User login
Suicide Prevention Grant Program Reauthorized
Suicide Prevention Grant Program Reauthorized
Community-based organizations that provide suicide-prevention services can now access about $52.5 million in US Department of Veterans Affairs (VA) grants. The grant is part of the 3-year Staff Sergeant Fox Suicide Prevention Grant Program, which honors Parker Gordon Fox, a sniper instructor at the U.S. Army Infantry School at Fort Benning, Georgia, who died by suicide in 2020. In consecutive Congressional hearings, lawmakers called for the reauthorization of the program to address gaps in VA care.
“It has been a game-changer for so many veterans,” Sen. Richard Blumenthal (D-CT) said.
The money provides or coordinates primarily nonclinical suicide prevention services, including outreach and linkage to VA and community resources. Services also may include baseline mental health screenings, case management and peer support, education on suicide risk, VA benefits assistance, and emergency clinical services.
Since its inception in 2022, the program has awarded $157.5 million to 95 organizations in 43 states, US territories, and tribal lands. Speaking before the House Committee on Veterans’ Affairs on May 15, VA Secretary Doug Collins praised the Fox program for bringing “different voices into the conversation,” but added it wasn’t enough. He noted that the veteran suicide rate has not changed since 2008, despite the VA annually spending $588 million on suicide prevention over the past few years.
In an op-ed, Russell Lemle, a senior policy analyst at the Veterans Healthcare Policy Institute, disputed Collins' characterization of veteran suicides. Between 2008 and 2022 (the last year for which complete data is available), US deaths by suicide increased 37% while the number of veteran deaths by suicide fell 2%. “This data collection was the single best part of the program,” he argued, calling for reauthorization to continue requiring data-targeted solutions.
According to a 2024 VA interim report on the Fox grant program, grantees had completed > 16,590 outreach contacts and engaged 3204 participants as of September 30, 2023. An additional 864 individuals were onboarding at the time of the report.
The current version of the grant program requires grantees to use validated tools, including the VA Data Collection Tool, and other assessments furnished by VA to determine the effectiveness of the suicide prevention services. They must also provide each participant with a satisfaction survey and submit periodic and annual financial and performance reports.
Despite the Trump administration’s cuts and cancellations to the federal workforce and federal programs, Collins told the Senate committee he is firmly on the side of working with community-based organizations like the Fox grant program to broaden the VA’s reach: “I want to use grants and programs like [the Fox grant program] to reach out beyond the scope of where we’re currently reaching, to say how can we actually touch the veteran that’s not being touched right now by these programs,” Collins said. “We’ve got to do better at using the grants, using our programs to go outside the normal bubble and use others to help get the word out.”
Grant applications are due in July and VA will choose awardees in September. Organizations can apply for grants worth up to $750,000 and may apply to renew awards from year to year throughout the length of the program.
Community-based organizations that provide suicide-prevention services can now access about $52.5 million in US Department of Veterans Affairs (VA) grants. The grant is part of the 3-year Staff Sergeant Fox Suicide Prevention Grant Program, which honors Parker Gordon Fox, a sniper instructor at the U.S. Army Infantry School at Fort Benning, Georgia, who died by suicide in 2020. In consecutive Congressional hearings, lawmakers called for the reauthorization of the program to address gaps in VA care.
“It has been a game-changer for so many veterans,” Sen. Richard Blumenthal (D-CT) said.
The money provides or coordinates primarily nonclinical suicide prevention services, including outreach and linkage to VA and community resources. Services also may include baseline mental health screenings, case management and peer support, education on suicide risk, VA benefits assistance, and emergency clinical services.
Since its inception in 2022, the program has awarded $157.5 million to 95 organizations in 43 states, US territories, and tribal lands. Speaking before the House Committee on Veterans’ Affairs on May 15, VA Secretary Doug Collins praised the Fox program for bringing “different voices into the conversation,” but added it wasn’t enough. He noted that the veteran suicide rate has not changed since 2008, despite the VA annually spending $588 million on suicide prevention over the past few years.
In an op-ed, Russell Lemle, a senior policy analyst at the Veterans Healthcare Policy Institute, disputed Collins' characterization of veteran suicides. Between 2008 and 2022 (the last year for which complete data is available), US deaths by suicide increased 37% while the number of veteran deaths by suicide fell 2%. “This data collection was the single best part of the program,” he argued, calling for reauthorization to continue requiring data-targeted solutions.
According to a 2024 VA interim report on the Fox grant program, grantees had completed > 16,590 outreach contacts and engaged 3204 participants as of September 30, 2023. An additional 864 individuals were onboarding at the time of the report.
The current version of the grant program requires grantees to use validated tools, including the VA Data Collection Tool, and other assessments furnished by VA to determine the effectiveness of the suicide prevention services. They must also provide each participant with a satisfaction survey and submit periodic and annual financial and performance reports.
Despite the Trump administration’s cuts and cancellations to the federal workforce and federal programs, Collins told the Senate committee he is firmly on the side of working with community-based organizations like the Fox grant program to broaden the VA’s reach: “I want to use grants and programs like [the Fox grant program] to reach out beyond the scope of where we’re currently reaching, to say how can we actually touch the veteran that’s not being touched right now by these programs,” Collins said. “We’ve got to do better at using the grants, using our programs to go outside the normal bubble and use others to help get the word out.”
Grant applications are due in July and VA will choose awardees in September. Organizations can apply for grants worth up to $750,000 and may apply to renew awards from year to year throughout the length of the program.
Community-based organizations that provide suicide-prevention services can now access about $52.5 million in US Department of Veterans Affairs (VA) grants. The grant is part of the 3-year Staff Sergeant Fox Suicide Prevention Grant Program, which honors Parker Gordon Fox, a sniper instructor at the U.S. Army Infantry School at Fort Benning, Georgia, who died by suicide in 2020. In consecutive Congressional hearings, lawmakers called for the reauthorization of the program to address gaps in VA care.
“It has been a game-changer for so many veterans,” Sen. Richard Blumenthal (D-CT) said.
The money provides or coordinates primarily nonclinical suicide prevention services, including outreach and linkage to VA and community resources. Services also may include baseline mental health screenings, case management and peer support, education on suicide risk, VA benefits assistance, and emergency clinical services.
Since its inception in 2022, the program has awarded $157.5 million to 95 organizations in 43 states, US territories, and tribal lands. Speaking before the House Committee on Veterans’ Affairs on May 15, VA Secretary Doug Collins praised the Fox program for bringing “different voices into the conversation,” but added it wasn’t enough. He noted that the veteran suicide rate has not changed since 2008, despite the VA annually spending $588 million on suicide prevention over the past few years.
In an op-ed, Russell Lemle, a senior policy analyst at the Veterans Healthcare Policy Institute, disputed Collins' characterization of veteran suicides. Between 2008 and 2022 (the last year for which complete data is available), US deaths by suicide increased 37% while the number of veteran deaths by suicide fell 2%. “This data collection was the single best part of the program,” he argued, calling for reauthorization to continue requiring data-targeted solutions.
According to a 2024 VA interim report on the Fox grant program, grantees had completed > 16,590 outreach contacts and engaged 3204 participants as of September 30, 2023. An additional 864 individuals were onboarding at the time of the report.
The current version of the grant program requires grantees to use validated tools, including the VA Data Collection Tool, and other assessments furnished by VA to determine the effectiveness of the suicide prevention services. They must also provide each participant with a satisfaction survey and submit periodic and annual financial and performance reports.
Despite the Trump administration’s cuts and cancellations to the federal workforce and federal programs, Collins told the Senate committee he is firmly on the side of working with community-based organizations like the Fox grant program to broaden the VA’s reach: “I want to use grants and programs like [the Fox grant program] to reach out beyond the scope of where we’re currently reaching, to say how can we actually touch the veteran that’s not being touched right now by these programs,” Collins said. “We’ve got to do better at using the grants, using our programs to go outside the normal bubble and use others to help get the word out.”
Grant applications are due in July and VA will choose awardees in September. Organizations can apply for grants worth up to $750,000 and may apply to renew awards from year to year throughout the length of the program.
Suicide Prevention Grant Program Reauthorized
Suicide Prevention Grant Program Reauthorized
Collins Lays Out Plans to Reduce VA by 15% in Congressional Hearings
Collins Lays Out Plans to Reduce VA by 15% in Senate Hearing
US Department of Veterans Affairs (VA) Secretary Doug Collins testified in US House of Representatives and US Senate committees hearings that bringing staff numbers down to fiscal year 2019 figures was simply a goal: “Our goal, as we look at it, as everything goes forward, is a 15% decrease,” he told the senators. “It’s a goal. You have to start somewhere.”
“It’s a process we’re going through and I’m not going to work out a process in front of a committee or anywhere else,” Collins testified in the Senate on May 6, adding that it would be “incompetence” or “malpractice” to do so before time. “[When] we’re doing something as large as we are in an organization as sensitive on this Hill, it would not be right for us to do that in public. It would not be right for us to just come out and say here’s everything that we got and then have everybody scared because in the end it may not be the final decision.”
“We’re going to come to the best possible decision we can for the veterans in this country so they can have a VA system that actually works,” Collins argued in the Senate. “The VA’s been an issue for a long time. We’re trying to not make it an issue anymore.”
Collins later told a House committee on May 15 that VA was conducting a thorough review of department structure and staffing across the enterpise. "Our goal is to increase productivity and efficiency and to eliminate waste and bureaucracy improving health care delivery and benefits to our veterans. We are going to maintain VA essential jobs like doctors and nurses and claims processors" but eliminate positions it deemed "nonmission-critical" and consolidating areas of "overlap and waste."
Senate ranking member Richard Blumenthal (D-CT) and Chairman Jerry Moran (R-KS) both placed an emphasis on accountability for responsible resizing at the hearing.
“The department is at a critical juncture,” Moran said. “Perhaps that’s always true, and I want to hear from you that the changes under way at the VA are backed by data, informed by veteran demand, focused on improving outcomes for men and women the VA serves, and will be carried out in close coordination with this committee, as well as with veterans, VA staff, and veteran organizations.” Moran stressed that cutting should be about right-sizing, done carefully, and while treating people “with gratitude and respect.”
Blumenthal was more direct in his criticism of the approach: “You cannot slash and trash the VA without eliminating those essential positions which provide access and availability of health care. It simply cannot be done,” he told Collins.
In response, Collins replied, “You have stated on several occasions already that I am saying we are going to fire 83,000 employees. That is wrong.” Collins insisted that the VA was “looking at a goal of how many employees we have and how many employees that are actually working in the front line taking care. I have doctors and nurses right now that do not see patients. Is that helping veteran health care?”
Collins defended the actions of the VA and spoke about challenges he was “constantly fighting” in the early weeks of his tenure. “We’ve been hit by a barrage of false rumors, innuendo, disinformation, speculation implying firing doctors and nurses, and forcing staff to work in closets and showers and that there’s chaos in the department, none of which have been backed up. Why? Because we canceled some contracts that worked for the VA that we should be doing in-house and we let go of less than one half of one percent of nonmission critical employees.”
The Trump Administration offered federal employees the option of resigning, which purportedly will go toward meeting the 15% target. NPR reported that VA employees have since shared data showing that 11,273 agency employees nationwide have applied for deferred resignation. Most of those employees are nurses (about 1300), medical support assistants (about 800), and social workers (about 300).
Collins stressed that the aim of restructuring was to protect veterans’ health care. By getting rid of DEI initiatives, the VA saved $14 million, which he said was redirected to veterans with disabilities who need prosthetics.
Sen. Bernie Sanders (D-VT) addressed concerns about the existing shortage of clinicians at the VA, asking Collins what he was doing to bring in more doctors, nurses, and social workers. In addition to moving doctors and nurses from nonpatient care to patient care, Collins said, he planned to work with Congress to make salaries more competitive.
But money and adding more employees are not always the solution, Collins said. For example, he said, the VA has been spending $588 million a year veteran suicide research, its top clinical priority. Yet, he said there has not been a significant decrease in veteran suicide rates since 2008.
The most recent VA suicide report, released in 2024, indicates suicide rates have remained steady since 2001. However, in 2022, the number of suicides among veterans (6407) was actually lower than in 12 of the previous 14 years.
According to media reports, congressional lawmakers, and union officials, Veteran Crisis Line (VCL) staff were among the 2400 probationary employees fired in February. In a Feb. 20 video, Collins accused Democrats of spreading lies and insisted no one who answered the phone was fired.
Later, in a letter to senators, Collins admitted that 24 VCL support staff were “erroneously” sent termination notices. The firings were later reversed, Collins said, and all VCL employees had been reinstated at the same position they previously held. “Ensuring the VCL is always accessible 24/7 is one of the department’s top priorities,” Collins insisted.
Collins shared his approval of keeping and expanding VA programs and studies on psychedelic treatments for patients with posttraumatic stress disorder and traumatic brain injury. He also spoke to the proposed 2026 budget calling for a $5.4 billion increase for the VA. If approved, that money would be targeted for medical care and homelessness.
US Department of Veterans Affairs (VA) Secretary Doug Collins testified in US House of Representatives and US Senate committees hearings that bringing staff numbers down to fiscal year 2019 figures was simply a goal: “Our goal, as we look at it, as everything goes forward, is a 15% decrease,” he told the senators. “It’s a goal. You have to start somewhere.”
“It’s a process we’re going through and I’m not going to work out a process in front of a committee or anywhere else,” Collins testified in the Senate on May 6, adding that it would be “incompetence” or “malpractice” to do so before time. “[When] we’re doing something as large as we are in an organization as sensitive on this Hill, it would not be right for us to do that in public. It would not be right for us to just come out and say here’s everything that we got and then have everybody scared because in the end it may not be the final decision.”
“We’re going to come to the best possible decision we can for the veterans in this country so they can have a VA system that actually works,” Collins argued in the Senate. “The VA’s been an issue for a long time. We’re trying to not make it an issue anymore.”
Collins later told a House committee on May 15 that VA was conducting a thorough review of department structure and staffing across the enterpise. "Our goal is to increase productivity and efficiency and to eliminate waste and bureaucracy improving health care delivery and benefits to our veterans. We are going to maintain VA essential jobs like doctors and nurses and claims processors" but eliminate positions it deemed "nonmission-critical" and consolidating areas of "overlap and waste."
Senate ranking member Richard Blumenthal (D-CT) and Chairman Jerry Moran (R-KS) both placed an emphasis on accountability for responsible resizing at the hearing.
“The department is at a critical juncture,” Moran said. “Perhaps that’s always true, and I want to hear from you that the changes under way at the VA are backed by data, informed by veteran demand, focused on improving outcomes for men and women the VA serves, and will be carried out in close coordination with this committee, as well as with veterans, VA staff, and veteran organizations.” Moran stressed that cutting should be about right-sizing, done carefully, and while treating people “with gratitude and respect.”
Blumenthal was more direct in his criticism of the approach: “You cannot slash and trash the VA without eliminating those essential positions which provide access and availability of health care. It simply cannot be done,” he told Collins.
In response, Collins replied, “You have stated on several occasions already that I am saying we are going to fire 83,000 employees. That is wrong.” Collins insisted that the VA was “looking at a goal of how many employees we have and how many employees that are actually working in the front line taking care. I have doctors and nurses right now that do not see patients. Is that helping veteran health care?”
Collins defended the actions of the VA and spoke about challenges he was “constantly fighting” in the early weeks of his tenure. “We’ve been hit by a barrage of false rumors, innuendo, disinformation, speculation implying firing doctors and nurses, and forcing staff to work in closets and showers and that there’s chaos in the department, none of which have been backed up. Why? Because we canceled some contracts that worked for the VA that we should be doing in-house and we let go of less than one half of one percent of nonmission critical employees.”
The Trump Administration offered federal employees the option of resigning, which purportedly will go toward meeting the 15% target. NPR reported that VA employees have since shared data showing that 11,273 agency employees nationwide have applied for deferred resignation. Most of those employees are nurses (about 1300), medical support assistants (about 800), and social workers (about 300).
Collins stressed that the aim of restructuring was to protect veterans’ health care. By getting rid of DEI initiatives, the VA saved $14 million, which he said was redirected to veterans with disabilities who need prosthetics.
Sen. Bernie Sanders (D-VT) addressed concerns about the existing shortage of clinicians at the VA, asking Collins what he was doing to bring in more doctors, nurses, and social workers. In addition to moving doctors and nurses from nonpatient care to patient care, Collins said, he planned to work with Congress to make salaries more competitive.
But money and adding more employees are not always the solution, Collins said. For example, he said, the VA has been spending $588 million a year veteran suicide research, its top clinical priority. Yet, he said there has not been a significant decrease in veteran suicide rates since 2008.
The most recent VA suicide report, released in 2024, indicates suicide rates have remained steady since 2001. However, in 2022, the number of suicides among veterans (6407) was actually lower than in 12 of the previous 14 years.
According to media reports, congressional lawmakers, and union officials, Veteran Crisis Line (VCL) staff were among the 2400 probationary employees fired in February. In a Feb. 20 video, Collins accused Democrats of spreading lies and insisted no one who answered the phone was fired.
Later, in a letter to senators, Collins admitted that 24 VCL support staff were “erroneously” sent termination notices. The firings were later reversed, Collins said, and all VCL employees had been reinstated at the same position they previously held. “Ensuring the VCL is always accessible 24/7 is one of the department’s top priorities,” Collins insisted.
Collins shared his approval of keeping and expanding VA programs and studies on psychedelic treatments for patients with posttraumatic stress disorder and traumatic brain injury. He also spoke to the proposed 2026 budget calling for a $5.4 billion increase for the VA. If approved, that money would be targeted for medical care and homelessness.
US Department of Veterans Affairs (VA) Secretary Doug Collins testified in US House of Representatives and US Senate committees hearings that bringing staff numbers down to fiscal year 2019 figures was simply a goal: “Our goal, as we look at it, as everything goes forward, is a 15% decrease,” he told the senators. “It’s a goal. You have to start somewhere.”
“It’s a process we’re going through and I’m not going to work out a process in front of a committee or anywhere else,” Collins testified in the Senate on May 6, adding that it would be “incompetence” or “malpractice” to do so before time. “[When] we’re doing something as large as we are in an organization as sensitive on this Hill, it would not be right for us to do that in public. It would not be right for us to just come out and say here’s everything that we got and then have everybody scared because in the end it may not be the final decision.”
“We’re going to come to the best possible decision we can for the veterans in this country so they can have a VA system that actually works,” Collins argued in the Senate. “The VA’s been an issue for a long time. We’re trying to not make it an issue anymore.”
Collins later told a House committee on May 15 that VA was conducting a thorough review of department structure and staffing across the enterpise. "Our goal is to increase productivity and efficiency and to eliminate waste and bureaucracy improving health care delivery and benefits to our veterans. We are going to maintain VA essential jobs like doctors and nurses and claims processors" but eliminate positions it deemed "nonmission-critical" and consolidating areas of "overlap and waste."
Senate ranking member Richard Blumenthal (D-CT) and Chairman Jerry Moran (R-KS) both placed an emphasis on accountability for responsible resizing at the hearing.
“The department is at a critical juncture,” Moran said. “Perhaps that’s always true, and I want to hear from you that the changes under way at the VA are backed by data, informed by veteran demand, focused on improving outcomes for men and women the VA serves, and will be carried out in close coordination with this committee, as well as with veterans, VA staff, and veteran organizations.” Moran stressed that cutting should be about right-sizing, done carefully, and while treating people “with gratitude and respect.”
Blumenthal was more direct in his criticism of the approach: “You cannot slash and trash the VA without eliminating those essential positions which provide access and availability of health care. It simply cannot be done,” he told Collins.
In response, Collins replied, “You have stated on several occasions already that I am saying we are going to fire 83,000 employees. That is wrong.” Collins insisted that the VA was “looking at a goal of how many employees we have and how many employees that are actually working in the front line taking care. I have doctors and nurses right now that do not see patients. Is that helping veteran health care?”
Collins defended the actions of the VA and spoke about challenges he was “constantly fighting” in the early weeks of his tenure. “We’ve been hit by a barrage of false rumors, innuendo, disinformation, speculation implying firing doctors and nurses, and forcing staff to work in closets and showers and that there’s chaos in the department, none of which have been backed up. Why? Because we canceled some contracts that worked for the VA that we should be doing in-house and we let go of less than one half of one percent of nonmission critical employees.”
The Trump Administration offered federal employees the option of resigning, which purportedly will go toward meeting the 15% target. NPR reported that VA employees have since shared data showing that 11,273 agency employees nationwide have applied for deferred resignation. Most of those employees are nurses (about 1300), medical support assistants (about 800), and social workers (about 300).
Collins stressed that the aim of restructuring was to protect veterans’ health care. By getting rid of DEI initiatives, the VA saved $14 million, which he said was redirected to veterans with disabilities who need prosthetics.
Sen. Bernie Sanders (D-VT) addressed concerns about the existing shortage of clinicians at the VA, asking Collins what he was doing to bring in more doctors, nurses, and social workers. In addition to moving doctors and nurses from nonpatient care to patient care, Collins said, he planned to work with Congress to make salaries more competitive.
But money and adding more employees are not always the solution, Collins said. For example, he said, the VA has been spending $588 million a year veteran suicide research, its top clinical priority. Yet, he said there has not been a significant decrease in veteran suicide rates since 2008.
The most recent VA suicide report, released in 2024, indicates suicide rates have remained steady since 2001. However, in 2022, the number of suicides among veterans (6407) was actually lower than in 12 of the previous 14 years.
According to media reports, congressional lawmakers, and union officials, Veteran Crisis Line (VCL) staff were among the 2400 probationary employees fired in February. In a Feb. 20 video, Collins accused Democrats of spreading lies and insisted no one who answered the phone was fired.
Later, in a letter to senators, Collins admitted that 24 VCL support staff were “erroneously” sent termination notices. The firings were later reversed, Collins said, and all VCL employees had been reinstated at the same position they previously held. “Ensuring the VCL is always accessible 24/7 is one of the department’s top priorities,” Collins insisted.
Collins shared his approval of keeping and expanding VA programs and studies on psychedelic treatments for patients with posttraumatic stress disorder and traumatic brain injury. He also spoke to the proposed 2026 budget calling for a $5.4 billion increase for the VA. If approved, that money would be targeted for medical care and homelessness.
Collins Lays Out Plans to Reduce VA by 15% in Senate Hearing
Collins Lays Out Plans to Reduce VA by 15% in Senate Hearing

Multiagent AI Systems in Health Care: Envisioning Next-Generation Intelligence
Artificial intelligence (AI) is rapidly evolving, with large language models (LLMs) marking a significant milestone in processing and generating human-like responses to natural language prompts. However, this advancement only signals the beginning of a more profound transformation in AI capabilities. The development of AI agents represents a new paradigm at the forefront of this evolution.
BACKGROUND
AI agents represent a leap forward from traditional LLM applications. While definitions may vary slightly among technology developers, the core concept remains: these agents are autonomous software entities designed to interact with their environment, make independent decisions, and execute tasks based on predefined goals.1-3 What sets AI agents apart is their combination of sophisticated components within structured architectures. At their core, AI agents incorporate an LLM for response generation, which is augmented by a suite of tools to optimize workflow and complete tasks, memory capabilities for personalized interactions, and autonomous reasoning. This combination allows AI agents to plan, create subtasks, gather information, and learn iteratively from their own experiences or other AI agents.
The true potential of this technology becomes apparent when multiple AI agents collaborate within multiagent AI systems. This concept introduces a new level of flexibility and capability in tackling complex tasks. Autogen, CrewAI, and LangChain offer various agent network configurations, including hierarchical, sequential, conditional, or even parallel task execution.4-6 This adaptability opens up a world of possibilities across various industries, but perhaps nowhere is the potential impact more exciting and profound than in health care.
AI agents in health care present an opportunity to revolutionize patient care, streamline administrative processes, and support complex clinical decision-making. This review examines 3 scenarios that illustrate the impact of AI agents in health care: a hypothetical sepsis management system, chronic disease management, and hospital patient flow optimization. This article will provide a detailed look at the technical implementation challenges, including the integration with existing health care IT systems, data privacy considerations, and the crucial role of explainable AI in maintaining trust and transparency.
It is challenging to implement AI agents in health care. Concerns include ensuring data quality and mitigating bias, seamlessly integrating these systems into existing clinical workflows, and navigating the complex ethical considerations that arise when deploying autonomous systems in health care. The integration with Internet of Things (IoT) devices for real-time patient data monitoring and the development of more sophisticated natural language interfaces to enhance future human-AI collaboration.
The adoption of AI agents in health care is only beginning, and it promises to be transformative. As AI continues to evolve, a comprehensive understanding of its applications, limitations, and ethical considerations is essential. This report provides a comprehensive overview of the current state, potential applications, and future directions of AI agents in health care, offering insights valuable to researchers, clinicians, and policymakers.
MultiAgent AI architecture
Sepsis Management
Despite advancements in broad-spectrum antibiotics, imaging, and life support systems, mortality rates associated with sepsis remain high. The complexity of optimizing care in clinical settings has hindered progress in managing sepsis. Previous attempts to develop predictive sepsis models have proven challenging.7 This report proposes a multiagent AI system designed to enhance comprehensive patient monitoring and care through coordinated AI-driven interventions.
Data Collection and Integration Agent. Powered by a controlled vocabulary to specify all data, the primary function for the data collection and integration agent is to clean, transform, and organize patient data from structured and unstructured sources. This agent prepares succinct summaries of consultant notes and formats data for human and machine consumption. All numerical data are presented graphically, including relevant historical data trends. The agent also digitally captures all orders in a structured format using a specified controlled vocabulary. This structured data feed supports the output of other agents, including documentation, treatment planning, and risk stratification, while also supplying the data structures for future training.
Diagnostic Agent. Critical illness is characterized by multiple abnormalities across a wide array of tests, ranging from plain chest X-ray, computed tomography (CT), blood cell composition, plasma chemistry, and microscopic evaluation of specimens. Additionally, life support parameters provide insights into disease severity and can inform management recommendations. These data offer a wide array of visual and numerical data to be used as input for computation, recommendation, and further training. For example, to evaluate fluid overload on chest X-rays or tissue histopathology slides, an AI agent can leverage deep learning models such as convolutional neural networks and vision transformers to analyze images like radiographs and histopathology slides.8,9 Recurrent neural networks or transformer models process sequential data like time-series vital signs. The agent also implements ensemble methods that combine multiple machine learning algorithms to enhance diagnostic accuracy.
Risk Stratification Agent. This assesses severity and predicts potential outcomes. Morbidity and mortality risks are calculated using an established scoring system and individualized based on the history of other agents’ conditional patients. These are presented graphically, with major risk factors highlighted for explainability.
Treatment Recommendation Agent. Using a reinforcement learning framework supplemented by up-to-date clinical guidelines, this system leverages historical data structured with standardized vocabulary to analyze patients with similar clinical features. Training is also conducted on the patient’s physiological data. All recommendations are presented via a dedicated user interface in a readable format, along with recommendations for editable, orderable items, references, and full-text snippets from previous research. Stop rules end computing if confidence in recommendations is too broad or no clear pathway can be computed with certainty, prompting human mitigation.
Resource Management Agent. This agent coordinates hospital resources using constraint programming techniques for optimal resource allocation, uses queueing theory models to predict and manage patient flow, and implements genetic algorithms for complex scheduling problems.10,11
Monitoring and Alert Agent. By tracking patients’ progress and alerting staff to changes, this agent uses anomaly detection algorithms to identify unusual patterns in patient data and implement time-series forecasting models, such as autoregressive integrated moving average and prophet, to predict future patient states. The agent also uses stream-processing techniques for real-time data analysis.12,13
Documentation and Reporting Agent. This agent maintains comprehensive medical records and generates reports. It employs advanced natural language processing techniques for automated report generation, uses advanced LLMs fine-tuned on medical corpora for narrative creation, and implements information-retrieval techniques to efficiently query patient records.
CLINICAL CASE STUDIES
To illustrate the functionality of a multiagent system, this report examines its application for managing sepsis. The data collection and integration agent continuously aggregates patient data from various sources, normalizing and timestamping it for consistent processing. The diagnostic agent analyzes this integrated data in real time, applying sepsis criteria and utilizing a deep learning model trained on a large sepsis dataset to detect subtle patterns.
The risk stratification agent calculates severity scores, such as the Sepsis-related Organ Failure Assessment (SOFA), quick SOFA (qSOFA), and Acute Physiology and Chronic Health Evaluation II, upon detecting a possible sepsis case.14 It predicts the likelihood of specific outcomes and estimates the potential trajectory of the patient’s condition for the next 24 to 48 hours. Based on this assessment, the treatment recommendation agent suggests an initial treatment plan, including appropriate antibiotics, fluid resuscitation protocols, and vasopressor recommendations, recommendations when indicated.
Concurrently, the resource management agent checks the availability of necessary resources and prioritizes allocation based on the severity. The monitoring agent tracks the patient’s response to interventions in real time, alerting the care team to any concerning changes or lack of expected improvement. Throughout this process, the documentation agent ensures that all actions, responses, and outcomes are meticulously recorded in a structured format and generates real-time updates for the patient’s electronic health record (EHR) and preparing summary reports for handoffs between care teams.
Administrative Workflow Support
Modern health care operations are resource-intensive, requiring coordination of advanced imaging, procedures, laboratory testing, and professional consultations.15 AI-powered health care administrative workflow systems are revolutionizing how medical facilities coordinate patient care. For patients with chronic cough, these systems seamlessly integrate scheduling, imaging, diagnostics, and follow-up care into a cohesive process that reduces administrative burden while improving patient outcomes. Through an intuitive interface and automated assistance, health care practitioners (HCPs) can track patient progress from initial consultation through diagnosis and treatment.
The process begins when an HCP enters a patient into the system, which triggers an automated CT scan scheduling system. The system considers factors like urgency, facility availability, and patient preferences to suggest optimal appointment times. Once imaging is complete, AI agents analyze the radiology reports, extract key findings, and generate structured summaries that highlight critical information such as “mild bronchial wall thickening with patchy ground-glass opacities” or “findings consistent with chronic bronchitis.”
Based on these findings, the system automatically generates evidence-based recommendations for follow-up care, such as pulmonology consultations or follow-up imaging in 3 months. These recommendations are presented to the ordering clinician, along with suggested appointment slots for specialist consultations. The system then manages the coordination of multiple appointments, ensuring each step in the patient’s care plan is properly sequenced and scheduled.
The entire process is monitored through a comprehensive dashboard that provides real-time updates on patient status, appointment schedules, and clinical recommendations. HCPs can track which patients require immediate attention, view upcoming appointments, and monitor the progress of ongoing care plans.
Multiagent AI Operation Optimization
Hospitals are complex entities that must function at different scales and respond in an agile, timely manner at all hours, deploying staff at various positions.16 A system of AI agents can receive signals from sensors monitoring foot traffic in the emergency department and trauma unit, as well as the availability of operating room staff, equipment, and intensive care unit beds. Smart sensors enable this monitoring through IoT networks. These networks benefit from advances in adaptive and consensus networking algorithms, along with recent advances in bioengineering and biocomputing.17
For example, in the case of imaging for suspected abdominal obstruction, an AI agent tasked with scheduling CTs could time the patient’s arrival based on acuity. Another AI agent could alert staff transporting the patient to the CT appointment, with the next location contingent on a clinical decision to proceed to the operating room. Yet another AI agent could summarize radiology interpretations and alert the surgery and anesthesia teams to a potential case, while others could notify operating room staff of equipment needs or reserve a bed. In this paradigm, AI agents facilitate more precise and timely communication between multiple staff members.
TECHNICAL IMPLEMENTATION
Large Language Models
Each agent uses a different LLM optimized for its specific task. For example, the diagnostic agent uses an LLM pretrained on a large corpus of biomedical literature and fine-tuned on a dataset of confirmed sepsis cases and their presentations.18 It implements few-shot learning techniques to adapt to rare or atypical presentations. The treatment recommendation agent also uses an LLM, employing a retrieval-augmented generation approach to access the latest clinical guidelines during inference. The documentation agent uses another advanced language model, fine-tuned on a large corpus of high-quality medical documentation, implementing controlled text generation techniques and utilizing a separate smaller model for real-time error checking and correction.
Interagent Quality Control
Agents learn from their own experience and the experience of other agents. They are equipped with user-defined rule-based and model-based systems for quality assurance, with clear stopping rules for human involvement and mitigation.
Sophisticated quality control measures bolster the system’s reliability, including ensemble techniques for result comparison, redundancy for critical tasks, and automatic human review for disagreements above a certain threshold. Each agent provides a calibrated confidence score with its output, used to weigh inputs in downstream tasks and trigger additional checks for low-confidence outputs.
A dedicated quality control agent monitors output from all agents, employing both supervised and unsupervised anomaly detection techniques. Feedback loops allow agents to evaluate the quality and utility of information received from other agents. The system implements a multiarmed bandit approach to dynamically adjust the influence of different agents based on their performance and periodically retrains agent models using federated learning techniques.19
Electronic Health Record Integration
Seamless EHR integration is crucial for practical implementation. The system has secure application programming interface access to various EHR platforms, implements OAuth 2.0 for authentication, and use HTTPS with perfect forward secrecy for all communications.20 It works with HL7 FHIR to ensure interoperability and uses SNOMED CT for clinical terminology to ensure semantic interoperability across different EHRs.21,22
The system implements a multilevel approval system for write-backs to EHRs, with different thresholds based on the information’s criticality. It uses digital signatures to ensure the integrity and nonrepudiation of AI-generated entries and implements blockchain technology to create an immutable and distributed ledger of all AI system actions.23
Decision Transparency
To ensure transparency in decision-making processes, the system applies techniques (eg, local interpretable model-agnostic explanations and Shapley additive explanations) to provide insights into agent decision-making processes.24-26 It provides customized visualizations for different stakeholders and allows users to explore alternative decision paths through what-if scenario modeling.27
The system provides calibrated confidence indicators for each recommendation or decision, implementing a novel confidence calibration agent that continuously monitors and adjusts confidence scores based on observed outcomes.
Continuous Learning and Adaptation
The system employs several techniques to remain current with evolving medical knowledge. Federated learning includes information from diverse datasets across multiple institutions without compromising patient privacy.28 A/B testing is used to safely deploy and compare new agent versions in controlled settings, implementing multiarmed bandit algorithms to efficiently explore new models while minimizing potential negative impacts. Human-in-the-loop learning and active learning techniques are used to incorporate feedback from HCPs and efficiently solicit expert input on the most informative data.29
CLINICAL IMPLICATIONS
The implementation of multiagent AI systems in health care has several potential benefits: enhanced diagnostic accuracy, personalized treatment, improved efficiency, continuous monitoring, and resource optimization. A recent review of AI sepsis predictive models exhibited superior results to standard clinical scoring methods like qSOFA.30 In oncology, such systems can result in more tailored treatments, enhancing outcomes.31 The implementation of an ambient dictation system can improve workflow and prevent HCP burnout.32
ETHICAL CONSIDERATIONS AND AI OVERSIGHT
Integrating AI agents into health care raises significant ethical considerations that must be carefully addressed to ensure equitable and effective care delivery. One primary concern involves cultural and linguistic competency, as AI systems may struggle with cultural nuances, idioms, and context-specific communication patterns. This becomes particularly challenging in regions with diverse ethnic populations or immigrant communities, where medical terminology may not have direct translations and cultural beliefs significantly influence health care decisions. AI systems also may inherit and amplify existing biases in health care delivery, whether through HCP bias reflected in training data, patient bias affecting acceptance of AI-assisted care, or demographic underrepresentation during system development.
AI agents present unique opportunities for improving health care access and outcomes through community engagement, though such initiatives require thoughtful implementation. Predictive analytics can identify high-risk individuals within communities who may benefit from preventive care, while analysis of social determinants of health can enable more targeted interventions. However, these capabilities must be balanced with privacy concerns and the risk of surveillance, particularly in communities that distrust health care institutions. The potential for AI to bridge health care gaps must be weighed against the need to maintain cultural sensitivity and community trust.
The governance and oversight of health care AI systems requires a multistakeholder approach with clear lines of responsibility and accountability. This includes involvement from government health care agencies, professional medical associations, ethics boards, and independent auditors, all working together to establish and enforce standards while monitoring system performance and addressing potential biases. Health care organizations must maintain transparent policies about AI use, implement regular monitoring and evaluation protocols, and establish precise mechanisms for patient feedback and grievance resolution. Ongoing assessment and adjustment of these systems, informed by community feedback and outcomes data, will be crucial for their ethical implementation, ensuring that AI agents complement, rather than replace, human judgment and cultural sensitivity.
FUTURE DIRECTIONS
Despite the potential benefits, implementing multiagent AI systems in health care faces significant challenges that require careful consideration. Beyond the fundamental issues such as data quality and bias mitigation, health care organizations struggle with fragmented systems, inconsistent data formats, and varying quality. Technical infrastructure requirements are substantial, particularly in rural or underserved areas that lack robust networks and cybersecurity. HCPs already face significant cognitive load and time pressures, making integrating AI agents into existing workflows particularly challenging. There is also the critical issue of transparency and interpretability, as health care decisions require clear reasoning and accountability that many black-box AI systems struggle to provide.
The legal landscape introduces another layer of complexity, particularly regarding liability, consent, and privacy questions. When AI agents contribute to medical decisions, establishing clear lines of responsibility becomes crucial. There are also serious concerns about algorithmic fairness and the potential for AI systems to perpetuate or amplify existing inequities. The cost of implementation remains a significant barrier, requiring substantial investment in technology, training, and ongoing maintenance while ensuring resources are not diverted from direct patient care. Moreover, HCPs may resist adoption due to concerns about job security, loss of autonomy, or skepticism about AI capabilities while paradoxically facing risks of overreliance on AI systems that could lead to the degradation of human clinical skills.
Addressing these challenges requires a multifaceted approach that combines technical solutions with organizational and policy changes. Health care organizations must implement rigorous data validation processes and interoperability standards while developing hybrid models that balance sophisticated AI capabilities with interpretable techniques. Extensive research and iterative design processes, with direct input from HCPs, are essential for successful integration. Establishing independent ethics boards to oversee system development and deployment, conducting multicenter randomized controlled trials, and creating clear regulatory frameworks will ensure safe and effective implementation. Success will ultimately depend on ongoing collaboration between technology developers, HCPs, policymakers, and patients, maintaining a steady focus on improving patient care and outcomes while carefully navigating the complex challenges of AI integration in health care.33-35
As multiagent AI systems in health care evolve, several exciting directions emerge. These include the integration of IoT and wearable devices, the development of more sophisticated natural language interfaces, and applying these systems to predictive maintenance of medical equipment.
CONCLUSIONS
The advent of multiagent AI systems in health care represents a paradigm shift in the approach to patient care, clinical decision making, and health care management. While these systems offer immense potential to transform health care delivery, their development and implementation must be guided by rigorous scientific validation, ethical considerations, and a patient-centered approach. The ultimate goal remains clear: harnessing the power of AI to improve patient outcomes, enhance the efficiency of health care delivery, and ultimately advance the health and well-being of patients.
