User login
Erythematous Pearly Papule on the Chest
Primary Cutaneous B-cell Lymphoma
Cutaneous B-cell lymphomas (CBCLs) are a diverse but rare group of cutaneous lymphoproliferative neoplasms that make up approximately 20% of the total number of hematolymphoid neoplasms primary to the skin.1 These lymphomas are comprised of neoplastic B cells in various stages of differentiation. As a whole, they are rare neoplasms that primarily involve the head, neck, trunk, arms, or legs.1 Clinically, patients present with nontender, compressible, solitary, red to violaceous papules or nodules. Most CBCLs are considered low-grade malignancies with nonaggressive behavior and excellent prognosis; however, the diffuse large B-cell lymphomas, including but not limited to intravascular and leg type; lymphomatoid granulomatosis; and B-cell lymphoblastic lymphoma can act more aggressively.1
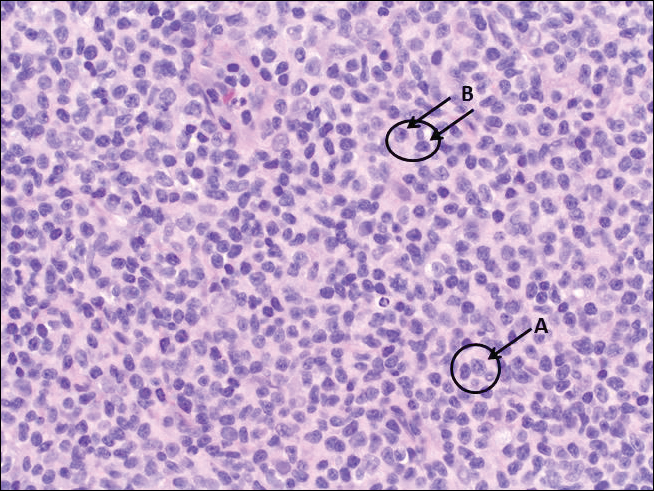
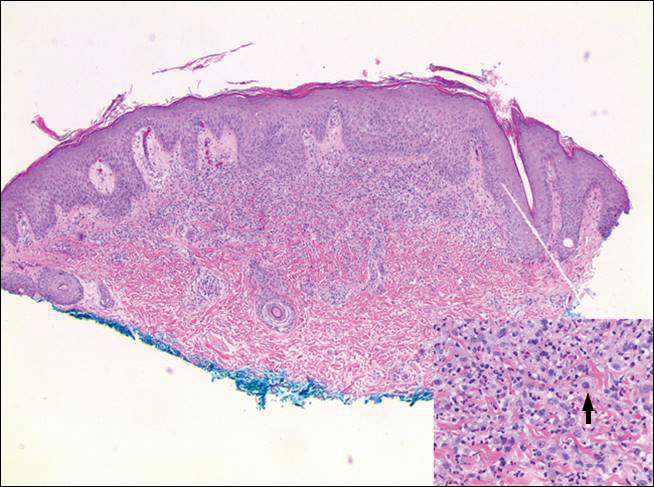
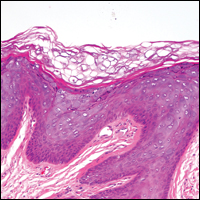
Histopathologic examination of primary CBCL generally reveals a relatively normal epidermis accompanied by a nodular to diffuse monomorphic lymphocytic cellular infiltrate in the dermis that can occasionally extend into the subcutaneous tissue (quiz image). Although not specific for CBCLs, oftentimes there is an acellular portion of the superficial papillary dermis known as a grenz zone that can serve as a histopathologic clue to the diagnosis of a cutaneous lymphoproliferative disorder. The list of malignant B-cell neoplasms is extensive (eg, cutaneous marginal zone B-cell lymphoma, primary cutaneous follicle center lymphoma, diffuse large B-cell lymphoma, intravascular large B-cell lymphoma), and few are seen in the skin.
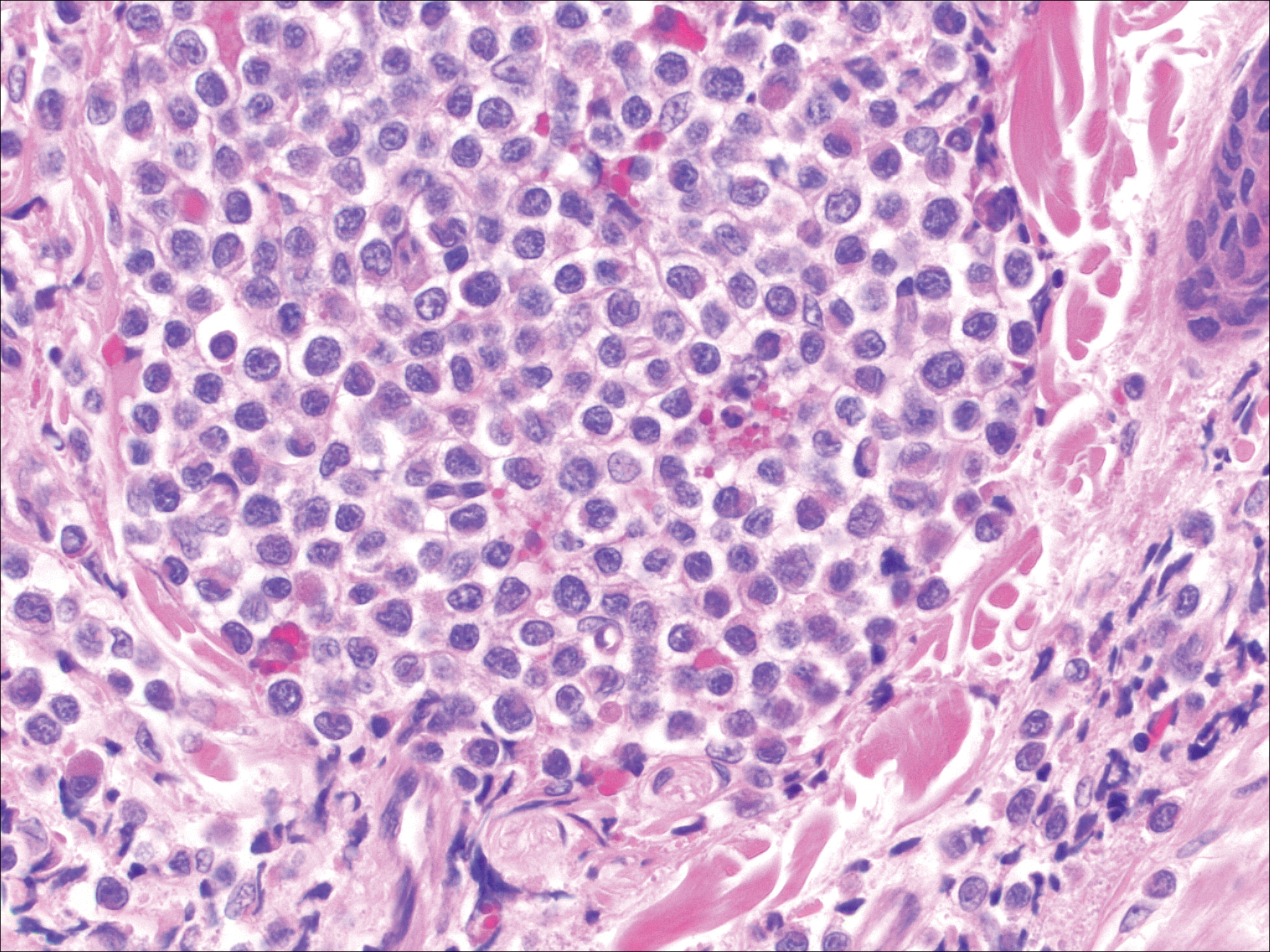
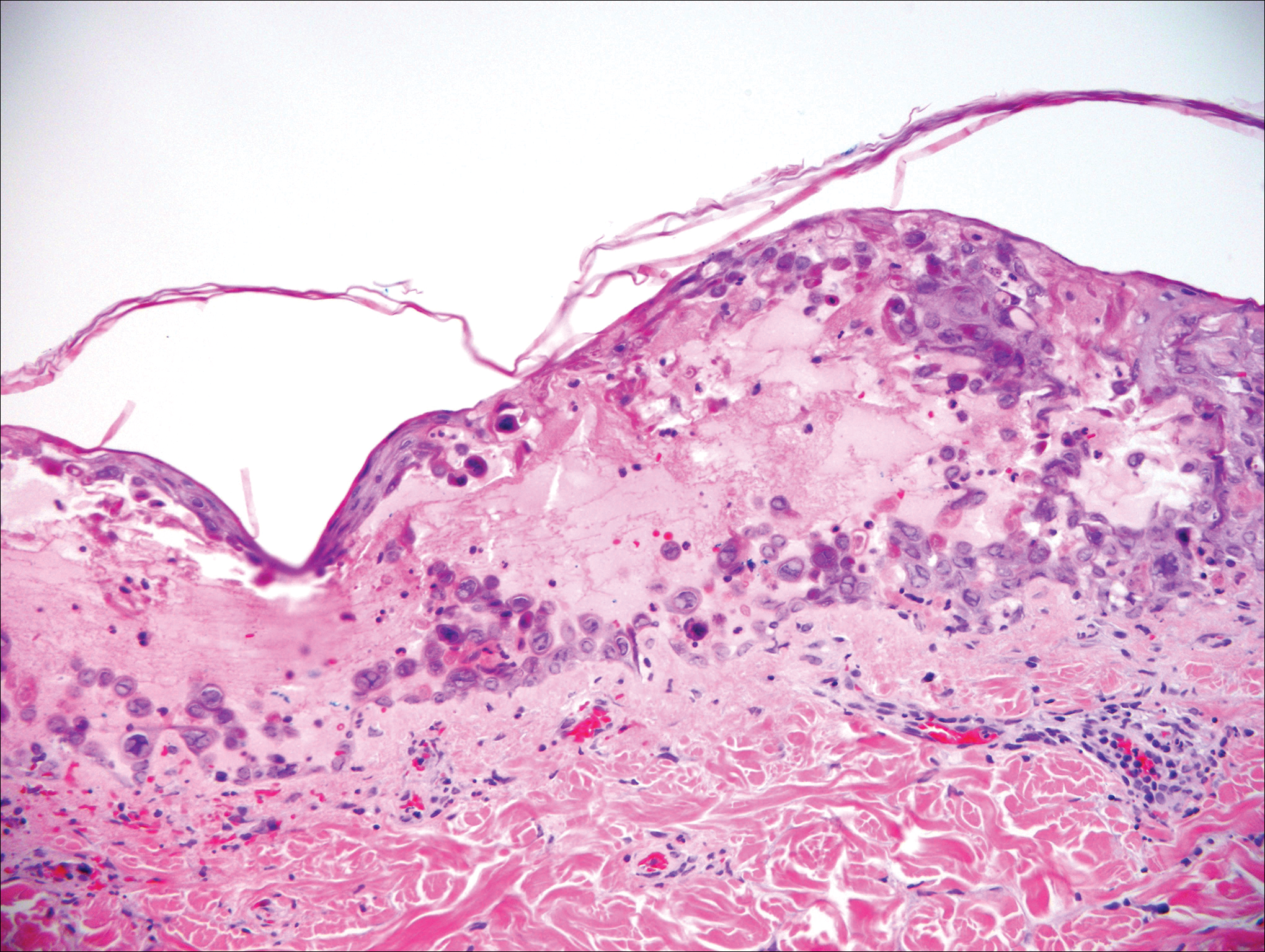
The most common type of CBCL is marginal zone B-cell lymphoma, which is considered to be a tumor of mucosa-associated (or skin-associated) lymphoid tissue. It is characterized by a monomorphous population of small mature lymphocytes showing characteristics of the B cells of the marginal zone of the lymph node. Some cells have the features of centrocytes/centroblasts (Figure 1) demonstrated by slightly irregular or indented nuclei and generous amounts of cytoplasm. Larger and more pleomorphic cells such as immunoblasts are similarly noted (Figure 1). The quiz image and Figure 1 demonstrate a cutaneous marginal zone B-cell lymphoma. A histomorphologic clue supporting a diagnosis of marginal zone B-cell lymphoma over reactive lymphoid hyperplasia is a B-cell predominate (B- to T-cell ratio of at least 3 to 1) infiltrate that is comprised of marginal zone-type cells. Immunohistochemistry demonstrating fewer differentiated B cells with light chain restriction may provide additional evidence that supports a clonal and potentially malignant process.
Erythematous to violaceous nodules on the head and neck of older individuals are characteristic of both granuloma faciale and CBCL. Histologically, granuloma faciale is characterized by a dense cellular infiltrate, often with a nodular outline, occupying the mid dermis.2 Granuloma faciale typically spares the immediate subepidermis and hair follicles, forming a grenz zone. The cellular infiltrate is polymorphic and consists of eosinophils and neutrophils with scattered plasma cells, mast cells, and lymphocytes in a vasculocentric distribution, eventually with chronic concentric fibrosis (Figure 2).
Leukemia cutis demonstrates a dermal infiltrate that contains atypical mononuclear cells (myeloblasts and myelocytes)(Figure 3).3 These markedly atypical mononuclear cells can have kidney bean-shaped nuclei and percolate through the dermal collagen, resembling single-file cells. They have increased nuclear to cytoplasmic ratios and occasionally have prominent nucleoli. Correlation with immunophenotypic and cytochemical studies is required for specific typing of the leukemic infiltrate.
Similar to primary CBCL, lymphomatoid papulosis (LyP) consists of erythematous papules or nodules that can occur anywhere on the body. In contrast to CBCL, the lesions of LyP classically self-resolve. However, approximately 10% to 20% of patients develop a malignant lymphoma, with mycosis fungoides, Hodgkin disease, and anaplastic large cell lymphoma being the most commonly associated.
Histologic examination of lesions of LyP classically demonstrates a wedge-shaped dermal infiltrate with variable epidermal changes (Figure 4). The wedge-shaped infiltrate is composed of large atypical cells. Three main types of lesions have been delineated: types A, B, and C. Type A is characterized by an increased number of cells with large vesicular nuclei with clumped chromatin, prominent nucleoli, and pronounced cytoplasm. Reed-Sternberg-like cells with an admixture of inflammatory cells including small lymphocytes, macrophages, neutrophils, and eosinophils also are present. Type B neoplastic cells vary in size and feature hyperchromatic, convoluted, or cerebriform nuclei. The infiltrate can be dense and bandlike with fewer cells resembling mycosis fungoides; type B LyP has neoplastic cells, not inflammatory cells. Finally, type C demonstrates solid sheets of large atypical cells resembling anaplastic large cell lymphoma. Immunohistochemically, the atypical cells often are CD4+ and CD8- with variable loss of pan-T-cell antigens. The atypical cells of types A and C express CD30 reactivity.4
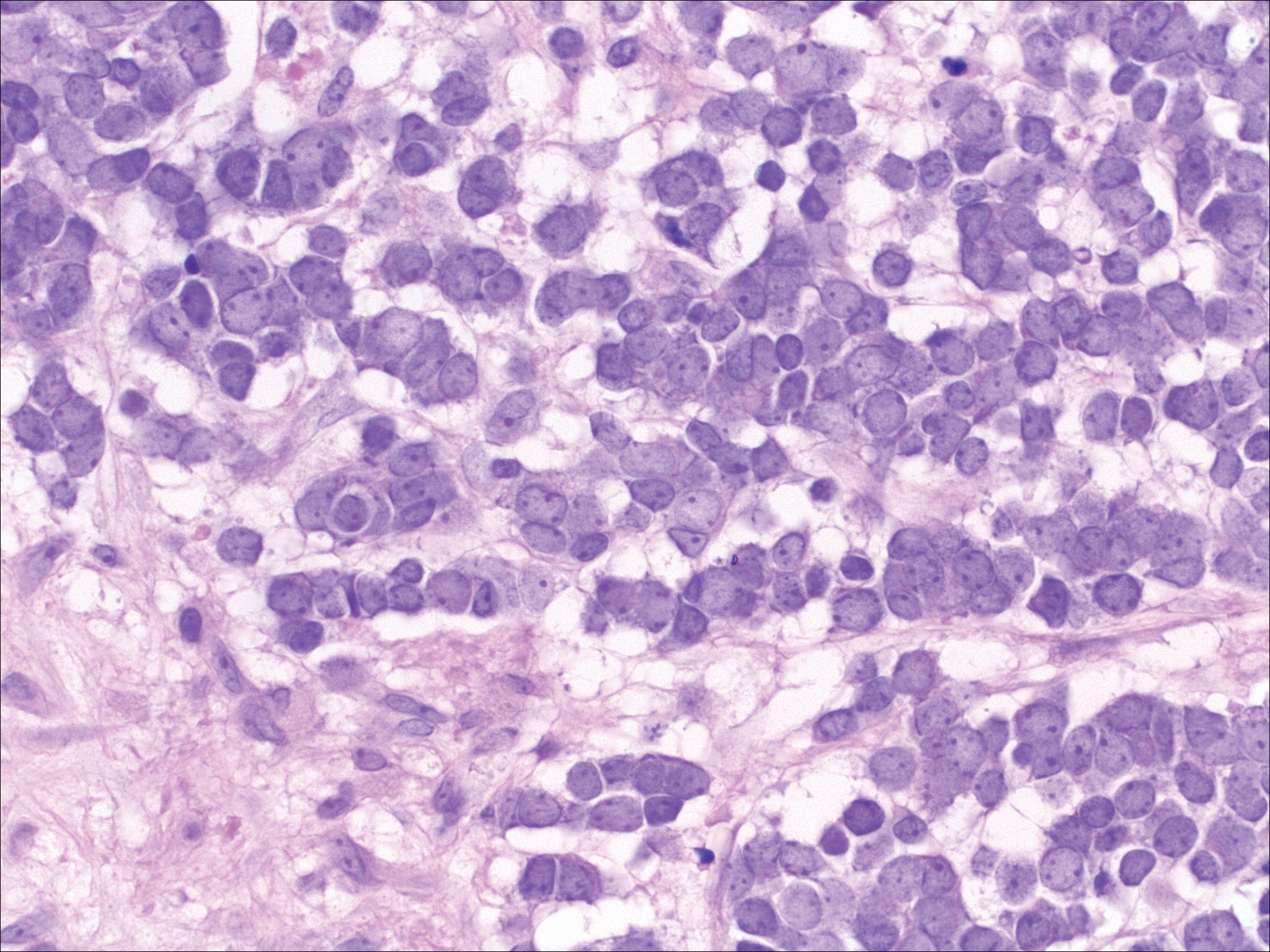
Merkel cell carcinoma (MCC) is a primary neuroendocrine carcinoma of the skin that usually arises on sun-exposed skin in elderly patients with lesions that histologically and clinically resemble cutaneous lymphoma.5 It classically is composed of small, round to oval, basophilic cells with a vesicular nucleus and multiple small nucleoli. Apoptotic cells and mitoses often are present.6 One key finding that helps to differentiate MCC from lymphoma is the presence of finely dispersed salt-and-pepper chromatin and molded nuclear contour in MCC (Figure 5).
Immunophenotyping is important in the differentiation of these diagnoses. The atypical cells of LyP are positive for CD3, CD4, and CD30 but are negative for CD8. However, in type B LyP, the large CD30+ cells seen in the other types are not commonly seen. In contrast, MCC expresses reactivity with cytokeratins, in particular cytokeratin 20 and CAM5.2, classically in a paranuclear dotlike pattern. In keeping with MCC's neuroendocrine differentiation, the tumor cells will demonstrate reactivity with synaptophysin, chromogranin, and CD56. The immunohistochemistry for leukemia cutis varies depending on the type of leukemia. Acute myelomonocytic leukemia is positive for myeloperoxidase, CD13, CD33, and CD68. The immunophenotype of these marginal zone lymphoma cells is as follows: positive for CD20, CD79a, and Bcl-2; negative for Bcl-6, CD5, CD10, CD23, and cyclin D1 (Bcl-1).7
- Olsen EA. Evaluation, diagnosis, and staging of cutaneous lymphoma. Dermatol Clin. 2015;33:643-654.
- Ortonne N, Wechsler J, Bagot M, et al. Granuloma faciale: a clinicopathologic study of 66 patients. J Am Acad Dermatol. 2005;53:1002-1009.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Wieser I, Wohlmuth C, Nunez CA, et al. Lymphomatoid papulosis in children and adolescents: a systematic review. Am J Clin Dermatol. 2016;17:319-327.
- Sibley RK, Dehner LP, Rosai J. Primary neuroendocrine (Merkel cell?) carcinoma of the skin: I. a clinicopathologic and ultrastructural study of 43 cases. Am J Surg Pathol. 1985;9:95-108.
- Frigerio B, Capella C, Eusebi V, et al. Merkel cell carcinoma of the skin: the structure and origin of normal Merkel cells. Histopathology. 1983;7:229-249.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
Primary Cutaneous B-cell Lymphoma
Cutaneous B-cell lymphomas (CBCLs) are a diverse but rare group of cutaneous lymphoproliferative neoplasms that make up approximately 20% of the total number of hematolymphoid neoplasms primary to the skin.1 These lymphomas are comprised of neoplastic B cells in various stages of differentiation. As a whole, they are rare neoplasms that primarily involve the head, neck, trunk, arms, or legs.1 Clinically, patients present with nontender, compressible, solitary, red to violaceous papules or nodules. Most CBCLs are considered low-grade malignancies with nonaggressive behavior and excellent prognosis; however, the diffuse large B-cell lymphomas, including but not limited to intravascular and leg type; lymphomatoid granulomatosis; and B-cell lymphoblastic lymphoma can act more aggressively.1
Histopathologic examination of primary CBCL generally reveals a relatively normal epidermis accompanied by a nodular to diffuse monomorphic lymphocytic cellular infiltrate in the dermis that can occasionally extend into the subcutaneous tissue (quiz image). Although not specific for CBCLs, oftentimes there is an acellular portion of the superficial papillary dermis known as a grenz zone that can serve as a histopathologic clue to the diagnosis of a cutaneous lymphoproliferative disorder. The list of malignant B-cell neoplasms is extensive (eg, cutaneous marginal zone B-cell lymphoma, primary cutaneous follicle center lymphoma, diffuse large B-cell lymphoma, intravascular large B-cell lymphoma), and few are seen in the skin.
The most common type of CBCL is marginal zone B-cell lymphoma, which is considered to be a tumor of mucosa-associated (or skin-associated) lymphoid tissue. It is characterized by a monomorphous population of small mature lymphocytes showing characteristics of the B cells of the marginal zone of the lymph node. Some cells have the features of centrocytes/centroblasts (Figure 1) demonstrated by slightly irregular or indented nuclei and generous amounts of cytoplasm. Larger and more pleomorphic cells such as immunoblasts are similarly noted (Figure 1). The quiz image and Figure 1 demonstrate a cutaneous marginal zone B-cell lymphoma. A histomorphologic clue supporting a diagnosis of marginal zone B-cell lymphoma over reactive lymphoid hyperplasia is a B-cell predominate (B- to T-cell ratio of at least 3 to 1) infiltrate that is comprised of marginal zone-type cells. Immunohistochemistry demonstrating fewer differentiated B cells with light chain restriction may provide additional evidence that supports a clonal and potentially malignant process.
Erythematous to violaceous nodules on the head and neck of older individuals are characteristic of both granuloma faciale and CBCL. Histologically, granuloma faciale is characterized by a dense cellular infiltrate, often with a nodular outline, occupying the mid dermis.2 Granuloma faciale typically spares the immediate subepidermis and hair follicles, forming a grenz zone. The cellular infiltrate is polymorphic and consists of eosinophils and neutrophils with scattered plasma cells, mast cells, and lymphocytes in a vasculocentric distribution, eventually with chronic concentric fibrosis (Figure 2).
Leukemia cutis demonstrates a dermal infiltrate that contains atypical mononuclear cells (myeloblasts and myelocytes)(Figure 3).3 These markedly atypical mononuclear cells can have kidney bean-shaped nuclei and percolate through the dermal collagen, resembling single-file cells. They have increased nuclear to cytoplasmic ratios and occasionally have prominent nucleoli. Correlation with immunophenotypic and cytochemical studies is required for specific typing of the leukemic infiltrate.
Similar to primary CBCL, lymphomatoid papulosis (LyP) consists of erythematous papules or nodules that can occur anywhere on the body. In contrast to CBCL, the lesions of LyP classically self-resolve. However, approximately 10% to 20% of patients develop a malignant lymphoma, with mycosis fungoides, Hodgkin disease, and anaplastic large cell lymphoma being the most commonly associated.
Histologic examination of lesions of LyP classically demonstrates a wedge-shaped dermal infiltrate with variable epidermal changes (Figure 4). The wedge-shaped infiltrate is composed of large atypical cells. Three main types of lesions have been delineated: types A, B, and C. Type A is characterized by an increased number of cells with large vesicular nuclei with clumped chromatin, prominent nucleoli, and pronounced cytoplasm. Reed-Sternberg-like cells with an admixture of inflammatory cells including small lymphocytes, macrophages, neutrophils, and eosinophils also are present. Type B neoplastic cells vary in size and feature hyperchromatic, convoluted, or cerebriform nuclei. The infiltrate can be dense and bandlike with fewer cells resembling mycosis fungoides; type B LyP has neoplastic cells, not inflammatory cells. Finally, type C demonstrates solid sheets of large atypical cells resembling anaplastic large cell lymphoma. Immunohistochemically, the atypical cells often are CD4+ and CD8- with variable loss of pan-T-cell antigens. The atypical cells of types A and C express CD30 reactivity.4
Merkel cell carcinoma (MCC) is a primary neuroendocrine carcinoma of the skin that usually arises on sun-exposed skin in elderly patients with lesions that histologically and clinically resemble cutaneous lymphoma.5 It classically is composed of small, round to oval, basophilic cells with a vesicular nucleus and multiple small nucleoli. Apoptotic cells and mitoses often are present.6 One key finding that helps to differentiate MCC from lymphoma is the presence of finely dispersed salt-and-pepper chromatin and molded nuclear contour in MCC (Figure 5).
Immunophenotyping is important in the differentiation of these diagnoses. The atypical cells of LyP are positive for CD3, CD4, and CD30 but are negative for CD8. However, in type B LyP, the large CD30+ cells seen in the other types are not commonly seen. In contrast, MCC expresses reactivity with cytokeratins, in particular cytokeratin 20 and CAM5.2, classically in a paranuclear dotlike pattern. In keeping with MCC's neuroendocrine differentiation, the tumor cells will demonstrate reactivity with synaptophysin, chromogranin, and CD56. The immunohistochemistry for leukemia cutis varies depending on the type of leukemia. Acute myelomonocytic leukemia is positive for myeloperoxidase, CD13, CD33, and CD68. The immunophenotype of these marginal zone lymphoma cells is as follows: positive for CD20, CD79a, and Bcl-2; negative for Bcl-6, CD5, CD10, CD23, and cyclin D1 (Bcl-1).7
Primary Cutaneous B-cell Lymphoma
Cutaneous B-cell lymphomas (CBCLs) are a diverse but rare group of cutaneous lymphoproliferative neoplasms that make up approximately 20% of the total number of hematolymphoid neoplasms primary to the skin.1 These lymphomas are comprised of neoplastic B cells in various stages of differentiation. As a whole, they are rare neoplasms that primarily involve the head, neck, trunk, arms, or legs.1 Clinically, patients present with nontender, compressible, solitary, red to violaceous papules or nodules. Most CBCLs are considered low-grade malignancies with nonaggressive behavior and excellent prognosis; however, the diffuse large B-cell lymphomas, including but not limited to intravascular and leg type; lymphomatoid granulomatosis; and B-cell lymphoblastic lymphoma can act more aggressively.1
Histopathologic examination of primary CBCL generally reveals a relatively normal epidermis accompanied by a nodular to diffuse monomorphic lymphocytic cellular infiltrate in the dermis that can occasionally extend into the subcutaneous tissue (quiz image). Although not specific for CBCLs, oftentimes there is an acellular portion of the superficial papillary dermis known as a grenz zone that can serve as a histopathologic clue to the diagnosis of a cutaneous lymphoproliferative disorder. The list of malignant B-cell neoplasms is extensive (eg, cutaneous marginal zone B-cell lymphoma, primary cutaneous follicle center lymphoma, diffuse large B-cell lymphoma, intravascular large B-cell lymphoma), and few are seen in the skin.
The most common type of CBCL is marginal zone B-cell lymphoma, which is considered to be a tumor of mucosa-associated (or skin-associated) lymphoid tissue. It is characterized by a monomorphous population of small mature lymphocytes showing characteristics of the B cells of the marginal zone of the lymph node. Some cells have the features of centrocytes/centroblasts (Figure 1) demonstrated by slightly irregular or indented nuclei and generous amounts of cytoplasm. Larger and more pleomorphic cells such as immunoblasts are similarly noted (Figure 1). The quiz image and Figure 1 demonstrate a cutaneous marginal zone B-cell lymphoma. A histomorphologic clue supporting a diagnosis of marginal zone B-cell lymphoma over reactive lymphoid hyperplasia is a B-cell predominate (B- to T-cell ratio of at least 3 to 1) infiltrate that is comprised of marginal zone-type cells. Immunohistochemistry demonstrating fewer differentiated B cells with light chain restriction may provide additional evidence that supports a clonal and potentially malignant process.
Erythematous to violaceous nodules on the head and neck of older individuals are characteristic of both granuloma faciale and CBCL. Histologically, granuloma faciale is characterized by a dense cellular infiltrate, often with a nodular outline, occupying the mid dermis.2 Granuloma faciale typically spares the immediate subepidermis and hair follicles, forming a grenz zone. The cellular infiltrate is polymorphic and consists of eosinophils and neutrophils with scattered plasma cells, mast cells, and lymphocytes in a vasculocentric distribution, eventually with chronic concentric fibrosis (Figure 2).
Leukemia cutis demonstrates a dermal infiltrate that contains atypical mononuclear cells (myeloblasts and myelocytes)(Figure 3).3 These markedly atypical mononuclear cells can have kidney bean-shaped nuclei and percolate through the dermal collagen, resembling single-file cells. They have increased nuclear to cytoplasmic ratios and occasionally have prominent nucleoli. Correlation with immunophenotypic and cytochemical studies is required for specific typing of the leukemic infiltrate.
