User login
Restless Legs Syndrome Among Veterans With Spinal Cord Lesions (FULL)
Spinal cord injuries (SCI) are common in veteran populations.1 Veterans with spinal cord injuries and disorders (SCI/D) also may have concurrent sleep disturbances. Spinal cord injury typically causes spasticity.2,3 Hypersensitivity of the flexor reflex pathways is believed to cause painful muscle spasms in patients with SCI.4 Neuropathic pain at or below the level of the lesion also is common.
Restless legs syndrome (RLS) is a common sleep disorder that affects sleep quality and can occur concomitantly with spinal cord lesions.5 In about 80% of RLS cases, involuntary movements of legs across hip, knee, and ankle joints during sleep, known as periodic limb movement during sleep (PLMS), occurs.6 Several studies showed increased prevalence of PLMS in patients with SCI, and some case reports suggest an increased prevalence of RLS in this population.7,8 One small study showed that 100% of patients with SCI had symptoms of RLS.6 Another study found that SCI could trigger PLMS.8
The pathophysiology of RLS and PLMS in patients with SCI is not fully understood, but case reports describing PLM in SCI patients points to a possible role of central pattern generators and the flexor reflex afferents in the pathophysiology of PLMS.9,10 Changes of the tissue microstructure in the midbrain and upper cervical spinal cord have been described in patients with RLS.11The objective of this study was to assess the prevalence of RLS in a veteran population with SCI/D and
Methods
The institutional review and ethical approval boards of the Minneapolis VA Health Care System approved the study. Within the VA system, 666 patients with SCI/D were identified using a national database. Of the 666 people, 316 were excluded, 199 were included, and 151 were deceased.
Patients aged between 18 and 65 years were included in the study. Charts of patients who had been discharged with the diagnosis of SCI from 2002 to 2008 were studied. All patients met the inclusion criteria of the International Restless Legs Syndrome Study Group diagnosis.
Exclusion criteria were as follows: Patients with evidence of brain pathology (eg, stroke), concurrent neurologic condition associated with RLS (Parkinson disease, spinocerebellar ataxia, peripheral neuropathy), concurrent psychiatric condition within the setting of treatment with dopamine antagonists, secondary causes of RLS (renal failure/uremia, iron deficiency, rheumatoid arthritis, and pregnancy) and a recent history of alcohol or drug misuse or current evidence of substance use of < 1 year.
A patient list was compiled that included the etiology of the SCI (vascular injury, multiple sclerosis [MS], trauma, unknown, and other), the level(s) and completeness of the SCI per radiology report, RLS pharmacotherapies, and pertinent medical history.
Axial T2-weighted images on magnetic resonance imaging (MRI) scans were retrospectively reviewed. Sagittal T1/T2-weighted and axial T2-weighted sequences were performed routinely on all patients with spinal cord lesions. The analysis included the extension of the lesion on both sagittal and axial distributions. The anatomic location of the cord lesion was categorized by the following: (1) pure gray matter (central cord); (2) white matter (dorsal [D], dorsolateral [DL], ventral [V], ventrolateral areas [VL]).
A questionnaire using standard diagnostic criteria for RLS was mailed to the 199 patients who met the inclusion criteria (Appendix A). 

All analyses were carried out using StataCorp STATA 13 (College Station, TX). Descriptive statistics were used. The analyses were carried out using chi-square and Fisher exact tests. Differences between the groups were considered statistically significant at P < .05. The data were analyzed to obtain point prevalence among patients with SCI, and comparisons were made among the different subgroups.
Results
Of the 162 patients who chose to participate in the study, the sleep specialists confirmed 31 (19%) to have RLS, 112 (69%) were confirmed negative for RLS, and an additional 19 (12%) screened positive for RLS but were not confirmed to have RLS by the sleep specialists (Figure 1). 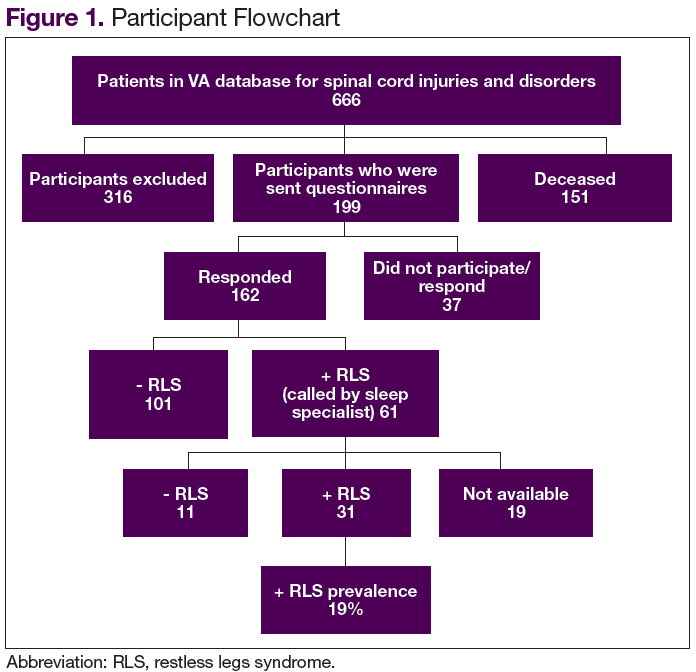
The etiology of SCI was subdivided into 4 groups: MS, trauma, vascular, and other/unknown. Within each group (– RLS vs + RLS), MS and trauma were the most common etiologies with 55% MS and 36% trauma in the + RLS group.
When comparing RLS among the spinal cord levels (cervical, thoracic, lumbar and cervical + thoracic), only the cervical + thoracic subgroup (18% + RLS vs 5% – RLS) showed a significant difference (Figure 2). 
There was no significant difference found with the prevalence of RLS in the axial plane of the spinal cord lesions (ventral/ventro-lateral/central cord vs dorsal/dorsolateral) or by the completeness of spinal cord lesions, P = .76. There was a higher prevalence of incomplete cord injury, however, within each subgroup of RLS.
The Mann-Whitney test was used to analyze the burden of disease in both groups (+ RLS vs – RLS). Moderate level of burden was most frequently reported with a higher prevalence within the + RLS group. Of those receiving treatment for RLS, 71% were + RLS vs 46% – RLS with a P value of .01. Symptoms of RLS after cord injury were 89% + RLS vs 55% – RLS with a P value of .03.
Discussion
This study represents one of the first studies to determine the prevalence of RLS in veterans with spinal cord disease. Research in this area is important to raise awareness of RLS among the veteran population with and without SCI and disorders. Restless legs syndrome often escapes diagnosis because of difficulty understanding the patient’s descriptions of their sensations. In addition, RLS may cause debilitating symptoms of sleep deprivation, daytime sleepiness, discomfort, and fatigue, which often results in decreased quality of life (QOL). Proper screening and treatment may improve QOL.
A study by Kumru and colleagues showed a similar rate of RLS in patients with SCI and RLS symptoms presented in the first year after SCI as did this study (18% vs 19%, respectively).4 In that study, RLS was more common in patients with lesions in lumbosacral area. Kumru and colleagues also showed that a dopaminergic medication improved symptoms of RLS in this population, whereas this study did not explore treatment outcomes.4
The pathogenesis of RLS is not fully known, but hereditary factors, iron metabolism, and the brain dopaminergic system are thought to be involved.11 It is hypothesized that spinal cord lesions allow the appearance of RLS symptoms and spinal leg movement generator by blocking descending inhibitory spinal pathways.12 One hypothesis is that damage to A11 nuclei (the main source of dopamine in the spinal cord or its diencephalospinal tract in animals) causes hyperexcitability of the spinal cord and leads to PLM and RLS symptoms.13 As the axons of A11 nuclei are present along the whole span of the spinal cord, SCI/D in patients with RLS might interrupt this dopaminergic tract and produce the RLS symptoms.
Limitations
This study included only veterans, so the prevalence may not apply to the nonveteran SCI population. Also, the population mainly was male, and there was no accurate information on race. Ferritin levels of the patients were not checked and is a major factor in RLS. The reported onset of RLS after the SCI could be due to recall bias.
Conclusion
The prevalence of RLS in veterans with SCI is above that reported in the general population (19% vs 10%, respectively). Furthermore, those with RLS have symptoms that often started after the SCI (suggesting causality) and required therapy due to their level of RLS symptom burden. A spectrum of severity of symptoms is present among those with RLS, with 83% having moderate-to-severe RLS affecting their QOL.
Although there was not a statistically significant relationship between RLS and spinal cord lesion level, there was a slightly higher prevalence of RLS at the cervical and thoracic levels, which may be relevant for future studies. There was no difference found between the RLS subgroups with respect to the location of the lesion within the spinal cord; however, a larger sample size may be needed to determine whether this would reach statistical significance. Prompt search for symptoms of RLS in veterans with SCI is warranted to provide adequate treatment to improve sleep health and QOL in this population.
1. Lasfargues JE, Custis D, Morrone F, Carswell J, Nguyen T. A model for estimating spinal cord injury prevalence in the United States. Paraplegia. 1995;33(2):62-68.
2. Sjölund BH. Pain and rehabilitation after spinal cord injury: the case of sensory spasticity? Brain Res Brain Res Rev. 2002;40(1-3):250-256.
3. Adams MM, Hicks AL. Spasticity after spinal cord injury. Spinal Cord. 2005;43(10):577-586.
4. Kumru H, Vidal J, Benito J, et al. Restless leg syndrome in patients with spinal cord injury. Parkinsonism Relat Disord. 2015;21(12):1461-1464.
5. Wilt TJ, MacDonald R, Ouellette J, et al. Pharmacologic therapy for primary restless legs syndrome: a systematic review and meta-analysis. JAMA Intern Med. 2013;173(7):496-505.
6. American Academy of Sleep Medicine. The International Classification of Sleep Disorders: Diagnostic and Coding Manual. (AASM ICSD-3). 3rd ed. Westchester, IL: American Academy of Sleep Medicine; 2014.
7. Telles SC, Alves RC, Chadi G. Periodic limb movements during sleep and restless legs syndrome in patients with ASIA A spinal cord injury. J Neurol Sci. 2011;303(1-2):119-123.
8. Telles SC, Alves RS, Chadi G. Spinal cord injury as a trigger to develop periodic leg movements during sleep: an evolutionary perspective. Arq Neuropsiquiatr. 2012;70(11):880-884.
9. Tings T, Baier PC, Paulus W, Trenkwalder C. Restless legs syndrome induced by impairment of sensory spinal pathways. J Neurol. 2003;250(4):499-500.
10. Paulus W, Trenkwalder C. Less is more: pathophysiology of dopaminergic-therapy-related augmentation in restless legs syndrome. Lancet Neurol. 2006;5(10):878-886.
11. Silber MH, Ehrenberg BL, Allen RP, et al; Medical Advisory Board of the Restless Legs Syndrome Foundation. An algorithm for the management of restless legs syndrome. Mayo Clin Proc. 2004;79(7):916-922.
12. Hartmann M, Pfister R, Pfadenhauer K. Restless legs syndrome associated with spinal cord lesions. J Neurol Neurosurg Psychiatry. 1999;66(5):688-689.
13. Clemens S, Rye D, Hochman S. Restless legs syndrome: revisiting the dopamine hypothesis from the spinal cord perspective. Neurology. 2006;67(1):125-130.
Spinal cord injuries (SCI) are common in veteran populations.1 Veterans with spinal cord injuries and disorders (SCI/D) also may have concurrent sleep disturbances. Spinal cord injury typically causes spasticity.2,3 Hypersensitivity of the flexor reflex pathways is believed to cause painful muscle spasms in patients with SCI.4 Neuropathic pain at or below the level of the lesion also is common.
Restless legs syndrome (RLS) is a common sleep disorder that affects sleep quality and can occur concomitantly with spinal cord lesions.5 In about 80% of RLS cases, involuntary movements of legs across hip, knee, and ankle joints during sleep, known as periodic limb movement during sleep (PLMS), occurs.6 Several studies showed increased prevalence of PLMS in patients with SCI, and some case reports suggest an increased prevalence of RLS in this population.7,8 One small study showed that 100% of patients with SCI had symptoms of RLS.6 Another study found that SCI could trigger PLMS.8
The pathophysiology of RLS and PLMS in patients with SCI is not fully understood, but case reports describing PLM in SCI patients points to a possible role of central pattern generators and the flexor reflex afferents in the pathophysiology of PLMS.9,10 Changes of the tissue microstructure in the midbrain and upper cervical spinal cord have been described in patients with RLS.11The objective of this study was to assess the prevalence of RLS in a veteran population with SCI/D and
Methods
The institutional review and ethical approval boards of the Minneapolis VA Health Care System approved the study. Within the VA system, 666 patients with SCI/D were identified using a national database. Of the 666 people, 316 were excluded, 199 were included, and 151 were deceased.
Patients aged between 18 and 65 years were included in the study. Charts of patients who had been discharged with the diagnosis of SCI from 2002 to 2008 were studied. All patients met the inclusion criteria of the International Restless Legs Syndrome Study Group diagnosis.
Exclusion criteria were as follows: Patients with evidence of brain pathology (eg, stroke), concurrent neurologic condition associated with RLS (Parkinson disease, spinocerebellar ataxia, peripheral neuropathy), concurrent psychiatric condition within the setting of treatment with dopamine antagonists, secondary causes of RLS (renal failure/uremia, iron deficiency, rheumatoid arthritis, and pregnancy) and a recent history of alcohol or drug misuse or current evidence of substance use of < 1 year.
A patient list was compiled that included the etiology of the SCI (vascular injury, multiple sclerosis [MS], trauma, unknown, and other), the level(s) and completeness of the SCI per radiology report, RLS pharmacotherapies, and pertinent medical history.
Axial T2-weighted images on magnetic resonance imaging (MRI) scans were retrospectively reviewed. Sagittal T1/T2-weighted and axial T2-weighted sequences were performed routinely on all patients with spinal cord lesions. The analysis included the extension of the lesion on both sagittal and axial distributions. The anatomic location of the cord lesion was categorized by the following: (1) pure gray matter (central cord); (2) white matter (dorsal [D], dorsolateral [DL], ventral [V], ventrolateral areas [VL]).
A questionnaire using standard diagnostic criteria for RLS was mailed to the 199 patients who met the inclusion criteria (Appendix A).
All analyses were carried out using StataCorp STATA 13 (College Station, TX). Descriptive statistics were used. The analyses were carried out using chi-square and Fisher exact tests. Differences between the groups were considered statistically significant at P < .05. The data were analyzed to obtain point prevalence among patients with SCI, and comparisons were made among the different subgroups.
Results
Of the 162 patients who chose to participate in the study, the sleep specialists confirmed 31 (19%) to have RLS, 112 (69%) were confirmed negative for RLS, and an additional 19 (12%) screened positive for RLS but were not confirmed to have RLS by the sleep specialists (Figure 1).
The etiology of SCI was subdivided into 4 groups: MS, trauma, vascular, and other/unknown. Within each group (– RLS vs + RLS), MS and trauma were the most common etiologies with 55% MS and 36% trauma in the + RLS group.
When comparing RLS among the spinal cord levels (cervical, thoracic, lumbar and cervical + thoracic), only the cervical + thoracic subgroup (18% + RLS vs 5% – RLS) showed a significant difference (Figure 2).
There was no significant difference found with the prevalence of RLS in the axial plane of the spinal cord lesions (ventral/ventro-lateral/central cord vs dorsal/dorsolateral) or by the completeness of spinal cord lesions, P = .76. There was a higher prevalence of incomplete cord injury, however, within each subgroup of RLS.
The Mann-Whitney test was used to analyze the burden of disease in both groups (+ RLS vs – RLS). Moderate level of burden was most frequently reported with a higher prevalence within the + RLS group. Of those receiving treatment for RLS, 71% were + RLS vs 46% – RLS with a P value of .01. Symptoms of RLS after cord injury were 89% + RLS vs 55% – RLS with a P value of .03.
Discussion
This study represents one of the first studies to determine the prevalence of RLS in veterans with spinal cord disease. Research in this area is important to raise awareness of RLS among the veteran population with and without SCI and disorders. Restless legs syndrome often escapes diagnosis because of difficulty understanding the patient’s descriptions of their sensations. In addition, RLS may cause debilitating symptoms of sleep deprivation, daytime sleepiness, discomfort, and fatigue, which often results in decreased quality of life (QOL). Proper screening and treatment may improve QOL.
A study by Kumru and colleagues showed a similar rate of RLS in patients with SCI and RLS symptoms presented in the first year after SCI as did this study (18% vs 19%, respectively).4 In that study, RLS was more common in patients with lesions in lumbosacral area. Kumru and colleagues also showed that a dopaminergic medication improved symptoms of RLS in this population, whereas this study did not explore treatment outcomes.4
The pathogenesis of RLS is not fully known, but hereditary factors, iron metabolism, and the brain dopaminergic system are thought to be involved.11 It is hypothesized that spinal cord lesions allow the appearance of RLS symptoms and spinal leg movement generator by blocking descending inhibitory spinal pathways.12 One hypothesis is that damage to A11 nuclei (the main source of dopamine in the spinal cord or its diencephalospinal tract in animals) causes hyperexcitability of the spinal cord and leads to PLM and RLS symptoms.13 As the axons of A11 nuclei are present along the whole span of the spinal cord, SCI/D in patients with RLS might interrupt this dopaminergic tract and produce the RLS symptoms.
Limitations
This study included only veterans, so the prevalence may not apply to the nonveteran SCI population. Also, the population mainly was male, and there was no accurate information on race. Ferritin levels of the patients were not checked and is a major factor in RLS. The reported onset of RLS after the SCI could be due to recall bias.
Conclusion
The prevalence of RLS in veterans with SCI is above that reported in the general population (19% vs 10%, respectively). Furthermore, those with RLS have symptoms that often started after the SCI (suggesting causality) and required therapy due to their level of RLS symptom burden. A spectrum of severity of symptoms is present among those with RLS, with 83% having moderate-to-severe RLS affecting their QOL.
Although there was not a statistically significant relationship between RLS and spinal cord lesion level, there was a slightly higher prevalence of RLS at the cervical and thoracic levels, which may be relevant for future studies. There was no difference found between the RLS subgroups with respect to the location of the lesion within the spinal cord; however, a larger sample size may be needed to determine whether this would reach statistical significance. Prompt search for symptoms of RLS in veterans with SCI is warranted to provide adequate treatment to improve sleep health and QOL in this population.
Spinal cord injuries (SCI) are common in veteran populations.1 Veterans with spinal cord injuries and disorders (SCI/D) also may have concurrent sleep disturbances. Spinal cord injury typically causes spasticity.2,3 Hypersensitivity of the flexor reflex pathways is believed to cause painful muscle spasms in patients with SCI.4 Neuropathic pain at or below the level of the lesion also is common.
Restless legs syndrome (RLS) is a common sleep disorder that affects sleep quality and can occur concomitantly with spinal cord lesions.5 In about 80% of RLS cases, involuntary movements of legs across hip, knee, and ankle joints during sleep, known as periodic limb movement during sleep (PLMS), occurs.6 Several studies showed increased prevalence of PLMS in patients with SCI, and some case reports suggest an increased prevalence of RLS in this population.7,8 One small study showed that 100% of patients with SCI had symptoms of RLS.6 Another study found that SCI could trigger PLMS.8
The pathophysiology of RLS and PLMS in patients with SCI is not fully understood, but case reports describing PLM in SCI patients points to a possible role of central pattern generators and the flexor reflex afferents in the pathophysiology of PLMS.9,10 Changes of the tissue microstructure in the midbrain and upper cervical spinal cord have been described in patients with RLS.11The objective of this study was to assess the prevalence of RLS in a veteran population with SCI/D and
Methods
The institutional review and ethical approval boards of the Minneapolis VA Health Care System approved the study. Within the VA system, 666 patients with SCI/D were identified using a national database. Of the 666 people, 316 were excluded, 199 were included, and 151 were deceased.
Patients aged between 18 and 65 years were included in the study. Charts of patients who had been discharged with the diagnosis of SCI from 2002 to 2008 were studied. All patients met the inclusion criteria of the International Restless Legs Syndrome Study Group diagnosis.
Exclusion criteria were as follows: Patients with evidence of brain pathology (eg, stroke), concurrent neurologic condition associated with RLS (Parkinson disease, spinocerebellar ataxia, peripheral neuropathy), concurrent psychiatric condition within the setting of treatment with dopamine antagonists, secondary causes of RLS (renal failure/uremia, iron deficiency, rheumatoid arthritis, and pregnancy) and a recent history of alcohol or drug misuse or current evidence of substance use of < 1 year.
A patient list was compiled that included the etiology of the SCI (vascular injury, multiple sclerosis [MS], trauma, unknown, and other), the level(s) and completeness of the SCI per radiology report, RLS pharmacotherapies, and pertinent medical history.
Axial T2-weighted images on magnetic resonance imaging (MRI) scans were retrospectively reviewed. Sagittal T1/T2-weighted and axial T2-weighted sequences were performed routinely on all patients with spinal cord lesions. The analysis included the extension of the lesion on both sagittal and axial distributions. The anatomic location of the cord lesion was categorized by the following: (1) pure gray matter (central cord); (2) white matter (dorsal [D], dorsolateral [DL], ventral [V], ventrolateral areas [VL]).
A questionnaire using standard diagnostic criteria for RLS was mailed to the 199 patients who met the inclusion criteria (Appendix A).
All analyses were carried out using StataCorp STATA 13 (College Station, TX). Descriptive statistics were used. The analyses were carried out using chi-square and Fisher exact tests. Differences between the groups were considered statistically significant at P < .05. The data were analyzed to obtain point prevalence among patients with SCI, and comparisons were made among the different subgroups.
Results
Of the 162 patients who chose to participate in the study, the sleep specialists confirmed 31 (19%) to have RLS, 112 (69%) were confirmed negative for RLS, and an additional 19 (12%) screened positive for RLS but were not confirmed to have RLS by the sleep specialists (Figure 1).
The etiology of SCI was subdivided into 4 groups: MS, trauma, vascular, and other/unknown. Within each group (– RLS vs + RLS), MS and trauma were the most common etiologies with 55% MS and 36% trauma in the + RLS group.
When comparing RLS among the spinal cord levels (cervical, thoracic, lumbar and cervical + thoracic), only the cervical + thoracic subgroup (18% + RLS vs 5% – RLS) showed a significant difference (Figure 2).
There was no significant difference found with the prevalence of RLS in the axial plane of the spinal cord lesions (ventral/ventro-lateral/central cord vs dorsal/dorsolateral) or by the completeness of spinal cord lesions, P = .76. There was a higher prevalence of incomplete cord injury, however, within each subgroup of RLS.
The Mann-Whitney test was used to analyze the burden of disease in both groups (+ RLS vs – RLS). Moderate level of burden was most frequently reported with a higher prevalence within the + RLS group. Of those receiving treatment for RLS, 71% were + RLS vs 46% – RLS with a P value of .01. Symptoms of RLS after cord injury were 89% + RLS vs 55% – RLS with a P value of .03.
Discussion
This study represents one of the first studies to determine the prevalence of RLS in veterans with spinal cord disease. Research in this area is important to raise awareness of RLS among the veteran population with and without SCI and disorders. Restless legs syndrome often escapes diagnosis because of difficulty understanding the patient’s descriptions of their sensations. In addition, RLS may cause debilitating symptoms of sleep deprivation, daytime sleepiness, discomfort, and fatigue, which often results in decreased quality of life (QOL). Proper screening and treatment may improve QOL.
A study by Kumru and colleagues showed a similar rate of RLS in patients with SCI and RLS symptoms presented in the first year after SCI as did this study (18% vs 19%, respectively).4 In that study, RLS was more common in patients with lesions in lumbosacral area. Kumru and colleagues also showed that a dopaminergic medication improved symptoms of RLS in this population, whereas this study did not explore treatment outcomes.4
The pathogenesis of RLS is not fully known, but hereditary factors, iron metabolism, and the brain dopaminergic system are thought to be involved.11 It is hypothesized that spinal cord lesions allow the appearance of RLS symptoms and spinal leg movement generator by blocking descending inhibitory spinal pathways.12 One hypothesis is that damage to A11 nuclei (the main source of dopamine in the spinal cord or its diencephalospinal tract in animals) causes hyperexcitability of the spinal cord and leads to PLM and RLS symptoms.13 As the axons of A11 nuclei are present along the whole span of the spinal cord, SCI/D in patients with RLS might interrupt this dopaminergic tract and produce the RLS symptoms.
Limitations
This study included only veterans, so the prevalence may not apply to the nonveteran SCI population. Also, the population mainly was male, and there was no accurate information on race. Ferritin levels of the patients were not checked and is a major factor in RLS. The reported onset of RLS after the SCI could be due to recall bias.
Conclusion
The prevalence of RLS in veterans with SCI is above that reported in the general population (19% vs 10%, respectively). Furthermore, those with RLS have symptoms that often started after the SCI (suggesting causality) and required therapy due to their level of RLS symptom burden. A spectrum of severity of symptoms is present among those with RLS, with 83% having moderate-to-severe RLS affecting their QOL.
Although there was not a statistically significant relationship between RLS and spinal cord lesion level, there was a slightly higher prevalence of RLS at the cervical and thoracic levels, which may be relevant for future studies. There was no difference found between the RLS subgroups with respect to the location of the lesion within the spinal cord; however, a larger sample size may be needed to determine whether this would reach statistical significance. Prompt search for symptoms of RLS in veterans with SCI is warranted to provide adequate treatment to improve sleep health and QOL in this population.
1. Lasfargues JE, Custis D, Morrone F, Carswell J, Nguyen T. A model for estimating spinal cord injury prevalence in the United States. Paraplegia. 1995;33(2):62-68.
2. Sjölund BH. Pain and rehabilitation after spinal cord injury: the case of sensory spasticity? Brain Res Brain Res Rev. 2002;40(1-3):250-256.
3. Adams MM, Hicks AL. Spasticity after spinal cord injury. Spinal Cord. 2005;43(10):577-586.
4. Kumru H, Vidal J, Benito J, et al. Restless leg syndrome in patients with spinal cord injury. Parkinsonism Relat Disord. 2015;21(12):1461-1464.
5. Wilt TJ, MacDonald R, Ouellette J, et al. Pharmacologic therapy for primary restless legs syndrome: a systematic review and meta-analysis. JAMA Intern Med. 2013;173(7):496-505.
6. American Academy of Sleep Medicine. The International Classification of Sleep Disorders: Diagnostic and Coding Manual. (AASM ICSD-3). 3rd ed. Westchester, IL: American Academy of Sleep Medicine; 2014.
7. Telles SC, Alves RC, Chadi G. Periodic limb movements during sleep and restless legs syndrome in patients with ASIA A spinal cord injury. J Neurol Sci. 2011;303(1-2):119-123.
8. Telles SC, Alves RS, Chadi G. Spinal cord injury as a trigger to develop periodic leg movements during sleep: an evolutionary perspective. Arq Neuropsiquiatr. 2012;70(11):880-884.
9. Tings T, Baier PC, Paulus W, Trenkwalder C. Restless legs syndrome induced by impairment of sensory spinal pathways. J Neurol. 2003;250(4):499-500.
10. Paulus W, Trenkwalder C. Less is more: pathophysiology of dopaminergic-therapy-related augmentation in restless legs syndrome. Lancet Neurol. 2006;5(10):878-886.
11. Silber MH, Ehrenberg BL, Allen RP, et al; Medical Advisory Board of the Restless Legs Syndrome Foundation. An algorithm for the management of restless legs syndrome. Mayo Clin Proc. 2004;79(7):916-922.
12. Hartmann M, Pfister R, Pfadenhauer K. Restless legs syndrome associated with spinal cord lesions. J Neurol Neurosurg Psychiatry. 1999;66(5):688-689.
13. Clemens S, Rye D, Hochman S. Restless legs syndrome: revisiting the dopamine hypothesis from the spinal cord perspective. Neurology. 2006;67(1):125-130.
1. Lasfargues JE, Custis D, Morrone F, Carswell J, Nguyen T. A model for estimating spinal cord injury prevalence in the United States. Paraplegia. 1995;33(2):62-68.
2. Sjölund BH. Pain and rehabilitation after spinal cord injury: the case of sensory spasticity? Brain Res Brain Res Rev. 2002;40(1-3):250-256.
3. Adams MM, Hicks AL. Spasticity after spinal cord injury. Spinal Cord. 2005;43(10):577-586.
4. Kumru H, Vidal J, Benito J, et al. Restless leg syndrome in patients with spinal cord injury. Parkinsonism Relat Disord. 2015;21(12):1461-1464.
5. Wilt TJ, MacDonald R, Ouellette J, et al. Pharmacologic therapy for primary restless legs syndrome: a systematic review and meta-analysis. JAMA Intern Med. 2013;173(7):496-505.
6. American Academy of Sleep Medicine. The International Classification of Sleep Disorders: Diagnostic and Coding Manual. (AASM ICSD-3). 3rd ed. Westchester, IL: American Academy of Sleep Medicine; 2014.
7. Telles SC, Alves RC, Chadi G. Periodic limb movements during sleep and restless legs syndrome in patients with ASIA A spinal cord injury. J Neurol Sci. 2011;303(1-2):119-123.
8. Telles SC, Alves RS, Chadi G. Spinal cord injury as a trigger to develop periodic leg movements during sleep: an evolutionary perspective. Arq Neuropsiquiatr. 2012;70(11):880-884.
9. Tings T, Baier PC, Paulus W, Trenkwalder C. Restless legs syndrome induced by impairment of sensory spinal pathways. J Neurol. 2003;250(4):499-500.
10. Paulus W, Trenkwalder C. Less is more: pathophysiology of dopaminergic-therapy-related augmentation in restless legs syndrome. Lancet Neurol. 2006;5(10):878-886.
11. Silber MH, Ehrenberg BL, Allen RP, et al; Medical Advisory Board of the Restless Legs Syndrome Foundation. An algorithm for the management of restless legs syndrome. Mayo Clin Proc. 2004;79(7):916-922.
12. Hartmann M, Pfister R, Pfadenhauer K. Restless legs syndrome associated with spinal cord lesions. J Neurol Neurosurg Psychiatry. 1999;66(5):688-689.
