User login
Verrucous Plaque on the Leg
Blastomycosis
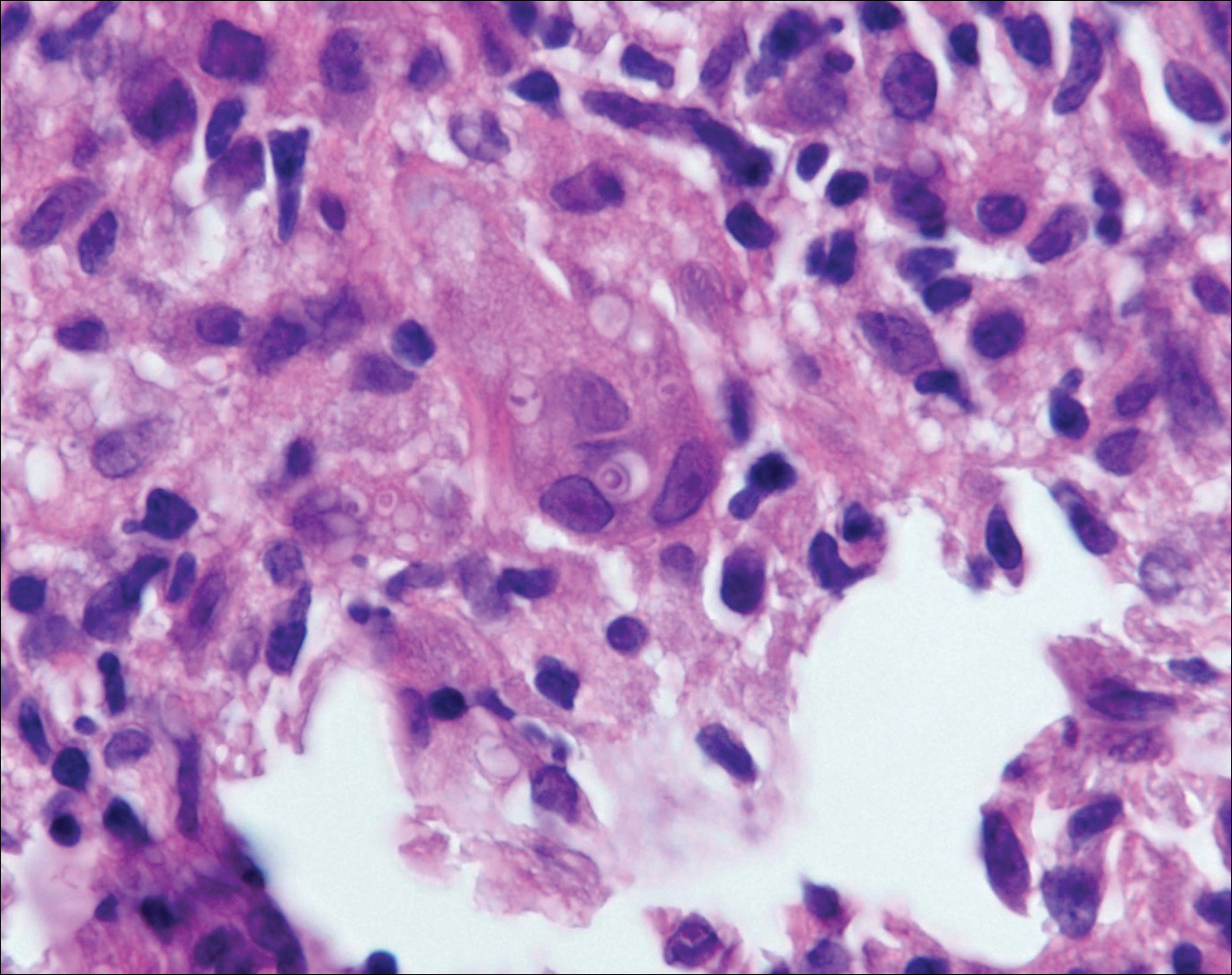
Blastomycosis is caused by Blastomyces dermatitidis, which is endemic in the Midwestern and southeastern United States where it occurs environmentally in wood and soil. Unlike many fungal infections, blastomycosis most often develops in immunocompetent hosts. Infection is usually acquired via inhalation,1 and cutaneous disease typically is secondary to pulmonary infection. Although not common, traumatic inoculation also can cause cutaneous blastomycosis. Skin lesions include crusted verrucous nodules and plaques with elevated borders.1,2 Histologic features include pseudoepitheliomatous hyperplasia with intraepidermal neutrophilic microabscesses (Figure 1), and a neutrophilic and granulomatous dermal infiltrate. Organisms often are found within histiocytes (quiz image) or small abscesses. The yeasts usually are 8 to 15 µm in diameter with a thick cell wall and occasionally display broad-based budding.
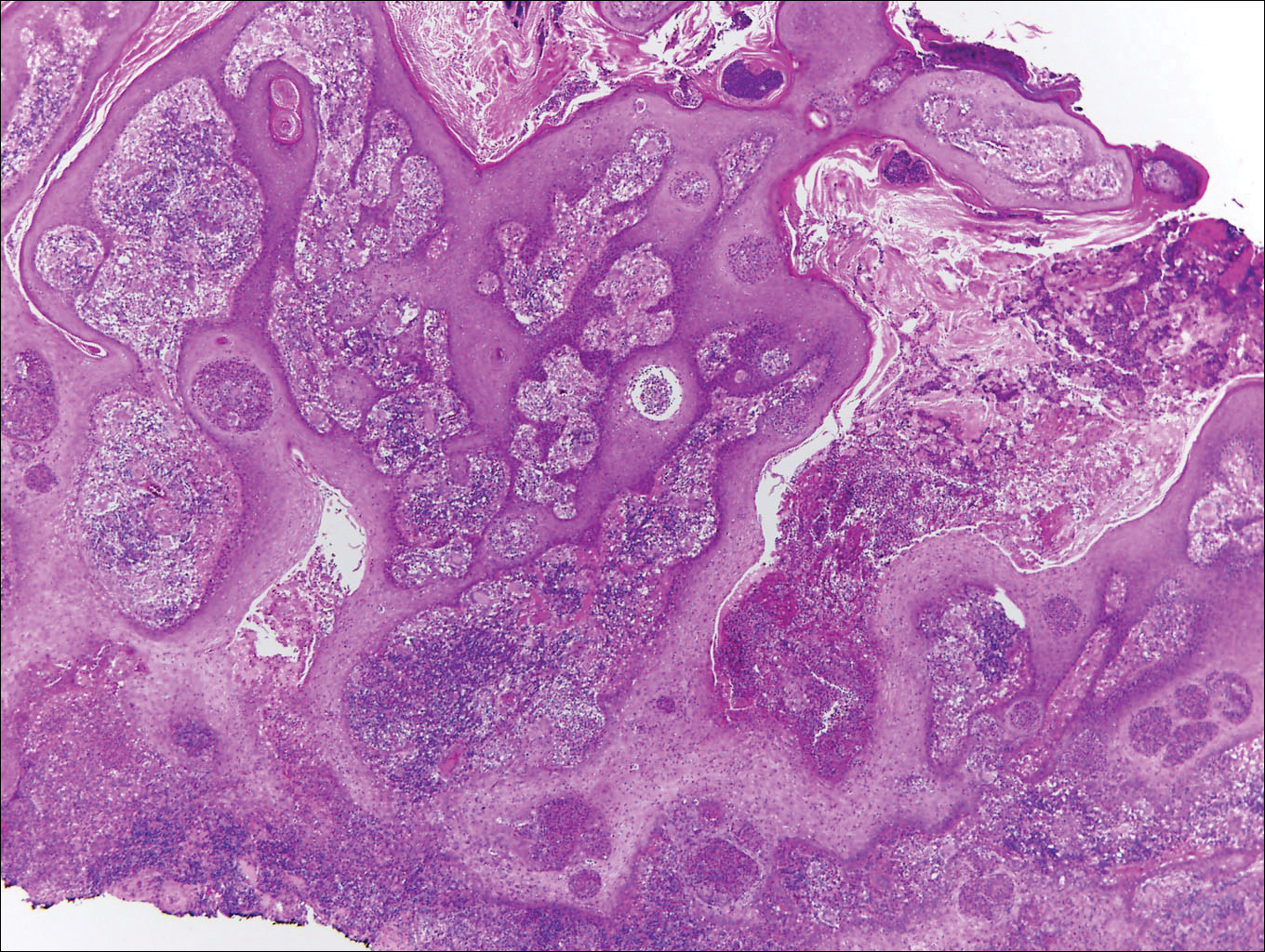
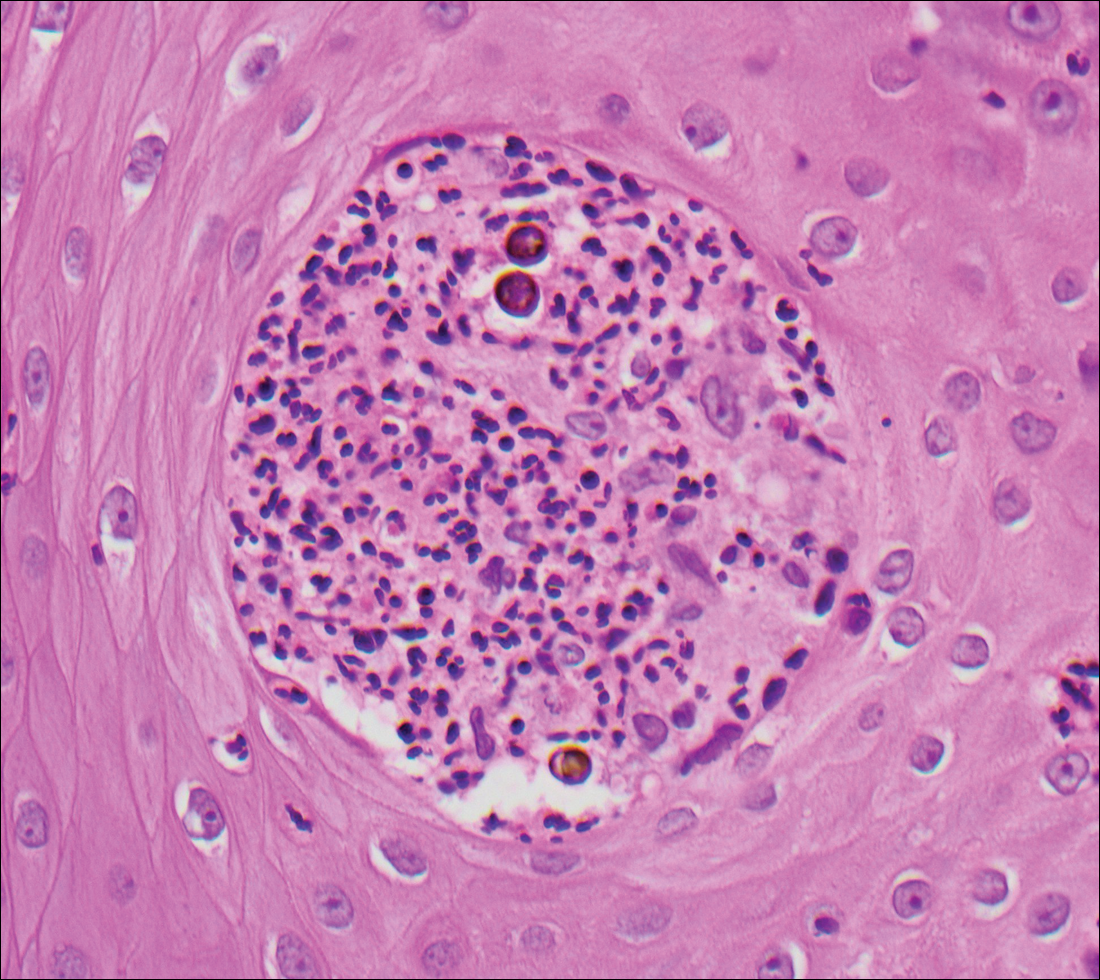
Chromoblastomycosis is caused by dematiaceous (pigmented) fungi, including Fonsecaea, Phialophora, Cladophialophora, and Rhinocladiella species,3 which are present in soil and vegetable debris in tropical and subtropical regions. Infection typically occurs in the foot or lower leg from traumatic inoculation, such as a thorn or splinter injury.2 Histologically, chromoblastomycosis is characterized by pseudoepitheliomatous hyperplasia; suppurative and granulomatous dermatitis; and sclerotic (Medlar) bodies, which are 5 to 12 µm in diameter, round, brown, sometimes septate cells resembling copper pennies (Figure 2).2
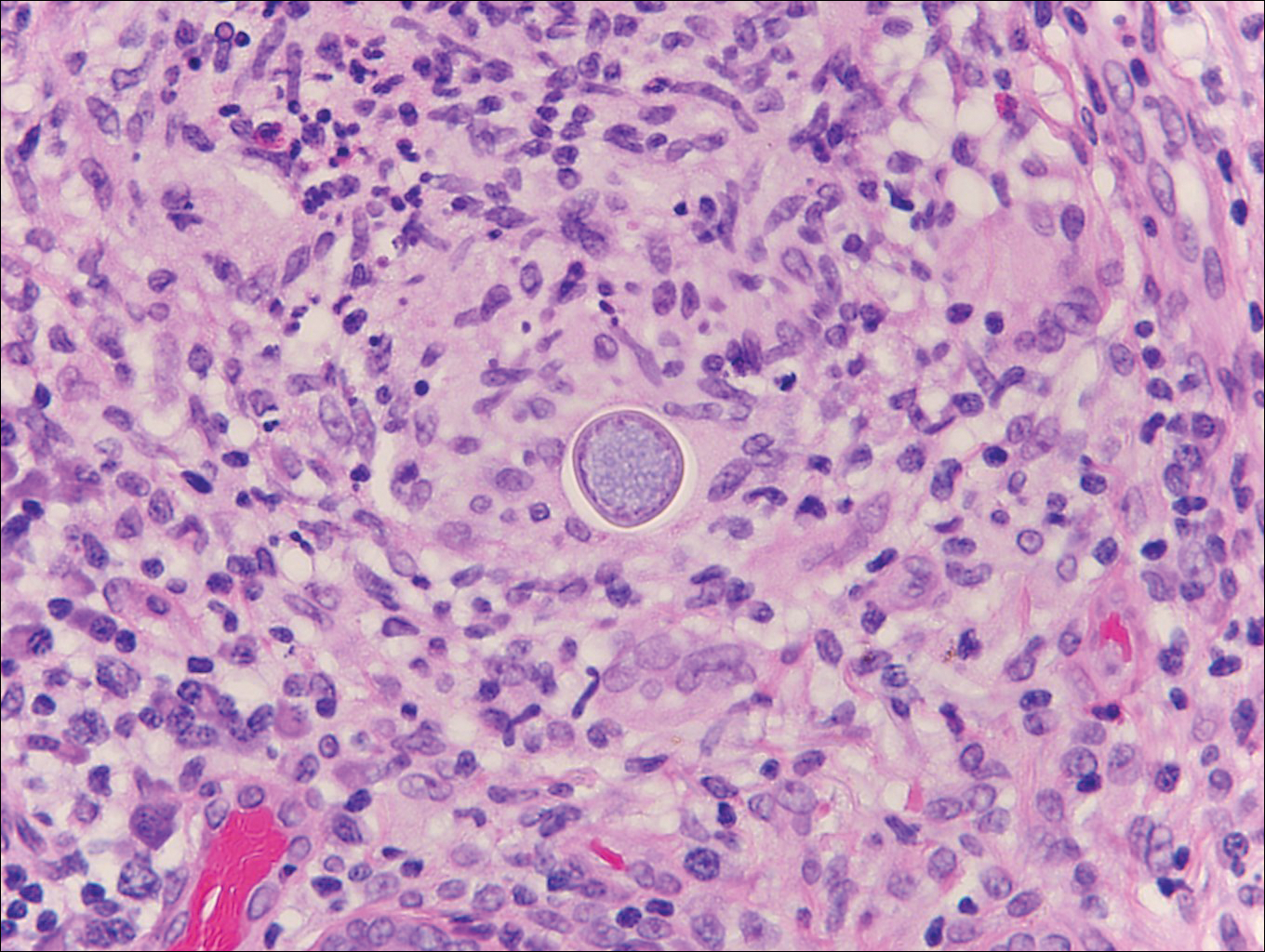
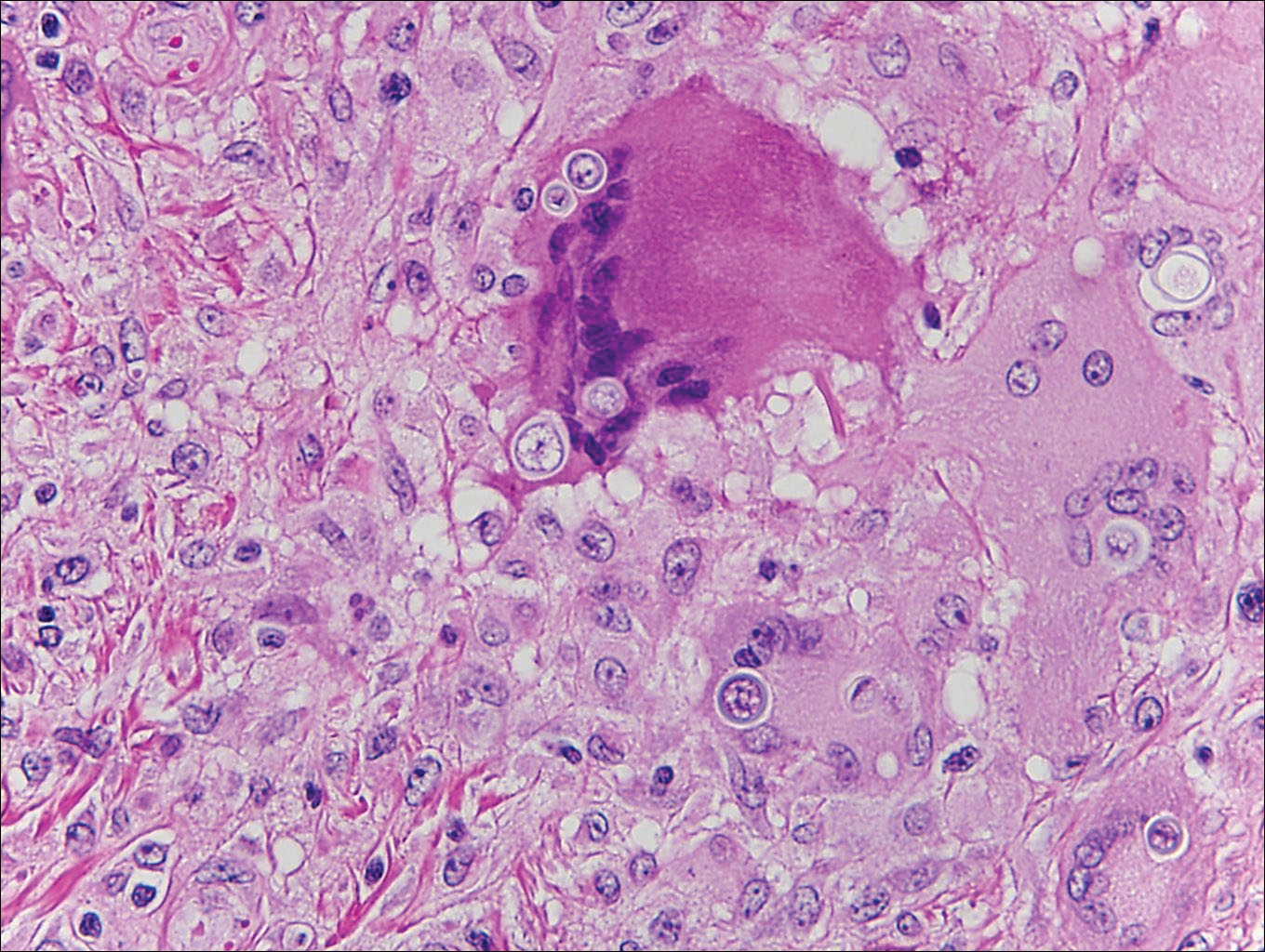
Coccidioidomycosis is caused by Coccidioides immitis, which is found in soil in the southwestern United States. Infection most often occurs via inhalation of airborne arthrospores.2 Cutaneous lesions occasionally are observed following dissemination or rarely following primary inoculation injury. They may present as papules, nodules, pustules, plaques, and ulcers, with the face being the most commonly affected site.1 Histologically, coccidioidomycosis is characterized by pseudoepitheliomatous hyperplasia, suppurative and granulomatous dermatitis, and large spherules (up to 100 µm in diameter) containing numerous small endospores (Figure 3).
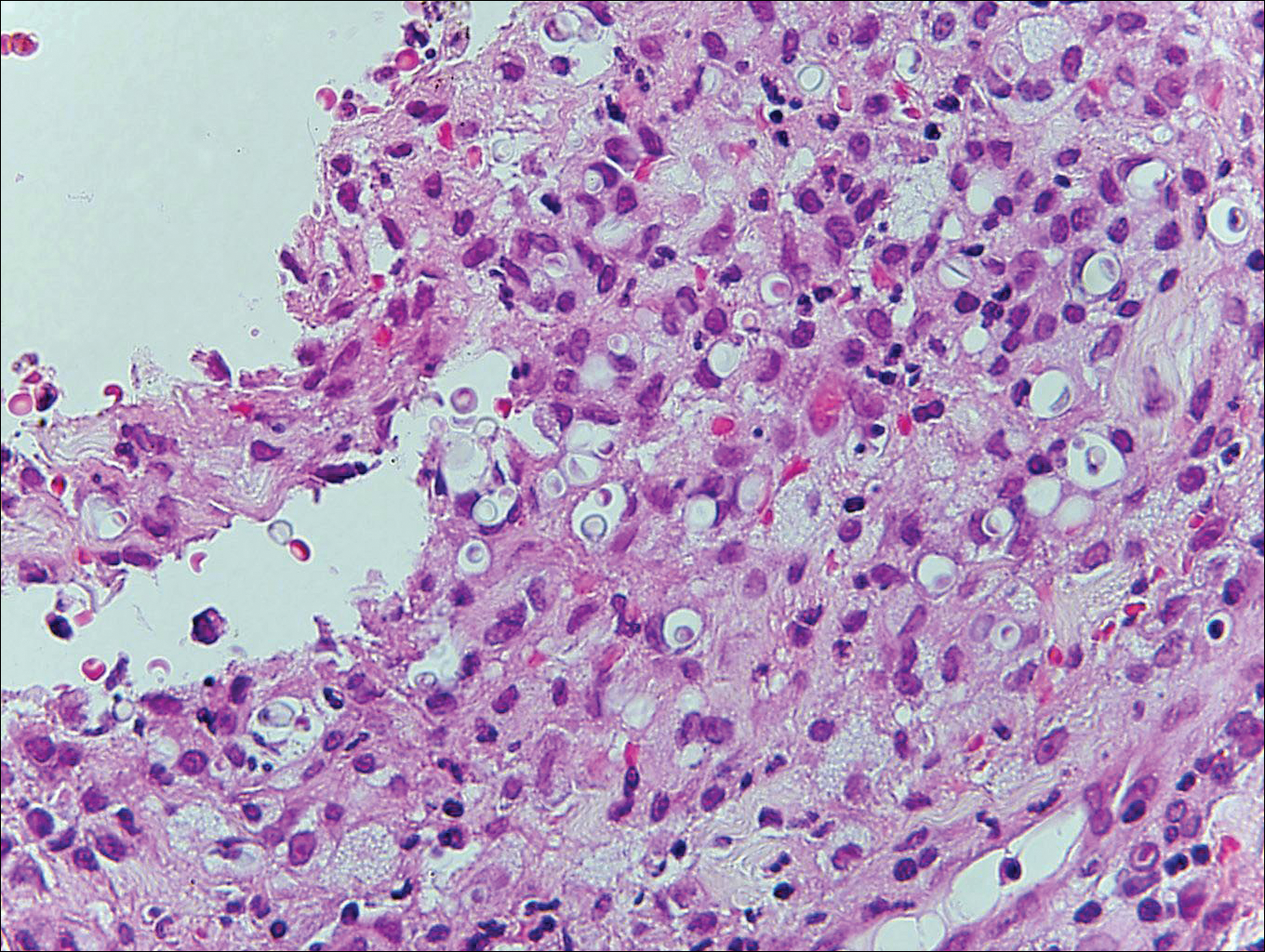
Cryptococcosis is caused by Cryptococcus neoformans, a fungus found in soil, fruit, and pigeon droppings throughout the world.2,3 The most common route of infection is via the respiratory tract. Systemic spread and central nervous system involvement may occur in immunocompromised hosts.2 Skin involvement is uncommon and may present on the head and neck with umbilicated papules, pustules, nodules, plaques, or ulcers. Histologically, Cryptococcus is a spherical yeast, often 4 to 20 µm in diameter. Replication is by narrow-based budding. A characteristic feature is a mucoid capsule, which retracts during processing, leaving a clear space around the yeast (Figure 4). When present, the mucoid capsule can be highlighted on mucicarmine or Alcian blue staining. Histologic variants of cryptococcosis include granulomatous (high host immune response), gelatinous (low host immune response), and suppurative types.3
Histoplasmosis is caused by Histoplasma capsulatum, which occurs in soil and bird and bat droppings, with exposure primarily via inhalation. Cutaneous histoplasmosis is almost always a feature of disseminated disease, which occurs most commonly in immunosuppressed individuals.1 Skin lesions may present as macules, papules, indurated plaques, ulcers, purpura, panniculitis, and subcutaneous nodules.2 Histologically, there is a granulomatous and neutrophilic infiltrate within the dermis and subcutis. Yeasts are small (2-4 µm in diameter) and are observed within the cytoplasm of macrophages (Figure 5) where they appear as basophilic dots, sometimes surrounded by an artifactual clear space (pseudocapsule).2
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier/Saunders; 2012.
- Schwarzenberger K, Werchniak A, Ko C. Requisites in Dermatology: General Dermatology. Philadelphia, PA: Elsevier/Saunders; 2009.
Blastomycosis
Blastomycosis is caused by Blastomyces dermatitidis, which is endemic in the Midwestern and southeastern United States where it occurs environmentally in wood and soil. Unlike many fungal infections, blastomycosis most often develops in immunocompetent hosts. Infection is usually acquired via inhalation,1 and cutaneous disease typically is secondary to pulmonary infection. Although not common, traumatic inoculation also can cause cutaneous blastomycosis. Skin lesions include crusted verrucous nodules and plaques with elevated borders.1,2 Histologic features include pseudoepitheliomatous hyperplasia with intraepidermal neutrophilic microabscesses (Figure 1), and a neutrophilic and granulomatous dermal infiltrate. Organisms often are found within histiocytes (quiz image) or small abscesses. The yeasts usually are 8 to 15 µm in diameter with a thick cell wall and occasionally display broad-based budding.
Chromoblastomycosis is caused by dematiaceous (pigmented) fungi, including Fonsecaea, Phialophora, Cladophialophora, and Rhinocladiella species,3 which are present in soil and vegetable debris in tropical and subtropical regions. Infection typically occurs in the foot or lower leg from traumatic inoculation, such as a thorn or splinter injury.2 Histologically, chromoblastomycosis is characterized by pseudoepitheliomatous hyperplasia; suppurative and granulomatous dermatitis; and sclerotic (Medlar) bodies, which are 5 to 12 µm in diameter, round, brown, sometimes septate cells resembling copper pennies (Figure 2).2
Coccidioidomycosis is caused by Coccidioides immitis, which is found in soil in the southwestern United States. Infection most often occurs via inhalation of airborne arthrospores.2 Cutaneous lesions occasionally are observed following dissemination or rarely following primary inoculation injury. They may present as papules, nodules, pustules, plaques, and ulcers, with the face being the most commonly affected site.1 Histologically, coccidioidomycosis is characterized by pseudoepitheliomatous hyperplasia, suppurative and granulomatous dermatitis, and large spherules (up to 100 µm in diameter) containing numerous small endospores (Figure 3).
Cryptococcosis is caused by Cryptococcus neoformans, a fungus found in soil, fruit, and pigeon droppings throughout the world.2,3 The most common route of infection is via the respiratory tract. Systemic spread and central nervous system involvement may occur in immunocompromised hosts.2 Skin involvement is uncommon and may present on the head and neck with umbilicated papules, pustules, nodules, plaques, or ulcers. Histologically, Cryptococcus is a spherical yeast, often 4 to 20 µm in diameter. Replication is by narrow-based budding. A characteristic feature is a mucoid capsule, which retracts during processing, leaving a clear space around the yeast (Figure 4). When present, the mucoid capsule can be highlighted on mucicarmine or Alcian blue staining. Histologic variants of cryptococcosis include granulomatous (high host immune response), gelatinous (low host immune response), and suppurative types.3
Histoplasmosis is caused by Histoplasma capsulatum, which occurs in soil and bird and bat droppings, with exposure primarily via inhalation. Cutaneous histoplasmosis is almost always a feature of disseminated disease, which occurs most commonly in immunosuppressed individuals.1 Skin lesions may present as macules, papules, indurated plaques, ulcers, purpura, panniculitis, and subcutaneous nodules.2 Histologically, there is a granulomatous and neutrophilic infiltrate within the dermis and subcutis. Yeasts are small (2-4 µm in diameter) and are observed within the cytoplasm of macrophages (Figure 5) where they appear as basophilic dots, sometimes surrounded by an artifactual clear space (pseudocapsule).2
Blastomycosis
Blastomycosis is caused by Blastomyces dermatitidis, which is endemic in the Midwestern and southeastern United States where it occurs environmentally in wood and soil. Unlike many fungal infections, blastomycosis most often develops in immunocompetent hosts. Infection is usually acquired via inhalation,1 and cutaneous disease typically is secondary to pulmonary infection. Although not common, traumatic inoculation also can cause cutaneous blastomycosis. Skin lesions include crusted verrucous nodules and plaques with elevated borders.1,2 Histologic features include pseudoepitheliomatous hyperplasia with intraepidermal neutrophilic microabscesses (Figure 1), and a neutrophilic and granulomatous dermal infiltrate. Organisms often are found within histiocytes (quiz image) or small abscesses. The yeasts usually are 8 to 15 µm in diameter with a thick cell wall and occasionally display broad-based budding.
Chromoblastomycosis is caused by dematiaceous (pigmented) fungi, including Fonsecaea, Phialophora, Cladophialophora, and Rhinocladiella species,3 which are present in soil and vegetable debris in tropical and subtropical regions. Infection typically occurs in the foot or lower leg from traumatic inoculation, such as a thorn or splinter injury.2 Histologically, chromoblastomycosis is characterized by pseudoepitheliomatous hyperplasia; suppurative and granulomatous dermatitis; and sclerotic (Medlar) bodies, which are 5 to 12 µm in diameter, round, brown, sometimes septate cells resembling copper pennies (Figure 2).2
Coccidioidomycosis is caused by Coccidioides immitis, which is found in soil in the southwestern United States. Infection most often occurs via inhalation of airborne arthrospores.2 Cutaneous lesions occasionally are observed following dissemination or rarely following primary inoculation injury. They may present as papules, nodules, pustules, plaques, and ulcers, with the face being the most commonly affected site.1 Histologically, coccidioidomycosis is characterized by pseudoepitheliomatous hyperplasia, suppurative and granulomatous dermatitis, and large spherules (up to 100 µm in diameter) containing numerous small endospores (Figure 3).
Cryptococcosis is caused by Cryptococcus neoformans, a fungus found in soil, fruit, and pigeon droppings throughout the world.2,3 The most common route of infection is via the respiratory tract. Systemic spread and central nervous system involvement may occur in immunocompromised hosts.2 Skin involvement is uncommon and may present on the head and neck with umbilicated papules, pustules, nodules, plaques, or ulcers. Histologically, Cryptococcus is a spherical yeast, often 4 to 20 µm in diameter. Replication is by narrow-based budding. A characteristic feature is a mucoid capsule, which retracts during processing, leaving a clear space around the yeast (Figure 4). When present, the mucoid capsule can be highlighted on mucicarmine or Alcian blue staining. Histologic variants of cryptococcosis include granulomatous (high host immune response), gelatinous (low host immune response), and suppurative types.3
Histoplasmosis is caused by Histoplasma capsulatum, which occurs in soil and bird and bat droppings, with exposure primarily via inhalation. Cutaneous histoplasmosis is almost always a feature of disseminated disease, which occurs most commonly in immunosuppressed individuals.1 Skin lesions may present as macules, papules, indurated plaques, ulcers, purpura, panniculitis, and subcutaneous nodules.2 Histologically, there is a granulomatous and neutrophilic infiltrate within the dermis and subcutis. Yeasts are small (2-4 µm in diameter) and are observed within the cytoplasm of macrophages (Figure 5) where they appear as basophilic dots, sometimes surrounded by an artifactual clear space (pseudocapsule).2
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier/Saunders; 2012.
- Schwarzenberger K, Werchniak A, Ko C. Requisites in Dermatology: General Dermatology. Philadelphia, PA: Elsevier/Saunders; 2009.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Calonje JE, Brenn T, Lazar AJ, et al. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier/Saunders; 2012.
- Schwarzenberger K, Werchniak A, Ko C. Requisites in Dermatology: General Dermatology. Philadelphia, PA: Elsevier/Saunders; 2009.
Aquatic Antagonists: Cutaneous Sea Urchin Spine Injury
Sea urchin injuries are commonly seen in coastal regions near both warm and cold salt water with frequent recreational water activities or fishing. Sea urchins belong to the class Echinoidea with approximately 600 species, of which roughly 80 are poisonous to humans.1,2 When a human comes in contact with a sea urchin, the spines of the sea urchin (made of calcium carbonate) can penetrate the skin and break off from the sea urchin, becoming embedded in the skin. Injuries from sea urchin spines are most commonly seen on the hands and feet, as the likelihood of contact with a sea urchin is greater on these sites. The severity of sea urchin spine injuries can vary widely, from minimal local trauma and pain to arthritis, synovitis, and occasionally systemic illness.1,3 It is important to recognize the wide variety of responses to sea urchin spine injuries and the impact of prompt treatment. Many published reports on injuries from sea urchin spines describe arthritis and synovitis from spines in the joints.1,2,4-6 Fewer reports discuss nonjoint injuries and the dermatologic aspects of sea urchin spine injuries.3,7,8 We pre-sent a case of a patient with a puncture injury from sea urchin spines that resulted in painful granulomas.
