User login
Localized Argyria With Pseudo-ochronosis
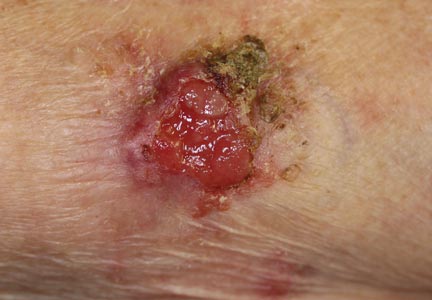
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
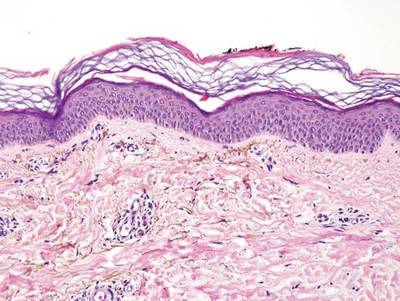
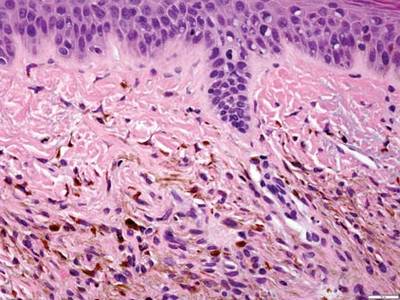
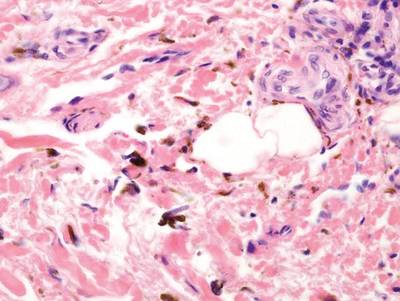
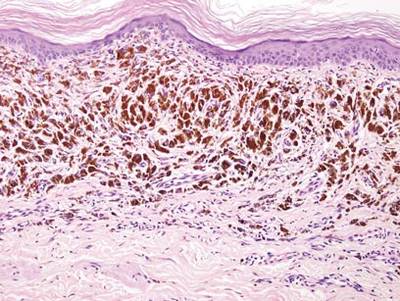
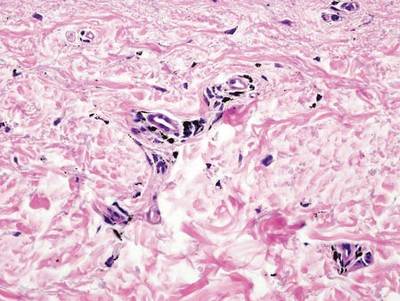
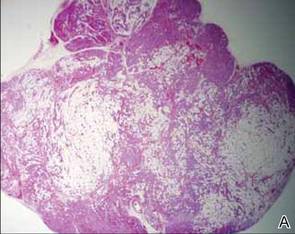
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.

A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.
A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.
A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
Autosomal-Dominant Familial Angiolipomatosis
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
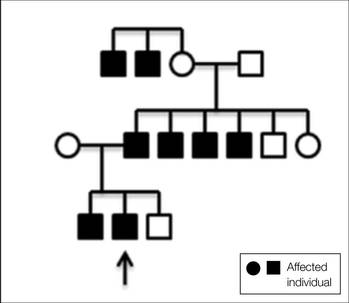
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
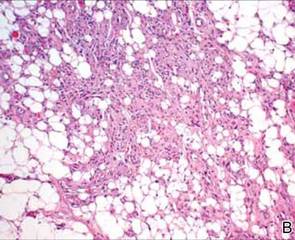
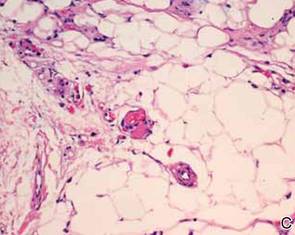
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
Practice Points
- Dermatologists should be familiar with the clinical and histological features of angiolipomas along with their potential inheritance patterns.
- Familial angiolipomatosis is a rare condition characterized by multiple angiolipomas that has been described as having an autosomal-recessive transmission pattern. Autosomal-dominant inheritance also may occur, as illustrated in the current case report.
- Awareness of the autosomal-dominant form of this entity is important to prevent its misdiagnosis as
neurofibromatosis type I, which has a similar family history and clinical presentation.
Multiple Firm Pink Papules and Nodules
The Diagnosis: Myeloid Leukemia Cutis
Leukemia cutis represents the infiltration of leukemic cells into the skin. It has been described in the setting of both myeloid and lymphoid leukemia. In the setting of acute myeloid leukemia, it has been reported to occur in 2% to 13% of patients overall,1,2 but it may occur in 31% of patients with the acute myelomonocytic or acute monocytic leukemia subtypes.3 Leukemia cutis is less common, with chronic myeloid leukemia occurring in 2.7% of patients in one study.4 In another study, 65% of patients with myeloid leukemia cutis had an acute myeloid leukemia.5
Myeloid leukemia cutis has been reported in patients aged 22 days to 90 years, with a median age of 62 years. There is a male predominance (1.4:1 ratio).5,6 The diagnosis of leukemia cutis is made concurrently with the diagnosis of leukemia in approximately 30% of cases, subsequent to the diagnosis of leukemia in approximately 60% of cases, and prior to the diagnosis of leukemia in approximately 10% of cases.5
Clinically, myeloid leukemia cutis presents as an asymptomatic solitary lesion in 23% of cases or as multiple lesions in 77% of cases. Lesions consist of pink to red to violaceous papules, nodules, and macules that are occasionally purpuric and involve any cutaneous surface.5
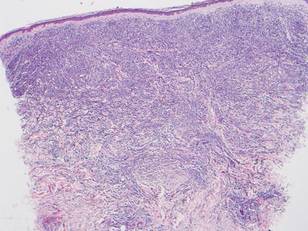
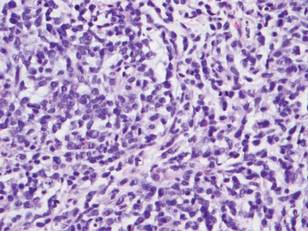
Histologically, the epidermis is unremarkable. Beneath a grenz zone within the dermis and usually extending into the subcutis there is a diffuse or nodular proliferation of neoplastic cells, often with perivascular and periadnexal accentuation and sometimes single filing of cells between collagen bundles (Figure 1). The cells are immature myeloid cells with irregular nuclear contours that may be indented or reniform (Figure 2). Nuclei contain finely dispersed chromatin with variably prominent nucleoli.5,6 Immunohistochemically, CD68 is positive in approximately 97% of cases, myeloperoxidase in 62%, and lysozyme in 85%. CD168, CD14, CD4, CD33, CD117, CD34, CD56, CD123, and CD303 are variably positive. CD3 and CD20, markers of lymphoid leukemia, are negative.5-8
Leukemia cutis in the setting of a myeloid leukemia portends a grave prognosis. In a series of 18 patients, 16 had additional extramedullary leukemia, including meningeal leukemia in 6 patients.2 Most patients with myeloid leukemia cutis die within an average of 1 to 8 months of diagnosis.9
- Boggs DR, Wintrobe MM, Cartwright GE. The acute leukemias. analysis of 322 cases and review of the literature. Medicine (Baltimore). 1962;41:163-225.
- Baer MR, Barcos M, Farrell H, et al. Acute myelogenous leukemia with leukemia cutis. eighteen cases seen between 1969 and 1986. Cancer. 1989;63:2192-2200.
- Straus DJ, Mertelsmann R, Koziner B, et al. The acute monocytic leukemias: multidisciplinary studies in 45 patients. Medicine (Baltimore). 1980;59:409-425.
- Rosenthal S, Canellos GP, DeVita VT Jr, et al. Characteristics of blast crisis in chronic granulocytic leukemia. Blood. 1977;49:705-714.
- Bénet C, Gomez A, Aguilar C, et al. Histologic and immunohistologic characterization of skin localization of myeloid disorders: a study of 173 cases. Am J Clin Pathol. 2011;135:278-290.
- Cronin DM, George TI, Sundram UN. An updated approach to the diagnosis of myeloid leukemia cutis. Am J Clin Pathol. 2009;132:101-110.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Su WP, Buechner SA, Li CY. Clinicopathologic correlations in leukemia cutis. J Am Acad Dermatol. 1984;11:121-128.
The Diagnosis: Myeloid Leukemia Cutis
Leukemia cutis represents the infiltration of leukemic cells into the skin. It has been described in the setting of both myeloid and lymphoid leukemia. In the setting of acute myeloid leukemia, it has been reported to occur in 2% to 13% of patients overall,1,2 but it may occur in 31% of patients with the acute myelomonocytic or acute monocytic leukemia subtypes.3 Leukemia cutis is less common, with chronic myeloid leukemia occurring in 2.7% of patients in one study.4 In another study, 65% of patients with myeloid leukemia cutis had an acute myeloid leukemia.5
Myeloid leukemia cutis has been reported in patients aged 22 days to 90 years, with a median age of 62 years. There is a male predominance (1.4:1 ratio).5,6 The diagnosis of leukemia cutis is made concurrently with the diagnosis of leukemia in approximately 30% of cases, subsequent to the diagnosis of leukemia in approximately 60% of cases, and prior to the diagnosis of leukemia in approximately 10% of cases.5
Clinically, myeloid leukemia cutis presents as an asymptomatic solitary lesion in 23% of cases or as multiple lesions in 77% of cases. Lesions consist of pink to red to violaceous papules, nodules, and macules that are occasionally purpuric and involve any cutaneous surface.5
Histologically, the epidermis is unremarkable. Beneath a grenz zone within the dermis and usually extending into the subcutis there is a diffuse or nodular proliferation of neoplastic cells, often with perivascular and periadnexal accentuation and sometimes single filing of cells between collagen bundles (Figure 1). The cells are immature myeloid cells with irregular nuclear contours that may be indented or reniform (Figure 2). Nuclei contain finely dispersed chromatin with variably prominent nucleoli.5,6 Immunohistochemically, CD68 is positive in approximately 97% of cases, myeloperoxidase in 62%, and lysozyme in 85%. CD168, CD14, CD4, CD33, CD117, CD34, CD56, CD123, and CD303 are variably positive. CD3 and CD20, markers of lymphoid leukemia, are negative.5-8
Leukemia cutis in the setting of a myeloid leukemia portends a grave prognosis. In a series of 18 patients, 16 had additional extramedullary leukemia, including meningeal leukemia in 6 patients.2 Most patients with myeloid leukemia cutis die within an average of 1 to 8 months of diagnosis.9
The Diagnosis: Myeloid Leukemia Cutis
Leukemia cutis represents the infiltration of leukemic cells into the skin. It has been described in the setting of both myeloid and lymphoid leukemia. In the setting of acute myeloid leukemia, it has been reported to occur in 2% to 13% of patients overall,1,2 but it may occur in 31% of patients with the acute myelomonocytic or acute monocytic leukemia subtypes.3 Leukemia cutis is less common, with chronic myeloid leukemia occurring in 2.7% of patients in one study.4 In another study, 65% of patients with myeloid leukemia cutis had an acute myeloid leukemia.5
Myeloid leukemia cutis has been reported in patients aged 22 days to 90 years, with a median age of 62 years. There is a male predominance (1.4:1 ratio).5,6 The diagnosis of leukemia cutis is made concurrently with the diagnosis of leukemia in approximately 30% of cases, subsequent to the diagnosis of leukemia in approximately 60% of cases, and prior to the diagnosis of leukemia in approximately 10% of cases.5
Clinically, myeloid leukemia cutis presents as an asymptomatic solitary lesion in 23% of cases or as multiple lesions in 77% of cases. Lesions consist of pink to red to violaceous papules, nodules, and macules that are occasionally purpuric and involve any cutaneous surface.5
Histologically, the epidermis is unremarkable. Beneath a grenz zone within the dermis and usually extending into the subcutis there is a diffuse or nodular proliferation of neoplastic cells, often with perivascular and periadnexal accentuation and sometimes single filing of cells between collagen bundles (Figure 1). The cells are immature myeloid cells with irregular nuclear contours that may be indented or reniform (Figure 2). Nuclei contain finely dispersed chromatin with variably prominent nucleoli.5,6 Immunohistochemically, CD68 is positive in approximately 97% of cases, myeloperoxidase in 62%, and lysozyme in 85%. CD168, CD14, CD4, CD33, CD117, CD34, CD56, CD123, and CD303 are variably positive. CD3 and CD20, markers of lymphoid leukemia, are negative.5-8
Leukemia cutis in the setting of a myeloid leukemia portends a grave prognosis. In a series of 18 patients, 16 had additional extramedullary leukemia, including meningeal leukemia in 6 patients.2 Most patients with myeloid leukemia cutis die within an average of 1 to 8 months of diagnosis.9
- Boggs DR, Wintrobe MM, Cartwright GE. The acute leukemias. analysis of 322 cases and review of the literature. Medicine (Baltimore). 1962;41:163-225.
- Baer MR, Barcos M, Farrell H, et al. Acute myelogenous leukemia with leukemia cutis. eighteen cases seen between 1969 and 1986. Cancer. 1989;63:2192-2200.
- Straus DJ, Mertelsmann R, Koziner B, et al. The acute monocytic leukemias: multidisciplinary studies in 45 patients. Medicine (Baltimore). 1980;59:409-425.
- Rosenthal S, Canellos GP, DeVita VT Jr, et al. Characteristics of blast crisis in chronic granulocytic leukemia. Blood. 1977;49:705-714.
- Bénet C, Gomez A, Aguilar C, et al. Histologic and immunohistologic characterization of skin localization of myeloid disorders: a study of 173 cases. Am J Clin Pathol. 2011;135:278-290.
- Cronin DM, George TI, Sundram UN. An updated approach to the diagnosis of myeloid leukemia cutis. Am J Clin Pathol. 2009;132:101-110.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Su WP, Buechner SA, Li CY. Clinicopathologic correlations in leukemia cutis. J Am Acad Dermatol. 1984;11:121-128.
- Boggs DR, Wintrobe MM, Cartwright GE. The acute leukemias. analysis of 322 cases and review of the literature. Medicine (Baltimore). 1962;41:163-225.
- Baer MR, Barcos M, Farrell H, et al. Acute myelogenous leukemia with leukemia cutis. eighteen cases seen between 1969 and 1986. Cancer. 1989;63:2192-2200.
- Straus DJ, Mertelsmann R, Koziner B, et al. The acute monocytic leukemias: multidisciplinary studies in 45 patients. Medicine (Baltimore). 1980;59:409-425.
- Rosenthal S, Canellos GP, DeVita VT Jr, et al. Characteristics of blast crisis in chronic granulocytic leukemia. Blood. 1977;49:705-714.
- Bénet C, Gomez A, Aguilar C, et al. Histologic and immunohistologic characterization of skin localization of myeloid disorders: a study of 173 cases. Am J Clin Pathol. 2011;135:278-290.
- Cronin DM, George TI, Sundram UN. An updated approach to the diagnosis of myeloid leukemia cutis. Am J Clin Pathol. 2009;132:101-110.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Kaddu S, Zenahlik P, Beham-Schmid C, et al. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999;40:966-978.
- Su WP, Buechner SA, Li CY. Clinicopathologic correlations in leukemia cutis. J Am Acad Dermatol. 1984;11:121-128.