13. Clemens S, Rye D, Hochman S. Restless legs syndrome: revisiting the dopamine hypothesis from the spinal cord perspective. Neurology. 2006;67(1):125-130.
USPSTF finds the evidence inconclusive for lead screening in young children, pregnant women
according to a recommendation from the U.S. Preventive Services Task Force.

Elevated blood lead levels are associated with potentially irreversible neurologic problems in children and with organ system impairment and adverse perinatal effects in pregnant women, according to the statement.
“Thus, the primary benefit of screening may be in preventing future exposures or exposure of others to environmental sources,” the task force members wrote in JAMA Pediatrics.
However, the task force issued I statements, meaning that “the current evidence is insufficient to assess the balance of benefits and harms of screening for elevated blood lead levels” in asymptomatic children aged 5 years and younger and in asymptomatic pregnant women.
The task force cited evidence that questionnaires and other clinical prediction tools are inaccurate at identifying elevated blood lead levels in asymptomatic children and pregnant women. In addition, the task force found adequate evidence that capillary blood testing identified elevated blood lead levels in children, but found inadequate evidence that treating elevated blood lead levels was effective in asymptomatic children aged 5 years and younger or in pregnant women.
In the evidence report accompanying the recommendation statement in JAMA Pediatrics, Amy G. Cantor, MD, MPH, of Oregon Health & Science University, Portland, and her colleagues reviewed data from a total of 24 studies including 11,433 individuals.
None of the studies evaluated the risks or benefits of blood lead screening in children. However, in three of four studies, capillary blood lead testing showed sensitivities ranging from 87% to 91% and specificities from 92% to 99%, based on a blood lead level cutoff of 10 mcg/dL or less.
“Evidence indicates that capillary sampling is slightly less sensitive than venous sampling, with comparable specificity,” Dr. Cantor and her colleagues wrote. “Both methods require confirmation.”
There is only limited evidence on whether intervening when children present with elevated blood lead levels results in better neurodevelopmental outcomes. One trial showed beneficial effects of dimercaptosuccinic acid chelation of lowering elevated blood lead levels (20-44 mcg/dL) at 1 year versus placebo, but no clear effect on longer term blood lead levels or neurodevelopmental outcomes, they reported.
For residential interventions, again evidence is limited and blood lead concentrations were not clearly affected. Evidence on calcium and iron interventions was poor quality and insufficient to tell if there was an effect on blood lead levels or clinical outcomes, Dr. Cantor and her colleagues wrote.
No studies of screening for elevated lead levels in pregnant women were identified, nor were studies of health outcomes after interventions to reduce blood lead levels in asymptomatic pregnant women, they noted.
Studies involving pregnant women were limited, and included data on the diagnostic accuracy of a clinical questionnaire and the effects of nutritional intervention during pregnancy, Dr. Cantor and her colleagues wrote.
“This update confirms there are no clear effects of interventions for lowering elevated blood levels in affected children or to improve neurodevelopmental outcomes,” they concluded. “Evidence to determine benefits and harms of screening or treating elevated lead levels during pregnancy remains extremely limited.”
The recommendation updates the last version issued in 2006. The USPSTF is supported by the Agency for Healthcare Research and Quality. The researchers for both articles reported no relevant financial disclosures.
SOURCE: Curry SJ et al. JAMA Pediatr. 2019 Apr 16. doi: 10.1001/jama.2019.3326; Cantor AG et al. JAMA Pediatr. 2019 Apr 16. doi: 10.1001/jama.2019.1004.
“The inconclusive findings of the new USPSTF [U.S. Preventive Services Task Force] recommendation does not mean that screening children for elevated lead levels is not necessary, nor does it shed light on whether screening should be targeted to children at high risk or whether it should be universally done,” Michael Weitzman, MD, wrote in an editorial in response to the USPSTF recommendations.
Dr. Weitzman noted that the recommendation is a consequence of the lack of quality studies on lead level screening, and wrote that, although the recommendations apply to asymptomatic children at both average risk and increased risk, the USPSTF does not recommend for or against screening or that screening be abandoned.
It is standard pediatric practice to counsel parents on lead exposure and screening for elevated blood lead levels in children aged 1-5 years, he wrote, adding that “the American Academy of Pediatrics, Bright Futures, the Centers for Disease Control and Prevention, and Medicaid all recommend universal blood lead screening or the screening of selected children believed to be at especially high risk of exposure at approximately age 1 and 2 years.”
More rigorous research is needed to make definitive recommendations, but in the meantime, clinicians should continue to work with local health departments, housing authorities, and schools to provide care for children with elevated lead levels while continuing with the screening practices recommended by the AAP and other organizations, and advocating for prevention of lead exposure, Dr. Weitzman wrote.
Dr. Weitzman is professor of pediatrics and professor of environmental medicine at New York University. This is a summary of the editorial Dr. Weitzman wrote to accompany the published USPSTF recommendation (JAMA Pediatr. 2019 Apr 16. doi:10.1001/jamapediatrics.2019.0855). He reported no relevant financial disclosures.
“The inconclusive findings of the new USPSTF [U.S. Preventive Services Task Force] recommendation does not mean that screening children for elevated lead levels is not necessary, nor does it shed light on whether screening should be targeted to children at high risk or whether it should be universally done,” Michael Weitzman, MD, wrote in an editorial in response to the USPSTF recommendations.
Dr. Weitzman noted that the recommendation is a consequence of the lack of quality studies on lead level screening, and wrote that, although the recommendations apply to asymptomatic children at both average risk and increased risk, the USPSTF does not recommend for or against screening or that screening be abandoned.
It is standard pediatric practice to counsel parents on lead exposure and screening for elevated blood lead levels in children aged 1-5 years, he wrote, adding that “the American Academy of Pediatrics, Bright Futures, the Centers for Disease Control and Prevention, and Medicaid all recommend universal blood lead screening or the screening of selected children believed to be at especially high risk of exposure at approximately age 1 and 2 years.”
More rigorous research is needed to make definitive recommendations, but in the meantime, clinicians should continue to work with local health departments, housing authorities, and schools to provide care for children with elevated lead levels while continuing with the screening practices recommended by the AAP and other organizations, and advocating for prevention of lead exposure, Dr. Weitzman wrote.
Dr. Weitzman is professor of pediatrics and professor of environmental medicine at New York University. This is a summary of the editorial Dr. Weitzman wrote to accompany the published USPSTF recommendation (JAMA Pediatr. 2019 Apr 16. doi:10.1001/jamapediatrics.2019.0855). He reported no relevant financial disclosures.
“The inconclusive findings of the new USPSTF [U.S. Preventive Services Task Force] recommendation does not mean that screening children for elevated lead levels is not necessary, nor does it shed light on whether screening should be targeted to children at high risk or whether it should be universally done,” Michael Weitzman, MD, wrote in an editorial in response to the USPSTF recommendations.
Dr. Weitzman noted that the recommendation is a consequence of the lack of quality studies on lead level screening, and wrote that, although the recommendations apply to asymptomatic children at both average risk and increased risk, the USPSTF does not recommend for or against screening or that screening be abandoned.
It is standard pediatric practice to counsel parents on lead exposure and screening for elevated blood lead levels in children aged 1-5 years, he wrote, adding that “the American Academy of Pediatrics, Bright Futures, the Centers for Disease Control and Prevention, and Medicaid all recommend universal blood lead screening or the screening of selected children believed to be at especially high risk of exposure at approximately age 1 and 2 years.”
More rigorous research is needed to make definitive recommendations, but in the meantime, clinicians should continue to work with local health departments, housing authorities, and schools to provide care for children with elevated lead levels while continuing with the screening practices recommended by the AAP and other organizations, and advocating for prevention of lead exposure, Dr. Weitzman wrote.
Dr. Weitzman is professor of pediatrics and professor of environmental medicine at New York University. This is a summary of the editorial Dr. Weitzman wrote to accompany the published USPSTF recommendation (JAMA Pediatr. 2019 Apr 16. doi:10.1001/jamapediatrics.2019.0855). He reported no relevant financial disclosures.
according to a recommendation from the U.S. Preventive Services Task Force.
Elevated blood lead levels are associated with potentially irreversible neurologic problems in children and with organ system impairment and adverse perinatal effects in pregnant women, according to the statement.
“Thus, the primary benefit of screening may be in preventing future exposures or exposure of others to environmental sources,” the task force members wrote in JAMA Pediatrics.
However, the task force issued I statements, meaning that “the current evidence is insufficient to assess the balance of benefits and harms of screening for elevated blood lead levels” in asymptomatic children aged 5 years and younger and in asymptomatic pregnant women.
The task force cited evidence that questionnaires and other clinical prediction tools are inaccurate at identifying elevated blood lead levels in asymptomatic children and pregnant women. In addition, the task force found adequate evidence that capillary blood testing identified elevated blood lead levels in children, but found inadequate evidence that treating elevated blood lead levels was effective in asymptomatic children aged 5 years and younger or in pregnant women.
In the evidence report accompanying the recommendation statement in JAMA Pediatrics, Amy G. Cantor, MD, MPH, of Oregon Health & Science University, Portland, and her colleagues reviewed data from a total of 24 studies including 11,433 individuals.
None of the studies evaluated the risks or benefits of blood lead screening in children. However, in three of four studies, capillary blood lead testing showed sensitivities ranging from 87% to 91% and specificities from 92% to 99%, based on a blood lead level cutoff of 10 mcg/dL or less.
“Evidence indicates that capillary sampling is slightly less sensitive than venous sampling, with comparable specificity,” Dr. Cantor and her colleagues wrote. “Both methods require confirmation.”
There is only limited evidence on whether intervening when children present with elevated blood lead levels results in better neurodevelopmental outcomes. One trial showed beneficial effects of dimercaptosuccinic acid chelation of lowering elevated blood lead levels (20-44 mcg/dL) at 1 year versus placebo, but no clear effect on longer term blood lead levels or neurodevelopmental outcomes, they reported.
For residential interventions, again evidence is limited and blood lead concentrations were not clearly affected. Evidence on calcium and iron interventions was poor quality and insufficient to tell if there was an effect on blood lead levels or clinical outcomes, Dr. Cantor and her colleagues wrote.
No studies of screening for elevated lead levels in pregnant women were identified, nor were studies of health outcomes after interventions to reduce blood lead levels in asymptomatic pregnant women, they noted.
Studies involving pregnant women were limited, and included data on the diagnostic accuracy of a clinical questionnaire and the effects of nutritional intervention during pregnancy, Dr. Cantor and her colleagues wrote.
“This update confirms there are no clear effects of interventions for lowering elevated blood levels in affected children or to improve neurodevelopmental outcomes,” they concluded. “Evidence to determine benefits and harms of screening or treating elevated lead levels during pregnancy remains extremely limited.”
The recommendation updates the last version issued in 2006. The USPSTF is supported by the Agency for Healthcare Research and Quality. The researchers for both articles reported no relevant financial disclosures.
SOURCE: Curry SJ et al. JAMA Pediatr. 2019 Apr 16. doi: 10.1001/jama.2019.3326; Cantor AG et al. JAMA Pediatr. 2019 Apr 16. doi: 10.1001/jama.2019.1004.
according to a recommendation from the U.S. Preventive Services Task Force.
Elevated blood lead levels are associated with potentially irreversible neurologic problems in children and with organ system impairment and adverse perinatal effects in pregnant women, according to the statement.
“Thus, the primary benefit of screening may be in preventing future exposures or exposure of others to environmental sources,” the task force members wrote in JAMA Pediatrics.
However, the task force issued I statements, meaning that “the current evidence is insufficient to assess the balance of benefits and harms of screening for elevated blood lead levels” in asymptomatic children aged 5 years and younger and in asymptomatic pregnant women.
The task force cited evidence that questionnaires and other clinical prediction tools are inaccurate at identifying elevated blood lead levels in asymptomatic children and pregnant women. In addition, the task force found adequate evidence that capillary blood testing identified elevated blood lead levels in children, but found inadequate evidence that treating elevated blood lead levels was effective in asymptomatic children aged 5 years and younger or in pregnant women.
In the evidence report accompanying the recommendation statement in JAMA Pediatrics, Amy G. Cantor, MD, MPH, of Oregon Health & Science University, Portland, and her colleagues reviewed data from a total of 24 studies including 11,433 individuals.
None of the studies evaluated the risks or benefits of blood lead screening in children. However, in three of four studies, capillary blood lead testing showed sensitivities ranging from 87% to 91% and specificities from 92% to 99%, based on a blood lead level cutoff of 10 mcg/dL or less.
“Evidence indicates that capillary sampling is slightly less sensitive than venous sampling, with comparable specificity,” Dr. Cantor and her colleagues wrote. “Both methods require confirmation.”
There is only limited evidence on whether intervening when children present with elevated blood lead levels results in better neurodevelopmental outcomes. One trial showed beneficial effects of dimercaptosuccinic acid chelation of lowering elevated blood lead levels (20-44 mcg/dL) at 1 year versus placebo, but no clear effect on longer term blood lead levels or neurodevelopmental outcomes, they reported.
For residential interventions, again evidence is limited and blood lead concentrations were not clearly affected. Evidence on calcium and iron interventions was poor quality and insufficient to tell if there was an effect on blood lead levels or clinical outcomes, Dr. Cantor and her colleagues wrote.
No studies of screening for elevated lead levels in pregnant women were identified, nor were studies of health outcomes after interventions to reduce blood lead levels in asymptomatic pregnant women, they noted.
Studies involving pregnant women were limited, and included data on the diagnostic accuracy of a clinical questionnaire and the effects of nutritional intervention during pregnancy, Dr. Cantor and her colleagues wrote.
“This update confirms there are no clear effects of interventions for lowering elevated blood levels in affected children or to improve neurodevelopmental outcomes,” they concluded. “Evidence to determine benefits and harms of screening or treating elevated lead levels during pregnancy remains extremely limited.”
The recommendation updates the last version issued in 2006. The USPSTF is supported by the Agency for Healthcare Research and Quality. The researchers for both articles reported no relevant financial disclosures.
SOURCE: Curry SJ et al. JAMA Pediatr. 2019 Apr 16. doi: 10.1001/jama.2019.3326; Cantor AG et al. JAMA Pediatr. 2019 Apr 16. doi: 10.1001/jama.2019.1004.
FROM JAMA PEDIATRICS
No clear winner for treating neuropathic pain
PHILADELPHIA – Nearly 7%-10% of the general population experiences neuropathic pain, but studies on treatments have not found a clear winner for reducing this “burning or electriclike pain,” explained Raymond Price, MD, during a presentation.

“It isn’t that exciting,” said Dr. Price, associate professor of neurology at the University of Pennsylvania, Philadelphia, in reference to his review of level 1-2 evidence for treatment of neuropathic pain that was presented in a study published in JAMA (2015 Nov 24;314[20]:2172-81). a few years ago. “On a scale of 1 to 10, you can reduce their pain scale by 1-2 points more than placebo,” he told his audience at the annual meeting of the American College of Physicians.
There are very limited head-to-head data as to which one is actually better,” he explained.
Given the absence of robust head-to-head trial data, Dr. Price tends to start a lot of patients on old, cheap medications like nortriptyline.
While there aren’t many head-to-head trials to guide treatment choice, the results of one prospective, randomized, open-label study of 333 patients with cryptogenic sensory polyneuropathy was presented by Barohn and colleagues at the 2018 annual meeting of the American Academy of Neurology, he said. In that study, somewhat higher efficacy rates were seen with duloxetine, a serotonin-noradrenaline reuptake inhibitor, and nortriptyline, a tricyclic antidepressant, compared with pregabalin, Dr. Price noted. Duloxetine and nortriptyline also had slightly better tolerability, as evidenced by a lower quit rate, compared with pregabalin, he added.
There was also a systematic review and meta-analysis (Lancet Neurol. 2015 Feb; 14[2]:162-73) conducted that determined the number needed to treat for neuropathic pain treatments, Dr. Price noted. In that paper, tricyclic antidepressants had a number needed to treat of 3.6, comparing favorably to 7.7 for pregabalin, 7.2 for gabapentin, and 6.4 for serotonin-noradrenaline reuptake inhibitors, mainly including duloxetine, said Dr. Price.
Regardless of the cause of neuropathic pain, the same general approach to treatment is taken, though most of the evidence comes from studies of patients with painful diabetic peripheral neuropathy or postherpetic neuralgia, he added.
For these patients, an adequate trial of a neuropathic pain treatment should be 6-12 weeks, reflecting the length of the intervention needed to demonstrate the efficacy of these agents, he said.
If that first drug doesn’t work, another can be tried, or multiple drugs can be tried together to see if the patient’s condition improves, he said.
Dr. Price reported no conflicts of interest.
SOURCE: Price R Internal Medicine 2019, Presentation MSFM 002.
PHILADELPHIA – Nearly 7%-10% of the general population experiences neuropathic pain, but studies on treatments have not found a clear winner for reducing this “burning or electriclike pain,” explained Raymond Price, MD, during a presentation.
“It isn’t that exciting,” said Dr. Price, associate professor of neurology at the University of Pennsylvania, Philadelphia, in reference to his review of level 1-2 evidence for treatment of neuropathic pain that was presented in a study published in JAMA (2015 Nov 24;314[20]:2172-81). a few years ago. “On a scale of 1 to 10, you can reduce their pain scale by 1-2 points more than placebo,” he told his audience at the annual meeting of the American College of Physicians.
There are very limited head-to-head data as to which one is actually better,” he explained.
Given the absence of robust head-to-head trial data, Dr. Price tends to start a lot of patients on old, cheap medications like nortriptyline.
While there aren’t many head-to-head trials to guide treatment choice, the results of one prospective, randomized, open-label study of 333 patients with cryptogenic sensory polyneuropathy was presented by Barohn and colleagues at the 2018 annual meeting of the American Academy of Neurology, he said. In that study, somewhat higher efficacy rates were seen with duloxetine, a serotonin-noradrenaline reuptake inhibitor, and nortriptyline, a tricyclic antidepressant, compared with pregabalin, Dr. Price noted. Duloxetine and nortriptyline also had slightly better tolerability, as evidenced by a lower quit rate, compared with pregabalin, he added.
There was also a systematic review and meta-analysis (Lancet Neurol. 2015 Feb; 14[2]:162-73) conducted that determined the number needed to treat for neuropathic pain treatments, Dr. Price noted. In that paper, tricyclic antidepressants had a number needed to treat of 3.6, comparing favorably to 7.7 for pregabalin, 7.2 for gabapentin, and 6.4 for serotonin-noradrenaline reuptake inhibitors, mainly including duloxetine, said Dr. Price.
Regardless of the cause of neuropathic pain, the same general approach to treatment is taken, though most of the evidence comes from studies of patients with painful diabetic peripheral neuropathy or postherpetic neuralgia, he added.
For these patients, an adequate trial of a neuropathic pain treatment should be 6-12 weeks, reflecting the length of the intervention needed to demonstrate the efficacy of these agents, he said.
If that first drug doesn’t work, another can be tried, or multiple drugs can be tried together to see if the patient’s condition improves, he said.
Dr. Price reported no conflicts of interest.
SOURCE: Price R Internal Medicine 2019, Presentation MSFM 002.
PHILADELPHIA – Nearly 7%-10% of the general population experiences neuropathic pain, but studies on treatments have not found a clear winner for reducing this “burning or electriclike pain,” explained Raymond Price, MD, during a presentation.
“It isn’t that exciting,” said Dr. Price, associate professor of neurology at the University of Pennsylvania, Philadelphia, in reference to his review of level 1-2 evidence for treatment of neuropathic pain that was presented in a study published in JAMA (2015 Nov 24;314[20]:2172-81). a few years ago. “On a scale of 1 to 10, you can reduce their pain scale by 1-2 points more than placebo,” he told his audience at the annual meeting of the American College of Physicians.
There are very limited head-to-head data as to which one is actually better,” he explained.
Given the absence of robust head-to-head trial data, Dr. Price tends to start a lot of patients on old, cheap medications like nortriptyline.
While there aren’t many head-to-head trials to guide treatment choice, the results of one prospective, randomized, open-label study of 333 patients with cryptogenic sensory polyneuropathy was presented by Barohn and colleagues at the 2018 annual meeting of the American Academy of Neurology, he said. In that study, somewhat higher efficacy rates were seen with duloxetine, a serotonin-noradrenaline reuptake inhibitor, and nortriptyline, a tricyclic antidepressant, compared with pregabalin, Dr. Price noted. Duloxetine and nortriptyline also had slightly better tolerability, as evidenced by a lower quit rate, compared with pregabalin, he added.
There was also a systematic review and meta-analysis (Lancet Neurol. 2015 Feb; 14[2]:162-73) conducted that determined the number needed to treat for neuropathic pain treatments, Dr. Price noted. In that paper, tricyclic antidepressants had a number needed to treat of 3.6, comparing favorably to 7.7 for pregabalin, 7.2 for gabapentin, and 6.4 for serotonin-noradrenaline reuptake inhibitors, mainly including duloxetine, said Dr. Price.
Regardless of the cause of neuropathic pain, the same general approach to treatment is taken, though most of the evidence comes from studies of patients with painful diabetic peripheral neuropathy or postherpetic neuralgia, he added.
For these patients, an adequate trial of a neuropathic pain treatment should be 6-12 weeks, reflecting the length of the intervention needed to demonstrate the efficacy of these agents, he said.
If that first drug doesn’t work, another can be tried, or multiple drugs can be tried together to see if the patient’s condition improves, he said.
Dr. Price reported no conflicts of interest.
SOURCE: Price R Internal Medicine 2019, Presentation MSFM 002.
AT INTERNAL MEDICINE 2019
A blood biomarker for MS: Coming to clinics soon?
DALLAS – Neurologists soon may use a blood biomarker of axonal damage to monitor patients with multiple sclerosis (MS) and guide treatment decisions, according to a lecture delivered at ACTRIMS Forum 2019.

said David Leppert, MD, senior research associate in the department of neurology at University Hopsital Basel (Switzerland).
Among patients with MS, blood NfL levels predict disability, brain volume loss, and spinal cord atrophy. In addition, studies have found that blood NfL decreases in response to disease-modifying therapy (DMT) and that second-line DMTs may decrease blood NfL more than first-line DMTs do.
The establishment of normative databases and reference biomarkers may allow neurologists to use NfL in their care of individual patients, Dr. Leppert predicted at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “I am very positive that we will make a breakthrough in the next 2 or 3 years for an individual use of neurofilaments,” he said.
Response to DMT
An analysis of blood NfL levels from patients with MS and from healthy controls in two phase 3 trials of fingolimod, FREEDOMS and TRANSFORMS, provides insights into NfL’s response to DMT (Neurology. 2019 Mar 5;92[10]:e1007-15). In FREEDOMS, which compared fingolimod with placebo, “fingolimod leads to a rapid decrease of neurofilament levels, close to normality, while placebo patients continue to have high levels,” said Dr. Leppert, a coauthor of the study.
TRANSFORMS compared interferon-beta and fingolimod. “The clinical experience that fingolimod is a more potent compound than interferon is actually reflected here by the NfL results,” Dr. Leppert said. “Both compounds lead to a decrease of neurofilaments – so, a decrease of neuronal damage – but one drug is more potent than the other one.”
Similarly, data from the observational EPIC study indicate that patients who do not receive DMT have a consistent increase in NfL levels, whereas those who receive platform therapies have a slight decrease in NfL and those who receive second-line therapies have a greater decrease, Dr. Leppert said.
Decades of research
For about 20 years, researchers have studied neurofilaments in cerebrospinal fluid (CSF) as a potential biomarker for MS and other diseases, including Alzheimer’s disease, amyotrophic lateral sclerosis, Parkinson’s disease, and head trauma.
“What prevented the emergence of NfL to clinical practice was the inability to measure it in blood because levels are 50-100 times lower [in blood] than in CSF,” Dr. Leppert said.
The development of single molecule array (SIMOA) technology enabled researchers to show a proper correlation between levels of NfL in CSF and those in blood, Dr. Leppert said. “That is now allowing repetitive testing in an accessible fluid compartment, meaning serum or plasma,” he said.
Compared with brain MRI, NfL may provide novel insights into MS disease activity. “MRI is measuring a structural deficit of the past,” Dr. Leppert said. “NfL is measuring online, real time what axonal damage is occurring.”
Correlation with outcomes
At the group level, patients with MS have higher levels of NfL, compared with controls, and levels are higher in patients with progressive forms of MS versus relapsing forms. “Levels increase dramatically in the wake of relapse,” he said.
Barro et al. found that patients with higher serum levels of NfL are more likely to experience Expanded Disability Status Scale (EDSS) worsening (Brain. 2018 Aug 1;141[8]:2382-91). Furthermore, MRI lesions are independently associated with serum NfL, and patients with higher levels of serum NfL have significantly greater average loss in brain volume and spinal cord volume over 5 years.
A treatment algorithm
NfL someday could be incorporated into MS treatment algorithms, Dr. Leppert suggested. For instance, if a patient has high levels of disease activity based on MRI or clinical grounds, then prescribe a high-efficacy therapy. If not, measure NfL. “If the levels are low, then you can be assured to use platform therapies or continue what the patient has. But if NfL levels are high, then you should choose high efficacious therapies or switch to high-efficacious therapies in the long run,” he said.
Limitations and next steps
Although NfL is a specific marker of neuronal damage, it is not specific for the cause of the damage. Further studies are needed to better understand NfL metabolism and confounding factors such as age. Reference biomarkers likely will be needed “to conceptualize whether the signal of NfL is due to acute disease or chronic disease,” Dr. Leppert said.
“We need to optimize the assay and come to a worldwide agreement on the platform. We need to have prospective studies, mainly to achieve regulatory acceptance. And we need to have a normative database” to determine which NfL values are pathologic at a particular age, he said.
Despite the biomarker’s potential, blood NfL levels will not replace clinical expertise. “Biomarkers cannot be of value without a clinical backing and a clinical evaluation,” Dr. Leppert said. “The idea that this will replace us or any other person who makes a clinical judgment is a big error. NfL will prevail as a biomarker. ... But interpretation of the clinical background is germane.”
Dr. Leppert has been an employee of pharmaceutical companies, most recently Novartis.
DALLAS – Neurologists soon may use a blood biomarker of axonal damage to monitor patients with multiple sclerosis (MS) and guide treatment decisions, according to a lecture delivered at ACTRIMS Forum 2019.
said David Leppert, MD, senior research associate in the department of neurology at University Hopsital Basel (Switzerland).
Among patients with MS, blood NfL levels predict disability, brain volume loss, and spinal cord atrophy. In addition, studies have found that blood NfL decreases in response to disease-modifying therapy (DMT) and that second-line DMTs may decrease blood NfL more than first-line DMTs do.
The establishment of normative databases and reference biomarkers may allow neurologists to use NfL in their care of individual patients, Dr. Leppert predicted at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “I am very positive that we will make a breakthrough in the next 2 or 3 years for an individual use of neurofilaments,” he said.
Response to DMT
An analysis of blood NfL levels from patients with MS and from healthy controls in two phase 3 trials of fingolimod, FREEDOMS and TRANSFORMS, provides insights into NfL’s response to DMT (Neurology. 2019 Mar 5;92[10]:e1007-15). In FREEDOMS, which compared fingolimod with placebo, “fingolimod leads to a rapid decrease of neurofilament levels, close to normality, while placebo patients continue to have high levels,” said Dr. Leppert, a coauthor of the study.
TRANSFORMS compared interferon-beta and fingolimod. “The clinical experience that fingolimod is a more potent compound than interferon is actually reflected here by the NfL results,” Dr. Leppert said. “Both compounds lead to a decrease of neurofilaments – so, a decrease of neuronal damage – but one drug is more potent than the other one.”
Similarly, data from the observational EPIC study indicate that patients who do not receive DMT have a consistent increase in NfL levels, whereas those who receive platform therapies have a slight decrease in NfL and those who receive second-line therapies have a greater decrease, Dr. Leppert said.
Decades of research
For about 20 years, researchers have studied neurofilaments in cerebrospinal fluid (CSF) as a potential biomarker for MS and other diseases, including Alzheimer’s disease, amyotrophic lateral sclerosis, Parkinson’s disease, and head trauma.
“What prevented the emergence of NfL to clinical practice was the inability to measure it in blood because levels are 50-100 times lower [in blood] than in CSF,” Dr. Leppert said.
The development of single molecule array (SIMOA) technology enabled researchers to show a proper correlation between levels of NfL in CSF and those in blood, Dr. Leppert said. “That is now allowing repetitive testing in an accessible fluid compartment, meaning serum or plasma,” he said.
Compared with brain MRI, NfL may provide novel insights into MS disease activity. “MRI is measuring a structural deficit of the past,” Dr. Leppert said. “NfL is measuring online, real time what axonal damage is occurring.”
Correlation with outcomes
At the group level, patients with MS have higher levels of NfL, compared with controls, and levels are higher in patients with progressive forms of MS versus relapsing forms. “Levels increase dramatically in the wake of relapse,” he said.
Barro et al. found that patients with higher serum levels of NfL are more likely to experience Expanded Disability Status Scale (EDSS) worsening (Brain. 2018 Aug 1;141[8]:2382-91). Furthermore, MRI lesions are independently associated with serum NfL, and patients with higher levels of serum NfL have significantly greater average loss in brain volume and spinal cord volume over 5 years.
A treatment algorithm
NfL someday could be incorporated into MS treatment algorithms, Dr. Leppert suggested. For instance, if a patient has high levels of disease activity based on MRI or clinical grounds, then prescribe a high-efficacy therapy. If not, measure NfL. “If the levels are low, then you can be assured to use platform therapies or continue what the patient has. But if NfL levels are high, then you should choose high efficacious therapies or switch to high-efficacious therapies in the long run,” he said.
Limitations and next steps
Although NfL is a specific marker of neuronal damage, it is not specific for the cause of the damage. Further studies are needed to better understand NfL metabolism and confounding factors such as age. Reference biomarkers likely will be needed “to conceptualize whether the signal of NfL is due to acute disease or chronic disease,” Dr. Leppert said.
“We need to optimize the assay and come to a worldwide agreement on the platform. We need to have prospective studies, mainly to achieve regulatory acceptance. And we need to have a normative database” to determine which NfL values are pathologic at a particular age, he said.
