User login
Dark Brown Hyperkeratotic Nodule on the Back
The Diagnosis: Seborrheic Keratosis-like Melanoma
Seborrheic keratosis (SK) is a benign neoplasm commonly encountered on the skin and frequently diagnosed by clinical examination alone. Seborrheic keratosis-like melanomas are melanomas that clinically or dermatoscopically resemble SKs and thus can be challenging to accurately diagnose. Melanomas can have a hyperkeratotic or verrucous appearance1-3 and can even exhibit dermatoscopic and microscopic features that are found in SKs such as comedolike openings and milialike cysts as well as acanthosis and pseudohorn cysts, respectively.2
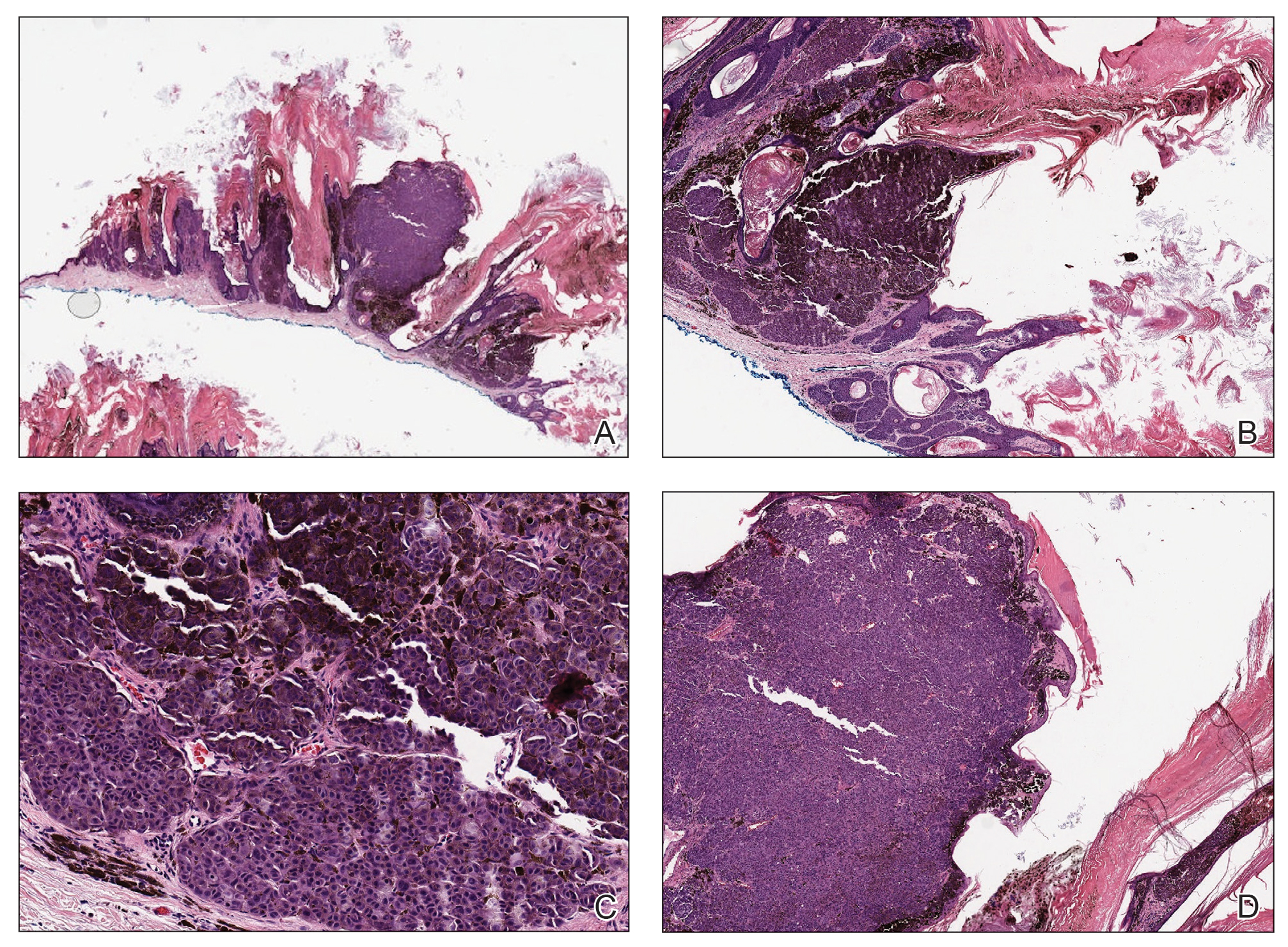
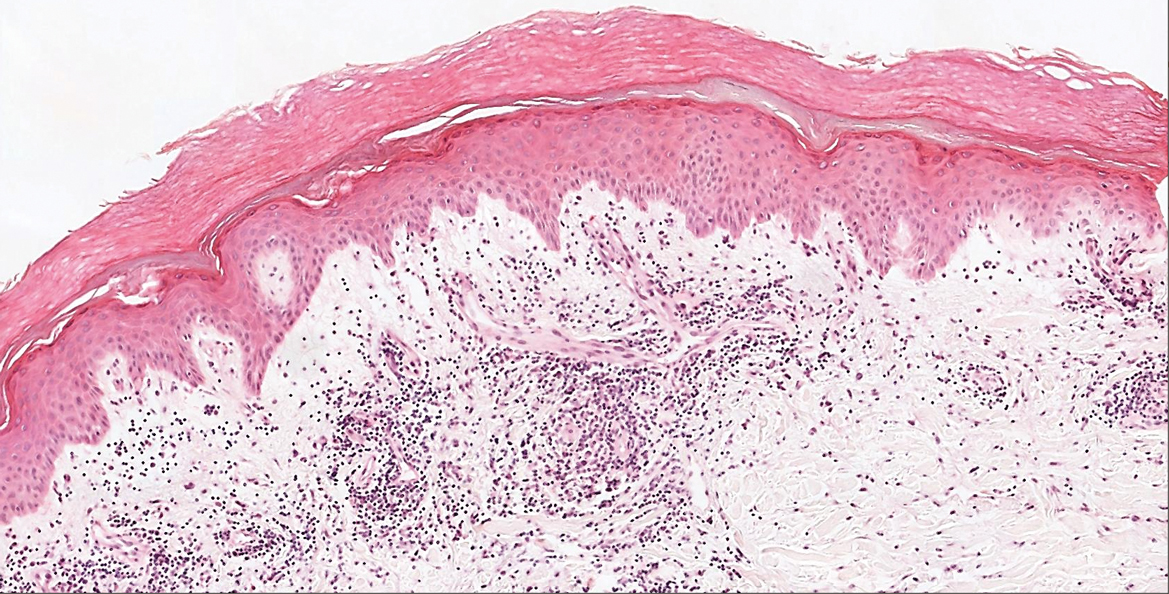
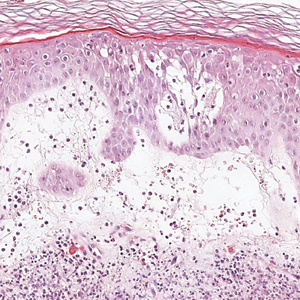
In our patient, histopathology revealed SK-like architecture with hyperorthokeratosis, papillomatosis, pseudohorn cyst formation, and basaloid acanthosis (Figure). However, within the lesion was an asymmetric proliferation of nested atypical melanocytes with melanin pigment production. The atypical melanocytes filled and expanded papillomatous projections without notable pagetoid growth and extended into the dermis. There was a background congenital nevus component. These findings were diagnostic of invasive malignant melanoma, extending to a Breslow depth of 5.5 mm. A follow-up sentinel lymph node biopsy was negative for metastatic melanoma. The clinical and histologic findings did not show melanoma in the surrounding skin to suggest colonization of an SK by an adjacent melanoma. The clinical history of a long-standing lesion in conjunction with a congenital nevus component on histology favored a diagnosis of melanoma arising in association with a congenital nevus with an SK-like architecture rather than arising in a preexisting SK or de novo melanoma.
Because our patient did not have multiple widespread SKs and reported rapid growth in the lesion in the last 6 months, there was concern for a malignant neoplasm. However, in patients with numerous SKs or areas of chronically sun-damaged skin, it can be difficult to identify suspicious lesions. It is important for clinicians to remain aware of SK-like melanomas and have a lower threshold for biopsy of any changing or symptomatic lesion that clinically resembles an SK. In our case, the history of change and the markedly different clinical appearance of the lesion in comparison to our patient's SKs prompted the biopsy. Criteria have been proposed to help differentiate these entities under dermoscopy, with melanoma showing the presence of the blue-black sign, pigment network, pseudopods or streaks, and/or the blue-white veil.4
Cutaneous metastases classically present as dermal nodules, plaques, or ulcers.5,6 A rare pigmented case of metastatic breast adenocarcinoma clinically mimicking melanoma has been reported.7 There is limited literature on the dermoscopic features of cutaneous metastases, but it appears that polymorphic vascular patterns are most common.5,8 The possibility of a metastatic melanoma involving an SK is a theoretical consideration, but there was no prior history of melanoma in our patient, and the histologic findings were consistent with primary melanoma. There was no histologic evidence of pigmented metastatic breast carcinoma or metastatic lung carcinoma.
Pigmented malignant hidroacanthoma simplex and pigmented porocarcinomas are rare malignant sweat gland tumors.9-11 Their benign counterparts are the more commonly encountered hidroacanthoma simplex (intraepidermal poroma) and poroma. Pigmented malignant hidroacanthoma simplex has been reported to clinically mimic an irritated SK.10 The histopathology of our case did not have features of malignant hidroacanthoma simplex or porocarcinoma. Pigmented squamous cell carcinoma is an uncommon variant of squamous cell carcinoma, and histopathology would reveal proliferation of atypical keratinocytes.12
- Saggini A, Cota C, Lora V, et al. Uncommon histopathological variants of malignant melanoma. part 2. Am J Dermatopathol. 2019;41:321-342.
- Klebanov N, Gunasekera N, Lin WM, et al. The clinical spectrum of cutaneous melanoma morphology. J Am Acad Dermatol. 2019;80:178-188.
- Tran PT, Truong AK, Munday W, et al. Verrucous melanoma masquerading as a seborrheic keratosis. Dermatol Online J. 2019;25:13030/qt1m07k7fm.
- Carrera C, Segura S, Aguilera P. Dermoscopic clues for diagnosing melanomas that resemble seborrheic keratosis. JAMA Dermatol. 2017;153:544-551.
- Strickley JD, Jenson AB, Jung JY. Cutaneous metastasis. Hematol Oncol Clin North Am. 2019;33:173-197.
- Chernoff KA, Marghoob AA, Lacouture ME. Dermoscopic findings in cutaneous metastases. JAMA Dermatol. 2014;150:429-433.
- Marti N, Molina I, Monteagudo C, et al. Cutaneous metastasis of breast carcinoma mimicking malignant melanoma in scalp. Dermatol Online J. 2008;14:12.
- Kelati A, Gallouj S. Dermoscopy of skin metastases from breast cancer: two case reports. J Med Case Rep. 2018;12:273.
- Ishida M, Hotta M, Kushima R, et al. A case of porocarcinoma arising in pigmented hidroacanthoma simplex with multiple lymph node, liver and bone metastases. J Cutan Pathol. 2011;38:227-231.
- Lee JY, Lin MH. Pigmented malignant hidroacanthoma simplex mimicking irritated seborrheic keratosis. J Cutan Pathol. 2006;33:705-708.
- Ueo T, Kashima K, Daa T, et al. Porocarcinoma arising in pigmented hidroacanthoma simplex. Am J Dermatopathol. 2005;27:500-503.
- Motta de Morais P, Schettini A, Rocha J, et al. Pigmented squamous cell carcinoma: case report and importance of differential diagnosis. An Bras Dermatol. 2018;93:96-98.
The Diagnosis: Seborrheic Keratosis-like Melanoma
Seborrheic keratosis (SK) is a benign neoplasm commonly encountered on the skin and frequently diagnosed by clinical examination alone. Seborrheic keratosis-like melanomas are melanomas that clinically or dermatoscopically resemble SKs and thus can be challenging to accurately diagnose. Melanomas can have a hyperkeratotic or verrucous appearance1-3 and can even exhibit dermatoscopic and microscopic features that are found in SKs such as comedolike openings and milialike cysts as well as acanthosis and pseudohorn cysts, respectively.2
In our patient, histopathology revealed SK-like architecture with hyperorthokeratosis, papillomatosis, pseudohorn cyst formation, and basaloid acanthosis (Figure). However, within the lesion was an asymmetric proliferation of nested atypical melanocytes with melanin pigment production. The atypical melanocytes filled and expanded papillomatous projections without notable pagetoid growth and extended into the dermis. There was a background congenital nevus component. These findings were diagnostic of invasive malignant melanoma, extending to a Breslow depth of 5.5 mm. A follow-up sentinel lymph node biopsy was negative for metastatic melanoma. The clinical and histologic findings did not show melanoma in the surrounding skin to suggest colonization of an SK by an adjacent melanoma. The clinical history of a long-standing lesion in conjunction with a congenital nevus component on histology favored a diagnosis of melanoma arising in association with a congenital nevus with an SK-like architecture rather than arising in a preexisting SK or de novo melanoma.
Because our patient did not have multiple widespread SKs and reported rapid growth in the lesion in the last 6 months, there was concern for a malignant neoplasm. However, in patients with numerous SKs or areas of chronically sun-damaged skin, it can be difficult to identify suspicious lesions. It is important for clinicians to remain aware of SK-like melanomas and have a lower threshold for biopsy of any changing or symptomatic lesion that clinically resembles an SK. In our case, the history of change and the markedly different clinical appearance of the lesion in comparison to our patient's SKs prompted the biopsy. Criteria have been proposed to help differentiate these entities under dermoscopy, with melanoma showing the presence of the blue-black sign, pigment network, pseudopods or streaks, and/or the blue-white veil.4
Cutaneous metastases classically present as dermal nodules, plaques, or ulcers.5,6 A rare pigmented case of metastatic breast adenocarcinoma clinically mimicking melanoma has been reported.7 There is limited literature on the dermoscopic features of cutaneous metastases, but it appears that polymorphic vascular patterns are most common.5,8 The possibility of a metastatic melanoma involving an SK is a theoretical consideration, but there was no prior history of melanoma in our patient, and the histologic findings were consistent with primary melanoma. There was no histologic evidence of pigmented metastatic breast carcinoma or metastatic lung carcinoma.
Pigmented malignant hidroacanthoma simplex and pigmented porocarcinomas are rare malignant sweat gland tumors.9-11 Their benign counterparts are the more commonly encountered hidroacanthoma simplex (intraepidermal poroma) and poroma. Pigmented malignant hidroacanthoma simplex has been reported to clinically mimic an irritated SK.10 The histopathology of our case did not have features of malignant hidroacanthoma simplex or porocarcinoma. Pigmented squamous cell carcinoma is an uncommon variant of squamous cell carcinoma, and histopathology would reveal proliferation of atypical keratinocytes.12
The Diagnosis: Seborrheic Keratosis-like Melanoma
Seborrheic keratosis (SK) is a benign neoplasm commonly encountered on the skin and frequently diagnosed by clinical examination alone. Seborrheic keratosis-like melanomas are melanomas that clinically or dermatoscopically resemble SKs and thus can be challenging to accurately diagnose. Melanomas can have a hyperkeratotic or verrucous appearance1-3 and can even exhibit dermatoscopic and microscopic features that are found in SKs such as comedolike openings and milialike cysts as well as acanthosis and pseudohorn cysts, respectively.2
In our patient, histopathology revealed SK-like architecture with hyperorthokeratosis, papillomatosis, pseudohorn cyst formation, and basaloid acanthosis (Figure). However, within the lesion was an asymmetric proliferation of nested atypical melanocytes with melanin pigment production. The atypical melanocytes filled and expanded papillomatous projections without notable pagetoid growth and extended into the dermis. There was a background congenital nevus component. These findings were diagnostic of invasive malignant melanoma, extending to a Breslow depth of 5.5 mm. A follow-up sentinel lymph node biopsy was negative for metastatic melanoma. The clinical and histologic findings did not show melanoma in the surrounding skin to suggest colonization of an SK by an adjacent melanoma. The clinical history of a long-standing lesion in conjunction with a congenital nevus component on histology favored a diagnosis of melanoma arising in association with a congenital nevus with an SK-like architecture rather than arising in a preexisting SK or de novo melanoma.
Because our patient did not have multiple widespread SKs and reported rapid growth in the lesion in the last 6 months, there was concern for a malignant neoplasm. However, in patients with numerous SKs or areas of chronically sun-damaged skin, it can be difficult to identify suspicious lesions. It is important for clinicians to remain aware of SK-like melanomas and have a lower threshold for biopsy of any changing or symptomatic lesion that clinically resembles an SK. In our case, the history of change and the markedly different clinical appearance of the lesion in comparison to our patient's SKs prompted the biopsy. Criteria have been proposed to help differentiate these entities under dermoscopy, with melanoma showing the presence of the blue-black sign, pigment network, pseudopods or streaks, and/or the blue-white veil.4
Cutaneous metastases classically present as dermal nodules, plaques, or ulcers.5,6 A rare pigmented case of metastatic breast adenocarcinoma clinically mimicking melanoma has been reported.7 There is limited literature on the dermoscopic features of cutaneous metastases, but it appears that polymorphic vascular patterns are most common.5,8 The possibility of a metastatic melanoma involving an SK is a theoretical consideration, but there was no prior history of melanoma in our patient, and the histologic findings were consistent with primary melanoma. There was no histologic evidence of pigmented metastatic breast carcinoma or metastatic lung carcinoma.
Pigmented malignant hidroacanthoma simplex and pigmented porocarcinomas are rare malignant sweat gland tumors.9-11 Their benign counterparts are the more commonly encountered hidroacanthoma simplex (intraepidermal poroma) and poroma. Pigmented malignant hidroacanthoma simplex has been reported to clinically mimic an irritated SK.10 The histopathology of our case did not have features of malignant hidroacanthoma simplex or porocarcinoma. Pigmented squamous cell carcinoma is an uncommon variant of squamous cell carcinoma, and histopathology would reveal proliferation of atypical keratinocytes.12
- Saggini A, Cota C, Lora V, et al. Uncommon histopathological variants of malignant melanoma. part 2. Am J Dermatopathol. 2019;41:321-342.
- Klebanov N, Gunasekera N, Lin WM, et al. The clinical spectrum of cutaneous melanoma morphology. J Am Acad Dermatol. 2019;80:178-188.
- Tran PT, Truong AK, Munday W, et al. Verrucous melanoma masquerading as a seborrheic keratosis. Dermatol Online J. 2019;25:13030/qt1m07k7fm.
- Carrera C, Segura S, Aguilera P. Dermoscopic clues for diagnosing melanomas that resemble seborrheic keratosis. JAMA Dermatol. 2017;153:544-551.
- Strickley JD, Jenson AB, Jung JY. Cutaneous metastasis. Hematol Oncol Clin North Am. 2019;33:173-197.
- Chernoff KA, Marghoob AA, Lacouture ME. Dermoscopic findings in cutaneous metastases. JAMA Dermatol. 2014;150:429-433.
- Marti N, Molina I, Monteagudo C, et al. Cutaneous metastasis of breast carcinoma mimicking malignant melanoma in scalp. Dermatol Online J. 2008;14:12.
- Kelati A, Gallouj S. Dermoscopy of skin metastases from breast cancer: two case reports. J Med Case Rep. 2018;12:273.
- Ishida M, Hotta M, Kushima R, et al. A case of porocarcinoma arising in pigmented hidroacanthoma simplex with multiple lymph node, liver and bone metastases. J Cutan Pathol. 2011;38:227-231.
- Lee JY, Lin MH. Pigmented malignant hidroacanthoma simplex mimicking irritated seborrheic keratosis. J Cutan Pathol. 2006;33:705-708.
- Ueo T, Kashima K, Daa T, et al. Porocarcinoma arising in pigmented hidroacanthoma simplex. Am J Dermatopathol. 2005;27:500-503.
- Motta de Morais P, Schettini A, Rocha J, et al. Pigmented squamous cell carcinoma: case report and importance of differential diagnosis. An Bras Dermatol. 2018;93:96-98.
- Saggini A, Cota C, Lora V, et al. Uncommon histopathological variants of malignant melanoma. part 2. Am J Dermatopathol. 2019;41:321-342.
- Klebanov N, Gunasekera N, Lin WM, et al. The clinical spectrum of cutaneous melanoma morphology. J Am Acad Dermatol. 2019;80:178-188.
- Tran PT, Truong AK, Munday W, et al. Verrucous melanoma masquerading as a seborrheic keratosis. Dermatol Online J. 2019;25:13030/qt1m07k7fm.
- Carrera C, Segura S, Aguilera P. Dermoscopic clues for diagnosing melanomas that resemble seborrheic keratosis. JAMA Dermatol. 2017;153:544-551.
- Strickley JD, Jenson AB, Jung JY. Cutaneous metastasis. Hematol Oncol Clin North Am. 2019;33:173-197.
- Chernoff KA, Marghoob AA, Lacouture ME. Dermoscopic findings in cutaneous metastases. JAMA Dermatol. 2014;150:429-433.
- Marti N, Molina I, Monteagudo C, et al. Cutaneous metastasis of breast carcinoma mimicking malignant melanoma in scalp. Dermatol Online J. 2008;14:12.
- Kelati A, Gallouj S. Dermoscopy of skin metastases from breast cancer: two case reports. J Med Case Rep. 2018;12:273.
- Ishida M, Hotta M, Kushima R, et al. A case of porocarcinoma arising in pigmented hidroacanthoma simplex with multiple lymph node, liver and bone metastases. J Cutan Pathol. 2011;38:227-231.
- Lee JY, Lin MH. Pigmented malignant hidroacanthoma simplex mimicking irritated seborrheic keratosis. J Cutan Pathol. 2006;33:705-708.
- Ueo T, Kashima K, Daa T, et al. Porocarcinoma arising in pigmented hidroacanthoma simplex. Am J Dermatopathol. 2005;27:500-503.
- Motta de Morais P, Schettini A, Rocha J, et al. Pigmented squamous cell carcinoma: case report and importance of differential diagnosis. An Bras Dermatol. 2018;93:96-98.
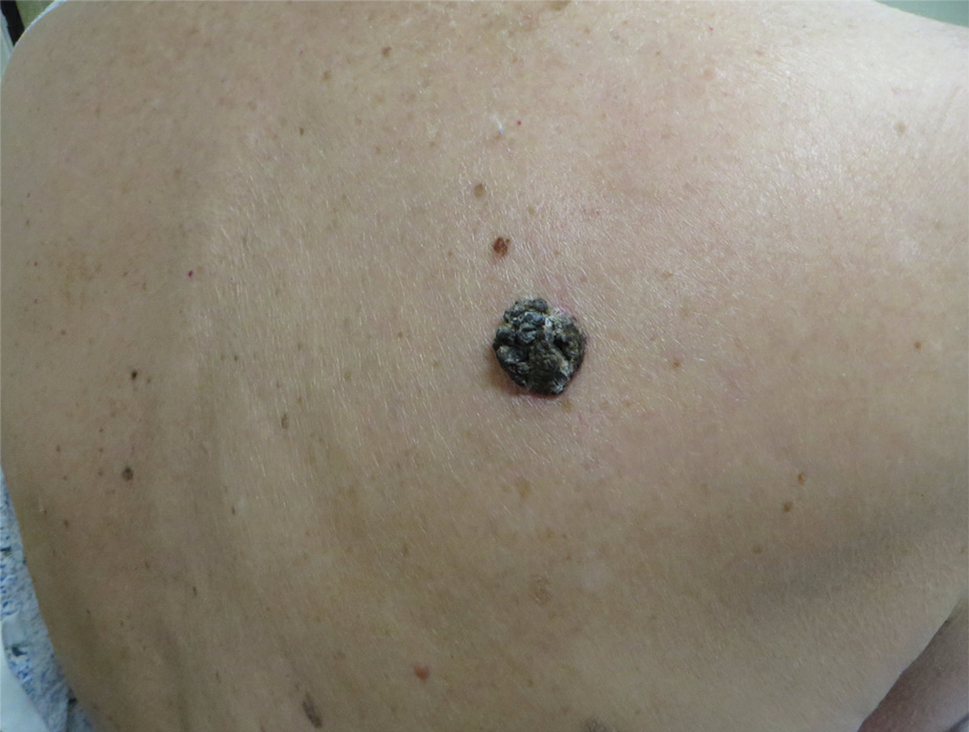
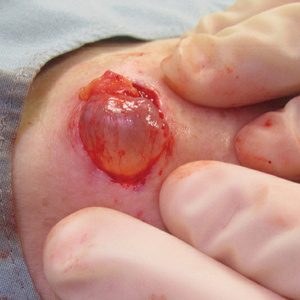
A 71-year-old woman presented with a persistent asymptomatic lesion on the right upper back that had recently increased in size and changed in color, shape, and texture. The lesion had been present for many years. Physical examination revealed a 1.5-cm, dark brown, hyperkeratotic nodule with no identifiable pigment network on dermatoscopy. The patient had no personal history of melanoma but did have a history of stage I non–small cell lung cancer. A review of systems was noncontributory. A shave biopsy of the lesion was performed.