Amazon Web Services, Inc. What are AI agents? Agents in artificial intelligence explained. Accessed April 7, 2025. https://aws.amazon.com/what-is/ai-agents/
Gutowska A. What are AI agents? IBM. Accessed April 7, 2025. https://www.ibm.com/think/topics/ai-agents
Agent AI. Microsoft Research. Accessed April 7, 2025. https://www.microsoft.com/en-us/research/project/agent-ai
Microsoft. AutoGen. Accessed April 7, 2025. https://microsoft.github.io/autogen/
Crew AI. The Leading Multi-Agent Platform. CrewAI. Accessed April 7, 2025. https://www.crewai.com/
LangChain. Accessed April 7, 2025. https://www.langchain.com/
Wong A, Otles E, Donnelly JP, et al. External validation of a widely implemented proprietary sepsis prediction model in hospitalized patients. JAMA Intern Med. 2021;181(8):1065-1070. doi:10.1001/jamainternmed.2021.2626
Willemink MJ, Roth HR, Sandfort V. Toward foundational deep learning models for medical imaging in the new era of transformer networks. Radiol Artif Intell. 2022;4(6):e210284. doi:10.1148/ryai.210284
Waqas A, Bui MM, Glassy EF, et al. Revolutionizing digital pathology with the power of generative artificial intelligence and foundation models. Lab Invest. 2023;103(11):100255. doi:10.1016/j.labinv.2023.100255
Moreno-Carrillo A, Arenas LMÁ, Fonseca JA, Caicedo CA, Tovar SV, Muñoz-Velandia OM. Application of queuing theory to optimize the triage process in a tertiary emergency care (“ER”) department. J Emerg Trauma Shock. 2019;12(4):268-273. doi:10.4103/JETS.JETS_42_19
Pongcharoen P, Hicks C, Braiden PM, Stewardson DJ. Determining optimum genetic algorithm parameters for scheduling the manufacturing and assembly of complex products. Int J Prod Econ. 2002;78(3):311-322. doi:10.1016/S0925-5273(02)00104-4
Sardar I, Akbar MA, Leiva V, Alsanad A, Mishra P. Machine learning and automatic ARIMA/Prophet models-based forecasting of COVID-19: methodology, evaluation, and case study in SAARC countries. Stoch Environ Res Risk Assess. 2023;37(1):345-359. doi:10.1007/s00477-022-02307-x
Samosir J, Indrawan-Santiago M, Haghighi PD. An evaluation of data stream processing systems for data driven applications. Procedia Comput Sci. 2016;80:439-449. doi:10.1016/j.procs.2016.05.322
Asmarawati TP, Suryantoro SD, Rosyid AN, et al. Predictive value of sequential organ failure assessment, quick sequential organ failure assessment, acute physiology and chronic health evaluation II, and new early warning signs scores estimate mortality of COVID-19 patients requiring intensive care unit. Indian J Crit Care Med. 2022;26(4):466-473. doi:10.5005/jp-journals-10071-24170
Khan S, Vandermorris A, Shepherd J, et al. Embracing uncertainty, managing complexity: applying complexity thinking principles to transformation efforts in healthcare systems. BMC Health Serv Res. 2018;18(1):192. doi:10.1186/s12913-018-2994-0
Plsek PE, Greenhalgh T. The challenge of complexity in health care. BMJ. 2001;323(7313):625-628. doi:10.1136/bmj.323.7313.625
Kouchaki S, Ding X, Sanei S. AI- and IoT-enabled solutions for healthcare. Sensors. 2024;24(8):2607. doi:10.3390/s24082607
Saab K, Tu T, Weng WH, et al. Capabilities of Gemini Models in Medicine. arXiv. doi:10.48550/arXiv.2404.18416
Villar SS, Bowden J, Wason J. Multi-armed bandit models for the optimal design of clinical trials: benefits and challenges. Stat Sci. 2015;30(2):199-215. doi:10.1214/14-STS504
Auth0. What is OAuth 2.0. Accessed April 7, 2025. https://auth0.com/intro-to-iam/what-is-oauth-2
HL7. Welcome to FHIR. Updated March 26, 2025. Accessed April 7, 2025. https://www.hl7.org/fhir/
SNOMED International. Accessed April 7, 2025. https://www.snomed.org
Hasselgren A, Kralevska K, Gligoroski D, Pedersen SA, Faxvaag A. Blockchain in healthcare and health sciences—a scoping review. Int J Med Inf. 2020;134:104040. doi:10.1016/j.ijmedinf.2019.104040
Ribeiro MT, Singh S, Guestrin C. “Why Should I Trust You?”: Explaining the predictions of any classifier. In: Proceedings of the 22nd ACM SIGKDD international conference on knowledge discovery and data mining. 2016:1135-1144. doi:10.1145/2939672.2939778
Ekanayake IU, Meddage DPP, Rathnayake U. A novel approach to explain the black-box nature of machine learning in compressive strength predictions of concrete using Shapley additive explanations (SHAP). Case Stud Constr Mater. 2022;16:e01059. doi:10.1016/j.cscm.2022.e01059
Alabi RO, Elmusrati M, Leivo I, Almangush A, Mäkitie AA. Machine learning explainability in nasopharyngeal cancer survival using LIME and SHAP. Sci Rep. 2023;13(1):8984. doi:10.1038/s41598-023-35795-0
Otto E, Culakova E, Meng S, et al. Overview of sankey flow diagrams: focusing on symptom trajectories in older adults with advanced cancer. J Geriatr Oncol. 2022;13(5):742-746. doi:10.1016/j.jgo.2021.12.017
Fereidooni H, Marchal S, Miettinen M, et al. SAFELearn: secure aggregation for private federated learning. In: 2021 IEEE security and privacy workshops (SPW). 2021:56-62. doi:10.1109/SPW53761.2021.00017
Linton DL, Pangle WM, Wyatt KH, Powell KN, Sherwood RE. Identifying key features of effective active learning: the effects of writing and peer discussion. Life Sci Educ. 2014;13(3):469-477. doi:10.1187/cbe.13-12-0242
Yang HS. Machine learning for sepsis prediction: prospects and challenges. Clin Chem. 2024;70(3):465-467. doi:10.1093/clinchem/hvae006
Liao J, Li X, Gan Y, et al. Artificial intelligence assists precision medicine in cancer treatment. Front Oncol. 2023;12. doi:10.3389/fonc.2022.998222
Tierney AA, Gayre G, Hoberman B, et al. Ambient artificial intelligence scribes to alleviate the burden of clinical documentation. NEJM Catal. 2024;5(3):CAT.23.0404. doi:10.1056/CAT.23.0404
Borkowski AA, Jakey CE, Thomas LB, Viswanadhan N, Mastorides SM. Establishing a hospital artificial intelligence committee to improve patient care. Fed Pract. 2022;39(8):334-336. doi:10.12788/fp.0299
Isaacks DB, Borkowski AA. Implementing trustworthy AI in VA high reliability health care organizations. Fed Pract.2024;41(2):40-43. doi:10.12788/fp.0454
Han R, Acosta JN, Shakeri Z, Ioannidis JPA, Topol EJ, Rajpurkar P. Randomized controlled trials evaluating artificial intelligence in clinical practice: a scoping review. Lancet Digit Health. 2024;6(5):e367-e373. doi:10.1016/S2589-7500(24)00047-5
Artificial intelligence (AI) is rapidly evolving, with large language models (LLMs) marking a significant milestone in processing and generating human-like responses to natural language prompts. However, this advancement only signals the beginning of a more profound transformation in AI capabilities. The development of AI agents represents a new paradigm at the forefront of this evolution.
BACKGROUND
AI agents represent a leap forward from traditional LLM applications. While definitions may vary slightly among technology developers, the core concept remains: these agents are autonomous software entities designed to interact with their environment, make independent decisions, and execute tasks based on predefined goals.1-3 What sets AI agents apart is their combination of sophisticated components within structured architectures. At their core, AI agents incorporate an LLM for response generation, which is augmented by a suite of tools to optimize workflow and complete tasks, memory capabilities for personalized interactions, and autonomous reasoning. This combination allows AI agents to plan, create subtasks, gather information, and learn iteratively from their own experiences or other AI agents.
The true potential of this technology becomes apparent when multiple AI agents collaborate within multiagent AI systems. This concept introduces a new level of flexibility and capability in tackling complex tasks. Autogen, CrewAI, and LangChain offer various agent network configurations, including hierarchical, sequential, conditional, or even parallel task execution.4-6 This adaptability opens up a world of possibilities across various industries, but perhaps nowhere is the potential impact more exciting and profound than in health care.
AI agents in health care present an opportunity to revolutionize patient care, streamline administrative processes, and support complex clinical decision-making. This review examines 3 scenarios that illustrate the impact of AI agents in health care: a hypothetical sepsis management system, chronic disease management, and hospital patient flow optimization. This article will provide a detailed look at the technical implementation challenges, including the integration with existing health care IT systems, data privacy considerations, and the crucial role of explainable AI in maintaining trust and transparency.
It is challenging to implement AI agents in health care. Concerns include ensuring data quality and mitigating bias, seamlessly integrating these systems into existing clinical workflows, and navigating the complex ethical considerations that arise when deploying autonomous systems in health care. The integration with Internet of Things (IoT) devices for real-time patient data monitoring and the development of more sophisticated natural language interfaces to enhance future human-AI collaboration.
The adoption of AI agents in health care is only beginning, and it promises to be transformative. As AI continues to evolve, a comprehensive understanding of its applications, limitations, and ethical considerations is essential. This report provides a comprehensive overview of the current state, potential applications, and future directions of AI agents in health care, offering insights valuable to researchers, clinicians, and policymakers.
MultiAgent AI architecture
Sepsis Management
Despite advancements in broad-spectrum antibiotics, imaging, and life support systems, mortality rates associated with sepsis remain high. The complexity of optimizing care in clinical settings has hindered progress in managing sepsis. Previous attempts to develop predictive sepsis models have proven challenging.7 This report proposes a multiagent AI system designed to enhance comprehensive patient monitoring and care through coordinated AI-driven interventions.
Data Collection and Integration Agent. Powered by a controlled vocabulary to specify all data, the primary function for the data collection and integration agent is to clean, transform, and organize patient data from structured and unstructured sources. This agent prepares succinct summaries of consultant notes and formats data for human and machine consumption. All numerical data are presented graphically, including relevant historical data trends. The agent also digitally captures all orders in a structured format using a specified controlled vocabulary. This structured data feed supports the output of other agents, including documentation, treatment planning, and risk stratification, while also supplying the data structures for future training.
Diagnostic Agent. Critical illness is characterized by multiple abnormalities across a wide array of tests, ranging from plain chest X-ray, computed tomography (CT), blood cell composition, plasma chemistry, and microscopic evaluation of specimens. Additionally, life support parameters provide insights into disease severity and can inform management recommendations. These data offer a wide array of visual and numerical data to be used as input for computation, recommendation, and further training. For example, to evaluate fluid overload on chest X-rays or tissue histopathology slides, an AI agent can leverage deep learning models such as convolutional neural networks and vision transformers to analyze images like radiographs and histopathology slides.8,9 Recurrent neural networks or transformer models process sequential data like time-series vital signs. The agent also implements ensemble methods that combine multiple machine learning algorithms to enhance diagnostic accuracy.
Risk Stratification Agent. This assesses severity and predicts potential outcomes. Morbidity and mortality risks are calculated using an established scoring system and individualized based on the history of other agents’ conditional patients. These are presented graphically, with major risk factors highlighted for explainability.
Treatment Recommendation Agent. Using a reinforcement learning framework supplemented by up-to-date clinical guidelines, this system leverages historical data structured with standardized vocabulary to analyze patients with similar clinical features. Training is also conducted on the patient’s physiological data. All recommendations are presented via a dedicated user interface in a readable format, along with recommendations for editable, orderable items, references, and full-text snippets from previous research. Stop rules end computing if confidence in recommendations is too broad or no clear pathway can be computed with certainty, prompting human mitigation.
Resource Management Agent. This agent coordinates hospital resources using constraint programming techniques for optimal resource allocation, uses queueing theory models to predict and manage patient flow, and implements genetic algorithms for complex scheduling problems.10,11
Monitoring and Alert Agent. By tracking patients’ progress and alerting staff to changes, this agent uses anomaly detection algorithms to identify unusual patterns in patient data and implement time-series forecasting models, such as autoregressive integrated moving average and prophet, to predict future patient states. The agent also uses stream-processing techniques for real-time data analysis.12,13
Documentation and Reporting Agent. This agent maintains comprehensive medical records and generates reports. It employs advanced natural language processing techniques for automated report generation, uses advanced LLMs fine-tuned on medical corpora for narrative creation, and implements information-retrieval techniques to efficiently query patient records.
CLINICAL CASE STUDIES
To illustrate the functionality of a multiagent system, this report examines its application for managing sepsis. The data collection and integration agent continuously aggregates patient data from various sources, normalizing and timestamping it for consistent processing. The diagnostic agent analyzes this integrated data in real time, applying sepsis criteria and utilizing a deep learning model trained on a large sepsis dataset to detect subtle patterns.
The risk stratification agent calculates severity scores, such as the Sepsis-related Organ Failure Assessment (SOFA), quick SOFA (qSOFA), and Acute Physiology and Chronic Health Evaluation II, upon detecting a possible sepsis case.14 It predicts the likelihood of specific outcomes and estimates the potential trajectory of the patient’s condition for the next 24 to 48 hours. Based on this assessment, the treatment recommendation agent suggests an initial treatment plan, including appropriate antibiotics, fluid resuscitation protocols, and vasopressor recommendations, recommendations when indicated.
Concurrently, the resource management agent checks the availability of necessary resources and prioritizes allocation based on the severity. The monitoring agent tracks the patient’s response to interventions in real time, alerting the care team to any concerning changes or lack of expected improvement. Throughout this process, the documentation agent ensures that all actions, responses, and outcomes are meticulously recorded in a structured format and generates real-time updates for the patient’s electronic health record (EHR) and preparing summary reports for handoffs between care teams.
Administrative Workflow Support
Modern health care operations are resource-intensive, requiring coordination of advanced imaging, procedures, laboratory testing, and professional consultations.15 AI-powered health care administrative workflow systems are revolutionizing how medical facilities coordinate patient care. For patients with chronic cough, these systems seamlessly integrate scheduling, imaging, diagnostics, and follow-up care into a cohesive process that reduces administrative burden while improving patient outcomes. Through an intuitive interface and automated assistance, health care practitioners (HCPs) can track patient progress from initial consultation through diagnosis and treatment.
The process begins when an HCP enters a patient into the system, which triggers an automated CT scan scheduling system. The system considers factors like urgency, facility availability, and patient preferences to suggest optimal appointment times. Once imaging is complete, AI agents analyze the radiology reports, extract key findings, and generate structured summaries that highlight critical information such as “mild bronchial wall thickening with patchy ground-glass opacities” or “findings consistent with chronic bronchitis.”
Based on these findings, the system automatically generates evidence-based recommendations for follow-up care, such as pulmonology consultations or follow-up imaging in 3 months. These recommendations are presented to the ordering clinician, along with suggested appointment slots for specialist consultations. The system then manages the coordination of multiple appointments, ensuring each step in the patient’s care plan is properly sequenced and scheduled.
The entire process is monitored through a comprehensive dashboard that provides real-time updates on patient status, appointment schedules, and clinical recommendations. HCPs can track which patients require immediate attention, view upcoming appointments, and monitor the progress of ongoing care plans.
Multiagent AI Operation Optimization
Hospitals are complex entities that must function at different scales and respond in an agile, timely manner at all hours, deploying staff at various positions.16 A system of AI agents can receive signals from sensors monitoring foot traffic in the emergency department and trauma unit, as well as the availability of operating room staff, equipment, and intensive care unit beds. Smart sensors enable this monitoring through IoT networks. These networks benefit from advances in adaptive and consensus networking algorithms, along with recent advances in bioengineering and biocomputing.17
For example, in the case of imaging for suspected abdominal obstruction, an AI agent tasked with scheduling CTs could time the patient’s arrival based on acuity. Another AI agent could alert staff transporting the patient to the CT appointment, with the next location contingent on a clinical decision to proceed to the operating room. Yet another AI agent could summarize radiology interpretations and alert the surgery and anesthesia teams to a potential case, while others could notify operating room staff of equipment needs or reserve a bed. In this paradigm, AI agents facilitate more precise and timely communication between multiple staff members.
TECHNICAL IMPLEMENTATION
Large Language Models
Each agent uses a different LLM optimized for its specific task. For example, the diagnostic agent uses an LLM pretrained on a large corpus of biomedical literature and fine-tuned on a dataset of confirmed sepsis cases and their presentations.18 It implements few-shot learning techniques to adapt to rare or atypical presentations. The treatment recommendation agent also uses an LLM, employing a retrieval-augmented generation approach to access the latest clinical guidelines during inference. The documentation agent uses another advanced language model, fine-tuned on a large corpus of high-quality medical documentation, implementing controlled text generation techniques and utilizing a separate smaller model for real-time error checking and correction.
Interagent Quality Control
Agents learn from their own experience and the experience of other agents. They are equipped with user-defined rule-based and model-based systems for quality assurance, with clear stopping rules for human involvement and mitigation.
Sophisticated quality control measures bolster the system’s reliability, including ensemble techniques for result comparison, redundancy for critical tasks, and automatic human review for disagreements above a certain threshold. Each agent provides a calibrated confidence score with its output, used to weigh inputs in downstream tasks and trigger additional checks for low-confidence outputs.
A dedicated quality control agent monitors output from all agents, employing both supervised and unsupervised anomaly detection techniques. Feedback loops allow agents to evaluate the quality and utility of information received from other agents. The system implements a multiarmed bandit approach to dynamically adjust the influence of different agents based on their performance and periodically retrains agent models using federated learning techniques.19
Electronic Health Record Integration
Seamless EHR integration is crucial for practical implementation. The system has secure application programming interface access to various EHR platforms, implements OAuth 2.0 for authentication, and use HTTPS with perfect forward secrecy for all communications.20 It works with HL7 FHIR to ensure interoperability and uses SNOMED CT for clinical terminology to ensure semantic interoperability across different EHRs.21,22
The system implements a multilevel approval system for write-backs to EHRs, with different thresholds based on the information’s criticality. It uses digital signatures to ensure the integrity and nonrepudiation of AI-generated entries and implements blockchain technology to create an immutable and distributed ledger of all AI system actions.23
Decision Transparency
To ensure transparency in decision-making processes, the system applies techniques (eg, local interpretable model-agnostic explanations and Shapley additive explanations) to provide insights into agent decision-making processes.24-26 It provides customized visualizations for different stakeholders and allows users to explore alternative decision paths through what-if scenario modeling.27
The system provides calibrated confidence indicators for each recommendation or decision, implementing a novel confidence calibration agent that continuously monitors and adjusts confidence scores based on observed outcomes.
Continuous Learning and Adaptation
The system employs several techniques to remain current with evolving medical knowledge. Federated learning includes information from diverse datasets across multiple institutions without compromising patient privacy.28 A/B testing is used to safely deploy and compare new agent versions in controlled settings, implementing multiarmed bandit algorithms to efficiently explore new models while minimizing potential negative impacts. Human-in-the-loop learning and active learning techniques are used to incorporate feedback from HCPs and efficiently solicit expert input on the most informative data.29
CLINICAL IMPLICATIONS
The implementation of multiagent AI systems in health care has several potential benefits: enhanced diagnostic accuracy, personalized treatment, improved efficiency, continuous monitoring, and resource optimization. A recent review of AI sepsis predictive models exhibited superior results to standard clinical scoring methods like qSOFA.30 In oncology, such systems can result in more tailored treatments, enhancing outcomes.31 The implementation of an ambient dictation system can improve workflow and prevent HCP burnout.32
ETHICAL CONSIDERATIONS AND AI OVERSIGHT
Integrating AI agents into health care raises significant ethical considerations that must be carefully addressed to ensure equitable and effective care delivery. One primary concern involves cultural and linguistic competency, as AI systems may struggle with cultural nuances, idioms, and context-specific communication patterns. This becomes particularly challenging in regions with diverse ethnic populations or immigrant communities, where medical terminology may not have direct translations and cultural beliefs significantly influence health care decisions. AI systems also may inherit and amplify existing biases in health care delivery, whether through HCP bias reflected in training data, patient bias affecting acceptance of AI-assisted care, or demographic underrepresentation during system development.
AI agents present unique opportunities for improving health care access and outcomes through community engagement, though such initiatives require thoughtful implementation. Predictive analytics can identify high-risk individuals within communities who may benefit from preventive care, while analysis of social determinants of health can enable more targeted interventions. However, these capabilities must be balanced with privacy concerns and the risk of surveillance, particularly in communities that distrust health care institutions. The potential for AI to bridge health care gaps must be weighed against the need to maintain cultural sensitivity and community trust.
The governance and oversight of health care AI systems requires a multistakeholder approach with clear lines of responsibility and accountability. This includes involvement from government health care agencies, professional medical associations, ethics boards, and independent auditors, all working together to establish and enforce standards while monitoring system performance and addressing potential biases. Health care organizations must maintain transparent policies about AI use, implement regular monitoring and evaluation protocols, and establish precise mechanisms for patient feedback and grievance resolution. Ongoing assessment and adjustment of these systems, informed by community feedback and outcomes data, will be crucial for their ethical implementation, ensuring that AI agents complement, rather than replace, human judgment and cultural sensitivity.
FUTURE DIRECTIONS
Despite the potential benefits, implementing multiagent AI systems in health care faces significant challenges that require careful consideration. Beyond the fundamental issues such as data quality and bias mitigation, health care organizations struggle with fragmented systems, inconsistent data formats, and varying quality. Technical infrastructure requirements are substantial, particularly in rural or underserved areas that lack robust networks and cybersecurity. HCPs already face significant cognitive load and time pressures, making integrating AI agents into existing workflows particularly challenging. There is also the critical issue of transparency and interpretability, as health care decisions require clear reasoning and accountability that many black-box AI systems struggle to provide.
The legal landscape introduces another layer of complexity, particularly regarding liability, consent, and privacy questions. When AI agents contribute to medical decisions, establishing clear lines of responsibility becomes crucial. There are also serious concerns about algorithmic fairness and the potential for AI systems to perpetuate or amplify existing inequities. The cost of implementation remains a significant barrier, requiring substantial investment in technology, training, and ongoing maintenance while ensuring resources are not diverted from direct patient care. Moreover, HCPs may resist adoption due to concerns about job security, loss of autonomy, or skepticism about AI capabilities while paradoxically facing risks of overreliance on AI systems that could lead to the degradation of human clinical skills.
Addressing these challenges requires a multifaceted approach that combines technical solutions with organizational and policy changes. Health care organizations must implement rigorous data validation processes and interoperability standards while developing hybrid models that balance sophisticated AI capabilities with interpretable techniques. Extensive research and iterative design processes, with direct input from HCPs, are essential for successful integration. Establishing independent ethics boards to oversee system development and deployment, conducting multicenter randomized controlled trials, and creating clear regulatory frameworks will ensure safe and effective implementation. Success will ultimately depend on ongoing collaboration between technology developers, HCPs, policymakers, and patients, maintaining a steady focus on improving patient care and outcomes while carefully navigating the complex challenges of AI integration in health care.33-35
As multiagent AI systems in health care evolve, several exciting directions emerge. These include the integration of IoT and wearable devices, the development of more sophisticated natural language interfaces, and applying these systems to predictive maintenance of medical equipment.
CONCLUSIONS
The advent of multiagent AI systems in health care represents a paradigm shift in the approach to patient care, clinical decision making, and health care management. While these systems offer immense potential to transform health care delivery, their development and implementation must be guided by rigorous scientific validation, ethical considerations, and a patient-centered approach. The ultimate goal remains clear: harnessing the power of AI to improve patient outcomes, enhance the efficiency of health care delivery, and ultimately advance the health and well-being of patients.
Artificial intelligence (AI) is rapidly evolving, with large language models (LLMs) marking a significant milestone in processing and generating human-like responses to natural language prompts. However, this advancement only signals the beginning of a more profound transformation in AI capabilities. The development of AI agents represents a new paradigm at the forefront of this evolution.
BACKGROUND
AI agents represent a leap forward from traditional LLM applications. While definitions may vary slightly among technology developers, the core concept remains: these agents are autonomous software entities designed to interact with their environment, make independent decisions, and execute tasks based on predefined goals.1-3 What sets AI agents apart is their combination of sophisticated components within structured architectures. At their core, AI agents incorporate an LLM for response generation, which is augmented by a suite of tools to optimize workflow and complete tasks, memory capabilities for personalized interactions, and autonomous reasoning. This combination allows AI agents to plan, create subtasks, gather information, and learn iteratively from their own experiences or other AI agents.
The true potential of this technology becomes apparent when multiple AI agents collaborate within multiagent AI systems. This concept introduces a new level of flexibility and capability in tackling complex tasks. Autogen, CrewAI, and LangChain offer various agent network configurations, including hierarchical, sequential, conditional, or even parallel task execution.4-6 This adaptability opens up a world of possibilities across various industries, but perhaps nowhere is the potential impact more exciting and profound than in health care.
AI agents in health care present an opportunity to revolutionize patient care, streamline administrative processes, and support complex clinical decision-making. This review examines 3 scenarios that illustrate the impact of AI agents in health care: a hypothetical sepsis management system, chronic disease management, and hospital patient flow optimization. This article will provide a detailed look at the technical implementation challenges, including the integration with existing health care IT systems, data privacy considerations, and the crucial role of explainable AI in maintaining trust and transparency.
It is challenging to implement AI agents in health care. Concerns include ensuring data quality and mitigating bias, seamlessly integrating these systems into existing clinical workflows, and navigating the complex ethical considerations that arise when deploying autonomous systems in health care. The integration with Internet of Things (IoT) devices for real-time patient data monitoring and the development of more sophisticated natural language interfaces to enhance future human-AI collaboration.
The adoption of AI agents in health care is only beginning, and it promises to be transformative. As AI continues to evolve, a comprehensive understanding of its applications, limitations, and ethical considerations is essential. This report provides a comprehensive overview of the current state, potential applications, and future directions of AI agents in health care, offering insights valuable to researchers, clinicians, and policymakers.
MultiAgent AI architecture
Sepsis Management
Despite advancements in broad-spectrum antibiotics, imaging, and life support systems, mortality rates associated with sepsis remain high. The complexity of optimizing care in clinical settings has hindered progress in managing sepsis. Previous attempts to develop predictive sepsis models have proven challenging.7 This report proposes a multiagent AI system designed to enhance comprehensive patient monitoring and care through coordinated AI-driven interventions.
Data Collection and Integration Agent. Powered by a controlled vocabulary to specify all data, the primary function for the data collection and integration agent is to clean, transform, and organize patient data from structured and unstructured sources. This agent prepares succinct summaries of consultant notes and formats data for human and machine consumption. All numerical data are presented graphically, including relevant historical data trends. The agent also digitally captures all orders in a structured format using a specified controlled vocabulary. This structured data feed supports the output of other agents, including documentation, treatment planning, and risk stratification, while also supplying the data structures for future training.
Diagnostic Agent. Critical illness is characterized by multiple abnormalities across a wide array of tests, ranging from plain chest X-ray, computed tomography (CT), blood cell composition, plasma chemistry, and microscopic evaluation of specimens. Additionally, life support parameters provide insights into disease severity and can inform management recommendations. These data offer a wide array of visual and numerical data to be used as input for computation, recommendation, and further training. For example, to evaluate fluid overload on chest X-rays or tissue histopathology slides, an AI agent can leverage deep learning models such as convolutional neural networks and vision transformers to analyze images like radiographs and histopathology slides.8,9 Recurrent neural networks or transformer models process sequential data like time-series vital signs. The agent also implements ensemble methods that combine multiple machine learning algorithms to enhance diagnostic accuracy.
Risk Stratification Agent. This assesses severity and predicts potential outcomes. Morbidity and mortality risks are calculated using an established scoring system and individualized based on the history of other agents’ conditional patients. These are presented graphically, with major risk factors highlighted for explainability.
Treatment Recommendation Agent. Using a reinforcement learning framework supplemented by up-to-date clinical guidelines, this system leverages historical data structured with standardized vocabulary to analyze patients with similar clinical features. Training is also conducted on the patient’s physiological data. All recommendations are presented via a dedicated user interface in a readable format, along with recommendations for editable, orderable items, references, and full-text snippets from previous research. Stop rules end computing if confidence in recommendations is too broad or no clear pathway can be computed with certainty, prompting human mitigation.
Resource Management Agent. This agent coordinates hospital resources using constraint programming techniques for optimal resource allocation, uses queueing theory models to predict and manage patient flow, and implements genetic algorithms for complex scheduling problems.10,11
Monitoring and Alert Agent. By tracking patients’ progress and alerting staff to changes, this agent uses anomaly detection algorithms to identify unusual patterns in patient data and implement time-series forecasting models, such as autoregressive integrated moving average and prophet, to predict future patient states. The agent also uses stream-processing techniques for real-time data analysis.12,13
Documentation and Reporting Agent. This agent maintains comprehensive medical records and generates reports. It employs advanced natural language processing techniques for automated report generation, uses advanced LLMs fine-tuned on medical corpora for narrative creation, and implements information-retrieval techniques to efficiently query patient records.
CLINICAL CASE STUDIES
To illustrate the functionality of a multiagent system, this report examines its application for managing sepsis. The data collection and integration agent continuously aggregates patient data from various sources, normalizing and timestamping it for consistent processing. The diagnostic agent analyzes this integrated data in real time, applying sepsis criteria and utilizing a deep learning model trained on a large sepsis dataset to detect subtle patterns.
The risk stratification agent calculates severity scores, such as the Sepsis-related Organ Failure Assessment (SOFA), quick SOFA (qSOFA), and Acute Physiology and Chronic Health Evaluation II, upon detecting a possible sepsis case.14 It predicts the likelihood of specific outcomes and estimates the potential trajectory of the patient’s condition for the next 24 to 48 hours. Based on this assessment, the treatment recommendation agent suggests an initial treatment plan, including appropriate antibiotics, fluid resuscitation protocols, and vasopressor recommendations, recommendations when indicated.
Concurrently, the resource management agent checks the availability of necessary resources and prioritizes allocation based on the severity. The monitoring agent tracks the patient’s response to interventions in real time, alerting the care team to any concerning changes or lack of expected improvement. Throughout this process, the documentation agent ensures that all actions, responses, and outcomes are meticulously recorded in a structured format and generates real-time updates for the patient’s electronic health record (EHR) and preparing summary reports for handoffs between care teams.
Administrative Workflow Support
Modern health care operations are resource-intensive, requiring coordination of advanced imaging, procedures, laboratory testing, and professional consultations.15 AI-powered health care administrative workflow systems are revolutionizing how medical facilities coordinate patient care. For patients with chronic cough, these systems seamlessly integrate scheduling, imaging, diagnostics, and follow-up care into a cohesive process that reduces administrative burden while improving patient outcomes. Through an intuitive interface and automated assistance, health care practitioners (HCPs) can track patient progress from initial consultation through diagnosis and treatment.
The process begins when an HCP enters a patient into the system, which triggers an automated CT scan scheduling system. The system considers factors like urgency, facility availability, and patient preferences to suggest optimal appointment times. Once imaging is complete, AI agents analyze the radiology reports, extract key findings, and generate structured summaries that highlight critical information such as “mild bronchial wall thickening with patchy ground-glass opacities” or “findings consistent with chronic bronchitis.”
Based on these findings, the system automatically generates evidence-based recommendations for follow-up care, such as pulmonology consultations or follow-up imaging in 3 months. These recommendations are presented to the ordering clinician, along with suggested appointment slots for specialist consultations. The system then manages the coordination of multiple appointments, ensuring each step in the patient’s care plan is properly sequenced and scheduled.
The entire process is monitored through a comprehensive dashboard that provides real-time updates on patient status, appointment schedules, and clinical recommendations. HCPs can track which patients require immediate attention, view upcoming appointments, and monitor the progress of ongoing care plans.
Multiagent AI Operation Optimization
Hospitals are complex entities that must function at different scales and respond in an agile, timely manner at all hours, deploying staff at various positions.16 A system of AI agents can receive signals from sensors monitoring foot traffic in the emergency department and trauma unit, as well as the availability of operating room staff, equipment, and intensive care unit beds. Smart sensors enable this monitoring through IoT networks. These networks benefit from advances in adaptive and consensus networking algorithms, along with recent advances in bioengineering and biocomputing.17
For example, in the case of imaging for suspected abdominal obstruction, an AI agent tasked with scheduling CTs could time the patient’s arrival based on acuity. Another AI agent could alert staff transporting the patient to the CT appointment, with the next location contingent on a clinical decision to proceed to the operating room. Yet another AI agent could summarize radiology interpretations and alert the surgery and anesthesia teams to a potential case, while others could notify operating room staff of equipment needs or reserve a bed. In this paradigm, AI agents facilitate more precise and timely communication between multiple staff members.
TECHNICAL IMPLEMENTATION
Large Language Models
Each agent uses a different LLM optimized for its specific task. For example, the diagnostic agent uses an LLM pretrained on a large corpus of biomedical literature and fine-tuned on a dataset of confirmed sepsis cases and their presentations.18 It implements few-shot learning techniques to adapt to rare or atypical presentations. The treatment recommendation agent also uses an LLM, employing a retrieval-augmented generation approach to access the latest clinical guidelines during inference. The documentation agent uses another advanced language model, fine-tuned on a large corpus of high-quality medical documentation, implementing controlled text generation techniques and utilizing a separate smaller model for real-time error checking and correction.
Interagent Quality Control
Agents learn from their own experience and the experience of other agents. They are equipped with user-defined rule-based and model-based systems for quality assurance, with clear stopping rules for human involvement and mitigation.
Sophisticated quality control measures bolster the system’s reliability, including ensemble techniques for result comparison, redundancy for critical tasks, and automatic human review for disagreements above a certain threshold. Each agent provides a calibrated confidence score with its output, used to weigh inputs in downstream tasks and trigger additional checks for low-confidence outputs.
A dedicated quality control agent monitors output from all agents, employing both supervised and unsupervised anomaly detection techniques. Feedback loops allow agents to evaluate the quality and utility of information received from other agents. The system implements a multiarmed bandit approach to dynamically adjust the influence of different agents based on their performance and periodically retrains agent models using federated learning techniques.19
Electronic Health Record Integration
Seamless EHR integration is crucial for practical implementation. The system has secure application programming interface access to various EHR platforms, implements OAuth 2.0 for authentication, and use HTTPS with perfect forward secrecy for all communications.20 It works with HL7 FHIR to ensure interoperability and uses SNOMED CT for clinical terminology to ensure semantic interoperability across different EHRs.21,22
The system implements a multilevel approval system for write-backs to EHRs, with different thresholds based on the information’s criticality. It uses digital signatures to ensure the integrity and nonrepudiation of AI-generated entries and implements blockchain technology to create an immutable and distributed ledger of all AI system actions.23
Decision Transparency
To ensure transparency in decision-making processes, the system applies techniques (eg, local interpretable model-agnostic explanations and Shapley additive explanations) to provide insights into agent decision-making processes.24-26 It provides customized visualizations for different stakeholders and allows users to explore alternative decision paths through what-if scenario modeling.27
The system provides calibrated confidence indicators for each recommendation or decision, implementing a novel confidence calibration agent that continuously monitors and adjusts confidence scores based on observed outcomes.
Continuous Learning and Adaptation
The system employs several techniques to remain current with evolving medical knowledge. Federated learning includes information from diverse datasets across multiple institutions without compromising patient privacy.28 A/B testing is used to safely deploy and compare new agent versions in controlled settings, implementing multiarmed bandit algorithms to efficiently explore new models while minimizing potential negative impacts. Human-in-the-loop learning and active learning techniques are used to incorporate feedback from HCPs and efficiently solicit expert input on the most informative data.29
CLINICAL IMPLICATIONS
The implementation of multiagent AI systems in health care has several potential benefits: enhanced diagnostic accuracy, personalized treatment, improved efficiency, continuous monitoring, and resource optimization. A recent review of AI sepsis predictive models exhibited superior results to standard clinical scoring methods like qSOFA.30 In oncology, such systems can result in more tailored treatments, enhancing outcomes.31 The implementation of an ambient dictation system can improve workflow and prevent HCP burnout.32
ETHICAL CONSIDERATIONS AND AI OVERSIGHT
Integrating AI agents into health care raises significant ethical considerations that must be carefully addressed to ensure equitable and effective care delivery. One primary concern involves cultural and linguistic competency, as AI systems may struggle with cultural nuances, idioms, and context-specific communication patterns. This becomes particularly challenging in regions with diverse ethnic populations or immigrant communities, where medical terminology may not have direct translations and cultural beliefs significantly influence health care decisions. AI systems also may inherit and amplify existing biases in health care delivery, whether through HCP bias reflected in training data, patient bias affecting acceptance of AI-assisted care, or demographic underrepresentation during system development.
AI agents present unique opportunities for improving health care access and outcomes through community engagement, though such initiatives require thoughtful implementation. Predictive analytics can identify high-risk individuals within communities who may benefit from preventive care, while analysis of social determinants of health can enable more targeted interventions. However, these capabilities must be balanced with privacy concerns and the risk of surveillance, particularly in communities that distrust health care institutions. The potential for AI to bridge health care gaps must be weighed against the need to maintain cultural sensitivity and community trust.
The governance and oversight of health care AI systems requires a multistakeholder approach with clear lines of responsibility and accountability. This includes involvement from government health care agencies, professional medical associations, ethics boards, and independent auditors, all working together to establish and enforce standards while monitoring system performance and addressing potential biases. Health care organizations must maintain transparent policies about AI use, implement regular monitoring and evaluation protocols, and establish precise mechanisms for patient feedback and grievance resolution. Ongoing assessment and adjustment of these systems, informed by community feedback and outcomes data, will be crucial for their ethical implementation, ensuring that AI agents complement, rather than replace, human judgment and cultural sensitivity.
FUTURE DIRECTIONS
Despite the potential benefits, implementing multiagent AI systems in health care faces significant challenges that require careful consideration. Beyond the fundamental issues such as data quality and bias mitigation, health care organizations struggle with fragmented systems, inconsistent data formats, and varying quality. Technical infrastructure requirements are substantial, particularly in rural or underserved areas that lack robust networks and cybersecurity. HCPs already face significant cognitive load and time pressures, making integrating AI agents into existing workflows particularly challenging. There is also the critical issue of transparency and interpretability, as health care decisions require clear reasoning and accountability that many black-box AI systems struggle to provide.
The legal landscape introduces another layer of complexity, particularly regarding liability, consent, and privacy questions. When AI agents contribute to medical decisions, establishing clear lines of responsibility becomes crucial. There are also serious concerns about algorithmic fairness and the potential for AI systems to perpetuate or amplify existing inequities. The cost of implementation remains a significant barrier, requiring substantial investment in technology, training, and ongoing maintenance while ensuring resources are not diverted from direct patient care. Moreover, HCPs may resist adoption due to concerns about job security, loss of autonomy, or skepticism about AI capabilities while paradoxically facing risks of overreliance on AI systems that could lead to the degradation of human clinical skills.
Addressing these challenges requires a multifaceted approach that combines technical solutions with organizational and policy changes. Health care organizations must implement rigorous data validation processes and interoperability standards while developing hybrid models that balance sophisticated AI capabilities with interpretable techniques. Extensive research and iterative design processes, with direct input from HCPs, are essential for successful integration. Establishing independent ethics boards to oversee system development and deployment, conducting multicenter randomized controlled trials, and creating clear regulatory frameworks will ensure safe and effective implementation. Success will ultimately depend on ongoing collaboration between technology developers, HCPs, policymakers, and patients, maintaining a steady focus on improving patient care and outcomes while carefully navigating the complex challenges of AI integration in health care.33-35
As multiagent AI systems in health care evolve, several exciting directions emerge. These include the integration of IoT and wearable devices, the development of more sophisticated natural language interfaces, and applying these systems to predictive maintenance of medical equipment.
CONCLUSIONS
The advent of multiagent AI systems in health care represents a paradigm shift in the approach to patient care, clinical decision making, and health care management. While these systems offer immense potential to transform health care delivery, their development and implementation must be guided by rigorous scientific validation, ethical considerations, and a patient-centered approach. The ultimate goal remains clear: harnessing the power of AI to improve patient outcomes, enhance the efficiency of health care delivery, and ultimately advance the health and well-being of patients.
