User login
Indurated Violaceous Lesions on the Face, Trunk, and Legs
The Diagnosis: Kaposi Sarcoma
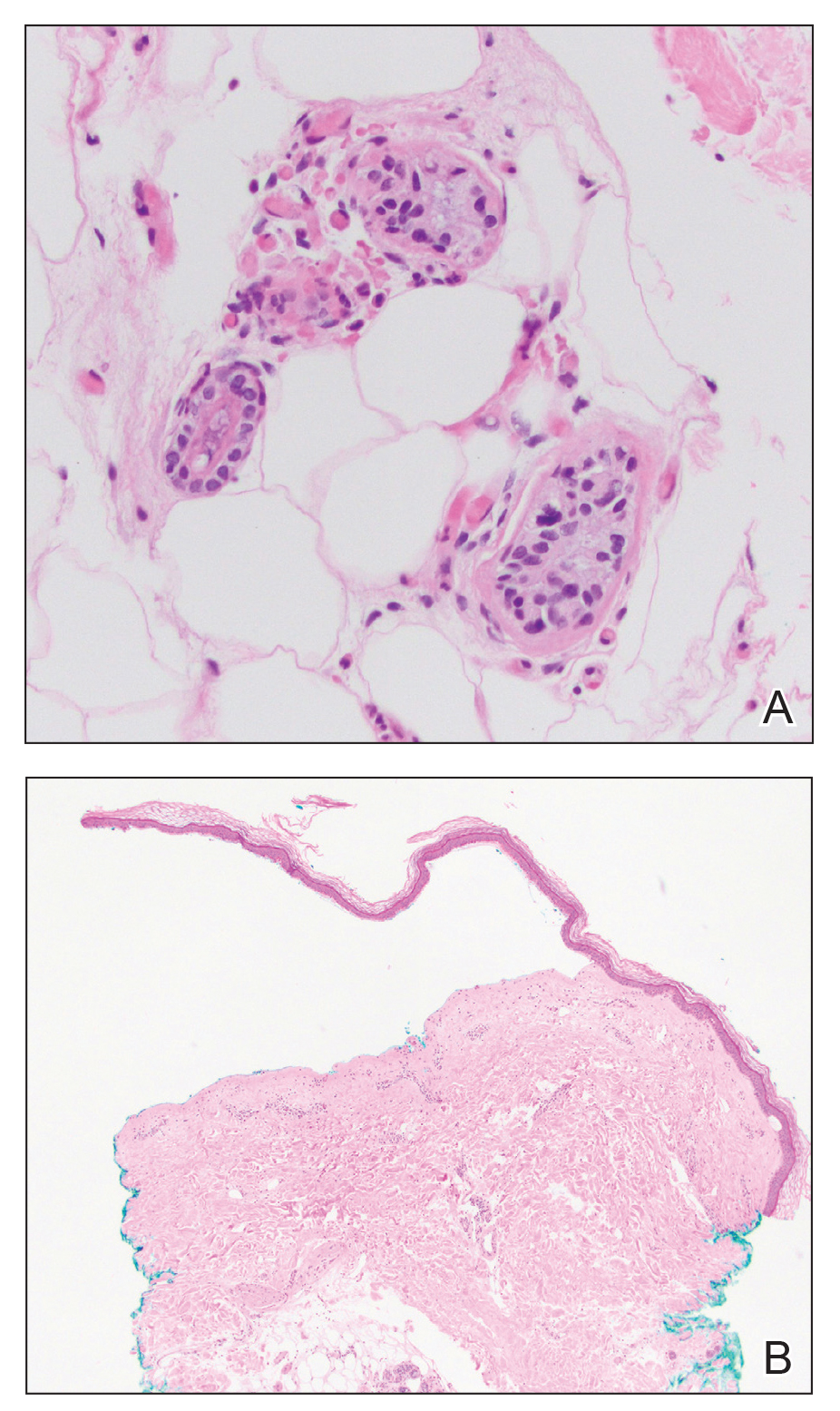
A punch biopsy of a lesion on the right side of the back revealed a diffuse, poorly circumscribed, spindle cell neoplasm of the papillary and reticular dermis with associated vascular and pseudovascular spaces distended by erythrocytes (Figure 1). Immunostaining was positive for human herpesvirus 8 (HHV-8)(Figure 2), ETS-related gene, CD31, and CD34 and negative for pan cytokeratin, confirming the diagnosis of Kaposi sarcoma (KS). Bacterial, fungal, and mycobacterial tissue cultures were negative. The patient was tested for HIV and referred to infectious disease and oncology. He subsequently was found to have HIV with a viral load greater than 1 million copies. He was started on antiretroviral therapy and Pneumocystis jirovecii pneumonia prophylaxis. Computed tomography of the chest, abdomen, and pelvis showed bilateral, multifocal, perihilar, flame-shaped consolidations suggestive of KS. The patient later disclosed having an intermittent dry cough of more than a year’s duration with occasional bright red blood per rectum after bowel movements. After workup, the patient was found to have cytomegalovirus esophagitis/gastritis and candidal esophagitis that were treated with valganciclovir and fluconazole, respectively.
.")
Kaposi sarcoma is an angioproliferative, AIDSdefining disease associated with HHV-8. There are 4 types of KS as defined by the populations they affect. AIDS-associated KS occurs in individuals with HIV, as seen in our patient. It often is accompanied by extensive mucocutaneous and visceral lesions, as well as systemic symptoms such as fever, weight loss, and diarrhea.1 Classic KS is a variant that presents in older men of Mediterranean, Eastern European, and South American descent. Cutaneous lesions typically are distributed on the lower extremities.2,3 Endemic (African) KS is seen in HIV-negative children and young adults in equatorial Africa. It most commonly affects the lower extremities or lymph nodes and usually follows a more aggressive course.2 Lastly, iatrogenic KS is associated with immunosuppressive medications or conditions, such as organ transplantation, chemotherapy, and rheumatologic disorders.3,4
.")
Kaposi sarcoma commonly presents as violaceous or dark red macules, patches, papules, plaques, and nodules on various parts of the body (Figure 3). Lesions typically begin as macules and progress into plaques or nodules. Our patient presented as a deceptively healthy young man with lesions at various stages of development. In addition to the skin and oral mucosa, the lungs, lymph nodes, and gastrointestinal tract commonly are involved in AIDS-associated KS.5 Patients may experience symptoms of internal involvement, including bleeding, hematochezia, odynophagia, or dyspnea.

The differential diagnosis includes conditions that can mimic KS, including bacillary angiomatosis, angioinvasive fungal disease, sarcoid, and other malignancies. A skin biopsy is the gold standard for definitive diagnosis of KS. Histopathology shows a vascular proliferation in the dermis and spindle cell proliferation.6 Kaposi sarcoma stains positively for factor VIII–related antigen, CD31, and CD34.2 Additionally, staining for HHV-8 gene products, such as latency-associated nuclear antigen 1, is helpful in differentiating KS from other conditions.7
In HIV-associated KS, the mainstay of treatment is initiation of highly active antiretroviral therapy. Typically, as the CD4 count rises with treatment, the tumor burden classic KS, effective treatment options include recurrent cryotherapy or intralesional chemotherapeutics, such as vincristine, for localized lesions; for widespread disease, pegylated liposomal doxorubicin or radiation have been found to be effective options. Lastly, for patients with iatrogenic KS, reducing immunosuppressive medications is a reasonable first step in management. If this does not yield adequate improvement, transitioning from calcineurin inhibitors (eg, cyclosporine) to proliferation signal inhibitors (eg, sirolimus) may lead to resolution.7
- Friedman-Kien AE, Saltzman BR. Clinical manifestations of classical, endemic African, and epidemic AIDS-associated Kaposi’s sarcoma. J Am Acad Dermatol. 1990;22:1237-1250.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Vangipuram R, Tyring SK. Epidemiology of Kaposi sarcoma: review and description of the nonepidemic variant. Int J Dermatol. 2019;58:538-542.
- Klepp O, Dahl O, Stenwig JT. Association of Kaposi’s sarcoma and prior immunosuppressive therapy. a 5‐year material of Kaposi’s sarcoma in Norway. Cancer. 1978;42:2626-2630.
- Lemlich G, Schwam L, Lebwohl M. Kaposi’s sarcoma and acquired immunodeficiency syndrome: postmortem findings in twenty-four cases. J Am Acad Dermatol. 1987;16:319-325.
- Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:10.
- Curtiss P, Strazzulla LC, Friedman-Kien AE. An update on Kaposi’s sarcoma: epidemiology, pathogenesis and treatment. Dermatol Ther. 2016;6:465-470.
The Diagnosis: Kaposi Sarcoma
A punch biopsy of a lesion on the right side of the back revealed a diffuse, poorly circumscribed, spindle cell neoplasm of the papillary and reticular dermis with associated vascular and pseudovascular spaces distended by erythrocytes (Figure 1). Immunostaining was positive for human herpesvirus 8 (HHV-8)(Figure 2), ETS-related gene, CD31, and CD34 and negative for pan cytokeratin, confirming the diagnosis of Kaposi sarcoma (KS). Bacterial, fungal, and mycobacterial tissue cultures were negative. The patient was tested for HIV and referred to infectious disease and oncology. He subsequently was found to have HIV with a viral load greater than 1 million copies. He was started on antiretroviral therapy and Pneumocystis jirovecii pneumonia prophylaxis. Computed tomography of the chest, abdomen, and pelvis showed bilateral, multifocal, perihilar, flame-shaped consolidations suggestive of KS. The patient later disclosed having an intermittent dry cough of more than a year’s duration with occasional bright red blood per rectum after bowel movements. After workup, the patient was found to have cytomegalovirus esophagitis/gastritis and candidal esophagitis that were treated with valganciclovir and fluconazole, respectively.
Kaposi sarcoma is an angioproliferative, AIDSdefining disease associated with HHV-8. There are 4 types of KS as defined by the populations they affect. AIDS-associated KS occurs in individuals with HIV, as seen in our patient. It often is accompanied by extensive mucocutaneous and visceral lesions, as well as systemic symptoms such as fever, weight loss, and diarrhea.1 Classic KS is a variant that presents in older men of Mediterranean, Eastern European, and South American descent. Cutaneous lesions typically are distributed on the lower extremities.2,3 Endemic (African) KS is seen in HIV-negative children and young adults in equatorial Africa. It most commonly affects the lower extremities or lymph nodes and usually follows a more aggressive course.2 Lastly, iatrogenic KS is associated with immunosuppressive medications or conditions, such as organ transplantation, chemotherapy, and rheumatologic disorders.3,4
Kaposi sarcoma commonly presents as violaceous or dark red macules, patches, papules, plaques, and nodules on various parts of the body (Figure 3). Lesions typically begin as macules and progress into plaques or nodules. Our patient presented as a deceptively healthy young man with lesions at various stages of development. In addition to the skin and oral mucosa, the lungs, lymph nodes, and gastrointestinal tract commonly are involved in AIDS-associated KS.5 Patients may experience symptoms of internal involvement, including bleeding, hematochezia, odynophagia, or dyspnea.
The differential diagnosis includes conditions that can mimic KS, including bacillary angiomatosis, angioinvasive fungal disease, sarcoid, and other malignancies. A skin biopsy is the gold standard for definitive diagnosis of KS. Histopathology shows a vascular proliferation in the dermis and spindle cell proliferation.6 Kaposi sarcoma stains positively for factor VIII–related antigen, CD31, and CD34.2 Additionally, staining for HHV-8 gene products, such as latency-associated nuclear antigen 1, is helpful in differentiating KS from other conditions.7
In HIV-associated KS, the mainstay of treatment is initiation of highly active antiretroviral therapy. Typically, as the CD4 count rises with treatment, the tumor burden classic KS, effective treatment options include recurrent cryotherapy or intralesional chemotherapeutics, such as vincristine, for localized lesions; for widespread disease, pegylated liposomal doxorubicin or radiation have been found to be effective options. Lastly, for patients with iatrogenic KS, reducing immunosuppressive medications is a reasonable first step in management. If this does not yield adequate improvement, transitioning from calcineurin inhibitors (eg, cyclosporine) to proliferation signal inhibitors (eg, sirolimus) may lead to resolution.7
The Diagnosis: Kaposi Sarcoma
A punch biopsy of a lesion on the right side of the back revealed a diffuse, poorly circumscribed, spindle cell neoplasm of the papillary and reticular dermis with associated vascular and pseudovascular spaces distended by erythrocytes (Figure 1). Immunostaining was positive for human herpesvirus 8 (HHV-8)(Figure 2), ETS-related gene, CD31, and CD34 and negative for pan cytokeratin, confirming the diagnosis of Kaposi sarcoma (KS). Bacterial, fungal, and mycobacterial tissue cultures were negative. The patient was tested for HIV and referred to infectious disease and oncology. He subsequently was found to have HIV with a viral load greater than 1 million copies. He was started on antiretroviral therapy and Pneumocystis jirovecii pneumonia prophylaxis. Computed tomography of the chest, abdomen, and pelvis showed bilateral, multifocal, perihilar, flame-shaped consolidations suggestive of KS. The patient later disclosed having an intermittent dry cough of more than a year’s duration with occasional bright red blood per rectum after bowel movements. After workup, the patient was found to have cytomegalovirus esophagitis/gastritis and candidal esophagitis that were treated with valganciclovir and fluconazole, respectively.
Kaposi sarcoma is an angioproliferative, AIDSdefining disease associated with HHV-8. There are 4 types of KS as defined by the populations they affect. AIDS-associated KS occurs in individuals with HIV, as seen in our patient. It often is accompanied by extensive mucocutaneous and visceral lesions, as well as systemic symptoms such as fever, weight loss, and diarrhea.1 Classic KS is a variant that presents in older men of Mediterranean, Eastern European, and South American descent. Cutaneous lesions typically are distributed on the lower extremities.2,3 Endemic (African) KS is seen in HIV-negative children and young adults in equatorial Africa. It most commonly affects the lower extremities or lymph nodes and usually follows a more aggressive course.2 Lastly, iatrogenic KS is associated with immunosuppressive medications or conditions, such as organ transplantation, chemotherapy, and rheumatologic disorders.3,4
Kaposi sarcoma commonly presents as violaceous or dark red macules, patches, papules, plaques, and nodules on various parts of the body (Figure 3). Lesions typically begin as macules and progress into plaques or nodules. Our patient presented as a deceptively healthy young man with lesions at various stages of development. In addition to the skin and oral mucosa, the lungs, lymph nodes, and gastrointestinal tract commonly are involved in AIDS-associated KS.5 Patients may experience symptoms of internal involvement, including bleeding, hematochezia, odynophagia, or dyspnea.
The differential diagnosis includes conditions that can mimic KS, including bacillary angiomatosis, angioinvasive fungal disease, sarcoid, and other malignancies. A skin biopsy is the gold standard for definitive diagnosis of KS. Histopathology shows a vascular proliferation in the dermis and spindle cell proliferation.6 Kaposi sarcoma stains positively for factor VIII–related antigen, CD31, and CD34.2 Additionally, staining for HHV-8 gene products, such as latency-associated nuclear antigen 1, is helpful in differentiating KS from other conditions.7
In HIV-associated KS, the mainstay of treatment is initiation of highly active antiretroviral therapy. Typically, as the CD4 count rises with treatment, the tumor burden classic KS, effective treatment options include recurrent cryotherapy or intralesional chemotherapeutics, such as vincristine, for localized lesions; for widespread disease, pegylated liposomal doxorubicin or radiation have been found to be effective options. Lastly, for patients with iatrogenic KS, reducing immunosuppressive medications is a reasonable first step in management. If this does not yield adequate improvement, transitioning from calcineurin inhibitors (eg, cyclosporine) to proliferation signal inhibitors (eg, sirolimus) may lead to resolution.7
- Friedman-Kien AE, Saltzman BR. Clinical manifestations of classical, endemic African, and epidemic AIDS-associated Kaposi’s sarcoma. J Am Acad Dermatol. 1990;22:1237-1250.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Vangipuram R, Tyring SK. Epidemiology of Kaposi sarcoma: review and description of the nonepidemic variant. Int J Dermatol. 2019;58:538-542.
- Klepp O, Dahl O, Stenwig JT. Association of Kaposi’s sarcoma and prior immunosuppressive therapy. a 5‐year material of Kaposi’s sarcoma in Norway. Cancer. 1978;42:2626-2630.
- Lemlich G, Schwam L, Lebwohl M. Kaposi’s sarcoma and acquired immunodeficiency syndrome: postmortem findings in twenty-four cases. J Am Acad Dermatol. 1987;16:319-325.
- Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:10.
- Curtiss P, Strazzulla LC, Friedman-Kien AE. An update on Kaposi’s sarcoma: epidemiology, pathogenesis and treatment. Dermatol Ther. 2016;6:465-470.
- Friedman-Kien AE, Saltzman BR. Clinical manifestations of classical, endemic African, and epidemic AIDS-associated Kaposi’s sarcoma. J Am Acad Dermatol. 1990;22:1237-1250.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Vangipuram R, Tyring SK. Epidemiology of Kaposi sarcoma: review and description of the nonepidemic variant. Int J Dermatol. 2019;58:538-542.
- Klepp O, Dahl O, Stenwig JT. Association of Kaposi’s sarcoma and prior immunosuppressive therapy. a 5‐year material of Kaposi’s sarcoma in Norway. Cancer. 1978;42:2626-2630.
- Lemlich G, Schwam L, Lebwohl M. Kaposi’s sarcoma and acquired immunodeficiency syndrome: postmortem findings in twenty-four cases. J Am Acad Dermatol. 1987;16:319-325.
- Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:10.
- Curtiss P, Strazzulla LC, Friedman-Kien AE. An update on Kaposi’s sarcoma: epidemiology, pathogenesis and treatment. Dermatol Ther. 2016;6:465-470.
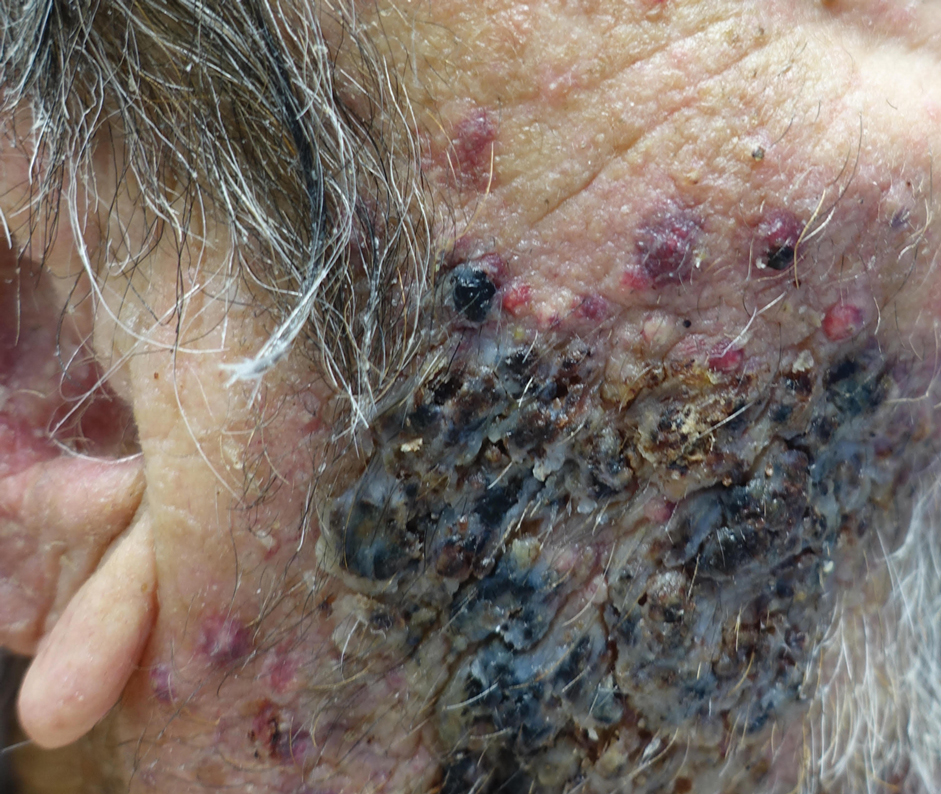
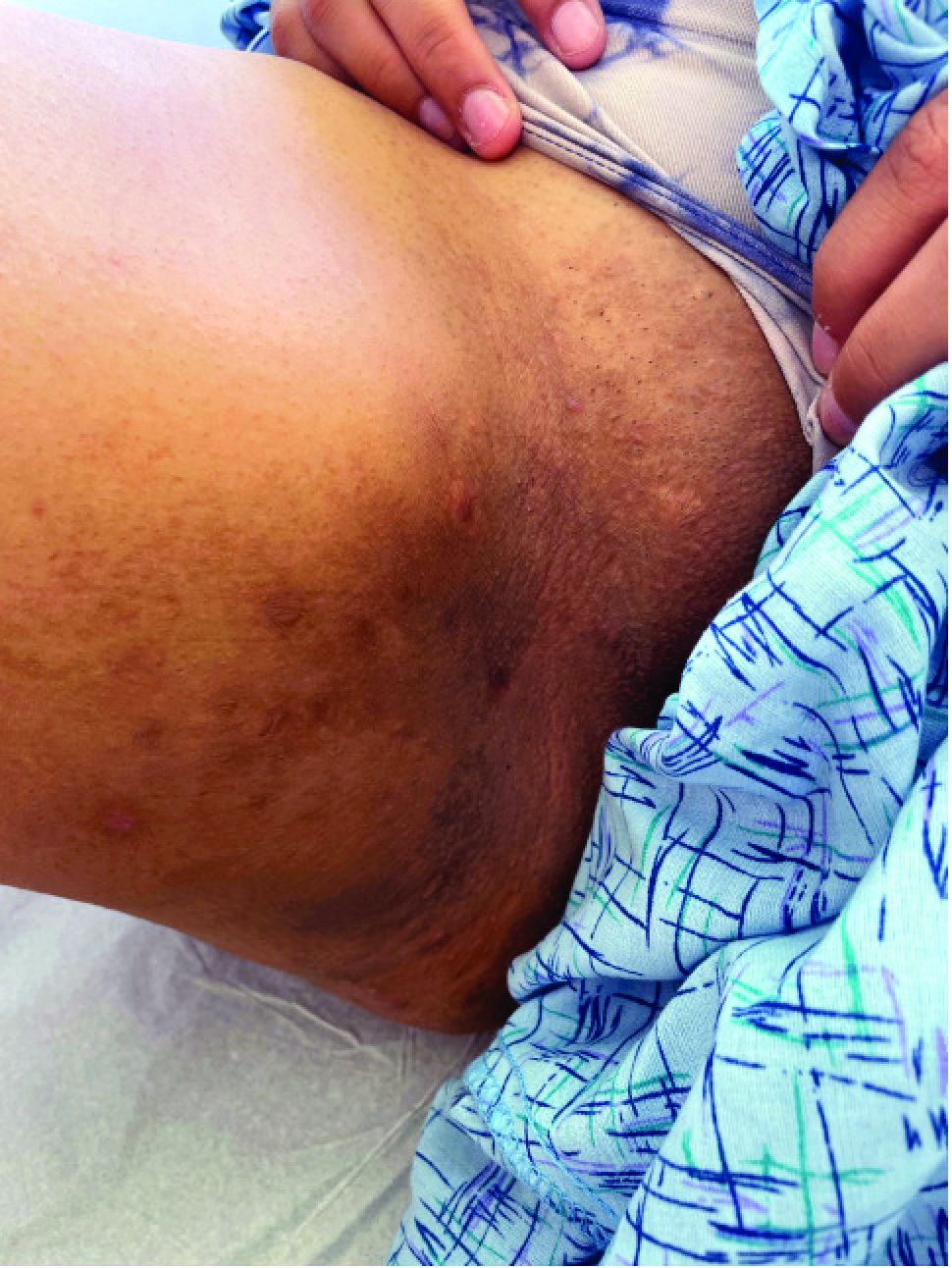
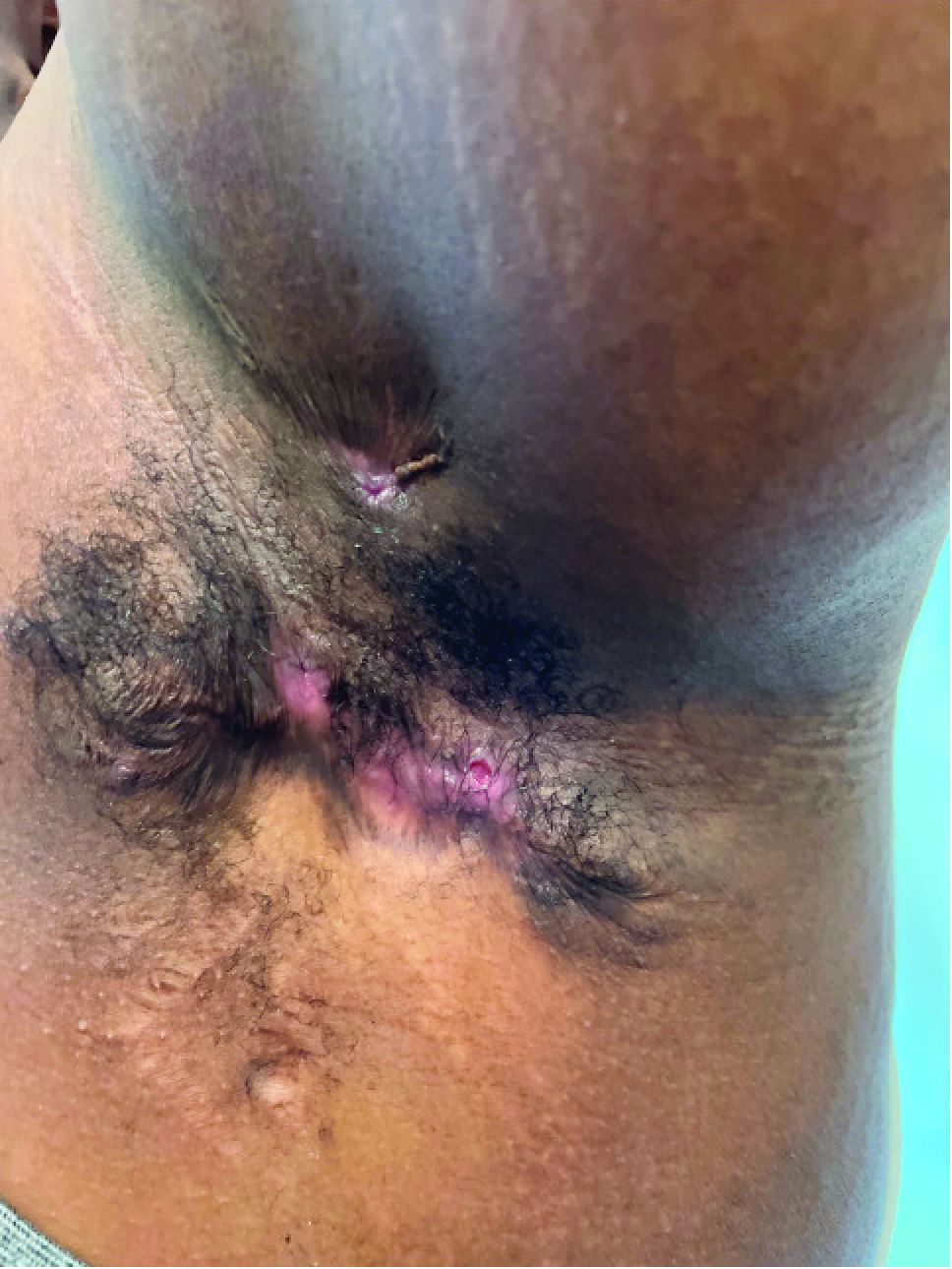
A 25-year-old man with no notable medical history presented to the dermatology clinic with growing selfdescribed cysts on the face, trunk, and legs of 6 months’ duration. The lesions started as bruiselike discolorations and progressed to become firm nodules and inflamed masses. Some were minimally itchy and sensitive to touch, but there was no history of bleeding or drainage. The patient denied any new or recent environmental or animal exposures, use of illicit drugs, or travel correlating with the rash onset. He denied any prior treatments. He reported being in his normal state of health and was not taking any medications. Physical examination revealed indurated, violaceous, purpuric subcutaneous nodules, plaques, and masses on the forehead, cheek (top), jaw, flank, axillae (bottom), and back.