Tender, Diffuse, Edematous, and Erythematous Papules on the Face, Neck, Chest, and Extremities
The Diagnosis: Sweet Syndrome
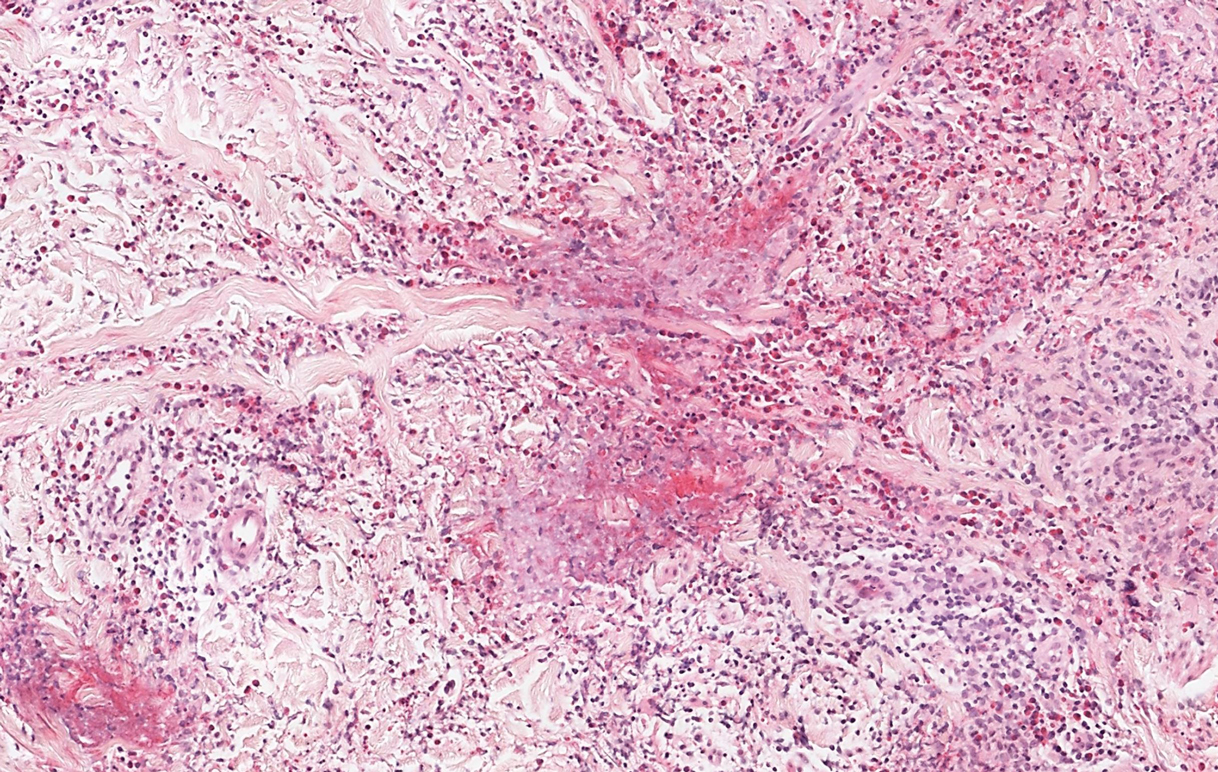
Sweet syndrome, alternatively known as acute febrile neutrophilic dermatosis, typically presents with variably tender, erythematous papules, plaques, or nodules in middle-aged adults.1 Systemic symptoms such as fever, fatigue, and arthralgia often accompany these cutaneous findings.1,2 Although the pathophysiology has not been fully elucidated, this syndrome frequently is associated with infections, especially upper respiratory illnesses; medications; and malignancies. Among cases of malignancy-associated Sweet syndrome, hematologic malignancies, particularly acute myeloid leukemia and myelodysplastic syndrome, are more common than solid organ malignancies.1,2 Sweet syndrome may precede the associated malignancy by several months; thus, patients without an identifiable trigger for Sweet syndrome should be closely followed.2 Treatment with systemic steroids typically is effective.1,3 Typical histologic features include papillary dermal edema and a brisk neutrophilic infiltrate in the superficial to mid dermis (quiz image).4 Overlying epidermal spongiosis with or without vesiculation also can be seen.4 Leukocytoclasia and endothelial swelling without fibrinoid necrosis are typical, though full-blown leukocytoclastic vasculitis can be seen.3,4 A histiocytoid variant also has been described in which the dermal infiltrate is composed of mononuclear cells reminiscent of histiocytes that are thought to be immature cells of myeloid origin. This variant histologically can simulate leukemia cutis.5
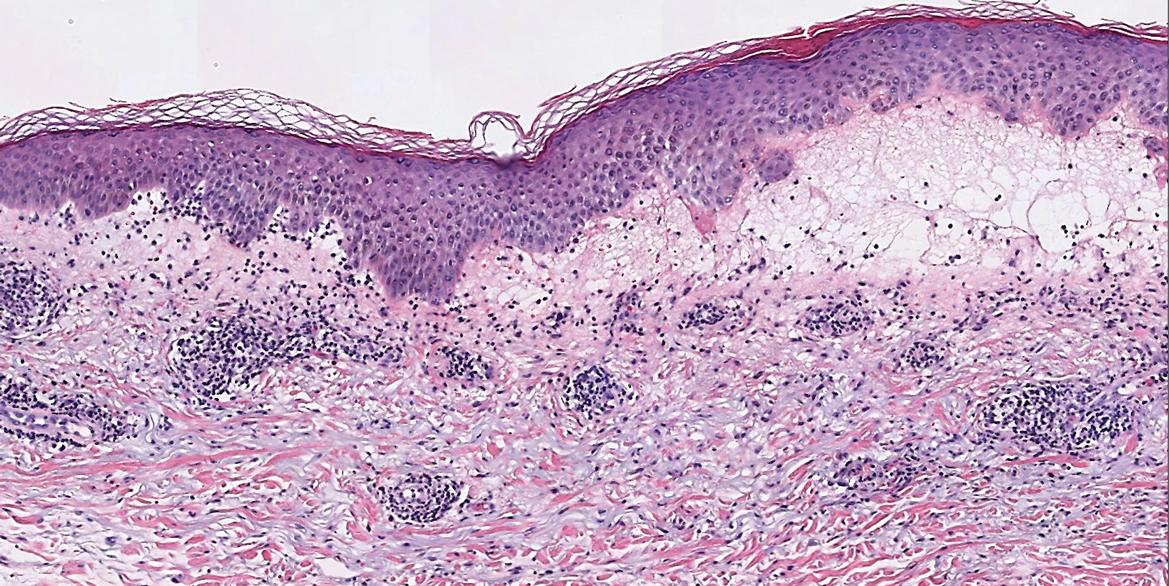
Perniosis, also known as chilblains, typically presents with red to violaceous macules or papules on acral sites, particularly the distal fingers and toes.6,7 It tends to affect young women more frequently than other demographic groups. Although the pathophysiology is not fully understood, perniosis is thought to represent an abnormal inflammatory response to cold environmental conditions. It can occur as an idiopathic disorder or in association with various systemic illnesses including lupus erythematosus.6,7 The typical histologic findings include papillary dermal edema and a lymphocytic infiltrate in the superficial to deep dermis, often with perivascular and perieccrine accentuation (Figure 1).3,6 Other less common microscopic findings include sparse keratinocyte necrosis, basal layer vacuolar change, swelling of endothelial cells, and lymphocytic vasculitis.6 The lesions typically resolve spontaneously within a few weeks, but in some cases they may be chronic.3
Polymorphous light eruption, a common photodermatosis induced by UV light exposure, typically presents in adolescence or early adulthood with a female predominance. Patients usually develop this pruritic rash on sun-exposed skin other than the face and dorsal aspects of the hands in the spring or early summer upon increased sun exposure after the winter season.3,8 Consistent sunlight exposure throughout the summer months results in decreased flares. Various cutaneous morphologies including papules, vesicles, and plaques can be seen.3,8 Histologic findings include papillary dermal edema and a perivascular lymphocytic infiltrate in the superficial to deep dermis (Figure 2).4
Tinea corporis, a superficial cutaneous dermatophyte infection, typically presents as annular scaly plaques with central clearing. Vesicles and pustules also can be seen.3 The diagnosis can be confirmed via fungal culture, identification of hyphae on microscopic examination of skin scrapings using potassium hydroxide, or cutaneous biopsy. Histologic clues to diagnosis include a "compact stratum corneum (either uniform or forming a layer beneath a basket weave stratum corneum), parakeratosis, mild spongiosis, and neutrophils in the stratum corneum" (Figure 3).9 Papillary dermal edema also may be present, though this finding less commonly is reported.9,10 Because fungal hyphae can be difficult to identify on hematoxylin and eosin-stained slides, special stains such as periodic acid-Schiff or Grocott-Gomori methenamine-silver may be helpful.9 These infections are managed with topical or oral antifungal medications.

Wells syndrome, also known as eosinophilic cellulitis, presents with an acute eruption that can clinically resemble bacterial cellulitis.3 It has been described in children and adults with various clinical morphologies including plaques, bullae, papulovesicles, and papulonodules. Peripheral eosinophilia may be present.11 The clinical lesions usually resolve spontaneously in a few weeks to months, but recurrences are typical.3,11 Histologic findings include papillary dermal edema with or without subepidermal bulla formation and epidermal spongiosis as well as a mixed inflammatory infiltrate with a predominance of eosinophils and flame figures (Figure 4).4 Flame figures are collagen fibers coated with major basic protein and other constituents of degranulated eosinophils.3 Although flame figures often are present in Wells syndrome, they are not specific to this condition.3,4 Some consider Wells syndrome an exaggerated reaction pattern rather than a specific entity.3
- Rochet N, Chavan R, Cappel M, et al. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol. 2013;69:557-564.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Elsevier; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. Elsevier Saunders; 2012.
- Alegría-Landa V, Rodríguez-Pinilla S, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Boada A, Bielsa I, Fernández-Figueras M, et al. Perniosis: clinical and histopathological analysis. Am J Dermatopathol. 2010;32:19-23.
- Takci Z, Vahaboglu G, Eksioglu H. Epidemiological patterns of perniosis, and its association with systemic disorder. Clin Exp Dermatol. 2012;37:844-849.
- Gruber-Wackernagel A, Byrne S, Wolf P. Polymorphous light eruption: clinic aspects and pathogenesis. Dermatol Clin. 2014;32:315-334.
- Elbendary A, Valdebran M, Gad A, et al. When to suspect tinea; a histopathologic study of 103 cases of PAS-positive tinea. J Cutan Pathol. 2016;46:852-857.
- Hoss D, Berke A, Kerr P, et al. Prominent papillary dermal edema in dermatophytosis (tinea corporis). J Cutan Pathol. 2010;37:237-242.
- Caputo R, Marzano A, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
The Diagnosis: Sweet Syndrome
Sweet syndrome, alternatively known as acute febrile neutrophilic dermatosis, typically presents with variably tender, erythematous papules, plaques, or nodules in middle-aged adults.1 Systemic symptoms such as fever, fatigue, and arthralgia often accompany these cutaneous findings.1,2 Although the pathophysiology has not been fully elucidated, this syndrome frequently is associated with infections, especially upper respiratory illnesses; medications; and malignancies. Among cases of malignancy-associated Sweet syndrome, hematologic malignancies, particularly acute myeloid leukemia and myelodysplastic syndrome, are more common than solid organ malignancies.1,2 Sweet syndrome may precede the associated malignancy by several months; thus, patients without an identifiable trigger for Sweet syndrome should be closely followed.2 Treatment with systemic steroids typically is effective.1,3 Typical histologic features include papillary dermal edema and a brisk neutrophilic infiltrate in the superficial to mid dermis (quiz image).4 Overlying epidermal spongiosis with or without vesiculation also can be seen.4 Leukocytoclasia and endothelial swelling without fibrinoid necrosis are typical, though full-blown leukocytoclastic vasculitis can be seen.3,4 A histiocytoid variant also has been described in which the dermal infiltrate is composed of mononuclear cells reminiscent of histiocytes that are thought to be immature cells of myeloid origin. This variant histologically can simulate leukemia cutis.5
Perniosis, also known as chilblains, typically presents with red to violaceous macules or papules on acral sites, particularly the distal fingers and toes.6,7 It tends to affect young women more frequently than other demographic groups. Although the pathophysiology is not fully understood, perniosis is thought to represent an abnormal inflammatory response to cold environmental conditions. It can occur as an idiopathic disorder or in association with various systemic illnesses including lupus erythematosus.6,7 The typical histologic findings include papillary dermal edema and a lymphocytic infiltrate in the superficial to deep dermis, often with perivascular and perieccrine accentuation (Figure 1).3,6 Other less common microscopic findings include sparse keratinocyte necrosis, basal layer vacuolar change, swelling of endothelial cells, and lymphocytic vasculitis.6 The lesions typically resolve spontaneously within a few weeks, but in some cases they may be chronic.3
Polymorphous light eruption, a common photodermatosis induced by UV light exposure, typically presents in adolescence or early adulthood with a female predominance. Patients usually develop this pruritic rash on sun-exposed skin other than the face and dorsal aspects of the hands in the spring or early summer upon increased sun exposure after the winter season.3,8 Consistent sunlight exposure throughout the summer months results in decreased flares. Various cutaneous morphologies including papules, vesicles, and plaques can be seen.3,8 Histologic findings include papillary dermal edema and a perivascular lymphocytic infiltrate in the superficial to deep dermis (Figure 2).4
Tinea corporis, a superficial cutaneous dermatophyte infection, typically presents as annular scaly plaques with central clearing. Vesicles and pustules also can be seen.3 The diagnosis can be confirmed via fungal culture, identification of hyphae on microscopic examination of skin scrapings using potassium hydroxide, or cutaneous biopsy. Histologic clues to diagnosis include a "compact stratum corneum (either uniform or forming a layer beneath a basket weave stratum corneum), parakeratosis, mild spongiosis, and neutrophils in the stratum corneum" (Figure 3).9 Papillary dermal edema also may be present, though this finding less commonly is reported.9,10 Because fungal hyphae can be difficult to identify on hematoxylin and eosin-stained slides, special stains such as periodic acid-Schiff or Grocott-Gomori methenamine-silver may be helpful.9 These infections are managed with topical or oral antifungal medications.
Wells syndrome, also known as eosinophilic cellulitis, presents with an acute eruption that can clinically resemble bacterial cellulitis.3 It has been described in children and adults with various clinical morphologies including plaques, bullae, papulovesicles, and papulonodules. Peripheral eosinophilia may be present.11 The clinical lesions usually resolve spontaneously in a few weeks to months, but recurrences are typical.3,11 Histologic findings include papillary dermal edema with or without subepidermal bulla formation and epidermal spongiosis as well as a mixed inflammatory infiltrate with a predominance of eosinophils and flame figures (Figure 4).4 Flame figures are collagen fibers coated with major basic protein and other constituents of degranulated eosinophils.3 Although flame figures often are present in Wells syndrome, they are not specific to this condition.3,4 Some consider Wells syndrome an exaggerated reaction pattern rather than a specific entity.3
The Diagnosis: Sweet Syndrome
Sweet syndrome, alternatively known as acute febrile neutrophilic dermatosis, typically presents with variably tender, erythematous papules, plaques, or nodules in middle-aged adults.1 Systemic symptoms such as fever, fatigue, and arthralgia often accompany these cutaneous findings.1,2 Although the pathophysiology has not been fully elucidated, this syndrome frequently is associated with infections, especially upper respiratory illnesses; medications; and malignancies. Among cases of malignancy-associated Sweet syndrome, hematologic malignancies, particularly acute myeloid leukemia and myelodysplastic syndrome, are more common than solid organ malignancies.1,2 Sweet syndrome may precede the associated malignancy by several months; thus, patients without an identifiable trigger for Sweet syndrome should be closely followed.2 Treatment with systemic steroids typically is effective.1,3 Typical histologic features include papillary dermal edema and a brisk neutrophilic infiltrate in the superficial to mid dermis (quiz image).4 Overlying epidermal spongiosis with or without vesiculation also can be seen.4 Leukocytoclasia and endothelial swelling without fibrinoid necrosis are typical, though full-blown leukocytoclastic vasculitis can be seen.3,4 A histiocytoid variant also has been described in which the dermal infiltrate is composed of mononuclear cells reminiscent of histiocytes that are thought to be immature cells of myeloid origin. This variant histologically can simulate leukemia cutis.5
Perniosis, also known as chilblains, typically presents with red to violaceous macules or papules on acral sites, particularly the distal fingers and toes.6,7 It tends to affect young women more frequently than other demographic groups. Although the pathophysiology is not fully understood, perniosis is thought to represent an abnormal inflammatory response to cold environmental conditions. It can occur as an idiopathic disorder or in association with various systemic illnesses including lupus erythematosus.6,7 The typical histologic findings include papillary dermal edema and a lymphocytic infiltrate in the superficial to deep dermis, often with perivascular and perieccrine accentuation (Figure 1).3,6 Other less common microscopic findings include sparse keratinocyte necrosis, basal layer vacuolar change, swelling of endothelial cells, and lymphocytic vasculitis.6 The lesions typically resolve spontaneously within a few weeks, but in some cases they may be chronic.3
Polymorphous light eruption, a common photodermatosis induced by UV light exposure, typically presents in adolescence or early adulthood with a female predominance. Patients usually develop this pruritic rash on sun-exposed skin other than the face and dorsal aspects of the hands in the spring or early summer upon increased sun exposure after the winter season.3,8 Consistent sunlight exposure throughout the summer months results in decreased flares. Various cutaneous morphologies including papules, vesicles, and plaques can be seen.3,8 Histologic findings include papillary dermal edema and a perivascular lymphocytic infiltrate in the superficial to deep dermis (Figure 2).4
Tinea corporis, a superficial cutaneous dermatophyte infection, typically presents as annular scaly plaques with central clearing. Vesicles and pustules also can be seen.3 The diagnosis can be confirmed via fungal culture, identification of hyphae on microscopic examination of skin scrapings using potassium hydroxide, or cutaneous biopsy. Histologic clues to diagnosis include a "compact stratum corneum (either uniform or forming a layer beneath a basket weave stratum corneum), parakeratosis, mild spongiosis, and neutrophils in the stratum corneum" (Figure 3).9 Papillary dermal edema also may be present, though this finding less commonly is reported.9,10 Because fungal hyphae can be difficult to identify on hematoxylin and eosin-stained slides, special stains such as periodic acid-Schiff or Grocott-Gomori methenamine-silver may be helpful.9 These infections are managed with topical or oral antifungal medications.
Wells syndrome, also known as eosinophilic cellulitis, presents with an acute eruption that can clinically resemble bacterial cellulitis.3 It has been described in children and adults with various clinical morphologies including plaques, bullae, papulovesicles, and papulonodules. Peripheral eosinophilia may be present.11 The clinical lesions usually resolve spontaneously in a few weeks to months, but recurrences are typical.3,11 Histologic findings include papillary dermal edema with or without subepidermal bulla formation and epidermal spongiosis as well as a mixed inflammatory infiltrate with a predominance of eosinophils and flame figures (Figure 4).4 Flame figures are collagen fibers coated with major basic protein and other constituents of degranulated eosinophils.3 Although flame figures often are present in Wells syndrome, they are not specific to this condition.3,4 Some consider Wells syndrome an exaggerated reaction pattern rather than a specific entity.3
- Rochet N, Chavan R, Cappel M, et al. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol. 2013;69:557-564.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Elsevier; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. Elsevier Saunders; 2012.
- Alegría-Landa V, Rodríguez-Pinilla S, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Boada A, Bielsa I, Fernández-Figueras M, et al. Perniosis: clinical and histopathological analysis. Am J Dermatopathol. 2010;32:19-23.
- Takci Z, Vahaboglu G, Eksioglu H. Epidemiological patterns of perniosis, and its association with systemic disorder. Clin Exp Dermatol. 2012;37:844-849.
- Gruber-Wackernagel A, Byrne S, Wolf P. Polymorphous light eruption: clinic aspects and pathogenesis. Dermatol Clin. 2014;32:315-334.
- Elbendary A, Valdebran M, Gad A, et al. When to suspect tinea; a histopathologic study of 103 cases of PAS-positive tinea. J Cutan Pathol. 2016;46:852-857.
- Hoss D, Berke A, Kerr P, et al. Prominent papillary dermal edema in dermatophytosis (tinea corporis). J Cutan Pathol. 2010;37:237-242.
- Caputo R, Marzano A, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Rochet N, Chavan R, Cappel M, et al. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol. 2013;69:557-564.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, et al. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41:741-746.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Elsevier; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. Elsevier Saunders; 2012.
- Alegría-Landa V, Rodríguez-Pinilla S, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Boada A, Bielsa I, Fernández-Figueras M, et al. Perniosis: clinical and histopathological analysis. Am J Dermatopathol. 2010;32:19-23.
- Takci Z, Vahaboglu G, Eksioglu H. Epidemiological patterns of perniosis, and its association with systemic disorder. Clin Exp Dermatol. 2012;37:844-849.
- Gruber-Wackernagel A, Byrne S, Wolf P. Polymorphous light eruption: clinic aspects and pathogenesis. Dermatol Clin. 2014;32:315-334.
- Elbendary A, Valdebran M, Gad A, et al. When to suspect tinea; a histopathologic study of 103 cases of PAS-positive tinea. J Cutan Pathol. 2016;46:852-857.
- Hoss D, Berke A, Kerr P, et al. Prominent papillary dermal edema in dermatophytosis (tinea corporis). J Cutan Pathol. 2010;37:237-242.
- Caputo R, Marzano A, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
A 62-year-old woman presented with a tender diffuse eruption of erythematous and edematous papules and plaques on the face, neck, chest, and extremities, some appearing vesiculopustular.
Asymptomatic Hemorrhagic Lesions in an Anemic Woman
The Diagnosis: Bullous Amyloidosis
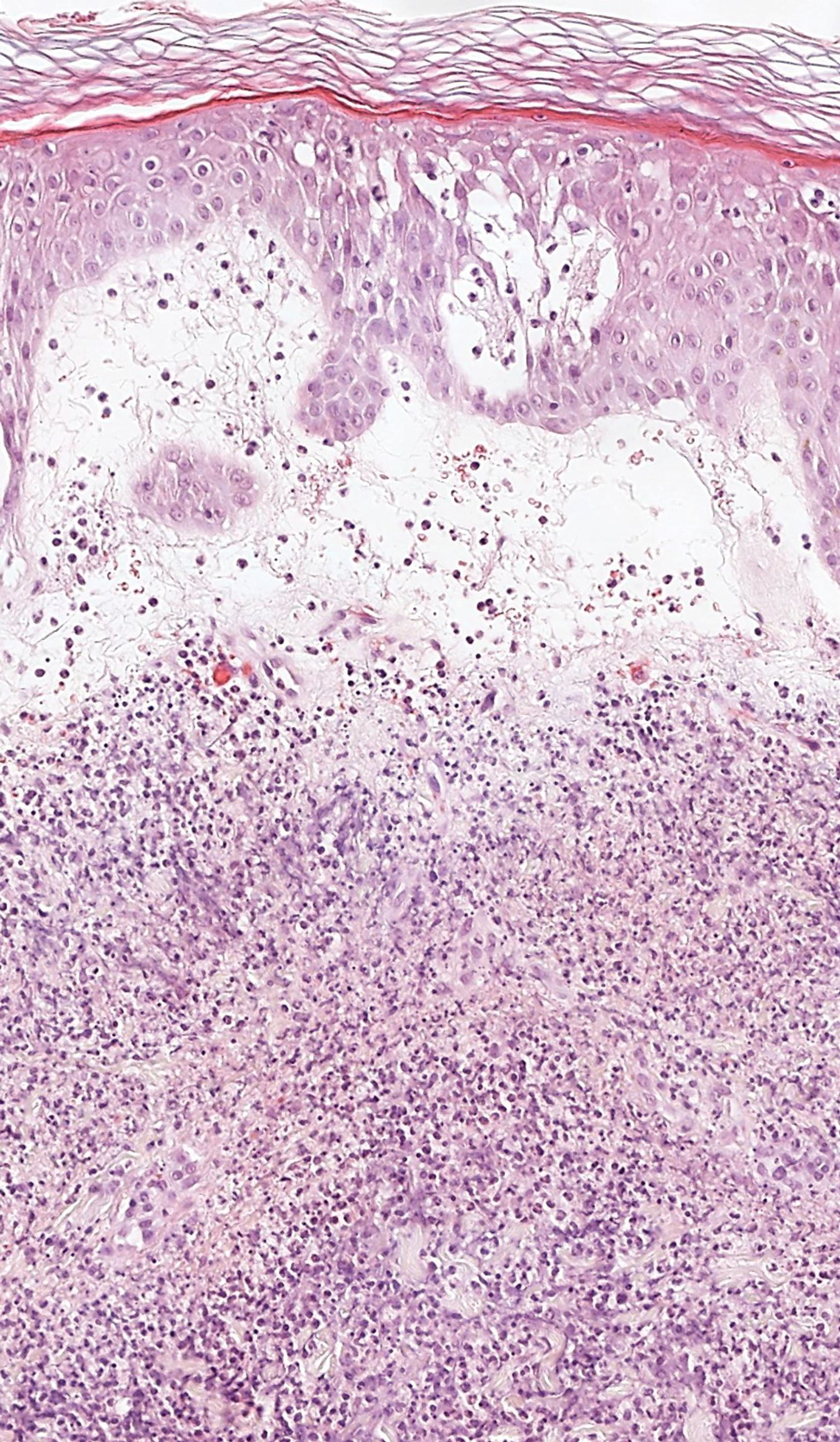
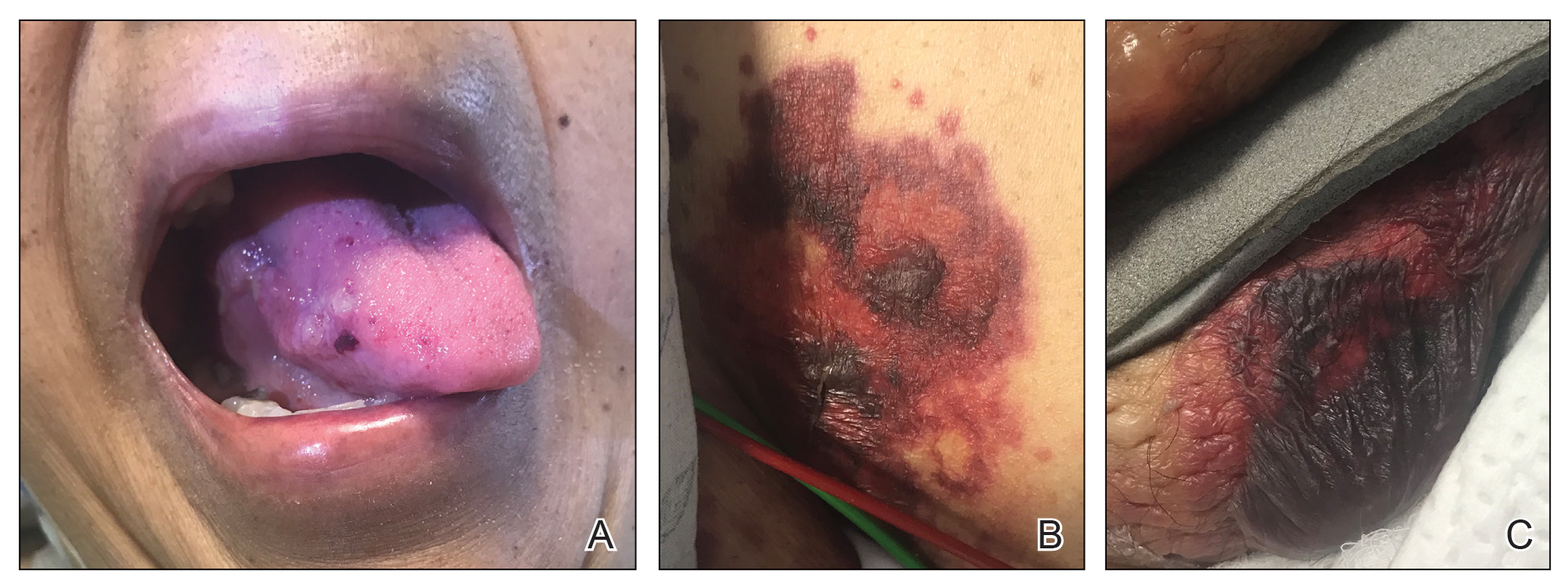
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
The Diagnosis: Bullous Amyloidosis
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
The Diagnosis: Bullous Amyloidosis
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
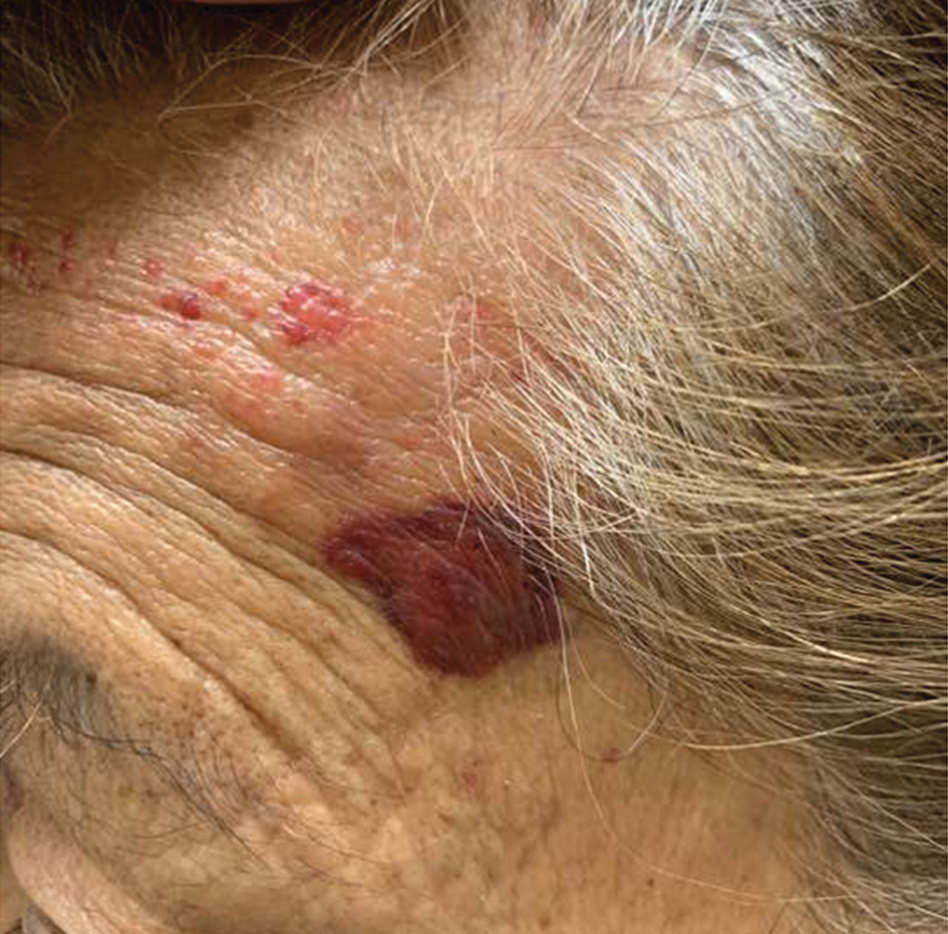
A 67-year-old woman with a medical history of type 2 diabetes mellitus, unspecified leukocytosis, and anemia presented to the dermatology clinic with asymptomatic hemorrhagic bullae on the face, chest, and tongue, as well as a large, tender, tense, hemorrhagic bulla on the groin of 3 to 4 months’ duration. A review of systems was negative for fever, chills, night sweats, malaise, shortness of breath, and dyspnea on exertion. A complete blood cell count showed mild leukocytosis, anemia, and thrombocytopenia. Her creatinine level was slightly elevated. Chest computed tomography showed early pulmonary fibrosis and coronary artery calcification. An echocardiogram showed diastolic dysfunction with moderate left ventricle thickening. A serum and urine electrophoresis demonstrated elevated free λ light chains with an M-spike. A punch biopsy was performed.