Amazon Web Services, Inc. What are AI agents? Agents in artificial intelligence explained. Accessed April 7, 2025. https://aws.amazon.com/what-is/ai-agents/
Gutowska A. What are AI agents? IBM. Accessed April 7, 2025. https://www.ibm.com/think/topics/ai-agents
Agent AI. Microsoft Research. Accessed April 7, 2025. https://www.microsoft.com/en-us/research/project/agent-ai
Microsoft. AutoGen. Accessed April 7, 2025. https://microsoft.github.io/autogen/
Crew AI. The Leading Multi-Agent Platform. CrewAI. Accessed April 7, 2025. https://www.crewai.com/
LangChain. Accessed April 7, 2025. https://www.langchain.com/
Wong A, Otles E, Donnelly JP, et al. External validation of a widely implemented proprietary sepsis prediction model in hospitalized patients. JAMA Intern Med. 2021;181(8):1065-1070. doi:10.1001/jamainternmed.2021.2626
Willemink MJ, Roth HR, Sandfort V. Toward foundational deep learning models for medical imaging in the new era of transformer networks. Radiol Artif Intell. 2022;4(6):e210284. doi:10.1148/ryai.210284
Waqas A, Bui MM, Glassy EF, et al. Revolutionizing digital pathology with the power of generative artificial intelligence and foundation models. Lab Invest. 2023;103(11):100255. doi:10.1016/j.labinv.2023.100255
Moreno-Carrillo A, Arenas LMÁ, Fonseca JA, Caicedo CA, Tovar SV, Muñoz-Velandia OM. Application of queuing theory to optimize the triage process in a tertiary emergency care (“ER”) department. J Emerg Trauma Shock. 2019;12(4):268-273. doi:10.4103/JETS.JETS_42_19
Pongcharoen P, Hicks C, Braiden PM, Stewardson DJ. Determining optimum genetic algorithm parameters for scheduling the manufacturing and assembly of complex products. Int J Prod Econ. 2002;78(3):311-322. doi:10.1016/S0925-5273(02)00104-4
Sardar I, Akbar MA, Leiva V, Alsanad A, Mishra P. Machine learning and automatic ARIMA/Prophet models-based forecasting of COVID-19: methodology, evaluation, and case study in SAARC countries. Stoch Environ Res Risk Assess. 2023;37(1):345-359. doi:10.1007/s00477-022-02307-x
Samosir J, Indrawan-Santiago M, Haghighi PD. An evaluation of data stream processing systems for data driven applications. Procedia Comput Sci. 2016;80:439-449. doi:10.1016/j.procs.2016.05.322
Asmarawati TP, Suryantoro SD, Rosyid AN, et al. Predictive value of sequential organ failure assessment, quick sequential organ failure assessment, acute physiology and chronic health evaluation II, and new early warning signs scores estimate mortality of COVID-19 patients requiring intensive care unit. Indian J Crit Care Med. 2022;26(4):466-473. doi:10.5005/jp-journals-10071-24170
Khan S, Vandermorris A, Shepherd J, et al. Embracing uncertainty, managing complexity: applying complexity thinking principles to transformation efforts in healthcare systems. BMC Health Serv Res. 2018;18(1):192. doi:10.1186/s12913-018-2994-0
Plsek PE, Greenhalgh T. The challenge of complexity in health care. BMJ. 2001;323(7313):625-628. doi:10.1136/bmj.323.7313.625
Kouchaki S, Ding X, Sanei S. AI- and IoT-enabled solutions for healthcare. Sensors. 2024;24(8):2607. doi:10.3390/s24082607
Saab K, Tu T, Weng WH, et al. Capabilities of Gemini Models in Medicine. arXiv. doi:10.48550/arXiv.2404.18416
Villar SS, Bowden J, Wason J. Multi-armed bandit models for the optimal design of clinical trials: benefits and challenges. Stat Sci. 2015;30(2):199-215. doi:10.1214/14-STS504
Auth0. What is OAuth 2.0. Accessed April 7, 2025. https://auth0.com/intro-to-iam/what-is-oauth-2
HL7. Welcome to FHIR. Updated March 26, 2025. Accessed April 7, 2025. https://www.hl7.org/fhir/
SNOMED International. Accessed April 7, 2025. https://www.snomed.org
Hasselgren A, Kralevska K, Gligoroski D, Pedersen SA, Faxvaag A. Blockchain in healthcare and health sciences—a scoping review. Int J Med Inf. 2020;134:104040. doi:10.1016/j.ijmedinf.2019.104040
Ribeiro MT, Singh S, Guestrin C. “Why Should I Trust You?”: Explaining the predictions of any classifier. In: Proceedings of the 22nd ACM SIGKDD international conference on knowledge discovery and data mining. 2016:1135-1144. doi:10.1145/2939672.2939778
Ekanayake IU, Meddage DPP, Rathnayake U. A novel approach to explain the black-box nature of machine learning in compressive strength predictions of concrete using Shapley additive explanations (SHAP). Case Stud Constr Mater. 2022;16:e01059. doi:10.1016/j.cscm.2022.e01059
Alabi RO, Elmusrati M, Leivo I, Almangush A, Mäkitie AA. Machine learning explainability in nasopharyngeal cancer survival using LIME and SHAP. Sci Rep. 2023;13(1):8984. doi:10.1038/s41598-023-35795-0
Otto E, Culakova E, Meng S, et al. Overview of sankey flow diagrams: focusing on symptom trajectories in older adults with advanced cancer. J Geriatr Oncol. 2022;13(5):742-746. doi:10.1016/j.jgo.2021.12.017
Fereidooni H, Marchal S, Miettinen M, et al. SAFELearn: secure aggregation for private federated learning. In: 2021 IEEE security and privacy workshops (SPW). 2021:56-62. doi:10.1109/SPW53761.2021.00017
Linton DL, Pangle WM, Wyatt KH, Powell KN, Sherwood RE. Identifying key features of effective active learning: the effects of writing and peer discussion. Life Sci Educ. 2014;13(3):469-477. doi:10.1187/cbe.13-12-0242
Yang HS. Machine learning for sepsis prediction: prospects and challenges. Clin Chem. 2024;70(3):465-467. doi:10.1093/clinchem/hvae006
Liao J, Li X, Gan Y, et al. Artificial intelligence assists precision medicine in cancer treatment. Front Oncol. 2023;12. doi:10.3389/fonc.2022.998222
Tierney AA, Gayre G, Hoberman B, et al. Ambient artificial intelligence scribes to alleviate the burden of clinical documentation. NEJM Catal. 2024;5(3):CAT.23.0404. doi:10.1056/CAT.23.0404
Borkowski AA, Jakey CE, Thomas LB, Viswanadhan N, Mastorides SM. Establishing a hospital artificial intelligence committee to improve patient care. Fed Pract. 2022;39(8):334-336. doi:10.12788/fp.0299
Isaacks DB, Borkowski AA. Implementing trustworthy AI in VA high reliability health care organizations. Fed Pract.2024;41(2):40-43. doi:10.12788/fp.0454
Han R, Acosta JN, Shakeri Z, Ioannidis JPA, Topol EJ, Rajpurkar P. Randomized controlled trials evaluating artificial intelligence in clinical practice: a scoping review. Lancet Digit Health. 2024;6(5):e367-e373. doi:10.1016/S2589-7500(24)00047-5
Amazon Web Services, Inc. What are AI agents? Agents in artificial intelligence explained. Accessed April 7, 2025. https://aws.amazon.com/what-is/ai-agents/
Gutowska A. What are AI agents? IBM. Accessed April 7, 2025. https://www.ibm.com/think/topics/ai-agents
Agent AI. Microsoft Research. Accessed April 7, 2025. https://www.microsoft.com/en-us/research/project/agent-ai
Microsoft. AutoGen. Accessed April 7, 2025. https://microsoft.github.io/autogen/
Crew AI. The Leading Multi-Agent Platform. CrewAI. Accessed April 7, 2025. https://www.crewai.com/
LangChain. Accessed April 7, 2025. https://www.langchain.com/
Wong A, Otles E, Donnelly JP, et al. External validation of a widely implemented proprietary sepsis prediction model in hospitalized patients. JAMA Intern Med. 2021;181(8):1065-1070. doi:10.1001/jamainternmed.2021.2626
Willemink MJ, Roth HR, Sandfort V. Toward foundational deep learning models for medical imaging in the new era of transformer networks. Radiol Artif Intell. 2022;4(6):e210284. doi:10.1148/ryai.210284
Waqas A, Bui MM, Glassy EF, et al. Revolutionizing digital pathology with the power of generative artificial intelligence and foundation models. Lab Invest. 2023;103(11):100255. doi:10.1016/j.labinv.2023.100255
Moreno-Carrillo A, Arenas LMÁ, Fonseca JA, Caicedo CA, Tovar SV, Muñoz-Velandia OM. Application of queuing theory to optimize the triage process in a tertiary emergency care (“ER”) department. J Emerg Trauma Shock. 2019;12(4):268-273. doi:10.4103/JETS.JETS_42_19
Pongcharoen P, Hicks C, Braiden PM, Stewardson DJ. Determining optimum genetic algorithm parameters for scheduling the manufacturing and assembly of complex products. Int J Prod Econ. 2002;78(3):311-322. doi:10.1016/S0925-5273(02)00104-4
Sardar I, Akbar MA, Leiva V, Alsanad A, Mishra P. Machine learning and automatic ARIMA/Prophet models-based forecasting of COVID-19: methodology, evaluation, and case study in SAARC countries. Stoch Environ Res Risk Assess. 2023;37(1):345-359. doi:10.1007/s00477-022-02307-x
Samosir J, Indrawan-Santiago M, Haghighi PD. An evaluation of data stream processing systems for data driven applications. Procedia Comput Sci. 2016;80:439-449. doi:10.1016/j.procs.2016.05.322
Asmarawati TP, Suryantoro SD, Rosyid AN, et al. Predictive value of sequential organ failure assessment, quick sequential organ failure assessment, acute physiology and chronic health evaluation II, and new early warning signs scores estimate mortality of COVID-19 patients requiring intensive care unit. Indian J Crit Care Med. 2022;26(4):466-473. doi:10.5005/jp-journals-10071-24170
Khan S, Vandermorris A, Shepherd J, et al. Embracing uncertainty, managing complexity: applying complexity thinking principles to transformation efforts in healthcare systems. BMC Health Serv Res. 2018;18(1):192. doi:10.1186/s12913-018-2994-0
Plsek PE, Greenhalgh T. The challenge of complexity in health care. BMJ. 2001;323(7313):625-628. doi:10.1136/bmj.323.7313.625
Kouchaki S, Ding X, Sanei S. AI- and IoT-enabled solutions for healthcare. Sensors. 2024;24(8):2607. doi:10.3390/s24082607
Saab K, Tu T, Weng WH, et al. Capabilities of Gemini Models in Medicine. arXiv. doi:10.48550/arXiv.2404.18416
Villar SS, Bowden J, Wason J. Multi-armed bandit models for the optimal design of clinical trials: benefits and challenges. Stat Sci. 2015;30(2):199-215. doi:10.1214/14-STS504
Auth0. What is OAuth 2.0. Accessed April 7, 2025. https://auth0.com/intro-to-iam/what-is-oauth-2
HL7. Welcome to FHIR. Updated March 26, 2025. Accessed April 7, 2025. https://www.hl7.org/fhir/
SNOMED International. Accessed April 7, 2025. https://www.snomed.org
Hasselgren A, Kralevska K, Gligoroski D, Pedersen SA, Faxvaag A. Blockchain in healthcare and health sciences—a scoping review. Int J Med Inf. 2020;134:104040. doi:10.1016/j.ijmedinf.2019.104040
Ribeiro MT, Singh S, Guestrin C. “Why Should I Trust You?”: Explaining the predictions of any classifier. In: Proceedings of the 22nd ACM SIGKDD international conference on knowledge discovery and data mining. 2016:1135-1144. doi:10.1145/2939672.2939778
Ekanayake IU, Meddage DPP, Rathnayake U. A novel approach to explain the black-box nature of machine learning in compressive strength predictions of concrete using Shapley additive explanations (SHAP). Case Stud Constr Mater. 2022;16:e01059. doi:10.1016/j.cscm.2022.e01059
Alabi RO, Elmusrati M, Leivo I, Almangush A, Mäkitie AA. Machine learning explainability in nasopharyngeal cancer survival using LIME and SHAP. Sci Rep. 2023;13(1):8984. doi:10.1038/s41598-023-35795-0
Otto E, Culakova E, Meng S, et al. Overview of sankey flow diagrams: focusing on symptom trajectories in older adults with advanced cancer. J Geriatr Oncol. 2022;13(5):742-746. doi:10.1016/j.jgo.2021.12.017
Fereidooni H, Marchal S, Miettinen M, et al. SAFELearn: secure aggregation for private federated learning. In: 2021 IEEE security and privacy workshops (SPW). 2021:56-62. doi:10.1109/SPW53761.2021.00017
Linton DL, Pangle WM, Wyatt KH, Powell KN, Sherwood RE. Identifying key features of effective active learning: the effects of writing and peer discussion. Life Sci Educ. 2014;13(3):469-477. doi:10.1187/cbe.13-12-0242
Yang HS. Machine learning for sepsis prediction: prospects and challenges. Clin Chem. 2024;70(3):465-467. doi:10.1093/clinchem/hvae006
Liao J, Li X, Gan Y, et al. Artificial intelligence assists precision medicine in cancer treatment. Front Oncol. 2023;12. doi:10.3389/fonc.2022.998222
Tierney AA, Gayre G, Hoberman B, et al. Ambient artificial intelligence scribes to alleviate the burden of clinical documentation. NEJM Catal. 2024;5(3):CAT.23.0404. doi:10.1056/CAT.23.0404
Borkowski AA, Jakey CE, Thomas LB, Viswanadhan N, Mastorides SM. Establishing a hospital artificial intelligence committee to improve patient care. Fed Pract. 2022;39(8):334-336. doi:10.12788/fp.0299
Isaacks DB, Borkowski AA. Implementing trustworthy AI in VA high reliability health care organizations. Fed Pract.2024;41(2):40-43. doi:10.12788/fp.0454
Han R, Acosta JN, Shakeri Z, Ioannidis JPA, Topol EJ, Rajpurkar P. Randomized controlled trials evaluating artificial intelligence in clinical practice: a scoping review. Lancet Digit Health. 2024;6(5):e367-e373. doi:10.1016/S2589-7500(24)00047-5

Community Care Radiation Oncology Cost Calculations for a VA Medical Center
Community Care Radiation Oncology Cost Calculations for a VA Medical Center
William Kissick’s description of health care’s iron triangle in 1994 still resonates. Access, quality, and cost will always come at the expense of the others.1 In 2018, Congress passed the VA MISSION Act, allowing patients to pursue community care options for extended waits (> 28 days) or longer distance drive times of > 60 minutes for specialty care services, such as radiation oncology. According to Albanese et al, the VA MISSION Act sought to address gaps in care for veterans living in rural and underserved areas.2 The Veterans Health Administration (VHA) continues to increase community care spending, with a 13.8% increase in fiscal year 2024 and an expected cost of > $40 billion for 2025.3 One could argue this pays for access for remote patients and quality when services are unavailable, making it a direct application of the iron triangle.
The VA MISSION Act also bolstered the expansion of existing community care department staff to expediently facilitate and coordinate care and payments.2 Cost management and monitoring have become critical in predicting future staff requirements, maintaining functionality, and ensuring patients receive optimal care. The VHA purchases care through partner networks and defines these bundled health care services as standard episodes of care (SEOCs), which are “clinically related health care services for a specific unique illness or medical condition… over a defined period of time.”4 Medicare publishes its rates quarterly, and outpatient procedure pricing is readily available online.5 Along these same lines, the US Department of Veterans Affairs (VA) publishes a current list of available procedures and associated Current Procedure Technology (CPT) codes that are covered under its VA fee schedule for community care.
Unique challenges persist when using this system to accurately account for radiation oncology expenditures. This study was based on the current practices at the Richard L. Roudebush VA Medical Center (RLRVAMC), a large 1a hospital. A detailed analysis reveals the contemporaneous cost of radiation oncology cancer care from October 1, 2021, through February 1, 2024, highlights the challenges in SEOC definition and duration, communication issues between RLRVAMC and purchase partners, inconsistencies in billing, erroneous payments, and difficulty of cost categorization.
METHODS
Community care radiation oncology-related costs were examined from October 1, 2021, to February 1, 2024 for RLRVAMC, 6 months prior to billing data extraction. Figure 1 shows a simple radiation oncology patient pathway with consultation or visit, simulation and planning, and treatment, with codes used to check billing. It illustrates the expected relationships between the VHA (radiation oncology, primary, and specialty care) and community care (clinicians and radiation oncology treatment sites).
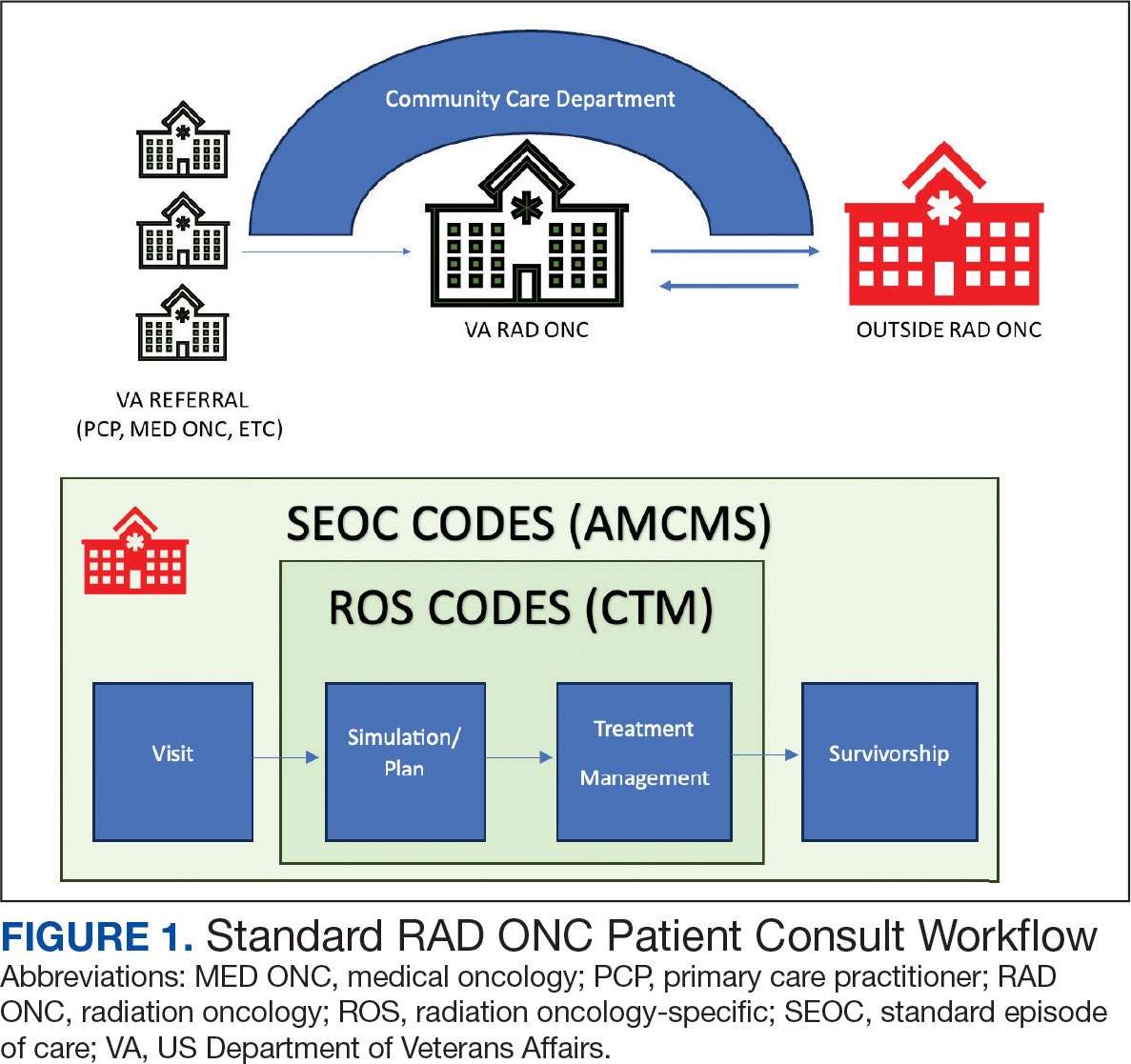
VHA standard operating procedures for a patient requesting community-based radiation oncology care require a board-certified radiation oncologist at RLRVAMC to review and approve the outside care request. Community care radiation oncology consultation data were accessed from the VA Corporate Data Warehouse (CDW) using Pyramid Analytics (V25.2). Nurses, physicians, and community care staff can add comments, forward consultations to other services, and mark them as complete or discontinued, when appropriate. Consultations not completed within 91 days are automatically discontinued. All community care requests from 2018 through 2024 were extracted; analysis began April 1, 2021, 6 months prior to the cost evaluation date of October 1, 2021.
An approved consultation is reviewed for eligibility by a nurse in the community care department and assigned an authorization number (a VA prefix followed by 12 digits). Billing codes are approved and organized by the community care networks, and all procedure codes should be captured and labeled under this number. The VAMC Community Care department obtains initial correspondence from the treating clinicians. Subsequent records from the treating radiation oncologist are expected to be scanned into the electronic health record and made accessible via the VA Joint Legacy Viewer (JLV) and Computerized Patient Record System (CPRS).
Radiation Oncology SEOC
The start date of the radiation oncology SEOC is determined by the community care nurse based on guidance established by the VA. It can be manually backdated or delayed, but current practice is to start at first visit or procedure code entry after approval from the VAMC Radiation Oncology department. Approved CPT codes from SEOC versions between October 1, 2021, and February 1, 2024, are in eAppendix 1 (available at doi:10.12788/fp.0585). These generally include 10 types of encounters, about 115 different laboratory tests, 115 imaging studies, 25 simulation and planning procedures, and 115 radiation treatment codes. The radiation oncology SEOCs during the study period had an approval duration of 180 days. Advanced Medical Cost Management Solutions software (AMCMS) is the VHA data analytics platform for community care medical service costs. AMCMS includes all individual CPT codes billed by specific radiation oncology SEOC versions. Data are refreshed monthly, and all charges were extracted on September 12, 2024, > 6 months after the final evaluated service date to allow for complete billing returns.6
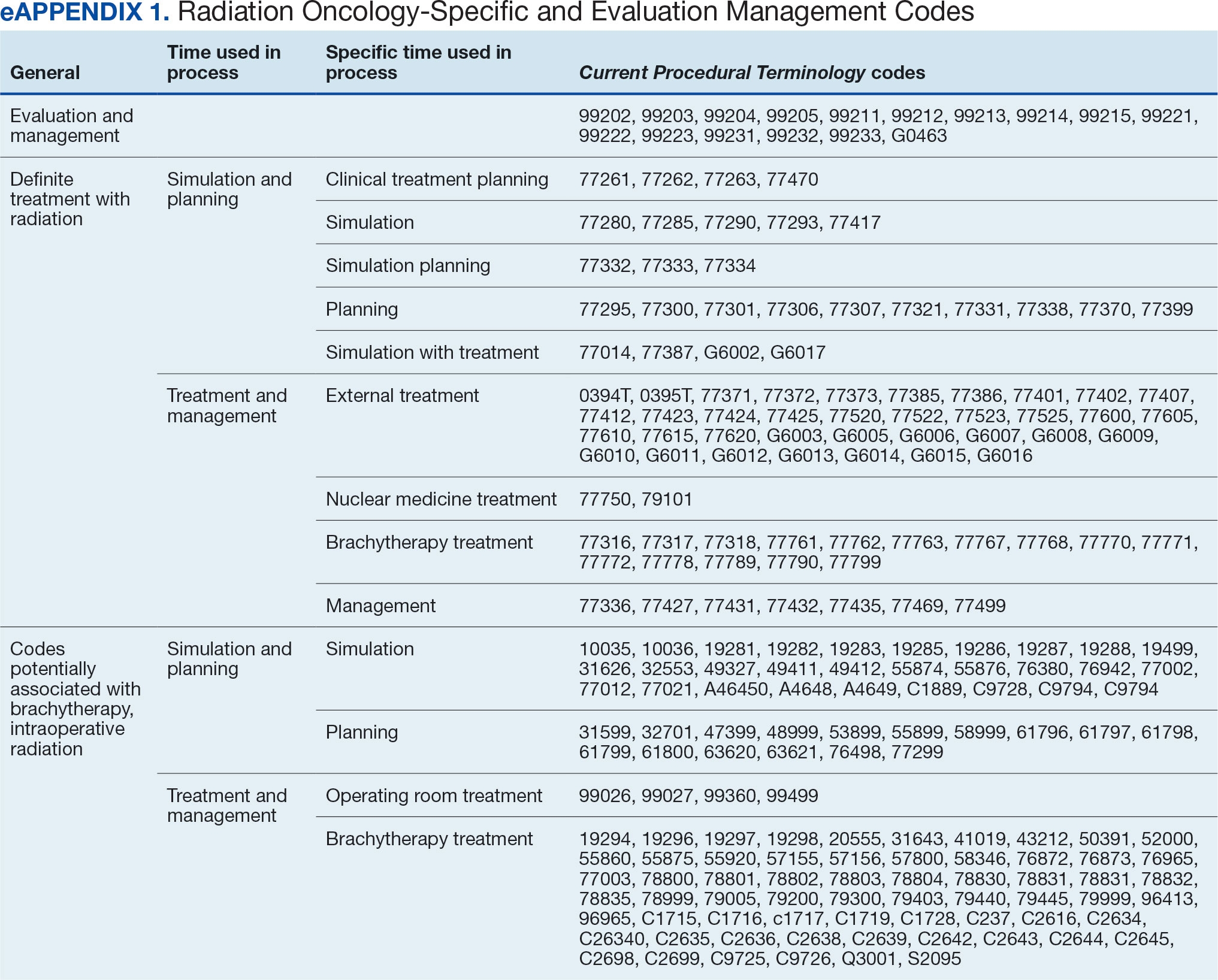
Radiation Oncology-Specific Costs
The VA Close to Me (CTM) program was used to find 84 specific radiation oncology CPT codes, nearly all within the 77.XXX or G6.XXX series, which included all radiation oncology-specific (ROS) codes (except visits accrued during consultation and return appointments). ROS costs are those that could not be performed by any other service and include procedures related to radiation oncology simulation, treatment planning, treatment delivery (with or without image guidance), and physician or physicist management. All ROS costs should be included in a patient’s radiation oncology SEOC. Other costs that may accompany operating room or brachytherapy administration did not follow a 77.XXX or G6.XXX pattern but were included in total radiation therapy operating costs.
Data obtained from AMCMS and CTM included patient name and identifier; CPT billed amount; CPT paid amount; dates of service; number of claims; International Classification of Diseases, Tenth Revision (ICD) diagnosis; and VA authorization numbers. Only CTM listed code modifiers. Only items categorized as paid were included in the analysis. Charges associated with discontinued consultations that had accrued costs also were included. Codes that were not directly related to ROS were separately characterized as other and further subcategorized.
Deep Dive Categorization
All scanned documents tagged to the community consultation were accessed and evaluated for completeness by a radiation oncologist (RS). The presence or absence of consultation notes and treatment summaries was evaluated based on necessity (ie, not needed for continuation of care or treatment was not given). In the absence of a specific completion summary or follow-up note detailing the treatment modality, number of fractions, and treatment sites, available documentation, including clinical notes and billing information, was used. Radical or curative therapies were identified as courses expected to eradicate disease, including stereotactic ablative radiotherapy to the brain, lung, liver, and other organs. Palliative therapies included whole-brain radiotherapy or other low-dose treatments. If the patient received the intended course, this was categorized as full. If incomplete, it was considered partial.
Billing Deviations
The complete document review allowed for close evaluation of paid therapy and identification of gaps in billing (eg, charges not found in extracted data that should have occurred) for external beam radiotherapy patients. Conversely, extra charges, such as an additional weekly treatment management charge (CPT code 77427), would be noted. Patients were expected to have the number of treatments specified in the summary, a clinical treatment planning code, and weekly treatment management notes from physicians and physicists every 5 fractions. Consultations and follow-up visits were expected to have 1 visit code; CPT codes 99205 and 99215, respectively, were used to estimate costs in their absence.
Costs were based on Medicare rates as of January 1 of the year in which they were accrued. 7-10 Duplicates were charges with the same code, date, billed quantity, and paid amounts for a given patient. These would always be considered erroneous. Medicare treatment costs for procedures such as intensity modulated radiotherapy (CPT code 77385 or 77386) are available on the Medicare website. When reviewing locality deviations for 77427, there was a maximum of 33% increase in Medicare rates. Therefore, for treatment codes, one would expect the range to be at least the Medicare rate and maximally 33% higher. These rates are negotiated with insurance companies, but this range was used for the purpose of reviewing and adjusting large data sets.
RESULTS
Since 2018, > 500 community care consults have been placed by radiation oncology for treatment in the community, with more following implementation of the VA MISSION Act. Use of radiation oncology community care services annually increased during the study period for this facility (Table 1, Figure 2). Of the 325 community care consults placed from October 1, 2021, to February 1, 2024, 248 radiation oncology SEOCs were recorded with charges for 181 patients (range, 1-5 SEOCs). Long drive time was the rationale for > 97% of patients directed to community care (Supplemental materials, available at doi:10.12788/fp.0585). Based on AMCMS data, $22.2 million was billed and $2.7 million was paid (20%) for 8747 CPT codes. Each community care interval cost the VA a median (range) of $5000 ($8-$168,000 (Figure 3).
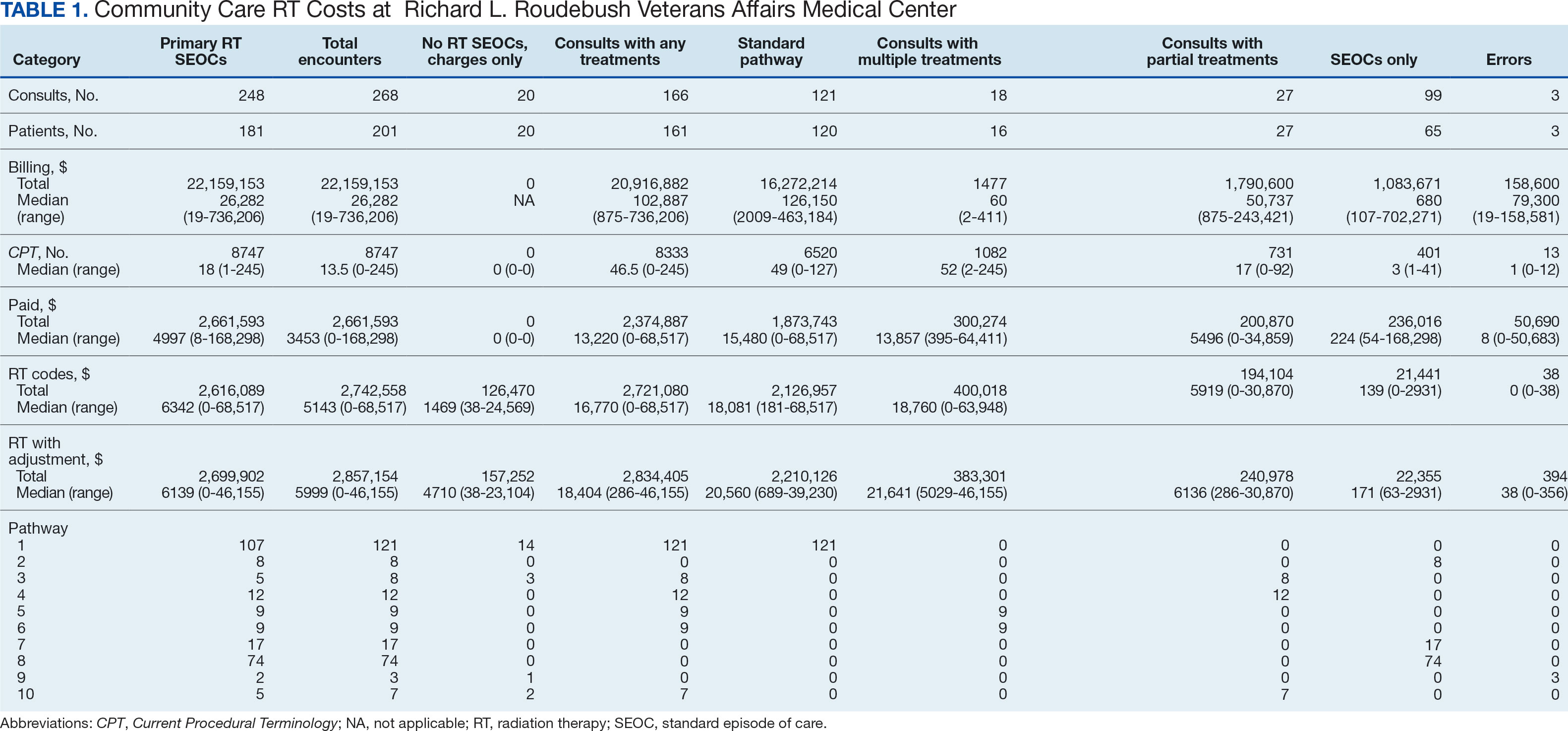
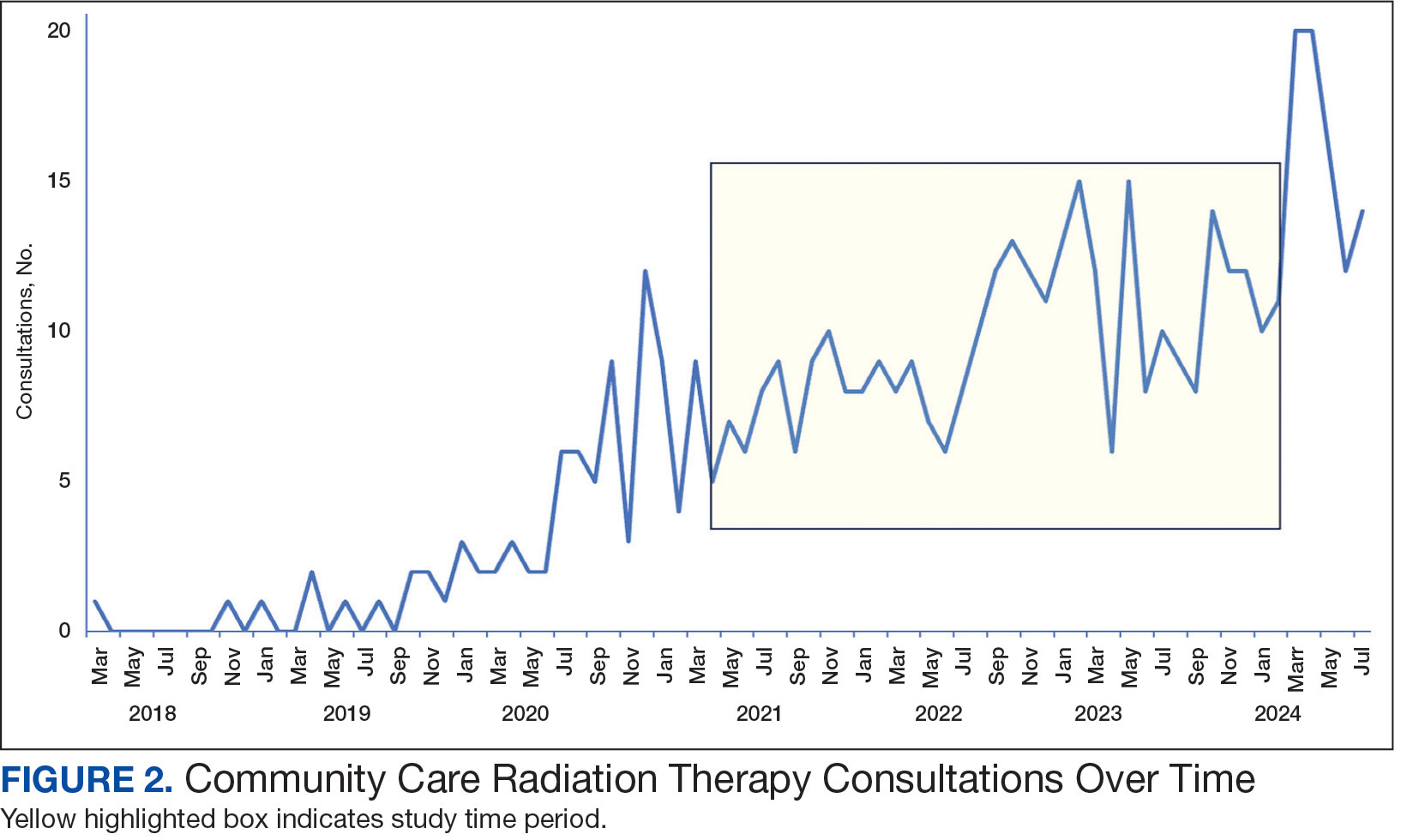
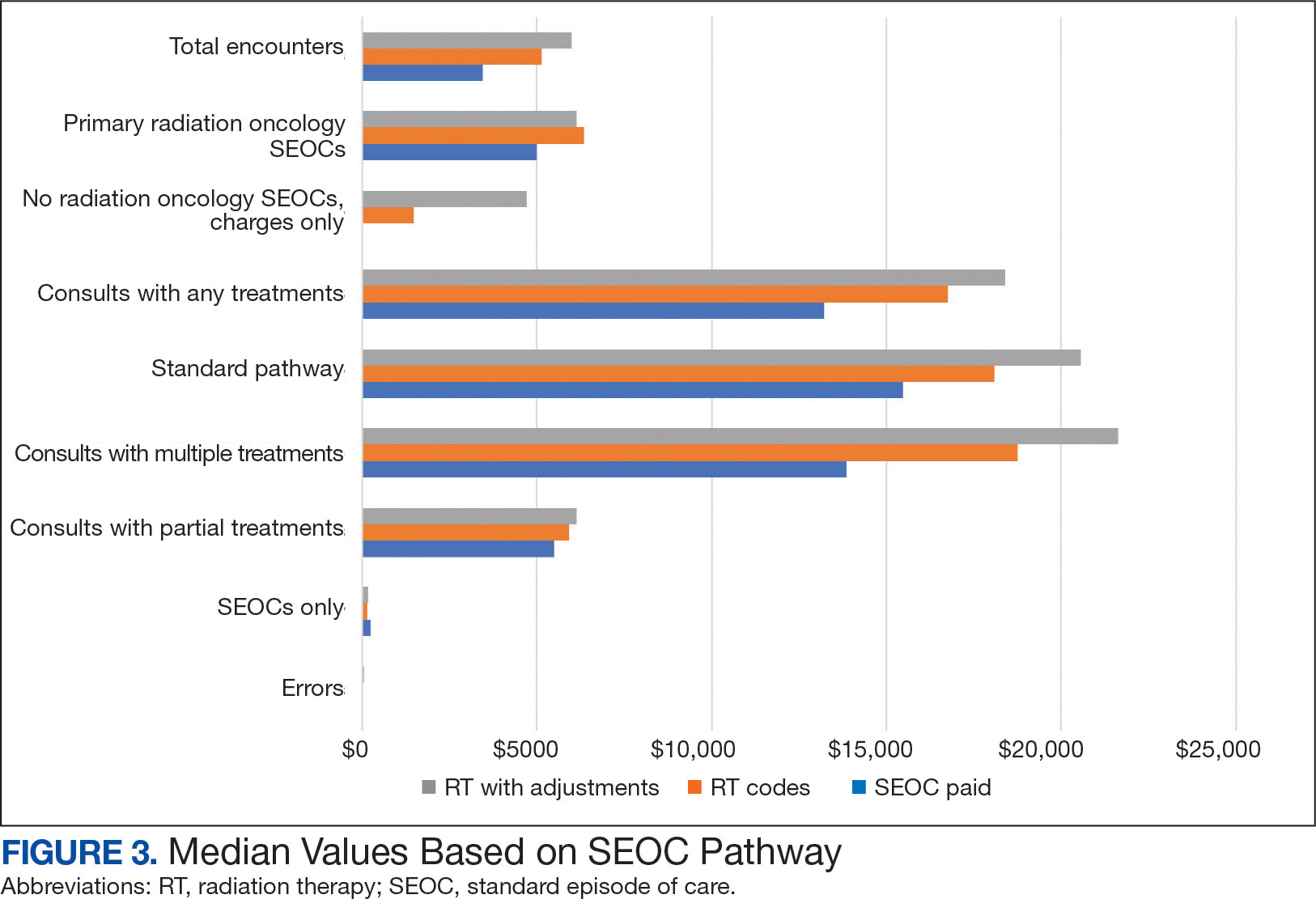
After reviewing ROS charges extracted from CTM, 20 additional patients had radiation oncology charges but did not have a radiation oncology SEOC for 268 episodes of care for 201 unique patients. In addition to the 20 patients who did not have a SEOC, 42 nonradiation oncology SEOCs contained 1148 radiation oncology codes, corresponding to almost $500,000 paid. Additional charges of about $416,000, which included biologic agents (eg, durvalumab, nivolumab), procedures (eg, mastectomies), and ambulance rides were inappropriately added to radiation oncology SEOCs.
