User login
Iododerma Following Exposure to Iodine: A Case of Explosive Acneform Eruption Overnight
To the Editor:
Iododerma is a rare dermatologic condition caused by exposure to iodinated contrast media, oral iodine suspensions, or topical povidone-iodine that can manifest as eruptive acneform lesions.1-3
A 27-year-old woman in septic shock presented for worsening facial lesions that showed no improvement on broad-spectrum antibiotics, antifungals, and antivirals. She initially presented to an outside hospital with abdominal pain and underwent computed tomography (CT) with intravenous (IV) iodinated contrast; 24 hours after this imaging study, the family reported the appearance of “explosive acne overnight.” The lesions first appeared as vegetative and acneform ulcerations on the face. A second abdominal CT scan with IV contrast was performed 4 days after the initial scan, given the concern for spontaneous bacterial peritonitis. Hours after the second study, the lesions progressed to involve the buccal mucosae, tongue, mucosal airway, and distal arms and legs. She became progressively disoriented and developed an altered mentation over the course of the following week. Due to progressive facial edema, she required intubation 5 days after the second CT scan.

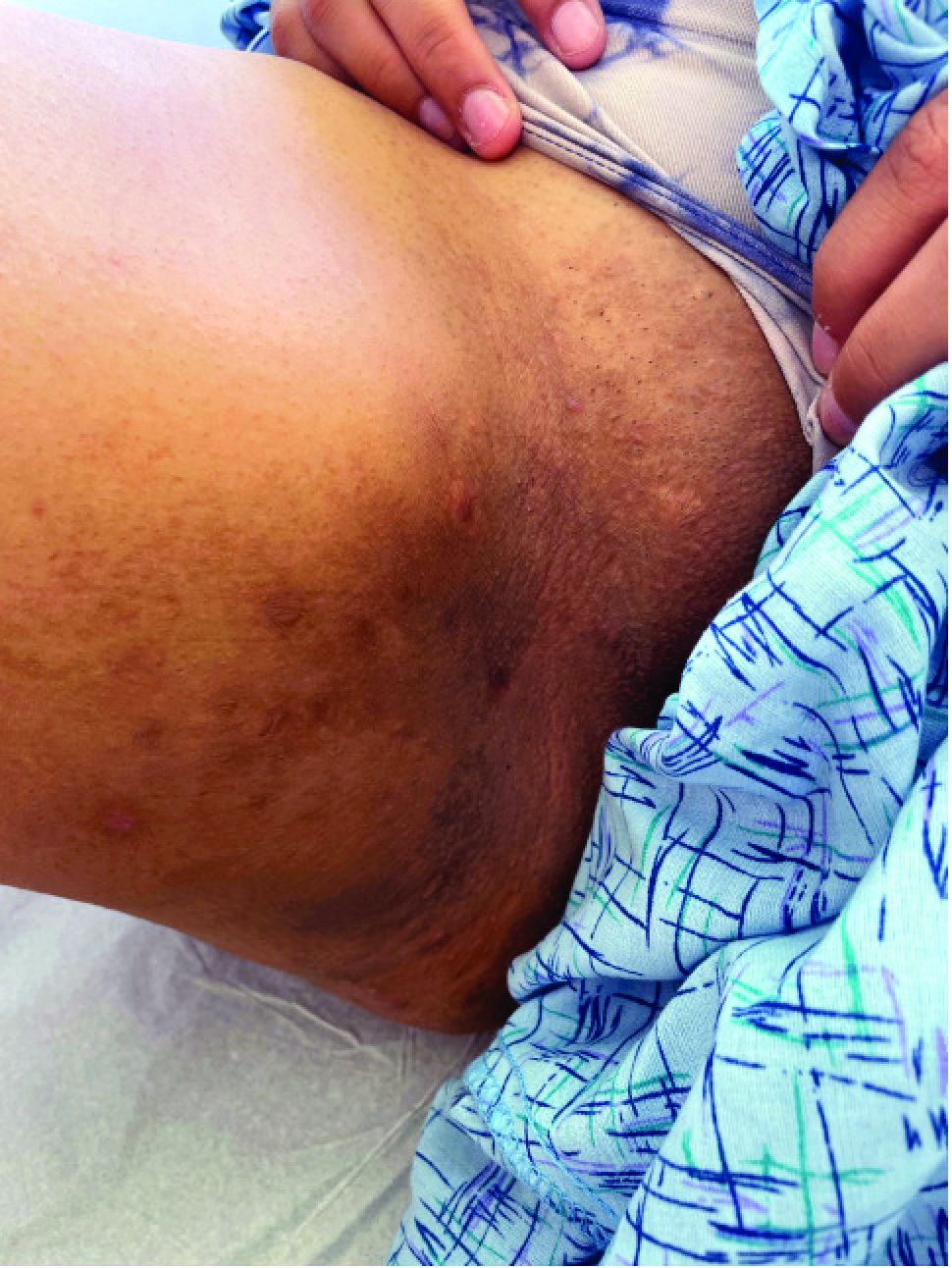
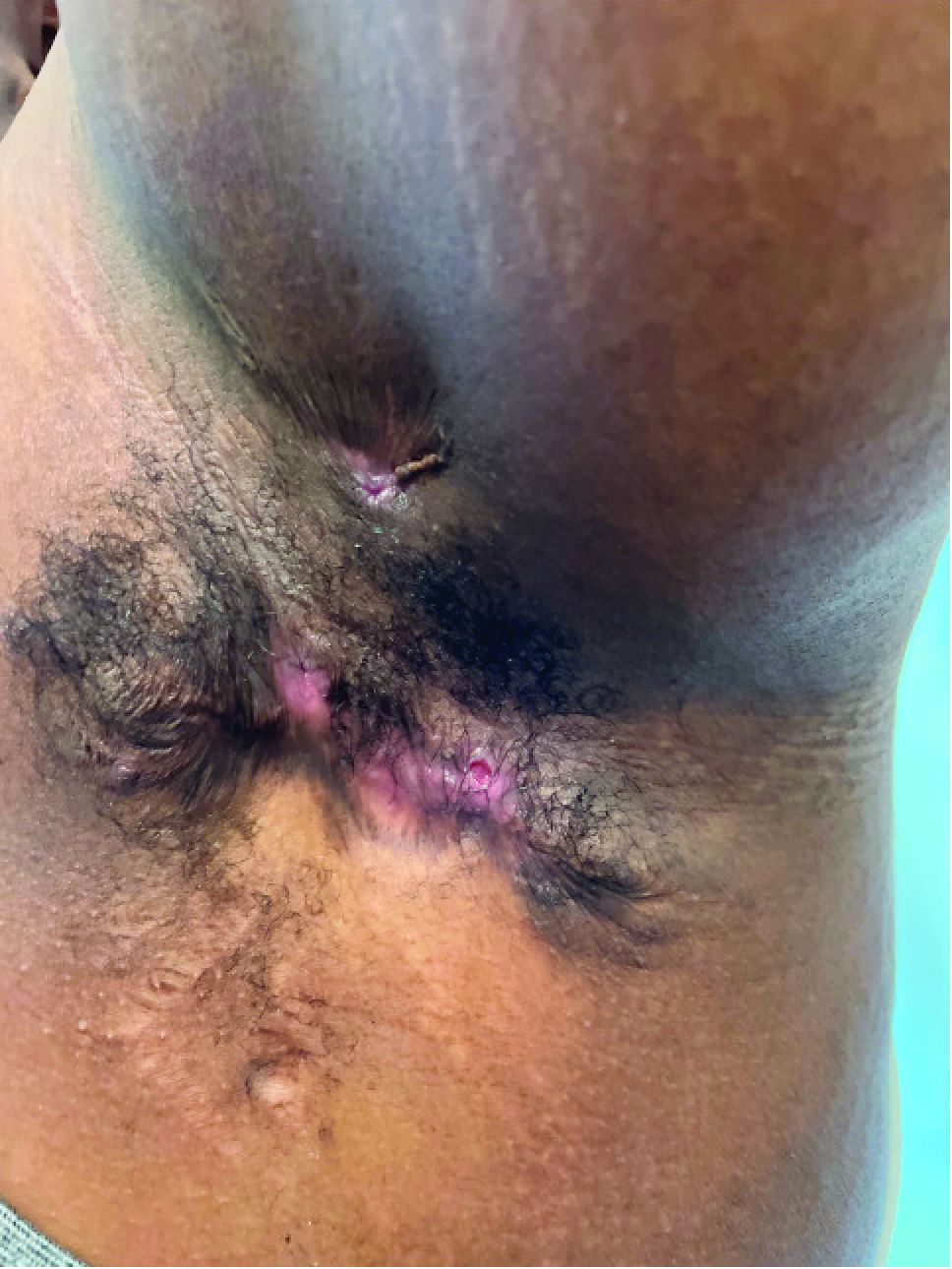
The patient had a medical history of end-stage renal disease secondary to crescenteric glomerulonephritis on peritoneal dialysis. Physical examination revealed numerous beefy-red, heaped-up, weepy, crusted nodules clustered on the face (Figure 1) and a few newer bullous-appearing lesions on the hands and feet. She had similar lesions involving the buccal mucosae and tongue with substantial facial edema. Infectious workup was notable for a positive skin culture growing methicillin-susceptible Staphylococcus aureus. All blood and tissue cultures as well as serologies for fungal and viral etiologies were negative. A tissue biopsy revealed necrosis with a neutrophilic infiltrate with mixed cell inflammation (Figure 2), and direct immunofluorescence was negative.

The patient initially was thought to be septic due to viral or bacterial infection. She was transferred from an outside hospital 7 days after the initial appearance of the acneform lesions, having already received IV contrast on 2 occasions within the first 48 hours of illness. Infectious disease was consulted and initiated broad-spectrum antiviral, antimicrobial, and antifungal therapy with acyclovir, linezolid, meropenem, and later micafungin without improvement. The diagnosis of iododerma ultimately was established based on the patient’s elevated urinary iodine levels with preceding iodine exposure in the context of renal failure. The preferential involvement of sebaceous areas and pathology findings were supportive of this diagnosis. Aggressive supportive measures including respiratory support, IV fluids, and dialysis were initiated. Topical iodine solutions, iodine-containing medications, and additional contrast subsequently were avoided. Despite these supportive measures, the patient died within 48 hours of admission from acute respiratory failure. Her autopsy attributed “septic complications of multifocal ulcerative cutaneous disease” as the anatomic cause of death.
Iododerma is an extremely rare neutrophilic dermatosis. The proposed mechanism of action involves a cell-mediated hypersensitivity reaction to iodine with induction of neutrophil degranulation.2 There have been documented cases with exposure to oral potassium iodide supplements, amiodarone, topical povidone-iodine, and IV iodinated contrast material.1-3 Iododerma typically presents 1 to 3 days after exposure to iodine. The most common source is IV radiocontrast. Diagnosis is based on the clinical presentation including acneform to vegetative nodular or bullous eruptions involving sebaceous areas in the context of recent iodine exposure. Elevated urinary iodine levels and histologic findings of neutrophilic infiltrate of the dermis support the diagnosis.3,4
Although there have been reported cases of iododerma in patients with normal renal function, patients with renal failure are much more susceptible due to the decreased clearance of iodine.5 The plasma half-life of radiocontrast is 23 hours in patients with end-stage renal disease vs 2 hours in patients with normal kidney function.3 Dosage adjustments for renal impairment have not been well studied, and no specific guidelines exist for the prevention of iododerma in patients with renal failure.
The first step in treating iododerma is to remove the offending iodine-containing agent. In most cases, cutaneous lesions resolve in 4 to 6 weeks after discontinuation of the source of iodine; however, there have been reported fatalities in the literature secondary to pulmonary edema in patients with iododerma.6,7 Despite the rarity and diagnostically challenging nature of iododerma, early recognition of this disease is crucial. Although our patient showed symptoms of iododerma after 1 dose of radiocontrast, she was not diagnosed at that time and received a second imaging study with contrast less than 48 hours later. These 2 consecutive exposures to iodine as well as the delayed diagnosis unfortunately resulted in rapid clinical deterioration.
The mainstay of therapy for iododerma includes avoidance of iodine-containing materials as soon as the diagnosis is suspected as well as supportive care. Patients have been successfully treated with systemic corticosteroids, with the addition of cyclosporine and hemodialysis in severe cases.3 Patients with a history of iododerma are advised to avoid iodine in their diet, in topical preparations, and in future imaging studies.8
- Aliagaoglu C, Turan H, Uslu E, et al. Iododerma following topical povidone-iodine application. Cutan Ocul Toxicol. 2013;32:339-340.
- Torkamani, N, Sinclair R. Iododerma in pregnancy secondary to iodinated multivitamins. Australas J Dermatol. 2015;56:235-236.
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast material. Br J Dermatol. 2014;170:1377-1379.
- Stavert R, Bunick CG, Modi B, et al. Vegetative plaques and hemorrhagic pustules. JAMA Dermatol. 2013;149:1231-1232.
- Rothman LR, Levender MM, Scharf MD, et al. Iododerma following serial computed tomography scans in a lung cancer patient. J Drugs Dermatol. 2013;12:574-576.
- Miranda-Romero A, Sánchez-Sambucety P, Gómez JE, et al. Vegetating iododerma with fatal outcome. Dermatology. 1999;198:295-297.
- Vailant L, Pengloan J, Blanchier D, et al. Iododerma and acute respiratory distress with leucocytoclastic vasculitis following the intravenous injection of contrast medium. Clin Exp Dermatol. 1990;15:232-233.
- Massé M, Flanaga V, Zhou LH. Use of topical povidone iodine resulting in an iododerma-like eruption. J Dermatol. 2008;35:744-747.
To the Editor:
Iododerma is a rare dermatologic condition caused by exposure to iodinated contrast media, oral iodine suspensions, or topical povidone-iodine that can manifest as eruptive acneform lesions.1-3
A 27-year-old woman in septic shock presented for worsening facial lesions that showed no improvement on broad-spectrum antibiotics, antifungals, and antivirals. She initially presented to an outside hospital with abdominal pain and underwent computed tomography (CT) with intravenous (IV) iodinated contrast; 24 hours after this imaging study, the family reported the appearance of “explosive acne overnight.” The lesions first appeared as vegetative and acneform ulcerations on the face. A second abdominal CT scan with IV contrast was performed 4 days after the initial scan, given the concern for spontaneous bacterial peritonitis. Hours after the second study, the lesions progressed to involve the buccal mucosae, tongue, mucosal airway, and distal arms and legs. She became progressively disoriented and developed an altered mentation over the course of the following week. Due to progressive facial edema, she required intubation 5 days after the second CT scan.
The patient had a medical history of end-stage renal disease secondary to crescenteric glomerulonephritis on peritoneal dialysis. Physical examination revealed numerous beefy-red, heaped-up, weepy, crusted nodules clustered on the face (Figure 1) and a few newer bullous-appearing lesions on the hands and feet. She had similar lesions involving the buccal mucosae and tongue with substantial facial edema. Infectious workup was notable for a positive skin culture growing methicillin-susceptible Staphylococcus aureus. All blood and tissue cultures as well as serologies for fungal and viral etiologies were negative. A tissue biopsy revealed necrosis with a neutrophilic infiltrate with mixed cell inflammation (Figure 2), and direct immunofluorescence was negative.
The patient initially was thought to be septic due to viral or bacterial infection. She was transferred from an outside hospital 7 days after the initial appearance of the acneform lesions, having already received IV contrast on 2 occasions within the first 48 hours of illness. Infectious disease was consulted and initiated broad-spectrum antiviral, antimicrobial, and antifungal therapy with acyclovir, linezolid, meropenem, and later micafungin without improvement. The diagnosis of iododerma ultimately was established based on the patient’s elevated urinary iodine levels with preceding iodine exposure in the context of renal failure. The preferential involvement of sebaceous areas and pathology findings were supportive of this diagnosis. Aggressive supportive measures including respiratory support, IV fluids, and dialysis were initiated. Topical iodine solutions, iodine-containing medications, and additional contrast subsequently were avoided. Despite these supportive measures, the patient died within 48 hours of admission from acute respiratory failure. Her autopsy attributed “septic complications of multifocal ulcerative cutaneous disease” as the anatomic cause of death.
Iododerma is an extremely rare neutrophilic dermatosis. The proposed mechanism of action involves a cell-mediated hypersensitivity reaction to iodine with induction of neutrophil degranulation.2 There have been documented cases with exposure to oral potassium iodide supplements, amiodarone, topical povidone-iodine, and IV iodinated contrast material.1-3 Iododerma typically presents 1 to 3 days after exposure to iodine. The most common source is IV radiocontrast. Diagnosis is based on the clinical presentation including acneform to vegetative nodular or bullous eruptions involving sebaceous areas in the context of recent iodine exposure. Elevated urinary iodine levels and histologic findings of neutrophilic infiltrate of the dermis support the diagnosis.3,4
Although there have been reported cases of iododerma in patients with normal renal function, patients with renal failure are much more susceptible due to the decreased clearance of iodine.5 The plasma half-life of radiocontrast is 23 hours in patients with end-stage renal disease vs 2 hours in patients with normal kidney function.3 Dosage adjustments for renal impairment have not been well studied, and no specific guidelines exist for the prevention of iododerma in patients with renal failure.
The first step in treating iododerma is to remove the offending iodine-containing agent. In most cases, cutaneous lesions resolve in 4 to 6 weeks after discontinuation of the source of iodine; however, there have been reported fatalities in the literature secondary to pulmonary edema in patients with iododerma.6,7 Despite the rarity and diagnostically challenging nature of iododerma, early recognition of this disease is crucial. Although our patient showed symptoms of iododerma after 1 dose of radiocontrast, she was not diagnosed at that time and received a second imaging study with contrast less than 48 hours later. These 2 consecutive exposures to iodine as well as the delayed diagnosis unfortunately resulted in rapid clinical deterioration.
The mainstay of therapy for iododerma includes avoidance of iodine-containing materials as soon as the diagnosis is suspected as well as supportive care. Patients have been successfully treated with systemic corticosteroids, with the addition of cyclosporine and hemodialysis in severe cases.3 Patients with a history of iododerma are advised to avoid iodine in their diet, in topical preparations, and in future imaging studies.8
To the Editor:
Iododerma is a rare dermatologic condition caused by exposure to iodinated contrast media, oral iodine suspensions, or topical povidone-iodine that can manifest as eruptive acneform lesions.1-3
A 27-year-old woman in septic shock presented for worsening facial lesions that showed no improvement on broad-spectrum antibiotics, antifungals, and antivirals. She initially presented to an outside hospital with abdominal pain and underwent computed tomography (CT) with intravenous (IV) iodinated contrast; 24 hours after this imaging study, the family reported the appearance of “explosive acne overnight.” The lesions first appeared as vegetative and acneform ulcerations on the face. A second abdominal CT scan with IV contrast was performed 4 days after the initial scan, given the concern for spontaneous bacterial peritonitis. Hours after the second study, the lesions progressed to involve the buccal mucosae, tongue, mucosal airway, and distal arms and legs. She became progressively disoriented and developed an altered mentation over the course of the following week. Due to progressive facial edema, she required intubation 5 days after the second CT scan.
The patient had a medical history of end-stage renal disease secondary to crescenteric glomerulonephritis on peritoneal dialysis. Physical examination revealed numerous beefy-red, heaped-up, weepy, crusted nodules clustered on the face (Figure 1) and a few newer bullous-appearing lesions on the hands and feet. She had similar lesions involving the buccal mucosae and tongue with substantial facial edema. Infectious workup was notable for a positive skin culture growing methicillin-susceptible Staphylococcus aureus. All blood and tissue cultures as well as serologies for fungal and viral etiologies were negative. A tissue biopsy revealed necrosis with a neutrophilic infiltrate with mixed cell inflammation (Figure 2), and direct immunofluorescence was negative.
The patient initially was thought to be septic due to viral or bacterial infection. She was transferred from an outside hospital 7 days after the initial appearance of the acneform lesions, having already received IV contrast on 2 occasions within the first 48 hours of illness. Infectious disease was consulted and initiated broad-spectrum antiviral, antimicrobial, and antifungal therapy with acyclovir, linezolid, meropenem, and later micafungin without improvement. The diagnosis of iododerma ultimately was established based on the patient’s elevated urinary iodine levels with preceding iodine exposure in the context of renal failure. The preferential involvement of sebaceous areas and pathology findings were supportive of this diagnosis. Aggressive supportive measures including respiratory support, IV fluids, and dialysis were initiated. Topical iodine solutions, iodine-containing medications, and additional contrast subsequently were avoided. Despite these supportive measures, the patient died within 48 hours of admission from acute respiratory failure. Her autopsy attributed “septic complications of multifocal ulcerative cutaneous disease” as the anatomic cause of death.
Iododerma is an extremely rare neutrophilic dermatosis. The proposed mechanism of action involves a cell-mediated hypersensitivity reaction to iodine with induction of neutrophil degranulation.2 There have been documented cases with exposure to oral potassium iodide supplements, amiodarone, topical povidone-iodine, and IV iodinated contrast material.1-3 Iododerma typically presents 1 to 3 days after exposure to iodine. The most common source is IV radiocontrast. Diagnosis is based on the clinical presentation including acneform to vegetative nodular or bullous eruptions involving sebaceous areas in the context of recent iodine exposure. Elevated urinary iodine levels and histologic findings of neutrophilic infiltrate of the dermis support the diagnosis.3,4
Although there have been reported cases of iododerma in patients with normal renal function, patients with renal failure are much more susceptible due to the decreased clearance of iodine.5 The plasma half-life of radiocontrast is 23 hours in patients with end-stage renal disease vs 2 hours in patients with normal kidney function.3 Dosage adjustments for renal impairment have not been well studied, and no specific guidelines exist for the prevention of iododerma in patients with renal failure.
The first step in treating iododerma is to remove the offending iodine-containing agent. In most cases, cutaneous lesions resolve in 4 to 6 weeks after discontinuation of the source of iodine; however, there have been reported fatalities in the literature secondary to pulmonary edema in patients with iododerma.6,7 Despite the rarity and diagnostically challenging nature of iododerma, early recognition of this disease is crucial. Although our patient showed symptoms of iododerma after 1 dose of radiocontrast, she was not diagnosed at that time and received a second imaging study with contrast less than 48 hours later. These 2 consecutive exposures to iodine as well as the delayed diagnosis unfortunately resulted in rapid clinical deterioration.
The mainstay of therapy for iododerma includes avoidance of iodine-containing materials as soon as the diagnosis is suspected as well as supportive care. Patients have been successfully treated with systemic corticosteroids, with the addition of cyclosporine and hemodialysis in severe cases.3 Patients with a history of iododerma are advised to avoid iodine in their diet, in topical preparations, and in future imaging studies.8
- Aliagaoglu C, Turan H, Uslu E, et al. Iododerma following topical povidone-iodine application. Cutan Ocul Toxicol. 2013;32:339-340.
- Torkamani, N, Sinclair R. Iododerma in pregnancy secondary to iodinated multivitamins. Australas J Dermatol. 2015;56:235-236.
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast material. Br J Dermatol. 2014;170:1377-1379.
- Stavert R, Bunick CG, Modi B, et al. Vegetative plaques and hemorrhagic pustules. JAMA Dermatol. 2013;149:1231-1232.
- Rothman LR, Levender MM, Scharf MD, et al. Iododerma following serial computed tomography scans in a lung cancer patient. J Drugs Dermatol. 2013;12:574-576.
- Miranda-Romero A, Sánchez-Sambucety P, Gómez JE, et al. Vegetating iododerma with fatal outcome. Dermatology. 1999;198:295-297.
- Vailant L, Pengloan J, Blanchier D, et al. Iododerma and acute respiratory distress with leucocytoclastic vasculitis following the intravenous injection of contrast medium. Clin Exp Dermatol. 1990;15:232-233.
- Massé M, Flanaga V, Zhou LH. Use of topical povidone iodine resulting in an iododerma-like eruption. J Dermatol. 2008;35:744-747.
- Aliagaoglu C, Turan H, Uslu E, et al. Iododerma following topical povidone-iodine application. Cutan Ocul Toxicol. 2013;32:339-340.
- Torkamani, N, Sinclair R. Iododerma in pregnancy secondary to iodinated multivitamins. Australas J Dermatol. 2015;56:235-236.
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast material. Br J Dermatol. 2014;170:1377-1379.
- Stavert R, Bunick CG, Modi B, et al. Vegetative plaques and hemorrhagic pustules. JAMA Dermatol. 2013;149:1231-1232.
- Rothman LR, Levender MM, Scharf MD, et al. Iododerma following serial computed tomography scans in a lung cancer patient. J Drugs Dermatol. 2013;12:574-576.
- Miranda-Romero A, Sánchez-Sambucety P, Gómez JE, et al. Vegetating iododerma with fatal outcome. Dermatology. 1999;198:295-297.
- Vailant L, Pengloan J, Blanchier D, et al. Iododerma and acute respiratory distress with leucocytoclastic vasculitis following the intravenous injection of contrast medium. Clin Exp Dermatol. 1990;15:232-233.
- Massé M, Flanaga V, Zhou LH. Use of topical povidone iodine resulting in an iododerma-like eruption. J Dermatol. 2008;35:744-747.
Practice Points
- Iododerma should be considered for patients who develop rapidly progressive, vegetative lesions, especially in those with renal failure. A thorough history should be obtained in these cases, focusing on medications and recent studies involving iodinated contrast.
- The most important first step in treating iododerma is to remove the iodine-containing agent to avoid continued exposure.
- Therapies for iododerma include supportive care, cyclosporine, systemic corticosteroids, and hemodialysis in severe cases.

