User login
Sick, or faking it?
CASE Vague symptoms; no clear etiology
Mr. W, age 53, presents to the emergency department (ED) describing acute mid-sternal chest pain (severity: 8 out of 10). His medical history is significant for pulmonary embolism and ascending aortic aneurysm in the context of Takayasu’s arteritis, an inflammatory condition of the large arterial blood vessels characterized by lesions that can lead to vascular stenosis, occlusion, or aneurysm. Takayasu’s arteritis is also known as pulseless disease due to the weak or absent pulses the condition produces.
A review of Mr. W’s medical records reveals that this is his 23rd visit to this hospital within a year; the year before that, he had 22 visits. At each of these previous visits, he had similar vague symptoms, including dizziness, chest pain, lightheadedness, fainting, bilateral knee weakness, and left-arm numbness/weakness, and no clear acute etiology for his reported symptoms. Each time, after the treating clinicians ruled out possible acute complications of a flare-up of Takayasu’s arteritis through a physical examination, laboratory tests, and imaging studies, Mr. W was discharged with recommendations that he follow-up with his primary care physician and specialists. At each discharge, he would leave the hospital with hesitation.
At this present visit, the ED physician recognizes Mr. W as someone who visits the ED often with no profound acute issues, and reviews the substantial medical records available to the hospital. He suspects Mr. W is feigning symptoms, and orders a psychiatric consultation.
EVALUATION Psychiatric interview and mental status exam
On examination, Mr. W is not in acute distress. Despite reporting an 8 out of 10 for chest pain severity, he displays no psychomotor agitation, and his pulse rate and blood pressure are within normal limits. He makes appropriate eye contact and describes his mood as “great.” He reports no problems with sleep, appetite, or disinterest in pleasurable activities, and denies being depressed or having any symptoms consistent with a mood disorder, anxiety disorder, or psychosis. He denies a history of panic attacks or excessive worrying that interferes with his sleep or activities of daily living. Additionally, Mr. W describes a stable, peaceful, and stress-free life within the limitations of his Takayasu’s arteritis, which he has been managing well since his diagnosis 6 years earlier.
Mr. W denies having any psychiatric symptoms, apprehensive feelings, or beliefs/fears that would be considered delusional, and he has no previous legal issues aside from an occasional driving citation. During the assessment, his affect remains broad and he denies having thoughts of suicide or homicide, or auditory or visual hallucinations.
Mr. W’s drug screen results are negative, and he denies using any illicit drugs. He uses only the medications that are prescribed by his clinicians. Overall, he seems to be a well-functioning individual. Mr. W reports that work is generally not stressful.
When the psychiatric team asks him about his frequent hospitalizations and ED visits, Mr. W is insistent that he is “just doing what my doctors said for me to do.” He repeats that he does not have any mental illness and did not see the point of seeing a psychiatrist.
")
In pursuit of collateral information, the psychiatry team accesses a regional medical record database that allows registered medical institutions and practices to track patients’ medical encounters within the region. According to this database, within approximately 5.5 years, Mr. W had 163 clinical encounters (ED visits and inpatient admissions) and 376 radiological studies in our region (Table 1 and Table 2).

[polldaddy:10394110]
Continue to: The authors' observations
The authors’ observations
The psychiatry team’s investigation of Mr. W’s medical records revealed the extent of his care-seeking behavior, and provided evidence for a diagnosis of factitious disorder.
Factitious disorder is an elusive psychiatric condition in which an individual chronically stimulates, induces, or aggravates illnesses to gain the status of being a patient. Although its exact cause has not been fully deciphered, it is seen mostly among individuals with knowledge of the workings of the medical field, such as a health care worker.1 Factitious disorder is taxing on the health care system, with an estimated cost in the thousands of dollars per patient visit.2 The condition has an estimated prevalence of 0.8% to 1.0% of patients seen by psychiatric consult services3 and is reported to be more prevalent among women than men.1 Its cardinal features include health care site hopping and hospital shopping, vagueness about the patient’s history and symptoms, and discrepancy among reported symptoms, the patient’s behaviors, and objective clinical findings.4,5 Although not all patients with factitious disorder have a legitimate medical reason for seeking care, some individuals with an established medical diagnosis use their condition as a tool to chronically seek care and play the sick role.
Factitious disorder should not be confused with malingering, which is differentiated by the patient’s search for a secondary gain, such as financial reward or avoiding jail; or conversion disorder, which is marked by true physical or neurologic symptoms and clinical findings triggered by psychological stressors. Patients with factitious disorder usually are cooperative during hospital stays and resume their normal daily routine shortly after discharge.4 In this case, Mr. W denied any psychiatric symptoms, apprehensive feelings, or beliefs or fears that would be considered delusional. He had no previous or pending legal issues, which ruled out malingering to avoid legal repercussions.
Mr. W’s presentation was complicated by his Takayasu’s arteritis diagnosis. Because Takayasu’s arteritis has a serious list of potential complications, ED physicians have a low threshold for ordering diagnostic studies for a patient with Takayasu’s arteritis who presents with a chief complaint of chest pain. In other words, when a patient with this condition presents to an acute setting (such as the ED) with chest pain, his/her chief complaint is taken with extreme seriousness. Conventional angiography is the standard diagnostic tool for Takayasu’s arteritis; CT angiography and magnetic resonance angiography are used for monitoring the disease’s progression.6
[polldaddy:10394113]
The authors’ observations
Currently, there are no FDA-approved treatments for factitious disorder, and patients with this condition generally are resistant to psychiatric and/or psychological care when discovered and offered treatment.7 Among those who consent to psychiatric care, psychoeducation, or psychotherapy, which have shown some efficacy for the condition, the dropout rate is high.8
Continue to: Although the instinctive approach...
Although the instinctive approach is to confront the patient once the deception has been uncovered, expert recommendations are contradictory. Some recommend confrontation as part of a treatment protocol,8 while others advise against such an approach.9
Because of how often patients with factitious disorder seek medical care, secondary iatrogenic consequences are possible. For example, for years, Mr. W has been unknowingly exposing himself to the iatrogenic consequences of the cumulative effect of diagnostic imaging for years. In 3 years alone, Mr. W had undergone an average of 125 diagnostic imaging studies per year—with and without contrast—and many unnecessary rounds of treatment with steroids and other interventions known to have secondary iatrogenic consequences.10 Excessive radiation exposure is known to be carcinogenic over time,10 and excessive use of steroids is associated with weight gain, physical habitus changes, and increased risk of infections.11 In addition, the renal effects of the contrast materials from repeated imaging studies over so many years on Mr. W’s future kidney function are unknown.
TREATMENT Psychoeducation and referral for psychotherapy
We counsel Mr. W about factitious disorder and the risks of excessive hospitalizations, and refer him for follow-up at our local psychiatric clinic, as well as for individual psychotherapy. Mr. W is discharged because his medical work-up does not reveal any significant acute medical issues.
The authors’ observations
Because of the poor insight associated with factitious disorder and the limited treatment options available, a patient with factitious disorder is unlikely to enter psychiatric treatment on his/her own. The prognosis for a patient with factitious disorder remains poor unless the patient is forced into treatment. More intervention-focused research is needed to help improve outcomes for patients with factitious disorder.
OUTCOME Failure to follow up
Mr. W fails to attend individual psychotherapy as recommended. According to our regional record database, Mr. W continues to present to other EDs regularly.
Continue to: Bottom Line
Bottom Line
A patient with factitious disorder stimulates, induces, or aggravates illnesses to gain the status of being a patient. Treatment options include psychiatric care, psychoeducation, or psychotherapy. However, due to poor insight, a patient with factitious disorder is unlikely to enter psychiatric treatment on his/her own.
Related Resources
- Yates GP, Feldman MD. Factitious disorder: a systematic review of 455 cases in the professional literature. Gen Hosp Psychiatry. 2016;41:20-28.
- Galli S, Tatu L, Bogousslavsky J, et al. Conversion, factitious disorder and malingering: a distinct pattern or a continuum? Front Neurol Neurosci. 2018;42:72-80.
1. Krahn LE, Li H, O’Connor MK. Patients who strive to be ill: factitious disorder with physical symptoms. Am J Psychiatry. 2003;160(6):1163-1168.
2. Hoertel N, Lavaud P, Le Strat Y, et al. Estimated cost of a factitious disorder with 6-year follow-up. Psychiatry Res. 2012;200(2):1077-1078.
3. Sadock BJ, Sadock VA, Ruiz P. Psychosomatic medicine; factitious disorder. In: Pataki CS, Sussman N, eds. Synopsis of psychiatry: Behavioral sciences/clinical psychiatry. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:34-45.
4 . Savino AC, Fordtran JS. Factitious disease: clinical lessons from case studies at Baylor University Medical Center. Proc (Bayl Univ Med Cent). 2006;19(3):195-208.
5. Burnel A. Recognition and management of factitious disorder. Prescriber. 2015;26(21):37-39.
6. Duftner C, Dejaco C, Sepriano A, et al. Imaging in diagnosis, outcome prediction and monitoring of large vessel vasculitis: a systematic literature review and meta-analysis informing the EULAR recommendations. RMD Open. 2018;4(1):e000612. doi: 10.1136/rmdopen-2017-000612.
7. Jafferany M, Khalid Z, McDonald KA, et al. Psychological aspects of factitious disorder. Prim Care Companion CNS Disord. 2018;20(1). doi: 10.4088/PCC.17nr02229.
8. Bolat N, Yalçin O. Factitious disorder presenting with stuttering in two adolescents: the importance of psychoeducation. Noro Psikiyatri Arsivi. 2017;54(1):87-89.
9. Eisendrath SJ. Factitious physical disorders. West J Med. 1994;160(2):177-179.
10. Sodickson A, Baeyens PF, Andriole KP, et al. Recurrent CT, cumulative radiation exposure, and associated radiation-induced cancer risks from CT of adults. Radiology. 2009;251(1):175-184.
11. Oray M, Abu Samra K, Ebrahimiadib N, et al. Long-term side effects of glucocorticoids. Expert Opin Drug Saf. 2016;15(4):457-465.
CASE Vague symptoms; no clear etiology
Mr. W, age 53, presents to the emergency department (ED) describing acute mid-sternal chest pain (severity: 8 out of 10). His medical history is significant for pulmonary embolism and ascending aortic aneurysm in the context of Takayasu’s arteritis, an inflammatory condition of the large arterial blood vessels characterized by lesions that can lead to vascular stenosis, occlusion, or aneurysm. Takayasu’s arteritis is also known as pulseless disease due to the weak or absent pulses the condition produces.
A review of Mr. W’s medical records reveals that this is his 23rd visit to this hospital within a year; the year before that, he had 22 visits. At each of these previous visits, he had similar vague symptoms, including dizziness, chest pain, lightheadedness, fainting, bilateral knee weakness, and left-arm numbness/weakness, and no clear acute etiology for his reported symptoms. Each time, after the treating clinicians ruled out possible acute complications of a flare-up of Takayasu’s arteritis through a physical examination, laboratory tests, and imaging studies, Mr. W was discharged with recommendations that he follow-up with his primary care physician and specialists. At each discharge, he would leave the hospital with hesitation.
At this present visit, the ED physician recognizes Mr. W as someone who visits the ED often with no profound acute issues, and reviews the substantial medical records available to the hospital. He suspects Mr. W is feigning symptoms, and orders a psychiatric consultation.
EVALUATION Psychiatric interview and mental status exam
On examination, Mr. W is not in acute distress. Despite reporting an 8 out of 10 for chest pain severity, he displays no psychomotor agitation, and his pulse rate and blood pressure are within normal limits. He makes appropriate eye contact and describes his mood as “great.” He reports no problems with sleep, appetite, or disinterest in pleasurable activities, and denies being depressed or having any symptoms consistent with a mood disorder, anxiety disorder, or psychosis. He denies a history of panic attacks or excessive worrying that interferes with his sleep or activities of daily living. Additionally, Mr. W describes a stable, peaceful, and stress-free life within the limitations of his Takayasu’s arteritis, which he has been managing well since his diagnosis 6 years earlier.
Mr. W denies having any psychiatric symptoms, apprehensive feelings, or beliefs/fears that would be considered delusional, and he has no previous legal issues aside from an occasional driving citation. During the assessment, his affect remains broad and he denies having thoughts of suicide or homicide, or auditory or visual hallucinations.
Mr. W’s drug screen results are negative, and he denies using any illicit drugs. He uses only the medications that are prescribed by his clinicians. Overall, he seems to be a well-functioning individual. Mr. W reports that work is generally not stressful.
When the psychiatric team asks him about his frequent hospitalizations and ED visits, Mr. W is insistent that he is “just doing what my doctors said for me to do.” He repeats that he does not have any mental illness and did not see the point of seeing a psychiatrist.
In pursuit of collateral information, the psychiatry team accesses a regional medical record database that allows registered medical institutions and practices to track patients’ medical encounters within the region. According to this database, within approximately 5.5 years, Mr. W had 163 clinical encounters (ED visits and inpatient admissions) and 376 radiological studies in our region (Table 1 and Table 2).
[polldaddy:10394110]
Continue to: The authors' observations
The authors’ observations
The psychiatry team’s investigation of Mr. W’s medical records revealed the extent of his care-seeking behavior, and provided evidence for a diagnosis of factitious disorder.
Factitious disorder is an elusive psychiatric condition in which an individual chronically stimulates, induces, or aggravates illnesses to gain the status of being a patient. Although its exact cause has not been fully deciphered, it is seen mostly among individuals with knowledge of the workings of the medical field, such as a health care worker.1 Factitious disorder is taxing on the health care system, with an estimated cost in the thousands of dollars per patient visit.2 The condition has an estimated prevalence of 0.8% to 1.0% of patients seen by psychiatric consult services3 and is reported to be more prevalent among women than men.1 Its cardinal features include health care site hopping and hospital shopping, vagueness about the patient’s history and symptoms, and discrepancy among reported symptoms, the patient’s behaviors, and objective clinical findings.4,5 Although not all patients with factitious disorder have a legitimate medical reason for seeking care, some individuals with an established medical diagnosis use their condition as a tool to chronically seek care and play the sick role.
Factitious disorder should not be confused with malingering, which is differentiated by the patient’s search for a secondary gain, such as financial reward or avoiding jail; or conversion disorder, which is marked by true physical or neurologic symptoms and clinical findings triggered by psychological stressors. Patients with factitious disorder usually are cooperative during hospital stays and resume their normal daily routine shortly after discharge.4 In this case, Mr. W denied any psychiatric symptoms, apprehensive feelings, or beliefs or fears that would be considered delusional. He had no previous or pending legal issues, which ruled out malingering to avoid legal repercussions.
Mr. W’s presentation was complicated by his Takayasu’s arteritis diagnosis. Because Takayasu’s arteritis has a serious list of potential complications, ED physicians have a low threshold for ordering diagnostic studies for a patient with Takayasu’s arteritis who presents with a chief complaint of chest pain. In other words, when a patient with this condition presents to an acute setting (such as the ED) with chest pain, his/her chief complaint is taken with extreme seriousness. Conventional angiography is the standard diagnostic tool for Takayasu’s arteritis; CT angiography and magnetic resonance angiography are used for monitoring the disease’s progression.6
[polldaddy:10394113]
The authors’ observations
Currently, there are no FDA-approved treatments for factitious disorder, and patients with this condition generally are resistant to psychiatric and/or psychological care when discovered and offered treatment.7 Among those who consent to psychiatric care, psychoeducation, or psychotherapy, which have shown some efficacy for the condition, the dropout rate is high.8
Continue to: Although the instinctive approach...
Although the instinctive approach is to confront the patient once the deception has been uncovered, expert recommendations are contradictory. Some recommend confrontation as part of a treatment protocol,8 while others advise against such an approach.9
Because of how often patients with factitious disorder seek medical care, secondary iatrogenic consequences are possible. For example, for years, Mr. W has been unknowingly exposing himself to the iatrogenic consequences of the cumulative effect of diagnostic imaging for years. In 3 years alone, Mr. W had undergone an average of 125 diagnostic imaging studies per year—with and without contrast—and many unnecessary rounds of treatment with steroids and other interventions known to have secondary iatrogenic consequences.10 Excessive radiation exposure is known to be carcinogenic over time,10 and excessive use of steroids is associated with weight gain, physical habitus changes, and increased risk of infections.11 In addition, the renal effects of the contrast materials from repeated imaging studies over so many years on Mr. W’s future kidney function are unknown.
TREATMENT Psychoeducation and referral for psychotherapy
We counsel Mr. W about factitious disorder and the risks of excessive hospitalizations, and refer him for follow-up at our local psychiatric clinic, as well as for individual psychotherapy. Mr. W is discharged because his medical work-up does not reveal any significant acute medical issues.
The authors’ observations
Because of the poor insight associated with factitious disorder and the limited treatment options available, a patient with factitious disorder is unlikely to enter psychiatric treatment on his/her own. The prognosis for a patient with factitious disorder remains poor unless the patient is forced into treatment. More intervention-focused research is needed to help improve outcomes for patients with factitious disorder.
OUTCOME Failure to follow up
Mr. W fails to attend individual psychotherapy as recommended. According to our regional record database, Mr. W continues to present to other EDs regularly.
Continue to: Bottom Line
Bottom Line
A patient with factitious disorder stimulates, induces, or aggravates illnesses to gain the status of being a patient. Treatment options include psychiatric care, psychoeducation, or psychotherapy. However, due to poor insight, a patient with factitious disorder is unlikely to enter psychiatric treatment on his/her own.
Related Resources
- Yates GP, Feldman MD. Factitious disorder: a systematic review of 455 cases in the professional literature. Gen Hosp Psychiatry. 2016;41:20-28.
- Galli S, Tatu L, Bogousslavsky J, et al. Conversion, factitious disorder and malingering: a distinct pattern or a continuum? Front Neurol Neurosci. 2018;42:72-80.
CASE Vague symptoms; no clear etiology
Mr. W, age 53, presents to the emergency department (ED) describing acute mid-sternal chest pain (severity: 8 out of 10). His medical history is significant for pulmonary embolism and ascending aortic aneurysm in the context of Takayasu’s arteritis, an inflammatory condition of the large arterial blood vessels characterized by lesions that can lead to vascular stenosis, occlusion, or aneurysm. Takayasu’s arteritis is also known as pulseless disease due to the weak or absent pulses the condition produces.
A review of Mr. W’s medical records reveals that this is his 23rd visit to this hospital within a year; the year before that, he had 22 visits. At each of these previous visits, he had similar vague symptoms, including dizziness, chest pain, lightheadedness, fainting, bilateral knee weakness, and left-arm numbness/weakness, and no clear acute etiology for his reported symptoms. Each time, after the treating clinicians ruled out possible acute complications of a flare-up of Takayasu’s arteritis through a physical examination, laboratory tests, and imaging studies, Mr. W was discharged with recommendations that he follow-up with his primary care physician and specialists. At each discharge, he would leave the hospital with hesitation.
At this present visit, the ED physician recognizes Mr. W as someone who visits the ED often with no profound acute issues, and reviews the substantial medical records available to the hospital. He suspects Mr. W is feigning symptoms, and orders a psychiatric consultation.
EVALUATION Psychiatric interview and mental status exam
On examination, Mr. W is not in acute distress. Despite reporting an 8 out of 10 for chest pain severity, he displays no psychomotor agitation, and his pulse rate and blood pressure are within normal limits. He makes appropriate eye contact and describes his mood as “great.” He reports no problems with sleep, appetite, or disinterest in pleasurable activities, and denies being depressed or having any symptoms consistent with a mood disorder, anxiety disorder, or psychosis. He denies a history of panic attacks or excessive worrying that interferes with his sleep or activities of daily living. Additionally, Mr. W describes a stable, peaceful, and stress-free life within the limitations of his Takayasu’s arteritis, which he has been managing well since his diagnosis 6 years earlier.
Mr. W denies having any psychiatric symptoms, apprehensive feelings, or beliefs/fears that would be considered delusional, and he has no previous legal issues aside from an occasional driving citation. During the assessment, his affect remains broad and he denies having thoughts of suicide or homicide, or auditory or visual hallucinations.
Mr. W’s drug screen results are negative, and he denies using any illicit drugs. He uses only the medications that are prescribed by his clinicians. Overall, he seems to be a well-functioning individual. Mr. W reports that work is generally not stressful.
When the psychiatric team asks him about his frequent hospitalizations and ED visits, Mr. W is insistent that he is “just doing what my doctors said for me to do.” He repeats that he does not have any mental illness and did not see the point of seeing a psychiatrist.
In pursuit of collateral information, the psychiatry team accesses a regional medical record database that allows registered medical institutions and practices to track patients’ medical encounters within the region. According to this database, within approximately 5.5 years, Mr. W had 163 clinical encounters (ED visits and inpatient admissions) and 376 radiological studies in our region (Table 1 and Table 2).
[polldaddy:10394110]
Continue to: The authors' observations
The authors’ observations
The psychiatry team’s investigation of Mr. W’s medical records revealed the extent of his care-seeking behavior, and provided evidence for a diagnosis of factitious disorder.
Factitious disorder is an elusive psychiatric condition in which an individual chronically stimulates, induces, or aggravates illnesses to gain the status of being a patient. Although its exact cause has not been fully deciphered, it is seen mostly among individuals with knowledge of the workings of the medical field, such as a health care worker.1 Factitious disorder is taxing on the health care system, with an estimated cost in the thousands of dollars per patient visit.2 The condition has an estimated prevalence of 0.8% to 1.0% of patients seen by psychiatric consult services3 and is reported to be more prevalent among women than men.1 Its cardinal features include health care site hopping and hospital shopping, vagueness about the patient’s history and symptoms, and discrepancy among reported symptoms, the patient’s behaviors, and objective clinical findings.4,5 Although not all patients with factitious disorder have a legitimate medical reason for seeking care, some individuals with an established medical diagnosis use their condition as a tool to chronically seek care and play the sick role.
Factitious disorder should not be confused with malingering, which is differentiated by the patient’s search for a secondary gain, such as financial reward or avoiding jail; or conversion disorder, which is marked by true physical or neurologic symptoms and clinical findings triggered by psychological stressors. Patients with factitious disorder usually are cooperative during hospital stays and resume their normal daily routine shortly after discharge.4 In this case, Mr. W denied any psychiatric symptoms, apprehensive feelings, or beliefs or fears that would be considered delusional. He had no previous or pending legal issues, which ruled out malingering to avoid legal repercussions.
Mr. W’s presentation was complicated by his Takayasu’s arteritis diagnosis. Because Takayasu’s arteritis has a serious list of potential complications, ED physicians have a low threshold for ordering diagnostic studies for a patient with Takayasu’s arteritis who presents with a chief complaint of chest pain. In other words, when a patient with this condition presents to an acute setting (such as the ED) with chest pain, his/her chief complaint is taken with extreme seriousness. Conventional angiography is the standard diagnostic tool for Takayasu’s arteritis; CT angiography and magnetic resonance angiography are used for monitoring the disease’s progression.6
[polldaddy:10394113]
The authors’ observations
Currently, there are no FDA-approved treatments for factitious disorder, and patients with this condition generally are resistant to psychiatric and/or psychological care when discovered and offered treatment.7 Among those who consent to psychiatric care, psychoeducation, or psychotherapy, which have shown some efficacy for the condition, the dropout rate is high.8
Continue to: Although the instinctive approach...
Although the instinctive approach is to confront the patient once the deception has been uncovered, expert recommendations are contradictory. Some recommend confrontation as part of a treatment protocol,8 while others advise against such an approach.9
Because of how often patients with factitious disorder seek medical care, secondary iatrogenic consequences are possible. For example, for years, Mr. W has been unknowingly exposing himself to the iatrogenic consequences of the cumulative effect of diagnostic imaging for years. In 3 years alone, Mr. W had undergone an average of 125 diagnostic imaging studies per year—with and without contrast—and many unnecessary rounds of treatment with steroids and other interventions known to have secondary iatrogenic consequences.10 Excessive radiation exposure is known to be carcinogenic over time,10 and excessive use of steroids is associated with weight gain, physical habitus changes, and increased risk of infections.11 In addition, the renal effects of the contrast materials from repeated imaging studies over so many years on Mr. W’s future kidney function are unknown.
TREATMENT Psychoeducation and referral for psychotherapy
We counsel Mr. W about factitious disorder and the risks of excessive hospitalizations, and refer him for follow-up at our local psychiatric clinic, as well as for individual psychotherapy. Mr. W is discharged because his medical work-up does not reveal any significant acute medical issues.
The authors’ observations
Because of the poor insight associated with factitious disorder and the limited treatment options available, a patient with factitious disorder is unlikely to enter psychiatric treatment on his/her own. The prognosis for a patient with factitious disorder remains poor unless the patient is forced into treatment. More intervention-focused research is needed to help improve outcomes for patients with factitious disorder.
OUTCOME Failure to follow up
Mr. W fails to attend individual psychotherapy as recommended. According to our regional record database, Mr. W continues to present to other EDs regularly.
Continue to: Bottom Line
Bottom Line
A patient with factitious disorder stimulates, induces, or aggravates illnesses to gain the status of being a patient. Treatment options include psychiatric care, psychoeducation, or psychotherapy. However, due to poor insight, a patient with factitious disorder is unlikely to enter psychiatric treatment on his/her own.
Related Resources
- Yates GP, Feldman MD. Factitious disorder: a systematic review of 455 cases in the professional literature. Gen Hosp Psychiatry. 2016;41:20-28.
- Galli S, Tatu L, Bogousslavsky J, et al. Conversion, factitious disorder and malingering: a distinct pattern or a continuum? Front Neurol Neurosci. 2018;42:72-80.
1. Krahn LE, Li H, O’Connor MK. Patients who strive to be ill: factitious disorder with physical symptoms. Am J Psychiatry. 2003;160(6):1163-1168.
2. Hoertel N, Lavaud P, Le Strat Y, et al. Estimated cost of a factitious disorder with 6-year follow-up. Psychiatry Res. 2012;200(2):1077-1078.
3. Sadock BJ, Sadock VA, Ruiz P. Psychosomatic medicine; factitious disorder. In: Pataki CS, Sussman N, eds. Synopsis of psychiatry: Behavioral sciences/clinical psychiatry. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:34-45.
4 . Savino AC, Fordtran JS. Factitious disease: clinical lessons from case studies at Baylor University Medical Center. Proc (Bayl Univ Med Cent). 2006;19(3):195-208.
5. Burnel A. Recognition and management of factitious disorder. Prescriber. 2015;26(21):37-39.
6. Duftner C, Dejaco C, Sepriano A, et al. Imaging in diagnosis, outcome prediction and monitoring of large vessel vasculitis: a systematic literature review and meta-analysis informing the EULAR recommendations. RMD Open. 2018;4(1):e000612. doi: 10.1136/rmdopen-2017-000612.
7. Jafferany M, Khalid Z, McDonald KA, et al. Psychological aspects of factitious disorder. Prim Care Companion CNS Disord. 2018;20(1). doi: 10.4088/PCC.17nr02229.
8. Bolat N, Yalçin O. Factitious disorder presenting with stuttering in two adolescents: the importance of psychoeducation. Noro Psikiyatri Arsivi. 2017;54(1):87-89.
9. Eisendrath SJ. Factitious physical disorders. West J Med. 1994;160(2):177-179.
10. Sodickson A, Baeyens PF, Andriole KP, et al. Recurrent CT, cumulative radiation exposure, and associated radiation-induced cancer risks from CT of adults. Radiology. 2009;251(1):175-184.
11. Oray M, Abu Samra K, Ebrahimiadib N, et al. Long-term side effects of glucocorticoids. Expert Opin Drug Saf. 2016;15(4):457-465.
1. Krahn LE, Li H, O’Connor MK. Patients who strive to be ill: factitious disorder with physical symptoms. Am J Psychiatry. 2003;160(6):1163-1168.
2. Hoertel N, Lavaud P, Le Strat Y, et al. Estimated cost of a factitious disorder with 6-year follow-up. Psychiatry Res. 2012;200(2):1077-1078.
3. Sadock BJ, Sadock VA, Ruiz P. Psychosomatic medicine; factitious disorder. In: Pataki CS, Sussman N, eds. Synopsis of psychiatry: Behavioral sciences/clinical psychiatry. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:34-45.
4 . Savino AC, Fordtran JS. Factitious disease: clinical lessons from case studies at Baylor University Medical Center. Proc (Bayl Univ Med Cent). 2006;19(3):195-208.
5. Burnel A. Recognition and management of factitious disorder. Prescriber. 2015;26(21):37-39.
6. Duftner C, Dejaco C, Sepriano A, et al. Imaging in diagnosis, outcome prediction and monitoring of large vessel vasculitis: a systematic literature review and meta-analysis informing the EULAR recommendations. RMD Open. 2018;4(1):e000612. doi: 10.1136/rmdopen-2017-000612.
7. Jafferany M, Khalid Z, McDonald KA, et al. Psychological aspects of factitious disorder. Prim Care Companion CNS Disord. 2018;20(1). doi: 10.4088/PCC.17nr02229.
8. Bolat N, Yalçin O. Factitious disorder presenting with stuttering in two adolescents: the importance of psychoeducation. Noro Psikiyatri Arsivi. 2017;54(1):87-89.
9. Eisendrath SJ. Factitious physical disorders. West J Med. 1994;160(2):177-179.
10. Sodickson A, Baeyens PF, Andriole KP, et al. Recurrent CT, cumulative radiation exposure, and associated radiation-induced cancer risks from CT of adults. Radiology. 2009;251(1):175-184.