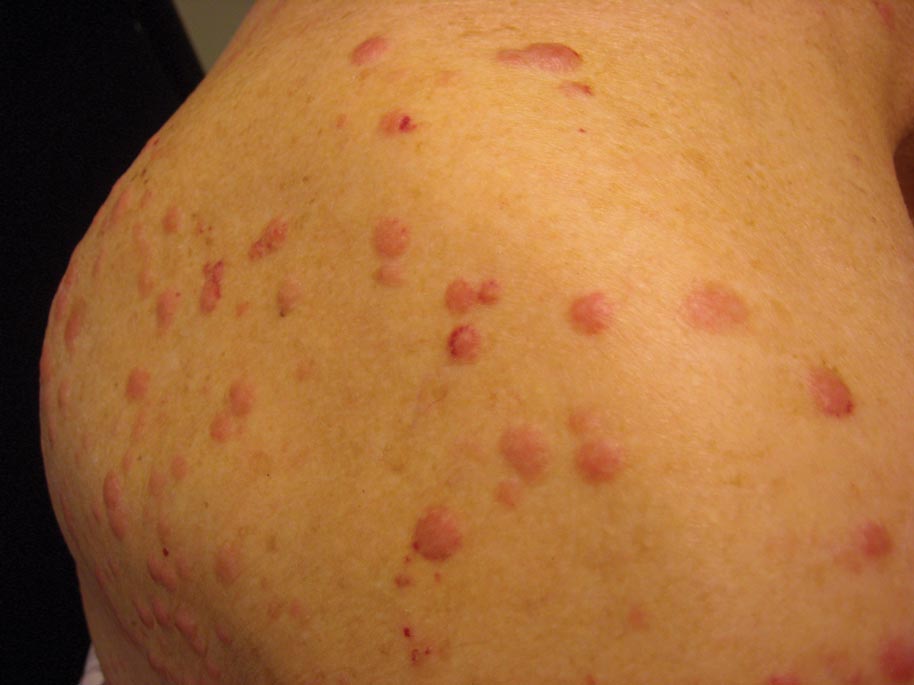
A 91-year-old man presented with numerous, scattered, asymptomatic, 3- to 9-mm, smooth, firm, pink papules and nodules involving the neck, trunk, and arms and legs of 1 week’s duration.
Stains and Smears: Resident Guide to Bedside Diagnostic Testing
Dermatologists are fortunate to specialize in the organ that is most accessible to evaluation. Although we use the physical examination to formulate the initial differential diagnosis, at times we must rely on ancillary tests to narrow down the diagnosis. Various bedside testing modalities—potassium hydroxide (KOH) preparation, Tzanck smear, mineral oil preparation, and Gram stain—are most useful in diagnosing infectious causes of cutaneous disease. This guide serves as a useful reference for residents on how to perform these tests and which conditions they can help diagnose. Several of these procedures have no standard protocol for performing them; the literature is littered with various methodologies, fixatives, and stains, and as such, this article will attempt to describe a technique that is convenient and quick to perform with readily available materials while still offering high diagnostic utility.
KOH Preparation
A standard in the armamentarium of a dermatologist, the KOH preparation is invaluable to diagnose fungal and yeast infections. Although there are many available preparations including varying concentrations of KOH, dimethyl sulfoxide, and various inks, the procedure is similar for all of them.1 The first step involves collecting the specimen, which can be scale from an active border of suspected cutaneous dermatophyte or Malassezia infection, debris from suspected candidiasis, or hair shafts plucked from an area of alopecia of presumed tinea capitis. A no. 15 blade can be used to scrape the specimen onto a microscope slide, though a second microscope slide can be used in lieu of a blade in patients who will not remain still, and then a coverslip is placed. Two drops of the KOH solution of your choice are then placed on opposite ends of the coverslip, allowing capillary action to spread the stain evenly. A paper towel can be folded in half and pushed down on the surface of the coverslip to spread the stain and soak up any excess, and this pressure also can help the KOH solution digest the keratin in the specimen. Briefly heating the underside of the slide (below boiling point) will help digest the keratin; this step is not necessary when you are using a KOH preparation with dimethyl sulfoxide. Although many dermatologists view the slide almost immediately, ideally at least 5 minutes should pass before it is read. Particularly thick specimens may require additional digestion time, so setting them aside for later review may help visualize infectious agents. In a busy clinic where an immediate diagnosis may not be requisite and a prescription can be called in pending the result, waiting to review the slide may be feasible.
Tzanck Smear
The Tzanck smear is a useful cytopathologic test in the rapid diagnosis of herpetic lesions, though it cannot differentiate between herpes simplex virus type 1, herpes simplex virus type 2, and varicella-zoster virus. It also has shown utility for rapid diagnosis of protean other dermatologic conditions including autoimmune blistering disorders, cutaneous malignancies, and other infectious processes, though it has been superseded by histopathology in most cases.2 An ideal sample is collected by scraping the base of a fresh blister with a no. 15 blade or a second microscope slide. The scrapings then are smeared onto another microscope slide and allowed to air-dry briefly. Then, Wright-Giemsa stain is dispensed to cover the sample and allowed to sit for 15 minutes before being washed off with sterile water. After air-drying, the sample is examined for the presence of clumped multinucleated giant cells, a feature that confirms herpetic infection and allows rapid initiation of antiviral medication.3
Mineral Oil Preparation
A mineral oil preparation has utility in diagnosing ectoparasitic infestation. In the case of scabies, a positive microscopic examination is diagnostic and requires no further testing, allowing for rapid initiation of therapy. This technique also is useful in diagnosing rosacea related to Demodex, which requires a treatment algorithm that differs from the classic papulopustular rosacea which it mimics.4
Mineral oil preparations can be rapidly performed and interpreted. Several drops of mineral oil are placed onto a microscope slide and a no. 15 blade is dipped into this oil prior to scraping the sample lesion. For scabies, a burrow is scraped repeatedly with the blade, and the debris is collected in the mineral oil. Occasionally, the mite can be dermoscopically visualized as a jet plane or arrowhead at the leading edge of a burrow; scraping should be focused in the vicinity of the mite.5 A coverslip is applied to the microscope slide and examination for the mite, egg casings, and scybala can be performed with microscopy.6 For Demodex infestation, a facial pustule can be expressed or several eyelash hairs can be plucked and suspended in mineral oil. Examination of this specimen is identical to scabies.
Gram Stain
The Gram stain is invaluable in classifying bacteria, and a properly performed test can narrow the identification of a causative organism based on cellular morphology. Although it is more technically complex than other bedside diagnostic maneuvers, it can be rapidly performed once the sequence of stains is mastered. The collected sample is smeared onto a glass slide and then briefly passed over a flame several times to heat-fix the specimen. Caution should be taken to avoid direct or prolonged flame contact with the underside of the slide. After fixation, the staining can be performed. First, crystal violet is instilled onto the slide and remains on for 30 seconds before being rinsed off with sink water. Then, Gram iodine is used for 30 seconds, followed by another rinse in water. Next, pour the decolorizer solution over the slide until the runoff is clear, and then rinse in water. Finally, flood with safranin counterstain for 30 seconds and give the slide a final rinse. After air-drying, it is ready to be interpreted.7
Final Thoughts
Although the modern dermatologist has access to biopsies, cultures, and sophisticated diagnostic techniques, it is important to remember these useful bedside tests. The ability to rapidly pin a diagnosis is particularly useful on the consultative service where critically ill patients can benefit from identification of a causative pathogen sooner rather than later. Residents should master these stains in their training, as this knowledge may prove to be invaluable in their careers.
1. Trozak DJ, Tennenhouse DJ, Russell JJ. Dermatology Skills for Primary Care: An Illustrated Guide. Totowa, NJ: Humana Press; 2006.
2. Kelly B, Shimoni T. Reintroducing the Tzanck smear. Am J Clin Dermatol. 2009;10:141-152.
3. Singhi M, Gupta L. Tzanck smear: a useful diagnostic tool. Indian J Dermatol Venereol Leprol. 2005;71:295.
4. Elston DM. Demodex mites: facts and controversies. Clin Dermatol. 2010;28:502-504.
5. Dupuy A, Dehen L, Bourrat E, et al. Accuracy of standard dermoscopy for diagnosing scabies. J Am Acad Dermatol. 2007;56:53-62.
6. Bolognia J, Schaffer J, Duncan K, et al. Dermatology Essentials. St. Louis, MO: Saunders Elsevier; 2014.
7. Ruocco E, Baroni A, Donnarumma G, et al. Diagnostic procedures in dermatology. Clin Dermatol. 2011;29:548-556.
Dermatologists are fortunate to specialize in the organ that is most accessible to evaluation. Although we use the physical examination to formulate the initial differential diagnosis, at times we must rely on ancillary tests to narrow down the diagnosis. Various bedside testing modalities—potassium hydroxide (KOH) preparation, Tzanck smear, mineral oil preparation, and Gram stain—are most useful in diagnosing infectious causes of cutaneous disease. This guide serves as a useful reference for residents on how to perform these tests and which conditions they can help diagnose. Several of these procedures have no standard protocol for performing them; the literature is littered with various methodologies, fixatives, and stains, and as such, this article will attempt to describe a technique that is convenient and quick to perform with readily available materials while still offering high diagnostic utility.
KOH Preparation
A standard in the armamentarium of a dermatologist, the KOH preparation is invaluable to diagnose fungal and yeast infections. Although there are many available preparations including varying concentrations of KOH, dimethyl sulfoxide, and various inks, the procedure is similar for all of them.1 The first step involves collecting the specimen, which can be scale from an active border of suspected cutaneous dermatophyte or Malassezia infection, debris from suspected candidiasis, or hair shafts plucked from an area of alopecia of presumed tinea capitis. A no. 15 blade can be used to scrape the specimen onto a microscope slide, though a second microscope slide can be used in lieu of a blade in patients who will not remain still, and then a coverslip is placed. Two drops of the KOH solution of your choice are then placed on opposite ends of the coverslip, allowing capillary action to spread the stain evenly. A paper towel can be folded in half and pushed down on the surface of the coverslip to spread the stain and soak up any excess, and this pressure also can help the KOH solution digest the keratin in the specimen. Briefly heating the underside of the slide (below boiling point) will help digest the keratin; this step is not necessary when you are using a KOH preparation with dimethyl sulfoxide. Although many dermatologists view the slide almost immediately, ideally at least 5 minutes should pass before it is read. Particularly thick specimens may require additional digestion time, so setting them aside for later review may help visualize infectious agents. In a busy clinic where an immediate diagnosis may not be requisite and a prescription can be called in pending the result, waiting to review the slide may be feasible.
Tzanck Smear
The Tzanck smear is a useful cytopathologic test in the rapid diagnosis of herpetic lesions, though it cannot differentiate between herpes simplex virus type 1, herpes simplex virus type 2, and varicella-zoster virus. It also has shown utility for rapid diagnosis of protean other dermatologic conditions including autoimmune blistering disorders, cutaneous malignancies, and other infectious processes, though it has been superseded by histopathology in most cases.2 An ideal sample is collected by scraping the base of a fresh blister with a no. 15 blade or a second microscope slide. The scrapings then are smeared onto another microscope slide and allowed to air-dry briefly. Then, Wright-Giemsa stain is dispensed to cover the sample and allowed to sit for 15 minutes before being washed off with sterile water. After air-drying, the sample is examined for the presence of clumped multinucleated giant cells, a feature that confirms herpetic infection and allows rapid initiation of antiviral medication.3
Mineral Oil Preparation
A mineral oil preparation has utility in diagnosing ectoparasitic infestation. In the case of scabies, a positive microscopic examination is diagnostic and requires no further testing, allowing for rapid initiation of therapy. This technique also is useful in diagnosing rosacea related to Demodex, which requires a treatment algorithm that differs from the classic papulopustular rosacea which it mimics.4
Mineral oil preparations can be rapidly performed and interpreted. Several drops of mineral oil are placed onto a microscope slide and a no. 15 blade is dipped into this oil prior to scraping the sample lesion. For scabies, a burrow is scraped repeatedly with the blade, and the debris is collected in the mineral oil. Occasionally, the mite can be dermoscopically visualized as a jet plane or arrowhead at the leading edge of a burrow; scraping should be focused in the vicinity of the mite.5 A coverslip is applied to the microscope slide and examination for the mite, egg casings, and scybala can be performed with microscopy.6 For Demodex infestation, a facial pustule can be expressed or several eyelash hairs can be plucked and suspended in mineral oil. Examination of this specimen is identical to scabies.
Gram Stain
The Gram stain is invaluable in classifying bacteria, and a properly performed test can narrow the identification of a causative organism based on cellular morphology. Although it is more technically complex than other bedside diagnostic maneuvers, it can be rapidly performed once the sequence of stains is mastered. The collected sample is smeared onto a glass slide and then briefly passed over a flame several times to heat-fix the specimen. Caution should be taken to avoid direct or prolonged flame contact with the underside of the slide. After fixation, the staining can be performed. First, crystal violet is instilled onto the slide and remains on for 30 seconds before being rinsed off with sink water. Then, Gram iodine is used for 30 seconds, followed by another rinse in water. Next, pour the decolorizer solution over the slide until the runoff is clear, and then rinse in water. Finally, flood with safranin counterstain for 30 seconds and give the slide a final rinse. After air-drying, it is ready to be interpreted.7
Final Thoughts
Although the modern dermatologist has access to biopsies, cultures, and sophisticated diagnostic techniques, it is important to remember these useful bedside tests. The ability to rapidly pin a diagnosis is particularly useful on the consultative service where critically ill patients can benefit from identification of a causative pathogen sooner rather than later. Residents should master these stains in their training, as this knowledge may prove to be invaluable in their careers.
Dermatologists are fortunate to specialize in the organ that is most accessible to evaluation. Although we use the physical examination to formulate the initial differential diagnosis, at times we must rely on ancillary tests to narrow down the diagnosis. Various bedside testing modalities—potassium hydroxide (KOH) preparation, Tzanck smear, mineral oil preparation, and Gram stain—are most useful in diagnosing infectious causes of cutaneous disease. This guide serves as a useful reference for residents on how to perform these tests and which conditions they can help diagnose. Several of these procedures have no standard protocol for performing them; the literature is littered with various methodologies, fixatives, and stains, and as such, this article will attempt to describe a technique that is convenient and quick to perform with readily available materials while still offering high diagnostic utility.
KOH Preparation
A standard in the armamentarium of a dermatologist, the KOH preparation is invaluable to diagnose fungal and yeast infections. Although there are many available preparations including varying concentrations of KOH, dimethyl sulfoxide, and various inks, the procedure is similar for all of them.1 The first step involves collecting the specimen, which can be scale from an active border of suspected cutaneous dermatophyte or Malassezia infection, debris from suspected candidiasis, or hair shafts plucked from an area of alopecia of presumed tinea capitis. A no. 15 blade can be used to scrape the specimen onto a microscope slide, though a second microscope slide can be used in lieu of a blade in patients who will not remain still, and then a coverslip is placed. Two drops of the KOH solution of your choice are then placed on opposite ends of the coverslip, allowing capillary action to spread the stain evenly. A paper towel can be folded in half and pushed down on the surface of the coverslip to spread the stain and soak up any excess, and this pressure also can help the KOH solution digest the keratin in the specimen. Briefly heating the underside of the slide (below boiling point) will help digest the keratin; this step is not necessary when you are using a KOH preparation with dimethyl sulfoxide. Although many dermatologists view the slide almost immediately, ideally at least 5 minutes should pass before it is read. Particularly thick specimens may require additional digestion time, so setting them aside for later review may help visualize infectious agents. In a busy clinic where an immediate diagnosis may not be requisite and a prescription can be called in pending the result, waiting to review the slide may be feasible.