Despite the biomarker’s potential, blood NfL levels will not replace clinical expertise. “Biomarkers cannot be of value without a clinical backing and a clinical evaluation,” Dr. Leppert said. “The idea that this will replace us or any other person who makes a clinical judgment is a big error. NfL will prevail as a biomarker. ... But interpretation of the clinical background is germane.”
Dr. Leppert has been an employee of pharmaceutical companies, most recently Novartis.
DALLAS – Neurologists soon may use a blood biomarker of axonal damage to monitor patients with multiple sclerosis (MS) and guide treatment decisions, according to a lecture delivered at ACTRIMS Forum 2019.
said David Leppert, MD, senior research associate in the department of neurology at University Hopsital Basel (Switzerland).
Among patients with MS, blood NfL levels predict disability, brain volume loss, and spinal cord atrophy. In addition, studies have found that blood NfL decreases in response to disease-modifying therapy (DMT) and that second-line DMTs may decrease blood NfL more than first-line DMTs do.
The establishment of normative databases and reference biomarkers may allow neurologists to use NfL in their care of individual patients, Dr. Leppert predicted at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “I am very positive that we will make a breakthrough in the next 2 or 3 years for an individual use of neurofilaments,” he said.
Response to DMT
An analysis of blood NfL levels from patients with MS and from healthy controls in two phase 3 trials of fingolimod, FREEDOMS and TRANSFORMS, provides insights into NfL’s response to DMT (Neurology. 2019 Mar 5;92[10]:e1007-15). In FREEDOMS, which compared fingolimod with placebo, “fingolimod leads to a rapid decrease of neurofilament levels, close to normality, while placebo patients continue to have high levels,” said Dr. Leppert, a coauthor of the study.
TRANSFORMS compared interferon-beta and fingolimod. “The clinical experience that fingolimod is a more potent compound than interferon is actually reflected here by the NfL results,” Dr. Leppert said. “Both compounds lead to a decrease of neurofilaments – so, a decrease of neuronal damage – but one drug is more potent than the other one.”
Similarly, data from the observational EPIC study indicate that patients who do not receive DMT have a consistent increase in NfL levels, whereas those who receive platform therapies have a slight decrease in NfL and those who receive second-line therapies have a greater decrease, Dr. Leppert said.
Decades of research
For about 20 years, researchers have studied neurofilaments in cerebrospinal fluid (CSF) as a potential biomarker for MS and other diseases, including Alzheimer’s disease, amyotrophic lateral sclerosis, Parkinson’s disease, and head trauma.
“What prevented the emergence of NfL to clinical practice was the inability to measure it in blood because levels are 50-100 times lower [in blood] than in CSF,” Dr. Leppert said.
The development of single molecule array (SIMOA) technology enabled researchers to show a proper correlation between levels of NfL in CSF and those in blood, Dr. Leppert said. “That is now allowing repetitive testing in an accessible fluid compartment, meaning serum or plasma,” he said.
Compared with brain MRI, NfL may provide novel insights into MS disease activity. “MRI is measuring a structural deficit of the past,” Dr. Leppert said. “NfL is measuring online, real time what axonal damage is occurring.”
Correlation with outcomes
At the group level, patients with MS have higher levels of NfL, compared with controls, and levels are higher in patients with progressive forms of MS versus relapsing forms. “Levels increase dramatically in the wake of relapse,” he said.
Barro et al. found that patients with higher serum levels of NfL are more likely to experience Expanded Disability Status Scale (EDSS) worsening (Brain. 2018 Aug 1;141[8]:2382-91). Furthermore, MRI lesions are independently associated with serum NfL, and patients with higher levels of serum NfL have significantly greater average loss in brain volume and spinal cord volume over 5 years.
A treatment algorithm
NfL someday could be incorporated into MS treatment algorithms, Dr. Leppert suggested. For instance, if a patient has high levels of disease activity based on MRI or clinical grounds, then prescribe a high-efficacy therapy. If not, measure NfL. “If the levels are low, then you can be assured to use platform therapies or continue what the patient has. But if NfL levels are high, then you should choose high efficacious therapies or switch to high-efficacious therapies in the long run,” he said.
Limitations and next steps
Although NfL is a specific marker of neuronal damage, it is not specific for the cause of the damage. Further studies are needed to better understand NfL metabolism and confounding factors such as age. Reference biomarkers likely will be needed “to conceptualize whether the signal of NfL is due to acute disease or chronic disease,” Dr. Leppert said.
“We need to optimize the assay and come to a worldwide agreement on the platform. We need to have prospective studies, mainly to achieve regulatory acceptance. And we need to have a normative database” to determine which NfL values are pathologic at a particular age, he said.
Despite the biomarker’s potential, blood NfL levels will not replace clinical expertise. “Biomarkers cannot be of value without a clinical backing and a clinical evaluation,” Dr. Leppert said. “The idea that this will replace us or any other person who makes a clinical judgment is a big error. NfL will prevail as a biomarker. ... But interpretation of the clinical background is germane.”
Dr. Leppert has been an employee of pharmaceutical companies, most recently Novartis.
EXPERT ANALYSIS FROM ACTRIMS FORUM 2019
Self-Management in Epilepsy Care: Untapped Opportunities (FULL)
Epilepsy is a chronic neurologic condition defined by recurrent seizures not provoked by an environmental or a reversible trigger. About 1% of the US population has an epilepsy diagnosis, and an even higher percentage of the world’s population has seizures.1 For the many US soldiers who sustain blast-and concussion-related injuries, posttraumatic epilepsy is a potential risk.2 Although the risk of epilepsy remains unknown, the Veterans Health Administration (VHA) prioritizes diagnosis and management of the condition. Fortunately, antiepileptic therapies are effective for most patients. About 65% of patients can be free of seizures with use of a single daily medication.3 Although the other 35% often experience refractory seizures, advanced medication regimens, surgical approaches, and innovative devices can effect improvement in some cases.
Increasingly, patients are urged to practice epilepsy self-management. The idea of self-managing epilepsy, which has existed for decades, is supported primarily by a theory of robust patient education intended to increase disease knowledge and improve decision making. Multiple formal self-management programs have been developed and academically tested for patients with epilepsy. In a 2013 report, the Institute of Medicine emphasized the importance of research on the effects of behavioral self-management interventions on health outcomes and quality of life for people with epilepsy. The report recommended improving and expanding educational opportunities for patients.4 Nevertheless, self-management programs have not found widespread traction in mainstream clinical use.
This article provides a review of chronic disease self-management with a focus on its application and study in epilepsy. The authors discuss self-management, including underlying theory, definitions, and various tools. The principal formal epilepsy programs that have been studied and published are highlighted and summarized. This review also includes a discussion of the potential barriers to successful implementation of these epilepsy programs along with emerging solutions and tools for addressing these barriers.
Self-Management Theory
Disease self-management originated in social cognitive theory, which addresses the cognitive, emotional, and behavioral aspects of behavior change and is relevant to managing chronic illness.5,6 Self-management of chronic illness is defined as the daily actions that people take to keep their illness under control, to minimize its impact on physical health status and functioning, and to cope with psychosocial sequelae.7 These actions include making informed decisions about care, performing activities intended to manage the condition, and applying the necessary skills to maintain adequate psychosocial functioning.7
Related to self-management is self-efficacy, people’s confidence in their ability to engage in these actions.7 Evidence-based self-management and self-efficacy strategies are recognized as central in managing a variety of chronic diseases by improving the medical, emotional, and social role that management demands of chronic conditions.8
Self-management and self-efficacy have been explored in patients with epilepsy for decades, with various approaches being developed, implemented, and tested. Findings of several historical studies discussed in this review indicate that patients with epilepsy and high levels of self-efficacy are more successful in performing self-care tasks.9 This growing body of evidence led to the establishment of the Managing Epilepsy Well network in 2007.10 The Centers for Disease Control and Prevention created the network to expand epilepsy self-management research. Since 2007, more research has been focused on the potential for online and mobile health approaches in supporting epilepsy self-management and on intervention studies evaluating e-tools.
Elements of Epilepsy Self-Management
The first element of an epilepsy-specific self-management program is formal education on the diagnosis, treatment, and psychosocial impact of epilepsy and on strategies for coping with it. This element usually includes tools for evaluating and understanding epilepsy, with the goal of empowering patients to become actively engaged in managing and coping with their epilepsy diagnosis. Medication adherence is key in the optimal management of epilepsy. This point is evident in the development of a validated metric for self-efficacy: the Epilepsy Self-Efficacy Scale (ESES).11 Of the 33 items on the ESES, 14 are devoted to aspects of medication management. Other crucial behavioral elements for epilepsy self-management relate to lifestyle issues, such as safety, diet, exercise, sleep, and stress management.
Various self-management programs have incorporated tracking systems for these lifestyle elements as well as epilepsy-specific measures, such as seizure frequency, duration, and type. In addition, social support is an important factor in chronic illness self-management. Results of several studies support the hypothesis that higher levels of social support, particularly disease- and regimen-specific support, are related to better self-management behaviors.12 An increasing number of formal epilepsy self-management programs include peer support platforms and peer navigator features in their suite of services.
Patient Education and Self-Management Programs
Over the past several decades, multiple research groups have developed, implemented, and tested formal self-management platforms for patients with epilepsy. Designs and results of prominent studies are summarized in the Table. 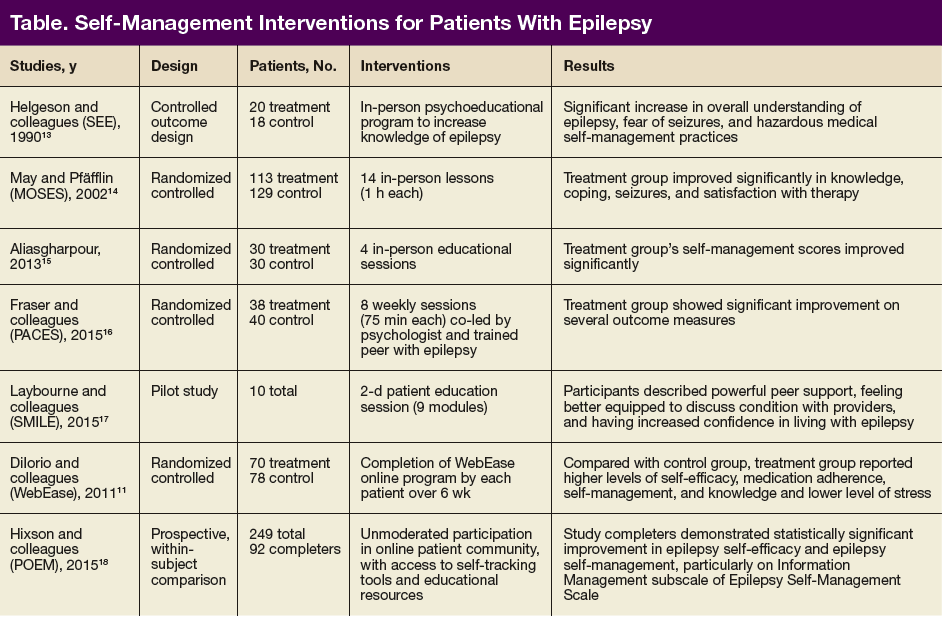
More recent programs also included a focus on peer-to-peer support and patient-driven content within the educational curriculum.16,17 In 2015, Hixson and colleagues used an entirely patient-driven online platform.18 Unlike the programs described thus far, this platform made educational modules available and did not require that patients complete them. Peer-to-peer support and self-tracking tools were prominently featured, and patients used them. In addition, this intervention focused exclusively on a group of US veterans with epilepsy.
Tools for Improving Self-Management
Self-management programs for patients with epilepsy historically have involved formalized programs conducted face-to-face with other patients, with professional moderators, and perhaps with caregivers. These programs depended entirely on in-person educational sessions and in-person support groups and were found to be very effective in improving self-management skills, though they were labor-intensive and logistically challenging for both practitioners and patients.
Since the advent of the Internet and mobile connectivity, many programs have incorporated the same elements in more accessible form. Educational content appears in live webinars and asynchronous video educational modules; the latter are attractive because patients and caregivers can access them independently at any time. Also readily available are tools for day-to-day self-management of medical conditions. These tools include mobile and online diaries for tracking seizure metrics and medication adherence reminder systems. Last, a variety of online and mobile disease-specific social networking platforms allow patients to connect directly to others without having to travel long distances to meet in-person. Although these digital solutions may not provide the exact experience offered by an in-person support group, the promise of superior accessibility creates an advantage in terms of accessibility and flexibility.
Self-Management in the Literature
In a recent review of care delivery and self-managementstrategies for adults with epilepsy, Bradley and colleagues analyzed 18 different studies of 16 separate interventions and concluded that 2 interventions, the specialist epilepsy nurse and self-management education, had some evidence of benefit. Four studies, detailed next, had the highest quality design, based on a focus on epilepsy self-management specifically, a prospective hypothesis-driven approach, and rigorous methodology.19
In 1990, Helgeson and colleagues evaluated Sepulveda Epilepsy Education, a 2-day in-person program designed to provide medical education and psychosocial therapy to patients with an epilepsy diagnosis.13 The program was based on the theory that having a better understanding of their epilepsy helps people cope with the condition. Medical, social, and emotional topics are covered. Medical topics include epilepsy and how it may change over time, as well as diagnosis, treatment, and first aid; social and emotional topics include coping with the psychological aspects of epilepsy, family, social aspects, and employment. In this small study (38 patients total), compared with the control group (18 patients), the treatment group (20 patients) demonstrated a significant reduction in the level of fear of death and brain damage caused by seizures, a significant decrease in hazardous medical self-management practices, and a significant decrease in misconceptions about epilepsy. The treatment group also increased their medication adherence, as determined by serum drug levels. In addition, statistically nonsignificant trends were shown by the treatment group toward improved emotional, interpersonal, and vocational functioning; improved adjustment to seizures; and improved overall psychosocial functioning.
In 2002, May and Pfäfflin evaluated the efficacy of the Modular Service Package Epilepsy (MOSES) educational program.14 This program was specifically developed to improve patient knowledge about epilepsy and its consequences and diagnostic and therapeutic measures, and to improve patient understanding of psychosocial and occupational problems. It was the first comprehensive program used in German-speaking countries. It had 9 modules: coping with epilepsy, epidemiology, basic knowledge, diagnostics, therapy, self-control, prognosis, psychosocial aspects, and network. To complete the program, patients work through about fourteen 1-hour lessons. The controlled, randomized study by May and Pfäfflin involved 242 patients (113 treatment, 129 control) aged 16 to 80 years. Patients in the treatment (MOSES) group demonstrated significant improvements in 2 of the 9 modules (knowledge, coping with epilepsy), had improved self-reported seizure outcomes, were more satisfied with therapy, experienced better tolerability of antiepileptic drugs with fewer adverse effects (AEs), and were highly satisfied with the program. The researchers concluded that educational programs, such as MOSES, should become a standard service for specialized epilepsy care.
Developed over many years, WebEase is an online epilepsy self-management program that supports education on medication, stress, and sleep management. In 2011, DiIorio and colleagues reported on a WebEase trial in which 194 patients were randomly assigned to either a treatment group (n = 96) or a wait-list control group (n = 96), and 2 were lost to follow up.11 After accounting for study criteria and study drop out, 70 participants completed the treatment arm, and 78 completed the control arm. The study measured the impact of the platform on multiple outcome metrics, including 3 behavioral areas of focus. At follow-up, self-reported levels of medication adherence were higher for patients in the treatment group than for those in the control group. Analyses also compared patients who completed WebEase modules with those who did not. Patients who completed at least some WebEase modules reported higher levels of self-efficacy, and a trend toward significance was found for medication adherence, perceived stress, self-management, and knowledge. The authors concluded that online tools that support epilepsy self-management could be effective.11
In 2015, Fraser and colleagues reported the results of the Program for Active Consumer Engagement in Self-Management in Epilepsy (PACES in Epilepsy), a consumer-generated self-management program.16 In the trial, 83 adults with chronic epilepsy were initially assigned either to an in-person intervention or to treatment as usual. After study drop outs, 38 patients remained in the intervention arm, with 40 in the control arm. In the intervention, 6 to 8 adults met for a 75-minute group session 1 evening per week for 8 weeks; these sessions were co-led by a psychologist and a trained peer with epilepsy. Topics included medical, psychosocial, cognitive, and self-management aspects of epilepsy, in addition to community integration and optimization of epilepsy-related communication. Outcomes were measured with various instruments, including the ESES, the Quality of Life in Epilepsy-31 (QOLIE-31), the Epilepsy Self-Management Scale (ESMS), the Patient Health Questionnaire-9, and the Generalized Anxiety Disorder-7. Each test was administered at baseline and after intervention. Outcomes were assessed immediately after program completion (8 weeks) and at follow-up 6 months later.
Findings suggested a substantial positive impact on epilepsy self-management capacities at program completion. In addition, benefit was sustained, particularly for epilepsy information management, over the 6 months after program completion. On the QOLIE-31 at 6 months, management of medication AEs also remained significantly improved, and fatigue management was improved at the P < .05 level. The researchers concluded that the PACES in Epilepsy program might have a more sustained impact on management of disability than on mood. They also noted that the effect was greater immediately after program completion than at 6 months. Patients gave the PACES program high satisfaction ratings.
Although these programs take slightly different approaches to epilepsy self-management, they have a similar focus: directed patient education. Furthermore, most of these programs are conducted in person, usually in a support group setting. In the WebEase trial, patients seem to have completed the online modules in a study setting, and a peer support component was not included. Overall, all programs successfully demonstrated various benefits for trial patients. These outcomes suggest that despite their subtle differences in approach, formal self-management programs are benefiting patients.
None of these platforms was designed for or specifically tested veterans with epilepsy. Although veterans theoretically would benefit from the same tools used by nonveterans, Iraq and Afghanistan veterans with epilepsy are more likely than are those without epilepsy to have mental and physical comorbidities and significantly higher mortality.2 Therefore, veterans potentially could benefit more from evidence-based chronic disease self-management programs designed to reduce physical and psychiatric comorbidities. Furthermore, programs that incorporate peer-to-peer support and direct links to VA care teams and mental health providers could be valuable.18
One research effort that directly addressed these issues is the Policy for Optimized Epilepsy Management (POEM) study, conducted by Hixson and colleagues in 2015.18 This study, not included in the review by Bradley and colleagues, used a purely online- and mobile-based social networking platform to promote self-management practices.19 Unlike the other programs described here, POEM did not require that patients view or attend formal educational seminars, though these seminars were available through the online platform for patient self-directed viewing. In addition, the intervention heavily promoted peer-to-peer engagement and disease tracking as means of increasing self-knowledge and activation. This study was unlike the other platforms in another way: It specifically focused on veterans with epilepsy, based on the idea that many veterans had a shared experience that would optimize a peer support approach.
The POEM investigators did not use a controlled design but found a significant benefit for both ESES and ESMS metrics on within-subject comparisons. Similar to the PACES in Epilepsy study, the POEM study found the highest benefit on the information management subscale of the ESMS.16 Practically speaking, this means patients were better able to use and manage digital and mobile information resources for controlling epilepsy. The POEM study results further reinforced the idea that epilepsy self-management programs are beneficial and expanded on earlier research to emphasize the value of peer support networks and digital interventions that can be used by patients at their convenience. These features provide greater access to more patients and maintain the crucial elements of peer-to-peer learning and counseling.
Implementation Barriers
Confirming the effectiveness of self-management programs is only the beginning of formal implementation and adoption. The real-world success of patient self-management programs has been documented for a few chronic diseases, including epilepsy. However, there is little research or commentary on lessons learned or on the challenges encountered with wide implementation of these programs.
Initial Setup and Sponsorship
To promote wider adoption, researchers should include commentary on initial setup, ongoing patient acceptance, and continual provider support. Many of the initial challenges in self-management programs involve a changing paradigm in the delivery and economics of health care. The transition to a more consumer-oriented health model with an emphasis on outcomes and patient-reported variables likely will support self-management strategies but is only slowly evolving. Many health care providers, hospitals, and payers may not be familiar with or have proper incentivizes to explore self-management tools even when proven effective.
More specifically, these epilepsy self-management programs are treatment adjuncts well suited to military and veteran health care systems. Self-management closely aligns with the overall VHA mission, vision, and values, including formal Department of Veteran Affairs (VA) goals and the MyVA priorities that collectively embrace improvement in access, a veteran-centric approach, and quality for improvement of the entire VA experience. Self-management platforms in the VA are recognized as empowering veterans and are thought to indirectly improve access to health care.20,21
The barriers of sponsorship and financial support likely will persist in the private health care sector but are less likely to significantly affect the VHA. Self-management programs have been researched and implemented for many health conditions across the VHA. For example, the VA Talent Management System course Patient Self-Management: Skill Building (TMS 6467) offers education and training to all clinical practitioners and managers involved in patient education and self-management activities for a variety of chronic medical conditions. Regarding epilepsy self-management more specifically, a patient brochure on the practice is distributed by the VHA Epilepsy Centers of Excellence (ECoEs) and an associated consortium.22 Last, a national provider educational lecture series has a corresponding patient and caregiver lecture set that emphasizes education and self-management behaviors.
Labor, Time, and Resource Needs
The most time-intensive aspect of designing self-management programs is developing the tool that allows clinicians and patients to work together. From a program perspective, the tool must be available and helpful not only to patients and specialists, but also to primary care providers. Tertiary-care centers usually accept the responsibility for program initiation, including patient recruitment, logistics coordination, and health care professional staffing. For epilepsy, the small pool of relevant specialists and centers limits the number of self-management education sessions that can be hosted and increases the need for complex travel and scheduling tasks. However, ECoE communication lines provide a basic infrastructure for collaboration and for development of tools that can be helpful to all clinicians treating veterans with epilepsy.23
Given the issues with coordinating the logistics of in-person programs at brick-and-mortar sites, this type of program may not be the best option for some patients and facilities. Alternative approaches, such as telehealth and asynchronous digital platforms, could expand access and increase convenience. Even though remotely administered programs may not be as powerful for some patients, the promise of scalable access supports consideration of these approaches.
Patient and Caregiver Logistics
Veterans with epilepsy may also have comorbid traumatic brain injury (TBI) and posttraumatic stress disorder, which can complicate self-managed care. In addition, many veterans live in rural areas and have limited travel options. All these factors challenge the success of epilepsy self-management programs. However, the network of ECoEs and associated consortium facilities can step up to deliver self-management tools and information.
The infrastructure of the VHA patient aligned care team (PACT) also contributes to the integration of self-management training. The PACT model takes a personalized, comprehensive, coordinated approach to promote team-based, veteran-centric care and actively partners with other VHA offices to incorporate alternative care services, including peer support and self-management platforms. The combination represents fertile ground for implementation and promotion of self-management tools in the VHA epilepsy population.
Health Care Economics
Given the uncertainties of the US health care economy, it is not surprising that many experts advocate a fundamental redesign of the health care team relationship and information infrastructure.24 This realignment includes partnering directly with patients and their families to encourage more reliance on self-management practices. Unfortunately, this approach does not lend itself to the well-entrenched business model on which most community medical practices are based. Health system leadership often must be convinced there are potential cost savings or a return on investment for new programs. As there is no consistent, comprehensive reimbursement policy for programs focused on self-management, health care systems must be creative and innovative when appraising the financial consequences of such programs.
Epilepsy remains a huge burden. In 2000, the annual total cost of epilepsy treatment in the US was $362 million for new patients and $2 billion for existing cases.25 Within the VHA, the occurrence of posttraumatic epilepsy among the increasing number of veterans with TBI contributes to the burden, and posttraumatic epilepsy and psychogenic nonepileptic seizures complicate treatment approaches. The incidence of comorbidities, including anxiety and depression, has been as high as 50%.23 Epilepsy health care programs are evaluating ways to validate their ability to minimize cost, improve access, and maintain quality of service. Integration of self-management should be included in these efforts.
The VHA represents a unique health care environment for testing and implementing self-management programs. Although the VHA is not immune to the traditional business models of medicine, it is less dependent on them, and it disproportionately cares for patients for long spans of time. From the health care team perspective, data indicate that ECoE physicians represent a high percentage of VHA epilepsy specialists but directly see only about 20% of veterans with an epilepsy or seizure-associated diagnosis. Therefore, future collaboration and connectivity of consortium sites can have a broader impact on self-management—highlighting the fact that concerted, scaled self-management programs have an important role in the VHA health care delivery system and should be promoted.26
Final Insights and Opportunities
Despite the barriers to adoption, formal epilepsy self-management programs are making gains in maturity and academic credibility. As the health care economy gradually shifts to more outcomes-based models, these offerings likely will become more valued, particularly by health care organizations focused on cost sharing, by large self-insuring employers, or organizations like the VHA where patients maintain a long-term relationship. Nevertheless, for the more resource-intensive, in-person self-management programs, adoption may remain constrained. Digital and mobile platforms should serve as more accessible entry points, with lower costs and more rapid scaling potential. Even though these online platforms may not have the same impact as intensive face-to-face programs, their scalability and constant accessibility should make them attractive, and the relatively modest cost of implementing self-guided programs should reduce barriers to adoption.
Integrated health care systems, such as the VHA and various European health systems, can serve as models for self-management implementation. Incorporating a live clinical implementation into parallel research efforts can continue to produce vital academic information on the real-world impact of these solutions, and this evidence in turn can be used to support policies that foster widespread adoption. More specifically, the ECoE model represents a clear opportunity to promote widespread implementation of self-management. The ECoEs are already publishing self-management materials that health care teams can use in patient counseling,and several self-care studies are being conducted within the network.22 In this model, compared with private sector health systems, ECoEs are well positioned to advance the use of formal self-management strategies.
The proposed epilepsy self-management model for ECoEs would be based on an iterative program that incorporates best practices from each of the research studies discussed earlier. With the publication of new research, successful self-management tools would be incorporated into the programs. From a curriculum perspective, educational platforms on medication adherence, seizure safety, and information/data management should be included. Evidence is increasing that peer support and use of licensed peer navigators should be incorporated as well. Last, flexible and asynchronous digital methods should be added to self-management platforms to maximize patient access. These features build on the growing body of evidence to maximize the likelihood of a successful and sustainable self-management strategy for patients with epilepsy.
Click here to read the digital edition.
1. Fiest KM, Sauro KM, Wiebe S, et al. Prevalence and incidence in epilepsy: a systematic review and meta-analysis of international studies. Neurology. 2017;88(3):296-303.
2. Pugh MJ, Van Cott AC, Amuan M, et al. Epilepsy among Iraq and Afghanistan war veterans—United States, 2002-2015. MMWR. 2016;65(44):1224-1227.
3. Kwan P, Brodie MJ. Effectiveness of first antiepileptic drug. Epilepsia. 2001;42(10):1255-1260.
4. Hesdorffer DC, Beck V, Begley CE, et al. Research implications of the Institute of Medicine report, Epilepsy Across the Spectrum: Promoting Health and Understanding. Epilepsia. 2013;54(2):207-216.
5. Bandura A. Social Foundations of Thought and Action: A Social Cognitive Theory. Englewood Cliffs, NJ: Prentice-Hall; 1986.
6. Bandura A. Social Learning Theory. Englewood Cliffs, NJ: Prentice-Hall; 1977.
7. Clark, NM, Becker MH, Janz NK, Lorig K, Rakowski W, Anderson L. Self-management of chronic disease by older adults. J Aging Health. 1991;3(1):3-27.
8. Ory MG, Ahn SM, Jiang L, et al. Successes of a national study of the Chronic Disease Self-Management Program: meeting the triple aim of health care reform. Med Care. 2013;51(11):992-998.
9. DiIorio C, Shafer PO, Letz R, Henry TR, Schomer DL, Yeager K; Project EASE Study Group. Behavioral, social and affective factors associated with self-efficacy for self-management among people with epilepsy. Epilepsy Behav. 2006;9(1):158-163.
10. Shegog R, Bamps YA, Patel A, et al. Managing Epilepsy Well: emerging e-tools for epilepsy self-management. Epilepsy Behav. 2013;29(1):133-140.
11. DiIorio C, Bamps Y, Walker ER, Escoffery C. Results of a research study evaluating WebEase, an online epilepsy self-management program. Epilepsy Behav. 2011;22(3):469-474.
12. Gallant MP. The influence of social support on chronic illness self-management: a review and directions for research. Health Educ Behav. 2003;30(2):170-195.
13. Helgeson DC, Mittan R, Tan SY, Chayasirisobhon S. Sepulveda Epilepsy Education: the efficacy of a psychoeducational treatment programme in treating medical and psychosocial aspects of epilepsy. Epilepsia. 1990;31(1):75-82.
14. May TW, Pfäfflin M. The efficacy of an educational treatment program for patients with epilepsy (MOSES): results of a controlled, randomized study. Modular Service Package Epilepsy. Epilepsia. 2002;43(5):539-549.
15. Aliasgharpour M, Dehgahn Nayeri N, Yadegary MA, Haghani H. Effects of an educational program on self-management in patients with epilepsy. Seizure. 2013;22(1):48-52.
16. Fraser RT, Johnson EK, Lashley S, et al. PACES in Epilepsy: results of a self-management randomized controlled trial. Epilepsia. 2015;56(8):1264-1274.
17. Laybourne AH, Morgan M, Watkins SH, Lawton R, Ridsdale L, Goldstein LH. Self-management for people with poorly controlled epilepsy: participants’ views of the UK self-management in epilepsy (SMILE) program. Epilepsy Behav. 2015;52(pt A):159-164.
18. Hixson JD, Barnes D, Parko K, et al. Patients optimizing epilepsy management via an online community: the POEM study. Neurology. 2015;85(2):129-136.
19. Bradley PM, Lindsay B, Fleeman N. Care delivery and self-management strategies for adults with epilepsy. Cochrane Database Syst Rev. 2016;2:CD006244.
20. Allicock M, Haynes-Maslow L, Carr C, et al. Training veterans to provide peer support in a weight-management program: MOVE! Prev Chronic Dis. 2013;10:E185.
21. Damush TM, Jackson GL, Powers BJ, et al. Implementing evidence-based patient self-management programs in the Veterans Health Administration: perspectives on delivery system design considerations. J Gen Intern Med. 2010;25(suppl 1):68-71.
22. Caraveo N, Chen S, Evrard C, Ozuna J; Epilepsy Centers of Excellence Nursing Workgroup. Self-management in epilepsy: a guide for healthcare professionals. https://www.epilepsy.va.gov/Library/Self-Management%20In%20Epilepsy.pdf. Published Winter 2015. Accessed February 26, 2018.
