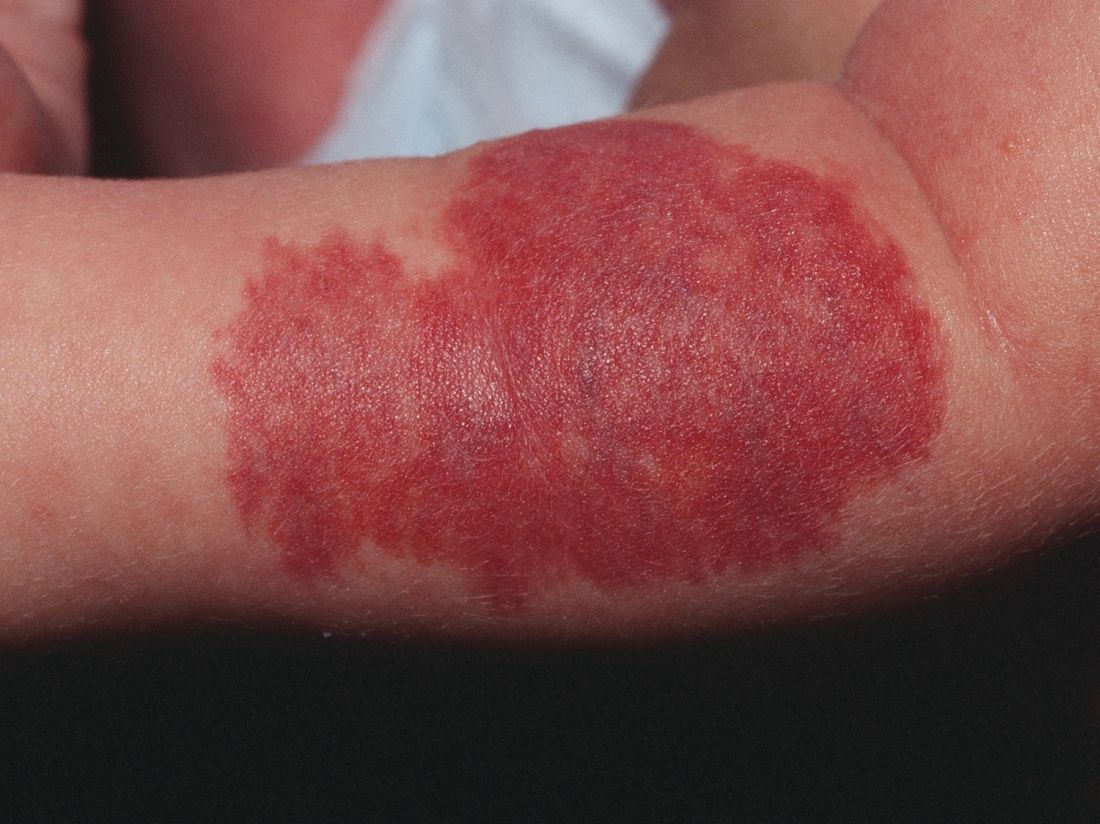
Among the 6% of patients included in the study who had a positive personal cardiac history, congenital heart disease was most common; coronary artery disease was most prevalent among the 41% with a positive family cardiac history. Baseline vital signs revealed no hypotension or bradycardia.
All patients prescribed propranolol were routinely screened with ECG prior to therapy during the study period. Baseline heart rate and blood pressure were observed for abnormalities; patients also were observed during follow-up for possible propranolol side effects.
A total of 43% of ECG screenings performed were found to be abnormal; left ventricular hypertrophy was the most common abnormality. Despite further cardiac evaluation of all but one patient with abnormal ECG, no contraindications to treatment were identified, Dr. Lund and her colleagues reported in Pediatric Dermatology.
Ultimately, 96% of patients observed started treatment with propranolol; of the remaining 4% who did not, the authors cited parental preference and lack of follow-up as the primary reasons for nontreatment.
The researchers found no association between reported side effects and abnormal ECG, a positive personal history of cardiac problems, or a positive family history of cardiac problems.
Dr. Lund and her associates suggested that future revision of the guidelines should emphasize the absence of significant positive predictive value of ECG abnormalities for treatment-related side effects.
The researchers reported no relevant financial disclosures.
SOURCE: Lund EB et al. Ped Dermatol. 2018. doi: 10.1111/pde.13508.
reported Emily B. Lund, MD, of the University of Chicago, and her associates.
This finding supports previously published studies that pretreatment ECG is not necessary, despite consensus guidelines published in 2013 that recommend ECG screening of high-risk infants presenting with below-normal heart rate, arrhythmia, or family history of either arrhythmia or congenital heart disease.
Among the 6% of patients included in the study who had a positive personal cardiac history, congenital heart disease was most common; coronary artery disease was most prevalent among the 41% with a positive family cardiac history. Baseline vital signs revealed no hypotension or bradycardia.
All patients prescribed propranolol were routinely screened with ECG prior to therapy during the study period. Baseline heart rate and blood pressure were observed for abnormalities; patients also were observed during follow-up for possible propranolol side effects.
A total of 43% of ECG screenings performed were found to be abnormal; left ventricular hypertrophy was the most common abnormality. Despite further cardiac evaluation of all but one patient with abnormal ECG, no contraindications to treatment were identified, Dr. Lund and her colleagues reported in Pediatric Dermatology.
Ultimately, 96% of patients observed started treatment with propranolol; of the remaining 4% who did not, the authors cited parental preference and lack of follow-up as the primary reasons for nontreatment.
The researchers found no association between reported side effects and abnormal ECG, a positive personal history of cardiac problems, or a positive family history of cardiac problems.
Dr. Lund and her associates suggested that future revision of the guidelines should emphasize the absence of significant positive predictive value of ECG abnormalities for treatment-related side effects.
The researchers reported no relevant financial disclosures.
SOURCE: Lund EB et al. Ped Dermatol. 2018. doi: 10.1111/pde.13508.
reported Emily B. Lund, MD, of the University of Chicago, and her associates.
This finding supports previously published studies that pretreatment ECG is not necessary, despite consensus guidelines published in 2013 that recommend ECG screening of high-risk infants presenting with below-normal heart rate, arrhythmia, or family history of either arrhythmia or congenital heart disease.
Among the 6% of patients included in the study who had a positive personal cardiac history, congenital heart disease was most common; coronary artery disease was most prevalent among the 41% with a positive family cardiac history. Baseline vital signs revealed no hypotension or bradycardia.
All patients prescribed propranolol were routinely screened with ECG prior to therapy during the study period. Baseline heart rate and blood pressure were observed for abnormalities; patients also were observed during follow-up for possible propranolol side effects.
A total of 43% of ECG screenings performed were found to be abnormal; left ventricular hypertrophy was the most common abnormality. Despite further cardiac evaluation of all but one patient with abnormal ECG, no contraindications to treatment were identified, Dr. Lund and her colleagues reported in Pediatric Dermatology.
Ultimately, 96% of patients observed started treatment with propranolol; of the remaining 4% who did not, the authors cited parental preference and lack of follow-up as the primary reasons for nontreatment.
The researchers found no association between reported side effects and abnormal ECG, a positive personal history of cardiac problems, or a positive family history of cardiac problems.
Dr. Lund and her associates suggested that future revision of the guidelines should emphasize the absence of significant positive predictive value of ECG abnormalities for treatment-related side effects.
The researchers reported no relevant financial disclosures.
SOURCE: Lund EB et al. Ped Dermatol. 2018. doi: 10.1111/pde.13508.
FROM PEDIATRIC DERMATOLOGY
Key clinical point: There was no association between side effects, abnormal ECG, and personal or family history of cardiac problems in children with infantile hemangioma who underwent propranolol therapy.
Major finding: Despite the fact that 43% of ECG screenings were abnormal, 96% of patients started treatment with propranolol, with no side effects related to abnormal ECG or to personal or family history of cardiac problems.
Study details: A retrospective chart review of 272 patients with infantile hemangioma.
Disclosures: The researchers reported no relevant financial disclosures.
Source: Lund EB et al. Ped Dermatol. 2018. doi: 10.1111/pde.13508.
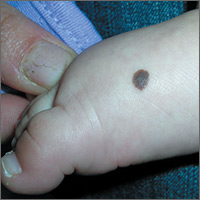
It's Just a Growth Spurt
A 38-year-old Latino man self-refers to dermatology for evaluation of a mass on his back that first appeared three years ago. Since then, it has grown steadily. There is no pain or discomfort associated with the lesion, and the patient claims to be quite healthy otherwise. There is no antecedent history for the affected area.
EXAMINATION
There is a subcutaneous, rubbery mass in the left infrascapular area. It measures 11 x 6 cm. Palpation reveals the lesion to be uniformly smooth and readily mobile. The overlying skin is free of abnormalities and increased warmth.

What is the diagnosis?
DISCUSSION
Lipomas are by far the most common soft-tissue tumor to affect humans and are totally benign. They typically measure 2 to 3 cm in diameter, but as this case demonstrates, they can grow much larger. While the rate of growth in this case was unusual, the location—and other features—are typical.
Lipomas are actual tumors, composed completely of adipose tissue contained in a thin, fragile, membranous capsule. Their tendency to develop can be hereditary, though most are spontaneous. They can manifest internally as well.
Superficial lipomas, which often manifest as multiple lesions on the arms and trunk, are usually easy to remove surgically. Lesions that are deeper and older or that appear on the face, however, often require considerable dissection to be freed from surrounding tissue. When excision is attempted, it is essential to remove the entire lesion to prevent recurrence. And, as always, the specimen must be sent for pathologic examination.
Patients often decide against surgery once they understand the issues. This is acceptable, but any deviation from the norm—such as pain, irregular surface texture, change in overlying skin, lack of mobility, or rapid growth—would constitute reasonable grounds for excision.
This man’s lesion likely extended down to the muscle fascia if not into the muscle itself. As a result, surgery would require general anesthesia and placement of a drain in the inferior portion of the wound, since such a large defect would likely invite a collection of blood and serum. For these reasons, he was referred to a general surgeon.
The differential for lipoma includes liposarcoma and angiolipoma. The latter are common and benign but become painful and are often more firm than normal. Histologically, they’re often indistinguishable from ordinary lipomas. Liposarcomas, when superficial, can imitate ordinary lipomas, but their surfaces tend to be more irregular and firm and the lesions themselves less mobile.
TAKE-HOME LEARNING POINTS
- Lipomas are the most common soft-tissue tumor encountered in outpatient practices.
- While the vast majority are benign and easy to remove surgically, most lipomas can be safely left alone.
- When excision is attempted, the entire lesion must be removed lest it regrow.
- Patients with larger, deeper lesions, or those in busy anatomical areas, should be referred to a general surgeon.
A 38-year-old Latino man self-refers to dermatology for evaluation of a mass on his back that first appeared three years ago. Since then, it has grown steadily. There is no pain or discomfort associated with the lesion, and the patient claims to be quite healthy otherwise. There is no antecedent history for the affected area.
EXAMINATION
There is a subcutaneous, rubbery mass in the left infrascapular area. It measures 11 x 6 cm. Palpation reveals the lesion to be uniformly smooth and readily mobile. The overlying skin is free of abnormalities and increased warmth.
What is the diagnosis?
DISCUSSION
Lipomas are by far the most common soft-tissue tumor to affect humans and are totally benign. They typically measure 2 to 3 cm in diameter, but as this case demonstrates, they can grow much larger. While the rate of growth in this case was unusual, the location—and other features—are typical.
Lipomas are actual tumors, composed completely of adipose tissue contained in a thin, fragile, membranous capsule. Their tendency to develop can be hereditary, though most are spontaneous. They can manifest internally as well.
Superficial lipomas, which often manifest as multiple lesions on the arms and trunk, are usually easy to remove surgically. Lesions that are deeper and older or that appear on the face, however, often require considerable dissection to be freed from surrounding tissue. When excision is attempted, it is essential to remove the entire lesion to prevent recurrence. And, as always, the specimen must be sent for pathologic examination.
Patients often decide against surgery once they understand the issues. This is acceptable, but any deviation from the norm—such as pain, irregular surface texture, change in overlying skin, lack of mobility, or rapid growth—would constitute reasonable grounds for excision.
This man’s lesion likely extended down to the muscle fascia if not into the muscle itself. As a result, surgery would require general anesthesia and placement of a drain in the inferior portion of the wound, since such a large defect would likely invite a collection of blood and serum. For these reasons, he was referred to a general surgeon.
The differential for lipoma includes liposarcoma and angiolipoma. The latter are common and benign but become painful and are often more firm than normal. Histologically, they’re often indistinguishable from ordinary lipomas. Liposarcomas, when superficial, can imitate ordinary lipomas, but their surfaces tend to be more irregular and firm and the lesions themselves less mobile.
TAKE-HOME LEARNING POINTS
- Lipomas are the most common soft-tissue tumor encountered in outpatient practices.
- While the vast majority are benign and easy to remove surgically, most lipomas can be safely left alone.
- When excision is attempted, the entire lesion must be removed lest it regrow.
- Patients with larger, deeper lesions, or those in busy anatomical areas, should be referred to a general surgeon.
A 38-year-old Latino man self-refers to dermatology for evaluation of a mass on his back that first appeared three years ago. Since then, it has grown steadily. There is no pain or discomfort associated with the lesion, and the patient claims to be quite healthy otherwise. There is no antecedent history for the affected area.