Similar to primary CBCL, lymphomatoid papulosis (LyP) consists of erythematous papules or nodules that can occur anywhere on the body. In contrast to CBCL, the lesions of LyP classically self-resolve. However, approximately 10% to 20% of patients develop a malignant lymphoma, with mycosis fungoides, Hodgkin disease, and anaplastic large cell lymphoma being the most commonly associated.
Histologic examination of lesions of LyP classically demonstrates a wedge-shaped dermal infiltrate with variable epidermal changes (Figure 4). The wedge-shaped infiltrate is composed of large atypical cells. Three main types of lesions have been delineated: types A, B, and C. Type A is characterized by an increased number of cells with large vesicular nuclei with clumped chromatin, prominent nucleoli, and pronounced cytoplasm. Reed-Sternberg-like cells with an admixture of inflammatory cells including small lymphocytes, macrophages, neutrophils, and eosinophils also are present. Type B neoplastic cells vary in size and feature hyperchromatic, convoluted, or cerebriform nuclei. The infiltrate can be dense and bandlike with fewer cells resembling mycosis fungoides; type B LyP has neoplastic cells, not inflammatory cells. Finally, type C demonstrates solid sheets of large atypical cells resembling anaplastic large cell lymphoma. Immunohistochemically, the atypical cells often are CD4+ and CD8- with variable loss of pan-T-cell antigens. The atypical cells of types A and C express CD30 reactivity.4
Merkel cell carcinoma (MCC) is a primary neuroendocrine carcinoma of the skin that usually arises on sun-exposed skin in elderly patients with lesions that histologically and clinically resemble cutaneous lymphoma.5 It classically is composed of small, round to oval, basophilic cells with a vesicular nucleus and multiple small nucleoli. Apoptotic cells and mitoses often are present.6 One key finding that helps to differentiate MCC from lymphoma is the presence of finely dispersed salt-and-pepper chromatin and molded nuclear contour in MCC (Figure 5).
Immunophenotyping is important in the differentiation of these diagnoses. The atypical cells of LyP are positive for CD3, CD4, and CD30 but are negative for CD8. However, in type B LyP, the large CD30+ cells seen in the other types are not commonly seen. In contrast, MCC expresses reactivity with cytokeratins, in particular cytokeratin 20 and CAM5.2, classically in a paranuclear dotlike pattern. In keeping with MCC's neuroendocrine differentiation, the tumor cells will demonstrate reactivity with synaptophysin, chromogranin, and CD56. The immunohistochemistry for leukemia cutis varies depending on the type of leukemia. Acute myelomonocytic leukemia is positive for myeloperoxidase, CD13, CD33, and CD68. The immunophenotype of these marginal zone lymphoma cells is as follows: positive for CD20, CD79a, and Bcl-2; negative for Bcl-6, CD5, CD10, CD23, and cyclin D1 (Bcl-1).7
- Olsen EA. Evaluation, diagnosis, and staging of cutaneous lymphoma. Dermatol Clin. 2015;33:643-654.
- Ortonne N, Wechsler J, Bagot M, et al. Granuloma faciale: a clinicopathologic study of 66 patients. J Am Acad Dermatol. 2005;53:1002-1009.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Wieser I, Wohlmuth C, Nunez CA, et al. Lymphomatoid papulosis in children and adolescents: a systematic review. Am J Clin Dermatol. 2016;17:319-327.
- Sibley RK, Dehner LP, Rosai J. Primary neuroendocrine (Merkel cell?) carcinoma of the skin: I. a clinicopathologic and ultrastructural study of 43 cases. Am J Surg Pathol. 1985;9:95-108.
- Frigerio B, Capella C, Eusebi V, et al. Merkel cell carcinoma of the skin: the structure and origin of normal Merkel cells. Histopathology. 1983;7:229-249.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
- Olsen EA. Evaluation, diagnosis, and staging of cutaneous lymphoma. Dermatol Clin. 2015;33:643-654.
- Ortonne N, Wechsler J, Bagot M, et al. Granuloma faciale: a clinicopathologic study of 66 patients. J Am Acad Dermatol. 2005;53:1002-1009.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Wieser I, Wohlmuth C, Nunez CA, et al. Lymphomatoid papulosis in children and adolescents: a systematic review. Am J Clin Dermatol. 2016;17:319-327.
- Sibley RK, Dehner LP, Rosai J. Primary neuroendocrine (Merkel cell?) carcinoma of the skin: I. a clinicopathologic and ultrastructural study of 43 cases. Am J Surg Pathol. 1985;9:95-108.
- Frigerio B, Capella C, Eusebi V, et al. Merkel cell carcinoma of the skin: the structure and origin of normal Merkel cells. Histopathology. 1983;7:229-249.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
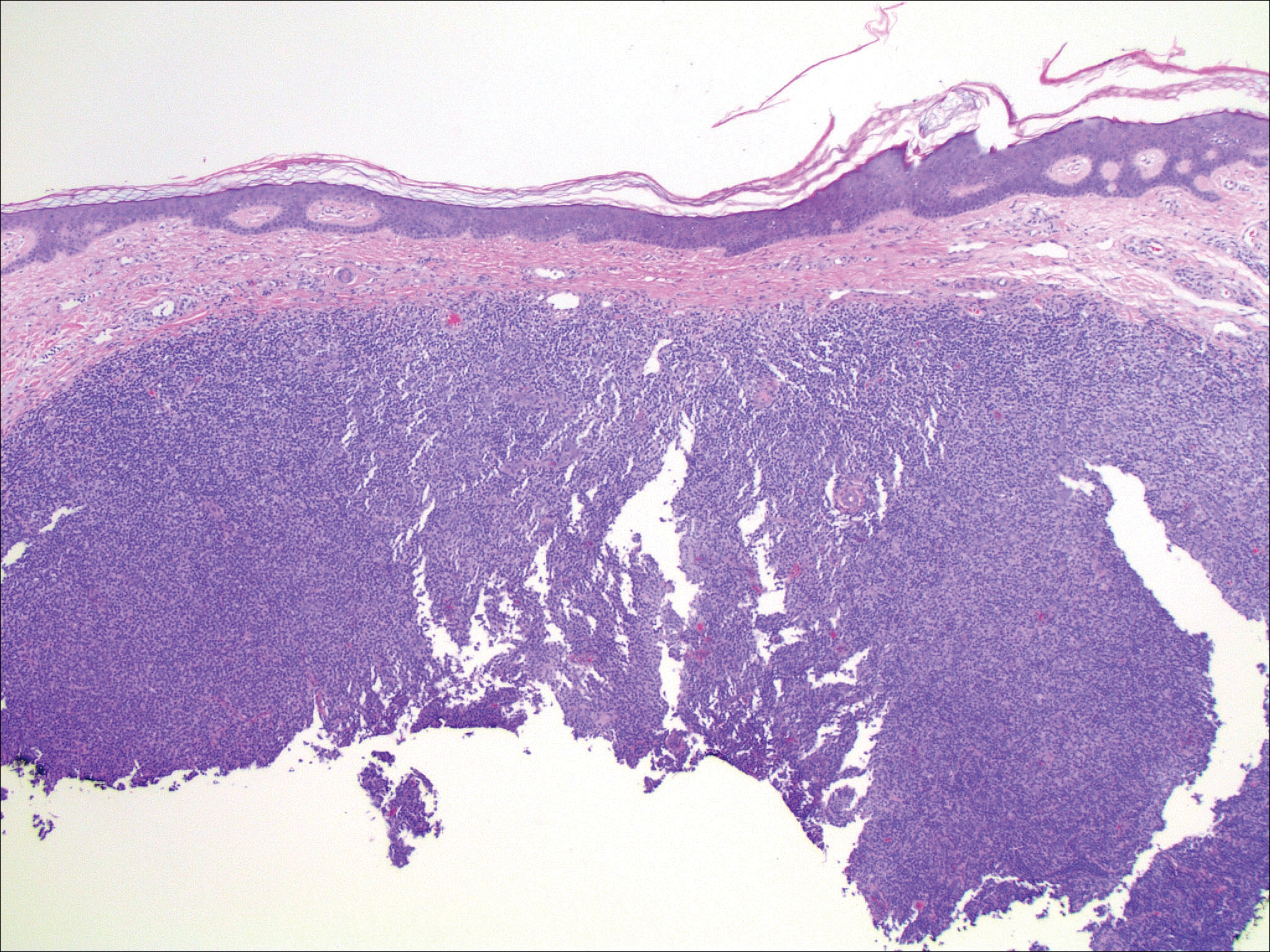
An 81-year-old man with a history of hyperthyroidism, paroxysmal atrial fibrillation, hypertension, and nonmelanoma skin cancer presented with an erythematous pearly papule on the right lateral chest of 1 year's duration. The patient reported no symptoms of pruritus, bleeding, or burning. He was otherwise asymptomatic, and a review of systems revealed no abnormalities. His current medications included aspirin, benazepril, finasteride, levothyroxine, tamsulosin, warfarin, and alprazolam. He denied any new medications, recent travel, or preceding trauma. He had a history of Agent Orange exposure. Physical examination revealed a 0.4-cm erythematous pearly papule on the right lateral chest. A shave biopsy was obtained.
Progressive Widespread Warty Skin Growths
Epidermodysplasia Verruciformis
Epidermodysplasia verruciformis (EV) is a rare hereditary disorder that predisposes affected individuals to widespread infection with various forms of human papillomavirus (HPV). It is inherited in an autosomal-recessive pattern.1 The first manifestations generally are seen in childhood. The clinical appearance of lesions can vary, at times mimicking other disease processes. Patients can present with flat wartlike papules resembling verruca plana distributed in sun-exposed areas. Another distinct presentation is multiple salmon-colored, hyperpigmented, or hypopigmented macules, papules, or plaques with overlying scale that can resemble tinea versicolor.1,2 A large percentage of patients will go on to develop actinic keratosis and squamous cell carcinoma by 40 years of age.2 The malignancies most commonly develop in sun-exposed areas, suggesting UV radiation as an important contributor to development along with HPV infection. Mutations in the EVER1 and EVER2 genes that code for transmembrane proteins on the endoplasmic reticulum that are involved in zinc transport lead to EV. The mutations lead to decreased zinc movement into the cytoplasm, which is thought to play a role in preventing HPV infection. The decreased protection against HPV leads to infections from both common subtypes and those that immunocompetent individuals would be resistant to, namely the β-genus HPV-5, -8, -9 and -20.1,2 Immunosuppressed individuals, such as those with human immunodeficiency virus/AIDS, also are at an increased risk for infection with these HPV subtypes and generally have similar clinical and histological presentations.1 It is important to promote sunscreen use for preventive care in patients with EV due to the increased risk for squamous cell carcinoma.
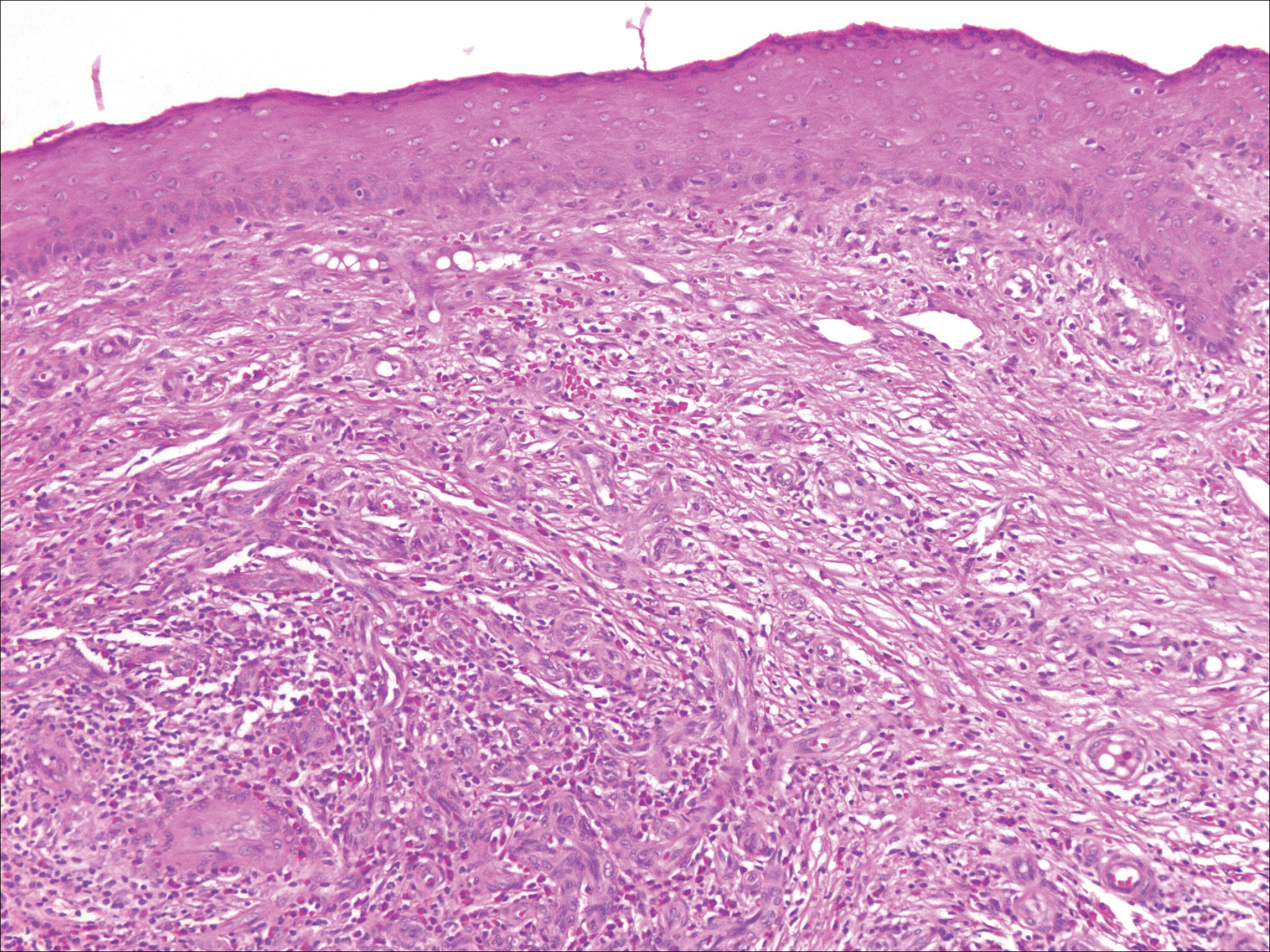
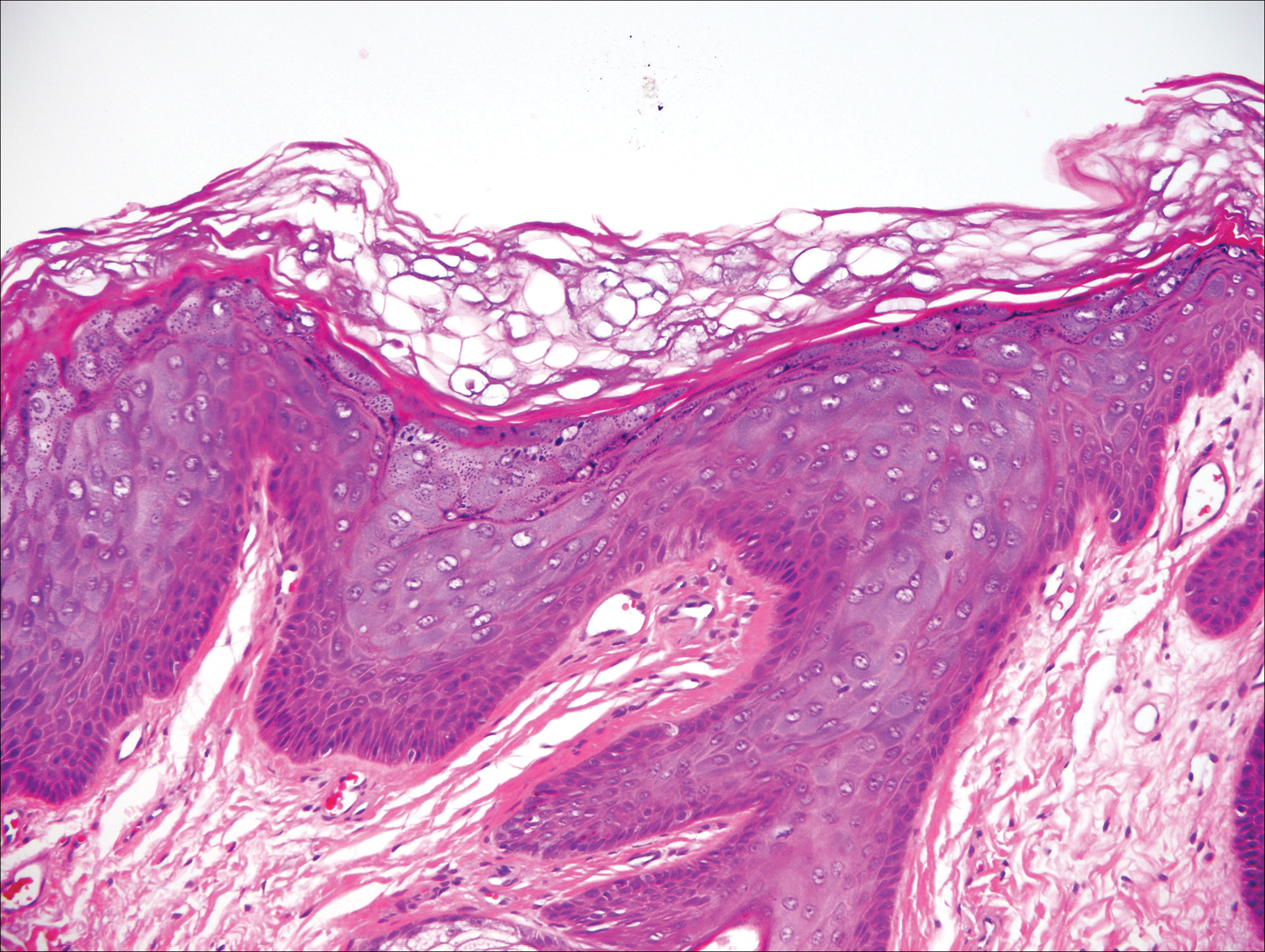
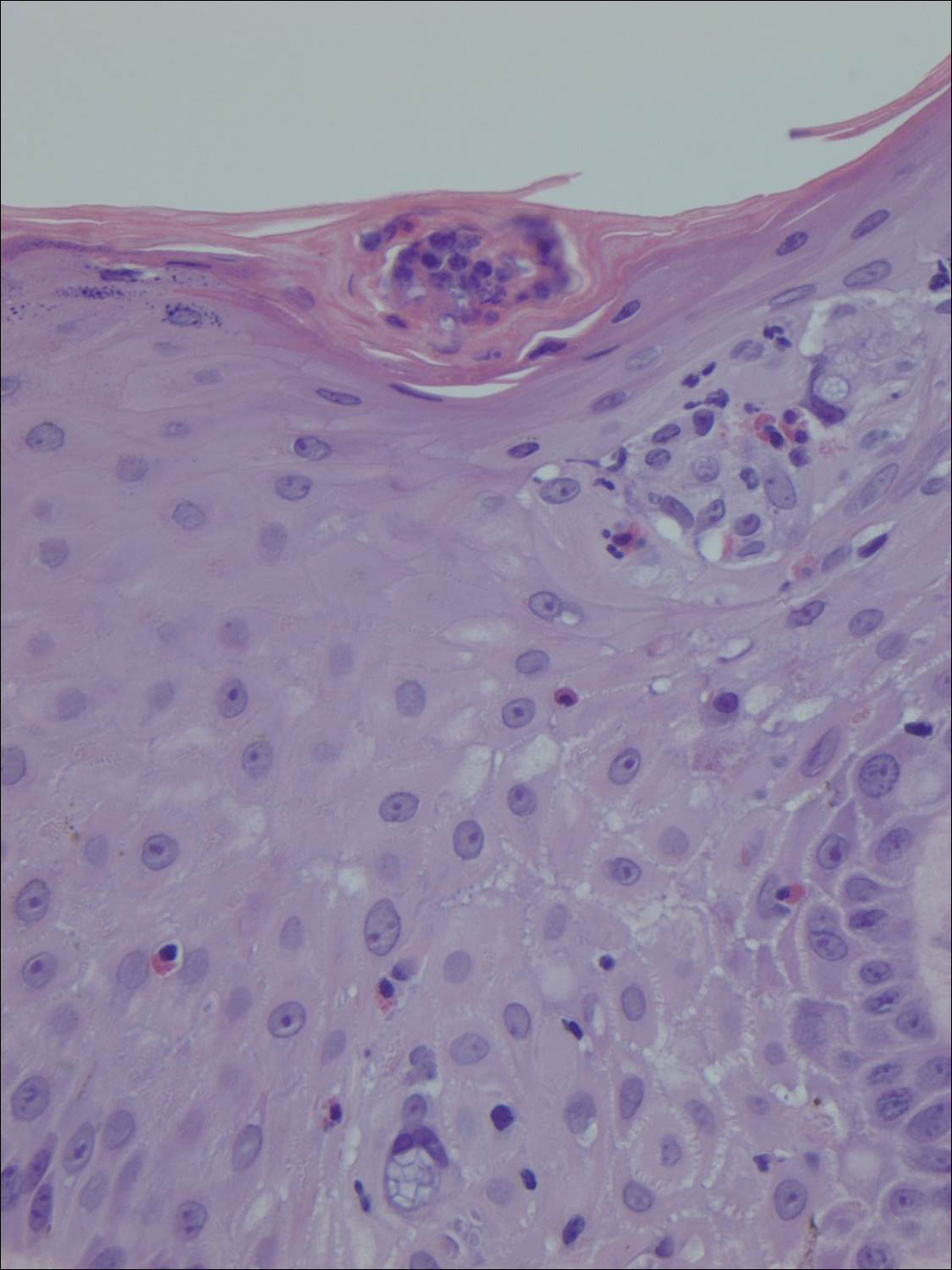
Histologically, the lesions in EV are composed of acanthosis and hyperkeratosis with keratinocytes arranged in clusters.1,3 There is orthokeratosis and parakeratosis.1 Scattered or clustered keratinocytes in the granular layer or upper stratum spinosum appear swollen with foamy blue-gray cytoplasm (quiz image and Figure 1).1,4 The keratinocytes may become atypical and progress to squamous cell carcinoma, particularly in sun-exposed regions. Cell differentiation becomes disorganized and nuclei become enlarged and hyperchromatic.1

Condyloma acuminatum will have pronounced acanthosis and hyperkeratosis with exophytic growth. Rounded parakeratosis is visible. The characteristic cell is the koilocyte, a keratinocyte that has an enlarged nucleus with areas of surrounding clearing, increased dark color in the nucleus, and wrinkled nuclear membrane (Figure 2).1,3 True koilocytes may be rare in condyloma acuminatum.4 Other distinct features include coarse hypergranulosis and a compact stratum corneum.

Herpesvirus lesions typically demonstrate ballooning degeneration of keratinocytes.1 They will become pale and fuse to form multinucleated giant cells, a feature not found in verruca. The nuclei will be slate gray with margination of the chromatin, which can be identified due to its increased basophilic appearance (Figure 3).1,4 Inclusion bodies can be found, but unlike molluscum contagiosum (MC), these bodies are intranuclear as opposed to cytoplasmic.1
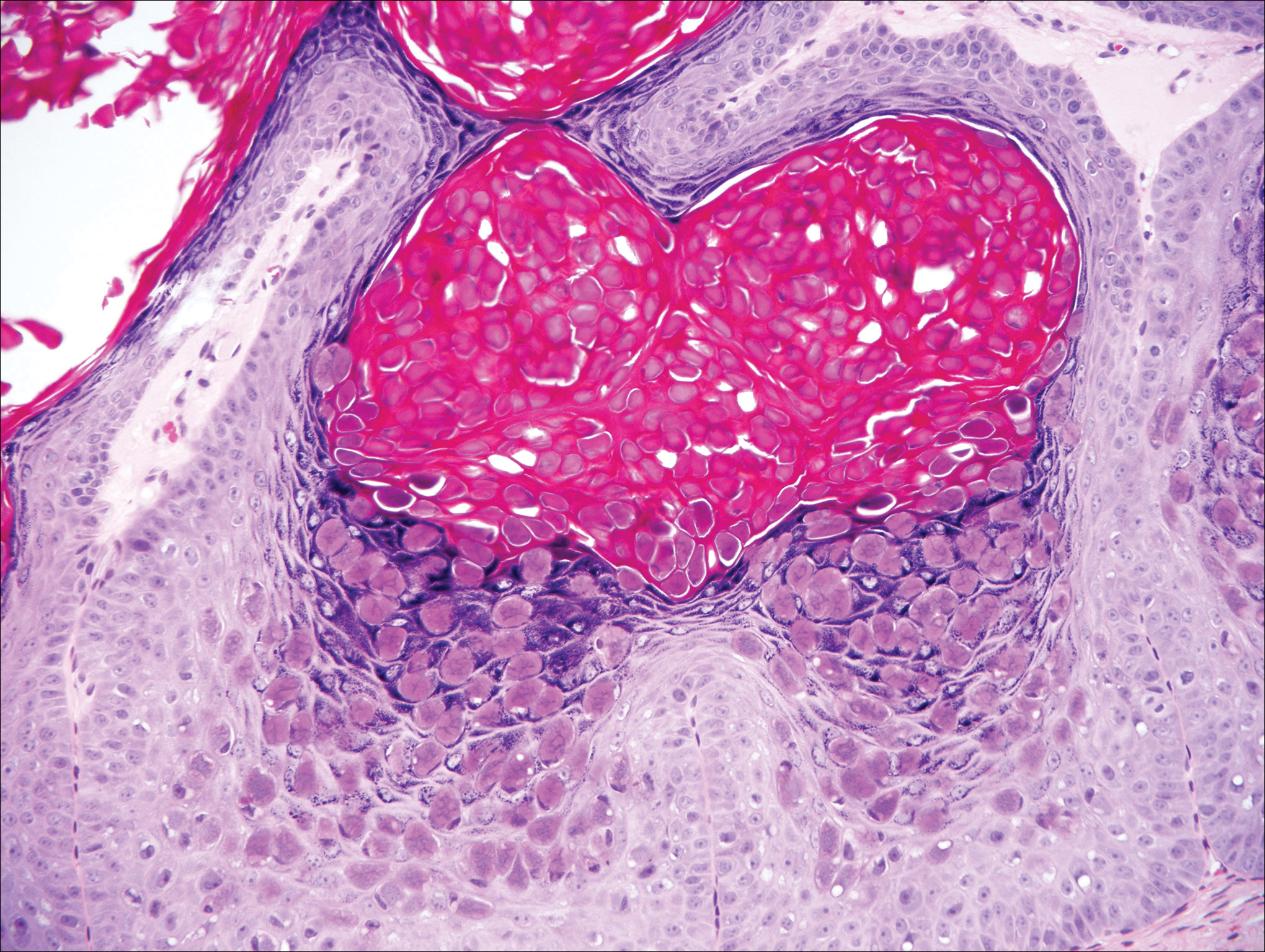
The telltale Henderson-Patterson (molluscum) bodies can identify MC histologically.4 Located within keratinocytes, these cytoplasmic inclusions can vary in both color and size as they mature. As the keratinocytes develop outward, the molluscum bodies grow larger and become more eosinophilic (Figure 4).1,4 Another feature of MC that can be used to differentiate it from EV is the scalloped borders located on lesions.4
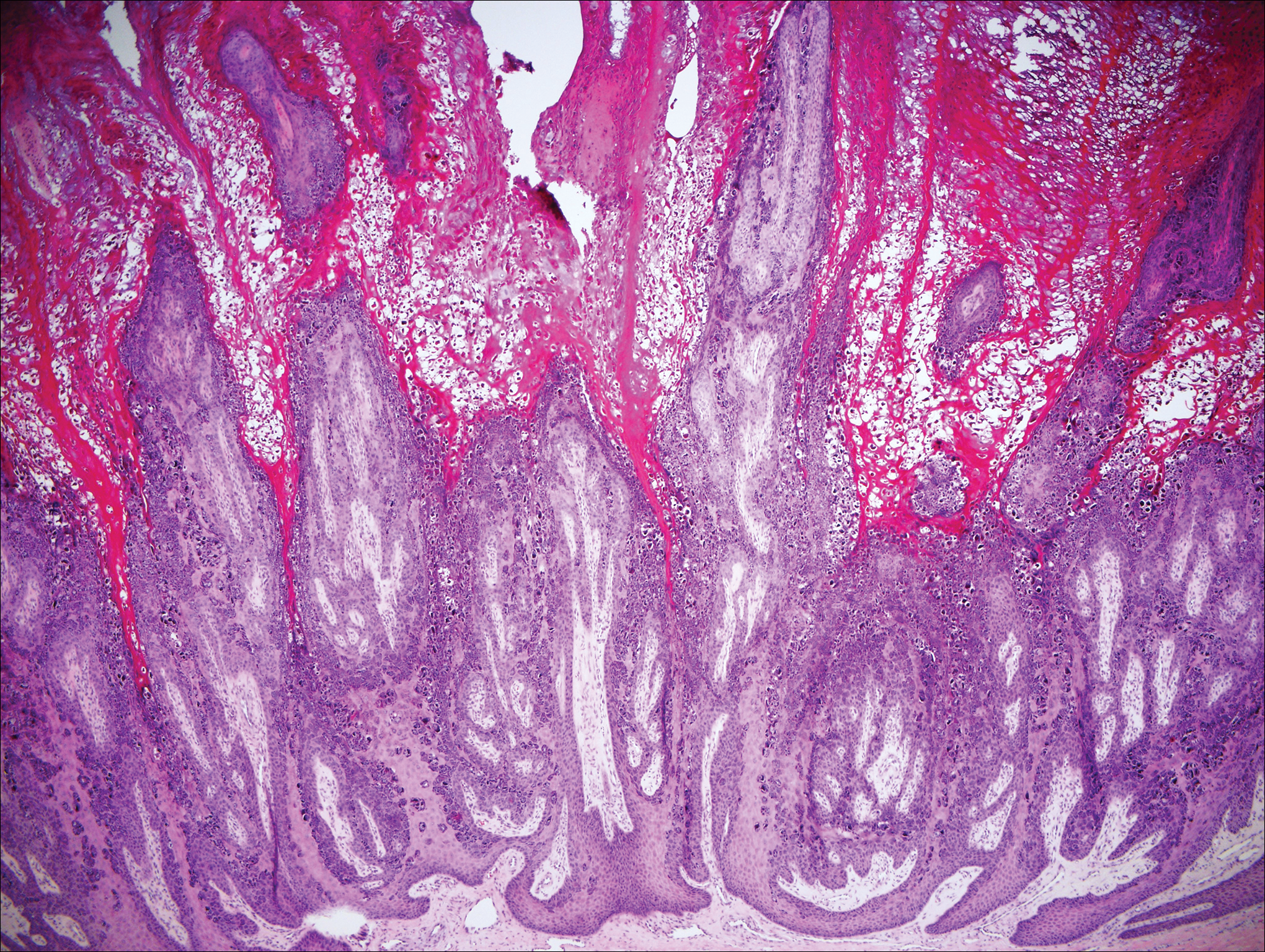
On histology, verruca vulgaris will have pronounced acanthosis with orthokeratosis and vertical tiers of parakeratosis.3,4 Growth is exophytic. The granular layer will have large irregular basophilic granules. Koilocytes may be seen. A distinctive feature is the papillomatosis with inward bending of rete ridges.3,4 It is common to see invasion of tortuous blood vessels into the exophytic projections.3 In myrmecia (palmoplantar warts) it is common to see thrombosis of these vessels and inclusions of red cytoplasmic bodies (Figure 5).1,4
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
- Hunzeker CM, Soldano AC, Prystowsky S. Epidermodysplasia verruciformis. Dermatology Online J. 2008;14:2.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Elston DM, Ko CJ, Ferringer T. Dermatopathology. Edinburgh, Scotland: Saunders/Elsevier; 2009.