11. Oray M, Abu Samra K, Ebrahimiadib N, et al. Long-term side effects of glucocorticoids. Expert Opin Drug Saf. 2016;15(4):457-465.

Alopecia areata linked to mental health disorders
Alopecia areata is associated with greater frequency of mental health disorders, according to a new analysis of U.S. hospitalizations.
Specifically, the analysis found, alopecia areata patients are at risk for any mental health disorder, anxiety disorders, attention-deficit/hyperactivity disorder, dementia, mood disorders, personality disorders, and suicide or intentionally self-inflicted injury. The report was published in the Journal of the American Academy of Dermatology.
The researchers worked with 87,053,155 adult and child records from the 2002-2012 National Inpatient Sample, which represents 20% of U.S. hospitalizations.
Overall, 5,605 patients had alopecia areata, which was the secondary diagnosis more than 99% of the time. Compared with inpatients without alopecia areata, those with the disorder were more likely to be younger (42.2 vs. 47.9 years; P less than .0001), female (61.7% vs. 58.6%; P = .0297), and uninsured (8.1% vs. 5.5%; P less than .0001). In addition, inpatients with alopecia areata had a greater frequency of mental health disorders (32.8% vs. 20.0%; P less than .0001) and were more likely to have a primary mental health diagnosis (5.5% vs. 2.2%; P less than .0001), reported Vivek Singam of Northwestern University, Chicago, and his associates.
Among 15 mental health or classes of disorders examined, alopecia areata patients were at greater risk in 13 of them. The only exceptions were delirium/dementia/amnestic/cognitive disorders and disorders diagnosed in infancy, childhood, or adolescence.
Alopecia areata patients with a mental health disorder had a mean hospital stay of 6.0 days (95% confidence interval, 5.4.-6.6) and hospitalization cost of $11,907 (95% CI, $10,312-$13,503).
Previous studies had shown similar relationships. However, previous studies showed lower risk of alopecia areata and schizophrenia and no increased risk of ADHD, compared with the current study’s findings. The authors could offer no explanation for those differences.
The strengths of the current analysis include its use of a large-scale, nationally representative cohort and its large sample size, as well its inclusion of a broad range of mental health disorders. Because of its cross-sectional design, the study could not establish the temporal relationship between alopecia areata and mental health disorders.
It is unclear whether psychosocial stress might cause or exacerbate alopecia areata, or whether alopecia areata can lead to or worsen mental health disorders.
The researchers called for additional studies to understand this relationship and potential mechanisms.
The Agency for Healthcare Research and Quality and the Dermatology Foundation funded the study. The researchers declared having no conflicts of interest.
SOURCE: Singam V et al. J Am Acad Dermatol. 2018 Aug 6. doi: 10.1016/j.jaad.2018.07.044.
Alopecia areata is associated with greater frequency of mental health disorders, according to a new analysis of U.S. hospitalizations.
Specifically, the analysis found, alopecia areata patients are at risk for any mental health disorder, anxiety disorders, attention-deficit/hyperactivity disorder, dementia, mood disorders, personality disorders, and suicide or intentionally self-inflicted injury. The report was published in the Journal of the American Academy of Dermatology.
The researchers worked with 87,053,155 adult and child records from the 2002-2012 National Inpatient Sample, which represents 20% of U.S. hospitalizations.
Overall, 5,605 patients had alopecia areata, which was the secondary diagnosis more than 99% of the time. Compared with inpatients without alopecia areata, those with the disorder were more likely to be younger (42.2 vs. 47.9 years; P less than .0001), female (61.7% vs. 58.6%; P = .0297), and uninsured (8.1% vs. 5.5%; P less than .0001). In addition, inpatients with alopecia areata had a greater frequency of mental health disorders (32.8% vs. 20.0%; P less than .0001) and were more likely to have a primary mental health diagnosis (5.5% vs. 2.2%; P less than .0001), reported Vivek Singam of Northwestern University, Chicago, and his associates.
Among 15 mental health or classes of disorders examined, alopecia areata patients were at greater risk in 13 of them. The only exceptions were delirium/dementia/amnestic/cognitive disorders and disorders diagnosed in infancy, childhood, or adolescence.
Alopecia areata patients with a mental health disorder had a mean hospital stay of 6.0 days (95% confidence interval, 5.4.-6.6) and hospitalization cost of $11,907 (95% CI, $10,312-$13,503).
Previous studies had shown similar relationships. However, previous studies showed lower risk of alopecia areata and schizophrenia and no increased risk of ADHD, compared with the current study’s findings. The authors could offer no explanation for those differences.
The strengths of the current analysis include its use of a large-scale, nationally representative cohort and its large sample size, as well its inclusion of a broad range of mental health disorders. Because of its cross-sectional design, the study could not establish the temporal relationship between alopecia areata and mental health disorders.
It is unclear whether psychosocial stress might cause or exacerbate alopecia areata, or whether alopecia areata can lead to or worsen mental health disorders.
The researchers called for additional studies to understand this relationship and potential mechanisms.
The Agency for Healthcare Research and Quality and the Dermatology Foundation funded the study. The researchers declared having no conflicts of interest.
SOURCE: Singam V et al. J Am Acad Dermatol. 2018 Aug 6. doi: 10.1016/j.jaad.2018.07.044.
Alopecia areata is associated with greater frequency of mental health disorders, according to a new analysis of U.S. hospitalizations.
Specifically, the analysis found, alopecia areata patients are at risk for any mental health disorder, anxiety disorders, attention-deficit/hyperactivity disorder, dementia, mood disorders, personality disorders, and suicide or intentionally self-inflicted injury. The report was published in the Journal of the American Academy of Dermatology.
The researchers worked with 87,053,155 adult and child records from the 2002-2012 National Inpatient Sample, which represents 20% of U.S. hospitalizations.
Overall, 5,605 patients had alopecia areata, which was the secondary diagnosis more than 99% of the time. Compared with inpatients without alopecia areata, those with the disorder were more likely to be younger (42.2 vs. 47.9 years; P less than .0001), female (61.7% vs. 58.6%; P = .0297), and uninsured (8.1% vs. 5.5%; P less than .0001). In addition, inpatients with alopecia areata had a greater frequency of mental health disorders (32.8% vs. 20.0%; P less than .0001) and were more likely to have a primary mental health diagnosis (5.5% vs. 2.2%; P less than .0001), reported Vivek Singam of Northwestern University, Chicago, and his associates.
Among 15 mental health or classes of disorders examined, alopecia areata patients were at greater risk in 13 of them. The only exceptions were delirium/dementia/amnestic/cognitive disorders and disorders diagnosed in infancy, childhood, or adolescence.
Alopecia areata patients with a mental health disorder had a mean hospital stay of 6.0 days (95% confidence interval, 5.4.-6.6) and hospitalization cost of $11,907 (95% CI, $10,312-$13,503).
Previous studies had shown similar relationships. However, previous studies showed lower risk of alopecia areata and schizophrenia and no increased risk of ADHD, compared with the current study’s findings. The authors could offer no explanation for those differences.
The strengths of the current analysis include its use of a large-scale, nationally representative cohort and its large sample size, as well its inclusion of a broad range of mental health disorders. Because of its cross-sectional design, the study could not establish the temporal relationship between alopecia areata and mental health disorders.
It is unclear whether psychosocial stress might cause or exacerbate alopecia areata, or whether alopecia areata can lead to or worsen mental health disorders.
The researchers called for additional studies to understand this relationship and potential mechanisms.
The Agency for Healthcare Research and Quality and the Dermatology Foundation funded the study. The researchers declared having no conflicts of interest.
SOURCE: Singam V et al. J Am Acad Dermatol. 2018 Aug 6. doi: 10.1016/j.jaad.2018.07.044.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Key clinical point: Alopecia areata patients should be monitored closely for mental health disorders.
Major finding: Overall, 32.8% of hospitalized alopecia areata patients had a mental health disorder, compared with 20.0% of controls.
Study details: Retrospective analysis of 87,053,155 U.S. adults and children.
Disclosures: The Agency for Healthcare Research & Quality and the Dermatology Foundation funded the study. The researchers declared having no conflicts of interest.
Source: Singam V et al. J Am Acad Dermatol. 2018 Aug 6. doi: 10.1016/j.jaad.2018.07.044.
VIDEO: Delusional parasitosis? Try these real solutions
SAN DIEGO – The path to successful treatment of patients with imagined skin disorders is paved with compassion, according to John Koo, MD, a dermatologist and psychiatrist with the University of California at San Francisco.
When a patient presents with delusional parasitosis -- horror stories about imagined infestations of parasites or bugs – the key to successful treatment is a positive attitude and validation, not denial, Dr. Koo said in a presentation at the annual meeting of the American Academy of Dermatology.
"I cannot afford to go in (the exam room) with a long face," he said. "If I go in and I’m not looking happy, things can deteriorate quickly. So I make sure I go in with the biggest smile on my face like I'm meeting my favorite Hollywood star."
"When I say something like 'It's like a living hell, isn't it,' patients are really touched, he said. The patient’s response is typically 'You're the first dermatologist to understand what I'm going through.' You cannot endorse their delusion, but you can endorse their suffering."
In our video interview, Dr. Koo delved into techniques for the successful work-up and evaluation of patients with delusional parasitosis, the varying degrees of the condition, medications used for treatment, and the prospects for eventual drug-free relief.
Dr. Koo reports no relevant financial disclosures.
SAN DIEGO – The path to successful treatment of patients with imagined skin disorders is paved with compassion, according to John Koo, MD, a dermatologist and psychiatrist with the University of California at San Francisco.
When a patient presents with delusional parasitosis -- horror stories about imagined infestations of parasites or bugs – the key to successful treatment is a positive attitude and validation, not denial, Dr. Koo said in a presentation at the annual meeting of the American Academy of Dermatology.
"I cannot afford to go in (the exam room) with a long face," he said. "If I go in and I’m not looking happy, things can deteriorate quickly. So I make sure I go in with the biggest smile on my face like I'm meeting my favorite Hollywood star."
"When I say something like 'It's like a living hell, isn't it,' patients are really touched, he said. The patient’s response is typically 'You're the first dermatologist to understand what I'm going through.' You cannot endorse their delusion, but you can endorse their suffering."
In our video interview, Dr. Koo delved into techniques for the successful work-up and evaluation of patients with delusional parasitosis, the varying degrees of the condition, medications used for treatment, and the prospects for eventual drug-free relief.
Dr. Koo reports no relevant financial disclosures.
SAN DIEGO – The path to successful treatment of patients with imagined skin disorders is paved with compassion, according to John Koo, MD, a dermatologist and psychiatrist with the University of California at San Francisco.
When a patient presents with delusional parasitosis -- horror stories about imagined infestations of parasites or bugs – the key to successful treatment is a positive attitude and validation, not denial, Dr. Koo said in a presentation at the annual meeting of the American Academy of Dermatology.
"I cannot afford to go in (the exam room) with a long face," he said. "If I go in and I’m not looking happy, things can deteriorate quickly. So I make sure I go in with the biggest smile on my face like I'm meeting my favorite Hollywood star."
"When I say something like 'It's like a living hell, isn't it,' patients are really touched, he said. The patient’s response is typically 'You're the first dermatologist to understand what I'm going through.' You cannot endorse their delusion, but you can endorse their suffering."
In our video interview, Dr. Koo delved into techniques for the successful work-up and evaluation of patients with delusional parasitosis, the varying degrees of the condition, medications used for treatment, and the prospects for eventual drug-free relief.
Dr. Koo reports no relevant financial disclosures.
REPORTING FROM AAD 18

The puzzling relationship between cholesterol and psychopathology
Cholesterol generally is regarded as a cardiovascular risk factor when elevated. However, numerous studies suggest that cholesterol levels—both high and low—may be associated with various psychiatric brai
The relationship between cholesterol and mental illness is fascinating, complex, and perplexing. Whether elevated or reduced, cholesterol’s effects can be deleterious or salutary, but the literature is riddled with conflicting reports. Physicians should measure their patients’ serum cholesterol levels not only to assess cardiovascular risk, but because cholesterol can be associated with certain neuropsychiatric disorders or may predict the lack of response to psychopharmacotherapy.2
The fact that lowering total cholesterol levels in people with hypercholesterolemia reduces the risk of coronary heart disease is indisputable. Large-scale cardiology clinical trials have shown a significant reduction in mortality from heart disease or stroke with cholesterol-lowering drugs (statins). However, the same trials found an uptick in “unnatural deaths,” mostly suicide or homicide.3 Those findings triggered numerous intriguing reports of the association between cholesterol levels and psychopathology.
Consider the following:
- Low cholesterol levels have been associated with depression, antisocial personality disorder, borderline personality disorder, and dissociative disorder.4
- High cholesterol levels have been associated with schizophrenia, obsessive-compulsive disorder, panic disorder, generalized anxiety disorder, and posttraumatic stress disorder.4
- Some studies suggest that high cholesterol levels are associated with better mental health, mental processing speed, social skills, responsibility, self-control, and self-awareness.5
- In the Clinical Antipsychotic Trials of Intervention Effectiveness schizophrenia study, better cognitive scores were found in patients with higher fasting cholesterol and triglyceride levels (H.A.N., unpublished data, 2017).
The brain is only 2% of body weight, but it contains 25% of the body’s cholesterol.6 Cholesterol is important for brain function and neurotransmission because neuroactive steroids (NASs) are synthesized from cholesterol and they modulate brain processes and interact with γ-aminobutyric acid, N-methyl-
Interestingly, both extremes in cholesterol levels represent a high risk for premature mortality.10 Hypercholesterolemia leads to early death from coronary artery disease. Studies that evaluated statins to lower cholesterol found increased mortality from suicide, accidents, and violence.11 Even without statin treatment, among persons with naturally low cholesterol, there is a significant increase in mortality from non-medical causes.12 However, some studies did not find an association between hypocholesterolemia and suicide.13,14
There also is some evidence that elevated cholesterol may play a role in dementia.15 Reducing cholesterol with statins decreases beta-amyloid in mice, while the opposite occurs with elevated cholesterol.2 Another possible mechanism by which high cholesterol worsens dementia is that neurodegeneration in Alzheimer’s disease (AD) breaks down neuronal cell membranes, which releases the neurotoxic metabolite of cholesterol (24-hydroxycholesterol), which leads to further neurodegeneration.16 Statins may decrease the production of 24-hydroxycholesterol in AD patients and slow down neurodegeneration.16
A large study of 4,444 consecutive patients in Taiwan found that those with low total cholesterol (<160 mg/dL) had higher scores of anxiety, phobia, psychoticism, and aggressive hostility.17 In the same study, women with low high-density lipoprotein cholesterol (<35 mg/dL) had significantly higher scores for depression, phobia, anxiety, interpersonal sensitivity, somatization, and aggressive hostility.17
Not surprisingly, low cholesterol has been proposed as a biomarker for mood dysregulation, depression, and suicidality,18 as well as a predictor of the depression severity and increased suicide risk.19 Clinical recovery in depression may be accompanied by a significant increase of total cholesterol20 but, interestingly, a decrease in cholesterol levels after treatment of mania. High cholesterol was reported to predict poorer response to selective serotonin reuptake inhibitors, and total cholesterol levels >200 mg/dL were associated with lack of response to fluoxetine and nortriptyline.2 Interestingly, clozapine, which elevates lipids, exerts a strong anti-suicide effect in schizophrenia and schizoaffective disorder, but that may not be the main reason for its efficacy in preventing suicide in patients with psychosis.
Cholesterol is an important lipid for brain function. At lower levels, it appears to be associated with depression, suicide, violence, anxiety, schizophrenia, and severe personality disorders (including antisocial personality disorder and borderline personality disorder). However, at high levels, it may improve cognition in schizophrenia and ameliorate the pace of AD and neurodegeneration. Psychiatrists should monitor patients for hypercholesterolemia and hypocholesterolemia, both of which are common among psychiatric patients. High levels may be genetic or the result of weight gain, hypercortisolemia, diabetes, or immune or inflammatory processes. Similarly, low levels may be genetic or secondary to statin therapy.
The bottom line: As psychiatric physicians, we should protect both the hearts and brains of our patients.
1. Hallahan B, Garland MR. Essential fatty acids and mental health. British J Psychiatry. 2005;186(4):275-277.
2. Papakostas GI, Ongür D, Iosifescu DV, et al. Cholesterol in mood and anxiety disorders: review of the literature and new hypotheses. Eur Neuropsychopharmacol. 2004;14(2):135-142.
3. Muldoon MF, Manuck SB, Matthews KA, et al. Lowering cholesterol concentrations and mortality: a quantitative review of primary prevention trials. BMJ. 1990;301(647):309-314.
4. Jakovljevic
5. Rogers PJ. A healthy body, a healthy mind: long-term impact of diet on mood and cognitive function. Pro Nutr Soc. 2001;60(1):135-143.
6. Björkhem I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med. 2006;260(6):493-508.
7. Tuem KB, Atey TM. Neuroactive steroids: receptor interactions and responses. Front Neurol. 2017;8:442.
8. Borroni MV, Vallés AS, Barrantes FJ. The lipid habitats of neurotransmitter receptors in the brain. Biochim Biophys Acta. 2016;1858(1):2662-2670.
9. Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci. 2003;60(6):1158-1171.
10. Graham I, Atar D, Borch-Johnsen K, et al; European Society of Cardiology (ESC); European Association for Cardiovascular Prevention and Rehabilitation (EACPR); Council on Cardiovascular Nursing; European Association for Study of Diabetes (EASD); International Diabetes Federation Europe (IDF-Europe); European Stroke Initiative (EUSI); Society of Behavioural Medicine (ISBM); European Society of Hypertension (ESH); WONCA Europe (European Society of General Practice/Family Medicine); European Heart Network (EHN); European Atherosclerosis Society (EAS). European guidelines on cardiovascular disease prevention in clinical practice: full text. Fourth Joint Task Force of the European Society of Cardiology and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of none societies and by invited experts). Eur J Cardiovasc Prev Rehabil. 2007;14(suppl 2):S1-S113.
11. Almeida-Montes LG, Valles-Sanchez V, Moreno-Aguilar J, et al. Relation of serum cholesterol, lipid, serotonin and tryptophan levels to severity of depression and to suicide attempts. J Psychiatry Neurosci. 2000;25(4):371-377.
12. Ryman A. Cholesterol, violent death, and mental disorder. BMJ. 1994;309(69525):421-422.
13. Wardle J. Cholesterol and psychological well-being. J Psychosom Res. 1995;39(5):549-562.
14. Irribarren C, Reed DM, Chen R, et al. Low serum cholesterol and mortality. Which is the cause and which is the effect? Circulation. 1995;92(9):2396-2403.
15. Stampfer MJ. Cardiovascular disease and Alzheimer’s disease: common links. J Intern Med. 2006;260(3):211-223.
16. Raffai RL, Weisgraber KH. Cholesterol: from heart attacks to Alzheimer’s disease. J Lipid Res. 2003;44(8):1423-1430.
17. Chen CC, Lu FH, Wu JS, et al. Correlation between serum lipid concentrations and psychological distress. Psychiatry Res. 2003;102(2):153-162.
18. Mössmer R, Mikova O, Koutsilieri E, et al. Consensus paper of the WFSBP Task Force on Biological Markers: biological markers in depression. World J Biol Psychiatry. 2007;8(3):141-174.
19. Papakostas GI, Petersen T, Sonawalla SB, et al. Serum cholesterol in treatment-resistant depression. Neuropsychobiology. 2003;47(3):146-151.
20. Gabriel A. Changes in plasma cholesterol in mood disorder patients: does treatment make a difference? J Affect Disord. 2007;99(1-3):273-278.
Cholesterol generally is regarded as a cardiovascular risk factor when elevated. However, numerous studies suggest that cholesterol levels—both high and low—may be associated with various psychiatric brai
The relationship between cholesterol and mental illness is fascinating, complex, and perplexing. Whether elevated or reduced, cholesterol’s effects can be deleterious or salutary, but the literature is riddled with conflicting reports. Physicians should measure their patients’ serum cholesterol levels not only to assess cardiovascular risk, but because cholesterol can be associated with certain neuropsychiatric disorders or may predict the lack of response to psychopharmacotherapy.2
The fact that lowering total cholesterol levels in people with hypercholesterolemia reduces the risk of coronary heart disease is indisputable. Large-scale cardiology clinical trials have shown a significant reduction in mortality from heart disease or stroke with cholesterol-lowering drugs (statins). However, the same trials found an uptick in “unnatural deaths,” mostly suicide or homicide.3 Those findings triggered numerous intriguing reports of the association between cholesterol levels and psychopathology.
Consider the following:
- Low cholesterol levels have been associated with depression, antisocial personality disorder, borderline personality disorder, and dissociative disorder.4
- High cholesterol levels have been associated with schizophrenia, obsessive-compulsive disorder, panic disorder, generalized anxiety disorder, and posttraumatic stress disorder.4
- Some studies suggest that high cholesterol levels are associated with better mental health, mental processing speed, social skills, responsibility, self-control, and self-awareness.5
- In the Clinical Antipsychotic Trials of Intervention Effectiveness schizophrenia study, better cognitive scores were found in patients with higher fasting cholesterol and triglyceride levels (H.A.N., unpublished data, 2017).
The brain is only 2% of body weight, but it contains 25% of the body’s cholesterol.6 Cholesterol is important for brain function and neurotransmission because neuroactive steroids (NASs) are synthesized from cholesterol and they modulate brain processes and interact with γ-aminobutyric acid, N-methyl-
Interestingly, both extremes in cholesterol levels represent a high risk for premature mortality.10 Hypercholesterolemia leads to early death from coronary artery disease. Studies that evaluated statins to lower cholesterol found increased mortality from suicide, accidents, and violence.11 Even without statin treatment, among persons with naturally low cholesterol, there is a significant increase in mortality from non-medical causes.12 However, some studies did not find an association between hypocholesterolemia and suicide.13,14
There also is some evidence that elevated cholesterol may play a role in dementia.15 Reducing cholesterol with statins decreases beta-amyloid in mice, while the opposite occurs with elevated cholesterol.2 Another possible mechanism by which high cholesterol worsens dementia is that neurodegeneration in Alzheimer’s disease (AD) breaks down neuronal cell membranes, which releases the neurotoxic metabolite of cholesterol (24-hydroxycholesterol), which leads to further neurodegeneration.16 Statins may decrease the production of 24-hydroxycholesterol in AD patients and slow down neurodegeneration.16
A large study of 4,444 consecutive patients in Taiwan found that those with low total cholesterol (<160 mg/dL) had higher scores of anxiety, phobia, psychoticism, and aggressive hostility.17 In the same study, women with low high-density lipoprotein cholesterol (<35 mg/dL) had significantly higher scores for depression, phobia, anxiety, interpersonal sensitivity, somatization, and aggressive hostility.17
Not surprisingly, low cholesterol has been proposed as a biomarker for mood dysregulation, depression, and suicidality,18 as well as a predictor of the depression severity and increased suicide risk.19 Clinical recovery in depression may be accompanied by a significant increase of total cholesterol20 but, interestingly, a decrease in cholesterol levels after treatment of mania. High cholesterol was reported to predict poorer response to selective serotonin reuptake inhibitors, and total cholesterol levels >200 mg/dL were associated with lack of response to fluoxetine and nortriptyline.2 Interestingly, clozapine, which elevates lipids, exerts a strong anti-suicide effect in schizophrenia and schizoaffective disorder, but that may not be the main reason for its efficacy in preventing suicide in patients with psychosis.
Cholesterol is an important lipid for brain function. At lower levels, it appears to be associated with depression, suicide, violence, anxiety, schizophrenia, and severe personality disorders (including antisocial personality disorder and borderline personality disorder). However, at high levels, it may improve cognition in schizophrenia and ameliorate the pace of AD and neurodegeneration. Psychiatrists should monitor patients for hypercholesterolemia and hypocholesterolemia, both of which are common among psychiatric patients. High levels may be genetic or the result of weight gain, hypercortisolemia, diabetes, or immune or inflammatory processes. Similarly, low levels may be genetic or secondary to statin therapy.
The bottom line: As psychiatric physicians, we should protect both the hearts and brains of our patients.
Cholesterol generally is regarded as a cardiovascular risk factor when elevated. However, numerous studies suggest that cholesterol levels—both high and low—may be associated with various psychiatric brai
The relationship between cholesterol and mental illness is fascinating, complex, and perplexing. Whether elevated or reduced, cholesterol’s effects can be deleterious or salutary, but the literature is riddled with conflicting reports. Physicians should measure their patients’ serum cholesterol levels not only to assess cardiovascular risk, but because cholesterol can be associated with certain neuropsychiatric disorders or may predict the lack of response to psychopharmacotherapy.2
The fact that lowering total cholesterol levels in people with hypercholesterolemia reduces the risk of coronary heart disease is indisputable. Large-scale cardiology clinical trials have shown a significant reduction in mortality from heart disease or stroke with cholesterol-lowering drugs (statins). However, the same trials found an uptick in “unnatural deaths,” mostly suicide or homicide.3 Those findings triggered numerous intriguing reports of the association between cholesterol levels and psychopathology.
Consider the following:
- Low cholesterol levels have been associated with depression, antisocial personality disorder, borderline personality disorder, and dissociative disorder.4
- High cholesterol levels have been associated with schizophrenia, obsessive-compulsive disorder, panic disorder, generalized anxiety disorder, and posttraumatic stress disorder.4
- Some studies suggest that high cholesterol levels are associated with better mental health, mental processing speed, social skills, responsibility, self-control, and self-awareness.5
- In the Clinical Antipsychotic Trials of Intervention Effectiveness schizophrenia study, better cognitive scores were found in patients with higher fasting cholesterol and triglyceride levels (H.A.N., unpublished data, 2017).
The brain is only 2% of body weight, but it contains 25% of the body’s cholesterol.6 Cholesterol is important for brain function and neurotransmission because neuroactive steroids (NASs) are synthesized from cholesterol and they modulate brain processes and interact with γ-aminobutyric acid, N-methyl-
Interestingly, both extremes in cholesterol levels represent a high risk for premature mortality.10 Hypercholesterolemia leads to early death from coronary artery disease. Studies that evaluated statins to lower cholesterol found increased mortality from suicide, accidents, and violence.11 Even without statin treatment, among persons with naturally low cholesterol, there is a significant increase in mortality from non-medical causes.12 However, some studies did not find an association between hypocholesterolemia and suicide.13,14
There also is some evidence that elevated cholesterol may play a role in dementia.15 Reducing cholesterol with statins decreases beta-amyloid in mice, while the opposite occurs with elevated cholesterol.2 Another possible mechanism by which high cholesterol worsens dementia is that neurodegeneration in Alzheimer’s disease (AD) breaks down neuronal cell membranes, which releases the neurotoxic metabolite of cholesterol (24-hydroxycholesterol), which leads to further neurodegeneration.16 Statins may decrease the production of 24-hydroxycholesterol in AD patients and slow down neurodegeneration.16
A large study of 4,444 consecutive patients in Taiwan found that those with low total cholesterol (<160 mg/dL) had higher scores of anxiety, phobia, psychoticism, and aggressive hostility.17 In the same study, women with low high-density lipoprotein cholesterol (<35 mg/dL) had significantly higher scores for depression, phobia, anxiety, interpersonal sensitivity, somatization, and aggressive hostility.17
Not surprisingly, low cholesterol has been proposed as a biomarker for mood dysregulation, depression, and suicidality,18 as well as a predictor of the depression severity and increased suicide risk.19 Clinical recovery in depression may be accompanied by a significant increase of total cholesterol20 but, interestingly, a decrease in cholesterol levels after treatment of mania. High cholesterol was reported to predict poorer response to selective serotonin reuptake inhibitors, and total cholesterol levels >200 mg/dL were associated with lack of response to fluoxetine and nortriptyline.2 Interestingly, clozapine, which elevates lipids, exerts a strong anti-suicide effect in schizophrenia and schizoaffective disorder, but that may not be the main reason for its efficacy in preventing suicide in patients with psychosis.
Cholesterol is an important lipid for brain function. At lower levels, it appears to be associated with depression, suicide, violence, anxiety, schizophrenia, and severe personality disorders (including antisocial personality disorder and borderline personality disorder). However, at high levels, it may improve cognition in schizophrenia and ameliorate the pace of AD and neurodegeneration. Psychiatrists should monitor patients for hypercholesterolemia and hypocholesterolemia, both of which are common among psychiatric patients. High levels may be genetic or the result of weight gain, hypercortisolemia, diabetes, or immune or inflammatory processes. Similarly, low levels may be genetic or secondary to statin therapy.
The bottom line: As psychiatric physicians, we should protect both the hearts and brains of our patients.
1. Hallahan B, Garland MR. Essential fatty acids and mental health. British J Psychiatry. 2005;186(4):275-277.
2. Papakostas GI, Ongür D, Iosifescu DV, et al. Cholesterol in mood and anxiety disorders: review of the literature and new hypotheses. Eur Neuropsychopharmacol. 2004;14(2):135-142.
3. Muldoon MF, Manuck SB, Matthews KA, et al. Lowering cholesterol concentrations and mortality: a quantitative review of primary prevention trials. BMJ. 1990;301(647):309-314.
4. Jakovljevic
5. Rogers PJ. A healthy body, a healthy mind: long-term impact of diet on mood and cognitive function. Pro Nutr Soc. 2001;60(1):135-143.
6. Björkhem I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med. 2006;260(6):493-508.
7. Tuem KB, Atey TM. Neuroactive steroids: receptor interactions and responses. Front Neurol. 2017;8:442.
8. Borroni MV, Vallés AS, Barrantes FJ. The lipid habitats of neurotransmitter receptors in the brain. Biochim Biophys Acta. 2016;1858(1):2662-2670.
9. Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci. 2003;60(6):1158-1171.
10. Graham I, Atar D, Borch-Johnsen K, et al; European Society of Cardiology (ESC); European Association for Cardiovascular Prevention and Rehabilitation (EACPR); Council on Cardiovascular Nursing; European Association for Study of Diabetes (EASD); International Diabetes Federation Europe (IDF-Europe); European Stroke Initiative (EUSI); Society of Behavioural Medicine (ISBM); European Society of Hypertension (ESH); WONCA Europe (European Society of General Practice/Family Medicine); European Heart Network (EHN); European Atherosclerosis Society (EAS). European guidelines on cardiovascular disease prevention in clinical practice: full text. Fourth Joint Task Force of the European Society of Cardiology and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of none societies and by invited experts). Eur J Cardiovasc Prev Rehabil. 2007;14(suppl 2):S1-S113.
11. Almeida-Montes LG, Valles-Sanchez V, Moreno-Aguilar J, et al. Relation of serum cholesterol, lipid, serotonin and tryptophan levels to severity of depression and to suicide attempts. J Psychiatry Neurosci. 2000;25(4):371-377.
12. Ryman A. Cholesterol, violent death, and mental disorder. BMJ. 1994;309(69525):421-422.
13. Wardle J. Cholesterol and psychological well-being. J Psychosom Res. 1995;39(5):549-562.
14. Irribarren C, Reed DM, Chen R, et al. Low serum cholesterol and mortality. Which is the cause and which is the effect? Circulation. 1995;92(9):2396-2403.
15. Stampfer MJ. Cardiovascular disease and Alzheimer’s disease: common links. J Intern Med. 2006;260(3):211-223.
16. Raffai RL, Weisgraber KH. Cholesterol: from heart attacks to Alzheimer’s disease. J Lipid Res. 2003;44(8):1423-1430.
17. Chen CC, Lu FH, Wu JS, et al. Correlation between serum lipid concentrations and psychological distress. Psychiatry Res. 2003;102(2):153-162.
18. Mössmer R, Mikova O, Koutsilieri E, et al. Consensus paper of the WFSBP Task Force on Biological Markers: biological markers in depression. World J Biol Psychiatry. 2007;8(3):141-174.
19. Papakostas GI, Petersen T, Sonawalla SB, et al. Serum cholesterol in treatment-resistant depression. Neuropsychobiology. 2003;47(3):146-151.
20. Gabriel A. Changes in plasma cholesterol in mood disorder patients: does treatment make a difference? J Affect Disord. 2007;99(1-3):273-278.
1. Hallahan B, Garland MR. Essential fatty acids and mental health. British J Psychiatry. 2005;186(4):275-277.
2. Papakostas GI, Ongür D, Iosifescu DV, et al. Cholesterol in mood and anxiety disorders: review of the literature and new hypotheses. Eur Neuropsychopharmacol. 2004;14(2):135-142.
3. Muldoon MF, Manuck SB, Matthews KA, et al. Lowering cholesterol concentrations and mortality: a quantitative review of primary prevention trials. BMJ. 1990;301(647):309-314.
4. Jakovljevic
5. Rogers PJ. A healthy body, a healthy mind: long-term impact of diet on mood and cognitive function. Pro Nutr Soc. 2001;60(1):135-143.
6. Björkhem I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med. 2006;260(6):493-508.
7. Tuem KB, Atey TM. Neuroactive steroids: receptor interactions and responses. Front Neurol. 2017;8:442.
8. Borroni MV, Vallés AS, Barrantes FJ. The lipid habitats of neurotransmitter receptors in the brain. Biochim Biophys Acta. 2016;1858(1):2662-2670.
9. Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci. 2003;60(6):1158-1171.
10. Graham I, Atar D, Borch-Johnsen K, et al; European Society of Cardiology (ESC); European Association for Cardiovascular Prevention and Rehabilitation (EACPR); Council on Cardiovascular Nursing; European Association for Study of Diabetes (EASD); International Diabetes Federation Europe (IDF-Europe); European Stroke Initiative (EUSI); Society of Behavioural Medicine (ISBM); European Society of Hypertension (ESH); WONCA Europe (European Society of General Practice/Family Medicine); European Heart Network (EHN); European Atherosclerosis Society (EAS). European guidelines on cardiovascular disease prevention in clinical practice: full text. Fourth Joint Task Force of the European Society of Cardiology and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of none societies and by invited experts). Eur J Cardiovasc Prev Rehabil. 2007;14(suppl 2):S1-S113.
11. Almeida-Montes LG, Valles-Sanchez V, Moreno-Aguilar J, et al. Relation of serum cholesterol, lipid, serotonin and tryptophan levels to severity of depression and to suicide attempts. J Psychiatry Neurosci. 2000;25(4):371-377.
12. Ryman A. Cholesterol, violent death, and mental disorder. BMJ. 1994;309(69525):421-422.
13. Wardle J. Cholesterol and psychological well-being. J Psychosom Res. 1995;39(5):549-562.
14. Irribarren C, Reed DM, Chen R, et al. Low serum cholesterol and mortality. Which is the cause and which is the effect? Circulation. 1995;92(9):2396-2403.
15. Stampfer MJ. Cardiovascular disease and Alzheimer’s disease: common links. J Intern Med. 2006;260(3):211-223.
16. Raffai RL, Weisgraber KH. Cholesterol: from heart attacks to Alzheimer’s disease. J Lipid Res. 2003;44(8):1423-1430.
17. Chen CC, Lu FH, Wu JS, et al. Correlation between serum lipid concentrations and psychological distress. Psychiatry Res. 2003;102(2):153-162.
18. Mössmer R, Mikova O, Koutsilieri E, et al. Consensus paper of the WFSBP Task Force on Biological Markers: biological markers in depression. World J Biol Psychiatry. 2007;8(3):141-174.
19. Papakostas GI, Petersen T, Sonawalla SB, et al. Serum cholesterol in treatment-resistant depression. Neuropsychobiology. 2003;47(3):146-151.
20. Gabriel A. Changes in plasma cholesterol in mood disorder patients: does treatment make a difference? J Affect Disord. 2007;99(1-3):273-278.

‘Self-anesthetizing’ to cope with grief
CASE Grieving, delusional
Mr. M, age 51, is brought to the emergency department (ED) because of new-onset delusions and decreased self-care over the last 2 weeks following the sudden death of his wife. He has become expansive and grandiose, with pressured speech, increased energy, and markedly reduced sleep. Mr. M is preoccupied with the idea that he is “the first to survive a human reboot process” and says that his and his wife’s bodies and brains had been “split apart.” Mr. M has limited his food and fluid intake and lost 15 lb within the past 2 to 3 weeks.
Mr. M has no history of any affective, psychotic, or other major mental disorders or treatment. He reports that he has regularly used Cannabis over the last 10 years, and a few years ago, he started occasionally using nitrous oxide (N2O). He says that in the week following his wife’s death, he used N2O almost daily and in copious amounts. In an attempt to “self-anesthetize” himself after his wife’s funeral, he isolated himself in his bedroom and used escalating amounts of Cannabis and N2O, while continually working on a book about their life together.
At first, Mr. M shows little emotion and describes his situation as “interesting and fascinating.” He mentions that he thinks he might have been “psychotic” the week after his wife’s death, but he shows no sustained insight and immediately relapses into psychotic thinking. Over several hours in the ED, he is tearful and sad about his wife’s death. Mr. M recalls a similar experience of grief after his mother died when he was a teenager, but at that time he did not abuse substances or have psychotic symptoms. He is fully alert, fully oriented, and has no significant deficits of attention or memory.
[polldaddy:9859135]
The authors’ observations
Grief was a precipitating event, but by itself grief cannot explain psychosis. Psychotic depression is a possibility, but Mr. M’s psychotic features are incongruent with his mood. Mania would be a diagnosis of exclusion. Mr. M had no prior history of major affective illness. Mr. M was abusing Cannabis, which might independently contribute to psychosis1; however, he had been using it recreationally for 10 years without psychiatric problems. N2O, however, can cause symptoms consistent with Mr. M’s presentation.
[polldaddy:9859140]
EVALUATION Laboratory tests
Mr. M’s physical examination is notable only for an elevated blood pressure of 196/120 mm Hg. Neurologic examination is normal. Toxicology is positive for cannabinoids and negative for amphetamines, cocaine, opiates, and phencyclidine. Chemistries are normal except for a potassium of 3.4 mEq/L (reference range, 3.7 to 5.2 mEq/L) and a blood urine nitrogen of 25 mg/dL (reference range, 6 to 20 mg/dL), which are consistent with reduced food and fluid intake. Mr. M shows no signs of anemia. Hematocrit is 42% and mean corpuscular volume is 90 fL. Syphilis screen is negative; a head CT scan is unremarkable.
The authors’ observations
N2O, also known as “laughing gas,” is routinely used by dentists and pediatric anesthesiologists, and has other medical uses. Some studies have examined an adjunctive use of N2O for pain control in the ED and during colonoscopies.3,4
In the 2013 U.S. National Survey on Drug Use and Health, 16% of respondents reported lifetime illicit use of N2O.5,6 It is readily available in tanks used in medicine and industry and in small dispensers called “whippits” that can be legally purchased. Acute effects of N2O include euphoric mood, numbness, feeling of warmth, dizziness, and auditory hallucinations.7 The anesthetic effects of N2O are linked to endogenous release of opiates, and recent research links its anxiolytic activity to the facilitation of GABAergic inhibitory and N-methyl-
Beginning with a 1960 report of a series of patients with “megaloblastic madness,”17 there have been calls for increased awareness of the potential for vitamin B12 deficiency–induced psychiatric disorders, even in the absence of other hematologic or neurologic sequelae that would alert clinicians of the deficiency. In a case series of 141 patients with a broad array of neurologic and psychiatric symptoms associated with vitamin B12 deficiency, 40 (28%) patients had no anemia or macrocytosis.2
Vitamin B12-responsive psychosis has been reported as the sole manifestation of illness, without associated neurologic or hematologic symptoms, in only a few case reports. Vitamin B12 levels in these cases ranged from 75 to 236 pg/mL (reference range, 160 to 950 pg/mL).18-20 In all of these cases, the vitamin B12 deficiency was traced to dietary causes. The clinical evaluation of suspected vitamin B12 deficiency is outlined in the Figure.21 Mr. M had used Cannabis recreationally for a long time, and his Cannabis use acutely escalated with use of N2O. Long-term use of Cannabis alone is a risk factor for psychotic illness.22 Combined abuse of Cannabis and N2O has been reported to provoke psychotic illness. In a case report of a 22-year-old male who was treated for paranoid delusions, using Cannabis and 100 cartridges of N2O daily was associated with low vitamin B12 and elevated homocysteine and methylmalonic acid levels.23
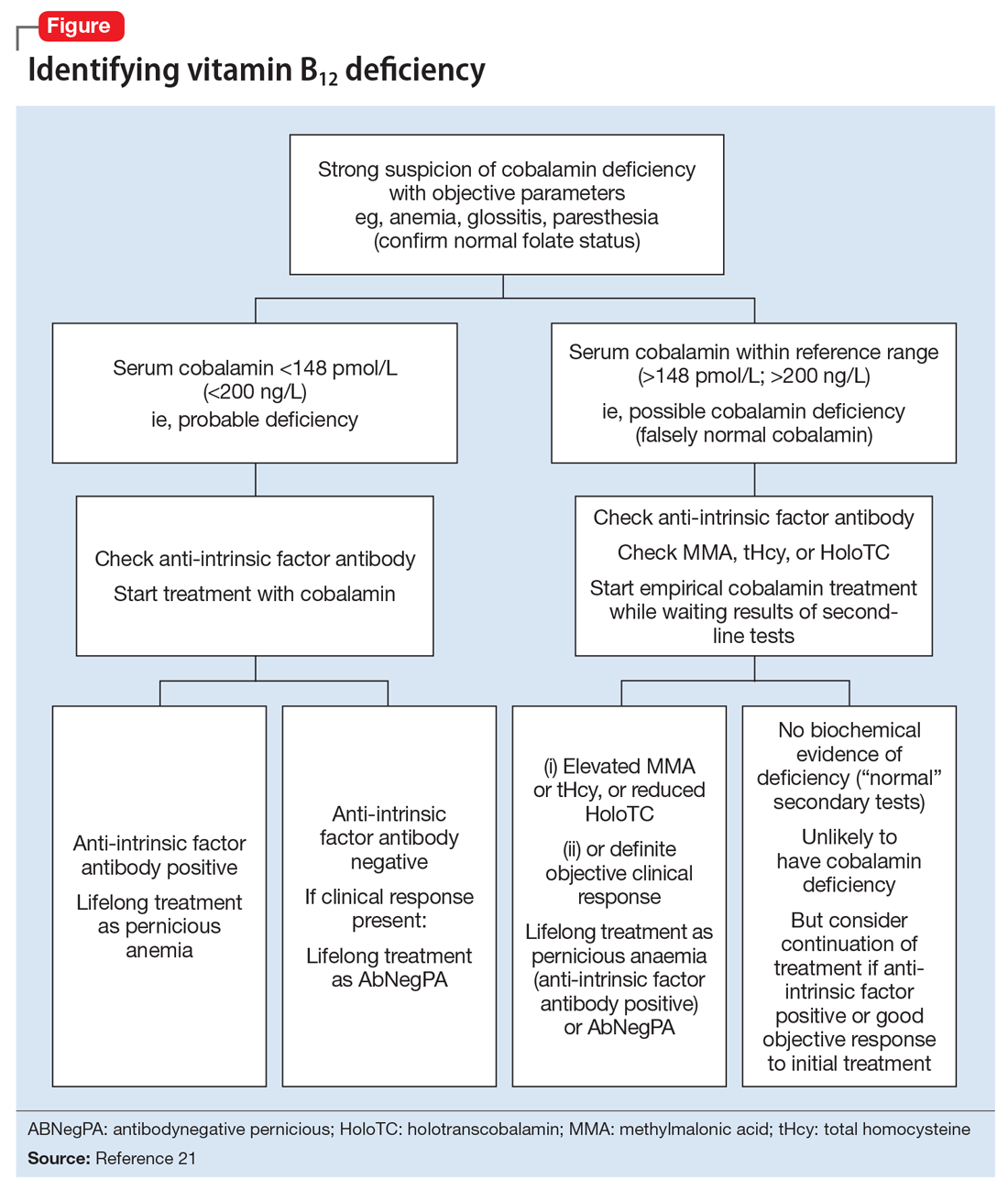
Cannabis use may have played a role in Mr. M’s escalating N2O use. In a study comparing 9 active Cannabis users with 9 non-using controls, users rated the subjective effects of N2O as more intense than non-users.24 In our patient’s case, Cannabis may have played a role in both sustaining his escalating N2O abuse and potentiating its psychotomimetic effects.
It also is possible that Mr. M may have been “self-medicating” his grief with N2O. In a recent placebo-controlled crossover trial of 20 patients with treatment-resistant depression, Nagele et al25 found a significant rapid and week-long antidepressant effect of subanesthetic N2O use. A model involving NMDA receptor activation has been proposed.25,26 Zorumski et al26 further reviewed possible antidepressant mechanisms of N2O. They compared N2O with ketamine as an NMDA receptor antagonist, but also noted its distinct effects on glutaminergic and GABAergic neurotransmitter systems as well as other receptors and channels.26 However, illicit use of N2O poses toxicity dangers and has no current indication for psychiatric treatment.
TREATMENT Supplementation
Mr. M is diagnosed with substance-induced psychotic disorder. His symptoms were precipitated by an acute increase in N2O use, which has been shown to cause vitamin B12 deficiency, which we consider was likely a primary contributor to his presentation. Other potential contributing factors are premorbid hyperthymic temperament, a possible propensity to psychotic thinking under stress, the sudden death of his wife, acute grief, the potentiating role of Cannabis, dehydration, and general malnutrition. The death of a loved one is associated with an increased risk of developing substance use disorders.27
During a 15-day psychiatric hospitalization, Mr. M is given olanzapine, increased to 15 mg/d and oral vitamin B12, 1,000 mcg/d for 4 days, then IM cyanocobalamin for 7 days. Mr. M’s symptoms steadily improve, with normalization of sleep and near-total resolution of delusions. On hospital Day 14, his vitamin B12 levels are within normal limits (844 pg/mL). At discharge, Mr. M shows residual mild grandiosity, with limited insight into his illness and what caused it, but frank delusional ideation has clearly receded. He still shows some signs of grief. Mr. M is advised to stop using Cannabis and N2O and about the potential consequences of continued use.
The authors’ observations
For patients with vitamin B12 deficiency, guidelines from the National Health Service in the United Kingdom and the British Society for Haematology recommend treatment with IM hydroxocobalamin, 1,000 IU, 3 times weekly, for 2 weeks.21,28 For patients with neurologic symptoms, the British National Foundation recommends treatment with IM hydroxocobalamin, 1,000 IU, on alternative days until there is no further improvement.21
This case is a reminder for clinicians to screen for inhalant use, specifically N2O, which can precipitate vitamin B12 deficiency with psychiatric symptoms as the only presenting concern. Clinicians should consider measuring vitamin B12 levels in psychiatric patients at risk of deficiency of this nutrient, including older adults, vegetarians, and those with alimentary disorders.29,30 Dietary sources of vitamin B12 include meat, milk, egg, fish, and shellfish.31 The body can store a total of 2 to 5 mg of vitamin B12; thus, it takes 2 to 5 years to develop vitamin B12 deficiency from malabsorption and can take as long as 20 years to develop vitamin B12 deficiency from vegetarianism.32 However, by chemically inactivating vitamin B12, N2O causes a rapid functional deficiency, as was seen in our patient.
OUTCOME Improved insight
At a 1-week follow-up appointment with a psychiatrist, Mr. M has no evident psychotic symptoms. He reports that he has not used Cannabis or N2O, and he discontinues olanzapine following this visit. Two weeks later, Mr. M shows no psychotic or affective symptoms other than grief, which is appropriately expressed. His insight has improved. He commits to not using Cannabis, N2O, or any other illicit substances. Mr. M is referred back to his long-standing primary care provider with the understanding that if any psychiatric symptoms recur he will see a psychiatrist again.
1. Semple DM, McIntosh AM, Lawrie SM. Cannabis as a risk factor for psychosis: systematic review. J Psychopharmacol. 2005;19(2):187-194.
2. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26)1720-1728.
3. Herres J, Chudnofsky CR, Manur R, et al. The use of inhaled nitrous oxide for analgesia in adult ED patients: a pilot study. Am J Emerg Med. 2016;34(2):269-273.
4. Aboumarzouk OM, Agarwal T, Syed Nong Chek SA, et al. Nitrous oxide for colonoscopy. Cochrane Database Syst Rev. 2011;(8):CD008506.
5. National Institute on Drug Abuse. Drug facts: inhalants. http://www.drugabuse.gov/publications/drugfacts/inhalants. Updated February 2017. Accessed September 30, 2017.
6. SAMHSA, Center for Behavioral Health Statistics and Quality, National Survey on Drug Use and Health 2012 and 2013: Table 1.88C. https://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabs2013.pdf. Published September 4, 2017. Accessed September 30, 2017.
7. Brouette T, Anton R. Clinical review of inhalants. Am J Addict. 2001;10(1):79-94.
8. Emmanouil DE, Quock RM. Advances in understanding the actions of nitrous oxide. Anesth Prog. 2007;54(1):9-18.
9. Garakani A, Jaffe RJ, Savla D, et al. Neurologic, psychiatric, and other medical manifestations of nitrous oxide abuse: a systematic review of the case literature. Am J Addict. 2016;25(5):358-369.
10. Hathout L, El-Saden S. Nitrous oxide-induced B12 deficiency myelopathy: perspectives on the clinical biochemistry of vitamin B12. J Neurol Sci. 2011;301(1-2):1-8.
11. van Tonder SV, Ruck A, van der Westhuyzen J, et al. Dissociation of methionine synthetase (EC 2.1.1.13) activity and impairment of DNA synthesis in fruit bats (Rousettus aegyptiacus) with nitrous oxide-induced vitamin B12 deficiency. Br J Nutr. 1986;55(1):187-192.
12. Schrier SL, Mentzer WC, Tirnauer JS. Diagnosis and treatment of vitamin B12 and folate deficiency. UpToDate. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-vitamin-b12-and-folate-deficiency. Updated September 30, 2011. Accessed September 8, 2015.
13. Sethi NK, Mullin P, Torgovnick J, et al. Nitrous oxide “whippit” abuse presenting with cobalamin responsive psychosis. J Med Toxicol. 2006;2(2):71-74.
14. Cousaert C, Heylens G, Audenaert K. Laughing gas abuse is no joke. An overview of the implications for psychiatric practice. Clin Neurol Neurosurg. 2013;115(7):859-862.
15. Brodsky L, Zuniga J. Nitrous oxide: a psychotogenic agent. Compr Psychiatry. 1975;16(2):185-188.
16. Wong SL, Harrison R, Mattman A, et al. Nitrous oxide (N2O)-induced acute psychosis. Can J Neurol Sci. 2014;41(5):672-674.
17. Smith AD. Megaloblastic madness. Br Med J. 1960;2(5216):1840-1845.
18. Masalha R, Chudakov B, Muhamad M, et al. Cobalamin-responsive psychosis as the sole manifestation of vitamin B12 deficiency. Isr Med Associ J. 2001;3(9):701-703.
19. Kuo SC, Yeh SB, Yeh YW, et al. Schizophrenia-like psychotic episode precipitated by cobalamin deficiency. Gen Hosp Psychiatry. 2009;31(6):586-588.
20. Raveendranathan D, Shiva L, Venkatasubramanian G, et al. Vitamin B12 deficiency masquerading as clozapine-resistant psychotic symptoms in schizophrenia. J Neuropsychiatry Clin Neurosci. 2013;25(2):E34-E35.
21. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513.
22. Moore THM, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet. 2007;370:319-328.
23. Garakani A, Welch AK, Jaffe RJ, et al. Psychosis and low cyanocobalamin in a patient abusing nitrous oxide and cannabis. Psychosomatics. 2014;55(6):715-719.
24. Yajnik S, Thapar P, Lichtor JL, et al. Effects of marijuana history on the subjective, psychomotor, and reinforcing effects of nitrous oxide in human. Drug Alcohol Depend. 1994;36(3):227-236.
25. Nagele P, Duma A, Kopec M, et al. Nitrous oxide for treatment-resistant major depression: a proof-of-concept trial. Biol Psychiatry. 2015;78(1):10-18.
26. Zorumski CF, Nagele P, Mennerick S, et al. Treatment-resistant major depression: rationale for NMDA receptors as targets and nitrous oxide as therapy. Front Psychiatry. 2015;6:172.
27. Shear MK. Clinical practice. Complicated grief. N Engl J Med. 2015;372(2):153-160.
28. Knechtli CJC, Crowe JN. Guidelines for the investigation & management of vitamin B12 deficiency. Royal United Hospital Bath, National Health Service. http://www.ruh.nhs.uk/For_Clinicians/departments_ruh/Pathology/documents/haematology/B12_-_advice_on_investigation_management.pdf. Accessed June 14, 2016.
29. Jayaram N, Rao MG, Narashima A, et al. Vitamin B12 levels and psychiatric symptomatology: a case series. J Neuropsychiatry Clin Neurosci. 2013;25(2):150-152.
30. Marks PW, Zukerberg LR. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 30-2004. A 37-year-old woman with paresthesias of the arms and legs. N Engl J Med. 2004;351(13):1333-1341.
31. Watanabe F. Vitamin B12 sources and bioavailablility. Exp Biol Med (Maywood). 2007;232(10):1266-1274.
32. Green R, Kinsella LJ. Current concepts in the diagnosis of cobalamin deficiency. Neurology. 1995;45(8):1435-1440.
CASE Grieving, delusional
Mr. M, age 51, is brought to the emergency department (ED) because of new-onset delusions and decreased self-care over the last 2 weeks following the sudden death of his wife. He has become expansive and grandiose, with pressured speech, increased energy, and markedly reduced sleep. Mr. M is preoccupied with the idea that he is “the first to survive a human reboot process” and says that his and his wife’s bodies and brains had been “split apart.” Mr. M has limited his food and fluid intake and lost 15 lb within the past 2 to 3 weeks.
Mr. M has no history of any affective, psychotic, or other major mental disorders or treatment. He reports that he has regularly used Cannabis over the last 10 years, and a few years ago, he started occasionally using nitrous oxide (N2O). He says that in the week following his wife’s death, he used N2O almost daily and in copious amounts. In an attempt to “self-anesthetize” himself after his wife’s funeral, he isolated himself in his bedroom and used escalating amounts of Cannabis and N2O, while continually working on a book about their life together.
At first, Mr. M shows little emotion and describes his situation as “interesting and fascinating.” He mentions that he thinks he might have been “psychotic” the week after his wife’s death, but he shows no sustained insight and immediately relapses into psychotic thinking. Over several hours in the ED, he is tearful and sad about his wife’s death. Mr. M recalls a similar experience of grief after his mother died when he was a teenager, but at that time he did not abuse substances or have psychotic symptoms. He is fully alert, fully oriented, and has no significant deficits of attention or memory.
[polldaddy:9859135]
The authors’ observations
Grief was a precipitating event, but by itself grief cannot explain psychosis. Psychotic depression is a possibility, but Mr. M’s psychotic features are incongruent with his mood. Mania would be a diagnosis of exclusion. Mr. M had no prior history of major affective illness. Mr. M was abusing Cannabis, which might independently contribute to psychosis1; however, he had been using it recreationally for 10 years without psychiatric problems. N2O, however, can cause symptoms consistent with Mr. M’s presentation.
[polldaddy:9859140]
EVALUATION Laboratory tests
Mr. M’s physical examination is notable only for an elevated blood pressure of 196/120 mm Hg. Neurologic examination is normal. Toxicology is positive for cannabinoids and negative for amphetamines, cocaine, opiates, and phencyclidine. Chemistries are normal except for a potassium of 3.4 mEq/L (reference range, 3.7 to 5.2 mEq/L) and a blood urine nitrogen of 25 mg/dL (reference range, 6 to 20 mg/dL), which are consistent with reduced food and fluid intake. Mr. M shows no signs of anemia. Hematocrit is 42% and mean corpuscular volume is 90 fL. Syphilis screen is negative; a head CT scan is unremarkable.
The authors’ observations
N2O, also known as “laughing gas,” is routinely used by dentists and pediatric anesthesiologists, and has other medical uses. Some studies have examined an adjunctive use of N2O for pain control in the ED and during colonoscopies.3,4
In the 2013 U.S. National Survey on Drug Use and Health, 16% of respondents reported lifetime illicit use of N2O.5,6 It is readily available in tanks used in medicine and industry and in small dispensers called “whippits” that can be legally purchased. Acute effects of N2O include euphoric mood, numbness, feeling of warmth, dizziness, and auditory hallucinations.7 The anesthetic effects of N2O are linked to endogenous release of opiates, and recent research links its anxiolytic activity to the facilitation of GABAergic inhibitory and N-methyl-
Beginning with a 1960 report of a series of patients with “megaloblastic madness,”17 there have been calls for increased awareness of the potential for vitamin B12 deficiency–induced psychiatric disorders, even in the absence of other hematologic or neurologic sequelae that would alert clinicians of the deficiency. In a case series of 141 patients with a broad array of neurologic and psychiatric symptoms associated with vitamin B12 deficiency, 40 (28%) patients had no anemia or macrocytosis.2
Vitamin B12-responsive psychosis has been reported as the sole manifestation of illness, without associated neurologic or hematologic symptoms, in only a few case reports. Vitamin B12 levels in these cases ranged from 75 to 236 pg/mL (reference range, 160 to 950 pg/mL).18-20 In all of these cases, the vitamin B12 deficiency was traced to dietary causes. The clinical evaluation of suspected vitamin B12 deficiency is outlined in the Figure.21 Mr. M had used Cannabis recreationally for a long time, and his Cannabis use acutely escalated with use of N2O. Long-term use of Cannabis alone is a risk factor for psychotic illness.22 Combined abuse of Cannabis and N2O has been reported to provoke psychotic illness. In a case report of a 22-year-old male who was treated for paranoid delusions, using Cannabis and 100 cartridges of N2O daily was associated with low vitamin B12 and elevated homocysteine and methylmalonic acid levels.23
Cannabis use may have played a role in Mr. M’s escalating N2O use. In a study comparing 9 active Cannabis users with 9 non-using controls, users rated the subjective effects of N2O as more intense than non-users.24 In our patient’s case, Cannabis may have played a role in both sustaining his escalating N2O abuse and potentiating its psychotomimetic effects.
It also is possible that Mr. M may have been “self-medicating” his grief with N2O. In a recent placebo-controlled crossover trial of 20 patients with treatment-resistant depression, Nagele et al25 found a significant rapid and week-long antidepressant effect of subanesthetic N2O use. A model involving NMDA receptor activation has been proposed.25,26 Zorumski et al26 further reviewed possible antidepressant mechanisms of N2O. They compared N2O with ketamine as an NMDA receptor antagonist, but also noted its distinct effects on glutaminergic and GABAergic neurotransmitter systems as well as other receptors and channels.26 However, illicit use of N2O poses toxicity dangers and has no current indication for psychiatric treatment.