EXAMINATION
There is a subcutaneous, rubbery mass in the left infrascapular area. It measures 11 x 6 cm. Palpation reveals the lesion to be uniformly smooth and readily mobile. The overlying skin is free of abnormalities and increased warmth.
What is the diagnosis?
DISCUSSION
Lipomas are by far the most common soft-tissue tumor to affect humans and are totally benign. They typically measure 2 to 3 cm in diameter, but as this case demonstrates, they can grow much larger. While the rate of growth in this case was unusual, the location—and other features—are typical.
Lipomas are actual tumors, composed completely of adipose tissue contained in a thin, fragile, membranous capsule. Their tendency to develop can be hereditary, though most are spontaneous. They can manifest internally as well.
Superficial lipomas, which often manifest as multiple lesions on the arms and trunk, are usually easy to remove surgically. Lesions that are deeper and older or that appear on the face, however, often require considerable dissection to be freed from surrounding tissue. When excision is attempted, it is essential to remove the entire lesion to prevent recurrence. And, as always, the specimen must be sent for pathologic examination.
Patients often decide against surgery once they understand the issues. This is acceptable, but any deviation from the norm—such as pain, irregular surface texture, change in overlying skin, lack of mobility, or rapid growth—would constitute reasonable grounds for excision.
This man’s lesion likely extended down to the muscle fascia if not into the muscle itself. As a result, surgery would require general anesthesia and placement of a drain in the inferior portion of the wound, since such a large defect would likely invite a collection of blood and serum. For these reasons, he was referred to a general surgeon.
The differential for lipoma includes liposarcoma and angiolipoma. The latter are common and benign but become painful and are often more firm than normal. Histologically, they’re often indistinguishable from ordinary lipomas. Liposarcomas, when superficial, can imitate ordinary lipomas, but their surfaces tend to be more irregular and firm and the lesions themselves less mobile.
TAKE-HOME LEARNING POINTS
- Lipomas are the most common soft-tissue tumor encountered in outpatient practices.
- While the vast majority are benign and easy to remove surgically, most lipomas can be safely left alone.
- When excision is attempted, the entire lesion must be removed lest it regrow.
- Patients with larger, deeper lesions, or those in busy anatomical areas, should be referred to a general surgeon.
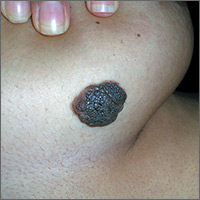
Growing mole on breast
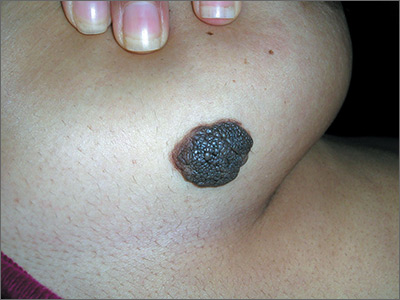
The FP recognized this lesion as a congenital nevus.
He was aware that nevi might become more raised during early adulthood, though this was not necessarily a sign of malignant degeneration. He looked at the nevus carefully and saw that it was relatively symmetrical with one predominant color and a light brown coloration on the left edge. (The patient stated it had always been this way.) The surface texture, which could be described as mamillated, was not unusual for congenital nevi. The FP examined the nevus using a dermatoscope and did not see any melanoma-specific structures.
The FP encouraged the patient to monitor the nevus and return for further evaluation if there were any changes or symptoms. He also offered her the option of a biopsy, but stated that it was not medically required. The patient noted that the changes of increased height of the congenital nevus had been very slow over the past 2 years, and she was willing to keep an eye on it. The patient returned in 6 months, and there were no visible changes to the congenital nevus.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Smith M. Congenital nevi. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:953-957.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/.
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com.
The FP recognized this lesion as a congenital nevus.
He was aware that nevi might become more raised during early adulthood, though this was not necessarily a sign of malignant degeneration. He looked at the nevus carefully and saw that it was relatively symmetrical with one predominant color and a light brown coloration on the left edge. (The patient stated it had always been this way.) The surface texture, which could be described as mamillated, was not unusual for congenital nevi. The FP examined the nevus using a dermatoscope and did not see any melanoma-specific structures.
The FP encouraged the patient to monitor the nevus and return for further evaluation if there were any changes or symptoms. He also offered her the option of a biopsy, but stated that it was not medically required. The patient noted that the changes of increased height of the congenital nevus had been very slow over the past 2 years, and she was willing to keep an eye on it. The patient returned in 6 months, and there were no visible changes to the congenital nevus.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Smith M. Congenital nevi. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:953-957.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/.
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com.
The FP recognized this lesion as a congenital nevus.
He was aware that nevi might become more raised during early adulthood, though this was not necessarily a sign of malignant degeneration. He looked at the nevus carefully and saw that it was relatively symmetrical with one predominant color and a light brown coloration on the left edge. (The patient stated it had always been this way.) The surface texture, which could be described as mamillated, was not unusual for congenital nevi. The FP examined the nevus using a dermatoscope and did not see any melanoma-specific structures.
The FP encouraged the patient to monitor the nevus and return for further evaluation if there were any changes or symptoms. He also offered her the option of a biopsy, but stated that it was not medically required. The patient noted that the changes of increased height of the congenital nevus had been very slow over the past 2 years, and she was willing to keep an eye on it. The patient returned in 6 months, and there were no visible changes to the congenital nevus.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Smith M. Congenital nevi. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:953-957.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/.
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com.
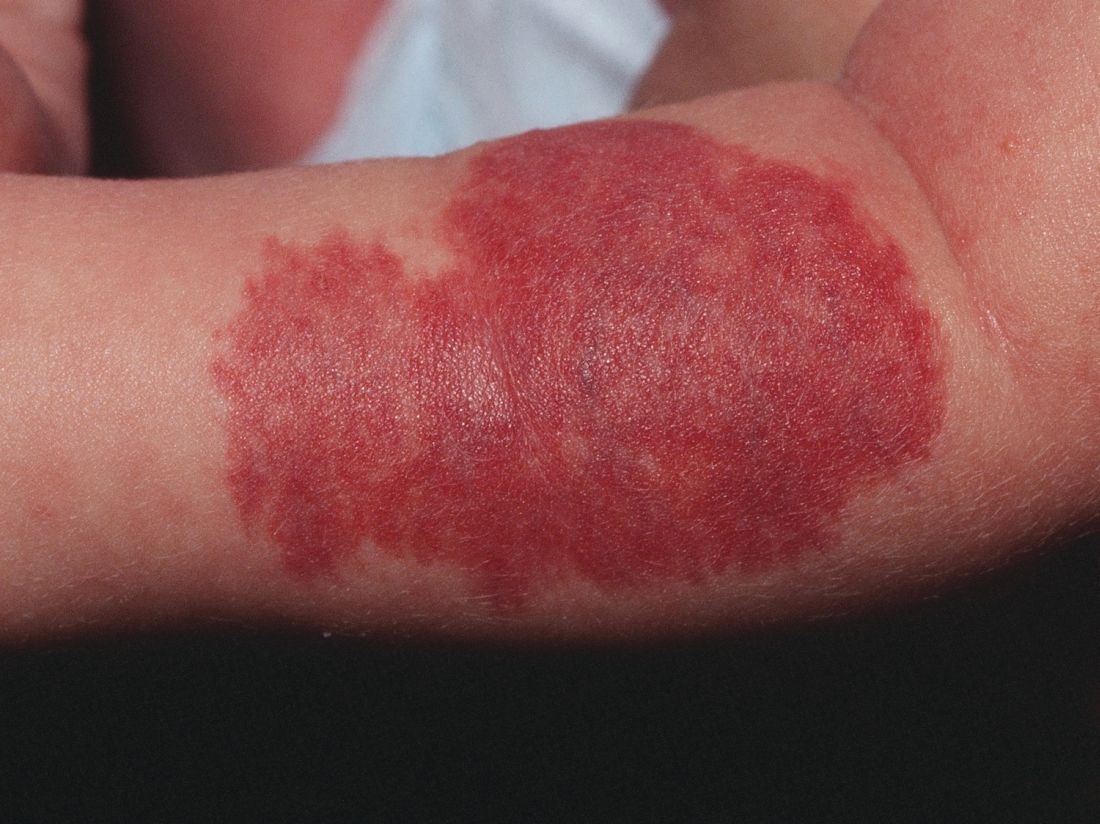
Unusual skin reactions to aluminum patch test seen in some children
Instead of the usual eczematous reaction confined to the test area, they exhibited erythema and papules distant from the test area.
In one girl, the reaction consisted of erythematous itchy papules across much of her back on day 2, and by day 4, there were even papules on her chest. In the other girl, erythema, infiltration, and occasional papules covered the whole patch area, and erythematous papules also were seen at some distance from the test area, Nadia Raison-Peyron, MD, of Saint-Eloi Hospital, Montpellier, France, and her colleagues, reported in Pediatric Dermatology.
Biopsies were not performed because of the girls’ young age.
SOURCE: Raison-Peyron N et al. Pediatr Dermatol. 2018. doi: 10.1111/pde.13481.
Instead of the usual eczematous reaction confined to the test area, they exhibited erythema and papules distant from the test area.
In one girl, the reaction consisted of erythematous itchy papules across much of her back on day 2, and by day 4, there were even papules on her chest. In the other girl, erythema, infiltration, and occasional papules covered the whole patch area, and erythematous papules also were seen at some distance from the test area, Nadia Raison-Peyron, MD, of Saint-Eloi Hospital, Montpellier, France, and her colleagues, reported in Pediatric Dermatology.
Biopsies were not performed because of the girls’ young age.
SOURCE: Raison-Peyron N et al. Pediatr Dermatol. 2018. doi: 10.1111/pde.13481.
Instead of the usual eczematous reaction confined to the test area, they exhibited erythema and papules distant from the test area.
In one girl, the reaction consisted of erythematous itchy papules across much of her back on day 2, and by day 4, there were even papules on her chest. In the other girl, erythema, infiltration, and occasional papules covered the whole patch area, and erythematous papules also were seen at some distance from the test area, Nadia Raison-Peyron, MD, of Saint-Eloi Hospital, Montpellier, France, and her colleagues, reported in Pediatric Dermatology.
Biopsies were not performed because of the girls’ young age.
SOURCE: Raison-Peyron N et al. Pediatr Dermatol. 2018. doi: 10.1111/pde.13481.
FROM PEDIATRIC DERMATOLOGY
The Clinical Pathophysiology of Chronic Systemic Sclerosis
Systemic sclerosis (SSc), also called scleroderma, is a rare but serious autoimmune connective tissue disease that has multiple fluctuating pathologic manifestations throughout its temporal course. Estimates have shown that the incidence is 10 to 20 cases per 1 million, and the prevalence is 4 to 253 cases per 1 million.1,2 Given the rarity of this incurable condition, it is vital that primary care providers (PCPs) are able to recognize its unique features early to limit and prevent acute and chronic complications. This case report discusses a patient’s journey with late-diagnosed scleroderma in order to convey these broad manifestations and what providers can do to manage it with their patients.
Case Presentation
Mr. P is a 60-year-old African American male with a history of hypertension, recurrent digital ulcers, pulmonary hypertension (PH), interstitial lung disease (ILD), kidney involvement, congestive heart failure (CHF), and gastroesophageal reflux disease (GERD). Mr. P’s workup began in his late 40s with resistant hypertension, resistant GERD, and multiple hospitalizations for hypertensive urgency. It was not until he was 54 years old that he was diagnosed with mixed connective tissue disorder with sclerodermatous predominance.
Review of systems throughout his medical examinations in his 50s were notable for skin tightening over his hands and shoulders, skin hypopigmentation over his scalp and face, and hair loss. Mr. P was found to have Raynaud phenomenon beginning with his original presentation and digital ulceration without complications of gangrene or autoamputation. Aggregate physical examinations were notable for digital ulceration, skin tightening/sclerodactyly, and telangiectasia. Serologic markers were notable for the following:
- Positive ANA (antinuclear antibody) with titer of 1:1,280/homogenous pattern;
- Positive anti-RNP (antiribonucleoprotein) with titer of 171.2;
- Positive anti-Scl-70 (antitopoisomerase I) with titer of 108.1;
- Positive anti-SM (anti-Smith antibody) with titer of 30.2;
- Positive anti-Ro (SSA) with titer of 107.6;
- Negative anti-La (SSB) with titer of 1.3;
- Negative anti-dsDNA (anti-double stranded DNA) with titer of 9; and
- Negative ACA (anticentromere antibody).