Patch Testing on Dupilumab: Reliable or Not?
In patients with persistent atopic dermatitis (AD) who are taking dupilumab, is there benefit of patch testing to determine if allergic contact dermatitis (ACD) also is contributing to their disease? Results of patch testing are likely be influenced by the immunomodulatory effects of dupilumab. Similar to the recommendation for patients to refrain from using topical or systemic corticosteroids for 1 week or more prior to patch testing to eliminate false negatives, we reviewed the literature to create practice guidelines for dermatologists regarding patch testing while a patient is taking dupilumab.
Pathophysiology and Pathomechanism
Dupilumab functions through the blockade of T helper 2 (TH2) cells; ACD is propagated through the T helper 1 (TH1) cellular pathway. However, patients with ACD that is unresponsive to allergen avoidance and traditional therapies, such as topical and oral corticosteroids, have responded to dupilumab. The more common reports of this responsiveness are with fragrances; multiple case series described patients with ACD to fragrance mix I1 and balsam of Peru1,2 who improved on dupilumab when other treatments failed. There also are reports of response when ACD was secondary to nickel,2,3p-phenylenediamine,1 Compositae,4 and non–formaldehyde-releasing preservatives (non-FRPs).5 Therefore, not all ACD is propagated through the TH1 cellular pathway.
As noted in these cases, ACD can be a response to an allergen whose pathogenesis involves the TH2 pathway or when patient characteristics favor a TH2 response. It has been suggested that AD patients are more susceptible to TH2-mediated contact sensitization to less-potent allergens, such as fragrances.6
Patch Test Results
Positive patch test results for allergens have been reported while patients are on dupilumab therapy, including a few studies in which results prior to starting dupilumab were compared with those while patients were on dupilumab therapy. In a retrospective chart review of 48 patients on dupilumab for AD with persistent disease, 23 patients were patch tested before and during dupilumab therapy. In these patients, the majority of contact allergies were persistent and only 10% (13/125) of patch test–positive results resolved on dupilumab therapy.7 Contact allergies that resolved included those to emulsifiers (propylene glycol, Amerchol L101 [lanolin-containing products found in cosmetics and other goods], dimethylaminopropylamine), fragrances (fragrance mix I, balsam of Peru), sunscreens (sulisobenzone, phenylbenzimidazole-5-sulfonic acid), and metals (vanadium chloride, phenylmercuric acetate).7 The following results observed in individual cases demonstrated conflicting findings: persistence of allergy to non-FRPs (methylisothiazolinone [MI]) but resolution of allergy to formaldehyde8; persistence of allergy to corticosteroids (budesonide and alclometasone)9; persistence of allergy to an antibiotic (neomycin sulfate) but resolution of allergies to a different antibiotic (bacitracin), glues (ethyl acrylate), bleach, and glutaraldehyde9; persistence of nickel allergy but resolution of allergies to fragrances (cinnamic aldehyde, balsam of Peru) and non-FRPs (methylchloroisothiazolinone or MI)10; and persistence of allergies to non-FRPs (MI) and FRPs (bronopol) but resolution of allergies to nickel, fragrances (hydroperoxides of linalool), and Compositae.11 Additional case reports of positive patch test results while on dupilumab but with no pretreatment results for comparison include allergies to rubber additives,12-14 nickel,14 textile dyes,14 cosmetic and hair care additives,12,14,15 corticosteroids,15 FRPs,15 fragrances,15,16 emulsifiers,16 and non-FRPs.17
An evident theme in the dupilumab patch-testing literature has been that results are variable and case specific: a given patient with ACD to an allergen will respond to dupilumab treatment and have subsequent negative patch testing, while another patient will not respond to dupilumab treatment and have persistent positive patch testing. This is likely because, in certain individuals, the allergen-immune system combination shifts ACD pathogenesis from a purely TH1 response to at least a partial TH2 response, thus allowing for benefit from dupilumab therapy. T helper 1 cell–mediated ACD should not be affected by dupilumab; therefore, reliable results can be elucidated from patch testing despite the drug.
Final Thoughts
We propose that AD patients with residual disease after taking dupilumab undergo patch testing. Positive results indicate allergens that are not inhibited by the drug. Patients will need to follow strict allergen avoidance to resolve this component of their disease; failure to improve might suggest the result was a nonrelevant positive.
If patch testing is negative, an alternative cause for residual disease must be sought. We do not recommend stopping dupilumab prior to patch testing to avoid a disease flare from AD or possible TH2-mediated ACD.
- Chipalkatti N, Lee N, Zancanaro P, et al. Dupilumab as a treatment for allergic contact dermatitis. Dermatitis. 2018;29:347-348. doi:10.1097/DER.0000000000000414
- Jacob SE, Sung CT, Machler BC. Dupilumab for systemic allergy syndrome with dermatitis. Dermatitis. 2019;30:164-167. doi:10.1097/DER.0000000000000446
- Joshi SR, Khan DA. Effective use of dupilumab in managing systemic allergic contact dermatitis. Dermatitis. 2018;29:282-284. doi:10.1097/DER.0000000000000409
- Ruge IF, Skov L, Zachariae C, et al. Dupilumab treatment in two patients with severe allergic contact dermatitis caused by sesquiterpene lactones. Contact Dermatitis. 2020:83;137-139. doi:10.1111/cod.13545
- Goldminz AM, Scheinman PL. A case series of dupilumab-treated allergic contact dermatitis patients. Dermatol Ther. 2018;31:e12701. doi:10.1111/dth.12701
- Kohli N, Nedorost S. Inflamed skin predisposes to sensitization to less potent allergens. J Am Acad Dermatol. 2016;75:312-317. doi:10.1016/j.jaad.2016.03.010
- Raffi J, Suresh R, Botto N, et al. The impact of dupilumab on patch testing and the prevalence of comorbid allergic contact dermatitis in recalcitrant atopic dermatitis: a retrospective chart review. J Am Acad Dermatol. 2020;82:132-138. doi:10.1016/j.jaad.2019.09.028
- Puza CJ, Atwater AR. Positive patch test reaction in a patient taking dupilumab. Dermatitis. 2018;29:89. doi:10.1097/DER.0000000000000346
- Suresh R, Murase JE. The role of expanded series patch testing in identifying causality of residual facial dermatitis following initiation of dupilumab therapy. JAAD Case Rep. 2018;4:899-904. doi:10.1016/j.jdcr.2018.08.027
- Stout M, Silverberg JI. Variable impact of dupilumab on patch testing results and allergic contact dermatitis in adults with atopic dermatitis. J Am Acad Dermatol. 2019;81:157-162. doi:10.1016/j.jaad.2019.03.020
- Raffi J, Botto N. Patch testing and allergen-specific inhibition in a patient taking dupilumab. JAMA Dermatol. 2019;155:120-121. doi:10.1001/jamadermatol.2018.4098
- Hoot JW, Douglas JD, Falo LD Jr. Patch testing in a patient on dupilumab. Dermatitis. 2018;29:164. doi:10.1097/DER.0000000000000357
- Crepy M-N, Nosbaum A, Bensefa-Colas L. Blocking type 2 inflammation by dupilumab does not control classic (type 1-driven) allergic contact dermatitis in chronic hand eczema. Contact Dermatitis. 2019;81:145-147. doi:10.1111/cod.13266
- Raffi J, Chen R, Botto N. Wide dye reactors. JAAD Case Rep. 2019;5:877-879. doi:10.1016/j.jdcr.2019.08.005
- Koblinski JE, Hamann D. Mixed occupational and iatrogenic allergic contact dermatitis in a hairdresser. Occup Med (Lond). 2020;70:523-526. doi:10.1093/occmed/kqaa152
- Raffi J, Suresh R, Fishman H, et al. Investigating the role of allergic contact dermatitis in residual ocular surface disease on dupilumab (ROSDD). Int J Womens Dermatol. 2019;5:308-313. doi:10.1016/j.ijwd.2019.10.001
- Zhu GA, Chen JK, Chiou A, et al. Repeat patch testing in a patient with allergic contact dermatitis improved on dupilumab. JAAD Case Rep. 2019;5:336-338. doi:10.1016/j.jdcr.2019.01.023
In patients with persistent atopic dermatitis (AD) who are taking dupilumab, is there benefit of patch testing to determine if allergic contact dermatitis (ACD) also is contributing to their disease? Results of patch testing are likely be influenced by the immunomodulatory effects of dupilumab. Similar to the recommendation for patients to refrain from using topical or systemic corticosteroids for 1 week or more prior to patch testing to eliminate false negatives, we reviewed the literature to create practice guidelines for dermatologists regarding patch testing while a patient is taking dupilumab.
Pathophysiology and Pathomechanism
Dupilumab functions through the blockade of T helper 2 (TH2) cells; ACD is propagated through the T helper 1 (TH1) cellular pathway. However, patients with ACD that is unresponsive to allergen avoidance and traditional therapies, such as topical and oral corticosteroids, have responded to dupilumab. The more common reports of this responsiveness are with fragrances; multiple case series described patients with ACD to fragrance mix I1 and balsam of Peru1,2 who improved on dupilumab when other treatments failed. There also are reports of response when ACD was secondary to nickel,2,3p-phenylenediamine,1 Compositae,4 and non–formaldehyde-releasing preservatives (non-FRPs).5 Therefore, not all ACD is propagated through the TH1 cellular pathway.
As noted in these cases, ACD can be a response to an allergen whose pathogenesis involves the TH2 pathway or when patient characteristics favor a TH2 response. It has been suggested that AD patients are more susceptible to TH2-mediated contact sensitization to less-potent allergens, such as fragrances.6
Patch Test Results
Positive patch test results for allergens have been reported while patients are on dupilumab therapy, including a few studies in which results prior to starting dupilumab were compared with those while patients were on dupilumab therapy. In a retrospective chart review of 48 patients on dupilumab for AD with persistent disease, 23 patients were patch tested before and during dupilumab therapy. In these patients, the majority of contact allergies were persistent and only 10% (13/125) of patch test–positive results resolved on dupilumab therapy.7 Contact allergies that resolved included those to emulsifiers (propylene glycol, Amerchol L101 [lanolin-containing products found in cosmetics and other goods], dimethylaminopropylamine), fragrances (fragrance mix I, balsam of Peru), sunscreens (sulisobenzone, phenylbenzimidazole-5-sulfonic acid), and metals (vanadium chloride, phenylmercuric acetate).7 The following results observed in individual cases demonstrated conflicting findings: persistence of allergy to non-FRPs (methylisothiazolinone [MI]) but resolution of allergy to formaldehyde8; persistence of allergy to corticosteroids (budesonide and alclometasone)9; persistence of allergy to an antibiotic (neomycin sulfate) but resolution of allergies to a different antibiotic (bacitracin), glues (ethyl acrylate), bleach, and glutaraldehyde9; persistence of nickel allergy but resolution of allergies to fragrances (cinnamic aldehyde, balsam of Peru) and non-FRPs (methylchloroisothiazolinone or MI)10; and persistence of allergies to non-FRPs (MI) and FRPs (bronopol) but resolution of allergies to nickel, fragrances (hydroperoxides of linalool), and Compositae.11 Additional case reports of positive patch test results while on dupilumab but with no pretreatment results for comparison include allergies to rubber additives,12-14 nickel,14 textile dyes,14 cosmetic and hair care additives,12,14,15 corticosteroids,15 FRPs,15 fragrances,15,16 emulsifiers,16 and non-FRPs.17
An evident theme in the dupilumab patch-testing literature has been that results are variable and case specific: a given patient with ACD to an allergen will respond to dupilumab treatment and have subsequent negative patch testing, while another patient will not respond to dupilumab treatment and have persistent positive patch testing. This is likely because, in certain individuals, the allergen-immune system combination shifts ACD pathogenesis from a purely TH1 response to at least a partial TH2 response, thus allowing for benefit from dupilumab therapy. T helper 1 cell–mediated ACD should not be affected by dupilumab; therefore, reliable results can be elucidated from patch testing despite the drug.
Final Thoughts
We propose that AD patients with residual disease after taking dupilumab undergo patch testing. Positive results indicate allergens that are not inhibited by the drug. Patients will need to follow strict allergen avoidance to resolve this component of their disease; failure to improve might suggest the result was a nonrelevant positive.
If patch testing is negative, an alternative cause for residual disease must be sought. We do not recommend stopping dupilumab prior to patch testing to avoid a disease flare from AD or possible TH2-mediated ACD.
In patients with persistent atopic dermatitis (AD) who are taking dupilumab, is there benefit of patch testing to determine if allergic contact dermatitis (ACD) also is contributing to their disease? Results of patch testing are likely be influenced by the immunomodulatory effects of dupilumab. Similar to the recommendation for patients to refrain from using topical or systemic corticosteroids for 1 week or more prior to patch testing to eliminate false negatives, we reviewed the literature to create practice guidelines for dermatologists regarding patch testing while a patient is taking dupilumab.
Pathophysiology and Pathomechanism
Dupilumab functions through the blockade of T helper 2 (TH2) cells; ACD is propagated through the T helper 1 (TH1) cellular pathway. However, patients with ACD that is unresponsive to allergen avoidance and traditional therapies, such as topical and oral corticosteroids, have responded to dupilumab. The more common reports of this responsiveness are with fragrances; multiple case series described patients with ACD to fragrance mix I1 and balsam of Peru1,2 who improved on dupilumab when other treatments failed. There also are reports of response when ACD was secondary to nickel,2,3p-phenylenediamine,1 Compositae,4 and non–formaldehyde-releasing preservatives (non-FRPs).5 Therefore, not all ACD is propagated through the TH1 cellular pathway.
As noted in these cases, ACD can be a response to an allergen whose pathogenesis involves the TH2 pathway or when patient characteristics favor a TH2 response. It has been suggested that AD patients are more susceptible to TH2-mediated contact sensitization to less-potent allergens, such as fragrances.6
Patch Test Results
Positive patch test results for allergens have been reported while patients are on dupilumab therapy, including a few studies in which results prior to starting dupilumab were compared with those while patients were on dupilumab therapy. In a retrospective chart review of 48 patients on dupilumab for AD with persistent disease, 23 patients were patch tested before and during dupilumab therapy. In these patients, the majority of contact allergies were persistent and only 10% (13/125) of patch test–positive results resolved on dupilumab therapy.7 Contact allergies that resolved included those to emulsifiers (propylene glycol, Amerchol L101 [lanolin-containing products found in cosmetics and other goods], dimethylaminopropylamine), fragrances (fragrance mix I, balsam of Peru), sunscreens (sulisobenzone, phenylbenzimidazole-5-sulfonic acid), and metals (vanadium chloride, phenylmercuric acetate).7 The following results observed in individual cases demonstrated conflicting findings: persistence of allergy to non-FRPs (methylisothiazolinone [MI]) but resolution of allergy to formaldehyde8; persistence of allergy to corticosteroids (budesonide and alclometasone)9; persistence of allergy to an antibiotic (neomycin sulfate) but resolution of allergies to a different antibiotic (bacitracin), glues (ethyl acrylate), bleach, and glutaraldehyde9; persistence of nickel allergy but resolution of allergies to fragrances (cinnamic aldehyde, balsam of Peru) and non-FRPs (methylchloroisothiazolinone or MI)10; and persistence of allergies to non-FRPs (MI) and FRPs (bronopol) but resolution of allergies to nickel, fragrances (hydroperoxides of linalool), and Compositae.11 Additional case reports of positive patch test results while on dupilumab but with no pretreatment results for comparison include allergies to rubber additives,12-14 nickel,14 textile dyes,14 cosmetic and hair care additives,12,14,15 corticosteroids,15 FRPs,15 fragrances,15,16 emulsifiers,16 and non-FRPs.17
An evident theme in the dupilumab patch-testing literature has been that results are variable and case specific: a given patient with ACD to an allergen will respond to dupilumab treatment and have subsequent negative patch testing, while another patient will not respond to dupilumab treatment and have persistent positive patch testing. This is likely because, in certain individuals, the allergen-immune system combination shifts ACD pathogenesis from a purely TH1 response to at least a partial TH2 response, thus allowing for benefit from dupilumab therapy. T helper 1 cell–mediated ACD should not be affected by dupilumab; therefore, reliable results can be elucidated from patch testing despite the drug.
Final Thoughts
We propose that AD patients with residual disease after taking dupilumab undergo patch testing. Positive results indicate allergens that are not inhibited by the drug. Patients will need to follow strict allergen avoidance to resolve this component of their disease; failure to improve might suggest the result was a nonrelevant positive.
If patch testing is negative, an alternative cause for residual disease must be sought. We do not recommend stopping dupilumab prior to patch testing to avoid a disease flare from AD or possible TH2-mediated ACD.
- Chipalkatti N, Lee N, Zancanaro P, et al. Dupilumab as a treatment for allergic contact dermatitis. Dermatitis. 2018;29:347-348. doi:10.1097/DER.0000000000000414
- Jacob SE, Sung CT, Machler BC. Dupilumab for systemic allergy syndrome with dermatitis. Dermatitis. 2019;30:164-167. doi:10.1097/DER.0000000000000446
- Joshi SR, Khan DA. Effective use of dupilumab in managing systemic allergic contact dermatitis. Dermatitis. 2018;29:282-284. doi:10.1097/DER.0000000000000409
- Ruge IF, Skov L, Zachariae C, et al. Dupilumab treatment in two patients with severe allergic contact dermatitis caused by sesquiterpene lactones. Contact Dermatitis. 2020:83;137-139. doi:10.1111/cod.13545
- Goldminz AM, Scheinman PL. A case series of dupilumab-treated allergic contact dermatitis patients. Dermatol Ther. 2018;31:e12701. doi:10.1111/dth.12701
- Kohli N, Nedorost S. Inflamed skin predisposes to sensitization to less potent allergens. J Am Acad Dermatol. 2016;75:312-317. doi:10.1016/j.jaad.2016.03.010
- Raffi J, Suresh R, Botto N, et al. The impact of dupilumab on patch testing and the prevalence of comorbid allergic contact dermatitis in recalcitrant atopic dermatitis: a retrospective chart review. J Am Acad Dermatol. 2020;82:132-138. doi:10.1016/j.jaad.2019.09.028
- Puza CJ, Atwater AR. Positive patch test reaction in a patient taking dupilumab. Dermatitis. 2018;29:89. doi:10.1097/DER.0000000000000346
- Suresh R, Murase JE. The role of expanded series patch testing in identifying causality of residual facial dermatitis following initiation of dupilumab therapy. JAAD Case Rep. 2018;4:899-904. doi:10.1016/j.jdcr.2018.08.027
- Stout M, Silverberg JI. Variable impact of dupilumab on patch testing results and allergic contact dermatitis in adults with atopic dermatitis. J Am Acad Dermatol. 2019;81:157-162. doi:10.1016/j.jaad.2019.03.020
- Raffi J, Botto N. Patch testing and allergen-specific inhibition in a patient taking dupilumab. JAMA Dermatol. 2019;155:120-121. doi:10.1001/jamadermatol.2018.4098
- Hoot JW, Douglas JD, Falo LD Jr. Patch testing in a patient on dupilumab. Dermatitis. 2018;29:164. doi:10.1097/DER.0000000000000357
- Crepy M-N, Nosbaum A, Bensefa-Colas L. Blocking type 2 inflammation by dupilumab does not control classic (type 1-driven) allergic contact dermatitis in chronic hand eczema. Contact Dermatitis. 2019;81:145-147. doi:10.1111/cod.13266
- Raffi J, Chen R, Botto N. Wide dye reactors. JAAD Case Rep. 2019;5:877-879. doi:10.1016/j.jdcr.2019.08.005
- Koblinski JE, Hamann D. Mixed occupational and iatrogenic allergic contact dermatitis in a hairdresser. Occup Med (Lond). 2020;70:523-526. doi:10.1093/occmed/kqaa152
- Raffi J, Suresh R, Fishman H, et al. Investigating the role of allergic contact dermatitis in residual ocular surface disease on dupilumab (ROSDD). Int J Womens Dermatol. 2019;5:308-313. doi:10.1016/j.ijwd.2019.10.001
- Zhu GA, Chen JK, Chiou A, et al. Repeat patch testing in a patient with allergic contact dermatitis improved on dupilumab. JAAD Case Rep. 2019;5:336-338. doi:10.1016/j.jdcr.2019.01.023
- Chipalkatti N, Lee N, Zancanaro P, et al. Dupilumab as a treatment for allergic contact dermatitis. Dermatitis. 2018;29:347-348. doi:10.1097/DER.0000000000000414
- Jacob SE, Sung CT, Machler BC. Dupilumab for systemic allergy syndrome with dermatitis. Dermatitis. 2019;30:164-167. doi:10.1097/DER.0000000000000446
- Joshi SR, Khan DA. Effective use of dupilumab in managing systemic allergic contact dermatitis. Dermatitis. 2018;29:282-284. doi:10.1097/DER.0000000000000409
- Ruge IF, Skov L, Zachariae C, et al. Dupilumab treatment in two patients with severe allergic contact dermatitis caused by sesquiterpene lactones. Contact Dermatitis. 2020:83;137-139. doi:10.1111/cod.13545
- Goldminz AM, Scheinman PL. A case series of dupilumab-treated allergic contact dermatitis patients. Dermatol Ther. 2018;31:e12701. doi:10.1111/dth.12701
- Kohli N, Nedorost S. Inflamed skin predisposes to sensitization to less potent allergens. J Am Acad Dermatol. 2016;75:312-317. doi:10.1016/j.jaad.2016.03.010
- Raffi J, Suresh R, Botto N, et al. The impact of dupilumab on patch testing and the prevalence of comorbid allergic contact dermatitis in recalcitrant atopic dermatitis: a retrospective chart review. J Am Acad Dermatol. 2020;82:132-138. doi:10.1016/j.jaad.2019.09.028
- Puza CJ, Atwater AR. Positive patch test reaction in a patient taking dupilumab. Dermatitis. 2018;29:89. doi:10.1097/DER.0000000000000346
- Suresh R, Murase JE. The role of expanded series patch testing in identifying causality of residual facial dermatitis following initiation of dupilumab therapy. JAAD Case Rep. 2018;4:899-904. doi:10.1016/j.jdcr.2018.08.027
- Stout M, Silverberg JI. Variable impact of dupilumab on patch testing results and allergic contact dermatitis in adults with atopic dermatitis. J Am Acad Dermatol. 2019;81:157-162. doi:10.1016/j.jaad.2019.03.020
- Raffi J, Botto N. Patch testing and allergen-specific inhibition in a patient taking dupilumab. JAMA Dermatol. 2019;155:120-121. doi:10.1001/jamadermatol.2018.4098
- Hoot JW, Douglas JD, Falo LD Jr. Patch testing in a patient on dupilumab. Dermatitis. 2018;29:164. doi:10.1097/DER.0000000000000357
- Crepy M-N, Nosbaum A, Bensefa-Colas L. Blocking type 2 inflammation by dupilumab does not control classic (type 1-driven) allergic contact dermatitis in chronic hand eczema. Contact Dermatitis. 2019;81:145-147. doi:10.1111/cod.13266
- Raffi J, Chen R, Botto N. Wide dye reactors. JAAD Case Rep. 2019;5:877-879. doi:10.1016/j.jdcr.2019.08.005
- Koblinski JE, Hamann D. Mixed occupational and iatrogenic allergic contact dermatitis in a hairdresser. Occup Med (Lond). 2020;70:523-526. doi:10.1093/occmed/kqaa152
- Raffi J, Suresh R, Fishman H, et al. Investigating the role of allergic contact dermatitis in residual ocular surface disease on dupilumab (ROSDD). Int J Womens Dermatol. 2019;5:308-313. doi:10.1016/j.ijwd.2019.10.001
- Zhu GA, Chen JK, Chiou A, et al. Repeat patch testing in a patient with allergic contact dermatitis improved on dupilumab. JAAD Case Rep. 2019;5:336-338. doi:10.1016/j.jdcr.2019.01.023
Practice Points
- Allergic contact dermatitis is an important diagnostic consideration in patients with refractory or persistent dermatitis.
- Patch testing is important to help determine a possible allergic contactant, but there is confusion about its accuracy in patients taking dupilumab.
- Patients with residual dermatitis while on dupilumab are likely to benefit from patch testing.
Leukemia Cutis Manifesting as Nonpalpable Purpura
To the Editor:
A 72-year-old man presented with symptomatic anemia and nonpalpable purpura of the legs, abdomen, and arms of 2 weeks’ duration (Figure 1). There were no associated perifollicular papules. Physical examination of the hair and gingiva were normal.