Blisters in a Comatose Elderly Woman
The Diagnosis: Coma Blisters
Histologic examination revealed pauci-inflammatory subepidermal blisters with swelling of eccrine cells, signaling impending gland necrosis (Figure). Direct immunofluorescence testing on perilesional skin was negative. These findings would be inconsistent for diagnoses of edema blisters (most commonly seen in patients with an acute exacerbation of chronic lower extremity edema), friction blisters (intraepidermal blisters seen on histopathology), and bullous pemphigoid (linear IgG and/or C3 staining along the basement membrane zone on direct immunofluorescence testing is characteristic). Although eccrine gland alterations have been seen in toxic epidermal necrolysis,1 the mucous membranes are involved in more than 90% of cases, making the diagnosis less likely. Furthermore, interface changes including prominent keratinocyte necrosis were not seen on histology.
Given the localized nature of the lesions in our patient and negative direct immunofluorescence studies, a diagnosis of coma blisters was made. Gentle wound care practices to the areas of denuded skin were implemented with complete resolution. The patient’s condition gradually improved, and she was extubated and discharged home.
Coma blisters are self-limited bullous lesions that have been reported in comatose patients as early as 1812 when Napoleon’s surgeon first noticed cutaneous blisters in comatose French soldiers being treated for carbon monoxide intoxication.2 Since then, barbiturate overdose has remained the most common association, but coma blisters have occurred in the absence of specific drug exposures. Clinically, erythematous or violaceous plaques typically appear within 24 hours of drug ingestion, and progression to large tense bullae usually occurs within 48 to 72 hours of unconsciousness.3 They characteristically occur in pressure-dependent areas, but reports have shown lesions in non–pressure-dependent areas, including the penis and mouth.1,4 Spontaneous resolution within 1 to 2 weeks is typical.5
The underlying pathogenesis remains controversial, as multiple mechanisms have been suggested, but clear causal evidence is lacking. The original proposition that direct effects of drug toxicity caused the cutaneous observations was later refuted after similar bullous lesions with eccrine gland necrosis were reported in comatose patients with neurologic conditions.6 It is largely accepted that pressure-induced local ischemia—proportional to the duration and amount of pressure—leads to tissue injury and is critical to the pathogenesis. During periods of ischemia, the most metabolically active tissues will undergo necrosis first; however, in eccrine glands, the earliest and most severe damage does not seem to occur in the most metabolically active cells.7 Additionally, this would not provide a viable explanation for coma blisters with eccrine gland necrosis developing in variable non–pressuredependent areas.
Moreover, drug- and non–drug-induced coma blisters can appear identically, but specific histopathologic differences have been reported. The most notable markers of non–drug-induced coma blisters are the absence of an inflammatory infiltrate in the epidermis and the presence of thrombosis in dermal vessels.8 Demonstration of necrotic changes in the secretory portion of the eccrine gland is considered the histopathologic hallmark for drug-induced coma blisters, but other findings can include subepidermal or intraepidermal bullae; perivascular infiltrates; and focal necrosis of the epidermis, dermis, subcutis, or epidermal appendages.6 Arteriolar wall necrosis and dermal inflammatory infiltrates also have been observed.7
Benzodiazepines have been widely prescribed and abused since their development, and overdose is much more common today than with barbiturates.9 Coma blisters rarely have been documented in the setting of isolated benzodiazepine overdose, and of the few cases, only one report implicated lorazepam as the causative agent.4,7 The characteristic finding of eccrine gland necrosis consistently was seen in our patient. This case not only emphasizes the need for greater awareness of the association between benzodiazepine overdose and coma blisters but also the importance of clinical context when considering diagnoses. It is essential to note that coma blisters themselves are nonspecific, and the diagnosis of drug-induced coma blisters warrants confirmatory toxicologic analysis.
- Ferreli C, Sulica VI, Aste N, et al. Drug-induced sweat gland necrosis in a non-comatose patient: a case presentation. J Eur Acad Dermatol Venereol. 2003;17:443-445.
- Larrey DJ. Memoires de Chirurgie Militaire et Campagnes. Smith and Buisson; 1812.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online J. 2012;18:10.
- Varma AJ, Fisher BK, Sarin MK. Diazepam-induced coma with bullae and eccrine sweat gland necrosis. Arch Intern Med. 1977;137:1207-1210.
- Rocha J, Pereira T, Ventura F, et al. Coma blisters. Case Rep Dermatol. 2009;1:66-70.
- Arndt KA, Mihm MC, Parrish JA. Bullae: a cutaneous sign of a variety of neurologic diseases. J Invest Dermatol. 1973;60:312-320.
- Sánchez Yus E, Requena L, Simón P. Histopathology of cutaneous changes in drug-induced coma. Am J Dermatopathol. 1993;15:208-216.
- Kato N, Ueno H, Mimura M. Histopathology of cutaneous changes in non-drug-induced coma. Am J Dermatopathol. 1996;18:344-350.
- Kang M, Ghassemzadeh S. Benzodiazepine Toxicity. StatPearls Publishing; 2018.
The Diagnosis: Coma Blisters
Histologic examination revealed pauci-inflammatory subepidermal blisters with swelling of eccrine cells, signaling impending gland necrosis (Figure). Direct immunofluorescence testing on perilesional skin was negative. These findings would be inconsistent for diagnoses of edema blisters (most commonly seen in patients with an acute exacerbation of chronic lower extremity edema), friction blisters (intraepidermal blisters seen on histopathology), and bullous pemphigoid (linear IgG and/or C3 staining along the basement membrane zone on direct immunofluorescence testing is characteristic). Although eccrine gland alterations have been seen in toxic epidermal necrolysis,1 the mucous membranes are involved in more than 90% of cases, making the diagnosis less likely. Furthermore, interface changes including prominent keratinocyte necrosis were not seen on histology.
Given the localized nature of the lesions in our patient and negative direct immunofluorescence studies, a diagnosis of coma blisters was made. Gentle wound care practices to the areas of denuded skin were implemented with complete resolution. The patient’s condition gradually improved, and she was extubated and discharged home.
Coma blisters are self-limited bullous lesions that have been reported in comatose patients as early as 1812 when Napoleon’s surgeon first noticed cutaneous blisters in comatose French soldiers being treated for carbon monoxide intoxication.2 Since then, barbiturate overdose has remained the most common association, but coma blisters have occurred in the absence of specific drug exposures. Clinically, erythematous or violaceous plaques typically appear within 24 hours of drug ingestion, and progression to large tense bullae usually occurs within 48 to 72 hours of unconsciousness.3 They characteristically occur in pressure-dependent areas, but reports have shown lesions in non–pressure-dependent areas, including the penis and mouth.1,4 Spontaneous resolution within 1 to 2 weeks is typical.5
The underlying pathogenesis remains controversial, as multiple mechanisms have been suggested, but clear causal evidence is lacking. The original proposition that direct effects of drug toxicity caused the cutaneous observations was later refuted after similar bullous lesions with eccrine gland necrosis were reported in comatose patients with neurologic conditions.6 It is largely accepted that pressure-induced local ischemia—proportional to the duration and amount of pressure—leads to tissue injury and is critical to the pathogenesis. During periods of ischemia, the most metabolically active tissues will undergo necrosis first; however, in eccrine glands, the earliest and most severe damage does not seem to occur in the most metabolically active cells.7 Additionally, this would not provide a viable explanation for coma blisters with eccrine gland necrosis developing in variable non–pressuredependent areas.
Moreover, drug- and non–drug-induced coma blisters can appear identically, but specific histopathologic differences have been reported. The most notable markers of non–drug-induced coma blisters are the absence of an inflammatory infiltrate in the epidermis and the presence of thrombosis in dermal vessels.8 Demonstration of necrotic changes in the secretory portion of the eccrine gland is considered the histopathologic hallmark for drug-induced coma blisters, but other findings can include subepidermal or intraepidermal bullae; perivascular infiltrates; and focal necrosis of the epidermis, dermis, subcutis, or epidermal appendages.6 Arteriolar wall necrosis and dermal inflammatory infiltrates also have been observed.7
Benzodiazepines have been widely prescribed and abused since their development, and overdose is much more common today than with barbiturates.9 Coma blisters rarely have been documented in the setting of isolated benzodiazepine overdose, and of the few cases, only one report implicated lorazepam as the causative agent.4,7 The characteristic finding of eccrine gland necrosis consistently was seen in our patient. This case not only emphasizes the need for greater awareness of the association between benzodiazepine overdose and coma blisters but also the importance of clinical context when considering diagnoses. It is essential to note that coma blisters themselves are nonspecific, and the diagnosis of drug-induced coma blisters warrants confirmatory toxicologic analysis.
The Diagnosis: Coma Blisters
Histologic examination revealed pauci-inflammatory subepidermal blisters with swelling of eccrine cells, signaling impending gland necrosis (Figure). Direct immunofluorescence testing on perilesional skin was negative. These findings would be inconsistent for diagnoses of edema blisters (most commonly seen in patients with an acute exacerbation of chronic lower extremity edema), friction blisters (intraepidermal blisters seen on histopathology), and bullous pemphigoid (linear IgG and/or C3 staining along the basement membrane zone on direct immunofluorescence testing is characteristic). Although eccrine gland alterations have been seen in toxic epidermal necrolysis,1 the mucous membranes are involved in more than 90% of cases, making the diagnosis less likely. Furthermore, interface changes including prominent keratinocyte necrosis were not seen on histology.
Given the localized nature of the lesions in our patient and negative direct immunofluorescence studies, a diagnosis of coma blisters was made. Gentle wound care practices to the areas of denuded skin were implemented with complete resolution. The patient’s condition gradually improved, and she was extubated and discharged home.
Coma blisters are self-limited bullous lesions that have been reported in comatose patients as early as 1812 when Napoleon’s surgeon first noticed cutaneous blisters in comatose French soldiers being treated for carbon monoxide intoxication.2 Since then, barbiturate overdose has remained the most common association, but coma blisters have occurred in the absence of specific drug exposures. Clinically, erythematous or violaceous plaques typically appear within 24 hours of drug ingestion, and progression to large tense bullae usually occurs within 48 to 72 hours of unconsciousness.3 They characteristically occur in pressure-dependent areas, but reports have shown lesions in non–pressure-dependent areas, including the penis and mouth.1,4 Spontaneous resolution within 1 to 2 weeks is typical.5
The underlying pathogenesis remains controversial, as multiple mechanisms have been suggested, but clear causal evidence is lacking. The original proposition that direct effects of drug toxicity caused the cutaneous observations was later refuted after similar bullous lesions with eccrine gland necrosis were reported in comatose patients with neurologic conditions.6 It is largely accepted that pressure-induced local ischemia—proportional to the duration and amount of pressure—leads to tissue injury and is critical to the pathogenesis. During periods of ischemia, the most metabolically active tissues will undergo necrosis first; however, in eccrine glands, the earliest and most severe damage does not seem to occur in the most metabolically active cells.7 Additionally, this would not provide a viable explanation for coma blisters with eccrine gland necrosis developing in variable non–pressuredependent areas.
Moreover, drug- and non–drug-induced coma blisters can appear identically, but specific histopathologic differences have been reported. The most notable markers of non–drug-induced coma blisters are the absence of an inflammatory infiltrate in the epidermis and the presence of thrombosis in dermal vessels.8 Demonstration of necrotic changes in the secretory portion of the eccrine gland is considered the histopathologic hallmark for drug-induced coma blisters, but other findings can include subepidermal or intraepidermal bullae; perivascular infiltrates; and focal necrosis of the epidermis, dermis, subcutis, or epidermal appendages.6 Arteriolar wall necrosis and dermal inflammatory infiltrates also have been observed.7
Benzodiazepines have been widely prescribed and abused since their development, and overdose is much more common today than with barbiturates.9 Coma blisters rarely have been documented in the setting of isolated benzodiazepine overdose, and of the few cases, only one report implicated lorazepam as the causative agent.4,7 The characteristic finding of eccrine gland necrosis consistently was seen in our patient. This case not only emphasizes the need for greater awareness of the association between benzodiazepine overdose and coma blisters but also the importance of clinical context when considering diagnoses. It is essential to note that coma blisters themselves are nonspecific, and the diagnosis of drug-induced coma blisters warrants confirmatory toxicologic analysis.
- Ferreli C, Sulica VI, Aste N, et al. Drug-induced sweat gland necrosis in a non-comatose patient: a case presentation. J Eur Acad Dermatol Venereol. 2003;17:443-445.
- Larrey DJ. Memoires de Chirurgie Militaire et Campagnes. Smith and Buisson; 1812.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online J. 2012;18:10.
- Varma AJ, Fisher BK, Sarin MK. Diazepam-induced coma with bullae and eccrine sweat gland necrosis. Arch Intern Med. 1977;137:1207-1210.
- Rocha J, Pereira T, Ventura F, et al. Coma blisters. Case Rep Dermatol. 2009;1:66-70.
- Arndt KA, Mihm MC, Parrish JA. Bullae: a cutaneous sign of a variety of neurologic diseases. J Invest Dermatol. 1973;60:312-320.
- Sánchez Yus E, Requena L, Simón P. Histopathology of cutaneous changes in drug-induced coma. Am J Dermatopathol. 1993;15:208-216.
- Kato N, Ueno H, Mimura M. Histopathology of cutaneous changes in non-drug-induced coma. Am J Dermatopathol. 1996;18:344-350.
- Kang M, Ghassemzadeh S. Benzodiazepine Toxicity. StatPearls Publishing; 2018.
- Ferreli C, Sulica VI, Aste N, et al. Drug-induced sweat gland necrosis in a non-comatose patient: a case presentation. J Eur Acad Dermatol Venereol. 2003;17:443-445.
- Larrey DJ. Memoires de Chirurgie Militaire et Campagnes. Smith and Buisson; 1812.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online J. 2012;18:10.
- Varma AJ, Fisher BK, Sarin MK. Diazepam-induced coma with bullae and eccrine sweat gland necrosis. Arch Intern Med. 1977;137:1207-1210.
- Rocha J, Pereira T, Ventura F, et al. Coma blisters. Case Rep Dermatol. 2009;1:66-70.
- Arndt KA, Mihm MC, Parrish JA. Bullae: a cutaneous sign of a variety of neurologic diseases. J Invest Dermatol. 1973;60:312-320.
- Sánchez Yus E, Requena L, Simón P. Histopathology of cutaneous changes in drug-induced coma. Am J Dermatopathol. 1993;15:208-216.
- Kato N, Ueno H, Mimura M. Histopathology of cutaneous changes in non-drug-induced coma. Am J Dermatopathol. 1996;18:344-350.
- Kang M, Ghassemzadeh S. Benzodiazepine Toxicity. StatPearls Publishing; 2018.
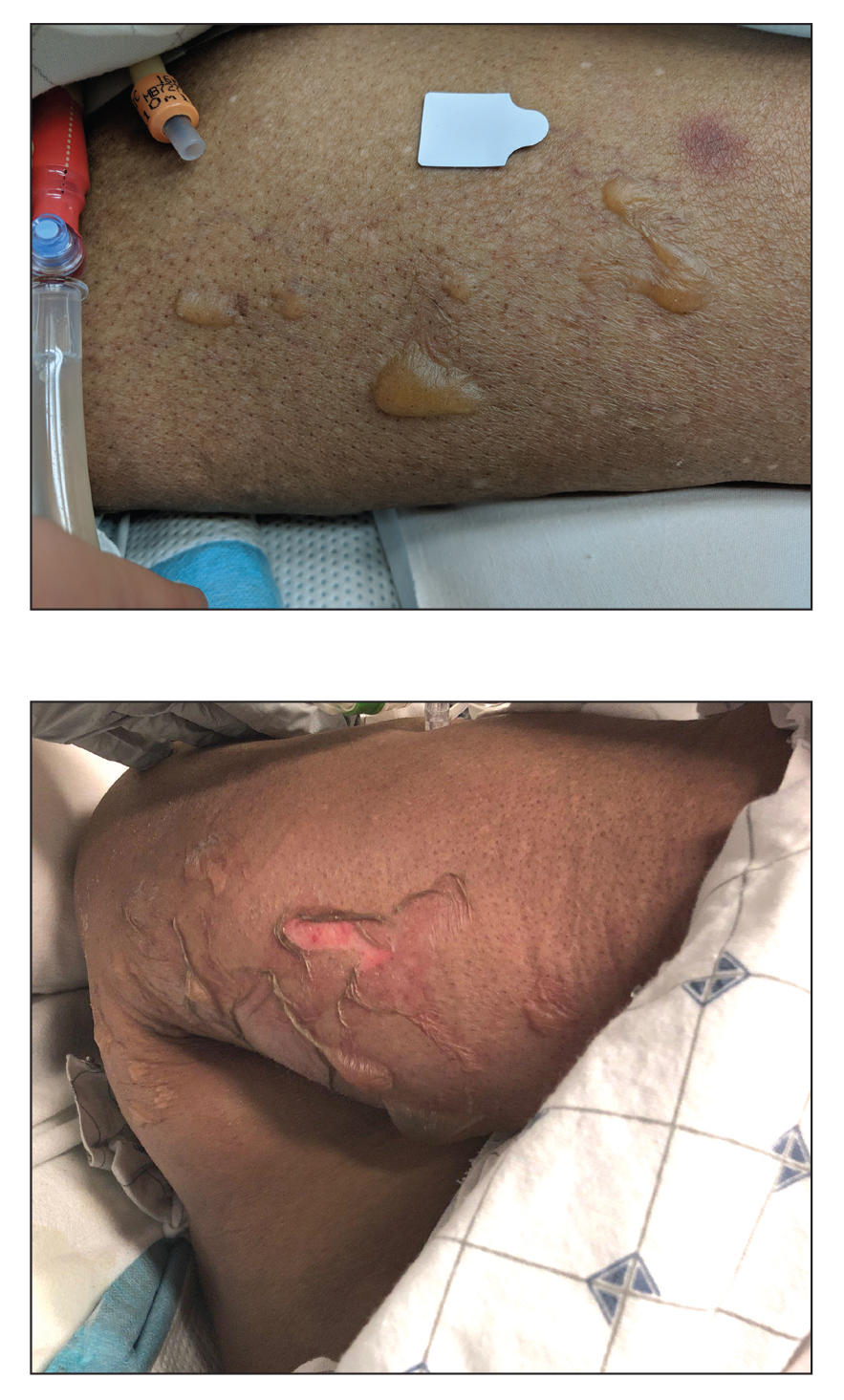
An 82-year-old woman presented to the emergency department after her daughter found her unconscious in the bathroom laying on her right side. Her medical history was notable for hypertension and asthma for which she was on losartan, furosemide, diltiazem, and albuterol. She recently had been prescribed lorazepam for insomnia and had started taking the medication 2 days prior. She underwent intubation and was noted to have flaccid, fluid-filled bullae on the right thigh (top) along with large areas of desquamation on the right lateral arm (bottom) with minimal surrounding erythema. There was no mucous membrane involvement. Urine toxicology was positive for benzodiazepines and negative for all other drugs, including barbiturates.
What is the diagnosis?
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that is becoming more recognized in children. It has a variable presentation, most commonly presenting as painful, recurrent cysts, abscesses, nodules, and/or pustules in classic locations with associated scarring and sinus tract formation.
The majority of patients present with bilateral lesions found most commonly in the axillae and inguinal folds.1 There are myriad other potential sites of involvement including the inframammary folds, inner thighs, buttocks, and groin.1 Diagnosis is made based on history and physical exam. There is a standard severity classification scheme called the Hurley score, which stratifies disease severity based on the presence of sinus tracts and extent of disease.1 HS is associated with comorbid conditions such as obesity, overweight, acne, and inflammatory bowel and joint disease.2 This painful, persistent condition is well documented to have a negative impact on quality of life in adult patients, and similar impairment has been found in pediatric patients.3,4

HS may be increasing in pediatric and adolescent patients, with recent studies showing onset coinciding most commonly with the onset of puberty.1,2 There is often a period of several years between symptom onset and diagnosis.1 A recent editorial highlighted the disparities that exist in HS, with disease more common in Black children and limited information about disease prevalence in Hispanic children.5
What’s the treatment plan?
HS is a difficult disease to treat, with few patients achieving remission and a significant proportion of patients with treatment-refractory disease.1 There are limited studies of HS treatment in pediatric patients. Topical and systemic antibiotic therapy are mainstays of HS treatment, with tetracyclines and a combination of clindamycin plus rifampin commonly used in adults and children alike. Topical therapies including topical antibiotics and antibacterial solutions are frequently used as adjunctive therapy.6 Adalimumab, a tumor necrosis factor receptor blocker, has been Food and Drug Administration approved for HS for ages 12 and up and is currently the only FDA-approved medication for HS in pediatric patients. Our patient was started on 100 mg doxycycline twice daily, with short-dose topical corticosteroids for symptom management of the most inflamed lesions.
What’s on the differential?
Acne conglobata
Acne conglobata is an uncommon, severe variant of acne vulgaris which arise in patients with a history of acne vulgaris and presents with comedones, cysts, abscesses, and scarring with possible drainage of pus. Lesions can present diffusely on the face, back, and body, including in the axillae, groin, and buttocks, and as such can be confused with HS.7
However, in contrast with HS, patients with acne conglobata will also develop disease in non–apocrine gland–bearing skin. This patient’s lack of preceding acne and restriction of lesions to the axillae, inguinal folds, and buttocks makes acne conglobata less likely.

