User login
Epidermolysis Bullosa Acquisita in Association With Mantle Cell Lymphoma
To the Editor:
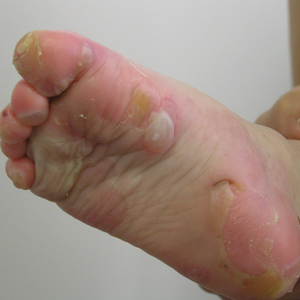
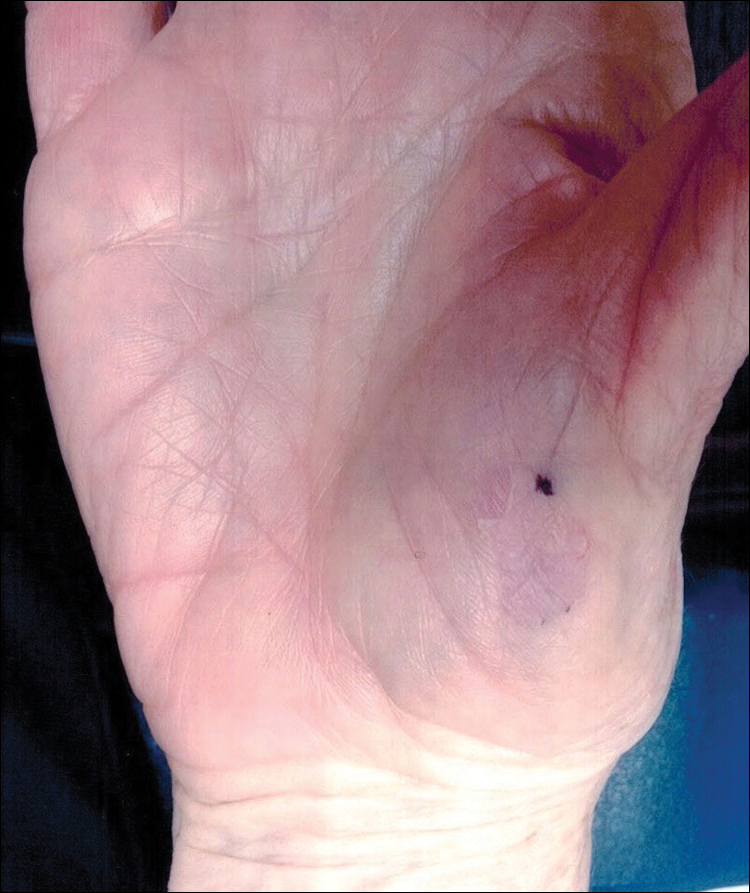
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
 and multiple bullae on the sole (B).")
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
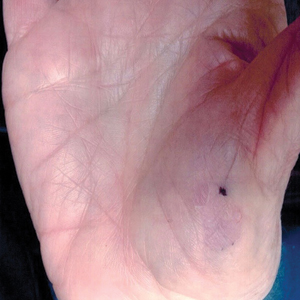
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
.")
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
To the Editor:
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
To the Editor:
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
Practice Points
- Epidermolysis bullosa acquisita (EBA) is an uncommon blistering disorder and few cases have been associated with malignancy.
- Diagnosis of EBA is challenging and requires exclusion of other blistering diseases.
Slow-growing, Asymptomatic, Annular Plaques on the Bilateral Palms
The Diagnosis: Circumscribed Palmar Hypokeratosis
Circumscribed palmar hypokeratosis is a rare, benign, acquired dermatosis that was first described by Pérez et al1 in 2002 and is characterized by annular plaques with an atrophic center and hyperkeratotic edges. Classically, the lesions present on the thenar and hypothenar eminences of the palms.2 The condition predominantly affects women (4:1 ratio), with a mean age of onset of 65 years.3
Although the pathogenesis of circumscribed palmar hypokeratosis is unknown, local trauma generally is considered to be the causative factor. Other hypotheses include human papillomaviruses 4 and 6 infection and primary abnormal keratinization in the epidermis.3 Immunohistochemical studies have demonstrated increased expression of keratin 16 and Ki-67 in cutaneous lesions, which is postulated to be responsible for keratinocyte fragility associated with epidermal hyperproliferation. Other reported cases have shown diminished keratin 9, keratin 2e, and connexin 26 expression, which normally are abundant in the acral epidermis. Abnormal expression of antigens associated with epidermal proliferation and differentiation also have been reported,3 suggesting that there is an altered regulation of the cutaneous desquamation process.
Histologically, circumscribed palmar hypokeratosis is characterized by an abrupt reduction in the stratum corneum (Figure), forming a step between the lesion and the perilesional normal skin.2,3 The clinical appearance of erythema is due to visualization of dermal blood circulation in the area of corneal thinning and is not a result of vasodilation. The dermis is uninvolved, and inflammation is absent. The differential diagnosis includes psoriasis, Bowen disease, porokeratosis, and dermatophytosis.3
(H&E, original magnification ×4). No notable inflammation was evident in the dermis (B)(H&E, original magnification ×10).")
Circumscribed palmar hypokeratosis is a chronic condition, and there are no known reports of development of malignancy. Treatment is not required but may include cryotherapy; topical therapy with corticosteroids, retinoids, urea, and calcipotriene; and photodynamic therapy. Circumscribed hypokeratosis should be included in the differential diagnosis of palmar lesions.
- Pérez A, Rütten A, Gold R, et al. Circumscribed palmar or plantar hypokeratosis: a distinctive epidermal malformation of the palms or soles. J Am Acad Dermatol. 2002;47:21-27.
- Mitkov M, Balagula Y, Lockshin B. Case report: circumscribed plantar hypokeratosis. Int J Dermatol. 2015;54:E203-E205.
- Rocha L, Nico M. Circumscribed palmoplantar hypokeratosis: report of two Brazilian cases. An Bras Dermatol. 2013;88:623-626.
The Diagnosis: Circumscribed Palmar Hypokeratosis
Circumscribed palmar hypokeratosis is a rare, benign, acquired dermatosis that was first described by Pérez et al1 in 2002 and is characterized by annular plaques with an atrophic center and hyperkeratotic edges. Classically, the lesions present on the thenar and hypothenar eminences of the palms.2 The condition predominantly affects women (4:1 ratio), with a mean age of onset of 65 years.3
Although the pathogenesis of circumscribed palmar hypokeratosis is unknown, local trauma generally is considered to be the causative factor. Other hypotheses include human papillomaviruses 4 and 6 infection and primary abnormal keratinization in the epidermis.3 Immunohistochemical studies have demonstrated increased expression of keratin 16 and Ki-67 in cutaneous lesions, which is postulated to be responsible for keratinocyte fragility associated with epidermal hyperproliferation. Other reported cases have shown diminished keratin 9, keratin 2e, and connexin 26 expression, which normally are abundant in the acral epidermis. Abnormal expression of antigens associated with epidermal proliferation and differentiation also have been reported,3 suggesting that there is an altered regulation of the cutaneous desquamation process.
Histologically, circumscribed palmar hypokeratosis is characterized by an abrupt reduction in the stratum corneum (Figure), forming a step between the lesion and the perilesional normal skin.2,3 The clinical appearance of erythema is due to visualization of dermal blood circulation in the area of corneal thinning and is not a result of vasodilation. The dermis is uninvolved, and inflammation is absent. The differential diagnosis includes psoriasis, Bowen disease, porokeratosis, and dermatophytosis.3
Circumscribed palmar hypokeratosis is a chronic condition, and there are no known reports of development of malignancy. Treatment is not required but may include cryotherapy; topical therapy with corticosteroids, retinoids, urea, and calcipotriene; and photodynamic therapy. Circumscribed hypokeratosis should be included in the differential diagnosis of palmar lesions.
The Diagnosis: Circumscribed Palmar Hypokeratosis
Circumscribed palmar hypokeratosis is a rare, benign, acquired dermatosis that was first described by Pérez et al1 in 2002 and is characterized by annular plaques with an atrophic center and hyperkeratotic edges. Classically, the lesions present on the thenar and hypothenar eminences of the palms.2 The condition predominantly affects women (4:1 ratio), with a mean age of onset of 65 years.3
Although the pathogenesis of circumscribed palmar hypokeratosis is unknown, local trauma generally is considered to be the causative factor. Other hypotheses include human papillomaviruses 4 and 6 infection and primary abnormal keratinization in the epidermis.3 Immunohistochemical studies have demonstrated increased expression of keratin 16 and Ki-67 in cutaneous lesions, which is postulated to be responsible for keratinocyte fragility associated with epidermal hyperproliferation. Other reported cases have shown diminished keratin 9, keratin 2e, and connexin 26 expression, which normally are abundant in the acral epidermis. Abnormal expression of antigens associated with epidermal proliferation and differentiation also have been reported,3 suggesting that there is an altered regulation of the cutaneous desquamation process.
Histologically, circumscribed palmar hypokeratosis is characterized by an abrupt reduction in the stratum corneum (Figure), forming a step between the lesion and the perilesional normal skin.2,3 The clinical appearance of erythema is due to visualization of dermal blood circulation in the area of corneal thinning and is not a result of vasodilation. The dermis is uninvolved, and inflammation is absent. The differential diagnosis includes psoriasis, Bowen disease, porokeratosis, and dermatophytosis.3
Circumscribed palmar hypokeratosis is a chronic condition, and there are no known reports of development of malignancy. Treatment is not required but may include cryotherapy; topical therapy with corticosteroids, retinoids, urea, and calcipotriene; and photodynamic therapy. Circumscribed hypokeratosis should be included in the differential diagnosis of palmar lesions.
- Pérez A, Rütten A, Gold R, et al. Circumscribed palmar or plantar hypokeratosis: a distinctive epidermal malformation of the palms or soles. J Am Acad Dermatol. 2002;47:21-27.
- Mitkov M, Balagula Y, Lockshin B. Case report: circumscribed plantar hypokeratosis. Int J Dermatol. 2015;54:E203-E205.
- Rocha L, Nico M. Circumscribed palmoplantar hypokeratosis: report of two Brazilian cases. An Bras Dermatol. 2013;88:623-626.
- Pérez A, Rütten A, Gold R, et al. Circumscribed palmar or plantar hypokeratosis: a distinctive epidermal malformation of the palms or soles. J Am Acad Dermatol. 2002;47:21-27.
- Mitkov M, Balagula Y, Lockshin B. Case report: circumscribed plantar hypokeratosis. Int J Dermatol. 2015;54:E203-E205.
- Rocha L, Nico M. Circumscribed palmoplantar hypokeratosis: report of two Brazilian cases. An Bras Dermatol. 2013;88:623-626.
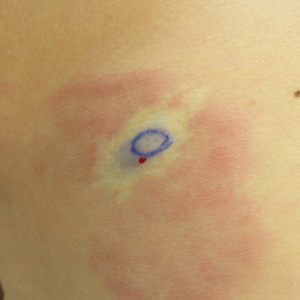
A 77-year-old woman presented with slow-growing, asymptomatic, annular plaques on the bilateral palms of many years' duration. There was no history of trauma or local infection. Prior treatment with over-the-counter creams was unsuccessful. A 3-mm punch biopsy of the lesion on the right palm was performed.
Painful Nonhealing Vulvar and Perianal Erosions
The Diagnosis: Cutaneous Crohn Disease
A punch biopsy of the vulvar skin revealed epidermal hyperplasia with moderate spongiosis and exocytosis of lymphocytes and neutrophils in the epidermis. A brisk mixed inflammatory infiltrate of epithelioid histiocytes, multinucleate foreign body-type giant cells, lymphocytes, plasma cells, neutrophils, and eosinophils in a granulomatous pattern also were present in the dermis (Figure). Periodic acid-Schiff and acid-fast bacillus stains were negative. Given the history of Crohn disease (CD) and the characteristic dermal noncaseating granulomas on histology, the patient was diagnosed with cutaneous CD.
(H&E, original magnification ×4) and mixed inflammatory granulomas (B)(H&E, original magnification ×40).")
Although the patient was offered a topical corticosteroid, she deferred topical therapy. Given the lack of response to adalimumab, the gastroenterology department switched the patient to a treatment of infliximab 5 mg/kg every 8 weeks. Azathioprine was discontinued and the patient was switched to intramuscular methotrexate 25 mg/mL weekly. Slow reepithelialization of the vulvar and perianal erosions occurred on this regimen.
Although CD has numerous cutaneous features, cutaneous CD, also known as metastatic CD, is the rarest cutaneous manifestation of CD.1 This disease process is characterized by noncaseating granulomatous cutaneous lesions that are not contiguous with the affected gastrointestinal tract.2 The pathogenesis of cutaneous CD is unknown. Young adults tend to be more predisposed to developing cutaneous CD, likely due to the age distribution of CD.3
Cutaneous CD commonly presents in patients with a well-established history of gastrointestinal CD but occasionally can be the presenting sign of CD.1 The most common sites of involvement are the legs, vulva, penis, trunk, face, and intertriginous areas. Cutaneous CD findings can be divided into 2 subgroups: genital and nongenital lesions. Genital findings involve ulceration, erythema, edema, and fissuring of the vulva, labia, clitoris, scrotum, penis, and perineum. Nongenital cutaneous manifestations include ulcers; erythematous papules, plaques, and nodules; abscesslike lesions; and lichenoid papules.4,5 The severity of cutaneous lesions does not correlate to the severity of gastrointestinal disease; however, colon involvement is more common in patients with cutaneous CD.6
Histologically, cutaneous CD presents as noncaseating granulomatous inflammation in the papillary and reticular dermis. These granulomas consist of epithelioid histiocytes and multinucleated giant cells with a lymphocytic infiltrate.5
Given the rarity of cutaneous CD, treatment approach is based on anecdotal evidence from case reports and case series. For a single lesion or localized disease, topical superpotent or intralesional steroids are recommended for initial therapy.3 Oral metronidazole also is an effective treatment and can be combined with topical or intralesional steroids.7 For disseminated disease, systemic corticosteroids have shown efficacy.3 Other reported treatment options include oral corticosteroids, sulfasalazine, azathioprine, 6-mercaptopurine, infliximab, and adalimumab. If monotherapy fails, combination therapy may be needed. Surgical debridement may be attempted if medical therapy fails but is complicated by wound dehiscence and disease recurrence.3
Although genital ulcers can be a presentation of Behçet disease and genital herpes infection, genital nodules and plaques are not typical for these 2 diseases. Also, the patient did not have oral ulcers, which is a common feature of Behçet disease. Genital sarcoidosis is extremely rare, and cutaneous CD was more likely given the patient's medical history. Finally, Jacquet dermatitis is more common in children, and patients with this condition typically have history of fecal and urinary incontinence.
