User login
Pediatric, adolescent migraine treatment and prevention guidelines are updated
Two new guidelines on the treatment and prevention of migraines in children and adolescents have been released by the American Academy of Neurology and the American Headache Society.
This update to the previous guidelines released by the American Academy of Neurology in 2004 reflects the expansion in pharmacologic and nonpharmacologic approaches during the last 15 years, Andrew D. Hershey, MD, PhD, director of the division of neurology at Cincinnati Children’s Hospital and a fellow of the American Academy of Neurology, said in an interview.
“There has also been an increase in the number of randomized controlled studies, which have allowed for a more robust statement on acute and preventive treatments to be made,” said Dr. Hershey, who is also a senior author for both guidelines.
The two reports focused on separate issues: One guideline outlined the options for treatment of acute migraine, and the second guideline summarized the available studies on the effectiveness of preventive medications for migraine in children and adolescents.
The guidelines recommend a physical examination and history to establish a specific headache diagnosis and afford a treatment that provides fast and complete pain relief. Treatment should be initiated as soon as a patient realizes an attack is occurring. Patients with signs of secondary headache should be evaluated by a neurologist or a headache specialist.
Studies support the use of ibuprofen and acetaminophen for pain relief in cases of acute migraine, but only some triptans (such as almotriptan, rizatriptan, sumatriptan/naproxen, and zolmitriptan nasal spray) are approved for use in adolescents. Specifically, sumatriptan/naproxen was shown to be effective when compared with placebo in studies with adolescents, whose headache symptoms resolved within 2 hours.
It may be necessary to try more than one triptan, the guidelines noted, because patients respond differently to medications. A failure to respond to one triptan does not necessarily mean that treatment with another triptan will be unsuccessful.
The guidelines also focused on patient and family education to improve medication safety and adherence. Lifestyle modification, avoidance of migraine triggers, creating good sleep habits, and staying hydrated can help reduce migraines. While no medications improved associated symptoms of migraines such as nausea or vomiting, triptans did show a benefit in reducing phonophobia and photophobia.
Evidence for pharmacologic prevention of migraines in children and adolescents is limited, according to the guidelines. In the 15 studies included in a literature review, there was not sufficient evidence to show preventive treatments, such as divalproex, onabotulinumtoxinA, amitriptyline, nimodipine, and flunarizine, were more effective than placebo at reducing the frequency of headaches. There was some evidence to show propranolol in children and topiramate and cinnarizine in children and adolescents can reduce headache frequency. Children and adolescents who received cognitive-behavioral therapy together with amitriptyline were more likely to have reduced frequency of headaches than were those who received amitriptyline with patient education.
“The consensus conclusion was that a multidisciplinary approach that combines acute treatments, preventive treatments, and healthy habits is likely to have the best outcomes,” said Dr. Hershey.
Dr. Hershey acknowledged the many gaps between what is clinically observed and what the studies in the guidelines demonstrated.
“One of the biggest questions is how to minimize the expectation response in the controlled studies,” he said. “Additionally, we are moving toward a better recognition of the mechanism by which the various treatments work in a genetic-based disease that is polygenic in nature” with up to 38 different gene polymorphisms identified to date.
The guidelines also do not address newer treatments, such as calcitonin gene–related peptide (CGRP) antibodies, CGRP antagonists, serotonin antagonists, and devices because there are as yet no studies of their effectiveness in children and adolescents.
“They have been studied in adults, so will be prone to the expectation response; but given the large number of diverse therapies, one can hope that many of the gaps can be filled,” said Dr. Hershey.
The American Academy of Neurology provided funding for development of the guidelines and reimbursed authors who served as subcommittee members for travel expenses and in-person meetings. The authors reported personal and institutional relationships in the form of advisory board memberships, investigator appointments, speakers bureau positions, research support, grants, honorariums, consultancies, and publishing royalties for pharmaceutical companies and other organizations.
SOURCES: Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008095. Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008105.
Two new guidelines on the treatment and prevention of migraines in children and adolescents have been released by the American Academy of Neurology and the American Headache Society.
This update to the previous guidelines released by the American Academy of Neurology in 2004 reflects the expansion in pharmacologic and nonpharmacologic approaches during the last 15 years, Andrew D. Hershey, MD, PhD, director of the division of neurology at Cincinnati Children’s Hospital and a fellow of the American Academy of Neurology, said in an interview.
“There has also been an increase in the number of randomized controlled studies, which have allowed for a more robust statement on acute and preventive treatments to be made,” said Dr. Hershey, who is also a senior author for both guidelines.
The two reports focused on separate issues: One guideline outlined the options for treatment of acute migraine, and the second guideline summarized the available studies on the effectiveness of preventive medications for migraine in children and adolescents.
The guidelines recommend a physical examination and history to establish a specific headache diagnosis and afford a treatment that provides fast and complete pain relief. Treatment should be initiated as soon as a patient realizes an attack is occurring. Patients with signs of secondary headache should be evaluated by a neurologist or a headache specialist.
Studies support the use of ibuprofen and acetaminophen for pain relief in cases of acute migraine, but only some triptans (such as almotriptan, rizatriptan, sumatriptan/naproxen, and zolmitriptan nasal spray) are approved for use in adolescents. Specifically, sumatriptan/naproxen was shown to be effective when compared with placebo in studies with adolescents, whose headache symptoms resolved within 2 hours.
It may be necessary to try more than one triptan, the guidelines noted, because patients respond differently to medications. A failure to respond to one triptan does not necessarily mean that treatment with another triptan will be unsuccessful.
The guidelines also focused on patient and family education to improve medication safety and adherence. Lifestyle modification, avoidance of migraine triggers, creating good sleep habits, and staying hydrated can help reduce migraines. While no medications improved associated symptoms of migraines such as nausea or vomiting, triptans did show a benefit in reducing phonophobia and photophobia.
Evidence for pharmacologic prevention of migraines in children and adolescents is limited, according to the guidelines. In the 15 studies included in a literature review, there was not sufficient evidence to show preventive treatments, such as divalproex, onabotulinumtoxinA, amitriptyline, nimodipine, and flunarizine, were more effective than placebo at reducing the frequency of headaches. There was some evidence to show propranolol in children and topiramate and cinnarizine in children and adolescents can reduce headache frequency. Children and adolescents who received cognitive-behavioral therapy together with amitriptyline were more likely to have reduced frequency of headaches than were those who received amitriptyline with patient education.
“The consensus conclusion was that a multidisciplinary approach that combines acute treatments, preventive treatments, and healthy habits is likely to have the best outcomes,” said Dr. Hershey.
Dr. Hershey acknowledged the many gaps between what is clinically observed and what the studies in the guidelines demonstrated.
“One of the biggest questions is how to minimize the expectation response in the controlled studies,” he said. “Additionally, we are moving toward a better recognition of the mechanism by which the various treatments work in a genetic-based disease that is polygenic in nature” with up to 38 different gene polymorphisms identified to date.
The guidelines also do not address newer treatments, such as calcitonin gene–related peptide (CGRP) antibodies, CGRP antagonists, serotonin antagonists, and devices because there are as yet no studies of their effectiveness in children and adolescents.
“They have been studied in adults, so will be prone to the expectation response; but given the large number of diverse therapies, one can hope that many of the gaps can be filled,” said Dr. Hershey.
The American Academy of Neurology provided funding for development of the guidelines and reimbursed authors who served as subcommittee members for travel expenses and in-person meetings. The authors reported personal and institutional relationships in the form of advisory board memberships, investigator appointments, speakers bureau positions, research support, grants, honorariums, consultancies, and publishing royalties for pharmaceutical companies and other organizations.
SOURCES: Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008095. Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008105.
Two new guidelines on the treatment and prevention of migraines in children and adolescents have been released by the American Academy of Neurology and the American Headache Society.
This update to the previous guidelines released by the American Academy of Neurology in 2004 reflects the expansion in pharmacologic and nonpharmacologic approaches during the last 15 years, Andrew D. Hershey, MD, PhD, director of the division of neurology at Cincinnati Children’s Hospital and a fellow of the American Academy of Neurology, said in an interview.
“There has also been an increase in the number of randomized controlled studies, which have allowed for a more robust statement on acute and preventive treatments to be made,” said Dr. Hershey, who is also a senior author for both guidelines.
The two reports focused on separate issues: One guideline outlined the options for treatment of acute migraine, and the second guideline summarized the available studies on the effectiveness of preventive medications for migraine in children and adolescents.
The guidelines recommend a physical examination and history to establish a specific headache diagnosis and afford a treatment that provides fast and complete pain relief. Treatment should be initiated as soon as a patient realizes an attack is occurring. Patients with signs of secondary headache should be evaluated by a neurologist or a headache specialist.
Studies support the use of ibuprofen and acetaminophen for pain relief in cases of acute migraine, but only some triptans (such as almotriptan, rizatriptan, sumatriptan/naproxen, and zolmitriptan nasal spray) are approved for use in adolescents. Specifically, sumatriptan/naproxen was shown to be effective when compared with placebo in studies with adolescents, whose headache symptoms resolved within 2 hours.
It may be necessary to try more than one triptan, the guidelines noted, because patients respond differently to medications. A failure to respond to one triptan does not necessarily mean that treatment with another triptan will be unsuccessful.
The guidelines also focused on patient and family education to improve medication safety and adherence. Lifestyle modification, avoidance of migraine triggers, creating good sleep habits, and staying hydrated can help reduce migraines. While no medications improved associated symptoms of migraines such as nausea or vomiting, triptans did show a benefit in reducing phonophobia and photophobia.
Evidence for pharmacologic prevention of migraines in children and adolescents is limited, according to the guidelines. In the 15 studies included in a literature review, there was not sufficient evidence to show preventive treatments, such as divalproex, onabotulinumtoxinA, amitriptyline, nimodipine, and flunarizine, were more effective than placebo at reducing the frequency of headaches. There was some evidence to show propranolol in children and topiramate and cinnarizine in children and adolescents can reduce headache frequency. Children and adolescents who received cognitive-behavioral therapy together with amitriptyline were more likely to have reduced frequency of headaches than were those who received amitriptyline with patient education.
“The consensus conclusion was that a multidisciplinary approach that combines acute treatments, preventive treatments, and healthy habits is likely to have the best outcomes,” said Dr. Hershey.
Dr. Hershey acknowledged the many gaps between what is clinically observed and what the studies in the guidelines demonstrated.
“One of the biggest questions is how to minimize the expectation response in the controlled studies,” he said. “Additionally, we are moving toward a better recognition of the mechanism by which the various treatments work in a genetic-based disease that is polygenic in nature” with up to 38 different gene polymorphisms identified to date.
The guidelines also do not address newer treatments, such as calcitonin gene–related peptide (CGRP) antibodies, CGRP antagonists, serotonin antagonists, and devices because there are as yet no studies of their effectiveness in children and adolescents.
“They have been studied in adults, so will be prone to the expectation response; but given the large number of diverse therapies, one can hope that many of the gaps can be filled,” said Dr. Hershey.
The American Academy of Neurology provided funding for development of the guidelines and reimbursed authors who served as subcommittee members for travel expenses and in-person meetings. The authors reported personal and institutional relationships in the form of advisory board memberships, investigator appointments, speakers bureau positions, research support, grants, honorariums, consultancies, and publishing royalties for pharmaceutical companies and other organizations.
SOURCES: Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008095. Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008105.
FROM NEUROLOGY
FDA approves drug combo to treat highly resistant TB
The U.S. Food and Drug Administration granted special approval to a new drug combo intended for the treatment of “a limited and specific population of adult patients with extensively drug resistant, treatment-intolerant or nonresponsive multidrug-resistant pulmonary” tuberculosis, according to an FDA news release.

The effectiveness of the combination treatment of pretomanid tablets with bedaquiline and linezolid was shown in a clinical study of patients with extensively drug-resistant, treatment-intolerant, or nonresponsive multidrug-resistant pulmonary tuberculosis of the lungs. Of 107 infected patients who were evaluated 6 months after the end of therapy, 95 (89%) were deemed successes, which significantly exceeded the historical success rates for treatment of extensively drug-resistant TB, the FDA reported. The trial is sponsored by the Global Alliance for TB Drug Development.
The most common adverse effects reported included peripheral neuropathy, anemia, nausea, vomiting, headache, increased liver enzymes, dyspepsia, rash, visual impairment, low blood sugar, and diarrhea, according to the release.
“Multidrug-resistant TB and extensively drug-resistant TB are public health threats due to limited treatment options. New treatments are important to meet patient national and global health needs,” stated FDA Principal Deputy Commissioner Amy Abernethy, MD, PhD, in the release. She also explained that the approval marked the second time a drug was approved under the “Limited Population Pathway for Antibacterial and Antifungal Drugs, a pathway advanced by Congress to spur development of drugs targeting infections that lack effective therapies.”
In 2016, the World Health Organization reported that there were an estimated 490,000 new cases of multidrug-resistant TB worldwide, with a smaller portion of cases of extensively drug-resistant TB, according to the release, demonstrating the need for new therapeutics.
SOURCE: U.S. Food and Drug Administration. Aug. 14, 2019. News release.
The U.S. Food and Drug Administration granted special approval to a new drug combo intended for the treatment of “a limited and specific population of adult patients with extensively drug resistant, treatment-intolerant or nonresponsive multidrug-resistant pulmonary” tuberculosis, according to an FDA news release.
The effectiveness of the combination treatment of pretomanid tablets with bedaquiline and linezolid was shown in a clinical study of patients with extensively drug-resistant, treatment-intolerant, or nonresponsive multidrug-resistant pulmonary tuberculosis of the lungs. Of 107 infected patients who were evaluated 6 months after the end of therapy, 95 (89%) were deemed successes, which significantly exceeded the historical success rates for treatment of extensively drug-resistant TB, the FDA reported. The trial is sponsored by the Global Alliance for TB Drug Development.
The most common adverse effects reported included peripheral neuropathy, anemia, nausea, vomiting, headache, increased liver enzymes, dyspepsia, rash, visual impairment, low blood sugar, and diarrhea, according to the release.
“Multidrug-resistant TB and extensively drug-resistant TB are public health threats due to limited treatment options. New treatments are important to meet patient national and global health needs,” stated FDA Principal Deputy Commissioner Amy Abernethy, MD, PhD, in the release. She also explained that the approval marked the second time a drug was approved under the “Limited Population Pathway for Antibacterial and Antifungal Drugs, a pathway advanced by Congress to spur development of drugs targeting infections that lack effective therapies.”
In 2016, the World Health Organization reported that there were an estimated 490,000 new cases of multidrug-resistant TB worldwide, with a smaller portion of cases of extensively drug-resistant TB, according to the release, demonstrating the need for new therapeutics.
SOURCE: U.S. Food and Drug Administration. Aug. 14, 2019. News release.
The U.S. Food and Drug Administration granted special approval to a new drug combo intended for the treatment of “a limited and specific population of adult patients with extensively drug resistant, treatment-intolerant or nonresponsive multidrug-resistant pulmonary” tuberculosis, according to an FDA news release.
The effectiveness of the combination treatment of pretomanid tablets with bedaquiline and linezolid was shown in a clinical study of patients with extensively drug-resistant, treatment-intolerant, or nonresponsive multidrug-resistant pulmonary tuberculosis of the lungs. Of 107 infected patients who were evaluated 6 months after the end of therapy, 95 (89%) were deemed successes, which significantly exceeded the historical success rates for treatment of extensively drug-resistant TB, the FDA reported. The trial is sponsored by the Global Alliance for TB Drug Development.
The most common adverse effects reported included peripheral neuropathy, anemia, nausea, vomiting, headache, increased liver enzymes, dyspepsia, rash, visual impairment, low blood sugar, and diarrhea, according to the release.
“Multidrug-resistant TB and extensively drug-resistant TB are public health threats due to limited treatment options. New treatments are important to meet patient national and global health needs,” stated FDA Principal Deputy Commissioner Amy Abernethy, MD, PhD, in the release. She also explained that the approval marked the second time a drug was approved under the “Limited Population Pathway for Antibacterial and Antifungal Drugs, a pathway advanced by Congress to spur development of drugs targeting infections that lack effective therapies.”
In 2016, the World Health Organization reported that there were an estimated 490,000 new cases of multidrug-resistant TB worldwide, with a smaller portion of cases of extensively drug-resistant TB, according to the release, demonstrating the need for new therapeutics.
SOURCE: U.S. Food and Drug Administration. Aug. 14, 2019. News release.
NEWS FROM THE FDA
Treatment of episodic cluster headache deviates from recommendations
PHILADELPHIA – , according to an analysis presented at the annual meeting of the American Headache Society.

Although consensus treatment guidelines do not exist for episodic cluster headache, treatment of this disorder did not follow many established recommendations that call for the use of preventive medications (e.g., MacGregor et al., 2010; Sarchielli et al., 2012; and May et al., 2006). Additional preventive medication options may be needed.
Patients with episodic cluster headache have several unilateral headache attacks per day. Little information is available to guide the selection of treatments for this population, and little is known about how available treatments are used in routine practice.
Analyzing cross-sectional survey data
To address this paucity of evidence, Jeffrey Scott Andrews, PharmD, a senior research scientist at Eli Lilly in Indianapolis, and colleagues examined data from the Adelphi 2017 Cluster Headache Disease Specific Programme, a large, international, cross-sectional survey. Physicians and patients in Germany, the United Kingdom, and the United States responded to the survey. Eligible physicians consulted with at least four patients with cluster headache per month, and eligible patients had a diagnosis of episodic cluster headache that was consistent with ICHD-3 beta criteria. Additional data were collected from all participants through questionnaires.
The analysis included 309 patients in Germany, 328 in the United Kingdom, and 375 in the United States. The average age of the patients was 40 years, and most of the patients were male. Less than 70% of patients reported working full time, which may indicate “the impact of this condition on work status,” said Dr. Andrews. Patients’ average number of attacks per day within an active period was 2.4. The two most commonly reported comorbidities were anxiety and depression. About 40% of cases of depression were reported to have occurred after the receipt of a diagnosis of cluster headache.
Use of inhaled oxygen was low
Most patients received acute treatments. The proportion of patients who received acute therapy only was 53% in Germany, 48% in the United Kingdom, and 43% in the United States. Approximately 34% of patients in Germany received a combination of acute and preventive therapy, compared with 37% in the United Kingdom and 42% in the United States. The proportion of patients who received preventive therapy only was 10% in Germany, 8% in the United Kingdom, and 12% in the United States.
The most commonly prescribed acute treatment, regardless of formulation, was sumatriptan. About 60% of patients received this medication. Less than one-third of patients used inhaled oxygen. Oxygen was prescribed more often in Germany (45%) and the United Kingdom (33%), compared with the United States (19%). U.S. patients face well-known obstacles in getting access to, and reimbursement for, oxygen, said Dr. Andrews. “That’s an area that deserves increased attention.” Zolmitriptan was the third most commonly prescribed acute medication.
Among prescriptions for sumatriptan, oral and injectable formulations were approximately equally common. Recommendations, however, indicate formulations with potentially fast onset of action. “The average duration of one of these attacks is between 15 and 180 minutes, so that certainly suggests that a formulation that gives you a faster onset of action might improve outcomes,” said Dr. Andrews. The use of injectable sumatriptan was lowest in the United States and highest in the United Kingdom.
“The most common decision regarding preventive treatment was [to give] no preventive treatment,” said Dr. Andrews. Verapamil was the most commonly prescribed preventive therapy (34% in Germany, 29% in the United States, and 25% in the United Kingdom), followed by topiramate, lithium, and valproate.
Nonadherence and noncompliance was common
Fewer U.K. patients (32%) reported taking their preventive therapy as advised, compared with German patients (60%) and U.S. patients (80%). Common reasons for noncompliance, regardless of location, were forgetfulness, the belief that a dose was not needed, and side effects. Most patients in the United Kingdom (60%) and the United States (54%) reported the need to take an extra dose of their acute medication to relieve pain symptoms, compared with 30% in Germany. Furthermore, 13% of U.S. patients indicated that they took extra doses all the time or nearly all the time, compared with 2% in Germany and 7% in the United Kingdom. Among patients who had discontinued a preventive treatment in the past, the most common reasons for discontinuation were lack of efficacy and problems with tolerability.
One limitation of the study was that the survey was not designed to represent the general cluster headache or treating physician populations fully. The data may reflect selection bias in favor of physicians who treat high volumes of patients and in favor of patients who frequently seek health care. In addition, the data were based on self-reports.
“Increased awareness and educational efforts that aim at promoting the need and benefit of the preventive treatment for these patients is warranted,” Dr. Andrews concluded.
Dr. Andrews is an employee of Eli Lilly, which funded the study.
SOURCE: Nichols R et al. AHS 2019. Abstract OR04.
PHILADELPHIA – , according to an analysis presented at the annual meeting of the American Headache Society.
Although consensus treatment guidelines do not exist for episodic cluster headache, treatment of this disorder did not follow many established recommendations that call for the use of preventive medications (e.g., MacGregor et al., 2010; Sarchielli et al., 2012; and May et al., 2006). Additional preventive medication options may be needed.
Patients with episodic cluster headache have several unilateral headache attacks per day. Little information is available to guide the selection of treatments for this population, and little is known about how available treatments are used in routine practice.
Analyzing cross-sectional survey data
To address this paucity of evidence, Jeffrey Scott Andrews, PharmD, a senior research scientist at Eli Lilly in Indianapolis, and colleagues examined data from the Adelphi 2017 Cluster Headache Disease Specific Programme, a large, international, cross-sectional survey. Physicians and patients in Germany, the United Kingdom, and the United States responded to the survey. Eligible physicians consulted with at least four patients with cluster headache per month, and eligible patients had a diagnosis of episodic cluster headache that was consistent with ICHD-3 beta criteria. Additional data were collected from all participants through questionnaires.
The analysis included 309 patients in Germany, 328 in the United Kingdom, and 375 in the United States. The average age of the patients was 40 years, and most of the patients were male. Less than 70% of patients reported working full time, which may indicate “the impact of this condition on work status,” said Dr. Andrews. Patients’ average number of attacks per day within an active period was 2.4. The two most commonly reported comorbidities were anxiety and depression. About 40% of cases of depression were reported to have occurred after the receipt of a diagnosis of cluster headache.
Use of inhaled oxygen was low
Most patients received acute treatments. The proportion of patients who received acute therapy only was 53% in Germany, 48% in the United Kingdom, and 43% in the United States. Approximately 34% of patients in Germany received a combination of acute and preventive therapy, compared with 37% in the United Kingdom and 42% in the United States. The proportion of patients who received preventive therapy only was 10% in Germany, 8% in the United Kingdom, and 12% in the United States.
The most commonly prescribed acute treatment, regardless of formulation, was sumatriptan. About 60% of patients received this medication. Less than one-third of patients used inhaled oxygen. Oxygen was prescribed more often in Germany (45%) and the United Kingdom (33%), compared with the United States (19%). U.S. patients face well-known obstacles in getting access to, and reimbursement for, oxygen, said Dr. Andrews. “That’s an area that deserves increased attention.” Zolmitriptan was the third most commonly prescribed acute medication.
Among prescriptions for sumatriptan, oral and injectable formulations were approximately equally common. Recommendations, however, indicate formulations with potentially fast onset of action. “The average duration of one of these attacks is between 15 and 180 minutes, so that certainly suggests that a formulation that gives you a faster onset of action might improve outcomes,” said Dr. Andrews. The use of injectable sumatriptan was lowest in the United States and highest in the United Kingdom.
“The most common decision regarding preventive treatment was [to give] no preventive treatment,” said Dr. Andrews. Verapamil was the most commonly prescribed preventive therapy (34% in Germany, 29% in the United States, and 25% in the United Kingdom), followed by topiramate, lithium, and valproate.
Nonadherence and noncompliance was common
Fewer U.K. patients (32%) reported taking their preventive therapy as advised, compared with German patients (60%) and U.S. patients (80%). Common reasons for noncompliance, regardless of location, were forgetfulness, the belief that a dose was not needed, and side effects. Most patients in the United Kingdom (60%) and the United States (54%) reported the need to take an extra dose of their acute medication to relieve pain symptoms, compared with 30% in Germany. Furthermore, 13% of U.S. patients indicated that they took extra doses all the time or nearly all the time, compared with 2% in Germany and 7% in the United Kingdom. Among patients who had discontinued a preventive treatment in the past, the most common reasons for discontinuation were lack of efficacy and problems with tolerability.
One limitation of the study was that the survey was not designed to represent the general cluster headache or treating physician populations fully. The data may reflect selection bias in favor of physicians who treat high volumes of patients and in favor of patients who frequently seek health care. In addition, the data were based on self-reports.
“Increased awareness and educational efforts that aim at promoting the need and benefit of the preventive treatment for these patients is warranted,” Dr. Andrews concluded.
Dr. Andrews is an employee of Eli Lilly, which funded the study.
SOURCE: Nichols R et al. AHS 2019. Abstract OR04.
PHILADELPHIA – , according to an analysis presented at the annual meeting of the American Headache Society.
Although consensus treatment guidelines do not exist for episodic cluster headache, treatment of this disorder did not follow many established recommendations that call for the use of preventive medications (e.g., MacGregor et al., 2010; Sarchielli et al., 2012; and May et al., 2006). Additional preventive medication options may be needed.
Patients with episodic cluster headache have several unilateral headache attacks per day. Little information is available to guide the selection of treatments for this population, and little is known about how available treatments are used in routine practice.
Analyzing cross-sectional survey data
To address this paucity of evidence, Jeffrey Scott Andrews, PharmD, a senior research scientist at Eli Lilly in Indianapolis, and colleagues examined data from the Adelphi 2017 Cluster Headache Disease Specific Programme, a large, international, cross-sectional survey. Physicians and patients in Germany, the United Kingdom, and the United States responded to the survey. Eligible physicians consulted with at least four patients with cluster headache per month, and eligible patients had a diagnosis of episodic cluster headache that was consistent with ICHD-3 beta criteria. Additional data were collected from all participants through questionnaires.
The analysis included 309 patients in Germany, 328 in the United Kingdom, and 375 in the United States. The average age of the patients was 40 years, and most of the patients were male. Less than 70% of patients reported working full time, which may indicate “the impact of this condition on work status,” said Dr. Andrews. Patients’ average number of attacks per day within an active period was 2.4. The two most commonly reported comorbidities were anxiety and depression. About 40% of cases of depression were reported to have occurred after the receipt of a diagnosis of cluster headache.
Use of inhaled oxygen was low
Most patients received acute treatments. The proportion of patients who received acute therapy only was 53% in Germany, 48% in the United Kingdom, and 43% in the United States. Approximately 34% of patients in Germany received a combination of acute and preventive therapy, compared with 37% in the United Kingdom and 42% in the United States. The proportion of patients who received preventive therapy only was 10% in Germany, 8% in the United Kingdom, and 12% in the United States.
The most commonly prescribed acute treatment, regardless of formulation, was sumatriptan. About 60% of patients received this medication. Less than one-third of patients used inhaled oxygen. Oxygen was prescribed more often in Germany (45%) and the United Kingdom (33%), compared with the United States (19%). U.S. patients face well-known obstacles in getting access to, and reimbursement for, oxygen, said Dr. Andrews. “That’s an area that deserves increased attention.” Zolmitriptan was the third most commonly prescribed acute medication.
Among prescriptions for sumatriptan, oral and injectable formulations were approximately equally common. Recommendations, however, indicate formulations with potentially fast onset of action. “The average duration of one of these attacks is between 15 and 180 minutes, so that certainly suggests that a formulation that gives you a faster onset of action might improve outcomes,” said Dr. Andrews. The use of injectable sumatriptan was lowest in the United States and highest in the United Kingdom.
“The most common decision regarding preventive treatment was [to give] no preventive treatment,” said Dr. Andrews. Verapamil was the most commonly prescribed preventive therapy (34% in Germany, 29% in the United States, and 25% in the United Kingdom), followed by topiramate, lithium, and valproate.
Nonadherence and noncompliance was common
Fewer U.K. patients (32%) reported taking their preventive therapy as advised, compared with German patients (60%) and U.S. patients (80%). Common reasons for noncompliance, regardless of location, were forgetfulness, the belief that a dose was not needed, and side effects. Most patients in the United Kingdom (60%) and the United States (54%) reported the need to take an extra dose of their acute medication to relieve pain symptoms, compared with 30% in Germany. Furthermore, 13% of U.S. patients indicated that they took extra doses all the time or nearly all the time, compared with 2% in Germany and 7% in the United Kingdom. Among patients who had discontinued a preventive treatment in the past, the most common reasons for discontinuation were lack of efficacy and problems with tolerability.
One limitation of the study was that the survey was not designed to represent the general cluster headache or treating physician populations fully. The data may reflect selection bias in favor of physicians who treat high volumes of patients and in favor of patients who frequently seek health care. In addition, the data were based on self-reports.
“Increased awareness and educational efforts that aim at promoting the need and benefit of the preventive treatment for these patients is warranted,” Dr. Andrews concluded.
Dr. Andrews is an employee of Eli Lilly, which funded the study.
SOURCE: Nichols R et al. AHS 2019. Abstract OR04.
REPORTING FROM AHS 2019
Translating the 2019 AAD-NPF Guidelines of Care for the Management of Psoriasis With Biologics to Clinical Practice
Psoriasis is a systemic immune-mediated disorder characterized by erythematous, scaly, well-demarcated plaques on the skin that affects approximately 3% of the world’s population.1 The disease is moderate to severe for approximately 1 in 6 individuals with psoriasis.2 These patients, particularly those with symptoms that are refractory to topical therapy and/or phototherapy, can benefit from the use of biologic agents, which are monoclonal antibodies and fusion proteins engineered to inhibit the action of cytokines that drive psoriatic inflammation.
In February 2019, the American Academy of Dermatology (AAD) and National Psoriasis Foundation (NPF) released an updated set of guidelines for the use of biologics in treating adult patients with psoriasis.3 The prior guidelines were released in 2008 when just 3 biologics—etanercept, infliximab, and adalimumab—were approved by the US Food and Drug Administration (FDA) for the management of psoriasis. These older recommendations were mostly based on studies of the efficacy and safety of biologics for patients with psoriatic arthritis.4 Over the last 11 years, 8 novel biologics have gained FDA approval, and numerous large phase 2 and phase 3 trials evaluating the risks and benefits of biologics have been conducted. The new guidelines contain considerably more detail and are based on evidence more specific to psoriasis rather than to psoriatic arthritis. Given the large repertoire of biologics available today and the increased amount of published research regarding each one, these guidelines may aid dermatologists in choosing the optimal biologic and managing therapy.
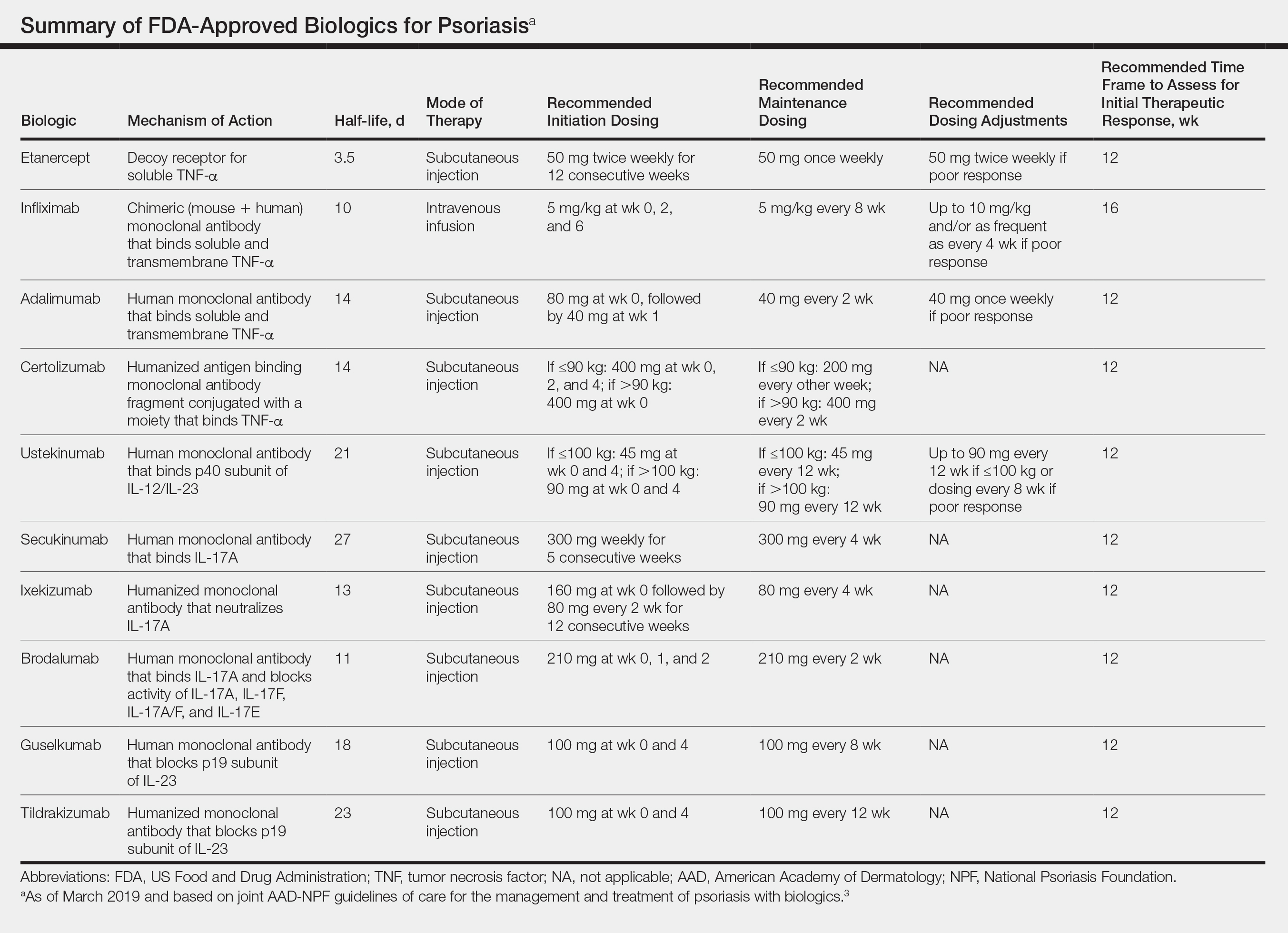
The AAD-NPF recommendations discuss the mechanism of action, efficacy, safety, and adverse events of the 10 biologics that have been FDA approved for the treatment of psoriasis as of March 2019, plus risankizumab, which was pending FDA approval at the time of publication and was later approved in April 2019. They also address dosing regimens, potential to combine biologics with other therapies, and different forms of psoriasis for which each may be effective.3 The purpose of this discussion is to present these guidelines in a condensed form to prescribers of biologic therapies and review the most clinically significant considerations during each step of treatment. Of note, we highlight only treatment of adult patients and do not discuss information relevant to risankizumab, as it was not FDA approved when the AAD-NPF guidelines were released.
Choosing a Biologic
Biologic therapy may be considered for patients with psoriasis that affects more than 3% of the body’s surface and is recalcitrant to localized therapies. There is no particular first-line biologic recommended for all patients with psoriasis; rather, choice of therapy should be individualized to the patient, considering factors such as body parts affected, comorbidities, lifestyle, and drug cost.
All 10 FDA-approved biologics (Table) have been ranked by the AAD and NPF as having grade A evidence for efficacy as monotherapy in the treatment of moderate to severe plaque-type psoriasis. Involvement of difficult-to-treat areas may be considered when choosing a specific therapy. The tumor necrosis factor α (TNF-α) inhibitors etanercept and adalimumab, the IL-17 inhibitor secukinumab, and the IL-23 inhibitor guselkumab have the greatest evidence for efficacy in treatment of nail disease. For scalp involvement, etanercept and guselkumab have the highest-quality evidence, and for palmoplantar disease, adalimumab, secukinumab, and guselkumab are considered the most effective. The TNF-α inhibitors are considered the optimal treatment option for concurrent psoriatic arthritis, though the IL-12/IL-23 inhibitor ustekinumab and the IL-17 inhibitors secukinumab and ixekizumab also have shown grade A evidence of efficacy. Of note, because TNF-α inhibitors received the earliest FDA approval, there is most evidence available for this class. Therapies with lower evidence quality for certain forms of psoriasis may show real-world effectiveness in individual patients, though more trials will be necessary to generate a body of evidence to change these clinical recommendations.
In pregnant women or those are anticipating pregnancy, certolizumab may be considered, as it is the only biologic shown to have minimal to no placental transfer. Other TNF-α inhibitors may undergo active placental transfer, particularly during the latter half of pregnancy,5 and the greatest theoretical risk of transfer occurs in the third trimester. Although these drugs may not directly harm the fetus, they do cause fetal immunosuppression for up to the first 3 months of life. All TNF-α inhibitors are considered safe during lactation. There are inadequate data regarding the safety of other classes of biologics during pregnancy and lactation.
Overweight and obese patients also require unique considerations when choosing a biologic. Infliximab is the only approved psoriasis biologic that utilizes proportional-to-weight dosing and hence may be particularly efficacious in patients with higher body mass. Ustekinumab dosing also takes patient weight into consideration; patients heavier than 100 kg should receive 90-mg doses at initiation and during maintenance compared to 45 mg for patients who weigh 100 kg or less. Other approved biologics also may be utilized in these patients but may require closer monitoring of treatment efficacy.
There are few serious contraindications for specific biologic therapies. Any history of allergic reaction to a particular therapy is an absolute contraindication to its use. In patients for whom IL-17 inhibitor treatment is being considered, inflammatory bowel disease (IBD) should be ruled out given the likelihood that IL-17 could reactivate or worsen IBD. Of note, TNF-α inhibitors and ustekinumab are approved therapies for patients with IBD and may be recommended in patients with comorbid psoriasis. Phase 2 and phase 3 trials have found no reactivation or worsening of IBD in patients with psoriasis who were treated with the IL-23 inhibitor tildrakizumab,6 and phase 2 trials of treatment of IBD with guselkumab are currently underway (ClinicalTrials.gov Identifier NCT03466411). In patients with New York Heart Association class III and class IV congestive heart failure or multiple sclerosis, initiation of TNF-α inhibitors should be avoided. Among 3 phase 3 trials encompassing nearly 3000 patients treated with the IL-17 inhibitor brodalumab, a total of 3 patients died by suicide7,8; hence, the FDA has issued a black box warning cautioning against use of this drug in patients with history of suicidal ideation or recent suicidal behavior. Although a causal relationship between brodalumab and suicide has not been well established,9 a thorough psychiatric history should be obtained in those initiating treatment with brodalumab.