While 77% of consultations were scanned into CPRS and JLV, only 54% of completion summaries were available with an estimated $115,000 in additional costs. The total adjusted costs was about $2.9 million. Almost 37% of SEOCs were for visits only. For the 166 SEOCs where patients received any radiation treatment or planning, the median cost was $18,000. Differences in SEOC pathways are shown in Figure 4. One hundred twenty-one SEOCs (45%) followed the standard pathway, with median SEOC costs of $15,500; when corrected for radiation-specific costs, the median cost increased to $18,000. When adjusted for billing irregularities, the median cost was $20,600. Ninety-nine SEOCs (37%) were for consultation/ follow-up visits only, with a median cost of $220. When omitting shared scans and nonradiation therapy costs and correcting for billing gaps, the median cost decreased to $170. A median of $9200 was paid per patient, with $12,900 for radiation therapy-specific costs and $13,300 adjusted for billing deviations. Narrowing to the 106 patients who received full, radical courses, the median SEOC, ROS, and adjusted radiation therapy costs increased to $19,400, $22,200, and $22,900, respectively (Table 2, Figure 5). Seventy-one SEOCs (26%) had already seen a radiation oncologist before the VA radiation oncology department was aware, and 49 SEOCs (18%) had retroactive approvals (Supplemental materials available at doi:10.12788/fp.0585).
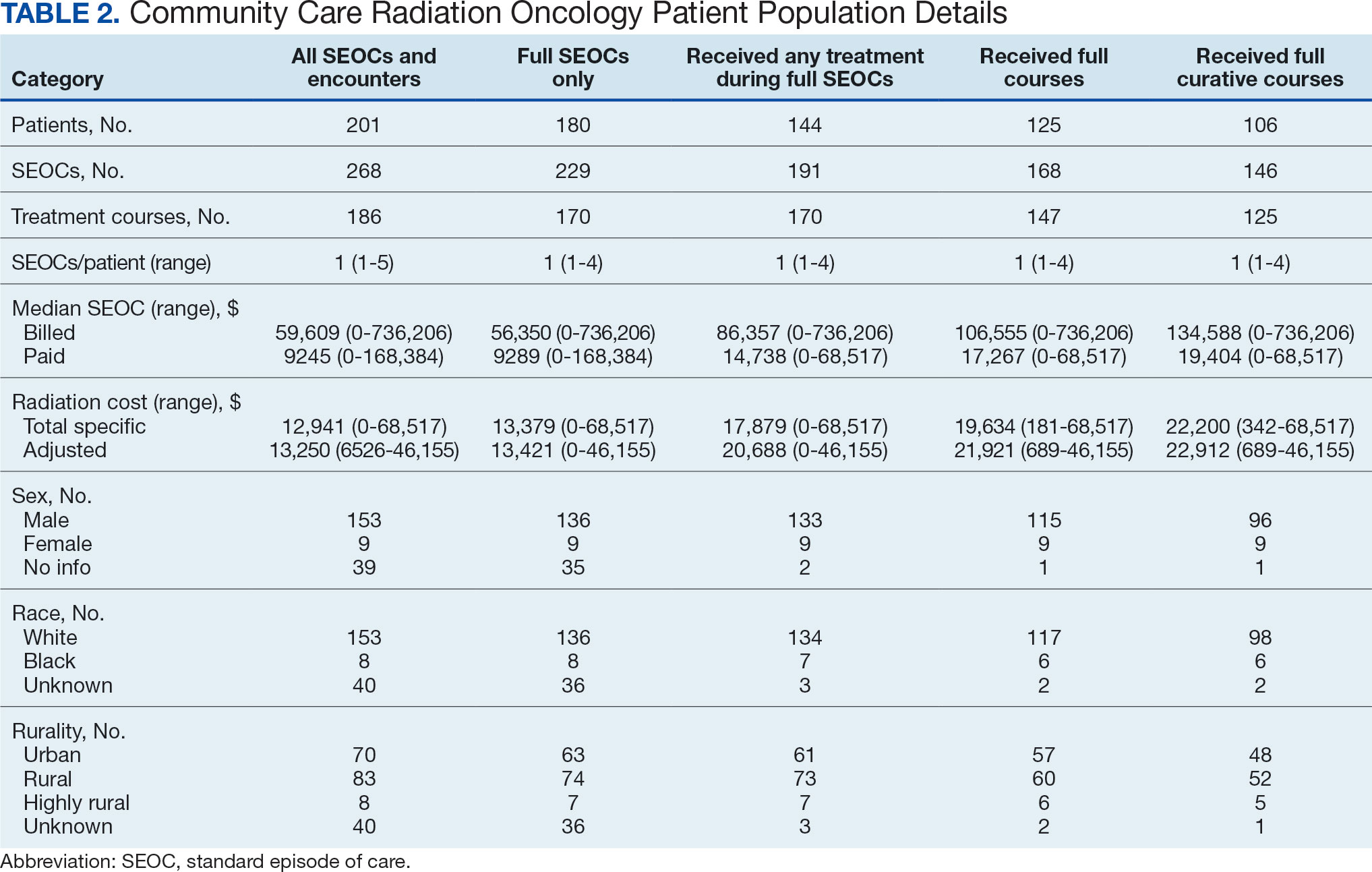
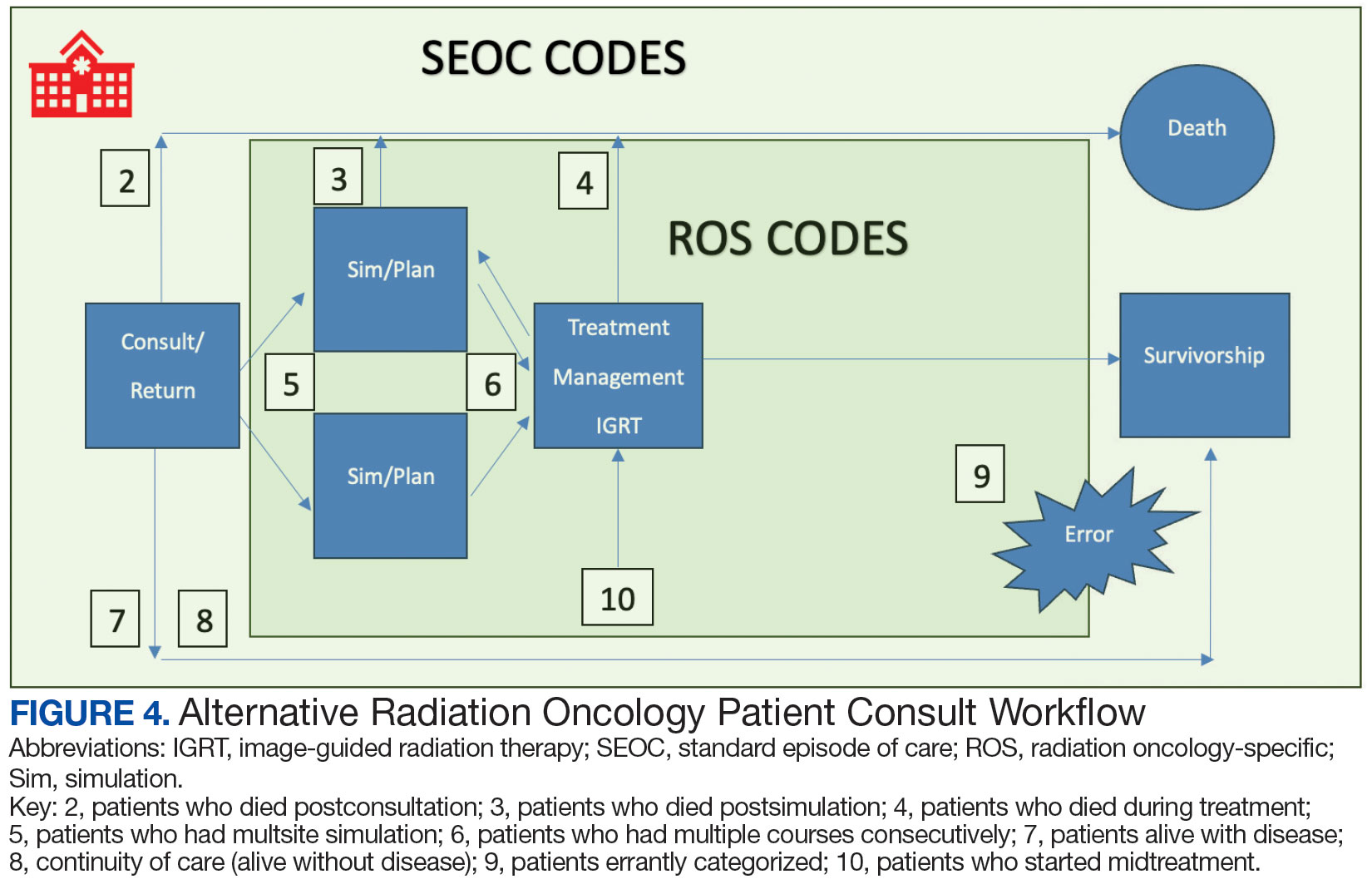
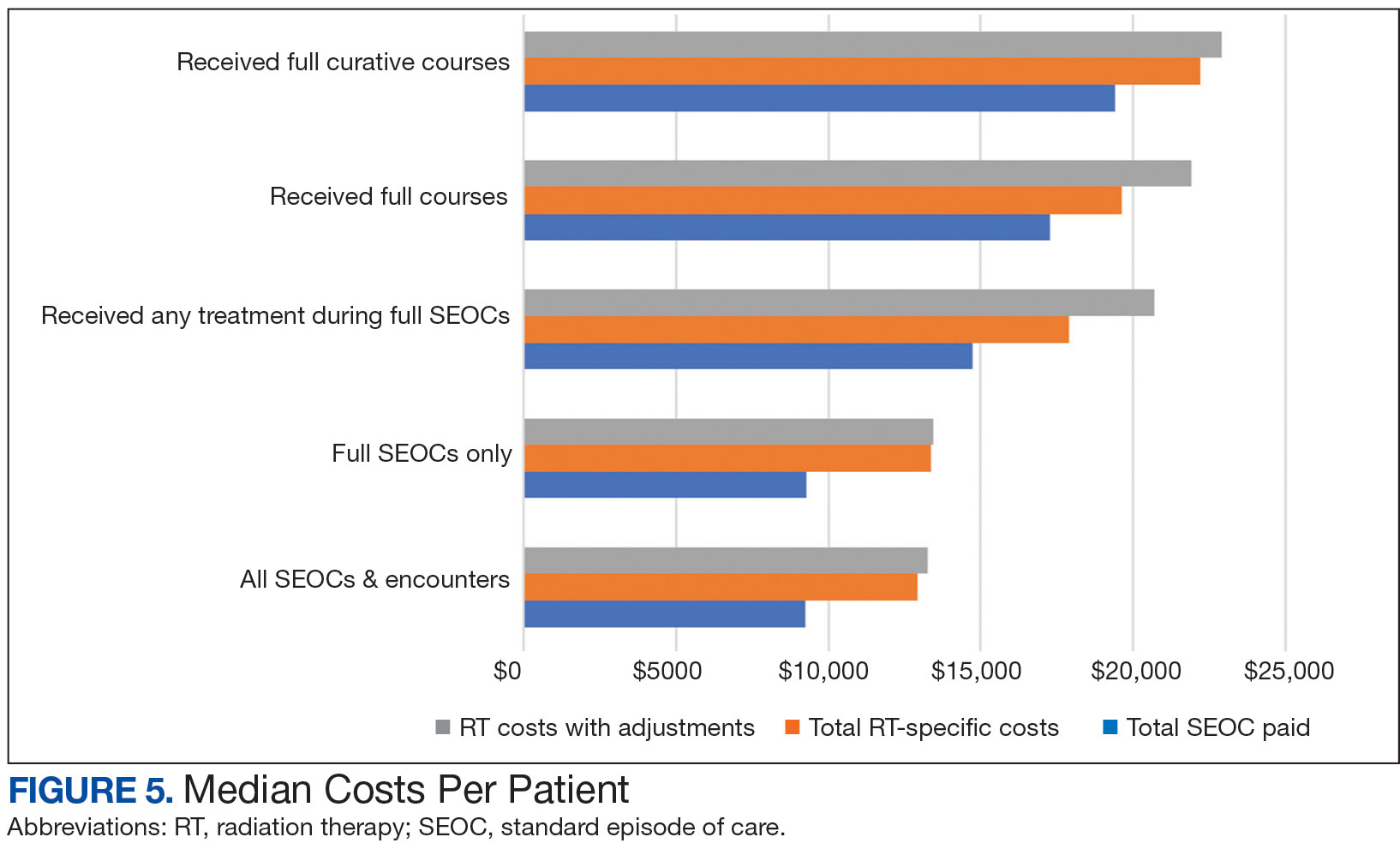
Every consultation charge was reviewed. A typical patient following the standard pathway (eAppendix 2, available at doi:10.12788/ fp.0585) exhibited a predictable pattern of consultation payment, simulation and planning, multiple radiation treatments interspersed with treatment management visits and a cone-down phase, and finishing with a follow-up visit. A less predictable case with excess CPT codes, gaps in charges, and an additional unexpected palliative course is shown in eAppendix 3 (available at doi:10.12788/fp.0585). Gaps occurred in 42% of SEOCs with missed bills costing as much as $12,000. For example, a patient with lung cancer had a treatment summary note for lung cancer after completion that showed the patient received 30 fractions of 2 Gy, a typical course. Only 10 treatment codes and 3 of 6 weekly treatment management codes were available. There was a gap of 20 volumetric modulated arc therapy treatments, 3 physics weekly status checks, 3 physician managements notes, and a computed tomography simulation charge.
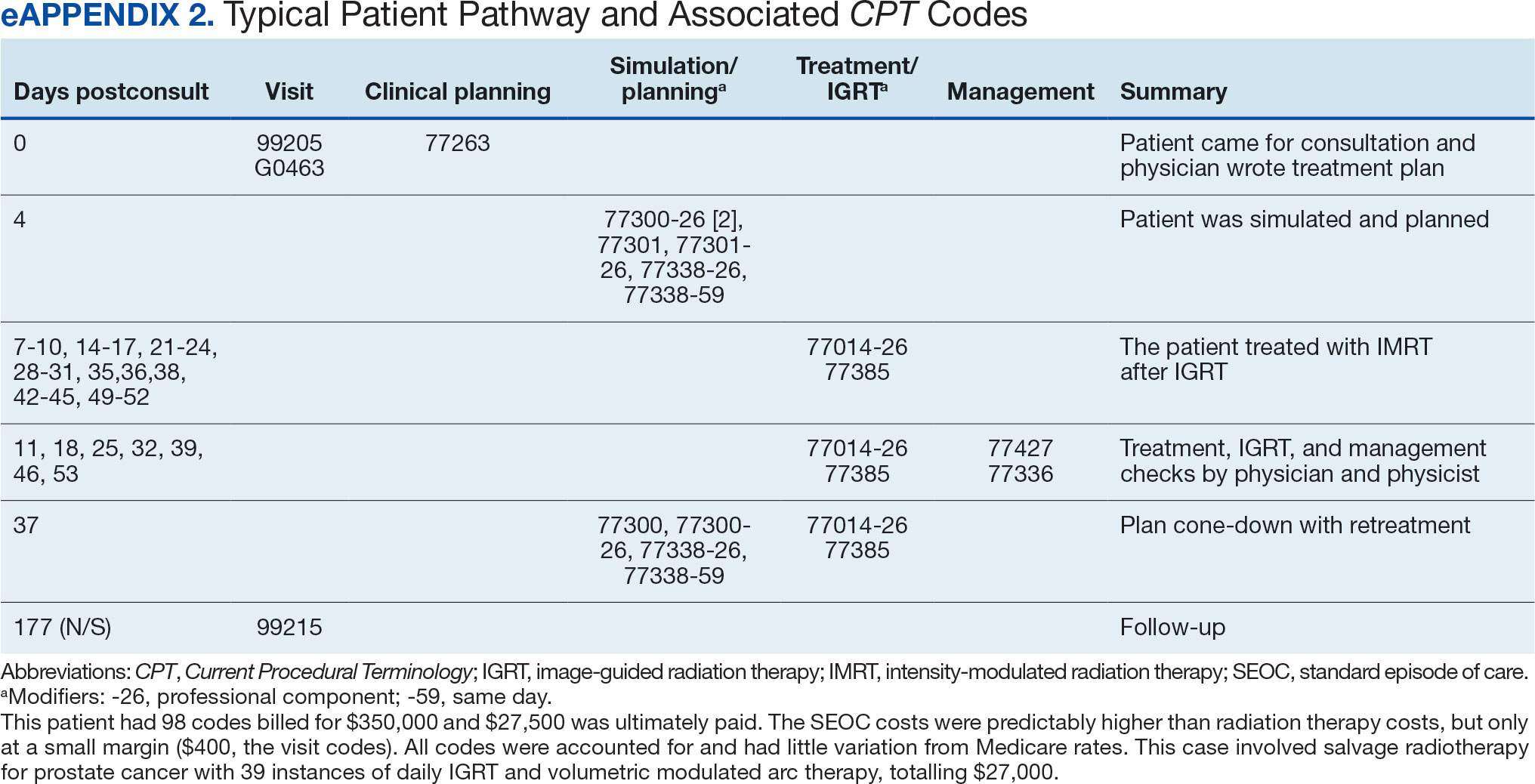

Between AMCMS and CTM, 10,005 CPT codes were evaluated; 1255 (12.5%) were unique to AMCMS (either related to the radiation oncology course, such as Evaluation and Management CPT codes or “other” unrelated codes) while 1158 (11.6%) were unique to CTM. Of the 7592 CPT codes shared between AMCMS and CTM, there was a discrepancy in 135 (1.8%); all were duplicates (CTM showed double payment while AMCMS showed $0 paid). The total CPT code costs came to $3.2 million with $560,000 unique to SEOCs and $500,000 unique to CTM. Treatment codes were the most common (33%) as shown in Table 3 and accounted for 55% of the cost ($1.8 million). About 700 CPT codes were considered “other,” typically for biologic therapeutic agents (Table 4 and eAppendix 4, available at doi:10.12788/fp.0585).
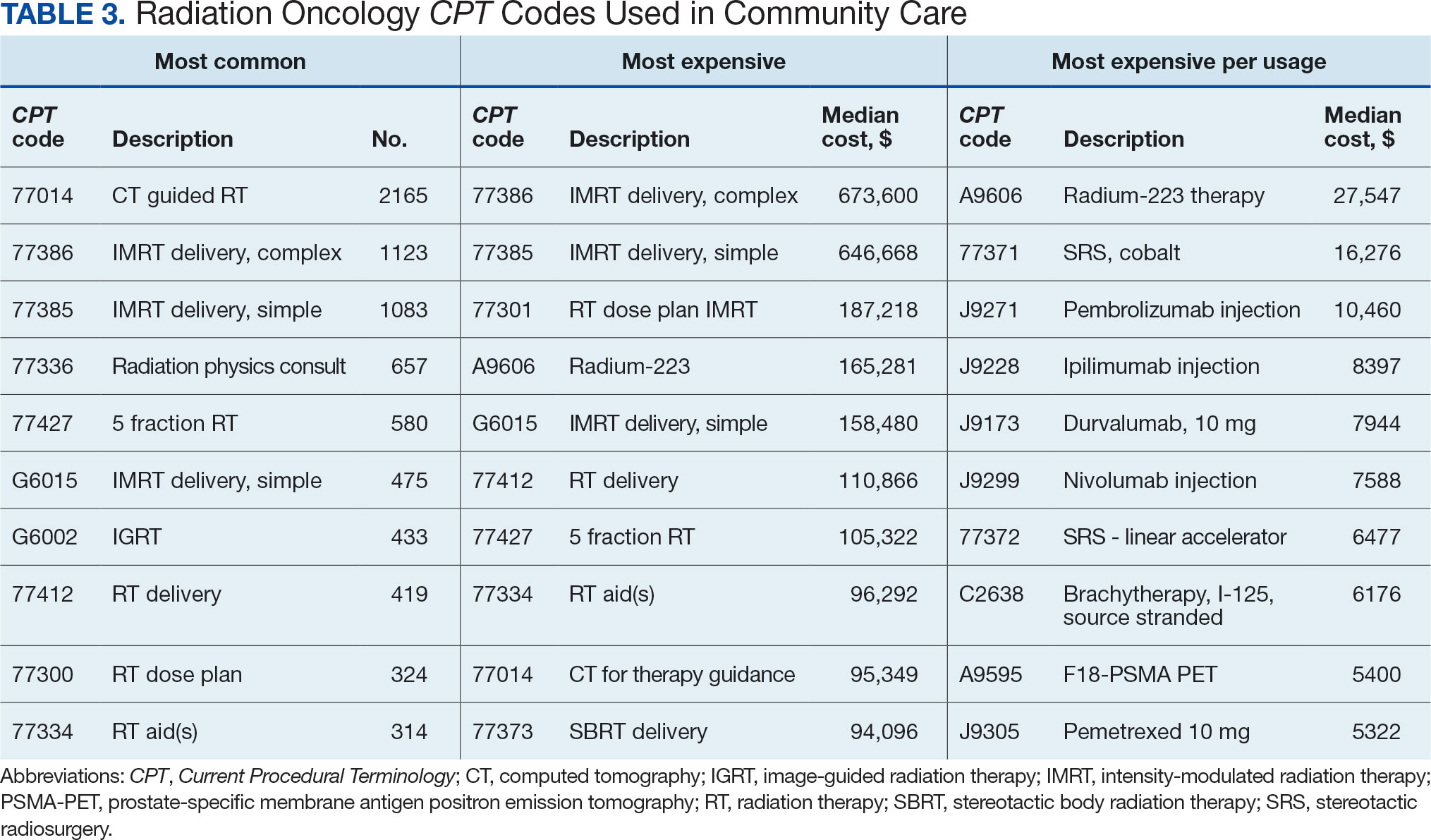
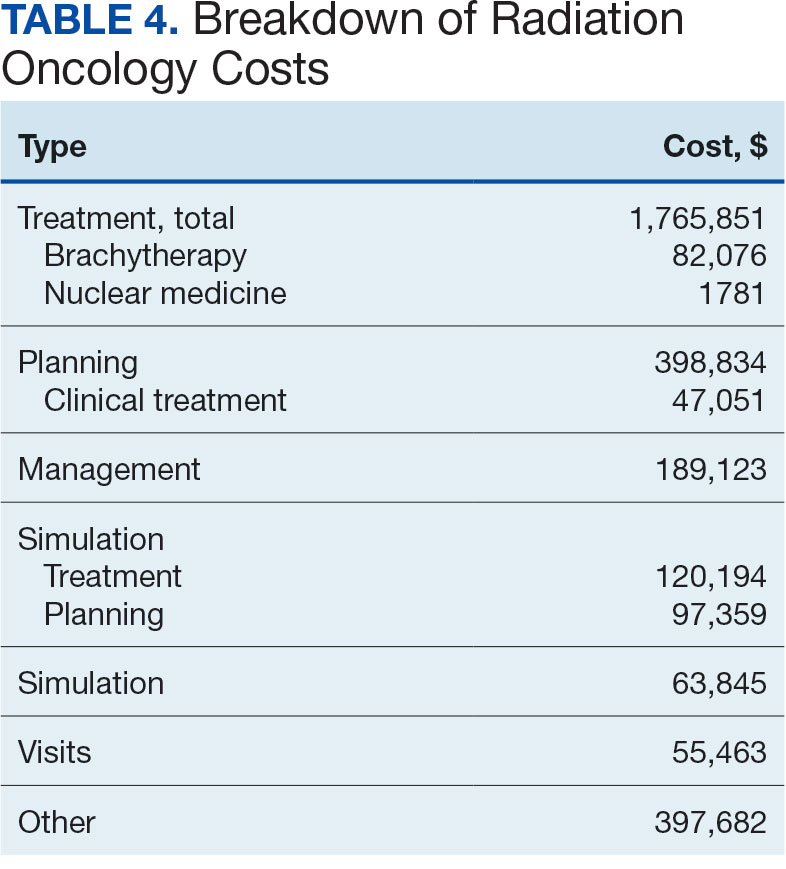

DISCUSSION
The current method of reporting radiation oncology costs used by VA is insufficient and misleading. Better data are needed to summarize purchased care costs to guide decisions about community care at the VA. Investigations into whether the extra costs for quality care (ie, expensive capital equipment, specialized staff, mandatory accreditations) are worthwhile if omitted at other facilities patients choose for their health care needs. No study has defined specialty care-specific costs by evaluating billing receipts from the CDW to answer the question. Kenamond et al highlight the need for radiation oncology for rural patients.11 Drive time was cited as the reason for community care referral for 97% of veterans, many of whom lived in rural locations. Of patients with rurality information who enrolled in community care, 57% came from rural or highly rural counties, and this ratio held for those who received full curative therapies. An executive administrator relying on AMCMS reports would see a median SEOC cost of $5000, but without ROS knowledge in coding, the administrator would miss many additional costs. For example, 2 patients who each had 5 SEOCs during the evaluated period, incurred a total cost of only $1800.
Additionally, an administrator could include miscategorized costs with significant ramifications. The 2 most expensive SEOCs were not typical radiation oncology treatments. A patient undergoing radium-223 dichloride therapy incurred charges exceeding $165,000, contributing disproportionately to the overall median cost analysis; this would normally be administered by the nuclear medicine department. Immunotherapy and chemotherapy are uniformly overseen by medical oncology services, but drug administration codes were still found in radiation oncology SEOCs. A patient (whose SEOC was discontinued but accrued charges) had an electrocardiogram interpretation for $8 as the SEOC cost; 3 other SEOCs continued to incur costs after being discontinued. There were 24 empty SEOCs for patients that had consults to the community, and 2 had notes stating treatment had been delivered yet there was no ROS costs or SEOC costs. Of the 268 encounters, 43% had some sort of billing irregularities (ie, missing treatment costs) that would be unlikely for a private practice to omit; it would be much more likely that the CDW miscategorized the payment despite confirmation of the 2 retrieval systems.
It would be inadvisable to make staffing decisions or forecast costs based on current SEOC reports without specialized curation. A simple yet effective improvement to the cost attribution process would be to restrict the analysis to encounters containing primary radiation treatment codes. This targeted approach allows more accurate identification of patients actively receiving radiation oncology treatment, while excluding those seen solely for consultations or follow-up visits. Implementing this refinement leads to a substantial increase in the median payment—from $5000 to $13,000—without requiring additional coding or data processing, thereby enhancing the accuracy of cost estimates with minimal effort.
Clarifying radiation oncology service costs requires addressing the time frame and services included, given laxity and interpretation of the SEOCs. VA community care departments have streamlined the reimbursement process at the expense of medical cost organization and accuracy; 86% of VA practitioners reported that ≥ 1 potential community health care partners had refused to work with the VA because of payment delays.12 Payments are contingent on correspondence from outside practices for community work. For radiation oncology, this includes the consultation but also critical radiation-related details of treatment, which were omitted nearly half the time. SEOC approval forms have many costly laboratory tests, imaging, and procedures that have little to do with radiation oncology cancer treatments but may be used in the workup and staging process; this creates noise when calculating radiation oncology fiscal cost.
The presumption that an episode of care equates to a completed radiation therapy course is incorrect; this occurs less than half of the time. An episode often refers to a return visit, or conversely, multiple treatment courses. As the patients’ medical homes are their VHA primary care practitioners, it would be particularly challenging to care for the patients without full treatment information, especially if adverse effects from therapy were to arise. As a tertiary specialty, radiation oncology does not seek out patients and are sent consultations from medical oncology, surgical, and medical oncologic specialties. Timesensitive processes such as workup, staging, and diagnosis often occur in parallel. This analysis revealed that patients see outside radiation oncologists prior to the VA. There are ≥ 100 patients who had radiation oncology codes without a radiation oncology SEOC or community care consultation, and in many cases, the consultation was placed after the patient was seen.
Given the lack of uniformity and standardization of patient traffic, the typical and expected pathways were insufficient to find the costs. Too many opportunities for errors and incorrect categorization of costs meant a different method would be necessary. Starting at the inception of the community care consult, only 1 diagnosis code can be entered. For patients with multiple diagnoses, one would not be able to tell what was treated without chart access. Radiation oncology consults come from primary and specialty care practitioners and nurses throughout the VA. Oftentimes, the referral would be solicited by the community radiation oncology clinic, diagnosing community specialty (ie, urology for a patient with prostate cancer), or indirectly from the patient through primary care. Many cases were retroactively approved as the veteran had already been consulted by the community care radiation oncologist. If the patient is drive-time eligible, it would be unlikely that they would leave and choose to return to the VA. There is no way for a facility VA service chief or administrator to mitigate VA community costs of care, especially as shown by the miscategorization of several codes. Database challenges exacerbate the issue: 1 patient changed her first and last name during this time frame, and 2 patients had the same name but different social security numbers. In order to strictly find costs between 2 discrete timepoints, 39 (15%) SEOCs were split and incomplete, and 6 SEOCs contained charges for 2 different patients. This was corrected, and all inadvertent charges were cancelled. Only 1 ICD code is allowed per community care consultation, so an investigation is required to find costs for patients with multiple sites of disease. Additionally, 5 of the patients marked for drive time were actually patients who received Gamma Knife and brachytherapy, services not available at the VA.
Hanks et al first attempted to calculate cost of radiation oncology services. External beam prostate cancer radiotherapy at 3 suburban California centers cost $6750 ($20,503 inflation adjusted) per patient before October 1984 and $5600 ($17,010 inflation adjusted) afterwards.13 According to the American Society for Radiation Oncology, Advocacy Radiation Oncology Case Rate Program Curative radiation courses should cost $20,000 to $30,000 and palliative courses should cost $10,000 to $15,000. These costs are consistent with totals demonstrated in this analysis and similar to the inflation-adjusted Hanks et al figures. Preliminary findings suggest that radiation treatment constituted more than half of the total expenditures, with a notable $4 million increase in adjusted cost compared to the Medicare rates, indicating significant variation. Direct comparisons with Medicaid or commercial payer rates remain unexplored.
Future Directions
During the study period, 201 patients received 186 courses of radiation therapy in the community, while 1014 patients were treated in-house for a total of 833 courses. A forthcoming analysis will directly compare the cost of in-house care with that of communitybased treatment, specifically breaking down expenditure differences by diagnosis. Future research should investigate strategies to align reimbursement with quality metrics, including the potential role of tertiary accreditation in incentivizing high-value care. Additional work is also warranted to assess patient out-ofpocket expenses across care settings and to benchmark VA reimbursement against Medicare, Medicaid, and private insurance rates. In any case, with the increasing possibility of fewer fractions for treatments such as stereotactic radiotherapy or palliative care therapy, there is a clear financial incentive to treat as frequently as allowed despite equal clinical outcomes.
CONCLUSIONS
Veterans increasingly choose to receive care closer to home if the option is available. In the VA iron triangle, cost comes at the expense of access but quantifying this has proved elusive in the cost accounting model currently used at the VA.1 The inclusion of all charges loosely associated with SEOCs significantly impairs the ability to conduct meaningful cost analyses. The current VA methodology not only introduces substantial noise into the data but also leads to a marked underestimation of the true cost of care delivered in community settings. Such misrepresentation risks driving policy decisions that could inappropriately reduce or eliminate in-house radiation oncology services. Categorizing costs effectively in the VA could assist in making managerial and administrative decisions and would prevent damaging service lines based on misleading or incorrect data. A system which differentiates between patients who have received any treatment codes vs those who have not would increase accuracy.
- Kissick W. Medicine’s Dilemmas: Infinite Needs Versus Finite Resources. 1st ed. Yale University Press; 1994.
- Albanese AP, Bope ET, Sanders KM, Bowman M. The VA MISSION Act of 2018: a potential game changer for rural GME expansion and veteran health care. J Rural Health. 2020;36(1):133-136. doi:10.1111/jrh.12360
- Office of Management and Budget (US). Budget of the United States Government, Fiscal Year 2025. Washington, DC: US Government Publishing Office; 2024. Available from: US Department of Veterans Affairs FY 2025 Budget Submission: Budget in Brief.
- US Department of Veterans Affairs. Veteran care claims. Accessed April 3, 2025. https://www.va.gov/COMMUNITYCARE/revenue-ops/Veteran-Care-Claims.asp
- US Centers for Medicare and Medicaid Services. Accessed April 3, 2025. Procedure price lookup https://www.medicare.gov/procedure-price-lookup
- US Department of Veterans Affairs. WellHive -Enterprise. Accessed April 3, 2025. https://department.va.gov/privacy/wp-content/uploads/sites/5/2023/05/FY23WellHiveEnterprisePIA.pdf
- US Centers for Medicare and Medicaid Services. RVU21a physician fee schedule, January 2021 release. Accessed April 3, 2025. https://www.cms.gov/medicaremedicare-fee-service-paymentphysicianfeeschedpfs-relative-value-files/rvu21a
- US Centers for Medicare and Medicaid Services. RVU22a physician fee schedule, January 2022 release. Accessed April 3, 2025. https://www.cms.gov/medicaremedicare-fee-service-paymentphysicianfeeschedpfs-relative-value-files/rvu22a
- US Centers for Medicare and Medicaid Services. RVU23a physician fee schedule, January 2023 release. Accessed April 3, 2025. https://www.cms.gov/medicare/medicare-fee-service-payment/physicianfeesched/pfs-relative-value-files/rvu23a
- US Centers for Medicare and Medicaid Services. RVU23a Medicare Physician Fee Schedule rates effective January 1, 2024, through March 8, 2024. Accessed on April 3, 2025. https://www.cms.gov/medicare/payment/fee-schedules/physician/pfs-relative-value-files/rvu24a
- Kenamond MC, Mourad WF, Randall ME, Kaushal A. No oncology patient left behind: challenges and solutions in rural radiation oncology. Lancet Reg Health Am. 2022;13:100289. doi:10.1016/j.lana.2022.100289
- Mattocks KM, Kroll-Desrosiers A, Kinney R, Elwy AR, Cunningham KJ, Mengeling MA. Understanding VA’s use of and relationships with community care providers under the MISSION Act. Med Care. 2021;59(Suppl 3):S252-S258. doi:10.1097/MLR.0000000000001545
- Hanks GE, Dunlap K. A comparison of the cost of various treatment methods for early cancer of the prostate. Int J Radiat Oncol Biol Phys. 1986;12(10):1879-1881. doi:10.1016/0360-3016(86)90334-2
- American Society of Radiation Oncology. Radiation oncology case rate program (ROCR). Accessed April 3, 2025. https://www.astro.org/advocacy/key-issues-8f3e5a3b76643265ee93287d79c4fc40/rocr
William Kissick’s description of health care’s iron triangle in 1994 still resonates. Access, quality, and cost will always come at the expense of the others.1 In 2018, Congress passed the VA MISSION Act, allowing patients to pursue community care options for extended waits (> 28 days) or longer distance drive times of > 60 minutes for specialty care services, such as radiation oncology. According to Albanese et al, the VA MISSION Act sought to address gaps in care for veterans living in rural and underserved areas.2 The Veterans Health Administration (VHA) continues to increase community care spending, with a 13.8% increase in fiscal year 2024 and an expected cost of > $40 billion for 2025.3 One could argue this pays for access for remote patients and quality when services are unavailable, making it a direct application of the iron triangle.
The VA MISSION Act also bolstered the expansion of existing community care department staff to expediently facilitate and coordinate care and payments.2 Cost management and monitoring have become critical in predicting future staff requirements, maintaining functionality, and ensuring patients receive optimal care. The VHA purchases care through partner networks and defines these bundled health care services as standard episodes of care (SEOCs), which are “clinically related health care services for a specific unique illness or medical condition… over a defined period of time.”4 Medicare publishes its rates quarterly, and outpatient procedure pricing is readily available online.5 Along these same lines, the US Department of Veterans Affairs (VA) publishes a current list of available procedures and associated Current Procedure Technology (CPT) codes that are covered under its VA fee schedule for community care.
Unique challenges persist when using this system to accurately account for radiation oncology expenditures. This study was based on the current practices at the Richard L. Roudebush VA Medical Center (RLRVAMC), a large 1a hospital. A detailed analysis reveals the contemporaneous cost of radiation oncology cancer care from October 1, 2021, through February 1, 2024, highlights the challenges in SEOC definition and duration, communication issues between RLRVAMC and purchase partners, inconsistencies in billing, erroneous payments, and difficulty of cost categorization.
METHODS
Community care radiation oncology-related costs were examined from October 1, 2021, to February 1, 2024 for RLRVAMC, 6 months prior to billing data extraction. Figure 1 shows a simple radiation oncology patient pathway with consultation or visit, simulation and planning, and treatment, with codes used to check billing. It illustrates the expected relationships between the VHA (radiation oncology, primary, and specialty care) and community care (clinicians and radiation oncology treatment sites).
VHA standard operating procedures for a patient requesting community-based radiation oncology care require a board-certified radiation oncologist at RLRVAMC to review and approve the outside care request. Community care radiation oncology consultation data were accessed from the VA Corporate Data Warehouse (CDW) using Pyramid Analytics (V25.2). Nurses, physicians, and community care staff can add comments, forward consultations to other services, and mark them as complete or discontinued, when appropriate. Consultations not completed within 91 days are automatically discontinued. All community care requests from 2018 through 2024 were extracted; analysis began April 1, 2021, 6 months prior to the cost evaluation date of October 1, 2021.
An approved consultation is reviewed for eligibility by a nurse in the community care department and assigned an authorization number (a VA prefix followed by 12 digits). Billing codes are approved and organized by the community care networks, and all procedure codes should be captured and labeled under this number. The VAMC Community Care department obtains initial correspondence from the treating clinicians. Subsequent records from the treating radiation oncologist are expected to be scanned into the electronic health record and made accessible via the VA Joint Legacy Viewer (JLV) and Computerized Patient Record System (CPRS).
Radiation Oncology SEOC
The start date of the radiation oncology SEOC is determined by the community care nurse based on guidance established by the VA. It can be manually backdated or delayed, but current practice is to start at first visit or procedure code entry after approval from the VAMC Radiation Oncology department. Approved CPT codes from SEOC versions between October 1, 2021, and February 1, 2024, are in eAppendix 1 (available at doi:10.12788/fp.0585). These generally include 10 types of encounters, about 115 different laboratory tests, 115 imaging studies, 25 simulation and planning procedures, and 115 radiation treatment codes. The radiation oncology SEOCs during the study period had an approval duration of 180 days. Advanced Medical Cost Management Solutions software (AMCMS) is the VHA data analytics platform for community care medical service costs. AMCMS includes all individual CPT codes billed by specific radiation oncology SEOC versions. Data are refreshed monthly, and all charges were extracted on September 12, 2024, > 6 months after the final evaluated service date to allow for complete billing returns.6
Radiation Oncology-Specific Costs
The VA Close to Me (CTM) program was used to find 84 specific radiation oncology CPT codes, nearly all within the 77.XXX or G6.XXX series, which included all radiation oncology-specific (ROS) codes (except visits accrued during consultation and return appointments). ROS costs are those that could not be performed by any other service and include procedures related to radiation oncology simulation, treatment planning, treatment delivery (with or without image guidance), and physician or physicist management. All ROS costs should be included in a patient’s radiation oncology SEOC. Other costs that may accompany operating room or brachytherapy administration did not follow a 77.XXX or G6.XXX pattern but were included in total radiation therapy operating costs.