- Teixeira M, Machado S, Lago P, et al. Cutaneous Crohn's disease. Int J Dermatol. 2006;45:1074-1076.
- Stingeni L, Neve D, Bassotti G, et al. Cutaneous Crohn's disease successfully treated with adalimumab [published online Sep 15, 2015]. J Eur Acad Dermatol Venerol. 2016;30:E72-E74.
- Kurtzman DJ, Jones T, Fangru L, et al. Metastatic Crohn's disease: a review and approach to therapy. J Am Acad Dermatol. 2014;71:804-813.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review [published online June 19, 2008]. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease, part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33.
- Abide JM. Metastatic Crohn disease: clearance with metronidazole. J Am Acad Dermatol. 2011;64:448-449.
The Diagnosis: Cutaneous Crohn Disease
A punch biopsy of the vulvar skin revealed epidermal hyperplasia with moderate spongiosis and exocytosis of lymphocytes and neutrophils in the epidermis. A brisk mixed inflammatory infiltrate of epithelioid histiocytes, multinucleate foreign body-type giant cells, lymphocytes, plasma cells, neutrophils, and eosinophils in a granulomatous pattern also were present in the dermis (Figure). Periodic acid-Schiff and acid-fast bacillus stains were negative. Given the history of Crohn disease (CD) and the characteristic dermal noncaseating granulomas on histology, the patient was diagnosed with cutaneous CD.
Although the patient was offered a topical corticosteroid, she deferred topical therapy. Given the lack of response to adalimumab, the gastroenterology department switched the patient to a treatment of infliximab 5 mg/kg every 8 weeks. Azathioprine was discontinued and the patient was switched to intramuscular methotrexate 25 mg/mL weekly. Slow reepithelialization of the vulvar and perianal erosions occurred on this regimen.
Although CD has numerous cutaneous features, cutaneous CD, also known as metastatic CD, is the rarest cutaneous manifestation of CD.1 This disease process is characterized by noncaseating granulomatous cutaneous lesions that are not contiguous with the affected gastrointestinal tract.2 The pathogenesis of cutaneous CD is unknown. Young adults tend to be more predisposed to developing cutaneous CD, likely due to the age distribution of CD.3
Cutaneous CD commonly presents in patients with a well-established history of gastrointestinal CD but occasionally can be the presenting sign of CD.1 The most common sites of involvement are the legs, vulva, penis, trunk, face, and intertriginous areas. Cutaneous CD findings can be divided into 2 subgroups: genital and nongenital lesions. Genital findings involve ulceration, erythema, edema, and fissuring of the vulva, labia, clitoris, scrotum, penis, and perineum. Nongenital cutaneous manifestations include ulcers; erythematous papules, plaques, and nodules; abscesslike lesions; and lichenoid papules.4,5 The severity of cutaneous lesions does not correlate to the severity of gastrointestinal disease; however, colon involvement is more common in patients with cutaneous CD.6
Histologically, cutaneous CD presents as noncaseating granulomatous inflammation in the papillary and reticular dermis. These granulomas consist of epithelioid histiocytes and multinucleated giant cells with a lymphocytic infiltrate.5
Given the rarity of cutaneous CD, treatment approach is based on anecdotal evidence from case reports and case series. For a single lesion or localized disease, topical superpotent or intralesional steroids are recommended for initial therapy.3 Oral metronidazole also is an effective treatment and can be combined with topical or intralesional steroids.7 For disseminated disease, systemic corticosteroids have shown efficacy.3 Other reported treatment options include oral corticosteroids, sulfasalazine, azathioprine, 6-mercaptopurine, infliximab, and adalimumab. If monotherapy fails, combination therapy may be needed. Surgical debridement may be attempted if medical therapy fails but is complicated by wound dehiscence and disease recurrence.3
Although genital ulcers can be a presentation of Behçet disease and genital herpes infection, genital nodules and plaques are not typical for these 2 diseases. Also, the patient did not have oral ulcers, which is a common feature of Behçet disease. Genital sarcoidosis is extremely rare, and cutaneous CD was more likely given the patient's medical history. Finally, Jacquet dermatitis is more common in children, and patients with this condition typically have history of fecal and urinary incontinence.
The Diagnosis: Cutaneous Crohn Disease
A punch biopsy of the vulvar skin revealed epidermal hyperplasia with moderate spongiosis and exocytosis of lymphocytes and neutrophils in the epidermis. A brisk mixed inflammatory infiltrate of epithelioid histiocytes, multinucleate foreign body-type giant cells, lymphocytes, plasma cells, neutrophils, and eosinophils in a granulomatous pattern also were present in the dermis (Figure). Periodic acid-Schiff and acid-fast bacillus stains were negative. Given the history of Crohn disease (CD) and the characteristic dermal noncaseating granulomas on histology, the patient was diagnosed with cutaneous CD.
Although the patient was offered a topical corticosteroid, she deferred topical therapy. Given the lack of response to adalimumab, the gastroenterology department switched the patient to a treatment of infliximab 5 mg/kg every 8 weeks. Azathioprine was discontinued and the patient was switched to intramuscular methotrexate 25 mg/mL weekly. Slow reepithelialization of the vulvar and perianal erosions occurred on this regimen.
Although CD has numerous cutaneous features, cutaneous CD, also known as metastatic CD, is the rarest cutaneous manifestation of CD.1 This disease process is characterized by noncaseating granulomatous cutaneous lesions that are not contiguous with the affected gastrointestinal tract.2 The pathogenesis of cutaneous CD is unknown. Young adults tend to be more predisposed to developing cutaneous CD, likely due to the age distribution of CD.3
Cutaneous CD commonly presents in patients with a well-established history of gastrointestinal CD but occasionally can be the presenting sign of CD.1 The most common sites of involvement are the legs, vulva, penis, trunk, face, and intertriginous areas. Cutaneous CD findings can be divided into 2 subgroups: genital and nongenital lesions. Genital findings involve ulceration, erythema, edema, and fissuring of the vulva, labia, clitoris, scrotum, penis, and perineum. Nongenital cutaneous manifestations include ulcers; erythematous papules, plaques, and nodules; abscesslike lesions; and lichenoid papules.4,5 The severity of cutaneous lesions does not correlate to the severity of gastrointestinal disease; however, colon involvement is more common in patients with cutaneous CD.6
Histologically, cutaneous CD presents as noncaseating granulomatous inflammation in the papillary and reticular dermis. These granulomas consist of epithelioid histiocytes and multinucleated giant cells with a lymphocytic infiltrate.5
Given the rarity of cutaneous CD, treatment approach is based on anecdotal evidence from case reports and case series. For a single lesion or localized disease, topical superpotent or intralesional steroids are recommended for initial therapy.3 Oral metronidazole also is an effective treatment and can be combined with topical or intralesional steroids.7 For disseminated disease, systemic corticosteroids have shown efficacy.3 Other reported treatment options include oral corticosteroids, sulfasalazine, azathioprine, 6-mercaptopurine, infliximab, and adalimumab. If monotherapy fails, combination therapy may be needed. Surgical debridement may be attempted if medical therapy fails but is complicated by wound dehiscence and disease recurrence.3
Although genital ulcers can be a presentation of Behçet disease and genital herpes infection, genital nodules and plaques are not typical for these 2 diseases. Also, the patient did not have oral ulcers, which is a common feature of Behçet disease. Genital sarcoidosis is extremely rare, and cutaneous CD was more likely given the patient's medical history. Finally, Jacquet dermatitis is more common in children, and patients with this condition typically have history of fecal and urinary incontinence.
- Teixeira M, Machado S, Lago P, et al. Cutaneous Crohn's disease. Int J Dermatol. 2006;45:1074-1076.
- Stingeni L, Neve D, Bassotti G, et al. Cutaneous Crohn's disease successfully treated with adalimumab [published online Sep 15, 2015]. J Eur Acad Dermatol Venerol. 2016;30:E72-E74.
- Kurtzman DJ, Jones T, Fangru L, et al. Metastatic Crohn's disease: a review and approach to therapy. J Am Acad Dermatol. 2014;71:804-813.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review [published online June 19, 2008]. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease, part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33.
- Abide JM. Metastatic Crohn disease: clearance with metronidazole. J Am Acad Dermatol. 2011;64:448-449.
- Teixeira M, Machado S, Lago P, et al. Cutaneous Crohn's disease. Int J Dermatol. 2006;45:1074-1076.
- Stingeni L, Neve D, Bassotti G, et al. Cutaneous Crohn's disease successfully treated with adalimumab [published online Sep 15, 2015]. J Eur Acad Dermatol Venerol. 2016;30:E72-E74.
- Kurtzman DJ, Jones T, Fangru L, et al. Metastatic Crohn's disease: a review and approach to therapy. J Am Acad Dermatol. 2014;71:804-813.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review [published online June 19, 2008]. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease, part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33.
- Abide JM. Metastatic Crohn disease: clearance with metronidazole. J Am Acad Dermatol. 2011;64:448-449.
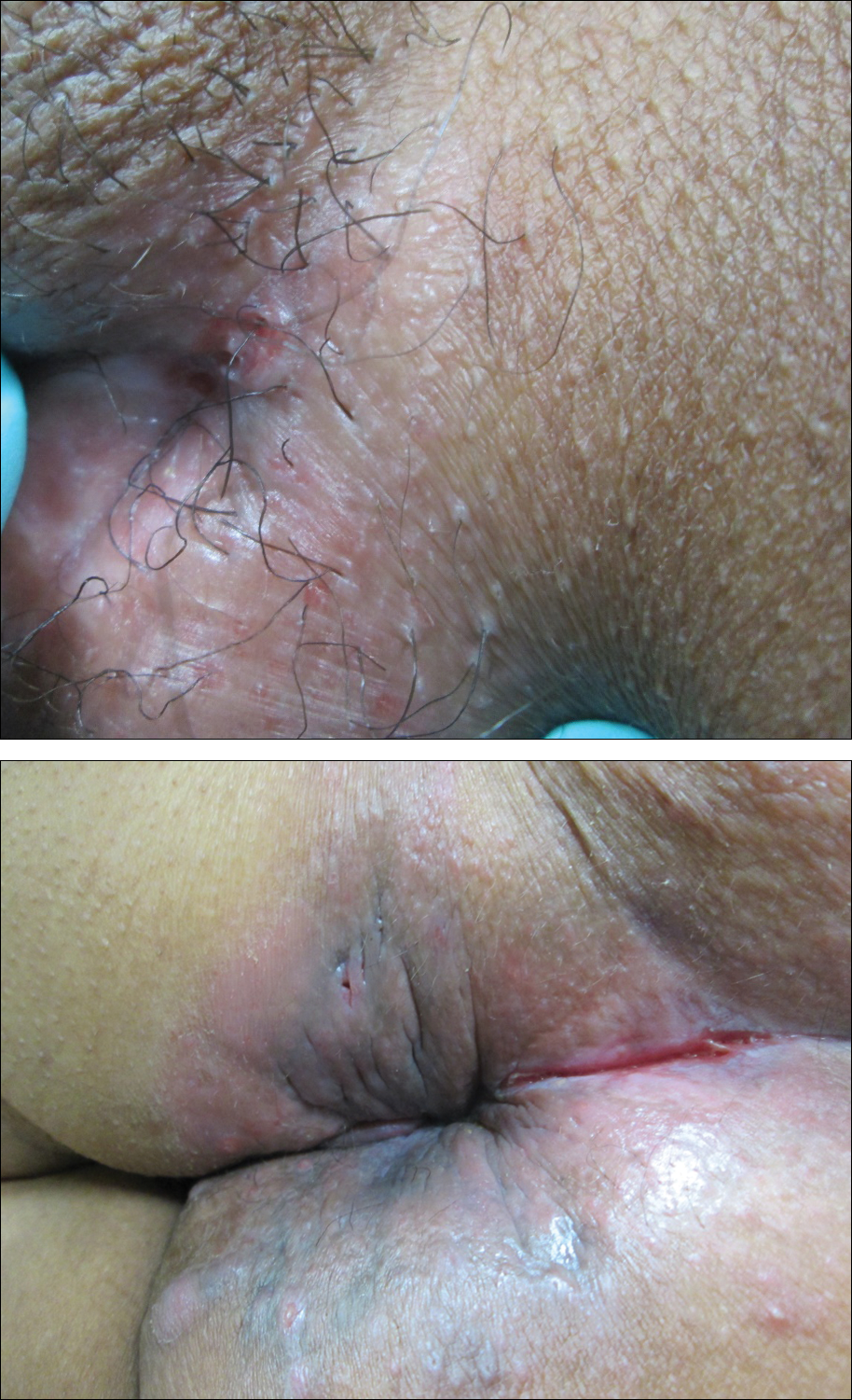
A 38-year-old woman with a history of Crohn disease presented with painful nonhealing vulvar and perianal erosions of 6 months' duration. The erosions developed 4 months after discontinuing adalimumab for a planned surgery. During this time, the patient also had an exacerbation of Crohn colitis and developed an anal fistula. Prior to this break in adalimumab, the patient's Crohn disease was well controlled on adalimumab 40 mg every 2 weeks, azathioprine 100 mg daily, and mesalamine 4.8 g daily. Despite restarting adalimumab and therapy with multiple antibiotics (ie, metronidazole, ciprofloxacin), the erosions persisted. On physical examination erythematous plaques and nodules were present at the vulvar (top) and perianal (bottom) skin. In addition, well-demarcated erosions measuring 20 mm and 80 mm were present on the vulvar and perianal skin, respectively. Human immunodeficiency virus screening and rapid plasma reagin were negative.