23. Rehman R, Kelly PR, Husain AM, Tran TT. Characteristics of veterans diagnosed with seizures within the Veterans Health Administration. J Rehabil Res Dev. 2015;52(7):751-762.
24. Merry MD. Healthcare’s need for revolutionary change. Quality Prog. 2003;36(9):31-35.
25. Halpern M, Rentz A, Murray M. Cost of illness of epilepsy in the US: comparison of patient-based and population-based estimates. Neuroepidemiology. 2000;19(2):87-99.
26. Kelly P, Chinta R. Do centers of excellence excel in patient outcomes?: Evidence from U.S. Veterans Health Administration Centers for Epilepsy. Int J Manage Excellence. 2015;4(3):529-538.
Epilepsy is a chronic neurologic condition defined by recurrent seizures not provoked by an environmental or a reversible trigger. About 1% of the US population has an epilepsy diagnosis, and an even higher percentage of the world’s population has seizures.1 For the many US soldiers who sustain blast-and concussion-related injuries, posttraumatic epilepsy is a potential risk.2 Although the risk of epilepsy remains unknown, the Veterans Health Administration (VHA) prioritizes diagnosis and management of the condition. Fortunately, antiepileptic therapies are effective for most patients. About 65% of patients can be free of seizures with use of a single daily medication.3 Although the other 35% often experience refractory seizures, advanced medication regimens, surgical approaches, and innovative devices can effect improvement in some cases.
Increasingly, patients are urged to practice epilepsy self-management. The idea of self-managing epilepsy, which has existed for decades, is supported primarily by a theory of robust patient education intended to increase disease knowledge and improve decision making. Multiple formal self-management programs have been developed and academically tested for patients with epilepsy. In a 2013 report, the Institute of Medicine emphasized the importance of research on the effects of behavioral self-management interventions on health outcomes and quality of life for people with epilepsy. The report recommended improving and expanding educational opportunities for patients.4 Nevertheless, self-management programs have not found widespread traction in mainstream clinical use.
This article provides a review of chronic disease self-management with a focus on its application and study in epilepsy. The authors discuss self-management, including underlying theory, definitions, and various tools. The principal formal epilepsy programs that have been studied and published are highlighted and summarized. This review also includes a discussion of the potential barriers to successful implementation of these epilepsy programs along with emerging solutions and tools for addressing these barriers.
Self-Management Theory
Disease self-management originated in social cognitive theory, which addresses the cognitive, emotional, and behavioral aspects of behavior change and is relevant to managing chronic illness.5,6 Self-management of chronic illness is defined as the daily actions that people take to keep their illness under control, to minimize its impact on physical health status and functioning, and to cope with psychosocial sequelae.7 These actions include making informed decisions about care, performing activities intended to manage the condition, and applying the necessary skills to maintain adequate psychosocial functioning.7
Related to self-management is self-efficacy, people’s confidence in their ability to engage in these actions.7 Evidence-based self-management and self-efficacy strategies are recognized as central in managing a variety of chronic diseases by improving the medical, emotional, and social role that management demands of chronic conditions.8
Self-management and self-efficacy have been explored in patients with epilepsy for decades, with various approaches being developed, implemented, and tested. Findings of several historical studies discussed in this review indicate that patients with epilepsy and high levels of self-efficacy are more successful in performing self-care tasks.9 This growing body of evidence led to the establishment of the Managing Epilepsy Well network in 2007.10 The Centers for Disease Control and Prevention created the network to expand epilepsy self-management research. Since 2007, more research has been focused on the potential for online and mobile health approaches in supporting epilepsy self-management and on intervention studies evaluating e-tools.
Elements of Epilepsy Self-Management
The first element of an epilepsy-specific self-management program is formal education on the diagnosis, treatment, and psychosocial impact of epilepsy and on strategies for coping with it. This element usually includes tools for evaluating and understanding epilepsy, with the goal of empowering patients to become actively engaged in managing and coping with their epilepsy diagnosis. Medication adherence is key in the optimal management of epilepsy. This point is evident in the development of a validated metric for self-efficacy: the Epilepsy Self-Efficacy Scale (ESES).11 Of the 33 items on the ESES, 14 are devoted to aspects of medication management. Other crucial behavioral elements for epilepsy self-management relate to lifestyle issues, such as safety, diet, exercise, sleep, and stress management.
Various self-management programs have incorporated tracking systems for these lifestyle elements as well as epilepsy-specific measures, such as seizure frequency, duration, and type. In addition, social support is an important factor in chronic illness self-management. Results of several studies support the hypothesis that higher levels of social support, particularly disease- and regimen-specific support, are related to better self-management behaviors.12 An increasing number of formal epilepsy self-management programs include peer support platforms and peer navigator features in their suite of services.
Patient Education and Self-Management Programs
Over the past several decades, multiple research groups have developed, implemented, and tested formal self-management platforms for patients with epilepsy. Designs and results of prominent studies are summarized in the Table.
More recent programs also included a focus on peer-to-peer support and patient-driven content within the educational curriculum.16,17 In 2015, Hixson and colleagues used an entirely patient-driven online platform.18 Unlike the programs described thus far, this platform made educational modules available and did not require that patients complete them. Peer-to-peer support and self-tracking tools were prominently featured, and patients used them. In addition, this intervention focused exclusively on a group of US veterans with epilepsy.
Tools for Improving Self-Management
Self-management programs for patients with epilepsy historically have involved formalized programs conducted face-to-face with other patients, with professional moderators, and perhaps with caregivers. These programs depended entirely on in-person educational sessions and in-person support groups and were found to be very effective in improving self-management skills, though they were labor-intensive and logistically challenging for both practitioners and patients.
Since the advent of the Internet and mobile connectivity, many programs have incorporated the same elements in more accessible form. Educational content appears in live webinars and asynchronous video educational modules; the latter are attractive because patients and caregivers can access them independently at any time. Also readily available are tools for day-to-day self-management of medical conditions. These tools include mobile and online diaries for tracking seizure metrics and medication adherence reminder systems. Last, a variety of online and mobile disease-specific social networking platforms allow patients to connect directly to others without having to travel long distances to meet in-person. Although these digital solutions may not provide the exact experience offered by an in-person support group, the promise of superior accessibility creates an advantage in terms of accessibility and flexibility.
Self-Management in the Literature
In a recent review of care delivery and self-managementstrategies for adults with epilepsy, Bradley and colleagues analyzed 18 different studies of 16 separate interventions and concluded that 2 interventions, the specialist epilepsy nurse and self-management education, had some evidence of benefit. Four studies, detailed next, had the highest quality design, based on a focus on epilepsy self-management specifically, a prospective hypothesis-driven approach, and rigorous methodology.19
In 1990, Helgeson and colleagues evaluated Sepulveda Epilepsy Education, a 2-day in-person program designed to provide medical education and psychosocial therapy to patients with an epilepsy diagnosis.13 The program was based on the theory that having a better understanding of their epilepsy helps people cope with the condition. Medical, social, and emotional topics are covered. Medical topics include epilepsy and how it may change over time, as well as diagnosis, treatment, and first aid; social and emotional topics include coping with the psychological aspects of epilepsy, family, social aspects, and employment. In this small study (38 patients total), compared with the control group (18 patients), the treatment group (20 patients) demonstrated a significant reduction in the level of fear of death and brain damage caused by seizures, a significant decrease in hazardous medical self-management practices, and a significant decrease in misconceptions about epilepsy. The treatment group also increased their medication adherence, as determined by serum drug levels. In addition, statistically nonsignificant trends were shown by the treatment group toward improved emotional, interpersonal, and vocational functioning; improved adjustment to seizures; and improved overall psychosocial functioning.
In 2002, May and Pfäfflin evaluated the efficacy of the Modular Service Package Epilepsy (MOSES) educational program.14 This program was specifically developed to improve patient knowledge about epilepsy and its consequences and diagnostic and therapeutic measures, and to improve patient understanding of psychosocial and occupational problems. It was the first comprehensive program used in German-speaking countries. It had 9 modules: coping with epilepsy, epidemiology, basic knowledge, diagnostics, therapy, self-control, prognosis, psychosocial aspects, and network. To complete the program, patients work through about fourteen 1-hour lessons. The controlled, randomized study by May and Pfäfflin involved 242 patients (113 treatment, 129 control) aged 16 to 80 years. Patients in the treatment (MOSES) group demonstrated significant improvements in 2 of the 9 modules (knowledge, coping with epilepsy), had improved self-reported seizure outcomes, were more satisfied with therapy, experienced better tolerability of antiepileptic drugs with fewer adverse effects (AEs), and were highly satisfied with the program. The researchers concluded that educational programs, such as MOSES, should become a standard service for specialized epilepsy care.
Developed over many years, WebEase is an online epilepsy self-management program that supports education on medication, stress, and sleep management. In 2011, DiIorio and colleagues reported on a WebEase trial in which 194 patients were randomly assigned to either a treatment group (n = 96) or a wait-list control group (n = 96), and 2 were lost to follow up.11 After accounting for study criteria and study drop out, 70 participants completed the treatment arm, and 78 completed the control arm. The study measured the impact of the platform on multiple outcome metrics, including 3 behavioral areas of focus. At follow-up, self-reported levels of medication adherence were higher for patients in the treatment group than for those in the control group. Analyses also compared patients who completed WebEase modules with those who did not. Patients who completed at least some WebEase modules reported higher levels of self-efficacy, and a trend toward significance was found for medication adherence, perceived stress, self-management, and knowledge. The authors concluded that online tools that support epilepsy self-management could be effective.11
In 2015, Fraser and colleagues reported the results of the Program for Active Consumer Engagement in Self-Management in Epilepsy (PACES in Epilepsy), a consumer-generated self-management program.16 In the trial, 83 adults with chronic epilepsy were initially assigned either to an in-person intervention or to treatment as usual. After study drop outs, 38 patients remained in the intervention arm, with 40 in the control arm. In the intervention, 6 to 8 adults met for a 75-minute group session 1 evening per week for 8 weeks; these sessions were co-led by a psychologist and a trained peer with epilepsy. Topics included medical, psychosocial, cognitive, and self-management aspects of epilepsy, in addition to community integration and optimization of epilepsy-related communication. Outcomes were measured with various instruments, including the ESES, the Quality of Life in Epilepsy-31 (QOLIE-31), the Epilepsy Self-Management Scale (ESMS), the Patient Health Questionnaire-9, and the Generalized Anxiety Disorder-7. Each test was administered at baseline and after intervention. Outcomes were assessed immediately after program completion (8 weeks) and at follow-up 6 months later.
Findings suggested a substantial positive impact on epilepsy self-management capacities at program completion. In addition, benefit was sustained, particularly for epilepsy information management, over the 6 months after program completion. On the QOLIE-31 at 6 months, management of medication AEs also remained significantly improved, and fatigue management was improved at the P < .05 level. The researchers concluded that the PACES in Epilepsy program might have a more sustained impact on management of disability than on mood. They also noted that the effect was greater immediately after program completion than at 6 months. Patients gave the PACES program high satisfaction ratings.
Although these programs take slightly different approaches to epilepsy self-management, they have a similar focus: directed patient education. Furthermore, most of these programs are conducted in person, usually in a support group setting. In the WebEase trial, patients seem to have completed the online modules in a study setting, and a peer support component was not included. Overall, all programs successfully demonstrated various benefits for trial patients. These outcomes suggest that despite their subtle differences in approach, formal self-management programs are benefiting patients.
None of these platforms was designed for or specifically tested veterans with epilepsy. Although veterans theoretically would benefit from the same tools used by nonveterans, Iraq and Afghanistan veterans with epilepsy are more likely than are those without epilepsy to have mental and physical comorbidities and significantly higher mortality.2 Therefore, veterans potentially could benefit more from evidence-based chronic disease self-management programs designed to reduce physical and psychiatric comorbidities. Furthermore, programs that incorporate peer-to-peer support and direct links to VA care teams and mental health providers could be valuable.18
One research effort that directly addressed these issues is the Policy for Optimized Epilepsy Management (POEM) study, conducted by Hixson and colleagues in 2015.18 This study, not included in the review by Bradley and colleagues, used a purely online- and mobile-based social networking platform to promote self-management practices.19 Unlike the other programs described here, POEM did not require that patients view or attend formal educational seminars, though these seminars were available through the online platform for patient self-directed viewing. In addition, the intervention heavily promoted peer-to-peer engagement and disease tracking as means of increasing self-knowledge and activation. This study was unlike the other platforms in another way: It specifically focused on veterans with epilepsy, based on the idea that many veterans had a shared experience that would optimize a peer support approach.
The POEM investigators did not use a controlled design but found a significant benefit for both ESES and ESMS metrics on within-subject comparisons. Similar to the PACES in Epilepsy study, the POEM study found the highest benefit on the information management subscale of the ESMS.16 Practically speaking, this means patients were better able to use and manage digital and mobile information resources for controlling epilepsy. The POEM study results further reinforced the idea that epilepsy self-management programs are beneficial and expanded on earlier research to emphasize the value of peer support networks and digital interventions that can be used by patients at their convenience. These features provide greater access to more patients and maintain the crucial elements of peer-to-peer learning and counseling.
Implementation Barriers
Confirming the effectiveness of self-management programs is only the beginning of formal implementation and adoption. The real-world success of patient self-management programs has been documented for a few chronic diseases, including epilepsy. However, there is little research or commentary on lessons learned or on the challenges encountered with wide implementation of these programs.
Initial Setup and Sponsorship
To promote wider adoption, researchers should include commentary on initial setup, ongoing patient acceptance, and continual provider support. Many of the initial challenges in self-management programs involve a changing paradigm in the delivery and economics of health care. The transition to a more consumer-oriented health model with an emphasis on outcomes and patient-reported variables likely will support self-management strategies but is only slowly evolving. Many health care providers, hospitals, and payers may not be familiar with or have proper incentivizes to explore self-management tools even when proven effective.
More specifically, these epilepsy self-management programs are treatment adjuncts well suited to military and veteran health care systems. Self-management closely aligns with the overall VHA mission, vision, and values, including formal Department of Veteran Affairs (VA) goals and the MyVA priorities that collectively embrace improvement in access, a veteran-centric approach, and quality for improvement of the entire VA experience. Self-management platforms in the VA are recognized as empowering veterans and are thought to indirectly improve access to health care.20,21
The barriers of sponsorship and financial support likely will persist in the private health care sector but are less likely to significantly affect the VHA. Self-management programs have been researched and implemented for many health conditions across the VHA. For example, the VA Talent Management System course Patient Self-Management: Skill Building (TMS 6467) offers education and training to all clinical practitioners and managers involved in patient education and self-management activities for a variety of chronic medical conditions. Regarding epilepsy self-management more specifically, a patient brochure on the practice is distributed by the VHA Epilepsy Centers of Excellence (ECoEs) and an associated consortium.22 Last, a national provider educational lecture series has a corresponding patient and caregiver lecture set that emphasizes education and self-management behaviors.
Labor, Time, and Resource Needs
The most time-intensive aspect of designing self-management programs is developing the tool that allows clinicians and patients to work together. From a program perspective, the tool must be available and helpful not only to patients and specialists, but also to primary care providers. Tertiary-care centers usually accept the responsibility for program initiation, including patient recruitment, logistics coordination, and health care professional staffing. For epilepsy, the small pool of relevant specialists and centers limits the number of self-management education sessions that can be hosted and increases the need for complex travel and scheduling tasks. However, ECoE communication lines provide a basic infrastructure for collaboration and for development of tools that can be helpful to all clinicians treating veterans with epilepsy.23
Given the issues with coordinating the logistics of in-person programs at brick-and-mortar sites, this type of program may not be the best option for some patients and facilities. Alternative approaches, such as telehealth and asynchronous digital platforms, could expand access and increase convenience. Even though remotely administered programs may not be as powerful for some patients, the promise of scalable access supports consideration of these approaches.
Patient and Caregiver Logistics
Veterans with epilepsy may also have comorbid traumatic brain injury (TBI) and posttraumatic stress disorder, which can complicate self-managed care. In addition, many veterans live in rural areas and have limited travel options. All these factors challenge the success of epilepsy self-management programs. However, the network of ECoEs and associated consortium facilities can step up to deliver self-management tools and information.
The infrastructure of the VHA patient aligned care team (PACT) also contributes to the integration of self-management training. The PACT model takes a personalized, comprehensive, coordinated approach to promote team-based, veteran-centric care and actively partners with other VHA offices to incorporate alternative care services, including peer support and self-management platforms. The combination represents fertile ground for implementation and promotion of self-management tools in the VHA epilepsy population.
Health Care Economics
Given the uncertainties of the US health care economy, it is not surprising that many experts advocate a fundamental redesign of the health care team relationship and information infrastructure.24 This realignment includes partnering directly with patients and their families to encourage more reliance on self-management practices. Unfortunately, this approach does not lend itself to the well-entrenched business model on which most community medical practices are based. Health system leadership often must be convinced there are potential cost savings or a return on investment for new programs. As there is no consistent, comprehensive reimbursement policy for programs focused on self-management, health care systems must be creative and innovative when appraising the financial consequences of such programs.
Epilepsy remains a huge burden. In 2000, the annual total cost of epilepsy treatment in the US was $362 million for new patients and $2 billion for existing cases.25 Within the VHA, the occurrence of posttraumatic epilepsy among the increasing number of veterans with TBI contributes to the burden, and posttraumatic epilepsy and psychogenic nonepileptic seizures complicate treatment approaches. The incidence of comorbidities, including anxiety and depression, has been as high as 50%.23 Epilepsy health care programs are evaluating ways to validate their ability to minimize cost, improve access, and maintain quality of service. Integration of self-management should be included in these efforts.
The VHA represents a unique health care environment for testing and implementing self-management programs. Although the VHA is not immune to the traditional business models of medicine, it is less dependent on them, and it disproportionately cares for patients for long spans of time. From the health care team perspective, data indicate that ECoE physicians represent a high percentage of VHA epilepsy specialists but directly see only about 20% of veterans with an epilepsy or seizure-associated diagnosis. Therefore, future collaboration and connectivity of consortium sites can have a broader impact on self-management—highlighting the fact that concerted, scaled self-management programs have an important role in the VHA health care delivery system and should be promoted.26
Final Insights and Opportunities
Despite the barriers to adoption, formal epilepsy self-management programs are making gains in maturity and academic credibility. As the health care economy gradually shifts to more outcomes-based models, these offerings likely will become more valued, particularly by health care organizations focused on cost sharing, by large self-insuring employers, or organizations like the VHA where patients maintain a long-term relationship. Nevertheless, for the more resource-intensive, in-person self-management programs, adoption may remain constrained. Digital and mobile platforms should serve as more accessible entry points, with lower costs and more rapid scaling potential. Even though these online platforms may not have the same impact as intensive face-to-face programs, their scalability and constant accessibility should make them attractive, and the relatively modest cost of implementing self-guided programs should reduce barriers to adoption.
Integrated health care systems, such as the VHA and various European health systems, can serve as models for self-management implementation. Incorporating a live clinical implementation into parallel research efforts can continue to produce vital academic information on the real-world impact of these solutions, and this evidence in turn can be used to support policies that foster widespread adoption. More specifically, the ECoE model represents a clear opportunity to promote widespread implementation of self-management. The ECoEs are already publishing self-management materials that health care teams can use in patient counseling,and several self-care studies are being conducted within the network.22 In this model, compared with private sector health systems, ECoEs are well positioned to advance the use of formal self-management strategies.
The proposed epilepsy self-management model for ECoEs would be based on an iterative program that incorporates best practices from each of the research studies discussed earlier. With the publication of new research, successful self-management tools would be incorporated into the programs. From a curriculum perspective, educational platforms on medication adherence, seizure safety, and information/data management should be included. Evidence is increasing that peer support and use of licensed peer navigators should be incorporated as well. Last, flexible and asynchronous digital methods should be added to self-management platforms to maximize patient access. These features build on the growing body of evidence to maximize the likelihood of a successful and sustainable self-management strategy for patients with epilepsy.
Click here to read the digital edition.
Epilepsy is a chronic neurologic condition defined by recurrent seizures not provoked by an environmental or a reversible trigger. About 1% of the US population has an epilepsy diagnosis, and an even higher percentage of the world’s population has seizures.1 For the many US soldiers who sustain blast-and concussion-related injuries, posttraumatic epilepsy is a potential risk.2 Although the risk of epilepsy remains unknown, the Veterans Health Administration (VHA) prioritizes diagnosis and management of the condition. Fortunately, antiepileptic therapies are effective for most patients. About 65% of patients can be free of seizures with use of a single daily medication.3 Although the other 35% often experience refractory seizures, advanced medication regimens, surgical approaches, and innovative devices can effect improvement in some cases.
Increasingly, patients are urged to practice epilepsy self-management. The idea of self-managing epilepsy, which has existed for decades, is supported primarily by a theory of robust patient education intended to increase disease knowledge and improve decision making. Multiple formal self-management programs have been developed and academically tested for patients with epilepsy. In a 2013 report, the Institute of Medicine emphasized the importance of research on the effects of behavioral self-management interventions on health outcomes and quality of life for people with epilepsy. The report recommended improving and expanding educational opportunities for patients.4 Nevertheless, self-management programs have not found widespread traction in mainstream clinical use.
This article provides a review of chronic disease self-management with a focus on its application and study in epilepsy. The authors discuss self-management, including underlying theory, definitions, and various tools. The principal formal epilepsy programs that have been studied and published are highlighted and summarized. This review also includes a discussion of the potential barriers to successful implementation of these epilepsy programs along with emerging solutions and tools for addressing these barriers.
Self-Management Theory
Disease self-management originated in social cognitive theory, which addresses the cognitive, emotional, and behavioral aspects of behavior change and is relevant to managing chronic illness.5,6 Self-management of chronic illness is defined as the daily actions that people take to keep their illness under control, to minimize its impact on physical health status and functioning, and to cope with psychosocial sequelae.7 These actions include making informed decisions about care, performing activities intended to manage the condition, and applying the necessary skills to maintain adequate psychosocial functioning.7
Related to self-management is self-efficacy, people’s confidence in their ability to engage in these actions.7 Evidence-based self-management and self-efficacy strategies are recognized as central in managing a variety of chronic diseases by improving the medical, emotional, and social role that management demands of chronic conditions.8
Self-management and self-efficacy have been explored in patients with epilepsy for decades, with various approaches being developed, implemented, and tested. Findings of several historical studies discussed in this review indicate that patients with epilepsy and high levels of self-efficacy are more successful in performing self-care tasks.9 This growing body of evidence led to the establishment of the Managing Epilepsy Well network in 2007.10 The Centers for Disease Control and Prevention created the network to expand epilepsy self-management research. Since 2007, more research has been focused on the potential for online and mobile health approaches in supporting epilepsy self-management and on intervention studies evaluating e-tools.
Elements of Epilepsy Self-Management
The first element of an epilepsy-specific self-management program is formal education on the diagnosis, treatment, and psychosocial impact of epilepsy and on strategies for coping with it. This element usually includes tools for evaluating and understanding epilepsy, with the goal of empowering patients to become actively engaged in managing and coping with their epilepsy diagnosis. Medication adherence is key in the optimal management of epilepsy. This point is evident in the development of a validated metric for self-efficacy: the Epilepsy Self-Efficacy Scale (ESES).11 Of the 33 items on the ESES, 14 are devoted to aspects of medication management. Other crucial behavioral elements for epilepsy self-management relate to lifestyle issues, such as safety, diet, exercise, sleep, and stress management.
Various self-management programs have incorporated tracking systems for these lifestyle elements as well as epilepsy-specific measures, such as seizure frequency, duration, and type. In addition, social support is an important factor in chronic illness self-management. Results of several studies support the hypothesis that higher levels of social support, particularly disease- and regimen-specific support, are related to better self-management behaviors.12 An increasing number of formal epilepsy self-management programs include peer support platforms and peer navigator features in their suite of services.
Patient Education and Self-Management Programs
Over the past several decades, multiple research groups have developed, implemented, and tested formal self-management platforms for patients with epilepsy. Designs and results of prominent studies are summarized in the Table.
More recent programs also included a focus on peer-to-peer support and patient-driven content within the educational curriculum.16,17 In 2015, Hixson and colleagues used an entirely patient-driven online platform.18 Unlike the programs described thus far, this platform made educational modules available and did not require that patients complete them. Peer-to-peer support and self-tracking tools were prominently featured, and patients used them. In addition, this intervention focused exclusively on a group of US veterans with epilepsy.
Tools for Improving Self-Management
Self-management programs for patients with epilepsy historically have involved formalized programs conducted face-to-face with other patients, with professional moderators, and perhaps with caregivers. These programs depended entirely on in-person educational sessions and in-person support groups and were found to be very effective in improving self-management skills, though they were labor-intensive and logistically challenging for both practitioners and patients.
Since the advent of the Internet and mobile connectivity, many programs have incorporated the same elements in more accessible form. Educational content appears in live webinars and asynchronous video educational modules; the latter are attractive because patients and caregivers can access them independently at any time. Also readily available are tools for day-to-day self-management of medical conditions. These tools include mobile and online diaries for tracking seizure metrics and medication adherence reminder systems. Last, a variety of online and mobile disease-specific social networking platforms allow patients to connect directly to others without having to travel long distances to meet in-person. Although these digital solutions may not provide the exact experience offered by an in-person support group, the promise of superior accessibility creates an advantage in terms of accessibility and flexibility.
Self-Management in the Literature
In a recent review of care delivery and self-managementstrategies for adults with epilepsy, Bradley and colleagues analyzed 18 different studies of 16 separate interventions and concluded that 2 interventions, the specialist epilepsy nurse and self-management education, had some evidence of benefit. Four studies, detailed next, had the highest quality design, based on a focus on epilepsy self-management specifically, a prospective hypothesis-driven approach, and rigorous methodology.19
In 1990, Helgeson and colleagues evaluated Sepulveda Epilepsy Education, a 2-day in-person program designed to provide medical education and psychosocial therapy to patients with an epilepsy diagnosis.13 The program was based on the theory that having a better understanding of their epilepsy helps people cope with the condition. Medical, social, and emotional topics are covered. Medical topics include epilepsy and how it may change over time, as well as diagnosis, treatment, and first aid; social and emotional topics include coping with the psychological aspects of epilepsy, family, social aspects, and employment. In this small study (38 patients total), compared with the control group (18 patients), the treatment group (20 patients) demonstrated a significant reduction in the level of fear of death and brain damage caused by seizures, a significant decrease in hazardous medical self-management practices, and a significant decrease in misconceptions about epilepsy. The treatment group also increased their medication adherence, as determined by serum drug levels. In addition, statistically nonsignificant trends were shown by the treatment group toward improved emotional, interpersonal, and vocational functioning; improved adjustment to seizures; and improved overall psychosocial functioning.
In 2002, May and Pfäfflin evaluated the efficacy of the Modular Service Package Epilepsy (MOSES) educational program.14 This program was specifically developed to improve patient knowledge about epilepsy and its consequences and diagnostic and therapeutic measures, and to improve patient understanding of psychosocial and occupational problems. It was the first comprehensive program used in German-speaking countries. It had 9 modules: coping with epilepsy, epidemiology, basic knowledge, diagnostics, therapy, self-control, prognosis, psychosocial aspects, and network. To complete the program, patients work through about fourteen 1-hour lessons. The controlled, randomized study by May and Pfäfflin involved 242 patients (113 treatment, 129 control) aged 16 to 80 years. Patients in the treatment (MOSES) group demonstrated significant improvements in 2 of the 9 modules (knowledge, coping with epilepsy), had improved self-reported seizure outcomes, were more satisfied with therapy, experienced better tolerability of antiepileptic drugs with fewer adverse effects (AEs), and were highly satisfied with the program. The researchers concluded that educational programs, such as MOSES, should become a standard service for specialized epilepsy care.
Developed over many years, WebEase is an online epilepsy self-management program that supports education on medication, stress, and sleep management. In 2011, DiIorio and colleagues reported on a WebEase trial in which 194 patients were randomly assigned to either a treatment group (n = 96) or a wait-list control group (n = 96), and 2 were lost to follow up.11 After accounting for study criteria and study drop out, 70 participants completed the treatment arm, and 78 completed the control arm. The study measured the impact of the platform on multiple outcome metrics, including 3 behavioral areas of focus. At follow-up, self-reported levels of medication adherence were higher for patients in the treatment group than for those in the control group. Analyses also compared patients who completed WebEase modules with those who did not. Patients who completed at least some WebEase modules reported higher levels of self-efficacy, and a trend toward significance was found for medication adherence, perceived stress, self-management, and knowledge. The authors concluded that online tools that support epilepsy self-management could be effective.11
In 2015, Fraser and colleagues reported the results of the Program for Active Consumer Engagement in Self-Management in Epilepsy (PACES in Epilepsy), a consumer-generated self-management program.16 In the trial, 83 adults with chronic epilepsy were initially assigned either to an in-person intervention or to treatment as usual. After study drop outs, 38 patients remained in the intervention arm, with 40 in the control arm. In the intervention, 6 to 8 adults met for a 75-minute group session 1 evening per week for 8 weeks; these sessions were co-led by a psychologist and a trained peer with epilepsy. Topics included medical, psychosocial, cognitive, and self-management aspects of epilepsy, in addition to community integration and optimization of epilepsy-related communication. Outcomes were measured with various instruments, including the ESES, the Quality of Life in Epilepsy-31 (QOLIE-31), the Epilepsy Self-Management Scale (ESMS), the Patient Health Questionnaire-9, and the Generalized Anxiety Disorder-7. Each test was administered at baseline and after intervention. Outcomes were assessed immediately after program completion (8 weeks) and at follow-up 6 months later.
Findings suggested a substantial positive impact on epilepsy self-management capacities at program completion. In addition, benefit was sustained, particularly for epilepsy information management, over the 6 months after program completion. On the QOLIE-31 at 6 months, management of medication AEs also remained significantly improved, and fatigue management was improved at the P < .05 level. The researchers concluded that the PACES in Epilepsy program might have a more sustained impact on management of disability than on mood. They also noted that the effect was greater immediately after program completion than at 6 months. Patients gave the PACES program high satisfaction ratings.