Isolated Perianal Erosive Lichen Planus: A Diagnostic Challenge
To the Editor:
Erosive lichen planus (LP) often is painful, debilitating, and resistant to topical therapy making it both a diagnostic and therapeutic challenge. We report the case of an elderly woman with isolated perianal erosive LP, a rare clinical manifestation. We also review cases of erosive perianal LP reported in the literature.
A 72-year-old woman was referred to our dermatology clinic for evaluation of multiple pruritic and painful perianal lesions of 1 year’s duration. The lesions had remained stable since onset, with no other reported lesions elsewhere on body, including the mucosae. Her medical history was notable for rheumatoid arthritis, osteoporosis, hypercholesterolemia, and hypertension. She was taking methotrexate, folic acid, abatacept, alendronate, atorvastatin, and lisinopril. The patient reported she had been using abatacept for 3 years and lisinopril for 2 years. Her primary care physician initially treated the lesions as hemorrhoids but referred her to a gastroenterologist when they failed to improve. Gastroenterology evaluated the patient, and a colonoscopy was performed with unremarkable results. Thus, she was referred to dermatology for further evaluation.
Physical examination revealed 2 tender, sharply defined, angulated erosions with irregular violaceous borders involving the perianal skin (Figure 1). A biopsy of one of the lesions was taken. Histopathologic examination revealed acanthosis of the epidermis with slight compact hyperkeratosis, scattered dyskeratotic keratinocytes, and a dense bandlike lymphohistiocytic infiltrate that obliterated the dermoepidermal junction (Figure 2). A diagnosis of perianal erosive LP was made. The patient was prescribed mometasone ointment 0.1% daily with notable improvement after 2 months.
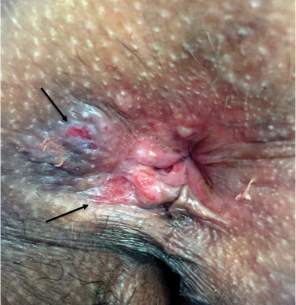
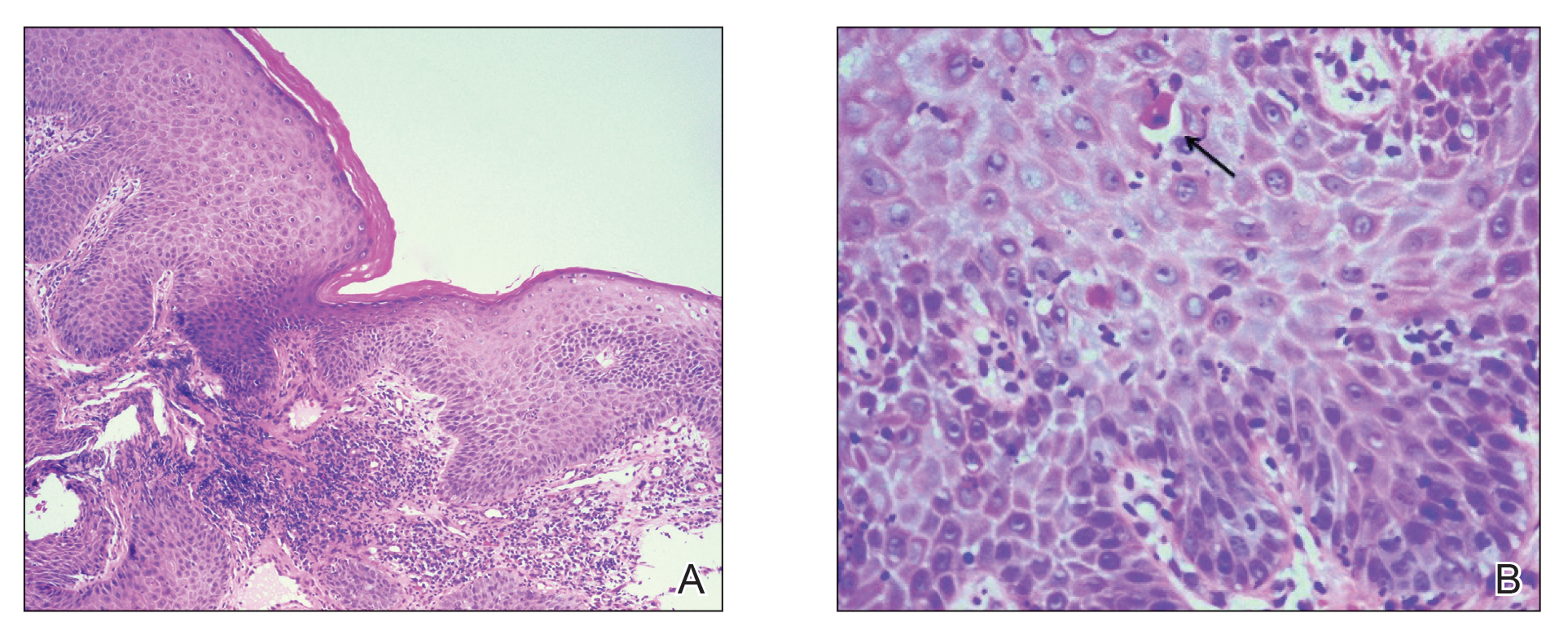
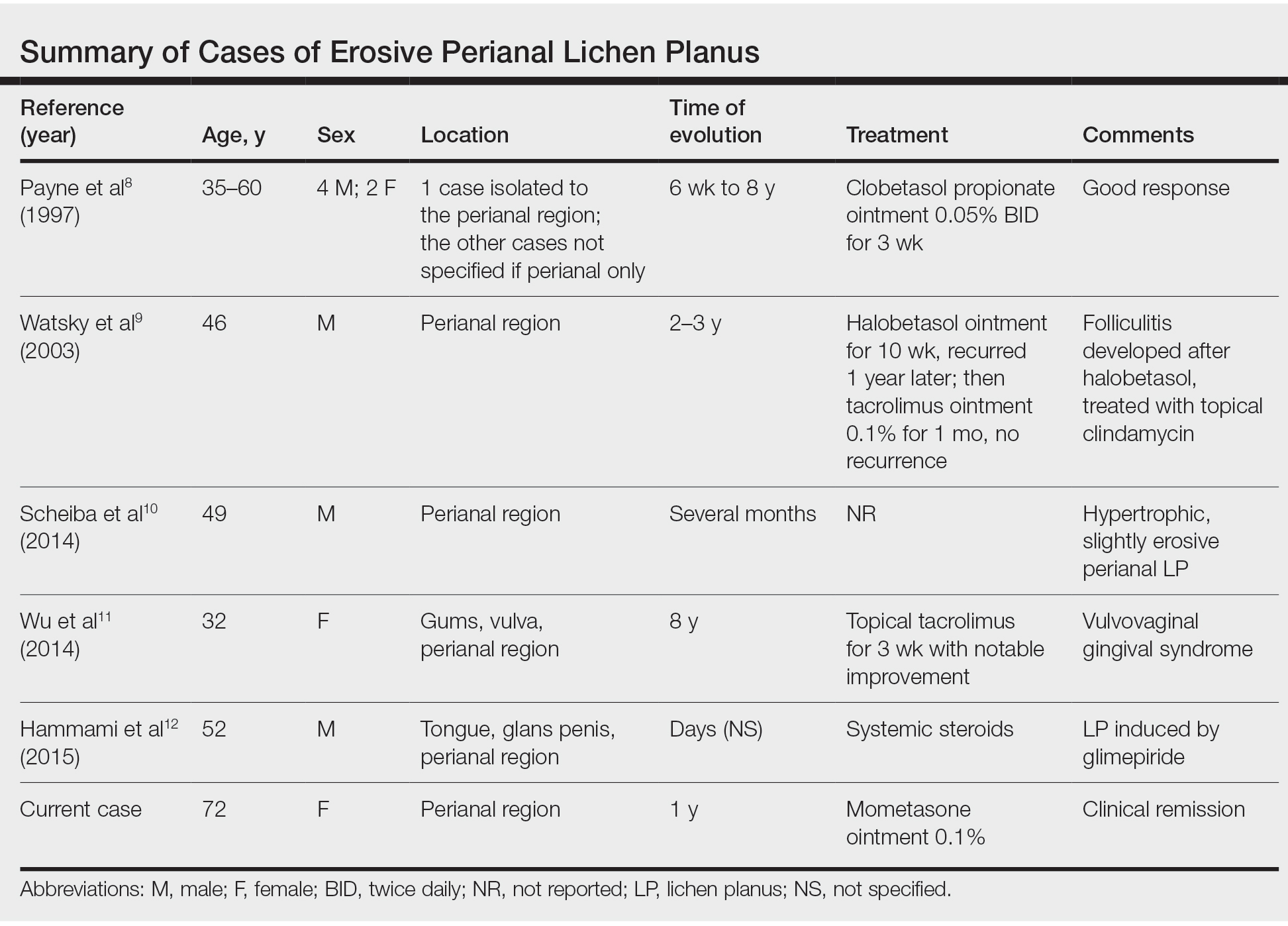
Erosive LP is an extremely rare variant of LP.1 It typically manifests as chronic painful erosions that often can progress to scarring, ulceration, and tissue destruction. Although erosive LP most commonly involves the mucosal surfaces of the genitalia and oral mucosa, it also has been reported in the palmoplantar skin, lacrimal duct, external auditory meatus, and esophagus.2-7 However, isolated perianal involvement is extremely rare. A PubMed search of articles indexed for MEDLINE using the terms erosive or ulcerative and lichen planus and perianal revealed 10 cases of perianal erosive LP, and weak data exist regarding therapy (Table).8-12 Of these cases, only 3 reported isolated perianal involvement.8-10 In most reported cases, perianal involvement manifested as extremely painful and occasionally pruritic, sharply angulated erosions and ulcers arising 0.5 to 3 cm from the anus with macerated, whitish, and violaceous borders. Most of the lesions occurred unilaterally, with only 1 case of bilateral perianal involvement.10
The differential diagnosis of perianal erosions is extensive and includes cutaneous Crohn disease, extramammary Paget disease, cutaneous malignancy, herpes simplex virus, cytomegalovirus, external hemorrhoids, lichen sclerosus, Behçet disease, lichen simplex chronicus, and drug-induced lichenoid reaction, among others. It is worth emphasizing infectious processes and cutaneous malignancies in light of our patient’s immunosuppression. Perianal cytomegalovirus has been reported in the literature in association with HIV, and it is a clinically challenging diagnosis.13 Cutaneous malignancy associated with the use of methotrexate also was considered in the differential diagnosis for our patient, given the increased risk for nonmelanoma skin cancer with the use of immunosuppresants.14
Along with a thorough patient history and physical examination, skin biopsy and clinicopathologic correlation are key to determine the exact etiology. Histologically, LP is characterized by a lichenoid interface dermatitis with a dense bandlike lymphohistiocytic infiltrate at the dermoepidermal junction. Other distinguishing factors include irregular acanthosis, hyperkeratosis, basal cell vacuolar degeneration, and Civatte bodies. Drug-induced LP is a possibility, but it is unclear if abatacept or lisinopril may have played a role in our patient. However, absence of eosinophils and parakeratosis suggested an idiopathic rather than drug-induced etiology. In 2016, Day et al2 published a clinicopathologic review of 60 cases of perianal lichenoid dermatoses in which only 17% of lesions were LP. Of note, 90% of perianal LP lesions were of the hypertrophic variant, and none were of the erosive variant, further supporting that our case represents a rare clinical manifestation of perianal LP.
Treatment of LP varies depending on the location and subtype of the lesions and is primarily aimed at improving symptoms. Topical corticosteroids are the standard treatment of LP; however, there is limited evidence regarding their efficacy for mucosal LP. Although randomized controlled trials assessing the efficacy of different interventions on oral erosive LP are available in the literature,15 there is a paucity of studies addressing this topic for genital or perianal LP. A review of the literature regarding perianal erosive LP suggests good response to high-potency topical steroids and calcineurin inhibitors with resolution of lesions within 3 to 4 weeks.11,15-18
Erosive LP is a painful variant that can cause erosions, ulcerations, and scarring. It rarely is seen in the perianal region alone and presents a diagnostic challenge. Treatment with high-potency topical steroid therapy seems to be effective in the few cases that have been reported as well as in our case. More comprehensive data from randomized controlled trials would be needed to evaluate their efficacy compared to other therapies.
- Rebora A. Erosive lichen planus: what is this? Dermatology. 2002;205:226-228; discussion 227.
- Day T, Bohl TG, Scurry J. Perianal lichen dermatoses: a review of 60 cases. Australas J Dermatol. 2016;57:210-215.
- Fox LP, Lightdale CJ, Grossman ME. Lichen planus of the esophagus: what dermatologists need to know. J Am Acad Dermatol. 2011;65:175-883.
- Holmstrup P, Thorn JJ, Rindum J, et al. Malignant development of lichen planus-affected oral mucosa. J Oral Pathol. 1988;17:219-225.
- Lewi, FM, Bogliatto F. Erosive vulval lichen planus—a diagnosis not to be missed: a clinical review. Eur J Obstet Gynecol Reprod Biol. 2013;171:214-219.
- Webber NK, Setterfield JF, Lewis FM, et al. Lacrimal canalicular duct scarring in patients with lichen planus. Arch Dermatol. 2012;148:224-227.
- Martin L, Moriniere S, Machet MC, et al. Bilateral conductive deafness related to erosive lichen planus. J Laryngol Otol. 1998;112:365-366.
- Payne CM, McPartlin JF, Hawley PR. Ulcerative perianal lichen planus. Br J Dermatol. 1997;136:479.
- Watsky KL. Erosive perianal lichen planus responsive to tacrolimus. Int J Dermatol. 2003;42:217-218.
- Scheiba N, Toberer F, Lenhard BH, et al. Erythema and erosions of the perianal region in a 49-year-old man. J Dtsch Dermatol Ges. 2014;12:162-165.
- Wu Y, Qiao J, Fang H. Syndrome in question. An Bras Dermatol. 2014;89:843-844.
- Hammami S, Ksouda K, Affes H, et al. Mucosal lichenoid drug reaction associated with glimepiride: a case report. Eur Rev Med Pharmacol Sci. 2015;19:2301-2302.
- Meyerle JH, Turiansky GW. Perianal ulcer in a patient with AIDS. Arch Dermatol. 2004;140:877-882.
- Scott FI, Mamtani R, Brensinger CM, et al. Risk of nonmelanoma skin cancer associated with the use of immunosuppressant and biologic agents in patients with a history of autoimmune disease and nonmelanoma skin cancer. JAMA Dermatol. 2016;152:164-172.
- Cheng S, Kirtschig G, Cooper S, et al. Interventions for erosive lichen planus affecting mucosal sites. Cochrane Database Syst Rev. 2012:Cd008092.
- Gunther S. Effect of retinoic acid in lichen planus of the genitalia and perianal region. Br J Vener Dis. 1973;49:553-554.
- Vente C, Reich K, Neumann C. Erosive mucosal lichen planus: response to topical treatment with tacrolimus. Br J Dermatol. 1999;140:338-342.
- Lonsdale-Eccles AA, Velangi S. Topical pimecrolimus in the treatment of genital lichen planus: a prospective case series. Br J Dermatol. 2005;153:390-394.
To the Editor:
Erosive lichen planus (LP) often is painful, debilitating, and resistant to topical therapy making it both a diagnostic and therapeutic challenge. We report the case of an elderly woman with isolated perianal erosive LP, a rare clinical manifestation. We also review cases of erosive perianal LP reported in the literature.
A 72-year-old woman was referred to our dermatology clinic for evaluation of multiple pruritic and painful perianal lesions of 1 year’s duration. The lesions had remained stable since onset, with no other reported lesions elsewhere on body, including the mucosae. Her medical history was notable for rheumatoid arthritis, osteoporosis, hypercholesterolemia, and hypertension. She was taking methotrexate, folic acid, abatacept, alendronate, atorvastatin, and lisinopril. The patient reported she had been using abatacept for 3 years and lisinopril for 2 years. Her primary care physician initially treated the lesions as hemorrhoids but referred her to a gastroenterologist when they failed to improve. Gastroenterology evaluated the patient, and a colonoscopy was performed with unremarkable results. Thus, she was referred to dermatology for further evaluation.
Physical examination revealed 2 tender, sharply defined, angulated erosions with irregular violaceous borders involving the perianal skin (Figure 1). A biopsy of one of the lesions was taken. Histopathologic examination revealed acanthosis of the epidermis with slight compact hyperkeratosis, scattered dyskeratotic keratinocytes, and a dense bandlike lymphohistiocytic infiltrate that obliterated the dermoepidermal junction (Figure 2). A diagnosis of perianal erosive LP was made. The patient was prescribed mometasone ointment 0.1% daily with notable improvement after 2 months.
Erosive LP is an extremely rare variant of LP.1 It typically manifests as chronic painful erosions that often can progress to scarring, ulceration, and tissue destruction. Although erosive LP most commonly involves the mucosal surfaces of the genitalia and oral mucosa, it also has been reported in the palmoplantar skin, lacrimal duct, external auditory meatus, and esophagus.2-7 However, isolated perianal involvement is extremely rare. A PubMed search of articles indexed for MEDLINE using the terms erosive or ulcerative and lichen planus and perianal revealed 10 cases of perianal erosive LP, and weak data exist regarding therapy (Table).8-12 Of these cases, only 3 reported isolated perianal involvement.8-10 In most reported cases, perianal involvement manifested as extremely painful and occasionally pruritic, sharply angulated erosions and ulcers arising 0.5 to 3 cm from the anus with macerated, whitish, and violaceous borders. Most of the lesions occurred unilaterally, with only 1 case of bilateral perianal involvement.10
The differential diagnosis of perianal erosions is extensive and includes cutaneous Crohn disease, extramammary Paget disease, cutaneous malignancy, herpes simplex virus, cytomegalovirus, external hemorrhoids, lichen sclerosus, Behçet disease, lichen simplex chronicus, and drug-induced lichenoid reaction, among others. It is worth emphasizing infectious processes and cutaneous malignancies in light of our patient’s immunosuppression. Perianal cytomegalovirus has been reported in the literature in association with HIV, and it is a clinically challenging diagnosis.13 Cutaneous malignancy associated with the use of methotrexate also was considered in the differential diagnosis for our patient, given the increased risk for nonmelanoma skin cancer with the use of immunosuppresants.14
Along with a thorough patient history and physical examination, skin biopsy and clinicopathologic correlation are key to determine the exact etiology. Histologically, LP is characterized by a lichenoid interface dermatitis with a dense bandlike lymphohistiocytic infiltrate at the dermoepidermal junction. Other distinguishing factors include irregular acanthosis, hyperkeratosis, basal cell vacuolar degeneration, and Civatte bodies. Drug-induced LP is a possibility, but it is unclear if abatacept or lisinopril may have played a role in our patient. However, absence of eosinophils and parakeratosis suggested an idiopathic rather than drug-induced etiology. In 2016, Day et al2 published a clinicopathologic review of 60 cases of perianal lichenoid dermatoses in which only 17% of lesions were LP. Of note, 90% of perianal LP lesions were of the hypertrophic variant, and none were of the erosive variant, further supporting that our case represents a rare clinical manifestation of perianal LP.
Treatment of LP varies depending on the location and subtype of the lesions and is primarily aimed at improving symptoms. Topical corticosteroids are the standard treatment of LP; however, there is limited evidence regarding their efficacy for mucosal LP. Although randomized controlled trials assessing the efficacy of different interventions on oral erosive LP are available in the literature,15 there is a paucity of studies addressing this topic for genital or perianal LP. A review of the literature regarding perianal erosive LP suggests good response to high-potency topical steroids and calcineurin inhibitors with resolution of lesions within 3 to 4 weeks.11,15-18
Erosive LP is a painful variant that can cause erosions, ulcerations, and scarring. It rarely is seen in the perianal region alone and presents a diagnostic challenge. Treatment with high-potency topical steroid therapy seems to be effective in the few cases that have been reported as well as in our case. More comprehensive data from randomized controlled trials would be needed to evaluate their efficacy compared to other therapies.
To the Editor:
Erosive lichen planus (LP) often is painful, debilitating, and resistant to topical therapy making it both a diagnostic and therapeutic challenge. We report the case of an elderly woman with isolated perianal erosive LP, a rare clinical manifestation. We also review cases of erosive perianal LP reported in the literature.
A 72-year-old woman was referred to our dermatology clinic for evaluation of multiple pruritic and painful perianal lesions of 1 year’s duration. The lesions had remained stable since onset, with no other reported lesions elsewhere on body, including the mucosae. Her medical history was notable for rheumatoid arthritis, osteoporosis, hypercholesterolemia, and hypertension. She was taking methotrexate, folic acid, abatacept, alendronate, atorvastatin, and lisinopril. The patient reported she had been using abatacept for 3 years and lisinopril for 2 years. Her primary care physician initially treated the lesions as hemorrhoids but referred her to a gastroenterologist when they failed to improve. Gastroenterology evaluated the patient, and a colonoscopy was performed with unremarkable results. Thus, she was referred to dermatology for further evaluation.
Physical examination revealed 2 tender, sharply defined, angulated erosions with irregular violaceous borders involving the perianal skin (Figure 1). A biopsy of one of the lesions was taken. Histopathologic examination revealed acanthosis of the epidermis with slight compact hyperkeratosis, scattered dyskeratotic keratinocytes, and a dense bandlike lymphohistiocytic infiltrate that obliterated the dermoepidermal junction (Figure 2). A diagnosis of perianal erosive LP was made. The patient was prescribed mometasone ointment 0.1% daily with notable improvement after 2 months.
Erosive LP is an extremely rare variant of LP.1 It typically manifests as chronic painful erosions that often can progress to scarring, ulceration, and tissue destruction. Although erosive LP most commonly involves the mucosal surfaces of the genitalia and oral mucosa, it also has been reported in the palmoplantar skin, lacrimal duct, external auditory meatus, and esophagus.2-7 However, isolated perianal involvement is extremely rare. A PubMed search of articles indexed for MEDLINE using the terms erosive or ulcerative and lichen planus and perianal revealed 10 cases of perianal erosive LP, and weak data exist regarding therapy (Table).8-12 Of these cases, only 3 reported isolated perianal involvement.8-10 In most reported cases, perianal involvement manifested as extremely painful and occasionally pruritic, sharply angulated erosions and ulcers arising 0.5 to 3 cm from the anus with macerated, whitish, and violaceous borders. Most of the lesions occurred unilaterally, with only 1 case of bilateral perianal involvement.10
The differential diagnosis of perianal erosions is extensive and includes cutaneous Crohn disease, extramammary Paget disease, cutaneous malignancy, herpes simplex virus, cytomegalovirus, external hemorrhoids, lichen sclerosus, Behçet disease, lichen simplex chronicus, and drug-induced lichenoid reaction, among others. It is worth emphasizing infectious processes and cutaneous malignancies in light of our patient’s immunosuppression. Perianal cytomegalovirus has been reported in the literature in association with HIV, and it is a clinically challenging diagnosis.13 Cutaneous malignancy associated with the use of methotrexate also was considered in the differential diagnosis for our patient, given the increased risk for nonmelanoma skin cancer with the use of immunosuppresants.14
Along with a thorough patient history and physical examination, skin biopsy and clinicopathologic correlation are key to determine the exact etiology. Histologically, LP is characterized by a lichenoid interface dermatitis with a dense bandlike lymphohistiocytic infiltrate at the dermoepidermal junction. Other distinguishing factors include irregular acanthosis, hyperkeratosis, basal cell vacuolar degeneration, and Civatte bodies. Drug-induced LP is a possibility, but it is unclear if abatacept or lisinopril may have played a role in our patient. However, absence of eosinophils and parakeratosis suggested an idiopathic rather than drug-induced etiology. In 2016, Day et al2 published a clinicopathologic review of 60 cases of perianal lichenoid dermatoses in which only 17% of lesions were LP. Of note, 90% of perianal LP lesions were of the hypertrophic variant, and none were of the erosive variant, further supporting that our case represents a rare clinical manifestation of perianal LP.
Treatment of LP varies depending on the location and subtype of the lesions and is primarily aimed at improving symptoms. Topical corticosteroids are the standard treatment of LP; however, there is limited evidence regarding their efficacy for mucosal LP. Although randomized controlled trials assessing the efficacy of different interventions on oral erosive LP are available in the literature,15 there is a paucity of studies addressing this topic for genital or perianal LP. A review of the literature regarding perianal erosive LP suggests good response to high-potency topical steroids and calcineurin inhibitors with resolution of lesions within 3 to 4 weeks.11,15-18
Erosive LP is a painful variant that can cause erosions, ulcerations, and scarring. It rarely is seen in the perianal region alone and presents a diagnostic challenge. Treatment with high-potency topical steroid therapy seems to be effective in the few cases that have been reported as well as in our case. More comprehensive data from randomized controlled trials would be needed to evaluate their efficacy compared to other therapies.
- Rebora A. Erosive lichen planus: what is this? Dermatology. 2002;205:226-228; discussion 227.
- Day T, Bohl TG, Scurry J. Perianal lichen dermatoses: a review of 60 cases. Australas J Dermatol. 2016;57:210-215.
- Fox LP, Lightdale CJ, Grossman ME. Lichen planus of the esophagus: what dermatologists need to know. J Am Acad Dermatol. 2011;65:175-883.
- Holmstrup P, Thorn JJ, Rindum J, et al. Malignant development of lichen planus-affected oral mucosa. J Oral Pathol. 1988;17:219-225.
- Lewi, FM, Bogliatto F. Erosive vulval lichen planus—a diagnosis not to be missed: a clinical review. Eur J Obstet Gynecol Reprod Biol. 2013;171:214-219.
- Webber NK, Setterfield JF, Lewis FM, et al. Lacrimal canalicular duct scarring in patients with lichen planus. Arch Dermatol. 2012;148:224-227.
- Martin L, Moriniere S, Machet MC, et al. Bilateral conductive deafness related to erosive lichen planus. J Laryngol Otol. 1998;112:365-366.
- Payne CM, McPartlin JF, Hawley PR. Ulcerative perianal lichen planus. Br J Dermatol. 1997;136:479.
- Watsky KL. Erosive perianal lichen planus responsive to tacrolimus. Int J Dermatol. 2003;42:217-218.
- Scheiba N, Toberer F, Lenhard BH, et al. Erythema and erosions of the perianal region in a 49-year-old man. J Dtsch Dermatol Ges. 2014;12:162-165.
- Wu Y, Qiao J, Fang H. Syndrome in question. An Bras Dermatol. 2014;89:843-844.
- Hammami S, Ksouda K, Affes H, et al. Mucosal lichenoid drug reaction associated with glimepiride: a case report. Eur Rev Med Pharmacol Sci. 2015;19:2301-2302.
- Meyerle JH, Turiansky GW. Perianal ulcer in a patient with AIDS. Arch Dermatol. 2004;140:877-882.
- Scott FI, Mamtani R, Brensinger CM, et al. Risk of nonmelanoma skin cancer associated with the use of immunosuppressant and biologic agents in patients with a history of autoimmune disease and nonmelanoma skin cancer. JAMA Dermatol. 2016;152:164-172.
- Cheng S, Kirtschig G, Cooper S, et al. Interventions for erosive lichen planus affecting mucosal sites. Cochrane Database Syst Rev. 2012:Cd008092.
- Gunther S. Effect of retinoic acid in lichen planus of the genitalia and perianal region. Br J Vener Dis. 1973;49:553-554.
- Vente C, Reich K, Neumann C. Erosive mucosal lichen planus: response to topical treatment with tacrolimus. Br J Dermatol. 1999;140:338-342.
- Lonsdale-Eccles AA, Velangi S. Topical pimecrolimus in the treatment of genital lichen planus: a prospective case series. Br J Dermatol. 2005;153:390-394.
- Rebora A. Erosive lichen planus: what is this? Dermatology. 2002;205:226-228; discussion 227.
- Day T, Bohl TG, Scurry J. Perianal lichen dermatoses: a review of 60 cases. Australas J Dermatol. 2016;57:210-215.
- Fox LP, Lightdale CJ, Grossman ME. Lichen planus of the esophagus: what dermatologists need to know. J Am Acad Dermatol. 2011;65:175-883.
- Holmstrup P, Thorn JJ, Rindum J, et al. Malignant development of lichen planus-affected oral mucosa. J Oral Pathol. 1988;17:219-225.