Scaly Annular and Concentric Plaques
The Diagnosis: Annular Psoriasis
Because the patient's history was nonconcordant with the clinical appearance, a 4-mm punch biopsy was performed from a lesion on the left hip. Hematoxylin and eosin-stained sections demonstrated mild irregular acanthosis of the epidermis with discrete mounds of parakeratin (Figure 1A). Higher power revealed numerous neutrophils entrapped within focal scale crusts (Figure 1B). Periodic acid-Schiff stain for fungus demonstrated no hyphal elements or yeast forms in the stratum corneum. These histopathology findings were consistent with the diagnosis of annular psoriasis.
(H&E, original magnification ×4) with neutrophils entrapped in thescale (B)(H&E, original magnification ×20).")
The manifestation of psoriasis may take many forms, ranging from classic plaques to pustular eruptions--either annular or generalized--and erythroderma. Primarily annular plaque-type psoriasis without pustules, however, remains an uncommon finding.1 Psoriatic plaques may become annular or arcuate with central clearing from partial treatment with topical medications, though our patient reported annular plaques prior to any treatment. His presentation was notably different than annular pustular psoriasis in that there were no pustules in the leading edge, and there was no trailing scale, which is typical of annular pustular psoriasis.
Topical triamcinolone prescribed at the initial presentation to the dermatology department helped with pruritus, but due to the large body surface area involved, methotrexate later was initiated. After a 10-mg test dose of methotrexate and titration to 15 mg weekly, dramatic improvement in the rash was noted after 8 weeks. As the rash resolved, only faint hyperpigmented patches remained (Figure 2).

Erythema gyratum repens is a rare paraneoplastic syndrome that presents with annular scaly plaques with concentric circles with a wood grain-like appearance. The borders can advance up to 1 cm daily and show nonspecific findings on histopathology.2 Due to the observation that approximately 80% of cases of erythema gyratum repens were associated with an underlying malignancy, most often of the lung,3 this diagnosis was entertained given our patient's clinical presentation.
Erythema annulare centrifugum (EAC) historically has been divided into 2 forms: superficial and deep.4 Both present with slowly expanding, annular, pink plaques. Superficial EAC demonstrates parakeratosis and trailing scale and has not been proven to be associated with other systemic diseases, while deep EAC has infiltrated borders without scale, and many cases of EAC may represent annular forms of tumid lupus.4 Inflammatory cells may cuff vessels tightly, resulting in so-called coat sleeve infiltrate in superficial EAC. Along with trailing scale, this finding suggests the diagnosis. It has been argued that EAC is not an entity on its own and should prompt evaluation for lupus erythematosus, dermatitis, hypersensitivity to tinea pedis, and Lyme disease in appropriate circumstances.5
Tinea corporis always should be considered when evaluating annular scaly plaques with central clearing. Diagnosis and treatment are straightforward when hyphae are found on microscopy of skin scrapings or seen on periodic acid-Schiff stains of formalin-fixed tissue. Tinea imbricata presents with an interesting morphology and appears more ornate or cerebriform than tinea corporis caused by Trichophyton rubrum. It is caused by infection with Trichophyton circumscriptum and occurs in certain regions in the South Pacific, Southeast Asia, and Central and South America, making the diagnosis within the United States unlikely for a patient who has not traveled to these areas.6
Erythema chronicum migrans is diagnostic of Lyme disease infection with Borrelia burgdorferi, and solitary lesions occur surrounding the site of a tick bite in the majority of patients. Only 20% of patients will develop multiple lesions consistent with erythema chronicum migrans due to multiple tick bites, spirochetemia, or lymphatic spread.7 Up to one-third of patients are unaware that they were bitten by a tick. In endemic areas, this diagnosis must be entertained in any patient presenting with an annular rash, as treatment may prevent notable morbidity.
- Guill C, Hoang M, Carder K. Primary annular plaque-type psoriasis. Pediatr Dermatol. 2005;22:15-18.
- Boyd A, Neldner K, Menter A. Erythema gyratum repens: a paraneoplastic eruption. J Am Acad Dermatol. 1992;26:757-762.
- Kawakami T, Saito R. Erythema gyratum repens unassociated with underlying malignancy. J Dermatol. 1995;22:587-589.
- Weyers W, Diaz-Cascajo C, Weyers I. Erythema annulare centrifugum: results of a clinicopathologic study of 73 patients. Am J Dermatopathol. 2003;25:451-462.
- Ziemer M, Eisendle K, Zelger B. New concepts on erythema annulare centrifugum: a clinical reaction pattern that does notrepresent a specific clinicopathological entity. Br J Dermatol. 2009;160:119-126.
- Bonifaz A, Vázquez-González D. Tinea imbricata in the Americas. Curr Opin Infect Dis. 2011;24:106-111.
- Müllegger R, Glatz M. Skin manifestations of Lyme borreliosis: diagnosis and management. Am J Clin Dermatol. 2008;9:355-368.
The Diagnosis: Annular Psoriasis
Because the patient's history was nonconcordant with the clinical appearance, a 4-mm punch biopsy was performed from a lesion on the left hip. Hematoxylin and eosin-stained sections demonstrated mild irregular acanthosis of the epidermis with discrete mounds of parakeratin (Figure 1A). Higher power revealed numerous neutrophils entrapped within focal scale crusts (Figure 1B). Periodic acid-Schiff stain for fungus demonstrated no hyphal elements or yeast forms in the stratum corneum. These histopathology findings were consistent with the diagnosis of annular psoriasis.
The manifestation of psoriasis may take many forms, ranging from classic plaques to pustular eruptions--either annular or generalized--and erythroderma. Primarily annular plaque-type psoriasis without pustules, however, remains an uncommon finding.1 Psoriatic plaques may become annular or arcuate with central clearing from partial treatment with topical medications, though our patient reported annular plaques prior to any treatment. His presentation was notably different than annular pustular psoriasis in that there were no pustules in the leading edge, and there was no trailing scale, which is typical of annular pustular psoriasis.
Topical triamcinolone prescribed at the initial presentation to the dermatology department helped with pruritus, but due to the large body surface area involved, methotrexate later was initiated. After a 10-mg test dose of methotrexate and titration to 15 mg weekly, dramatic improvement in the rash was noted after 8 weeks. As the rash resolved, only faint hyperpigmented patches remained (Figure 2).
Erythema gyratum repens is a rare paraneoplastic syndrome that presents with annular scaly plaques with concentric circles with a wood grain-like appearance. The borders can advance up to 1 cm daily and show nonspecific findings on histopathology.2 Due to the observation that approximately 80% of cases of erythema gyratum repens were associated with an underlying malignancy, most often of the lung,3 this diagnosis was entertained given our patient's clinical presentation.
Erythema annulare centrifugum (EAC) historically has been divided into 2 forms: superficial and deep.4 Both present with slowly expanding, annular, pink plaques. Superficial EAC demonstrates parakeratosis and trailing scale and has not been proven to be associated with other systemic diseases, while deep EAC has infiltrated borders without scale, and many cases of EAC may represent annular forms of tumid lupus.4 Inflammatory cells may cuff vessels tightly, resulting in so-called coat sleeve infiltrate in superficial EAC. Along with trailing scale, this finding suggests the diagnosis. It has been argued that EAC is not an entity on its own and should prompt evaluation for lupus erythematosus, dermatitis, hypersensitivity to tinea pedis, and Lyme disease in appropriate circumstances.5
Tinea corporis always should be considered when evaluating annular scaly plaques with central clearing. Diagnosis and treatment are straightforward when hyphae are found on microscopy of skin scrapings or seen on periodic acid-Schiff stains of formalin-fixed tissue. Tinea imbricata presents with an interesting morphology and appears more ornate or cerebriform than tinea corporis caused by Trichophyton rubrum. It is caused by infection with Trichophyton circumscriptum and occurs in certain regions in the South Pacific, Southeast Asia, and Central and South America, making the diagnosis within the United States unlikely for a patient who has not traveled to these areas.6
Erythema chronicum migrans is diagnostic of Lyme disease infection with Borrelia burgdorferi, and solitary lesions occur surrounding the site of a tick bite in the majority of patients. Only 20% of patients will develop multiple lesions consistent with erythema chronicum migrans due to multiple tick bites, spirochetemia, or lymphatic spread.7 Up to one-third of patients are unaware that they were bitten by a tick. In endemic areas, this diagnosis must be entertained in any patient presenting with an annular rash, as treatment may prevent notable morbidity.
The Diagnosis: Annular Psoriasis
Because the patient's history was nonconcordant with the clinical appearance, a 4-mm punch biopsy was performed from a lesion on the left hip. Hematoxylin and eosin-stained sections demonstrated mild irregular acanthosis of the epidermis with discrete mounds of parakeratin (Figure 1A). Higher power revealed numerous neutrophils entrapped within focal scale crusts (Figure 1B). Periodic acid-Schiff stain for fungus demonstrated no hyphal elements or yeast forms in the stratum corneum. These histopathology findings were consistent with the diagnosis of annular psoriasis.
The manifestation of psoriasis may take many forms, ranging from classic plaques to pustular eruptions--either annular or generalized--and erythroderma. Primarily annular plaque-type psoriasis without pustules, however, remains an uncommon finding.1 Psoriatic plaques may become annular or arcuate with central clearing from partial treatment with topical medications, though our patient reported annular plaques prior to any treatment. His presentation was notably different than annular pustular psoriasis in that there were no pustules in the leading edge, and there was no trailing scale, which is typical of annular pustular psoriasis.
Topical triamcinolone prescribed at the initial presentation to the dermatology department helped with pruritus, but due to the large body surface area involved, methotrexate later was initiated. After a 10-mg test dose of methotrexate and titration to 15 mg weekly, dramatic improvement in the rash was noted after 8 weeks. As the rash resolved, only faint hyperpigmented patches remained (Figure 2).
Erythema gyratum repens is a rare paraneoplastic syndrome that presents with annular scaly plaques with concentric circles with a wood grain-like appearance. The borders can advance up to 1 cm daily and show nonspecific findings on histopathology.2 Due to the observation that approximately 80% of cases of erythema gyratum repens were associated with an underlying malignancy, most often of the lung,3 this diagnosis was entertained given our patient's clinical presentation.
Erythema annulare centrifugum (EAC) historically has been divided into 2 forms: superficial and deep.4 Both present with slowly expanding, annular, pink plaques. Superficial EAC demonstrates parakeratosis and trailing scale and has not been proven to be associated with other systemic diseases, while deep EAC has infiltrated borders without scale, and many cases of EAC may represent annular forms of tumid lupus.4 Inflammatory cells may cuff vessels tightly, resulting in so-called coat sleeve infiltrate in superficial EAC. Along with trailing scale, this finding suggests the diagnosis. It has been argued that EAC is not an entity on its own and should prompt evaluation for lupus erythematosus, dermatitis, hypersensitivity to tinea pedis, and Lyme disease in appropriate circumstances.5
Tinea corporis always should be considered when evaluating annular scaly plaques with central clearing. Diagnosis and treatment are straightforward when hyphae are found on microscopy of skin scrapings or seen on periodic acid-Schiff stains of formalin-fixed tissue. Tinea imbricata presents with an interesting morphology and appears more ornate or cerebriform than tinea corporis caused by Trichophyton rubrum. It is caused by infection with Trichophyton circumscriptum and occurs in certain regions in the South Pacific, Southeast Asia, and Central and South America, making the diagnosis within the United States unlikely for a patient who has not traveled to these areas.6
Erythema chronicum migrans is diagnostic of Lyme disease infection with Borrelia burgdorferi, and solitary lesions occur surrounding the site of a tick bite in the majority of patients. Only 20% of patients will develop multiple lesions consistent with erythema chronicum migrans due to multiple tick bites, spirochetemia, or lymphatic spread.7 Up to one-third of patients are unaware that they were bitten by a tick. In endemic areas, this diagnosis must be entertained in any patient presenting with an annular rash, as treatment may prevent notable morbidity.
- Guill C, Hoang M, Carder K. Primary annular plaque-type psoriasis. Pediatr Dermatol. 2005;22:15-18.
- Boyd A, Neldner K, Menter A. Erythema gyratum repens: a paraneoplastic eruption. J Am Acad Dermatol. 1992;26:757-762.
- Kawakami T, Saito R. Erythema gyratum repens unassociated with underlying malignancy. J Dermatol. 1995;22:587-589.
- Weyers W, Diaz-Cascajo C, Weyers I. Erythema annulare centrifugum: results of a clinicopathologic study of 73 patients. Am J Dermatopathol. 2003;25:451-462.
- Ziemer M, Eisendle K, Zelger B. New concepts on erythema annulare centrifugum: a clinical reaction pattern that does notrepresent a specific clinicopathological entity. Br J Dermatol. 2009;160:119-126.
- Bonifaz A, Vázquez-González D. Tinea imbricata in the Americas. Curr Opin Infect Dis. 2011;24:106-111.
- Müllegger R, Glatz M. Skin manifestations of Lyme borreliosis: diagnosis and management. Am J Clin Dermatol. 2008;9:355-368.
- Guill C, Hoang M, Carder K. Primary annular plaque-type psoriasis. Pediatr Dermatol. 2005;22:15-18.
- Boyd A, Neldner K, Menter A. Erythema gyratum repens: a paraneoplastic eruption. J Am Acad Dermatol. 1992;26:757-762.
- Kawakami T, Saito R. Erythema gyratum repens unassociated with underlying malignancy. J Dermatol. 1995;22:587-589.
- Weyers W, Diaz-Cascajo C, Weyers I. Erythema annulare centrifugum: results of a clinicopathologic study of 73 patients. Am J Dermatopathol. 2003;25:451-462.