Epidermal inclusion cyst
Epidermal inclusion cyst (EIC) is a common cutaneous cyst, presenting as a well-circumscribed nodule(s) with a central punctum. If not excised, lesions can sometimes become infected and painful.8 In contrast with HS, EIC presents only uncommonly as multiple lesions arising in different areas, and spontaneous drainage is uncommon. Our patient’s development of multiple draining lesions makes this diagnosis unlikely.
Furunculosis
Furunculosis is a common bacterial infection of the skin, presenting with inflammatory nodules or pustules centered around the hair follicle. Lesions may commonly present at sites of skin trauma and are found most frequently on the extremities.9 Though furunculosis lesions may drain pus and can coalesce to form larger “carbuncles,” our patient’s presence of significant scarring and lack of extremity involvement makes HS more likely.
Recurrent MRSA abscesses
Methicillin-resistant Staphylococcus aureus skin and soft-tissue infections are not uncommon in the pediatric population, with presentation of infection ranging from cellulitis to fluid-containing abscesses.10 Recurrent abscesses may be seen in MRSA infection, however in this patient the presence of draining, scarring lesions in multiple locations typical for HS over time is more consistent with a diagnosis of HS.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Appiah is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Appiah have no relevant financial disclosures.
References
1. Liy-Wong C et al. JAMA Dermatol. 2021;157(4):385-91.
2. Choi E et al. J Am Acad Dermatol. 2022;86(1):140-7.
3. Machado MO et al. JAMA Dermatol. 2019;155(8):939-45.
4. McAndrew R et al. J Am Acad Dermatol. 2021;84(3):829-30.
5. Kirby JS and Zaenglein AL. JAMA Dermatol. 2021;157(4):379-80.
6. Alikhan A et al. J Am Acad Dermatol. 2019;81(1):91-101.
7. Greydanus DE et al. Dis Mon. 2021;67(4):101103.
8. Weir CB, St. Hilaire NJ. Epidermal Inclusion Cyst, in “StatPearls.” Treasure Island, Fla: StatPearls Publishing, 2021.
9. Atanaskova N and Tomecki KJ. Dermatol Clin. 2010;28(3):479-87.
10. Papastefan ST et al. J Surg Res. 2019;242:70-7.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that is becoming more recognized in children. It has a variable presentation, most commonly presenting as painful, recurrent cysts, abscesses, nodules, and/or pustules in classic locations with associated scarring and sinus tract formation.
The majority of patients present with bilateral lesions found most commonly in the axillae and inguinal folds.1 There are myriad other potential sites of involvement including the inframammary folds, inner thighs, buttocks, and groin.1 Diagnosis is made based on history and physical exam. There is a standard severity classification scheme called the Hurley score, which stratifies disease severity based on the presence of sinus tracts and extent of disease.1 HS is associated with comorbid conditions such as obesity, overweight, acne, and inflammatory bowel and joint disease.2 This painful, persistent condition is well documented to have a negative impact on quality of life in adult patients, and similar impairment has been found in pediatric patients.3,4
HS may be increasing in pediatric and adolescent patients, with recent studies showing onset coinciding most commonly with the onset of puberty.1,2 There is often a period of several years between symptom onset and diagnosis.1 A recent editorial highlighted the disparities that exist in HS, with disease more common in Black children and limited information about disease prevalence in Hispanic children.5
What’s the treatment plan?
HS is a difficult disease to treat, with few patients achieving remission and a significant proportion of patients with treatment-refractory disease.1 There are limited studies of HS treatment in pediatric patients. Topical and systemic antibiotic therapy are mainstays of HS treatment, with tetracyclines and a combination of clindamycin plus rifampin commonly used in adults and children alike. Topical therapies including topical antibiotics and antibacterial solutions are frequently used as adjunctive therapy.6 Adalimumab, a tumor necrosis factor receptor blocker, has been Food and Drug Administration approved for HS for ages 12 and up and is currently the only FDA-approved medication for HS in pediatric patients. Our patient was started on 100 mg doxycycline twice daily, with short-dose topical corticosteroids for symptom management of the most inflamed lesions.
What’s on the differential?
Acne conglobata
Acne conglobata is an uncommon, severe variant of acne vulgaris which arise in patients with a history of acne vulgaris and presents with comedones, cysts, abscesses, and scarring with possible drainage of pus. Lesions can present diffusely on the face, back, and body, including in the axillae, groin, and buttocks, and as such can be confused with HS.7
However, in contrast with HS, patients with acne conglobata will also develop disease in non–apocrine gland–bearing skin. This patient’s lack of preceding acne and restriction of lesions to the axillae, inguinal folds, and buttocks makes acne conglobata less likely.
Epidermal inclusion cyst
Epidermal inclusion cyst (EIC) is a common cutaneous cyst, presenting as a well-circumscribed nodule(s) with a central punctum. If not excised, lesions can sometimes become infected and painful.8 In contrast with HS, EIC presents only uncommonly as multiple lesions arising in different areas, and spontaneous drainage is uncommon. Our patient’s development of multiple draining lesions makes this diagnosis unlikely.
Furunculosis
Furunculosis is a common bacterial infection of the skin, presenting with inflammatory nodules or pustules centered around the hair follicle. Lesions may commonly present at sites of skin trauma and are found most frequently on the extremities.9 Though furunculosis lesions may drain pus and can coalesce to form larger “carbuncles,” our patient’s presence of significant scarring and lack of extremity involvement makes HS more likely.
Recurrent MRSA abscesses
Methicillin-resistant Staphylococcus aureus skin and soft-tissue infections are not uncommon in the pediatric population, with presentation of infection ranging from cellulitis to fluid-containing abscesses.10 Recurrent abscesses may be seen in MRSA infection, however in this patient the presence of draining, scarring lesions in multiple locations typical for HS over time is more consistent with a diagnosis of HS.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Appiah is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Appiah have no relevant financial disclosures.
References
1. Liy-Wong C et al. JAMA Dermatol. 2021;157(4):385-91.
2. Choi E et al. J Am Acad Dermatol. 2022;86(1):140-7.
3. Machado MO et al. JAMA Dermatol. 2019;155(8):939-45.
4. McAndrew R et al. J Am Acad Dermatol. 2021;84(3):829-30.
5. Kirby JS and Zaenglein AL. JAMA Dermatol. 2021;157(4):379-80.
6. Alikhan A et al. J Am Acad Dermatol. 2019;81(1):91-101.
7. Greydanus DE et al. Dis Mon. 2021;67(4):101103.
8. Weir CB, St. Hilaire NJ. Epidermal Inclusion Cyst, in “StatPearls.” Treasure Island, Fla: StatPearls Publishing, 2021.
9. Atanaskova N and Tomecki KJ. Dermatol Clin. 2010;28(3):479-87.
10. Papastefan ST et al. J Surg Res. 2019;242:70-7.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that is becoming more recognized in children. It has a variable presentation, most commonly presenting as painful, recurrent cysts, abscesses, nodules, and/or pustules in classic locations with associated scarring and sinus tract formation.
The majority of patients present with bilateral lesions found most commonly in the axillae and inguinal folds.1 There are myriad other potential sites of involvement including the inframammary folds, inner thighs, buttocks, and groin.1 Diagnosis is made based on history and physical exam. There is a standard severity classification scheme called the Hurley score, which stratifies disease severity based on the presence of sinus tracts and extent of disease.1 HS is associated with comorbid conditions such as obesity, overweight, acne, and inflammatory bowel and joint disease.2 This painful, persistent condition is well documented to have a negative impact on quality of life in adult patients, and similar impairment has been found in pediatric patients.3,4
HS may be increasing in pediatric and adolescent patients, with recent studies showing onset coinciding most commonly with the onset of puberty.1,2 There is often a period of several years between symptom onset and diagnosis.1 A recent editorial highlighted the disparities that exist in HS, with disease more common in Black children and limited information about disease prevalence in Hispanic children.5
What’s the treatment plan?
HS is a difficult disease to treat, with few patients achieving remission and a significant proportion of patients with treatment-refractory disease.1 There are limited studies of HS treatment in pediatric patients. Topical and systemic antibiotic therapy are mainstays of HS treatment, with tetracyclines and a combination of clindamycin plus rifampin commonly used in adults and children alike. Topical therapies including topical antibiotics and antibacterial solutions are frequently used as adjunctive therapy.6 Adalimumab, a tumor necrosis factor receptor blocker, has been Food and Drug Administration approved for HS for ages 12 and up and is currently the only FDA-approved medication for HS in pediatric patients. Our patient was started on 100 mg doxycycline twice daily, with short-dose topical corticosteroids for symptom management of the most inflamed lesions.
What’s on the differential?
Acne conglobata
Acne conglobata is an uncommon, severe variant of acne vulgaris which arise in patients with a history of acne vulgaris and presents with comedones, cysts, abscesses, and scarring with possible drainage of pus. Lesions can present diffusely on the face, back, and body, including in the axillae, groin, and buttocks, and as such can be confused with HS.7
However, in contrast with HS, patients with acne conglobata will also develop disease in non–apocrine gland–bearing skin. This patient’s lack of preceding acne and restriction of lesions to the axillae, inguinal folds, and buttocks makes acne conglobata less likely.
Epidermal inclusion cyst
Epidermal inclusion cyst (EIC) is a common cutaneous cyst, presenting as a well-circumscribed nodule(s) with a central punctum. If not excised, lesions can sometimes become infected and painful.8 In contrast with HS, EIC presents only uncommonly as multiple lesions arising in different areas, and spontaneous drainage is uncommon. Our patient’s development of multiple draining lesions makes this diagnosis unlikely.
Furunculosis
Furunculosis is a common bacterial infection of the skin, presenting with inflammatory nodules or pustules centered around the hair follicle. Lesions may commonly present at sites of skin trauma and are found most frequently on the extremities.9 Though furunculosis lesions may drain pus and can coalesce to form larger “carbuncles,” our patient’s presence of significant scarring and lack of extremity involvement makes HS more likely.
Recurrent MRSA abscesses
Methicillin-resistant Staphylococcus aureus skin and soft-tissue infections are not uncommon in the pediatric population, with presentation of infection ranging from cellulitis to fluid-containing abscesses.10 Recurrent abscesses may be seen in MRSA infection, however in this patient the presence of draining, scarring lesions in multiple locations typical for HS over time is more consistent with a diagnosis of HS.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Appiah is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Appiah have no relevant financial disclosures.
References
1. Liy-Wong C et al. JAMA Dermatol. 2021;157(4):385-91.
2. Choi E et al. J Am Acad Dermatol. 2022;86(1):140-7.
3. Machado MO et al. JAMA Dermatol. 2019;155(8):939-45.
4. McAndrew R et al. J Am Acad Dermatol. 2021;84(3):829-30.
5. Kirby JS and Zaenglein AL. JAMA Dermatol. 2021;157(4):379-80.
6. Alikhan A et al. J Am Acad Dermatol. 2019;81(1):91-101.
7. Greydanus DE et al. Dis Mon. 2021;67(4):101103.
8. Weir CB, St. Hilaire NJ. Epidermal Inclusion Cyst, in “StatPearls.” Treasure Island, Fla: StatPearls Publishing, 2021.
9. Atanaskova N and Tomecki KJ. Dermatol Clin. 2010;28(3):479-87.
10. Papastefan ST et al. J Surg Res. 2019;242:70-7.
Enlarging Nodule on the Back
The Diagnosis: Cutaneous Myxoma
Microscopic analysis showed features of cutaneous myxoma (quiz images). The epidermis was essentially unremarkable. Stellate to spindle cells with bland nuclear chromatin were present in the dermis with abundant pools of myxoid stroma. Colloidal iron staining highlighted the markedly increased dermal mucin.
Cutaneous myxomas (also referred to as superficial angiomyxomas) are rare, well-demarcated tumors of the dermis and subcutis.1,2 They can present as solitary, fleshcolored nodules on the trunk, lower extremities, head, or neck, and they often measure between 1 and 5 cm.2,3 Histologically, cutaneous myxomas are hypocellular with some stellate fibroblasts, occasional epithelial structures, and an abundant myxoid stroma, with notable thinwalled small blood vessels.2,4 These lesions contain pools of mucin and are positive for mesenchymal mucin stains such as colloidal iron and Alcian blue.1 Moreover, perivascular neutrophils are a distinguishing characteristic of cutaneous myxomas.4
Multiple cutaneous myxomas should raise concern for Carney complex,1,5 a genodermatologic syndrome that arises due to a mutation in the protein kinase CAMP-dependent type I regulatory subunit alpha gene, PRKAR1A, on chromosome 2.1,5 Additional cutaneous manifestations include blue nevi, lentigines, and café-aulait macules.5 Carney complex also is known for endocrine overactivity and cardiac myxomas, which can cause serious embolic complications.1
Recommended management is complete excision with close follow-up, as these lesions may recur in up to one-third of cases. Although there is a potential for recurrence, metastases are uncommon.3 Even without recurrence in the presenting location, follow-up should include screening for manifestations of Carney complex.1,3
The clinical and histological differential for cutaneous myxoma may include nerve sheath myxoma or neurofibroma. A nerve sheath myxoma is a dermal tumor that manifests as a solitary, flesh-colored nodule, measuring less than 2 cm. These lesions commonly present on the head, neck, and upper body.6 Cutaneous myxomas can grow larger than 2 cm, but these two lesions have a great deal of overlap in their other features.3,6 Thus, histology can be used to distinguish them.

Nerve sheath myxomas are circumscribed nonencapsulated tumors of the dermis composed of multilobular aggregates of spindle to epithelioid cells in a mucinous matrix (Figure 1). Clefts often are present around the cell aggregates. Despite previously being termed myxoid neurothekeomas, nerve sheath myxomas are S-100 positive, whereas cellular neurothekeomas are S-100 negative and likely not of neural origin. Cutaneous myxomas, in contrast to nerve sheath myxomas, are S-100 negative. Nerve sheath myxomas are more cellular and lack the characteristic mucin pools compared with cutaneous myxomas.1,2,6 Neurofibromas frequently are flesh colored and pedunculated, as was the lesion in our patient, yet they are vastly different microscopically. The stroma of neurofibromas can vary, but cellularity typically is greater than a cutaneous myxoma and consists of increased numbers of bland spindle cells with wavy nuclei (Schwann cells) and fibrillar cytoplasm as well as mast cells and fibroblasts (Figure 2). Neurofibromas stain positively for S-100 and SOX-10 (Sry-related HMg-box 10).2,7 In addition to café-au-lait macules, axillary freckling, optic gliomas, and positive family history, neurofibromas are associated with neurofibromatosis type 1, which is linked to a defect in a tumor suppressor gene that codes for neurofibromin.7

Nodular fasciitis is a self-limited myofibroblastic neoplasm that contains fusion genes, with the most common being myosin-9–ubiquitin specific peptidase 6, MYH9-USP6, which leads to overexpression of USP6. Nodular fasciitis presents as a solitary, rapidly enlarging nodule affecting the subcutaneous tissue, muscles, or fascia.8,9 It usually presents in the third or fourth decades of life.8 The arms are the most common location in adults, while the most commonly affected site in children is the head or neck. Histopathology reveals a characteristic tissue culture pattern with a proliferation of plump spindle and stellate fibroblasts as well as myofibroblasts (Figure 3). Early lesions have haphazard spindle cells with a proliferation of small blood vessels and extravasated erythrocytes. Despite increased mitotic figures, cellular atypia is rare. The fibroblasts and myofibroblasts react positively for vimentin and muscle-specific actin.8 This lesion is highly cellular comparatively and notably lacks the perivascular neutrophils and epithelial structures that would be expected in a cutaneous myxoma.4,8

Spindle cell lipomas, solitary subcutaneous masses commonly presenting on the upper back in middle-aged men, also can mimic cutaneous myxomas.4 Histologically, these lesions may contain short bundles of spindle cells arranged in a school of fish–like pattern, mature adipocytes, or myxoid stroma and characteristic CD34 positivity (Figure 4). Spindle cell lipomas often will present with ropey collagen, which can easily distinguish them from cutaneous myxomas.4

- Lanjewar DN, Bhatia VO, Lanjewar SD, et al. Cutaneous myxoma: an important clue to Carney complex. Indian J Pathol Microbiol. 2014;57:460-462.
- Choi HJ, Kim YJ, Yim JH, et al. Unusual presentation of solitary cutaneous myxoma. J Eur Acad Dermatol Venereol. 2007;21:403-404. doi:10.1111/j.1468-3083.2006.01881.x
- Kura MM, Jindal SR. Solitary superficial acral angiomyxoma: an infrequently reported soft tissue tumor. Indian J Dermatol. 2014;59:1-3. doi:10.4103/0019-5154.139893
- Zou Y, Billings SD. Myxoid cutaneous tumors: a review. J Cutan Pathol. 2016;43:903-918.
- Sarfo A, Helm K, Flamm A. Cutaneous myxomas and a psammomatous melanotic schwannoma in a patient with Carney complex. J Cutan Pathol. 2019;46:93-96. doi:10.1111/cup.13385
- Gill P, Abi Daoud MS. Multiple cellular neurothekeomas in a middleaged woman including the lower extremity: a case report and review of the current literature. J Cutan Pathol. 2019;46:67-73. doi:10.1111/ cup.13366
- Ohgaki H, Kim Y, Steinbach JP. Nervous system tumors associated with familial tumor syndromes. Curr Opin Neurol. 2010;23:583-591. doi:10.1097/WCO.0b013e3283405b5f
- Luna A, Molinari L, Bollea Garlatti LA, et al. Nodular fasciitis, a forgotten entity. Int J Dermatol. 2019;58:190-193. doi:10.1111/ijd.14219
- Patel N, Chrisinger J, Demicco E, et al. USP6 activation in nodular fasciitis by promoter-swapping gene fusions. Mod Pathol. 2017; 30:1577-1588.
The Diagnosis: Cutaneous Myxoma
Microscopic analysis showed features of cutaneous myxoma (quiz images). The epidermis was essentially unremarkable. Stellate to spindle cells with bland nuclear chromatin were present in the dermis with abundant pools of myxoid stroma. Colloidal iron staining highlighted the markedly increased dermal mucin.
Cutaneous myxomas (also referred to as superficial angiomyxomas) are rare, well-demarcated tumors of the dermis and subcutis.1,2 They can present as solitary, fleshcolored nodules on the trunk, lower extremities, head, or neck, and they often measure between 1 and 5 cm.2,3 Histologically, cutaneous myxomas are hypocellular with some stellate fibroblasts, occasional epithelial structures, and an abundant myxoid stroma, with notable thinwalled small blood vessels.2,4 These lesions contain pools of mucin and are positive for mesenchymal mucin stains such as colloidal iron and Alcian blue.1 Moreover, perivascular neutrophils are a distinguishing characteristic of cutaneous myxomas.4
Multiple cutaneous myxomas should raise concern for Carney complex,1,5 a genodermatologic syndrome that arises due to a mutation in the protein kinase CAMP-dependent type I regulatory subunit alpha gene, PRKAR1A, on chromosome 2.1,5 Additional cutaneous manifestations include blue nevi, lentigines, and café-aulait macules.5 Carney complex also is known for endocrine overactivity and cardiac myxomas, which can cause serious embolic complications.1
Recommended management is complete excision with close follow-up, as these lesions may recur in up to one-third of cases. Although there is a potential for recurrence, metastases are uncommon.3 Even without recurrence in the presenting location, follow-up should include screening for manifestations of Carney complex.1,3
The clinical and histological differential for cutaneous myxoma may include nerve sheath myxoma or neurofibroma. A nerve sheath myxoma is a dermal tumor that manifests as a solitary, flesh-colored nodule, measuring less than 2 cm. These lesions commonly present on the head, neck, and upper body.6 Cutaneous myxomas can grow larger than 2 cm, but these two lesions have a great deal of overlap in their other features.3,6 Thus, histology can be used to distinguish them.
Nerve sheath myxomas are circumscribed nonencapsulated tumors of the dermis composed of multilobular aggregates of spindle to epithelioid cells in a mucinous matrix (Figure 1). Clefts often are present around the cell aggregates. Despite previously being termed myxoid neurothekeomas, nerve sheath myxomas are S-100 positive, whereas cellular neurothekeomas are S-100 negative and likely not of neural origin. Cutaneous myxomas, in contrast to nerve sheath myxomas, are S-100 negative. Nerve sheath myxomas are more cellular and lack the characteristic mucin pools compared with cutaneous myxomas.1,2,6 Neurofibromas frequently are flesh colored and pedunculated, as was the lesion in our patient, yet they are vastly different microscopically. The stroma of neurofibromas can vary, but cellularity typically is greater than a cutaneous myxoma and consists of increased numbers of bland spindle cells with wavy nuclei (Schwann cells) and fibrillar cytoplasm as well as mast cells and fibroblasts (Figure 2). Neurofibromas stain positively for S-100 and SOX-10 (Sry-related HMg-box 10).2,7 In addition to café-au-lait macules, axillary freckling, optic gliomas, and positive family history, neurofibromas are associated with neurofibromatosis type 1, which is linked to a defect in a tumor suppressor gene that codes for neurofibromin.7
Nodular fasciitis is a self-limited myofibroblastic neoplasm that contains fusion genes, with the most common being myosin-9–ubiquitin specific peptidase 6, MYH9-USP6, which leads to overexpression of USP6. Nodular fasciitis presents as a solitary, rapidly enlarging nodule affecting the subcutaneous tissue, muscles, or fascia.8,9 It usually presents in the third or fourth decades of life.8 The arms are the most common location in adults, while the most commonly affected site in children is the head or neck. Histopathology reveals a characteristic tissue culture pattern with a proliferation of plump spindle and stellate fibroblasts as well as myofibroblasts (Figure 3). Early lesions have haphazard spindle cells with a proliferation of small blood vessels and extravasated erythrocytes. Despite increased mitotic figures, cellular atypia is rare. The fibroblasts and myofibroblasts react positively for vimentin and muscle-specific actin.8 This lesion is highly cellular comparatively and notably lacks the perivascular neutrophils and epithelial structures that would be expected in a cutaneous myxoma.4,8
Spindle cell lipomas, solitary subcutaneous masses commonly presenting on the upper back in middle-aged men, also can mimic cutaneous myxomas.4 Histologically, these lesions may contain short bundles of spindle cells arranged in a school of fish–like pattern, mature adipocytes, or myxoid stroma and characteristic CD34 positivity (Figure 4). Spindle cell lipomas often will present with ropey collagen, which can easily distinguish them from cutaneous myxomas.4
The Diagnosis: Cutaneous Myxoma
Microscopic analysis showed features of cutaneous myxoma (quiz images). The epidermis was essentially unremarkable. Stellate to spindle cells with bland nuclear chromatin were present in the dermis with abundant pools of myxoid stroma. Colloidal iron staining highlighted the markedly increased dermal mucin.
Cutaneous myxomas (also referred to as superficial angiomyxomas) are rare, well-demarcated tumors of the dermis and subcutis.1,2 They can present as solitary, fleshcolored nodules on the trunk, lower extremities, head, or neck, and they often measure between 1 and 5 cm.2,3 Histologically, cutaneous myxomas are hypocellular with some stellate fibroblasts, occasional epithelial structures, and an abundant myxoid stroma, with notable thinwalled small blood vessels.2,4 These lesions contain pools of mucin and are positive for mesenchymal mucin stains such as colloidal iron and Alcian blue.1 Moreover, perivascular neutrophils are a distinguishing characteristic of cutaneous myxomas.4
Multiple cutaneous myxomas should raise concern for Carney complex,1,5 a genodermatologic syndrome that arises due to a mutation in the protein kinase CAMP-dependent type I regulatory subunit alpha gene, PRKAR1A, on chromosome 2.1,5 Additional cutaneous manifestations include blue nevi, lentigines, and café-aulait macules.5 Carney complex also is known for endocrine overactivity and cardiac myxomas, which can cause serious embolic complications.1
Recommended management is complete excision with close follow-up, as these lesions may recur in up to one-third of cases. Although there is a potential for recurrence, metastases are uncommon.3 Even without recurrence in the presenting location, follow-up should include screening for manifestations of Carney complex.1,3
The clinical and histological differential for cutaneous myxoma may include nerve sheath myxoma or neurofibroma. A nerve sheath myxoma is a dermal tumor that manifests as a solitary, flesh-colored nodule, measuring less than 2 cm. These lesions commonly present on the head, neck, and upper body.6 Cutaneous myxomas can grow larger than 2 cm, but these two lesions have a great deal of overlap in their other features.3,6 Thus, histology can be used to distinguish them.
Nerve sheath myxomas are circumscribed nonencapsulated tumors of the dermis composed of multilobular aggregates of spindle to epithelioid cells in a mucinous matrix (Figure 1). Clefts often are present around the cell aggregates. Despite previously being termed myxoid neurothekeomas, nerve sheath myxomas are S-100 positive, whereas cellular neurothekeomas are S-100 negative and likely not of neural origin. Cutaneous myxomas, in contrast to nerve sheath myxomas, are S-100 negative. Nerve sheath myxomas are more cellular and lack the characteristic mucin pools compared with cutaneous myxomas.1,2,6 Neurofibromas frequently are flesh colored and pedunculated, as was the lesion in our patient, yet they are vastly different microscopically. The stroma of neurofibromas can vary, but cellularity typically is greater than a cutaneous myxoma and consists of increased numbers of bland spindle cells with wavy nuclei (Schwann cells) and fibrillar cytoplasm as well as mast cells and fibroblasts (Figure 2). Neurofibromas stain positively for S-100 and SOX-10 (Sry-related HMg-box 10).2,7 In addition to café-au-lait macules, axillary freckling, optic gliomas, and positive family history, neurofibromas are associated with neurofibromatosis type 1, which is linked to a defect in a tumor suppressor gene that codes for neurofibromin.7
Nodular fasciitis is a self-limited myofibroblastic neoplasm that contains fusion genes, with the most common being myosin-9–ubiquitin specific peptidase 6, MYH9-USP6, which leads to overexpression of USP6. Nodular fasciitis presents as a solitary, rapidly enlarging nodule affecting the subcutaneous tissue, muscles, or fascia.8,9 It usually presents in the third or fourth decades of life.8 The arms are the most common location in adults, while the most commonly affected site in children is the head or neck. Histopathology reveals a characteristic tissue culture pattern with a proliferation of plump spindle and stellate fibroblasts as well as myofibroblasts (Figure 3). Early lesions have haphazard spindle cells with a proliferation of small blood vessels and extravasated erythrocytes. Despite increased mitotic figures, cellular atypia is rare. The fibroblasts and myofibroblasts react positively for vimentin and muscle-specific actin.8 This lesion is highly cellular comparatively and notably lacks the perivascular neutrophils and epithelial structures that would be expected in a cutaneous myxoma.4,8
Spindle cell lipomas, solitary subcutaneous masses commonly presenting on the upper back in middle-aged men, also can mimic cutaneous myxomas.4 Histologically, these lesions may contain short bundles of spindle cells arranged in a school of fish–like pattern, mature adipocytes, or myxoid stroma and characteristic CD34 positivity (Figure 4). Spindle cell lipomas often will present with ropey collagen, which can easily distinguish them from cutaneous myxomas.4
- Lanjewar DN, Bhatia VO, Lanjewar SD, et al. Cutaneous myxoma: an important clue to Carney complex. Indian J Pathol Microbiol. 2014;57:460-462.
- Choi HJ, Kim YJ, Yim JH, et al. Unusual presentation of solitary cutaneous myxoma. J Eur Acad Dermatol Venereol. 2007;21:403-404. doi:10.1111/j.1468-3083.2006.01881.x
- Kura MM, Jindal SR. Solitary superficial acral angiomyxoma: an infrequently reported soft tissue tumor. Indian J Dermatol. 2014;59:1-3. doi:10.4103/0019-5154.139893
- Zou Y, Billings SD. Myxoid cutaneous tumors: a review. J Cutan Pathol. 2016;43:903-918.
- Sarfo A, Helm K, Flamm A. Cutaneous myxomas and a psammomatous melanotic schwannoma in a patient with Carney complex. J Cutan Pathol. 2019;46:93-96. doi:10.1111/cup.13385
- Gill P, Abi Daoud MS. Multiple cellular neurothekeomas in a middleaged woman including the lower extremity: a case report and review of the current literature. J Cutan Pathol. 2019;46:67-73. doi:10.1111/ cup.13366
- Ohgaki H, Kim Y, Steinbach JP. Nervous system tumors associated with familial tumor syndromes. Curr Opin Neurol. 2010;23:583-591. doi:10.1097/WCO.0b013e3283405b5f
- Luna A, Molinari L, Bollea Garlatti LA, et al. Nodular fasciitis, a forgotten entity. Int J Dermatol. 2019;58:190-193. doi:10.1111/ijd.14219
- Patel N, Chrisinger J, Demicco E, et al. USP6 activation in nodular fasciitis by promoter-swapping gene fusions. Mod Pathol. 2017; 30:1577-1588.
- Lanjewar DN, Bhatia VO, Lanjewar SD, et al. Cutaneous myxoma: an important clue to Carney complex. Indian J Pathol Microbiol. 2014;57:460-462.
- Choi HJ, Kim YJ, Yim JH, et al. Unusual presentation of solitary cutaneous myxoma. J Eur Acad Dermatol Venereol. 2007;21:403-404. doi:10.1111/j.1468-3083.2006.01881.x
- Kura MM, Jindal SR. Solitary superficial acral angiomyxoma: an infrequently reported soft tissue tumor. Indian J Dermatol. 2014;59:1-3. doi:10.4103/0019-5154.139893
- Zou Y, Billings SD. Myxoid cutaneous tumors: a review. J Cutan Pathol. 2016;43:903-918.
- Sarfo A, Helm K, Flamm A. Cutaneous myxomas and a psammomatous melanotic schwannoma in a patient with Carney complex. J Cutan Pathol. 2019;46:93-96. doi:10.1111/cup.13385
- Gill P, Abi Daoud MS. Multiple cellular neurothekeomas in a middleaged woman including the lower extremity: a case report and review of the current literature. J Cutan Pathol. 2019;46:67-73. doi:10.1111/ cup.13366
- Ohgaki H, Kim Y, Steinbach JP. Nervous system tumors associated with familial tumor syndromes. Curr Opin Neurol. 2010;23:583-591. doi:10.1097/WCO.0b013e3283405b5f
- Luna A, Molinari L, Bollea Garlatti LA, et al. Nodular fasciitis, a forgotten entity. Int J Dermatol. 2019;58:190-193. doi:10.1111/ijd.14219
- Patel N, Chrisinger J, Demicco E, et al. USP6 activation in nodular fasciitis by promoter-swapping gene fusions. Mod Pathol. 2017; 30:1577-1588.
A 43-year-old man with an unremarkable medical history presented to our clinic with an enlarging painful nodule on the upper back that was present for years without bleeding or ulceration. He denied prior treatment or any similar lesions. Physical examination was notable for a 2×1.5-cm, pedunculated, flesh-colored nodule on the left upper back. A shave excision of the lesion was performed.



Erythematous Indurated Nodule on the Forehead
The Diagnosis: Dermatofibrosarcoma Protuberans
Histopathologic examination showed a dermal tumor composed of spindle cells in a storiform arrangement (Figure 1). Immunohistochemistry demonstrated positive CD34 staining of the tumoral cells (Figure 2). Clinical review, histopathologic examination, and immunohistochemistry confirmed a diagnosis of dermatofibrosarcoma protuberans (DFSP). The patient underwent Mohs micrographic surgery (MMS) with clear margins after 3 stages, followed by repair with a rotation flap. No evidence of recurrence was found at 4-year follow-up.

