User login
FDA concerned about e-cigs/seizures in youth
the agency announced April 3.

Between 2010 and early 2019, the FDA and poison control centers received 35 reports of seizures that mentioned the use of e-cigarettes. Most reports involved youth or young adults, and the reports have increased slightly since June 2018, the announcement says.
“We want to be clear that we don’t yet know if there’s a direct relationship between the use of e-cigarettes and a risk of seizure,” said FDA Commissioner Scott Gottlieb, MD, and Principal Deputy Commissioner Amy Abernethy, MD, PhD, in a statement. “We believe these 35 cases warrant scientific investigation into whether there is in fact a connection.”
In addition, the FDA is trying to determine whether any e-cigarette product-specific factors may be associated with the risk of seizures.
Seizures have been reported after a few puffs or up to 1 day after e-cigarette use and among first-time and experienced users. A few patients had a prior history of seizures or also used other substances, such as marijuana or amphetamines.
“While 35 cases may not seem like much compared to the total number of people using e-cigarettes, we are nonetheless concerned by these reported cases. We also recognized that not all of the cases may be reported,” Dr. Gottlieb and Dr. Abernethy said.
Although seizures are known side effects of nicotine toxicity and have been reported in the context of intentional or accidental swallowing of e-cigarette liquid, the voluntary reports of seizures occurring with vaping could represent a new safety issue, the FDA said.
The agency encouraged people to report cases via an online safety reporting portal. It also provided redacted case reports that involve vaping and seizures.
the agency announced April 3.
Between 2010 and early 2019, the FDA and poison control centers received 35 reports of seizures that mentioned the use of e-cigarettes. Most reports involved youth or young adults, and the reports have increased slightly since June 2018, the announcement says.
“We want to be clear that we don’t yet know if there’s a direct relationship between the use of e-cigarettes and a risk of seizure,” said FDA Commissioner Scott Gottlieb, MD, and Principal Deputy Commissioner Amy Abernethy, MD, PhD, in a statement. “We believe these 35 cases warrant scientific investigation into whether there is in fact a connection.”
In addition, the FDA is trying to determine whether any e-cigarette product-specific factors may be associated with the risk of seizures.
Seizures have been reported after a few puffs or up to 1 day after e-cigarette use and among first-time and experienced users. A few patients had a prior history of seizures or also used other substances, such as marijuana or amphetamines.
“While 35 cases may not seem like much compared to the total number of people using e-cigarettes, we are nonetheless concerned by these reported cases. We also recognized that not all of the cases may be reported,” Dr. Gottlieb and Dr. Abernethy said.
Although seizures are known side effects of nicotine toxicity and have been reported in the context of intentional or accidental swallowing of e-cigarette liquid, the voluntary reports of seizures occurring with vaping could represent a new safety issue, the FDA said.
The agency encouraged people to report cases via an online safety reporting portal. It also provided redacted case reports that involve vaping and seizures.
the agency announced April 3.
Between 2010 and early 2019, the FDA and poison control centers received 35 reports of seizures that mentioned the use of e-cigarettes. Most reports involved youth or young adults, and the reports have increased slightly since June 2018, the announcement says.
“We want to be clear that we don’t yet know if there’s a direct relationship between the use of e-cigarettes and a risk of seizure,” said FDA Commissioner Scott Gottlieb, MD, and Principal Deputy Commissioner Amy Abernethy, MD, PhD, in a statement. “We believe these 35 cases warrant scientific investigation into whether there is in fact a connection.”
In addition, the FDA is trying to determine whether any e-cigarette product-specific factors may be associated with the risk of seizures.
Seizures have been reported after a few puffs or up to 1 day after e-cigarette use and among first-time and experienced users. A few patients had a prior history of seizures or also used other substances, such as marijuana or amphetamines.
“While 35 cases may not seem like much compared to the total number of people using e-cigarettes, we are nonetheless concerned by these reported cases. We also recognized that not all of the cases may be reported,” Dr. Gottlieb and Dr. Abernethy said.
Although seizures are known side effects of nicotine toxicity and have been reported in the context of intentional or accidental swallowing of e-cigarette liquid, the voluntary reports of seizures occurring with vaping could represent a new safety issue, the FDA said.
The agency encouraged people to report cases via an online safety reporting portal. It also provided redacted case reports that involve vaping and seizures.
Rituximab does not improve fatigue symptoms of ME/CFS
according to results from the phase 3 RituxME trial.

“The lack of clinical effect of B-cell depletion in this trial weakens the case for an important role of B lymphocytes in ME/CFS but does not exclude an immunologic basis,” Øystein Fluge, MD, PhD, of the department of oncology and medical physics at Haukeland University Hospital in Bergen, Norway, and his colleagues wrote April 1 in Annals of Internal Medicine.
The investigators noted that the basis for testing the effects of a B-cell–depleting intervention on clinical symptoms in patients with ME/CFS came from observations of its potential benefit in a subgroup of patients in previous studies. Dr. Fluge and his colleagues performed a three-patient case series in their own group that found benefit for patients who received rituximab for treatment of CFS (BMC Neurol. 2009 May 8;9:28. doi: 10.1186/1471-2377-9-28). A phase 2 trial of 30 patients with CFS also performed by his own group found improved fatigue scores in 66.7% of patients in the rituximab group, compared with placebo (PLOS One. 2011 Oct 19. doi: 10.1371/journal.pone.0026358).
In the double-blinded RituxME trial, 151 patients with ME/CFS from four university hospitals and one general hospital in Norway were recruited and randomized to receive infusions of rituximab (n = 77) or placebo (n = 74). The patients were aged 18-65 years old and had the disease ranging from 2 years to 15 years. Patients reported and rated their ME/CFS symptoms at baseline as well as completed forms for the SF-36, Hospital Anxiety and Depression Scale, Fatigue Severity Scale, and modified DePaul Symptom Questionnaire out to 24 months. The rituximab group received two infusions at 500 mg/m2 across body surface area at 2 weeks apart. They then received 500-mg maintenance infusions at 3 months, 6 months, 9 months, and 12 months where they also self-reported changes in ME/CFS symptoms.
There were no significant differences between groups regarding fatigue score at any follow-up period, with an average between-group difference of 0.02 at 24 months (95% confidence interval, –0.27 to 0.31). The overall response rate was 26% with rituximab and 35% with placebo. Dr. Fluge and his colleagues also noted no significant differences regarding SF-36 scores, function level, and fatigue severity between groups.
Adverse event rates were higher in the rituximab group (63 patients; 82%) than in the placebo group (48 patients; 65%), and these were more often attributed to treatment for those taking rituximab (26 patients [34%]) than for placebo (12 patients [16%]). Adverse events requiring hospitalization also occurred more often among those taking rituximab (31 events in 20 patients [26%]) than for those who took placebo (16 events in 14 patients [19%]).
Some of the weaknesses of the trial included its use of self-referral and self-reported symptom scores, which might have been subject to recall bias. In commenting on the difference in outcome between the phase 3 trial and others, Dr. Fluge and his associates said the discrepancy could potentially be high expectations in the placebo group, unknown factors surrounding symptom variation in ME/CFS patients, and unknown patient selection effects.
“[T]he negative outcome of RituxME should spur research to assess patient subgroups and further elucidate disease mechanisms, of which recently disclosed impairment of energy metabolism may be important,” Dr. Fluge and his coauthors wrote.
The trial was funded by grants to the researchers from the Norwegian Research Council, the Norwegian Regional Health Trusts, the MEandYou Foundation, the Norwegian ME Association, and the legacy of Torstein Hereid.
SOURCE: Fluge Ø et al. Ann Intern Med. 2019 Apr 1. doi: 10.7326/M18-1451
The RituxME trial’s results weaken the case for the use of rituximab to treat myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), but there are opportunities to test other treatments targeting immunologic abnormalities in ME/CFS, Peter C. Rowe, MD, of Johns Hopkins University, Baltimore, wrote in a related editorial.

“Persons with ME/CFS often meet criteria for several comorbid conditions, each of which could flare during a trial, possibly obscuring a true beneficial effect of an intervention,” Dr. Rowe wrote.
Trials with open treatment periods, in which ME/CFS patients all receive rituximab and then are grouped based on nontargeted conditions, could be warranted to “allow better control” of these conditions. Other trial designs could include randomizing patients to continue or discontinue therapy for responders, he added.
“The profound level of impaired function of affected individuals warrants a new commitment to hypothesis-driven clinical trials that incorporate and expand on the methodological sophistication of the rituximab trial,” Dr. Rowe wrote.
Dr. Rowe is with Johns Hopkins University, Baltimore. These comments summarize his editorial in response to Fluge et al. (Ann Intern Med. 2019 Apr 1. doi: 10.7326/M19-0643). Dr. Rowe reported receiving grants from the National Institutes of Health and is a scientific advisory board member for Solve ME/CFS, all outside the submitted work.
The RituxME trial’s results weaken the case for the use of rituximab to treat myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), but there are opportunities to test other treatments targeting immunologic abnormalities in ME/CFS, Peter C. Rowe, MD, of Johns Hopkins University, Baltimore, wrote in a related editorial.
“Persons with ME/CFS often meet criteria for several comorbid conditions, each of which could flare during a trial, possibly obscuring a true beneficial effect of an intervention,” Dr. Rowe wrote.
Trials with open treatment periods, in which ME/CFS patients all receive rituximab and then are grouped based on nontargeted conditions, could be warranted to “allow better control” of these conditions. Other trial designs could include randomizing patients to continue or discontinue therapy for responders, he added.
“The profound level of impaired function of affected individuals warrants a new commitment to hypothesis-driven clinical trials that incorporate and expand on the methodological sophistication of the rituximab trial,” Dr. Rowe wrote.
Dr. Rowe is with Johns Hopkins University, Baltimore. These comments summarize his editorial in response to Fluge et al. (Ann Intern Med. 2019 Apr 1. doi: 10.7326/M19-0643). Dr. Rowe reported receiving grants from the National Institutes of Health and is a scientific advisory board member for Solve ME/CFS, all outside the submitted work.
The RituxME trial’s results weaken the case for the use of rituximab to treat myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), but there are opportunities to test other treatments targeting immunologic abnormalities in ME/CFS, Peter C. Rowe, MD, of Johns Hopkins University, Baltimore, wrote in a related editorial.
“Persons with ME/CFS often meet criteria for several comorbid conditions, each of which could flare during a trial, possibly obscuring a true beneficial effect of an intervention,” Dr. Rowe wrote.
Trials with open treatment periods, in which ME/CFS patients all receive rituximab and then are grouped based on nontargeted conditions, could be warranted to “allow better control” of these conditions. Other trial designs could include randomizing patients to continue or discontinue therapy for responders, he added.
“The profound level of impaired function of affected individuals warrants a new commitment to hypothesis-driven clinical trials that incorporate and expand on the methodological sophistication of the rituximab trial,” Dr. Rowe wrote.
Dr. Rowe is with Johns Hopkins University, Baltimore. These comments summarize his editorial in response to Fluge et al. (Ann Intern Med. 2019 Apr 1. doi: 10.7326/M19-0643). Dr. Rowe reported receiving grants from the National Institutes of Health and is a scientific advisory board member for Solve ME/CFS, all outside the submitted work.
according to results from the phase 3 RituxME trial.
“The lack of clinical effect of B-cell depletion in this trial weakens the case for an important role of B lymphocytes in ME/CFS but does not exclude an immunologic basis,” Øystein Fluge, MD, PhD, of the department of oncology and medical physics at Haukeland University Hospital in Bergen, Norway, and his colleagues wrote April 1 in Annals of Internal Medicine.
The investigators noted that the basis for testing the effects of a B-cell–depleting intervention on clinical symptoms in patients with ME/CFS came from observations of its potential benefit in a subgroup of patients in previous studies. Dr. Fluge and his colleagues performed a three-patient case series in their own group that found benefit for patients who received rituximab for treatment of CFS (BMC Neurol. 2009 May 8;9:28. doi: 10.1186/1471-2377-9-28). A phase 2 trial of 30 patients with CFS also performed by his own group found improved fatigue scores in 66.7% of patients in the rituximab group, compared with placebo (PLOS One. 2011 Oct 19. doi: 10.1371/journal.pone.0026358).
In the double-blinded RituxME trial, 151 patients with ME/CFS from four university hospitals and one general hospital in Norway were recruited and randomized to receive infusions of rituximab (n = 77) or placebo (n = 74). The patients were aged 18-65 years old and had the disease ranging from 2 years to 15 years. Patients reported and rated their ME/CFS symptoms at baseline as well as completed forms for the SF-36, Hospital Anxiety and Depression Scale, Fatigue Severity Scale, and modified DePaul Symptom Questionnaire out to 24 months. The rituximab group received two infusions at 500 mg/m2 across body surface area at 2 weeks apart. They then received 500-mg maintenance infusions at 3 months, 6 months, 9 months, and 12 months where they also self-reported changes in ME/CFS symptoms.
There were no significant differences between groups regarding fatigue score at any follow-up period, with an average between-group difference of 0.02 at 24 months (95% confidence interval, –0.27 to 0.31). The overall response rate was 26% with rituximab and 35% with placebo. Dr. Fluge and his colleagues also noted no significant differences regarding SF-36 scores, function level, and fatigue severity between groups.
Adverse event rates were higher in the rituximab group (63 patients; 82%) than in the placebo group (48 patients; 65%), and these were more often attributed to treatment for those taking rituximab (26 patients [34%]) than for placebo (12 patients [16%]). Adverse events requiring hospitalization also occurred more often among those taking rituximab (31 events in 20 patients [26%]) than for those who took placebo (16 events in 14 patients [19%]).
Some of the weaknesses of the trial included its use of self-referral and self-reported symptom scores, which might have been subject to recall bias. In commenting on the difference in outcome between the phase 3 trial and others, Dr. Fluge and his associates said the discrepancy could potentially be high expectations in the placebo group, unknown factors surrounding symptom variation in ME/CFS patients, and unknown patient selection effects.
“[T]he negative outcome of RituxME should spur research to assess patient subgroups and further elucidate disease mechanisms, of which recently disclosed impairment of energy metabolism may be important,” Dr. Fluge and his coauthors wrote.
The trial was funded by grants to the researchers from the Norwegian Research Council, the Norwegian Regional Health Trusts, the MEandYou Foundation, the Norwegian ME Association, and the legacy of Torstein Hereid.
SOURCE: Fluge Ø et al. Ann Intern Med. 2019 Apr 1. doi: 10.7326/M18-1451
according to results from the phase 3 RituxME trial.
“The lack of clinical effect of B-cell depletion in this trial weakens the case for an important role of B lymphocytes in ME/CFS but does not exclude an immunologic basis,” Øystein Fluge, MD, PhD, of the department of oncology and medical physics at Haukeland University Hospital in Bergen, Norway, and his colleagues wrote April 1 in Annals of Internal Medicine.
The investigators noted that the basis for testing the effects of a B-cell–depleting intervention on clinical symptoms in patients with ME/CFS came from observations of its potential benefit in a subgroup of patients in previous studies. Dr. Fluge and his colleagues performed a three-patient case series in their own group that found benefit for patients who received rituximab for treatment of CFS (BMC Neurol. 2009 May 8;9:28. doi: 10.1186/1471-2377-9-28). A phase 2 trial of 30 patients with CFS also performed by his own group found improved fatigue scores in 66.7% of patients in the rituximab group, compared with placebo (PLOS One. 2011 Oct 19. doi: 10.1371/journal.pone.0026358).
In the double-blinded RituxME trial, 151 patients with ME/CFS from four university hospitals and one general hospital in Norway were recruited and randomized to receive infusions of rituximab (n = 77) or placebo (n = 74). The patients were aged 18-65 years old and had the disease ranging from 2 years to 15 years. Patients reported and rated their ME/CFS symptoms at baseline as well as completed forms for the SF-36, Hospital Anxiety and Depression Scale, Fatigue Severity Scale, and modified DePaul Symptom Questionnaire out to 24 months. The rituximab group received two infusions at 500 mg/m2 across body surface area at 2 weeks apart. They then received 500-mg maintenance infusions at 3 months, 6 months, 9 months, and 12 months where they also self-reported changes in ME/CFS symptoms.
There were no significant differences between groups regarding fatigue score at any follow-up period, with an average between-group difference of 0.02 at 24 months (95% confidence interval, –0.27 to 0.31). The overall response rate was 26% with rituximab and 35% with placebo. Dr. Fluge and his colleagues also noted no significant differences regarding SF-36 scores, function level, and fatigue severity between groups.
Adverse event rates were higher in the rituximab group (63 patients; 82%) than in the placebo group (48 patients; 65%), and these were more often attributed to treatment for those taking rituximab (26 patients [34%]) than for placebo (12 patients [16%]). Adverse events requiring hospitalization also occurred more often among those taking rituximab (31 events in 20 patients [26%]) than for those who took placebo (16 events in 14 patients [19%]).
Some of the weaknesses of the trial included its use of self-referral and self-reported symptom scores, which might have been subject to recall bias. In commenting on the difference in outcome between the phase 3 trial and others, Dr. Fluge and his associates said the discrepancy could potentially be high expectations in the placebo group, unknown factors surrounding symptom variation in ME/CFS patients, and unknown patient selection effects.
“[T]he negative outcome of RituxME should spur research to assess patient subgroups and further elucidate disease mechanisms, of which recently disclosed impairment of energy metabolism may be important,” Dr. Fluge and his coauthors wrote.
The trial was funded by grants to the researchers from the Norwegian Research Council, the Norwegian Regional Health Trusts, the MEandYou Foundation, the Norwegian ME Association, and the legacy of Torstein Hereid.
SOURCE: Fluge Ø et al. Ann Intern Med. 2019 Apr 1. doi: 10.7326/M18-1451
FROM ANNALS OF INTERNAL MEDICINE
FDA approves Mavenclad for treatment of relapsing MS
including relapsing/remitting and active secondary progressive disease.

The drug’s manufacturer, EMD Serono, said in a press release that cladribine is the first short-course oral therapy for such patients, and its use is generally recommended for patients who have had an inadequate response to, or are unable to tolerate, an alternate drug indicated for the treatment of MS. Cladribine is not recommended for use in patients with clinically isolated syndrome.
The agency’s decision is based on results from a clinical trial of 1,326 patients with relapsing MS who had experienced at least one relapse in the previous 12 months. Patients who received cladribine had significantly fewer relapses than did those who received placebo; the progression of disability was also significantly reduced in the cladribine group, compared with placebo, according to the FDA’s announcement.
The most common adverse events associated with cladribine include upper respiratory tract infections, headache, and decreased lymphocyte counts. In addition, the medication must be dispensed with a patient medication guide because the label includes a boxed warning for increased risk of malignancy and fetal harm. Other warnings include a risk for decreased lymphocyte count, hematologic toxicity and bone marrow suppression, and graft-versus-host-disease.