Early transthoracic echocardiograms revealed an ejection fraction (EF) initially at 55% with evidence of left ventricular hypertrophy. Following treatment with phosphodiesterase-5 inhibitors (PDE-5 inhibitors) and endothelin-1 antagonists for pulmonary hypertension, serial transthoracic echocardiograms showed improvement in his EF. 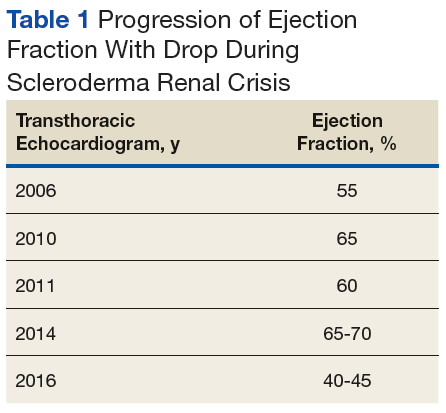
A chest X-ray did not show signs of ILD, but a subsequent high-resolution computed tomography (HRCT) scan was consistent with chronic ILD with a main pulmonary artery diameter of 3 cm (Figure 1). 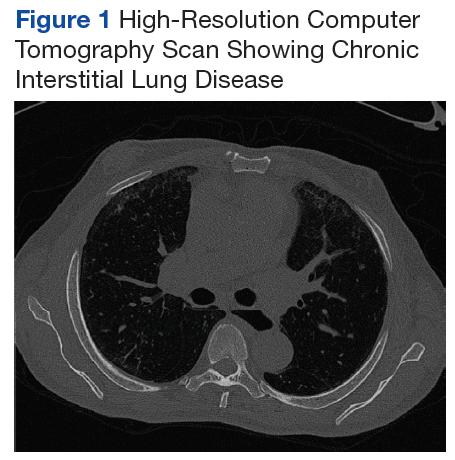
In subsequent years, Mr. P was hospitalized several times, secondary to digit pain, ulcerations, and osteomyelitis. His first episode was 1 year after his scleroderma diagnosis, when he was hospitalized for 6 days for complications of SSc and finger pain. The following year, he had a 3-day hospitalization for hypertensive urgency and right third-digit osteomyelitis, treated initially with IV fluids, levofloxacin, and vancomycin, and then ceftaroline for 1 month. Throughout the next 6 years, Mr. P presented multiple times with fingertip ulcerations and was followed in the Infectious Disease clinic for recurrent osteomyelitis. He found some relief with systemic antibiotics, including augmentin, minocycline, moxifloxacin, and doxycycline.
At age 59, he was hospitalized for scleroderma renal crisis (SRC). Early in his disease, his kidney function was normal, but the SRC was discovered after an abrupt rise in his blood pressure (BP) and an increase in serum creatinine (SCr) from 1.2 mg/dL at baseline to 3.06 mg/dL. The presence of brown granular cast in his urine prompted a renal biopsy that showed thrombotic microangiopathy with schistocytes. Mr. P was started on captopril and remained stable with outpatient follow-up for this renal complication.
Discussion
Initial presentation of SSc can occur along a spectrum of its pathophysiology. A more severe presentation, like the one seen in Mr. P, seems to occur more frequently in African American patients relative to white patients.3 Differentiating between the 2 types—diffuse vs limited SSc—is vital to managing patients and disease progression. Limited-type SSc is more common (60%) and less severe with slower progression than is diffuse SSc. 
Diffuse-type SSc (35%) includes features such as skin thickening and tightening, ILD, SRC, tendon friction rubs (palpable crepitus over tendons), and skin pigment changes.4 The specific involvement of the renal and cardiopulmonary systems accounts for the higher mortality rates in the diffuse-type.5
Many patients with SSc require periodic hospitalizations throughout their life for the acute complications of the disease. Hospitalized patients often range from age 45 to 64 years and are more often female. However, of hospitalized patients with SSc, in-hospital death rates are higher among men.3,6 Although these rates have decreased as the pathogenesis of SSc has become better understood, it is important to note that in-hospital mortality in 1995 for all patients with SSc was 7.1% and mean length of stay was 7.5 days, and in 2002 to 2003, 6.3% and 6.6 days, respectively.3 Though the burden of this disease has decreased, mortality and hospitalizations continue to persist at high rates. Understanding the pathogenesis, progression, and treatments of SSc are essential to aiding patients with this diagnosis.
Skin Involvement
A common finding and presentation for patients with SSc is related to skin involvement. Common patient complaints and exam findings include calcinosis along extensor tendons and digits, Raynaud phenomenon (seen in more than 95% of patients), sclerodactyly, telangiectasias, hyper/hypopigmentation, and pruritus.4 These findings are useful in diagnosing and monitoring patients for disease progression.
Many of the listed skin manifestations affect patients’ quality of life (QOL) but are not directly associated with mortality. However, a common and feared complication includes skin ulcers and osteomyelitis, seen in 48% and 7.7% of patients, respectively.7 Digit ulcers, areas with loss of dermis and epidermis distal to the proximal interphalangeal joints (Figures 2A and 2B), are significant because they parallel a more rapid progression of internal organ involvement.8 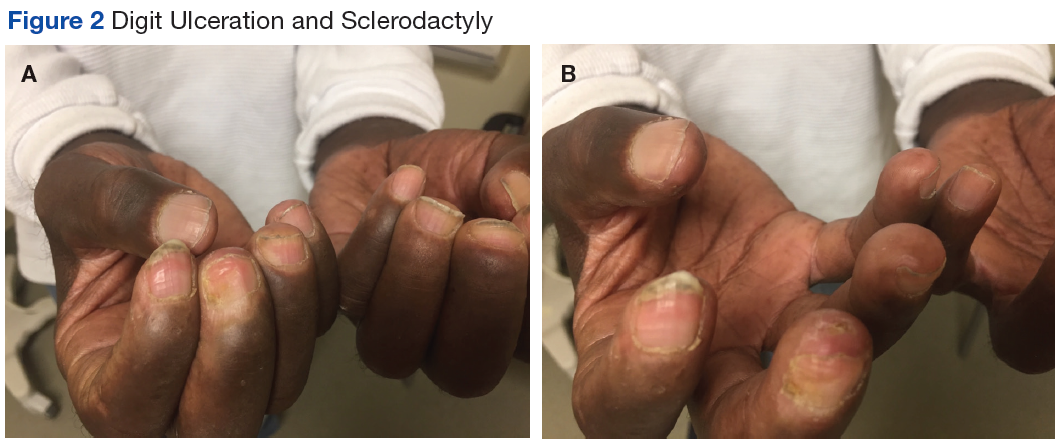
Mr. P required multiple hospitalizations and antibiotic regimens for painful digit ulcers complicated by osteomyelitis (Figure 3). 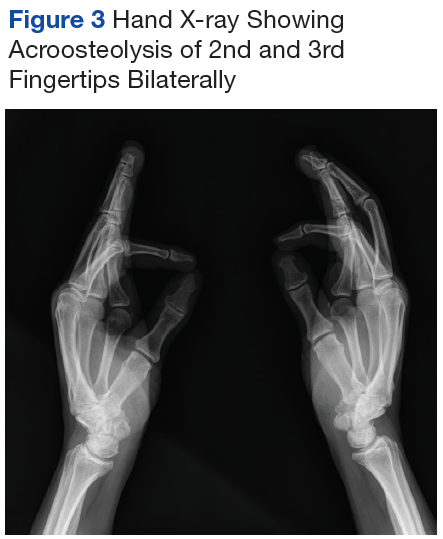
Treatment usually is aimed at infections that complicate these skin ulcers and are based on site-specific cultures. Preventive measures are aimed at the risk factors associated with digit ulcers, including decreased whole-body warmth, direct trauma to digits, smoking, and vasoconstrictors (eg, cocaine, sympathomimetics).8 Some patients may prevent ulcers by using D-penicillamine, mycophenolate mofetil, and cyclophosphamide, although definitive treatment has not been found.4 Calcium channel blockers, PDE-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues also have been used to reduce the severity of Raynaud phenomenon attacks and to decrease the number of digital ulcers (in addition to their beneficial effects on pulmonary involvement).8 Additional pharmacologic agents that have been linked to an improvement in Raynaud phenomenon and digital ulcers include statins, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, intravenous N-acetylcysteine, vitamin E gel, and surgical options (eg, revascularization and sympathectomy).8
Pulmonary Involvement
Systemic sclerosis can lead to 2 complications in the lungs, both present in Mr. P: PAH (mean pulmonary arterial pressure > 25 mm Hg) and ILD, which together make lung involvement the leading cause of death for these patients. Limited-type SSc usually is restricted to PAH, whereas diffuse-type SSc usually leads to ILD.4
On diagnosis of SSc, an HRCT is indicated to assess the degree of lung involvement. Mr. P’s HRCT showed a 3-cm main pulmonary artery, suggestive of PAH, in addition to evidence of ILD seen on the chest X-ray (Figure 4). 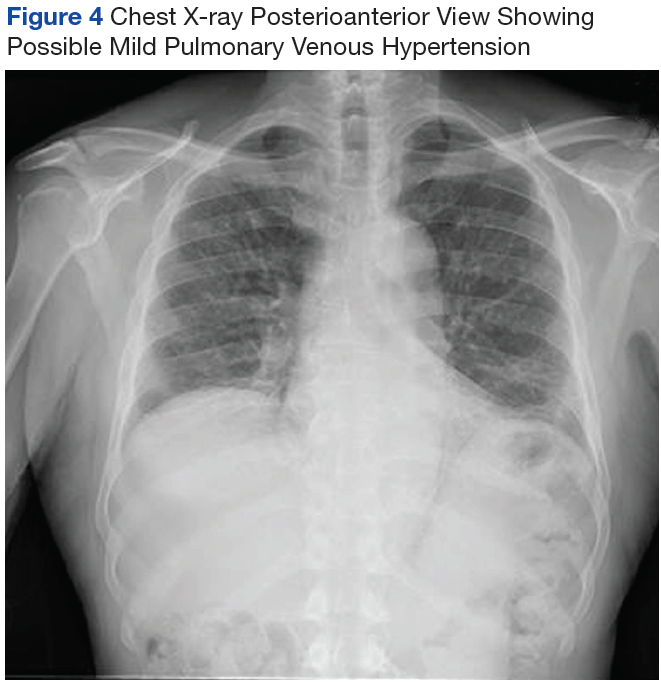
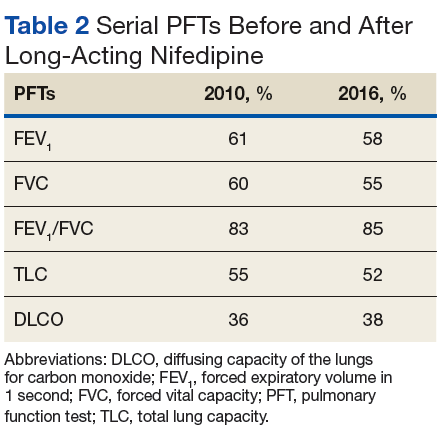
Pulmonary arterial hypertension is a common finding in patients with SSc and carries a severe prognosis. Risk factors for the development of PAH, when not present on initial diagnosis, include limited-type SSc; late age of onset; Raynaud phenomenon; decreased DLCO, FVC/DLCO < 1.6; increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) serum levels; and the presence of antibodies.9 Patients with both SSc and PAH have a 50% to 87% 1-year survival, whereas patients with idiopathic PAH without connective tissue disease have an 88% 1-year survival.9
Many patients with lung involvement are asymptomatic, but some have findings of crackles and interstitial thickening on chest X-ray that can progress to cyanosis and right heart failure (cor pulmonale).10 On discovery of lung involvement (Figure 5), it is important to follow up with a PFT, primarily because the outcomes and prognosis for patients with SSc are correlated with the presenting severity of ILD and the subsequent progression of their DLCO.4,10 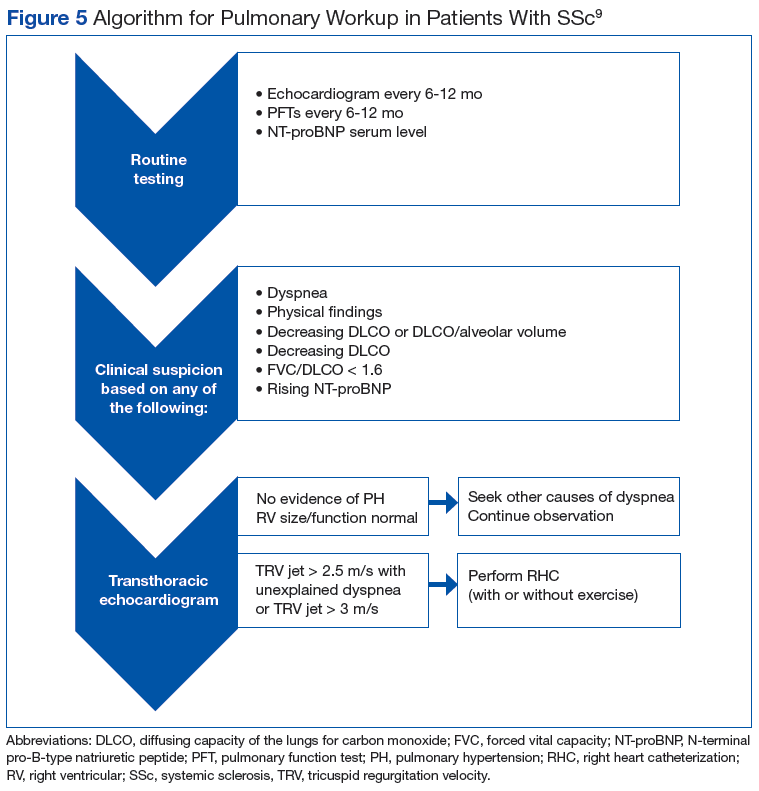
Treatment is directed at PAH and ILD separately. For PAH, a PDE-5 inhibitor, such as tadalafil or sildenafil, and an endothelin-1 receptor antagonist, such as ambrisentan or bosentan, are indicated. Other PAH treatments include diuretics and prostacyclin analogues (eg, epoprostenol, treprostinil, or iloprost) in addition to warfarin if patients have a history of thrombotic events.4 Mr. P’s pulmonologists deferred treatment with prostacyclin analogues given the potential for adverse effects with endothelin-1 receptor inhibitors, PDE-5 inhibitors, and calcium-channel blockers, although combination studies with bosentan and inhaled iloprost have shown promise.11
Like the skin manifestations of SSc, ILD lacks definitive treatment to prevent disease progression. However, some patients benefit from cyclophosphamide, followed by mycophenolate mofetil—which is usually better tolerated—azathioprine, haematopoietic stem cell transplant as rescue therapy, and lung transplant for life-saving treatment.10
Renal Involvement
Renal involvement in patients with SSc can have profound effects on QOL. Even without clinical renal involvement, glomerular filtration rate (GFR) usually decreases with the progression of vascular damage correlated with age and disease duration.12 Patients with a history of digital ulcers, like Mr. P, usually have a lower GFR than that of patients without digital ulcers.12 Monitoring renal function is vital in caring for these patients, because SRC occurs in 2% to 15% of all patients with SSc.13,14
Scleroderma renal crisis is generally defined as an abruptly elevated BP (> 140/90 mm Hgor > 30 mm Hg rise from baseline) with acute renal failure (elevated SCr) and decreased urine output.14 Given the rarity of SSc in the population, diagnosis of SRC requires high clinical suspicion. In the case of Mr. P, a workup involving serum analysis, urinalysis, and renal biopsy allowed for a definitive diagnosis. A renal biopsy can show microangiopathic hemolytic anemia and confirm SRC, although it may not be necessary in patients with known SSc presenting with new hypertension, rising creatinine, and unremarkable urine sediment on microscopy.14,15
Although an acute SRC can be difficult to predict, monitoring renal function and attention to key factors can assist in discovering this SSc complication. Scleroderma renal crisis usually occurs within the first 4 years of SSc diagnosis, often paralleling rapid progression of skin thickening and tightening with higher rates in both African Americans and males.13,14 Additional predictive factors include diffuse skin involvement, rapid progression of skin involvement, positive anti-RNA polymerase III antibodies, new anemia, new cardiac events (eg, pericardial effusion, pericarditis, left ventricular insufficiency), CHF, tendon friction rubs, arthritis, and recent (within 3 months) high-dose glucocorticoid use.4,13-15
The presentation of SRC can be nonspecific, often resembling findings related to acute kidney injury. Patients may report malaise, fatigue, fever, headache, seizure, blurred vision, or dyspnea.13 Clinical parametersto examine include systolic BP > 140 mm Hgand/or diastolic BP > 90 mm Hg (or an abrupt rise of > 30 or > 20, respectively), SCr increase by 50%+ or > 120% of the upper normal limit, proteinuria at 2+, high protein:creatinine ratio, hematuria > 2+ or > 10 red blood cells (RBCs), platelets < 100,000/mm3, and hypertensive encephalopathy.13 Mr. P presented with fatigue, dyspnea, an abrupt rise in BP (156/96 mm Hg), foamy urine, bilateral lower extremity edema (3.9 g/dL albumin), 2+ RBCs, and a SCr of 2.46 mg/dL on admission (a 205% increase from his last baseline of 1.2 mg/dL).