TREATMENT Supplementation
Mr. M is diagnosed with substance-induced psychotic disorder. His symptoms were precipitated by an acute increase in N2O use, which has been shown to cause vitamin B12 deficiency, which we consider was likely a primary contributor to his presentation. Other potential contributing factors are premorbid hyperthymic temperament, a possible propensity to psychotic thinking under stress, the sudden death of his wife, acute grief, the potentiating role of Cannabis, dehydration, and general malnutrition. The death of a loved one is associated with an increased risk of developing substance use disorders.27
During a 15-day psychiatric hospitalization, Mr. M is given olanzapine, increased to 15 mg/d and oral vitamin B12, 1,000 mcg/d for 4 days, then IM cyanocobalamin for 7 days. Mr. M’s symptoms steadily improve, with normalization of sleep and near-total resolution of delusions. On hospital Day 14, his vitamin B12 levels are within normal limits (844 pg/mL). At discharge, Mr. M shows residual mild grandiosity, with limited insight into his illness and what caused it, but frank delusional ideation has clearly receded. He still shows some signs of grief. Mr. M is advised to stop using Cannabis and N2O and about the potential consequences of continued use.
The authors’ observations
For patients with vitamin B12 deficiency, guidelines from the National Health Service in the United Kingdom and the British Society for Haematology recommend treatment with IM hydroxocobalamin, 1,000 IU, 3 times weekly, for 2 weeks.21,28 For patients with neurologic symptoms, the British National Foundation recommends treatment with IM hydroxocobalamin, 1,000 IU, on alternative days until there is no further improvement.21
This case is a reminder for clinicians to screen for inhalant use, specifically N2O, which can precipitate vitamin B12 deficiency with psychiatric symptoms as the only presenting concern. Clinicians should consider measuring vitamin B12 levels in psychiatric patients at risk of deficiency of this nutrient, including older adults, vegetarians, and those with alimentary disorders.29,30 Dietary sources of vitamin B12 include meat, milk, egg, fish, and shellfish.31 The body can store a total of 2 to 5 mg of vitamin B12; thus, it takes 2 to 5 years to develop vitamin B12 deficiency from malabsorption and can take as long as 20 years to develop vitamin B12 deficiency from vegetarianism.32 However, by chemically inactivating vitamin B12, N2O causes a rapid functional deficiency, as was seen in our patient.
OUTCOME Improved insight
At a 1-week follow-up appointment with a psychiatrist, Mr. M has no evident psychotic symptoms. He reports that he has not used Cannabis or N2O, and he discontinues olanzapine following this visit. Two weeks later, Mr. M shows no psychotic or affective symptoms other than grief, which is appropriately expressed. His insight has improved. He commits to not using Cannabis, N2O, or any other illicit substances. Mr. M is referred back to his long-standing primary care provider with the understanding that if any psychiatric symptoms recur he will see a psychiatrist again.
CASE Grieving, delusional
Mr. M, age 51, is brought to the emergency department (ED) because of new-onset delusions and decreased self-care over the last 2 weeks following the sudden death of his wife. He has become expansive and grandiose, with pressured speech, increased energy, and markedly reduced sleep. Mr. M is preoccupied with the idea that he is “the first to survive a human reboot process” and says that his and his wife’s bodies and brains had been “split apart.” Mr. M has limited his food and fluid intake and lost 15 lb within the past 2 to 3 weeks.
Mr. M has no history of any affective, psychotic, or other major mental disorders or treatment. He reports that he has regularly used Cannabis over the last 10 years, and a few years ago, he started occasionally using nitrous oxide (N2O). He says that in the week following his wife’s death, he used N2O almost daily and in copious amounts. In an attempt to “self-anesthetize” himself after his wife’s funeral, he isolated himself in his bedroom and used escalating amounts of Cannabis and N2O, while continually working on a book about their life together.
At first, Mr. M shows little emotion and describes his situation as “interesting and fascinating.” He mentions that he thinks he might have been “psychotic” the week after his wife’s death, but he shows no sustained insight and immediately relapses into psychotic thinking. Over several hours in the ED, he is tearful and sad about his wife’s death. Mr. M recalls a similar experience of grief after his mother died when he was a teenager, but at that time he did not abuse substances or have psychotic symptoms. He is fully alert, fully oriented, and has no significant deficits of attention or memory.
[polldaddy:9859135]
The authors’ observations
Grief was a precipitating event, but by itself grief cannot explain psychosis. Psychotic depression is a possibility, but Mr. M’s psychotic features are incongruent with his mood. Mania would be a diagnosis of exclusion. Mr. M had no prior history of major affective illness. Mr. M was abusing Cannabis, which might independently contribute to psychosis1; however, he had been using it recreationally for 10 years without psychiatric problems. N2O, however, can cause symptoms consistent with Mr. M’s presentation.
[polldaddy:9859140]
EVALUATION Laboratory tests
Mr. M’s physical examination is notable only for an elevated blood pressure of 196/120 mm Hg. Neurologic examination is normal. Toxicology is positive for cannabinoids and negative for amphetamines, cocaine, opiates, and phencyclidine. Chemistries are normal except for a potassium of 3.4 mEq/L (reference range, 3.7 to 5.2 mEq/L) and a blood urine nitrogen of 25 mg/dL (reference range, 6 to 20 mg/dL), which are consistent with reduced food and fluid intake. Mr. M shows no signs of anemia. Hematocrit is 42% and mean corpuscular volume is 90 fL. Syphilis screen is negative; a head CT scan is unremarkable.
The authors’ observations
N2O, also known as “laughing gas,” is routinely used by dentists and pediatric anesthesiologists, and has other medical uses. Some studies have examined an adjunctive use of N2O for pain control in the ED and during colonoscopies.3,4
In the 2013 U.S. National Survey on Drug Use and Health, 16% of respondents reported lifetime illicit use of N2O.5,6 It is readily available in tanks used in medicine and industry and in small dispensers called “whippits” that can be legally purchased. Acute effects of N2O include euphoric mood, numbness, feeling of warmth, dizziness, and auditory hallucinations.7 The anesthetic effects of N2O are linked to endogenous release of opiates, and recent research links its anxiolytic activity to the facilitation of GABAergic inhibitory and N-methyl-
Beginning with a 1960 report of a series of patients with “megaloblastic madness,”17 there have been calls for increased awareness of the potential for vitamin B12 deficiency–induced psychiatric disorders, even in the absence of other hematologic or neurologic sequelae that would alert clinicians of the deficiency. In a case series of 141 patients with a broad array of neurologic and psychiatric symptoms associated with vitamin B12 deficiency, 40 (28%) patients had no anemia or macrocytosis.2
Vitamin B12-responsive psychosis has been reported as the sole manifestation of illness, without associated neurologic or hematologic symptoms, in only a few case reports. Vitamin B12 levels in these cases ranged from 75 to 236 pg/mL (reference range, 160 to 950 pg/mL).18-20 In all of these cases, the vitamin B12 deficiency was traced to dietary causes. The clinical evaluation of suspected vitamin B12 deficiency is outlined in the Figure.21 Mr. M had used Cannabis recreationally for a long time, and his Cannabis use acutely escalated with use of N2O. Long-term use of Cannabis alone is a risk factor for psychotic illness.22 Combined abuse of Cannabis and N2O has been reported to provoke psychotic illness. In a case report of a 22-year-old male who was treated for paranoid delusions, using Cannabis and 100 cartridges of N2O daily was associated with low vitamin B12 and elevated homocysteine and methylmalonic acid levels.23
Cannabis use may have played a role in Mr. M’s escalating N2O use. In a study comparing 9 active Cannabis users with 9 non-using controls, users rated the subjective effects of N2O as more intense than non-users.24 In our patient’s case, Cannabis may have played a role in both sustaining his escalating N2O abuse and potentiating its psychotomimetic effects.
It also is possible that Mr. M may have been “self-medicating” his grief with N2O. In a recent placebo-controlled crossover trial of 20 patients with treatment-resistant depression, Nagele et al25 found a significant rapid and week-long antidepressant effect of subanesthetic N2O use. A model involving NMDA receptor activation has been proposed.25,26 Zorumski et al26 further reviewed possible antidepressant mechanisms of N2O. They compared N2O with ketamine as an NMDA receptor antagonist, but also noted its distinct effects on glutaminergic and GABAergic neurotransmitter systems as well as other receptors and channels.26 However, illicit use of N2O poses toxicity dangers and has no current indication for psychiatric treatment.
TREATMENT Supplementation
Mr. M is diagnosed with substance-induced psychotic disorder. His symptoms were precipitated by an acute increase in N2O use, which has been shown to cause vitamin B12 deficiency, which we consider was likely a primary contributor to his presentation. Other potential contributing factors are premorbid hyperthymic temperament, a possible propensity to psychotic thinking under stress, the sudden death of his wife, acute grief, the potentiating role of Cannabis, dehydration, and general malnutrition. The death of a loved one is associated with an increased risk of developing substance use disorders.27
During a 15-day psychiatric hospitalization, Mr. M is given olanzapine, increased to 15 mg/d and oral vitamin B12, 1,000 mcg/d for 4 days, then IM cyanocobalamin for 7 days. Mr. M’s symptoms steadily improve, with normalization of sleep and near-total resolution of delusions. On hospital Day 14, his vitamin B12 levels are within normal limits (844 pg/mL). At discharge, Mr. M shows residual mild grandiosity, with limited insight into his illness and what caused it, but frank delusional ideation has clearly receded. He still shows some signs of grief. Mr. M is advised to stop using Cannabis and N2O and about the potential consequences of continued use.
The authors’ observations
For patients with vitamin B12 deficiency, guidelines from the National Health Service in the United Kingdom and the British Society for Haematology recommend treatment with IM hydroxocobalamin, 1,000 IU, 3 times weekly, for 2 weeks.21,28 For patients with neurologic symptoms, the British National Foundation recommends treatment with IM hydroxocobalamin, 1,000 IU, on alternative days until there is no further improvement.21
This case is a reminder for clinicians to screen for inhalant use, specifically N2O, which can precipitate vitamin B12 deficiency with psychiatric symptoms as the only presenting concern. Clinicians should consider measuring vitamin B12 levels in psychiatric patients at risk of deficiency of this nutrient, including older adults, vegetarians, and those with alimentary disorders.29,30 Dietary sources of vitamin B12 include meat, milk, egg, fish, and shellfish.31 The body can store a total of 2 to 5 mg of vitamin B12; thus, it takes 2 to 5 years to develop vitamin B12 deficiency from malabsorption and can take as long as 20 years to develop vitamin B12 deficiency from vegetarianism.32 However, by chemically inactivating vitamin B12, N2O causes a rapid functional deficiency, as was seen in our patient.
OUTCOME Improved insight
At a 1-week follow-up appointment with a psychiatrist, Mr. M has no evident psychotic symptoms. He reports that he has not used Cannabis or N2O, and he discontinues olanzapine following this visit. Two weeks later, Mr. M shows no psychotic or affective symptoms other than grief, which is appropriately expressed. His insight has improved. He commits to not using Cannabis, N2O, or any other illicit substances. Mr. M is referred back to his long-standing primary care provider with the understanding that if any psychiatric symptoms recur he will see a psychiatrist again.
1. Semple DM, McIntosh AM, Lawrie SM. Cannabis as a risk factor for psychosis: systematic review. J Psychopharmacol. 2005;19(2):187-194.
2. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26)1720-1728.
3. Herres J, Chudnofsky CR, Manur R, et al. The use of inhaled nitrous oxide for analgesia in adult ED patients: a pilot study. Am J Emerg Med. 2016;34(2):269-273.
4. Aboumarzouk OM, Agarwal T, Syed Nong Chek SA, et al. Nitrous oxide for colonoscopy. Cochrane Database Syst Rev. 2011;(8):CD008506.
5. National Institute on Drug Abuse. Drug facts: inhalants. http://www.drugabuse.gov/publications/drugfacts/inhalants. Updated February 2017. Accessed September 30, 2017.
6. SAMHSA, Center for Behavioral Health Statistics and Quality, National Survey on Drug Use and Health 2012 and 2013: Table 1.88C. https://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabs2013.pdf. Published September 4, 2017. Accessed September 30, 2017.
7. Brouette T, Anton R. Clinical review of inhalants. Am J Addict. 2001;10(1):79-94.
8. Emmanouil DE, Quock RM. Advances in understanding the actions of nitrous oxide. Anesth Prog. 2007;54(1):9-18.
9. Garakani A, Jaffe RJ, Savla D, et al. Neurologic, psychiatric, and other medical manifestations of nitrous oxide abuse: a systematic review of the case literature. Am J Addict. 2016;25(5):358-369.
10. Hathout L, El-Saden S. Nitrous oxide-induced B12 deficiency myelopathy: perspectives on the clinical biochemistry of vitamin B12. J Neurol Sci. 2011;301(1-2):1-8.
11. van Tonder SV, Ruck A, van der Westhuyzen J, et al. Dissociation of methionine synthetase (EC 2.1.1.13) activity and impairment of DNA synthesis in fruit bats (Rousettus aegyptiacus) with nitrous oxide-induced vitamin B12 deficiency. Br J Nutr. 1986;55(1):187-192.
12. Schrier SL, Mentzer WC, Tirnauer JS. Diagnosis and treatment of vitamin B12 and folate deficiency. UpToDate. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-vitamin-b12-and-folate-deficiency. Updated September 30, 2011. Accessed September 8, 2015.
13. Sethi NK, Mullin P, Torgovnick J, et al. Nitrous oxide “whippit” abuse presenting with cobalamin responsive psychosis. J Med Toxicol. 2006;2(2):71-74.
14. Cousaert C, Heylens G, Audenaert K. Laughing gas abuse is no joke. An overview of the implications for psychiatric practice. Clin Neurol Neurosurg. 2013;115(7):859-862.
15. Brodsky L, Zuniga J. Nitrous oxide: a psychotogenic agent. Compr Psychiatry. 1975;16(2):185-188.
16. Wong SL, Harrison R, Mattman A, et al. Nitrous oxide (N2O)-induced acute psychosis. Can J Neurol Sci. 2014;41(5):672-674.
17. Smith AD. Megaloblastic madness. Br Med J. 1960;2(5216):1840-1845.
18. Masalha R, Chudakov B, Muhamad M, et al. Cobalamin-responsive psychosis as the sole manifestation of vitamin B12 deficiency. Isr Med Associ J. 2001;3(9):701-703.
19. Kuo SC, Yeh SB, Yeh YW, et al. Schizophrenia-like psychotic episode precipitated by cobalamin deficiency. Gen Hosp Psychiatry. 2009;31(6):586-588.
20. Raveendranathan D, Shiva L, Venkatasubramanian G, et al. Vitamin B12 deficiency masquerading as clozapine-resistant psychotic symptoms in schizophrenia. J Neuropsychiatry Clin Neurosci. 2013;25(2):E34-E35.
21. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513.
22. Moore THM, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet. 2007;370:319-328.
23. Garakani A, Welch AK, Jaffe RJ, et al. Psychosis and low cyanocobalamin in a patient abusing nitrous oxide and cannabis. Psychosomatics. 2014;55(6):715-719.
24. Yajnik S, Thapar P, Lichtor JL, et al. Effects of marijuana history on the subjective, psychomotor, and reinforcing effects of nitrous oxide in human. Drug Alcohol Depend. 1994;36(3):227-236.
25. Nagele P, Duma A, Kopec M, et al. Nitrous oxide for treatment-resistant major depression: a proof-of-concept trial. Biol Psychiatry. 2015;78(1):10-18.
26. Zorumski CF, Nagele P, Mennerick S, et al. Treatment-resistant major depression: rationale for NMDA receptors as targets and nitrous oxide as therapy. Front Psychiatry. 2015;6:172.
27. Shear MK. Clinical practice. Complicated grief. N Engl J Med. 2015;372(2):153-160.
28. Knechtli CJC, Crowe JN. Guidelines for the investigation & management of vitamin B12 deficiency. Royal United Hospital Bath, National Health Service. http://www.ruh.nhs.uk/For_Clinicians/departments_ruh/Pathology/documents/haematology/B12_-_advice_on_investigation_management.pdf. Accessed June 14, 2016.
29. Jayaram N, Rao MG, Narashima A, et al. Vitamin B12 levels and psychiatric symptomatology: a case series. J Neuropsychiatry Clin Neurosci. 2013;25(2):150-152.
30. Marks PW, Zukerberg LR. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 30-2004. A 37-year-old woman with paresthesias of the arms and legs. N Engl J Med. 2004;351(13):1333-1341.
31. Watanabe F. Vitamin B12 sources and bioavailablility. Exp Biol Med (Maywood). 2007;232(10):1266-1274.
32. Green R, Kinsella LJ. Current concepts in the diagnosis of cobalamin deficiency. Neurology. 1995;45(8):1435-1440.
1. Semple DM, McIntosh AM, Lawrie SM. Cannabis as a risk factor for psychosis: systematic review. J Psychopharmacol. 2005;19(2):187-194.
2. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26)1720-1728.
3. Herres J, Chudnofsky CR, Manur R, et al. The use of inhaled nitrous oxide for analgesia in adult ED patients: a pilot study. Am J Emerg Med. 2016;34(2):269-273.
4. Aboumarzouk OM, Agarwal T, Syed Nong Chek SA, et al. Nitrous oxide for colonoscopy. Cochrane Database Syst Rev. 2011;(8):CD008506.
5. National Institute on Drug Abuse. Drug facts: inhalants. http://www.drugabuse.gov/publications/drugfacts/inhalants. Updated February 2017. Accessed September 30, 2017.
6. SAMHSA, Center for Behavioral Health Statistics and Quality, National Survey on Drug Use and Health 2012 and 2013: Table 1.88C. https://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabs2013.pdf. Published September 4, 2017. Accessed September 30, 2017.
7. Brouette T, Anton R. Clinical review of inhalants. Am J Addict. 2001;10(1):79-94.
8. Emmanouil DE, Quock RM. Advances in understanding the actions of nitrous oxide. Anesth Prog. 2007;54(1):9-18.
9. Garakani A, Jaffe RJ, Savla D, et al. Neurologic, psychiatric, and other medical manifestations of nitrous oxide abuse: a systematic review of the case literature. Am J Addict. 2016;25(5):358-369.
10. Hathout L, El-Saden S. Nitrous oxide-induced B12 deficiency myelopathy: perspectives on the clinical biochemistry of vitamin B12. J Neurol Sci. 2011;301(1-2):1-8.
11. van Tonder SV, Ruck A, van der Westhuyzen J, et al. Dissociation of methionine synthetase (EC 2.1.1.13) activity and impairment of DNA synthesis in fruit bats (Rousettus aegyptiacus) with nitrous oxide-induced vitamin B12 deficiency. Br J Nutr. 1986;55(1):187-192.
12. Schrier SL, Mentzer WC, Tirnauer JS. Diagnosis and treatment of vitamin B12 and folate deficiency. UpToDate. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-vitamin-b12-and-folate-deficiency. Updated September 30, 2011. Accessed September 8, 2015.
13. Sethi NK, Mullin P, Torgovnick J, et al. Nitrous oxide “whippit” abuse presenting with cobalamin responsive psychosis. J Med Toxicol. 2006;2(2):71-74.
14. Cousaert C, Heylens G, Audenaert K. Laughing gas abuse is no joke. An overview of the implications for psychiatric practice. Clin Neurol Neurosurg. 2013;115(7):859-862.
15. Brodsky L, Zuniga J. Nitrous oxide: a psychotogenic agent. Compr Psychiatry. 1975;16(2):185-188.
16. Wong SL, Harrison R, Mattman A, et al. Nitrous oxide (N2O)-induced acute psychosis. Can J Neurol Sci. 2014;41(5):672-674.
17. Smith AD. Megaloblastic madness. Br Med J. 1960;2(5216):1840-1845.
18. Masalha R, Chudakov B, Muhamad M, et al. Cobalamin-responsive psychosis as the sole manifestation of vitamin B12 deficiency. Isr Med Associ J. 2001;3(9):701-703.
19. Kuo SC, Yeh SB, Yeh YW, et al. Schizophrenia-like psychotic episode precipitated by cobalamin deficiency. Gen Hosp Psychiatry. 2009;31(6):586-588.
20. Raveendranathan D, Shiva L, Venkatasubramanian G, et al. Vitamin B12 deficiency masquerading as clozapine-resistant psychotic symptoms in schizophrenia. J Neuropsychiatry Clin Neurosci. 2013;25(2):E34-E35.
21. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513.
22. Moore THM, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet. 2007;370:319-328.
23. Garakani A, Welch AK, Jaffe RJ, et al. Psychosis and low cyanocobalamin in a patient abusing nitrous oxide and cannabis. Psychosomatics. 2014;55(6):715-719.
24. Yajnik S, Thapar P, Lichtor JL, et al. Effects of marijuana history on the subjective, psychomotor, and reinforcing effects of nitrous oxide in human. Drug Alcohol Depend. 1994;36(3):227-236.
25. Nagele P, Duma A, Kopec M, et al. Nitrous oxide for treatment-resistant major depression: a proof-of-concept trial. Biol Psychiatry. 2015;78(1):10-18.
26. Zorumski CF, Nagele P, Mennerick S, et al. Treatment-resistant major depression: rationale for NMDA receptors as targets and nitrous oxide as therapy. Front Psychiatry. 2015;6:172.
27. Shear MK. Clinical practice. Complicated grief. N Engl J Med. 2015;372(2):153-160.
28. Knechtli CJC, Crowe JN. Guidelines for the investigation & management of vitamin B12 deficiency. Royal United Hospital Bath, National Health Service. http://www.ruh.nhs.uk/For_Clinicians/departments_ruh/Pathology/documents/haematology/B12_-_advice_on_investigation_management.pdf. Accessed June 14, 2016.
29. Jayaram N, Rao MG, Narashima A, et al. Vitamin B12 levels and psychiatric symptomatology: a case series. J Neuropsychiatry Clin Neurosci. 2013;25(2):150-152.
30. Marks PW, Zukerberg LR. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 30-2004. A 37-year-old woman with paresthesias of the arms and legs. N Engl J Med. 2004;351(13):1333-1341.
31. Watanabe F. Vitamin B12 sources and bioavailablility. Exp Biol Med (Maywood). 2007;232(10):1266-1274.
32. Green R, Kinsella LJ. Current concepts in the diagnosis of cobalamin deficiency. Neurology. 1995;45(8):1435-1440.
Deutetrabenazine for tardive dyskinesia
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
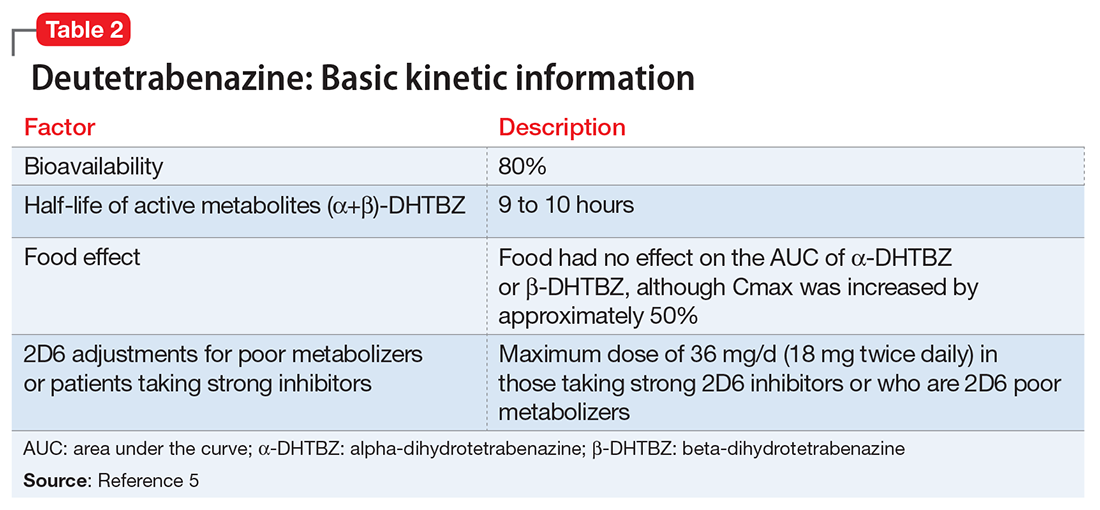
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
The etiology of premenstrual dysphoric disorder: 5 interwoven pieces
In an age when psychiatry strives to identify the biologic causes of disease, studying endocrine-related mood disorders is particularly intriguing. DSM-5 defines premenstrual dysphoric disorder (PMDD) as a depressive disorder, with a 12-month prevalence ranging from 1.8% to 5.8% among women who menstruate.1-3 Factors that differentiate PMDD from other affective disorders include etiology, duration, and temporal relationship with the menstrual cycle.
PMDD is a disorder of consistent yet intermittent change in mental health and functionality. Therefore, it may be underdiagnosed and consequently undertreated if a psychiatric evaluation does not coincide with symptom occurrence or if patients do not understand that intermittent symptoms are treatable.
This article summarizes what is known about the etiology of PMDD. Although there are several treatments for PMDD, many women experience adverse effects or incomplete effectiveness. Further understanding of this disorder may lead to more efficacious treatments. Additionally, understanding the pathophysiology of PMDD might shed a light on the etiology of other disorders that are temporally related to reproductive life changes, such as pregnancy-, postpartum-, or menopause-related affective dysregulation.
Making the diagnosis
The diagnosis of PMDD is made when a patient has at least 5 of 11 specific symptoms that occur during the week before onset of menses, improve within a few days after the onset of menses (shown as the “PMDD Hazard Zone” in Figure 1), and are minimal or absent post-menses.3 Symptoms should be tracked prospectively for at least 2 menstrual cycles in order to confirm the diagnosis (one must be an affective symptom and another must be a behavioral/cognitive symptom).3
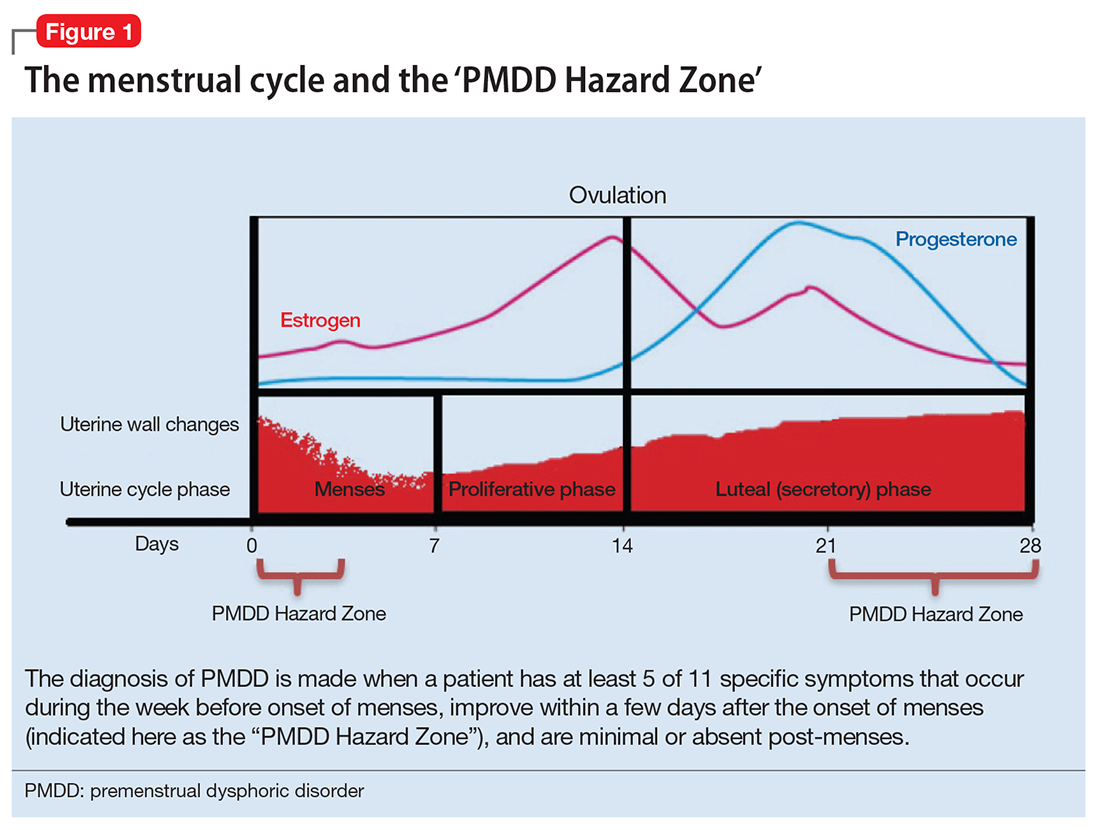
The affective symptoms are:
- lability of affect (eg, sudden sadness, tearfulness, or sensitivity to rejection)
- irritability, anger, or increased interpersonal conflicts
- depressed mood, hopelessness, or self- deprecating thoughts
- anxiety or tension, feeling “keyed up” or “on edge.”
The behavioral/cognitive symptoms are:
- decreased interest in usual activities (eg, work, hobbies, friends, school)
- difficulty concentrating
- lethargy, low energy, easy fatigability
- change in appetite, overeating, food cravings
- hypersomnia or insomnia
- feeling overwhelmed or out of control
- physical symptoms (breast tenderness or swelling, headache, joint or muscle pain, bloating, weight gain).