“We are committed to supporting the development of safe and effective treatments for patients with multiple sclerosis. The approval of Mavenclad represents an additional option for patients who have tried another treatment without success,” Billy Dunn, MD, director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research, said in the announcement.
The approved dose of cladribine is 3.5 mg/kg body weight over 2 years, administered as one treatment course of 1.75 mg/kg per year, each consisting of 2 treatment weeks. Additional courses of cladribine are not to be administered because retreatment with cladribine during years 3 and 4 may further increase the risk of malignancy. The safety and efficacy of reinitiating cladribine more than 2 years after completing two treatment courses has not been studied, according to EMD Serono.
Cladribine is approved in more than 50 other countries and was approved for use in the European Union in August 2017.
including relapsing/remitting and active secondary progressive disease.
The drug’s manufacturer, EMD Serono, said in a press release that cladribine is the first short-course oral therapy for such patients, and its use is generally recommended for patients who have had an inadequate response to, or are unable to tolerate, an alternate drug indicated for the treatment of MS. Cladribine is not recommended for use in patients with clinically isolated syndrome.
The agency’s decision is based on results from a clinical trial of 1,326 patients with relapsing MS who had experienced at least one relapse in the previous 12 months. Patients who received cladribine had significantly fewer relapses than did those who received placebo; the progression of disability was also significantly reduced in the cladribine group, compared with placebo, according to the FDA’s announcement.
The most common adverse events associated with cladribine include upper respiratory tract infections, headache, and decreased lymphocyte counts. In addition, the medication must be dispensed with a patient medication guide because the label includes a boxed warning for increased risk of malignancy and fetal harm. Other warnings include a risk for decreased lymphocyte count, hematologic toxicity and bone marrow suppression, and graft-versus-host-disease.
“We are committed to supporting the development of safe and effective treatments for patients with multiple sclerosis. The approval of Mavenclad represents an additional option for patients who have tried another treatment without success,” Billy Dunn, MD, director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research, said in the announcement.
The approved dose of cladribine is 3.5 mg/kg body weight over 2 years, administered as one treatment course of 1.75 mg/kg per year, each consisting of 2 treatment weeks. Additional courses of cladribine are not to be administered because retreatment with cladribine during years 3 and 4 may further increase the risk of malignancy. The safety and efficacy of reinitiating cladribine more than 2 years after completing two treatment courses has not been studied, according to EMD Serono.
Cladribine is approved in more than 50 other countries and was approved for use in the European Union in August 2017.
including relapsing/remitting and active secondary progressive disease.
The drug’s manufacturer, EMD Serono, said in a press release that cladribine is the first short-course oral therapy for such patients, and its use is generally recommended for patients who have had an inadequate response to, or are unable to tolerate, an alternate drug indicated for the treatment of MS. Cladribine is not recommended for use in patients with clinically isolated syndrome.
The agency’s decision is based on results from a clinical trial of 1,326 patients with relapsing MS who had experienced at least one relapse in the previous 12 months. Patients who received cladribine had significantly fewer relapses than did those who received placebo; the progression of disability was also significantly reduced in the cladribine group, compared with placebo, according to the FDA’s announcement.
The most common adverse events associated with cladribine include upper respiratory tract infections, headache, and decreased lymphocyte counts. In addition, the medication must be dispensed with a patient medication guide because the label includes a boxed warning for increased risk of malignancy and fetal harm. Other warnings include a risk for decreased lymphocyte count, hematologic toxicity and bone marrow suppression, and graft-versus-host-disease.
“We are committed to supporting the development of safe and effective treatments for patients with multiple sclerosis. The approval of Mavenclad represents an additional option for patients who have tried another treatment without success,” Billy Dunn, MD, director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research, said in the announcement.
The approved dose of cladribine is 3.5 mg/kg body weight over 2 years, administered as one treatment course of 1.75 mg/kg per year, each consisting of 2 treatment weeks. Additional courses of cladribine are not to be administered because retreatment with cladribine during years 3 and 4 may further increase the risk of malignancy. The safety and efficacy of reinitiating cladribine more than 2 years after completing two treatment courses has not been studied, according to EMD Serono.
Cladribine is approved in more than 50 other countries and was approved for use in the European Union in August 2017.
Valproate, topiramate prescribed in young women despite known teratogenicity risks
results of a retrospective analysis suggest.

Topiramate, linked to increased risk of cleft palate and smaller-than-gestational-age newborns, was among the top three antiepileptic drugs (AEDs) prescribed to women 15-44 years of age in the population-based cohort study.
Valproate, linked to increases in both anatomic and behavioral teratogenicity, was less often prescribed, but nevertheless still prescribed in a considerable proportion of patients in the study, which looked at U.S. commercial, Medicare, and Medicaid claims data from 2009 to 2013.
Presence of comorbidities could be influencing whether or not a woman of childbearing age receives one of these AEDs, the investigators said. Specifically, they found valproate more often prescribed for women with epilepsy who also had mood or anxiety and dissociative disorder, while topiramate was more often prescribed in women with headaches or migraines.
Taken together, these findings suggest a lack of awareness of the teratogenic risks of valproate and topiramate, said the investigators, led by Hyunmi Kim, MD, PhD, MPH, of the department of neurology at Stanford (Calif.) University.
“To improve current practice, knowledge of the teratogenicity of certain AEDs should be disseminated to health care professionals and patients,” they wrote. The report is in JAMA Neurology.
The findings of Dr. Kim and her colleagues were based on data for 46,767 women of childbearing age: 8,003 incident (new) cases with a mean age of 27 years, and 38,764 prevalent cases with a mean age of 30 years.
Topiramate was the second- or third-most prescribed AED in the analyses, alongside levetiracetam and lamotrigine. In particular, topiramate prescriptions were found in incident cases receiving first-line monotherapy (15%), prevalent cases receiving first-line monotherapy (13%), and prevalent cases receiving polytherapy (29%).
Valproate was the fifth-most prescribed AED for incident and prevalent cases receiving first-line monotherapy (5% and 10%, respectively), and came in fourth place among prevalent cases receiving polytherapy (22%).
The somewhat lower rate of valproate prescriptions tracks with other recent analyses showing that valproate use decreased among women of childbearing age following recommendations against its use during pregnancy, according to Dr. Kim and her coauthors.
However, topiramate is another story: “Although the magnitude of risk and range of adverse reproductive outcomes associated with topiramate use appear substantially less than those associated with valproate, some reduction in the use of topiramate in this population might be expected after evidence emerged in 2008 of its association with cleft palate,” they said in their report.
UCB Pharma sponsored this study. Study authors reported disclosures related to UCB Pharma, Biogen, Eisai, SK Life Science, Brain Sentinel, UCB Pharma, and the University of Alabama at Birmingham.
SOURCE: Kim H et al. JAMA Neurol. 2019 Apr 1. doi: 10.1001/jamaneurol.2019.0447.
results of a retrospective analysis suggest.
Topiramate, linked to increased risk of cleft palate and smaller-than-gestational-age newborns, was among the top three antiepileptic drugs (AEDs) prescribed to women 15-44 years of age in the population-based cohort study.
Valproate, linked to increases in both anatomic and behavioral teratogenicity, was less often prescribed, but nevertheless still prescribed in a considerable proportion of patients in the study, which looked at U.S. commercial, Medicare, and Medicaid claims data from 2009 to 2013.
Presence of comorbidities could be influencing whether or not a woman of childbearing age receives one of these AEDs, the investigators said. Specifically, they found valproate more often prescribed for women with epilepsy who also had mood or anxiety and dissociative disorder, while topiramate was more often prescribed in women with headaches or migraines.
Taken together, these findings suggest a lack of awareness of the teratogenic risks of valproate and topiramate, said the investigators, led by Hyunmi Kim, MD, PhD, MPH, of the department of neurology at Stanford (Calif.) University.
“To improve current practice, knowledge of the teratogenicity of certain AEDs should be disseminated to health care professionals and patients,” they wrote. The report is in JAMA Neurology.
The findings of Dr. Kim and her colleagues were based on data for 46,767 women of childbearing age: 8,003 incident (new) cases with a mean age of 27 years, and 38,764 prevalent cases with a mean age of 30 years.
Topiramate was the second- or third-most prescribed AED in the analyses, alongside levetiracetam and lamotrigine. In particular, topiramate prescriptions were found in incident cases receiving first-line monotherapy (15%), prevalent cases receiving first-line monotherapy (13%), and prevalent cases receiving polytherapy (29%).
Valproate was the fifth-most prescribed AED for incident and prevalent cases receiving first-line monotherapy (5% and 10%, respectively), and came in fourth place among prevalent cases receiving polytherapy (22%).
The somewhat lower rate of valproate prescriptions tracks with other recent analyses showing that valproate use decreased among women of childbearing age following recommendations against its use during pregnancy, according to Dr. Kim and her coauthors.
However, topiramate is another story: “Although the magnitude of risk and range of adverse reproductive outcomes associated with topiramate use appear substantially less than those associated with valproate, some reduction in the use of topiramate in this population might be expected after evidence emerged in 2008 of its association with cleft palate,” they said in their report.
UCB Pharma sponsored this study. Study authors reported disclosures related to UCB Pharma, Biogen, Eisai, SK Life Science, Brain Sentinel, UCB Pharma, and the University of Alabama at Birmingham.
SOURCE: Kim H et al. JAMA Neurol. 2019 Apr 1. doi: 10.1001/jamaneurol.2019.0447.
results of a retrospective analysis suggest.
Topiramate, linked to increased risk of cleft palate and smaller-than-gestational-age newborns, was among the top three antiepileptic drugs (AEDs) prescribed to women 15-44 years of age in the population-based cohort study.
Valproate, linked to increases in both anatomic and behavioral teratogenicity, was less often prescribed, but nevertheless still prescribed in a considerable proportion of patients in the study, which looked at U.S. commercial, Medicare, and Medicaid claims data from 2009 to 2013.
Presence of comorbidities could be influencing whether or not a woman of childbearing age receives one of these AEDs, the investigators said. Specifically, they found valproate more often prescribed for women with epilepsy who also had mood or anxiety and dissociative disorder, while topiramate was more often prescribed in women with headaches or migraines.
Taken together, these findings suggest a lack of awareness of the teratogenic risks of valproate and topiramate, said the investigators, led by Hyunmi Kim, MD, PhD, MPH, of the department of neurology at Stanford (Calif.) University.
“To improve current practice, knowledge of the teratogenicity of certain AEDs should be disseminated to health care professionals and patients,” they wrote. The report is in JAMA Neurology.
The findings of Dr. Kim and her colleagues were based on data for 46,767 women of childbearing age: 8,003 incident (new) cases with a mean age of 27 years, and 38,764 prevalent cases with a mean age of 30 years.
Topiramate was the second- or third-most prescribed AED in the analyses, alongside levetiracetam and lamotrigine. In particular, topiramate prescriptions were found in incident cases receiving first-line monotherapy (15%), prevalent cases receiving first-line monotherapy (13%), and prevalent cases receiving polytherapy (29%).
Valproate was the fifth-most prescribed AED for incident and prevalent cases receiving first-line monotherapy (5% and 10%, respectively), and came in fourth place among prevalent cases receiving polytherapy (22%).
The somewhat lower rate of valproate prescriptions tracks with other recent analyses showing that valproate use decreased among women of childbearing age following recommendations against its use during pregnancy, according to Dr. Kim and her coauthors.
However, topiramate is another story: “Although the magnitude of risk and range of adverse reproductive outcomes associated with topiramate use appear substantially less than those associated with valproate, some reduction in the use of topiramate in this population might be expected after evidence emerged in 2008 of its association with cleft palate,” they said in their report.
UCB Pharma sponsored this study. Study authors reported disclosures related to UCB Pharma, Biogen, Eisai, SK Life Science, Brain Sentinel, UCB Pharma, and the University of Alabama at Birmingham.
SOURCE: Kim H et al. JAMA Neurol. 2019 Apr 1. doi: 10.1001/jamaneurol.2019.0447.
FROM JAMA NEUROLOGY
Key clinical point: Both valproate and topiramate are prescribed relatively often in women of childbearing age despite known teratogenic risks.
Major finding: Topiramate was the second- or third-most prescribed AED in the analyses. Valproate was the fifth-most prescribed AED for incident and prevalent cases receiving first-line monotherapy.
Study details: Retrospective cohort study including nearly 47,000 women of childbearing age enrolled in claims databases between 2009 and 2013.
Disclosures: UCB Pharma sponsored the study. Study authors reported disclosures related to UCB Pharma, Biogen, Eisai, SK Life Science, Brain Sentinel, UCB Pharma, and the University of Alabama at Birmingham.
Source: Kim H et al. JAMA Neurol. 2019 Apr 1. doi: 10.1001/jamaneurol.2019.0447.
Gastroparesis in a patient with diabetic ketoacidosis
A 40-year-old man with type 1 diabetes mellitus and recurrent renal calculi presented to the emergency department with nausea, vomiting, and abdominal pain for the past day. He had been checking his blood glucose level regularly, and it had usually been within the normal range until 2 or 3 days previously, when he stopped taking his insulin because he ran out and could not afford to buy more.
He said he initially vomited clear mucus but then had 2 episodes of black vomit. His abdominal pain was diffuse but more intense in his flanks. He said he had never had nausea or vomiting before this episode.
In the emergency department, his heart rate was 136 beats per minute and respiratory rate 24 breaths per minute. He appeared to be in mild distress, and physical examination revealed a distended abdomen, decreased bowel sounds on auscultation, tympanic sound elicited by percussion, and diffuse abdominal tenderness to palpation without rebound tenderness or rigidity. His blood glucose level was 993 mg/dL, and his anion gap was 36 mmol/L.
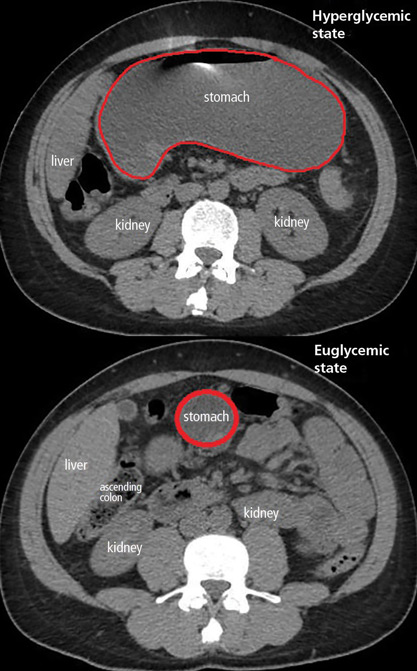
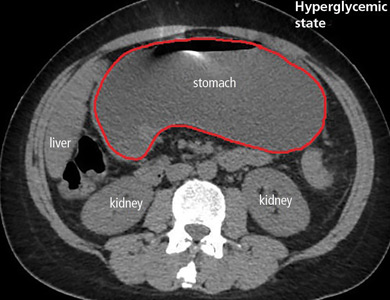
The patient was treated with hydration, insulin, and a nasogastric tube to relieve the pressure. The following day, his symptoms had significantly improved, his abdomen was less distended, his bowel sounds had returned, and his plasma glucose levels were in the normal range. The nasogastric tube was removed after he started to have bowel movements; he was given liquids by mouth and eventually solid food. Since his condition had significantly improved and he had started to have bowel movements, no follow-up imaging was done. The next day, he was symptom-free, his laboratory values were normal, and he was discharged home.
GASTROPARESIS
Gastroparesis is defined by delayed gastric emptying in the absence of a mechanical obstruction, with symptoms of nausea, vomiting, bloating, and abdominal pain. Most commonly it is idiopathic or caused by long-standing uncontrolled diabetes.
Diabetic gastroparesis is thought to result from impaired neural control of gastric function. Damage to the pacemaker interstitial cells of Cajal and underlying smooth muscle may be contributing factors.1 It is usually chronic, with a mean duration of symptoms of 26.5 months.2 However, acute gastroparesis can occur after an acute elevation in the plasma glucose concentration, which can affect gastric sensory and motor function3 via relaxation of the proximal stomach, decrease in antral pressure waves, and increase in pyloric pressure waves.4
Patients with diabetic ketoacidosis often present with symptoms similar to those of gastroparesis, including nausea, vomiting, and abdominal pain.5 But acute gastroparesis can coexist with diabetic ketoacidosis, as in our patient, and the gastroparesis can go undiagnosed, since imaging studies are not routinely done for diabetic ketoacidosis unless there is another reason—as in our patient.
More study is needed to answer questions on long-term outcomes for patients presenting with acute gastroparesis: Do they develop chronic gastroparesis? And is there is a correlation with progression of neuropathy?
The diagnosis usually requires a high level of suspicion in patients with nausea, vomiting, fullness, abdominal pain, and bloating; exclusion of gastric outlet obstruction by a mass or antral stenosis; and evidence of delayed gastric emptying. Gastric outlet obstruction can be ruled out by endoscopy, abdominal CT, or magnetic resonance enterography. Delayed gastric emptying can be quantified with scintigraphy and endoscopy. In our patient, gastroparesis was diagnosed on the basis of the clinical symptoms and CT findings.
Treatment is usually directed at symptoms, with better glycemic control and dietary modification for moderate cases, and prokinetics and a gastrostomy tube for severe cases.
TAKE-HOME POINTS
- Gastroparesis is usually chronic but can present acutely with acute severe hyperglycemia.
- Gastrointestinal tract motor function is affected by plasma glucose levels and can change over brief intervals.
- Diabetic ketoacidosis symptoms can mask acute gastroparesis, as imaging studies are not routinely done.
- Acute gastroparesis can be diagnosed clinically along with abdominal CT or endoscopy to rule out gastric outlet obstruction.
- Acute gastroparesis caused by diabetic ketoacidosis can resolve promptly with tight control of plasma glucose levels, anion gap closing, and nasogastric tube placement.
- Parkman HP, Hasler WL, Fisher RS; American Gastroenterological Association. American Gastroenterological Association technical review on the diagnosis and treatment of gastroparesis. Gastroenterology 2004; 127(5):1592–1622. pmid:15521026
- Dudekula A, O’Connell M, Bielefeldt K. Hospitalizations and testing in gastroparesis. J Gastroenterol Hepatol 2011; 26(8):1275–1282. doi:10.1111/j.1440-1746.2011.06735.x
- Fraser RJ, Horowitz M, Maddox AF, Harding PE, Chatterton BE, Dent J. Hyperglycaemia slows gastric emptying in type 1 (insulin-dependent) diabetes mellitus. Diabetologia 1990; 33(11):675–680. pmid:2076799
- Mearin F, Malagelada JR. Gastroparesis and dyspepsia in patients with diabetes mellitus. Eur J Gastroenterol Hepatol 1995; 7(8):717–723. pmid:7496857
- Malone ML, Gennis V, Goodwin JS. Characteristics of diabetic ketoacidosis in older versus younger adults. J Am Geriatr Soc 1992; 40(11):1100–1104. pmid:1401693
A 40-year-old man with type 1 diabetes mellitus and recurrent renal calculi presented to the emergency department with nausea, vomiting, and abdominal pain for the past day. He had been checking his blood glucose level regularly, and it had usually been within the normal range until 2 or 3 days previously, when he stopped taking his insulin because he ran out and could not afford to buy more.
