User login
Recalcitrant Solitary Erythematous Scaly Patch on the Foot
The Diagnosis: Pagetoid Reticulosis
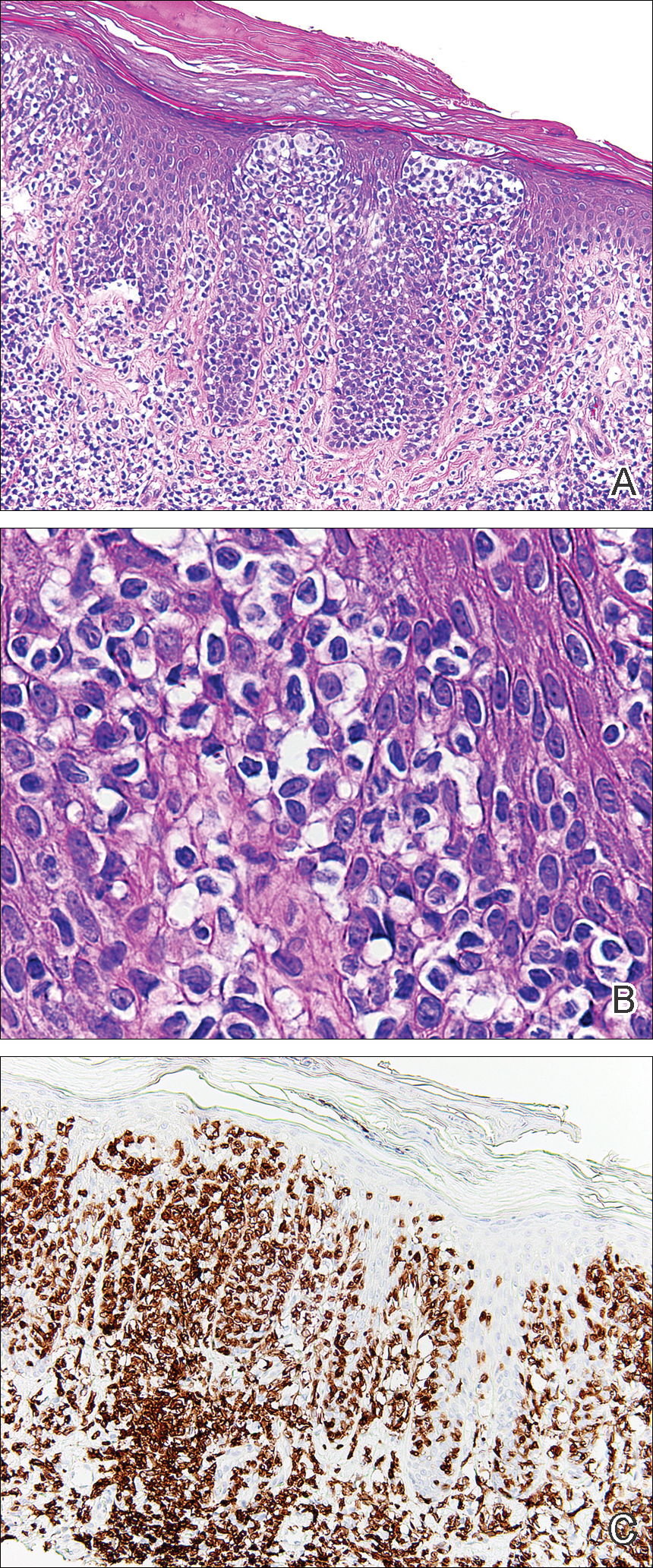
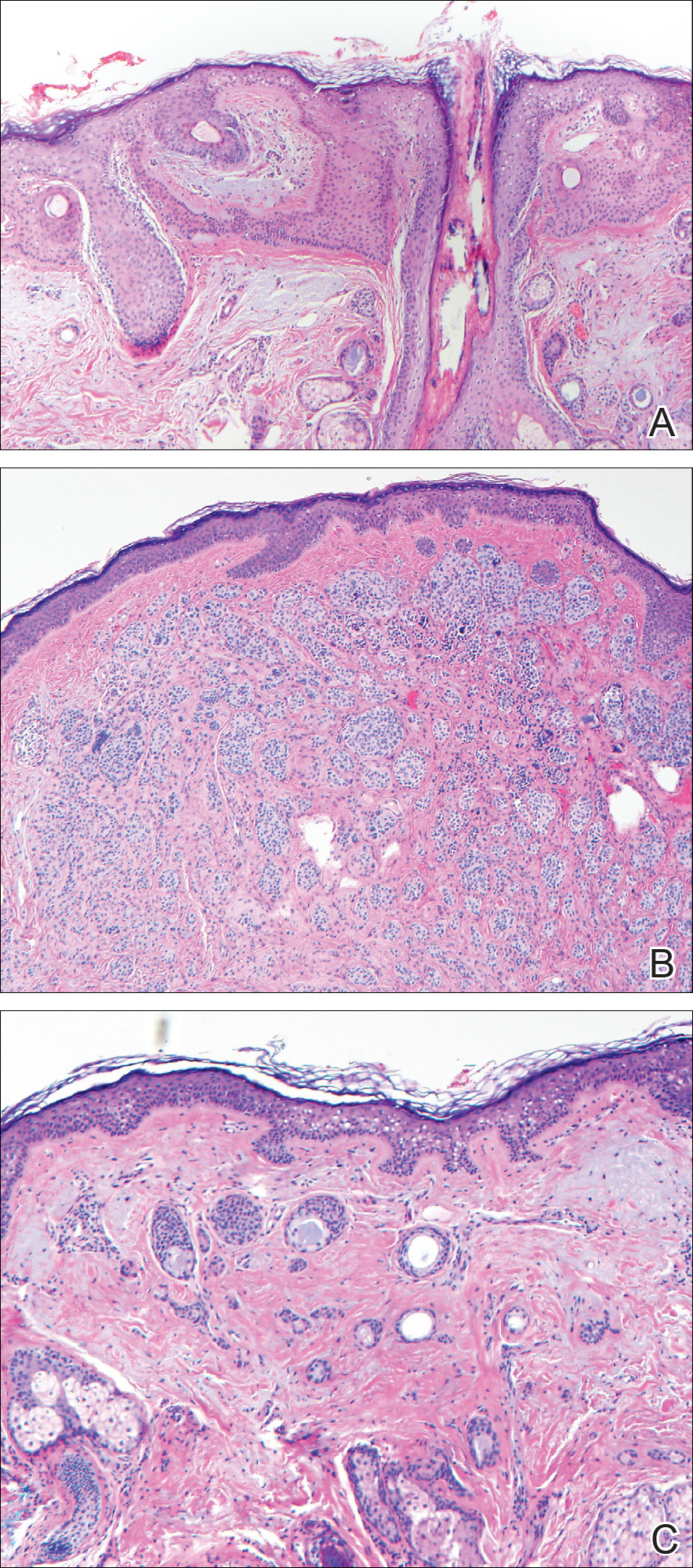
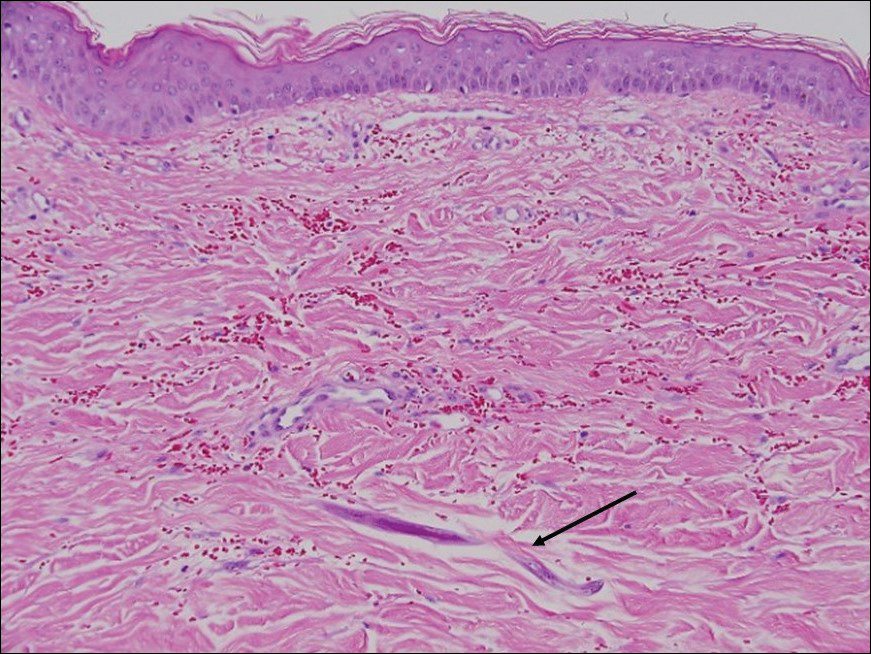
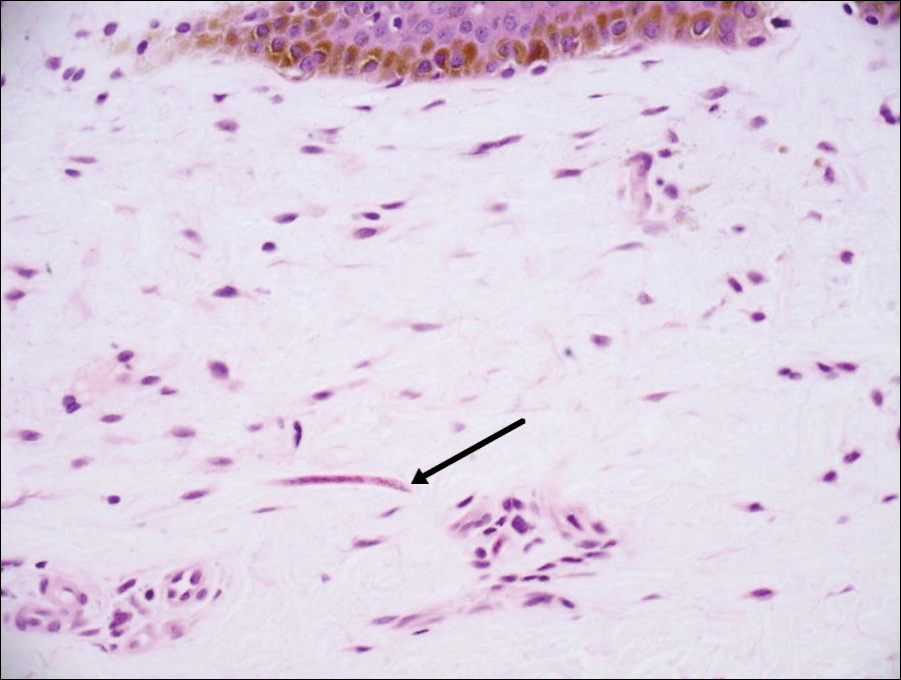
Histopathologic examination demonstrated a dense infiltrate and psoriasiform pattern epidermal hyperplasia (Figure, A). There was conspicuous epidermotropism of moderately enlarged, hyperchromatic lymphocytes. Intraepidermal lymphocytes were slightly larger, darker, and more convoluted than those in the subjacent dermis (Figure, B). These cells exhibited CD3+ T-cell differentiation with an abnormal CD4-CD7-CD8- phenotype (Figure, C). The histopathologic finding of atypical epidermotropic T-cell infiltrate was compatible with a rare variant of mycosis fungoides known as pagetoid reticulosis (PR). After discussing the diagnosis and treatment options, the patient elected to begin with a conservative approach to therapy. We prescribed fluocinonide ointment 0.05% twice daily under occlusion. At 1 month follow-up, the patient experienced marked improvement of the erythema and scaling of the lesion.
Pagetoid reticulosis is a primary cutaneous T-cell lymphoma that has been categorized as an indolent localized variant of mycosis fungoides. This rare skin disorder was originally described by Woringer and Kolopp in 19391 and was further renamed in 1973 by Braun-Falco et al.2 At that time the term pagetoid reticulosis was introduced due to similarities in histopathologic findings seen in Paget disease of the nipple. Two variants of the disease have been described since then: the localized type and the disseminated type. The localized type, also known as Woringer-Kolopp disease (WKD), typically presents as a persistent, sharply localized, scaly patch that slowly expands over several years. The lesion is classically located on the extensor surface of the hand or foot and often is asymptomatic. Due to the benign presentation, WKD can easily be confused with much more common diseases, such as psoriasis or fungal infections, resulting in a substantial delay in the diagnosis. The patient will often report a medical history notable for frequent office visits and numerous failed therapies. Even though it is exceedingly uncommon, these findings should prompt the practitioner to add WKD to their differential. The disseminated type of PR (also known as Ketron-Goodman disease) is characterized by diffuse cutaneous involvement, carries a much more progressive course, and often leads to a poor outcome.3 The histopathologic features of WKD and Ketron-Goodman disease are identical, and the 2 types are distinguished on clinical grounds alone.
Histopathologic features of PR are unique and often distinct in comparison to mycosis fungoides. Pagetoid reticulosis often is described as epidermal hyperplasia with parakeratosis, prominent acanthosis, and excessive epidermotropism of atypical lymphocytes scattered throughout the epidermis.3 The distinct pattern of epidermotropism seen in PR is the characteristic finding. Review of immunocytochemistry from reported cases has shown that CD marker expression of neoplastic T cells in PR can be variable in nature.4 Although it is known that immunophenotyping can be useful in diagnosing and distinguishing PR from other types of primary cutaneous T-cell lymphoma, the clinical significance of the observed phenotypic variation remains a mystery. As of now, it appears to be prognostically irrelevant.5
There are numerous therapeutic options available for PR. Depending on the size and extent of the disease, surgical excision and radiotherapy may be an option and are the most effective.6 For patients who are not good candidates or opt out of these options, there are various pharmacotherapies that also have proven to work. Traditional therapies include topical corticosteroids, corticosteroid injections, and phototherapy. However, more recent trials with retinoids, such as alitretinoin or bexarotene, appear to offer a promising therapeutic approach.7
Pagetoid reticulosis is a true malignant lymphoma of T-cell lineage, but it typically carries an excellent prognosis. Rare cases have been reported to progress to disseminated lymphoma.8 Therefore, long-term follow-up for a patient diagnosed with PR is recommended.
- Woringer FR, Kolopp P. Lésion érythémato-squameuse polycyclique de l'avant-bras évoluantdepuis 6 ans chez un garçonnet de 13 ans. Ann Dermatol Venereol. 1939;10:945-948.
- Braun-Falco O, Marghescu S, Wolff HH. Pagetoid reticulosis--Woringer-Kolopp's disease [in German]. Hautarzt. 1973;24:11-21.
- Haghighi B, Smoller BR, Leboit PE, et al. Pagetoid reticulosis (Woringer-Kolopp disease): an immunophenotypic, molecular, and clinicopathologic study. Mod Pathol. 2000;13:502-510.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Mourtzinos N, Puri PK, Wang G, et al. CD4/CD8 double negative pagetoid reticulosis: a case report and literature review. J Cutan Pathol. 2010;37:491-496.
- Lee J, Viakhireva N, Cesca C, et al. Clinicopathologic features and treatment outcomes in Woringer-Kolopp disease. J Am Acad Dermatol. 2008;59:706-712.
- Schmitz L, Bierhoff E, Dirschka T. Alitretinoin: an effective treatment option for pagetoid reticulosis. J Dtsch Dermatol Ges. 2013;11:1194-1195.
- Ioannides G, Engel MF, Rywlin AM. Woringer-Kolopp disease (pagetoid reticulosis). Am J Dermatopathol. 1983;5:153-158.
The Diagnosis: Pagetoid Reticulosis
Histopathologic examination demonstrated a dense infiltrate and psoriasiform pattern epidermal hyperplasia (Figure, A). There was conspicuous epidermotropism of moderately enlarged, hyperchromatic lymphocytes. Intraepidermal lymphocytes were slightly larger, darker, and more convoluted than those in the subjacent dermis (Figure, B). These cells exhibited CD3+ T-cell differentiation with an abnormal CD4-CD7-CD8- phenotype (Figure, C). The histopathologic finding of atypical epidermotropic T-cell infiltrate was compatible with a rare variant of mycosis fungoides known as pagetoid reticulosis (PR). After discussing the diagnosis and treatment options, the patient elected to begin with a conservative approach to therapy. We prescribed fluocinonide ointment 0.05% twice daily under occlusion. At 1 month follow-up, the patient experienced marked improvement of the erythema and scaling of the lesion.
Pagetoid reticulosis is a primary cutaneous T-cell lymphoma that has been categorized as an indolent localized variant of mycosis fungoides. This rare skin disorder was originally described by Woringer and Kolopp in 19391 and was further renamed in 1973 by Braun-Falco et al.2 At that time the term pagetoid reticulosis was introduced due to similarities in histopathologic findings seen in Paget disease of the nipple. Two variants of the disease have been described since then: the localized type and the disseminated type. The localized type, also known as Woringer-Kolopp disease (WKD), typically presents as a persistent, sharply localized, scaly patch that slowly expands over several years. The lesion is classically located on the extensor surface of the hand or foot and often is asymptomatic. Due to the benign presentation, WKD can easily be confused with much more common diseases, such as psoriasis or fungal infections, resulting in a substantial delay in the diagnosis. The patient will often report a medical history notable for frequent office visits and numerous failed therapies. Even though it is exceedingly uncommon, these findings should prompt the practitioner to add WKD to their differential. The disseminated type of PR (also known as Ketron-Goodman disease) is characterized by diffuse cutaneous involvement, carries a much more progressive course, and often leads to a poor outcome.3 The histopathologic features of WKD and Ketron-Goodman disease are identical, and the 2 types are distinguished on clinical grounds alone.
Histopathologic features of PR are unique and often distinct in comparison to mycosis fungoides. Pagetoid reticulosis often is described as epidermal hyperplasia with parakeratosis, prominent acanthosis, and excessive epidermotropism of atypical lymphocytes scattered throughout the epidermis.3 The distinct pattern of epidermotropism seen in PR is the characteristic finding. Review of immunocytochemistry from reported cases has shown that CD marker expression of neoplastic T cells in PR can be variable in nature.4 Although it is known that immunophenotyping can be useful in diagnosing and distinguishing PR from other types of primary cutaneous T-cell lymphoma, the clinical significance of the observed phenotypic variation remains a mystery. As of now, it appears to be prognostically irrelevant.5
There are numerous therapeutic options available for PR. Depending on the size and extent of the disease, surgical excision and radiotherapy may be an option and are the most effective.6 For patients who are not good candidates or opt out of these options, there are various pharmacotherapies that also have proven to work. Traditional therapies include topical corticosteroids, corticosteroid injections, and phototherapy. However, more recent trials with retinoids, such as alitretinoin or bexarotene, appear to offer a promising therapeutic approach.7
Pagetoid reticulosis is a true malignant lymphoma of T-cell lineage, but it typically carries an excellent prognosis. Rare cases have been reported to progress to disseminated lymphoma.8 Therefore, long-term follow-up for a patient diagnosed with PR is recommended.
The Diagnosis: Pagetoid Reticulosis
Histopathologic examination demonstrated a dense infiltrate and psoriasiform pattern epidermal hyperplasia (Figure, A). There was conspicuous epidermotropism of moderately enlarged, hyperchromatic lymphocytes. Intraepidermal lymphocytes were slightly larger, darker, and more convoluted than those in the subjacent dermis (Figure, B). These cells exhibited CD3+ T-cell differentiation with an abnormal CD4-CD7-CD8- phenotype (Figure, C). The histopathologic finding of atypical epidermotropic T-cell infiltrate was compatible with a rare variant of mycosis fungoides known as pagetoid reticulosis (PR). After discussing the diagnosis and treatment options, the patient elected to begin with a conservative approach to therapy. We prescribed fluocinonide ointment 0.05% twice daily under occlusion. At 1 month follow-up, the patient experienced marked improvement of the erythema and scaling of the lesion.
Pagetoid reticulosis is a primary cutaneous T-cell lymphoma that has been categorized as an indolent localized variant of mycosis fungoides. This rare skin disorder was originally described by Woringer and Kolopp in 19391 and was further renamed in 1973 by Braun-Falco et al.2 At that time the term pagetoid reticulosis was introduced due to similarities in histopathologic findings seen in Paget disease of the nipple. Two variants of the disease have been described since then: the localized type and the disseminated type. The localized type, also known as Woringer-Kolopp disease (WKD), typically presents as a persistent, sharply localized, scaly patch that slowly expands over several years. The lesion is classically located on the extensor surface of the hand or foot and often is asymptomatic. Due to the benign presentation, WKD can easily be confused with much more common diseases, such as psoriasis or fungal infections, resulting in a substantial delay in the diagnosis. The patient will often report a medical history notable for frequent office visits and numerous failed therapies. Even though it is exceedingly uncommon, these findings should prompt the practitioner to add WKD to their differential. The disseminated type of PR (also known as Ketron-Goodman disease) is characterized by diffuse cutaneous involvement, carries a much more progressive course, and often leads to a poor outcome.3 The histopathologic features of WKD and Ketron-Goodman disease are identical, and the 2 types are distinguished on clinical grounds alone.
Histopathologic features of PR are unique and often distinct in comparison to mycosis fungoides. Pagetoid reticulosis often is described as epidermal hyperplasia with parakeratosis, prominent acanthosis, and excessive epidermotropism of atypical lymphocytes scattered throughout the epidermis.3 The distinct pattern of epidermotropism seen in PR is the characteristic finding. Review of immunocytochemistry from reported cases has shown that CD marker expression of neoplastic T cells in PR can be variable in nature.4 Although it is known that immunophenotyping can be useful in diagnosing and distinguishing PR from other types of primary cutaneous T-cell lymphoma, the clinical significance of the observed phenotypic variation remains a mystery. As of now, it appears to be prognostically irrelevant.5
There are numerous therapeutic options available for PR. Depending on the size and extent of the disease, surgical excision and radiotherapy may be an option and are the most effective.6 For patients who are not good candidates or opt out of these options, there are various pharmacotherapies that also have proven to work. Traditional therapies include topical corticosteroids, corticosteroid injections, and phototherapy. However, more recent trials with retinoids, such as alitretinoin or bexarotene, appear to offer a promising therapeutic approach.7
Pagetoid reticulosis is a true malignant lymphoma of T-cell lineage, but it typically carries an excellent prognosis. Rare cases have been reported to progress to disseminated lymphoma.8 Therefore, long-term follow-up for a patient diagnosed with PR is recommended.
- Woringer FR, Kolopp P. Lésion érythémato-squameuse polycyclique de l'avant-bras évoluantdepuis 6 ans chez un garçonnet de 13 ans. Ann Dermatol Venereol. 1939;10:945-948.
- Braun-Falco O, Marghescu S, Wolff HH. Pagetoid reticulosis--Woringer-Kolopp's disease [in German]. Hautarzt. 1973;24:11-21.
- Haghighi B, Smoller BR, Leboit PE, et al. Pagetoid reticulosis (Woringer-Kolopp disease): an immunophenotypic, molecular, and clinicopathologic study. Mod Pathol. 2000;13:502-510.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Mourtzinos N, Puri PK, Wang G, et al. CD4/CD8 double negative pagetoid reticulosis: a case report and literature review. J Cutan Pathol. 2010;37:491-496.
- Lee J, Viakhireva N, Cesca C, et al. Clinicopathologic features and treatment outcomes in Woringer-Kolopp disease. J Am Acad Dermatol. 2008;59:706-712.
- Schmitz L, Bierhoff E, Dirschka T. Alitretinoin: an effective treatment option for pagetoid reticulosis. J Dtsch Dermatol Ges. 2013;11:1194-1195.
- Ioannides G, Engel MF, Rywlin AM. Woringer-Kolopp disease (pagetoid reticulosis). Am J Dermatopathol. 1983;5:153-158.
- Woringer FR, Kolopp P. Lésion érythémato-squameuse polycyclique de l'avant-bras évoluantdepuis 6 ans chez un garçonnet de 13 ans. Ann Dermatol Venereol. 1939;10:945-948.
- Braun-Falco O, Marghescu S, Wolff HH. Pagetoid reticulosis--Woringer-Kolopp's disease [in German]. Hautarzt. 1973;24:11-21.
- Haghighi B, Smoller BR, Leboit PE, et al. Pagetoid reticulosis (Woringer-Kolopp disease): an immunophenotypic, molecular, and clinicopathologic study. Mod Pathol. 2000;13:502-510.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Mourtzinos N, Puri PK, Wang G, et al. CD4/CD8 double negative pagetoid reticulosis: a case report and literature review. J Cutan Pathol. 2010;37:491-496.
- Lee J, Viakhireva N, Cesca C, et al. Clinicopathologic features and treatment outcomes in Woringer-Kolopp disease. J Am Acad Dermatol. 2008;59:706-712.
- Schmitz L, Bierhoff E, Dirschka T. Alitretinoin: an effective treatment option for pagetoid reticulosis. J Dtsch Dermatol Ges. 2013;11:1194-1195.
- Ioannides G, Engel MF, Rywlin AM. Woringer-Kolopp disease (pagetoid reticulosis). Am J Dermatopathol. 1983;5:153-158.
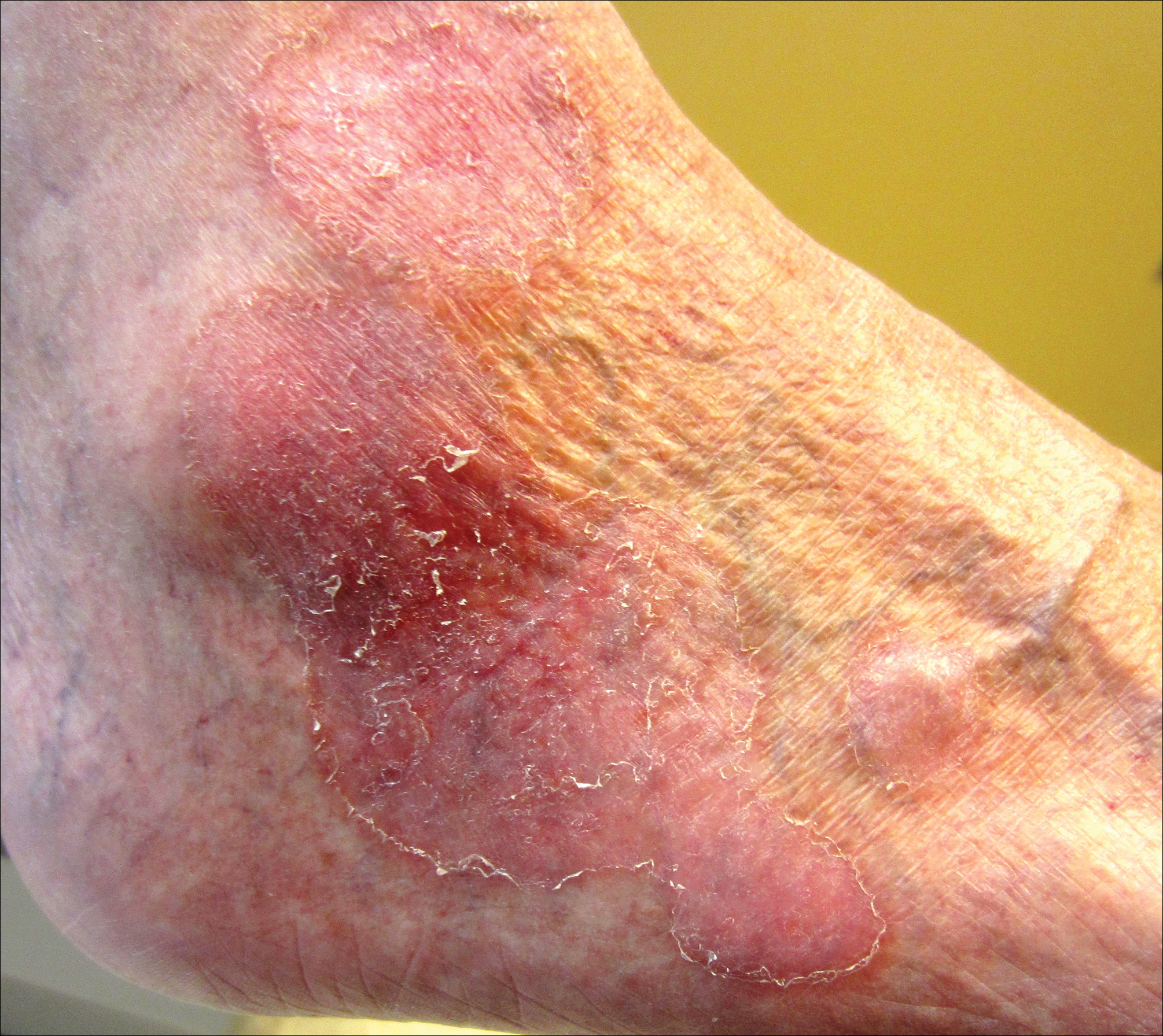
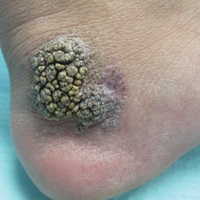
An 80-year-old man with a history of malignant melanoma and squamous cell carcinoma presented to the dermatology clinic with a chronic rash of 20 years' duration on the right ankle that extended to the instep of the right foot. His medical history was notable for hypertension and hyperlipidemia. Family history was unremarkable. The patient described the rash as red and scaly but denied associated pain or pruritus. Over the last 2 to 3 years he had tried treating the affected area with petroleum jelly, topical and oral antifungals, and mild topical steroids with minimal improvement. Complete review of systems was performed and was negative other than some mild constipation. Physical examination revealed an erythematous scaly patch on the dorsal aspect of the right ankle. Potassium hydroxide preparation and fungal culture swab yielded negative results, and a shave biopsy was performed.