Data obtained from AMCMS and CTM included patient name and identifier; CPT billed amount; CPT paid amount; dates of service; number of claims; International Classification of Diseases, Tenth Revision (ICD) diagnosis; and VA authorization numbers. Only CTM listed code modifiers. Only items categorized as paid were included in the analysis. Charges associated with discontinued consultations that had accrued costs also were included. Codes that were not directly related to ROS were separately characterized as other and further subcategorized.
Deep Dive Categorization
All scanned documents tagged to the community consultation were accessed and evaluated for completeness by a radiation oncologist (RS). The presence or absence of consultation notes and treatment summaries was evaluated based on necessity (ie, not needed for continuation of care or treatment was not given). In the absence of a specific completion summary or follow-up note detailing the treatment modality, number of fractions, and treatment sites, available documentation, including clinical notes and billing information, was used. Radical or curative therapies were identified as courses expected to eradicate disease, including stereotactic ablative radiotherapy to the brain, lung, liver, and other organs. Palliative therapies included whole-brain radiotherapy or other low-dose treatments. If the patient received the intended course, this was categorized as full. If incomplete, it was considered partial.
Billing Deviations
The complete document review allowed for close evaluation of paid therapy and identification of gaps in billing (eg, charges not found in extracted data that should have occurred) for external beam radiotherapy patients. Conversely, extra charges, such as an additional weekly treatment management charge (CPT code 77427), would be noted. Patients were expected to have the number of treatments specified in the summary, a clinical treatment planning code, and weekly treatment management notes from physicians and physicists every 5 fractions. Consultations and follow-up visits were expected to have 1 visit code; CPT codes 99205 and 99215, respectively, were used to estimate costs in their absence.
Costs were based on Medicare rates as of January 1 of the year in which they were accrued. 7-10 Duplicates were charges with the same code, date, billed quantity, and paid amounts for a given patient. These would always be considered erroneous. Medicare treatment costs for procedures such as intensity modulated radiotherapy (CPT code 77385 or 77386) are available on the Medicare website. When reviewing locality deviations for 77427, there was a maximum of 33% increase in Medicare rates. Therefore, for treatment codes, one would expect the range to be at least the Medicare rate and maximally 33% higher. These rates are negotiated with insurance companies, but this range was used for the purpose of reviewing and adjusting large data sets.
RESULTS
Since 2018, > 500 community care consults have been placed by radiation oncology for treatment in the community, with more following implementation of the VA MISSION Act. Use of radiation oncology community care services annually increased during the study period for this facility (Table 1, Figure 2). Of the 325 community care consults placed from October 1, 2021, to February 1, 2024, 248 radiation oncology SEOCs were recorded with charges for 181 patients (range, 1-5 SEOCs). Long drive time was the rationale for > 97% of patients directed to community care (Supplemental materials, available at doi:10.12788/fp.0585). Based on AMCMS data, $22.2 million was billed and $2.7 million was paid (20%) for 8747 CPT codes. Each community care interval cost the VA a median (range) of $5000 ($8-$168,000 (Figure 3).
After reviewing ROS charges extracted from CTM, 20 additional patients had radiation oncology charges but did not have a radiation oncology SEOC for 268 episodes of care for 201 unique patients. In addition to the 20 patients who did not have a SEOC, 42 nonradiation oncology SEOCs contained 1148 radiation oncology codes, corresponding to almost $500,000 paid. Additional charges of about $416,000, which included biologic agents (eg, durvalumab, nivolumab), procedures (eg, mastectomies), and ambulance rides were inappropriately added to radiation oncology SEOCs.
While 77% of consultations were scanned into CPRS and JLV, only 54% of completion summaries were available with an estimated $115,000 in additional costs. The total adjusted costs was about $2.9 million. Almost 37% of SEOCs were for visits only. For the 166 SEOCs where patients received any radiation treatment or planning, the median cost was $18,000. Differences in SEOC pathways are shown in Figure 4. One hundred twenty-one SEOCs (45%) followed the standard pathway, with median SEOC costs of $15,500; when corrected for radiation-specific costs, the median cost increased to $18,000. When adjusted for billing irregularities, the median cost was $20,600. Ninety-nine SEOCs (37%) were for consultation/ follow-up visits only, with a median cost of $220. When omitting shared scans and nonradiation therapy costs and correcting for billing gaps, the median cost decreased to $170. A median of $9200 was paid per patient, with $12,900 for radiation therapy-specific costs and $13,300 adjusted for billing deviations. Narrowing to the 106 patients who received full, radical courses, the median SEOC, ROS, and adjusted radiation therapy costs increased to $19,400, $22,200, and $22,900, respectively (Table 2, Figure 5). Seventy-one SEOCs (26%) had already seen a radiation oncologist before the VA radiation oncology department was aware, and 49 SEOCs (18%) had retroactive approvals (Supplemental materials available at doi:10.12788/fp.0585).
Every consultation charge was reviewed. A typical patient following the standard pathway (eAppendix 2, available at doi:10.12788/ fp.0585) exhibited a predictable pattern of consultation payment, simulation and planning, multiple radiation treatments interspersed with treatment management visits and a cone-down phase, and finishing with a follow-up visit. A less predictable case with excess CPT codes, gaps in charges, and an additional unexpected palliative course is shown in eAppendix 3 (available at doi:10.12788/fp.0585). Gaps occurred in 42% of SEOCs with missed bills costing as much as $12,000. For example, a patient with lung cancer had a treatment summary note for lung cancer after completion that showed the patient received 30 fractions of 2 Gy, a typical course. Only 10 treatment codes and 3 of 6 weekly treatment management codes were available. There was a gap of 20 volumetric modulated arc therapy treatments, 3 physics weekly status checks, 3 physician managements notes, and a computed tomography simulation charge.
Between AMCMS and CTM, 10,005 CPT codes were evaluated; 1255 (12.5%) were unique to AMCMS (either related to the radiation oncology course, such as Evaluation and Management CPT codes or “other” unrelated codes) while 1158 (11.6%) were unique to CTM. Of the 7592 CPT codes shared between AMCMS and CTM, there was a discrepancy in 135 (1.8%); all were duplicates (CTM showed double payment while AMCMS showed $0 paid). The total CPT code costs came to $3.2 million with $560,000 unique to SEOCs and $500,000 unique to CTM. Treatment codes were the most common (33%) as shown in Table 3 and accounted for 55% of the cost ($1.8 million). About 700 CPT codes were considered “other,” typically for biologic therapeutic agents (Table 4 and eAppendix 4, available at doi:10.12788/fp.0585).
DISCUSSION
The current method of reporting radiation oncology costs used by VA is insufficient and misleading. Better data are needed to summarize purchased care costs to guide decisions about community care at the VA. Investigations into whether the extra costs for quality care (ie, expensive capital equipment, specialized staff, mandatory accreditations) are worthwhile if omitted at other facilities patients choose for their health care needs. No study has defined specialty care-specific costs by evaluating billing receipts from the CDW to answer the question. Kenamond et al highlight the need for radiation oncology for rural patients.11 Drive time was cited as the reason for community care referral for 97% of veterans, many of whom lived in rural locations. Of patients with rurality information who enrolled in community care, 57% came from rural or highly rural counties, and this ratio held for those who received full curative therapies. An executive administrator relying on AMCMS reports would see a median SEOC cost of $5000, but without ROS knowledge in coding, the administrator would miss many additional costs. For example, 2 patients who each had 5 SEOCs during the evaluated period, incurred a total cost of only $1800.
Additionally, an administrator could include miscategorized costs with significant ramifications. The 2 most expensive SEOCs were not typical radiation oncology treatments. A patient undergoing radium-223 dichloride therapy incurred charges exceeding $165,000, contributing disproportionately to the overall median cost analysis; this would normally be administered by the nuclear medicine department. Immunotherapy and chemotherapy are uniformly overseen by medical oncology services, but drug administration codes were still found in radiation oncology SEOCs. A patient (whose SEOC was discontinued but accrued charges) had an electrocardiogram interpretation for $8 as the SEOC cost; 3 other SEOCs continued to incur costs after being discontinued. There were 24 empty SEOCs for patients that had consults to the community, and 2 had notes stating treatment had been delivered yet there was no ROS costs or SEOC costs. Of the 268 encounters, 43% had some sort of billing irregularities (ie, missing treatment costs) that would be unlikely for a private practice to omit; it would be much more likely that the CDW miscategorized the payment despite confirmation of the 2 retrieval systems.
It would be inadvisable to make staffing decisions or forecast costs based on current SEOC reports without specialized curation. A simple yet effective improvement to the cost attribution process would be to restrict the analysis to encounters containing primary radiation treatment codes. This targeted approach allows more accurate identification of patients actively receiving radiation oncology treatment, while excluding those seen solely for consultations or follow-up visits. Implementing this refinement leads to a substantial increase in the median payment—from $5000 to $13,000—without requiring additional coding or data processing, thereby enhancing the accuracy of cost estimates with minimal effort.
Clarifying radiation oncology service costs requires addressing the time frame and services included, given laxity and interpretation of the SEOCs. VA community care departments have streamlined the reimbursement process at the expense of medical cost organization and accuracy; 86% of VA practitioners reported that ≥ 1 potential community health care partners had refused to work with the VA because of payment delays.12 Payments are contingent on correspondence from outside practices for community work. For radiation oncology, this includes the consultation but also critical radiation-related details of treatment, which were omitted nearly half the time. SEOC approval forms have many costly laboratory tests, imaging, and procedures that have little to do with radiation oncology cancer treatments but may be used in the workup and staging process; this creates noise when calculating radiation oncology fiscal cost.
The presumption that an episode of care equates to a completed radiation therapy course is incorrect; this occurs less than half of the time. An episode often refers to a return visit, or conversely, multiple treatment courses. As the patients’ medical homes are their VHA primary care practitioners, it would be particularly challenging to care for the patients without full treatment information, especially if adverse effects from therapy were to arise. As a tertiary specialty, radiation oncology does not seek out patients and are sent consultations from medical oncology, surgical, and medical oncologic specialties. Timesensitive processes such as workup, staging, and diagnosis often occur in parallel. This analysis revealed that patients see outside radiation oncologists prior to the VA. There are ≥ 100 patients who had radiation oncology codes without a radiation oncology SEOC or community care consultation, and in many cases, the consultation was placed after the patient was seen.
Given the lack of uniformity and standardization of patient traffic, the typical and expected pathways were insufficient to find the costs. Too many opportunities for errors and incorrect categorization of costs meant a different method would be necessary. Starting at the inception of the community care consult, only 1 diagnosis code can be entered. For patients with multiple diagnoses, one would not be able to tell what was treated without chart access. Radiation oncology consults come from primary and specialty care practitioners and nurses throughout the VA. Oftentimes, the referral would be solicited by the community radiation oncology clinic, diagnosing community specialty (ie, urology for a patient with prostate cancer), or indirectly from the patient through primary care. Many cases were retroactively approved as the veteran had already been consulted by the community care radiation oncologist. If the patient is drive-time eligible, it would be unlikely that they would leave and choose to return to the VA. There is no way for a facility VA service chief or administrator to mitigate VA community costs of care, especially as shown by the miscategorization of several codes. Database challenges exacerbate the issue: 1 patient changed her first and last name during this time frame, and 2 patients had the same name but different social security numbers. In order to strictly find costs between 2 discrete timepoints, 39 (15%) SEOCs were split and incomplete, and 6 SEOCs contained charges for 2 different patients. This was corrected, and all inadvertent charges were cancelled. Only 1 ICD code is allowed per community care consultation, so an investigation is required to find costs for patients with multiple sites of disease. Additionally, 5 of the patients marked for drive time were actually patients who received Gamma Knife and brachytherapy, services not available at the VA.
Hanks et al first attempted to calculate cost of radiation oncology services. External beam prostate cancer radiotherapy at 3 suburban California centers cost $6750 ($20,503 inflation adjusted) per patient before October 1984 and $5600 ($17,010 inflation adjusted) afterwards.13 According to the American Society for Radiation Oncology, Advocacy Radiation Oncology Case Rate Program Curative radiation courses should cost $20,000 to $30,000 and palliative courses should cost $10,000 to $15,000. These costs are consistent with totals demonstrated in this analysis and similar to the inflation-adjusted Hanks et al figures. Preliminary findings suggest that radiation treatment constituted more than half of the total expenditures, with a notable $4 million increase in adjusted cost compared to the Medicare rates, indicating significant variation. Direct comparisons with Medicaid or commercial payer rates remain unexplored.
Future Directions
During the study period, 201 patients received 186 courses of radiation therapy in the community, while 1014 patients were treated in-house for a total of 833 courses. A forthcoming analysis will directly compare the cost of in-house care with that of communitybased treatment, specifically breaking down expenditure differences by diagnosis. Future research should investigate strategies to align reimbursement with quality metrics, including the potential role of tertiary accreditation in incentivizing high-value care. Additional work is also warranted to assess patient out-ofpocket expenses across care settings and to benchmark VA reimbursement against Medicare, Medicaid, and private insurance rates. In any case, with the increasing possibility of fewer fractions for treatments such as stereotactic radiotherapy or palliative care therapy, there is a clear financial incentive to treat as frequently as allowed despite equal clinical outcomes.
CONCLUSIONS
Veterans increasingly choose to receive care closer to home if the option is available. In the VA iron triangle, cost comes at the expense of access but quantifying this has proved elusive in the cost accounting model currently used at the VA.1 The inclusion of all charges loosely associated with SEOCs significantly impairs the ability to conduct meaningful cost analyses. The current VA methodology not only introduces substantial noise into the data but also leads to a marked underestimation of the true cost of care delivered in community settings. Such misrepresentation risks driving policy decisions that could inappropriately reduce or eliminate in-house radiation oncology services. Categorizing costs effectively in the VA could assist in making managerial and administrative decisions and would prevent damaging service lines based on misleading or incorrect data. A system which differentiates between patients who have received any treatment codes vs those who have not would increase accuracy.
William Kissick’s description of health care’s iron triangle in 1994 still resonates. Access, quality, and cost will always come at the expense of the others.1 In 2018, Congress passed the VA MISSION Act, allowing patients to pursue community care options for extended waits (> 28 days) or longer distance drive times of > 60 minutes for specialty care services, such as radiation oncology. According to Albanese et al, the VA MISSION Act sought to address gaps in care for veterans living in rural and underserved areas.2 The Veterans Health Administration (VHA) continues to increase community care spending, with a 13.8% increase in fiscal year 2024 and an expected cost of > $40 billion for 2025.3 One could argue this pays for access for remote patients and quality when services are unavailable, making it a direct application of the iron triangle.
The VA MISSION Act also bolstered the expansion of existing community care department staff to expediently facilitate and coordinate care and payments.2 Cost management and monitoring have become critical in predicting future staff requirements, maintaining functionality, and ensuring patients receive optimal care. The VHA purchases care through partner networks and defines these bundled health care services as standard episodes of care (SEOCs), which are “clinically related health care services for a specific unique illness or medical condition… over a defined period of time.”4 Medicare publishes its rates quarterly, and outpatient procedure pricing is readily available online.5 Along these same lines, the US Department of Veterans Affairs (VA) publishes a current list of available procedures and associated Current Procedure Technology (CPT) codes that are covered under its VA fee schedule for community care.
Unique challenges persist when using this system to accurately account for radiation oncology expenditures. This study was based on the current practices at the Richard L. Roudebush VA Medical Center (RLRVAMC), a large 1a hospital. A detailed analysis reveals the contemporaneous cost of radiation oncology cancer care from October 1, 2021, through February 1, 2024, highlights the challenges in SEOC definition and duration, communication issues between RLRVAMC and purchase partners, inconsistencies in billing, erroneous payments, and difficulty of cost categorization.
METHODS
Community care radiation oncology-related costs were examined from October 1, 2021, to February 1, 2024 for RLRVAMC, 6 months prior to billing data extraction. Figure 1 shows a simple radiation oncology patient pathway with consultation or visit, simulation and planning, and treatment, with codes used to check billing. It illustrates the expected relationships between the VHA (radiation oncology, primary, and specialty care) and community care (clinicians and radiation oncology treatment sites).
VHA standard operating procedures for a patient requesting community-based radiation oncology care require a board-certified radiation oncologist at RLRVAMC to review and approve the outside care request. Community care radiation oncology consultation data were accessed from the VA Corporate Data Warehouse (CDW) using Pyramid Analytics (V25.2). Nurses, physicians, and community care staff can add comments, forward consultations to other services, and mark them as complete or discontinued, when appropriate. Consultations not completed within 91 days are automatically discontinued. All community care requests from 2018 through 2024 were extracted; analysis began April 1, 2021, 6 months prior to the cost evaluation date of October 1, 2021.
An approved consultation is reviewed for eligibility by a nurse in the community care department and assigned an authorization number (a VA prefix followed by 12 digits). Billing codes are approved and organized by the community care networks, and all procedure codes should be captured and labeled under this number. The VAMC Community Care department obtains initial correspondence from the treating clinicians. Subsequent records from the treating radiation oncologist are expected to be scanned into the electronic health record and made accessible via the VA Joint Legacy Viewer (JLV) and Computerized Patient Record System (CPRS).
Radiation Oncology SEOC
The start date of the radiation oncology SEOC is determined by the community care nurse based on guidance established by the VA. It can be manually backdated or delayed, but current practice is to start at first visit or procedure code entry after approval from the VAMC Radiation Oncology department. Approved CPT codes from SEOC versions between October 1, 2021, and February 1, 2024, are in eAppendix 1 (available at doi:10.12788/fp.0585). These generally include 10 types of encounters, about 115 different laboratory tests, 115 imaging studies, 25 simulation and planning procedures, and 115 radiation treatment codes. The radiation oncology SEOCs during the study period had an approval duration of 180 days. Advanced Medical Cost Management Solutions software (AMCMS) is the VHA data analytics platform for community care medical service costs. AMCMS includes all individual CPT codes billed by specific radiation oncology SEOC versions. Data are refreshed monthly, and all charges were extracted on September 12, 2024, > 6 months after the final evaluated service date to allow for complete billing returns.6
Radiation Oncology-Specific Costs
The VA Close to Me (CTM) program was used to find 84 specific radiation oncology CPT codes, nearly all within the 77.XXX or G6.XXX series, which included all radiation oncology-specific (ROS) codes (except visits accrued during consultation and return appointments). ROS costs are those that could not be performed by any other service and include procedures related to radiation oncology simulation, treatment planning, treatment delivery (with or without image guidance), and physician or physicist management. All ROS costs should be included in a patient’s radiation oncology SEOC. Other costs that may accompany operating room or brachytherapy administration did not follow a 77.XXX or G6.XXX pattern but were included in total radiation therapy operating costs.
Data obtained from AMCMS and CTM included patient name and identifier; CPT billed amount; CPT paid amount; dates of service; number of claims; International Classification of Diseases, Tenth Revision (ICD) diagnosis; and VA authorization numbers. Only CTM listed code modifiers. Only items categorized as paid were included in the analysis. Charges associated with discontinued consultations that had accrued costs also were included. Codes that were not directly related to ROS were separately characterized as other and further subcategorized.
Deep Dive Categorization
All scanned documents tagged to the community consultation were accessed and evaluated for completeness by a radiation oncologist (RS). The presence or absence of consultation notes and treatment summaries was evaluated based on necessity (ie, not needed for continuation of care or treatment was not given). In the absence of a specific completion summary or follow-up note detailing the treatment modality, number of fractions, and treatment sites, available documentation, including clinical notes and billing information, was used. Radical or curative therapies were identified as courses expected to eradicate disease, including stereotactic ablative radiotherapy to the brain, lung, liver, and other organs. Palliative therapies included whole-brain radiotherapy or other low-dose treatments. If the patient received the intended course, this was categorized as full. If incomplete, it was considered partial.
Billing Deviations
The complete document review allowed for close evaluation of paid therapy and identification of gaps in billing (eg, charges not found in extracted data that should have occurred) for external beam radiotherapy patients. Conversely, extra charges, such as an additional weekly treatment management charge (CPT code 77427), would be noted. Patients were expected to have the number of treatments specified in the summary, a clinical treatment planning code, and weekly treatment management notes from physicians and physicists every 5 fractions. Consultations and follow-up visits were expected to have 1 visit code; CPT codes 99205 and 99215, respectively, were used to estimate costs in their absence.
Costs were based on Medicare rates as of January 1 of the year in which they were accrued. 7-10 Duplicates were charges with the same code, date, billed quantity, and paid amounts for a given patient. These would always be considered erroneous. Medicare treatment costs for procedures such as intensity modulated radiotherapy (CPT code 77385 or 77386) are available on the Medicare website. When reviewing locality deviations for 77427, there was a maximum of 33% increase in Medicare rates. Therefore, for treatment codes, one would expect the range to be at least the Medicare rate and maximally 33% higher. These rates are negotiated with insurance companies, but this range was used for the purpose of reviewing and adjusting large data sets.
RESULTS
Since 2018, > 500 community care consults have been placed by radiation oncology for treatment in the community, with more following implementation of the VA MISSION Act. Use of radiation oncology community care services annually increased during the study period for this facility (Table 1, Figure 2). Of the 325 community care consults placed from October 1, 2021, to February 1, 2024, 248 radiation oncology SEOCs were recorded with charges for 181 patients (range, 1-5 SEOCs). Long drive time was the rationale for > 97% of patients directed to community care (Supplemental materials, available at doi:10.12788/fp.0585). Based on AMCMS data, $22.2 million was billed and $2.7 million was paid (20%) for 8747 CPT codes. Each community care interval cost the VA a median (range) of $5000 ($8-$168,000 (Figure 3).
After reviewing ROS charges extracted from CTM, 20 additional patients had radiation oncology charges but did not have a radiation oncology SEOC for 268 episodes of care for 201 unique patients. In addition to the 20 patients who did not have a SEOC, 42 nonradiation oncology SEOCs contained 1148 radiation oncology codes, corresponding to almost $500,000 paid. Additional charges of about $416,000, which included biologic agents (eg, durvalumab, nivolumab), procedures (eg, mastectomies), and ambulance rides were inappropriately added to radiation oncology SEOCs.
While 77% of consultations were scanned into CPRS and JLV, only 54% of completion summaries were available with an estimated $115,000 in additional costs. The total adjusted costs was about $2.9 million. Almost 37% of SEOCs were for visits only. For the 166 SEOCs where patients received any radiation treatment or planning, the median cost was $18,000. Differences in SEOC pathways are shown in Figure 4. One hundred twenty-one SEOCs (45%) followed the standard pathway, with median SEOC costs of $15,500; when corrected for radiation-specific costs, the median cost increased to $18,000. When adjusted for billing irregularities, the median cost was $20,600. Ninety-nine SEOCs (37%) were for consultation/ follow-up visits only, with a median cost of $220. When omitting shared scans and nonradiation therapy costs and correcting for billing gaps, the median cost decreased to $170. A median of $9200 was paid per patient, with $12,900 for radiation therapy-specific costs and $13,300 adjusted for billing deviations. Narrowing to the 106 patients who received full, radical courses, the median SEOC, ROS, and adjusted radiation therapy costs increased to $19,400, $22,200, and $22,900, respectively (Table 2, Figure 5). Seventy-one SEOCs (26%) had already seen a radiation oncologist before the VA radiation oncology department was aware, and 49 SEOCs (18%) had retroactive approvals (Supplemental materials available at doi:10.12788/fp.0585).
Every consultation charge was reviewed. A typical patient following the standard pathway (eAppendix 2, available at doi:10.12788/ fp.0585) exhibited a predictable pattern of consultation payment, simulation and planning, multiple radiation treatments interspersed with treatment management visits and a cone-down phase, and finishing with a follow-up visit. A less predictable case with excess CPT codes, gaps in charges, and an additional unexpected palliative course is shown in eAppendix 3 (available at doi:10.12788/fp.0585). Gaps occurred in 42% of SEOCs with missed bills costing as much as $12,000. For example, a patient with lung cancer had a treatment summary note for lung cancer after completion that showed the patient received 30 fractions of 2 Gy, a typical course. Only 10 treatment codes and 3 of 6 weekly treatment management codes were available. There was a gap of 20 volumetric modulated arc therapy treatments, 3 physics weekly status checks, 3 physician managements notes, and a computed tomography simulation charge.
Between AMCMS and CTM, 10,005 CPT codes were evaluated; 1255 (12.5%) were unique to AMCMS (either related to the radiation oncology course, such as Evaluation and Management CPT codes or “other” unrelated codes) while 1158 (11.6%) were unique to CTM. Of the 7592 CPT codes shared between AMCMS and CTM, there was a discrepancy in 135 (1.8%); all were duplicates (CTM showed double payment while AMCMS showed $0 paid). The total CPT code costs came to $3.2 million with $560,000 unique to SEOCs and $500,000 unique to CTM. Treatment codes were the most common (33%) as shown in Table 3 and accounted for 55% of the cost ($1.8 million). About 700 CPT codes were considered “other,” typically for biologic therapeutic agents (Table 4 and eAppendix 4, available at doi:10.12788/fp.0585).
DISCUSSION
The current method of reporting radiation oncology costs used by VA is insufficient and misleading. Better data are needed to summarize purchased care costs to guide decisions about community care at the VA. Investigations into whether the extra costs for quality care (ie, expensive capital equipment, specialized staff, mandatory accreditations) are worthwhile if omitted at other facilities patients choose for their health care needs. No study has defined specialty care-specific costs by evaluating billing receipts from the CDW to answer the question. Kenamond et al highlight the need for radiation oncology for rural patients.11 Drive time was cited as the reason for community care referral for 97% of veterans, many of whom lived in rural locations. Of patients with rurality information who enrolled in community care, 57% came from rural or highly rural counties, and this ratio held for those who received full curative therapies. An executive administrator relying on AMCMS reports would see a median SEOC cost of $5000, but without ROS knowledge in coding, the administrator would miss many additional costs. For example, 2 patients who each had 5 SEOCs during the evaluated period, incurred a total cost of only $1800.
Additionally, an administrator could include miscategorized costs with significant ramifications. The 2 most expensive SEOCs were not typical radiation oncology treatments. A patient undergoing radium-223 dichloride therapy incurred charges exceeding $165,000, contributing disproportionately to the overall median cost analysis; this would normally be administered by the nuclear medicine department. Immunotherapy and chemotherapy are uniformly overseen by medical oncology services, but drug administration codes were still found in radiation oncology SEOCs. A patient (whose SEOC was discontinued but accrued charges) had an electrocardiogram interpretation for $8 as the SEOC cost; 3 other SEOCs continued to incur costs after being discontinued. There were 24 empty SEOCs for patients that had consults to the community, and 2 had notes stating treatment had been delivered yet there was no ROS costs or SEOC costs. Of the 268 encounters, 43% had some sort of billing irregularities (ie, missing treatment costs) that would be unlikely for a private practice to omit; it would be much more likely that the CDW miscategorized the payment despite confirmation of the 2 retrieval systems.
It would be inadvisable to make staffing decisions or forecast costs based on current SEOC reports without specialized curation. A simple yet effective improvement to the cost attribution process would be to restrict the analysis to encounters containing primary radiation treatment codes. This targeted approach allows more accurate identification of patients actively receiving radiation oncology treatment, while excluding those seen solely for consultations or follow-up visits. Implementing this refinement leads to a substantial increase in the median payment—from $5000 to $13,000—without requiring additional coding or data processing, thereby enhancing the accuracy of cost estimates with minimal effort.
Clarifying radiation oncology service costs requires addressing the time frame and services included, given laxity and interpretation of the SEOCs. VA community care departments have streamlined the reimbursement process at the expense of medical cost organization and accuracy; 86% of VA practitioners reported that ≥ 1 potential community health care partners had refused to work with the VA because of payment delays.12 Payments are contingent on correspondence from outside practices for community work. For radiation oncology, this includes the consultation but also critical radiation-related details of treatment, which were omitted nearly half the time. SEOC approval forms have many costly laboratory tests, imaging, and procedures that have little to do with radiation oncology cancer treatments but may be used in the workup and staging process; this creates noise when calculating radiation oncology fiscal cost.
The presumption that an episode of care equates to a completed radiation therapy course is incorrect; this occurs less than half of the time. An episode often refers to a return visit, or conversely, multiple treatment courses. As the patients’ medical homes are their VHA primary care practitioners, it would be particularly challenging to care for the patients without full treatment information, especially if adverse effects from therapy were to arise. As a tertiary specialty, radiation oncology does not seek out patients and are sent consultations from medical oncology, surgical, and medical oncologic specialties. Timesensitive processes such as workup, staging, and diagnosis often occur in parallel. This analysis revealed that patients see outside radiation oncologists prior to the VA. There are ≥ 100 patients who had radiation oncology codes without a radiation oncology SEOC or community care consultation, and in many cases, the consultation was placed after the patient was seen.
Given the lack of uniformity and standardization of patient traffic, the typical and expected pathways were insufficient to find the costs. Too many opportunities for errors and incorrect categorization of costs meant a different method would be necessary. Starting at the inception of the community care consult, only 1 diagnosis code can be entered. For patients with multiple diagnoses, one would not be able to tell what was treated without chart access. Radiation oncology consults come from primary and specialty care practitioners and nurses throughout the VA. Oftentimes, the referral would be solicited by the community radiation oncology clinic, diagnosing community specialty (ie, urology for a patient with prostate cancer), or indirectly from the patient through primary care. Many cases were retroactively approved as the veteran had already been consulted by the community care radiation oncologist. If the patient is drive-time eligible, it would be unlikely that they would leave and choose to return to the VA. There is no way for a facility VA service chief or administrator to mitigate VA community costs of care, especially as shown by the miscategorization of several codes. Database challenges exacerbate the issue: 1 patient changed her first and last name during this time frame, and 2 patients had the same name but different social security numbers. In order to strictly find costs between 2 discrete timepoints, 39 (15%) SEOCs were split and incomplete, and 6 SEOCs contained charges for 2 different patients. This was corrected, and all inadvertent charges were cancelled. Only 1 ICD code is allowed per community care consultation, so an investigation is required to find costs for patients with multiple sites of disease. Additionally, 5 of the patients marked for drive time were actually patients who received Gamma Knife and brachytherapy, services not available at the VA.
Hanks et al first attempted to calculate cost of radiation oncology services. External beam prostate cancer radiotherapy at 3 suburban California centers cost $6750 ($20,503 inflation adjusted) per patient before October 1984 and $5600 ($17,010 inflation adjusted) afterwards.13 According to the American Society for Radiation Oncology, Advocacy Radiation Oncology Case Rate Program Curative radiation courses should cost $20,000 to $30,000 and palliative courses should cost $10,000 to $15,000. These costs are consistent with totals demonstrated in this analysis and similar to the inflation-adjusted Hanks et al figures. Preliminary findings suggest that radiation treatment constituted more than half of the total expenditures, with a notable $4 million increase in adjusted cost compared to the Medicare rates, indicating significant variation. Direct comparisons with Medicaid or commercial payer rates remain unexplored.
Future Directions
During the study period, 201 patients received 186 courses of radiation therapy in the community, while 1014 patients were treated in-house for a total of 833 courses. A forthcoming analysis will directly compare the cost of in-house care with that of communitybased treatment, specifically breaking down expenditure differences by diagnosis. Future research should investigate strategies to align reimbursement with quality metrics, including the potential role of tertiary accreditation in incentivizing high-value care. Additional work is also warranted to assess patient out-ofpocket expenses across care settings and to benchmark VA reimbursement against Medicare, Medicaid, and private insurance rates. In any case, with the increasing possibility of fewer fractions for treatments such as stereotactic radiotherapy or palliative care therapy, there is a clear financial incentive to treat as frequently as allowed despite equal clinical outcomes.
CONCLUSIONS
Veterans increasingly choose to receive care closer to home if the option is available. In the VA iron triangle, cost comes at the expense of access but quantifying this has proved elusive in the cost accounting model currently used at the VA.1 The inclusion of all charges loosely associated with SEOCs significantly impairs the ability to conduct meaningful cost analyses. The current VA methodology not only introduces substantial noise into the data but also leads to a marked underestimation of the true cost of care delivered in community settings. Such misrepresentation risks driving policy decisions that could inappropriately reduce or eliminate in-house radiation oncology services. Categorizing costs effectively in the VA could assist in making managerial and administrative decisions and would prevent damaging service lines based on misleading or incorrect data. A system which differentiates between patients who have received any treatment codes vs those who have not would increase accuracy.
- Kissick W. Medicine’s Dilemmas: Infinite Needs Versus Finite Resources. 1st ed. Yale University Press; 1994.
- Albanese AP, Bope ET, Sanders KM, Bowman M. The VA MISSION Act of 2018: a potential game changer for rural GME expansion and veteran health care. J Rural Health. 2020;36(1):133-136. doi:10.1111/jrh.12360
- Office of Management and Budget (US). Budget of the United States Government, Fiscal Year 2025. Washington, DC: US Government Publishing Office; 2024. Available from: US Department of Veterans Affairs FY 2025 Budget Submission: Budget in Brief.
- US Department of Veterans Affairs. Veteran care claims. Accessed April 3, 2025. https://www.va.gov/COMMUNITYCARE/revenue-ops/Veteran-Care-Claims.asp
- US Centers for Medicare and Medicaid Services. Accessed April 3, 2025. Procedure price lookup https://www.medicare.gov/procedure-price-lookup
- US Department of Veterans Affairs. WellHive -Enterprise. Accessed April 3, 2025. https://department.va.gov/privacy/wp-content/uploads/sites/5/2023/05/FY23WellHiveEnterprisePIA.pdf
- US Centers for Medicare and Medicaid Services. RVU21a physician fee schedule, January 2021 release. Accessed April 3, 2025. https://www.cms.gov/medicaremedicare-fee-service-paymentphysicianfeeschedpfs-relative-value-files/rvu21a
- US Centers for Medicare and Medicaid Services. RVU22a physician fee schedule, January 2022 release. Accessed April 3, 2025. https://www.cms.gov/medicaremedicare-fee-service-paymentphysicianfeeschedpfs-relative-value-files/rvu22a
- US Centers for Medicare and Medicaid Services. RVU23a physician fee schedule, January 2023 release. Accessed April 3, 2025. https://www.cms.gov/medicare/medicare-fee-service-payment/physicianfeesched/pfs-relative-value-files/rvu23a
- US Centers for Medicare and Medicaid Services. RVU23a Medicare Physician Fee Schedule rates effective January 1, 2024, through March 8, 2024. Accessed on April 3, 2025. https://www.cms.gov/medicare/payment/fee-schedules/physician/pfs-relative-value-files/rvu24a
- Kenamond MC, Mourad WF, Randall ME, Kaushal A. No oncology patient left behind: challenges and solutions in rural radiation oncology. Lancet Reg Health Am. 2022;13:100289. doi:10.1016/j.lana.2022.100289
- Mattocks KM, Kroll-Desrosiers A, Kinney R, Elwy AR, Cunningham KJ, Mengeling MA. Understanding VA’s use of and relationships with community care providers under the MISSION Act. Med Care. 2021;59(Suppl 3):S252-S258. doi:10.1097/MLR.0000000000001545
- Hanks GE, Dunlap K. A comparison of the cost of various treatment methods for early cancer of the prostate. Int J Radiat Oncol Biol Phys. 1986;12(10):1879-1881. doi:10.1016/0360-3016(86)90334-2
- American Society of Radiation Oncology. Radiation oncology case rate program (ROCR). Accessed April 3, 2025. https://www.astro.org/advocacy/key-issues-8f3e5a3b76643265ee93287d79c4fc40/rocr
- Kissick W. Medicine’s Dilemmas: Infinite Needs Versus Finite Resources. 1st ed. Yale University Press; 1994.
- Albanese AP, Bope ET, Sanders KM, Bowman M. The VA MISSION Act of 2018: a potential game changer for rural GME expansion and veteran health care. J Rural Health. 2020;36(1):133-136. doi:10.1111/jrh.12360
- Office of Management and Budget (US). Budget of the United States Government, Fiscal Year 2025. Washington, DC: US Government Publishing Office; 2024. Available from: US Department of Veterans Affairs FY 2025 Budget Submission: Budget in Brief.
- US Department of Veterans Affairs. Veteran care claims. Accessed April 3, 2025. https://www.va.gov/COMMUNITYCARE/revenue-ops/Veteran-Care-Claims.asp
- US Centers for Medicare and Medicaid Services. Accessed April 3, 2025. Procedure price lookup https://www.medicare.gov/procedure-price-lookup
- US Department of Veterans Affairs. WellHive -Enterprise. Accessed April 3, 2025. https://department.va.gov/privacy/wp-content/uploads/sites/5/2023/05/FY23WellHiveEnterprisePIA.pdf
- US Centers for Medicare and Medicaid Services. RVU21a physician fee schedule, January 2021 release. Accessed April 3, 2025. https://www.cms.gov/medicaremedicare-fee-service-paymentphysicianfeeschedpfs-relative-value-files/rvu21a
- US Centers for Medicare and Medicaid Services. RVU22a physician fee schedule, January 2022 release. Accessed April 3, 2025. https://www.cms.gov/medicaremedicare-fee-service-paymentphysicianfeeschedpfs-relative-value-files/rvu22a
- US Centers for Medicare and Medicaid Services. RVU23a physician fee schedule, January 2023 release. Accessed April 3, 2025. https://www.cms.gov/medicare/medicare-fee-service-payment/physicianfeesched/pfs-relative-value-files/rvu23a
- US Centers for Medicare and Medicaid Services. RVU23a Medicare Physician Fee Schedule rates effective January 1, 2024, through March 8, 2024. Accessed on April 3, 2025. https://www.cms.gov/medicare/payment/fee-schedules/physician/pfs-relative-value-files/rvu24a
- Kenamond MC, Mourad WF, Randall ME, Kaushal A. No oncology patient left behind: challenges and solutions in rural radiation oncology. Lancet Reg Health Am. 2022;13:100289. doi:10.1016/j.lana.2022.100289
- Mattocks KM, Kroll-Desrosiers A, Kinney R, Elwy AR, Cunningham KJ, Mengeling MA. Understanding VA’s use of and relationships with community care providers under the MISSION Act. Med Care. 2021;59(Suppl 3):S252-S258. doi:10.1097/MLR.0000000000001545
- Hanks GE, Dunlap K. A comparison of the cost of various treatment methods for early cancer of the prostate. Int J Radiat Oncol Biol Phys. 1986;12(10):1879-1881. doi:10.1016/0360-3016(86)90334-2
- American Society of Radiation Oncology. Radiation oncology case rate program (ROCR). Accessed April 3, 2025. https://www.astro.org/advocacy/key-issues-8f3e5a3b76643265ee93287d79c4fc40/rocr
Community Care Radiation Oncology Cost Calculations for a VA Medical Center
Community Care Radiation Oncology Cost Calculations for a VA Medical Center
Clinicians Should Have Private Spaces for Telehealth According to VA Memo
US Department of Veterans Affairs (VA) officials are insisting that when remote telehealth clinicians return to an office setting, they must have private workspaces “that foster trusted, confidential, and therapeutic relationships with veterans,” according to an April internal memo reported on by NPR.