The patient’s medical history was notable for a poorly differentiated pancreatic adenocarcinoma (pT3N1M0) resected 7 months prior using a Whipple operation (pancreaticoduodenectomy). Adjuvant therapy consisted of 5 cycles of intravenous gemcitabine and paclitaxel. Treatment was discontinued 1 month prior due to progressive weight loss and the presence of new liver metastases on computed tomography. There was no recent history of corticosteroid, antiplatelet, or anticoagulant use. The patient had no known history of trauma at the affected sites.
The patient’s laboratory workup revealed the following results: hemoglobin, 5.5 g/dL (reference range, 13–18 g/dL); platelets, 128×109/L (reference range, 150–400×109/L); total white blood cell count (24.0×109/L [reference range, 4.0–11.0×109/L]), consisting of neutrophils (2.4×109/L [reference range, 2.0–7.5×109/L]), lymphocytes (3.1×109/L [reference range, 1.5–4.0×109/L]), and monocytes (18.5×109/L [reference range, 0.2–0.8×109/L]). Fibrinogen, activated partial thromboplastin time, and prothrombin time were within reference range. Results of a bone marrow biopsy showed 64% blasts. The lactate dehydrogenase level was 286 U/L (reference range, 135–220 U/L) and CA-19-9 antigen was 238 U/mL (reference range, 0–39 U/mL).

Results from a skin punch biopsy from the right leg showed a normal epidermis and papillary dermis. The reticular dermis was expanded by a diffuse cellular infiltrate with dermal edema and separation of collagen bundles (Figure 2), which consisted of small cells with irregular, cleaved, and notched nuclei, containing a variable amount of eosinophilic cytoplasm. Mitotic figures were present (Figure 3). There was no evidence of vasculitis, and Congo red stain for amyloid was negative. These atypical cells were positive for the leukocyte common antigen, favoring a hematopoietic infiltrate (Figure 4). Other positive markers included CD4 (associated with helper T cells, and mature and immature monocytes), CD68 (a monocyte/macrophage marker), and CD56 (associated with natural killer cells, myeloma, acute myeloid leukemia [AML], and neuroendocrine tumors). The cells were negative for CD3 (T-cell lineage–specific antigen), CD5 (marker of T cells and a subset of IgM-secreting B cells), CD34 (early hematopoietic marker), and CD20 (B-cell marker). Other negative myeloid markers included myeloperoxidase, CD117, and CD138. These findings suggested leukemic cell recruitment at the site of a reactive infiltrate. The patient completed 2 cycles of intravenous azacitidine with little response and subsequently opted for palliative measures.
 and numerous notched nuclei")
Nonpalpable purpura has a broad differential diagnosis including primary and secondary thrombocytopenia; coagulopathies, including vitamin K deficiency, specific clotting factor deficiencies, and amyloid-related purpura; genetic or acquired collagen disorders, including vitamin C deficiency; and eruptions induced by drugs and herbal remedies.

Leukemia cutis is a relatively rare cause of purpura and is defined as cutaneous infiltration by neoplastic leucocytes.1 It most commonly is associated with AML and complicates approximately 5% to 15%of all adult cases.2 Cutaneous involvement occurs predominantly in monocytic variants; acute myelomonocytic leukemia and acute monocytic leukemia may arise in up to 50% of these cases.3 The clinical presentation may vary from papules, nodules, and plaques to rarer manifestations including purpura. A leukemic infiltrate often is associated with sites of inflammation, such as infection or ulceration,4 though there was no reported history of any known triggering events in our patient. Lesions usually involve the legs, followed by the arms, back, chest, scalp, and face.4 One-third of cases coincide with systemic symptoms, and approximately 10% precede bone marrow or peripheral blood involvement, referred to as aleukemic leukemia. The remainder of cases arise following an established diagnosis of systemic leukemia.5 Leukemia cutis is considered a marker of poor prognosis in AML,4,6 requiring treatment for the underlying systemic disease. Our case also was complicated by a concurrent pancreatic malignancy and relatively advanced age, which limited the feasibility of further treatment.
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In: Weedon D, ed. Skin Pathology. 2nd ed. Churchill Livingstone; 2002:1118-1120.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Paydas S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Shaikh BS, Frantz E, Lookingbill DP. Histologically proven leukemia cutis carries a poor prognosis in acute nonlymphocytic leukemia. Cutis. 1987;39:57-60.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
To the Editor:
A 72-year-old man presented with symptomatic anemia and nonpalpable purpura of the legs, abdomen, and arms of 2 weeks’ duration (Figure 1). There were no associated perifollicular papules. Physical examination of the hair and gingiva were normal.
The patient’s medical history was notable for a poorly differentiated pancreatic adenocarcinoma (pT3N1M0) resected 7 months prior using a Whipple operation (pancreaticoduodenectomy). Adjuvant therapy consisted of 5 cycles of intravenous gemcitabine and paclitaxel. Treatment was discontinued 1 month prior due to progressive weight loss and the presence of new liver metastases on computed tomography. There was no recent history of corticosteroid, antiplatelet, or anticoagulant use. The patient had no known history of trauma at the affected sites.
The patient’s laboratory workup revealed the following results: hemoglobin, 5.5 g/dL (reference range, 13–18 g/dL); platelets, 128×109/L (reference range, 150–400×109/L); total white blood cell count (24.0×109/L [reference range, 4.0–11.0×109/L]), consisting of neutrophils (2.4×109/L [reference range, 2.0–7.5×109/L]), lymphocytes (3.1×109/L [reference range, 1.5–4.0×109/L]), and monocytes (18.5×109/L [reference range, 0.2–0.8×109/L]). Fibrinogen, activated partial thromboplastin time, and prothrombin time were within reference range. Results of a bone marrow biopsy showed 64% blasts. The lactate dehydrogenase level was 286 U/L (reference range, 135–220 U/L) and CA-19-9 antigen was 238 U/mL (reference range, 0–39 U/mL).
Results from a skin punch biopsy from the right leg showed a normal epidermis and papillary dermis. The reticular dermis was expanded by a diffuse cellular infiltrate with dermal edema and separation of collagen bundles (Figure 2), which consisted of small cells with irregular, cleaved, and notched nuclei, containing a variable amount of eosinophilic cytoplasm. Mitotic figures were present (Figure 3). There was no evidence of vasculitis, and Congo red stain for amyloid was negative. These atypical cells were positive for the leukocyte common antigen, favoring a hematopoietic infiltrate (Figure 4). Other positive markers included CD4 (associated with helper T cells, and mature and immature monocytes), CD68 (a monocyte/macrophage marker), and CD56 (associated with natural killer cells, myeloma, acute myeloid leukemia [AML], and neuroendocrine tumors). The cells were negative for CD3 (T-cell lineage–specific antigen), CD5 (marker of T cells and a subset of IgM-secreting B cells), CD34 (early hematopoietic marker), and CD20 (B-cell marker). Other negative myeloid markers included myeloperoxidase, CD117, and CD138. These findings suggested leukemic cell recruitment at the site of a reactive infiltrate. The patient completed 2 cycles of intravenous azacitidine with little response and subsequently opted for palliative measures.
Nonpalpable purpura has a broad differential diagnosis including primary and secondary thrombocytopenia; coagulopathies, including vitamin K deficiency, specific clotting factor deficiencies, and amyloid-related purpura; genetic or acquired collagen disorders, including vitamin C deficiency; and eruptions induced by drugs and herbal remedies.
Leukemia cutis is a relatively rare cause of purpura and is defined as cutaneous infiltration by neoplastic leucocytes.1 It most commonly is associated with AML and complicates approximately 5% to 15%of all adult cases.2 Cutaneous involvement occurs predominantly in monocytic variants; acute myelomonocytic leukemia and acute monocytic leukemia may arise in up to 50% of these cases.3 The clinical presentation may vary from papules, nodules, and plaques to rarer manifestations including purpura. A leukemic infiltrate often is associated with sites of inflammation, such as infection or ulceration,4 though there was no reported history of any known triggering events in our patient. Lesions usually involve the legs, followed by the arms, back, chest, scalp, and face.4 One-third of cases coincide with systemic symptoms, and approximately 10% precede bone marrow or peripheral blood involvement, referred to as aleukemic leukemia. The remainder of cases arise following an established diagnosis of systemic leukemia.5 Leukemia cutis is considered a marker of poor prognosis in AML,4,6 requiring treatment for the underlying systemic disease. Our case also was complicated by a concurrent pancreatic malignancy and relatively advanced age, which limited the feasibility of further treatment.
To the Editor:
A 72-year-old man presented with symptomatic anemia and nonpalpable purpura of the legs, abdomen, and arms of 2 weeks’ duration (Figure 1). There were no associated perifollicular papules. Physical examination of the hair and gingiva were normal.
The patient’s medical history was notable for a poorly differentiated pancreatic adenocarcinoma (pT3N1M0) resected 7 months prior using a Whipple operation (pancreaticoduodenectomy). Adjuvant therapy consisted of 5 cycles of intravenous gemcitabine and paclitaxel. Treatment was discontinued 1 month prior due to progressive weight loss and the presence of new liver metastases on computed tomography. There was no recent history of corticosteroid, antiplatelet, or anticoagulant use. The patient had no known history of trauma at the affected sites.
The patient’s laboratory workup revealed the following results: hemoglobin, 5.5 g/dL (reference range, 13–18 g/dL); platelets, 128×109/L (reference range, 150–400×109/L); total white blood cell count (24.0×109/L [reference range, 4.0–11.0×109/L]), consisting of neutrophils (2.4×109/L [reference range, 2.0–7.5×109/L]), lymphocytes (3.1×109/L [reference range, 1.5–4.0×109/L]), and monocytes (18.5×109/L [reference range, 0.2–0.8×109/L]). Fibrinogen, activated partial thromboplastin time, and prothrombin time were within reference range. Results of a bone marrow biopsy showed 64% blasts. The lactate dehydrogenase level was 286 U/L (reference range, 135–220 U/L) and CA-19-9 antigen was 238 U/mL (reference range, 0–39 U/mL).
Results from a skin punch biopsy from the right leg showed a normal epidermis and papillary dermis. The reticular dermis was expanded by a diffuse cellular infiltrate with dermal edema and separation of collagen bundles (Figure 2), which consisted of small cells with irregular, cleaved, and notched nuclei, containing a variable amount of eosinophilic cytoplasm. Mitotic figures were present (Figure 3). There was no evidence of vasculitis, and Congo red stain for amyloid was negative. These atypical cells were positive for the leukocyte common antigen, favoring a hematopoietic infiltrate (Figure 4). Other positive markers included CD4 (associated with helper T cells, and mature and immature monocytes), CD68 (a monocyte/macrophage marker), and CD56 (associated with natural killer cells, myeloma, acute myeloid leukemia [AML], and neuroendocrine tumors). The cells were negative for CD3 (T-cell lineage–specific antigen), CD5 (marker of T cells and a subset of IgM-secreting B cells), CD34 (early hematopoietic marker), and CD20 (B-cell marker). Other negative myeloid markers included myeloperoxidase, CD117, and CD138. These findings suggested leukemic cell recruitment at the site of a reactive infiltrate. The patient completed 2 cycles of intravenous azacitidine with little response and subsequently opted for palliative measures.
Nonpalpable purpura has a broad differential diagnosis including primary and secondary thrombocytopenia; coagulopathies, including vitamin K deficiency, specific clotting factor deficiencies, and amyloid-related purpura; genetic or acquired collagen disorders, including vitamin C deficiency; and eruptions induced by drugs and herbal remedies.
Leukemia cutis is a relatively rare cause of purpura and is defined as cutaneous infiltration by neoplastic leucocytes.1 It most commonly is associated with AML and complicates approximately 5% to 15%of all adult cases.2 Cutaneous involvement occurs predominantly in monocytic variants; acute myelomonocytic leukemia and acute monocytic leukemia may arise in up to 50% of these cases.3 The clinical presentation may vary from papules, nodules, and plaques to rarer manifestations including purpura. A leukemic infiltrate often is associated with sites of inflammation, such as infection or ulceration,4 though there was no reported history of any known triggering events in our patient. Lesions usually involve the legs, followed by the arms, back, chest, scalp, and face.4 One-third of cases coincide with systemic symptoms, and approximately 10% precede bone marrow or peripheral blood involvement, referred to as aleukemic leukemia. The remainder of cases arise following an established diagnosis of systemic leukemia.5 Leukemia cutis is considered a marker of poor prognosis in AML,4,6 requiring treatment for the underlying systemic disease. Our case also was complicated by a concurrent pancreatic malignancy and relatively advanced age, which limited the feasibility of further treatment.
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In: Weedon D, ed. Skin Pathology. 2nd ed. Churchill Livingstone; 2002:1118-1120.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Paydas S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Shaikh BS, Frantz E, Lookingbill DP. Histologically proven leukemia cutis carries a poor prognosis in acute nonlymphocytic leukemia. Cutis. 1987;39:57-60.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In: Weedon D, ed. Skin Pathology. 2nd ed. Churchill Livingstone; 2002:1118-1120.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Paydas S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Shaikh BS, Frantz E, Lookingbill DP. Histologically proven leukemia cutis carries a poor prognosis in acute nonlymphocytic leukemia. Cutis. 1987;39:57-60.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
Practice Points
- Leukemia cutis complicates 5% to 15% of all cases of acute myeloid leukemia (AML) in adults.
- The appearance of leukemia cutis may be highly variable. Therefore, it should be included in the differential diagnosis for any cutaneous presentation in patients with an existing diagnosis or high likelihood of AML.
- Leukemic infiltrates are associated with sites of inflammation.

Treatment of Elephantiasic Pretibial Myxedema With Rituximab Therapy
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
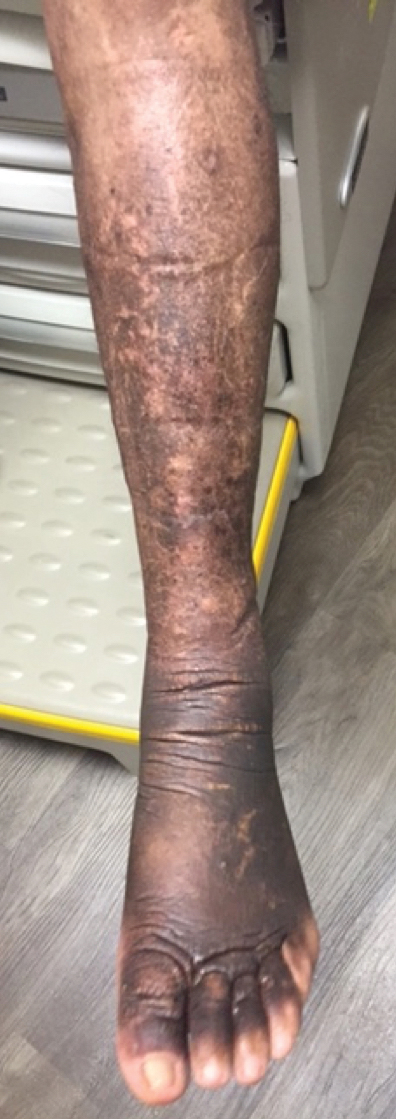
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.

The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
. B, Colloidal iron staining highlighted the prominent mucin within the dermis")
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.
The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.
The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
Practice Points
- Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet.
- Although many therapeutic modalities have been described for the management of the elephantiasis variant of PTM, few treatments have shown notable efficacy.
- Rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.

Graft-vs-host Disease and Toxic Epidermal Necrolysis Following Hematopoietic Stem Cell Transplantation
To the Editor:
Acute graft-vs-host disease (GVHD) remains a limitation to hematopoietic stem cell transplantation (HSCT) in 20% to 50% of patients after transplant. Furthermore, failed treatment with corticosteroids is frequent and portends a poor prognosis.1 Toxic epidermal necrolysis (TEN) is an epidermolytic skin disorder thought to represent an adverse drug reaction, though its pathogenesis remains unclear. Severe forms of acute GVHD can mimic TEN clinically and histologically. Both can present with widespread cutaneous and mucosal bullae, erosions, and desquamation. Toxic epidermal necrolysis in the context of allogeneic hematopoietic stem cell transplantation is extremely rare, with almost 100% mortality in adult patients. Features that favor acute GVHD over TEN include diarrhea, elevation in bilirubin level, and chimerism.2 However, these features might be absent, posing a therapeutic dilemma, as current treatment preferences for each of these entities differ.
Growing evidence supports the use of anti–tumor necrosis factor (TNF) α drugs for the treatment of TEN. Success has been reported with both anti–TNF-α monoclonal antibodies as well as the soluble fusion protein etanercept.3,4 The use of TNF-α inhibitors in acute GVHD remains anecdotal.

A 58-year-old man (patient 1) with a history of acute myelogenous leukemia presented with a pruritic morbilliform eruption 28 days after HSCT. There was no desquamation or mucosal involvement and the biopsy obtained was histologically suggestive of grade 2 acute GVHD. His immunosuppressive regimen included sirolimus and cyclophosphamide. He was receiving trimethoprim-sulfamethoxazole (TMP-SMX), voriconazole, and acyclovir for infectious prophylaxis. At the time of presentation, he was treated with high-dose systemic steroids (prednisone 2 mg/kg/d) for acute GVHD with partial improvement. Upon tapering of the steroids 3 weeks after initiating TMP-SMX and 1 week after initiating voriconazole, he developed painful desquamation and erosions involving 95% of the body surface area (BSA), necessitating admission to the local burn unit (Figure 1). Biopsies demonstrated full-thickness epidermal necrosis with subepidermal blistering and interface dermatitis (Figure 2). No gastrointestinal tract involvement of acute GVHD was noted. The patient was a 100% donor chimera, supporting the diagnosis of acute GVHD; however, the patient and donor carried the HLA-C*06:02 allele, which previously has been described in association with TMP-SMX–related Stevens-Johnson syndrome/TEN.5 In addition, causality assessment using the algorithm of drug causality for epidermal necrolysis indicated TMP-SMX as a probable cause and voriconazole as a possible cause. The diagnosis of TEN with a SCORe of Toxic Epidermal Necrosis (SCORTEN) of 4 in the setting of acute GVHD was favored, though grade 4 acute GVHD could not be excluded. Trimethoprim-sulfamethoxazole was discontinued, and voriconazole was changed to posaconazole. He received supportive care along with 1 dose of 25-mg subcutaneous etanercept and 3 days of intravenous immunoglobulin (IVIG). Skin re-epithelialization was complete by 3 weeks. At 4 weeks, the patient developed a new asymptomatic erythematous eruption. Biopsies demonstrated changes of acute and chronic GVHD (Figure 3) that resolved with up-titration of sirolimus. The patient remained hospitalized for 96 days and continued to follow up with his transplant team as well as ophthalmology and dermatology. He died 2 years after HSCT.

A 67-year-old woman (patient 2) with high-grade myelodysplastic syndrome presented with an erythematous morbilliform eruption on the torso on day 20 after a matched unrelated HSCT that histologically was consistent with grade 2 GVHD (Figure 4). She had been receiving sirolimus and tacrolimus for GVHD prophylaxis. Infectious prophylaxis included acyclovir, pentamidine, micafungin, and TMP-SMX. Despite high-dose systemic steroids, the rash progressed and ultimately involved 80% BSA. A positive Nikolsky sign was noted involving 21% BSA (Figure 5), in addition to oral and genital mucosal ulcers. She denied nausea, vomiting, fever, or diarrhea. Chimerism studies were negative. Trimethoprim-sulfamethoxazole was discontinued, and she was transferred to a burn unit. Biopsies showed full-thickness epidermal necrosis. A diagnosis of TEN with a SCORTEN of 4 in the setting of acute GVHD was favored; grade 4 acute GVHD could not be excluded. Steroids were discontinued. Because laboratory studies indicated IgA deficiency, IVIG was not considered as a systemic option for therapy. The patient received 1 dose of infliximab (5 mg/kg). Cyclophosphamide 1600 mg weekly was added for GVHD therapy. The wounds progressively healed, and 2 weeks into her admission she was noted to have only 3% BSA with denuded skin. The patient was transferred to the cancer treatment center for further management of the malignancy. Unfortunately, after 2 months she died due to ischemic colitis that was confirmed on autopsy.

Graft-vs-host disease and TEN are rare, life-threatening complications seen in patients with allogeneic HSCT.2 Graft-vs-host disease and TEN share clinicopathologic characteristics and effector immune mechanisms, largely the substantial role of T-cell activation and tissue destruction, which occur through mediators such as TNF-α.6-8

Given the sparse lymphocytic infiltrate, keratinocyte death in TEN is thought to result from soluble molecules, including TNF-α and TNF-related apoptosis-inducing ligand.9 Tumor necrosis factor α has been identified in blister fluid, biopsy specimens, and serum of patients with TEN. Tumor necrosis factor α increases the expression of keratinocyte-inducible nitric oxide synthase, which upregulates keratinocyte Fas ligand expression and subsequent Fas- and caspase-8–mediated keratinocyte cell death.10