- Lewi, FM, Bogliatto F. Erosive vulval lichen planus—a diagnosis not to be missed: a clinical review. Eur J Obstet Gynecol Reprod Biol. 2013;171:214-219.
- Webber NK, Setterfield JF, Lewis FM, et al. Lacrimal canalicular duct scarring in patients with lichen planus. Arch Dermatol. 2012;148:224-227.
- Martin L, Moriniere S, Machet MC, et al. Bilateral conductive deafness related to erosive lichen planus. J Laryngol Otol. 1998;112:365-366.
- Payne CM, McPartlin JF, Hawley PR. Ulcerative perianal lichen planus. Br J Dermatol. 1997;136:479.
- Watsky KL. Erosive perianal lichen planus responsive to tacrolimus. Int J Dermatol. 2003;42:217-218.
- Scheiba N, Toberer F, Lenhard BH, et al. Erythema and erosions of the perianal region in a 49-year-old man. J Dtsch Dermatol Ges. 2014;12:162-165.
- Wu Y, Qiao J, Fang H. Syndrome in question. An Bras Dermatol. 2014;89:843-844.
- Hammami S, Ksouda K, Affes H, et al. Mucosal lichenoid drug reaction associated with glimepiride: a case report. Eur Rev Med Pharmacol Sci. 2015;19:2301-2302.
- Meyerle JH, Turiansky GW. Perianal ulcer in a patient with AIDS. Arch Dermatol. 2004;140:877-882.
- Scott FI, Mamtani R, Brensinger CM, et al. Risk of nonmelanoma skin cancer associated with the use of immunosuppressant and biologic agents in patients with a history of autoimmune disease and nonmelanoma skin cancer. JAMA Dermatol. 2016;152:164-172.
- Cheng S, Kirtschig G, Cooper S, et al. Interventions for erosive lichen planus affecting mucosal sites. Cochrane Database Syst Rev. 2012:Cd008092.
- Gunther S. Effect of retinoic acid in lichen planus of the genitalia and perianal region. Br J Vener Dis. 1973;49:553-554.
- Vente C, Reich K, Neumann C. Erosive mucosal lichen planus: response to topical treatment with tacrolimus. Br J Dermatol. 1999;140:338-342.
- Lonsdale-Eccles AA, Velangi S. Topical pimecrolimus in the treatment of genital lichen planus: a prospective case series. Br J Dermatol. 2005;153:390-394.
Practice Points
- Erosive lichen planus (LP) is an underrecognized variant of LP presenting with painful erosions, ulcerations, and scarring.
- Although rare, perianal erosive LP should be included in the differential diagnosis of perianal erosions.
- Treatment with high-potency steroids is an effective therapeutic option resulting in notable improvement.
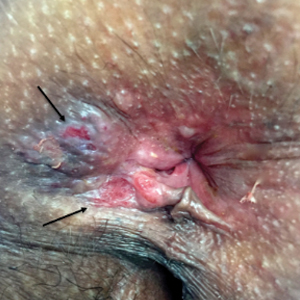
Asymptomatic Discolored Lesions on the Groin
The Diagnosis: Lichen Planus Pigmentosus-Inversus
Histopathologic examination revealed hyperkeratosis with dense, bandlike, lymphocytic inflammation at the dermoepidermal junction with associated melanin-containing macrophages in the papillary dermis (Figure 1). The physical examination and histopathology were consistent with a diagnosis of lichen planus pigmentosus-inversus (LPPI). Treatment was discussed with the patient, with options including phototherapy, tacrolimus, or a high-dose steroid. Given that the lesions were asymptomatic and not bothersome, the patient denied treatment and agreed to routine follow-up.
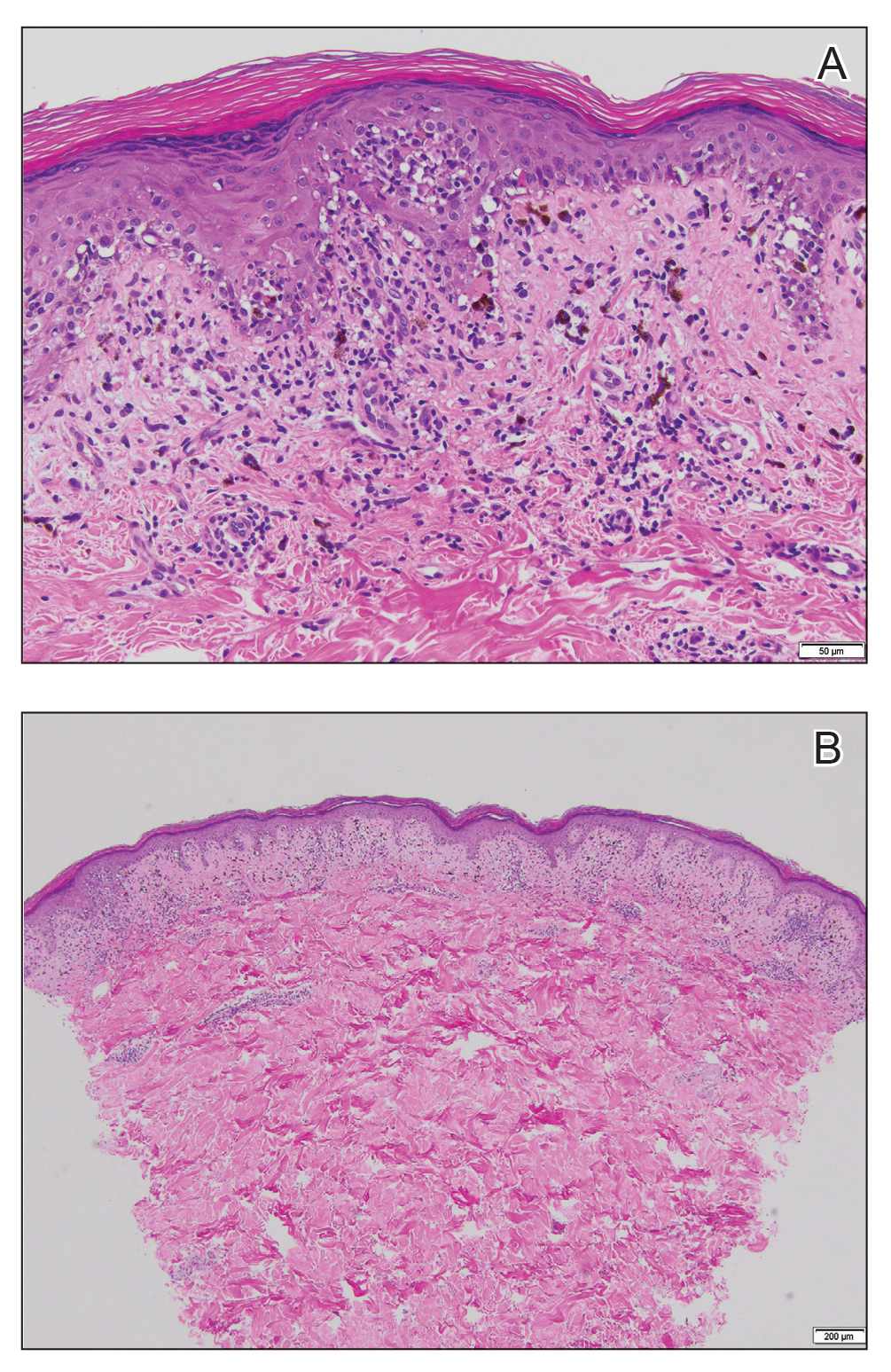
The first case of LPPI was reported in 20011; since then, approximately 100 cases have been reported in the literature.2 A rare variant of lichen planus, LPPI predominantly occurs in middle-aged women.2,3 Lichen planus pigmentosus-inversus is characterized by well-circumscribed, brown macules confined to non-sun-exposed intertriginous areas such as the axillae and groin.2 Although the rash remains localized, multiple lesions could arise in the same area, such as the groin as seen in our patient (Figure 2). Unlike in lichen planus, the oral mucosa, nails, and scalp are not affected. Furthermore, pruritus typically is absent in most cases of LPPI.2,4 Histopathologic findings include an atrophic epidermis with lichenoid infiltrates of lymphocytes and histocytes as well as substantial pigmentary incontinence with melanin-containing macrophages in the papillary dermis.4,5
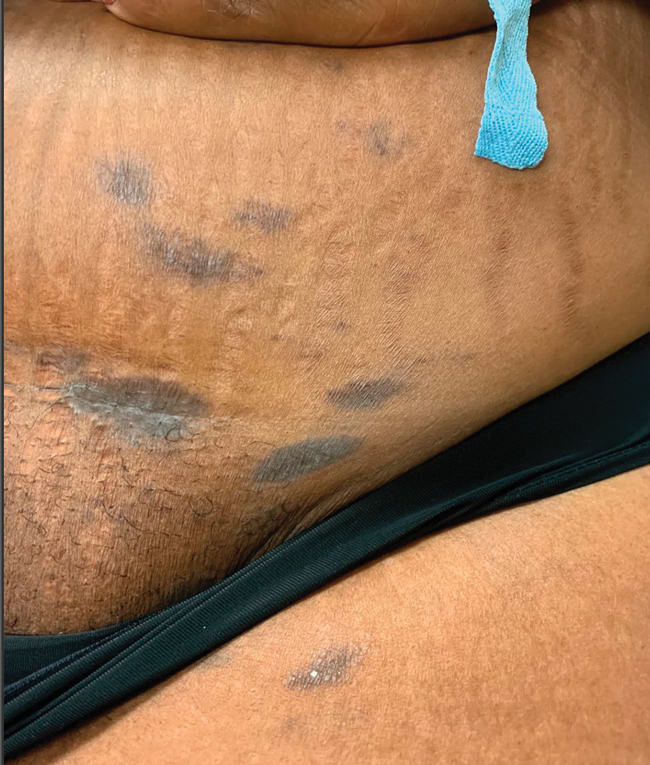
Given the gender, age, and clinical features of our patient, this case represents a classic scenario of LPPI. It currently is unknown if ethnicity plays a role in the disorder. Lichen planus pigmentosus-inversus initially was thought to be more prevalent in White patients; however, studies have been reported in individuals with darker skin.1,2
The main differential diagnosis includes erythema dyschromicum perstans, postinflammatory hyperpigmentation, and lichen planus. Although erythema dyschromicum perstans develops in individuals with darker skin, lesions are restricted to the upper torso and limbs.2-4 In both lichen planus and lichen actinicus, skin findings primarily develop in sun-exposed areas, such as the face, neck, and hands.4,6 Given the negative history of trauma, postinflammatory hyperpigmentation was unlikely in our patient. Furthermore, a distinguishing characteristic of LPPI is the deposition of melanin deep within the dermal layer.3
Lesions developing in nonexposed intertriginous skin makes LPPI unique and distinguishes it from other more common conditions. The lesions commonly are hyperpigmented and are not as pruritic as other lichen-associated conditions. Lichen planus pigmentosus-inversus often persists for months, and the rash generally is resistant to treatment.2,5 Topical tacrolimus and high-dose steroids may improve symptoms, though results have varied substantially. In addition, some cases have resolved spontaneously.1,4,6,7 Because LPPI is asymptomatic and benign, spontaneous resolution and routine care is a reasonable treatment strategy. Some cases have supported this strategy as safe and high-value care.2
- Mohamed M, Korbi M, Hammedi F, et al. Lichen planus pigmentosus inversus: a series of 10 Tunisian patients. Int J Dermatol. 2016;55:1088-1091.
- Lichen planus pigmentosus-inversus: a rare variant of lichen planus. J Am Acad Dermatol. 2015;72(suppl 1):AB239. https://doi.org /10.1016/j.jaad.2015.02.959
- Chen S, Sun W, Zhou G, et al. Lichen planus pigmentosus-inversus: report of three Chinese cases and review of the published work. J Dermatol. 2015;42:77-80.
- Tabanlıoǧlu-Onan D, Íncel-Uysal P, Öktem A, et al. Lichen planus pigmentosus-inversus: a peculiar variant of lichen planus. Dermatologica Sinica. 2017;35:210-212.
- Barros HR, Almeida JR, Mattos e Dinato SL, et al. Lichen planus pigmentosus inversus. An Bras Dermatol. 2013;88(6 suppl 1):146-149.
- Bennàssar A, Mas A, Julià M, et al. Annular plaques in the skin folds: 4 cases of lichen planus pigmentosus-inversus [in Spanish]. Actas Dermosifiliogr. 2009;100:602-605.
- Ghorbel HH, Badri T, Ben Brahim E, et al. Lichen planus pigmentosus inversus. Indian J Dermatol Venereol Leprol. 2014;80:580.
The Diagnosis: Lichen Planus Pigmentosus-Inversus
Histopathologic examination revealed hyperkeratosis with dense, bandlike, lymphocytic inflammation at the dermoepidermal junction with associated melanin-containing macrophages in the papillary dermis (Figure 1). The physical examination and histopathology were consistent with a diagnosis of lichen planus pigmentosus-inversus (LPPI). Treatment was discussed with the patient, with options including phototherapy, tacrolimus, or a high-dose steroid. Given that the lesions were asymptomatic and not bothersome, the patient denied treatment and agreed to routine follow-up.
The first case of LPPI was reported in 20011; since then, approximately 100 cases have been reported in the literature.2 A rare variant of lichen planus, LPPI predominantly occurs in middle-aged women.2,3 Lichen planus pigmentosus-inversus is characterized by well-circumscribed, brown macules confined to non-sun-exposed intertriginous areas such as the axillae and groin.2 Although the rash remains localized, multiple lesions could arise in the same area, such as the groin as seen in our patient (Figure 2). Unlike in lichen planus, the oral mucosa, nails, and scalp are not affected. Furthermore, pruritus typically is absent in most cases of LPPI.2,4 Histopathologic findings include an atrophic epidermis with lichenoid infiltrates of lymphocytes and histocytes as well as substantial pigmentary incontinence with melanin-containing macrophages in the papillary dermis.4,5
Given the gender, age, and clinical features of our patient, this case represents a classic scenario of LPPI. It currently is unknown if ethnicity plays a role in the disorder. Lichen planus pigmentosus-inversus initially was thought to be more prevalent in White patients; however, studies have been reported in individuals with darker skin.1,2
The main differential diagnosis includes erythema dyschromicum perstans, postinflammatory hyperpigmentation, and lichen planus. Although erythema dyschromicum perstans develops in individuals with darker skin, lesions are restricted to the upper torso and limbs.2-4 In both lichen planus and lichen actinicus, skin findings primarily develop in sun-exposed areas, such as the face, neck, and hands.4,6 Given the negative history of trauma, postinflammatory hyperpigmentation was unlikely in our patient. Furthermore, a distinguishing characteristic of LPPI is the deposition of melanin deep within the dermal layer.3
Lesions developing in nonexposed intertriginous skin makes LPPI unique and distinguishes it from other more common conditions. The lesions commonly are hyperpigmented and are not as pruritic as other lichen-associated conditions. Lichen planus pigmentosus-inversus often persists for months, and the rash generally is resistant to treatment.2,5 Topical tacrolimus and high-dose steroids may improve symptoms, though results have varied substantially. In addition, some cases have resolved spontaneously.1,4,6,7 Because LPPI is asymptomatic and benign, spontaneous resolution and routine care is a reasonable treatment strategy. Some cases have supported this strategy as safe and high-value care.2
The Diagnosis: Lichen Planus Pigmentosus-Inversus
Histopathologic examination revealed hyperkeratosis with dense, bandlike, lymphocytic inflammation at the dermoepidermal junction with associated melanin-containing macrophages in the papillary dermis (Figure 1). The physical examination and histopathology were consistent with a diagnosis of lichen planus pigmentosus-inversus (LPPI). Treatment was discussed with the patient, with options including phototherapy, tacrolimus, or a high-dose steroid. Given that the lesions were asymptomatic and not bothersome, the patient denied treatment and agreed to routine follow-up.
The first case of LPPI was reported in 20011; since then, approximately 100 cases have been reported in the literature.2 A rare variant of lichen planus, LPPI predominantly occurs in middle-aged women.2,3 Lichen planus pigmentosus-inversus is characterized by well-circumscribed, brown macules confined to non-sun-exposed intertriginous areas such as the axillae and groin.2 Although the rash remains localized, multiple lesions could arise in the same area, such as the groin as seen in our patient (Figure 2). Unlike in lichen planus, the oral mucosa, nails, and scalp are not affected. Furthermore, pruritus typically is absent in most cases of LPPI.2,4 Histopathologic findings include an atrophic epidermis with lichenoid infiltrates of lymphocytes and histocytes as well as substantial pigmentary incontinence with melanin-containing macrophages in the papillary dermis.4,5
Given the gender, age, and clinical features of our patient, this case represents a classic scenario of LPPI. It currently is unknown if ethnicity plays a role in the disorder. Lichen planus pigmentosus-inversus initially was thought to be more prevalent in White patients; however, studies have been reported in individuals with darker skin.1,2
The main differential diagnosis includes erythema dyschromicum perstans, postinflammatory hyperpigmentation, and lichen planus. Although erythema dyschromicum perstans develops in individuals with darker skin, lesions are restricted to the upper torso and limbs.2-4 In both lichen planus and lichen actinicus, skin findings primarily develop in sun-exposed areas, such as the face, neck, and hands.4,6 Given the negative history of trauma, postinflammatory hyperpigmentation was unlikely in our patient. Furthermore, a distinguishing characteristic of LPPI is the deposition of melanin deep within the dermal layer.3
Lesions developing in nonexposed intertriginous skin makes LPPI unique and distinguishes it from other more common conditions. The lesions commonly are hyperpigmented and are not as pruritic as other lichen-associated conditions. Lichen planus pigmentosus-inversus often persists for months, and the rash generally is resistant to treatment.2,5 Topical tacrolimus and high-dose steroids may improve symptoms, though results have varied substantially. In addition, some cases have resolved spontaneously.1,4,6,7 Because LPPI is asymptomatic and benign, spontaneous resolution and routine care is a reasonable treatment strategy. Some cases have supported this strategy as safe and high-value care.2
- Mohamed M, Korbi M, Hammedi F, et al. Lichen planus pigmentosus inversus: a series of 10 Tunisian patients. Int J Dermatol. 2016;55:1088-1091.
- Lichen planus pigmentosus-inversus: a rare variant of lichen planus. J Am Acad Dermatol. 2015;72(suppl 1):AB239. https://doi.org /10.1016/j.jaad.2015.02.959
- Chen S, Sun W, Zhou G, et al. Lichen planus pigmentosus-inversus: report of three Chinese cases and review of the published work. J Dermatol. 2015;42:77-80.
- Tabanlıoǧlu-Onan D, Íncel-Uysal P, Öktem A, et al. Lichen planus pigmentosus-inversus: a peculiar variant of lichen planus. Dermatologica Sinica. 2017;35:210-212.
- Barros HR, Almeida JR, Mattos e Dinato SL, et al. Lichen planus pigmentosus inversus. An Bras Dermatol. 2013;88(6 suppl 1):146-149.
- Bennàssar A, Mas A, Julià M, et al. Annular plaques in the skin folds: 4 cases of lichen planus pigmentosus-inversus [in Spanish]. Actas Dermosifiliogr. 2009;100:602-605.
- Ghorbel HH, Badri T, Ben Brahim E, et al. Lichen planus pigmentosus inversus. Indian J Dermatol Venereol Leprol. 2014;80:580.
- Mohamed M, Korbi M, Hammedi F, et al. Lichen planus pigmentosus inversus: a series of 10 Tunisian patients. Int J Dermatol. 2016;55:1088-1091.
- Lichen planus pigmentosus-inversus: a rare variant of lichen planus. J Am Acad Dermatol. 2015;72(suppl 1):AB239. https://doi.org /10.1016/j.jaad.2015.02.959
- Chen S, Sun W, Zhou G, et al. Lichen planus pigmentosus-inversus: report of three Chinese cases and review of the published work. J Dermatol. 2015;42:77-80.
- Tabanlıoǧlu-Onan D, Íncel-Uysal P, Öktem A, et al. Lichen planus pigmentosus-inversus: a peculiar variant of lichen planus. Dermatologica Sinica. 2017;35:210-212.
- Barros HR, Almeida JR, Mattos e Dinato SL, et al. Lichen planus pigmentosus inversus. An Bras Dermatol. 2013;88(6 suppl 1):146-149.
- Bennàssar A, Mas A, Julià M, et al. Annular plaques in the skin folds: 4 cases of lichen planus pigmentosus-inversus [in Spanish]. Actas Dermosifiliogr. 2009;100:602-605.
- Ghorbel HH, Badri T, Ben Brahim E, et al. Lichen planus pigmentosus inversus. Indian J Dermatol Venereol Leprol. 2014;80:580.
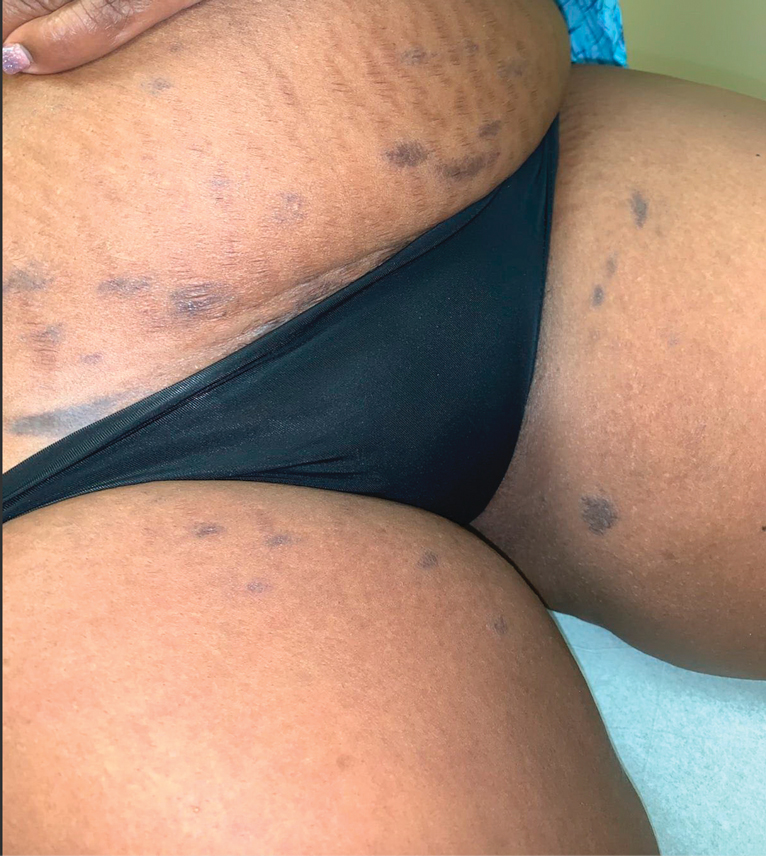
A 45-year-old African American woman presented with an asymptomatic rash that had worsened over the month prior to presentation. It initially began on the upper thighs and then spread to the abdomen, groin, and buttocks. The rash was mildly pruritic and had grown both in size and number of lesions. She had not tried any new over-the-counter medications. Her medical history was notable for late-stage breast cancer diagnosed 4 years prior that was treated with radiation and neoadjuvant NeoPACT—carboplatin, docetaxel, and pembrolizumab. One year prior to presentation, she underwent a lumpectomy that was complicated by gas gangrene of the finger. She has been in remission since the surgery. Physical examination at the current presentation was remarkable for multiple well-circumscribed, hyperpigmented macules on the medial thighs, lower abdomen, and buttocks. Syphilis antibody screening was negative.
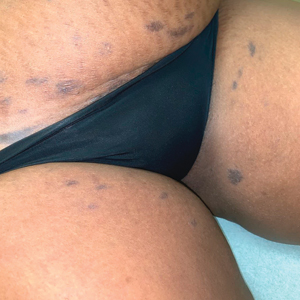
Cutaneous Cholesterol Embolization to the Lower Trunk: An Underrecognized Presentation
To the Editor:
A 65-year-old man with severe atherosclerotic disease developed multiple painful eschars on the lower abdomen, thighs, sacrum, and perineum. He initially presented with myocardial ischemia and claudication and underwent 3 cardiac catheterizations as well as stenting of the superficial femoral artery. Within 2 weeks, he developed exquisitely tender nodules on the lower abdomen, clinically presumed to be sites of enoxaparin injections. These lesions gradually expanded and ulcerated to involve the sacrum, buttock, perineum, and upper thighs (Figure 1). Two punch biopsies from ulcerated skin taken 10 days apart demonstrated necrosis of skin and subcutaneous fat without evidence of vasculitis, vasculopathy, emboli, or notable inflammation despite examination of multiple levels of all submitted tissue. A definitive cause for the ulcerations remained elusive with development of new lesions. A third incisional biopsy of a newly developed, nonulcerated, subcutaneous nodule performed 8 weeks after presentation revealed multiple cholesterol emboli (Figure 2). He was treated with warfarin and clopidogrel bisulfate as well as local wound care. The lesions slowly resolved over the next 4 to 6 months.
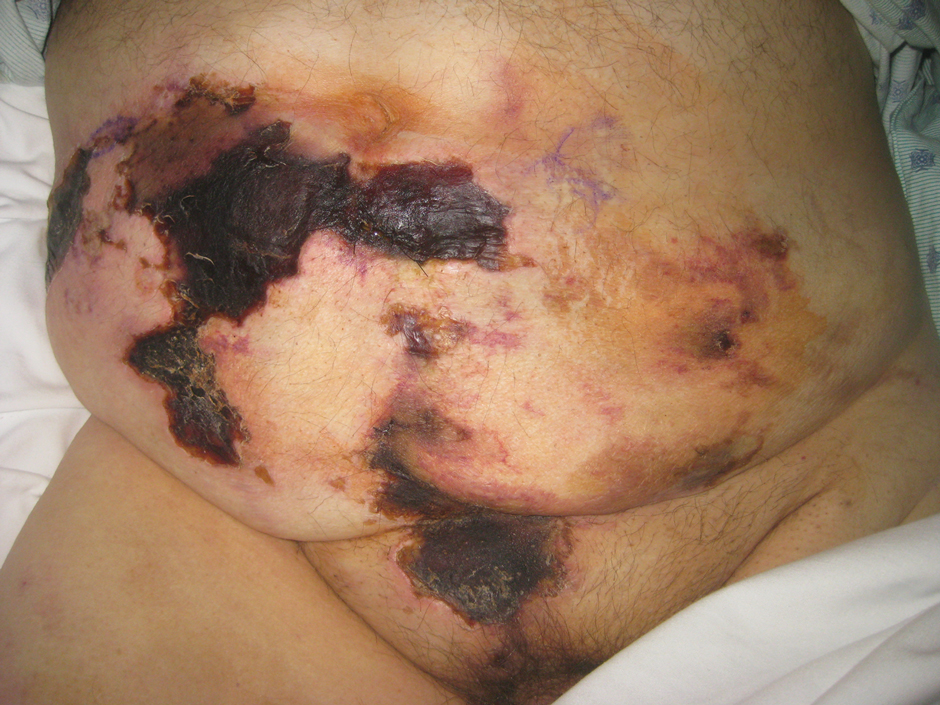
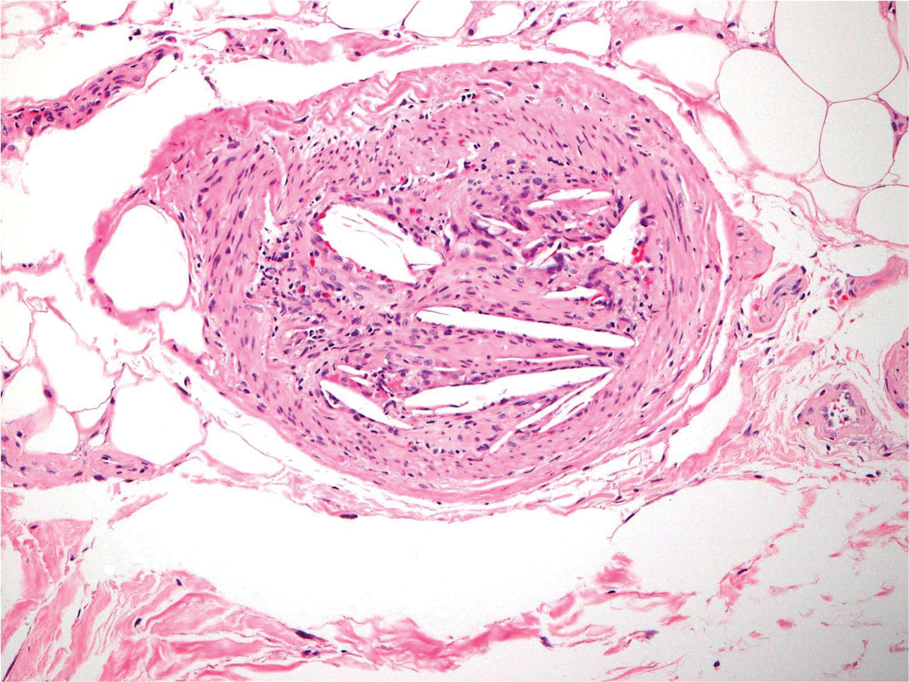
Cholesterol embolization syndrome occurs when disrupted atherosclerotic plaques embolize from large proximal arteries to more distal arterioles, resulting in ischemic damage to 1 or more organ systems.1 It can occur spontaneously but often is a consequence of thrombolytic therapy, anticoagulation, and angioinvasive procedures.2,3 Cutaneous manifestations include livedo reticularis, retiform purpura, nodules, and gangrene. Although livedo reticularis may extend from the legs to the trunk, gangrenous lesions predominantly involve the distal digits.