Epidermodysplasia Verruciformis
Epidermodysplasia verruciformis (EV) is a rare hereditary disorder that predisposes affected individuals to widespread infection with various forms of human papillomavirus (HPV). It is inherited in an autosomal-recessive pattern.1 The first manifestations generally are seen in childhood. The clinical appearance of lesions can vary, at times mimicking other disease processes. Patients can present with flat wartlike papules resembling verruca plana distributed in sun-exposed areas. Another distinct presentation is multiple salmon-colored, hyperpigmented, or hypopigmented macules, papules, or plaques with overlying scale that can resemble tinea versicolor.1,2 A large percentage of patients will go on to develop actinic keratosis and squamous cell carcinoma by 40 years of age.2 The malignancies most commonly develop in sun-exposed areas, suggesting UV radiation as an important contributor to development along with HPV infection. Mutations in the EVER1 and EVER2 genes that code for transmembrane proteins on the endoplasmic reticulum that are involved in zinc transport lead to EV. The mutations lead to decreased zinc movement into the cytoplasm, which is thought to play a role in preventing HPV infection. The decreased protection against HPV leads to infections from both common subtypes and those that immunocompetent individuals would be resistant to, namely the β-genus HPV-5, -8, -9 and -20.1,2 Immunosuppressed individuals, such as those with human immunodeficiency virus/AIDS, also are at an increased risk for infection with these HPV subtypes and generally have similar clinical and histological presentations.1 It is important to promote sunscreen use for preventive care in patients with EV due to the increased risk for squamous cell carcinoma.
Histologically, the lesions in EV are composed of acanthosis and hyperkeratosis with keratinocytes arranged in clusters.1,3 There is orthokeratosis and parakeratosis.1 Scattered or clustered keratinocytes in the granular layer or upper stratum spinosum appear swollen with foamy blue-gray cytoplasm (quiz image and Figure 1).1,4 The keratinocytes may become atypical and progress to squamous cell carcinoma, particularly in sun-exposed regions. Cell differentiation becomes disorganized and nuclei become enlarged and hyperchromatic.1
Condyloma acuminatum will have pronounced acanthosis and hyperkeratosis with exophytic growth. Rounded parakeratosis is visible. The characteristic cell is the koilocyte, a keratinocyte that has an enlarged nucleus with areas of surrounding clearing, increased dark color in the nucleus, and wrinkled nuclear membrane (Figure 2).1,3 True koilocytes may be rare in condyloma acuminatum.4 Other distinct features include coarse hypergranulosis and a compact stratum corneum.
Herpesvirus lesions typically demonstrate ballooning degeneration of keratinocytes.1 They will become pale and fuse to form multinucleated giant cells, a feature not found in verruca. The nuclei will be slate gray with margination of the chromatin, which can be identified due to its increased basophilic appearance (Figure 3).1,4 Inclusion bodies can be found, but unlike molluscum contagiosum (MC), these bodies are intranuclear as opposed to cytoplasmic.1
The telltale Henderson-Patterson (molluscum) bodies can identify MC histologically.4 Located within keratinocytes, these cytoplasmic inclusions can vary in both color and size as they mature. As the keratinocytes develop outward, the molluscum bodies grow larger and become more eosinophilic (Figure 4).1,4 Another feature of MC that can be used to differentiate it from EV is the scalloped borders located on lesions.4
On histology, verruca vulgaris will have pronounced acanthosis with orthokeratosis and vertical tiers of parakeratosis.3,4 Growth is exophytic. The granular layer will have large irregular basophilic granules. Koilocytes may be seen. A distinctive feature is the papillomatosis with inward bending of rete ridges.3,4 It is common to see invasion of tortuous blood vessels into the exophytic projections.3 In myrmecia (palmoplantar warts) it is common to see thrombosis of these vessels and inclusions of red cytoplasmic bodies (Figure 5).1,4
Epidermodysplasia Verruciformis
Epidermodysplasia verruciformis (EV) is a rare hereditary disorder that predisposes affected individuals to widespread infection with various forms of human papillomavirus (HPV). It is inherited in an autosomal-recessive pattern.1 The first manifestations generally are seen in childhood. The clinical appearance of lesions can vary, at times mimicking other disease processes. Patients can present with flat wartlike papules resembling verruca plana distributed in sun-exposed areas. Another distinct presentation is multiple salmon-colored, hyperpigmented, or hypopigmented macules, papules, or plaques with overlying scale that can resemble tinea versicolor.1,2 A large percentage of patients will go on to develop actinic keratosis and squamous cell carcinoma by 40 years of age.2 The malignancies most commonly develop in sun-exposed areas, suggesting UV radiation as an important contributor to development along with HPV infection. Mutations in the EVER1 and EVER2 genes that code for transmembrane proteins on the endoplasmic reticulum that are involved in zinc transport lead to EV. The mutations lead to decreased zinc movement into the cytoplasm, which is thought to play a role in preventing HPV infection. The decreased protection against HPV leads to infections from both common subtypes and those that immunocompetent individuals would be resistant to, namely the β-genus HPV-5, -8, -9 and -20.1,2 Immunosuppressed individuals, such as those with human immunodeficiency virus/AIDS, also are at an increased risk for infection with these HPV subtypes and generally have similar clinical and histological presentations.1 It is important to promote sunscreen use for preventive care in patients with EV due to the increased risk for squamous cell carcinoma.
Histologically, the lesions in EV are composed of acanthosis and hyperkeratosis with keratinocytes arranged in clusters.1,3 There is orthokeratosis and parakeratosis.1 Scattered or clustered keratinocytes in the granular layer or upper stratum spinosum appear swollen with foamy blue-gray cytoplasm (quiz image and Figure 1).1,4 The keratinocytes may become atypical and progress to squamous cell carcinoma, particularly in sun-exposed regions. Cell differentiation becomes disorganized and nuclei become enlarged and hyperchromatic.1
Condyloma acuminatum will have pronounced acanthosis and hyperkeratosis with exophytic growth. Rounded parakeratosis is visible. The characteristic cell is the koilocyte, a keratinocyte that has an enlarged nucleus with areas of surrounding clearing, increased dark color in the nucleus, and wrinkled nuclear membrane (Figure 2).1,3 True koilocytes may be rare in condyloma acuminatum.4 Other distinct features include coarse hypergranulosis and a compact stratum corneum.
Herpesvirus lesions typically demonstrate ballooning degeneration of keratinocytes.1 They will become pale and fuse to form multinucleated giant cells, a feature not found in verruca. The nuclei will be slate gray with margination of the chromatin, which can be identified due to its increased basophilic appearance (Figure 3).1,4 Inclusion bodies can be found, but unlike molluscum contagiosum (MC), these bodies are intranuclear as opposed to cytoplasmic.1
The telltale Henderson-Patterson (molluscum) bodies can identify MC histologically.4 Located within keratinocytes, these cytoplasmic inclusions can vary in both color and size as they mature. As the keratinocytes develop outward, the molluscum bodies grow larger and become more eosinophilic (Figure 4).1,4 Another feature of MC that can be used to differentiate it from EV is the scalloped borders located on lesions.4
On histology, verruca vulgaris will have pronounced acanthosis with orthokeratosis and vertical tiers of parakeratosis.3,4 Growth is exophytic. The granular layer will have large irregular basophilic granules. Koilocytes may be seen. A distinctive feature is the papillomatosis with inward bending of rete ridges.3,4 It is common to see invasion of tortuous blood vessels into the exophytic projections.3 In myrmecia (palmoplantar warts) it is common to see thrombosis of these vessels and inclusions of red cytoplasmic bodies (Figure 5).1,4
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
- Hunzeker CM, Soldano AC, Prystowsky S. Epidermodysplasia verruciformis. Dermatology Online J. 2008;14:2.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Elston DM, Ko CJ, Ferringer T. Dermatopathology. Edinburgh, Scotland: Saunders/Elsevier; 2009.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
- Hunzeker CM, Soldano AC, Prystowsky S. Epidermodysplasia verruciformis. Dermatology Online J. 2008;14:2.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Elston DM, Ko CJ, Ferringer T. Dermatopathology. Edinburgh, Scotland: Saunders/Elsevier; 2009.
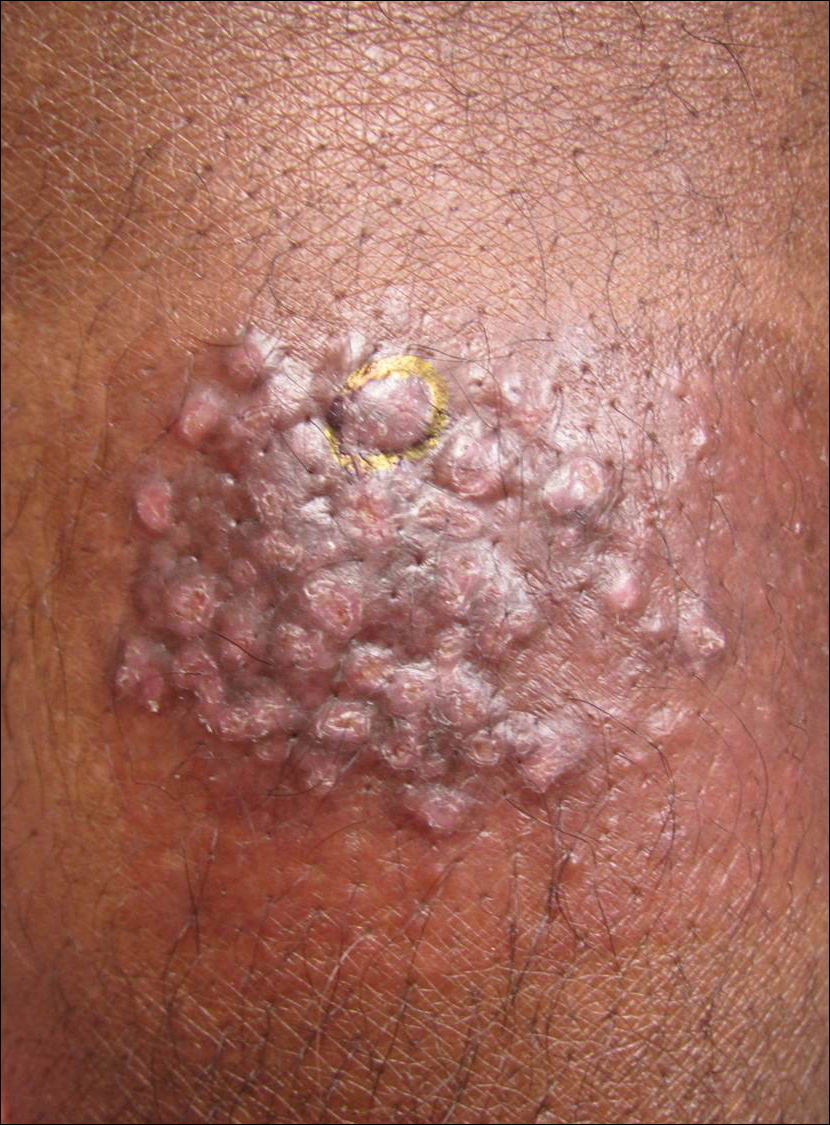
A 33-year-old man presented with progressive widespread warty skin growths that had been present since 6 years of age. Physical examination revealed numerous verrucous papules on the face and neck along with verrucous, tan-pink papules and plaques diffusely scattered on the trunk, arms, and legs. A biopsy of a lesion on the neck was performed.
Expanding Pruritic Plaque on the Forearm
The Diagnosis: Cutaneous Protothecosis
A 4-mm punch biopsy of the plaque on the right forearm was performed. The biopsy showed chronic inflammation with prominent histiocytes, foreign body giant cells, plasma cells, and abundant eosinophils (Figure 1). Grocott-Gomori methenamine-silver stain demonstrated abundant soccer ball-like or floretlike sporangia that were 3 to 11 μm, consistent with a diagnosis of protothecosis (Figure 2).

Cutaneous protothecosis is an infection caused by chlorophyll-lacking algae of the genus Prototheca.1 It is ubiquitous in nature and can be isolated from various reservoirs such as trees, grass, water, and food sources.2 Protothecosis is present worldwide and in the United States; it is most prevalent in the Southeast. Prototheca species are rare but often endemic in cattle and can cause bovine mastitis and enteritis.3 However, they are rare opportunistic infections in humans.
The pathogenesis of cutaneous protothecosis is largely unknown.4 However, most infections are thought to be caused by traumatic inoculation into subcutaneous tissues.1,2 The majority of cases occur in patients older than 30 years. To date, approximately 160 cases have been reported in the literature worldwide.5 There are 3 main species of Prototheca, but almost all human infections are caused by Prototheca wickerhamii.2 Clinically, most patients with protothecosis present with cutaneous findings, but olecranon bursitis and systemic forms also have been reported.1
Risk factors for protothecosis include immunosuppression, most often due to steroids, in addition to malignancies, diabetes mellitus, and certain occupations.1 The presentation can be variable from papules and plaques to even herpetiform appearances.4 Protothecosis usually affects the skin and soft tissues of exposed areas such as the extremities or the face.6 Diagnosis largely is made on detection of characteristic floretlike sporangia with a prominent cell wall on histopathological examination. Prototheca wickerhamii specifically produces a morula form of sporangia with endospores arranged symmetrically, giving it a characteristic soccer ball appearance.2
Treatment of protothecosis is difficult and remains controversial.1 There are no established protothecosis treatment protocols or guidelines due to the small number of cases.7 In vitro studies have demonstrated sensitivity to amphotericin B and various azoles as well as a wide range of antibiotics.1 Olecranon bursitis and small skin lesions can be treated by surgical excision. All other Prototheca infections require systemic treatment with azoles or intravenous amphotericin B for immunocompromised patients or those with disseminated disease.5 However, failure to respond to medical management often occurs, requiring surgical excision.1,6
Our patient was treated with a 3-month course of voriconazole but therapy failed and the plaque continued to expand. The patient underwent a wide excision that was repaired with a partial-thickness skin graft. Rebiopsy of the papule adjacent to the skin graft showed no further recurrence.
In conclusion, protothecosis generally is not clinically suspected and patients are subjected to various treatments without adequate results. A definitive diagnosis easily can be established with a skin biopsy, which can direct timely and appropriate treatment.
- Lass-Flörl C, Mary A. Human protothecosis. Clin Microbiol Rev. 2007;20:230-242.
- Mayorga J, Barba-Gómez JF, Verduzco-Martínez AP, et al. Protothecosis. Clin Dermatol. 2012;30:432-436.
- Jensen HE, Aalbaek B, Bloch B, et al. Bovine mammary protothecosis due to Prototheca zopfii. Med Mycol. 1998;36:89-95.
- Boyd AS, Langley M, King LE Jr. Cutaneous manifestations of Prototheca infections. J Am Acad Dermatol. 1995;32:758-764.
- Todd JR, King JW, Oberle A, et al. Protothecosis: report of a case with 20-year follow-up, and review of previously published cases. Med Mycol. 2012;50:673-689.
- Hightower KD, Messina JL. Cutaneous protothecosis: a case report and review of the literature. Cutis. 2007;80:129-131.
- Yamada N, Yoshida Y, Ohsawa T, et al. A case of cutaneous protothecosis successfully treated with local thermal therapy as an adjunct to itraconazole therapy in an immunocompromised host. Med Mycol. 2010;48:643-646.
The Diagnosis: Cutaneous Protothecosis
A 4-mm punch biopsy of the plaque on the right forearm was performed. The biopsy showed chronic inflammation with prominent histiocytes, foreign body giant cells, plasma cells, and abundant eosinophils (Figure 1). Grocott-Gomori methenamine-silver stain demonstrated abundant soccer ball-like or floretlike sporangia that were 3 to 11 μm, consistent with a diagnosis of protothecosis (Figure 2).
Cutaneous protothecosis is an infection caused by chlorophyll-lacking algae of the genus Prototheca.1 It is ubiquitous in nature and can be isolated from various reservoirs such as trees, grass, water, and food sources.2 Protothecosis is present worldwide and in the United States; it is most prevalent in the Southeast. Prototheca species are rare but often endemic in cattle and can cause bovine mastitis and enteritis.3 However, they are rare opportunistic infections in humans.
The pathogenesis of cutaneous protothecosis is largely unknown.4 However, most infections are thought to be caused by traumatic inoculation into subcutaneous tissues.1,2 The majority of cases occur in patients older than 30 years. To date, approximately 160 cases have been reported in the literature worldwide.5 There are 3 main species of Prototheca, but almost all human infections are caused by Prototheca wickerhamii.2 Clinically, most patients with protothecosis present with cutaneous findings, but olecranon bursitis and systemic forms also have been reported.1
Risk factors for protothecosis include immunosuppression, most often due to steroids, in addition to malignancies, diabetes mellitus, and certain occupations.1 The presentation can be variable from papules and plaques to even herpetiform appearances.4 Protothecosis usually affects the skin and soft tissues of exposed areas such as the extremities or the face.6 Diagnosis largely is made on detection of characteristic floretlike sporangia with a prominent cell wall on histopathological examination. Prototheca wickerhamii specifically produces a morula form of sporangia with endospores arranged symmetrically, giving it a characteristic soccer ball appearance.2
Treatment of protothecosis is difficult and remains controversial.1 There are no established protothecosis treatment protocols or guidelines due to the small number of cases.7 In vitro studies have demonstrated sensitivity to amphotericin B and various azoles as well as a wide range of antibiotics.1 Olecranon bursitis and small skin lesions can be treated by surgical excision. All other Prototheca infections require systemic treatment with azoles or intravenous amphotericin B for immunocompromised patients or those with disseminated disease.5 However, failure to respond to medical management often occurs, requiring surgical excision.1,6
Our patient was treated with a 3-month course of voriconazole but therapy failed and the plaque continued to expand. The patient underwent a wide excision that was repaired with a partial-thickness skin graft. Rebiopsy of the papule adjacent to the skin graft showed no further recurrence.
In conclusion, protothecosis generally is not clinically suspected and patients are subjected to various treatments without adequate results. A definitive diagnosis easily can be established with a skin biopsy, which can direct timely and appropriate treatment.
The Diagnosis: Cutaneous Protothecosis
A 4-mm punch biopsy of the plaque on the right forearm was performed. The biopsy showed chronic inflammation with prominent histiocytes, foreign body giant cells, plasma cells, and abundant eosinophils (Figure 1). Grocott-Gomori methenamine-silver stain demonstrated abundant soccer ball-like or floretlike sporangia that were 3 to 11 μm, consistent with a diagnosis of protothecosis (Figure 2).
Cutaneous protothecosis is an infection caused by chlorophyll-lacking algae of the genus Prototheca.1 It is ubiquitous in nature and can be isolated from various reservoirs such as trees, grass, water, and food sources.2 Protothecosis is present worldwide and in the United States; it is most prevalent in the Southeast. Prototheca species are rare but often endemic in cattle and can cause bovine mastitis and enteritis.3 However, they are rare opportunistic infections in humans.
The pathogenesis of cutaneous protothecosis is largely unknown.4 However, most infections are thought to be caused by traumatic inoculation into subcutaneous tissues.1,2 The majority of cases occur in patients older than 30 years. To date, approximately 160 cases have been reported in the literature worldwide.5 There are 3 main species of Prototheca, but almost all human infections are caused by Prototheca wickerhamii.2 Clinically, most patients with protothecosis present with cutaneous findings, but olecranon bursitis and systemic forms also have been reported.1
Risk factors for protothecosis include immunosuppression, most often due to steroids, in addition to malignancies, diabetes mellitus, and certain occupations.1 The presentation can be variable from papules and plaques to even herpetiform appearances.4 Protothecosis usually affects the skin and soft tissues of exposed areas such as the extremities or the face.6 Diagnosis largely is made on detection of characteristic floretlike sporangia with a prominent cell wall on histopathological examination. Prototheca wickerhamii specifically produces a morula form of sporangia with endospores arranged symmetrically, giving it a characteristic soccer ball appearance.2
Treatment of protothecosis is difficult and remains controversial.1 There are no established protothecosis treatment protocols or guidelines due to the small number of cases.7 In vitro studies have demonstrated sensitivity to amphotericin B and various azoles as well as a wide range of antibiotics.1 Olecranon bursitis and small skin lesions can be treated by surgical excision. All other Prototheca infections require systemic treatment with azoles or intravenous amphotericin B for immunocompromised patients or those with disseminated disease.5 However, failure to respond to medical management often occurs, requiring surgical excision.1,6
Our patient was treated with a 3-month course of voriconazole but therapy failed and the plaque continued to expand. The patient underwent a wide excision that was repaired with a partial-thickness skin graft. Rebiopsy of the papule adjacent to the skin graft showed no further recurrence.
In conclusion, protothecosis generally is not clinically suspected and patients are subjected to various treatments without adequate results. A definitive diagnosis easily can be established with a skin biopsy, which can direct timely and appropriate treatment.
- Lass-Flörl C, Mary A. Human protothecosis. Clin Microbiol Rev. 2007;20:230-242.
- Mayorga J, Barba-Gómez JF, Verduzco-Martínez AP, et al. Protothecosis. Clin Dermatol. 2012;30:432-436.
- Jensen HE, Aalbaek B, Bloch B, et al. Bovine mammary protothecosis due to Prototheca zopfii. Med Mycol. 1998;36:89-95.
- Boyd AS, Langley M, King LE Jr. Cutaneous manifestations of Prototheca infections. J Am Acad Dermatol. 1995;32:758-764.
- Todd JR, King JW, Oberle A, et al. Protothecosis: report of a case with 20-year follow-up, and review of previously published cases. Med Mycol. 2012;50:673-689.
- Hightower KD, Messina JL. Cutaneous protothecosis: a case report and review of the literature. Cutis. 2007;80:129-131.
- Yamada N, Yoshida Y, Ohsawa T, et al. A case of cutaneous protothecosis successfully treated with local thermal therapy as an adjunct to itraconazole therapy in an immunocompromised host. Med Mycol. 2010;48:643-646.
- Lass-Flörl C, Mary A. Human protothecosis. Clin Microbiol Rev. 2007;20:230-242.
- Mayorga J, Barba-Gómez JF, Verduzco-Martínez AP, et al. Protothecosis. Clin Dermatol. 2012;30:432-436.
- Jensen HE, Aalbaek B, Bloch B, et al. Bovine mammary protothecosis due to Prototheca zopfii. Med Mycol. 1998;36:89-95.
- Boyd AS, Langley M, King LE Jr. Cutaneous manifestations of Prototheca infections. J Am Acad Dermatol. 1995;32:758-764.
- Todd JR, King JW, Oberle A, et al. Protothecosis: report of a case with 20-year follow-up, and review of previously published cases. Med Mycol. 2012;50:673-689.
- Hightower KD, Messina JL. Cutaneous protothecosis: a case report and review of the literature. Cutis. 2007;80:129-131.
- Yamada N, Yoshida Y, Ohsawa T, et al. A case of cutaneous protothecosis successfully treated with local thermal therapy as an adjunct to itraconazole therapy in an immunocompromised host. Med Mycol. 2010;48:643-646.
A 66-year-old male firefighter initially presented to the emergency department with an expanding pruritic plaque on the dorsal aspect of the right forearm. The patient recalled the appearance of a single 3-mm papule shortly after doing yardwork in Biloxi, Mississippi. He remembered getting wet grass on the arms, which he later washed off without any notable trauma. The single papule grew into a larger plaque over the next month. In the emergency department he was treated with sulfamethoxazole-trimethoprim, mupirocin, and clotrimazole without response. He was referred to the dermatology department 6 months later and was noted to have multiple 3- to 4-mm papules that coalesced into a 4-cm lichenified plaque with surrounding erythema on the right forearm. His medical history was notable for type 2 diabetes mellitus, hypertension, and hyperlipidemia. The remainder of the physical examination and review of systems was negative.
Progressive Papular Eruption on the Face and Groin
The Diagnosis: Xanthoma Disseminatum
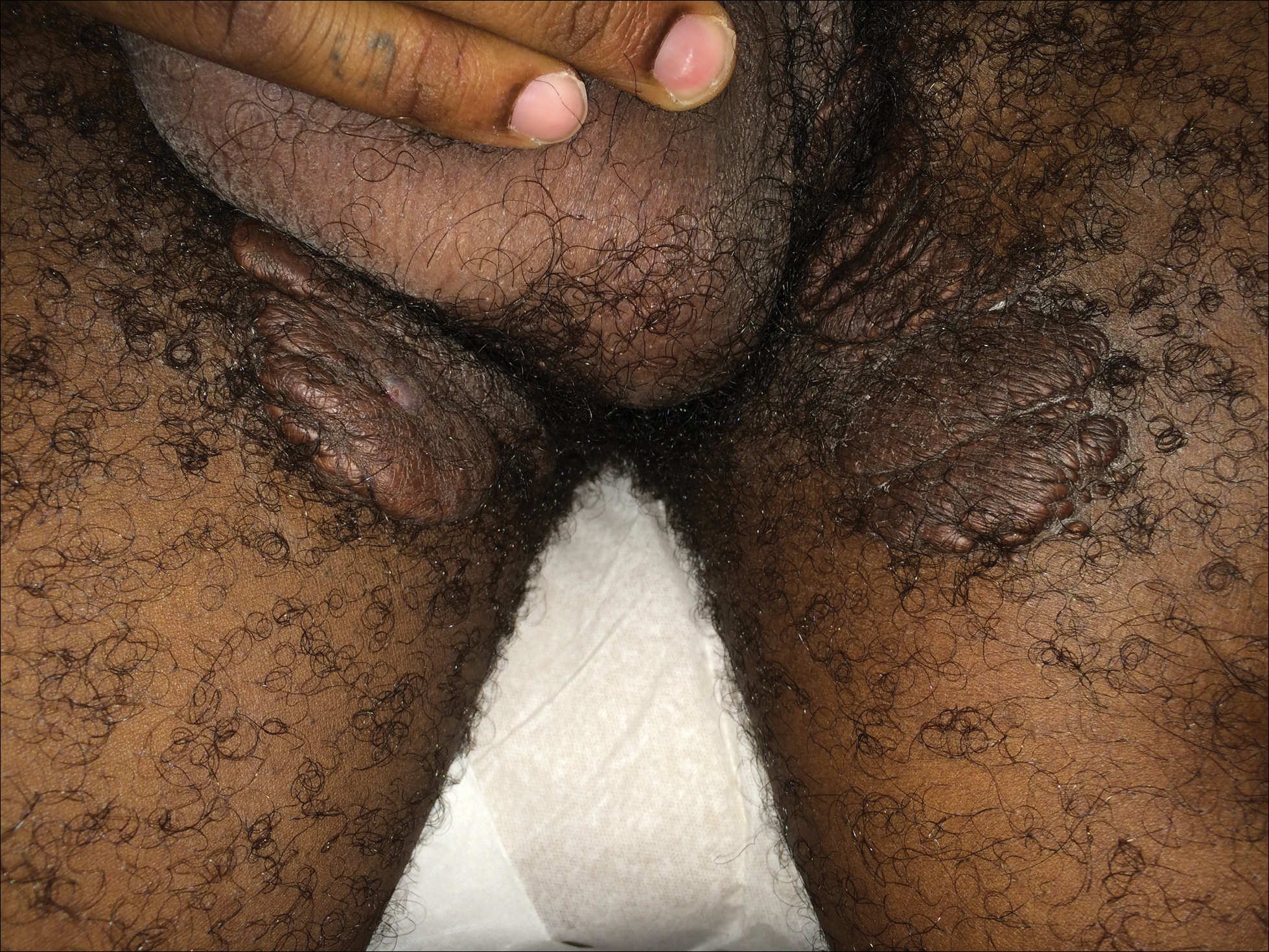
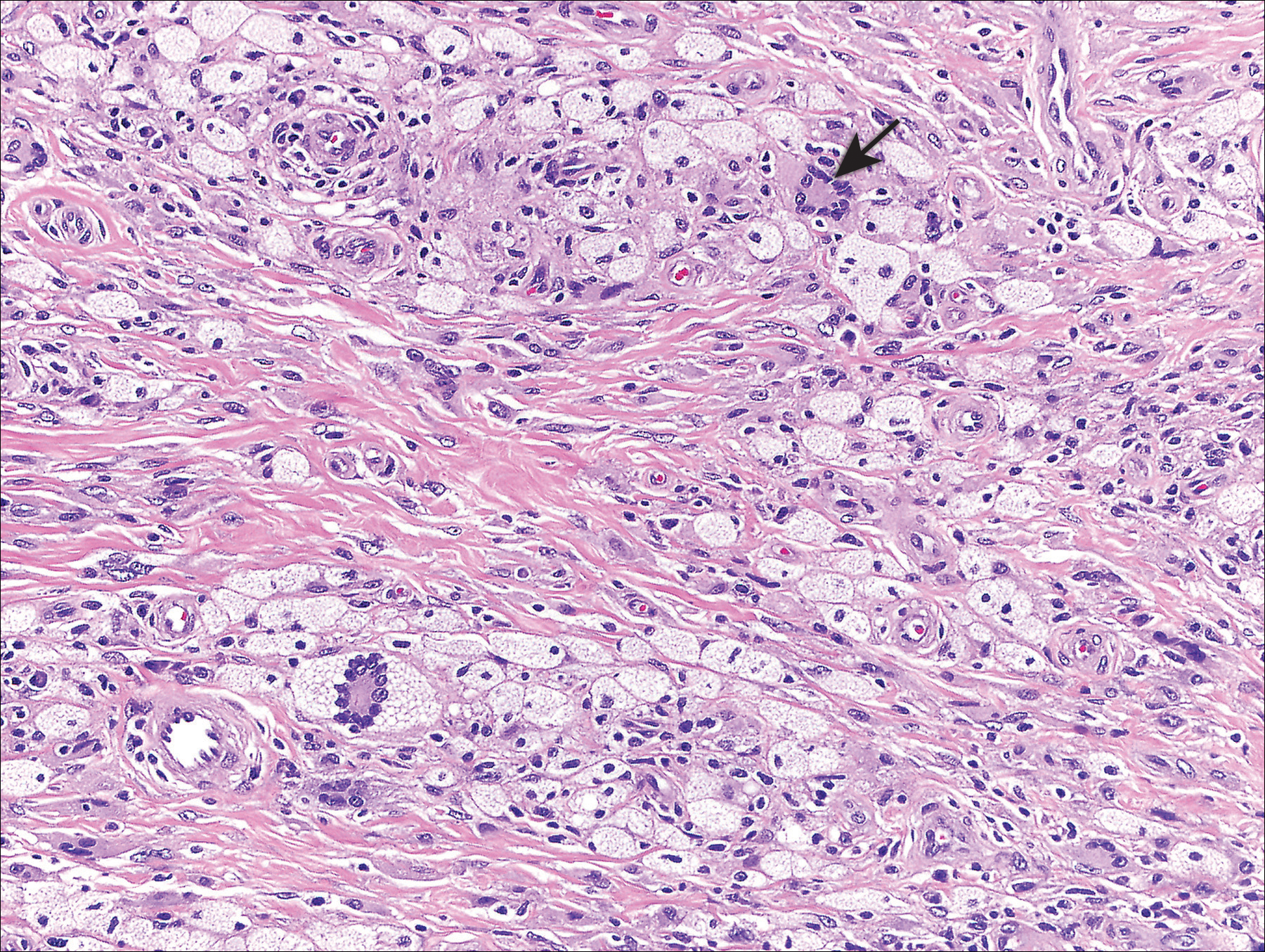
Genital examination revealed approximately 1.5×3-cm soft, yellow-pink plaques extending from the bilateral inguinal folds to the proximal medial thighs (Figure 1). There was no mucosal, axillary, extensor extremity, or palmoplantar involvement. Histopathologic examination of a biopsy from a plaque on the left side of the lower abdomen revealed sheets of foamy histiocytes distributed throughout a fibrotic dermis. Both mononucleated and multinucleated histiocytes were present, including many Touton giant cells (Figure 2). A patchy infiltrate of lymphocytes and rare eosinophils also was noted. The histiocytes labeled with factor XIIIa but not with S-100. Laboratory tests were performed with the following pertinent findings: low-density lipoprotein, 150 mg/dL (reference range, <130 mg/dL); high-density lipoprotein, 30 mg/dL (reference range, >40 mg/dL). Total cholesterol and triglyceride levels were within reference range, and complete blood cell count and basic metabolic panel were normal.