Tzanck Smear
The Tzanck smear is a useful cytopathologic test in the rapid diagnosis of herpetic lesions, though it cannot differentiate between herpes simplex virus type 1, herpes simplex virus type 2, and varicella-zoster virus. It also has shown utility for rapid diagnosis of protean other dermatologic conditions including autoimmune blistering disorders, cutaneous malignancies, and other infectious processes, though it has been superseded by histopathology in most cases.2 An ideal sample is collected by scraping the base of a fresh blister with a no. 15 blade or a second microscope slide. The scrapings then are smeared onto another microscope slide and allowed to air-dry briefly. Then, Wright-Giemsa stain is dispensed to cover the sample and allowed to sit for 15 minutes before being washed off with sterile water. After air-drying, the sample is examined for the presence of clumped multinucleated giant cells, a feature that confirms herpetic infection and allows rapid initiation of antiviral medication.3
Mineral Oil Preparation
A mineral oil preparation has utility in diagnosing ectoparasitic infestation. In the case of scabies, a positive microscopic examination is diagnostic and requires no further testing, allowing for rapid initiation of therapy. This technique also is useful in diagnosing rosacea related to Demodex, which requires a treatment algorithm that differs from the classic papulopustular rosacea which it mimics.4
Mineral oil preparations can be rapidly performed and interpreted. Several drops of mineral oil are placed onto a microscope slide and a no. 15 blade is dipped into this oil prior to scraping the sample lesion. For scabies, a burrow is scraped repeatedly with the blade, and the debris is collected in the mineral oil. Occasionally, the mite can be dermoscopically visualized as a jet plane or arrowhead at the leading edge of a burrow; scraping should be focused in the vicinity of the mite.5 A coverslip is applied to the microscope slide and examination for the mite, egg casings, and scybala can be performed with microscopy.6 For Demodex infestation, a facial pustule can be expressed or several eyelash hairs can be plucked and suspended in mineral oil. Examination of this specimen is identical to scabies.
Gram Stain
The Gram stain is invaluable in classifying bacteria, and a properly performed test can narrow the identification of a causative organism based on cellular morphology. Although it is more technically complex than other bedside diagnostic maneuvers, it can be rapidly performed once the sequence of stains is mastered. The collected sample is smeared onto a glass slide and then briefly passed over a flame several times to heat-fix the specimen. Caution should be taken to avoid direct or prolonged flame contact with the underside of the slide. After fixation, the staining can be performed. First, crystal violet is instilled onto the slide and remains on for 30 seconds before being rinsed off with sink water. Then, Gram iodine is used for 30 seconds, followed by another rinse in water. Next, pour the decolorizer solution over the slide until the runoff is clear, and then rinse in water. Finally, flood with safranin counterstain for 30 seconds and give the slide a final rinse. After air-drying, it is ready to be interpreted.7
Final Thoughts
Although the modern dermatologist has access to biopsies, cultures, and sophisticated diagnostic techniques, it is important to remember these useful bedside tests. The ability to rapidly pin a diagnosis is particularly useful on the consultative service where critically ill patients can benefit from identification of a causative pathogen sooner rather than later. Residents should master these stains in their training, as this knowledge may prove to be invaluable in their careers.
1. Trozak DJ, Tennenhouse DJ, Russell JJ. Dermatology Skills for Primary Care: An Illustrated Guide. Totowa, NJ: Humana Press; 2006.
2. Kelly B, Shimoni T. Reintroducing the Tzanck smear. Am J Clin Dermatol. 2009;10:141-152.
3. Singhi M, Gupta L. Tzanck smear: a useful diagnostic tool. Indian J Dermatol Venereol Leprol. 2005;71:295.
4. Elston DM. Demodex mites: facts and controversies. Clin Dermatol. 2010;28:502-504.
5. Dupuy A, Dehen L, Bourrat E, et al. Accuracy of standard dermoscopy for diagnosing scabies. J Am Acad Dermatol. 2007;56:53-62.
6. Bolognia J, Schaffer J, Duncan K, et al. Dermatology Essentials. St. Louis, MO: Saunders Elsevier; 2014.
7. Ruocco E, Baroni A, Donnarumma G, et al. Diagnostic procedures in dermatology. Clin Dermatol. 2011;29:548-556.
1. Trozak DJ, Tennenhouse DJ, Russell JJ. Dermatology Skills for Primary Care: An Illustrated Guide. Totowa, NJ: Humana Press; 2006.
2. Kelly B, Shimoni T. Reintroducing the Tzanck smear. Am J Clin Dermatol. 2009;10:141-152.
3. Singhi M, Gupta L. Tzanck smear: a useful diagnostic tool. Indian J Dermatol Venereol Leprol. 2005;71:295.
4. Elston DM. Demodex mites: facts and controversies. Clin Dermatol. 2010;28:502-504.
5. Dupuy A, Dehen L, Bourrat E, et al. Accuracy of standard dermoscopy for diagnosing scabies. J Am Acad Dermatol. 2007;56:53-62.
6. Bolognia J, Schaffer J, Duncan K, et al. Dermatology Essentials. St. Louis, MO: Saunders Elsevier; 2014.
7. Ruocco E, Baroni A, Donnarumma G, et al. Diagnostic procedures in dermatology. Clin Dermatol. 2011;29:548-556.

Granular Cell Tumor
Granular cell tumors (GCTs) tend to present as solitary nodules, not uncommonly affecting the dorsum of the tongue but also involving the skin, breasts, and internal organs.1 Cutaneous GCTs typically present as 0.5- to 3-cm firm nodules with a verrucous or eroded surface.2 They most commonly present in dark-skinned, middle-aged women but have been reported in all age groups and in both sexes.3 Multiple GCTs are reported in up to 25% of cases, rarely in association with LEOPARD syndrome (consisting of lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, and deafness).4 Granular cell tumors generally are benign with a metastatic rate of approximately 3%.2
Granular cell tumors are histopathologically characterized by sheets of large polygonal cells with small, round, central nuclei; cytoplasm that is eosinophilic, coarse, and granular, as well as periodic acid–Schiff positive and diastase resistant; and distinct cytoplasmic membranes (Figure 1). Pustulo-ovoid bodies of Milian often generally appear as larger eosinophilic granules surrounded by a clear halo (Figure 2).5 Increased mitotic activity, a high nuclear-cytoplasmic ratio, pleomorphism, and necrosis suggest malignancy.6
|
|
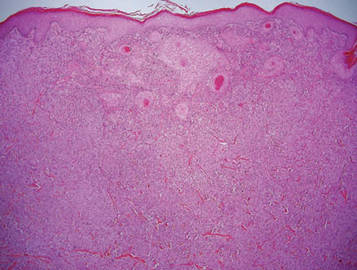
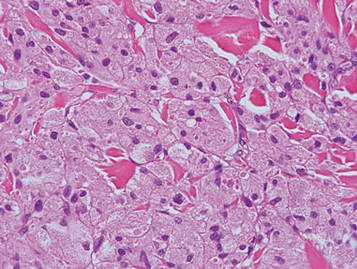
Lepromatous leprosy is characterized by sheets of histiocytes with vacuolated cytoplasm, some with clumped amphophilic bacilli known as globi (Figure 3). Mastocytoma can be distinguished from GCTs by the “fried egg” appearance of the mast cells (Figure 4). Although mast cells have a pale granular cytoplasm, they are smaller and lack pustulo-ovoid bodies and the polygonal shape of GCT cells. Reticulohistiocytoma, on the other hand, has two-toned dusty rose ground glass histiocytes (Figure 5), and xanthelasma can be distinguished histologically from GCT by the presence of a foamy rather than granular cytoplasm (Figure 6).
|
|
|
|
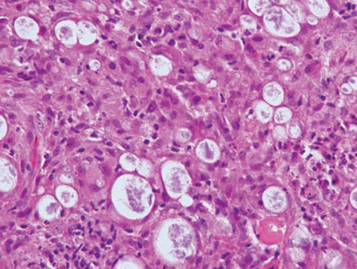

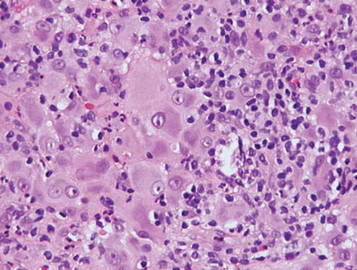

1. Elston DM, Ko C, Ferringer TC, et al, eds. Dermatopathology: Requisites in Dermatology. Philadelphia, PA: Saunders Elsevier; 2009.
2. Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
3. van de Loo S, Thunnissen E, Postmus P, et al. Granular cell tumor of the oral cavity; a case series including a case of metachronous occurrence in the tongue and the lung [published online ahead of print June 1, 2014]. Med Oral Patol Oral Cir Bucal. doi:10.4317/medoral.19867.
4. Schrader KA, Nelson TN, De Luca A, et al. Multiple granular cell tumors are an associated feature of LEOPARD syndrome caused by mutation in PTPN11. Clin Genet. 2009;75:185-189.
5. Epstein DS, Pashaei S, Hunt E Jr, et al. Pustulo-ovoid bodies of Milian in granular cell tumors. J Cutan Pathol. 2007;34:405-409.
6. Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
Granular cell tumors (GCTs) tend to present as solitary nodules, not uncommonly affecting the dorsum of the tongue but also involving the skin, breasts, and internal organs.1 Cutaneous GCTs typically present as 0.5- to 3-cm firm nodules with a verrucous or eroded surface.2 They most commonly present in dark-skinned, middle-aged women but have been reported in all age groups and in both sexes.3 Multiple GCTs are reported in up to 25% of cases, rarely in association with LEOPARD syndrome (consisting of lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, and deafness).4 Granular cell tumors generally are benign with a metastatic rate of approximately 3%.2
Granular cell tumors are histopathologically characterized by sheets of large polygonal cells with small, round, central nuclei; cytoplasm that is eosinophilic, coarse, and granular, as well as periodic acid–Schiff positive and diastase resistant; and distinct cytoplasmic membranes (Figure 1). Pustulo-ovoid bodies of Milian often generally appear as larger eosinophilic granules surrounded by a clear halo (Figure 2).5 Increased mitotic activity, a high nuclear-cytoplasmic ratio, pleomorphism, and necrosis suggest malignancy.6
|
|
Lepromatous leprosy is characterized by sheets of histiocytes with vacuolated cytoplasm, some with clumped amphophilic bacilli known as globi (Figure 3). Mastocytoma can be distinguished from GCTs by the “fried egg” appearance of the mast cells (Figure 4). Although mast cells have a pale granular cytoplasm, they are smaller and lack pustulo-ovoid bodies and the polygonal shape of GCT cells. Reticulohistiocytoma, on the other hand, has two-toned dusty rose ground glass histiocytes (Figure 5), and xanthelasma can be distinguished histologically from GCT by the presence of a foamy rather than granular cytoplasm (Figure 6).
|
|
|
|
Granular cell tumors (GCTs) tend to present as solitary nodules, not uncommonly affecting the dorsum of the tongue but also involving the skin, breasts, and internal organs.1 Cutaneous GCTs typically present as 0.5- to 3-cm firm nodules with a verrucous or eroded surface.2 They most commonly present in dark-skinned, middle-aged women but have been reported in all age groups and in both sexes.3 Multiple GCTs are reported in up to 25% of cases, rarely in association with LEOPARD syndrome (consisting of lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, and deafness).4 Granular cell tumors generally are benign with a metastatic rate of approximately 3%.2
Granular cell tumors are histopathologically characterized by sheets of large polygonal cells with small, round, central nuclei; cytoplasm that is eosinophilic, coarse, and granular, as well as periodic acid–Schiff positive and diastase resistant; and distinct cytoplasmic membranes (Figure 1). Pustulo-ovoid bodies of Milian often generally appear as larger eosinophilic granules surrounded by a clear halo (Figure 2).5 Increased mitotic activity, a high nuclear-cytoplasmic ratio, pleomorphism, and necrosis suggest malignancy.6
|
|
Lepromatous leprosy is characterized by sheets of histiocytes with vacuolated cytoplasm, some with clumped amphophilic bacilli known as globi (Figure 3). Mastocytoma can be distinguished from GCTs by the “fried egg” appearance of the mast cells (Figure 4). Although mast cells have a pale granular cytoplasm, they are smaller and lack pustulo-ovoid bodies and the polygonal shape of GCT cells. Reticulohistiocytoma, on the other hand, has two-toned dusty rose ground glass histiocytes (Figure 5), and xanthelasma can be distinguished histologically from GCT by the presence of a foamy rather than granular cytoplasm (Figure 6).
|
|
|
|
1. Elston DM, Ko C, Ferringer TC, et al, eds. Dermatopathology: Requisites in Dermatology. Philadelphia, PA: Saunders Elsevier; 2009.
2. Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
3. van de Loo S, Thunnissen E, Postmus P, et al. Granular cell tumor of the oral cavity; a case series including a case of metachronous occurrence in the tongue and the lung [published online ahead of print June 1, 2014]. Med Oral Patol Oral Cir Bucal. doi:10.4317/medoral.19867.
4. Schrader KA, Nelson TN, De Luca A, et al. Multiple granular cell tumors are an associated feature of LEOPARD syndrome caused by mutation in PTPN11. Clin Genet. 2009;75:185-189.
5. Epstein DS, Pashaei S, Hunt E Jr, et al. Pustulo-ovoid bodies of Milian in granular cell tumors. J Cutan Pathol. 2007;34:405-409.
6. Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
1. Elston DM, Ko C, Ferringer TC, et al, eds. Dermatopathology: Requisites in Dermatology. Philadelphia, PA: Saunders Elsevier; 2009.
2. Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
3. van de Loo S, Thunnissen E, Postmus P, et al. Granular cell tumor of the oral cavity; a case series including a case of metachronous occurrence in the tongue and the lung [published online ahead of print June 1, 2014]. Med Oral Patol Oral Cir Bucal. doi:10.4317/medoral.19867.
4. Schrader KA, Nelson TN, De Luca A, et al. Multiple granular cell tumors are an associated feature of LEOPARD syndrome caused by mutation in PTPN11. Clin Genet. 2009;75:185-189.
5. Epstein DS, Pashaei S, Hunt E Jr, et al. Pustulo-ovoid bodies of Milian in granular cell tumors. J Cutan Pathol. 2007;34:405-409.
6. Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
Nodular Extramammary Paget Disease With Fibroepitheliomatous Hyperplasia
Extramammary Paget disease (EMPD) is an uncommon neoplasm that most commonly occurs in the anogenital region but can arise in any area of the skin or mucosa.1 On clinical examination, EMPD typically presents as a sharply demarcated, erythematous, eczematoid, weeping lesion with varying degrees of induration; it rarely presents as a palpable mass or evenly raised nodule.2 Microscopically, it may be accompanied by varying degrees of epidermal hyperplasia.1 In particular, fibroepitheliomatous hyperplasia contains lacy strands of squamous epithelium resembling fibroepithelioma of Pinkus.3 We report a case of EMPD in a 90-year-old man who presented with a verrucous nodule in the pubic area that histologically demonstrated fibroepitheliomatous hyperplasia with lacy strands of squamous epithelium.