Treatments have had a large impact on the mortality rates of SRC. Following the introduction of ACE inhibitors, mortality from SRC has decreased from 76% to < 10% over recent decades.13 In addition to improving survival, these medications also have improved hospitalization outcomes for these patients.3 Captopril is often the medication of choice given its rapid onset and short duration of action relative to other medications in the same class, such as benazepril, enalapril, fosinopril, lisinopril, quinapril, and ramipril.4,13 Current clinical trials are assessing the specific renal benefits of endothelin-1 receptor antagonists, which often are used for pulmonary and skin involvement.14 For patients presenting with acute SRC and uremia, dialysis may be necessary (typically predicted by elevated NT-proBNP serum levels), while the decision for transplantation may be indicated at least 2 years after SRC onset and resolution.4,13-15
Once the acute crisis resolves, it is important to discuss prognosis with patients. The 1-, 2-, 3-, 5-, and 10-year survival rates are 82%, 74%, 71%, 59%, and 47%, respectively; however, when looking at only male patients at 10 years, the survival rate is 17%.13 Prevention of SRC can be addressed with daily BP checks and advising patients to seek medical care if they notice a consecutive 2-day abrupt rise.13
Cardiac Involvement
Systemic sclerosis has significant effects on patients’ heart function. The introduction of ACE inhibitors has shifted mortality in SSc patients from predominantly SRC to cardiac causes.6 Cardiac involvement can occur through a range of processes, including abnormal cardiac conduction, CHF, diastolic dysfunction, mitral valve nodular thickening, and pericardial effusion.3 Even though patients often present with skin findings, an initial cardiac workup is crucial to understanding disease progression and patient prognosis. The severity of the cutaneous manifestations often predicts the degree of diastolic dysfunction.16
Clinical evidence of cardiac involvement is seen in 20% to 25% of patients with SSc and is associated with a 70% mortality at 5 years when symptoms are evident.16 Additionally, right ventricular dysfunction at presentation is the strongest marker for all-mortality prognosis, representing the degree of pulmonary involvement, and includes findings such as progressive shortness of breath and systemic edema.9
Given the increasing survival of patients with SSc, cardiac involvement is becoming more evident and prominent. Direct treatments for cardiac manifestations are based on the causative feature, namely, focusing on pulmonary and renal involvement, which can be assessed with periodic echocardiograms evaluating left ventricular EF.
Gastrointestinal Involvement
Along with skin manifestations, the gastrointestinal (GI) involvement of SSc can have a significant impact on patients’ QOL without direct contribution to mortality. One of Mr. P’s earliest symptoms that led to a diagnosis of SSc was GERD, which caused a chronic cough, dental erosions, esophageal erosions, duodenal ulcers, dysphagia, abdominal pain, halitosis, pharyngitis, and weight loss. Esophageal involvement occurs in up to 96% of patients with SSc and can include motility abnormalities (eg, strictures and/or muscle dysfunction), lower esophageal sphincter abnormalities, and Barrett esophagus.17
Additional symptoms of the GI system linked with SSc can occur anywhere along the GI tract and include gastric antral vascular ectasia, causing GI bleeds and pernicious anemia, gastroparesis, bacterial overgrowth, intestinal malabsorption, pseudo-obstruction due to hypomotility, fecal incontinence due to anorectal involvement, and rarely, primary biliary cirrhosis.4,17 Decreased mobility of the oral aperture secondary to skin thickening and tightening also can contribute to malnutrition by decreasing oral intake.
Treatments are supportive and target symptom relief. Chronic treatment of GERD is often necessary and includes antacids, histamine-2 receptor blockers, and proton pump inhibitors.4 Other medications that can help with symptom relief include motility agents (such as metoclopramide, domperidone, prucalopride, tegaserod, and macrolides), osmotic laxatives, and ursodeoxycholic acid for primary biliary cirrhosis.17 Surgical intervention should be considered depending on the severity and progression of involvement within the GI tract. Behavioral changes that can improve patient symptoms include facial grimacing and other mouth-stretching exercises, frequent smaller meals followed by maintaining a vertical posture, and high fiber diets.17
Conclusion
Systemic sclerosis is an autoimmune and connective tissue disease with a pathophysiology that can manifest throughout the body. The organ systems that impact patient outcomes include skin, pulmonary, renal, cardiac, and GI. Primary care providers caring for patients diagnosed with SSc should monitor acute management and disease progression in all these systems. Important acute events that can impact morbidity, mortality, and/or QOL include Raynaud phenomenon, SRC, and pericardial effusion. Chronic manifestations that may be present on diagnosis of SSc or may develop while a patient is under a provider’s care include sclerodactyly, tendon calcinosis, PAH, ILD, chronic kidney injury, chronic cardiac damage, GERD, and esophageal dysmotility. While this discussion serves as a pertinent overview of patients with SSc, it is summative, and providers are encouraged to seek a stronger understanding of both the common and rarer manifestations within each of their patients.
1. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778-799.
2. Silman AG. Scleroderma. In: Epidemiology of the Rheumatic Diseases. 2nd ed. Silman AJ, Hochberg MC, eds. Oxford, UK: Oxford University Press. 1993:chap 8.
3. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford). 2007;46(12):1808-1813.
4. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78(8):961-968.
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809-1815.
6. Piga M, Casula L, Sanna S, et al. Population-based analysis of hospitalizations for patients with systemic sclerosis in a West-European region over the period 2001-2012. Rheumatol Int. 2016;36(1):73-81.
7. Giuggioli D, Manfredi A, Colaci M, Lumetti F, Ferri C. Osteomyelitis complicating scleroderma digital ulcers. Clin Rheumatol. 2013;32(5):623-627.
8. Schiopu E, Impens AJ, Phillips K. Digital ischemia in scleroderma spectrum of diseases. Int J Rheumatol. 2010;2010:923743.
9. Hassoun PM. Therapies for scleroderma-related pulmonary arterial hypertension. Expert Rev Respir Med. 2009;3(2):187-196.
10. Giacomelli R, Liakouli V, Berardicurti O, et al. Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatol Int. 2017;37(6):853-863.
11. McLaughlin V, Humbert M, Coghlan G, Nash P, Steen V. Pulmonary arterial hypertension: the most devastating vascular complication of systemic sclerosis. Rheumatology (Oxford). 2009; (suppl 3):iii25-iii31.
12. Gigante A, Barbano B, Granata G, et al. Evaluation of estimated glomerular filtration rate and clinical variables in systemic sclerosis patients. Clin Nephrol. 2016;85(6):326-331.
13. Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthritis Rheum. 2015;44(6):687-694.
14. Woodworth TG, Suliman YA, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678-691.
15. Mouthon L, Bérezné A, Bussone G, Noël LH, Villiger PM, Guillevin L. Scleroderma renal crisis: a rare but severe complication of systemic sclerosis. Clin Rev Allergy Immunol. 2011;40(2):84-91.
16. Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008;34(1):181-90.
17. Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol. 2013;19(41):7062-7068.
Systemic sclerosis (SSc), also called scleroderma, is a rare but serious autoimmune connective tissue disease that has multiple fluctuating pathologic manifestations throughout its temporal course. Estimates have shown that the incidence is 10 to 20 cases per 1 million, and the prevalence is 4 to 253 cases per 1 million.1,2 Given the rarity of this incurable condition, it is vital that primary care providers (PCPs) are able to recognize its unique features early to limit and prevent acute and chronic complications. This case report discusses a patient’s journey with late-diagnosed scleroderma in order to convey these broad manifestations and what providers can do to manage it with their patients.
Case Presentation
Mr. P is a 60-year-old African American male with a history of hypertension, recurrent digital ulcers, pulmonary hypertension (PH), interstitial lung disease (ILD), kidney involvement, congestive heart failure (CHF), and gastroesophageal reflux disease (GERD). Mr. P’s workup began in his late 40s with resistant hypertension, resistant GERD, and multiple hospitalizations for hypertensive urgency. It was not until he was 54 years old that he was diagnosed with mixed connective tissue disorder with sclerodermatous predominance.
Review of systems throughout his medical examinations in his 50s were notable for skin tightening over his hands and shoulders, skin hypopigmentation over his scalp and face, and hair loss. Mr. P was found to have Raynaud phenomenon beginning with his original presentation and digital ulceration without complications of gangrene or autoamputation. Aggregate physical examinations were notable for digital ulceration, skin tightening/sclerodactyly, and telangiectasia. Serologic markers were notable for the following:
- Positive ANA (antinuclear antibody) with titer of 1:1,280/homogenous pattern;
- Positive anti-RNP (antiribonucleoprotein) with titer of 171.2;
- Positive anti-Scl-70 (antitopoisomerase I) with titer of 108.1;
- Positive anti-SM (anti-Smith antibody) with titer of 30.2;
- Positive anti-Ro (SSA) with titer of 107.6;
- Negative anti-La (SSB) with titer of 1.3;
- Negative anti-dsDNA (anti-double stranded DNA) with titer of 9; and
- Negative ACA (anticentromere antibody).
Early transthoracic echocardiograms revealed an ejection fraction (EF) initially at 55% with evidence of left ventricular hypertrophy. Following treatment with phosphodiesterase-5 inhibitors (PDE-5 inhibitors) and endothelin-1 antagonists for pulmonary hypertension, serial transthoracic echocardiograms showed improvement in his EF.
A chest X-ray did not show signs of ILD, but a subsequent high-resolution computed tomography (HRCT) scan was consistent with chronic ILD with a main pulmonary artery diameter of 3 cm (Figure 1).
In subsequent years, Mr. P was hospitalized several times, secondary to digit pain, ulcerations, and osteomyelitis. His first episode was 1 year after his scleroderma diagnosis, when he was hospitalized for 6 days for complications of SSc and finger pain. The following year, he had a 3-day hospitalization for hypertensive urgency and right third-digit osteomyelitis, treated initially with IV fluids, levofloxacin, and vancomycin, and then ceftaroline for 1 month. Throughout the next 6 years, Mr. P presented multiple times with fingertip ulcerations and was followed in the Infectious Disease clinic for recurrent osteomyelitis. He found some relief with systemic antibiotics, including augmentin, minocycline, moxifloxacin, and doxycycline.
At age 59, he was hospitalized for scleroderma renal crisis (SRC). Early in his disease, his kidney function was normal, but the SRC was discovered after an abrupt rise in his blood pressure (BP) and an increase in serum creatinine (SCr) from 1.2 mg/dL at baseline to 3.06 mg/dL. The presence of brown granular cast in his urine prompted a renal biopsy that showed thrombotic microangiopathy with schistocytes. Mr. P was started on captopril and remained stable with outpatient follow-up for this renal complication.