Initiation of Therapy
Prior to initiating biologic therapy, it is important to obtain a complete blood cell count, complete metabolic panel, tuberculosis testing, and hepatitis B virus (HBV) and hepatitis C virus serologies. Testing for human immunodeficiency virus may be pursued at the clinician’s discretion. It is important to address any positive or concerning results prior to starting biologics. In patients with active infections, therapy may be initiated alongside guidance from an infectious disease specialist. Those with a positive purified protein derivative test, T-SPOT test, or QuantiFERON-TB Gold test must be referred for chest radiographs to rule out active tuberculosis. Patients with active HBV infection should receive appropriate referral to initiate antiviral therapy as well as core antibody testing, and those with active hepatitis C virus infection may only receive biologics under the combined discretion of a dermatologist and an appropriate specialist. Patients with human immunodeficiency virus must concurrently receive highly active antiretroviral therapy, show normal CD4+ T-cell count and undetectable viral load, and have no recent history of opportunistic infection.
Therapy should be commenced using specific dosing regimens, which are unique for each biologic (Table). Patients also must be educated on routine follow-up to assess treatment response and tolerability.
Assessment and Optimization of Treatment Response
Patients taking biologics may experience primary treatment failure, defined as lack of response to therapy from initiation. One predisposing factor may be increased body mass; patients who are overweight and obese are less likely to respond to standard regimens of TNF-α inhibitors and 45-mg dosing of ustekinumab. In most cases, however, the cause of primary nonresponse is unpredictable. For patients in whom therapy has failed within the recommended initial time frame (Table), dose escalation or shortening of dosing intervals may be pursued. Recommended dosing adjustments are outlined in the Table. Alternatively, patients may be switched to a different biologic.
If desired effectiveness is not reached with biologic monotherapy, topical corticosteroids, topical vitamin D analogues, or narrowband UVB light therapy may be concurrently used for difficult-to-treat areas. Evidence for safety and effectiveness of systemic adjuncts to biologics is moderate to low, warranting caution with their use. Methotrexate, cyclosporine, and apremilast have synergistic effects with biologics, though they may increase the risk for immunosuppression-related complications. Acitretin, an oral retinoid, likely is the most reasonable systemic adjunct to biologics because of its lack of immunosuppressive properties.
In patients with a suboptimal response to biologics, particularly those taking therapies that require frequent dosing, poor compliance should be considered.10 These patients may be switched to a biologic with less-frequent maintenance dosing (Table). Ustekinumab and tildrakizumab may be the best options for optimizing compliance, as they require dosing only once every 12 weeks after administration of loading doses.
Secondary treatment failure is diminished efficacy of treatment following successful initial response despite no changes in regimen. The best-known factor contributing to secondary nonresponse to biologics is the development of antidrug antibodies (ADAs), a phenomenon known as immunogenicity. The development of efficacy-limiting ADAs has been observed in response to most biologics, though ADAs against etanercept and guselkumab do not limit therapeutic response. Patients taking adalimumab and infliximab have particularly well-documented efficacy-limiting immunogenicity, and those who develop ADAs to infliximab are considered more prone to developing infusion reactions. Methotrexate, which limits antibody formation, may concomitantly be prescribed in patients who experience secondary treatment failure. It should be considered in all patients taking infliximab to increase efficacy and tolerability of therapy.
Considerations During Active Therapy
In addition to monitoring adherence and response to regimens, dermatologists must be heavily involved in counseling patients regarding the risks and adverse effects associated with these therapies. During maintenance therapy with biologics, patients must follow up with the prescriber at minimum every 3 to 6 months to evaluate for continued efficacy of treatment, extent of side effects, and effects of treatment on overall health and quality of life. Given the immunosuppressive effects of biologics, annual testing for tuberculosis should be considered in high-risk individuals. In those who are considered at low risk, tuberculosis testing may be done at the discretion of the dermatologist. In those with a history of HBV infection, HBV serologies should be pursued routinely given the risk for reactivation.
Annual screening for nonmelanoma skin cancer should be performed in all patients taking biologics. Tumor necrosis factor α inhibitor therapy in particular confers an elevated risk for cutaneous squamous cell carcinoma, especially in patients who are immunosuppressed at baseline and those with history of UV phototherapy. Use of acitretin alongside TNF-α inhibitors or ustekinumab may prevent squamous cell carcinoma formation in high-risk patients.
Because infliximab treatment poses an elevated risk of liver injury,11 liver function tests should be repeated 3 months following initiation of treatment and then every 6 to 12 months subsequently if results are normal. Periodic assessment of suicidal ideation is recommended in patients on brodalumab therapy, which may necessitate more frequent follow-up visits and potentially psychiatry referrals in certain patients. Patients taking IL-17 inhibitors, particularly those who are concurrently taking methotrexate, are at increased risk for developing mucocutaneous Candida infections; these patients should be monitored for such infections and treated appropriately.12
It is additionally important for prescribing dermatologists to ensure that patients on biologics are following up with their general providers to receive timely age-appropriate preventative screenings and vaccines. Inactivated vaccinations may be administered during therapy with any biologic; however, live vaccinations may induce systemic infection in those who are immunocompromised, which theoretically includes individuals taking biologic agents, though incidence data in this patient population are scarce.13 Some experts believe that administration of live vaccines warrants temporary discontinuation of biologic therapy for 2 to 3 half-lives before and after vaccination (Table). Others recommend stopping treatment at least 4 weeks before and until 2 weeks after vaccination. For patients taking biologics with half-lives greater than 20 days, which would theoretically require stopping the drug 2 months prior to vaccination, the benefit of vaccination should be weighed against the risk of prolonged discontinuation of therapy. Until recently, this recommendation was particularly important, as a live herpes zoster vaccination was recommended by the Centers for Disease Control and Prevention for adults older than 60 years. In 2017, a new inactivated herpes zoster vaccine was introduced and is now the preferred vaccine for all patients older than 50 years.14 It is especially important that patients on biologics receive this vaccine to avoid temporary drug discontinuation.
Evidence that any particular class of biologics increases risk for solid tumors or lymphoreticular malignancy is limited. One case-control analysis reported that more than 12 months of treatment with TNF-α inhibitors may increase risk for malignancy; however, the confidence interval reported hardly allows for statistical significance.15 Another retrospective cohort study found no elevated incidence of cancer in patients on TNF-α inhibitors compared to nonbiologic comparators.16 Ustekinumab was shown to confer no increased risk for malignancy in 1 large study,15 but no large studies have been conducted for other classes of drugs. Given the limited and inconclusive evidence available, the guidelines recommend that age-appropriate cancer screenings recommended for the general population should be pursued in patients taking biologics.
Surgery while taking biologics may lead to stress-induced augmentation of immunosuppression, resulting in elevated risk of infection.17 Low-risk surgeries that do not warrant discontinuation of treatment include endoscopic, ophthalmologic, dermatologic, orthopedic, and breast procedures. In patients preparing for elective surgery in which respiratory, gastrointestinal, or genitourinary tracts will be entered, biologics may be discontinued at least 3 half-lives (Table) prior to surgery if the dermatologist and surgeon collaboratively deem that risk of infection outweighs benefit of continued therapy.18 Therapy may be resumed within 1 to 2 weeks postoperatively if there are no surgical complications.
Switching Biologics
Changing therapy to another biologic should be considered if there is no response to treatment or the patient experiences adverse effects while taking a particular biologic. Because evidence is limited regarding the ideal time frame between discontinuation of a prior medication and initiation of a new biologic, this interval should be determined at the discretion of the provider based on the patient’s disease severity and response to prior treatment. For individuals who experience primary or secondary treatment failure while maintaining appropriate dosing and treatment compliance, switching to a different biologic is recommended to maximize treatment response.19 Changing therapy to a biologic within the same class is generally effective,20 and switching to a biologic with another mechanism of action should be considered if a class-specific adverse effect is the major reason for altering the regimen. Nonetheless, some patients may be unresponsive to biologic changes. Further research is necessary to determine which biologics may be most effective when previously used biologics have failed and particular factors that may predispose patients to biologic unresponsiveness.
Resuming Biologic Treatment Following Cessation
In cases where therapy is discontinued for any reason, it may be necessary to repeat initiation dosing when resuming treatment. In patients with severe or flaring disease or if more than 3 to 4 half-lives have passed since the most recent dose, it may be necessary to restart therapy with the loading dose (Table). Unfortunately, restarting therapy may preclude some patients from experiencing the maximal response that they attained prior to cessation. In such cases, switching biologic therapy to a different class may prove beneficial.
Final Thoughts
These recommendations contain valuable information that will assist dermatologists when initiating biologics and managing outcomes of their psoriasis patients. It is, however, crucial to bear in mind that these guidelines serve as merely a tool. Given the paucity of comprehensive research, particularly regarding some of the more recently approved therapies, there are many questions that are unanswered within the guidelines. Their utility for each individual patient situation is therefore limited, and clinical judgement may outweigh the information presented. The recommendations nevertheless provide a pivotal and unprecedented framework that promotes discourse among patients, dermatologists, and other providers to optimize the efficacy of biologic therapy for psoriasis.
- Michalek IM, Loring B, John SM. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol. 2017;31:205-212.
- Kurd SK, Gelfand JM. The prevalence of previously diagnosed and undiagnosed psoriasis in US adults: results from NHANES 2003-2004. J Am Acad Dermatol. 2009;60:218-224.
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics [published online February 13, 2019]. J Am Acad Dermatol. 2019;80:1029-1072.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- Förger F, Villiger PM. Treatment of rheumatoid arthritis during pregnancy: present and future. Expert Rev Clin Immunol. 2016;12:937-944.
- Gooderham M, Elewski B, Pariser D, et al. Incidence of serious gastrointestinal events and inflammatory bowel disease among tildrakizumab-treated patients with moderate-to-severe plaque psoriasis: data from 3 large randomized clinical trials [abstract]. J Am Acad Dermatol. 2018;79(suppl 1):AB166.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-328.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016;175:273-286
- Beck KM, Koo J. Brodalumab for the treatment of plaque psoriasis: up-to-date. Expert Opin Biol Ther. 2019;19:287-292.
- Fouéré S, Adjadj L, Pawin H. How patients experience psoriasis: results from a European survey. J Eur Acad Dermatol Venereol. 2005;19(suppl 3):2-6.
- Björnsson ES, Bergmann OM, Björnsson HK, et al. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 2013;144:1419-1425, 1425.e1-3; quiz e19-20.
- Saunte DM, Mrowietz U, Puig L, et al. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177:47-62.
- Huber F, Ehrensperger B, Hatz C, et al. Safety of live vaccines on immunosuppressive or immunomodulatory therapy—a retrospective study in three Swiss Travel Clinics [published online January 1, 2018]. J Travel Med. doi:10.1093/jtm/tax082.
- Dooling KL, Guo A, Patel M, et al. Recommendations of the Advisory Committee on Immunization Practices for Use of Herpes Zoster Vaccines. MMWR Morb Mortal Wkly Rep. 2018;67:103-108.
- Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77:845-854.e5.
- Haynes K, Beukelman T, Curtis JR, et al. Tumor necrosis factor α inhibitor therapy and cancer risk in chronic immune-mediated diseases. Arthritis Rheum. 2013;65:48-58.
- Fabiano A, De Simone C, Gisondi P, et al. Management of patients with psoriasis treated with biologic drugs needing a surgical treatment. Drug Dev Res. 2014;75(suppl 1):S24-S26.
- Choi YM, Debbaneh M, Weinberg JM, et al. From the Medical Board of the National Psoriasis Foundation: perioperative management of systemic immunomodulatory agents in patients with psoriasis and psoriatic arthritis. J Am Acad Dermatol. 2016;75:798-805.e7.
- Honda H, Umezawa Y, Kikuchi S, et al. Switching of biologics in psoriasis: reasons and results. J Dermatol. 2017;44:1015-1019.
- Bracke S, Lambert J. Viewpoint on handling anti-TNF failure in psoriasis. Arch Dermatol Res. 2013;305:945-950.
Psoriasis is a systemic immune-mediated disorder characterized by erythematous, scaly, well-demarcated plaques on the skin that affects approximately 3% of the world’s population.1 The disease is moderate to severe for approximately 1 in 6 individuals with psoriasis.2 These patients, particularly those with symptoms that are refractory to topical therapy and/or phototherapy, can benefit from the use of biologic agents, which are monoclonal antibodies and fusion proteins engineered to inhibit the action of cytokines that drive psoriatic inflammation.
In February 2019, the American Academy of Dermatology (AAD) and National Psoriasis Foundation (NPF) released an updated set of guidelines for the use of biologics in treating adult patients with psoriasis.3 The prior guidelines were released in 2008 when just 3 biologics—etanercept, infliximab, and adalimumab—were approved by the US Food and Drug Administration (FDA) for the management of psoriasis. These older recommendations were mostly based on studies of the efficacy and safety of biologics for patients with psoriatic arthritis.4 Over the last 11 years, 8 novel biologics have gained FDA approval, and numerous large phase 2 and phase 3 trials evaluating the risks and benefits of biologics have been conducted. The new guidelines contain considerably more detail and are based on evidence more specific to psoriasis rather than to psoriatic arthritis. Given the large repertoire of biologics available today and the increased amount of published research regarding each one, these guidelines may aid dermatologists in choosing the optimal biologic and managing therapy.
The AAD-NPF recommendations discuss the mechanism of action, efficacy, safety, and adverse events of the 10 biologics that have been FDA approved for the treatment of psoriasis as of March 2019, plus risankizumab, which was pending FDA approval at the time of publication and was later approved in April 2019. They also address dosing regimens, potential to combine biologics with other therapies, and different forms of psoriasis for which each may be effective.3 The purpose of this discussion is to present these guidelines in a condensed form to prescribers of biologic therapies and review the most clinically significant considerations during each step of treatment. Of note, we highlight only treatment of adult patients and do not discuss information relevant to risankizumab, as it was not FDA approved when the AAD-NPF guidelines were released.
Choosing a Biologic
Biologic therapy may be considered for patients with psoriasis that affects more than 3% of the body’s surface and is recalcitrant to localized therapies. There is no particular first-line biologic recommended for all patients with psoriasis; rather, choice of therapy should be individualized to the patient, considering factors such as body parts affected, comorbidities, lifestyle, and drug cost.
All 10 FDA-approved biologics (Table) have been ranked by the AAD and NPF as having grade A evidence for efficacy as monotherapy in the treatment of moderate to severe plaque-type psoriasis. Involvement of difficult-to-treat areas may be considered when choosing a specific therapy. The tumor necrosis factor α (TNF-α) inhibitors etanercept and adalimumab, the IL-17 inhibitor secukinumab, and the IL-23 inhibitor guselkumab have the greatest evidence for efficacy in treatment of nail disease. For scalp involvement, etanercept and guselkumab have the highest-quality evidence, and for palmoplantar disease, adalimumab, secukinumab, and guselkumab are considered the most effective. The TNF-α inhibitors are considered the optimal treatment option for concurrent psoriatic arthritis, though the IL-12/IL-23 inhibitor ustekinumab and the IL-17 inhibitors secukinumab and ixekizumab also have shown grade A evidence of efficacy. Of note, because TNF-α inhibitors received the earliest FDA approval, there is most evidence available for this class. Therapies with lower evidence quality for certain forms of psoriasis may show real-world effectiveness in individual patients, though more trials will be necessary to generate a body of evidence to change these clinical recommendations.
In pregnant women or those are anticipating pregnancy, certolizumab may be considered, as it is the only biologic shown to have minimal to no placental transfer. Other TNF-α inhibitors may undergo active placental transfer, particularly during the latter half of pregnancy,5 and the greatest theoretical risk of transfer occurs in the third trimester. Although these drugs may not directly harm the fetus, they do cause fetal immunosuppression for up to the first 3 months of life. All TNF-α inhibitors are considered safe during lactation. There are inadequate data regarding the safety of other classes of biologics during pregnancy and lactation.
Overweight and obese patients also require unique considerations when choosing a biologic. Infliximab is the only approved psoriasis biologic that utilizes proportional-to-weight dosing and hence may be particularly efficacious in patients with higher body mass. Ustekinumab dosing also takes patient weight into consideration; patients heavier than 100 kg should receive 90-mg doses at initiation and during maintenance compared to 45 mg for patients who weigh 100 kg or less. Other approved biologics also may be utilized in these patients but may require closer monitoring of treatment efficacy.
There are few serious contraindications for specific biologic therapies. Any history of allergic reaction to a particular therapy is an absolute contraindication to its use. In patients for whom IL-17 inhibitor treatment is being considered, inflammatory bowel disease (IBD) should be ruled out given the likelihood that IL-17 could reactivate or worsen IBD. Of note, TNF-α inhibitors and ustekinumab are approved therapies for patients with IBD and may be recommended in patients with comorbid psoriasis. Phase 2 and phase 3 trials have found no reactivation or worsening of IBD in patients with psoriasis who were treated with the IL-23 inhibitor tildrakizumab,6 and phase 2 trials of treatment of IBD with guselkumab are currently underway (ClinicalTrials.gov Identifier NCT03466411). In patients with New York Heart Association class III and class IV congestive heart failure or multiple sclerosis, initiation of TNF-α inhibitors should be avoided. Among 3 phase 3 trials encompassing nearly 3000 patients treated with the IL-17 inhibitor brodalumab, a total of 3 patients died by suicide7,8; hence, the FDA has issued a black box warning cautioning against use of this drug in patients with history of suicidal ideation or recent suicidal behavior. Although a causal relationship between brodalumab and suicide has not been well established,9 a thorough psychiatric history should be obtained in those initiating treatment with brodalumab.
Initiation of Therapy
Prior to initiating biologic therapy, it is important to obtain a complete blood cell count, complete metabolic panel, tuberculosis testing, and hepatitis B virus (HBV) and hepatitis C virus serologies. Testing for human immunodeficiency virus may be pursued at the clinician’s discretion. It is important to address any positive or concerning results prior to starting biologics. In patients with active infections, therapy may be initiated alongside guidance from an infectious disease specialist. Those with a positive purified protein derivative test, T-SPOT test, or QuantiFERON-TB Gold test must be referred for chest radiographs to rule out active tuberculosis. Patients with active HBV infection should receive appropriate referral to initiate antiviral therapy as well as core antibody testing, and those with active hepatitis C virus infection may only receive biologics under the combined discretion of a dermatologist and an appropriate specialist. Patients with human immunodeficiency virus must concurrently receive highly active antiretroviral therapy, show normal CD4+ T-cell count and undetectable viral load, and have no recent history of opportunistic infection.
Therapy should be commenced using specific dosing regimens, which are unique for each biologic (Table). Patients also must be educated on routine follow-up to assess treatment response and tolerability.
Assessment and Optimization of Treatment Response
Patients taking biologics may experience primary treatment failure, defined as lack of response to therapy from initiation. One predisposing factor may be increased body mass; patients who are overweight and obese are less likely to respond to standard regimens of TNF-α inhibitors and 45-mg dosing of ustekinumab. In most cases, however, the cause of primary nonresponse is unpredictable. For patients in whom therapy has failed within the recommended initial time frame (Table), dose escalation or shortening of dosing intervals may be pursued. Recommended dosing adjustments are outlined in the Table. Alternatively, patients may be switched to a different biologic.
If desired effectiveness is not reached with biologic monotherapy, topical corticosteroids, topical vitamin D analogues, or narrowband UVB light therapy may be concurrently used for difficult-to-treat areas. Evidence for safety and effectiveness of systemic adjuncts to biologics is moderate to low, warranting caution with their use. Methotrexate, cyclosporine, and apremilast have synergistic effects with biologics, though they may increase the risk for immunosuppression-related complications. Acitretin, an oral retinoid, likely is the most reasonable systemic adjunct to biologics because of its lack of immunosuppressive properties.
In patients with a suboptimal response to biologics, particularly those taking therapies that require frequent dosing, poor compliance should be considered.10 These patients may be switched to a biologic with less-frequent maintenance dosing (Table). Ustekinumab and tildrakizumab may be the best options for optimizing compliance, as they require dosing only once every 12 weeks after administration of loading doses.
Secondary treatment failure is diminished efficacy of treatment following successful initial response despite no changes in regimen. The best-known factor contributing to secondary nonresponse to biologics is the development of antidrug antibodies (ADAs), a phenomenon known as immunogenicity. The development of efficacy-limiting ADAs has been observed in response to most biologics, though ADAs against etanercept and guselkumab do not limit therapeutic response. Patients taking adalimumab and infliximab have particularly well-documented efficacy-limiting immunogenicity, and those who develop ADAs to infliximab are considered more prone to developing infusion reactions. Methotrexate, which limits antibody formation, may concomitantly be prescribed in patients who experience secondary treatment failure. It should be considered in all patients taking infliximab to increase efficacy and tolerability of therapy.
Considerations During Active Therapy
In addition to monitoring adherence and response to regimens, dermatologists must be heavily involved in counseling patients regarding the risks and adverse effects associated with these therapies. During maintenance therapy with biologics, patients must follow up with the prescriber at minimum every 3 to 6 months to evaluate for continued efficacy of treatment, extent of side effects, and effects of treatment on overall health and quality of life. Given the immunosuppressive effects of biologics, annual testing for tuberculosis should be considered in high-risk individuals. In those who are considered at low risk, tuberculosis testing may be done at the discretion of the dermatologist. In those with a history of HBV infection, HBV serologies should be pursued routinely given the risk for reactivation.
Annual screening for nonmelanoma skin cancer should be performed in all patients taking biologics. Tumor necrosis factor α inhibitor therapy in particular confers an elevated risk for cutaneous squamous cell carcinoma, especially in patients who are immunosuppressed at baseline and those with history of UV phototherapy. Use of acitretin alongside TNF-α inhibitors or ustekinumab may prevent squamous cell carcinoma formation in high-risk patients.
Because infliximab treatment poses an elevated risk of liver injury,11 liver function tests should be repeated 3 months following initiation of treatment and then every 6 to 12 months subsequently if results are normal. Periodic assessment of suicidal ideation is recommended in patients on brodalumab therapy, which may necessitate more frequent follow-up visits and potentially psychiatry referrals in certain patients. Patients taking IL-17 inhibitors, particularly those who are concurrently taking methotrexate, are at increased risk for developing mucocutaneous Candida infections; these patients should be monitored for such infections and treated appropriately.12
It is additionally important for prescribing dermatologists to ensure that patients on biologics are following up with their general providers to receive timely age-appropriate preventative screenings and vaccines. Inactivated vaccinations may be administered during therapy with any biologic; however, live vaccinations may induce systemic infection in those who are immunocompromised, which theoretically includes individuals taking biologic agents, though incidence data in this patient population are scarce.13 Some experts believe that administration of live vaccines warrants temporary discontinuation of biologic therapy for 2 to 3 half-lives before and after vaccination (Table). Others recommend stopping treatment at least 4 weeks before and until 2 weeks after vaccination. For patients taking biologics with half-lives greater than 20 days, which would theoretically require stopping the drug 2 months prior to vaccination, the benefit of vaccination should be weighed against the risk of prolonged discontinuation of therapy. Until recently, this recommendation was particularly important, as a live herpes zoster vaccination was recommended by the Centers for Disease Control and Prevention for adults older than 60 years. In 2017, a new inactivated herpes zoster vaccine was introduced and is now the preferred vaccine for all patients older than 50 years.14 It is especially important that patients on biologics receive this vaccine to avoid temporary drug discontinuation.
Evidence that any particular class of biologics increases risk for solid tumors or lymphoreticular malignancy is limited. One case-control analysis reported that more than 12 months of treatment with TNF-α inhibitors may increase risk for malignancy; however, the confidence interval reported hardly allows for statistical significance.15 Another retrospective cohort study found no elevated incidence of cancer in patients on TNF-α inhibitors compared to nonbiologic comparators.16 Ustekinumab was shown to confer no increased risk for malignancy in 1 large study,15 but no large studies have been conducted for other classes of drugs. Given the limited and inconclusive evidence available, the guidelines recommend that age-appropriate cancer screenings recommended for the general population should be pursued in patients taking biologics.
Surgery while taking biologics may lead to stress-induced augmentation of immunosuppression, resulting in elevated risk of infection.17 Low-risk surgeries that do not warrant discontinuation of treatment include endoscopic, ophthalmologic, dermatologic, orthopedic, and breast procedures. In patients preparing for elective surgery in which respiratory, gastrointestinal, or genitourinary tracts will be entered, biologics may be discontinued at least 3 half-lives (Table) prior to surgery if the dermatologist and surgeon collaboratively deem that risk of infection outweighs benefit of continued therapy.18 Therapy may be resumed within 1 to 2 weeks postoperatively if there are no surgical complications.
Switching Biologics
Changing therapy to another biologic should be considered if there is no response to treatment or the patient experiences adverse effects while taking a particular biologic. Because evidence is limited regarding the ideal time frame between discontinuation of a prior medication and initiation of a new biologic, this interval should be determined at the discretion of the provider based on the patient’s disease severity and response to prior treatment. For individuals who experience primary or secondary treatment failure while maintaining appropriate dosing and treatment compliance, switching to a different biologic is recommended to maximize treatment response.19 Changing therapy to a biologic within the same class is generally effective,20 and switching to a biologic with another mechanism of action should be considered if a class-specific adverse effect is the major reason for altering the regimen. Nonetheless, some patients may be unresponsive to biologic changes. Further research is necessary to determine which biologics may be most effective when previously used biologics have failed and particular factors that may predispose patients to biologic unresponsiveness.
Resuming Biologic Treatment Following Cessation
In cases where therapy is discontinued for any reason, it may be necessary to repeat initiation dosing when resuming treatment. In patients with severe or flaring disease or if more than 3 to 4 half-lives have passed since the most recent dose, it may be necessary to restart therapy with the loading dose (Table). Unfortunately, restarting therapy may preclude some patients from experiencing the maximal response that they attained prior to cessation. In such cases, switching biologic therapy to a different class may prove beneficial.
Final Thoughts
These recommendations contain valuable information that will assist dermatologists when initiating biologics and managing outcomes of their psoriasis patients. It is, however, crucial to bear in mind that these guidelines serve as merely a tool. Given the paucity of comprehensive research, particularly regarding some of the more recently approved therapies, there are many questions that are unanswered within the guidelines. Their utility for each individual patient situation is therefore limited, and clinical judgement may outweigh the information presented. The recommendations nevertheless provide a pivotal and unprecedented framework that promotes discourse among patients, dermatologists, and other providers to optimize the efficacy of biologic therapy for psoriasis.
Psoriasis is a systemic immune-mediated disorder characterized by erythematous, scaly, well-demarcated plaques on the skin that affects approximately 3% of the world’s population.1 The disease is moderate to severe for approximately 1 in 6 individuals with psoriasis.2 These patients, particularly those with symptoms that are refractory to topical therapy and/or phototherapy, can benefit from the use of biologic agents, which are monoclonal antibodies and fusion proteins engineered to inhibit the action of cytokines that drive psoriatic inflammation.
In February 2019, the American Academy of Dermatology (AAD) and National Psoriasis Foundation (NPF) released an updated set of guidelines for the use of biologics in treating adult patients with psoriasis.3 The prior guidelines were released in 2008 when just 3 biologics—etanercept, infliximab, and adalimumab—were approved by the US Food and Drug Administration (FDA) for the management of psoriasis. These older recommendations were mostly based on studies of the efficacy and safety of biologics for patients with psoriatic arthritis.4 Over the last 11 years, 8 novel biologics have gained FDA approval, and numerous large phase 2 and phase 3 trials evaluating the risks and benefits of biologics have been conducted. The new guidelines contain considerably more detail and are based on evidence more specific to psoriasis rather than to psoriatic arthritis. Given the large repertoire of biologics available today and the increased amount of published research regarding each one, these guidelines may aid dermatologists in choosing the optimal biologic and managing therapy.
The AAD-NPF recommendations discuss the mechanism of action, efficacy, safety, and adverse events of the 10 biologics that have been FDA approved for the treatment of psoriasis as of March 2019, plus risankizumab, which was pending FDA approval at the time of publication and was later approved in April 2019. They also address dosing regimens, potential to combine biologics with other therapies, and different forms of psoriasis for which each may be effective.3 The purpose of this discussion is to present these guidelines in a condensed form to prescribers of biologic therapies and review the most clinically significant considerations during each step of treatment. Of note, we highlight only treatment of adult patients and do not discuss information relevant to risankizumab, as it was not FDA approved when the AAD-NPF guidelines were released.
Choosing a Biologic
Biologic therapy may be considered for patients with psoriasis that affects more than 3% of the body’s surface and is recalcitrant to localized therapies. There is no particular first-line biologic recommended for all patients with psoriasis; rather, choice of therapy should be individualized to the patient, considering factors such as body parts affected, comorbidities, lifestyle, and drug cost.
All 10 FDA-approved biologics (Table) have been ranked by the AAD and NPF as having grade A evidence for efficacy as monotherapy in the treatment of moderate to severe plaque-type psoriasis. Involvement of difficult-to-treat areas may be considered when choosing a specific therapy. The tumor necrosis factor α (TNF-α) inhibitors etanercept and adalimumab, the IL-17 inhibitor secukinumab, and the IL-23 inhibitor guselkumab have the greatest evidence for efficacy in treatment of nail disease. For scalp involvement, etanercept and guselkumab have the highest-quality evidence, and for palmoplantar disease, adalimumab, secukinumab, and guselkumab are considered the most effective. The TNF-α inhibitors are considered the optimal treatment option for concurrent psoriatic arthritis, though the IL-12/IL-23 inhibitor ustekinumab and the IL-17 inhibitors secukinumab and ixekizumab also have shown grade A evidence of efficacy. Of note, because TNF-α inhibitors received the earliest FDA approval, there is most evidence available for this class. Therapies with lower evidence quality for certain forms of psoriasis may show real-world effectiveness in individual patients, though more trials will be necessary to generate a body of evidence to change these clinical recommendations.
In pregnant women or those are anticipating pregnancy, certolizumab may be considered, as it is the only biologic shown to have minimal to no placental transfer. Other TNF-α inhibitors may undergo active placental transfer, particularly during the latter half of pregnancy,5 and the greatest theoretical risk of transfer occurs in the third trimester. Although these drugs may not directly harm the fetus, they do cause fetal immunosuppression for up to the first 3 months of life. All TNF-α inhibitors are considered safe during lactation. There are inadequate data regarding the safety of other classes of biologics during pregnancy and lactation.
Overweight and obese patients also require unique considerations when choosing a biologic. Infliximab is the only approved psoriasis biologic that utilizes proportional-to-weight dosing and hence may be particularly efficacious in patients with higher body mass. Ustekinumab dosing also takes patient weight into consideration; patients heavier than 100 kg should receive 90-mg doses at initiation and during maintenance compared to 45 mg for patients who weigh 100 kg or less. Other approved biologics also may be utilized in these patients but may require closer monitoring of treatment efficacy.
There are few serious contraindications for specific biologic therapies. Any history of allergic reaction to a particular therapy is an absolute contraindication to its use. In patients for whom IL-17 inhibitor treatment is being considered, inflammatory bowel disease (IBD) should be ruled out given the likelihood that IL-17 could reactivate or worsen IBD. Of note, TNF-α inhibitors and ustekinumab are approved therapies for patients with IBD and may be recommended in patients with comorbid psoriasis. Phase 2 and phase 3 trials have found no reactivation or worsening of IBD in patients with psoriasis who were treated with the IL-23 inhibitor tildrakizumab,6 and phase 2 trials of treatment of IBD with guselkumab are currently underway (ClinicalTrials.gov Identifier NCT03466411). In patients with New York Heart Association class III and class IV congestive heart failure or multiple sclerosis, initiation of TNF-α inhibitors should be avoided. Among 3 phase 3 trials encompassing nearly 3000 patients treated with the IL-17 inhibitor brodalumab, a total of 3 patients died by suicide7,8; hence, the FDA has issued a black box warning cautioning against use of this drug in patients with history of suicidal ideation or recent suicidal behavior. Although a causal relationship between brodalumab and suicide has not been well established,9 a thorough psychiatric history should be obtained in those initiating treatment with brodalumab.
Initiation of Therapy
Prior to initiating biologic therapy, it is important to obtain a complete blood cell count, complete metabolic panel, tuberculosis testing, and hepatitis B virus (HBV) and hepatitis C virus serologies. Testing for human immunodeficiency virus may be pursued at the clinician’s discretion. It is important to address any positive or concerning results prior to starting biologics. In patients with active infections, therapy may be initiated alongside guidance from an infectious disease specialist. Those with a positive purified protein derivative test, T-SPOT test, or QuantiFERON-TB Gold test must be referred for chest radiographs to rule out active tuberculosis. Patients with active HBV infection should receive appropriate referral to initiate antiviral therapy as well as core antibody testing, and those with active hepatitis C virus infection may only receive biologics under the combined discretion of a dermatologist and an appropriate specialist. Patients with human immunodeficiency virus must concurrently receive highly active antiretroviral therapy, show normal CD4+ T-cell count and undetectable viral load, and have no recent history of opportunistic infection.
Therapy should be commenced using specific dosing regimens, which are unique for each biologic (Table). Patients also must be educated on routine follow-up to assess treatment response and tolerability.
Assessment and Optimization of Treatment Response
Patients taking biologics may experience primary treatment failure, defined as lack of response to therapy from initiation. One predisposing factor may be increased body mass; patients who are overweight and obese are less likely to respond to standard regimens of TNF-α inhibitors and 45-mg dosing of ustekinumab. In most cases, however, the cause of primary nonresponse is unpredictable. For patients in whom therapy has failed within the recommended initial time frame (Table), dose escalation or shortening of dosing intervals may be pursued. Recommended dosing adjustments are outlined in the Table. Alternatively, patients may be switched to a different biologic.
If desired effectiveness is not reached with biologic monotherapy, topical corticosteroids, topical vitamin D analogues, or narrowband UVB light therapy may be concurrently used for difficult-to-treat areas. Evidence for safety and effectiveness of systemic adjuncts to biologics is moderate to low, warranting caution with their use. Methotrexate, cyclosporine, and apremilast have synergistic effects with biologics, though they may increase the risk for immunosuppression-related complications. Acitretin, an oral retinoid, likely is the most reasonable systemic adjunct to biologics because of its lack of immunosuppressive properties.
In patients with a suboptimal response to biologics, particularly those taking therapies that require frequent dosing, poor compliance should be considered.10 These patients may be switched to a biologic with less-frequent maintenance dosing (Table). Ustekinumab and tildrakizumab may be the best options for optimizing compliance, as they require dosing only once every 12 weeks after administration of loading doses.
Secondary treatment failure is diminished efficacy of treatment following successful initial response despite no changes in regimen. The best-known factor contributing to secondary nonresponse to biologics is the development of antidrug antibodies (ADAs), a phenomenon known as immunogenicity. The development of efficacy-limiting ADAs has been observed in response to most biologics, though ADAs against etanercept and guselkumab do not limit therapeutic response. Patients taking adalimumab and infliximab have particularly well-documented efficacy-limiting immunogenicity, and those who develop ADAs to infliximab are considered more prone to developing infusion reactions. Methotrexate, which limits antibody formation, may concomitantly be prescribed in patients who experience secondary treatment failure. It should be considered in all patients taking infliximab to increase efficacy and tolerability of therapy.
Considerations During Active Therapy
In addition to monitoring adherence and response to regimens, dermatologists must be heavily involved in counseling patients regarding the risks and adverse effects associated with these therapies. During maintenance therapy with biologics, patients must follow up with the prescriber at minimum every 3 to 6 months to evaluate for continued efficacy of treatment, extent of side effects, and effects of treatment on overall health and quality of life. Given the immunosuppressive effects of biologics, annual testing for tuberculosis should be considered in high-risk individuals. In those who are considered at low risk, tuberculosis testing may be done at the discretion of the dermatologist. In those with a history of HBV infection, HBV serologies should be pursued routinely given the risk for reactivation.
Annual screening for nonmelanoma skin cancer should be performed in all patients taking biologics. Tumor necrosis factor α inhibitor therapy in particular confers an elevated risk for cutaneous squamous cell carcinoma, especially in patients who are immunosuppressed at baseline and those with history of UV phototherapy. Use of acitretin alongside TNF-α inhibitors or ustekinumab may prevent squamous cell carcinoma formation in high-risk patients.
Because infliximab treatment poses an elevated risk of liver injury,11 liver function tests should be repeated 3 months following initiation of treatment and then every 6 to 12 months subsequently if results are normal. Periodic assessment of suicidal ideation is recommended in patients on brodalumab therapy, which may necessitate more frequent follow-up visits and potentially psychiatry referrals in certain patients. Patients taking IL-17 inhibitors, particularly those who are concurrently taking methotrexate, are at increased risk for developing mucocutaneous Candida infections; these patients should be monitored for such infections and treated appropriately.12
It is additionally important for prescribing dermatologists to ensure that patients on biologics are following up with their general providers to receive timely age-appropriate preventative screenings and vaccines. Inactivated vaccinations may be administered during therapy with any biologic; however, live vaccinations may induce systemic infection in those who are immunocompromised, which theoretically includes individuals taking biologic agents, though incidence data in this patient population are scarce.13 Some experts believe that administration of live vaccines warrants temporary discontinuation of biologic therapy for 2 to 3 half-lives before and after vaccination (Table). Others recommend stopping treatment at least 4 weeks before and until 2 weeks after vaccination. For patients taking biologics with half-lives greater than 20 days, which would theoretically require stopping the drug 2 months prior to vaccination, the benefit of vaccination should be weighed against the risk of prolonged discontinuation of therapy. Until recently, this recommendation was particularly important, as a live herpes zoster vaccination was recommended by the Centers for Disease Control and Prevention for adults older than 60 years. In 2017, a new inactivated herpes zoster vaccine was introduced and is now the preferred vaccine for all patients older than 50 years.14 It is especially important that patients on biologics receive this vaccine to avoid temporary drug discontinuation.