This case illustrates the challenge in diagnosis of cholesterol emboli, both clinically and histologically. Cutaneous lesions are morphologically variable and often occur with systemic manifestations, mimicking numerous conditions.1 Lower extremity involvement is a well-known occurrence in cholesterol embolization (ie, blue toe syndrome); however, periumbilical and lumbosacral lesions have not been emphasized in the dermatologic or peripheral vascular literature. Our patient’s initial diagnosis was enoxaparin necrosis at abdominal injection sites; however, this unusual distribution of lesions was ultimately determined to be the consequence of cholesterol embolization from the inferior epigastric and superficial external pudendal arteries at the time of stenting of the superficial femoral artery. Proximal truncal involvement should be recognized as an atypical but important cutaneous manifestation to facilitate timely diagnosis and treatment.4,5
Our patient’s course also highlights the potential need for multiple biopsies. Although the gold standard for diagnosis is histologic confirmation, a negative biopsy does not always exclude cholesterol emboli, and one should have a low threshold to perform additional biopsies in the appropriate clinical setting.
- Fine MJ, Kapoor W, Falanga V. Cholesterol crystal embolization: a review of 221 cases in the English literature. Angiology. 1987;38:769-784.
- Fukumoto Y, Tsutsui H, Tsuchihashi M, et al. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol. 2003;42:211-216.
- Karalis DG, Chandrasekaran K, Victor MF, et al. Recognition and embolic potential of intraaortic atherosclerotic debris. J Am Coll Cardiol. 1991;17:73.
- Zaytsev P, Miller K, Pellettiere EV. Cutaneous cholesterol emboli with infarction clinically mimicking heparin necrosis—a case report. Angiology. 1986;37:471-476.
- Erdim M, Tezel E, Biskin N. A case of skin necrosis as a result of cholesterol crystal embolisation. J Plast Reconstr Aesthet Surg. 2006;59:429-432.
To the Editor:
A 65-year-old man with severe atherosclerotic disease developed multiple painful eschars on the lower abdomen, thighs, sacrum, and perineum. He initially presented with myocardial ischemia and claudication and underwent 3 cardiac catheterizations as well as stenting of the superficial femoral artery. Within 2 weeks, he developed exquisitely tender nodules on the lower abdomen, clinically presumed to be sites of enoxaparin injections. These lesions gradually expanded and ulcerated to involve the sacrum, buttock, perineum, and upper thighs (Figure 1). Two punch biopsies from ulcerated skin taken 10 days apart demonstrated necrosis of skin and subcutaneous fat without evidence of vasculitis, vasculopathy, emboli, or notable inflammation despite examination of multiple levels of all submitted tissue. A definitive cause for the ulcerations remained elusive with development of new lesions. A third incisional biopsy of a newly developed, nonulcerated, subcutaneous nodule performed 8 weeks after presentation revealed multiple cholesterol emboli (Figure 2). He was treated with warfarin and clopidogrel bisulfate as well as local wound care. The lesions slowly resolved over the next 4 to 6 months.
Cholesterol embolization syndrome occurs when disrupted atherosclerotic plaques embolize from large proximal arteries to more distal arterioles, resulting in ischemic damage to 1 or more organ systems.1 It can occur spontaneously but often is a consequence of thrombolytic therapy, anticoagulation, and angioinvasive procedures.2,3 Cutaneous manifestations include livedo reticularis, retiform purpura, nodules, and gangrene. Although livedo reticularis may extend from the legs to the trunk, gangrenous lesions predominantly involve the distal digits.
This case illustrates the challenge in diagnosis of cholesterol emboli, both clinically and histologically. Cutaneous lesions are morphologically variable and often occur with systemic manifestations, mimicking numerous conditions.1 Lower extremity involvement is a well-known occurrence in cholesterol embolization (ie, blue toe syndrome); however, periumbilical and lumbosacral lesions have not been emphasized in the dermatologic or peripheral vascular literature. Our patient’s initial diagnosis was enoxaparin necrosis at abdominal injection sites; however, this unusual distribution of lesions was ultimately determined to be the consequence of cholesterol embolization from the inferior epigastric and superficial external pudendal arteries at the time of stenting of the superficial femoral artery. Proximal truncal involvement should be recognized as an atypical but important cutaneous manifestation to facilitate timely diagnosis and treatment.4,5
Our patient’s course also highlights the potential need for multiple biopsies. Although the gold standard for diagnosis is histologic confirmation, a negative biopsy does not always exclude cholesterol emboli, and one should have a low threshold to perform additional biopsies in the appropriate clinical setting.
To the Editor:
A 65-year-old man with severe atherosclerotic disease developed multiple painful eschars on the lower abdomen, thighs, sacrum, and perineum. He initially presented with myocardial ischemia and claudication and underwent 3 cardiac catheterizations as well as stenting of the superficial femoral artery. Within 2 weeks, he developed exquisitely tender nodules on the lower abdomen, clinically presumed to be sites of enoxaparin injections. These lesions gradually expanded and ulcerated to involve the sacrum, buttock, perineum, and upper thighs (Figure 1). Two punch biopsies from ulcerated skin taken 10 days apart demonstrated necrosis of skin and subcutaneous fat without evidence of vasculitis, vasculopathy, emboli, or notable inflammation despite examination of multiple levels of all submitted tissue. A definitive cause for the ulcerations remained elusive with development of new lesions. A third incisional biopsy of a newly developed, nonulcerated, subcutaneous nodule performed 8 weeks after presentation revealed multiple cholesterol emboli (Figure 2). He was treated with warfarin and clopidogrel bisulfate as well as local wound care. The lesions slowly resolved over the next 4 to 6 months.
Cholesterol embolization syndrome occurs when disrupted atherosclerotic plaques embolize from large proximal arteries to more distal arterioles, resulting in ischemic damage to 1 or more organ systems.1 It can occur spontaneously but often is a consequence of thrombolytic therapy, anticoagulation, and angioinvasive procedures.2,3 Cutaneous manifestations include livedo reticularis, retiform purpura, nodules, and gangrene. Although livedo reticularis may extend from the legs to the trunk, gangrenous lesions predominantly involve the distal digits.
This case illustrates the challenge in diagnosis of cholesterol emboli, both clinically and histologically. Cutaneous lesions are morphologically variable and often occur with systemic manifestations, mimicking numerous conditions.1 Lower extremity involvement is a well-known occurrence in cholesterol embolization (ie, blue toe syndrome); however, periumbilical and lumbosacral lesions have not been emphasized in the dermatologic or peripheral vascular literature. Our patient’s initial diagnosis was enoxaparin necrosis at abdominal injection sites; however, this unusual distribution of lesions was ultimately determined to be the consequence of cholesterol embolization from the inferior epigastric and superficial external pudendal arteries at the time of stenting of the superficial femoral artery. Proximal truncal involvement should be recognized as an atypical but important cutaneous manifestation to facilitate timely diagnosis and treatment.4,5
Our patient’s course also highlights the potential need for multiple biopsies. Although the gold standard for diagnosis is histologic confirmation, a negative biopsy does not always exclude cholesterol emboli, and one should have a low threshold to perform additional biopsies in the appropriate clinical setting.
- Fine MJ, Kapoor W, Falanga V. Cholesterol crystal embolization: a review of 221 cases in the English literature. Angiology. 1987;38:769-784.
- Fukumoto Y, Tsutsui H, Tsuchihashi M, et al. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol. 2003;42:211-216.
- Karalis DG, Chandrasekaran K, Victor MF, et al. Recognition and embolic potential of intraaortic atherosclerotic debris. J Am Coll Cardiol. 1991;17:73.
- Zaytsev P, Miller K, Pellettiere EV. Cutaneous cholesterol emboli with infarction clinically mimicking heparin necrosis—a case report. Angiology. 1986;37:471-476.
- Erdim M, Tezel E, Biskin N. A case of skin necrosis as a result of cholesterol crystal embolisation. J Plast Reconstr Aesthet Surg. 2006;59:429-432.
- Fine MJ, Kapoor W, Falanga V. Cholesterol crystal embolization: a review of 221 cases in the English literature. Angiology. 1987;38:769-784.
- Fukumoto Y, Tsutsui H, Tsuchihashi M, et al. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol. 2003;42:211-216.
- Karalis DG, Chandrasekaran K, Victor MF, et al. Recognition and embolic potential of intraaortic atherosclerotic debris. J Am Coll Cardiol. 1991;17:73.
- Zaytsev P, Miller K, Pellettiere EV. Cutaneous cholesterol emboli with infarction clinically mimicking heparin necrosis—a case report. Angiology. 1986;37:471-476.
- Erdim M, Tezel E, Biskin N. A case of skin necrosis as a result of cholesterol crystal embolisation. J Plast Reconstr Aesthet Surg. 2006;59:429-432.
Practice Points
- Cholesterol embolization may occur in proximal locations, and index of suspicion should be high in patients who are at risk.
- Several biopsies may be necessary to make a diagnosis of cholesterol emboli.

Progressive Telangiectatic Rash
The Diagnosis: Cutaneous Collagenous Vasculopathy
Cutaneous collagenous vasculopathy (CCV) is an idiopathic microangiopathy of the small vessels in the superficial dermis. A condition first identified by Salama and Rosenthal1 in 2000, CCV likely is underreported, as its clinical mimickers are not routinely biopsied.2 It presents as asymptomatic telangiectatic macules, initially on the lower extremities and often spreading to the trunk. Cutaneous collagenous vasculopathy often is seen in middle-aged adults, and most patients have comorbidities such as hypertension, diabetes mellitus, or cardiovascular disease. The exact etiology of this disease is unknown.3,4
Histopathologically, CCV is characterized by dilated superficial vessels with thickened eosinophilic walls. The eosinophilic material is composed of hyalinized type IV collagen, which is periodic acid-Schiff positive and diastase resistant (Figure 1).3,4 Stains for amyloid are negative.
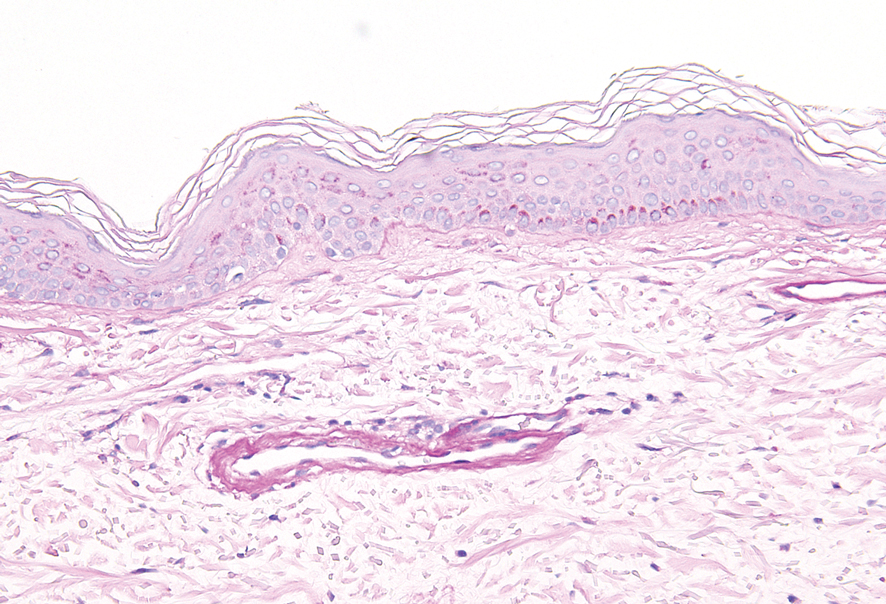
Generalized essential telangiectasia (GET) is a condition that presents with symmetric, blanchable, erythematous telangiectases.5 These lesions can occur alone or can accompany systemic diseases. Similar to CCV, the telangiectases tend to begin on the legs before gradually spreading to the trunk; however, this process more often is seen in females and occurs at an earlier age. Unlike CCV, GET can occur on mucosal surfaces, with cases of conjunctival and oral involvement reported.6 Generalized essential telangiectasia usually is a diagnosis of exclusion.7,8 It is thought that many CCV lesions have been misclassified clinically as GET, which highlights the importance of biopsy. Microscopically, GET is distinct from CCV in that the superficial dermis lacks thick-walled vessels.5,7 Although usually not associated with systemic diseases or progressive morbidity, treatment options are limited.8
Livedoid vasculopathy, also known as atrophie blanche, is caused by fibrin thrombi occlusion of dermal vessels. Clinically, patients have recurrent telangiectatic papules and painful ulcers on the lower extremities that gradually heal, leaving behind white stellate scars. It is caused by an underlying prothrombotic state with a superimposed inflammatory response.9 Livedoid vasculopathy primarily affects middle-aged women, and many patients have comorbidities such as scleroderma or systemic lupus erythematosus. Histologically, the epidermis often is ulcerated, and thrombi are visualized within small vessels. Eosinophilic fibrinoid material is deposited in vessel walls, including but not confined to vessels at the base of the epidermal ulcer (Figure 2). The fibrinoid material is periodic acid-Schiff positive and diastase resistant and can be highlighted with immunofluorescence, which may help to distinguish this entity from CCV.1,9 As the disease progresses, vessels are diffusely hyalinized, and there is epidermal atrophy and dermal sclerosis. Treatment options include antiplatelet and fibrinolytic drugs with a multidisciplinary approach to resolve pain and scarring.9
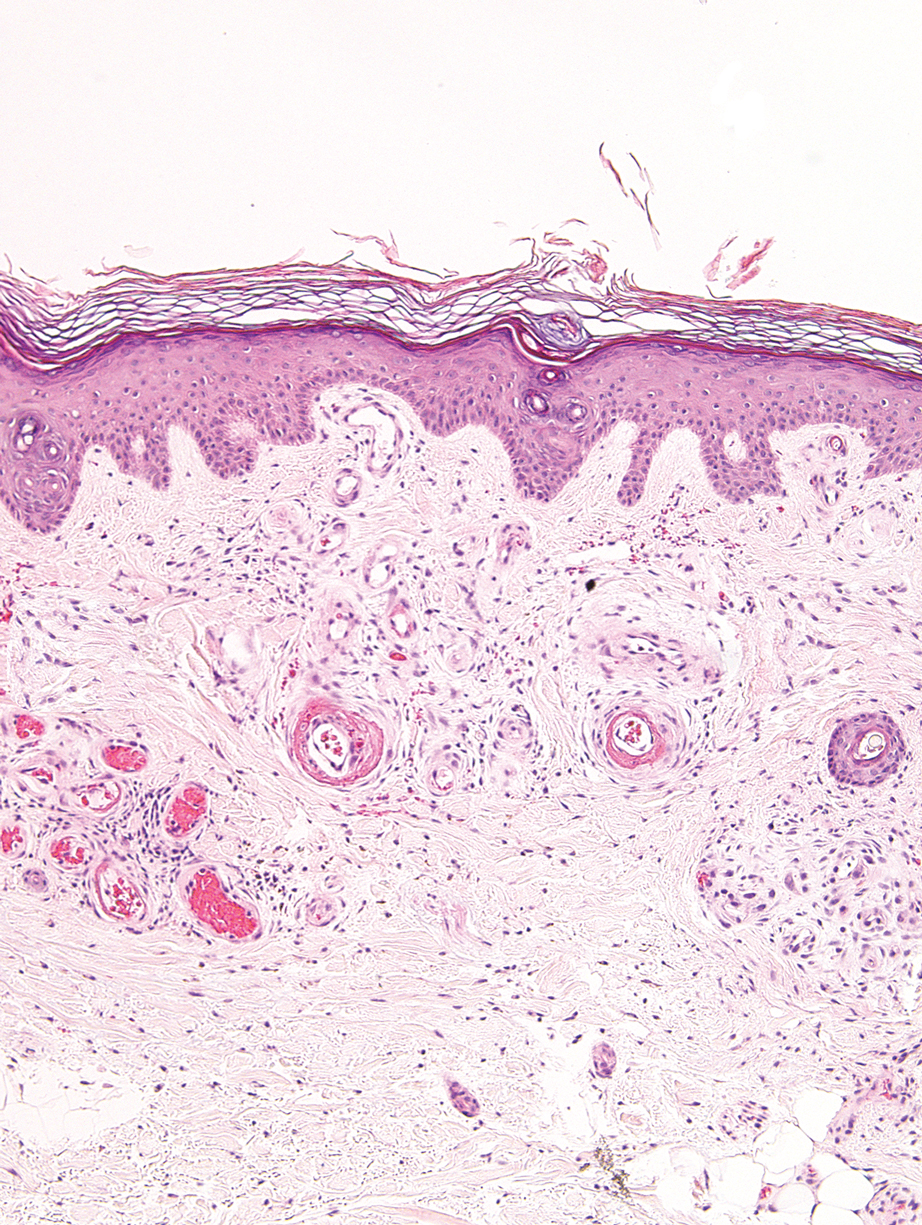
Primary systemic amyloidosis is a rare condition, and cutaneous manifestations are seen in approximately one-third of affected individuals. Amyloid deposition results in waxy papules that predominantly affect the face and periorbital areas but also may occur on the neck, flexural areas, and genitalia.5 Because the amyloid deposits also can be found within vessel walls, hemorrhagic lesions may occur. Microscopically, amorphous eosinophilic material can be found within the vessel walls, similar to CCV (Figure 3A); however, when stained with Congo red, cutaneous amyloidosis shows waxy red-orange material involving the vessel walls and exhibits apple green birefringence under polarization (Figure 3B).10 Amyloid also will be negative for type IV collagen, fibronectin, and laminin, whereas CCV will be positive.5
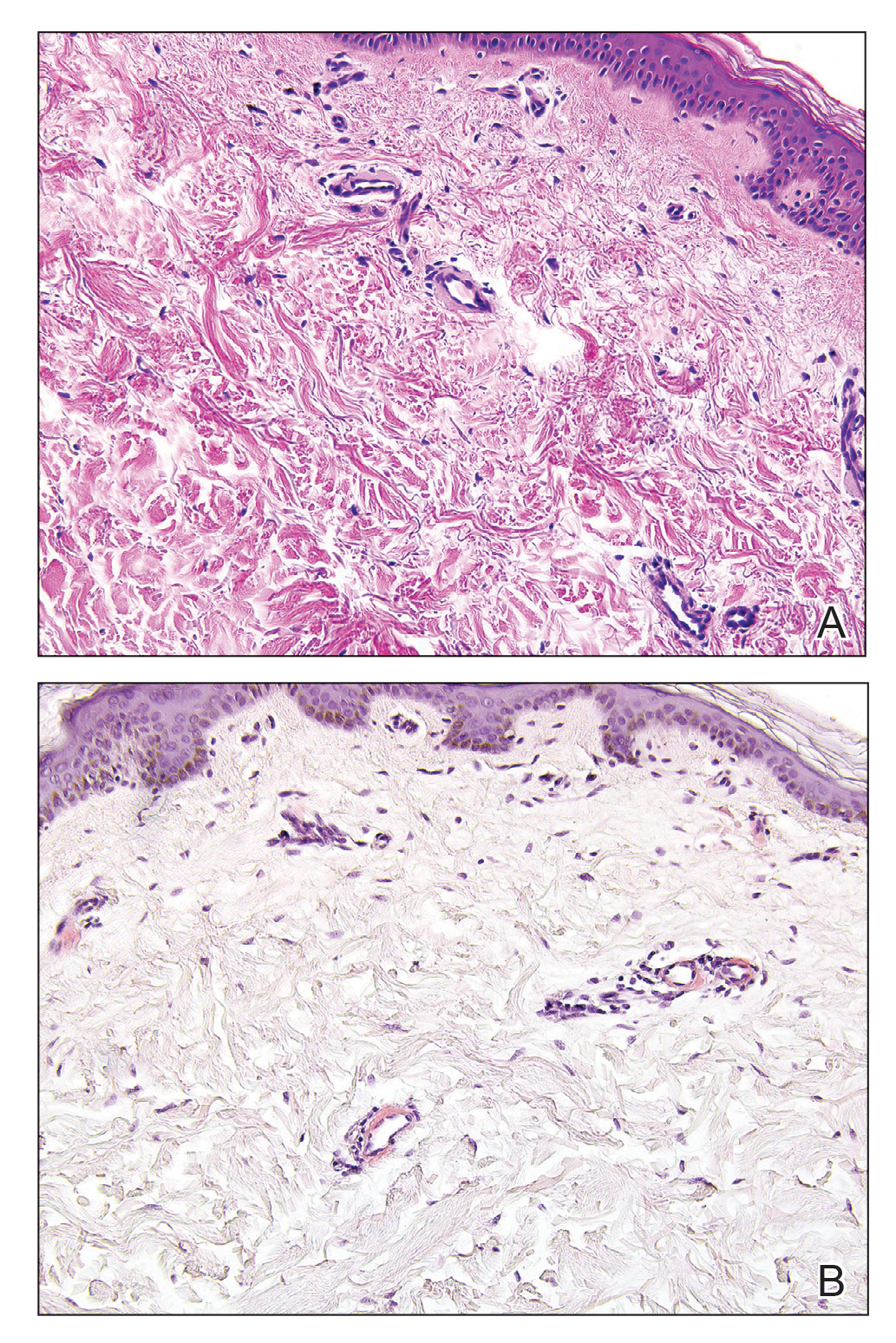
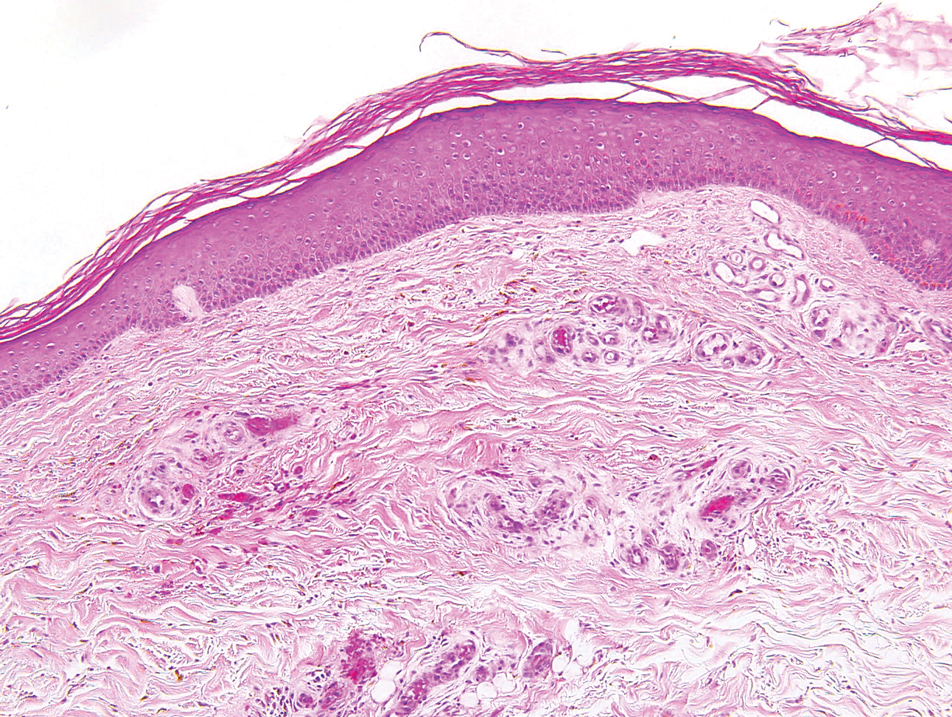
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Sartori DS, Almeida HL Jr, Dorn TV, et al. Cutaneous collagenous vasculopathy: light and transmission electron microscopy. An Bras Dermatol. 2019;94:211-213.
- Brady BG, Ortleb M, Boyd AS, et al. Cutaneous collagenous vasculopathy. J Clin Aesthet Dermatol. 2015;8:49-52.
- Patterson JW, ed. Vascular tumors. Weedon's Skin Pathology. 4th ed. Churchill Livingstone/Elsevier; 2016:1069-1115.
- Knöpfel N, Martín-Santiago A, Saus C, et al. Extensive acquired telangiectasias: comparison of generalized essential telangiectasia and cutaneous collagenous vasculopathy. Actas Dermosifiliogr. 2017;108:E21-E26.
- Karimkhani C, Boyers LN, Olivere J, et al. Cutaneous collagenous vasculopathy. Cutis. 2019;103:E7-E8.
- McGrae JD, Winkelmann RK. Generalized essential telangiectasia: report of a clinical and histochemical study of 13 patients with acquired cutaneous lesions. JAMA. 1963;185:909-913.
- Vasudeva B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478.
- Ko CJ, Barr RJ. Color--pink. In: Ko CJ, Barr RJ, eds. Dermatopathology: Diagnosis by First Impression. 3rd ed. Wiley; 2016:303-322.
- Clark ML, McGuinness AE, Vidal CI. Cutaneous collagenous vasculopathy: a unique case with positive direct immunofluorescence findings. Am J Dermatopathol. 2019;41:77-79.
The Diagnosis: Cutaneous Collagenous Vasculopathy
Cutaneous collagenous vasculopathy (CCV) is an idiopathic microangiopathy of the small vessels in the superficial dermis. A condition first identified by Salama and Rosenthal1 in 2000, CCV likely is underreported, as its clinical mimickers are not routinely biopsied.2 It presents as asymptomatic telangiectatic macules, initially on the lower extremities and often spreading to the trunk. Cutaneous collagenous vasculopathy often is seen in middle-aged adults, and most patients have comorbidities such as hypertension, diabetes mellitus, or cardiovascular disease. The exact etiology of this disease is unknown.3,4
Histopathologically, CCV is characterized by dilated superficial vessels with thickened eosinophilic walls. The eosinophilic material is composed of hyalinized type IV collagen, which is periodic acid-Schiff positive and diastase resistant (Figure 1).3,4 Stains for amyloid are negative.
Generalized essential telangiectasia (GET) is a condition that presents with symmetric, blanchable, erythematous telangiectases.5 These lesions can occur alone or can accompany systemic diseases. Similar to CCV, the telangiectases tend to begin on the legs before gradually spreading to the trunk; however, this process more often is seen in females and occurs at an earlier age. Unlike CCV, GET can occur on mucosal surfaces, with cases of conjunctival and oral involvement reported.6 Generalized essential telangiectasia usually is a diagnosis of exclusion.7,8 It is thought that many CCV lesions have been misclassified clinically as GET, which highlights the importance of biopsy. Microscopically, GET is distinct from CCV in that the superficial dermis lacks thick-walled vessels.5,7 Although usually not associated with systemic diseases or progressive morbidity, treatment options are limited.8
Livedoid vasculopathy, also known as atrophie blanche, is caused by fibrin thrombi occlusion of dermal vessels. Clinically, patients have recurrent telangiectatic papules and painful ulcers on the lower extremities that gradually heal, leaving behind white stellate scars. It is caused by an underlying prothrombotic state with a superimposed inflammatory response.9 Livedoid vasculopathy primarily affects middle-aged women, and many patients have comorbidities such as scleroderma or systemic lupus erythematosus. Histologically, the epidermis often is ulcerated, and thrombi are visualized within small vessels. Eosinophilic fibrinoid material is deposited in vessel walls, including but not confined to vessels at the base of the epidermal ulcer (Figure 2). The fibrinoid material is periodic acid-Schiff positive and diastase resistant and can be highlighted with immunofluorescence, which may help to distinguish this entity from CCV.1,9 As the disease progresses, vessels are diffusely hyalinized, and there is epidermal atrophy and dermal sclerosis. Treatment options include antiplatelet and fibrinolytic drugs with a multidisciplinary approach to resolve pain and scarring.9
Primary systemic amyloidosis is a rare condition, and cutaneous manifestations are seen in approximately one-third of affected individuals. Amyloid deposition results in waxy papules that predominantly affect the face and periorbital areas but also may occur on the neck, flexural areas, and genitalia.5 Because the amyloid deposits also can be found within vessel walls, hemorrhagic lesions may occur. Microscopically, amorphous eosinophilic material can be found within the vessel walls, similar to CCV (Figure 3A); however, when stained with Congo red, cutaneous amyloidosis shows waxy red-orange material involving the vessel walls and exhibits apple green birefringence under polarization (Figure 3B).10 Amyloid also will be negative for type IV collagen, fibronectin, and laminin, whereas CCV will be positive.5
The Diagnosis: Cutaneous Collagenous Vasculopathy
Cutaneous collagenous vasculopathy (CCV) is an idiopathic microangiopathy of the small vessels in the superficial dermis. A condition first identified by Salama and Rosenthal1 in 2000, CCV likely is underreported, as its clinical mimickers are not routinely biopsied.2 It presents as asymptomatic telangiectatic macules, initially on the lower extremities and often spreading to the trunk. Cutaneous collagenous vasculopathy often is seen in middle-aged adults, and most patients have comorbidities such as hypertension, diabetes mellitus, or cardiovascular disease. The exact etiology of this disease is unknown.3,4
Histopathologically, CCV is characterized by dilated superficial vessels with thickened eosinophilic walls. The eosinophilic material is composed of hyalinized type IV collagen, which is periodic acid-Schiff positive and diastase resistant (Figure 1).3,4 Stains for amyloid are negative.