He said he initially vomited clear mucus but then had 2 episodes of black vomit. His abdominal pain was diffuse but more intense in his flanks. He said he had never had nausea or vomiting before this episode.
In the emergency department, his heart rate was 136 beats per minute and respiratory rate 24 breaths per minute. He appeared to be in mild distress, and physical examination revealed a distended abdomen, decreased bowel sounds on auscultation, tympanic sound elicited by percussion, and diffuse abdominal tenderness to palpation without rebound tenderness or rigidity. His blood glucose level was 993 mg/dL, and his anion gap was 36 mmol/L.
The patient was treated with hydration, insulin, and a nasogastric tube to relieve the pressure. The following day, his symptoms had significantly improved, his abdomen was less distended, his bowel sounds had returned, and his plasma glucose levels were in the normal range. The nasogastric tube was removed after he started to have bowel movements; he was given liquids by mouth and eventually solid food. Since his condition had significantly improved and he had started to have bowel movements, no follow-up imaging was done. The next day, he was symptom-free, his laboratory values were normal, and he was discharged home.
GASTROPARESIS
Gastroparesis is defined by delayed gastric emptying in the absence of a mechanical obstruction, with symptoms of nausea, vomiting, bloating, and abdominal pain. Most commonly it is idiopathic or caused by long-standing uncontrolled diabetes.
Diabetic gastroparesis is thought to result from impaired neural control of gastric function. Damage to the pacemaker interstitial cells of Cajal and underlying smooth muscle may be contributing factors.1 It is usually chronic, with a mean duration of symptoms of 26.5 months.2 However, acute gastroparesis can occur after an acute elevation in the plasma glucose concentration, which can affect gastric sensory and motor function3 via relaxation of the proximal stomach, decrease in antral pressure waves, and increase in pyloric pressure waves.4
Patients with diabetic ketoacidosis often present with symptoms similar to those of gastroparesis, including nausea, vomiting, and abdominal pain.5 But acute gastroparesis can coexist with diabetic ketoacidosis, as in our patient, and the gastroparesis can go undiagnosed, since imaging studies are not routinely done for diabetic ketoacidosis unless there is another reason—as in our patient.
More study is needed to answer questions on long-term outcomes for patients presenting with acute gastroparesis: Do they develop chronic gastroparesis? And is there is a correlation with progression of neuropathy?
The diagnosis usually requires a high level of suspicion in patients with nausea, vomiting, fullness, abdominal pain, and bloating; exclusion of gastric outlet obstruction by a mass or antral stenosis; and evidence of delayed gastric emptying. Gastric outlet obstruction can be ruled out by endoscopy, abdominal CT, or magnetic resonance enterography. Delayed gastric emptying can be quantified with scintigraphy and endoscopy. In our patient, gastroparesis was diagnosed on the basis of the clinical symptoms and CT findings.
Treatment is usually directed at symptoms, with better glycemic control and dietary modification for moderate cases, and prokinetics and a gastrostomy tube for severe cases.
TAKE-HOME POINTS
- Gastroparesis is usually chronic but can present acutely with acute severe hyperglycemia.
- Gastrointestinal tract motor function is affected by plasma glucose levels and can change over brief intervals.
- Diabetic ketoacidosis symptoms can mask acute gastroparesis, as imaging studies are not routinely done.
- Acute gastroparesis can be diagnosed clinically along with abdominal CT or endoscopy to rule out gastric outlet obstruction.
- Acute gastroparesis caused by diabetic ketoacidosis can resolve promptly with tight control of plasma glucose levels, anion gap closing, and nasogastric tube placement.
A 40-year-old man with type 1 diabetes mellitus and recurrent renal calculi presented to the emergency department with nausea, vomiting, and abdominal pain for the past day. He had been checking his blood glucose level regularly, and it had usually been within the normal range until 2 or 3 days previously, when he stopped taking his insulin because he ran out and could not afford to buy more.
He said he initially vomited clear mucus but then had 2 episodes of black vomit. His abdominal pain was diffuse but more intense in his flanks. He said he had never had nausea or vomiting before this episode.
In the emergency department, his heart rate was 136 beats per minute and respiratory rate 24 breaths per minute. He appeared to be in mild distress, and physical examination revealed a distended abdomen, decreased bowel sounds on auscultation, tympanic sound elicited by percussion, and diffuse abdominal tenderness to palpation without rebound tenderness or rigidity. His blood glucose level was 993 mg/dL, and his anion gap was 36 mmol/L.
The patient was treated with hydration, insulin, and a nasogastric tube to relieve the pressure. The following day, his symptoms had significantly improved, his abdomen was less distended, his bowel sounds had returned, and his plasma glucose levels were in the normal range. The nasogastric tube was removed after he started to have bowel movements; he was given liquids by mouth and eventually solid food. Since his condition had significantly improved and he had started to have bowel movements, no follow-up imaging was done. The next day, he was symptom-free, his laboratory values were normal, and he was discharged home.
GASTROPARESIS
Gastroparesis is defined by delayed gastric emptying in the absence of a mechanical obstruction, with symptoms of nausea, vomiting, bloating, and abdominal pain. Most commonly it is idiopathic or caused by long-standing uncontrolled diabetes.
Diabetic gastroparesis is thought to result from impaired neural control of gastric function. Damage to the pacemaker interstitial cells of Cajal and underlying smooth muscle may be contributing factors.1 It is usually chronic, with a mean duration of symptoms of 26.5 months.2 However, acute gastroparesis can occur after an acute elevation in the plasma glucose concentration, which can affect gastric sensory and motor function3 via relaxation of the proximal stomach, decrease in antral pressure waves, and increase in pyloric pressure waves.4
Patients with diabetic ketoacidosis often present with symptoms similar to those of gastroparesis, including nausea, vomiting, and abdominal pain.5 But acute gastroparesis can coexist with diabetic ketoacidosis, as in our patient, and the gastroparesis can go undiagnosed, since imaging studies are not routinely done for diabetic ketoacidosis unless there is another reason—as in our patient.
More study is needed to answer questions on long-term outcomes for patients presenting with acute gastroparesis: Do they develop chronic gastroparesis? And is there is a correlation with progression of neuropathy?
The diagnosis usually requires a high level of suspicion in patients with nausea, vomiting, fullness, abdominal pain, and bloating; exclusion of gastric outlet obstruction by a mass or antral stenosis; and evidence of delayed gastric emptying. Gastric outlet obstruction can be ruled out by endoscopy, abdominal CT, or magnetic resonance enterography. Delayed gastric emptying can be quantified with scintigraphy and endoscopy. In our patient, gastroparesis was diagnosed on the basis of the clinical symptoms and CT findings.
Treatment is usually directed at symptoms, with better glycemic control and dietary modification for moderate cases, and prokinetics and a gastrostomy tube for severe cases.
TAKE-HOME POINTS
- Gastroparesis is usually chronic but can present acutely with acute severe hyperglycemia.
- Gastrointestinal tract motor function is affected by plasma glucose levels and can change over brief intervals.
- Diabetic ketoacidosis symptoms can mask acute gastroparesis, as imaging studies are not routinely done.
- Acute gastroparesis can be diagnosed clinically along with abdominal CT or endoscopy to rule out gastric outlet obstruction.
- Acute gastroparesis caused by diabetic ketoacidosis can resolve promptly with tight control of plasma glucose levels, anion gap closing, and nasogastric tube placement.
- Parkman HP, Hasler WL, Fisher RS; American Gastroenterological Association. American Gastroenterological Association technical review on the diagnosis and treatment of gastroparesis. Gastroenterology 2004; 127(5):1592–1622. pmid:15521026
- Dudekula A, O’Connell M, Bielefeldt K. Hospitalizations and testing in gastroparesis. J Gastroenterol Hepatol 2011; 26(8):1275–1282. doi:10.1111/j.1440-1746.2011.06735.x
- Fraser RJ, Horowitz M, Maddox AF, Harding PE, Chatterton BE, Dent J. Hyperglycaemia slows gastric emptying in type 1 (insulin-dependent) diabetes mellitus. Diabetologia 1990; 33(11):675–680. pmid:2076799
- Mearin F, Malagelada JR. Gastroparesis and dyspepsia in patients with diabetes mellitus. Eur J Gastroenterol Hepatol 1995; 7(8):717–723. pmid:7496857
- Malone ML, Gennis V, Goodwin JS. Characteristics of diabetic ketoacidosis in older versus younger adults. J Am Geriatr Soc 1992; 40(11):1100–1104. pmid:1401693
- Parkman HP, Hasler WL, Fisher RS; American Gastroenterological Association. American Gastroenterological Association technical review on the diagnosis and treatment of gastroparesis. Gastroenterology 2004; 127(5):1592–1622. pmid:15521026
- Dudekula A, O’Connell M, Bielefeldt K. Hospitalizations and testing in gastroparesis. J Gastroenterol Hepatol 2011; 26(8):1275–1282. doi:10.1111/j.1440-1746.2011.06735.x
- Fraser RJ, Horowitz M, Maddox AF, Harding PE, Chatterton BE, Dent J. Hyperglycaemia slows gastric emptying in type 1 (insulin-dependent) diabetes mellitus. Diabetologia 1990; 33(11):675–680. pmid:2076799
- Mearin F, Malagelada JR. Gastroparesis and dyspepsia in patients with diabetes mellitus. Eur J Gastroenterol Hepatol 1995; 7(8):717–723. pmid:7496857
- Malone ML, Gennis V, Goodwin JS. Characteristics of diabetic ketoacidosis in older versus younger adults. J Am Geriatr Soc 1992; 40(11):1100–1104. pmid:1401693
Is neuroimaging necessary to evaluate syncope?
A 40-year-old woman with a history of hypertension, who was recently started on a diuretic, presents to the emergency department after a witnessed syncopal event. She reports a prodrome of lightheadedness, nausea, and darkening of her vision that occurred a few seconds after standing, followed by loss of consciousness. She had a complete, spontaneous recovery after 10 seconds, but upon arousal she noticed she had lost bladder control.
Her blood pressure is 120/80 mm Hg supine, 110/70 mm Hg sitting, and 90/60 mm Hg standing. She has no focal neurologic deficits. The cardiac examination is normal, without murmurs, and electrocardiography shows sinus tachycardia (heart rate 110 bpm) without other abnormalities. Results of laboratory testing are unremarkable.
Should you order neuroimaging to evaluate for syncope?
DEFINITIONS, CLASSIFICATIONS
Syncope is an abrupt loss of consciousness due to transient global cerebral hypoperfusion, with a concomitant loss of postural tone and rapid, spontaneous recovery.1 Recovery from syncope is characterized by immediate restoration of orientation and normal behavior, although the period after recovery may be accompanied by fatigue.2
The European Society of Cardiology2 has classified syncope into 3 main categories: reflex (neurally mediated) syncope, syncope due to orthostatic hypotension, and cardiac syncope. Determining the cause is critical, as this determines the prognosis.
KEYS TO THE EVALUATION
According to the 2017 American College of Cardiology/American Heart Association (ACC/AHA) and the 2009 European Society of Cardiology guidelines, the evaluation of syncope should include a thorough history, taken from the patient and witnesses, and a complete physical examination. This can identify the cause of syncope in up to 50% of cases and differentiate between cardiac and noncardiac causes. Features that point to cardiac syncope include age older than 60, male sex, known heart disease, brief prodrome, syncope during exertion or when supine, first syncopal event, family history of sudden cardiac death, and abnormal physical examination.1
Features that suggest noncardiac syncope are young age; syncope only when standing; recurrent syncope; a prodrome of nausea, vomiting, and a warm sensation; and triggers such as dehydration, pain, distressful stimulus, cough, laugh micturition, defecation, and swallowing.1
Electrocardiography should follow the history and physical examination. When done at presentation, electrocardiography is diagnostic in only about 5% of cases. However, given the importance of the diagnosis, it remains an essential part of the initial evaluation of syncope.3
If a clear cause of syncope is identified at this point, no further workup is needed, and the cause of syncope should be addressed.1 If the cause is still unclear, the ACC/AHA guidelines recommend further evaluation based on the clinical presentation and risk stratification.
WHEN TO PURSUE ADDITIONAL TESTING
Routine use of additional testing is costly; tests should be ordered on the basis of their potential diagnostic and prognostic value. Additional evaluation should follow a stepwise approach and can include targeted blood work, autonomic nerve evaluation, tilt-table testing, transthoracic echocardiography, stress testing, electrocardiographic monitoring, and electrophysiologic testing.1

Syncope is rarely a manifestation of neurologic disease, yet 11% to 58% of patients with a first episode of uncomplicated syncope undergo extensive neuroimaging with magnetic resonance imaging, computed tomography, electroencephalography (EEG), and carotid ultrasonography.4 Evidence suggests that routine neurologic testing is of limited value given its low diagnostic yield and high cost.
Epilepsy is the most common neurologic cause of loss of consciousness but is estimated to account for less than 5% of patients with syncope.5 A thorough and thoughtful neurologic history and examination is often enough to distinguish between syncope, convulsive syncope, epileptic convulsions, and pseudosyncope.
In syncope, the loss of consciousness usually occurs 30 seconds to several minutes after standing. It presents with or without a prodrome (warmth, palpitations, and diaphoresis) and can be relieved with supine positioning. True loss of consciousness usually lasts less than a minute and is accompanied by loss of postural tone, with little or no fatigue in the recovery period.6
Conversely, in convulsive syncope, the prodrome can include pallor and diaphoresis. Loss of consciousness lasts about 30 seconds but is accompanied by fixed gaze, upward eye deviation, nuchal rigidity, tonic spasms, myoclonic jerks, tonic-clonic convulsions, and oral automatisms.6
Pseudosyncope is characterized by a prodrome of lightheadedness, shortness of breath, chest pain, and tingling sensations, followed by episodes of apparent loss of consciousness that last longer than several minutes and occur multiple times a day. During these episodes, patients purposefully try to avoid trauma when they lose consciousness, and almost always keep their eyes closed, in contrast to syncopal episodes, when the eyes are open and glassy.7
ROLE OF ELECTROENCEPHALOGRAPHY
If the diagnosis remains unclear after the history and neurologic examination, EEG is recommended (class IIa, ie, reasonable, can be useful) during tilt-table testing, as it can help differentiate syncope, pseudosyncope, and epilepsy.1
In an epileptic convulsion, EEG shows epileptiform discharges, whereas in syncope, it shows diffuse brainwave slowing with delta waves and a flatline pattern. In pseudosyncope and psychogenic nonepileptic seizures, EEG shows normal activity.8
Routine EEG is not recommended if there are no specific neurologic signs of epilepsy or if the history and neurologic examination indicate syncope or pseudosyncope.1
Structural brain disease does not typically present with transient global cerebral hypoperfusion resulting in syncope, so magnetic resonance imaging and computed tomography have a low diagnostic yield. Studies have revealed that for the 11% to 58% of patients who undergo neuroimaging, it establishes a diagnosis in only 0.2% to 1%.9 For this reason and in view of their high cost, these imaging tests should not be routinely ordered in the evaluation of syncope.4,10 Similarly, carotid artery imaging should not be routinely ordered if there is no focal neurologic finding suggesting unilateral ischemia.10
CASE CONTINUED
In our 40-year-old patient, the history suggests dehydration, as she recently started taking a diuretic. Thus, laboratory testing is reasonable.
Loss of bladder control is often interpreted as a red flag for neurologic disease, but syncope can often present with urinary incontinence. Urinary incontinence may also occur in epileptic seizure and in nonepileptic events such as syncope. A pooled analysis by Brigo et al11 determined that urinary incontinence had no value in distinguishing between epilepsy and syncope. Therefore, this physical finding should not incline the clinician to one diagnosis or the other.
Given our patient’s presentation, findings on physical examination, and absence of focal neurologic deficits, she should not undergo neuroimaging for syncope evaluation. The more likely cause of her syncope is orthostatic intolerance (orthostatic hypotension or vasovagal syncope) in the setting of intravascular volume depletion, likely secondary to diuretic use. Obtaining orthostatic vital signs is mandatory, and this confirms the diagnosis.
- Shen WK, Sheldon RS, Benditt DG, et al. 2017 ACC/AHA/HRS guideline for the evaluation and management of patients with syncope: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2017; 70(5):e39–e110. doi:10.1016/j.jacc.2017.03.003
- Task Force for the Diagnosis and Management of Syncope; European Society of Cardiology (ESC); European Heart Rhythm Association (EHRA); Heart Failure Association (HFA); Heart Rhythm Society (HRS), Moya A, Sutton R, Ammirati F, et al. Guidelines for the diagnosis and management of syncope (version 2009). Eur Heart J 2009; 30(21):2631–2671. doi:10.1093/eurheartj/ehp298
- Mehlsen J, Kaijer MN, Mehlsen AB. Autonomic and electrocardiographic changes in cardioinhibitory syncope. Europace 2008; 10(1):91–95. doi:10.1093/europace/eum237
- Goyal N, Donnino MW, Vachhani R, Bajwa R, Ahmad T, Otero R. The utility of head computed tomography in the emergency department evaluation of syncope. Intern Emerg Med 2006; 1(2):148–150. pmid:17111790
- Kapoor WN, Karpf M, Wieand S, Peterson JR, Levey GS. A prospective evaluation and follow-up of patients with syncope. N Engl J Med 1983; 309(4):197–204. doi:10.1056/NEJM198307283090401
- Sheldon R. How to differentiate syncope from seizure. Cardiol Clin 2015; 33(3):377–385. doi:10.1016/j.ccl.2015.04.006
- Raj V, Rowe AA, Fleisch SB, Paranjape SY, Arain AM, Nicolson SE. Psychogenic pseudosyncope: diagnosis and management. Auton Neurosci 2014; 184:66–72. doi:10.1016/j.autneu.2014.05.003
- Mecarelli O, Pulitano P, Vicenzini E, Vanacore N, Accornero N, De Marinis M. Observations on EEG patterns in neurally-mediated syncope: an inspective and quantitative study. Neurophysiol Clin 2004; 34(5):203–207. doi:10.1016/j.neucli.2004.09.004
- Johnson PC, Ammar H, Zohdy W, Fouda R, Govindu R. Yield of diagnostic tests and its impact on cost in adult patients with syncope presenting to a community hospital. South Med J 2014; 107(11):707–714. doi:10.14423/SMJ.0000000000000184
- Sclafani JJ, My J, Zacher LL, Eckart RE. Intensive education on evidence-based evaluation of syncope increases sudden death risk stratification but fails to reduce use of neuroimaging. Arch Intern Med 2010; 170(13):1150–1154. doi:10.1001/archinternmed.2010.205
- Brigo F, Nardone R Ausserer H, et al. The diagnostic value of urinary incontinence in the differential diagnosis of seizures. Seizure 2013; 22(2):85–90. doi:10.1016/j.seizure.2012.10.011
A 40-year-old woman with a history of hypertension, who was recently started on a diuretic, presents to the emergency department after a witnessed syncopal event. She reports a prodrome of lightheadedness, nausea, and darkening of her vision that occurred a few seconds after standing, followed by loss of consciousness. She had a complete, spontaneous recovery after 10 seconds, but upon arousal she noticed she had lost bladder control.