- Ziemer M, Eisendle K, Zelger B. New concepts on erythema annulare centrifugum: a clinical reaction pattern that does notrepresent a specific clinicopathological entity. Br J Dermatol. 2009;160:119-126.
- Bonifaz A, Vázquez-González D. Tinea imbricata in the Americas. Curr Opin Infect Dis. 2011;24:106-111.
- Müllegger R, Glatz M. Skin manifestations of Lyme borreliosis: diagnosis and management. Am J Clin Dermatol. 2008;9:355-368.
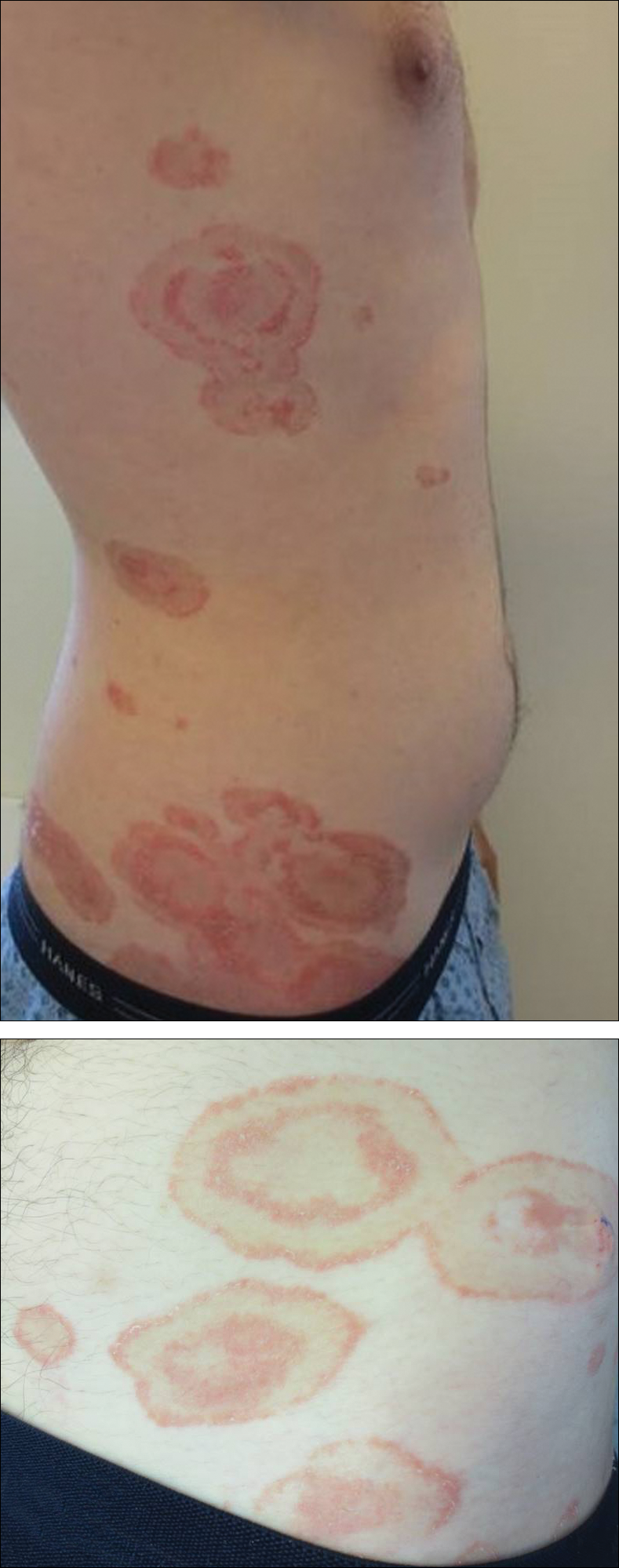
A healthy 23-year-old man presented for evaluation of an enlarging annular pruritic rash of 1.5 years' duration. Treatment with ciclopirox cream 0.77%, calcipotriene cream 0.005%, tacrolimus ointment 0.1%, fluticasone cream 0.05%, and halobetasol cream 0.05% prescribed by an outside physician provided only modest temporary improvement. The patient reported no history of travel outside of western New York, camping, tick bites, or medications. He denied any joint swelling or morning stiffness. Physical examination revealed multiple 4- to 6-cm pink, annular, scaly plaques with central clearing on the abdomen (top) and thighs. A few 1-cm pink scaly patches were present on the back (bottom), and few 2- to 3-mm pink scaly papules were noted on the extensor aspects of the elbows and forearms. A potassium hydroxide examination revealed no hyphal elements or yeast forms.
Idiopathic Eruptive Macular Pigmentation With Papillomatosis
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).

.")
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
Practice Points
- Idiopathic eruptive macular pigmentation with papillomatosis is a rare disorder that most frequently affects children and young adults.
- Idiopathic eruptive macular pigmentation with papillomatosis is characterized by asymptomatic, brownish, hyperpigmented macules involving the neck, trunk, arms, and legs.
- The disorder is important to consider in the differential diagnosis of asymptomatic pigmentary disorders to avoid unnecessary treatment because the disease is self-limiting and resolves over weeks to years.
Sweet Syndrome With Aseptic Splenic Abscesses and Multiple Myeloma
To the Editor:
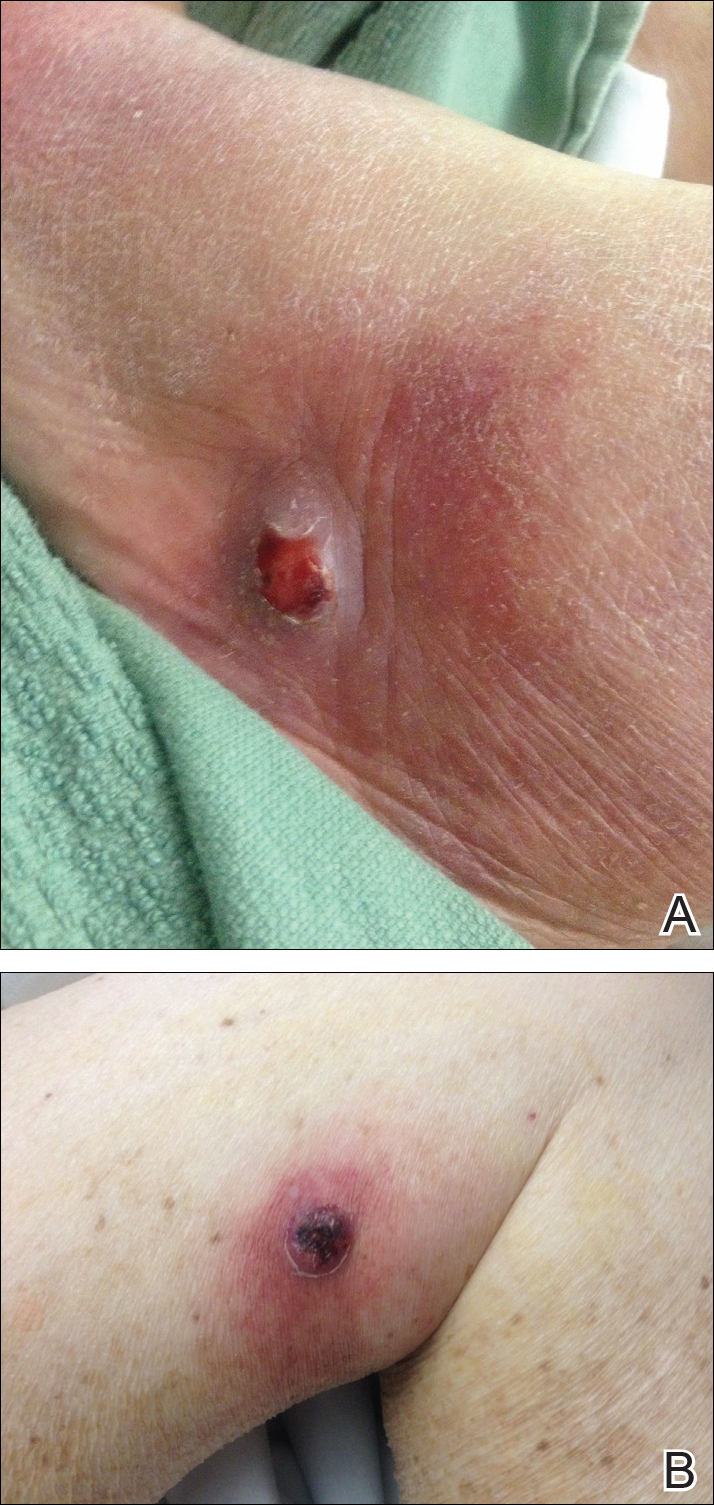
An 84-year-old man was admitted to the hospital with 5 erythematous cutaneous nodules of several days’ duration on the legs ranging in size from 1.0 to 1.5 cm. Upon admission, the patient also had a chest radiograph suspicious for pneumonia. The patient had received sulfamethoxazole/trimethoprim for a urinary tract infection as an outpatient 5 days prior to presentation, but he stopped the medication due to the appearance of the cutaneous nodules. Of note, the patient also reported unintentional weight loss of 15 pounds over the last few months.
New nodules had developed at a rate of 1 to 2 lesions daily in the 3 days prior to presentation and continued to develop after admission to the hospital. The nodules appeared as tender, erythematous lesions that evolved to form pustules and developed overlying crusts in later stages (Figure 1). They were limited to the arms and legs, primarily involving the lower legs. There was no evidence of oral or ocular involvement. A hemoglobin count of 10.9 g/dL (reference range, 14.0–17.5 g/dL), white blood cell count of 8.8×109/L (reference range, 4.5–11.0×109/L), and erythrocyte sedimentation rate of 69 mm/h (reference range, 0–20 mm/h) were noted on admission.
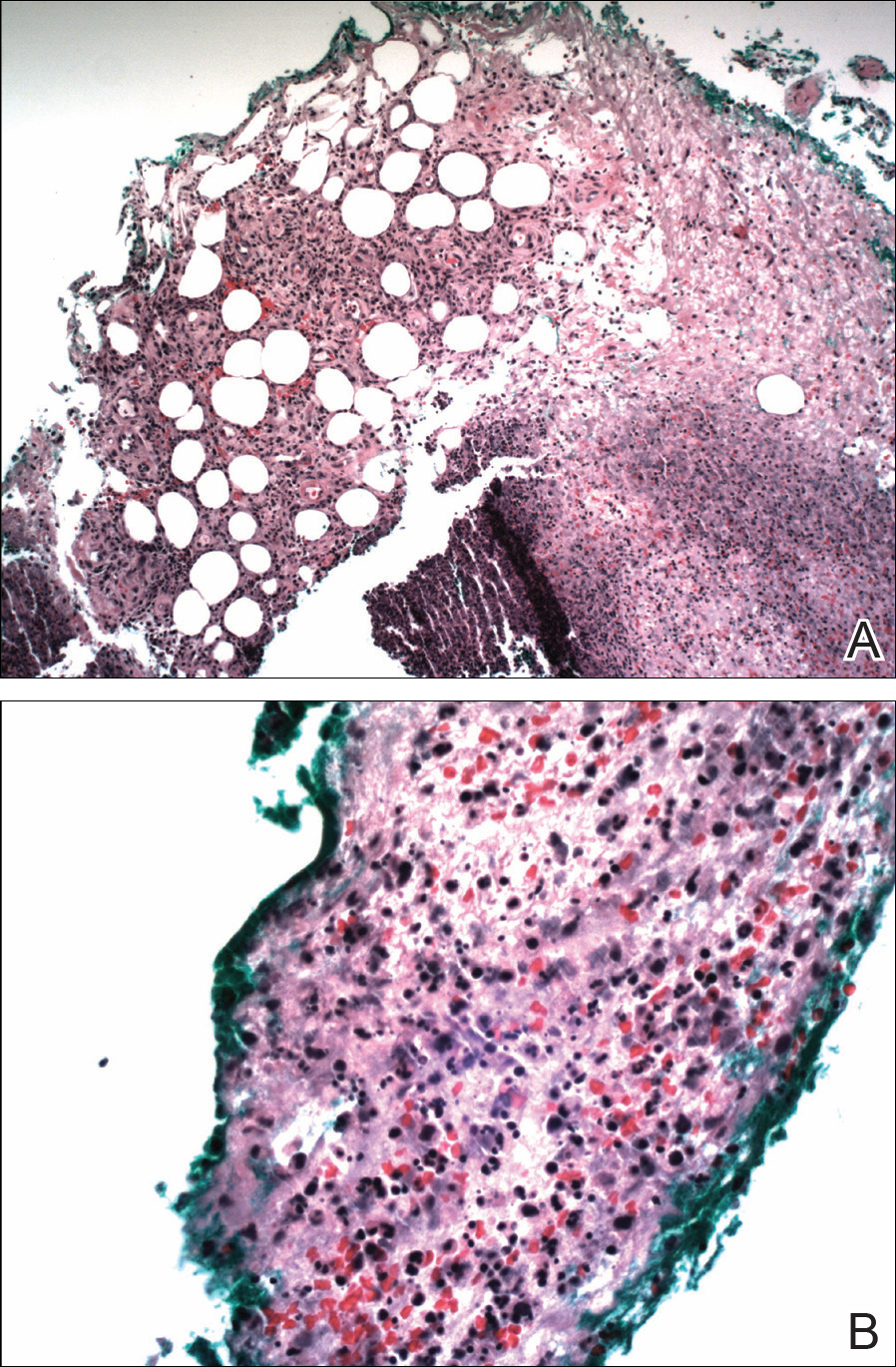
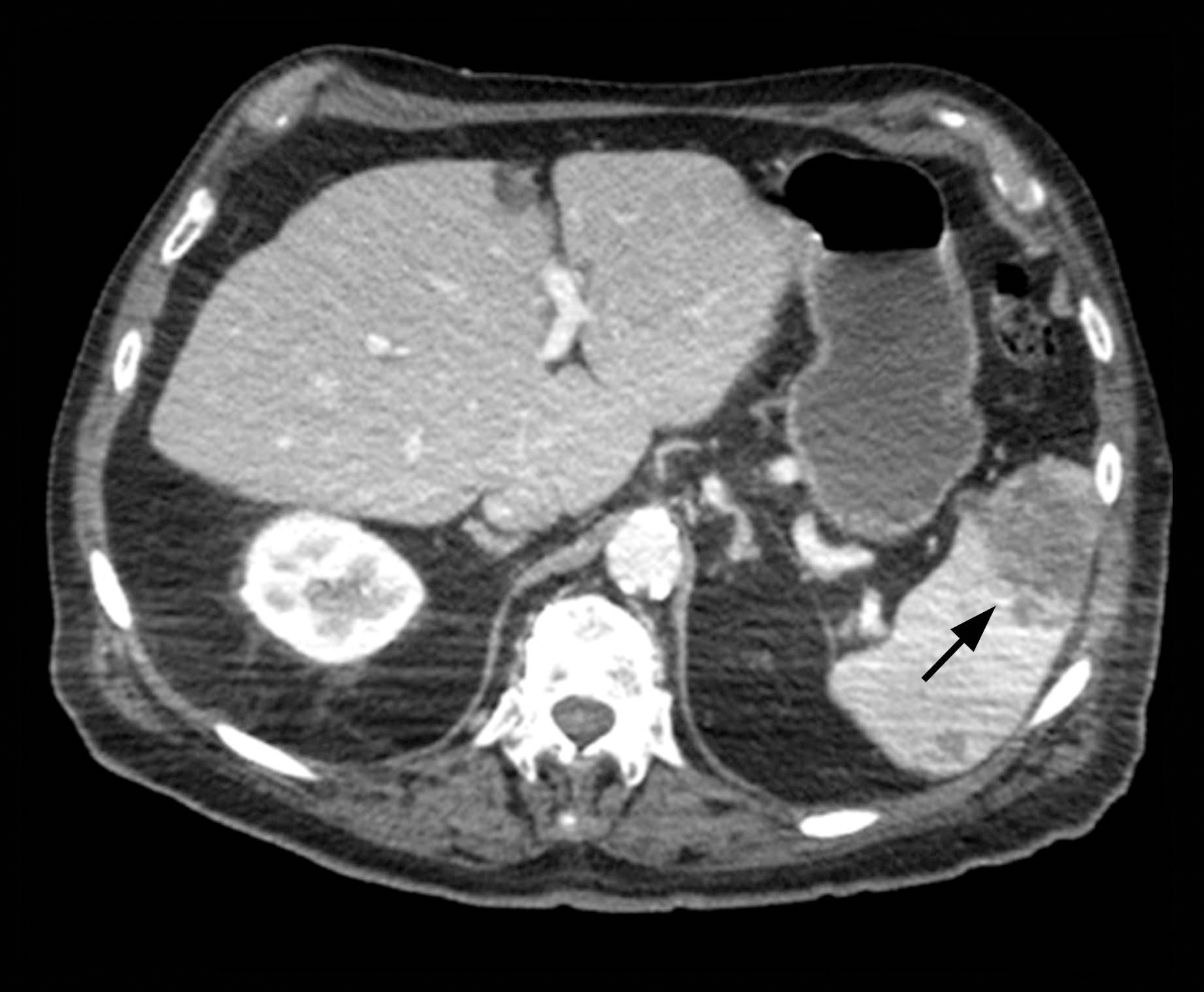
The patient was started on ceftriaxone and azithromycin for suspected pneumonia. The differential diagnosis for the cutaneous nodules included lymphoma, acid-fast bacilli (AFB) infection, deep fungal infection, pyoderma gangrenosum, Sweet syndrome (SS), panniculitis, erythema elevatum diutinum, and polyarteritis nodosa. A punch biopsy of a nodule on the left foot was performed. Histopathology demonstrated a neutrophilic panniculitis (Figure 2) with an epidermal abscess. No vasculitis was identified, and periodic acid–Schiff and AFB staining of the skin biopsy were negative. These findings were consistent with SS. Computed tomography scans of the chest, abdomen, and pelvis, which were completed early in the course of hospitalization due to concern for underlying malignancy, revealed pericardial and pleural effusions as well as cystic lesions in the lungs, spleen, kidneys, and prostate, with the largest lesion on the spleen measuring 5.6×4.8 cm (Figure 3). Computed tomography scanning was negative for areas of consolidation in the lungs. A splenic biopsy was performed by an interventional radiologist during the patient's hospitalization that identified an aseptic, neutrophilic process. Fungal, bacterial, and AFB cultures of the splenic tissue and cystic contents were negative. Bilateral pleural effusions also were identified, and a thoracentesis was performed. The pleural fluid indicated rare mesothelial cells in the background of acute inflammation with no growth of the bacterial, fungal, or AFB cultures.
Due to the association of hematologic malignances with SS, a bone marrow biopsy was performed, which revealed multiple myeloma. Serum protein electrophoresis demonstrated monoclonal gammopathy of κ light chains. During the course of his hospitalization, new skin lesions continued to develop on the hands, face, and trunk. The patient was discharged from the hospital shortly after diagnosis to receive outpatient treatment for multiple myeloma with lenalidomide and dexamethasone. Upon follow-up with the patient’s family via telephone 3 weeks into treatment, his son confirmed that the nodules were resolving.
Our case could be consistent with either drug-induced or malignancy-associated SS. Sweet syndrome initially was described in 1964 in 8 female patients with leukocytosis and cutaneous plaques infiltrated by neutrophils.1 The skin lesions typically are red and painful, ranging in size from 0.5 cm to 12.0 cm, and can last weeks to years if not treated.2 Variations of skin lesions include bullous and pustular morphologies.3
Diagnostic criteria for SS have been established.4 Both of the major criteria must be met as well as 2 of 4 minor criteria. Major criteria include abrupt onset of tender erythematous plaques and nodules; secondly, a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis must be seen on histopathology. Minor criteria include pyrexia, association with underlying condition (malignancy, pregnancy, drug exposure, inflammatory disorder), responsiveness to systemic steroids, and abnormal laboratory values (erythrocyte sedimentation rate, white blood cell count, C-reactive protein, neutrophilia).4
Sweet syndrome can be divided into 3 classifications: classical or idiopathic, drug-induced, or malignancy-associated.4 Classical SS most commonly is seen in middle-aged women after an upper respiratory or gastrointestinal infection. Drug-induced SS most often is associated with granulocyte-stimulating factor colony therapy4; however, it has been associated with use of trimethoprim/sulfamethoxazole.5 Malignancy-associated SS most commonly is seen in individuals with hematologic malignancy, specifically acute myeloid leukemia. Although its association with multiple myeloma is not as frequent, cases of malignancy-associated SS identifying this association have been reported.6,7 Mucosal involvement in the form of aphthouslike lesions more frequently is seen in malignancy-associated SS.8 Differing from classical SS, which has a female predilection of around 4:1, the malignancy-associated disorder has a 1:1 female-to-male ratio.4