Dermatofibrosarcoma protuberans is a rare low-grade sarcoma of fibroblast origin with an annual incidence of 0.8 to 5 cases per million individuals.1 It typically presents in patients aged 30 to 50 years on the trunk, scalp, or proximal extremities as an asymptomatic, flesh-colored, erythematous or brown, indurated plaque or nodule.2 Due to its variable presentation, these lesions often may be misdiagnosed as lipomas or epidermoid cysts, preventing proper targeted treatment. Therefore, suspicious enlarging indurated nodules require a lower threshold for biopsy.1

A definitive diagnosis of DFSP is achieved after a biopsy and histopathologic evaluation. Hematoxylin and eosin staining typically shows diffuse infiltration of the dermis and the subcutaneous fat by densely packed, cytologic, relatively uniform, spindle-shaped tumor cells arranged in a characteristic storiform shape. Tumor cells are spread along the septae of the subcutaneous fatty tissue.3 Immunohistochemistry is characterized by positive CD34 and negative factor XIIIa, with rare exceptions.
The differential diagnosis includes lipoma, epidermoid cyst, plexiform fibrohistiocytic tumor, and malignant peripheral nerve sheath tumor.3 Positive CD34 immunostaining, negative S-100 staining, and a storiform pattern of spindle cells can assist in differentiating DFSP from these possible differential diagnoses; lesions of these other entities are characterized by different pathologic findings. Lipomas are composed of fat tissue, epidermoid cysts have epithelial-lined cysts filled with keratin, plexiform fibrohistiocytic tumors have plexiform rays of fibrous tissue extending into fat with negative CD34 staining, and malignant peripheral nerve sheath tumors have fleshy variegated masses involving the peripheral nerve trunks with partial S-100 staining.4-7 Additional evaluation to confirm DFSP can be accomplished by analysis of tumor samples by fluorescence in situ hybridization or reverse transcriptase–polymerase chain reaction to detect chromosomal translocations and fusion gene transcripts, as chromosomal translocations may be found in more than 90% of cases.3
Early diagnosis of DFSP is beneficial, as it can help prevent recurrence as well as metastasis. Studies have attempted to document the risk for recurrence as well as metastasis based on characteristic features and treatment strategies of DFSP. In a study of 186 patients, 3 had metastatic disease to the lungs, the most common site of metastasis.8 These 3 patients had fibrosarcomatous transformation within DFSP, emphasizing the importance of detailing this finding early in the diagnosis, as it was characterized by a higher degree of cellularity, cytologic atypia, mitotic activity, and negative CD34 immunostaining.9 In patients with suspected metastasis, lymph node ultrasonography, chest radiography, and computed tomography may be utilized.3
When treating DFSP, the goal is complete removal of the tumor with clear margins. Mohs micrographic surgery, modified MMS, and wide local excision (WLE) with 2- to 4-cm margins are appropriate treatment options, though MMS is the treatment of choice. A study comparing MMS and WLE demonstrated 3% and 30.8% recurrence rates, respectively.8 In MMS, complete margin evaluation on microscopy is performed after each stage to ensure negative surgical margins. The presence of positive surgical margins elicits continued resection until the margins are clear.10,11
Other treatment modalities may be considered for patients with DFSP. Molecular therapy with imatinib, an oral tyrosine kinase inhibitor targeting platelet-derived growth factor–regulated expression, can be utilized for inoperable tumors; however, additional clinical trials are required to ensure efficacy.3 Surgical removal of the possible remaining tumor is still recommended after molecular therapy. Radiotherapy is an additional method of treatment that may be used for inoperable tumors.3
Dermatofibrosarcoma protuberans is a rare lowgrade sarcoma of fibroblast origin that typically does not metastasize but often has notable subclinical extension and recurrence. Differentiating DFSP from other tumors often may be difficult. A protuberant, flesh-colored, slowgrowing, and asymptomatic lesion often may be confused with lipomas or epidermoid cysts; therefore, biopsies with immunohistostaining for suspicious lesions is required.12 Mohs micrographic surgery has evolved as the treatment of choice for this tumor, though WLE and new targeted molecular therapies still are considered. Proper diagnosis and treatment of DFSP is paramount in preventing future morbidity.
- Benoit A, Aycock J, Milam D, et al. Dermatofibrosarcoma protuberans of the forehead with extensive subclinical spread. Dermatol Surg. 2016;42:261-264. doi:10.1097/DSS.0000000000000604
- Khachemoune A, Barkoe D, Braun M, et al. Dermatofibrosarcoma protuberans of the forehead and scalp with involvement of the outer calvarial plate: multistaged repair with the use of skin expanders. Dermatol Surg. 2005;31:115-119. doi:10.1111/j.1524-4725.2005.31021
- Saiag P, Grob J-J, Lebbe C, et al. Diagnosis and treatment of dermatofibrosarcoma protuberans. European consensus-based interdisciplinary guideline. Eur J Cancer. 2015;51:2604-2608. doi:10.1016/j.ejca.2015.06.108
- Charifa A, Badri T. Lipomas, pathology. StatPearls. StatPearls Publishing; 2020.
- Zito PM, Scharf R. Cyst, epidermoid (sebaceous cyst). StatPearls. StatPearls Publishing; 2020.
- Taher A, Pushpanathan C. Plexiform fibrohistiocytic tumor: a brief review. Arch Pathol Lab Med. 2007;131:1135-1138. doi:10.5858 /2007-131-1135-PFTABR
- Rodriguez FJ, Folpe AL, Giannini C, et al. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295-319. doi:10.1007 /s00401-012-0954-z
- Lowe GC, Onajin O, Baum CL, et al. A comparison of Mohs micrographic surgery and wide local excision for treatment of dermatofibrosarcoma protuberans with long-term follow-up: the Mayo Clinic experience. Dermatol Surg. 2017;43:98-106. doi:10.1097/DSS.0000000000000910
- Rouhani P, Fletcher CDM, Devesa SS, et al. Cutaneous soft tissue sarcoma incidence patterns in the U.S.: an analysis of 12,114 cases. Cancer. 2008;113:616-627. doi:10.1002/cncr.23571
- Ratner D, Thomas CO, Johnson TM, et al. Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans. results of a multiinstitutional series with an analysis of the extent of microscopic spread. J Am Acad Dermatol. 1997;37:600-613. doi:10.1016/s0190 -9622(97)70179-8
- Buck DW, Kim JYS, Alam M, et al. Multidisciplinary approach to the management of dermatofibrosarcoma protuberans. J Am Acad Dermatol. 2012;67:861-866. doi:10.1016/j.jaad.2012.01.039
- Shih P-Y, Chen C-H, Kuo T-T, et al. Deep dermatofibrosarcoma protuberans: a pitfall in the ultrasonographic diagnosis of lipoma -like subcutaneous lesions. Dermatologica Sinica. 2010;28:32-35. doi:10.1016/S1027-8117(10)60005-5
The Diagnosis: Dermatofibrosarcoma Protuberans
Histopathologic examination showed a dermal tumor composed of spindle cells in a storiform arrangement (Figure 1). Immunohistochemistry demonstrated positive CD34 staining of the tumoral cells (Figure 2). Clinical review, histopathologic examination, and immunohistochemistry confirmed a diagnosis of dermatofibrosarcoma protuberans (DFSP). The patient underwent Mohs micrographic surgery (MMS) with clear margins after 3 stages, followed by repair with a rotation flap. No evidence of recurrence was found at 4-year follow-up.
Dermatofibrosarcoma protuberans is a rare low-grade sarcoma of fibroblast origin with an annual incidence of 0.8 to 5 cases per million individuals.1 It typically presents in patients aged 30 to 50 years on the trunk, scalp, or proximal extremities as an asymptomatic, flesh-colored, erythematous or brown, indurated plaque or nodule.2 Due to its variable presentation, these lesions often may be misdiagnosed as lipomas or epidermoid cysts, preventing proper targeted treatment. Therefore, suspicious enlarging indurated nodules require a lower threshold for biopsy.1
A definitive diagnosis of DFSP is achieved after a biopsy and histopathologic evaluation. Hematoxylin and eosin staining typically shows diffuse infiltration of the dermis and the subcutaneous fat by densely packed, cytologic, relatively uniform, spindle-shaped tumor cells arranged in a characteristic storiform shape. Tumor cells are spread along the septae of the subcutaneous fatty tissue.3 Immunohistochemistry is characterized by positive CD34 and negative factor XIIIa, with rare exceptions.
The differential diagnosis includes lipoma, epidermoid cyst, plexiform fibrohistiocytic tumor, and malignant peripheral nerve sheath tumor.3 Positive CD34 immunostaining, negative S-100 staining, and a storiform pattern of spindle cells can assist in differentiating DFSP from these possible differential diagnoses; lesions of these other entities are characterized by different pathologic findings. Lipomas are composed of fat tissue, epidermoid cysts have epithelial-lined cysts filled with keratin, plexiform fibrohistiocytic tumors have plexiform rays of fibrous tissue extending into fat with negative CD34 staining, and malignant peripheral nerve sheath tumors have fleshy variegated masses involving the peripheral nerve trunks with partial S-100 staining.4-7 Additional evaluation to confirm DFSP can be accomplished by analysis of tumor samples by fluorescence in situ hybridization or reverse transcriptase–polymerase chain reaction to detect chromosomal translocations and fusion gene transcripts, as chromosomal translocations may be found in more than 90% of cases.3
Early diagnosis of DFSP is beneficial, as it can help prevent recurrence as well as metastasis. Studies have attempted to document the risk for recurrence as well as metastasis based on characteristic features and treatment strategies of DFSP. In a study of 186 patients, 3 had metastatic disease to the lungs, the most common site of metastasis.8 These 3 patients had fibrosarcomatous transformation within DFSP, emphasizing the importance of detailing this finding early in the diagnosis, as it was characterized by a higher degree of cellularity, cytologic atypia, mitotic activity, and negative CD34 immunostaining.9 In patients with suspected metastasis, lymph node ultrasonography, chest radiography, and computed tomography may be utilized.3
When treating DFSP, the goal is complete removal of the tumor with clear margins. Mohs micrographic surgery, modified MMS, and wide local excision (WLE) with 2- to 4-cm margins are appropriate treatment options, though MMS is the treatment of choice. A study comparing MMS and WLE demonstrated 3% and 30.8% recurrence rates, respectively.8 In MMS, complete margin evaluation on microscopy is performed after each stage to ensure negative surgical margins. The presence of positive surgical margins elicits continued resection until the margins are clear.10,11
Other treatment modalities may be considered for patients with DFSP. Molecular therapy with imatinib, an oral tyrosine kinase inhibitor targeting platelet-derived growth factor–regulated expression, can be utilized for inoperable tumors; however, additional clinical trials are required to ensure efficacy.3 Surgical removal of the possible remaining tumor is still recommended after molecular therapy. Radiotherapy is an additional method of treatment that may be used for inoperable tumors.3
Dermatofibrosarcoma protuberans is a rare lowgrade sarcoma of fibroblast origin that typically does not metastasize but often has notable subclinical extension and recurrence. Differentiating DFSP from other tumors often may be difficult. A protuberant, flesh-colored, slowgrowing, and asymptomatic lesion often may be confused with lipomas or epidermoid cysts; therefore, biopsies with immunohistostaining for suspicious lesions is required.12 Mohs micrographic surgery has evolved as the treatment of choice for this tumor, though WLE and new targeted molecular therapies still are considered. Proper diagnosis and treatment of DFSP is paramount in preventing future morbidity.
The Diagnosis: Dermatofibrosarcoma Protuberans
Histopathologic examination showed a dermal tumor composed of spindle cells in a storiform arrangement (Figure 1). Immunohistochemistry demonstrated positive CD34 staining of the tumoral cells (Figure 2). Clinical review, histopathologic examination, and immunohistochemistry confirmed a diagnosis of dermatofibrosarcoma protuberans (DFSP). The patient underwent Mohs micrographic surgery (MMS) with clear margins after 3 stages, followed by repair with a rotation flap. No evidence of recurrence was found at 4-year follow-up.
Dermatofibrosarcoma protuberans is a rare low-grade sarcoma of fibroblast origin with an annual incidence of 0.8 to 5 cases per million individuals.1 It typically presents in patients aged 30 to 50 years on the trunk, scalp, or proximal extremities as an asymptomatic, flesh-colored, erythematous or brown, indurated plaque or nodule.2 Due to its variable presentation, these lesions often may be misdiagnosed as lipomas or epidermoid cysts, preventing proper targeted treatment. Therefore, suspicious enlarging indurated nodules require a lower threshold for biopsy.1
A definitive diagnosis of DFSP is achieved after a biopsy and histopathologic evaluation. Hematoxylin and eosin staining typically shows diffuse infiltration of the dermis and the subcutaneous fat by densely packed, cytologic, relatively uniform, spindle-shaped tumor cells arranged in a characteristic storiform shape. Tumor cells are spread along the septae of the subcutaneous fatty tissue.3 Immunohistochemistry is characterized by positive CD34 and negative factor XIIIa, with rare exceptions.
The differential diagnosis includes lipoma, epidermoid cyst, plexiform fibrohistiocytic tumor, and malignant peripheral nerve sheath tumor.3 Positive CD34 immunostaining, negative S-100 staining, and a storiform pattern of spindle cells can assist in differentiating DFSP from these possible differential diagnoses; lesions of these other entities are characterized by different pathologic findings. Lipomas are composed of fat tissue, epidermoid cysts have epithelial-lined cysts filled with keratin, plexiform fibrohistiocytic tumors have plexiform rays of fibrous tissue extending into fat with negative CD34 staining, and malignant peripheral nerve sheath tumors have fleshy variegated masses involving the peripheral nerve trunks with partial S-100 staining.4-7 Additional evaluation to confirm DFSP can be accomplished by analysis of tumor samples by fluorescence in situ hybridization or reverse transcriptase–polymerase chain reaction to detect chromosomal translocations and fusion gene transcripts, as chromosomal translocations may be found in more than 90% of cases.3
Early diagnosis of DFSP is beneficial, as it can help prevent recurrence as well as metastasis. Studies have attempted to document the risk for recurrence as well as metastasis based on characteristic features and treatment strategies of DFSP. In a study of 186 patients, 3 had metastatic disease to the lungs, the most common site of metastasis.8 These 3 patients had fibrosarcomatous transformation within DFSP, emphasizing the importance of detailing this finding early in the diagnosis, as it was characterized by a higher degree of cellularity, cytologic atypia, mitotic activity, and negative CD34 immunostaining.9 In patients with suspected metastasis, lymph node ultrasonography, chest radiography, and computed tomography may be utilized.3
When treating DFSP, the goal is complete removal of the tumor with clear margins. Mohs micrographic surgery, modified MMS, and wide local excision (WLE) with 2- to 4-cm margins are appropriate treatment options, though MMS is the treatment of choice. A study comparing MMS and WLE demonstrated 3% and 30.8% recurrence rates, respectively.8 In MMS, complete margin evaluation on microscopy is performed after each stage to ensure negative surgical margins. The presence of positive surgical margins elicits continued resection until the margins are clear.10,11
Other treatment modalities may be considered for patients with DFSP. Molecular therapy with imatinib, an oral tyrosine kinase inhibitor targeting platelet-derived growth factor–regulated expression, can be utilized for inoperable tumors; however, additional clinical trials are required to ensure efficacy.3 Surgical removal of the possible remaining tumor is still recommended after molecular therapy. Radiotherapy is an additional method of treatment that may be used for inoperable tumors.3
Dermatofibrosarcoma protuberans is a rare lowgrade sarcoma of fibroblast origin that typically does not metastasize but often has notable subclinical extension and recurrence. Differentiating DFSP from other tumors often may be difficult. A protuberant, flesh-colored, slowgrowing, and asymptomatic lesion often may be confused with lipomas or epidermoid cysts; therefore, biopsies with immunohistostaining for suspicious lesions is required.12 Mohs micrographic surgery has evolved as the treatment of choice for this tumor, though WLE and new targeted molecular therapies still are considered. Proper diagnosis and treatment of DFSP is paramount in preventing future morbidity.
- Benoit A, Aycock J, Milam D, et al. Dermatofibrosarcoma protuberans of the forehead with extensive subclinical spread. Dermatol Surg. 2016;42:261-264. doi:10.1097/DSS.0000000000000604
- Khachemoune A, Barkoe D, Braun M, et al. Dermatofibrosarcoma protuberans of the forehead and scalp with involvement of the outer calvarial plate: multistaged repair with the use of skin expanders. Dermatol Surg. 2005;31:115-119. doi:10.1111/j.1524-4725.2005.31021
- Saiag P, Grob J-J, Lebbe C, et al. Diagnosis and treatment of dermatofibrosarcoma protuberans. European consensus-based interdisciplinary guideline. Eur J Cancer. 2015;51:2604-2608. doi:10.1016/j.ejca.2015.06.108
- Charifa A, Badri T. Lipomas, pathology. StatPearls. StatPearls Publishing; 2020.
- Zito PM, Scharf R. Cyst, epidermoid (sebaceous cyst). StatPearls. StatPearls Publishing; 2020.
- Taher A, Pushpanathan C. Plexiform fibrohistiocytic tumor: a brief review. Arch Pathol Lab Med. 2007;131:1135-1138. doi:10.5858 /2007-131-1135-PFTABR
- Rodriguez FJ, Folpe AL, Giannini C, et al. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295-319. doi:10.1007 /s00401-012-0954-z
- Lowe GC, Onajin O, Baum CL, et al. A comparison of Mohs micrographic surgery and wide local excision for treatment of dermatofibrosarcoma protuberans with long-term follow-up: the Mayo Clinic experience. Dermatol Surg. 2017;43:98-106. doi:10.1097/DSS.0000000000000910
- Rouhani P, Fletcher CDM, Devesa SS, et al. Cutaneous soft tissue sarcoma incidence patterns in the U.S.: an analysis of 12,114 cases. Cancer. 2008;113:616-627. doi:10.1002/cncr.23571
- Ratner D, Thomas CO, Johnson TM, et al. Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans. results of a multiinstitutional series with an analysis of the extent of microscopic spread. J Am Acad Dermatol. 1997;37:600-613. doi:10.1016/s0190 -9622(97)70179-8
- Buck DW, Kim JYS, Alam M, et al. Multidisciplinary approach to the management of dermatofibrosarcoma protuberans. J Am Acad Dermatol. 2012;67:861-866. doi:10.1016/j.jaad.2012.01.039
- Shih P-Y, Chen C-H, Kuo T-T, et al. Deep dermatofibrosarcoma protuberans: a pitfall in the ultrasonographic diagnosis of lipoma -like subcutaneous lesions. Dermatologica Sinica. 2010;28:32-35. doi:10.1016/S1027-8117(10)60005-5
- Benoit A, Aycock J, Milam D, et al. Dermatofibrosarcoma protuberans of the forehead with extensive subclinical spread. Dermatol Surg. 2016;42:261-264. doi:10.1097/DSS.0000000000000604
- Khachemoune A, Barkoe D, Braun M, et al. Dermatofibrosarcoma protuberans of the forehead and scalp with involvement of the outer calvarial plate: multistaged repair with the use of skin expanders. Dermatol Surg. 2005;31:115-119. doi:10.1111/j.1524-4725.2005.31021
- Saiag P, Grob J-J, Lebbe C, et al. Diagnosis and treatment of dermatofibrosarcoma protuberans. European consensus-based interdisciplinary guideline. Eur J Cancer. 2015;51:2604-2608. doi:10.1016/j.ejca.2015.06.108
- Charifa A, Badri T. Lipomas, pathology. StatPearls. StatPearls Publishing; 2020.
- Zito PM, Scharf R. Cyst, epidermoid (sebaceous cyst). StatPearls. StatPearls Publishing; 2020.
- Taher A, Pushpanathan C. Plexiform fibrohistiocytic tumor: a brief review. Arch Pathol Lab Med. 2007;131:1135-1138. doi:10.5858 /2007-131-1135-PFTABR
- Rodriguez FJ, Folpe AL, Giannini C, et al. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295-319. doi:10.1007 /s00401-012-0954-z
- Lowe GC, Onajin O, Baum CL, et al. A comparison of Mohs micrographic surgery and wide local excision for treatment of dermatofibrosarcoma protuberans with long-term follow-up: the Mayo Clinic experience. Dermatol Surg. 2017;43:98-106. doi:10.1097/DSS.0000000000000910
- Rouhani P, Fletcher CDM, Devesa SS, et al. Cutaneous soft tissue sarcoma incidence patterns in the U.S.: an analysis of 12,114 cases. Cancer. 2008;113:616-627. doi:10.1002/cncr.23571
- Ratner D, Thomas CO, Johnson TM, et al. Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans. results of a multiinstitutional series with an analysis of the extent of microscopic spread. J Am Acad Dermatol. 1997;37:600-613. doi:10.1016/s0190 -9622(97)70179-8
- Buck DW, Kim JYS, Alam M, et al. Multidisciplinary approach to the management of dermatofibrosarcoma protuberans. J Am Acad Dermatol. 2012;67:861-866. doi:10.1016/j.jaad.2012.01.039
- Shih P-Y, Chen C-H, Kuo T-T, et al. Deep dermatofibrosarcoma protuberans: a pitfall in the ultrasonographic diagnosis of lipoma -like subcutaneous lesions. Dermatologica Sinica. 2010;28:32-35. doi:10.1016/S1027-8117(10)60005-5
A 39-year-old man presented with an enlarging asymptomatic nodule on the forehead of more than 3 years’ duration. Physical examination revealed a 3.4×2.3-cm, indurated, firm, erythematous nodule on the frontotemporal scalp. The patient denied any history of trauma to the area.


Febrile Ulceronecrotic Mucha-Habermann Disease: A Rare Form of Pityriasis Lichenoides et Varioliformis Acuta
To the Editor:
Pityriasis lichenoides is a papulosquamous dermatologic disorder that is characterized by recurrent papules.1 There is a spectrum of disease in pityriasis lichenoides that includes pityriasis lichenoides et varioliformis acuta (PLEVA) at one end and pityriasis lichenoides chronica at the other. Pityriasis lichenoides et varioliformis acuta is more common in younger individuals and is characterized by erythematous papules that often crust; these lesions resolve over weeks. The lesions of pityriasis lichenoides chronica are characteristically scaly, pink to red-brown papules that tend to resolve over months.1
Histologically, PLEVA exhibits parakeratosis, interface dermatitis, and a wedge-shaped infiltrate.1 Necrotic keratinocytes and extravasated erythrocytes also are common features. Additionally, monoclonal T cells may be present in the infiltrate.1
Febrile ulceronecrotic Mucha-Habermann disease (FUMHD) is a rare and severe variant of PLEVA. Febrile ulceronecrotic Mucha-Habermann disease is characterized by ulceronecrotic lesions, fever, and systemic symptoms.2 Herein, we present a case of FUMHD.

A 57-year-old man presented with an eruption of painful lesions involving the face, trunk, arms, legs, and genitalia of 1 month’s duration. The patient denied oral and ocular involvement. He had soreness and swelling of the arms and legs. A prior 12-day course of prednisone prescribed by a community dermatologist failed to improve the rash. A biopsy performed by a community dermatologist was nondiagnostic. The patient denied fever but did report chills. He had no preceding illness and was not taking new medications. On physical examination, the patient was afebrile and normotensive with innumerable deep-seated pustules and crusted ulcerations on the face, palms, soles, trunk, extremities, and penis (Figures 1 and 2). There was a background morbilliform eruption on the trunk. The ocular and oral mucosae were spared. The upper and lower extremities had pitting edema.

The patient’s alanine aminotransaminase and aspartate aminotransaminase levels were elevated at 55 and 51 U/L, respectively. His white blood cell count was within reference range; however, there was an elevated absolute neutrophil count (8.7×103/μL). No eosinophilia was noted. Laboratory evaluation showed a positive antimitochondrial antibody, and magnetic resonance imaging showed evidence of steatohepatitis. Punch biopsies from both the morbilliform eruption and a deep-seated pustule showed epidermal necrosis, parakeratosis, necrotic keratinocytes, and a lichenoid infiltrate of lymphocytes at the dermoepidermal interface. In the dermis, there was a wedge-shaped superficial and deep, perivascular infiltrate with extravasated erythrocytes (Figures 3 and 4). Tissue Gram stain was negative for bacteria. Varicella-zoster virus and herpes simplex virus immunostains were negative. Direct immunofluorescence showed colloid bodies, as can be seen in lichenoid dermatitis.

At the next clinic visit, the patient reported a fever of 39.4 °C. After reviewing the patient’s histopathology and clinical picture, along with the presence of fever, a final diagnosis of FUMHD was made. The patient was started on an oral regimen of prednisone 80 mg once daily, minocycline 100 mg twice daily, and methotrexate 15 mg weekly. Unna boots (specialized compression wraps) with triamcinolone acetonide ointment 0.1% were placed weekly until the leg edema and ulcerations healed. He was maintained on methotrexate 15 mg weekly and 5 to 10 mg of prednisone once daily. The patient demonstrated residual scarring, with only rare new papulonodules that did not ulcerate when attempts were made to taper his medications. He was followed for nearly 3 years, with a recurrence of symptoms 2 years and 3 months after initial presentation to the academic dermatology clinic.