Case Report
A 90-year-old man presented with asymptomatic, well-demarcated, erythematous plaques in the pubic area of 5 years’ duration, along with a 3.0×2.5-cm nodule on the left side of the pubic area (Figure 1). Laboratory test results including a complete blood cell count, blood chemistry, and routine urinalysis were within reference range. Punch biopsies were taken from each plaque and nodule, as marked with arrows in Figure 1. Histopathologically, the plaques were seen to contain a number of large round cells with abundant pale cytoplasm and pleomorphic hyperchromatic nuclei that were present at various levels of the epidermis where they formed nests and clusters but did not extend into the dermis (Figures 2A and 2B). The nodule contained lacy strands of squamous epithelium extending from the epidermis to the mid dermis as well as many glandular structures (Figures 2C and 2D). The cells in the epidermis stained positively with periodic acid–Schiff (PAS), carcinoembryonic antigen (CEA), and cytokeratin 7 (Figure 2E). We also tested for S-100 protein to rule out malignant melanoma, which was negative.
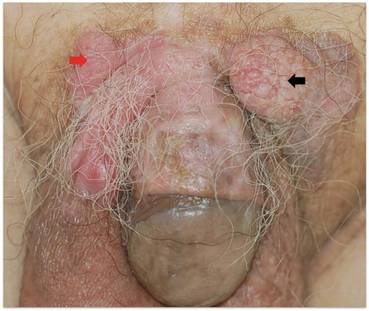
Based on both the clinical and histological features, a diagnosis of EMPD with fibroepitheliomatous hyperplasia was made. It was recommended that the patient undergo further evaluation and treatment; he declined due to his financial situation and was subsequently lost to follow-up.
Comment
Clinically, EMPD usually presents as a patch of macular erythema, an erythematous eruption, or erythematous papules and plaques.4 The palpable nodule seen in our patient is not a common presentation of EMPD. Pruritus is the most common symptom of EMPD, occurring in 70% of patients.5 Other symptoms include burning, irritation, pain, tenderness, bleeding, and swelling. Ten percent of EMPD cases are asymptomatic.5
Histologically, Paget cells primarily involve the epidermis where they usually form clusters or solid nests. In more than 90% of EMPD cases, the Paget cells contain cytoplasmic mucin that stains positively with mucicarmine and PAS. Immunohistochemical staining for cytokeratin 7, gross cystic disease fluid protein-15, S-100 protein, and CEA sometimes may be needed to differentiate from mimickers such as Bowen disease and superficial spreading melanoma.6 In our patient, the tumor cells stained positive for cytokeratin 7, CEA, and PAS. Malignant melanoma was ruled out with a test for S-100 protein.
|
|
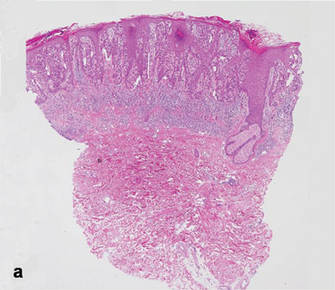
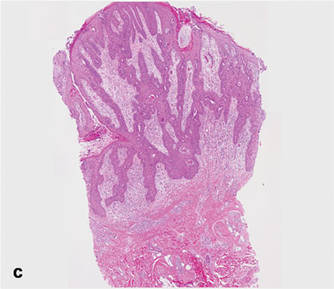
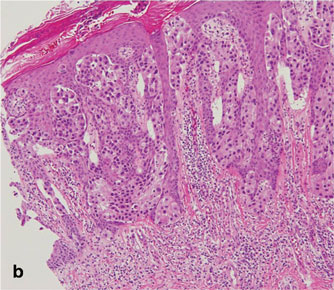
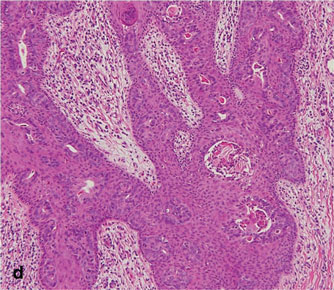
Extramammary Paget disease often is associated with epidermal hyperplasia, which can be classified as squamous, papillomatous, or fibroepitheliomatous.3 Microscopically, squamous hyperplasia is characterized by prominent thickening of the epidermis from diffuse plaquelike hyperplasia and is usually associated with hyperkeratosis. Papillomatous hyperplasia has an exophytic papillary or verrucous architecture and is associated with parakeratosis. Fibroepitheliomatous, or fibroepitheliomalike, hyperplasia generally consists of a discrete, broad, elevated plaque or nodule produced by hyperplasia of keratinocytes that form lacy strands of squamous epithelium.3 The biphasic pattern of proliferating epidermis and entrapped dermis simulates a so-called fibroepithelioma. Paget cells can be seen within the lacy strands of epidermal columns and in the acanthotic surface component.2 The finding of fibroepitheliomatous hyperplasia in anogenital skin should prompt a search for the diagnostic Paget cells to eliminate a fibroepithelioma of Pinkus variant of basal cell carcinoma, though the latter is uncommon and rarely occurs at this site.7
Of the 3 types of epidermal hyperplasia, our case demonstrated the fibroepitheliomatous type. There may be some relationship between EMPD and fibroepitheliomatous hyperplasia because most reported cases of EMPD with fibroepitheliomatous hyperplasia have occurred in the anogenital region. Also, epidermal hyperplasia is more frequent in anogenital Paget disease than in axillary Paget disease.8
Conclusion
Our case showed the unique finding of a verrucous nodular EMPD lesion in which peculiar histological features presented as extensions of the tumor cells forming lacy strands of squamous epithelium from the epidermis to the mid dermis as well as many glandular structures.
1. Lloyd J, Flanagan AM. Mammary and extramammary Paget’s disease. J Clin Pathol. 2000;53:742-749.
2. Billings SD, Roth LM. Pseudoinvasive, nodular extramam-mary Paget’s disease of the vulva. Arch Pathol Lab Med. 1998;122:471-474.
3. Brainard JA, Hart WR. Proliferative epidermal lesions associated with anogenital Paget’s disease. Am J Surg Pathol. 2000;24:543-552.
4. Neuhaus IM, Grekin RC. Mammary and extramammary Paget disease. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. Vol 1. 7th ed. New York, NY: McGraw-Hill; 2008:1094-1098.
5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Kim JC, Kim HC, Jeong CS, et al. Extramammary Paget’s disease with aggressive behavior: a report of two cases. J Korean Med Sci. 1999;14:223-226.
7. Rahbari H, Mehregan AH. Basal cell epitheliomas in usual and unusual sites. J Cutan Pathol. 1979;6:425-431.
8. Ishida-Yamamoto A, Sato K, Wada T, et al. Fibroepithelioma-like changes occurring in perianal Paget’s disease with rectal mucinous carcinoma: case report and review of 49 cases of extramammary Paget’s disease. J Cutan Pathol. 2002;29:185-189.
Extramammary Paget disease (EMPD) is an uncommon neoplasm that most commonly occurs in the anogenital region but can arise in any area of the skin or mucosa.1 On clinical examination, EMPD typically presents as a sharply demarcated, erythematous, eczematoid, weeping lesion with varying degrees of induration; it rarely presents as a palpable mass or evenly raised nodule.2 Microscopically, it may be accompanied by varying degrees of epidermal hyperplasia.1 In particular, fibroepitheliomatous hyperplasia contains lacy strands of squamous epithelium resembling fibroepithelioma of Pinkus.3 We report a case of EMPD in a 90-year-old man who presented with a verrucous nodule in the pubic area that histologically demonstrated fibroepitheliomatous hyperplasia with lacy strands of squamous epithelium.
Case Report
A 90-year-old man presented with asymptomatic, well-demarcated, erythematous plaques in the pubic area of 5 years’ duration, along with a 3.0×2.5-cm nodule on the left side of the pubic area (Figure 1). Laboratory test results including a complete blood cell count, blood chemistry, and routine urinalysis were within reference range. Punch biopsies were taken from each plaque and nodule, as marked with arrows in Figure 1. Histopathologically, the plaques were seen to contain a number of large round cells with abundant pale cytoplasm and pleomorphic hyperchromatic nuclei that were present at various levels of the epidermis where they formed nests and clusters but did not extend into the dermis (Figures 2A and 2B). The nodule contained lacy strands of squamous epithelium extending from the epidermis to the mid dermis as well as many glandular structures (Figures 2C and 2D). The cells in the epidermis stained positively with periodic acid–Schiff (PAS), carcinoembryonic antigen (CEA), and cytokeratin 7 (Figure 2E). We also tested for S-100 protein to rule out malignant melanoma, which was negative.
Based on both the clinical and histological features, a diagnosis of EMPD with fibroepitheliomatous hyperplasia was made. It was recommended that the patient undergo further evaluation and treatment; he declined due to his financial situation and was subsequently lost to follow-up.
Comment
Clinically, EMPD usually presents as a patch of macular erythema, an erythematous eruption, or erythematous papules and plaques.4 The palpable nodule seen in our patient is not a common presentation of EMPD. Pruritus is the most common symptom of EMPD, occurring in 70% of patients.5 Other symptoms include burning, irritation, pain, tenderness, bleeding, and swelling. Ten percent of EMPD cases are asymptomatic.5
Histologically, Paget cells primarily involve the epidermis where they usually form clusters or solid nests. In more than 90% of EMPD cases, the Paget cells contain cytoplasmic mucin that stains positively with mucicarmine and PAS. Immunohistochemical staining for cytokeratin 7, gross cystic disease fluid protein-15, S-100 protein, and CEA sometimes may be needed to differentiate from mimickers such as Bowen disease and superficial spreading melanoma.6 In our patient, the tumor cells stained positive for cytokeratin 7, CEA, and PAS. Malignant melanoma was ruled out with a test for S-100 protein.
|
|
Extramammary Paget disease often is associated with epidermal hyperplasia, which can be classified as squamous, papillomatous, or fibroepitheliomatous.3 Microscopically, squamous hyperplasia is characterized by prominent thickening of the epidermis from diffuse plaquelike hyperplasia and is usually associated with hyperkeratosis. Papillomatous hyperplasia has an exophytic papillary or verrucous architecture and is associated with parakeratosis. Fibroepitheliomatous, or fibroepitheliomalike, hyperplasia generally consists of a discrete, broad, elevated plaque or nodule produced by hyperplasia of keratinocytes that form lacy strands of squamous epithelium.3 The biphasic pattern of proliferating epidermis and entrapped dermis simulates a so-called fibroepithelioma. Paget cells can be seen within the lacy strands of epidermal columns and in the acanthotic surface component.2 The finding of fibroepitheliomatous hyperplasia in anogenital skin should prompt a search for the diagnostic Paget cells to eliminate a fibroepithelioma of Pinkus variant of basal cell carcinoma, though the latter is uncommon and rarely occurs at this site.7
Of the 3 types of epidermal hyperplasia, our case demonstrated the fibroepitheliomatous type. There may be some relationship between EMPD and fibroepitheliomatous hyperplasia because most reported cases of EMPD with fibroepitheliomatous hyperplasia have occurred in the anogenital region. Also, epidermal hyperplasia is more frequent in anogenital Paget disease than in axillary Paget disease.8
Conclusion
Our case showed the unique finding of a verrucous nodular EMPD lesion in which peculiar histological features presented as extensions of the tumor cells forming lacy strands of squamous epithelium from the epidermis to the mid dermis as well as many glandular structures.
Extramammary Paget disease (EMPD) is an uncommon neoplasm that most commonly occurs in the anogenital region but can arise in any area of the skin or mucosa.1 On clinical examination, EMPD typically presents as a sharply demarcated, erythematous, eczematoid, weeping lesion with varying degrees of induration; it rarely presents as a palpable mass or evenly raised nodule.2 Microscopically, it may be accompanied by varying degrees of epidermal hyperplasia.1 In particular, fibroepitheliomatous hyperplasia contains lacy strands of squamous epithelium resembling fibroepithelioma of Pinkus.3 We report a case of EMPD in a 90-year-old man who presented with a verrucous nodule in the pubic area that histologically demonstrated fibroepitheliomatous hyperplasia with lacy strands of squamous epithelium.
Case Report
A 90-year-old man presented with asymptomatic, well-demarcated, erythematous plaques in the pubic area of 5 years’ duration, along with a 3.0×2.5-cm nodule on the left side of the pubic area (Figure 1). Laboratory test results including a complete blood cell count, blood chemistry, and routine urinalysis were within reference range. Punch biopsies were taken from each plaque and nodule, as marked with arrows in Figure 1. Histopathologically, the plaques were seen to contain a number of large round cells with abundant pale cytoplasm and pleomorphic hyperchromatic nuclei that were present at various levels of the epidermis where they formed nests and clusters but did not extend into the dermis (Figures 2A and 2B). The nodule contained lacy strands of squamous epithelium extending from the epidermis to the mid dermis as well as many glandular structures (Figures 2C and 2D). The cells in the epidermis stained positively with periodic acid–Schiff (PAS), carcinoembryonic antigen (CEA), and cytokeratin 7 (Figure 2E). We also tested for S-100 protein to rule out malignant melanoma, which was negative.
Based on both the clinical and histological features, a diagnosis of EMPD with fibroepitheliomatous hyperplasia was made. It was recommended that the patient undergo further evaluation and treatment; he declined due to his financial situation and was subsequently lost to follow-up.
Comment
Clinically, EMPD usually presents as a patch of macular erythema, an erythematous eruption, or erythematous papules and plaques.4 The palpable nodule seen in our patient is not a common presentation of EMPD. Pruritus is the most common symptom of EMPD, occurring in 70% of patients.5 Other symptoms include burning, irritation, pain, tenderness, bleeding, and swelling. Ten percent of EMPD cases are asymptomatic.5
Histologically, Paget cells primarily involve the epidermis where they usually form clusters or solid nests. In more than 90% of EMPD cases, the Paget cells contain cytoplasmic mucin that stains positively with mucicarmine and PAS. Immunohistochemical staining for cytokeratin 7, gross cystic disease fluid protein-15, S-100 protein, and CEA sometimes may be needed to differentiate from mimickers such as Bowen disease and superficial spreading melanoma.6 In our patient, the tumor cells stained positive for cytokeratin 7, CEA, and PAS. Malignant melanoma was ruled out with a test for S-100 protein.
|
|
Extramammary Paget disease often is associated with epidermal hyperplasia, which can be classified as squamous, papillomatous, or fibroepitheliomatous.3 Microscopically, squamous hyperplasia is characterized by prominent thickening of the epidermis from diffuse plaquelike hyperplasia and is usually associated with hyperkeratosis. Papillomatous hyperplasia has an exophytic papillary or verrucous architecture and is associated with parakeratosis. Fibroepitheliomatous, or fibroepitheliomalike, hyperplasia generally consists of a discrete, broad, elevated plaque or nodule produced by hyperplasia of keratinocytes that form lacy strands of squamous epithelium.3 The biphasic pattern of proliferating epidermis and entrapped dermis simulates a so-called fibroepithelioma. Paget cells can be seen within the lacy strands of epidermal columns and in the acanthotic surface component.2 The finding of fibroepitheliomatous hyperplasia in anogenital skin should prompt a search for the diagnostic Paget cells to eliminate a fibroepithelioma of Pinkus variant of basal cell carcinoma, though the latter is uncommon and rarely occurs at this site.7
Of the 3 types of epidermal hyperplasia, our case demonstrated the fibroepitheliomatous type. There may be some relationship between EMPD and fibroepitheliomatous hyperplasia because most reported cases of EMPD with fibroepitheliomatous hyperplasia have occurred in the anogenital region. Also, epidermal hyperplasia is more frequent in anogenital Paget disease than in axillary Paget disease.8
Conclusion
Our case showed the unique finding of a verrucous nodular EMPD lesion in which peculiar histological features presented as extensions of the tumor cells forming lacy strands of squamous epithelium from the epidermis to the mid dermis as well as many glandular structures.