Expanding Uses of Propranolol in Dermatology
Since the serendipitous discovery of expedited involution of infantile hemangiomas (IHs) with propranolol in 2008,1 current research has proliferated to discern the mechanism of action of beta-blockers in the care of IHs. Propranolol is a nonselective beta-blocker with a structure similar to catecholamines and thus competes for β-adrenergic receptors. Blocking β1-receptors is cardioselective, leading to decreased heart rate and myocardial contractility, while blocking β2-receptors leads to inhibition of smooth muscle relaxation and decreased glycogenolysis. The endothelial cells of IH express β2-adrenergic receptors; the mechanistic role of propranolol in these lesions is surmised to be due to vasoconstriction, decreased angiogenesis through inhibition of vascular endothelial growth factor, and subsequent endothelial cell apoptosis.2
After this breakthrough finding, a subsequent novel development was made when an ophthalmologist demonstrated that timolol, a topical beta-blocker, could be utilized to expedite IH involution and prevent ocular complications such as amblyopia secondary to the mass effect of the lesion. Guo and Ni3 prescribed the commercially available ophthalmologic solution of timolol maleate 0.5% for twice-daily use for 5 weeks. Remarkable reduction in the periorbital IH without rebound phenomenon was observed.3 A recent multicenter retrospective cohort of more than 700 patients with IH were treated with topical timolol with a 70% success rate, corresponding to 10% improvement from baseline; this study highlights the efficacy of timolol while confirming the safety of the medication.4
Systemic beta-blockers for IH have been used predominately for critical sites such as the nasal tip, lip, ear, perineum, and periocular area; ulcerated lesions or those that may be prone to leave a fibrofatty tissue residue after involution also have been targeted. Contraindications for use include premature infants younger than 5 weeks, infants weighing less than 2 kg, history of asthma or bronchospasm, heart rate less than 80 beats per minute, blood pressure less than 50/30 mm Hg, or hypersensitivity to the medication.5 Current guidelines for propranolol initiation vary; some dermatologists consult cardiology prior to initiation, while others perform routine vitals and an indication-driven electrocardiogram as needed based on family history of cardiac disease, maternal history of connective tissue disease, congenital heart block, or abnormal vital signs.
Given the demonstrated long-term safety of propranolol and the acceptable side-effect profile, the use of beta-blockers for IH has become increasingly mainstream. Three randomized controlled trials (RCTs) have evaluated the efficacy and minimal adverse effects of propranolol for IH. The first RCT evaluated 40 patients who received either placebo or propranolol 2 mg/kg daily (divided into 3 doses) for 6 months; IH growth stopped by week 4 in the treatment group and the largest volume difference in IH was seen at week 12.6 Léauté-Labrèze et al7 demonstrated that propranolol could be given earlier to patients and at higher doses; the treatment group included 7 patients at 3 mg/kg daily of propranolol for 15 days, followed by 15 additional days of 4 mg/kg daily of propranolol. A statistically significant (P=.004) decrease in IH volume, quantified by use of ultrasonography, was exhibited by the propranolol group.7 Lastly, the largest RCT (N=456) established the efficacy of propranolol 3 mg/kg daily for 6 months with a 60% successful treatment rate compared to 4% for patients receiving placebo.8
Given the efficacy of propranolol for IH, other investigators have experimented with nonselective beta-blockers for other dermatologic conditions. In addition to second-line use for flushing, hyperhidrosis, and adrenergic urticaria, the future of propranolol is expanding for vascular lesions in particular.9 Chow et al10 highlighted a case of progressive angiosarcoma of the scalp that responded to propranolol hydrochloride therapy at 40 mg 3 times daily with extensive regression; propranolol was given in addition to chemotherapy and radiation. The tumor was biopsied before and after propranolol therapy and exhibited a 34% decrease in the proliferative index (Ki-67).10 Interestingly, Chisholm et al11 evaluated the expression of β-adrenergic expression in 141 vascular lesions; endothelial cell expression of β2-adrenergic receptors was found positive in 100% of IHs, 67% of kaposiform hemangioendotheliomas, 41% of angiosarcomas, 50% of pyogenic granulomas, and 75% of Kaposi sarcomas, to name merely a few studied lesions.
These data have spurred physicians to further seek beta-blocker dermatologic use in specific patient populations. For example, Meseguer-Yebra et al12 employed timolol solution 0.5% twice daily for 12 weeks for 2 human immunodeficiency virus–negative patients with limited Kaposi sarcoma of the right thigh and foot; no clinical evidence of recurrence was seen at 20 months, and one of the patients had a subsequent biopsy performed with negative human herpesvirus 8 staining after therapy. In the pediatric arena, topical timolol has been used for both port-wine stains and pyogenic granulomas.13-15 Two lesions of pyogenic granulomas on the scalp of a child were treated with timolol ophthalmic solution 0.5% under occlusion for 4 weeks with resolution.15 Propranolol also has been utilized as adjunctive therapy for aggressive pediatric vascular lesions such as kaposiform hemangioendothelioma with promising results and additionally reducing the duration of therapy needed with vincristine.2
In summary, propranolol and timolol have made an indelible impression on the field of pediatric dermatology and have demonstrated a burgeoning role in the dermatologic arena. The use of nonselective beta-blockers for the management of vascular lesions can serve as adjunctive or monotherapy for certain patient populations. The relatively low adverse risk profile of propranolol makes it a versatile tool to use both systemically and topically. Although the authors of the study assessing the β2-adrenergic expression in vascular lesions admittedly stated that the positivity of the receptors does not necessarily correlate with therapeutic management, it is an interesting subject area with much potential in the future.11 This review serves to illuminate the expanding role of beta-blockers in dermatology.
- Léauté-Labrèze C, Dumas de la Roque E, Hubiche T, et al. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358:2649-2651.
- Hermans DJ, van Beynum IM, van der Vijver RJ, et al. Kaposiform hemangioendothelioma with Kasabach-Merritt syndrome: a new indication for propranolol treatment. J Pediatr Hematol Oncol. 2011;33:E171-E173.
- Guo S, Ni N. Topical treatment for capillary hemangioma of the eyelid using beta-blocker solution. Arch Ophthalmol. 2010;128:255-256.
- Püttgen K, Lucky A, Adams D, et al. Topical timolol maleate treatment of infantile hemangiomas. Pediatrics. 2016;138:3.
- Drolet BA, Frommelt PC, Chamlin SL, et al. Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics. 2013;131:128-140.
- Hogeling M, Adams S, Wargon O. A randomized controlled trial of propranolol for infantile hemangiomas [published online July 25, 2011]. Pediatrics. 2011;128:E259-E266.
- Léauté-Labrèze C, Dumas de la Roque E, Nacka F, et al. Doubleblind randomized pilot trial evaluating the efficacy of oral propranolol on infantile haemangiomas in infants < 4 months of age. Br J Dermatol. 2013;169:181-183.
- Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J, et al. A randomized, controlled trial of oral propranolol in infantile hemangioma. N Engl J Med. 2015;372:735-746.
- Shelley WB, Shelley ED. Adrenergic urticaria: a new form of stress induced hives. Lancet. 1985;2:1031-1033.
- Chow W, Amaya CN, Rains S, et al. Growth attenuation of cutaneous angiosarcoma with propranolol-mediated β-blockade. JAMA Dermatol. 2015;151:1226-1229.
- Chisholm KM, Chang KW, Truong MT, et al. β-adrenergic receptor expression in vascular tumors. Mod Pathol. 2012;25:1446-1451.
- Meseguer-Yebra C, Cardeñoso-Álvarez, ME, Bordel-Gómez MT, et al. Successful treatment of classic Kaposi sarcoma with topical timolol: report of two cases. Br J Dermatol. 2015;173:860-862.
- Passeron T, Maza A, Fontas E, et al. Treatment of port wine stains and pulsed dye laser and topical timolol: a multicenter randomized controlled trial. Br J Dermatol. 2014;170:1350-1353.
- Wine LL, Goff KL, Lam JM, et al. Treatment of pediatric pyogenic granulomas using β-adrenergic receptor antagonist. Pediatr Dermatol. 2014;31:203-207.
- Knöpfel N, Escudero-Góngora Mdel M, Bauzà A, et al. Timolol for the treatment of pyogenic granuloma (PG) in children. J Am Acad Dermatol. 2016;75:E105-E106.
Since the serendipitous discovery of expedited involution of infantile hemangiomas (IHs) with propranolol in 2008,1 current research has proliferated to discern the mechanism of action of beta-blockers in the care of IHs. Propranolol is a nonselective beta-blocker with a structure similar to catecholamines and thus competes for β-adrenergic receptors. Blocking β1-receptors is cardioselective, leading to decreased heart rate and myocardial contractility, while blocking β2-receptors leads to inhibition of smooth muscle relaxation and decreased glycogenolysis. The endothelial cells of IH express β2-adrenergic receptors; the mechanistic role of propranolol in these lesions is surmised to be due to vasoconstriction, decreased angiogenesis through inhibition of vascular endothelial growth factor, and subsequent endothelial cell apoptosis.2
After this breakthrough finding, a subsequent novel development was made when an ophthalmologist demonstrated that timolol, a topical beta-blocker, could be utilized to expedite IH involution and prevent ocular complications such as amblyopia secondary to the mass effect of the lesion. Guo and Ni3 prescribed the commercially available ophthalmologic solution of timolol maleate 0.5% for twice-daily use for 5 weeks. Remarkable reduction in the periorbital IH without rebound phenomenon was observed.3 A recent multicenter retrospective cohort of more than 700 patients with IH were treated with topical timolol with a 70% success rate, corresponding to 10% improvement from baseline; this study highlights the efficacy of timolol while confirming the safety of the medication.4
Systemic beta-blockers for IH have been used predominately for critical sites such as the nasal tip, lip, ear, perineum, and periocular area; ulcerated lesions or those that may be prone to leave a fibrofatty tissue residue after involution also have been targeted. Contraindications for use include premature infants younger than 5 weeks, infants weighing less than 2 kg, history of asthma or bronchospasm, heart rate less than 80 beats per minute, blood pressure less than 50/30 mm Hg, or hypersensitivity to the medication.5 Current guidelines for propranolol initiation vary; some dermatologists consult cardiology prior to initiation, while others perform routine vitals and an indication-driven electrocardiogram as needed based on family history of cardiac disease, maternal history of connective tissue disease, congenital heart block, or abnormal vital signs.
Given the demonstrated long-term safety of propranolol and the acceptable side-effect profile, the use of beta-blockers for IH has become increasingly mainstream. Three randomized controlled trials (RCTs) have evaluated the efficacy and minimal adverse effects of propranolol for IH. The first RCT evaluated 40 patients who received either placebo or propranolol 2 mg/kg daily (divided into 3 doses) for 6 months; IH growth stopped by week 4 in the treatment group and the largest volume difference in IH was seen at week 12.6 Léauté-Labrèze et al7 demonstrated that propranolol could be given earlier to patients and at higher doses; the treatment group included 7 patients at 3 mg/kg daily of propranolol for 15 days, followed by 15 additional days of 4 mg/kg daily of propranolol. A statistically significant (P=.004) decrease in IH volume, quantified by use of ultrasonography, was exhibited by the propranolol group.7 Lastly, the largest RCT (N=456) established the efficacy of propranolol 3 mg/kg daily for 6 months with a 60% successful treatment rate compared to 4% for patients receiving placebo.8
Given the efficacy of propranolol for IH, other investigators have experimented with nonselective beta-blockers for other dermatologic conditions. In addition to second-line use for flushing, hyperhidrosis, and adrenergic urticaria, the future of propranolol is expanding for vascular lesions in particular.9 Chow et al10 highlighted a case of progressive angiosarcoma of the scalp that responded to propranolol hydrochloride therapy at 40 mg 3 times daily with extensive regression; propranolol was given in addition to chemotherapy and radiation. The tumor was biopsied before and after propranolol therapy and exhibited a 34% decrease in the proliferative index (Ki-67).10 Interestingly, Chisholm et al11 evaluated the expression of β-adrenergic expression in 141 vascular lesions; endothelial cell expression of β2-adrenergic receptors was found positive in 100% of IHs, 67% of kaposiform hemangioendotheliomas, 41% of angiosarcomas, 50% of pyogenic granulomas, and 75% of Kaposi sarcomas, to name merely a few studied lesions.
These data have spurred physicians to further seek beta-blocker dermatologic use in specific patient populations. For example, Meseguer-Yebra et al12 employed timolol solution 0.5% twice daily for 12 weeks for 2 human immunodeficiency virus–negative patients with limited Kaposi sarcoma of the right thigh and foot; no clinical evidence of recurrence was seen at 20 months, and one of the patients had a subsequent biopsy performed with negative human herpesvirus 8 staining after therapy. In the pediatric arena, topical timolol has been used for both port-wine stains and pyogenic granulomas.13-15 Two lesions of pyogenic granulomas on the scalp of a child were treated with timolol ophthalmic solution 0.5% under occlusion for 4 weeks with resolution.15 Propranolol also has been utilized as adjunctive therapy for aggressive pediatric vascular lesions such as kaposiform hemangioendothelioma with promising results and additionally reducing the duration of therapy needed with vincristine.2
In summary, propranolol and timolol have made an indelible impression on the field of pediatric dermatology and have demonstrated a burgeoning role in the dermatologic arena. The use of nonselective beta-blockers for the management of vascular lesions can serve as adjunctive or monotherapy for certain patient populations. The relatively low adverse risk profile of propranolol makes it a versatile tool to use both systemically and topically. Although the authors of the study assessing the β2-adrenergic expression in vascular lesions admittedly stated that the positivity of the receptors does not necessarily correlate with therapeutic management, it is an interesting subject area with much potential in the future.11 This review serves to illuminate the expanding role of beta-blockers in dermatology.
Since the serendipitous discovery of expedited involution of infantile hemangiomas (IHs) with propranolol in 2008,1 current research has proliferated to discern the mechanism of action of beta-blockers in the care of IHs. Propranolol is a nonselective beta-blocker with a structure similar to catecholamines and thus competes for β-adrenergic receptors. Blocking β1-receptors is cardioselective, leading to decreased heart rate and myocardial contractility, while blocking β2-receptors leads to inhibition of smooth muscle relaxation and decreased glycogenolysis. The endothelial cells of IH express β2-adrenergic receptors; the mechanistic role of propranolol in these lesions is surmised to be due to vasoconstriction, decreased angiogenesis through inhibition of vascular endothelial growth factor, and subsequent endothelial cell apoptosis.2
After this breakthrough finding, a subsequent novel development was made when an ophthalmologist demonstrated that timolol, a topical beta-blocker, could be utilized to expedite IH involution and prevent ocular complications such as amblyopia secondary to the mass effect of the lesion. Guo and Ni3 prescribed the commercially available ophthalmologic solution of timolol maleate 0.5% for twice-daily use for 5 weeks. Remarkable reduction in the periorbital IH without rebound phenomenon was observed.3 A recent multicenter retrospective cohort of more than 700 patients with IH were treated with topical timolol with a 70% success rate, corresponding to 10% improvement from baseline; this study highlights the efficacy of timolol while confirming the safety of the medication.4
Systemic beta-blockers for IH have been used predominately for critical sites such as the nasal tip, lip, ear, perineum, and periocular area; ulcerated lesions or those that may be prone to leave a fibrofatty tissue residue after involution also have been targeted. Contraindications for use include premature infants younger than 5 weeks, infants weighing less than 2 kg, history of asthma or bronchospasm, heart rate less than 80 beats per minute, blood pressure less than 50/30 mm Hg, or hypersensitivity to the medication.5 Current guidelines for propranolol initiation vary; some dermatologists consult cardiology prior to initiation, while others perform routine vitals and an indication-driven electrocardiogram as needed based on family history of cardiac disease, maternal history of connective tissue disease, congenital heart block, or abnormal vital signs.
Given the demonstrated long-term safety of propranolol and the acceptable side-effect profile, the use of beta-blockers for IH has become increasingly mainstream. Three randomized controlled trials (RCTs) have evaluated the efficacy and minimal adverse effects of propranolol for IH. The first RCT evaluated 40 patients who received either placebo or propranolol 2 mg/kg daily (divided into 3 doses) for 6 months; IH growth stopped by week 4 in the treatment group and the largest volume difference in IH was seen at week 12.6 Léauté-Labrèze et al7 demonstrated that propranolol could be given earlier to patients and at higher doses; the treatment group included 7 patients at 3 mg/kg daily of propranolol for 15 days, followed by 15 additional days of 4 mg/kg daily of propranolol. A statistically significant (P=.004) decrease in IH volume, quantified by use of ultrasonography, was exhibited by the propranolol group.7 Lastly, the largest RCT (N=456) established the efficacy of propranolol 3 mg/kg daily for 6 months with a 60% successful treatment rate compared to 4% for patients receiving placebo.8
Given the efficacy of propranolol for IH, other investigators have experimented with nonselective beta-blockers for other dermatologic conditions. In addition to second-line use for flushing, hyperhidrosis, and adrenergic urticaria, the future of propranolol is expanding for vascular lesions in particular.9 Chow et al10 highlighted a case of progressive angiosarcoma of the scalp that responded to propranolol hydrochloride therapy at 40 mg 3 times daily with extensive regression; propranolol was given in addition to chemotherapy and radiation. The tumor was biopsied before and after propranolol therapy and exhibited a 34% decrease in the proliferative index (Ki-67).10 Interestingly, Chisholm et al11 evaluated the expression of β-adrenergic expression in 141 vascular lesions; endothelial cell expression of β2-adrenergic receptors was found positive in 100% of IHs, 67% of kaposiform hemangioendotheliomas, 41% of angiosarcomas, 50% of pyogenic granulomas, and 75% of Kaposi sarcomas, to name merely a few studied lesions.
These data have spurred physicians to further seek beta-blocker dermatologic use in specific patient populations. For example, Meseguer-Yebra et al12 employed timolol solution 0.5% twice daily for 12 weeks for 2 human immunodeficiency virus–negative patients with limited Kaposi sarcoma of the right thigh and foot; no clinical evidence of recurrence was seen at 20 months, and one of the patients had a subsequent biopsy performed with negative human herpesvirus 8 staining after therapy. In the pediatric arena, topical timolol has been used for both port-wine stains and pyogenic granulomas.13-15 Two lesions of pyogenic granulomas on the scalp of a child were treated with timolol ophthalmic solution 0.5% under occlusion for 4 weeks with resolution.15 Propranolol also has been utilized as adjunctive therapy for aggressive pediatric vascular lesions such as kaposiform hemangioendothelioma with promising results and additionally reducing the duration of therapy needed with vincristine.2
In summary, propranolol and timolol have made an indelible impression on the field of pediatric dermatology and have demonstrated a burgeoning role in the dermatologic arena. The use of nonselective beta-blockers for the management of vascular lesions can serve as adjunctive or monotherapy for certain patient populations. The relatively low adverse risk profile of propranolol makes it a versatile tool to use both systemically and topically. Although the authors of the study assessing the β2-adrenergic expression in vascular lesions admittedly stated that the positivity of the receptors does not necessarily correlate with therapeutic management, it is an interesting subject area with much potential in the future.11 This review serves to illuminate the expanding role of beta-blockers in dermatology.
- Léauté-Labrèze C, Dumas de la Roque E, Hubiche T, et al. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358:2649-2651.
- Hermans DJ, van Beynum IM, van der Vijver RJ, et al. Kaposiform hemangioendothelioma with Kasabach-Merritt syndrome: a new indication for propranolol treatment. J Pediatr Hematol Oncol. 2011;33:E171-E173.
- Guo S, Ni N. Topical treatment for capillary hemangioma of the eyelid using beta-blocker solution. Arch Ophthalmol. 2010;128:255-256.
- Püttgen K, Lucky A, Adams D, et al. Topical timolol maleate treatment of infantile hemangiomas. Pediatrics. 2016;138:3.
- Drolet BA, Frommelt PC, Chamlin SL, et al. Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics. 2013;131:128-140.
- Hogeling M, Adams S, Wargon O. A randomized controlled trial of propranolol for infantile hemangiomas [published online July 25, 2011]. Pediatrics. 2011;128:E259-E266.
- Léauté-Labrèze C, Dumas de la Roque E, Nacka F, et al. Doubleblind randomized pilot trial evaluating the efficacy of oral propranolol on infantile haemangiomas in infants < 4 months of age. Br J Dermatol. 2013;169:181-183.
- Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J, et al. A randomized, controlled trial of oral propranolol in infantile hemangioma. N Engl J Med. 2015;372:735-746.
- Shelley WB, Shelley ED. Adrenergic urticaria: a new form of stress induced hives. Lancet. 1985;2:1031-1033.
- Chow W, Amaya CN, Rains S, et al. Growth attenuation of cutaneous angiosarcoma with propranolol-mediated β-blockade. JAMA Dermatol. 2015;151:1226-1229.
- Chisholm KM, Chang KW, Truong MT, et al. β-adrenergic receptor expression in vascular tumors. Mod Pathol. 2012;25:1446-1451.
- Meseguer-Yebra C, Cardeñoso-Álvarez, ME, Bordel-Gómez MT, et al. Successful treatment of classic Kaposi sarcoma with topical timolol: report of two cases. Br J Dermatol. 2015;173:860-862.
- Passeron T, Maza A, Fontas E, et al. Treatment of port wine stains and pulsed dye laser and topical timolol: a multicenter randomized controlled trial. Br J Dermatol. 2014;170:1350-1353.
- Wine LL, Goff KL, Lam JM, et al. Treatment of pediatric pyogenic granulomas using β-adrenergic receptor antagonist. Pediatr Dermatol. 2014;31:203-207.
- Knöpfel N, Escudero-Góngora Mdel M, Bauzà A, et al. Timolol for the treatment of pyogenic granuloma (PG) in children. J Am Acad Dermatol. 2016;75:E105-E106.
- Léauté-Labrèze C, Dumas de la Roque E, Hubiche T, et al. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358:2649-2651.
- Hermans DJ, van Beynum IM, van der Vijver RJ, et al. Kaposiform hemangioendothelioma with Kasabach-Merritt syndrome: a new indication for propranolol treatment. J Pediatr Hematol Oncol. 2011;33:E171-E173.
- Guo S, Ni N. Topical treatment for capillary hemangioma of the eyelid using beta-blocker solution. Arch Ophthalmol. 2010;128:255-256.
- Püttgen K, Lucky A, Adams D, et al. Topical timolol maleate treatment of infantile hemangiomas. Pediatrics. 2016;138:3.
- Drolet BA, Frommelt PC, Chamlin SL, et al. Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics. 2013;131:128-140.
- Hogeling M, Adams S, Wargon O. A randomized controlled trial of propranolol for infantile hemangiomas [published online July 25, 2011]. Pediatrics. 2011;128:E259-E266.
- Léauté-Labrèze C, Dumas de la Roque E, Nacka F, et al. Doubleblind randomized pilot trial evaluating the efficacy of oral propranolol on infantile haemangiomas in infants < 4 months of age. Br J Dermatol. 2013;169:181-183.
- Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J, et al. A randomized, controlled trial of oral propranolol in infantile hemangioma. N Engl J Med. 2015;372:735-746.
- Shelley WB, Shelley ED. Adrenergic urticaria: a new form of stress induced hives. Lancet. 1985;2:1031-1033.
- Chow W, Amaya CN, Rains S, et al. Growth attenuation of cutaneous angiosarcoma with propranolol-mediated β-blockade. JAMA Dermatol. 2015;151:1226-1229.
- Chisholm KM, Chang KW, Truong MT, et al. β-adrenergic receptor expression in vascular tumors. Mod Pathol. 2012;25:1446-1451.
- Meseguer-Yebra C, Cardeñoso-Álvarez, ME, Bordel-Gómez MT, et al. Successful treatment of classic Kaposi sarcoma with topical timolol: report of two cases. Br J Dermatol. 2015;173:860-862.
- Passeron T, Maza A, Fontas E, et al. Treatment of port wine stains and pulsed dye laser and topical timolol: a multicenter randomized controlled trial. Br J Dermatol. 2014;170:1350-1353.
- Wine LL, Goff KL, Lam JM, et al. Treatment of pediatric pyogenic granulomas using β-adrenergic receptor antagonist. Pediatr Dermatol. 2014;31:203-207.
- Knöpfel N, Escudero-Góngora Mdel M, Bauzà A, et al. Timolol for the treatment of pyogenic granuloma (PG) in children. J Am Acad Dermatol. 2016;75:E105-E106.

Flesh-Colored Nodule With Underlying Sclerotic Plaque
The Diagnosis: Collision Tumor
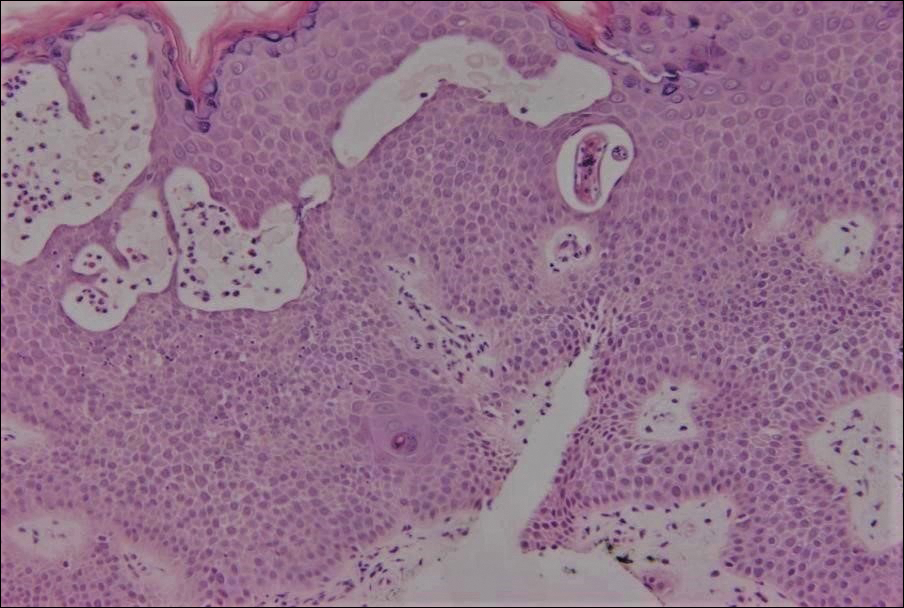
Excisional biopsy and histopathological examination demonstrated a collision tumor composed of a benign intradermal melanocytic nevus, tumor of follicular infundibulum, and an underlying sclerosing epithelial neoplasm, with a differential diagnosis of desmoplastic trichoepithelioma, morpheaform basal cell carcinoma, and microcystic adnexal carcinoma (Figure).
Common acquired melanocytic nevus presents clinically as a macule, papule, or nodule with smooth regular borders. The pigmented variant displays an evenly distributed pigment on the lesion. Intradermal melanocytic nevus often presents as a flesh-colored nodule, as in our case. Histopathologically, benign intradermal nevus typically is composed of a proliferation of melanocytes that exhibit dispersion as they go deeper in the dermis and maturation that manifests as melanocytes becoming smaller and more spindled in the deeper portions of the lesion.1 These 2 characteristics plus the bland cytology seen in the present case confirm the benign characteristic of this lesion (Figure, B).