Acute GVHD results from donor lymphocyte activation after infusion into damaged recipient tissues that previously have been radiated or chemoablated. Mismatches in histocompatibility complexes between donor cells and recipient tissue antigens serve as the initial trigger for immune activation. Activation of antigen-presenting cells followed by activation, proliferation, differentiation, and migration of donor T cells ultimately results in destruction of the target tissue.11 Immune mediators, such as TNF-α and lymphotoxin α (another member of the TNF superfamily), play a nonredundant role in the pathogenesis of GVHD.12
Current treatment strategies for severe acute GVHD and TEN differ. In North America, high-dose IVIG frequently is used as first-line systemic therapy, while high-dose systemic corticosteroids rarely are used.13 Studies have demonstrated successful use of anti–TNF-α drugs for the treatment of TEN.3,4 Moreover, etanercept has shown to effectively inhibit lymphotoxin α.14 Similarly, TNF inhibition in the management of steroid-refractory acute GVHD has been successful.1 These studies coupled with the underlying immune mechanisms that both diseases share encouraged initiating a trial of anti–TNF-α therapy in our patients.
Patient 1 merits further discussion because he was both a 100% donor chimera as well as a carrier of an human leukocyte antigen susceptibility candidate allele to TMP-SMX. Historical features of his presentation are consistent with either steroid-refractory GVHD or TEN superimposed on acute GVHD. His initial presentation of the more typical macular exanthem of cutaneous acute GVHD was both biopsy proven and supported by clinical improvement with steroid therapy, which was later followed by a robust blistering mucocutaneous presentation approximately 3 weeks after the administration of TMP-SMX and 1 week after initiating voriconazole that improved with IVIG and etanercept.
It is difficult to determine if TEN represents a continuum or result of the underlying drivers of acute GVHD vs a drug reaction. Although there is insufficient evidence to establish a clear-cut diagnosis of TEN, these cases illustrate the need for better diagnostic techniques to allow differentiation between TEN and grade 4 acute GVHD, and in the context of uncertainty, TNF-α inhibition poses a viable therapeutic strategy for these 2 often lethal conditions. Our cases do unequivocally indicate the benefit of this therapeutic modality, add to the current body of literature supporting the use of TNF-α inhibitors in patients such as ours without an official TEN diagnosis, and may guide future investigative efforts.
- Couriel DR, Saliba R, de Lima M, et al. A phase III study of infliximab and corticosteroids for the initial treatment of acute graft-versus-host disease. Biol Blood Marrow Transplant. 2009;15:1555-1562.
- Jeanmonod P, Hubbuch M, Grünhage F, et al. Graft-versus-host disease or toxic epidermal necrolysis: diagnostic dilemma after liver transplantation. Transpl Infect Dis. 2012;14:422-426.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Scott-Lang V, Tidman M, McKay D. Toxic epidermal necrolysis in a child successfully treated with infliximab. Pediatr Dermatol. 2014;31:532-534.
- Kingpin T, Mahasirimongkol S, Konyoung P, et al. Candidate HLA genes for prediction of co-trimoxazole-induced severe cutaneous reactions. Pharmacogenet Genomics. 2015;25:402-411.
- Correia O, Delgado L, Barbosa IL, et al. Increased interleukin 10, tumor necrosis factor alpha, and interleukin 6 levels in blister fluid of toxic epidermal necrolysis. J Am Acad Dermatol. 2002;47:58-62.
- French LE, Tschopp J. Fas-mediated cell death in toxic epidermal necrolysis and graft-versus-host disease: potential for therapeutic inhibition. Schweiz Med Wochenschr. 2000;130:1656-1661.
- Downey A, Jackson C, Harun N, et al. Toxic epidermal necrolysis: review of pathogenesis and management. J Am Acad Dermatol. 2012;66:995-1003.
- de Araujo E, Dessirier V, Laprée G, et al. Death ligand TRAIL, secreted by CD1a+ and CD14+ cells in blister fluids, is involved in killing keratinocytes in toxic epidermal necrolysis. Exp Dermatol. 2011;20:107-112.
- Viard-Leveugle I, Gaide O, Jankovic D, et al. TNF-α and IFN-γ are potential inducers of Fas-mediated keratinocyte apoptosis through activation of inducible nitric oxide synthase in toxic epidermal necrolysis. J Invest Dermatol. 2013;133:489-498.
- Choi SW, Levine JE, Ferrara JL. Pathogenesis and management of graft-versus-host disease. Immunol Allergy Clin North Am. 2010;30:75-101.
- Markey KA, Burman AC, Banovic T, et al. Soluble lymphotoxin is an important effector molecule in GVHD and GVL. Blood. 2010;115:122-132.
- Dodiuk-Gad RP, Olteanu C, Jeschke MG, et al. Treatment of toxic epidermal necrolysis in North America. J Am Acad Dermatol. 2015;73:876-877.
- Tracey D, Klareskog L, Sasso EH, et al. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244-279.
To the Editor:
Acute graft-vs-host disease (GVHD) remains a limitation to hematopoietic stem cell transplantation (HSCT) in 20% to 50% of patients after transplant. Furthermore, failed treatment with corticosteroids is frequent and portends a poor prognosis.1 Toxic epidermal necrolysis (TEN) is an epidermolytic skin disorder thought to represent an adverse drug reaction, though its pathogenesis remains unclear. Severe forms of acute GVHD can mimic TEN clinically and histologically. Both can present with widespread cutaneous and mucosal bullae, erosions, and desquamation. Toxic epidermal necrolysis in the context of allogeneic hematopoietic stem cell transplantation is extremely rare, with almost 100% mortality in adult patients. Features that favor acute GVHD over TEN include diarrhea, elevation in bilirubin level, and chimerism.2 However, these features might be absent, posing a therapeutic dilemma, as current treatment preferences for each of these entities differ.
Growing evidence supports the use of anti–tumor necrosis factor (TNF) α drugs for the treatment of TEN. Success has been reported with both anti–TNF-α monoclonal antibodies as well as the soluble fusion protein etanercept.3,4 The use of TNF-α inhibitors in acute GVHD remains anecdotal.
A 58-year-old man (patient 1) with a history of acute myelogenous leukemia presented with a pruritic morbilliform eruption 28 days after HSCT. There was no desquamation or mucosal involvement and the biopsy obtained was histologically suggestive of grade 2 acute GVHD. His immunosuppressive regimen included sirolimus and cyclophosphamide. He was receiving trimethoprim-sulfamethoxazole (TMP-SMX), voriconazole, and acyclovir for infectious prophylaxis. At the time of presentation, he was treated with high-dose systemic steroids (prednisone 2 mg/kg/d) for acute GVHD with partial improvement. Upon tapering of the steroids 3 weeks after initiating TMP-SMX and 1 week after initiating voriconazole, he developed painful desquamation and erosions involving 95% of the body surface area (BSA), necessitating admission to the local burn unit (Figure 1). Biopsies demonstrated full-thickness epidermal necrosis with subepidermal blistering and interface dermatitis (Figure 2). No gastrointestinal tract involvement of acute GVHD was noted. The patient was a 100% donor chimera, supporting the diagnosis of acute GVHD; however, the patient and donor carried the HLA-C*06:02 allele, which previously has been described in association with TMP-SMX–related Stevens-Johnson syndrome/TEN.5 In addition, causality assessment using the algorithm of drug causality for epidermal necrolysis indicated TMP-SMX as a probable cause and voriconazole as a possible cause. The diagnosis of TEN with a SCORe of Toxic Epidermal Necrosis (SCORTEN) of 4 in the setting of acute GVHD was favored, though grade 4 acute GVHD could not be excluded. Trimethoprim-sulfamethoxazole was discontinued, and voriconazole was changed to posaconazole. He received supportive care along with 1 dose of 25-mg subcutaneous etanercept and 3 days of intravenous immunoglobulin (IVIG). Skin re-epithelialization was complete by 3 weeks. At 4 weeks, the patient developed a new asymptomatic erythematous eruption. Biopsies demonstrated changes of acute and chronic GVHD (Figure 3) that resolved with up-titration of sirolimus. The patient remained hospitalized for 96 days and continued to follow up with his transplant team as well as ophthalmology and dermatology. He died 2 years after HSCT.
A 67-year-old woman (patient 2) with high-grade myelodysplastic syndrome presented with an erythematous morbilliform eruption on the torso on day 20 after a matched unrelated HSCT that histologically was consistent with grade 2 GVHD (Figure 4). She had been receiving sirolimus and tacrolimus for GVHD prophylaxis. Infectious prophylaxis included acyclovir, pentamidine, micafungin, and TMP-SMX. Despite high-dose systemic steroids, the rash progressed and ultimately involved 80% BSA. A positive Nikolsky sign was noted involving 21% BSA (Figure 5), in addition to oral and genital mucosal ulcers. She denied nausea, vomiting, fever, or diarrhea. Chimerism studies were negative. Trimethoprim-sulfamethoxazole was discontinued, and she was transferred to a burn unit. Biopsies showed full-thickness epidermal necrosis. A diagnosis of TEN with a SCORTEN of 4 in the setting of acute GVHD was favored; grade 4 acute GVHD could not be excluded. Steroids were discontinued. Because laboratory studies indicated IgA deficiency, IVIG was not considered as a systemic option for therapy. The patient received 1 dose of infliximab (5 mg/kg). Cyclophosphamide 1600 mg weekly was added for GVHD therapy. The wounds progressively healed, and 2 weeks into her admission she was noted to have only 3% BSA with denuded skin. The patient was transferred to the cancer treatment center for further management of the malignancy. Unfortunately, after 2 months she died due to ischemic colitis that was confirmed on autopsy.
Graft-vs-host disease and TEN are rare, life-threatening complications seen in patients with allogeneic HSCT.2 Graft-vs-host disease and TEN share clinicopathologic characteristics and effector immune mechanisms, largely the substantial role of T-cell activation and tissue destruction, which occur through mediators such as TNF-α.6-8
Given the sparse lymphocytic infiltrate, keratinocyte death in TEN is thought to result from soluble molecules, including TNF-α and TNF-related apoptosis-inducing ligand.9 Tumor necrosis factor α has been identified in blister fluid, biopsy specimens, and serum of patients with TEN. Tumor necrosis factor α increases the expression of keratinocyte-inducible nitric oxide synthase, which upregulates keratinocyte Fas ligand expression and subsequent Fas- and caspase-8–mediated keratinocyte cell death.10
Acute GVHD results from donor lymphocyte activation after infusion into damaged recipient tissues that previously have been radiated or chemoablated. Mismatches in histocompatibility complexes between donor cells and recipient tissue antigens serve as the initial trigger for immune activation. Activation of antigen-presenting cells followed by activation, proliferation, differentiation, and migration of donor T cells ultimately results in destruction of the target tissue.11 Immune mediators, such as TNF-α and lymphotoxin α (another member of the TNF superfamily), play a nonredundant role in the pathogenesis of GVHD.12
Current treatment strategies for severe acute GVHD and TEN differ. In North America, high-dose IVIG frequently is used as first-line systemic therapy, while high-dose systemic corticosteroids rarely are used.13 Studies have demonstrated successful use of anti–TNF-α drugs for the treatment of TEN.3,4 Moreover, etanercept has shown to effectively inhibit lymphotoxin α.14 Similarly, TNF inhibition in the management of steroid-refractory acute GVHD has been successful.1 These studies coupled with the underlying immune mechanisms that both diseases share encouraged initiating a trial of anti–TNF-α therapy in our patients.
Patient 1 merits further discussion because he was both a 100% donor chimera as well as a carrier of an human leukocyte antigen susceptibility candidate allele to TMP-SMX. Historical features of his presentation are consistent with either steroid-refractory GVHD or TEN superimposed on acute GVHD. His initial presentation of the more typical macular exanthem of cutaneous acute GVHD was both biopsy proven and supported by clinical improvement with steroid therapy, which was later followed by a robust blistering mucocutaneous presentation approximately 3 weeks after the administration of TMP-SMX and 1 week after initiating voriconazole that improved with IVIG and etanercept.
It is difficult to determine if TEN represents a continuum or result of the underlying drivers of acute GVHD vs a drug reaction. Although there is insufficient evidence to establish a clear-cut diagnosis of TEN, these cases illustrate the need for better diagnostic techniques to allow differentiation between TEN and grade 4 acute GVHD, and in the context of uncertainty, TNF-α inhibition poses a viable therapeutic strategy for these 2 often lethal conditions. Our cases do unequivocally indicate the benefit of this therapeutic modality, add to the current body of literature supporting the use of TNF-α inhibitors in patients such as ours without an official TEN diagnosis, and may guide future investigative efforts.
To the Editor:
Acute graft-vs-host disease (GVHD) remains a limitation to hematopoietic stem cell transplantation (HSCT) in 20% to 50% of patients after transplant. Furthermore, failed treatment with corticosteroids is frequent and portends a poor prognosis.1 Toxic epidermal necrolysis (TEN) is an epidermolytic skin disorder thought to represent an adverse drug reaction, though its pathogenesis remains unclear. Severe forms of acute GVHD can mimic TEN clinically and histologically. Both can present with widespread cutaneous and mucosal bullae, erosions, and desquamation. Toxic epidermal necrolysis in the context of allogeneic hematopoietic stem cell transplantation is extremely rare, with almost 100% mortality in adult patients. Features that favor acute GVHD over TEN include diarrhea, elevation in bilirubin level, and chimerism.2 However, these features might be absent, posing a therapeutic dilemma, as current treatment preferences for each of these entities differ.
Growing evidence supports the use of anti–tumor necrosis factor (TNF) α drugs for the treatment of TEN. Success has been reported with both anti–TNF-α monoclonal antibodies as well as the soluble fusion protein etanercept.3,4 The use of TNF-α inhibitors in acute GVHD remains anecdotal.
A 58-year-old man (patient 1) with a history of acute myelogenous leukemia presented with a pruritic morbilliform eruption 28 days after HSCT. There was no desquamation or mucosal involvement and the biopsy obtained was histologically suggestive of grade 2 acute GVHD. His immunosuppressive regimen included sirolimus and cyclophosphamide. He was receiving trimethoprim-sulfamethoxazole (TMP-SMX), voriconazole, and acyclovir for infectious prophylaxis. At the time of presentation, he was treated with high-dose systemic steroids (prednisone 2 mg/kg/d) for acute GVHD with partial improvement. Upon tapering of the steroids 3 weeks after initiating TMP-SMX and 1 week after initiating voriconazole, he developed painful desquamation and erosions involving 95% of the body surface area (BSA), necessitating admission to the local burn unit (Figure 1). Biopsies demonstrated full-thickness epidermal necrosis with subepidermal blistering and interface dermatitis (Figure 2). No gastrointestinal tract involvement of acute GVHD was noted. The patient was a 100% donor chimera, supporting the diagnosis of acute GVHD; however, the patient and donor carried the HLA-C*06:02 allele, which previously has been described in association with TMP-SMX–related Stevens-Johnson syndrome/TEN.5 In addition, causality assessment using the algorithm of drug causality for epidermal necrolysis indicated TMP-SMX as a probable cause and voriconazole as a possible cause. The diagnosis of TEN with a SCORe of Toxic Epidermal Necrosis (SCORTEN) of 4 in the setting of acute GVHD was favored, though grade 4 acute GVHD could not be excluded. Trimethoprim-sulfamethoxazole was discontinued, and voriconazole was changed to posaconazole. He received supportive care along with 1 dose of 25-mg subcutaneous etanercept and 3 days of intravenous immunoglobulin (IVIG). Skin re-epithelialization was complete by 3 weeks. At 4 weeks, the patient developed a new asymptomatic erythematous eruption. Biopsies demonstrated changes of acute and chronic GVHD (Figure 3) that resolved with up-titration of sirolimus. The patient remained hospitalized for 96 days and continued to follow up with his transplant team as well as ophthalmology and dermatology. He died 2 years after HSCT.
A 67-year-old woman (patient 2) with high-grade myelodysplastic syndrome presented with an erythematous morbilliform eruption on the torso on day 20 after a matched unrelated HSCT that histologically was consistent with grade 2 GVHD (Figure 4). She had been receiving sirolimus and tacrolimus for GVHD prophylaxis. Infectious prophylaxis included acyclovir, pentamidine, micafungin, and TMP-SMX. Despite high-dose systemic steroids, the rash progressed and ultimately involved 80% BSA. A positive Nikolsky sign was noted involving 21% BSA (Figure 5), in addition to oral and genital mucosal ulcers. She denied nausea, vomiting, fever, or diarrhea. Chimerism studies were negative. Trimethoprim-sulfamethoxazole was discontinued, and she was transferred to a burn unit. Biopsies showed full-thickness epidermal necrosis. A diagnosis of TEN with a SCORTEN of 4 in the setting of acute GVHD was favored; grade 4 acute GVHD could not be excluded. Steroids were discontinued. Because laboratory studies indicated IgA deficiency, IVIG was not considered as a systemic option for therapy. The patient received 1 dose of infliximab (5 mg/kg). Cyclophosphamide 1600 mg weekly was added for GVHD therapy. The wounds progressively healed, and 2 weeks into her admission she was noted to have only 3% BSA with denuded skin. The patient was transferred to the cancer treatment center for further management of the malignancy. Unfortunately, after 2 months she died due to ischemic colitis that was confirmed on autopsy.
Graft-vs-host disease and TEN are rare, life-threatening complications seen in patients with allogeneic HSCT.2 Graft-vs-host disease and TEN share clinicopathologic characteristics and effector immune mechanisms, largely the substantial role of T-cell activation and tissue destruction, which occur through mediators such as TNF-α.6-8
Given the sparse lymphocytic infiltrate, keratinocyte death in TEN is thought to result from soluble molecules, including TNF-α and TNF-related apoptosis-inducing ligand.9 Tumor necrosis factor α has been identified in blister fluid, biopsy specimens, and serum of patients with TEN. Tumor necrosis factor α increases the expression of keratinocyte-inducible nitric oxide synthase, which upregulates keratinocyte Fas ligand expression and subsequent Fas- and caspase-8–mediated keratinocyte cell death.10
Acute GVHD results from donor lymphocyte activation after infusion into damaged recipient tissues that previously have been radiated or chemoablated. Mismatches in histocompatibility complexes between donor cells and recipient tissue antigens serve as the initial trigger for immune activation. Activation of antigen-presenting cells followed by activation, proliferation, differentiation, and migration of donor T cells ultimately results in destruction of the target tissue.11 Immune mediators, such as TNF-α and lymphotoxin α (another member of the TNF superfamily), play a nonredundant role in the pathogenesis of GVHD.12
Current treatment strategies for severe acute GVHD and TEN differ. In North America, high-dose IVIG frequently is used as first-line systemic therapy, while high-dose systemic corticosteroids rarely are used.13 Studies have demonstrated successful use of anti–TNF-α drugs for the treatment of TEN.3,4 Moreover, etanercept has shown to effectively inhibit lymphotoxin α.14 Similarly, TNF inhibition in the management of steroid-refractory acute GVHD has been successful.1 These studies coupled with the underlying immune mechanisms that both diseases share encouraged initiating a trial of anti–TNF-α therapy in our patients.
Patient 1 merits further discussion because he was both a 100% donor chimera as well as a carrier of an human leukocyte antigen susceptibility candidate allele to TMP-SMX. Historical features of his presentation are consistent with either steroid-refractory GVHD or TEN superimposed on acute GVHD. His initial presentation of the more typical macular exanthem of cutaneous acute GVHD was both biopsy proven and supported by clinical improvement with steroid therapy, which was later followed by a robust blistering mucocutaneous presentation approximately 3 weeks after the administration of TMP-SMX and 1 week after initiating voriconazole that improved with IVIG and etanercept.
It is difficult to determine if TEN represents a continuum or result of the underlying drivers of acute GVHD vs a drug reaction. Although there is insufficient evidence to establish a clear-cut diagnosis of TEN, these cases illustrate the need for better diagnostic techniques to allow differentiation between TEN and grade 4 acute GVHD, and in the context of uncertainty, TNF-α inhibition poses a viable therapeutic strategy for these 2 often lethal conditions. Our cases do unequivocally indicate the benefit of this therapeutic modality, add to the current body of literature supporting the use of TNF-α inhibitors in patients such as ours without an official TEN diagnosis, and may guide future investigative efforts.
- Couriel DR, Saliba R, de Lima M, et al. A phase III study of infliximab and corticosteroids for the initial treatment of acute graft-versus-host disease. Biol Blood Marrow Transplant. 2009;15:1555-1562.
- Jeanmonod P, Hubbuch M, Grünhage F, et al. Graft-versus-host disease or toxic epidermal necrolysis: diagnostic dilemma after liver transplantation. Transpl Infect Dis. 2012;14:422-426.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Scott-Lang V, Tidman M, McKay D. Toxic epidermal necrolysis in a child successfully treated with infliximab. Pediatr Dermatol. 2014;31:532-534.
- Kingpin T, Mahasirimongkol S, Konyoung P, et al. Candidate HLA genes for prediction of co-trimoxazole-induced severe cutaneous reactions. Pharmacogenet Genomics. 2015;25:402-411.
- Correia O, Delgado L, Barbosa IL, et al. Increased interleukin 10, tumor necrosis factor alpha, and interleukin 6 levels in blister fluid of toxic epidermal necrolysis. J Am Acad Dermatol. 2002;47:58-62.
- French LE, Tschopp J. Fas-mediated cell death in toxic epidermal necrolysis and graft-versus-host disease: potential for therapeutic inhibition. Schweiz Med Wochenschr. 2000;130:1656-1661.
- Downey A, Jackson C, Harun N, et al. Toxic epidermal necrolysis: review of pathogenesis and management. J Am Acad Dermatol. 2012;66:995-1003.
- de Araujo E, Dessirier V, Laprée G, et al. Death ligand TRAIL, secreted by CD1a+ and CD14+ cells in blister fluids, is involved in killing keratinocytes in toxic epidermal necrolysis. Exp Dermatol. 2011;20:107-112.
- Viard-Leveugle I, Gaide O, Jankovic D, et al. TNF-α and IFN-γ are potential inducers of Fas-mediated keratinocyte apoptosis through activation of inducible nitric oxide synthase in toxic epidermal necrolysis. J Invest Dermatol. 2013;133:489-498.
- Choi SW, Levine JE, Ferrara JL. Pathogenesis and management of graft-versus-host disease. Immunol Allergy Clin North Am. 2010;30:75-101.
- Markey KA, Burman AC, Banovic T, et al. Soluble lymphotoxin is an important effector molecule in GVHD and GVL. Blood. 2010;115:122-132.
- Dodiuk-Gad RP, Olteanu C, Jeschke MG, et al. Treatment of toxic epidermal necrolysis in North America. J Am Acad Dermatol. 2015;73:876-877.
- Tracey D, Klareskog L, Sasso EH, et al. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244-279.
- Couriel DR, Saliba R, de Lima M, et al. A phase III study of infliximab and corticosteroids for the initial treatment of acute graft-versus-host disease. Biol Blood Marrow Transplant. 2009;15:1555-1562.
- Jeanmonod P, Hubbuch M, Grünhage F, et al. Graft-versus-host disease or toxic epidermal necrolysis: diagnostic dilemma after liver transplantation. Transpl Infect Dis. 2012;14:422-426.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Scott-Lang V, Tidman M, McKay D. Toxic epidermal necrolysis in a child successfully treated with infliximab. Pediatr Dermatol. 2014;31:532-534.
- Kingpin T, Mahasirimongkol S, Konyoung P, et al. Candidate HLA genes for prediction of co-trimoxazole-induced severe cutaneous reactions. Pharmacogenet Genomics. 2015;25:402-411.
- Correia O, Delgado L, Barbosa IL, et al. Increased interleukin 10, tumor necrosis factor alpha, and interleukin 6 levels in blister fluid of toxic epidermal necrolysis. J Am Acad Dermatol. 2002;47:58-62.
- French LE, Tschopp J. Fas-mediated cell death in toxic epidermal necrolysis and graft-versus-host disease: potential for therapeutic inhibition. Schweiz Med Wochenschr. 2000;130:1656-1661.
- Downey A, Jackson C, Harun N, et al. Toxic epidermal necrolysis: review of pathogenesis and management. J Am Acad Dermatol. 2012;66:995-1003.
- de Araujo E, Dessirier V, Laprée G, et al. Death ligand TRAIL, secreted by CD1a+ and CD14+ cells in blister fluids, is involved in killing keratinocytes in toxic epidermal necrolysis. Exp Dermatol. 2011;20:107-112.
- Viard-Leveugle I, Gaide O, Jankovic D, et al. TNF-α and IFN-γ are potential inducers of Fas-mediated keratinocyte apoptosis through activation of inducible nitric oxide synthase in toxic epidermal necrolysis. J Invest Dermatol. 2013;133:489-498.
- Choi SW, Levine JE, Ferrara JL. Pathogenesis and management of graft-versus-host disease. Immunol Allergy Clin North Am. 2010;30:75-101.
- Markey KA, Burman AC, Banovic T, et al. Soluble lymphotoxin is an important effector molecule in GVHD and GVL. Blood. 2010;115:122-132.
- Dodiuk-Gad RP, Olteanu C, Jeschke MG, et al. Treatment of toxic epidermal necrolysis in North America. J Am Acad Dermatol. 2015;73:876-877.
- Tracey D, Klareskog L, Sasso EH, et al. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244-279.
Practice Points
- Graft-vs-host disease (GVHD) and toxic epidermal necrolysis (TEN) are rare life-threatening complications seen in patients with allogeneic hematopoietic stem cell transplantation.
- Although mild acute GVHD easily is distinguished from TEN, severe acute GVHD and TEN share overlapping features and present a diagnostic challenge.
- Therapeutic decisions and associated outcomes hinge on accurate diagnosis, as high-dose systemic corticosteroids have been associated with higher mortality rates in TEN.