Xanthoma disseminatum (XD)(also known as Montgomery syndrome) is a rare, nonfamilial, normolipemic non-Langerhans cell histiocytosis characterized by extensive lipid deposition in the skin, mucous membranes, and internal organs. The pathogenesis of XD is poorly understood, but it may represent a macrophage-mediated reactive process triggered by superantigens.1
Xanthoma disseminatum most commonly affects males aged 5 to 25 years.2 Clinically, it is characterized by red-brown to yellowish papules and plaques symmetrically distributed over the eyelids, trunk, face, and proximal extremities. There is a predilection for involvement of flexural and intertriginous surfaces and tendency for extension along Langer lines. Extracutaneous involvement can be a notable cause of morbidity and mortality, underscoring the importance of distinguishing XD from other clinically similar xanthomatoses. Mucous membrane involvement occurs in 40% to 60% of patients.3 The oropharynx, larynx, and corneal and conjunctival membranes are most commonly affected, resulting in dysphagia, dysphonia or dyspnea, and visual impairment, respectively. Symptoms of internal organ involvement can be manifold, including pain or limited mobility secondary to osteolytic bone lesions or muscle or synovial membrane involvement, as well as seizures, strabismus, and cerebellar ataxia due to central nervous system lesions.2-4 Approximately 40% of patients develop diabetes insipidus secondary to involvement of the pituitary meninges.3
The differential diagnosis of XD includes juvenile xanthogranuloma, papular xanthomas, eruptive xanthomas, generalized eruptive histiocytosis, progressive nodular histiocytosis, multicentric reticulohistiocytosis, eruptive syringomas, sarcoidosis, and Langerhans cell histiocytosis; the latter should be considered, especially when there is concomitant diabetes insipidus.5 Laboratory studies typically are unremarkable. Although the majority of patients are normolipemic, rates of hyperlipemia within this group are comparable to the general population, occasionally rendering it difficult for the clinician to distinguish XD from hyperlipemic xanthomatoses. As such, diagnosis and differentiation from other xanthomatous processes rests on clinicopathological correlation. Histopathology reveals dermal collections of histiocytes, some with foamy cytoplasm, that range in appearance from spindled to scalloped to Touton-like. Early histopathology demonstrates scalloped macrophages with few foamy cells; a mixture of foamy cells, scalloped cells, inflammatory cells, and Touton and foreign body giant cells is characteristic of late lesions. Immunohistochemistry stains positive for non-Langerhans cell surface markers CD68 and factor XIIIa. Electron microscopy demonstrates dense and myeloid bodies, cholesterol crystals, and lipid vacuoles.5
Three subtypes of XD have been described based on the distinct clinical courses that have been observed in patients: a common, persistent, cutaneous form; a self-limited form with spontaneous resolution; and a progressive subtype with internal organ involvement. No consistently efficacious therapies have been identified, but isolated case reports attest to the efficacy of various agents, including azathioprine, clofibrate, cyclophosphamide, glucocorticoids, chlorambucil, and combination or monotherapy with lipid-lowering agents.3,5,6 Surgical resection, cryotherapy, radiotherapy, and CO2 laser therapy may offer some temporary benefit but do not alter the typically relapsing course of the disease.7,8 Remission and long-term control of lesions was reported with use of 2-chlorodeoxyadenosine, a purine nucleoside analogue, for 5 of 8 patients in a case series.3
- Zelger B, Cerio R, Orchard G, et al. Histologic and immunohistochemical study comparing xanthoma disseminatum and histiocytosis X. Arch Dermatol. 1992;128:1207-1212.
- Mahajan V, Sharma A, Chauhan P, et al. Xanthoma disseminatum: a red herring xanthomatosis. Indian J Dermatol Venereol Leprol. 2013;79:253-254.
- Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol. 2011;147:459-464.
- Weiss N, Keller C. Xanthoma disseminatum: a rare normolipemic xanthomatosis. Clin Investig. 1993;71:233-238.
- Park HY, Cho DH, Joe DH, et al. A case of xanthoma disseminatum with spontaneous resolution over 10 years: review of the literature on long-term follow-up [published online May 26, 2011]. Dermatology. 2011;222:236-243.
- Kim SM, Waters P, Vincent A, et al. Sjogren's syndrome myelopathy: spinal cord involvement in Sjogren's syndrome might be a manifestation of neuromyelitis optica. Mult Scler. 2009;15:1062-1068.
- Eisendle K, Linder D, Ratzinger G, et al. Inflammation and lipid accumulation in xanthoma disseminatum: therapeutic considerations. J Am Acad Dermatol. 2008;58(2 suppl):S47-S49.
- Kim JY, Jung HD, Choe YS, et al. A case of xanthoma disseminatum accentuating over the eyelids. Ann Dermatol. 2010;22:353-357.
The Diagnosis: Xanthoma Disseminatum
Genital examination revealed approximately 1.5×3-cm soft, yellow-pink plaques extending from the bilateral inguinal folds to the proximal medial thighs (Figure 1). There was no mucosal, axillary, extensor extremity, or palmoplantar involvement. Histopathologic examination of a biopsy from a plaque on the left side of the lower abdomen revealed sheets of foamy histiocytes distributed throughout a fibrotic dermis. Both mononucleated and multinucleated histiocytes were present, including many Touton giant cells (Figure 2). A patchy infiltrate of lymphocytes and rare eosinophils also was noted. The histiocytes labeled with factor XIIIa but not with S-100. Laboratory tests were performed with the following pertinent findings: low-density lipoprotein, 150 mg/dL (reference range, <130 mg/dL); high-density lipoprotein, 30 mg/dL (reference range, >40 mg/dL). Total cholesterol and triglyceride levels were within reference range, and complete blood cell count and basic metabolic panel were normal.
Xanthoma disseminatum (XD)(also known as Montgomery syndrome) is a rare, nonfamilial, normolipemic non-Langerhans cell histiocytosis characterized by extensive lipid deposition in the skin, mucous membranes, and internal organs. The pathogenesis of XD is poorly understood, but it may represent a macrophage-mediated reactive process triggered by superantigens.1
Xanthoma disseminatum most commonly affects males aged 5 to 25 years.2 Clinically, it is characterized by red-brown to yellowish papules and plaques symmetrically distributed over the eyelids, trunk, face, and proximal extremities. There is a predilection for involvement of flexural and intertriginous surfaces and tendency for extension along Langer lines. Extracutaneous involvement can be a notable cause of morbidity and mortality, underscoring the importance of distinguishing XD from other clinically similar xanthomatoses. Mucous membrane involvement occurs in 40% to 60% of patients.3 The oropharynx, larynx, and corneal and conjunctival membranes are most commonly affected, resulting in dysphagia, dysphonia or dyspnea, and visual impairment, respectively. Symptoms of internal organ involvement can be manifold, including pain or limited mobility secondary to osteolytic bone lesions or muscle or synovial membrane involvement, as well as seizures, strabismus, and cerebellar ataxia due to central nervous system lesions.2-4 Approximately 40% of patients develop diabetes insipidus secondary to involvement of the pituitary meninges.3
The differential diagnosis of XD includes juvenile xanthogranuloma, papular xanthomas, eruptive xanthomas, generalized eruptive histiocytosis, progressive nodular histiocytosis, multicentric reticulohistiocytosis, eruptive syringomas, sarcoidosis, and Langerhans cell histiocytosis; the latter should be considered, especially when there is concomitant diabetes insipidus.5 Laboratory studies typically are unremarkable. Although the majority of patients are normolipemic, rates of hyperlipemia within this group are comparable to the general population, occasionally rendering it difficult for the clinician to distinguish XD from hyperlipemic xanthomatoses. As such, diagnosis and differentiation from other xanthomatous processes rests on clinicopathological correlation. Histopathology reveals dermal collections of histiocytes, some with foamy cytoplasm, that range in appearance from spindled to scalloped to Touton-like. Early histopathology demonstrates scalloped macrophages with few foamy cells; a mixture of foamy cells, scalloped cells, inflammatory cells, and Touton and foreign body giant cells is characteristic of late lesions. Immunohistochemistry stains positive for non-Langerhans cell surface markers CD68 and factor XIIIa. Electron microscopy demonstrates dense and myeloid bodies, cholesterol crystals, and lipid vacuoles.5
Three subtypes of XD have been described based on the distinct clinical courses that have been observed in patients: a common, persistent, cutaneous form; a self-limited form with spontaneous resolution; and a progressive subtype with internal organ involvement. No consistently efficacious therapies have been identified, but isolated case reports attest to the efficacy of various agents, including azathioprine, clofibrate, cyclophosphamide, glucocorticoids, chlorambucil, and combination or monotherapy with lipid-lowering agents.3,5,6 Surgical resection, cryotherapy, radiotherapy, and CO2 laser therapy may offer some temporary benefit but do not alter the typically relapsing course of the disease.7,8 Remission and long-term control of lesions was reported with use of 2-chlorodeoxyadenosine, a purine nucleoside analogue, for 5 of 8 patients in a case series.3
The Diagnosis: Xanthoma Disseminatum
Genital examination revealed approximately 1.5×3-cm soft, yellow-pink plaques extending from the bilateral inguinal folds to the proximal medial thighs (Figure 1). There was no mucosal, axillary, extensor extremity, or palmoplantar involvement. Histopathologic examination of a biopsy from a plaque on the left side of the lower abdomen revealed sheets of foamy histiocytes distributed throughout a fibrotic dermis. Both mononucleated and multinucleated histiocytes were present, including many Touton giant cells (Figure 2). A patchy infiltrate of lymphocytes and rare eosinophils also was noted. The histiocytes labeled with factor XIIIa but not with S-100. Laboratory tests were performed with the following pertinent findings: low-density lipoprotein, 150 mg/dL (reference range, <130 mg/dL); high-density lipoprotein, 30 mg/dL (reference range, >40 mg/dL). Total cholesterol and triglyceride levels were within reference range, and complete blood cell count and basic metabolic panel were normal.
Xanthoma disseminatum (XD)(also known as Montgomery syndrome) is a rare, nonfamilial, normolipemic non-Langerhans cell histiocytosis characterized by extensive lipid deposition in the skin, mucous membranes, and internal organs. The pathogenesis of XD is poorly understood, but it may represent a macrophage-mediated reactive process triggered by superantigens.1
Xanthoma disseminatum most commonly affects males aged 5 to 25 years.2 Clinically, it is characterized by red-brown to yellowish papules and plaques symmetrically distributed over the eyelids, trunk, face, and proximal extremities. There is a predilection for involvement of flexural and intertriginous surfaces and tendency for extension along Langer lines. Extracutaneous involvement can be a notable cause of morbidity and mortality, underscoring the importance of distinguishing XD from other clinically similar xanthomatoses. Mucous membrane involvement occurs in 40% to 60% of patients.3 The oropharynx, larynx, and corneal and conjunctival membranes are most commonly affected, resulting in dysphagia, dysphonia or dyspnea, and visual impairment, respectively. Symptoms of internal organ involvement can be manifold, including pain or limited mobility secondary to osteolytic bone lesions or muscle or synovial membrane involvement, as well as seizures, strabismus, and cerebellar ataxia due to central nervous system lesions.2-4 Approximately 40% of patients develop diabetes insipidus secondary to involvement of the pituitary meninges.3
The differential diagnosis of XD includes juvenile xanthogranuloma, papular xanthomas, eruptive xanthomas, generalized eruptive histiocytosis, progressive nodular histiocytosis, multicentric reticulohistiocytosis, eruptive syringomas, sarcoidosis, and Langerhans cell histiocytosis; the latter should be considered, especially when there is concomitant diabetes insipidus.5 Laboratory studies typically are unremarkable. Although the majority of patients are normolipemic, rates of hyperlipemia within this group are comparable to the general population, occasionally rendering it difficult for the clinician to distinguish XD from hyperlipemic xanthomatoses. As such, diagnosis and differentiation from other xanthomatous processes rests on clinicopathological correlation. Histopathology reveals dermal collections of histiocytes, some with foamy cytoplasm, that range in appearance from spindled to scalloped to Touton-like. Early histopathology demonstrates scalloped macrophages with few foamy cells; a mixture of foamy cells, scalloped cells, inflammatory cells, and Touton and foreign body giant cells is characteristic of late lesions. Immunohistochemistry stains positive for non-Langerhans cell surface markers CD68 and factor XIIIa. Electron microscopy demonstrates dense and myeloid bodies, cholesterol crystals, and lipid vacuoles.5
Three subtypes of XD have been described based on the distinct clinical courses that have been observed in patients: a common, persistent, cutaneous form; a self-limited form with spontaneous resolution; and a progressive subtype with internal organ involvement. No consistently efficacious therapies have been identified, but isolated case reports attest to the efficacy of various agents, including azathioprine, clofibrate, cyclophosphamide, glucocorticoids, chlorambucil, and combination or monotherapy with lipid-lowering agents.3,5,6 Surgical resection, cryotherapy, radiotherapy, and CO2 laser therapy may offer some temporary benefit but do not alter the typically relapsing course of the disease.7,8 Remission and long-term control of lesions was reported with use of 2-chlorodeoxyadenosine, a purine nucleoside analogue, for 5 of 8 patients in a case series.3
- Zelger B, Cerio R, Orchard G, et al. Histologic and immunohistochemical study comparing xanthoma disseminatum and histiocytosis X. Arch Dermatol. 1992;128:1207-1212.
- Mahajan V, Sharma A, Chauhan P, et al. Xanthoma disseminatum: a red herring xanthomatosis. Indian J Dermatol Venereol Leprol. 2013;79:253-254.
- Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol. 2011;147:459-464.
- Weiss N, Keller C. Xanthoma disseminatum: a rare normolipemic xanthomatosis. Clin Investig. 1993;71:233-238.
- Park HY, Cho DH, Joe DH, et al. A case of xanthoma disseminatum with spontaneous resolution over 10 years: review of the literature on long-term follow-up [published online May 26, 2011]. Dermatology. 2011;222:236-243.
- Kim SM, Waters P, Vincent A, et al. Sjogren's syndrome myelopathy: spinal cord involvement in Sjogren's syndrome might be a manifestation of neuromyelitis optica. Mult Scler. 2009;15:1062-1068.
- Eisendle K, Linder D, Ratzinger G, et al. Inflammation and lipid accumulation in xanthoma disseminatum: therapeutic considerations. J Am Acad Dermatol. 2008;58(2 suppl):S47-S49.
- Kim JY, Jung HD, Choe YS, et al. A case of xanthoma disseminatum accentuating over the eyelids. Ann Dermatol. 2010;22:353-357.
- Zelger B, Cerio R, Orchard G, et al. Histologic and immunohistochemical study comparing xanthoma disseminatum and histiocytosis X. Arch Dermatol. 1992;128:1207-1212.
- Mahajan V, Sharma A, Chauhan P, et al. Xanthoma disseminatum: a red herring xanthomatosis. Indian J Dermatol Venereol Leprol. 2013;79:253-254.
- Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol. 2011;147:459-464.
- Weiss N, Keller C. Xanthoma disseminatum: a rare normolipemic xanthomatosis. Clin Investig. 1993;71:233-238.
- Park HY, Cho DH, Joe DH, et al. A case of xanthoma disseminatum with spontaneous resolution over 10 years: review of the literature on long-term follow-up [published online May 26, 2011]. Dermatology. 2011;222:236-243.
- Kim SM, Waters P, Vincent A, et al. Sjogren's syndrome myelopathy: spinal cord involvement in Sjogren's syndrome might be a manifestation of neuromyelitis optica. Mult Scler. 2009;15:1062-1068.
- Eisendle K, Linder D, Ratzinger G, et al. Inflammation and lipid accumulation in xanthoma disseminatum: therapeutic considerations. J Am Acad Dermatol. 2008;58(2 suppl):S47-S49.
- Kim JY, Jung HD, Choe YS, et al. A case of xanthoma disseminatum accentuating over the eyelids. Ann Dermatol. 2010;22:353-357.

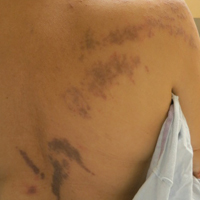
A 28-year-old man presented for evaluation of numerous papules on the face and groin that first appeared in adolescence and had been increasing in size and number over the last several years. The lesions occasionally were pruritic. Review of systems was noncontributory. His medical history was notable for asthma, and there were no affected family members. Physical examination revealed numerous symmetrically distributed, soft, yellow-pink, 1- to 5-mm papules coalescing into plaques on the bilateral malar cheeks extending to the medial canthi and the maxillary, mandibular, zygomatic, and submental regions, as well as the bilateral external auditory meatus.
Hyperpigmented Papules and Plaques
The Diagnosis: Persistent Still Disease
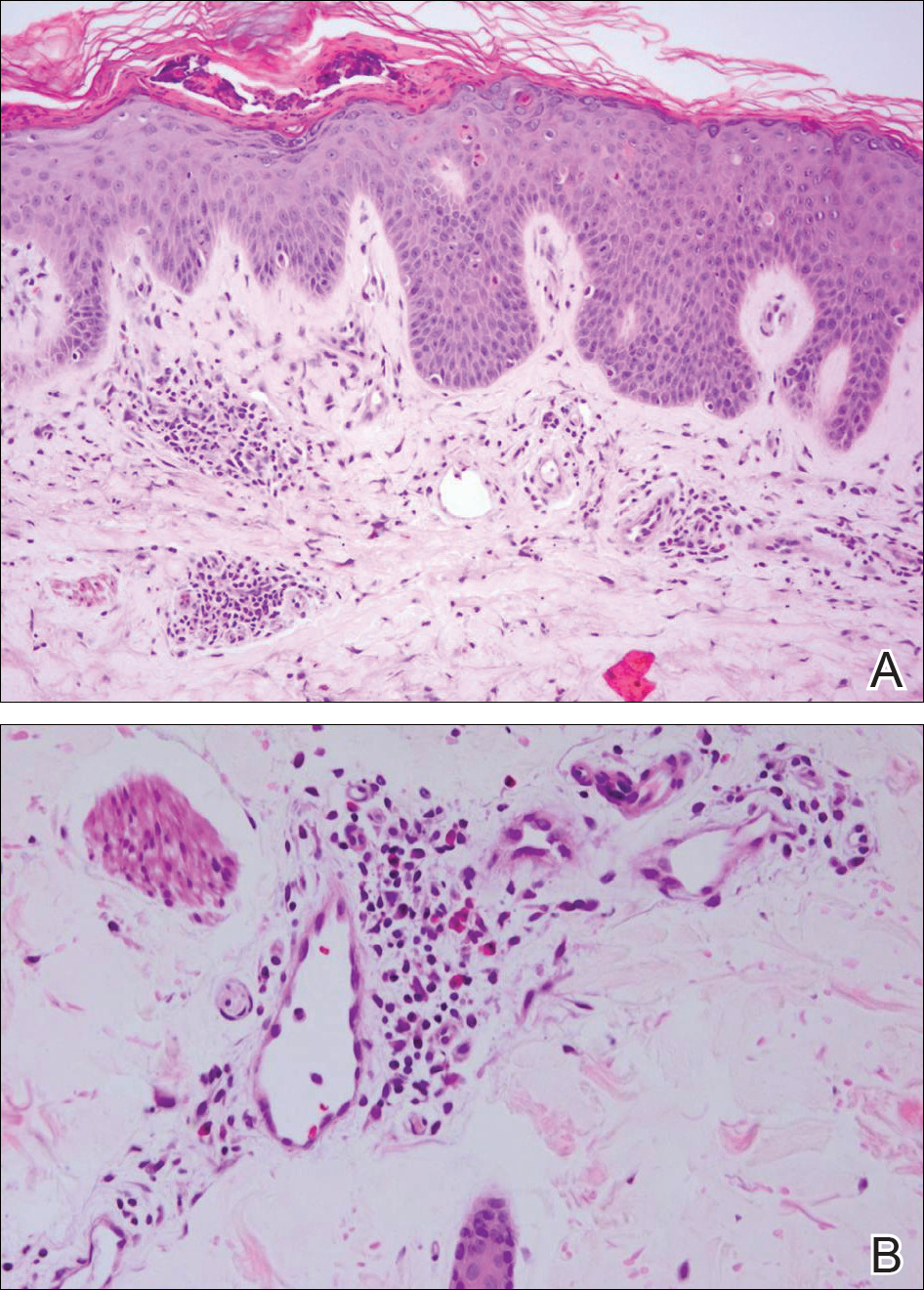
At the time of presentation, the patient had not taken systemic medications for a year. Laboratory studies revealed leukocytosis with neutrophilia and a serum ferritin level of 5493 ng/mL (reference range, 15-200 ng/mL). Rheumatoid factor and antinuclear antibody serologies were within reference range. Microbiologic workup was negative. Lymph node and bone marrow biopsies were negative for a lymphoproliferative disorder. Skin biopsies were performed on the back and forearm. Histologic evaluation revealed orthokeratosis, slight acanthosis, and dyskeratosis confined to the upper layers of the epidermis without evidence of interface dermatitis. There was a mixed perivascular infiltrate composed of lymphocytes and neutrophils with no attendant vasculitic change (Figure).
The patient was discharged on prednisone and seen for outpatient follow-up weeks later. Six weeks later, the cutaneous eruption remained unchanged. The patient was unable to start other systemic medications due to lack of insurance and ineligibility for the local patient-assistance program; he was subsequently lost to follow-up.
Adult-onset Still disease is a rare, systemic, inflammatory condition with a broad spectrum of clinical presentations.1-3 Still disease affects all age groups, and children with Still disease (<16 years) usually have a concurrent diagnosis of juvenile idiopathic arthritis (formerly known as juvenile rheumatoid arthritis).1,2,4 Still disease preferentially affects adolescents and adults aged 16 to 35 years, with more than 75% of new cases occurring in this age range.1 Worldwide, the incidence and prevalence of Still disease is disputed with no conclusive rates established.1,3
Still disease is characterized by 4 cardinal signs: high spiking fevers (temperature, ≥39°C); leukocytosis with a predominance of neutrophils (≥10,000 cells/mm3 with ≥80% neutrophils); arthralgia or arthritis; and an evanescent, nonpruritic, salmon-colored morbilliform eruption of the skin, typically on the trunk or extremities.2 Histologic evaluation of the classic Still disease eruption displays perivascular inflammation of the superficial dermis with infiltration by lymphocytes and histiocytes.3
In 1992, major and minor diagnostic criteria were established for adult-onset Still disease. For diagnosis, patients must meet 5 criteria, including 2 major criteria.5 Major criteria include arthralgia or arthritis present for more than 2 weeks, fever (temperature, >39°C) for at least 1 week, the classic Still disease morbilliform eruption (ie, salmon colored, evanescent, morbilliform), and leukocytosis with more than 80% neutrophils. Minor criteria include sore throat, lymphadenopathy and/or splenomegaly, negative rheumatoid factor and antinuclear antibody serologies, and abnormal liver function (defined as elevated transaminases).5 Although not included in the diagnostic criteria, there have been reports of elevated serum ferritin levels in patients with Still disease, a finding that potentially is useful in distinguishing between active and inactive rheumatic conditions.6,7
Several case reports have described persistent Still disease, a subtype of Still disease in which patients present with brown-red, persistent, pruritic macules, papules, and plaques that are widespread and oddly shaped.8,9 Histologically, this subtype is characterized by necrotic keratinocytes in the epidermis and dermal perivascular inflammation composed of neutrophils and lymphocytes.10 This histology differs from classic Still disease in that the latter typically does not have superficial epidermal dyskeratosis. Our case is consistent with reports of persistent Still disease.
Although the etiology of Still disease remains to be elucidated, HLA-B17, -B18, -B35, and -DR2 have been associated with the disease.3 Furthermore, helper T cell TH1, IL-2, IFN-γ, and tumor necrosis factor α have been implicated in disease pathology, enabling the use of newer targeted pharmacologic therapies. Canakinumab, an IL-1β inhibitor, has been found to improve arthritis, fever, and rash in patients with Still disease.11 These findings are particularly encouraging for patients who have not experienced improvement with traditional antirheumatic drugs, such as our patient who was not steroid responsive.3
Although a salmon-colored, evanescent, morbilliform eruption in the context of other systemic signs and symptoms readily evokes consideration of Still disease, the less common fixed cutaneous eruption seen in our case may evade accurate diagnosis. Our case aims to increase awareness of this unusual and rare subtype of the cutaneous eruption of Still disease, as a timely diagnosis may prevent potentially life-threatening sequelae including cardiopulmonary disease and respiratory failure.3,5,9
- Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still's disease [published online October 11, 2005]. Ann Rheum Dis. 2006;65:564-572.
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22:773-792.
- Bagnari V, Colina M, Ciancio G, et al. Adult-onset Still's disease. Rheumatol Int. 2010;30:855-862.
- Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767-778.
- Yamaguchi M, Ohta A, Tsunematsu, T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424-430.
- Van Reeth C, Le Moel G, Lasne Y, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still's disease. J Rheumatol. 1994;21:890-895.
- Novak S, Anic F, Luke-Vrbanic TS. Extremely high serum ferritin levels as a main diagnostic tool of adult-onset Still's disease. Rheumatol Int. 2012;32:1091-1094.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still's disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still's disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor in refractory adult-onset Still's disease. Sem Arthritis Rheum. 2012;42:201-205.
The Diagnosis: Persistent Still Disease
At the time of presentation, the patient had not taken systemic medications for a year. Laboratory studies revealed leukocytosis with neutrophilia and a serum ferritin level of 5493 ng/mL (reference range, 15-200 ng/mL). Rheumatoid factor and antinuclear antibody serologies were within reference range. Microbiologic workup was negative. Lymph node and bone marrow biopsies were negative for a lymphoproliferative disorder. Skin biopsies were performed on the back and forearm. Histologic evaluation revealed orthokeratosis, slight acanthosis, and dyskeratosis confined to the upper layers of the epidermis without evidence of interface dermatitis. There was a mixed perivascular infiltrate composed of lymphocytes and neutrophils with no attendant vasculitic change (Figure).
The patient was discharged on prednisone and seen for outpatient follow-up weeks later. Six weeks later, the cutaneous eruption remained unchanged. The patient was unable to start other systemic medications due to lack of insurance and ineligibility for the local patient-assistance program; he was subsequently lost to follow-up.