Although these programs take slightly different approaches to epilepsy self-management, they have a similar focus: directed patient education. Furthermore, most of these programs are conducted in person, usually in a support group setting. In the WebEase trial, patients seem to have completed the online modules in a study setting, and a peer support component was not included. Overall, all programs successfully demonstrated various benefits for trial patients. These outcomes suggest that despite their subtle differences in approach, formal self-management programs are benefiting patients.
None of these platforms was designed for or specifically tested veterans with epilepsy. Although veterans theoretically would benefit from the same tools used by nonveterans, Iraq and Afghanistan veterans with epilepsy are more likely than are those without epilepsy to have mental and physical comorbidities and significantly higher mortality.2 Therefore, veterans potentially could benefit more from evidence-based chronic disease self-management programs designed to reduce physical and psychiatric comorbidities. Furthermore, programs that incorporate peer-to-peer support and direct links to VA care teams and mental health providers could be valuable.18
One research effort that directly addressed these issues is the Policy for Optimized Epilepsy Management (POEM) study, conducted by Hixson and colleagues in 2015.18 This study, not included in the review by Bradley and colleagues, used a purely online- and mobile-based social networking platform to promote self-management practices.19 Unlike the other programs described here, POEM did not require that patients view or attend formal educational seminars, though these seminars were available through the online platform for patient self-directed viewing. In addition, the intervention heavily promoted peer-to-peer engagement and disease tracking as means of increasing self-knowledge and activation. This study was unlike the other platforms in another way: It specifically focused on veterans with epilepsy, based on the idea that many veterans had a shared experience that would optimize a peer support approach.
The POEM investigators did not use a controlled design but found a significant benefit for both ESES and ESMS metrics on within-subject comparisons. Similar to the PACES in Epilepsy study, the POEM study found the highest benefit on the information management subscale of the ESMS.16 Practically speaking, this means patients were better able to use and manage digital and mobile information resources for controlling epilepsy. The POEM study results further reinforced the idea that epilepsy self-management programs are beneficial and expanded on earlier research to emphasize the value of peer support networks and digital interventions that can be used by patients at their convenience. These features provide greater access to more patients and maintain the crucial elements of peer-to-peer learning and counseling.
Implementation Barriers
Confirming the effectiveness of self-management programs is only the beginning of formal implementation and adoption. The real-world success of patient self-management programs has been documented for a few chronic diseases, including epilepsy. However, there is little research or commentary on lessons learned or on the challenges encountered with wide implementation of these programs.
Initial Setup and Sponsorship
To promote wider adoption, researchers should include commentary on initial setup, ongoing patient acceptance, and continual provider support. Many of the initial challenges in self-management programs involve a changing paradigm in the delivery and economics of health care. The transition to a more consumer-oriented health model with an emphasis on outcomes and patient-reported variables likely will support self-management strategies but is only slowly evolving. Many health care providers, hospitals, and payers may not be familiar with or have proper incentivizes to explore self-management tools even when proven effective.
More specifically, these epilepsy self-management programs are treatment adjuncts well suited to military and veteran health care systems. Self-management closely aligns with the overall VHA mission, vision, and values, including formal Department of Veteran Affairs (VA) goals and the MyVA priorities that collectively embrace improvement in access, a veteran-centric approach, and quality for improvement of the entire VA experience. Self-management platforms in the VA are recognized as empowering veterans and are thought to indirectly improve access to health care.20,21
The barriers of sponsorship and financial support likely will persist in the private health care sector but are less likely to significantly affect the VHA. Self-management programs have been researched and implemented for many health conditions across the VHA. For example, the VA Talent Management System course Patient Self-Management: Skill Building (TMS 6467) offers education and training to all clinical practitioners and managers involved in patient education and self-management activities for a variety of chronic medical conditions. Regarding epilepsy self-management more specifically, a patient brochure on the practice is distributed by the VHA Epilepsy Centers of Excellence (ECoEs) and an associated consortium.22 Last, a national provider educational lecture series has a corresponding patient and caregiver lecture set that emphasizes education and self-management behaviors.
Labor, Time, and Resource Needs
The most time-intensive aspect of designing self-management programs is developing the tool that allows clinicians and patients to work together. From a program perspective, the tool must be available and helpful not only to patients and specialists, but also to primary care providers. Tertiary-care centers usually accept the responsibility for program initiation, including patient recruitment, logistics coordination, and health care professional staffing. For epilepsy, the small pool of relevant specialists and centers limits the number of self-management education sessions that can be hosted and increases the need for complex travel and scheduling tasks. However, ECoE communication lines provide a basic infrastructure for collaboration and for development of tools that can be helpful to all clinicians treating veterans with epilepsy.23
Given the issues with coordinating the logistics of in-person programs at brick-and-mortar sites, this type of program may not be the best option for some patients and facilities. Alternative approaches, such as telehealth and asynchronous digital platforms, could expand access and increase convenience. Even though remotely administered programs may not be as powerful for some patients, the promise of scalable access supports consideration of these approaches.
Patient and Caregiver Logistics
Veterans with epilepsy may also have comorbid traumatic brain injury (TBI) and posttraumatic stress disorder, which can complicate self-managed care. In addition, many veterans live in rural areas and have limited travel options. All these factors challenge the success of epilepsy self-management programs. However, the network of ECoEs and associated consortium facilities can step up to deliver self-management tools and information.
The infrastructure of the VHA patient aligned care team (PACT) also contributes to the integration of self-management training. The PACT model takes a personalized, comprehensive, coordinated approach to promote team-based, veteran-centric care and actively partners with other VHA offices to incorporate alternative care services, including peer support and self-management platforms. The combination represents fertile ground for implementation and promotion of self-management tools in the VHA epilepsy population.
Health Care Economics
Given the uncertainties of the US health care economy, it is not surprising that many experts advocate a fundamental redesign of the health care team relationship and information infrastructure.24 This realignment includes partnering directly with patients and their families to encourage more reliance on self-management practices. Unfortunately, this approach does not lend itself to the well-entrenched business model on which most community medical practices are based. Health system leadership often must be convinced there are potential cost savings or a return on investment for new programs. As there is no consistent, comprehensive reimbursement policy for programs focused on self-management, health care systems must be creative and innovative when appraising the financial consequences of such programs.
Epilepsy remains a huge burden. In 2000, the annual total cost of epilepsy treatment in the US was $362 million for new patients and $2 billion for existing cases.25 Within the VHA, the occurrence of posttraumatic epilepsy among the increasing number of veterans with TBI contributes to the burden, and posttraumatic epilepsy and psychogenic nonepileptic seizures complicate treatment approaches. The incidence of comorbidities, including anxiety and depression, has been as high as 50%.23 Epilepsy health care programs are evaluating ways to validate their ability to minimize cost, improve access, and maintain quality of service. Integration of self-management should be included in these efforts.
The VHA represents a unique health care environment for testing and implementing self-management programs. Although the VHA is not immune to the traditional business models of medicine, it is less dependent on them, and it disproportionately cares for patients for long spans of time. From the health care team perspective, data indicate that ECoE physicians represent a high percentage of VHA epilepsy specialists but directly see only about 20% of veterans with an epilepsy or seizure-associated diagnosis. Therefore, future collaboration and connectivity of consortium sites can have a broader impact on self-management—highlighting the fact that concerted, scaled self-management programs have an important role in the VHA health care delivery system and should be promoted.26
Final Insights and Opportunities
Despite the barriers to adoption, formal epilepsy self-management programs are making gains in maturity and academic credibility. As the health care economy gradually shifts to more outcomes-based models, these offerings likely will become more valued, particularly by health care organizations focused on cost sharing, by large self-insuring employers, or organizations like the VHA where patients maintain a long-term relationship. Nevertheless, for the more resource-intensive, in-person self-management programs, adoption may remain constrained. Digital and mobile platforms should serve as more accessible entry points, with lower costs and more rapid scaling potential. Even though these online platforms may not have the same impact as intensive face-to-face programs, their scalability and constant accessibility should make them attractive, and the relatively modest cost of implementing self-guided programs should reduce barriers to adoption.
Integrated health care systems, such as the VHA and various European health systems, can serve as models for self-management implementation. Incorporating a live clinical implementation into parallel research efforts can continue to produce vital academic information on the real-world impact of these solutions, and this evidence in turn can be used to support policies that foster widespread adoption. More specifically, the ECoE model represents a clear opportunity to promote widespread implementation of self-management. The ECoEs are already publishing self-management materials that health care teams can use in patient counseling,and several self-care studies are being conducted within the network.22 In this model, compared with private sector health systems, ECoEs are well positioned to advance the use of formal self-management strategies.
The proposed epilepsy self-management model for ECoEs would be based on an iterative program that incorporates best practices from each of the research studies discussed earlier. With the publication of new research, successful self-management tools would be incorporated into the programs. From a curriculum perspective, educational platforms on medication adherence, seizure safety, and information/data management should be included. Evidence is increasing that peer support and use of licensed peer navigators should be incorporated as well. Last, flexible and asynchronous digital methods should be added to self-management platforms to maximize patient access. These features build on the growing body of evidence to maximize the likelihood of a successful and sustainable self-management strategy for patients with epilepsy.
Click here to read the digital edition.
1. Fiest KM, Sauro KM, Wiebe S, et al. Prevalence and incidence in epilepsy: a systematic review and meta-analysis of international studies. Neurology. 2017;88(3):296-303.
2. Pugh MJ, Van Cott AC, Amuan M, et al. Epilepsy among Iraq and Afghanistan war veterans—United States, 2002-2015. MMWR. 2016;65(44):1224-1227.
3. Kwan P, Brodie MJ. Effectiveness of first antiepileptic drug. Epilepsia. 2001;42(10):1255-1260.
4. Hesdorffer DC, Beck V, Begley CE, et al. Research implications of the Institute of Medicine report, Epilepsy Across the Spectrum: Promoting Health and Understanding. Epilepsia. 2013;54(2):207-216.
5. Bandura A. Social Foundations of Thought and Action: A Social Cognitive Theory. Englewood Cliffs, NJ: Prentice-Hall; 1986.
6. Bandura A. Social Learning Theory. Englewood Cliffs, NJ: Prentice-Hall; 1977.
7. Clark, NM, Becker MH, Janz NK, Lorig K, Rakowski W, Anderson L. Self-management of chronic disease by older adults. J Aging Health. 1991;3(1):3-27.
8. Ory MG, Ahn SM, Jiang L, et al. Successes of a national study of the Chronic Disease Self-Management Program: meeting the triple aim of health care reform. Med Care. 2013;51(11):992-998.
9. DiIorio C, Shafer PO, Letz R, Henry TR, Schomer DL, Yeager K; Project EASE Study Group. Behavioral, social and affective factors associated with self-efficacy for self-management among people with epilepsy. Epilepsy Behav. 2006;9(1):158-163.
10. Shegog R, Bamps YA, Patel A, et al. Managing Epilepsy Well: emerging e-tools for epilepsy self-management. Epilepsy Behav. 2013;29(1):133-140.
11. DiIorio C, Bamps Y, Walker ER, Escoffery C. Results of a research study evaluating WebEase, an online epilepsy self-management program. Epilepsy Behav. 2011;22(3):469-474.
12. Gallant MP. The influence of social support on chronic illness self-management: a review and directions for research. Health Educ Behav. 2003;30(2):170-195.
13. Helgeson DC, Mittan R, Tan SY, Chayasirisobhon S. Sepulveda Epilepsy Education: the efficacy of a psychoeducational treatment programme in treating medical and psychosocial aspects of epilepsy. Epilepsia. 1990;31(1):75-82.
14. May TW, Pfäfflin M. The efficacy of an educational treatment program for patients with epilepsy (MOSES): results of a controlled, randomized study. Modular Service Package Epilepsy. Epilepsia. 2002;43(5):539-549.
15. Aliasgharpour M, Dehgahn Nayeri N, Yadegary MA, Haghani H. Effects of an educational program on self-management in patients with epilepsy. Seizure. 2013;22(1):48-52.
16. Fraser RT, Johnson EK, Lashley S, et al. PACES in Epilepsy: results of a self-management randomized controlled trial. Epilepsia. 2015;56(8):1264-1274.
17. Laybourne AH, Morgan M, Watkins SH, Lawton R, Ridsdale L, Goldstein LH. Self-management for people with poorly controlled epilepsy: participants’ views of the UK self-management in epilepsy (SMILE) program. Epilepsy Behav. 2015;52(pt A):159-164.
18. Hixson JD, Barnes D, Parko K, et al. Patients optimizing epilepsy management via an online community: the POEM study. Neurology. 2015;85(2):129-136.
19. Bradley PM, Lindsay B, Fleeman N. Care delivery and self-management strategies for adults with epilepsy. Cochrane Database Syst Rev. 2016;2:CD006244.
20. Allicock M, Haynes-Maslow L, Carr C, et al. Training veterans to provide peer support in a weight-management program: MOVE! Prev Chronic Dis. 2013;10:E185.
21. Damush TM, Jackson GL, Powers BJ, et al. Implementing evidence-based patient self-management programs in the Veterans Health Administration: perspectives on delivery system design considerations. J Gen Intern Med. 2010;25(suppl 1):68-71.
22. Caraveo N, Chen S, Evrard C, Ozuna J; Epilepsy Centers of Excellence Nursing Workgroup. Self-management in epilepsy: a guide for healthcare professionals. https://www.epilepsy.va.gov/Library/Self-Management%20In%20Epilepsy.pdf. Published Winter 2015. Accessed February 26, 2018.
23. Rehman R, Kelly PR, Husain AM, Tran TT. Characteristics of veterans diagnosed with seizures within the Veterans Health Administration. J Rehabil Res Dev. 2015;52(7):751-762.
24. Merry MD. Healthcare’s need for revolutionary change. Quality Prog. 2003;36(9):31-35.
25. Halpern M, Rentz A, Murray M. Cost of illness of epilepsy in the US: comparison of patient-based and population-based estimates. Neuroepidemiology. 2000;19(2):87-99.
26. Kelly P, Chinta R. Do centers of excellence excel in patient outcomes?: Evidence from U.S. Veterans Health Administration Centers for Epilepsy. Int J Manage Excellence. 2015;4(3):529-538.
1. Fiest KM, Sauro KM, Wiebe S, et al. Prevalence and incidence in epilepsy: a systematic review and meta-analysis of international studies. Neurology. 2017;88(3):296-303.
2. Pugh MJ, Van Cott AC, Amuan M, et al. Epilepsy among Iraq and Afghanistan war veterans—United States, 2002-2015. MMWR. 2016;65(44):1224-1227.
3. Kwan P, Brodie MJ. Effectiveness of first antiepileptic drug. Epilepsia. 2001;42(10):1255-1260.
4. Hesdorffer DC, Beck V, Begley CE, et al. Research implications of the Institute of Medicine report, Epilepsy Across the Spectrum: Promoting Health and Understanding. Epilepsia. 2013;54(2):207-216.
5. Bandura A. Social Foundations of Thought and Action: A Social Cognitive Theory. Englewood Cliffs, NJ: Prentice-Hall; 1986.
6. Bandura A. Social Learning Theory. Englewood Cliffs, NJ: Prentice-Hall; 1977.
7. Clark, NM, Becker MH, Janz NK, Lorig K, Rakowski W, Anderson L. Self-management of chronic disease by older adults. J Aging Health. 1991;3(1):3-27.
8. Ory MG, Ahn SM, Jiang L, et al. Successes of a national study of the Chronic Disease Self-Management Program: meeting the triple aim of health care reform. Med Care. 2013;51(11):992-998.
9. DiIorio C, Shafer PO, Letz R, Henry TR, Schomer DL, Yeager K; Project EASE Study Group. Behavioral, social and affective factors associated with self-efficacy for self-management among people with epilepsy. Epilepsy Behav. 2006;9(1):158-163.
10. Shegog R, Bamps YA, Patel A, et al. Managing Epilepsy Well: emerging e-tools for epilepsy self-management. Epilepsy Behav. 2013;29(1):133-140.
11. DiIorio C, Bamps Y, Walker ER, Escoffery C. Results of a research study evaluating WebEase, an online epilepsy self-management program. Epilepsy Behav. 2011;22(3):469-474.
12. Gallant MP. The influence of social support on chronic illness self-management: a review and directions for research. Health Educ Behav. 2003;30(2):170-195.
13. Helgeson DC, Mittan R, Tan SY, Chayasirisobhon S. Sepulveda Epilepsy Education: the efficacy of a psychoeducational treatment programme in treating medical and psychosocial aspects of epilepsy. Epilepsia. 1990;31(1):75-82.
14. May TW, Pfäfflin M. The efficacy of an educational treatment program for patients with epilepsy (MOSES): results of a controlled, randomized study. Modular Service Package Epilepsy. Epilepsia. 2002;43(5):539-549.
15. Aliasgharpour M, Dehgahn Nayeri N, Yadegary MA, Haghani H. Effects of an educational program on self-management in patients with epilepsy. Seizure. 2013;22(1):48-52.
16. Fraser RT, Johnson EK, Lashley S, et al. PACES in Epilepsy: results of a self-management randomized controlled trial. Epilepsia. 2015;56(8):1264-1274.
17. Laybourne AH, Morgan M, Watkins SH, Lawton R, Ridsdale L, Goldstein LH. Self-management for people with poorly controlled epilepsy: participants’ views of the UK self-management in epilepsy (SMILE) program. Epilepsy Behav. 2015;52(pt A):159-164.
18. Hixson JD, Barnes D, Parko K, et al. Patients optimizing epilepsy management via an online community: the POEM study. Neurology. 2015;85(2):129-136.
19. Bradley PM, Lindsay B, Fleeman N. Care delivery and self-management strategies for adults with epilepsy. Cochrane Database Syst Rev. 2016;2:CD006244.
20. Allicock M, Haynes-Maslow L, Carr C, et al. Training veterans to provide peer support in a weight-management program: MOVE! Prev Chronic Dis. 2013;10:E185.
21. Damush TM, Jackson GL, Powers BJ, et al. Implementing evidence-based patient self-management programs in the Veterans Health Administration: perspectives on delivery system design considerations. J Gen Intern Med. 2010;25(suppl 1):68-71.
22. Caraveo N, Chen S, Evrard C, Ozuna J; Epilepsy Centers of Excellence Nursing Workgroup. Self-management in epilepsy: a guide for healthcare professionals. https://www.epilepsy.va.gov/Library/Self-Management%20In%20Epilepsy.pdf. Published Winter 2015. Accessed February 26, 2018.
23. Rehman R, Kelly PR, Husain AM, Tran TT. Characteristics of veterans diagnosed with seizures within the Veterans Health Administration. J Rehabil Res Dev. 2015;52(7):751-762.
24. Merry MD. Healthcare’s need for revolutionary change. Quality Prog. 2003;36(9):31-35.
25. Halpern M, Rentz A, Murray M. Cost of illness of epilepsy in the US: comparison of patient-based and population-based estimates. Neuroepidemiology. 2000;19(2):87-99.
26. Kelly P, Chinta R. Do centers of excellence excel in patient outcomes?: Evidence from U.S. Veterans Health Administration Centers for Epilepsy. Int J Manage Excellence. 2015;4(3):529-538.
BACE-1 inhibition worsens cognition in patients with prodromal Alzheimer’s disease
More bad news for Alzheimer’s research. Two more BACE inhibitors fall far short of the finish line.
Declining cognitive scores, falls, suicidal ideation, and liver enzyme abnormalities were all seen in clinical trials.
The news doesn’t bode well for the therapeutic target of BACE (beta-site APP cleaving enzyme) inhibition. BACE is one of the enzymes that trims the amyloid precursor protein (APP). Inhibiting it does reduce the amount of toxic amyloid-beta in cerebrospinal fluid, and amyloid plaque in the brain. But none of these molecules has shown clinical benefit in dementia patients, whether their disease is mild, or moderate or – now – prodromal. And it is now apparent that inhibiting BACE also produces serious off-target problems.
“BACE-1 inhibition certainly seemed to have a sound rationale assuming the basis for amyloid’s role in Alzheimer’s disease pathogenesis is a gain of toxicity,” Richard J. Caselli, MD, of the Mayo Clinic, Rochester, Minn., said in an interview. “That APP is important for Alzheimer’s pathogenesis still seems clear but whether amyloid-beta toxicity is the driving force is no longer clear. Further, interruption of the APP system disrupts more than amyloid-beta peptide, possibly explaining the adverse cognitive effects of BACE-1 inhibition shown exhibited now by three different BACE-1 inhibitors.”
Verubecestat
Researchers got their first dose of bad news regarding verubecestat at the 2017 Clinical Trials in Alzheimer’s Disease meeting. There, Michael F. Egan, MD, Merck’s associate vice president of clinical neuroscience, discussed the molecule’s failure to slow cognitive decline in patients with mild to moderate Alzheimer’s disease. There was plenty of biomarker evidence that the drug did block amyloid-beta production, but there also was a plethora of concerning adverse events, Dr. Egan said in an interview.
However, verubecestat still was being pursued in patients with prodromal Alzheimer’s disease. In February, Merck stopped the trial after a futility analysis and announced that the company was terminating studies of verubecestat in that population as well. In the April 11 issue of the New England Journal of Medicine, Dr. Egan and his colleagues report the full extent of verubecestat’s failure in prodromal patients, and the accompanying adverse events.
At the time of termination, 1,454 patients had been enrolled. Of these, 485 received 12 mg/day, 484 received 40 mg/day, and 485 received placebo. About half of each group completed 104 weeks of treatment in the study, which was designed to extend up to 5 years.
The primary outcome was change in the Clinical Dementia Rating Scale–Sum of Boxes score (CDR-SB). Seven secondary outcomes examined other cognitive and functional end points, along with changes in hippocampal volume on MRI and amyloid burden as determined in PET imaging.
Not only did verubecestat fail to slow cognitive decline, it appeared to exacerbate it. The mean change on the CDR-SB was 1.65 in the 12-mg group, 2.02 in the 40-mg group, and 1.58 in the placebo group, favoring placebo.
“In an exploratory analysis according to time point, scores on the CDR-SB were also higher [signifying more impairment of cognition and daily functioning] in the 40-mg group than in the placebo group at 13, 26, and 52 weeks ... suggesting but not confirming the possibility of worse performance at these earlier time points in the high-dose verubecestat group,” the investigators said.
Verubecestat also was associated with more conversions to Alzheimer’s disease. Per 100 patient-years, the Alzheimer’s disease event rates were 24.5 in the 12-mg group, 25.5 in the 40-mg group, and 19.3 in the placebo group. Compared with placebo, those taking 12-mg doses were 30% more likely to develop Alzheimer’s disease and those taking 40-mg doses were 38% more likely. The findings suggest that “verubecestat may have accelerated the progression to diagnosis of dementia due to Alzheimer’s disease,” the investigators said.
The negative impact of verubecestat was apparent quite early in the study. “In exploratory analyses, both dose levels of verubecestat were associated with poorer outcomes on the [Composite Cognition Score-3 Domain] and the ADAS-Cog [Alzheimer’s Disease Assessment Scale–Cognitive Subscale] measures of cognition that, relative to placebo, appeared worse at week 13 and did not appear to progress thereafter.”
Results of the secondary end points, including the ADAS-Cog and the Mini-Mental State Exam, also indicated that verubecestat may have worsened cognitive performance.
Imaging outcomes were positive, however. Hippocampal volume was 6,448 mL in the 12-mg group, 6,469 mL in the 40-mg group, and 6,435 mL in the placebo group. Brain amyloid increased in the placebo group, as expected, and decreased in the active groups. The small group of patients who underwent cerebrospinal fluid sampling showed reductions of more than 60% in amyloid-beta and soluble APP-beta associated with verubecestat. These results show that the molecule was indeed hitting its intended target, but that doing so was not clinically beneficial.
Adverse events were more common in the verubecestat groups. These included rash, dermatitis, urticaria, sleep disturbance, weight loss, and cough. Hair coloring changed in 2.5% of patients in the 12-mg group and 5% of the 40-mg group, but in none of the subjects taking placebo.
Patients taking verubecestat were more likely to sustain falls and injuries and to express suicidal ideation.
The results of this trial differ from the study of verubecestat in mild to moderate Alzheimer’s disease, the investigators noted. Those patients did not decline cognitively as did those with prodromal disease.
“Patients at an earlier stage of the disease may be more sensitive to the effects of substantial BACE-1 inhibition, perhaps because of a role of BACE-1 in normal synaptic function. It is also possible that the effects of verubecestat are due to inhibition of BACE-2,” they said.
Atabecestat
In a research letter in the same issue of the New England Journal of Medicine (2019 Apr 11;380:1483-5), David Henley, MD, senior director of Janssen’s Alzheimer’s clinical development core, released similarly negative results from an interim analysis of EARLY (Efficacy and Safety Study of Atabecestat in Participants Who Are Asymptomatic at Risk for Developing Alzheimer’s Dementia) trial, a randomized study of the BACE-1 inhibitor candidate, atabecestat.
The phase 2 trial enrolled 557 patients with prodromal Alzheimer’s disease. The primary cognitive end point was change from baseline in the Preclinical Alzheimer’s Cognitive Composite (PACC) score.
This trial was discontinued in May 2018 because of liver-related adverse events, although safety follow-up continues. The research letter did not disclose details of the hepatic events, but a company press release from May 2018 referred to them in a general sense.
“Elevations of liver enzymes, which were serious in nature, have been observed in some study participants who received the Janssen BACE inhibitor, atabecestat. After a thorough evaluation of all available liver safety data from our studies, Janssen has concluded that the benefit-risk ratio is no longer favorable to continue development of atabecestat for people who have late-onset preclinical stage Alzheimer’s disease.”
Patients in EARLY were randomized to 5 mg, 25 mg, or placebo. As in the verubecestat trial, those randomized to placebo did better. The mean changes from baseline in the PACC score were −1.44 in the 25-mg group, −0.58 in the 5-mg group, and −0.32 in the placebo group.
“At month 6, the difference between the 25-mg group and the placebo group was −1.12 and the difference between the 5-mg group and the placebo group was −0.26, favoring placebo over the higher dose,” the authors said.
This theme reemerged in a secondary end point, the Repeatable Battery for the Assessment of Neuropsychological Status. The 25-mg group declined 3.58 points more than placebo, and the 5-mg group, 1.43 points more.
Adverse events were more common in the active groups and included depression, effects on sleep and dreams, and anxiety.
“The differences in cognitive performance between the groups are of uncertain clinical significance; however, given similar findings favoring placebo over BACE-1 inhibitors in other trial, we are communicating this potential signal of worsening cognitive function in the treated groups,” Dr. Henley said.
SOURCE: Egan MF et al. N Engl J Med. 2019 Apr 11;380:1408-20.
“Some trials fail because the experimental treatment proves to be no different than a control or standard intervention,” David Knopman, MD, wrote in an accompanying editorial (N Engl J Med. 2019 Apr 11;380:1476-8). “Others fail because of unacceptable side effects. In this issue of the Journal, an article by Egan et al. and a letter to the editor by Henley et al. (N Engl J Med. 2019 Apr 11;380:1483-5) describe a third reason for failure – a treatment worsens the target symptoms.
Certainly, beta-site amyloid precursor protein-cleaving enzyme 1 (BACE-1) inhibition makes sense when viewed in the light of the current understanding of Alzheimer’s disease neuropathology. The amyloid cascade hypothesis holds that toxic amyloid-beta fragments accumulate in the brain, form dense neuritic plaques, and lead to neuronal death and cognitive decline.
“The model is rooted in the inseparability of Alzheimer’s disease from abundant amyloid-beta pathologic features,” Dr. Knopman wrote. But, “Over the past 2 decades, the amyloid-beta–lowering strategy has been put to the test in trials of antiamyloid antibodies, none of which have been successful.”
Therefore, hitting amyloid at the source – the transmembrane cleavage domain – seemed important and, potentially, effective. But three BACE inhibitors (verubecestat, atabecestat, and lanabecestat) have shown similarly negative cognitive effects. “Together, these results suggest that preserved BACE-1 activity may be critical to normal synaptic functions. These observations place a limitation on how amyloid-beta lowering can be accomplished.”
It is possible that decreasing the level of BACE inhibition might ameliorate off-target effects and neuronal compromise but still be enough to reduce the generation of toxic amyloid-beta fragments, Dr. Knopman said. But, “Adjustments in the dose to a narrow window of BACE-1 inhibition would be difficult to accomplish in a clinical trial until there are peripheral biomarkers that reflect the activity of the agent in the brain.”
Thus far, most of the studied antiamyloid drugs have indeed reduced amyloid-beta levels, but none of those reductions affected cognition. A rethinking of amyloid-beta’s place in dementia progression may be in order.
“The dissociation between amyloid-beta lowering and cognitive benefits with both BACE-1 inhibition and antiamyloid antibody therapy is troubling. To be blunt, amyloid-beta lowering seems to be an ineffective approach, and it is time to focus on other targets to move therapeutics for Alzheimer’s disease forward.”
Dr. Knopman is a clinical neurologist and Alzheimer’s researcher at the Mayo Clinic, Rochester, Minn.
“Some trials fail because the experimental treatment proves to be no different than a control or standard intervention,” David Knopman, MD, wrote in an accompanying editorial (N Engl J Med. 2019 Apr 11;380:1476-8). “Others fail because of unacceptable side effects. In this issue of the Journal, an article by Egan et al. and a letter to the editor by Henley et al. (N Engl J Med. 2019 Apr 11;380:1483-5) describe a third reason for failure – a treatment worsens the target symptoms.
Certainly, beta-site amyloid precursor protein-cleaving enzyme 1 (BACE-1) inhibition makes sense when viewed in the light of the current understanding of Alzheimer’s disease neuropathology. The amyloid cascade hypothesis holds that toxic amyloid-beta fragments accumulate in the brain, form dense neuritic plaques, and lead to neuronal death and cognitive decline.
“The model is rooted in the inseparability of Alzheimer’s disease from abundant amyloid-beta pathologic features,” Dr. Knopman wrote. But, “Over the past 2 decades, the amyloid-beta–lowering strategy has been put to the test in trials of antiamyloid antibodies, none of which have been successful.”
Therefore, hitting amyloid at the source – the transmembrane cleavage domain – seemed important and, potentially, effective. But three BACE inhibitors (verubecestat, atabecestat, and lanabecestat) have shown similarly negative cognitive effects. “Together, these results suggest that preserved BACE-1 activity may be critical to normal synaptic functions. These observations place a limitation on how amyloid-beta lowering can be accomplished.”