Case Report
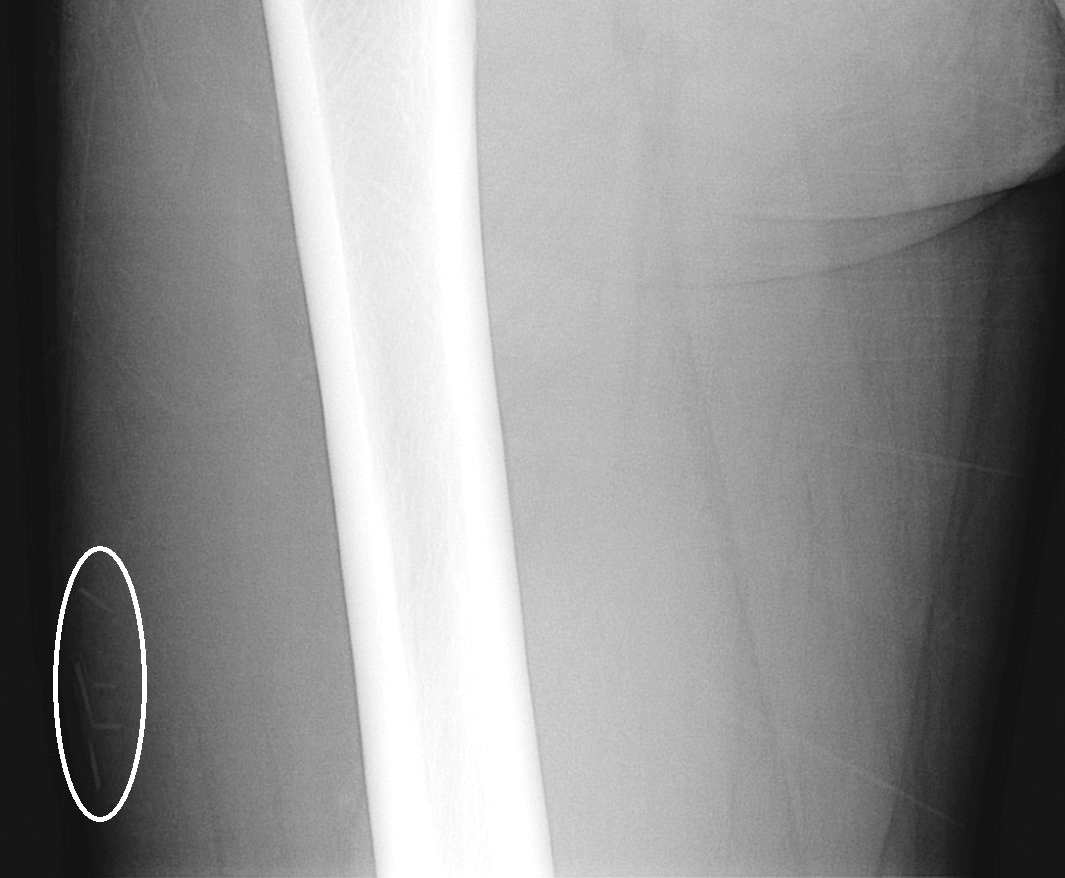
A 29-year-old otherwise healthy man was referred to our dermatology clinic by the university student health center due to continued pain in the right thigh. Five weeks prior to presentation to the student health center, the patient had fallen on a sea urchin while snorkeling in Hawaii. Sea urchin spines became lodged in the right thigh, some of which were removed in a local medical clinic in Hawaii. He was given oral antibiotics prior to his return home. A plain film radiograph of the affected area ordered by the student health center showed several punctate and linear densities in the lateral aspect of the right mid thigh (Figure 1). These findings were consistent with sea urchin spines within the superficial soft tissues of the lateral thigh.
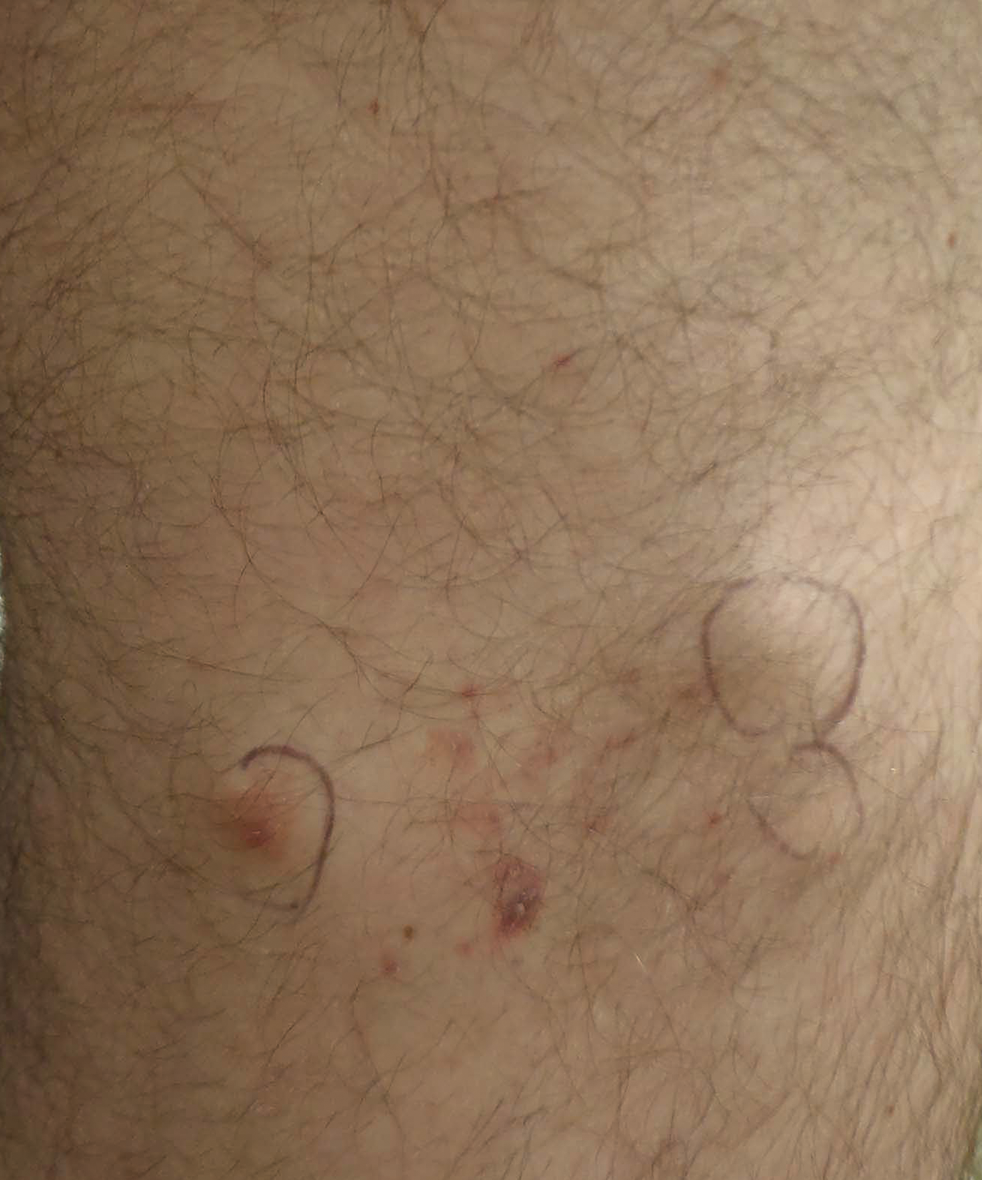
At the time of presentation to our dermatology clinic, the patient reported sharp intermittent pain localized to the right thigh. The patient denied any fever, chills, or pain in the joints. On physical examination, there were several firm nodules on the right thigh, ranging from 4 to 20 mm in diameter (Figure 2). The nodules were tender to palpation with some surrounding edema. Drainage was not noted. Several scars were visible at sites of the original puncture injuries and removal of the spines.
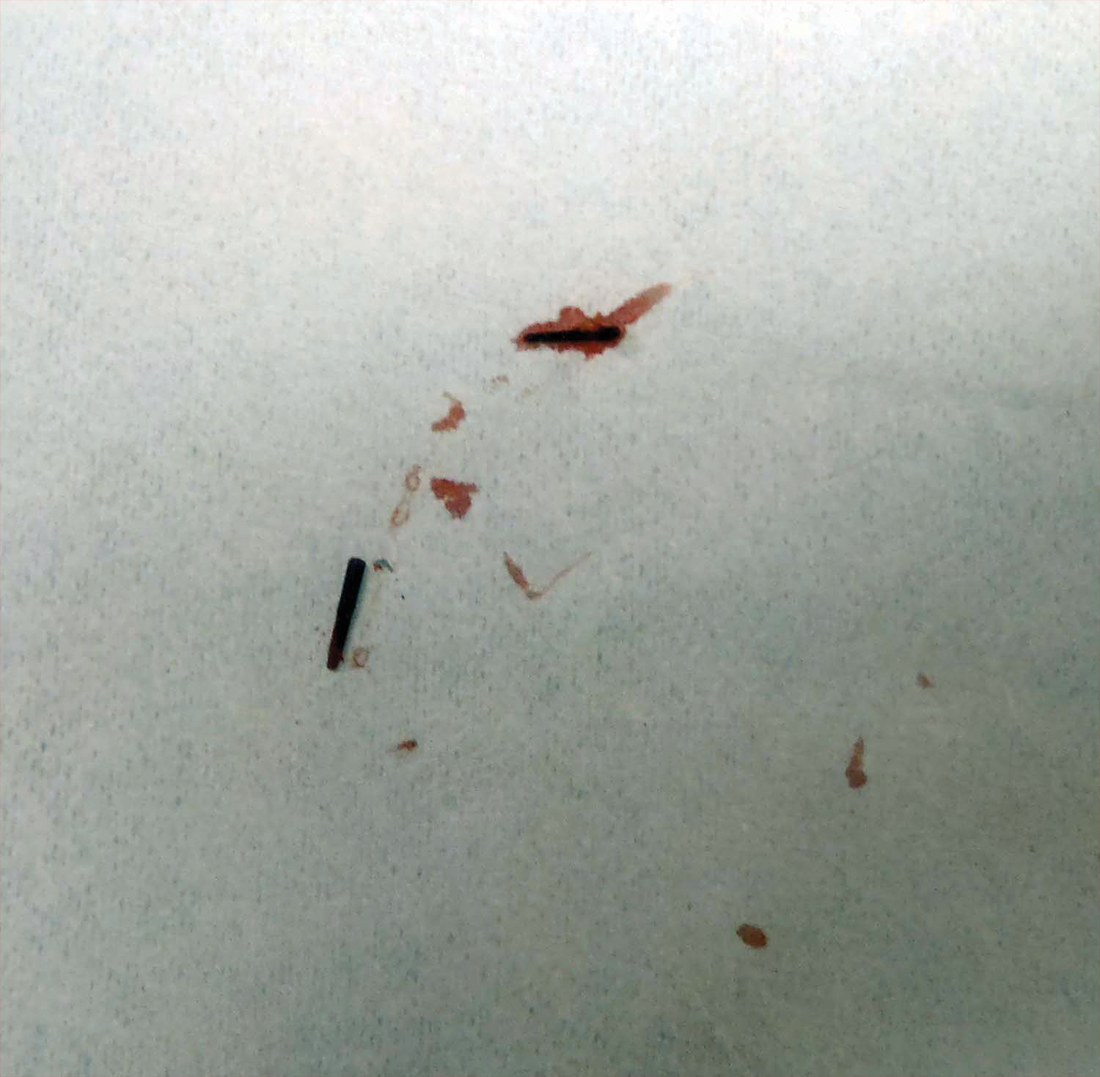
Two 6-mm punch biopsies were performed on representative nodules on the right thigh for histopathologic examination. Along with the biopsy tissue, firm, brown-black, linear foreign bodies consistent with sea urchin spines were extracted with forceps (Figure 3). Histopathologic examination revealed a dense, diffuse, mixed inflammatory cell infiltrate in the dermis predominantly composed of lymphocytes, histiocytes, and numerous eosinophils. Proliferation of small vessels was noted. In one of the biopsies, small fragments of necrotic tissue were present. These findings were consistent with granulomatous inflammation and granulation tissue due to a foreign body.
At the time of suture removal 2 weeks later, the biopsied areas were well healed with minimal erythema. The patient reported decreased pain in the involved areas. He was not seen in clinic again due to resolution of the nodules and associated pain.
Comment
Sea urchin spine injuries are commonly seen in coastal regions with frequent participation in recreational and occupational water activities. A wide variety of responses can be seen in sea urchin spine injuries. There generally are 2 types of cutaneous reaction patterns to sea urchin spines: a primary initial reaction and a secondary delayed/granulomatous reaction. When the spines initially penetrate the skin, the primary initial reaction consists of sharp localized pain that worsens with applied pressure. In addition to pain, bleeding, erythema, edema, and myalgia can occur.3 These symptoms typically subside a few hours after complete removal of the spines from the skin.6 If some spines remain in the skin, a secondary delayed/granulomatous reaction can occur, which can lead to the formation of granulomas that can manifest as nodules or papules and can be diffuse.
Many patients may think their painful encounter with a sea urchin was just an unfortunate event, but depending on the location of the injury, more serious extracutaneous reactions and chronic symptoms may occur. Some cases have described the development of arthritis and synovitis from the implantation of spines into joints.1,2,4-6 Other extracutaneous complications include neuropathy and paresthesia, local bone destruction, radiating pain, muscular weakness, and hypotension.3
The severity of the injury also can depend on the sea urchin species and the number of spines implanted. There are approximately 80 poisonous sea urchin species possessing toxins in venomous spines, resulting in edema and change in the leukocyte-endothelial interaction.9 Substances identified in the spines include proteins, steroids, serotonin, histamine, and glycosides.3,9 The number of spines implanted, particularly the number of venomous spines, can lead to more severe complications. Penetration of 15 or more venomous spines can commonly lead to extracutaneous symptoms.3 Another concern, irrespective of species type, is the potential for secondary infection associated with the spine penetration or implantation into the skin. Mycobacterium marinum infections have been reported in some sea urchin granulomas,10 as well as fungal infection, bacterial infection, and tetanus.3
The diagnosis of sea urchin spine injuries starts with a thorough history and physical examination. A positive history of sea urchin contact suggests the diagnosis, and radiographs can be useful to find the location of the spine(s), especially if there are no visible nodules on the skin. However, small fragments of spine may not be completely observed on plain radiographs. Any signs or symptoms of infection should prompt a culture for confirmation and guidance for management. Cutaneous biopsies can be helpful for both diagnosis confirmation and symptomatic relief. Reported cases have described granulomatous reactions in the vast majority of the histologic specimens, with necrosis an additional common finding.7,8 Sea urchin granulomas can be of varying types, the majority being foreign-body and sarcoid types.3,6,7
Treatment of sea urchin spine injuries primarily involves removal of the spines by a physician. Patients may soak the affected areas in warm water prior to the removal of the spines to aid in pain relief. Surgical removal with local anesthesia and cutaneous extraction is a common treatment method, and more extensive surgical removal of the spines is another option, especially in areas around the joints.2 The use of liquid nitrogen or skin punch biopsy also have been described as possible methods to remove the spines.11,12
Conclusion
Sea urchin spine injuries can result in a wide range of cutaneous and systemic complications. Prompt diagnosis and treatment to remove the sea urchin spines can lessen the associated pain and is important in the prevention of more serious complications.
- Liram N, Gomori M, Perouansky M. Sea urchin puncture resulting in PIP joint synovial arthritis: case report and MRI study. J Travel Med. 2000;7:43-45.
- Dahl WJ, Jebson P, Louis DS. Sea urchin injuries to the hand: a case report and review of the literature. Iowa Orthop J. 2010;30:153-156.
- Rossetto AL, de Macedo Mora J, Haddad Junior V. Sea urchin granuloma. Rev Inst Med Trop Sao Paulo. 2006;48:303-306.
- Ahmad R, McCann PA, Barakat M, et al. Sea urchin spine injuries of the hand. J Hand Surg Eur Vol. 2008;33:670-671.
- Schefflein J, Umans H, Ellenbogen D, et al. Sea urchin spine arthritis in the foot. Skeletal Radiol. 2012;41:1327-1331.
- Wada T, Soma T, Gaman K, et al. Sea urchin spine arthritis of the hand. J Hand Surg. 2008;33:398-401.
- Suárez-Peñaranda JM, Vieites B, Del Río E, et al. Histopathologic and immunohistochemical features of sea urchin granulomas. J Cutan Pathol. 2013;40:550-556.
- De La Torre C, Toribio J. Sea-urchin granuloma: histologic profile. a pathologic study of 50 biopsies. J Cutan Pathol. 2001;28:223-228.
- Sciani JM, Zychar BC, Gonçalves LR, et al. Pro-inflammatory effects of the aqueous extract of Echinometra lucunter sea urchin spines. Exp Biol Med (Maywood). 2011;236:277-280.
- De la Torre C, Vega A, Carracedo A, et al. Identification of Mycobacterium marinum in sea-urchin granulomas. Br J Dermatol. 2001;145:114-116.
- Gargus MD, Morohashi DK. A sea-urchin spine chilling remedy. N Engl J Med. 2012;367:1867-1868.
- Sjøberg T, de Weerd L. The usefulness of a skin biopsy punch to remove sea urchin spines. ANZ J Surg. 2010;80:383.
Sea urchin injuries are commonly seen in coastal regions near both warm and cold salt water with frequent recreational water activities or fishing. Sea urchins belong to the class Echinoidea with approximately 600 species, of which roughly 80 are poisonous to humans.1,2 When a human comes in contact with a sea urchin, the spines of the sea urchin (made of calcium carbonate) can penetrate the skin and break off from the sea urchin, becoming embedded in the skin. Injuries from sea urchin spines are most commonly seen on the hands and feet, as the likelihood of contact with a sea urchin is greater on these sites. The severity of sea urchin spine injuries can vary widely, from minimal local trauma and pain to arthritis, synovitis, and occasionally systemic illness.1,3 It is important to recognize the wide variety of responses to sea urchin spine injuries and the impact of prompt treatment. Many published reports on injuries from sea urchin spines describe arthritis and synovitis from spines in the joints.1,2,4-6 Fewer reports discuss nonjoint injuries and the dermatologic aspects of sea urchin spine injuries.3,7,8 We pre-sent a case of a patient with a puncture injury from sea urchin spines that resulted in painful granulomas.
Case Report
A 29-year-old otherwise healthy man was referred to our dermatology clinic by the university student health center due to continued pain in the right thigh. Five weeks prior to presentation to the student health center, the patient had fallen on a sea urchin while snorkeling in Hawaii. Sea urchin spines became lodged in the right thigh, some of which were removed in a local medical clinic in Hawaii. He was given oral antibiotics prior to his return home. A plain film radiograph of the affected area ordered by the student health center showed several punctate and linear densities in the lateral aspect of the right mid thigh (Figure 1). These findings were consistent with sea urchin spines within the superficial soft tissues of the lateral thigh.
At the time of presentation to our dermatology clinic, the patient reported sharp intermittent pain localized to the right thigh. The patient denied any fever, chills, or pain in the joints. On physical examination, there were several firm nodules on the right thigh, ranging from 4 to 20 mm in diameter (Figure 2). The nodules were tender to palpation with some surrounding edema. Drainage was not noted. Several scars were visible at sites of the original puncture injuries and removal of the spines.
Two 6-mm punch biopsies were performed on representative nodules on the right thigh for histopathologic examination. Along with the biopsy tissue, firm, brown-black, linear foreign bodies consistent with sea urchin spines were extracted with forceps (Figure 3). Histopathologic examination revealed a dense, diffuse, mixed inflammatory cell infiltrate in the dermis predominantly composed of lymphocytes, histiocytes, and numerous eosinophils. Proliferation of small vessels was noted. In one of the biopsies, small fragments of necrotic tissue were present. These findings were consistent with granulomatous inflammation and granulation tissue due to a foreign body.
At the time of suture removal 2 weeks later, the biopsied areas were well healed with minimal erythema. The patient reported decreased pain in the involved areas. He was not seen in clinic again due to resolution of the nodules and associated pain.
Comment
Sea urchin spine injuries are commonly seen in coastal regions with frequent participation in recreational and occupational water activities. A wide variety of responses can be seen in sea urchin spine injuries. There generally are 2 types of cutaneous reaction patterns to sea urchin spines: a primary initial reaction and a secondary delayed/granulomatous reaction. When the spines initially penetrate the skin, the primary initial reaction consists of sharp localized pain that worsens with applied pressure. In addition to pain, bleeding, erythema, edema, and myalgia can occur.3 These symptoms typically subside a few hours after complete removal of the spines from the skin.6 If some spines remain in the skin, a secondary delayed/granulomatous reaction can occur, which can lead to the formation of granulomas that can manifest as nodules or papules and can be diffuse.
Many patients may think their painful encounter with a sea urchin was just an unfortunate event, but depending on the location of the injury, more serious extracutaneous reactions and chronic symptoms may occur. Some cases have described the development of arthritis and synovitis from the implantation of spines into joints.1,2,4-6 Other extracutaneous complications include neuropathy and paresthesia, local bone destruction, radiating pain, muscular weakness, and hypotension.3
The severity of the injury also can depend on the sea urchin species and the number of spines implanted. There are approximately 80 poisonous sea urchin species possessing toxins in venomous spines, resulting in edema and change in the leukocyte-endothelial interaction.9 Substances identified in the spines include proteins, steroids, serotonin, histamine, and glycosides.3,9 The number of spines implanted, particularly the number of venomous spines, can lead to more severe complications. Penetration of 15 or more venomous spines can commonly lead to extracutaneous symptoms.3 Another concern, irrespective of species type, is the potential for secondary infection associated with the spine penetration or implantation into the skin. Mycobacterium marinum infections have been reported in some sea urchin granulomas,10 as well as fungal infection, bacterial infection, and tetanus.3
The diagnosis of sea urchin spine injuries starts with a thorough history and physical examination. A positive history of sea urchin contact suggests the diagnosis, and radiographs can be useful to find the location of the spine(s), especially if there are no visible nodules on the skin. However, small fragments of spine may not be completely observed on plain radiographs. Any signs or symptoms of infection should prompt a culture for confirmation and guidance for management. Cutaneous biopsies can be helpful for both diagnosis confirmation and symptomatic relief. Reported cases have described granulomatous reactions in the vast majority of the histologic specimens, with necrosis an additional common finding.7,8 Sea urchin granulomas can be of varying types, the majority being foreign-body and sarcoid types.3,6,7
Treatment of sea urchin spine injuries primarily involves removal of the spines by a physician. Patients may soak the affected areas in warm water prior to the removal of the spines to aid in pain relief. Surgical removal with local anesthesia and cutaneous extraction is a common treatment method, and more extensive surgical removal of the spines is another option, especially in areas around the joints.2 The use of liquid nitrogen or skin punch biopsy also have been described as possible methods to remove the spines.11,12
Conclusion
Sea urchin spine injuries can result in a wide range of cutaneous and systemic complications. Prompt diagnosis and treatment to remove the sea urchin spines can lessen the associated pain and is important in the prevention of more serious complications.
Sea urchin injuries are commonly seen in coastal regions near both warm and cold salt water with frequent recreational water activities or fishing. Sea urchins belong to the class Echinoidea with approximately 600 species, of which roughly 80 are poisonous to humans.1,2 When a human comes in contact with a sea urchin, the spines of the sea urchin (made of calcium carbonate) can penetrate the skin and break off from the sea urchin, becoming embedded in the skin. Injuries from sea urchin spines are most commonly seen on the hands and feet, as the likelihood of contact with a sea urchin is greater on these sites. The severity of sea urchin spine injuries can vary widely, from minimal local trauma and pain to arthritis, synovitis, and occasionally systemic illness.1,3 It is important to recognize the wide variety of responses to sea urchin spine injuries and the impact of prompt treatment. Many published reports on injuries from sea urchin spines describe arthritis and synovitis from spines in the joints.1,2,4-6 Fewer reports discuss nonjoint injuries and the dermatologic aspects of sea urchin spine injuries.3,7,8 We pre-sent a case of a patient with a puncture injury from sea urchin spines that resulted in painful granulomas.
Case Report
A 29-year-old otherwise healthy man was referred to our dermatology clinic by the university student health center due to continued pain in the right thigh. Five weeks prior to presentation to the student health center, the patient had fallen on a sea urchin while snorkeling in Hawaii. Sea urchin spines became lodged in the right thigh, some of which were removed in a local medical clinic in Hawaii. He was given oral antibiotics prior to his return home. A plain film radiograph of the affected area ordered by the student health center showed several punctate and linear densities in the lateral aspect of the right mid thigh (Figure 1). These findings were consistent with sea urchin spines within the superficial soft tissues of the lateral thigh.
At the time of presentation to our dermatology clinic, the patient reported sharp intermittent pain localized to the right thigh. The patient denied any fever, chills, or pain in the joints. On physical examination, there were several firm nodules on the right thigh, ranging from 4 to 20 mm in diameter (Figure 2). The nodules were tender to palpation with some surrounding edema. Drainage was not noted. Several scars were visible at sites of the original puncture injuries and removal of the spines.
Two 6-mm punch biopsies were performed on representative nodules on the right thigh for histopathologic examination. Along with the biopsy tissue, firm, brown-black, linear foreign bodies consistent with sea urchin spines were extracted with forceps (Figure 3). Histopathologic examination revealed a dense, diffuse, mixed inflammatory cell infiltrate in the dermis predominantly composed of lymphocytes, histiocytes, and numerous eosinophils. Proliferation of small vessels was noted. In one of the biopsies, small fragments of necrotic tissue were present. These findings were consistent with granulomatous inflammation and granulation tissue due to a foreign body.
At the time of suture removal 2 weeks later, the biopsied areas were well healed with minimal erythema. The patient reported decreased pain in the involved areas. He was not seen in clinic again due to resolution of the nodules and associated pain.
Comment
Sea urchin spine injuries are commonly seen in coastal regions with frequent participation in recreational and occupational water activities. A wide variety of responses can be seen in sea urchin spine injuries. There generally are 2 types of cutaneous reaction patterns to sea urchin spines: a primary initial reaction and a secondary delayed/granulomatous reaction. When the spines initially penetrate the skin, the primary initial reaction consists of sharp localized pain that worsens with applied pressure. In addition to pain, bleeding, erythema, edema, and myalgia can occur.3 These symptoms typically subside a few hours after complete removal of the spines from the skin.6 If some spines remain in the skin, a secondary delayed/granulomatous reaction can occur, which can lead to the formation of granulomas that can manifest as nodules or papules and can be diffuse.
Many patients may think their painful encounter with a sea urchin was just an unfortunate event, but depending on the location of the injury, more serious extracutaneous reactions and chronic symptoms may occur. Some cases have described the development of arthritis and synovitis from the implantation of spines into joints.1,2,4-6 Other extracutaneous complications include neuropathy and paresthesia, local bone destruction, radiating pain, muscular weakness, and hypotension.3
The severity of the injury also can depend on the sea urchin species and the number of spines implanted. There are approximately 80 poisonous sea urchin species possessing toxins in venomous spines, resulting in edema and change in the leukocyte-endothelial interaction.9 Substances identified in the spines include proteins, steroids, serotonin, histamine, and glycosides.3,9 The number of spines implanted, particularly the number of venomous spines, can lead to more severe complications. Penetration of 15 or more venomous spines can commonly lead to extracutaneous symptoms.3 Another concern, irrespective of species type, is the potential for secondary infection associated with the spine penetration or implantation into the skin. Mycobacterium marinum infections have been reported in some sea urchin granulomas,10 as well as fungal infection, bacterial infection, and tetanus.3
The diagnosis of sea urchin spine injuries starts with a thorough history and physical examination. A positive history of sea urchin contact suggests the diagnosis, and radiographs can be useful to find the location of the spine(s), especially if there are no visible nodules on the skin. However, small fragments of spine may not be completely observed on plain radiographs. Any signs or symptoms of infection should prompt a culture for confirmation and guidance for management. Cutaneous biopsies can be helpful for both diagnosis confirmation and symptomatic relief. Reported cases have described granulomatous reactions in the vast majority of the histologic specimens, with necrosis an additional common finding.7,8 Sea urchin granulomas can be of varying types, the majority being foreign-body and sarcoid types.3,6,7
Treatment of sea urchin spine injuries primarily involves removal of the spines by a physician. Patients may soak the affected areas in warm water prior to the removal of the spines to aid in pain relief. Surgical removal with local anesthesia and cutaneous extraction is a common treatment method, and more extensive surgical removal of the spines is another option, especially in areas around the joints.2 The use of liquid nitrogen or skin punch biopsy also have been described as possible methods to remove the spines.11,12
Conclusion
Sea urchin spine injuries can result in a wide range of cutaneous and systemic complications. Prompt diagnosis and treatment to remove the sea urchin spines can lessen the associated pain and is important in the prevention of more serious complications.
- Liram N, Gomori M, Perouansky M. Sea urchin puncture resulting in PIP joint synovial arthritis: case report and MRI study. J Travel Med. 2000;7:43-45.
- Dahl WJ, Jebson P, Louis DS. Sea urchin injuries to the hand: a case report and review of the literature. Iowa Orthop J. 2010;30:153-156.
- Rossetto AL, de Macedo Mora J, Haddad Junior V. Sea urchin granuloma. Rev Inst Med Trop Sao Paulo. 2006;48:303-306.
- Ahmad R, McCann PA, Barakat M, et al. Sea urchin spine injuries of the hand. J Hand Surg Eur Vol. 2008;33:670-671.
- Schefflein J, Umans H, Ellenbogen D, et al. Sea urchin spine arthritis in the foot. Skeletal Radiol. 2012;41:1327-1331.
- Wada T, Soma T, Gaman K, et al. Sea urchin spine arthritis of the hand. J Hand Surg. 2008;33:398-401.
- Suárez-Peñaranda JM, Vieites B, Del Río E, et al. Histopathologic and immunohistochemical features of sea urchin granulomas. J Cutan Pathol. 2013;40:550-556.
- De La Torre C, Toribio J. Sea-urchin granuloma: histologic profile. a pathologic study of 50 biopsies. J Cutan Pathol. 2001;28:223-228.
- Sciani JM, Zychar BC, Gonçalves LR, et al. Pro-inflammatory effects of the aqueous extract of Echinometra lucunter sea urchin spines. Exp Biol Med (Maywood). 2011;236:277-280.
- De la Torre C, Vega A, Carracedo A, et al. Identification of Mycobacterium marinum in sea-urchin granulomas. Br J Dermatol. 2001;145:114-116.
- Gargus MD, Morohashi DK. A sea-urchin spine chilling remedy. N Engl J Med. 2012;367:1867-1868.
- Sjøberg T, de Weerd L. The usefulness of a skin biopsy punch to remove sea urchin spines. ANZ J Surg. 2010;80:383.
- Liram N, Gomori M, Perouansky M. Sea urchin puncture resulting in PIP joint synovial arthritis: case report and MRI study. J Travel Med. 2000;7:43-45.
- Dahl WJ, Jebson P, Louis DS. Sea urchin injuries to the hand: a case report and review of the literature. Iowa Orthop J. 2010;30:153-156.
- Rossetto AL, de Macedo Mora J, Haddad Junior V. Sea urchin granuloma. Rev Inst Med Trop Sao Paulo. 2006;48:303-306.
- Ahmad R, McCann PA, Barakat M, et al. Sea urchin spine injuries of the hand. J Hand Surg Eur Vol. 2008;33:670-671.
- Schefflein J, Umans H, Ellenbogen D, et al. Sea urchin spine arthritis in the foot. Skeletal Radiol. 2012;41:1327-1331.
- Wada T, Soma T, Gaman K, et al. Sea urchin spine arthritis of the hand. J Hand Surg. 2008;33:398-401.
- Suárez-Peñaranda JM, Vieites B, Del Río E, et al. Histopathologic and immunohistochemical features of sea urchin granulomas. J Cutan Pathol. 2013;40:550-556.
- De La Torre C, Toribio J. Sea-urchin granuloma: histologic profile. a pathologic study of 50 biopsies. J Cutan Pathol. 2001;28:223-228.
- Sciani JM, Zychar BC, Gonçalves LR, et al. Pro-inflammatory effects of the aqueous extract of Echinometra lucunter sea urchin spines. Exp Biol Med (Maywood). 2011;236:277-280.
- De la Torre C, Vega A, Carracedo A, et al. Identification of Mycobacterium marinum in sea-urchin granulomas. Br J Dermatol. 2001;145:114-116.
- Gargus MD, Morohashi DK. A sea-urchin spine chilling remedy. N Engl J Med. 2012;367:1867-1868.
- Sjøberg T, de Weerd L. The usefulness of a skin biopsy punch to remove sea urchin spines. ANZ J Surg. 2010;80:383.
Practice Points
- Radiographic imaging may aid in the identification of sea urchin spines, especially if there are no visible or palpable skin nodules.
- Treatment of sea urchin spine injuries typically involves surgical removal of the spines with local anesthesia and cutaneous extraction.
- Prompt extraction of sea urchin spines can improve pain symptoms and decrease the likelihood of granuloma formation, infection, and extracutaneous complications.
Necrotic Lesion of the Ear
The Diagnosis: Chondrodermatitis Nodularis Chronica Helicis
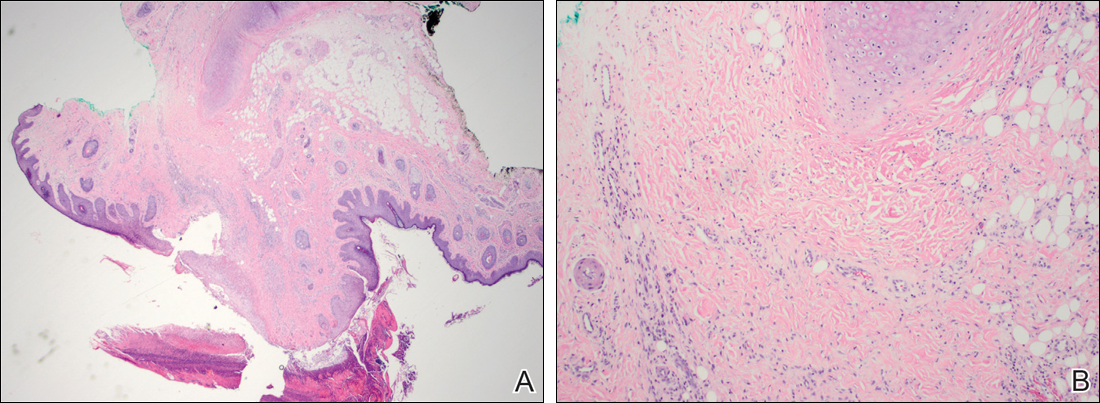
Histopathologic examination revealed focal epidermal erosion and ulceration directly overlying the hyaline cartilage with degenerative changes (Figure). The dermis was relatively noninflamed with fibroplasia of the vasculature. The blood vessels indirectly beneath the ulceration were found to be unremarkable with no indications of fibrinoid necrosis, vasculitis, or the presence of thrombi. The patient was informed of the diagnosis, at which point she reported that she slept on the right side. The excisional biopsy site healed well without recurrence of chondrodermatitis nodularis chronica helicis (CNH).