1. Lloyd J, Flanagan AM. Mammary and extramammary Paget’s disease. J Clin Pathol. 2000;53:742-749.
2. Billings SD, Roth LM. Pseudoinvasive, nodular extramam-mary Paget’s disease of the vulva. Arch Pathol Lab Med. 1998;122:471-474.
3. Brainard JA, Hart WR. Proliferative epidermal lesions associated with anogenital Paget’s disease. Am J Surg Pathol. 2000;24:543-552.
4. Neuhaus IM, Grekin RC. Mammary and extramammary Paget disease. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. Vol 1. 7th ed. New York, NY: McGraw-Hill; 2008:1094-1098.
5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Kim JC, Kim HC, Jeong CS, et al. Extramammary Paget’s disease with aggressive behavior: a report of two cases. J Korean Med Sci. 1999;14:223-226.
7. Rahbari H, Mehregan AH. Basal cell epitheliomas in usual and unusual sites. J Cutan Pathol. 1979;6:425-431.
8. Ishida-Yamamoto A, Sato K, Wada T, et al. Fibroepithelioma-like changes occurring in perianal Paget’s disease with rectal mucinous carcinoma: case report and review of 49 cases of extramammary Paget’s disease. J Cutan Pathol. 2002;29:185-189.
1. Lloyd J, Flanagan AM. Mammary and extramammary Paget’s disease. J Clin Pathol. 2000;53:742-749.
2. Billings SD, Roth LM. Pseudoinvasive, nodular extramam-mary Paget’s disease of the vulva. Arch Pathol Lab Med. 1998;122:471-474.
3. Brainard JA, Hart WR. Proliferative epidermal lesions associated with anogenital Paget’s disease. Am J Surg Pathol. 2000;24:543-552.
4. Neuhaus IM, Grekin RC. Mammary and extramammary Paget disease. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. Vol 1. 7th ed. New York, NY: McGraw-Hill; 2008:1094-1098.
5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Kim JC, Kim HC, Jeong CS, et al. Extramammary Paget’s disease with aggressive behavior: a report of two cases. J Korean Med Sci. 1999;14:223-226.
7. Rahbari H, Mehregan AH. Basal cell epitheliomas in usual and unusual sites. J Cutan Pathol. 1979;6:425-431.
8. Ishida-Yamamoto A, Sato K, Wada T, et al. Fibroepithelioma-like changes occurring in perianal Paget’s disease with rectal mucinous carcinoma: case report and review of 49 cases of extramammary Paget’s disease. J Cutan Pathol. 2002;29:185-189.
- Extramammary Paget disease (EMPD) should be considered in the clinical differential diagnosis of verrucous nodules in the pubic area.
- Histopathologically, EMPD in the anogenital area could show fibroepitheliomatous hyperplasia with lacy strands of squamous epithelium.
Hypopigmented Facial Papules on the Cheeks
The Diagnosis: Tumor of the Follicular Infundibulum
Histopathologic findings from a facial papule in our patient revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (Figure). There was no atypia. Gomori methenamine-silver and periodic acid–Schiff stains for fungi were negative. The combined clinical presentation and histopathologic findings supported the diagnosis of multiple tumor of the follicular infundibulum (TFI).
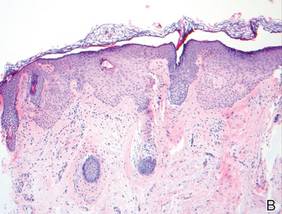
Tumor of the follicular infundibulum was diagnosed based on a biopsy from the right cheek that revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (A and B)(H&E, original magnifications ×40 and ×100). |
Tumor of the follicular infundibulum is an uncommon benign neoplasm that was first described in 1961 by Mehregan and Butler.1 The reported frequency is 10 per 100,000 biopsies.2 The majority of cases have been reported as solitary lesions, and multiple TFI are rare.3 Tumor of the follicular infundibulum affects middle-aged and elderly individuals with a female predominance.4 Multiple lesions generally range in number from 10 to 20, but there are few reports of more than 100 lesions.2,3,5,6 The solitary tumors often are initially misdiagnosed as basal cell carcinomas (BCCs) or seborrheic keratosis. Multiple TFI have been described variably as hypopigmented, flesh-colored and pink, flat and slightly depressed macules and thin papules. Sites of predilection include the scalp, face, neck, and upper trunk.2,3,5
There is no histopathologic difference between solitary and multiple TFI. Tumor of the follicular infundibulum displays a characteristic pale platelike proliferation of keratinocytes within the upper dermis attached to the overlying epidermis. The proliferating cells stain positive with periodic acid–Schiff, diastase-digestible glycogen is present in the cells at the base of the tumor, and a thickened network or brushlike pattern of elastic fibers surrounds the periphery of the tumor.1 Tumor of the follicular infundibulum is occasionally discovered incidentally on biopsy and has been observed in the margin of wide excisions of a variety of neoplasms including BCC.7 Based on the close association of TFI and BCC in the same specimens, Weyers et al7 concluded that TFI may be a nonaggressive type of BCC. Cribier and Grosshans2 reported 2 cases of TFI overlying a nevus sebaceous and a fibroma.
Treatment of TFI includes topical keratolytics, topical retinoic acid,5 imiquimod,8 topical steroids, and oral etretinate,6 all of which result in minimal improvement or incomplete resolution. Destructive treatments include cryotherapy, curettage, electrosurgery, laser ablation, and surgical excision, but all may lead to an unacceptable cosmetic result.
1. Mehregan AH, Butler JD. A tumor of follicular infundibulum. Arch Dermatol. 1961;83:78-81.
2. Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
3. Kolenik SA 3rd, Bolognia JL, Castiglione FM Jr, et al. Multiple tumors of the follicular infundibulum. Int J Dermatol. 1996;35:282-284.
4. Ackerman AB, Reddy VB, Soyer HP. Neoplasms With Follicular Differentiation. New York, NY: Ardor Scribendi; 2001.
5. Kossard S, Finley AG, Poyzer K, et al. Eruptive infundibulomas. J Am Acad Dermatol. 1989;21:361-366.
6. Schnitzler L, Civatte J, Robin F, et al. Multiple tumors of the follicular infundibulum with basocellular degeneration. apropos of a case [in French]. Ann Dermatol Venereol. 1987;114:551-556.
7. Weyers W, Horster S, Diaz-Cascajo C. Tumor of follicular infundibulum is basal cell carcinoma. Am J Dermatopathol. 2009;31:634-641.
8. Martin JE, Hsu M, Wang LC. An unusual clinical presentation of multiple tumors of the follicular infundibulum. J Am Acad Dermatol. 2009;60:885-886.
The Diagnosis: Tumor of the Follicular Infundibulum
Histopathologic findings from a facial papule in our patient revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (Figure). There was no atypia. Gomori methenamine-silver and periodic acid–Schiff stains for fungi were negative. The combined clinical presentation and histopathologic findings supported the diagnosis of multiple tumor of the follicular infundibulum (TFI).
|
Tumor of the follicular infundibulum was diagnosed based on a biopsy from the right cheek that revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (A and B)(H&E, original magnifications ×40 and ×100). |
Tumor of the follicular infundibulum is an uncommon benign neoplasm that was first described in 1961 by Mehregan and Butler.1 The reported frequency is 10 per 100,000 biopsies.2 The majority of cases have been reported as solitary lesions, and multiple TFI are rare.3 Tumor of the follicular infundibulum affects middle-aged and elderly individuals with a female predominance.4 Multiple lesions generally range in number from 10 to 20, but there are few reports of more than 100 lesions.2,3,5,6 The solitary tumors often are initially misdiagnosed as basal cell carcinomas (BCCs) or seborrheic keratosis. Multiple TFI have been described variably as hypopigmented, flesh-colored and pink, flat and slightly depressed macules and thin papules. Sites of predilection include the scalp, face, neck, and upper trunk.2,3,5
There is no histopathologic difference between solitary and multiple TFI. Tumor of the follicular infundibulum displays a characteristic pale platelike proliferation of keratinocytes within the upper dermis attached to the overlying epidermis. The proliferating cells stain positive with periodic acid–Schiff, diastase-digestible glycogen is present in the cells at the base of the tumor, and a thickened network or brushlike pattern of elastic fibers surrounds the periphery of the tumor.1 Tumor of the follicular infundibulum is occasionally discovered incidentally on biopsy and has been observed in the margin of wide excisions of a variety of neoplasms including BCC.7 Based on the close association of TFI and BCC in the same specimens, Weyers et al7 concluded that TFI may be a nonaggressive type of BCC. Cribier and Grosshans2 reported 2 cases of TFI overlying a nevus sebaceous and a fibroma.
Treatment of TFI includes topical keratolytics, topical retinoic acid,5 imiquimod,8 topical steroids, and oral etretinate,6 all of which result in minimal improvement or incomplete resolution. Destructive treatments include cryotherapy, curettage, electrosurgery, laser ablation, and surgical excision, but all may lead to an unacceptable cosmetic result.
The Diagnosis: Tumor of the Follicular Infundibulum
Histopathologic findings from a facial papule in our patient revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (Figure). There was no atypia. Gomori methenamine-silver and periodic acid–Schiff stains for fungi were negative. The combined clinical presentation and histopathologic findings supported the diagnosis of multiple tumor of the follicular infundibulum (TFI).
|
Tumor of the follicular infundibulum was diagnosed based on a biopsy from the right cheek that revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (A and B)(H&E, original magnifications ×40 and ×100). |
Tumor of the follicular infundibulum is an uncommon benign neoplasm that was first described in 1961 by Mehregan and Butler.1 The reported frequency is 10 per 100,000 biopsies.2 The majority of cases have been reported as solitary lesions, and multiple TFI are rare.3 Tumor of the follicular infundibulum affects middle-aged and elderly individuals with a female predominance.4 Multiple lesions generally range in number from 10 to 20, but there are few reports of more than 100 lesions.2,3,5,6 The solitary tumors often are initially misdiagnosed as basal cell carcinomas (BCCs) or seborrheic keratosis. Multiple TFI have been described variably as hypopigmented, flesh-colored and pink, flat and slightly depressed macules and thin papules. Sites of predilection include the scalp, face, neck, and upper trunk.2,3,5
There is no histopathologic difference between solitary and multiple TFI. Tumor of the follicular infundibulum displays a characteristic pale platelike proliferation of keratinocytes within the upper dermis attached to the overlying epidermis. The proliferating cells stain positive with periodic acid–Schiff, diastase-digestible glycogen is present in the cells at the base of the tumor, and a thickened network or brushlike pattern of elastic fibers surrounds the periphery of the tumor.1 Tumor of the follicular infundibulum is occasionally discovered incidentally on biopsy and has been observed in the margin of wide excisions of a variety of neoplasms including BCC.7 Based on the close association of TFI and BCC in the same specimens, Weyers et al7 concluded that TFI may be a nonaggressive type of BCC. Cribier and Grosshans2 reported 2 cases of TFI overlying a nevus sebaceous and a fibroma.
Treatment of TFI includes topical keratolytics, topical retinoic acid,5 imiquimod,8 topical steroids, and oral etretinate,6 all of which result in minimal improvement or incomplete resolution. Destructive treatments include cryotherapy, curettage, electrosurgery, laser ablation, and surgical excision, but all may lead to an unacceptable cosmetic result.
1. Mehregan AH, Butler JD. A tumor of follicular infundibulum. Arch Dermatol. 1961;83:78-81.
2. Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
3. Kolenik SA 3rd, Bolognia JL, Castiglione FM Jr, et al. Multiple tumors of the follicular infundibulum. Int J Dermatol. 1996;35:282-284.
4. Ackerman AB, Reddy VB, Soyer HP. Neoplasms With Follicular Differentiation. New York, NY: Ardor Scribendi; 2001.
5. Kossard S, Finley AG, Poyzer K, et al. Eruptive infundibulomas. J Am Acad Dermatol. 1989;21:361-366.
6. Schnitzler L, Civatte J, Robin F, et al. Multiple tumors of the follicular infundibulum with basocellular degeneration. apropos of a case [in French]. Ann Dermatol Venereol. 1987;114:551-556.
7. Weyers W, Horster S, Diaz-Cascajo C. Tumor of follicular infundibulum is basal cell carcinoma. Am J Dermatopathol. 2009;31:634-641.
8. Martin JE, Hsu M, Wang LC. An unusual clinical presentation of multiple tumors of the follicular infundibulum. J Am Acad Dermatol. 2009;60:885-886.
1. Mehregan AH, Butler JD. A tumor of follicular infundibulum. Arch Dermatol. 1961;83:78-81.
2. Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
3. Kolenik SA 3rd, Bolognia JL, Castiglione FM Jr, et al. Multiple tumors of the follicular infundibulum. Int J Dermatol. 1996;35:282-284.
4. Ackerman AB, Reddy VB, Soyer HP. Neoplasms With Follicular Differentiation. New York, NY: Ardor Scribendi; 2001.
5. Kossard S, Finley AG, Poyzer K, et al. Eruptive infundibulomas. J Am Acad Dermatol. 1989;21:361-366.
6. Schnitzler L, Civatte J, Robin F, et al. Multiple tumors of the follicular infundibulum with basocellular degeneration. apropos of a case [in French]. Ann Dermatol Venereol. 1987;114:551-556.
7. Weyers W, Horster S, Diaz-Cascajo C. Tumor of follicular infundibulum is basal cell carcinoma. Am J Dermatopathol. 2009;31:634-641.
8. Martin JE, Hsu M, Wang LC. An unusual clinical presentation of multiple tumors of the follicular infundibulum. J Am Acad Dermatol. 2009;60:885-886.
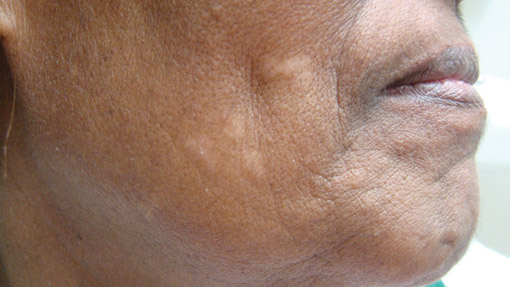
A 73-year-old woman presented with multiple mildly pruritic, hypopigmented, thin papules involving both cheeks of 5 months’ duration. The patient had no improvement with ketoconazole cream 2% and hydrocortisone cream 1% used daily for 1 month for presumed tinea versicolor. Physical examination revealed 10 ill-defined, 2- to 5-mm, round and oval, smooth hypopigmented, slightly raised papules located on the lower aspect of both cheeks.
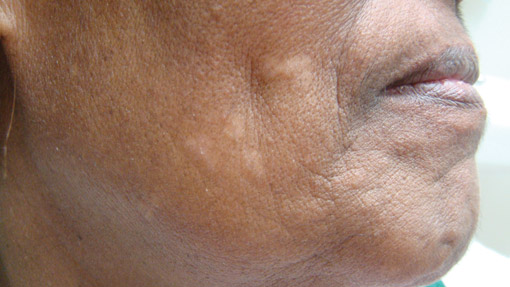
Sulfur Spring Dermatitis
Sulfur spring dermatitis is characterized by multiple punched-out erosions and pits. In prior case reports, patients often presented with painful swollen lesions that developed within 24 hours of bathing in hot sulfur springs.1 Because spa therapy and thermal spring baths are common in modern society, dermatologists should be aware of sulfur spring dermatitis as a potential adverse effect.