Her blood pressure is 120/80 mm Hg supine, 110/70 mm Hg sitting, and 90/60 mm Hg standing. She has no focal neurologic deficits. The cardiac examination is normal, without murmurs, and electrocardiography shows sinus tachycardia (heart rate 110 bpm) without other abnormalities. Results of laboratory testing are unremarkable.
Should you order neuroimaging to evaluate for syncope?
DEFINITIONS, CLASSIFICATIONS
Syncope is an abrupt loss of consciousness due to transient global cerebral hypoperfusion, with a concomitant loss of postural tone and rapid, spontaneous recovery.1 Recovery from syncope is characterized by immediate restoration of orientation and normal behavior, although the period after recovery may be accompanied by fatigue.2
The European Society of Cardiology2 has classified syncope into 3 main categories: reflex (neurally mediated) syncope, syncope due to orthostatic hypotension, and cardiac syncope. Determining the cause is critical, as this determines the prognosis.
KEYS TO THE EVALUATION
According to the 2017 American College of Cardiology/American Heart Association (ACC/AHA) and the 2009 European Society of Cardiology guidelines, the evaluation of syncope should include a thorough history, taken from the patient and witnesses, and a complete physical examination. This can identify the cause of syncope in up to 50% of cases and differentiate between cardiac and noncardiac causes. Features that point to cardiac syncope include age older than 60, male sex, known heart disease, brief prodrome, syncope during exertion or when supine, first syncopal event, family history of sudden cardiac death, and abnormal physical examination.1
Features that suggest noncardiac syncope are young age; syncope only when standing; recurrent syncope; a prodrome of nausea, vomiting, and a warm sensation; and triggers such as dehydration, pain, distressful stimulus, cough, laugh micturition, defecation, and swallowing.1
Electrocardiography should follow the history and physical examination. When done at presentation, electrocardiography is diagnostic in only about 5% of cases. However, given the importance of the diagnosis, it remains an essential part of the initial evaluation of syncope.3
If a clear cause of syncope is identified at this point, no further workup is needed, and the cause of syncope should be addressed.1 If the cause is still unclear, the ACC/AHA guidelines recommend further evaluation based on the clinical presentation and risk stratification.
WHEN TO PURSUE ADDITIONAL TESTING
Routine use of additional testing is costly; tests should be ordered on the basis of their potential diagnostic and prognostic value. Additional evaluation should follow a stepwise approach and can include targeted blood work, autonomic nerve evaluation, tilt-table testing, transthoracic echocardiography, stress testing, electrocardiographic monitoring, and electrophysiologic testing.1
Syncope is rarely a manifestation of neurologic disease, yet 11% to 58% of patients with a first episode of uncomplicated syncope undergo extensive neuroimaging with magnetic resonance imaging, computed tomography, electroencephalography (EEG), and carotid ultrasonography.4 Evidence suggests that routine neurologic testing is of limited value given its low diagnostic yield and high cost.
Epilepsy is the most common neurologic cause of loss of consciousness but is estimated to account for less than 5% of patients with syncope.5 A thorough and thoughtful neurologic history and examination is often enough to distinguish between syncope, convulsive syncope, epileptic convulsions, and pseudosyncope.
In syncope, the loss of consciousness usually occurs 30 seconds to several minutes after standing. It presents with or without a prodrome (warmth, palpitations, and diaphoresis) and can be relieved with supine positioning. True loss of consciousness usually lasts less than a minute and is accompanied by loss of postural tone, with little or no fatigue in the recovery period.6
Conversely, in convulsive syncope, the prodrome can include pallor and diaphoresis. Loss of consciousness lasts about 30 seconds but is accompanied by fixed gaze, upward eye deviation, nuchal rigidity, tonic spasms, myoclonic jerks, tonic-clonic convulsions, and oral automatisms.6
Pseudosyncope is characterized by a prodrome of lightheadedness, shortness of breath, chest pain, and tingling sensations, followed by episodes of apparent loss of consciousness that last longer than several minutes and occur multiple times a day. During these episodes, patients purposefully try to avoid trauma when they lose consciousness, and almost always keep their eyes closed, in contrast to syncopal episodes, when the eyes are open and glassy.7
ROLE OF ELECTROENCEPHALOGRAPHY
If the diagnosis remains unclear after the history and neurologic examination, EEG is recommended (class IIa, ie, reasonable, can be useful) during tilt-table testing, as it can help differentiate syncope, pseudosyncope, and epilepsy.1
In an epileptic convulsion, EEG shows epileptiform discharges, whereas in syncope, it shows diffuse brainwave slowing with delta waves and a flatline pattern. In pseudosyncope and psychogenic nonepileptic seizures, EEG shows normal activity.8
Routine EEG is not recommended if there are no specific neurologic signs of epilepsy or if the history and neurologic examination indicate syncope or pseudosyncope.1
Structural brain disease does not typically present with transient global cerebral hypoperfusion resulting in syncope, so magnetic resonance imaging and computed tomography have a low diagnostic yield. Studies have revealed that for the 11% to 58% of patients who undergo neuroimaging, it establishes a diagnosis in only 0.2% to 1%.9 For this reason and in view of their high cost, these imaging tests should not be routinely ordered in the evaluation of syncope.4,10 Similarly, carotid artery imaging should not be routinely ordered if there is no focal neurologic finding suggesting unilateral ischemia.10
CASE CONTINUED
In our 40-year-old patient, the history suggests dehydration, as she recently started taking a diuretic. Thus, laboratory testing is reasonable.
Loss of bladder control is often interpreted as a red flag for neurologic disease, but syncope can often present with urinary incontinence. Urinary incontinence may also occur in epileptic seizure and in nonepileptic events such as syncope. A pooled analysis by Brigo et al11 determined that urinary incontinence had no value in distinguishing between epilepsy and syncope. Therefore, this physical finding should not incline the clinician to one diagnosis or the other.
Given our patient’s presentation, findings on physical examination, and absence of focal neurologic deficits, she should not undergo neuroimaging for syncope evaluation. The more likely cause of her syncope is orthostatic intolerance (orthostatic hypotension or vasovagal syncope) in the setting of intravascular volume depletion, likely secondary to diuretic use. Obtaining orthostatic vital signs is mandatory, and this confirms the diagnosis.
A 40-year-old woman with a history of hypertension, who was recently started on a diuretic, presents to the emergency department after a witnessed syncopal event. She reports a prodrome of lightheadedness, nausea, and darkening of her vision that occurred a few seconds after standing, followed by loss of consciousness. She had a complete, spontaneous recovery after 10 seconds, but upon arousal she noticed she had lost bladder control.
Her blood pressure is 120/80 mm Hg supine, 110/70 mm Hg sitting, and 90/60 mm Hg standing. She has no focal neurologic deficits. The cardiac examination is normal, without murmurs, and electrocardiography shows sinus tachycardia (heart rate 110 bpm) without other abnormalities. Results of laboratory testing are unremarkable.
Should you order neuroimaging to evaluate for syncope?
DEFINITIONS, CLASSIFICATIONS
Syncope is an abrupt loss of consciousness due to transient global cerebral hypoperfusion, with a concomitant loss of postural tone and rapid, spontaneous recovery.1 Recovery from syncope is characterized by immediate restoration of orientation and normal behavior, although the period after recovery may be accompanied by fatigue.2
The European Society of Cardiology2 has classified syncope into 3 main categories: reflex (neurally mediated) syncope, syncope due to orthostatic hypotension, and cardiac syncope. Determining the cause is critical, as this determines the prognosis.
KEYS TO THE EVALUATION
According to the 2017 American College of Cardiology/American Heart Association (ACC/AHA) and the 2009 European Society of Cardiology guidelines, the evaluation of syncope should include a thorough history, taken from the patient and witnesses, and a complete physical examination. This can identify the cause of syncope in up to 50% of cases and differentiate between cardiac and noncardiac causes. Features that point to cardiac syncope include age older than 60, male sex, known heart disease, brief prodrome, syncope during exertion or when supine, first syncopal event, family history of sudden cardiac death, and abnormal physical examination.1
Features that suggest noncardiac syncope are young age; syncope only when standing; recurrent syncope; a prodrome of nausea, vomiting, and a warm sensation; and triggers such as dehydration, pain, distressful stimulus, cough, laugh micturition, defecation, and swallowing.1
Electrocardiography should follow the history and physical examination. When done at presentation, electrocardiography is diagnostic in only about 5% of cases. However, given the importance of the diagnosis, it remains an essential part of the initial evaluation of syncope.3
If a clear cause of syncope is identified at this point, no further workup is needed, and the cause of syncope should be addressed.1 If the cause is still unclear, the ACC/AHA guidelines recommend further evaluation based on the clinical presentation and risk stratification.
WHEN TO PURSUE ADDITIONAL TESTING
Routine use of additional testing is costly; tests should be ordered on the basis of their potential diagnostic and prognostic value. Additional evaluation should follow a stepwise approach and can include targeted blood work, autonomic nerve evaluation, tilt-table testing, transthoracic echocardiography, stress testing, electrocardiographic monitoring, and electrophysiologic testing.1
Syncope is rarely a manifestation of neurologic disease, yet 11% to 58% of patients with a first episode of uncomplicated syncope undergo extensive neuroimaging with magnetic resonance imaging, computed tomography, electroencephalography (EEG), and carotid ultrasonography.4 Evidence suggests that routine neurologic testing is of limited value given its low diagnostic yield and high cost.
Epilepsy is the most common neurologic cause of loss of consciousness but is estimated to account for less than 5% of patients with syncope.5 A thorough and thoughtful neurologic history and examination is often enough to distinguish between syncope, convulsive syncope, epileptic convulsions, and pseudosyncope.
In syncope, the loss of consciousness usually occurs 30 seconds to several minutes after standing. It presents with or without a prodrome (warmth, palpitations, and diaphoresis) and can be relieved with supine positioning. True loss of consciousness usually lasts less than a minute and is accompanied by loss of postural tone, with little or no fatigue in the recovery period.6
Conversely, in convulsive syncope, the prodrome can include pallor and diaphoresis. Loss of consciousness lasts about 30 seconds but is accompanied by fixed gaze, upward eye deviation, nuchal rigidity, tonic spasms, myoclonic jerks, tonic-clonic convulsions, and oral automatisms.6
Pseudosyncope is characterized by a prodrome of lightheadedness, shortness of breath, chest pain, and tingling sensations, followed by episodes of apparent loss of consciousness that last longer than several minutes and occur multiple times a day. During these episodes, patients purposefully try to avoid trauma when they lose consciousness, and almost always keep their eyes closed, in contrast to syncopal episodes, when the eyes are open and glassy.7
ROLE OF ELECTROENCEPHALOGRAPHY
If the diagnosis remains unclear after the history and neurologic examination, EEG is recommended (class IIa, ie, reasonable, can be useful) during tilt-table testing, as it can help differentiate syncope, pseudosyncope, and epilepsy.1
In an epileptic convulsion, EEG shows epileptiform discharges, whereas in syncope, it shows diffuse brainwave slowing with delta waves and a flatline pattern. In pseudosyncope and psychogenic nonepileptic seizures, EEG shows normal activity.8
Routine EEG is not recommended if there are no specific neurologic signs of epilepsy or if the history and neurologic examination indicate syncope or pseudosyncope.1
Structural brain disease does not typically present with transient global cerebral hypoperfusion resulting in syncope, so magnetic resonance imaging and computed tomography have a low diagnostic yield. Studies have revealed that for the 11% to 58% of patients who undergo neuroimaging, it establishes a diagnosis in only 0.2% to 1%.9 For this reason and in view of their high cost, these imaging tests should not be routinely ordered in the evaluation of syncope.4,10 Similarly, carotid artery imaging should not be routinely ordered if there is no focal neurologic finding suggesting unilateral ischemia.10
CASE CONTINUED
In our 40-year-old patient, the history suggests dehydration, as she recently started taking a diuretic. Thus, laboratory testing is reasonable.
Loss of bladder control is often interpreted as a red flag for neurologic disease, but syncope can often present with urinary incontinence. Urinary incontinence may also occur in epileptic seizure and in nonepileptic events such as syncope. A pooled analysis by Brigo et al11 determined that urinary incontinence had no value in distinguishing between epilepsy and syncope. Therefore, this physical finding should not incline the clinician to one diagnosis or the other.
Given our patient’s presentation, findings on physical examination, and absence of focal neurologic deficits, she should not undergo neuroimaging for syncope evaluation. The more likely cause of her syncope is orthostatic intolerance (orthostatic hypotension or vasovagal syncope) in the setting of intravascular volume depletion, likely secondary to diuretic use. Obtaining orthostatic vital signs is mandatory, and this confirms the diagnosis.
- Shen WK, Sheldon RS, Benditt DG, et al. 2017 ACC/AHA/HRS guideline for the evaluation and management of patients with syncope: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2017; 70(5):e39–e110. doi:10.1016/j.jacc.2017.03.003
- Task Force for the Diagnosis and Management of Syncope; European Society of Cardiology (ESC); European Heart Rhythm Association (EHRA); Heart Failure Association (HFA); Heart Rhythm Society (HRS), Moya A, Sutton R, Ammirati F, et al. Guidelines for the diagnosis and management of syncope (version 2009). Eur Heart J 2009; 30(21):2631–2671. doi:10.1093/eurheartj/ehp298
- Mehlsen J, Kaijer MN, Mehlsen AB. Autonomic and electrocardiographic changes in cardioinhibitory syncope. Europace 2008; 10(1):91–95. doi:10.1093/europace/eum237
- Goyal N, Donnino MW, Vachhani R, Bajwa R, Ahmad T, Otero R. The utility of head computed tomography in the emergency department evaluation of syncope. Intern Emerg Med 2006; 1(2):148–150. pmid:17111790
- Kapoor WN, Karpf M, Wieand S, Peterson JR, Levey GS. A prospective evaluation and follow-up of patients with syncope. N Engl J Med 1983; 309(4):197–204. doi:10.1056/NEJM198307283090401
- Sheldon R. How to differentiate syncope from seizure. Cardiol Clin 2015; 33(3):377–385. doi:10.1016/j.ccl.2015.04.006
- Raj V, Rowe AA, Fleisch SB, Paranjape SY, Arain AM, Nicolson SE. Psychogenic pseudosyncope: diagnosis and management. Auton Neurosci 2014; 184:66–72. doi:10.1016/j.autneu.2014.05.003
- Mecarelli O, Pulitano P, Vicenzini E, Vanacore N, Accornero N, De Marinis M. Observations on EEG patterns in neurally-mediated syncope: an inspective and quantitative study. Neurophysiol Clin 2004; 34(5):203–207. doi:10.1016/j.neucli.2004.09.004
- Johnson PC, Ammar H, Zohdy W, Fouda R, Govindu R. Yield of diagnostic tests and its impact on cost in adult patients with syncope presenting to a community hospital. South Med J 2014; 107(11):707–714. doi:10.14423/SMJ.0000000000000184
- Sclafani JJ, My J, Zacher LL, Eckart RE. Intensive education on evidence-based evaluation of syncope increases sudden death risk stratification but fails to reduce use of neuroimaging. Arch Intern Med 2010; 170(13):1150–1154. doi:10.1001/archinternmed.2010.205
- Brigo F, Nardone R Ausserer H, et al. The diagnostic value of urinary incontinence in the differential diagnosis of seizures. Seizure 2013; 22(2):85–90. doi:10.1016/j.seizure.2012.10.011
- Shen WK, Sheldon RS, Benditt DG, et al. 2017 ACC/AHA/HRS guideline for the evaluation and management of patients with syncope: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2017; 70(5):e39–e110. doi:10.1016/j.jacc.2017.03.003
- Task Force for the Diagnosis and Management of Syncope; European Society of Cardiology (ESC); European Heart Rhythm Association (EHRA); Heart Failure Association (HFA); Heart Rhythm Society (HRS), Moya A, Sutton R, Ammirati F, et al. Guidelines for the diagnosis and management of syncope (version 2009). Eur Heart J 2009; 30(21):2631–2671. doi:10.1093/eurheartj/ehp298
- Mehlsen J, Kaijer MN, Mehlsen AB. Autonomic and electrocardiographic changes in cardioinhibitory syncope. Europace 2008; 10(1):91–95. doi:10.1093/europace/eum237
- Goyal N, Donnino MW, Vachhani R, Bajwa R, Ahmad T, Otero R. The utility of head computed tomography in the emergency department evaluation of syncope. Intern Emerg Med 2006; 1(2):148–150. pmid:17111790
- Kapoor WN, Karpf M, Wieand S, Peterson JR, Levey GS. A prospective evaluation and follow-up of patients with syncope. N Engl J Med 1983; 309(4):197–204. doi:10.1056/NEJM198307283090401
- Sheldon R. How to differentiate syncope from seizure. Cardiol Clin 2015; 33(3):377–385. doi:10.1016/j.ccl.2015.04.006
- Raj V, Rowe AA, Fleisch SB, Paranjape SY, Arain AM, Nicolson SE. Psychogenic pseudosyncope: diagnosis and management. Auton Neurosci 2014; 184:66–72. doi:10.1016/j.autneu.2014.05.003
- Mecarelli O, Pulitano P, Vicenzini E, Vanacore N, Accornero N, De Marinis M. Observations on EEG patterns in neurally-mediated syncope: an inspective and quantitative study. Neurophysiol Clin 2004; 34(5):203–207. doi:10.1016/j.neucli.2004.09.004
- Johnson PC, Ammar H, Zohdy W, Fouda R, Govindu R. Yield of diagnostic tests and its impact on cost in adult patients with syncope presenting to a community hospital. South Med J 2014; 107(11):707–714. doi:10.14423/SMJ.0000000000000184
- Sclafani JJ, My J, Zacher LL, Eckart RE. Intensive education on evidence-based evaluation of syncope increases sudden death risk stratification but fails to reduce use of neuroimaging. Arch Intern Med 2010; 170(13):1150–1154. doi:10.1001/archinternmed.2010.205
- Brigo F, Nardone R Ausserer H, et al. The diagnostic value of urinary incontinence in the differential diagnosis of seizures. Seizure 2013; 22(2):85–90. doi:10.1016/j.seizure.2012.10.011
The angry disciple
CASE Disorganized thoughts and grandiose delusions
Mr. J, age 54, presents to the psychiatric emergency department (ED) with agitation and disruptive behavior. He claims that he is “the son of Jesus Christ” and has to travel to the Middle East to be baptized. Mr. J is irritable, shouting, and threatening staff members. He receives
The next day, he is calm and cooperative, but continues to express the same religious delusions. Mr. J is admitted to the psychiatric inpatient unit for further evaluation.