Chondrodermatitis nodularis chronica helicis, also known as clavus helicis, is a benign, usually solitary, painful lesion. Historically, it was first described in 1915 by Winkler1 and in the 1960s the most common documented cases were attributed to the headpieces of telephone operators and nuns.2 In the early 2000s, cell phones were determined to be a growing cause.3 Chondrodermatitis nodularis chronica helicis is most commonly found on the helix with the antihelix being affected less often.4 The condition is more common in men, with a male to female ratio being reported as high as 10:1. Possible causes of this disorder stem from damage to cartilage associated with pressure, sun exposure, cold temperatures, and microvascular disease. Additionally, some researchers have hypothesized that the cartilaginous damage resulting from solar elastosis and minor trauma leaves a susceptibility to CNH. This disorder usually presents as a small, exquisitely tender nodule that may ulcerate and crust.4 Chondrodermatitis nodularis chronica helicis may be mistaken for basal cell carcinoma, squamous cell carcinoma, actinic keratosis, and weathering nodules, though CNH tends to be more painful.
The diagnosis of CNH often is clinical but may require a skin biopsy. Histopathology of CNH shows a benign inflammatory lesion with an acanthotic hyperkeratotic epidermis that may be ulcerated. A primarily lymphocytic infiltrate usually is observed with variable presence of histiocytes and neutrophils. Cartilaginous changes range from simple perichondral thickening to notable areas of degeneration with calcification and ossification.4
Although the diagnosis of CNH often is straightforward, the remarkable necrosis present in our case made for an interesting differential diagnosis. Pernio, cryoglobulinemia, and levamisole-induced vasculopathy were all considered. Pernio, caused by cold-induced vasoconstriction and hypoxemia, classically presents as erythematous lesions with a symmetrical distribution on acral sites.5 Cryoglobulinemia involves proteins that precipitate at cold temperatures causing damage via an occlusive vasculopathy or an immune complex-mediated vasculitis. The presence of cryoglobulinemia is strongly associated with concomitant hepatitis C virus infection.6 Ulcerated and purpuric lesions of cryoglobulinemia may become necrotic. Levamisole is a veterinary antihelminthic drug and common cocaine contaminant, often added to cocaine as a cutting agent. Levamisole-induced vasculopathy favors acral sites and often is noted on the ears as purpuric patches, sometimes with necrosis.7
Several therapies for CNH have been reported with variable effectiveness.8 First-line treatments are the use of pressure-relieving devices including a doughnut-shaped pillow during sleep and intralesional corticosteroids.9 Surgical treatments including cryotherapy, simple excision, electrodesiccation and curettage, wedge resection with helical rim advancement flap, punch and graft technique, and CO2 laser have been tried.8 Photodynamic therapy and topical nitroglycerine also have shown to be of benefit.8,9
Our case of CNH is unique because of the remarkable degree of necrosis present on clinical examination. Chondrodermatitis nodularis chronica helicis with such an impressive necrotic presentation is rare. We speculate that the patient's underlying hypercoagulable state may have contributed to the dramatic presentation. It is important to keep CNH in mind when evaluating any necrotic lesion on the ear.
- Winkler M. Knötcehnformige Erkrankung am helix. chondrodermatitis nodularis chronic helicis. Arch für Dermatologie und Syphilis. 1915;121:278-285.
- Barker L, Young AW, Sachs W. Chondrodermatitis of the ears: a differential study of nodules of the helix and antihelix. Arch Dermatol. 1960;81:15-25.
- Elgart M. Cell phone chondrodermatitis. Arch Dermatol. 2000;136:1568.
- Cribier B, Scrivener Y, Peltre B. Neural hyperplasia in chondrodermatitis nodularis chronica helicis. J Am Acad Dermatol. 2006;55:844-848.
- King JM, Plotner AN, Adams BB. Perniosis induced by a cold-therapy system. Arch Dermatol. 2012;148:1101-1102.
- Berk DR, Mallory SB, Keeffe EB, et al. Dermatologic disorders associated with chronic hepatitis C: effect of interferon therapy. Clin Gastroenterol Hepatol. 2007;5:142-151.
- Hennings C, Miller J. Illicit drugs: what dermatologists need to know. J Am Acad Dermatol. 2013;69:135-142.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;64:531-536.
- Gilaberte Y, Frias M, Pérez-Lorenz J. Chondrodermatitis nodularis helicis successfully treated with photodynamic therapy. Arch Dermatol. 2010;146:1080-1082.
The Diagnosis: Chondrodermatitis Nodularis Chronica Helicis
Histopathologic examination revealed focal epidermal erosion and ulceration directly overlying the hyaline cartilage with degenerative changes (Figure). The dermis was relatively noninflamed with fibroplasia of the vasculature. The blood vessels indirectly beneath the ulceration were found to be unremarkable with no indications of fibrinoid necrosis, vasculitis, or the presence of thrombi. The patient was informed of the diagnosis, at which point she reported that she slept on the right side. The excisional biopsy site healed well without recurrence of chondrodermatitis nodularis chronica helicis (CNH).
Chondrodermatitis nodularis chronica helicis, also known as clavus helicis, is a benign, usually solitary, painful lesion. Historically, it was first described in 1915 by Winkler1 and in the 1960s the most common documented cases were attributed to the headpieces of telephone operators and nuns.2 In the early 2000s, cell phones were determined to be a growing cause.3 Chondrodermatitis nodularis chronica helicis is most commonly found on the helix with the antihelix being affected less often.4 The condition is more common in men, with a male to female ratio being reported as high as 10:1. Possible causes of this disorder stem from damage to cartilage associated with pressure, sun exposure, cold temperatures, and microvascular disease. Additionally, some researchers have hypothesized that the cartilaginous damage resulting from solar elastosis and minor trauma leaves a susceptibility to CNH. This disorder usually presents as a small, exquisitely tender nodule that may ulcerate and crust.4 Chondrodermatitis nodularis chronica helicis may be mistaken for basal cell carcinoma, squamous cell carcinoma, actinic keratosis, and weathering nodules, though CNH tends to be more painful.
The diagnosis of CNH often is clinical but may require a skin biopsy. Histopathology of CNH shows a benign inflammatory lesion with an acanthotic hyperkeratotic epidermis that may be ulcerated. A primarily lymphocytic infiltrate usually is observed with variable presence of histiocytes and neutrophils. Cartilaginous changes range from simple perichondral thickening to notable areas of degeneration with calcification and ossification.4
Although the diagnosis of CNH often is straightforward, the remarkable necrosis present in our case made for an interesting differential diagnosis. Pernio, cryoglobulinemia, and levamisole-induced vasculopathy were all considered. Pernio, caused by cold-induced vasoconstriction and hypoxemia, classically presents as erythematous lesions with a symmetrical distribution on acral sites.5 Cryoglobulinemia involves proteins that precipitate at cold temperatures causing damage via an occlusive vasculopathy or an immune complex-mediated vasculitis. The presence of cryoglobulinemia is strongly associated with concomitant hepatitis C virus infection.6 Ulcerated and purpuric lesions of cryoglobulinemia may become necrotic. Levamisole is a veterinary antihelminthic drug and common cocaine contaminant, often added to cocaine as a cutting agent. Levamisole-induced vasculopathy favors acral sites and often is noted on the ears as purpuric patches, sometimes with necrosis.7
Several therapies for CNH have been reported with variable effectiveness.8 First-line treatments are the use of pressure-relieving devices including a doughnut-shaped pillow during sleep and intralesional corticosteroids.9 Surgical treatments including cryotherapy, simple excision, electrodesiccation and curettage, wedge resection with helical rim advancement flap, punch and graft technique, and CO2 laser have been tried.8 Photodynamic therapy and topical nitroglycerine also have shown to be of benefit.8,9
Our case of CNH is unique because of the remarkable degree of necrosis present on clinical examination. Chondrodermatitis nodularis chronica helicis with such an impressive necrotic presentation is rare. We speculate that the patient's underlying hypercoagulable state may have contributed to the dramatic presentation. It is important to keep CNH in mind when evaluating any necrotic lesion on the ear.
The Diagnosis: Chondrodermatitis Nodularis Chronica Helicis
Histopathologic examination revealed focal epidermal erosion and ulceration directly overlying the hyaline cartilage with degenerative changes (Figure). The dermis was relatively noninflamed with fibroplasia of the vasculature. The blood vessels indirectly beneath the ulceration were found to be unremarkable with no indications of fibrinoid necrosis, vasculitis, or the presence of thrombi. The patient was informed of the diagnosis, at which point she reported that she slept on the right side. The excisional biopsy site healed well without recurrence of chondrodermatitis nodularis chronica helicis (CNH).
Chondrodermatitis nodularis chronica helicis, also known as clavus helicis, is a benign, usually solitary, painful lesion. Historically, it was first described in 1915 by Winkler1 and in the 1960s the most common documented cases were attributed to the headpieces of telephone operators and nuns.2 In the early 2000s, cell phones were determined to be a growing cause.3 Chondrodermatitis nodularis chronica helicis is most commonly found on the helix with the antihelix being affected less often.4 The condition is more common in men, with a male to female ratio being reported as high as 10:1. Possible causes of this disorder stem from damage to cartilage associated with pressure, sun exposure, cold temperatures, and microvascular disease. Additionally, some researchers have hypothesized that the cartilaginous damage resulting from solar elastosis and minor trauma leaves a susceptibility to CNH. This disorder usually presents as a small, exquisitely tender nodule that may ulcerate and crust.4 Chondrodermatitis nodularis chronica helicis may be mistaken for basal cell carcinoma, squamous cell carcinoma, actinic keratosis, and weathering nodules, though CNH tends to be more painful.
The diagnosis of CNH often is clinical but may require a skin biopsy. Histopathology of CNH shows a benign inflammatory lesion with an acanthotic hyperkeratotic epidermis that may be ulcerated. A primarily lymphocytic infiltrate usually is observed with variable presence of histiocytes and neutrophils. Cartilaginous changes range from simple perichondral thickening to notable areas of degeneration with calcification and ossification.4
Although the diagnosis of CNH often is straightforward, the remarkable necrosis present in our case made for an interesting differential diagnosis. Pernio, cryoglobulinemia, and levamisole-induced vasculopathy were all considered. Pernio, caused by cold-induced vasoconstriction and hypoxemia, classically presents as erythematous lesions with a symmetrical distribution on acral sites.5 Cryoglobulinemia involves proteins that precipitate at cold temperatures causing damage via an occlusive vasculopathy or an immune complex-mediated vasculitis. The presence of cryoglobulinemia is strongly associated with concomitant hepatitis C virus infection.6 Ulcerated and purpuric lesions of cryoglobulinemia may become necrotic. Levamisole is a veterinary antihelminthic drug and common cocaine contaminant, often added to cocaine as a cutting agent. Levamisole-induced vasculopathy favors acral sites and often is noted on the ears as purpuric patches, sometimes with necrosis.7
Several therapies for CNH have been reported with variable effectiveness.8 First-line treatments are the use of pressure-relieving devices including a doughnut-shaped pillow during sleep and intralesional corticosteroids.9 Surgical treatments including cryotherapy, simple excision, electrodesiccation and curettage, wedge resection with helical rim advancement flap, punch and graft technique, and CO2 laser have been tried.8 Photodynamic therapy and topical nitroglycerine also have shown to be of benefit.8,9
Our case of CNH is unique because of the remarkable degree of necrosis present on clinical examination. Chondrodermatitis nodularis chronica helicis with such an impressive necrotic presentation is rare. We speculate that the patient's underlying hypercoagulable state may have contributed to the dramatic presentation. It is important to keep CNH in mind when evaluating any necrotic lesion on the ear.
- Winkler M. Knötcehnformige Erkrankung am helix. chondrodermatitis nodularis chronic helicis. Arch für Dermatologie und Syphilis. 1915;121:278-285.
- Barker L, Young AW, Sachs W. Chondrodermatitis of the ears: a differential study of nodules of the helix and antihelix. Arch Dermatol. 1960;81:15-25.
- Elgart M. Cell phone chondrodermatitis. Arch Dermatol. 2000;136:1568.
- Cribier B, Scrivener Y, Peltre B. Neural hyperplasia in chondrodermatitis nodularis chronica helicis. J Am Acad Dermatol. 2006;55:844-848.
- King JM, Plotner AN, Adams BB. Perniosis induced by a cold-therapy system. Arch Dermatol. 2012;148:1101-1102.
- Berk DR, Mallory SB, Keeffe EB, et al. Dermatologic disorders associated with chronic hepatitis C: effect of interferon therapy. Clin Gastroenterol Hepatol. 2007;5:142-151.
- Hennings C, Miller J. Illicit drugs: what dermatologists need to know. J Am Acad Dermatol. 2013;69:135-142.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;64:531-536.
- Gilaberte Y, Frias M, Pérez-Lorenz J. Chondrodermatitis nodularis helicis successfully treated with photodynamic therapy. Arch Dermatol. 2010;146:1080-1082.
- Winkler M. Knötcehnformige Erkrankung am helix. chondrodermatitis nodularis chronic helicis. Arch für Dermatologie und Syphilis. 1915;121:278-285.
- Barker L, Young AW, Sachs W. Chondrodermatitis of the ears: a differential study of nodules of the helix and antihelix. Arch Dermatol. 1960;81:15-25.
- Elgart M. Cell phone chondrodermatitis. Arch Dermatol. 2000;136:1568.
- Cribier B, Scrivener Y, Peltre B. Neural hyperplasia in chondrodermatitis nodularis chronica helicis. J Am Acad Dermatol. 2006;55:844-848.
- King JM, Plotner AN, Adams BB. Perniosis induced by a cold-therapy system. Arch Dermatol. 2012;148:1101-1102.
- Berk DR, Mallory SB, Keeffe EB, et al. Dermatologic disorders associated with chronic hepatitis C: effect of interferon therapy. Clin Gastroenterol Hepatol. 2007;5:142-151.
- Hennings C, Miller J. Illicit drugs: what dermatologists need to know. J Am Acad Dermatol. 2013;69:135-142.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;64:531-536.
- Gilaberte Y, Frias M, Pérez-Lorenz J. Chondrodermatitis nodularis helicis successfully treated with photodynamic therapy. Arch Dermatol. 2010;146:1080-1082.
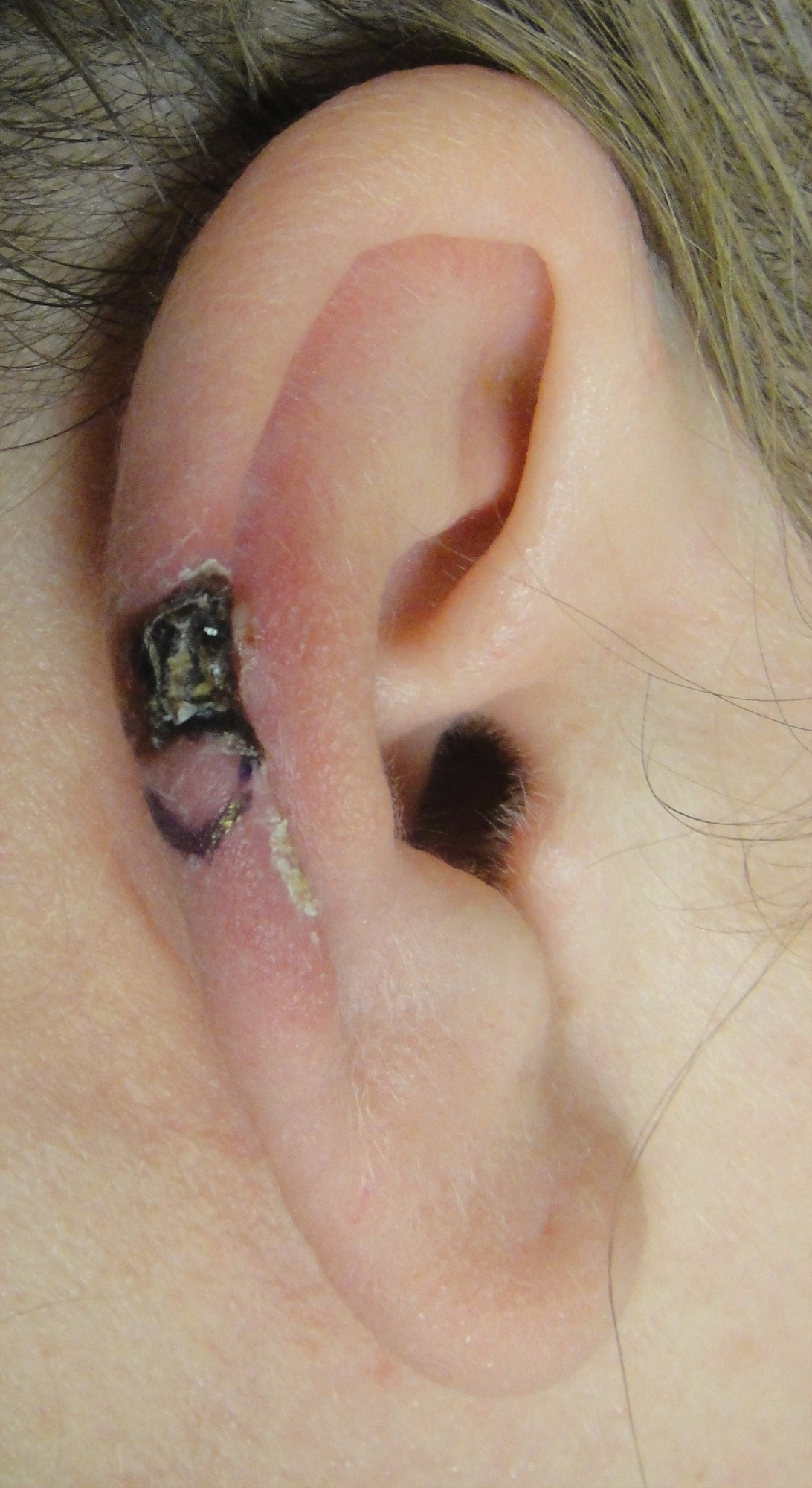
A 43-year-old woman presented with a painful necrotic lesion on the right ear of 1 month's duration. She denied trauma to the ear and had no other skin lesions elsewhere on the body. A course of doxycycline prior to presentation did not result in improvement. Her medical history was remarkable for diabetes mellitus, deep vein thrombosis, depression, and gastroesophageal reflux disease. She had been taking warfarin regularly for years. She denied using recreational drugs. On physical examination, the right ear demonstrated a 6-mm necrotic area with surrounding tender erythema. Examinations of the left ear, face, and legs were normal. An excisional biopsy was performed.
Crusted Plaque in the Umbilicus
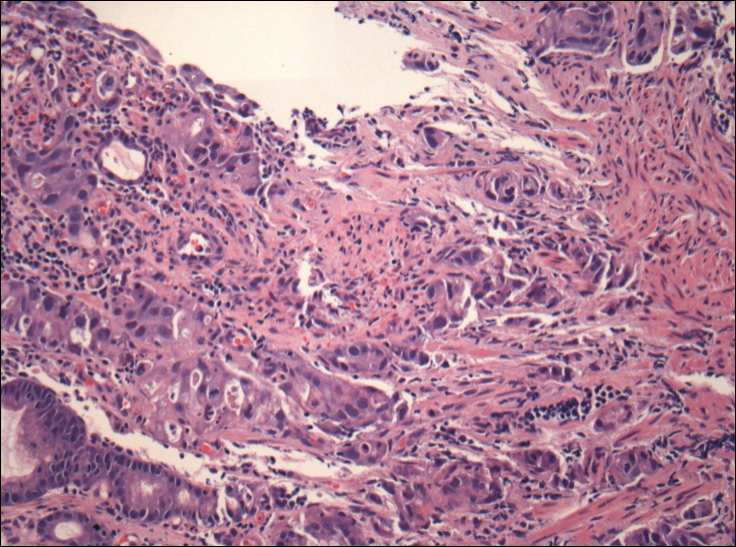
The Diagnosis: Sister Mary Joseph Nodule
The umbilical skin biopsy revealed a moderately differentiated adenocarcinoma (Figure) that was positive for cytokeratin 20 and CDX2 and negative for cytokeratin 7 and transcription termination factor 1. The patient subsequently underwent computed tomography of the abdomen and pelvis, which showed multiple soft-tissue nodules on the greater omentum, a soft-tissue density at the umbilicus, and thickening of the gastric mucosa. An upper endoscopy was then performed, which revealed a large fungating ulcerated mass in the stomach. Biopsy of this mass showed an invasive moderately differentiated adenocarcinoma, which was ERBB2 (formerly HER2) negative. Histopathologically, these pleomorphic glands looked similar to the glands seen in the original skin biopsy. With this diagnosis of metastatic gastric adenocarcinoma, our patient chose palliative chemotherapy but declined precipitously and died 2 months after the initial skin biopsy of the umbilical lesion.
When encountering a patient with an umbilical lesion, it is important to consider benign and malignant lesions in the differential diagnosis. A benign lesion may include scar, cyst, pyogenic granuloma, hemangioma, umbilical hernia, endometriosis, polyp, abscess, or the presence of an omphalith.1 Inflammatory dermatoses such as psoriasis or eczema also should be considered. Malignant lesions could be either primary or secondary, with metastatic disease being the most common.2 Sister Mary Joseph nodule (SMJN) is the eponymgiven to an umbilical lesion representing metastatic disease. Sister Mary Joseph was a nurse and surgical assistant to Dr. William Mayo in Rochester, Minnesota, in what is now known as the Mayo Clinic. She is credited to be the first to observe and note the association between an umbilical nodule and intra-abdominal malignancy. Metastasis to the umbilicus is thought to occur by way of contiguous, hematogenous, lymphatic, or direct spread through embryologic remnants from primary cancers of nearby gastrointestinal or pelvic viscera. It is a rare cutaneous sign of internal malignancy, with an estimated prevalence of 1% to 3%.3 The most common primary cancer is gastric adenocarcinoma, though cases of metastasis from pancreatic, endometrial, and less commonly hematopoietic or supradiaphragmatic cancers have been reported.4 It is more common in women, likely due to the addition of gynecologic malignancies.1
The use of dermoscopy has been advocated as an adjuvant tool in delineating benign and malignant umbilical lesions when an atypical polymorphous vascular pattern indicating neovascularization has been observed with neoplastic growth.5 Once a suspicious umbilical lesion is identified, the first step should be to obtain a skin biopsy or to use fine needle aspiration for cytology.6 Biopsy is especially relevant in the background of cancer history because SMJN may present with cancer recurrence.3 Once one of these is obtained, histological and immunohistochemical analysis will guide further workup and diagnosis of the umbilical lesion.
The importance of reviewing such cases lies in the variable presentation of cutaneous metastases such as SMJN and the grim prognosis that accompanies this finding. It presents as a firm indurated plaque or nodule that may present with systemic symptoms suggestive of malignancy, though in 30% of cases it is the sole initial sign.7 The nodule may be painful if ulcerated or fissured. Bloody, serous, or purulent discharge may be present. After diagnosis of an SMJN, most patients succumb to the disease within 12 months. Thus, it is vital for dermatologists to investigate umbilical lesions with great caution and a high index of suspicion.
- Chalya PL, Mabula JB, Rambau PF, et al. Sister Mary Joseph's nodule at a University teaching hospital in northwestern Tanzania: a retrospective review of 34 cases. World J Surg Oncol. 2013;11:151.
- Papalas JA, Selim MA. Metastatic vs primary malignant neoplasms affecting the umbilicus: clinicopathologic features of 77 tumors. Ann Diagn Pathol. 2011;15:237-242.
- Palaniappan M, Jose WM, Mehta A, et al. Umbilical metastasis: a case series of four Sister Joseph nodules from four different visceral malignancies. Curr Oncol. 2010;17:78-81.
- Zhang YL, Selvaggi SM. Metastatic islet cell carcinoma to the umbilicus: diagnosis by fine-needle aspiration. Diagn Cytopathol. 2003;29:91-94.
- Mun JH, Kim JM, Ko HC, et al. Dermoscopy of a Sister Mary Joseph nodule. J Am Acad Dermatol. 2013;68:e190-e192.
- Handa U, Garg S, Mohan H. Fine-needle aspiration of Sister Mary Joseph's (paraumbilical) nodules. Diagn Cytopathol. 2008;36:348-350.
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273.
The Diagnosis: Sister Mary Joseph Nodule
The umbilical skin biopsy revealed a moderately differentiated adenocarcinoma (Figure) that was positive for cytokeratin 20 and CDX2 and negative for cytokeratin 7 and transcription termination factor 1. The patient subsequently underwent computed tomography of the abdomen and pelvis, which showed multiple soft-tissue nodules on the greater omentum, a soft-tissue density at the umbilicus, and thickening of the gastric mucosa. An upper endoscopy was then performed, which revealed a large fungating ulcerated mass in the stomach. Biopsy of this mass showed an invasive moderately differentiated adenocarcinoma, which was ERBB2 (formerly HER2) negative. Histopathologically, these pleomorphic glands looked similar to the glands seen in the original skin biopsy. With this diagnosis of metastatic gastric adenocarcinoma, our patient chose palliative chemotherapy but declined precipitously and died 2 months after the initial skin biopsy of the umbilical lesion.
When encountering a patient with an umbilical lesion, it is important to consider benign and malignant lesions in the differential diagnosis. A benign lesion may include scar, cyst, pyogenic granuloma, hemangioma, umbilical hernia, endometriosis, polyp, abscess, or the presence of an omphalith.1 Inflammatory dermatoses such as psoriasis or eczema also should be considered. Malignant lesions could be either primary or secondary, with metastatic disease being the most common.2 Sister Mary Joseph nodule (SMJN) is the eponymgiven to an umbilical lesion representing metastatic disease. Sister Mary Joseph was a nurse and surgical assistant to Dr. William Mayo in Rochester, Minnesota, in what is now known as the Mayo Clinic. She is credited to be the first to observe and note the association between an umbilical nodule and intra-abdominal malignancy. Metastasis to the umbilicus is thought to occur by way of contiguous, hematogenous, lymphatic, or direct spread through embryologic remnants from primary cancers of nearby gastrointestinal or pelvic viscera. It is a rare cutaneous sign of internal malignancy, with an estimated prevalence of 1% to 3%.3 The most common primary cancer is gastric adenocarcinoma, though cases of metastasis from pancreatic, endometrial, and less commonly hematopoietic or supradiaphragmatic cancers have been reported.4 It is more common in women, likely due to the addition of gynecologic malignancies.1
The use of dermoscopy has been advocated as an adjuvant tool in delineating benign and malignant umbilical lesions when an atypical polymorphous vascular pattern indicating neovascularization has been observed with neoplastic growth.5 Once a suspicious umbilical lesion is identified, the first step should be to obtain a skin biopsy or to use fine needle aspiration for cytology.6 Biopsy is especially relevant in the background of cancer history because SMJN may present with cancer recurrence.3 Once one of these is obtained, histological and immunohistochemical analysis will guide further workup and diagnosis of the umbilical lesion.
The importance of reviewing such cases lies in the variable presentation of cutaneous metastases such as SMJN and the grim prognosis that accompanies this finding. It presents as a firm indurated plaque or nodule that may present with systemic symptoms suggestive of malignancy, though in 30% of cases it is the sole initial sign.7 The nodule may be painful if ulcerated or fissured. Bloody, serous, or purulent discharge may be present. After diagnosis of an SMJN, most patients succumb to the disease within 12 months. Thus, it is vital for dermatologists to investigate umbilical lesions with great caution and a high index of suspicion.
The Diagnosis: Sister Mary Joseph Nodule
The umbilical skin biopsy revealed a moderately differentiated adenocarcinoma (Figure) that was positive for cytokeratin 20 and CDX2 and negative for cytokeratin 7 and transcription termination factor 1. The patient subsequently underwent computed tomography of the abdomen and pelvis, which showed multiple soft-tissue nodules on the greater omentum, a soft-tissue density at the umbilicus, and thickening of the gastric mucosa. An upper endoscopy was then performed, which revealed a large fungating ulcerated mass in the stomach. Biopsy of this mass showed an invasive moderately differentiated adenocarcinoma, which was ERBB2 (formerly HER2) negative. Histopathologically, these pleomorphic glands looked similar to the glands seen in the original skin biopsy. With this diagnosis of metastatic gastric adenocarcinoma, our patient chose palliative chemotherapy but declined precipitously and died 2 months after the initial skin biopsy of the umbilical lesion.
When encountering a patient with an umbilical lesion, it is important to consider benign and malignant lesions in the differential diagnosis. A benign lesion may include scar, cyst, pyogenic granuloma, hemangioma, umbilical hernia, endometriosis, polyp, abscess, or the presence of an omphalith.1 Inflammatory dermatoses such as psoriasis or eczema also should be considered. Malignant lesions could be either primary or secondary, with metastatic disease being the most common.2 Sister Mary Joseph nodule (SMJN) is the eponymgiven to an umbilical lesion representing metastatic disease. Sister Mary Joseph was a nurse and surgical assistant to Dr. William Mayo in Rochester, Minnesota, in what is now known as the Mayo Clinic. She is credited to be the first to observe and note the association between an umbilical nodule and intra-abdominal malignancy. Metastasis to the umbilicus is thought to occur by way of contiguous, hematogenous, lymphatic, or direct spread through embryologic remnants from primary cancers of nearby gastrointestinal or pelvic viscera. It is a rare cutaneous sign of internal malignancy, with an estimated prevalence of 1% to 3%.3 The most common primary cancer is gastric adenocarcinoma, though cases of metastasis from pancreatic, endometrial, and less commonly hematopoietic or supradiaphragmatic cancers have been reported.4 It is more common in women, likely due to the addition of gynecologic malignancies.1
The use of dermoscopy has been advocated as an adjuvant tool in delineating benign and malignant umbilical lesions when an atypical polymorphous vascular pattern indicating neovascularization has been observed with neoplastic growth.5 Once a suspicious umbilical lesion is identified, the first step should be to obtain a skin biopsy or to use fine needle aspiration for cytology.6 Biopsy is especially relevant in the background of cancer history because SMJN may present with cancer recurrence.3 Once one of these is obtained, histological and immunohistochemical analysis will guide further workup and diagnosis of the umbilical lesion.
The importance of reviewing such cases lies in the variable presentation of cutaneous metastases such as SMJN and the grim prognosis that accompanies this finding. It presents as a firm indurated plaque or nodule that may present with systemic symptoms suggestive of malignancy, though in 30% of cases it is the sole initial sign.7 The nodule may be painful if ulcerated or fissured. Bloody, serous, or purulent discharge may be present. After diagnosis of an SMJN, most patients succumb to the disease within 12 months. Thus, it is vital for dermatologists to investigate umbilical lesions with great caution and a high index of suspicion.
- Chalya PL, Mabula JB, Rambau PF, et al. Sister Mary Joseph's nodule at a University teaching hospital in northwestern Tanzania: a retrospective review of 34 cases. World J Surg Oncol. 2013;11:151.
- Papalas JA, Selim MA. Metastatic vs primary malignant neoplasms affecting the umbilicus: clinicopathologic features of 77 tumors. Ann Diagn Pathol. 2011;15:237-242.
- Palaniappan M, Jose WM, Mehta A, et al. Umbilical metastasis: a case series of four Sister Joseph nodules from four different visceral malignancies. Curr Oncol. 2010;17:78-81.
- Zhang YL, Selvaggi SM. Metastatic islet cell carcinoma to the umbilicus: diagnosis by fine-needle aspiration. Diagn Cytopathol. 2003;29:91-94.
- Mun JH, Kim JM, Ko HC, et al. Dermoscopy of a Sister Mary Joseph nodule. J Am Acad Dermatol. 2013;68:e190-e192.
- Handa U, Garg S, Mohan H. Fine-needle aspiration of Sister Mary Joseph's (paraumbilical) nodules. Diagn Cytopathol. 2008;36:348-350.
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273.
- Chalya PL, Mabula JB, Rambau PF, et al. Sister Mary Joseph's nodule at a University teaching hospital in northwestern Tanzania: a retrospective review of 34 cases. World J Surg Oncol. 2013;11:151.
- Papalas JA, Selim MA. Metastatic vs primary malignant neoplasms affecting the umbilicus: clinicopathologic features of 77 tumors. Ann Diagn Pathol. 2011;15:237-242.
- Palaniappan M, Jose WM, Mehta A, et al. Umbilical metastasis: a case series of four Sister Joseph nodules from four different visceral malignancies. Curr Oncol. 2010;17:78-81.
- Zhang YL, Selvaggi SM. Metastatic islet cell carcinoma to the umbilicus: diagnosis by fine-needle aspiration. Diagn Cytopathol. 2003;29:91-94.
- Mun JH, Kim JM, Ko HC, et al. Dermoscopy of a Sister Mary Joseph nodule. J Am Acad Dermatol. 2013;68:e190-e192.
- Handa U, Garg S, Mohan H. Fine-needle aspiration of Sister Mary Joseph's (paraumbilical) nodules. Diagn Cytopathol. 2008;36:348-350.
- Abu-Hilal M, Newman JS. Sister Mary Joseph and her nodule: historical and clinical perspective. Am J Med Sci. 2009;337:271-273.
A 74-year-old man presented to our outpatient dermatology clinic with an asymptomatic umbilical lesion of unknown duration. The patient believed the lesion was a scar resulting from a prior laparoscopic repair of an umbilical hernia. However, the patient reported epigastric abdominal pain and diarrhea of 1 month's duration that he believed was due to the stomach flu. The patient denied fever, chills, loss of appetite, or weight loss. History was remarkable for hypertension, hyperlipidemia, coronary artery disease, chronic kidney disease, and emphysema. The patient had a surgical history of percutaneous transluminal coronary angioplasty in addition to the laparoscopic umbilical hernia repair. The patient's medications included pantoprazole, ondansetron, diphenoxylate-atropine as needed, amlodipine, lisinopril-hydrochlorothiazide, simvastatin, and aspirin. Physical examination revealed a 1×2-cm pink, nodular, firm plaque with crust at the umbilicus that was tender on palpation. A shave biopsy of the umbilicus was performed and sent for both pathological and immunohistochemical analysis.
Blaschkoid Unilateral Patch on the Chest
The Diagnosis: Lichen Striatus
Lichen striatus (LS) is an acquired and self-limited linear inflammatory dermatosis that most frequently occurs in children and less commonly in adults.1-3 Clinically, it is characterized by the sudden onset of an eruption consisting of slightly pigmented, erythematous, flat-topped papules with minimal scaling. These papules quickly coalesce to form a linear band that extends along a limb, the trunk, or the face, within Blaschko lines.1,4 In the adult form, patients tend to experience more diffuse lesions as well as severe pruritus with higher rates of relapse. It occasionally manifests in a dermatomal manner.1
The differential diagnosis includes other linear acquired inflammatory dermatoses such as blaschkitis, lichen planus, inflammatory linear verrucous epidermal nevus, and psoriasis. Blaschkitis has been described as a rare dermatosis that occurs along the Blaschko lines, affecting adults preferentially over children. Controversy exists whether blaschkitis and lichen striatus are the same disease or 2 separate entities.5 Clinically, both blaschkitis and lichen striatus can present with multiple linear papules and vesicles predominantly on the trunk. In blaschkitis, there is a predilection for males, with an older mean age at onset of 40 years.5 Lesions quickly resolve over months with frequent relapse compared to lichen striatus, which can persist for months to years.