In the majority of cases of SS, the neutrophilic infiltrate is in the papillary and upper reticular dermis; however, if the neutrophilic infiltrate is predominately in the subcutaneous tissue (known as subcutaneous SS), there is a strong association with malignancy.9 The histopathology in our case demonstrated a neutrophilic infiltrate in the subcutaneous tissue.
Fever is the most common systemic manifestation of SS and is present in 54% to 65% of patients.8,10 Besides the skin, the most common site affected is the eye, with 13% to 75% of patients reporting ocular involvement, usually conjunctivitis.4,10 Although infrequent, extracutaneous SS has been identified in the bones, central nervous system, kidneys, heart, liver, spleen, lungs, ears, eyes, and intestines.4 A case of SS with splenic involvement in the form of sterile abscesses also was reported.11 This case was related to parvovirus B19.
Sweet syndrome is a condition characterized by tender, erythematous cutaneous lesions with histopathology demonstrating neutrophilic infiltrate in the absence of vasculitis. We report a case of suspected extracutaneous SS in the form of splenic cysts in a patient whose SS was associated with malignancy and/or drug ingestion.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome and malignancy. Am J Med. 1987;82:1220-1226.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2002;41:182-184.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Belhadjali H, Chaabane S, Njim L, et al. Sweet’s syndrome associated with multiple myeloma. Acta Dermatovenerol Alp Pannonica Adriat. 2008;17:31-33.
- Bayer-Garner IB, Cottler-Fox M, Smoller BR. Sweet syndrome in multiple myeloma: a series of six cases. J Cutan Pathol. 2003;30:261-264.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556; quiz 557-560.
- Neoh CY, Tan AW, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentation and associations. Br J Dermatol. 2007;156:480-485.
- Fortna RR, Toporcer M, Elder DE, et al. A case of sweet syndrome with spleen and lymph node involvement preceded by parvovirus B19 infection, and review of the literature on extracutaneous Sweet syndrome. Am J Dermatopathol. 2010;32:621-627.
To the Editor:
An 84-year-old man was admitted to the hospital with 5 erythematous cutaneous nodules of several days’ duration on the legs ranging in size from 1.0 to 1.5 cm. Upon admission, the patient also had a chest radiograph suspicious for pneumonia. The patient had received sulfamethoxazole/trimethoprim for a urinary tract infection as an outpatient 5 days prior to presentation, but he stopped the medication due to the appearance of the cutaneous nodules. Of note, the patient also reported unintentional weight loss of 15 pounds over the last few months.
New nodules had developed at a rate of 1 to 2 lesions daily in the 3 days prior to presentation and continued to develop after admission to the hospital. The nodules appeared as tender, erythematous lesions that evolved to form pustules and developed overlying crusts in later stages (Figure 1). They were limited to the arms and legs, primarily involving the lower legs. There was no evidence of oral or ocular involvement. A hemoglobin count of 10.9 g/dL (reference range, 14.0–17.5 g/dL), white blood cell count of 8.8×109/L (reference range, 4.5–11.0×109/L), and erythrocyte sedimentation rate of 69 mm/h (reference range, 0–20 mm/h) were noted on admission.
The patient was started on ceftriaxone and azithromycin for suspected pneumonia. The differential diagnosis for the cutaneous nodules included lymphoma, acid-fast bacilli (AFB) infection, deep fungal infection, pyoderma gangrenosum, Sweet syndrome (SS), panniculitis, erythema elevatum diutinum, and polyarteritis nodosa. A punch biopsy of a nodule on the left foot was performed. Histopathology demonstrated a neutrophilic panniculitis (Figure 2) with an epidermal abscess. No vasculitis was identified, and periodic acid–Schiff and AFB staining of the skin biopsy were negative. These findings were consistent with SS. Computed tomography scans of the chest, abdomen, and pelvis, which were completed early in the course of hospitalization due to concern for underlying malignancy, revealed pericardial and pleural effusions as well as cystic lesions in the lungs, spleen, kidneys, and prostate, with the largest lesion on the spleen measuring 5.6×4.8 cm (Figure 3). Computed tomography scanning was negative for areas of consolidation in the lungs. A splenic biopsy was performed by an interventional radiologist during the patient's hospitalization that identified an aseptic, neutrophilic process. Fungal, bacterial, and AFB cultures of the splenic tissue and cystic contents were negative. Bilateral pleural effusions also were identified, and a thoracentesis was performed. The pleural fluid indicated rare mesothelial cells in the background of acute inflammation with no growth of the bacterial, fungal, or AFB cultures.
Due to the association of hematologic malignances with SS, a bone marrow biopsy was performed, which revealed multiple myeloma. Serum protein electrophoresis demonstrated monoclonal gammopathy of κ light chains. During the course of his hospitalization, new skin lesions continued to develop on the hands, face, and trunk. The patient was discharged from the hospital shortly after diagnosis to receive outpatient treatment for multiple myeloma with lenalidomide and dexamethasone. Upon follow-up with the patient’s family via telephone 3 weeks into treatment, his son confirmed that the nodules were resolving.
Our case could be consistent with either drug-induced or malignancy-associated SS. Sweet syndrome initially was described in 1964 in 8 female patients with leukocytosis and cutaneous plaques infiltrated by neutrophils.1 The skin lesions typically are red and painful, ranging in size from 0.5 cm to 12.0 cm, and can last weeks to years if not treated.2 Variations of skin lesions include bullous and pustular morphologies.3
Diagnostic criteria for SS have been established.4 Both of the major criteria must be met as well as 2 of 4 minor criteria. Major criteria include abrupt onset of tender erythematous plaques and nodules; secondly, a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis must be seen on histopathology. Minor criteria include pyrexia, association with underlying condition (malignancy, pregnancy, drug exposure, inflammatory disorder), responsiveness to systemic steroids, and abnormal laboratory values (erythrocyte sedimentation rate, white blood cell count, C-reactive protein, neutrophilia).4
Sweet syndrome can be divided into 3 classifications: classical or idiopathic, drug-induced, or malignancy-associated.4 Classical SS most commonly is seen in middle-aged women after an upper respiratory or gastrointestinal infection. Drug-induced SS most often is associated with granulocyte-stimulating factor colony therapy4; however, it has been associated with use of trimethoprim/sulfamethoxazole.5 Malignancy-associated SS most commonly is seen in individuals with hematologic malignancy, specifically acute myeloid leukemia. Although its association with multiple myeloma is not as frequent, cases of malignancy-associated SS identifying this association have been reported.6,7 Mucosal involvement in the form of aphthouslike lesions more frequently is seen in malignancy-associated SS.8 Differing from classical SS, which has a female predilection of around 4:1, the malignancy-associated disorder has a 1:1 female-to-male ratio.4
In the majority of cases of SS, the neutrophilic infiltrate is in the papillary and upper reticular dermis; however, if the neutrophilic infiltrate is predominately in the subcutaneous tissue (known as subcutaneous SS), there is a strong association with malignancy.9 The histopathology in our case demonstrated a neutrophilic infiltrate in the subcutaneous tissue.