Light Brown and Pink Macule on the Upper Arm
The Diagnosis: Desmoplastic Spitz Nevus
Desmoplastic Spitz nevus is a rare variant of Spitz nevus that commonly presents as a red to brown papule on the head, neck, or extremities. It is pertinent to review the histologic features of this neoplasm, as it can be confused with other more sinister entities such as spitzoid melanoma. Histologically, there is a dermal infiltrate of melanocytes containing eosinophilic cytoplasm and vesicular nuclei. Junctional involvement is rare, and there should be no pagetoid spread.1 This entity features abundant stromal fibrosis formed by dense collagen bundles, low cellular density, and polygonal-shaped melanocytes, which helps to differentiate it from melanoma.2,3 In a retrospective study comparing the characteristics of desmoplastic Spitz nevi with desmoplastic melanoma, desmoplastic Spitz nevi histologically were more symmetric and circumscribed with greater melanocytic maturation and adnexal structure involvement.3 Although this entity demonstrates maturation from the superficial to the deep dermis, it also may feature deep dermal vascular proliferation.4 S-100 and SRY-related HMG box 10, SOX-10, are noted to be positive in desmoplastic Spitz nevi, which can help to differentiate it from nonmelanocytic entities (Figure 1).
 positivity (original magnification ×40).")
Although spitzoid lesions can be ambiguous and difficult even for experts to classify, spitzoid melanoma tends to have a high Breslow thickness, high cell density, marked atypia, and an increased nucleus to cytoplasm ratio.5 Additionally, desmoplastic melanoma was found to more often display “melanocytic junctional nests associated with discohesive cells, variations in size and shape of the nests, lentiginous melanocytic proliferation, actinic elastosis, pagetoid spread, dermal mitosis, perineural involvement and brisk inflammatory infiltrate.”3 Given the challenge of histologically separating desmoplastic Spitz nevi from melanoma, immunostaining can be useful. For example, Hilliard et al6 used a p16 antibody to differentiate desmoplastic Spitz nevi from desmoplastic melanoma, finding that most desmoplastic melanomas (81.8%; n=11) were negative for p16, whereas all desmoplastic Spitz nevi were at least moderately positive. However, another study re-evaluated the utility of p16 in desmoplastic melanoma and found that 72.7% (16/22) were at least focally reactive for the immunostain.7 Thus, caution must be exercised when using p16.
PReferentially expressed Antigen in MElanoma (PRAME) is a newer nuclear immunohistochemical marker that tends to be positive in melanomas and negative in nevi. Desmoplastic Spitz nevi would be expected to be negative for PRAME, while desmoplastic melanoma may be positive; however, this marker seems to be less effective in desmoplastic melanoma than in most other subtypes of the malignancy. In one study, only 35% (n=20) of desmoplastic melanomas were positive for PRAME.8 Likewise, another study showed that some benign Spitz nevi may diffusely express PRAME.9 As such, PRAME should be used prudently.
For cases in which immunohistochemistry is equivocal, molecular testing may aid in differentiating Spitz nevi from melanoma. For example, comparative genomic hybridization has revealed an increased copy number of chromosome 11p in approximately 20% of Spitz nevi cases10; this finding is not seen in melanoma. Mutation analyses of HRas proto-oncogene, GTPase, HRAS; B-Raf proto-oncogene, serine/threonine kinase, BRAF; and NRAS proto-oncogene, GTPase, NRAS, also have shown some promise in distinguishing spitzoid lesions from melanoma, but these analyses may be oversimplified.11 Fluorescence in situ hybridization (FISH) is another diagnostic modality that has been studied to differentiate benign nevi from melanoma. One study challenged the utility of FISH, reporting 7 of 15 desmoplastic melanomas tested positive compared to 0 of 15 sclerotic melanocytic nevi.12 Thus, negative FISH cannot reliably rule out melanoma. Ultimately, a combination of immunostains along with FISH or another genetic study would prove to be most effective in ruling out melanoma in difficult cases. Even then, a dermatopathologist may be faced with a degree of uncertainty.
Cellular blue nevi predominantly affect adults younger than 40 years and commonly are seen on the buttocks.13 This benign neoplasm demonstrates areas that are distinctly sclerotic as well as those that are cellular in nature.14 This entity demonstrates a well-circumscribed dermal growth pattern with 2 main populations of cells. The sclerotic portion of the cellular blue nevus mimics that of the blue nevus in that it is noted superficially with irregular margins. The cellular aspect of the nevus features spindle cells contained within well-circumscribed nodules (Figure 2). Stromal melanophages are not uncommon, and some can be observed adjacent to nerve fibers. Although this blue nevus variant displays features of the common blue nevus, its melanocytes track along adnexal and neurovascular structures similar to the deep penetrating nevus and the desmoplastic Spitz nevus. However, these melanocytes are variable in morphology and can appear on a spectrum spanning from pale and lightly pigmented to clear.15

The breast is the most common site of origin of tumor metastasis to the skin. These cutaneous metastases can vary in both their clinical and histological presentations. For example, cutaneous metastatic breast adenocarcinoma often can present clinically as pink-violaceous papules and plaques on the breast or on other parts of the body. Histologically, it can demonstrate a varying degree of patterns such as collagen infiltration by single cells, cords, tubules, and sheets of atypical cells (Figure 3) that can be observed together in areas of mucin or can form glandular structures.16 Metastatic breast carcinoma is noted to be positive for gross cystic disease fluid protein-15, estrogen receptor, and cytokeratin 7, which can help differentiate this entity from other tumors of glandular origin.16 Although rare, primary melanoma of the breast has been reported in the literature.17,18 These malignant melanocytic lesions easily could be differentiated from other breast tumors such as adenocarcinoma using immunohistochemical staining patterns.

Deep penetrating nevi most often are observed clinically as blue, brown, or black papules or nodules on the head or neck.19 Histologically, this lesion features a wedge-shaped infiltrate of deep dermal melanocytes with oval nuclei. It commonly extends to the reticular dermis or further into the subcutis (Figure 4).20,21 This neoplasm frequently tracks along adnexal and neurovascular structures, resulting in a plexiform appearance.22 The adnexal involvement of deep penetrating nevi is a shared feature with desmoplastic Spitz nevi. The presence of any number of melanophages is characteristic of this lesion.23 Lastly, there is a well-documented association between β-catenin mutations and deep penetrating nevi.24 Multicentric reticulohistiocytosis (MRH) is a rare form of non-Langerhans cell histiocytosis that has the pathognomonic clinical finding of pink-red papules (coral beading) with a predilection for acral surfaces. Histology of affected skin reveals a dermal infiltrate of ground glass as well as eosinophilic histiocytes that most often stain positive for CD68 and human alveolar macrophage 56 but negative for S-100 and CD1a (Figure 5).25 Although MRH is rare, negative staining for S-100 could serve as a useful diagnostic clue to differentiate it from other entities that are positive for S-100, such as the desmoplastic Spitz nevus. Arthritis mutilans is a potential complication of MRH, but a reported association with an underlying malignancy is seen in approximately 25% of cases.26 Thus, the cutaneous, rheumatologic, and oncologic implications of this disease help to distinguish it from other differential diagnoses that may be considered.


- Luzar B, Bastian BC, North JP, et al. Melanocytic nevi. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin. 5th ed. Elsevier; 2020:1275-1280.
- Busam KJ, Gerami P. Spitz nevi. In: Busam KJ, Gerami P, Scolyer RA, eds. Pathology of Melanocytic Tumors. Elsevier; 2019:37-60.
- Nojavan H, Cribier B, Mehregan DR. Desmoplastic Spitz nevus: a histopathological review and comparison with desmoplastic melanoma [in French]. Ann Dermatol Venereol. 2009;136:689-695.
- Tomizawa K. Desmoplastic Spitz nevus showing vascular proliferation more prominently in the deep portion. Am J Dermatopathol. 2002;24:184-185.
- Requena C, Botella R, Nagore E, et al. Characteristics of spitzoid melanoma and clues for differential diagnosis with Spitz nevus. Am J Dermatopathol. 2012;34:478-486.
- Hilliard NJ, Krahl D, Sellheyer K. p16 expression differentiates between desmoplastic Spitz nevus and desmoplastic melanoma. J Cutan Pathol. 2009;36:753-759.
- Blokhin E, Pulitzer M, Busam KJ. Immunohistochemical expression of p16 in desmoplastic melanoma. J Cutan Pathol. 2013;40:796-800.
- Lezcano C, Jungbluth AA, Nehal KS, et al. PRAME expression in melanocytic tumors. Am J Surg Pathol. 2018;42:1456-1465.
- Raghavan SS, Wang JY, Kwok S, et al. PRAME expression in melanocytic proliferations with intermediate histopathologic or spitzoid features. J Cutan Pathol. 2020;47:1123-1131.
- Bauer J, Bastian BC. DNA copy number changes in the diagnosis of melanocytic tumors [in German]. Pathologe. 2007;28:464-473.
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part I. background and diagnoses. J Am Acad Dermatol. 2011;65:1073-1084.
- Gerami P, Beilfuss B, Haghighat Z, et al. Fluorescence in situ hybridization as an ancillary method for the distinction of desmoplastic melanomas from sclerosing melanocytic nevi. J Cutan Pathol. 2011;38:329-334.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017; 37:401-415.
- Rodriguez HA, Ackerman LV. Cellular blue nevus. clinicopathologic study of forty-five cases. Cancer. 1968;21:393-405.
- Phadke PA, Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2011;31:345-358.
- Ko CJ. Metastatic tumors and simulators. In: Elston DM, Ferringer T, eds. Dermatopathology. 3rd ed. Elsevier Limited; 2019:496-504.
- Drueppel D, Schultheis B, Solass W, et al. Primary malignant melanoma of the breast: case report and review of the literature. Anticancer Res. 2015;35:1709-1713.
- Kurul S, Tas¸ F, Büyükbabani N, et al. Different manifestations of malignant melanoma in the breast: a report of 12 cases and a review of the literature. Jpn J Clin Oncol. 2005;35:202-206.
- Strazzula L, Senna MM, Yasuda M, et al. The deep penetrating nevus. J Am Acad Dermatol. 2014;71:1234-1240.
- Mehregan DA, Mehregan AH. Deep penetrating nevus. Arch Dermatol. 1993;129:328-331.
- Robson A, Morley-Quante M, Hempel H, et al. Deep penetrating naevus: clinicopathological study of 31 cases with further delineation of histological features allowing distinction from other pigmented benign melanocytic lesions and melanoma. Histopathology. 2003;43:529-537.
- Luzar B, Calonje E. Deep penetrating nevus: a review. Arch Pathol Lab Med. 2011;135:321-326.
- Cooper PH. Deep penetrating (plexiform spindle cell) nevus. a frequent participant in combined nevus. J Cutan Pathol. 1992;19:172-180.
- de la Fouchardière A, Caillot C, Jacquemus J, et al. β-Catenin nuclear expression discriminates deep penetrating nevi from other cutaneous melanocytic tumors. Virchows Arch. 2019;474:539-550.
- Gorman JD, Danning C, Schumacher HR, et al. Multicentric reticulohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum. 2000;43:930-938.
- Selmi C, Greenspan A, Huntley A, et al. Multicentric reticulohistiocytosis: a critical review. Curr Rheumatol Rep. 2015;17:511.
The Diagnosis: Desmoplastic Spitz Nevus
Desmoplastic Spitz nevus is a rare variant of Spitz nevus that commonly presents as a red to brown papule on the head, neck, or extremities. It is pertinent to review the histologic features of this neoplasm, as it can be confused with other more sinister entities such as spitzoid melanoma. Histologically, there is a dermal infiltrate of melanocytes containing eosinophilic cytoplasm and vesicular nuclei. Junctional involvement is rare, and there should be no pagetoid spread.1 This entity features abundant stromal fibrosis formed by dense collagen bundles, low cellular density, and polygonal-shaped melanocytes, which helps to differentiate it from melanoma.2,3 In a retrospective study comparing the characteristics of desmoplastic Spitz nevi with desmoplastic melanoma, desmoplastic Spitz nevi histologically were more symmetric and circumscribed with greater melanocytic maturation and adnexal structure involvement.3 Although this entity demonstrates maturation from the superficial to the deep dermis, it also may feature deep dermal vascular proliferation.4 S-100 and SRY-related HMG box 10, SOX-10, are noted to be positive in desmoplastic Spitz nevi, which can help to differentiate it from nonmelanocytic entities (Figure 1).
Although spitzoid lesions can be ambiguous and difficult even for experts to classify, spitzoid melanoma tends to have a high Breslow thickness, high cell density, marked atypia, and an increased nucleus to cytoplasm ratio.5 Additionally, desmoplastic melanoma was found to more often display “melanocytic junctional nests associated with discohesive cells, variations in size and shape of the nests, lentiginous melanocytic proliferation, actinic elastosis, pagetoid spread, dermal mitosis, perineural involvement and brisk inflammatory infiltrate.”3 Given the challenge of histologically separating desmoplastic Spitz nevi from melanoma, immunostaining can be useful. For example, Hilliard et al6 used a p16 antibody to differentiate desmoplastic Spitz nevi from desmoplastic melanoma, finding that most desmoplastic melanomas (81.8%; n=11) were negative for p16, whereas all desmoplastic Spitz nevi were at least moderately positive. However, another study re-evaluated the utility of p16 in desmoplastic melanoma and found that 72.7% (16/22) were at least focally reactive for the immunostain.7 Thus, caution must be exercised when using p16.
PReferentially expressed Antigen in MElanoma (PRAME) is a newer nuclear immunohistochemical marker that tends to be positive in melanomas and negative in nevi. Desmoplastic Spitz nevi would be expected to be negative for PRAME, while desmoplastic melanoma may be positive; however, this marker seems to be less effective in desmoplastic melanoma than in most other subtypes of the malignancy. In one study, only 35% (n=20) of desmoplastic melanomas were positive for PRAME.8 Likewise, another study showed that some benign Spitz nevi may diffusely express PRAME.9 As such, PRAME should be used prudently.
For cases in which immunohistochemistry is equivocal, molecular testing may aid in differentiating Spitz nevi from melanoma. For example, comparative genomic hybridization has revealed an increased copy number of chromosome 11p in approximately 20% of Spitz nevi cases10; this finding is not seen in melanoma. Mutation analyses of HRas proto-oncogene, GTPase, HRAS; B-Raf proto-oncogene, serine/threonine kinase, BRAF; and NRAS proto-oncogene, GTPase, NRAS, also have shown some promise in distinguishing spitzoid lesions from melanoma, but these analyses may be oversimplified.11 Fluorescence in situ hybridization (FISH) is another diagnostic modality that has been studied to differentiate benign nevi from melanoma. One study challenged the utility of FISH, reporting 7 of 15 desmoplastic melanomas tested positive compared to 0 of 15 sclerotic melanocytic nevi.12 Thus, negative FISH cannot reliably rule out melanoma. Ultimately, a combination of immunostains along with FISH or another genetic study would prove to be most effective in ruling out melanoma in difficult cases. Even then, a dermatopathologist may be faced with a degree of uncertainty.
Cellular blue nevi predominantly affect adults younger than 40 years and commonly are seen on the buttocks.13 This benign neoplasm demonstrates areas that are distinctly sclerotic as well as those that are cellular in nature.14 This entity demonstrates a well-circumscribed dermal growth pattern with 2 main populations of cells. The sclerotic portion of the cellular blue nevus mimics that of the blue nevus in that it is noted superficially with irregular margins. The cellular aspect of the nevus features spindle cells contained within well-circumscribed nodules (Figure 2). Stromal melanophages are not uncommon, and some can be observed adjacent to nerve fibers. Although this blue nevus variant displays features of the common blue nevus, its melanocytes track along adnexal and neurovascular structures similar to the deep penetrating nevus and the desmoplastic Spitz nevus. However, these melanocytes are variable in morphology and can appear on a spectrum spanning from pale and lightly pigmented to clear.15
The breast is the most common site of origin of tumor metastasis to the skin. These cutaneous metastases can vary in both their clinical and histological presentations. For example, cutaneous metastatic breast adenocarcinoma often can present clinically as pink-violaceous papules and plaques on the breast or on other parts of the body. Histologically, it can demonstrate a varying degree of patterns such as collagen infiltration by single cells, cords, tubules, and sheets of atypical cells (Figure 3) that can be observed together in areas of mucin or can form glandular structures.16 Metastatic breast carcinoma is noted to be positive for gross cystic disease fluid protein-15, estrogen receptor, and cytokeratin 7, which can help differentiate this entity from other tumors of glandular origin.16 Although rare, primary melanoma of the breast has been reported in the literature.17,18 These malignant melanocytic lesions easily could be differentiated from other breast tumors such as adenocarcinoma using immunohistochemical staining patterns.
Deep penetrating nevi most often are observed clinically as blue, brown, or black papules or nodules on the head or neck.19 Histologically, this lesion features a wedge-shaped infiltrate of deep dermal melanocytes with oval nuclei. It commonly extends to the reticular dermis or further into the subcutis (Figure 4).20,21 This neoplasm frequently tracks along adnexal and neurovascular structures, resulting in a plexiform appearance.22 The adnexal involvement of deep penetrating nevi is a shared feature with desmoplastic Spitz nevi. The presence of any number of melanophages is characteristic of this lesion.23 Lastly, there is a well-documented association between β-catenin mutations and deep penetrating nevi.24 Multicentric reticulohistiocytosis (MRH) is a rare form of non-Langerhans cell histiocytosis that has the pathognomonic clinical finding of pink-red papules (coral beading) with a predilection for acral surfaces. Histology of affected skin reveals a dermal infiltrate of ground glass as well as eosinophilic histiocytes that most often stain positive for CD68 and human alveolar macrophage 56 but negative for S-100 and CD1a (Figure 5).25 Although MRH is rare, negative staining for S-100 could serve as a useful diagnostic clue to differentiate it from other entities that are positive for S-100, such as the desmoplastic Spitz nevus. Arthritis mutilans is a potential complication of MRH, but a reported association with an underlying malignancy is seen in approximately 25% of cases.26 Thus, the cutaneous, rheumatologic, and oncologic implications of this disease help to distinguish it from other differential diagnoses that may be considered.
The Diagnosis: Desmoplastic Spitz Nevus
Desmoplastic Spitz nevus is a rare variant of Spitz nevus that commonly presents as a red to brown papule on the head, neck, or extremities. It is pertinent to review the histologic features of this neoplasm, as it can be confused with other more sinister entities such as spitzoid melanoma. Histologically, there is a dermal infiltrate of melanocytes containing eosinophilic cytoplasm and vesicular nuclei. Junctional involvement is rare, and there should be no pagetoid spread.1 This entity features abundant stromal fibrosis formed by dense collagen bundles, low cellular density, and polygonal-shaped melanocytes, which helps to differentiate it from melanoma.2,3 In a retrospective study comparing the characteristics of desmoplastic Spitz nevi with desmoplastic melanoma, desmoplastic Spitz nevi histologically were more symmetric and circumscribed with greater melanocytic maturation and adnexal structure involvement.3 Although this entity demonstrates maturation from the superficial to the deep dermis, it also may feature deep dermal vascular proliferation.4 S-100 and SRY-related HMG box 10, SOX-10, are noted to be positive in desmoplastic Spitz nevi, which can help to differentiate it from nonmelanocytic entities (Figure 1).
Although spitzoid lesions can be ambiguous and difficult even for experts to classify, spitzoid melanoma tends to have a high Breslow thickness, high cell density, marked atypia, and an increased nucleus to cytoplasm ratio.5 Additionally, desmoplastic melanoma was found to more often display “melanocytic junctional nests associated with discohesive cells, variations in size and shape of the nests, lentiginous melanocytic proliferation, actinic elastosis, pagetoid spread, dermal mitosis, perineural involvement and brisk inflammatory infiltrate.”3 Given the challenge of histologically separating desmoplastic Spitz nevi from melanoma, immunostaining can be useful. For example, Hilliard et al6 used a p16 antibody to differentiate desmoplastic Spitz nevi from desmoplastic melanoma, finding that most desmoplastic melanomas (81.8%; n=11) were negative for p16, whereas all desmoplastic Spitz nevi were at least moderately positive. However, another study re-evaluated the utility of p16 in desmoplastic melanoma and found that 72.7% (16/22) were at least focally reactive for the immunostain.7 Thus, caution must be exercised when using p16.
PReferentially expressed Antigen in MElanoma (PRAME) is a newer nuclear immunohistochemical marker that tends to be positive in melanomas and negative in nevi. Desmoplastic Spitz nevi would be expected to be negative for PRAME, while desmoplastic melanoma may be positive; however, this marker seems to be less effective in desmoplastic melanoma than in most other subtypes of the malignancy. In one study, only 35% (n=20) of desmoplastic melanomas were positive for PRAME.8 Likewise, another study showed that some benign Spitz nevi may diffusely express PRAME.9 As such, PRAME should be used prudently.
For cases in which immunohistochemistry is equivocal, molecular testing may aid in differentiating Spitz nevi from melanoma. For example, comparative genomic hybridization has revealed an increased copy number of chromosome 11p in approximately 20% of Spitz nevi cases10; this finding is not seen in melanoma. Mutation analyses of HRas proto-oncogene, GTPase, HRAS; B-Raf proto-oncogene, serine/threonine kinase, BRAF; and NRAS proto-oncogene, GTPase, NRAS, also have shown some promise in distinguishing spitzoid lesions from melanoma, but these analyses may be oversimplified.11 Fluorescence in situ hybridization (FISH) is another diagnostic modality that has been studied to differentiate benign nevi from melanoma. One study challenged the utility of FISH, reporting 7 of 15 desmoplastic melanomas tested positive compared to 0 of 15 sclerotic melanocytic nevi.12 Thus, negative FISH cannot reliably rule out melanoma. Ultimately, a combination of immunostains along with FISH or another genetic study would prove to be most effective in ruling out melanoma in difficult cases. Even then, a dermatopathologist may be faced with a degree of uncertainty.
Cellular blue nevi predominantly affect adults younger than 40 years and commonly are seen on the buttocks.13 This benign neoplasm demonstrates areas that are distinctly sclerotic as well as those that are cellular in nature.14 This entity demonstrates a well-circumscribed dermal growth pattern with 2 main populations of cells. The sclerotic portion of the cellular blue nevus mimics that of the blue nevus in that it is noted superficially with irregular margins. The cellular aspect of the nevus features spindle cells contained within well-circumscribed nodules (Figure 2). Stromal melanophages are not uncommon, and some can be observed adjacent to nerve fibers. Although this blue nevus variant displays features of the common blue nevus, its melanocytes track along adnexal and neurovascular structures similar to the deep penetrating nevus and the desmoplastic Spitz nevus. However, these melanocytes are variable in morphology and can appear on a spectrum spanning from pale and lightly pigmented to clear.15
The breast is the most common site of origin of tumor metastasis to the skin. These cutaneous metastases can vary in both their clinical and histological presentations. For example, cutaneous metastatic breast adenocarcinoma often can present clinically as pink-violaceous papules and plaques on the breast or on other parts of the body. Histologically, it can demonstrate a varying degree of patterns such as collagen infiltration by single cells, cords, tubules, and sheets of atypical cells (Figure 3) that can be observed together in areas of mucin or can form glandular structures.16 Metastatic breast carcinoma is noted to be positive for gross cystic disease fluid protein-15, estrogen receptor, and cytokeratin 7, which can help differentiate this entity from other tumors of glandular origin.16 Although rare, primary melanoma of the breast has been reported in the literature.17,18 These malignant melanocytic lesions easily could be differentiated from other breast tumors such as adenocarcinoma using immunohistochemical staining patterns.
Deep penetrating nevi most often are observed clinically as blue, brown, or black papules or nodules on the head or neck.19 Histologically, this lesion features a wedge-shaped infiltrate of deep dermal melanocytes with oval nuclei. It commonly extends to the reticular dermis or further into the subcutis (Figure 4).20,21 This neoplasm frequently tracks along adnexal and neurovascular structures, resulting in a plexiform appearance.22 The adnexal involvement of deep penetrating nevi is a shared feature with desmoplastic Spitz nevi. The presence of any number of melanophages is characteristic of this lesion.23 Lastly, there is a well-documented association between β-catenin mutations and deep penetrating nevi.24 Multicentric reticulohistiocytosis (MRH) is a rare form of non-Langerhans cell histiocytosis that has the pathognomonic clinical finding of pink-red papules (coral beading) with a predilection for acral surfaces. Histology of affected skin reveals a dermal infiltrate of ground glass as well as eosinophilic histiocytes that most often stain positive for CD68 and human alveolar macrophage 56 but negative for S-100 and CD1a (Figure 5).25 Although MRH is rare, negative staining for S-100 could serve as a useful diagnostic clue to differentiate it from other entities that are positive for S-100, such as the desmoplastic Spitz nevus. Arthritis mutilans is a potential complication of MRH, but a reported association with an underlying malignancy is seen in approximately 25% of cases.26 Thus, the cutaneous, rheumatologic, and oncologic implications of this disease help to distinguish it from other differential diagnoses that may be considered.
- Luzar B, Bastian BC, North JP, et al. Melanocytic nevi. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin. 5th ed. Elsevier; 2020:1275-1280.
- Busam KJ, Gerami P. Spitz nevi. In: Busam KJ, Gerami P, Scolyer RA, eds. Pathology of Melanocytic Tumors. Elsevier; 2019:37-60.
- Nojavan H, Cribier B, Mehregan DR. Desmoplastic Spitz nevus: a histopathological review and comparison with desmoplastic melanoma [in French]. Ann Dermatol Venereol. 2009;136:689-695.
- Tomizawa K. Desmoplastic Spitz nevus showing vascular proliferation more prominently in the deep portion. Am J Dermatopathol. 2002;24:184-185.
- Requena C, Botella R, Nagore E, et al. Characteristics of spitzoid melanoma and clues for differential diagnosis with Spitz nevus. Am J Dermatopathol. 2012;34:478-486.
- Hilliard NJ, Krahl D, Sellheyer K. p16 expression differentiates between desmoplastic Spitz nevus and desmoplastic melanoma. J Cutan Pathol. 2009;36:753-759.
- Blokhin E, Pulitzer M, Busam KJ. Immunohistochemical expression of p16 in desmoplastic melanoma. J Cutan Pathol. 2013;40:796-800.
- Lezcano C, Jungbluth AA, Nehal KS, et al. PRAME expression in melanocytic tumors. Am J Surg Pathol. 2018;42:1456-1465.
- Raghavan SS, Wang JY, Kwok S, et al. PRAME expression in melanocytic proliferations with intermediate histopathologic or spitzoid features. J Cutan Pathol. 2020;47:1123-1131.
- Bauer J, Bastian BC. DNA copy number changes in the diagnosis of melanocytic tumors [in German]. Pathologe. 2007;28:464-473.
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part I. background and diagnoses. J Am Acad Dermatol. 2011;65:1073-1084.
- Gerami P, Beilfuss B, Haghighat Z, et al. Fluorescence in situ hybridization as an ancillary method for the distinction of desmoplastic melanomas from sclerosing melanocytic nevi. J Cutan Pathol. 2011;38:329-334.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017; 37:401-415.
- Rodriguez HA, Ackerman LV. Cellular blue nevus. clinicopathologic study of forty-five cases. Cancer. 1968;21:393-405.
- Phadke PA, Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2011;31:345-358.
- Ko CJ. Metastatic tumors and simulators. In: Elston DM, Ferringer T, eds. Dermatopathology. 3rd ed. Elsevier Limited; 2019:496-504.
- Drueppel D, Schultheis B, Solass W, et al. Primary malignant melanoma of the breast: case report and review of the literature. Anticancer Res. 2015;35:1709-1713.
- Kurul S, Tas¸ F, Büyükbabani N, et al. Different manifestations of malignant melanoma in the breast: a report of 12 cases and a review of the literature. Jpn J Clin Oncol. 2005;35:202-206.
- Strazzula L, Senna MM, Yasuda M, et al. The deep penetrating nevus. J Am Acad Dermatol. 2014;71:1234-1240.
- Mehregan DA, Mehregan AH. Deep penetrating nevus. Arch Dermatol. 1993;129:328-331.
- Robson A, Morley-Quante M, Hempel H, et al. Deep penetrating naevus: clinicopathological study of 31 cases with further delineation of histological features allowing distinction from other pigmented benign melanocytic lesions and melanoma. Histopathology. 2003;43:529-537.
- Luzar B, Calonje E. Deep penetrating nevus: a review. Arch Pathol Lab Med. 2011;135:321-326.
- Cooper PH. Deep penetrating (plexiform spindle cell) nevus. a frequent participant in combined nevus. J Cutan Pathol. 1992;19:172-180.
- de la Fouchardière A, Caillot C, Jacquemus J, et al. β-Catenin nuclear expression discriminates deep penetrating nevi from other cutaneous melanocytic tumors. Virchows Arch. 2019;474:539-550.
- Gorman JD, Danning C, Schumacher HR, et al. Multicentric reticulohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum. 2000;43:930-938.
- Selmi C, Greenspan A, Huntley A, et al. Multicentric reticulohistiocytosis: a critical review. Curr Rheumatol Rep. 2015;17:511.
- Luzar B, Bastian BC, North JP, et al. Melanocytic nevi. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin. 5th ed. Elsevier; 2020:1275-1280.
- Busam KJ, Gerami P. Spitz nevi. In: Busam KJ, Gerami P, Scolyer RA, eds. Pathology of Melanocytic Tumors. Elsevier; 2019:37-60.
- Nojavan H, Cribier B, Mehregan DR. Desmoplastic Spitz nevus: a histopathological review and comparison with desmoplastic melanoma [in French]. Ann Dermatol Venereol. 2009;136:689-695.
- Tomizawa K. Desmoplastic Spitz nevus showing vascular proliferation more prominently in the deep portion. Am J Dermatopathol. 2002;24:184-185.
- Requena C, Botella R, Nagore E, et al. Characteristics of spitzoid melanoma and clues for differential diagnosis with Spitz nevus. Am J Dermatopathol. 2012;34:478-486.
- Hilliard NJ, Krahl D, Sellheyer K. p16 expression differentiates between desmoplastic Spitz nevus and desmoplastic melanoma. J Cutan Pathol. 2009;36:753-759.
- Blokhin E, Pulitzer M, Busam KJ. Immunohistochemical expression of p16 in desmoplastic melanoma. J Cutan Pathol. 2013;40:796-800.
- Lezcano C, Jungbluth AA, Nehal KS, et al. PRAME expression in melanocytic tumors. Am J Surg Pathol. 2018;42:1456-1465.
- Raghavan SS, Wang JY, Kwok S, et al. PRAME expression in melanocytic proliferations with intermediate histopathologic or spitzoid features. J Cutan Pathol. 2020;47:1123-1131.
- Bauer J, Bastian BC. DNA copy number changes in the diagnosis of melanocytic tumors [in German]. Pathologe. 2007;28:464-473.
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part I. background and diagnoses. J Am Acad Dermatol. 2011;65:1073-1084.
- Gerami P, Beilfuss B, Haghighat Z, et al. Fluorescence in situ hybridization as an ancillary method for the distinction of desmoplastic melanomas from sclerosing melanocytic nevi. J Cutan Pathol. 2011;38:329-334.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017; 37:401-415.
- Rodriguez HA, Ackerman LV. Cellular blue nevus. clinicopathologic study of forty-five cases. Cancer. 1968;21:393-405.
- Phadke PA, Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2011;31:345-358.
- Ko CJ. Metastatic tumors and simulators. In: Elston DM, Ferringer T, eds. Dermatopathology. 3rd ed. Elsevier Limited; 2019:496-504.
- Drueppel D, Schultheis B, Solass W, et al. Primary malignant melanoma of the breast: case report and review of the literature. Anticancer Res. 2015;35:1709-1713.
- Kurul S, Tas¸ F, Büyükbabani N, et al. Different manifestations of malignant melanoma in the breast: a report of 12 cases and a review of the literature. Jpn J Clin Oncol. 2005;35:202-206.
- Strazzula L, Senna MM, Yasuda M, et al. The deep penetrating nevus. J Am Acad Dermatol. 2014;71:1234-1240.
- Mehregan DA, Mehregan AH. Deep penetrating nevus. Arch Dermatol. 1993;129:328-331.
- Robson A, Morley-Quante M, Hempel H, et al. Deep penetrating naevus: clinicopathological study of 31 cases with further delineation of histological features allowing distinction from other pigmented benign melanocytic lesions and melanoma. Histopathology. 2003;43:529-537.
- Luzar B, Calonje E. Deep penetrating nevus: a review. Arch Pathol Lab Med. 2011;135:321-326.
- Cooper PH. Deep penetrating (plexiform spindle cell) nevus. a frequent participant in combined nevus. J Cutan Pathol. 1992;19:172-180.
- de la Fouchardière A, Caillot C, Jacquemus J, et al. β-Catenin nuclear expression discriminates deep penetrating nevi from other cutaneous melanocytic tumors. Virchows Arch. 2019;474:539-550.
- Gorman JD, Danning C, Schumacher HR, et al. Multicentric reticulohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum. 2000;43:930-938.
- Selmi C, Greenspan A, Huntley A, et al. Multicentric reticulohistiocytosis: a critical review. Curr Rheumatol Rep. 2015;17:511.
A 37-year-old woman with a history of fibrocystic breast disease and a family history of breast cancer presented with a light brown macule on the right upper arm of 10 years’ duration. The patient first noticed this macule 10 years prior; however, within the last 4 months she noticed a small amount of homogenous darkening and occasional pruritus. Physical examination revealed a 4.0-mm, light brown and pink macule on the right upper arm. Dermoscopy showed a homogenous pigment network with reticular lines and branched streaks centrally. No crystalline structures, milky red globules, or pseudopods were appreciated. A tangential shave biopsy was obtained and submitted for hematoxylin and eosin staining.
.")
.")
Disseminated Erythematous-Violet Edematous Plaques and Necrotic Nodules
The Diagnosis: Histiocytoid Sweet Syndrome
The patient was admitted for clinical study and treatment monitoring. During the first 72 hours of admittance, the lesions and general malaise further developed along with C-reactive protein elevation (126 mg/L). Administration of intravenous prednisone at a dosage of 1 mg/kg daily was accompanied by substantial improvement after 1 week of treatment, with subsequent follow-up and outpatient monitoring. An underlying neoplasia was ruled out after review of medical history, physical examination, complete blood cell count, chest radiography, abdominal ultrasonography, colonoscopy, and bone marrow aspiration.