Discussion
Initial presentation of SSc can occur along a spectrum of its pathophysiology. A more severe presentation, like the one seen in Mr. P, seems to occur more frequently in African American patients relative to white patients.3 Differentiating between the 2 types—diffuse vs limited SSc—is vital to managing patients and disease progression. Limited-type SSc is more common (60%) and less severe with slower progression than is diffuse SSc.
Diffuse-type SSc (35%) includes features such as skin thickening and tightening, ILD, SRC, tendon friction rubs (palpable crepitus over tendons), and skin pigment changes.4 The specific involvement of the renal and cardiopulmonary systems accounts for the higher mortality rates in the diffuse-type.5
Many patients with SSc require periodic hospitalizations throughout their life for the acute complications of the disease. Hospitalized patients often range from age 45 to 64 years and are more often female. However, of hospitalized patients with SSc, in-hospital death rates are higher among men.3,6 Although these rates have decreased as the pathogenesis of SSc has become better understood, it is important to note that in-hospital mortality in 1995 for all patients with SSc was 7.1% and mean length of stay was 7.5 days, and in 2002 to 2003, 6.3% and 6.6 days, respectively.3 Though the burden of this disease has decreased, mortality and hospitalizations continue to persist at high rates. Understanding the pathogenesis, progression, and treatments of SSc are essential to aiding patients with this diagnosis.
Skin Involvement
A common finding and presentation for patients with SSc is related to skin involvement. Common patient complaints and exam findings include calcinosis along extensor tendons and digits, Raynaud phenomenon (seen in more than 95% of patients), sclerodactyly, telangiectasias, hyper/hypopigmentation, and pruritus.4 These findings are useful in diagnosing and monitoring patients for disease progression.
Many of the listed skin manifestations affect patients’ quality of life (QOL) but are not directly associated with mortality. However, a common and feared complication includes skin ulcers and osteomyelitis, seen in 48% and 7.7% of patients, respectively.7 Digit ulcers, areas with loss of dermis and epidermis distal to the proximal interphalangeal joints (Figures 2A and 2B), are significant because they parallel a more rapid progression of internal organ involvement.8
Mr. P required multiple hospitalizations and antibiotic regimens for painful digit ulcers complicated by osteomyelitis (Figure 3).
Treatment usually is aimed at infections that complicate these skin ulcers and are based on site-specific cultures. Preventive measures are aimed at the risk factors associated with digit ulcers, including decreased whole-body warmth, direct trauma to digits, smoking, and vasoconstrictors (eg, cocaine, sympathomimetics).8 Some patients may prevent ulcers by using D-penicillamine, mycophenolate mofetil, and cyclophosphamide, although definitive treatment has not been found.4 Calcium channel blockers, PDE-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues also have been used to reduce the severity of Raynaud phenomenon attacks and to decrease the number of digital ulcers (in addition to their beneficial effects on pulmonary involvement).8 Additional pharmacologic agents that have been linked to an improvement in Raynaud phenomenon and digital ulcers include statins, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, intravenous N-acetylcysteine, vitamin E gel, and surgical options (eg, revascularization and sympathectomy).8
Pulmonary Involvement
Systemic sclerosis can lead to 2 complications in the lungs, both present in Mr. P: PAH (mean pulmonary arterial pressure > 25 mm Hg) and ILD, which together make lung involvement the leading cause of death for these patients. Limited-type SSc usually is restricted to PAH, whereas diffuse-type SSc usually leads to ILD.4
On diagnosis of SSc, an HRCT is indicated to assess the degree of lung involvement. Mr. P’s HRCT showed a 3-cm main pulmonary artery, suggestive of PAH, in addition to evidence of ILD seen on the chest X-ray (Figure 4).
Pulmonary arterial hypertension is a common finding in patients with SSc and carries a severe prognosis. Risk factors for the development of PAH, when not present on initial diagnosis, include limited-type SSc; late age of onset; Raynaud phenomenon; decreased DLCO, FVC/DLCO < 1.6; increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) serum levels; and the presence of antibodies.9 Patients with both SSc and PAH have a 50% to 87% 1-year survival, whereas patients with idiopathic PAH without connective tissue disease have an 88% 1-year survival.9
Many patients with lung involvement are asymptomatic, but some have findings of crackles and interstitial thickening on chest X-ray that can progress to cyanosis and right heart failure (cor pulmonale).10 On discovery of lung involvement (Figure 5), it is important to follow up with a PFT, primarily because the outcomes and prognosis for patients with SSc are correlated with the presenting severity of ILD and the subsequent progression of their DLCO.4,10
Treatment is directed at PAH and ILD separately. For PAH, a PDE-5 inhibitor, such as tadalafil or sildenafil, and an endothelin-1 receptor antagonist, such as ambrisentan or bosentan, are indicated. Other PAH treatments include diuretics and prostacyclin analogues (eg, epoprostenol, treprostinil, or iloprost) in addition to warfarin if patients have a history of thrombotic events.4 Mr. P’s pulmonologists deferred treatment with prostacyclin analogues given the potential for adverse effects with endothelin-1 receptor inhibitors, PDE-5 inhibitors, and calcium-channel blockers, although combination studies with bosentan and inhaled iloprost have shown promise.11
Like the skin manifestations of SSc, ILD lacks definitive treatment to prevent disease progression. However, some patients benefit from cyclophosphamide, followed by mycophenolate mofetil—which is usually better tolerated—azathioprine, haematopoietic stem cell transplant as rescue therapy, and lung transplant for life-saving treatment.10
Renal Involvement
Renal involvement in patients with SSc can have profound effects on QOL. Even without clinical renal involvement, glomerular filtration rate (GFR) usually decreases with the progression of vascular damage correlated with age and disease duration.12 Patients with a history of digital ulcers, like Mr. P, usually have a lower GFR than that of patients without digital ulcers.12 Monitoring renal function is vital in caring for these patients, because SRC occurs in 2% to 15% of all patients with SSc.13,14
Scleroderma renal crisis is generally defined as an abruptly elevated BP (> 140/90 mm Hgor > 30 mm Hg rise from baseline) with acute renal failure (elevated SCr) and decreased urine output.14 Given the rarity of SSc in the population, diagnosis of SRC requires high clinical suspicion. In the case of Mr. P, a workup involving serum analysis, urinalysis, and renal biopsy allowed for a definitive diagnosis. A renal biopsy can show microangiopathic hemolytic anemia and confirm SRC, although it may not be necessary in patients with known SSc presenting with new hypertension, rising creatinine, and unremarkable urine sediment on microscopy.14,15
Although an acute SRC can be difficult to predict, monitoring renal function and attention to key factors can assist in discovering this SSc complication. Scleroderma renal crisis usually occurs within the first 4 years of SSc diagnosis, often paralleling rapid progression of skin thickening and tightening with higher rates in both African Americans and males.13,14 Additional predictive factors include diffuse skin involvement, rapid progression of skin involvement, positive anti-RNA polymerase III antibodies, new anemia, new cardiac events (eg, pericardial effusion, pericarditis, left ventricular insufficiency), CHF, tendon friction rubs, arthritis, and recent (within 3 months) high-dose glucocorticoid use.4,13-15
The presentation of SRC can be nonspecific, often resembling findings related to acute kidney injury. Patients may report malaise, fatigue, fever, headache, seizure, blurred vision, or dyspnea.13 Clinical parametersto examine include systolic BP > 140 mm Hgand/or diastolic BP > 90 mm Hg (or an abrupt rise of > 30 or > 20, respectively), SCr increase by 50%+ or > 120% of the upper normal limit, proteinuria at 2+, high protein:creatinine ratio, hematuria > 2+ or > 10 red blood cells (RBCs), platelets < 100,000/mm3, and hypertensive encephalopathy.13 Mr. P presented with fatigue, dyspnea, an abrupt rise in BP (156/96 mm Hg), foamy urine, bilateral lower extremity edema (3.9 g/dL albumin), 2+ RBCs, and a SCr of 2.46 mg/dL on admission (a 205% increase from his last baseline of 1.2 mg/dL).
Treatments have had a large impact on the mortality rates of SRC. Following the introduction of ACE inhibitors, mortality from SRC has decreased from 76% to < 10% over recent decades.13 In addition to improving survival, these medications also have improved hospitalization outcomes for these patients.3 Captopril is often the medication of choice given its rapid onset and short duration of action relative to other medications in the same class, such as benazepril, enalapril, fosinopril, lisinopril, quinapril, and ramipril.4,13 Current clinical trials are assessing the specific renal benefits of endothelin-1 receptor antagonists, which often are used for pulmonary and skin involvement.14 For patients presenting with acute SRC and uremia, dialysis may be necessary (typically predicted by elevated NT-proBNP serum levels), while the decision for transplantation may be indicated at least 2 years after SRC onset and resolution.4,13-15
Once the acute crisis resolves, it is important to discuss prognosis with patients. The 1-, 2-, 3-, 5-, and 10-year survival rates are 82%, 74%, 71%, 59%, and 47%, respectively; however, when looking at only male patients at 10 years, the survival rate is 17%.13 Prevention of SRC can be addressed with daily BP checks and advising patients to seek medical care if they notice a consecutive 2-day abrupt rise.13
Cardiac Involvement
Systemic sclerosis has significant effects on patients’ heart function. The introduction of ACE inhibitors has shifted mortality in SSc patients from predominantly SRC to cardiac causes.6 Cardiac involvement can occur through a range of processes, including abnormal cardiac conduction, CHF, diastolic dysfunction, mitral valve nodular thickening, and pericardial effusion.3 Even though patients often present with skin findings, an initial cardiac workup is crucial to understanding disease progression and patient prognosis. The severity of the cutaneous manifestations often predicts the degree of diastolic dysfunction.16
Clinical evidence of cardiac involvement is seen in 20% to 25% of patients with SSc and is associated with a 70% mortality at 5 years when symptoms are evident.16 Additionally, right ventricular dysfunction at presentation is the strongest marker for all-mortality prognosis, representing the degree of pulmonary involvement, and includes findings such as progressive shortness of breath and systemic edema.9
Given the increasing survival of patients with SSc, cardiac involvement is becoming more evident and prominent. Direct treatments for cardiac manifestations are based on the causative feature, namely, focusing on pulmonary and renal involvement, which can be assessed with periodic echocardiograms evaluating left ventricular EF.
Gastrointestinal Involvement
Along with skin manifestations, the gastrointestinal (GI) involvement of SSc can have a significant impact on patients’ QOL without direct contribution to mortality. One of Mr. P’s earliest symptoms that led to a diagnosis of SSc was GERD, which caused a chronic cough, dental erosions, esophageal erosions, duodenal ulcers, dysphagia, abdominal pain, halitosis, pharyngitis, and weight loss. Esophageal involvement occurs in up to 96% of patients with SSc and can include motility abnormalities (eg, strictures and/or muscle dysfunction), lower esophageal sphincter abnormalities, and Barrett esophagus.17
Additional symptoms of the GI system linked with SSc can occur anywhere along the GI tract and include gastric antral vascular ectasia, causing GI bleeds and pernicious anemia, gastroparesis, bacterial overgrowth, intestinal malabsorption, pseudo-obstruction due to hypomotility, fecal incontinence due to anorectal involvement, and rarely, primary biliary cirrhosis.4,17 Decreased mobility of the oral aperture secondary to skin thickening and tightening also can contribute to malnutrition by decreasing oral intake.
Treatments are supportive and target symptom relief. Chronic treatment of GERD is often necessary and includes antacids, histamine-2 receptor blockers, and proton pump inhibitors.4 Other medications that can help with symptom relief include motility agents (such as metoclopramide, domperidone, prucalopride, tegaserod, and macrolides), osmotic laxatives, and ursodeoxycholic acid for primary biliary cirrhosis.17 Surgical intervention should be considered depending on the severity and progression of involvement within the GI tract. Behavioral changes that can improve patient symptoms include facial grimacing and other mouth-stretching exercises, frequent smaller meals followed by maintaining a vertical posture, and high fiber diets.17
Conclusion
Systemic sclerosis is an autoimmune and connective tissue disease with a pathophysiology that can manifest throughout the body. The organ systems that impact patient outcomes include skin, pulmonary, renal, cardiac, and GI. Primary care providers caring for patients diagnosed with SSc should monitor acute management and disease progression in all these systems. Important acute events that can impact morbidity, mortality, and/or QOL include Raynaud phenomenon, SRC, and pericardial effusion. Chronic manifestations that may be present on diagnosis of SSc or may develop while a patient is under a provider’s care include sclerodactyly, tendon calcinosis, PAH, ILD, chronic kidney injury, chronic cardiac damage, GERD, and esophageal dysmotility. While this discussion serves as a pertinent overview of patients with SSc, it is summative, and providers are encouraged to seek a stronger understanding of both the common and rarer manifestations within each of their patients.
Systemic sclerosis (SSc), also called scleroderma, is a rare but serious autoimmune connective tissue disease that has multiple fluctuating pathologic manifestations throughout its temporal course. Estimates have shown that the incidence is 10 to 20 cases per 1 million, and the prevalence is 4 to 253 cases per 1 million.1,2 Given the rarity of this incurable condition, it is vital that primary care providers (PCPs) are able to recognize its unique features early to limit and prevent acute and chronic complications. This case report discusses a patient’s journey with late-diagnosed scleroderma in order to convey these broad manifestations and what providers can do to manage it with their patients.