In addition to the benign intradermal melanocytic nevus, an adjacent tumor of follicular infundibulum was noted. Tumor of follicular infundibulum is a rare adnexal tumor. It occurs frequently on the head and neck and shows some female predominance.2,3 Multiple lesions and eruptive lesions are rare forms that also have been reported.4 Histopathologically, the tumor demonstrates an epithelial plate that is present in the papillary dermis and is connected to the epidermis at multiple points with attachment to the follicular outer root sheath. Peripheral palisading is characteristically present above an eosinophilic basement membrane (Figure, A). Rare reports have documented sebaceous and eccrine differentiation.5,6
Tumor of follicular infundibulum has been reported to be associated with other tumors. Organoid nevus (nevus sebaceous), trichilemmal tumor, and fibroma have been reported to occur as a collision tumor with tumor of follicular infundibulum. An association with Cowden disease also has been described.7 Biopsies that represent partial samples should be interpreted cautiously, as step sections can reveal basal cell carcinoma.
The term sclerosing epithelial neoplasm describes tumors that share a paisley tielike epithelial pattern and sclerotic stroma. Small specimens often require clinicopathologic correlation (Figure, C). The differential diagnosis includes morpheaform basal cell carcinoma, desmoplastic trichoepithelioma, and microcystic adnexal carcinoma. A panel of stains using Ber-EP4, PHLDA1, cytokeratin 15, and cytokeratin 19 has been proposed to help differentiate these entities.8 CD34 and cytokeratin 20 also have been used with varying success in small specimens.9,10
- Ferringer T, Peckham S, Ko CJ, et al. Melanocytic neoplasms. In: Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:105-109.
- Headington JT. Tumors of the hair follicle. Am J Pathol. 1976;85:480-505.
- Davis DA, Cohen PR. Hair follicle nevus: case report and review of the literature. Pediatr Dermatol. 1996;13:135-138.
- Ikeda S, Kawada J, Yaguchi H, et al. A case of unilateral, systematized linear hair follicle nevi associated with epidermal nevus-like lesions. Dermatology. 2003;206:172-174.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Mahalingam M, Bhawan J, Finn R, et al. Tumor of the follicular infundibulum with sebaceous differentiation. J Cutan Pathol. 2001;28:314-317.
- Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
- Sellheyer K, Nelson P, Kutzner H, et al. The immunohistochemical differential diagnosis of microcystic adnexal carcinoma, desmoplastic trichoepithelioma and morpheaform basal cell carcinoma using BerEP4 and stem cell markers. J Cutan Pathol. 2013;40:363-370.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
The Diagnosis: Collision Tumor
Excisional biopsy and histopathological examination demonstrated a collision tumor composed of a benign intradermal melanocytic nevus, tumor of follicular infundibulum, and an underlying sclerosing epithelial neoplasm, with a differential diagnosis of desmoplastic trichoepithelioma, morpheaform basal cell carcinoma, and microcystic adnexal carcinoma (Figure).
Common acquired melanocytic nevus presents clinically as a macule, papule, or nodule with smooth regular borders. The pigmented variant displays an evenly distributed pigment on the lesion. Intradermal melanocytic nevus often presents as a flesh-colored nodule, as in our case. Histopathologically, benign intradermal nevus typically is composed of a proliferation of melanocytes that exhibit dispersion as they go deeper in the dermis and maturation that manifests as melanocytes becoming smaller and more spindled in the deeper portions of the lesion.1 These 2 characteristics plus the bland cytology seen in the present case confirm the benign characteristic of this lesion (Figure, B).
In addition to the benign intradermal melanocytic nevus, an adjacent tumor of follicular infundibulum was noted. Tumor of follicular infundibulum is a rare adnexal tumor. It occurs frequently on the head and neck and shows some female predominance.2,3 Multiple lesions and eruptive lesions are rare forms that also have been reported.4 Histopathologically, the tumor demonstrates an epithelial plate that is present in the papillary dermis and is connected to the epidermis at multiple points with attachment to the follicular outer root sheath. Peripheral palisading is characteristically present above an eosinophilic basement membrane (Figure, A). Rare reports have documented sebaceous and eccrine differentiation.5,6
Tumor of follicular infundibulum has been reported to be associated with other tumors. Organoid nevus (nevus sebaceous), trichilemmal tumor, and fibroma have been reported to occur as a collision tumor with tumor of follicular infundibulum. An association with Cowden disease also has been described.7 Biopsies that represent partial samples should be interpreted cautiously, as step sections can reveal basal cell carcinoma.
The term sclerosing epithelial neoplasm describes tumors that share a paisley tielike epithelial pattern and sclerotic stroma. Small specimens often require clinicopathologic correlation (Figure, C). The differential diagnosis includes morpheaform basal cell carcinoma, desmoplastic trichoepithelioma, and microcystic adnexal carcinoma. A panel of stains using Ber-EP4, PHLDA1, cytokeratin 15, and cytokeratin 19 has been proposed to help differentiate these entities.8 CD34 and cytokeratin 20 also have been used with varying success in small specimens.9,10
The Diagnosis: Collision Tumor
Excisional biopsy and histopathological examination demonstrated a collision tumor composed of a benign intradermal melanocytic nevus, tumor of follicular infundibulum, and an underlying sclerosing epithelial neoplasm, with a differential diagnosis of desmoplastic trichoepithelioma, morpheaform basal cell carcinoma, and microcystic adnexal carcinoma (Figure).
Common acquired melanocytic nevus presents clinically as a macule, papule, or nodule with smooth regular borders. The pigmented variant displays an evenly distributed pigment on the lesion. Intradermal melanocytic nevus often presents as a flesh-colored nodule, as in our case. Histopathologically, benign intradermal nevus typically is composed of a proliferation of melanocytes that exhibit dispersion as they go deeper in the dermis and maturation that manifests as melanocytes becoming smaller and more spindled in the deeper portions of the lesion.1 These 2 characteristics plus the bland cytology seen in the present case confirm the benign characteristic of this lesion (Figure, B).
In addition to the benign intradermal melanocytic nevus, an adjacent tumor of follicular infundibulum was noted. Tumor of follicular infundibulum is a rare adnexal tumor. It occurs frequently on the head and neck and shows some female predominance.2,3 Multiple lesions and eruptive lesions are rare forms that also have been reported.4 Histopathologically, the tumor demonstrates an epithelial plate that is present in the papillary dermis and is connected to the epidermis at multiple points with attachment to the follicular outer root sheath. Peripheral palisading is characteristically present above an eosinophilic basement membrane (Figure, A). Rare reports have documented sebaceous and eccrine differentiation.5,6
Tumor of follicular infundibulum has been reported to be associated with other tumors. Organoid nevus (nevus sebaceous), trichilemmal tumor, and fibroma have been reported to occur as a collision tumor with tumor of follicular infundibulum. An association with Cowden disease also has been described.7 Biopsies that represent partial samples should be interpreted cautiously, as step sections can reveal basal cell carcinoma.
The term sclerosing epithelial neoplasm describes tumors that share a paisley tielike epithelial pattern and sclerotic stroma. Small specimens often require clinicopathologic correlation (Figure, C). The differential diagnosis includes morpheaform basal cell carcinoma, desmoplastic trichoepithelioma, and microcystic adnexal carcinoma. A panel of stains using Ber-EP4, PHLDA1, cytokeratin 15, and cytokeratin 19 has been proposed to help differentiate these entities.8 CD34 and cytokeratin 20 also have been used with varying success in small specimens.9,10
- Ferringer T, Peckham S, Ko CJ, et al. Melanocytic neoplasms. In: Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:105-109.
- Headington JT. Tumors of the hair follicle. Am J Pathol. 1976;85:480-505.
- Davis DA, Cohen PR. Hair follicle nevus: case report and review of the literature. Pediatr Dermatol. 1996;13:135-138.
- Ikeda S, Kawada J, Yaguchi H, et al. A case of unilateral, systematized linear hair follicle nevi associated with epidermal nevus-like lesions. Dermatology. 2003;206:172-174.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Mahalingam M, Bhawan J, Finn R, et al. Tumor of the follicular infundibulum with sebaceous differentiation. J Cutan Pathol. 2001;28:314-317.
- Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
- Sellheyer K, Nelson P, Kutzner H, et al. The immunohistochemical differential diagnosis of microcystic adnexal carcinoma, desmoplastic trichoepithelioma and morpheaform basal cell carcinoma using BerEP4 and stem cell markers. J Cutan Pathol. 2013;40:363-370.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
- Ferringer T, Peckham S, Ko CJ, et al. Melanocytic neoplasms. In: Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:105-109.
- Headington JT. Tumors of the hair follicle. Am J Pathol. 1976;85:480-505.
- Davis DA, Cohen PR. Hair follicle nevus: case report and review of the literature. Pediatr Dermatol. 1996;13:135-138.
- Ikeda S, Kawada J, Yaguchi H, et al. A case of unilateral, systematized linear hair follicle nevi associated with epidermal nevus-like lesions. Dermatology. 2003;206:172-174.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Mahalingam M, Bhawan J, Finn R, et al. Tumor of the follicular infundibulum with sebaceous differentiation. J Cutan Pathol. 2001;28:314-317.
- Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
- Sellheyer K, Nelson P, Kutzner H, et al. The immunohistochemical differential diagnosis of microcystic adnexal carcinoma, desmoplastic trichoepithelioma and morpheaform basal cell carcinoma using BerEP4 and stem cell markers. J Cutan Pathol. 2013;40:363-370.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
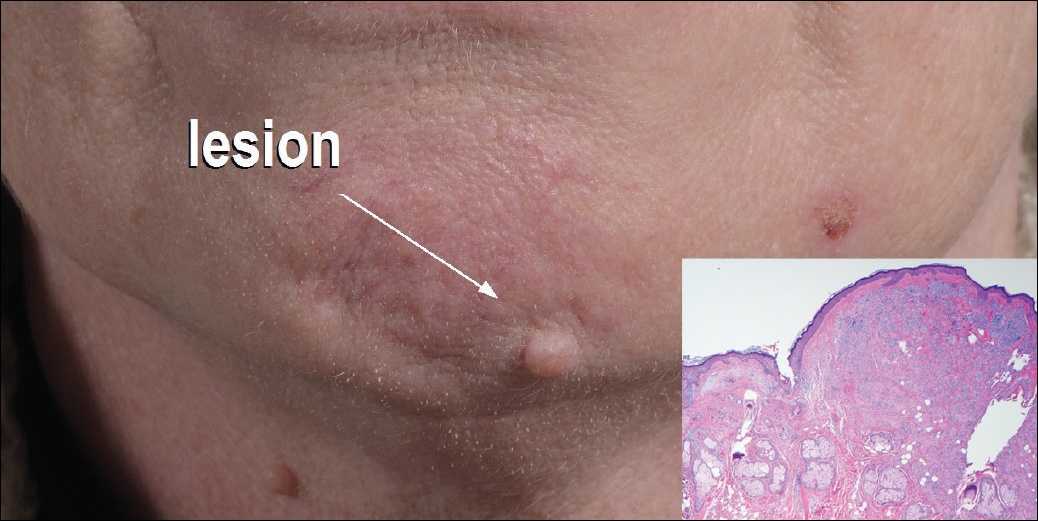
A 54-year-old man presented with a flesh-colored lesion on the chin. The nodule measured 0.6 cm in diameter. There was an underlying sclerotic plaque with indistinct borders.
Pruritic Rash on the Buttock
Cutaneous Larva Migrans
Cutaneous larva migrans (CLM) is caused by the larval migration of animal hookworms. Ancylostoma braziliense, Ancylostoma ceylanicum, and Ancylostoma caninum are the species most commonly associated with the disease. The hookworm is endemic to tropical and subtropical climates in areas such as Africa, Southeast Asia, South America, and the southeastern United States.1 Although cats and dogs are most commonly affected, humans can be infected if they are exposed to sand or soil containing hookworm larvae, often due to contamination from animal feces.2 Cutaneous larva migrans is characterized by pruritic erythematous papules and linear or serpiginous, reddish brown, elevated tracks most commonly appearing on the feet, buttocks, thighs, and lower legs; however, lesions can appear anywhere. In human hosts, the larvae travel in the epidermis and are unable to invade the dermis; it is speculated that they lack the collagenase enzymes required to penetrate the basement membrane before invading the dermis.2
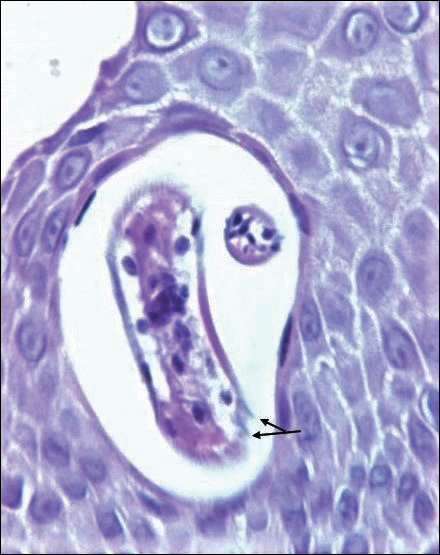
On histopathology, there typically are small cavities in the epidermis corresponding to the track of the larvae.3 There often is a spongiotic dermatitis with a mixed inflammatory infiltrate following the larvae with scattered eosinophils. The migrating larvae may be up to 1 mm in size and have bilateral double alae, or winglike projections, on the side of the body (Figure 1).4 The larvae are difficult to find on histopathology because they often travel beyond the areas that demonstrate clinical findings. The diagnosis of CLM is mostly clinical, but if a biopsy is performed, the specimen should be taken ahead of the track.
Disseminated strongyloidiasis is caused by Strongyloides stercoralis. When filariform larvae migrate out of the intestinal tract into the skin, they can cause an urticarial rash and serpiginous patterns on the skin that can move 5 to 15 cm per hour, a clinical condition known as larva currens. In immunocompromised individuals, there can be hyperinfection with diffuse petechial thumbprint purpura seen clinically, which characteristically radiate from the periumbilical area.1 On pathology, there may be numerous larvae found between the dermal collagen bundles, measuring 9 to 15 µm in diameter. Rarely, they also can be found in small blood vessels.3 They often are accompanied by extravasated red blood cells in the tissues (Figure 2).
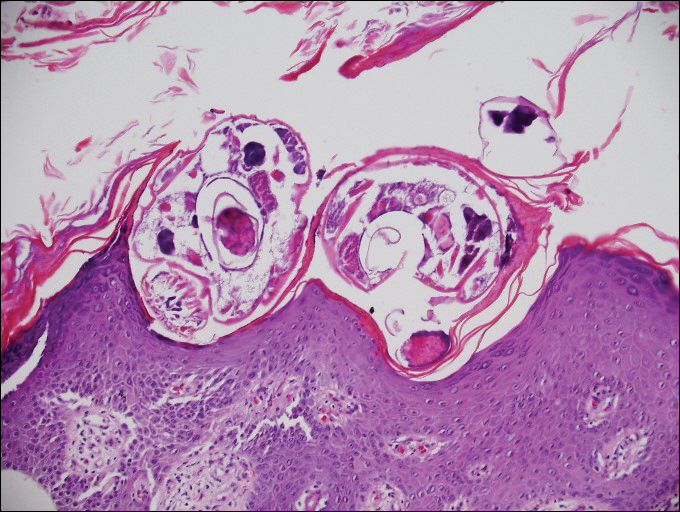
Myiasis represents the largest pathogen in the differential diagnosis for CLM. In myiasis, fly larvae will infest human tissue, usually by forming a small cavity in the dermis or subcutaneous tissue. The larvae are visible to the human eye and can be up to several centimeters in length. In the skin, the histology of myiasis usually is accompanied by a heavy mixed inflammatory cell infiltrate with many eosinophils. Fragments of the larvae are seen encased by a thick chitinous cuticle with widely spaced spines or pigmented setae (Figure 3) on the surface of the cuticle.5 Layers of striated muscle and internal organs may be seen beneath the cuticle.3

Onchocerciasis, or river blindness, is a parasitic disease caused by Onchocerca volvulus that is most often seen in sub-Saharan Africa. It may cause the skin finding of an onchocercoma, a subcutaneous nodule made up of Onchocerca nematodes. However, when the filaria disseminate, it may cause onchocerciasis with cutaneous findings of an eczematous dermatitis with itching and lichenification.1 In onchocercal dermatitis, microfilariae may be found in the dermis and there may be a mild dermal chronic inflammatory infiltrate with eosinophils.3 These microfilariae are smaller than Strongyloides larvae (Figure 4).
Sarcoptes scabiei are mites that are pathologically found limited to the stratum corneum. There often is a spongiotic dermatitis as the mite travels with an accompanying mixed cell inflammatory infiltrate with many eosinophils. One or more mites may be seen with or without eggs and excreta or scybala (Figure 5). Pink pigtails may be seen connected to the stratum corneum, representing egg fragments or casings left behind after the mite hatches.3 The female mite measures up to 0.4 mm in length.3
- Lupi O, Downing C, Lee M, et al. Mucocutaneous manifestations of helminth infections. J Am Acad Dermatol. 2015;73:929-944.
- James WD, Berger T, Elston D. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2016.
- Patterson J. Weedon's Skin Pathology. 4th ed. London, England: Churchill Livingstone Elsevier; 2016.
- Milner D. Diagnostic Pathology: Infectious Diseases. Philadelphia, PA: Elsevier; 2015.
- Ferringer T, Peckham S, Ko CJ, et al. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2013.
Cutaneous Larva Migrans
Cutaneous larva migrans (CLM) is caused by the larval migration of animal hookworms. Ancylostoma braziliense, Ancylostoma ceylanicum, and Ancylostoma caninum are the species most commonly associated with the disease. The hookworm is endemic to tropical and subtropical climates in areas such as Africa, Southeast Asia, South America, and the southeastern United States.1 Although cats and dogs are most commonly affected, humans can be infected if they are exposed to sand or soil containing hookworm larvae, often due to contamination from animal feces.2 Cutaneous larva migrans is characterized by pruritic erythematous papules and linear or serpiginous, reddish brown, elevated tracks most commonly appearing on the feet, buttocks, thighs, and lower legs; however, lesions can appear anywhere. In human hosts, the larvae travel in the epidermis and are unable to invade the dermis; it is speculated that they lack the collagenase enzymes required to penetrate the basement membrane before invading the dermis.2
On histopathology, there typically are small cavities in the epidermis corresponding to the track of the larvae.3 There often is a spongiotic dermatitis with a mixed inflammatory infiltrate following the larvae with scattered eosinophils. The migrating larvae may be up to 1 mm in size and have bilateral double alae, or winglike projections, on the side of the body (Figure 1).4 The larvae are difficult to find on histopathology because they often travel beyond the areas that demonstrate clinical findings. The diagnosis of CLM is mostly clinical, but if a biopsy is performed, the specimen should be taken ahead of the track.
Disseminated strongyloidiasis is caused by Strongyloides stercoralis. When filariform larvae migrate out of the intestinal tract into the skin, they can cause an urticarial rash and serpiginous patterns on the skin that can move 5 to 15 cm per hour, a clinical condition known as larva currens. In immunocompromised individuals, there can be hyperinfection with diffuse petechial thumbprint purpura seen clinically, which characteristically radiate from the periumbilical area.1 On pathology, there may be numerous larvae found between the dermal collagen bundles, measuring 9 to 15 µm in diameter. Rarely, they also can be found in small blood vessels.3 They often are accompanied by extravasated red blood cells in the tissues (Figure 2).
Myiasis represents the largest pathogen in the differential diagnosis for CLM. In myiasis, fly larvae will infest human tissue, usually by forming a small cavity in the dermis or subcutaneous tissue. The larvae are visible to the human eye and can be up to several centimeters in length. In the skin, the histology of myiasis usually is accompanied by a heavy mixed inflammatory cell infiltrate with many eosinophils. Fragments of the larvae are seen encased by a thick chitinous cuticle with widely spaced spines or pigmented setae (Figure 3) on the surface of the cuticle.5 Layers of striated muscle and internal organs may be seen beneath the cuticle.3
Onchocerciasis, or river blindness, is a parasitic disease caused by Onchocerca volvulus that is most often seen in sub-Saharan Africa. It may cause the skin finding of an onchocercoma, a subcutaneous nodule made up of Onchocerca nematodes. However, when the filaria disseminate, it may cause onchocerciasis with cutaneous findings of an eczematous dermatitis with itching and lichenification.1 In onchocercal dermatitis, microfilariae may be found in the dermis and there may be a mild dermal chronic inflammatory infiltrate with eosinophils.3 These microfilariae are smaller than Strongyloides larvae (Figure 4).
Sarcoptes scabiei are mites that are pathologically found limited to the stratum corneum. There often is a spongiotic dermatitis as the mite travels with an accompanying mixed cell inflammatory infiltrate with many eosinophils. One or more mites may be seen with or without eggs and excreta or scybala (Figure 5). Pink pigtails may be seen connected to the stratum corneum, representing egg fragments or casings left behind after the mite hatches.3 The female mite measures up to 0.4 mm in length.3
Cutaneous Larva Migrans
Cutaneous larva migrans (CLM) is caused by the larval migration of animal hookworms. Ancylostoma braziliense, Ancylostoma ceylanicum, and Ancylostoma caninum are the species most commonly associated with the disease. The hookworm is endemic to tropical and subtropical climates in areas such as Africa, Southeast Asia, South America, and the southeastern United States.1 Although cats and dogs are most commonly affected, humans can be infected if they are exposed to sand or soil containing hookworm larvae, often due to contamination from animal feces.2 Cutaneous larva migrans is characterized by pruritic erythematous papules and linear or serpiginous, reddish brown, elevated tracks most commonly appearing on the feet, buttocks, thighs, and lower legs; however, lesions can appear anywhere. In human hosts, the larvae travel in the epidermis and are unable to invade the dermis; it is speculated that they lack the collagenase enzymes required to penetrate the basement membrane before invading the dermis.2
On histopathology, there typically are small cavities in the epidermis corresponding to the track of the larvae.3 There often is a spongiotic dermatitis with a mixed inflammatory infiltrate following the larvae with scattered eosinophils. The migrating larvae may be up to 1 mm in size and have bilateral double alae, or winglike projections, on the side of the body (Figure 1).4 The larvae are difficult to find on histopathology because they often travel beyond the areas that demonstrate clinical findings. The diagnosis of CLM is mostly clinical, but if a biopsy is performed, the specimen should be taken ahead of the track.
Disseminated strongyloidiasis is caused by Strongyloides stercoralis. When filariform larvae migrate out of the intestinal tract into the skin, they can cause an urticarial rash and serpiginous patterns on the skin that can move 5 to 15 cm per hour, a clinical condition known as larva currens. In immunocompromised individuals, there can be hyperinfection with diffuse petechial thumbprint purpura seen clinically, which characteristically radiate from the periumbilical area.1 On pathology, there may be numerous larvae found between the dermal collagen bundles, measuring 9 to 15 µm in diameter. Rarely, they also can be found in small blood vessels.3 They often are accompanied by extravasated red blood cells in the tissues (Figure 2).
Myiasis represents the largest pathogen in the differential diagnosis for CLM. In myiasis, fly larvae will infest human tissue, usually by forming a small cavity in the dermis or subcutaneous tissue. The larvae are visible to the human eye and can be up to several centimeters in length. In the skin, the histology of myiasis usually is accompanied by a heavy mixed inflammatory cell infiltrate with many eosinophils. Fragments of the larvae are seen encased by a thick chitinous cuticle with widely spaced spines or pigmented setae (Figure 3) on the surface of the cuticle.5 Layers of striated muscle and internal organs may be seen beneath the cuticle.3
Onchocerciasis, or river blindness, is a parasitic disease caused by Onchocerca volvulus that is most often seen in sub-Saharan Africa. It may cause the skin finding of an onchocercoma, a subcutaneous nodule made up of Onchocerca nematodes. However, when the filaria disseminate, it may cause onchocerciasis with cutaneous findings of an eczematous dermatitis with itching and lichenification.1 In onchocercal dermatitis, microfilariae may be found in the dermis and there may be a mild dermal chronic inflammatory infiltrate with eosinophils.3 These microfilariae are smaller than Strongyloides larvae (Figure 4).
Sarcoptes scabiei are mites that are pathologically found limited to the stratum corneum. There often is a spongiotic dermatitis as the mite travels with an accompanying mixed cell inflammatory infiltrate with many eosinophils. One or more mites may be seen with or without eggs and excreta or scybala (Figure 5). Pink pigtails may be seen connected to the stratum corneum, representing egg fragments or casings left behind after the mite hatches.3 The female mite measures up to 0.4 mm in length.3
- Lupi O, Downing C, Lee M, et al. Mucocutaneous manifestations of helminth infections. J Am Acad Dermatol. 2015;73:929-944.
- James WD, Berger T, Elston D. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2016.
- Patterson J. Weedon's Skin Pathology. 4th ed. London, England: Churchill Livingstone Elsevier; 2016.
- Milner D. Diagnostic Pathology: Infectious Diseases. Philadelphia, PA: Elsevier; 2015.
- Ferringer T, Peckham S, Ko CJ, et al. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Lupi O, Downing C, Lee M, et al. Mucocutaneous manifestations of helminth infections. J Am Acad Dermatol. 2015;73:929-944.
- James WD, Berger T, Elston D. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2016.
- Patterson J. Weedon's Skin Pathology. 4th ed. London, England: Churchill Livingstone Elsevier; 2016.
- Milner D. Diagnostic Pathology: Infectious Diseases. Philadelphia, PA: Elsevier; 2015.
- Ferringer T, Peckham S, Ko CJ, et al. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2013.
An 18-year-old man presented with a several-week history of an expanding pruritic serpiginous and linear eruption on the buttock. The patient recently had spent some time vacationing at the beach in the southeastern United States. Physical examination revealed erythematous linear papules and serpiginous raised tracks on the buttock. A biopsy of the lesion was performed.