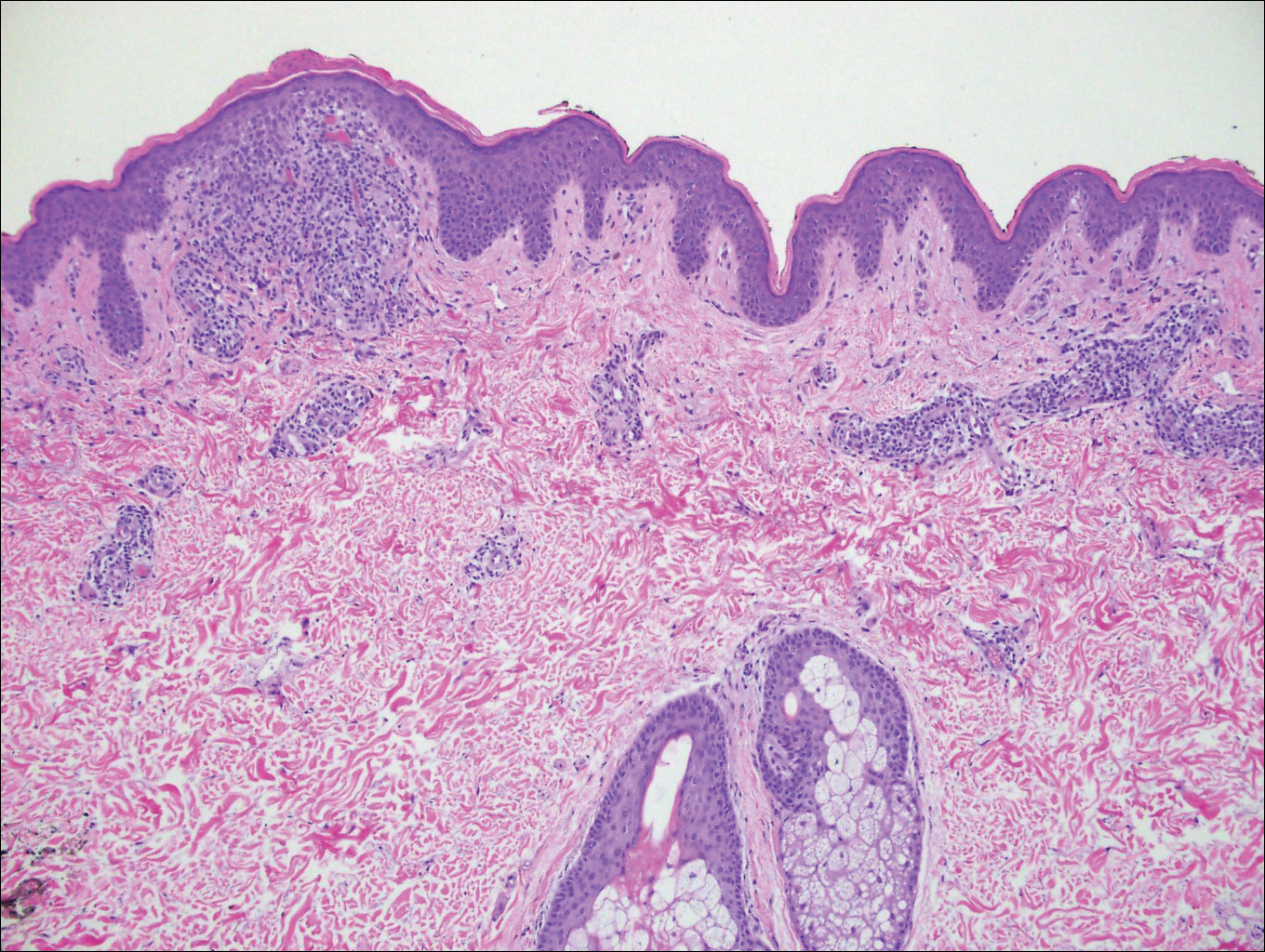
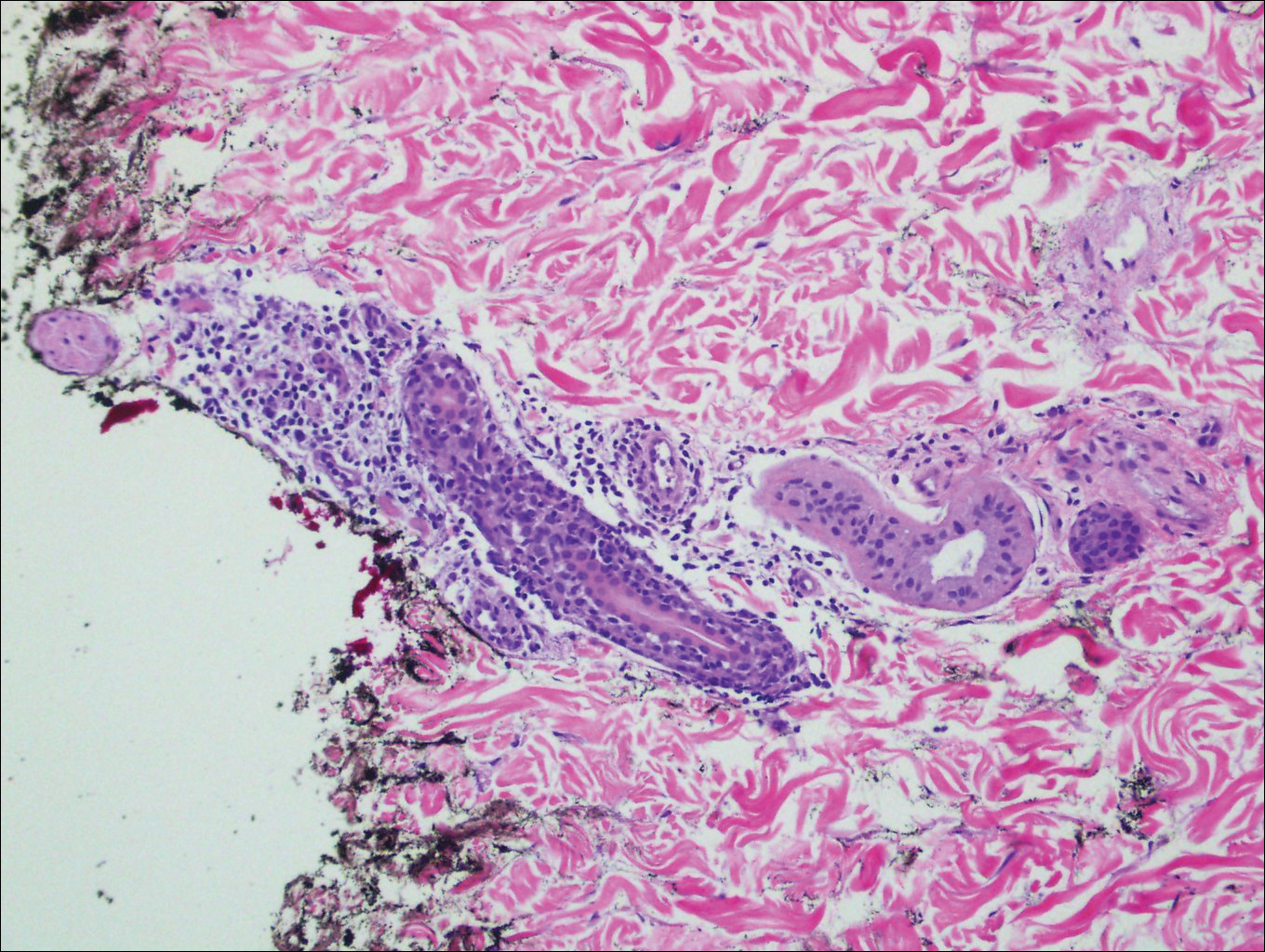
Histopathologically, blaschkitis demonstrates spongiosis, usually without involvement of the adnexal structures. Lichenoid and spongiotic changes with adnexal extension are the hallmark features of lichen striatus. In our patient, biopsy showed several dense bandlike foci of lymphohistiocytic infiltrates along the dermoepidermal junction with spongiosis, basal cell liquefactive degeneration, and pigmentary incontinence (Figure 1). The focal areas were surfaced by parakeratotic and orthohyperkeratotic scale. Deep dermal perivascular and periadnexal extension was present (Figure 2). Periodic acid-Schiff stain was negative for fungi.
The pathogenesis of lichen striatus is not entirely understood, but it has been postulated that trauma, vaccinations, or viral infections may induce loss of immunologic tolerance to keratinocytes.1 This loss of tolerance can result in a T cell-mediated autoimmune reaction against malpighian cells, which show genetic mosaicism and are arranged along Blaschko lines.1,3 Familial cases also have been reported, suggesting that there may be an epigenetic mosaicism that contributes to this group of skin diseases.6,7
Lichen striatus tends to resolve on its own after approximately 6 to 9 months.8 Treatment typically consists of application of topical corticosteroids.1 Cases also have been successfully treated with tacrolimus and pimecrolimus.1,8 Our patient was treated with a midpotency topical steroid with improvement of the appearance but not complete resolution.
- Campanati A, Brandozzi G, Giangiacomi M, et al. Lichen striatus in adults and pimecrolimus: open, off-label clinical study. Int J Dermatol. 2008;47:732-736.
- Lee DY, Kim S, Kim CR, et al. Lichen striatus in an adult treated by a short course of low-dose systemic corticosteroid. J Dermatol. 2011;38:298-299.
- Hofer T. Lichen striatus in adults or "adult blaschkitis"? there is no need for a new naming. Dermatology. 2003;207:89-92.
- Shepherd V, Lun K, Strutton G. Lichen striatus in an adult following trauma. Australas J Dermatol. 2005;46:25-28.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis reappraisal of the concept of blaschkolinear dermatoses. Br J Dermatol. 2011;164:257-262.
- Yaosaka M, Sawamura D, Iitoyo M, et al. Lichen striatus affecting a mother and her son. J Am Acad Dermatol. 2005;53:352-353.
- Jackson R. The lines of Blaschko: a review and reconsideration: observations of the cause of certain unusual linear conditions of the skin. Br J Dermatol. 1976;95:349-360.
- Sorgentini C, Allevato MA, Dahbar M, et al. Lichen striatus in an adult: successful treatment with tacrolimus. Br J Dermatol. 2004;150:776-777.
The Diagnosis: Lichen Striatus
Lichen striatus (LS) is an acquired and self-limited linear inflammatory dermatosis that most frequently occurs in children and less commonly in adults.1-3 Clinically, it is characterized by the sudden onset of an eruption consisting of slightly pigmented, erythematous, flat-topped papules with minimal scaling. These papules quickly coalesce to form a linear band that extends along a limb, the trunk, or the face, within Blaschko lines.1,4 In the adult form, patients tend to experience more diffuse lesions as well as severe pruritus with higher rates of relapse. It occasionally manifests in a dermatomal manner.1
The differential diagnosis includes other linear acquired inflammatory dermatoses such as blaschkitis, lichen planus, inflammatory linear verrucous epidermal nevus, and psoriasis. Blaschkitis has been described as a rare dermatosis that occurs along the Blaschko lines, affecting adults preferentially over children. Controversy exists whether blaschkitis and lichen striatus are the same disease or 2 separate entities.5 Clinically, both blaschkitis and lichen striatus can present with multiple linear papules and vesicles predominantly on the trunk. In blaschkitis, there is a predilection for males, with an older mean age at onset of 40 years.5 Lesions quickly resolve over months with frequent relapse compared to lichen striatus, which can persist for months to years.
Histopathologically, blaschkitis demonstrates spongiosis, usually without involvement of the adnexal structures. Lichenoid and spongiotic changes with adnexal extension are the hallmark features of lichen striatus. In our patient, biopsy showed several dense bandlike foci of lymphohistiocytic infiltrates along the dermoepidermal junction with spongiosis, basal cell liquefactive degeneration, and pigmentary incontinence (Figure 1). The focal areas were surfaced by parakeratotic and orthohyperkeratotic scale. Deep dermal perivascular and periadnexal extension was present (Figure 2). Periodic acid-Schiff stain was negative for fungi.
The pathogenesis of lichen striatus is not entirely understood, but it has been postulated that trauma, vaccinations, or viral infections may induce loss of immunologic tolerance to keratinocytes.1 This loss of tolerance can result in a T cell-mediated autoimmune reaction against malpighian cells, which show genetic mosaicism and are arranged along Blaschko lines.1,3 Familial cases also have been reported, suggesting that there may be an epigenetic mosaicism that contributes to this group of skin diseases.6,7
Lichen striatus tends to resolve on its own after approximately 6 to 9 months.8 Treatment typically consists of application of topical corticosteroids.1 Cases also have been successfully treated with tacrolimus and pimecrolimus.1,8 Our patient was treated with a midpotency topical steroid with improvement of the appearance but not complete resolution.
The Diagnosis: Lichen Striatus
Lichen striatus (LS) is an acquired and self-limited linear inflammatory dermatosis that most frequently occurs in children and less commonly in adults.1-3 Clinically, it is characterized by the sudden onset of an eruption consisting of slightly pigmented, erythematous, flat-topped papules with minimal scaling. These papules quickly coalesce to form a linear band that extends along a limb, the trunk, or the face, within Blaschko lines.1,4 In the adult form, patients tend to experience more diffuse lesions as well as severe pruritus with higher rates of relapse. It occasionally manifests in a dermatomal manner.1
The differential diagnosis includes other linear acquired inflammatory dermatoses such as blaschkitis, lichen planus, inflammatory linear verrucous epidermal nevus, and psoriasis. Blaschkitis has been described as a rare dermatosis that occurs along the Blaschko lines, affecting adults preferentially over children. Controversy exists whether blaschkitis and lichen striatus are the same disease or 2 separate entities.5 Clinically, both blaschkitis and lichen striatus can present with multiple linear papules and vesicles predominantly on the trunk. In blaschkitis, there is a predilection for males, with an older mean age at onset of 40 years.5 Lesions quickly resolve over months with frequent relapse compared to lichen striatus, which can persist for months to years.
Histopathologically, blaschkitis demonstrates spongiosis, usually without involvement of the adnexal structures. Lichenoid and spongiotic changes with adnexal extension are the hallmark features of lichen striatus. In our patient, biopsy showed several dense bandlike foci of lymphohistiocytic infiltrates along the dermoepidermal junction with spongiosis, basal cell liquefactive degeneration, and pigmentary incontinence (Figure 1). The focal areas were surfaced by parakeratotic and orthohyperkeratotic scale. Deep dermal perivascular and periadnexal extension was present (Figure 2). Periodic acid-Schiff stain was negative for fungi.
The pathogenesis of lichen striatus is not entirely understood, but it has been postulated that trauma, vaccinations, or viral infections may induce loss of immunologic tolerance to keratinocytes.1 This loss of tolerance can result in a T cell-mediated autoimmune reaction against malpighian cells, which show genetic mosaicism and are arranged along Blaschko lines.1,3 Familial cases also have been reported, suggesting that there may be an epigenetic mosaicism that contributes to this group of skin diseases.6,7
Lichen striatus tends to resolve on its own after approximately 6 to 9 months.8 Treatment typically consists of application of topical corticosteroids.1 Cases also have been successfully treated with tacrolimus and pimecrolimus.1,8 Our patient was treated with a midpotency topical steroid with improvement of the appearance but not complete resolution.
- Campanati A, Brandozzi G, Giangiacomi M, et al. Lichen striatus in adults and pimecrolimus: open, off-label clinical study. Int J Dermatol. 2008;47:732-736.
- Lee DY, Kim S, Kim CR, et al. Lichen striatus in an adult treated by a short course of low-dose systemic corticosteroid. J Dermatol. 2011;38:298-299.
- Hofer T. Lichen striatus in adults or "adult blaschkitis"? there is no need for a new naming. Dermatology. 2003;207:89-92.
- Shepherd V, Lun K, Strutton G. Lichen striatus in an adult following trauma. Australas J Dermatol. 2005;46:25-28.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis reappraisal of the concept of blaschkolinear dermatoses. Br J Dermatol. 2011;164:257-262.
- Yaosaka M, Sawamura D, Iitoyo M, et al. Lichen striatus affecting a mother and her son. J Am Acad Dermatol. 2005;53:352-353.
- Jackson R. The lines of Blaschko: a review and reconsideration: observations of the cause of certain unusual linear conditions of the skin. Br J Dermatol. 1976;95:349-360.
- Sorgentini C, Allevato MA, Dahbar M, et al. Lichen striatus in an adult: successful treatment with tacrolimus. Br J Dermatol. 2004;150:776-777.
- Campanati A, Brandozzi G, Giangiacomi M, et al. Lichen striatus in adults and pimecrolimus: open, off-label clinical study. Int J Dermatol. 2008;47:732-736.
- Lee DY, Kim S, Kim CR, et al. Lichen striatus in an adult treated by a short course of low-dose systemic corticosteroid. J Dermatol. 2011;38:298-299.
- Hofer T. Lichen striatus in adults or "adult blaschkitis"? there is no need for a new naming. Dermatology. 2003;207:89-92.
- Shepherd V, Lun K, Strutton G. Lichen striatus in an adult following trauma. Australas J Dermatol. 2005;46:25-28.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis reappraisal of the concept of blaschkolinear dermatoses. Br J Dermatol. 2011;164:257-262.
- Yaosaka M, Sawamura D, Iitoyo M, et al. Lichen striatus affecting a mother and her son. J Am Acad Dermatol. 2005;53:352-353.
- Jackson R. The lines of Blaschko: a review and reconsideration: observations of the cause of certain unusual linear conditions of the skin. Br J Dermatol. 1976;95:349-360.
- Sorgentini C, Allevato MA, Dahbar M, et al. Lichen striatus in an adult: successful treatment with tacrolimus. Br J Dermatol. 2004;150:776-777.
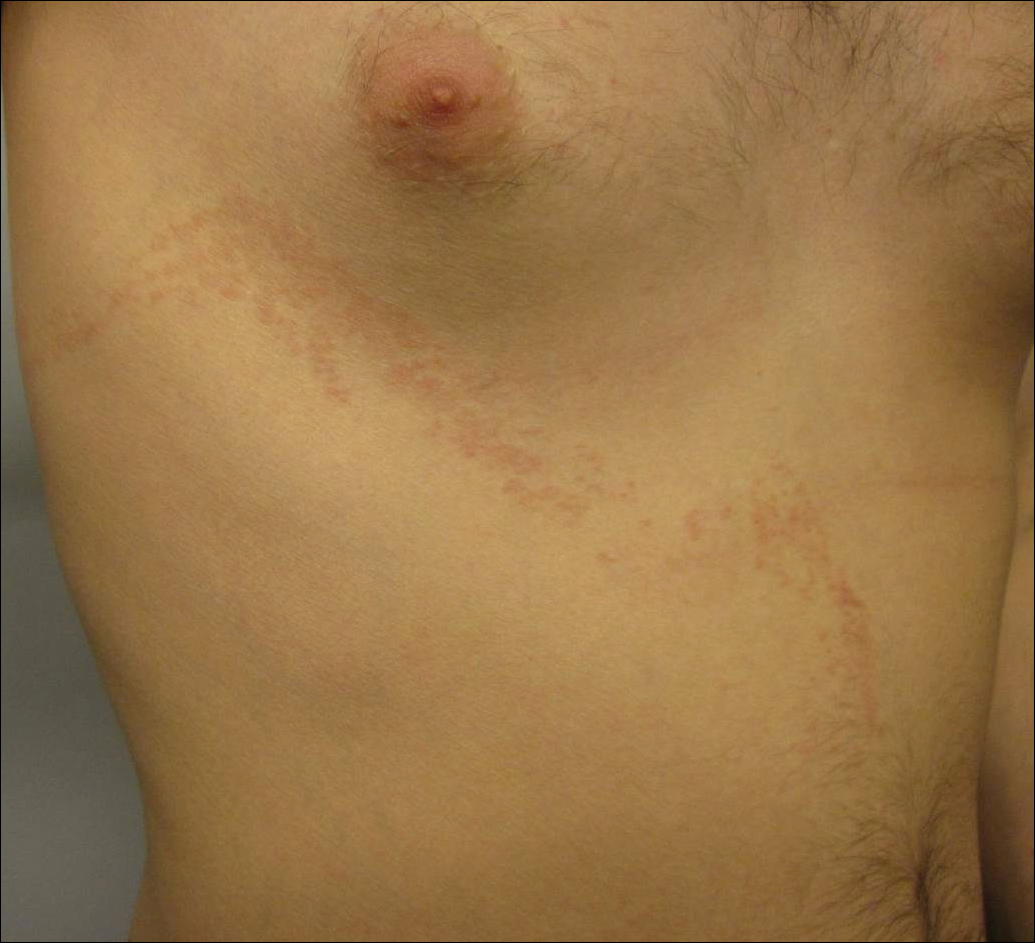
A 26-year-old man presented with erythematous, scaly, grouped papules along the right side of the chest of 3 weeks' duration, extending to the flank following a blaschkoid distribution on the right side of the chest and not crossing the midline. He reported occasional irritation but otherwise was asymptomatic. His medical history was unremarkable and he was not taking any medications. He also denied trauma to the area.
Epidermodysplasia Verruciformis and the Risk for Malignancy
To the Editor:
Epidermodysplasia verruciformis (EV) is a rare autosomal-recessive genodermatosis characterized by widespread infection with specific strains of human papillomavirus (HPV). Patients with EV have a unique susceptibility to acquire HPV due to defects in cellular immunity to the presenting antigens.1 These defects may be related to mutations of the EVER genes or due to acquisition of an immunosuppressive condition.2,3 Infections with HPV-3 and HPV-10 do not lead to the development of malignancies. However, infection with HPV-5, HPV-8, and HPV-14 can lead to the development of nonmelanoma skin cancers, usually squamous cell carcinomas (SCCs), in approximately 60% of patients.3,4 This viral condition lasts throughout the patient’s lifetime and presents as tinea versicolor–like macules and patches. These lesions may be confused with seborrheic keratosis or verruca plana.5 Lesions typically are hypopigmented but occasionally may be hyperpigmented or erythematous. They often are found on the trunk, but lesions on the face, arms, palms, legs, and soles have been reported.5 Mucous membranes are always spared. Epidermodysplasia verruciformis often presents in childhood, except in cases related to acquired immunosuppression. The condition has no sex or racial predilection and no geographical preference.5
A 7-year-old boy (Fitzpatrick skin type V) presented with an asymptomatic rash on the trunk (Figure 1), dorsal aspect of the hands, and forehead. The lesions first appeared 5 years prior on the upper back and upper chest and had recently spread to the forehead and frontal aspect of the scalp. The patient had a history of myelomeningocele, which was corrected at birth with surgical placement of a ventriculoperitoneal shunt. The patient was otherwise healthy and met all appropriate developmental milestones for his age group. Family history revealed consanguinity of the patient’s paternal grandparents who were first cousins. The patient’s mother denied any other family member having similar rashes or lesions.
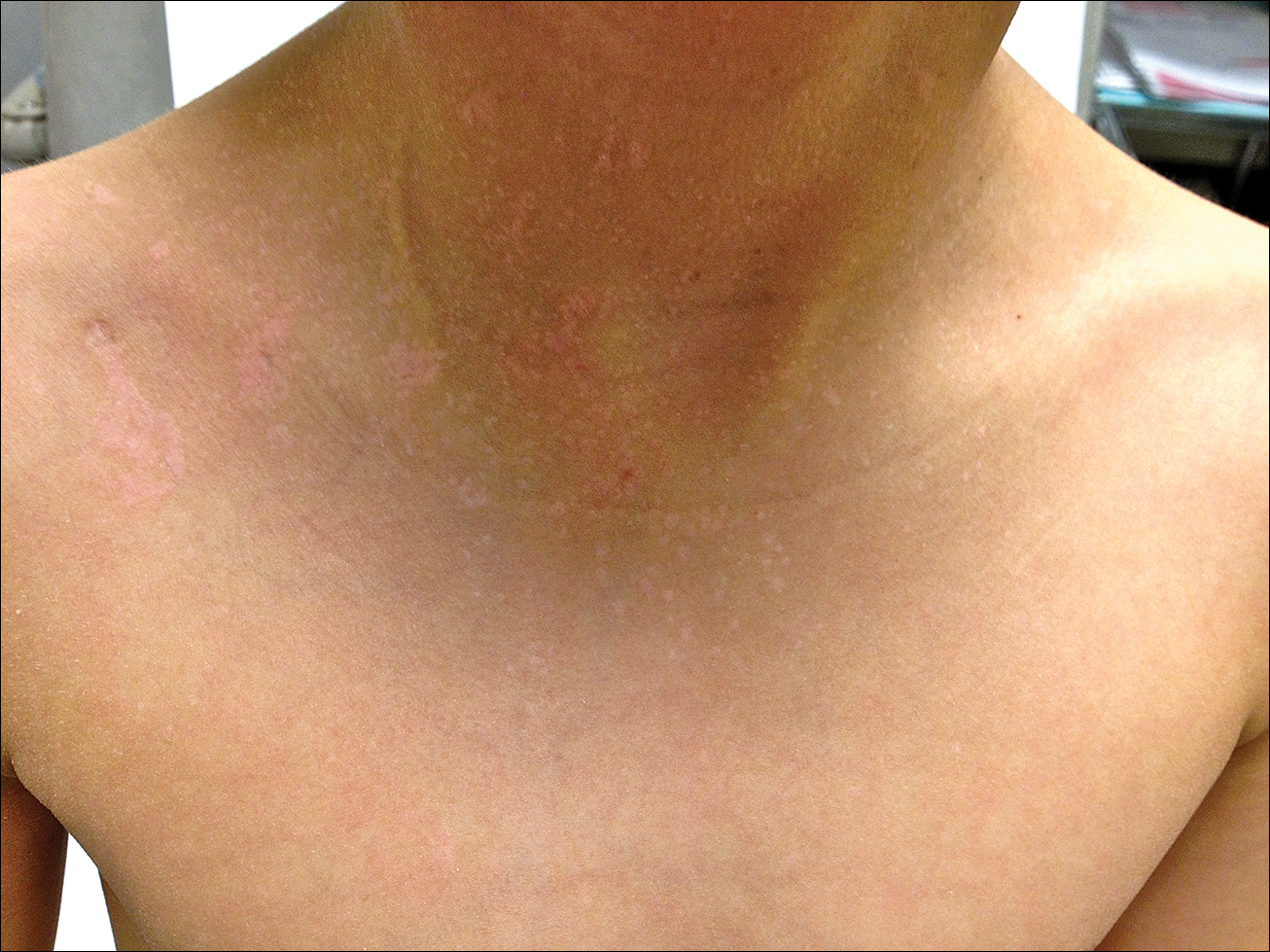
The patient had been treated for pityriasis versicolor on and off for 2 years by another dermatologist. His mother reported faithfully applying ketoconazole cream twice daily for several months with no improvement. She also reported using topical steroids, which did not provide any benefit. The patient and mother denied any associated pruritus, bleeding, burning, or physical discomfort.
Skin examination revealed diffuse, flat, polymorphous, hypopigmented and salmon-colored hyperkeratotic macules and patches with mild scaling on the upper region of the anterior aspect of the chest and upper back (Figure 2A). Additionally, the patient had an extensive number of lesions on the forehead and frontal aspect of the scalp (Figure 2B).

A shave biopsy demonstrated a thick basket weave stratum corneum, koilocytes, and large pale keratinocytes with characteristic blue cytoplasm. These findings were characteristic for EV.
At the patient’s 3-month follow-up visit, he again denied any symptoms associated with the lesions and reported that the appearance was diminishing in severity. On examination there was no evidence of SCC. The mother was advised to discontinue all topical treatments for the patient and return to the office every 3 to 6 months for regular skin surveillance. The mother was further advised to protect the patient from UV radiation with sunscreen and sun-protective clothing.
Epidermodysplasia verruciformis was first reported by Lewandowsky and Lutz6 in 1922. This rare condition often presents in childhood and is characterized by a persistent HPV infection and an autosomal-recessive inheritance pattern. Reports in the literature frequently involve kindreds. Often, patients with EV have a family history of first-degree or second-degree consanguinity.7
The clinical presentation of EV often resembles a pityriasis versicolor–like eruption. However, pityriasis versicolor is less commonly seen in childhood and is more prevalent in patients aged 21 to 30 years, likely due to increased sebum production and changing hormone levels. Furthermore, it is unusual to see pityriasis versicolor affect the face and scalp.8 Lesions of EV vary from hypopigmented and pinkish red macules to confluent patches and hyperkeratotic verrucalike lesions.3 Clinical characteristics also may include dyschromic patches; lesions that resemble flat warts on the trunk, face, and distal arms; and/or lesions that appear similar to seborrheic keratoses on the dorsal aspect of the hands.9,10
Mutations of the EVER gene downregulate a cell’s ability to adequately attack the HPV antigens.11 Although some patients with EV are found to have mutations of the EVER1 and EVER2 genes, a notable portion of patients with EV lack these mutations. Three other causes of EV include acquisition of immunosuppressive conditions including lymphoma, solid organ transplant, and human immunodeficiency virus. If one suspects autosomal-recessive inheritance of EV, genetic testing such as polymerase chain reaction DNA fragment analysis can be performed to determine if there are mutations on the EVER1 or EVER2 genes.12
The inability of patients with EV to mount an immune response to multiple types of HPV increases the risk for developing cutaneous malignancies.7 Additionally, it is known that UV radiation diminishes skin cell immunity, and the combination of EV and UV radiation further increases the risk for developing SCCs.11 The development of nonmelanoma skin cancers usually occurs on sun-exposed skin 20 to 30 years after the onset of lesions, with the highest occurrence of SCCs presenting in the fourth decade of life.1
Protection from UV light exposure is critical to reduce the risk for malignancy. Treatment options for EV lesions have included topical imiquimod 5%, 5-fluorouracil, oral isotretinoin, and intralesional interferon alfa, but patients are often refractory to these interventions. Curettage, surgical excision, electrosurgery, and laser ablation can be effective for individual lesions but carry a greater risk for scarring.1 Photodynamic therapy with aminolevulinic acid and blue light represents a promising option that deserves further study.
Epidermodysplasia verruciformis should be considered as a differential diagnosis in all patients presenting with disseminated lesions resembling pityriasis versicolor that are unresponsive to treatment. A biopsy will help to establish the diagnosis. Patients should minimize sun exposure and report any skin lesions that are changing in appearance.
- Hoffner MV, Camacho FM. Surgical treatment of epidermodysplasia verruciformis. Dermatol Surg. 2010;36:363-367.
- McDermott D, Gammon B, Snijders P. Autosomal dominant epidermodysplasia verruciformis lacking a known EVER1 or EVER2 mutation. Pediatr Dermatol. 2009;26:306-310.
- Patel T, Morrison K, Rady P, et al. Epidermodysplasia verruciformis and susceptibility to HPV. Dis Markers. 2010;29:199-206.
- Hultgren TL, Srinivasan SK, DiMaio DJ. Epidermodysplasia verruciformis occurring in a patient with human immunodeficiency virus: a case report. Cutis. 2007;79:308-311.
- Oliveira W, Netu C, Rady P, et al. Clinical aspects of epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2003;17:394-398.
- Lewandowsky F, Lutz W. Ein Fall einer bisher nicht beschriebenen Hauterkrankung (epidermodysplasia verruciformis). Arch Dermatol Syphilol. 1922;141:193-203.
- Prystowsky S, Herndon J, Freeman R, et al. Epidermodysplasia verruciformis. Am J Dis Child. 1976;130:437-440.
- Kyriakis KP, Terzoudi S, Palamaras I, et al. Pityriasis versicolor prevalence by age and gender. Mycoses. 2006;49:517-518.
- Nuovo G, Ishag M. The histologic spectrum of epidermodysplasia verruciformis. Am J Surg Pathol. 2000;24:1400-1406.
- Jacobelli S, Laude H, Carlotti A, et al. Epidermodysplasia verruciformis in human immunodeficiency virus-infected patients: a marker of human papillomavirus-related disorders not affected by antiretroviral therapy. Arch Dermatol. 2011;147:590-596.
- Rogers HD, MacGregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:316-320.
- Gober MD, Rady PL, He Q, et al. Novel homozygous frameshift mutation of EVER1 gene in an epidermodysplasia verruciformis patient. J Invest Dermatol. 2007;127:817-820.
To the Editor:
Epidermodysplasia verruciformis (EV) is a rare autosomal-recessive genodermatosis characterized by widespread infection with specific strains of human papillomavirus (HPV). Patients with EV have a unique susceptibility to acquire HPV due to defects in cellular immunity to the presenting antigens.1 These defects may be related to mutations of the EVER genes or due to acquisition of an immunosuppressive condition.2,3 Infections with HPV-3 and HPV-10 do not lead to the development of malignancies. However, infection with HPV-5, HPV-8, and HPV-14 can lead to the development of nonmelanoma skin cancers, usually squamous cell carcinomas (SCCs), in approximately 60% of patients.3,4 This viral condition lasts throughout the patient’s lifetime and presents as tinea versicolor–like macules and patches. These lesions may be confused with seborrheic keratosis or verruca plana.5 Lesions typically are hypopigmented but occasionally may be hyperpigmented or erythematous. They often are found on the trunk, but lesions on the face, arms, palms, legs, and soles have been reported.5 Mucous membranes are always spared. Epidermodysplasia verruciformis often presents in childhood, except in cases related to acquired immunosuppression. The condition has no sex or racial predilection and no geographical preference.5
A 7-year-old boy (Fitzpatrick skin type V) presented with an asymptomatic rash on the trunk (Figure 1), dorsal aspect of the hands, and forehead. The lesions first appeared 5 years prior on the upper back and upper chest and had recently spread to the forehead and frontal aspect of the scalp. The patient had a history of myelomeningocele, which was corrected at birth with surgical placement of a ventriculoperitoneal shunt. The patient was otherwise healthy and met all appropriate developmental milestones for his age group. Family history revealed consanguinity of the patient’s paternal grandparents who were first cousins. The patient’s mother denied any other family member having similar rashes or lesions.
The patient had been treated for pityriasis versicolor on and off for 2 years by another dermatologist. His mother reported faithfully applying ketoconazole cream twice daily for several months with no improvement. She also reported using topical steroids, which did not provide any benefit. The patient and mother denied any associated pruritus, bleeding, burning, or physical discomfort.
Skin examination revealed diffuse, flat, polymorphous, hypopigmented and salmon-colored hyperkeratotic macules and patches with mild scaling on the upper region of the anterior aspect of the chest and upper back (Figure 2A). Additionally, the patient had an extensive number of lesions on the forehead and frontal aspect of the scalp (Figure 2B).