Generalized essential telangiectasia (GET) is a condition that presents with symmetric, blanchable, erythematous telangiectases.5 These lesions can occur alone or can accompany systemic diseases. Similar to CCV, the telangiectases tend to begin on the legs before gradually spreading to the trunk; however, this process more often is seen in females and occurs at an earlier age. Unlike CCV, GET can occur on mucosal surfaces, with cases of conjunctival and oral involvement reported.6 Generalized essential telangiectasia usually is a diagnosis of exclusion.7,8 It is thought that many CCV lesions have been misclassified clinically as GET, which highlights the importance of biopsy. Microscopically, GET is distinct from CCV in that the superficial dermis lacks thick-walled vessels.5,7 Although usually not associated with systemic diseases or progressive morbidity, treatment options are limited.8
Livedoid vasculopathy, also known as atrophie blanche, is caused by fibrin thrombi occlusion of dermal vessels. Clinically, patients have recurrent telangiectatic papules and painful ulcers on the lower extremities that gradually heal, leaving behind white stellate scars. It is caused by an underlying prothrombotic state with a superimposed inflammatory response.9 Livedoid vasculopathy primarily affects middle-aged women, and many patients have comorbidities such as scleroderma or systemic lupus erythematosus. Histologically, the epidermis often is ulcerated, and thrombi are visualized within small vessels. Eosinophilic fibrinoid material is deposited in vessel walls, including but not confined to vessels at the base of the epidermal ulcer (Figure 2). The fibrinoid material is periodic acid-Schiff positive and diastase resistant and can be highlighted with immunofluorescence, which may help to distinguish this entity from CCV.1,9 As the disease progresses, vessels are diffusely hyalinized, and there is epidermal atrophy and dermal sclerosis. Treatment options include antiplatelet and fibrinolytic drugs with a multidisciplinary approach to resolve pain and scarring.9
Primary systemic amyloidosis is a rare condition, and cutaneous manifestations are seen in approximately one-third of affected individuals. Amyloid deposition results in waxy papules that predominantly affect the face and periorbital areas but also may occur on the neck, flexural areas, and genitalia.5 Because the amyloid deposits also can be found within vessel walls, hemorrhagic lesions may occur. Microscopically, amorphous eosinophilic material can be found within the vessel walls, similar to CCV (Figure 3A); however, when stained with Congo red, cutaneous amyloidosis shows waxy red-orange material involving the vessel walls and exhibits apple green birefringence under polarization (Figure 3B).10 Amyloid also will be negative for type IV collagen, fibronectin, and laminin, whereas CCV will be positive.5
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Sartori DS, Almeida HL Jr, Dorn TV, et al. Cutaneous collagenous vasculopathy: light and transmission electron microscopy. An Bras Dermatol. 2019;94:211-213.
- Brady BG, Ortleb M, Boyd AS, et al. Cutaneous collagenous vasculopathy. J Clin Aesthet Dermatol. 2015;8:49-52.
- Patterson JW, ed. Vascular tumors. Weedon's Skin Pathology. 4th ed. Churchill Livingstone/Elsevier; 2016:1069-1115.
- Knöpfel N, Martín-Santiago A, Saus C, et al. Extensive acquired telangiectasias: comparison of generalized essential telangiectasia and cutaneous collagenous vasculopathy. Actas Dermosifiliogr. 2017;108:E21-E26.
- Karimkhani C, Boyers LN, Olivere J, et al. Cutaneous collagenous vasculopathy. Cutis. 2019;103:E7-E8.
- McGrae JD, Winkelmann RK. Generalized essential telangiectasia: report of a clinical and histochemical study of 13 patients with acquired cutaneous lesions. JAMA. 1963;185:909-913.
- Vasudeva B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478.
- Ko CJ, Barr RJ. Color--pink. In: Ko CJ, Barr RJ, eds. Dermatopathology: Diagnosis by First Impression. 3rd ed. Wiley; 2016:303-322.
- Clark ML, McGuinness AE, Vidal CI. Cutaneous collagenous vasculopathy: a unique case with positive direct immunofluorescence findings. Am J Dermatopathol. 2019;41:77-79.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Sartori DS, Almeida HL Jr, Dorn TV, et al. Cutaneous collagenous vasculopathy: light and transmission electron microscopy. An Bras Dermatol. 2019;94:211-213.
- Brady BG, Ortleb M, Boyd AS, et al. Cutaneous collagenous vasculopathy. J Clin Aesthet Dermatol. 2015;8:49-52.
- Patterson JW, ed. Vascular tumors. Weedon's Skin Pathology. 4th ed. Churchill Livingstone/Elsevier; 2016:1069-1115.
- Knöpfel N, Martín-Santiago A, Saus C, et al. Extensive acquired telangiectasias: comparison of generalized essential telangiectasia and cutaneous collagenous vasculopathy. Actas Dermosifiliogr. 2017;108:E21-E26.
- Karimkhani C, Boyers LN, Olivere J, et al. Cutaneous collagenous vasculopathy. Cutis. 2019;103:E7-E8.
- McGrae JD, Winkelmann RK. Generalized essential telangiectasia: report of a clinical and histochemical study of 13 patients with acquired cutaneous lesions. JAMA. 1963;185:909-913.
- Vasudeva B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478.
- Ko CJ, Barr RJ. Color--pink. In: Ko CJ, Barr RJ, eds. Dermatopathology: Diagnosis by First Impression. 3rd ed. Wiley; 2016:303-322.
- Clark ML, McGuinness AE, Vidal CI. Cutaneous collagenous vasculopathy: a unique case with positive direct immunofluorescence findings. Am J Dermatopathol. 2019;41:77-79.
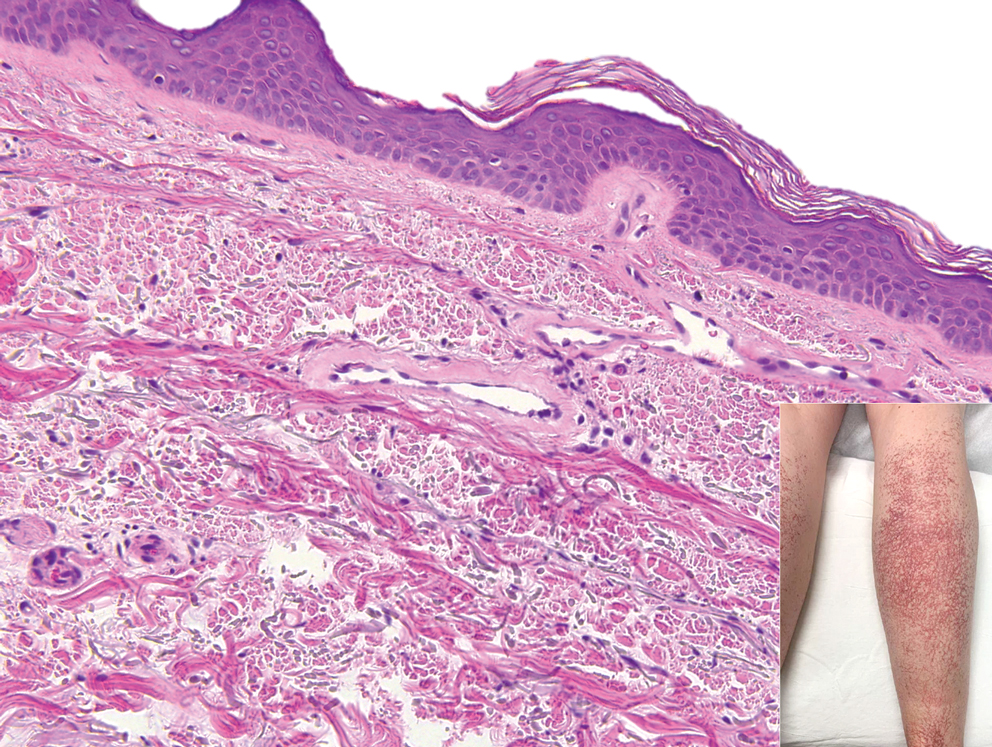
A 54-year-old woman presented with purple-red vessels on the lower legs of 15 years’ duration with gradual proximal progression to involve the thighs, breasts, and forearms. A punch biopsy of the inner thigh was obtained for histopathologic evaluation.
Comparison of Shave and Punch Biopsy Utilization Among Dermatology Practices
In 2019, the 2 Current Procedural Terminology (CPT) codes for skin biopsies (11100 and 11101) were replaced with 6 new CPT codes that specify biopsy technique and associated procedural complexity. 1,2 Prior to the coding changes, all biopsies were reimbursed at the same payment level, but a punch biopsy (11104; national nonfacility Medicare payment, $133.29) is now reimbursed more than a shave biopsy (11102; national nonfacility Medicare payment, $106.42). 3 We sought to evaluate whether the decrease in reimbursement for shave biopsies and concurrent increase in reimbursement for punch biopsies led to a shift from shave to punch biopsy utilization.
Methods
We examined shave and punch biopsies submitted for pathologic examination at Brigham and Women’s Hospital, Massachusetts General Hospital, and Massachusetts General Physician’s Organization (all in Boston, Massachusetts), and Penn Medicine, University of Pennsylvania Health System (Philadelphia, Pennsylvania), in May 2018 vs May 2019 (four months after new codes were implemented). This study was approved by Partners HealthCare (Boston, Massachusetts) and the University of Pennsylvania institutional review boards.
We included shave and punch biopsies of skin performed by physician dermatologists and mid-level providers (ie, physician assistants, nurse practitioners) at dermatology practices. All biopsies performed by a technique other than shave or punch, unspecified biopsy type, consultation cases, nonskin biopsies (eg, mucosa), and biopsies performed at nondermatology practices were excluded. We also excluded biopsies by providers who were not present during both study periods to account for provider mix.
Statistical Analysis
To evaluate for changes in the ratio of shave to punch biopsy utilization over time, we performed χ2 tests. Because care practices may differ between private and academic settings as well as between physicians and mid-level providers, we performed subgroup analyses by practice setting and provider type.4
Results
We identified 11,785 biopsies (12.11% of which were punch) submitted for pathologic examination in May 2018 compared to 11,291 biopsies (12.08% of which were punch) in May 2019 (Table). The overall use of punch biopsies relative to shave biopsies did not change between the years. There was a relative decrease in punch biopsy use among academic practices (−1.88%; P=.032) and a relative increase in punch biopsy use among private practices (+0.90%; P=.043). Provider type was not associated with differing utilization of biopsy type.
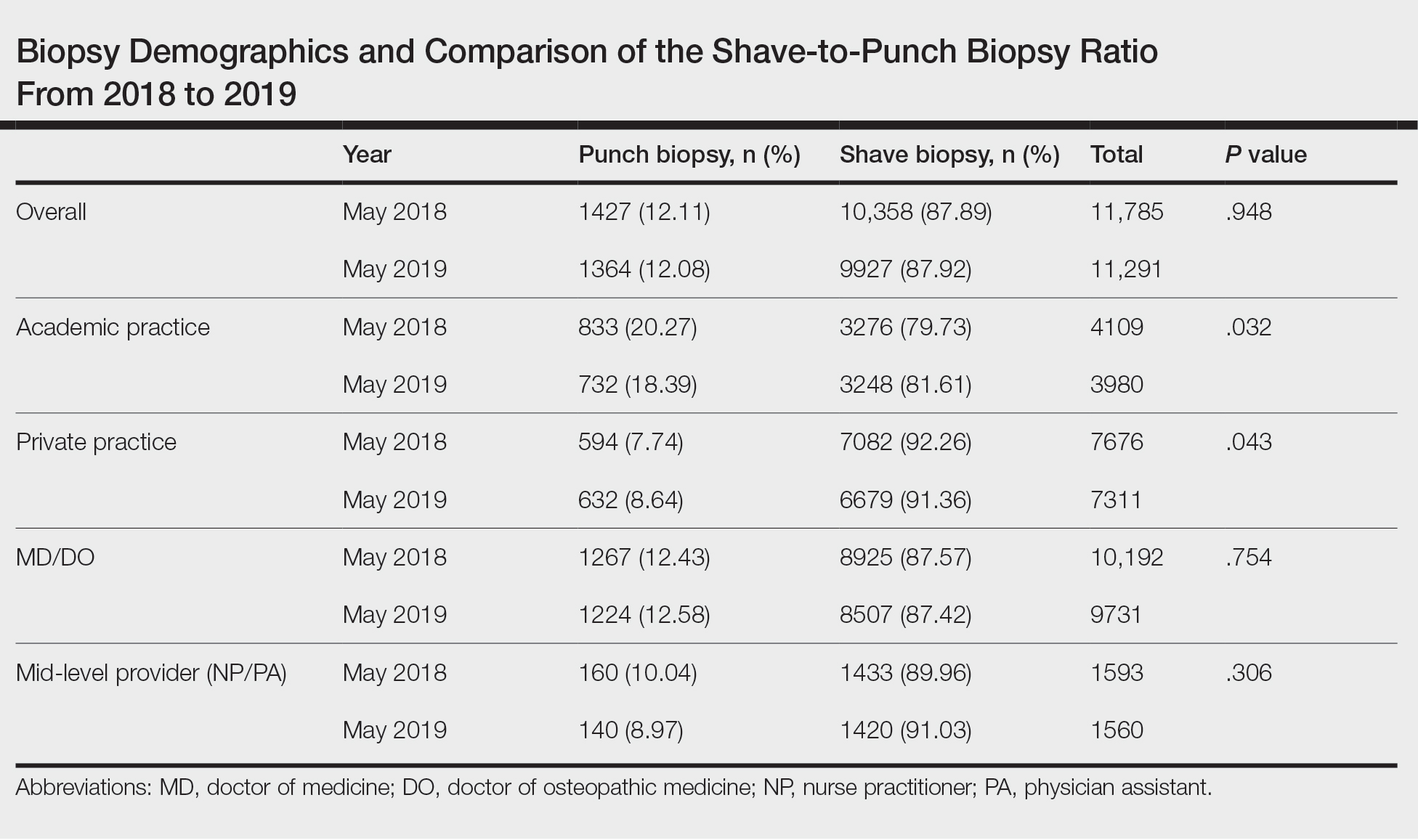
Comment
Overall, there was not a considerable shift in utilization behavior from shave to punch biopsies after the introduction of new coding changes. However, our study does demonstrate a small yet significant increase in punch biopsy utilization among private practices, and a decrease among academic practices. Although the change in biopsy utilization behavior is small in magnitude, it may have a substantial impact when extrapolated to behavior across the entire United States.
We were unable to assess additional factors, such as clinical diagnosis, body site, and cosmetic concerns, that may impact biopsy type selection in this study. Although we included multiple study sites to improve generalizability, our findings may not be representative of all biopsies performed in the dermatology setting. The baseline difference in relative punch biopsy use in academic vs private practices may reflect differences in patient populations and chief concerns, but assuming these features are stable over a 1-year time period, our findings should remain valid. Future studies should focus on qualitative evaluations of physician decision-making and evaluate whether similar trends persist over time.
Conclusion
Skin biopsy utilization trends among differing practice and provider types should continue to be monitored to assess for longitudinal trends in utilization within the context of updated billing codes and associated reimbursements.
- Grider D. 2019 CPT® coding for skin biopsies. ICD10 monitor website. September 17, 2018. Updated January 7, 2019. Accessed February 17, 2021. https://www.icd10monitor.com/2019-cpt-coding-for-skin-biopsies 2.
- Tongdee E, Siegel DM, Markowitz O. New diagnostic procedure codes and reimbursement. Cutis. 2019;103:208-211.
- Search the physician fee schedule. Centers for Medicare & Medicaid Services website. Updated January 20, 2021. Accessed February 17, 2021. https://www.cms.gov/medicare/physician-fee-schedule/search
- Zhang M, Zippin J, Kaffenberger B. Trends and scope of dermatology procedures billed by advanced practice professionals from 2012 through 2015. JAMA Dermatol. 2018;154:1040-1044.
In 2019, the 2 Current Procedural Terminology (CPT) codes for skin biopsies (11100 and 11101) were replaced with 6 new CPT codes that specify biopsy technique and associated procedural complexity. 1,2 Prior to the coding changes, all biopsies were reimbursed at the same payment level, but a punch biopsy (11104; national nonfacility Medicare payment, $133.29) is now reimbursed more than a shave biopsy (11102; national nonfacility Medicare payment, $106.42). 3 We sought to evaluate whether the decrease in reimbursement for shave biopsies and concurrent increase in reimbursement for punch biopsies led to a shift from shave to punch biopsy utilization.
Methods
We examined shave and punch biopsies submitted for pathologic examination at Brigham and Women’s Hospital, Massachusetts General Hospital, and Massachusetts General Physician’s Organization (all in Boston, Massachusetts), and Penn Medicine, University of Pennsylvania Health System (Philadelphia, Pennsylvania), in May 2018 vs May 2019 (four months after new codes were implemented). This study was approved by Partners HealthCare (Boston, Massachusetts) and the University of Pennsylvania institutional review boards.
We included shave and punch biopsies of skin performed by physician dermatologists and mid-level providers (ie, physician assistants, nurse practitioners) at dermatology practices. All biopsies performed by a technique other than shave or punch, unspecified biopsy type, consultation cases, nonskin biopsies (eg, mucosa), and biopsies performed at nondermatology practices were excluded. We also excluded biopsies by providers who were not present during both study periods to account for provider mix.
Statistical Analysis
To evaluate for changes in the ratio of shave to punch biopsy utilization over time, we performed χ2 tests. Because care practices may differ between private and academic settings as well as between physicians and mid-level providers, we performed subgroup analyses by practice setting and provider type.4
Results
We identified 11,785 biopsies (12.11% of which were punch) submitted for pathologic examination in May 2018 compared to 11,291 biopsies (12.08% of which were punch) in May 2019 (Table). The overall use of punch biopsies relative to shave biopsies did not change between the years. There was a relative decrease in punch biopsy use among academic practices (−1.88%; P=.032) and a relative increase in punch biopsy use among private practices (+0.90%; P=.043). Provider type was not associated with differing utilization of biopsy type.
Comment
Overall, there was not a considerable shift in utilization behavior from shave to punch biopsies after the introduction of new coding changes. However, our study does demonstrate a small yet significant increase in punch biopsy utilization among private practices, and a decrease among academic practices. Although the change in biopsy utilization behavior is small in magnitude, it may have a substantial impact when extrapolated to behavior across the entire United States.
We were unable to assess additional factors, such as clinical diagnosis, body site, and cosmetic concerns, that may impact biopsy type selection in this study. Although we included multiple study sites to improve generalizability, our findings may not be representative of all biopsies performed in the dermatology setting. The baseline difference in relative punch biopsy use in academic vs private practices may reflect differences in patient populations and chief concerns, but assuming these features are stable over a 1-year time period, our findings should remain valid. Future studies should focus on qualitative evaluations of physician decision-making and evaluate whether similar trends persist over time.
Conclusion
Skin biopsy utilization trends among differing practice and provider types should continue to be monitored to assess for longitudinal trends in utilization within the context of updated billing codes and associated reimbursements.
In 2019, the 2 Current Procedural Terminology (CPT) codes for skin biopsies (11100 and 11101) were replaced with 6 new CPT codes that specify biopsy technique and associated procedural complexity. 1,2 Prior to the coding changes, all biopsies were reimbursed at the same payment level, but a punch biopsy (11104; national nonfacility Medicare payment, $133.29) is now reimbursed more than a shave biopsy (11102; national nonfacility Medicare payment, $106.42). 3 We sought to evaluate whether the decrease in reimbursement for shave biopsies and concurrent increase in reimbursement for punch biopsies led to a shift from shave to punch biopsy utilization.
Methods
We examined shave and punch biopsies submitted for pathologic examination at Brigham and Women’s Hospital, Massachusetts General Hospital, and Massachusetts General Physician’s Organization (all in Boston, Massachusetts), and Penn Medicine, University of Pennsylvania Health System (Philadelphia, Pennsylvania), in May 2018 vs May 2019 (four months after new codes were implemented). This study was approved by Partners HealthCare (Boston, Massachusetts) and the University of Pennsylvania institutional review boards.
We included shave and punch biopsies of skin performed by physician dermatologists and mid-level providers (ie, physician assistants, nurse practitioners) at dermatology practices. All biopsies performed by a technique other than shave or punch, unspecified biopsy type, consultation cases, nonskin biopsies (eg, mucosa), and biopsies performed at nondermatology practices were excluded. We also excluded biopsies by providers who were not present during both study periods to account for provider mix.
Statistical Analysis
To evaluate for changes in the ratio of shave to punch biopsy utilization over time, we performed χ2 tests. Because care practices may differ between private and academic settings as well as between physicians and mid-level providers, we performed subgroup analyses by practice setting and provider type.4
Results
We identified 11,785 biopsies (12.11% of which were punch) submitted for pathologic examination in May 2018 compared to 11,291 biopsies (12.08% of which were punch) in May 2019 (Table). The overall use of punch biopsies relative to shave biopsies did not change between the years. There was a relative decrease in punch biopsy use among academic practices (−1.88%; P=.032) and a relative increase in punch biopsy use among private practices (+0.90%; P=.043). Provider type was not associated with differing utilization of biopsy type.
Comment
Overall, there was not a considerable shift in utilization behavior from shave to punch biopsies after the introduction of new coding changes. However, our study does demonstrate a small yet significant increase in punch biopsy utilization among private practices, and a decrease among academic practices. Although the change in biopsy utilization behavior is small in magnitude, it may have a substantial impact when extrapolated to behavior across the entire United States.
We were unable to assess additional factors, such as clinical diagnosis, body site, and cosmetic concerns, that may impact biopsy type selection in this study. Although we included multiple study sites to improve generalizability, our findings may not be representative of all biopsies performed in the dermatology setting. The baseline difference in relative punch biopsy use in academic vs private practices may reflect differences in patient populations and chief concerns, but assuming these features are stable over a 1-year time period, our findings should remain valid. Future studies should focus on qualitative evaluations of physician decision-making and evaluate whether similar trends persist over time.
Conclusion
Skin biopsy utilization trends among differing practice and provider types should continue to be monitored to assess for longitudinal trends in utilization within the context of updated billing codes and associated reimbursements.
- Grider D. 2019 CPT® coding for skin biopsies. ICD10 monitor website. September 17, 2018. Updated January 7, 2019. Accessed February 17, 2021. https://www.icd10monitor.com/2019-cpt-coding-for-skin-biopsies 2.
- Tongdee E, Siegel DM, Markowitz O. New diagnostic procedure codes and reimbursement. Cutis. 2019;103:208-211.
- Search the physician fee schedule. Centers for Medicare & Medicaid Services website. Updated January 20, 2021. Accessed February 17, 2021. https://www.cms.gov/medicare/physician-fee-schedule/search
- Zhang M, Zippin J, Kaffenberger B. Trends and scope of dermatology procedures billed by advanced practice professionals from 2012 through 2015. JAMA Dermatol. 2018;154:1040-1044.
- Grider D. 2019 CPT® coding for skin biopsies. ICD10 monitor website. September 17, 2018. Updated January 7, 2019. Accessed February 17, 2021. https://www.icd10monitor.com/2019-cpt-coding-for-skin-biopsies 2.
- Tongdee E, Siegel DM, Markowitz O. New diagnostic procedure codes and reimbursement. Cutis. 2019;103:208-211.
- Search the physician fee schedule. Centers for Medicare & Medicaid Services website. Updated January 20, 2021. Accessed February 17, 2021. https://www.cms.gov/medicare/physician-fee-schedule/search
- Zhang M, Zippin J, Kaffenberger B. Trends and scope of dermatology procedures billed by advanced practice professionals from 2012 through 2015. JAMA Dermatol. 2018;154:1040-1044.
Practice Points
- Dermatologists should be aware that skin biopsy billing codes and reimbursements were changed in 2019 to reflect their level of complexity, which may impact how often each type of biopsy is used.
- Even small shifts in biopsy utilization behavior among dermatologists in the context of reimbursement changes can have a large impact on net reimbursements.
Hyperkeratotic Nummular Plaques on the Upper Trunk
The Diagnosis: Extragenital Lichen Sclerosus Et Atrophicus
Histopathologic evaluation revealed hyperkeratosis, follicular plugging, epidermal atrophy, and homogenization of papillary dermal collagen with an underlying lymphocytic infiltrate (Figure 1). Direct immunofluorescence of a plaque with a superimposed bulla was negative for deposition of C3, IgG, IgA, IgM, or fibrinogen. Accordingly, clinicopathologic correlation supported a diagnosis of extragenital lichen sclerosus et atrophicus (LSA). Of note, the patient's history of genital irritation was due to genital LSA that preceded the extragenital manifestations.