On the unit, Mr. J is pleasant and cooperative, but tangential in thought process. He reports he was “saved” by God 4 years ago, and that God communicates with him through music. Despite this, he denies having auditory or visual hallucinations.
Approximately 3 months earlier, Mr. J had stopped working and left his home and family in another state to pursue his “mission” of being baptized in the Middle East. Mr. J has been homeless since then. Despite that, he reports that his mood is “great” and denies any recent changes in mood, sleep, appetite, energy level, or psychomotor agitation. Although no formal cognitive testing is performed, Mr. J is alert and oriented to person, place, and time with intact remote and recent memory, no language deficits, and no lapses in concentration or attention throughout interview.
Mr. J says he has been drinking alcohol regularly throughout his adult life, often a few times per week, up to “a case and a half” of beer at times. He claims he’s had multiple periods of sobriety but denies having experienced withdrawal symptoms during those times. Mr. J reports 1 prior psychiatric hospitalization 25 years ago after attempting suicide by overdose following the loss of a loved one. At that time, he was diagnosed with posttraumatic stress disorder (PTSD). During this admission, he denies having any symptoms of PTSD or periods of mania or depression, and he has not undergone psychiatric treatment since he had been diagnosed with PTSD. He denies any family history of psychiatric illness as well as any medical comorbidities or medication use.
[polldaddy:10279202]
The authors’ observations
Mr. J’s presentation had a wide differential diagnosis (Table 1). The initial agitation Mr. J displayed in the psychiatric ED was likely secondary to acute alcohol intoxication, given that he was subsequently pleasant, calm, and cooperative after the alcohol was metabolized. Despite this, Mr. J continued to demonstrate delusions of a religious and somewhat grandiose nature with tangential thought processes, which made substance-induced psychosis less likely to be the sole diagnosis. Although it is possible to develop psychotic symptoms due to severe alcohol withdrawal (alcoholic hallucinosis), Mr. J’s vital signs remained stable, and he demonstrated no other signs or symptoms of withdrawal throughout his hospitalization. His presentation also did not fit that of delirium tremens because he was not confused or disoriented, and did not demonstrate perceptual disturbance.

While delusions were the most prominent feature of Mr. J’s apparent psychosis, the presence of disorganized thought processes and impaired functioning, as evidenced by Mr. J’s unemployment and recent homelessness, were more consistent with a primary psychotic disorder than a delusional disorder.1
Continue to: Mr. J began to exhibit...
Mr. J began to exhibit these psychotic symptoms in his early 50s; because the average age of onset of schizophrenia for males is approximately age 20 to 25, the likelihood of his presentation being the result of a primary psychotic disorder was low.1 Although less common, it was possible that Mr. J had developed late-onset schizophrenia, where the first episode typically occurs after approximately age 40 to 45. Mr. J also described that he was in a “great” mood but had grandiose delusions and had made recent impulsive decisions, which suggests there was a possible mood component to his presentation and a potential diagnosis of schizoaffective disorder or bipolar disorder with psychotic symptoms. However, before any of these diagnoses could be made, a medical or neurologic condition that could cause his symptoms needed to be investigated and ruled out. Further collateral information regarding Mr. J’s history and timeline of symptoms was required.
EVALUATION Family history reveals clues
All laboratory studies completed during Mr. J’s hospitalization are unremarkable, including complete blood count, basic metabolic panel, hepatic function panel, gamma-glutamyl transferase test, magnesium, phosphate, thyroid-stimulating hormone, vitamin B12, thiamine, folate, urinalysis, and urine drug screen. Mr. J does not undergo any head imaging.
Mr. J has not been in touch with his family since leaving his home approximately 3 months before he presented to the ED, and he gives consent for the inpatient team to attempt to contact them. One week into hospitalization, Mr. J’s sibling informs the team of a family history of genetically confirmed Huntington’s disease (HD), with psychiatric symptoms preceding the onset of motor symptoms in multiple first-degree relatives. His family says that before Mr. J first developed delusions 4 years ago, he had not exhibited any psychotic symptoms during periods of alcohol use or sobriety.
Mr. J does not demonstrate any overt movement symptoms on the unit and denies noting any rigidity, change in gait, or abnormal/uncontrolled movements. The inpatient psychiatric team consults neurology and a full neurologic evaluation is performed. The results are unremarkable outside of his psychiatric symptoms; specifically, Mr. J does not demonstrate even subtle motor signs or cognitive impairment. Given Mr. J’s family history, unremarkable lab findings, and age at presentation, the neurology team and inpatient psychiatry team suspect that his psychosis is likely an early presentation of HD.
[polldaddy:10279212]
The authors’ observations
Genetics of Huntington’s disease
Huntington’s disease is an autosomal dominant neurodegenerative disorder caused by expansion of cytosine-adenine-guanine (CAG) trinucleotide repeats within the Huntingtin (HTT) gene on chromosome 4, which codes for the huntingtin protein.2,3 While the function of “normal” huntingtin protein is not fully understood, it is known that CAG repeat expansion in the HTT gene of >35 repeats codes for a mutant huntingtin protein.2,3 The mutant huntingtin protein causes progressive neuronal loss in the basal ganglia and striatum, resulting in the clinical Huntington’s phenotype.3 Notably, the patient’s age at disease onset is inversely correlated with the number of repeats. For example, expansions of approximately 40 to 50 CAG repeats often result in adult-onset HD, while expansions of >60 repeats are typically associated with juvenile-onset HD (before age 20). CAG repeat lengths of approximately 36 to 39 demonstrate reduced penetrance, with some individuals developing symptomatic HD while others do not.2 Instability of the CAG repeat expansion can result in genetic “anticipation,” wherein repeat length increases between generations, causing earlier age of onset in affected offspring. Genetic anticipation in HD occurs more frequently in paternal transmission—approximately 80% to 90% of juvenile HD cases are inherited paternally, at times with the number of CAG repeats exceeding 200.3
Continue to: Psychiatric manifestations of Huntington's disease
Psychiatric manifestations of Huntington’s disease
Huntington’s disease is characterized by motor, cognitive, and behavioral disturbances (Table 22,4). Motor symptoms include a characteristic and well-recognized chorea, often predominating earlier in HD, that progresses to rigidity, spasticity, and bradykinesia later in the disease course.2 Cognitive impairments develop in a similar progressive manner and can often precede the onset of motor symptoms, beginning with early executive dysfunction. Thinking often becomes more rigid and less efficient, causing difficulty with multi-tasking and concentration, and often progressing to subcortical dementia.2

Psychiatric symptoms have long been recognized as a feature of HD; the estimated lifetime prevalence in patients with HD ranges from approximately 33% to 76%.4 Depressed mood, anxiety, irritability, and apathy are the most commonly reported symptoms, while a smaller percentage of patients with HD can experience obsessive-compulsive disorder (10% to 52%) or psychotic symptoms (3% to 11%).4 A more specific schizophrenia-like psychosis occurs in approximately 3% to 6% of patients, and often is a paranoid type.5,6 Positive psychotic symptoms, such as hallucinations and delusions, typically become less overt as HD progresses and cognitive impairments worsen.7
Although the onset of motor symptoms leads to diagnosis in the majority of patients with HD, many patients present with psychiatric symptoms—most commonly depression—prior to motor symptoms.8 An increasing body of literature details instances of psychosis preceding motor symptom onset by up to 10 years.6,9-12 In many of these cases, the patient has a family history of HD-associated psychosis. Family history is a major risk factor for HD-associated psychosis, as is early-onset HD.7,9
TREATMENT Antipsychotics result in some improvement
On Day 1 or 2, Mr. J is started on risperidone, 1 mg twice daily, to manage his symptoms. He shows incremental improvement in thought organization. Although his religious and grandiose delusions persist, they become less fixed, and he is able to take the team’s suggestion that he reconnect with his family.
Mr. J is aware of his family history of HD and acknowledges that multiple relatives had early psychiatric manifestations of HD. Despite this, he still has difficulty recognizing any connection between other family members’ presentation and his own. The psychiatry and neurology teams discuss the process, ethics, and implications of genetic testing for HD with Mr. J; however, he is ambivalent regarding genetic testing, and states he would consider it after discussing it with his family.
Continue to: The neurology team recommends...
The neurology team recommends against imaging for Mr. J because HD-related changes are not typically seen until later in the disease progression. On Day 9, they recommend changing from risperidone to quetiapine (50 mg every night at bedtime) due to evidence of its effectiveness specifically for treating behavioral symptoms of HD.13
While receiving quetiapine, Mr. J experiences significant drowsiness. Because he had experienced improvement in thought organization while he was receiving risperidone, he is switched back to risperidone.
[polldaddy:10279220]
The authors’ observations
Currently, no treatments are available to prevent the development or progression of HD. However, symptomatic treatment of motor and behavioral disturbances can lead to functional improvement and improved quality of life for individuals affected by HD.
There are no extensive clinical trials to date, but multiple case reports and studies have shown second-generation antipsychotics (SGAs), including quetiapine, olanzapine,
OUTCOME Discharge despite persistent delusions
Mr. J’s religious and grandiose delusions continue throughout hospitalization despite treatment with antipsychotics. However, because he remains calm and cooperative and demonstrates improvement in thought organization, he is deemed safe for discharge and instructed to continue risperidone. The team coordinates with Mr. J’s family to arrange transportation home and outpatient neurology follow-up.
Bottom Line
Psychiatric manifestations, including psychosis, are prominent symptoms of Huntington’s disease (HD) and may precede the onset of more readily recognized motor symptoms. This poses a diagnostic challenge, and clinicians should remain cognizant of this possibility, especially in patients with a family history of HD-associated psychosis.
Related Resources
- Huntington’s Disease Society of America. http://hdsa.org.
- National Institute of Neurological Disorders and Stroke. Huntington’s disease information page: What research is being done? https://www.ninds.nih.gov/Disorders/All-Disorders/Huntingtons-Disease-Information-Page.
- Scher LM. How to target psychiatric symptoms of Huntington’s disease. Current Psychiatry. 2012;11(9):34-39.
Drug Brand Names
Aripiprazole • Abilify
Clozapine • Clozaril
Haloperidol • Haldol
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
2. Novak MJ, Tabrizi SJ. Huntington’s disease: clinical presentation and treatment. Int Rev Neurobiol. 2011;98:297-323.
3. Reiner A, Dragatsis I, Dietrich P. Genetics and neuropathology of Huntington’s disease. Int Rev Neurobiol. 2011;98:325-372.
4. van Duijn E, Kingma EM, Van der mast RC. Psychopathology in verified Huntington’s disease gene carriers. J Neuropsychiatry Clin Neurosci. 2007;19(4):441-448.
5. Naarding P, Kremer HP, Zitman FG. Huntington’s disease: a review of the literature on prevalence and treatment of neuropsychiatric phenomena. Eur Psychiatry. 2001;16(8):439-445.
6. Xu C, Yogaratnam J, Tan N, et al. Psychosis, treatment emergent extrapyramidal events, and subsequent onset of Huntington’s disease: a case report and review of the literature. Clin Psychopharmacol Neurosci. 2016;14(3):302-304.
7. Mendez MF. Huntington’s disease: update and review of neuropsychiatric aspects. Int J Psychiatry Med. 1994;24(3):189-208.
8. Di Maio L, Squitieri F, Napolitano G, et al. Onset symptoms in 510 patients with Huntington’s disease. J Med Genet. 1993;30(4):289-292.
9. Jauhar S, Ritchie S. Psychiatric and behavioural manifestations of Huntington’s disease. Adv Psychiatr Treat. 2010;16(3):168-175.
10. Nagel M, Rumpf HJ, Kasten M. Acute psychosis in a verified Huntington disease gene carrier with subtle motor signs: psychiatric criteria should be considered for the diagnosis. Gen Hosp Psychiatry. 2014;36(3):361.e3-e4. doi: 10.1016/j.genhosppsych.2014.01.008.
11. Corrêa BB, Xavier M, Guimarães J. Association of Huntington’s disease and schizophrenia-like psychosis in a Huntington’s disease pedigree. Clin Pract Epidemiol Ment Health. 2006;2:1.
12. Ding J, Gadit AM. Psychosis with Huntington’s disease: role of antipsychotic medications. BMJ Case Rep. 2014: bcr2013202625. doi: 10.1136/bcr-2013-202625.
13. Alpay M, Koroshetz WJ. Quetiapine in the treatment of behavioral disturbances in patients with Huntington’s disease. Psychosomatics. 2006;47(1):70-72.
14. Duff K, Beglinger LJ, O’Rourke ME, et al. Risperidone and the treatment of psychiatric, motor, and cognitive symptoms in Huntington’s disease. Ann Clin Psychiatry. 2008;20(1):1-3.
15. Paleacu D, Anca M, Giladi N. Olanzapine in Huntington’s disease. Acta Neurol Scand. 2002;105(6):441-444.
16. Lin W, Chou Y. Aripiprazole effects on psychosis and chorea in a patient with Huntington’s disease. Am J Psychiatry. 2008;165(9):1207-1208.
17. van Vugt JP, Siesling S, Vergeer M, et al. Clozapine versus placebo in Huntington’s disease: a double blind randomized comparative study. J Neurol Neurosurg Psychiatr. 1997;63(1):35-39.
CASE Disorganized thoughts and grandiose delusions
Mr. J, age 54, presents to the psychiatric emergency department (ED) with agitation and disruptive behavior. He claims that he is “the son of Jesus Christ” and has to travel to the Middle East to be baptized. Mr. J is irritable, shouting, and threatening staff members. He receives
The next day, he is calm and cooperative, but continues to express the same religious delusions. Mr. J is admitted to the psychiatric inpatient unit for further evaluation.
On the unit, Mr. J is pleasant and cooperative, but tangential in thought process. He reports he was “saved” by God 4 years ago, and that God communicates with him through music. Despite this, he denies having auditory or visual hallucinations.
Approximately 3 months earlier, Mr. J had stopped working and left his home and family in another state to pursue his “mission” of being baptized in the Middle East. Mr. J has been homeless since then. Despite that, he reports that his mood is “great” and denies any recent changes in mood, sleep, appetite, energy level, or psychomotor agitation. Although no formal cognitive testing is performed, Mr. J is alert and oriented to person, place, and time with intact remote and recent memory, no language deficits, and no lapses in concentration or attention throughout interview.
Mr. J says he has been drinking alcohol regularly throughout his adult life, often a few times per week, up to “a case and a half” of beer at times. He claims he’s had multiple periods of sobriety but denies having experienced withdrawal symptoms during those times. Mr. J reports 1 prior psychiatric hospitalization 25 years ago after attempting suicide by overdose following the loss of a loved one. At that time, he was diagnosed with posttraumatic stress disorder (PTSD). During this admission, he denies having any symptoms of PTSD or periods of mania or depression, and he has not undergone psychiatric treatment since he had been diagnosed with PTSD. He denies any family history of psychiatric illness as well as any medical comorbidities or medication use.
[polldaddy:10279202]
The authors’ observations
Mr. J’s presentation had a wide differential diagnosis (Table 1). The initial agitation Mr. J displayed in the psychiatric ED was likely secondary to acute alcohol intoxication, given that he was subsequently pleasant, calm, and cooperative after the alcohol was metabolized. Despite this, Mr. J continued to demonstrate delusions of a religious and somewhat grandiose nature with tangential thought processes, which made substance-induced psychosis less likely to be the sole diagnosis. Although it is possible to develop psychotic symptoms due to severe alcohol withdrawal (alcoholic hallucinosis), Mr. J’s vital signs remained stable, and he demonstrated no other signs or symptoms of withdrawal throughout his hospitalization. His presentation also did not fit that of delirium tremens because he was not confused or disoriented, and did not demonstrate perceptual disturbance.
While delusions were the most prominent feature of Mr. J’s apparent psychosis, the presence of disorganized thought processes and impaired functioning, as evidenced by Mr. J’s unemployment and recent homelessness, were more consistent with a primary psychotic disorder than a delusional disorder.1
Continue to: Mr. J began to exhibit...
Mr. J began to exhibit these psychotic symptoms in his early 50s; because the average age of onset of schizophrenia for males is approximately age 20 to 25, the likelihood of his presentation being the result of a primary psychotic disorder was low.1 Although less common, it was possible that Mr. J had developed late-onset schizophrenia, where the first episode typically occurs after approximately age 40 to 45. Mr. J also described that he was in a “great” mood but had grandiose delusions and had made recent impulsive decisions, which suggests there was a possible mood component to his presentation and a potential diagnosis of schizoaffective disorder or bipolar disorder with psychotic symptoms. However, before any of these diagnoses could be made, a medical or neurologic condition that could cause his symptoms needed to be investigated and ruled out. Further collateral information regarding Mr. J’s history and timeline of symptoms was required.
EVALUATION Family history reveals clues
All laboratory studies completed during Mr. J’s hospitalization are unremarkable, including complete blood count, basic metabolic panel, hepatic function panel, gamma-glutamyl transferase test, magnesium, phosphate, thyroid-stimulating hormone, vitamin B12, thiamine, folate, urinalysis, and urine drug screen. Mr. J does not undergo any head imaging.
Mr. J has not been in touch with his family since leaving his home approximately 3 months before he presented to the ED, and he gives consent for the inpatient team to attempt to contact them. One week into hospitalization, Mr. J’s sibling informs the team of a family history of genetically confirmed Huntington’s disease (HD), with psychiatric symptoms preceding the onset of motor symptoms in multiple first-degree relatives. His family says that before Mr. J first developed delusions 4 years ago, he had not exhibited any psychotic symptoms during periods of alcohol use or sobriety.
Mr. J does not demonstrate any overt movement symptoms on the unit and denies noting any rigidity, change in gait, or abnormal/uncontrolled movements. The inpatient psychiatric team consults neurology and a full neurologic evaluation is performed. The results are unremarkable outside of his psychiatric symptoms; specifically, Mr. J does not demonstrate even subtle motor signs or cognitive impairment. Given Mr. J’s family history, unremarkable lab findings, and age at presentation, the neurology team and inpatient psychiatry team suspect that his psychosis is likely an early presentation of HD.