The return-to-office mandate followed a Trump Administration executive order in February indicated that mental health clinicians at the US Department of Veterans Affairs (VA) must physically return to their workplace by May 5. For some, the deadline came as early as April 14; however, that order, like many others, may now be being revised or reconsidered due to concerns that have been raised. Many mental health clinicians were hired specifically to work remotely. They worried there would simply not be enough space for them, particularly to provide confidential counseling.
Millions of veterans use telehealth to access VA care. More than 98% of VA mental health clinicians have conducted ≥ 1 video visit to screen and treat patients for anxiety, depression, posttraumatic stress disorder, and more. Telehealth has been particularly important for veterans living in rural communities.
The April VA memo stipulated that “spaces used to deliver synchronous telehealth services should offer the same level of privacy and therapeutic environment applicable to an in-person visit in the same space.”
Therapists, patients, advocacy groups, and lawmakers have expressed concern about the potential impacts the policy change could have on patient care for veterans and, above all, about what it could mean for privacy. On Mar. 27, the American Psychological Association issued a statement noting that the change “resulted in providers being asked to conduct sensitive therapy sessions in open office environments, cubicles, or shared spaces that fail to meet basic confidentiality and privacy requirements for the delivery of mental health care services.”
Twenty Democrats in the House of Representatives sent a letter to VA Secretary Doug Collins expressing concern with the return to office policy. According to the letter a VA social worker supervisor reported managing their caseload while sharing a 100 ft2 shower space with another supervisor. It also reported that Clinical Resource Hub employees were being told to report to buildings where federal employees from other agencies work. “We have heard from countless stakeholders, veterans, and Department of Veterans Affairs (VA) employees that by carrying out President Trump’s blanket return-to-office policy your administration is damaging veteran and employee trust in VA, disrupting and impeding veterans’ access to care, and creating untenable and inefficient conditions for both veterans and the VA workforce,” the letter stated.
“This is a clear violation of veterans’ privacy and VA’s obligation to protect veterans’ private health information, and risks violation of the Health Insurance Portability and Accountability Act (HIPAA),” the letter added.
The lawmakers noted that, as of March 10, the VA was exempting Veterans Crisis Line workers, most of whom had been working remotely for the past 5 years, responding to more than 10 million calls, texts, and chats. That move, they said, indicated “that you understand there will be negative impacts to veterans’ care due to the return-to-office order and that these must be mitigated.”
VA spokesperson Peter Kasperowicz called the privacy concerns “nonsensical” and blamed “fear mongering from the media.” The VA, he said, “is no longer a place where the status quo for employees is to simply phone it in from home.” He also claimed that “the small number of employees who are desperate to avoid returning to the office will do more to drive away staff and patients than VA’s commonsense return-to-office policy ever will.”
VA care, he said, would continue uninterrupted and the “VA will ensure that employees have a workspace that is appropriate for the work they do.”
US Department of Veterans Affairs (VA) officials are insisting that when remote telehealth clinicians return to an office setting, they must have private workspaces “that foster trusted, confidential, and therapeutic relationships with veterans,” according to an April internal memo reported on by NPR.
The return-to-office mandate followed a Trump Administration executive order in February indicated that mental health clinicians at the US Department of Veterans Affairs (VA) must physically return to their workplace by May 5. For some, the deadline came as early as April 14; however, that order, like many others, may now be being revised or reconsidered due to concerns that have been raised. Many mental health clinicians were hired specifically to work remotely. They worried there would simply not be enough space for them, particularly to provide confidential counseling.
Millions of veterans use telehealth to access VA care. More than 98% of VA mental health clinicians have conducted ≥ 1 video visit to screen and treat patients for anxiety, depression, posttraumatic stress disorder, and more. Telehealth has been particularly important for veterans living in rural communities.
The April VA memo stipulated that “spaces used to deliver synchronous telehealth services should offer the same level of privacy and therapeutic environment applicable to an in-person visit in the same space.”
Therapists, patients, advocacy groups, and lawmakers have expressed concern about the potential impacts the policy change could have on patient care for veterans and, above all, about what it could mean for privacy. On Mar. 27, the American Psychological Association issued a statement noting that the change “resulted in providers being asked to conduct sensitive therapy sessions in open office environments, cubicles, or shared spaces that fail to meet basic confidentiality and privacy requirements for the delivery of mental health care services.”
Twenty Democrats in the House of Representatives sent a letter to VA Secretary Doug Collins expressing concern with the return to office policy. According to the letter a VA social worker supervisor reported managing their caseload while sharing a 100 ft2 shower space with another supervisor. It also reported that Clinical Resource Hub employees were being told to report to buildings where federal employees from other agencies work. “We have heard from countless stakeholders, veterans, and Department of Veterans Affairs (VA) employees that by carrying out President Trump’s blanket return-to-office policy your administration is damaging veteran and employee trust in VA, disrupting and impeding veterans’ access to care, and creating untenable and inefficient conditions for both veterans and the VA workforce,” the letter stated.
“This is a clear violation of veterans’ privacy and VA’s obligation to protect veterans’ private health information, and risks violation of the Health Insurance Portability and Accountability Act (HIPAA),” the letter added.
The lawmakers noted that, as of March 10, the VA was exempting Veterans Crisis Line workers, most of whom had been working remotely for the past 5 years, responding to more than 10 million calls, texts, and chats. That move, they said, indicated “that you understand there will be negative impacts to veterans’ care due to the return-to-office order and that these must be mitigated.”
VA spokesperson Peter Kasperowicz called the privacy concerns “nonsensical” and blamed “fear mongering from the media.” The VA, he said, “is no longer a place where the status quo for employees is to simply phone it in from home.” He also claimed that “the small number of employees who are desperate to avoid returning to the office will do more to drive away staff and patients than VA’s commonsense return-to-office policy ever will.”
VA care, he said, would continue uninterrupted and the “VA will ensure that employees have a workspace that is appropriate for the work they do.”
US Department of Veterans Affairs (VA) officials are insisting that when remote telehealth clinicians return to an office setting, they must have private workspaces “that foster trusted, confidential, and therapeutic relationships with veterans,” according to an April internal memo reported on by NPR.
The return-to-office mandate followed a Trump Administration executive order in February indicated that mental health clinicians at the US Department of Veterans Affairs (VA) must physically return to their workplace by May 5. For some, the deadline came as early as April 14; however, that order, like many others, may now be being revised or reconsidered due to concerns that have been raised. Many mental health clinicians were hired specifically to work remotely. They worried there would simply not be enough space for them, particularly to provide confidential counseling.
Millions of veterans use telehealth to access VA care. More than 98% of VA mental health clinicians have conducted ≥ 1 video visit to screen and treat patients for anxiety, depression, posttraumatic stress disorder, and more. Telehealth has been particularly important for veterans living in rural communities.
The April VA memo stipulated that “spaces used to deliver synchronous telehealth services should offer the same level of privacy and therapeutic environment applicable to an in-person visit in the same space.”
Therapists, patients, advocacy groups, and lawmakers have expressed concern about the potential impacts the policy change could have on patient care for veterans and, above all, about what it could mean for privacy. On Mar. 27, the American Psychological Association issued a statement noting that the change “resulted in providers being asked to conduct sensitive therapy sessions in open office environments, cubicles, or shared spaces that fail to meet basic confidentiality and privacy requirements for the delivery of mental health care services.”
Twenty Democrats in the House of Representatives sent a letter to VA Secretary Doug Collins expressing concern with the return to office policy. According to the letter a VA social worker supervisor reported managing their caseload while sharing a 100 ft2 shower space with another supervisor. It also reported that Clinical Resource Hub employees were being told to report to buildings where federal employees from other agencies work. “We have heard from countless stakeholders, veterans, and Department of Veterans Affairs (VA) employees that by carrying out President Trump’s blanket return-to-office policy your administration is damaging veteran and employee trust in VA, disrupting and impeding veterans’ access to care, and creating untenable and inefficient conditions for both veterans and the VA workforce,” the letter stated.
“This is a clear violation of veterans’ privacy and VA’s obligation to protect veterans’ private health information, and risks violation of the Health Insurance Portability and Accountability Act (HIPAA),” the letter added.
The lawmakers noted that, as of March 10, the VA was exempting Veterans Crisis Line workers, most of whom had been working remotely for the past 5 years, responding to more than 10 million calls, texts, and chats. That move, they said, indicated “that you understand there will be negative impacts to veterans’ care due to the return-to-office order and that these must be mitigated.”
VA spokesperson Peter Kasperowicz called the privacy concerns “nonsensical” and blamed “fear mongering from the media.” The VA, he said, “is no longer a place where the status quo for employees is to simply phone it in from home.” He also claimed that “the small number of employees who are desperate to avoid returning to the office will do more to drive away staff and patients than VA’s commonsense return-to-office policy ever will.”
VA care, he said, would continue uninterrupted and the “VA will ensure that employees have a workspace that is appropriate for the work they do.”
Achieving Psychological Safety in High Reliability Organizations
Achieving Psychological Safety in High Reliability Organizations
Worldwide, health care is becoming increasingly complex as a result of greater clinical workforce demands, expanded roles and responsibilities, health care system mergers, stakeholder calls for new capabilities, and digital transformation. 1,2These increasing demands has prompted many health care institutions to place greater focus on the psychological safety of their workforce, particularly in high reliability organizations (HROs). Building a robust foundation for high reliability in health care requires the presence of psychological safety—that is, staff members at all levels of the organization must feel comfortable speaking up when they have questions or concerns.3,4 Psychological safety can improve the safety and quality of patient care but has not reached its full potential in health care.5,6 However, there are strategies that promote the widespread implementation of psychological safety in health care organizations.3-6
PSYCHOLOGICAL SAFETY
The concept of psychological safety in organizational behavior originated in 1965 when Edgar Schein and Warren Bennis, leaders in organizational psychology and management, published their reflections on the importance of psychological safety in helping individuals feel secure in the work environment.5-7 Psychological safety in the workplace is foundational to staff members feeling comfortable asking questions or expressing concerns without fear of negative consequences.8,9 It supports both individual and team efforts to raise safety concerns and report near misses and adverse events so that similar events can be averted in the future.9 Patients aren’t the only ones who benefit; psychological safety has also been found to promote job satisfaction and employee well-being.10
THE VETERANS HEALTH ADMINISTRATION JOURNEY
Achieving psychological safety is by no means an easy or comfortable process. As with any organizational change, a multipronged approach offers the best chance of success.6,9 When the Veterans Health Administration (VHA) began its incremental, enterprise-wide journey to high reliability in 2019, 3 cohorts were identified. In February 2019, 18 US Department of Veterans Affairs (VA) medical centers (VAMCs) (cohort 1) began the process of becoming HROs. Cohort 2 followed in October 2020 and included 54 VAMC. Finally, in October 2021, 67 additional VAMCs (cohort 3) started the process.2 During cohort 2, the VA Providence Healthcare System (VAPHCS) decided to emphasize psychological safety at the start of the journey to becoming an HRO. This system is part of the VA New England Healthcare System (VISN 1), which includes VAMCs and clinics in Connecticut, Maine, Massachusetts, New Hampshire, Rhode Island, and Vermont.11 Soon thereafter, the VA Bedford Healthcare System and the VA Connecticut Healthcare System adopted similar strategies. Since then, other VAMCs have also adopted this approach. These collective experiences identified 4 useful strategies for achieving psychological safety: leadership engagement, open communication, education and training, and accountability.
Leadership Engagement
Health care organization leaders play a critical role in making psychological safety happen—especially in complex and constantly changing environments, such as HROs.4 Leaders behaviors are consistently linked to the perception of psychological safety at the individual, team, and organizational levels.8 It is especially important to have leaders who recognize the views of individuals and team members and encourage staff participation in discussions to gain additional perspectives.7,8,12 Psychological safety can also be facilitated when leaders are visible, approachable, and communicative.4,7-9
Organizational practices, policies, and processes (eg, reporting adverse events without the fear of negative consequences) are also important ways that leaders can establish and sustain psychological safety. On a more granular level, leaders can enhance psychological safety by promoting and acknowledging individuals who speak up, regularly asking staff about safety concerns, highlighting “good catches” when harm is avoided, and using staff feedback to initiate improvements.4,7,13Finally, in the authors’ experience, psychological safety requires clear commitment from leaders at all levels of an organization. Communication should be bidirectional, and leaders should close the proverbial “loop” with feedback and timely follow-up. This encourages and reinforces staff engagement and speaking up behaviors.2,4,7,13
Open Communication
Promoting an environment of open communication, where all individuals and teams feel empowered to speak up with questions, concerns, and recommendations—regardless of position within the organization—is critical to psychological safety.4,6,9 Open communication is especially critical when processes and systems are constantly changing and advancing as a result of new information and technology.9 Promoting open, bidirectional communication during the delivery of patient care can be accomplished with huddles, tiered safety huddles, leader rounding for high reliability, and time-outs.2,4,6 These opportunities allow team members to discuss concerns, identify resources that support safe, high-quality care; reflect on successes and opportunities for improvement; and circle back on concerns.2,6 Open communication in psychologically safe environments empowers staff to raise patient care concerns and is instrumental for improving patient safety, increasing staff job satisfaction, and decreasing turnover.6,14
Education and Training
Education and training for all staff—from the frontline to the executive level—are essential to successfully implementing the principles and practices of psychological safety.5-7 VHA training covers many topics, including the origins, benefits, and implementation strategies of psychological safety (Table). Role-playing simulation is an effective teaching format, providing staff with opportunities to practice techniques for raising concerns or share feedback in a controlled environment.6 In addition, education should be ongoing; it helps leaders and staff members feel competent and confident when implementing psychological safety across the health care organization.6,10

Accountability
The final critical strategy for achieving psychological safety is accountability. It is the responsibility of all leadership—from senior leaders to clinical and nonclinical managers—to create a culture of shared accountability.5 But first, expectations must be set. Leadership must establish well-defined behavioral expectations that align with the organization’s values. Understanding behavioral expectations will help to ensure that employees know what achievement looks like, as well as how they are being held accountable for their individual actions.4,5,7 In practical terms, this means ensuring that staff members have the skills and resources to achieve goals and expectations, providing performance feedback in a timely manner, and including expectations in annual performance evaluations (as they are in the VHA).
Consistency is key. Accountability should be the expectation across all levels and services of the health care organization. No staff member should be exempt from promoting a psychologically safe work environment. Compliance with behavioral expectations should be monitored and if a person’s actions are not consistent with expectations, the situation will need to be addressed. Interventions will depend on the type, severity, and frequency of the problematic behaviors. Depending on an organization’s policies and practices, courses of action can range from feedback counseling to employment termination.5
A practical matter in ensuring accountability is implementing a psychologically safe process for reporting concerns. Staff members must feel comfortable reporting behavioral concerns without fear of retaliation, negative judgment, or consequences from peers and supervisors. One method for doing this is to create a confidential, centralized process for reporting concerns.5
First-Hand Results
VAPHCS has seen the results of implementing the strategies outlined here. For example, VAPHCS has observed a 45% increase in the use of the patient safety reporting system that logs medical errors and near-misses. In addition, there have been improvements in levels of psychological safety and patient safety reported in the annual VHA All Employee Survey, which is conducted annually to gauge workplace satisfaction, culture, climate, turnover, supervisory behaviors, and general workplace perceptions. VAPHCS has shown consistent improvements in 12 patient safety elements scored on a 5-point scale (1, very dissatisfied; 5, very satisfied) (Figure). Notably, employee ratings of error prevention discussed increased from 4.0 in 2022 to 4.3 in 2024. Data collection and analysis are ongoing; more comprehensive findings will be published in the future.
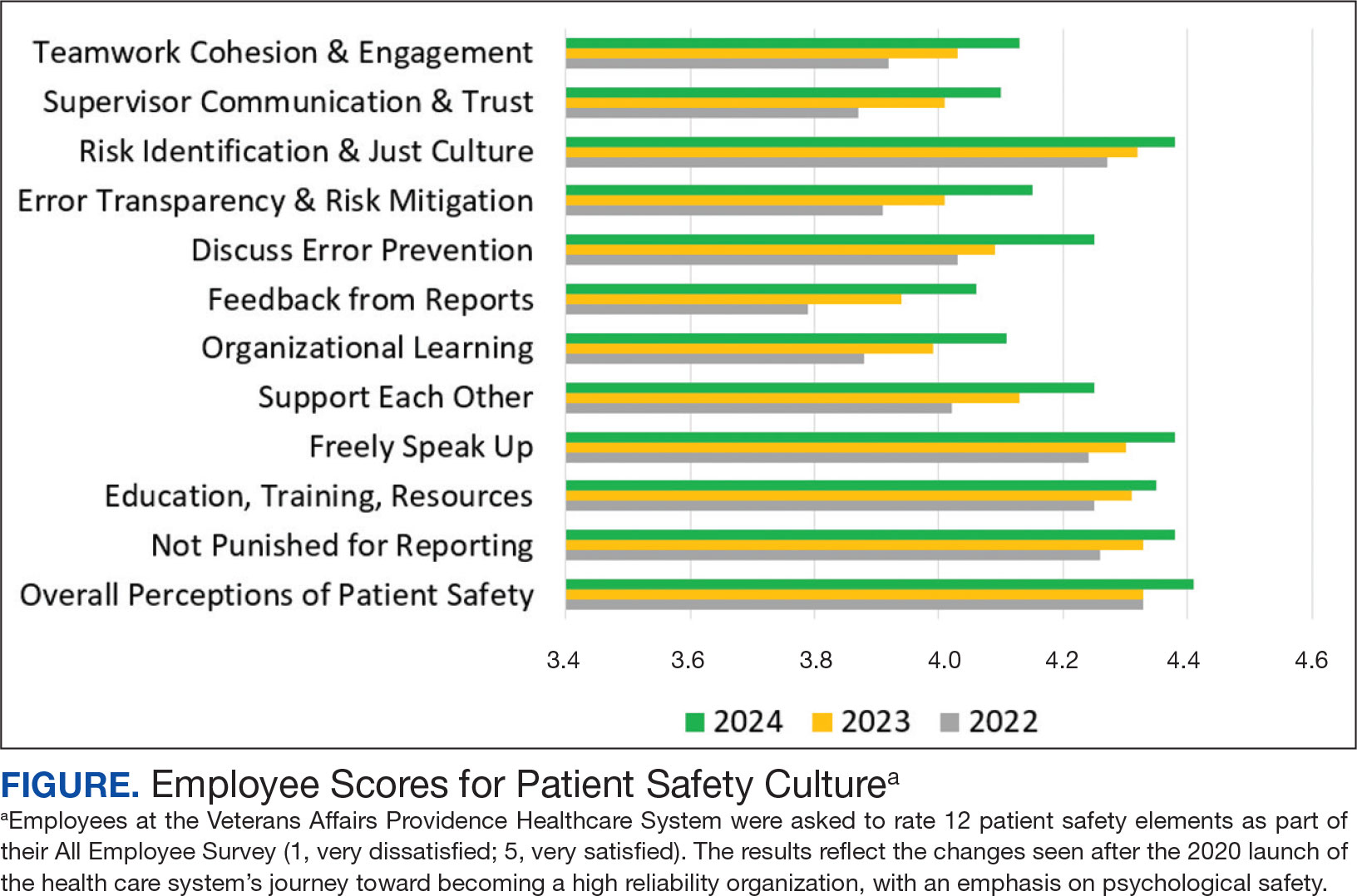
CONCLUSIONS
Health care organizations are increasingly recognizing the importance of psychologically safe workplaces in order to provide safe, high-quality patient care. Psychological safety is a critical tool for empowering staff to raise concerns, ask tough questions, challenge the status quo, and share new ideas for providing health care services. While psychological safety has been slowly adopted in health care, it’s clear that evidence-based strategies can make psychological safety a reality.
- Spanos S, Leask E, Patel R, Datyner M, Loh E, Braithwaite J. Healthcare leaders navigating complexity: A scoping review of key trends in future roles and competencies. BMC Med Educ. 2024;24(1):720. doi:10.1186/s12909-024-05689-4
- Murray JS, Baghdadi A, Dannenberg W, Crews P, Walsh ND. The role of high reliability organization foundational practices in building a culture of safety. Fed Pract. 2024;41(7):214-221. doi:10.12788/fp.0486
- Bransby DP, Kerrissey M, Edmondson AC. Paradise lost (and restored?): a study of psychological safety over time. Acad Manag Discov. Published online March 14, 2024. doi:10.5465/amd.2023.0084
- Murray JS, Kelly S, Hanover C. Promoting psychological safety in healthcare organizations. Mil Med. 2022;187(7-8):808-810. doi:10.1093/milmed/usac041
- Jamal N, Young VN, Shapiro J, Brenner MJ, Schmalbach CE. Patient safety/quality improvement primer, part IV: Psychological safety-drivers to outcomes and well-being. Otolaryngol Head Neck Surg. 2023;168(4):881-888. doi:10.1177/01945998221126966
- Sarofim M. Psychological safety in medicine: What is it, and who cares? Med J Aust. 2024;220(8):398-399. doi:10.5694/mja2.52263
- Edmondson AC, Bransby DP. Psychological safety comes of age: Observed themes in an established literature. Annu Rev Organ Psychol Organ Behav. 2023;10:55-78. doi.org/10.1146/annurev-orgpsych-120920-055217
- Kumar S. Psychological safety: What it is, why teams need it, and how to make it flourish. Chest. 2024; 165(4):942-949. doi:10.1016/j.chest.2023.11.016
- Hallam KT, Popovic N, Karimi L. Identifying the key elements of psychologically safe workplaces in healthcare settings. Brain Sci. 2023;13(10):1450. doi:10.3390/brainsci13101450
- Grailey KE, Murray E, Reader T, Brett SJ. The presence and potential impact of psychological safety in the healthcare setting: an evidence synthesis. BMC Health Serv Res. 2021;21(1):773. doi:10.1186/s12913-021-06740-6
- US Department of Veterans Affairs. VISN 1: VA New England Healthcare System. Accessed March 25, 2025. https://department.va.gov/integrated-service-networks/visn-01
- Brimhall KC, Tsai CY, Eckardt R, Dionne S, Yang B, Sharp A. The effects of leadership for self-worth, inclusion, trust, and psychological safety on medical error reporting. Health Care Manage Rev. 2023;48(2):120-129. doi:10.1097/HMR.0000000000000358
- Adair KC, Heath A, Frye MA, et al. The Psychological Safety Scale of the Safety, Communication, Operational, Reliability, and Engagement (SCORE) Survey: a brief, diagnostic, and actionable metric for the ability to speak up in healthcare settings. J Patient Saf. 2022;18(6):513-520. doi:10.1097/PTS.0000000000001048
- Cho H, Steege LM, Arsenault Knudsen ÉN. Psychological safety, communication openness, nurse job outcomes, and patient safety in hospital nurses. Res Nurs Health. 2023;46(4):445-453.
- Practical Tool 2: 5 minute psychological safety audit. Accessed March 25, 2025. https://www.educationsupport.org.uk/media/jlnf3cju/practical-tool-2-psychological-safety-audit.pdf
Worldwide, health care is becoming increasingly complex as a result of greater clinical workforce demands, expanded roles and responsibilities, health care system mergers, stakeholder calls for new capabilities, and digital transformation. 1,2These increasing demands has prompted many health care institutions to place greater focus on the psychological safety of their workforce, particularly in high reliability organizations (HROs). Building a robust foundation for high reliability in health care requires the presence of psychological safety—that is, staff members at all levels of the organization must feel comfortable speaking up when they have questions or concerns.3,4 Psychological safety can improve the safety and quality of patient care but has not reached its full potential in health care.5,6 However, there are strategies that promote the widespread implementation of psychological safety in health care organizations.3-6
PSYCHOLOGICAL SAFETY
The concept of psychological safety in organizational behavior originated in 1965 when Edgar Schein and Warren Bennis, leaders in organizational psychology and management, published their reflections on the importance of psychological safety in helping individuals feel secure in the work environment.5-7 Psychological safety in the workplace is foundational to staff members feeling comfortable asking questions or expressing concerns without fear of negative consequences.8,9 It supports both individual and team efforts to raise safety concerns and report near misses and adverse events so that similar events can be averted in the future.9 Patients aren’t the only ones who benefit; psychological safety has also been found to promote job satisfaction and employee well-being.10
THE VETERANS HEALTH ADMINISTRATION JOURNEY
Achieving psychological safety is by no means an easy or comfortable process. As with any organizational change, a multipronged approach offers the best chance of success.6,9 When the Veterans Health Administration (VHA) began its incremental, enterprise-wide journey to high reliability in 2019, 3 cohorts were identified. In February 2019, 18 US Department of Veterans Affairs (VA) medical centers (VAMCs) (cohort 1) began the process of becoming HROs. Cohort 2 followed in October 2020 and included 54 VAMC. Finally, in October 2021, 67 additional VAMCs (cohort 3) started the process.2 During cohort 2, the VA Providence Healthcare System (VAPHCS) decided to emphasize psychological safety at the start of the journey to becoming an HRO. This system is part of the VA New England Healthcare System (VISN 1), which includes VAMCs and clinics in Connecticut, Maine, Massachusetts, New Hampshire, Rhode Island, and Vermont.11 Soon thereafter, the VA Bedford Healthcare System and the VA Connecticut Healthcare System adopted similar strategies. Since then, other VAMCs have also adopted this approach. These collective experiences identified 4 useful strategies for achieving psychological safety: leadership engagement, open communication, education and training, and accountability.
Leadership Engagement
Health care organization leaders play a critical role in making psychological safety happen—especially in complex and constantly changing environments, such as HROs.4 Leaders behaviors are consistently linked to the perception of psychological safety at the individual, team, and organizational levels.8 It is especially important to have leaders who recognize the views of individuals and team members and encourage staff participation in discussions to gain additional perspectives.7,8,12 Psychological safety can also be facilitated when leaders are visible, approachable, and communicative.4,7-9
Organizational practices, policies, and processes (eg, reporting adverse events without the fear of negative consequences) are also important ways that leaders can establish and sustain psychological safety. On a more granular level, leaders can enhance psychological safety by promoting and acknowledging individuals who speak up, regularly asking staff about safety concerns, highlighting “good catches” when harm is avoided, and using staff feedback to initiate improvements.4,7,13Finally, in the authors’ experience, psychological safety requires clear commitment from leaders at all levels of an organization. Communication should be bidirectional, and leaders should close the proverbial “loop” with feedback and timely follow-up. This encourages and reinforces staff engagement and speaking up behaviors.2,4,7,13
Open Communication
Promoting an environment of open communication, where all individuals and teams feel empowered to speak up with questions, concerns, and recommendations—regardless of position within the organization—is critical to psychological safety.4,6,9 Open communication is especially critical when processes and systems are constantly changing and advancing as a result of new information and technology.9 Promoting open, bidirectional communication during the delivery of patient care can be accomplished with huddles, tiered safety huddles, leader rounding for high reliability, and time-outs.2,4,6 These opportunities allow team members to discuss concerns, identify resources that support safe, high-quality care; reflect on successes and opportunities for improvement; and circle back on concerns.2,6 Open communication in psychologically safe environments empowers staff to raise patient care concerns and is instrumental for improving patient safety, increasing staff job satisfaction, and decreasing turnover.6,14
Education and Training
Education and training for all staff—from the frontline to the executive level—are essential to successfully implementing the principles and practices of psychological safety.5-7 VHA training covers many topics, including the origins, benefits, and implementation strategies of psychological safety (Table). Role-playing simulation is an effective teaching format, providing staff with opportunities to practice techniques for raising concerns or share feedback in a controlled environment.6 In addition, education should be ongoing; it helps leaders and staff members feel competent and confident when implementing psychological safety across the health care organization.6,10
Accountability
The final critical strategy for achieving psychological safety is accountability. It is the responsibility of all leadership—from senior leaders to clinical and nonclinical managers—to create a culture of shared accountability.5 But first, expectations must be set. Leadership must establish well-defined behavioral expectations that align with the organization’s values. Understanding behavioral expectations will help to ensure that employees know what achievement looks like, as well as how they are being held accountable for their individual actions.4,5,7 In practical terms, this means ensuring that staff members have the skills and resources to achieve goals and expectations, providing performance feedback in a timely manner, and including expectations in annual performance evaluations (as they are in the VHA).
Consistency is key. Accountability should be the expectation across all levels and services of the health care organization. No staff member should be exempt from promoting a psychologically safe work environment. Compliance with behavioral expectations should be monitored and if a person’s actions are not consistent with expectations, the situation will need to be addressed. Interventions will depend on the type, severity, and frequency of the problematic behaviors. Depending on an organization’s policies and practices, courses of action can range from feedback counseling to employment termination.5
A practical matter in ensuring accountability is implementing a psychologically safe process for reporting concerns. Staff members must feel comfortable reporting behavioral concerns without fear of retaliation, negative judgment, or consequences from peers and supervisors. One method for doing this is to create a confidential, centralized process for reporting concerns.5
First-Hand Results
VAPHCS has seen the results of implementing the strategies outlined here. For example, VAPHCS has observed a 45% increase in the use of the patient safety reporting system that logs medical errors and near-misses. In addition, there have been improvements in levels of psychological safety and patient safety reported in the annual VHA All Employee Survey, which is conducted annually to gauge workplace satisfaction, culture, climate, turnover, supervisory behaviors, and general workplace perceptions. VAPHCS has shown consistent improvements in 12 patient safety elements scored on a 5-point scale (1, very dissatisfied; 5, very satisfied) (Figure). Notably, employee ratings of error prevention discussed increased from 4.0 in 2022 to 4.3 in 2024. Data collection and analysis are ongoing; more comprehensive findings will be published in the future.
CONCLUSIONS
Health care organizations are increasingly recognizing the importance of psychologically safe workplaces in order to provide safe, high-quality patient care. Psychological safety is a critical tool for empowering staff to raise concerns, ask tough questions, challenge the status quo, and share new ideas for providing health care services. While psychological safety has been slowly adopted in health care, it’s clear that evidence-based strategies can make psychological safety a reality.
Worldwide, health care is becoming increasingly complex as a result of greater clinical workforce demands, expanded roles and responsibilities, health care system mergers, stakeholder calls for new capabilities, and digital transformation. 1,2These increasing demands has prompted many health care institutions to place greater focus on the psychological safety of their workforce, particularly in high reliability organizations (HROs). Building a robust foundation for high reliability in health care requires the presence of psychological safety—that is, staff members at all levels of the organization must feel comfortable speaking up when they have questions or concerns.3,4 Psychological safety can improve the safety and quality of patient care but has not reached its full potential in health care.5,6 However, there are strategies that promote the widespread implementation of psychological safety in health care organizations.3-6
PSYCHOLOGICAL SAFETY
The concept of psychological safety in organizational behavior originated in 1965 when Edgar Schein and Warren Bennis, leaders in organizational psychology and management, published their reflections on the importance of psychological safety in helping individuals feel secure in the work environment.5-7 Psychological safety in the workplace is foundational to staff members feeling comfortable asking questions or expressing concerns without fear of negative consequences.8,9 It supports both individual and team efforts to raise safety concerns and report near misses and adverse events so that similar events can be averted in the future.9 Patients aren’t the only ones who benefit; psychological safety has also been found to promote job satisfaction and employee well-being.10
THE VETERANS HEALTH ADMINISTRATION JOURNEY
Achieving psychological safety is by no means an easy or comfortable process. As with any organizational change, a multipronged approach offers the best chance of success.6,9 When the Veterans Health Administration (VHA) began its incremental, enterprise-wide journey to high reliability in 2019, 3 cohorts were identified. In February 2019, 18 US Department of Veterans Affairs (VA) medical centers (VAMCs) (cohort 1) began the process of becoming HROs. Cohort 2 followed in October 2020 and included 54 VAMC. Finally, in October 2021, 67 additional VAMCs (cohort 3) started the process.2 During cohort 2, the VA Providence Healthcare System (VAPHCS) decided to emphasize psychological safety at the start of the journey to becoming an HRO. This system is part of the VA New England Healthcare System (VISN 1), which includes VAMCs and clinics in Connecticut, Maine, Massachusetts, New Hampshire, Rhode Island, and Vermont.11 Soon thereafter, the VA Bedford Healthcare System and the VA Connecticut Healthcare System adopted similar strategies. Since then, other VAMCs have also adopted this approach. These collective experiences identified 4 useful strategies for achieving psychological safety: leadership engagement, open communication, education and training, and accountability.
Leadership Engagement
Health care organization leaders play a critical role in making psychological safety happen—especially in complex and constantly changing environments, such as HROs.4 Leaders behaviors are consistently linked to the perception of psychological safety at the individual, team, and organizational levels.8 It is especially important to have leaders who recognize the views of individuals and team members and encourage staff participation in discussions to gain additional perspectives.7,8,12 Psychological safety can also be facilitated when leaders are visible, approachable, and communicative.4,7-9
Organizational practices, policies, and processes (eg, reporting adverse events without the fear of negative consequences) are also important ways that leaders can establish and sustain psychological safety. On a more granular level, leaders can enhance psychological safety by promoting and acknowledging individuals who speak up, regularly asking staff about safety concerns, highlighting “good catches” when harm is avoided, and using staff feedback to initiate improvements.4,7,13Finally, in the authors’ experience, psychological safety requires clear commitment from leaders at all levels of an organization. Communication should be bidirectional, and leaders should close the proverbial “loop” with feedback and timely follow-up. This encourages and reinforces staff engagement and speaking up behaviors.2,4,7,13
Open Communication
Promoting an environment of open communication, where all individuals and teams feel empowered to speak up with questions, concerns, and recommendations—regardless of position within the organization—is critical to psychological safety.4,6,9 Open communication is especially critical when processes and systems are constantly changing and advancing as a result of new information and technology.9 Promoting open, bidirectional communication during the delivery of patient care can be accomplished with huddles, tiered safety huddles, leader rounding for high reliability, and time-outs.2,4,6 These opportunities allow team members to discuss concerns, identify resources that support safe, high-quality care; reflect on successes and opportunities for improvement; and circle back on concerns.2,6 Open communication in psychologically safe environments empowers staff to raise patient care concerns and is instrumental for improving patient safety, increasing staff job satisfaction, and decreasing turnover.6,14
Education and Training
Education and training for all staff—from the frontline to the executive level—are essential to successfully implementing the principles and practices of psychological safety.5-7 VHA training covers many topics, including the origins, benefits, and implementation strategies of psychological safety (Table). Role-playing simulation is an effective teaching format, providing staff with opportunities to practice techniques for raising concerns or share feedback in a controlled environment.6 In addition, education should be ongoing; it helps leaders and staff members feel competent and confident when implementing psychological safety across the health care organization.6,10
Accountability
The final critical strategy for achieving psychological safety is accountability. It is the responsibility of all leadership—from senior leaders to clinical and nonclinical managers—to create a culture of shared accountability.5 But first, expectations must be set. Leadership must establish well-defined behavioral expectations that align with the organization’s values. Understanding behavioral expectations will help to ensure that employees know what achievement looks like, as well as how they are being held accountable for their individual actions.4,5,7 In practical terms, this means ensuring that staff members have the skills and resources to achieve goals and expectations, providing performance feedback in a timely manner, and including expectations in annual performance evaluations (as they are in the VHA).
Consistency is key. Accountability should be the expectation across all levels and services of the health care organization. No staff member should be exempt from promoting a psychologically safe work environment. Compliance with behavioral expectations should be monitored and if a person’s actions are not consistent with expectations, the situation will need to be addressed. Interventions will depend on the type, severity, and frequency of the problematic behaviors. Depending on an organization’s policies and practices, courses of action can range from feedback counseling to employment termination.5
A practical matter in ensuring accountability is implementing a psychologically safe process for reporting concerns. Staff members must feel comfortable reporting behavioral concerns without fear of retaliation, negative judgment, or consequences from peers and supervisors. One method for doing this is to create a confidential, centralized process for reporting concerns.5
First-Hand Results
VAPHCS has seen the results of implementing the strategies outlined here. For example, VAPHCS has observed a 45% increase in the use of the patient safety reporting system that logs medical errors and near-misses. In addition, there have been improvements in levels of psychological safety and patient safety reported in the annual VHA All Employee Survey, which is conducted annually to gauge workplace satisfaction, culture, climate, turnover, supervisory behaviors, and general workplace perceptions. VAPHCS has shown consistent improvements in 12 patient safety elements scored on a 5-point scale (1, very dissatisfied; 5, very satisfied) (Figure). Notably, employee ratings of error prevention discussed increased from 4.0 in 2022 to 4.3 in 2024. Data collection and analysis are ongoing; more comprehensive findings will be published in the future.
CONCLUSIONS
Health care organizations are increasingly recognizing the importance of psychologically safe workplaces in order to provide safe, high-quality patient care. Psychological safety is a critical tool for empowering staff to raise concerns, ask tough questions, challenge the status quo, and share new ideas for providing health care services. While psychological safety has been slowly adopted in health care, it’s clear that evidence-based strategies can make psychological safety a reality.
- Spanos S, Leask E, Patel R, Datyner M, Loh E, Braithwaite J. Healthcare leaders navigating complexity: A scoping review of key trends in future roles and competencies. BMC Med Educ. 2024;24(1):720. doi:10.1186/s12909-024-05689-4
- Murray JS, Baghdadi A, Dannenberg W, Crews P, Walsh ND. The role of high reliability organization foundational practices in building a culture of safety. Fed Pract. 2024;41(7):214-221. doi:10.12788/fp.0486
- Bransby DP, Kerrissey M, Edmondson AC. Paradise lost (and restored?): a study of psychological safety over time. Acad Manag Discov. Published online March 14, 2024. doi:10.5465/amd.2023.0084
- Murray JS, Kelly S, Hanover C. Promoting psychological safety in healthcare organizations. Mil Med. 2022;187(7-8):808-810. doi:10.1093/milmed/usac041
- Jamal N, Young VN, Shapiro J, Brenner MJ, Schmalbach CE. Patient safety/quality improvement primer, part IV: Psychological safety-drivers to outcomes and well-being. Otolaryngol Head Neck Surg. 2023;168(4):881-888. doi:10.1177/01945998221126966
- Sarofim M. Psychological safety in medicine: What is it, and who cares? Med J Aust. 2024;220(8):398-399. doi:10.5694/mja2.52263
- Edmondson AC, Bransby DP. Psychological safety comes of age: Observed themes in an established literature. Annu Rev Organ Psychol Organ Behav. 2023;10:55-78. doi.org/10.1146/annurev-orgpsych-120920-055217
- Kumar S. Psychological safety: What it is, why teams need it, and how to make it flourish. Chest. 2024; 165(4):942-949. doi:10.1016/j.chest.2023.11.016
- Hallam KT, Popovic N, Karimi L. Identifying the key elements of psychologically safe workplaces in healthcare settings. Brain Sci. 2023;13(10):1450. doi:10.3390/brainsci13101450
- Grailey KE, Murray E, Reader T, Brett SJ. The presence and potential impact of psychological safety in the healthcare setting: an evidence synthesis. BMC Health Serv Res. 2021;21(1):773. doi:10.1186/s12913-021-06740-6
- US Department of Veterans Affairs. VISN 1: VA New England Healthcare System. Accessed March 25, 2025. https://department.va.gov/integrated-service-networks/visn-01
- Brimhall KC, Tsai CY, Eckardt R, Dionne S, Yang B, Sharp A. The effects of leadership for self-worth, inclusion, trust, and psychological safety on medical error reporting. Health Care Manage Rev. 2023;48(2):120-129. doi:10.1097/HMR.0000000000000358
- Adair KC, Heath A, Frye MA, et al. The Psychological Safety Scale of the Safety, Communication, Operational, Reliability, and Engagement (SCORE) Survey: a brief, diagnostic, and actionable metric for the ability to speak up in healthcare settings. J Patient Saf. 2022;18(6):513-520. doi:10.1097/PTS.0000000000001048
- Cho H, Steege LM, Arsenault Knudsen ÉN. Psychological safety, communication openness, nurse job outcomes, and patient safety in hospital nurses. Res Nurs Health. 2023;46(4):445-453.