Ruling out premenstrual exacerbation (PME). Perhaps the most common cause for misdiagnosis of PMDD is failing to rule out PME of another underlying or comorbid condition (Figure 2). In many women who have a primary mood or anxiety disorder, the late luteal phase is a vulnerable time. A patient might be coping with untreated anxiety, for example, but the symptoms become unbearable the week before menstruation begins, which is likely when she seeks help. At this stage, a diagnosis of PMDD should be provisional at best. Often, PME is treated by treating the underlying condition. Therefore, a full diagnostic psychiatric interview is important to first rule out other underlying psychiatric disorders. PMDD is diagnosed if the premenstrual symptoms persist for 2 consecutive months after treating the suspected mood or anxiety disorder. Patients can use one of many PMDD daily symptom charts available online. Alternatively, they can use a cycle-tracking mobile phone application to correlate their symptoms with their cycle and share this information with their providers.
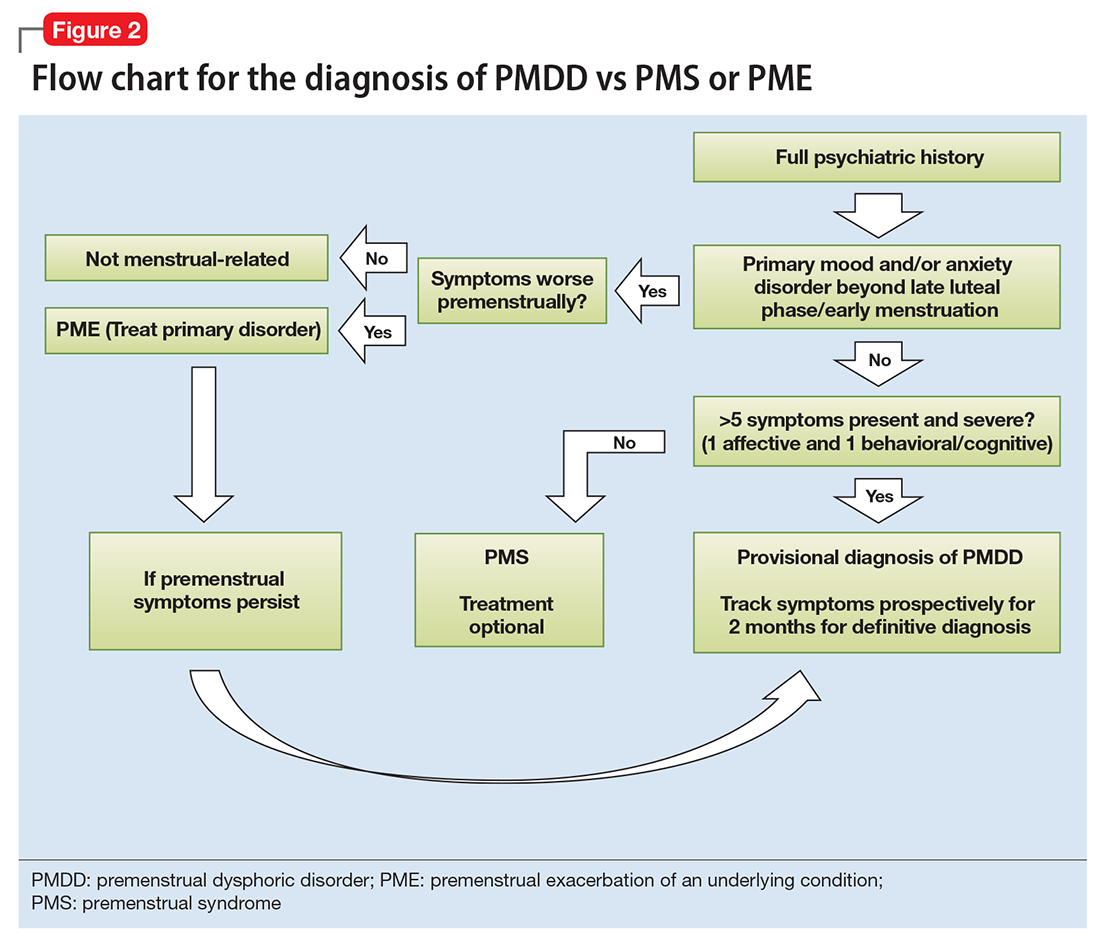
Consider these 5 interwoven pieces
The many variables that contribute to the pathophysiology of PMDD overlap and should be considered connecting pieces in the puzzle that is the etiology of this disorder (Figure 3). In reviewing the literature, we have identified 5 topics likely to be major contributors to this disorder:
- genetic susceptibility
- progesterone and allopregnanolone (ALLO)
- estrogen, serotonin, and brain-derived neurotrophic factor (BDNF)
- putative brain structural and functional differences
- further involvement of the hypothalamic–pituitary–adrenal (HPA) axis and hypothalamic–pituitary–gonadal (HPG) axis: trauma, resiliency, and inflammation.
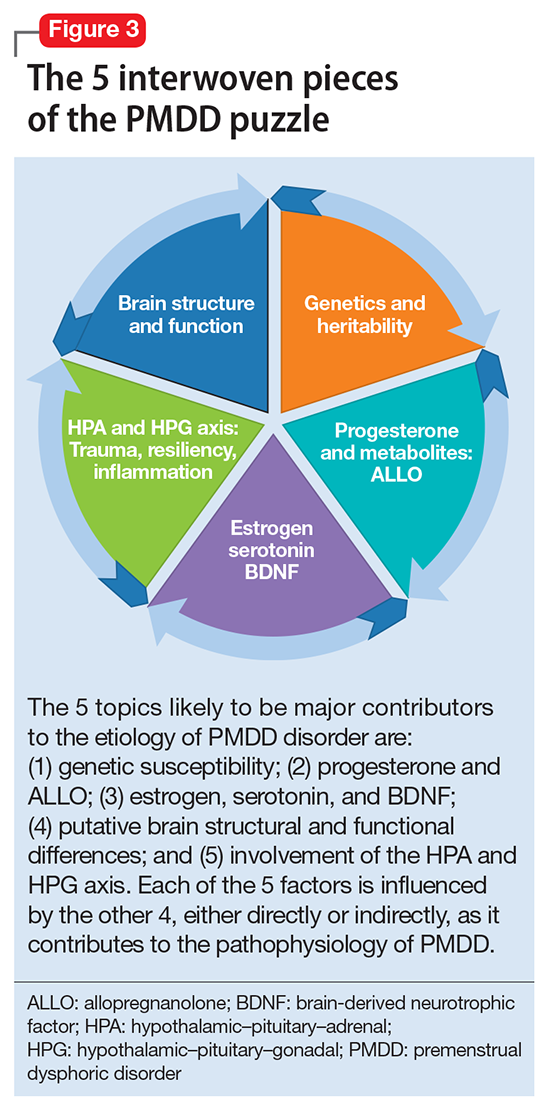
Genetic susceptibility. PMDD is thought to have a heritability range between 30% to 80%.3 This is demonstrated by family and twin studies4-7 and specific genetic studies.8 The involvement of genetics means an underlying neurobiologic pathophysiology is in place.
Estrogen receptor alpha (ESR1) gene. Huo et al8 found an associated variation in ESR1 in women with PMDD compared with controls. They speculated that because ESR1 is important for arousal, if dysfunctional, this gene could be implicated in somatic as well as affective and cognitive deficits in PMDD patients. In another study, investigators reported a relationship between PMDD and heritable personality traits, as well as a link between these traits and ESR1 polymorphic variants.1 They suggested that personality traits (independent of affective state) might be used to distinguish patients with PMDD from controls.1
Studies on serotonin gene polymorphism and serotonin transporter genotype. Although a study of serotonin gene polymorphism did not find an association between serotonin1A gene polymorphism and PMDD, it did show that the presence of at least 1 C allele was associated with a 2.5-fold increased risk of PMDD.9 Another study did not find an association between the serotonin transporter genotype 5-HTTLPR and PMDD.10 However, it showed lower frontocingulate cortex activation during the luteal phase of PMDD patients compared with controls, suggesting that PMDD is linked to impaired frontocingulate cortex activation induced by emotions during the luteal phase.10
Seasonal affective disorder (SAD) and PMDD have shared clinical features. A polymorphism in the serotonin transporter promoter gene 5-HTTLPR has been associated with SAD. One study found that patients with comorbid SAD and PMDD are genetically more vulnerable to comorbid affective disorders compared with patients who have SAD only.11
Progesterone and ALLO. Chronic exposure to progesterone and ALLO (a main progesterone metabolite) and rapid withdrawal from ovarian hormones may play a role in the etiology of PMDD. Much like alcohol or benzodiazepines, ALLO is a potent positive allosteric modulator of GABAA receptors and has sedative, anesthetic, and anxiolytic properties. In times of acute stress, increased ALLO is known to provide relief.12,13 However, in women with PMDD, this typical ALLO increase might not occur.14
Patients with PMDD have been reported to have decreased levels of ALLO in the luteal phase.15-17 In one study, women with highly symptomatic PMDD had lower levels of ALLO compared with women with less symptomatic PMDD.14 A gonadotropin-releasing hormone challenge study showed the increase in ALLO response was less in PMDD patients compared with controls.17 Luteal-phase ALLO concentrations are reported to be lower in women with premenstrual syndrome (PMS), a milder form of PMDD.14,17
The efficacy of selective serotonin reuptake inhibitors (SSRIs) for treating PMDD could be the result of the interaction of these medications with neuroactive steroids,18 possibly because SSRIs enhance the sensitivity of GABAA receptors or promote the formation of more ALLO (Figure 4).19-21
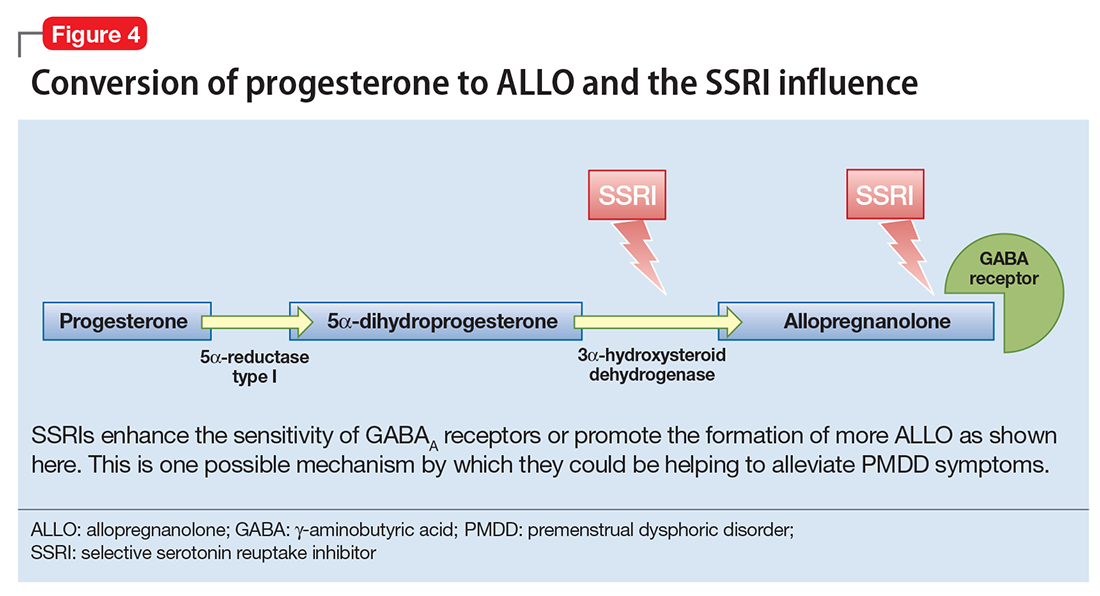
Estrogen, serotonin, and BDNF. Estrogen affects multiple neurotransmitter systems that regulate mood, cognition, sleep, and eating.22 Studying estrogen in context of PMDD is important because women with PMDD can have low mood, specific food cravings, and impaired cognitive function.
Estrogen–serotonin interactions are thought to be involved in hormone-related mood disorders such as perimenopausal depression and PMDD.23,24 However, the nature of their relationship is not yet fully understood. Ovariectomized animals have shown estrogen-induced changes related to serotonin metabolism, binding, and transmission in the regions of the brain involved in regulation of affect and cognition. Research in menopausal women also has provided some support for this interaction.24
Positron emission tomography studies in humans have found increased cortical serotonin binding modulated by levels of estrogen, similar to those previously seen in rat studies.24-27 One study showed an increased binding potential of serotonin in the cerebral cortex with estrogen treatment. This study further showed an even greater binding potential with estrogen plus progesterone, signaling a synergistic effect of the 2 hormones.28
SSRIs are an effective treatment for the irritability, anxiety, and mood swings of PMDD.29-30 Although the exact mechanism of action is unknown, the serotonergic properties are certainly of primary attention. For some PMDD patients, SSRIs work within hours to days, as opposed to days or weeks for patients with depression or anxiety, which suggests a separate or co-occurring mechanism of action is in place. In a double-blind, placebo-controlled crossover study, researchers administered the serotonin receptor antagonist metergoline to women with PMDD whose symptoms had remitted during treatment with fluoxetine and a group of healthy controls who were not receiving any medication.31 The women with PMDD experienced a return of symptoms 24 hours after treatment with metergoline but not with placebo; the controls experienced no mood changes.31
BDNF is a neurotransmitter linked to estrogen and likely related to PMDD. BDNF is critical for neurogenesis and is expressed in brain regions involved in learning and memory and also affects regulation.32 BDNF levels are increased by serotonergic antidepressants, affected by estradiol, and have cyclicity throughout the menstrual cycle.33-35
Putative brain structural and functional differences. Imaging studies have suggested differences in brain structure in women with PMDD, with a focus on the amygdala and the prefrontal cortex. Women with PMDD have greater gray matter volume in the posterior cerebellum,36 greater gray matter density of hippocampal cortex, and lower gray matter density in the parahippocampal cortex.37
Some studies have shown a functional variability of the amygdala’s response to stress in women with PMDD vs healthy controls.38,39 A proton magnetic resonance spectroscopy (1H-MRS) study of the displays the possibility of an altered GABAergic function in patients with PMDD.40
Patients will PMDD have enhanced dorsolateral prefrontal cortex reactivity when anticipating negative stimuli (but not to the actual exposure) during the luteal phase. A positive correlation between this reactivity and progesterone levels also was observed.41 Some researchers have suggested that prefrontal cortex dysfunction may be a risk factor for PMDD.42
HPA axis and HPG axis: Trauma, resiliency, inflammation. Altered cortisol levels (higher during the luteal phase43 and lower during times of stress14,44) suggest a possibly altered HPA axis in some women with PMDD. However, studies on this topic have been few and inconsistent.
Dysregulation of the HPG axis could cause vasomotor symptoms, sleep dysregulation, and mood symptoms during menopause; women with PMDD can also experience these symptoms. The influence of estrogen and progesterone on mood is also highly dependent on this axis.
Ultimately, the interplay between the HPA axis and the HPG axis is important. One study found that women with PMDD who had high serum ALLO levels (HPG-related) had blunted nocturnal cortisol levels (HPA-related) compared with healthy controls who had low ALLO levels.45
Significant stress and trauma exposure have been associated with PMDD. A study of 3,968 women found a history of trauma and PTSD were independently associated with PMDD.46 Another study of approximately 3,000 women found a strong correlation between abuse and PMS.47 However, a third study found no correlations between PMDD and trauma.48
Patients with a predisposition to PMDD may be more vulnerable to develop a posttraumatic stress-related disorder, perhaps due to decreased biologic resiliency. For example, the startle response (hypervigilance) has been shown to be different in women with PMDD. One study suggested that suboptimal production of premenstrual ALLO may lead to increased arousal and increased stress reactivity to psychosocial or environmental triggers.49
The possible role of inflammation in PMDD deserves further investigation. The luteal phase entails an increase in the production of proinflammatory markers.50,51 A 10-fold increase in progesterone is correlated with a 20% to 23% increase in C-reactive protein levels.52,53 Women with inflammatory diseases (eg, gingivitis or irritable bowel syndrome) show worsening of symptoms prior to menstruation.54-56 One study found increased levels of proinflammatory markers in women with PMDD compared with controls.57
Putting together the 5 pieces of the puzzle
Because PMDD is heritable, it must have an underlying neurobiologic pathophysiology. Brain imaging studies show differences in structure and function in women with PMDD across the menstrual cycle. Conversion of progesterone to ALLO and the GABAergic influence of this metabolite is a topic of interest in current research. Similarly, the role of estrogen and its connection to serotonin and other neurotransmitters such as BDNF have been implicated.
The link between a history of stress, trauma, and PMDD raises the question of biologic resiliency and illness in these patients, as it connects to the HPA and HPG axis and production of inflammatory stress hormones and steroid hormones and their metabolites. PMDD can be conceptualized as variable sensitivity to hormonal response to stress,58 thus contextualizing biochemical and psychological resiliency.
Further research is needed to clarify the possibility of a shared pathophysiology between endocrine-related mood disorders such as postpartum depression (PPD) and PMDD because current research is controversial.59,60 In PPD, women who are exposed to high levels of progesterone and estrogen during pregnancy (just like in the mid-luteal phase) have a sudden drop in these hormones postpartum.
The ‘withdrawal theory.’ The affective symptoms of PMDD resolve almost instantaneously after the start of menstruation. Perhaps this type of immediate relief is akin to substance use disorders and symptoms of withdrawal. It could be that reinstatement of a certain amount of gonadal steroids in the follicular phase of the cycle diminishes a withdrawal-like response to these steroids.
Currently, the main leading theory is that PMDD is a result of “an abnormal response to normal hormonal changes.”61 A new study also has shown that the change in estradiol/progesterone levels (vs the steady state) was associated with PMDD symptoms.62 Thinking of PMDD as a disorder of withdrawal offers an alternative (yet complementary) perspective to the current theory: PMDD may be caused by the absence or diminishing of the above-named hormones and their metabolites in the late luteal phase (in the context of developed “tolerance” during the early- to mid-luteal phase).
Considering the interplay between neurotransmitters and neurosteroids, both a “serotonin withdrawal theory” (caused by a drop in steroid hormones) and a “GABAergic withdrawal theory” (due to the decline in progesterone) could be proposed. This theory would be supported by the fact that SSRIs seem to mitigate symptoms of PMDD as well as the genetic association between PMDD and ESR1. It is more than likely that the “withdrawal” is caused by the interactions between estrogen-serotonin, progesterone-ALLO, and GABA receptors, and the complementary fashion in which progesterone and estrogen influence each other.
1. Miller A, Vo H, Huo L, et al. Estrogen receptor alpha (ESR-1) associations with psychological traits in women with PMDD and controls. J Psychiatr Res. 2010;44(12):788-794.
2. Epperson CN, Steiner M, Hartlage SA, et al. Premenstrual dysphoric disorder: evidence for a new category for DSM-5. Am J Psychiatry. 2012;169(5):465-475.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Wilson CA, Turner CW, Keye WR Jr. Firstborn adolescent daughters and mothers with and without premenstrual syndrome: a comparison. J Adolesc Health. 1991;12(2):130-137.
5. Kendler KS, Silberg JL, Neale MC, et al. Genetic and environmental factors in the aetiology of menstrual, premenstrual and neurotic symptoms: a population-based twin study. Psychol Med. 1992;22(1):85-100.
6. Condon JT. The premenstrual syndrome: a twin study. Br J Psychiatry. 1993;162:481-486.
7. Kendler KS, Karkowski LM, Corey LA, et al. Longitudinal population-based twin study of retrospectively reported premenstrual symptoms and lifetime major depression. Am J Psychiatry. 1998;155(9):1234-1240.
8. Huo L, Straub RE, Roca C, et al. Risk for premenstrual dysphoric disorder is associated with genetic variation in ESR1, the estrogen receptor alpha gene. Biol Psychiatry. 2007;62(8):925-933.
9. Dhingra V, Magnay JL, O’Brien PM, et al. Serotonin receptor 1A C(-1019)G polymorphism associated with premenstrual dysphoric disorder. Obstet Gynecol. 2007;110(4):788-792.
10. Comasco E, Hahn A, Ganger S, et al. Emotional fronto-cingulate cortex activation and brain derived neurotrophic factor polymorphism in premenstrual dysphoric disorder. Hum Brain Mapp. 2014;35(9):4450-4458.
11. Praschak-Rieder N, Willeit M, Winkler D, et al. Role of family history and 5-HTTLPR polymorphism in female seasonal affective disorder patients with and without premenstrual dysphoric disorder. Eur Neuropsychopharmacol. 2002;12(2):129-134.
12. Klatzkin RR, Morrow AL, Light KC, et al. Associations of histories of depression and PMDD diagnosis with allopregnanolone concentrations following the oral administration of micronized progesterone. Psychoneuroendocrinology. 2006;31(10):1208-1219.
13. Crowley SK, Girdler SS. Neurosteroid, GABAergic and hypothalamic pituitary adrenal (HPA) axis regulation: what is the current state of knowledge in humans? Psychopharmacology (Berl). 2014;231(17):3619-3634.
14. Girdler SS, Straneva PA, Light KC, et al. Allopregnanolone levels and reactivity to mental stress in premenstrual dysphoric disorder. Biol Psychiatry. 2001;49(9):788-797.
15. Rapkin AJ, Morgan M, Goldman L, et al. Progesterone metabolite allopregnanolone in women with premenstrual syndrome. Obstet Gynecol. 1997;90(5):709-714.
16. Bicíková M, Dibbelt L, Hill M, et al. Allopregnanolone in women with premenstrual syndrome. Horm Metab Res. 1998;30(4):227-230.
17. Monteleone P, Luisi S, Tonetti A, et al. Allopregnanolone concentrations and premenstrual syndrome. Eur J Endocrinol. 2000;142(3):269-273.
18. Steiner M, Steinberg S, Stewart D, et al. Fluoxetine in the treatment of premenstrual dysphoria. Canadian Fluoxetine/Premenstrual Dysphoria Collaborative Study Group. N Engl J Med. 1995;332(23):1529-1534.
19. Sundström I, Bäckström T. Citalopram increases pregnanolone sensitivity in patients with premenstrual syndrome: an open trial. Psychoneuroendocrinology. 1998;23(1):73-88.
20. Griffin LD, Mellon SH. Selective serotonin reuptake inhibitors directly alter activity of neurosteroidogenic enzymes. Proc Natl Acad Sci U S A. 1999;96(23):13512-13517.
21. Trauger JW, Jiang A, Stearns BA, et al. Kinetics of allopregnanolone formation catalyzed by human 3 alpha-hydroxysteroid dehydrogenase type III (AKR1C2). Biochemistry. 2002;41(45):13451-13459.
22. Shanmugan S, Epperson CN. Estrogen and the prefrontal cortex: towards a new understanding of estrogen’s effects on executive functions in the menopause transition. Hum Brain Mapp. 2014;35(3):847-865.
23. Rubinow DR, Schmidt PJ, Roca CA. Estrogen-serotonin interactions: implications for affective regulation. Biol Psychiatry. 1998;44(9):839-850.
24. Amin Z, Canli T, Epperson CN. Effect of estrogen-serotonin interactions on mood and cognition. Behav Cogn Neurosci Rev. 2005;4(1):43-58.
25. Cyr M, Bossé R, Di Paolo T. Gonadal hormones modulate 5-hydroxytryptamine2A receptors: emphasis on the rat frontal cortex. Neuroscience. 1998;83(3):829-836.
26. Fink G, Sumner BE, Rosie R, et al. Estrogen control of central neurotransmission: effect on mood, mental state, and memory. Cell Mol Neurobiol. 1996;16(3):325-344.
27. Sumner BE, Grant KE, Rosie R, et al. Effects of tamoxifen on serotonin transporter and 5-hydroxytryptamine(2A) receptor binding sites and mRNA levels in the brain of ovariectomized rats with or without acute estradiol replacement. Brain Res Mol Brain Res. 1999;73(1-2):119-128.
28. Moses-Kolko EL, Berga SL, Greer PJ, et al. Widespread increases of cortical serotonin type 2A receptor availability after hormone therapy in euthymic postmenopausal women. Fertil Steril. 2003;80(3):554-559.
29. Su TP, Schmidt PJ, Danaceau MA, et al. Fluoxetine in the treatment of premenstrual dysphoria. Neuropsychopharmacology. 1997;16(5):346-356.
30. Steinberg EM, Cardoso GM, Martinez PE, et al. Rapid response to fluoxetine in women with premenstrual dysphoric disorder. Depress Anxiety. 2012;29(6):531-540.
31. Roca CA, Schmidt PJ, Smith MJ, et al. Effects of metergoline on symptoms in women with premenstrual dysphoric disorder. Am J Psychiatry. 2002;159(11):1876-1881.
32. Gray JD, Milner TA, McEwen BS. Dynamic plasticity: the role of glucocorticoids, brain-derived neurotrophic factor and other trophic factors. Neuroscience. 2013;239:214-227.
33. Carbone DL, Handa RJ. Sex and stress hormone influences on the expression and activity of brain-derived neurotrophic factor. Neuroscience. 2013;239:295-303.
34. Pilar-Cuéllar F, Vidal R, Pazos A. Subchronic treatment with fluoxetine and ketanserin increases hippocampal brain-derived neurotrophic factor, β-catenin and antidepressant-like effects. Br J Pharmacol. 2012;165(4b):1046-1057.
35. Deuschle M, Gilles M, Scharnholz B, et al. Changes of serum concentrations of brain-derived neurotrophic factor (BDNF) during treatment with venlafaxine and mirtazapine: role of medication and response to treatment. Pharmacopsychiatry. 2013;46(2):54-58.
36. Berman SM, London ED, Morgan M, et al. Elevated gray matter volume of the emotional cerebellum in women with premenstrual dysphoric disorder. J Affect Disord. 2013;146(2):266-271.
37. Jeong HG, Ham BJ, Yeo HB, et al. Gray matter abnormalities in patients with premenstrual dysphoric disorder: an optimized voxel-based morphometry. J Affect Disord. 2012;140(3):260-267.
38. Protopopescu X, Tuescher O, Pan H, et al. Toward a functional neuroanatomy of premenstrual dysphoric disorder. J Affect Disord. 2008;108(1-2):87-94.
39. Gingnell M, Morell A, Bannbers E, et al. Menstrual cycle effects on amygdala reactivity to emotional stimulation in premenstrual dysphoric disorder. Horm Behav. 2012;62(4):400-406.
40. Epperson CN, Haga K, Mason GF, et al. Cortical gamma-aminobutyric acid levels across the menstrual cycle in healthy women and those with premenstrual dysphoric disorder: a proton magnetic resonance spectroscopy study. Arch Gen Psychiatry. 2002;59(9):851-858.
41. Gingnell M, Bannbers E, Wikström J, et al. Premenstrual dysphoric disorder and prefrontal reactivity during anticipation of emotional stimuli. Eur Neuropsychopharmacol. 2013;23(11):1474-1483.
42. Baller EB, Wei SM, Kohn PD, et al. Abnormalities of dorsolateral prefrontal function in women with premenstrual dysphoric disorder: a multimodal neuroimaging study. Am J Psychiatry. 2013;170(3):305-314.
43. Rasgon N, McGuire M, Tanavoli S, et al. Neuroendocrine response to an intravenous L-tryptophan challenge in women with premenstrual syndrome. Fertil Steril. 2000;73(1):144-149.
44. Huang Y, Zhou R, Wu M, et al. Premenstrual syndrome is associated with blunted cortisol reactivity to the TSST. Stress. 2015;18(2):160-168.
45. Segebladh B, Bannbers E, Moby L, et al. Allopregnanolone serum concentrations and diurnal cortisol secretion in women with premenstrual dysphoric disorder. Arch Womens Ment Health. 2013;16(2):131-137.
46. Pilver CE, Levy BR, Libby DJ, et al. Posttraumatic stress disorder and trauma characteristics are correlates of premenstrual dysphoric disorder. Arch Womens Ment Health. 2011;14(5):383-393.
47. Bertone-Johnson ER, Whitcomb BW, Missmer SA, et al. Early life emotional, physical, and sexual abuse and the development of premenstrual syndrome: a longitudinal study. J Womens Health (Larchmt). 2014;23(9):729-739.
48. Segebladh B, Bannbers E, Kask K, et al. Prevalence of violence exposure in women with premenstrual dysphoric disorder in comparison with other gynecological patients and asymptomatic controls. Acta Obstet Gynecol Scand. 2011;90(7):746-752.
49. Kask K, Gulinello M, Bäckström T, et al. Patients with premenstrual dysphoric disorder have increased startle response across both cycle phases and lower levels of prepulse inhibition during the late luteal phase of the menstrual cycle. Neuropsychopharmacology. 2008;33(9):2283-2290.
50. O’Brien SM, Fitzgerald P, Scully P, et al. Impact of gender and menstrual cycle phase on plasma cytokine concentrations. Neuroimmunomodulation. 2007;14(2):84-90.
51. Northoff H, Symons S, Zieker D, et al. Gender- and menstrual phase dependent regulation of inflammatory gene expression in response to aerobic exercise. Exerc Immunol Rev. 2008;14:86-103.
52. Gaskins AJ, Wilchesky M, Mumford SL, et al. Endogenous reproductive hormones and C-reactive protein across the menstrual cycle: the BioCycle Study. Am J Epidemiol. 2012;175(5):423-431.
53. Wander K, Brindle E, O’Connor KA. C-reactive protein across the menstrual cycle. Am J Phys Anthropol. 2008;136(2):138-146.
54. Jane ZY, Chang CC, Lin HK, et al. The association between the exacerbation of irritable bowel syndrome and menstrual symptoms in young Taiwanese women. Gastroenterol Nurs. 2011;34(4):277-286.