Febrile ulceronecrotic Mucha-Habermann disease is a rare and severe variant of PLEVA that can present with the rapid appearance of necrotic skin lesions, fever, and systemic manifestations, including pulmonary, gastrointestinal, central nervous system, cardiac, hematologic, and rheumatologic symptoms.2-4 The evolution from PLEVA to FUMHD ranges from days to weeks, and patientsrarely can have an initial presentation of FUMHD.2 The duration of illness has been reported to be 1 to 24 months5; however, the length of illness still remains unclear, as many studies of FUMHD are case reports with limited follow-up. Our patient had a disease duration of at least 27 months. The lesions of FUMHD usually are generalized with flexural prominence, and mucosal involvement occurs in approximately one-quarter of cases. Hypertrophic scarring may be seen after the ulcerated lesions heal.2 The incidence of FUMHD is higher in men than in women, and it is more common in younger individuals.2,6 There have been reported fatalities associated with FUMHD, mostly in adults.2,4
The clinical differential diagnosis for PLEVA includes disseminated herpes zoster, varicella-zoster virus or coxsackievirus infections, lymphomatoid papulosis, angiodestructive lymphoma such as extranodal natural killer/T-cell lymphoma, drug eruption, arthropod bite, erythema multiforme, ecthyma, ecthyma gangrenosum, necrotic folliculitis, and cutaneous small vessel vasculitis. To differentiate between these diagnoses and PLEVA or FUMHD, it is important to take a strong clinical history. For example, for varicella-zoster virus and coxsackievirus infections, exposure history to the viruses and vaccination history for varicella-zoster virus can help elucidate the diagnosis.
Skin biopsy can help differentiate between these entities and PLEVA or FUMHD. The histopathology of a nonulcerated lesion of FUMHD shows parakeratosis, spongiosis, and lymphocyte exocytosis, as well as lymphocytic vasculitis—findings commonly seen in PLEVA. With the ulceronecrotic lesions of FUMHD, epidermal necrosis and ulceration can be seen microscopically.2 Although skin biopsy is not absolutely necessary for making the diagnosis of PLEVA, it can be helpful.3 However, given the dramatic and extreme clinical impression with an extensive differential diagnosis that includes disorders ranging from infectious to neoplastic, biopsy of FUMHD with clinicopathologic correlation often is required.
It is important to avoid biopsying ulcerated lesions of FUMHD, as the histopathologic findings are more likely to be nonspecific. Additionally, nonspecific features often are seen with immunohistochemistry; abnormal laboratory testing may be seen in FUMHD, but there is no specific test to diagnose FUMHD.2 Finally, a predominantly CD8+ cell infiltrate was seen in 4 of 6 cases of FUMHD, with 2 cases showing a mixed infiltrate of CD8+ and CD4+ cells.5,7-10
Although no unified diagnostic criterion exists for FUMHD, Nofal et al2 proposed criteria comprised of constant features, which are found in every case of FUMHD and can confirm the diagnosis alone, and variable features to help ensure that cases of FUMHD are not missed. The constant features include fever, acute onset of generalized ulceronecrotic papules and plaques, a course that is rapid and progressive (without a tendency for spontaneous resolution), and histopathology that is consistent with PLEVA. The variable features include history of PLEVA, involvement of mucous membranes, and systemic involvement.2
No single unifying treatment modality for all cases of FUMHD has been described. Immunosuppressive drugs (eg, systemic steroids, methotrexate), antibiotics, antivirals, phototherapy, intravenous immunoglobulin, and dapsone have been tried in patients with FUMHD.2 Combination therapy with an oral medication such as erythromycin or methotrexate and psoralen plus UVA may be effective for FUMHD.3 Additionally, some authors believe that patients with FUMHD should be treated similar to burn victims with intensive supportive care.2
The etiology of PLEVA is unknown, but it is presumed to be associated with an effector cytotoxic T-cell response to either an infectious agent or a drug.11
Four cases of FUMHD with monoclonality have been reported,4,7,8 and some researchers propose that FUMHD may be a subset of cutaneous T-cell lymphoma.7 However, 2 other cases of FUMHD did not show monoclonality of T cells,5,18 suggesting that FUMHD may represent an inflammatory disorder, rather than a lymphoproliferative process of T cells.18 Given the controversy surrounding the clonality of FUMHD, T-cell gene rearrangement studies were not performed in our case.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other papulosquamous disorders. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:68-69.
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738.
- Milligan A, Johnston GA. Pityriasis lichenoides et varioliformis acuta. In: Lebwohl MG, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease, Comprehensive Therapeutic Strategies. 4th ed. Saunders; 2013:580-582.
- Miyamoto T, Takayama N, Kitada S, et al. Febrile ulceronecrotic Mucha-Habermann disease: a case report and a review of the literature. J Clin Pathol. 2003;56:795-797.
- Meziane L, Caudron A, Dhaille F, et al. Febrile ulceronecrotic Mucha-Habermann disease: treatment with infliximab and intravenous immunoglobulins and review of the literature. Dermatology. 2012;225:344-348.
- Robinson AB, Stein LD. Miscellaneous conditions associated with arthritis. In: Kliegman RM, Stanton BF, St. Geme JW III, et al, eds. Nelson Textbook of Pediatrics. 19th ed. W.B. Saunders Company; 2011:880.
- Cozzio A, Hafner J, Kempf W, et al. Febrile ulceronecrotic Mucha-Habermann disease with clonality: a cutaneous T-cell lymphoma entity? J Am Acad Dermatol. 2004;51:1014-1017.
- Tsianakas A, Hoeger PH. Transition of pityriasis lichenoides et varioliformis acuta to febrile ulceronecrotic Mucha-Habermann disease is associated with elevated serum tumour necrosis factor-alpha. Br J Dermatol. 2005;152:794-799.
- Yanaba K, Ito M, Sasaki H, et al. A case of febrile ulceronecrotic Mucha-Habermann disease requiring debridement of necrotic skin and epidermal autograft. Br J Dermatol. 2002;147:1249-1253.
- Lode HN, Döring P, Lauenstein P, et al. Febrile ulceronecrotic Mucha-Habermann disease following suspected hemorrhagic chickenpox infection in a 20-month-old boy. Infection. 2015;43:583-588.
- Tomasini D, Tomasini CF, Cerri A, et al. Pityriasis lichenoides: a cytotoxic T-cell-mediated skin disorder: evidence of human parvovirus B19 DNA in nine cases. J Cutan Pathol. 2004;31:531-538.
- Weiss LM, Wood GS, Ellisen LW, et al. Clonal T-cell populations in pityriasis lichenoides et varioliformis acuta (Mucha-Habermann disease). Am J Pathol. 1987;126:417-421.
- Dereure O, Levi E, Kadin ME. T-cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
- Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
- Fortson JS, Schroeter AL, Esterly NB. Cutaneous T-cell lymphoma (parapsoriasis en plaque): an association with pityriasis lichenoides et varioliformis acuta in young children. Arch Dermatol. 1990;126:1449-1453.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Cutaneous T-cell lymphoma. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:958.
- Kim JE, Yun WJ, Mun SK, et al. Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica: comparison of lesional T-cell subsets and investigation of viral associations. J Cutan Pathol. 2011;38:649-656.
- López-Estebaran´z JL, Vanaclocha F, Gil R, et al. Febrile ulceronecrotic Mucha-Habermann disease. J Am Acad Dermatol. 1993;29(5, pt 2):903-906.
To the Editor:
Pityriasis lichenoides is a papulosquamous dermatologic disorder that is characterized by recurrent papules.1 There is a spectrum of disease in pityriasis lichenoides that includes pityriasis lichenoides et varioliformis acuta (PLEVA) at one end and pityriasis lichenoides chronica at the other. Pityriasis lichenoides et varioliformis acuta is more common in younger individuals and is characterized by erythematous papules that often crust; these lesions resolve over weeks. The lesions of pityriasis lichenoides chronica are characteristically scaly, pink to red-brown papules that tend to resolve over months.1
Histologically, PLEVA exhibits parakeratosis, interface dermatitis, and a wedge-shaped infiltrate.1 Necrotic keratinocytes and extravasated erythrocytes also are common features. Additionally, monoclonal T cells may be present in the infiltrate.1
Febrile ulceronecrotic Mucha-Habermann disease (FUMHD) is a rare and severe variant of PLEVA. Febrile ulceronecrotic Mucha-Habermann disease is characterized by ulceronecrotic lesions, fever, and systemic symptoms.2 Herein, we present a case of FUMHD.
A 57-year-old man presented with an eruption of painful lesions involving the face, trunk, arms, legs, and genitalia of 1 month’s duration. The patient denied oral and ocular involvement. He had soreness and swelling of the arms and legs. A prior 12-day course of prednisone prescribed by a community dermatologist failed to improve the rash. A biopsy performed by a community dermatologist was nondiagnostic. The patient denied fever but did report chills. He had no preceding illness and was not taking new medications. On physical examination, the patient was afebrile and normotensive with innumerable deep-seated pustules and crusted ulcerations on the face, palms, soles, trunk, extremities, and penis (Figures 1 and 2). There was a background morbilliform eruption on the trunk. The ocular and oral mucosae were spared. The upper and lower extremities had pitting edema.
The patient’s alanine aminotransaminase and aspartate aminotransaminase levels were elevated at 55 and 51 U/L, respectively. His white blood cell count was within reference range; however, there was an elevated absolute neutrophil count (8.7×103/μL). No eosinophilia was noted. Laboratory evaluation showed a positive antimitochondrial antibody, and magnetic resonance imaging showed evidence of steatohepatitis. Punch biopsies from both the morbilliform eruption and a deep-seated pustule showed epidermal necrosis, parakeratosis, necrotic keratinocytes, and a lichenoid infiltrate of lymphocytes at the dermoepidermal interface. In the dermis, there was a wedge-shaped superficial and deep, perivascular infiltrate with extravasated erythrocytes (Figures 3 and 4). Tissue Gram stain was negative for bacteria. Varicella-zoster virus and herpes simplex virus immunostains were negative. Direct immunofluorescence showed colloid bodies, as can be seen in lichenoid dermatitis.
At the next clinic visit, the patient reported a fever of 39.4 °C. After reviewing the patient’s histopathology and clinical picture, along with the presence of fever, a final diagnosis of FUMHD was made. The patient was started on an oral regimen of prednisone 80 mg once daily, minocycline 100 mg twice daily, and methotrexate 15 mg weekly. Unna boots (specialized compression wraps) with triamcinolone acetonide ointment 0.1% were placed weekly until the leg edema and ulcerations healed. He was maintained on methotrexate 15 mg weekly and 5 to 10 mg of prednisone once daily. The patient demonstrated residual scarring, with only rare new papulonodules that did not ulcerate when attempts were made to taper his medications. He was followed for nearly 3 years, with a recurrence of symptoms 2 years and 3 months after initial presentation to the academic dermatology clinic.
Febrile ulceronecrotic Mucha-Habermann disease is a rare and severe variant of PLEVA that can present with the rapid appearance of necrotic skin lesions, fever, and systemic manifestations, including pulmonary, gastrointestinal, central nervous system, cardiac, hematologic, and rheumatologic symptoms.2-4 The evolution from PLEVA to FUMHD ranges from days to weeks, and patientsrarely can have an initial presentation of FUMHD.2 The duration of illness has been reported to be 1 to 24 months5; however, the length of illness still remains unclear, as many studies of FUMHD are case reports with limited follow-up. Our patient had a disease duration of at least 27 months. The lesions of FUMHD usually are generalized with flexural prominence, and mucosal involvement occurs in approximately one-quarter of cases. Hypertrophic scarring may be seen after the ulcerated lesions heal.2 The incidence of FUMHD is higher in men than in women, and it is more common in younger individuals.2,6 There have been reported fatalities associated with FUMHD, mostly in adults.2,4
The clinical differential diagnosis for PLEVA includes disseminated herpes zoster, varicella-zoster virus or coxsackievirus infections, lymphomatoid papulosis, angiodestructive lymphoma such as extranodal natural killer/T-cell lymphoma, drug eruption, arthropod bite, erythema multiforme, ecthyma, ecthyma gangrenosum, necrotic folliculitis, and cutaneous small vessel vasculitis. To differentiate between these diagnoses and PLEVA or FUMHD, it is important to take a strong clinical history. For example, for varicella-zoster virus and coxsackievirus infections, exposure history to the viruses and vaccination history for varicella-zoster virus can help elucidate the diagnosis.
Skin biopsy can help differentiate between these entities and PLEVA or FUMHD. The histopathology of a nonulcerated lesion of FUMHD shows parakeratosis, spongiosis, and lymphocyte exocytosis, as well as lymphocytic vasculitis—findings commonly seen in PLEVA. With the ulceronecrotic lesions of FUMHD, epidermal necrosis and ulceration can be seen microscopically.2 Although skin biopsy is not absolutely necessary for making the diagnosis of PLEVA, it can be helpful.3 However, given the dramatic and extreme clinical impression with an extensive differential diagnosis that includes disorders ranging from infectious to neoplastic, biopsy of FUMHD with clinicopathologic correlation often is required.
It is important to avoid biopsying ulcerated lesions of FUMHD, as the histopathologic findings are more likely to be nonspecific. Additionally, nonspecific features often are seen with immunohistochemistry; abnormal laboratory testing may be seen in FUMHD, but there is no specific test to diagnose FUMHD.2 Finally, a predominantly CD8+ cell infiltrate was seen in 4 of 6 cases of FUMHD, with 2 cases showing a mixed infiltrate of CD8+ and CD4+ cells.5,7-10
Although no unified diagnostic criterion exists for FUMHD, Nofal et al2 proposed criteria comprised of constant features, which are found in every case of FUMHD and can confirm the diagnosis alone, and variable features to help ensure that cases of FUMHD are not missed. The constant features include fever, acute onset of generalized ulceronecrotic papules and plaques, a course that is rapid and progressive (without a tendency for spontaneous resolution), and histopathology that is consistent with PLEVA. The variable features include history of PLEVA, involvement of mucous membranes, and systemic involvement.2
No single unifying treatment modality for all cases of FUMHD has been described. Immunosuppressive drugs (eg, systemic steroids, methotrexate), antibiotics, antivirals, phototherapy, intravenous immunoglobulin, and dapsone have been tried in patients with FUMHD.2 Combination therapy with an oral medication such as erythromycin or methotrexate and psoralen plus UVA may be effective for FUMHD.3 Additionally, some authors believe that patients with FUMHD should be treated similar to burn victims with intensive supportive care.2
The etiology of PLEVA is unknown, but it is presumed to be associated with an effector cytotoxic T-cell response to either an infectious agent or a drug.11
Four cases of FUMHD with monoclonality have been reported,4,7,8 and some researchers propose that FUMHD may be a subset of cutaneous T-cell lymphoma.7 However, 2 other cases of FUMHD did not show monoclonality of T cells,5,18 suggesting that FUMHD may represent an inflammatory disorder, rather than a lymphoproliferative process of T cells.18 Given the controversy surrounding the clonality of FUMHD, T-cell gene rearrangement studies were not performed in our case.
To the Editor:
Pityriasis lichenoides is a papulosquamous dermatologic disorder that is characterized by recurrent papules.1 There is a spectrum of disease in pityriasis lichenoides that includes pityriasis lichenoides et varioliformis acuta (PLEVA) at one end and pityriasis lichenoides chronica at the other. Pityriasis lichenoides et varioliformis acuta is more common in younger individuals and is characterized by erythematous papules that often crust; these lesions resolve over weeks. The lesions of pityriasis lichenoides chronica are characteristically scaly, pink to red-brown papules that tend to resolve over months.1
Histologically, PLEVA exhibits parakeratosis, interface dermatitis, and a wedge-shaped infiltrate.1 Necrotic keratinocytes and extravasated erythrocytes also are common features. Additionally, monoclonal T cells may be present in the infiltrate.1
Febrile ulceronecrotic Mucha-Habermann disease (FUMHD) is a rare and severe variant of PLEVA. Febrile ulceronecrotic Mucha-Habermann disease is characterized by ulceronecrotic lesions, fever, and systemic symptoms.2 Herein, we present a case of FUMHD.
A 57-year-old man presented with an eruption of painful lesions involving the face, trunk, arms, legs, and genitalia of 1 month’s duration. The patient denied oral and ocular involvement. He had soreness and swelling of the arms and legs. A prior 12-day course of prednisone prescribed by a community dermatologist failed to improve the rash. A biopsy performed by a community dermatologist was nondiagnostic. The patient denied fever but did report chills. He had no preceding illness and was not taking new medications. On physical examination, the patient was afebrile and normotensive with innumerable deep-seated pustules and crusted ulcerations on the face, palms, soles, trunk, extremities, and penis (Figures 1 and 2). There was a background morbilliform eruption on the trunk. The ocular and oral mucosae were spared. The upper and lower extremities had pitting edema.
The patient’s alanine aminotransaminase and aspartate aminotransaminase levels were elevated at 55 and 51 U/L, respectively. His white blood cell count was within reference range; however, there was an elevated absolute neutrophil count (8.7×103/μL). No eosinophilia was noted. Laboratory evaluation showed a positive antimitochondrial antibody, and magnetic resonance imaging showed evidence of steatohepatitis. Punch biopsies from both the morbilliform eruption and a deep-seated pustule showed epidermal necrosis, parakeratosis, necrotic keratinocytes, and a lichenoid infiltrate of lymphocytes at the dermoepidermal interface. In the dermis, there was a wedge-shaped superficial and deep, perivascular infiltrate with extravasated erythrocytes (Figures 3 and 4). Tissue Gram stain was negative for bacteria. Varicella-zoster virus and herpes simplex virus immunostains were negative. Direct immunofluorescence showed colloid bodies, as can be seen in lichenoid dermatitis.
At the next clinic visit, the patient reported a fever of 39.4 °C. After reviewing the patient’s histopathology and clinical picture, along with the presence of fever, a final diagnosis of FUMHD was made. The patient was started on an oral regimen of prednisone 80 mg once daily, minocycline 100 mg twice daily, and methotrexate 15 mg weekly. Unna boots (specialized compression wraps) with triamcinolone acetonide ointment 0.1% were placed weekly until the leg edema and ulcerations healed. He was maintained on methotrexate 15 mg weekly and 5 to 10 mg of prednisone once daily. The patient demonstrated residual scarring, with only rare new papulonodules that did not ulcerate when attempts were made to taper his medications. He was followed for nearly 3 years, with a recurrence of symptoms 2 years and 3 months after initial presentation to the academic dermatology clinic.
Febrile ulceronecrotic Mucha-Habermann disease is a rare and severe variant of PLEVA that can present with the rapid appearance of necrotic skin lesions, fever, and systemic manifestations, including pulmonary, gastrointestinal, central nervous system, cardiac, hematologic, and rheumatologic symptoms.2-4 The evolution from PLEVA to FUMHD ranges from days to weeks, and patientsrarely can have an initial presentation of FUMHD.2 The duration of illness has been reported to be 1 to 24 months5; however, the length of illness still remains unclear, as many studies of FUMHD are case reports with limited follow-up. Our patient had a disease duration of at least 27 months. The lesions of FUMHD usually are generalized with flexural prominence, and mucosal involvement occurs in approximately one-quarter of cases. Hypertrophic scarring may be seen after the ulcerated lesions heal.2 The incidence of FUMHD is higher in men than in women, and it is more common in younger individuals.2,6 There have been reported fatalities associated with FUMHD, mostly in adults.2,4
The clinical differential diagnosis for PLEVA includes disseminated herpes zoster, varicella-zoster virus or coxsackievirus infections, lymphomatoid papulosis, angiodestructive lymphoma such as extranodal natural killer/T-cell lymphoma, drug eruption, arthropod bite, erythema multiforme, ecthyma, ecthyma gangrenosum, necrotic folliculitis, and cutaneous small vessel vasculitis. To differentiate between these diagnoses and PLEVA or FUMHD, it is important to take a strong clinical history. For example, for varicella-zoster virus and coxsackievirus infections, exposure history to the viruses and vaccination history for varicella-zoster virus can help elucidate the diagnosis.
Skin biopsy can help differentiate between these entities and PLEVA or FUMHD. The histopathology of a nonulcerated lesion of FUMHD shows parakeratosis, spongiosis, and lymphocyte exocytosis, as well as lymphocytic vasculitis—findings commonly seen in PLEVA. With the ulceronecrotic lesions of FUMHD, epidermal necrosis and ulceration can be seen microscopically.2 Although skin biopsy is not absolutely necessary for making the diagnosis of PLEVA, it can be helpful.3 However, given the dramatic and extreme clinical impression with an extensive differential diagnosis that includes disorders ranging from infectious to neoplastic, biopsy of FUMHD with clinicopathologic correlation often is required.
It is important to avoid biopsying ulcerated lesions of FUMHD, as the histopathologic findings are more likely to be nonspecific. Additionally, nonspecific features often are seen with immunohistochemistry; abnormal laboratory testing may be seen in FUMHD, but there is no specific test to diagnose FUMHD.2 Finally, a predominantly CD8+ cell infiltrate was seen in 4 of 6 cases of FUMHD, with 2 cases showing a mixed infiltrate of CD8+ and CD4+ cells.5,7-10
Although no unified diagnostic criterion exists for FUMHD, Nofal et al2 proposed criteria comprised of constant features, which are found in every case of FUMHD and can confirm the diagnosis alone, and variable features to help ensure that cases of FUMHD are not missed. The constant features include fever, acute onset of generalized ulceronecrotic papules and plaques, a course that is rapid and progressive (without a tendency for spontaneous resolution), and histopathology that is consistent with PLEVA. The variable features include history of PLEVA, involvement of mucous membranes, and systemic involvement.2
No single unifying treatment modality for all cases of FUMHD has been described. Immunosuppressive drugs (eg, systemic steroids, methotrexate), antibiotics, antivirals, phototherapy, intravenous immunoglobulin, and dapsone have been tried in patients with FUMHD.2 Combination therapy with an oral medication such as erythromycin or methotrexate and psoralen plus UVA may be effective for FUMHD.3 Additionally, some authors believe that patients with FUMHD should be treated similar to burn victims with intensive supportive care.2
The etiology of PLEVA is unknown, but it is presumed to be associated with an effector cytotoxic T-cell response to either an infectious agent or a drug.11
Four cases of FUMHD with monoclonality have been reported,4,7,8 and some researchers propose that FUMHD may be a subset of cutaneous T-cell lymphoma.7 However, 2 other cases of FUMHD did not show monoclonality of T cells,5,18 suggesting that FUMHD may represent an inflammatory disorder, rather than a lymphoproliferative process of T cells.18 Given the controversy surrounding the clonality of FUMHD, T-cell gene rearrangement studies were not performed in our case.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other papulosquamous disorders. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:68-69.
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738.
- Milligan A, Johnston GA. Pityriasis lichenoides et varioliformis acuta. In: Lebwohl MG, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease, Comprehensive Therapeutic Strategies. 4th ed. Saunders; 2013:580-582.
- Miyamoto T, Takayama N, Kitada S, et al. Febrile ulceronecrotic Mucha-Habermann disease: a case report and a review of the literature. J Clin Pathol. 2003;56:795-797.
- Meziane L, Caudron A, Dhaille F, et al. Febrile ulceronecrotic Mucha-Habermann disease: treatment with infliximab and intravenous immunoglobulins and review of the literature. Dermatology. 2012;225:344-348.
- Robinson AB, Stein LD. Miscellaneous conditions associated with arthritis. In: Kliegman RM, Stanton BF, St. Geme JW III, et al, eds. Nelson Textbook of Pediatrics. 19th ed. W.B. Saunders Company; 2011:880.
- Cozzio A, Hafner J, Kempf W, et al. Febrile ulceronecrotic Mucha-Habermann disease with clonality: a cutaneous T-cell lymphoma entity? J Am Acad Dermatol. 2004;51:1014-1017.
- Tsianakas A, Hoeger PH. Transition of pityriasis lichenoides et varioliformis acuta to febrile ulceronecrotic Mucha-Habermann disease is associated with elevated serum tumour necrosis factor-alpha. Br J Dermatol. 2005;152:794-799.
- Yanaba K, Ito M, Sasaki H, et al. A case of febrile ulceronecrotic Mucha-Habermann disease requiring debridement of necrotic skin and epidermal autograft. Br J Dermatol. 2002;147:1249-1253.
- Lode HN, Döring P, Lauenstein P, et al. Febrile ulceronecrotic Mucha-Habermann disease following suspected hemorrhagic chickenpox infection in a 20-month-old boy. Infection. 2015;43:583-588.
- Tomasini D, Tomasini CF, Cerri A, et al. Pityriasis lichenoides: a cytotoxic T-cell-mediated skin disorder: evidence of human parvovirus B19 DNA in nine cases. J Cutan Pathol. 2004;31:531-538.
- Weiss LM, Wood GS, Ellisen LW, et al. Clonal T-cell populations in pityriasis lichenoides et varioliformis acuta (Mucha-Habermann disease). Am J Pathol. 1987;126:417-421.
- Dereure O, Levi E, Kadin ME. T-cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
- Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
- Fortson JS, Schroeter AL, Esterly NB. Cutaneous T-cell lymphoma (parapsoriasis en plaque): an association with pityriasis lichenoides et varioliformis acuta in young children. Arch Dermatol. 1990;126:1449-1453.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Cutaneous T-cell lymphoma. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:958.
- Kim JE, Yun WJ, Mun SK, et al. Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica: comparison of lesional T-cell subsets and investigation of viral associations. J Cutan Pathol. 2011;38:649-656.
- López-Estebaran´z JL, Vanaclocha F, Gil R, et al. Febrile ulceronecrotic Mucha-Habermann disease. J Am Acad Dermatol. 1993;29(5, pt 2):903-906.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Other papulosquamous disorders. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:68-69.
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738.
- Milligan A, Johnston GA. Pityriasis lichenoides et varioliformis acuta. In: Lebwohl MG, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease, Comprehensive Therapeutic Strategies. 4th ed. Saunders; 2013:580-582.
- Miyamoto T, Takayama N, Kitada S, et al. Febrile ulceronecrotic Mucha-Habermann disease: a case report and a review of the literature. J Clin Pathol. 2003;56:795-797.
- Meziane L, Caudron A, Dhaille F, et al. Febrile ulceronecrotic Mucha-Habermann disease: treatment with infliximab and intravenous immunoglobulins and review of the literature. Dermatology. 2012;225:344-348.
- Robinson AB, Stein LD. Miscellaneous conditions associated with arthritis. In: Kliegman RM, Stanton BF, St. Geme JW III, et al, eds. Nelson Textbook of Pediatrics. 19th ed. W.B. Saunders Company; 2011:880.
- Cozzio A, Hafner J, Kempf W, et al. Febrile ulceronecrotic Mucha-Habermann disease with clonality: a cutaneous T-cell lymphoma entity? J Am Acad Dermatol. 2004;51:1014-1017.
- Tsianakas A, Hoeger PH. Transition of pityriasis lichenoides et varioliformis acuta to febrile ulceronecrotic Mucha-Habermann disease is associated with elevated serum tumour necrosis factor-alpha. Br J Dermatol. 2005;152:794-799.
- Yanaba K, Ito M, Sasaki H, et al. A case of febrile ulceronecrotic Mucha-Habermann disease requiring debridement of necrotic skin and epidermal autograft. Br J Dermatol. 2002;147:1249-1253.
- Lode HN, Döring P, Lauenstein P, et al. Febrile ulceronecrotic Mucha-Habermann disease following suspected hemorrhagic chickenpox infection in a 20-month-old boy. Infection. 2015;43:583-588.
- Tomasini D, Tomasini CF, Cerri A, et al. Pityriasis lichenoides: a cytotoxic T-cell-mediated skin disorder: evidence of human parvovirus B19 DNA in nine cases. J Cutan Pathol. 2004;31:531-538.
- Weiss LM, Wood GS, Ellisen LW, et al. Clonal T-cell populations in pityriasis lichenoides et varioliformis acuta (Mucha-Habermann disease). Am J Pathol. 1987;126:417-421.
- Dereure O, Levi E, Kadin ME. T-cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
- Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
- Fortson JS, Schroeter AL, Esterly NB. Cutaneous T-cell lymphoma (parapsoriasis en plaque): an association with pityriasis lichenoides et varioliformis acuta in young children. Arch Dermatol. 1990;126:1449-1453.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Cutaneous T-cell lymphoma. In: Bolognia JL, Schaffer JV, Duncan KO, et al, eds. Dermatology Essentials. Elsevier Saunders; 2014:958.
- Kim JE, Yun WJ, Mun SK, et al. Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica: comparison of lesional T-cell subsets and investigation of viral associations. J Cutan Pathol. 2011;38:649-656.
- López-Estebaran´z JL, Vanaclocha F, Gil R, et al. Febrile ulceronecrotic Mucha-Habermann disease. J Am Acad Dermatol. 1993;29(5, pt 2):903-906.
Practice Points
- Febrile ulceronecrotic Mucha-Habermann disease (FUMHD) is a rare variant of pityriasis lichenoides et varioliformis acuta, characterized by ulceronecrotic lesions, fever, and systemic symptoms.
- A variety of treatments including immunosuppressive drugs (eg, systemic steroids, methotrexate), antibiotics, antivirals, phototherapy, intravenous immunoglobulin, and dapsone have been used in patients with FUMHD.