Fever is the most common systemic manifestation of SS and is present in 54% to 65% of patients.8,10 Besides the skin, the most common site affected is the eye, with 13% to 75% of patients reporting ocular involvement, usually conjunctivitis.4,10 Although infrequent, extracutaneous SS has been identified in the bones, central nervous system, kidneys, heart, liver, spleen, lungs, ears, eyes, and intestines.4 A case of SS with splenic involvement in the form of sterile abscesses also was reported.11 This case was related to parvovirus B19.
Sweet syndrome is a condition characterized by tender, erythematous cutaneous lesions with histopathology demonstrating neutrophilic infiltrate in the absence of vasculitis. We report a case of suspected extracutaneous SS in the form of splenic cysts in a patient whose SS was associated with malignancy and/or drug ingestion.
To the Editor:
An 84-year-old man was admitted to the hospital with 5 erythematous cutaneous nodules of several days’ duration on the legs ranging in size from 1.0 to 1.5 cm. Upon admission, the patient also had a chest radiograph suspicious for pneumonia. The patient had received sulfamethoxazole/trimethoprim for a urinary tract infection as an outpatient 5 days prior to presentation, but he stopped the medication due to the appearance of the cutaneous nodules. Of note, the patient also reported unintentional weight loss of 15 pounds over the last few months.
New nodules had developed at a rate of 1 to 2 lesions daily in the 3 days prior to presentation and continued to develop after admission to the hospital. The nodules appeared as tender, erythematous lesions that evolved to form pustules and developed overlying crusts in later stages (Figure 1). They were limited to the arms and legs, primarily involving the lower legs. There was no evidence of oral or ocular involvement. A hemoglobin count of 10.9 g/dL (reference range, 14.0–17.5 g/dL), white blood cell count of 8.8×109/L (reference range, 4.5–11.0×109/L), and erythrocyte sedimentation rate of 69 mm/h (reference range, 0–20 mm/h) were noted on admission.
The patient was started on ceftriaxone and azithromycin for suspected pneumonia. The differential diagnosis for the cutaneous nodules included lymphoma, acid-fast bacilli (AFB) infection, deep fungal infection, pyoderma gangrenosum, Sweet syndrome (SS), panniculitis, erythema elevatum diutinum, and polyarteritis nodosa. A punch biopsy of a nodule on the left foot was performed. Histopathology demonstrated a neutrophilic panniculitis (Figure 2) with an epidermal abscess. No vasculitis was identified, and periodic acid–Schiff and AFB staining of the skin biopsy were negative. These findings were consistent with SS. Computed tomography scans of the chest, abdomen, and pelvis, which were completed early in the course of hospitalization due to concern for underlying malignancy, revealed pericardial and pleural effusions as well as cystic lesions in the lungs, spleen, kidneys, and prostate, with the largest lesion on the spleen measuring 5.6×4.8 cm (Figure 3). Computed tomography scanning was negative for areas of consolidation in the lungs. A splenic biopsy was performed by an interventional radiologist during the patient's hospitalization that identified an aseptic, neutrophilic process. Fungal, bacterial, and AFB cultures of the splenic tissue and cystic contents were negative. Bilateral pleural effusions also were identified, and a thoracentesis was performed. The pleural fluid indicated rare mesothelial cells in the background of acute inflammation with no growth of the bacterial, fungal, or AFB cultures.
Due to the association of hematologic malignances with SS, a bone marrow biopsy was performed, which revealed multiple myeloma. Serum protein electrophoresis demonstrated monoclonal gammopathy of κ light chains. During the course of his hospitalization, new skin lesions continued to develop on the hands, face, and trunk. The patient was discharged from the hospital shortly after diagnosis to receive outpatient treatment for multiple myeloma with lenalidomide and dexamethasone. Upon follow-up with the patient’s family via telephone 3 weeks into treatment, his son confirmed that the nodules were resolving.
Our case could be consistent with either drug-induced or malignancy-associated SS. Sweet syndrome initially was described in 1964 in 8 female patients with leukocytosis and cutaneous plaques infiltrated by neutrophils.1 The skin lesions typically are red and painful, ranging in size from 0.5 cm to 12.0 cm, and can last weeks to years if not treated.2 Variations of skin lesions include bullous and pustular morphologies.3
Diagnostic criteria for SS have been established.4 Both of the major criteria must be met as well as 2 of 4 minor criteria. Major criteria include abrupt onset of tender erythematous plaques and nodules; secondly, a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis must be seen on histopathology. Minor criteria include pyrexia, association with underlying condition (malignancy, pregnancy, drug exposure, inflammatory disorder), responsiveness to systemic steroids, and abnormal laboratory values (erythrocyte sedimentation rate, white blood cell count, C-reactive protein, neutrophilia).4
Sweet syndrome can be divided into 3 classifications: classical or idiopathic, drug-induced, or malignancy-associated.4 Classical SS most commonly is seen in middle-aged women after an upper respiratory or gastrointestinal infection. Drug-induced SS most often is associated with granulocyte-stimulating factor colony therapy4; however, it has been associated with use of trimethoprim/sulfamethoxazole.5 Malignancy-associated SS most commonly is seen in individuals with hematologic malignancy, specifically acute myeloid leukemia. Although its association with multiple myeloma is not as frequent, cases of malignancy-associated SS identifying this association have been reported.6,7 Mucosal involvement in the form of aphthouslike lesions more frequently is seen in malignancy-associated SS.8 Differing from classical SS, which has a female predilection of around 4:1, the malignancy-associated disorder has a 1:1 female-to-male ratio.4
In the majority of cases of SS, the neutrophilic infiltrate is in the papillary and upper reticular dermis; however, if the neutrophilic infiltrate is predominately in the subcutaneous tissue (known as subcutaneous SS), there is a strong association with malignancy.9 The histopathology in our case demonstrated a neutrophilic infiltrate in the subcutaneous tissue.
Fever is the most common systemic manifestation of SS and is present in 54% to 65% of patients.8,10 Besides the skin, the most common site affected is the eye, with 13% to 75% of patients reporting ocular involvement, usually conjunctivitis.4,10 Although infrequent, extracutaneous SS has been identified in the bones, central nervous system, kidneys, heart, liver, spleen, lungs, ears, eyes, and intestines.4 A case of SS with splenic involvement in the form of sterile abscesses also was reported.11 This case was related to parvovirus B19.
Sweet syndrome is a condition characterized by tender, erythematous cutaneous lesions with histopathology demonstrating neutrophilic infiltrate in the absence of vasculitis. We report a case of suspected extracutaneous SS in the form of splenic cysts in a patient whose SS was associated with malignancy and/or drug ingestion.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome and malignancy. Am J Med. 1987;82:1220-1226.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2002;41:182-184.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Belhadjali H, Chaabane S, Njim L, et al. Sweet’s syndrome associated with multiple myeloma. Acta Dermatovenerol Alp Pannonica Adriat. 2008;17:31-33.