Case Presentation
Mr. P is a 60-year-old African American male with a history of hypertension, recurrent digital ulcers, pulmonary hypertension (PH), interstitial lung disease (ILD), kidney involvement, congestive heart failure (CHF), and gastroesophageal reflux disease (GERD). Mr. P’s workup began in his late 40s with resistant hypertension, resistant GERD, and multiple hospitalizations for hypertensive urgency. It was not until he was 54 years old that he was diagnosed with mixed connective tissue disorder with sclerodermatous predominance.
Review of systems throughout his medical examinations in his 50s were notable for skin tightening over his hands and shoulders, skin hypopigmentation over his scalp and face, and hair loss. Mr. P was found to have Raynaud phenomenon beginning with his original presentation and digital ulceration without complications of gangrene or autoamputation. Aggregate physical examinations were notable for digital ulceration, skin tightening/sclerodactyly, and telangiectasia. Serologic markers were notable for the following:
- Positive ANA (antinuclear antibody) with titer of 1:1,280/homogenous pattern;
- Positive anti-RNP (antiribonucleoprotein) with titer of 171.2;
- Positive anti-Scl-70 (antitopoisomerase I) with titer of 108.1;
- Positive anti-SM (anti-Smith antibody) with titer of 30.2;
- Positive anti-Ro (SSA) with titer of 107.6;
- Negative anti-La (SSB) with titer of 1.3;
- Negative anti-dsDNA (anti-double stranded DNA) with titer of 9; and
- Negative ACA (anticentromere antibody).
Early transthoracic echocardiograms revealed an ejection fraction (EF) initially at 55% with evidence of left ventricular hypertrophy. Following treatment with phosphodiesterase-5 inhibitors (PDE-5 inhibitors) and endothelin-1 antagonists for pulmonary hypertension, serial transthoracic echocardiograms showed improvement in his EF.
A chest X-ray did not show signs of ILD, but a subsequent high-resolution computed tomography (HRCT) scan was consistent with chronic ILD with a main pulmonary artery diameter of 3 cm (Figure 1).
In subsequent years, Mr. P was hospitalized several times, secondary to digit pain, ulcerations, and osteomyelitis. His first episode was 1 year after his scleroderma diagnosis, when he was hospitalized for 6 days for complications of SSc and finger pain. The following year, he had a 3-day hospitalization for hypertensive urgency and right third-digit osteomyelitis, treated initially with IV fluids, levofloxacin, and vancomycin, and then ceftaroline for 1 month. Throughout the next 6 years, Mr. P presented multiple times with fingertip ulcerations and was followed in the Infectious Disease clinic for recurrent osteomyelitis. He found some relief with systemic antibiotics, including augmentin, minocycline, moxifloxacin, and doxycycline.
At age 59, he was hospitalized for scleroderma renal crisis (SRC). Early in his disease, his kidney function was normal, but the SRC was discovered after an abrupt rise in his blood pressure (BP) and an increase in serum creatinine (SCr) from 1.2 mg/dL at baseline to 3.06 mg/dL. The presence of brown granular cast in his urine prompted a renal biopsy that showed thrombotic microangiopathy with schistocytes. Mr. P was started on captopril and remained stable with outpatient follow-up for this renal complication.
Discussion
Initial presentation of SSc can occur along a spectrum of its pathophysiology. A more severe presentation, like the one seen in Mr. P, seems to occur more frequently in African American patients relative to white patients.3 Differentiating between the 2 types—diffuse vs limited SSc—is vital to managing patients and disease progression. Limited-type SSc is more common (60%) and less severe with slower progression than is diffuse SSc.
Diffuse-type SSc (35%) includes features such as skin thickening and tightening, ILD, SRC, tendon friction rubs (palpable crepitus over tendons), and skin pigment changes.4 The specific involvement of the renal and cardiopulmonary systems accounts for the higher mortality rates in the diffuse-type.5
Many patients with SSc require periodic hospitalizations throughout their life for the acute complications of the disease. Hospitalized patients often range from age 45 to 64 years and are more often female. However, of hospitalized patients with SSc, in-hospital death rates are higher among men.3,6 Although these rates have decreased as the pathogenesis of SSc has become better understood, it is important to note that in-hospital mortality in 1995 for all patients with SSc was 7.1% and mean length of stay was 7.5 days, and in 2002 to 2003, 6.3% and 6.6 days, respectively.3 Though the burden of this disease has decreased, mortality and hospitalizations continue to persist at high rates. Understanding the pathogenesis, progression, and treatments of SSc are essential to aiding patients with this diagnosis.
Skin Involvement
A common finding and presentation for patients with SSc is related to skin involvement. Common patient complaints and exam findings include calcinosis along extensor tendons and digits, Raynaud phenomenon (seen in more than 95% of patients), sclerodactyly, telangiectasias, hyper/hypopigmentation, and pruritus.4 These findings are useful in diagnosing and monitoring patients for disease progression.
Many of the listed skin manifestations affect patients’ quality of life (QOL) but are not directly associated with mortality. However, a common and feared complication includes skin ulcers and osteomyelitis, seen in 48% and 7.7% of patients, respectively.7 Digit ulcers, areas with loss of dermis and epidermis distal to the proximal interphalangeal joints (Figures 2A and 2B), are significant because they parallel a more rapid progression of internal organ involvement.8
Mr. P required multiple hospitalizations and antibiotic regimens for painful digit ulcers complicated by osteomyelitis (Figure 3).
Treatment usually is aimed at infections that complicate these skin ulcers and are based on site-specific cultures. Preventive measures are aimed at the risk factors associated with digit ulcers, including decreased whole-body warmth, direct trauma to digits, smoking, and vasoconstrictors (eg, cocaine, sympathomimetics).8 Some patients may prevent ulcers by using D-penicillamine, mycophenolate mofetil, and cyclophosphamide, although definitive treatment has not been found.4 Calcium channel blockers, PDE-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues also have been used to reduce the severity of Raynaud phenomenon attacks and to decrease the number of digital ulcers (in addition to their beneficial effects on pulmonary involvement).8 Additional pharmacologic agents that have been linked to an improvement in Raynaud phenomenon and digital ulcers include statins, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, intravenous N-acetylcysteine, vitamin E gel, and surgical options (eg, revascularization and sympathectomy).8
Pulmonary Involvement
Systemic sclerosis can lead to 2 complications in the lungs, both present in Mr. P: PAH (mean pulmonary arterial pressure > 25 mm Hg) and ILD, which together make lung involvement the leading cause of death for these patients. Limited-type SSc usually is restricted to PAH, whereas diffuse-type SSc usually leads to ILD.4
On diagnosis of SSc, an HRCT is indicated to assess the degree of lung involvement. Mr. P’s HRCT showed a 3-cm main pulmonary artery, suggestive of PAH, in addition to evidence of ILD seen on the chest X-ray (Figure 4).
Pulmonary arterial hypertension is a common finding in patients with SSc and carries a severe prognosis. Risk factors for the development of PAH, when not present on initial diagnosis, include limited-type SSc; late age of onset; Raynaud phenomenon; decreased DLCO, FVC/DLCO < 1.6; increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) serum levels; and the presence of antibodies.9 Patients with both SSc and PAH have a 50% to 87% 1-year survival, whereas patients with idiopathic PAH without connective tissue disease have an 88% 1-year survival.9
Many patients with lung involvement are asymptomatic, but some have findings of crackles and interstitial thickening on chest X-ray that can progress to cyanosis and right heart failure (cor pulmonale).10 On discovery of lung involvement (Figure 5), it is important to follow up with a PFT, primarily because the outcomes and prognosis for patients with SSc are correlated with the presenting severity of ILD and the subsequent progression of their DLCO.4,10
Treatment is directed at PAH and ILD separately. For PAH, a PDE-5 inhibitor, such as tadalafil or sildenafil, and an endothelin-1 receptor antagonist, such as ambrisentan or bosentan, are indicated. Other PAH treatments include diuretics and prostacyclin analogues (eg, epoprostenol, treprostinil, or iloprost) in addition to warfarin if patients have a history of thrombotic events.4 Mr. P’s pulmonologists deferred treatment with prostacyclin analogues given the potential for adverse effects with endothelin-1 receptor inhibitors, PDE-5 inhibitors, and calcium-channel blockers, although combination studies with bosentan and inhaled iloprost have shown promise.11
Like the skin manifestations of SSc, ILD lacks definitive treatment to prevent disease progression. However, some patients benefit from cyclophosphamide, followed by mycophenolate mofetil—which is usually better tolerated—azathioprine, haematopoietic stem cell transplant as rescue therapy, and lung transplant for life-saving treatment.10
Renal Involvement
Renal involvement in patients with SSc can have profound effects on QOL. Even without clinical renal involvement, glomerular filtration rate (GFR) usually decreases with the progression of vascular damage correlated with age and disease duration.12 Patients with a history of digital ulcers, like Mr. P, usually have a lower GFR than that of patients without digital ulcers.12 Monitoring renal function is vital in caring for these patients, because SRC occurs in 2% to 15% of all patients with SSc.13,14
Scleroderma renal crisis is generally defined as an abruptly elevated BP (> 140/90 mm Hgor > 30 mm Hg rise from baseline) with acute renal failure (elevated SCr) and decreased urine output.14 Given the rarity of SSc in the population, diagnosis of SRC requires high clinical suspicion. In the case of Mr. P, a workup involving serum analysis, urinalysis, and renal biopsy allowed for a definitive diagnosis. A renal biopsy can show microangiopathic hemolytic anemia and confirm SRC, although it may not be necessary in patients with known SSc presenting with new hypertension, rising creatinine, and unremarkable urine sediment on microscopy.14,15
Although an acute SRC can be difficult to predict, monitoring renal function and attention to key factors can assist in discovering this SSc complication. Scleroderma renal crisis usually occurs within the first 4 years of SSc diagnosis, often paralleling rapid progression of skin thickening and tightening with higher rates in both African Americans and males.13,14 Additional predictive factors include diffuse skin involvement, rapid progression of skin involvement, positive anti-RNA polymerase III antibodies, new anemia, new cardiac events (eg, pericardial effusion, pericarditis, left ventricular insufficiency), CHF, tendon friction rubs, arthritis, and recent (within 3 months) high-dose glucocorticoid use.4,13-15
The presentation of SRC can be nonspecific, often resembling findings related to acute kidney injury. Patients may report malaise, fatigue, fever, headache, seizure, blurred vision, or dyspnea.13 Clinical parametersto examine include systolic BP > 140 mm Hgand/or diastolic BP > 90 mm Hg (or an abrupt rise of > 30 or > 20, respectively), SCr increase by 50%+ or > 120% of the upper normal limit, proteinuria at 2+, high protein:creatinine ratio, hematuria > 2+ or > 10 red blood cells (RBCs), platelets < 100,000/mm3, and hypertensive encephalopathy.13 Mr. P presented with fatigue, dyspnea, an abrupt rise in BP (156/96 mm Hg), foamy urine, bilateral lower extremity edema (3.9 g/dL albumin), 2+ RBCs, and a SCr of 2.46 mg/dL on admission (a 205% increase from his last baseline of 1.2 mg/dL).
Treatments have had a large impact on the mortality rates of SRC. Following the introduction of ACE inhibitors, mortality from SRC has decreased from 76% to < 10% over recent decades.13 In addition to improving survival, these medications also have improved hospitalization outcomes for these patients.3 Captopril is often the medication of choice given its rapid onset and short duration of action relative to other medications in the same class, such as benazepril, enalapril, fosinopril, lisinopril, quinapril, and ramipril.4,13 Current clinical trials are assessing the specific renal benefits of endothelin-1 receptor antagonists, which often are used for pulmonary and skin involvement.14 For patients presenting with acute SRC and uremia, dialysis may be necessary (typically predicted by elevated NT-proBNP serum levels), while the decision for transplantation may be indicated at least 2 years after SRC onset and resolution.4,13-15
Once the acute crisis resolves, it is important to discuss prognosis with patients. The 1-, 2-, 3-, 5-, and 10-year survival rates are 82%, 74%, 71%, 59%, and 47%, respectively; however, when looking at only male patients at 10 years, the survival rate is 17%.13 Prevention of SRC can be addressed with daily BP checks and advising patients to seek medical care if they notice a consecutive 2-day abrupt rise.13
Cardiac Involvement
Systemic sclerosis has significant effects on patients’ heart function. The introduction of ACE inhibitors has shifted mortality in SSc patients from predominantly SRC to cardiac causes.6 Cardiac involvement can occur through a range of processes, including abnormal cardiac conduction, CHF, diastolic dysfunction, mitral valve nodular thickening, and pericardial effusion.3 Even though patients often present with skin findings, an initial cardiac workup is crucial to understanding disease progression and patient prognosis. The severity of the cutaneous manifestations often predicts the degree of diastolic dysfunction.16
Clinical evidence of cardiac involvement is seen in 20% to 25% of patients with SSc and is associated with a 70% mortality at 5 years when symptoms are evident.16 Additionally, right ventricular dysfunction at presentation is the strongest marker for all-mortality prognosis, representing the degree of pulmonary involvement, and includes findings such as progressive shortness of breath and systemic edema.9
Given the increasing survival of patients with SSc, cardiac involvement is becoming more evident and prominent. Direct treatments for cardiac manifestations are based on the causative feature, namely, focusing on pulmonary and renal involvement, which can be assessed with periodic echocardiograms evaluating left ventricular EF.