Adult-onset Still disease is a rare, systemic, inflammatory condition with a broad spectrum of clinical presentations.1-3 Still disease affects all age groups, and children with Still disease (<16 years) usually have a concurrent diagnosis of juvenile idiopathic arthritis (formerly known as juvenile rheumatoid arthritis).1,2,4 Still disease preferentially affects adolescents and adults aged 16 to 35 years, with more than 75% of new cases occurring in this age range.1 Worldwide, the incidence and prevalence of Still disease is disputed with no conclusive rates established.1,3
Still disease is characterized by 4 cardinal signs: high spiking fevers (temperature, ≥39°C); leukocytosis with a predominance of neutrophils (≥10,000 cells/mm3 with ≥80% neutrophils); arthralgia or arthritis; and an evanescent, nonpruritic, salmon-colored morbilliform eruption of the skin, typically on the trunk or extremities.2 Histologic evaluation of the classic Still disease eruption displays perivascular inflammation of the superficial dermis with infiltration by lymphocytes and histiocytes.3
In 1992, major and minor diagnostic criteria were established for adult-onset Still disease. For diagnosis, patients must meet 5 criteria, including 2 major criteria.5 Major criteria include arthralgia or arthritis present for more than 2 weeks, fever (temperature, >39°C) for at least 1 week, the classic Still disease morbilliform eruption (ie, salmon colored, evanescent, morbilliform), and leukocytosis with more than 80% neutrophils. Minor criteria include sore throat, lymphadenopathy and/or splenomegaly, negative rheumatoid factor and antinuclear antibody serologies, and abnormal liver function (defined as elevated transaminases).5 Although not included in the diagnostic criteria, there have been reports of elevated serum ferritin levels in patients with Still disease, a finding that potentially is useful in distinguishing between active and inactive rheumatic conditions.6,7
Several case reports have described persistent Still disease, a subtype of Still disease in which patients present with brown-red, persistent, pruritic macules, papules, and plaques that are widespread and oddly shaped.8,9 Histologically, this subtype is characterized by necrotic keratinocytes in the epidermis and dermal perivascular inflammation composed of neutrophils and lymphocytes.10 This histology differs from classic Still disease in that the latter typically does not have superficial epidermal dyskeratosis. Our case is consistent with reports of persistent Still disease.
Although the etiology of Still disease remains to be elucidated, HLA-B17, -B18, -B35, and -DR2 have been associated with the disease.3 Furthermore, helper T cell TH1, IL-2, IFN-γ, and tumor necrosis factor α have been implicated in disease pathology, enabling the use of newer targeted pharmacologic therapies. Canakinumab, an IL-1β inhibitor, has been found to improve arthritis, fever, and rash in patients with Still disease.11 These findings are particularly encouraging for patients who have not experienced improvement with traditional antirheumatic drugs, such as our patient who was not steroid responsive.3
Although a salmon-colored, evanescent, morbilliform eruption in the context of other systemic signs and symptoms readily evokes consideration of Still disease, the less common fixed cutaneous eruption seen in our case may evade accurate diagnosis. Our case aims to increase awareness of this unusual and rare subtype of the cutaneous eruption of Still disease, as a timely diagnosis may prevent potentially life-threatening sequelae including cardiopulmonary disease and respiratory failure.3,5,9
The Diagnosis: Persistent Still Disease
At the time of presentation, the patient had not taken systemic medications for a year. Laboratory studies revealed leukocytosis with neutrophilia and a serum ferritin level of 5493 ng/mL (reference range, 15-200 ng/mL). Rheumatoid factor and antinuclear antibody serologies were within reference range. Microbiologic workup was negative. Lymph node and bone marrow biopsies were negative for a lymphoproliferative disorder. Skin biopsies were performed on the back and forearm. Histologic evaluation revealed orthokeratosis, slight acanthosis, and dyskeratosis confined to the upper layers of the epidermis without evidence of interface dermatitis. There was a mixed perivascular infiltrate composed of lymphocytes and neutrophils with no attendant vasculitic change (Figure).
The patient was discharged on prednisone and seen for outpatient follow-up weeks later. Six weeks later, the cutaneous eruption remained unchanged. The patient was unable to start other systemic medications due to lack of insurance and ineligibility for the local patient-assistance program; he was subsequently lost to follow-up.
Adult-onset Still disease is a rare, systemic, inflammatory condition with a broad spectrum of clinical presentations.1-3 Still disease affects all age groups, and children with Still disease (<16 years) usually have a concurrent diagnosis of juvenile idiopathic arthritis (formerly known as juvenile rheumatoid arthritis).1,2,4 Still disease preferentially affects adolescents and adults aged 16 to 35 years, with more than 75% of new cases occurring in this age range.1 Worldwide, the incidence and prevalence of Still disease is disputed with no conclusive rates established.1,3
Still disease is characterized by 4 cardinal signs: high spiking fevers (temperature, ≥39°C); leukocytosis with a predominance of neutrophils (≥10,000 cells/mm3 with ≥80% neutrophils); arthralgia or arthritis; and an evanescent, nonpruritic, salmon-colored morbilliform eruption of the skin, typically on the trunk or extremities.2 Histologic evaluation of the classic Still disease eruption displays perivascular inflammation of the superficial dermis with infiltration by lymphocytes and histiocytes.3
In 1992, major and minor diagnostic criteria were established for adult-onset Still disease. For diagnosis, patients must meet 5 criteria, including 2 major criteria.5 Major criteria include arthralgia or arthritis present for more than 2 weeks, fever (temperature, >39°C) for at least 1 week, the classic Still disease morbilliform eruption (ie, salmon colored, evanescent, morbilliform), and leukocytosis with more than 80% neutrophils. Minor criteria include sore throat, lymphadenopathy and/or splenomegaly, negative rheumatoid factor and antinuclear antibody serologies, and abnormal liver function (defined as elevated transaminases).5 Although not included in the diagnostic criteria, there have been reports of elevated serum ferritin levels in patients with Still disease, a finding that potentially is useful in distinguishing between active and inactive rheumatic conditions.6,7
Several case reports have described persistent Still disease, a subtype of Still disease in which patients present with brown-red, persistent, pruritic macules, papules, and plaques that are widespread and oddly shaped.8,9 Histologically, this subtype is characterized by necrotic keratinocytes in the epidermis and dermal perivascular inflammation composed of neutrophils and lymphocytes.10 This histology differs from classic Still disease in that the latter typically does not have superficial epidermal dyskeratosis. Our case is consistent with reports of persistent Still disease.
Although the etiology of Still disease remains to be elucidated, HLA-B17, -B18, -B35, and -DR2 have been associated with the disease.3 Furthermore, helper T cell TH1, IL-2, IFN-γ, and tumor necrosis factor α have been implicated in disease pathology, enabling the use of newer targeted pharmacologic therapies. Canakinumab, an IL-1β inhibitor, has been found to improve arthritis, fever, and rash in patients with Still disease.11 These findings are particularly encouraging for patients who have not experienced improvement with traditional antirheumatic drugs, such as our patient who was not steroid responsive.3
Although a salmon-colored, evanescent, morbilliform eruption in the context of other systemic signs and symptoms readily evokes consideration of Still disease, the less common fixed cutaneous eruption seen in our case may evade accurate diagnosis. Our case aims to increase awareness of this unusual and rare subtype of the cutaneous eruption of Still disease, as a timely diagnosis may prevent potentially life-threatening sequelae including cardiopulmonary disease and respiratory failure.3,5,9
- Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still's disease [published online October 11, 2005]. Ann Rheum Dis. 2006;65:564-572.
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22:773-792.
- Bagnari V, Colina M, Ciancio G, et al. Adult-onset Still's disease. Rheumatol Int. 2010;30:855-862.
- Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767-778.
- Yamaguchi M, Ohta A, Tsunematsu, T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424-430.
- Van Reeth C, Le Moel G, Lasne Y, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still's disease. J Rheumatol. 1994;21:890-895.
- Novak S, Anic F, Luke-Vrbanic TS. Extremely high serum ferritin levels as a main diagnostic tool of adult-onset Still's disease. Rheumatol Int. 2012;32:1091-1094.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still's disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still's disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor in refractory adult-onset Still's disease. Sem Arthritis Rheum. 2012;42:201-205.
- Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still's disease [published online October 11, 2005]. Ann Rheum Dis. 2006;65:564-572.
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22:773-792.
- Bagnari V, Colina M, Ciancio G, et al. Adult-onset Still's disease. Rheumatol Int. 2010;30:855-862.
- Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767-778.
- Yamaguchi M, Ohta A, Tsunematsu, T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424-430.
- Van Reeth C, Le Moel G, Lasne Y, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still's disease. J Rheumatol. 1994;21:890-895.
- Novak S, Anic F, Luke-Vrbanic TS. Extremely high serum ferritin levels as a main diagnostic tool of adult-onset Still's disease. Rheumatol Int. 2012;32:1091-1094.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still's disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still's disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor in refractory adult-onset Still's disease. Sem Arthritis Rheum. 2012;42:201-205.
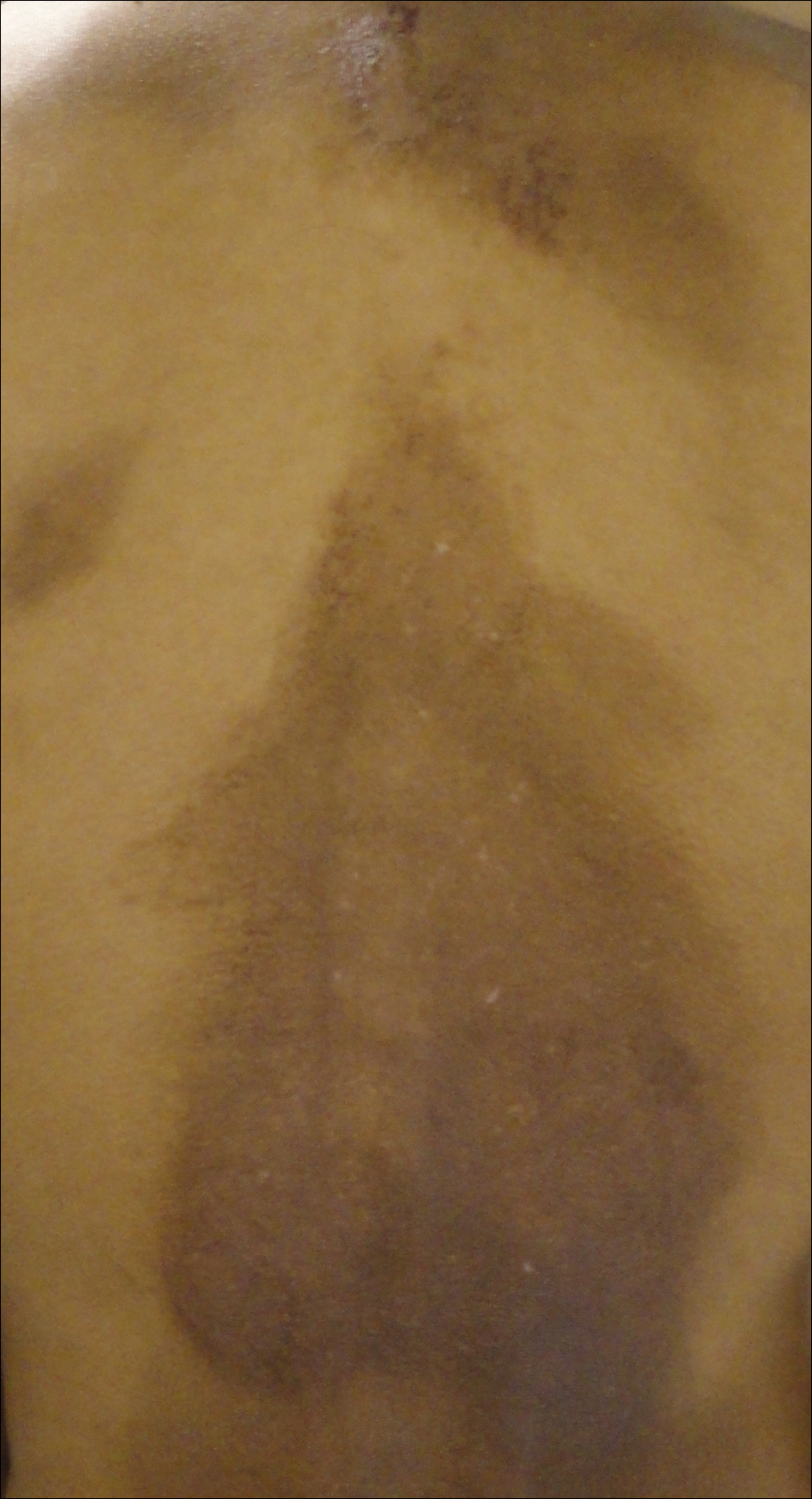
A 25-year-old Hispanic man with a history of juvenile idiopathic arthritis was admitted with a high-grade fever (temperature, >38.9°C) and diffuse nonlocalized abdominal pain of 2 days' duration. Physical examination revealed tachycardia, axillary lymphadenopathy, and hepatosplenomegaly. Cutaneous findings consisted of striking hyperpigmented patches on the chest and back, and hyperpigmented scaly lichenoid papules and plaques on the upper and lower extremities. The plaques on the lower extremities exhibited koebnerization. The patient reported that the eruption initially presented at 16 years of age as pruritic papules on the legs, which gradually spread to involve the arms, chest, and back. Prior treatments of juvenile idiopathic arthritis included prednisone, methotrexate, infliximab, and etanercept, though they were intermittent and temporary. Over time, the cutaneous eruption evolved into its current morphology and distribution, with periods of clearance observed while receiving systemic medications.
Coding Changes for 2017
All physicians will see changes in reimbursement in 2017. A new president with a new agenda makes for an interesting time ahead for health care in the United States. However, in this time of flux, there is one constant: the Final Rule, an informal term for the annual update on how the Medicare system will function and how much you will get paid for what you do.1 The document is 393 pages and outlines what is new in the Medicare system, with lots of supplements giving granular details about physician work, overhead, and supply and labor costs. In this column, I have taken the liberty of dissecting the Final Rule for you and to bring attention to its high and low points for dermatologists.
Changes in Relative Value Units
The conversion factor has gone up, meaning you will be paid a bit more this year for what you do; it is not enough to account for inflation or the increasing cost of unfunded mandates, but it is better than nothing. Although the conversion factor was $35.8043 in 2016, it increased by more than 0.2% on January 1, 2017, to $35.8887.1 How is this conversion factor calculated? We go up 0.5% due to MACRA (Medicare Access and CHIP Reauthorization Act), down 0.013% due to budget neutrality, down 0.07% due to multiple procedure payment reduction changes, and down another 0.18% due to the misvalued code target.1 The misvalued code target is related to targets established by statute for 2016 to 2018 and payment rates are reduced across the board if they are not met.
If payments suffer from reductions in work value, they may not happen all at once. If the Centers for Medicare & Medicaid Services (CMS) reduce total relative value units (RVUs) by more than 20%, reductions will take place over at least 2 years with a single year drop maximum of 19%.1 Unfortunately, such limits do not apply to revised codes, which can take as big a hit as the CMS cares to make.
Changes to Global Periods
In 2015, we learned that 10- and 90-day global periods would be eliminated in 2017 and 2018, respectively, with great concern on the part of the government about the number and level of evaluation and management services embedded in these codes. The implementation of global policy elimination was prohibited by MACRA and the CMS was required to develop and implement a process to gather data on services furnished in the global period from a representative sample of physicians, which they will use to value surgical services beginningin 2019.1 The CMS decided to capture this data with a new set of time-based G codes (which would be onerous for all practicing physicians), not just the unlucky folks who were to be the sample mandated under MACRA.2 During the comment period, it became obvious to the CMS that this concept was flawed for many reasons and it decided to hold a town hall meeting at the CMS headquarters on August 25, 2016, on data collection on resources used in furnishing global services in which 90 minutes of live testimony in the morning was followed by another 90 minutes by telephone in the afternoon.3 This meeting, which I attended, resulted in the CMS changing the all-practitioner reporting program to a specified sample with others allowed to opt in. Practitioners in groups of less than 10 are exempt, and only physicians in Florida, Kentucky, Louisiana, Nevada, New Jersey, North Dakota, Ohio, Oregon, and Rhode Island must capture data beginning in July 2017.1 These data only have to be captured on codes that are used by more than 100 practitioners and are furnished at least 10,000 times or have allowed charges of greater than $10,000,000 annually. If you are lucky enough to live in one of the testing states, you must start on July 1 but can start before July 1 if you wish. Practitioners in smaller practices or in other geographic areas are encouraged to report data if feasible but are not required to do so. Current Procedural Terminology (CPT) code 99024 will be used for reporting postoperative services rather than the proposed onerous set of G codes, and reporting will not be required for preoperative visits included in the global package or for services not related to the patient’s visit.
Changes to Chronic Care Management
There are new and modified chronic care management codes that are not of use to you unless you are the primary provider for the patient and you and the patient meet multiple stringent requirements.4 The patient must have multiple illnesses, use multiple medications, be unable to perform activities of daily living, require a caregiver, and/or have repeat admissions or emergency department visits. Typical adult patients who receive complex chronic care management services are treated with 3 or more prescription medications and may be receiving other types of therapeutic interventions (eg, physical therapy, occupational therapy). Typical pediatric patients receive 3 or more therapeutic interventions (eg, medications, nutritional support, respiratory therapy). All patients have 2 or more chronic continuous or episodic health conditions that are expected to last at least 12 months or until the death of the patient and place the patient at serious risk for death, acute exacerbation/decompensation, or functional decline.4
Changes to Moderate Sedation Codes
The economic value of providing moderate sedation (eg, drug-induced depression of consciousness during which patients respond purposefully to verbal commands, either alone or accompanied by light tactile stimulation) used to be embedded in a variety of CPT codes, which is no longer the case in 2017. Diazepam or similar drugs swallowed or dissolved under the tongue are not included. The new CPT codes 99151, 99152, 99153, 99155, 99156, and 99157 are not to be used to report administration of medications for pain control or minimal sedation (anxiolysis). An independent trained observer, an individual who is qualified to monitor the patient during the procedure and who has no other duties (eg, assisting at surgery) during the procedure, must be present. If you are thinking of using these codes, read the entire section in the CPT manual,4 check your state laws, and consult your malpractice carrier and perhaps even your health care attorney.
Changes to Nail Procedure Codes
Current Procedural Terminology code 11752 (excision of nail and nail matrix, partial or complete [eg, ingrown or deformed nail], for permanent removal; with amputation of tuft of distal phalanx) is now gone, while base code 11750 remains. If you are doing nail surgery and removing underlying bone, instead use code 26236 (partial excision [craterization, saucerization, or diaphysectomy] bone [eg, osteomyelitis]; distal phalanx of finger), 28124 (partial excision [craterization, saucerization, sequestrectomy, or diaphysectomy] bone [eg, osteomyelitis or bossing]; phalanx of toe), or other codes in the same section of the CPT manual if they more precisely describe the procedure performed.
Changes to Slide Consultation Codes
The slide consultation codes 88321 (consultation and report on referred slides prepared elsewhere), 88323 (consultation and report on referred material requiring preparation of slides), and 88325 (consultation, comprehensive, with review of records and specimens, with report on referred material) were revalued this year, with the first 2 showing no change but the latter showing an increase in value from 2.50 to 2.85 RVUs.1 None are meant to be routine. If you have every slide looked at by someone else for “quality assurance reasons,” the consultation is not reportable. If you use these consultation codes too often, the CMS might have concerns about fraud and abuse. Visit http://data.cms.gov to see how you compare to your peers.
Changes to Reflectance Confocal Microscopy Codes
Reflectance confocal microscopy had new codes for 2016, which were carrier priced, and in 2017 they have real RVUs per the CMS. The payments for these codes have a national average reimbursement of $161.85 for 96931 (reflectance confocal microscopy for cellular and subcellular imaging of skin; image acquisition and interpretation and report, first lesion), $104.80 for 96932 (image acquisition only, first lesion), and $45.94 for 96933 (interpretation and report only, first lesion).5 The respective add-on codes have values of $83.26 for 96934 (image acquisition and interpretation and report, each additional lesion [list separately in addition to code for primary procedure]), $35.17 for 96935 (image acquisition only, each additional lesion [list separately in addition to code for primary procedure]), and $43.78 for 96936 (interpretation and report only, each additional lesion [list separately in addition to code for primary procedure]).
Other Coding Changes
There are a whole bunch of new codes in the “Genomic Sequencing Procedures and Other Molecular Multianalyte Assays” (MMAAs) section of CPT. The important thing for you to remember is these codes are for the laboratory performing the assay to report, not the physician ordering it. There is a new Appendix O for proprietary laboratory analysis MMAAs, including those that do not have a Category I code. These MMAAs are identified in Appendix O by a 4-digit number followed by the letter M.4
There are some revisions to psychotherapy codes 90832 to 90847. These codes are outside our scope of practice and should only be used by psychiatrists, social workers, psychologists, or other appropriate mental health workers.
Final Thoughts
It has not been a breakout year for telehealth and we still do not have payment for store-and-forward teledermatology, except in a few designated rural areas. With the advent of the rhetoric we have heard after the presidential election, any speculation on what will happen to the brave new world of the merit-based incentive payment system, alternative payment models, and other regulations are anyone’s guess.
- Medicare Program; Revisions to Payment Policies Under the Physician Fee Schedule and Other Revisions to Part B for CY 2017; Medicare Advantage Bid Pricing Data Release; Medicare Advantage and Part D Medical Loss Ratio Data Release; Medicare Advantage Provider Network Requirements; Expansion of Medicare Diabetes Prevention Program Model; Medicare Shared Savings Program Requirements. Fed Regist. 2016;81(220):80170-80562. To be codified at 42 CFR § 405, 410, 411, 414, 417, 422, 423, 424, 425, and 460.
- Siegel DM. The Proposed Rule and payments for 2017: the good, the bad, and the ugly. Cutis. 2016;98:245-248.
- Data collection on resources used in furnishing global services town hall CY 2017 Medicare physician fee schedule Proposed Rule. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/Downloads/CY2017-PFS-FR-Townhall.pdf. Published August 25, 2016. Accessed January 4, 2017.
- Current Procedural Terminology 2017, Professional Edition. Chicago, IL: American Medical Association; 2016.
- Addendum B—relative value units and related information used in CY 2017 final rule. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/Downloads/CY2017-PFS-FR-Addenda.zip. Accessed January 23, 2017.
All physicians will see changes in reimbursement in 2017. A new president with a new agenda makes for an interesting time ahead for health care in the United States. However, in this time of flux, there is one constant: the Final Rule, an informal term for the annual update on how the Medicare system will function and how much you will get paid for what you do.1 The document is 393 pages and outlines what is new in the Medicare system, with lots of supplements giving granular details about physician work, overhead, and supply and labor costs. In this column, I have taken the liberty of dissecting the Final Rule for you and to bring attention to its high and low points for dermatologists.
Changes in Relative Value Units
The conversion factor has gone up, meaning you will be paid a bit more this year for what you do; it is not enough to account for inflation or the increasing cost of unfunded mandates, but it is better than nothing. Although the conversion factor was $35.8043 in 2016, it increased by more than 0.2% on January 1, 2017, to $35.8887.1 How is this conversion factor calculated? We go up 0.5% due to MACRA (Medicare Access and CHIP Reauthorization Act), down 0.013% due to budget neutrality, down 0.07% due to multiple procedure payment reduction changes, and down another 0.18% due to the misvalued code target.1 The misvalued code target is related to targets established by statute for 2016 to 2018 and payment rates are reduced across the board if they are not met.
If payments suffer from reductions in work value, they may not happen all at once. If the Centers for Medicare & Medicaid Services (CMS) reduce total relative value units (RVUs) by more than 20%, reductions will take place over at least 2 years with a single year drop maximum of 19%.1 Unfortunately, such limits do not apply to revised codes, which can take as big a hit as the CMS cares to make.
Changes to Global Periods
In 2015, we learned that 10- and 90-day global periods would be eliminated in 2017 and 2018, respectively, with great concern on the part of the government about the number and level of evaluation and management services embedded in these codes. The implementation of global policy elimination was prohibited by MACRA and the CMS was required to develop and implement a process to gather data on services furnished in the global period from a representative sample of physicians, which they will use to value surgical services beginningin 2019.1 The CMS decided to capture this data with a new set of time-based G codes (which would be onerous for all practicing physicians), not just the unlucky folks who were to be the sample mandated under MACRA.2 During the comment period, it became obvious to the CMS that this concept was flawed for many reasons and it decided to hold a town hall meeting at the CMS headquarters on August 25, 2016, on data collection on resources used in furnishing global services in which 90 minutes of live testimony in the morning was followed by another 90 minutes by telephone in the afternoon.3 This meeting, which I attended, resulted in the CMS changing the all-practitioner reporting program to a specified sample with others allowed to opt in. Practitioners in groups of less than 10 are exempt, and only physicians in Florida, Kentucky, Louisiana, Nevada, New Jersey, North Dakota, Ohio, Oregon, and Rhode Island must capture data beginning in July 2017.1 These data only have to be captured on codes that are used by more than 100 practitioners and are furnished at least 10,000 times or have allowed charges of greater than $10,000,000 annually. If you are lucky enough to live in one of the testing states, you must start on July 1 but can start before July 1 if you wish. Practitioners in smaller practices or in other geographic areas are encouraged to report data if feasible but are not required to do so. Current Procedural Terminology (CPT) code 99024 will be used for reporting postoperative services rather than the proposed onerous set of G codes, and reporting will not be required for preoperative visits included in the global package or for services not related to the patient’s visit.
Changes to Chronic Care Management
There are new and modified chronic care management codes that are not of use to you unless you are the primary provider for the patient and you and the patient meet multiple stringent requirements.4 The patient must have multiple illnesses, use multiple medications, be unable to perform activities of daily living, require a caregiver, and/or have repeat admissions or emergency department visits. Typical adult patients who receive complex chronic care management services are treated with 3 or more prescription medications and may be receiving other types of therapeutic interventions (eg, physical therapy, occupational therapy). Typical pediatric patients receive 3 or more therapeutic interventions (eg, medications, nutritional support, respiratory therapy). All patients have 2 or more chronic continuous or episodic health conditions that are expected to last at least 12 months or until the death of the patient and place the patient at serious risk for death, acute exacerbation/decompensation, or functional decline.4
Changes to Moderate Sedation Codes
The economic value of providing moderate sedation (eg, drug-induced depression of consciousness during which patients respond purposefully to verbal commands, either alone or accompanied by light tactile stimulation) used to be embedded in a variety of CPT codes, which is no longer the case in 2017. Diazepam or similar drugs swallowed or dissolved under the tongue are not included. The new CPT codes 99151, 99152, 99153, 99155, 99156, and 99157 are not to be used to report administration of medications for pain control or minimal sedation (anxiolysis). An independent trained observer, an individual who is qualified to monitor the patient during the procedure and who has no other duties (eg, assisting at surgery) during the procedure, must be present. If you are thinking of using these codes, read the entire section in the CPT manual,4 check your state laws, and consult your malpractice carrier and perhaps even your health care attorney.
Changes to Nail Procedure Codes
Current Procedural Terminology code 11752 (excision of nail and nail matrix, partial or complete [eg, ingrown or deformed nail], for permanent removal; with amputation of tuft of distal phalanx) is now gone, while base code 11750 remains. If you are doing nail surgery and removing underlying bone, instead use code 26236 (partial excision [craterization, saucerization, or diaphysectomy] bone [eg, osteomyelitis]; distal phalanx of finger), 28124 (partial excision [craterization, saucerization, sequestrectomy, or diaphysectomy] bone [eg, osteomyelitis or bossing]; phalanx of toe), or other codes in the same section of the CPT manual if they more precisely describe the procedure performed.
Changes to Slide Consultation Codes
The slide consultation codes 88321 (consultation and report on referred slides prepared elsewhere), 88323 (consultation and report on referred material requiring preparation of slides), and 88325 (consultation, comprehensive, with review of records and specimens, with report on referred material) were revalued this year, with the first 2 showing no change but the latter showing an increase in value from 2.50 to 2.85 RVUs.1 None are meant to be routine. If you have every slide looked at by someone else for “quality assurance reasons,” the consultation is not reportable. If you use these consultation codes too often, the CMS might have concerns about fraud and abuse. Visit http://data.cms.gov to see how you compare to your peers.
Changes to Reflectance Confocal Microscopy Codes
Reflectance confocal microscopy had new codes for 2016, which were carrier priced, and in 2017 they have real RVUs per the CMS. The payments for these codes have a national average reimbursement of $161.85 for 96931 (reflectance confocal microscopy for cellular and subcellular imaging of skin; image acquisition and interpretation and report, first lesion), $104.80 for 96932 (image acquisition only, first lesion), and $45.94 for 96933 (interpretation and report only, first lesion).5 The respective add-on codes have values of $83.26 for 96934 (image acquisition and interpretation and report, each additional lesion [list separately in addition to code for primary procedure]), $35.17 for 96935 (image acquisition only, each additional lesion [list separately in addition to code for primary procedure]), and $43.78 for 96936 (interpretation and report only, each additional lesion [list separately in addition to code for primary procedure]).
Other Coding Changes
There are a whole bunch of new codes in the “Genomic Sequencing Procedures and Other Molecular Multianalyte Assays” (MMAAs) section of CPT. The important thing for you to remember is these codes are for the laboratory performing the assay to report, not the physician ordering it. There is a new Appendix O for proprietary laboratory analysis MMAAs, including those that do not have a Category I code. These MMAAs are identified in Appendix O by a 4-digit number followed by the letter M.4
There are some revisions to psychotherapy codes 90832 to 90847. These codes are outside our scope of practice and should only be used by psychiatrists, social workers, psychologists, or other appropriate mental health workers.
Final Thoughts
It has not been a breakout year for telehealth and we still do not have payment for store-and-forward teledermatology, except in a few designated rural areas. With the advent of the rhetoric we have heard after the presidential election, any speculation on what will happen to the brave new world of the merit-based incentive payment system, alternative payment models, and other regulations are anyone’s guess.
All physicians will see changes in reimbursement in 2017. A new president with a new agenda makes for an interesting time ahead for health care in the United States. However, in this time of flux, there is one constant: the Final Rule, an informal term for the annual update on how the Medicare system will function and how much you will get paid for what you do.1 The document is 393 pages and outlines what is new in the Medicare system, with lots of supplements giving granular details about physician work, overhead, and supply and labor costs. In this column, I have taken the liberty of dissecting the Final Rule for you and to bring attention to its high and low points for dermatologists.