Eruptive Melanocytic Nevi During Azathioprine Therapy for Antisynthetase Syndrome
Case Report
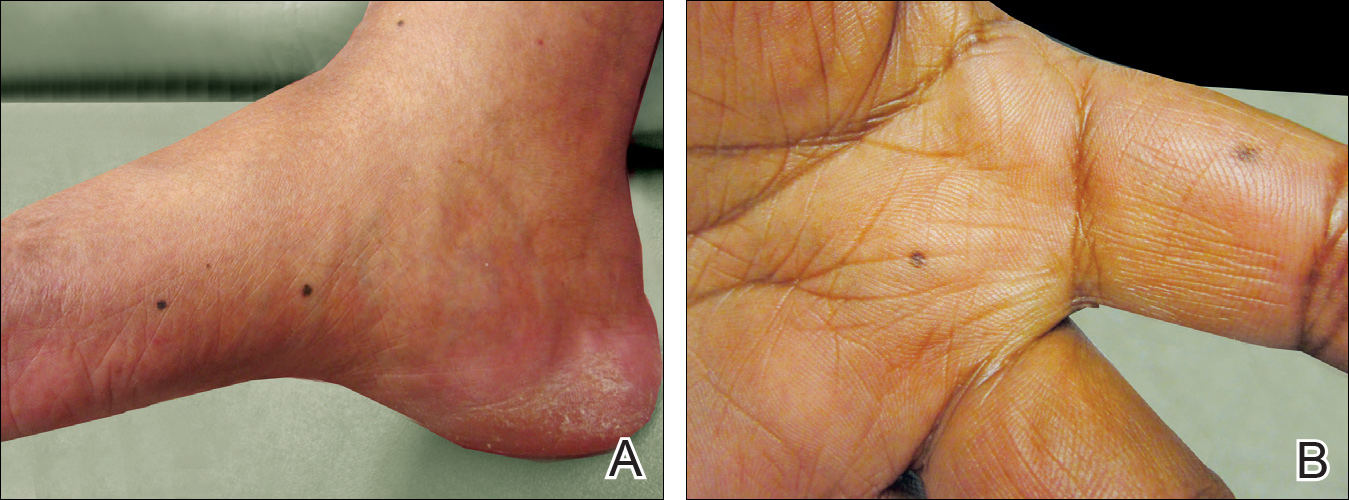
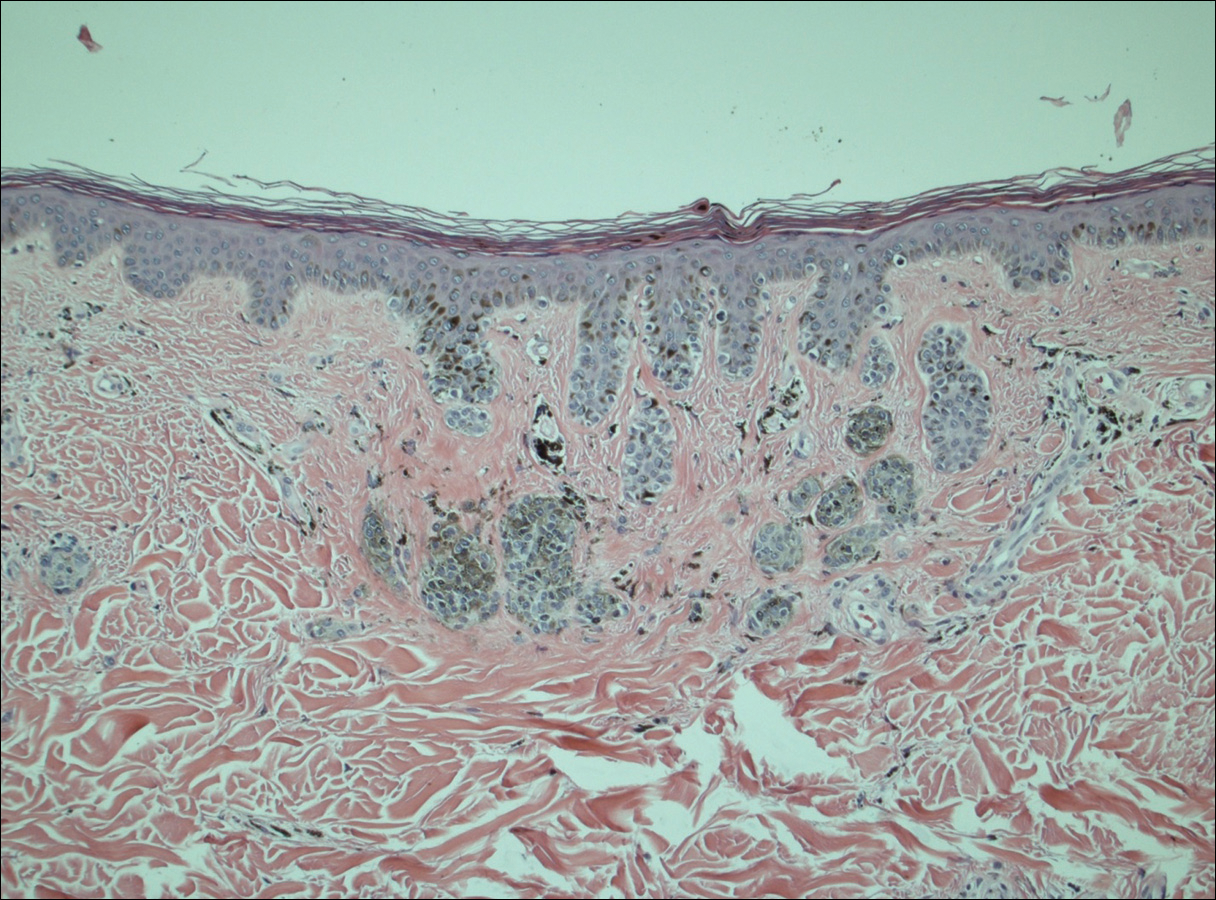
A 50-year-old man with a history of antisynthetase syndrome (positive for anti–Jo-1 polymyositis with interstitial lung disease) and sarcoidosis presented for evaluation of numerous new moles. The lesions had developed on the trunk, arms, legs, hands, and feet approximately 3 weeks after starting azathioprine 100 mg once daily for pulmonary and muscular involvement of antisynthetase syndrome. He denied any preceding cutaneous inflammation or sunburns. He had no personal or family history of skin cancer, and no family members had multiple nevi. Physical examination revealed 30 to 40 benign-appearing, 2- to 5-mm, hyperpigmented macules scattered on the medial aspect of the right foot (Figure 1A), left palm (Figure 1B), back, abdomen, chest, arms, and legs. A larger, somewhat asymmetric, irregularly bordered, and irregularly pigmented macule was noted on the left side of the upper back. A punch biopsy of the lesion revealed a benign, mildly atypical lentiginous compound nevus (Figure 2). Pathology confirmed that the lesions represented eruptive melanocytic nevi (EMN). The patient continued azathioprine therapy and was followed with regular full-body skin examinations. Mycophenolate mofetil was suggested as an alternative therapy, if clinically appropriate, though this change has not been made by the patient’s rheumatologists.
Comment
A PubMed search of articles indexed for MEDLINE using the search terms eruptive melanocytic nevi and azathioprine revealed 14 cases of EMN in the setting of azathioprine therapy, either during azathioprine monotherapy or in combination with other immunosuppressants, including systemic corticosteroids, biologics, and cyclosporine (Table).1-5 The majority of these cases occurred in renal transplant patients,1 with 3 additional cases reported in the setting of Crohn disease,2,3,5 and another in a patient with myasthenia gravis.4 Patients ranged in age from 8 to 42 years (mean age, 22 years), with lesions developing a few months to up to 7 years after starting therapy. When specified, the reported lesions typically were small, ranging from 1 to 3 mm in size, and developed rapidly over a couple of months with a predilection for the palms, soles, and trunk. Although dysplastic nevi were described in only 2 patients, melanomas were not detected.
Various hypotheses have sought to explain the largely unknown etiology of EMN. Bovenschen et al3 suggested that immunocompromised patients have diminished immune surveillance in the skin, which allows for unchecked proliferation of melanocytes. Specifically, immune suppression may induce melanocyte-stimulating hormone or melanoma growth stimulatory activity, with composition-specific growth in skin at the palms and soles.3,4 The preferential growth on the palms and soles suggests that those regions may have special sensitivity to melanocyte-stimulating hormone.4 Woodhouse and Maytin6 postulated that the increased density of eccrine sweat glands in the palms and soles as well as the absence of pilosebaceous units and apocrine glands and plentiful Pacinian and Meissner corpuscles may allow for a unique response to circulating melanocytic growth factors. Another hypothesis suggests the presence of genetic factors that allow subclinical nests of nevus cells to form, which become clinical eruptions following chemotherapy or immunosuppressive therapy.3 Azathioprine also has been suggested to induce various transcription factors that play a critical role in differentiation and proliferation of melanocytic stem cells, which leads to the formation of nevi.4 Our case and others similar to it implore that further studies be done to determine the molecular mechanism driving this phenomenon and whether a specific genetic predisposition exists that lowers the threshold for rapid proliferation of melanocytes given an immunosuppressed status.2
The risk for melanoma development in cases of EMN is unknown. Although our review of the literature did not reveal any melanomas reported in cases attributed to azathioprine, a theoretical risk exists given the established associations between melanoma and immunosuppression as well as increased numbers of nevi.6 Accordingly, these patients should be followed with regular skin examinations and biopsies of atypical-appearing lesions as indicated.2,3,5 Braun et al4 also suggested the discontinuance of azathioprine and switch to mycophenolic acid, which has not been noted to cause such eruptions; this drug was recommended in our case.
- Alaibac M, Piaserico S, Rossi CR, et al. Eruptive melanocytic nevi in patients with renal allografts: report of 10 cases with dermoscopic findings. J Am Acad Dermatol. 2003;49:1020-1022.
- Belloni FA, Piaserico S, Zattra E, et al. Dermoscopic features of eruptive melanocytic naevi in an adult patient receiving immunosuppressive therapy for Crohn’s disease. Melanoma Res. 2005;15:223-224.
- Bovenschen HJ, Tjioe M, Vermaat H, et al. Induction of eruptive benign melanocytic naevi by immune suppressive agents, including biologicals. Br J Dermatol. 2006;154:880-884.
- Braun SA, Helbig D, Frank J, et al. Eruptive melanocytic nevi during azathioprine therapy in myasthenia gravis [in German]. Hautarzt. 2012;63:756-759.
- Wonders J, De Boer N, Van Weyenberg S. Spot diagnosis: eruptive melanocytic naevi during azathioprine therapy in Crohn’s disease [published online March 6, 2012]. J Crohns Colitis. 2012;6:636.
- Woodhouse J, Maytin EV. Eruptive nevi of the palms and soles. J Am Acad Dermatol. 2005;52(5 suppl 1):S96-S100.
Case Report
A 50-year-old man with a history of antisynthetase syndrome (positive for anti–Jo-1 polymyositis with interstitial lung disease) and sarcoidosis presented for evaluation of numerous new moles. The lesions had developed on the trunk, arms, legs, hands, and feet approximately 3 weeks after starting azathioprine 100 mg once daily for pulmonary and muscular involvement of antisynthetase syndrome. He denied any preceding cutaneous inflammation or sunburns. He had no personal or family history of skin cancer, and no family members had multiple nevi. Physical examination revealed 30 to 40 benign-appearing, 2- to 5-mm, hyperpigmented macules scattered on the medial aspect of the right foot (Figure 1A), left palm (Figure 1B), back, abdomen, chest, arms, and legs. A larger, somewhat asymmetric, irregularly bordered, and irregularly pigmented macule was noted on the left side of the upper back. A punch biopsy of the lesion revealed a benign, mildly atypical lentiginous compound nevus (Figure 2). Pathology confirmed that the lesions represented eruptive melanocytic nevi (EMN). The patient continued azathioprine therapy and was followed with regular full-body skin examinations. Mycophenolate mofetil was suggested as an alternative therapy, if clinically appropriate, though this change has not been made by the patient’s rheumatologists.
Comment
A PubMed search of articles indexed for MEDLINE using the search terms eruptive melanocytic nevi and azathioprine revealed 14 cases of EMN in the setting of azathioprine therapy, either during azathioprine monotherapy or in combination with other immunosuppressants, including systemic corticosteroids, biologics, and cyclosporine (Table).1-5 The majority of these cases occurred in renal transplant patients,1 with 3 additional cases reported in the setting of Crohn disease,2,3,5 and another in a patient with myasthenia gravis.4 Patients ranged in age from 8 to 42 years (mean age, 22 years), with lesions developing a few months to up to 7 years after starting therapy. When specified, the reported lesions typically were small, ranging from 1 to 3 mm in size, and developed rapidly over a couple of months with a predilection for the palms, soles, and trunk. Although dysplastic nevi were described in only 2 patients, melanomas were not detected.
Various hypotheses have sought to explain the largely unknown etiology of EMN. Bovenschen et al3 suggested that immunocompromised patients have diminished immune surveillance in the skin, which allows for unchecked proliferation of melanocytes. Specifically, immune suppression may induce melanocyte-stimulating hormone or melanoma growth stimulatory activity, with composition-specific growth in skin at the palms and soles.3,4 The preferential growth on the palms and soles suggests that those regions may have special sensitivity to melanocyte-stimulating hormone.4 Woodhouse and Maytin6 postulated that the increased density of eccrine sweat glands in the palms and soles as well as the absence of pilosebaceous units and apocrine glands and plentiful Pacinian and Meissner corpuscles may allow for a unique response to circulating melanocytic growth factors. Another hypothesis suggests the presence of genetic factors that allow subclinical nests of nevus cells to form, which become clinical eruptions following chemotherapy or immunosuppressive therapy.3 Azathioprine also has been suggested to induce various transcription factors that play a critical role in differentiation and proliferation of melanocytic stem cells, which leads to the formation of nevi.4 Our case and others similar to it implore that further studies be done to determine the molecular mechanism driving this phenomenon and whether a specific genetic predisposition exists that lowers the threshold for rapid proliferation of melanocytes given an immunosuppressed status.2
The risk for melanoma development in cases of EMN is unknown. Although our review of the literature did not reveal any melanomas reported in cases attributed to azathioprine, a theoretical risk exists given the established associations between melanoma and immunosuppression as well as increased numbers of nevi.6 Accordingly, these patients should be followed with regular skin examinations and biopsies of atypical-appearing lesions as indicated.2,3,5 Braun et al4 also suggested the discontinuance of azathioprine and switch to mycophenolic acid, which has not been noted to cause such eruptions; this drug was recommended in our case.
Case Report
A 50-year-old man with a history of antisynthetase syndrome (positive for anti–Jo-1 polymyositis with interstitial lung disease) and sarcoidosis presented for evaluation of numerous new moles. The lesions had developed on the trunk, arms, legs, hands, and feet approximately 3 weeks after starting azathioprine 100 mg once daily for pulmonary and muscular involvement of antisynthetase syndrome. He denied any preceding cutaneous inflammation or sunburns. He had no personal or family history of skin cancer, and no family members had multiple nevi. Physical examination revealed 30 to 40 benign-appearing, 2- to 5-mm, hyperpigmented macules scattered on the medial aspect of the right foot (Figure 1A), left palm (Figure 1B), back, abdomen, chest, arms, and legs. A larger, somewhat asymmetric, irregularly bordered, and irregularly pigmented macule was noted on the left side of the upper back. A punch biopsy of the lesion revealed a benign, mildly atypical lentiginous compound nevus (Figure 2). Pathology confirmed that the lesions represented eruptive melanocytic nevi (EMN). The patient continued azathioprine therapy and was followed with regular full-body skin examinations. Mycophenolate mofetil was suggested as an alternative therapy, if clinically appropriate, though this change has not been made by the patient’s rheumatologists.
Comment
A PubMed search of articles indexed for MEDLINE using the search terms eruptive melanocytic nevi and azathioprine revealed 14 cases of EMN in the setting of azathioprine therapy, either during azathioprine monotherapy or in combination with other immunosuppressants, including systemic corticosteroids, biologics, and cyclosporine (Table).1-5 The majority of these cases occurred in renal transplant patients,1 with 3 additional cases reported in the setting of Crohn disease,2,3,5 and another in a patient with myasthenia gravis.4 Patients ranged in age from 8 to 42 years (mean age, 22 years), with lesions developing a few months to up to 7 years after starting therapy. When specified, the reported lesions typically were small, ranging from 1 to 3 mm in size, and developed rapidly over a couple of months with a predilection for the palms, soles, and trunk. Although dysplastic nevi were described in only 2 patients, melanomas were not detected.
Various hypotheses have sought to explain the largely unknown etiology of EMN. Bovenschen et al3 suggested that immunocompromised patients have diminished immune surveillance in the skin, which allows for unchecked proliferation of melanocytes. Specifically, immune suppression may induce melanocyte-stimulating hormone or melanoma growth stimulatory activity, with composition-specific growth in skin at the palms and soles.3,4 The preferential growth on the palms and soles suggests that those regions may have special sensitivity to melanocyte-stimulating hormone.4 Woodhouse and Maytin6 postulated that the increased density of eccrine sweat glands in the palms and soles as well as the absence of pilosebaceous units and apocrine glands and plentiful Pacinian and Meissner corpuscles may allow for a unique response to circulating melanocytic growth factors. Another hypothesis suggests the presence of genetic factors that allow subclinical nests of nevus cells to form, which become clinical eruptions following chemotherapy or immunosuppressive therapy.3 Azathioprine also has been suggested to induce various transcription factors that play a critical role in differentiation and proliferation of melanocytic stem cells, which leads to the formation of nevi.4 Our case and others similar to it implore that further studies be done to determine the molecular mechanism driving this phenomenon and whether a specific genetic predisposition exists that lowers the threshold for rapid proliferation of melanocytes given an immunosuppressed status.2
The risk for melanoma development in cases of EMN is unknown. Although our review of the literature did not reveal any melanomas reported in cases attributed to azathioprine, a theoretical risk exists given the established associations between melanoma and immunosuppression as well as increased numbers of nevi.6 Accordingly, these patients should be followed with regular skin examinations and biopsies of atypical-appearing lesions as indicated.2,3,5 Braun et al4 also suggested the discontinuance of azathioprine and switch to mycophenolic acid, which has not been noted to cause such eruptions; this drug was recommended in our case.
- Alaibac M, Piaserico S, Rossi CR, et al. Eruptive melanocytic nevi in patients with renal allografts: report of 10 cases with dermoscopic findings. J Am Acad Dermatol. 2003;49:1020-1022.
- Belloni FA, Piaserico S, Zattra E, et al. Dermoscopic features of eruptive melanocytic naevi in an adult patient receiving immunosuppressive therapy for Crohn’s disease. Melanoma Res. 2005;15:223-224.
- Bovenschen HJ, Tjioe M, Vermaat H, et al. Induction of eruptive benign melanocytic naevi by immune suppressive agents, including biologicals. Br J Dermatol. 2006;154:880-884.
- Braun SA, Helbig D, Frank J, et al. Eruptive melanocytic nevi during azathioprine therapy in myasthenia gravis [in German]. Hautarzt. 2012;63:756-759.
- Wonders J, De Boer N, Van Weyenberg S. Spot diagnosis: eruptive melanocytic naevi during azathioprine therapy in Crohn’s disease [published online March 6, 2012]. J Crohns Colitis. 2012;6:636.
- Woodhouse J, Maytin EV. Eruptive nevi of the palms and soles. J Am Acad Dermatol. 2005;52(5 suppl 1):S96-S100.
- Alaibac M, Piaserico S, Rossi CR, et al. Eruptive melanocytic nevi in patients with renal allografts: report of 10 cases with dermoscopic findings. J Am Acad Dermatol. 2003;49:1020-1022.
- Belloni FA, Piaserico S, Zattra E, et al. Dermoscopic features of eruptive melanocytic naevi in an adult patient receiving immunosuppressive therapy for Crohn’s disease. Melanoma Res. 2005;15:223-224.
- Bovenschen HJ, Tjioe M, Vermaat H, et al. Induction of eruptive benign melanocytic naevi by immune suppressive agents, including biologicals. Br J Dermatol. 2006;154:880-884.
- Braun SA, Helbig D, Frank J, et al. Eruptive melanocytic nevi during azathioprine therapy in myasthenia gravis [in German]. Hautarzt. 2012;63:756-759.
- Wonders J, De Boer N, Van Weyenberg S. Spot diagnosis: eruptive melanocytic naevi during azathioprine therapy in Crohn’s disease [published online March 6, 2012]. J Crohns Colitis. 2012;6:636.
- Woodhouse J, Maytin EV. Eruptive nevi of the palms and soles. J Am Acad Dermatol. 2005;52(5 suppl 1):S96-S100.
Practice Points
- A theoretical risk exists in the setting of eruptive melanocytic nevi (EMN) given the established associations between melanoma and immunosuppression as well as increased numbers of nevi.
- Follow patients with EMN with regular skin examinations and biopsies of atypical-appearing lesions given the increased risk for melanoma in this population.
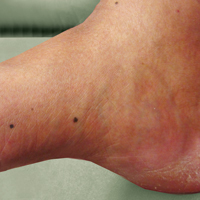
Collagenous and Elastotic Marginal Plaques of the Hands
To the Editor:
Collagenous and elastotic marginal plaques of the hands (CEMPHs) has several names including degenerative collagenous plaques of the hands, keratoelastoidosis marginalis, and digital papular calcific elastosis. This rare disorder is an acquired, slowly progressive, asymptomatic, dermal connective tissue abnormality that is underrecognized and underdiagnosed. Clinical presentation includes hyperkeratotic translucent papules arranged linearly on the radial aspect of the hands.
A 74-year-old woman described having "rough hands" of more than 20 years' duration. She presented with 4-cm wide longitudinal, erythematous, firm, depressed plaques along the lateral edge of the second finger and extending to the medial thumb in both hands (Figure 1). She had attempted multiple treatments by her primary care physician, including topical and oral medications unknown to the patient and light therapy, all without benefit over a period of several years. We have attempted salicylic acid 40%, clobetasol cream 0.05%, and emollient creams containing α-hydroxy acid. At best the condition fluctuated between a subtle raised scale at the edge to smooth and occasionally more red-pink, seemingly unrelated to any treatments.
The patient did not have plaques elsewhere on the body, and notably, the feet were clear. She did not have a history of repeated trauma to the hands and did not engage in manual labor. She denied excessive sun exposure, though she had Fitzpatrick skin type III and a history of multiple precancers and nonmelanoma skin cancers 7 years prior to presentation.
Histology of CEMPH reveals a hyperkeratotic epidermis with an avascular and acellular replacement of the superficial reticular dermis by haphazardly arranged, thickened collagen fibers (Figure 2A-2C). Collagen fibers were oriented perpendicularly to the epidermal surface. Intervening amorphous basophilic elastotic masses were present in the upper dermis with occasional calcification and degenerative elastic fibers (Figure 2D).

Collagenous and elastotic marginal plaques of the hands is a chronic, asymptomatic, sclerotic skin disorder described in a 1960 case series of 5 patients reported by Burks et al.1 Although it has many names, the most common is CEMPH. Collagenous and elastotic marginal plaques of the hands most often presents in white men aged 50 to 60 years.2 Patients typically are asymptomatic with plaques limited to the junction of the palmar and dorsal surfaces of the hands with only minimal intermittent stiffness around the flexor creases. Lesions begin as discrete yellow papules that coalesce to form hyperkeratotic linear plaques with occasional telangiectasia.3
The etiology of CEMPH is attributed to collagen and elastin degeneration by chronic actinic damage, pressure, or trauma.4,5 The 3 stages of degeneration include an initial linear padded stage, an intermediate padded plaque stage, and an advanced padded hyperkeratotic plaque stage.4 Vascular compromise is seen from the enlarged and fused thickened collagen and elastic fibers that in turn lead to ischemic changes, hyperkeratosis with epidermal atrophy, and papillary dermis telangiectasia. Absence or weak expression of keratins 14 and 10 and strong expression of keratin 16 have been reported in the epidermis of CEMPH patients.4
Collagenous and elastotic marginal plaques of the hands do not have a specific treatment, as it is a benign, slowly progressive condition. Several treatments such as laser therapy, high-potency topical corticosteroids, topical tazarotene and tretinoin, oral isotretinoin, and cryotherapy have been tried with little long-term success.4 Moisturizing may help reduce fissuring, and patients are advised to avoid the sun and repeated trauma to the hands.
The differential diagnosis of CEMPH is summarized in the Table. Two genodermatoses—acrokeratoelastoidosis of Costa and focal acral hyperkeratosis—clinically resemble CEMPH. Acrokeratoelastoidosis of Costa is an autosomal-dominant condition that occurs without trauma in children and young adults. Histopathology shows orthokeratotic hyperkeratosis due to an overproduction of filaggrin in the granular layer of the epidermis. The reticular dermis shows basophilic, thick, curled and fragmented elastic fibers with dilated capillaries that can be seen with Weigert elastic, Verhoeff-van Gieson, or orcein stains. Focal acral hyperkeratosis occurs on the hands and feet, predominantly in black patients. On histology, the epidermis shows a characteristic orthohyperkeratosis, moderate acanthosis, and slight hypergranulosis with no dermal involvment.6
Chronic hyperkeratotic eczematous dermatitis is another common entity in the differential characterized by hyperkeratotic plaques that scale and fissure. Biopsy demonstrates a spongiotic acanthotic epidermis.7,8
Psoriasis of the hands, specifically hyperkeratotic palmoplantar psoriasis, is associated with manual labor, similar to CEMPH. Histology shows epidermal hyperplasia; regular acanthosis; loss of the granular skin layer with prominent dermal capillaries; and a mixed dermal infiltrate of lymphocytes, macrophages, and neutrophils.9 Hyperkeratotic palmoplantar lichen planus presents with pruritic papules in the third and fifth decades of life. Histologically, hyperkeratosis, acanthosis, and wedge-shaped hypergranulosis with a lichenoid lymphocytic infiltration at the dermoepidermal junction is seen.10
Palmoplantar keratodermas due to inflammatory reactive dermatoses include callosities that develop in response to repeated trauma or friction on the skin. On histology, there is prominent hyperkeratosis and acanthosis with moderate papillomatosis.11 Drug-related palmoplantar keratodermas such as those from arsenic exposure can lead to multiple, irregular, verrucous, keratotic, and pigmented lesions on the palms and soles. Histologically, atypical keratinocytes are seen in the epidermis with thick hyperkeratosis and vacuolated cells without solar elastosis.12
In conclusion, CEMPH is an underdiagnosed and underrecognized condition characterized by asymptomatic hyperkeratotic linear plaques along the medial aspect of the thumb and radial aspect of the index finger. It is important to keep CEMPH in mind when dealing with occupational cases of repeated long-term trauma or pressure to the hands as well as excessive sun exposure. It also is imperative to separate it from other diseases and avoid misdiagnosing this degenerative collagenous and elastotic disease as a malignant lesion.
- Burks JW, Wise LJ, Clark WH. Degenerative collagenous plaques of the hands. Arch Dermatol. 1960;82:362-366.
- Jordaan HF, Rossouw DJ. Digital papular calcific elastosis: a histopathological, histochemical and ultrastructural study of 20 patients. J Cutan Pathol. 1990;17:358-370.
- Mortimore RJ, Conrad RJ. Collagenous and elastotic marginal plaques of the hands. Australas J Dermatol. 2001;42:211-213.
- Tieu KD, Satter EK. Thickened plaques on the hands. Collagenous and elastotic marginal plaques of the hands (CEMPH). Arch Dermatol. 2011;147:499-504.
- Todd D, Al-Aboosi M, Hameed O, et al. The role of UV light in the pathogenesis of digital papular calcific elastosis. Arch Dermatol. 2001;137:379-381.
- Mengesha YM, Kayal JD, Swerlick RA. Keratoelastoidosis marginalis. J Cutan Med Surg. 2002;6:23-25.
- MacKee MG, Lewis MG. Keratolysis exfoliativa and the mosaic fungus. Arch Dermatol. 1931;23:445-447.
- Walling HW, Swick BL, Storrs FJ, et al. Frictional hyperkeratotic hand dermatitis responding to Grenz ray therapy. Contact Dermatitis. 2008;58:49-51.
- Farley E, Masrour S, McKey J, et al. Palmoplantar psoriasis: a phenotypical and clinical review with introduction of a new quality-of-life assessment tool. J Am Acad Dermatol. 2009;60:1024-1031.
- Rotunda AM, Craft N, Haley JC. Hyperkeratotic plaques on the palms and soles. palmoplantar lichen planus, hyperkeratotic variant. Arch Dermatol. 2004;140:1275-1280.
- Unal VS, Sevin A, Dayican A. Palmar callus formation as a result of mechanical trauma during sailing. Plast Reconstr Surg. 2005;115:2161-2162.
- Cöl M, Cöl C, Soran A, et al. Arsenic-related Bowen's disease, palmar keratosis, and skin cancer. Environ Health Perspect. 1999;107:687-689.
To the Editor:
Collagenous and elastotic marginal plaques of the hands (CEMPHs) has several names including degenerative collagenous plaques of the hands, keratoelastoidosis marginalis, and digital papular calcific elastosis. This rare disorder is an acquired, slowly progressive, asymptomatic, dermal connective tissue abnormality that is underrecognized and underdiagnosed. Clinical presentation includes hyperkeratotic translucent papules arranged linearly on the radial aspect of the hands.
A 74-year-old woman described having "rough hands" of more than 20 years' duration. She presented with 4-cm wide longitudinal, erythematous, firm, depressed plaques along the lateral edge of the second finger and extending to the medial thumb in both hands (Figure 1). She had attempted multiple treatments by her primary care physician, including topical and oral medications unknown to the patient and light therapy, all without benefit over a period of several years. We have attempted salicylic acid 40%, clobetasol cream 0.05%, and emollient creams containing α-hydroxy acid. At best the condition fluctuated between a subtle raised scale at the edge to smooth and occasionally more red-pink, seemingly unrelated to any treatments.
The patient did not have plaques elsewhere on the body, and notably, the feet were clear. She did not have a history of repeated trauma to the hands and did not engage in manual labor. She denied excessive sun exposure, though she had Fitzpatrick skin type III and a history of multiple precancers and nonmelanoma skin cancers 7 years prior to presentation.
Histology of CEMPH reveals a hyperkeratotic epidermis with an avascular and acellular replacement of the superficial reticular dermis by haphazardly arranged, thickened collagen fibers (Figure 2A-2C). Collagen fibers were oriented perpendicularly to the epidermal surface. Intervening amorphous basophilic elastotic masses were present in the upper dermis with occasional calcification and degenerative elastic fibers (Figure 2D).