Gastrointestinal Involvement
Along with skin manifestations, the gastrointestinal (GI) involvement of SSc can have a significant impact on patients’ QOL without direct contribution to mortality. One of Mr. P’s earliest symptoms that led to a diagnosis of SSc was GERD, which caused a chronic cough, dental erosions, esophageal erosions, duodenal ulcers, dysphagia, abdominal pain, halitosis, pharyngitis, and weight loss. Esophageal involvement occurs in up to 96% of patients with SSc and can include motility abnormalities (eg, strictures and/or muscle dysfunction), lower esophageal sphincter abnormalities, and Barrett esophagus.17
Additional symptoms of the GI system linked with SSc can occur anywhere along the GI tract and include gastric antral vascular ectasia, causing GI bleeds and pernicious anemia, gastroparesis, bacterial overgrowth, intestinal malabsorption, pseudo-obstruction due to hypomotility, fecal incontinence due to anorectal involvement, and rarely, primary biliary cirrhosis.4,17 Decreased mobility of the oral aperture secondary to skin thickening and tightening also can contribute to malnutrition by decreasing oral intake.
Treatments are supportive and target symptom relief. Chronic treatment of GERD is often necessary and includes antacids, histamine-2 receptor blockers, and proton pump inhibitors.4 Other medications that can help with symptom relief include motility agents (such as metoclopramide, domperidone, prucalopride, tegaserod, and macrolides), osmotic laxatives, and ursodeoxycholic acid for primary biliary cirrhosis.17 Surgical intervention should be considered depending on the severity and progression of involvement within the GI tract. Behavioral changes that can improve patient symptoms include facial grimacing and other mouth-stretching exercises, frequent smaller meals followed by maintaining a vertical posture, and high fiber diets.17
Conclusion
Systemic sclerosis is an autoimmune and connective tissue disease with a pathophysiology that can manifest throughout the body. The organ systems that impact patient outcomes include skin, pulmonary, renal, cardiac, and GI. Primary care providers caring for patients diagnosed with SSc should monitor acute management and disease progression in all these systems. Important acute events that can impact morbidity, mortality, and/or QOL include Raynaud phenomenon, SRC, and pericardial effusion. Chronic manifestations that may be present on diagnosis of SSc or may develop while a patient is under a provider’s care include sclerodactyly, tendon calcinosis, PAH, ILD, chronic kidney injury, chronic cardiac damage, GERD, and esophageal dysmotility. While this discussion serves as a pertinent overview of patients with SSc, it is summative, and providers are encouraged to seek a stronger understanding of both the common and rarer manifestations within each of their patients.
1. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778-799.
2. Silman AG. Scleroderma. In: Epidemiology of the Rheumatic Diseases. 2nd ed. Silman AJ, Hochberg MC, eds. Oxford, UK: Oxford University Press. 1993:chap 8.
3. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford). 2007;46(12):1808-1813.
4. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78(8):961-968.
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809-1815.
6. Piga M, Casula L, Sanna S, et al. Population-based analysis of hospitalizations for patients with systemic sclerosis in a West-European region over the period 2001-2012. Rheumatol Int. 2016;36(1):73-81.
7. Giuggioli D, Manfredi A, Colaci M, Lumetti F, Ferri C. Osteomyelitis complicating scleroderma digital ulcers. Clin Rheumatol. 2013;32(5):623-627.
8. Schiopu E, Impens AJ, Phillips K. Digital ischemia in scleroderma spectrum of diseases. Int J Rheumatol. 2010;2010:923743.
9. Hassoun PM. Therapies for scleroderma-related pulmonary arterial hypertension. Expert Rev Respir Med. 2009;3(2):187-196.
10. Giacomelli R, Liakouli V, Berardicurti O, et al. Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatol Int. 2017;37(6):853-863.
11. McLaughlin V, Humbert M, Coghlan G, Nash P, Steen V. Pulmonary arterial hypertension: the most devastating vascular complication of systemic sclerosis. Rheumatology (Oxford). 2009; (suppl 3):iii25-iii31.
12. Gigante A, Barbano B, Granata G, et al. Evaluation of estimated glomerular filtration rate and clinical variables in systemic sclerosis patients. Clin Nephrol. 2016;85(6):326-331.
13. Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthritis Rheum. 2015;44(6):687-694.
14. Woodworth TG, Suliman YA, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678-691.
15. Mouthon L, Bérezné A, Bussone G, Noël LH, Villiger PM, Guillevin L. Scleroderma renal crisis: a rare but severe complication of systemic sclerosis. Clin Rev Allergy Immunol. 2011;40(2):84-91.
16. Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008;34(1):181-90.
17. Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol. 2013;19(41):7062-7068.
1. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778-799.
2. Silman AG. Scleroderma. In: Epidemiology of the Rheumatic Diseases. 2nd ed. Silman AJ, Hochberg MC, eds. Oxford, UK: Oxford University Press. 1993:chap 8.
3. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford). 2007;46(12):1808-1813.
4. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78(8):961-968.
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809-1815.
6. Piga M, Casula L, Sanna S, et al. Population-based analysis of hospitalizations for patients with systemic sclerosis in a West-European region over the period 2001-2012. Rheumatol Int. 2016;36(1):73-81.
7. Giuggioli D, Manfredi A, Colaci M, Lumetti F, Ferri C. Osteomyelitis complicating scleroderma digital ulcers. Clin Rheumatol. 2013;32(5):623-627.
8. Schiopu E, Impens AJ, Phillips K. Digital ischemia in scleroderma spectrum of diseases. Int J Rheumatol. 2010;2010:923743.
9. Hassoun PM. Therapies for scleroderma-related pulmonary arterial hypertension. Expert Rev Respir Med. 2009;3(2):187-196.
10. Giacomelli R, Liakouli V, Berardicurti O, et al. Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatol Int. 2017;37(6):853-863.
11. McLaughlin V, Humbert M, Coghlan G, Nash P, Steen V. Pulmonary arterial hypertension: the most devastating vascular complication of systemic sclerosis. Rheumatology (Oxford). 2009; (suppl 3):iii25-iii31.
12. Gigante A, Barbano B, Granata G, et al. Evaluation of estimated glomerular filtration rate and clinical variables in systemic sclerosis patients. Clin Nephrol. 2016;85(6):326-331.
13. Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthritis Rheum. 2015;44(6):687-694.
14. Woodworth TG, Suliman YA, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678-691.
15. Mouthon L, Bérezné A, Bussone G, Noël LH, Villiger PM, Guillevin L. Scleroderma renal crisis: a rare but severe complication of systemic sclerosis. Clin Rev Allergy Immunol. 2011;40(2):84-91.
16. Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008;34(1):181-90.
17. Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol. 2013;19(41):7062-7068.
Slime is not sublime: It may cause hand dermatitis
A young, otherwise healthy 9-year-old girl was evaluated for pruritic hand dermatitis which lasted 5 months after exposure to homemade slime. Physical exam revealed erythematous, scaly plaques on the palmar surfaces of her hands; her fingernails had onychomadesis and longitudinal ridging. Despite frequent emolliation, her dermatitis persisted. She was then treated empirically for scabies and for culture-positive Staphylococcus aureus infection, which required a full round of cephalexin and mupirocin ointment. This also did not alleviate the dermatitis. A combination of homemade borax-containing slime avoidance, brief course of high-dose corticosteroids, and frequent bland emollients was prescribed because the dermatitis was assumed to be caused by an irritant.
After review of this case and evaluation of other children with hand dermatitis, Julia K. Gittler, MD, of Columbia University, New York, and her colleagues have made a case that “slime” and new-onset hand dermatitis may be linked.
SOURCE: Gittler JK et al. J Pediatr. 2018 May 3. doi: 10.1016/j.jpeds.2018.03.064 .
A young, otherwise healthy 9-year-old girl was evaluated for pruritic hand dermatitis which lasted 5 months after exposure to homemade slime. Physical exam revealed erythematous, scaly plaques on the palmar surfaces of her hands; her fingernails had onychomadesis and longitudinal ridging. Despite frequent emolliation, her dermatitis persisted. She was then treated empirically for scabies and for culture-positive Staphylococcus aureus infection, which required a full round of cephalexin and mupirocin ointment. This also did not alleviate the dermatitis. A combination of homemade borax-containing slime avoidance, brief course of high-dose corticosteroids, and frequent bland emollients was prescribed because the dermatitis was assumed to be caused by an irritant.
After review of this case and evaluation of other children with hand dermatitis, Julia K. Gittler, MD, of Columbia University, New York, and her colleagues have made a case that “slime” and new-onset hand dermatitis may be linked.
SOURCE: Gittler JK et al. J Pediatr. 2018 May 3. doi: 10.1016/j.jpeds.2018.03.064 .
A young, otherwise healthy 9-year-old girl was evaluated for pruritic hand dermatitis which lasted 5 months after exposure to homemade slime. Physical exam revealed erythematous, scaly plaques on the palmar surfaces of her hands; her fingernails had onychomadesis and longitudinal ridging. Despite frequent emolliation, her dermatitis persisted. She was then treated empirically for scabies and for culture-positive Staphylococcus aureus infection, which required a full round of cephalexin and mupirocin ointment. This also did not alleviate the dermatitis. A combination of homemade borax-containing slime avoidance, brief course of high-dose corticosteroids, and frequent bland emollients was prescribed because the dermatitis was assumed to be caused by an irritant.
After review of this case and evaluation of other children with hand dermatitis, Julia K. Gittler, MD, of Columbia University, New York, and her colleagues have made a case that “slime” and new-onset hand dermatitis may be linked.
SOURCE: Gittler JK et al. J Pediatr. 2018 May 3. doi: 10.1016/j.jpeds.2018.03.064 .
FROM THE JOURNAL OF PEDIATRICS
Collagen remodeling observed after laser treatment in EB patient
DALLAS – Fractional led to considerable clinical improvement, including thickening of the dermis, results from a case report showed.
“We have so much more to learn about how the laser treatments are modifying these intricate pathways,” lead study author Samantha Schneider, MD, said in an interview following the annual conference of the American Society for Laser Medicine and Surgery. “But, our project suggests that patients with genetic blistering diseases may benefit from fractional laser therapy in combination with topical PLLA. This may be a good option, particularly for patients who are looking for more therapeutic options for slowly healing wounds and have exhausted other more conventional treatment modalities.”

Drawing from this previous work, Dr. Schneider and her associates hypothesized that fractional ablative laser treatment and topical PLLA might help a 27-year-old RDEB patient with revertant mosaicism who presented for management of large, nonhealing erosions on her upper back and posterior neck, complicated by frequent Staphylococcus infections. Over a 2-year period the researchers administered 15 fractional CO2 laser treatments with a single-pulse, nonoverlapping technique with settings of 15 mJ of energy and 15% density. They immediately applied concentrated topical PLLA to the treated area and obtained punch biopsy specimens from treated and untreated affected skin and clinically normal-appearing skin after the seventh treatment for histopathologic and immunohistologic examination.
Since the time of treatment, the patient reported marked improvement with a decreased number of erosions, as well as decreased pain. In addition, the hematoxylin and eosin slides showed increased collagen I (mature collagen) in the treated sample, “which suggests that we may be inducing a type of neocollagenesis, which is exciting particularly if it seems to work for patients with genetic alterations in collagen,” Dr. Schneider said. “Additionally, the indirect immunofluorescence [IIF] showed increased collagen VII, which is absent in the patient’s untreated skin. This was truly surprising and warrants more investigation as to how we may be affecting patients’ biology with this combination treatment.”
She acknowledged that more studies are required to confirm the findings. “Furthermore, we did not examine the fractional laser therapy and the topical PLLA independently so we cannot say whether the effect is synergistic or due primarily to one modality versus the other,” she noted. “Lastly, the IIF interpretation was challenging particularly in the untreated skin due to the epidermal detachment and edge staining. However, when viewed in comparison to the treated skin, we noted increased collagen VII in the treated sample.”
Dr. Schneider reported having no relevant disclosures.
DALLAS – Fractional led to considerable clinical improvement, including thickening of the dermis, results from a case report showed.
“We have so much more to learn about how the laser treatments are modifying these intricate pathways,” lead study author Samantha Schneider, MD, said in an interview following the annual conference of the American Society for Laser Medicine and Surgery. “But, our project suggests that patients with genetic blistering diseases may benefit from fractional laser therapy in combination with topical PLLA. This may be a good option, particularly for patients who are looking for more therapeutic options for slowly healing wounds and have exhausted other more conventional treatment modalities.”
Drawing from this previous work, Dr. Schneider and her associates hypothesized that fractional ablative laser treatment and topical PLLA might help a 27-year-old RDEB patient with revertant mosaicism who presented for management of large, nonhealing erosions on her upper back and posterior neck, complicated by frequent Staphylococcus infections. Over a 2-year period the researchers administered 15 fractional CO2 laser treatments with a single-pulse, nonoverlapping technique with settings of 15 mJ of energy and 15% density. They immediately applied concentrated topical PLLA to the treated area and obtained punch biopsy specimens from treated and untreated affected skin and clinically normal-appearing skin after the seventh treatment for histopathologic and immunohistologic examination.
Since the time of treatment, the patient reported marked improvement with a decreased number of erosions, as well as decreased pain. In addition, the hematoxylin and eosin slides showed increased collagen I (mature collagen) in the treated sample, “which suggests that we may be inducing a type of neocollagenesis, which is exciting particularly if it seems to work for patients with genetic alterations in collagen,” Dr. Schneider said. “Additionally, the indirect immunofluorescence [IIF] showed increased collagen VII, which is absent in the patient’s untreated skin. This was truly surprising and warrants more investigation as to how we may be affecting patients’ biology with this combination treatment.”