A shave biopsy demonstrated a thick basket weave stratum corneum, koilocytes, and large pale keratinocytes with characteristic blue cytoplasm. These findings were characteristic for EV.
At the patient’s 3-month follow-up visit, he again denied any symptoms associated with the lesions and reported that the appearance was diminishing in severity. On examination there was no evidence of SCC. The mother was advised to discontinue all topical treatments for the patient and return to the office every 3 to 6 months for regular skin surveillance. The mother was further advised to protect the patient from UV radiation with sunscreen and sun-protective clothing.
Epidermodysplasia verruciformis was first reported by Lewandowsky and Lutz6 in 1922. This rare condition often presents in childhood and is characterized by a persistent HPV infection and an autosomal-recessive inheritance pattern. Reports in the literature frequently involve kindreds. Often, patients with EV have a family history of first-degree or second-degree consanguinity.7
The clinical presentation of EV often resembles a pityriasis versicolor–like eruption. However, pityriasis versicolor is less commonly seen in childhood and is more prevalent in patients aged 21 to 30 years, likely due to increased sebum production and changing hormone levels. Furthermore, it is unusual to see pityriasis versicolor affect the face and scalp.8 Lesions of EV vary from hypopigmented and pinkish red macules to confluent patches and hyperkeratotic verrucalike lesions.3 Clinical characteristics also may include dyschromic patches; lesions that resemble flat warts on the trunk, face, and distal arms; and/or lesions that appear similar to seborrheic keratoses on the dorsal aspect of the hands.9,10
Mutations of the EVER gene downregulate a cell’s ability to adequately attack the HPV antigens.11 Although some patients with EV are found to have mutations of the EVER1 and EVER2 genes, a notable portion of patients with EV lack these mutations. Three other causes of EV include acquisition of immunosuppressive conditions including lymphoma, solid organ transplant, and human immunodeficiency virus. If one suspects autosomal-recessive inheritance of EV, genetic testing such as polymerase chain reaction DNA fragment analysis can be performed to determine if there are mutations on the EVER1 or EVER2 genes.12
The inability of patients with EV to mount an immune response to multiple types of HPV increases the risk for developing cutaneous malignancies.7 Additionally, it is known that UV radiation diminishes skin cell immunity, and the combination of EV and UV radiation further increases the risk for developing SCCs.11 The development of nonmelanoma skin cancers usually occurs on sun-exposed skin 20 to 30 years after the onset of lesions, with the highest occurrence of SCCs presenting in the fourth decade of life.1
Protection from UV light exposure is critical to reduce the risk for malignancy. Treatment options for EV lesions have included topical imiquimod 5%, 5-fluorouracil, oral isotretinoin, and intralesional interferon alfa, but patients are often refractory to these interventions. Curettage, surgical excision, electrosurgery, and laser ablation can be effective for individual lesions but carry a greater risk for scarring.1 Photodynamic therapy with aminolevulinic acid and blue light represents a promising option that deserves further study.
Epidermodysplasia verruciformis should be considered as a differential diagnosis in all patients presenting with disseminated lesions resembling pityriasis versicolor that are unresponsive to treatment. A biopsy will help to establish the diagnosis. Patients should minimize sun exposure and report any skin lesions that are changing in appearance.
To the Editor:
Epidermodysplasia verruciformis (EV) is a rare autosomal-recessive genodermatosis characterized by widespread infection with specific strains of human papillomavirus (HPV). Patients with EV have a unique susceptibility to acquire HPV due to defects in cellular immunity to the presenting antigens.1 These defects may be related to mutations of the EVER genes or due to acquisition of an immunosuppressive condition.2,3 Infections with HPV-3 and HPV-10 do not lead to the development of malignancies. However, infection with HPV-5, HPV-8, and HPV-14 can lead to the development of nonmelanoma skin cancers, usually squamous cell carcinomas (SCCs), in approximately 60% of patients.3,4 This viral condition lasts throughout the patient’s lifetime and presents as tinea versicolor–like macules and patches. These lesions may be confused with seborrheic keratosis or verruca plana.5 Lesions typically are hypopigmented but occasionally may be hyperpigmented or erythematous. They often are found on the trunk, but lesions on the face, arms, palms, legs, and soles have been reported.5 Mucous membranes are always spared. Epidermodysplasia verruciformis often presents in childhood, except in cases related to acquired immunosuppression. The condition has no sex or racial predilection and no geographical preference.5
A 7-year-old boy (Fitzpatrick skin type V) presented with an asymptomatic rash on the trunk (Figure 1), dorsal aspect of the hands, and forehead. The lesions first appeared 5 years prior on the upper back and upper chest and had recently spread to the forehead and frontal aspect of the scalp. The patient had a history of myelomeningocele, which was corrected at birth with surgical placement of a ventriculoperitoneal shunt. The patient was otherwise healthy and met all appropriate developmental milestones for his age group. Family history revealed consanguinity of the patient’s paternal grandparents who were first cousins. The patient’s mother denied any other family member having similar rashes or lesions.
The patient had been treated for pityriasis versicolor on and off for 2 years by another dermatologist. His mother reported faithfully applying ketoconazole cream twice daily for several months with no improvement. She also reported using topical steroids, which did not provide any benefit. The patient and mother denied any associated pruritus, bleeding, burning, or physical discomfort.
Skin examination revealed diffuse, flat, polymorphous, hypopigmented and salmon-colored hyperkeratotic macules and patches with mild scaling on the upper region of the anterior aspect of the chest and upper back (Figure 2A). Additionally, the patient had an extensive number of lesions on the forehead and frontal aspect of the scalp (Figure 2B).
A shave biopsy demonstrated a thick basket weave stratum corneum, koilocytes, and large pale keratinocytes with characteristic blue cytoplasm. These findings were characteristic for EV.
At the patient’s 3-month follow-up visit, he again denied any symptoms associated with the lesions and reported that the appearance was diminishing in severity. On examination there was no evidence of SCC. The mother was advised to discontinue all topical treatments for the patient and return to the office every 3 to 6 months for regular skin surveillance. The mother was further advised to protect the patient from UV radiation with sunscreen and sun-protective clothing.
Epidermodysplasia verruciformis was first reported by Lewandowsky and Lutz6 in 1922. This rare condition often presents in childhood and is characterized by a persistent HPV infection and an autosomal-recessive inheritance pattern. Reports in the literature frequently involve kindreds. Often, patients with EV have a family history of first-degree or second-degree consanguinity.7
The clinical presentation of EV often resembles a pityriasis versicolor–like eruption. However, pityriasis versicolor is less commonly seen in childhood and is more prevalent in patients aged 21 to 30 years, likely due to increased sebum production and changing hormone levels. Furthermore, it is unusual to see pityriasis versicolor affect the face and scalp.8 Lesions of EV vary from hypopigmented and pinkish red macules to confluent patches and hyperkeratotic verrucalike lesions.3 Clinical characteristics also may include dyschromic patches; lesions that resemble flat warts on the trunk, face, and distal arms; and/or lesions that appear similar to seborrheic keratoses on the dorsal aspect of the hands.9,10
Mutations of the EVER gene downregulate a cell’s ability to adequately attack the HPV antigens.11 Although some patients with EV are found to have mutations of the EVER1 and EVER2 genes, a notable portion of patients with EV lack these mutations. Three other causes of EV include acquisition of immunosuppressive conditions including lymphoma, solid organ transplant, and human immunodeficiency virus. If one suspects autosomal-recessive inheritance of EV, genetic testing such as polymerase chain reaction DNA fragment analysis can be performed to determine if there are mutations on the EVER1 or EVER2 genes.12
The inability of patients with EV to mount an immune response to multiple types of HPV increases the risk for developing cutaneous malignancies.7 Additionally, it is known that UV radiation diminishes skin cell immunity, and the combination of EV and UV radiation further increases the risk for developing SCCs.11 The development of nonmelanoma skin cancers usually occurs on sun-exposed skin 20 to 30 years after the onset of lesions, with the highest occurrence of SCCs presenting in the fourth decade of life.1
Protection from UV light exposure is critical to reduce the risk for malignancy. Treatment options for EV lesions have included topical imiquimod 5%, 5-fluorouracil, oral isotretinoin, and intralesional interferon alfa, but patients are often refractory to these interventions. Curettage, surgical excision, electrosurgery, and laser ablation can be effective for individual lesions but carry a greater risk for scarring.1 Photodynamic therapy with aminolevulinic acid and blue light represents a promising option that deserves further study.
Epidermodysplasia verruciformis should be considered as a differential diagnosis in all patients presenting with disseminated lesions resembling pityriasis versicolor that are unresponsive to treatment. A biopsy will help to establish the diagnosis. Patients should minimize sun exposure and report any skin lesions that are changing in appearance.
- Hoffner MV, Camacho FM. Surgical treatment of epidermodysplasia verruciformis. Dermatol Surg. 2010;36:363-367.
- McDermott D, Gammon B, Snijders P. Autosomal dominant epidermodysplasia verruciformis lacking a known EVER1 or EVER2 mutation. Pediatr Dermatol. 2009;26:306-310.
- Patel T, Morrison K, Rady P, et al. Epidermodysplasia verruciformis and susceptibility to HPV. Dis Markers. 2010;29:199-206.
- Hultgren TL, Srinivasan SK, DiMaio DJ. Epidermodysplasia verruciformis occurring in a patient with human immunodeficiency virus: a case report. Cutis. 2007;79:308-311.
- Oliveira W, Netu C, Rady P, et al. Clinical aspects of epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2003;17:394-398.
- Lewandowsky F, Lutz W. Ein Fall einer bisher nicht beschriebenen Hauterkrankung (epidermodysplasia verruciformis). Arch Dermatol Syphilol. 1922;141:193-203.
- Prystowsky S, Herndon J, Freeman R, et al. Epidermodysplasia verruciformis. Am J Dis Child. 1976;130:437-440.
- Kyriakis KP, Terzoudi S, Palamaras I, et al. Pityriasis versicolor prevalence by age and gender. Mycoses. 2006;49:517-518.
- Nuovo G, Ishag M. The histologic spectrum of epidermodysplasia verruciformis. Am J Surg Pathol. 2000;24:1400-1406.
- Jacobelli S, Laude H, Carlotti A, et al. Epidermodysplasia verruciformis in human immunodeficiency virus-infected patients: a marker of human papillomavirus-related disorders not affected by antiretroviral therapy. Arch Dermatol. 2011;147:590-596.
- Rogers HD, MacGregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:316-320.
- Gober MD, Rady PL, He Q, et al. Novel homozygous frameshift mutation of EVER1 gene in an epidermodysplasia verruciformis patient. J Invest Dermatol. 2007;127:817-820.
- Hoffner MV, Camacho FM. Surgical treatment of epidermodysplasia verruciformis. Dermatol Surg. 2010;36:363-367.
- McDermott D, Gammon B, Snijders P. Autosomal dominant epidermodysplasia verruciformis lacking a known EVER1 or EVER2 mutation. Pediatr Dermatol. 2009;26:306-310.
- Patel T, Morrison K, Rady P, et al. Epidermodysplasia verruciformis and susceptibility to HPV. Dis Markers. 2010;29:199-206.
- Hultgren TL, Srinivasan SK, DiMaio DJ. Epidermodysplasia verruciformis occurring in a patient with human immunodeficiency virus: a case report. Cutis. 2007;79:308-311.
- Oliveira W, Netu C, Rady P, et al. Clinical aspects of epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2003;17:394-398.
- Lewandowsky F, Lutz W. Ein Fall einer bisher nicht beschriebenen Hauterkrankung (epidermodysplasia verruciformis). Arch Dermatol Syphilol. 1922;141:193-203.
- Prystowsky S, Herndon J, Freeman R, et al. Epidermodysplasia verruciformis. Am J Dis Child. 1976;130:437-440.
- Kyriakis KP, Terzoudi S, Palamaras I, et al. Pityriasis versicolor prevalence by age and gender. Mycoses. 2006;49:517-518.
- Nuovo G, Ishag M. The histologic spectrum of epidermodysplasia verruciformis. Am J Surg Pathol. 2000;24:1400-1406.
- Jacobelli S, Laude H, Carlotti A, et al. Epidermodysplasia verruciformis in human immunodeficiency virus-infected patients: a marker of human papillomavirus-related disorders not affected by antiretroviral therapy. Arch Dermatol. 2011;147:590-596.
- Rogers HD, MacGregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:316-320.
- Gober MD, Rady PL, He Q, et al. Novel homozygous frameshift mutation of EVER1 gene in an epidermodysplasia verruciformis patient. J Invest Dermatol. 2007;127:817-820.
Practice Points
- Epidermodysplasia verruciformis (EV) is a rare genodermatosis that usually presents in early childhood and presents as verrucous papules and plaques most commonly on the skin of the head, neck, and upper extremities. It often is misdiagnosed at pityriasis versicolor.
- Mutations of the EVER1 and EVER2 genes have been identified as a source for developing EV.
- Epidermodysplasia verruciformis produces wartlike lesions in individuals who have a unique susceptibility to acquiring the human papillomavirus and early onset of nonmelanoma skin cancers, most commonly squamous cell carcinomas related to viral oncogenesis.
- Avoidance and protection from UV exposure is a critical component of treatment plans for patients with EV.
Diagnosis of a Rapidly Growing Preauricular Nodule: Chondroid Syringoma or Pleomorphic Adenoma?
To the Editor:
Chondroid syringoma is a rare benign mixed tumor that originates from the sweat glands, typically presenting with both epithelial and mesenchymal components.1 It differs from pleomorphic adenoma, which arises from salivary glands. The surgical approach for complete excision is different for the 2 tumors; therefore, definitive diagnosis is important. For chondroid syringoma, total excision is recommended,2 while for pleomorphic adenoma, extracapsular dissection or superficial parotidectomy is commonly indicated. We report a case of a preauricular nodule at presentation and highlight the importance of differentiating a chondroid syringoma from a pleomorphic adenoma. This case is unique because of the anatomic location of the nodule, making diagnosis difficult because the tumor was abutting the parotid gland and a biopsy included normal salivary gland cells.
A 19-year-old man with a history of moderate acne on the shoulders, back, and face presented with a rapidly growing, painless nodule on right preauricular region of 6 months’ duration. The nodule was originally thought to be acne related and monitored, as the patient was asymptomatic. On examination the patient was found to have a firm, fixed, nontender, subcutaneous nodule overlying the right temporomandibular joint just anterior to the right tragus (Figure 1). Laboratory results were unremarkable. Computed tomography showed a subcutaneous nonaggressive-appearing soft-tissue mass measuring 16×17×12 mm just anterior and inferior to the external auditory canal cartilaginous segment with no bony abnormalities. The patient was initially treated with incomplete excision of the area for a presumed sebaceous cyst; 2 months later, an abnormal biopsy prompted a complete excisional biopsy.
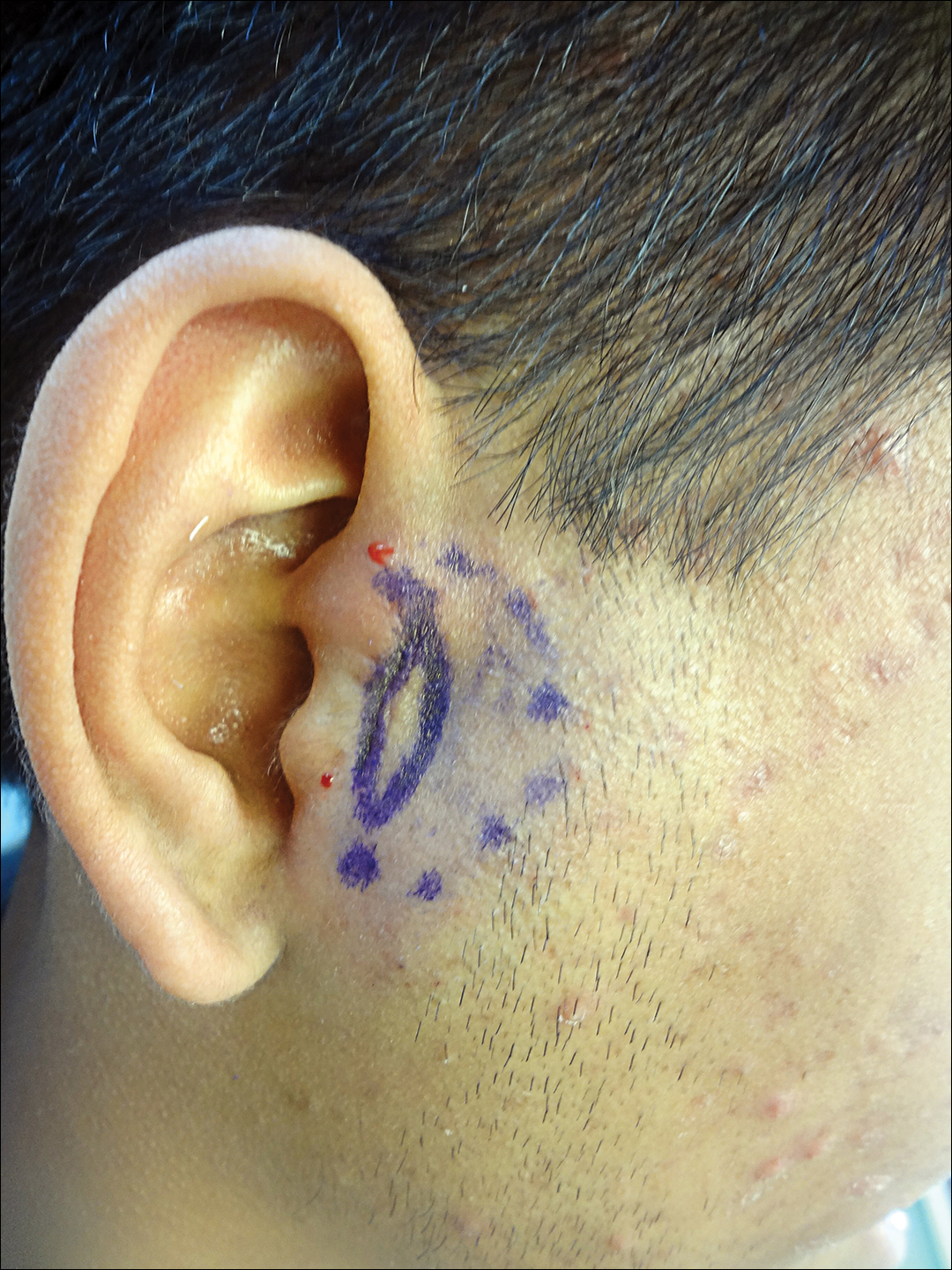
Histologically, the initial incomplete excision biopsy revealed myxoid and chondroid tissue with glandular elements and adjacent lymph node with strong positivity for S-100 protein and moderate positivity for glial fibrillary acid protein, consistent with chondroid syringoma (Figure 2). Histological findings on second excision biopsy performed 2 months later showed tumor cells surrounded by normal salivary gland cells; therefore, it was difficult to define the origin of this tumor. Subsequent magnetic resonance imaging showed no evidence of the tumor and normal parotid gland borders.
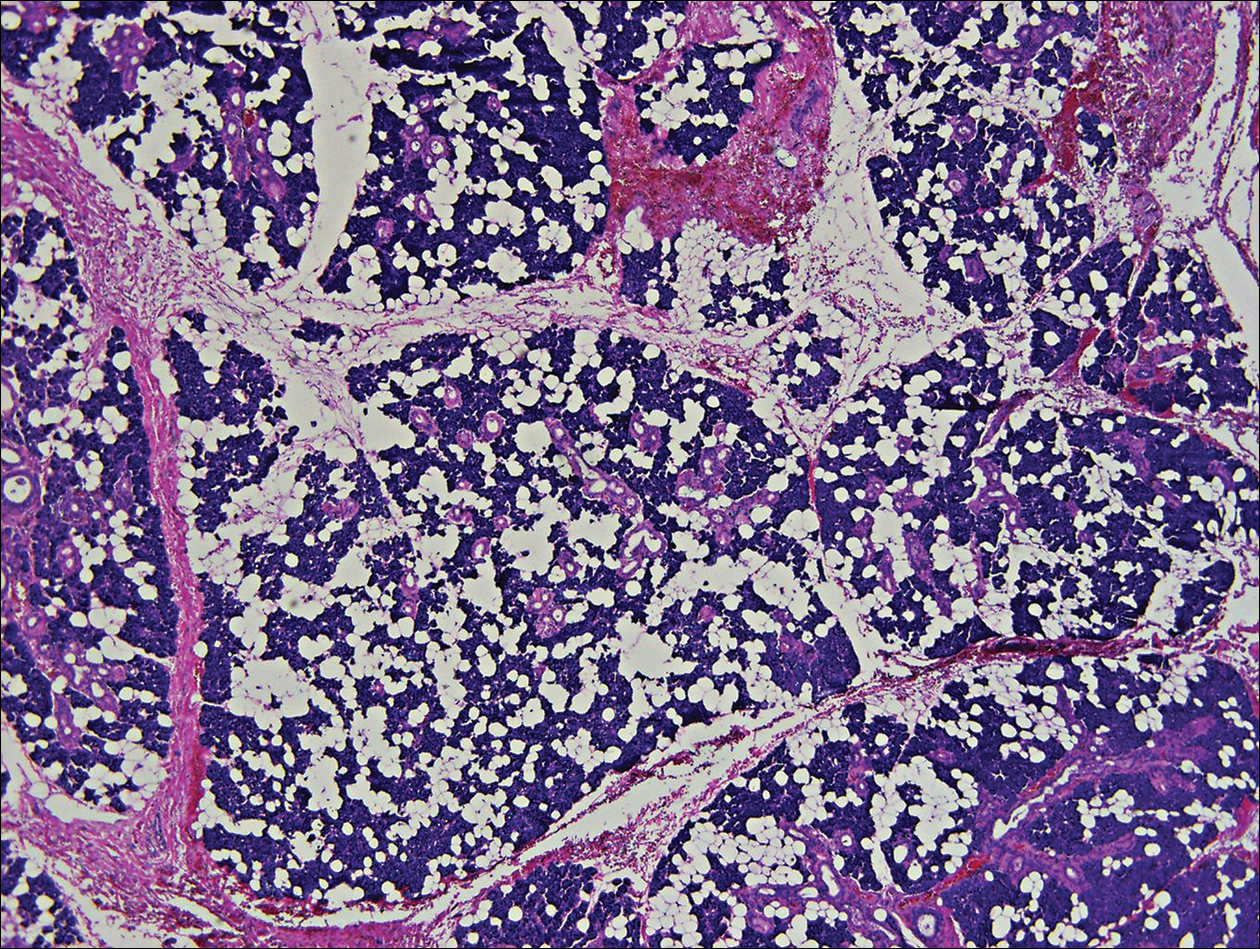
Chondroid syringoma is a rare nonmelanoma skin tumor of the head and neck, mostly benign in nature but with malignant potential. Predominantly, it presents in males as an asymptomatic, slow-growing, nontender nodule.2 Malignant chondroid syringomas are more rare, typically appear on the trunk and legs of females, and present as rapidly growing hard nodules. They can arise de novo or from a preexisting chondroid syringoma and can metastasize.3,4
Clinically and histologically, chondroid syringoma resembles a pleomorphic adenoma. Its diagnosis is dependent on the clinical location to exclude origin in a salivary gland.5 Folliculosebaceous and myoepithelial differentiation within the tumor has been reported.6 Immunocytochemistry is the same in both types and is used to identify 2 prominent components—epithelial and mesenchymal—found in both chondroid syringoma and pleomorphic adenoma. Immunocytochemistry differentiates the epithelial component, which expresses cytokeratin, epithelial membrane antigen, and carcinoembryonic antigen. In contrast, the mesenchymal component expresses S-100, vimentin, and neuron‐specific enolase, and less often glial fibrillary acidic protein, smooth muscle actin, calponin, or p63.5,7,8 Identification of both layers is a distinctive trait of both tumors, rendering it apart from other conditions in the differential diagnosis.5
Typical treatment options include excision, electrodesiccation, dermabrasion, and argon or CO2 laser. Total excision is recommended if there is a benign tumor and complete excision is a cure.2 One case of recurrent benign chondroid syringoma was treated by Mohs micrographic surgery on the eyebrow9; however, Mohs surgery was not recommended in our case due to concerns of spread if malignant as well as an unknown tumor depth, as these tumors have a tendency for deep infiltration.
Due to its anatomical location and presentation as an anterior preauricular mass, it was difficult to differentiate between chondroid syringoma from sweat gland origin and pleomorphic adenoma from the salivary gland. As seen in our case, it is important for physicians to be aware of the differential diagnosis for mixed tumors because it can have a notable effect on the type of surgical therapy and follow-up management.
- Hirsch P, Helwig EB. Chondroid syringoma. Arch Dermatol. 1961;84:835-847.
- Turhan-Haktanir N, Sahin O, Bukulmez A, et al. Chondroid syringoma in a child. Pediatr Dermatol. 2007;24:505-507.
- Mathiasen RA, Rasgon BM, Rumore G. Malignant chondroid syringoma of the face: a first reported case. Otolaryngol Head Neck Surg. 2005;133:305-307.
- Barnett MD, Wallack MK, Zuretti A, et al. Recurrent malignant chondroid syringoma of the foot: a case report and review of the literature. Am J Clin Oncol. 2000;23:227-232.
- Dubb M, Michelow P. Cytologic features of chondroid syringoma in fine needle aspiration biopsies a report of 3 cases. Acta Cytol. 2010;54:183-186.
- Rauso R, Santagata M, Tartaro G, et al. Chondroid syringoma: rare tumor of orofacial region. Minerva Stomatol. 2009;58:383-388.
- Metzler G, Schaumburg-Lever G, Hornstein O, et al. Malignant chondroid syringoma: immunohistopathology. Am J Dermatopathol. 1996;18:83-89.
- Argenyi ZB, Balogh K, Goeken JA. Immunohistochemical characterization of chondroid syringomas. Am J Clin Pathol. 1988;90:662-669.
- Walls AC, Deng A, Geist DE. Mohs micrographic surgery for recurrent chondroid syringoma of the eyebrow. Dermatol Surg. 2012;38:800-802.
To the Editor:
Chondroid syringoma is a rare benign mixed tumor that originates from the sweat glands, typically presenting with both epithelial and mesenchymal components.1 It differs from pleomorphic adenoma, which arises from salivary glands. The surgical approach for complete excision is different for the 2 tumors; therefore, definitive diagnosis is important. For chondroid syringoma, total excision is recommended,2 while for pleomorphic adenoma, extracapsular dissection or superficial parotidectomy is commonly indicated. We report a case of a preauricular nodule at presentation and highlight the importance of differentiating a chondroid syringoma from a pleomorphic adenoma. This case is unique because of the anatomic location of the nodule, making diagnosis difficult because the tumor was abutting the parotid gland and a biopsy included normal salivary gland cells.
A 19-year-old man with a history of moderate acne on the shoulders, back, and face presented with a rapidly growing, painless nodule on right preauricular region of 6 months’ duration. The nodule was originally thought to be acne related and monitored, as the patient was asymptomatic. On examination the patient was found to have a firm, fixed, nontender, subcutaneous nodule overlying the right temporomandibular joint just anterior to the right tragus (Figure 1). Laboratory results were unremarkable. Computed tomography showed a subcutaneous nonaggressive-appearing soft-tissue mass measuring 16×17×12 mm just anterior and inferior to the external auditory canal cartilaginous segment with no bony abnormalities. The patient was initially treated with incomplete excision of the area for a presumed sebaceous cyst; 2 months later, an abnormal biopsy prompted a complete excisional biopsy.
Histologically, the initial incomplete excision biopsy revealed myxoid and chondroid tissue with glandular elements and adjacent lymph node with strong positivity for S-100 protein and moderate positivity for glial fibrillary acid protein, consistent with chondroid syringoma (Figure 2). Histological findings on second excision biopsy performed 2 months later showed tumor cells surrounded by normal salivary gland cells; therefore, it was difficult to define the origin of this tumor. Subsequent magnetic resonance imaging showed no evidence of the tumor and normal parotid gland borders.
Chondroid syringoma is a rare nonmelanoma skin tumor of the head and neck, mostly benign in nature but with malignant potential. Predominantly, it presents in males as an asymptomatic, slow-growing, nontender nodule.2 Malignant chondroid syringomas are more rare, typically appear on the trunk and legs of females, and present as rapidly growing hard nodules. They can arise de novo or from a preexisting chondroid syringoma and can metastasize.3,4
Clinically and histologically, chondroid syringoma resembles a pleomorphic adenoma. Its diagnosis is dependent on the clinical location to exclude origin in a salivary gland.5 Folliculosebaceous and myoepithelial differentiation within the tumor has been reported.6 Immunocytochemistry is the same in both types and is used to identify 2 prominent components—epithelial and mesenchymal—found in both chondroid syringoma and pleomorphic adenoma. Immunocytochemistry differentiates the epithelial component, which expresses cytokeratin, epithelial membrane antigen, and carcinoembryonic antigen. In contrast, the mesenchymal component expresses S-100, vimentin, and neuron‐specific enolase, and less often glial fibrillary acidic protein, smooth muscle actin, calponin, or p63.5,7,8 Identification of both layers is a distinctive trait of both tumors, rendering it apart from other conditions in the differential diagnosis.5
Typical treatment options include excision, electrodesiccation, dermabrasion, and argon or CO2 laser. Total excision is recommended if there is a benign tumor and complete excision is a cure.2 One case of recurrent benign chondroid syringoma was treated by Mohs micrographic surgery on the eyebrow9; however, Mohs surgery was not recommended in our case due to concerns of spread if malignant as well as an unknown tumor depth, as these tumors have a tendency for deep infiltration.
Due to its anatomical location and presentation as an anterior preauricular mass, it was difficult to differentiate between chondroid syringoma from sweat gland origin and pleomorphic adenoma from the salivary gland. As seen in our case, it is important for physicians to be aware of the differential diagnosis for mixed tumors because it can have a notable effect on the type of surgical therapy and follow-up management.
To the Editor:
Chondroid syringoma is a rare benign mixed tumor that originates from the sweat glands, typically presenting with both epithelial and mesenchymal components.1 It differs from pleomorphic adenoma, which arises from salivary glands. The surgical approach for complete excision is different for the 2 tumors; therefore, definitive diagnosis is important. For chondroid syringoma, total excision is recommended,2 while for pleomorphic adenoma, extracapsular dissection or superficial parotidectomy is commonly indicated. We report a case of a preauricular nodule at presentation and highlight the importance of differentiating a chondroid syringoma from a pleomorphic adenoma. This case is unique because of the anatomic location of the nodule, making diagnosis difficult because the tumor was abutting the parotid gland and a biopsy included normal salivary gland cells.
A 19-year-old man with a history of moderate acne on the shoulders, back, and face presented with a rapidly growing, painless nodule on right preauricular region of 6 months’ duration. The nodule was originally thought to be acne related and monitored, as the patient was asymptomatic. On examination the patient was found to have a firm, fixed, nontender, subcutaneous nodule overlying the right temporomandibular joint just anterior to the right tragus (Figure 1). Laboratory results were unremarkable. Computed tomography showed a subcutaneous nonaggressive-appearing soft-tissue mass measuring 16×17×12 mm just anterior and inferior to the external auditory canal cartilaginous segment with no bony abnormalities. The patient was initially treated with incomplete excision of the area for a presumed sebaceous cyst; 2 months later, an abnormal biopsy prompted a complete excisional biopsy.
Histologically, the initial incomplete excision biopsy revealed myxoid and chondroid tissue with glandular elements and adjacent lymph node with strong positivity for S-100 protein and moderate positivity for glial fibrillary acid protein, consistent with chondroid syringoma (Figure 2). Histological findings on second excision biopsy performed 2 months later showed tumor cells surrounded by normal salivary gland cells; therefore, it was difficult to define the origin of this tumor. Subsequent magnetic resonance imaging showed no evidence of the tumor and normal parotid gland borders.
Chondroid syringoma is a rare nonmelanoma skin tumor of the head and neck, mostly benign in nature but with malignant potential. Predominantly, it presents in males as an asymptomatic, slow-growing, nontender nodule.2 Malignant chondroid syringomas are more rare, typically appear on the trunk and legs of females, and present as rapidly growing hard nodules. They can arise de novo or from a preexisting chondroid syringoma and can metastasize.3,4
Clinically and histologically, chondroid syringoma resembles a pleomorphic adenoma. Its diagnosis is dependent on the clinical location to exclude origin in a salivary gland.5 Folliculosebaceous and myoepithelial differentiation within the tumor has been reported.6 Immunocytochemistry is the same in both types and is used to identify 2 prominent components—epithelial and mesenchymal—found in both chondroid syringoma and pleomorphic adenoma. Immunocytochemistry differentiates the epithelial component, which expresses cytokeratin, epithelial membrane antigen, and carcinoembryonic antigen. In contrast, the mesenchymal component expresses S-100, vimentin, and neuron‐specific enolase, and less often glial fibrillary acidic protein, smooth muscle actin, calponin, or p63.5,7,8 Identification of both layers is a distinctive trait of both tumors, rendering it apart from other conditions in the differential diagnosis.5
Typical treatment options include excision, electrodesiccation, dermabrasion, and argon or CO2 laser. Total excision is recommended if there is a benign tumor and complete excision is a cure.2 One case of recurrent benign chondroid syringoma was treated by Mohs micrographic surgery on the eyebrow9; however, Mohs surgery was not recommended in our case due to concerns of spread if malignant as well as an unknown tumor depth, as these tumors have a tendency for deep infiltration.
Due to its anatomical location and presentation as an anterior preauricular mass, it was difficult to differentiate between chondroid syringoma from sweat gland origin and pleomorphic adenoma from the salivary gland. As seen in our case, it is important for physicians to be aware of the differential diagnosis for mixed tumors because it can have a notable effect on the type of surgical therapy and follow-up management.
- Hirsch P, Helwig EB. Chondroid syringoma. Arch Dermatol. 1961;84:835-847.
- Turhan-Haktanir N, Sahin O, Bukulmez A, et al. Chondroid syringoma in a child. Pediatr Dermatol. 2007;24:505-507.
- Mathiasen RA, Rasgon BM, Rumore G. Malignant chondroid syringoma of the face: a first reported case. Otolaryngol Head Neck Surg. 2005;133:305-307.
- Barnett MD, Wallack MK, Zuretti A, et al. Recurrent malignant chondroid syringoma of the foot: a case report and review of the literature. Am J Clin Oncol. 2000;23:227-232.
- Dubb M, Michelow P. Cytologic features of chondroid syringoma in fine needle aspiration biopsies a report of 3 cases. Acta Cytol. 2010;54:183-186.
- Rauso R, Santagata M, Tartaro G, et al. Chondroid syringoma: rare tumor of orofacial region. Minerva Stomatol. 2009;58:383-388.
- Metzler G, Schaumburg-Lever G, Hornstein O, et al. Malignant chondroid syringoma: immunohistopathology. Am J Dermatopathol. 1996;18:83-89.