A 4-mm skin biopsy was performed from a lesion on the neck (Figure 1). Histology revealed a dermis with prominent edema alongside superficial, deep, and periadnexal perivascular inflammatory infiltrates, as well as predominant lymphocytes and cells with a histiocytoid profile (Figure 2). These findings were accompanied by isolated neutrophil foci. The absence of leukocytoclastic vasculitis was noted. Immunohistochemistry demonstrated that the histiocyte population was positive for myeloperoxidase and CD68, which categorized them as immature cells of myeloid origin (Figure 3). Clinical and histopathologic findings led to a definitive diagnosis of histiocytoid Sweet syndrome (SS). Sweet syndrome consists of a neutrophilic dermatosis profile. Clinically, it manifests as a sudden onset of painful nodules and plaques accompanied by fever, malaise, and leukocytosis.

Histiocytoid SS is a rare histologic variant of SS initially described by Requena et al1 in 2005. In histiocytoid SS, the main inflammatory infiltrates are promyelocytes and myelocytes.2 Immunohistochemistry shows positivity for myeloperoxidase, CD15, CD43, CD45, CD68, MAC-386, and HAM56.1 The diagnosis is determined by exclusion after adequate clinical and histopathologic correlation, which also should exclude other diagnoses such as leukemia cutis and interstitial granulomatous dermatitis.3 Histiocytoid SS may be related to an increased risk for underlying malignancy. Haber et al4 performed a systematic review in which they concluded that approximately 40% of patients newly diagnosed with histiocytoid SS subsequently were diagnosed or already were diagnosed with a hematologic or solid cancer vs 21% in the classical neutrophilic infiltrate of SS (NSS). Histiocytoid SS more commonly was associated with myelodysplastic syndrome (46% vs 2.5% in NSS) and hematologic malignancies (42.5% vs 25% in SS).

The initial differential diagnoses include inflammatory dermatoses, infections, neoplasms, and systemic diseases. In exudative erythema multiforme, early lesions are composed of typical target lesions with mucosal involvement in 25% to 60% of patients.5 Erythema elevatum diutinum is a chronic dermatosis characterized by asymptomatic papules and red-violet nodules. The most characteristic histologic finding is leukocytoclastic vasculitis.6 The absence of vasculitis is part of the major diagnostic criteria for SS.7 Wells syndrome is associated with general malaise, and edematous and erythematous-violet plaques or nodules appear on the limbs; however, it frequently is associated with eosinophilia in peripheral blood, and histology shows that the main cell population of the inflammatory infiltrate also is eosinophilic.8 Painful, superficial, and erosive blisters appear preferentially on the face and backs of the arms in bullous pyoderma gangrenosum. It usually is not associated with the typical systemic manifestations of SS (ie, fever, arthralgia, damage to target organs). On histopathology, the neutrophilic infiltrate is accompanied by subepidermal vesicles.9
Histiocytoid SS responds dramatically to corticosteroids. Other first-line treatments that avoid use of corticosteroids are colchicine, dapsone, and potassium iodide. Multiple treatments were attempted in our patient, including corticosteroids, methotrexate, dapsone, colchicine, and anakinra. Despite patients responding well to treatment, a possible underlying neoplasm, most frequently of hematologic origin, must be excluded.10
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842. doi:10.1001/archderm.141.7.834
- Alegría-Landa V, Rodríguez-Pinilla SM, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659. doi:10.1001/jamadermatol.2016.6092
- Llamas-Velasco M, Concha-Garzón MJ, Fraga J, et al. Histiocytoid Sweet syndrome related to bortezomib: a mimicker of cutaneous infiltration by myeloma. Indian J Dermatol Venereol Leprol. 2015; 81:305-306. doi:10.4103/0378-6323.152743
- Haber R, Feghali J, El Gemayel M. Risk of malignancy in histiocytoid Sweet syndrome: a systematic review and reappraisal [published online February 21, 2020]. J Am Acad Dermatol. 2020;83:661-663. doi:10.1016/j.jaad.2020.02.048
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902. doi:10.1111/j.1365-4632.2011.05348.x
- Newburger J, Schmieder GJ. Erythema elevatum diutinum. StatPearls. StatPearls Publishing; 2021. http://www.ncbi.nlm.nih.gov /books/NBK448069/
- Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37:167-174.
- Weins AB, Biedermann T, Weiss T, et al. Wells syndrome. J Dtsch Dermatol Ges. 2016;14:989-993. doi:10.1111/ddg.13132
- Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34:395-409; quiz 410-412. doi:10.1016/s0190-9622(96)90428-4
- Villarreal-Villarreal CD, Ocampo-Candiani J, Villarreal-Martínez A. Sweet syndrome: a review and update. Actas Dermosifiliogr. 2016;107:369-378. doi:10.1016/j.ad.2015.12.001
The Diagnosis: Histiocytoid Sweet Syndrome
The patient was admitted for clinical study and treatment monitoring. During the first 72 hours of admittance, the lesions and general malaise further developed along with C-reactive protein elevation (126 mg/L). Administration of intravenous prednisone at a dosage of 1 mg/kg daily was accompanied by substantial improvement after 1 week of treatment, with subsequent follow-up and outpatient monitoring. An underlying neoplasia was ruled out after review of medical history, physical examination, complete blood cell count, chest radiography, abdominal ultrasonography, colonoscopy, and bone marrow aspiration.
A 4-mm skin biopsy was performed from a lesion on the neck (Figure 1). Histology revealed a dermis with prominent edema alongside superficial, deep, and periadnexal perivascular inflammatory infiltrates, as well as predominant lymphocytes and cells with a histiocytoid profile (Figure 2). These findings were accompanied by isolated neutrophil foci. The absence of leukocytoclastic vasculitis was noted. Immunohistochemistry demonstrated that the histiocyte population was positive for myeloperoxidase and CD68, which categorized them as immature cells of myeloid origin (Figure 3). Clinical and histopathologic findings led to a definitive diagnosis of histiocytoid Sweet syndrome (SS). Sweet syndrome consists of a neutrophilic dermatosis profile. Clinically, it manifests as a sudden onset of painful nodules and plaques accompanied by fever, malaise, and leukocytosis.
Histiocytoid SS is a rare histologic variant of SS initially described by Requena et al1 in 2005. In histiocytoid SS, the main inflammatory infiltrates are promyelocytes and myelocytes.2 Immunohistochemistry shows positivity for myeloperoxidase, CD15, CD43, CD45, CD68, MAC-386, and HAM56.1 The diagnosis is determined by exclusion after adequate clinical and histopathologic correlation, which also should exclude other diagnoses such as leukemia cutis and interstitial granulomatous dermatitis.3 Histiocytoid SS may be related to an increased risk for underlying malignancy. Haber et al4 performed a systematic review in which they concluded that approximately 40% of patients newly diagnosed with histiocytoid SS subsequently were diagnosed or already were diagnosed with a hematologic or solid cancer vs 21% in the classical neutrophilic infiltrate of SS (NSS). Histiocytoid SS more commonly was associated with myelodysplastic syndrome (46% vs 2.5% in NSS) and hematologic malignancies (42.5% vs 25% in SS).
The initial differential diagnoses include inflammatory dermatoses, infections, neoplasms, and systemic diseases. In exudative erythema multiforme, early lesions are composed of typical target lesions with mucosal involvement in 25% to 60% of patients.5 Erythema elevatum diutinum is a chronic dermatosis characterized by asymptomatic papules and red-violet nodules. The most characteristic histologic finding is leukocytoclastic vasculitis.6 The absence of vasculitis is part of the major diagnostic criteria for SS.7 Wells syndrome is associated with general malaise, and edematous and erythematous-violet plaques or nodules appear on the limbs; however, it frequently is associated with eosinophilia in peripheral blood, and histology shows that the main cell population of the inflammatory infiltrate also is eosinophilic.8 Painful, superficial, and erosive blisters appear preferentially on the face and backs of the arms in bullous pyoderma gangrenosum. It usually is not associated with the typical systemic manifestations of SS (ie, fever, arthralgia, damage to target organs). On histopathology, the neutrophilic infiltrate is accompanied by subepidermal vesicles.9
Histiocytoid SS responds dramatically to corticosteroids. Other first-line treatments that avoid use of corticosteroids are colchicine, dapsone, and potassium iodide. Multiple treatments were attempted in our patient, including corticosteroids, methotrexate, dapsone, colchicine, and anakinra. Despite patients responding well to treatment, a possible underlying neoplasm, most frequently of hematologic origin, must be excluded.10
The Diagnosis: Histiocytoid Sweet Syndrome
The patient was admitted for clinical study and treatment monitoring. During the first 72 hours of admittance, the lesions and general malaise further developed along with C-reactive protein elevation (126 mg/L). Administration of intravenous prednisone at a dosage of 1 mg/kg daily was accompanied by substantial improvement after 1 week of treatment, with subsequent follow-up and outpatient monitoring. An underlying neoplasia was ruled out after review of medical history, physical examination, complete blood cell count, chest radiography, abdominal ultrasonography, colonoscopy, and bone marrow aspiration.
A 4-mm skin biopsy was performed from a lesion on the neck (Figure 1). Histology revealed a dermis with prominent edema alongside superficial, deep, and periadnexal perivascular inflammatory infiltrates, as well as predominant lymphocytes and cells with a histiocytoid profile (Figure 2). These findings were accompanied by isolated neutrophil foci. The absence of leukocytoclastic vasculitis was noted. Immunohistochemistry demonstrated that the histiocyte population was positive for myeloperoxidase and CD68, which categorized them as immature cells of myeloid origin (Figure 3). Clinical and histopathologic findings led to a definitive diagnosis of histiocytoid Sweet syndrome (SS). Sweet syndrome consists of a neutrophilic dermatosis profile. Clinically, it manifests as a sudden onset of painful nodules and plaques accompanied by fever, malaise, and leukocytosis.
Histiocytoid SS is a rare histologic variant of SS initially described by Requena et al1 in 2005. In histiocytoid SS, the main inflammatory infiltrates are promyelocytes and myelocytes.2 Immunohistochemistry shows positivity for myeloperoxidase, CD15, CD43, CD45, CD68, MAC-386, and HAM56.1 The diagnosis is determined by exclusion after adequate clinical and histopathologic correlation, which also should exclude other diagnoses such as leukemia cutis and interstitial granulomatous dermatitis.3 Histiocytoid SS may be related to an increased risk for underlying malignancy. Haber et al4 performed a systematic review in which they concluded that approximately 40% of patients newly diagnosed with histiocytoid SS subsequently were diagnosed or already were diagnosed with a hematologic or solid cancer vs 21% in the classical neutrophilic infiltrate of SS (NSS). Histiocytoid SS more commonly was associated with myelodysplastic syndrome (46% vs 2.5% in NSS) and hematologic malignancies (42.5% vs 25% in SS).
The initial differential diagnoses include inflammatory dermatoses, infections, neoplasms, and systemic diseases. In exudative erythema multiforme, early lesions are composed of typical target lesions with mucosal involvement in 25% to 60% of patients.5 Erythema elevatum diutinum is a chronic dermatosis characterized by asymptomatic papules and red-violet nodules. The most characteristic histologic finding is leukocytoclastic vasculitis.6 The absence of vasculitis is part of the major diagnostic criteria for SS.7 Wells syndrome is associated with general malaise, and edematous and erythematous-violet plaques or nodules appear on the limbs; however, it frequently is associated with eosinophilia in peripheral blood, and histology shows that the main cell population of the inflammatory infiltrate also is eosinophilic.8 Painful, superficial, and erosive blisters appear preferentially on the face and backs of the arms in bullous pyoderma gangrenosum. It usually is not associated with the typical systemic manifestations of SS (ie, fever, arthralgia, damage to target organs). On histopathology, the neutrophilic infiltrate is accompanied by subepidermal vesicles.9
Histiocytoid SS responds dramatically to corticosteroids. Other first-line treatments that avoid use of corticosteroids are colchicine, dapsone, and potassium iodide. Multiple treatments were attempted in our patient, including corticosteroids, methotrexate, dapsone, colchicine, and anakinra. Despite patients responding well to treatment, a possible underlying neoplasm, most frequently of hematologic origin, must be excluded.10
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842. doi:10.1001/archderm.141.7.834
- Alegría-Landa V, Rodríguez-Pinilla SM, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659. doi:10.1001/jamadermatol.2016.6092
- Llamas-Velasco M, Concha-Garzón MJ, Fraga J, et al. Histiocytoid Sweet syndrome related to bortezomib: a mimicker of cutaneous infiltration by myeloma. Indian J Dermatol Venereol Leprol. 2015; 81:305-306. doi:10.4103/0378-6323.152743
- Haber R, Feghali J, El Gemayel M. Risk of malignancy in histiocytoid Sweet syndrome: a systematic review and reappraisal [published online February 21, 2020]. J Am Acad Dermatol. 2020;83:661-663. doi:10.1016/j.jaad.2020.02.048
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902. doi:10.1111/j.1365-4632.2011.05348.x
- Newburger J, Schmieder GJ. Erythema elevatum diutinum. StatPearls. StatPearls Publishing; 2021. http://www.ncbi.nlm.nih.gov /books/NBK448069/
- Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37:167-174.
- Weins AB, Biedermann T, Weiss T, et al. Wells syndrome. J Dtsch Dermatol Ges. 2016;14:989-993. doi:10.1111/ddg.13132
- Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34:395-409; quiz 410-412. doi:10.1016/s0190-9622(96)90428-4
- Villarreal-Villarreal CD, Ocampo-Candiani J, Villarreal-Martínez A. Sweet syndrome: a review and update. Actas Dermosifiliogr. 2016;107:369-378. doi:10.1016/j.ad.2015.12.001
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842. doi:10.1001/archderm.141.7.834
- Alegría-Landa V, Rodríguez-Pinilla SM, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659. doi:10.1001/jamadermatol.2016.6092
- Llamas-Velasco M, Concha-Garzón MJ, Fraga J, et al. Histiocytoid Sweet syndrome related to bortezomib: a mimicker of cutaneous infiltration by myeloma. Indian J Dermatol Venereol Leprol. 2015; 81:305-306. doi:10.4103/0378-6323.152743
- Haber R, Feghali J, El Gemayel M. Risk of malignancy in histiocytoid Sweet syndrome: a systematic review and reappraisal [published online February 21, 2020]. J Am Acad Dermatol. 2020;83:661-663. doi:10.1016/j.jaad.2020.02.048
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902. doi:10.1111/j.1365-4632.2011.05348.x
- Newburger J, Schmieder GJ. Erythema elevatum diutinum. StatPearls. StatPearls Publishing; 2021. http://www.ncbi.nlm.nih.gov /books/NBK448069/
- Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37:167-174.
- Weins AB, Biedermann T, Weiss T, et al. Wells syndrome. J Dtsch Dermatol Ges. 2016;14:989-993. doi:10.1111/ddg.13132
- Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34:395-409; quiz 410-412. doi:10.1016/s0190-9622(96)90428-4
- Villarreal-Villarreal CD, Ocampo-Candiani J, Villarreal-Martínez A. Sweet syndrome: a review and update. Actas Dermosifiliogr. 2016;107:369-378. doi:10.1016/j.ad.2015.12.001
A 53-year-old man presented to the emergency department with a fever and painful skin lesions of 2 days’ duration. He reported a medical history of an upper respiratory infection 4 weeks prior. Physical examination was notable for erythematous-violet edematous papules, necrotic lesions, and pseudovesicles located on the face (top), head, neck, arms, and legs (bottom). Hemorrhagic splinters were evidenced in multiple nail sections. Urgent blood work revealed microcytic anemia (hemoglobin, 12.6 g/dL [reference range, 14.0–17.5 g/dL]) and elevated C-reactive protein (58 mg/L [reference range, 0.0–5.0 mg/L]).


Indurated Violaceous Lesions on the Face, Trunk, and Legs
The Diagnosis: Kaposi Sarcoma
A punch biopsy of a lesion on the right side of the back revealed a diffuse, poorly circumscribed, spindle cell neoplasm of the papillary and reticular dermis with associated vascular and pseudovascular spaces distended by erythrocytes (Figure 1). Immunostaining was positive for human herpesvirus 8 (HHV-8)(Figure 2), ETS-related gene, CD31, and CD34 and negative for pan cytokeratin, confirming the diagnosis of Kaposi sarcoma (KS). Bacterial, fungal, and mycobacterial tissue cultures were negative. The patient was tested for HIV and referred to infectious disease and oncology. He subsequently was found to have HIV with a viral load greater than 1 million copies. He was started on antiretroviral therapy and Pneumocystis jirovecii pneumonia prophylaxis. Computed tomography of the chest, abdomen, and pelvis showed bilateral, multifocal, perihilar, flame-shaped consolidations suggestive of KS. The patient later disclosed having an intermittent dry cough of more than a year’s duration with occasional bright red blood per rectum after bowel movements. After workup, the patient was found to have cytomegalovirus esophagitis/gastritis and candidal esophagitis that were treated with valganciclovir and fluconazole, respectively.
.")
Kaposi sarcoma is an angioproliferative, AIDSdefining disease associated with HHV-8. There are 4 types of KS as defined by the populations they affect. AIDS-associated KS occurs in individuals with HIV, as seen in our patient. It often is accompanied by extensive mucocutaneous and visceral lesions, as well as systemic symptoms such as fever, weight loss, and diarrhea.1 Classic KS is a variant that presents in older men of Mediterranean, Eastern European, and South American descent. Cutaneous lesions typically are distributed on the lower extremities.2,3 Endemic (African) KS is seen in HIV-negative children and young adults in equatorial Africa. It most commonly affects the lower extremities or lymph nodes and usually follows a more aggressive course.2 Lastly, iatrogenic KS is associated with immunosuppressive medications or conditions, such as organ transplantation, chemotherapy, and rheumatologic disorders.3,4
.")
Kaposi sarcoma commonly presents as violaceous or dark red macules, patches, papules, plaques, and nodules on various parts of the body (Figure 3). Lesions typically begin as macules and progress into plaques or nodules. Our patient presented as a deceptively healthy young man with lesions at various stages of development. In addition to the skin and oral mucosa, the lungs, lymph nodes, and gastrointestinal tract commonly are involved in AIDS-associated KS.5 Patients may experience symptoms of internal involvement, including bleeding, hematochezia, odynophagia, or dyspnea.