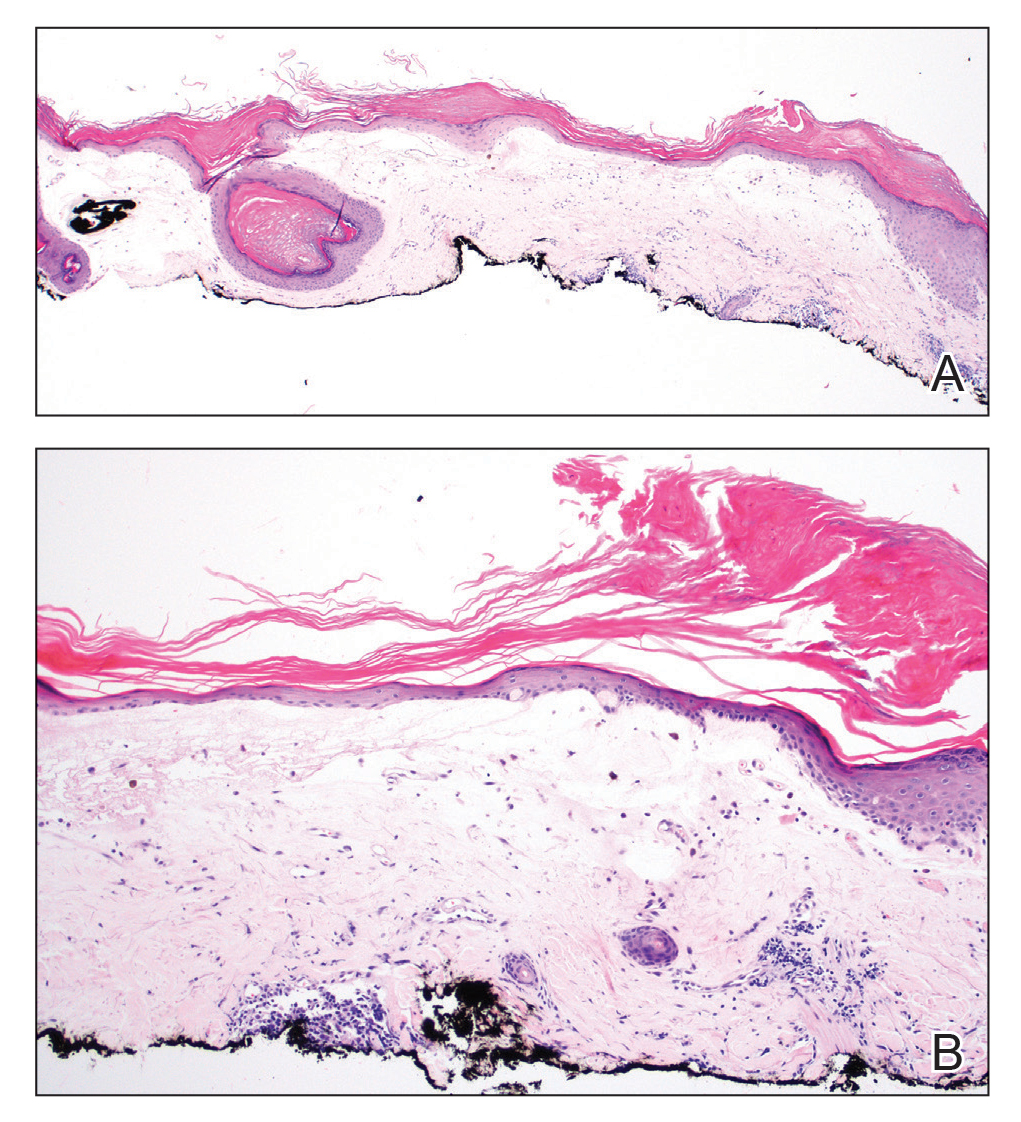
Lichen sclerosus et atrophicus is an inflammatory dermatosis that typically presents as atrophic white papules of the anogenital area that coalesce into pruritic plaques; the exact etiology remains to be elucidated, yet various circulating autoantibodies have been identified, suggesting a role for autoimmunity.1,2 Lichen sclerosus et atrophicus is more common in women than in men, with a bimodal peak in the age of onset affecting postmenopausal and prepubertal populations.1 In women, affected areas include the labia minora and majora, clitoris, perineum, and perianal skin; LSA spares the mucosal surfaces of the vagina and cervix.2 In men, uncircumscribed genital skin more commonly is affected. Involvement is localized to the foreskin and glans with occasional urethral involvement.2
In contrast, extragenital LSA tends to present as asymptomatic papules and plaques that develop atrophy with time, involving the back, shoulders, neck, chest, thighs, axillae, and flexural wrists2,3; an erythematous rim often is present,4 and hyperkeratosis with follicular plugging may be prominent.5 Our patient's case emphasizes the predilection of plaques for the chest and intermammary skin (Figure 2A). Approximately 15% of LSA cases have extragenital involvement, and extragenital-limited disease accounts for roughly 5% of cases.6,7 Unlike genital LSA, extragenital disease has not been associated with an increased risk for squamous cell carcinoma.1 Bullae formation within plaques of genital or extragenital LSA has been reported3,8 and is exemplified in our patient (Figure 2B). Intralesional bullae formation likely is due to a combination of internal and external factors, mainly the inability to withstand shear forces due to an atrophic epidermis with basal vacuolar injury overlying an edematous papillary dermis with altered collagen.8 Dermatoscopic findings may aid in recognizing extragenital LSA9,10; our patient's plaques demonstrated the characteristic findings of comedolike openings, structureless white areas, and pink borders (Figure 3).
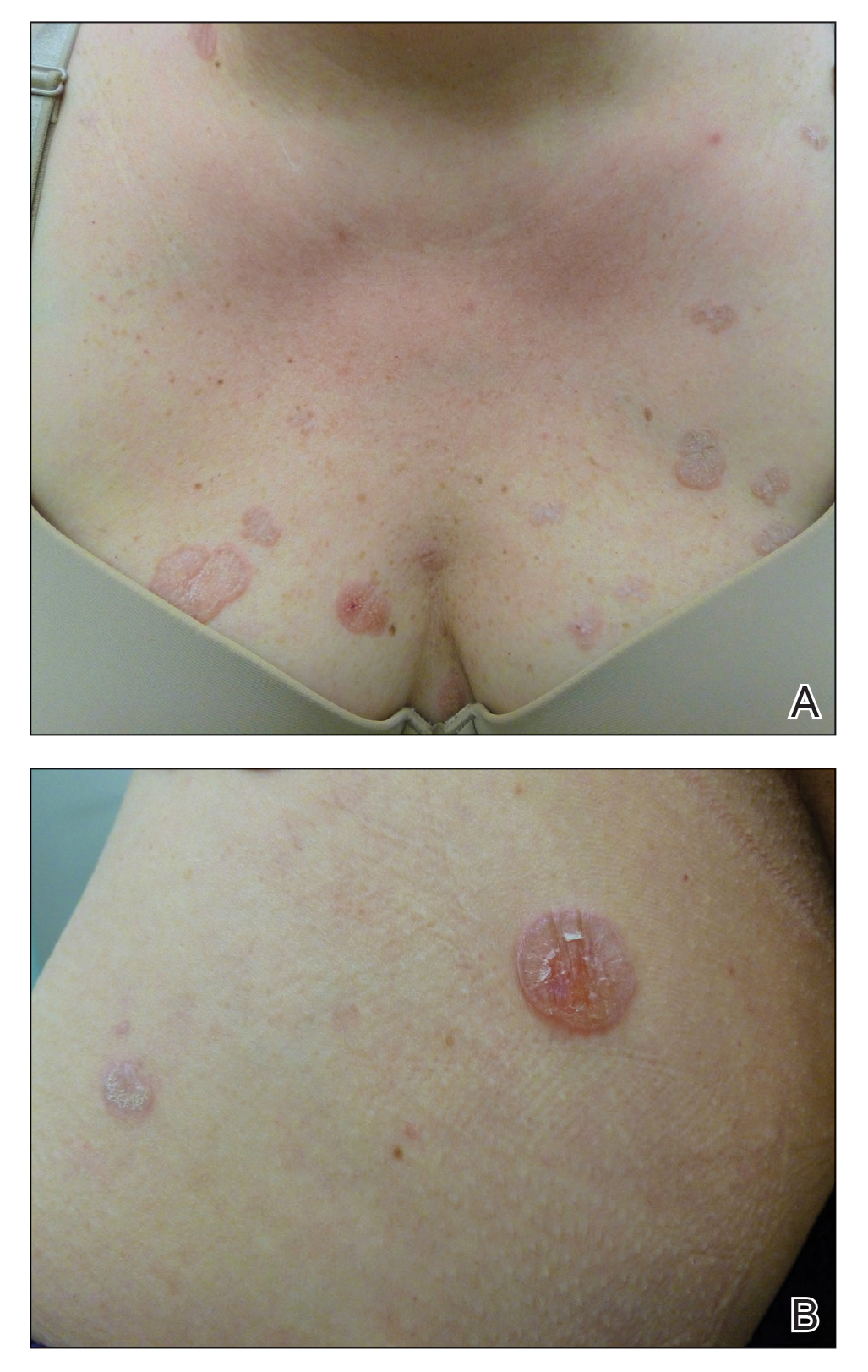
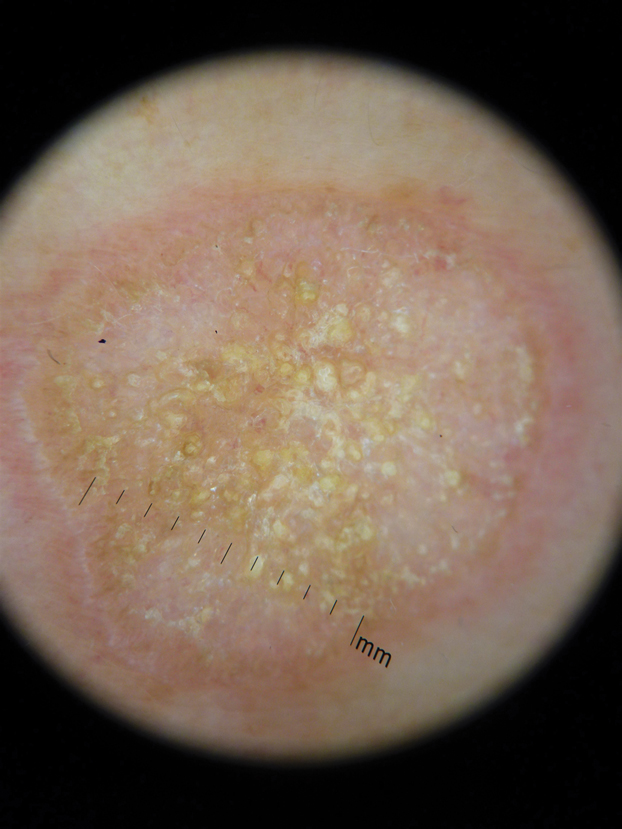
The clinical differential diagnosis for well-demarcated, pink, scaly plaques is broad. Nummular eczema usually presents as coin-shaped eczematous plaques on the dorsal aspects of the hands or lower extremities, and histology shows epidermal spongiosis.11 Nummular eczema may be considered due to the striking round morphology of various plaques, yet our patient's presentation was better served by a consideration of several papulosquamous disorders.
Lichen planus (LP) presents as intensely pruritic, violaceous, polygonal, flat-topped papules with overlying reticular white lines, or Wickham striae, that favor the flexural wrists, lower back, and lower extremities. Lichen planus also may have oral and genital mucosal involvement. Similar to LSA, LP is more common in women and preferentially affects the postmenopausal population.12 Additionally, hypertrophic LP may obscure Wickham striae and mimic extragenital LSA; distinguishing features of hypertrophic LP are intense pruritus and a predilection for the shins. Histology is defined by orthohyperkeratosis, hypergranulosis, sawtooth acanthosis, and vacuolar degeneration of the basal layer with Civatte bodies or dyskeratotic basal keratinocytes overlying a characteristic bandlike infiltrate of lymphocytes.12
Lichen simplex chronicus (LSC) is characterized by intense pruritus and presents as hyperkeratotic plaques with a predilection for accessible regions such as the posterior neck and extremities.13 The striking annular demarcation of this case makes LSC unlikely. Comparable to LSA and LP, LSC also may present with both genital and extragenital findings. Histology of LSC is characterized by irregular acanthosis or thickening of the epidermis with vertical streaking of collagen and vascular bundles of the papillary dermis.13
Subacute cutaneous lupus erythematosus (SCLE) is important to consider for a new papulosquamous eruption with a predilection for the sun-exposed skin of a middle-aged woman. The presence of papules on the volar wrist and history of genital irritation, however, make this entity less likely. Similar to LSA, histologic examination of SCLE reveals epidermal atrophy, basal layer degeneration, and papillary dermal edema with lymphocytic inflammation. However, SCLE lacks the band of inflammation underlying pale homogenized papillary dermal collagen, the most distinguishing feature of LSA; instead, SCLE shows superficial and deep perivascular and periadnexal lymphocytes and mucin in the dermis.14
Lichen sclerosus et atrophicus may be chronic and progressive in nature or cycle through remissions and relapses.2 Treatment is not curative, and management is directed to alleviating symptoms and preventing the progression of disease. First-line management of extragenital LSA is potent topical steroids.1 Adjuvant topical calcineurin inhibitors may be used as steroid-sparing agents.2 Phototherapy is a second-line therapy and even narrowband UVB phototherapy has demonstrated efficacy in managing extragenital LSA.15,16 Our patient was started on mometasone ointment and calcipotriene cream with slight improvement after a 6-month trial. Ongoing management is focused on optimizing application of topical therapies.
- Powell JJ, Wojnarowska F. Lichen sclerosus. Lancet. 1999;353:1777-1783.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus. Am J Clin Dermatol. 2013;14:27-47.
- Meffert JJ, Davis BM, Grimwood RE. Lichen sclerosus. J Am Acad Dermatol. 1995;32:393-416.
- Surkan M, Hull P. A case of lichen sclerosus et atrophicus with distinct erythematous borders. J Cutan Med Surg. 2015;19:600-603.
- Kimura A, Kambe N, Satoh T, et al. Follicular keratosis and bullous formation are typical signs of extragenital lichen sclerosus. J Dermatol. 2011;38:834-836.
- Meyrick Thomas RH, Ridley CM, McGibbon DH, et al. Lichen sclerosus et atrophicus and autoimmunity: a study of 350 women. Br J Dermatol. 1988;118:41-46.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Hallel-Halevy D, Grunwald MH, Yerushalmi J, et al. Bullous lichen sclerosus et atrophicus. J Am Acad Dermatol. 1998;39:500-501.
- Garrido-Ríos AA, Álvarez-Garrido H, Sanz-Muñoz C, et al. Dermoscopy of extragenital lichen sclerosus. Arch Dermatol. 2009;145:1468.
- Larre Borges A, Tiodorovic-Zivkovic D, Lallas A, et al. Clinical, dermoscopic and histopathologic features of genital and extragenital lichen sclerosus. J Eur Acad Dermatol Venereol. 2013;27:1433-1439.
- Rudikoff D. Differential diagnosis of round or discoid lesions. Clin Dermatol. 2011;29:489-497.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Shaffer B, Beerman H. Lichen simplex chronicus and its variants: a discussion of certain psychodynamic mechanisms and clinical and histopathologic correlations. AMA Arch Derm Syphilol. 1951;64:340-351.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. Am J Clin Dermatol. 2009;10:365-381.
- Sauder MB, Linzon-Smith J, Beecker J. Extragenital bullous lichen sclerosus. J Am Acad Dermatol. 2014;71:981-984.
- Colbert RL, Chiang MP, Carlin CS, et al. Progressive extragenital lichen sclerosus successfully treated with narrowband UV-B phototherapy. Arch Dermatol. 2007;143:19-20.
The Diagnosis: Extragenital Lichen Sclerosus Et Atrophicus
Histopathologic evaluation revealed hyperkeratosis, follicular plugging, epidermal atrophy, and homogenization of papillary dermal collagen with an underlying lymphocytic infiltrate (Figure 1). Direct immunofluorescence of a plaque with a superimposed bulla was negative for deposition of C3, IgG, IgA, IgM, or fibrinogen. Accordingly, clinicopathologic correlation supported a diagnosis of extragenital lichen sclerosus et atrophicus (LSA). Of note, the patient's history of genital irritation was due to genital LSA that preceded the extragenital manifestations.
Lichen sclerosus et atrophicus is an inflammatory dermatosis that typically presents as atrophic white papules of the anogenital area that coalesce into pruritic plaques; the exact etiology remains to be elucidated, yet various circulating autoantibodies have been identified, suggesting a role for autoimmunity.1,2 Lichen sclerosus et atrophicus is more common in women than in men, with a bimodal peak in the age of onset affecting postmenopausal and prepubertal populations.1 In women, affected areas include the labia minora and majora, clitoris, perineum, and perianal skin; LSA spares the mucosal surfaces of the vagina and cervix.2 In men, uncircumscribed genital skin more commonly is affected. Involvement is localized to the foreskin and glans with occasional urethral involvement.2
In contrast, extragenital LSA tends to present as asymptomatic papules and plaques that develop atrophy with time, involving the back, shoulders, neck, chest, thighs, axillae, and flexural wrists2,3; an erythematous rim often is present,4 and hyperkeratosis with follicular plugging may be prominent.5 Our patient's case emphasizes the predilection of plaques for the chest and intermammary skin (Figure 2A). Approximately 15% of LSA cases have extragenital involvement, and extragenital-limited disease accounts for roughly 5% of cases.6,7 Unlike genital LSA, extragenital disease has not been associated with an increased risk for squamous cell carcinoma.1 Bullae formation within plaques of genital or extragenital LSA has been reported3,8 and is exemplified in our patient (Figure 2B). Intralesional bullae formation likely is due to a combination of internal and external factors, mainly the inability to withstand shear forces due to an atrophic epidermis with basal vacuolar injury overlying an edematous papillary dermis with altered collagen.8 Dermatoscopic findings may aid in recognizing extragenital LSA9,10; our patient's plaques demonstrated the characteristic findings of comedolike openings, structureless white areas, and pink borders (Figure 3).
The clinical differential diagnosis for well-demarcated, pink, scaly plaques is broad. Nummular eczema usually presents as coin-shaped eczematous plaques on the dorsal aspects of the hands or lower extremities, and histology shows epidermal spongiosis.11 Nummular eczema may be considered due to the striking round morphology of various plaques, yet our patient's presentation was better served by a consideration of several papulosquamous disorders.
Lichen planus (LP) presents as intensely pruritic, violaceous, polygonal, flat-topped papules with overlying reticular white lines, or Wickham striae, that favor the flexural wrists, lower back, and lower extremities. Lichen planus also may have oral and genital mucosal involvement. Similar to LSA, LP is more common in women and preferentially affects the postmenopausal population.12 Additionally, hypertrophic LP may obscure Wickham striae and mimic extragenital LSA; distinguishing features of hypertrophic LP are intense pruritus and a predilection for the shins. Histology is defined by orthohyperkeratosis, hypergranulosis, sawtooth acanthosis, and vacuolar degeneration of the basal layer with Civatte bodies or dyskeratotic basal keratinocytes overlying a characteristic bandlike infiltrate of lymphocytes.12
Lichen simplex chronicus (LSC) is characterized by intense pruritus and presents as hyperkeratotic plaques with a predilection for accessible regions such as the posterior neck and extremities.13 The striking annular demarcation of this case makes LSC unlikely. Comparable to LSA and LP, LSC also may present with both genital and extragenital findings. Histology of LSC is characterized by irregular acanthosis or thickening of the epidermis with vertical streaking of collagen and vascular bundles of the papillary dermis.13
Subacute cutaneous lupus erythematosus (SCLE) is important to consider for a new papulosquamous eruption with a predilection for the sun-exposed skin of a middle-aged woman. The presence of papules on the volar wrist and history of genital irritation, however, make this entity less likely. Similar to LSA, histologic examination of SCLE reveals epidermal atrophy, basal layer degeneration, and papillary dermal edema with lymphocytic inflammation. However, SCLE lacks the band of inflammation underlying pale homogenized papillary dermal collagen, the most distinguishing feature of LSA; instead, SCLE shows superficial and deep perivascular and periadnexal lymphocytes and mucin in the dermis.14
Lichen sclerosus et atrophicus may be chronic and progressive in nature or cycle through remissions and relapses.2 Treatment is not curative, and management is directed to alleviating symptoms and preventing the progression of disease. First-line management of extragenital LSA is potent topical steroids.1 Adjuvant topical calcineurin inhibitors may be used as steroid-sparing agents.2 Phototherapy is a second-line therapy and even narrowband UVB phototherapy has demonstrated efficacy in managing extragenital LSA.15,16 Our patient was started on mometasone ointment and calcipotriene cream with slight improvement after a 6-month trial. Ongoing management is focused on optimizing application of topical therapies.
The Diagnosis: Extragenital Lichen Sclerosus Et Atrophicus
Histopathologic evaluation revealed hyperkeratosis, follicular plugging, epidermal atrophy, and homogenization of papillary dermal collagen with an underlying lymphocytic infiltrate (Figure 1). Direct immunofluorescence of a plaque with a superimposed bulla was negative for deposition of C3, IgG, IgA, IgM, or fibrinogen. Accordingly, clinicopathologic correlation supported a diagnosis of extragenital lichen sclerosus et atrophicus (LSA). Of note, the patient's history of genital irritation was due to genital LSA that preceded the extragenital manifestations.
Lichen sclerosus et atrophicus is an inflammatory dermatosis that typically presents as atrophic white papules of the anogenital area that coalesce into pruritic plaques; the exact etiology remains to be elucidated, yet various circulating autoantibodies have been identified, suggesting a role for autoimmunity.1,2 Lichen sclerosus et atrophicus is more common in women than in men, with a bimodal peak in the age of onset affecting postmenopausal and prepubertal populations.1 In women, affected areas include the labia minora and majora, clitoris, perineum, and perianal skin; LSA spares the mucosal surfaces of the vagina and cervix.2 In men, uncircumscribed genital skin more commonly is affected. Involvement is localized to the foreskin and glans with occasional urethral involvement.2
In contrast, extragenital LSA tends to present as asymptomatic papules and plaques that develop atrophy with time, involving the back, shoulders, neck, chest, thighs, axillae, and flexural wrists2,3; an erythematous rim often is present,4 and hyperkeratosis with follicular plugging may be prominent.5 Our patient's case emphasizes the predilection of plaques for the chest and intermammary skin (Figure 2A). Approximately 15% of LSA cases have extragenital involvement, and extragenital-limited disease accounts for roughly 5% of cases.6,7 Unlike genital LSA, extragenital disease has not been associated with an increased risk for squamous cell carcinoma.1 Bullae formation within plaques of genital or extragenital LSA has been reported3,8 and is exemplified in our patient (Figure 2B). Intralesional bullae formation likely is due to a combination of internal and external factors, mainly the inability to withstand shear forces due to an atrophic epidermis with basal vacuolar injury overlying an edematous papillary dermis with altered collagen.8 Dermatoscopic findings may aid in recognizing extragenital LSA9,10; our patient's plaques demonstrated the characteristic findings of comedolike openings, structureless white areas, and pink borders (Figure 3).
The clinical differential diagnosis for well-demarcated, pink, scaly plaques is broad. Nummular eczema usually presents as coin-shaped eczematous plaques on the dorsal aspects of the hands or lower extremities, and histology shows epidermal spongiosis.11 Nummular eczema may be considered due to the striking round morphology of various plaques, yet our patient's presentation was better served by a consideration of several papulosquamous disorders.
Lichen planus (LP) presents as intensely pruritic, violaceous, polygonal, flat-topped papules with overlying reticular white lines, or Wickham striae, that favor the flexural wrists, lower back, and lower extremities. Lichen planus also may have oral and genital mucosal involvement. Similar to LSA, LP is more common in women and preferentially affects the postmenopausal population.12 Additionally, hypertrophic LP may obscure Wickham striae and mimic extragenital LSA; distinguishing features of hypertrophic LP are intense pruritus and a predilection for the shins. Histology is defined by orthohyperkeratosis, hypergranulosis, sawtooth acanthosis, and vacuolar degeneration of the basal layer with Civatte bodies or dyskeratotic basal keratinocytes overlying a characteristic bandlike infiltrate of lymphocytes.12
Lichen simplex chronicus (LSC) is characterized by intense pruritus and presents as hyperkeratotic plaques with a predilection for accessible regions such as the posterior neck and extremities.13 The striking annular demarcation of this case makes LSC unlikely. Comparable to LSA and LP, LSC also may present with both genital and extragenital findings. Histology of LSC is characterized by irregular acanthosis or thickening of the epidermis with vertical streaking of collagen and vascular bundles of the papillary dermis.13
Subacute cutaneous lupus erythematosus (SCLE) is important to consider for a new papulosquamous eruption with a predilection for the sun-exposed skin of a middle-aged woman. The presence of papules on the volar wrist and history of genital irritation, however, make this entity less likely. Similar to LSA, histologic examination of SCLE reveals epidermal atrophy, basal layer degeneration, and papillary dermal edema with lymphocytic inflammation. However, SCLE lacks the band of inflammation underlying pale homogenized papillary dermal collagen, the most distinguishing feature of LSA; instead, SCLE shows superficial and deep perivascular and periadnexal lymphocytes and mucin in the dermis.14
Lichen sclerosus et atrophicus may be chronic and progressive in nature or cycle through remissions and relapses.2 Treatment is not curative, and management is directed to alleviating symptoms and preventing the progression of disease. First-line management of extragenital LSA is potent topical steroids.1 Adjuvant topical calcineurin inhibitors may be used as steroid-sparing agents.2 Phototherapy is a second-line therapy and even narrowband UVB phototherapy has demonstrated efficacy in managing extragenital LSA.15,16 Our patient was started on mometasone ointment and calcipotriene cream with slight improvement after a 6-month trial. Ongoing management is focused on optimizing application of topical therapies.
- Powell JJ, Wojnarowska F. Lichen sclerosus. Lancet. 1999;353:1777-1783.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus. Am J Clin Dermatol. 2013;14:27-47.
- Meffert JJ, Davis BM, Grimwood RE. Lichen sclerosus. J Am Acad Dermatol. 1995;32:393-416.
- Surkan M, Hull P. A case of lichen sclerosus et atrophicus with distinct erythematous borders. J Cutan Med Surg. 2015;19:600-603.
- Kimura A, Kambe N, Satoh T, et al. Follicular keratosis and bullous formation are typical signs of extragenital lichen sclerosus. J Dermatol. 2011;38:834-836.
- Meyrick Thomas RH, Ridley CM, McGibbon DH, et al. Lichen sclerosus et atrophicus and autoimmunity: a study of 350 women. Br J Dermatol. 1988;118:41-46.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Hallel-Halevy D, Grunwald MH, Yerushalmi J, et al. Bullous lichen sclerosus et atrophicus. J Am Acad Dermatol. 1998;39:500-501.
- Garrido-Ríos AA, Álvarez-Garrido H, Sanz-Muñoz C, et al. Dermoscopy of extragenital lichen sclerosus. Arch Dermatol. 2009;145:1468.
- Larre Borges A, Tiodorovic-Zivkovic D, Lallas A, et al. Clinical, dermoscopic and histopathologic features of genital and extragenital lichen sclerosus. J Eur Acad Dermatol Venereol. 2013;27:1433-1439.
- Rudikoff D. Differential diagnosis of round or discoid lesions. Clin Dermatol. 2011;29:489-497.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Shaffer B, Beerman H. Lichen simplex chronicus and its variants: a discussion of certain psychodynamic mechanisms and clinical and histopathologic correlations. AMA Arch Derm Syphilol. 1951;64:340-351.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. Am J Clin Dermatol. 2009;10:365-381.
- Sauder MB, Linzon-Smith J, Beecker J. Extragenital bullous lichen sclerosus. J Am Acad Dermatol. 2014;71:981-984.
- Colbert RL, Chiang MP, Carlin CS, et al. Progressive extragenital lichen sclerosus successfully treated with narrowband UV-B phototherapy. Arch Dermatol. 2007;143:19-20.
- Powell JJ, Wojnarowska F. Lichen sclerosus. Lancet. 1999;353:1777-1783.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus. Am J Clin Dermatol. 2013;14:27-47.
- Meffert JJ, Davis BM, Grimwood RE. Lichen sclerosus. J Am Acad Dermatol. 1995;32:393-416.
- Surkan M, Hull P. A case of lichen sclerosus et atrophicus with distinct erythematous borders. J Cutan Med Surg. 2015;19:600-603.
- Kimura A, Kambe N, Satoh T, et al. Follicular keratosis and bullous formation are typical signs of extragenital lichen sclerosus. J Dermatol. 2011;38:834-836.
- Meyrick Thomas RH, Ridley CM, McGibbon DH, et al. Lichen sclerosus et atrophicus and autoimmunity: a study of 350 women. Br J Dermatol. 1988;118:41-46.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Hallel-Halevy D, Grunwald MH, Yerushalmi J, et al. Bullous lichen sclerosus et atrophicus. J Am Acad Dermatol. 1998;39:500-501.
- Garrido-Ríos AA, Álvarez-Garrido H, Sanz-Muñoz C, et al. Dermoscopy of extragenital lichen sclerosus. Arch Dermatol. 2009;145:1468.
- Larre Borges A, Tiodorovic-Zivkovic D, Lallas A, et al. Clinical, dermoscopic and histopathologic features of genital and extragenital lichen sclerosus. J Eur Acad Dermatol Venereol. 2013;27:1433-1439.
- Rudikoff D. Differential diagnosis of round or discoid lesions. Clin Dermatol. 2011;29:489-497.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Shaffer B, Beerman H. Lichen simplex chronicus and its variants: a discussion of certain psychodynamic mechanisms and clinical and histopathologic correlations. AMA Arch Derm Syphilol. 1951;64:340-351.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. Am J Clin Dermatol. 2009;10:365-381.
- Sauder MB, Linzon-Smith J, Beecker J. Extragenital bullous lichen sclerosus. J Am Acad Dermatol. 2014;71:981-984.
- Colbert RL, Chiang MP, Carlin CS, et al. Progressive extragenital lichen sclerosus successfully treated with narrowband UV-B phototherapy. Arch Dermatol. 2007;143:19-20.