- Argenyi ZB, Balogh K, Goeken JA. Immunohistochemical characterization of chondroid syringomas. Am J Clin Pathol. 1988;90:662-669.
- Walls AC, Deng A, Geist DE. Mohs micrographic surgery for recurrent chondroid syringoma of the eyebrow. Dermatol Surg. 2012;38:800-802.
- Hirsch P, Helwig EB. Chondroid syringoma. Arch Dermatol. 1961;84:835-847.
- Turhan-Haktanir N, Sahin O, Bukulmez A, et al. Chondroid syringoma in a child. Pediatr Dermatol. 2007;24:505-507.
- Mathiasen RA, Rasgon BM, Rumore G. Malignant chondroid syringoma of the face: a first reported case. Otolaryngol Head Neck Surg. 2005;133:305-307.
- Barnett MD, Wallack MK, Zuretti A, et al. Recurrent malignant chondroid syringoma of the foot: a case report and review of the literature. Am J Clin Oncol. 2000;23:227-232.
- Dubb M, Michelow P. Cytologic features of chondroid syringoma in fine needle aspiration biopsies a report of 3 cases. Acta Cytol. 2010;54:183-186.
- Rauso R, Santagata M, Tartaro G, et al. Chondroid syringoma: rare tumor of orofacial region. Minerva Stomatol. 2009;58:383-388.
- Metzler G, Schaumburg-Lever G, Hornstein O, et al. Malignant chondroid syringoma: immunohistopathology. Am J Dermatopathol. 1996;18:83-89.
- Argenyi ZB, Balogh K, Goeken JA. Immunohistochemical characterization of chondroid syringomas. Am J Clin Pathol. 1988;90:662-669.
- Walls AC, Deng A, Geist DE. Mohs micrographic surgery for recurrent chondroid syringoma of the eyebrow. Dermatol Surg. 2012;38:800-802.
Practice Points
- Clinically and histologically, pleomorphic adenomas and chondroid syringoma both have identical presentations. Differentiation can be determined by knowing where the mixed tumor originated.
- Both lesions warrant different surgical management techniques. Pleomorphic adenoma requires extracapsular dissection or superficial parotidectomy, while complete excision is recommended for chondroid syringoma.
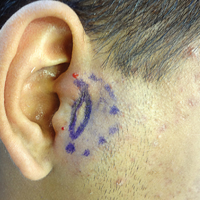
Bullous Pemphigoid Associated With a Lymphoepithelial Cyst of the Pancreas
Bullous pemphigoid (BP) is an acquired, autoimmune, subepidermal blistering disease that is more common in elderly patients.1 An association with internal neoplasms and BP has been established; however, there is debate regarding the precise nature of the relationship.2 Several gastrointestinal tract tumors have been associated with BP, including adenocarcinoma of the colon, adenosquamous cell carcinoma and adenocarcinoma of the stomach, adenocarcinoma of the rectum, and liver and bile duct malignancies.3-5 Association with pancreatic neoplasms (eg, carcinoma of the pancreas) rarely has been reported.5-7 We present an unusual case of a lymphoepithelial cyst of the pancreas in a patient with BP.
Case Report
A 67-year-old man presented with erythematous crusted plaques and pink scars over the chest, back, arms, and legs (Figure 1). A 1.5-cm tense bullous lesion was observed on the right knee. The patient’s medical history was notable for biopsy-proven BP of 8 months’ duration as well as diabetes mellitus and hypothyroidism. The patient was being followed by his surgeon for a 1.9-cm soft-tissue lesion in the pancreatic tail and was awaiting surgical excision at the time of the current presentation. The pancreatic lesion was discovered incidentally on magnetic resonance imaging performed following urologic concerns. At the current presentation, the patient’s medications included nifedipine, hydralazine, metoprolol, torsemide, aspirin, levothyroxine, atorvastatin, insulin lispro, and insulin glargine. He had previously been treated for BP with prednisone at a maximum dosage of 60 mg daily, clobetasol propionate cream 0.05%, and mupirocin ointment 2% without improvement. Because of substantial weight gain and poorly controlled diabetes, prednisone was discontinued.
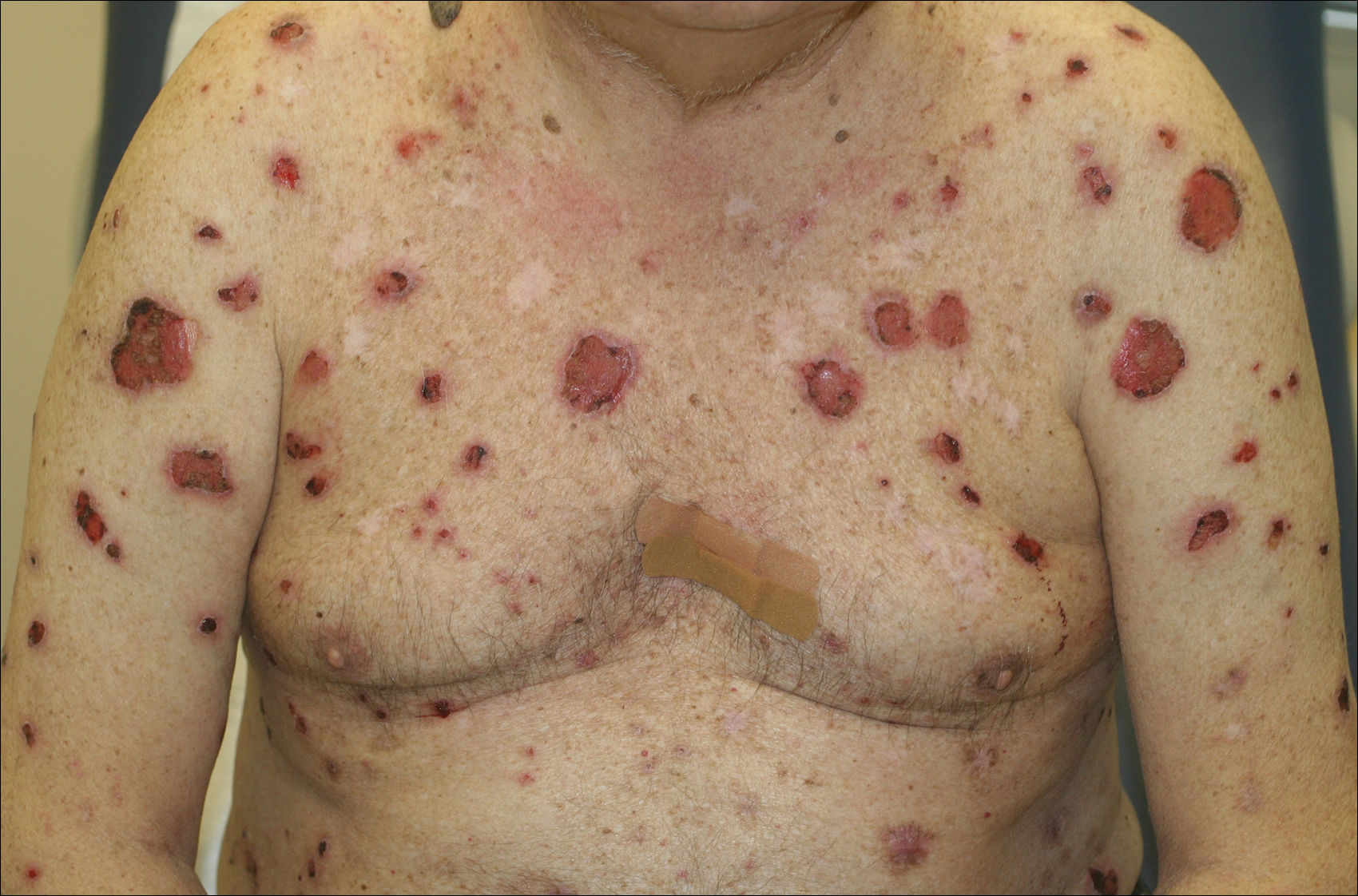
Bullous pemphigoid had been diagnosed histopathologically by a prior dermatologist. Hematoxylin and eosin staining demonstrated a subepidermal separation with eosinophils within the perivascular infiltrate (Figure 2). Direct immunofluorescence was noted in a linear pattern at the dermoepidermal junction with IgG and C3. Bullous pemphigoid antigen antibodies 1 and 2 were obtained via enzyme-linked immunosorbent assay with a positive BP-1 antigen antibody of 19 U/mL (positive, >15 U/mL) and a normal BP-2 antigen antibody of less than 9 U/mL (reference range, <9 U/mL). The glucagon level was elevated at 245 pg/mL (reference range, ≤134 pg/mL).
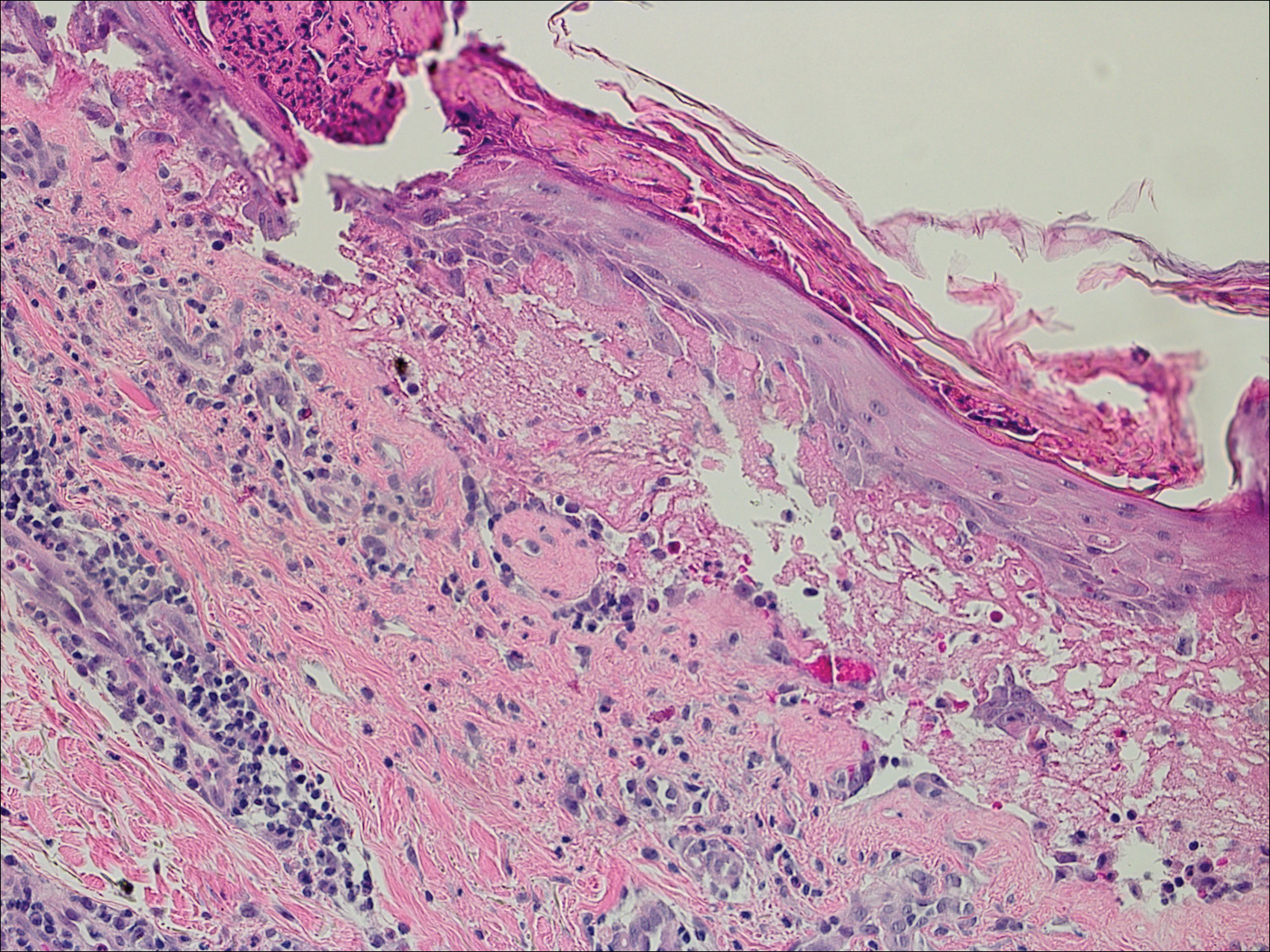
The patient was prescribed minocycline 100 mg twice daily and niacinamide 500 mg 3 times daily. Topical treatment with clobetasol and mupirocin was continued. One month later, the patient returned with an increase in disease activity. Changes to his therapeutic regimen were deferred until after excision of the pancreatic lesion based on the decision not to start immunosuppressive therapy until the precise nature of the pancreatic lesion was determined.
The patient underwent excision of the pancreatic lesion approximately 3 months later, which proved to be a benign lymphoepithelial cyst of the pancreas. Histology of the cyst consisted of dense fibrous tissue with a squamous epithelial lining focally infiltrated by lymphocytes (Figure 3A). Immunoperoxidase staining of the cyst revealed focal linear areas of C3d staining along the basement membrane of the stratified squamous epithelium (Figure 3B).
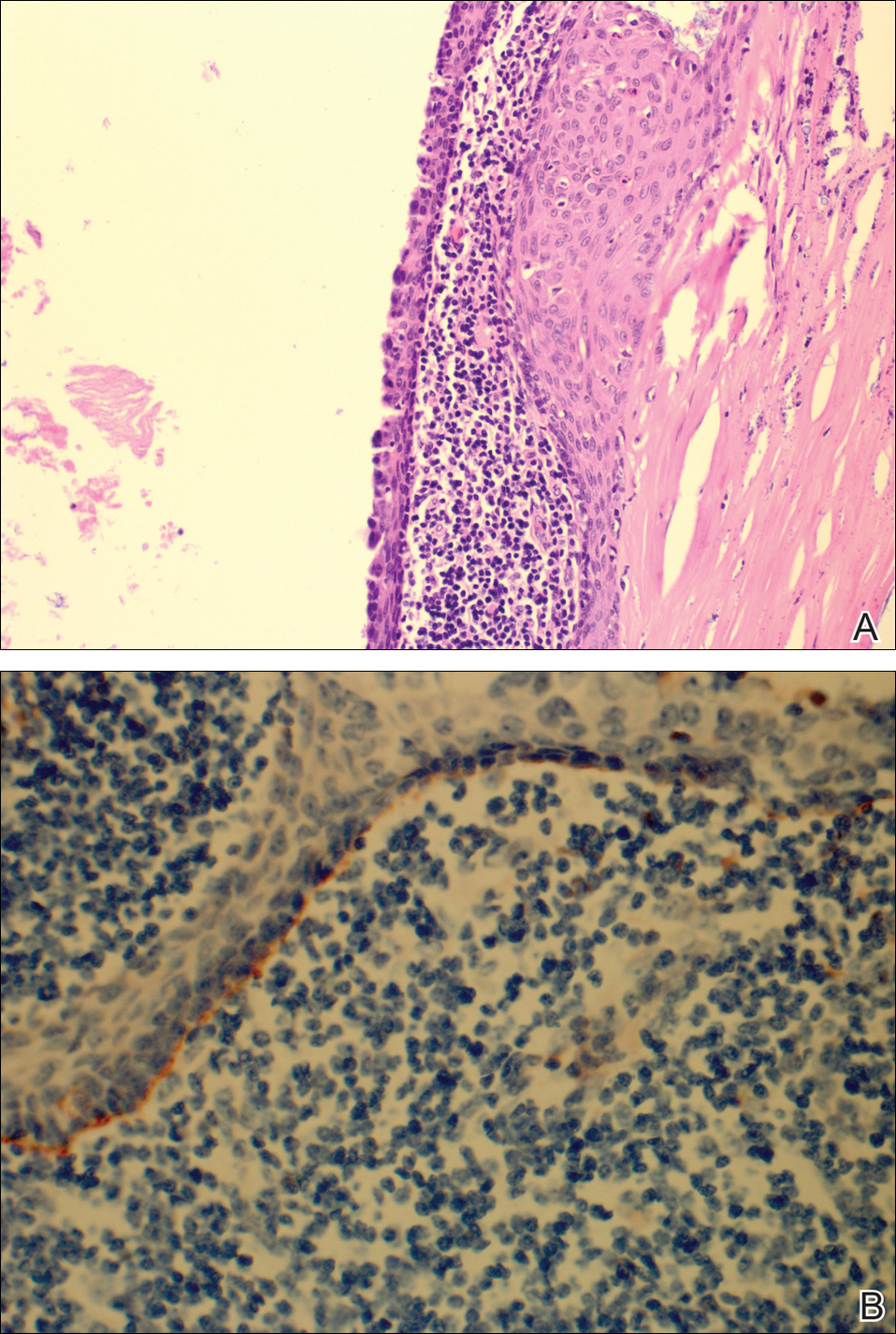
The patient stated that his skin started to improve virtually immediately following the excision without systemic treatment for BP. On follow-up examination 3 weeks postoperatively, no bullae were observed and there was a notable decrease in erythematous crusted plaques (Figure 4).
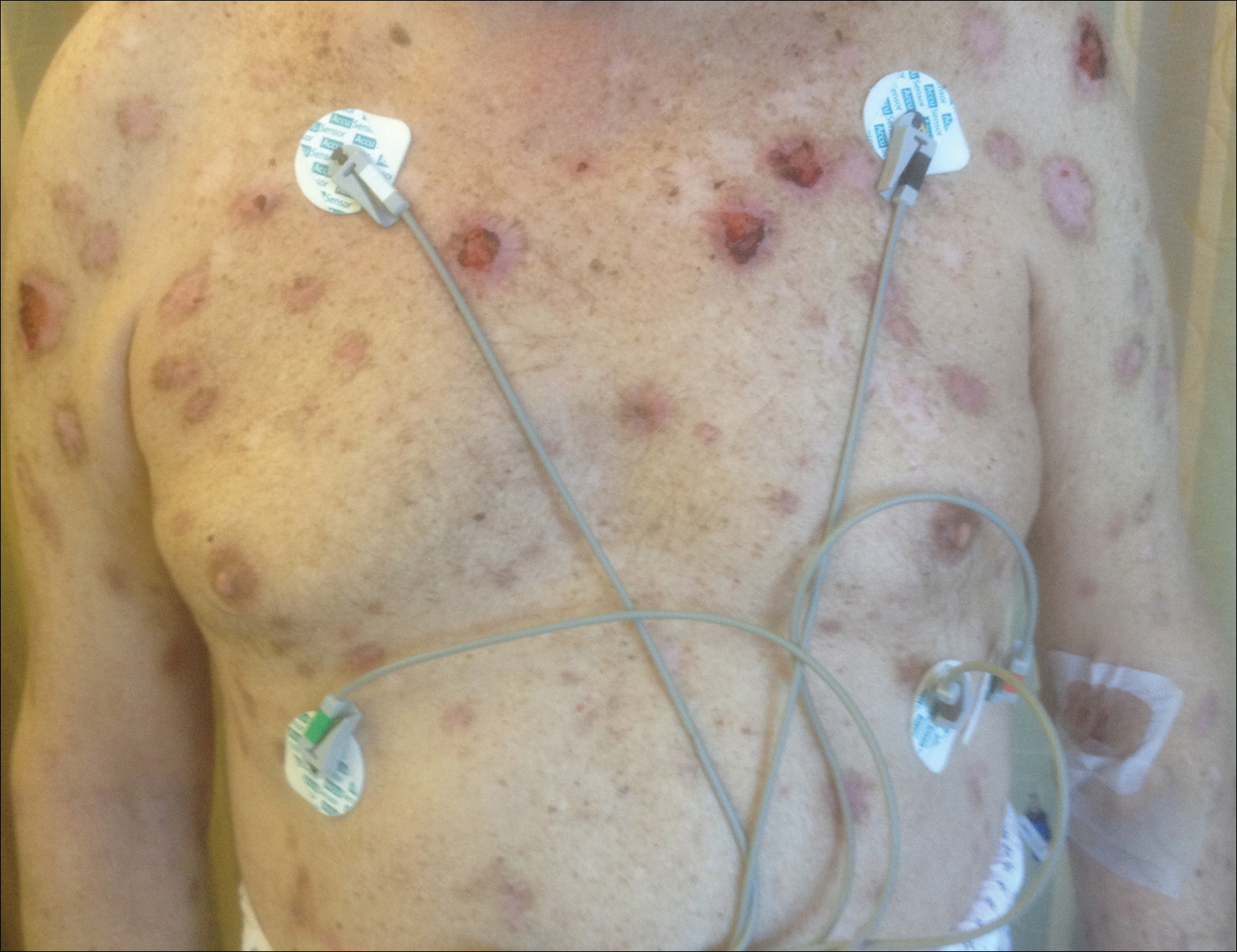
Comment
Paraneoplastic BP has been documented; however, lymphoepithelial cysts of the pancreas in association with BP are rare. We propose that the lymphoepithelial cyst of the pancreas provided the immunologic stimulus for the development of cutaneous BP based on the observation that our patient’s condition remarkably improved with resection of the tumor.
There are fewer than 100 cases of lymphoepithelial cysts of the pancreas reported in the literature.8 The histologic appearance is consistent with a true cyst exhibiting a well-differentiated stratified squamous epithelium, often with keratinization, surrounded by lymphoid tissue. These tumors are most commonly seen in middle-aged men and are frequently found incidentally,8-10 as was the case with our patient. Although histologically similar, lymphoepithelial cysts of the pancreas are considered distinct from lymphoepithelial cysts of the parotid gland or head and neck region.10 Lymphoepithelial cysts of the pancreas are unrelated to elevated glucagon levels; it is likely that our patient’s glucagon levels were associated with his history of diabetes.11
The diagnosis of BP is characteristically confirmed by direct immunofluorescence. Although it was performed for our patient’s cutaneous lesions, it was not obtained for the lymphoepithelial cyst of the pancreas. Once the diagnosis of the lymphoepithelial cyst of the pancreas was established, as direct immunofluorescence could not be performed in formalin-fixed tissue, immunoperoxidase staining with C3d was obtained. C3 has a well-established role in activation of complement and as a marker in BP. Deposition of C3d is a result of deactivation of C3b, a cleavage product of C3. In a study of 6 autoimmune blistering disorders that included 32 patients with BP, Pfaltz et al12 found positive immunoperoxidase staining for C3d in 31 of 32 patients, which translated to a sensitivity of 97%, a positive predictive value of 100%, and a negative predictive value of 98% among the blistering diseases being studied. Similarly, Magro and Dyrsen13 had positive staining of C3d in 17 of 17 (100%) patients with BP.
In theory, any process that involves deposition of C3 should be positive for C3d on immunoperoxidase staining. Other dermatologic inflammatory conditions stain positively with C3d, such as systemic lupus erythematosus, discoid lupus erythematosus, subacute cutaneous lupus erythematosus, and dermatomysositis.13 The staining for these diseases correlates with the site of the associated inflammatory component seen on hematoxylin and eosin staining. The staining of C3d along the basement membrane of stratified squamous epithelium in the lymphoepithelial cyst of the pancreas seen in our patient closely resembles the staining seen in cutaneous BP.
A proposed mechanism for BP in our patient would be exposure of BP-1 antigen in the pancreatic cyst leading to antibody recognition and C3 deposition along the basement membrane in the cyst, as evidenced by C3d immunoperoxidase staining. The IgG and C3 deposition along the cutaneous basement membrane would then represent a systemic response to the antigen exposure in the cyst. Thus, the lymphoepithelial cyst provided the immunologic stimulus for the development of the cutaneous BP. This theory is based on the observation of our patient’s rapid improvement without a change in his treatment regimen immediately after surgical excision of the cyst.
Despite the plausibility of our hypothesis, several questions remain regarding the validity of our assumptions. Although sensitive for C3 deposition, C3d immunoperoxidase staining is not specific for BP. If the proposed mechanism for causation is true, one might have expected that a subepithelial cleft along the basement membrane of the pancreatic cyst would be observed, which was not seen. A repeat BP antigen antibody was not obtained, which would have been helpful in determining if there was clearance of the antibody that would have correlated with the clinical resolution of the BP lesions.
Conclusion
Our case suggests that paraneoplastic BP is a genuine entity. Indeed, the primary tumor itself may be the immunologic stimulus in the development of BP. Recalcitrant BP should raise the question of a neoplastic process that is exposing the BP antigen. If a thorough review of systems accompanied by corroborating laboratory studies suggests a neoplastic process, the suspect lesion should be further evaluated and surgically excised if clinically indicated. Further evaluation of neoplasms with advanced staining methods may aid in establishing the causative nature of tumors in the development of BP.
Acknowledgments
We are grateful to John Stanley, MD, and Aimee Payne, MD (both from Philadelphia, Pennsylvania), for theirinsights into this case.
- Charneux J, Lorin J, Vitry F, et al. Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:286-291.
- Patel M, Sniha AA, Gilbert E. Bullous pemphigoid associated with renal cell carcinoma and invasive squamous cell carcinoma. J Drugs Dermatol. 2012;11:234-238.
- Song HJ, Han SH, Hong WK, et al. Paraneoplastic bullous pemphigoid: clinical disease activity correlated with enzyme-linked immunosorbent assay index for NC16A domain of BP180. J Dermatol. 2009;36:66-68.
- Muramatsu T, Iida T, Tada H, et al. Bullous pemphigoid associated with internal malignancies: identification of 180-kDa antigen by Western immunoblotting. Br J Dermatol. 1996;135:782-784.
- Ogawa H, Sakuma M, Morioka S, et al. The incidence of internal malignancies in pemphigus and bullous pemphigoid in Japan. J Dermatol Sci. 1995;9:136-141.
- Boyd RV. Pemphigoid and carcinoma of the pancreas. Br Med J. 1964;1:1092.
- Eustace S, Morrow G, O’Loughlin S, et al. The role of computed tomography and sonography in acute bullous pemphigoid. Ir J Med Sci. 1993;162:401-404.
- Clemente G, Sarno G, De Rose AM, et al. Lymphoepithelial cyst of the pancreas: case report and review of the literature. Acta Gastroenterol Belg. 2011;74:343-346.
- Frezza E, Wachtel MS. Lymphoepithelial cyst of the pancreas tail. case report and review of the literature. JOP. 2008;9:46-49.
- Basturk O, Coban I, Adsay NV. Pancreatic cysts: pathologic classification, differential diagnosis and clinical implications. Arch Pathol Lab Med. 2009;133:423-438.
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4-12.
- Pfaltz K, Mertz K, Rose C, et al. C3d immunohistochemistry on formalin-fixed tissue is a valuable tool in the diagnosis of bullous pemphigoid of the skin. J Cutan Pathol. 2010;37:654-658.
- Magro CM, Dyrsen ME. The use of C3d and C4d immunohistochemistry on formalin-fixed tissue as a diagnostic adjunct in the assessment of inflammatory skin disease. J Am Acad Dermatol. 2008;59:822-833.
Bullous pemphigoid (BP) is an acquired, autoimmune, subepidermal blistering disease that is more common in elderly patients.1 An association with internal neoplasms and BP has been established; however, there is debate regarding the precise nature of the relationship.2 Several gastrointestinal tract tumors have been associated with BP, including adenocarcinoma of the colon, adenosquamous cell carcinoma and adenocarcinoma of the stomach, adenocarcinoma of the rectum, and liver and bile duct malignancies.3-5 Association with pancreatic neoplasms (eg, carcinoma of the pancreas) rarely has been reported.5-7 We present an unusual case of a lymphoepithelial cyst of the pancreas in a patient with BP.
Case Report
A 67-year-old man presented with erythematous crusted plaques and pink scars over the chest, back, arms, and legs (Figure 1). A 1.5-cm tense bullous lesion was observed on the right knee. The patient’s medical history was notable for biopsy-proven BP of 8 months’ duration as well as diabetes mellitus and hypothyroidism. The patient was being followed by his surgeon for a 1.9-cm soft-tissue lesion in the pancreatic tail and was awaiting surgical excision at the time of the current presentation. The pancreatic lesion was discovered incidentally on magnetic resonance imaging performed following urologic concerns. At the current presentation, the patient’s medications included nifedipine, hydralazine, metoprolol, torsemide, aspirin, levothyroxine, atorvastatin, insulin lispro, and insulin glargine. He had previously been treated for BP with prednisone at a maximum dosage of 60 mg daily, clobetasol propionate cream 0.05%, and mupirocin ointment 2% without improvement. Because of substantial weight gain and poorly controlled diabetes, prednisone was discontinued.
Bullous pemphigoid had been diagnosed histopathologically by a prior dermatologist. Hematoxylin and eosin staining demonstrated a subepidermal separation with eosinophils within the perivascular infiltrate (Figure 2). Direct immunofluorescence was noted in a linear pattern at the dermoepidermal junction with IgG and C3. Bullous pemphigoid antigen antibodies 1 and 2 were obtained via enzyme-linked immunosorbent assay with a positive BP-1 antigen antibody of 19 U/mL (positive, >15 U/mL) and a normal BP-2 antigen antibody of less than 9 U/mL (reference range, <9 U/mL). The glucagon level was elevated at 245 pg/mL (reference range, ≤134 pg/mL).
The patient was prescribed minocycline 100 mg twice daily and niacinamide 500 mg 3 times daily. Topical treatment with clobetasol and mupirocin was continued. One month later, the patient returned with an increase in disease activity. Changes to his therapeutic regimen were deferred until after excision of the pancreatic lesion based on the decision not to start immunosuppressive therapy until the precise nature of the pancreatic lesion was determined.
The patient underwent excision of the pancreatic lesion approximately 3 months later, which proved to be a benign lymphoepithelial cyst of the pancreas. Histology of the cyst consisted of dense fibrous tissue with a squamous epithelial lining focally infiltrated by lymphocytes (Figure 3A). Immunoperoxidase staining of the cyst revealed focal linear areas of C3d staining along the basement membrane of the stratified squamous epithelium (Figure 3B).
The patient stated that his skin started to improve virtually immediately following the excision without systemic treatment for BP. On follow-up examination 3 weeks postoperatively, no bullae were observed and there was a notable decrease in erythematous crusted plaques (Figure 4).
Comment
Paraneoplastic BP has been documented; however, lymphoepithelial cysts of the pancreas in association with BP are rare. We propose that the lymphoepithelial cyst of the pancreas provided the immunologic stimulus for the development of cutaneous BP based on the observation that our patient’s condition remarkably improved with resection of the tumor.
There are fewer than 100 cases of lymphoepithelial cysts of the pancreas reported in the literature.8 The histologic appearance is consistent with a true cyst exhibiting a well-differentiated stratified squamous epithelium, often with keratinization, surrounded by lymphoid tissue. These tumors are most commonly seen in middle-aged men and are frequently found incidentally,8-10 as was the case with our patient. Although histologically similar, lymphoepithelial cysts of the pancreas are considered distinct from lymphoepithelial cysts of the parotid gland or head and neck region.10 Lymphoepithelial cysts of the pancreas are unrelated to elevated glucagon levels; it is likely that our patient’s glucagon levels were associated with his history of diabetes.11
The diagnosis of BP is characteristically confirmed by direct immunofluorescence. Although it was performed for our patient’s cutaneous lesions, it was not obtained for the lymphoepithelial cyst of the pancreas. Once the diagnosis of the lymphoepithelial cyst of the pancreas was established, as direct immunofluorescence could not be performed in formalin-fixed tissue, immunoperoxidase staining with C3d was obtained. C3 has a well-established role in activation of complement and as a marker in BP. Deposition of C3d is a result of deactivation of C3b, a cleavage product of C3. In a study of 6 autoimmune blistering disorders that included 32 patients with BP, Pfaltz et al12 found positive immunoperoxidase staining for C3d in 31 of 32 patients, which translated to a sensitivity of 97%, a positive predictive value of 100%, and a negative predictive value of 98% among the blistering diseases being studied. Similarly, Magro and Dyrsen13 had positive staining of C3d in 17 of 17 (100%) patients with BP.
In theory, any process that involves deposition of C3 should be positive for C3d on immunoperoxidase staining. Other dermatologic inflammatory conditions stain positively with C3d, such as systemic lupus erythematosus, discoid lupus erythematosus, subacute cutaneous lupus erythematosus, and dermatomysositis.13 The staining for these diseases correlates with the site of the associated inflammatory component seen on hematoxylin and eosin staining. The staining of C3d along the basement membrane of stratified squamous epithelium in the lymphoepithelial cyst of the pancreas seen in our patient closely resembles the staining seen in cutaneous BP.
A proposed mechanism for BP in our patient would be exposure of BP-1 antigen in the pancreatic cyst leading to antibody recognition and C3 deposition along the basement membrane in the cyst, as evidenced by C3d immunoperoxidase staining. The IgG and C3 deposition along the cutaneous basement membrane would then represent a systemic response to the antigen exposure in the cyst. Thus, the lymphoepithelial cyst provided the immunologic stimulus for the development of the cutaneous BP. This theory is based on the observation of our patient’s rapid improvement without a change in his treatment regimen immediately after surgical excision of the cyst.
Despite the plausibility of our hypothesis, several questions remain regarding the validity of our assumptions. Although sensitive for C3 deposition, C3d immunoperoxidase staining is not specific for BP. If the proposed mechanism for causation is true, one might have expected that a subepithelial cleft along the basement membrane of the pancreatic cyst would be observed, which was not seen. A repeat BP antigen antibody was not obtained, which would have been helpful in determining if there was clearance of the antibody that would have correlated with the clinical resolution of the BP lesions.
Conclusion
Our case suggests that paraneoplastic BP is a genuine entity. Indeed, the primary tumor itself may be the immunologic stimulus in the development of BP. Recalcitrant BP should raise the question of a neoplastic process that is exposing the BP antigen. If a thorough review of systems accompanied by corroborating laboratory studies suggests a neoplastic process, the suspect lesion should be further evaluated and surgically excised if clinically indicated. Further evaluation of neoplasms with advanced staining methods may aid in establishing the causative nature of tumors in the development of BP.
Acknowledgments
We are grateful to John Stanley, MD, and Aimee Payne, MD (both from Philadelphia, Pennsylvania), for theirinsights into this case.
Bullous pemphigoid (BP) is an acquired, autoimmune, subepidermal blistering disease that is more common in elderly patients.1 An association with internal neoplasms and BP has been established; however, there is debate regarding the precise nature of the relationship.2 Several gastrointestinal tract tumors have been associated with BP, including adenocarcinoma of the colon, adenosquamous cell carcinoma and adenocarcinoma of the stomach, adenocarcinoma of the rectum, and liver and bile duct malignancies.3-5 Association with pancreatic neoplasms (eg, carcinoma of the pancreas) rarely has been reported.5-7 We present an unusual case of a lymphoepithelial cyst of the pancreas in a patient with BP.