Changes in Relative Value Units
The conversion factor has gone up, meaning you will be paid a bit more this year for what you do; it is not enough to account for inflation or the increasing cost of unfunded mandates, but it is better than nothing. Although the conversion factor was $35.8043 in 2016, it increased by more than 0.2% on January 1, 2017, to $35.8887.1 How is this conversion factor calculated? We go up 0.5% due to MACRA (Medicare Access and CHIP Reauthorization Act), down 0.013% due to budget neutrality, down 0.07% due to multiple procedure payment reduction changes, and down another 0.18% due to the misvalued code target.1 The misvalued code target is related to targets established by statute for 2016 to 2018 and payment rates are reduced across the board if they are not met.
If payments suffer from reductions in work value, they may not happen all at once. If the Centers for Medicare & Medicaid Services (CMS) reduce total relative value units (RVUs) by more than 20%, reductions will take place over at least 2 years with a single year drop maximum of 19%.1 Unfortunately, such limits do not apply to revised codes, which can take as big a hit as the CMS cares to make.
Changes to Global Periods
In 2015, we learned that 10- and 90-day global periods would be eliminated in 2017 and 2018, respectively, with great concern on the part of the government about the number and level of evaluation and management services embedded in these codes. The implementation of global policy elimination was prohibited by MACRA and the CMS was required to develop and implement a process to gather data on services furnished in the global period from a representative sample of physicians, which they will use to value surgical services beginningin 2019.1 The CMS decided to capture this data with a new set of time-based G codes (which would be onerous for all practicing physicians), not just the unlucky folks who were to be the sample mandated under MACRA.2 During the comment period, it became obvious to the CMS that this concept was flawed for many reasons and it decided to hold a town hall meeting at the CMS headquarters on August 25, 2016, on data collection on resources used in furnishing global services in which 90 minutes of live testimony in the morning was followed by another 90 minutes by telephone in the afternoon.3 This meeting, which I attended, resulted in the CMS changing the all-practitioner reporting program to a specified sample with others allowed to opt in. Practitioners in groups of less than 10 are exempt, and only physicians in Florida, Kentucky, Louisiana, Nevada, New Jersey, North Dakota, Ohio, Oregon, and Rhode Island must capture data beginning in July 2017.1 These data only have to be captured on codes that are used by more than 100 practitioners and are furnished at least 10,000 times or have allowed charges of greater than $10,000,000 annually. If you are lucky enough to live in one of the testing states, you must start on July 1 but can start before July 1 if you wish. Practitioners in smaller practices or in other geographic areas are encouraged to report data if feasible but are not required to do so. Current Procedural Terminology (CPT) code 99024 will be used for reporting postoperative services rather than the proposed onerous set of G codes, and reporting will not be required for preoperative visits included in the global package or for services not related to the patient’s visit.
Changes to Chronic Care Management
There are new and modified chronic care management codes that are not of use to you unless you are the primary provider for the patient and you and the patient meet multiple stringent requirements.4 The patient must have multiple illnesses, use multiple medications, be unable to perform activities of daily living, require a caregiver, and/or have repeat admissions or emergency department visits. Typical adult patients who receive complex chronic care management services are treated with 3 or more prescription medications and may be receiving other types of therapeutic interventions (eg, physical therapy, occupational therapy). Typical pediatric patients receive 3 or more therapeutic interventions (eg, medications, nutritional support, respiratory therapy). All patients have 2 or more chronic continuous or episodic health conditions that are expected to last at least 12 months or until the death of the patient and place the patient at serious risk for death, acute exacerbation/decompensation, or functional decline.4
Changes to Moderate Sedation Codes
The economic value of providing moderate sedation (eg, drug-induced depression of consciousness during which patients respond purposefully to verbal commands, either alone or accompanied by light tactile stimulation) used to be embedded in a variety of CPT codes, which is no longer the case in 2017. Diazepam or similar drugs swallowed or dissolved under the tongue are not included. The new CPT codes 99151, 99152, 99153, 99155, 99156, and 99157 are not to be used to report administration of medications for pain control or minimal sedation (anxiolysis). An independent trained observer, an individual who is qualified to monitor the patient during the procedure and who has no other duties (eg, assisting at surgery) during the procedure, must be present. If you are thinking of using these codes, read the entire section in the CPT manual,4 check your state laws, and consult your malpractice carrier and perhaps even your health care attorney.
Changes to Nail Procedure Codes
Current Procedural Terminology code 11752 (excision of nail and nail matrix, partial or complete [eg, ingrown or deformed nail], for permanent removal; with amputation of tuft of distal phalanx) is now gone, while base code 11750 remains. If you are doing nail surgery and removing underlying bone, instead use code 26236 (partial excision [craterization, saucerization, or diaphysectomy] bone [eg, osteomyelitis]; distal phalanx of finger), 28124 (partial excision [craterization, saucerization, sequestrectomy, or diaphysectomy] bone [eg, osteomyelitis or bossing]; phalanx of toe), or other codes in the same section of the CPT manual if they more precisely describe the procedure performed.
Changes to Slide Consultation Codes
The slide consultation codes 88321 (consultation and report on referred slides prepared elsewhere), 88323 (consultation and report on referred material requiring preparation of slides), and 88325 (consultation, comprehensive, with review of records and specimens, with report on referred material) were revalued this year, with the first 2 showing no change but the latter showing an increase in value from 2.50 to 2.85 RVUs.1 None are meant to be routine. If you have every slide looked at by someone else for “quality assurance reasons,” the consultation is not reportable. If you use these consultation codes too often, the CMS might have concerns about fraud and abuse. Visit http://data.cms.gov to see how you compare to your peers.
Changes to Reflectance Confocal Microscopy Codes
Reflectance confocal microscopy had new codes for 2016, which were carrier priced, and in 2017 they have real RVUs per the CMS. The payments for these codes have a national average reimbursement of $161.85 for 96931 (reflectance confocal microscopy for cellular and subcellular imaging of skin; image acquisition and interpretation and report, first lesion), $104.80 for 96932 (image acquisition only, first lesion), and $45.94 for 96933 (interpretation and report only, first lesion).5 The respective add-on codes have values of $83.26 for 96934 (image acquisition and interpretation and report, each additional lesion [list separately in addition to code for primary procedure]), $35.17 for 96935 (image acquisition only, each additional lesion [list separately in addition to code for primary procedure]), and $43.78 for 96936 (interpretation and report only, each additional lesion [list separately in addition to code for primary procedure]).
Other Coding Changes
There are a whole bunch of new codes in the “Genomic Sequencing Procedures and Other Molecular Multianalyte Assays” (MMAAs) section of CPT. The important thing for you to remember is these codes are for the laboratory performing the assay to report, not the physician ordering it. There is a new Appendix O for proprietary laboratory analysis MMAAs, including those that do not have a Category I code. These MMAAs are identified in Appendix O by a 4-digit number followed by the letter M.4
There are some revisions to psychotherapy codes 90832 to 90847. These codes are outside our scope of practice and should only be used by psychiatrists, social workers, psychologists, or other appropriate mental health workers.
Final Thoughts
It has not been a breakout year for telehealth and we still do not have payment for store-and-forward teledermatology, except in a few designated rural areas. With the advent of the rhetoric we have heard after the presidential election, any speculation on what will happen to the brave new world of the merit-based incentive payment system, alternative payment models, and other regulations are anyone’s guess.
- Medicare Program; Revisions to Payment Policies Under the Physician Fee Schedule and Other Revisions to Part B for CY 2017; Medicare Advantage Bid Pricing Data Release; Medicare Advantage and Part D Medical Loss Ratio Data Release; Medicare Advantage Provider Network Requirements; Expansion of Medicare Diabetes Prevention Program Model; Medicare Shared Savings Program Requirements. Fed Regist. 2016;81(220):80170-80562. To be codified at 42 CFR § 405, 410, 411, 414, 417, 422, 423, 424, 425, and 460.
- Siegel DM. The Proposed Rule and payments for 2017: the good, the bad, and the ugly. Cutis. 2016;98:245-248.
- Data collection on resources used in furnishing global services town hall CY 2017 Medicare physician fee schedule Proposed Rule. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/Downloads/CY2017-PFS-FR-Townhall.pdf. Published August 25, 2016. Accessed January 4, 2017.
- Current Procedural Terminology 2017, Professional Edition. Chicago, IL: American Medical Association; 2016.
- Addendum B—relative value units and related information used in CY 2017 final rule. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/Downloads/CY2017-PFS-FR-Addenda.zip. Accessed January 23, 2017.
- Medicare Program; Revisions to Payment Policies Under the Physician Fee Schedule and Other Revisions to Part B for CY 2017; Medicare Advantage Bid Pricing Data Release; Medicare Advantage and Part D Medical Loss Ratio Data Release; Medicare Advantage Provider Network Requirements; Expansion of Medicare Diabetes Prevention Program Model; Medicare Shared Savings Program Requirements. Fed Regist. 2016;81(220):80170-80562. To be codified at 42 CFR § 405, 410, 411, 414, 417, 422, 423, 424, 425, and 460.
- Siegel DM. The Proposed Rule and payments for 2017: the good, the bad, and the ugly. Cutis. 2016;98:245-248.
- Data collection on resources used in furnishing global services town hall CY 2017 Medicare physician fee schedule Proposed Rule. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/Downloads/CY2017-PFS-FR-Townhall.pdf. Published August 25, 2016. Accessed January 4, 2017.
- Current Procedural Terminology 2017, Professional Edition. Chicago, IL: American Medical Association; 2016.
- Addendum B—relative value units and related information used in CY 2017 final rule. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/Downloads/CY2017-PFS-FR-Addenda.zip. Accessed January 23, 2017.
Practice Points
- The conversion factor has increased more than 0.2%, which means you will be paid a bit more this year.
- Review Current Procedural Terminology codes carefully for pain control or moderate sedation as well as nail surgery and slide consultation.
- Reflectance confocal microscopy now has relative value units assigned by the Centers for Medicare & Medicaid Services.

Localized Pemphigus Foliaceus
To the Editor:
Pemphigus foliaceus is a rare autoimmune blistering disorder that typically presents with crusted scaly erosions in a seborrheic distribution. We describe a case of pemphigus foliaceus localized to the right cheek of 10 years’ duration that spread to other areas. With a PubMed search of articles indexed for MEDLINE yielding only 14 cases of localized pemphigus foliaceus (Table), it represents an extremely rare entity that often is a diagnostic challenge and may be a harbinger for disseminated disease months to years after the inciting lesion appears.
A 51-year-old woman presented with an asymptomatic cutaneous eruption that had remained localized to the right cheek for 10 years before it increased in size and new lesions developed on the left cheek, chest, and upper back. No inciting factors, such as contactants, insect bites, infections, medications, or recent travel were identified. On physical examination a well-demarcated, hypertrophic, verrucouslike plaque with central pink atrophy and exfoliative scale involved the right malar and submalar regions but spared the mucocutaneous junctions of the face (Figure 1). Subtle dark brown papules, some with overlying scale, speckled the left cheek, right jawline, chest, and upper back. The oral cavity was clear.
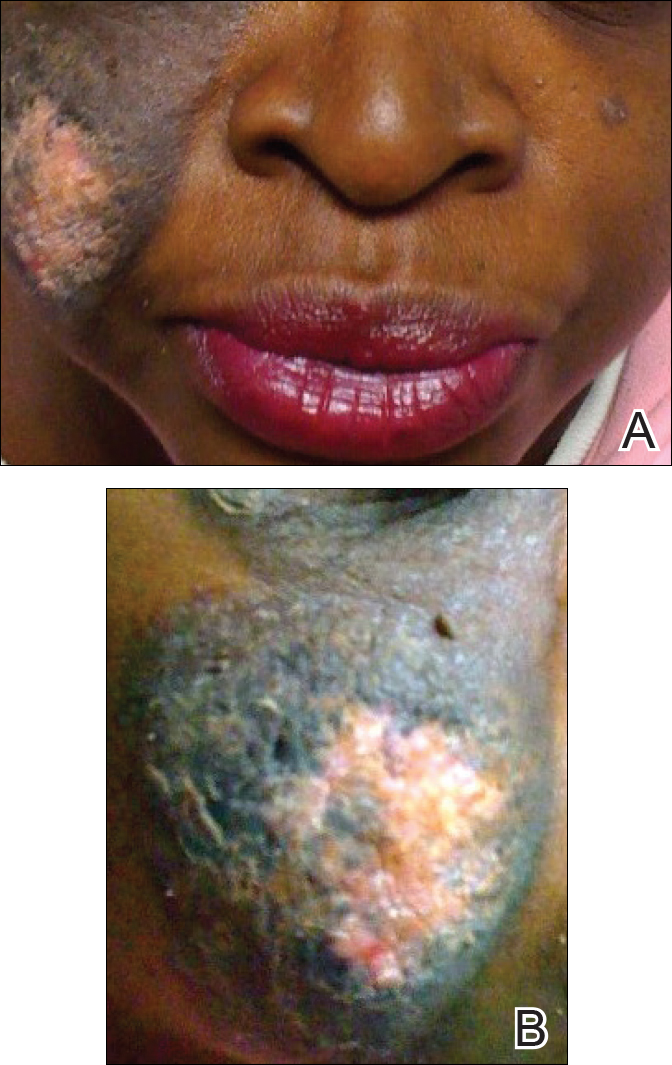
Leading differentials included hypertrophic discoid lupus erythematosus and pemphigus vegetans. Other considerations included sarcoidosis, granuloma faciale, lupus vulgaris, disseminated coccidioidomycosis or blastomycosis, and squamous cell carcinoma.
An initial biopsy revealed a lymphocytic lichenoid dermatitis with epidermal hyperplasia and scattered eosinophils for which the following differentials were provided: insect bite, hypertrophic lichen planus, prurigo nodularis superimposed on rosacea, and allergic contact dermatitis. Under these histologic diagnoses, tacrolimus ointment 0.03%, topical mid-potency corticosteroid, and a combination of oral doxycycline and metronidazole gel 1% were prescribed but failed to ameliorate her condition.
Because the clinical differentials were vast and noncorrelative with the original pathology, additional biopsies were performed: one from the edge of the large malar plaque, which was transected for hematoxylin and eosin (H&E) and tissue cultures; one perilesional to the large malar plaque for direct immunofluorescence (DIF); and one from the papule on the right jawline for H&E. Tissue cultures were negative for fungal and mycobacterial organisms. Both specimens submitted for H&E showed the prominent epidermal hyperplasia and lymphocytic dermal infiltrate noted on the original H&E but also demonstrated intragranular acantholysis (Figure 2). The DIF revealed intercellular IgG and C3 deposition throughout the epidermis (Figure 3). Indirect immunofluorescence was negative, but enzyme-linked immunosorbent assay detected circulating antidesmoglein-1 but not antidesmoglein-3 autoantibodies. Other serologies including antinuclear antibody, anti–double-stranded DNA, antihistone, anti–Sjögren syndrome A, and anti–Sjögren syndrome B antibodies were negative.
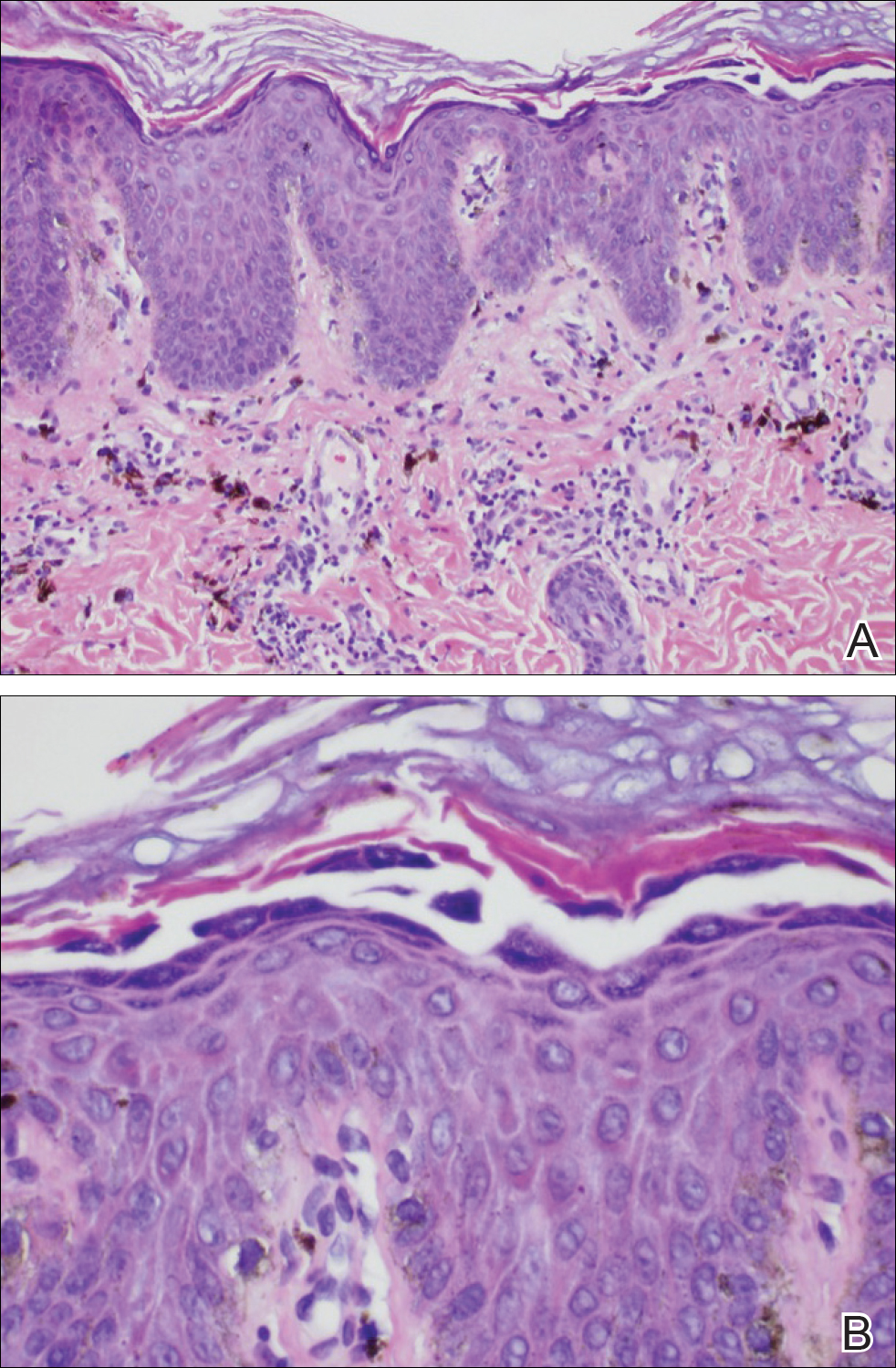

The diagnosis of localized pemphigus foliaceus was made and management with oral prednisone and mycophenolate mofetil resulted in improvement within weeks.
Localized pemphigus foliaceus is extremely rare with only 14 cases reported in the literature (Table).1-10 Its diagnosis is challenging, as the clinical presentation simulates various entities and the histological features and serological markers are difficult to capture.
Localized pemphigus foliaceus typically presents as an isolated, erythematous, scaly, crusted plaque involving the nose, cheek, or scalp and may mimic several conditions including contact dermatitis, seborrheic dermatitis, rosacea, cutaneous sarcoidosis, discoid lupus erythematosus, lupus vulgaris, impetigo contagiosa, solar keratosis, and nonmelanoma skin cancer.1-10
The predilection for sun-exposed areas suggests UV radiation may induce binding of antidesmoglein-1 autoantibodies with subsequent cytokine-mediated inflammation and acantholysis at these sites.11-13 Similarly, the immunomodulatory agent imiquimod has been reported to induce pemphigus foliaceus at its application sites.6
When pemphigus foliaceus is clinically discernible, the histology and DIF are in accordance with the clinical diagnosis 53.8% of the time.13 In cases of localized pemphigus foliaceus in which the diagnosis is more elusive, many biopsies often are needed to capture the characteristic intragranular acantholysis; this feature often is so subtle that unless the diagnosis is suspected, it is underappreciated or undetectable. In chronic lesions, it may be masked by secondary changes such as acanthosis, hyperkeratosis, and parakeratosis.14
In pemphigus foliaceus, detection of circulating antidesmoglein-1 autoantibodies by enzyme-linked immunosorbent assay is slightly more sensitive and specific compared to indirect immunofluorescence, but both correlate with disease activity.15,16 The low or absent autoantibody titers in localized pemphigus foliaceus may reflect its limited involvement, but dissemination of the disease with subsequent elevation of autoantibody titers may occur months to years after initial presentation,1,2,9 as was the case with our patient.
The majority of localized pemphigus foliaceus cases require systemic prednisone, sometimes in conjunction with nonsteroidal immunosuppressants or topical high-potency corticosteroids.1-3,5,6,8-10 One case was efficaciously managed with tacrolimus ointment 0.1%.7
Localized pemphigus foliaceus is a rare and challenging entity that must be a diagnostic consideration for any chronic focal plaque on the face or scalp, as it may herald disseminated disease.
- Paramsothy Y, Lawrence CM. “Tin-tack” sign in localized pemphigus foliaceus. Br J Dermatol. 1987;116:127-129.
- Newton JA, McGibbon DH, Monk B, et al. Pemphigus foliaceus localized to the nose. Br J Dermatol. 1988;118:303-312.
- Koide M, Kokura N, Takano N. Pemphigus foliaceus localized on the face [in Japanese]. Jpn J Dermatol. 1989;97:1262.
- Yamamoto S, Kanekura T, Gushi A, et al. A case of localized pemphigus foliaceus. J Dermatol. 1996;23:893-895.
- Kishibe M, Kinouchi M, Ishida-Yamamoto A, et al. Pemphigus foliaceus localized to the nose. Clin Exp Dermatol. 2003;28:560-562.
- Lin R, Ladd DJ, Powell DJ, et al. Localized pemphigus foliaceus induced by topical imiquimod treatment. Arch Dermatol. 2004;140:889-890.
- Termeer CC, Technau K, Augustin M, et al. Topical tacrolimus (Protopic) for the treatment of a localized pemphigus foliaceus. J Eur Acad Dermatol Venereol. 2004;18:636-637.
- Zaraa I, El Euch D, Kort R, et al. Localized pemphigus: a report of three cases. Int J Dermatol 2010;49:715-716.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- Maderal AD, Miner A, Nousari C, et al. Localized pemphigus foliaceus with unilateral facial involvement. Actas Dermosifiliogr. 2014;105:413-417.
- Cram DL, Winkelmann RK. Ultraviolet-induced acantholysis in pemphigus. Arch Dermatol. 1965;92:7-13.
- Kano Y, Shimosegawa M, Mizukawa Y, et al. Pemphigus foliaceus induced by exposure to sunlight. Dermatology. 2000;201:132-138.
- Lebe B, Gül Nıflıoğlu G, Seyrek S, et al. Evaluation of clinical and histopathologic/direct immunofluorescence diagnosis in autoimmune vesiculobullous dermatitis: utility of direct immunofluorescence. Turk Patoloji Derg. 2012;28:11-16.
- Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29:432-436.
- Ishii K, Amagai M, Hall RP, et al. Characterization of autoantibodies in pemphigus using antigen specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010-2017.
- Ng PP, Thng ST, Mohamed K, et al. Comparison of desmoglein ELISA and indirect immunofluorescence using two substrates (monkey esophagus and normal human skin) in the diagnosis of pemphigus. Australas J Dermatol. 2005;46:239-241.
To the Editor:
Pemphigus foliaceus is a rare autoimmune blistering disorder that typically presents with crusted scaly erosions in a seborrheic distribution. We describe a case of pemphigus foliaceus localized to the right cheek of 10 years’ duration that spread to other areas. With a PubMed search of articles indexed for MEDLINE yielding only 14 cases of localized pemphigus foliaceus (Table), it represents an extremely rare entity that often is a diagnostic challenge and may be a harbinger for disseminated disease months to years after the inciting lesion appears.
A 51-year-old woman presented with an asymptomatic cutaneous eruption that had remained localized to the right cheek for 10 years before it increased in size and new lesions developed on the left cheek, chest, and upper back. No inciting factors, such as contactants, insect bites, infections, medications, or recent travel were identified. On physical examination a well-demarcated, hypertrophic, verrucouslike plaque with central pink atrophy and exfoliative scale involved the right malar and submalar regions but spared the mucocutaneous junctions of the face (Figure 1). Subtle dark brown papules, some with overlying scale, speckled the left cheek, right jawline, chest, and upper back. The oral cavity was clear.
Leading differentials included hypertrophic discoid lupus erythematosus and pemphigus vegetans. Other considerations included sarcoidosis, granuloma faciale, lupus vulgaris, disseminated coccidioidomycosis or blastomycosis, and squamous cell carcinoma.
An initial biopsy revealed a lymphocytic lichenoid dermatitis with epidermal hyperplasia and scattered eosinophils for which the following differentials were provided: insect bite, hypertrophic lichen planus, prurigo nodularis superimposed on rosacea, and allergic contact dermatitis. Under these histologic diagnoses, tacrolimus ointment 0.03%, topical mid-potency corticosteroid, and a combination of oral doxycycline and metronidazole gel 1% were prescribed but failed to ameliorate her condition.
Because the clinical differentials were vast and noncorrelative with the original pathology, additional biopsies were performed: one from the edge of the large malar plaque, which was transected for hematoxylin and eosin (H&E) and tissue cultures; one perilesional to the large malar plaque for direct immunofluorescence (DIF); and one from the papule on the right jawline for H&E. Tissue cultures were negative for fungal and mycobacterial organisms. Both specimens submitted for H&E showed the prominent epidermal hyperplasia and lymphocytic dermal infiltrate noted on the original H&E but also demonstrated intragranular acantholysis (Figure 2). The DIF revealed intercellular IgG and C3 deposition throughout the epidermis (Figure 3). Indirect immunofluorescence was negative, but enzyme-linked immunosorbent assay detected circulating antidesmoglein-1 but not antidesmoglein-3 autoantibodies. Other serologies including antinuclear antibody, anti–double-stranded DNA, antihistone, anti–Sjögren syndrome A, and anti–Sjögren syndrome B antibodies were negative.
The diagnosis of localized pemphigus foliaceus was made and management with oral prednisone and mycophenolate mofetil resulted in improvement within weeks.
Localized pemphigus foliaceus is extremely rare with only 14 cases reported in the literature (Table).1-10 Its diagnosis is challenging, as the clinical presentation simulates various entities and the histological features and serological markers are difficult to capture.
Localized pemphigus foliaceus typically presents as an isolated, erythematous, scaly, crusted plaque involving the nose, cheek, or scalp and may mimic several conditions including contact dermatitis, seborrheic dermatitis, rosacea, cutaneous sarcoidosis, discoid lupus erythematosus, lupus vulgaris, impetigo contagiosa, solar keratosis, and nonmelanoma skin cancer.1-10
The predilection for sun-exposed areas suggests UV radiation may induce binding of antidesmoglein-1 autoantibodies with subsequent cytokine-mediated inflammation and acantholysis at these sites.11-13 Similarly, the immunomodulatory agent imiquimod has been reported to induce pemphigus foliaceus at its application sites.6
When pemphigus foliaceus is clinically discernible, the histology and DIF are in accordance with the clinical diagnosis 53.8% of the time.13 In cases of localized pemphigus foliaceus in which the diagnosis is more elusive, many biopsies often are needed to capture the characteristic intragranular acantholysis; this feature often is so subtle that unless the diagnosis is suspected, it is underappreciated or undetectable. In chronic lesions, it may be masked by secondary changes such as acanthosis, hyperkeratosis, and parakeratosis.14
In pemphigus foliaceus, detection of circulating antidesmoglein-1 autoantibodies by enzyme-linked immunosorbent assay is slightly more sensitive and specific compared to indirect immunofluorescence, but both correlate with disease activity.15,16 The low or absent autoantibody titers in localized pemphigus foliaceus may reflect its limited involvement, but dissemination of the disease with subsequent elevation of autoantibody titers may occur months to years after initial presentation,1,2,9 as was the case with our patient.
The majority of localized pemphigus foliaceus cases require systemic prednisone, sometimes in conjunction with nonsteroidal immunosuppressants or topical high-potency corticosteroids.1-3,5,6,8-10 One case was efficaciously managed with tacrolimus ointment 0.1%.7
Localized pemphigus foliaceus is a rare and challenging entity that must be a diagnostic consideration for any chronic focal plaque on the face or scalp, as it may herald disseminated disease.
To the Editor:
Pemphigus foliaceus is a rare autoimmune blistering disorder that typically presents with crusted scaly erosions in a seborrheic distribution. We describe a case of pemphigus foliaceus localized to the right cheek of 10 years’ duration that spread to other areas. With a PubMed search of articles indexed for MEDLINE yielding only 14 cases of localized pemphigus foliaceus (Table), it represents an extremely rare entity that often is a diagnostic challenge and may be a harbinger for disseminated disease months to years after the inciting lesion appears.
A 51-year-old woman presented with an asymptomatic cutaneous eruption that had remained localized to the right cheek for 10 years before it increased in size and new lesions developed on the left cheek, chest, and upper back. No inciting factors, such as contactants, insect bites, infections, medications, or recent travel were identified. On physical examination a well-demarcated, hypertrophic, verrucouslike plaque with central pink atrophy and exfoliative scale involved the right malar and submalar regions but spared the mucocutaneous junctions of the face (Figure 1). Subtle dark brown papules, some with overlying scale, speckled the left cheek, right jawline, chest, and upper back. The oral cavity was clear.
Leading differentials included hypertrophic discoid lupus erythematosus and pemphigus vegetans. Other considerations included sarcoidosis, granuloma faciale, lupus vulgaris, disseminated coccidioidomycosis or blastomycosis, and squamous cell carcinoma.
An initial biopsy revealed a lymphocytic lichenoid dermatitis with epidermal hyperplasia and scattered eosinophils for which the following differentials were provided: insect bite, hypertrophic lichen planus, prurigo nodularis superimposed on rosacea, and allergic contact dermatitis. Under these histologic diagnoses, tacrolimus ointment 0.03%, topical mid-potency corticosteroid, and a combination of oral doxycycline and metronidazole gel 1% were prescribed but failed to ameliorate her condition.