Evidence that any particular class of biologics increases risk for solid tumors or lymphoreticular malignancy is limited. One case-control analysis reported that more than 12 months of treatment with TNF-α inhibitors may increase risk for malignancy; however, the confidence interval reported hardly allows for statistical significance.15 Another retrospective cohort study found no elevated incidence of cancer in patients on TNF-α inhibitors compared to nonbiologic comparators.16 Ustekinumab was shown to confer no increased risk for malignancy in 1 large study,15 but no large studies have been conducted for other classes of drugs. Given the limited and inconclusive evidence available, the guidelines recommend that age-appropriate cancer screenings recommended for the general population should be pursued in patients taking biologics.
Surgery while taking biologics may lead to stress-induced augmentation of immunosuppression, resulting in elevated risk of infection.17 Low-risk surgeries that do not warrant discontinuation of treatment include endoscopic, ophthalmologic, dermatologic, orthopedic, and breast procedures. In patients preparing for elective surgery in which respiratory, gastrointestinal, or genitourinary tracts will be entered, biologics may be discontinued at least 3 half-lives (Table) prior to surgery if the dermatologist and surgeon collaboratively deem that risk of infection outweighs benefit of continued therapy.18 Therapy may be resumed within 1 to 2 weeks postoperatively if there are no surgical complications.
Switching Biologics
Changing therapy to another biologic should be considered if there is no response to treatment or the patient experiences adverse effects while taking a particular biologic. Because evidence is limited regarding the ideal time frame between discontinuation of a prior medication and initiation of a new biologic, this interval should be determined at the discretion of the provider based on the patient’s disease severity and response to prior treatment. For individuals who experience primary or secondary treatment failure while maintaining appropriate dosing and treatment compliance, switching to a different biologic is recommended to maximize treatment response.19 Changing therapy to a biologic within the same class is generally effective,20 and switching to a biologic with another mechanism of action should be considered if a class-specific adverse effect is the major reason for altering the regimen. Nonetheless, some patients may be unresponsive to biologic changes. Further research is necessary to determine which biologics may be most effective when previously used biologics have failed and particular factors that may predispose patients to biologic unresponsiveness.
Resuming Biologic Treatment Following Cessation
In cases where therapy is discontinued for any reason, it may be necessary to repeat initiation dosing when resuming treatment. In patients with severe or flaring disease or if more than 3 to 4 half-lives have passed since the most recent dose, it may be necessary to restart therapy with the loading dose (Table). Unfortunately, restarting therapy may preclude some patients from experiencing the maximal response that they attained prior to cessation. In such cases, switching biologic therapy to a different class may prove beneficial.
Final Thoughts
These recommendations contain valuable information that will assist dermatologists when initiating biologics and managing outcomes of their psoriasis patients. It is, however, crucial to bear in mind that these guidelines serve as merely a tool. Given the paucity of comprehensive research, particularly regarding some of the more recently approved therapies, there are many questions that are unanswered within the guidelines. Their utility for each individual patient situation is therefore limited, and clinical judgement may outweigh the information presented. The recommendations nevertheless provide a pivotal and unprecedented framework that promotes discourse among patients, dermatologists, and other providers to optimize the efficacy of biologic therapy for psoriasis.
- Michalek IM, Loring B, John SM. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol. 2017;31:205-212.
- Kurd SK, Gelfand JM. The prevalence of previously diagnosed and undiagnosed psoriasis in US adults: results from NHANES 2003-2004. J Am Acad Dermatol. 2009;60:218-224.
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics [published online February 13, 2019]. J Am Acad Dermatol. 2019;80:1029-1072.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- Förger F, Villiger PM. Treatment of rheumatoid arthritis during pregnancy: present and future. Expert Rev Clin Immunol. 2016;12:937-944.
- Gooderham M, Elewski B, Pariser D, et al. Incidence of serious gastrointestinal events and inflammatory bowel disease among tildrakizumab-treated patients with moderate-to-severe plaque psoriasis: data from 3 large randomized clinical trials [abstract]. J Am Acad Dermatol. 2018;79(suppl 1):AB166.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-328.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016;175:273-286
- Beck KM, Koo J. Brodalumab for the treatment of plaque psoriasis: up-to-date. Expert Opin Biol Ther. 2019;19:287-292.
- Fouéré S, Adjadj L, Pawin H. How patients experience psoriasis: results from a European survey. J Eur Acad Dermatol Venereol. 2005;19(suppl 3):2-6.
- Björnsson ES, Bergmann OM, Björnsson HK, et al. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 2013;144:1419-1425, 1425.e1-3; quiz e19-20.
- Saunte DM, Mrowietz U, Puig L, et al. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177:47-62.
- Huber F, Ehrensperger B, Hatz C, et al. Safety of live vaccines on immunosuppressive or immunomodulatory therapy—a retrospective study in three Swiss Travel Clinics [published online January 1, 2018]. J Travel Med. doi:10.1093/jtm/tax082.
- Dooling KL, Guo A, Patel M, et al. Recommendations of the Advisory Committee on Immunization Practices for Use of Herpes Zoster Vaccines. MMWR Morb Mortal Wkly Rep. 2018;67:103-108.
- Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77:845-854.e5.
- Haynes K, Beukelman T, Curtis JR, et al. Tumor necrosis factor α inhibitor therapy and cancer risk in chronic immune-mediated diseases. Arthritis Rheum. 2013;65:48-58.
- Fabiano A, De Simone C, Gisondi P, et al. Management of patients with psoriasis treated with biologic drugs needing a surgical treatment. Drug Dev Res. 2014;75(suppl 1):S24-S26.
- Choi YM, Debbaneh M, Weinberg JM, et al. From the Medical Board of the National Psoriasis Foundation: perioperative management of systemic immunomodulatory agents in patients with psoriasis and psoriatic arthritis. J Am Acad Dermatol. 2016;75:798-805.e7.
- Honda H, Umezawa Y, Kikuchi S, et al. Switching of biologics in psoriasis: reasons and results. J Dermatol. 2017;44:1015-1019.
- Bracke S, Lambert J. Viewpoint on handling anti-TNF failure in psoriasis. Arch Dermatol Res. 2013;305:945-950.
- Michalek IM, Loring B, John SM. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol. 2017;31:205-212.
- Kurd SK, Gelfand JM. The prevalence of previously diagnosed and undiagnosed psoriasis in US adults: results from NHANES 2003-2004. J Am Acad Dermatol. 2009;60:218-224.
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics [published online February 13, 2019]. J Am Acad Dermatol. 2019;80:1029-1072.
- Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
- Förger F, Villiger PM. Treatment of rheumatoid arthritis during pregnancy: present and future. Expert Rev Clin Immunol. 2016;12:937-944.
- Gooderham M, Elewski B, Pariser D, et al. Incidence of serious gastrointestinal events and inflammatory bowel disease among tildrakizumab-treated patients with moderate-to-severe plaque psoriasis: data from 3 large randomized clinical trials [abstract]. J Am Acad Dermatol. 2018;79(suppl 1):AB166.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-328.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016;175:273-286
- Beck KM, Koo J. Brodalumab for the treatment of plaque psoriasis: up-to-date. Expert Opin Biol Ther. 2019;19:287-292.
- Fouéré S, Adjadj L, Pawin H. How patients experience psoriasis: results from a European survey. J Eur Acad Dermatol Venereol. 2005;19(suppl 3):2-6.
- Björnsson ES, Bergmann OM, Björnsson HK, et al. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 2013;144:1419-1425, 1425.e1-3; quiz e19-20.
- Saunte DM, Mrowietz U, Puig L, et al. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177:47-62.
- Huber F, Ehrensperger B, Hatz C, et al. Safety of live vaccines on immunosuppressive or immunomodulatory therapy—a retrospective study in three Swiss Travel Clinics [published online January 1, 2018]. J Travel Med. doi:10.1093/jtm/tax082.
- Dooling KL, Guo A, Patel M, et al. Recommendations of the Advisory Committee on Immunization Practices for Use of Herpes Zoster Vaccines. MMWR Morb Mortal Wkly Rep. 2018;67:103-108.
- Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77:845-854.e5.
- Haynes K, Beukelman T, Curtis JR, et al. Tumor necrosis factor α inhibitor therapy and cancer risk in chronic immune-mediated diseases. Arthritis Rheum. 2013;65:48-58.
- Fabiano A, De Simone C, Gisondi P, et al. Management of patients with psoriasis treated with biologic drugs needing a surgical treatment. Drug Dev Res. 2014;75(suppl 1):S24-S26.
- Choi YM, Debbaneh M, Weinberg JM, et al. From the Medical Board of the National Psoriasis Foundation: perioperative management of systemic immunomodulatory agents in patients with psoriasis and psoriatic arthritis. J Am Acad Dermatol. 2016;75:798-805.e7.
- Honda H, Umezawa Y, Kikuchi S, et al. Switching of biologics in psoriasis: reasons and results. J Dermatol. 2017;44:1015-1019.
- Bracke S, Lambert J. Viewpoint on handling anti-TNF failure in psoriasis. Arch Dermatol Res. 2013;305:945-950.
Practice Points
- There are currently 11 biologics approved for psoriasis, but there is no first-line or optimalbiologic. The choice must be made using clinical judgment based on a variety of medical and social factors.
- Frequent assessment for efficacy of and adverse events due to biologic therapy is warranted, as lack of response, loss of response, or severe side effects may warrant addition of concurrent therapies or switching to a different biologic.
- There are important considerations to make when immunizing and planning for surgery in patients on biologics.
Oral semaglutide monotherapy delivers HbA1c improvements in type 2 diabetes
Oral semaglutide monotherapy was superior to placebo for improving glycated hemoglobin (HbA1c) levels at all doses tested in adults with type 2 diabetes who had been previously insufficiently managed with diet and exercise, according to findings from a global, randomized trial.
The drug also showed dose-dependent weight loss, with a statistically significant effect on body weight, compared with placebo, at higher doses.
To date, the glucagon-like peptide–1 receptor agonist has been available as weekly subcutaneous shots for patients with type 2 diabetes, and in that form they have been shown to be effective in reducing HbA1c, inducing weight loss, and lowering the risk of cardiovascular events in patients with cardiovascular disease or those who are at high risk for it, wrote Vanita R. Aroda, MD, of Brigham and Women’s Hospital, Boston, and colleagues. The report is in Diabetes Care.
The novel oral semaglutide tablet is designed to enhance medication absorption, and the pharmacokinetics and dosage were established in phase 2 studies, they noted.
In the phase 3 Peptide Innovation for Early Diabetes Treatment 1 (PIONEER 1) study, Dr. Aroda and colleagues randomized 703 adults with type 2 diabetes to receive either 3 mg, 7 mg, or 14 mg of oral semaglutide daily, or placebo. The average age of the patients was 55 years, about half were women, and the average baseline HbA1c was 8.0% (64 mmol/mol). The primary endpoint was change in HbA1c level from baseline to week 26, and the secondary endpoint was change in body weight over the same period.
After 26 weeks of once-daily treatment, patients in semaglutide group showed significant reductions in HbA1c from baseline with all three doses: –0.6% (3 mg), –0.9% (7 mg), and –1.1% (14 mg), with P less than .001 for all, based on an intention-to-treat analysis. Similar results occurred using an on-treatment analysis, with differences of –0.7%, –1.2%, and –1.4%, respectively, for the three doses.
In addition, patients in all dose groups achieved the secondary endpoint of reduction in body weight, compared with placebo, from baseline to 26 weeks based on both types of analyses. “Significantly more patients achieved body weight loss of at least 5% with oral semaglutide at 7 mg and 14 mg, compared with placebo,” Dr. Aroda and colleagues wrote (intention-to-treat: –0.1 for 3 mg daily [P = .87], –0.9 for 7 mg [P = .09], –2.3 for 14 mg [P less than .001]; and on-treatment: –0.2 for 3 mg [P = .71], –1.0 for 7 mg [P = .01], –2.6 for 14 mg [P less than .001]).
The overall incidence of adverse events and serious adverse events was similar in the treatment and placebo groups, with the most frequent being nausea and diarrhea. No deaths occurred among patients on the medication.
The findings were limited by several factors, including a patient population that had a relatively short duration of diabetes (mean, 3.5 years) and that the oral semaglutide was used as first-line monotherapy, without first using metformin, the researchers noted. However, oral semaglutide “achieved clinically meaningful and superior glucose lowering,” compared with placebo, at all three doses, they wrote.
“Ongoing additional studies in the PIONEER program will further define the effect when used in combination with other glucose-lowering therapies and in other populations of interest, such as those with high cardiovascular risk or renal impairment,” they emphasized
Novo Nordisk funded the study. The lead author disclosed relationships with Novo Nordisk, and several coauthors disclosed relationships with or employment by the company.
SOURCE: Aroda VR et al. Diabetes Care. 2019 Jul. doi: 10.2337/dc19-0749.
Oral semaglutide monotherapy was superior to placebo for improving glycated hemoglobin (HbA1c) levels at all doses tested in adults with type 2 diabetes who had been previously insufficiently managed with diet and exercise, according to findings from a global, randomized trial.
The drug also showed dose-dependent weight loss, with a statistically significant effect on body weight, compared with placebo, at higher doses.
To date, the glucagon-like peptide–1 receptor agonist has been available as weekly subcutaneous shots for patients with type 2 diabetes, and in that form they have been shown to be effective in reducing HbA1c, inducing weight loss, and lowering the risk of cardiovascular events in patients with cardiovascular disease or those who are at high risk for it, wrote Vanita R. Aroda, MD, of Brigham and Women’s Hospital, Boston, and colleagues. The report is in Diabetes Care.
The novel oral semaglutide tablet is designed to enhance medication absorption, and the pharmacokinetics and dosage were established in phase 2 studies, they noted.
In the phase 3 Peptide Innovation for Early Diabetes Treatment 1 (PIONEER 1) study, Dr. Aroda and colleagues randomized 703 adults with type 2 diabetes to receive either 3 mg, 7 mg, or 14 mg of oral semaglutide daily, or placebo. The average age of the patients was 55 years, about half were women, and the average baseline HbA1c was 8.0% (64 mmol/mol). The primary endpoint was change in HbA1c level from baseline to week 26, and the secondary endpoint was change in body weight over the same period.
After 26 weeks of once-daily treatment, patients in semaglutide group showed significant reductions in HbA1c from baseline with all three doses: –0.6% (3 mg), –0.9% (7 mg), and –1.1% (14 mg), with P less than .001 for all, based on an intention-to-treat analysis. Similar results occurred using an on-treatment analysis, with differences of –0.7%, –1.2%, and –1.4%, respectively, for the three doses.
In addition, patients in all dose groups achieved the secondary endpoint of reduction in body weight, compared with placebo, from baseline to 26 weeks based on both types of analyses. “Significantly more patients achieved body weight loss of at least 5% with oral semaglutide at 7 mg and 14 mg, compared with placebo,” Dr. Aroda and colleagues wrote (intention-to-treat: –0.1 for 3 mg daily [P = .87], –0.9 for 7 mg [P = .09], –2.3 for 14 mg [P less than .001]; and on-treatment: –0.2 for 3 mg [P = .71], –1.0 for 7 mg [P = .01], –2.6 for 14 mg [P less than .001]).
The overall incidence of adverse events and serious adverse events was similar in the treatment and placebo groups, with the most frequent being nausea and diarrhea. No deaths occurred among patients on the medication.
The findings were limited by several factors, including a patient population that had a relatively short duration of diabetes (mean, 3.5 years) and that the oral semaglutide was used as first-line monotherapy, without first using metformin, the researchers noted. However, oral semaglutide “achieved clinically meaningful and superior glucose lowering,” compared with placebo, at all three doses, they wrote.
“Ongoing additional studies in the PIONEER program will further define the effect when used in combination with other glucose-lowering therapies and in other populations of interest, such as those with high cardiovascular risk or renal impairment,” they emphasized
Novo Nordisk funded the study. The lead author disclosed relationships with Novo Nordisk, and several coauthors disclosed relationships with or employment by the company.
SOURCE: Aroda VR et al. Diabetes Care. 2019 Jul. doi: 10.2337/dc19-0749.
Oral semaglutide monotherapy was superior to placebo for improving glycated hemoglobin (HbA1c) levels at all doses tested in adults with type 2 diabetes who had been previously insufficiently managed with diet and exercise, according to findings from a global, randomized trial.
The drug also showed dose-dependent weight loss, with a statistically significant effect on body weight, compared with placebo, at higher doses.
To date, the glucagon-like peptide–1 receptor agonist has been available as weekly subcutaneous shots for patients with type 2 diabetes, and in that form they have been shown to be effective in reducing HbA1c, inducing weight loss, and lowering the risk of cardiovascular events in patients with cardiovascular disease or those who are at high risk for it, wrote Vanita R. Aroda, MD, of Brigham and Women’s Hospital, Boston, and colleagues. The report is in Diabetes Care.
The novel oral semaglutide tablet is designed to enhance medication absorption, and the pharmacokinetics and dosage were established in phase 2 studies, they noted.
In the phase 3 Peptide Innovation for Early Diabetes Treatment 1 (PIONEER 1) study, Dr. Aroda and colleagues randomized 703 adults with type 2 diabetes to receive either 3 mg, 7 mg, or 14 mg of oral semaglutide daily, or placebo. The average age of the patients was 55 years, about half were women, and the average baseline HbA1c was 8.0% (64 mmol/mol). The primary endpoint was change in HbA1c level from baseline to week 26, and the secondary endpoint was change in body weight over the same period.
After 26 weeks of once-daily treatment, patients in semaglutide group showed significant reductions in HbA1c from baseline with all three doses: –0.6% (3 mg), –0.9% (7 mg), and –1.1% (14 mg), with P less than .001 for all, based on an intention-to-treat analysis. Similar results occurred using an on-treatment analysis, with differences of –0.7%, –1.2%, and –1.4%, respectively, for the three doses.
In addition, patients in all dose groups achieved the secondary endpoint of reduction in body weight, compared with placebo, from baseline to 26 weeks based on both types of analyses. “Significantly more patients achieved body weight loss of at least 5% with oral semaglutide at 7 mg and 14 mg, compared with placebo,” Dr. Aroda and colleagues wrote (intention-to-treat: –0.1 for 3 mg daily [P = .87], –0.9 for 7 mg [P = .09], –2.3 for 14 mg [P less than .001]; and on-treatment: –0.2 for 3 mg [P = .71], –1.0 for 7 mg [P = .01], –2.6 for 14 mg [P less than .001]).
The overall incidence of adverse events and serious adverse events was similar in the treatment and placebo groups, with the most frequent being nausea and diarrhea. No deaths occurred among patients on the medication.
The findings were limited by several factors, including a patient population that had a relatively short duration of diabetes (mean, 3.5 years) and that the oral semaglutide was used as first-line monotherapy, without first using metformin, the researchers noted. However, oral semaglutide “achieved clinically meaningful and superior glucose lowering,” compared with placebo, at all three doses, they wrote.
“Ongoing additional studies in the PIONEER program will further define the effect when used in combination with other glucose-lowering therapies and in other populations of interest, such as those with high cardiovascular risk or renal impairment,” they emphasized
Novo Nordisk funded the study. The lead author disclosed relationships with Novo Nordisk, and several coauthors disclosed relationships with or employment by the company.
SOURCE: Aroda VR et al. Diabetes Care. 2019 Jul. doi: 10.2337/dc19-0749.
FROM DIABETES CARE
Aspirin interacts with epigenetics to influence breast cancer mortality
The impact of prediagnosis aspirin use on mortality in women with breast cancer is significantly tied to epigenetic changes in certain breast cancer-related genes, investigators reported.
While studies have shown aspirin reduces the risk of breast cancer development, there is limited and inconsistent data on the effect of aspirin on prognosis and mortality after a diagnosis of breast cancer, Tengteng Wang, PhD, from the department of epidemiology at the University of North Carolina at Chapel Hill and coauthors wrote in Cancer.
To address this, they analyzed data from 1,508 women who had a first diagnosis of primary breast cancer and were involved in the Long Island Breast Cancer Study Project; they then looked at the women’s methylation status, which is a mechanism of epigenetic change.
Around one in five participants reported ever using aspirin, and the analysis showed that ever use of aspirin was associated with an overall 13% decrease in breast cancer–specific mortality.
However researchers saw significant interactions between aspirin use and LINE-1 methylation status – which is a marker of methylation of genetic elements that play key roles in maintaining genomic stability – and breast cancer–specific genes.
They found that aspirin use in women with LINE-1 hypomethylation was associated with a risk of breast cancer–specific mortality that was 45% higher than that of nonusers (P = .05).
Compared with nonusers, aspirin users with methylated tumor BRCA1 promoter had significant 16% higher breast cancer mortality (P = .04) and 67% higher all-cause mortality (P = .02). However the study showed aspirin did not affect mortality in women with unmethylated BRCA1 promoter.
Among women with the PR breast cancer gene, aspirin use by those with methylation of the PR promoter was associated with a 63% higher breast cancer–specific mortality, but methylation showed no statistically significant effect on all-cause mortality, compared with nonusers.
The study found no significant change when they restricted the analysis to receptor-positive or invasive breast cancer, and the associations remained consistent even after adjusting for global methylation.
“Our findings suggest that the association between aspirin use and mortality after breast cancer may depend on methylation profiles and warrant further investigation,” the authors wrote. “These findings, if confirmed, may provide new biological insights into the association between aspirin use and breast cancer prognosis, may affect clinical decision making by identifying a subgroup of patients with breast cancer using epigenetic markers for whom prediagnosis aspirin use affects subsequent mortality, and may help refine risk-reduction strategies to improve survival among women with breast cancer.”
The study was partly supported by the National Institutes of Health. One author declared personal fees from the private sector outside the submitted work.
SOURCE: Wang T et al. Cancer. 2019 Aug 12. doi: 10.1002/cncr.32364.
This study offers new insights into the intersection of epigenetics, prediagnosis aspirin use, and breast cancer survival at a time when there is an urgent need to understand why some women respond differently to treatment and to find cost-effective therapies for the disease.
Epigenetics is a promising avenue of investigation because epigenetic shifts, such as DNA methylation, that impact the genes responsible for cell behavior and DNA damage and repair are known to contribute to and exacerbate cancer. These epigenetic signatures could act as biomarkers for risk in cancer and also aid with more effective treatment approaches. For example, aspirin is known to affect DNA methylation at certain sites in colon cancer, hence this study’s hypothesis that pre–cancer diagnosis aspirin use would interact with epigenetic signatures and influence breast cancer outcomes.
Kristen M. C. Malecki, PhD, is from the department of population health sciences in the School of Medicine and Public Health at the University of Wisconsin, Madison. The comments are adapted from an accompanying editorial (Cancer. 2019 Aug 12. doi: 10.1002/cncr.32365). Dr. Malecki declared support from the National Institutes of Health, National Institute for Environmental Health Sciences Breast Cancer, and the Environment Research Program.
This study offers new insights into the intersection of epigenetics, prediagnosis aspirin use, and breast cancer survival at a time when there is an urgent need to understand why some women respond differently to treatment and to find cost-effective therapies for the disease.
Epigenetics is a promising avenue of investigation because epigenetic shifts, such as DNA methylation, that impact the genes responsible for cell behavior and DNA damage and repair are known to contribute to and exacerbate cancer. These epigenetic signatures could act as biomarkers for risk in cancer and also aid with more effective treatment approaches. For example, aspirin is known to affect DNA methylation at certain sites in colon cancer, hence this study’s hypothesis that pre–cancer diagnosis aspirin use would interact with epigenetic signatures and influence breast cancer outcomes.
Kristen M. C. Malecki, PhD, is from the department of population health sciences in the School of Medicine and Public Health at the University of Wisconsin, Madison. The comments are adapted from an accompanying editorial (Cancer. 2019 Aug 12. doi: 10.1002/cncr.32365). Dr. Malecki declared support from the National Institutes of Health, National Institute for Environmental Health Sciences Breast Cancer, and the Environment Research Program.
This study offers new insights into the intersection of epigenetics, prediagnosis aspirin use, and breast cancer survival at a time when there is an urgent need to understand why some women respond differently to treatment and to find cost-effective therapies for the disease.
Epigenetics is a promising avenue of investigation because epigenetic shifts, such as DNA methylation, that impact the genes responsible for cell behavior and DNA damage and repair are known to contribute to and exacerbate cancer. These epigenetic signatures could act as biomarkers for risk in cancer and also aid with more effective treatment approaches. For example, aspirin is known to affect DNA methylation at certain sites in colon cancer, hence this study’s hypothesis that pre–cancer diagnosis aspirin use would interact with epigenetic signatures and influence breast cancer outcomes.
Kristen M. C. Malecki, PhD, is from the department of population health sciences in the School of Medicine and Public Health at the University of Wisconsin, Madison. The comments are adapted from an accompanying editorial (Cancer. 2019 Aug 12. doi: 10.1002/cncr.32365). Dr. Malecki declared support from the National Institutes of Health, National Institute for Environmental Health Sciences Breast Cancer, and the Environment Research Program.
The impact of prediagnosis aspirin use on mortality in women with breast cancer is significantly tied to epigenetic changes in certain breast cancer-related genes, investigators reported.
While studies have shown aspirin reduces the risk of breast cancer development, there is limited and inconsistent data on the effect of aspirin on prognosis and mortality after a diagnosis of breast cancer, Tengteng Wang, PhD, from the department of epidemiology at the University of North Carolina at Chapel Hill and coauthors wrote in Cancer.
To address this, they analyzed data from 1,508 women who had a first diagnosis of primary breast cancer and were involved in the Long Island Breast Cancer Study Project; they then looked at the women’s methylation status, which is a mechanism of epigenetic change.
Around one in five participants reported ever using aspirin, and the analysis showed that ever use of aspirin was associated with an overall 13% decrease in breast cancer–specific mortality.
However researchers saw significant interactions between aspirin use and LINE-1 methylation status – which is a marker of methylation of genetic elements that play key roles in maintaining genomic stability – and breast cancer–specific genes.
They found that aspirin use in women with LINE-1 hypomethylation was associated with a risk of breast cancer–specific mortality that was 45% higher than that of nonusers (P = .05).
Compared with nonusers, aspirin users with methylated tumor BRCA1 promoter had significant 16% higher breast cancer mortality (P = .04) and 67% higher all-cause mortality (P = .02). However the study showed aspirin did not affect mortality in women with unmethylated BRCA1 promoter.
Among women with the PR breast cancer gene, aspirin use by those with methylation of the PR promoter was associated with a 63% higher breast cancer–specific mortality, but methylation showed no statistically significant effect on all-cause mortality, compared with nonusers.
The study found no significant change when they restricted the analysis to receptor-positive or invasive breast cancer, and the associations remained consistent even after adjusting for global methylation.
“Our findings suggest that the association between aspirin use and mortality after breast cancer may depend on methylation profiles and warrant further investigation,” the authors wrote. “These findings, if confirmed, may provide new biological insights into the association between aspirin use and breast cancer prognosis, may affect clinical decision making by identifying a subgroup of patients with breast cancer using epigenetic markers for whom prediagnosis aspirin use affects subsequent mortality, and may help refine risk-reduction strategies to improve survival among women with breast cancer.”
The study was partly supported by the National Institutes of Health. One author declared personal fees from the private sector outside the submitted work.
SOURCE: Wang T et al. Cancer. 2019 Aug 12. doi: 10.1002/cncr.32364.
The impact of prediagnosis aspirin use on mortality in women with breast cancer is significantly tied to epigenetic changes in certain breast cancer-related genes, investigators reported.
While studies have shown aspirin reduces the risk of breast cancer development, there is limited and inconsistent data on the effect of aspirin on prognosis and mortality after a diagnosis of breast cancer, Tengteng Wang, PhD, from the department of epidemiology at the University of North Carolina at Chapel Hill and coauthors wrote in Cancer.
To address this, they analyzed data from 1,508 women who had a first diagnosis of primary breast cancer and were involved in the Long Island Breast Cancer Study Project; they then looked at the women’s methylation status, which is a mechanism of epigenetic change.
Around one in five participants reported ever using aspirin, and the analysis showed that ever use of aspirin was associated with an overall 13% decrease in breast cancer–specific mortality.
However researchers saw significant interactions between aspirin use and LINE-1 methylation status – which is a marker of methylation of genetic elements that play key roles in maintaining genomic stability – and breast cancer–specific genes.
They found that aspirin use in women with LINE-1 hypomethylation was associated with a risk of breast cancer–specific mortality that was 45% higher than that of nonusers (P = .05).
Compared with nonusers, aspirin users with methylated tumor BRCA1 promoter had significant 16% higher breast cancer mortality (P = .04) and 67% higher all-cause mortality (P = .02). However the study showed aspirin did not affect mortality in women with unmethylated BRCA1 promoter.
Among women with the PR breast cancer gene, aspirin use by those with methylation of the PR promoter was associated with a 63% higher breast cancer–specific mortality, but methylation showed no statistically significant effect on all-cause mortality, compared with nonusers.
The study found no significant change when they restricted the analysis to receptor-positive or invasive breast cancer, and the associations remained consistent even after adjusting for global methylation.
“Our findings suggest that the association between aspirin use and mortality after breast cancer may depend on methylation profiles and warrant further investigation,” the authors wrote. “These findings, if confirmed, may provide new biological insights into the association between aspirin use and breast cancer prognosis, may affect clinical decision making by identifying a subgroup of patients with breast cancer using epigenetic markers for whom prediagnosis aspirin use affects subsequent mortality, and may help refine risk-reduction strategies to improve survival among women with breast cancer.”
The study was partly supported by the National Institutes of Health. One author declared personal fees from the private sector outside the submitted work.
SOURCE: Wang T et al. Cancer. 2019 Aug 12. doi: 10.1002/cncr.32364.
FROM CANCER
Quarterly intravenous eptinezumab prevents migraine
PHILADELPHIA – An intravenous formulation of a calcitonin gene–related peptide inhibitor monoclonal antibody showed efficacy for preventing chronic migraine headaches for 3 months in a dose-ranging, phase 3 trial with 1,072 patients.
In a separate study with 669 patients, a single IV dose of the antibody, eptinezumab, also significantly reduced the incidence of episodic migraine headaches during 3 months of follow-up, compared with placebo. And in both the chronic and episodic migraine studies a similar 3-month effect resulted from a second IV dose of the humanized antibody that binds the calcitonin gene–related peptide (CGRP) ligand, thereby blocking the pathway, Laszlo L. Mechtler, MD, and his associates reported in a poster at the annual meeting of the American Headache Society.
Eptinezumab follows the therapeutic approach already used by three Food and Drug Administration–approved monoclonal antibody drugs that cut migraine headache recurrences by blocking the CGRP pathway by binding either the peptide ligand or its receptor: erenumab-aooe (Aimovig), fremanezumab-vfrm (Ajovy), and galcanezumab-gnlm (Emgality). Eptinezumab differs from the three approved CGRP antibodies by using an IV route of administration – the other three are delivered by subcutaneous injection – and by a 3-month dosing interval. Both erenumab-aooe and galcanezumab-gnlm are labeled for monthly administration only, while fremanezumab-vfrm is labeled for both monthly and once every 3 months dosing schedules.
The PROMISE-1 (A Multicenter Assessment of ALD403 in Frequent Episodic Migraine) trial randomized 669 patients with episodic migraine (defined as 4-14 headache days/month with at least 4 classifiable as migraine headache days) at 87 centers mostly in the United States and with some in Georgia. The PROMISE-2 (Evaluation of ALD403 (Eptinezumab) in the Prevention of Chronic Migraine) trial randomized 1,072 patients with chronic migraine (defined as a history of 15-26 headache days/month and with at least 8 of the days involving a migraine headache) at any of 145 study sites, many in the United States, in several countries.
In PROMISE-1, patients could receive as many as four serial infusions every 3 months, and up to two serial infusions in PROMISE-2, but the primary endpoint in both studies was the change in monthly migraine count from baseline during the 3 months following the first dosage.
Among patients with chronic migraine in PROMISE-2, the average monthly migraine number fell by 8.2 migraine days/month, compared with an average 5.6 monthly migraine days drop from baseline among placebo patients, which was a statistically significant difference for the higher dosage of eptinezumab tested, 300 mg. A 100-mg dose linked with an average 7.7 migraine days/month reduction, also a statistically significant difference from the placebo patients, reported Dr. Mechtler, professor of neurology at the State University of New York at Buffalo and medical director of the Dent Neurologic Institute in Buffalo, and his associates.
Among patients with episodic migraine in PROMISE-1, the 300-mg dosage cut monthly migraines by an average 4.3 migraine headache days/month, compared with 3.2 in the placebo group, a statistically significant difference. Among patients who received the 100-mg dosage, the average cut was 3.9 migraine headache days/month, also a statistically significant difference from the placebo controls.
The researchers included no safety findings in their report, but in an interview Dr. Mechtler said that eptinezumab showed an excellent safety profile that was consistent with what’s been previously reported for the approved agents from this class. He cited the safety of the drugs in the class as a major feature of their clinical utility.
PROMISE-1 and PROMISE-2 were sponsored by Alder BioPharmaceuticals, the company developing eptinezumab. Dr. Mechtler has been a speaker on behalf of Allergan, Amgen/Novartis, Boston Biomedical, Promius, Avanir, and Teva, and he has received research funding from Allergan, Autonomic Technologies, Boston Biomedical, and Teva.
SOURCE: Mechtler LL et al. Headache. 2019 June;59[S1]:34, Abstract P12
PHILADELPHIA – An intravenous formulation of a calcitonin gene–related peptide inhibitor monoclonal antibody showed efficacy for preventing chronic migraine headaches for 3 months in a dose-ranging, phase 3 trial with 1,072 patients.
In a separate study with 669 patients, a single IV dose of the antibody, eptinezumab, also significantly reduced the incidence of episodic migraine headaches during 3 months of follow-up, compared with placebo. And in both the chronic and episodic migraine studies a similar 3-month effect resulted from a second IV dose of the humanized antibody that binds the calcitonin gene–related peptide (CGRP) ligand, thereby blocking the pathway, Laszlo L. Mechtler, MD, and his associates reported in a poster at the annual meeting of the American Headache Society.
Eptinezumab follows the therapeutic approach already used by three Food and Drug Administration–approved monoclonal antibody drugs that cut migraine headache recurrences by blocking the CGRP pathway by binding either the peptide ligand or its receptor: erenumab-aooe (Aimovig), fremanezumab-vfrm (Ajovy), and galcanezumab-gnlm (Emgality). Eptinezumab differs from the three approved CGRP antibodies by using an IV route of administration – the other three are delivered by subcutaneous injection – and by a 3-month dosing interval. Both erenumab-aooe and galcanezumab-gnlm are labeled for monthly administration only, while fremanezumab-vfrm is labeled for both monthly and once every 3 months dosing schedules.
The PROMISE-1 (A Multicenter Assessment of ALD403 in Frequent Episodic Migraine) trial randomized 669 patients with episodic migraine (defined as 4-14 headache days/month with at least 4 classifiable as migraine headache days) at 87 centers mostly in the United States and with some in Georgia. The PROMISE-2 (Evaluation of ALD403 (Eptinezumab) in the Prevention of Chronic Migraine) trial randomized 1,072 patients with chronic migraine (defined as a history of 15-26 headache days/month and with at least 8 of the days involving a migraine headache) at any of 145 study sites, many in the United States, in several countries.
In PROMISE-1, patients could receive as many as four serial infusions every 3 months, and up to two serial infusions in PROMISE-2, but the primary endpoint in both studies was the change in monthly migraine count from baseline during the 3 months following the first dosage.
Among patients with chronic migraine in PROMISE-2, the average monthly migraine number fell by 8.2 migraine days/month, compared with an average 5.6 monthly migraine days drop from baseline among placebo patients, which was a statistically significant difference for the higher dosage of eptinezumab tested, 300 mg. A 100-mg dose linked with an average 7.7 migraine days/month reduction, also a statistically significant difference from the placebo patients, reported Dr. Mechtler, professor of neurology at the State University of New York at Buffalo and medical director of the Dent Neurologic Institute in Buffalo, and his associates.
Among patients with episodic migraine in PROMISE-1, the 300-mg dosage cut monthly migraines by an average 4.3 migraine headache days/month, compared with 3.2 in the placebo group, a statistically significant difference. Among patients who received the 100-mg dosage, the average cut was 3.9 migraine headache days/month, also a statistically significant difference from the placebo controls.
The researchers included no safety findings in their report, but in an interview Dr. Mechtler said that eptinezumab showed an excellent safety profile that was consistent with what’s been previously reported for the approved agents from this class. He cited the safety of the drugs in the class as a major feature of their clinical utility.
PROMISE-1 and PROMISE-2 were sponsored by Alder BioPharmaceuticals, the company developing eptinezumab. Dr. Mechtler has been a speaker on behalf of Allergan, Amgen/Novartis, Boston Biomedical, Promius, Avanir, and Teva, and he has received research funding from Allergan, Autonomic Technologies, Boston Biomedical, and Teva.
SOURCE: Mechtler LL et al. Headache. 2019 June;59[S1]:34, Abstract P12
PHILADELPHIA – An intravenous formulation of a calcitonin gene–related peptide inhibitor monoclonal antibody showed efficacy for preventing chronic migraine headaches for 3 months in a dose-ranging, phase 3 trial with 1,072 patients.
In a separate study with 669 patients, a single IV dose of the antibody, eptinezumab, also significantly reduced the incidence of episodic migraine headaches during 3 months of follow-up, compared with placebo. And in both the chronic and episodic migraine studies a similar 3-month effect resulted from a second IV dose of the humanized antibody that binds the calcitonin gene–related peptide (CGRP) ligand, thereby blocking the pathway, Laszlo L. Mechtler, MD, and his associates reported in a poster at the annual meeting of the American Headache Society.