She acknowledged that more studies are required to confirm the findings. “Furthermore, we did not examine the fractional laser therapy and the topical PLLA independently so we cannot say whether the effect is synergistic or due primarily to one modality versus the other,” she noted. “Lastly, the IIF interpretation was challenging particularly in the untreated skin due to the epidermal detachment and edge staining. However, when viewed in comparison to the treated skin, we noted increased collagen VII in the treated sample.”
Dr. Schneider reported having no relevant disclosures.
DALLAS – Fractional led to considerable clinical improvement, including thickening of the dermis, results from a case report showed.
“We have so much more to learn about how the laser treatments are modifying these intricate pathways,” lead study author Samantha Schneider, MD, said in an interview following the annual conference of the American Society for Laser Medicine and Surgery. “But, our project suggests that patients with genetic blistering diseases may benefit from fractional laser therapy in combination with topical PLLA. This may be a good option, particularly for patients who are looking for more therapeutic options for slowly healing wounds and have exhausted other more conventional treatment modalities.”
Drawing from this previous work, Dr. Schneider and her associates hypothesized that fractional ablative laser treatment and topical PLLA might help a 27-year-old RDEB patient with revertant mosaicism who presented for management of large, nonhealing erosions on her upper back and posterior neck, complicated by frequent Staphylococcus infections. Over a 2-year period the researchers administered 15 fractional CO2 laser treatments with a single-pulse, nonoverlapping technique with settings of 15 mJ of energy and 15% density. They immediately applied concentrated topical PLLA to the treated area and obtained punch biopsy specimens from treated and untreated affected skin and clinically normal-appearing skin after the seventh treatment for histopathologic and immunohistologic examination.
Since the time of treatment, the patient reported marked improvement with a decreased number of erosions, as well as decreased pain. In addition, the hematoxylin and eosin slides showed increased collagen I (mature collagen) in the treated sample, “which suggests that we may be inducing a type of neocollagenesis, which is exciting particularly if it seems to work for patients with genetic alterations in collagen,” Dr. Schneider said. “Additionally, the indirect immunofluorescence [IIF] showed increased collagen VII, which is absent in the patient’s untreated skin. This was truly surprising and warrants more investigation as to how we may be affecting patients’ biology with this combination treatment.”
She acknowledged that more studies are required to confirm the findings. “Furthermore, we did not examine the fractional laser therapy and the topical PLLA independently so we cannot say whether the effect is synergistic or due primarily to one modality versus the other,” she noted. “Lastly, the IIF interpretation was challenging particularly in the untreated skin due to the epidermal detachment and edge staining. However, when viewed in comparison to the treated skin, we noted increased collagen VII in the treated sample.”
Dr. Schneider reported having no relevant disclosures.
Key clinical point: Fractional ablative laser treatment combined with poly-L-lactic acid may aid in the care of certain patients with recessive dystrophic epidermolysis bullosa.
Major finding: Since the time of treatment, the patient reported marked improvement with a decreased number of erosions as well as decreased pain.
Study details: A case report of a 27-year-old recessive dystrophic epidermolysis bullosa patient with revertant mosaicism.
Disclosures: Dr. Schneider reported having no financial disclosures.
Topical corticosteroid-retinoid combination effective in moderate to severe psoriasis
The combination of a topical corticosteroid and a topical retinoid for the treatment of plaque psoriasis resulted in significant improvements in clinical signs, in two multicenter, double-blind, vehicle-controlled phase 3 studies.
In the two studies, investigators randomized a total of 418 or vehicle lotion, applied once a day to affected areas. After 8 weeks of treatment, 35.8% of adults in the first study and 45.3% of those in the second study had achieved the primary outcome of at least a two-grade improvement in the Investigator’s Global Assessment score and reaching “clear” or “almost clear,” compared with 7.0% and 12.5%, respectively, of patients treated with the vehicle (P less than .001). The report was published online in the Journal of the American Academy of Dermatology.
At 8 weeks, reduction in erythema was achieved by 44.2% and 49.6% of patients in the treatment arms, compared with 10% and 18.7% of patients in the control arms. Plaque elevation was reduced in 59.3% and 59.7% of patients in the treatment arms, compared with 17.9% and 21.3% of patients in the control arms; and scaling was reduced in 59.4% and 62.9% of those on treatment, compared with 20.6% and 21.0%, respectively. All differences between the treatment and control groups were statistically significant (P less than .001).
Participants who received the treatment also reported significantly lower scores for itching, dryness, and burning or stinging compared with those who received the vehicle lotion.
Dr. Linda Stein Gold of Henry Ford Hospital in Detroit, and her coauthors, wrote that while clinical studies have established the benefit of using a topical corticosteroid as an adjunct to tazarotene for plaque psoriasis, data on their combined use was limited. This combination “was consistently more effective than vehicle in achieving treatment success; effectively reducing affected area and psoriasis signs at the target lesion, and improving QoL [quality of life],” they wrote.
Most patients maintained these improvements over the 4-week posttreatment period.
Patients who received the halobetasol propionate/tazarotene lotion reported more adverse events than did those who received the control lotion, but most were mild to moderate and included contact dermatitis (6.3%), pruritus (2.2%) and application site pain (2.6%). Three serious adverse events were not related to treatment.
The studies were funded by Dow Pharmaceutical Sciences, a division of Valeant Pharmaceuticals North America. Four authors disclosed advisory, consultancy and speaking positions and other funding from the pharmaceutical industry, including with Valeant Pharmaceuticals. Five authors are employees of the company.
SOURCE: Gold L et al. J Am Acad Dermatol. 2018 Mar 31. pii: S0190-9622(18)30494-8. doi: 10.1016/j.jaad.2018.03.040.
The combination of a topical corticosteroid and a topical retinoid for the treatment of plaque psoriasis resulted in significant improvements in clinical signs, in two multicenter, double-blind, vehicle-controlled phase 3 studies.
In the two studies, investigators randomized a total of 418 or vehicle lotion, applied once a day to affected areas. After 8 weeks of treatment, 35.8% of adults in the first study and 45.3% of those in the second study had achieved the primary outcome of at least a two-grade improvement in the Investigator’s Global Assessment score and reaching “clear” or “almost clear,” compared with 7.0% and 12.5%, respectively, of patients treated with the vehicle (P less than .001). The report was published online in the Journal of the American Academy of Dermatology.
At 8 weeks, reduction in erythema was achieved by 44.2% and 49.6% of patients in the treatment arms, compared with 10% and 18.7% of patients in the control arms. Plaque elevation was reduced in 59.3% and 59.7% of patients in the treatment arms, compared with 17.9% and 21.3% of patients in the control arms; and scaling was reduced in 59.4% and 62.9% of those on treatment, compared with 20.6% and 21.0%, respectively. All differences between the treatment and control groups were statistically significant (P less than .001).
Participants who received the treatment also reported significantly lower scores for itching, dryness, and burning or stinging compared with those who received the vehicle lotion.
Dr. Linda Stein Gold of Henry Ford Hospital in Detroit, and her coauthors, wrote that while clinical studies have established the benefit of using a topical corticosteroid as an adjunct to tazarotene for plaque psoriasis, data on their combined use was limited. This combination “was consistently more effective than vehicle in achieving treatment success; effectively reducing affected area and psoriasis signs at the target lesion, and improving QoL [quality of life],” they wrote.
Most patients maintained these improvements over the 4-week posttreatment period.
Patients who received the halobetasol propionate/tazarotene lotion reported more adverse events than did those who received the control lotion, but most were mild to moderate and included contact dermatitis (6.3%), pruritus (2.2%) and application site pain (2.6%). Three serious adverse events were not related to treatment.
The studies were funded by Dow Pharmaceutical Sciences, a division of Valeant Pharmaceuticals North America. Four authors disclosed advisory, consultancy and speaking positions and other funding from the pharmaceutical industry, including with Valeant Pharmaceuticals. Five authors are employees of the company.
SOURCE: Gold L et al. J Am Acad Dermatol. 2018 Mar 31. pii: S0190-9622(18)30494-8. doi: 10.1016/j.jaad.2018.03.040.
The combination of a topical corticosteroid and a topical retinoid for the treatment of plaque psoriasis resulted in significant improvements in clinical signs, in two multicenter, double-blind, vehicle-controlled phase 3 studies.
In the two studies, investigators randomized a total of 418 or vehicle lotion, applied once a day to affected areas. After 8 weeks of treatment, 35.8% of adults in the first study and 45.3% of those in the second study had achieved the primary outcome of at least a two-grade improvement in the Investigator’s Global Assessment score and reaching “clear” or “almost clear,” compared with 7.0% and 12.5%, respectively, of patients treated with the vehicle (P less than .001). The report was published online in the Journal of the American Academy of Dermatology.
At 8 weeks, reduction in erythema was achieved by 44.2% and 49.6% of patients in the treatment arms, compared with 10% and 18.7% of patients in the control arms. Plaque elevation was reduced in 59.3% and 59.7% of patients in the treatment arms, compared with 17.9% and 21.3% of patients in the control arms; and scaling was reduced in 59.4% and 62.9% of those on treatment, compared with 20.6% and 21.0%, respectively. All differences between the treatment and control groups were statistically significant (P less than .001).
Participants who received the treatment also reported significantly lower scores for itching, dryness, and burning or stinging compared with those who received the vehicle lotion.
Dr. Linda Stein Gold of Henry Ford Hospital in Detroit, and her coauthors, wrote that while clinical studies have established the benefit of using a topical corticosteroid as an adjunct to tazarotene for plaque psoriasis, data on their combined use was limited. This combination “was consistently more effective than vehicle in achieving treatment success; effectively reducing affected area and psoriasis signs at the target lesion, and improving QoL [quality of life],” they wrote.
Most patients maintained these improvements over the 4-week posttreatment period.
Patients who received the halobetasol propionate/tazarotene lotion reported more adverse events than did those who received the control lotion, but most were mild to moderate and included contact dermatitis (6.3%), pruritus (2.2%) and application site pain (2.6%). Three serious adverse events were not related to treatment.
The studies were funded by Dow Pharmaceutical Sciences, a division of Valeant Pharmaceuticals North America. Four authors disclosed advisory, consultancy and speaking positions and other funding from the pharmaceutical industry, including with Valeant Pharmaceuticals. Five authors are employees of the company.
SOURCE: Gold L et al. J Am Acad Dermatol. 2018 Mar 31. pii: S0190-9622(18)30494-8. doi: 10.1016/j.jaad.2018.03.040.
FROM JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Key clinical point: Topical halobetasol propionate and tazarotene can significantly improve plaque psoriasis symptoms.
Major finding: Nearly half of adults treated with topical halobetasol propionate and tazarotene were “clear” or “almost clear” after 8 weeks.
Study details: Two multicenter, double-blind, vehicle-controlled phase 3 studies of 418 adults with psoriasis.
Disclosures: The studies were funded by Dow Pharmaceutical Sciences, a division of Valeant Pharmaceuticals North America. Four authors disclosed advisory, consultancy and speaking positions and other funding from the pharmaceutical industry, including Valeant Pharmaceuticals. Five authors are employees of the company.
Source: Gold L et al. J Am Acad Dermatol. 2018 Mar 31. pii: S0190-9622(18)30494-8. doi: 10.1016/j.jaad.2018.03.040.
MDedge Daily News: How to handle opioid constipation
Bath emollients are a washout for childhood eczema. Does warfarin cause acute kidney injury? And there may be a new option for postpartum depression.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
Bath emollients are a washout for childhood eczema. Does warfarin cause acute kidney injury? And there may be a new option for postpartum depression.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
Bath emollients are a washout for childhood eczema. Does warfarin cause acute kidney injury? And there may be a new option for postpartum depression.
Listen to the MDedge Daily News podcast for all the details on today’s top news.

Brown spot on right foot

The FP recognized this as a benign congenital nevus.
While most congenital nevi are visible at birth, there are some that appear in the first year of life and are known as tardive congenital nevi. The FP used a dermatoscope to look at this nevus and found that its features were benign.
The parents wondered whether this needed to be removed to prevent it from becoming skin cancer in the future. The FP reassured them that the risk of melanoma from this one nevus was too small to warrant a prophylactic surgical excision. The parents agreed to the standard 6-month immunizations.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Smith, M. Congenital nevi. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:953-957.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/.
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com.
The FP recognized this as a benign congenital nevus.
While most congenital nevi are visible at birth, there are some that appear in the first year of life and are known as tardive congenital nevi. The FP used a dermatoscope to look at this nevus and found that its features were benign.
The parents wondered whether this needed to be removed to prevent it from becoming skin cancer in the future. The FP reassured them that the risk of melanoma from this one nevus was too small to warrant a prophylactic surgical excision. The parents agreed to the standard 6-month immunizations.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Smith, M. Congenital nevi. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:953-957.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/.
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com.
The FP recognized this as a benign congenital nevus.
While most congenital nevi are visible at birth, there are some that appear in the first year of life and are known as tardive congenital nevi. The FP used a dermatoscope to look at this nevus and found that its features were benign.
The parents wondered whether this needed to be removed to prevent it from becoming skin cancer in the future. The FP reassured them that the risk of melanoma from this one nevus was too small to warrant a prophylactic surgical excision. The parents agreed to the standard 6-month immunizations.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Smith, M. Congenital nevi. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:953-957.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/.
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com.