[polldaddy:10279212]
The authors’ observations
Genetics of Huntington’s disease
Huntington’s disease is an autosomal dominant neurodegenerative disorder caused by expansion of cytosine-adenine-guanine (CAG) trinucleotide repeats within the Huntingtin (HTT) gene on chromosome 4, which codes for the huntingtin protein.2,3 While the function of “normal” huntingtin protein is not fully understood, it is known that CAG repeat expansion in the HTT gene of >35 repeats codes for a mutant huntingtin protein.2,3 The mutant huntingtin protein causes progressive neuronal loss in the basal ganglia and striatum, resulting in the clinical Huntington’s phenotype.3 Notably, the patient’s age at disease onset is inversely correlated with the number of repeats. For example, expansions of approximately 40 to 50 CAG repeats often result in adult-onset HD, while expansions of >60 repeats are typically associated with juvenile-onset HD (before age 20). CAG repeat lengths of approximately 36 to 39 demonstrate reduced penetrance, with some individuals developing symptomatic HD while others do not.2 Instability of the CAG repeat expansion can result in genetic “anticipation,” wherein repeat length increases between generations, causing earlier age of onset in affected offspring. Genetic anticipation in HD occurs more frequently in paternal transmission—approximately 80% to 90% of juvenile HD cases are inherited paternally, at times with the number of CAG repeats exceeding 200.3
Continue to: Psychiatric manifestations of Huntington's disease
Psychiatric manifestations of Huntington’s disease
Huntington’s disease is characterized by motor, cognitive, and behavioral disturbances (Table 22,4). Motor symptoms include a characteristic and well-recognized chorea, often predominating earlier in HD, that progresses to rigidity, spasticity, and bradykinesia later in the disease course.2 Cognitive impairments develop in a similar progressive manner and can often precede the onset of motor symptoms, beginning with early executive dysfunction. Thinking often becomes more rigid and less efficient, causing difficulty with multi-tasking and concentration, and often progressing to subcortical dementia.2
Psychiatric symptoms have long been recognized as a feature of HD; the estimated lifetime prevalence in patients with HD ranges from approximately 33% to 76%.4 Depressed mood, anxiety, irritability, and apathy are the most commonly reported symptoms, while a smaller percentage of patients with HD can experience obsessive-compulsive disorder (10% to 52%) or psychotic symptoms (3% to 11%).4 A more specific schizophrenia-like psychosis occurs in approximately 3% to 6% of patients, and often is a paranoid type.5,6 Positive psychotic symptoms, such as hallucinations and delusions, typically become less overt as HD progresses and cognitive impairments worsen.7
Although the onset of motor symptoms leads to diagnosis in the majority of patients with HD, many patients present with psychiatric symptoms—most commonly depression—prior to motor symptoms.8 An increasing body of literature details instances of psychosis preceding motor symptom onset by up to 10 years.6,9-12 In many of these cases, the patient has a family history of HD-associated psychosis. Family history is a major risk factor for HD-associated psychosis, as is early-onset HD.7,9
TREATMENT Antipsychotics result in some improvement
On Day 1 or 2, Mr. J is started on risperidone, 1 mg twice daily, to manage his symptoms. He shows incremental improvement in thought organization. Although his religious and grandiose delusions persist, they become less fixed, and he is able to take the team’s suggestion that he reconnect with his family.
Mr. J is aware of his family history of HD and acknowledges that multiple relatives had early psychiatric manifestations of HD. Despite this, he still has difficulty recognizing any connection between other family members’ presentation and his own. The psychiatry and neurology teams discuss the process, ethics, and implications of genetic testing for HD with Mr. J; however, he is ambivalent regarding genetic testing, and states he would consider it after discussing it with his family.
Continue to: The neurology team recommends...
The neurology team recommends against imaging for Mr. J because HD-related changes are not typically seen until later in the disease progression. On Day 9, they recommend changing from risperidone to quetiapine (50 mg every night at bedtime) due to evidence of its effectiveness specifically for treating behavioral symptoms of HD.13
While receiving quetiapine, Mr. J experiences significant drowsiness. Because he had experienced improvement in thought organization while he was receiving risperidone, he is switched back to risperidone.
[polldaddy:10279220]
The authors’ observations
Currently, no treatments are available to prevent the development or progression of HD. However, symptomatic treatment of motor and behavioral disturbances can lead to functional improvement and improved quality of life for individuals affected by HD.
There are no extensive clinical trials to date, but multiple case reports and studies have shown second-generation antipsychotics (SGAs), including quetiapine, olanzapine,
OUTCOME Discharge despite persistent delusions
Mr. J’s religious and grandiose delusions continue throughout hospitalization despite treatment with antipsychotics. However, because he remains calm and cooperative and demonstrates improvement in thought organization, he is deemed safe for discharge and instructed to continue risperidone. The team coordinates with Mr. J’s family to arrange transportation home and outpatient neurology follow-up.
Bottom Line
Psychiatric manifestations, including psychosis, are prominent symptoms of Huntington’s disease (HD) and may precede the onset of more readily recognized motor symptoms. This poses a diagnostic challenge, and clinicians should remain cognizant of this possibility, especially in patients with a family history of HD-associated psychosis.
Related Resources
- Huntington’s Disease Society of America. http://hdsa.org.
- National Institute of Neurological Disorders and Stroke. Huntington’s disease information page: What research is being done? https://www.ninds.nih.gov/Disorders/All-Disorders/Huntingtons-Disease-Information-Page.
- Scher LM. How to target psychiatric symptoms of Huntington’s disease. Current Psychiatry. 2012;11(9):34-39.
Drug Brand Names
Aripiprazole • Abilify
Clozapine • Clozaril
Haloperidol • Haldol
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
CASE Disorganized thoughts and grandiose delusions
Mr. J, age 54, presents to the psychiatric emergency department (ED) with agitation and disruptive behavior. He claims that he is “the son of Jesus Christ” and has to travel to the Middle East to be baptized. Mr. J is irritable, shouting, and threatening staff members. He receives
The next day, he is calm and cooperative, but continues to express the same religious delusions. Mr. J is admitted to the psychiatric inpatient unit for further evaluation.
On the unit, Mr. J is pleasant and cooperative, but tangential in thought process. He reports he was “saved” by God 4 years ago, and that God communicates with him through music. Despite this, he denies having auditory or visual hallucinations.
Approximately 3 months earlier, Mr. J had stopped working and left his home and family in another state to pursue his “mission” of being baptized in the Middle East. Mr. J has been homeless since then. Despite that, he reports that his mood is “great” and denies any recent changes in mood, sleep, appetite, energy level, or psychomotor agitation. Although no formal cognitive testing is performed, Mr. J is alert and oriented to person, place, and time with intact remote and recent memory, no language deficits, and no lapses in concentration or attention throughout interview.
Mr. J says he has been drinking alcohol regularly throughout his adult life, often a few times per week, up to “a case and a half” of beer at times. He claims he’s had multiple periods of sobriety but denies having experienced withdrawal symptoms during those times. Mr. J reports 1 prior psychiatric hospitalization 25 years ago after attempting suicide by overdose following the loss of a loved one. At that time, he was diagnosed with posttraumatic stress disorder (PTSD). During this admission, he denies having any symptoms of PTSD or periods of mania or depression, and he has not undergone psychiatric treatment since he had been diagnosed with PTSD. He denies any family history of psychiatric illness as well as any medical comorbidities or medication use.
[polldaddy:10279202]
The authors’ observations
Mr. J’s presentation had a wide differential diagnosis (Table 1). The initial agitation Mr. J displayed in the psychiatric ED was likely secondary to acute alcohol intoxication, given that he was subsequently pleasant, calm, and cooperative after the alcohol was metabolized. Despite this, Mr. J continued to demonstrate delusions of a religious and somewhat grandiose nature with tangential thought processes, which made substance-induced psychosis less likely to be the sole diagnosis. Although it is possible to develop psychotic symptoms due to severe alcohol withdrawal (alcoholic hallucinosis), Mr. J’s vital signs remained stable, and he demonstrated no other signs or symptoms of withdrawal throughout his hospitalization. His presentation also did not fit that of delirium tremens because he was not confused or disoriented, and did not demonstrate perceptual disturbance.
While delusions were the most prominent feature of Mr. J’s apparent psychosis, the presence of disorganized thought processes and impaired functioning, as evidenced by Mr. J’s unemployment and recent homelessness, were more consistent with a primary psychotic disorder than a delusional disorder.1
Continue to: Mr. J began to exhibit...
Mr. J began to exhibit these psychotic symptoms in his early 50s; because the average age of onset of schizophrenia for males is approximately age 20 to 25, the likelihood of his presentation being the result of a primary psychotic disorder was low.1 Although less common, it was possible that Mr. J had developed late-onset schizophrenia, where the first episode typically occurs after approximately age 40 to 45. Mr. J also described that he was in a “great” mood but had grandiose delusions and had made recent impulsive decisions, which suggests there was a possible mood component to his presentation and a potential diagnosis of schizoaffective disorder or bipolar disorder with psychotic symptoms. However, before any of these diagnoses could be made, a medical or neurologic condition that could cause his symptoms needed to be investigated and ruled out. Further collateral information regarding Mr. J’s history and timeline of symptoms was required.
EVALUATION Family history reveals clues
All laboratory studies completed during Mr. J’s hospitalization are unremarkable, including complete blood count, basic metabolic panel, hepatic function panel, gamma-glutamyl transferase test, magnesium, phosphate, thyroid-stimulating hormone, vitamin B12, thiamine, folate, urinalysis, and urine drug screen. Mr. J does not undergo any head imaging.
Mr. J has not been in touch with his family since leaving his home approximately 3 months before he presented to the ED, and he gives consent for the inpatient team to attempt to contact them. One week into hospitalization, Mr. J’s sibling informs the team of a family history of genetically confirmed Huntington’s disease (HD), with psychiatric symptoms preceding the onset of motor symptoms in multiple first-degree relatives. His family says that before Mr. J first developed delusions 4 years ago, he had not exhibited any psychotic symptoms during periods of alcohol use or sobriety.
Mr. J does not demonstrate any overt movement symptoms on the unit and denies noting any rigidity, change in gait, or abnormal/uncontrolled movements. The inpatient psychiatric team consults neurology and a full neurologic evaluation is performed. The results are unremarkable outside of his psychiatric symptoms; specifically, Mr. J does not demonstrate even subtle motor signs or cognitive impairment. Given Mr. J’s family history, unremarkable lab findings, and age at presentation, the neurology team and inpatient psychiatry team suspect that his psychosis is likely an early presentation of HD.
[polldaddy:10279212]
The authors’ observations
Genetics of Huntington’s disease
Huntington’s disease is an autosomal dominant neurodegenerative disorder caused by expansion of cytosine-adenine-guanine (CAG) trinucleotide repeats within the Huntingtin (HTT) gene on chromosome 4, which codes for the huntingtin protein.2,3 While the function of “normal” huntingtin protein is not fully understood, it is known that CAG repeat expansion in the HTT gene of >35 repeats codes for a mutant huntingtin protein.2,3 The mutant huntingtin protein causes progressive neuronal loss in the basal ganglia and striatum, resulting in the clinical Huntington’s phenotype.3 Notably, the patient’s age at disease onset is inversely correlated with the number of repeats. For example, expansions of approximately 40 to 50 CAG repeats often result in adult-onset HD, while expansions of >60 repeats are typically associated with juvenile-onset HD (before age 20). CAG repeat lengths of approximately 36 to 39 demonstrate reduced penetrance, with some individuals developing symptomatic HD while others do not.2 Instability of the CAG repeat expansion can result in genetic “anticipation,” wherein repeat length increases between generations, causing earlier age of onset in affected offspring. Genetic anticipation in HD occurs more frequently in paternal transmission—approximately 80% to 90% of juvenile HD cases are inherited paternally, at times with the number of CAG repeats exceeding 200.3
Continue to: Psychiatric manifestations of Huntington's disease
Psychiatric manifestations of Huntington’s disease
Huntington’s disease is characterized by motor, cognitive, and behavioral disturbances (Table 22,4). Motor symptoms include a characteristic and well-recognized chorea, often predominating earlier in HD, that progresses to rigidity, spasticity, and bradykinesia later in the disease course.2 Cognitive impairments develop in a similar progressive manner and can often precede the onset of motor symptoms, beginning with early executive dysfunction. Thinking often becomes more rigid and less efficient, causing difficulty with multi-tasking and concentration, and often progressing to subcortical dementia.2
Psychiatric symptoms have long been recognized as a feature of HD; the estimated lifetime prevalence in patients with HD ranges from approximately 33% to 76%.4 Depressed mood, anxiety, irritability, and apathy are the most commonly reported symptoms, while a smaller percentage of patients with HD can experience obsessive-compulsive disorder (10% to 52%) or psychotic symptoms (3% to 11%).4 A more specific schizophrenia-like psychosis occurs in approximately 3% to 6% of patients, and often is a paranoid type.5,6 Positive psychotic symptoms, such as hallucinations and delusions, typically become less overt as HD progresses and cognitive impairments worsen.7
Although the onset of motor symptoms leads to diagnosis in the majority of patients with HD, many patients present with psychiatric symptoms—most commonly depression—prior to motor symptoms.8 An increasing body of literature details instances of psychosis preceding motor symptom onset by up to 10 years.6,9-12 In many of these cases, the patient has a family history of HD-associated psychosis. Family history is a major risk factor for HD-associated psychosis, as is early-onset HD.7,9
TREATMENT Antipsychotics result in some improvement
On Day 1 or 2, Mr. J is started on risperidone, 1 mg twice daily, to manage his symptoms. He shows incremental improvement in thought organization. Although his religious and grandiose delusions persist, they become less fixed, and he is able to take the team’s suggestion that he reconnect with his family.
Mr. J is aware of his family history of HD and acknowledges that multiple relatives had early psychiatric manifestations of HD. Despite this, he still has difficulty recognizing any connection between other family members’ presentation and his own. The psychiatry and neurology teams discuss the process, ethics, and implications of genetic testing for HD with Mr. J; however, he is ambivalent regarding genetic testing, and states he would consider it after discussing it with his family.
Continue to: The neurology team recommends...
The neurology team recommends against imaging for Mr. J because HD-related changes are not typically seen until later in the disease progression. On Day 9, they recommend changing from risperidone to quetiapine (50 mg every night at bedtime) due to evidence of its effectiveness specifically for treating behavioral symptoms of HD.13
While receiving quetiapine, Mr. J experiences significant drowsiness. Because he had experienced improvement in thought organization while he was receiving risperidone, he is switched back to risperidone.
[polldaddy:10279220]
The authors’ observations
Currently, no treatments are available to prevent the development or progression of HD. However, symptomatic treatment of motor and behavioral disturbances can lead to functional improvement and improved quality of life for individuals affected by HD.
There are no extensive clinical trials to date, but multiple case reports and studies have shown second-generation antipsychotics (SGAs), including quetiapine, olanzapine,
OUTCOME Discharge despite persistent delusions
Mr. J’s religious and grandiose delusions continue throughout hospitalization despite treatment with antipsychotics. However, because he remains calm and cooperative and demonstrates improvement in thought organization, he is deemed safe for discharge and instructed to continue risperidone. The team coordinates with Mr. J’s family to arrange transportation home and outpatient neurology follow-up.
Bottom Line
Psychiatric manifestations, including psychosis, are prominent symptoms of Huntington’s disease (HD) and may precede the onset of more readily recognized motor symptoms. This poses a diagnostic challenge, and clinicians should remain cognizant of this possibility, especially in patients with a family history of HD-associated psychosis.
Related Resources
- Huntington’s Disease Society of America. http://hdsa.org.
- National Institute of Neurological Disorders and Stroke. Huntington’s disease information page: What research is being done? https://www.ninds.nih.gov/Disorders/All-Disorders/Huntingtons-Disease-Information-Page.
- Scher LM. How to target psychiatric symptoms of Huntington’s disease. Current Psychiatry. 2012;11(9):34-39.
Drug Brand Names
Aripiprazole • Abilify
Clozapine • Clozaril
Haloperidol • Haldol
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
2. Novak MJ, Tabrizi SJ. Huntington’s disease: clinical presentation and treatment. Int Rev Neurobiol. 2011;98:297-323.
3. Reiner A, Dragatsis I, Dietrich P. Genetics and neuropathology of Huntington’s disease. Int Rev Neurobiol. 2011;98:325-372.
4. van Duijn E, Kingma EM, Van der mast RC. Psychopathology in verified Huntington’s disease gene carriers. J Neuropsychiatry Clin Neurosci. 2007;19(4):441-448.
5. Naarding P, Kremer HP, Zitman FG. Huntington’s disease: a review of the literature on prevalence and treatment of neuropsychiatric phenomena. Eur Psychiatry. 2001;16(8):439-445.
6. Xu C, Yogaratnam J, Tan N, et al. Psychosis, treatment emergent extrapyramidal events, and subsequent onset of Huntington’s disease: a case report and review of the literature. Clin Psychopharmacol Neurosci. 2016;14(3):302-304.
7. Mendez MF. Huntington’s disease: update and review of neuropsychiatric aspects. Int J Psychiatry Med. 1994;24(3):189-208.
8. Di Maio L, Squitieri F, Napolitano G, et al. Onset symptoms in 510 patients with Huntington’s disease. J Med Genet. 1993;30(4):289-292.
9. Jauhar S, Ritchie S. Psychiatric and behavioural manifestations of Huntington’s disease. Adv Psychiatr Treat. 2010;16(3):168-175.
10. Nagel M, Rumpf HJ, Kasten M. Acute psychosis in a verified Huntington disease gene carrier with subtle motor signs: psychiatric criteria should be considered for the diagnosis. Gen Hosp Psychiatry. 2014;36(3):361.e3-e4. doi: 10.1016/j.genhosppsych.2014.01.008.
11. Corrêa BB, Xavier M, Guimarães J. Association of Huntington’s disease and schizophrenia-like psychosis in a Huntington’s disease pedigree. Clin Pract Epidemiol Ment Health. 2006;2:1.
12. Ding J, Gadit AM. Psychosis with Huntington’s disease: role of antipsychotic medications. BMJ Case Rep. 2014: bcr2013202625. doi: 10.1136/bcr-2013-202625.
13. Alpay M, Koroshetz WJ. Quetiapine in the treatment of behavioral disturbances in patients with Huntington’s disease. Psychosomatics. 2006;47(1):70-72.
14. Duff K, Beglinger LJ, O’Rourke ME, et al. Risperidone and the treatment of psychiatric, motor, and cognitive symptoms in Huntington’s disease. Ann Clin Psychiatry. 2008;20(1):1-3.
15. Paleacu D, Anca M, Giladi N. Olanzapine in Huntington’s disease. Acta Neurol Scand. 2002;105(6):441-444.
16. Lin W, Chou Y. Aripiprazole effects on psychosis and chorea in a patient with Huntington’s disease. Am J Psychiatry. 2008;165(9):1207-1208.