Eptinezumab follows the therapeutic approach already used by three Food and Drug Administration–approved monoclonal antibody drugs that cut migraine headache recurrences by blocking the CGRP pathway by binding either the peptide ligand or its receptor: erenumab-aooe (Aimovig), fremanezumab-vfrm (Ajovy), and galcanezumab-gnlm (Emgality). Eptinezumab differs from the three approved CGRP antibodies by using an IV route of administration – the other three are delivered by subcutaneous injection – and by a 3-month dosing interval. Both erenumab-aooe and galcanezumab-gnlm are labeled for monthly administration only, while fremanezumab-vfrm is labeled for both monthly and once every 3 months dosing schedules.
The PROMISE-1 (A Multicenter Assessment of ALD403 in Frequent Episodic Migraine) trial randomized 669 patients with episodic migraine (defined as 4-14 headache days/month with at least 4 classifiable as migraine headache days) at 87 centers mostly in the United States and with some in Georgia. The PROMISE-2 (Evaluation of ALD403 (Eptinezumab) in the Prevention of Chronic Migraine) trial randomized 1,072 patients with chronic migraine (defined as a history of 15-26 headache days/month and with at least 8 of the days involving a migraine headache) at any of 145 study sites, many in the United States, in several countries.
In PROMISE-1, patients could receive as many as four serial infusions every 3 months, and up to two serial infusions in PROMISE-2, but the primary endpoint in both studies was the change in monthly migraine count from baseline during the 3 months following the first dosage.
Among patients with chronic migraine in PROMISE-2, the average monthly migraine number fell by 8.2 migraine days/month, compared with an average 5.6 monthly migraine days drop from baseline among placebo patients, which was a statistically significant difference for the higher dosage of eptinezumab tested, 300 mg. A 100-mg dose linked with an average 7.7 migraine days/month reduction, also a statistically significant difference from the placebo patients, reported Dr. Mechtler, professor of neurology at the State University of New York at Buffalo and medical director of the Dent Neurologic Institute in Buffalo, and his associates.
Among patients with episodic migraine in PROMISE-1, the 300-mg dosage cut monthly migraines by an average 4.3 migraine headache days/month, compared with 3.2 in the placebo group, a statistically significant difference. Among patients who received the 100-mg dosage, the average cut was 3.9 migraine headache days/month, also a statistically significant difference from the placebo controls.
The researchers included no safety findings in their report, but in an interview Dr. Mechtler said that eptinezumab showed an excellent safety profile that was consistent with what’s been previously reported for the approved agents from this class. He cited the safety of the drugs in the class as a major feature of their clinical utility.
PROMISE-1 and PROMISE-2 were sponsored by Alder BioPharmaceuticals, the company developing eptinezumab. Dr. Mechtler has been a speaker on behalf of Allergan, Amgen/Novartis, Boston Biomedical, Promius, Avanir, and Teva, and he has received research funding from Allergan, Autonomic Technologies, Boston Biomedical, and Teva.
SOURCE: Mechtler LL et al. Headache. 2019 June;59[S1]:34, Abstract P12
REPORTING FROM AHS 2019

Hospital slashes S. aureus vancomycin resistance
LJUBLJANA, SLOVENIA – , Johannes Huebner, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
He presented a retrospective analysis of S. aureus isolates obtained from 540 patients at the Dr. von Hauner Children’s Hospital, Munich, from 2002 to 2017. All were either newly identified methicillin-resistant S. aureus (MRSA) or specimens from bacteremic children with invasive MRSA or methicillin-sensitive S. aureus (MSSA). The strains were tested for vancomycin resistance and minimum inhibitory concentration (MIC). The results from the 200 isolates obtained from 2002 to 2009 were then compared to the 340 specimens from 2010 to 2017, when antibiotic stewardship programs rose to the fore at the pediatric hospital.
All samples proved to be vancomycin sensitive. The further good news was there was absolutely no evidence of the worrisome vancomycin MIC creep that has been described at some centers. On the contrary, the MIC was significantly lower in the later samples, at 0.99 mcg/mL, compared with 1.11 mcg/mL in the earlier period. Moreover, the prevalence of heterogeneous glycopeptide-intermediate S. aureus (hGISA) – a phenotype that has been associated with increased rates of treatment failure – improved from 25% in the earlier period to 6% during the later period, reported Dr. Huebner, head of the division of pediatric infectious diseases at the children’s hospital, part of the University of Munich.
Vancomycin MICs weren’t significantly different between the MRSA and MSSA samples.
Based upon this favorable institutional experience, vancomycin remains the first-line treatment for suspected severe gram-positive cocci infections as well as proven infections involving MRSA at Dr. von Hauner Children’s Hospital.
These vancomycin MIC and hGISA data underscore the importance of periodically monitoring local S. aureus antimicrobial susceptibilities, which, as in this case, can differ from the broader global trends. The vancomycin MIC creep issue hadn’t been studied previously in German hospitals, according to Dr. Huebner.
He and his coworkers have published details of the elements of pediatric antibiotic stewardship programs they have found to be most effective (Infection. 2017 Aug;45[4]:493-504) as well as a systematic review of studies on the favorable economic impact of such programs (J Hosp Infect. 2019 Aug;102[4]:369-376).
Dr. Huebner reported having no financial conflicts regarding his study, which was conducted free of commercial support.
LJUBLJANA, SLOVENIA – , Johannes Huebner, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
He presented a retrospective analysis of S. aureus isolates obtained from 540 patients at the Dr. von Hauner Children’s Hospital, Munich, from 2002 to 2017. All were either newly identified methicillin-resistant S. aureus (MRSA) or specimens from bacteremic children with invasive MRSA or methicillin-sensitive S. aureus (MSSA). The strains were tested for vancomycin resistance and minimum inhibitory concentration (MIC). The results from the 200 isolates obtained from 2002 to 2009 were then compared to the 340 specimens from 2010 to 2017, when antibiotic stewardship programs rose to the fore at the pediatric hospital.
All samples proved to be vancomycin sensitive. The further good news was there was absolutely no evidence of the worrisome vancomycin MIC creep that has been described at some centers. On the contrary, the MIC was significantly lower in the later samples, at 0.99 mcg/mL, compared with 1.11 mcg/mL in the earlier period. Moreover, the prevalence of heterogeneous glycopeptide-intermediate S. aureus (hGISA) – a phenotype that has been associated with increased rates of treatment failure – improved from 25% in the earlier period to 6% during the later period, reported Dr. Huebner, head of the division of pediatric infectious diseases at the children’s hospital, part of the University of Munich.
Vancomycin MICs weren’t significantly different between the MRSA and MSSA samples.
Based upon this favorable institutional experience, vancomycin remains the first-line treatment for suspected severe gram-positive cocci infections as well as proven infections involving MRSA at Dr. von Hauner Children’s Hospital.
These vancomycin MIC and hGISA data underscore the importance of periodically monitoring local S. aureus antimicrobial susceptibilities, which, as in this case, can differ from the broader global trends. The vancomycin MIC creep issue hadn’t been studied previously in German hospitals, according to Dr. Huebner.
He and his coworkers have published details of the elements of pediatric antibiotic stewardship programs they have found to be most effective (Infection. 2017 Aug;45[4]:493-504) as well as a systematic review of studies on the favorable economic impact of such programs (J Hosp Infect. 2019 Aug;102[4]:369-376).
Dr. Huebner reported having no financial conflicts regarding his study, which was conducted free of commercial support.
LJUBLJANA, SLOVENIA – , Johannes Huebner, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
He presented a retrospective analysis of S. aureus isolates obtained from 540 patients at the Dr. von Hauner Children’s Hospital, Munich, from 2002 to 2017. All were either newly identified methicillin-resistant S. aureus (MRSA) or specimens from bacteremic children with invasive MRSA or methicillin-sensitive S. aureus (MSSA). The strains were tested for vancomycin resistance and minimum inhibitory concentration (MIC). The results from the 200 isolates obtained from 2002 to 2009 were then compared to the 340 specimens from 2010 to 2017, when antibiotic stewardship programs rose to the fore at the pediatric hospital.
All samples proved to be vancomycin sensitive. The further good news was there was absolutely no evidence of the worrisome vancomycin MIC creep that has been described at some centers. On the contrary, the MIC was significantly lower in the later samples, at 0.99 mcg/mL, compared with 1.11 mcg/mL in the earlier period. Moreover, the prevalence of heterogeneous glycopeptide-intermediate S. aureus (hGISA) – a phenotype that has been associated with increased rates of treatment failure – improved from 25% in the earlier period to 6% during the later period, reported Dr. Huebner, head of the division of pediatric infectious diseases at the children’s hospital, part of the University of Munich.
Vancomycin MICs weren’t significantly different between the MRSA and MSSA samples.
Based upon this favorable institutional experience, vancomycin remains the first-line treatment for suspected severe gram-positive cocci infections as well as proven infections involving MRSA at Dr. von Hauner Children’s Hospital.
These vancomycin MIC and hGISA data underscore the importance of periodically monitoring local S. aureus antimicrobial susceptibilities, which, as in this case, can differ from the broader global trends. The vancomycin MIC creep issue hadn’t been studied previously in German hospitals, according to Dr. Huebner.
He and his coworkers have published details of the elements of pediatric antibiotic stewardship programs they have found to be most effective (Infection. 2017 Aug;45[4]:493-504) as well as a systematic review of studies on the favorable economic impact of such programs (J Hosp Infect. 2019 Aug;102[4]:369-376).
Dr. Huebner reported having no financial conflicts regarding his study, which was conducted free of commercial support.
REPORTING FROM ESPID 2019
Key clinical point: Staphylococcus aureus vancomycin MIC creep is reversible through dedicated antimicrobial stewardship.
Major finding: The prevalence of hGISA in MRSA and MSSA specimens improved from 25% during 2002-2009 to 6% during 2010-2017 at one German tertiary children’s hospital.
Study details: This was a retrospective single-center analysis of vancomycin resistance trends over time in 540 S. aureus specimens gathered in 2002-2017.
Disclosures: The presenter reported having no financial conflicts regarding this study, which was conducted free of commercial support.
Rheumatoid Arthritis: Therapeutic Strategies After Inadequate Response to Initial TNF Inhibitor Therapy
From the University of Iowa Hospitals and Clinics, Iowa City, IA.
Abstract
- Objective: To discuss the variability in response to tumor necrosis factor inhibitors (TNFis) observed in patients with rheumatoid arthritis (RA) and discuss therapeutic options for patients who do not respond to initial TNFi therapy.
- Methods: Review of the literature.
- Results: Optimal treatment of RA aims at achieving and then maintaining remission or low disease activity. In a patient with an inadequate response to initial biologic therapy, several therapeutic options exist. Current evidence supports TNFi dose escalation for only infliximab; optimization of concurrent conventional synthetic disease-modifying antirheumatic drug (csDMARD) or switching to a different csDMARD are other options. Cycling (switching to an alternative TNFi) and swapping (switching to a therapy with a different mode of action) strategies are other alternate approaches supported by many observational studies. While no head-to-head trials exist directly comparing the 2 strategies, data suggest superiority of the swapping strategy over the cycling approach. Also, several studies have shown that switching to a drug with a different mechanism of action is associated with higher treatment persistence and lower health care costs than TNFi cycling.
- Conclusion: Physicians have a growing list of treatment options to help their patients with RA achieve disease remission. The choice of best treatment for a given patient needs to be individualized, keeping in mind other factors, including comorbidities.
Keywords: biologics; rheumatoid arthritis; swapping strategy; cycling strategy; TNF inhibitors.
Following the discovery of tumor necrosis factor (TNF) as a proinflammatory cytokine 30 years ago, the use of TNF antagonists has revolutionized the treatment of rheumatoid arthritis (RA). Although TNF inhibitors (TNFIs) are frequently used as a first-line biologic disease-modifying antirheumatic drug (bDMARD), they are not uniformly efficacious in achieving remission in all patients with RA. This article highlights the reasons for such variability in observed response and discusses therapeutic options for patients who do not respond to TNFi therapy.
Case Presentation
A 60-year-old woman is evaluated in the clinic for complaints of pain in her hands, morning stiffness lasting 2 hours, and swelling in her wrists, all of which have been ongoing for 3 months. Physical exam reveals evidence of active inflammation, with synovitis in her second, third, and fourth metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints bilaterally, swelling over both wrists, and a weak grip. Inflammatory markers are elevated, and rheumatoid factor and anti-cyclic citrullinated peptide (anti-CCP) are both positive at high titer. Radiographs reveal evidence of small erosions at the third and fourth MCPs and PIPs bilaterally and periarticular osteopenia. The patient is diagnosed with seropositive, erosive RA based on history, physical exam, laboratory studies, and imaging. She is started on 20 mg of prednisone for acute treatment of her symptoms along with methotrexate, and, initially, her symptoms are well controlled. A few months after starting treatment, she develops voluminous diarrhea that necessitates cessation of methotrexate. Leflunomide also causes similar symptoms. The combination of sulfasalazine and hydroxychloroquine does not adequately control her symptoms, and ongoing use of low-dose glucocorticoids is required to improve functionality in all joints. Using the treat-to-target (T2T) strategy, adalimumab is initiated. However, she continues to report persistent swelling and pain and still requests oral glucocorticoids to help decrease inflammation. The 28-joint Disease Activity Score (DAS28) is 4.8, suggestive of moderate disease activity.
Why are TNFi agents sometimes ineffective?
The introduction of monoclonal antibodies and fusion proteins to block TNF and other cytokines was a remarkable development in the treatment of RA that revolutionized patient care. Despite the efficacy of TNFis, clinical response to these agents is not universal and only some patients achieve complete remission. In targeting the eventual goal of remission or low disease activity in patients with RA, the concept of “TNF failure” becomes extremely relevant. These inadequate responses to anti-TNF therapy may be due to primary failures, or complete lack of clinical response after initiation of the bDMARD, and secondary failures, or the loss of initially achieved clinical response to therapy. Other reasons for discontinuation of a given TNFi include partial disease control and intolerance to the medication (possible injection-site or infusion reactions). Keystone and Kavanaugh1 divided causes of failure of TNF agents into 2 broad categories: perceptual (related to natural variations in disease course like hormonal variation and physical and emotional stress) and pathophysiological failures (genetic variations, high body mass index, concomitant cigarette use).
Another important consideration in patients treated with a TNFi is the consequent formation of anti-drug antibodies (ADAs). TNFi agents are immunogenic and normally elicit an immune response. The appearance of such ADAs may reduce the bioavailability of free drug, resulting in a decreased clinical response,2 or may lead to serious adverse effects.
How common is discontinuation of the first TNFi?
Several studies have reported that the prevalence of primary failure, secondary failure, and intolerance to TNFis ranges from 30% to 40%.3-6 Female sex,7 concurrent prednisone use,8 high disease activity scores,6,8,9 and the absence of treatment with low-dose methotrexate7,8 have all been shown to be negative predictors of bDMARD retention and response.10
Are there any factors that predict TNFi failure?
There are no specific parameters to accurately predict responses to TNFI therapy.11 Several clinical and molecular biomarkers in synovium (initial TNF levels, macrophages, T cells)12 and peripheral blood (serum myeloid-related protein 8 and 14 complex levels,13 prealbumin, platelet factor 4, and S100A12)14 have been described as predictors of clinical response to TNFis, but their utility in clinical practice has not been established and the use of these markers has not yet been incorporated into clinical guidelines.
How is disease activity measured in patients with RA?
In 2010 an international expert consensus panel published treatment recommendations for RA that emphasized a T2T strategy of individualizing and escalating treatment to achieve the lowest disease activity or remission. In clinical practice, numerous tools are available to measure RA disease activity. Herein, we mention several that are most commonly used in clinical practice.
DAS28 combines single activity measures into an overall continuous measure of disease activity and has been endorsed by both the American College of Rheumatology (ACR) and European League Against Rheumatism (EULAR). It includes a 28-swollen joint count (SJC), 28-tender joint count (TJC), erythrocyte sedimentation rate (ESR; can also be calculated using C-reactive protein [CRP]), and a patient global assessment (PtGA). The cut-offs used for DAS28 interpretation are as follows: remission (< 2.6), low (≥ 2.6 but ≤ 3.2), moderate (> 3.2 but ≤ 5.1), or high (> 5.1).15 Some of the difficulties in using DAS28 in daily clinical practice include the need for a lab value and the time needed to perform the joint counts. Note also that due to the inclusion of ESR, which is influenced by age and other factors, DAS28 may underestimate remission in the elderly.
Another measure of RA disease activity is the Simplified Disease Activity Index (SDAI), which includes 28 SJC, 28 TJC, PtGA, provider global assessment (PrGA), and CRP in mg/dL. The level of disease activity using the SDAI is interpreted as: remission (SDAI ≤ 3.3), low (≥ 3.4 but ≤ 11), moderate (> 11 but ≤ 26), or high (> 26). The advantage of the SDAI is that a calculator or computer is not required for calculations. Another measure, the Clinical Disease Activity Index (CDAI), includes a 28 SJC, 28 TJC, PtGA, and PrGA. Because a laboratory value is not needed to calculate the CDAI, it is well-suited for use in clinical practice. When using the CDAI, the level of disease activity can be defined as remission (CDAI ≤ 2.8), low (> 2.8 but ≤ 10), moderate (> 10 but ≤ 22), or high (> 22). Again, as with the SDAI, a calculator or computer is not needed for calculations.
What are the alternative treatment options after first biologic failure?
In patients who have failed treatment with an initial biologic, usually a TNFi, the treating rheumatologist has the following options (Figure), with the best treatment strategy being driven by individualized patient and disease-related factors (Table 1 and Table 2):
- TNFi dose escalation
- Trial of an alternate TNFi agent (the “cycling” strategy)
- Optimization of therapy conjoined with a conventional synthetic DMARD (csDMARD)
- Use of a non-TNF biologic or targeted synthetic DMARD (the “swapping” strategy)
. DMARD, disease-modifying antirheumatic drug; Jakinibs; Janus kinase inhibitors; TNF, tumor necrosis factor.")
If all the listed strategies fail, the next step can be the addition of short-term, low-dose glucocorticoid therapy.

TNFi Dose Escalation
The available data have demonstrated the safety, efficacy, and cost-effectiveness of dose escalation in patients with RA receiving infliximab.16-18 The ATTRACT trial first demonstrated this, with greater clinical and radiographic improvements in those with higher trough serum concentrations, suggesting that doses higher than 3 mg/kg or more frequent than every 8 weeks may be needed for full response in some patients.19

There is a lack of studies in RA patients to determine the most effective dose escalation strategy. A study in patients with Crohn disease showed that intensification to 10 mg/kg every 8 weeks (dose doubling) was at least as effective as 5 mg/kg every 4 weeks (halving interval) at 12 months.16 Due to greater patient and administration convenience of dose-doubling, this strategy may be preferred.17 A starting dose of 10 mg/kg every 8 weeks is not routinely recommended due to an increased risk of serious infection; these adverse events were not found when the dose was gradually increased, as clinically indicated, starting at 3 mg/kg.19,20 Further studies are needed to explore this approach in RA patients.
These results, however, have not been replicated with other TNFi agents. No significant clinical improvements were identified with etanercept 50 mg twice weekly,21 adalimumab 40 mg every week in the PREMIER trial,18 or certolizumab 400 mg every other week in an open-label extension phase of the RAPID 1 study.22 A Japanese study found significantly worse clinical outcomes with dose escalation of golimumab.23 Conversely, 2 studies found clinical benefits after escalating the tocilizumab dose, the first a real-world review from the Consortium of Rheumatology Researchers of North America (CORRONA) registry using the intravenous formulation,24 and the other the BREVACTA study utilizing subcutaneous tocilizumab.25 No studies to date have been published on dose escalation of abatacept in patients with RA who respond poorly. Overall, previous studies support dose escalation in individuals being treated with infliximab to improve clinical outcomes, but additional studies are needed for other bDMARDs.
Trial of an Alternate TNF Agent: The “Cycling” Strategy
Per the ACR/EULAR26,27 guidelines, all approved bDMARDs may be used without hierarchical positioning. However, after the failure of a TNFi agent, these guidelines do not provide specific advice about a preference between the “cycling” strategy (switching to an alternative TNFi) and “swapping” strategy (switching to a therapy with a different mode of action). Cycling might work for several reasons, including differences in the agents’ molecular structure, immunological mechanism of action, immunogenicity, and pharmacokinetics.28-30 The cycling strategy is a well-established approach adopted by more than 94% of practicing rheumatologists, according to a national survey,31 and its efficacy is supported by trials and additional observational studies.32-35
The greater clinical effectiveness of switching to infliximab compared with continuing with etanercept in patients with inadequate response to etanercept (n = 28) was suggested in the open-label OPPOSITE trial.36 Data from the GO-AFTER trial37 suggests that a greater proportion of patients with RA refractory to adalimumab, etanercept, or infliximab who were treated with golimumab achieved an ACR20 and ACR50 response compared with patients who received placebo, and this response persisted through 5 years.38 More recently, certolizumab pegol and adalimumab were compared head-to-head in the EXXELERATE trial.39 The results of this trial revealed the adequate efficacy of cycling to another TNFi after primary insufficient response to the first.
In studies from Finland and Sweden,35,40 it has been observed that a better response is achieved in patients in whom TNF failure was initially due to secondary failure or intolerance rather than primary failure. A post-hoc analysis of the results of the GO-AFTER trial41 and from a few observational studies35,40,42 revealed that switching from one TNFi to another, especially from a monoclonal antibody to a soluble receptor, was often more beneficial for RA patients than switching from a soluble receptor to a monoclonal antibody.
Optimization of Therapy Conjoined with csDMARDs
Methotrexate is one of the oldest and most effective csDMARDs available for the treatment of RA.43 The 2016 EULAR guidelines recommend the addition of methotrexate and/or other csDMARDs to potentiate the effect of bDMARDs.26 In the case of TNFi therapy, the observed synergistic effect between the monoclonal antibody and methotrexate may be explained by sustained suppression of ADA formation.44 In the TEMPO,45 PREMIER,18 and GO-BEFORE46 trials, the addition of methotrexate led to improved clinical and radiological outcomes in patients treated with etanercept, adalimumab, and golimumab,47 respectively. These findings were also demonstrated in several registries, where significant improvement in clinical response and retention rate of the TNFi agents was noted. Results have been replicated with non-TNFi bDMARDs, including abatacept48,49 and rituximab.50 Patients treated with interleukin (IL)-6 inhibitors in combination with methotrexate have shown significantly less radiographic progression compared to those treated with tocilizumab alone and those treated with monotherapy tocilizumab versus monotherapy methotrexate.51,52 Results possibly favor the use of IL-6 inhibitors alone in those who cannot tolerate or have contraindications to methotrexate.
An open prospective study by Cohen et al added methotrexate to the treatment regimens of individuals on bDMARD monotherapy with a primary failure and found favorable changes in ACR20 and DAS28 scores at 3 and 12 months and therapeutic biological response (ESR, CRP) at 3 months.53 Unlike monotherapy, in these situations methotrexate is known to be efficacious even at a lower dose, possibly at 7.5 mg to 10 mg per week. Some studies have shown that methotrexate administered parenterally may be more efficacious than when given orally.54-58
In clinical trials and observational studies, leflunomide, sulfasalazine, and hydroxychloroquine have been used as alternate csDMARDs added to the treatment regimen.59-62 There are, however, only 2 trials comparing the efficacy of methotrexate with that of other csDMARDs as concomitant treatment in patients with inadequate response to TNFi therapy. The RABBIT trial found a slight decrease in effectiveness with concomitant TNFi and leflunomide compared to TNFi/methotrexate, but overall each group had similar EULAR responses at 24 months.63 A study by De Stefano et al found comparable ACR20 and DAS28 responses among individuals receiving TNFis with methotrexate or leflunomide.61
The “Swapping” Strategy
The efficacy of the swapping strategy has been shown in 3 randomized clinical trials demonstrating the superiority of abatacept, tocilizumab, and rituximab in the treatment of individuals with RA refractory to TNFis. Tocilizumab was studied in the RADIATE64 trial, which involved 499 patients with inadequate response to 1 or more TNFi agents. The primary endpoint (24-week ACR20) was achieved by 50.0%, 30.4%, and 10.1% of patients in the 8 mg/kg, 4 mg/kg, and control groups, respectively (P < 0.001 for both tocilizumab groups versus placebo). The utility of abatacept as second-line therapy after initial TNF failure was evaluated in the ATTAIN65 study. Participants with an inadequate response to etanercept or infliximab were randomly assigned to receive either abatacept or placebo. ACR50 response rates after 6 months of treatment were 20.3% with abatacept and 3.8% with placebo (P < 0.001). The SWITCH-RA study,66 an observational study, compared rituximab to TNFis in 1112 participants with inadequate response to initial anti-TNF therapy. At 6 months, mean change in DAS28 was small but significantly greater for the rituximab group (–1.5 vs –1.1; P = 0.007). The difference in response rates was greatest among seropositive patients. These data suggest that rituximab has efficacy following TNFi failure, particularly for seropositive patients. Additionally, REFLEX67 is the sole randomized controlled trial in patients with insufficient response to TNFis that showed significant prevention of radiographic progression at week 56 in patients on rituximab compared to placebo (mean change from baseline in total Genant-modified Sharp score, 1.00 vs 2.31, respectively; P = 0.005).
One study randomly assigned 399 patients with active RA who had inadequate response to prior TNFi therapy to tofacitinib68 (5 mg twice daily or 10 mg twice daily) or placebo, both with methotrexate.6 After 3 months of treatment, ACR20 response rates (41.7% for 5 mg, 28.1% for 10 mg, 24.4% for placebo) and DAS28 remission rates (6.7% for 5 mg, 8.8% for 10 mg, 1.7% for placebo) were significantly greater among patients treated with tofacitinib compared to those treated with placebo. More recently, the RA-BEACON trial69 demonstrated a consistent, beneficial treatment effect of baricitinib in patients with insufficient response to 1 or more TNFis. In this trial, 527 patients with an inadequate response to bDMARDs were randomly assigned to receive baricitinib 2 mg or 4 mg daily or placebo for 24 weeks. A higher proportion of patients receiving baricitinib 4 mg had an ACR20 response at week 12 compared with those treated with placebo (55% vs 27%, P < 0.001), and patients receiving the 4-mg dose had significant improvements from baseline in DAS28 and Health Assessment Questionnaire–Disability Index scores (P < 0.001 for both comparisons).
To Cycle or to Swap?
Several observational studies (SCQM-RA,70 STURE,71 BSRBR,72 Favalli,43 MIRAR,73 SWITCH-RA,74 ROC72) have clearly demonstrated that the swapping strategy is favored over the cycling strategy. In the ROC study,72 patients were randomly assigned (based on physician discretion) to receive a non-TNF biologic or a TNFi. More patients in the non-TNF group than in the TNFi group showed low disease activity at week 24 (45% vs 28%; odds ratio [OR], 2.09; 95% confidence interval [CI], 1.27-3.43; P = 0.004) and at week 52 (41% vs 23%; OR, 2.26; 95% CI, 1.33-3.86; P = 0.003). The authors concluded that in patients having an insufficient response to TNFi therapy, a non-TNF biologic agent may be more effective than a second TNFi drug. Only a few studies75-77 have demonstrated similar results between the 2 strategies. Overall, the available evidence seems to suggest the superiority of the swapping over the cycling strategy.
An important clinical pearl to keep in mind is that both swapping and cycling strategies might theoretically increase the risk of infection; however, limited evidence is reported in the literature. In a large retrospective analysis78 of data on 4332 RA patients from a large US claims database, patients who had cycled between TNFi agents had a 30% to 40% increased risk of infection compared to patients treated with rituximab. Patients on infliximab had a 62% higher hazard of severe infections, and this has also been reported in an observational study.79 In another study,70 41% of 201 patients with RA followed between 1999 and 2013 who swapped to abatacept/rituximab or tocilizumab developed adverse events, as compared to 59% of those who switched to a second TNFi.
What are recent trends in the use of bDMARDs?
Currently, there are no specific guidelines or biomarkers available to facilitate selection of specific treatment from among the classes of biologics. With the development of several new drugs and regulatory approval of baricitinib, physicians now have several biologic options to treat patients. A recent large time-trend study80 deriving data from more than 200,000 patients with RA showed that etanercept remains the most frequently used agent for the treatment of RA; it also showed that the use of adalimumab and infliximab is decreasing, and that the use of newer agents, especially abatacept, golimumab, and certolizumab, has considerably risen in recent years. In this study, abatacept, rituximab, certolizumab, golimumab, tocilizumab, and tofacitinib accounted for 13.2%, 13.8%, 6.9%, 11.9%, and 7.5% switches from first TNFi therapy.
Jin et al81 studied factors associated with the choice of bDMARD for initial and subsequent use. They found that patients with commercial insurance had an 87% higher likelihood of initiating a bDMARD. In the Medicaid subgroup, African Americans had lower odds of initiating and switching bDMARDs than non-Hispanic whites. Prior use of steroids and nonbiologic DMARDs predicted both bDMARD initiation and subsequent switching. Etanercept, adalimumab, and infliximab were the most commonly used first- and second-line bDMARDS; patients on anakinra and golimumab were most likely to be switched to other bDMARDs.
Which treatment strategy is the most cost-effective?
Several studies have reported better treatment persistence rates among patients who are treated with the swapping strategy compared to the cycling strategy. In a retrospective analysis of claims data,82 the authors examined treatment persistence and health care costs in patients switching to biologics with a different mechanism of action or cycling to another TNFi. The mean cost was significantly lower among patients treated using the swapping strategy than among the TNFi cyclers, both for the total cost of care for RA and for the total cost of the targeted DMARDs in the first year after the change in therapy. The authors concluded that switching to a drug with a different mechanism of action is associated with higher treatment persistence and lower health care costs than TNFi cycling.
What about biosimilars?
Biosimilars are copies of already licensed biologics that are very similar to the biologics, but are made by different sponsors using independently derived cell lines and separately developed manufacturing processes.83 Regarding biosimilar use, EULAR26 states that biosimilar bDMARDs approved by the European Medicines Agency or US Food and Drug Administration have similar efficacy and safety as the originator bDMARDs, and recommends them as preferred agents if they are indeed appreciably cheaper than originator or other bDMARDs.
What are the novel treatment targets in RA?
New therapeutics for RA continue to be developed. One of the new agents is peficitinib (ASP015K), an oral, once-daily Janus kinase (Jak) inhibitor targeting Jak-1, Jak-2, and tyrosine kinase-2, with moderate selectivity for Jak-3. In a phase 2b trial, 100-mg and 150-mg doses of peficitinib achieved a statistically significant ACR20 response (48.3% and 56.3%) compared to placebo (29.4%) at 12 weeks.84
Given the benefit of targeting TNF-α and IL-17 in RA, a novel molecule (ABT-122) that targets both human TNF and IL-17 has been developed. Two phase 1 studies85 showed that dual neutralization of TNF and IL-17 with ABT-122 has characteristics acceptable for further exploration of therapeutic potential of this agent in TNF- and IL-17A–driven immune-mediated inflammatory diseases. Another novel drug is mavrilimumab, a human monoclonal antibody that targets granulocyte–macrophage colony-stimulating factor receptor α. A recent studyshowed that long-term treatment with mavrilimumab maintained response and was well-tolerated, with no increased incidence of treatment-emergent adverse events.86
Namilumab (AMG203) is an immunoglobulin G1 monoclonal antibody that binds with high affinity to the GM-CSF ligand. In a phase 1b, randomized, double-blind study (PRIORA)87 to assess namilumab in treating active, mild-to-moderate RA, significant improvement was seen in the DAS28-CRP score with namilumab (150 and 300 mg groups combined) compared with placebo at day 43 (P = 0.0117) and also 8 weeks after last dosing at day 99 (P = 0.0154). Adverse events were similar across different doses of namilumab and placebo, and included nasopharyngitis and exacerbation/worsening of RA. Another drug showing promise in RA is fosdagrocorat (PF-04171327), a potential dissociated agonist of the glucocorticoid receptor. A multicenter, double-blind, parallel-group, active- and placebo-controlled phase 2 study randomly assigned 86 patients to receive fosdagrocorat 10 mg, fosdagrocorat 25 mg, prednisone 5 mg, or placebo, all with stable background methotrexate therapy.88 Both fosdagrocorat doses demonstrated efficacy in improving signs and symptoms in RA patients, with manageable adverse events.
Case Conclusion
There are several available treatment options for the case patient. Based on the PREMIER trial, solely increasing the dose of adalimumab is unlikely to provide a therapeutic benefit. Adding low-dose methotrexate (possibly via a parenteral route because of patient-reported gastrointestinal discomfort) might provide some synergistic and therapeutic effect. However, because of primary failure with TNFi therapy, she may benefit from the initiation of a biologic with a different mechanism of action (ie, swapping strategy). Therapeutic options include tocilizumab, abatacept, rituximab, and the Jak inhibitors (tofacitinib and baricitinib).
Summary
The optimal treatment of RA aims at achieving, and then maintaining, remission or a low disease activity. The choice of best treatment must be individualized to the patient, keeping in mind other factors, including comorbidities like fibromyalgia, history of diverticulitis (prior to use of tocilizumab), history of chronic obstructive pulmonary disease (prior to the use of abatacept), malignancy, and the presence of risk factors for infections (age, diabetes, chronic bronchitis). In a patient with inadequate response to initial biologic therapy, several options exist for the rheumatologist. Current evidence supports TNFi dose escalation for only infliximab; optimization of concurrent csDMARD or switching to a different csDMARD are other options. Cycling and swapping are other alternate approaches supported by many observational studies. While no head-to-head trials exist comparing the 2 strategies, data suggest superiority of the swapping strategy over the cycling approach. With the continuing development of novel therapeutics in RA, physicians have a growing list of treatment options to help their patients achieve disease remission.
Corresponding author: Namrata Singh, MD, 200 Hawkins Drive, Iowa City, IA 52242.
Financial disclosures: None.
1. Keystone ED, Kavanaugh KA. What to do with TNF failures. Expert Opin Drug Saf. 2005;4:149-155.
2. Kalden JR, Schulze-Koops H. Immunogenicity and loss of response to TNF inhibitors: implications for rheumatoid arthritis treatment. Nat Rev Rheumatol. 2017;13:707-718.
3. Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340:253-259.
4. Keystone EC, Kavanaugh AF, Sharp JT, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004;50(5):1400-1411.
5. Lipsky PE, van der Heijde DM, St Clair EW, et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000;343:1594-1602.
6. Finckh A, Simard JF, Gabay C, et al. Evidence for differential acquired drug resistance to anti-tumour necrosis factor agents in rheumatoid arthritis. Ann Rheum Dis. 2006;65:746-752.
7. Souto A, Maneiro JR, Gomez-Reino JJ. Rate of discontinuation and drug survival of biologic therapies in rheumatoid arthritis: a systematic review and meta-analysis of drug registries and health care databases. Rheumatology. 2016;55:523-534.
8. Hetland ML, Christensen IJ, Tarp U, et al. Direct comparison of treatment responses, remission rates, and drug adherence in patients with rheumatoid arthritis treated with adalimumab, etanercept, or infliximab: results from eight years of surveillance of clinical practice in the nationwide Danish DANBIO registry. Arthritis Rheum. 2010;62:22-32.
9. Gabay C, Riek M, Scherer A, et al. Effectiveness of biologic DMARDs in monotherapy versus in combination with synthetic DMARDs in rheumatoid arthritis: data from the Swiss Clinical Quality Management Registry. Rheumatology. 2015;54(9):1664-1672.
10. Ebina K, Hashimoto M, Yamamoto W, et al. Drug retention and discontinuation reasons between seven biologics in patients with rheumatoid arthritis-The ANSWER cohort study. PloS One. 2018;13:e0194130.
11. Wijbrandts CA, Tak PP. Prediction of response to targeted treatment in rheumatoid arthritis. Mayo Clin Proc. 2017;92:1129-1143.
12. Ulfgren AK, Andersson U, Engstrom M, et al. Systemic anti-tumor necrosis factor alpha therapy in rheumatoid arthritis down-regulates synovial tumor necrosis factor alpha synthesis. Arthritis Rheum. 2000;43:2391-2396.
13. Choi IY, Gerlag DM, Herenius MJ, et al. MRP8/14 serum levels as a strong predictor of response to biological treatments in patients with rheumatoid arthritis. Ann Rheum Dis. 2015;74:499-505.
14. Nguyen MVC, Baillet A, Romand X, et al. Prealbumin, platelet factor 4 and S100A12 combination at baseline predicts good response to TNF alpha inhibitors in rheumatoid arthritis. Joint Bone Spine. 2019;86:195-201.
15. Anderson JK, Zimmerman L, Caplan L, Michaud K. Measures of rheumatoid arthritis disease activity: Patient (PtGA) and Provider (PrGA) Global Assessment of Disease Activity, Disease Activity Score (DAS) and Disease Activity Score with 28-Joint Counts (DAS28), Simplified Disease Activity Index (SDAI), Clinical Disease Activity Index (CDAI), Patient Activity Score (PAS) and Patient Activity Score-II (PASII), Routine Assessment of Patient Index Data (RAPID), Rheumatoid Arthritis Disease Activity Index (RADAI) and Rheumatoid Arthritis Disease Activity Index-5 (RADAI-5), Chronic Arthritis Systemic Index (CASI), Patient-Based Disease Activity Score With ESR (PDAS1) and Patient-Based Disease Activity Score without ESR (PDAS2), and Mean Overall Index for Rheumatoid Arthritis (MOI-RA). Arthritis Care Res. 2011;63(suppl 11):S14-S36.
16. Katz L, Gisbert JP, Manoogian B, et al. Doubling the infliximab dose versus halving the infusion intervals in Crohn’s disease patients with loss of response. Inflamm Bowel Dis. 2012;18:2026-2033.
17. Durez P, Van den Bosch F, Corluy L, et al. A dose adjustment in patients with rheumatoid arthritis not optimally responding to a standard dose of infliximab of 3 mg/kg every 8 weeks can be effective: a Belgian prospective study. Rheumatology. 2005;44:465-468.
18. Breedveld FC, Weisman MH, Kavanaugh AF, et al. The PREMIER study: A multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54:26-37.