Because the clinical differentials were vast and noncorrelative with the original pathology, additional biopsies were performed: one from the edge of the large malar plaque, which was transected for hematoxylin and eosin (H&E) and tissue cultures; one perilesional to the large malar plaque for direct immunofluorescence (DIF); and one from the papule on the right jawline for H&E. Tissue cultures were negative for fungal and mycobacterial organisms. Both specimens submitted for H&E showed the prominent epidermal hyperplasia and lymphocytic dermal infiltrate noted on the original H&E but also demonstrated intragranular acantholysis (Figure 2). The DIF revealed intercellular IgG and C3 deposition throughout the epidermis (Figure 3). Indirect immunofluorescence was negative, but enzyme-linked immunosorbent assay detected circulating antidesmoglein-1 but not antidesmoglein-3 autoantibodies. Other serologies including antinuclear antibody, anti–double-stranded DNA, antihistone, anti–Sjögren syndrome A, and anti–Sjögren syndrome B antibodies were negative.
The diagnosis of localized pemphigus foliaceus was made and management with oral prednisone and mycophenolate mofetil resulted in improvement within weeks.
Localized pemphigus foliaceus is extremely rare with only 14 cases reported in the literature (Table).1-10 Its diagnosis is challenging, as the clinical presentation simulates various entities and the histological features and serological markers are difficult to capture.
Localized pemphigus foliaceus typically presents as an isolated, erythematous, scaly, crusted plaque involving the nose, cheek, or scalp and may mimic several conditions including contact dermatitis, seborrheic dermatitis, rosacea, cutaneous sarcoidosis, discoid lupus erythematosus, lupus vulgaris, impetigo contagiosa, solar keratosis, and nonmelanoma skin cancer.1-10
The predilection for sun-exposed areas suggests UV radiation may induce binding of antidesmoglein-1 autoantibodies with subsequent cytokine-mediated inflammation and acantholysis at these sites.11-13 Similarly, the immunomodulatory agent imiquimod has been reported to induce pemphigus foliaceus at its application sites.6
When pemphigus foliaceus is clinically discernible, the histology and DIF are in accordance with the clinical diagnosis 53.8% of the time.13 In cases of localized pemphigus foliaceus in which the diagnosis is more elusive, many biopsies often are needed to capture the characteristic intragranular acantholysis; this feature often is so subtle that unless the diagnosis is suspected, it is underappreciated or undetectable. In chronic lesions, it may be masked by secondary changes such as acanthosis, hyperkeratosis, and parakeratosis.14
In pemphigus foliaceus, detection of circulating antidesmoglein-1 autoantibodies by enzyme-linked immunosorbent assay is slightly more sensitive and specific compared to indirect immunofluorescence, but both correlate with disease activity.15,16 The low or absent autoantibody titers in localized pemphigus foliaceus may reflect its limited involvement, but dissemination of the disease with subsequent elevation of autoantibody titers may occur months to years after initial presentation,1,2,9 as was the case with our patient.
The majority of localized pemphigus foliaceus cases require systemic prednisone, sometimes in conjunction with nonsteroidal immunosuppressants or topical high-potency corticosteroids.1-3,5,6,8-10 One case was efficaciously managed with tacrolimus ointment 0.1%.7
Localized pemphigus foliaceus is a rare and challenging entity that must be a diagnostic consideration for any chronic focal plaque on the face or scalp, as it may herald disseminated disease.
- Paramsothy Y, Lawrence CM. “Tin-tack” sign in localized pemphigus foliaceus. Br J Dermatol. 1987;116:127-129.
- Newton JA, McGibbon DH, Monk B, et al. Pemphigus foliaceus localized to the nose. Br J Dermatol. 1988;118:303-312.
- Koide M, Kokura N, Takano N. Pemphigus foliaceus localized on the face [in Japanese]. Jpn J Dermatol. 1989;97:1262.
- Yamamoto S, Kanekura T, Gushi A, et al. A case of localized pemphigus foliaceus. J Dermatol. 1996;23:893-895.
- Kishibe M, Kinouchi M, Ishida-Yamamoto A, et al. Pemphigus foliaceus localized to the nose. Clin Exp Dermatol. 2003;28:560-562.
- Lin R, Ladd DJ, Powell DJ, et al. Localized pemphigus foliaceus induced by topical imiquimod treatment. Arch Dermatol. 2004;140:889-890.
- Termeer CC, Technau K, Augustin M, et al. Topical tacrolimus (Protopic) for the treatment of a localized pemphigus foliaceus. J Eur Acad Dermatol Venereol. 2004;18:636-637.
- Zaraa I, El Euch D, Kort R, et al. Localized pemphigus: a report of three cases. Int J Dermatol 2010;49:715-716.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- Maderal AD, Miner A, Nousari C, et al. Localized pemphigus foliaceus with unilateral facial involvement. Actas Dermosifiliogr. 2014;105:413-417.
- Cram DL, Winkelmann RK. Ultraviolet-induced acantholysis in pemphigus. Arch Dermatol. 1965;92:7-13.
- Kano Y, Shimosegawa M, Mizukawa Y, et al. Pemphigus foliaceus induced by exposure to sunlight. Dermatology. 2000;201:132-138.
- Lebe B, Gül Nıflıoğlu G, Seyrek S, et al. Evaluation of clinical and histopathologic/direct immunofluorescence diagnosis in autoimmune vesiculobullous dermatitis: utility of direct immunofluorescence. Turk Patoloji Derg. 2012;28:11-16.
- Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29:432-436.
- Ishii K, Amagai M, Hall RP, et al. Characterization of autoantibodies in pemphigus using antigen specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010-2017.
- Ng PP, Thng ST, Mohamed K, et al. Comparison of desmoglein ELISA and indirect immunofluorescence using two substrates (monkey esophagus and normal human skin) in the diagnosis of pemphigus. Australas J Dermatol. 2005;46:239-241.
- Paramsothy Y, Lawrence CM. “Tin-tack” sign in localized pemphigus foliaceus. Br J Dermatol. 1987;116:127-129.
- Newton JA, McGibbon DH, Monk B, et al. Pemphigus foliaceus localized to the nose. Br J Dermatol. 1988;118:303-312.
- Koide M, Kokura N, Takano N. Pemphigus foliaceus localized on the face [in Japanese]. Jpn J Dermatol. 1989;97:1262.
- Yamamoto S, Kanekura T, Gushi A, et al. A case of localized pemphigus foliaceus. J Dermatol. 1996;23:893-895.
- Kishibe M, Kinouchi M, Ishida-Yamamoto A, et al. Pemphigus foliaceus localized to the nose. Clin Exp Dermatol. 2003;28:560-562.
- Lin R, Ladd DJ, Powell DJ, et al. Localized pemphigus foliaceus induced by topical imiquimod treatment. Arch Dermatol. 2004;140:889-890.
- Termeer CC, Technau K, Augustin M, et al. Topical tacrolimus (Protopic) for the treatment of a localized pemphigus foliaceus. J Eur Acad Dermatol Venereol. 2004;18:636-637.
- Zaraa I, El Euch D, Kort R, et al. Localized pemphigus: a report of three cases. Int J Dermatol 2010;49:715-716.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- Maderal AD, Miner A, Nousari C, et al. Localized pemphigus foliaceus with unilateral facial involvement. Actas Dermosifiliogr. 2014;105:413-417.
- Cram DL, Winkelmann RK. Ultraviolet-induced acantholysis in pemphigus. Arch Dermatol. 1965;92:7-13.
- Kano Y, Shimosegawa M, Mizukawa Y, et al. Pemphigus foliaceus induced by exposure to sunlight. Dermatology. 2000;201:132-138.
- Lebe B, Gül Nıflıoğlu G, Seyrek S, et al. Evaluation of clinical and histopathologic/direct immunofluorescence diagnosis in autoimmune vesiculobullous dermatitis: utility of direct immunofluorescence. Turk Patoloji Derg. 2012;28:11-16.
- Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29:432-436.
- Ishii K, Amagai M, Hall RP, et al. Characterization of autoantibodies in pemphigus using antigen specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010-2017.
- Ng PP, Thng ST, Mohamed K, et al. Comparison of desmoglein ELISA and indirect immunofluorescence using two substrates (monkey esophagus and normal human skin) in the diagnosis of pemphigus. Australas J Dermatol. 2005;46:239-241.
Practice Points
- The diagnosis of pemphigus foliceus is challenging, as the clinical presentation simulates various entities.
- Clinicopathological correlation is important. If pathology and other diagnostics do not support clinical findings, trust your clinical assessment and consider repeating or adjusting the workup.
Red-Blue Nodule on the Scalp
Metastatic Clear Cell Renal Cell Carcinoma
The differential diagnosis of cutaneous neoplasms with clear cells is broad. Clear cell features can be seen in primary tumors arising from the epidermis and cutaneous adnexa as well as in mesenchymal and melanocytic neoplasms. Furthermore, metastatic disease should be considered in the histologic differential diagnosis, as many visceral malignancies have clear cell features. This patient was subsequently found to have a large renal mass with metastasis to the lungs, spleen, and bone. The histologic findings support the diagnosis of metastatic clear cell renal cell carcinoma (RCC) to the skin.
Approximately 30% of patients with clear cell RCC present with metastatic disease with approximately 8% of those involving the skin.1,2 Cutaneous RCC metastases show a predilection for the head, especially the scalp. The clinical presentation is variable, but there often is a history of a rapidly growing brown, black, or purple nodule or plaque. A thorough review of the patient's history should be conducted if metastatic RCC is in the differential diagnosis, as it has been reported to occur up to 20 years after initial diagnosis.3
Histologically, clear cell RCC (quiz image) is composed of nests of tumor cells with clear cytoplasm and centrally located nuclei with prominent nucleoli. The clear cell features result from abundant cytoplasmic glycogen and lipid but may not be present in every case. One of the most important histologic features is the presence of delicate branching blood vessels (Figure 1). Numerous extravasated red blood cells also may be present. Positive immunohistochemical staining for PAX8, CD10, and RCC antigens support the diagnosis.4
Balloon cell nevi (Figure 2) most commonly occur on the head and neck in adolescents and young adults but clinically are indistinguishable from other banal nevi. The nevus cells are large with foamy to finely vacuolated cytoplasm and lack atypia. The clear cell change is the result of melanosome degeneration and may be extensive. The presence of melanin pigment, nests of typical nevus cells, and positive staining with MART-1 can help distinguish the tumor from xanthomas and RCC.5
Clear cell hidradenoma (Figure 3) is a well-circumscribed tumor of sweat gland origin that arises in the dermis. The architecture usually is solid, cystic, or a combination of both. The cytology is classically bland with poroid, squamoid, or clear cell morphology. Clear cells that are positive on periodic acid-Schiff staining predominate in up to one-third of cases. Carcinoembryonic antigen and epithelial membrane antigen can be used to highlight the eosinophilic cuticles of ducts within solid areas.6

Sebaceous carcinoma (Figure 4) most frequently arises in a periorbital distribution, although extraocular lesions are known to occur. Histologically, there is a proliferation of both mature sebocytes and basaloid cells in the dermis, occasionally involving the epidermis. The mature sebocytes demonstrate clear cell features with foamy to vacuolated cytoplasm and large nuclei with scalloped borders. The clear cells may vary greatly in number and often are sparse in poorly differentiated tumors in which pleomorphic basaloid cells may predominate. The basaloid cells may resemble those of squamous or basal cell carcinoma, leading to a diagnostic dilemma in some cases. Special staining with Sudan black B and oil red O highlights the cytoplasmic lipid but must be performed on frozen section specimens. Although not entirely specific, immunohistochemical expression of epithelial membrane antigen, androgen receptor, and membranous vesicular adipophilin staining in sebaceous carcinoma can assist in the diagnosis.7
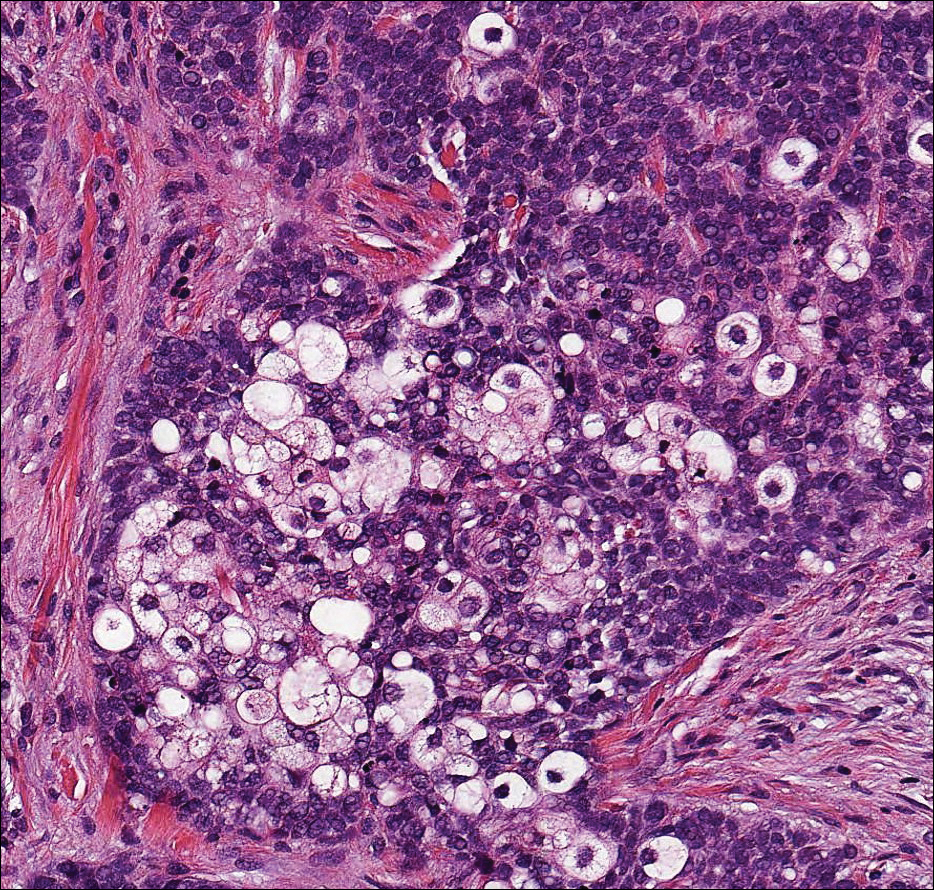
Cutaneous xanthomas (Figure 5) may arise in patients of any age and represent deposition of lipid-laden macrophages. Classification often is dependent on the clinical presentation; however, some subtypes demonstrate unique morphologic features (eg, verruciform xanthomas). Xanthomas classically arise in association with elevated serum lipids, but they also may occur in normolipemic patients. Individuals with Erdheim-Chester disease have an increased propensity to develop xanthelasma. Similarly, plane xanthomas have been associated with monoclonal gammopathy. Histologically, xanthomas are characterized by sheets of foamy macrophages within the dermis and subcutis. Positive immunohistochemical staining for CD68 highlighting the histiocytic nature of the cells and the absence of a delicate vascular network aid in the differentiation from RCC.
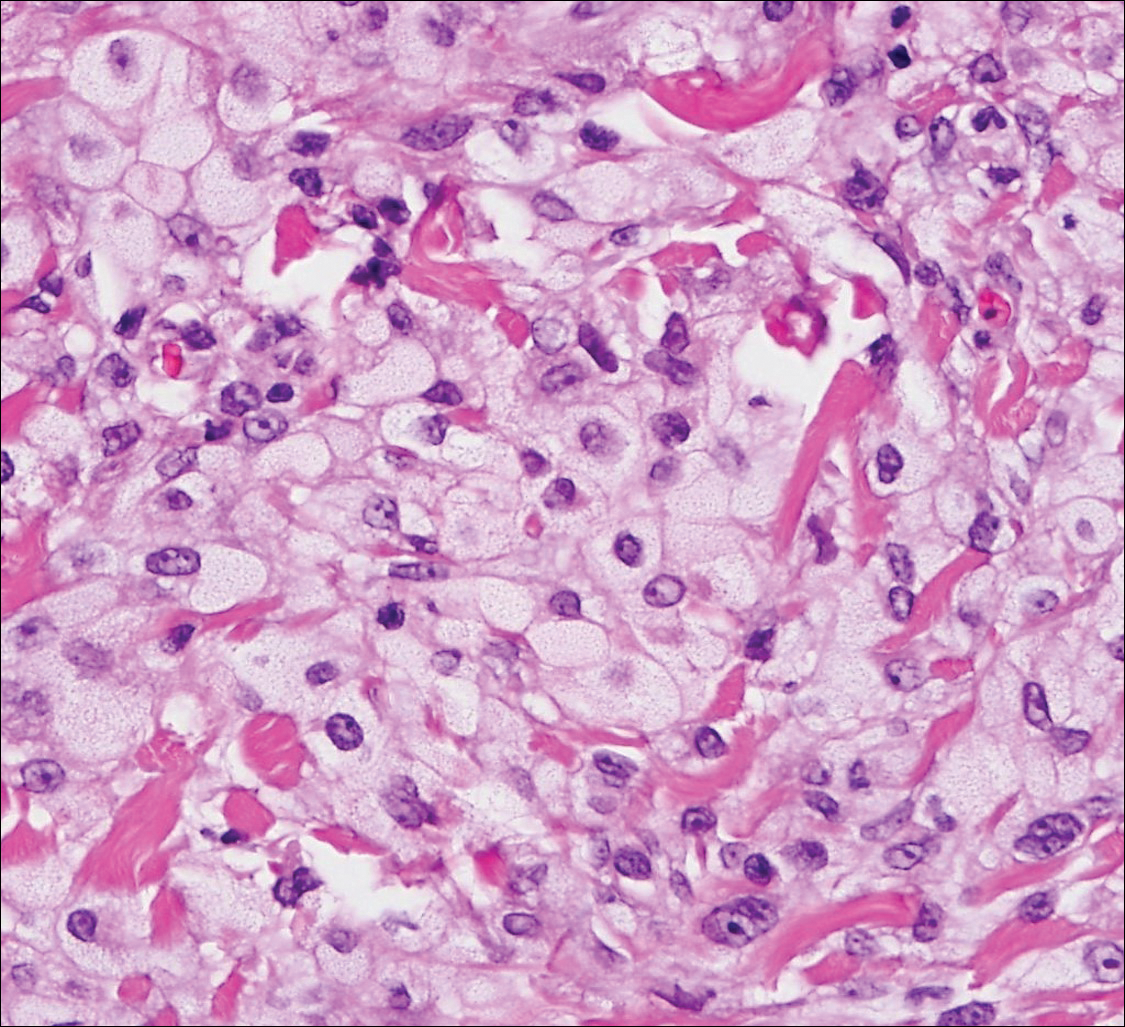
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Lin F, Prichard J. Handbook of Practical Immunohistochemistry: Frequently Asked Questions. 2nd ed. New York, NY: Springer; 2015.
- McKee PH, Calonje E. Diagnostic Atlas of Melanocytic Pathology. Edinburgh, Scotland: Mosby/Elsevier; 2009.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Ansai S, Takeichi H, Arase S, et al. Sebaceous carcinoma: an immunohistochemical reappraisal. Am J Dermatopathol. 2011;33:579-587.
Metastatic Clear Cell Renal Cell Carcinoma
The differential diagnosis of cutaneous neoplasms with clear cells is broad. Clear cell features can be seen in primary tumors arising from the epidermis and cutaneous adnexa as well as in mesenchymal and melanocytic neoplasms. Furthermore, metastatic disease should be considered in the histologic differential diagnosis, as many visceral malignancies have clear cell features. This patient was subsequently found to have a large renal mass with metastasis to the lungs, spleen, and bone. The histologic findings support the diagnosis of metastatic clear cell renal cell carcinoma (RCC) to the skin.
Approximately 30% of patients with clear cell RCC present with metastatic disease with approximately 8% of those involving the skin.1,2 Cutaneous RCC metastases show a predilection for the head, especially the scalp. The clinical presentation is variable, but there often is a history of a rapidly growing brown, black, or purple nodule or plaque. A thorough review of the patient's history should be conducted if metastatic RCC is in the differential diagnosis, as it has been reported to occur up to 20 years after initial diagnosis.3
Histologically, clear cell RCC (quiz image) is composed of nests of tumor cells with clear cytoplasm and centrally located nuclei with prominent nucleoli. The clear cell features result from abundant cytoplasmic glycogen and lipid but may not be present in every case. One of the most important histologic features is the presence of delicate branching blood vessels (Figure 1). Numerous extravasated red blood cells also may be present. Positive immunohistochemical staining for PAX8, CD10, and RCC antigens support the diagnosis.4
Balloon cell nevi (Figure 2) most commonly occur on the head and neck in adolescents and young adults but clinically are indistinguishable from other banal nevi. The nevus cells are large with foamy to finely vacuolated cytoplasm and lack atypia. The clear cell change is the result of melanosome degeneration and may be extensive. The presence of melanin pigment, nests of typical nevus cells, and positive staining with MART-1 can help distinguish the tumor from xanthomas and RCC.5
Clear cell hidradenoma (Figure 3) is a well-circumscribed tumor of sweat gland origin that arises in the dermis. The architecture usually is solid, cystic, or a combination of both. The cytology is classically bland with poroid, squamoid, or clear cell morphology. Clear cells that are positive on periodic acid-Schiff staining predominate in up to one-third of cases. Carcinoembryonic antigen and epithelial membrane antigen can be used to highlight the eosinophilic cuticles of ducts within solid areas.6
Sebaceous carcinoma (Figure 4) most frequently arises in a periorbital distribution, although extraocular lesions are known to occur. Histologically, there is a proliferation of both mature sebocytes and basaloid cells in the dermis, occasionally involving the epidermis. The mature sebocytes demonstrate clear cell features with foamy to vacuolated cytoplasm and large nuclei with scalloped borders. The clear cells may vary greatly in number and often are sparse in poorly differentiated tumors in which pleomorphic basaloid cells may predominate. The basaloid cells may resemble those of squamous or basal cell carcinoma, leading to a diagnostic dilemma in some cases. Special staining with Sudan black B and oil red O highlights the cytoplasmic lipid but must be performed on frozen section specimens. Although not entirely specific, immunohistochemical expression of epithelial membrane antigen, androgen receptor, and membranous vesicular adipophilin staining in sebaceous carcinoma can assist in the diagnosis.7
Cutaneous xanthomas (Figure 5) may arise in patients of any age and represent deposition of lipid-laden macrophages. Classification often is dependent on the clinical presentation; however, some subtypes demonstrate unique morphologic features (eg, verruciform xanthomas). Xanthomas classically arise in association with elevated serum lipids, but they also may occur in normolipemic patients. Individuals with Erdheim-Chester disease have an increased propensity to develop xanthelasma. Similarly, plane xanthomas have been associated with monoclonal gammopathy. Histologically, xanthomas are characterized by sheets of foamy macrophages within the dermis and subcutis. Positive immunohistochemical staining for CD68 highlighting the histiocytic nature of the cells and the absence of a delicate vascular network aid in the differentiation from RCC.
Metastatic Clear Cell Renal Cell Carcinoma
The differential diagnosis of cutaneous neoplasms with clear cells is broad. Clear cell features can be seen in primary tumors arising from the epidermis and cutaneous adnexa as well as in mesenchymal and melanocytic neoplasms. Furthermore, metastatic disease should be considered in the histologic differential diagnosis, as many visceral malignancies have clear cell features. This patient was subsequently found to have a large renal mass with metastasis to the lungs, spleen, and bone. The histologic findings support the diagnosis of metastatic clear cell renal cell carcinoma (RCC) to the skin.
Approximately 30% of patients with clear cell RCC present with metastatic disease with approximately 8% of those involving the skin.1,2 Cutaneous RCC metastases show a predilection for the head, especially the scalp. The clinical presentation is variable, but there often is a history of a rapidly growing brown, black, or purple nodule or plaque. A thorough review of the patient's history should be conducted if metastatic RCC is in the differential diagnosis, as it has been reported to occur up to 20 years after initial diagnosis.3
Histologically, clear cell RCC (quiz image) is composed of nests of tumor cells with clear cytoplasm and centrally located nuclei with prominent nucleoli. The clear cell features result from abundant cytoplasmic glycogen and lipid but may not be present in every case. One of the most important histologic features is the presence of delicate branching blood vessels (Figure 1). Numerous extravasated red blood cells also may be present. Positive immunohistochemical staining for PAX8, CD10, and RCC antigens support the diagnosis.4
Balloon cell nevi (Figure 2) most commonly occur on the head and neck in adolescents and young adults but clinically are indistinguishable from other banal nevi. The nevus cells are large with foamy to finely vacuolated cytoplasm and lack atypia. The clear cell change is the result of melanosome degeneration and may be extensive. The presence of melanin pigment, nests of typical nevus cells, and positive staining with MART-1 can help distinguish the tumor from xanthomas and RCC.5
Clear cell hidradenoma (Figure 3) is a well-circumscribed tumor of sweat gland origin that arises in the dermis. The architecture usually is solid, cystic, or a combination of both. The cytology is classically bland with poroid, squamoid, or clear cell morphology. Clear cells that are positive on periodic acid-Schiff staining predominate in up to one-third of cases. Carcinoembryonic antigen and epithelial membrane antigen can be used to highlight the eosinophilic cuticles of ducts within solid areas.6
Sebaceous carcinoma (Figure 4) most frequently arises in a periorbital distribution, although extraocular lesions are known to occur. Histologically, there is a proliferation of both mature sebocytes and basaloid cells in the dermis, occasionally involving the epidermis. The mature sebocytes demonstrate clear cell features with foamy to vacuolated cytoplasm and large nuclei with scalloped borders. The clear cells may vary greatly in number and often are sparse in poorly differentiated tumors in which pleomorphic basaloid cells may predominate. The basaloid cells may resemble those of squamous or basal cell carcinoma, leading to a diagnostic dilemma in some cases. Special staining with Sudan black B and oil red O highlights the cytoplasmic lipid but must be performed on frozen section specimens. Although not entirely specific, immunohistochemical expression of epithelial membrane antigen, androgen receptor, and membranous vesicular adipophilin staining in sebaceous carcinoma can assist in the diagnosis.7
Cutaneous xanthomas (Figure 5) may arise in patients of any age and represent deposition of lipid-laden macrophages. Classification often is dependent on the clinical presentation; however, some subtypes demonstrate unique morphologic features (eg, verruciform xanthomas). Xanthomas classically arise in association with elevated serum lipids, but they also may occur in normolipemic patients. Individuals with Erdheim-Chester disease have an increased propensity to develop xanthelasma. Similarly, plane xanthomas have been associated with monoclonal gammopathy. Histologically, xanthomas are characterized by sheets of foamy macrophages within the dermis and subcutis. Positive immunohistochemical staining for CD68 highlighting the histiocytic nature of the cells and the absence of a delicate vascular network aid in the differentiation from RCC.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Lin F, Prichard J. Handbook of Practical Immunohistochemistry: Frequently Asked Questions. 2nd ed. New York, NY: Springer; 2015.
- McKee PH, Calonje E. Diagnostic Atlas of Melanocytic Pathology. Edinburgh, Scotland: Mosby/Elsevier; 2009.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Ansai S, Takeichi H, Arase S, et al. Sebaceous carcinoma: an immunohistochemical reappraisal. Am J Dermatopathol. 2011;33:579-587.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Lin F, Prichard J. Handbook of Practical Immunohistochemistry: Frequently Asked Questions. 2nd ed. New York, NY: Springer; 2015.
- McKee PH, Calonje E. Diagnostic Atlas of Melanocytic Pathology. Edinburgh, Scotland: Mosby/Elsevier; 2009.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Ansai S, Takeichi H, Arase S, et al. Sebaceous carcinoma: an immunohistochemical reappraisal. Am J Dermatopathol. 2011;33:579-587.
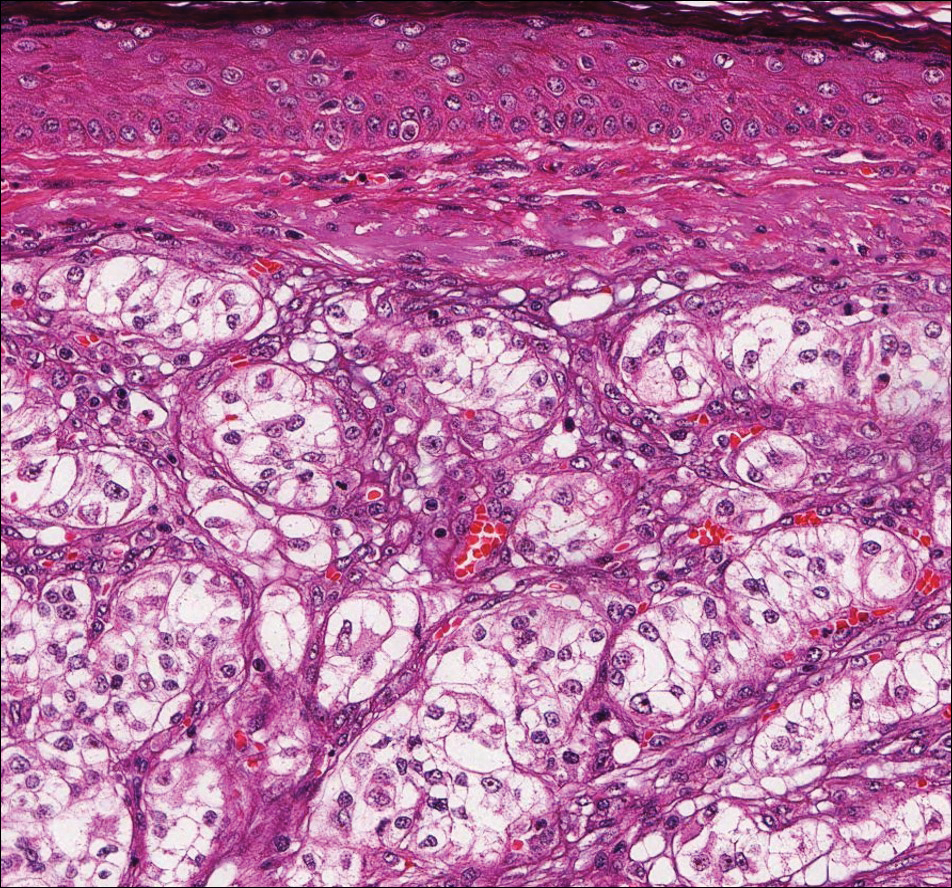
A 59-year-old man presented with a 1.5×1.0-cm asymptomatic, smooth, red-blue nodule on the left parietal scalp. The nodule had been rapidly enlarging over the last 3 weeks. After resection, the cut surface was golden yellow and focally hemorrhagic.