It is possible that decreasing the level of BACE inhibition might ameliorate off-target effects and neuronal compromise but still be enough to reduce the generation of toxic amyloid-beta fragments, Dr. Knopman said. But, “Adjustments in the dose to a narrow window of BACE-1 inhibition would be difficult to accomplish in a clinical trial until there are peripheral biomarkers that reflect the activity of the agent in the brain.”
Thus far, most of the studied antiamyloid drugs have indeed reduced amyloid-beta levels, but none of those reductions affected cognition. A rethinking of amyloid-beta’s place in dementia progression may be in order.
“The dissociation between amyloid-beta lowering and cognitive benefits with both BACE-1 inhibition and antiamyloid antibody therapy is troubling. To be blunt, amyloid-beta lowering seems to be an ineffective approach, and it is time to focus on other targets to move therapeutics for Alzheimer’s disease forward.”
Dr. Knopman is a clinical neurologist and Alzheimer’s researcher at the Mayo Clinic, Rochester, Minn.
“Some trials fail because the experimental treatment proves to be no different than a control or standard intervention,” David Knopman, MD, wrote in an accompanying editorial (N Engl J Med. 2019 Apr 11;380:1476-8). “Others fail because of unacceptable side effects. In this issue of the Journal, an article by Egan et al. and a letter to the editor by Henley et al. (N Engl J Med. 2019 Apr 11;380:1483-5) describe a third reason for failure – a treatment worsens the target symptoms.
Certainly, beta-site amyloid precursor protein-cleaving enzyme 1 (BACE-1) inhibition makes sense when viewed in the light of the current understanding of Alzheimer’s disease neuropathology. The amyloid cascade hypothesis holds that toxic amyloid-beta fragments accumulate in the brain, form dense neuritic plaques, and lead to neuronal death and cognitive decline.
“The model is rooted in the inseparability of Alzheimer’s disease from abundant amyloid-beta pathologic features,” Dr. Knopman wrote. But, “Over the past 2 decades, the amyloid-beta–lowering strategy has been put to the test in trials of antiamyloid antibodies, none of which have been successful.”
Therefore, hitting amyloid at the source – the transmembrane cleavage domain – seemed important and, potentially, effective. But three BACE inhibitors (verubecestat, atabecestat, and lanabecestat) have shown similarly negative cognitive effects. “Together, these results suggest that preserved BACE-1 activity may be critical to normal synaptic functions. These observations place a limitation on how amyloid-beta lowering can be accomplished.”
It is possible that decreasing the level of BACE inhibition might ameliorate off-target effects and neuronal compromise but still be enough to reduce the generation of toxic amyloid-beta fragments, Dr. Knopman said. But, “Adjustments in the dose to a narrow window of BACE-1 inhibition would be difficult to accomplish in a clinical trial until there are peripheral biomarkers that reflect the activity of the agent in the brain.”
Thus far, most of the studied antiamyloid drugs have indeed reduced amyloid-beta levels, but none of those reductions affected cognition. A rethinking of amyloid-beta’s place in dementia progression may be in order.
“The dissociation between amyloid-beta lowering and cognitive benefits with both BACE-1 inhibition and antiamyloid antibody therapy is troubling. To be blunt, amyloid-beta lowering seems to be an ineffective approach, and it is time to focus on other targets to move therapeutics for Alzheimer’s disease forward.”
Dr. Knopman is a clinical neurologist and Alzheimer’s researcher at the Mayo Clinic, Rochester, Minn.
More bad news for Alzheimer’s research. Two more BACE inhibitors fall far short of the finish line.
Declining cognitive scores, falls, suicidal ideation, and liver enzyme abnormalities were all seen in clinical trials.
The news doesn’t bode well for the therapeutic target of BACE (beta-site APP cleaving enzyme) inhibition. BACE is one of the enzymes that trims the amyloid precursor protein (APP). Inhibiting it does reduce the amount of toxic amyloid-beta in cerebrospinal fluid, and amyloid plaque in the brain. But none of these molecules has shown clinical benefit in dementia patients, whether their disease is mild, or moderate or – now – prodromal. And it is now apparent that inhibiting BACE also produces serious off-target problems.
“BACE-1 inhibition certainly seemed to have a sound rationale assuming the basis for amyloid’s role in Alzheimer’s disease pathogenesis is a gain of toxicity,” Richard J. Caselli, MD, of the Mayo Clinic, Rochester, Minn., said in an interview. “That APP is important for Alzheimer’s pathogenesis still seems clear but whether amyloid-beta toxicity is the driving force is no longer clear. Further, interruption of the APP system disrupts more than amyloid-beta peptide, possibly explaining the adverse cognitive effects of BACE-1 inhibition shown exhibited now by three different BACE-1 inhibitors.”
Verubecestat
Researchers got their first dose of bad news regarding verubecestat at the 2017 Clinical Trials in Alzheimer’s Disease meeting. There, Michael F. Egan, MD, Merck’s associate vice president of clinical neuroscience, discussed the molecule’s failure to slow cognitive decline in patients with mild to moderate Alzheimer’s disease. There was plenty of biomarker evidence that the drug did block amyloid-beta production, but there also was a plethora of concerning adverse events, Dr. Egan said in an interview.
However, verubecestat still was being pursued in patients with prodromal Alzheimer’s disease. In February, Merck stopped the trial after a futility analysis and announced that the company was terminating studies of verubecestat in that population as well. In the April 11 issue of the New England Journal of Medicine, Dr. Egan and his colleagues report the full extent of verubecestat’s failure in prodromal patients, and the accompanying adverse events.
At the time of termination, 1,454 patients had been enrolled. Of these, 485 received 12 mg/day, 484 received 40 mg/day, and 485 received placebo. About half of each group completed 104 weeks of treatment in the study, which was designed to extend up to 5 years.
The primary outcome was change in the Clinical Dementia Rating Scale–Sum of Boxes score (CDR-SB). Seven secondary outcomes examined other cognitive and functional end points, along with changes in hippocampal volume on MRI and amyloid burden as determined in PET imaging.
Not only did verubecestat fail to slow cognitive decline, it appeared to exacerbate it. The mean change on the CDR-SB was 1.65 in the 12-mg group, 2.02 in the 40-mg group, and 1.58 in the placebo group, favoring placebo.
“In an exploratory analysis according to time point, scores on the CDR-SB were also higher [signifying more impairment of cognition and daily functioning] in the 40-mg group than in the placebo group at 13, 26, and 52 weeks ... suggesting but not confirming the possibility of worse performance at these earlier time points in the high-dose verubecestat group,” the investigators said.
Verubecestat also was associated with more conversions to Alzheimer’s disease. Per 100 patient-years, the Alzheimer’s disease event rates were 24.5 in the 12-mg group, 25.5 in the 40-mg group, and 19.3 in the placebo group. Compared with placebo, those taking 12-mg doses were 30% more likely to develop Alzheimer’s disease and those taking 40-mg doses were 38% more likely. The findings suggest that “verubecestat may have accelerated the progression to diagnosis of dementia due to Alzheimer’s disease,” the investigators said.
The negative impact of verubecestat was apparent quite early in the study. “In exploratory analyses, both dose levels of verubecestat were associated with poorer outcomes on the [Composite Cognition Score-3 Domain] and the ADAS-Cog [Alzheimer’s Disease Assessment Scale–Cognitive Subscale] measures of cognition that, relative to placebo, appeared worse at week 13 and did not appear to progress thereafter.”
Results of the secondary end points, including the ADAS-Cog and the Mini-Mental State Exam, also indicated that verubecestat may have worsened cognitive performance.
Imaging outcomes were positive, however. Hippocampal volume was 6,448 mL in the 12-mg group, 6,469 mL in the 40-mg group, and 6,435 mL in the placebo group. Brain amyloid increased in the placebo group, as expected, and decreased in the active groups. The small group of patients who underwent cerebrospinal fluid sampling showed reductions of more than 60% in amyloid-beta and soluble APP-beta associated with verubecestat. These results show that the molecule was indeed hitting its intended target, but that doing so was not clinically beneficial.
Adverse events were more common in the verubecestat groups. These included rash, dermatitis, urticaria, sleep disturbance, weight loss, and cough. Hair coloring changed in 2.5% of patients in the 12-mg group and 5% of the 40-mg group, but in none of the subjects taking placebo.
Patients taking verubecestat were more likely to sustain falls and injuries and to express suicidal ideation.
The results of this trial differ from the study of verubecestat in mild to moderate Alzheimer’s disease, the investigators noted. Those patients did not decline cognitively as did those with prodromal disease.
“Patients at an earlier stage of the disease may be more sensitive to the effects of substantial BACE-1 inhibition, perhaps because of a role of BACE-1 in normal synaptic function. It is also possible that the effects of verubecestat are due to inhibition of BACE-2,” they said.
Atabecestat
In a research letter in the same issue of the New England Journal of Medicine (2019 Apr 11;380:1483-5), David Henley, MD, senior director of Janssen’s Alzheimer’s clinical development core, released similarly negative results from an interim analysis of EARLY (Efficacy and Safety Study of Atabecestat in Participants Who Are Asymptomatic at Risk for Developing Alzheimer’s Dementia) trial, a randomized study of the BACE-1 inhibitor candidate, atabecestat.
The phase 2 trial enrolled 557 patients with prodromal Alzheimer’s disease. The primary cognitive end point was change from baseline in the Preclinical Alzheimer’s Cognitive Composite (PACC) score.
This trial was discontinued in May 2018 because of liver-related adverse events, although safety follow-up continues. The research letter did not disclose details of the hepatic events, but a company press release from May 2018 referred to them in a general sense.
“Elevations of liver enzymes, which were serious in nature, have been observed in some study participants who received the Janssen BACE inhibitor, atabecestat. After a thorough evaluation of all available liver safety data from our studies, Janssen has concluded that the benefit-risk ratio is no longer favorable to continue development of atabecestat for people who have late-onset preclinical stage Alzheimer’s disease.”
Patients in EARLY were randomized to 5 mg, 25 mg, or placebo. As in the verubecestat trial, those randomized to placebo did better. The mean changes from baseline in the PACC score were −1.44 in the 25-mg group, −0.58 in the 5-mg group, and −0.32 in the placebo group.
“At month 6, the difference between the 25-mg group and the placebo group was −1.12 and the difference between the 5-mg group and the placebo group was −0.26, favoring placebo over the higher dose,” the authors said.
This theme reemerged in a secondary end point, the Repeatable Battery for the Assessment of Neuropsychological Status. The 25-mg group declined 3.58 points more than placebo, and the 5-mg group, 1.43 points more.
Adverse events were more common in the active groups and included depression, effects on sleep and dreams, and anxiety.
“The differences in cognitive performance between the groups are of uncertain clinical significance; however, given similar findings favoring placebo over BACE-1 inhibitors in other trial, we are communicating this potential signal of worsening cognitive function in the treated groups,” Dr. Henley said.
SOURCE: Egan MF et al. N Engl J Med. 2019 Apr 11;380:1408-20.
More bad news for Alzheimer’s research. Two more BACE inhibitors fall far short of the finish line.
Declining cognitive scores, falls, suicidal ideation, and liver enzyme abnormalities were all seen in clinical trials.
The news doesn’t bode well for the therapeutic target of BACE (beta-site APP cleaving enzyme) inhibition. BACE is one of the enzymes that trims the amyloid precursor protein (APP). Inhibiting it does reduce the amount of toxic amyloid-beta in cerebrospinal fluid, and amyloid plaque in the brain. But none of these molecules has shown clinical benefit in dementia patients, whether their disease is mild, or moderate or – now – prodromal. And it is now apparent that inhibiting BACE also produces serious off-target problems.
“BACE-1 inhibition certainly seemed to have a sound rationale assuming the basis for amyloid’s role in Alzheimer’s disease pathogenesis is a gain of toxicity,” Richard J. Caselli, MD, of the Mayo Clinic, Rochester, Minn., said in an interview. “That APP is important for Alzheimer’s pathogenesis still seems clear but whether amyloid-beta toxicity is the driving force is no longer clear. Further, interruption of the APP system disrupts more than amyloid-beta peptide, possibly explaining the adverse cognitive effects of BACE-1 inhibition shown exhibited now by three different BACE-1 inhibitors.”
Verubecestat
Researchers got their first dose of bad news regarding verubecestat at the 2017 Clinical Trials in Alzheimer’s Disease meeting. There, Michael F. Egan, MD, Merck’s associate vice president of clinical neuroscience, discussed the molecule’s failure to slow cognitive decline in patients with mild to moderate Alzheimer’s disease. There was plenty of biomarker evidence that the drug did block amyloid-beta production, but there also was a plethora of concerning adverse events, Dr. Egan said in an interview.
However, verubecestat still was being pursued in patients with prodromal Alzheimer’s disease. In February, Merck stopped the trial after a futility analysis and announced that the company was terminating studies of verubecestat in that population as well. In the April 11 issue of the New England Journal of Medicine, Dr. Egan and his colleagues report the full extent of verubecestat’s failure in prodromal patients, and the accompanying adverse events.
At the time of termination, 1,454 patients had been enrolled. Of these, 485 received 12 mg/day, 484 received 40 mg/day, and 485 received placebo. About half of each group completed 104 weeks of treatment in the study, which was designed to extend up to 5 years.
The primary outcome was change in the Clinical Dementia Rating Scale–Sum of Boxes score (CDR-SB). Seven secondary outcomes examined other cognitive and functional end points, along with changes in hippocampal volume on MRI and amyloid burden as determined in PET imaging.
Not only did verubecestat fail to slow cognitive decline, it appeared to exacerbate it. The mean change on the CDR-SB was 1.65 in the 12-mg group, 2.02 in the 40-mg group, and 1.58 in the placebo group, favoring placebo.
“In an exploratory analysis according to time point, scores on the CDR-SB were also higher [signifying more impairment of cognition and daily functioning] in the 40-mg group than in the placebo group at 13, 26, and 52 weeks ... suggesting but not confirming the possibility of worse performance at these earlier time points in the high-dose verubecestat group,” the investigators said.
Verubecestat also was associated with more conversions to Alzheimer’s disease. Per 100 patient-years, the Alzheimer’s disease event rates were 24.5 in the 12-mg group, 25.5 in the 40-mg group, and 19.3 in the placebo group. Compared with placebo, those taking 12-mg doses were 30% more likely to develop Alzheimer’s disease and those taking 40-mg doses were 38% more likely. The findings suggest that “verubecestat may have accelerated the progression to diagnosis of dementia due to Alzheimer’s disease,” the investigators said.
The negative impact of verubecestat was apparent quite early in the study. “In exploratory analyses, both dose levels of verubecestat were associated with poorer outcomes on the [Composite Cognition Score-3 Domain] and the ADAS-Cog [Alzheimer’s Disease Assessment Scale–Cognitive Subscale] measures of cognition that, relative to placebo, appeared worse at week 13 and did not appear to progress thereafter.”
Results of the secondary end points, including the ADAS-Cog and the Mini-Mental State Exam, also indicated that verubecestat may have worsened cognitive performance.
Imaging outcomes were positive, however. Hippocampal volume was 6,448 mL in the 12-mg group, 6,469 mL in the 40-mg group, and 6,435 mL in the placebo group. Brain amyloid increased in the placebo group, as expected, and decreased in the active groups. The small group of patients who underwent cerebrospinal fluid sampling showed reductions of more than 60% in amyloid-beta and soluble APP-beta associated with verubecestat. These results show that the molecule was indeed hitting its intended target, but that doing so was not clinically beneficial.
Adverse events were more common in the verubecestat groups. These included rash, dermatitis, urticaria, sleep disturbance, weight loss, and cough. Hair coloring changed in 2.5% of patients in the 12-mg group and 5% of the 40-mg group, but in none of the subjects taking placebo.
Patients taking verubecestat were more likely to sustain falls and injuries and to express suicidal ideation.
The results of this trial differ from the study of verubecestat in mild to moderate Alzheimer’s disease, the investigators noted. Those patients did not decline cognitively as did those with prodromal disease.
“Patients at an earlier stage of the disease may be more sensitive to the effects of substantial BACE-1 inhibition, perhaps because of a role of BACE-1 in normal synaptic function. It is also possible that the effects of verubecestat are due to inhibition of BACE-2,” they said.
Atabecestat
In a research letter in the same issue of the New England Journal of Medicine (2019 Apr 11;380:1483-5), David Henley, MD, senior director of Janssen’s Alzheimer’s clinical development core, released similarly negative results from an interim analysis of EARLY (Efficacy and Safety Study of Atabecestat in Participants Who Are Asymptomatic at Risk for Developing Alzheimer’s Dementia) trial, a randomized study of the BACE-1 inhibitor candidate, atabecestat.
The phase 2 trial enrolled 557 patients with prodromal Alzheimer’s disease. The primary cognitive end point was change from baseline in the Preclinical Alzheimer’s Cognitive Composite (PACC) score.
This trial was discontinued in May 2018 because of liver-related adverse events, although safety follow-up continues. The research letter did not disclose details of the hepatic events, but a company press release from May 2018 referred to them in a general sense.
“Elevations of liver enzymes, which were serious in nature, have been observed in some study participants who received the Janssen BACE inhibitor, atabecestat. After a thorough evaluation of all available liver safety data from our studies, Janssen has concluded that the benefit-risk ratio is no longer favorable to continue development of atabecestat for people who have late-onset preclinical stage Alzheimer’s disease.”
Patients in EARLY were randomized to 5 mg, 25 mg, or placebo. As in the verubecestat trial, those randomized to placebo did better. The mean changes from baseline in the PACC score were −1.44 in the 25-mg group, −0.58 in the 5-mg group, and −0.32 in the placebo group.
“At month 6, the difference between the 25-mg group and the placebo group was −1.12 and the difference between the 5-mg group and the placebo group was −0.26, favoring placebo over the higher dose,” the authors said.
This theme reemerged in a secondary end point, the Repeatable Battery for the Assessment of Neuropsychological Status. The 25-mg group declined 3.58 points more than placebo, and the 5-mg group, 1.43 points more.
Adverse events were more common in the active groups and included depression, effects on sleep and dreams, and anxiety.
“The differences in cognitive performance between the groups are of uncertain clinical significance; however, given similar findings favoring placebo over BACE-1 inhibitors in other trial, we are communicating this potential signal of worsening cognitive function in the treated groups,” Dr. Henley said.
SOURCE: Egan MF et al. N Engl J Med. 2019 Apr 11;380:1408-20.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
FDA to expand opioid labeling with instructions on proper tapering
The Food and Drug Administration is making changes to opioid analgesic labeling to give better information to clinicians on how to properly taper patients dependent on opioid use, according to Douglas Throckmorton, MD, deputy director for regulatory programs in the FDA’s Center for Drug Evaluation and Research.

, such as withdrawal symptoms, uncontrolled pain, and suicide. Both the FDA and the Centers for Disease Control and Prevention offer guidelines on how to properly taper opioids, Dr. Throckmorton said, but more needs to be done to ensure that patients are being provided with the correct advice and care.
The changes to the labels will include expanded information to health care clinicians and are intended to be used when both the clinician and patient have agreed to reduce the opioid dosage. When this is discussed, factors that should be considered include the dose of the drug, the duration of treatment, the type of pain being treated, and the physical and psychological attributes of the patient.
Other actions the FDA is pursuing to combat opioid use disorder include working with the National Academies of Sciences, Engineering, and Medicine on guidelines for the proper opioid analgesic prescribing for acute pain resulting from specific conditions or procedures, and advancing policies that make immediate-release opioid formulations available in fixed-quantity packaging for 1 or 2 days.
“The FDA remains committed to addressing the opioid crisis on all fronts, with a significant focus on decreasing unnecessary exposure to opioids and preventing new addiction; supporting the treatment of those with opioid use disorder; fostering the development of novel pain treatment therapies and opioids more resistant to abuse and misuse; and taking action against those involved in the illegal importation and sale of opioids,” Dr. Throckmorton said.
Find the full statement by Dr. Throckmorton on the FDA website.
The Food and Drug Administration is making changes to opioid analgesic labeling to give better information to clinicians on how to properly taper patients dependent on opioid use, according to Douglas Throckmorton, MD, deputy director for regulatory programs in the FDA’s Center for Drug Evaluation and Research.
, such as withdrawal symptoms, uncontrolled pain, and suicide. Both the FDA and the Centers for Disease Control and Prevention offer guidelines on how to properly taper opioids, Dr. Throckmorton said, but more needs to be done to ensure that patients are being provided with the correct advice and care.
The changes to the labels will include expanded information to health care clinicians and are intended to be used when both the clinician and patient have agreed to reduce the opioid dosage. When this is discussed, factors that should be considered include the dose of the drug, the duration of treatment, the type of pain being treated, and the physical and psychological attributes of the patient.
Other actions the FDA is pursuing to combat opioid use disorder include working with the National Academies of Sciences, Engineering, and Medicine on guidelines for the proper opioid analgesic prescribing for acute pain resulting from specific conditions or procedures, and advancing policies that make immediate-release opioid formulations available in fixed-quantity packaging for 1 or 2 days.
“The FDA remains committed to addressing the opioid crisis on all fronts, with a significant focus on decreasing unnecessary exposure to opioids and preventing new addiction; supporting the treatment of those with opioid use disorder; fostering the development of novel pain treatment therapies and opioids more resistant to abuse and misuse; and taking action against those involved in the illegal importation and sale of opioids,” Dr. Throckmorton said.
Find the full statement by Dr. Throckmorton on the FDA website.
The Food and Drug Administration is making changes to opioid analgesic labeling to give better information to clinicians on how to properly taper patients dependent on opioid use, according to Douglas Throckmorton, MD, deputy director for regulatory programs in the FDA’s Center for Drug Evaluation and Research.
, such as withdrawal symptoms, uncontrolled pain, and suicide. Both the FDA and the Centers for Disease Control and Prevention offer guidelines on how to properly taper opioids, Dr. Throckmorton said, but more needs to be done to ensure that patients are being provided with the correct advice and care.
The changes to the labels will include expanded information to health care clinicians and are intended to be used when both the clinician and patient have agreed to reduce the opioid dosage. When this is discussed, factors that should be considered include the dose of the drug, the duration of treatment, the type of pain being treated, and the physical and psychological attributes of the patient.
Other actions the FDA is pursuing to combat opioid use disorder include working with the National Academies of Sciences, Engineering, and Medicine on guidelines for the proper opioid analgesic prescribing for acute pain resulting from specific conditions or procedures, and advancing policies that make immediate-release opioid formulations available in fixed-quantity packaging for 1 or 2 days.
“The FDA remains committed to addressing the opioid crisis on all fronts, with a significant focus on decreasing unnecessary exposure to opioids and preventing new addiction; supporting the treatment of those with opioid use disorder; fostering the development of novel pain treatment therapies and opioids more resistant to abuse and misuse; and taking action against those involved in the illegal importation and sale of opioids,” Dr. Throckmorton said.
Find the full statement by Dr. Throckmorton on the FDA website.
Acute Encephalopathy Following Hyperbaric Oxygen Therapy in a Patient on Metronidazole
Altered mental status (AMS) is a common presentation to the emergency department (ED) for older patients and is often due to underlying drug-associated adverse effects (AEs), medical or psychiatric illness, or neurologic disease. EDs often have protocols for diagnosing and managing AMS to assess the underlying etiology. A formal assessment with a full history and physical examination is paramount to diagnosing the cause of AMS.
Oral metronidazole is a commonly used antibiotic for anaerobic bacterial infections and Clostridium difficile-associated diarrhea and colitis.1Metronidazole produces cytotoxic intermediates that cause DNA strand breakage and destabilization, resulting in bactericidal activity in host cells.2Common AEs include gastrointestinal symptoms such as nausea, vomiting, and diarrhea; less common AEs can involve the nervous system and include seizures, peripheral neuropathy, dizziness, ataxia, and encephalopathy.3,4A pattern of magnetic resonance image (MRI) abnormalities typically located at the cerebellar dentate nucleus midbrain, dorsal pons, medulla, and splenium of the corpus callosum have been associated with metronidazole usage.5
Hyperbaric oxygen therapy (HBOT) is a treatment modality used as the primary therapy for decompression sickness, arterial gas embolism, and carbon monoxide poisoning. HBOT is used as adjuvant therapy for osteonecrosis caused by radiation or bisphosphonate use.6,7 HBOT increases the partial pressure of oxygen in plasma and increases the amount of oxygen delivered to tissues throughout the body.8Hyperoxia, defined as an elevated partial pressure of oxygen leading to excess oxygenation to tissues and organs, increases production of reactive oxygen and nitrogen species, which are signaling factors in a variety of pathways that stimulate angiogenesis.8 AEs of HBOT include barotrauma-related injuries and oxygen toxicity, such as respiratory distress or central nervous system (CNS) symptoms.9 Severe CNS AEs occur in 1% to 2% of patients undergoing therapy and manifest as generalized tonic-clonic seizures, typically in patients with preexisting neurologic disorders, brain injury, or lowered seizure threshold.7,8,10 There have been no documented incidences of HBOT inducing acute encephalopathy.
Case Presentation
A 63-year-old male smoker with no history of alcohol use presented to the ED with an acute onset of lightheadedness, confusion, and poor coordination following his second HBOT for radiation-induced osteonecrosis of the mandible. The patient reported chronic, slowly progressive pain and numbness of the feet that began 4 years earlier. He noted marked worsening of pain and difficulty standing and walking 3 to 4 months prior to presentation.
Ten years prior, the patient was diagnosed with cancer of the right tonsil. A tonsillectomy with wide margins was performed, followed by 35 rounds of radiation treatment and 2 rounds of chemotherapy with cisplatin.
In May 2017, the patient presented with a lump in the right cheek that was diagnosed as osteonecrosis of the mandible. An oral surgeon prescribed metronidazole 500 mg qid and amoxicillin 500 mg tid. The patient was adherent until presentation in November 2017. Following lack of improvement of the osteonecrosis from antibiotic therapy, oral surgery was planned, and the patient was referred for HBOT with a planned 20 HBOT preoperative treatments and 10 postoperative treatments.
Following his first 2-hour HBOT treatment on November 13, 2017, the patient complained of light-headedness, confusion, and incoordination. While driving on a familiar route to his home, he collided with a tree that was 6 feet from the curb. The patient attempted to drive another vehicle later that day, resulting in a second motor vehicle accident. There was no significant injury reported in either accident.
His partner described the patient’s episode of disorientation lasting 6 to 8 hours, during which he “looked drunk” and was unable to sit in a chair without falling. The following morning, the patient had improved mental status but had not returned to baseline. His second HBOT treatment took place that day, and again, the patient acutely experienced light-headedness and confusion following completion. Therapy was suspended, and the patient was referred to the ED for further evaluation. Mild facial asymmetry without weakness, decreased sensation from toes to knees bilaterally, and absent Achilles reflexes bilaterally were found on neurologic examination. He exhibited past-pointing on finger-to-nose testing bilaterally. He was able to ambulate independently, but he could not perform tandem gait.
An MRI of the brain showed abnormal T2 hyperintensity found bilaterally at the dentate nuclei and inferior colliculi. The splenium of the corpus callosum also showed mild involvement with hyperintense lesions. Laboratory tests of the patient’s complete blood count; comprehensive metabolic panel; vitamins B1, B6, B12; and folic acid levels had no notable abnormalities and were within normal limits.
Metronidazole and HBOT therapy were discontinued, and all of the patient’s symptoms resolved within 2 weeks. A repeat examination and MRI performed 1 month later showed resolution of all the patient’s clinical findings and MRI abnormalities. HBOT was resumed without the recurrence of previously described symptoms.
Discussion
This patient’s encephalopathic symptoms correlate temporally with the onset of HBOT. There is no medical literature suggesting a relationship between HBOT and encephalopathic symptoms with MRI abnormalities, and in fact, some studies suggest HBOT as a treatment for hypoxic-ischemic encephalopathy in neonates.11 This led us to believe that the HBOT may have exacerbated some underlying condition, evidenced by the specific MRI findings of T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the dentate nuclei and inferior colliculi (Figures 1 and 2). 
Differential diagnoses for T2 hyperintense lesions in the dentate nuclei include metronidazole toxicity, acute Wernicke encephalopathy (WE), and methyl bromide intoxication. Diseases that would have presented in infancy with similar MRI findings (Canavan disease, maple-syrup urine disease, and glutaric aciduria type 1) were not considered plausible.12-14 
Despite his denial of alcohol use, the patient was at risk for malnutrition secondary to his mandibular lesion and difficulty eating. Clinically, he presented with episodes of confusion and ataxia, consistent with 2 of the classic triad of symptoms of WE (no ocular abnormalities noted on exam). Typical MRI findings in WE include signal intensity alterations (including T2 hyperintensities) in the medial thalami, mammillary bodies, collicular bodies, and periaqueductal and periventricular regions.14,15 Atypical MRI findings in WE include symmetric signal intensity changes in the cerebellum, dentate nuclei, caudate nuclei, red nuclei, cranial nerve nuclei, and splenium.14 Of note, atypical MRI findings were more common in patients without alcohol use disorders and WE, and typical MRI findings were more common in patients with alcohol use disorders.14 However, this patient’s report of no alcohol use and the serum thiamine level being within normal limits (173 nmol/L; range 78-185 nmol/L) made acute WE less likely than metronidonazale-induced encephalopathy (MIE).
The most common neurologic AE of metronidazole is distal symmetric sensory polyneuropathy, which also can have motor or autonomic features.16,17 While our patient had a history of peripheral neuropathy, he noted marked worsening of foot pain 3 months after initiating metronidazole therapy. A potential mechanism involves metronidazole or its cytotoxic intermediates binding neuronal ribonucleic acids, thus inhibiting protein synthesis and resulting in degenerative neuronal changes and reversible axonal swelling (as opposed to the DNA interference attributed to the drug’s mechanism of bactericidal action).18 Neuropathies may result from prolonged high-dose metronidazole therapy (cumulative dose > 42 g),3 but they also have been seen in short-term use of high dosages.17
CNS AEs are much rarer and are thought to be associated with metronidazole’s ability to cross the blood-brain barrier. These patients present as a toxic encephalopathy with cerebellar dysfunction (dysarthria, ataxia) as the most common presentation, followed by AMS and seizures.4 Our patient presented with acute confusion and ataxia. Animal studies suggest that γ-aminobutyric acid (GABA) receptor modulation in the cerebellar and vestibular systems may contribute to this neurotoxicity, but no definitive mechanism of injury has been found.19
On MRI, MIE most commonly presents with hyperintense lesions in the bilateral cerebellar dentate nucleus on T2-weighted and FLAIR images.5,20 The midbrain, dorsal pons, medulla, and corpus callosum also can show increased signal intensity.5 This AE does not seem to be dose- or duration-dependent, and most cases report complete or partial resolution of symptoms following discontinuation of the drug, though this is not absolute.4,13,21 The patient’s MRI findings were highly consistent with MIE (Figure 2).