19. St Clair EW, Wagner CL, Fasanmade AA, et al. The relationship of serum infliximab concentrations to clinical improvement in rheumatoid arthritis: results from ATTRACT, a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46:1451-1459.
20. Rahman MU, Strusberg I, Geusens P, et al. Double-blinded infliximab dose escalation in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66:1233-1238.
21. Weinblatt ME, Schiff MH, Ruderman EM, et al. Efficacy and safety of etanercept 50 mg twice a week in patients with rheumatoid arthritis who had a suboptimal response to etanercept 50 mg once a week: results of a multicenter, randomized, double-blind, active drug-controlled study. Arthritis Rheum. 2008;58:1921-1930.
22. Curtis JR, Chen L, Luijtens K, et al. Dose escalation of certolizumab pegol from 200 mg to 400 mg every other week provides no additional efficacy in rheumatoid arthritis: an analysis of individual patient-level data. Arthritis Rheum. 2011;63:2203-2208.
23. Okazaki M, Kobayashi H, Ishii Y, et al. Real-world treatment patterns for golimumab and concomitant medications in Japanese rheumatoid arthritis patients. Rheumatol Ther. 2018;5:185-201.
24. Pappas DA, John A, Curtis JR, et al. Dosing of intravenous tocilizumab in a real-world setting of rheumatoid arthritis: analyses from the Corrona Registry. Rheumatol Ther. 2016;3:103-115.
25. Kivitz A, Olech E, Borofsky MA, et al. Two-year efficacy and safety of subcutaneous tocilizumab in combination with disease-modifying antirheumatic drugs including escalation to weekly dosing in rheumatoid arthritis. J Rheumatol. 2018;45:456-464.
26. Smolen JS, Landewe R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76:960-977.
27. Singh JA, Saag KG, Bridges SL Jr, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res. 2016;68:1-25.
28. Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006;11:81-88.
29. Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:493-507.
30. Navarro Coy NC, Brown S, Bosworth A, et al. The ‘Switch’ study protocol: a randomised-controlled trial of switching to an alternative tumour-necrosis factor (TNF)-inhibitor drug or abatacept or rituximab in patients with rheumatoid arthritis who have failed an initial TNF-inhibitor drug. BMC Musculoskelet Disord. 2014;15:452.
31. Kamal KM, Madhavan SS, Hornsby JA, et al. Use of tumor necrosis factor inhibitors in rheumatoid arthritis: a national survey of practicing United States rheumatologists. Joint Bone Spine. 2006;73:718-724.
32. Gomez-Reino JJ, Carmona L, BIOBADASER Group. Switching TNF antagonists in patients with chronic arthritis: an observational study of 488 patients over a four-year period. Arthritis Res Ther. 2006;8:R29.
33. Caporali R, Sarzi-Puttini P, Atzeni F, et al. Switching TNF-alpha antagonists in rheumatoid arthritis: The experience of the LORHEN registry. Autoimmun Rev. 2010;9:465-469.
34. Iannone F, Trotta F, Monteccuco C, et al. Etanercept maintains the clinical benefit achieved by infliximab in patients with rheumatoid arthritis who discontinued infliximab because of side effects. Ann Rheum Dis. 2007;66:249-252.
35. Virkki LM, Valleala H, Takakubo Y, et al. Outcomes of switching anti-TNF drugs in rheumatoid arthritis—a study based on observational data from the Finnish Register of Biological Treatment (ROB-FIN). Clin Rheumatol. 2011;30:1447-1454.
36. Furst DE, Gaylis N, Bray V, et al. Open-label, pilot protocol of patients with rheumatoid arthritis who switch to infliximab after an incomplete response to etanercept: the opposite study. Ann Rheum Dis. 2007;66:893-899.
37. Smolen JS, Kay J, Doyle MK, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor alpha inhibitors (GO-AFTER study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet. 2009;374:210-221.
38. Smolen JS, Kay J, Doyle M, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumor necrosis factor α inhibitors: findings with up to five years of treatment in the multicenter, randomized, double-blind, placebo-controlled, phase 3 GO-AFTER study. Arthritis Res Ther. 2015;17:14.
39. Smolen JS, Burmester G-R, Combe B, et al. Head-to-head comparison of certolizumab pegol versus adalimumab in rheumatoid arthritis: 2-year efficacy and safety results from the randomised EXXELERATE study. Lancet. 2016;388:2763-2774.
40. Chatzidionysiou K, Askling J, Eriksson J, et al. Effectiveness of TNF inhibitor switch in RA: results from the national Swedish register. Ann Rheum Dis. 2015;74:890.
41. Smolen JS, Kay J, Doyle M, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumor necrosis factor alpha inhibitors: findings with up to five years of treatment in the multicenter, randomized, double-blind, placebo-controlled, phase 3 GO-AFTER study. Arthritis Res Ther. 2015;17:14.
42. Lequerré T, Farran É, Ménard J-F, et al. Switching from an anti-TNF monoclonal antibody to soluble TNF-receptor yields better results than vice versa: An observational retrospective study of 72 rheumatoid arthritis switchers. Joint Bone Spine. 2015;82:330-337.
43. Favalli EG, Biggioggero M, Meroni PL. Methotrexate for the treatment of rheumatoid arthritis in the biologic era: Still an “anchor” drug? Autoimmun Rev. 2014;13:1102-1108.
44. Kalden JR, Schulze-Koops H. Immunogenicity and loss of response to TNF inhibitors: implications for rheumatoid arthritis treatment. Nat Rev Rheumatol. 2017;13:707-718.
45. Klareskog L, van der Heijde D, de Jager JP, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet. 2004;363:675-681.
46. Emery P, Fleischmann RM, Strusberg I, et al. Efficacy and safety of subcutaneous golimumab in methotrexate-naive patients with rheumatoid arthritis: five-year results of a randomized clinical trial. Arthritis Care Res. 2016;68:744-752.
47. Emery P, Fleischmann RM, Moreland LW, et al. Golimumab, a human anti-tumor necrosis factor alpha monoclonal antibody, injected subcutaneously every four weeks in methotrexate-naive patients with active rheumatoid arthritis: twenty-four-week results of a phase III, multicenter, randomized, double-blind, placebo-controlled study of golimumab before methotrexate as first-line therapy for early-onset rheumatoid arthritis. Arthritis Rheum. 2009;60:2272-2283.
48. Emery P, Burmester GR, Bykerk VP, et al. Evaluating drug-free remission with abatacept in early rheumatoid arthritis: results from the phase 3b, multicentre, randomised, active-controlled AVERT study of 24 months, with a 12-month, double-blind treatment period. Ann Rheum Dis. 2015;74:19-26.
49. Westhovens R, Robles M, Ximenes AC, et al. Clinical efficacy and safety of abatacept in methotrexate-naive patients with early rheumatoid arthritis and poor prognostic factors. Ann Rheum Dis. 2009;68:1870-1877.
50. Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793-2806.
51. Burmester GR, Rigby WF, van Vollenhoven RF, et al. Tocilizumab in early progressive rheumatoid arthritis: FUNCTION, a randomised controlled trial. Ann Rheum Dis. 2016;75:1081-1091.
52. Bijlsma JWJ, Welsing PMJ, Woodworth TG, et al. Early rheumatoid arthritis treated with tocilizumab, methotrexate, or their combination (U-Act-Early): a multicentre, randomised, double-blind, double-dummy, strategy trial. Lancet. 2016;388:343-355.
53. Cohen JD, Zaltni S, Kaiser MJ, et al. Secondary addition of methotrexate to partial responders to etanercept alone is effective in severe rheumatoid arthritis. Ann Rheum Dis. 2004;63:209-210.
54. Hamilton RA, Kremer JM. Why intramuscular methotrexate may be more efficacious than oral dosing in patients with rheumatoid arthritis. Br J Rheumatol. 1997;36:86-90.
55. Hoekstra M, Haagsma C, Neef C, et al. Bioavailability of higher dose methotrexate comparing oral and subcutaneous administration in patients with rheumatoid arthritis. J Rheumatol. 2004;31:645-648.
56. Herman RA, Veng-Pedersen P, Hoffman J, et al. Pharmacokinetics of low-dose methotrexate in rheumatoid arthritis patients. J Pharm Sci. 1989;78:165-171.
57. Schiff MH, Jaffe JS, Freundlich B. Head-to-head, randomised, crossover study of oral versus subcutaneous methotrexate in patients with rheumatoid arthritis: drug-exposure limitations of oral methotrexate at doses ± 15 mg may be overcome with subcutaneous administration. Ann Rheum Dis. 2014;73:1549-1551.
58. Hazlewood GS, Thorne JC, Pope JE, et al. The comparative effectiveness of oral versus subcutaneous methotrexate for the treatment of early rheumatoid arthritis. Ann Rheum Dis. 2016;75:1003-1008.
59. O’Dell JR, Petersen K, Leff R, et al. Etanercept in combination with sulfasalazine, hydroxychloroquine, or gold in the treatment of rheumatoid arthritis. J Rheumatol. 2006;33:213-218.
60. Finckh A, Dehler S, Gabay C. The effectiveness of leflunomide as a co-therapy of tumour necrosis factor inhibitors in rheumatoid arthritis: a population-based study. Ann Rheum Dis. 2009;68:33-39.
61. De Stefano R, Frati E, Nargi F, et al. Comparison of combination therapies in the treatment of rheumatoid arthritis: leflunomide-anti-TNF-alpha versus methotrexate-anti-TNF-alpha. Clin Rheumatol. 2010;29:517-524.
62. Combe B, Codreanu C, Fiocco U, et al. Etanercept and sulfasalazine, alone and combined, in patients with active rheumatoid arthritis despite receiving sulfasalazine: a double-blind comparison. Ann Rheum Dis. 2006;65:1357-1362.
63. Strangfeld A, Hierse F, Kekow J, et al. Comparative effectiveness of tumour necrosis factor α inhibitors in combination with either methotrexate or leflunomide. Ann Rheum Dis. 2009;68:1856.
64. Emery P, Keystone E, Tony HP, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67:1516.
65. Genovese MC, Becker J-C, Schiff M, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor α inhibition. N Engl J Med. 2005;353:1114-1123.
66. Emery P, Gottenberg JE, Rubbert-Roth A, et al. Rituximab versus an alternative TNF inhibitor in patients with rheumatoid arthritis who failed to respond to a single previous TNF inhibitor: SWITCH-RA, a global, observational, comparative effectiveness study. Ann Rheum Dis. 2015;74:979-984.
67. Keystone E, Emery P, Peterfy CG, et al. Rituximab inhibits structural joint damage in patients with rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitor therapies. Ann Rheum Dis. 2009;68:216.
68. Burmester GR, Blanco R, Charles-Schoeman C, et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381:451-460.
69. Genovese MC, Kremer J, Zamani O, et al. Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med. 2016;374:1243-1252.
70. Favalli EG, Biggioggero M, Marchesoni A, Meroni PL. Survival on treatment with second-line biologic therapy: a cohort study comparing cycling and swap strategies. Rheumatology. 2014;53:1664-1668.
71. Harrold LR, Reed GW, Solomon DH, et al. Comparative effectiveness of abatacept versus tocilizumab in rheumatoid arthritis patients with prior TNFi exposure in the US Corrona registry. Arthritis Res Ther. 2016;18:280.
72. Gottenberg J, Brocq O, Perdriger A, et al. Non–TNF-targeted biologic vs a second anti-TNF drug to treat rheumatoid arthritis in patients with insufficient response to a first anti-TNF drug: A randomized clinical trial. JAMA. 2016;316:1172-1180.
73. Pascart T, Philippe P, Drumez E, et al. Comparative efficacy of tocilizumab, abatacept and rituximab after non-TNF inhibitor failure: results from a multicentre study. Int J Rheum Dis. 2016;19:1093-1102.
74. Akiyama M, Kaneko Y, Kondo H, Takeuchi T. Comparison of the clinical effectiveness of tumour necrosis factor inhibitors and abatacept after insufficient response to tocilizumab in patients with rheumatoid arthritis. Clin Rheumatol. 2016;35:2829-2834.
75. Schoels M, Aletaha D, Smolen JS, Wong JB. Comparative effectiveness and safety of biological treatment options after tumour necrosis factor α inhibitor failure in rheumatoid arthritis: systematic review and indirect pairwise meta-analysis. Ann Rheum Dis. 2012;71:1303.
76. Soliman MM, Hyrich KL, Lunt M, et al. Rituximab or a second anti-tumor necrosis factor therapy for rheumatoid arthritis patients who have failed their first anti-tumor necrosis factor therapy? Comparative analysis from the British Society for Rheumatology Biologics Register. Arthritis Care Res. 2012;64:1108-1115.
77. Chatzidionysiou K, Vollenhoven RF. Rituximab versus anti-TNF in patients who previously failed one TNF inhibitor in an observational cohort. Scand J Rheumatol. 2013;42:190-195.
78. Johnston SS, Turpcu A, Shi N, et al. Risk of infections in rheumatoid arthritis patients switching from anti-TNF agents to rituximab, abatacept, or another anti-TNF agent, a retrospective administrative claims analysis. Semim Arthritis Rheum. 2013;43:39-47.
79. Curtis JR, Xie F, Chen L, et al. The comparative risk of serious infections among rheumatoid arthritis patients starting or switching biological agents. Ann Rheum Dis. 2011;70:1401.
80. Desai RJ, Solomon DH, Jin Y, et al. Temporal trends in use of biologic DMARDs for rheumatoid arthritis in the United States: a cohort study of publicly and privately insured patients. J Manag Care Spec Pharm. 2017;23:809-814.
81. Jin Y, Desai RJ, Liu J, et al. Factors associated with initial or subsequent choice of biologic disease-modifying antirheumatic drugs for treatment of rheumatoid arthritis. Arthritis Res Ther. 2017;19:159.
82. Bonafede MMK, McMorrow D, Proudfoot C, et al. Treatment persistence and healthcare costs among patients with rheumatoid arthritis after a change in targeted therapy. Am Health Drug Benefits. 2018;11:192-202.
83. US Food and Drug Administration. Biosimilars are safe, effective treatment options. www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeuticbiologicapplications/biosimilars/. Accessed November 9, 2018.
84. Genovese MC, Greenwald M, Codding C, et al. Peficitinib, a JAK inhibitor, in combination with limited conventional synthetic disease-modifying antirheumatic drugs in the treatment of moderate-to-severe rheumatoid arthritis. Arthritis Rheumatol. 2017;69:932-942.
85. Fleischmann RM, Wagner F, Kivitz AJ, et al. Safety, tolerability, and pharmacodynamics of ABT-122, a tumor necrosis factor- and interleukin-17-targeted dual variable domain immunoglobulin, in patients with rheumatoid arthritis. Arthritis Rheumatol. 2017;69:2283-2291.
86. Burmester GR, McInnes IB, Kremer JM, et al. Mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor alpha monoclonal antibody: long-term safety and efficacy in patients with rheumatoid arthritis. Arthritis Rheumatol. 2018;70:679-689.
87. Huizinga TW, Batalov A, Stoilov R, et al. Phase 1b randomized, double-blind study of namilumab, an anti-granulocyte macrophage colony-stimulating factor monoclonal antibody, in mild-to-moderate rheumatoid arthritis. Arthritis Res Ther. 2017;19:53.
88. Stock T, Fleishaker D, Wang X, et al. Improved disease activity with fosdagrocorat (PF-04171327), a partial agonist of the glucocorticoid receptor, in patients with rheumatoid arthritis: a Phase 2 randomized study. Int J Rheum Dis. 2017;20:960-970.
89. Orencia [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2013.
90. Humira[package insert]. North Chicago, IL: AbbVie; 2012.
91. Kineret [package insert]. Stockholm, Sweden: Sobi; 2012.
92. Olumiant [package insert]. Indianapolis, IN: Lilly USA, LLC; 2018.
93. Cimzia [package insert]. Smyrna, GA: UCB, Inc; 2008.
94. Enbrel [package insert]. Thousand Oaks, CA: Immunex Corporation; 1998.
95. Simponi [package insert]. Horsham, PA: Janssen Biotech, Inc; 2009.
96. Remicade [package insert]. Horsham, PA: Janssen Biotech, Inc; 1998.
97. Rituxan [package insert]. South San Francisco, CA: Genetech, Inc; 1997.
98. Kevzara [package insert]. Bridgewater, NJ: Sanofi-Aventis US LLC; 2018.
99. Actemra [package insert]. South San Francisco, CA: Genentech, Inc; 2013.
100. Xeljanz [package insert]. New York, NY: Pfizer Inc; 2016.
From the University of Iowa Hospitals and Clinics, Iowa City, IA.
Abstract
- Objective: To discuss the variability in response to tumor necrosis factor inhibitors (TNFis) observed in patients with rheumatoid arthritis (RA) and discuss therapeutic options for patients who do not respond to initial TNFi therapy.
- Methods: Review of the literature.
- Results: Optimal treatment of RA aims at achieving and then maintaining remission or low disease activity. In a patient with an inadequate response to initial biologic therapy, several therapeutic options exist. Current evidence supports TNFi dose escalation for only infliximab; optimization of concurrent conventional synthetic disease-modifying antirheumatic drug (csDMARD) or switching to a different csDMARD are other options. Cycling (switching to an alternative TNFi) and swapping (switching to a therapy with a different mode of action) strategies are other alternate approaches supported by many observational studies. While no head-to-head trials exist directly comparing the 2 strategies, data suggest superiority of the swapping strategy over the cycling approach. Also, several studies have shown that switching to a drug with a different mechanism of action is associated with higher treatment persistence and lower health care costs than TNFi cycling.
- Conclusion: Physicians have a growing list of treatment options to help their patients with RA achieve disease remission. The choice of best treatment for a given patient needs to be individualized, keeping in mind other factors, including comorbidities.
Keywords: biologics; rheumatoid arthritis; swapping strategy; cycling strategy; TNF inhibitors.
Following the discovery of tumor necrosis factor (TNF) as a proinflammatory cytokine 30 years ago, the use of TNF antagonists has revolutionized the treatment of rheumatoid arthritis (RA). Although TNF inhibitors (TNFIs) are frequently used as a first-line biologic disease-modifying antirheumatic drug (bDMARD), they are not uniformly efficacious in achieving remission in all patients with RA. This article highlights the reasons for such variability in observed response and discusses therapeutic options for patients who do not respond to TNFi therapy.
Case Presentation
A 60-year-old woman is evaluated in the clinic for complaints of pain in her hands, morning stiffness lasting 2 hours, and swelling in her wrists, all of which have been ongoing for 3 months. Physical exam reveals evidence of active inflammation, with synovitis in her second, third, and fourth metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints bilaterally, swelling over both wrists, and a weak grip. Inflammatory markers are elevated, and rheumatoid factor and anti-cyclic citrullinated peptide (anti-CCP) are both positive at high titer. Radiographs reveal evidence of small erosions at the third and fourth MCPs and PIPs bilaterally and periarticular osteopenia. The patient is diagnosed with seropositive, erosive RA based on history, physical exam, laboratory studies, and imaging. She is started on 20 mg of prednisone for acute treatment of her symptoms along with methotrexate, and, initially, her symptoms are well controlled. A few months after starting treatment, she develops voluminous diarrhea that necessitates cessation of methotrexate. Leflunomide also causes similar symptoms. The combination of sulfasalazine and hydroxychloroquine does not adequately control her symptoms, and ongoing use of low-dose glucocorticoids is required to improve functionality in all joints. Using the treat-to-target (T2T) strategy, adalimumab is initiated. However, she continues to report persistent swelling and pain and still requests oral glucocorticoids to help decrease inflammation. The 28-joint Disease Activity Score (DAS28) is 4.8, suggestive of moderate disease activity.
Why are TNFi agents sometimes ineffective?
The introduction of monoclonal antibodies and fusion proteins to block TNF and other cytokines was a remarkable development in the treatment of RA that revolutionized patient care. Despite the efficacy of TNFis, clinical response to these agents is not universal and only some patients achieve complete remission. In targeting the eventual goal of remission or low disease activity in patients with RA, the concept of “TNF failure” becomes extremely relevant. These inadequate responses to anti-TNF therapy may be due to primary failures, or complete lack of clinical response after initiation of the bDMARD, and secondary failures, or the loss of initially achieved clinical response to therapy. Other reasons for discontinuation of a given TNFi include partial disease control and intolerance to the medication (possible injection-site or infusion reactions). Keystone and Kavanaugh1 divided causes of failure of TNF agents into 2 broad categories: perceptual (related to natural variations in disease course like hormonal variation and physical and emotional stress) and pathophysiological failures (genetic variations, high body mass index, concomitant cigarette use).
Another important consideration in patients treated with a TNFi is the consequent formation of anti-drug antibodies (ADAs). TNFi agents are immunogenic and normally elicit an immune response. The appearance of such ADAs may reduce the bioavailability of free drug, resulting in a decreased clinical response,2 or may lead to serious adverse effects.
How common is discontinuation of the first TNFi?
Several studies have reported that the prevalence of primary failure, secondary failure, and intolerance to TNFis ranges from 30% to 40%.3-6 Female sex,7 concurrent prednisone use,8 high disease activity scores,6,8,9 and the absence of treatment with low-dose methotrexate7,8 have all been shown to be negative predictors of bDMARD retention and response.10
Are there any factors that predict TNFi failure?
There are no specific parameters to accurately predict responses to TNFI therapy.11 Several clinical and molecular biomarkers in synovium (initial TNF levels, macrophages, T cells)12 and peripheral blood (serum myeloid-related protein 8 and 14 complex levels,13 prealbumin, platelet factor 4, and S100A12)14 have been described as predictors of clinical response to TNFis, but their utility in clinical practice has not been established and the use of these markers has not yet been incorporated into clinical guidelines.
How is disease activity measured in patients with RA?
In 2010 an international expert consensus panel published treatment recommendations for RA that emphasized a T2T strategy of individualizing and escalating treatment to achieve the lowest disease activity or remission. In clinical practice, numerous tools are available to measure RA disease activity. Herein, we mention several that are most commonly used in clinical practice.
DAS28 combines single activity measures into an overall continuous measure of disease activity and has been endorsed by both the American College of Rheumatology (ACR) and European League Against Rheumatism (EULAR). It includes a 28-swollen joint count (SJC), 28-tender joint count (TJC), erythrocyte sedimentation rate (ESR; can also be calculated using C-reactive protein [CRP]), and a patient global assessment (PtGA). The cut-offs used for DAS28 interpretation are as follows: remission (< 2.6), low (≥ 2.6 but ≤ 3.2), moderate (> 3.2 but ≤ 5.1), or high (> 5.1).15 Some of the difficulties in using DAS28 in daily clinical practice include the need for a lab value and the time needed to perform the joint counts. Note also that due to the inclusion of ESR, which is influenced by age and other factors, DAS28 may underestimate remission in the elderly.
Another measure of RA disease activity is the Simplified Disease Activity Index (SDAI), which includes 28 SJC, 28 TJC, PtGA, provider global assessment (PrGA), and CRP in mg/dL. The level of disease activity using the SDAI is interpreted as: remission (SDAI ≤ 3.3), low (≥ 3.4 but ≤ 11), moderate (> 11 but ≤ 26), or high (> 26). The advantage of the SDAI is that a calculator or computer is not required for calculations. Another measure, the Clinical Disease Activity Index (CDAI), includes a 28 SJC, 28 TJC, PtGA, and PrGA. Because a laboratory value is not needed to calculate the CDAI, it is well-suited for use in clinical practice. When using the CDAI, the level of disease activity can be defined as remission (CDAI ≤ 2.8), low (> 2.8 but ≤ 10), moderate (> 10 but ≤ 22), or high (> 22). Again, as with the SDAI, a calculator or computer is not needed for calculations.
What are the alternative treatment options after first biologic failure?
In patients who have failed treatment with an initial biologic, usually a TNFi, the treating rheumatologist has the following options (Figure), with the best treatment strategy being driven by individualized patient and disease-related factors (Table 1 and Table 2):
- TNFi dose escalation
- Trial of an alternate TNFi agent (the “cycling” strategy)
- Optimization of therapy conjoined with a conventional synthetic DMARD (csDMARD)
- Use of a non-TNF biologic or targeted synthetic DMARD (the “swapping” strategy)
If all the listed strategies fail, the next step can be the addition of short-term, low-dose glucocorticoid therapy.
TNFi Dose Escalation
The available data have demonstrated the safety, efficacy, and cost-effectiveness of dose escalation in patients with RA receiving infliximab.16-18 The ATTRACT trial first demonstrated this, with greater clinical and radiographic improvements in those with higher trough serum concentrations, suggesting that doses higher than 3 mg/kg or more frequent than every 8 weeks may be needed for full response in some patients.19
There is a lack of studies in RA patients to determine the most effective dose escalation strategy. A study in patients with Crohn disease showed that intensification to 10 mg/kg every 8 weeks (dose doubling) was at least as effective as 5 mg/kg every 4 weeks (halving interval) at 12 months.16 Due to greater patient and administration convenience of dose-doubling, this strategy may be preferred.17 A starting dose of 10 mg/kg every 8 weeks is not routinely recommended due to an increased risk of serious infection; these adverse events were not found when the dose was gradually increased, as clinically indicated, starting at 3 mg/kg.19,20 Further studies are needed to explore this approach in RA patients.
These results, however, have not been replicated with other TNFi agents. No significant clinical improvements were identified with etanercept 50 mg twice weekly,21 adalimumab 40 mg every week in the PREMIER trial,18 or certolizumab 400 mg every other week in an open-label extension phase of the RAPID 1 study.22 A Japanese study found significantly worse clinical outcomes with dose escalation of golimumab.23 Conversely, 2 studies found clinical benefits after escalating the tocilizumab dose, the first a real-world review from the Consortium of Rheumatology Researchers of North America (CORRONA) registry using the intravenous formulation,24 and the other the BREVACTA study utilizing subcutaneous tocilizumab.25 No studies to date have been published on dose escalation of abatacept in patients with RA who respond poorly. Overall, previous studies support dose escalation in individuals being treated with infliximab to improve clinical outcomes, but additional studies are needed for other bDMARDs.
Trial of an Alternate TNF Agent: The “Cycling” Strategy
Per the ACR/EULAR26,27 guidelines, all approved bDMARDs may be used without hierarchical positioning. However, after the failure of a TNFi agent, these guidelines do not provide specific advice about a preference between the “cycling” strategy (switching to an alternative TNFi) and “swapping” strategy (switching to a therapy with a different mode of action). Cycling might work for several reasons, including differences in the agents’ molecular structure, immunological mechanism of action, immunogenicity, and pharmacokinetics.28-30 The cycling strategy is a well-established approach adopted by more than 94% of practicing rheumatologists, according to a national survey,31 and its efficacy is supported by trials and additional observational studies.32-35
The greater clinical effectiveness of switching to infliximab compared with continuing with etanercept in patients with inadequate response to etanercept (n = 28) was suggested in the open-label OPPOSITE trial.36 Data from the GO-AFTER trial37 suggests that a greater proportion of patients with RA refractory to adalimumab, etanercept, or infliximab who were treated with golimumab achieved an ACR20 and ACR50 response compared with patients who received placebo, and this response persisted through 5 years.38 More recently, certolizumab pegol and adalimumab were compared head-to-head in the EXXELERATE trial.39 The results of this trial revealed the adequate efficacy of cycling to another TNFi after primary insufficient response to the first.
In studies from Finland and Sweden,35,40 it has been observed that a better response is achieved in patients in whom TNF failure was initially due to secondary failure or intolerance rather than primary failure. A post-hoc analysis of the results of the GO-AFTER trial41 and from a few observational studies35,40,42 revealed that switching from one TNFi to another, especially from a monoclonal antibody to a soluble receptor, was often more beneficial for RA patients than switching from a soluble receptor to a monoclonal antibody.
Optimization of Therapy Conjoined with csDMARDs
Methotrexate is one of the oldest and most effective csDMARDs available for the treatment of RA.43 The 2016 EULAR guidelines recommend the addition of methotrexate and/or other csDMARDs to potentiate the effect of bDMARDs.26 In the case of TNFi therapy, the observed synergistic effect between the monoclonal antibody and methotrexate may be explained by sustained suppression of ADA formation.44 In the TEMPO,45 PREMIER,18 and GO-BEFORE46 trials, the addition of methotrexate led to improved clinical and radiological outcomes in patients treated with etanercept, adalimumab, and golimumab,47 respectively. These findings were also demonstrated in several registries, where significant improvement in clinical response and retention rate of the TNFi agents was noted. Results have been replicated with non-TNFi bDMARDs, including abatacept48,49 and rituximab.50 Patients treated with interleukin (IL)-6 inhibitors in combination with methotrexate have shown significantly less radiographic progression compared to those treated with tocilizumab alone and those treated with monotherapy tocilizumab versus monotherapy methotrexate.51,52 Results possibly favor the use of IL-6 inhibitors alone in those who cannot tolerate or have contraindications to methotrexate.
An open prospective study by Cohen et al added methotrexate to the treatment regimens of individuals on bDMARD monotherapy with a primary failure and found favorable changes in ACR20 and DAS28 scores at 3 and 12 months and therapeutic biological response (ESR, CRP) at 3 months.53 Unlike monotherapy, in these situations methotrexate is known to be efficacious even at a lower dose, possibly at 7.5 mg to 10 mg per week. Some studies have shown that methotrexate administered parenterally may be more efficacious than when given orally.54-58
In clinical trials and observational studies, leflunomide, sulfasalazine, and hydroxychloroquine have been used as alternate csDMARDs added to the treatment regimen.59-62 There are, however, only 2 trials comparing the efficacy of methotrexate with that of other csDMARDs as concomitant treatment in patients with inadequate response to TNFi therapy. The RABBIT trial found a slight decrease in effectiveness with concomitant TNFi and leflunomide compared to TNFi/methotrexate, but overall each group had similar EULAR responses at 24 months.63 A study by De Stefano et al found comparable ACR20 and DAS28 responses among individuals receiving TNFis with methotrexate or leflunomide.61
The “Swapping” Strategy
The efficacy of the swapping strategy has been shown in 3 randomized clinical trials demonstrating the superiority of abatacept, tocilizumab, and rituximab in the treatment of individuals with RA refractory to TNFis. Tocilizumab was studied in the RADIATE64 trial, which involved 499 patients with inadequate response to 1 or more TNFi agents. The primary endpoint (24-week ACR20) was achieved by 50.0%, 30.4%, and 10.1% of patients in the 8 mg/kg, 4 mg/kg, and control groups, respectively (P < 0.001 for both tocilizumab groups versus placebo). The utility of abatacept as second-line therapy after initial TNF failure was evaluated in the ATTAIN65 study. Participants with an inadequate response to etanercept or infliximab were randomly assigned to receive either abatacept or placebo. ACR50 response rates after 6 months of treatment were 20.3% with abatacept and 3.8% with placebo (P < 0.001). The SWITCH-RA study,66 an observational study, compared rituximab to TNFis in 1112 participants with inadequate response to initial anti-TNF therapy. At 6 months, mean change in DAS28 was small but significantly greater for the rituximab group (–1.5 vs –1.1; P = 0.007). The difference in response rates was greatest among seropositive patients. These data suggest that rituximab has efficacy following TNFi failure, particularly for seropositive patients. Additionally, REFLEX67 is the sole randomized controlled trial in patients with insufficient response to TNFis that showed significant prevention of radiographic progression at week 56 in patients on rituximab compared to placebo (mean change from baseline in total Genant-modified Sharp score, 1.00 vs 2.31, respectively; P = 0.005).
One study randomly assigned 399 patients with active RA who had inadequate response to prior TNFi therapy to tofacitinib68 (5 mg twice daily or 10 mg twice daily) or placebo, both with methotrexate.6 After 3 months of treatment, ACR20 response rates (41.7% for 5 mg, 28.1% for 10 mg, 24.4% for placebo) and DAS28 remission rates (6.7% for 5 mg, 8.8% for 10 mg, 1.7% for placebo) were significantly greater among patients treated with tofacitinib compared to those treated with placebo. More recently, the RA-BEACON trial69 demonstrated a consistent, beneficial treatment effect of baricitinib in patients with insufficient response to 1 or more TNFis. In this trial, 527 patients with an inadequate response to bDMARDs were randomly assigned to receive baricitinib 2 mg or 4 mg daily or placebo for 24 weeks. A higher proportion of patients receiving baricitinib 4 mg had an ACR20 response at week 12 compared with those treated with placebo (55% vs 27%, P < 0.001), and patients receiving the 4-mg dose had significant improvements from baseline in DAS28 and Health Assessment Questionnaire–Disability Index scores (P < 0.001 for both comparisons).
To Cycle or to Swap?
Several observational studies (SCQM-RA,70 STURE,71 BSRBR,72 Favalli,43 MIRAR,73 SWITCH-RA,74 ROC72) have clearly demonstrated that the swapping strategy is favored over the cycling strategy. In the ROC study,72 patients were randomly assigned (based on physician discretion) to receive a non-TNF biologic or a TNFi. More patients in the non-TNF group than in the TNFi group showed low disease activity at week 24 (45% vs 28%; odds ratio [OR], 2.09; 95% confidence interval [CI], 1.27-3.43; P = 0.004) and at week 52 (41% vs 23%; OR, 2.26; 95% CI, 1.33-3.86; P = 0.003). The authors concluded that in patients having an insufficient response to TNFi therapy, a non-TNF biologic agent may be more effective than a second TNFi drug. Only a few studies75-77 have demonstrated similar results between the 2 strategies. Overall, the available evidence seems to suggest the superiority of the swapping over the cycling strategy.
An important clinical pearl to keep in mind is that both swapping and cycling strategies might theoretically increase the risk of infection; however, limited evidence is reported in the literature. In a large retrospective analysis78 of data on 4332 RA patients from a large US claims database, patients who had cycled between TNFi agents had a 30% to 40% increased risk of infection compared to patients treated with rituximab. Patients on infliximab had a 62% higher hazard of severe infections, and this has also been reported in an observational study.79 In another study,70 41% of 201 patients with RA followed between 1999 and 2013 who swapped to abatacept/rituximab or tocilizumab developed adverse events, as compared to 59% of those who switched to a second TNFi.
What are recent trends in the use of bDMARDs?
Currently, there are no specific guidelines or biomarkers available to facilitate selection of specific treatment from among the classes of biologics. With the development of several new drugs and regulatory approval of baricitinib, physicians now have several biologic options to treat patients. A recent large time-trend study80 deriving data from more than 200,000 patients with RA showed that etanercept remains the most frequently used agent for the treatment of RA; it also showed that the use of adalimumab and infliximab is decreasing, and that the use of newer agents, especially abatacept, golimumab, and certolizumab, has considerably risen in recent years. In this study, abatacept, rituximab, certolizumab, golimumab, tocilizumab, and tofacitinib accounted for 13.2%, 13.8%, 6.9%, 11.9%, and 7.5% switches from first TNFi therapy.
Jin et al81 studied factors associated with the choice of bDMARD for initial and subsequent use. They found that patients with commercial insurance had an 87% higher likelihood of initiating a bDMARD. In the Medicaid subgroup, African Americans had lower odds of initiating and switching bDMARDs than non-Hispanic whites. Prior use of steroids and nonbiologic DMARDs predicted both bDMARD initiation and subsequent switching. Etanercept, adalimumab, and infliximab were the most commonly used first- and second-line bDMARDS; patients on anakinra and golimumab were most likely to be switched to other bDMARDs.
Which treatment strategy is the most cost-effective?
Several studies have reported better treatment persistence rates among patients who are treated with the swapping strategy compared to the cycling strategy. In a retrospective analysis of claims data,82 the authors examined treatment persistence and health care costs in patients switching to biologics with a different mechanism of action or cycling to another TNFi. The mean cost was significantly lower among patients treated using the swapping strategy than among the TNFi cyclers, both for the total cost of care for RA and for the total cost of the targeted DMARDs in the first year after the change in therapy. The authors concluded that switching to a drug with a different mechanism of action is associated with higher treatment persistence and lower health care costs than TNFi cycling.
What about biosimilars?
Biosimilars are copies of already licensed biologics that are very similar to the biologics, but are made by different sponsors using independently derived cell lines and separately developed manufacturing processes.83 Regarding biosimilar use, EULAR26 states that biosimilar bDMARDs approved by the European Medicines Agency or US Food and Drug Administration have similar efficacy and safety as the originator bDMARDs, and recommends them as preferred agents if they are indeed appreciably cheaper than originator or other bDMARDs.
What are the novel treatment targets in RA?
New therapeutics for RA continue to be developed. One of the new agents is peficitinib (ASP015K), an oral, once-daily Janus kinase (Jak) inhibitor targeting Jak-1, Jak-2, and tyrosine kinase-2, with moderate selectivity for Jak-3. In a phase 2b trial, 100-mg and 150-mg doses of peficitinib achieved a statistically significant ACR20 response (48.3% and 56.3%) compared to placebo (29.4%) at 12 weeks.84
Given the benefit of targeting TNF-α and IL-17 in RA, a novel molecule (ABT-122) that targets both human TNF and IL-17 has been developed. Two phase 1 studies85 showed that dual neutralization of TNF and IL-17 with ABT-122 has characteristics acceptable for further exploration of therapeutic potential of this agent in TNF- and IL-17A–driven immune-mediated inflammatory diseases. Another novel drug is mavrilimumab, a human monoclonal antibody that targets granulocyte–macrophage colony-stimulating factor receptor α. A recent studyshowed that long-term treatment with mavrilimumab maintained response and was well-tolerated, with no increased incidence of treatment-emergent adverse events.86
Namilumab (AMG203) is an immunoglobulin G1 monoclonal antibody that binds with high affinity to the GM-CSF ligand. In a phase 1b, randomized, double-blind study (PRIORA)87 to assess namilumab in treating active, mild-to-moderate RA, significant improvement was seen in the DAS28-CRP score with namilumab (150 and 300 mg groups combined) compared with placebo at day 43 (P = 0.0117) and also 8 weeks after last dosing at day 99 (P = 0.0154). Adverse events were similar across different doses of namilumab and placebo, and included nasopharyngitis and exacerbation/worsening of RA. Another drug showing promise in RA is fosdagrocorat (PF-04171327), a potential dissociated agonist of the glucocorticoid receptor. A multicenter, double-blind, parallel-group, active- and placebo-controlled phase 2 study randomly assigned 86 patients to receive fosdagrocorat 10 mg, fosdagrocorat 25 mg, prednisone 5 mg, or placebo, all with stable background methotrexate therapy.88 Both fosdagrocorat doses demonstrated efficacy in improving signs and symptoms in RA patients, with manageable adverse events.