- Practical Tool 2: 5 minute psychological safety audit. Accessed March 25, 2025. https://www.educationsupport.org.uk/media/jlnf3cju/practical-tool-2-psychological-safety-audit.pdf
- Spanos S, Leask E, Patel R, Datyner M, Loh E, Braithwaite J. Healthcare leaders navigating complexity: A scoping review of key trends in future roles and competencies. BMC Med Educ. 2024;24(1):720. doi:10.1186/s12909-024-05689-4
- Murray JS, Baghdadi A, Dannenberg W, Crews P, Walsh ND. The role of high reliability organization foundational practices in building a culture of safety. Fed Pract. 2024;41(7):214-221. doi:10.12788/fp.0486
- Bransby DP, Kerrissey M, Edmondson AC. Paradise lost (and restored?): a study of psychological safety over time. Acad Manag Discov. Published online March 14, 2024. doi:10.5465/amd.2023.0084
- Murray JS, Kelly S, Hanover C. Promoting psychological safety in healthcare organizations. Mil Med. 2022;187(7-8):808-810. doi:10.1093/milmed/usac041
- Jamal N, Young VN, Shapiro J, Brenner MJ, Schmalbach CE. Patient safety/quality improvement primer, part IV: Psychological safety-drivers to outcomes and well-being. Otolaryngol Head Neck Surg. 2023;168(4):881-888. doi:10.1177/01945998221126966
- Sarofim M. Psychological safety in medicine: What is it, and who cares? Med J Aust. 2024;220(8):398-399. doi:10.5694/mja2.52263
- Edmondson AC, Bransby DP. Psychological safety comes of age: Observed themes in an established literature. Annu Rev Organ Psychol Organ Behav. 2023;10:55-78. doi.org/10.1146/annurev-orgpsych-120920-055217
- Kumar S. Psychological safety: What it is, why teams need it, and how to make it flourish. Chest. 2024; 165(4):942-949. doi:10.1016/j.chest.2023.11.016
- Hallam KT, Popovic N, Karimi L. Identifying the key elements of psychologically safe workplaces in healthcare settings. Brain Sci. 2023;13(10):1450. doi:10.3390/brainsci13101450
- Grailey KE, Murray E, Reader T, Brett SJ. The presence and potential impact of psychological safety in the healthcare setting: an evidence synthesis. BMC Health Serv Res. 2021;21(1):773. doi:10.1186/s12913-021-06740-6
- US Department of Veterans Affairs. VISN 1: VA New England Healthcare System. Accessed March 25, 2025. https://department.va.gov/integrated-service-networks/visn-01
- Brimhall KC, Tsai CY, Eckardt R, Dionne S, Yang B, Sharp A. The effects of leadership for self-worth, inclusion, trust, and psychological safety on medical error reporting. Health Care Manage Rev. 2023;48(2):120-129. doi:10.1097/HMR.0000000000000358
- Adair KC, Heath A, Frye MA, et al. The Psychological Safety Scale of the Safety, Communication, Operational, Reliability, and Engagement (SCORE) Survey: a brief, diagnostic, and actionable metric for the ability to speak up in healthcare settings. J Patient Saf. 2022;18(6):513-520. doi:10.1097/PTS.0000000000001048
- Cho H, Steege LM, Arsenault Knudsen ÉN. Psychological safety, communication openness, nurse job outcomes, and patient safety in hospital nurses. Res Nurs Health. 2023;46(4):445-453.
- Practical Tool 2: 5 minute psychological safety audit. Accessed March 25, 2025. https://www.educationsupport.org.uk/media/jlnf3cju/practical-tool-2-psychological-safety-audit.pdf
Achieving Psychological Safety in High Reliability Organizations
Achieving Psychological Safety in High Reliability Organizations
HHS Cuts Thousands of Jobs, Eliminates Entire Services
On March 27, Health and Human Services (HHS) Secretary Robert F. Kennedy Jr. said he planned to cut about 10,000 full-time jobs from the department in a sweeping “reorganization.” Less than a week later, the reduction in force (RIF) notifications were sent out, and in the very early hours of April 1, hundreds of employees found themselves locked out from their offices, often so abruptly their belongings were left behind.
Most affected employees were told they would be placed on administrative leave; some were told to continue working until they can hand off their duties but they would be formally separated on June 2. Many of the email RIF notifications used the recommended wording provided by the US Office of Personnel Management: “This RIF action does not reflect directly on your service, performance, or conduct.”
"The Trump Administration has launched an unprecedented attack on the federal health workforce," said House Energy and Commerce Committee Ranking Member Frank Pallone, Jr. (D-NJ), during an oversight and investigations hearing on medical device technology and cybersecurity.
The cuts in personnel and programs are broad and deep, and touch every aspect of public health. Alzheimer’s disease programs are being eliminated, measles vaccine clinics are being shuttered, and tuberculosis, HIV prevention, and cancer research are being stalled. A Reddit thread for RIF notices from HHS employees had nearly 750 postings, suggesting a broad cross-section of individuals and departments had received them.
Secretary Kennedy stated the layoffs and restructuring will save $1.8 billion a year. “We aren’t just reducing bureaucratic sprawl," he said in a statement. "We are realigning the organization with its core mission and our new priorities in reversing the chronic disease epidemic.” On the social platform X, Kennedy acknowledged, “This will be a painful period for HHS.”
Entire offices devoted to Freedom of Information Act-related requests, communications, and human resources were also shut down, according to multiple reports.
The agency's 28 divisions will be reformatted into a “new, unified entity” of 15 divisions—the Administration for a Healthy America, or AHA, aimed at carrying out Kennedy's “Make America Healthy Again” agenda. The AHA will include the Substance Abuse and Mental Health Services Administration (SAMHSA), the Agency for Toxic Substances and Disease Registry, and the National Institute for Occupational Safety and Health. The Administration for Community Living's functions will shift into the Centers for Medicare and Medicaid Services, the Administration for Children and Families, and the Assistant Secretary for Planning and Evaluation (ASPE). ASPE will be combined with the Agency for Health Research and Quality into the Office of Strategy
“This centralization,” HHS says, “will improve coordination of health resources for low-income Americans and will focus on areas including, Primary Care, Maternal and Child Health, Mental Health, Environmental Health, HIV/AIDS, and Workforce development.”
US Food and Drug Administration
An estimated 3500 full-time FDA employees are expected to receive RIF notices. The agency said reductions will not affect drug, medical device, or food reviewers or inspectors.
Politico spoke with fired employees on condition of anonymity. According to them, Dr. Peter Stein, director of the FDA Office of New Drugs (OND), was let go. The policy office inside of OND was also eliminated. Another top FDA regulator, Dr. Brian King, the director of the Center for Tobacco Products (CTP), was placed on administrative leave, according to an email sent to his staff and obtained by Politico. “I encourage you to hold your heads high and never compromise the guiding tenets that CTP has held dear since its inception,” King wrote in the email to his staff. “We obeyed the law. We followed the science. We told the truth.”
Julie Tierney, who was recently elevated to acting director of the FDA Center for Biologics Evaluation and Research, according to an agency website, was also placed on administrative leave, according to 2 people familiar with the decision. The FDA Office of Strategic Programs, including its director, Sridhar Mantha, has been completely shuttered. Mantha cochaired the Artificial Intelligence (AI) Council at the Center for Drug Evaluation and Research (CDER), which helped develop policy around AI use in drug development and assisted the FDA in using AI internally.
Centers for Disease Control and Prevention
About 2400 CDC employees are expected to receive RIF notices. According to Government Executive, the National Institute for Occupational Safety and Health (NIOSH) sustained more than one-third of the cuts at CDC. About 80% of the 1100 employees at the institute were laid off, including its director and deputy director. An HHS letter to a labor union said about 185 NIOSH employees would be let go in just the Morgantown, W. Va., location. However, NIOSH is apparently slated to be part of the newly created AHA.
Other layoffs hit the National Center for Chronic Disease Prevention and Health Promotion; National Center for Injury Prevention and Control; the National Center for HIV, Viral Hepatitis, STD, and Tuberculosis Prevention; the Global Health Center; the National Center on Birth Defects and Developmental Disabilities; and the National Center for Environmental Health. Two sources familiar with the firings said the Office on Smoking and Health was eliminated. The Administration for Strategic Preparedness and Response, currently part of the US Public Health Service, will move to the CDC.
A compensation program for employees who developed cancer due to radiation exposure while working for the federal government was also eliminated. Similarly, a national registry that tracks rates of cancer among firefighters was cut. One employee said NIOSH laid off veterinarians despite the bureau having laboratory animals that need care.
National Institutes of Health
NIH will lose 1200 employees, due to "centralizing” procurement, human resources, and communications across its 27 institutes and centers. According to the employees who spoke with Politico, scientists were also targeted, including National Institute of Nursing Research Director Shannon Zenk; National Institute of Child Health and Human Development Director Diana Bianchi; Emily Erbelding, who leads the Division of Microbiology and Infectious Diseases at National Institutes of Allergy and Infectious Diseases; and National Institute on Minority Health and Health Disparities Director Eliseo Pérez-Stable. National Institute of Allergy and Infectious Diseases Director Jeanne Marrazzo, who replaced Anthony Fauci, was also put on leave.
In his “welcome” email to staff, the new NIH director, Dr. Jay Bhattacharya, wrote: “I recognize that I am joining NIH at a time of tremendous change. Every inch of the federal government is under scrutiny—and NIH is not exempt. These reductions in the workforce will have a profound impact on key NIH administrative functions, including communications, legislative affairs, procurement, and human resources, and will require an entirely new approach to how we carry them out.”
Deep Cuts at Other HHS Agencies
As many as 500 to 600 people were let go at the Health Resource and Services Administration (HRSA). Its Bureau of Primary Health Care, which oversees the national network of health care centers that collectively provide care to 31 million people, was “severely impacted,” and the agency lost much of its regional staff, according to an article in Government Executive. “This will have an enormous impact on the program and viability of health centers,” an HRSA employee said.
About 50% of the nearly 900 SAMHSA employees were laid off and its 10 regional offices were closed. SAMHSA will be “hamstrung for data,” according to an agency employee, who added contracts may be cut en masse due the departure of the contract management staff. They added that even if funding remains for the agency, the support systems for grantees were being decimated.
More than 800 people lost their jobs at the CDER, according to an official who was laid off; this part of the agency had around 6,000 employees before the cuts.
Indian Health Service
The IHS offers a rare bright spot. Although it was also in line for massive cuts, it has been spared, for now. According to a statement emailed to Native News Online, Secretary Kennedy said the Trump administration intends to prioritize the IHS.
“The Indian Health Service has always been treated as the redheaded stepchild at HHS,” Secretary Kennedy wrote. “My father often complained that IHS was chronically understaffed and underfunded. President Trump wants me to rectify this sad history. Indians suffer at the highest level of chronic disease of any demographic. IHS will be a priority over the next 4 years. President Trump wants me to end the chronic disease epidemic beginning in Indian country.”
March layoffs that had been announced for 1000 IHS employees were rescinded.
“We can confirm the layoffs were rescinded thanks at least in part to advocacy by the many Tribal organizations,” a spokesperson for the National Indian Health Board told Native News Online.
In fact, top career executives across the department are now being offered reassignments to the IHS, which employees must accept to keep their jobs. One executive who received the offer told Native News Online that no details on positions or location were provided, and they doubted that everyone who got such a notice would ultimately be matched to a suitable position.
"Streamline the Agency"
The dramatic actions at HHS were not unexpected. In fact, employees had been in an unsettling limbo since Kennedy was appointed Secretary, not knowing when the axe would fall, or where, or on whom. Kennedy, when describing the restructuring plans, said, “We're going to streamline our agency and eliminate the redundancies and invite everyone to align behind a simple, bold mission. I want every HHS employee to wake up every morning asking themselves, ‘What can I do to restore American Health?’ I want to empower everyone in the HHS family to have a sense of purpose and pride and a sense of personal agency and responsibility to this larger goal.”
“The FDA as we've known it is finished,” Dr. Robert M. Califf, who served as FDA commissioner twice, wrote on LinkedIn. In an interview with CNN, Califf said he was dismayed to see how federal workers were being treated.
“This is a sad and inhumane way to treat people,” he said. “It’s different when you’re a company and you’re out of money and you can’t pay people, but the federal government can pay people and do things in an orderly, respectful fashion—and not have them end up in line trying to get to work and have their badges not work as a way to fire them.”
But the fired HHS employees aren’t the only ones who will bear the brunt of the cuts. “Today’s announcement is not just a restructuring of the Department of Health and Human Services. It is a catastrophe for the health care of every American,” Senator Ed Markey (D-MA) said in a press briefing.
Calling the cuts “a recipe for disaster,” former CDC director Tom Frieden said, “[Secretary] Kennedy claims that health care services will not be harmed by the dramatic downsizing, but he is wrong, and everyone who is paying any attention knows it.”
Senators Bill Cassidy (R-LA) and Bernie Sanders (I-VT), of the Senate Health, Education, Labor, and Pensions Committee, announced Tuesday that they were inviting Kennedy to a hearing April 10 about the restructuring of HHS. “This will be a good opportunity,” Cassidy said in a statement, “for him to set the record straight and speak to the goals, structure and benefits of the proposed reorganization.”
On March 27, Health and Human Services (HHS) Secretary Robert F. Kennedy Jr. said he planned to cut about 10,000 full-time jobs from the department in a sweeping “reorganization.” Less than a week later, the reduction in force (RIF) notifications were sent out, and in the very early hours of April 1, hundreds of employees found themselves locked out from their offices, often so abruptly their belongings were left behind.
Most affected employees were told they would be placed on administrative leave; some were told to continue working until they can hand off their duties but they would be formally separated on June 2. Many of the email RIF notifications used the recommended wording provided by the US Office of Personnel Management: “This RIF action does not reflect directly on your service, performance, or conduct.”
"The Trump Administration has launched an unprecedented attack on the federal health workforce," said House Energy and Commerce Committee Ranking Member Frank Pallone, Jr. (D-NJ), during an oversight and investigations hearing on medical device technology and cybersecurity.
The cuts in personnel and programs are broad and deep, and touch every aspect of public health. Alzheimer’s disease programs are being eliminated, measles vaccine clinics are being shuttered, and tuberculosis, HIV prevention, and cancer research are being stalled. A Reddit thread for RIF notices from HHS employees had nearly 750 postings, suggesting a broad cross-section of individuals and departments had received them.
Secretary Kennedy stated the layoffs and restructuring will save $1.8 billion a year. “We aren’t just reducing bureaucratic sprawl," he said in a statement. "We are realigning the organization with its core mission and our new priorities in reversing the chronic disease epidemic.” On the social platform X, Kennedy acknowledged, “This will be a painful period for HHS.”
Entire offices devoted to Freedom of Information Act-related requests, communications, and human resources were also shut down, according to multiple reports.
The agency's 28 divisions will be reformatted into a “new, unified entity” of 15 divisions—the Administration for a Healthy America, or AHA, aimed at carrying out Kennedy's “Make America Healthy Again” agenda. The AHA will include the Substance Abuse and Mental Health Services Administration (SAMHSA), the Agency for Toxic Substances and Disease Registry, and the National Institute for Occupational Safety and Health. The Administration for Community Living's functions will shift into the Centers for Medicare and Medicaid Services, the Administration for Children and Families, and the Assistant Secretary for Planning and Evaluation (ASPE). ASPE will be combined with the Agency for Health Research and Quality into the Office of Strategy
“This centralization,” HHS says, “will improve coordination of health resources for low-income Americans and will focus on areas including, Primary Care, Maternal and Child Health, Mental Health, Environmental Health, HIV/AIDS, and Workforce development.”
US Food and Drug Administration
An estimated 3500 full-time FDA employees are expected to receive RIF notices. The agency said reductions will not affect drug, medical device, or food reviewers or inspectors.
Politico spoke with fired employees on condition of anonymity. According to them, Dr. Peter Stein, director of the FDA Office of New Drugs (OND), was let go. The policy office inside of OND was also eliminated. Another top FDA regulator, Dr. Brian King, the director of the Center for Tobacco Products (CTP), was placed on administrative leave, according to an email sent to his staff and obtained by Politico. “I encourage you to hold your heads high and never compromise the guiding tenets that CTP has held dear since its inception,” King wrote in the email to his staff. “We obeyed the law. We followed the science. We told the truth.”
Julie Tierney, who was recently elevated to acting director of the FDA Center for Biologics Evaluation and Research, according to an agency website, was also placed on administrative leave, according to 2 people familiar with the decision. The FDA Office of Strategic Programs, including its director, Sridhar Mantha, has been completely shuttered. Mantha cochaired the Artificial Intelligence (AI) Council at the Center for Drug Evaluation and Research (CDER), which helped develop policy around AI use in drug development and assisted the FDA in using AI internally.
Centers for Disease Control and Prevention
About 2400 CDC employees are expected to receive RIF notices. According to Government Executive, the National Institute for Occupational Safety and Health (NIOSH) sustained more than one-third of the cuts at CDC. About 80% of the 1100 employees at the institute were laid off, including its director and deputy director. An HHS letter to a labor union said about 185 NIOSH employees would be let go in just the Morgantown, W. Va., location. However, NIOSH is apparently slated to be part of the newly created AHA.
Other layoffs hit the National Center for Chronic Disease Prevention and Health Promotion; National Center for Injury Prevention and Control; the National Center for HIV, Viral Hepatitis, STD, and Tuberculosis Prevention; the Global Health Center; the National Center on Birth Defects and Developmental Disabilities; and the National Center for Environmental Health. Two sources familiar with the firings said the Office on Smoking and Health was eliminated. The Administration for Strategic Preparedness and Response, currently part of the US Public Health Service, will move to the CDC.
A compensation program for employees who developed cancer due to radiation exposure while working for the federal government was also eliminated. Similarly, a national registry that tracks rates of cancer among firefighters was cut. One employee said NIOSH laid off veterinarians despite the bureau having laboratory animals that need care.
National Institutes of Health
NIH will lose 1200 employees, due to "centralizing” procurement, human resources, and communications across its 27 institutes and centers. According to the employees who spoke with Politico, scientists were also targeted, including National Institute of Nursing Research Director Shannon Zenk; National Institute of Child Health and Human Development Director Diana Bianchi; Emily Erbelding, who leads the Division of Microbiology and Infectious Diseases at National Institutes of Allergy and Infectious Diseases; and National Institute on Minority Health and Health Disparities Director Eliseo Pérez-Stable. National Institute of Allergy and Infectious Diseases Director Jeanne Marrazzo, who replaced Anthony Fauci, was also put on leave.
In his “welcome” email to staff, the new NIH director, Dr. Jay Bhattacharya, wrote: “I recognize that I am joining NIH at a time of tremendous change. Every inch of the federal government is under scrutiny—and NIH is not exempt. These reductions in the workforce will have a profound impact on key NIH administrative functions, including communications, legislative affairs, procurement, and human resources, and will require an entirely new approach to how we carry them out.”
Deep Cuts at Other HHS Agencies
As many as 500 to 600 people were let go at the Health Resource and Services Administration (HRSA). Its Bureau of Primary Health Care, which oversees the national network of health care centers that collectively provide care to 31 million people, was “severely impacted,” and the agency lost much of its regional staff, according to an article in Government Executive. “This will have an enormous impact on the program and viability of health centers,” an HRSA employee said.
About 50% of the nearly 900 SAMHSA employees were laid off and its 10 regional offices were closed. SAMHSA will be “hamstrung for data,” according to an agency employee, who added contracts may be cut en masse due the departure of the contract management staff. They added that even if funding remains for the agency, the support systems for grantees were being decimated.
More than 800 people lost their jobs at the CDER, according to an official who was laid off; this part of the agency had around 6,000 employees before the cuts.
Indian Health Service
The IHS offers a rare bright spot. Although it was also in line for massive cuts, it has been spared, for now. According to a statement emailed to Native News Online, Secretary Kennedy said the Trump administration intends to prioritize the IHS.
“The Indian Health Service has always been treated as the redheaded stepchild at HHS,” Secretary Kennedy wrote. “My father often complained that IHS was chronically understaffed and underfunded. President Trump wants me to rectify this sad history. Indians suffer at the highest level of chronic disease of any demographic. IHS will be a priority over the next 4 years. President Trump wants me to end the chronic disease epidemic beginning in Indian country.”
March layoffs that had been announced for 1000 IHS employees were rescinded.
“We can confirm the layoffs were rescinded thanks at least in part to advocacy by the many Tribal organizations,” a spokesperson for the National Indian Health Board told Native News Online.
In fact, top career executives across the department are now being offered reassignments to the IHS, which employees must accept to keep their jobs. One executive who received the offer told Native News Online that no details on positions or location were provided, and they doubted that everyone who got such a notice would ultimately be matched to a suitable position.
"Streamline the Agency"
The dramatic actions at HHS were not unexpected. In fact, employees had been in an unsettling limbo since Kennedy was appointed Secretary, not knowing when the axe would fall, or where, or on whom. Kennedy, when describing the restructuring plans, said, “We're going to streamline our agency and eliminate the redundancies and invite everyone to align behind a simple, bold mission. I want every HHS employee to wake up every morning asking themselves, ‘What can I do to restore American Health?’ I want to empower everyone in the HHS family to have a sense of purpose and pride and a sense of personal agency and responsibility to this larger goal.”
“The FDA as we've known it is finished,” Dr. Robert M. Califf, who served as FDA commissioner twice, wrote on LinkedIn. In an interview with CNN, Califf said he was dismayed to see how federal workers were being treated.
“This is a sad and inhumane way to treat people,” he said. “It’s different when you’re a company and you’re out of money and you can’t pay people, but the federal government can pay people and do things in an orderly, respectful fashion—and not have them end up in line trying to get to work and have their badges not work as a way to fire them.”
But the fired HHS employees aren’t the only ones who will bear the brunt of the cuts. “Today’s announcement is not just a restructuring of the Department of Health and Human Services. It is a catastrophe for the health care of every American,” Senator Ed Markey (D-MA) said in a press briefing.
Calling the cuts “a recipe for disaster,” former CDC director Tom Frieden said, “[Secretary] Kennedy claims that health care services will not be harmed by the dramatic downsizing, but he is wrong, and everyone who is paying any attention knows it.”
Senators Bill Cassidy (R-LA) and Bernie Sanders (I-VT), of the Senate Health, Education, Labor, and Pensions Committee, announced Tuesday that they were inviting Kennedy to a hearing April 10 about the restructuring of HHS. “This will be a good opportunity,” Cassidy said in a statement, “for him to set the record straight and speak to the goals, structure and benefits of the proposed reorganization.”
On March 27, Health and Human Services (HHS) Secretary Robert F. Kennedy Jr. said he planned to cut about 10,000 full-time jobs from the department in a sweeping “reorganization.” Less than a week later, the reduction in force (RIF) notifications were sent out, and in the very early hours of April 1, hundreds of employees found themselves locked out from their offices, often so abruptly their belongings were left behind.
Most affected employees were told they would be placed on administrative leave; some were told to continue working until they can hand off their duties but they would be formally separated on June 2. Many of the email RIF notifications used the recommended wording provided by the US Office of Personnel Management: “This RIF action does not reflect directly on your service, performance, or conduct.”
"The Trump Administration has launched an unprecedented attack on the federal health workforce," said House Energy and Commerce Committee Ranking Member Frank Pallone, Jr. (D-NJ), during an oversight and investigations hearing on medical device technology and cybersecurity.
The cuts in personnel and programs are broad and deep, and touch every aspect of public health. Alzheimer’s disease programs are being eliminated, measles vaccine clinics are being shuttered, and tuberculosis, HIV prevention, and cancer research are being stalled. A Reddit thread for RIF notices from HHS employees had nearly 750 postings, suggesting a broad cross-section of individuals and departments had received them.
Secretary Kennedy stated the layoffs and restructuring will save $1.8 billion a year. “We aren’t just reducing bureaucratic sprawl," he said in a statement. "We are realigning the organization with its core mission and our new priorities in reversing the chronic disease epidemic.” On the social platform X, Kennedy acknowledged, “This will be a painful period for HHS.”
Entire offices devoted to Freedom of Information Act-related requests, communications, and human resources were also shut down, according to multiple reports.
The agency's 28 divisions will be reformatted into a “new, unified entity” of 15 divisions—the Administration for a Healthy America, or AHA, aimed at carrying out Kennedy's “Make America Healthy Again” agenda. The AHA will include the Substance Abuse and Mental Health Services Administration (SAMHSA), the Agency for Toxic Substances and Disease Registry, and the National Institute for Occupational Safety and Health. The Administration for Community Living's functions will shift into the Centers for Medicare and Medicaid Services, the Administration for Children and Families, and the Assistant Secretary for Planning and Evaluation (ASPE). ASPE will be combined with the Agency for Health Research and Quality into the Office of Strategy
“This centralization,” HHS says, “will improve coordination of health resources for low-income Americans and will focus on areas including, Primary Care, Maternal and Child Health, Mental Health, Environmental Health, HIV/AIDS, and Workforce development.”
US Food and Drug Administration
An estimated 3500 full-time FDA employees are expected to receive RIF notices. The agency said reductions will not affect drug, medical device, or food reviewers or inspectors.
Politico spoke with fired employees on condition of anonymity. According to them, Dr. Peter Stein, director of the FDA Office of New Drugs (OND), was let go. The policy office inside of OND was also eliminated. Another top FDA regulator, Dr. Brian King, the director of the Center for Tobacco Products (CTP), was placed on administrative leave, according to an email sent to his staff and obtained by Politico. “I encourage you to hold your heads high and never compromise the guiding tenets that CTP has held dear since its inception,” King wrote in the email to his staff. “We obeyed the law. We followed the science. We told the truth.”
Julie Tierney, who was recently elevated to acting director of the FDA Center for Biologics Evaluation and Research, according to an agency website, was also placed on administrative leave, according to 2 people familiar with the decision. The FDA Office of Strategic Programs, including its director, Sridhar Mantha, has been completely shuttered. Mantha cochaired the Artificial Intelligence (AI) Council at the Center for Drug Evaluation and Research (CDER), which helped develop policy around AI use in drug development and assisted the FDA in using AI internally.
Centers for Disease Control and Prevention
About 2400 CDC employees are expected to receive RIF notices. According to Government Executive, the National Institute for Occupational Safety and Health (NIOSH) sustained more than one-third of the cuts at CDC. About 80% of the 1100 employees at the institute were laid off, including its director and deputy director. An HHS letter to a labor union said about 185 NIOSH employees would be let go in just the Morgantown, W. Va., location. However, NIOSH is apparently slated to be part of the newly created AHA.
Other layoffs hit the National Center for Chronic Disease Prevention and Health Promotion; National Center for Injury Prevention and Control; the National Center for HIV, Viral Hepatitis, STD, and Tuberculosis Prevention; the Global Health Center; the National Center on Birth Defects and Developmental Disabilities; and the National Center for Environmental Health. Two sources familiar with the firings said the Office on Smoking and Health was eliminated. The Administration for Strategic Preparedness and Response, currently part of the US Public Health Service, will move to the CDC.
A compensation program for employees who developed cancer due to radiation exposure while working for the federal government was also eliminated. Similarly, a national registry that tracks rates of cancer among firefighters was cut. One employee said NIOSH laid off veterinarians despite the bureau having laboratory animals that need care.
National Institutes of Health
NIH will lose 1200 employees, due to "centralizing” procurement, human resources, and communications across its 27 institutes and centers. According to the employees who spoke with Politico, scientists were also targeted, including National Institute of Nursing Research Director Shannon Zenk; National Institute of Child Health and Human Development Director Diana Bianchi; Emily Erbelding, who leads the Division of Microbiology and Infectious Diseases at National Institutes of Allergy and Infectious Diseases; and National Institute on Minority Health and Health Disparities Director Eliseo Pérez-Stable. National Institute of Allergy and Infectious Diseases Director Jeanne Marrazzo, who replaced Anthony Fauci, was also put on leave.
In his “welcome” email to staff, the new NIH director, Dr. Jay Bhattacharya, wrote: “I recognize that I am joining NIH at a time of tremendous change. Every inch of the federal government is under scrutiny—and NIH is not exempt. These reductions in the workforce will have a profound impact on key NIH administrative functions, including communications, legislative affairs, procurement, and human resources, and will require an entirely new approach to how we carry them out.”
Deep Cuts at Other HHS Agencies
As many as 500 to 600 people were let go at the Health Resource and Services Administration (HRSA). Its Bureau of Primary Health Care, which oversees the national network of health care centers that collectively provide care to 31 million people, was “severely impacted,” and the agency lost much of its regional staff, according to an article in Government Executive. “This will have an enormous impact on the program and viability of health centers,” an HRSA employee said.
About 50% of the nearly 900 SAMHSA employees were laid off and its 10 regional offices were closed. SAMHSA will be “hamstrung for data,” according to an agency employee, who added contracts may be cut en masse due the departure of the contract management staff. They added that even if funding remains for the agency, the support systems for grantees were being decimated.
More than 800 people lost their jobs at the CDER, according to an official who was laid off; this part of the agency had around 6,000 employees before the cuts.
Indian Health Service
The IHS offers a rare bright spot. Although it was also in line for massive cuts, it has been spared, for now. According to a statement emailed to Native News Online, Secretary Kennedy said the Trump administration intends to prioritize the IHS.
“The Indian Health Service has always been treated as the redheaded stepchild at HHS,” Secretary Kennedy wrote. “My father often complained that IHS was chronically understaffed and underfunded. President Trump wants me to rectify this sad history. Indians suffer at the highest level of chronic disease of any demographic. IHS will be a priority over the next 4 years. President Trump wants me to end the chronic disease epidemic beginning in Indian country.”
March layoffs that had been announced for 1000 IHS employees were rescinded.
“We can confirm the layoffs were rescinded thanks at least in part to advocacy by the many Tribal organizations,” a spokesperson for the National Indian Health Board told Native News Online.
In fact, top career executives across the department are now being offered reassignments to the IHS, which employees must accept to keep their jobs. One executive who received the offer told Native News Online that no details on positions or location were provided, and they doubted that everyone who got such a notice would ultimately be matched to a suitable position.
"Streamline the Agency"
The dramatic actions at HHS were not unexpected. In fact, employees had been in an unsettling limbo since Kennedy was appointed Secretary, not knowing when the axe would fall, or where, or on whom. Kennedy, when describing the restructuring plans, said, “We're going to streamline our agency and eliminate the redundancies and invite everyone to align behind a simple, bold mission. I want every HHS employee to wake up every morning asking themselves, ‘What can I do to restore American Health?’ I want to empower everyone in the HHS family to have a sense of purpose and pride and a sense of personal agency and responsibility to this larger goal.”
“The FDA as we've known it is finished,” Dr. Robert M. Califf, who served as FDA commissioner twice, wrote on LinkedIn. In an interview with CNN, Califf said he was dismayed to see how federal workers were being treated.
“This is a sad and inhumane way to treat people,” he said. “It’s different when you’re a company and you’re out of money and you can’t pay people, but the federal government can pay people and do things in an orderly, respectful fashion—and not have them end up in line trying to get to work and have their badges not work as a way to fire them.”
But the fired HHS employees aren’t the only ones who will bear the brunt of the cuts. “Today’s announcement is not just a restructuring of the Department of Health and Human Services. It is a catastrophe for the health care of every American,” Senator Ed Markey (D-MA) said in a press briefing.
Calling the cuts “a recipe for disaster,” former CDC director Tom Frieden said, “[Secretary] Kennedy claims that health care services will not be harmed by the dramatic downsizing, but he is wrong, and everyone who is paying any attention knows it.”
Senators Bill Cassidy (R-LA) and Bernie Sanders (I-VT), of the Senate Health, Education, Labor, and Pensions Committee, announced Tuesday that they were inviting Kennedy to a hearing April 10 about the restructuring of HHS. “This will be a good opportunity,” Cassidy said in a statement, “for him to set the record straight and speak to the goals, structure and benefits of the proposed reorganization.”

VA Shake-up Disrupts Mental Health Services for Some US Veterans
SAN FRANCISCO (Reuters) — Joey Cortez, who served 24 years in the US Air Force, had been waiting since August to see a mental health specialist from the Department of Veterans’ Affairs, when he experienced a fresh jolt of anxiety.
Cortez was fired last month from his human resources job at the agency - one of about 2400 employees who lost their jobs at Veterans’ Affairs (VA) in the first wave of President Donald Trump’s efforts to shrink the federal workforce.
“Once the firings happened and I was terminated, I started having panic attacks to the point where I black out,” Cortez, who suffers from post-traumatic stress disorder, told Reuters. The layoff is also making it harder to maintain his sobriety, as a recovering alcoholic.
“Not a day has gone by since I was fired that I haven’t thought about picking up a bottle,” said Cortez.
After losing his job, Cortez asked the VA to expedite his wait for a therapist and was told there was no record of his request, he said. After a month of calls to the agency, he got an appointment for this August, one year after he started the process. Then the VA offered him an appointment next week because another patient had canceled.
The VA provides health care to 9.31 million US veterans at hundreds of medical centers, clinics, and nursing homes across the country.
It also faces complex problems.
“The VA has bloat. There are redundancies. There are places where we have questioned the administration of care and asked, does it need to be the way it is?” Pat Murray, the legislative director for the Veterans of Foreign Wars, which represents Americans who have fought overseas, said in an interview.
The Trump administration plans additional cuts to the VA of more than 80,000 personnel, according to an internal memo obtained by Reuters. The agency has also announced it is phasing out telework.
Reuters spoke to nine current and former VA employees in California, Oregon, Texas, and the Washington D.C. area who said the changes were further disrupting some mental health services and fueling anxieties among those who provide and rely on them.
The VA employees — who include six mental health professionals and three people in leadership positions — described cancellations of some in-person and telehealth appointments; confusion over staffing of a crisis hot-line; and professionals conducting telehealth visits in makeshift meeting rooms inside VA buildings.
They spoke on the condition of anonymity, because they were not authorized to speak with the media.
STAFFING SHORTAGES
A former employee at the VA’s Office of Inspector General, who is also a veteran, said any future large-scale staffing cuts would likely worsen shortages and impact the quality of care.
“There’s no way to take a scalpel and do it appropriately that quickly,” he said.
VA spokesperson Peter Kasperowicz told Reuters mental health professionals, such as psychologists and social workers, were not included in February’s staffing cuts, and the agency is working to recruit mental health providers and improve wait times.
He did not specify how many support staff for these providers had been affected.
Last week, two federal judges ordered the VA and other federal agencies to reinstate thousands of fired probationary workers. Cortez’s pay was reinstated but he was told not to return to work.
The Veterans Health Administration, the branch of the VA that provides healthcare, has experienced severe staffing shortages since 2015, especially among mental health professionals, according to an OIG report last year.
Veterans often benefit from specialized services to treat anxiety, trauma, depression and substance abuse. The proportion of veterans receiving mental health services rose to 31% in 2022 from 20% in 2007, according to the VA. Suicide among veterans is twice the rate of Americans overall.
The VFW’s Murray said his organization supports a thorough review of the VA’s mental health services, but it needs to be done carefully, “not with a chainsaw.”
‘THE MOOD IS SO LOW’
In recent years, the agency had encouraged remote work to help expand access to telehealth services and reduce wait times, especially in rural areas where recruiting providers is difficult.
The VA’s Kasperowicz said that, while providers will need to return to VA facilities, veterans will be able to access telehealth appointments.
He did not directly address questions about why mental health providers needed to return to the office.
“The VA will make accommodations as needed to ensure employees have enough space to work and will always ensure that Veterans’ access to benefits and services remains uninterrupted as employees return to in-person work,” Kasperowicz said.
In the last few weeks, demand for services among veterans who are VA employees has also risen, one of the mental health professionals, a social worker, told Reuters. A quarter of VA employees are veterans.
The social worker said he is meeting with two to three VA employees a week who are seeking access to mental health care, citing stress and the fear that they will lose their jobs.
“People are calling out sick. People are ill with stress and worry. The mood is so low.”
A mental health supervisor in California described scrambling to cover the caseload of a remote worker who had to cancel appointments with more than a dozen veterans, because she could not access a VA facility.
VA employees in the Washington area and in Oregon said mental health professionals were unsure if they were allowed to answer calls from the VA’s crisis hot-line if they were not physically in an office, because they had been instructed not to conduct work outside of a facility.
“People are nervous to be on-call,” said a supervisor of mental health providers in the Washington area. “The system is under a lot of duress.”
The VA told Reuters that crisis line workers are exempt from the return-to-office policy, and that staff continue to respond quickly to nearly 3000 calls daily.
Therapists returning to the office are struggling to find private meeting rooms at some VA facilities, according to four of the mental health professionals interviewed by Reuters.
They described medical and mental health professionals converting closets and conference rooms into offices to comply with the mandate to conduct telehealth visits from VA facilities. They expressed concerns that the crowded rooms could violate patient privacy rights.
“We are scrambling to find space,” said a provider in California. “Veterans are going without until we can find spaces for these providers.”
Reuters was unable to independently verify the accounts of overcrowding. Kasperowicz said the agency’s “policy is to bring as many employees back to the office as space permits.”
(Reporting by Robin Respaut in San Francisco; additional reporting by Julia Harte in New York and Gabriella Borter in D.C.; Editing by Michele Gershberg and Suzanne Goldenberg)
SAN FRANCISCO (Reuters) — Joey Cortez, who served 24 years in the US Air Force, had been waiting since August to see a mental health specialist from the Department of Veterans’ Affairs, when he experienced a fresh jolt of anxiety.
Cortez was fired last month from his human resources job at the agency - one of about 2400 employees who lost their jobs at Veterans’ Affairs (VA) in the first wave of President Donald Trump’s efforts to shrink the federal workforce.
“Once the firings happened and I was terminated, I started having panic attacks to the point where I black out,” Cortez, who suffers from post-traumatic stress disorder, told Reuters. The layoff is also making it harder to maintain his sobriety, as a recovering alcoholic.
“Not a day has gone by since I was fired that I haven’t thought about picking up a bottle,” said Cortez.
After losing his job, Cortez asked the VA to expedite his wait for a therapist and was told there was no record of his request, he said. After a month of calls to the agency, he got an appointment for this August, one year after he started the process. Then the VA offered him an appointment next week because another patient had canceled.
The VA provides health care to 9.31 million US veterans at hundreds of medical centers, clinics, and nursing homes across the country.
It also faces complex problems.
“The VA has bloat. There are redundancies. There are places where we have questioned the administration of care and asked, does it need to be the way it is?” Pat Murray, the legislative director for the Veterans of Foreign Wars, which represents Americans who have fought overseas, said in an interview.
The Trump administration plans additional cuts to the VA of more than 80,000 personnel, according to an internal memo obtained by Reuters. The agency has also announced it is phasing out telework.
Reuters spoke to nine current and former VA employees in California, Oregon, Texas, and the Washington D.C. area who said the changes were further disrupting some mental health services and fueling anxieties among those who provide and rely on them.
The VA employees — who include six mental health professionals and three people in leadership positions — described cancellations of some in-person and telehealth appointments; confusion over staffing of a crisis hot-line; and professionals conducting telehealth visits in makeshift meeting rooms inside VA buildings.
They spoke on the condition of anonymity, because they were not authorized to speak with the media.
STAFFING SHORTAGES
A former employee at the VA’s Office of Inspector General, who is also a veteran, said any future large-scale staffing cuts would likely worsen shortages and impact the quality of care.
“There’s no way to take a scalpel and do it appropriately that quickly,” he said.
VA spokesperson Peter Kasperowicz told Reuters mental health professionals, such as psychologists and social workers, were not included in February’s staffing cuts, and the agency is working to recruit mental health providers and improve wait times.
He did not specify how many support staff for these providers had been affected.
Last week, two federal judges ordered the VA and other federal agencies to reinstate thousands of fired probationary workers. Cortez’s pay was reinstated but he was told not to return to work.
The Veterans Health Administration, the branch of the VA that provides healthcare, has experienced severe staffing shortages since 2015, especially among mental health professionals, according to an OIG report last year.