55. Kane SV, Sable K, Hanauer SB. The menstrual cycle and its effect on inflammatory bowel disease and irritable bowel syndrome: a prevalence study. Am J Gastroenterol. 1998;93(10):1867-1872.
56. Shourie V, Dwarakanath CD, Prashanth GV, et al. The effect of menstrual cycle on periodontal health - a clinical and microbiological study. Oral Health Prev Dent. 2012;10(2):185-192.
57. Hantsoo L, Epperson CN. Premenstrual dysphoric disorder: epidemiology and treatment. Curr Psychiatry Rep. 2015;17(11):87.
58. Maeng LY, Milad MR. Sex differences in anxiety disorders: Interactions between fear, stress, and gonadal hormones. Horm Behav. 2015;76:106-117.
59. Lee YJ, Yi SW, Ju DH, et al. Correlation between postpartum depression and premenstrual dysphoric disorder: single center study. Obstet Gynecol Sci. 2015;58(5):353-358.
60. Kepple AL, Lee EE, Haq N, et al. History of postpartum depression in a clinic-based sample of women with premenstrual dysphoric disorder. J Clin Psychiatry. 2016;77(4):e415-e420.
61. Schmidt PJ, Nieman LK, Danaceau MA, et al. Differential behavioral effects of gonadal steroids in women with and in those without premenstrual syndrome. N Engl J Med. 1998;338(4):209-216.
62. Schmidt PJ, Martinez PE, Nieman LK, et al. Premenstrual dysphoric disorder symptoms following ovarian suppression: Triggered by change in ovarian steroid levels but not continuous stable levels. Am J Psychiatry. [published online April 21, 2017]. doi: 10.1176/appi.ajp.2017.16101113.
In an age when psychiatry strives to identify the biologic causes of disease, studying endocrine-related mood disorders is particularly intriguing. DSM-5 defines premenstrual dysphoric disorder (PMDD) as a depressive disorder, with a 12-month prevalence ranging from 1.8% to 5.8% among women who menstruate.1-3 Factors that differentiate PMDD from other affective disorders include etiology, duration, and temporal relationship with the menstrual cycle.
PMDD is a disorder of consistent yet intermittent change in mental health and functionality. Therefore, it may be underdiagnosed and consequently undertreated if a psychiatric evaluation does not coincide with symptom occurrence or if patients do not understand that intermittent symptoms are treatable.
This article summarizes what is known about the etiology of PMDD. Although there are several treatments for PMDD, many women experience adverse effects or incomplete effectiveness. Further understanding of this disorder may lead to more efficacious treatments. Additionally, understanding the pathophysiology of PMDD might shed a light on the etiology of other disorders that are temporally related to reproductive life changes, such as pregnancy-, postpartum-, or menopause-related affective dysregulation.
Making the diagnosis
The diagnosis of PMDD is made when a patient has at least 5 of 11 specific symptoms that occur during the week before onset of menses, improve within a few days after the onset of menses (shown as the “PMDD Hazard Zone” in Figure 1), and are minimal or absent post-menses.3 Symptoms should be tracked prospectively for at least 2 menstrual cycles in order to confirm the diagnosis (one must be an affective symptom and another must be a behavioral/cognitive symptom).3
The affective symptoms are:
- lability of affect (eg, sudden sadness, tearfulness, or sensitivity to rejection)
- irritability, anger, or increased interpersonal conflicts
- depressed mood, hopelessness, or self- deprecating thoughts
- anxiety or tension, feeling “keyed up” or “on edge.”
The behavioral/cognitive symptoms are:
- decreased interest in usual activities (eg, work, hobbies, friends, school)
- difficulty concentrating
- lethargy, low energy, easy fatigability
- change in appetite, overeating, food cravings
- hypersomnia or insomnia
- feeling overwhelmed or out of control
- physical symptoms (breast tenderness or swelling, headache, joint or muscle pain, bloating, weight gain).
Ruling out premenstrual exacerbation (PME). Perhaps the most common cause for misdiagnosis of PMDD is failing to rule out PME of another underlying or comorbid condition (Figure 2). In many women who have a primary mood or anxiety disorder, the late luteal phase is a vulnerable time. A patient might be coping with untreated anxiety, for example, but the symptoms become unbearable the week before menstruation begins, which is likely when she seeks help. At this stage, a diagnosis of PMDD should be provisional at best. Often, PME is treated by treating the underlying condition. Therefore, a full diagnostic psychiatric interview is important to first rule out other underlying psychiatric disorders. PMDD is diagnosed if the premenstrual symptoms persist for 2 consecutive months after treating the suspected mood or anxiety disorder. Patients can use one of many PMDD daily symptom charts available online. Alternatively, they can use a cycle-tracking mobile phone application to correlate their symptoms with their cycle and share this information with their providers.
Consider these 5 interwoven pieces
The many variables that contribute to the pathophysiology of PMDD overlap and should be considered connecting pieces in the puzzle that is the etiology of this disorder (Figure 3). In reviewing the literature, we have identified 5 topics likely to be major contributors to this disorder:
- genetic susceptibility
- progesterone and allopregnanolone (ALLO)
- estrogen, serotonin, and brain-derived neurotrophic factor (BDNF)
- putative brain structural and functional differences
- further involvement of the hypothalamic–pituitary–adrenal (HPA) axis and hypothalamic–pituitary–gonadal (HPG) axis: trauma, resiliency, and inflammation.
Genetic susceptibility. PMDD is thought to have a heritability range between 30% to 80%.3 This is demonstrated by family and twin studies4-7 and specific genetic studies.8 The involvement of genetics means an underlying neurobiologic pathophysiology is in place.
Estrogen receptor alpha (ESR1) gene. Huo et al8 found an associated variation in ESR1 in women with PMDD compared with controls. They speculated that because ESR1 is important for arousal, if dysfunctional, this gene could be implicated in somatic as well as affective and cognitive deficits in PMDD patients. In another study, investigators reported a relationship between PMDD and heritable personality traits, as well as a link between these traits and ESR1 polymorphic variants.1 They suggested that personality traits (independent of affective state) might be used to distinguish patients with PMDD from controls.1
Studies on serotonin gene polymorphism and serotonin transporter genotype. Although a study of serotonin gene polymorphism did not find an association between serotonin1A gene polymorphism and PMDD, it did show that the presence of at least 1 C allele was associated with a 2.5-fold increased risk of PMDD.9 Another study did not find an association between the serotonin transporter genotype 5-HTTLPR and PMDD.10 However, it showed lower frontocingulate cortex activation during the luteal phase of PMDD patients compared with controls, suggesting that PMDD is linked to impaired frontocingulate cortex activation induced by emotions during the luteal phase.10
Seasonal affective disorder (SAD) and PMDD have shared clinical features. A polymorphism in the serotonin transporter promoter gene 5-HTTLPR has been associated with SAD. One study found that patients with comorbid SAD and PMDD are genetically more vulnerable to comorbid affective disorders compared with patients who have SAD only.11
Progesterone and ALLO. Chronic exposure to progesterone and ALLO (a main progesterone metabolite) and rapid withdrawal from ovarian hormones may play a role in the etiology of PMDD. Much like alcohol or benzodiazepines, ALLO is a potent positive allosteric modulator of GABAA receptors and has sedative, anesthetic, and anxiolytic properties. In times of acute stress, increased ALLO is known to provide relief.12,13 However, in women with PMDD, this typical ALLO increase might not occur.14
Patients with PMDD have been reported to have decreased levels of ALLO in the luteal phase.15-17 In one study, women with highly symptomatic PMDD had lower levels of ALLO compared with women with less symptomatic PMDD.14 A gonadotropin-releasing hormone challenge study showed the increase in ALLO response was less in PMDD patients compared with controls.17 Luteal-phase ALLO concentrations are reported to be lower in women with premenstrual syndrome (PMS), a milder form of PMDD.14,17
The efficacy of selective serotonin reuptake inhibitors (SSRIs) for treating PMDD could be the result of the interaction of these medications with neuroactive steroids,18 possibly because SSRIs enhance the sensitivity of GABAA receptors or promote the formation of more ALLO (Figure 4).19-21
Estrogen, serotonin, and BDNF. Estrogen affects multiple neurotransmitter systems that regulate mood, cognition, sleep, and eating.22 Studying estrogen in context of PMDD is important because women with PMDD can have low mood, specific food cravings, and impaired cognitive function.
Estrogen–serotonin interactions are thought to be involved in hormone-related mood disorders such as perimenopausal depression and PMDD.23,24 However, the nature of their relationship is not yet fully understood. Ovariectomized animals have shown estrogen-induced changes related to serotonin metabolism, binding, and transmission in the regions of the brain involved in regulation of affect and cognition. Research in menopausal women also has provided some support for this interaction.24
Positron emission tomography studies in humans have found increased cortical serotonin binding modulated by levels of estrogen, similar to those previously seen in rat studies.24-27 One study showed an increased binding potential of serotonin in the cerebral cortex with estrogen treatment. This study further showed an even greater binding potential with estrogen plus progesterone, signaling a synergistic effect of the 2 hormones.28
SSRIs are an effective treatment for the irritability, anxiety, and mood swings of PMDD.29-30 Although the exact mechanism of action is unknown, the serotonergic properties are certainly of primary attention. For some PMDD patients, SSRIs work within hours to days, as opposed to days or weeks for patients with depression or anxiety, which suggests a separate or co-occurring mechanism of action is in place. In a double-blind, placebo-controlled crossover study, researchers administered the serotonin receptor antagonist metergoline to women with PMDD whose symptoms had remitted during treatment with fluoxetine and a group of healthy controls who were not receiving any medication.31 The women with PMDD experienced a return of symptoms 24 hours after treatment with metergoline but not with placebo; the controls experienced no mood changes.31
BDNF is a neurotransmitter linked to estrogen and likely related to PMDD. BDNF is critical for neurogenesis and is expressed in brain regions involved in learning and memory and also affects regulation.32 BDNF levels are increased by serotonergic antidepressants, affected by estradiol, and have cyclicity throughout the menstrual cycle.33-35
Putative brain structural and functional differences. Imaging studies have suggested differences in brain structure in women with PMDD, with a focus on the amygdala and the prefrontal cortex. Women with PMDD have greater gray matter volume in the posterior cerebellum,36 greater gray matter density of hippocampal cortex, and lower gray matter density in the parahippocampal cortex.37
Some studies have shown a functional variability of the amygdala’s response to stress in women with PMDD vs healthy controls.38,39 A proton magnetic resonance spectroscopy (1H-MRS) study of the displays the possibility of an altered GABAergic function in patients with PMDD.40
Patients will PMDD have enhanced dorsolateral prefrontal cortex reactivity when anticipating negative stimuli (but not to the actual exposure) during the luteal phase. A positive correlation between this reactivity and progesterone levels also was observed.41 Some researchers have suggested that prefrontal cortex dysfunction may be a risk factor for PMDD.42
HPA axis and HPG axis: Trauma, resiliency, inflammation. Altered cortisol levels (higher during the luteal phase43 and lower during times of stress14,44) suggest a possibly altered HPA axis in some women with PMDD. However, studies on this topic have been few and inconsistent.
Dysregulation of the HPG axis could cause vasomotor symptoms, sleep dysregulation, and mood symptoms during menopause; women with PMDD can also experience these symptoms. The influence of estrogen and progesterone on mood is also highly dependent on this axis.
Ultimately, the interplay between the HPA axis and the HPG axis is important. One study found that women with PMDD who had high serum ALLO levels (HPG-related) had blunted nocturnal cortisol levels (HPA-related) compared with healthy controls who had low ALLO levels.45
Significant stress and trauma exposure have been associated with PMDD. A study of 3,968 women found a history of trauma and PTSD were independently associated with PMDD.46 Another study of approximately 3,000 women found a strong correlation between abuse and PMS.47 However, a third study found no correlations between PMDD and trauma.48
Patients with a predisposition to PMDD may be more vulnerable to develop a posttraumatic stress-related disorder, perhaps due to decreased biologic resiliency. For example, the startle response (hypervigilance) has been shown to be different in women with PMDD. One study suggested that suboptimal production of premenstrual ALLO may lead to increased arousal and increased stress reactivity to psychosocial or environmental triggers.49
The possible role of inflammation in PMDD deserves further investigation. The luteal phase entails an increase in the production of proinflammatory markers.50,51 A 10-fold increase in progesterone is correlated with a 20% to 23% increase in C-reactive protein levels.52,53 Women with inflammatory diseases (eg, gingivitis or irritable bowel syndrome) show worsening of symptoms prior to menstruation.54-56 One study found increased levels of proinflammatory markers in women with PMDD compared with controls.57
Putting together the 5 pieces of the puzzle
Because PMDD is heritable, it must have an underlying neurobiologic pathophysiology. Brain imaging studies show differences in structure and function in women with PMDD across the menstrual cycle. Conversion of progesterone to ALLO and the GABAergic influence of this metabolite is a topic of interest in current research. Similarly, the role of estrogen and its connection to serotonin and other neurotransmitters such as BDNF have been implicated.
The link between a history of stress, trauma, and PMDD raises the question of biologic resiliency and illness in these patients, as it connects to the HPA and HPG axis and production of inflammatory stress hormones and steroid hormones and their metabolites. PMDD can be conceptualized as variable sensitivity to hormonal response to stress,58 thus contextualizing biochemical and psychological resiliency.
Further research is needed to clarify the possibility of a shared pathophysiology between endocrine-related mood disorders such as postpartum depression (PPD) and PMDD because current research is controversial.59,60 In PPD, women who are exposed to high levels of progesterone and estrogen during pregnancy (just like in the mid-luteal phase) have a sudden drop in these hormones postpartum.
The ‘withdrawal theory.’ The affective symptoms of PMDD resolve almost instantaneously after the start of menstruation. Perhaps this type of immediate relief is akin to substance use disorders and symptoms of withdrawal. It could be that reinstatement of a certain amount of gonadal steroids in the follicular phase of the cycle diminishes a withdrawal-like response to these steroids.
Currently, the main leading theory is that PMDD is a result of “an abnormal response to normal hormonal changes.”61 A new study also has shown that the change in estradiol/progesterone levels (vs the steady state) was associated with PMDD symptoms.62 Thinking of PMDD as a disorder of withdrawal offers an alternative (yet complementary) perspective to the current theory: PMDD may be caused by the absence or diminishing of the above-named hormones and their metabolites in the late luteal phase (in the context of developed “tolerance” during the early- to mid-luteal phase).
Considering the interplay between neurotransmitters and neurosteroids, both a “serotonin withdrawal theory” (caused by a drop in steroid hormones) and a “GABAergic withdrawal theory” (due to the decline in progesterone) could be proposed. This theory would be supported by the fact that SSRIs seem to mitigate symptoms of PMDD as well as the genetic association between PMDD and ESR1. It is more than likely that the “withdrawal” is caused by the interactions between estrogen-serotonin, progesterone-ALLO, and GABA receptors, and the complementary fashion in which progesterone and estrogen influence each other.
In an age when psychiatry strives to identify the biologic causes of disease, studying endocrine-related mood disorders is particularly intriguing. DSM-5 defines premenstrual dysphoric disorder (PMDD) as a depressive disorder, with a 12-month prevalence ranging from 1.8% to 5.8% among women who menstruate.1-3 Factors that differentiate PMDD from other affective disorders include etiology, duration, and temporal relationship with the menstrual cycle.
PMDD is a disorder of consistent yet intermittent change in mental health and functionality. Therefore, it may be underdiagnosed and consequently undertreated if a psychiatric evaluation does not coincide with symptom occurrence or if patients do not understand that intermittent symptoms are treatable.
This article summarizes what is known about the etiology of PMDD. Although there are several treatments for PMDD, many women experience adverse effects or incomplete effectiveness. Further understanding of this disorder may lead to more efficacious treatments. Additionally, understanding the pathophysiology of PMDD might shed a light on the etiology of other disorders that are temporally related to reproductive life changes, such as pregnancy-, postpartum-, or menopause-related affective dysregulation.
Making the diagnosis
The diagnosis of PMDD is made when a patient has at least 5 of 11 specific symptoms that occur during the week before onset of menses, improve within a few days after the onset of menses (shown as the “PMDD Hazard Zone” in Figure 1), and are minimal or absent post-menses.3 Symptoms should be tracked prospectively for at least 2 menstrual cycles in order to confirm the diagnosis (one must be an affective symptom and another must be a behavioral/cognitive symptom).3
The affective symptoms are:
- lability of affect (eg, sudden sadness, tearfulness, or sensitivity to rejection)
- irritability, anger, or increased interpersonal conflicts
- depressed mood, hopelessness, or self- deprecating thoughts
- anxiety or tension, feeling “keyed up” or “on edge.”
The behavioral/cognitive symptoms are:
- decreased interest in usual activities (eg, work, hobbies, friends, school)
- difficulty concentrating
- lethargy, low energy, easy fatigability
- change in appetite, overeating, food cravings
- hypersomnia or insomnia
- feeling overwhelmed or out of control
- physical symptoms (breast tenderness or swelling, headache, joint or muscle pain, bloating, weight gain).
Ruling out premenstrual exacerbation (PME). Perhaps the most common cause for misdiagnosis of PMDD is failing to rule out PME of another underlying or comorbid condition (Figure 2). In many women who have a primary mood or anxiety disorder, the late luteal phase is a vulnerable time. A patient might be coping with untreated anxiety, for example, but the symptoms become unbearable the week before menstruation begins, which is likely when she seeks help. At this stage, a diagnosis of PMDD should be provisional at best. Often, PME is treated by treating the underlying condition. Therefore, a full diagnostic psychiatric interview is important to first rule out other underlying psychiatric disorders. PMDD is diagnosed if the premenstrual symptoms persist for 2 consecutive months after treating the suspected mood or anxiety disorder. Patients can use one of many PMDD daily symptom charts available online. Alternatively, they can use a cycle-tracking mobile phone application to correlate their symptoms with their cycle and share this information with their providers.
Consider these 5 interwoven pieces
The many variables that contribute to the pathophysiology of PMDD overlap and should be considered connecting pieces in the puzzle that is the etiology of this disorder (Figure 3). In reviewing the literature, we have identified 5 topics likely to be major contributors to this disorder:
- genetic susceptibility
- progesterone and allopregnanolone (ALLO)
- estrogen, serotonin, and brain-derived neurotrophic factor (BDNF)
- putative brain structural and functional differences
- further involvement of the hypothalamic–pituitary–adrenal (HPA) axis and hypothalamic–pituitary–gonadal (HPG) axis: trauma, resiliency, and inflammation.
Genetic susceptibility. PMDD is thought to have a heritability range between 30% to 80%.3 This is demonstrated by family and twin studies4-7 and specific genetic studies.8 The involvement of genetics means an underlying neurobiologic pathophysiology is in place.
Estrogen receptor alpha (ESR1) gene. Huo et al8 found an associated variation in ESR1 in women with PMDD compared with controls. They speculated that because ESR1 is important for arousal, if dysfunctional, this gene could be implicated in somatic as well as affective and cognitive deficits in PMDD patients. In another study, investigators reported a relationship between PMDD and heritable personality traits, as well as a link between these traits and ESR1 polymorphic variants.1 They suggested that personality traits (independent of affective state) might be used to distinguish patients with PMDD from controls.1
Studies on serotonin gene polymorphism and serotonin transporter genotype. Although a study of serotonin gene polymorphism did not find an association between serotonin1A gene polymorphism and PMDD, it did show that the presence of at least 1 C allele was associated with a 2.5-fold increased risk of PMDD.9 Another study did not find an association between the serotonin transporter genotype 5-HTTLPR and PMDD.10 However, it showed lower frontocingulate cortex activation during the luteal phase of PMDD patients compared with controls, suggesting that PMDD is linked to impaired frontocingulate cortex activation induced by emotions during the luteal phase.10
Seasonal affective disorder (SAD) and PMDD have shared clinical features. A polymorphism in the serotonin transporter promoter gene 5-HTTLPR has been associated with SAD. One study found that patients with comorbid SAD and PMDD are genetically more vulnerable to comorbid affective disorders compared with patients who have SAD only.11
Progesterone and ALLO. Chronic exposure to progesterone and ALLO (a main progesterone metabolite) and rapid withdrawal from ovarian hormones may play a role in the etiology of PMDD. Much like alcohol or benzodiazepines, ALLO is a potent positive allosteric modulator of GABAA receptors and has sedative, anesthetic, and anxiolytic properties. In times of acute stress, increased ALLO is known to provide relief.12,13 However, in women with PMDD, this typical ALLO increase might not occur.14
Patients with PMDD have been reported to have decreased levels of ALLO in the luteal phase.15-17 In one study, women with highly symptomatic PMDD had lower levels of ALLO compared with women with less symptomatic PMDD.14 A gonadotropin-releasing hormone challenge study showed the increase in ALLO response was less in PMDD patients compared with controls.17 Luteal-phase ALLO concentrations are reported to be lower in women with premenstrual syndrome (PMS), a milder form of PMDD.14,17
The efficacy of selective serotonin reuptake inhibitors (SSRIs) for treating PMDD could be the result of the interaction of these medications with neuroactive steroids,18 possibly because SSRIs enhance the sensitivity of GABAA receptors or promote the formation of more ALLO (Figure 4).19-21
Estrogen, serotonin, and BDNF. Estrogen affects multiple neurotransmitter systems that regulate mood, cognition, sleep, and eating.22 Studying estrogen in context of PMDD is important because women with PMDD can have low mood, specific food cravings, and impaired cognitive function.
Estrogen–serotonin interactions are thought to be involved in hormone-related mood disorders such as perimenopausal depression and PMDD.23,24 However, the nature of their relationship is not yet fully understood. Ovariectomized animals have shown estrogen-induced changes related to serotonin metabolism, binding, and transmission in the regions of the brain involved in regulation of affect and cognition. Research in menopausal women also has provided some support for this interaction.24
Positron emission tomography studies in humans have found increased cortical serotonin binding modulated by levels of estrogen, similar to those previously seen in rat studies.24-27 One study showed an increased binding potential of serotonin in the cerebral cortex with estrogen treatment. This study further showed an even greater binding potential with estrogen plus progesterone, signaling a synergistic effect of the 2 hormones.28
SSRIs are an effective treatment for the irritability, anxiety, and mood swings of PMDD.29-30 Although the exact mechanism of action is unknown, the serotonergic properties are certainly of primary attention. For some PMDD patients, SSRIs work within hours to days, as opposed to days or weeks for patients with depression or anxiety, which suggests a separate or co-occurring mechanism of action is in place. In a double-blind, placebo-controlled crossover study, researchers administered the serotonin receptor antagonist metergoline to women with PMDD whose symptoms had remitted during treatment with fluoxetine and a group of healthy controls who were not receiving any medication.31 The women with PMDD experienced a return of symptoms 24 hours after treatment with metergoline but not with placebo; the controls experienced no mood changes.31
BDNF is a neurotransmitter linked to estrogen and likely related to PMDD. BDNF is critical for neurogenesis and is expressed in brain regions involved in learning and memory and also affects regulation.32 BDNF levels are increased by serotonergic antidepressants, affected by estradiol, and have cyclicity throughout the menstrual cycle.33-35
Putative brain structural and functional differences. Imaging studies have suggested differences in brain structure in women with PMDD, with a focus on the amygdala and the prefrontal cortex. Women with PMDD have greater gray matter volume in the posterior cerebellum,36 greater gray matter density of hippocampal cortex, and lower gray matter density in the parahippocampal cortex.37
Some studies have shown a functional variability of the amygdala’s response to stress in women with PMDD vs healthy controls.38,39 A proton magnetic resonance spectroscopy (1H-MRS) study of the displays the possibility of an altered GABAergic function in patients with PMDD.40
Patients will PMDD have enhanced dorsolateral prefrontal cortex reactivity when anticipating negative stimuli (but not to the actual exposure) during the luteal phase. A positive correlation between this reactivity and progesterone levels also was observed.41 Some researchers have suggested that prefrontal cortex dysfunction may be a risk factor for PMDD.42
HPA axis and HPG axis: Trauma, resiliency, inflammation. Altered cortisol levels (higher during the luteal phase43 and lower during times of stress14,44) suggest a possibly altered HPA axis in some women with PMDD. However, studies on this topic have been few and inconsistent.
Dysregulation of the HPG axis could cause vasomotor symptoms, sleep dysregulation, and mood symptoms during menopause; women with PMDD can also experience these symptoms. The influence of estrogen and progesterone on mood is also highly dependent on this axis.
Ultimately, the interplay between the HPA axis and the HPG axis is important. One study found that women with PMDD who had high serum ALLO levels (HPG-related) had blunted nocturnal cortisol levels (HPA-related) compared with healthy controls who had low ALLO levels.45
Significant stress and trauma exposure have been associated with PMDD. A study of 3,968 women found a history of trauma and PTSD were independently associated with PMDD.46 Another study of approximately 3,000 women found a strong correlation between abuse and PMS.47 However, a third study found no correlations between PMDD and trauma.48
Patients with a predisposition to PMDD may be more vulnerable to develop a posttraumatic stress-related disorder, perhaps due to decreased biologic resiliency. For example, the startle response (hypervigilance) has been shown to be different in women with PMDD. One study suggested that suboptimal production of premenstrual ALLO may lead to increased arousal and increased stress reactivity to psychosocial or environmental triggers.49
The possible role of inflammation in PMDD deserves further investigation. The luteal phase entails an increase in the production of proinflammatory markers.50,51 A 10-fold increase in progesterone is correlated with a 20% to 23% increase in C-reactive protein levels.52,53 Women with inflammatory diseases (eg, gingivitis or irritable bowel syndrome) show worsening of symptoms prior to menstruation.54-56 One study found increased levels of proinflammatory markers in women with PMDD compared with controls.57
Putting together the 5 pieces of the puzzle
Because PMDD is heritable, it must have an underlying neurobiologic pathophysiology. Brain imaging studies show differences in structure and function in women with PMDD across the menstrual cycle. Conversion of progesterone to ALLO and the GABAergic influence of this metabolite is a topic of interest in current research. Similarly, the role of estrogen and its connection to serotonin and other neurotransmitters such as BDNF have been implicated.
The link between a history of stress, trauma, and PMDD raises the question of biologic resiliency and illness in these patients, as it connects to the HPA and HPG axis and production of inflammatory stress hormones and steroid hormones and their metabolites. PMDD can be conceptualized as variable sensitivity to hormonal response to stress,58 thus contextualizing biochemical and psychological resiliency.
Further research is needed to clarify the possibility of a shared pathophysiology between endocrine-related mood disorders such as postpartum depression (PPD) and PMDD because current research is controversial.59,60 In PPD, women who are exposed to high levels of progesterone and estrogen during pregnancy (just like in the mid-luteal phase) have a sudden drop in these hormones postpartum.
The ‘withdrawal theory.’ The affective symptoms of PMDD resolve almost instantaneously after the start of menstruation. Perhaps this type of immediate relief is akin to substance use disorders and symptoms of withdrawal. It could be that reinstatement of a certain amount of gonadal steroids in the follicular phase of the cycle diminishes a withdrawal-like response to these steroids.
Currently, the main leading theory is that PMDD is a result of “an abnormal response to normal hormonal changes.”61 A new study also has shown that the change in estradiol/progesterone levels (vs the steady state) was associated with PMDD symptoms.62 Thinking of PMDD as a disorder of withdrawal offers an alternative (yet complementary) perspective to the current theory: PMDD may be caused by the absence or diminishing of the above-named hormones and their metabolites in the late luteal phase (in the context of developed “tolerance” during the early- to mid-luteal phase).
Considering the interplay between neurotransmitters and neurosteroids, both a “serotonin withdrawal theory” (caused by a drop in steroid hormones) and a “GABAergic withdrawal theory” (due to the decline in progesterone) could be proposed. This theory would be supported by the fact that SSRIs seem to mitigate symptoms of PMDD as well as the genetic association between PMDD and ESR1. It is more than likely that the “withdrawal” is caused by the interactions between estrogen-serotonin, progesterone-ALLO, and GABA receptors, and the complementary fashion in which progesterone and estrogen influence each other.
1. Miller A, Vo H, Huo L, et al. Estrogen receptor alpha (ESR-1) associations with psychological traits in women with PMDD and controls. J Psychiatr Res. 2010;44(12):788-794.
2. Epperson CN, Steiner M, Hartlage SA, et al. Premenstrual dysphoric disorder: evidence for a new category for DSM-5. Am J Psychiatry. 2012;169(5):465-475.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Wilson CA, Turner CW, Keye WR Jr. Firstborn adolescent daughters and mothers with and without premenstrual syndrome: a comparison. J Adolesc Health. 1991;12(2):130-137.
5. Kendler KS, Silberg JL, Neale MC, et al. Genetic and environmental factors in the aetiology of menstrual, premenstrual and neurotic symptoms: a population-based twin study. Psychol Med. 1992;22(1):85-100.
6. Condon JT. The premenstrual syndrome: a twin study. Br J Psychiatry. 1993;162:481-486.
7. Kendler KS, Karkowski LM, Corey LA, et al. Longitudinal population-based twin study of retrospectively reported premenstrual symptoms and lifetime major depression. Am J Psychiatry. 1998;155(9):1234-1240.
8. Huo L, Straub RE, Roca C, et al. Risk for premenstrual dysphoric disorder is associated with genetic variation in ESR1, the estrogen receptor alpha gene. Biol Psychiatry. 2007;62(8):925-933.
9. Dhingra V, Magnay JL, O’Brien PM, et al. Serotonin receptor 1A C(-1019)G polymorphism associated with premenstrual dysphoric disorder. Obstet Gynecol. 2007;110(4):788-792.