Case Conclusion
There are several available treatment options for the case patient. Based on the PREMIER trial, solely increasing the dose of adalimumab is unlikely to provide a therapeutic benefit. Adding low-dose methotrexate (possibly via a parenteral route because of patient-reported gastrointestinal discomfort) might provide some synergistic and therapeutic effect. However, because of primary failure with TNFi therapy, she may benefit from the initiation of a biologic with a different mechanism of action (ie, swapping strategy). Therapeutic options include tocilizumab, abatacept, rituximab, and the Jak inhibitors (tofacitinib and baricitinib).
Summary
The optimal treatment of RA aims at achieving, and then maintaining, remission or a low disease activity. The choice of best treatment must be individualized to the patient, keeping in mind other factors, including comorbidities like fibromyalgia, history of diverticulitis (prior to use of tocilizumab), history of chronic obstructive pulmonary disease (prior to the use of abatacept), malignancy, and the presence of risk factors for infections (age, diabetes, chronic bronchitis). In a patient with inadequate response to initial biologic therapy, several options exist for the rheumatologist. Current evidence supports TNFi dose escalation for only infliximab; optimization of concurrent csDMARD or switching to a different csDMARD are other options. Cycling and swapping are other alternate approaches supported by many observational studies. While no head-to-head trials exist comparing the 2 strategies, data suggest superiority of the swapping strategy over the cycling approach. With the continuing development of novel therapeutics in RA, physicians have a growing list of treatment options to help their patients achieve disease remission.
Corresponding author: Namrata Singh, MD, 200 Hawkins Drive, Iowa City, IA 52242.
Financial disclosures: None.
From the University of Iowa Hospitals and Clinics, Iowa City, IA.
Abstract
- Objective: To discuss the variability in response to tumor necrosis factor inhibitors (TNFis) observed in patients with rheumatoid arthritis (RA) and discuss therapeutic options for patients who do not respond to initial TNFi therapy.
- Methods: Review of the literature.
- Results: Optimal treatment of RA aims at achieving and then maintaining remission or low disease activity. In a patient with an inadequate response to initial biologic therapy, several therapeutic options exist. Current evidence supports TNFi dose escalation for only infliximab; optimization of concurrent conventional synthetic disease-modifying antirheumatic drug (csDMARD) or switching to a different csDMARD are other options. Cycling (switching to an alternative TNFi) and swapping (switching to a therapy with a different mode of action) strategies are other alternate approaches supported by many observational studies. While no head-to-head trials exist directly comparing the 2 strategies, data suggest superiority of the swapping strategy over the cycling approach. Also, several studies have shown that switching to a drug with a different mechanism of action is associated with higher treatment persistence and lower health care costs than TNFi cycling.
- Conclusion: Physicians have a growing list of treatment options to help their patients with RA achieve disease remission. The choice of best treatment for a given patient needs to be individualized, keeping in mind other factors, including comorbidities.
Keywords: biologics; rheumatoid arthritis; swapping strategy; cycling strategy; TNF inhibitors.
Following the discovery of tumor necrosis factor (TNF) as a proinflammatory cytokine 30 years ago, the use of TNF antagonists has revolutionized the treatment of rheumatoid arthritis (RA). Although TNF inhibitors (TNFIs) are frequently used as a first-line biologic disease-modifying antirheumatic drug (bDMARD), they are not uniformly efficacious in achieving remission in all patients with RA. This article highlights the reasons for such variability in observed response and discusses therapeutic options for patients who do not respond to TNFi therapy.
Case Presentation
A 60-year-old woman is evaluated in the clinic for complaints of pain in her hands, morning stiffness lasting 2 hours, and swelling in her wrists, all of which have been ongoing for 3 months. Physical exam reveals evidence of active inflammation, with synovitis in her second, third, and fourth metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints bilaterally, swelling over both wrists, and a weak grip. Inflammatory markers are elevated, and rheumatoid factor and anti-cyclic citrullinated peptide (anti-CCP) are both positive at high titer. Radiographs reveal evidence of small erosions at the third and fourth MCPs and PIPs bilaterally and periarticular osteopenia. The patient is diagnosed with seropositive, erosive RA based on history, physical exam, laboratory studies, and imaging. She is started on 20 mg of prednisone for acute treatment of her symptoms along with methotrexate, and, initially, her symptoms are well controlled. A few months after starting treatment, she develops voluminous diarrhea that necessitates cessation of methotrexate. Leflunomide also causes similar symptoms. The combination of sulfasalazine and hydroxychloroquine does not adequately control her symptoms, and ongoing use of low-dose glucocorticoids is required to improve functionality in all joints. Using the treat-to-target (T2T) strategy, adalimumab is initiated. However, she continues to report persistent swelling and pain and still requests oral glucocorticoids to help decrease inflammation. The 28-joint Disease Activity Score (DAS28) is 4.8, suggestive of moderate disease activity.
Why are TNFi agents sometimes ineffective?
The introduction of monoclonal antibodies and fusion proteins to block TNF and other cytokines was a remarkable development in the treatment of RA that revolutionized patient care. Despite the efficacy of TNFis, clinical response to these agents is not universal and only some patients achieve complete remission. In targeting the eventual goal of remission or low disease activity in patients with RA, the concept of “TNF failure” becomes extremely relevant. These inadequate responses to anti-TNF therapy may be due to primary failures, or complete lack of clinical response after initiation of the bDMARD, and secondary failures, or the loss of initially achieved clinical response to therapy. Other reasons for discontinuation of a given TNFi include partial disease control and intolerance to the medication (possible injection-site or infusion reactions). Keystone and Kavanaugh1 divided causes of failure of TNF agents into 2 broad categories: perceptual (related to natural variations in disease course like hormonal variation and physical and emotional stress) and pathophysiological failures (genetic variations, high body mass index, concomitant cigarette use).
Another important consideration in patients treated with a TNFi is the consequent formation of anti-drug antibodies (ADAs). TNFi agents are immunogenic and normally elicit an immune response. The appearance of such ADAs may reduce the bioavailability of free drug, resulting in a decreased clinical response,2 or may lead to serious adverse effects.
How common is discontinuation of the first TNFi?
Several studies have reported that the prevalence of primary failure, secondary failure, and intolerance to TNFis ranges from 30% to 40%.3-6 Female sex,7 concurrent prednisone use,8 high disease activity scores,6,8,9 and the absence of treatment with low-dose methotrexate7,8 have all been shown to be negative predictors of bDMARD retention and response.10
Are there any factors that predict TNFi failure?
There are no specific parameters to accurately predict responses to TNFI therapy.11 Several clinical and molecular biomarkers in synovium (initial TNF levels, macrophages, T cells)12 and peripheral blood (serum myeloid-related protein 8 and 14 complex levels,13 prealbumin, platelet factor 4, and S100A12)14 have been described as predictors of clinical response to TNFis, but their utility in clinical practice has not been established and the use of these markers has not yet been incorporated into clinical guidelines.
How is disease activity measured in patients with RA?
In 2010 an international expert consensus panel published treatment recommendations for RA that emphasized a T2T strategy of individualizing and escalating treatment to achieve the lowest disease activity or remission. In clinical practice, numerous tools are available to measure RA disease activity. Herein, we mention several that are most commonly used in clinical practice.
DAS28 combines single activity measures into an overall continuous measure of disease activity and has been endorsed by both the American College of Rheumatology (ACR) and European League Against Rheumatism (EULAR). It includes a 28-swollen joint count (SJC), 28-tender joint count (TJC), erythrocyte sedimentation rate (ESR; can also be calculated using C-reactive protein [CRP]), and a patient global assessment (PtGA). The cut-offs used for DAS28 interpretation are as follows: remission (< 2.6), low (≥ 2.6 but ≤ 3.2), moderate (> 3.2 but ≤ 5.1), or high (> 5.1).15 Some of the difficulties in using DAS28 in daily clinical practice include the need for a lab value and the time needed to perform the joint counts. Note also that due to the inclusion of ESR, which is influenced by age and other factors, DAS28 may underestimate remission in the elderly.
Another measure of RA disease activity is the Simplified Disease Activity Index (SDAI), which includes 28 SJC, 28 TJC, PtGA, provider global assessment (PrGA), and CRP in mg/dL. The level of disease activity using the SDAI is interpreted as: remission (SDAI ≤ 3.3), low (≥ 3.4 but ≤ 11), moderate (> 11 but ≤ 26), or high (> 26). The advantage of the SDAI is that a calculator or computer is not required for calculations. Another measure, the Clinical Disease Activity Index (CDAI), includes a 28 SJC, 28 TJC, PtGA, and PrGA. Because a laboratory value is not needed to calculate the CDAI, it is well-suited for use in clinical practice. When using the CDAI, the level of disease activity can be defined as remission (CDAI ≤ 2.8), low (> 2.8 but ≤ 10), moderate (> 10 but ≤ 22), or high (> 22). Again, as with the SDAI, a calculator or computer is not needed for calculations.
What are the alternative treatment options after first biologic failure?
In patients who have failed treatment with an initial biologic, usually a TNFi, the treating rheumatologist has the following options (Figure), with the best treatment strategy being driven by individualized patient and disease-related factors (Table 1 and Table 2):
- TNFi dose escalation
- Trial of an alternate TNFi agent (the “cycling” strategy)
- Optimization of therapy conjoined with a conventional synthetic DMARD (csDMARD)
- Use of a non-TNF biologic or targeted synthetic DMARD (the “swapping” strategy)
If all the listed strategies fail, the next step can be the addition of short-term, low-dose glucocorticoid therapy.
TNFi Dose Escalation
The available data have demonstrated the safety, efficacy, and cost-effectiveness of dose escalation in patients with RA receiving infliximab.16-18 The ATTRACT trial first demonstrated this, with greater clinical and radiographic improvements in those with higher trough serum concentrations, suggesting that doses higher than 3 mg/kg or more frequent than every 8 weeks may be needed for full response in some patients.19
There is a lack of studies in RA patients to determine the most effective dose escalation strategy. A study in patients with Crohn disease showed that intensification to 10 mg/kg every 8 weeks (dose doubling) was at least as effective as 5 mg/kg every 4 weeks (halving interval) at 12 months.16 Due to greater patient and administration convenience of dose-doubling, this strategy may be preferred.17 A starting dose of 10 mg/kg every 8 weeks is not routinely recommended due to an increased risk of serious infection; these adverse events were not found when the dose was gradually increased, as clinically indicated, starting at 3 mg/kg.19,20 Further studies are needed to explore this approach in RA patients.
These results, however, have not been replicated with other TNFi agents. No significant clinical improvements were identified with etanercept 50 mg twice weekly,21 adalimumab 40 mg every week in the PREMIER trial,18 or certolizumab 400 mg every other week in an open-label extension phase of the RAPID 1 study.22 A Japanese study found significantly worse clinical outcomes with dose escalation of golimumab.23 Conversely, 2 studies found clinical benefits after escalating the tocilizumab dose, the first a real-world review from the Consortium of Rheumatology Researchers of North America (CORRONA) registry using the intravenous formulation,24 and the other the BREVACTA study utilizing subcutaneous tocilizumab.25 No studies to date have been published on dose escalation of abatacept in patients with RA who respond poorly. Overall, previous studies support dose escalation in individuals being treated with infliximab to improve clinical outcomes, but additional studies are needed for other bDMARDs.
Trial of an Alternate TNF Agent: The “Cycling” Strategy
Per the ACR/EULAR26,27 guidelines, all approved bDMARDs may be used without hierarchical positioning. However, after the failure of a TNFi agent, these guidelines do not provide specific advice about a preference between the “cycling” strategy (switching to an alternative TNFi) and “swapping” strategy (switching to a therapy with a different mode of action). Cycling might work for several reasons, including differences in the agents’ molecular structure, immunological mechanism of action, immunogenicity, and pharmacokinetics.28-30 The cycling strategy is a well-established approach adopted by more than 94% of practicing rheumatologists, according to a national survey,31 and its efficacy is supported by trials and additional observational studies.32-35
The greater clinical effectiveness of switching to infliximab compared with continuing with etanercept in patients with inadequate response to etanercept (n = 28) was suggested in the open-label OPPOSITE trial.36 Data from the GO-AFTER trial37 suggests that a greater proportion of patients with RA refractory to adalimumab, etanercept, or infliximab who were treated with golimumab achieved an ACR20 and ACR50 response compared with patients who received placebo, and this response persisted through 5 years.38 More recently, certolizumab pegol and adalimumab were compared head-to-head in the EXXELERATE trial.39 The results of this trial revealed the adequate efficacy of cycling to another TNFi after primary insufficient response to the first.
In studies from Finland and Sweden,35,40 it has been observed that a better response is achieved in patients in whom TNF failure was initially due to secondary failure or intolerance rather than primary failure. A post-hoc analysis of the results of the GO-AFTER trial41 and from a few observational studies35,40,42 revealed that switching from one TNFi to another, especially from a monoclonal antibody to a soluble receptor, was often more beneficial for RA patients than switching from a soluble receptor to a monoclonal antibody.
Optimization of Therapy Conjoined with csDMARDs
Methotrexate is one of the oldest and most effective csDMARDs available for the treatment of RA.43 The 2016 EULAR guidelines recommend the addition of methotrexate and/or other csDMARDs to potentiate the effect of bDMARDs.26 In the case of TNFi therapy, the observed synergistic effect between the monoclonal antibody and methotrexate may be explained by sustained suppression of ADA formation.44 In the TEMPO,45 PREMIER,18 and GO-BEFORE46 trials, the addition of methotrexate led to improved clinical and radiological outcomes in patients treated with etanercept, adalimumab, and golimumab,47 respectively. These findings were also demonstrated in several registries, where significant improvement in clinical response and retention rate of the TNFi agents was noted. Results have been replicated with non-TNFi bDMARDs, including abatacept48,49 and rituximab.50 Patients treated with interleukin (IL)-6 inhibitors in combination with methotrexate have shown significantly less radiographic progression compared to those treated with tocilizumab alone and those treated with monotherapy tocilizumab versus monotherapy methotrexate.51,52 Results possibly favor the use of IL-6 inhibitors alone in those who cannot tolerate or have contraindications to methotrexate.
An open prospective study by Cohen et al added methotrexate to the treatment regimens of individuals on bDMARD monotherapy with a primary failure and found favorable changes in ACR20 and DAS28 scores at 3 and 12 months and therapeutic biological response (ESR, CRP) at 3 months.53 Unlike monotherapy, in these situations methotrexate is known to be efficacious even at a lower dose, possibly at 7.5 mg to 10 mg per week. Some studies have shown that methotrexate administered parenterally may be more efficacious than when given orally.54-58
In clinical trials and observational studies, leflunomide, sulfasalazine, and hydroxychloroquine have been used as alternate csDMARDs added to the treatment regimen.59-62 There are, however, only 2 trials comparing the efficacy of methotrexate with that of other csDMARDs as concomitant treatment in patients with inadequate response to TNFi therapy. The RABBIT trial found a slight decrease in effectiveness with concomitant TNFi and leflunomide compared to TNFi/methotrexate, but overall each group had similar EULAR responses at 24 months.63 A study by De Stefano et al found comparable ACR20 and DAS28 responses among individuals receiving TNFis with methotrexate or leflunomide.61
The “Swapping” Strategy
The efficacy of the swapping strategy has been shown in 3 randomized clinical trials demonstrating the superiority of abatacept, tocilizumab, and rituximab in the treatment of individuals with RA refractory to TNFis. Tocilizumab was studied in the RADIATE64 trial, which involved 499 patients with inadequate response to 1 or more TNFi agents. The primary endpoint (24-week ACR20) was achieved by 50.0%, 30.4%, and 10.1% of patients in the 8 mg/kg, 4 mg/kg, and control groups, respectively (P < 0.001 for both tocilizumab groups versus placebo). The utility of abatacept as second-line therapy after initial TNF failure was evaluated in the ATTAIN65 study. Participants with an inadequate response to etanercept or infliximab were randomly assigned to receive either abatacept or placebo. ACR50 response rates after 6 months of treatment were 20.3% with abatacept and 3.8% with placebo (P < 0.001). The SWITCH-RA study,66 an observational study, compared rituximab to TNFis in 1112 participants with inadequate response to initial anti-TNF therapy. At 6 months, mean change in DAS28 was small but significantly greater for the rituximab group (–1.5 vs –1.1; P = 0.007). The difference in response rates was greatest among seropositive patients. These data suggest that rituximab has efficacy following TNFi failure, particularly for seropositive patients. Additionally, REFLEX67 is the sole randomized controlled trial in patients with insufficient response to TNFis that showed significant prevention of radiographic progression at week 56 in patients on rituximab compared to placebo (mean change from baseline in total Genant-modified Sharp score, 1.00 vs 2.31, respectively; P = 0.005).
One study randomly assigned 399 patients with active RA who had inadequate response to prior TNFi therapy to tofacitinib68 (5 mg twice daily or 10 mg twice daily) or placebo, both with methotrexate.6 After 3 months of treatment, ACR20 response rates (41.7% for 5 mg, 28.1% for 10 mg, 24.4% for placebo) and DAS28 remission rates (6.7% for 5 mg, 8.8% for 10 mg, 1.7% for placebo) were significantly greater among patients treated with tofacitinib compared to those treated with placebo. More recently, the RA-BEACON trial69 demonstrated a consistent, beneficial treatment effect of baricitinib in patients with insufficient response to 1 or more TNFis. In this trial, 527 patients with an inadequate response to bDMARDs were randomly assigned to receive baricitinib 2 mg or 4 mg daily or placebo for 24 weeks. A higher proportion of patients receiving baricitinib 4 mg had an ACR20 response at week 12 compared with those treated with placebo (55% vs 27%, P < 0.001), and patients receiving the 4-mg dose had significant improvements from baseline in DAS28 and Health Assessment Questionnaire–Disability Index scores (P < 0.001 for both comparisons).
To Cycle or to Swap?
Several observational studies (SCQM-RA,70 STURE,71 BSRBR,72 Favalli,43 MIRAR,73 SWITCH-RA,74 ROC72) have clearly demonstrated that the swapping strategy is favored over the cycling strategy. In the ROC study,72 patients were randomly assigned (based on physician discretion) to receive a non-TNF biologic or a TNFi. More patients in the non-TNF group than in the TNFi group showed low disease activity at week 24 (45% vs 28%; odds ratio [OR], 2.09; 95% confidence interval [CI], 1.27-3.43; P = 0.004) and at week 52 (41% vs 23%; OR, 2.26; 95% CI, 1.33-3.86; P = 0.003). The authors concluded that in patients having an insufficient response to TNFi therapy, a non-TNF biologic agent may be more effective than a second TNFi drug. Only a few studies75-77 have demonstrated similar results between the 2 strategies. Overall, the available evidence seems to suggest the superiority of the swapping over the cycling strategy.
An important clinical pearl to keep in mind is that both swapping and cycling strategies might theoretically increase the risk of infection; however, limited evidence is reported in the literature. In a large retrospective analysis78 of data on 4332 RA patients from a large US claims database, patients who had cycled between TNFi agents had a 30% to 40% increased risk of infection compared to patients treated with rituximab. Patients on infliximab had a 62% higher hazard of severe infections, and this has also been reported in an observational study.79 In another study,70 41% of 201 patients with RA followed between 1999 and 2013 who swapped to abatacept/rituximab or tocilizumab developed adverse events, as compared to 59% of those who switched to a second TNFi.
What are recent trends in the use of bDMARDs?
Currently, there are no specific guidelines or biomarkers available to facilitate selection of specific treatment from among the classes of biologics. With the development of several new drugs and regulatory approval of baricitinib, physicians now have several biologic options to treat patients. A recent large time-trend study80 deriving data from more than 200,000 patients with RA showed that etanercept remains the most frequently used agent for the treatment of RA; it also showed that the use of adalimumab and infliximab is decreasing, and that the use of newer agents, especially abatacept, golimumab, and certolizumab, has considerably risen in recent years. In this study, abatacept, rituximab, certolizumab, golimumab, tocilizumab, and tofacitinib accounted for 13.2%, 13.8%, 6.9%, 11.9%, and 7.5% switches from first TNFi therapy.
Jin et al81 studied factors associated with the choice of bDMARD for initial and subsequent use. They found that patients with commercial insurance had an 87% higher likelihood of initiating a bDMARD. In the Medicaid subgroup, African Americans had lower odds of initiating and switching bDMARDs than non-Hispanic whites. Prior use of steroids and nonbiologic DMARDs predicted both bDMARD initiation and subsequent switching. Etanercept, adalimumab, and infliximab were the most commonly used first- and second-line bDMARDS; patients on anakinra and golimumab were most likely to be switched to other bDMARDs.
Which treatment strategy is the most cost-effective?
Several studies have reported better treatment persistence rates among patients who are treated with the swapping strategy compared to the cycling strategy. In a retrospective analysis of claims data,82 the authors examined treatment persistence and health care costs in patients switching to biologics with a different mechanism of action or cycling to another TNFi. The mean cost was significantly lower among patients treated using the swapping strategy than among the TNFi cyclers, both for the total cost of care for RA and for the total cost of the targeted DMARDs in the first year after the change in therapy. The authors concluded that switching to a drug with a different mechanism of action is associated with higher treatment persistence and lower health care costs than TNFi cycling.
What about biosimilars?
Biosimilars are copies of already licensed biologics that are very similar to the biologics, but are made by different sponsors using independently derived cell lines and separately developed manufacturing processes.83 Regarding biosimilar use, EULAR26 states that biosimilar bDMARDs approved by the European Medicines Agency or US Food and Drug Administration have similar efficacy and safety as the originator bDMARDs, and recommends them as preferred agents if they are indeed appreciably cheaper than originator or other bDMARDs.
What are the novel treatment targets in RA?
New therapeutics for RA continue to be developed. One of the new agents is peficitinib (ASP015K), an oral, once-daily Janus kinase (Jak) inhibitor targeting Jak-1, Jak-2, and tyrosine kinase-2, with moderate selectivity for Jak-3. In a phase 2b trial, 100-mg and 150-mg doses of peficitinib achieved a statistically significant ACR20 response (48.3% and 56.3%) compared to placebo (29.4%) at 12 weeks.84
Given the benefit of targeting TNF-α and IL-17 in RA, a novel molecule (ABT-122) that targets both human TNF and IL-17 has been developed. Two phase 1 studies85 showed that dual neutralization of TNF and IL-17 with ABT-122 has characteristics acceptable for further exploration of therapeutic potential of this agent in TNF- and IL-17A–driven immune-mediated inflammatory diseases. Another novel drug is mavrilimumab, a human monoclonal antibody that targets granulocyte–macrophage colony-stimulating factor receptor α. A recent studyshowed that long-term treatment with mavrilimumab maintained response and was well-tolerated, with no increased incidence of treatment-emergent adverse events.86
Namilumab (AMG203) is an immunoglobulin G1 monoclonal antibody that binds with high affinity to the GM-CSF ligand. In a phase 1b, randomized, double-blind study (PRIORA)87 to assess namilumab in treating active, mild-to-moderate RA, significant improvement was seen in the DAS28-CRP score with namilumab (150 and 300 mg groups combined) compared with placebo at day 43 (P = 0.0117) and also 8 weeks after last dosing at day 99 (P = 0.0154). Adverse events were similar across different doses of namilumab and placebo, and included nasopharyngitis and exacerbation/worsening of RA. Another drug showing promise in RA is fosdagrocorat (PF-04171327), a potential dissociated agonist of the glucocorticoid receptor. A multicenter, double-blind, parallel-group, active- and placebo-controlled phase 2 study randomly assigned 86 patients to receive fosdagrocorat 10 mg, fosdagrocorat 25 mg, prednisone 5 mg, or placebo, all with stable background methotrexate therapy.88 Both fosdagrocorat doses demonstrated efficacy in improving signs and symptoms in RA patients, with manageable adverse events.
Case Conclusion
There are several available treatment options for the case patient. Based on the PREMIER trial, solely increasing the dose of adalimumab is unlikely to provide a therapeutic benefit. Adding low-dose methotrexate (possibly via a parenteral route because of patient-reported gastrointestinal discomfort) might provide some synergistic and therapeutic effect. However, because of primary failure with TNFi therapy, she may benefit from the initiation of a biologic with a different mechanism of action (ie, swapping strategy). Therapeutic options include tocilizumab, abatacept, rituximab, and the Jak inhibitors (tofacitinib and baricitinib).
Summary
The optimal treatment of RA aims at achieving, and then maintaining, remission or a low disease activity. The choice of best treatment must be individualized to the patient, keeping in mind other factors, including comorbidities like fibromyalgia, history of diverticulitis (prior to use of tocilizumab), history of chronic obstructive pulmonary disease (prior to the use of abatacept), malignancy, and the presence of risk factors for infections (age, diabetes, chronic bronchitis). In a patient with inadequate response to initial biologic therapy, several options exist for the rheumatologist. Current evidence supports TNFi dose escalation for only infliximab; optimization of concurrent csDMARD or switching to a different csDMARD are other options. Cycling and swapping are other alternate approaches supported by many observational studies. While no head-to-head trials exist comparing the 2 strategies, data suggest superiority of the swapping strategy over the cycling approach. With the continuing development of novel therapeutics in RA, physicians have a growing list of treatment options to help their patients achieve disease remission.
Corresponding author: Namrata Singh, MD, 200 Hawkins Drive, Iowa City, IA 52242.
Financial disclosures: None.
1. Keystone ED, Kavanaugh KA. What to do with TNF failures. Expert Opin Drug Saf. 2005;4:149-155.
2. Kalden JR, Schulze-Koops H. Immunogenicity and loss of response to TNF inhibitors: implications for rheumatoid arthritis treatment. Nat Rev Rheumatol. 2017;13:707-718.
3. Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340:253-259.
4. Keystone EC, Kavanaugh AF, Sharp JT, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004;50(5):1400-1411.
5. Lipsky PE, van der Heijde DM, St Clair EW, et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000;343:1594-1602.
6. Finckh A, Simard JF, Gabay C, et al. Evidence for differential acquired drug resistance to anti-tumour necrosis factor agents in rheumatoid arthritis. Ann Rheum Dis. 2006;65:746-752.
7. Souto A, Maneiro JR, Gomez-Reino JJ. Rate of discontinuation and drug survival of biologic therapies in rheumatoid arthritis: a systematic review and meta-analysis of drug registries and health care databases. Rheumatology. 2016;55:523-534.
8. Hetland ML, Christensen IJ, Tarp U, et al. Direct comparison of treatment responses, remission rates, and drug adherence in patients with rheumatoid arthritis treated with adalimumab, etanercept, or infliximab: results from eight years of surveillance of clinical practice in the nationwide Danish DANBIO registry. Arthritis Rheum. 2010;62:22-32.
9. Gabay C, Riek M, Scherer A, et al. Effectiveness of biologic DMARDs in monotherapy versus in combination with synthetic DMARDs in rheumatoid arthritis: data from the Swiss Clinical Quality Management Registry. Rheumatology. 2015;54(9):1664-1672.
10. Ebina K, Hashimoto M, Yamamoto W, et al. Drug retention and discontinuation reasons between seven biologics in patients with rheumatoid arthritis-The ANSWER cohort study. PloS One. 2018;13:e0194130.
11. Wijbrandts CA, Tak PP. Prediction of response to targeted treatment in rheumatoid arthritis. Mayo Clin Proc. 2017;92:1129-1143.
12. Ulfgren AK, Andersson U, Engstrom M, et al. Systemic anti-tumor necrosis factor alpha therapy in rheumatoid arthritis down-regulates synovial tumor necrosis factor alpha synthesis. Arthritis Rheum. 2000;43:2391-2396.
13. Choi IY, Gerlag DM, Herenius MJ, et al. MRP8/14 serum levels as a strong predictor of response to biological treatments in patients with rheumatoid arthritis. Ann Rheum Dis. 2015;74:499-505.
14. Nguyen MVC, Baillet A, Romand X, et al. Prealbumin, platelet factor 4 and S100A12 combination at baseline predicts good response to TNF alpha inhibitors in rheumatoid arthritis. Joint Bone Spine. 2019;86:195-201.
15. Anderson JK, Zimmerman L, Caplan L, Michaud K. Measures of rheumatoid arthritis disease activity: Patient (PtGA) and Provider (PrGA) Global Assessment of Disease Activity, Disease Activity Score (DAS) and Disease Activity Score with 28-Joint Counts (DAS28), Simplified Disease Activity Index (SDAI), Clinical Disease Activity Index (CDAI), Patient Activity Score (PAS) and Patient Activity Score-II (PASII), Routine Assessment of Patient Index Data (RAPID), Rheumatoid Arthritis Disease Activity Index (RADAI) and Rheumatoid Arthritis Disease Activity Index-5 (RADAI-5), Chronic Arthritis Systemic Index (CASI), Patient-Based Disease Activity Score With ESR (PDAS1) and Patient-Based Disease Activity Score without ESR (PDAS2), and Mean Overall Index for Rheumatoid Arthritis (MOI-RA). Arthritis Care Res. 2011;63(suppl 11):S14-S36.
16. Katz L, Gisbert JP, Manoogian B, et al. Doubling the infliximab dose versus halving the infusion intervals in Crohn’s disease patients with loss of response. Inflamm Bowel Dis. 2012;18:2026-2033.
17. Durez P, Van den Bosch F, Corluy L, et al. A dose adjustment in patients with rheumatoid arthritis not optimally responding to a standard dose of infliximab of 3 mg/kg every 8 weeks can be effective: a Belgian prospective study. Rheumatology. 2005;44:465-468.
18. Breedveld FC, Weisman MH, Kavanaugh AF, et al. The PREMIER study: A multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54:26-37.
19. St Clair EW, Wagner CL, Fasanmade AA, et al. The relationship of serum infliximab concentrations to clinical improvement in rheumatoid arthritis: results from ATTRACT, a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46:1451-1459.
20. Rahman MU, Strusberg I, Geusens P, et al. Double-blinded infliximab dose escalation in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66:1233-1238.
21. Weinblatt ME, Schiff MH, Ruderman EM, et al. Efficacy and safety of etanercept 50 mg twice a week in patients with rheumatoid arthritis who had a suboptimal response to etanercept 50 mg once a week: results of a multicenter, randomized, double-blind, active drug-controlled study. Arthritis Rheum. 2008;58:1921-1930.
22. Curtis JR, Chen L, Luijtens K, et al. Dose escalation of certolizumab pegol from 200 mg to 400 mg every other week provides no additional efficacy in rheumatoid arthritis: an analysis of individual patient-level data. Arthritis Rheum. 2011;63:2203-2208.
23. Okazaki M, Kobayashi H, Ishii Y, et al. Real-world treatment patterns for golimumab and concomitant medications in Japanese rheumatoid arthritis patients. Rheumatol Ther. 2018;5:185-201.
24. Pappas DA, John A, Curtis JR, et al. Dosing of intravenous tocilizumab in a real-world setting of rheumatoid arthritis: analyses from the Corrona Registry. Rheumatol Ther. 2016;3:103-115.
25. Kivitz A, Olech E, Borofsky MA, et al. Two-year efficacy and safety of subcutaneous tocilizumab in combination with disease-modifying antirheumatic drugs including escalation to weekly dosing in rheumatoid arthritis. J Rheumatol. 2018;45:456-464.
26. Smolen JS, Landewe R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76:960-977.
27. Singh JA, Saag KG, Bridges SL Jr, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res. 2016;68:1-25.
28. Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006;11:81-88.
29. Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:493-507.
30. Navarro Coy NC, Brown S, Bosworth A, et al. The ‘Switch’ study protocol: a randomised-controlled trial of switching to an alternative tumour-necrosis factor (TNF)-inhibitor drug or abatacept or rituximab in patients with rheumatoid arthritis who have failed an initial TNF-inhibitor drug. BMC Musculoskelet Disord. 2014;15:452.
31. Kamal KM, Madhavan SS, Hornsby JA, et al. Use of tumor necrosis factor inhibitors in rheumatoid arthritis: a national survey of practicing United States rheumatologists. Joint Bone Spine. 2006;73:718-724.
32. Gomez-Reino JJ, Carmona L, BIOBADASER Group. Switching TNF antagonists in patients with chronic arthritis: an observational study of 488 patients over a four-year period. Arthritis Res Ther. 2006;8:R29.
33. Caporali R, Sarzi-Puttini P, Atzeni F, et al. Switching TNF-alpha antagonists in rheumatoid arthritis: The experience of the LORHEN registry. Autoimmun Rev. 2010;9:465-469.
34. Iannone F, Trotta F, Monteccuco C, et al. Etanercept maintains the clinical benefit achieved by infliximab in patients with rheumatoid arthritis who discontinued infliximab because of side effects. Ann Rheum Dis. 2007;66:249-252.
35. Virkki LM, Valleala H, Takakubo Y, et al. Outcomes of switching anti-TNF drugs in rheumatoid arthritis—a study based on observational data from the Finnish Register of Biological Treatment (ROB-FIN). Clin Rheumatol. 2011;30:1447-1454.
36. Furst DE, Gaylis N, Bray V, et al. Open-label, pilot protocol of patients with rheumatoid arthritis who switch to infliximab after an incomplete response to etanercept: the opposite study. Ann Rheum Dis. 2007;66:893-899.
37. Smolen JS, Kay J, Doyle MK, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor alpha inhibitors (GO-AFTER study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet. 2009;374:210-221.
38. Smolen JS, Kay J, Doyle M, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumor necrosis factor α inhibitors: findings with up to five years of treatment in the multicenter, randomized, double-blind, placebo-controlled, phase 3 GO-AFTER study. Arthritis Res Ther. 2015;17:14.
39. Smolen JS, Burmester G-R, Combe B, et al. Head-to-head comparison of certolizumab pegol versus adalimumab in rheumatoid arthritis: 2-year efficacy and safety results from the randomised EXXELERATE study. Lancet. 2016;388:2763-2774.
40. Chatzidionysiou K, Askling J, Eriksson J, et al. Effectiveness of TNF inhibitor switch in RA: results from the national Swedish register. Ann Rheum Dis. 2015;74:890.
41. Smolen JS, Kay J, Doyle M, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumor necrosis factor alpha inhibitors: findings with up to five years of treatment in the multicenter, randomized, double-blind, placebo-controlled, phase 3 GO-AFTER study. Arthritis Res Ther. 2015;17:14.
42. Lequerré T, Farran É, Ménard J-F, et al. Switching from an anti-TNF monoclonal antibody to soluble TNF-receptor yields better results than vice versa: An observational retrospective study of 72 rheumatoid arthritis switchers. Joint Bone Spine. 2015;82:330-337.
43. Favalli EG, Biggioggero M, Meroni PL. Methotrexate for the treatment of rheumatoid arthritis in the biologic era: Still an “anchor” drug? Autoimmun Rev. 2014;13:1102-1108.
44. Kalden JR, Schulze-Koops H. Immunogenicity and loss of response to TNF inhibitors: implications for rheumatoid arthritis treatment. Nat Rev Rheumatol. 2017;13:707-718.
45. Klareskog L, van der Heijde D, de Jager JP, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet. 2004;363:675-681.
46. Emery P, Fleischmann RM, Strusberg I, et al. Efficacy and safety of subcutaneous golimumab in methotrexate-naive patients with rheumatoid arthritis: five-year results of a randomized clinical trial. Arthritis Care Res. 2016;68:744-752.
47. Emery P, Fleischmann RM, Moreland LW, et al. Golimumab, a human anti-tumor necrosis factor alpha monoclonal antibody, injected subcutaneously every four weeks in methotrexate-naive patients with active rheumatoid arthritis: twenty-four-week results of a phase III, multicenter, randomized, double-blind, placebo-controlled study of golimumab before methotrexate as first-line therapy for early-onset rheumatoid arthritis. Arthritis Rheum. 2009;60:2272-2283.
48. Emery P, Burmester GR, Bykerk VP, et al. Evaluating drug-free remission with abatacept in early rheumatoid arthritis: results from the phase 3b, multicentre, randomised, active-controlled AVERT study of 24 months, with a 12-month, double-blind treatment period. Ann Rheum Dis. 2015;74:19-26.
49. Westhovens R, Robles M, Ximenes AC, et al. Clinical efficacy and safety of abatacept in methotrexate-naive patients with early rheumatoid arthritis and poor prognostic factors. Ann Rheum Dis. 2009;68:1870-1877.
50. Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793-2806.
51. Burmester GR, Rigby WF, van Vollenhoven RF, et al. Tocilizumab in early progressive rheumatoid arthritis: FUNCTION, a randomised controlled trial. Ann Rheum Dis. 2016;75:1081-1091.
52. Bijlsma JWJ, Welsing PMJ, Woodworth TG, et al. Early rheumatoid arthritis treated with tocilizumab, methotrexate, or their combination (U-Act-Early): a multicentre, randomised, double-blind, double-dummy, strategy trial. Lancet. 2016;388:343-355.
53. Cohen JD, Zaltni S, Kaiser MJ, et al. Secondary addition of methotrexate to partial responders to etanercept alone is effective in severe rheumatoid arthritis. Ann Rheum Dis. 2004;63:209-210.
54. Hamilton RA, Kremer JM. Why intramuscular methotrexate may be more efficacious than oral dosing in patients with rheumatoid arthritis. Br J Rheumatol. 1997;36:86-90.
55. Hoekstra M, Haagsma C, Neef C, et al. Bioavailability of higher dose methotrexate comparing oral and subcutaneous administration in patients with rheumatoid arthritis. J Rheumatol. 2004;31:645-648.
56. Herman RA, Veng-Pedersen P, Hoffman J, et al. Pharmacokinetics of low-dose methotrexate in rheumatoid arthritis patients. J Pharm Sci. 1989;78:165-171.