Clinicians Should Retain the Ability to Choose a Pathologist
As employers search for ways to reduce the cost of providing health care to their employees, there is a growing trend toward narrowed provider networks and exclusive laboratory contracts. In the case of clinical pathology, some of these choices make sense from the employer’s perspective. A complete blood cell count or comprehensive metabolic panel is done on a machine and the result is much the same regardless of the laboratory. So why not have all laboratory tests performed by the lowest bidder?
Laboratories vary in quality and anatomic pathology services are different from blood tests. Each slide must be interpreted by a physician and skill in the interpretation of skin specimens varies widely. Dermatopathology was one of the first subspecialties to be recognized within pathology, as it requires a high level of expertise. Clinicopathological correlation often is key to the accurate interpretation of a specimen. The stakes are high, and a delay in diagnosis of melanoma remains one of the most serious errors in medicine and one of the most common causes for litigation in dermatology.
The accurate interpretation of skin biopsy specimens becomes especially difficult when inadequate or misleading clinical information accompanies the specimen. A study of 589 biopsies submitted by primary care physicians and reported by general pathologists demonstrated a 6.5% error rate. False-negative errors were the most common, but false-positives also were observed.1 A study of pigmented lesions referred to the University of California, San Francisco, demonstrated a discordance rate of 14.3%.2 The degree of discordance would be expected to vary based on the range of diagnoses included in each study.
Board-certified dermatopathologists have varying areas of expertise and there is notable subjectivity in the interpretation of biopsy specimens. In the case of problematic pigmented lesions such as atypical Spitz nevi, there can be low interobserver agreement even among the experts in categorizing lesions as malignant versus nonmalignant (κ=0.30).3 The low concordance among expert dermatopathologists demonstrates that light microscopic features alone often are inadequate for diagnosis. Advanced studies, including immunohistochemical stains, can help to clarify the diagnosis. In the case of atypical Spitz tumors, the contribution of special stains to the final diagnosis is statistically similar to that of hematoxylin and eosin sections and age, suggesting that nothing alone is sufficiently reliable to establish a definitive diagnosis in every case.4 Although helpful, these studies are costly, and savings obtained by sending cases to the lowest bidder can evaporate quickly. Costs are higher when factoring in molecular studies, which can run upwards of $3000 per slide; the cost of litigation related to incorrect diagnoses; or the human costs of an incorrect diagnosis.
As a group, dermatopathologists are highly skilled in the interpretation of skin specimens, but challenging lesions are common in the routine practice of dermatopathology. A study of 1249 pigmented melanocytic lesions demonstrated substantial agreement among expert dermatopathologists for less problematic lesions, though agreement was greater for patients 40 years and older (κ=0.67) than for younger patients (κ=0.49). Agreement was lower for patients with atypical mole syndrome (κ=0.31).5 These discrepancies occur despite the fact that there is good interobserver reproducibility for grading of individual histological features such as asymmetry, circumscription, irregular confluent nests, single melanocytes predominating, absence of maturation, suprabasal melanocytes, symmetrical melanin, deep melanin, cytological atypia, mitoses, dermal lymphocytic infiltrate, and necrosis.6 These results indicate that accurate diagnoses cannot be reliably established simply by grading a list of histological features. Accurate diagnosis requires complex pattern recognition and integration of findings. Conflicting criteria often are present and an accurate interpretation requires considerable judgment as to which features are significant and which are not.
Separation of sebaceous adenoma, sebaceoma, and well-differentiated sebaceous carcinoma is another challenging area, and interobserver consensus can be as low as 11%,7 which suggests notable subjectivity in the criteria for diagnosis of nonmelanocytic tumors and emphasizes the importance of communication between the dermatopathologist and clinician when determining how to manage an ambiguous lesion. The interpretation of inflammatory skin diseases, alopecia, and lymphoid proliferations also can be problematic, and expert consultation often is required.
All dermatologists receive substantial training in dermatopathology, which puts them in an excellent position to interpret ambiguous findings in the context of the clinical presentation. Sometimes the dermatologist who has seen the clinical presentation can be in the best position to make the diagnosis. All clinicians must be wary of bias and an objective set of eyes often can be helpful. Communication is crucial to ensure appropriate care for each patient, and policies that restrict the choice of pathologist can be damaging.
- Trotter MJ, Bruecks AK. Interpretation of skin biopsies by general pathologists: diagnostic discrepancy rate measured by blinded review. Arch Pathol Lab Med. 2003;127:1489-1492.
- Shoo BA, Sagebiel RW, Kashani-Sabet M. Discordance in the histopathologic diagnosis of melanoma at a melanoma referral center [published online March 19, 2010]. J Am Acad Dermatol. 2010;62:751-756.
- Gerami P, Busam K, Cochran A, et al. Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathol. 2014;38:934-940.
- Puri PK, Ferringer TC, Tyler WB, et al. Statistical analysis of the concordance of immunohistochemical stains with the final diagnosis in spitzoid neoplasms. Am J Dermatopathol. 2011;33:72-77.
- Braun RP, Gutkowicz-Krusin D, Rabinovitz H, et al. Agreement of dermatopathologists in the evaluation of clinically difficult melanocytic lesions: how golden is the ‘gold standard’? Dermatology. 2012;224:51-58.
- Urso C, Rongioletti F, Innocenzi D, et al. Interobserver reproducibility of histological features in cutaneous malignant melanoma. J Clin Pathol. 2005;58:1194-1198.
- Harvey NT, Budgeon CA, Leecy T, et al. Interobserver variability in the diagnosis of circumscribed sebaceous neoplasms of the skin. Pathology. 2013;45:581-586.
As employers search for ways to reduce the cost of providing health care to their employees, there is a growing trend toward narrowed provider networks and exclusive laboratory contracts. In the case of clinical pathology, some of these choices make sense from the employer’s perspective. A complete blood cell count or comprehensive metabolic panel is done on a machine and the result is much the same regardless of the laboratory. So why not have all laboratory tests performed by the lowest bidder?
Laboratories vary in quality and anatomic pathology services are different from blood tests. Each slide must be interpreted by a physician and skill in the interpretation of skin specimens varies widely. Dermatopathology was one of the first subspecialties to be recognized within pathology, as it requires a high level of expertise. Clinicopathological correlation often is key to the accurate interpretation of a specimen. The stakes are high, and a delay in diagnosis of melanoma remains one of the most serious errors in medicine and one of the most common causes for litigation in dermatology.
The accurate interpretation of skin biopsy specimens becomes especially difficult when inadequate or misleading clinical information accompanies the specimen. A study of 589 biopsies submitted by primary care physicians and reported by general pathologists demonstrated a 6.5% error rate. False-negative errors were the most common, but false-positives also were observed.1 A study of pigmented lesions referred to the University of California, San Francisco, demonstrated a discordance rate of 14.3%.2 The degree of discordance would be expected to vary based on the range of diagnoses included in each study.
Board-certified dermatopathologists have varying areas of expertise and there is notable subjectivity in the interpretation of biopsy specimens. In the case of problematic pigmented lesions such as atypical Spitz nevi, there can be low interobserver agreement even among the experts in categorizing lesions as malignant versus nonmalignant (κ=0.30).3 The low concordance among expert dermatopathologists demonstrates that light microscopic features alone often are inadequate for diagnosis. Advanced studies, including immunohistochemical stains, can help to clarify the diagnosis. In the case of atypical Spitz tumors, the contribution of special stains to the final diagnosis is statistically similar to that of hematoxylin and eosin sections and age, suggesting that nothing alone is sufficiently reliable to establish a definitive diagnosis in every case.4 Although helpful, these studies are costly, and savings obtained by sending cases to the lowest bidder can evaporate quickly. Costs are higher when factoring in molecular studies, which can run upwards of $3000 per slide; the cost of litigation related to incorrect diagnoses; or the human costs of an incorrect diagnosis.
As a group, dermatopathologists are highly skilled in the interpretation of skin specimens, but challenging lesions are common in the routine practice of dermatopathology. A study of 1249 pigmented melanocytic lesions demonstrated substantial agreement among expert dermatopathologists for less problematic lesions, though agreement was greater for patients 40 years and older (κ=0.67) than for younger patients (κ=0.49). Agreement was lower for patients with atypical mole syndrome (κ=0.31).5 These discrepancies occur despite the fact that there is good interobserver reproducibility for grading of individual histological features such as asymmetry, circumscription, irregular confluent nests, single melanocytes predominating, absence of maturation, suprabasal melanocytes, symmetrical melanin, deep melanin, cytological atypia, mitoses, dermal lymphocytic infiltrate, and necrosis.6 These results indicate that accurate diagnoses cannot be reliably established simply by grading a list of histological features. Accurate diagnosis requires complex pattern recognition and integration of findings. Conflicting criteria often are present and an accurate interpretation requires considerable judgment as to which features are significant and which are not.
Separation of sebaceous adenoma, sebaceoma, and well-differentiated sebaceous carcinoma is another challenging area, and interobserver consensus can be as low as 11%,7 which suggests notable subjectivity in the criteria for diagnosis of nonmelanocytic tumors and emphasizes the importance of communication between the dermatopathologist and clinician when determining how to manage an ambiguous lesion. The interpretation of inflammatory skin diseases, alopecia, and lymphoid proliferations also can be problematic, and expert consultation often is required.
All dermatologists receive substantial training in dermatopathology, which puts them in an excellent position to interpret ambiguous findings in the context of the clinical presentation. Sometimes the dermatologist who has seen the clinical presentation can be in the best position to make the diagnosis. All clinicians must be wary of bias and an objective set of eyes often can be helpful. Communication is crucial to ensure appropriate care for each patient, and policies that restrict the choice of pathologist can be damaging.
As employers search for ways to reduce the cost of providing health care to their employees, there is a growing trend toward narrowed provider networks and exclusive laboratory contracts. In the case of clinical pathology, some of these choices make sense from the employer’s perspective. A complete blood cell count or comprehensive metabolic panel is done on a machine and the result is much the same regardless of the laboratory. So why not have all laboratory tests performed by the lowest bidder?
Laboratories vary in quality and anatomic pathology services are different from blood tests. Each slide must be interpreted by a physician and skill in the interpretation of skin specimens varies widely. Dermatopathology was one of the first subspecialties to be recognized within pathology, as it requires a high level of expertise. Clinicopathological correlation often is key to the accurate interpretation of a specimen. The stakes are high, and a delay in diagnosis of melanoma remains one of the most serious errors in medicine and one of the most common causes for litigation in dermatology.
The accurate interpretation of skin biopsy specimens becomes especially difficult when inadequate or misleading clinical information accompanies the specimen. A study of 589 biopsies submitted by primary care physicians and reported by general pathologists demonstrated a 6.5% error rate. False-negative errors were the most common, but false-positives also were observed.1 A study of pigmented lesions referred to the University of California, San Francisco, demonstrated a discordance rate of 14.3%.2 The degree of discordance would be expected to vary based on the range of diagnoses included in each study.
Board-certified dermatopathologists have varying areas of expertise and there is notable subjectivity in the interpretation of biopsy specimens. In the case of problematic pigmented lesions such as atypical Spitz nevi, there can be low interobserver agreement even among the experts in categorizing lesions as malignant versus nonmalignant (κ=0.30).3 The low concordance among expert dermatopathologists demonstrates that light microscopic features alone often are inadequate for diagnosis. Advanced studies, including immunohistochemical stains, can help to clarify the diagnosis. In the case of atypical Spitz tumors, the contribution of special stains to the final diagnosis is statistically similar to that of hematoxylin and eosin sections and age, suggesting that nothing alone is sufficiently reliable to establish a definitive diagnosis in every case.4 Although helpful, these studies are costly, and savings obtained by sending cases to the lowest bidder can evaporate quickly. Costs are higher when factoring in molecular studies, which can run upwards of $3000 per slide; the cost of litigation related to incorrect diagnoses; or the human costs of an incorrect diagnosis.
As a group, dermatopathologists are highly skilled in the interpretation of skin specimens, but challenging lesions are common in the routine practice of dermatopathology. A study of 1249 pigmented melanocytic lesions demonstrated substantial agreement among expert dermatopathologists for less problematic lesions, though agreement was greater for patients 40 years and older (κ=0.67) than for younger patients (κ=0.49). Agreement was lower for patients with atypical mole syndrome (κ=0.31).5 These discrepancies occur despite the fact that there is good interobserver reproducibility for grading of individual histological features such as asymmetry, circumscription, irregular confluent nests, single melanocytes predominating, absence of maturation, suprabasal melanocytes, symmetrical melanin, deep melanin, cytological atypia, mitoses, dermal lymphocytic infiltrate, and necrosis.6 These results indicate that accurate diagnoses cannot be reliably established simply by grading a list of histological features. Accurate diagnosis requires complex pattern recognition and integration of findings. Conflicting criteria often are present and an accurate interpretation requires considerable judgment as to which features are significant and which are not.
Separation of sebaceous adenoma, sebaceoma, and well-differentiated sebaceous carcinoma is another challenging area, and interobserver consensus can be as low as 11%,7 which suggests notable subjectivity in the criteria for diagnosis of nonmelanocytic tumors and emphasizes the importance of communication between the dermatopathologist and clinician when determining how to manage an ambiguous lesion. The interpretation of inflammatory skin diseases, alopecia, and lymphoid proliferations also can be problematic, and expert consultation often is required.
All dermatologists receive substantial training in dermatopathology, which puts them in an excellent position to interpret ambiguous findings in the context of the clinical presentation. Sometimes the dermatologist who has seen the clinical presentation can be in the best position to make the diagnosis. All clinicians must be wary of bias and an objective set of eyes often can be helpful. Communication is crucial to ensure appropriate care for each patient, and policies that restrict the choice of pathologist can be damaging.
- Trotter MJ, Bruecks AK. Interpretation of skin biopsies by general pathologists: diagnostic discrepancy rate measured by blinded review. Arch Pathol Lab Med. 2003;127:1489-1492.
- Shoo BA, Sagebiel RW, Kashani-Sabet M. Discordance in the histopathologic diagnosis of melanoma at a melanoma referral center [published online March 19, 2010]. J Am Acad Dermatol. 2010;62:751-756.
- Gerami P, Busam K, Cochran A, et al. Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathol. 2014;38:934-940.
- Puri PK, Ferringer TC, Tyler WB, et al. Statistical analysis of the concordance of immunohistochemical stains with the final diagnosis in spitzoid neoplasms. Am J Dermatopathol. 2011;33:72-77.
- Braun RP, Gutkowicz-Krusin D, Rabinovitz H, et al. Agreement of dermatopathologists in the evaluation of clinically difficult melanocytic lesions: how golden is the ‘gold standard’? Dermatology. 2012;224:51-58.
- Urso C, Rongioletti F, Innocenzi D, et al. Interobserver reproducibility of histological features in cutaneous malignant melanoma. J Clin Pathol. 2005;58:1194-1198.
- Harvey NT, Budgeon CA, Leecy T, et al. Interobserver variability in the diagnosis of circumscribed sebaceous neoplasms of the skin. Pathology. 2013;45:581-586.
- Trotter MJ, Bruecks AK. Interpretation of skin biopsies by general pathologists: diagnostic discrepancy rate measured by blinded review. Arch Pathol Lab Med. 2003;127:1489-1492.
- Shoo BA, Sagebiel RW, Kashani-Sabet M. Discordance in the histopathologic diagnosis of melanoma at a melanoma referral center [published online March 19, 2010]. J Am Acad Dermatol. 2010;62:751-756.
- Gerami P, Busam K, Cochran A, et al. Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathol. 2014;38:934-940.
- Puri PK, Ferringer TC, Tyler WB, et al. Statistical analysis of the concordance of immunohistochemical stains with the final diagnosis in spitzoid neoplasms. Am J Dermatopathol. 2011;33:72-77.
- Braun RP, Gutkowicz-Krusin D, Rabinovitz H, et al. Agreement of dermatopathologists in the evaluation of clinically difficult melanocytic lesions: how golden is the ‘gold standard’? Dermatology. 2012;224:51-58.
- Urso C, Rongioletti F, Innocenzi D, et al. Interobserver reproducibility of histological features in cutaneous malignant melanoma. J Clin Pathol. 2005;58:1194-1198.
- Harvey NT, Budgeon CA, Leecy T, et al. Interobserver variability in the diagnosis of circumscribed sebaceous neoplasms of the skin. Pathology. 2013;45:581-586.

Purple Curvilinear Papules on the Back
The Diagnosis: Blaschkoid Graft-vs-host Disease
The patient had a history of myelodysplastic syndrome and underwent a bone marrow transplant 1 year prior to presentation. She had acute graft-vs-host disease (GVHD) 6 weeks following the transplant, which resolved with high-dose prednisone followed by UVB phototherapy. Skin biopsy demonstrated lichenoid dermatitis with vacuolar degeneration, dyskeratosis, and prominent pigment incontinence (Figure). Based on these findings and her clinical presentation, a diagnosis of blaschkoid GVHD was made.

Although acute GVHD is the result of immunocompetent donor T cells recognizing host tissues as foreign and initiating an immune response, the pathophysiology of chronic GVHD is not well understood.1,2 Theories for disease pathogenesis in chronic GVHD suggest an underlying autoimmune and/or alloreactive process.2-5 The skin often is the first organ affected in acute GVHD, and patients generally present with a pruritic morbilliform eruption that begins on the trunk and spreads to the rest of the body.1,2 Cutaneous manifestations of chronic GVHD may be protean. Lesions can resemble systemic sclerosis or morphea, lichen planus, psoriasis, ichthyosis, and many other conditions.2
The differential diagnosis of linear dermatoses includes herpes zoster, contact dermatitis, lichen striatus (blaschkitis), nevus unius lateris, inflammatory linear verrucous epidermal nevus, and incontinentia pigmenti.6,7 Lichen planus-like chronic GVHD occurring in a linear distribution has been described.6-14 Distinction between dermatomal and blaschkoid processes is diagnostically important. In the case of GVHD, dermatomal distribution may suggest an association between GVHD and prior herpes simplex virus or varicella-zoster virus infection.6,8 Herpesvirus may alter surface antigens of keratinocytes, rendering them targets of donor lymphocytes, and antibodies to viral particles may cross-react with host keratinocyte HLA antigens. It also is possible that dermatomal GVHD may simply be a type of isomorphic response (Köbner phenomenon).8
When cutaneous GVHD follows Blaschko lines, other mechanisms appear to be at play.9-14 It is plausible that these patients have an underlying genetic mosaicism, perhaps the result of a postzygotic mutation, that results in a daughter cell population that expresses surface antigens different from those of the primary cell population found elsewhere in the skin. Donor lymphocytes may selectively react to this mosaic population, leading to the clinical picture of chronic GVHD oriented along Blaschko lines.10,11,13,14
In conclusion, lichenoid linear GVHD following Blaschko lines is an uncommon presentation of chronic GVHD that highlights the heterogeneity of this disease and should be considered in the appropriate clinical setting.
- Ferrara JL, Levine JE, Reddy P, et al. Graft-versus-host disease. Lancet. 2009;373:1550-1561.
- Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012;66:515.e1-515.e18; quiz 533-534.
- Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389-396.
- Shimada M, Onizuka M, Machida S, et al. Association of autoimmune disease-related gene polymorphisms with chronic graft-versus-host disease. Br J Haematol. 2007;139:458-463.
- Zhang C, Todorov I, Zhang Z, et al. Donor CD4+ T and B cells in transplants induce chronic graft-versus-host disease with autoimmune manifestations. Blood. 2006;107:2993-3001.
- Freemer CS, Farmer ER, Corio RL, et al. Lichenoid chronic graft-vs-host disease occurring in a dermatomal distribution. Arch Dermatol. 1994;130:70-72.
- Kikuchi A, Okamoto S, Takahashi S, et al. Linear chronic cutaneous graft-versus-host disease. J Am Acad Dermatol. 1997;37:1004-1006.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Kennedy FE, Hilari H, Ferrer B, et al. Lichenoid chronic graft-vs-host disease following Blaschko lines. ActasDermosifiliogr. 2014;105:89-92.
- Lee SW, Kim YC, Lee E, et al. Linear lichenoid graft versus host disease: an unusual configuration following Blaschko's lines. J Dermatol. 2006;33:583-584.
- Beers B, Kalish RS, Kaye VN, et al. Unilateral linear lichenoid eruption after bone marrow transplantation: an unmasking of tolerance to an abnormal keratinocyte clone? J Am Acad Dermatol. 1993;28(5, pt 2):888-892.
- Wilson B, Lockman D. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130(9):1206-1208.
- Reisfeld PL. Lichenoid chronic graft-vs-host disease. Arch Dermatol. 1994;130:1207-1208.
- Vassallo C, Derlino F, Ripamonti F, et al. Lichenoid cutaneous chronic GvHD following Blaschko lines. Int J Dermatol. 2014;53:473-475.
The Diagnosis: Blaschkoid Graft-vs-host Disease
The patient had a history of myelodysplastic syndrome and underwent a bone marrow transplant 1 year prior to presentation. She had acute graft-vs-host disease (GVHD) 6 weeks following the transplant, which resolved with high-dose prednisone followed by UVB phototherapy. Skin biopsy demonstrated lichenoid dermatitis with vacuolar degeneration, dyskeratosis, and prominent pigment incontinence (Figure). Based on these findings and her clinical presentation, a diagnosis of blaschkoid GVHD was made.
Although acute GVHD is the result of immunocompetent donor T cells recognizing host tissues as foreign and initiating an immune response, the pathophysiology of chronic GVHD is not well understood.1,2 Theories for disease pathogenesis in chronic GVHD suggest an underlying autoimmune and/or alloreactive process.2-5 The skin often is the first organ affected in acute GVHD, and patients generally present with a pruritic morbilliform eruption that begins on the trunk and spreads to the rest of the body.1,2 Cutaneous manifestations of chronic GVHD may be protean. Lesions can resemble systemic sclerosis or morphea, lichen planus, psoriasis, ichthyosis, and many other conditions.2
The differential diagnosis of linear dermatoses includes herpes zoster, contact dermatitis, lichen striatus (blaschkitis), nevus unius lateris, inflammatory linear verrucous epidermal nevus, and incontinentia pigmenti.6,7 Lichen planus-like chronic GVHD occurring in a linear distribution has been described.6-14 Distinction between dermatomal and blaschkoid processes is diagnostically important. In the case of GVHD, dermatomal distribution may suggest an association between GVHD and prior herpes simplex virus or varicella-zoster virus infection.6,8 Herpesvirus may alter surface antigens of keratinocytes, rendering them targets of donor lymphocytes, and antibodies to viral particles may cross-react with host keratinocyte HLA antigens. It also is possible that dermatomal GVHD may simply be a type of isomorphic response (Köbner phenomenon).8
When cutaneous GVHD follows Blaschko lines, other mechanisms appear to be at play.9-14 It is plausible that these patients have an underlying genetic mosaicism, perhaps the result of a postzygotic mutation, that results in a daughter cell population that expresses surface antigens different from those of the primary cell population found elsewhere in the skin. Donor lymphocytes may selectively react to this mosaic population, leading to the clinical picture of chronic GVHD oriented along Blaschko lines.10,11,13,14
In conclusion, lichenoid linear GVHD following Blaschko lines is an uncommon presentation of chronic GVHD that highlights the heterogeneity of this disease and should be considered in the appropriate clinical setting.
The Diagnosis: Blaschkoid Graft-vs-host Disease
The patient had a history of myelodysplastic syndrome and underwent a bone marrow transplant 1 year prior to presentation. She had acute graft-vs-host disease (GVHD) 6 weeks following the transplant, which resolved with high-dose prednisone followed by UVB phototherapy. Skin biopsy demonstrated lichenoid dermatitis with vacuolar degeneration, dyskeratosis, and prominent pigment incontinence (Figure). Based on these findings and her clinical presentation, a diagnosis of blaschkoid GVHD was made.
Although acute GVHD is the result of immunocompetent donor T cells recognizing host tissues as foreign and initiating an immune response, the pathophysiology of chronic GVHD is not well understood.1,2 Theories for disease pathogenesis in chronic GVHD suggest an underlying autoimmune and/or alloreactive process.2-5 The skin often is the first organ affected in acute GVHD, and patients generally present with a pruritic morbilliform eruption that begins on the trunk and spreads to the rest of the body.1,2 Cutaneous manifestations of chronic GVHD may be protean. Lesions can resemble systemic sclerosis or morphea, lichen planus, psoriasis, ichthyosis, and many other conditions.2
The differential diagnosis of linear dermatoses includes herpes zoster, contact dermatitis, lichen striatus (blaschkitis), nevus unius lateris, inflammatory linear verrucous epidermal nevus, and incontinentia pigmenti.6,7 Lichen planus-like chronic GVHD occurring in a linear distribution has been described.6-14 Distinction between dermatomal and blaschkoid processes is diagnostically important. In the case of GVHD, dermatomal distribution may suggest an association between GVHD and prior herpes simplex virus or varicella-zoster virus infection.6,8 Herpesvirus may alter surface antigens of keratinocytes, rendering them targets of donor lymphocytes, and antibodies to viral particles may cross-react with host keratinocyte HLA antigens. It also is possible that dermatomal GVHD may simply be a type of isomorphic response (Köbner phenomenon).8
When cutaneous GVHD follows Blaschko lines, other mechanisms appear to be at play.9-14 It is plausible that these patients have an underlying genetic mosaicism, perhaps the result of a postzygotic mutation, that results in a daughter cell population that expresses surface antigens different from those of the primary cell population found elsewhere in the skin. Donor lymphocytes may selectively react to this mosaic population, leading to the clinical picture of chronic GVHD oriented along Blaschko lines.10,11,13,14
In conclusion, lichenoid linear GVHD following Blaschko lines is an uncommon presentation of chronic GVHD that highlights the heterogeneity of this disease and should be considered in the appropriate clinical setting.
- Ferrara JL, Levine JE, Reddy P, et al. Graft-versus-host disease. Lancet. 2009;373:1550-1561.
- Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012;66:515.e1-515.e18; quiz 533-534.
- Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389-396.
- Shimada M, Onizuka M, Machida S, et al. Association of autoimmune disease-related gene polymorphisms with chronic graft-versus-host disease. Br J Haematol. 2007;139:458-463.
- Zhang C, Todorov I, Zhang Z, et al. Donor CD4+ T and B cells in transplants induce chronic graft-versus-host disease with autoimmune manifestations. Blood. 2006;107:2993-3001.
- Freemer CS, Farmer ER, Corio RL, et al. Lichenoid chronic graft-vs-host disease occurring in a dermatomal distribution. Arch Dermatol. 1994;130:70-72.
- Kikuchi A, Okamoto S, Takahashi S, et al. Linear chronic cutaneous graft-versus-host disease. J Am Acad Dermatol. 1997;37:1004-1006.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Kennedy FE, Hilari H, Ferrer B, et al. Lichenoid chronic graft-vs-host disease following Blaschko lines. ActasDermosifiliogr. 2014;105:89-92.
- Lee SW, Kim YC, Lee E, et al. Linear lichenoid graft versus host disease: an unusual configuration following Blaschko's lines. J Dermatol. 2006;33:583-584.
- Beers B, Kalish RS, Kaye VN, et al. Unilateral linear lichenoid eruption after bone marrow transplantation: an unmasking of tolerance to an abnormal keratinocyte clone? J Am Acad Dermatol. 1993;28(5, pt 2):888-892.
- Wilson B, Lockman D. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130(9):1206-1208.
- Reisfeld PL. Lichenoid chronic graft-vs-host disease. Arch Dermatol. 1994;130:1207-1208.
- Vassallo C, Derlino F, Ripamonti F, et al. Lichenoid cutaneous chronic GvHD following Blaschko lines. Int J Dermatol. 2014;53:473-475.
- Ferrara JL, Levine JE, Reddy P, et al. Graft-versus-host disease. Lancet. 2009;373:1550-1561.
- Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012;66:515.e1-515.e18; quiz 533-534.
- Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389-396.
- Shimada M, Onizuka M, Machida S, et al. Association of autoimmune disease-related gene polymorphisms with chronic graft-versus-host disease. Br J Haematol. 2007;139:458-463.
- Zhang C, Todorov I, Zhang Z, et al. Donor CD4+ T and B cells in transplants induce chronic graft-versus-host disease with autoimmune manifestations. Blood. 2006;107:2993-3001.
- Freemer CS, Farmer ER, Corio RL, et al. Lichenoid chronic graft-vs-host disease occurring in a dermatomal distribution. Arch Dermatol. 1994;130:70-72.
- Kikuchi A, Okamoto S, Takahashi S, et al. Linear chronic cutaneous graft-versus-host disease. J Am Acad Dermatol. 1997;37:1004-1006.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Kennedy FE, Hilari H, Ferrer B, et al. Lichenoid chronic graft-vs-host disease following Blaschko lines. ActasDermosifiliogr. 2014;105:89-92.
- Lee SW, Kim YC, Lee E, et al. Linear lichenoid graft versus host disease: an unusual configuration following Blaschko's lines. J Dermatol. 2006;33:583-584.
- Beers B, Kalish RS, Kaye VN, et al. Unilateral linear lichenoid eruption after bone marrow transplantation: an unmasking of tolerance to an abnormal keratinocyte clone? J Am Acad Dermatol. 1993;28(5, pt 2):888-892.
- Wilson B, Lockman D. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130(9):1206-1208.
- Reisfeld PL. Lichenoid chronic graft-vs-host disease. Arch Dermatol. 1994;130:1207-1208.
- Vassallo C, Derlino F, Ripamonti F, et al. Lichenoid cutaneous chronic GvHD following Blaschko lines. Int J Dermatol. 2014;53:473-475.
A 56-year-old woman with a history of bone marrow transplant presented for evaluation of a nonpruritic rash of 3 months' duration. Physical examination revealed confluent purple-colored and hyperpigmented papules localized to the back and right arm in a curvilinear pattern. Laboratory results were notable for mildly elevated aspartate transaminase and alanine transaminase levels.