The differential diagnosis includes conditions that can mimic KS, including bacillary angiomatosis, angioinvasive fungal disease, sarcoid, and other malignancies. A skin biopsy is the gold standard for definitive diagnosis of KS. Histopathology shows a vascular proliferation in the dermis and spindle cell proliferation.6 Kaposi sarcoma stains positively for factor VIII–related antigen, CD31, and CD34.2 Additionally, staining for HHV-8 gene products, such as latency-associated nuclear antigen 1, is helpful in differentiating KS from other conditions.7
In HIV-associated KS, the mainstay of treatment is initiation of highly active antiretroviral therapy. Typically, as the CD4 count rises with treatment, the tumor burden classic KS, effective treatment options include recurrent cryotherapy or intralesional chemotherapeutics, such as vincristine, for localized lesions; for widespread disease, pegylated liposomal doxorubicin or radiation have been found to be effective options. Lastly, for patients with iatrogenic KS, reducing immunosuppressive medications is a reasonable first step in management. If this does not yield adequate improvement, transitioning from calcineurin inhibitors (eg, cyclosporine) to proliferation signal inhibitors (eg, sirolimus) may lead to resolution.7
- Friedman-Kien AE, Saltzman BR. Clinical manifestations of classical, endemic African, and epidemic AIDS-associated Kaposi’s sarcoma. J Am Acad Dermatol. 1990;22:1237-1250.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Vangipuram R, Tyring SK. Epidemiology of Kaposi sarcoma: review and description of the nonepidemic variant. Int J Dermatol. 2019;58:538-542.
- Klepp O, Dahl O, Stenwig JT. Association of Kaposi’s sarcoma and prior immunosuppressive therapy. a 5‐year material of Kaposi’s sarcoma in Norway. Cancer. 1978;42:2626-2630.
- Lemlich G, Schwam L, Lebwohl M. Kaposi’s sarcoma and acquired immunodeficiency syndrome: postmortem findings in twenty-four cases. J Am Acad Dermatol. 1987;16:319-325.
- Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:10.
- Curtiss P, Strazzulla LC, Friedman-Kien AE. An update on Kaposi’s sarcoma: epidemiology, pathogenesis and treatment. Dermatol Ther. 2016;6:465-470.
The Diagnosis: Kaposi Sarcoma
A punch biopsy of a lesion on the right side of the back revealed a diffuse, poorly circumscribed, spindle cell neoplasm of the papillary and reticular dermis with associated vascular and pseudovascular spaces distended by erythrocytes (Figure 1). Immunostaining was positive for human herpesvirus 8 (HHV-8)(Figure 2), ETS-related gene, CD31, and CD34 and negative for pan cytokeratin, confirming the diagnosis of Kaposi sarcoma (KS). Bacterial, fungal, and mycobacterial tissue cultures were negative. The patient was tested for HIV and referred to infectious disease and oncology. He subsequently was found to have HIV with a viral load greater than 1 million copies. He was started on antiretroviral therapy and Pneumocystis jirovecii pneumonia prophylaxis. Computed tomography of the chest, abdomen, and pelvis showed bilateral, multifocal, perihilar, flame-shaped consolidations suggestive of KS. The patient later disclosed having an intermittent dry cough of more than a year’s duration with occasional bright red blood per rectum after bowel movements. After workup, the patient was found to have cytomegalovirus esophagitis/gastritis and candidal esophagitis that were treated with valganciclovir and fluconazole, respectively.
Kaposi sarcoma is an angioproliferative, AIDSdefining disease associated with HHV-8. There are 4 types of KS as defined by the populations they affect. AIDS-associated KS occurs in individuals with HIV, as seen in our patient. It often is accompanied by extensive mucocutaneous and visceral lesions, as well as systemic symptoms such as fever, weight loss, and diarrhea.1 Classic KS is a variant that presents in older men of Mediterranean, Eastern European, and South American descent. Cutaneous lesions typically are distributed on the lower extremities.2,3 Endemic (African) KS is seen in HIV-negative children and young adults in equatorial Africa. It most commonly affects the lower extremities or lymph nodes and usually follows a more aggressive course.2 Lastly, iatrogenic KS is associated with immunosuppressive medications or conditions, such as organ transplantation, chemotherapy, and rheumatologic disorders.3,4
Kaposi sarcoma commonly presents as violaceous or dark red macules, patches, papules, plaques, and nodules on various parts of the body (Figure 3). Lesions typically begin as macules and progress into plaques or nodules. Our patient presented as a deceptively healthy young man with lesions at various stages of development. In addition to the skin and oral mucosa, the lungs, lymph nodes, and gastrointestinal tract commonly are involved in AIDS-associated KS.5 Patients may experience symptoms of internal involvement, including bleeding, hematochezia, odynophagia, or dyspnea.
The differential diagnosis includes conditions that can mimic KS, including bacillary angiomatosis, angioinvasive fungal disease, sarcoid, and other malignancies. A skin biopsy is the gold standard for definitive diagnosis of KS. Histopathology shows a vascular proliferation in the dermis and spindle cell proliferation.6 Kaposi sarcoma stains positively for factor VIII–related antigen, CD31, and CD34.2 Additionally, staining for HHV-8 gene products, such as latency-associated nuclear antigen 1, is helpful in differentiating KS from other conditions.7
In HIV-associated KS, the mainstay of treatment is initiation of highly active antiretroviral therapy. Typically, as the CD4 count rises with treatment, the tumor burden classic KS, effective treatment options include recurrent cryotherapy or intralesional chemotherapeutics, such as vincristine, for localized lesions; for widespread disease, pegylated liposomal doxorubicin or radiation have been found to be effective options. Lastly, for patients with iatrogenic KS, reducing immunosuppressive medications is a reasonable first step in management. If this does not yield adequate improvement, transitioning from calcineurin inhibitors (eg, cyclosporine) to proliferation signal inhibitors (eg, sirolimus) may lead to resolution.7
The Diagnosis: Kaposi Sarcoma
A punch biopsy of a lesion on the right side of the back revealed a diffuse, poorly circumscribed, spindle cell neoplasm of the papillary and reticular dermis with associated vascular and pseudovascular spaces distended by erythrocytes (Figure 1). Immunostaining was positive for human herpesvirus 8 (HHV-8)(Figure 2), ETS-related gene, CD31, and CD34 and negative for pan cytokeratin, confirming the diagnosis of Kaposi sarcoma (KS). Bacterial, fungal, and mycobacterial tissue cultures were negative. The patient was tested for HIV and referred to infectious disease and oncology. He subsequently was found to have HIV with a viral load greater than 1 million copies. He was started on antiretroviral therapy and Pneumocystis jirovecii pneumonia prophylaxis. Computed tomography of the chest, abdomen, and pelvis showed bilateral, multifocal, perihilar, flame-shaped consolidations suggestive of KS. The patient later disclosed having an intermittent dry cough of more than a year’s duration with occasional bright red blood per rectum after bowel movements. After workup, the patient was found to have cytomegalovirus esophagitis/gastritis and candidal esophagitis that were treated with valganciclovir and fluconazole, respectively.
Kaposi sarcoma is an angioproliferative, AIDSdefining disease associated with HHV-8. There are 4 types of KS as defined by the populations they affect. AIDS-associated KS occurs in individuals with HIV, as seen in our patient. It often is accompanied by extensive mucocutaneous and visceral lesions, as well as systemic symptoms such as fever, weight loss, and diarrhea.1 Classic KS is a variant that presents in older men of Mediterranean, Eastern European, and South American descent. Cutaneous lesions typically are distributed on the lower extremities.2,3 Endemic (African) KS is seen in HIV-negative children and young adults in equatorial Africa. It most commonly affects the lower extremities or lymph nodes and usually follows a more aggressive course.2 Lastly, iatrogenic KS is associated with immunosuppressive medications or conditions, such as organ transplantation, chemotherapy, and rheumatologic disorders.3,4
Kaposi sarcoma commonly presents as violaceous or dark red macules, patches, papules, plaques, and nodules on various parts of the body (Figure 3). Lesions typically begin as macules and progress into plaques or nodules. Our patient presented as a deceptively healthy young man with lesions at various stages of development. In addition to the skin and oral mucosa, the lungs, lymph nodes, and gastrointestinal tract commonly are involved in AIDS-associated KS.5 Patients may experience symptoms of internal involvement, including bleeding, hematochezia, odynophagia, or dyspnea.
The differential diagnosis includes conditions that can mimic KS, including bacillary angiomatosis, angioinvasive fungal disease, sarcoid, and other malignancies. A skin biopsy is the gold standard for definitive diagnosis of KS. Histopathology shows a vascular proliferation in the dermis and spindle cell proliferation.6 Kaposi sarcoma stains positively for factor VIII–related antigen, CD31, and CD34.2 Additionally, staining for HHV-8 gene products, such as latency-associated nuclear antigen 1, is helpful in differentiating KS from other conditions.7
In HIV-associated KS, the mainstay of treatment is initiation of highly active antiretroviral therapy. Typically, as the CD4 count rises with treatment, the tumor burden classic KS, effective treatment options include recurrent cryotherapy or intralesional chemotherapeutics, such as vincristine, for localized lesions; for widespread disease, pegylated liposomal doxorubicin or radiation have been found to be effective options. Lastly, for patients with iatrogenic KS, reducing immunosuppressive medications is a reasonable first step in management. If this does not yield adequate improvement, transitioning from calcineurin inhibitors (eg, cyclosporine) to proliferation signal inhibitors (eg, sirolimus) may lead to resolution.7
- Friedman-Kien AE, Saltzman BR. Clinical manifestations of classical, endemic African, and epidemic AIDS-associated Kaposi’s sarcoma. J Am Acad Dermatol. 1990;22:1237-1250.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Vangipuram R, Tyring SK. Epidemiology of Kaposi sarcoma: review and description of the nonepidemic variant. Int J Dermatol. 2019;58:538-542.
- Klepp O, Dahl O, Stenwig JT. Association of Kaposi’s sarcoma and prior immunosuppressive therapy. a 5‐year material of Kaposi’s sarcoma in Norway. Cancer. 1978;42:2626-2630.
- Lemlich G, Schwam L, Lebwohl M. Kaposi’s sarcoma and acquired immunodeficiency syndrome: postmortem findings in twenty-four cases. J Am Acad Dermatol. 1987;16:319-325.
- Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:10.
- Curtiss P, Strazzulla LC, Friedman-Kien AE. An update on Kaposi’s sarcoma: epidemiology, pathogenesis and treatment. Dermatol Ther. 2016;6:465-470.
- Friedman-Kien AE, Saltzman BR. Clinical manifestations of classical, endemic African, and epidemic AIDS-associated Kaposi’s sarcoma. J Am Acad Dermatol. 1990;22:1237-1250.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
- Vangipuram R, Tyring SK. Epidemiology of Kaposi sarcoma: review and description of the nonepidemic variant. Int J Dermatol. 2019;58:538-542.
- Klepp O, Dahl O, Stenwig JT. Association of Kaposi’s sarcoma and prior immunosuppressive therapy. a 5‐year material of Kaposi’s sarcoma in Norway. Cancer. 1978;42:2626-2630.
- Lemlich G, Schwam L, Lebwohl M. Kaposi’s sarcoma and acquired immunodeficiency syndrome: postmortem findings in twenty-four cases. J Am Acad Dermatol. 1987;16:319-325.
- Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:10.
- Curtiss P, Strazzulla LC, Friedman-Kien AE. An update on Kaposi’s sarcoma: epidemiology, pathogenesis and treatment. Dermatol Ther. 2016;6:465-470.
A 25-year-old man with no notable medical history presented to the dermatology clinic with growing selfdescribed cysts on the face, trunk, and legs of 6 months’ duration. The lesions started as bruiselike discolorations and progressed to become firm nodules and inflamed masses. Some were minimally itchy and sensitive to touch, but there was no history of bleeding or drainage. The patient denied any new or recent environmental or animal exposures, use of illicit drugs, or travel correlating with the rash onset. He denied any prior treatments. He reported being in his normal state of health and was not taking any medications. Physical examination revealed indurated, violaceous, purpuric subcutaneous nodules, plaques, and masses on the forehead, cheek (top), jaw, flank, axillae (bottom), and back.