57. Schiff MH, Jaffe JS, Freundlich B. Head-to-head, randomised, crossover study of oral versus subcutaneous methotrexate in patients with rheumatoid arthritis: drug-exposure limitations of oral methotrexate at doses ± 15 mg may be overcome with subcutaneous administration. Ann Rheum Dis. 2014;73:1549-1551.
58. Hazlewood GS, Thorne JC, Pope JE, et al. The comparative effectiveness of oral versus subcutaneous methotrexate for the treatment of early rheumatoid arthritis. Ann Rheum Dis. 2016;75:1003-1008.
59. O’Dell JR, Petersen K, Leff R, et al. Etanercept in combination with sulfasalazine, hydroxychloroquine, or gold in the treatment of rheumatoid arthritis. J Rheumatol. 2006;33:213-218.
60. Finckh A, Dehler S, Gabay C. The effectiveness of leflunomide as a co-therapy of tumour necrosis factor inhibitors in rheumatoid arthritis: a population-based study. Ann Rheum Dis. 2009;68:33-39.
61. De Stefano R, Frati E, Nargi F, et al. Comparison of combination therapies in the treatment of rheumatoid arthritis: leflunomide-anti-TNF-alpha versus methotrexate-anti-TNF-alpha. Clin Rheumatol. 2010;29:517-524.
62. Combe B, Codreanu C, Fiocco U, et al. Etanercept and sulfasalazine, alone and combined, in patients with active rheumatoid arthritis despite receiving sulfasalazine: a double-blind comparison. Ann Rheum Dis. 2006;65:1357-1362.
63. Strangfeld A, Hierse F, Kekow J, et al. Comparative effectiveness of tumour necrosis factor α inhibitors in combination with either methotrexate or leflunomide. Ann Rheum Dis. 2009;68:1856.
64. Emery P, Keystone E, Tony HP, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67:1516.
65. Genovese MC, Becker J-C, Schiff M, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor α inhibition. N Engl J Med. 2005;353:1114-1123.
66. Emery P, Gottenberg JE, Rubbert-Roth A, et al. Rituximab versus an alternative TNF inhibitor in patients with rheumatoid arthritis who failed to respond to a single previous TNF inhibitor: SWITCH-RA, a global, observational, comparative effectiveness study. Ann Rheum Dis. 2015;74:979-984.
67. Keystone E, Emery P, Peterfy CG, et al. Rituximab inhibits structural joint damage in patients with rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitor therapies. Ann Rheum Dis. 2009;68:216.
68. Burmester GR, Blanco R, Charles-Schoeman C, et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381:451-460.
69. Genovese MC, Kremer J, Zamani O, et al. Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med. 2016;374:1243-1252.
70. Favalli EG, Biggioggero M, Marchesoni A, Meroni PL. Survival on treatment with second-line biologic therapy: a cohort study comparing cycling and swap strategies. Rheumatology. 2014;53:1664-1668.
71. Harrold LR, Reed GW, Solomon DH, et al. Comparative effectiveness of abatacept versus tocilizumab in rheumatoid arthritis patients with prior TNFi exposure in the US Corrona registry. Arthritis Res Ther. 2016;18:280.
72. Gottenberg J, Brocq O, Perdriger A, et al. Non–TNF-targeted biologic vs a second anti-TNF drug to treat rheumatoid arthritis in patients with insufficient response to a first anti-TNF drug: A randomized clinical trial. JAMA. 2016;316:1172-1180.
73. Pascart T, Philippe P, Drumez E, et al. Comparative efficacy of tocilizumab, abatacept and rituximab after non-TNF inhibitor failure: results from a multicentre study. Int J Rheum Dis. 2016;19:1093-1102.
74. Akiyama M, Kaneko Y, Kondo H, Takeuchi T. Comparison of the clinical effectiveness of tumour necrosis factor inhibitors and abatacept after insufficient response to tocilizumab in patients with rheumatoid arthritis. Clin Rheumatol. 2016;35:2829-2834.
75. Schoels M, Aletaha D, Smolen JS, Wong JB. Comparative effectiveness and safety of biological treatment options after tumour necrosis factor α inhibitor failure in rheumatoid arthritis: systematic review and indirect pairwise meta-analysis. Ann Rheum Dis. 2012;71:1303.
76. Soliman MM, Hyrich KL, Lunt M, et al. Rituximab or a second anti-tumor necrosis factor therapy for rheumatoid arthritis patients who have failed their first anti-tumor necrosis factor therapy? Comparative analysis from the British Society for Rheumatology Biologics Register. Arthritis Care Res. 2012;64:1108-1115.
77. Chatzidionysiou K, Vollenhoven RF. Rituximab versus anti-TNF in patients who previously failed one TNF inhibitor in an observational cohort. Scand J Rheumatol. 2013;42:190-195.
78. Johnston SS, Turpcu A, Shi N, et al. Risk of infections in rheumatoid arthritis patients switching from anti-TNF agents to rituximab, abatacept, or another anti-TNF agent, a retrospective administrative claims analysis. Semim Arthritis Rheum. 2013;43:39-47.
79. Curtis JR, Xie F, Chen L, et al. The comparative risk of serious infections among rheumatoid arthritis patients starting or switching biological agents. Ann Rheum Dis. 2011;70:1401.
80. Desai RJ, Solomon DH, Jin Y, et al. Temporal trends in use of biologic DMARDs for rheumatoid arthritis in the United States: a cohort study of publicly and privately insured patients. J Manag Care Spec Pharm. 2017;23:809-814.
81. Jin Y, Desai RJ, Liu J, et al. Factors associated with initial or subsequent choice of biologic disease-modifying antirheumatic drugs for treatment of rheumatoid arthritis. Arthritis Res Ther. 2017;19:159.
82. Bonafede MMK, McMorrow D, Proudfoot C, et al. Treatment persistence and healthcare costs among patients with rheumatoid arthritis after a change in targeted therapy. Am Health Drug Benefits. 2018;11:192-202.
83. US Food and Drug Administration. Biosimilars are safe, effective treatment options. www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeuticbiologicapplications/biosimilars/. Accessed November 9, 2018.
84. Genovese MC, Greenwald M, Codding C, et al. Peficitinib, a JAK inhibitor, in combination with limited conventional synthetic disease-modifying antirheumatic drugs in the treatment of moderate-to-severe rheumatoid arthritis. Arthritis Rheumatol. 2017;69:932-942.
85. Fleischmann RM, Wagner F, Kivitz AJ, et al. Safety, tolerability, and pharmacodynamics of ABT-122, a tumor necrosis factor- and interleukin-17-targeted dual variable domain immunoglobulin, in patients with rheumatoid arthritis. Arthritis Rheumatol. 2017;69:2283-2291.
86. Burmester GR, McInnes IB, Kremer JM, et al. Mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor alpha monoclonal antibody: long-term safety and efficacy in patients with rheumatoid arthritis. Arthritis Rheumatol. 2018;70:679-689.
87. Huizinga TW, Batalov A, Stoilov R, et al. Phase 1b randomized, double-blind study of namilumab, an anti-granulocyte macrophage colony-stimulating factor monoclonal antibody, in mild-to-moderate rheumatoid arthritis. Arthritis Res Ther. 2017;19:53.
88. Stock T, Fleishaker D, Wang X, et al. Improved disease activity with fosdagrocorat (PF-04171327), a partial agonist of the glucocorticoid receptor, in patients with rheumatoid arthritis: a Phase 2 randomized study. Int J Rheum Dis. 2017;20:960-970.
89. Orencia [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2013.
90. Humira[package insert]. North Chicago, IL: AbbVie; 2012.
91. Kineret [package insert]. Stockholm, Sweden: Sobi; 2012.
92. Olumiant [package insert]. Indianapolis, IN: Lilly USA, LLC; 2018.
93. Cimzia [package insert]. Smyrna, GA: UCB, Inc; 2008.
94. Enbrel [package insert]. Thousand Oaks, CA: Immunex Corporation; 1998.
95. Simponi [package insert]. Horsham, PA: Janssen Biotech, Inc; 2009.
96. Remicade [package insert]. Horsham, PA: Janssen Biotech, Inc; 1998.
97. Rituxan [package insert]. South San Francisco, CA: Genetech, Inc; 1997.
98. Kevzara [package insert]. Bridgewater, NJ: Sanofi-Aventis US LLC; 2018.
99. Actemra [package insert]. South San Francisco, CA: Genentech, Inc; 2013.
100. Xeljanz [package insert]. New York, NY: Pfizer Inc; 2016.
1. Keystone ED, Kavanaugh KA. What to do with TNF failures. Expert Opin Drug Saf. 2005;4:149-155.
2. Kalden JR, Schulze-Koops H. Immunogenicity and loss of response to TNF inhibitors: implications for rheumatoid arthritis treatment. Nat Rev Rheumatol. 2017;13:707-718.
3. Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340:253-259.
4. Keystone EC, Kavanaugh AF, Sharp JT, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004;50(5):1400-1411.
5. Lipsky PE, van der Heijde DM, St Clair EW, et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000;343:1594-1602.
6. Finckh A, Simard JF, Gabay C, et al. Evidence for differential acquired drug resistance to anti-tumour necrosis factor agents in rheumatoid arthritis. Ann Rheum Dis. 2006;65:746-752.
7. Souto A, Maneiro JR, Gomez-Reino JJ. Rate of discontinuation and drug survival of biologic therapies in rheumatoid arthritis: a systematic review and meta-analysis of drug registries and health care databases. Rheumatology. 2016;55:523-534.
8. Hetland ML, Christensen IJ, Tarp U, et al. Direct comparison of treatment responses, remission rates, and drug adherence in patients with rheumatoid arthritis treated with adalimumab, etanercept, or infliximab: results from eight years of surveillance of clinical practice in the nationwide Danish DANBIO registry. Arthritis Rheum. 2010;62:22-32.
9. Gabay C, Riek M, Scherer A, et al. Effectiveness of biologic DMARDs in monotherapy versus in combination with synthetic DMARDs in rheumatoid arthritis: data from the Swiss Clinical Quality Management Registry. Rheumatology. 2015;54(9):1664-1672.
10. Ebina K, Hashimoto M, Yamamoto W, et al. Drug retention and discontinuation reasons between seven biologics in patients with rheumatoid arthritis-The ANSWER cohort study. PloS One. 2018;13:e0194130.
11. Wijbrandts CA, Tak PP. Prediction of response to targeted treatment in rheumatoid arthritis. Mayo Clin Proc. 2017;92:1129-1143.
12. Ulfgren AK, Andersson U, Engstrom M, et al. Systemic anti-tumor necrosis factor alpha therapy in rheumatoid arthritis down-regulates synovial tumor necrosis factor alpha synthesis. Arthritis Rheum. 2000;43:2391-2396.
13. Choi IY, Gerlag DM, Herenius MJ, et al. MRP8/14 serum levels as a strong predictor of response to biological treatments in patients with rheumatoid arthritis. Ann Rheum Dis. 2015;74:499-505.
14. Nguyen MVC, Baillet A, Romand X, et al. Prealbumin, platelet factor 4 and S100A12 combination at baseline predicts good response to TNF alpha inhibitors in rheumatoid arthritis. Joint Bone Spine. 2019;86:195-201.
15. Anderson JK, Zimmerman L, Caplan L, Michaud K. Measures of rheumatoid arthritis disease activity: Patient (PtGA) and Provider (PrGA) Global Assessment of Disease Activity, Disease Activity Score (DAS) and Disease Activity Score with 28-Joint Counts (DAS28), Simplified Disease Activity Index (SDAI), Clinical Disease Activity Index (CDAI), Patient Activity Score (PAS) and Patient Activity Score-II (PASII), Routine Assessment of Patient Index Data (RAPID), Rheumatoid Arthritis Disease Activity Index (RADAI) and Rheumatoid Arthritis Disease Activity Index-5 (RADAI-5), Chronic Arthritis Systemic Index (CASI), Patient-Based Disease Activity Score With ESR (PDAS1) and Patient-Based Disease Activity Score without ESR (PDAS2), and Mean Overall Index for Rheumatoid Arthritis (MOI-RA). Arthritis Care Res. 2011;63(suppl 11):S14-S36.
16. Katz L, Gisbert JP, Manoogian B, et al. Doubling the infliximab dose versus halving the infusion intervals in Crohn’s disease patients with loss of response. Inflamm Bowel Dis. 2012;18:2026-2033.
17. Durez P, Van den Bosch F, Corluy L, et al. A dose adjustment in patients with rheumatoid arthritis not optimally responding to a standard dose of infliximab of 3 mg/kg every 8 weeks can be effective: a Belgian prospective study. Rheumatology. 2005;44:465-468.
18. Breedveld FC, Weisman MH, Kavanaugh AF, et al. The PREMIER study: A multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54:26-37.
19. St Clair EW, Wagner CL, Fasanmade AA, et al. The relationship of serum infliximab concentrations to clinical improvement in rheumatoid arthritis: results from ATTRACT, a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46:1451-1459.
20. Rahman MU, Strusberg I, Geusens P, et al. Double-blinded infliximab dose escalation in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66:1233-1238.
21. Weinblatt ME, Schiff MH, Ruderman EM, et al. Efficacy and safety of etanercept 50 mg twice a week in patients with rheumatoid arthritis who had a suboptimal response to etanercept 50 mg once a week: results of a multicenter, randomized, double-blind, active drug-controlled study. Arthritis Rheum. 2008;58:1921-1930.
22. Curtis JR, Chen L, Luijtens K, et al. Dose escalation of certolizumab pegol from 200 mg to 400 mg every other week provides no additional efficacy in rheumatoid arthritis: an analysis of individual patient-level data. Arthritis Rheum. 2011;63:2203-2208.
23. Okazaki M, Kobayashi H, Ishii Y, et al. Real-world treatment patterns for golimumab and concomitant medications in Japanese rheumatoid arthritis patients. Rheumatol Ther. 2018;5:185-201.
24. Pappas DA, John A, Curtis JR, et al. Dosing of intravenous tocilizumab in a real-world setting of rheumatoid arthritis: analyses from the Corrona Registry. Rheumatol Ther. 2016;3:103-115.
25. Kivitz A, Olech E, Borofsky MA, et al. Two-year efficacy and safety of subcutaneous tocilizumab in combination with disease-modifying antirheumatic drugs including escalation to weekly dosing in rheumatoid arthritis. J Rheumatol. 2018;45:456-464.
26. Smolen JS, Landewe R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76:960-977.
27. Singh JA, Saag KG, Bridges SL Jr, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res. 2016;68:1-25.
28. Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006;11:81-88.
29. Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:493-507.
30. Navarro Coy NC, Brown S, Bosworth A, et al. The ‘Switch’ study protocol: a randomised-controlled trial of switching to an alternative tumour-necrosis factor (TNF)-inhibitor drug or abatacept or rituximab in patients with rheumatoid arthritis who have failed an initial TNF-inhibitor drug. BMC Musculoskelet Disord. 2014;15:452.
31. Kamal KM, Madhavan SS, Hornsby JA, et al. Use of tumor necrosis factor inhibitors in rheumatoid arthritis: a national survey of practicing United States rheumatologists. Joint Bone Spine. 2006;73:718-724.
32. Gomez-Reino JJ, Carmona L, BIOBADASER Group. Switching TNF antagonists in patients with chronic arthritis: an observational study of 488 patients over a four-year period. Arthritis Res Ther. 2006;8:R29.
33. Caporali R, Sarzi-Puttini P, Atzeni F, et al. Switching TNF-alpha antagonists in rheumatoid arthritis: The experience of the LORHEN registry. Autoimmun Rev. 2010;9:465-469.
34. Iannone F, Trotta F, Monteccuco C, et al. Etanercept maintains the clinical benefit achieved by infliximab in patients with rheumatoid arthritis who discontinued infliximab because of side effects. Ann Rheum Dis. 2007;66:249-252.
35. Virkki LM, Valleala H, Takakubo Y, et al. Outcomes of switching anti-TNF drugs in rheumatoid arthritis—a study based on observational data from the Finnish Register of Biological Treatment (ROB-FIN). Clin Rheumatol. 2011;30:1447-1454.
36. Furst DE, Gaylis N, Bray V, et al. Open-label, pilot protocol of patients with rheumatoid arthritis who switch to infliximab after an incomplete response to etanercept: the opposite study. Ann Rheum Dis. 2007;66:893-899.
37. Smolen JS, Kay J, Doyle MK, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor alpha inhibitors (GO-AFTER study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet. 2009;374:210-221.
38. Smolen JS, Kay J, Doyle M, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumor necrosis factor α inhibitors: findings with up to five years of treatment in the multicenter, randomized, double-blind, placebo-controlled, phase 3 GO-AFTER study. Arthritis Res Ther. 2015;17:14.
39. Smolen JS, Burmester G-R, Combe B, et al. Head-to-head comparison of certolizumab pegol versus adalimumab in rheumatoid arthritis: 2-year efficacy and safety results from the randomised EXXELERATE study. Lancet. 2016;388:2763-2774.
40. Chatzidionysiou K, Askling J, Eriksson J, et al. Effectiveness of TNF inhibitor switch in RA: results from the national Swedish register. Ann Rheum Dis. 2015;74:890.
41. Smolen JS, Kay J, Doyle M, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumor necrosis factor alpha inhibitors: findings with up to five years of treatment in the multicenter, randomized, double-blind, placebo-controlled, phase 3 GO-AFTER study. Arthritis Res Ther. 2015;17:14.
42. Lequerré T, Farran É, Ménard J-F, et al. Switching from an anti-TNF monoclonal antibody to soluble TNF-receptor yields better results than vice versa: An observational retrospective study of 72 rheumatoid arthritis switchers. Joint Bone Spine. 2015;82:330-337.
43. Favalli EG, Biggioggero M, Meroni PL. Methotrexate for the treatment of rheumatoid arthritis in the biologic era: Still an “anchor” drug? Autoimmun Rev. 2014;13:1102-1108.
44. Kalden JR, Schulze-Koops H. Immunogenicity and loss of response to TNF inhibitors: implications for rheumatoid arthritis treatment. Nat Rev Rheumatol. 2017;13:707-718.
45. Klareskog L, van der Heijde D, de Jager JP, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet. 2004;363:675-681.
46. Emery P, Fleischmann RM, Strusberg I, et al. Efficacy and safety of subcutaneous golimumab in methotrexate-naive patients with rheumatoid arthritis: five-year results of a randomized clinical trial. Arthritis Care Res. 2016;68:744-752.
47. Emery P, Fleischmann RM, Moreland LW, et al. Golimumab, a human anti-tumor necrosis factor alpha monoclonal antibody, injected subcutaneously every four weeks in methotrexate-naive patients with active rheumatoid arthritis: twenty-four-week results of a phase III, multicenter, randomized, double-blind, placebo-controlled study of golimumab before methotrexate as first-line therapy for early-onset rheumatoid arthritis. Arthritis Rheum. 2009;60:2272-2283.
48. Emery P, Burmester GR, Bykerk VP, et al. Evaluating drug-free remission with abatacept in early rheumatoid arthritis: results from the phase 3b, multicentre, randomised, active-controlled AVERT study of 24 months, with a 12-month, double-blind treatment period. Ann Rheum Dis. 2015;74:19-26.
49. Westhovens R, Robles M, Ximenes AC, et al. Clinical efficacy and safety of abatacept in methotrexate-naive patients with early rheumatoid arthritis and poor prognostic factors. Ann Rheum Dis. 2009;68:1870-1877.
50. Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793-2806.
51. Burmester GR, Rigby WF, van Vollenhoven RF, et al. Tocilizumab in early progressive rheumatoid arthritis: FUNCTION, a randomised controlled trial. Ann Rheum Dis. 2016;75:1081-1091.
52. Bijlsma JWJ, Welsing PMJ, Woodworth TG, et al. Early rheumatoid arthritis treated with tocilizumab, methotrexate, or their combination (U-Act-Early): a multicentre, randomised, double-blind, double-dummy, strategy trial. Lancet. 2016;388:343-355.
53. Cohen JD, Zaltni S, Kaiser MJ, et al. Secondary addition of methotrexate to partial responders to etanercept alone is effective in severe rheumatoid arthritis. Ann Rheum Dis. 2004;63:209-210.
54. Hamilton RA, Kremer JM. Why intramuscular methotrexate may be more efficacious than oral dosing in patients with rheumatoid arthritis. Br J Rheumatol. 1997;36:86-90.
55. Hoekstra M, Haagsma C, Neef C, et al. Bioavailability of higher dose methotrexate comparing oral and subcutaneous administration in patients with rheumatoid arthritis. J Rheumatol. 2004;31:645-648.
56. Herman RA, Veng-Pedersen P, Hoffman J, et al. Pharmacokinetics of low-dose methotrexate in rheumatoid arthritis patients. J Pharm Sci. 1989;78:165-171.
57. Schiff MH, Jaffe JS, Freundlich B. Head-to-head, randomised, crossover study of oral versus subcutaneous methotrexate in patients with rheumatoid arthritis: drug-exposure limitations of oral methotrexate at doses ± 15 mg may be overcome with subcutaneous administration. Ann Rheum Dis. 2014;73:1549-1551.
58. Hazlewood GS, Thorne JC, Pope JE, et al. The comparative effectiveness of oral versus subcutaneous methotrexate for the treatment of early rheumatoid arthritis. Ann Rheum Dis. 2016;75:1003-1008.
59. O’Dell JR, Petersen K, Leff R, et al. Etanercept in combination with sulfasalazine, hydroxychloroquine, or gold in the treatment of rheumatoid arthritis. J Rheumatol. 2006;33:213-218.
60. Finckh A, Dehler S, Gabay C. The effectiveness of leflunomide as a co-therapy of tumour necrosis factor inhibitors in rheumatoid arthritis: a population-based study. Ann Rheum Dis. 2009;68:33-39.
61. De Stefano R, Frati E, Nargi F, et al. Comparison of combination therapies in the treatment of rheumatoid arthritis: leflunomide-anti-TNF-alpha versus methotrexate-anti-TNF-alpha. Clin Rheumatol. 2010;29:517-524.
62. Combe B, Codreanu C, Fiocco U, et al. Etanercept and sulfasalazine, alone and combined, in patients with active rheumatoid arthritis despite receiving sulfasalazine: a double-blind comparison. Ann Rheum Dis. 2006;65:1357-1362.
63. Strangfeld A, Hierse F, Kekow J, et al. Comparative effectiveness of tumour necrosis factor α inhibitors in combination with either methotrexate or leflunomide. Ann Rheum Dis. 2009;68:1856.
64. Emery P, Keystone E, Tony HP, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67:1516.
65. Genovese MC, Becker J-C, Schiff M, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor α inhibition. N Engl J Med. 2005;353:1114-1123.
66. Emery P, Gottenberg JE, Rubbert-Roth A, et al. Rituximab versus an alternative TNF inhibitor in patients with rheumatoid arthritis who failed to respond to a single previous TNF inhibitor: SWITCH-RA, a global, observational, comparative effectiveness study. Ann Rheum Dis. 2015;74:979-984.
67. Keystone E, Emery P, Peterfy CG, et al. Rituximab inhibits structural joint damage in patients with rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitor therapies. Ann Rheum Dis. 2009;68:216.
68. Burmester GR, Blanco R, Charles-Schoeman C, et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381:451-460.
69. Genovese MC, Kremer J, Zamani O, et al. Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med. 2016;374:1243-1252.
70. Favalli EG, Biggioggero M, Marchesoni A, Meroni PL. Survival on treatment with second-line biologic therapy: a cohort study comparing cycling and swap strategies. Rheumatology. 2014;53:1664-1668.
71. Harrold LR, Reed GW, Solomon DH, et al. Comparative effectiveness of abatacept versus tocilizumab in rheumatoid arthritis patients with prior TNFi exposure in the US Corrona registry. Arthritis Res Ther. 2016;18:280.
72. Gottenberg J, Brocq O, Perdriger A, et al. Non–TNF-targeted biologic vs a second anti-TNF drug to treat rheumatoid arthritis in patients with insufficient response to a first anti-TNF drug: A randomized clinical trial. JAMA. 2016;316:1172-1180.
73. Pascart T, Philippe P, Drumez E, et al. Comparative efficacy of tocilizumab, abatacept and rituximab after non-TNF inhibitor failure: results from a multicentre study. Int J Rheum Dis. 2016;19:1093-1102.
74. Akiyama M, Kaneko Y, Kondo H, Takeuchi T. Comparison of the clinical effectiveness of tumour necrosis factor inhibitors and abatacept after insufficient response to tocilizumab in patients with rheumatoid arthritis. Clin Rheumatol. 2016;35:2829-2834.
75. Schoels M, Aletaha D, Smolen JS, Wong JB. Comparative effectiveness and safety of biological treatment options after tumour necrosis factor α inhibitor failure in rheumatoid arthritis: systematic review and indirect pairwise meta-analysis. Ann Rheum Dis. 2012;71:1303.
76. Soliman MM, Hyrich KL, Lunt M, et al. Rituximab or a second anti-tumor necrosis factor therapy for rheumatoid arthritis patients who have failed their first anti-tumor necrosis factor therapy? Comparative analysis from the British Society for Rheumatology Biologics Register. Arthritis Care Res. 2012;64:1108-1115.
77. Chatzidionysiou K, Vollenhoven RF. Rituximab versus anti-TNF in patients who previously failed one TNF inhibitor in an observational cohort. Scand J Rheumatol. 2013;42:190-195.
78. Johnston SS, Turpcu A, Shi N, et al. Risk of infections in rheumatoid arthritis patients switching from anti-TNF agents to rituximab, abatacept, or another anti-TNF agent, a retrospective administrative claims analysis. Semim Arthritis Rheum. 2013;43:39-47.
79. Curtis JR, Xie F, Chen L, et al. The comparative risk of serious infections among rheumatoid arthritis patients starting or switching biological agents. Ann Rheum Dis. 2011;70:1401.
80. Desai RJ, Solomon DH, Jin Y, et al. Temporal trends in use of biologic DMARDs for rheumatoid arthritis in the United States: a cohort study of publicly and privately insured patients. J Manag Care Spec Pharm. 2017;23:809-814.
81. Jin Y, Desai RJ, Liu J, et al. Factors associated with initial or subsequent choice of biologic disease-modifying antirheumatic drugs for treatment of rheumatoid arthritis. Arthritis Res Ther. 2017;19:159.
82. Bonafede MMK, McMorrow D, Proudfoot C, et al. Treatment persistence and healthcare costs among patients with rheumatoid arthritis after a change in targeted therapy. Am Health Drug Benefits. 2018;11:192-202.
83. US Food and Drug Administration. Biosimilars are safe, effective treatment options. www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeuticbiologicapplications/biosimilars/. Accessed November 9, 2018.
84. Genovese MC, Greenwald M, Codding C, et al. Peficitinib, a JAK inhibitor, in combination with limited conventional synthetic disease-modifying antirheumatic drugs in the treatment of moderate-to-severe rheumatoid arthritis. Arthritis Rheumatol. 2017;69:932-942.
85. Fleischmann RM, Wagner F, Kivitz AJ, et al. Safety, tolerability, and pharmacodynamics of ABT-122, a tumor necrosis factor- and interleukin-17-targeted dual variable domain immunoglobulin, in patients with rheumatoid arthritis. Arthritis Rheumatol. 2017;69:2283-2291.
86. Burmester GR, McInnes IB, Kremer JM, et al. Mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor alpha monoclonal antibody: long-term safety and efficacy in patients with rheumatoid arthritis. Arthritis Rheumatol. 2018;70:679-689.
87. Huizinga TW, Batalov A, Stoilov R, et al. Phase 1b randomized, double-blind study of namilumab, an anti-granulocyte macrophage colony-stimulating factor monoclonal antibody, in mild-to-moderate rheumatoid arthritis. Arthritis Res Ther. 2017;19:53.
88. Stock T, Fleishaker D, Wang X, et al. Improved disease activity with fosdagrocorat (PF-04171327), a partial agonist of the glucocorticoid receptor, in patients with rheumatoid arthritis: a Phase 2 randomized study. Int J Rheum Dis. 2017;20:960-970.
89. Orencia [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2013.
90. Humira[package insert]. North Chicago, IL: AbbVie; 2012.
91. Kineret [package insert]. Stockholm, Sweden: Sobi; 2012.
92. Olumiant [package insert]. Indianapolis, IN: Lilly USA, LLC; 2018.
93. Cimzia [package insert]. Smyrna, GA: UCB, Inc; 2008.
94. Enbrel [package insert]. Thousand Oaks, CA: Immunex Corporation; 1998.
95. Simponi [package insert]. Horsham, PA: Janssen Biotech, Inc; 2009.
96. Remicade [package insert]. Horsham, PA: Janssen Biotech, Inc; 1998.
97. Rituxan [package insert]. South San Francisco, CA: Genetech, Inc; 1997.
98. Kevzara [package insert]. Bridgewater, NJ: Sanofi-Aventis US LLC; 2018.
99. Actemra [package insert]. South San Francisco, CA: Genentech, Inc; 2013.
100. Xeljanz [package insert]. New York, NY: Pfizer Inc; 2016.
Decreasing Treatment of Asymptomatic Bacteriuria: An Interprofessional Approach to Antibiotic Stewardship
From the Mayo Clinic, Rochester, MN.
Abstract
- Objective: Asymptomatic bacteriuria (ASB) denotes asymptomatic carriage of bacteria within the urinary tract and does not require treatment in most patient populations. Unnecessary antimicrobial treatment has several consequences, including promotion of antimicrobial resistance, potential for medication adverse effects, and risk for Clostridiodes difficile infection. The aim of this quality improvement effort was to decrease both the unnecessary ordering of urine culture studies and unnecessary treatment of ASB.
- Methods: This is a single-center study of patients who received care on 3 internal medicine units at a large, academic medical center. We sought to determine the impact of information technology and educational interventions to decrease both inappropriate urine culture ordering and treatment of ASB. Data from included patients were collected over 3 1-month time periods: baseline, post-information technology intervention, and post-educational intervention.
- Results: There was a reduction in the percentage of patients who received antibiotics for ASB in the post-education intervention period as compared to baseline (35% vs 42%). The proportion of total urine cultures ordered by internal medicine clinicians did not change after an information technology intervention to redesign the computerized physician order entry screen for urine cultures.
- Conclusion: Educational interventions are effective ways to reduce rates of inappropriate treatment of ASB in patients admitted to internal medicine services.
Keywords: asymptomatic bacteriuria, UTI, information technology, education, quality.
Asymptomatic bacteriuria (ASB) is a common condition in which bacteria are recovered from a urine culture (UC) in patients without symptoms suggestive of urinary tract infection (UTI), with no pathologic consequences to most patients who are not treated.1,2 Patients with ASB do not exhibit symptoms of a UTI such as dysuria, increased frequency of urination, increased urgency, suprapubic tenderness, or costovertebral pain. Treatment with antibiotics is not indicated for most patients with ASB.1,3 According to the Infectious Diseases Society of America (IDSA), screening for bacteriuria and treatment for positive results is only indicated during pregnancy and prior to urologic procedures with anticipated breach of the mucosal lining.1
An estimated 20% to 52% of patients in hospital settings receive inappropriate treatment with antibiotics for ASB.4 Unnecessary prescribing of antibiotics has several negative consequences, including increased rates of antibiotic resistance, Clostridioides difficile infection, and medication adverse events, as well as increased health care costs.2,5 Antimicrobial stewardship programs to improve judicious use of antimicrobials are paramount to reducing these consequences, and their importance is heightened with recent requirements for antimicrobial stewardship put forth by The Joint Commission and the Centers for Medicare & Medicaid Services.6,7
A previous review of UC and antimicrobial use in patients for purposes of quality improvement at our institution over a 2-month period showed that of 59 patients with positive UCs, 47 patients (80%) did not have documented symptoms of a UTI. Of these 47 patients with ASB, 29 (61.7%) received antimicrobial treatment unnecessarily (unpublished data). We convened a group of clinicians and nonclinicians representing the areas of infectious disease, pharmacy, microbiology, statistics, and hospital internal medicine (IM) to examine the unnecessary treatment of ASB in our institution. Our objective was to address 2 antimicrobial stewardship issues: inappropriate UC ordering and unnecessary use of antibiotics to treat ASB. Our aim was to reduce the inappropriate ordering of UCs and to reduce treatment of ASB.
Methods
Setting
The study was conducted on 3 IM nursing units with a total of 83 beds at a large tertiary care academic medical center in the midwestern United States, and was approved by the organization’s Institutional Review Board.
Participants
We included all non-pregnant patients aged 18 years or older who received care from an IM primary service. These patients were admitted directly to an IM team through the emergency department (ED) or transferred to an IM team after an initial stay in the intensive care unit.
Data Source
Microbiology laboratory reports generated from the electronic health record were used to identify all patients with a collected UC sample who received care from an IM service prior to discharge. Urine samples were collected by midstream catch or catheterization. Data on urine Gram stain and urine dipstick were not included. Henceforth, the phrase “urine culture order” indicates that a UC was both ordered and performed. Data reports were generated for the month of August 2016 to determine the baseline number of UCs ordered. Charts of patients with positive UCs were reviewed to determine if antibiotics were started for the positive UC and whether the patient had signs or symptoms consistent with a UTI. If antibiotics were started in the absence of signs or symptoms to support a UTI, the patient was determined to have been unnecessarily treated for ASB. Reports were then generated for the month after each intervention was implemented, with the same chart review undertaken for positive UCs. Bacteriuria was defined in our study as the presence of microbial growth greater than 10,000 CFU/mL in UC.
Interventions
Initial analysis by our study group determined that lack of electronic clinical decision support (CDS) at the point of care and provider knowledge gaps in interpreting positive UCs were the 2 main contributors to unnecessary UC orders and unnecessary treatment of positive UCs, respectively. We reviewed the work of other groups who reported interventions to decrease treatment of ASB, ranging from educational presentations to pocket cards and treatment algorithms.8-13 We hypothesized that there would be a decrease in UC orders with CDS embedded in the computerized order entry screen, and that we would decrease unnecessary treatment of positive UCs by educating clinicians on indications for appropriate antibiotic prescribing in the setting of a positive UC.
Information technology intervention. The first intervention implemented involved redesign of the UC ordering screen in the computerized physician order entry (CPOE) system. This intervention went live hospital-wide, including the IM floors, intensive care units, and all other areas except the ED, on February 1, 2017 (Figure 1). The ordering screen required the prescriber to select from a list of appropriate indications for ordering a UC, including urine frequency, urgency, or dysuria; unexplained suprapubic or flank pain; fever in patients without another recognized cause; screening obtained prior to urologic procedure; or screening during pregnancy. An additional message advised prescribers to avoid ordering the culture if the patient had malodorous or cloudy urine, pyuria without urinary symptoms, or had an alternative cause of fever. Before we implemented the information technology (IT) intervention, there had been no specific point-of-care guidance on UC ordering.

Educational intervention. The second intervention, driven by clinical pharmacists, involved active and passive education of prescribers specifically designed to address unnecessary treatment of ASB. The IT intervention with CDS for UC ordering remained live. Presentations designed by the study group summarizing the appropriate indications for ordering a UC, distinguishing ASB from UTI, and discouraging treatment of ASB were delivered via a variety of routes by clinical pharmacists to nurses, nurse practitioners, physician assistants, pharmacists, medical residents, and staff physicians providing care to patients on the 3 IM units over a 1-month period in March 2017. The presentations contained the same basic content, but the information was delivered to target each specific audience group.
Medical residents received a 10-minute live presentation during a conference. Nurse practitioners, physician assistants, and staff physicians received a presentation via email, and highlights of the presentation were delivered by clinical pharmacists at their respective monthly group meetings. A handout was presented to nursing staff at nursing huddles, and presentation slides were distributed by email. Educational posters were posted in the medical resident workrooms, nursing breakrooms, and staff bathrooms on the units.
Outcome Measurements
The endpoints of interest were the percentage of patients with positive UCs unnecessarily treated for ASB before and after each intervention and the number of UCs ordered at baseline and after implementation of each intervention. Counterbalance measures assessed included the incidence of UTI, pyelonephritis, or urosepsis within 7 days of positive UC for patients who did not receive antibiotic treatment for ASB.
Results
Data from a total of 270 cultures were examined from IM nursing units. A total of 117 UCs were ordered during the baseline period before interventions were implemented. For a period of 1 month following activation of the IT intervention, 73 UCs were ordered. For a period of 1 month following the educational interventions, 80 UCs were ordered. Of these, 61 (52%) UCs were positive at baseline, 37 (51%) after the IT intervention, and 41 (51%) after the educational intervention. Patient characteristics were similar between the 3 groups (Table); 64.7% of patients were female in their early to mid-seventies. The majority of UCs were ordered by providers in the ED in all 3 periods examined (51%-70%). The percentage of patients who received antibiotics prior to UC for another indication (including bacteriuria) in the baseline, post-IT intervention, and post-education intervention groups were 30%, 27%, and 45%, respectively.

The study outcomes are summarized in Figure 2. Among patients with positive cultures, there was not a reduction in inappropriate treatment of ASB compared to baseline after the IT intervention (48% vs 42%). Following the education intervention, there was a reduction in unnecessary ASB treatment as compared both to baseline (35% vs 42%) and to post-IT intervention (35% vs 48%). There was no difference between the 3 study periods in the percentage of total UCs ordered by IM clinicians. The counterbalance measure showed that 1 patient who did not receive antibiotics within 7 days of a positive UC developed pyelonephritis, UTI, or sepsis due to a UTI in each intervention group.
 ordered and cases of asymptomatic bacteriuria (ASB) treated at baseline and after interventions. UTI, urinary tract infection.")
Discussion
The results of this study demonstrate the role of multimodal interventions in antimicrobial stewardship and add to the growing body of evidence supporting the work of antimicrobial stewardship programs. Our multidisciplinary study group and multipronged intervention follow recent guideline recommendations for antimicrobial stewardship program interventions against unnecessary treatment of ASB.14 Initial analysis by our study group determined lack of CDS at the point of care and provider knowledge gaps in interpreting positive UCs as the 2 main contributors to unnecessary UC orders and unnecessary treatment of positive UCs in our local practice culture. The IT component of our intervention was intended to provide CDS for ordering UCs, and the education component focused on informing clinicians’ treatment decisions for positive UCs.