- Bayer-Garner IB, Cottler-Fox M, Smoller BR. Sweet syndrome in multiple myeloma: a series of six cases. J Cutan Pathol. 2003;30:261-264.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556; quiz 557-560.
- Neoh CY, Tan AW, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentation and associations. Br J Dermatol. 2007;156:480-485.
- Fortna RR, Toporcer M, Elder DE, et al. A case of sweet syndrome with spleen and lymph node involvement preceded by parvovirus B19 infection, and review of the literature on extracutaneous Sweet syndrome. Am J Dermatopathol. 2010;32:621-627.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome and malignancy. Am J Med. 1987;82:1220-1226.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2002;41:182-184.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Belhadjali H, Chaabane S, Njim L, et al. Sweet’s syndrome associated with multiple myeloma. Acta Dermatovenerol Alp Pannonica Adriat. 2008;17:31-33.
- Bayer-Garner IB, Cottler-Fox M, Smoller BR. Sweet syndrome in multiple myeloma: a series of six cases. J Cutan Pathol. 2003;30:261-264.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556; quiz 557-560.
- Neoh CY, Tan AW, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentation and associations. Br J Dermatol. 2007;156:480-485.
- Fortna RR, Toporcer M, Elder DE, et al. A case of sweet syndrome with spleen and lymph node involvement preceded by parvovirus B19 infection, and review of the literature on extracutaneous Sweet syndrome. Am J Dermatopathol. 2010;32:621-627.
Practice Points
- Sweet syndrome (SS), also known as acute febrile neutrophilic dermatosis, is an inflammatory process characterized by a diffuse dermal neutrophilic infiltrate in the absence of vasculitis.
- A diagnosis of SS warrants further investigation due to its association with malignancy, especially hematologic malignancy.
- Other organs in SS also may have aseptic involvement.
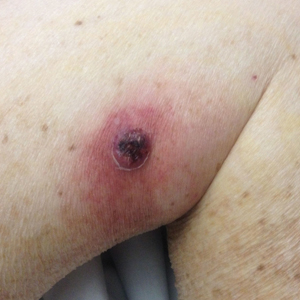
Red-Brown Patches in the Groin
The Diagnosis: Erythrasma
Erythrasma usually involves intertriginous areas (eg, axillae, groin, inframammary area). Patients present with well-demarcated, minimally scaly, red-brown patches. The interdigital web space of the toes also can be involved with macerated white plaques, often with coexistent dermatophyte infection. Corynebacterium minutissimum, the bacteria responsible for erythrasma, produces coproporphyrin type III, which emits coral red fluorescence under Wood lamp examination.1 Bathing may result in removal of the porphyrin and result in a false-negative finding. Potassium hydroxide preparation of skin scrapings can show chains of bacilli. Biopsy appears relatively normal at low power but reveals compact orthokeratosis with coccobacilli and filamentous organisms in the superficial stratum corneum (quiz image). When not obvious on hematoxylin and eosin-stained sections, the organisms are Gram-positive and also are seen with periodic acid-Schiff (PAS) and methenamine silver stains. Unlike fungal hyphae, these organisms are thinner and nonrefractile. Inflammation typically is minimal. Due to the subtle histologic findings at low power, erythrasma is considered one of the invisible dermatoses.2 The differential diagnosis of these inconspicuous dermatoses that appear normal at first glance can be approached in a stepwise fashion starting in the stratum corneum, followed by the granular layer, basal layer, dermal papillae, dermal inflammatory cells, dermal connective tissue, and eccrine glands, and should consider each of the following diagnoses: candidiasis, dermatophytosis, ichthyosis vulgaris, vitiligo, macular amyloid, urticaria, telangiectasia macularis eruptiva perstans, connective tissue nevus, and argyria.2
Candidiasis, most commonly caused by Candida albicans, usually involves the oral cavity (eg, thrush, median rhomboid glossitis, angular cheilitis), intertriginous zones, nail fold (paronychia), genital areas (eg, vulvovaginitis, balanitis), and diaper area.3 The web space between the third and fourth fingers (erosio interdigitalis blastomycetica) can be involved in patients whose hands are frequently in water. Intertriginous candidiasis presents with bright red, sometimes erosive patches with satellite lesions. Spores and mycelia (filamentous forms) are noted on potassium hydroxide preparation of skin scrapings. Histologically, the epidermis often is acanthotic, mildly spongiotic, and contains groups of neutrophils in the superficial layers. The mnemonic device for diseases with clusters of neutrophils in the stratum corneum is PTICSS (psoriasis, tinea, impetigo, candida, seborrheic dermatitis, syphilis).2 Yeast, pseudohyphae, and even true hyphae can be seen in the stratum corneum with hematoxylin and eosin-stained sections and PAS. The filamentous forms tend to be vertically oriented in relation to the skin surface (Figure 1) compared to dermatophyte hyphae that tend to be parallel to the surface.2
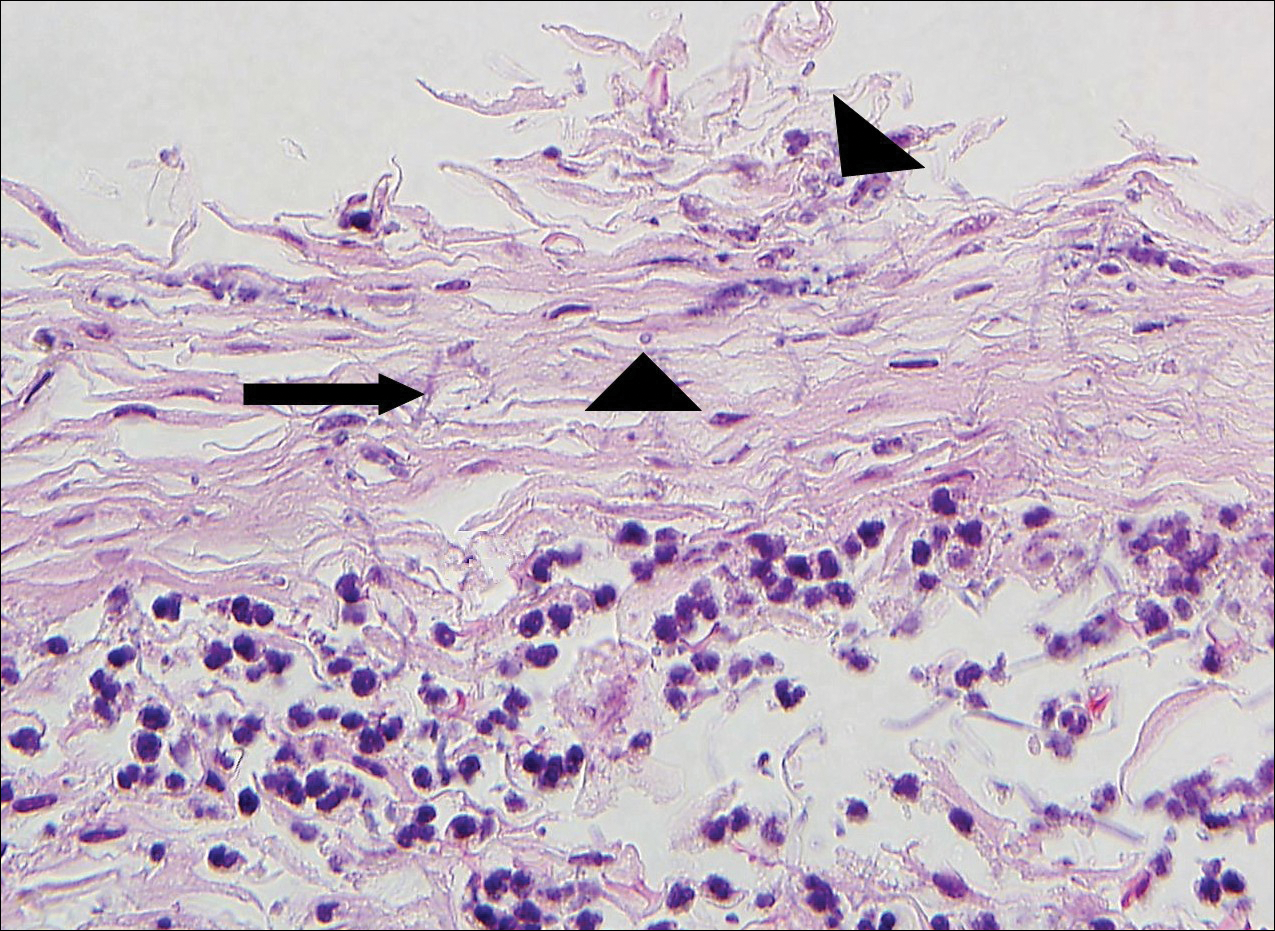
Pitted keratolysis is a superficial bacterial infection involving the soles of the feet. The classic clinical findings are shallow 1- to 2-mm pits in clusters that can coalesce on pressure-bearing areas. Hyperhidrosis, malodor, and maceration commonly are associated. Microscopic examination reveals clusters of small cocci and filamentous bacteria located in the dell or pit of a thick compact orthokeratotic stratum corneum of acral skin with no notable inflammatory infiltrate (Figure 2).2 Special stains such as Gram, methenamine silver, or PAS can assist in visualization of the organisms. Pitted keratolysis is caused by Dermatophilus congolensis and Kytococcus sedentarius (formerly Micrococcus sedentarius), which produce keratinolytic enzymes causing the defect in the stratum corneum.3
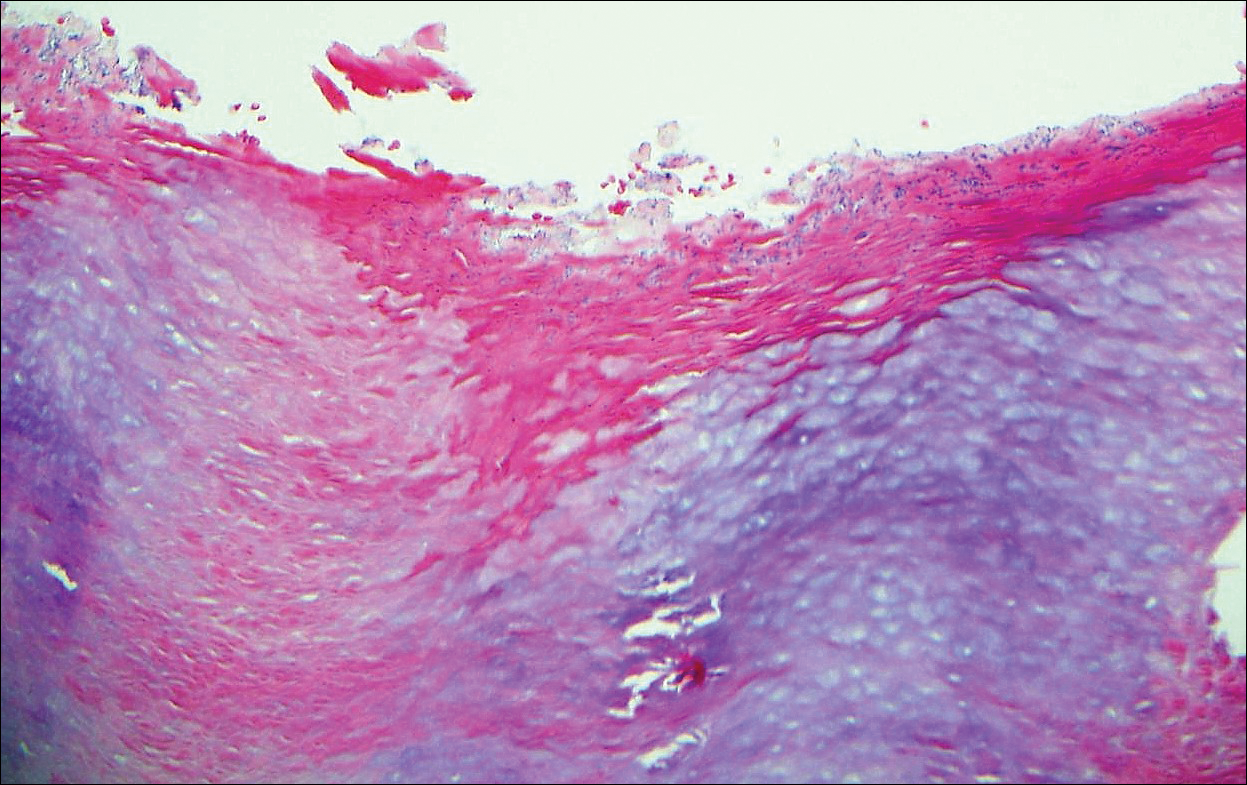
Tinea cruris, also known as jock itch and ringworm of the groin, presents with advancing pruritic, circinate, erythematous, scaling patches with central clearing on the inner thighs and crural folds. Similar to tinea pedis, Trichophyton rubrum is the most common dermatophyte to cause tinea cruris.4 Potassium hydroxide preparation of skin scrapings from the advancing border show fungal hyphae that cross the keratin cell borders. The histopathology of dermatophyte infections can be subtle and resemble normal skin before close inspection of the stratum corneum, which can show compact orthokeratosis, neutrophils, or "sandwich sign" where hyphae are sandwiched between an upper basket weave layer and a lower compact cornified layer (orthokeratotic or parakeratotic)(Figure 3).1 The presence of these patterns in the stratum corneum should result in performance of PAS to highlight obscure hyphae.
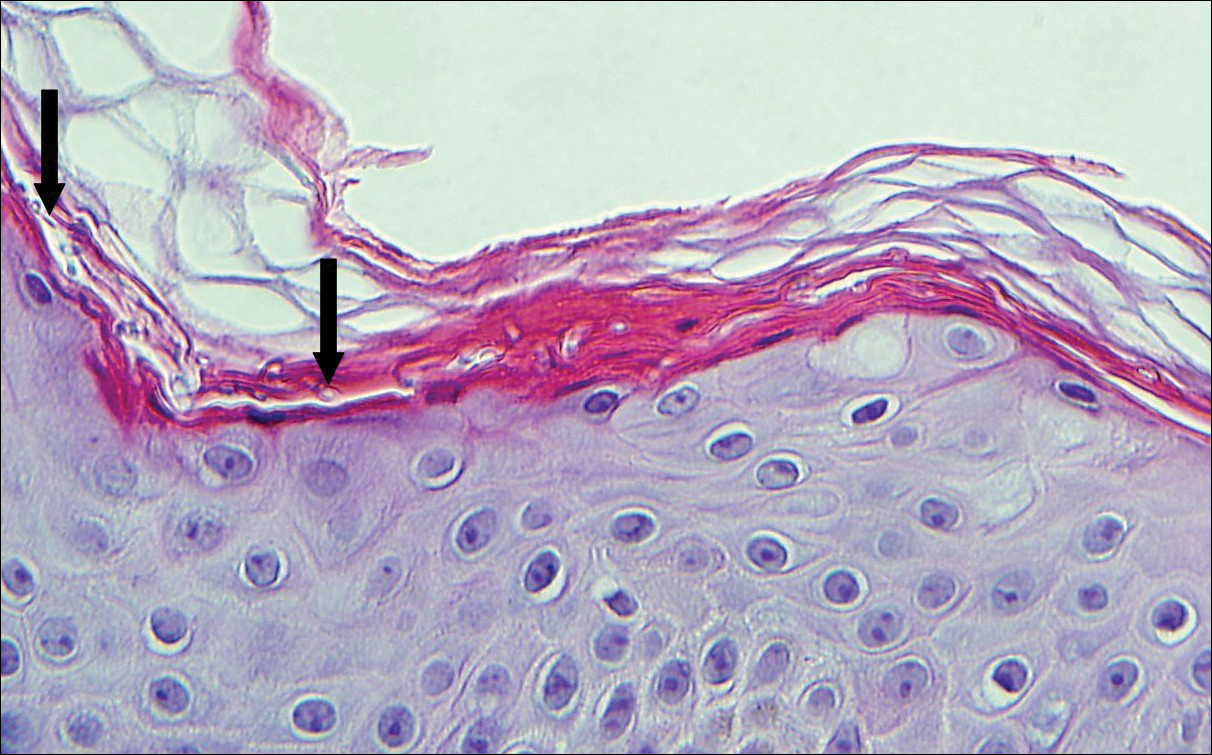
Tinea versicolor, also called pityriasis versicolor, usually presents with hypopigmented or less commonly hyperpigmented circular patches that coalesce on the upper trunk and shoulders. There is a fine fluffy scale that is most notable after scraping the skin for a potassium hydroxide preparation, which shows "spaghetti and meatballs" (hyphae and spores). Tinea versicolor typically is caused by the mycelial phase of the lipophilic yeast Malassezia globosae.3 Histologically, there are yeast and short septate hyphae scattered in a loose basket weave hyperkeratotic stratum corneum with minimal or no inflammation (Figure 4). On occasion, PAS is required for identification.
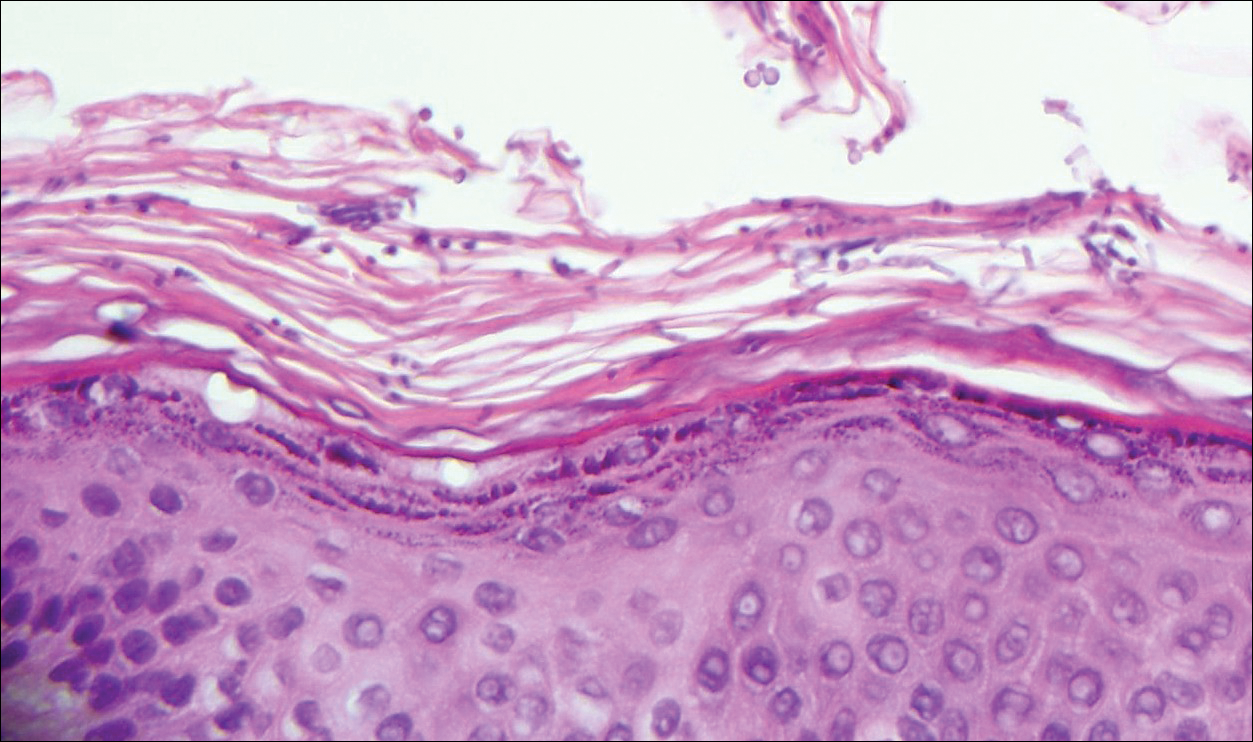
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Bolognia JL, Shaffer JV, Cerroni L, eds. Dermatolology. 4th ed. China: Elsevier; 2018.