Blisters in a Comatose Elderly Woman
The Diagnosis: Coma Blisters
Histologic examination revealed pauci-inflammatory subepidermal blisters with swelling of eccrine cells, signaling impending gland necrosis (Figure). Direct immunofluorescence testing on perilesional skin was negative. These findings would be inconsistent for diagnoses of edema blisters (most commonly seen in patients with an acute exacerbation of chronic lower extremity edema), friction blisters (intraepidermal blisters seen on histopathology), and bullous pemphigoid (linear IgG and/or C3 staining along the basement membrane zone on direct immunofluorescence testing is characteristic). Although eccrine gland alterations have been seen in toxic epidermal necrolysis,1 the mucous membranes are involved in more than 90% of cases, making the diagnosis less likely. Furthermore, interface changes including prominent keratinocyte necrosis were not seen on histology.
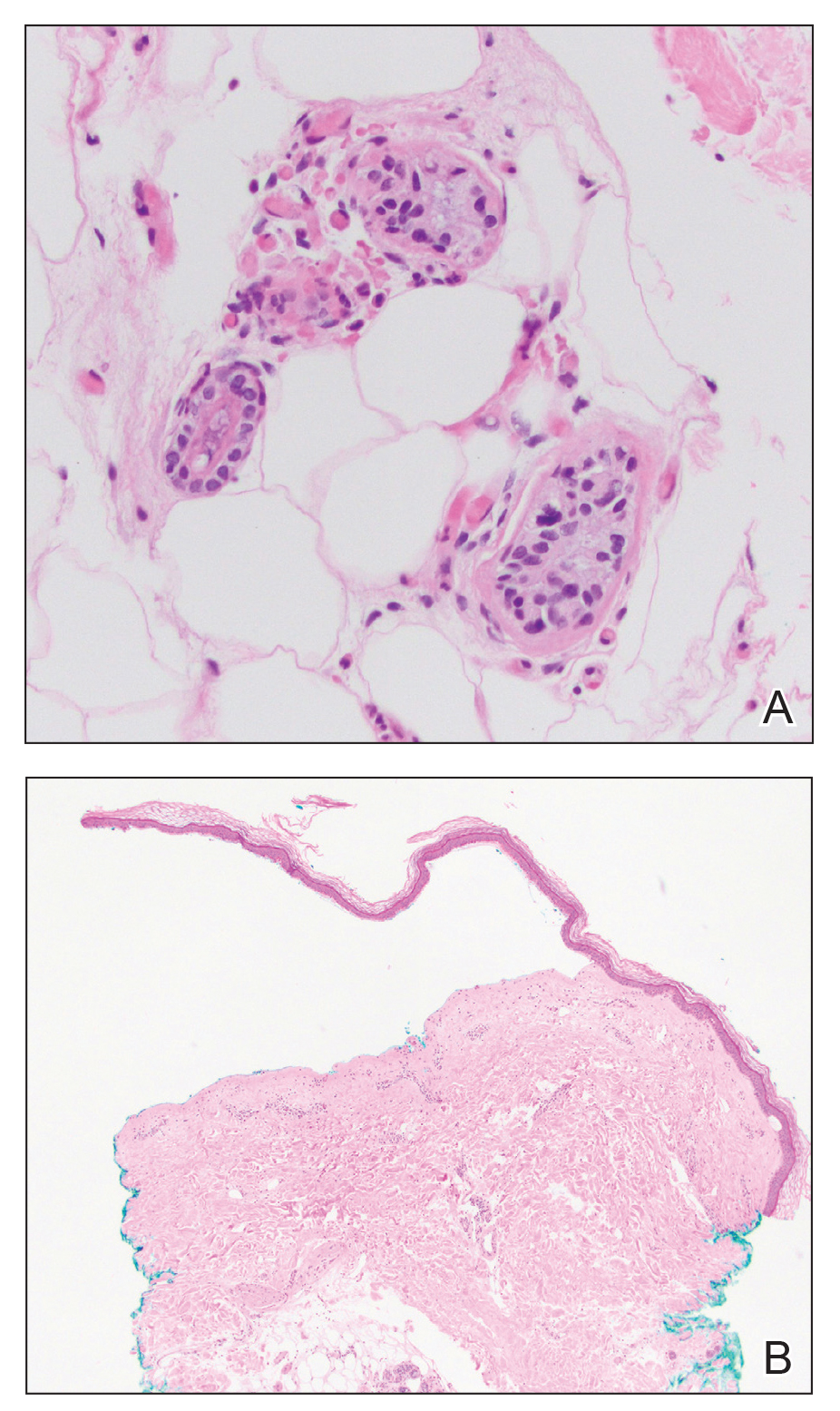
Given the localized nature of the lesions in our patient and negative direct immunofluorescence studies, a diagnosis of coma blisters was made. Gentle wound care practices to the areas of denuded skin were implemented with complete resolution. The patient’s condition gradually improved, and she was extubated and discharged home.
Coma blisters are self-limited bullous lesions that have been reported in comatose patients as early as 1812 when Napoleon’s surgeon first noticed cutaneous blisters in comatose French soldiers being treated for carbon monoxide intoxication.2 Since then, barbiturate overdose has remained the most common association, but coma blisters have occurred in the absence of specific drug exposures. Clinically, erythematous or violaceous plaques typically appear within 24 hours of drug ingestion, and progression to large tense bullae usually occurs within 48 to 72 hours of unconsciousness.3 They characteristically occur in pressure-dependent areas, but reports have shown lesions in non–pressure-dependent areas, including the penis and mouth.1,4 Spontaneous resolution within 1 to 2 weeks is typical.5
The underlying pathogenesis remains controversial, as multiple mechanisms have been suggested, but clear causal evidence is lacking. The original proposition that direct effects of drug toxicity caused the cutaneous observations was later refuted after similar bullous lesions with eccrine gland necrosis were reported in comatose patients with neurologic conditions.6 It is largely accepted that pressure-induced local ischemia—proportional to the duration and amount of pressure—leads to tissue injury and is critical to the pathogenesis. During periods of ischemia, the most metabolically active tissues will undergo necrosis first; however, in eccrine glands, the earliest and most severe damage does not seem to occur in the most metabolically active cells.7 Additionally, this would not provide a viable explanation for coma blisters with eccrine gland necrosis developing in variable non–pressuredependent areas.
Moreover, drug- and non–drug-induced coma blisters can appear identically, but specific histopathologic differences have been reported. The most notable markers of non–drug-induced coma blisters are the absence of an inflammatory infiltrate in the epidermis and the presence of thrombosis in dermal vessels.8 Demonstration of necrotic changes in the secretory portion of the eccrine gland is considered the histopathologic hallmark for drug-induced coma blisters, but other findings can include subepidermal or intraepidermal bullae; perivascular infiltrates; and focal necrosis of the epidermis, dermis, subcutis, or epidermal appendages.6 Arteriolar wall necrosis and dermal inflammatory infiltrates also have been observed.7
Benzodiazepines have been widely prescribed and abused since their development, and overdose is much more common today than with barbiturates.9 Coma blisters rarely have been documented in the setting of isolated benzodiazepine overdose, and of the few cases, only one report implicated lorazepam as the causative agent.4,7 The characteristic finding of eccrine gland necrosis consistently was seen in our patient. This case not only emphasizes the need for greater awareness of the association between benzodiazepine overdose and coma blisters but also the importance of clinical context when considering diagnoses. It is essential to note that coma blisters themselves are nonspecific, and the diagnosis of drug-induced coma blisters warrants confirmatory toxicologic analysis.
- Ferreli C, Sulica VI, Aste N, et al. Drug-induced sweat gland necrosis in a non-comatose patient: a case presentation. J Eur Acad Dermatol Venereol. 2003;17:443-445.
- Larrey DJ. Memoires de Chirurgie Militaire et Campagnes. Smith and Buisson; 1812.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online J. 2012;18:10.
- Varma AJ, Fisher BK, Sarin MK. Diazepam-induced coma with bullae and eccrine sweat gland necrosis. Arch Intern Med. 1977;137:1207-1210.
- Rocha J, Pereira T, Ventura F, et al. Coma blisters. Case Rep Dermatol. 2009;1:66-70.
- Arndt KA, Mihm MC, Parrish JA. Bullae: a cutaneous sign of a variety of neurologic diseases. J Invest Dermatol. 1973;60:312-320.
- Sánchez Yus E, Requena L, Simón P. Histopathology of cutaneous changes in drug-induced coma. Am J Dermatopathol. 1993;15:208-216.
- Kato N, Ueno H, Mimura M. Histopathology of cutaneous changes in non-drug-induced coma. Am J Dermatopathol. 1996;18:344-350.
- Kang M, Ghassemzadeh S. Benzodiazepine Toxicity. StatPearls Publishing; 2018.
The Diagnosis: Coma Blisters
Histologic examination revealed pauci-inflammatory subepidermal blisters with swelling of eccrine cells, signaling impending gland necrosis (Figure). Direct immunofluorescence testing on perilesional skin was negative. These findings would be inconsistent for diagnoses of edema blisters (most commonly seen in patients with an acute exacerbation of chronic lower extremity edema), friction blisters (intraepidermal blisters seen on histopathology), and bullous pemphigoid (linear IgG and/or C3 staining along the basement membrane zone on direct immunofluorescence testing is characteristic). Although eccrine gland alterations have been seen in toxic epidermal necrolysis,1 the mucous membranes are involved in more than 90% of cases, making the diagnosis less likely. Furthermore, interface changes including prominent keratinocyte necrosis were not seen on histology.
Given the localized nature of the lesions in our patient and negative direct immunofluorescence studies, a diagnosis of coma blisters was made. Gentle wound care practices to the areas of denuded skin were implemented with complete resolution. The patient’s condition gradually improved, and she was extubated and discharged home.
Coma blisters are self-limited bullous lesions that have been reported in comatose patients as early as 1812 when Napoleon’s surgeon first noticed cutaneous blisters in comatose French soldiers being treated for carbon monoxide intoxication.2 Since then, barbiturate overdose has remained the most common association, but coma blisters have occurred in the absence of specific drug exposures. Clinically, erythematous or violaceous plaques typically appear within 24 hours of drug ingestion, and progression to large tense bullae usually occurs within 48 to 72 hours of unconsciousness.3 They characteristically occur in pressure-dependent areas, but reports have shown lesions in non–pressure-dependent areas, including the penis and mouth.1,4 Spontaneous resolution within 1 to 2 weeks is typical.5
The underlying pathogenesis remains controversial, as multiple mechanisms have been suggested, but clear causal evidence is lacking. The original proposition that direct effects of drug toxicity caused the cutaneous observations was later refuted after similar bullous lesions with eccrine gland necrosis were reported in comatose patients with neurologic conditions.6 It is largely accepted that pressure-induced local ischemia—proportional to the duration and amount of pressure—leads to tissue injury and is critical to the pathogenesis. During periods of ischemia, the most metabolically active tissues will undergo necrosis first; however, in eccrine glands, the earliest and most severe damage does not seem to occur in the most metabolically active cells.7 Additionally, this would not provide a viable explanation for coma blisters with eccrine gland necrosis developing in variable non–pressuredependent areas.
Moreover, drug- and non–drug-induced coma blisters can appear identically, but specific histopathologic differences have been reported. The most notable markers of non–drug-induced coma blisters are the absence of an inflammatory infiltrate in the epidermis and the presence of thrombosis in dermal vessels.8 Demonstration of necrotic changes in the secretory portion of the eccrine gland is considered the histopathologic hallmark for drug-induced coma blisters, but other findings can include subepidermal or intraepidermal bullae; perivascular infiltrates; and focal necrosis of the epidermis, dermis, subcutis, or epidermal appendages.6 Arteriolar wall necrosis and dermal inflammatory infiltrates also have been observed.7
Benzodiazepines have been widely prescribed and abused since their development, and overdose is much more common today than with barbiturates.9 Coma blisters rarely have been documented in the setting of isolated benzodiazepine overdose, and of the few cases, only one report implicated lorazepam as the causative agent.4,7 The characteristic finding of eccrine gland necrosis consistently was seen in our patient. This case not only emphasizes the need for greater awareness of the association between benzodiazepine overdose and coma blisters but also the importance of clinical context when considering diagnoses. It is essential to note that coma blisters themselves are nonspecific, and the diagnosis of drug-induced coma blisters warrants confirmatory toxicologic analysis.
The Diagnosis: Coma Blisters
Histologic examination revealed pauci-inflammatory subepidermal blisters with swelling of eccrine cells, signaling impending gland necrosis (Figure). Direct immunofluorescence testing on perilesional skin was negative. These findings would be inconsistent for diagnoses of edema blisters (most commonly seen in patients with an acute exacerbation of chronic lower extremity edema), friction blisters (intraepidermal blisters seen on histopathology), and bullous pemphigoid (linear IgG and/or C3 staining along the basement membrane zone on direct immunofluorescence testing is characteristic). Although eccrine gland alterations have been seen in toxic epidermal necrolysis,1 the mucous membranes are involved in more than 90% of cases, making the diagnosis less likely. Furthermore, interface changes including prominent keratinocyte necrosis were not seen on histology.
Given the localized nature of the lesions in our patient and negative direct immunofluorescence studies, a diagnosis of coma blisters was made. Gentle wound care practices to the areas of denuded skin were implemented with complete resolution. The patient’s condition gradually improved, and she was extubated and discharged home.
Coma blisters are self-limited bullous lesions that have been reported in comatose patients as early as 1812 when Napoleon’s surgeon first noticed cutaneous blisters in comatose French soldiers being treated for carbon monoxide intoxication.2 Since then, barbiturate overdose has remained the most common association, but coma blisters have occurred in the absence of specific drug exposures. Clinically, erythematous or violaceous plaques typically appear within 24 hours of drug ingestion, and progression to large tense bullae usually occurs within 48 to 72 hours of unconsciousness.3 They characteristically occur in pressure-dependent areas, but reports have shown lesions in non–pressure-dependent areas, including the penis and mouth.1,4 Spontaneous resolution within 1 to 2 weeks is typical.5
The underlying pathogenesis remains controversial, as multiple mechanisms have been suggested, but clear causal evidence is lacking. The original proposition that direct effects of drug toxicity caused the cutaneous observations was later refuted after similar bullous lesions with eccrine gland necrosis were reported in comatose patients with neurologic conditions.6 It is largely accepted that pressure-induced local ischemia—proportional to the duration and amount of pressure—leads to tissue injury and is critical to the pathogenesis. During periods of ischemia, the most metabolically active tissues will undergo necrosis first; however, in eccrine glands, the earliest and most severe damage does not seem to occur in the most metabolically active cells.7 Additionally, this would not provide a viable explanation for coma blisters with eccrine gland necrosis developing in variable non–pressuredependent areas.
Moreover, drug- and non–drug-induced coma blisters can appear identically, but specific histopathologic differences have been reported. The most notable markers of non–drug-induced coma blisters are the absence of an inflammatory infiltrate in the epidermis and the presence of thrombosis in dermal vessels.8 Demonstration of necrotic changes in the secretory portion of the eccrine gland is considered the histopathologic hallmark for drug-induced coma blisters, but other findings can include subepidermal or intraepidermal bullae; perivascular infiltrates; and focal necrosis of the epidermis, dermis, subcutis, or epidermal appendages.6 Arteriolar wall necrosis and dermal inflammatory infiltrates also have been observed.7
Benzodiazepines have been widely prescribed and abused since their development, and overdose is much more common today than with barbiturates.9 Coma blisters rarely have been documented in the setting of isolated benzodiazepine overdose, and of the few cases, only one report implicated lorazepam as the causative agent.4,7 The characteristic finding of eccrine gland necrosis consistently was seen in our patient. This case not only emphasizes the need for greater awareness of the association between benzodiazepine overdose and coma blisters but also the importance of clinical context when considering diagnoses. It is essential to note that coma blisters themselves are nonspecific, and the diagnosis of drug-induced coma blisters warrants confirmatory toxicologic analysis.
- Ferreli C, Sulica VI, Aste N, et al. Drug-induced sweat gland necrosis in a non-comatose patient: a case presentation. J Eur Acad Dermatol Venereol. 2003;17:443-445.
- Larrey DJ. Memoires de Chirurgie Militaire et Campagnes. Smith and Buisson; 1812.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online J. 2012;18:10.
- Varma AJ, Fisher BK, Sarin MK. Diazepam-induced coma with bullae and eccrine sweat gland necrosis. Arch Intern Med. 1977;137:1207-1210.
- Rocha J, Pereira T, Ventura F, et al. Coma blisters. Case Rep Dermatol. 2009;1:66-70.
- Arndt KA, Mihm MC, Parrish JA. Bullae: a cutaneous sign of a variety of neurologic diseases. J Invest Dermatol. 1973;60:312-320.
- Sánchez Yus E, Requena L, Simón P. Histopathology of cutaneous changes in drug-induced coma. Am J Dermatopathol. 1993;15:208-216.
- Kato N, Ueno H, Mimura M. Histopathology of cutaneous changes in non-drug-induced coma. Am J Dermatopathol. 1996;18:344-350.
- Kang M, Ghassemzadeh S. Benzodiazepine Toxicity. StatPearls Publishing; 2018.
- Ferreli C, Sulica VI, Aste N, et al. Drug-induced sweat gland necrosis in a non-comatose patient: a case presentation. J Eur Acad Dermatol Venereol. 2003;17:443-445.
- Larrey DJ. Memoires de Chirurgie Militaire et Campagnes. Smith and Buisson; 1812.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online J. 2012;18:10.
- Varma AJ, Fisher BK, Sarin MK. Diazepam-induced coma with bullae and eccrine sweat gland necrosis. Arch Intern Med. 1977;137:1207-1210.
- Rocha J, Pereira T, Ventura F, et al. Coma blisters. Case Rep Dermatol. 2009;1:66-70.
- Arndt KA, Mihm MC, Parrish JA. Bullae: a cutaneous sign of a variety of neurologic diseases. J Invest Dermatol. 1973;60:312-320.
- Sánchez Yus E, Requena L, Simón P. Histopathology of cutaneous changes in drug-induced coma. Am J Dermatopathol. 1993;15:208-216.
- Kato N, Ueno H, Mimura M. Histopathology of cutaneous changes in non-drug-induced coma. Am J Dermatopathol. 1996;18:344-350.
- Kang M, Ghassemzadeh S. Benzodiazepine Toxicity. StatPearls Publishing; 2018.
An 82-year-old woman presented to the emergency department after her daughter found her unconscious in the bathroom laying on her right side. Her medical history was notable for hypertension and asthma for which she was on losartan, furosemide, diltiazem, and albuterol. She recently had been prescribed lorazepam for insomnia and had started taking the medication 2 days prior. She underwent intubation and was noted to have flaccid, fluid-filled bullae on the right thigh (top) along with large areas of desquamation on the right lateral arm (bottom) with minimal surrounding erythema. There was no mucous membrane involvement. Urine toxicology was positive for benzodiazepines and negative for all other drugs, including barbiturates.
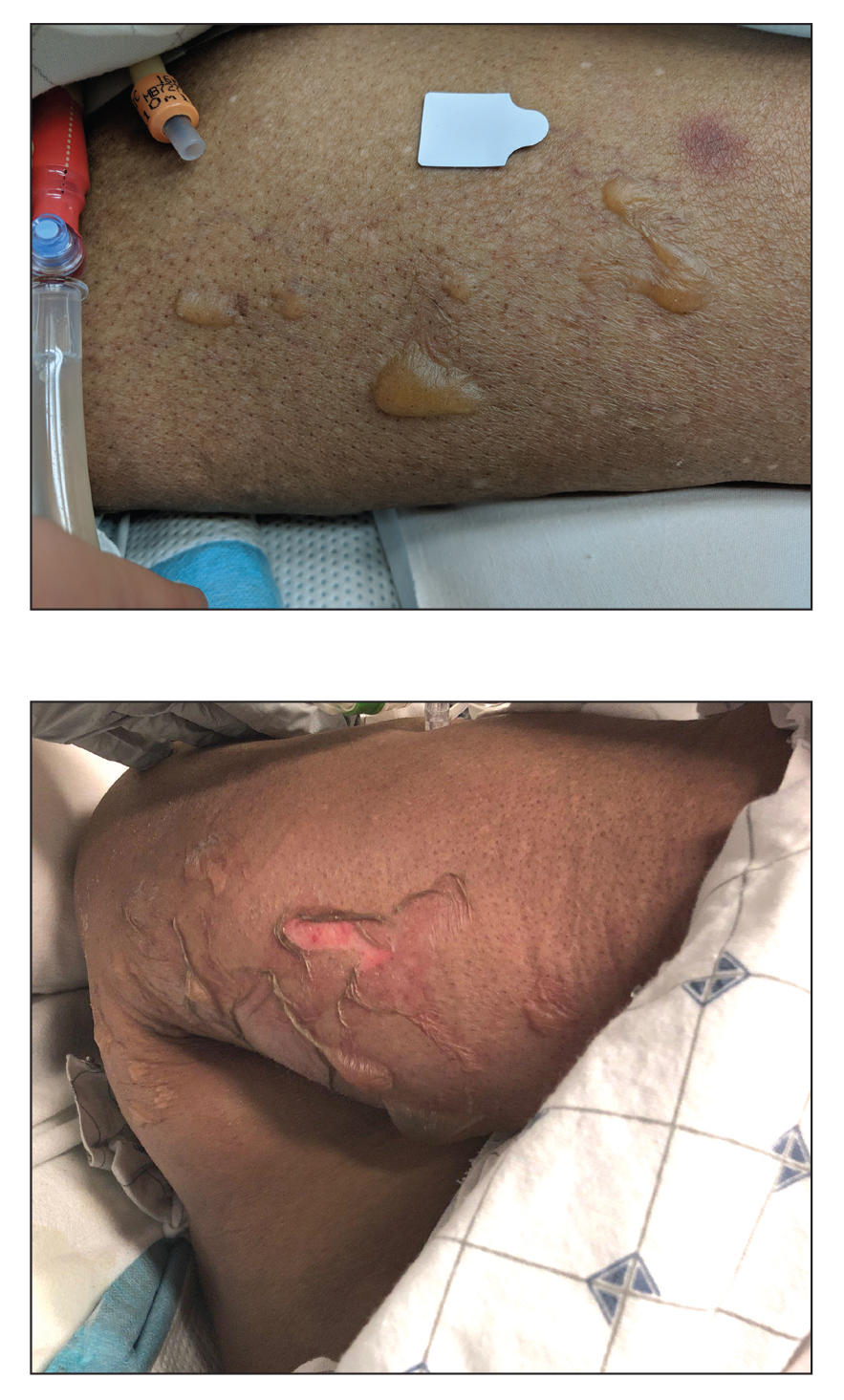
What is the diagnosis?
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that is becoming more recognized in children. It has a variable presentation, most commonly presenting as painful, recurrent cysts, abscesses, nodules, and/or pustules in classic locations with associated scarring and sinus tract formation.
The majority of patients present with bilateral lesions found most commonly in the axillae and inguinal folds.1 There are myriad other potential sites of involvement including the inframammary folds, inner thighs, buttocks, and groin.1 Diagnosis is made based on history and physical exam. There is a standard severity classification scheme called the Hurley score, which stratifies disease severity based on the presence of sinus tracts and extent of disease.1 HS is associated with comorbid conditions such as obesity, overweight, acne, and inflammatory bowel and joint disease.2 This painful, persistent condition is well documented to have a negative impact on quality of life in adult patients, and similar impairment has been found in pediatric patients.3,4
HS may be increasing in pediatric and adolescent patients, with recent studies showing onset coinciding most commonly with the onset of puberty.1,2 There is often a period of several years between symptom onset and diagnosis.1 A recent editorial highlighted the disparities that exist in HS, with disease more common in Black children and limited information about disease prevalence in Hispanic children.5
What’s the treatment plan?
HS is a difficult disease to treat, with few patients achieving remission and a significant proportion of patients with treatment-refractory disease.1 There are limited studies of HS treatment in pediatric patients. Topical and systemic antibiotic therapy are mainstays of HS treatment, with tetracyclines and a combination of clindamycin plus rifampin commonly used in adults and children alike. Topical therapies including topical antibiotics and antibacterial solutions are frequently used as adjunctive therapy.6 Adalimumab, a tumor necrosis factor receptor blocker, has been Food and Drug Administration approved for HS for ages 12 and up and is currently the only FDA-approved medication for HS in pediatric patients. Our patient was started on 100 mg doxycycline twice daily, with short-dose topical corticosteroids for symptom management of the most inflamed lesions.
What’s on the differential?
Acne conglobata
Acne conglobata is an uncommon, severe variant of acne vulgaris which arise in patients with a history of acne vulgaris and presents with comedones, cysts, abscesses, and scarring with possible drainage of pus. Lesions can present diffusely on the face, back, and body, including in the axillae, groin, and buttocks, and as such can be confused with HS.7
However, in contrast with HS, patients with acne conglobata will also develop disease in non–apocrine gland–bearing skin. This patient’s lack of preceding acne and restriction of lesions to the axillae, inguinal folds, and buttocks makes acne conglobata less likely.
Epidermal inclusion cyst
Epidermal inclusion cyst (EIC) is a common cutaneous cyst, presenting as a well-circumscribed nodule(s) with a central punctum. If not excised, lesions can sometimes become infected and painful.8 In contrast with HS, EIC presents only uncommonly as multiple lesions arising in different areas, and spontaneous drainage is uncommon. Our patient’s development of multiple draining lesions makes this diagnosis unlikely.
Furunculosis
Furunculosis is a common bacterial infection of the skin, presenting with inflammatory nodules or pustules centered around the hair follicle. Lesions may commonly present at sites of skin trauma and are found most frequently on the extremities.9 Though furunculosis lesions may drain pus and can coalesce to form larger “carbuncles,” our patient’s presence of significant scarring and lack of extremity involvement makes HS more likely.
Recurrent MRSA abscesses
Methicillin-resistant Staphylococcus aureus skin and soft-tissue infections are not uncommon in the pediatric population, with presentation of infection ranging from cellulitis to fluid-containing abscesses.10 Recurrent abscesses may be seen in MRSA infection, however in this patient the presence of draining, scarring lesions in multiple locations typical for HS over time is more consistent with a diagnosis of HS.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Appiah is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Appiah have no relevant financial disclosures.
References
1. Liy-Wong C et al. JAMA Dermatol. 2021;157(4):385-91.
2. Choi E et al. J Am Acad Dermatol. 2022;86(1):140-7.
3. Machado MO et al. JAMA Dermatol. 2019;155(8):939-45.
4. McAndrew R et al. J Am Acad Dermatol. 2021;84(3):829-30.
5. Kirby JS and Zaenglein AL. JAMA Dermatol. 2021;157(4):379-80.
6. Alikhan A et al. J Am Acad Dermatol. 2019;81(1):91-101.
7. Greydanus DE et al. Dis Mon. 2021;67(4):101103.
8. Weir CB, St. Hilaire NJ. Epidermal Inclusion Cyst, in “StatPearls.” Treasure Island, Fla: StatPearls Publishing, 2021.
9. Atanaskova N and Tomecki KJ. Dermatol Clin. 2010;28(3):479-87.
10. Papastefan ST et al. J Surg Res. 2019;242:70-7.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that is becoming more recognized in children. It has a variable presentation, most commonly presenting as painful, recurrent cysts, abscesses, nodules, and/or pustules in classic locations with associated scarring and sinus tract formation.
The majority of patients present with bilateral lesions found most commonly in the axillae and inguinal folds.1 There are myriad other potential sites of involvement including the inframammary folds, inner thighs, buttocks, and groin.1 Diagnosis is made based on history and physical exam. There is a standard severity classification scheme called the Hurley score, which stratifies disease severity based on the presence of sinus tracts and extent of disease.1 HS is associated with comorbid conditions such as obesity, overweight, acne, and inflammatory bowel and joint disease.2 This painful, persistent condition is well documented to have a negative impact on quality of life in adult patients, and similar impairment has been found in pediatric patients.3,4
HS may be increasing in pediatric and adolescent patients, with recent studies showing onset coinciding most commonly with the onset of puberty.1,2 There is often a period of several years between symptom onset and diagnosis.1 A recent editorial highlighted the disparities that exist in HS, with disease more common in Black children and limited information about disease prevalence in Hispanic children.5
What’s the treatment plan?
HS is a difficult disease to treat, with few patients achieving remission and a significant proportion of patients with treatment-refractory disease.1 There are limited studies of HS treatment in pediatric patients. Topical and systemic antibiotic therapy are mainstays of HS treatment, with tetracyclines and a combination of clindamycin plus rifampin commonly used in adults and children alike. Topical therapies including topical antibiotics and antibacterial solutions are frequently used as adjunctive therapy.6 Adalimumab, a tumor necrosis factor receptor blocker, has been Food and Drug Administration approved for HS for ages 12 and up and is currently the only FDA-approved medication for HS in pediatric patients. Our patient was started on 100 mg doxycycline twice daily, with short-dose topical corticosteroids for symptom management of the most inflamed lesions.
What’s on the differential?
Acne conglobata
Acne conglobata is an uncommon, severe variant of acne vulgaris which arise in patients with a history of acne vulgaris and presents with comedones, cysts, abscesses, and scarring with possible drainage of pus. Lesions can present diffusely on the face, back, and body, including in the axillae, groin, and buttocks, and as such can be confused with HS.7
However, in contrast with HS, patients with acne conglobata will also develop disease in non–apocrine gland–bearing skin. This patient’s lack of preceding acne and restriction of lesions to the axillae, inguinal folds, and buttocks makes acne conglobata less likely.
Epidermal inclusion cyst
Epidermal inclusion cyst (EIC) is a common cutaneous cyst, presenting as a well-circumscribed nodule(s) with a central punctum. If not excised, lesions can sometimes become infected and painful.8 In contrast with HS, EIC presents only uncommonly as multiple lesions arising in different areas, and spontaneous drainage is uncommon. Our patient’s development of multiple draining lesions makes this diagnosis unlikely.
Furunculosis
Furunculosis is a common bacterial infection of the skin, presenting with inflammatory nodules or pustules centered around the hair follicle. Lesions may commonly present at sites of skin trauma and are found most frequently on the extremities.9 Though furunculosis lesions may drain pus and can coalesce to form larger “carbuncles,” our patient’s presence of significant scarring and lack of extremity involvement makes HS more likely.
Recurrent MRSA abscesses
Methicillin-resistant Staphylococcus aureus skin and soft-tissue infections are not uncommon in the pediatric population, with presentation of infection ranging from cellulitis to fluid-containing abscesses.10 Recurrent abscesses may be seen in MRSA infection, however in this patient the presence of draining, scarring lesions in multiple locations typical for HS over time is more consistent with a diagnosis of HS.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Appiah is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Appiah have no relevant financial disclosures.
References
1. Liy-Wong C et al. JAMA Dermatol. 2021;157(4):385-91.
2. Choi E et al. J Am Acad Dermatol. 2022;86(1):140-7.
3. Machado MO et al. JAMA Dermatol. 2019;155(8):939-45.
4. McAndrew R et al. J Am Acad Dermatol. 2021;84(3):829-30.
5. Kirby JS and Zaenglein AL. JAMA Dermatol. 2021;157(4):379-80.
6. Alikhan A et al. J Am Acad Dermatol. 2019;81(1):91-101.
7. Greydanus DE et al. Dis Mon. 2021;67(4):101103.
8. Weir CB, St. Hilaire NJ. Epidermal Inclusion Cyst, in “StatPearls.” Treasure Island, Fla: StatPearls Publishing, 2021.
9. Atanaskova N and Tomecki KJ. Dermatol Clin. 2010;28(3):479-87.
10. Papastefan ST et al. J Surg Res. 2019;242:70-7.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that is becoming more recognized in children. It has a variable presentation, most commonly presenting as painful, recurrent cysts, abscesses, nodules, and/or pustules in classic locations with associated scarring and sinus tract formation.
The majority of patients present with bilateral lesions found most commonly in the axillae and inguinal folds.1 There are myriad other potential sites of involvement including the inframammary folds, inner thighs, buttocks, and groin.1 Diagnosis is made based on history and physical exam. There is a standard severity classification scheme called the Hurley score, which stratifies disease severity based on the presence of sinus tracts and extent of disease.1 HS is associated with comorbid conditions such as obesity, overweight, acne, and inflammatory bowel and joint disease.2 This painful, persistent condition is well documented to have a negative impact on quality of life in adult patients, and similar impairment has been found in pediatric patients.3,4
HS may be increasing in pediatric and adolescent patients, with recent studies showing onset coinciding most commonly with the onset of puberty.1,2 There is often a period of several years between symptom onset and diagnosis.1 A recent editorial highlighted the disparities that exist in HS, with disease more common in Black children and limited information about disease prevalence in Hispanic children.5
What’s the treatment plan?
HS is a difficult disease to treat, with few patients achieving remission and a significant proportion of patients with treatment-refractory disease.1 There are limited studies of HS treatment in pediatric patients. Topical and systemic antibiotic therapy are mainstays of HS treatment, with tetracyclines and a combination of clindamycin plus rifampin commonly used in adults and children alike. Topical therapies including topical antibiotics and antibacterial solutions are frequently used as adjunctive therapy.6 Adalimumab, a tumor necrosis factor receptor blocker, has been Food and Drug Administration approved for HS for ages 12 and up and is currently the only FDA-approved medication for HS in pediatric patients. Our patient was started on 100 mg doxycycline twice daily, with short-dose topical corticosteroids for symptom management of the most inflamed lesions.
What’s on the differential?
Acne conglobata
Acne conglobata is an uncommon, severe variant of acne vulgaris which arise in patients with a history of acne vulgaris and presents with comedones, cysts, abscesses, and scarring with possible drainage of pus. Lesions can present diffusely on the face, back, and body, including in the axillae, groin, and buttocks, and as such can be confused with HS.7
However, in contrast with HS, patients with acne conglobata will also develop disease in non–apocrine gland–bearing skin. This patient’s lack of preceding acne and restriction of lesions to the axillae, inguinal folds, and buttocks makes acne conglobata less likely.
Epidermal inclusion cyst
Epidermal inclusion cyst (EIC) is a common cutaneous cyst, presenting as a well-circumscribed nodule(s) with a central punctum. If not excised, lesions can sometimes become infected and painful.8 In contrast with HS, EIC presents only uncommonly as multiple lesions arising in different areas, and spontaneous drainage is uncommon. Our patient’s development of multiple draining lesions makes this diagnosis unlikely.
Furunculosis
Furunculosis is a common bacterial infection of the skin, presenting with inflammatory nodules or pustules centered around the hair follicle. Lesions may commonly present at sites of skin trauma and are found most frequently on the extremities.9 Though furunculosis lesions may drain pus and can coalesce to form larger “carbuncles,” our patient’s presence of significant scarring and lack of extremity involvement makes HS more likely.
Recurrent MRSA abscesses
Methicillin-resistant Staphylococcus aureus skin and soft-tissue infections are not uncommon in the pediatric population, with presentation of infection ranging from cellulitis to fluid-containing abscesses.10 Recurrent abscesses may be seen in MRSA infection, however in this patient the presence of draining, scarring lesions in multiple locations typical for HS over time is more consistent with a diagnosis of HS.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Appiah is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Appiah have no relevant financial disclosures.
References
1. Liy-Wong C et al. JAMA Dermatol. 2021;157(4):385-91.
2. Choi E et al. J Am Acad Dermatol. 2022;86(1):140-7.
3. Machado MO et al. JAMA Dermatol. 2019;155(8):939-45.
4. McAndrew R et al. J Am Acad Dermatol. 2021;84(3):829-30.
5. Kirby JS and Zaenglein AL. JAMA Dermatol. 2021;157(4):379-80.
6. Alikhan A et al. J Am Acad Dermatol. 2019;81(1):91-101.
7. Greydanus DE et al. Dis Mon. 2021;67(4):101103.
8. Weir CB, St. Hilaire NJ. Epidermal Inclusion Cyst, in “StatPearls.” Treasure Island, Fla: StatPearls Publishing, 2021.
9. Atanaskova N and Tomecki KJ. Dermatol Clin. 2010;28(3):479-87.
10. Papastefan ST et al. J Surg Res. 2019;242:70-7.