Collagenous and elastotic marginal plaques of the hands is a chronic, asymptomatic, sclerotic skin disorder described in a 1960 case series of 5 patients reported by Burks et al.1 Although it has many names, the most common is CEMPH. Collagenous and elastotic marginal plaques of the hands most often presents in white men aged 50 to 60 years.2 Patients typically are asymptomatic with plaques limited to the junction of the palmar and dorsal surfaces of the hands with only minimal intermittent stiffness around the flexor creases. Lesions begin as discrete yellow papules that coalesce to form hyperkeratotic linear plaques with occasional telangiectasia.3
The etiology of CEMPH is attributed to collagen and elastin degeneration by chronic actinic damage, pressure, or trauma.4,5 The 3 stages of degeneration include an initial linear padded stage, an intermediate padded plaque stage, and an advanced padded hyperkeratotic plaque stage.4 Vascular compromise is seen from the enlarged and fused thickened collagen and elastic fibers that in turn lead to ischemic changes, hyperkeratosis with epidermal atrophy, and papillary dermis telangiectasia. Absence or weak expression of keratins 14 and 10 and strong expression of keratin 16 have been reported in the epidermis of CEMPH patients.4
Collagenous and elastotic marginal plaques of the hands do not have a specific treatment, as it is a benign, slowly progressive condition. Several treatments such as laser therapy, high-potency topical corticosteroids, topical tazarotene and tretinoin, oral isotretinoin, and cryotherapy have been tried with little long-term success.4 Moisturizing may help reduce fissuring, and patients are advised to avoid the sun and repeated trauma to the hands.
The differential diagnosis of CEMPH is summarized in the Table. Two genodermatoses—acrokeratoelastoidosis of Costa and focal acral hyperkeratosis—clinically resemble CEMPH. Acrokeratoelastoidosis of Costa is an autosomal-dominant condition that occurs without trauma in children and young adults. Histopathology shows orthokeratotic hyperkeratosis due to an overproduction of filaggrin in the granular layer of the epidermis. The reticular dermis shows basophilic, thick, curled and fragmented elastic fibers with dilated capillaries that can be seen with Weigert elastic, Verhoeff-van Gieson, or orcein stains. Focal acral hyperkeratosis occurs on the hands and feet, predominantly in black patients. On histology, the epidermis shows a characteristic orthohyperkeratosis, moderate acanthosis, and slight hypergranulosis with no dermal involvment.6
Chronic hyperkeratotic eczematous dermatitis is another common entity in the differential characterized by hyperkeratotic plaques that scale and fissure. Biopsy demonstrates a spongiotic acanthotic epidermis.7,8
Psoriasis of the hands, specifically hyperkeratotic palmoplantar psoriasis, is associated with manual labor, similar to CEMPH. Histology shows epidermal hyperplasia; regular acanthosis; loss of the granular skin layer with prominent dermal capillaries; and a mixed dermal infiltrate of lymphocytes, macrophages, and neutrophils.9 Hyperkeratotic palmoplantar lichen planus presents with pruritic papules in the third and fifth decades of life. Histologically, hyperkeratosis, acanthosis, and wedge-shaped hypergranulosis with a lichenoid lymphocytic infiltration at the dermoepidermal junction is seen.10
Palmoplantar keratodermas due to inflammatory reactive dermatoses include callosities that develop in response to repeated trauma or friction on the skin. On histology, there is prominent hyperkeratosis and acanthosis with moderate papillomatosis.11 Drug-related palmoplantar keratodermas such as those from arsenic exposure can lead to multiple, irregular, verrucous, keratotic, and pigmented lesions on the palms and soles. Histologically, atypical keratinocytes are seen in the epidermis with thick hyperkeratosis and vacuolated cells without solar elastosis.12
In conclusion, CEMPH is an underdiagnosed and underrecognized condition characterized by asymptomatic hyperkeratotic linear plaques along the medial aspect of the thumb and radial aspect of the index finger. It is important to keep CEMPH in mind when dealing with occupational cases of repeated long-term trauma or pressure to the hands as well as excessive sun exposure. It also is imperative to separate it from other diseases and avoid misdiagnosing this degenerative collagenous and elastotic disease as a malignant lesion.
To the Editor:
Collagenous and elastotic marginal plaques of the hands (CEMPHs) has several names including degenerative collagenous plaques of the hands, keratoelastoidosis marginalis, and digital papular calcific elastosis. This rare disorder is an acquired, slowly progressive, asymptomatic, dermal connective tissue abnormality that is underrecognized and underdiagnosed. Clinical presentation includes hyperkeratotic translucent papules arranged linearly on the radial aspect of the hands.
A 74-year-old woman described having "rough hands" of more than 20 years' duration. She presented with 4-cm wide longitudinal, erythematous, firm, depressed plaques along the lateral edge of the second finger and extending to the medial thumb in both hands (Figure 1). She had attempted multiple treatments by her primary care physician, including topical and oral medications unknown to the patient and light therapy, all without benefit over a period of several years. We have attempted salicylic acid 40%, clobetasol cream 0.05%, and emollient creams containing α-hydroxy acid. At best the condition fluctuated between a subtle raised scale at the edge to smooth and occasionally more red-pink, seemingly unrelated to any treatments.
The patient did not have plaques elsewhere on the body, and notably, the feet were clear. She did not have a history of repeated trauma to the hands and did not engage in manual labor. She denied excessive sun exposure, though she had Fitzpatrick skin type III and a history of multiple precancers and nonmelanoma skin cancers 7 years prior to presentation.
Histology of CEMPH reveals a hyperkeratotic epidermis with an avascular and acellular replacement of the superficial reticular dermis by haphazardly arranged, thickened collagen fibers (Figure 2A-2C). Collagen fibers were oriented perpendicularly to the epidermal surface. Intervening amorphous basophilic elastotic masses were present in the upper dermis with occasional calcification and degenerative elastic fibers (Figure 2D).
Collagenous and elastotic marginal plaques of the hands is a chronic, asymptomatic, sclerotic skin disorder described in a 1960 case series of 5 patients reported by Burks et al.1 Although it has many names, the most common is CEMPH. Collagenous and elastotic marginal plaques of the hands most often presents in white men aged 50 to 60 years.2 Patients typically are asymptomatic with plaques limited to the junction of the palmar and dorsal surfaces of the hands with only minimal intermittent stiffness around the flexor creases. Lesions begin as discrete yellow papules that coalesce to form hyperkeratotic linear plaques with occasional telangiectasia.3
The etiology of CEMPH is attributed to collagen and elastin degeneration by chronic actinic damage, pressure, or trauma.4,5 The 3 stages of degeneration include an initial linear padded stage, an intermediate padded plaque stage, and an advanced padded hyperkeratotic plaque stage.4 Vascular compromise is seen from the enlarged and fused thickened collagen and elastic fibers that in turn lead to ischemic changes, hyperkeratosis with epidermal atrophy, and papillary dermis telangiectasia. Absence or weak expression of keratins 14 and 10 and strong expression of keratin 16 have been reported in the epidermis of CEMPH patients.4
Collagenous and elastotic marginal plaques of the hands do not have a specific treatment, as it is a benign, slowly progressive condition. Several treatments such as laser therapy, high-potency topical corticosteroids, topical tazarotene and tretinoin, oral isotretinoin, and cryotherapy have been tried with little long-term success.4 Moisturizing may help reduce fissuring, and patients are advised to avoid the sun and repeated trauma to the hands.
The differential diagnosis of CEMPH is summarized in the Table. Two genodermatoses—acrokeratoelastoidosis of Costa and focal acral hyperkeratosis—clinically resemble CEMPH. Acrokeratoelastoidosis of Costa is an autosomal-dominant condition that occurs without trauma in children and young adults. Histopathology shows orthokeratotic hyperkeratosis due to an overproduction of filaggrin in the granular layer of the epidermis. The reticular dermis shows basophilic, thick, curled and fragmented elastic fibers with dilated capillaries that can be seen with Weigert elastic, Verhoeff-van Gieson, or orcein stains. Focal acral hyperkeratosis occurs on the hands and feet, predominantly in black patients. On histology, the epidermis shows a characteristic orthohyperkeratosis, moderate acanthosis, and slight hypergranulosis with no dermal involvment.6
Chronic hyperkeratotic eczematous dermatitis is another common entity in the differential characterized by hyperkeratotic plaques that scale and fissure. Biopsy demonstrates a spongiotic acanthotic epidermis.7,8
Psoriasis of the hands, specifically hyperkeratotic palmoplantar psoriasis, is associated with manual labor, similar to CEMPH. Histology shows epidermal hyperplasia; regular acanthosis; loss of the granular skin layer with prominent dermal capillaries; and a mixed dermal infiltrate of lymphocytes, macrophages, and neutrophils.9 Hyperkeratotic palmoplantar lichen planus presents with pruritic papules in the third and fifth decades of life. Histologically, hyperkeratosis, acanthosis, and wedge-shaped hypergranulosis with a lichenoid lymphocytic infiltration at the dermoepidermal junction is seen.10
Palmoplantar keratodermas due to inflammatory reactive dermatoses include callosities that develop in response to repeated trauma or friction on the skin. On histology, there is prominent hyperkeratosis and acanthosis with moderate papillomatosis.11 Drug-related palmoplantar keratodermas such as those from arsenic exposure can lead to multiple, irregular, verrucous, keratotic, and pigmented lesions on the palms and soles. Histologically, atypical keratinocytes are seen in the epidermis with thick hyperkeratosis and vacuolated cells without solar elastosis.12
In conclusion, CEMPH is an underdiagnosed and underrecognized condition characterized by asymptomatic hyperkeratotic linear plaques along the medial aspect of the thumb and radial aspect of the index finger. It is important to keep CEMPH in mind when dealing with occupational cases of repeated long-term trauma or pressure to the hands as well as excessive sun exposure. It also is imperative to separate it from other diseases and avoid misdiagnosing this degenerative collagenous and elastotic disease as a malignant lesion.
- Burks JW, Wise LJ, Clark WH. Degenerative collagenous plaques of the hands. Arch Dermatol. 1960;82:362-366.
- Jordaan HF, Rossouw DJ. Digital papular calcific elastosis: a histopathological, histochemical and ultrastructural study of 20 patients. J Cutan Pathol. 1990;17:358-370.
- Mortimore RJ, Conrad RJ. Collagenous and elastotic marginal plaques of the hands. Australas J Dermatol. 2001;42:211-213.
- Tieu KD, Satter EK. Thickened plaques on the hands. Collagenous and elastotic marginal plaques of the hands (CEMPH). Arch Dermatol. 2011;147:499-504.
- Todd D, Al-Aboosi M, Hameed O, et al. The role of UV light in the pathogenesis of digital papular calcific elastosis. Arch Dermatol. 2001;137:379-381.
- Mengesha YM, Kayal JD, Swerlick RA. Keratoelastoidosis marginalis. J Cutan Med Surg. 2002;6:23-25.
- MacKee MG, Lewis MG. Keratolysis exfoliativa and the mosaic fungus. Arch Dermatol. 1931;23:445-447.
- Walling HW, Swick BL, Storrs FJ, et al. Frictional hyperkeratotic hand dermatitis responding to Grenz ray therapy. Contact Dermatitis. 2008;58:49-51.
- Farley E, Masrour S, McKey J, et al. Palmoplantar psoriasis: a phenotypical and clinical review with introduction of a new quality-of-life assessment tool. J Am Acad Dermatol. 2009;60:1024-1031.
- Rotunda AM, Craft N, Haley JC. Hyperkeratotic plaques on the palms and soles. palmoplantar lichen planus, hyperkeratotic variant. Arch Dermatol. 2004;140:1275-1280.
- Unal VS, Sevin A, Dayican A. Palmar callus formation as a result of mechanical trauma during sailing. Plast Reconstr Surg. 2005;115:2161-2162.
- Cöl M, Cöl C, Soran A, et al. Arsenic-related Bowen's disease, palmar keratosis, and skin cancer. Environ Health Perspect. 1999;107:687-689.
- Burks JW, Wise LJ, Clark WH. Degenerative collagenous plaques of the hands. Arch Dermatol. 1960;82:362-366.
- Jordaan HF, Rossouw DJ. Digital papular calcific elastosis: a histopathological, histochemical and ultrastructural study of 20 patients. J Cutan Pathol. 1990;17:358-370.
- Mortimore RJ, Conrad RJ. Collagenous and elastotic marginal plaques of the hands. Australas J Dermatol. 2001;42:211-213.
- Tieu KD, Satter EK. Thickened plaques on the hands. Collagenous and elastotic marginal plaques of the hands (CEMPH). Arch Dermatol. 2011;147:499-504.
- Todd D, Al-Aboosi M, Hameed O, et al. The role of UV light in the pathogenesis of digital papular calcific elastosis. Arch Dermatol. 2001;137:379-381.
- Mengesha YM, Kayal JD, Swerlick RA. Keratoelastoidosis marginalis. J Cutan Med Surg. 2002;6:23-25.
- MacKee MG, Lewis MG. Keratolysis exfoliativa and the mosaic fungus. Arch Dermatol. 1931;23:445-447.
- Walling HW, Swick BL, Storrs FJ, et al. Frictional hyperkeratotic hand dermatitis responding to Grenz ray therapy. Contact Dermatitis. 2008;58:49-51.
- Farley E, Masrour S, McKey J, et al. Palmoplantar psoriasis: a phenotypical and clinical review with introduction of a new quality-of-life assessment tool. J Am Acad Dermatol. 2009;60:1024-1031.
- Rotunda AM, Craft N, Haley JC. Hyperkeratotic plaques on the palms and soles. palmoplantar lichen planus, hyperkeratotic variant. Arch Dermatol. 2004;140:1275-1280.
- Unal VS, Sevin A, Dayican A. Palmar callus formation as a result of mechanical trauma during sailing. Plast Reconstr Surg. 2005;115:2161-2162.
- Cöl M, Cöl C, Soran A, et al. Arsenic-related Bowen's disease, palmar keratosis, and skin cancer. Environ Health Perspect. 1999;107:687-689.
Practice Points
- The etiology of collagenous and elastotic marginal plaques of the hands (CEMPHs) is attributed to collagen and elastin degeneration by chronic actinic damage, pressure, or trauma.
- It is important to keep CEMPH in mind when dealing with occupational cases of repeated long-term trauma or pressure to the hands as well as excessive sun exposure. It should be separated from other diseases and avoid being misdiagnosed as a malignant lesion.
Bluish Gray Hyperpigmentation on the Face and Neck
The Diagnosis: Erythema Dyschromicum Perstans
Erythema dyschromicum perstans (EDP), also referred to as ashy dermatosis, was first described by Ramirez1 in 1957 who labeled the patients los cenicientos (the ashen ones). It preferentially affects women in the second decade of life; however, patients of all ages can be affected, with reported cases occurring in children as young as 2 years of age.2 Most patients have Fitzpatrick skin type IV, mainly Amerindian, Hispanic South Asian, and Southwest Asian; however, there are cases reported worldwide.3 A genetic predisposition is proposed, as major histocompatibility complex genes associated with HLA-DR4⁎0407 are frequent in Mexican patients with ashy dermatosis and in the Amerindian population.4
The etiology of EDP is unknown. Various contributing factors have been reported including alimentary, occupational, and climatic factors,5,6 yet none have been conclusively demonstrated. High expression of CD36 (thrombospondin receptor not found in normal skin) in spinous and granular layers, CD94 (cytotoxic cell marker) in the basal cell layer and in the inflammatory dermal infiltrate,7 and focal keratinocytic expression of intercellular adhesion molecule I (CD54) in the active lesions of EDP, as well as the absence of these findings in normal skin, suggests an immunologic role in the development of the disease.8
Erythema dyschromicum perstans presents clinically with blue-gray hyperpigmented macules varying in size and shape and developing symmetrically in both sun-exposed and sun-protected areas of the face, neck, trunk, arms, and sometimes the dorsal hands (Figures 1 and 2). Notable sparing of the palms, soles, scalp, and mucous membranes occurs.
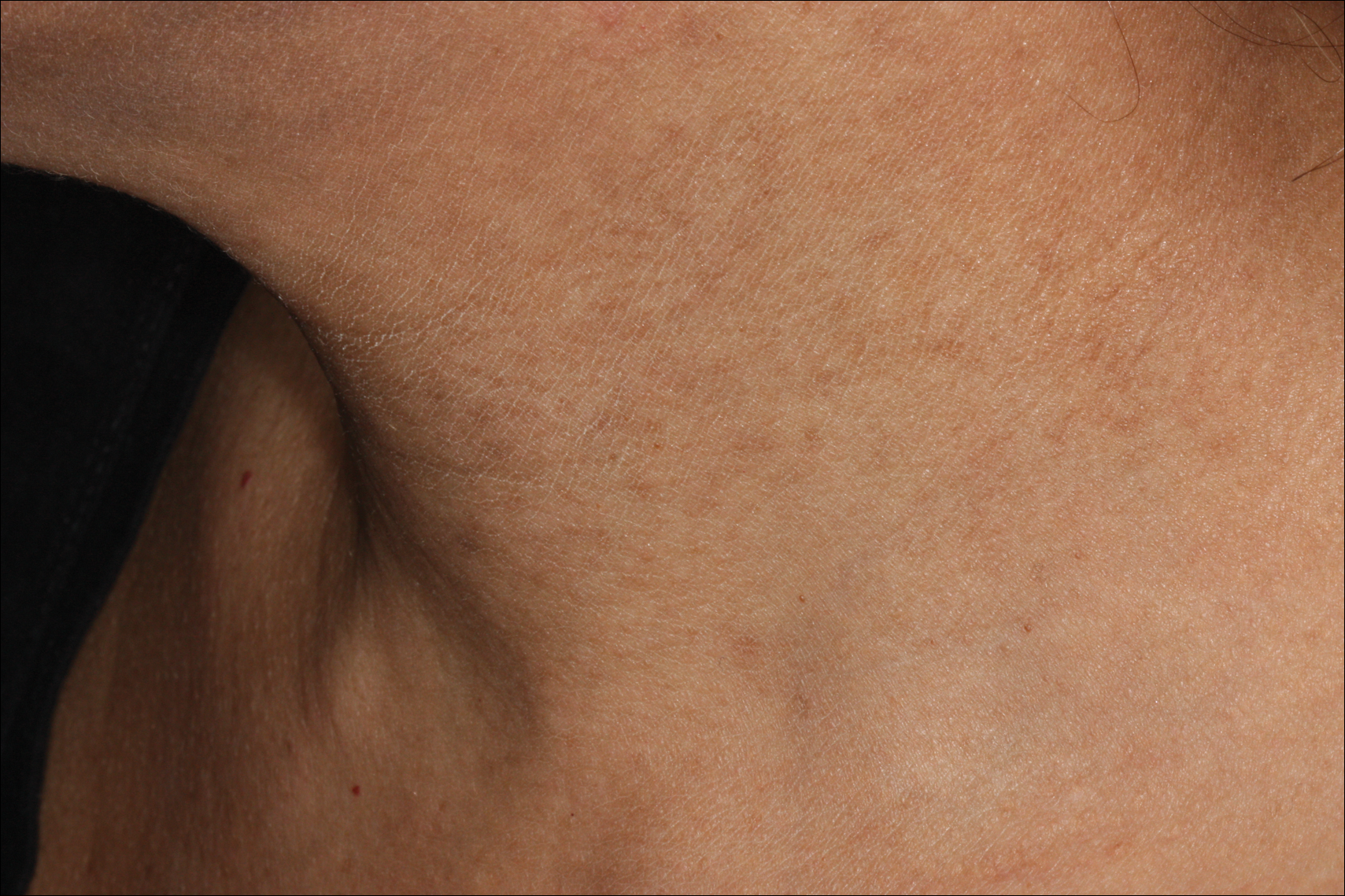
Occasionally, in the early active stage of the disease, elevated erythematous borders are noted surrounding the hyperpigmented macules. Eventually a hypopigmented halo develops after a prolonged duration of disease.9 The eruption typically is chronic and asymptomatic, though some cases may be pruritic.10
Histopathologically, the early lesions of EDP with an erythematous active border reveal lichenoid dermatitis with basal vacuolar change and occasional Civatte bodies. A mild to moderate perivascular lymphohistiocytic infiltrate admixed with melanophages can be seen in the papillary dermis (Figure 3). In older lesions, the inflammatory infiltrate is sparse, and pigment incontinence consistent with postinflammatory pigmentation is prominent, though melanophages extending deep into the reticular dermis may aid in distinguishing EDP from other causes of postinflammatory pigment alteration.7,11
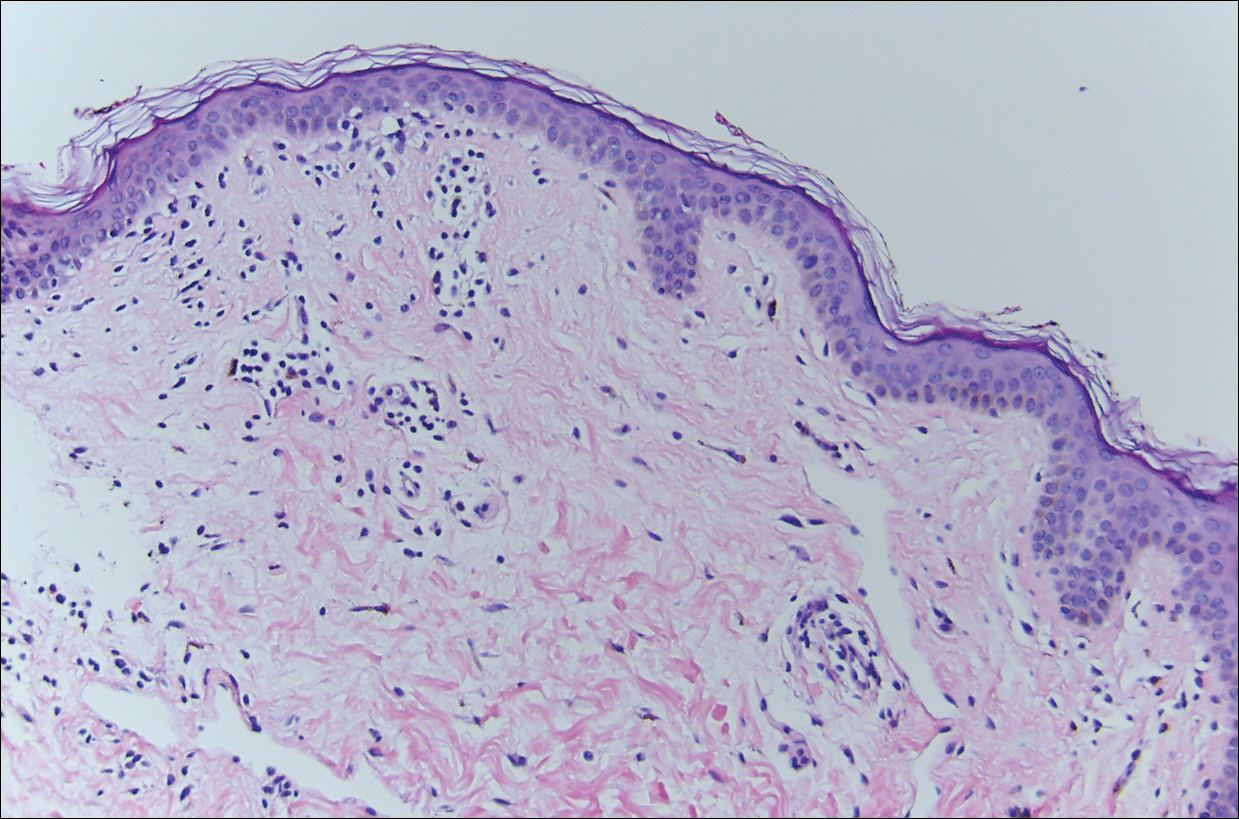
Erythema dyschromicum perstans and lichen planus pigmentosus (LPP) may be indistinguishable histopathologically and may both be variants of lichen planus actinicus. Lichen planus pigmentosus often differs from EDP in that it presents with brown-black macules and patches often on the face and flexural areas. A subset of cases of LPP also may have mucous membrane involvement. The erythematous border that characterizes the active lesion of EDP is characteristically absent in LPP. In addition, pruritus often is reported with LPP. Direct immunofluorescence is not a beneficial tool in distinguishing the entities.12
Other differential diagnoses of predominantly facial hyperpigmentation include a lichenoid drug eruption; drug-induced hyperpigmentation (deposition disorder); postinflammatory hyperpigmentation following atopic dermatitis; contact dermatitis or photosensitivity reaction; early pinta; and cutaneous findings of systemic diseases manifesting with diffuse hyperpigmentation such as lupus erythematosus, dermatomyositis, hemochromatosis, and Addison disease. A detailed history including medication use, thorough clinical examination, and careful histopathologic evaluation will help distinguish these conditions.
Chrysiasis is a rare bluish to slate gray discoloration of the skin that predominantly occurs in sun-exposed areas. It is caused by chronic use of gold salts, which have been used to treat rheumatoid arthritis. UV light may contribute to induce the uptake of gold and subsequently stimulate tyrosinase activity.13 Histologic features of chrysiasis include dermal and perivascular gold deposition within the macrophages and endothelial cells as well as extracellular granules. It demonstrates an orange-red birefringence on fluorescent microscopy.14,15
Minocycline-induced hyperpigmentation is a well-recognized side effect of this drug. It is dose dependent and appears as a blue-black pigmentation that most frequently affects the shins, ankles, and arms.16 Three distinct types were documented: abnormal discoloration of the skin that has been linked to deposition of pigmented metabolites of minocycline producing blue-black pigmentation at the site of scarring or prior inflammation (type 1); blue-gray pigmentation affecting normal skin, mainly the legs (type 2); and elevated levels of melanin on the sun-exposed areas producing dirty skin syndrome (type 3).17,18
Topical and systemic corticosteroids, UV light therapy, oral dapsone, griseofulvin, retinoids, and clofazimine are reported as treatment options for ashy dermatosis, though results typically are disappointing.7
- Ramirez CO. Los cenicientos: problema clinica. In: Memoria del Primer Congresso Centroamericano de Dermatologica, December 5-8, 1957. San Salvador, El Salvador; 1957:122-130.
- Lee SJ, Chung KY. Erythema dyschromicum perstans in early childhood. J Dermatol. 1999;26:119-121.
- Homez-Chacin, Barroso C. On the etiopathogenic of the erythema dyschromicum perstans: possibility of a melanosis neurocutaneous. Dermatol Venez. 1996;4:149-151.
- Correa MC, Memije EV, Vargas-Alarcon G, et al. HLA-DR association with the genetic susceptibility to develop ashy dermatosis in Mexican Mestizo patients [published online November 20, 2006]. J Am Acad Dermatol. 2007;56:617-620.
- Jablonska S. Ingestion of ammonium nitrate as a possible cause of erythema dyschromicum perstans (ashy dermatosis). Dermatologica. 1975;150:287-291.
- Stevenson JR, Miura M. Erythema dyschromicum perstans (ashy dermatosis). Arch Dermatol. 1966;94:196-199.
- Baranda L, Torres-Alvarez B, Cortes-Franco R, et al. Involvement of cell adhesion and activation molecules in the pathogenesis of erythema dyschromicum perstans (ashy dermatitis). the effect of clofazimine therapy. Arch Dermatol. 1997;133:325-329.
- Vasquez-Ochoa LA, Isaza-Guzman DM, Orozco-Mora B, et al. Immunopathologic study of erythema dyschromicum perstans (ashy dermatosis). Int J Dermatol. 2006;45:937-941.
- Convit J, Kerdel-Vegas F, Roderiguez G. Erythema dyschromicum perstans: a hiltherto undescribed skin disease. J Invest Dermatol. 1961;36:457-462.
- Ono S, Miyachi Y, Kabashima K. Ashy dermatosis with prior pruritic and scaling skin lesions. J Dermatol. 2012;39:1103-1104.
- Sanchez NP, Pathak MA, Sato SS, et al. Circumscribed dermal melaninoses: classification, light, histochemical, and electron microscopic studies on three patients with the erythema dyschromicum perstans type. Int J Dermatol. 1982;21:25-32.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Ahmed SV, Sajjan R. Chrysiasis: a gold "curse!" [published online May 21, 2009]. BMJ Case Rep. 2009;2009.
- Fiscus V, Hankinson A, Alweis R. Minocycline-induced hyperpigmentation. J Community Hosp Intern Med Perspect. 2014;4. doi:10.3402/jchimp.v4.24063.
- Cox AJ, Marich KW. Gold in the dermis following gold therapy for rheumatoid arthritis. Arch Dermatol. 1973;108:655-657.