Conclusion
This patient’s highly specific MRI findings, neurologic examination consistent with confusion, ataxia, length-dependent sensory neuropathy, and 360-g cumulative dose of metronidazole over the previous 6 months suggest he experienced MIE. The mechanism of how HBOT precipitated the patient’s altered mental status, incoordination, and worsening of peripheral neuropathy is unknown. Although encephalopathy with MRI abnormalities as described is not a reported AE of HBOT, it may be unrecognized. It is possible that without HBOT the patient would have remained asymptomatic apart from his peripheral neuropathy.
We propose HBOT may exacerbate or increase the risk of a patient developing MIE. Our patient was able to safely resume HBOT after metronidazole was discontinued, suggesting that the combination was the causation for the development of encephalopathy. We do not believe any similar cases have been reported.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541.
2. Edwards DI. The action of metronidazole on DNA. J Antimicrob Chemother. 1977;3(1):43-48.
3. Goolsby TA, Jakeman B, Gaynes RP. Clinical relevance of metronidazole and peripheral neuropathy: a systematic review of the literature. Int J Antimicrob Agents. 2018;51(3):319-325.
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systematic review. Clin Neuropharmacol. 2011;34(6):241-247.
5. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658.
6. Ceponis P, Keilman C, Guerry C, Freiberger JJ. Hyperbaric oxygen therapy and osteonecrosis. Oral Dis. 2017;23(2):141-151.
7. Leach R, Rees P, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317(7166):1140-1143.
8. Thom SR. Hyperbaric oxygen–its mechanisms and efficacy. Plastic Reconstr Surg. 2011;127(suppl 1):131S-141S.
9. Plafki C, Peters P, Almeling M, Welslau W, Busch R. Complications and side effects of hyperbaric oxygen therapy. Aviation Space Environ Med. 2000;71(2):119-124.
10. Hadanny A, Meir O, Bechor Y, Fishlev G, Bergan J, Efrati S. Seizures during hyperbaric oxygen therapy: retrospective analysis of 62,614 treatment sessions. Undersea Hyperb Med. 2016;43(1):21-28.
11. Liu Z, Xiong T, Meads C. Clinical effectiveness of treatment with hyperbaric oxygen for neonatal hypoxic-ischaemic encephalopathy: systematic review of Chinese literature. BMJ. 2006;333(7564):374.
12. Bond KM, Brinjikji W, Eckel LJ, Kallmes DF, McDonald RJ, Carr CM. Dentate update: imaging features of entities that affect the dentate nucleus. AJNR Am J Neuroradiol. 2017;38(8):1467-1474.
13. Agarwal A, Kanekar S, Sabat S, Thamburaj K. Metronidazole-induced cerebellar toxicity. Neurol Int. 2016;8(1):6365.
14. Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501-508.
15. Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170-180.
16. Hobson-Webb LD, Roach ES, Donofrio PD. Metronidazole: newly recognized cause of autonomic neuropathy. J Child Neurol. 2006;21(5):429-431.
17. Nath Chaurasia R. Rapid onset metronidazole induced sensory neuropathy: case series and review of literature. Int J Neurorehabilitation. 2015;02:152.
18. Bradley WG, Karlsson IJ, Rassol CG. Metronidazole neuropathy. Br Med J. 1977;2(6087):610-611.
19. Evans J, Levesque D, Knowles K, Longshore R, Plummer S. Diazepam as a treatment for metronidazole toxicosis in dogs: a retrospective study of 21 cases. J Vet Intern Med. 2003;17(3):304-310.
20. Farmakiotis D, Zeluff B. Images in clinical medicine. Metronidazole-associated encephalopathy. N Engl J Med. 2016;374(15):1465.
21. Hobbs K, Stern-Nezer S, Buckwalter MS, Fischbein N, Finley Caulfield A. Metronidazole-induced encephalopathy: not always a reversible situation. Neurocrit Care. 2015;22(3):429-436.
Altered mental status (AMS) is a common presentation to the emergency department (ED) for older patients and is often due to underlying drug-associated adverse effects (AEs), medical or psychiatric illness, or neurologic disease. EDs often have protocols for diagnosing and managing AMS to assess the underlying etiology. A formal assessment with a full history and physical examination is paramount to diagnosing the cause of AMS.
Oral metronidazole is a commonly used antibiotic for anaerobic bacterial infections and Clostridium difficile-associated diarrhea and colitis.1Metronidazole produces cytotoxic intermediates that cause DNA strand breakage and destabilization, resulting in bactericidal activity in host cells.2Common AEs include gastrointestinal symptoms such as nausea, vomiting, and diarrhea; less common AEs can involve the nervous system and include seizures, peripheral neuropathy, dizziness, ataxia, and encephalopathy.3,4A pattern of magnetic resonance image (MRI) abnormalities typically located at the cerebellar dentate nucleus midbrain, dorsal pons, medulla, and splenium of the corpus callosum have been associated with metronidazole usage.5
Hyperbaric oxygen therapy (HBOT) is a treatment modality used as the primary therapy for decompression sickness, arterial gas embolism, and carbon monoxide poisoning. HBOT is used as adjuvant therapy for osteonecrosis caused by radiation or bisphosphonate use.6,7 HBOT increases the partial pressure of oxygen in plasma and increases the amount of oxygen delivered to tissues throughout the body.8Hyperoxia, defined as an elevated partial pressure of oxygen leading to excess oxygenation to tissues and organs, increases production of reactive oxygen and nitrogen species, which are signaling factors in a variety of pathways that stimulate angiogenesis.8 AEs of HBOT include barotrauma-related injuries and oxygen toxicity, such as respiratory distress or central nervous system (CNS) symptoms.9 Severe CNS AEs occur in 1% to 2% of patients undergoing therapy and manifest as generalized tonic-clonic seizures, typically in patients with preexisting neurologic disorders, brain injury, or lowered seizure threshold.7,8,10 There have been no documented incidences of HBOT inducing acute encephalopathy.
Case Presentation
A 63-year-old male smoker with no history of alcohol use presented to the ED with an acute onset of lightheadedness, confusion, and poor coordination following his second HBOT for radiation-induced osteonecrosis of the mandible. The patient reported chronic, slowly progressive pain and numbness of the feet that began 4 years earlier. He noted marked worsening of pain and difficulty standing and walking 3 to 4 months prior to presentation.
Ten years prior, the patient was diagnosed with cancer of the right tonsil. A tonsillectomy with wide margins was performed, followed by 35 rounds of radiation treatment and 2 rounds of chemotherapy with cisplatin.
In May 2017, the patient presented with a lump in the right cheek that was diagnosed as osteonecrosis of the mandible. An oral surgeon prescribed metronidazole 500 mg qid and amoxicillin 500 mg tid. The patient was adherent until presentation in November 2017. Following lack of improvement of the osteonecrosis from antibiotic therapy, oral surgery was planned, and the patient was referred for HBOT with a planned 20 HBOT preoperative treatments and 10 postoperative treatments.
Following his first 2-hour HBOT treatment on November 13, 2017, the patient complained of light-headedness, confusion, and incoordination. While driving on a familiar route to his home, he collided with a tree that was 6 feet from the curb. The patient attempted to drive another vehicle later that day, resulting in a second motor vehicle accident. There was no significant injury reported in either accident.
His partner described the patient’s episode of disorientation lasting 6 to 8 hours, during which he “looked drunk” and was unable to sit in a chair without falling. The following morning, the patient had improved mental status but had not returned to baseline. His second HBOT treatment took place that day, and again, the patient acutely experienced light-headedness and confusion following completion. Therapy was suspended, and the patient was referred to the ED for further evaluation. Mild facial asymmetry without weakness, decreased sensation from toes to knees bilaterally, and absent Achilles reflexes bilaterally were found on neurologic examination. He exhibited past-pointing on finger-to-nose testing bilaterally. He was able to ambulate independently, but he could not perform tandem gait.
An MRI of the brain showed abnormal T2 hyperintensity found bilaterally at the dentate nuclei and inferior colliculi. The splenium of the corpus callosum also showed mild involvement with hyperintense lesions. Laboratory tests of the patient’s complete blood count; comprehensive metabolic panel; vitamins B1, B6, B12; and folic acid levels had no notable abnormalities and were within normal limits.
Metronidazole and HBOT therapy were discontinued, and all of the patient’s symptoms resolved within 2 weeks. A repeat examination and MRI performed 1 month later showed resolution of all the patient’s clinical findings and MRI abnormalities. HBOT was resumed without the recurrence of previously described symptoms.
Discussion
This patient’s encephalopathic symptoms correlate temporally with the onset of HBOT. There is no medical literature suggesting a relationship between HBOT and encephalopathic symptoms with MRI abnormalities, and in fact, some studies suggest HBOT as a treatment for hypoxic-ischemic encephalopathy in neonates.11 This led us to believe that the HBOT may have exacerbated some underlying condition, evidenced by the specific MRI findings of T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the dentate nuclei and inferior colliculi (Figures 1 and 2).
Differential diagnoses for T2 hyperintense lesions in the dentate nuclei include metronidazole toxicity, acute Wernicke encephalopathy (WE), and methyl bromide intoxication. Diseases that would have presented in infancy with similar MRI findings (Canavan disease, maple-syrup urine disease, and glutaric aciduria type 1) were not considered plausible.12-14
Despite his denial of alcohol use, the patient was at risk for malnutrition secondary to his mandibular lesion and difficulty eating. Clinically, he presented with episodes of confusion and ataxia, consistent with 2 of the classic triad of symptoms of WE (no ocular abnormalities noted on exam). Typical MRI findings in WE include signal intensity alterations (including T2 hyperintensities) in the medial thalami, mammillary bodies, collicular bodies, and periaqueductal and periventricular regions.14,15 Atypical MRI findings in WE include symmetric signal intensity changes in the cerebellum, dentate nuclei, caudate nuclei, red nuclei, cranial nerve nuclei, and splenium.14 Of note, atypical MRI findings were more common in patients without alcohol use disorders and WE, and typical MRI findings were more common in patients with alcohol use disorders.14 However, this patient’s report of no alcohol use and the serum thiamine level being within normal limits (173 nmol/L; range 78-185 nmol/L) made acute WE less likely than metronidonazale-induced encephalopathy (MIE).
The most common neurologic AE of metronidazole is distal symmetric sensory polyneuropathy, which also can have motor or autonomic features.16,17 While our patient had a history of peripheral neuropathy, he noted marked worsening of foot pain 3 months after initiating metronidazole therapy. A potential mechanism involves metronidazole or its cytotoxic intermediates binding neuronal ribonucleic acids, thus inhibiting protein synthesis and resulting in degenerative neuronal changes and reversible axonal swelling (as opposed to the DNA interference attributed to the drug’s mechanism of bactericidal action).18 Neuropathies may result from prolonged high-dose metronidazole therapy (cumulative dose > 42 g),3 but they also have been seen in short-term use of high dosages.17
CNS AEs are much rarer and are thought to be associated with metronidazole’s ability to cross the blood-brain barrier. These patients present as a toxic encephalopathy with cerebellar dysfunction (dysarthria, ataxia) as the most common presentation, followed by AMS and seizures.4 Our patient presented with acute confusion and ataxia. Animal studies suggest that γ-aminobutyric acid (GABA) receptor modulation in the cerebellar and vestibular systems may contribute to this neurotoxicity, but no definitive mechanism of injury has been found.19
On MRI, MIE most commonly presents with hyperintense lesions in the bilateral cerebellar dentate nucleus on T2-weighted and FLAIR images.5,20 The midbrain, dorsal pons, medulla, and corpus callosum also can show increased signal intensity.5 This AE does not seem to be dose- or duration-dependent, and most cases report complete or partial resolution of symptoms following discontinuation of the drug, though this is not absolute.4,13,21 The patient’s MRI findings were highly consistent with MIE (Figure 2).
Conclusion
This patient’s highly specific MRI findings, neurologic examination consistent with confusion, ataxia, length-dependent sensory neuropathy, and 360-g cumulative dose of metronidazole over the previous 6 months suggest he experienced MIE. The mechanism of how HBOT precipitated the patient’s altered mental status, incoordination, and worsening of peripheral neuropathy is unknown. Although encephalopathy with MRI abnormalities as described is not a reported AE of HBOT, it may be unrecognized. It is possible that without HBOT the patient would have remained asymptomatic apart from his peripheral neuropathy.
We propose HBOT may exacerbate or increase the risk of a patient developing MIE. Our patient was able to safely resume HBOT after metronidazole was discontinued, suggesting that the combination was the causation for the development of encephalopathy. We do not believe any similar cases have been reported.
Altered mental status (AMS) is a common presentation to the emergency department (ED) for older patients and is often due to underlying drug-associated adverse effects (AEs), medical or psychiatric illness, or neurologic disease. EDs often have protocols for diagnosing and managing AMS to assess the underlying etiology. A formal assessment with a full history and physical examination is paramount to diagnosing the cause of AMS.
Oral metronidazole is a commonly used antibiotic for anaerobic bacterial infections and Clostridium difficile-associated diarrhea and colitis.1Metronidazole produces cytotoxic intermediates that cause DNA strand breakage and destabilization, resulting in bactericidal activity in host cells.2Common AEs include gastrointestinal symptoms such as nausea, vomiting, and diarrhea; less common AEs can involve the nervous system and include seizures, peripheral neuropathy, dizziness, ataxia, and encephalopathy.3,4A pattern of magnetic resonance image (MRI) abnormalities typically located at the cerebellar dentate nucleus midbrain, dorsal pons, medulla, and splenium of the corpus callosum have been associated with metronidazole usage.5
Hyperbaric oxygen therapy (HBOT) is a treatment modality used as the primary therapy for decompression sickness, arterial gas embolism, and carbon monoxide poisoning. HBOT is used as adjuvant therapy for osteonecrosis caused by radiation or bisphosphonate use.6,7 HBOT increases the partial pressure of oxygen in plasma and increases the amount of oxygen delivered to tissues throughout the body.8Hyperoxia, defined as an elevated partial pressure of oxygen leading to excess oxygenation to tissues and organs, increases production of reactive oxygen and nitrogen species, which are signaling factors in a variety of pathways that stimulate angiogenesis.8 AEs of HBOT include barotrauma-related injuries and oxygen toxicity, such as respiratory distress or central nervous system (CNS) symptoms.9 Severe CNS AEs occur in 1% to 2% of patients undergoing therapy and manifest as generalized tonic-clonic seizures, typically in patients with preexisting neurologic disorders, brain injury, or lowered seizure threshold.7,8,10 There have been no documented incidences of HBOT inducing acute encephalopathy.
Case Presentation
A 63-year-old male smoker with no history of alcohol use presented to the ED with an acute onset of lightheadedness, confusion, and poor coordination following his second HBOT for radiation-induced osteonecrosis of the mandible. The patient reported chronic, slowly progressive pain and numbness of the feet that began 4 years earlier. He noted marked worsening of pain and difficulty standing and walking 3 to 4 months prior to presentation.
Ten years prior, the patient was diagnosed with cancer of the right tonsil. A tonsillectomy with wide margins was performed, followed by 35 rounds of radiation treatment and 2 rounds of chemotherapy with cisplatin.
In May 2017, the patient presented with a lump in the right cheek that was diagnosed as osteonecrosis of the mandible. An oral surgeon prescribed metronidazole 500 mg qid and amoxicillin 500 mg tid. The patient was adherent until presentation in November 2017. Following lack of improvement of the osteonecrosis from antibiotic therapy, oral surgery was planned, and the patient was referred for HBOT with a planned 20 HBOT preoperative treatments and 10 postoperative treatments.
Following his first 2-hour HBOT treatment on November 13, 2017, the patient complained of light-headedness, confusion, and incoordination. While driving on a familiar route to his home, he collided with a tree that was 6 feet from the curb. The patient attempted to drive another vehicle later that day, resulting in a second motor vehicle accident. There was no significant injury reported in either accident.
His partner described the patient’s episode of disorientation lasting 6 to 8 hours, during which he “looked drunk” and was unable to sit in a chair without falling. The following morning, the patient had improved mental status but had not returned to baseline. His second HBOT treatment took place that day, and again, the patient acutely experienced light-headedness and confusion following completion. Therapy was suspended, and the patient was referred to the ED for further evaluation. Mild facial asymmetry without weakness, decreased sensation from toes to knees bilaterally, and absent Achilles reflexes bilaterally were found on neurologic examination. He exhibited past-pointing on finger-to-nose testing bilaterally. He was able to ambulate independently, but he could not perform tandem gait.
An MRI of the brain showed abnormal T2 hyperintensity found bilaterally at the dentate nuclei and inferior colliculi. The splenium of the corpus callosum also showed mild involvement with hyperintense lesions. Laboratory tests of the patient’s complete blood count; comprehensive metabolic panel; vitamins B1, B6, B12; and folic acid levels had no notable abnormalities and were within normal limits.
Metronidazole and HBOT therapy were discontinued, and all of the patient’s symptoms resolved within 2 weeks. A repeat examination and MRI performed 1 month later showed resolution of all the patient’s clinical findings and MRI abnormalities. HBOT was resumed without the recurrence of previously described symptoms.
Discussion
This patient’s encephalopathic symptoms correlate temporally with the onset of HBOT. There is no medical literature suggesting a relationship between HBOT and encephalopathic symptoms with MRI abnormalities, and in fact, some studies suggest HBOT as a treatment for hypoxic-ischemic encephalopathy in neonates.11 This led us to believe that the HBOT may have exacerbated some underlying condition, evidenced by the specific MRI findings of T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the dentate nuclei and inferior colliculi (Figures 1 and 2).
Differential diagnoses for T2 hyperintense lesions in the dentate nuclei include metronidazole toxicity, acute Wernicke encephalopathy (WE), and methyl bromide intoxication. Diseases that would have presented in infancy with similar MRI findings (Canavan disease, maple-syrup urine disease, and glutaric aciduria type 1) were not considered plausible.12-14
Despite his denial of alcohol use, the patient was at risk for malnutrition secondary to his mandibular lesion and difficulty eating. Clinically, he presented with episodes of confusion and ataxia, consistent with 2 of the classic triad of symptoms of WE (no ocular abnormalities noted on exam). Typical MRI findings in WE include signal intensity alterations (including T2 hyperintensities) in the medial thalami, mammillary bodies, collicular bodies, and periaqueductal and periventricular regions.14,15 Atypical MRI findings in WE include symmetric signal intensity changes in the cerebellum, dentate nuclei, caudate nuclei, red nuclei, cranial nerve nuclei, and splenium.14 Of note, atypical MRI findings were more common in patients without alcohol use disorders and WE, and typical MRI findings were more common in patients with alcohol use disorders.14 However, this patient’s report of no alcohol use and the serum thiamine level being within normal limits (173 nmol/L; range 78-185 nmol/L) made acute WE less likely than metronidonazale-induced encephalopathy (MIE).
The most common neurologic AE of metronidazole is distal symmetric sensory polyneuropathy, which also can have motor or autonomic features.16,17 While our patient had a history of peripheral neuropathy, he noted marked worsening of foot pain 3 months after initiating metronidazole therapy. A potential mechanism involves metronidazole or its cytotoxic intermediates binding neuronal ribonucleic acids, thus inhibiting protein synthesis and resulting in degenerative neuronal changes and reversible axonal swelling (as opposed to the DNA interference attributed to the drug’s mechanism of bactericidal action).18 Neuropathies may result from prolonged high-dose metronidazole therapy (cumulative dose > 42 g),3 but they also have been seen in short-term use of high dosages.17
CNS AEs are much rarer and are thought to be associated with metronidazole’s ability to cross the blood-brain barrier. These patients present as a toxic encephalopathy with cerebellar dysfunction (dysarthria, ataxia) as the most common presentation, followed by AMS and seizures.4 Our patient presented with acute confusion and ataxia. Animal studies suggest that γ-aminobutyric acid (GABA) receptor modulation in the cerebellar and vestibular systems may contribute to this neurotoxicity, but no definitive mechanism of injury has been found.19
On MRI, MIE most commonly presents with hyperintense lesions in the bilateral cerebellar dentate nucleus on T2-weighted and FLAIR images.5,20 The midbrain, dorsal pons, medulla, and corpus callosum also can show increased signal intensity.5 This AE does not seem to be dose- or duration-dependent, and most cases report complete or partial resolution of symptoms following discontinuation of the drug, though this is not absolute.4,13,21 The patient’s MRI findings were highly consistent with MIE (Figure 2).
Conclusion
This patient’s highly specific MRI findings, neurologic examination consistent with confusion, ataxia, length-dependent sensory neuropathy, and 360-g cumulative dose of metronidazole over the previous 6 months suggest he experienced MIE. The mechanism of how HBOT precipitated the patient’s altered mental status, incoordination, and worsening of peripheral neuropathy is unknown. Although encephalopathy with MRI abnormalities as described is not a reported AE of HBOT, it may be unrecognized. It is possible that without HBOT the patient would have remained asymptomatic apart from his peripheral neuropathy.
We propose HBOT may exacerbate or increase the risk of a patient developing MIE. Our patient was able to safely resume HBOT after metronidazole was discontinued, suggesting that the combination was the causation for the development of encephalopathy. We do not believe any similar cases have been reported.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541.
2. Edwards DI. The action of metronidazole on DNA. J Antimicrob Chemother. 1977;3(1):43-48.
3. Goolsby TA, Jakeman B, Gaynes RP. Clinical relevance of metronidazole and peripheral neuropathy: a systematic review of the literature. Int J Antimicrob Agents. 2018;51(3):319-325.
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systematic review. Clin Neuropharmacol. 2011;34(6):241-247.
5. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658.
6. Ceponis P, Keilman C, Guerry C, Freiberger JJ. Hyperbaric oxygen therapy and osteonecrosis. Oral Dis. 2017;23(2):141-151.
7. Leach R, Rees P, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317(7166):1140-1143.
8. Thom SR. Hyperbaric oxygen–its mechanisms and efficacy. Plastic Reconstr Surg. 2011;127(suppl 1):131S-141S.
9. Plafki C, Peters P, Almeling M, Welslau W, Busch R. Complications and side effects of hyperbaric oxygen therapy. Aviation Space Environ Med. 2000;71(2):119-124.
10. Hadanny A, Meir O, Bechor Y, Fishlev G, Bergan J, Efrati S. Seizures during hyperbaric oxygen therapy: retrospective analysis of 62,614 treatment sessions. Undersea Hyperb Med. 2016;43(1):21-28.
11. Liu Z, Xiong T, Meads C. Clinical effectiveness of treatment with hyperbaric oxygen for neonatal hypoxic-ischaemic encephalopathy: systematic review of Chinese literature. BMJ. 2006;333(7564):374.
12. Bond KM, Brinjikji W, Eckel LJ, Kallmes DF, McDonald RJ, Carr CM. Dentate update: imaging features of entities that affect the dentate nucleus. AJNR Am J Neuroradiol. 2017;38(8):1467-1474.
13. Agarwal A, Kanekar S, Sabat S, Thamburaj K. Metronidazole-induced cerebellar toxicity. Neurol Int. 2016;8(1):6365.
14. Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501-508.
15. Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170-180.
16. Hobson-Webb LD, Roach ES, Donofrio PD. Metronidazole: newly recognized cause of autonomic neuropathy. J Child Neurol. 2006;21(5):429-431.
17. Nath Chaurasia R. Rapid onset metronidazole induced sensory neuropathy: case series and review of literature. Int J Neurorehabilitation. 2015;02:152.
18. Bradley WG, Karlsson IJ, Rassol CG. Metronidazole neuropathy. Br Med J. 1977;2(6087):610-611.
19. Evans J, Levesque D, Knowles K, Longshore R, Plummer S. Diazepam as a treatment for metronidazole toxicosis in dogs: a retrospective study of 21 cases. J Vet Intern Med. 2003;17(3):304-310.
20. Farmakiotis D, Zeluff B. Images in clinical medicine. Metronidazole-associated encephalopathy. N Engl J Med. 2016;374(15):1465.
21. Hobbs K, Stern-Nezer S, Buckwalter MS, Fischbein N, Finley Caulfield A. Metronidazole-induced encephalopathy: not always a reversible situation. Neurocrit Care. 2015;22(3):429-436.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541.
2. Edwards DI. The action of metronidazole on DNA. J Antimicrob Chemother. 1977;3(1):43-48.
3. Goolsby TA, Jakeman B, Gaynes RP. Clinical relevance of metronidazole and peripheral neuropathy: a systematic review of the literature. Int J Antimicrob Agents. 2018;51(3):319-325.
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systematic review. Clin Neuropharmacol. 2011;34(6):241-247.
5. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658.
6. Ceponis P, Keilman C, Guerry C, Freiberger JJ. Hyperbaric oxygen therapy and osteonecrosis. Oral Dis. 2017;23(2):141-151.
7. Leach R, Rees P, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317(7166):1140-1143.
8. Thom SR. Hyperbaric oxygen–its mechanisms and efficacy. Plastic Reconstr Surg. 2011;127(suppl 1):131S-141S.
9. Plafki C, Peters P, Almeling M, Welslau W, Busch R. Complications and side effects of hyperbaric oxygen therapy. Aviation Space Environ Med. 2000;71(2):119-124.
10. Hadanny A, Meir O, Bechor Y, Fishlev G, Bergan J, Efrati S. Seizures during hyperbaric oxygen therapy: retrospective analysis of 62,614 treatment sessions. Undersea Hyperb Med. 2016;43(1):21-28.
11. Liu Z, Xiong T, Meads C. Clinical effectiveness of treatment with hyperbaric oxygen for neonatal hypoxic-ischaemic encephalopathy: systematic review of Chinese literature. BMJ. 2006;333(7564):374.
12. Bond KM, Brinjikji W, Eckel LJ, Kallmes DF, McDonald RJ, Carr CM. Dentate update: imaging features of entities that affect the dentate nucleus. AJNR Am J Neuroradiol. 2017;38(8):1467-1474.
13. Agarwal A, Kanekar S, Sabat S, Thamburaj K. Metronidazole-induced cerebellar toxicity. Neurol Int. 2016;8(1):6365.
14. Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501-508.
15. Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170-180.
16. Hobson-Webb LD, Roach ES, Donofrio PD. Metronidazole: newly recognized cause of autonomic neuropathy. J Child Neurol. 2006;21(5):429-431.
17. Nath Chaurasia R. Rapid onset metronidazole induced sensory neuropathy: case series and review of literature. Int J Neurorehabilitation. 2015;02:152.
18. Bradley WG, Karlsson IJ, Rassol CG. Metronidazole neuropathy. Br Med J. 1977;2(6087):610-611.
19. Evans J, Levesque D, Knowles K, Longshore R, Plummer S. Diazepam as a treatment for metronidazole toxicosis in dogs: a retrospective study of 21 cases. J Vet Intern Med. 2003;17(3):304-310.
20. Farmakiotis D, Zeluff B. Images in clinical medicine. Metronidazole-associated encephalopathy. N Engl J Med. 2016;374(15):1465.
21. Hobbs K, Stern-Nezer S, Buckwalter MS, Fischbein N, Finley Caulfield A. Metronidazole-induced encephalopathy: not always a reversible situation. Neurocrit Care. 2015;22(3):429-436.
Mindfulness yoga reduced stress and motor symptoms in patients with Parkinson’s disease
Among patients with mild or moderate Parkinson’s disease, mindfulness yoga was as effective as stretching and resistance training in improving motor function and mobility, a randomized trial found.
In addition, mindfulness yoga reduced anxiety and depressive symptoms and increased spiritual well-being and health-related quality of life more than stretching and resistance training, researchers reported in JAMA Neurology.
Although guidelines support exercise for patients with Parkinson’s disease, investigators had not examined whether yoga is superior to conventional exercise for stress and symptom management in this patient population. Jojo Y. Y. Kwok, PhD, a research assistant professor of nursing at the University of Hong Kong, and her colleagues conducted an assessor-masked, randomized trial that included 138 adults with idiopathic Parkinson’s disease who were able to stand on their own and walk with or without an assistive device. The trial was conducted at 4 community rehabilitation centers in Hong Kong between December 1, 2016, and May 31, 2017. Participants were randomized to 8 weeks of mindfulness yoga delivered weekly in 90-minute group sessions (71) or stretching and resistance training delivered in weekly 60-minute group sessions (67).
The primary outcomes was psychological distress in terms of anxiety and depressive symptoms assessed with the Hospital Anxiety and Depression Scale (HADS). Secondary outcomes included motor symptom severity, mobility, spiritual well-being in terms of perceived hardship and equanimity, and health-related quality of life. The researchers assessed patients at baseline, 8 weeks, and 20 weeks.
The average age of the participants was 63.7 years; 65 (47.1%) were men. Generalized estimating equation analyses found that patients in the yoga group had significantly better outcomes, including for anxiety (time-by-group interaction, beta, –1.79 at 8 weeks and –2.05 at 20 weeks), and depressive symptoms (beta, –2.75 at 8 weeks and –2.75 at 20 weeks). These improvements were considered “statistically and clinically significant, the authors wrote. There were no significant improvements in anxiety or depressive symptoms in the stretching and resistance training group at the different time points.
Outcomes in the yoga group were also better with regards to disease-specific health-related quality of life (beta, –7.77 at 8 weeks and –7.99 at 20 weeks). Those who were in the mindfulness yoga group also had greater improvements in measures of perceived hardship and equanimity, compared with the stretching and resistance training group.
Referring to the improved psychological outcomes in the yoga group, the authors wrote, “these benefits were remarkable because the participants who received the [mindfulness yoga] intervention attended a mean of only 6 sessions.”