Case Report
A healthy 65-year-old man presented with painful skin lesions on the legs that developed after bathing for 25 minutes in a hot sulfur spring 1 day prior. The patient had no history of dermatologic disease. He reported a 10-year history of bathing in a hot sulfur spring for 20 minutes every 3 days in the winter. This time, he bathed 5 minutes longer than usual. No skin condition was noted prior to bathing, but he reported feeling a tickling sensation and scratching the legs while he was immersed in the water. One hour after bathing, he noted confluent, punched-out, round ulcers with peripheral erythema on the thighs and shins (Figure 1).
|
|
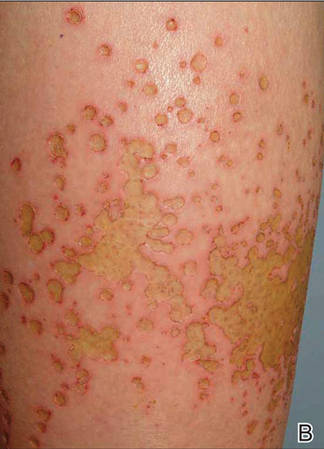
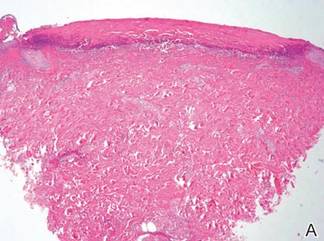
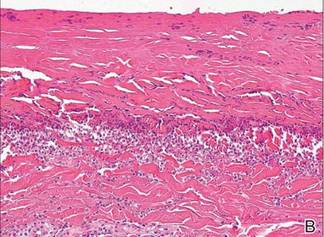
A skin biopsy revealed sharply demarcated, homogeneous coagulation necrosis of the epidermis. Many neutrophils were present under the necrosis (Figure 2). Periodic acid–Schiff and acid-fast stains were negative for infectious organisms, and a skin tissue culture yielded negative results. Intensive wound care was started with nitrofurazone ointment 0.2%. The ulcers healed gradually in the following months with scar formation and hyperpigmentation.
Comment
Thermal sulfur baths are a form of balneotherapy promoted in many cultures for improvement of skin conditions; however, certain uncommon skin problems may occur after bathing in hot sulfur springs.2 In particular, sulfur spring dermatitis is a potential adverse effect.
Thermal sulfur water is known to exert anti-inflammatory, keratoplastic, and antipruriginous effects. As a result, it often is used in many cultures as an alternative treatment of various skin conditions.2-4 Moreover, thermal sulfur baths are popular in northeastern Asian countries for their effects on mental health.5 Hot springs in northern Taiwan, which contain large amounts of hydrogen sulfide, sulfate, and sulfur differ from other thermal springs in that they are rather acidic in nature and release geothermal energy from volcanic activity.6 In addition to hot sulfur springs, there are neutral salt and CO2 springs in Taiwan.5 However, spring dermatitis has only been associated with bathing in hot sulfur springs due to high concentrations of hydrogen sulfide that break down keratin and cause dissolution of the stratum corneum.7
The incidence of sulfur spring dermatitis is unknown. Although the largest known case series reported 44 cases occurring within a decade in Taiwan,1 it is rarely seen in our daily practice. Previously reported cases of sulfur spring dermatitis noted clinical findings of swelling of the affected area followed by punched-out erosions with surrounding erythema. Most lesions gradually healed with dry brownish crusts. A patch test with sulfur spring water and sulfur compounds showed negative results; therefore, the mechanism is unlikely to be allergic reaction.1 The clinical differential diagnosis includes factitious ulcers as well as viral and fungal infections. A tissue culture should be performed to exclude infectious conditions.
This characteristic skin disease does not present in all individuals after bathing in hot sulfur springs. Lesions may present anywhere on the body with a predilection for skin folds, including the penis and scrotum. Preexisting skin conditions such as pruritus and xerosis are considered to be contributing factors. The possible etiology of sulfur spring dermatitis may be acid irritation from the unstable amount of soluble sulfur in the water, which is enhanced by the heat.1 In our patient, no prior skin disease was noted, but he scratched the skin on the thighs while bathing, which may have contributed to the development of lesions in this area rather than in the skin folds.
The skin biopsy specimen demonstrated epidermal coagulation necrosis, mild superficial dermal damage, and preservation of the pilosebaceous appendages. The ulcers were painful during healing and resolved with scarring and hyperpigmentation. The histopathologic findings and clinical course in our patient were similar to cases of superficial second-degree burns.8 It is possible that the keratoplastic effect of sulfur at high concentrations along with thermal water caused the skin condition.
Conclusion
Individuals who engage in thermal sulfur baths should be aware of potential adverse effects such as sulfur spring dermatitis, especially those with preexisting skin disorders.
1. Sun CC, Sue MS. Sulfur spring dermatitis. Contact Dermatitis. 1995;32:31-34.
2. Matz H, Orion E, Wolf R. Balneotherapy in dermatology. Dermatol Ther. 2003;16:132-140.
3. Leslie KS, Millington GW, Levell NJ. Sulphur and skin: from Satan to Saddam! J Cosmet Dermatol. 2004;3:94-98.
4. Millikan LE. Unapproved treatments or indications in dermatology: physical therapy including balneotherapy. Clin Dermatol. 2000;18:125-129.
5. Nirei H, Furuno K, Kusuda T. Medical geology in Japan. In: Selinus O, Finkelman RB, Centeno JA, eds. Medical Geology: A Regional Synthesis. New York, NY: Springer; 2010:329-354.
6. Liu CM, Song SR, Chen YL, et al. Characteristics and origins of hot springs in the Tatun Volcano Group in northern Taiwan. Terr Atmos Ocean Sci. 2011;22:475-489.
7. Lin AN, Reimer RJ, Carter DM. Sulfur revisited. J Am Acad Dermatol. 1988;18:553-558.
8. Weedon D. Reaction to physical agents. In: Weedon D. Weedon’s Skin Pathology. 3rd ed. London, England: Churchill Livingstone, Elsevier Health; 2010:525-540.
Sulfur spring dermatitis is characterized by multiple punched-out erosions and pits. In prior case reports, patients often presented with painful swollen lesions that developed within 24 hours of bathing in hot sulfur springs.1 Because spa therapy and thermal spring baths are common in modern society, dermatologists should be aware of sulfur spring dermatitis as a potential adverse effect.
Case Report
A healthy 65-year-old man presented with painful skin lesions on the legs that developed after bathing for 25 minutes in a hot sulfur spring 1 day prior. The patient had no history of dermatologic disease. He reported a 10-year history of bathing in a hot sulfur spring for 20 minutes every 3 days in the winter. This time, he bathed 5 minutes longer than usual. No skin condition was noted prior to bathing, but he reported feeling a tickling sensation and scratching the legs while he was immersed in the water. One hour after bathing, he noted confluent, punched-out, round ulcers with peripheral erythema on the thighs and shins (Figure 1).
|
|
A skin biopsy revealed sharply demarcated, homogeneous coagulation necrosis of the epidermis. Many neutrophils were present under the necrosis (Figure 2). Periodic acid–Schiff and acid-fast stains were negative for infectious organisms, and a skin tissue culture yielded negative results. Intensive wound care was started with nitrofurazone ointment 0.2%. The ulcers healed gradually in the following months with scar formation and hyperpigmentation.
Comment
Thermal sulfur baths are a form of balneotherapy promoted in many cultures for improvement of skin conditions; however, certain uncommon skin problems may occur after bathing in hot sulfur springs.2 In particular, sulfur spring dermatitis is a potential adverse effect.
Thermal sulfur water is known to exert anti-inflammatory, keratoplastic, and antipruriginous effects. As a result, it often is used in many cultures as an alternative treatment of various skin conditions.2-4 Moreover, thermal sulfur baths are popular in northeastern Asian countries for their effects on mental health.5 Hot springs in northern Taiwan, which contain large amounts of hydrogen sulfide, sulfate, and sulfur differ from other thermal springs in that they are rather acidic in nature and release geothermal energy from volcanic activity.6 In addition to hot sulfur springs, there are neutral salt and CO2 springs in Taiwan.5 However, spring dermatitis has only been associated with bathing in hot sulfur springs due to high concentrations of hydrogen sulfide that break down keratin and cause dissolution of the stratum corneum.7
The incidence of sulfur spring dermatitis is unknown. Although the largest known case series reported 44 cases occurring within a decade in Taiwan,1 it is rarely seen in our daily practice. Previously reported cases of sulfur spring dermatitis noted clinical findings of swelling of the affected area followed by punched-out erosions with surrounding erythema. Most lesions gradually healed with dry brownish crusts. A patch test with sulfur spring water and sulfur compounds showed negative results; therefore, the mechanism is unlikely to be allergic reaction.1 The clinical differential diagnosis includes factitious ulcers as well as viral and fungal infections. A tissue culture should be performed to exclude infectious conditions.
This characteristic skin disease does not present in all individuals after bathing in hot sulfur springs. Lesions may present anywhere on the body with a predilection for skin folds, including the penis and scrotum. Preexisting skin conditions such as pruritus and xerosis are considered to be contributing factors. The possible etiology of sulfur spring dermatitis may be acid irritation from the unstable amount of soluble sulfur in the water, which is enhanced by the heat.1 In our patient, no prior skin disease was noted, but he scratched the skin on the thighs while bathing, which may have contributed to the development of lesions in this area rather than in the skin folds.
The skin biopsy specimen demonstrated epidermal coagulation necrosis, mild superficial dermal damage, and preservation of the pilosebaceous appendages. The ulcers were painful during healing and resolved with scarring and hyperpigmentation. The histopathologic findings and clinical course in our patient were similar to cases of superficial second-degree burns.8 It is possible that the keratoplastic effect of sulfur at high concentrations along with thermal water caused the skin condition.
Conclusion
Individuals who engage in thermal sulfur baths should be aware of potential adverse effects such as sulfur spring dermatitis, especially those with preexisting skin disorders.
Sulfur spring dermatitis is characterized by multiple punched-out erosions and pits. In prior case reports, patients often presented with painful swollen lesions that developed within 24 hours of bathing in hot sulfur springs.1 Because spa therapy and thermal spring baths are common in modern society, dermatologists should be aware of sulfur spring dermatitis as a potential adverse effect.
Case Report
A healthy 65-year-old man presented with painful skin lesions on the legs that developed after bathing for 25 minutes in a hot sulfur spring 1 day prior. The patient had no history of dermatologic disease. He reported a 10-year history of bathing in a hot sulfur spring for 20 minutes every 3 days in the winter. This time, he bathed 5 minutes longer than usual. No skin condition was noted prior to bathing, but he reported feeling a tickling sensation and scratching the legs while he was immersed in the water. One hour after bathing, he noted confluent, punched-out, round ulcers with peripheral erythema on the thighs and shins (Figure 1).
|
|
A skin biopsy revealed sharply demarcated, homogeneous coagulation necrosis of the epidermis. Many neutrophils were present under the necrosis (Figure 2). Periodic acid–Schiff and acid-fast stains were negative for infectious organisms, and a skin tissue culture yielded negative results. Intensive wound care was started with nitrofurazone ointment 0.2%. The ulcers healed gradually in the following months with scar formation and hyperpigmentation.
Comment
Thermal sulfur baths are a form of balneotherapy promoted in many cultures for improvement of skin conditions; however, certain uncommon skin problems may occur after bathing in hot sulfur springs.2 In particular, sulfur spring dermatitis is a potential adverse effect.
Thermal sulfur water is known to exert anti-inflammatory, keratoplastic, and antipruriginous effects. As a result, it often is used in many cultures as an alternative treatment of various skin conditions.2-4 Moreover, thermal sulfur baths are popular in northeastern Asian countries for their effects on mental health.5 Hot springs in northern Taiwan, which contain large amounts of hydrogen sulfide, sulfate, and sulfur differ from other thermal springs in that they are rather acidic in nature and release geothermal energy from volcanic activity.6 In addition to hot sulfur springs, there are neutral salt and CO2 springs in Taiwan.5 However, spring dermatitis has only been associated with bathing in hot sulfur springs due to high concentrations of hydrogen sulfide that break down keratin and cause dissolution of the stratum corneum.7
The incidence of sulfur spring dermatitis is unknown. Although the largest known case series reported 44 cases occurring within a decade in Taiwan,1 it is rarely seen in our daily practice. Previously reported cases of sulfur spring dermatitis noted clinical findings of swelling of the affected area followed by punched-out erosions with surrounding erythema. Most lesions gradually healed with dry brownish crusts. A patch test with sulfur spring water and sulfur compounds showed negative results; therefore, the mechanism is unlikely to be allergic reaction.1 The clinical differential diagnosis includes factitious ulcers as well as viral and fungal infections. A tissue culture should be performed to exclude infectious conditions.
This characteristic skin disease does not present in all individuals after bathing in hot sulfur springs. Lesions may present anywhere on the body with a predilection for skin folds, including the penis and scrotum. Preexisting skin conditions such as pruritus and xerosis are considered to be contributing factors. The possible etiology of sulfur spring dermatitis may be acid irritation from the unstable amount of soluble sulfur in the water, which is enhanced by the heat.1 In our patient, no prior skin disease was noted, but he scratched the skin on the thighs while bathing, which may have contributed to the development of lesions in this area rather than in the skin folds.
The skin biopsy specimen demonstrated epidermal coagulation necrosis, mild superficial dermal damage, and preservation of the pilosebaceous appendages. The ulcers were painful during healing and resolved with scarring and hyperpigmentation. The histopathologic findings and clinical course in our patient were similar to cases of superficial second-degree burns.8 It is possible that the keratoplastic effect of sulfur at high concentrations along with thermal water caused the skin condition.
Conclusion
Individuals who engage in thermal sulfur baths should be aware of potential adverse effects such as sulfur spring dermatitis, especially those with preexisting skin disorders.
1. Sun CC, Sue MS. Sulfur spring dermatitis. Contact Dermatitis. 1995;32:31-34.
2. Matz H, Orion E, Wolf R. Balneotherapy in dermatology. Dermatol Ther. 2003;16:132-140.
3. Leslie KS, Millington GW, Levell NJ. Sulphur and skin: from Satan to Saddam! J Cosmet Dermatol. 2004;3:94-98.
4. Millikan LE. Unapproved treatments or indications in dermatology: physical therapy including balneotherapy. Clin Dermatol. 2000;18:125-129.
5. Nirei H, Furuno K, Kusuda T. Medical geology in Japan. In: Selinus O, Finkelman RB, Centeno JA, eds. Medical Geology: A Regional Synthesis. New York, NY: Springer; 2010:329-354.
6. Liu CM, Song SR, Chen YL, et al. Characteristics and origins of hot springs in the Tatun Volcano Group in northern Taiwan. Terr Atmos Ocean Sci. 2011;22:475-489.
7. Lin AN, Reimer RJ, Carter DM. Sulfur revisited. J Am Acad Dermatol. 1988;18:553-558.
8. Weedon D. Reaction to physical agents. In: Weedon D. Weedon’s Skin Pathology. 3rd ed. London, England: Churchill Livingstone, Elsevier Health; 2010:525-540.
1. Sun CC, Sue MS. Sulfur spring dermatitis. Contact Dermatitis. 1995;32:31-34.
2. Matz H, Orion E, Wolf R. Balneotherapy in dermatology. Dermatol Ther. 2003;16:132-140.
3. Leslie KS, Millington GW, Levell NJ. Sulphur and skin: from Satan to Saddam! J Cosmet Dermatol. 2004;3:94-98.
4. Millikan LE. Unapproved treatments or indications in dermatology: physical therapy including balneotherapy. Clin Dermatol. 2000;18:125-129.