- al-Talib RK, Wright DH, Theaker JM. Orange-red birefringence of gold particles in paraffin wax embedded sections: an aid to the diagnosis of chrysiasis. Histopathology. 1994;24:176-178.
- Meyer AJ, Nahass GT. Hyperpigmented patches on the dorsa of the feet. minocycline pigmentation. Arch Dermatol. 1995;131:1447-1450.
- Bayne-Poorman M, Shubrook J. Bluish pigmentation of face and sclera. J Fam Pract. 2010;59:519-522.
The Diagnosis: Erythema Dyschromicum Perstans
Erythema dyschromicum perstans (EDP), also referred to as ashy dermatosis, was first described by Ramirez1 in 1957 who labeled the patients los cenicientos (the ashen ones). It preferentially affects women in the second decade of life; however, patients of all ages can be affected, with reported cases occurring in children as young as 2 years of age.2 Most patients have Fitzpatrick skin type IV, mainly Amerindian, Hispanic South Asian, and Southwest Asian; however, there are cases reported worldwide.3 A genetic predisposition is proposed, as major histocompatibility complex genes associated with HLA-DR4⁎0407 are frequent in Mexican patients with ashy dermatosis and in the Amerindian population.4
The etiology of EDP is unknown. Various contributing factors have been reported including alimentary, occupational, and climatic factors,5,6 yet none have been conclusively demonstrated. High expression of CD36 (thrombospondin receptor not found in normal skin) in spinous and granular layers, CD94 (cytotoxic cell marker) in the basal cell layer and in the inflammatory dermal infiltrate,7 and focal keratinocytic expression of intercellular adhesion molecule I (CD54) in the active lesions of EDP, as well as the absence of these findings in normal skin, suggests an immunologic role in the development of the disease.8
Erythema dyschromicum perstans presents clinically with blue-gray hyperpigmented macules varying in size and shape and developing symmetrically in both sun-exposed and sun-protected areas of the face, neck, trunk, arms, and sometimes the dorsal hands (Figures 1 and 2). Notable sparing of the palms, soles, scalp, and mucous membranes occurs.
Occasionally, in the early active stage of the disease, elevated erythematous borders are noted surrounding the hyperpigmented macules. Eventually a hypopigmented halo develops after a prolonged duration of disease.9 The eruption typically is chronic and asymptomatic, though some cases may be pruritic.10
Histopathologically, the early lesions of EDP with an erythematous active border reveal lichenoid dermatitis with basal vacuolar change and occasional Civatte bodies. A mild to moderate perivascular lymphohistiocytic infiltrate admixed with melanophages can be seen in the papillary dermis (Figure 3). In older lesions, the inflammatory infiltrate is sparse, and pigment incontinence consistent with postinflammatory pigmentation is prominent, though melanophages extending deep into the reticular dermis may aid in distinguishing EDP from other causes of postinflammatory pigment alteration.7,11
Erythema dyschromicum perstans and lichen planus pigmentosus (LPP) may be indistinguishable histopathologically and may both be variants of lichen planus actinicus. Lichen planus pigmentosus often differs from EDP in that it presents with brown-black macules and patches often on the face and flexural areas. A subset of cases of LPP also may have mucous membrane involvement. The erythematous border that characterizes the active lesion of EDP is characteristically absent in LPP. In addition, pruritus often is reported with LPP. Direct immunofluorescence is not a beneficial tool in distinguishing the entities.12
Other differential diagnoses of predominantly facial hyperpigmentation include a lichenoid drug eruption; drug-induced hyperpigmentation (deposition disorder); postinflammatory hyperpigmentation following atopic dermatitis; contact dermatitis or photosensitivity reaction; early pinta; and cutaneous findings of systemic diseases manifesting with diffuse hyperpigmentation such as lupus erythematosus, dermatomyositis, hemochromatosis, and Addison disease. A detailed history including medication use, thorough clinical examination, and careful histopathologic evaluation will help distinguish these conditions.
Chrysiasis is a rare bluish to slate gray discoloration of the skin that predominantly occurs in sun-exposed areas. It is caused by chronic use of gold salts, which have been used to treat rheumatoid arthritis. UV light may contribute to induce the uptake of gold and subsequently stimulate tyrosinase activity.13 Histologic features of chrysiasis include dermal and perivascular gold deposition within the macrophages and endothelial cells as well as extracellular granules. It demonstrates an orange-red birefringence on fluorescent microscopy.14,15
Minocycline-induced hyperpigmentation is a well-recognized side effect of this drug. It is dose dependent and appears as a blue-black pigmentation that most frequently affects the shins, ankles, and arms.16 Three distinct types were documented: abnormal discoloration of the skin that has been linked to deposition of pigmented metabolites of minocycline producing blue-black pigmentation at the site of scarring or prior inflammation (type 1); blue-gray pigmentation affecting normal skin, mainly the legs (type 2); and elevated levels of melanin on the sun-exposed areas producing dirty skin syndrome (type 3).17,18
Topical and systemic corticosteroids, UV light therapy, oral dapsone, griseofulvin, retinoids, and clofazimine are reported as treatment options for ashy dermatosis, though results typically are disappointing.7
The Diagnosis: Erythema Dyschromicum Perstans
Erythema dyschromicum perstans (EDP), also referred to as ashy dermatosis, was first described by Ramirez1 in 1957 who labeled the patients los cenicientos (the ashen ones). It preferentially affects women in the second decade of life; however, patients of all ages can be affected, with reported cases occurring in children as young as 2 years of age.2 Most patients have Fitzpatrick skin type IV, mainly Amerindian, Hispanic South Asian, and Southwest Asian; however, there are cases reported worldwide.3 A genetic predisposition is proposed, as major histocompatibility complex genes associated with HLA-DR4⁎0407 are frequent in Mexican patients with ashy dermatosis and in the Amerindian population.4
The etiology of EDP is unknown. Various contributing factors have been reported including alimentary, occupational, and climatic factors,5,6 yet none have been conclusively demonstrated. High expression of CD36 (thrombospondin receptor not found in normal skin) in spinous and granular layers, CD94 (cytotoxic cell marker) in the basal cell layer and in the inflammatory dermal infiltrate,7 and focal keratinocytic expression of intercellular adhesion molecule I (CD54) in the active lesions of EDP, as well as the absence of these findings in normal skin, suggests an immunologic role in the development of the disease.8
Erythema dyschromicum perstans presents clinically with blue-gray hyperpigmented macules varying in size and shape and developing symmetrically in both sun-exposed and sun-protected areas of the face, neck, trunk, arms, and sometimes the dorsal hands (Figures 1 and 2). Notable sparing of the palms, soles, scalp, and mucous membranes occurs.
Occasionally, in the early active stage of the disease, elevated erythematous borders are noted surrounding the hyperpigmented macules. Eventually a hypopigmented halo develops after a prolonged duration of disease.9 The eruption typically is chronic and asymptomatic, though some cases may be pruritic.10
Histopathologically, the early lesions of EDP with an erythematous active border reveal lichenoid dermatitis with basal vacuolar change and occasional Civatte bodies. A mild to moderate perivascular lymphohistiocytic infiltrate admixed with melanophages can be seen in the papillary dermis (Figure 3). In older lesions, the inflammatory infiltrate is sparse, and pigment incontinence consistent with postinflammatory pigmentation is prominent, though melanophages extending deep into the reticular dermis may aid in distinguishing EDP from other causes of postinflammatory pigment alteration.7,11
Erythema dyschromicum perstans and lichen planus pigmentosus (LPP) may be indistinguishable histopathologically and may both be variants of lichen planus actinicus. Lichen planus pigmentosus often differs from EDP in that it presents with brown-black macules and patches often on the face and flexural areas. A subset of cases of LPP also may have mucous membrane involvement. The erythematous border that characterizes the active lesion of EDP is characteristically absent in LPP. In addition, pruritus often is reported with LPP. Direct immunofluorescence is not a beneficial tool in distinguishing the entities.12
Other differential diagnoses of predominantly facial hyperpigmentation include a lichenoid drug eruption; drug-induced hyperpigmentation (deposition disorder); postinflammatory hyperpigmentation following atopic dermatitis; contact dermatitis or photosensitivity reaction; early pinta; and cutaneous findings of systemic diseases manifesting with diffuse hyperpigmentation such as lupus erythematosus, dermatomyositis, hemochromatosis, and Addison disease. A detailed history including medication use, thorough clinical examination, and careful histopathologic evaluation will help distinguish these conditions.
Chrysiasis is a rare bluish to slate gray discoloration of the skin that predominantly occurs in sun-exposed areas. It is caused by chronic use of gold salts, which have been used to treat rheumatoid arthritis. UV light may contribute to induce the uptake of gold and subsequently stimulate tyrosinase activity.13 Histologic features of chrysiasis include dermal and perivascular gold deposition within the macrophages and endothelial cells as well as extracellular granules. It demonstrates an orange-red birefringence on fluorescent microscopy.14,15
Minocycline-induced hyperpigmentation is a well-recognized side effect of this drug. It is dose dependent and appears as a blue-black pigmentation that most frequently affects the shins, ankles, and arms.16 Three distinct types were documented: abnormal discoloration of the skin that has been linked to deposition of pigmented metabolites of minocycline producing blue-black pigmentation at the site of scarring or prior inflammation (type 1); blue-gray pigmentation affecting normal skin, mainly the legs (type 2); and elevated levels of melanin on the sun-exposed areas producing dirty skin syndrome (type 3).17,18
Topical and systemic corticosteroids, UV light therapy, oral dapsone, griseofulvin, retinoids, and clofazimine are reported as treatment options for ashy dermatosis, though results typically are disappointing.7
- Ramirez CO. Los cenicientos: problema clinica. In: Memoria del Primer Congresso Centroamericano de Dermatologica, December 5-8, 1957. San Salvador, El Salvador; 1957:122-130.
- Lee SJ, Chung KY. Erythema dyschromicum perstans in early childhood. J Dermatol. 1999;26:119-121.
- Homez-Chacin, Barroso C. On the etiopathogenic of the erythema dyschromicum perstans: possibility of a melanosis neurocutaneous. Dermatol Venez. 1996;4:149-151.
- Correa MC, Memije EV, Vargas-Alarcon G, et al. HLA-DR association with the genetic susceptibility to develop ashy dermatosis in Mexican Mestizo patients [published online November 20, 2006]. J Am Acad Dermatol. 2007;56:617-620.
- Jablonska S. Ingestion of ammonium nitrate as a possible cause of erythema dyschromicum perstans (ashy dermatosis). Dermatologica. 1975;150:287-291.
- Stevenson JR, Miura M. Erythema dyschromicum perstans (ashy dermatosis). Arch Dermatol. 1966;94:196-199.
- Baranda L, Torres-Alvarez B, Cortes-Franco R, et al. Involvement of cell adhesion and activation molecules in the pathogenesis of erythema dyschromicum perstans (ashy dermatitis). the effect of clofazimine therapy. Arch Dermatol. 1997;133:325-329.
- Vasquez-Ochoa LA, Isaza-Guzman DM, Orozco-Mora B, et al. Immunopathologic study of erythema dyschromicum perstans (ashy dermatosis). Int J Dermatol. 2006;45:937-941.
- Convit J, Kerdel-Vegas F, Roderiguez G. Erythema dyschromicum perstans: a hiltherto undescribed skin disease. J Invest Dermatol. 1961;36:457-462.
- Ono S, Miyachi Y, Kabashima K. Ashy dermatosis with prior pruritic and scaling skin lesions. J Dermatol. 2012;39:1103-1104.
- Sanchez NP, Pathak MA, Sato SS, et al. Circumscribed dermal melaninoses: classification, light, histochemical, and electron microscopic studies on three patients with the erythema dyschromicum perstans type. Int J Dermatol. 1982;21:25-32.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Ahmed SV, Sajjan R. Chrysiasis: a gold "curse!" [published online May 21, 2009]. BMJ Case Rep. 2009;2009.
- Fiscus V, Hankinson A, Alweis R. Minocycline-induced hyperpigmentation. J Community Hosp Intern Med Perspect. 2014;4. doi:10.3402/jchimp.v4.24063.
- Cox AJ, Marich KW. Gold in the dermis following gold therapy for rheumatoid arthritis. Arch Dermatol. 1973;108:655-657.
- al-Talib RK, Wright DH, Theaker JM. Orange-red birefringence of gold particles in paraffin wax embedded sections: an aid to the diagnosis of chrysiasis. Histopathology. 1994;24:176-178.
- Meyer AJ, Nahass GT. Hyperpigmented patches on the dorsa of the feet. minocycline pigmentation. Arch Dermatol. 1995;131:1447-1450.
- Bayne-Poorman M, Shubrook J. Bluish pigmentation of face and sclera. J Fam Pract. 2010;59:519-522.
- Ramirez CO. Los cenicientos: problema clinica. In: Memoria del Primer Congresso Centroamericano de Dermatologica, December 5-8, 1957. San Salvador, El Salvador; 1957:122-130.
- Lee SJ, Chung KY. Erythema dyschromicum perstans in early childhood. J Dermatol. 1999;26:119-121.
- Homez-Chacin, Barroso C. On the etiopathogenic of the erythema dyschromicum perstans: possibility of a melanosis neurocutaneous. Dermatol Venez. 1996;4:149-151.
- Correa MC, Memije EV, Vargas-Alarcon G, et al. HLA-DR association with the genetic susceptibility to develop ashy dermatosis in Mexican Mestizo patients [published online November 20, 2006]. J Am Acad Dermatol. 2007;56:617-620.
- Jablonska S. Ingestion of ammonium nitrate as a possible cause of erythema dyschromicum perstans (ashy dermatosis). Dermatologica. 1975;150:287-291.
- Stevenson JR, Miura M. Erythema dyschromicum perstans (ashy dermatosis). Arch Dermatol. 1966;94:196-199.
- Baranda L, Torres-Alvarez B, Cortes-Franco R, et al. Involvement of cell adhesion and activation molecules in the pathogenesis of erythema dyschromicum perstans (ashy dermatitis). the effect of clofazimine therapy. Arch Dermatol. 1997;133:325-329.
- Vasquez-Ochoa LA, Isaza-Guzman DM, Orozco-Mora B, et al. Immunopathologic study of erythema dyschromicum perstans (ashy dermatosis). Int J Dermatol. 2006;45:937-941.
- Convit J, Kerdel-Vegas F, Roderiguez G. Erythema dyschromicum perstans: a hiltherto undescribed skin disease. J Invest Dermatol. 1961;36:457-462.
- Ono S, Miyachi Y, Kabashima K. Ashy dermatosis with prior pruritic and scaling skin lesions. J Dermatol. 2012;39:1103-1104.
- Sanchez NP, Pathak MA, Sato SS, et al. Circumscribed dermal melaninoses: classification, light, histochemical, and electron microscopic studies on three patients with the erythema dyschromicum perstans type. Int J Dermatol. 1982;21:25-32.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Ahmed SV, Sajjan R. Chrysiasis: a gold "curse!" [published online May 21, 2009]. BMJ Case Rep. 2009;2009.
- Fiscus V, Hankinson A, Alweis R. Minocycline-induced hyperpigmentation. J Community Hosp Intern Med Perspect. 2014;4. doi:10.3402/jchimp.v4.24063.
- Cox AJ, Marich KW. Gold in the dermis following gold therapy for rheumatoid arthritis. Arch Dermatol. 1973;108:655-657.
- al-Talib RK, Wright DH, Theaker JM. Orange-red birefringence of gold particles in paraffin wax embedded sections: an aid to the diagnosis of chrysiasis. Histopathology. 1994;24:176-178.
- Meyer AJ, Nahass GT. Hyperpigmented patches on the dorsa of the feet. minocycline pigmentation. Arch Dermatol. 1995;131:1447-1450.
- Bayne-Poorman M, Shubrook J. Bluish pigmentation of face and sclera. J Fam Pract. 2010;59:519-522.
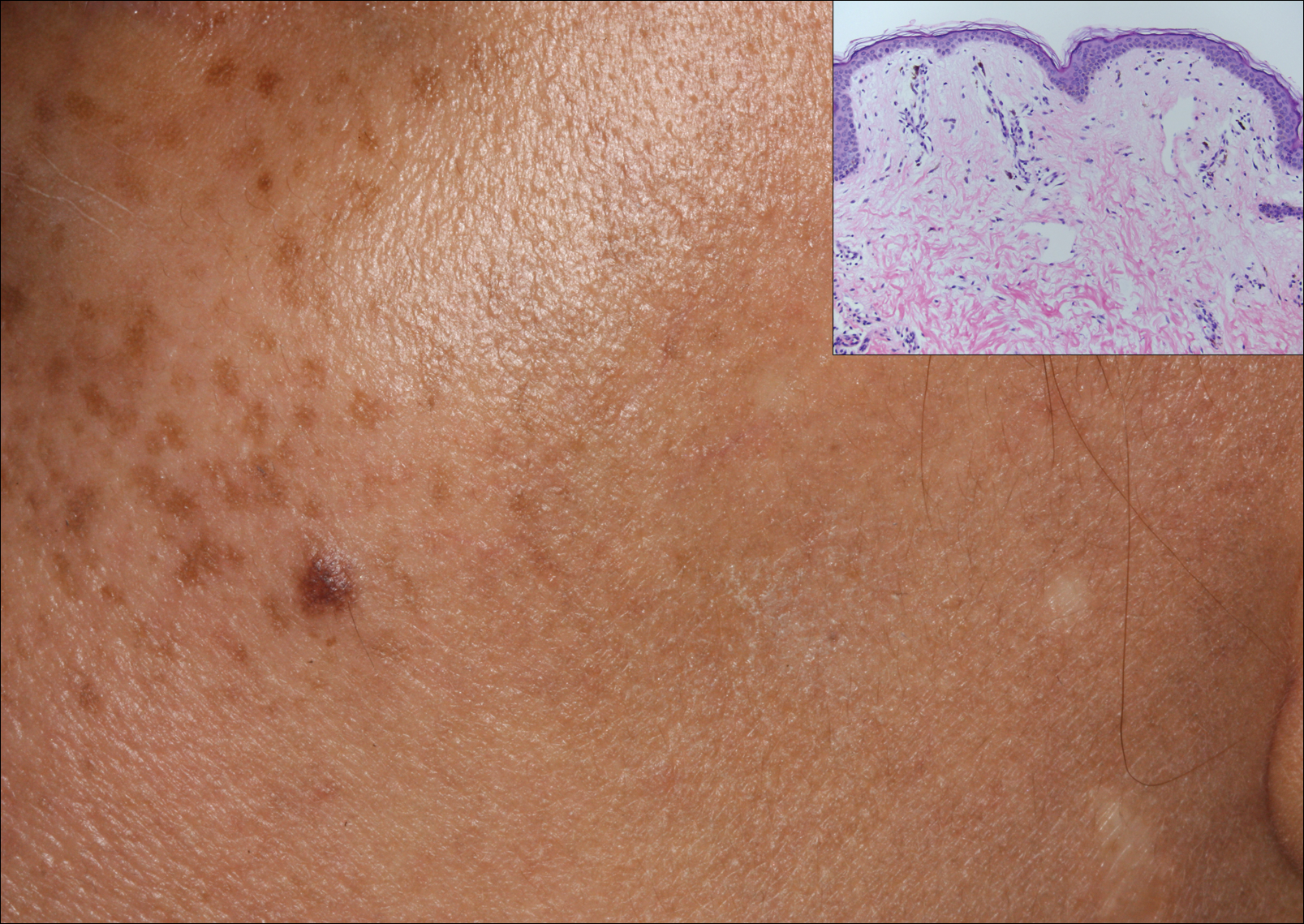
A middle-aged woman with Fitzpatrick skin type IV was evaluated for progressive hyperpigmentation of several months' duration involving the neck, jawline, both sides of the face, and forehead. The lesions were mildly pruritic. She denied contact with any new substance and there was no history of an eruption preceding the hyperpigmentation. Medical history included chronic anemia that was managed with iron supplementation. On physical examination, blue-gray nonscaly macules and patches were observed distributed symmetrically on the neck, jawline, sides of the face, and forehead. Microscopic examination of 2 shave biopsies revealed subtle vacuolar interface dermatitis with mild perivascular lymphocytic infiltrate and dermal melanophages (inset).
Postoperative Henoch-Schönlein Purpura
To the Editor:
A 57-year-old man with a history of type 2 diabetes mellitus and hypertension was hospitalized for heart disease resulting in an aortic valve replacement and multiple-vessel bypass grafting. He experienced a stormy septic postoperative course during which he developed numerous palpable purplish plaques (Figure 1). The lesions were bilateral and more heavily involved the lower legs and buttocks. The head and neck remained free of skin lesions. Additionally, the patient reported a bilateral burning sensation from the knees to the feet.
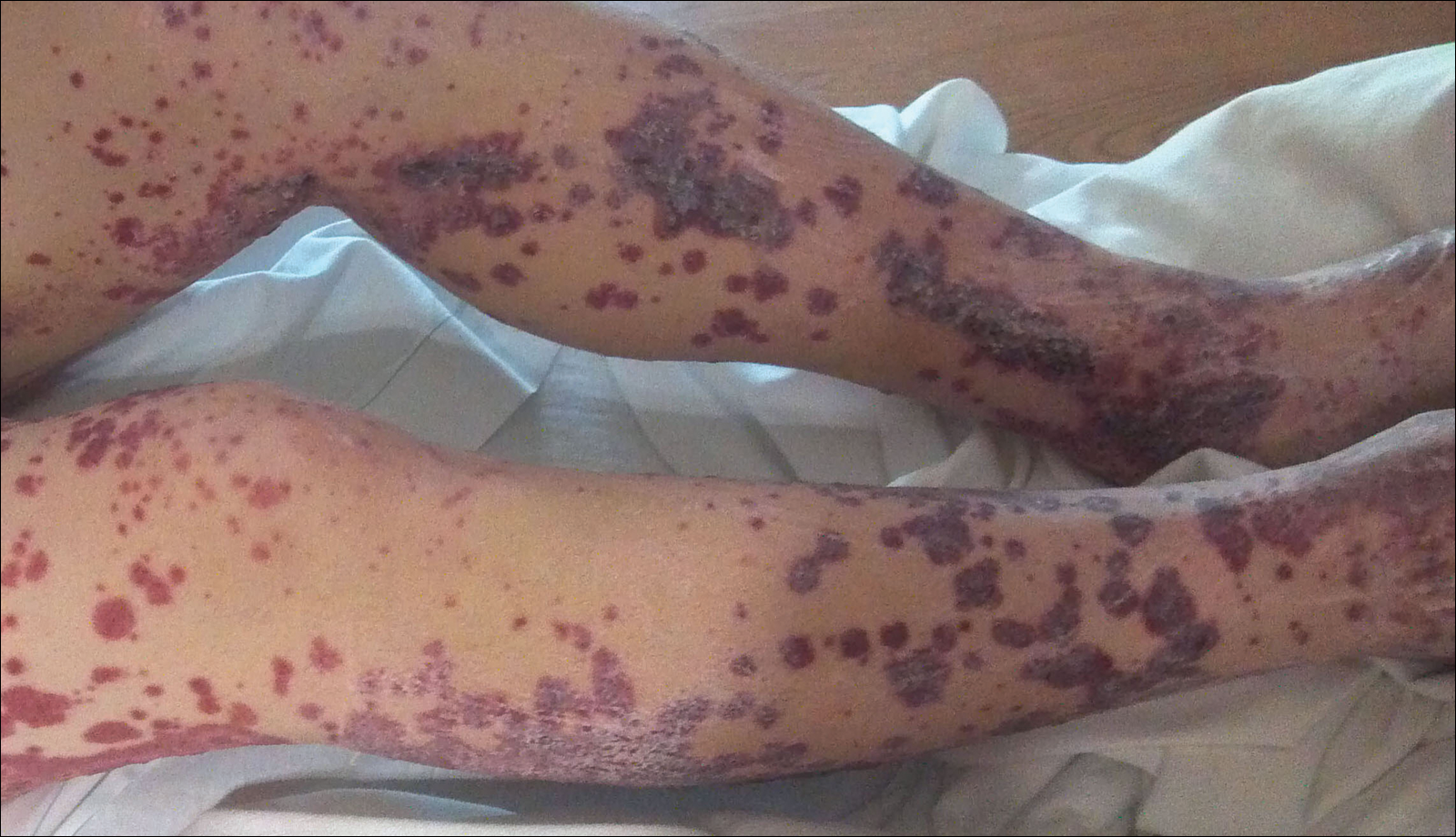
Punch biopsies of lesions from the right upper arm were obtained. Hematoxylin and eosin staining revealed neutrophilic-predominant small vessel vasculitis (Figure 2A) with the upper dermal location more heavily involved, as demonstrated by involvement of a superficial vascular plexus (Figures 2B and 2C) that was consistent with Henoch-Schönlein purpura (HSP). The diagnosis later was confirmed with immunofluorescence. Direct immunofluorescence revealed granular IgA deposition around the superficial vascular plexus (Figure 3). No IgG, IgM, C3, C5b-9 complement complex, or fibrinogen deposition was seen. Additionally, periodic acid-Schiff staining failed to show microorganisms, thrombi, or intravascular hyaline material.
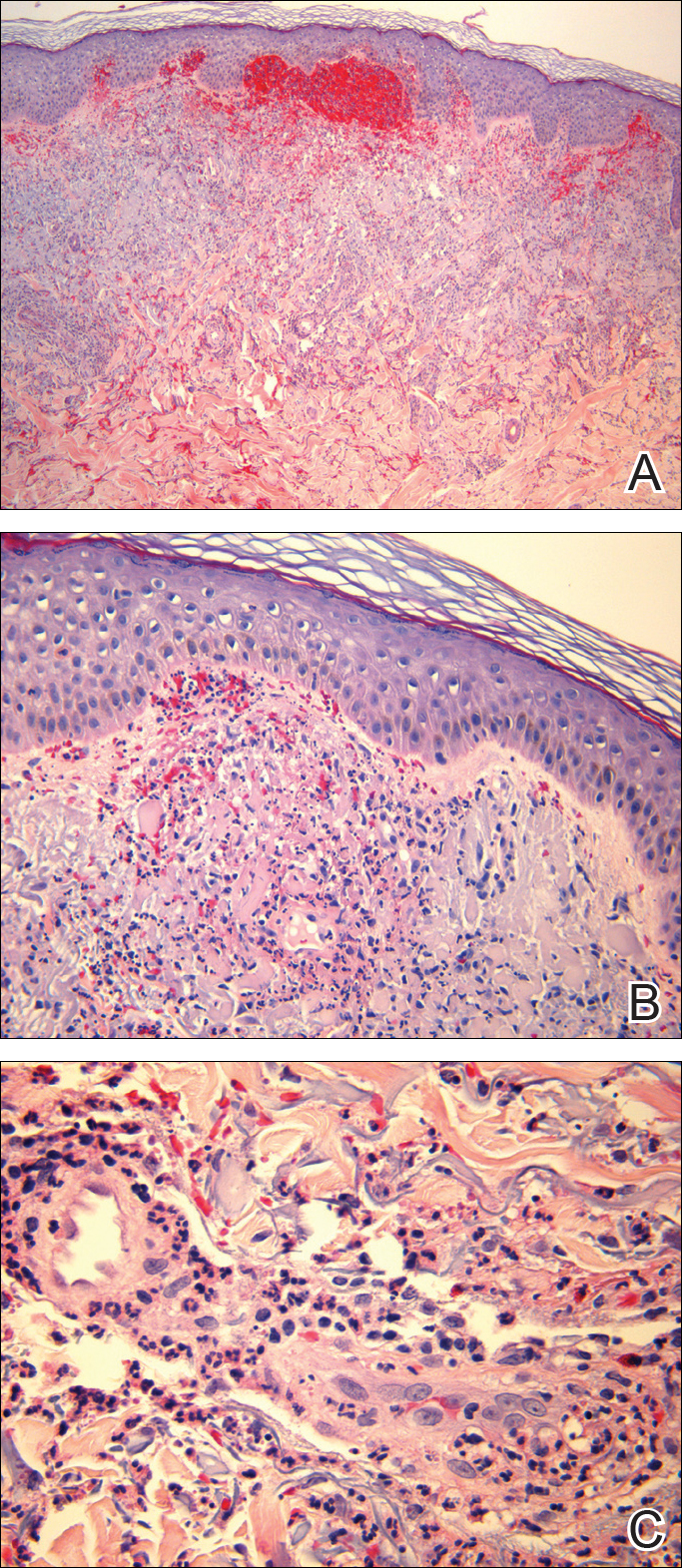
At our initial consultation, we observed an ill-appearing afebrile man with purplish plaques. Our impression was that he had vasculitis and not warfarin necrosis, which had been suspected by the cardiovascular team. The burning sensation noted by the patient lent credence to our vasculitic diagnosis. Proteinuria and hematuria were present; however, the values for blood urea nitrogen, creatinine, and glomerular filtration rate all remained within reference range. His signs and symptoms responded dramatically to prednisone. He remains on 1 mg of prednisone daily and a nephrologist continues to monitor renal function as an outpatient.