It has been suggested that the type of stewardship intervention that is most effective fits the specific needs and resources of an institution.14,15 And although the IDSA does not recommend education as a stand-alone intervention,16 we found it to be an effective intervention for our clinicians in our work environment. However, since the CPOE guidance was in place during the educational study periods, it is possible that the effect was due to a combination of these 2 approaches. Our pre-intervention ASB treatment rates were consistent with a recent meta-analysis in which the rate of inappropriate treatment of ASB was 45%.17 This meta-analysis found educational and organizational interventions led to a mean absolute risk reduction of 33%. After the education intervention, we saw a 7% decrease in unnecessary treatment of ASB compared to baseline, and a 13% decrease compared to the month just prior to the educational intervention.
Lessons learned from our work included how clear review of local processes can inform quality improvement interventions. For instance, we initially hypothesized that IM clinicians would benefit from point-of-care CDS guidance, but such guidance used alone without educational interventions was not supported by the results. We also determined that the majority of UCs from patients on general medicine units were ordered by ED providers. This revealed an opportunity to implement similar interventions in the ED, as this was the initial point of contact for many of these patients.
As with any clinical intervention, the anticipated benefits should be weighed against potential harm. Using counterbalance measures, we found there was minimal risk in the occurrence of UTI, pyelonephritis, or sepsis if clinicians avoided treating ASB. This finding is consistent with IDSA guideline recommendations and other studies that suggest that withholding treatment for asymptomatic bacteriuria does not lead to worse outcomes.1
This study has several limitations. Data were obtained through review of the electronic health record and therefore documentation may be incomplete. Also, antimicrobials for empiric coverage or treatment for other infections (eg, pneumonia, sepsis) may have confounded our results, as empirical antimicrobials were given to 27% to 45% of patients prior to UC. This was a quality improvement project carried out over defined time intervals, and thus our sample size was limited and not adequately powered to show statistical significance. Additionally, given the bundling of interventions, it is difficult to determine the impact of each intervention independently. Although CDS for UC ordering may not have influenced ordering, it is possible that the IT intervention raised awareness of ASB and influenced treatment practices.
Conclusion
Our work supports the principles of antibiotic stewardship as brought forth by IDSA.16 This work was the effort of a multidisciplinary team, which aligns with recommendations by Daniel and colleagues, published after our study had ended, for reducing overtreatment of ASB.14 Additionally, our study results provided valuable information for our institution. Although improvements in management of ASB were modest, the success of provider education and identification of other work areas and clinicians to target for future intervention were helpful in consideration of further studies. This work will also aid us in developing an expected effect size for future studies. We plan to provide ongoing education for IM providers as well as education in the ED to target providers who make first contact with patients admitted to inpatient services. In addition, the CPOE UC ordering screen message will continue to be used hospital-wide and will be expanded to the ED ordering system. Our interventions, experiences, and challenges may be used by other institutions to design effective antimicrobial stewardship interventions directed towards reducing rates of inappropriate ASB treatment.
Corresponding author: Prasanna P. Narayanan, PharmD, 200 First Street SW, Rochester, MN 55905; [email protected].
Financial disclosures: None.
1. Nicolle LE, Gupta K, Bradley SF, et al. Clinical practice guideline for the management of asymptomatic bacteriuria: 2019 update by the Infectious Diseases Society of America. Clin Infect Dis. 2019;68:e83–75.
2. Trautner BW, Grigoryan L, Petersen NJ, et al. Effectiveness of an antimicrobial stewardship approach for urinary catheter-associated asymptomatic bacteriuria. JAMA Intern Med. 2015;175:1120-1127.
3. Horan TC, Andrus M, Dudeck MA. CDC/NHSN surveillance definition of health care-associated infection and criteria for specific types of infections in the acute care setting. Am J Infect Control. 2008;36:309-332.
4. Trautner BW. Asymptomatic bacteriuria: when the treatment is worse than the disease. Nat Rev Urol. 2011;9:85-93.
5. Costelloe C, Metcalfe C, Lovering A, et al. Effect of antibiotic prescribing in primary care on antimicrobial resistance in individual patients: systematic review and meta-analysis. BMJ. 2010;340:c2096.
6. The Joint Commission. Prepublication Requirements: New antimicrobial stewardship standard. Jun 22, 2016. www.jointcommission.org/assets/1/6/HAP-CAH_Antimicrobial_Prepub.pdf. Accessed January 24, 2019.
7. Federal Register. Medicare and Medicaid Programs; Hospital and Critical Access Hospital (CAH) Changes to Promote Innovation, Flexibility, and Improvement in Patient Care.Centers for Medicare & Medicaid Services. June 16, 2016. CMS-3295-P
8. Hartley SE, Kuhn L, Valley S, et al. Evaluating a hospitalist-based intervention to decrease unnecessary antimicrobial use in patients with asymptomatic bacteriuria. Infect Control Hosp Epidemiol. 2016;37:1044-1051.
9. Pavese P, Saurel N, Labarere J, et al. Does an educational session with an infectious diseases physician reduce the use of inappropriate antibiotic therapy for inpatients with positive urine culture results? A controlled before-and-after study. Infect Control Hosp Epidemiol. 2009;30:596-599.
10. Kelley D, Aaronson P, Poon E, et al. Evaluation of an antimicrobial stewardship approach to minimize overuse of antibiotics in patients with asymptomatic bacteriuria. Infect Control Hosp Epidemiol. 2014;35:193-195.
11. Chowdhury F, Sarkar K, Branche A, et al. Preventing the inappropriate treatment of asymptomatic bacteriuria at a community teaching hospital. J Community Hosp Intern Med Perspect. 2012;2.
12. Bonnal C, Baune B, Mion M, et al. Bacteriuria in a geriatric hospital: impact of an antibiotic improvement program. J Am Med Dir Assoc. 2008;9:605-609.
13. Linares LA, Thornton DJ, Strymish J, et al. Electronic memorandum decreases unnecessary antimicrobial use for asymptomatic bacteriuria and culture-negative pyuria. Infect Control Hosp Epidemiol. 2011;32:644-648.
14. Daniel M, Keller S, Mozafarihashjin M, et al. An implementation guide to reducing overtreatment of asymptomatic bacteriuria. JAMA Intern Med. 2018;178:271-276.
15. Redwood R, Knobloch MJ, Pellegrini DC, et al. Reducing unnecessary culturing: a systems approach to evaluating urine culture ordering and collection practices among nurses in two acute care settings. Antimicrob Resist Infect Control. 2018;7:4.
16. Barlam TF, Cosgrove SE, Abbo LM, et al. Implementing an antibiotic stewardship program: guidelines by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America. Clin Infect Dis. 2016;62:e51–e7.
17. Flokas ME, Andreatos N, Alevizakos M, et al. Inappropriate management of asymptomatic patients with positive urine cultures: a systematic review and meta-analysis. Open Forum Infect Dis. 2017;4:1-10.
From the Mayo Clinic, Rochester, MN.
Abstract
- Objective: Asymptomatic bacteriuria (ASB) denotes asymptomatic carriage of bacteria within the urinary tract and does not require treatment in most patient populations. Unnecessary antimicrobial treatment has several consequences, including promotion of antimicrobial resistance, potential for medication adverse effects, and risk for Clostridiodes difficile infection. The aim of this quality improvement effort was to decrease both the unnecessary ordering of urine culture studies and unnecessary treatment of ASB.
- Methods: This is a single-center study of patients who received care on 3 internal medicine units at a large, academic medical center. We sought to determine the impact of information technology and educational interventions to decrease both inappropriate urine culture ordering and treatment of ASB. Data from included patients were collected over 3 1-month time periods: baseline, post-information technology intervention, and post-educational intervention.
- Results: There was a reduction in the percentage of patients who received antibiotics for ASB in the post-education intervention period as compared to baseline (35% vs 42%). The proportion of total urine cultures ordered by internal medicine clinicians did not change after an information technology intervention to redesign the computerized physician order entry screen for urine cultures.
- Conclusion: Educational interventions are effective ways to reduce rates of inappropriate treatment of ASB in patients admitted to internal medicine services.
Keywords: asymptomatic bacteriuria, UTI, information technology, education, quality.
Asymptomatic bacteriuria (ASB) is a common condition in which bacteria are recovered from a urine culture (UC) in patients without symptoms suggestive of urinary tract infection (UTI), with no pathologic consequences to most patients who are not treated.1,2 Patients with ASB do not exhibit symptoms of a UTI such as dysuria, increased frequency of urination, increased urgency, suprapubic tenderness, or costovertebral pain. Treatment with antibiotics is not indicated for most patients with ASB.1,3 According to the Infectious Diseases Society of America (IDSA), screening for bacteriuria and treatment for positive results is only indicated during pregnancy and prior to urologic procedures with anticipated breach of the mucosal lining.1
An estimated 20% to 52% of patients in hospital settings receive inappropriate treatment with antibiotics for ASB.4 Unnecessary prescribing of antibiotics has several negative consequences, including increased rates of antibiotic resistance, Clostridioides difficile infection, and medication adverse events, as well as increased health care costs.2,5 Antimicrobial stewardship programs to improve judicious use of antimicrobials are paramount to reducing these consequences, and their importance is heightened with recent requirements for antimicrobial stewardship put forth by The Joint Commission and the Centers for Medicare & Medicaid Services.6,7
A previous review of UC and antimicrobial use in patients for purposes of quality improvement at our institution over a 2-month period showed that of 59 patients with positive UCs, 47 patients (80%) did not have documented symptoms of a UTI. Of these 47 patients with ASB, 29 (61.7%) received antimicrobial treatment unnecessarily (unpublished data). We convened a group of clinicians and nonclinicians representing the areas of infectious disease, pharmacy, microbiology, statistics, and hospital internal medicine (IM) to examine the unnecessary treatment of ASB in our institution. Our objective was to address 2 antimicrobial stewardship issues: inappropriate UC ordering and unnecessary use of antibiotics to treat ASB. Our aim was to reduce the inappropriate ordering of UCs and to reduce treatment of ASB.
Methods
Setting
The study was conducted on 3 IM nursing units with a total of 83 beds at a large tertiary care academic medical center in the midwestern United States, and was approved by the organization’s Institutional Review Board.
Participants
We included all non-pregnant patients aged 18 years or older who received care from an IM primary service. These patients were admitted directly to an IM team through the emergency department (ED) or transferred to an IM team after an initial stay in the intensive care unit.
Data Source
Microbiology laboratory reports generated from the electronic health record were used to identify all patients with a collected UC sample who received care from an IM service prior to discharge. Urine samples were collected by midstream catch or catheterization. Data on urine Gram stain and urine dipstick were not included. Henceforth, the phrase “urine culture order” indicates that a UC was both ordered and performed. Data reports were generated for the month of August 2016 to determine the baseline number of UCs ordered. Charts of patients with positive UCs were reviewed to determine if antibiotics were started for the positive UC and whether the patient had signs or symptoms consistent with a UTI. If antibiotics were started in the absence of signs or symptoms to support a UTI, the patient was determined to have been unnecessarily treated for ASB. Reports were then generated for the month after each intervention was implemented, with the same chart review undertaken for positive UCs. Bacteriuria was defined in our study as the presence of microbial growth greater than 10,000 CFU/mL in UC.
Interventions
Initial analysis by our study group determined that lack of electronic clinical decision support (CDS) at the point of care and provider knowledge gaps in interpreting positive UCs were the 2 main contributors to unnecessary UC orders and unnecessary treatment of positive UCs, respectively. We reviewed the work of other groups who reported interventions to decrease treatment of ASB, ranging from educational presentations to pocket cards and treatment algorithms.8-13 We hypothesized that there would be a decrease in UC orders with CDS embedded in the computerized order entry screen, and that we would decrease unnecessary treatment of positive UCs by educating clinicians on indications for appropriate antibiotic prescribing in the setting of a positive UC.
Information technology intervention. The first intervention implemented involved redesign of the UC ordering screen in the computerized physician order entry (CPOE) system. This intervention went live hospital-wide, including the IM floors, intensive care units, and all other areas except the ED, on February 1, 2017 (Figure 1). The ordering screen required the prescriber to select from a list of appropriate indications for ordering a UC, including urine frequency, urgency, or dysuria; unexplained suprapubic or flank pain; fever in patients without another recognized cause; screening obtained prior to urologic procedure; or screening during pregnancy. An additional message advised prescribers to avoid ordering the culture if the patient had malodorous or cloudy urine, pyuria without urinary symptoms, or had an alternative cause of fever. Before we implemented the information technology (IT) intervention, there had been no specific point-of-care guidance on UC ordering.
Educational intervention. The second intervention, driven by clinical pharmacists, involved active and passive education of prescribers specifically designed to address unnecessary treatment of ASB. The IT intervention with CDS for UC ordering remained live. Presentations designed by the study group summarizing the appropriate indications for ordering a UC, distinguishing ASB from UTI, and discouraging treatment of ASB were delivered via a variety of routes by clinical pharmacists to nurses, nurse practitioners, physician assistants, pharmacists, medical residents, and staff physicians providing care to patients on the 3 IM units over a 1-month period in March 2017. The presentations contained the same basic content, but the information was delivered to target each specific audience group.
Medical residents received a 10-minute live presentation during a conference. Nurse practitioners, physician assistants, and staff physicians received a presentation via email, and highlights of the presentation were delivered by clinical pharmacists at their respective monthly group meetings. A handout was presented to nursing staff at nursing huddles, and presentation slides were distributed by email. Educational posters were posted in the medical resident workrooms, nursing breakrooms, and staff bathrooms on the units.
Outcome Measurements
The endpoints of interest were the percentage of patients with positive UCs unnecessarily treated for ASB before and after each intervention and the number of UCs ordered at baseline and after implementation of each intervention. Counterbalance measures assessed included the incidence of UTI, pyelonephritis, or urosepsis within 7 days of positive UC for patients who did not receive antibiotic treatment for ASB.
Results
Data from a total of 270 cultures were examined from IM nursing units. A total of 117 UCs were ordered during the baseline period before interventions were implemented. For a period of 1 month following activation of the IT intervention, 73 UCs were ordered. For a period of 1 month following the educational interventions, 80 UCs were ordered. Of these, 61 (52%) UCs were positive at baseline, 37 (51%) after the IT intervention, and 41 (51%) after the educational intervention. Patient characteristics were similar between the 3 groups (Table); 64.7% of patients were female in their early to mid-seventies. The majority of UCs were ordered by providers in the ED in all 3 periods examined (51%-70%). The percentage of patients who received antibiotics prior to UC for another indication (including bacteriuria) in the baseline, post-IT intervention, and post-education intervention groups were 30%, 27%, and 45%, respectively.
The study outcomes are summarized in Figure 2. Among patients with positive cultures, there was not a reduction in inappropriate treatment of ASB compared to baseline after the IT intervention (48% vs 42%). Following the education intervention, there was a reduction in unnecessary ASB treatment as compared both to baseline (35% vs 42%) and to post-IT intervention (35% vs 48%). There was no difference between the 3 study periods in the percentage of total UCs ordered by IM clinicians. The counterbalance measure showed that 1 patient who did not receive antibiotics within 7 days of a positive UC developed pyelonephritis, UTI, or sepsis due to a UTI in each intervention group.
Discussion
The results of this study demonstrate the role of multimodal interventions in antimicrobial stewardship and add to the growing body of evidence supporting the work of antimicrobial stewardship programs. Our multidisciplinary study group and multipronged intervention follow recent guideline recommendations for antimicrobial stewardship program interventions against unnecessary treatment of ASB.14 Initial analysis by our study group determined lack of CDS at the point of care and provider knowledge gaps in interpreting positive UCs as the 2 main contributors to unnecessary UC orders and unnecessary treatment of positive UCs in our local practice culture. The IT component of our intervention was intended to provide CDS for ordering UCs, and the education component focused on informing clinicians’ treatment decisions for positive UCs.
It has been suggested that the type of stewardship intervention that is most effective fits the specific needs and resources of an institution.14,15 And although the IDSA does not recommend education as a stand-alone intervention,16 we found it to be an effective intervention for our clinicians in our work environment. However, since the CPOE guidance was in place during the educational study periods, it is possible that the effect was due to a combination of these 2 approaches. Our pre-intervention ASB treatment rates were consistent with a recent meta-analysis in which the rate of inappropriate treatment of ASB was 45%.17 This meta-analysis found educational and organizational interventions led to a mean absolute risk reduction of 33%. After the education intervention, we saw a 7% decrease in unnecessary treatment of ASB compared to baseline, and a 13% decrease compared to the month just prior to the educational intervention.
Lessons learned from our work included how clear review of local processes can inform quality improvement interventions. For instance, we initially hypothesized that IM clinicians would benefit from point-of-care CDS guidance, but such guidance used alone without educational interventions was not supported by the results. We also determined that the majority of UCs from patients on general medicine units were ordered by ED providers. This revealed an opportunity to implement similar interventions in the ED, as this was the initial point of contact for many of these patients.
As with any clinical intervention, the anticipated benefits should be weighed against potential harm. Using counterbalance measures, we found there was minimal risk in the occurrence of UTI, pyelonephritis, or sepsis if clinicians avoided treating ASB. This finding is consistent with IDSA guideline recommendations and other studies that suggest that withholding treatment for asymptomatic bacteriuria does not lead to worse outcomes.1
This study has several limitations. Data were obtained through review of the electronic health record and therefore documentation may be incomplete. Also, antimicrobials for empiric coverage or treatment for other infections (eg, pneumonia, sepsis) may have confounded our results, as empirical antimicrobials were given to 27% to 45% of patients prior to UC. This was a quality improvement project carried out over defined time intervals, and thus our sample size was limited and not adequately powered to show statistical significance. Additionally, given the bundling of interventions, it is difficult to determine the impact of each intervention independently. Although CDS for UC ordering may not have influenced ordering, it is possible that the IT intervention raised awareness of ASB and influenced treatment practices.
Conclusion
Our work supports the principles of antibiotic stewardship as brought forth by IDSA.16 This work was the effort of a multidisciplinary team, which aligns with recommendations by Daniel and colleagues, published after our study had ended, for reducing overtreatment of ASB.14 Additionally, our study results provided valuable information for our institution. Although improvements in management of ASB were modest, the success of provider education and identification of other work areas and clinicians to target for future intervention were helpful in consideration of further studies. This work will also aid us in developing an expected effect size for future studies. We plan to provide ongoing education for IM providers as well as education in the ED to target providers who make first contact with patients admitted to inpatient services. In addition, the CPOE UC ordering screen message will continue to be used hospital-wide and will be expanded to the ED ordering system. Our interventions, experiences, and challenges may be used by other institutions to design effective antimicrobial stewardship interventions directed towards reducing rates of inappropriate ASB treatment.
Corresponding author: Prasanna P. Narayanan, PharmD, 200 First Street SW, Rochester, MN 55905; [email protected].
Financial disclosures: None.
From the Mayo Clinic, Rochester, MN.
Abstract
- Objective: Asymptomatic bacteriuria (ASB) denotes asymptomatic carriage of bacteria within the urinary tract and does not require treatment in most patient populations. Unnecessary antimicrobial treatment has several consequences, including promotion of antimicrobial resistance, potential for medication adverse effects, and risk for Clostridiodes difficile infection. The aim of this quality improvement effort was to decrease both the unnecessary ordering of urine culture studies and unnecessary treatment of ASB.
- Methods: This is a single-center study of patients who received care on 3 internal medicine units at a large, academic medical center. We sought to determine the impact of information technology and educational interventions to decrease both inappropriate urine culture ordering and treatment of ASB. Data from included patients were collected over 3 1-month time periods: baseline, post-information technology intervention, and post-educational intervention.
- Results: There was a reduction in the percentage of patients who received antibiotics for ASB in the post-education intervention period as compared to baseline (35% vs 42%). The proportion of total urine cultures ordered by internal medicine clinicians did not change after an information technology intervention to redesign the computerized physician order entry screen for urine cultures.
- Conclusion: Educational interventions are effective ways to reduce rates of inappropriate treatment of ASB in patients admitted to internal medicine services.
Keywords: asymptomatic bacteriuria, UTI, information technology, education, quality.
Asymptomatic bacteriuria (ASB) is a common condition in which bacteria are recovered from a urine culture (UC) in patients without symptoms suggestive of urinary tract infection (UTI), with no pathologic consequences to most patients who are not treated.1,2 Patients with ASB do not exhibit symptoms of a UTI such as dysuria, increased frequency of urination, increased urgency, suprapubic tenderness, or costovertebral pain. Treatment with antibiotics is not indicated for most patients with ASB.1,3 According to the Infectious Diseases Society of America (IDSA), screening for bacteriuria and treatment for positive results is only indicated during pregnancy and prior to urologic procedures with anticipated breach of the mucosal lining.1
An estimated 20% to 52% of patients in hospital settings receive inappropriate treatment with antibiotics for ASB.4 Unnecessary prescribing of antibiotics has several negative consequences, including increased rates of antibiotic resistance, Clostridioides difficile infection, and medication adverse events, as well as increased health care costs.2,5 Antimicrobial stewardship programs to improve judicious use of antimicrobials are paramount to reducing these consequences, and their importance is heightened with recent requirements for antimicrobial stewardship put forth by The Joint Commission and the Centers for Medicare & Medicaid Services.6,7
A previous review of UC and antimicrobial use in patients for purposes of quality improvement at our institution over a 2-month period showed that of 59 patients with positive UCs, 47 patients (80%) did not have documented symptoms of a UTI. Of these 47 patients with ASB, 29 (61.7%) received antimicrobial treatment unnecessarily (unpublished data). We convened a group of clinicians and nonclinicians representing the areas of infectious disease, pharmacy, microbiology, statistics, and hospital internal medicine (IM) to examine the unnecessary treatment of ASB in our institution. Our objective was to address 2 antimicrobial stewardship issues: inappropriate UC ordering and unnecessary use of antibiotics to treat ASB. Our aim was to reduce the inappropriate ordering of UCs and to reduce treatment of ASB.
Methods
Setting
The study was conducted on 3 IM nursing units with a total of 83 beds at a large tertiary care academic medical center in the midwestern United States, and was approved by the organization’s Institutional Review Board.
Participants
We included all non-pregnant patients aged 18 years or older who received care from an IM primary service. These patients were admitted directly to an IM team through the emergency department (ED) or transferred to an IM team after an initial stay in the intensive care unit.
Data Source
Microbiology laboratory reports generated from the electronic health record were used to identify all patients with a collected UC sample who received care from an IM service prior to discharge. Urine samples were collected by midstream catch or catheterization. Data on urine Gram stain and urine dipstick were not included. Henceforth, the phrase “urine culture order” indicates that a UC was both ordered and performed. Data reports were generated for the month of August 2016 to determine the baseline number of UCs ordered. Charts of patients with positive UCs were reviewed to determine if antibiotics were started for the positive UC and whether the patient had signs or symptoms consistent with a UTI. If antibiotics were started in the absence of signs or symptoms to support a UTI, the patient was determined to have been unnecessarily treated for ASB. Reports were then generated for the month after each intervention was implemented, with the same chart review undertaken for positive UCs. Bacteriuria was defined in our study as the presence of microbial growth greater than 10,000 CFU/mL in UC.
Interventions
Initial analysis by our study group determined that lack of electronic clinical decision support (CDS) at the point of care and provider knowledge gaps in interpreting positive UCs were the 2 main contributors to unnecessary UC orders and unnecessary treatment of positive UCs, respectively. We reviewed the work of other groups who reported interventions to decrease treatment of ASB, ranging from educational presentations to pocket cards and treatment algorithms.8-13 We hypothesized that there would be a decrease in UC orders with CDS embedded in the computerized order entry screen, and that we would decrease unnecessary treatment of positive UCs by educating clinicians on indications for appropriate antibiotic prescribing in the setting of a positive UC.
Information technology intervention. The first intervention implemented involved redesign of the UC ordering screen in the computerized physician order entry (CPOE) system. This intervention went live hospital-wide, including the IM floors, intensive care units, and all other areas except the ED, on February 1, 2017 (Figure 1). The ordering screen required the prescriber to select from a list of appropriate indications for ordering a UC, including urine frequency, urgency, or dysuria; unexplained suprapubic or flank pain; fever in patients without another recognized cause; screening obtained prior to urologic procedure; or screening during pregnancy. An additional message advised prescribers to avoid ordering the culture if the patient had malodorous or cloudy urine, pyuria without urinary symptoms, or had an alternative cause of fever. Before we implemented the information technology (IT) intervention, there had been no specific point-of-care guidance on UC ordering.
Educational intervention. The second intervention, driven by clinical pharmacists, involved active and passive education of prescribers specifically designed to address unnecessary treatment of ASB. The IT intervention with CDS for UC ordering remained live. Presentations designed by the study group summarizing the appropriate indications for ordering a UC, distinguishing ASB from UTI, and discouraging treatment of ASB were delivered via a variety of routes by clinical pharmacists to nurses, nurse practitioners, physician assistants, pharmacists, medical residents, and staff physicians providing care to patients on the 3 IM units over a 1-month period in March 2017. The presentations contained the same basic content, but the information was delivered to target each specific audience group.
Medical residents received a 10-minute live presentation during a conference. Nurse practitioners, physician assistants, and staff physicians received a presentation via email, and highlights of the presentation were delivered by clinical pharmacists at their respective monthly group meetings. A handout was presented to nursing staff at nursing huddles, and presentation slides were distributed by email. Educational posters were posted in the medical resident workrooms, nursing breakrooms, and staff bathrooms on the units.
Outcome Measurements
The endpoints of interest were the percentage of patients with positive UCs unnecessarily treated for ASB before and after each intervention and the number of UCs ordered at baseline and after implementation of each intervention. Counterbalance measures assessed included the incidence of UTI, pyelonephritis, or urosepsis within 7 days of positive UC for patients who did not receive antibiotic treatment for ASB.
Results
Data from a total of 270 cultures were examined from IM nursing units. A total of 117 UCs were ordered during the baseline period before interventions were implemented. For a period of 1 month following activation of the IT intervention, 73 UCs were ordered. For a period of 1 month following the educational interventions, 80 UCs were ordered. Of these, 61 (52%) UCs were positive at baseline, 37 (51%) after the IT intervention, and 41 (51%) after the educational intervention. Patient characteristics were similar between the 3 groups (Table); 64.7% of patients were female in their early to mid-seventies. The majority of UCs were ordered by providers in the ED in all 3 periods examined (51%-70%). The percentage of patients who received antibiotics prior to UC for another indication (including bacteriuria) in the baseline, post-IT intervention, and post-education intervention groups were 30%, 27%, and 45%, respectively.
The study outcomes are summarized in Figure 2. Among patients with positive cultures, there was not a reduction in inappropriate treatment of ASB compared to baseline after the IT intervention (48% vs 42%). Following the education intervention, there was a reduction in unnecessary ASB treatment as compared both to baseline (35% vs 42%) and to post-IT intervention (35% vs 48%). There was no difference between the 3 study periods in the percentage of total UCs ordered by IM clinicians. The counterbalance measure showed that 1 patient who did not receive antibiotics within 7 days of a positive UC developed pyelonephritis, UTI, or sepsis due to a UTI in each intervention group.
Discussion
The results of this study demonstrate the role of multimodal interventions in antimicrobial stewardship and add to the growing body of evidence supporting the work of antimicrobial stewardship programs. Our multidisciplinary study group and multipronged intervention follow recent guideline recommendations for antimicrobial stewardship program interventions against unnecessary treatment of ASB.14 Initial analysis by our study group determined lack of CDS at the point of care and provider knowledge gaps in interpreting positive UCs as the 2 main contributors to unnecessary UC orders and unnecessary treatment of positive UCs in our local practice culture. The IT component of our intervention was intended to provide CDS for ordering UCs, and the education component focused on informing clinicians’ treatment decisions for positive UCs.
It has been suggested that the type of stewardship intervention that is most effective fits the specific needs and resources of an institution.14,15 And although the IDSA does not recommend education as a stand-alone intervention,16 we found it to be an effective intervention for our clinicians in our work environment. However, since the CPOE guidance was in place during the educational study periods, it is possible that the effect was due to a combination of these 2 approaches. Our pre-intervention ASB treatment rates were consistent with a recent meta-analysis in which the rate of inappropriate treatment of ASB was 45%.17 This meta-analysis found educational and organizational interventions led to a mean absolute risk reduction of 33%. After the education intervention, we saw a 7% decrease in unnecessary treatment of ASB compared to baseline, and a 13% decrease compared to the month just prior to the educational intervention.
Lessons learned from our work included how clear review of local processes can inform quality improvement interventions. For instance, we initially hypothesized that IM clinicians would benefit from point-of-care CDS guidance, but such guidance used alone without educational interventions was not supported by the results. We also determined that the majority of UCs from patients on general medicine units were ordered by ED providers. This revealed an opportunity to implement similar interventions in the ED, as this was the initial point of contact for many of these patients.
As with any clinical intervention, the anticipated benefits should be weighed against potential harm. Using counterbalance measures, we found there was minimal risk in the occurrence of UTI, pyelonephritis, or sepsis if clinicians avoided treating ASB. This finding is consistent with IDSA guideline recommendations and other studies that suggest that withholding treatment for asymptomatic bacteriuria does not lead to worse outcomes.1
This study has several limitations. Data were obtained through review of the electronic health record and therefore documentation may be incomplete. Also, antimicrobials for empiric coverage or treatment for other infections (eg, pneumonia, sepsis) may have confounded our results, as empirical antimicrobials were given to 27% to 45% of patients prior to UC. This was a quality improvement project carried out over defined time intervals, and thus our sample size was limited and not adequately powered to show statistical significance. Additionally, given the bundling of interventions, it is difficult to determine the impact of each intervention independently. Although CDS for UC ordering may not have influenced ordering, it is possible that the IT intervention raised awareness of ASB and influenced treatment practices.
Conclusion
Our work supports the principles of antibiotic stewardship as brought forth by IDSA.16 This work was the effort of a multidisciplinary team, which aligns with recommendations by Daniel and colleagues, published after our study had ended, for reducing overtreatment of ASB.14 Additionally, our study results provided valuable information for our institution. Although improvements in management of ASB were modest, the success of provider education and identification of other work areas and clinicians to target for future intervention were helpful in consideration of further studies. This work will also aid us in developing an expected effect size for future studies. We plan to provide ongoing education for IM providers as well as education in the ED to target providers who make first contact with patients admitted to inpatient services. In addition, the CPOE UC ordering screen message will continue to be used hospital-wide and will be expanded to the ED ordering system. Our interventions, experiences, and challenges may be used by other institutions to design effective antimicrobial stewardship interventions directed towards reducing rates of inappropriate ASB treatment.
Corresponding author: Prasanna P. Narayanan, PharmD, 200 First Street SW, Rochester, MN 55905; [email protected].
Financial disclosures: None.
1. Nicolle LE, Gupta K, Bradley SF, et al. Clinical practice guideline for the management of asymptomatic bacteriuria: 2019 update by the Infectious Diseases Society of America. Clin Infect Dis. 2019;68:e83–75.
2. Trautner BW, Grigoryan L, Petersen NJ, et al. Effectiveness of an antimicrobial stewardship approach for urinary catheter-associated asymptomatic bacteriuria. JAMA Intern Med. 2015;175:1120-1127.
3. Horan TC, Andrus M, Dudeck MA. CDC/NHSN surveillance definition of health care-associated infection and criteria for specific types of infections in the acute care setting. Am J Infect Control. 2008;36:309-332.
4. Trautner BW. Asymptomatic bacteriuria: when the treatment is worse than the disease. Nat Rev Urol. 2011;9:85-93.
5. Costelloe C, Metcalfe C, Lovering A, et al. Effect of antibiotic prescribing in primary care on antimicrobial resistance in individual patients: systematic review and meta-analysis. BMJ. 2010;340:c2096.
6. The Joint Commission. Prepublication Requirements: New antimicrobial stewardship standard. Jun 22, 2016. www.jointcommission.org/assets/1/6/HAP-CAH_Antimicrobial_Prepub.pdf. Accessed January 24, 2019.
7. Federal Register. Medicare and Medicaid Programs; Hospital and Critical Access Hospital (CAH) Changes to Promote Innovation, Flexibility, and Improvement in Patient Care.Centers for Medicare & Medicaid Services. June 16, 2016. CMS-3295-P
8. Hartley SE, Kuhn L, Valley S, et al. Evaluating a hospitalist-based intervention to decrease unnecessary antimicrobial use in patients with asymptomatic bacteriuria. Infect Control Hosp Epidemiol. 2016;37:1044-1051.
9. Pavese P, Saurel N, Labarere J, et al. Does an educational session with an infectious diseases physician reduce the use of inappropriate antibiotic therapy for inpatients with positive urine culture results? A controlled before-and-after study. Infect Control Hosp Epidemiol. 2009;30:596-599.
10. Kelley D, Aaronson P, Poon E, et al. Evaluation of an antimicrobial stewardship approach to minimize overuse of antibiotics in patients with asymptomatic bacteriuria. Infect Control Hosp Epidemiol. 2014;35:193-195.
11. Chowdhury F, Sarkar K, Branche A, et al. Preventing the inappropriate treatment of asymptomatic bacteriuria at a community teaching hospital. J Community Hosp Intern Med Perspect. 2012;2.
12. Bonnal C, Baune B, Mion M, et al. Bacteriuria in a geriatric hospital: impact of an antibiotic improvement program. J Am Med Dir Assoc. 2008;9:605-609.
13. Linares LA, Thornton DJ, Strymish J, et al. Electronic memorandum decreases unnecessary antimicrobial use for asymptomatic bacteriuria and culture-negative pyuria. Infect Control Hosp Epidemiol. 2011;32:644-648.
14. Daniel M, Keller S, Mozafarihashjin M, et al. An implementation guide to reducing overtreatment of asymptomatic bacteriuria. JAMA Intern Med. 2018;178:271-276.
15. Redwood R, Knobloch MJ, Pellegrini DC, et al. Reducing unnecessary culturing: a systems approach to evaluating urine culture ordering and collection practices among nurses in two acute care settings. Antimicrob Resist Infect Control. 2018;7:4.
16. Barlam TF, Cosgrove SE, Abbo LM, et al. Implementing an antibiotic stewardship program: guidelines by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America. Clin Infect Dis. 2016;62:e51–e7.
17. Flokas ME, Andreatos N, Alevizakos M, et al. Inappropriate management of asymptomatic patients with positive urine cultures: a systematic review and meta-analysis. Open Forum Infect Dis. 2017;4:1-10.
1. Nicolle LE, Gupta K, Bradley SF, et al. Clinical practice guideline for the management of asymptomatic bacteriuria: 2019 update by the Infectious Diseases Society of America. Clin Infect Dis. 2019;68:e83–75.
2. Trautner BW, Grigoryan L, Petersen NJ, et al. Effectiveness of an antimicrobial stewardship approach for urinary catheter-associated asymptomatic bacteriuria. JAMA Intern Med. 2015;175:1120-1127.
3. Horan TC, Andrus M, Dudeck MA. CDC/NHSN surveillance definition of health care-associated infection and criteria for specific types of infections in the acute care setting. Am J Infect Control. 2008;36:309-332.
4. Trautner BW. Asymptomatic bacteriuria: when the treatment is worse than the disease. Nat Rev Urol. 2011;9:85-93.
5. Costelloe C, Metcalfe C, Lovering A, et al. Effect of antibiotic prescribing in primary care on antimicrobial resistance in individual patients: systematic review and meta-analysis. BMJ. 2010;340:c2096.
6. The Joint Commission. Prepublication Requirements: New antimicrobial stewardship standard. Jun 22, 2016. www.jointcommission.org/assets/1/6/HAP-CAH_Antimicrobial_Prepub.pdf. Accessed January 24, 2019.
7. Federal Register. Medicare and Medicaid Programs; Hospital and Critical Access Hospital (CAH) Changes to Promote Innovation, Flexibility, and Improvement in Patient Care.Centers for Medicare & Medicaid Services. June 16, 2016. CMS-3295-P
8. Hartley SE, Kuhn L, Valley S, et al. Evaluating a hospitalist-based intervention to decrease unnecessary antimicrobial use in patients with asymptomatic bacteriuria. Infect Control Hosp Epidemiol. 2016;37:1044-1051.
9. Pavese P, Saurel N, Labarere J, et al. Does an educational session with an infectious diseases physician reduce the use of inappropriate antibiotic therapy for inpatients with positive urine culture results? A controlled before-and-after study. Infect Control Hosp Epidemiol. 2009;30:596-599.
10. Kelley D, Aaronson P, Poon E, et al. Evaluation of an antimicrobial stewardship approach to minimize overuse of antibiotics in patients with asymptomatic bacteriuria. Infect Control Hosp Epidemiol. 2014;35:193-195.
11. Chowdhury F, Sarkar K, Branche A, et al. Preventing the inappropriate treatment of asymptomatic bacteriuria at a community teaching hospital. J Community Hosp Intern Med Perspect. 2012;2.
12. Bonnal C, Baune B, Mion M, et al. Bacteriuria in a geriatric hospital: impact of an antibiotic improvement program. J Am Med Dir Assoc. 2008;9:605-609.
13. Linares LA, Thornton DJ, Strymish J, et al. Electronic memorandum decreases unnecessary antimicrobial use for asymptomatic bacteriuria and culture-negative pyuria. Infect Control Hosp Epidemiol. 2011;32:644-648.
14. Daniel M, Keller S, Mozafarihashjin M, et al. An implementation guide to reducing overtreatment of asymptomatic bacteriuria. JAMA Intern Med. 2018;178:271-276.
15. Redwood R, Knobloch MJ, Pellegrini DC, et al. Reducing unnecessary culturing: a systems approach to evaluating urine culture ordering and collection practices among nurses in two acute care settings. Antimicrob Resist Infect Control. 2018;7:4.
16. Barlam TF, Cosgrove SE, Abbo LM, et al. Implementing an antibiotic stewardship program: guidelines by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America. Clin Infect Dis. 2016;62:e51–e7.
17. Flokas ME, Andreatos N, Alevizakos M, et al. Inappropriate management of asymptomatic patients with positive urine cultures: a systematic review and meta-analysis. Open Forum Infect Dis. 2017;4:1-10.