There were significant reductions in motor symptoms in both groups, which were significantly higher among those undergoing stretching, but the differences in the mean scores between the two groups were “clinically insignificant,” they wrote.
Three participants in the yoga group and 2 in the control group reported temporary mild knee pain. No serious adverse events were reported.
Expectation bias, selection bias, and the dropout rates of 15.2% at 8 weeks and 18.8% at 20 weeks are limitations of the study, the authors noted.
“These findings suggest that ,” Dr. Kwok and her colleagues concluded. “Considering that PD is not only a physically limiting condition but also a psychologically distressing life event, health care professionals should adopt a holistic approach in PD rehabilitation. Future rehabilitation programs could consider integrating mindfulness skills into physical therapy to enhance the holistic well-being of people with neurodegenerative conditions.”
The trial was supported by the Professional Development Fund of the Association of Hong Kong Nursing Staff. The authors had no disclosures.
SOURCE: Kwok JYY et al. JAMA Neurol. 2019 Apr 8. doi: 10.1001/jamaneurol.2019.0534.
Among patients with mild or moderate Parkinson’s disease, mindfulness yoga was as effective as stretching and resistance training in improving motor function and mobility, a randomized trial found.
In addition, mindfulness yoga reduced anxiety and depressive symptoms and increased spiritual well-being and health-related quality of life more than stretching and resistance training, researchers reported in JAMA Neurology.
Although guidelines support exercise for patients with Parkinson’s disease, investigators had not examined whether yoga is superior to conventional exercise for stress and symptom management in this patient population. Jojo Y. Y. Kwok, PhD, a research assistant professor of nursing at the University of Hong Kong, and her colleagues conducted an assessor-masked, randomized trial that included 138 adults with idiopathic Parkinson’s disease who were able to stand on their own and walk with or without an assistive device. The trial was conducted at 4 community rehabilitation centers in Hong Kong between December 1, 2016, and May 31, 2017. Participants were randomized to 8 weeks of mindfulness yoga delivered weekly in 90-minute group sessions (71) or stretching and resistance training delivered in weekly 60-minute group sessions (67).
The primary outcomes was psychological distress in terms of anxiety and depressive symptoms assessed with the Hospital Anxiety and Depression Scale (HADS). Secondary outcomes included motor symptom severity, mobility, spiritual well-being in terms of perceived hardship and equanimity, and health-related quality of life. The researchers assessed patients at baseline, 8 weeks, and 20 weeks.
The average age of the participants was 63.7 years; 65 (47.1%) were men. Generalized estimating equation analyses found that patients in the yoga group had significantly better outcomes, including for anxiety (time-by-group interaction, beta, –1.79 at 8 weeks and –2.05 at 20 weeks), and depressive symptoms (beta, –2.75 at 8 weeks and –2.75 at 20 weeks). These improvements were considered “statistically and clinically significant, the authors wrote. There were no significant improvements in anxiety or depressive symptoms in the stretching and resistance training group at the different time points.
Outcomes in the yoga group were also better with regards to disease-specific health-related quality of life (beta, –7.77 at 8 weeks and –7.99 at 20 weeks). Those who were in the mindfulness yoga group also had greater improvements in measures of perceived hardship and equanimity, compared with the stretching and resistance training group.
Referring to the improved psychological outcomes in the yoga group, the authors wrote, “these benefits were remarkable because the participants who received the [mindfulness yoga] intervention attended a mean of only 6 sessions.”
There were significant reductions in motor symptoms in both groups, which were significantly higher among those undergoing stretching, but the differences in the mean scores between the two groups were “clinically insignificant,” they wrote.
Three participants in the yoga group and 2 in the control group reported temporary mild knee pain. No serious adverse events were reported.
Expectation bias, selection bias, and the dropout rates of 15.2% at 8 weeks and 18.8% at 20 weeks are limitations of the study, the authors noted.
“These findings suggest that ,” Dr. Kwok and her colleagues concluded. “Considering that PD is not only a physically limiting condition but also a psychologically distressing life event, health care professionals should adopt a holistic approach in PD rehabilitation. Future rehabilitation programs could consider integrating mindfulness skills into physical therapy to enhance the holistic well-being of people with neurodegenerative conditions.”
The trial was supported by the Professional Development Fund of the Association of Hong Kong Nursing Staff. The authors had no disclosures.
SOURCE: Kwok JYY et al. JAMA Neurol. 2019 Apr 8. doi: 10.1001/jamaneurol.2019.0534.
Among patients with mild or moderate Parkinson’s disease, mindfulness yoga was as effective as stretching and resistance training in improving motor function and mobility, a randomized trial found.
In addition, mindfulness yoga reduced anxiety and depressive symptoms and increased spiritual well-being and health-related quality of life more than stretching and resistance training, researchers reported in JAMA Neurology.
Although guidelines support exercise for patients with Parkinson’s disease, investigators had not examined whether yoga is superior to conventional exercise for stress and symptom management in this patient population. Jojo Y. Y. Kwok, PhD, a research assistant professor of nursing at the University of Hong Kong, and her colleagues conducted an assessor-masked, randomized trial that included 138 adults with idiopathic Parkinson’s disease who were able to stand on their own and walk with or without an assistive device. The trial was conducted at 4 community rehabilitation centers in Hong Kong between December 1, 2016, and May 31, 2017. Participants were randomized to 8 weeks of mindfulness yoga delivered weekly in 90-minute group sessions (71) or stretching and resistance training delivered in weekly 60-minute group sessions (67).
The primary outcomes was psychological distress in terms of anxiety and depressive symptoms assessed with the Hospital Anxiety and Depression Scale (HADS). Secondary outcomes included motor symptom severity, mobility, spiritual well-being in terms of perceived hardship and equanimity, and health-related quality of life. The researchers assessed patients at baseline, 8 weeks, and 20 weeks.
The average age of the participants was 63.7 years; 65 (47.1%) were men. Generalized estimating equation analyses found that patients in the yoga group had significantly better outcomes, including for anxiety (time-by-group interaction, beta, –1.79 at 8 weeks and –2.05 at 20 weeks), and depressive symptoms (beta, –2.75 at 8 weeks and –2.75 at 20 weeks). These improvements were considered “statistically and clinically significant, the authors wrote. There were no significant improvements in anxiety or depressive symptoms in the stretching and resistance training group at the different time points.
Outcomes in the yoga group were also better with regards to disease-specific health-related quality of life (beta, –7.77 at 8 weeks and –7.99 at 20 weeks). Those who were in the mindfulness yoga group also had greater improvements in measures of perceived hardship and equanimity, compared with the stretching and resistance training group.
Referring to the improved psychological outcomes in the yoga group, the authors wrote, “these benefits were remarkable because the participants who received the [mindfulness yoga] intervention attended a mean of only 6 sessions.”
There were significant reductions in motor symptoms in both groups, which were significantly higher among those undergoing stretching, but the differences in the mean scores between the two groups were “clinically insignificant,” they wrote.
Three participants in the yoga group and 2 in the control group reported temporary mild knee pain. No serious adverse events were reported.
Expectation bias, selection bias, and the dropout rates of 15.2% at 8 weeks and 18.8% at 20 weeks are limitations of the study, the authors noted.
“These findings suggest that ,” Dr. Kwok and her colleagues concluded. “Considering that PD is not only a physically limiting condition but also a psychologically distressing life event, health care professionals should adopt a holistic approach in PD rehabilitation. Future rehabilitation programs could consider integrating mindfulness skills into physical therapy to enhance the holistic well-being of people with neurodegenerative conditions.”
The trial was supported by the Professional Development Fund of the Association of Hong Kong Nursing Staff. The authors had no disclosures.
SOURCE: Kwok JYY et al. JAMA Neurol. 2019 Apr 8. doi: 10.1001/jamaneurol.2019.0534.
FROM JAMA NEUROLOGY
Daily headaches • associated nausea • obesity • Dx?
THE CASE
A 22-year-old woman presented to our office complaining of headaches that started 6 weeks earlier. Initially the headache was throbbing, nonpositional, infrequent, and intermittent, lasting 15 to 45 minutes, often starting in the neck and migrating towards the right frontotemporal region. During the week prior to presentation, the headaches became daily and constant, with brief periods of relief after the patient took ibuprofen 400 mg 4 times a day as needed. The patient reported associated nausea, a sensation of pressure changes in the ears, and intermittent dimming of vision in the right eye (sometimes independent of headache). The patient denied photophobia and phonophobia. Her only medication was an oral contraceptive pill (OCP). She had no prior history of headaches.
Physical examination showed a blood pressure of 148/66 mm Hg, body mass index of 44.38, muscle tenderness in the neck and upper back, and no focal neurological findings. Funduscopic examination was unsuccessful. A working diagnosis of atypical migraine was made, but because of unilateral visual disturbance the patient was referred to Ophthalmology for further evaluation. The following day, ophthalmological consultation found bilateral papilledema and the patient was admitted to our hospitalist service via the Emergency Department. She subsequently was referred to inpatient Neurology.
THE DIAGNOSIS
Magnetic resonance imaging (MRI) of the brain and orbits with and without contrast was unremarkable. Magnetic resonance venography (MRV) with contrast of the brain showed possible stenosis at the junction of the transverse and sigmoid sinuses but no mass lesion nor venous sinus thrombosis. Lumbar puncture (LP) revealed an opening pressure of 650 mm H20 (reference range, 60–250 mm H2O).1 A diagnosis of idiopathic intracranial hypertension (IIH) was made.

DISCUSSION
IIH, previously known as pseudotumor cerebri and benign intracranial hypertension, is defined by signs and symptoms of elevated intracranial pressure (ICP) without obvious cause on neuroimaging (TABLE 12-5). It is well documented that IIH is consequential and can result in vision loss and intractable chronic headaches.5,6 Older terms such as pseudotumor cerebri and benign intracranial hypertension are therefore no longer recommended because they are considered misleading and not reflective of the severity of potential injury caused by the condition3,4,6 IIH is considered a diagnosis of exclusion requiring certain criteria to be met (TABLE 22). Although the etiology of IIH is unclear, associations have been made between IIH and various medications and conditions2-5,7 (TABLE 33,5).

Classically, IIH affects women who are obese and of childbearing age, but studies have shown that this condition also can affect men and children—albeit less frequently.3,5-7 The incidence of IIH in the general population is between 0.03 to 2.36/100,000 people per year, but in women, the incidence is 0.65 to 4.65/100,000 per year.6 Furthermore, females who are obese have an incidence of 2.7 to 19.3/100,000 per year.6

Headache is the most common symptom of IIH. Unfortunately, the differential diagnosis of headache is vast; thus, a careful history is needed to narrow the field3,5-7 (TABLE 42). Associated symptoms of transient visual changes, pulsatile tinnitus, neck and back pain, nausea, vomiting, photo/phonophobia, and findings of abducens nerve palsy or papilledema—while nonspecific— should raise suspicion for elevated ICP and IIH, especially in women who are obese.2-8 Once IIH is suspected, an urgent diagnosis and treatment is necessary to prevent permanent vision loss.3,4,6

Headache with findings of papilledema warrants neuroimaging, preferably with MRI, to rule out intracranial mass and hydrocephalus.1,2,5 MRV also is recommended to assess for intracranial venous thrombosis, an alternate cause for papilledema and increased ICP.1,2,4,5
Continue to: Recently, a classification of IIH...
Recently, a classification of IIH without papilledema has been acknowledged by the International Headache Society.2,8 Specific MRI findings have been suggested to help make this diagnosis5,9 (TABLE 55).

TREATMENT FOR IIH CAN BE MEDICAL OR SURGICAL
Medications associated with IIH should be discontinued.7 The first-line medication for IIH is acetazolamide, a carbonic anhydrase inhibitor that works in the choroid plexus to decrease cerebrospinal fluid (CSF) production and thus, lower ICP.3,6 An adult dose of 1 to 2 g/day3,4,6 is tolerated well, but can be increased to 4 g/day,10 if necessary. Weight loss via diet and exercise or bariatric surgery has been shown to be effective in patients who are obese and have been given a diagnosis of IIH.3,4
Topiramate also has been suggested as a treatment option, based on its usefulness in weight loss and because of its action as a weak carbonic anhydrase inhibitor.3,6 Also, LP has therapeutic merit—although relief is only short-term.3,6 Patients who fail medical therapy and have intractable headache or progressive visual loss appear to benefit from optic nerve sheath fenestration.3,7,8
Our patient experienced notable improvement in her headache after LP. Her OCP was discontinued, a diuretic regimen started, and weight loss counseling was provided. Prior to discharge, the patient was seen by a neuro-ophthalmologist for perimetry, a visual field test that assesses for acute vision loss and establishes a baseline for follow-up monitoring of vision.7
THE TAKEAWAY
Headache is a common condition that may be challenging to correctly diagnose. A thorough history and neurological examination, including fundoscopy, are essential in the evaluation of headache and suspected IIH. In the primary care setting, limited time, lack of mydriatic agents, suboptimal lighting, and practitioner inexperience may pose challenges for funduscopic examination. Ophthalmoscopes incorporating new technology to expand and magnify the examiner’s field of view may facilitate this exam.11 A global rise in the prevalence of obesity underscores a need for primary care providers to be compulsive about their clinical evaluation when symptoms suspicious of IIH are present. Lastly, if IIH cannot be ruled out confidently, recommend a prompt evaluation by an ophthalmologist.
CORRESPONDENCE
Aarti Paltoo, MD, MSc, CCFP, Peel Village Medical Center, 28 Rambler Drive, Brampton, Ontario L6W 1E2 Canada; [email protected]
1. Lee SC, Lueck CJ. Cerebrospinal fluid pressure in adults. J Neuroophthalmol. 2014;34:278-283.
2. International Headache Society. Idiopathic intracranial hypertension. The International Classification of Headache Disorders. 2nd ed. Oxford, UK: Blackwell Publishing; 2003:1-232.
3. Biousse V, Bruce BB, Newman NJ. Update on the pathophysiology and management of idiopathic intracranial hypertension. J Neurol Neurosurg Psychiatry. 2012;83:488-494.
4. Mollan SP, Markey KA, Benzimra JD, et al. A practical approach to diagnosis, assessment and management of idiopathic intracranial hypertension. Pract Neurol. 2014;14:380-390.
5. Friedman DI, Liu GT, Digre KB. Revised diagnostic criteria for the pseudotumor cerebri syndrome in adults and children. Neurology. 2013;81:1159-1165.
6. Julayanont P, Karukote A, Ruthirago D, et al. Idiopathic intracranial hypertension: ongoing clinical challenges and future prospects. J Pain Res. 2016;9:87-99.
7. Friedman DI, Digre KB. Headache medicine meets neuro-ophthalmology: exam techniques and challenging cases. Headache. 2013;53:703-716.
8. Digre KB, Nakamoto BK, Warner JE, et al. A comparison of idiopathic intracranaial hypertension with and without papilledema. Headache. 2009;49:185-193.
9. Digre KB. Imaging characteristics of IIH: are they reliable? Cephalagia. 2013;33:1067-1069.
10. Horton J. Acetazolamide for pseudotumor cerebri--evidence from the NORDIC trial. JAMA. 2014;311:1618-1619.
11. Petrushkin H, Barsam A, Mavrakakis M, et al. Optic disc assessment in the emergency department: a comparative study between the PanOptic and direct ophthalmoscopes. Emerg Med J. 2012;29:1007-1008.
THE CASE
A 22-year-old woman presented to our office complaining of headaches that started 6 weeks earlier. Initially the headache was throbbing, nonpositional, infrequent, and intermittent, lasting 15 to 45 minutes, often starting in the neck and migrating towards the right frontotemporal region. During the week prior to presentation, the headaches became daily and constant, with brief periods of relief after the patient took ibuprofen 400 mg 4 times a day as needed. The patient reported associated nausea, a sensation of pressure changes in the ears, and intermittent dimming of vision in the right eye (sometimes independent of headache). The patient denied photophobia and phonophobia. Her only medication was an oral contraceptive pill (OCP). She had no prior history of headaches.
Physical examination showed a blood pressure of 148/66 mm Hg, body mass index of 44.38, muscle tenderness in the neck and upper back, and no focal neurological findings. Funduscopic examination was unsuccessful. A working diagnosis of atypical migraine was made, but because of unilateral visual disturbance the patient was referred to Ophthalmology for further evaluation. The following day, ophthalmological consultation found bilateral papilledema and the patient was admitted to our hospitalist service via the Emergency Department. She subsequently was referred to inpatient Neurology.
THE DIAGNOSIS
Magnetic resonance imaging (MRI) of the brain and orbits with and without contrast was unremarkable. Magnetic resonance venography (MRV) with contrast of the brain showed possible stenosis at the junction of the transverse and sigmoid sinuses but no mass lesion nor venous sinus thrombosis. Lumbar puncture (LP) revealed an opening pressure of 650 mm H20 (reference range, 60–250 mm H2O).1 A diagnosis of idiopathic intracranial hypertension (IIH) was made.
DISCUSSION
IIH, previously known as pseudotumor cerebri and benign intracranial hypertension, is defined by signs and symptoms of elevated intracranial pressure (ICP) without obvious cause on neuroimaging (TABLE 12-5). It is well documented that IIH is consequential and can result in vision loss and intractable chronic headaches.5,6 Older terms such as pseudotumor cerebri and benign intracranial hypertension are therefore no longer recommended because they are considered misleading and not reflective of the severity of potential injury caused by the condition3,4,6 IIH is considered a diagnosis of exclusion requiring certain criteria to be met (TABLE 22). Although the etiology of IIH is unclear, associations have been made between IIH and various medications and conditions2-5,7 (TABLE 33,5).
Classically, IIH affects women who are obese and of childbearing age, but studies have shown that this condition also can affect men and children—albeit less frequently.3,5-7 The incidence of IIH in the general population is between 0.03 to 2.36/100,000 people per year, but in women, the incidence is 0.65 to 4.65/100,000 per year.6 Furthermore, females who are obese have an incidence of 2.7 to 19.3/100,000 per year.6
Headache is the most common symptom of IIH. Unfortunately, the differential diagnosis of headache is vast; thus, a careful history is needed to narrow the field3,5-7 (TABLE 42). Associated symptoms of transient visual changes, pulsatile tinnitus, neck and back pain, nausea, vomiting, photo/phonophobia, and findings of abducens nerve palsy or papilledema—while nonspecific— should raise suspicion for elevated ICP and IIH, especially in women who are obese.2-8 Once IIH is suspected, an urgent diagnosis and treatment is necessary to prevent permanent vision loss.3,4,6
Headache with findings of papilledema warrants neuroimaging, preferably with MRI, to rule out intracranial mass and hydrocephalus.1,2,5 MRV also is recommended to assess for intracranial venous thrombosis, an alternate cause for papilledema and increased ICP.1,2,4,5
Continue to: Recently, a classification of IIH...
Recently, a classification of IIH without papilledema has been acknowledged by the International Headache Society.2,8 Specific MRI findings have been suggested to help make this diagnosis5,9 (TABLE 55).
TREATMENT FOR IIH CAN BE MEDICAL OR SURGICAL
Medications associated with IIH should be discontinued.7 The first-line medication for IIH is acetazolamide, a carbonic anhydrase inhibitor that works in the choroid plexus to decrease cerebrospinal fluid (CSF) production and thus, lower ICP.3,6 An adult dose of 1 to 2 g/day3,4,6 is tolerated well, but can be increased to 4 g/day,10 if necessary. Weight loss via diet and exercise or bariatric surgery has been shown to be effective in patients who are obese and have been given a diagnosis of IIH.3,4
Topiramate also has been suggested as a treatment option, based on its usefulness in weight loss and because of its action as a weak carbonic anhydrase inhibitor.3,6 Also, LP has therapeutic merit—although relief is only short-term.3,6 Patients who fail medical therapy and have intractable headache or progressive visual loss appear to benefit from optic nerve sheath fenestration.3,7,8
Our patient experienced notable improvement in her headache after LP. Her OCP was discontinued, a diuretic regimen started, and weight loss counseling was provided. Prior to discharge, the patient was seen by a neuro-ophthalmologist for perimetry, a visual field test that assesses for acute vision loss and establishes a baseline for follow-up monitoring of vision.7
THE TAKEAWAY
Headache is a common condition that may be challenging to correctly diagnose. A thorough history and neurological examination, including fundoscopy, are essential in the evaluation of headache and suspected IIH. In the primary care setting, limited time, lack of mydriatic agents, suboptimal lighting, and practitioner inexperience may pose challenges for funduscopic examination. Ophthalmoscopes incorporating new technology to expand and magnify the examiner’s field of view may facilitate this exam.11 A global rise in the prevalence of obesity underscores a need for primary care providers to be compulsive about their clinical evaluation when symptoms suspicious of IIH are present. Lastly, if IIH cannot be ruled out confidently, recommend a prompt evaluation by an ophthalmologist.
CORRESPONDENCE
Aarti Paltoo, MD, MSc, CCFP, Peel Village Medical Center, 28 Rambler Drive, Brampton, Ontario L6W 1E2 Canada; [email protected]
THE CASE
A 22-year-old woman presented to our office complaining of headaches that started 6 weeks earlier. Initially the headache was throbbing, nonpositional, infrequent, and intermittent, lasting 15 to 45 minutes, often starting in the neck and migrating towards the right frontotemporal region. During the week prior to presentation, the headaches became daily and constant, with brief periods of relief after the patient took ibuprofen 400 mg 4 times a day as needed. The patient reported associated nausea, a sensation of pressure changes in the ears, and intermittent dimming of vision in the right eye (sometimes independent of headache). The patient denied photophobia and phonophobia. Her only medication was an oral contraceptive pill (OCP). She had no prior history of headaches.
Physical examination showed a blood pressure of 148/66 mm Hg, body mass index of 44.38, muscle tenderness in the neck and upper back, and no focal neurological findings. Funduscopic examination was unsuccessful. A working diagnosis of atypical migraine was made, but because of unilateral visual disturbance the patient was referred to Ophthalmology for further evaluation. The following day, ophthalmological consultation found bilateral papilledema and the patient was admitted to our hospitalist service via the Emergency Department. She subsequently was referred to inpatient Neurology.
THE DIAGNOSIS
Magnetic resonance imaging (MRI) of the brain and orbits with and without contrast was unremarkable. Magnetic resonance venography (MRV) with contrast of the brain showed possible stenosis at the junction of the transverse and sigmoid sinuses but no mass lesion nor venous sinus thrombosis. Lumbar puncture (LP) revealed an opening pressure of 650 mm H20 (reference range, 60–250 mm H2O).1 A diagnosis of idiopathic intracranial hypertension (IIH) was made.
DISCUSSION
IIH, previously known as pseudotumor cerebri and benign intracranial hypertension, is defined by signs and symptoms of elevated intracranial pressure (ICP) without obvious cause on neuroimaging (TABLE 12-5). It is well documented that IIH is consequential and can result in vision loss and intractable chronic headaches.5,6 Older terms such as pseudotumor cerebri and benign intracranial hypertension are therefore no longer recommended because they are considered misleading and not reflective of the severity of potential injury caused by the condition3,4,6 IIH is considered a diagnosis of exclusion requiring certain criteria to be met (TABLE 22). Although the etiology of IIH is unclear, associations have been made between IIH and various medications and conditions2-5,7 (TABLE 33,5).
Classically, IIH affects women who are obese and of childbearing age, but studies have shown that this condition also can affect men and children—albeit less frequently.3,5-7 The incidence of IIH in the general population is between 0.03 to 2.36/100,000 people per year, but in women, the incidence is 0.65 to 4.65/100,000 per year.6 Furthermore, females who are obese have an incidence of 2.7 to 19.3/100,000 per year.6
Headache is the most common symptom of IIH. Unfortunately, the differential diagnosis of headache is vast; thus, a careful history is needed to narrow the field3,5-7 (TABLE 42). Associated symptoms of transient visual changes, pulsatile tinnitus, neck and back pain, nausea, vomiting, photo/phonophobia, and findings of abducens nerve palsy or papilledema—while nonspecific— should raise suspicion for elevated ICP and IIH, especially in women who are obese.2-8 Once IIH is suspected, an urgent diagnosis and treatment is necessary to prevent permanent vision loss.3,4,6
Headache with findings of papilledema warrants neuroimaging, preferably with MRI, to rule out intracranial mass and hydrocephalus.1,2,5 MRV also is recommended to assess for intracranial venous thrombosis, an alternate cause for papilledema and increased ICP.1,2,4,5
Continue to: Recently, a classification of IIH...
Recently, a classification of IIH without papilledema has been acknowledged by the International Headache Society.2,8 Specific MRI findings have been suggested to help make this diagnosis5,9 (TABLE 55).
TREATMENT FOR IIH CAN BE MEDICAL OR SURGICAL
Medications associated with IIH should be discontinued.7 The first-line medication for IIH is acetazolamide, a carbonic anhydrase inhibitor that works in the choroid plexus to decrease cerebrospinal fluid (CSF) production and thus, lower ICP.3,6 An adult dose of 1 to 2 g/day3,4,6 is tolerated well, but can be increased to 4 g/day,10 if necessary. Weight loss via diet and exercise or bariatric surgery has been shown to be effective in patients who are obese and have been given a diagnosis of IIH.3,4
Topiramate also has been suggested as a treatment option, based on its usefulness in weight loss and because of its action as a weak carbonic anhydrase inhibitor.3,6 Also, LP has therapeutic merit—although relief is only short-term.3,6 Patients who fail medical therapy and have intractable headache or progressive visual loss appear to benefit from optic nerve sheath fenestration.3,7,8
Our patient experienced notable improvement in her headache after LP. Her OCP was discontinued, a diuretic regimen started, and weight loss counseling was provided. Prior to discharge, the patient was seen by a neuro-ophthalmologist for perimetry, a visual field test that assesses for acute vision loss and establishes a baseline for follow-up monitoring of vision.7
THE TAKEAWAY
Headache is a common condition that may be challenging to correctly diagnose. A thorough history and neurological examination, including fundoscopy, are essential in the evaluation of headache and suspected IIH. In the primary care setting, limited time, lack of mydriatic agents, suboptimal lighting, and practitioner inexperience may pose challenges for funduscopic examination. Ophthalmoscopes incorporating new technology to expand and magnify the examiner’s field of view may facilitate this exam.11 A global rise in the prevalence of obesity underscores a need for primary care providers to be compulsive about their clinical evaluation when symptoms suspicious of IIH are present. Lastly, if IIH cannot be ruled out confidently, recommend a prompt evaluation by an ophthalmologist.
CORRESPONDENCE
Aarti Paltoo, MD, MSc, CCFP, Peel Village Medical Center, 28 Rambler Drive, Brampton, Ontario L6W 1E2 Canada; [email protected]
1. Lee SC, Lueck CJ. Cerebrospinal fluid pressure in adults. J Neuroophthalmol. 2014;34:278-283.
2. International Headache Society. Idiopathic intracranial hypertension. The International Classification of Headache Disorders. 2nd ed. Oxford, UK: Blackwell Publishing; 2003:1-232.
3. Biousse V, Bruce BB, Newman NJ. Update on the pathophysiology and management of idiopathic intracranial hypertension. J Neurol Neurosurg Psychiatry. 2012;83:488-494.
4. Mollan SP, Markey KA, Benzimra JD, et al. A practical approach to diagnosis, assessment and management of idiopathic intracranial hypertension. Pract Neurol. 2014;14:380-390.
5. Friedman DI, Liu GT, Digre KB. Revised diagnostic criteria for the pseudotumor cerebri syndrome in adults and children. Neurology. 2013;81:1159-1165.
6. Julayanont P, Karukote A, Ruthirago D, et al. Idiopathic intracranial hypertension: ongoing clinical challenges and future prospects. J Pain Res. 2016;9:87-99.
7. Friedman DI, Digre KB. Headache medicine meets neuro-ophthalmology: exam techniques and challenging cases. Headache. 2013;53:703-716.
8. Digre KB, Nakamoto BK, Warner JE, et al. A comparison of idiopathic intracranaial hypertension with and without papilledema. Headache. 2009;49:185-193.
9. Digre KB. Imaging characteristics of IIH: are they reliable? Cephalagia. 2013;33:1067-1069.
10. Horton J. Acetazolamide for pseudotumor cerebri--evidence from the NORDIC trial. JAMA. 2014;311:1618-1619.
11. Petrushkin H, Barsam A, Mavrakakis M, et al. Optic disc assessment in the emergency department: a comparative study between the PanOptic and direct ophthalmoscopes. Emerg Med J. 2012;29:1007-1008.
1. Lee SC, Lueck CJ. Cerebrospinal fluid pressure in adults. J Neuroophthalmol. 2014;34:278-283.
2. International Headache Society. Idiopathic intracranial hypertension. The International Classification of Headache Disorders. 2nd ed. Oxford, UK: Blackwell Publishing; 2003:1-232.
3. Biousse V, Bruce BB, Newman NJ. Update on the pathophysiology and management of idiopathic intracranial hypertension. J Neurol Neurosurg Psychiatry. 2012;83:488-494.
4. Mollan SP, Markey KA, Benzimra JD, et al. A practical approach to diagnosis, assessment and management of idiopathic intracranial hypertension. Pract Neurol. 2014;14:380-390.
5. Friedman DI, Liu GT, Digre KB. Revised diagnostic criteria for the pseudotumor cerebri syndrome in adults and children. Neurology. 2013;81:1159-1165.
6. Julayanont P, Karukote A, Ruthirago D, et al. Idiopathic intracranial hypertension: ongoing clinical challenges and future prospects. J Pain Res. 2016;9:87-99.
7. Friedman DI, Digre KB. Headache medicine meets neuro-ophthalmology: exam techniques and challenging cases. Headache. 2013;53:703-716.
8. Digre KB, Nakamoto BK, Warner JE, et al. A comparison of idiopathic intracranaial hypertension with and without papilledema. Headache. 2009;49:185-193.
9. Digre KB. Imaging characteristics of IIH: are they reliable? Cephalagia. 2013;33:1067-1069.
10. Horton J. Acetazolamide for pseudotumor cerebri--evidence from the NORDIC trial. JAMA. 2014;311:1618-1619.
11. Petrushkin H, Barsam A, Mavrakakis M, et al. Optic disc assessment in the emergency department: a comparative study between the PanOptic and direct ophthalmoscopes. Emerg Med J. 2012;29:1007-1008.