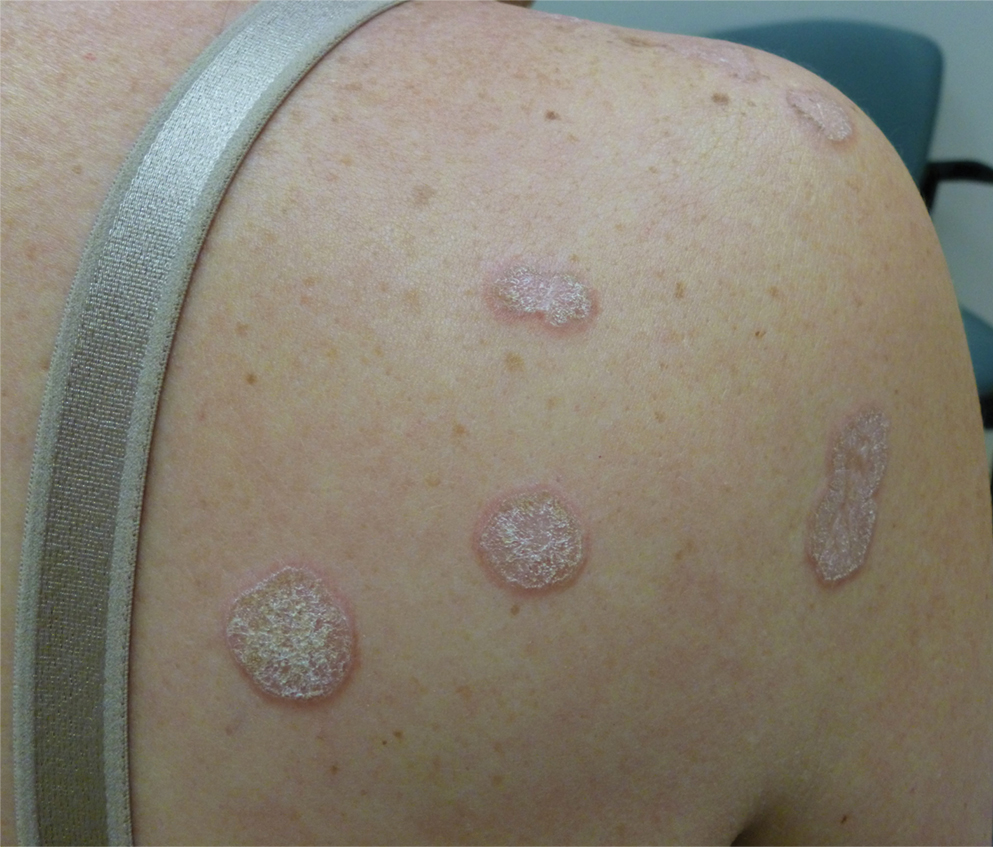
A 48-year-old woman with a history of type 2 diabetes mellitus and nonalcoholic steatohepatitis presented with papules and plaques on the upper trunk, proximal extremities, and volar wrists. Clear fluid–filled bullae occasionally developed within the plaques and subsequently ruptured and healed. Aside from intermittent lesion tenderness and irritation with the formation and rupture of the bullae, the patient’s plaques were asymptomatic, and she specifically denied pruritus. A review of systems revealed a history of genital irritation evaluated by a gynecologist; nystatin–triamcinolone cream 0.1% applied as needed provided relief. The patient denied any recent flares or any new or changing oral mucosa findings or symptoms, preceding medications, or family history of similar lesions. Physical examination revealed well-demarcated, round, pink plaques with keratotic scale scattered across the upper trunk and central chest. The bilateral volar wrists were surfaced by well-circumscribed, thin, pink to violaceous, hyperkeratotic papules.
Painless Mobile Nodule on the Shoulder
The Diagnosis: Cutaneous Metaplastic Synovial Cyst
Gross examination of the excised nodule revealed a 2.5×1.2×1.0-cm, intact, gray-white, thin-walled, smooth-lined nodule filled with clear mucinouslike material. Hematoxylin and eosin-stained sections demonstrated a dermal-based cystlike structure composed of a lining of connective tissue with hyalinized material and fibrin as well as spindle and epithelioid cells with a mild mixed inflammatory infiltrate (Figure). These histopathologic findings led to the diagnosis of cutaneous metaplastic synovial cyst (CMSC).
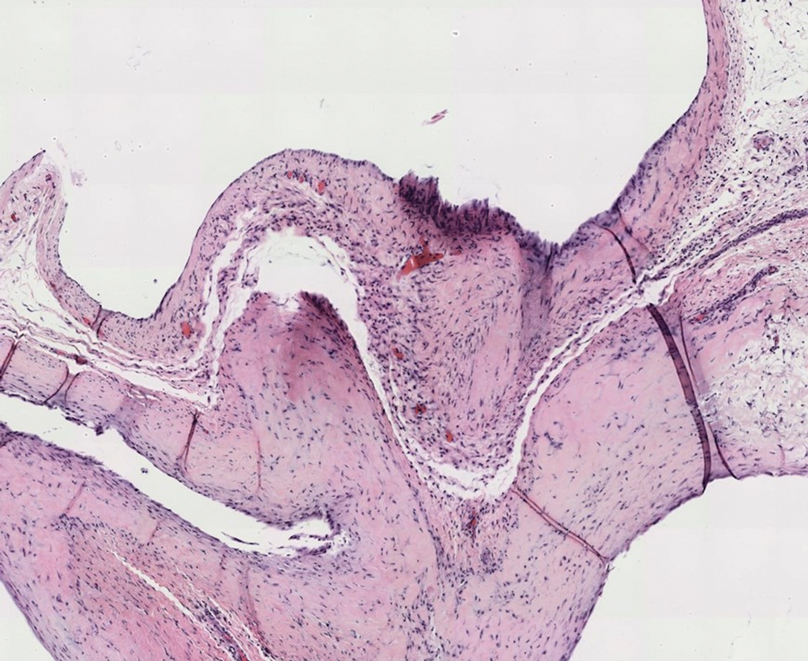
Cutaneous metaplastic synovial cyst, also known as synovial metaplasia of the skin, is an uncommon benign cystic lesion that was first reported by Gonzalez et al1 in 1987. Histologically, CMSC lacks an epithelial lining and therefore is not a true cyst but rather a pseudocyst.2 Clinically, the lesion typically presents as a solitary subcutaneous nodule that may be tender or painless. In a literature review of CMSC cases performed by Fukuyama et al,3 distribution of reported cases according to body site varied; however, limbs were found to be the most commonly involved area. A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the term cutaneous metaplastic synovial cyst revealed at least 37 cases reported in the English-language literature,3-9 including our present case. The pathogenesis remains uncertain; however, a majority of previously reported cases of CMSC characteristically have been associated with a pre-existing lesion, with most presentations developing at surgical scar sites secondary to operation or trauma.5 Relative tissue fragility secondary to rheumatoid arthritis10 and Ehlers-Danlos syndrome9,11,12 has been linked to CMSC in some documented reports, while a minority of cases report no antecedent events triggering formation of the lesion.13-15
As evidenced by our patient, CMSC clinically mimics several other benign entities; histopathologic examination is necessary to confirm the diagnosis. Although nodular hidradenoma also may clinically present as a solitary firm intradermal nodule, microscopy reveals a dermal-based lobulated tumor containing cystic spaces and solid areas composed of basophilic polyhedral cells and round glycogen-filled clear cells.16 Epidermoid cysts are differentiated from CMSC by the presence of a cyst wall lining composed of stratified squamous epithelium and associated laminated keratin within the lumen,17 which corresponds to its pearly white appearance on gross examination. Cutaneous ciliated cysts predominantly occur on the lower extremities of young women and are lined by simple cuboidal or columnar ciliated cells that resemble müllerian epithelium.18 Similar to CMSC, ganglion cysts are pseudocysts that lack a true epithelial lining but differ in appearance due to their mucin-filled synovial-lined sac.19 Additionally, ganglion cysts most often occur on the dorsal and volar aspects of the wrist.
Excisional biopsy is indicated as the preferred treatment of CMSC, given the lesion's benign behavior and low recurrence rate.6 Our case highlights this rare entity and reinforces its inclusion in the differential diagnosis of subcutaneous mobile nodules, especially in the setting of prior tissue injury secondary to trauma, surgical procedures, or conditions such as rheumatoid arthritis or Ehlers-Danlos syndrome. Unlike most previously reported cases, our patient reported no preceding tissue injury associated with formation of the lesion, and she was largely asymptomatic on presentation. Considering the limited number of CMSC cases demonstrated in the literature, it is important to continue reporting new cases to better understand characteristics and presentations of this uncommon lesion.
- Gonzalez JG, Ghiselli RW, Santa Cruz DJ. Synovial metaplasia of the skin. Am J Surg Pathol. 1987;11:343-350.
- Calonje E, Brenn T, Lazar A, et al. Cutaneous cysts. In: Calonje E, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 5th ed. Elsevier Limited; 2020:1680-1697.
- Fukuyama M, Sato Y, Hayakawa J, et al. Cutaneous metaplastic synovial cyst: case report and literature review from the dermatological point of view. Keio J Med. 2016;66:9-13.
- Karaytug K, Kapicioglu M, Can N, et al. Unprecedented recurrence of carpal tunnel syndrome by metaplastic synovial cyst in the carpal tunnel. Acta Orthop Traumatol Turc. 2019;53:230-232.
- Martelli SJ, Silveira FM, Carvalho PH, et al. Asymptomatic subcutaneous swelling of lower face. Oral Surg Oral Med Oral Pathol Oral Radiol. 2019;128:101-105.
- Majdi M, Saffar H, Ghanadan A. Cutaneous metaplastic synovial cyst: a case report. Iran J Pathol. 2016;11:423-426.
- Ramachandra S, Rao L, Al-Kindi M. Cutaneous metaplastic synovial cyst. Sultan Qaboos Univ Med J. 2016;16:E117-E118.
- Heidarian A, Xie Q, Banihashemi A. Cutaneous metaplastic synovial cyst presenting as an axillary mass after modified mastectomy and adjuvant radiotherapy. Am J Clin Pathol. 2016;146:S2.
- Fernandez-Flores A, Barja-Lopez JM. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome. J Cutan Pathol. 2020;47:729-733.
- Choonhakarn C, Tang S. Cutaneous metaplastic synovial cyst. J Dermatol. 2003;30:480-484.
- Guala A, Viglio S, Ottinetti A, et al. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome: report of a second case. Am J Dermatopathol. 2008;30:59-61.
- Nieto S, Buezo GF, Jones-Caballero M, et al. Cutaneous metaplastic synovial cyst in an Ehlers-Danlos patient. Am J Dermatopathol. 1997;19:407-410.
- Goiriz R, Rios-Buceta L, Alonso-Perez A, et al. Cutaneous metaplastic synovial cyst. J Am Acad Dermatol. 2005;53:180-181.
- Kim BC, Choi WJ, Park EJ, et al. Cutaneous metaplastic synovial cyst of the first metatarsal head area. Ann Dermatol. 2011;23(suppl 2):S165-S168.
- Yang HC, Tsai YJ, Hu SL, et al. Cutaneous metaplastic synovial cyst--a case report and review of literature. Dermatol Sinica. 2003;21:275-279.
- Kataria SP, Singh G, Batra A, et al. Nodular hidradenoma: a series of five cases in male subjects and review of literature. Adv Cytol Pathol. 2018;3:46-47.
- Mohamed Haflah N, Mohd Kassim A, Hassan Shukur M. Giant epidermoid cyst of the thigh. Malays Orthop J. 2011;5:17-19.
- Torisu-Itakura H, Itakura E, Horiuchi R, et al. Cutaneous ciliated cyst on the leg of a woman of menopausal age. Acta Derm Venereol. 2009;89:323-324.
- Fullen DR. Cysts and sinuses. In: Busam K, ed. Dermatopathology. Saunders; 2010:300-330.
The Diagnosis: Cutaneous Metaplastic Synovial Cyst
Gross examination of the excised nodule revealed a 2.5×1.2×1.0-cm, intact, gray-white, thin-walled, smooth-lined nodule filled with clear mucinouslike material. Hematoxylin and eosin-stained sections demonstrated a dermal-based cystlike structure composed of a lining of connective tissue with hyalinized material and fibrin as well as spindle and epithelioid cells with a mild mixed inflammatory infiltrate (Figure). These histopathologic findings led to the diagnosis of cutaneous metaplastic synovial cyst (CMSC).
Cutaneous metaplastic synovial cyst, also known as synovial metaplasia of the skin, is an uncommon benign cystic lesion that was first reported by Gonzalez et al1 in 1987. Histologically, CMSC lacks an epithelial lining and therefore is not a true cyst but rather a pseudocyst.2 Clinically, the lesion typically presents as a solitary subcutaneous nodule that may be tender or painless. In a literature review of CMSC cases performed by Fukuyama et al,3 distribution of reported cases according to body site varied; however, limbs were found to be the most commonly involved area. A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the term cutaneous metaplastic synovial cyst revealed at least 37 cases reported in the English-language literature,3-9 including our present case. The pathogenesis remains uncertain; however, a majority of previously reported cases of CMSC characteristically have been associated with a pre-existing lesion, with most presentations developing at surgical scar sites secondary to operation or trauma.5 Relative tissue fragility secondary to rheumatoid arthritis10 and Ehlers-Danlos syndrome9,11,12 has been linked to CMSC in some documented reports, while a minority of cases report no antecedent events triggering formation of the lesion.13-15
As evidenced by our patient, CMSC clinically mimics several other benign entities; histopathologic examination is necessary to confirm the diagnosis. Although nodular hidradenoma also may clinically present as a solitary firm intradermal nodule, microscopy reveals a dermal-based lobulated tumor containing cystic spaces and solid areas composed of basophilic polyhedral cells and round glycogen-filled clear cells.16 Epidermoid cysts are differentiated from CMSC by the presence of a cyst wall lining composed of stratified squamous epithelium and associated laminated keratin within the lumen,17 which corresponds to its pearly white appearance on gross examination. Cutaneous ciliated cysts predominantly occur on the lower extremities of young women and are lined by simple cuboidal or columnar ciliated cells that resemble müllerian epithelium.18 Similar to CMSC, ganglion cysts are pseudocysts that lack a true epithelial lining but differ in appearance due to their mucin-filled synovial-lined sac.19 Additionally, ganglion cysts most often occur on the dorsal and volar aspects of the wrist.
Excisional biopsy is indicated as the preferred treatment of CMSC, given the lesion's benign behavior and low recurrence rate.6 Our case highlights this rare entity and reinforces its inclusion in the differential diagnosis of subcutaneous mobile nodules, especially in the setting of prior tissue injury secondary to trauma, surgical procedures, or conditions such as rheumatoid arthritis or Ehlers-Danlos syndrome. Unlike most previously reported cases, our patient reported no preceding tissue injury associated with formation of the lesion, and she was largely asymptomatic on presentation. Considering the limited number of CMSC cases demonstrated in the literature, it is important to continue reporting new cases to better understand characteristics and presentations of this uncommon lesion.
The Diagnosis: Cutaneous Metaplastic Synovial Cyst
Gross examination of the excised nodule revealed a 2.5×1.2×1.0-cm, intact, gray-white, thin-walled, smooth-lined nodule filled with clear mucinouslike material. Hematoxylin and eosin-stained sections demonstrated a dermal-based cystlike structure composed of a lining of connective tissue with hyalinized material and fibrin as well as spindle and epithelioid cells with a mild mixed inflammatory infiltrate (Figure). These histopathologic findings led to the diagnosis of cutaneous metaplastic synovial cyst (CMSC).
Cutaneous metaplastic synovial cyst, also known as synovial metaplasia of the skin, is an uncommon benign cystic lesion that was first reported by Gonzalez et al1 in 1987. Histologically, CMSC lacks an epithelial lining and therefore is not a true cyst but rather a pseudocyst.2 Clinically, the lesion typically presents as a solitary subcutaneous nodule that may be tender or painless. In a literature review of CMSC cases performed by Fukuyama et al,3 distribution of reported cases according to body site varied; however, limbs were found to be the most commonly involved area. A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the term cutaneous metaplastic synovial cyst revealed at least 37 cases reported in the English-language literature,3-9 including our present case. The pathogenesis remains uncertain; however, a majority of previously reported cases of CMSC characteristically have been associated with a pre-existing lesion, with most presentations developing at surgical scar sites secondary to operation or trauma.5 Relative tissue fragility secondary to rheumatoid arthritis10 and Ehlers-Danlos syndrome9,11,12 has been linked to CMSC in some documented reports, while a minority of cases report no antecedent events triggering formation of the lesion.13-15
As evidenced by our patient, CMSC clinically mimics several other benign entities; histopathologic examination is necessary to confirm the diagnosis. Although nodular hidradenoma also may clinically present as a solitary firm intradermal nodule, microscopy reveals a dermal-based lobulated tumor containing cystic spaces and solid areas composed of basophilic polyhedral cells and round glycogen-filled clear cells.16 Epidermoid cysts are differentiated from CMSC by the presence of a cyst wall lining composed of stratified squamous epithelium and associated laminated keratin within the lumen,17 which corresponds to its pearly white appearance on gross examination. Cutaneous ciliated cysts predominantly occur on the lower extremities of young women and are lined by simple cuboidal or columnar ciliated cells that resemble müllerian epithelium.18 Similar to CMSC, ganglion cysts are pseudocysts that lack a true epithelial lining but differ in appearance due to their mucin-filled synovial-lined sac.19 Additionally, ganglion cysts most often occur on the dorsal and volar aspects of the wrist.
Excisional biopsy is indicated as the preferred treatment of CMSC, given the lesion's benign behavior and low recurrence rate.6 Our case highlights this rare entity and reinforces its inclusion in the differential diagnosis of subcutaneous mobile nodules, especially in the setting of prior tissue injury secondary to trauma, surgical procedures, or conditions such as rheumatoid arthritis or Ehlers-Danlos syndrome. Unlike most previously reported cases, our patient reported no preceding tissue injury associated with formation of the lesion, and she was largely asymptomatic on presentation. Considering the limited number of CMSC cases demonstrated in the literature, it is important to continue reporting new cases to better understand characteristics and presentations of this uncommon lesion.
- Gonzalez JG, Ghiselli RW, Santa Cruz DJ. Synovial metaplasia of the skin. Am J Surg Pathol. 1987;11:343-350.
- Calonje E, Brenn T, Lazar A, et al. Cutaneous cysts. In: Calonje E, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 5th ed. Elsevier Limited; 2020:1680-1697.
- Fukuyama M, Sato Y, Hayakawa J, et al. Cutaneous metaplastic synovial cyst: case report and literature review from the dermatological point of view. Keio J Med. 2016;66:9-13.
- Karaytug K, Kapicioglu M, Can N, et al. Unprecedented recurrence of carpal tunnel syndrome by metaplastic synovial cyst in the carpal tunnel. Acta Orthop Traumatol Turc. 2019;53:230-232.
- Martelli SJ, Silveira FM, Carvalho PH, et al. Asymptomatic subcutaneous swelling of lower face. Oral Surg Oral Med Oral Pathol Oral Radiol. 2019;128:101-105.
- Majdi M, Saffar H, Ghanadan A. Cutaneous metaplastic synovial cyst: a case report. Iran J Pathol. 2016;11:423-426.
- Ramachandra S, Rao L, Al-Kindi M. Cutaneous metaplastic synovial cyst. Sultan Qaboos Univ Med J. 2016;16:E117-E118.
- Heidarian A, Xie Q, Banihashemi A. Cutaneous metaplastic synovial cyst presenting as an axillary mass after modified mastectomy and adjuvant radiotherapy. Am J Clin Pathol. 2016;146:S2.
- Fernandez-Flores A, Barja-Lopez JM. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome. J Cutan Pathol. 2020;47:729-733.
- Choonhakarn C, Tang S. Cutaneous metaplastic synovial cyst. J Dermatol. 2003;30:480-484.
- Guala A, Viglio S, Ottinetti A, et al. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome: report of a second case. Am J Dermatopathol. 2008;30:59-61.
- Nieto S, Buezo GF, Jones-Caballero M, et al. Cutaneous metaplastic synovial cyst in an Ehlers-Danlos patient. Am J Dermatopathol. 1997;19:407-410.
- Goiriz R, Rios-Buceta L, Alonso-Perez A, et al. Cutaneous metaplastic synovial cyst. J Am Acad Dermatol. 2005;53:180-181.
- Kim BC, Choi WJ, Park EJ, et al. Cutaneous metaplastic synovial cyst of the first metatarsal head area. Ann Dermatol. 2011;23(suppl 2):S165-S168.
- Yang HC, Tsai YJ, Hu SL, et al. Cutaneous metaplastic synovial cyst--a case report and review of literature. Dermatol Sinica. 2003;21:275-279.
- Kataria SP, Singh G, Batra A, et al. Nodular hidradenoma: a series of five cases in male subjects and review of literature. Adv Cytol Pathol. 2018;3:46-47.
- Mohamed Haflah N, Mohd Kassim A, Hassan Shukur M. Giant epidermoid cyst of the thigh. Malays Orthop J. 2011;5:17-19.
- Torisu-Itakura H, Itakura E, Horiuchi R, et al. Cutaneous ciliated cyst on the leg of a woman of menopausal age. Acta Derm Venereol. 2009;89:323-324.
- Fullen DR. Cysts and sinuses. In: Busam K, ed. Dermatopathology. Saunders; 2010:300-330.
- Gonzalez JG, Ghiselli RW, Santa Cruz DJ. Synovial metaplasia of the skin. Am J Surg Pathol. 1987;11:343-350.
- Calonje E, Brenn T, Lazar A, et al. Cutaneous cysts. In: Calonje E, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 5th ed. Elsevier Limited; 2020:1680-1697.
- Fukuyama M, Sato Y, Hayakawa J, et al. Cutaneous metaplastic synovial cyst: case report and literature review from the dermatological point of view. Keio J Med. 2016;66:9-13.
- Karaytug K, Kapicioglu M, Can N, et al. Unprecedented recurrence of carpal tunnel syndrome by metaplastic synovial cyst in the carpal tunnel. Acta Orthop Traumatol Turc. 2019;53:230-232.
- Martelli SJ, Silveira FM, Carvalho PH, et al. Asymptomatic subcutaneous swelling of lower face. Oral Surg Oral Med Oral Pathol Oral Radiol. 2019;128:101-105.
- Majdi M, Saffar H, Ghanadan A. Cutaneous metaplastic synovial cyst: a case report. Iran J Pathol. 2016;11:423-426.
- Ramachandra S, Rao L, Al-Kindi M. Cutaneous metaplastic synovial cyst. Sultan Qaboos Univ Med J. 2016;16:E117-E118.
- Heidarian A, Xie Q, Banihashemi A. Cutaneous metaplastic synovial cyst presenting as an axillary mass after modified mastectomy and adjuvant radiotherapy. Am J Clin Pathol. 2016;146:S2.
- Fernandez-Flores A, Barja-Lopez JM. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome. J Cutan Pathol. 2020;47:729-733.
- Choonhakarn C, Tang S. Cutaneous metaplastic synovial cyst. J Dermatol. 2003;30:480-484.
- Guala A, Viglio S, Ottinetti A, et al. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome: report of a second case. Am J Dermatopathol. 2008;30:59-61.
- Nieto S, Buezo GF, Jones-Caballero M, et al. Cutaneous metaplastic synovial cyst in an Ehlers-Danlos patient. Am J Dermatopathol. 1997;19:407-410.
- Goiriz R, Rios-Buceta L, Alonso-Perez A, et al. Cutaneous metaplastic synovial cyst. J Am Acad Dermatol. 2005;53:180-181.
- Kim BC, Choi WJ, Park EJ, et al. Cutaneous metaplastic synovial cyst of the first metatarsal head area. Ann Dermatol. 2011;23(suppl 2):S165-S168.
- Yang HC, Tsai YJ, Hu SL, et al. Cutaneous metaplastic synovial cyst--a case report and review of literature. Dermatol Sinica. 2003;21:275-279.
- Kataria SP, Singh G, Batra A, et al. Nodular hidradenoma: a series of five cases in male subjects and review of literature. Adv Cytol Pathol. 2018;3:46-47.
- Mohamed Haflah N, Mohd Kassim A, Hassan Shukur M. Giant epidermoid cyst of the thigh. Malays Orthop J. 2011;5:17-19.
- Torisu-Itakura H, Itakura E, Horiuchi R, et al. Cutaneous ciliated cyst on the leg of a woman of menopausal age. Acta Derm Venereol. 2009;89:323-324.
- Fullen DR. Cysts and sinuses. In: Busam K, ed. Dermatopathology. Saunders; 2010:300-330.
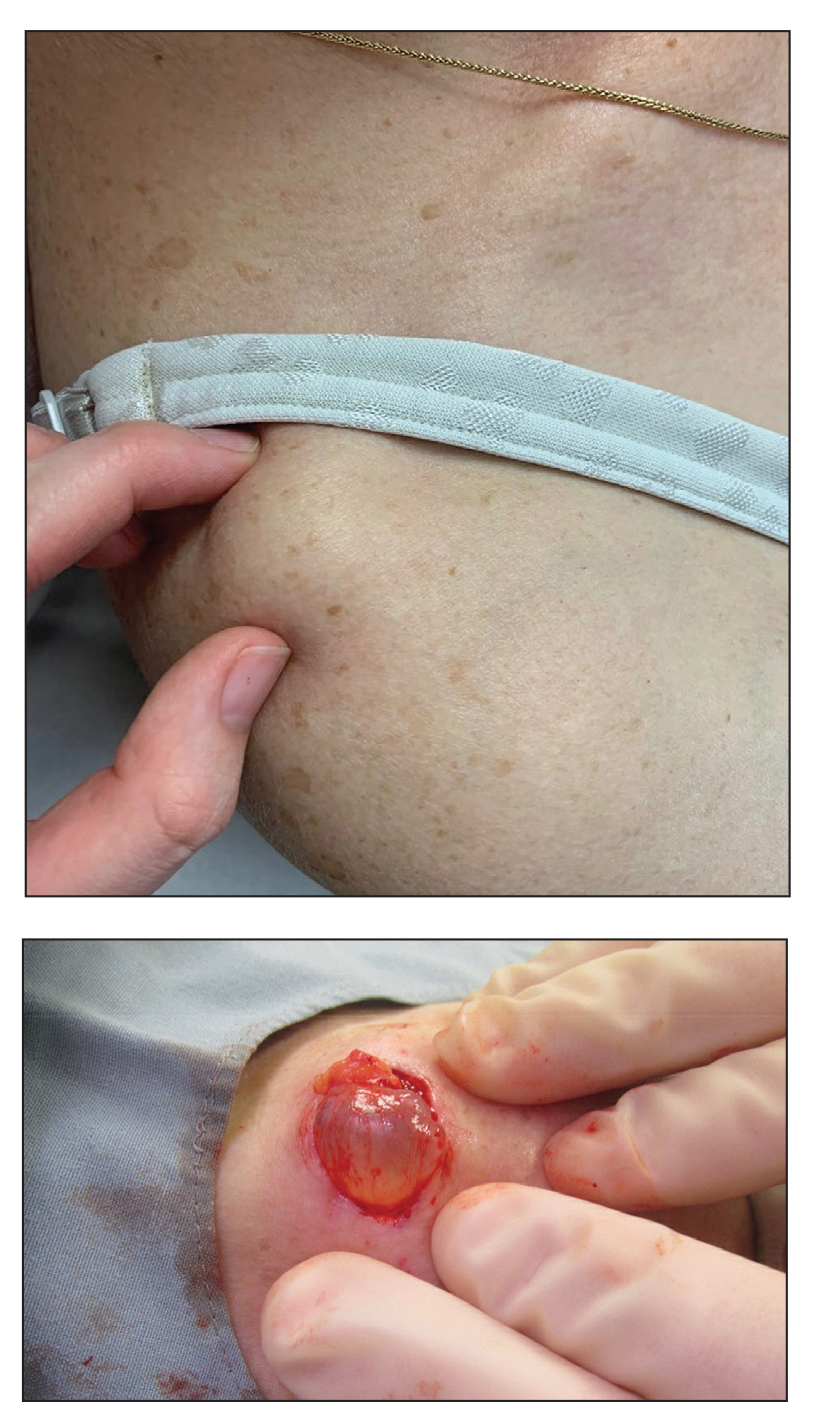
A 70-year-old woman presented to the outpatient dermatology clinic with an acute-onset lesion on the right shoulder. She first noticed a “cyst” developing in the area approximately 3 weeks prior but noted that it may have been present longer. The lesion was bothersome when her undergarments rubbed against it, but she otherwise denied pain, increase in size, or drainage from the site. Her medical history was remarkable for a proliferating trichilemmal tumor on the right parietal scalp treated with Mohs surgery approximately 13 years prior to presentation. She had no personal or family history of skin cancer. Physical examination revealed a 2.5-cm, mobile, nontender, flesh-colored subcutaneous nodule on the right shoulder (top); no ulceration, bleeding, or drainage was present. The surrounding skin demonstrated no clinical changes. The patient was scheduled for outpatient surgical excision of the nodule, which initially was suspected to be a lipoma. During the excision, a translucent cystlike nodule (bottom) was gently dissected and sent for histopathologic examination.