10. Comasco E, Hahn A, Ganger S, et al. Emotional fronto-cingulate cortex activation and brain derived neurotrophic factor polymorphism in premenstrual dysphoric disorder. Hum Brain Mapp. 2014;35(9):4450-4458.
11. Praschak-Rieder N, Willeit M, Winkler D, et al. Role of family history and 5-HTTLPR polymorphism in female seasonal affective disorder patients with and without premenstrual dysphoric disorder. Eur Neuropsychopharmacol. 2002;12(2):129-134.
12. Klatzkin RR, Morrow AL, Light KC, et al. Associations of histories of depression and PMDD diagnosis with allopregnanolone concentrations following the oral administration of micronized progesterone. Psychoneuroendocrinology. 2006;31(10):1208-1219.
13. Crowley SK, Girdler SS. Neurosteroid, GABAergic and hypothalamic pituitary adrenal (HPA) axis regulation: what is the current state of knowledge in humans? Psychopharmacology (Berl). 2014;231(17):3619-3634.
14. Girdler SS, Straneva PA, Light KC, et al. Allopregnanolone levels and reactivity to mental stress in premenstrual dysphoric disorder. Biol Psychiatry. 2001;49(9):788-797.
15. Rapkin AJ, Morgan M, Goldman L, et al. Progesterone metabolite allopregnanolone in women with premenstrual syndrome. Obstet Gynecol. 1997;90(5):709-714.
16. Bicíková M, Dibbelt L, Hill M, et al. Allopregnanolone in women with premenstrual syndrome. Horm Metab Res. 1998;30(4):227-230.
17. Monteleone P, Luisi S, Tonetti A, et al. Allopregnanolone concentrations and premenstrual syndrome. Eur J Endocrinol. 2000;142(3):269-273.
18. Steiner M, Steinberg S, Stewart D, et al. Fluoxetine in the treatment of premenstrual dysphoria. Canadian Fluoxetine/Premenstrual Dysphoria Collaborative Study Group. N Engl J Med. 1995;332(23):1529-1534.
19. Sundström I, Bäckström T. Citalopram increases pregnanolone sensitivity in patients with premenstrual syndrome: an open trial. Psychoneuroendocrinology. 1998;23(1):73-88.
20. Griffin LD, Mellon SH. Selective serotonin reuptake inhibitors directly alter activity of neurosteroidogenic enzymes. Proc Natl Acad Sci U S A. 1999;96(23):13512-13517.
21. Trauger JW, Jiang A, Stearns BA, et al. Kinetics of allopregnanolone formation catalyzed by human 3 alpha-hydroxysteroid dehydrogenase type III (AKR1C2). Biochemistry. 2002;41(45):13451-13459.
22. Shanmugan S, Epperson CN. Estrogen and the prefrontal cortex: towards a new understanding of estrogen’s effects on executive functions in the menopause transition. Hum Brain Mapp. 2014;35(3):847-865.
23. Rubinow DR, Schmidt PJ, Roca CA. Estrogen-serotonin interactions: implications for affective regulation. Biol Psychiatry. 1998;44(9):839-850.
24. Amin Z, Canli T, Epperson CN. Effect of estrogen-serotonin interactions on mood and cognition. Behav Cogn Neurosci Rev. 2005;4(1):43-58.
25. Cyr M, Bossé R, Di Paolo T. Gonadal hormones modulate 5-hydroxytryptamine2A receptors: emphasis on the rat frontal cortex. Neuroscience. 1998;83(3):829-836.
26. Fink G, Sumner BE, Rosie R, et al. Estrogen control of central neurotransmission: effect on mood, mental state, and memory. Cell Mol Neurobiol. 1996;16(3):325-344.
27. Sumner BE, Grant KE, Rosie R, et al. Effects of tamoxifen on serotonin transporter and 5-hydroxytryptamine(2A) receptor binding sites and mRNA levels in the brain of ovariectomized rats with or without acute estradiol replacement. Brain Res Mol Brain Res. 1999;73(1-2):119-128.
28. Moses-Kolko EL, Berga SL, Greer PJ, et al. Widespread increases of cortical serotonin type 2A receptor availability after hormone therapy in euthymic postmenopausal women. Fertil Steril. 2003;80(3):554-559.
29. Su TP, Schmidt PJ, Danaceau MA, et al. Fluoxetine in the treatment of premenstrual dysphoria. Neuropsychopharmacology. 1997;16(5):346-356.
30. Steinberg EM, Cardoso GM, Martinez PE, et al. Rapid response to fluoxetine in women with premenstrual dysphoric disorder. Depress Anxiety. 2012;29(6):531-540.
31. Roca CA, Schmidt PJ, Smith MJ, et al. Effects of metergoline on symptoms in women with premenstrual dysphoric disorder. Am J Psychiatry. 2002;159(11):1876-1881.
32. Gray JD, Milner TA, McEwen BS. Dynamic plasticity: the role of glucocorticoids, brain-derived neurotrophic factor and other trophic factors. Neuroscience. 2013;239:214-227.
33. Carbone DL, Handa RJ. Sex and stress hormone influences on the expression and activity of brain-derived neurotrophic factor. Neuroscience. 2013;239:295-303.
34. Pilar-Cuéllar F, Vidal R, Pazos A. Subchronic treatment with fluoxetine and ketanserin increases hippocampal brain-derived neurotrophic factor, β-catenin and antidepressant-like effects. Br J Pharmacol. 2012;165(4b):1046-1057.
35. Deuschle M, Gilles M, Scharnholz B, et al. Changes of serum concentrations of brain-derived neurotrophic factor (BDNF) during treatment with venlafaxine and mirtazapine: role of medication and response to treatment. Pharmacopsychiatry. 2013;46(2):54-58.
36. Berman SM, London ED, Morgan M, et al. Elevated gray matter volume of the emotional cerebellum in women with premenstrual dysphoric disorder. J Affect Disord. 2013;146(2):266-271.
37. Jeong HG, Ham BJ, Yeo HB, et al. Gray matter abnormalities in patients with premenstrual dysphoric disorder: an optimized voxel-based morphometry. J Affect Disord. 2012;140(3):260-267.
38. Protopopescu X, Tuescher O, Pan H, et al. Toward a functional neuroanatomy of premenstrual dysphoric disorder. J Affect Disord. 2008;108(1-2):87-94.
39. Gingnell M, Morell A, Bannbers E, et al. Menstrual cycle effects on amygdala reactivity to emotional stimulation in premenstrual dysphoric disorder. Horm Behav. 2012;62(4):400-406.
40. Epperson CN, Haga K, Mason GF, et al. Cortical gamma-aminobutyric acid levels across the menstrual cycle in healthy women and those with premenstrual dysphoric disorder: a proton magnetic resonance spectroscopy study. Arch Gen Psychiatry. 2002;59(9):851-858.
41. Gingnell M, Bannbers E, Wikström J, et al. Premenstrual dysphoric disorder and prefrontal reactivity during anticipation of emotional stimuli. Eur Neuropsychopharmacol. 2013;23(11):1474-1483.
42. Baller EB, Wei SM, Kohn PD, et al. Abnormalities of dorsolateral prefrontal function in women with premenstrual dysphoric disorder: a multimodal neuroimaging study. Am J Psychiatry. 2013;170(3):305-314.
43. Rasgon N, McGuire M, Tanavoli S, et al. Neuroendocrine response to an intravenous L-tryptophan challenge in women with premenstrual syndrome. Fertil Steril. 2000;73(1):144-149.
44. Huang Y, Zhou R, Wu M, et al. Premenstrual syndrome is associated with blunted cortisol reactivity to the TSST. Stress. 2015;18(2):160-168.
45. Segebladh B, Bannbers E, Moby L, et al. Allopregnanolone serum concentrations and diurnal cortisol secretion in women with premenstrual dysphoric disorder. Arch Womens Ment Health. 2013;16(2):131-137.
46. Pilver CE, Levy BR, Libby DJ, et al. Posttraumatic stress disorder and trauma characteristics are correlates of premenstrual dysphoric disorder. Arch Womens Ment Health. 2011;14(5):383-393.
47. Bertone-Johnson ER, Whitcomb BW, Missmer SA, et al. Early life emotional, physical, and sexual abuse and the development of premenstrual syndrome: a longitudinal study. J Womens Health (Larchmt). 2014;23(9):729-739.
48. Segebladh B, Bannbers E, Kask K, et al. Prevalence of violence exposure in women with premenstrual dysphoric disorder in comparison with other gynecological patients and asymptomatic controls. Acta Obstet Gynecol Scand. 2011;90(7):746-752.
49. Kask K, Gulinello M, Bäckström T, et al. Patients with premenstrual dysphoric disorder have increased startle response across both cycle phases and lower levels of prepulse inhibition during the late luteal phase of the menstrual cycle. Neuropsychopharmacology. 2008;33(9):2283-2290.
50. O’Brien SM, Fitzgerald P, Scully P, et al. Impact of gender and menstrual cycle phase on plasma cytokine concentrations. Neuroimmunomodulation. 2007;14(2):84-90.
51. Northoff H, Symons S, Zieker D, et al. Gender- and menstrual phase dependent regulation of inflammatory gene expression in response to aerobic exercise. Exerc Immunol Rev. 2008;14:86-103.
52. Gaskins AJ, Wilchesky M, Mumford SL, et al. Endogenous reproductive hormones and C-reactive protein across the menstrual cycle: the BioCycle Study. Am J Epidemiol. 2012;175(5):423-431.
53. Wander K, Brindle E, O’Connor KA. C-reactive protein across the menstrual cycle. Am J Phys Anthropol. 2008;136(2):138-146.
54. Jane ZY, Chang CC, Lin HK, et al. The association between the exacerbation of irritable bowel syndrome and menstrual symptoms in young Taiwanese women. Gastroenterol Nurs. 2011;34(4):277-286.
55. Kane SV, Sable K, Hanauer SB. The menstrual cycle and its effect on inflammatory bowel disease and irritable bowel syndrome: a prevalence study. Am J Gastroenterol. 1998;93(10):1867-1872.
56. Shourie V, Dwarakanath CD, Prashanth GV, et al. The effect of menstrual cycle on periodontal health - a clinical and microbiological study. Oral Health Prev Dent. 2012;10(2):185-192.
57. Hantsoo L, Epperson CN. Premenstrual dysphoric disorder: epidemiology and treatment. Curr Psychiatry Rep. 2015;17(11):87.
58. Maeng LY, Milad MR. Sex differences in anxiety disorders: Interactions between fear, stress, and gonadal hormones. Horm Behav. 2015;76:106-117.
59. Lee YJ, Yi SW, Ju DH, et al. Correlation between postpartum depression and premenstrual dysphoric disorder: single center study. Obstet Gynecol Sci. 2015;58(5):353-358.
60. Kepple AL, Lee EE, Haq N, et al. History of postpartum depression in a clinic-based sample of women with premenstrual dysphoric disorder. J Clin Psychiatry. 2016;77(4):e415-e420.
61. Schmidt PJ, Nieman LK, Danaceau MA, et al. Differential behavioral effects of gonadal steroids in women with and in those without premenstrual syndrome. N Engl J Med. 1998;338(4):209-216.
62. Schmidt PJ, Martinez PE, Nieman LK, et al. Premenstrual dysphoric disorder symptoms following ovarian suppression: Triggered by change in ovarian steroid levels but not continuous stable levels. Am J Psychiatry. [published online April 21, 2017]. doi: 10.1176/appi.ajp.2017.16101113.
1. Miller A, Vo H, Huo L, et al. Estrogen receptor alpha (ESR-1) associations with psychological traits in women with PMDD and controls. J Psychiatr Res. 2010;44(12):788-794.
2. Epperson CN, Steiner M, Hartlage SA, et al. Premenstrual dysphoric disorder: evidence for a new category for DSM-5. Am J Psychiatry. 2012;169(5):465-475.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Wilson CA, Turner CW, Keye WR Jr. Firstborn adolescent daughters and mothers with and without premenstrual syndrome: a comparison. J Adolesc Health. 1991;12(2):130-137.
5. Kendler KS, Silberg JL, Neale MC, et al. Genetic and environmental factors in the aetiology of menstrual, premenstrual and neurotic symptoms: a population-based twin study. Psychol Med. 1992;22(1):85-100.
6. Condon JT. The premenstrual syndrome: a twin study. Br J Psychiatry. 1993;162:481-486.
7. Kendler KS, Karkowski LM, Corey LA, et al. Longitudinal population-based twin study of retrospectively reported premenstrual symptoms and lifetime major depression. Am J Psychiatry. 1998;155(9):1234-1240.
8. Huo L, Straub RE, Roca C, et al. Risk for premenstrual dysphoric disorder is associated with genetic variation in ESR1, the estrogen receptor alpha gene. Biol Psychiatry. 2007;62(8):925-933.
9. Dhingra V, Magnay JL, O’Brien PM, et al. Serotonin receptor 1A C(-1019)G polymorphism associated with premenstrual dysphoric disorder. Obstet Gynecol. 2007;110(4):788-792.
10. Comasco E, Hahn A, Ganger S, et al. Emotional fronto-cingulate cortex activation and brain derived neurotrophic factor polymorphism in premenstrual dysphoric disorder. Hum Brain Mapp. 2014;35(9):4450-4458.
11. Praschak-Rieder N, Willeit M, Winkler D, et al. Role of family history and 5-HTTLPR polymorphism in female seasonal affective disorder patients with and without premenstrual dysphoric disorder. Eur Neuropsychopharmacol. 2002;12(2):129-134.
12. Klatzkin RR, Morrow AL, Light KC, et al. Associations of histories of depression and PMDD diagnosis with allopregnanolone concentrations following the oral administration of micronized progesterone. Psychoneuroendocrinology. 2006;31(10):1208-1219.
13. Crowley SK, Girdler SS. Neurosteroid, GABAergic and hypothalamic pituitary adrenal (HPA) axis regulation: what is the current state of knowledge in humans? Psychopharmacology (Berl). 2014;231(17):3619-3634.
14. Girdler SS, Straneva PA, Light KC, et al. Allopregnanolone levels and reactivity to mental stress in premenstrual dysphoric disorder. Biol Psychiatry. 2001;49(9):788-797.
15. Rapkin AJ, Morgan M, Goldman L, et al. Progesterone metabolite allopregnanolone in women with premenstrual syndrome. Obstet Gynecol. 1997;90(5):709-714.
16. Bicíková M, Dibbelt L, Hill M, et al. Allopregnanolone in women with premenstrual syndrome. Horm Metab Res. 1998;30(4):227-230.
17. Monteleone P, Luisi S, Tonetti A, et al. Allopregnanolone concentrations and premenstrual syndrome. Eur J Endocrinol. 2000;142(3):269-273.
18. Steiner M, Steinberg S, Stewart D, et al. Fluoxetine in the treatment of premenstrual dysphoria. Canadian Fluoxetine/Premenstrual Dysphoria Collaborative Study Group. N Engl J Med. 1995;332(23):1529-1534.
19. Sundström I, Bäckström T. Citalopram increases pregnanolone sensitivity in patients with premenstrual syndrome: an open trial. Psychoneuroendocrinology. 1998;23(1):73-88.
20. Griffin LD, Mellon SH. Selective serotonin reuptake inhibitors directly alter activity of neurosteroidogenic enzymes. Proc Natl Acad Sci U S A. 1999;96(23):13512-13517.
21. Trauger JW, Jiang A, Stearns BA, et al. Kinetics of allopregnanolone formation catalyzed by human 3 alpha-hydroxysteroid dehydrogenase type III (AKR1C2). Biochemistry. 2002;41(45):13451-13459.
22. Shanmugan S, Epperson CN. Estrogen and the prefrontal cortex: towards a new understanding of estrogen’s effects on executive functions in the menopause transition. Hum Brain Mapp. 2014;35(3):847-865.
23. Rubinow DR, Schmidt PJ, Roca CA. Estrogen-serotonin interactions: implications for affective regulation. Biol Psychiatry. 1998;44(9):839-850.
24. Amin Z, Canli T, Epperson CN. Effect of estrogen-serotonin interactions on mood and cognition. Behav Cogn Neurosci Rev. 2005;4(1):43-58.
25. Cyr M, Bossé R, Di Paolo T. Gonadal hormones modulate 5-hydroxytryptamine2A receptors: emphasis on the rat frontal cortex. Neuroscience. 1998;83(3):829-836.
26. Fink G, Sumner BE, Rosie R, et al. Estrogen control of central neurotransmission: effect on mood, mental state, and memory. Cell Mol Neurobiol. 1996;16(3):325-344.
27. Sumner BE, Grant KE, Rosie R, et al. Effects of tamoxifen on serotonin transporter and 5-hydroxytryptamine(2A) receptor binding sites and mRNA levels in the brain of ovariectomized rats with or without acute estradiol replacement. Brain Res Mol Brain Res. 1999;73(1-2):119-128.
28. Moses-Kolko EL, Berga SL, Greer PJ, et al. Widespread increases of cortical serotonin type 2A receptor availability after hormone therapy in euthymic postmenopausal women. Fertil Steril. 2003;80(3):554-559.
29. Su TP, Schmidt PJ, Danaceau MA, et al. Fluoxetine in the treatment of premenstrual dysphoria. Neuropsychopharmacology. 1997;16(5):346-356.
30. Steinberg EM, Cardoso GM, Martinez PE, et al. Rapid response to fluoxetine in women with premenstrual dysphoric disorder. Depress Anxiety. 2012;29(6):531-540.
31. Roca CA, Schmidt PJ, Smith MJ, et al. Effects of metergoline on symptoms in women with premenstrual dysphoric disorder. Am J Psychiatry. 2002;159(11):1876-1881.
32. Gray JD, Milner TA, McEwen BS. Dynamic plasticity: the role of glucocorticoids, brain-derived neurotrophic factor and other trophic factors. Neuroscience. 2013;239:214-227.
33. Carbone DL, Handa RJ. Sex and stress hormone influences on the expression and activity of brain-derived neurotrophic factor. Neuroscience. 2013;239:295-303.
34. Pilar-Cuéllar F, Vidal R, Pazos A. Subchronic treatment with fluoxetine and ketanserin increases hippocampal brain-derived neurotrophic factor, β-catenin and antidepressant-like effects. Br J Pharmacol. 2012;165(4b):1046-1057.
35. Deuschle M, Gilles M, Scharnholz B, et al. Changes of serum concentrations of brain-derived neurotrophic factor (BDNF) during treatment with venlafaxine and mirtazapine: role of medication and response to treatment. Pharmacopsychiatry. 2013;46(2):54-58.
36. Berman SM, London ED, Morgan M, et al. Elevated gray matter volume of the emotional cerebellum in women with premenstrual dysphoric disorder. J Affect Disord. 2013;146(2):266-271.
37. Jeong HG, Ham BJ, Yeo HB, et al. Gray matter abnormalities in patients with premenstrual dysphoric disorder: an optimized voxel-based morphometry. J Affect Disord. 2012;140(3):260-267.
38. Protopopescu X, Tuescher O, Pan H, et al. Toward a functional neuroanatomy of premenstrual dysphoric disorder. J Affect Disord. 2008;108(1-2):87-94.
39. Gingnell M, Morell A, Bannbers E, et al. Menstrual cycle effects on amygdala reactivity to emotional stimulation in premenstrual dysphoric disorder. Horm Behav. 2012;62(4):400-406.
40. Epperson CN, Haga K, Mason GF, et al. Cortical gamma-aminobutyric acid levels across the menstrual cycle in healthy women and those with premenstrual dysphoric disorder: a proton magnetic resonance spectroscopy study. Arch Gen Psychiatry. 2002;59(9):851-858.
41. Gingnell M, Bannbers E, Wikström J, et al. Premenstrual dysphoric disorder and prefrontal reactivity during anticipation of emotional stimuli. Eur Neuropsychopharmacol. 2013;23(11):1474-1483.
42. Baller EB, Wei SM, Kohn PD, et al. Abnormalities of dorsolateral prefrontal function in women with premenstrual dysphoric disorder: a multimodal neuroimaging study. Am J Psychiatry. 2013;170(3):305-314.
43. Rasgon N, McGuire M, Tanavoli S, et al. Neuroendocrine response to an intravenous L-tryptophan challenge in women with premenstrual syndrome. Fertil Steril. 2000;73(1):144-149.
44. Huang Y, Zhou R, Wu M, et al. Premenstrual syndrome is associated with blunted cortisol reactivity to the TSST. Stress. 2015;18(2):160-168.
45. Segebladh B, Bannbers E, Moby L, et al. Allopregnanolone serum concentrations and diurnal cortisol secretion in women with premenstrual dysphoric disorder. Arch Womens Ment Health. 2013;16(2):131-137.
46. Pilver CE, Levy BR, Libby DJ, et al. Posttraumatic stress disorder and trauma characteristics are correlates of premenstrual dysphoric disorder. Arch Womens Ment Health. 2011;14(5):383-393.
47. Bertone-Johnson ER, Whitcomb BW, Missmer SA, et al. Early life emotional, physical, and sexual abuse and the development of premenstrual syndrome: a longitudinal study. J Womens Health (Larchmt). 2014;23(9):729-739.
48. Segebladh B, Bannbers E, Kask K, et al. Prevalence of violence exposure in women with premenstrual dysphoric disorder in comparison with other gynecological patients and asymptomatic controls. Acta Obstet Gynecol Scand. 2011;90(7):746-752.
49. Kask K, Gulinello M, Bäckström T, et al. Patients with premenstrual dysphoric disorder have increased startle response across both cycle phases and lower levels of prepulse inhibition during the late luteal phase of the menstrual cycle. Neuropsychopharmacology. 2008;33(9):2283-2290.
50. O’Brien SM, Fitzgerald P, Scully P, et al. Impact of gender and menstrual cycle phase on plasma cytokine concentrations. Neuroimmunomodulation. 2007;14(2):84-90.
51. Northoff H, Symons S, Zieker D, et al. Gender- and menstrual phase dependent regulation of inflammatory gene expression in response to aerobic exercise. Exerc Immunol Rev. 2008;14:86-103.
52. Gaskins AJ, Wilchesky M, Mumford SL, et al. Endogenous reproductive hormones and C-reactive protein across the menstrual cycle: the BioCycle Study. Am J Epidemiol. 2012;175(5):423-431.
53. Wander K, Brindle E, O’Connor KA. C-reactive protein across the menstrual cycle. Am J Phys Anthropol. 2008;136(2):138-146.
54. Jane ZY, Chang CC, Lin HK, et al. The association between the exacerbation of irritable bowel syndrome and menstrual symptoms in young Taiwanese women. Gastroenterol Nurs. 2011;34(4):277-286.
55. Kane SV, Sable K, Hanauer SB. The menstrual cycle and its effect on inflammatory bowel disease and irritable bowel syndrome: a prevalence study. Am J Gastroenterol. 1998;93(10):1867-1872.
56. Shourie V, Dwarakanath CD, Prashanth GV, et al. The effect of menstrual cycle on periodontal health - a clinical and microbiological study. Oral Health Prev Dent. 2012;10(2):185-192.
57. Hantsoo L, Epperson CN. Premenstrual dysphoric disorder: epidemiology and treatment. Curr Psychiatry Rep. 2015;17(11):87.
58. Maeng LY, Milad MR. Sex differences in anxiety disorders: Interactions between fear, stress, and gonadal hormones. Horm Behav. 2015;76:106-117.
59. Lee YJ, Yi SW, Ju DH, et al. Correlation between postpartum depression and premenstrual dysphoric disorder: single center study. Obstet Gynecol Sci. 2015;58(5):353-358.
60. Kepple AL, Lee EE, Haq N, et al. History of postpartum depression in a clinic-based sample of women with premenstrual dysphoric disorder. J Clin Psychiatry. 2016;77(4):e415-e420.
61. Schmidt PJ, Nieman LK, Danaceau MA, et al. Differential behavioral effects of gonadal steroids in women with and in those without premenstrual syndrome. N Engl J Med. 1998;338(4):209-216.
62. Schmidt PJ, Martinez PE, Nieman LK, et al. Premenstrual dysphoric disorder symptoms following ovarian suppression: Triggered by change in ovarian steroid levels but not continuous stable levels. Am J Psychiatry. [published online April 21, 2017]. doi: 10.1176/appi.ajp.2017.16101113.

Premenstrual dysphoric disorder

A shot in the arm: Boost your knowledge about immunizations for psychiatric patients
Patients with chronic, severe mental illness live much shorter lives than the general population. The 25-year loss in life expectancy for people with chronic mental illness has been attributed to higher rates of cardiovascular disease driven by increased smoking, obesity, poverty, and poor nutrition.1 These individuals also face the added burden of struggling with a psychiatric condition that often interferes with their ability to make optimal preventative health decisions, including staying up to date on vaccinations.2 A recent review from Toronto, Canada, found that the influenza vaccination rates among homeless adults with mental illness—a population at high risk of respiratory illness—was only 6.7% compared with 31.1% for the general population of Ontario.3
Mental health professionals may serve as the only contacts to offer medical care to this vulnerable population, leading some psychiatric leaders to advocate that psychiatrists be considered primary care providers within accountable care organizations. Because most vaccines are easily available, mental health professionals should know about key immunizations to guide their patients accordingly.
In the United States, approximately 45,000 adults die annually from vaccine-preventable diseases, the majority from influenza.4 When combined with the most recent Adult Immunization Schedule and general recommendations adapted from the CDC,5,6 the mnemonic ARM SHOT allows for a quick assessment of risk factors to guide administration and education about most vaccinations (Table 1). ARM SHOT involves assessing the following components of an individual’s health status and living arrangements to determine one’s risk of contracting communicable diseases:
- Age
- Risk of exposure
- Medical conditions (comorbidities)
- Substance use history
- HIV status or other immunocompromised states
- Occupancy, or living arrangements
- Tobacco use.

We recommend keeping a copy of the Adult Immunization Schedule (age ≥19) and/or the immunization schedule for children and adolescents (age ≤18) close for quick reference. Here, we provide a case and then explore how each component of the ARM SHOT mnemonic applies in decision-making.
Case Evaluating risk, assess needs
Ms. W, age 24, has bipolar I disorder, most recently manic with psychotic features. She presents for follow-up in clinic after a 5-day hospitalization for mania and comorbid alcohol use disorder. Her medical comorbidities include asthma and active tobacco use. She is taking lurasidone, 20 mg/d, and lithium, 900 mg/d. Her case manager is working to place Ms. W in a residential substance use disorder treatment program. Ms. W is on a waiting list to establish care with a primary care physician and has a history of poor engagement with medical services in general; prior attempts to place her with a primary care physician failed.
In advance of Ms. W’s transfer to a residential treatment facility, you have been asked to place a Mantoux screening test for tuberculosis (purified protein derivative), which raises the important question about her susceptibility to infectious diseases in general. To protect Ms. W from preventable diseases for which vaccines are available, you review the ARM SHOT mnemonic to broadly assess her candidacy for vaccinations.
Age
Age may be the most important determinant of a patient’s need for vaccination (Table 2). The CDC immunization schedules account for age-specific risks for diseases, complications, and responses to vaccination (Figure 1).6
Influenza vaccination. Adults can have an intramuscular or intradermal inactivated influenza vaccination yearly in the fall or winter, unless they have an allergy to a vaccine component such as egg protein. Those with such an allergy can receive a recombinant influenza vaccine. Until the 2016 to 2017 flu season, an intranasal mist of live, attenuated influenza vaccine was available to healthy, non-pregnant women, ages 2 to 49, without high-risk medical conditions. However, the CDC dropped its recommendation for this vaccine because data showed it did not effectively prevent the flu.7 Individuals age ≥65 can receive either the standard- or high-dose inactivated influenza vaccination. The latter contains 4 times the amount of antigen with the intention of triggering a stronger immune response in older adults.
Pneumonia immunization. All patients age ≥65 should receive vaccinations for Streptococcus pneumoniae and its variants in the form of one 13-valent pneumococcal conjugate vaccine and, at least 1 year later, one 23-valent pneumococcal polysaccharide vaccine (PPSV23). Immunization reduces the morbidity and mortality from pneumococcal illness by decreasing the burden of a pneumonia, bacteremia, or meningitis infection. Adults, ages 19 to 64, with a chronic disease (referred to as “special populations” in CDC tables), such as diabetes, heart or lung disease, alcoholism, or cirrhosis, or those who smoke cigarettes, should receive PPSV23 with a second dose administered at least 5 years after the first. The CDC recommends a 1-time re-vaccination at age 65 for patients if >5 years have passed since the last PPSV23 and if the patient was younger than age 65 at the time of primary vaccine for S. pneumoniae. This can be a rather tricky clinical situation; the health care provider should verify a patient’s immunization history to ensure that she (he) is receiving only necessary vaccines. However, when the history cannot be verified, err on the side of inclusion, because risks are minimal.
Shingles vaccination. Adults age ≥60 who are not immunocompromised should receive a single dose of live attenuated vaccine from varicella-zoster virus (VZV) to limit the risk of shingles from a prior chickenpox infection. The vaccine is approximately 66.5% effective at preventing postherpetic neuralgia for up to 4.9 years. Individuals as young as age 50 may have the vaccine because the risk of herpes zoster radically increases from then on,8 although most insurers only cover VZV vaccination after age 60.