Case Report
A 67-year-old man presented with erythematous crusted plaques and pink scars over the chest, back, arms, and legs (Figure 1). A 1.5-cm tense bullous lesion was observed on the right knee. The patient’s medical history was notable for biopsy-proven BP of 8 months’ duration as well as diabetes mellitus and hypothyroidism. The patient was being followed by his surgeon for a 1.9-cm soft-tissue lesion in the pancreatic tail and was awaiting surgical excision at the time of the current presentation. The pancreatic lesion was discovered incidentally on magnetic resonance imaging performed following urologic concerns. At the current presentation, the patient’s medications included nifedipine, hydralazine, metoprolol, torsemide, aspirin, levothyroxine, atorvastatin, insulin lispro, and insulin glargine. He had previously been treated for BP with prednisone at a maximum dosage of 60 mg daily, clobetasol propionate cream 0.05%, and mupirocin ointment 2% without improvement. Because of substantial weight gain and poorly controlled diabetes, prednisone was discontinued.
Bullous pemphigoid had been diagnosed histopathologically by a prior dermatologist. Hematoxylin and eosin staining demonstrated a subepidermal separation with eosinophils within the perivascular infiltrate (Figure 2). Direct immunofluorescence was noted in a linear pattern at the dermoepidermal junction with IgG and C3. Bullous pemphigoid antigen antibodies 1 and 2 were obtained via enzyme-linked immunosorbent assay with a positive BP-1 antigen antibody of 19 U/mL (positive, >15 U/mL) and a normal BP-2 antigen antibody of less than 9 U/mL (reference range, <9 U/mL). The glucagon level was elevated at 245 pg/mL (reference range, ≤134 pg/mL).
The patient was prescribed minocycline 100 mg twice daily and niacinamide 500 mg 3 times daily. Topical treatment with clobetasol and mupirocin was continued. One month later, the patient returned with an increase in disease activity. Changes to his therapeutic regimen were deferred until after excision of the pancreatic lesion based on the decision not to start immunosuppressive therapy until the precise nature of the pancreatic lesion was determined.
The patient underwent excision of the pancreatic lesion approximately 3 months later, which proved to be a benign lymphoepithelial cyst of the pancreas. Histology of the cyst consisted of dense fibrous tissue with a squamous epithelial lining focally infiltrated by lymphocytes (Figure 3A). Immunoperoxidase staining of the cyst revealed focal linear areas of C3d staining along the basement membrane of the stratified squamous epithelium (Figure 3B).
The patient stated that his skin started to improve virtually immediately following the excision without systemic treatment for BP. On follow-up examination 3 weeks postoperatively, no bullae were observed and there was a notable decrease in erythematous crusted plaques (Figure 4).
Comment
Paraneoplastic BP has been documented; however, lymphoepithelial cysts of the pancreas in association with BP are rare. We propose that the lymphoepithelial cyst of the pancreas provided the immunologic stimulus for the development of cutaneous BP based on the observation that our patient’s condition remarkably improved with resection of the tumor.
There are fewer than 100 cases of lymphoepithelial cysts of the pancreas reported in the literature.8 The histologic appearance is consistent with a true cyst exhibiting a well-differentiated stratified squamous epithelium, often with keratinization, surrounded by lymphoid tissue. These tumors are most commonly seen in middle-aged men and are frequently found incidentally,8-10 as was the case with our patient. Although histologically similar, lymphoepithelial cysts of the pancreas are considered distinct from lymphoepithelial cysts of the parotid gland or head and neck region.10 Lymphoepithelial cysts of the pancreas are unrelated to elevated glucagon levels; it is likely that our patient’s glucagon levels were associated with his history of diabetes.11
The diagnosis of BP is characteristically confirmed by direct immunofluorescence. Although it was performed for our patient’s cutaneous lesions, it was not obtained for the lymphoepithelial cyst of the pancreas. Once the diagnosis of the lymphoepithelial cyst of the pancreas was established, as direct immunofluorescence could not be performed in formalin-fixed tissue, immunoperoxidase staining with C3d was obtained. C3 has a well-established role in activation of complement and as a marker in BP. Deposition of C3d is a result of deactivation of C3b, a cleavage product of C3. In a study of 6 autoimmune blistering disorders that included 32 patients with BP, Pfaltz et al12 found positive immunoperoxidase staining for C3d in 31 of 32 patients, which translated to a sensitivity of 97%, a positive predictive value of 100%, and a negative predictive value of 98% among the blistering diseases being studied. Similarly, Magro and Dyrsen13 had positive staining of C3d in 17 of 17 (100%) patients with BP.
In theory, any process that involves deposition of C3 should be positive for C3d on immunoperoxidase staining. Other dermatologic inflammatory conditions stain positively with C3d, such as systemic lupus erythematosus, discoid lupus erythematosus, subacute cutaneous lupus erythematosus, and dermatomysositis.13 The staining for these diseases correlates with the site of the associated inflammatory component seen on hematoxylin and eosin staining. The staining of C3d along the basement membrane of stratified squamous epithelium in the lymphoepithelial cyst of the pancreas seen in our patient closely resembles the staining seen in cutaneous BP.
A proposed mechanism for BP in our patient would be exposure of BP-1 antigen in the pancreatic cyst leading to antibody recognition and C3 deposition along the basement membrane in the cyst, as evidenced by C3d immunoperoxidase staining. The IgG and C3 deposition along the cutaneous basement membrane would then represent a systemic response to the antigen exposure in the cyst. Thus, the lymphoepithelial cyst provided the immunologic stimulus for the development of the cutaneous BP. This theory is based on the observation of our patient’s rapid improvement without a change in his treatment regimen immediately after surgical excision of the cyst.
Despite the plausibility of our hypothesis, several questions remain regarding the validity of our assumptions. Although sensitive for C3 deposition, C3d immunoperoxidase staining is not specific for BP. If the proposed mechanism for causation is true, one might have expected that a subepithelial cleft along the basement membrane of the pancreatic cyst would be observed, which was not seen. A repeat BP antigen antibody was not obtained, which would have been helpful in determining if there was clearance of the antibody that would have correlated with the clinical resolution of the BP lesions.
Conclusion
Our case suggests that paraneoplastic BP is a genuine entity. Indeed, the primary tumor itself may be the immunologic stimulus in the development of BP. Recalcitrant BP should raise the question of a neoplastic process that is exposing the BP antigen. If a thorough review of systems accompanied by corroborating laboratory studies suggests a neoplastic process, the suspect lesion should be further evaluated and surgically excised if clinically indicated. Further evaluation of neoplasms with advanced staining methods may aid in establishing the causative nature of tumors in the development of BP.
Acknowledgments
We are grateful to John Stanley, MD, and Aimee Payne, MD (both from Philadelphia, Pennsylvania), for theirinsights into this case.
- Charneux J, Lorin J, Vitry F, et al. Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:286-291.
- Patel M, Sniha AA, Gilbert E. Bullous pemphigoid associated with renal cell carcinoma and invasive squamous cell carcinoma. J Drugs Dermatol. 2012;11:234-238.
- Song HJ, Han SH, Hong WK, et al. Paraneoplastic bullous pemphigoid: clinical disease activity correlated with enzyme-linked immunosorbent assay index for NC16A domain of BP180. J Dermatol. 2009;36:66-68.
- Muramatsu T, Iida T, Tada H, et al. Bullous pemphigoid associated with internal malignancies: identification of 180-kDa antigen by Western immunoblotting. Br J Dermatol. 1996;135:782-784.
- Ogawa H, Sakuma M, Morioka S, et al. The incidence of internal malignancies in pemphigus and bullous pemphigoid in Japan. J Dermatol Sci. 1995;9:136-141.
- Boyd RV. Pemphigoid and carcinoma of the pancreas. Br Med J. 1964;1:1092.
- Eustace S, Morrow G, O’Loughlin S, et al. The role of computed tomography and sonography in acute bullous pemphigoid. Ir J Med Sci. 1993;162:401-404.
- Clemente G, Sarno G, De Rose AM, et al. Lymphoepithelial cyst of the pancreas: case report and review of the literature. Acta Gastroenterol Belg. 2011;74:343-346.
- Frezza E, Wachtel MS. Lymphoepithelial cyst of the pancreas tail. case report and review of the literature. JOP. 2008;9:46-49.
- Basturk O, Coban I, Adsay NV. Pancreatic cysts: pathologic classification, differential diagnosis and clinical implications. Arch Pathol Lab Med. 2009;133:423-438.
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4-12.
- Pfaltz K, Mertz K, Rose C, et al. C3d immunohistochemistry on formalin-fixed tissue is a valuable tool in the diagnosis of bullous pemphigoid of the skin. J Cutan Pathol. 2010;37:654-658.
- Magro CM, Dyrsen ME. The use of C3d and C4d immunohistochemistry on formalin-fixed tissue as a diagnostic adjunct in the assessment of inflammatory skin disease. J Am Acad Dermatol. 2008;59:822-833.
- Charneux J, Lorin J, Vitry F, et al. Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:286-291.
- Patel M, Sniha AA, Gilbert E. Bullous pemphigoid associated with renal cell carcinoma and invasive squamous cell carcinoma. J Drugs Dermatol. 2012;11:234-238.
- Song HJ, Han SH, Hong WK, et al. Paraneoplastic bullous pemphigoid: clinical disease activity correlated with enzyme-linked immunosorbent assay index for NC16A domain of BP180. J Dermatol. 2009;36:66-68.
- Muramatsu T, Iida T, Tada H, et al. Bullous pemphigoid associated with internal malignancies: identification of 180-kDa antigen by Western immunoblotting. Br J Dermatol. 1996;135:782-784.
- Ogawa H, Sakuma M, Morioka S, et al. The incidence of internal malignancies in pemphigus and bullous pemphigoid in Japan. J Dermatol Sci. 1995;9:136-141.
- Boyd RV. Pemphigoid and carcinoma of the pancreas. Br Med J. 1964;1:1092.
- Eustace S, Morrow G, O’Loughlin S, et al. The role of computed tomography and sonography in acute bullous pemphigoid. Ir J Med Sci. 1993;162:401-404.
- Clemente G, Sarno G, De Rose AM, et al. Lymphoepithelial cyst of the pancreas: case report and review of the literature. Acta Gastroenterol Belg. 2011;74:343-346.
- Frezza E, Wachtel MS. Lymphoepithelial cyst of the pancreas tail. case report and review of the literature. JOP. 2008;9:46-49.
- Basturk O, Coban I, Adsay NV. Pancreatic cysts: pathologic classification, differential diagnosis and clinical implications. Arch Pathol Lab Med. 2009;133:423-438.
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4-12.
- Pfaltz K, Mertz K, Rose C, et al. C3d immunohistochemistry on formalin-fixed tissue is a valuable tool in the diagnosis of bullous pemphigoid of the skin. J Cutan Pathol. 2010;37:654-658.
- Magro CM, Dyrsen ME. The use of C3d and C4d immunohistochemistry on formalin-fixed tissue as a diagnostic adjunct in the assessment of inflammatory skin disease. J Am Acad Dermatol. 2008;59:822-833.
Pruritic Papules on the Scalp and Arms
Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) that occurs mostly in adults with a male predilection. The disease clinically favors the head and neck. Patients commonly present with pruritic papules that often are grouped, alopecia, and frequent secondary bacterial infections. Less commonly patients present with acneiform lesions and mucinorrhea. Patients often experience more pruritus in FMF than in classic MF, which can provide a good means of assessing disease activity. Disease-specific 5-year survival is approximately 70% to 80%, which is worse than classic plaque-stage MF and similar to tumor-stage MF.1
Treatment of FMF differs from classic MF in that the lesions are less responsive to skin-targeted therapies due to the perifollicular nature of dermal infiltrates. Superficial skin lesions can be treated with psoralen plus UVA (PUVA) therapy. Other options include PUVA in combination with interferon alfa or retinoids and local radiotherapy for solitary thick tumors; however, in patients who have more infiltrative skin lesions or had PUVA therapy that failed, total skin electron beam therapy may be required.2
On histologic examination, there typically is perivascular and periadnexal localization of dermal infiltrates with varied involvement of the follicular epithelium and damage to hair follicles by atypical small, medium, and large hyperchromatic lymphocytes with cerebriform nuclei. Mucinous degeneration of the follicular epithelium can be seen, as highlighted on Alcian blue staining, and a mixed infiltrate of eosinophils and plasma cells often is present (quiz image and Figure 1). Frequent sparing of the epidermis is noteworthy.2-4 In most cases, the neoplastic T lymphocytes are characterized by a CD3+CD4+CD8-immunophenotype as is seen in classic MF. Sometimes an admixture of CD30+ blast cells is seen.1
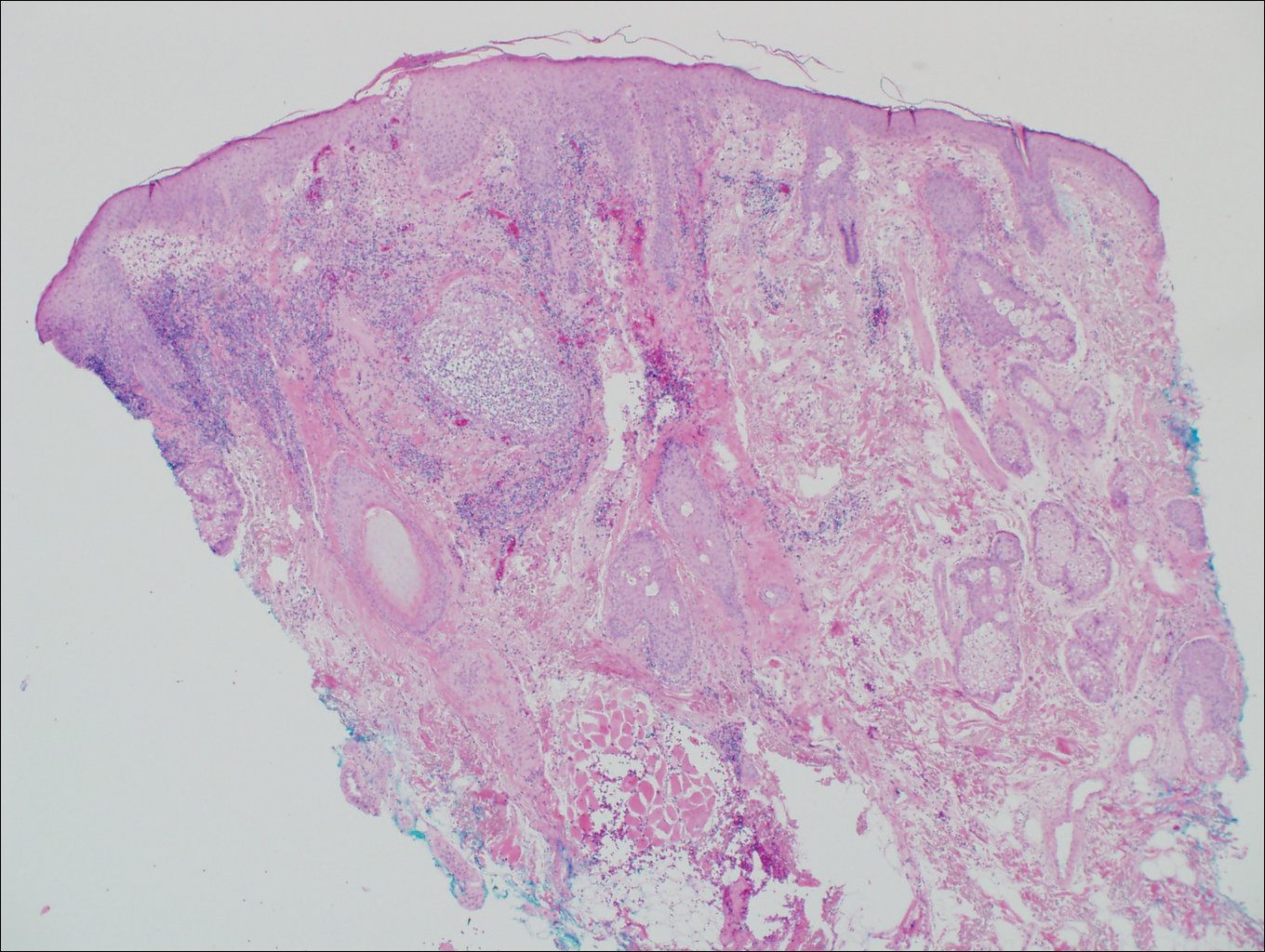
Histologic differential considerations for FMF include eosinophilic pustular folliculitis (EPF), primary follicular mucinosis, lupus erythematosus, and pityrosporum folliculitis.
Eosinophilic pustular folliculitis has several clinical subtypes, such as classic Ofuji disease and immunosuppression-associated EPF secondary to human immunodeficiency virus. Histologically, EPF is characterized by spongiosis of the hair follicle epithelium with exocytosis of a mixed infiltrate of lymphocytes and eosinophils extending from the sebaceous gland and its duct to the infundibulum with formation of hallmark eosinophilic pustules (Figure 2). Infiltration of neutrophils in inflamed lesions generally is seen. Eosinophilic pustular folliculitis is an important differential for FMF, as follicular mucinosis has been observed in lesions of EPF.5 Both EPF and FMF can exhibit eosinophils and lymphocytes in the upper dermis, spongiosis of the hair follicle epithelium, and mucinous degeneration of follicles,6 though lymphocytic atypia and relatively fewer eosinophils are suggestive of the latter.
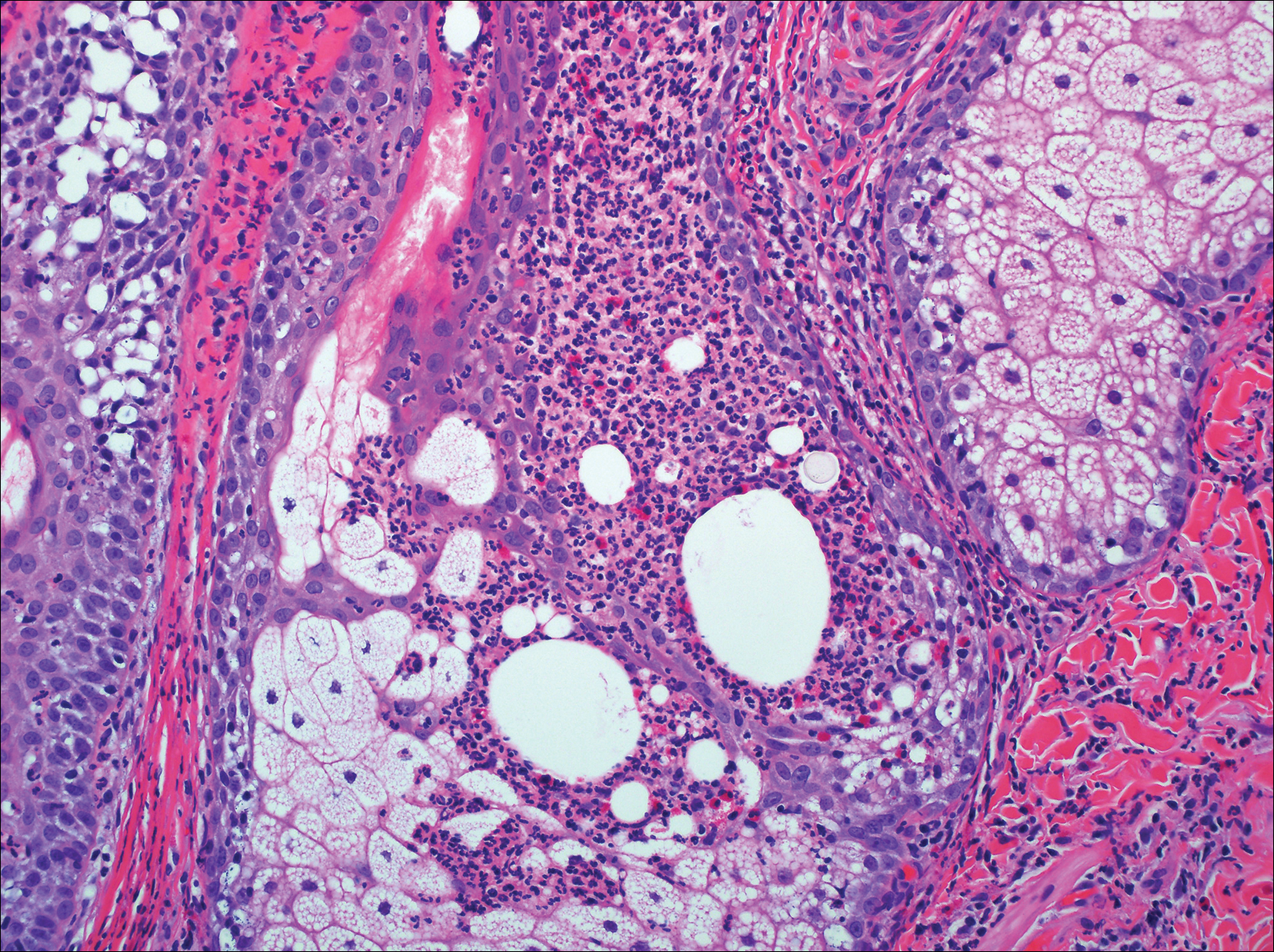
Primary follicular mucinosis (PFM) tends to occur as a solitary lesion in younger female patients in contrast to the multiple lesions that typically appear in older male patients with FMF. Histologically, PFM usually manifests as large, cystic, mucin-filled spaces and polyclonal perivascular and periadnexal lymphocytic infiltrate without notable cellular atypia or epidermotropism (Figure 3). Because follicular mucinosis is a common feature of FMF, its distinction from PFM can be challenging and often is aided by the absence of cellular atypia and relatively mild lymphocytic infiltrate in the latter.7
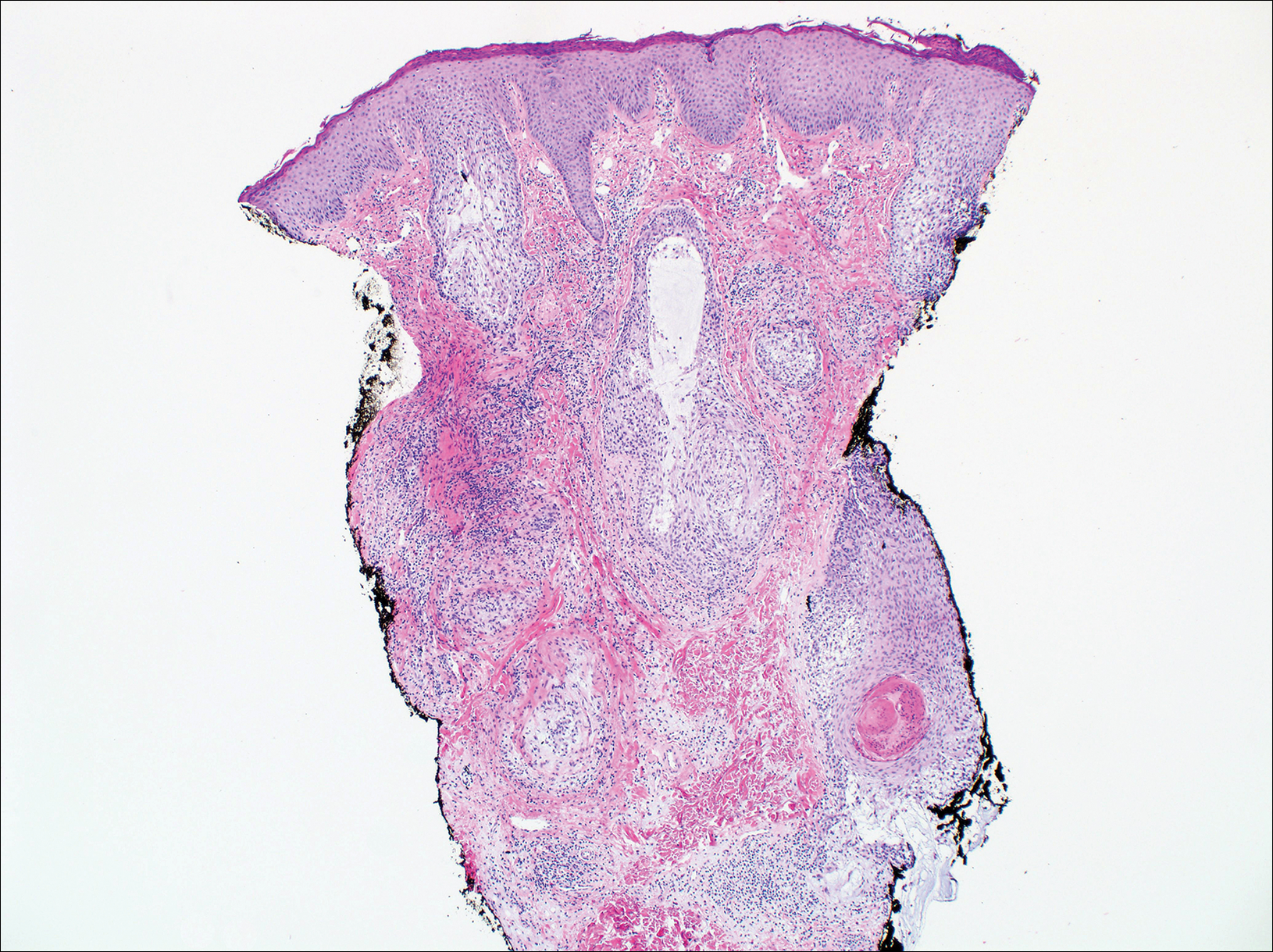
Cutaneous lupus erythematosus with its characteristic folliculocentric lymphocytic infiltration and associated dermal mucin also qualifies as a potential differential possibility for FMF; however, the perivascular and periadnexal pattern of lymphocytic infiltration as well as the localization of mucin to the reticular dermal interstitium8,9 are key histopathologic distinctions (Figure 4). Furthermore, although the histologic presentation of lupus erythematosus can be variable, it also classically shows interface dermatitis, basement membrane thickening, and follicular plugging.
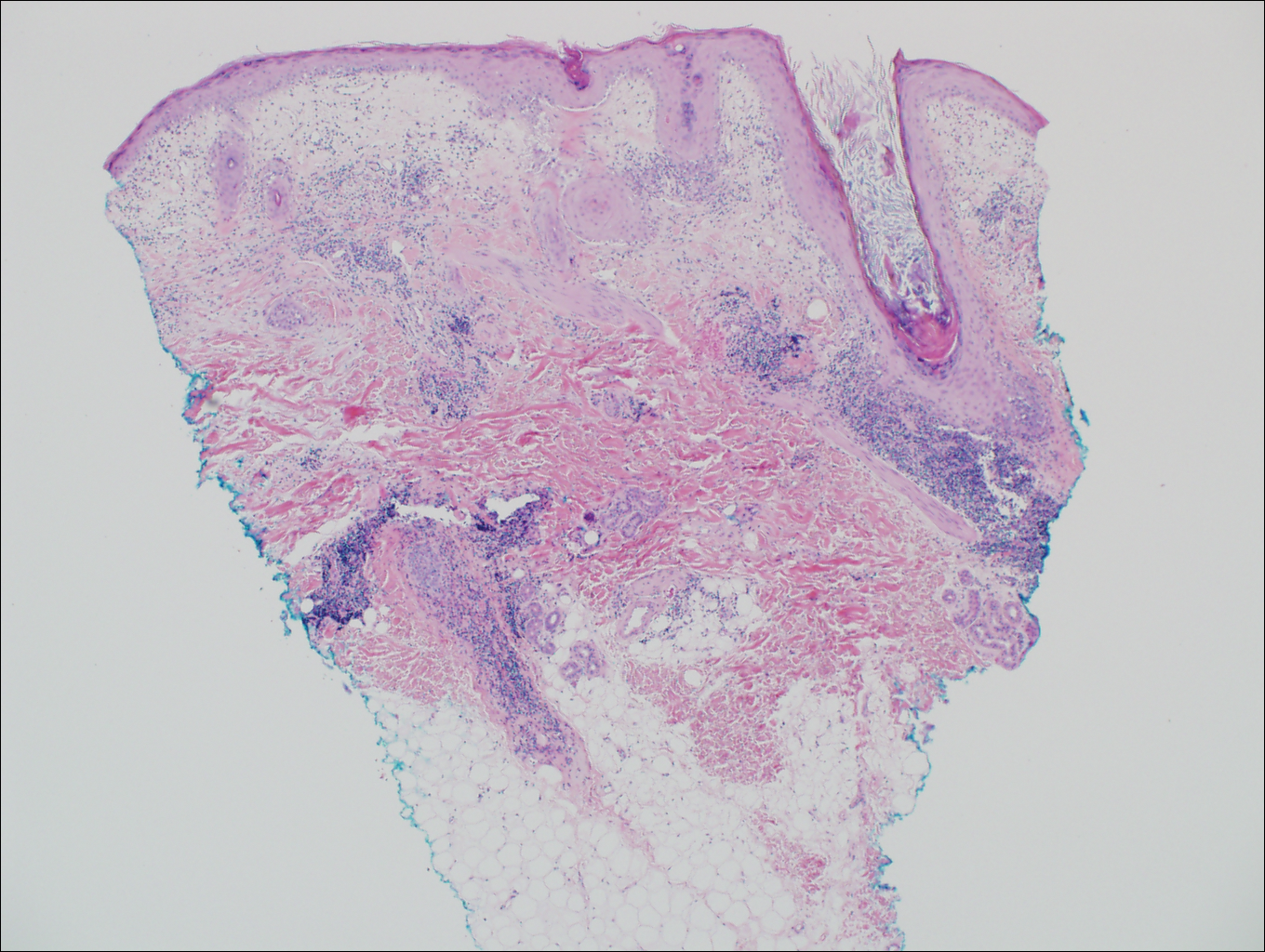
Pityrosporum folliculitis is the most common cause of fungal folliculitis and is caused by the Malassezia species. On histology, there typically is an unremarkable epithelium with plugged follicles and suppurative folliculitis. Serial sections of the biopsy specimen often are required to identify dilated, follicle-containing, budding yeast cells (Figure 5). Organisms are located predominantly within the infundibulum and orifice of follicular lumen, are positive for periodic acid-Schiff, and are diastase resistant.10
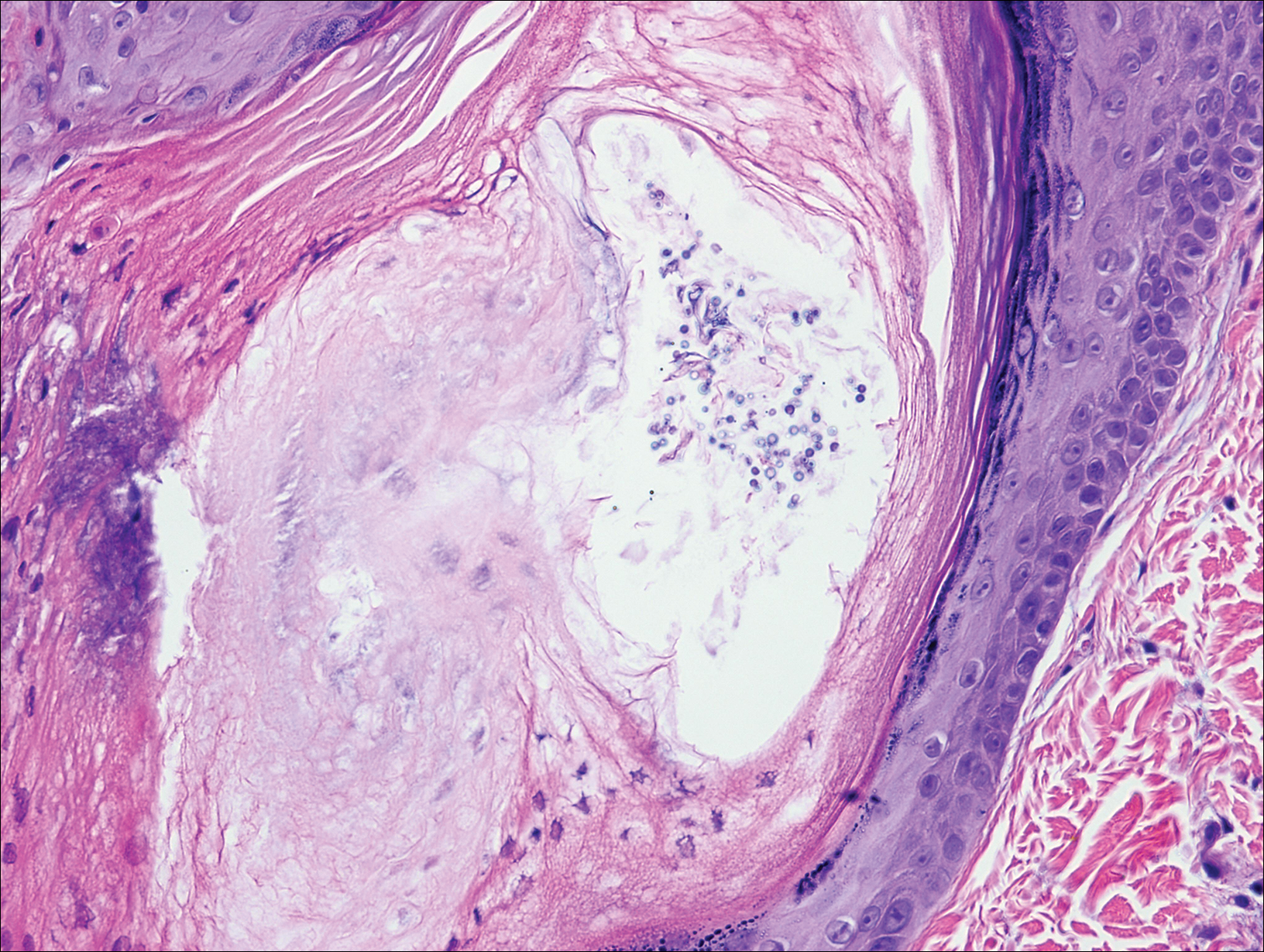
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides, a distinct disease entity with or without associated follicular mucinosis. a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198.
- DeBloom J 2nd, Severson J, Gaspari A, et al. Follicular mycosis fungoides: a case report and review of the literature. J Cutan Pathol. 2001;28:318-324.
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525-530.
- Fujiyama T, Tokura Y. Clinical and histopathological differential diagnosis of eosinophilic pustular folliculitis. J Dermatol. 2013;40:419-423.
- Lee JY, Tsai YM, Sheu HM. Ofuji's disease with follicular mucinosis and its differential diagnosis from alopecia mucinosa. J Cutan Pathol. 2003;30:307-313.
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19.
- Vincent JG, Chan MP. Specificity of dermal mucin in the diagnosis of lupus erythematosus: comparison with other dermatitides and normal skin. J Cutan Pathol. 2015;42:722-729.
- Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol. 1996;135:355-362.
- Durdu M, Ilkit M. First step in the differential diagnosis of folliculitis: cytology. Crit Rev Microbiol. 2013;39:9-25.
Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) that occurs mostly in adults with a male predilection. The disease clinically favors the head and neck. Patients commonly present with pruritic papules that often are grouped, alopecia, and frequent secondary bacterial infections. Less commonly patients present with acneiform lesions and mucinorrhea. Patients often experience more pruritus in FMF than in classic MF, which can provide a good means of assessing disease activity. Disease-specific 5-year survival is approximately 70% to 80%, which is worse than classic plaque-stage MF and similar to tumor-stage MF.1
Treatment of FMF differs from classic MF in that the lesions are less responsive to skin-targeted therapies due to the perifollicular nature of dermal infiltrates. Superficial skin lesions can be treated with psoralen plus UVA (PUVA) therapy. Other options include PUVA in combination with interferon alfa or retinoids and local radiotherapy for solitary thick tumors; however, in patients who have more infiltrative skin lesions or had PUVA therapy that failed, total skin electron beam therapy may be required.2
On histologic examination, there typically is perivascular and periadnexal localization of dermal infiltrates with varied involvement of the follicular epithelium and damage to hair follicles by atypical small, medium, and large hyperchromatic lymphocytes with cerebriform nuclei. Mucinous degeneration of the follicular epithelium can be seen, as highlighted on Alcian blue staining, and a mixed infiltrate of eosinophils and plasma cells often is present (quiz image and Figure 1). Frequent sparing of the epidermis is noteworthy.2-4 In most cases, the neoplastic T lymphocytes are characterized by a CD3+CD4+CD8-immunophenotype as is seen in classic MF. Sometimes an admixture of CD30+ blast cells is seen.1
Histologic differential considerations for FMF include eosinophilic pustular folliculitis (EPF), primary follicular mucinosis, lupus erythematosus, and pityrosporum folliculitis.
Eosinophilic pustular folliculitis has several clinical subtypes, such as classic Ofuji disease and immunosuppression-associated EPF secondary to human immunodeficiency virus. Histologically, EPF is characterized by spongiosis of the hair follicle epithelium with exocytosis of a mixed infiltrate of lymphocytes and eosinophils extending from the sebaceous gland and its duct to the infundibulum with formation of hallmark eosinophilic pustules (Figure 2). Infiltration of neutrophils in inflamed lesions generally is seen. Eosinophilic pustular folliculitis is an important differential for FMF, as follicular mucinosis has been observed in lesions of EPF.5 Both EPF and FMF can exhibit eosinophils and lymphocytes in the upper dermis, spongiosis of the hair follicle epithelium, and mucinous degeneration of follicles,6 though lymphocytic atypia and relatively fewer eosinophils are suggestive of the latter.
Primary follicular mucinosis (PFM) tends to occur as a solitary lesion in younger female patients in contrast to the multiple lesions that typically appear in older male patients with FMF. Histologically, PFM usually manifests as large, cystic, mucin-filled spaces and polyclonal perivascular and periadnexal lymphocytic infiltrate without notable cellular atypia or epidermotropism (Figure 3). Because follicular mucinosis is a common feature of FMF, its distinction from PFM can be challenging and often is aided by the absence of cellular atypia and relatively mild lymphocytic infiltrate in the latter.7
Cutaneous lupus erythematosus with its characteristic folliculocentric lymphocytic infiltration and associated dermal mucin also qualifies as a potential differential possibility for FMF; however, the perivascular and periadnexal pattern of lymphocytic infiltration as well as the localization of mucin to the reticular dermal interstitium8,9 are key histopathologic distinctions (Figure 4). Furthermore, although the histologic presentation of lupus erythematosus can be variable, it also classically shows interface dermatitis, basement membrane thickening, and follicular plugging.
Pityrosporum folliculitis is the most common cause of fungal folliculitis and is caused by the Malassezia species. On histology, there typically is an unremarkable epithelium with plugged follicles and suppurative folliculitis. Serial sections of the biopsy specimen often are required to identify dilated, follicle-containing, budding yeast cells (Figure 5). Organisms are located predominantly within the infundibulum and orifice of follicular lumen, are positive for periodic acid-Schiff, and are diastase resistant.10
Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) that occurs mostly in adults with a male predilection. The disease clinically favors the head and neck. Patients commonly present with pruritic papules that often are grouped, alopecia, and frequent secondary bacterial infections. Less commonly patients present with acneiform lesions and mucinorrhea. Patients often experience more pruritus in FMF than in classic MF, which can provide a good means of assessing disease activity. Disease-specific 5-year survival is approximately 70% to 80%, which is worse than classic plaque-stage MF and similar to tumor-stage MF.1
Treatment of FMF differs from classic MF in that the lesions are less responsive to skin-targeted therapies due to the perifollicular nature of dermal infiltrates. Superficial skin lesions can be treated with psoralen plus UVA (PUVA) therapy. Other options include PUVA in combination with interferon alfa or retinoids and local radiotherapy for solitary thick tumors; however, in patients who have more infiltrative skin lesions or had PUVA therapy that failed, total skin electron beam therapy may be required.2
On histologic examination, there typically is perivascular and periadnexal localization of dermal infiltrates with varied involvement of the follicular epithelium and damage to hair follicles by atypical small, medium, and large hyperchromatic lymphocytes with cerebriform nuclei. Mucinous degeneration of the follicular epithelium can be seen, as highlighted on Alcian blue staining, and a mixed infiltrate of eosinophils and plasma cells often is present (quiz image and Figure 1). Frequent sparing of the epidermis is noteworthy.2-4 In most cases, the neoplastic T lymphocytes are characterized by a CD3+CD4+CD8-immunophenotype as is seen in classic MF. Sometimes an admixture of CD30+ blast cells is seen.1
Histologic differential considerations for FMF include eosinophilic pustular folliculitis (EPF), primary follicular mucinosis, lupus erythematosus, and pityrosporum folliculitis.
Eosinophilic pustular folliculitis has several clinical subtypes, such as classic Ofuji disease and immunosuppression-associated EPF secondary to human immunodeficiency virus. Histologically, EPF is characterized by spongiosis of the hair follicle epithelium with exocytosis of a mixed infiltrate of lymphocytes and eosinophils extending from the sebaceous gland and its duct to the infundibulum with formation of hallmark eosinophilic pustules (Figure 2). Infiltration of neutrophils in inflamed lesions generally is seen. Eosinophilic pustular folliculitis is an important differential for FMF, as follicular mucinosis has been observed in lesions of EPF.5 Both EPF and FMF can exhibit eosinophils and lymphocytes in the upper dermis, spongiosis of the hair follicle epithelium, and mucinous degeneration of follicles,6 though lymphocytic atypia and relatively fewer eosinophils are suggestive of the latter.
Primary follicular mucinosis (PFM) tends to occur as a solitary lesion in younger female patients in contrast to the multiple lesions that typically appear in older male patients with FMF. Histologically, PFM usually manifests as large, cystic, mucin-filled spaces and polyclonal perivascular and periadnexal lymphocytic infiltrate without notable cellular atypia or epidermotropism (Figure 3). Because follicular mucinosis is a common feature of FMF, its distinction from PFM can be challenging and often is aided by the absence of cellular atypia and relatively mild lymphocytic infiltrate in the latter.7
Cutaneous lupus erythematosus with its characteristic folliculocentric lymphocytic infiltration and associated dermal mucin also qualifies as a potential differential possibility for FMF; however, the perivascular and periadnexal pattern of lymphocytic infiltration as well as the localization of mucin to the reticular dermal interstitium8,9 are key histopathologic distinctions (Figure 4). Furthermore, although the histologic presentation of lupus erythematosus can be variable, it also classically shows interface dermatitis, basement membrane thickening, and follicular plugging.
Pityrosporum folliculitis is the most common cause of fungal folliculitis and is caused by the Malassezia species. On histology, there typically is an unremarkable epithelium with plugged follicles and suppurative folliculitis. Serial sections of the biopsy specimen often are required to identify dilated, follicle-containing, budding yeast cells (Figure 5). Organisms are located predominantly within the infundibulum and orifice of follicular lumen, are positive for periodic acid-Schiff, and are diastase resistant.10
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides, a distinct disease entity with or without associated follicular mucinosis. a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198.
- DeBloom J 2nd, Severson J, Gaspari A, et al. Follicular mycosis fungoides: a case report and review of the literature. J Cutan Pathol. 2001;28:318-324.
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525-530.
- Fujiyama T, Tokura Y. Clinical and histopathological differential diagnosis of eosinophilic pustular folliculitis. J Dermatol. 2013;40:419-423.
- Lee JY, Tsai YM, Sheu HM. Ofuji's disease with follicular mucinosis and its differential diagnosis from alopecia mucinosa. J Cutan Pathol. 2003;30:307-313.
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19.
- Vincent JG, Chan MP. Specificity of dermal mucin in the diagnosis of lupus erythematosus: comparison with other dermatitides and normal skin. J Cutan Pathol. 2015;42:722-729.
- Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol. 1996;135:355-362.
- Durdu M, Ilkit M. First step in the differential diagnosis of folliculitis: cytology. Crit Rev Microbiol. 2013;39:9-25.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides, a distinct disease entity with or without associated follicular mucinosis. a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198.
- DeBloom J 2nd, Severson J, Gaspari A, et al. Follicular mycosis fungoides: a case report and review of the literature. J Cutan Pathol. 2001;28:318-324.
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525-530.
- Fujiyama T, Tokura Y. Clinical and histopathological differential diagnosis of eosinophilic pustular folliculitis. J Dermatol. 2013;40:419-423.
- Lee JY, Tsai YM, Sheu HM. Ofuji's disease with follicular mucinosis and its differential diagnosis from alopecia mucinosa. J Cutan Pathol. 2003;30:307-313.
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19.
- Vincent JG, Chan MP. Specificity of dermal mucin in the diagnosis of lupus erythematosus: comparison with other dermatitides and normal skin. J Cutan Pathol. 2015;42:722-729.
- Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol. 1996;135:355-362.
- Durdu M, Ilkit M. First step in the differential diagnosis of folliculitis: cytology. Crit Rev Microbiol. 2013;39:9-25.
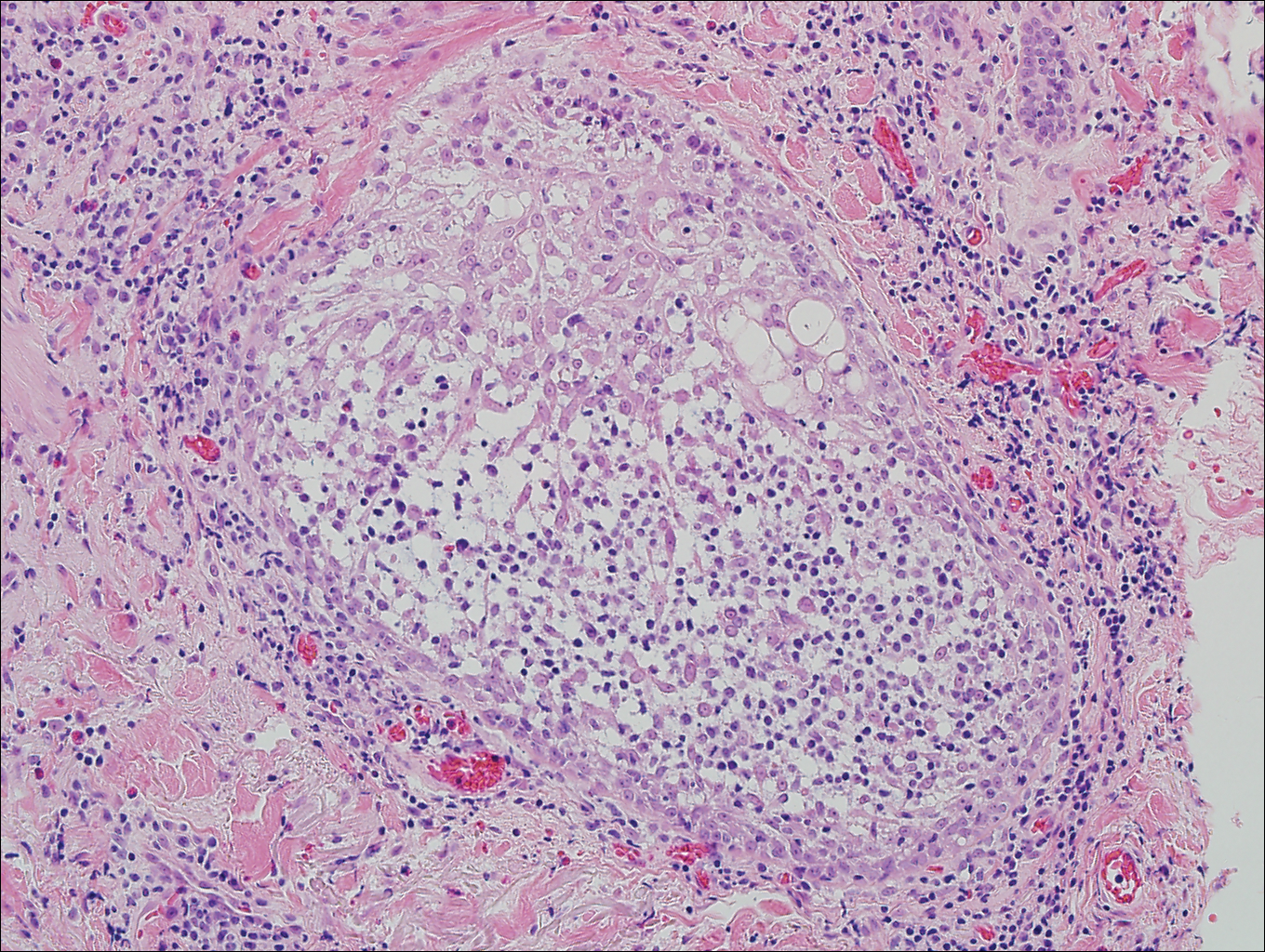
A 60-year-old man presented with a 3-month history of itchy bumps on the scalp and arms. He also noticed some patches of hair loss in these areas. He had no history of other skin conditions and was otherwise healthy with no other medical comorbidities.
The Proposed Rule and Payments for 2017: The Good, the Bad, and the Ugly
Just as Charlie Brown looks forward to the coming of the Great Pumpkin each Halloween, those of us who dance in the minefields of payment policy await the publication of the Proposed Rule, more formally known as the “Revisions to Payment Policies under the Physician Fee Schedule and Other Revisions to Part B for CY 2017.”1,2 You could read the entire tome—a mere 316 pages (excluding the hundreds of pages of granular supplement data discussed in the last few columns)—or simply read what I have outlined as the good, the bad, and the ugly for the Proposed Rule for 2017.
The Good
In 2017, dermatology will increase its share of the pie by 1% to $3.505 billion of a total $89.467 billion expected to be expended for physician services.1 The effect on individual providers will vary by geographic location and practice mix. Half is from the 0.5% increase that has come to all physicians across the board as mandated by the Medicare Access and CHIP Reauthorization Act (MACRA).3
Current Procedural Terminology (CPT) codes for reflectance confocal microscopy (96931–96936) will have Centers for Medicare & Medicaid Services valuations beginning in 2017, and individuals performing this service should be able to report it and be paid for their efforts.1 The values are below the American Medical Association/Specialty Society Relative Value Scale Update Committee (RUC) recommendations.
The Bad
Payment rates for 2017 will be based on a conversion factor of 35.7751,1 a drop from the 2016 conversion factor of 35.8043. Cuts will be made for some specialties. Gastroenterology, nephrology, neurosurgery, radiology, urology, and radiation therapy centers will take a 1% hit; ophthalmology, pathology, and vascular surgery will take 2% cuts; and interventional radiology will lose 7%.1 A special case within dermatology and pathology is a 15% cut to the technical component of slide preparation for CPT code 883054 due to a redefinition of the valuation of eosin stains.2 While the accuracy and precision of the value of these practice expense inputs can be debated, the government by definition makes the rules and involved specialties had an opportunity to appeal this change through the comment process that ended on September 6, 2016. The government can take comments into account, but substantial changes usually are not made from the Proposed Rule to the Final Rule, which usually arrives around the beginning of November; however, in an election year, the Final Rule can be a few weeks late.
The Ugly
The government will increase its unfunded mandates with the creation of new Medicare G codes (global services codes) that will allow the government to track the provision of postoperative care for all 010 and 090 global service periods (Table 1). The codes look mostly at time and do not clearly take into account the severity or complexity of the conditions being cared for and will be reported on claim forms as an unfunded mandate with more confusion and cost.1 Because not all claim-paying intermediaries are likely to have these G codes smoothly set up in their systems, there will still be a cost to filing the claim. Unless changes occur in the Final Rule, which is unlikely, there will be no payment for the time and effort of submitting these claims. The goal of the US Government is to hone in on postoperative services and parse them down so they can cut payments wherever possible beginning in 2019.1 Everyone wants to save money, from the consumer5 to the payer, and the ultimate payer is playing hardball. Additional validation efforts likely will lower physician fee-for-service payments further.

The US Government also is taking a shot at what they call “misvalued services” that have not had recent refinement within the RUC process.1 The work list for 2017 includes a number of 000 global period codes where additional evaluation and management services are reported using modifier -25, which implies a substantial, separately identifiable cognitive service performed by the same physician on the day of a procedure above and beyond other services provided or beyond the usual preservice and postservice care associated with the procedure that was performed. Although codes such as biopsies (11100 and 11101) and premalignant destructions (17000–17004) have an adjustment built in and dermatologists who provide services on the same day are actually penalized for the multiple built-in reductions that are already additive, the government is concerned that 19% of the 000 global services were billed more than 50% of the time with an evaluation and management code with modifier -25. Eighty-three codes met the criteria for which the government believes it may be overpaying1; the codes of interest to dermatology are shown in Table 2.1

The refinement of global periods will be an ongoing exercise through 2017, and beyond, with results likely to play an important role in the 2019 fee schedule. These global period reviews combined with some Stark law refinement relating the leasing of space at market rates while disallowing the landlord physician from receiving patient referrals from the tenant may also affect practitioner income.1,6 I never cease to be amazed that former Congressman Fortney Hillman “Pete” Stark (D), who has an antikickback scheme that keeps expanding, never went after the banking and brokerage industries. The founder of the $1.1 billion Security National Bank, a small bank in Walnut Creek, California,7 never focused on regulating banks. In his 40-year congressional career, he decided physicians make better targets. His efforts have not helped physicians but have helped lawyers, as he is quick to acknowledge.8
Final Thoughts
I end this column with an appeal to the dermatologists of America. Go to the American Academy of Dermatology Association Political Action Committee website (https://skinpac.org/), the home page for the only political action committee that represents the dermatology specialty, and consider making a donation. Emergency medicine physicians created the “Giving a Shift” campaign, which is a donation to their national political action committee of one shift’s earnings, and most of us could easily donate a half day’s income, as the only way to potentially change the increasingly onerous burdens on practitioners is through political action. As we say at RUC meetings, you can eat lunch or be lunch. The choice is yours.
- Medicare Program; Revisions to Payment Policies Under the Physician Fee Schedule and Other Revisions to Part B for CY 2017; Medicare Advantage Pricing Data Release; Medicare Advantage and Part D Medical Low Ratio Data Release; Medicare Advantage Provider Network Requirements; Expansion of Medicare Diabetes Prevention Program Model. Fed Regist. 2016;81(136):46162-46476. To be codified at 42 CFR §405, 410, 411, et al. https://www.gpo.gov/fdsys/pkg/FR-2016-07-15/pdf/2016-16097.pdf. Accessed September 7, 2016.
- Revisions to Payment Policies under the Physician Fee Schedule and Other Revisions to Part B for CY 2017. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/PFS-Federal-Regulation-Notices-Items/CMS-1654-P.html. Accessed September 7, 2016.
- Text of the Medicare Access and CHIP Reauthorization Act of 2015. GovTrack website. https://www.govtrack.us/congress/bills/114/hr2/text. Accessed September 9, 2016.
- Kaplan KJ. Proposed Medicare 2017 reimbursement schedule whacks biopsy payments; digital pathology payments up. Digital Pathology Blog website. http://tissuepathology.com/2016/07/20/proposed-medicare-2017-reimbursement-schedule-whacks-biopsy-payments-digital-pathology-payments-up/#ixzz4HEqBLgzu. Published July 20, 2016. Accessed September 7, 2016.
- Abelson R. Cost, not choice, is top concern of health insurance customers. The New York Times. http://www.nytimes.com/2016/08/13/business/cost-not-choice-is-top-concern-of-health-insurance-customers.html?_r=0. Published August 12, 2016. Accessed September 7, 2016.
- Stark Law website. http://starklaw.org/. Accessed September 7, 2016.
- Pete Stark. Freedom From Religion website. https://ffrf.org/news/day/dayitems/item/14800-pete-stark. Accessed September 19, 2016.
- Adamy J. Pete Stark: Law regulating doctors mostly helped lawyers. The Wall Street Journal. October 22, 2014. http://blogs.wsj.com/washwire/2014/10/22/pete-stark-law-regulating-doctors-mostly-helped-lawyers/. Accessed September 19, 2016.
Just as Charlie Brown looks forward to the coming of the Great Pumpkin each Halloween, those of us who dance in the minefields of payment policy await the publication of the Proposed Rule, more formally known as the “Revisions to Payment Policies under the Physician Fee Schedule and Other Revisions to Part B for CY 2017.”1,2 You could read the entire tome—a mere 316 pages (excluding the hundreds of pages of granular supplement data discussed in the last few columns)—or simply read what I have outlined as the good, the bad, and the ugly for the Proposed Rule for 2017.
The Good
In 2017, dermatology will increase its share of the pie by 1% to $3.505 billion of a total $89.467 billion expected to be expended for physician services.1 The effect on individual providers will vary by geographic location and practice mix. Half is from the 0.5% increase that has come to all physicians across the board as mandated by the Medicare Access and CHIP Reauthorization Act (MACRA).3
Current Procedural Terminology (CPT) codes for reflectance confocal microscopy (96931–96936) will have Centers for Medicare & Medicaid Services valuations beginning in 2017, and individuals performing this service should be able to report it and be paid for their efforts.1 The values are below the American Medical Association/Specialty Society Relative Value Scale Update Committee (RUC) recommendations.
The Bad
Payment rates for 2017 will be based on a conversion factor of 35.7751,1 a drop from the 2016 conversion factor of 35.8043. Cuts will be made for some specialties. Gastroenterology, nephrology, neurosurgery, radiology, urology, and radiation therapy centers will take a 1% hit; ophthalmology, pathology, and vascular surgery will take 2% cuts; and interventional radiology will lose 7%.1 A special case within dermatology and pathology is a 15% cut to the technical component of slide preparation for CPT code 883054 due to a redefinition of the valuation of eosin stains.2 While the accuracy and precision of the value of these practice expense inputs can be debated, the government by definition makes the rules and involved specialties had an opportunity to appeal this change through the comment process that ended on September 6, 2016. The government can take comments into account, but substantial changes usually are not made from the Proposed Rule to the Final Rule, which usually arrives around the beginning of November; however, in an election year, the Final Rule can be a few weeks late.
The Ugly
The government will increase its unfunded mandates with the creation of new Medicare G codes (global services codes) that will allow the government to track the provision of postoperative care for all 010 and 090 global service periods (Table 1). The codes look mostly at time and do not clearly take into account the severity or complexity of the conditions being cared for and will be reported on claim forms as an unfunded mandate with more confusion and cost.1 Because not all claim-paying intermediaries are likely to have these G codes smoothly set up in their systems, there will still be a cost to filing the claim. Unless changes occur in the Final Rule, which is unlikely, there will be no payment for the time and effort of submitting these claims. The goal of the US Government is to hone in on postoperative services and parse them down so they can cut payments wherever possible beginning in 2019.1 Everyone wants to save money, from the consumer5 to the payer, and the ultimate payer is playing hardball. Additional validation efforts likely will lower physician fee-for-service payments further.
The US Government also is taking a shot at what they call “misvalued services” that have not had recent refinement within the RUC process.1 The work list for 2017 includes a number of 000 global period codes where additional evaluation and management services are reported using modifier -25, which implies a substantial, separately identifiable cognitive service performed by the same physician on the day of a procedure above and beyond other services provided or beyond the usual preservice and postservice care associated with the procedure that was performed. Although codes such as biopsies (11100 and 11101) and premalignant destructions (17000–17004) have an adjustment built in and dermatologists who provide services on the same day are actually penalized for the multiple built-in reductions that are already additive, the government is concerned that 19% of the 000 global services were billed more than 50% of the time with an evaluation and management code with modifier -25. Eighty-three codes met the criteria for which the government believes it may be overpaying1; the codes of interest to dermatology are shown in Table 2.1
The refinement of global periods will be an ongoing exercise through 2017, and beyond, with results likely to play an important role in the 2019 fee schedule. These global period reviews combined with some Stark law refinement relating the leasing of space at market rates while disallowing the landlord physician from receiving patient referrals from the tenant may also affect practitioner income.1,6 I never cease to be amazed that former Congressman Fortney Hillman “Pete” Stark (D), who has an antikickback scheme that keeps expanding, never went after the banking and brokerage industries. The founder of the $1.1 billion Security National Bank, a small bank in Walnut Creek, California,7 never focused on regulating banks. In his 40-year congressional career, he decided physicians make better targets. His efforts have not helped physicians but have helped lawyers, as he is quick to acknowledge.8
Final Thoughts
I end this column with an appeal to the dermatologists of America. Go to the American Academy of Dermatology Association Political Action Committee website (https://skinpac.org/), the home page for the only political action committee that represents the dermatology specialty, and consider making a donation. Emergency medicine physicians created the “Giving a Shift” campaign, which is a donation to their national political action committee of one shift’s earnings, and most of us could easily donate a half day’s income, as the only way to potentially change the increasingly onerous burdens on practitioners is through political action. As we say at RUC meetings, you can eat lunch or be lunch. The choice is yours.
Just as Charlie Brown looks forward to the coming of the Great Pumpkin each Halloween, those of us who dance in the minefields of payment policy await the publication of the Proposed Rule, more formally known as the “Revisions to Payment Policies under the Physician Fee Schedule and Other Revisions to Part B for CY 2017.”1,2 You could read the entire tome—a mere 316 pages (excluding the hundreds of pages of granular supplement data discussed in the last few columns)—or simply read what I have outlined as the good, the bad, and the ugly for the Proposed Rule for 2017.
The Good
In 2017, dermatology will increase its share of the pie by 1% to $3.505 billion of a total $89.467 billion expected to be expended for physician services.1 The effect on individual providers will vary by geographic location and practice mix. Half is from the 0.5% increase that has come to all physicians across the board as mandated by the Medicare Access and CHIP Reauthorization Act (MACRA).3
Current Procedural Terminology (CPT) codes for reflectance confocal microscopy (96931–96936) will have Centers for Medicare & Medicaid Services valuations beginning in 2017, and individuals performing this service should be able to report it and be paid for their efforts.1 The values are below the American Medical Association/Specialty Society Relative Value Scale Update Committee (RUC) recommendations.
The Bad
Payment rates for 2017 will be based on a conversion factor of 35.7751,1 a drop from the 2016 conversion factor of 35.8043. Cuts will be made for some specialties. Gastroenterology, nephrology, neurosurgery, radiology, urology, and radiation therapy centers will take a 1% hit; ophthalmology, pathology, and vascular surgery will take 2% cuts; and interventional radiology will lose 7%.1 A special case within dermatology and pathology is a 15% cut to the technical component of slide preparation for CPT code 883054 due to a redefinition of the valuation of eosin stains.2 While the accuracy and precision of the value of these practice expense inputs can be debated, the government by definition makes the rules and involved specialties had an opportunity to appeal this change through the comment process that ended on September 6, 2016. The government can take comments into account, but substantial changes usually are not made from the Proposed Rule to the Final Rule, which usually arrives around the beginning of November; however, in an election year, the Final Rule can be a few weeks late.
The Ugly
The government will increase its unfunded mandates with the creation of new Medicare G codes (global services codes) that will allow the government to track the provision of postoperative care for all 010 and 090 global service periods (Table 1). The codes look mostly at time and do not clearly take into account the severity or complexity of the conditions being cared for and will be reported on claim forms as an unfunded mandate with more confusion and cost.1 Because not all claim-paying intermediaries are likely to have these G codes smoothly set up in their systems, there will still be a cost to filing the claim. Unless changes occur in the Final Rule, which is unlikely, there will be no payment for the time and effort of submitting these claims. The goal of the US Government is to hone in on postoperative services and parse them down so they can cut payments wherever possible beginning in 2019.1 Everyone wants to save money, from the consumer5 to the payer, and the ultimate payer is playing hardball. Additional validation efforts likely will lower physician fee-for-service payments further.
The US Government also is taking a shot at what they call “misvalued services” that have not had recent refinement within the RUC process.1 The work list for 2017 includes a number of 000 global period codes where additional evaluation and management services are reported using modifier -25, which implies a substantial, separately identifiable cognitive service performed by the same physician on the day of a procedure above and beyond other services provided or beyond the usual preservice and postservice care associated with the procedure that was performed. Although codes such as biopsies (11100 and 11101) and premalignant destructions (17000–17004) have an adjustment built in and dermatologists who provide services on the same day are actually penalized for the multiple built-in reductions that are already additive, the government is concerned that 19% of the 000 global services were billed more than 50% of the time with an evaluation and management code with modifier -25. Eighty-three codes met the criteria for which the government believes it may be overpaying1; the codes of interest to dermatology are shown in Table 2.1
The refinement of global periods will be an ongoing exercise through 2017, and beyond, with results likely to play an important role in the 2019 fee schedule. These global period reviews combined with some Stark law refinement relating the leasing of space at market rates while disallowing the landlord physician from receiving patient referrals from the tenant may also affect practitioner income.1,6 I never cease to be amazed that former Congressman Fortney Hillman “Pete” Stark (D), who has an antikickback scheme that keeps expanding, never went after the banking and brokerage industries. The founder of the $1.1 billion Security National Bank, a small bank in Walnut Creek, California,7 never focused on regulating banks. In his 40-year congressional career, he decided physicians make better targets. His efforts have not helped physicians but have helped lawyers, as he is quick to acknowledge.8
Final Thoughts
I end this column with an appeal to the dermatologists of America. Go to the American Academy of Dermatology Association Political Action Committee website (https://skinpac.org/), the home page for the only political action committee that represents the dermatology specialty, and consider making a donation. Emergency medicine physicians created the “Giving a Shift” campaign, which is a donation to their national political action committee of one shift’s earnings, and most of us could easily donate a half day’s income, as the only way to potentially change the increasingly onerous burdens on practitioners is through political action. As we say at RUC meetings, you can eat lunch or be lunch. The choice is yours.
- Medicare Program; Revisions to Payment Policies Under the Physician Fee Schedule and Other Revisions to Part B for CY 2017; Medicare Advantage Pricing Data Release; Medicare Advantage and Part D Medical Low Ratio Data Release; Medicare Advantage Provider Network Requirements; Expansion of Medicare Diabetes Prevention Program Model. Fed Regist. 2016;81(136):46162-46476. To be codified at 42 CFR §405, 410, 411, et al. https://www.gpo.gov/fdsys/pkg/FR-2016-07-15/pdf/2016-16097.pdf. Accessed September 7, 2016.
- Revisions to Payment Policies under the Physician Fee Schedule and Other Revisions to Part B for CY 2017. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/PFS-Federal-Regulation-Notices-Items/CMS-1654-P.html. Accessed September 7, 2016.
- Text of the Medicare Access and CHIP Reauthorization Act of 2015. GovTrack website. https://www.govtrack.us/congress/bills/114/hr2/text. Accessed September 9, 2016.
- Kaplan KJ. Proposed Medicare 2017 reimbursement schedule whacks biopsy payments; digital pathology payments up. Digital Pathology Blog website. http://tissuepathology.com/2016/07/20/proposed-medicare-2017-reimbursement-schedule-whacks-biopsy-payments-digital-pathology-payments-up/#ixzz4HEqBLgzu. Published July 20, 2016. Accessed September 7, 2016.
- Abelson R. Cost, not choice, is top concern of health insurance customers. The New York Times. http://www.nytimes.com/2016/08/13/business/cost-not-choice-is-top-concern-of-health-insurance-customers.html?_r=0. Published August 12, 2016. Accessed September 7, 2016.
- Stark Law website. http://starklaw.org/. Accessed September 7, 2016.
- Pete Stark. Freedom From Religion website. https://ffrf.org/news/day/dayitems/item/14800-pete-stark. Accessed September 19, 2016.
- Adamy J. Pete Stark: Law regulating doctors mostly helped lawyers. The Wall Street Journal. October 22, 2014. http://blogs.wsj.com/washwire/2014/10/22/pete-stark-law-regulating-doctors-mostly-helped-lawyers/. Accessed September 19, 2016.
- Medicare Program; Revisions to Payment Policies Under the Physician Fee Schedule and Other Revisions to Part B for CY 2017; Medicare Advantage Pricing Data Release; Medicare Advantage and Part D Medical Low Ratio Data Release; Medicare Advantage Provider Network Requirements; Expansion of Medicare Diabetes Prevention Program Model. Fed Regist. 2016;81(136):46162-46476. To be codified at 42 CFR §405, 410, 411, et al. https://www.gpo.gov/fdsys/pkg/FR-2016-07-15/pdf/2016-16097.pdf. Accessed September 7, 2016.
- Revisions to Payment Policies under the Physician Fee Schedule and Other Revisions to Part B for CY 2017. Centers for Medicare & Medicaid Services website. https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/PhysicianFeeSched/PFS-Federal-Regulation-Notices-Items/CMS-1654-P.html. Accessed September 7, 2016.
- Text of the Medicare Access and CHIP Reauthorization Act of 2015. GovTrack website. https://www.govtrack.us/congress/bills/114/hr2/text. Accessed September 9, 2016.
- Kaplan KJ. Proposed Medicare 2017 reimbursement schedule whacks biopsy payments; digital pathology payments up. Digital Pathology Blog website. http://tissuepathology.com/2016/07/20/proposed-medicare-2017-reimbursement-schedule-whacks-biopsy-payments-digital-pathology-payments-up/#ixzz4HEqBLgzu. Published July 20, 2016. Accessed September 7, 2016.
- Abelson R. Cost, not choice, is top concern of health insurance customers. The New York Times. http://www.nytimes.com/2016/08/13/business/cost-not-choice-is-top-concern-of-health-insurance-customers.html?_r=0. Published August 12, 2016. Accessed September 7, 2016.
- Stark Law website. http://starklaw.org/. Accessed September 7, 2016.
- Pete Stark. Freedom From Religion website. https://ffrf.org/news/day/dayitems/item/14800-pete-stark. Accessed September 19, 2016.
- Adamy J. Pete Stark: Law regulating doctors mostly helped lawyers. The Wall Street Journal. October 22, 2014. http://blogs.wsj.com/washwire/2014/10/22/pete-stark-law-regulating-doctors-mostly-helped-lawyers/. Accessed September 19, 2016.
Practice Points
- The Proposed Rule outlines the probable payment levels for calendar year 2017.
- The rule also announces how the Medicare Access and CHIP Reauthorization Act (MACRA) may be implemented.