Veterans often benefit from specialized services to treat anxiety, trauma, depression and substance abuse. The proportion of veterans receiving mental health services rose to 31% in 2022 from 20% in 2007, according to the VA. Suicide among veterans is twice the rate of Americans overall.
The VFW’s Murray said his organization supports a thorough review of the VA’s mental health services, but it needs to be done carefully, “not with a chainsaw.”
‘THE MOOD IS SO LOW’
In recent years, the agency had encouraged remote work to help expand access to telehealth services and reduce wait times, especially in rural areas where recruiting providers is difficult.
The VA’s Kasperowicz said that, while providers will need to return to VA facilities, veterans will be able to access telehealth appointments.
He did not directly address questions about why mental health providers needed to return to the office.
“The VA will make accommodations as needed to ensure employees have enough space to work and will always ensure that Veterans’ access to benefits and services remains uninterrupted as employees return to in-person work,” Kasperowicz said.
In the last few weeks, demand for services among veterans who are VA employees has also risen, one of the mental health professionals, a social worker, told Reuters. A quarter of VA employees are veterans.
The social worker said he is meeting with two to three VA employees a week who are seeking access to mental health care, citing stress and the fear that they will lose their jobs.
“People are calling out sick. People are ill with stress and worry. The mood is so low.”
A mental health supervisor in California described scrambling to cover the caseload of a remote worker who had to cancel appointments with more than a dozen veterans, because she could not access a VA facility.
VA employees in the Washington area and in Oregon said mental health professionals were unsure if they were allowed to answer calls from the VA’s crisis hot-line if they were not physically in an office, because they had been instructed not to conduct work outside of a facility.
“People are nervous to be on-call,” said a supervisor of mental health providers in the Washington area. “The system is under a lot of duress.”
The VA told Reuters that crisis line workers are exempt from the return-to-office policy, and that staff continue to respond quickly to nearly 3000 calls daily.
Therapists returning to the office are struggling to find private meeting rooms at some VA facilities, according to four of the mental health professionals interviewed by Reuters.
They described medical and mental health professionals converting closets and conference rooms into offices to comply with the mandate to conduct telehealth visits from VA facilities. They expressed concerns that the crowded rooms could violate patient privacy rights.
“We are scrambling to find space,” said a provider in California. “Veterans are going without until we can find spaces for these providers.”
Reuters was unable to independently verify the accounts of overcrowding. Kasperowicz said the agency’s “policy is to bring as many employees back to the office as space permits.”
(Reporting by Robin Respaut in San Francisco; additional reporting by Julia Harte in New York and Gabriella Borter in D.C.; Editing by Michele Gershberg and Suzanne Goldenberg)
SAN FRANCISCO (Reuters) — Joey Cortez, who served 24 years in the US Air Force, had been waiting since August to see a mental health specialist from the Department of Veterans’ Affairs, when he experienced a fresh jolt of anxiety.
Cortez was fired last month from his human resources job at the agency - one of about 2400 employees who lost their jobs at Veterans’ Affairs (VA) in the first wave of President Donald Trump’s efforts to shrink the federal workforce.
“Once the firings happened and I was terminated, I started having panic attacks to the point where I black out,” Cortez, who suffers from post-traumatic stress disorder, told Reuters. The layoff is also making it harder to maintain his sobriety, as a recovering alcoholic.
“Not a day has gone by since I was fired that I haven’t thought about picking up a bottle,” said Cortez.
After losing his job, Cortez asked the VA to expedite his wait for a therapist and was told there was no record of his request, he said. After a month of calls to the agency, he got an appointment for this August, one year after he started the process. Then the VA offered him an appointment next week because another patient had canceled.
The VA provides health care to 9.31 million US veterans at hundreds of medical centers, clinics, and nursing homes across the country.
It also faces complex problems.
“The VA has bloat. There are redundancies. There are places where we have questioned the administration of care and asked, does it need to be the way it is?” Pat Murray, the legislative director for the Veterans of Foreign Wars, which represents Americans who have fought overseas, said in an interview.
The Trump administration plans additional cuts to the VA of more than 80,000 personnel, according to an internal memo obtained by Reuters. The agency has also announced it is phasing out telework.
Reuters spoke to nine current and former VA employees in California, Oregon, Texas, and the Washington D.C. area who said the changes were further disrupting some mental health services and fueling anxieties among those who provide and rely on them.
The VA employees — who include six mental health professionals and three people in leadership positions — described cancellations of some in-person and telehealth appointments; confusion over staffing of a crisis hot-line; and professionals conducting telehealth visits in makeshift meeting rooms inside VA buildings.
They spoke on the condition of anonymity, because they were not authorized to speak with the media.
STAFFING SHORTAGES
A former employee at the VA’s Office of Inspector General, who is also a veteran, said any future large-scale staffing cuts would likely worsen shortages and impact the quality of care.
“There’s no way to take a scalpel and do it appropriately that quickly,” he said.
VA spokesperson Peter Kasperowicz told Reuters mental health professionals, such as psychologists and social workers, were not included in February’s staffing cuts, and the agency is working to recruit mental health providers and improve wait times.
He did not specify how many support staff for these providers had been affected.
Last week, two federal judges ordered the VA and other federal agencies to reinstate thousands of fired probationary workers. Cortez’s pay was reinstated but he was told not to return to work.
The Veterans Health Administration, the branch of the VA that provides healthcare, has experienced severe staffing shortages since 2015, especially among mental health professionals, according to an OIG report last year.
Veterans often benefit from specialized services to treat anxiety, trauma, depression and substance abuse. The proportion of veterans receiving mental health services rose to 31% in 2022 from 20% in 2007, according to the VA. Suicide among veterans is twice the rate of Americans overall.
The VFW’s Murray said his organization supports a thorough review of the VA’s mental health services, but it needs to be done carefully, “not with a chainsaw.”
‘THE MOOD IS SO LOW’
In recent years, the agency had encouraged remote work to help expand access to telehealth services and reduce wait times, especially in rural areas where recruiting providers is difficult.
The VA’s Kasperowicz said that, while providers will need to return to VA facilities, veterans will be able to access telehealth appointments.
He did not directly address questions about why mental health providers needed to return to the office.
“The VA will make accommodations as needed to ensure employees have enough space to work and will always ensure that Veterans’ access to benefits and services remains uninterrupted as employees return to in-person work,” Kasperowicz said.
In the last few weeks, demand for services among veterans who are VA employees has also risen, one of the mental health professionals, a social worker, told Reuters. A quarter of VA employees are veterans.
The social worker said he is meeting with two to three VA employees a week who are seeking access to mental health care, citing stress and the fear that they will lose their jobs.
“People are calling out sick. People are ill with stress and worry. The mood is so low.”
A mental health supervisor in California described scrambling to cover the caseload of a remote worker who had to cancel appointments with more than a dozen veterans, because she could not access a VA facility.
VA employees in the Washington area and in Oregon said mental health professionals were unsure if they were allowed to answer calls from the VA’s crisis hot-line if they were not physically in an office, because they had been instructed not to conduct work outside of a facility.
“People are nervous to be on-call,” said a supervisor of mental health providers in the Washington area. “The system is under a lot of duress.”
The VA told Reuters that crisis line workers are exempt from the return-to-office policy, and that staff continue to respond quickly to nearly 3000 calls daily.
Therapists returning to the office are struggling to find private meeting rooms at some VA facilities, according to four of the mental health professionals interviewed by Reuters.
They described medical and mental health professionals converting closets and conference rooms into offices to comply with the mandate to conduct telehealth visits from VA facilities. They expressed concerns that the crowded rooms could violate patient privacy rights.
“We are scrambling to find space,” said a provider in California. “Veterans are going without until we can find spaces for these providers.”
Reuters was unable to independently verify the accounts of overcrowding. Kasperowicz said the agency’s “policy is to bring as many employees back to the office as space permits.”
(Reporting by Robin Respaut in San Francisco; additional reporting by Julia Harte in New York and Gabriella Borter in D.C.; Editing by Michele Gershberg and Suzanne Goldenberg)
The Unholy Trinity: Unlawful Prescriptions, False Claims, and Dangerous Drugs
The Unholy Trinity: Unlawful Prescriptions, False Claims, and Dangerous Drugs
Express Scripts, the contractor that manages the pharmacy benefit for Tricare, the military health insurance program, announced in 2021 that after a 5-year absence, CVS Pharmacy was once more in the network. In 2023, CVS had the largest profits of any pharmacy chain in the United States, about $159 billion, and generated a quarter of the overall revenue of the US pharmacy industry.1 Tricare officials heralded the return of CVS as a move that would offer US Department of Defense (DoD) beneficiaries more competitive prices, convenient access, and overall quality.2
DOJ Files Lawsuit Against CVS
In December 2024, the US Department of Justice (DoJ) filed a lawsuit alleging that CVS violated both the Controlled Substances Act (CSA) and the False Claims Act (FCA).3,4 The United States ex rel. Estright v Health Corporation, et al, filed in Rhode Island, charged that CVS “routinely” and “knowingly” filled invalid prescriptions for controlled substances violating the CSA and then billed federal health care programs for payment for these prescriptions, a breach of the FCA.5 The DoJ alleged that CVS pharmacies and pharmacists filled prescriptions for controlled substances that (1) lacked a legitimate medical purpose; (2) were not legally valid; and/or (3) were not issued in the usual course of medical practice. 6 CVS contests the charges and issued an official response, stating that it disputes the allegations as false, plans to disprove them in litigation, and has nonetheless fully cooperated with the investigation.7
The allegations involved prescriptions for drugs like opioids and benzodiazepines, primary culprits in the American overdose epidemic.8 The complaint notes that the prescriptions were early refills in excessive quantities and included what has been called the “holy trinity” of dangerous medications: opioids, benzodiazepines, and muscle relaxants. 5,8 Even worse (if that is possible), as the complaint outlines, CVS had access to data from both inside and outside the company that these prescriptions came from notorious pill mills and were hence unlawful and yet continued to fill them, leading the DoJ to file the more serious charge that the corporation “knowingly” violated the CSA and “prioritized profits over safety in dispensing controlled substances.”5,6
The Unholy Trinity
The infamous members of what I prefer to call the “unholy trinity” are a benzodiazepine, often alprazolam, an opioid, and the muscle relaxant carisoprodol. The combination amplifies each agent’s independent risk of respiratory depression. The latter is a schedule IV medication with an active metabolite, meprobamate, that also has this adverse effect. All 3 drugs have high abuse potential and, when combined, increase the risk of fatal overdose. The colloquial name holy trinity derives from the synergistic euphoria experienced when taking this triple cocktail of sedative agents.9 This pharmacological recipe for disaster is the house specialty of pill mills: infamous storefront practices that generate high profits and exploit persons with chronic pain and addiction by handing out controlled substances with little clinical assessment and even less oversight.10
When the Means Become the End
The DoJ allegations suggest that the violations resulted from “corporate-mandated performance metrics, incentive compensation, and staffing policies that prioritized corporate profits over patient safety.”6 If the allegations are true, why would a company reinvited by Tricare to serve the nation’s heroes seemingly engage in illegal practices? While CVS has not responded in court, their statement argued that “too often, we have seen government agencies and trial lawyers question the good-faith decisions made by pharmacists while a patient waits at the pharmacy counter, often in pain.”6
The DoJ complaint offers a cautionary warning for the US health care system, which is increasingly being micromanaged in the pursuit of efficiency. Like many practitioners in and out of the federal system, I get a cold chill when I read the word productivity. “CVS pharmacists described working at CVS as ‘soul crushing’ because it was impossible to meet the company’s expectations,” the complaint alleges, because “CVS set staffing levels so low that it was impossible for pharmacists to comply with their legal obligations and meet CVS’s demanding metrics.”5 Did top-down mandates drive the alleged activities by imposing unattainable performance metrics on pharmacists, offering incentives that encouraged and rewarded corner-cutting, and refusing to fund sufficient staffing to ensure patient safety? This may be what happens when the means (efficiency) become the end rather than a mechanism to achieve the goal of more accessible, affordable, high-quality health care.
Ethically, what is most concerning is that leadership intentionally “deprived its pharmacists of crucial information” about specific practitioners known to engage in illegal prescribing practices.6 CVS did not provide pharmacists with “information about prescribers’ prescribing habits that CVS routinely collected and reviewed at the corporate level,” and even removed prescriber blocks that were implemented at Target pharmacies before it was acquired by CVS.5 The first element of informed consent is providing patients with adequate information upon which to decide whether to accept or decline treatment. 11 In this situation, however, CVS allegedly prevented “pharmacists from warning one another about certain prescribers.”6
If true, the company deprived frontline pharmacists of the information they needed to safely and responsibly dispense medications: “The practices alleged contributed to the opioid crisis and opioid-related deaths, and today’s complaint seeks to hold CVS accountable for its misconduct.”6 Though the cost in human life that may have resulted from CSA violations must absolutely and always outweigh financial considerations, the economic damage to Tricare from fraudulent billing and the betrayal of its fiduciary responsinility cannot be underestimated.
A Corporate Morality Play
CVS is not the only company, nor is pharmacy the only industry in health care, that has been the subject of watchdog agency lawsuits or variegated forms of wrongdoing, including violations of the CSA and FCA.10,12 As of this writing, the DoJ case against CVS has not been heard, much less adjudicated in a court of law. It is ironic that both the DoJ claims and the CVS rebuttal describe the manifest conflict of obligation that pharmacists confront between protecting their livelihood and safeguarding patients’ lives as suggested in the epigraph that has been attributed to the 19th-century British physician and medical educator Peter Mere Latham. It is a dilemma that a growing number of health care practitioners face daily in a vocation becoming increasingly commercialized. It is all too easy for an individual physician, nurse, or pharmacist to feel hopeless and helpless before the behemoth might of a large and looming entity. Yet, it was a whistleblower whose moral courage led to the DoJ investigation and subsequent charges.13 We must all never doubt the power of a committed person of conscience to withstand the pressure to mutate medications into poison and stand up for the principles of our professions and inspire a community of colleagues to follow their example.
- Fein AJ. The Top U.S. pharmacy markets of 2023: market shares and revenues at the biggest chains and PBMs. Drug Channels. March 12, 2024. Accessed February 24, 2025. https://www.drugchannels.net/2024/03/the-top-15-us-pharmacies-of-2023-market.html
- Jowers K. CVS returns to the military Tricare network. Walmart’s out. Military Times. October 18, 2021. Accessed February 24, 2025. https://www.militarytimes.com/pay-benefits/mil-money/2021/10/28/cvs-returns-to-the-military-tricare-pharmacy-network-walmarts-out/
- False Claims, 31 USC § 3729 (2009). Accessed February 24, 2025. https://www.govinfo.gov/content/pkg/USCODE-2011-title31/pdf/USCODE-2011-title31-subtitleIII-chap37-subchapIII-sec3729.pdf
- Drug Abuse Prevention and Control, Control and Enforcement, 21 USC 13 § 801 (2022). Accessed February 24, 2025. https://www.govinfo.gov/app/details/USCODE-2021-title21/USCODE-2021-title21-chap13-subchapI-partA-sec801
- United States ex rel. Estright v Health Corporation, et al. Accessed February 26, 2025. https://www.justice.gov/archives/opa/media/1381111/dl
- US Department of Justice. Justice Department files nationwide lawsuit alleging CVS knowingly dispensed controlled substances in violation of the Controlled Substances ACT and the False Claims Act. News release. December 18, 2024. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/justice-department-files-nationwide-lawsuit-alleging-cvs-knowingly-dispensed-controlled
- CVS Health. CVS Health statement regarding the U.S. Department of Justice’s lawsuit against CVS pharmacy. News release. December 18, 2024. Accessed February 24, 2025. https://www.cvshealth.com/impact/healthy-community/our-opioid-response.html
- Park TW, Saitz R, Ganoczy D, Ilgen MA, Bohnert AS. Benzodiazepine prescribing patterns and deaths from drug overdose among US veterans receiving opioid analgesics: case-cohort study. BMJ. 2015;350:h2698. doi:10.1136/bmj.h2698
- Wang Y, Delcher C, Li Y, Goldberger BA, Reisfield GM. Overlapping prescriptions of opioids, benzodiazepines, and carisoprodol: “Holy Trinity” prescribing in the state of Florida. Drug Alcohol Depend. 2019;205:107693. doi:10.1016/j.drugalcdep.2019.107693
- Wolf AA. The perfect storm: opioid risks and ‘The Holy Trinity’. Pharmacy Times. September 24, 2014. Accessed February 24, 2025. https://www.pharmacytimes.com/view/the-perfect-storm-opioid-risks-and-the-holy-trinity
- The meaning and justification of informed consent. In: Beauchamp TL, Childress JF. Principles of Biomedical Ethics. Eighth Edition. Oxford University Press; 2019:118-123.
- US Department of Justice. OptumRX agrees to pay $20M to resolve allegations that it filled certain opioid prescriptions in violation of the Controlled Substances Act. News release. June 27, 2024. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/optumrx-agrees-pay-20m-resolve-allegations-it-filled-certain-opioid-prescriptions-violation
- US Department of Justice. False Claims Act settlements and judgments exceed $2.9B in fiscal year 2024. News release. January 15, 2025. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/false-claims-act-settlements-and-judgments-exceed-29b-fiscal-year-2024
Express Scripts, the contractor that manages the pharmacy benefit for Tricare, the military health insurance program, announced in 2021 that after a 5-year absence, CVS Pharmacy was once more in the network. In 2023, CVS had the largest profits of any pharmacy chain in the United States, about $159 billion, and generated a quarter of the overall revenue of the US pharmacy industry.1 Tricare officials heralded the return of CVS as a move that would offer US Department of Defense (DoD) beneficiaries more competitive prices, convenient access, and overall quality.2
DOJ Files Lawsuit Against CVS
In December 2024, the US Department of Justice (DoJ) filed a lawsuit alleging that CVS violated both the Controlled Substances Act (CSA) and the False Claims Act (FCA).3,4 The United States ex rel. Estright v Health Corporation, et al, filed in Rhode Island, charged that CVS “routinely” and “knowingly” filled invalid prescriptions for controlled substances violating the CSA and then billed federal health care programs for payment for these prescriptions, a breach of the FCA.5 The DoJ alleged that CVS pharmacies and pharmacists filled prescriptions for controlled substances that (1) lacked a legitimate medical purpose; (2) were not legally valid; and/or (3) were not issued in the usual course of medical practice. 6 CVS contests the charges and issued an official response, stating that it disputes the allegations as false, plans to disprove them in litigation, and has nonetheless fully cooperated with the investigation.7
The allegations involved prescriptions for drugs like opioids and benzodiazepines, primary culprits in the American overdose epidemic.8 The complaint notes that the prescriptions were early refills in excessive quantities and included what has been called the “holy trinity” of dangerous medications: opioids, benzodiazepines, and muscle relaxants. 5,8 Even worse (if that is possible), as the complaint outlines, CVS had access to data from both inside and outside the company that these prescriptions came from notorious pill mills and were hence unlawful and yet continued to fill them, leading the DoJ to file the more serious charge that the corporation “knowingly” violated the CSA and “prioritized profits over safety in dispensing controlled substances.”5,6
The Unholy Trinity
The infamous members of what I prefer to call the “unholy trinity” are a benzodiazepine, often alprazolam, an opioid, and the muscle relaxant carisoprodol. The combination amplifies each agent’s independent risk of respiratory depression. The latter is a schedule IV medication with an active metabolite, meprobamate, that also has this adverse effect. All 3 drugs have high abuse potential and, when combined, increase the risk of fatal overdose. The colloquial name holy trinity derives from the synergistic euphoria experienced when taking this triple cocktail of sedative agents.9 This pharmacological recipe for disaster is the house specialty of pill mills: infamous storefront practices that generate high profits and exploit persons with chronic pain and addiction by handing out controlled substances with little clinical assessment and even less oversight.10
When the Means Become the End
The DoJ allegations suggest that the violations resulted from “corporate-mandated performance metrics, incentive compensation, and staffing policies that prioritized corporate profits over patient safety.”6 If the allegations are true, why would a company reinvited by Tricare to serve the nation’s heroes seemingly engage in illegal practices? While CVS has not responded in court, their statement argued that “too often, we have seen government agencies and trial lawyers question the good-faith decisions made by pharmacists while a patient waits at the pharmacy counter, often in pain.”6
The DoJ complaint offers a cautionary warning for the US health care system, which is increasingly being micromanaged in the pursuit of efficiency. Like many practitioners in and out of the federal system, I get a cold chill when I read the word productivity. “CVS pharmacists described working at CVS as ‘soul crushing’ because it was impossible to meet the company’s expectations,” the complaint alleges, because “CVS set staffing levels so low that it was impossible for pharmacists to comply with their legal obligations and meet CVS’s demanding metrics.”5 Did top-down mandates drive the alleged activities by imposing unattainable performance metrics on pharmacists, offering incentives that encouraged and rewarded corner-cutting, and refusing to fund sufficient staffing to ensure patient safety? This may be what happens when the means (efficiency) become the end rather than a mechanism to achieve the goal of more accessible, affordable, high-quality health care.
Ethically, what is most concerning is that leadership intentionally “deprived its pharmacists of crucial information” about specific practitioners known to engage in illegal prescribing practices.6 CVS did not provide pharmacists with “information about prescribers’ prescribing habits that CVS routinely collected and reviewed at the corporate level,” and even removed prescriber blocks that were implemented at Target pharmacies before it was acquired by CVS.5 The first element of informed consent is providing patients with adequate information upon which to decide whether to accept or decline treatment. 11 In this situation, however, CVS allegedly prevented “pharmacists from warning one another about certain prescribers.”6
If true, the company deprived frontline pharmacists of the information they needed to safely and responsibly dispense medications: “The practices alleged contributed to the opioid crisis and opioid-related deaths, and today’s complaint seeks to hold CVS accountable for its misconduct.”6 Though the cost in human life that may have resulted from CSA violations must absolutely and always outweigh financial considerations, the economic damage to Tricare from fraudulent billing and the betrayal of its fiduciary responsinility cannot be underestimated.
A Corporate Morality Play
CVS is not the only company, nor is pharmacy the only industry in health care, that has been the subject of watchdog agency lawsuits or variegated forms of wrongdoing, including violations of the CSA and FCA.10,12 As of this writing, the DoJ case against CVS has not been heard, much less adjudicated in a court of law. It is ironic that both the DoJ claims and the CVS rebuttal describe the manifest conflict of obligation that pharmacists confront between protecting their livelihood and safeguarding patients’ lives as suggested in the epigraph that has been attributed to the 19th-century British physician and medical educator Peter Mere Latham. It is a dilemma that a growing number of health care practitioners face daily in a vocation becoming increasingly commercialized. It is all too easy for an individual physician, nurse, or pharmacist to feel hopeless and helpless before the behemoth might of a large and looming entity. Yet, it was a whistleblower whose moral courage led to the DoJ investigation and subsequent charges.13 We must all never doubt the power of a committed person of conscience to withstand the pressure to mutate medications into poison and stand up for the principles of our professions and inspire a community of colleagues to follow their example.
Express Scripts, the contractor that manages the pharmacy benefit for Tricare, the military health insurance program, announced in 2021 that after a 5-year absence, CVS Pharmacy was once more in the network. In 2023, CVS had the largest profits of any pharmacy chain in the United States, about $159 billion, and generated a quarter of the overall revenue of the US pharmacy industry.1 Tricare officials heralded the return of CVS as a move that would offer US Department of Defense (DoD) beneficiaries more competitive prices, convenient access, and overall quality.2
DOJ Files Lawsuit Against CVS
In December 2024, the US Department of Justice (DoJ) filed a lawsuit alleging that CVS violated both the Controlled Substances Act (CSA) and the False Claims Act (FCA).3,4 The United States ex rel. Estright v Health Corporation, et al, filed in Rhode Island, charged that CVS “routinely” and “knowingly” filled invalid prescriptions for controlled substances violating the CSA and then billed federal health care programs for payment for these prescriptions, a breach of the FCA.5 The DoJ alleged that CVS pharmacies and pharmacists filled prescriptions for controlled substances that (1) lacked a legitimate medical purpose; (2) were not legally valid; and/or (3) were not issued in the usual course of medical practice. 6 CVS contests the charges and issued an official response, stating that it disputes the allegations as false, plans to disprove them in litigation, and has nonetheless fully cooperated with the investigation.7
The allegations involved prescriptions for drugs like opioids and benzodiazepines, primary culprits in the American overdose epidemic.8 The complaint notes that the prescriptions were early refills in excessive quantities and included what has been called the “holy trinity” of dangerous medications: opioids, benzodiazepines, and muscle relaxants. 5,8 Even worse (if that is possible), as the complaint outlines, CVS had access to data from both inside and outside the company that these prescriptions came from notorious pill mills and were hence unlawful and yet continued to fill them, leading the DoJ to file the more serious charge that the corporation “knowingly” violated the CSA and “prioritized profits over safety in dispensing controlled substances.”5,6
The Unholy Trinity
The infamous members of what I prefer to call the “unholy trinity” are a benzodiazepine, often alprazolam, an opioid, and the muscle relaxant carisoprodol. The combination amplifies each agent’s independent risk of respiratory depression. The latter is a schedule IV medication with an active metabolite, meprobamate, that also has this adverse effect. All 3 drugs have high abuse potential and, when combined, increase the risk of fatal overdose. The colloquial name holy trinity derives from the synergistic euphoria experienced when taking this triple cocktail of sedative agents.9 This pharmacological recipe for disaster is the house specialty of pill mills: infamous storefront practices that generate high profits and exploit persons with chronic pain and addiction by handing out controlled substances with little clinical assessment and even less oversight.10
When the Means Become the End
The DoJ allegations suggest that the violations resulted from “corporate-mandated performance metrics, incentive compensation, and staffing policies that prioritized corporate profits over patient safety.”6 If the allegations are true, why would a company reinvited by Tricare to serve the nation’s heroes seemingly engage in illegal practices? While CVS has not responded in court, their statement argued that “too often, we have seen government agencies and trial lawyers question the good-faith decisions made by pharmacists while a patient waits at the pharmacy counter, often in pain.”6
The DoJ complaint offers a cautionary warning for the US health care system, which is increasingly being micromanaged in the pursuit of efficiency. Like many practitioners in and out of the federal system, I get a cold chill when I read the word productivity. “CVS pharmacists described working at CVS as ‘soul crushing’ because it was impossible to meet the company’s expectations,” the complaint alleges, because “CVS set staffing levels so low that it was impossible for pharmacists to comply with their legal obligations and meet CVS’s demanding metrics.”5 Did top-down mandates drive the alleged activities by imposing unattainable performance metrics on pharmacists, offering incentives that encouraged and rewarded corner-cutting, and refusing to fund sufficient staffing to ensure patient safety? This may be what happens when the means (efficiency) become the end rather than a mechanism to achieve the goal of more accessible, affordable, high-quality health care.
Ethically, what is most concerning is that leadership intentionally “deprived its pharmacists of crucial information” about specific practitioners known to engage in illegal prescribing practices.6 CVS did not provide pharmacists with “information about prescribers’ prescribing habits that CVS routinely collected and reviewed at the corporate level,” and even removed prescriber blocks that were implemented at Target pharmacies before it was acquired by CVS.5 The first element of informed consent is providing patients with adequate information upon which to decide whether to accept or decline treatment. 11 In this situation, however, CVS allegedly prevented “pharmacists from warning one another about certain prescribers.”6
If true, the company deprived frontline pharmacists of the information they needed to safely and responsibly dispense medications: “The practices alleged contributed to the opioid crisis and opioid-related deaths, and today’s complaint seeks to hold CVS accountable for its misconduct.”6 Though the cost in human life that may have resulted from CSA violations must absolutely and always outweigh financial considerations, the economic damage to Tricare from fraudulent billing and the betrayal of its fiduciary responsinility cannot be underestimated.
A Corporate Morality Play
CVS is not the only company, nor is pharmacy the only industry in health care, that has been the subject of watchdog agency lawsuits or variegated forms of wrongdoing, including violations of the CSA and FCA.10,12 As of this writing, the DoJ case against CVS has not been heard, much less adjudicated in a court of law. It is ironic that both the DoJ claims and the CVS rebuttal describe the manifest conflict of obligation that pharmacists confront between protecting their livelihood and safeguarding patients’ lives as suggested in the epigraph that has been attributed to the 19th-century British physician and medical educator Peter Mere Latham. It is a dilemma that a growing number of health care practitioners face daily in a vocation becoming increasingly commercialized. It is all too easy for an individual physician, nurse, or pharmacist to feel hopeless and helpless before the behemoth might of a large and looming entity. Yet, it was a whistleblower whose moral courage led to the DoJ investigation and subsequent charges.13 We must all never doubt the power of a committed person of conscience to withstand the pressure to mutate medications into poison and stand up for the principles of our professions and inspire a community of colleagues to follow their example.
- Fein AJ. The Top U.S. pharmacy markets of 2023: market shares and revenues at the biggest chains and PBMs. Drug Channels. March 12, 2024. Accessed February 24, 2025. https://www.drugchannels.net/2024/03/the-top-15-us-pharmacies-of-2023-market.html
- Jowers K. CVS returns to the military Tricare network. Walmart’s out. Military Times. October 18, 2021. Accessed February 24, 2025. https://www.militarytimes.com/pay-benefits/mil-money/2021/10/28/cvs-returns-to-the-military-tricare-pharmacy-network-walmarts-out/
- False Claims, 31 USC § 3729 (2009). Accessed February 24, 2025. https://www.govinfo.gov/content/pkg/USCODE-2011-title31/pdf/USCODE-2011-title31-subtitleIII-chap37-subchapIII-sec3729.pdf
- Drug Abuse Prevention and Control, Control and Enforcement, 21 USC 13 § 801 (2022). Accessed February 24, 2025. https://www.govinfo.gov/app/details/USCODE-2021-title21/USCODE-2021-title21-chap13-subchapI-partA-sec801
- United States ex rel. Estright v Health Corporation, et al. Accessed February 26, 2025. https://www.justice.gov/archives/opa/media/1381111/dl
- US Department of Justice. Justice Department files nationwide lawsuit alleging CVS knowingly dispensed controlled substances in violation of the Controlled Substances ACT and the False Claims Act. News release. December 18, 2024. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/justice-department-files-nationwide-lawsuit-alleging-cvs-knowingly-dispensed-controlled
- CVS Health. CVS Health statement regarding the U.S. Department of Justice’s lawsuit against CVS pharmacy. News release. December 18, 2024. Accessed February 24, 2025. https://www.cvshealth.com/impact/healthy-community/our-opioid-response.html
- Park TW, Saitz R, Ganoczy D, Ilgen MA, Bohnert AS. Benzodiazepine prescribing patterns and deaths from drug overdose among US veterans receiving opioid analgesics: case-cohort study. BMJ. 2015;350:h2698. doi:10.1136/bmj.h2698
- Wang Y, Delcher C, Li Y, Goldberger BA, Reisfield GM. Overlapping prescriptions of opioids, benzodiazepines, and carisoprodol: “Holy Trinity” prescribing in the state of Florida. Drug Alcohol Depend. 2019;205:107693. doi:10.1016/j.drugalcdep.2019.107693
- Wolf AA. The perfect storm: opioid risks and ‘The Holy Trinity’. Pharmacy Times. September 24, 2014. Accessed February 24, 2025. https://www.pharmacytimes.com/view/the-perfect-storm-opioid-risks-and-the-holy-trinity
- The meaning and justification of informed consent. In: Beauchamp TL, Childress JF. Principles of Biomedical Ethics. Eighth Edition. Oxford University Press; 2019:118-123.
- US Department of Justice. OptumRX agrees to pay $20M to resolve allegations that it filled certain opioid prescriptions in violation of the Controlled Substances Act. News release. June 27, 2024. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/optumrx-agrees-pay-20m-resolve-allegations-it-filled-certain-opioid-prescriptions-violation
- US Department of Justice. False Claims Act settlements and judgments exceed $2.9B in fiscal year 2024. News release. January 15, 2025. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/false-claims-act-settlements-and-judgments-exceed-29b-fiscal-year-2024
- Fein AJ. The Top U.S. pharmacy markets of 2023: market shares and revenues at the biggest chains and PBMs. Drug Channels. March 12, 2024. Accessed February 24, 2025. https://www.drugchannels.net/2024/03/the-top-15-us-pharmacies-of-2023-market.html
- Jowers K. CVS returns to the military Tricare network. Walmart’s out. Military Times. October 18, 2021. Accessed February 24, 2025. https://www.militarytimes.com/pay-benefits/mil-money/2021/10/28/cvs-returns-to-the-military-tricare-pharmacy-network-walmarts-out/
- False Claims, 31 USC § 3729 (2009). Accessed February 24, 2025. https://www.govinfo.gov/content/pkg/USCODE-2011-title31/pdf/USCODE-2011-title31-subtitleIII-chap37-subchapIII-sec3729.pdf
- Drug Abuse Prevention and Control, Control and Enforcement, 21 USC 13 § 801 (2022). Accessed February 24, 2025. https://www.govinfo.gov/app/details/USCODE-2021-title21/USCODE-2021-title21-chap13-subchapI-partA-sec801
- United States ex rel. Estright v Health Corporation, et al. Accessed February 26, 2025. https://www.justice.gov/archives/opa/media/1381111/dl
- US Department of Justice. Justice Department files nationwide lawsuit alleging CVS knowingly dispensed controlled substances in violation of the Controlled Substances ACT and the False Claims Act. News release. December 18, 2024. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/justice-department-files-nationwide-lawsuit-alleging-cvs-knowingly-dispensed-controlled
- CVS Health. CVS Health statement regarding the U.S. Department of Justice’s lawsuit against CVS pharmacy. News release. December 18, 2024. Accessed February 24, 2025. https://www.cvshealth.com/impact/healthy-community/our-opioid-response.html
- Park TW, Saitz R, Ganoczy D, Ilgen MA, Bohnert AS. Benzodiazepine prescribing patterns and deaths from drug overdose among US veterans receiving opioid analgesics: case-cohort study. BMJ. 2015;350:h2698. doi:10.1136/bmj.h2698
- Wang Y, Delcher C, Li Y, Goldberger BA, Reisfield GM. Overlapping prescriptions of opioids, benzodiazepines, and carisoprodol: “Holy Trinity” prescribing in the state of Florida. Drug Alcohol Depend. 2019;205:107693. doi:10.1016/j.drugalcdep.2019.107693
- Wolf AA. The perfect storm: opioid risks and ‘The Holy Trinity’. Pharmacy Times. September 24, 2014. Accessed February 24, 2025. https://www.pharmacytimes.com/view/the-perfect-storm-opioid-risks-and-the-holy-trinity
- The meaning and justification of informed consent. In: Beauchamp TL, Childress JF. Principles of Biomedical Ethics. Eighth Edition. Oxford University Press; 2019:118-123.
- US Department of Justice. OptumRX agrees to pay $20M to resolve allegations that it filled certain opioid prescriptions in violation of the Controlled Substances Act. News release. June 27, 2024. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/optumrx-agrees-pay-20m-resolve-allegations-it-filled-certain-opioid-prescriptions-violation
- US Department of Justice. False Claims Act settlements and judgments exceed $2.9B in fiscal year 2024. News release. January 15, 2025. Accessed February 24, 2025. https://www.justice.gov/archives/opa/pr/false-claims-act-settlements-and-judgments-exceed-29b-fiscal-year-2024
The Unholy Trinity: Unlawful Prescriptions, False Claims, and Dangerous Drugs
The Unholy Trinity: Unlawful Prescriptions, False Claims, and Dangerous Drugs
AVAHO Implores VA Secretary Collins to Use Caution Amid Rapid Changes
The Association of VA Hematology/Oncology outlined its concerns over “unintended consequences” to recent changes at the US Department of Veterans Affairs (VA) in a March 3, 2025, letter to Secretary Doug A. Collins. “Indiscriminate cuts to contracts and personnel could have unforeseen consequences in many research areas within the VA, so we implore scrutiny,” the letter warns.
“We have already seen specific examples this past week of swift contract cuts impairing the VA’s ability to implement research protocols, process and report pharmacogenomic results, management of Electronic Health Record Modernization (EHRM) council workgroups, and execute new oncology services through the Close to Me initiative,” AVAHO Executive Director Julie Lawson said.
As Lawson noted, the return-to-office order for the staff of the Clinical Resource Hubs (CRHs) and the National Tele-Oncology programs could significantly impair their ability to function. Both departments have been fully remote since their start and are key elements of VA care for rural veterans. In fiscal year 2024, > 500,000 veterans received > 1.4 million CRH encounters. Nearly 20,000 veterans have utilized the National Tele-Oncology program in > 80,000 cancer-care encounters.
“We have significant concern that a blanket return to office of these fully remote programs, without an adequate plan for office space, teleworking equipment, and clinical and administrative support could have significant disruption and impairment in their delivery of care, negatively impacting veteran outcomes,” Lawson said.
AVAHO also strongly urged Collins to continue VA investment in clinical trials specifically and research in general: "To implement and execute research, there must be an adequte system in place to support these research programs."
The Association of VA Hematology/Oncology outlined its concerns over “unintended consequences” to recent changes at the US Department of Veterans Affairs (VA) in a March 3, 2025, letter to Secretary Doug A. Collins. “Indiscriminate cuts to contracts and personnel could have unforeseen consequences in many research areas within the VA, so we implore scrutiny,” the letter warns.
“We have already seen specific examples this past week of swift contract cuts impairing the VA’s ability to implement research protocols, process and report pharmacogenomic results, management of Electronic Health Record Modernization (EHRM) council workgroups, and execute new oncology services through the Close to Me initiative,” AVAHO Executive Director Julie Lawson said.
As Lawson noted, the return-to-office order for the staff of the Clinical Resource Hubs (CRHs) and the National Tele-Oncology programs could significantly impair their ability to function. Both departments have been fully remote since their start and are key elements of VA care for rural veterans. In fiscal year 2024, > 500,000 veterans received > 1.4 million CRH encounters. Nearly 20,000 veterans have utilized the National Tele-Oncology program in > 80,000 cancer-care encounters.
“We have significant concern that a blanket return to office of these fully remote programs, without an adequate plan for office space, teleworking equipment, and clinical and administrative support could have significant disruption and impairment in their delivery of care, negatively impacting veteran outcomes,” Lawson said.
AVAHO also strongly urged Collins to continue VA investment in clinical trials specifically and research in general: "To implement and execute research, there must be an adequte system in place to support these research programs."
The Association of VA Hematology/Oncology outlined its concerns over “unintended consequences” to recent changes at the US Department of Veterans Affairs (VA) in a March 3, 2025, letter to Secretary Doug A. Collins. “Indiscriminate cuts to contracts and personnel could have unforeseen consequences in many research areas within the VA, so we implore scrutiny,” the letter warns.
“We have already seen specific examples this past week of swift contract cuts impairing the VA’s ability to implement research protocols, process and report pharmacogenomic results, management of Electronic Health Record Modernization (EHRM) council workgroups, and execute new oncology services through the Close to Me initiative,” AVAHO Executive Director Julie Lawson said.
As Lawson noted, the return-to-office order for the staff of the Clinical Resource Hubs (CRHs) and the National Tele-Oncology programs could significantly impair their ability to function. Both departments have been fully remote since their start and are key elements of VA care for rural veterans. In fiscal year 2024, > 500,000 veterans received > 1.4 million CRH encounters. Nearly 20,000 veterans have utilized the National Tele-Oncology program in > 80,000 cancer-care encounters.
“We have significant concern that a blanket return to office of these fully remote programs, without an adequate plan for office space, teleworking equipment, and clinical and administrative support could have significant disruption and impairment in their delivery of care, negatively impacting veteran outcomes,” Lawson said.
AVAHO also strongly urged Collins to continue VA investment in clinical trials specifically and research in general: "To implement and execute research, there must be an adequte system in place to support these research programs."