Henoch-Schönlein purpura is a systemic leukocytoclastic vasculitis involving small vessels. The small vessel vasculitis is associated with IgA antigen-antibody complex deposition in areas throughout the body. Palpable purpura typically is seen on the skin, which characteristically involves dependent areas such as the legs and the buttocks. Lesions normally are present bilaterally in a symmetric distribution. Initially, the lesions develop as erythematous macules that progress to purple, nonblanching, palpable, and purpuric plaques.1 Henoch-Schönlein purpura most commonly involves the skin; however, other locations for the immune complexes include the gastrointestinal tract, joints, and kidneys.2 The cause for the body's immunogenic deposition response is unknown in a majority of cases.
Henoch-Schönlein purpura most commonly is seen in the pediatric population with a predilection for males.3 The incidence in the pediatric population is 13.5 to 20 per 100,000 children per year; HSP is more rare in adults.4-6 Henoch-Schönlein purpura most often is a self-limiting disease that requires only supportive treatment. The signs and symptoms last 4 to 6 weeks in most patients and resolve completely in 94% of children and 89% of adults.7 Renal involvement carries a worse prognosis. Adult patients have a higher incidence of renal involvement, renal insufficiency, and subsequent progression to end-stage renal disease.3,8-10 In a study by Hung et al8 of 65 children and 22 adult HSP patients, 12 adults presented with renal involvement in which hematuria or proteinuria were present. Of them, 6 progressed to renal insufficiency (defined as having a plasma creatinine concentration>1.2 mg/dL).8 Fogazzi et al11 reported similar findings; 8 of 16 patients affected with HSP progressed to renal insufficiency with creatinine clearances ranging from 31 to 60 mL/min, and 3 patients required chronic dialysis. Pillebout et al9 evaluated 250 adults with HSP and 32% reached renal insufficiency with creatinine clearances of less than 50 mL/min, with 11% of patients developing end-stage renal disease. The degree of hematuria and/or proteinuria has been shown to be an effective prognostic indicator.9,10 Coppo et al10 found a similar prognosis among children and adults with HSP-related nephritis.
Our patient described the burning sensation as occurring bilaterally from the knees down to the feet, which provided an additional clue that small vessel vasculitis was involved, as occluded blood vessels can cause ischemia to nerves and perivascular involvement can affect nearby neural structures. Sais et al12 demonstrated that paresthesia in the setting of HSP was a risk factor for systemic involvement. Of note, our patient's paresthesia lasted only several days.
The cause of HSP is not always as evident in the adult population as in the pediatric population. Early diagnosis of HSP in adults may allow for the proper instatement of treatment to deter long-term renal complications. Follow-up with urinalysis is recommended because a small percentage of patients have a late progression to renal failure.13,14
Because the dermatologists involved in this case knew where and what types of biopsies to perform, a correct diagnosis was obtained quickly, allowing for the correct therapeutic intervention. After the diagnosis of HSP is made in an adult, nephrology should be consulted early in the treatment course.
- Rai A, Nast C, Adler S. Henoch-Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Helander SD, De Castro FR, Gibson LE. Henoch-Schönlein purpura: clinicopathologic correlation of cutaneous vascular IgA deposits and the relationship to leukocytoclastic vasculitis. Acta Derm Venereol. 1995;75:125-129.
- Garcia-Porrua C, Calvino MC, Llorca J, et al. Henoch-Schönlein purpura in children and adults: clinical differences in a defined population. Semin Arthritis Rheum. 2002;32:149-156.
- Stewart M, Savage JM, Bell B, et al. Long term renal prognosis of Henoch-Schönlein purpura in an unselected childhood population. Eur J Pediatr. 1988;147:113-115.
- Watts RA, Scott DG. Epidemiology of the vasculitides. Semin Respir Crit Care. 2004;25:455-464.
- Gardner-Medwin JM, Dolezalova P, Cummins C, et al. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. 2002;360:1197-1202.
- Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, et al. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum. 1997;40:859-864.
- Hung SP, Yang YH, Lin YT, et al. Clinical manifestations and outcomes of Henoch-Schönlein purpura: comparison between adults and children. Pediatr Neonatol. 2009;50:162-168.
- Pillebout E, Thervet E, Hill G, et al. Henoch-Schönlein purpura in adults: outcomes and prognostic factors. J Am Soc Nephrol. 2002;13:1271-1278.
- Coppo R, Mazzucco G, Cagnoli L, et al. Long-term prognosis of Henoch-Schönlein nephritis in adults and children. Italian Group of Renal Immunopathology collaborative study on Henoch-Schönlein purpura. Nephrol Dial Transplant. 1997;12:2277-2283.
- Fogazzi GB, Pasquali S, Moriggi M, et al. Long-term outcome of Schönlein-Henoch nephritis in the adult. Clin Nephrol. 1989;31:60-66.
- Sais G, Vidaller A, Jucgla A. Prognostic factors in leukocytoclastic vasculitis. a clinicopathologic study of 160 patients. Arch Dermatol. 1998;134:309-315.
- Kraft DM, McKee D, Scott C. Henoch-Schönlein purpura: a review. Am Fam Physician. 1998;58:405-408.
- Narchi H. Risk of long-term renal impairment and duration of follow up recommended for Henoch-Schönlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child. 2005;90:916-920.
To the Editor:
A 57-year-old man with a history of type 2 diabetes mellitus and hypertension was hospitalized for heart disease resulting in an aortic valve replacement and multiple-vessel bypass grafting. He experienced a stormy septic postoperative course during which he developed numerous palpable purplish plaques (Figure 1). The lesions were bilateral and more heavily involved the lower legs and buttocks. The head and neck remained free of skin lesions. Additionally, the patient reported a bilateral burning sensation from the knees to the feet.
Punch biopsies of lesions from the right upper arm were obtained. Hematoxylin and eosin staining revealed neutrophilic-predominant small vessel vasculitis (Figure 2A) with the upper dermal location more heavily involved, as demonstrated by involvement of a superficial vascular plexus (Figures 2B and 2C) that was consistent with Henoch-Schönlein purpura (HSP). The diagnosis later was confirmed with immunofluorescence. Direct immunofluorescence revealed granular IgA deposition around the superficial vascular plexus (Figure 3). No IgG, IgM, C3, C5b-9 complement complex, or fibrinogen deposition was seen. Additionally, periodic acid-Schiff staining failed to show microorganisms, thrombi, or intravascular hyaline material.
At our initial consultation, we observed an ill-appearing afebrile man with purplish plaques. Our impression was that he had vasculitis and not warfarin necrosis, which had been suspected by the cardiovascular team. The burning sensation noted by the patient lent credence to our vasculitic diagnosis. Proteinuria and hematuria were present; however, the values for blood urea nitrogen, creatinine, and glomerular filtration rate all remained within reference range. His signs and symptoms responded dramatically to prednisone. He remains on 1 mg of prednisone daily and a nephrologist continues to monitor renal function as an outpatient.
Henoch-Schönlein purpura is a systemic leukocytoclastic vasculitis involving small vessels. The small vessel vasculitis is associated with IgA antigen-antibody complex deposition in areas throughout the body. Palpable purpura typically is seen on the skin, which characteristically involves dependent areas such as the legs and the buttocks. Lesions normally are present bilaterally in a symmetric distribution. Initially, the lesions develop as erythematous macules that progress to purple, nonblanching, palpable, and purpuric plaques.1 Henoch-Schönlein purpura most commonly involves the skin; however, other locations for the immune complexes include the gastrointestinal tract, joints, and kidneys.2 The cause for the body's immunogenic deposition response is unknown in a majority of cases.
Henoch-Schönlein purpura most commonly is seen in the pediatric population with a predilection for males.3 The incidence in the pediatric population is 13.5 to 20 per 100,000 children per year; HSP is more rare in adults.4-6 Henoch-Schönlein purpura most often is a self-limiting disease that requires only supportive treatment. The signs and symptoms last 4 to 6 weeks in most patients and resolve completely in 94% of children and 89% of adults.7 Renal involvement carries a worse prognosis. Adult patients have a higher incidence of renal involvement, renal insufficiency, and subsequent progression to end-stage renal disease.3,8-10 In a study by Hung et al8 of 65 children and 22 adult HSP patients, 12 adults presented with renal involvement in which hematuria or proteinuria were present. Of them, 6 progressed to renal insufficiency (defined as having a plasma creatinine concentration>1.2 mg/dL).8 Fogazzi et al11 reported similar findings; 8 of 16 patients affected with HSP progressed to renal insufficiency with creatinine clearances ranging from 31 to 60 mL/min, and 3 patients required chronic dialysis. Pillebout et al9 evaluated 250 adults with HSP and 32% reached renal insufficiency with creatinine clearances of less than 50 mL/min, with 11% of patients developing end-stage renal disease. The degree of hematuria and/or proteinuria has been shown to be an effective prognostic indicator.9,10 Coppo et al10 found a similar prognosis among children and adults with HSP-related nephritis.
Our patient described the burning sensation as occurring bilaterally from the knees down to the feet, which provided an additional clue that small vessel vasculitis was involved, as occluded blood vessels can cause ischemia to nerves and perivascular involvement can affect nearby neural structures. Sais et al12 demonstrated that paresthesia in the setting of HSP was a risk factor for systemic involvement. Of note, our patient's paresthesia lasted only several days.
The cause of HSP is not always as evident in the adult population as in the pediatric population. Early diagnosis of HSP in adults may allow for the proper instatement of treatment to deter long-term renal complications. Follow-up with urinalysis is recommended because a small percentage of patients have a late progression to renal failure.13,14
Because the dermatologists involved in this case knew where and what types of biopsies to perform, a correct diagnosis was obtained quickly, allowing for the correct therapeutic intervention. After the diagnosis of HSP is made in an adult, nephrology should be consulted early in the treatment course.
To the Editor:
A 57-year-old man with a history of type 2 diabetes mellitus and hypertension was hospitalized for heart disease resulting in an aortic valve replacement and multiple-vessel bypass grafting. He experienced a stormy septic postoperative course during which he developed numerous palpable purplish plaques (Figure 1). The lesions were bilateral and more heavily involved the lower legs and buttocks. The head and neck remained free of skin lesions. Additionally, the patient reported a bilateral burning sensation from the knees to the feet.
Punch biopsies of lesions from the right upper arm were obtained. Hematoxylin and eosin staining revealed neutrophilic-predominant small vessel vasculitis (Figure 2A) with the upper dermal location more heavily involved, as demonstrated by involvement of a superficial vascular plexus (Figures 2B and 2C) that was consistent with Henoch-Schönlein purpura (HSP). The diagnosis later was confirmed with immunofluorescence. Direct immunofluorescence revealed granular IgA deposition around the superficial vascular plexus (Figure 3). No IgG, IgM, C3, C5b-9 complement complex, or fibrinogen deposition was seen. Additionally, periodic acid-Schiff staining failed to show microorganisms, thrombi, or intravascular hyaline material.
At our initial consultation, we observed an ill-appearing afebrile man with purplish plaques. Our impression was that he had vasculitis and not warfarin necrosis, which had been suspected by the cardiovascular team. The burning sensation noted by the patient lent credence to our vasculitic diagnosis. Proteinuria and hematuria were present; however, the values for blood urea nitrogen, creatinine, and glomerular filtration rate all remained within reference range. His signs and symptoms responded dramatically to prednisone. He remains on 1 mg of prednisone daily and a nephrologist continues to monitor renal function as an outpatient.
Henoch-Schönlein purpura is a systemic leukocytoclastic vasculitis involving small vessels. The small vessel vasculitis is associated with IgA antigen-antibody complex deposition in areas throughout the body. Palpable purpura typically is seen on the skin, which characteristically involves dependent areas such as the legs and the buttocks. Lesions normally are present bilaterally in a symmetric distribution. Initially, the lesions develop as erythematous macules that progress to purple, nonblanching, palpable, and purpuric plaques.1 Henoch-Schönlein purpura most commonly involves the skin; however, other locations for the immune complexes include the gastrointestinal tract, joints, and kidneys.2 The cause for the body's immunogenic deposition response is unknown in a majority of cases.
Henoch-Schönlein purpura most commonly is seen in the pediatric population with a predilection for males.3 The incidence in the pediatric population is 13.5 to 20 per 100,000 children per year; HSP is more rare in adults.4-6 Henoch-Schönlein purpura most often is a self-limiting disease that requires only supportive treatment. The signs and symptoms last 4 to 6 weeks in most patients and resolve completely in 94% of children and 89% of adults.7 Renal involvement carries a worse prognosis. Adult patients have a higher incidence of renal involvement, renal insufficiency, and subsequent progression to end-stage renal disease.3,8-10 In a study by Hung et al8 of 65 children and 22 adult HSP patients, 12 adults presented with renal involvement in which hematuria or proteinuria were present. Of them, 6 progressed to renal insufficiency (defined as having a plasma creatinine concentration>1.2 mg/dL).8 Fogazzi et al11 reported similar findings; 8 of 16 patients affected with HSP progressed to renal insufficiency with creatinine clearances ranging from 31 to 60 mL/min, and 3 patients required chronic dialysis. Pillebout et al9 evaluated 250 adults with HSP and 32% reached renal insufficiency with creatinine clearances of less than 50 mL/min, with 11% of patients developing end-stage renal disease. The degree of hematuria and/or proteinuria has been shown to be an effective prognostic indicator.9,10 Coppo et al10 found a similar prognosis among children and adults with HSP-related nephritis.
Our patient described the burning sensation as occurring bilaterally from the knees down to the feet, which provided an additional clue that small vessel vasculitis was involved, as occluded blood vessels can cause ischemia to nerves and perivascular involvement can affect nearby neural structures. Sais et al12 demonstrated that paresthesia in the setting of HSP was a risk factor for systemic involvement. Of note, our patient's paresthesia lasted only several days.
The cause of HSP is not always as evident in the adult population as in the pediatric population. Early diagnosis of HSP in adults may allow for the proper instatement of treatment to deter long-term renal complications. Follow-up with urinalysis is recommended because a small percentage of patients have a late progression to renal failure.13,14
Because the dermatologists involved in this case knew where and what types of biopsies to perform, a correct diagnosis was obtained quickly, allowing for the correct therapeutic intervention. After the diagnosis of HSP is made in an adult, nephrology should be consulted early in the treatment course.
- Rai A, Nast C, Adler S. Henoch-Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Helander SD, De Castro FR, Gibson LE. Henoch-Schönlein purpura: clinicopathologic correlation of cutaneous vascular IgA deposits and the relationship to leukocytoclastic vasculitis. Acta Derm Venereol. 1995;75:125-129.
- Garcia-Porrua C, Calvino MC, Llorca J, et al. Henoch-Schönlein purpura in children and adults: clinical differences in a defined population. Semin Arthritis Rheum. 2002;32:149-156.
- Stewart M, Savage JM, Bell B, et al. Long term renal prognosis of Henoch-Schönlein purpura in an unselected childhood population. Eur J Pediatr. 1988;147:113-115.
- Watts RA, Scott DG. Epidemiology of the vasculitides. Semin Respir Crit Care. 2004;25:455-464.
- Gardner-Medwin JM, Dolezalova P, Cummins C, et al. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. 2002;360:1197-1202.
- Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, et al. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum. 1997;40:859-864.
- Hung SP, Yang YH, Lin YT, et al. Clinical manifestations and outcomes of Henoch-Schönlein purpura: comparison between adults and children. Pediatr Neonatol. 2009;50:162-168.
- Pillebout E, Thervet E, Hill G, et al. Henoch-Schönlein purpura in adults: outcomes and prognostic factors. J Am Soc Nephrol. 2002;13:1271-1278.
- Coppo R, Mazzucco G, Cagnoli L, et al. Long-term prognosis of Henoch-Schönlein nephritis in adults and children. Italian Group of Renal Immunopathology collaborative study on Henoch-Schönlein purpura. Nephrol Dial Transplant. 1997;12:2277-2283.
- Fogazzi GB, Pasquali S, Moriggi M, et al. Long-term outcome of Schönlein-Henoch nephritis in the adult. Clin Nephrol. 1989;31:60-66.
- Sais G, Vidaller A, Jucgla A. Prognostic factors in leukocytoclastic vasculitis. a clinicopathologic study of 160 patients. Arch Dermatol. 1998;134:309-315.
- Kraft DM, McKee D, Scott C. Henoch-Schönlein purpura: a review. Am Fam Physician. 1998;58:405-408.
- Narchi H. Risk of long-term renal impairment and duration of follow up recommended for Henoch-Schönlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child. 2005;90:916-920.
- Rai A, Nast C, Adler S. Henoch-Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Helander SD, De Castro FR, Gibson LE. Henoch-Schönlein purpura: clinicopathologic correlation of cutaneous vascular IgA deposits and the relationship to leukocytoclastic vasculitis. Acta Derm Venereol. 1995;75:125-129.
- Garcia-Porrua C, Calvino MC, Llorca J, et al. Henoch-Schönlein purpura in children and adults: clinical differences in a defined population. Semin Arthritis Rheum. 2002;32:149-156.
- Stewart M, Savage JM, Bell B, et al. Long term renal prognosis of Henoch-Schönlein purpura in an unselected childhood population. Eur J Pediatr. 1988;147:113-115.
- Watts RA, Scott DG. Epidemiology of the vasculitides. Semin Respir Crit Care. 2004;25:455-464.
- Gardner-Medwin JM, Dolezalova P, Cummins C, et al. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. 2002;360:1197-1202.
- Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, et al. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum. 1997;40:859-864.
- Hung SP, Yang YH, Lin YT, et al. Clinical manifestations and outcomes of Henoch-Schönlein purpura: comparison between adults and children. Pediatr Neonatol. 2009;50:162-168.
- Pillebout E, Thervet E, Hill G, et al. Henoch-Schönlein purpura in adults: outcomes and prognostic factors. J Am Soc Nephrol. 2002;13:1271-1278.
- Coppo R, Mazzucco G, Cagnoli L, et al. Long-term prognosis of Henoch-Schönlein nephritis in adults and children. Italian Group of Renal Immunopathology collaborative study on Henoch-Schönlein purpura. Nephrol Dial Transplant. 1997;12:2277-2283.
- Fogazzi GB, Pasquali S, Moriggi M, et al. Long-term outcome of Schönlein-Henoch nephritis in the adult. Clin Nephrol. 1989;31:60-66.
- Sais G, Vidaller A, Jucgla A. Prognostic factors in leukocytoclastic vasculitis. a clinicopathologic study of 160 patients. Arch Dermatol. 1998;134:309-315.
- Kraft DM, McKee D, Scott C. Henoch-Schönlein purpura: a review. Am Fam Physician. 1998;58:405-408.
- Narchi H. Risk of long-term renal impairment and duration of follow up recommended for Henoch-Schönlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child. 2005;90:916-920.
Practice Points
- Henoch-Schönlein purpura is a multidisciplinary problem.
- Henoch-Schönlein purpura is an IgA-mediated disorder that is more common in children and has a more severe course in adults.
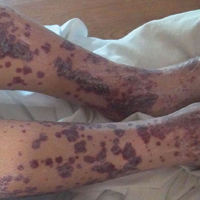
Recalcitrant Hyperkeratotic Plaques
The Diagnosis: Hypertrophic Lupus Erythematosus
Physical examination at initial presentation revealed well-demarcated, 2- to 3-cm plaques with scale distributed most extensively on the elbows and shins with lesser involvement of the chest and abdomen. After treatment with topical steroids, adalimumab, methotrexate, and narrowband UVB phototherapy, new annular, erythematous, and edematous lesions began to appear on the chest and abdomen (Figure 1). These new lesions appeared less hyperkeratotic than the older ones.
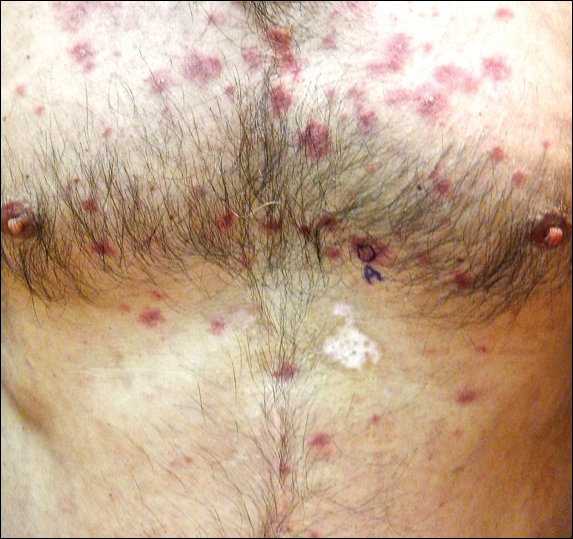
Biopsy of a hyperkeratotic lesion from the patient's arm revealed marked hyperkeratosis, parakeratosis, epidermal hyperplasia, focal vacuolar change, solar elastosis, and transepidermal elastotic elimination (Figure 2A). A second biopsy performed on a newer chest lesion revealed interface changes, degeneration of the basal layer, follicular plugging, and dermal mucin (Figure 2B). Serology revealed an antinuclear antibody (ANA) titer of 1:1280 (reference range, <1:40 dilution) and hemoglobin of 11.5 g/dL (reference range, 14.0-17.5 g/dL). On the basis of clinical, histologic, and serologic findings, hypertrophic lupus erythematosus (LE) was diagnosed. The patient was treated with oral prednisone, which resulted in rapid improvement.
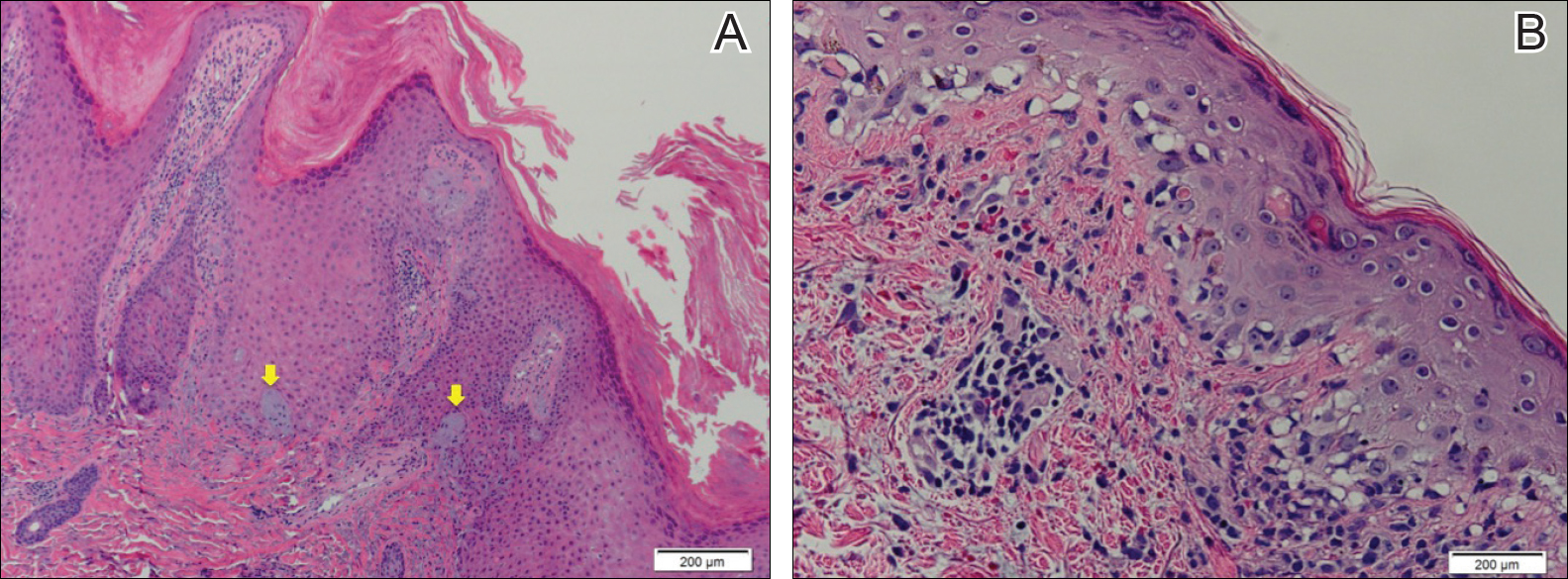
Hypertrophic LE is a rare subset of chronic cutaneous lupus first described by Behcet1 in 1942. Lesions are identified as verrucous keratotic plaques with a characteristic erythematous indurated border.2 Patients predominantly are middle-aged women with lesions distributed on sun-exposed areas. Most often, hypertrophic LE is seen in association with the classic lesions of discoid LE; however, patients may present exclusively with the cutaneous manifestations of hypertrophic LE. More rarely, as seen in this case, hypertrophic LE may present in conjunction with systemic features.3 The diagnosis of systemic LE requires 4 of the following criteria be fulfilled: malar rash; discoid rash; photosensitivity; oral ulcers; arthritis; cardiopulmonary serositis; renal involvement; positive ANA titer; and neurologic, hematologic, or immunologic disorders.4 Our patient qualified for discoid rash, photosensitivity, cardiopulmonary involvement with mitral valve defects and pulmonary pleuritis, hematologic disorder (anemia), and a positive ANA titer. Furthermore, in patients with only cutaneous discoid LE, serology generally reveals negative or low-titer ANA and negative anti-Ro antibodies.5
Hypertrophic LE is characterized histologically by irregular epidermal hyperplasia in association with features of classic cutaneous LE. Distinctive features of cutaneous LE include interface changes, follicular plugging, dermal mucin, and angiocentric lymphocytic inflammation.6 Notably, additional biopsies of the less hyperkeratotic lesions on our patient's chest and abdomen were performed, which revealed classic cutaneous LE features (Figure 2B).
Hypertrophic LE has 2 histological variants: lichen planus-like and keratoacanthoma (KA)-like patterns. Most cases are described as lichen planus-like, with a dense bandlike infiltrate in association with irregular epidermal hyperplasia, vacuolar interface changes, and reactive squamous atypia.5 In contrast, the less common KA-like lesions consist of a keratinous center with vigorous squamous epithelial proliferation.6
Clinically, hypertrophic LE may resemble hypertrophic psoriasis, lichen planus, KA, or squamous cell carcinoma (SCC). Due to the presence of pseudocarcinomatous hyperplasia, the histopathologic differential includes hypertrophic lichen planus, SCC, KA, and deep fungal infections. However, these other diseases lack the classic features of cutaneous LE, which include interface changes, follicular plugging, dermal mucin, and perivascular lymphocytic inflammation. Additionally, transepidermal elastotic elimination (Figure 2A) helps distinguish hypertrophic LE from other diagnoses.7 One of the most important tasks is distinguishing hypertrophic LE from SCC. Hypertrophic LE does not typically display eosinophil infiltrates, which differentiates it from SCC and KA. Additionally, studies report that CD123 positivity can be useful.6 Positive plasmacytoid dendritic cells are abundant at the dermoepidermal junction in hypertrophic LE, while only single or rare clusters of CD123+ cells are seen in SCC.8 Also, SCC has been found to arise in long-standing cutaneous LE lesions including both discoid and hypertrophic LE. Therefore, clinical and sometimes histological follow-up is required.
Hypertrophic LE often is challenging to treat and frequently is resistant to antimalarial drugs. The primary goals of treatment involve reducing inflammatory infiltrate and minimizing hyperkeratinization. Topical corticosteroids and calcineurin inhibitors often are inadequate as monotherapy due to reduced penetrance through the thick lesions; however, intralesional corticosteroids may be beneficial in patients with localized disease.9 Unfortunately, topical or intralesional treatments are impractical in patients with extensive lesions, as seen in our patient, in which case systemic corticosteroids can be beneficial.