Indurated Mass on the Right Central Back
The Diagnosis: Actinomycetoma
Histopathology revealed evidence of an actinomycete organism within the suppuration, consistent with actinomycosis (quiz image [inset]). Given the clinical presentation and histopathologic findings, our patient was diagnosed with actinomycetoma.
Actinomycetoma is an indolent, progressive, subcutaneous infection characterized by a well-known clinical triad of tumefaction/subcutaneous mass, draining sinuses, and an exudate containing grains on microscopy. The sinus tracts are formed from the chronic infectious process that destroys tissue, creating tunnels. This infectious disease of soft tissue is a clinical subset of mycetoma, which is categorized as eumycetoma (fungal) and actinomycetoma (bacterial). Actinomycetoma resembles the behavior of insidious and chronic fungal infections; however, most mycetoma infections are bacterial.1,2 Actinomycetoma may be confused with actinomycosis, which is caused by Actinomycoses species, commensal organisms commonly located on the teeth and oral mucosa in association with other microorganisms that may pathogenically cause cervicofacial actinomycosis.3,4 Actinomycetoma can be caused by Nocardia, Streptomyces, and Actinomadura. 2,5 The foot is the most common location of involvement followed by the thoracic region. It is more common in tropical or equatorial locations and may be contracted through exposure to soil or wood.5 Mycetoma is considered a neglected tropical disease by the World Health Organization.1 In tropical countries, this disease may go undiagnosed or untreated for so long that surgical amputation may be the only effective treatment.
Actinomycetoma commonly is identifiable by direct microscopy, Gram stain, or bacterial culture, with Gram stain being more sensitive than bacterial culture.3 It is important to indicate the suspected organism to the microbiology laboratory because common bacterial pathogens are detected within 24 to 48 hours, but the causative microorganism in actinomycetoma may require up to 4 weeks for culture,2 leading to possible false negatives due to inadequate culture time.3 Histopathology of actinomycotic infections will demonstrate granulomatous inflammation, focal suppuration, and the presence of grains (ie, a colony of filamentous bacteria in a stellate shaped mass)(quiz image [inset]).
The gold standard of treatment is trimethoprim-sulfamethoxazole for up to several years.4,5 Amoxicillin–clavulanic acid, dapsone, amikacin, streptomycin, and beta-lactams have been used successfully.2,5 The treatment course is dependent on clinical severity and location of the disease. The cure rate with appropriate antibiotics can be as high as 90%,2,5 and thus surgical intervention can be avoided.
In the differential, cutaneous tuberculosis would show tuberculoid granulomas with epithelioid histiocytes with possible caseation on histopathology, typically alongside positive tuberculosis screening. Botryomycosis has a similar clinical presentation of a swollen or indurated lesion with draining sinus tracts, but it less commonly occurs on the trunk. Histopathology also is a close mimic of actinomycetoma with a small grain inside a suppurative infiltrate; however, it has no filamentous bacteria. A foreign body reaction would not histologically present with suppuration or grains, and draining sinuses typically would not be seen on clinical presentation. Sarcoma is a neoplastic process and most commonly would show a proliferation of cells with soft tissue or bone origin on histopathology and not primarily an inflammatory cell process.6
- Verma P, Jha A. Mycetoma: reviewing a neglected disease. Clin Exp Dermatol. 2019;44:123-129.
- Valour F, Sénéchal A, Dupieux C, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Resist. 2014;7:183-197.
- Bennhoff DF. Actinomycosis: diagnostic and therapeutic considerations and a review of 32 cases. Laryngoscope. 1984;94:1198-1217.
- Welsh O, Vera-Cabrera L, Welsh E, et al. Actinomycetoma and advances in its treatment. Clin Dermatol. 2012;30:372-381.
- Arenas R, Fernandez Martinez RF, Torres-Guerrero E, et al. Actinomycetoma: an update on diagnosis and treatment. Cutis. 2017;99:E11-E15.
- Weedon D. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier; 2010.
The Diagnosis: Actinomycetoma
Histopathology revealed evidence of an actinomycete organism within the suppuration, consistent with actinomycosis (quiz image [inset]). Given the clinical presentation and histopathologic findings, our patient was diagnosed with actinomycetoma.
Actinomycetoma is an indolent, progressive, subcutaneous infection characterized by a well-known clinical triad of tumefaction/subcutaneous mass, draining sinuses, and an exudate containing grains on microscopy. The sinus tracts are formed from the chronic infectious process that destroys tissue, creating tunnels. This infectious disease of soft tissue is a clinical subset of mycetoma, which is categorized as eumycetoma (fungal) and actinomycetoma (bacterial). Actinomycetoma resembles the behavior of insidious and chronic fungal infections; however, most mycetoma infections are bacterial.1,2 Actinomycetoma may be confused with actinomycosis, which is caused by Actinomycoses species, commensal organisms commonly located on the teeth and oral mucosa in association with other microorganisms that may pathogenically cause cervicofacial actinomycosis.3,4 Actinomycetoma can be caused by Nocardia, Streptomyces, and Actinomadura. 2,5 The foot is the most common location of involvement followed by the thoracic region. It is more common in tropical or equatorial locations and may be contracted through exposure to soil or wood.5 Mycetoma is considered a neglected tropical disease by the World Health Organization.1 In tropical countries, this disease may go undiagnosed or untreated for so long that surgical amputation may be the only effective treatment.
Actinomycetoma commonly is identifiable by direct microscopy, Gram stain, or bacterial culture, with Gram stain being more sensitive than bacterial culture.3 It is important to indicate the suspected organism to the microbiology laboratory because common bacterial pathogens are detected within 24 to 48 hours, but the causative microorganism in actinomycetoma may require up to 4 weeks for culture,2 leading to possible false negatives due to inadequate culture time.3 Histopathology of actinomycotic infections will demonstrate granulomatous inflammation, focal suppuration, and the presence of grains (ie, a colony of filamentous bacteria in a stellate shaped mass)(quiz image [inset]).
The gold standard of treatment is trimethoprim-sulfamethoxazole for up to several years.4,5 Amoxicillin–clavulanic acid, dapsone, amikacin, streptomycin, and beta-lactams have been used successfully.2,5 The treatment course is dependent on clinical severity and location of the disease. The cure rate with appropriate antibiotics can be as high as 90%,2,5 and thus surgical intervention can be avoided.
In the differential, cutaneous tuberculosis would show tuberculoid granulomas with epithelioid histiocytes with possible caseation on histopathology, typically alongside positive tuberculosis screening. Botryomycosis has a similar clinical presentation of a swollen or indurated lesion with draining sinus tracts, but it less commonly occurs on the trunk. Histopathology also is a close mimic of actinomycetoma with a small grain inside a suppurative infiltrate; however, it has no filamentous bacteria. A foreign body reaction would not histologically present with suppuration or grains, and draining sinuses typically would not be seen on clinical presentation. Sarcoma is a neoplastic process and most commonly would show a proliferation of cells with soft tissue or bone origin on histopathology and not primarily an inflammatory cell process.6
The Diagnosis: Actinomycetoma
Histopathology revealed evidence of an actinomycete organism within the suppuration, consistent with actinomycosis (quiz image [inset]). Given the clinical presentation and histopathologic findings, our patient was diagnosed with actinomycetoma.
Actinomycetoma is an indolent, progressive, subcutaneous infection characterized by a well-known clinical triad of tumefaction/subcutaneous mass, draining sinuses, and an exudate containing grains on microscopy. The sinus tracts are formed from the chronic infectious process that destroys tissue, creating tunnels. This infectious disease of soft tissue is a clinical subset of mycetoma, which is categorized as eumycetoma (fungal) and actinomycetoma (bacterial). Actinomycetoma resembles the behavior of insidious and chronic fungal infections; however, most mycetoma infections are bacterial.1,2 Actinomycetoma may be confused with actinomycosis, which is caused by Actinomycoses species, commensal organisms commonly located on the teeth and oral mucosa in association with other microorganisms that may pathogenically cause cervicofacial actinomycosis.3,4 Actinomycetoma can be caused by Nocardia, Streptomyces, and Actinomadura. 2,5 The foot is the most common location of involvement followed by the thoracic region. It is more common in tropical or equatorial locations and may be contracted through exposure to soil or wood.5 Mycetoma is considered a neglected tropical disease by the World Health Organization.1 In tropical countries, this disease may go undiagnosed or untreated for so long that surgical amputation may be the only effective treatment.
Actinomycetoma commonly is identifiable by direct microscopy, Gram stain, or bacterial culture, with Gram stain being more sensitive than bacterial culture.3 It is important to indicate the suspected organism to the microbiology laboratory because common bacterial pathogens are detected within 24 to 48 hours, but the causative microorganism in actinomycetoma may require up to 4 weeks for culture,2 leading to possible false negatives due to inadequate culture time.3 Histopathology of actinomycotic infections will demonstrate granulomatous inflammation, focal suppuration, and the presence of grains (ie, a colony of filamentous bacteria in a stellate shaped mass)(quiz image [inset]).
The gold standard of treatment is trimethoprim-sulfamethoxazole for up to several years.4,5 Amoxicillin–clavulanic acid, dapsone, amikacin, streptomycin, and beta-lactams have been used successfully.2,5 The treatment course is dependent on clinical severity and location of the disease. The cure rate with appropriate antibiotics can be as high as 90%,2,5 and thus surgical intervention can be avoided.
In the differential, cutaneous tuberculosis would show tuberculoid granulomas with epithelioid histiocytes with possible caseation on histopathology, typically alongside positive tuberculosis screening. Botryomycosis has a similar clinical presentation of a swollen or indurated lesion with draining sinus tracts, but it less commonly occurs on the trunk. Histopathology also is a close mimic of actinomycetoma with a small grain inside a suppurative infiltrate; however, it has no filamentous bacteria. A foreign body reaction would not histologically present with suppuration or grains, and draining sinuses typically would not be seen on clinical presentation. Sarcoma is a neoplastic process and most commonly would show a proliferation of cells with soft tissue or bone origin on histopathology and not primarily an inflammatory cell process.6
- Verma P, Jha A. Mycetoma: reviewing a neglected disease. Clin Exp Dermatol. 2019;44:123-129.
- Valour F, Sénéchal A, Dupieux C, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Resist. 2014;7:183-197.
- Bennhoff DF. Actinomycosis: diagnostic and therapeutic considerations and a review of 32 cases. Laryngoscope. 1984;94:1198-1217.
- Welsh O, Vera-Cabrera L, Welsh E, et al. Actinomycetoma and advances in its treatment. Clin Dermatol. 2012;30:372-381.
- Arenas R, Fernandez Martinez RF, Torres-Guerrero E, et al. Actinomycetoma: an update on diagnosis and treatment. Cutis. 2017;99:E11-E15.
- Weedon D. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier; 2010.
- Verma P, Jha A. Mycetoma: reviewing a neglected disease. Clin Exp Dermatol. 2019;44:123-129.
- Valour F, Sénéchal A, Dupieux C, et al. Actinomycosis: etiology, clinical features, diagnosis, treatment, and management. Infect Drug Resist. 2014;7:183-197.
- Bennhoff DF. Actinomycosis: diagnostic and therapeutic considerations and a review of 32 cases. Laryngoscope. 1984;94:1198-1217.
- Welsh O, Vera-Cabrera L, Welsh E, et al. Actinomycetoma and advances in its treatment. Clin Dermatol. 2012;30:372-381.
- Arenas R, Fernandez Martinez RF, Torres-Guerrero E, et al. Actinomycetoma: an update on diagnosis and treatment. Cutis. 2017;99:E11-E15.
- Weedon D. Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier; 2010.
A 26-year-old Guatemalan man who was a former carpenter presented with an indurated, nontender, nonpruritic, subcutaneous mass on the right central back with multiple draining sinus tracts on the surface and several depressed circular atrophic scars on the periphery of the mass. He noticed that the lesion began as a pustule 1.5 years prior and gradually enlarged. He denied any trauma, insect bites, fever, chills, headaches, weight loss, or travel history (he relocated to the United States 3.5 years ago) prior to the skin eruption. A biopsy was performed by an outside dermatologist 1 year prior to the current presentation, with a diagnosis of Pityrosporum folliculitis. Throughout his clinical course, treatment with oral antifungals, oral doxycycline, and topical clindamycin all failed. The mass was removed by plastic surgery 1 year prior.
A tissue biopsy for histology and culture was obtained at presentation to our institution. Laboratory findings showed that the basic metabolic panel was within reference range. Chest radiography indicated no active disease. A tuberculosis screening was negative. A bacterial culture of the lesion identified no growth after 48 hours. Our tissue biopsy revealed fibrosing granulation tissue, but the surgical pathology from a prior mass excision revealed sinus tracts with suppuration, evidence of scarring, foreign body giant cell reaction, and a characteristic finding (inset: H&E, original magnification ×200).