17. van Vugt JP, Siesling S, Vergeer M, et al. Clozapine versus placebo in Huntington’s disease: a double blind randomized comparative study. J Neurol Neurosurg Psychiatr. 1997;63(1):35-39.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
2. Novak MJ, Tabrizi SJ. Huntington’s disease: clinical presentation and treatment. Int Rev Neurobiol. 2011;98:297-323.
3. Reiner A, Dragatsis I, Dietrich P. Genetics and neuropathology of Huntington’s disease. Int Rev Neurobiol. 2011;98:325-372.
4. van Duijn E, Kingma EM, Van der mast RC. Psychopathology in verified Huntington’s disease gene carriers. J Neuropsychiatry Clin Neurosci. 2007;19(4):441-448.
5. Naarding P, Kremer HP, Zitman FG. Huntington’s disease: a review of the literature on prevalence and treatment of neuropsychiatric phenomena. Eur Psychiatry. 2001;16(8):439-445.
6. Xu C, Yogaratnam J, Tan N, et al. Psychosis, treatment emergent extrapyramidal events, and subsequent onset of Huntington’s disease: a case report and review of the literature. Clin Psychopharmacol Neurosci. 2016;14(3):302-304.
7. Mendez MF. Huntington’s disease: update and review of neuropsychiatric aspects. Int J Psychiatry Med. 1994;24(3):189-208.
8. Di Maio L, Squitieri F, Napolitano G, et al. Onset symptoms in 510 patients with Huntington’s disease. J Med Genet. 1993;30(4):289-292.
9. Jauhar S, Ritchie S. Psychiatric and behavioural manifestations of Huntington’s disease. Adv Psychiatr Treat. 2010;16(3):168-175.
10. Nagel M, Rumpf HJ, Kasten M. Acute psychosis in a verified Huntington disease gene carrier with subtle motor signs: psychiatric criteria should be considered for the diagnosis. Gen Hosp Psychiatry. 2014;36(3):361.e3-e4. doi: 10.1016/j.genhosppsych.2014.01.008.
11. Corrêa BB, Xavier M, Guimarães J. Association of Huntington’s disease and schizophrenia-like psychosis in a Huntington’s disease pedigree. Clin Pract Epidemiol Ment Health. 2006;2:1.
12. Ding J, Gadit AM. Psychosis with Huntington’s disease: role of antipsychotic medications. BMJ Case Rep. 2014: bcr2013202625. doi: 10.1136/bcr-2013-202625.
13. Alpay M, Koroshetz WJ. Quetiapine in the treatment of behavioral disturbances in patients with Huntington’s disease. Psychosomatics. 2006;47(1):70-72.
14. Duff K, Beglinger LJ, O’Rourke ME, et al. Risperidone and the treatment of psychiatric, motor, and cognitive symptoms in Huntington’s disease. Ann Clin Psychiatry. 2008;20(1):1-3.
15. Paleacu D, Anca M, Giladi N. Olanzapine in Huntington’s disease. Acta Neurol Scand. 2002;105(6):441-444.
16. Lin W, Chou Y. Aripiprazole effects on psychosis and chorea in a patient with Huntington’s disease. Am J Psychiatry. 2008;165(9):1207-1208.
17. van Vugt JP, Siesling S, Vergeer M, et al. Clozapine versus placebo in Huntington’s disease: a double blind randomized comparative study. J Neurol Neurosurg Psychiatr. 1997;63(1):35-39.

When Diet Is an Emergency
At age 35, a woman underwent Roux-en-Y gastric bypass surgery. About 1 month later, she began vomiting and became unable to keep down any food or liquids. She was admitted to the hospital.
Two days after her admission, a dietitian evaluated the patient and recommended that she receive total parenteral nutrition (TPN). However, the attending physician did not order TPN during the patient’s 12-day hospital stay. As a result, the patient experienced vitamin deficiencies, including low thiamine. The patient developed symptoms of neurologic complications but was discharged.
Within 1 week, she was readmitted with the same symptoms, as well as signs of delirium and reduced level of consciousness. Her mental state continued to decline, and she became comatose for a period of time.
The patient now has Wernicke encephalopathy, which she alleged was caused by a lack of thiamine. She has no short-term memory, is wheelchair bound, and lives in a nursing home.
VERDICT
The jury found in favor of the plaintiff, awarding her $14,285,505.86 in damages, including $133,202 for loss of past earning capacity, $888,429 for loss of earning capacity, and $13,263,874.86 for medical care expenses.
COMMENTARY
It is foolish to think of diet as ancillary to medicine. While we often consider the long-term health implications of diet—obesity, atherosclerosis—we may overlook the urgent and emergent conditions that can result from a patient’s diet.
A familiar example is hypoglycemia. We associate it with agents used to treat diabetes. But it also can occur in the context of renal failure, tumor, severe infection, alcohol, or starvation. Similarly, thiamine deficiency would be an obvious consideration in a patient who presented in a coma or with altered mental status. But, as this case shows, thiamine deficiency can sneak up on you.
Continue to: In this case...
In this case, the patient’s altered structural anatomy rendered her more susceptible to thiamine deficiency, which was ultimately found to be causally related to the physician’s failure to order TPN. This raises an important issue in the management of patients who have had bariatric procedures.
This plaintiff had Roux-en-Y gastric bypass, a significant procedure that short circuits a sizable portion of the stomach and about 75 to 100 cm of the small intestine. This surgery carries long-term risks including bowel obstruction, hernias, ulcers, dumping syndrome, low blood sugar, and malnutrition. The last of these can manifest as low levels of B12, folate, thiamine, iron, calcium, and vitamin D.
Roux-en-Y bypass requires adherence to dietary recommendations, lifelong vitamin/mineral supplementation, and follow-up compliance. Patients who have had bariatric procedures are at increased risk for complications—which also raise malpractice risks. Clinicians must be aware that patients who have had a bariatric procedure have altered anatomy. We must take steps to understand the nature of those alterations and how they impact the present clinical picture.
In this case, the altered anatomy in combination with the failure to order TPN resulted in Wernicke encephalopathy—a condition caused by a biochemical lesion that occurs after stores of B vitamin are exhausted. Classic Wernicke encephalopathy is advertised as a triad of ophthalmoplegia, ataxia, and confusion, but only 10% of patients will demonstrate a true triad.
Wernicke encephalopathy typically occurs in the setting of alcoholism. However, certain other conditions can cause it, including recurrent dialysis, uremia, hyperemesis, thyrotoxicosis, cancer, AIDS, and starvation. It may be caused by surgical GI changes (eg, gastric bypass and banding) and nonsurgical GI causes (eg, pancreatitis, liver dysfunction, chronic diarrhea, celiac disease, and Crohn disease).
Continue to: Prognosis depends on...
Prognosis depends on how quickly the condition is recognized. Prompt treatment can lead to cure. But treatment delays (or lack of treatment) give the disorder the opportunity to progress, and memory and learning impairment may not completely resolve. Mortality in those untreated is 10% to 20%. And 80% of untreated or undertreated patients will develop Korsakoff psychosis, with severe retrograde and anterograde amnesia, disorientation, and emotional changes.
These factors make Wernicke encephalopathy a medical malpractice risk. If the diagnosis is missed, the damage is clear, substantial, and irreversible—as seen in this unfortunate case. This plaintiff’s attorney had a dream of a closing argument to make: Her client will suffer the effects of brain damage, robbing her of her life, her memory, and her very personality. This damage could have been prevented—not through use of an experimental new procedure or an investigational drug, but through use of a simple and readily available vitamin.
Worse still, cases such as this can involve a punitive element. Jurors would be invited to conclude that treating clinicians ignored the patient and left her to starve in her own bed. Always act in the patient’s best interest—and be attuned to situations that may evolve into claims that the patient was abandoned, neglected, or ignored.
Finally, you must address anything in the patient’s written record that is contrary to your plan. The fact pattern makes clear that a nutritional evaluation was obtained 2 days after the plaintiff’s admission. The dietitian recommended TPN. The record is not clear on why the physician did not order it. If you plan to take an action at odds with a prior observation or recommendation, be sure to clearly explain the rationale supporting your course of treatment. If you perform a risk-benefit analysis that leads you to a different conclusion, document that in the record—preferably with a second opinion from another clinician who supports your decision to deviate from the recommendation.
IN SUMMARY
Nutrition can be critically important. Make sure you consider both short- and long-term consequences of nutritional deficiencies. Bariatric surgery patients have altered anatomy, so be cautious with them. Consider the possibility of thiamine encephalopathy—which can be devastating—when the setting is suggestive. And make sure that all recommendations from other clinicians recorded in the patient’s chart are acted on. If you select a course of treatment that departs from prior recommendation, make clear your risk-benefit analysis and consider obtaining a second opinion in support of your decision.
At age 35, a woman underwent Roux-en-Y gastric bypass surgery. About 1 month later, she began vomiting and became unable to keep down any food or liquids. She was admitted to the hospital.
Two days after her admission, a dietitian evaluated the patient and recommended that she receive total parenteral nutrition (TPN). However, the attending physician did not order TPN during the patient’s 12-day hospital stay. As a result, the patient experienced vitamin deficiencies, including low thiamine. The patient developed symptoms of neurologic complications but was discharged.
Within 1 week, she was readmitted with the same symptoms, as well as signs of delirium and reduced level of consciousness. Her mental state continued to decline, and she became comatose for a period of time.
The patient now has Wernicke encephalopathy, which she alleged was caused by a lack of thiamine. She has no short-term memory, is wheelchair bound, and lives in a nursing home.
VERDICT
The jury found in favor of the plaintiff, awarding her $14,285,505.86 in damages, including $133,202 for loss of past earning capacity, $888,429 for loss of earning capacity, and $13,263,874.86 for medical care expenses.
COMMENTARY
It is foolish to think of diet as ancillary to medicine. While we often consider the long-term health implications of diet—obesity, atherosclerosis—we may overlook the urgent and emergent conditions that can result from a patient’s diet.
A familiar example is hypoglycemia. We associate it with agents used to treat diabetes. But it also can occur in the context of renal failure, tumor, severe infection, alcohol, or starvation. Similarly, thiamine deficiency would be an obvious consideration in a patient who presented in a coma or with altered mental status. But, as this case shows, thiamine deficiency can sneak up on you.
Continue to: In this case...
In this case, the patient’s altered structural anatomy rendered her more susceptible to thiamine deficiency, which was ultimately found to be causally related to the physician’s failure to order TPN. This raises an important issue in the management of patients who have had bariatric procedures.
This plaintiff had Roux-en-Y gastric bypass, a significant procedure that short circuits a sizable portion of the stomach and about 75 to 100 cm of the small intestine. This surgery carries long-term risks including bowel obstruction, hernias, ulcers, dumping syndrome, low blood sugar, and malnutrition. The last of these can manifest as low levels of B12, folate, thiamine, iron, calcium, and vitamin D.
Roux-en-Y bypass requires adherence to dietary recommendations, lifelong vitamin/mineral supplementation, and follow-up compliance. Patients who have had bariatric procedures are at increased risk for complications—which also raise malpractice risks. Clinicians must be aware that patients who have had a bariatric procedure have altered anatomy. We must take steps to understand the nature of those alterations and how they impact the present clinical picture.
In this case, the altered anatomy in combination with the failure to order TPN resulted in Wernicke encephalopathy—a condition caused by a biochemical lesion that occurs after stores of B vitamin are exhausted. Classic Wernicke encephalopathy is advertised as a triad of ophthalmoplegia, ataxia, and confusion, but only 10% of patients will demonstrate a true triad.
Wernicke encephalopathy typically occurs in the setting of alcoholism. However, certain other conditions can cause it, including recurrent dialysis, uremia, hyperemesis, thyrotoxicosis, cancer, AIDS, and starvation. It may be caused by surgical GI changes (eg, gastric bypass and banding) and nonsurgical GI causes (eg, pancreatitis, liver dysfunction, chronic diarrhea, celiac disease, and Crohn disease).
Continue to: Prognosis depends on...
Prognosis depends on how quickly the condition is recognized. Prompt treatment can lead to cure. But treatment delays (or lack of treatment) give the disorder the opportunity to progress, and memory and learning impairment may not completely resolve. Mortality in those untreated is 10% to 20%. And 80% of untreated or undertreated patients will develop Korsakoff psychosis, with severe retrograde and anterograde amnesia, disorientation, and emotional changes.
These factors make Wernicke encephalopathy a medical malpractice risk. If the diagnosis is missed, the damage is clear, substantial, and irreversible—as seen in this unfortunate case. This plaintiff’s attorney had a dream of a closing argument to make: Her client will suffer the effects of brain damage, robbing her of her life, her memory, and her very personality. This damage could have been prevented—not through use of an experimental new procedure or an investigational drug, but through use of a simple and readily available vitamin.
Worse still, cases such as this can involve a punitive element. Jurors would be invited to conclude that treating clinicians ignored the patient and left her to starve in her own bed. Always act in the patient’s best interest—and be attuned to situations that may evolve into claims that the patient was abandoned, neglected, or ignored.
Finally, you must address anything in the patient’s written record that is contrary to your plan. The fact pattern makes clear that a nutritional evaluation was obtained 2 days after the plaintiff’s admission. The dietitian recommended TPN. The record is not clear on why the physician did not order it. If you plan to take an action at odds with a prior observation or recommendation, be sure to clearly explain the rationale supporting your course of treatment. If you perform a risk-benefit analysis that leads you to a different conclusion, document that in the record—preferably with a second opinion from another clinician who supports your decision to deviate from the recommendation.
IN SUMMARY
Nutrition can be critically important. Make sure you consider both short- and long-term consequences of nutritional deficiencies. Bariatric surgery patients have altered anatomy, so be cautious with them. Consider the possibility of thiamine encephalopathy—which can be devastating—when the setting is suggestive. And make sure that all recommendations from other clinicians recorded in the patient’s chart are acted on. If you select a course of treatment that departs from prior recommendation, make clear your risk-benefit analysis and consider obtaining a second opinion in support of your decision.
At age 35, a woman underwent Roux-en-Y gastric bypass surgery. About 1 month later, she began vomiting and became unable to keep down any food or liquids. She was admitted to the hospital.
Two days after her admission, a dietitian evaluated the patient and recommended that she receive total parenteral nutrition (TPN). However, the attending physician did not order TPN during the patient’s 12-day hospital stay. As a result, the patient experienced vitamin deficiencies, including low thiamine. The patient developed symptoms of neurologic complications but was discharged.
Within 1 week, she was readmitted with the same symptoms, as well as signs of delirium and reduced level of consciousness. Her mental state continued to decline, and she became comatose for a period of time.
The patient now has Wernicke encephalopathy, which she alleged was caused by a lack of thiamine. She has no short-term memory, is wheelchair bound, and lives in a nursing home.
VERDICT
The jury found in favor of the plaintiff, awarding her $14,285,505.86 in damages, including $133,202 for loss of past earning capacity, $888,429 for loss of earning capacity, and $13,263,874.86 for medical care expenses.
COMMENTARY
It is foolish to think of diet as ancillary to medicine. While we often consider the long-term health implications of diet—obesity, atherosclerosis—we may overlook the urgent and emergent conditions that can result from a patient’s diet.
A familiar example is hypoglycemia. We associate it with agents used to treat diabetes. But it also can occur in the context of renal failure, tumor, severe infection, alcohol, or starvation. Similarly, thiamine deficiency would be an obvious consideration in a patient who presented in a coma or with altered mental status. But, as this case shows, thiamine deficiency can sneak up on you.
Continue to: In this case...
In this case, the patient’s altered structural anatomy rendered her more susceptible to thiamine deficiency, which was ultimately found to be causally related to the physician’s failure to order TPN. This raises an important issue in the management of patients who have had bariatric procedures.
This plaintiff had Roux-en-Y gastric bypass, a significant procedure that short circuits a sizable portion of the stomach and about 75 to 100 cm of the small intestine. This surgery carries long-term risks including bowel obstruction, hernias, ulcers, dumping syndrome, low blood sugar, and malnutrition. The last of these can manifest as low levels of B12, folate, thiamine, iron, calcium, and vitamin D.
Roux-en-Y bypass requires adherence to dietary recommendations, lifelong vitamin/mineral supplementation, and follow-up compliance. Patients who have had bariatric procedures are at increased risk for complications—which also raise malpractice risks. Clinicians must be aware that patients who have had a bariatric procedure have altered anatomy. We must take steps to understand the nature of those alterations and how they impact the present clinical picture.
In this case, the altered anatomy in combination with the failure to order TPN resulted in Wernicke encephalopathy—a condition caused by a biochemical lesion that occurs after stores of B vitamin are exhausted. Classic Wernicke encephalopathy is advertised as a triad of ophthalmoplegia, ataxia, and confusion, but only 10% of patients will demonstrate a true triad.
Wernicke encephalopathy typically occurs in the setting of alcoholism. However, certain other conditions can cause it, including recurrent dialysis, uremia, hyperemesis, thyrotoxicosis, cancer, AIDS, and starvation. It may be caused by surgical GI changes (eg, gastric bypass and banding) and nonsurgical GI causes (eg, pancreatitis, liver dysfunction, chronic diarrhea, celiac disease, and Crohn disease).
Continue to: Prognosis depends on...
Prognosis depends on how quickly the condition is recognized. Prompt treatment can lead to cure. But treatment delays (or lack of treatment) give the disorder the opportunity to progress, and memory and learning impairment may not completely resolve. Mortality in those untreated is 10% to 20%. And 80% of untreated or undertreated patients will develop Korsakoff psychosis, with severe retrograde and anterograde amnesia, disorientation, and emotional changes.
These factors make Wernicke encephalopathy a medical malpractice risk. If the diagnosis is missed, the damage is clear, substantial, and irreversible—as seen in this unfortunate case. This plaintiff’s attorney had a dream of a closing argument to make: Her client will suffer the effects of brain damage, robbing her of her life, her memory, and her very personality. This damage could have been prevented—not through use of an experimental new procedure or an investigational drug, but through use of a simple and readily available vitamin.
Worse still, cases such as this can involve a punitive element. Jurors would be invited to conclude that treating clinicians ignored the patient and left her to starve in her own bed. Always act in the patient’s best interest—and be attuned to situations that may evolve into claims that the patient was abandoned, neglected, or ignored.
Finally, you must address anything in the patient’s written record that is contrary to your plan. The fact pattern makes clear that a nutritional evaluation was obtained 2 days after the plaintiff’s admission. The dietitian recommended TPN. The record is not clear on why the physician did not order it. If you plan to take an action at odds with a prior observation or recommendation, be sure to clearly explain the rationale supporting your course of treatment. If you perform a risk-benefit analysis that leads you to a different conclusion, document that in the record—preferably with a second opinion from another clinician who supports your decision to deviate from the recommendation.