The Diagnosis: Erythrasma
Erythrasma usually involves intertriginous areas (eg, axillae, groin, inframammary area). Patients present with well-demarcated, minimally scaly, red-brown patches. The interdigital web space of the toes also can be involved with macerated white plaques, often with coexistent dermatophyte infection. Corynebacterium minutissimum, the bacteria responsible for erythrasma, produces coproporphyrin type III, which emits coral red fluorescence under Wood lamp examination.1 Bathing may result in removal of the porphyrin and result in a false-negative finding. Potassium hydroxide preparation of skin scrapings can show chains of bacilli. Biopsy appears relatively normal at low power but reveals compact orthokeratosis with coccobacilli and filamentous organisms in the superficial stratum corneum (quiz image). When not obvious on hematoxylin and eosin-stained sections, the organisms are Gram-positive and also are seen with periodic acid-Schiff (PAS) and methenamine silver stains. Unlike fungal hyphae, these organisms are thinner and nonrefractile. Inflammation typically is minimal. Due to the subtle histologic findings at low power, erythrasma is considered one of the invisible dermatoses.2 The differential diagnosis of these inconspicuous dermatoses that appear normal at first glance can be approached in a stepwise fashion starting in the stratum corneum, followed by the granular layer, basal layer, dermal papillae, dermal inflammatory cells, dermal connective tissue, and eccrine glands, and should consider each of the following diagnoses: candidiasis, dermatophytosis, ichthyosis vulgaris, vitiligo, macular amyloid, urticaria, telangiectasia macularis eruptiva perstans, connective tissue nevus, and argyria.2
Candidiasis, most commonly caused by Candida albicans, usually involves the oral cavity (eg, thrush, median rhomboid glossitis, angular cheilitis), intertriginous zones, nail fold (paronychia), genital areas (eg, vulvovaginitis, balanitis), and diaper area.3 The web space between the third and fourth fingers (erosio interdigitalis blastomycetica) can be involved in patients whose hands are frequently in water. Intertriginous candidiasis presents with bright red, sometimes erosive patches with satellite lesions. Spores and mycelia (filamentous forms) are noted on potassium hydroxide preparation of skin scrapings. Histologically, the epidermis often is acanthotic, mildly spongiotic, and contains groups of neutrophils in the superficial layers. The mnemonic device for diseases with clusters of neutrophils in the stratum corneum is PTICSS (psoriasis, tinea, impetigo, candida, seborrheic dermatitis, syphilis).2 Yeast, pseudohyphae, and even true hyphae can be seen in the stratum corneum with hematoxylin and eosin-stained sections and PAS. The filamentous forms tend to be vertically oriented in relation to the skin surface (Figure 1) compared to dermatophyte hyphae that tend to be parallel to the surface.2
Pitted keratolysis is a superficial bacterial infection involving the soles of the feet. The classic clinical findings are shallow 1- to 2-mm pits in clusters that can coalesce on pressure-bearing areas. Hyperhidrosis, malodor, and maceration commonly are associated. Microscopic examination reveals clusters of small cocci and filamentous bacteria located in the dell or pit of a thick compact orthokeratotic stratum corneum of acral skin with no notable inflammatory infiltrate (Figure 2).2 Special stains such as Gram, methenamine silver, or PAS can assist in visualization of the organisms. Pitted keratolysis is caused by Dermatophilus congolensis and Kytococcus sedentarius (formerly Micrococcus sedentarius), which produce keratinolytic enzymes causing the defect in the stratum corneum.3
Tinea cruris, also known as jock itch and ringworm of the groin, presents with advancing pruritic, circinate, erythematous, scaling patches with central clearing on the inner thighs and crural folds. Similar to tinea pedis, Trichophyton rubrum is the most common dermatophyte to cause tinea cruris.4 Potassium hydroxide preparation of skin scrapings from the advancing border show fungal hyphae that cross the keratin cell borders. The histopathology of dermatophyte infections can be subtle and resemble normal skin before close inspection of the stratum corneum, which can show compact orthokeratosis, neutrophils, or "sandwich sign" where hyphae are sandwiched between an upper basket weave layer and a lower compact cornified layer (orthokeratotic or parakeratotic)(Figure 3).1 The presence of these patterns in the stratum corneum should result in performance of PAS to highlight obscure hyphae.
Tinea versicolor, also called pityriasis versicolor, usually presents with hypopigmented or less commonly hyperpigmented circular patches that coalesce on the upper trunk and shoulders. There is a fine fluffy scale that is most notable after scraping the skin for a potassium hydroxide preparation, which shows "spaghetti and meatballs" (hyphae and spores). Tinea versicolor typically is caused by the mycelial phase of the lipophilic yeast Malassezia globosae.3 Histologically, there are yeast and short septate hyphae scattered in a loose basket weave hyperkeratotic stratum corneum with minimal or no inflammation (Figure 4). On occasion, PAS is required for identification.
The Diagnosis: Erythrasma
Erythrasma usually involves intertriginous areas (eg, axillae, groin, inframammary area). Patients present with well-demarcated, minimally scaly, red-brown patches. The interdigital web space of the toes also can be involved with macerated white plaques, often with coexistent dermatophyte infection. Corynebacterium minutissimum, the bacteria responsible for erythrasma, produces coproporphyrin type III, which emits coral red fluorescence under Wood lamp examination.1 Bathing may result in removal of the porphyrin and result in a false-negative finding. Potassium hydroxide preparation of skin scrapings can show chains of bacilli. Biopsy appears relatively normal at low power but reveals compact orthokeratosis with coccobacilli and filamentous organisms in the superficial stratum corneum (quiz image). When not obvious on hematoxylin and eosin-stained sections, the organisms are Gram-positive and also are seen with periodic acid-Schiff (PAS) and methenamine silver stains. Unlike fungal hyphae, these organisms are thinner and nonrefractile. Inflammation typically is minimal. Due to the subtle histologic findings at low power, erythrasma is considered one of the invisible dermatoses.2 The differential diagnosis of these inconspicuous dermatoses that appear normal at first glance can be approached in a stepwise fashion starting in the stratum corneum, followed by the granular layer, basal layer, dermal papillae, dermal inflammatory cells, dermal connective tissue, and eccrine glands, and should consider each of the following diagnoses: candidiasis, dermatophytosis, ichthyosis vulgaris, vitiligo, macular amyloid, urticaria, telangiectasia macularis eruptiva perstans, connective tissue nevus, and argyria.2
Candidiasis, most commonly caused by Candida albicans, usually involves the oral cavity (eg, thrush, median rhomboid glossitis, angular cheilitis), intertriginous zones, nail fold (paronychia), genital areas (eg, vulvovaginitis, balanitis), and diaper area.3 The web space between the third and fourth fingers (erosio interdigitalis blastomycetica) can be involved in patients whose hands are frequently in water. Intertriginous candidiasis presents with bright red, sometimes erosive patches with satellite lesions. Spores and mycelia (filamentous forms) are noted on potassium hydroxide preparation of skin scrapings. Histologically, the epidermis often is acanthotic, mildly spongiotic, and contains groups of neutrophils in the superficial layers. The mnemonic device for diseases with clusters of neutrophils in the stratum corneum is PTICSS (psoriasis, tinea, impetigo, candida, seborrheic dermatitis, syphilis).2 Yeast, pseudohyphae, and even true hyphae can be seen in the stratum corneum with hematoxylin and eosin-stained sections and PAS. The filamentous forms tend to be vertically oriented in relation to the skin surface (Figure 1) compared to dermatophyte hyphae that tend to be parallel to the surface.2
Pitted keratolysis is a superficial bacterial infection involving the soles of the feet. The classic clinical findings are shallow 1- to 2-mm pits in clusters that can coalesce on pressure-bearing areas. Hyperhidrosis, malodor, and maceration commonly are associated. Microscopic examination reveals clusters of small cocci and filamentous bacteria located in the dell or pit of a thick compact orthokeratotic stratum corneum of acral skin with no notable inflammatory infiltrate (Figure 2).2 Special stains such as Gram, methenamine silver, or PAS can assist in visualization of the organisms. Pitted keratolysis is caused by Dermatophilus congolensis and Kytococcus sedentarius (formerly Micrococcus sedentarius), which produce keratinolytic enzymes causing the defect in the stratum corneum.3
Tinea cruris, also known as jock itch and ringworm of the groin, presents with advancing pruritic, circinate, erythematous, scaling patches with central clearing on the inner thighs and crural folds. Similar to tinea pedis, Trichophyton rubrum is the most common dermatophyte to cause tinea cruris.4 Potassium hydroxide preparation of skin scrapings from the advancing border show fungal hyphae that cross the keratin cell borders. The histopathology of dermatophyte infections can be subtle and resemble normal skin before close inspection of the stratum corneum, which can show compact orthokeratosis, neutrophils, or "sandwich sign" where hyphae are sandwiched between an upper basket weave layer and a lower compact cornified layer (orthokeratotic or parakeratotic)(Figure 3).1 The presence of these patterns in the stratum corneum should result in performance of PAS to highlight obscure hyphae.
Tinea versicolor, also called pityriasis versicolor, usually presents with hypopigmented or less commonly hyperpigmented circular patches that coalesce on the upper trunk and shoulders. There is a fine fluffy scale that is most notable after scraping the skin for a potassium hydroxide preparation, which shows "spaghetti and meatballs" (hyphae and spores). Tinea versicolor typically is caused by the mycelial phase of the lipophilic yeast Malassezia globosae.3 Histologically, there are yeast and short septate hyphae scattered in a loose basket weave hyperkeratotic stratum corneum with minimal or no inflammation (Figure 4). On occasion, PAS is required for identification.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Bolognia JL, Shaffer JV, Cerroni L, eds. Dermatolology. 4th ed. China: Elsevier; 2018.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Bolognia JL, Shaffer JV, Cerroni L, eds. Dermatolology. 4th ed. China: Elsevier; 2018.

A 66-year-old man presented with reddish arciform patches in the inguinal area.
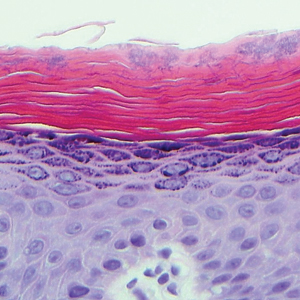
Beltlike Lichen Planus Pigmentosus Complicated With Focal Amyloidosis
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.

Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.

.")
.")
.")
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.
Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.
Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
Practice Points
- Lichen planus pigmentosus can present in a unique beltlike distribution pattern.
- Focal amyloidosis due to chronic friction and scratching cannot be excluded from the differential diagnosis.
Acute Generalized Exanthematous Pustulosis Caused by Pantoprazole
To the Editor:
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
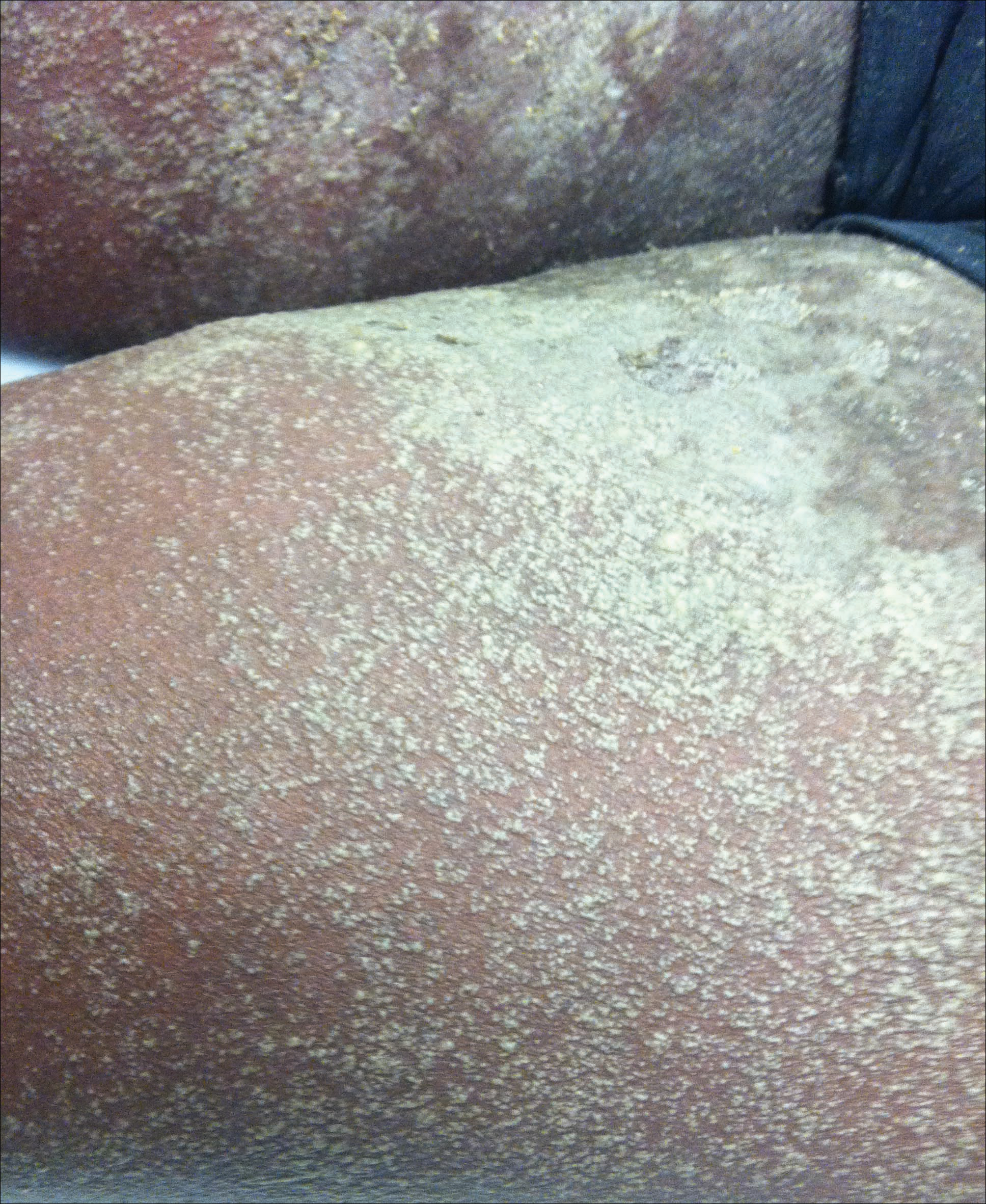
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.

Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
To the Editor:
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.
Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
To the Editor:
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.
Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
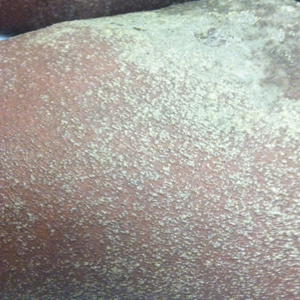
Interstitial Granulomatous Dermatitis and Palisaded Neutrophilic Granulomatous Dermatitis
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.

.")
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.

.")
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
Practice Points
- The clinical features of interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis exist on a spectrum, and these is considerable overlap between the features of these 2 clinicopathologic entities.
- Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis may respond to systemic steroids or treatment of the underlying systemic disease. Some cases spontaneously resolve.