Topical retinoids also have been found to be highly effective.10 Specifically, retinoids such as acitretin and isotretinoin, in some cases combined with antimalarial drugs, are effective in reducing the keratinization of these lesions. Successful treatment also has been reported with ustekinumab, thalidomide, mycophenolate mofetil, and pulsed dye laser.11 As in other types of cutaneous LE, hyperkeratotic LE is photosensitive; avoidance of prolonged sun exposure should be advised.8
- Bechet PE. Lupus erythematosus hypertrophicus et profundus. Arch Derm Syphilol. 1942;45:33-39.
- Bernardi M, Bahrami S, Callen JP. Hypertrophic lupus erythematous complicating long-standing systemic lupus erythematous. Lupus. 2011;20:549-550.
- Spann CR, Callen JP, Klein JB, et al. Clinical, serologic and immunogenetic studies in patients with chronic cutaneous (discoid) lupus erythematosus who have verrucous and/or hypertrophic skin lesions. J Rheumatol. 1988;15:256-261.
- Yu C, Gershwin E, Chang C. Diagnostic criteria for systemic lupus erythematosus: a critical review [published online January 21, 2014]. J Autoimmun. 2014;48-49:10-13.
- Provost TT. The relationship between discoid and systemic lupus erythematous. Arch Dermatol. 1994;130:1308-1310.
- Arps DP, Patel RM. Cutaneous hypertrophic lupus erythematous: a challenging histopathologic diagnosis in the absence of clinical information. Arch Pathol Lab Med. 2013;137:1205-1210.
- Daldon PE, De Souza EM, Cintra ML. Hypertrophic lupus erythematous: a clinicopathological study of 14 cases. J Cutan Pathol. 2003;30:443-448.
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophiclupus erythematous: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. issues in diagnosis and treatment. Am J Clin Dermatol. 2009;10:366-381.
- Al-Mutairi N, Rijhwani M, Nour-Eldin O. Hypertrophic lupus erythematosus treated successfully with acitretin as monotherapy. J Dermatol. 2005;32:482-486.
- Winchester D, Duffin KC, Hansen C. Response to ustekinumab in a patient with both severe psoriasis and hypertrophic cutaneous lupus. Lupus. 2012;12:1007-1010.
The Diagnosis: Hypertrophic Lupus Erythematosus
Physical examination at initial presentation revealed well-demarcated, 2- to 3-cm plaques with scale distributed most extensively on the elbows and shins with lesser involvement of the chest and abdomen. After treatment with topical steroids, adalimumab, methotrexate, and narrowband UVB phototherapy, new annular, erythematous, and edematous lesions began to appear on the chest and abdomen (Figure 1). These new lesions appeared less hyperkeratotic than the older ones.
Biopsy of a hyperkeratotic lesion from the patient's arm revealed marked hyperkeratosis, parakeratosis, epidermal hyperplasia, focal vacuolar change, solar elastosis, and transepidermal elastotic elimination (Figure 2A). A second biopsy performed on a newer chest lesion revealed interface changes, degeneration of the basal layer, follicular plugging, and dermal mucin (Figure 2B). Serology revealed an antinuclear antibody (ANA) titer of 1:1280 (reference range, <1:40 dilution) and hemoglobin of 11.5 g/dL (reference range, 14.0-17.5 g/dL). On the basis of clinical, histologic, and serologic findings, hypertrophic lupus erythematosus (LE) was diagnosed. The patient was treated with oral prednisone, which resulted in rapid improvement.
Hypertrophic LE is a rare subset of chronic cutaneous lupus first described by Behcet1 in 1942. Lesions are identified as verrucous keratotic plaques with a characteristic erythematous indurated border.2 Patients predominantly are middle-aged women with lesions distributed on sun-exposed areas. Most often, hypertrophic LE is seen in association with the classic lesions of discoid LE; however, patients may present exclusively with the cutaneous manifestations of hypertrophic LE. More rarely, as seen in this case, hypertrophic LE may present in conjunction with systemic features.3 The diagnosis of systemic LE requires 4 of the following criteria be fulfilled: malar rash; discoid rash; photosensitivity; oral ulcers; arthritis; cardiopulmonary serositis; renal involvement; positive ANA titer; and neurologic, hematologic, or immunologic disorders.4 Our patient qualified for discoid rash, photosensitivity, cardiopulmonary involvement with mitral valve defects and pulmonary pleuritis, hematologic disorder (anemia), and a positive ANA titer. Furthermore, in patients with only cutaneous discoid LE, serology generally reveals negative or low-titer ANA and negative anti-Ro antibodies.5
Hypertrophic LE is characterized histologically by irregular epidermal hyperplasia in association with features of classic cutaneous LE. Distinctive features of cutaneous LE include interface changes, follicular plugging, dermal mucin, and angiocentric lymphocytic inflammation.6 Notably, additional biopsies of the less hyperkeratotic lesions on our patient's chest and abdomen were performed, which revealed classic cutaneous LE features (Figure 2B).
Hypertrophic LE has 2 histological variants: lichen planus-like and keratoacanthoma (KA)-like patterns. Most cases are described as lichen planus-like, with a dense bandlike infiltrate in association with irregular epidermal hyperplasia, vacuolar interface changes, and reactive squamous atypia.5 In contrast, the less common KA-like lesions consist of a keratinous center with vigorous squamous epithelial proliferation.6
Clinically, hypertrophic LE may resemble hypertrophic psoriasis, lichen planus, KA, or squamous cell carcinoma (SCC). Due to the presence of pseudocarcinomatous hyperplasia, the histopathologic differential includes hypertrophic lichen planus, SCC, KA, and deep fungal infections. However, these other diseases lack the classic features of cutaneous LE, which include interface changes, follicular plugging, dermal mucin, and perivascular lymphocytic inflammation. Additionally, transepidermal elastotic elimination (Figure 2A) helps distinguish hypertrophic LE from other diagnoses.7 One of the most important tasks is distinguishing hypertrophic LE from SCC. Hypertrophic LE does not typically display eosinophil infiltrates, which differentiates it from SCC and KA. Additionally, studies report that CD123 positivity can be useful.6 Positive plasmacytoid dendritic cells are abundant at the dermoepidermal junction in hypertrophic LE, while only single or rare clusters of CD123+ cells are seen in SCC.8 Also, SCC has been found to arise in long-standing cutaneous LE lesions including both discoid and hypertrophic LE. Therefore, clinical and sometimes histological follow-up is required.
Hypertrophic LE often is challenging to treat and frequently is resistant to antimalarial drugs. The primary goals of treatment involve reducing inflammatory infiltrate and minimizing hyperkeratinization. Topical corticosteroids and calcineurin inhibitors often are inadequate as monotherapy due to reduced penetrance through the thick lesions; however, intralesional corticosteroids may be beneficial in patients with localized disease.9 Unfortunately, topical or intralesional treatments are impractical in patients with extensive lesions, as seen in our patient, in which case systemic corticosteroids can be beneficial.
Topical retinoids also have been found to be highly effective.10 Specifically, retinoids such as acitretin and isotretinoin, in some cases combined with antimalarial drugs, are effective in reducing the keratinization of these lesions. Successful treatment also has been reported with ustekinumab, thalidomide, mycophenolate mofetil, and pulsed dye laser.11 As in other types of cutaneous LE, hyperkeratotic LE is photosensitive; avoidance of prolonged sun exposure should be advised.8
The Diagnosis: Hypertrophic Lupus Erythematosus
Physical examination at initial presentation revealed well-demarcated, 2- to 3-cm plaques with scale distributed most extensively on the elbows and shins with lesser involvement of the chest and abdomen. After treatment with topical steroids, adalimumab, methotrexate, and narrowband UVB phototherapy, new annular, erythematous, and edematous lesions began to appear on the chest and abdomen (Figure 1). These new lesions appeared less hyperkeratotic than the older ones.
Biopsy of a hyperkeratotic lesion from the patient's arm revealed marked hyperkeratosis, parakeratosis, epidermal hyperplasia, focal vacuolar change, solar elastosis, and transepidermal elastotic elimination (Figure 2A). A second biopsy performed on a newer chest lesion revealed interface changes, degeneration of the basal layer, follicular plugging, and dermal mucin (Figure 2B). Serology revealed an antinuclear antibody (ANA) titer of 1:1280 (reference range, <1:40 dilution) and hemoglobin of 11.5 g/dL (reference range, 14.0-17.5 g/dL). On the basis of clinical, histologic, and serologic findings, hypertrophic lupus erythematosus (LE) was diagnosed. The patient was treated with oral prednisone, which resulted in rapid improvement.
Hypertrophic LE is a rare subset of chronic cutaneous lupus first described by Behcet1 in 1942. Lesions are identified as verrucous keratotic plaques with a characteristic erythematous indurated border.2 Patients predominantly are middle-aged women with lesions distributed on sun-exposed areas. Most often, hypertrophic LE is seen in association with the classic lesions of discoid LE; however, patients may present exclusively with the cutaneous manifestations of hypertrophic LE. More rarely, as seen in this case, hypertrophic LE may present in conjunction with systemic features.3 The diagnosis of systemic LE requires 4 of the following criteria be fulfilled: malar rash; discoid rash; photosensitivity; oral ulcers; arthritis; cardiopulmonary serositis; renal involvement; positive ANA titer; and neurologic, hematologic, or immunologic disorders.4 Our patient qualified for discoid rash, photosensitivity, cardiopulmonary involvement with mitral valve defects and pulmonary pleuritis, hematologic disorder (anemia), and a positive ANA titer. Furthermore, in patients with only cutaneous discoid LE, serology generally reveals negative or low-titer ANA and negative anti-Ro antibodies.5
Hypertrophic LE is characterized histologically by irregular epidermal hyperplasia in association with features of classic cutaneous LE. Distinctive features of cutaneous LE include interface changes, follicular plugging, dermal mucin, and angiocentric lymphocytic inflammation.6 Notably, additional biopsies of the less hyperkeratotic lesions on our patient's chest and abdomen were performed, which revealed classic cutaneous LE features (Figure 2B).
Hypertrophic LE has 2 histological variants: lichen planus-like and keratoacanthoma (KA)-like patterns. Most cases are described as lichen planus-like, with a dense bandlike infiltrate in association with irregular epidermal hyperplasia, vacuolar interface changes, and reactive squamous atypia.5 In contrast, the less common KA-like lesions consist of a keratinous center with vigorous squamous epithelial proliferation.6
Clinically, hypertrophic LE may resemble hypertrophic psoriasis, lichen planus, KA, or squamous cell carcinoma (SCC). Due to the presence of pseudocarcinomatous hyperplasia, the histopathologic differential includes hypertrophic lichen planus, SCC, KA, and deep fungal infections. However, these other diseases lack the classic features of cutaneous LE, which include interface changes, follicular plugging, dermal mucin, and perivascular lymphocytic inflammation. Additionally, transepidermal elastotic elimination (Figure 2A) helps distinguish hypertrophic LE from other diagnoses.7 One of the most important tasks is distinguishing hypertrophic LE from SCC. Hypertrophic LE does not typically display eosinophil infiltrates, which differentiates it from SCC and KA. Additionally, studies report that CD123 positivity can be useful.6 Positive plasmacytoid dendritic cells are abundant at the dermoepidermal junction in hypertrophic LE, while only single or rare clusters of CD123+ cells are seen in SCC.8 Also, SCC has been found to arise in long-standing cutaneous LE lesions including both discoid and hypertrophic LE. Therefore, clinical and sometimes histological follow-up is required.
Hypertrophic LE often is challenging to treat and frequently is resistant to antimalarial drugs. The primary goals of treatment involve reducing inflammatory infiltrate and minimizing hyperkeratinization. Topical corticosteroids and calcineurin inhibitors often are inadequate as monotherapy due to reduced penetrance through the thick lesions; however, intralesional corticosteroids may be beneficial in patients with localized disease.9 Unfortunately, topical or intralesional treatments are impractical in patients with extensive lesions, as seen in our patient, in which case systemic corticosteroids can be beneficial.
Topical retinoids also have been found to be highly effective.10 Specifically, retinoids such as acitretin and isotretinoin, in some cases combined with antimalarial drugs, are effective in reducing the keratinization of these lesions. Successful treatment also has been reported with ustekinumab, thalidomide, mycophenolate mofetil, and pulsed dye laser.11 As in other types of cutaneous LE, hyperkeratotic LE is photosensitive; avoidance of prolonged sun exposure should be advised.8
- Bechet PE. Lupus erythematosus hypertrophicus et profundus. Arch Derm Syphilol. 1942;45:33-39.
- Bernardi M, Bahrami S, Callen JP. Hypertrophic lupus erythematous complicating long-standing systemic lupus erythematous. Lupus. 2011;20:549-550.
- Spann CR, Callen JP, Klein JB, et al. Clinical, serologic and immunogenetic studies in patients with chronic cutaneous (discoid) lupus erythematosus who have verrucous and/or hypertrophic skin lesions. J Rheumatol. 1988;15:256-261.
- Yu C, Gershwin E, Chang C. Diagnostic criteria for systemic lupus erythematosus: a critical review [published online January 21, 2014]. J Autoimmun. 2014;48-49:10-13.
- Provost TT. The relationship between discoid and systemic lupus erythematous. Arch Dermatol. 1994;130:1308-1310.
- Arps DP, Patel RM. Cutaneous hypertrophic lupus erythematous: a challenging histopathologic diagnosis in the absence of clinical information. Arch Pathol Lab Med. 2013;137:1205-1210.
- Daldon PE, De Souza EM, Cintra ML. Hypertrophic lupus erythematous: a clinicopathological study of 14 cases. J Cutan Pathol. 2003;30:443-448.
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophiclupus erythematous: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. issues in diagnosis and treatment. Am J Clin Dermatol. 2009;10:366-381.
- Al-Mutairi N, Rijhwani M, Nour-Eldin O. Hypertrophic lupus erythematosus treated successfully with acitretin as monotherapy. J Dermatol. 2005;32:482-486.
- Winchester D, Duffin KC, Hansen C. Response to ustekinumab in a patient with both severe psoriasis and hypertrophic cutaneous lupus. Lupus. 2012;12:1007-1010.
- Bechet PE. Lupus erythematosus hypertrophicus et profundus. Arch Derm Syphilol. 1942;45:33-39.
- Bernardi M, Bahrami S, Callen JP. Hypertrophic lupus erythematous complicating long-standing systemic lupus erythematous. Lupus. 2011;20:549-550.
- Spann CR, Callen JP, Klein JB, et al. Clinical, serologic and immunogenetic studies in patients with chronic cutaneous (discoid) lupus erythematosus who have verrucous and/or hypertrophic skin lesions. J Rheumatol. 1988;15:256-261.
- Yu C, Gershwin E, Chang C. Diagnostic criteria for systemic lupus erythematosus: a critical review [published online January 21, 2014]. J Autoimmun. 2014;48-49:10-13.
- Provost TT. The relationship between discoid and systemic lupus erythematous. Arch Dermatol. 1994;130:1308-1310.
- Arps DP, Patel RM. Cutaneous hypertrophic lupus erythematous: a challenging histopathologic diagnosis in the absence of clinical information. Arch Pathol Lab Med. 2013;137:1205-1210.
- Daldon PE, De Souza EM, Cintra ML. Hypertrophic lupus erythematous: a clinicopathological study of 14 cases. J Cutan Pathol. 2003;30:443-448.
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophiclupus erythematous: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. issues in diagnosis and treatment. Am J Clin Dermatol. 2009;10:366-381.
- Al-Mutairi N, Rijhwani M, Nour-Eldin O. Hypertrophic lupus erythematosus treated successfully with acitretin as monotherapy. J Dermatol. 2005;32:482-486.
- Winchester D, Duffin KC, Hansen C. Response to ustekinumab in a patient with both severe psoriasis and hypertrophic cutaneous lupus. Lupus. 2012;12:1007-1010.
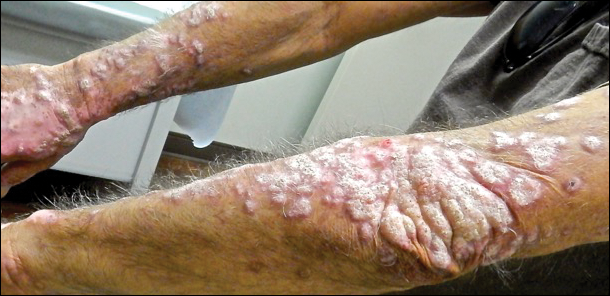
A 53-year-old man presented with a persistent, hyperkeratotic, pruritic rash on the arms, chest, and abdomen. The patient was treated for presumed psoriasis for 9 months by a primary care physician. However, despite an extensive treatment history, which included topical steroids, adalimumab, methotrexate, and narrowband UVB phototherapy, his condition worsened, and new erythematous and edematous lesions with no scale appeared on the back and chest. The patient's history also was notable for splenic rupture and mitral valve defects for which he was maintained on warfarin. In addition, he was evaluated by an allergist for new-onset dyspnea and treated with prednisone, which subsequently resulted in partial resolution of the skin lesions.
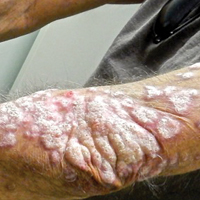
Friable Warty Plaque on the Heel
The Diagnosis: Verrucous Hemangioma
Verrucous hemangioma (VH) is a rare vascular anomaly that has not been definitively delineated as a malformation or a tumor, as it has features of both. Verrucous hemangioma presents at birth as a compressible soft mass with a red violaceous hue favoring the legs.1,2 Over time VH will develop a warty, friable, and keratotic surface that can begin to evolve as early as 6 months or as late as 34 years of age.3 Verrucous hemangioma does not involute and tends to grow proportionally with the patient. Thus, VH classically has been considered a vascular malformation.
On histopathology VH shows collections of uniform, thin-walled vessels with a multilamellated basement membrane throughout the dermis, similar to an infantile hemangioma (IH). These lesions extend deep into the subcutaneous tissue and often involve the underlying fascia. The papillary dermis has large ectatic vessels, while the epidermis displays verrucous hyperkeratosis, papillomatosis, and irregular acanthosis without viral change (Figure).4,5 The superficial component can resemble an angiokeratoma; however, VH is differentiated by a deeper component that is often larger in size and has a more protracted clinical course.
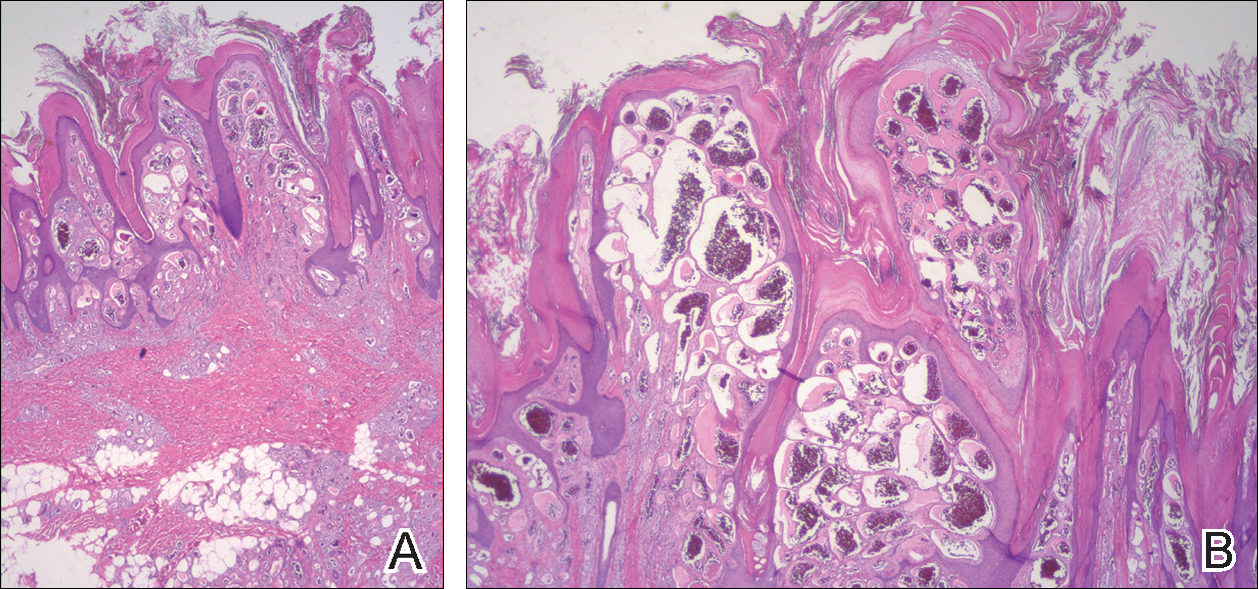
Similar to IH, immunohistochemical studies have shown that VH expresses Wilms tumor 1 and glucose transporter 1 but is negative for D2-40.4 These findings suggest that VH is a vascular tumor rather than a vascular malformation, as was previously reported.6 Additional research has shown that the immunohistochemical staining profile of VH is nearly identical to IH, which has led to postulation that VH may be of placental mesodermal origin, as has been hypothesized for IH.5
Due to its deep infiltration and tendency for recurrence, VH is most effectively treated with wide local excision.3,6-8 Preoperative planning with magnetic resonance imaging may be indicated. Although laser monotherapy and other local destructive therapies have been largely unsuccessful, postsurgical laser therapy with CO2 lasers as well as dual pulsed dye laser and Nd:YAG laser have shown promise in preventing recurrence.3
- Tennant LB, Mulliken JB, Perez-Atayde AR, et al. Verrucous hemangioma revisited. Pediatr Dermatol. 2006;23:208-215.
- Koc M, Kavala M, Kocatür E, et al. An unusual vascular tumor: verrucous hemangioma. Dermatol Online J. 2009;15:7.
- Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919; discussion 920.
- Trindade F, Torrelo A, Requena L, et al. An immunohistochemical study of verrucous hemangiomas. J Cutan Pathol. 2013;40:472-476.
- Laing EL, Brasch HD, Steel R, et al. Verrucous hemangioma expresses primitive markers. J Cutan Pathol. 2013;40:391-396.
- Mankani MH, Dufresne CR. Verrucous malformations: their presentation and management. Ann Plast Surg. 2000;45:31-36.
- Clairwood MQ, Bruckner AL, Dadras SS. Verrucous hemangioma: a report of two cases and review of the literature. J Cutan Pathol. 2011;38:740-746.
- Segura Palacios JM, Boixeda P, Rocha J, et al. Laser treatment for verrucous hemangioma. Laser Med Sci. 2012;27:681-684.
The Diagnosis: Verrucous Hemangioma
Verrucous hemangioma (VH) is a rare vascular anomaly that has not been definitively delineated as a malformation or a tumor, as it has features of both. Verrucous hemangioma presents at birth as a compressible soft mass with a red violaceous hue favoring the legs.1,2 Over time VH will develop a warty, friable, and keratotic surface that can begin to evolve as early as 6 months or as late as 34 years of age.3 Verrucous hemangioma does not involute and tends to grow proportionally with the patient. Thus, VH classically has been considered a vascular malformation.
On histopathology VH shows collections of uniform, thin-walled vessels with a multilamellated basement membrane throughout the dermis, similar to an infantile hemangioma (IH). These lesions extend deep into the subcutaneous tissue and often involve the underlying fascia. The papillary dermis has large ectatic vessels, while the epidermis displays verrucous hyperkeratosis, papillomatosis, and irregular acanthosis without viral change (Figure).4,5 The superficial component can resemble an angiokeratoma; however, VH is differentiated by a deeper component that is often larger in size and has a more protracted clinical course.
Similar to IH, immunohistochemical studies have shown that VH expresses Wilms tumor 1 and glucose transporter 1 but is negative for D2-40.4 These findings suggest that VH is a vascular tumor rather than a vascular malformation, as was previously reported.6 Additional research has shown that the immunohistochemical staining profile of VH is nearly identical to IH, which has led to postulation that VH may be of placental mesodermal origin, as has been hypothesized for IH.5
Due to its deep infiltration and tendency for recurrence, VH is most effectively treated with wide local excision.3,6-8 Preoperative planning with magnetic resonance imaging may be indicated. Although laser monotherapy and other local destructive therapies have been largely unsuccessful, postsurgical laser therapy with CO2 lasers as well as dual pulsed dye laser and Nd:YAG laser have shown promise in preventing recurrence.3
The Diagnosis: Verrucous Hemangioma
Verrucous hemangioma (VH) is a rare vascular anomaly that has not been definitively delineated as a malformation or a tumor, as it has features of both. Verrucous hemangioma presents at birth as a compressible soft mass with a red violaceous hue favoring the legs.1,2 Over time VH will develop a warty, friable, and keratotic surface that can begin to evolve as early as 6 months or as late as 34 years of age.3 Verrucous hemangioma does not involute and tends to grow proportionally with the patient. Thus, VH classically has been considered a vascular malformation.
On histopathology VH shows collections of uniform, thin-walled vessels with a multilamellated basement membrane throughout the dermis, similar to an infantile hemangioma (IH). These lesions extend deep into the subcutaneous tissue and often involve the underlying fascia. The papillary dermis has large ectatic vessels, while the epidermis displays verrucous hyperkeratosis, papillomatosis, and irregular acanthosis without viral change (Figure).4,5 The superficial component can resemble an angiokeratoma; however, VH is differentiated by a deeper component that is often larger in size and has a more protracted clinical course.
Similar to IH, immunohistochemical studies have shown that VH expresses Wilms tumor 1 and glucose transporter 1 but is negative for D2-40.4 These findings suggest that VH is a vascular tumor rather than a vascular malformation, as was previously reported.6 Additional research has shown that the immunohistochemical staining profile of VH is nearly identical to IH, which has led to postulation that VH may be of placental mesodermal origin, as has been hypothesized for IH.5
Due to its deep infiltration and tendency for recurrence, VH is most effectively treated with wide local excision.3,6-8 Preoperative planning with magnetic resonance imaging may be indicated. Although laser monotherapy and other local destructive therapies have been largely unsuccessful, postsurgical laser therapy with CO2 lasers as well as dual pulsed dye laser and Nd:YAG laser have shown promise in preventing recurrence.3
- Tennant LB, Mulliken JB, Perez-Atayde AR, et al. Verrucous hemangioma revisited. Pediatr Dermatol. 2006;23:208-215.
- Koc M, Kavala M, Kocatür E, et al. An unusual vascular tumor: verrucous hemangioma. Dermatol Online J. 2009;15:7.
- Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919; discussion 920.
- Trindade F, Torrelo A, Requena L, et al. An immunohistochemical study of verrucous hemangiomas. J Cutan Pathol. 2013;40:472-476.
- Laing EL, Brasch HD, Steel R, et al. Verrucous hemangioma expresses primitive markers. J Cutan Pathol. 2013;40:391-396.
- Mankani MH, Dufresne CR. Verrucous malformations: their presentation and management. Ann Plast Surg. 2000;45:31-36.
- Clairwood MQ, Bruckner AL, Dadras SS. Verrucous hemangioma: a report of two cases and review of the literature. J Cutan Pathol. 2011;38:740-746.
- Segura Palacios JM, Boixeda P, Rocha J, et al. Laser treatment for verrucous hemangioma. Laser Med Sci. 2012;27:681-684.
- Tennant LB, Mulliken JB, Perez-Atayde AR, et al. Verrucous hemangioma revisited. Pediatr Dermatol. 2006;23:208-215.
- Koc M, Kavala M, Kocatür E, et al. An unusual vascular tumor: verrucous hemangioma. Dermatol Online J. 2009;15:7.
- Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919; discussion 920.
- Trindade F, Torrelo A, Requena L, et al. An immunohistochemical study of verrucous hemangiomas. J Cutan Pathol. 2013;40:472-476.
- Laing EL, Brasch HD, Steel R, et al. Verrucous hemangioma expresses primitive markers. J Cutan Pathol. 2013;40:391-396.
- Mankani MH, Dufresne CR. Verrucous malformations: their presentation and management. Ann Plast Surg. 2000;45:31-36.
- Clairwood MQ, Bruckner AL, Dadras SS. Verrucous hemangioma: a report of two cases and review of the literature. J Cutan Pathol. 2011;38:740-746.
- Segura Palacios JM, Boixeda P, Rocha J, et al. Laser treatment for verrucous hemangioma. Laser Med Sci. 2012;27:681-684.
A 31-year-old man presented with a large friable and warty plaque on the left heel. He recalled that the lesion had been present since birth as a flat red birthmark that grew proportionally with him. Throughout his adolescence its surface became increasingly rough and bumpy. The patient described receiving laser treatment twice in his early 20s without notable improvement. He wanted the lesion removed because it was easily traumatized, resulting in bleeding, pain, and infection. The patient reported being otherwise healthy.