5. Nirei H, Furuno K, Kusuda T. Medical geology in Japan. In: Selinus O, Finkelman RB, Centeno JA, eds. Medical Geology: A Regional Synthesis. New York, NY: Springer; 2010:329-354.
6. Liu CM, Song SR, Chen YL, et al. Characteristics and origins of hot springs in the Tatun Volcano Group in northern Taiwan. Terr Atmos Ocean Sci. 2011;22:475-489.
7. Lin AN, Reimer RJ, Carter DM. Sulfur revisited. J Am Acad Dermatol. 1988;18:553-558.
8. Weedon D. Reaction to physical agents. In: Weedon D. Weedon’s Skin Pathology. 3rd ed. London, England: Churchill Livingstone, Elsevier Health; 2010:525-540.
Practice Points
- The clinical findings of sulfur spring dermatitis are similar to those of a superficial second-degree burn.
- Careful evaluation of the patient’s clinical history and recognition of characteristic findings are important for correct diagnosis.
- Patients with preexisting skin disorders who engage in thermal sulfur baths should be aware of the potential adverse effect of sulfur spring dermatitis.
Clear Cell Fibrous Papule
A fibrous papule is a common benign lesion that usually presents in adults on the face, especially on the lower portion of the nose. It typically presents as a small (2–5 mm), asymptomatic, flesh-colored, dome-shaped lesion that is firm and nontender. Several histopathologic variants of fibrous papules have been described, including clear cell, granular, epithelioid, hypercellular, pleomorphic, pigmented, and inflammatory.1 Clear cell fibrous papules are exceedingly rare. On microscopic examination the epidermis may be normal or show some degree of hyperkeratosis and parakeratosis, erosion, ulceration, or crust. The basal layer may show an increase of melanin. The dermis is expanded by a proliferation of clear cells arranged in sheets, clusters, or as single cells (Figure 1). The clear cells show variation in size and shape. The nuclei are small and round without pleomorphism, hyperchromasia, or mitoses. The nuclei may be centrally located or eccentrically displaced by a large intracytoplasmic vacuole (Figure 2). Some clear cells may exhibit finely vacuolated cytoplasm with nuclear scalloping. The surrounding stroma usually consists of sclerotic collagen and dilated blood vessels (Figure 3). Extravasated red blood cells may be present focally. Patchy lymphocytic infiltrates may be found in the stroma at the periphery of the lesion. Periodic acid–Schiff and mucicarmine staining of the clear cells is negative. On immunohistochemistry, the clear cells are diffusely positive for vimentin and negative for cytokeratin AE1/AE3, epithelial membrane antigen, carcinoembryonic antigen, and HMB-45 (human melanoma black 45).2,3 The clear cells often are positive for CD68, factor XIIIa, and NKI/C3 (anti-CD63) but also may be negative. The S-100 protein often is negative but may be focally positive.
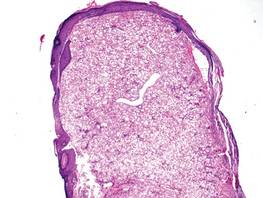
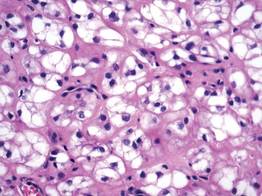
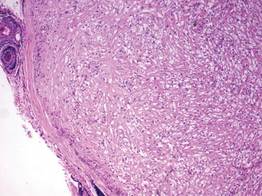
The differential diagnosis for clear cell fibrous papules is broad but reasonably includes balloon cell nevus, clear cell hidradenoma, and cutaneous metastasis of clear cell (conventional) renal cell carcinoma (ccRCC). Balloon cell malignant melanoma is not considered strongly in the differential diagnosis because it usually exhibits invasive growth, cytologic atypia, and mitoses, all of which are not characteristic morphologic features of clear cell fibrous papules.
A balloon cell nevus may be difficult to distinguish from a clear cell fibrous papule on routine hematoxylin and eosin staining (Figure 4); however, the nuclei of a balloon cell nevus tend to be more rounded and centrally located. Any junctional nesting or nests of conventional nevus cells in the dermis also help differentiate a balloon cell nevus from a clear cell fibrous papule. Diffusely positive immunostaining for S-100 protein also is indicative of a balloon cell nevus.
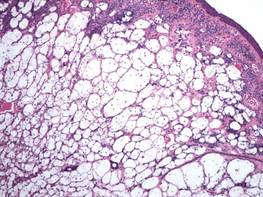
Clear cell hidradenoma consists predominantly of cells with clear cytoplasm and small dark nuclei that may closely mimic a clear cell fibrous papule (Figure 5) but often shows a second population of cells with more vesicular nuclei and dark eosinophilic cytoplasm. Cystic spaces containing hyaline material and foci of squamoid change are common, along with occasional tubular lumina that may be prominent or inconspicuous. Further, the tumor cells of clear cell hidradenoma show positive immunostaining for epithelial markers (eg, cytokeratin AE1/AE3, CAM5.2).
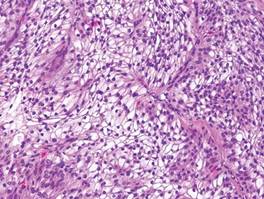
Cutaneous metastasis of ccRCC is rare and usually presents clinically as a larger lesion than a clear cell fibrous papule. The cells of ccRCC have moderate to abundant clear cytoplasm and nuclei with varying degrees of pleomorphism (Figure 6). Periodic acid–Schiff staining demonstrates intracytoplasmic glycogen. The stroma is abundantly vascular and extravasated blood cells are frequently observed. On immunohistochemistry, the tumor cells of ccRCC stain positively for cytokeratin AE1/AE3, CAM5.2, epithelial membrane antigen, CD10, and vimentin.
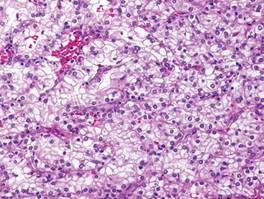
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Chiang YY, Tsai HH, Lee WR, et al. Clear cell fibrous papule: report of a case mimicking a balloon cell nevus. J Cutan Pathol. 2009;36:381-384.
- Lee AN, Stein SL, Cohen LM. Clear cell fibrous papule with NKI/C3 expression: clinical and histologic features in six cases. Am J Dermatopathol. 2005;27:296-300.
A fibrous papule is a common benign lesion that usually presents in adults on the face, especially on the lower portion of the nose. It typically presents as a small (2–5 mm), asymptomatic, flesh-colored, dome-shaped lesion that is firm and nontender. Several histopathologic variants of fibrous papules have been described, including clear cell, granular, epithelioid, hypercellular, pleomorphic, pigmented, and inflammatory.1 Clear cell fibrous papules are exceedingly rare. On microscopic examination the epidermis may be normal or show some degree of hyperkeratosis and parakeratosis, erosion, ulceration, or crust. The basal layer may show an increase of melanin. The dermis is expanded by a proliferation of clear cells arranged in sheets, clusters, or as single cells (Figure 1). The clear cells show variation in size and shape. The nuclei are small and round without pleomorphism, hyperchromasia, or mitoses. The nuclei may be centrally located or eccentrically displaced by a large intracytoplasmic vacuole (Figure 2). Some clear cells may exhibit finely vacuolated cytoplasm with nuclear scalloping. The surrounding stroma usually consists of sclerotic collagen and dilated blood vessels (Figure 3). Extravasated red blood cells may be present focally. Patchy lymphocytic infiltrates may be found in the stroma at the periphery of the lesion. Periodic acid–Schiff and mucicarmine staining of the clear cells is negative. On immunohistochemistry, the clear cells are diffusely positive for vimentin and negative for cytokeratin AE1/AE3, epithelial membrane antigen, carcinoembryonic antigen, and HMB-45 (human melanoma black 45).2,3 The clear cells often are positive for CD68, factor XIIIa, and NKI/C3 (anti-CD63) but also may be negative. The S-100 protein often is negative but may be focally positive.
The differential diagnosis for clear cell fibrous papules is broad but reasonably includes balloon cell nevus, clear cell hidradenoma, and cutaneous metastasis of clear cell (conventional) renal cell carcinoma (ccRCC). Balloon cell malignant melanoma is not considered strongly in the differential diagnosis because it usually exhibits invasive growth, cytologic atypia, and mitoses, all of which are not characteristic morphologic features of clear cell fibrous papules.
A balloon cell nevus may be difficult to distinguish from a clear cell fibrous papule on routine hematoxylin and eosin staining (Figure 4); however, the nuclei of a balloon cell nevus tend to be more rounded and centrally located. Any junctional nesting or nests of conventional nevus cells in the dermis also help differentiate a balloon cell nevus from a clear cell fibrous papule. Diffusely positive immunostaining for S-100 protein also is indicative of a balloon cell nevus.
Clear cell hidradenoma consists predominantly of cells with clear cytoplasm and small dark nuclei that may closely mimic a clear cell fibrous papule (Figure 5) but often shows a second population of cells with more vesicular nuclei and dark eosinophilic cytoplasm. Cystic spaces containing hyaline material and foci of squamoid change are common, along with occasional tubular lumina that may be prominent or inconspicuous. Further, the tumor cells of clear cell hidradenoma show positive immunostaining for epithelial markers (eg, cytokeratin AE1/AE3, CAM5.2).
Cutaneous metastasis of ccRCC is rare and usually presents clinically as a larger lesion than a clear cell fibrous papule. The cells of ccRCC have moderate to abundant clear cytoplasm and nuclei with varying degrees of pleomorphism (Figure 6). Periodic acid–Schiff staining demonstrates intracytoplasmic glycogen. The stroma is abundantly vascular and extravasated blood cells are frequently observed. On immunohistochemistry, the tumor cells of ccRCC stain positively for cytokeratin AE1/AE3, CAM5.2, epithelial membrane antigen, CD10, and vimentin.
A fibrous papule is a common benign lesion that usually presents in adults on the face, especially on the lower portion of the nose. It typically presents as a small (2–5 mm), asymptomatic, flesh-colored, dome-shaped lesion that is firm and nontender. Several histopathologic variants of fibrous papules have been described, including clear cell, granular, epithelioid, hypercellular, pleomorphic, pigmented, and inflammatory.1 Clear cell fibrous papules are exceedingly rare. On microscopic examination the epidermis may be normal or show some degree of hyperkeratosis and parakeratosis, erosion, ulceration, or crust. The basal layer may show an increase of melanin. The dermis is expanded by a proliferation of clear cells arranged in sheets, clusters, or as single cells (Figure 1). The clear cells show variation in size and shape. The nuclei are small and round without pleomorphism, hyperchromasia, or mitoses. The nuclei may be centrally located or eccentrically displaced by a large intracytoplasmic vacuole (Figure 2). Some clear cells may exhibit finely vacuolated cytoplasm with nuclear scalloping. The surrounding stroma usually consists of sclerotic collagen and dilated blood vessels (Figure 3). Extravasated red blood cells may be present focally. Patchy lymphocytic infiltrates may be found in the stroma at the periphery of the lesion. Periodic acid–Schiff and mucicarmine staining of the clear cells is negative. On immunohistochemistry, the clear cells are diffusely positive for vimentin and negative for cytokeratin AE1/AE3, epithelial membrane antigen, carcinoembryonic antigen, and HMB-45 (human melanoma black 45).2,3 The clear cells often are positive for CD68, factor XIIIa, and NKI/C3 (anti-CD63) but also may be negative. The S-100 protein often is negative but may be focally positive.
The differential diagnosis for clear cell fibrous papules is broad but reasonably includes balloon cell nevus, clear cell hidradenoma, and cutaneous metastasis of clear cell (conventional) renal cell carcinoma (ccRCC). Balloon cell malignant melanoma is not considered strongly in the differential diagnosis because it usually exhibits invasive growth, cytologic atypia, and mitoses, all of which are not characteristic morphologic features of clear cell fibrous papules.
A balloon cell nevus may be difficult to distinguish from a clear cell fibrous papule on routine hematoxylin and eosin staining (Figure 4); however, the nuclei of a balloon cell nevus tend to be more rounded and centrally located. Any junctional nesting or nests of conventional nevus cells in the dermis also help differentiate a balloon cell nevus from a clear cell fibrous papule. Diffusely positive immunostaining for S-100 protein also is indicative of a balloon cell nevus.
Clear cell hidradenoma consists predominantly of cells with clear cytoplasm and small dark nuclei that may closely mimic a clear cell fibrous papule (Figure 5) but often shows a second population of cells with more vesicular nuclei and dark eosinophilic cytoplasm. Cystic spaces containing hyaline material and foci of squamoid change are common, along with occasional tubular lumina that may be prominent or inconspicuous. Further, the tumor cells of clear cell hidradenoma show positive immunostaining for epithelial markers (eg, cytokeratin AE1/AE3, CAM5.2).
Cutaneous metastasis of ccRCC is rare and usually presents clinically as a larger lesion than a clear cell fibrous papule. The cells of ccRCC have moderate to abundant clear cytoplasm and nuclei with varying degrees of pleomorphism (Figure 6). Periodic acid–Schiff staining demonstrates intracytoplasmic glycogen. The stroma is abundantly vascular and extravasated blood cells are frequently observed. On immunohistochemistry, the tumor cells of ccRCC stain positively for cytokeratin AE1/AE3, CAM5.2, epithelial membrane antigen, CD10, and vimentin.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Chiang YY, Tsai HH, Lee WR, et al. Clear cell fibrous papule: report of a case mimicking a balloon cell nevus. J Cutan Pathol. 2009;36:381-384.
- Lee AN, Stein SL, Cohen LM. Clear cell fibrous papule with NKI/C3 expression: clinical and histologic features in six cases. Am J Dermatopathol. 2005;27:296-300.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Chiang YY, Tsai HH, Lee WR, et al. Clear cell fibrous papule: report of a case mimicking a balloon cell nevus. J Cutan Pathol. 2009;36:381-384.
- Lee AN, Stein SL, Cohen LM. Clear cell fibrous papule with NKI/C3 expression: clinical and histologic features in six cases. Am J Dermatopathol. 2005;27:296-300.
Cutaneous Adenosquamous Carcinoma: A Rare Neoplasm With Biphasic Differentiation
Case Report
An 85-year-old woman presented with a painless red plaque on the right bicep of 5 years’ duration. The patient had not seen a physician in the last 63 years and had unsuccessfully attempted to treat the plaque by occlusion with an adhesive bandage. A review of systems was negative for pain, pruritus, bleeding, fever, unexplained weight loss, and night sweats. Physical examination revealed a raised, 2×4×1-cm, red, nontender, ulcerated plaque with slight exudate and gelatinous texture on the right bicep (Figure 1). Full-body skin examination revealed erythema and swelling of the right wrist and forearm consistent with cellulitis as well as tinea pedis and onychomycosis of the toenails of both feet.