Pruritic Eruption on the Trunk and Extremities
THE DIAGNOSIS:
Acquired Perforating Disorder of Renal Disease
A papule with the central plug removed left a pitlike depression, representing Kyrle disease (Figure 1). A punch biopsy of the left forearm revealed epidermal hyperplasia (Figure 2A) surrounding a keratin plug that contained degenerated basophilic material (Figure 2B), confirming the diagnosis of acquired perforating disorder of renal disease (APDRD), classically described as Kyrle disease.
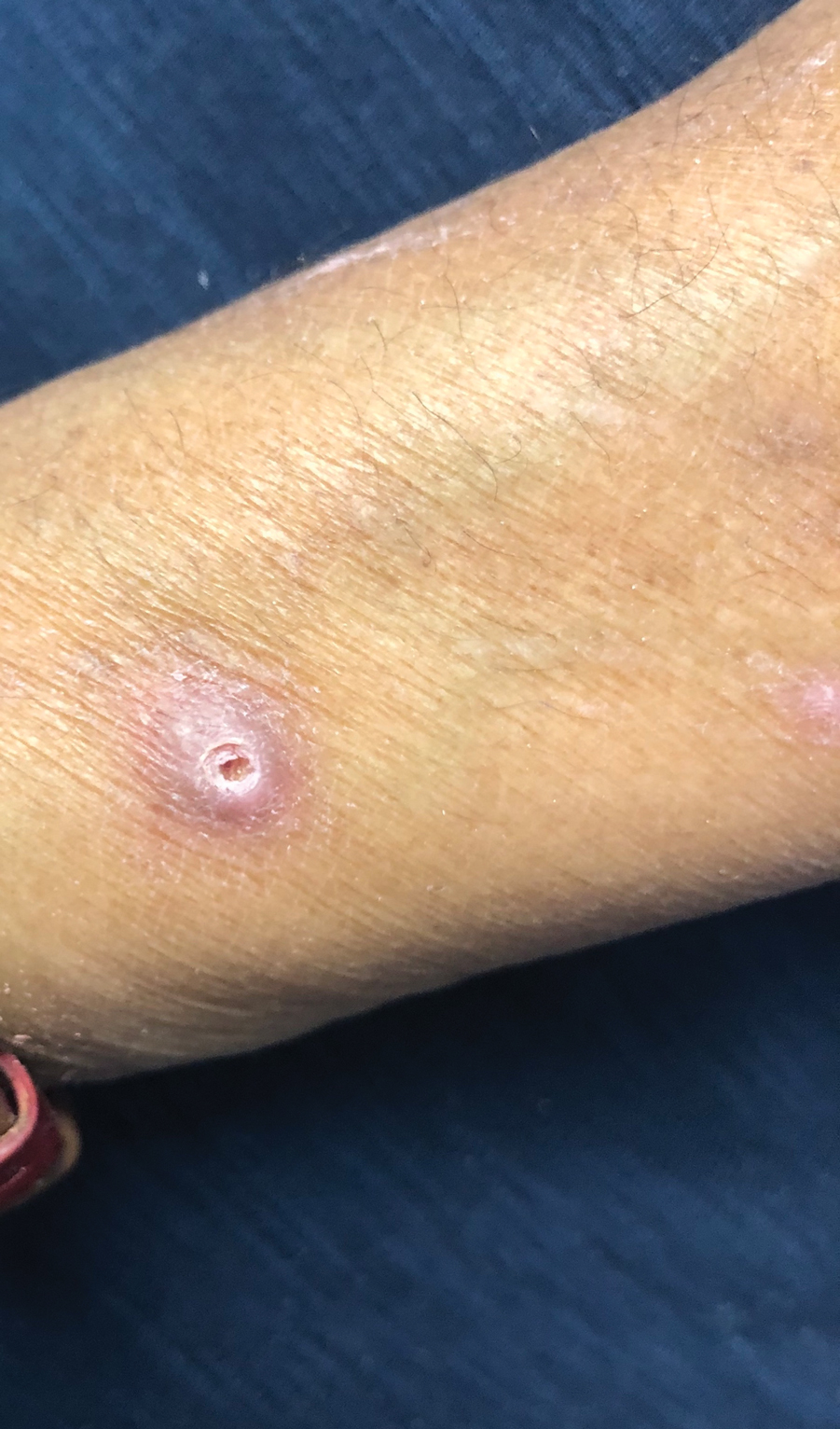
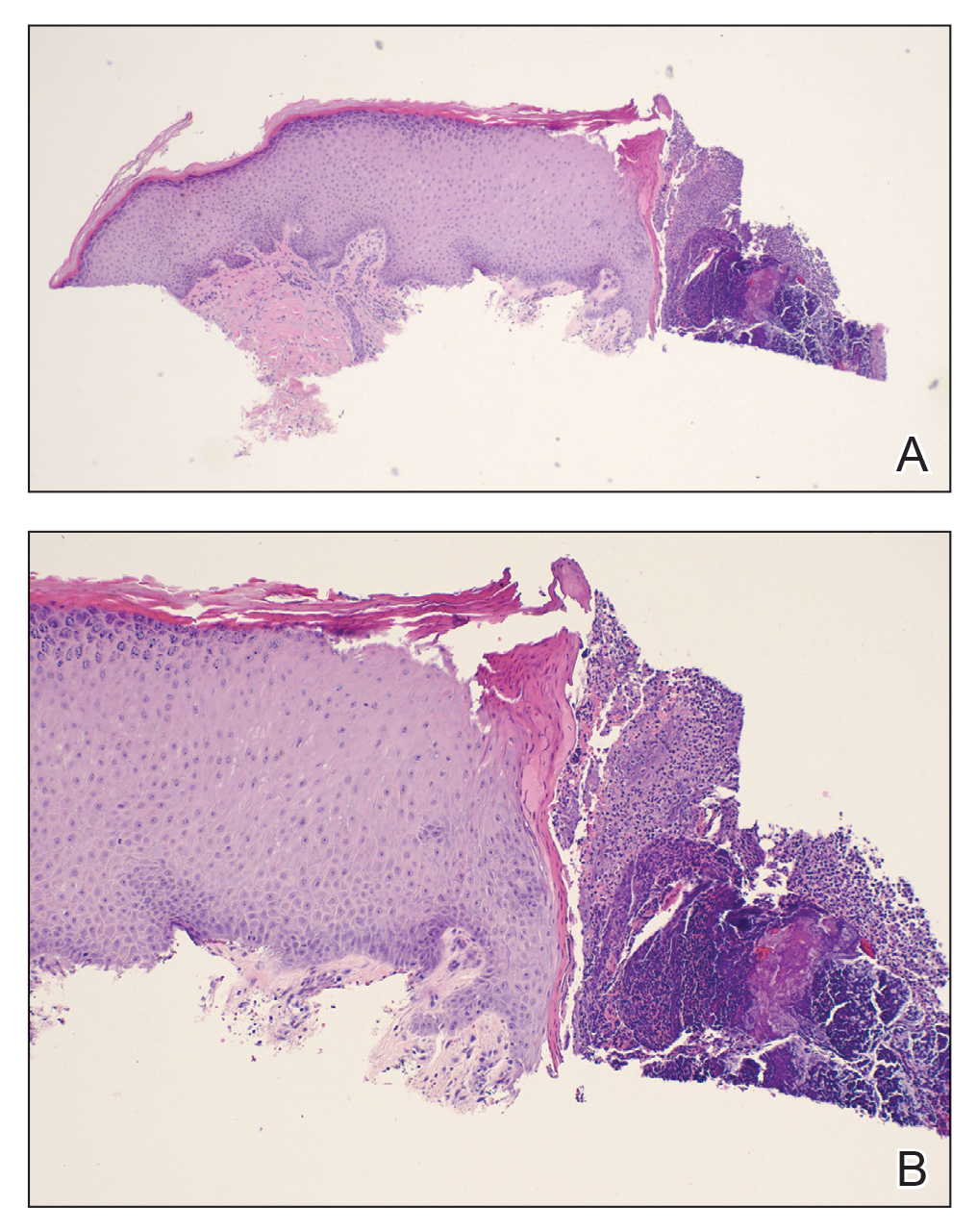
Acquired perforating disorder of renal disease is an uncommon condition in the general population. It is associated with systemic disease, commonly diabetes mellitus and chronic renal failure, and is seen in up to 10% patients receiving hemodialysis.1 The underlying etiology and pathogenesis of APDRD remains unknown. It has been proposed to be a variant of prurigo nodularis, representing end-stage excoriated folliculitis.1 Given that most cases appear in patients with systemic disease and metabolic abnormalities, APDRD also has been classified under the spectrum of acquired perforating dermatoses, a group of disorders defined by transepithelial elimination of dermal connective tissue. Elevated levels of serum and tissue fibronectin, uremia, and hyperphosphatemia have been observed in patients with APDRD.1,2 Fibronectin stimulates epithelial migration and proliferation and may lead to expulsion of keratin. Furthermore, dermal deposition of excess urea and/or phosphate could initiate transepithelial elimination of material. Alternative hypotheses implicate abnormal keratinization or an imbalance between the rates of epidermal proliferation/ differentiation and keratin production, whereby keratin production outpaces the former. Keratin deposited within the dermis subsequently elicits an inflammatory response along with alterations in the local dermis and connective tissue. These components become intermixed and are extruded through the plug opening.3 Lastly, immune dysregulation resulting from systemic disease could contribute to APDRD through increased expression of IL-31, a cytokine thought to play a role in several pruritic inflammatory skin diseases.4
Although standardized treatment guidelines for APDRD have not been established, the mainstay of therapy is control of the underlying systemic disorder. Intense pruritus and repeated scratching may contribute to microtrauma and subsequent koebnerization of new lesions.3 Thus, ameliorating pruritus can provide both symptomatic relief and prevent the development of new lesions. Retinoids, UV light, oral antibiotics, antihistamines, corticosteroids, keratolytic agents, and immunosuppressants (eg, allopurinol, tacrolimus) have shown some benefit.4
The differential diagnoses for APDRD include arthropod hypersensitivity reactions, eruptive keratoacanthomas, keratosis pilaris, and prurigo nodularis. Arthropod hypersensitivity reactions are seen in patients with a history of a bite or sting from arthropods such as bees, fleas, mites, ticks, and spiders. These reactions cause symptoms of pain, burning, or pruritus and present heterogeneously. They can be edematous and appear as single or multiple papules, pustules, plaques, vesicles, and/or bullae. A central punctum or crusting also may be present. Eruptive keratoacanthomas are seen in Grzybowski syndrome and Ferguson-Smith disease. Grzybowski syndrome arises in the fifth to seventh decades of life and is characterized by the eruptive onset of hundreds to thousands of pruritic, dome-shaped, follicular papules with or without central keratin plugs. Ectropion, mucosal lesions, and masklike facies are other clinical characteristics of Grzybowski syndrome. Ferguson-Smith disease begins in the second decade of life. The eruption of multiple keratoacanthomas and/or squamous cell carcinomas occurs in crops, rapidly growing over 2 to 4 weeks, and then self-resolves. This disease is inherited in an autosomal-dominant manner and is associated with chromosome 9q22. Keratosis pilaris is a benign condition of follicular hyperkeratosis that can appear in any age group and usually is absent of symptoms. It is not associated with any systemic disease. Clinically, the condition appears as folliculocentric keratotic papules with varying degrees of perifollicular erythema located along the extensor surfaces. Keratosis pilaris and APDRD share features of a follicular hyperkeratosis and dilated infundibulum; however, perforation is absent in keratosis pilaris. Lastly, prurigo nodularis is another intensely pruritic dermatosis associated with renal disease that presents as papulonodules on the extensor surfaces of the arms and legs. A biopsy can help to distinguish prurigo nodularis from APDRD.
- Rice AS, Zedek D. Kyrle disease. StatPearls [internet]. StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK532886/
- McKinley-Grant L, Peebles J. Renal disease. In: Kelly A, Taylor SC, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. McGraw-Hill; 2016
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Forouzandeh M, Stratman S, Yosipovitch G. The treatment of Kyrle’s disease: a systematic review. J Eur Acad Dermatol Venereol. 2020;34:1457-1463. doi:10.1111/jdv.16182
THE DIAGNOSIS:
Acquired Perforating Disorder of Renal Disease
A papule with the central plug removed left a pitlike depression, representing Kyrle disease (Figure 1). A punch biopsy of the left forearm revealed epidermal hyperplasia (Figure 2A) surrounding a keratin plug that contained degenerated basophilic material (Figure 2B), confirming the diagnosis of acquired perforating disorder of renal disease (APDRD), classically described as Kyrle disease.
Acquired perforating disorder of renal disease is an uncommon condition in the general population. It is associated with systemic disease, commonly diabetes mellitus and chronic renal failure, and is seen in up to 10% patients receiving hemodialysis.1 The underlying etiology and pathogenesis of APDRD remains unknown. It has been proposed to be a variant of prurigo nodularis, representing end-stage excoriated folliculitis.1 Given that most cases appear in patients with systemic disease and metabolic abnormalities, APDRD also has been classified under the spectrum of acquired perforating dermatoses, a group of disorders defined by transepithelial elimination of dermal connective tissue. Elevated levels of serum and tissue fibronectin, uremia, and hyperphosphatemia have been observed in patients with APDRD.1,2 Fibronectin stimulates epithelial migration and proliferation and may lead to expulsion of keratin. Furthermore, dermal deposition of excess urea and/or phosphate could initiate transepithelial elimination of material. Alternative hypotheses implicate abnormal keratinization or an imbalance between the rates of epidermal proliferation/ differentiation and keratin production, whereby keratin production outpaces the former. Keratin deposited within the dermis subsequently elicits an inflammatory response along with alterations in the local dermis and connective tissue. These components become intermixed and are extruded through the plug opening.3 Lastly, immune dysregulation resulting from systemic disease could contribute to APDRD through increased expression of IL-31, a cytokine thought to play a role in several pruritic inflammatory skin diseases.4
Although standardized treatment guidelines for APDRD have not been established, the mainstay of therapy is control of the underlying systemic disorder. Intense pruritus and repeated scratching may contribute to microtrauma and subsequent koebnerization of new lesions.3 Thus, ameliorating pruritus can provide both symptomatic relief and prevent the development of new lesions. Retinoids, UV light, oral antibiotics, antihistamines, corticosteroids, keratolytic agents, and immunosuppressants (eg, allopurinol, tacrolimus) have shown some benefit.4
The differential diagnoses for APDRD include arthropod hypersensitivity reactions, eruptive keratoacanthomas, keratosis pilaris, and prurigo nodularis. Arthropod hypersensitivity reactions are seen in patients with a history of a bite or sting from arthropods such as bees, fleas, mites, ticks, and spiders. These reactions cause symptoms of pain, burning, or pruritus and present heterogeneously. They can be edematous and appear as single or multiple papules, pustules, plaques, vesicles, and/or bullae. A central punctum or crusting also may be present. Eruptive keratoacanthomas are seen in Grzybowski syndrome and Ferguson-Smith disease. Grzybowski syndrome arises in the fifth to seventh decades of life and is characterized by the eruptive onset of hundreds to thousands of pruritic, dome-shaped, follicular papules with or without central keratin plugs. Ectropion, mucosal lesions, and masklike facies are other clinical characteristics of Grzybowski syndrome. Ferguson-Smith disease begins in the second decade of life. The eruption of multiple keratoacanthomas and/or squamous cell carcinomas occurs in crops, rapidly growing over 2 to 4 weeks, and then self-resolves. This disease is inherited in an autosomal-dominant manner and is associated with chromosome 9q22. Keratosis pilaris is a benign condition of follicular hyperkeratosis that can appear in any age group and usually is absent of symptoms. It is not associated with any systemic disease. Clinically, the condition appears as folliculocentric keratotic papules with varying degrees of perifollicular erythema located along the extensor surfaces. Keratosis pilaris and APDRD share features of a follicular hyperkeratosis and dilated infundibulum; however, perforation is absent in keratosis pilaris. Lastly, prurigo nodularis is another intensely pruritic dermatosis associated with renal disease that presents as papulonodules on the extensor surfaces of the arms and legs. A biopsy can help to distinguish prurigo nodularis from APDRD.
THE DIAGNOSIS:
Acquired Perforating Disorder of Renal Disease
A papule with the central plug removed left a pitlike depression, representing Kyrle disease (Figure 1). A punch biopsy of the left forearm revealed epidermal hyperplasia (Figure 2A) surrounding a keratin plug that contained degenerated basophilic material (Figure 2B), confirming the diagnosis of acquired perforating disorder of renal disease (APDRD), classically described as Kyrle disease.
Acquired perforating disorder of renal disease is an uncommon condition in the general population. It is associated with systemic disease, commonly diabetes mellitus and chronic renal failure, and is seen in up to 10% patients receiving hemodialysis.1 The underlying etiology and pathogenesis of APDRD remains unknown. It has been proposed to be a variant of prurigo nodularis, representing end-stage excoriated folliculitis.1 Given that most cases appear in patients with systemic disease and metabolic abnormalities, APDRD also has been classified under the spectrum of acquired perforating dermatoses, a group of disorders defined by transepithelial elimination of dermal connective tissue. Elevated levels of serum and tissue fibronectin, uremia, and hyperphosphatemia have been observed in patients with APDRD.1,2 Fibronectin stimulates epithelial migration and proliferation and may lead to expulsion of keratin. Furthermore, dermal deposition of excess urea and/or phosphate could initiate transepithelial elimination of material. Alternative hypotheses implicate abnormal keratinization or an imbalance between the rates of epidermal proliferation/ differentiation and keratin production, whereby keratin production outpaces the former. Keratin deposited within the dermis subsequently elicits an inflammatory response along with alterations in the local dermis and connective tissue. These components become intermixed and are extruded through the plug opening.3 Lastly, immune dysregulation resulting from systemic disease could contribute to APDRD through increased expression of IL-31, a cytokine thought to play a role in several pruritic inflammatory skin diseases.4
Although standardized treatment guidelines for APDRD have not been established, the mainstay of therapy is control of the underlying systemic disorder. Intense pruritus and repeated scratching may contribute to microtrauma and subsequent koebnerization of new lesions.3 Thus, ameliorating pruritus can provide both symptomatic relief and prevent the development of new lesions. Retinoids, UV light, oral antibiotics, antihistamines, corticosteroids, keratolytic agents, and immunosuppressants (eg, allopurinol, tacrolimus) have shown some benefit.4
The differential diagnoses for APDRD include arthropod hypersensitivity reactions, eruptive keratoacanthomas, keratosis pilaris, and prurigo nodularis. Arthropod hypersensitivity reactions are seen in patients with a history of a bite or sting from arthropods such as bees, fleas, mites, ticks, and spiders. These reactions cause symptoms of pain, burning, or pruritus and present heterogeneously. They can be edematous and appear as single or multiple papules, pustules, plaques, vesicles, and/or bullae. A central punctum or crusting also may be present. Eruptive keratoacanthomas are seen in Grzybowski syndrome and Ferguson-Smith disease. Grzybowski syndrome arises in the fifth to seventh decades of life and is characterized by the eruptive onset of hundreds to thousands of pruritic, dome-shaped, follicular papules with or without central keratin plugs. Ectropion, mucosal lesions, and masklike facies are other clinical characteristics of Grzybowski syndrome. Ferguson-Smith disease begins in the second decade of life. The eruption of multiple keratoacanthomas and/or squamous cell carcinomas occurs in crops, rapidly growing over 2 to 4 weeks, and then self-resolves. This disease is inherited in an autosomal-dominant manner and is associated with chromosome 9q22. Keratosis pilaris is a benign condition of follicular hyperkeratosis that can appear in any age group and usually is absent of symptoms. It is not associated with any systemic disease. Clinically, the condition appears as folliculocentric keratotic papules with varying degrees of perifollicular erythema located along the extensor surfaces. Keratosis pilaris and APDRD share features of a follicular hyperkeratosis and dilated infundibulum; however, perforation is absent in keratosis pilaris. Lastly, prurigo nodularis is another intensely pruritic dermatosis associated with renal disease that presents as papulonodules on the extensor surfaces of the arms and legs. A biopsy can help to distinguish prurigo nodularis from APDRD.
- Rice AS, Zedek D. Kyrle disease. StatPearls [internet]. StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK532886/
- McKinley-Grant L, Peebles J. Renal disease. In: Kelly A, Taylor SC, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. McGraw-Hill; 2016
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Forouzandeh M, Stratman S, Yosipovitch G. The treatment of Kyrle’s disease: a systematic review. J Eur Acad Dermatol Venereol. 2020;34:1457-1463. doi:10.1111/jdv.16182
- Rice AS, Zedek D. Kyrle disease. StatPearls [internet]. StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK532886/
- McKinley-Grant L, Peebles J. Renal disease. In: Kelly A, Taylor SC, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. McGraw-Hill; 2016
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Forouzandeh M, Stratman S, Yosipovitch G. The treatment of Kyrle’s disease: a systematic review. J Eur Acad Dermatol Venereol. 2020;34:1457-1463. doi:10.1111/jdv.16182
A 74-year-old woman with a 30-year history of type 2 diabetes mellitus presented to our dermatology clinic with a pruritic eruption on the trunk, arms, and legs of 2 months’ duration. Several over-the-counter moisturizers had been used without improvement, and the pruritus was notably impacting her sleep. Physical examination revealed discrete, hyperkeratotic, predominantly follicular, eruptive papules with hyperkeratotic plugs diffusely distributed on the trunk, arms, and legs.
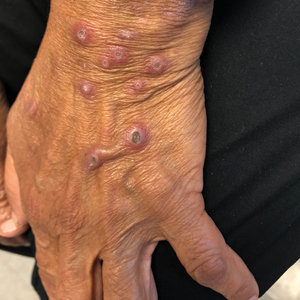
Zosteriform Eruption on the Chest and Abdomen
THE DIAGNOSIS:
Cutaneous Metastatic Mesothelioma
Biopsies of the larger erythematous papules revealed an infiltrate of atypical tumor cells with mitoses (Figure 1) that were immunoreactive for calretinin (Figure 2) and lacked nuclear BRCA1 associated protein-1, BAP1, expression (not shown). The patient’s prior mesothelioma was re-reviewed, and the cutaneous tumor cells were similar to the primary mesothelioma. A diagnosis of cutaneous metastatic mesothelioma (CMM) was made.
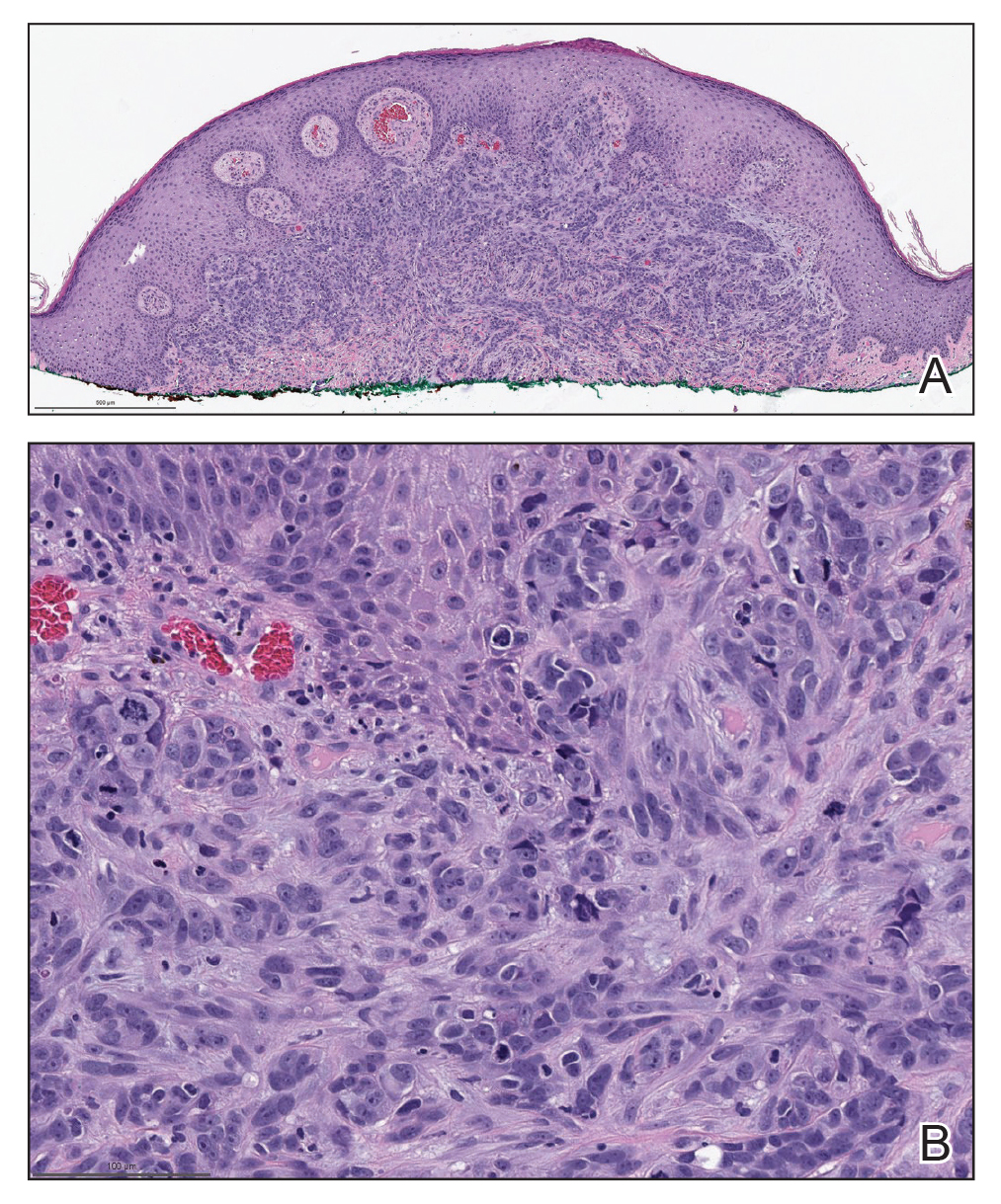
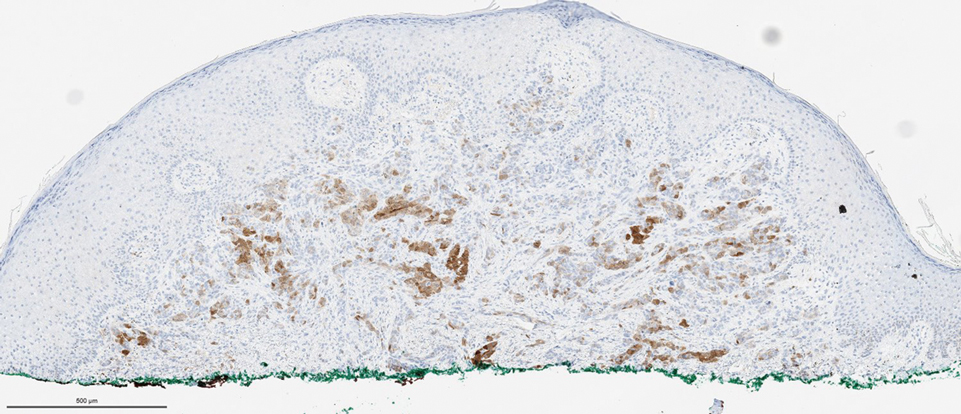
Mesothelioma is a rare neoplasm arising from the pleura, pericardium, peritoneum, and tunica vaginalis,1 with an estimated annual incidence of 2500 cases.2 The predominant risk factor for the development of pleural mesothelioma is asbestos exposure, which has been identified in up to 90% of cases. Mesothelioma can give rise to local and less frequently distant hematogenous metastases. Cutaneous involvement of mesothelioma is rare.3 More than 80% of CMM cases are attributed to seeding the skin at procedure sites or by direct infiltration of scars. Distant CMM is rare and typically presents as subcutaneous nodules.4 Few cases of inflammatory CMM have been published,1,4,5 with even fewer mimicking herpes zoster infection (HZI), as seen in our patient.
The most specific stain for mesothelioma is calretinin, which strongly and diffusely stains both the nucleus and cytoplasm. Other markers include Wilms tumor 1, cytokeratin 5/6, thrombomodulin, and HBME-1. Immunohistochemistry to detect the loss of BAP1 staining in the nucleus is important for differentiating between mesothelioma and mesothelial hyperplasia.3
Cutaneous metastases occur in 0.7% to 9% of patients with internal malignant disease. Most commonly, cutaneous metastases present as cutaneous nodules, though other reported inflammatory presentations include erysipeloides, generalized erythematous patches, telangiectasia, and zosteriform distributions.6 Zosteriform distributions are particularly rare and most commonly are due to breast carcinomas or lymphomas. The mechanism of zosteriform metastasis is unknown, but theories include tumoral spread along vessels, invasion of the thoracic perineural sheaths, localized spread of tumor cells from a surgical site, or a Koebner-like reaction at the site of an existing HZI. Regardless of primary tumor type or presentation, cutaneous metastasis is a poor prognostic sign, with survival rates varying based on primary tumor type.7
Other differential diagnoses include herpes zoster granulomatous dermatitis, radiation recall dermatitis, cutaneous Rosai-Dorfman disease, and zosteriform lichen planus, all of which have been reported after HZI.8-10 Herpes zoster granulomatous dermatitis typically presents weeks to years after acute HZI with erythematous to violaceous papules and plaques at the site of the prior HZI. A biopsy reveals interstitial granulomatous dermatitis and multinucleated giant cells.8 Radiation recall dermatitis is a cutaneous inflammatory reaction limited to regions of prior radiation exposure after the administration of a triggering medication. Radiation recall dermatitis can present days to many years after the completion of treatment.9 Although the eruption in our patient was at the site of prior radiation, the pathologic and clinical presentation was not consistent with radiation recall dermatitis. Cutaneous Rosai-Dorfman disease is a non-Langerhans cell histiocytosis that may present as either solitary or numerous papules, plaques, or nodules and has been reported to occur after HZI. Biopsy reveals a diffuse dermal histiocytic infiltration with plasma cells and lymphocytes. In contrast to metastatic disease, mitoses and nuclear atypia are rare in cutaneous RosaiDorfman disease.11 Lichen planus is an inflammatory disease of unknown etiology presenting as flat-topped, violaceous, pruritic papules12 that may present in a zosteriform pattern.13
Although it is uncommon, metastatic spread should be considered in patients with known malignancy presenting with zosteriform eruptions.2 Our patient remained on treatment with immunotherapy, as he was unable to undergo additional radiation and had failed multiple other lines of therapy. He died 3 months after presentation.
- Klebanov N, Reddy BY, Husain S, et al. Cutaneous presentation of mesothelioma with a sarcomatoid transformation. Am J Dermatopathol. 2018;40:378-382.
- Patel SC, Dowell JE. Modern management of malignant pleural mesothelioma. Lung Cancer (Auckl). 2016;7:63-72.
- Ward RE, Ali SA, Kuhar M. Epithelioid malignant mesothelioma metastatic to the skin: a case report and review of the literature. J Cutan Pathol. 2017;44:1057-1063.
- Prieto VG, Kenet BJ, Varghese M. Malignant mesothelioma metastatic to the skin, presenting as inflammatory carcinoma. Am J Dermatopathol. 1997;19:261-265.
- Gaudy-Marqueste C, Dales JP, Collet-Villette AM, et al. Cutaneous metastasis of pleural mesothelioma: two cases [in French]. Ann Dermatol Venereol. 2003;130:455-459.
- Chiang A, Salomon N, Gaikwad R, et al. A case of cutaneous metastasis mimicking herpes zoster rash. IDCases. 2018;12:167-168.
- Thomaidou E, Armon G, Klapholz L, et al. Zosteriform cutaneous metastases. Clin Exp Dermatol. 2018;43:734-736.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Carrasco L, Pastor MA, Izquierdo MJ, et al. Drug eruption secondary to acyclovir with recall phenomenon in a dermatome previously affected by herpes zoster. Clin Exp Dermatol. 2002;27:132-134.
- Malviya N, Marzuka A, Maamed-Tayeb M, et al. Cutaneous involvement of pre-existing Rosai-Dorfman disease via post-herpetic isotopic response. J Cutan Pathol. 2016;43:1211-1214.
- Fang S, Chen AJ. Facial cutaneous Rosai-Dorfman disease: a case report and literature review. Exp Ther Med. 2015;9:1389-1392.
- Le Cleach L, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;366:723-732.
- Fink-Puches R, Hofmann-Wellenhof R, Smolle J. Zosteriform lichen planus. Dermatology. 1996;192:375-377.
THE DIAGNOSIS:
Cutaneous Metastatic Mesothelioma
Biopsies of the larger erythematous papules revealed an infiltrate of atypical tumor cells with mitoses (Figure 1) that were immunoreactive for calretinin (Figure 2) and lacked nuclear BRCA1 associated protein-1, BAP1, expression (not shown). The patient’s prior mesothelioma was re-reviewed, and the cutaneous tumor cells were similar to the primary mesothelioma. A diagnosis of cutaneous metastatic mesothelioma (CMM) was made.
Mesothelioma is a rare neoplasm arising from the pleura, pericardium, peritoneum, and tunica vaginalis,1 with an estimated annual incidence of 2500 cases.2 The predominant risk factor for the development of pleural mesothelioma is asbestos exposure, which has been identified in up to 90% of cases. Mesothelioma can give rise to local and less frequently distant hematogenous metastases. Cutaneous involvement of mesothelioma is rare.3 More than 80% of CMM cases are attributed to seeding the skin at procedure sites or by direct infiltration of scars. Distant CMM is rare and typically presents as subcutaneous nodules.4 Few cases of inflammatory CMM have been published,1,4,5 with even fewer mimicking herpes zoster infection (HZI), as seen in our patient.
The most specific stain for mesothelioma is calretinin, which strongly and diffusely stains both the nucleus and cytoplasm. Other markers include Wilms tumor 1, cytokeratin 5/6, thrombomodulin, and HBME-1. Immunohistochemistry to detect the loss of BAP1 staining in the nucleus is important for differentiating between mesothelioma and mesothelial hyperplasia.3
Cutaneous metastases occur in 0.7% to 9% of patients with internal malignant disease. Most commonly, cutaneous metastases present as cutaneous nodules, though other reported inflammatory presentations include erysipeloides, generalized erythematous patches, telangiectasia, and zosteriform distributions.6 Zosteriform distributions are particularly rare and most commonly are due to breast carcinomas or lymphomas. The mechanism of zosteriform metastasis is unknown, but theories include tumoral spread along vessels, invasion of the thoracic perineural sheaths, localized spread of tumor cells from a surgical site, or a Koebner-like reaction at the site of an existing HZI. Regardless of primary tumor type or presentation, cutaneous metastasis is a poor prognostic sign, with survival rates varying based on primary tumor type.7
Other differential diagnoses include herpes zoster granulomatous dermatitis, radiation recall dermatitis, cutaneous Rosai-Dorfman disease, and zosteriform lichen planus, all of which have been reported after HZI.8-10 Herpes zoster granulomatous dermatitis typically presents weeks to years after acute HZI with erythematous to violaceous papules and plaques at the site of the prior HZI. A biopsy reveals interstitial granulomatous dermatitis and multinucleated giant cells.8 Radiation recall dermatitis is a cutaneous inflammatory reaction limited to regions of prior radiation exposure after the administration of a triggering medication. Radiation recall dermatitis can present days to many years after the completion of treatment.9 Although the eruption in our patient was at the site of prior radiation, the pathologic and clinical presentation was not consistent with radiation recall dermatitis. Cutaneous Rosai-Dorfman disease is a non-Langerhans cell histiocytosis that may present as either solitary or numerous papules, plaques, or nodules and has been reported to occur after HZI. Biopsy reveals a diffuse dermal histiocytic infiltration with plasma cells and lymphocytes. In contrast to metastatic disease, mitoses and nuclear atypia are rare in cutaneous RosaiDorfman disease.11 Lichen planus is an inflammatory disease of unknown etiology presenting as flat-topped, violaceous, pruritic papules12 that may present in a zosteriform pattern.13
Although it is uncommon, metastatic spread should be considered in patients with known malignancy presenting with zosteriform eruptions.2 Our patient remained on treatment with immunotherapy, as he was unable to undergo additional radiation and had failed multiple other lines of therapy. He died 3 months after presentation.