IN SUMMARY
Nutrition can be critically important. Make sure you consider both short- and long-term consequences of nutritional deficiencies. Bariatric surgery patients have altered anatomy, so be cautious with them. Consider the possibility of thiamine encephalopathy—which can be devastating—when the setting is suggestive. And make sure that all recommendations from other clinicians recorded in the patient’s chart are acted on. If you select a course of treatment that departs from prior recommendation, make clear your risk-benefit analysis and consider obtaining a second opinion in support of your decision.

FDA approves siponimod for relapsing forms of MS
(MS), including clinically isolated syndrome, relapsing-remitting MS, and active secondary progressive MS.

Siponimod is a selective sphingosine 1-phosphate (S1P) receptor modulator that binds to S1P1 and S1P5 receptors. Its binding to the S1P1 receptor prevents lymphocytes from leaving the lymph nodes, which contributes to the treatment’s anti-inflammatory effects. Its binding to the S1P5 and S1P1 subreceptors on oligodendrocytes and astrocytes is intended to promote remyelination and prevent inflammation.
The treatment’s approval is based on the results of the phase 3 EXPAND study, according to the agency’s March 26 announcement. This randomized, double-blind study compared siponimod with placebo among 1,651 patients with secondary progressive MS. At baseline, the population’s mean age was 48 years, and mean disease duration was approximately 16 years. More than half the study population had a median Expanded Disability Status Scale score of 6.0 and relied on a walking aid.
Siponimod reduced the risk of 3-month confirmed disability progression (CDP) by 21%, compared with placebo (P = .013). Among participants with relapse activity in the 2 years prior to screening, siponimod reduced the risk of this outcome by 33%, compared with placebo (P = .0100). Siponimod delayed the risk of 6-month CDP by 26%, compared with placebo (P = .0058) and reduced the annualized relapse rate by 55%. In addition, the data suggested beneficial effects of siponimod on cognition, MRI disease activity, and brain volume loss. Siponimod did not provide significant improvements in patients with nonactive secondary progressive MS.
Common adverse events included headache, hypertension, and transaminase increase. The FDA requires siponimod to be dispensed with a medication guide that describes the treatment’s associated risks of infection, macular edema, decreased heart rate, and impaired lung function.
Novartis manufactures the drug. The company expects the drug to be available within 1 week, according to its press release.
(MS), including clinically isolated syndrome, relapsing-remitting MS, and active secondary progressive MS.
Siponimod is a selective sphingosine 1-phosphate (S1P) receptor modulator that binds to S1P1 and S1P5 receptors. Its binding to the S1P1 receptor prevents lymphocytes from leaving the lymph nodes, which contributes to the treatment’s anti-inflammatory effects. Its binding to the S1P5 and S1P1 subreceptors on oligodendrocytes and astrocytes is intended to promote remyelination and prevent inflammation.
The treatment’s approval is based on the results of the phase 3 EXPAND study, according to the agency’s March 26 announcement. This randomized, double-blind study compared siponimod with placebo among 1,651 patients with secondary progressive MS. At baseline, the population’s mean age was 48 years, and mean disease duration was approximately 16 years. More than half the study population had a median Expanded Disability Status Scale score of 6.0 and relied on a walking aid.
Siponimod reduced the risk of 3-month confirmed disability progression (CDP) by 21%, compared with placebo (P = .013). Among participants with relapse activity in the 2 years prior to screening, siponimod reduced the risk of this outcome by 33%, compared with placebo (P = .0100). Siponimod delayed the risk of 6-month CDP by 26%, compared with placebo (P = .0058) and reduced the annualized relapse rate by 55%. In addition, the data suggested beneficial effects of siponimod on cognition, MRI disease activity, and brain volume loss. Siponimod did not provide significant improvements in patients with nonactive secondary progressive MS.
Common adverse events included headache, hypertension, and transaminase increase. The FDA requires siponimod to be dispensed with a medication guide that describes the treatment’s associated risks of infection, macular edema, decreased heart rate, and impaired lung function.
Novartis manufactures the drug. The company expects the drug to be available within 1 week, according to its press release.
(MS), including clinically isolated syndrome, relapsing-remitting MS, and active secondary progressive MS.
Siponimod is a selective sphingosine 1-phosphate (S1P) receptor modulator that binds to S1P1 and S1P5 receptors. Its binding to the S1P1 receptor prevents lymphocytes from leaving the lymph nodes, which contributes to the treatment’s anti-inflammatory effects. Its binding to the S1P5 and S1P1 subreceptors on oligodendrocytes and astrocytes is intended to promote remyelination and prevent inflammation.
The treatment’s approval is based on the results of the phase 3 EXPAND study, according to the agency’s March 26 announcement. This randomized, double-blind study compared siponimod with placebo among 1,651 patients with secondary progressive MS. At baseline, the population’s mean age was 48 years, and mean disease duration was approximately 16 years. More than half the study population had a median Expanded Disability Status Scale score of 6.0 and relied on a walking aid.
Siponimod reduced the risk of 3-month confirmed disability progression (CDP) by 21%, compared with placebo (P = .013). Among participants with relapse activity in the 2 years prior to screening, siponimod reduced the risk of this outcome by 33%, compared with placebo (P = .0100). Siponimod delayed the risk of 6-month CDP by 26%, compared with placebo (P = .0058) and reduced the annualized relapse rate by 55%. In addition, the data suggested beneficial effects of siponimod on cognition, MRI disease activity, and brain volume loss. Siponimod did not provide significant improvements in patients with nonactive secondary progressive MS.
Common adverse events included headache, hypertension, and transaminase increase. The FDA requires siponimod to be dispensed with a medication guide that describes the treatment’s associated risks of infection, macular edema, decreased heart rate, and impaired lung function.
Novartis manufactures the drug. The company expects the drug to be available within 1 week, according to its press release.
Study launched to further evaluate the central vein sign in MS
DALLAS –
At the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis, Daniel Ontaneda, MD, said that up to 20% of individuals referred for a diagnosis of multiple sclerosis (MS) are incorrectly diagnosed with the disease, and about two-thirds of misdiagnosed patients are exposed to unnecessary and sometimes life-threatening risks associated with disease-modifying therapies. “MRI is a sensitive tool for diagnosis of MS and is an integral component of the diagnostic criteria for MS,” said Dr. Ontaneda, a neurologist at the Cleveland Clinic Mellen Center for Multiple Sclerosis Treatment and Research. “However, there are problems with its implementation. Approximately half of individuals referred to an MS clinic present with atypical symptoms [fatigue, cognitive disturbance, pain] and not typical syndromes [unilateral optic neuritis, brain stem syndromes, partial myelitis]. Increasing diagnostic sensitivity may have come at the price of decreased specificity. MRI criteria have a specificity of 32% for dissemination in space and 42% for dissemination in time.”
While misdiagnosis appears to be mainly caused by overinterpretation of abnormal MRI findings, the central vein sign (CVS) is an effective method to overcome such challenges. Recent studies have demonstrated that CVS may help to identify MS, as 85% of white matter lesions in MS have a central vein, compared with only 8% of small vessel ischemic disease, 34% of migraine, and 14% of other inflammatory or autoimmune diseases.
“We think there is a significant and unmet need for more specific and accurate diagnostic tests to facilitate early confirmation of a diagnosis of MS,” Dr. Ontaneda said. “We propose a prospective evaluation of the central vein sign, which we hypothesize will reduce misdiagnosis, hasten early diagnosis, and simplify clinical decision making.”
With funding from the Race to Erase MS Foundation, he and his associates have designed CAVS-MS (Central Vein Sign in MS), a multicenter, prospective, observational trial being conducted at 10 sites. The first phase of the study is a cross-sectional pilot at the 10 sites. The primary objective is to establish the contrast-to-noise ratio of lesion to normal-appearing white matter and central vein to lesion across the 10 sites using 3-tesla FLAIR imaging in subjects with a clinical or radiologic suspicion of MS. The secondary objectives are to investigate the difference in contrast-to-noise ratio identified in the primary objective between pre- and postcontrast FLAIR imaging to identify whether gadolinium injection improves central vein detection, to determine the reproducibility of different methods for detection of positive CVS across sites, and to determine the sensitivity and specificity of the different methods for the diagnosis of MS, compared with the McDonald 2010 MS criteria.
The study population will consist of 100 individuals referred to an MS center based on clinical or radiologic suspicion of MS; 30 participants are currently enrolled. The 10 sites include the Cleveland Clinic; Johns Hopkins University, Baltimore; the University of California, San Francisco; the University of Texas, Houston; the University of Toronto; the University of Vermont, Burlington; the University of Southern California, Los Angeles; Cedars-Sinai Medical Center, Los Angeles; Yale University, New Haven, Conn.; and the University of Pennsylvania, Philadelphia.
CAVS-MS includes development of a software platform for rating of central veins through an imaging software partner, QMENTA. “We are going to have the individual clinicians at each site rate the lesions, so we will have information from 10 different raters,” Dr. Ontaneda said. The study will be coordinated at the Cleveland Clinic, central image analysis will be conducted at the National Institutes of Health, and statistical analysis will be performed at the University of Pennsylvania.
The researchers also hope to perform a prospective study with three objectives. The first is to determine if incorporation of CVS for the diagnosis of MS improves diagnostic accuracy and hastens diagnosis in individuals presenting with typical first clinical events. The second objective “is to determine if incorporation of CVS for the diagnosis of MS improves specificity among individuals presenting with atypical syndromes,” Dr. Ontaneda said. “The third aim is to look at central vein volume as a predictor of clinical/MRI disease activity associated with disability in MS.”
He concluded his remarks by describing the CVS as “a tool that offers promise both for increasing specificity and perhaps enabling earlier diagnosis of MS. Studies will determine if the central vein sign can be incorporated into the diagnostic criteria. The NIH is working with MRI manufacturers to make sequences available for disseminated clinical use.”
Dr. Ontaneda reported that he has received grant support from the National Institutes of Health, the Race to Erase MS Foundation, the Patient-Centered Outcomes Research Institute, the National Multiple Sclerosis Society, Genentech, Genzyme, and Novartis. He has also received consulting fees from Biogen, Genentech, and Novartis.
DALLAS –
At the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis, Daniel Ontaneda, MD, said that up to 20% of individuals referred for a diagnosis of multiple sclerosis (MS) are incorrectly diagnosed with the disease, and about two-thirds of misdiagnosed patients are exposed to unnecessary and sometimes life-threatening risks associated with disease-modifying therapies. “MRI is a sensitive tool for diagnosis of MS and is an integral component of the diagnostic criteria for MS,” said Dr. Ontaneda, a neurologist at the Cleveland Clinic Mellen Center for Multiple Sclerosis Treatment and Research. “However, there are problems with its implementation. Approximately half of individuals referred to an MS clinic present with atypical symptoms [fatigue, cognitive disturbance, pain] and not typical syndromes [unilateral optic neuritis, brain stem syndromes, partial myelitis]. Increasing diagnostic sensitivity may have come at the price of decreased specificity. MRI criteria have a specificity of 32% for dissemination in space and 42% for dissemination in time.”
While misdiagnosis appears to be mainly caused by overinterpretation of abnormal MRI findings, the central vein sign (CVS) is an effective method to overcome such challenges. Recent studies have demonstrated that CVS may help to identify MS, as 85% of white matter lesions in MS have a central vein, compared with only 8% of small vessel ischemic disease, 34% of migraine, and 14% of other inflammatory or autoimmune diseases.
“We think there is a significant and unmet need for more specific and accurate diagnostic tests to facilitate early confirmation of a diagnosis of MS,” Dr. Ontaneda said. “We propose a prospective evaluation of the central vein sign, which we hypothesize will reduce misdiagnosis, hasten early diagnosis, and simplify clinical decision making.”
With funding from the Race to Erase MS Foundation, he and his associates have designed CAVS-MS (Central Vein Sign in MS), a multicenter, prospective, observational trial being conducted at 10 sites. The first phase of the study is a cross-sectional pilot at the 10 sites. The primary objective is to establish the contrast-to-noise ratio of lesion to normal-appearing white matter and central vein to lesion across the 10 sites using 3-tesla FLAIR imaging in subjects with a clinical or radiologic suspicion of MS. The secondary objectives are to investigate the difference in contrast-to-noise ratio identified in the primary objective between pre- and postcontrast FLAIR imaging to identify whether gadolinium injection improves central vein detection, to determine the reproducibility of different methods for detection of positive CVS across sites, and to determine the sensitivity and specificity of the different methods for the diagnosis of MS, compared with the McDonald 2010 MS criteria.
The study population will consist of 100 individuals referred to an MS center based on clinical or radiologic suspicion of MS; 30 participants are currently enrolled. The 10 sites include the Cleveland Clinic; Johns Hopkins University, Baltimore; the University of California, San Francisco; the University of Texas, Houston; the University of Toronto; the University of Vermont, Burlington; the University of Southern California, Los Angeles; Cedars-Sinai Medical Center, Los Angeles; Yale University, New Haven, Conn.; and the University of Pennsylvania, Philadelphia.
CAVS-MS includes development of a software platform for rating of central veins through an imaging software partner, QMENTA. “We are going to have the individual clinicians at each site rate the lesions, so we will have information from 10 different raters,” Dr. Ontaneda said. The study will be coordinated at the Cleveland Clinic, central image analysis will be conducted at the National Institutes of Health, and statistical analysis will be performed at the University of Pennsylvania.
The researchers also hope to perform a prospective study with three objectives. The first is to determine if incorporation of CVS for the diagnosis of MS improves diagnostic accuracy and hastens diagnosis in individuals presenting with typical first clinical events. The second objective “is to determine if incorporation of CVS for the diagnosis of MS improves specificity among individuals presenting with atypical syndromes,” Dr. Ontaneda said. “The third aim is to look at central vein volume as a predictor of clinical/MRI disease activity associated with disability in MS.”
He concluded his remarks by describing the CVS as “a tool that offers promise both for increasing specificity and perhaps enabling earlier diagnosis of MS. Studies will determine if the central vein sign can be incorporated into the diagnostic criteria. The NIH is working with MRI manufacturers to make sequences available for disseminated clinical use.”
Dr. Ontaneda reported that he has received grant support from the National Institutes of Health, the Race to Erase MS Foundation, the Patient-Centered Outcomes Research Institute, the National Multiple Sclerosis Society, Genentech, Genzyme, and Novartis. He has also received consulting fees from Biogen, Genentech, and Novartis.
DALLAS –
At the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis, Daniel Ontaneda, MD, said that up to 20% of individuals referred for a diagnosis of multiple sclerosis (MS) are incorrectly diagnosed with the disease, and about two-thirds of misdiagnosed patients are exposed to unnecessary and sometimes life-threatening risks associated with disease-modifying therapies. “MRI is a sensitive tool for diagnosis of MS and is an integral component of the diagnostic criteria for MS,” said Dr. Ontaneda, a neurologist at the Cleveland Clinic Mellen Center for Multiple Sclerosis Treatment and Research. “However, there are problems with its implementation. Approximately half of individuals referred to an MS clinic present with atypical symptoms [fatigue, cognitive disturbance, pain] and not typical syndromes [unilateral optic neuritis, brain stem syndromes, partial myelitis]. Increasing diagnostic sensitivity may have come at the price of decreased specificity. MRI criteria have a specificity of 32% for dissemination in space and 42% for dissemination in time.”
While misdiagnosis appears to be mainly caused by overinterpretation of abnormal MRI findings, the central vein sign (CVS) is an effective method to overcome such challenges. Recent studies have demonstrated that CVS may help to identify MS, as 85% of white matter lesions in MS have a central vein, compared with only 8% of small vessel ischemic disease, 34% of migraine, and 14% of other inflammatory or autoimmune diseases.
“We think there is a significant and unmet need for more specific and accurate diagnostic tests to facilitate early confirmation of a diagnosis of MS,” Dr. Ontaneda said. “We propose a prospective evaluation of the central vein sign, which we hypothesize will reduce misdiagnosis, hasten early diagnosis, and simplify clinical decision making.”
With funding from the Race to Erase MS Foundation, he and his associates have designed CAVS-MS (Central Vein Sign in MS), a multicenter, prospective, observational trial being conducted at 10 sites. The first phase of the study is a cross-sectional pilot at the 10 sites. The primary objective is to establish the contrast-to-noise ratio of lesion to normal-appearing white matter and central vein to lesion across the 10 sites using 3-tesla FLAIR imaging in subjects with a clinical or radiologic suspicion of MS. The secondary objectives are to investigate the difference in contrast-to-noise ratio identified in the primary objective between pre- and postcontrast FLAIR imaging to identify whether gadolinium injection improves central vein detection, to determine the reproducibility of different methods for detection of positive CVS across sites, and to determine the sensitivity and specificity of the different methods for the diagnosis of MS, compared with the McDonald 2010 MS criteria.
The study population will consist of 100 individuals referred to an MS center based on clinical or radiologic suspicion of MS; 30 participants are currently enrolled. The 10 sites include the Cleveland Clinic; Johns Hopkins University, Baltimore; the University of California, San Francisco; the University of Texas, Houston; the University of Toronto; the University of Vermont, Burlington; the University of Southern California, Los Angeles; Cedars-Sinai Medical Center, Los Angeles; Yale University, New Haven, Conn.; and the University of Pennsylvania, Philadelphia.
CAVS-MS includes development of a software platform for rating of central veins through an imaging software partner, QMENTA. “We are going to have the individual clinicians at each site rate the lesions, so we will have information from 10 different raters,” Dr. Ontaneda said. The study will be coordinated at the Cleveland Clinic, central image analysis will be conducted at the National Institutes of Health, and statistical analysis will be performed at the University of Pennsylvania.
The researchers also hope to perform a prospective study with three objectives. The first is to determine if incorporation of CVS for the diagnosis of MS improves diagnostic accuracy and hastens diagnosis in individuals presenting with typical first clinical events. The second objective “is to determine if incorporation of CVS for the diagnosis of MS improves specificity among individuals presenting with atypical syndromes,” Dr. Ontaneda said. “The third aim is to look at central vein volume as a predictor of clinical/MRI disease activity associated with disability in MS.”
He concluded his remarks by describing the CVS as “a tool that offers promise both for increasing specificity and perhaps enabling earlier diagnosis of MS. Studies will determine if the central vein sign can be incorporated into the diagnostic criteria. The NIH is working with MRI manufacturers to make sequences available for disseminated clinical use.”
Dr. Ontaneda reported that he has received grant support from the National Institutes of Health, the Race to Erase MS Foundation, the Patient-Centered Outcomes Research Institute, the National Multiple Sclerosis Society, Genentech, Genzyme, and Novartis. He has also received consulting fees from Biogen, Genentech, and Novartis.
EXPERT ANALYSIS FROM ACTRIMS FORUM 2019