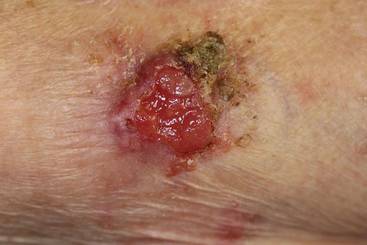
Figure 1. A raised, red, nontender, ulcerated plaque with slight exudate and gelatinous texture on the right bicep. 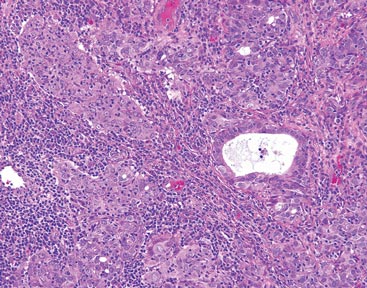
Figure 2. The tumor was comprised of gland-forming cells and exhibited crowding, pleomorphism, enlarged hyperchromatic nuclei, and mitotic division figures (H&E, original magnification ×100). 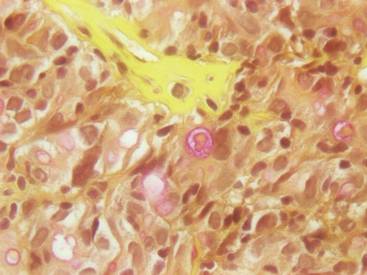
Figure 3. Mucicarmine staining highlighted sialomucin within the glandular component (original magnification ×400). |
Hematoxylin and eosin as well as mucicarmine staining of a shave biopsy from the lesion demonstrated an invasive epithelial neoplasm comprised of squamoid and gland-forming cells broadly attached to the epidermis, which suggested a primary cutaneous origin (Figures 2 and 3). The tumor cells were arranged in infiltrating cords and nests; they exhibited crowding, pleomorphism, enlarged hyperchromatic nuclei, and mitotic division figures. Epithelial mucin (sialomucin) within the glandular component was highlighted on mucicarmine staining. The gland-forming segment of the tumor was strongly positive for cytokeratin (CK) 7. Gastrointestinal tumors were excluded on negative CDX2 and CK20 staining, pulmonary and thyroid tumors were excluded on negative thyroid transcription factor 1 staining, and endometrial and ovarian tumors were excluded with negative estrogen receptor staining. On physical examination the breasts were soft, nontender, and without deformity. A chest radiograph demonstrated normal heart size and pulmonary vasculature with mild bibasilar atelectasis and no areas of consolidation. Given these clinical findings along with a negative history of cancer and negative estrogen receptor staining, breast cancer was excluded from the differential diagnosis, and the diagnosis of cASC was made. The tumor was excised using Mohs micrographic surgery and was free of recurrence at 6- and 12-month follow-up.
Comment
Primary cutaneous adenosquamous carcinoma (cASC) is an aggressive subtype of squamous cell carcinoma that was first described in 1985.1 It typically presents as an erythematous, indurated, keratotic papule or plaque with a predilection for the face, scalp, and upper extremities of immunocompromised individuals and elderly men.2,3 Biopsies generally demonstrate a malignant epithelial neoplasm arising from the epidermis and exhibiting squamous and glandular differentiation. The glandular segment usually is indistinguishable from adenocarcinoma and can be highlighted on CK7, carcinoembryonic antigen, mucicarmine, and periodic acid–Schiff staining. The squamous segment typically is indistinguishable from squamous cell carcinoma and shows aberrant keratinization and intercellular bridges. Tumors often are deeply invasive, poorly differentiated, and associated with a desmoplastic stromal reaction. Local recurrence rates are between 22% and 26%,4 but metastasis is rare. Surgical excision is the mainstay of therapy. When clear margins cannot be obtained using Mohs micrographic surgery, adjuvant external beam radiation therapy and epidermal growth factor receptor inhibitors can be used to treat locally recurrent cASCs.2
The differential diagnosis for cASC includes cutaneous mucoepidermoid carcinoma, cutaneous acantholytic squamous cell carcinoma, and cutaneous manifestations of metastatic visceral adenosquamous carcinoma. Mucoepidermoid carcinoma sometimes is used interchangeably with cASC in the literature, but it is a different cutaneous neoplasm that forms goblet cells, intermediate cells, and squamous cells. It is considered the cutaneous analogue of salivary gland mucoepidermoid carcinoma and does not exhibit the anaplasia, stromal desmoplasia, and aggressive course of cASC.5 The acantholytic subtype of squamous cell carcinoma forms glandlike spaces due to poor adhesion between keratinocytes, but the glandlike spaces do not form mucin or stain positive for CK7 or carcinoembryonic antigen. Adenosquamous carcinomas are well recognized in the lungs, breasts, genitourinary tract, pancreas, and gastroenteric system. Visceral tumor metastasis to the skin should be excluded by appropriate screening.
Conclusion
Although cASCs are not commonly encountered in clinical practice, accurate diagnosis of these lesions is important due to their potentially aggressive behavior. Misdiagnosis and improper treatment could be attributed to lack of awareness of this type of lesion.
1. Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin-and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
2. Fu JM, McCalmont T, Siegrid YS. Adenosquamous carcinoma of the skin: a case series. Arch Dermatol. 2009;145:1152-1158.
3. Ko JK, Leffel DJ, McNiff JM. Adenosquamous carcinoma: a report of 9 cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
4. Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
5. Riedlinger WF, Hurley MY, Dehner LP, et al. Muco-epidermoid carcinoma of the skin: a distinct entity from adenosquamous carcinoma: a case study with a review of the literature. Am J Surg Pathol. 2005;29:131-135.
Case Report
An 85-year-old woman presented with a painless red plaque on the right bicep of 5 years’ duration. The patient had not seen a physician in the last 63 years and had unsuccessfully attempted to treat the plaque by occlusion with an adhesive bandage. A review of systems was negative for pain, pruritus, bleeding, fever, unexplained weight loss, and night sweats. Physical examination revealed a raised, 2×4×1-cm, red, nontender, ulcerated plaque with slight exudate and gelatinous texture on the right bicep (Figure 1). Full-body skin examination revealed erythema and swelling of the right wrist and forearm consistent with cellulitis as well as tinea pedis and onychomycosis of the toenails of both feet.
|
Figure 1. A raised, red, nontender, ulcerated plaque with slight exudate and gelatinous texture on the right bicep.
Figure 2. The tumor was comprised of gland-forming cells and exhibited crowding, pleomorphism, enlarged hyperchromatic nuclei, and mitotic division figures (H&E, original magnification ×100).
Figure 3. Mucicarmine staining highlighted sialomucin within the glandular component (original magnification ×400). |
Hematoxylin and eosin as well as mucicarmine staining of a shave biopsy from the lesion demonstrated an invasive epithelial neoplasm comprised of squamoid and gland-forming cells broadly attached to the epidermis, which suggested a primary cutaneous origin (Figures 2 and 3). The tumor cells were arranged in infiltrating cords and nests; they exhibited crowding, pleomorphism, enlarged hyperchromatic nuclei, and mitotic division figures. Epithelial mucin (sialomucin) within the glandular component was highlighted on mucicarmine staining. The gland-forming segment of the tumor was strongly positive for cytokeratin (CK) 7. Gastrointestinal tumors were excluded on negative CDX2 and CK20 staining, pulmonary and thyroid tumors were excluded on negative thyroid transcription factor 1 staining, and endometrial and ovarian tumors were excluded with negative estrogen receptor staining. On physical examination the breasts were soft, nontender, and without deformity. A chest radiograph demonstrated normal heart size and pulmonary vasculature with mild bibasilar atelectasis and no areas of consolidation. Given these clinical findings along with a negative history of cancer and negative estrogen receptor staining, breast cancer was excluded from the differential diagnosis, and the diagnosis of cASC was made. The tumor was excised using Mohs micrographic surgery and was free of recurrence at 6- and 12-month follow-up.
Comment
Primary cutaneous adenosquamous carcinoma (cASC) is an aggressive subtype of squamous cell carcinoma that was first described in 1985.1 It typically presents as an erythematous, indurated, keratotic papule or plaque with a predilection for the face, scalp, and upper extremities of immunocompromised individuals and elderly men.2,3 Biopsies generally demonstrate a malignant epithelial neoplasm arising from the epidermis and exhibiting squamous and glandular differentiation. The glandular segment usually is indistinguishable from adenocarcinoma and can be highlighted on CK7, carcinoembryonic antigen, mucicarmine, and periodic acid–Schiff staining. The squamous segment typically is indistinguishable from squamous cell carcinoma and shows aberrant keratinization and intercellular bridges. Tumors often are deeply invasive, poorly differentiated, and associated with a desmoplastic stromal reaction. Local recurrence rates are between 22% and 26%,4 but metastasis is rare. Surgical excision is the mainstay of therapy. When clear margins cannot be obtained using Mohs micrographic surgery, adjuvant external beam radiation therapy and epidermal growth factor receptor inhibitors can be used to treat locally recurrent cASCs.2
The differential diagnosis for cASC includes cutaneous mucoepidermoid carcinoma, cutaneous acantholytic squamous cell carcinoma, and cutaneous manifestations of metastatic visceral adenosquamous carcinoma. Mucoepidermoid carcinoma sometimes is used interchangeably with cASC in the literature, but it is a different cutaneous neoplasm that forms goblet cells, intermediate cells, and squamous cells. It is considered the cutaneous analogue of salivary gland mucoepidermoid carcinoma and does not exhibit the anaplasia, stromal desmoplasia, and aggressive course of cASC.5 The acantholytic subtype of squamous cell carcinoma forms glandlike spaces due to poor adhesion between keratinocytes, but the glandlike spaces do not form mucin or stain positive for CK7 or carcinoembryonic antigen. Adenosquamous carcinomas are well recognized in the lungs, breasts, genitourinary tract, pancreas, and gastroenteric system. Visceral tumor metastasis to the skin should be excluded by appropriate screening.
Conclusion
Although cASCs are not commonly encountered in clinical practice, accurate diagnosis of these lesions is important due to their potentially aggressive behavior. Misdiagnosis and improper treatment could be attributed to lack of awareness of this type of lesion.
Case Report
An 85-year-old woman presented with a painless red plaque on the right bicep of 5 years’ duration. The patient had not seen a physician in the last 63 years and had unsuccessfully attempted to treat the plaque by occlusion with an adhesive bandage. A review of systems was negative for pain, pruritus, bleeding, fever, unexplained weight loss, and night sweats. Physical examination revealed a raised, 2×4×1-cm, red, nontender, ulcerated plaque with slight exudate and gelatinous texture on the right bicep (Figure 1). Full-body skin examination revealed erythema and swelling of the right wrist and forearm consistent with cellulitis as well as tinea pedis and onychomycosis of the toenails of both feet.
|
Figure 1. A raised, red, nontender, ulcerated plaque with slight exudate and gelatinous texture on the right bicep.
Figure 2. The tumor was comprised of gland-forming cells and exhibited crowding, pleomorphism, enlarged hyperchromatic nuclei, and mitotic division figures (H&E, original magnification ×100).
Figure 3. Mucicarmine staining highlighted sialomucin within the glandular component (original magnification ×400). |
Hematoxylin and eosin as well as mucicarmine staining of a shave biopsy from the lesion demonstrated an invasive epithelial neoplasm comprised of squamoid and gland-forming cells broadly attached to the epidermis, which suggested a primary cutaneous origin (Figures 2 and 3). The tumor cells were arranged in infiltrating cords and nests; they exhibited crowding, pleomorphism, enlarged hyperchromatic nuclei, and mitotic division figures. Epithelial mucin (sialomucin) within the glandular component was highlighted on mucicarmine staining. The gland-forming segment of the tumor was strongly positive for cytokeratin (CK) 7. Gastrointestinal tumors were excluded on negative CDX2 and CK20 staining, pulmonary and thyroid tumors were excluded on negative thyroid transcription factor 1 staining, and endometrial and ovarian tumors were excluded with negative estrogen receptor staining. On physical examination the breasts were soft, nontender, and without deformity. A chest radiograph demonstrated normal heart size and pulmonary vasculature with mild bibasilar atelectasis and no areas of consolidation. Given these clinical findings along with a negative history of cancer and negative estrogen receptor staining, breast cancer was excluded from the differential diagnosis, and the diagnosis of cASC was made. The tumor was excised using Mohs micrographic surgery and was free of recurrence at 6- and 12-month follow-up.
Comment
Primary cutaneous adenosquamous carcinoma (cASC) is an aggressive subtype of squamous cell carcinoma that was first described in 1985.1 It typically presents as an erythematous, indurated, keratotic papule or plaque with a predilection for the face, scalp, and upper extremities of immunocompromised individuals and elderly men.2,3 Biopsies generally demonstrate a malignant epithelial neoplasm arising from the epidermis and exhibiting squamous and glandular differentiation. The glandular segment usually is indistinguishable from adenocarcinoma and can be highlighted on CK7, carcinoembryonic antigen, mucicarmine, and periodic acid–Schiff staining. The squamous segment typically is indistinguishable from squamous cell carcinoma and shows aberrant keratinization and intercellular bridges. Tumors often are deeply invasive, poorly differentiated, and associated with a desmoplastic stromal reaction. Local recurrence rates are between 22% and 26%,4 but metastasis is rare. Surgical excision is the mainstay of therapy. When clear margins cannot be obtained using Mohs micrographic surgery, adjuvant external beam radiation therapy and epidermal growth factor receptor inhibitors can be used to treat locally recurrent cASCs.2
The differential diagnosis for cASC includes cutaneous mucoepidermoid carcinoma, cutaneous acantholytic squamous cell carcinoma, and cutaneous manifestations of metastatic visceral adenosquamous carcinoma. Mucoepidermoid carcinoma sometimes is used interchangeably with cASC in the literature, but it is a different cutaneous neoplasm that forms goblet cells, intermediate cells, and squamous cells. It is considered the cutaneous analogue of salivary gland mucoepidermoid carcinoma and does not exhibit the anaplasia, stromal desmoplasia, and aggressive course of cASC.5 The acantholytic subtype of squamous cell carcinoma forms glandlike spaces due to poor adhesion between keratinocytes, but the glandlike spaces do not form mucin or stain positive for CK7 or carcinoembryonic antigen. Adenosquamous carcinomas are well recognized in the lungs, breasts, genitourinary tract, pancreas, and gastroenteric system. Visceral tumor metastasis to the skin should be excluded by appropriate screening.
Conclusion
Although cASCs are not commonly encountered in clinical practice, accurate diagnosis of these lesions is important due to their potentially aggressive behavior. Misdiagnosis and improper treatment could be attributed to lack of awareness of this type of lesion.
1. Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin-and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
2. Fu JM, McCalmont T, Siegrid YS. Adenosquamous carcinoma of the skin: a case series. Arch Dermatol. 2009;145:1152-1158.
3. Ko JK, Leffel DJ, McNiff JM. Adenosquamous carcinoma: a report of 9 cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
4. Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
5. Riedlinger WF, Hurley MY, Dehner LP, et al. Muco-epidermoid carcinoma of the skin: a distinct entity from adenosquamous carcinoma: a case study with a review of the literature. Am J Surg Pathol. 2005;29:131-135.
1. Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin-and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
2. Fu JM, McCalmont T, Siegrid YS. Adenosquamous carcinoma of the skin: a case series. Arch Dermatol. 2009;145:1152-1158.
3. Ko JK, Leffel DJ, McNiff JM. Adenosquamous carcinoma: a report of 9 cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
4. Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
5. Riedlinger WF, Hurley MY, Dehner LP, et al. Muco-epidermoid carcinoma of the skin: a distinct entity from adenosquamous carcinoma: a case study with a review of the literature. Am J Surg Pathol. 2005;29:131-135.
Practice Points
- Cutaneous adenosquamous carcinoma (cASC) is an extremely rare malignant neoplasm with histologic similarities to both squamous cell carcinoma and adenocarcinoma.
- Mohs micrographic surgery for excision is recommended; however, adjuvant external beam radiation therapy and epidermal growth factor receptor inhibitors also have been used to treat locally recurrent cASCs.