THE DIAGNOSIS:
Cutaneous Metastatic Mesothelioma
Biopsies of the larger erythematous papules revealed an infiltrate of atypical tumor cells with mitoses (Figure 1) that were immunoreactive for calretinin (Figure 2) and lacked nuclear BRCA1 associated protein-1, BAP1, expression (not shown). The patient’s prior mesothelioma was re-reviewed, and the cutaneous tumor cells were similar to the primary mesothelioma. A diagnosis of cutaneous metastatic mesothelioma (CMM) was made.
Mesothelioma is a rare neoplasm arising from the pleura, pericardium, peritoneum, and tunica vaginalis,1 with an estimated annual incidence of 2500 cases.2 The predominant risk factor for the development of pleural mesothelioma is asbestos exposure, which has been identified in up to 90% of cases. Mesothelioma can give rise to local and less frequently distant hematogenous metastases. Cutaneous involvement of mesothelioma is rare.3 More than 80% of CMM cases are attributed to seeding the skin at procedure sites or by direct infiltration of scars. Distant CMM is rare and typically presents as subcutaneous nodules.4 Few cases of inflammatory CMM have been published,1,4,5 with even fewer mimicking herpes zoster infection (HZI), as seen in our patient.
The most specific stain for mesothelioma is calretinin, which strongly and diffusely stains both the nucleus and cytoplasm. Other markers include Wilms tumor 1, cytokeratin 5/6, thrombomodulin, and HBME-1. Immunohistochemistry to detect the loss of BAP1 staining in the nucleus is important for differentiating between mesothelioma and mesothelial hyperplasia.3
Cutaneous metastases occur in 0.7% to 9% of patients with internal malignant disease. Most commonly, cutaneous metastases present as cutaneous nodules, though other reported inflammatory presentations include erysipeloides, generalized erythematous patches, telangiectasia, and zosteriform distributions.6 Zosteriform distributions are particularly rare and most commonly are due to breast carcinomas or lymphomas. The mechanism of zosteriform metastasis is unknown, but theories include tumoral spread along vessels, invasion of the thoracic perineural sheaths, localized spread of tumor cells from a surgical site, or a Koebner-like reaction at the site of an existing HZI. Regardless of primary tumor type or presentation, cutaneous metastasis is a poor prognostic sign, with survival rates varying based on primary tumor type.7
Other differential diagnoses include herpes zoster granulomatous dermatitis, radiation recall dermatitis, cutaneous Rosai-Dorfman disease, and zosteriform lichen planus, all of which have been reported after HZI.8-10 Herpes zoster granulomatous dermatitis typically presents weeks to years after acute HZI with erythematous to violaceous papules and plaques at the site of the prior HZI. A biopsy reveals interstitial granulomatous dermatitis and multinucleated giant cells.8 Radiation recall dermatitis is a cutaneous inflammatory reaction limited to regions of prior radiation exposure after the administration of a triggering medication. Radiation recall dermatitis can present days to many years after the completion of treatment.9 Although the eruption in our patient was at the site of prior radiation, the pathologic and clinical presentation was not consistent with radiation recall dermatitis. Cutaneous Rosai-Dorfman disease is a non-Langerhans cell histiocytosis that may present as either solitary or numerous papules, plaques, or nodules and has been reported to occur after HZI. Biopsy reveals a diffuse dermal histiocytic infiltration with plasma cells and lymphocytes. In contrast to metastatic disease, mitoses and nuclear atypia are rare in cutaneous RosaiDorfman disease.11 Lichen planus is an inflammatory disease of unknown etiology presenting as flat-topped, violaceous, pruritic papules12 that may present in a zosteriform pattern.13
Although it is uncommon, metastatic spread should be considered in patients with known malignancy presenting with zosteriform eruptions.2 Our patient remained on treatment with immunotherapy, as he was unable to undergo additional radiation and had failed multiple other lines of therapy. He died 3 months after presentation.
- Klebanov N, Reddy BY, Husain S, et al. Cutaneous presentation of mesothelioma with a sarcomatoid transformation. Am J Dermatopathol. 2018;40:378-382.
- Patel SC, Dowell JE. Modern management of malignant pleural mesothelioma. Lung Cancer (Auckl). 2016;7:63-72.
- Ward RE, Ali SA, Kuhar M. Epithelioid malignant mesothelioma metastatic to the skin: a case report and review of the literature. J Cutan Pathol. 2017;44:1057-1063.
- Prieto VG, Kenet BJ, Varghese M. Malignant mesothelioma metastatic to the skin, presenting as inflammatory carcinoma. Am J Dermatopathol. 1997;19:261-265.
- Gaudy-Marqueste C, Dales JP, Collet-Villette AM, et al. Cutaneous metastasis of pleural mesothelioma: two cases [in French]. Ann Dermatol Venereol. 2003;130:455-459.
- Chiang A, Salomon N, Gaikwad R, et al. A case of cutaneous metastasis mimicking herpes zoster rash. IDCases. 2018;12:167-168.
- Thomaidou E, Armon G, Klapholz L, et al. Zosteriform cutaneous metastases. Clin Exp Dermatol. 2018;43:734-736.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Carrasco L, Pastor MA, Izquierdo MJ, et al. Drug eruption secondary to acyclovir with recall phenomenon in a dermatome previously affected by herpes zoster. Clin Exp Dermatol. 2002;27:132-134.
- Malviya N, Marzuka A, Maamed-Tayeb M, et al. Cutaneous involvement of pre-existing Rosai-Dorfman disease via post-herpetic isotopic response. J Cutan Pathol. 2016;43:1211-1214.
- Fang S, Chen AJ. Facial cutaneous Rosai-Dorfman disease: a case report and literature review. Exp Ther Med. 2015;9:1389-1392.
- Le Cleach L, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;366:723-732.
- Fink-Puches R, Hofmann-Wellenhof R, Smolle J. Zosteriform lichen planus. Dermatology. 1996;192:375-377.
- Klebanov N, Reddy BY, Husain S, et al. Cutaneous presentation of mesothelioma with a sarcomatoid transformation. Am J Dermatopathol. 2018;40:378-382.
- Patel SC, Dowell JE. Modern management of malignant pleural mesothelioma. Lung Cancer (Auckl). 2016;7:63-72.
- Ward RE, Ali SA, Kuhar M. Epithelioid malignant mesothelioma metastatic to the skin: a case report and review of the literature. J Cutan Pathol. 2017;44:1057-1063.
- Prieto VG, Kenet BJ, Varghese M. Malignant mesothelioma metastatic to the skin, presenting as inflammatory carcinoma. Am J Dermatopathol. 1997;19:261-265.
- Gaudy-Marqueste C, Dales JP, Collet-Villette AM, et al. Cutaneous metastasis of pleural mesothelioma: two cases [in French]. Ann Dermatol Venereol. 2003;130:455-459.
- Chiang A, Salomon N, Gaikwad R, et al. A case of cutaneous metastasis mimicking herpes zoster rash. IDCases. 2018;12:167-168.
- Thomaidou E, Armon G, Klapholz L, et al. Zosteriform cutaneous metastases. Clin Exp Dermatol. 2018;43:734-736.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Carrasco L, Pastor MA, Izquierdo MJ, et al. Drug eruption secondary to acyclovir with recall phenomenon in a dermatome previously affected by herpes zoster. Clin Exp Dermatol. 2002;27:132-134.
- Malviya N, Marzuka A, Maamed-Tayeb M, et al. Cutaneous involvement of pre-existing Rosai-Dorfman disease via post-herpetic isotopic response. J Cutan Pathol. 2016;43:1211-1214.
- Fang S, Chen AJ. Facial cutaneous Rosai-Dorfman disease: a case report and literature review. Exp Ther Med. 2015;9:1389-1392.
- Le Cleach L, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;366:723-732.
- Fink-Puches R, Hofmann-Wellenhof R, Smolle J. Zosteriform lichen planus. Dermatology. 1996;192:375-377.
A 50-year-old man presented with erythematous macules and papules with a dermatomal distribution on the left thoracic region with associated pain of 3 weeks’ duration. The lesions persisted after treatment for herpes zoster. His medical history was notable for mesothelioma that was diagnosed 6 years prior and was treated with ipilimumab and nivolumab following multiple lines of chemotherapy and investigational agents, left thoracotomy, extrapleural pneumonectomy, diaphragmatic reconstruction, and left chest radiation. His medical history also included Hodgkin lymphoma diagnosed 36 years prior that was treated with an appendectomy, splenectomy, systemic chemotherapy, and radiation. Three weeks prior to the current presentation, he was treated by oncology with valacyclovir 1 g 3 times daily for 7 days for presumed herpes zoster without improvement. Physical examination revealed the absence of vesicles, as well as firm, 1- to 6-mm, erythematous papules and plaques, including a few outside of the most affected dermatomes.
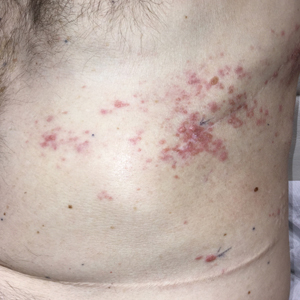
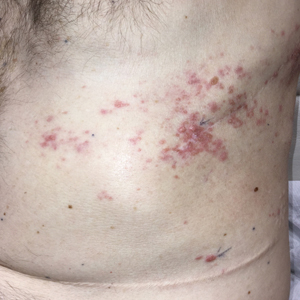
Vegetative Plaques on the Face
THE DIAGNOSIS: Vegetative Majocchi Granuloma
A biopsy and tissue culture showed acute dermal inflammation with granulomatous features and numerous fungal hyphae within the stratum corneum (Figure 1A), which were confirmed on GrocottGomori methenamine-silver staining (Figure 1B). Gram and Fite stains were negative for bacteria. A tissue culture speciated Trichophyton rubrum, which led to a diagnosis of deep dermatophyte infection (Majocchi granuloma) with a highly unusual clinical presentation of vegetative plaques. Predisposing factors included treatment with topical corticosteroids and possibly poor health and nutritional status at baseline. Our patient was treated with fluconazole 200 mg daily for 6 weeks, with near resolution of lesions at 3-week follow-up (Figure 2).
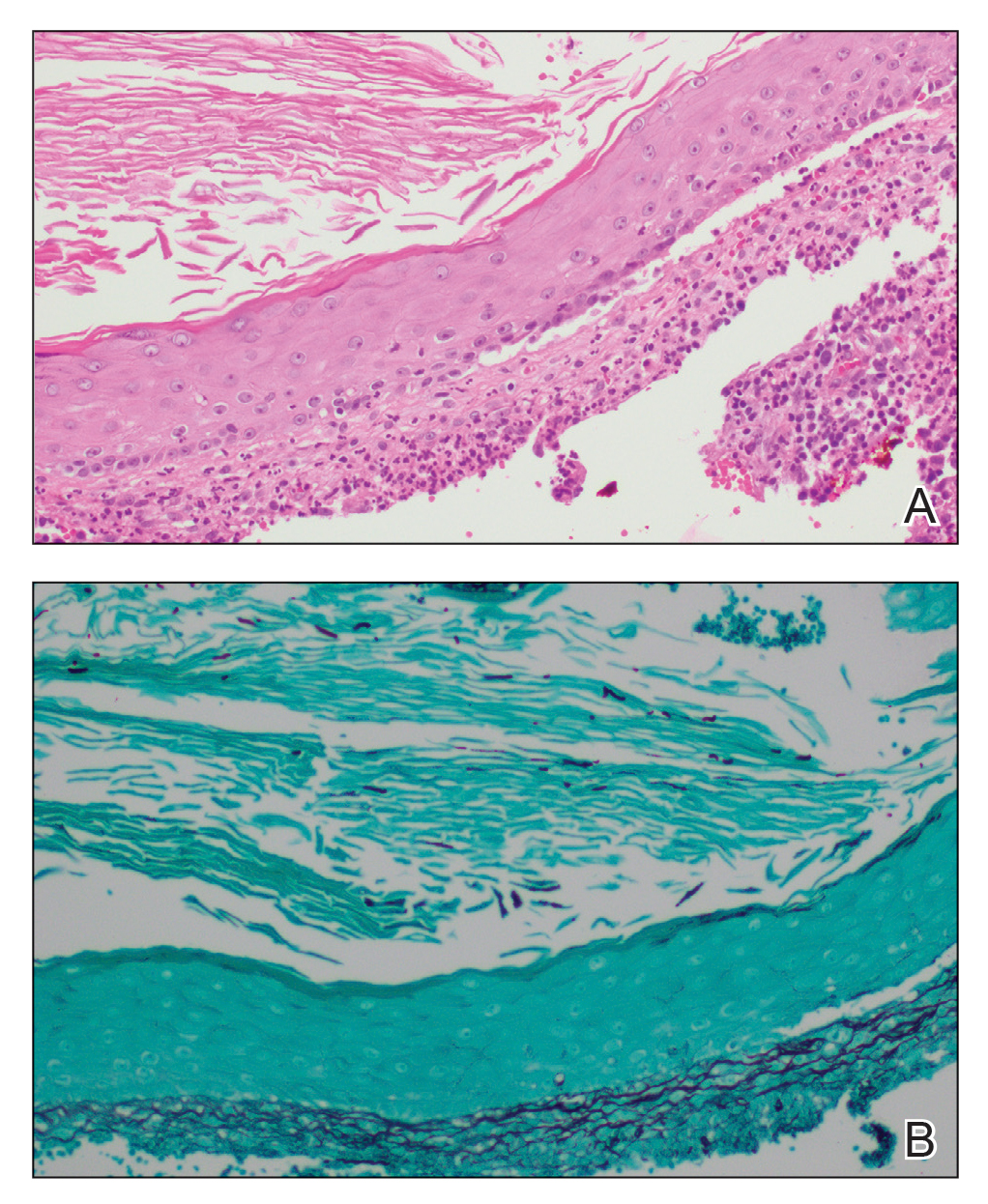
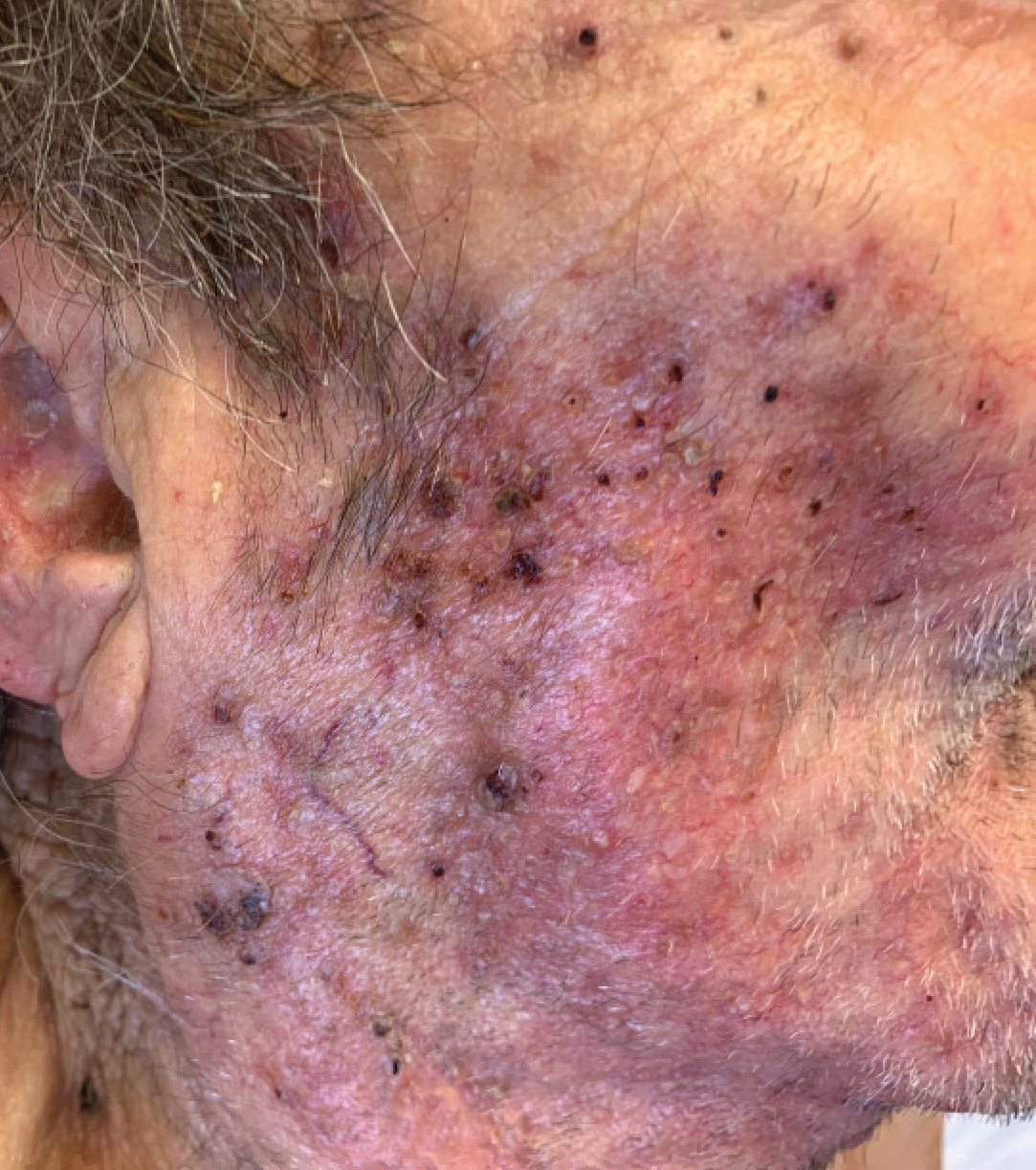
Dermatophytes are a common cause of superficial skin infections. The classic morphology consists of an annular scaly plaque; however, a wide variety of presentations have been observed (eg, verrucous, vesicular, pustular, granulomatous). Therefore, dermatophyte infections often mimic other dermatologic conditions, including atopic dermatitis, rosacea, psoriasis, bacterial abscess, erythema gyratum repens, lupus, granuloma annulare, cutaneous lymphoma, Hailey-Hailey disease, scarring alopecia, and syphilis.1
Notably, when dermatophytes grow downward along hair follicles causing deeper infection, disruption of the follicular wall can lead to an excessive inflammatory response with granulomatous features.2 Risk factors include cutaneous trauma, long-standing infection, immunocompromise, and treatment with topical corticosteroids.3 This disease evolution clinically appears as a nodule or infiltrated plaque, often without scale. The most well-known example is a kerion on the scalp. Elsewhere on the body, lesions often are termed Majocchi granulomas.2
Vegetative plaques, as seen in our patient, are a highly unusual morphology for deep tinea infection. Guanziroli et al4 reported a case of vegetative lesions on the forearm of a 67-year-old immunocompromised man that were successfully treated with a 3-month course of oral terbinafine after Trichophyton verrucosum was isolated. Skorepova et al5 reported a case of pyoderma vegetans triggered by recurrent Trichophyton mentagrophytes on the dorsal hands of a 64-year-old man with immunoglobulin deficiency of unknown etiology. The lesions were successfully treated with a prolonged course of doxycycline, topical triamcinolone, and intravenous immunoglobulin following 2 initial courses of terbinafine.
The differential diagnosis for vegetative lesions includes pemphigus vegetans, a vegetative variant of pyoderma gangrenosum; halogenoderma; and a variety of infections, including dimorphic fungi (histoplasmosis, blastomycosis), blastomycosislike pyoderma (bacterial), and candidiasis.6 These conditions usually can be distinguished based on histopathology. Clinically, pemphigus vegetans presents with pustules and vegetative lesions, as in our patient, but usually is more diffuse and favors the intertriginous areas. Histology likely would reveal foci of acantholysis and eosinophils. Vegetative pyoderma gangrenosum favors the trunk, particularly in sites of surgical trauma. In our patient, no lesions were present near the abdominal surgical sites, and there was no antecedent cribriform ulceration. Halogenoderma was a strong initial consideration given the localization, presence of large pustules, and history of numerous contrast computed tomography studies; however, our patient’s iodine levels were normal. Infectious etiologies including dimorphic fungi and blastomycosislike pyoderma generally are not restricted to the head and neck, and tissue culture helps exclude them. Vegetative lesions may occur in the setting of other infections, and tissue culture may be necessary to differentiate them if histopathology is not suggestive.
Deep dermatophyte infections require treatment with oral antifungals, as topicals do not penetrate adequately into the hair follicles. Exact regimens vary, but generally oral terbinafine or an oral azole (except ketoconazole) is administered for 2 to 6 weeks, with immunocompromise necessitating longer courses.
We present a rare case of vegetative Majocchi granuloma secondary to T rubrum infection. A dermatophyte infection should be included in the differential for vegetative lesions, especially in dense hair-bearing areas such as the beard. Treatment generally is straightforward with oral antifungals.
- Atzori L, Pau M, Aste N, et al. Dermatophyte infections mimicking other skin diseases: a 154-person case survey of tinea atypica in the district of Cagliari (Italy). Int J Dermatol. 2012;51:410-415.
- Ilkit M, Durdu M, Karakas M. Majocchi’s granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
- Jevremovic L, Ilijin I, Kostic K, et al. Pyoderma vegetans—a case report. Serbian J Dermatol Venereol. 2017;9:22-28.
- Guanziroli E, Pavia G, Guttadauro A, et al. Deep dermatophytosis caused by Trichophyton verrucosum in an immunosuppressed patient: successful outcome with terbinafine. Mycopathologia. 2019;184:543-545.
- Skorepová M, Stuchlík D. Chronic pyoderma vegetans triggered by Trichophyton mentagrophytes. Mycoses. 2006;49:143-144.
- Reinholz M, Hermans C, Dietrich A, et al. A case of cutaneous vegetating candidiasis in a patient with keratitis-ichthyosis-deafness syndrome. J Eur Acad Dermatol Venereol. 2016;30:537-539.
THE DIAGNOSIS: Vegetative Majocchi Granuloma
A biopsy and tissue culture showed acute dermal inflammation with granulomatous features and numerous fungal hyphae within the stratum corneum (Figure 1A), which were confirmed on GrocottGomori methenamine-silver staining (Figure 1B). Gram and Fite stains were negative for bacteria. A tissue culture speciated Trichophyton rubrum, which led to a diagnosis of deep dermatophyte infection (Majocchi granuloma) with a highly unusual clinical presentation of vegetative plaques. Predisposing factors included treatment with topical corticosteroids and possibly poor health and nutritional status at baseline. Our patient was treated with fluconazole 200 mg daily for 6 weeks, with near resolution of lesions at 3-week follow-up (Figure 2).
Dermatophytes are a common cause of superficial skin infections. The classic morphology consists of an annular scaly plaque; however, a wide variety of presentations have been observed (eg, verrucous, vesicular, pustular, granulomatous). Therefore, dermatophyte infections often mimic other dermatologic conditions, including atopic dermatitis, rosacea, psoriasis, bacterial abscess, erythema gyratum repens, lupus, granuloma annulare, cutaneous lymphoma, Hailey-Hailey disease, scarring alopecia, and syphilis.1
Notably, when dermatophytes grow downward along hair follicles causing deeper infection, disruption of the follicular wall can lead to an excessive inflammatory response with granulomatous features.2 Risk factors include cutaneous trauma, long-standing infection, immunocompromise, and treatment with topical corticosteroids.3 This disease evolution clinically appears as a nodule or infiltrated plaque, often without scale. The most well-known example is a kerion on the scalp. Elsewhere on the body, lesions often are termed Majocchi granulomas.2
Vegetative plaques, as seen in our patient, are a highly unusual morphology for deep tinea infection. Guanziroli et al4 reported a case of vegetative lesions on the forearm of a 67-year-old immunocompromised man that were successfully treated with a 3-month course of oral terbinafine after Trichophyton verrucosum was isolated. Skorepova et al5 reported a case of pyoderma vegetans triggered by recurrent Trichophyton mentagrophytes on the dorsal hands of a 64-year-old man with immunoglobulin deficiency of unknown etiology. The lesions were successfully treated with a prolonged course of doxycycline, topical triamcinolone, and intravenous immunoglobulin following 2 initial courses of terbinafine.
The differential diagnosis for vegetative lesions includes pemphigus vegetans, a vegetative variant of pyoderma gangrenosum; halogenoderma; and a variety of infections, including dimorphic fungi (histoplasmosis, blastomycosis), blastomycosislike pyoderma (bacterial), and candidiasis.6 These conditions usually can be distinguished based on histopathology. Clinically, pemphigus vegetans presents with pustules and vegetative lesions, as in our patient, but usually is more diffuse and favors the intertriginous areas. Histology likely would reveal foci of acantholysis and eosinophils. Vegetative pyoderma gangrenosum favors the trunk, particularly in sites of surgical trauma. In our patient, no lesions were present near the abdominal surgical sites, and there was no antecedent cribriform ulceration. Halogenoderma was a strong initial consideration given the localization, presence of large pustules, and history of numerous contrast computed tomography studies; however, our patient’s iodine levels were normal. Infectious etiologies including dimorphic fungi and blastomycosislike pyoderma generally are not restricted to the head and neck, and tissue culture helps exclude them. Vegetative lesions may occur in the setting of other infections, and tissue culture may be necessary to differentiate them if histopathology is not suggestive.
Deep dermatophyte infections require treatment with oral antifungals, as topicals do not penetrate adequately into the hair follicles. Exact regimens vary, but generally oral terbinafine or an oral azole (except ketoconazole) is administered for 2 to 6 weeks, with immunocompromise necessitating longer courses.
We present a rare case of vegetative Majocchi granuloma secondary to T rubrum infection. A dermatophyte infection should be included in the differential for vegetative lesions, especially in dense hair-bearing areas such as the beard. Treatment generally is straightforward with oral antifungals.
THE DIAGNOSIS: Vegetative Majocchi Granuloma
A biopsy and tissue culture showed acute dermal inflammation with granulomatous features and numerous fungal hyphae within the stratum corneum (Figure 1A), which were confirmed on GrocottGomori methenamine-silver staining (Figure 1B). Gram and Fite stains were negative for bacteria. A tissue culture speciated Trichophyton rubrum, which led to a diagnosis of deep dermatophyte infection (Majocchi granuloma) with a highly unusual clinical presentation of vegetative plaques. Predisposing factors included treatment with topical corticosteroids and possibly poor health and nutritional status at baseline. Our patient was treated with fluconazole 200 mg daily for 6 weeks, with near resolution of lesions at 3-week follow-up (Figure 2).
Dermatophytes are a common cause of superficial skin infections. The classic morphology consists of an annular scaly plaque; however, a wide variety of presentations have been observed (eg, verrucous, vesicular, pustular, granulomatous). Therefore, dermatophyte infections often mimic other dermatologic conditions, including atopic dermatitis, rosacea, psoriasis, bacterial abscess, erythema gyratum repens, lupus, granuloma annulare, cutaneous lymphoma, Hailey-Hailey disease, scarring alopecia, and syphilis.1
Notably, when dermatophytes grow downward along hair follicles causing deeper infection, disruption of the follicular wall can lead to an excessive inflammatory response with granulomatous features.2 Risk factors include cutaneous trauma, long-standing infection, immunocompromise, and treatment with topical corticosteroids.3 This disease evolution clinically appears as a nodule or infiltrated plaque, often without scale. The most well-known example is a kerion on the scalp. Elsewhere on the body, lesions often are termed Majocchi granulomas.2
Vegetative plaques, as seen in our patient, are a highly unusual morphology for deep tinea infection. Guanziroli et al4 reported a case of vegetative lesions on the forearm of a 67-year-old immunocompromised man that were successfully treated with a 3-month course of oral terbinafine after Trichophyton verrucosum was isolated. Skorepova et al5 reported a case of pyoderma vegetans triggered by recurrent Trichophyton mentagrophytes on the dorsal hands of a 64-year-old man with immunoglobulin deficiency of unknown etiology. The lesions were successfully treated with a prolonged course of doxycycline, topical triamcinolone, and intravenous immunoglobulin following 2 initial courses of terbinafine.
The differential diagnosis for vegetative lesions includes pemphigus vegetans, a vegetative variant of pyoderma gangrenosum; halogenoderma; and a variety of infections, including dimorphic fungi (histoplasmosis, blastomycosis), blastomycosislike pyoderma (bacterial), and candidiasis.6 These conditions usually can be distinguished based on histopathology. Clinically, pemphigus vegetans presents with pustules and vegetative lesions, as in our patient, but usually is more diffuse and favors the intertriginous areas. Histology likely would reveal foci of acantholysis and eosinophils. Vegetative pyoderma gangrenosum favors the trunk, particularly in sites of surgical trauma. In our patient, no lesions were present near the abdominal surgical sites, and there was no antecedent cribriform ulceration. Halogenoderma was a strong initial consideration given the localization, presence of large pustules, and history of numerous contrast computed tomography studies; however, our patient’s iodine levels were normal. Infectious etiologies including dimorphic fungi and blastomycosislike pyoderma generally are not restricted to the head and neck, and tissue culture helps exclude them. Vegetative lesions may occur in the setting of other infections, and tissue culture may be necessary to differentiate them if histopathology is not suggestive.
Deep dermatophyte infections require treatment with oral antifungals, as topicals do not penetrate adequately into the hair follicles. Exact regimens vary, but generally oral terbinafine or an oral azole (except ketoconazole) is administered for 2 to 6 weeks, with immunocompromise necessitating longer courses.
We present a rare case of vegetative Majocchi granuloma secondary to T rubrum infection. A dermatophyte infection should be included in the differential for vegetative lesions, especially in dense hair-bearing areas such as the beard. Treatment generally is straightforward with oral antifungals.
- Atzori L, Pau M, Aste N, et al. Dermatophyte infections mimicking other skin diseases: a 154-person case survey of tinea atypica in the district of Cagliari (Italy). Int J Dermatol. 2012;51:410-415.
- Ilkit M, Durdu M, Karakas M. Majocchi’s granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
- Jevremovic L, Ilijin I, Kostic K, et al. Pyoderma vegetans—a case report. Serbian J Dermatol Venereol. 2017;9:22-28.
- Guanziroli E, Pavia G, Guttadauro A, et al. Deep dermatophytosis caused by Trichophyton verrucosum in an immunosuppressed patient: successful outcome with terbinafine. Mycopathologia. 2019;184:543-545.
- Skorepová M, Stuchlík D. Chronic pyoderma vegetans triggered by Trichophyton mentagrophytes. Mycoses. 2006;49:143-144.
- Reinholz M, Hermans C, Dietrich A, et al. A case of cutaneous vegetating candidiasis in a patient with keratitis-ichthyosis-deafness syndrome. J Eur Acad Dermatol Venereol. 2016;30:537-539.
- Atzori L, Pau M, Aste N, et al. Dermatophyte infections mimicking other skin diseases: a 154-person case survey of tinea atypica in the district of Cagliari (Italy). Int J Dermatol. 2012;51:410-415.
- Ilkit M, Durdu M, Karakas M. Majocchi’s granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
- Jevremovic L, Ilijin I, Kostic K, et al. Pyoderma vegetans—a case report. Serbian J Dermatol Venereol. 2017;9:22-28.
- Guanziroli E, Pavia G, Guttadauro A, et al. Deep dermatophytosis caused by Trichophyton verrucosum in an immunosuppressed patient: successful outcome with terbinafine. Mycopathologia. 2019;184:543-545.
- Skorepová M, Stuchlík D. Chronic pyoderma vegetans triggered by Trichophyton mentagrophytes. Mycoses. 2006;49:143-144.
- Reinholz M, Hermans C, Dietrich A, et al. A case of cutaneous vegetating candidiasis in a patient with keratitis-ichthyosis-deafness syndrome. J Eur Acad Dermatol Venereol. 2016;30:537-539.
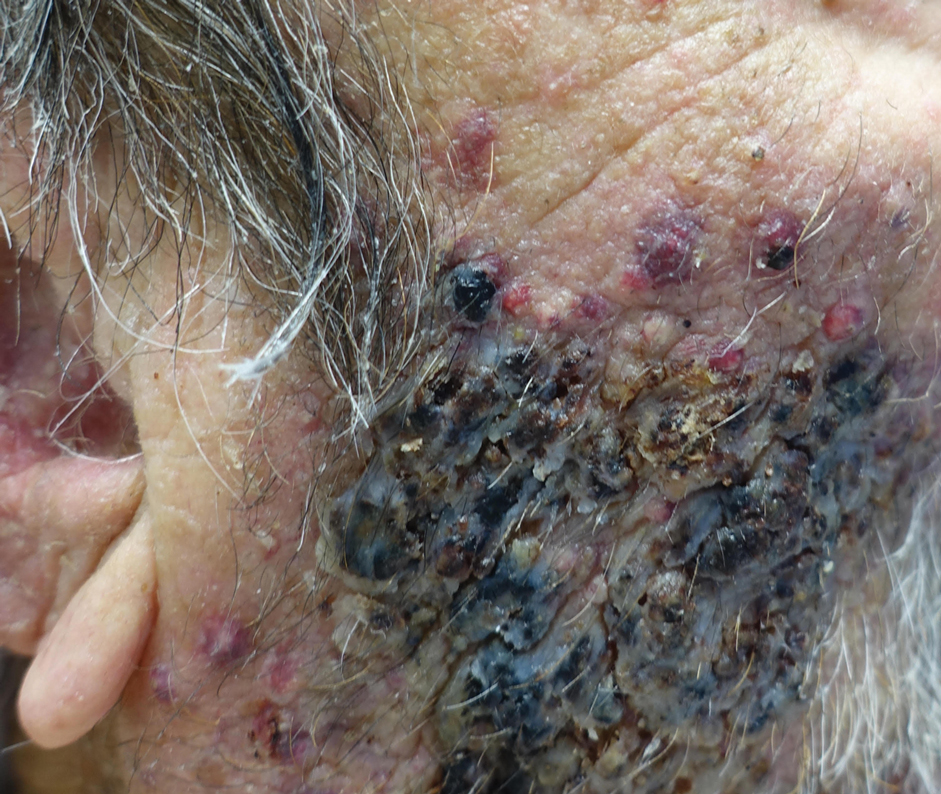
An 86-year-old man was admitted to the hospital for sigmoid colon perforation secondary to ischemic colitis. His medical history consisted of sequelae from atherosclerotic vascular disease. He had no known personal or family history of skin disease. His bowel perforation was surgically repaired, and his clinical status was stabilized, enabling transfer to a transitional care hospital. His course was complicated by delayed healing of the midline abdominal surgical wounds, leading to multiple computed tomography studies with iodinated contrast. One week following arrival at the transitional care hospital, he was noted to have a pustular rash on the face. He was empirically treated with topical steroids, mupirocin, and sulfacetamide. The rash did not improve, and the appearance changed, at which point dermatology was consulted. On evaluation, the patient was afebrile with a normal white blood cell count. Physical examination revealed gray-brown, moist, vegetative plaques on the cheeks with a few large pustules as well as similar-appearing lesions on the neck and upper chest. Attempted removal of a portion of the plaque left an erosion.