Tetanus, diphtheria, and acellular pertussis (Tdap) vaccine. All adults should complete the 3-dose primary vaccination series for tetanus, diphtheria, and pertussis (also known as whooping cough) and this should include 1 dose of Tdap. Administration of the primary series is staged so that the second dose is given 4 weeks after the initial dose and the final dose 6 to 12 months after the first dose. After receiving the primary series, adults should receive a tetanus-diphtheria booster dose every 10 years. For adults ages 19 to 64, the Advisory Committee on Immunization Practices (ACIP) recommends 1 dose of Tdap in place of a booster vaccination to decrease the transmission risk of pertussis to vulnerable persons, especially children.
Human papillomavirus (HPV) immunization. The ACIP recommendation9 has been for children to receive routine vaccination for the 4 major strains of HPV—strains 6, 11, 16, and 18—starting at ages 11 to 12 to confer protection from HPV-associated diseases, such as genital warts, oropharyngeal cancer, and anal cancer; cancers of the cervix, vulva, and vagina in women; and penile cancer in men. Ideally, the vaccines are administered prior to HPV exposure from sexual contact. The quadrivalent HPV vaccine is safe and is administered as a 3-dose series, with the second and third doses given 2 and 6 months, respectively, after the initial dose. Adolescent girls also have the option of a bivalent HPV vaccine.
In 2016, the FDA approved a 9-valent HPV vaccine, a simpler 2-dose schedule for children ages 9 to 14 (2 doses at least 6 months apart). Leading cancer centers have endorsed this vaccine based on strong comparative data with the 3-dose regimen.10 For those not previously vaccinated, the HPV vaccine is available for women ages 13 to 26 and for men ages 13 to 21 (although men ages 22 to 26 can receive the vaccine, and it is recommended for men who have sex with men [MSM]). Women do not require Papanicolaou, serum pregnancy, HPV DNA, or HPV antibody tests prior to vaccination. If a woman becomes pregnant, remaining doses of the vaccine should be postponed until after delivery. Women still need to follow recommendations for cervical cancer screening because the HPV vaccine does not cover all genital strains of the virus. For sexually active individuals who might have HPV or genital warts, immunization has no clinical effect except to prevent other HPV strains.
Measles, mumps, and rubella (MMR) vaccine. All adults should receive, at minimum, 1 dose of MMR vaccination unless serological immunity can be verified or if contraindicated. Two doses of the vaccine are recommended for students attending post-high school institutions, health care personnel, and international travelers because they are at higher risk for exposure and transmission of measles and mumps. Individuals born before 1957 are considered immune to measles and mumps. A measles outbreak from December 2014 to February 201511 highlighted the importance of maintaining one’s immunity status for MMR.
Case continued
Based on Ms. W’s age, she should be offered vaccinations for influenza and opportunities to receive vaccinations for HPV, Tdap (the primary series, a Tdap or Td booster), and MMR, if appropriate and not completed previously.

Risk of exposure

Certain behaviors will increase the risk of exposure to and transmission of diseases communicable by blood and other bodily fluids (Table 3). These behaviors include needle injections (eg, during use of illicit drugs) and sexual activity with multiple partners, including MSM or promiscuity/impulsivity during a manic episode. A common consequence of risky behaviors is comorbid infection of HIV and viral hepatitis for those with substance use disorder or those who engage in high-risk sexual practices.12,13
Hepatitis B virus (HBV) immunization. Vaccination is one of the most effective ways to prevent HBV infection, which is why it is offered to all health care workers. HBV immunization is a 3-dose series in which the second and third doses are given 1 and 6 months after the initial doses, respectively. In addition to certain medical risk factors or conditions that indicate HBV vaccination, people should be offered the vaccine if they are in a higher risk occupation, travel, are of Asian or Pacific Islander ethnicity from an endemic area, or have any present or suspected sexually transmitted diseases.
Hepatitis A virus (HAV) vaccination. HAV is transmitted via fecal–oral routes, often from contaminated water or food, or through household or sexual contact with an infected person. Individuals should receive the HAV vaccine if they use illicit drugs by any route of administration, work with primates infected with HAV, travel to countries with unknown or high rates of HAV, or have chronic liver disease (ie, hepatitis, alcohol use disorder, or non-alcoholic fatty liver disease) or clotting deficiencies. The CDC Health Information for International Travel, commonly called the “Yellow Book,” publishes vaccination recommendations for those who plan travel to specific countries.14
Case continued
Ms. W’s history of mania (if such episodes included increased sexual activity) and substance use would make her a candidate for the HBV and HAV vaccinations and could also strengthen our previous recommendation that she receive the HPV vaccination.
Medical conditions
Patients with certain medical conditions may have difficulty fighting infections or become more susceptible to morbidity and mortality from coinfection with vaccine-preventable illnesses. Secondary effects of psychotropic medications that may carry implications for vaccine recommendations (eg, risk of agranulocytosis and impaired cell-medicated immunity with mirtazapine and clozapine or renal impairment from lithium use) are of particular concern in psychiatric patients.2
To help care for these patients, the CDC has developed a “medical conditions” schedule (Figure 2). This schedule makes vaccination recommendations for those with a weakened immune system, including patients with HIV, chronic obstructive pulmonary disease (COPD), diabetes, hepatitis, asplenia, end-stage renal disease, cardiac disease, and pregnancy.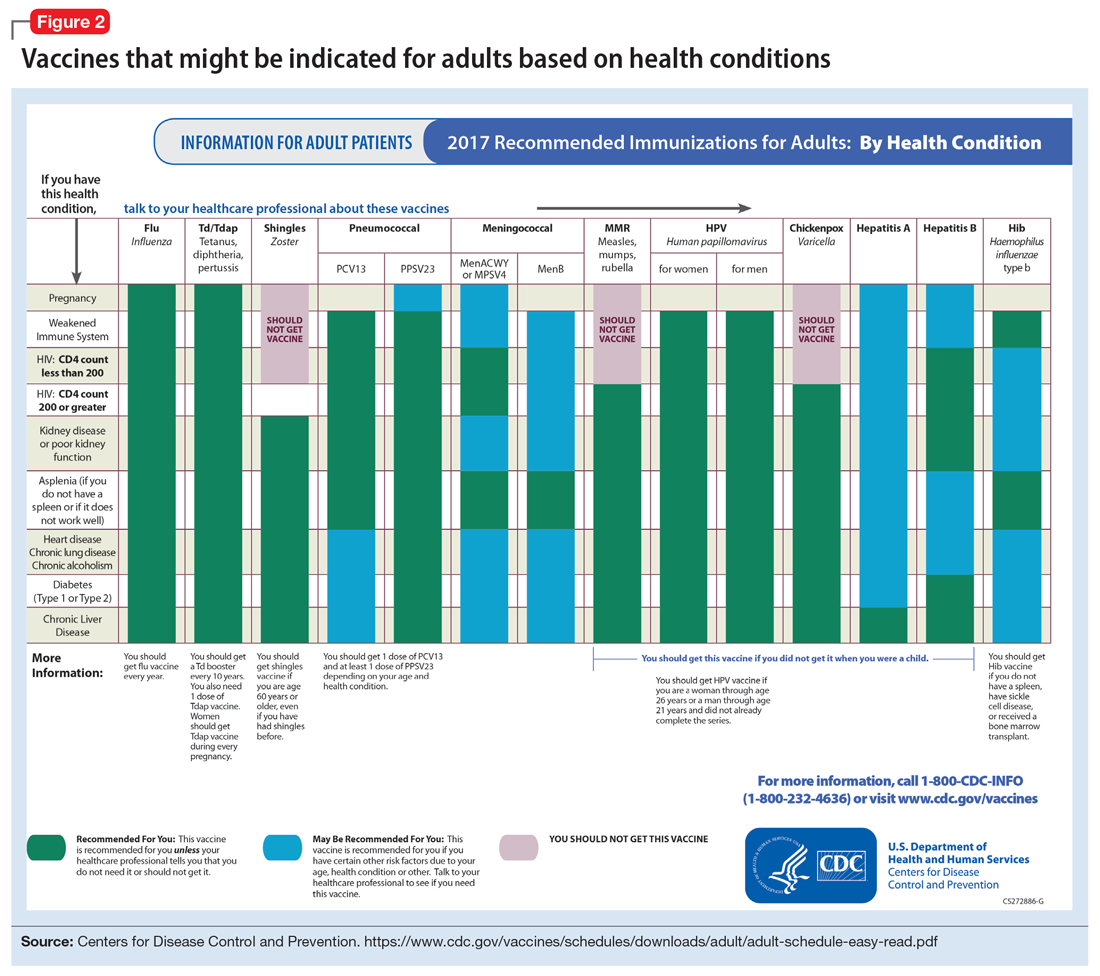
Because patients with psychiatric illness face a greater risk of heart disease and diabetes, these conditions may warrant special reference on the schedule. The increased cardiometabolic risk factors in these patients may be due in part to genetics, socioeconomic status, lifestyle behaviors, and medications to treat their mental illness (eg, antipsychotics). Patients with bipolar disorder or schizophrenia in particular tend to have higher rates of COPD (mainly from chronic bronchitis) and asthma than the general population.12 Pay special attention to the indications schedule for those with chronic lung disease, especially patients who continue to smoke cigarettes.
Case continued
Because of Ms. W’s asthma, the CDC schedule recommends ensuring she is up to date on her influenza, pneumococcal, and Tdap vaccinations.
Substance use
Patients with combined psychiatric and substance use disorders (“dual diagnosis”) have lower rates of receiving preventive care than patients with either condition alone.15 Substance use can be behaviorally disinhibiting, leading to increased risk of exposures from sexual contact or other risky activities. The use of illicit substances can provide a nidus for infection depending on the route of administration and can result in negative effects on organ systems, compromising one’s ability to ward off infection.
Patients who use any illicit drugs, regardless of the method of delivery, should be recommended for HAV vaccination. For those with alcohol use disorder and/or chronic liver disease, and/or seeking treatment for substance use, hepatitis B screening and vaccination is recommended.
Case continued
From a substance use perspective, discussion of vaccination status for both hepatitis A and B would be important for Ms. W.
HIV or immunocompromised
Persons with severe mental illness have high rates of HIV, with almost 8 times the risk of exposure, compared with the general population due to myriad reasons, including greater rates of substance abuse, higher risk sexual behavior, and lack of awareness of HIV transmission.12,13 Patients with mental illness are also at risk of leukopenia and agranulocytosis from certain drugs used to treat their conditions, such as clozapine.
Pregnancy is a challenge for women with mental illness because of the pharmacologic risk and immune-system compromise to the mother and baby. A pregnant woman who has HIV with a CD4 count <200, or has a weakened immune system from an organ transplant or a similar condition, is a candidate for certain vaccines based on the Adult Immunization Schedule (Figure 2). However, these patients should avoid live vaccines, such as the intranasal mist of live influenza, MMR, VZV, and varicella, to avoid illness from these inoculations.
Case continued
Ms. W should undergo testing for pregnancy and HIV (and preferably other sexually transmitted infections per general preventive health guidelines) before receiving any live vaccinations.
Occupancy
Aside from direct transmission of bodily fluids, infectious diseases also can spread through droplets/secretions from the throat and respiratory tract. Close quarters or lengthy contact enhances communicability by droplets, and therefore people who reside in a communal living space (eg, individuals in substance use treatment facilities or those who reside in a nursing home) are most susceptible.
The bacterial disease Neisseria meningitidis (meningococcus) can spread through droplets and can cause pneumonia, bacteremia, and meningitis. Vaccination is indicated, and in some states is mandated, for college students who live in residence halls and missed routine vaccination by age 16. Meningococcus conjugate vaccine is administered in 2 doses; each dose may be given at least 2 months apart for those with HIV, asplenia, or persistent complement-related disorders. A single dose may be recommended for travelers to areas where meningococcal disease is hyperendemic or epidemic, military recruits, or microbiologists. For those age ≥55 and older, meningococcal polysaccharide vaccine is recommended over meningococcal conjugate vaccine.
Influenza, MMR, diphtheria, pertussis, and pneumococcus also spread through droplet contact.
Case continued
If Ms. W had not previously received the meningococcus vaccine as part of adolescent immunizations, she could benefit from this vaccine because she plans to enter a residential substance use disorder treatment program.
Tobacco use
Patients with psychiatric illness are twice as likely to smoke compared with the general population.16 Adult smokers, especially those with chronic lung disease, are at higher risk for influenza and pneumococcal-related illness; they should be vaccinated against these illnesses regardless of age (as discussed in the “Age” section).
Case continued
Because she smokes, Ms. W should receive counseling on vaccinations, such as influenza and pneumonia, to lessen her risk of respiratory illnesses and downstream sepsis.
Conclusion
Ms. W’s case represents an unfortunately all-too-common scenario where her multifaceted biopsychosocial circumstances place her at high risk for vaccine-preventable conditions. Her weight is recorded and laboratory work ordered to evaluate her pregnancy status, blood counts, lipids, complete metabolic panel, lithium level, and HIV status. Fortunately, she had received her series of MMR, meningococcal, and Tdap vaccinations when she was younger. Influenza, HPV, HAV, HBV, and pneumococcal vaccinations were all recommended to her, all of which can be given on the same day (HAV and HBV often are available as a combined vaccine). Ms. W receives a renewal of her psychiatric medications and counseling on healthy living habits (eg, diet and exercise, quitting tobacco and alcohol use, and safe sex practices) and the importance of immunizations.
Vaccination is 1 of the 10 great public health achievements of the 20th century when one considers how immunization of vaccine-preventable diseases has reduced morbidity, mortality, and health-associated costs.17 As mental health professionals, we can help pass on the direct and indirect benefits of immunizations to an often underserved and undertreated population to help improve their health outcomes and quality of life.
1. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
2. Raj YP, Lloyd L. Adult immunizations. In: McCarron RM, Xiong GL, Keenan GR, et al, eds. Preventive medical care in psychiatry. Arlington, VA: American Psychiatric Publishing. 2015;215-227.
3. Young S, Dosani N, Whisler A, et al. Influenza vaccination rates among homeless adults with mental illness in Toronto. J Prim Care Community Health. 2015;6(3):211-214.
4. Kroger AT, Atkinson WL, Marcues EK, et al; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). General recommendations on immunization: recommendations on the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2006;55(RR-15):1-48.
5. Centers for Disease Control and Prevention. Recommended Adult Immunization by Vaccine and Age Group. http://www.cdc.gov/vaccines/schedules/hcp/adult.html. Updated February 27, 2017. Accessed February 1, 2017.
6. National Center for Immunization and Respiratory Diseases. General recommendations on immunization—recommendations on the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2011;60(2):1-64.
7. Centers for Disease Control and Prevention. ACIP votes down use of LAIV for 2016-2017 flu season. https://www.cdc.gov/media/releases/2016/s0622-laiv-flu.html. Updated June 22, 2016. Accessed February 1, 2017.
8. Hales CM, Harpaz, R, Ortega-Sanchez I, et al; Centers for Disease Control and Prevention. Update on recommendations for use of herpes zoster vaccine. MMWR Morb Mortal Wkly Rep. 2014;63(33):729-731.
9. Petrosky E, Bocchini Jr JA, Hariri S, et al; Centers for Disease Control and Prevention (CDC). Use of 9-valent human papillomavirus (HPV) vaccine: updated HPV vaccine recommendations of the advisory committee on immunization practices. MMWR Morb Mortal Wkly Rep. 2015;64(11)300-304.
10. Iversen OE, Miranda MJ, Ulied A, et al. Immunogenicity of the 9-valent HPV vaccine using 2-dose regimens in girls and boys vs a 3-dose regimen in women. JAMA. 2016;316(22):2411-2421.
11. Zipprich J, Winter K, Hacker J, et al; Centers for Disease Control and Prevention (CDC). Measles outbreak—California, December 2014-February 2015. MMWR Morb Mortal Wkly Rep. 2015;64(6):153-154.
12. De Hert M, Correll CU, Bobes J, et al. Physical illness in patients with severe mental disorders. I. Prevalence, impact of medications and disparities in health care. World Psychiatry. 2011;10(1):52-77.
13. Rosenberg SD, Goodman LA, Osher FC, et al. Prevalence of HIV, hepatitis B, and hepatitis C in people with severe mental illness. Am J Public Health. 2001;91(1):31-37.
14. Centers for Disease for Control and Prevention. CDC yellow book 2018: health information for international travel. New York, NY: Oxford University Press; 2017.
15. Druss BG, Rosenheck RA, Desai MM, et al. Quality of preventive medical care for patients with mental disorders. Med Care. 2002;40(2):129-136.
16. Lasser K, Boyd J, Woolhandler S, et al. Smoking and mental illness: a population-based prevalence study. JAMA. 2000;284(20):2606-2610.
17. Centers for Disease Control and Prevention (CDC). Ten great public health achievements—United States, 2001-2010. MMWR Morb Mortal Wkly Rep. 2011;60(19);619-623.
Patients with chronic, severe mental illness live much shorter lives than the general population. The 25-year loss in life expectancy for people with chronic mental illness has been attributed to higher rates of cardiovascular disease driven by increased smoking, obesity, poverty, and poor nutrition.1 These individuals also face the added burden of struggling with a psychiatric condition that often interferes with their ability to make optimal preventative health decisions, including staying up to date on vaccinations.2 A recent review from Toronto, Canada, found that the influenza vaccination rates among homeless adults with mental illness—a population at high risk of respiratory illness—was only 6.7% compared with 31.1% for the general population of Ontario.3
Mental health professionals may serve as the only contacts to offer medical care to this vulnerable population, leading some psychiatric leaders to advocate that psychiatrists be considered primary care providers within accountable care organizations. Because most vaccines are easily available, mental health professionals should know about key immunizations to guide their patients accordingly.
In the United States, approximately 45,000 adults die annually from vaccine-preventable diseases, the majority from influenza.4 When combined with the most recent Adult Immunization Schedule and general recommendations adapted from the CDC,5,6 the mnemonic ARM SHOT allows for a quick assessment of risk factors to guide administration and education about most vaccinations (Table 1). ARM SHOT involves assessing the following components of an individual’s health status and living arrangements to determine one’s risk of contracting communicable diseases:
- Age
- Risk of exposure
- Medical conditions (comorbidities)
- Substance use history
- HIV status or other immunocompromised states
- Occupancy, or living arrangements
- Tobacco use.
We recommend keeping a copy of the Adult Immunization Schedule (age ≥19) and/or the immunization schedule for children and adolescents (age ≤18) close for quick reference. Here, we provide a case and then explore how each component of the ARM SHOT mnemonic applies in decision-making.
Case Evaluating risk, assess needs
Ms. W, age 24, has bipolar I disorder, most recently manic with psychotic features. She presents for follow-up in clinic after a 5-day hospitalization for mania and comorbid alcohol use disorder. Her medical comorbidities include asthma and active tobacco use. She is taking lurasidone, 20 mg/d, and lithium, 900 mg/d. Her case manager is working to place Ms. W in a residential substance use disorder treatment program. Ms. W is on a waiting list to establish care with a primary care physician and has a history of poor engagement with medical services in general; prior attempts to place her with a primary care physician failed.
In advance of Ms. W’s transfer to a residential treatment facility, you have been asked to place a Mantoux screening test for tuberculosis (purified protein derivative), which raises the important question about her susceptibility to infectious diseases in general. To protect Ms. W from preventable diseases for which vaccines are available, you review the ARM SHOT mnemonic to broadly assess her candidacy for vaccinations.
Age
Age may be the most important determinant of a patient’s need for vaccination (Table 2). The CDC immunization schedules account for age-specific risks for diseases, complications, and responses to vaccination (Figure 1).6
Influenza vaccination. Adults can have an intramuscular or intradermal inactivated influenza vaccination yearly in the fall or winter, unless they have an allergy to a vaccine component such as egg protein. Those with such an allergy can receive a recombinant influenza vaccine. Until the 2016 to 2017 flu season, an intranasal mist of live, attenuated influenza vaccine was available to healthy, non-pregnant women, ages 2 to 49, without high-risk medical conditions. However, the CDC dropped its recommendation for this vaccine because data showed it did not effectively prevent the flu.7 Individuals age ≥65 can receive either the standard- or high-dose inactivated influenza vaccination. The latter contains 4 times the amount of antigen with the intention of triggering a stronger immune response in older adults.
Pneumonia immunization. All patients age ≥65 should receive vaccinations for Streptococcus pneumoniae and its variants in the form of one 13-valent pneumococcal conjugate vaccine and, at least 1 year later, one 23-valent pneumococcal polysaccharide vaccine (PPSV23). Immunization reduces the morbidity and mortality from pneumococcal illness by decreasing the burden of a pneumonia, bacteremia, or meningitis infection. Adults, ages 19 to 64, with a chronic disease (referred to as “special populations” in CDC tables), such as diabetes, heart or lung disease, alcoholism, or cirrhosis, or those who smoke cigarettes, should receive PPSV23 with a second dose administered at least 5 years after the first. The CDC recommends a 1-time re-vaccination at age 65 for patients if >5 years have passed since the last PPSV23 and if the patient was younger than age 65 at the time of primary vaccine for S. pneumoniae. This can be a rather tricky clinical situation; the health care provider should verify a patient’s immunization history to ensure that she (he) is receiving only necessary vaccines. However, when the history cannot be verified, err on the side of inclusion, because risks are minimal.
Shingles vaccination. Adults age ≥60 who are not immunocompromised should receive a single dose of live attenuated vaccine from varicella-zoster virus (VZV) to limit the risk of shingles from a prior chickenpox infection. The vaccine is approximately 66.5% effective at preventing postherpetic neuralgia for up to 4.9 years. Individuals as young as age 50 may have the vaccine because the risk of herpes zoster radically increases from then on,8 although most insurers only cover VZV vaccination after age 60.
Tetanus, diphtheria, and acellular pertussis (Tdap) vaccine. All adults should complete the 3-dose primary vaccination series for tetanus, diphtheria, and pertussis (also known as whooping cough) and this should include 1 dose of Tdap. Administration of the primary series is staged so that the second dose is given 4 weeks after the initial dose and the final dose 6 to 12 months after the first dose. After receiving the primary series, adults should receive a tetanus-diphtheria booster dose every 10 years. For adults ages 19 to 64, the Advisory Committee on Immunization Practices (ACIP) recommends 1 dose of Tdap in place of a booster vaccination to decrease the transmission risk of pertussis to vulnerable persons, especially children.
Human papillomavirus (HPV) immunization. The ACIP recommendation9 has been for children to receive routine vaccination for the 4 major strains of HPV—strains 6, 11, 16, and 18—starting at ages 11 to 12 to confer protection from HPV-associated diseases, such as genital warts, oropharyngeal cancer, and anal cancer; cancers of the cervix, vulva, and vagina in women; and penile cancer in men. Ideally, the vaccines are administered prior to HPV exposure from sexual contact. The quadrivalent HPV vaccine is safe and is administered as a 3-dose series, with the second and third doses given 2 and 6 months, respectively, after the initial dose. Adolescent girls also have the option of a bivalent HPV vaccine.
In 2016, the FDA approved a 9-valent HPV vaccine, a simpler 2-dose schedule for children ages 9 to 14 (2 doses at least 6 months apart). Leading cancer centers have endorsed this vaccine based on strong comparative data with the 3-dose regimen.10 For those not previously vaccinated, the HPV vaccine is available for women ages 13 to 26 and for men ages 13 to 21 (although men ages 22 to 26 can receive the vaccine, and it is recommended for men who have sex with men [MSM]). Women do not require Papanicolaou, serum pregnancy, HPV DNA, or HPV antibody tests prior to vaccination. If a woman becomes pregnant, remaining doses of the vaccine should be postponed until after delivery. Women still need to follow recommendations for cervical cancer screening because the HPV vaccine does not cover all genital strains of the virus. For sexually active individuals who might have HPV or genital warts, immunization has no clinical effect except to prevent other HPV strains.
Measles, mumps, and rubella (MMR) vaccine. All adults should receive, at minimum, 1 dose of MMR vaccination unless serological immunity can be verified or if contraindicated. Two doses of the vaccine are recommended for students attending post-high school institutions, health care personnel, and international travelers because they are at higher risk for exposure and transmission of measles and mumps. Individuals born before 1957 are considered immune to measles and mumps. A measles outbreak from December 2014 to February 201511 highlighted the importance of maintaining one’s immunity status for MMR.
Case continued
Based on Ms. W’s age, she should be offered vaccinations for influenza and opportunities to receive vaccinations for HPV, Tdap (the primary series, a Tdap or Td booster), and MMR, if appropriate and not completed previously.
Risk of exposure
Certain behaviors will increase the risk of exposure to and transmission of diseases communicable by blood and other bodily fluids (Table 3). These behaviors include needle injections (eg, during use of illicit drugs) and sexual activity with multiple partners, including MSM or promiscuity/impulsivity during a manic episode. A common consequence of risky behaviors is comorbid infection of HIV and viral hepatitis for those with substance use disorder or those who engage in high-risk sexual practices.12,13
Hepatitis B virus (HBV) immunization. Vaccination is one of the most effective ways to prevent HBV infection, which is why it is offered to all health care workers. HBV immunization is a 3-dose series in which the second and third doses are given 1 and 6 months after the initial doses, respectively. In addition to certain medical risk factors or conditions that indicate HBV vaccination, people should be offered the vaccine if they are in a higher risk occupation, travel, are of Asian or Pacific Islander ethnicity from an endemic area, or have any present or suspected sexually transmitted diseases.
Hepatitis A virus (HAV) vaccination. HAV is transmitted via fecal–oral routes, often from contaminated water or food, or through household or sexual contact with an infected person. Individuals should receive the HAV vaccine if they use illicit drugs by any route of administration, work with primates infected with HAV, travel to countries with unknown or high rates of HAV, or have chronic liver disease (ie, hepatitis, alcohol use disorder, or non-alcoholic fatty liver disease) or clotting deficiencies. The CDC Health Information for International Travel, commonly called the “Yellow Book,” publishes vaccination recommendations for those who plan travel to specific countries.14
Case continued
Ms. W’s history of mania (if such episodes included increased sexual activity) and substance use would make her a candidate for the HBV and HAV vaccinations and could also strengthen our previous recommendation that she receive the HPV vaccination.
Medical conditions
Patients with certain medical conditions may have difficulty fighting infections or become more susceptible to morbidity and mortality from coinfection with vaccine-preventable illnesses. Secondary effects of psychotropic medications that may carry implications for vaccine recommendations (eg, risk of agranulocytosis and impaired cell-medicated immunity with mirtazapine and clozapine or renal impairment from lithium use) are of particular concern in psychiatric patients.2
To help care for these patients, the CDC has developed a “medical conditions” schedule (Figure 2). This schedule makes vaccination recommendations for those with a weakened immune system, including patients with HIV, chronic obstructive pulmonary disease (COPD), diabetes, hepatitis, asplenia, end-stage renal disease, cardiac disease, and pregnancy.
Because patients with psychiatric illness face a greater risk of heart disease and diabetes, these conditions may warrant special reference on the schedule. The increased cardiometabolic risk factors in these patients may be due in part to genetics, socioeconomic status, lifestyle behaviors, and medications to treat their mental illness (eg, antipsychotics). Patients with bipolar disorder or schizophrenia in particular tend to have higher rates of COPD (mainly from chronic bronchitis) and asthma than the general population.12 Pay special attention to the indications schedule for those with chronic lung disease, especially patients who continue to smoke cigarettes.
Case continued
Because of Ms. W’s asthma, the CDC schedule recommends ensuring she is up to date on her influenza, pneumococcal, and Tdap vaccinations.
Substance use
Patients with combined psychiatric and substance use disorders (“dual diagnosis”) have lower rates of receiving preventive care than patients with either condition alone.15 Substance use can be behaviorally disinhibiting, leading to increased risk of exposures from sexual contact or other risky activities. The use of illicit substances can provide a nidus for infection depending on the route of administration and can result in negative effects on organ systems, compromising one’s ability to ward off infection.
Patients who use any illicit drugs, regardless of the method of delivery, should be recommended for HAV vaccination. For those with alcohol use disorder and/or chronic liver disease, and/or seeking treatment for substance use, hepatitis B screening and vaccination is recommended.
Case continued
From a substance use perspective, discussion of vaccination status for both hepatitis A and B would be important for Ms. W.
HIV or immunocompromised
Persons with severe mental illness have high rates of HIV, with almost 8 times the risk of exposure, compared with the general population due to myriad reasons, including greater rates of substance abuse, higher risk sexual behavior, and lack of awareness of HIV transmission.12,13 Patients with mental illness are also at risk of leukopenia and agranulocytosis from certain drugs used to treat their conditions, such as clozapine.
Pregnancy is a challenge for women with mental illness because of the pharmacologic risk and immune-system compromise to the mother and baby. A pregnant woman who has HIV with a CD4 count <200, or has a weakened immune system from an organ transplant or a similar condition, is a candidate for certain vaccines based on the Adult Immunization Schedule (Figure 2). However, these patients should avoid live vaccines, such as the intranasal mist of live influenza, MMR, VZV, and varicella, to avoid illness from these inoculations.
Case continued
Ms. W should undergo testing for pregnancy and HIV (and preferably other sexually transmitted infections per general preventive health guidelines) before receiving any live vaccinations.
Occupancy
Aside from direct transmission of bodily fluids, infectious diseases also can spread through droplets/secretions from the throat and respiratory tract. Close quarters or lengthy contact enhances communicability by droplets, and therefore people who reside in a communal living space (eg, individuals in substance use treatment facilities or those who reside in a nursing home) are most susceptible.
The bacterial disease Neisseria meningitidis (meningococcus) can spread through droplets and can cause pneumonia, bacteremia, and meningitis. Vaccination is indicated, and in some states is mandated, for college students who live in residence halls and missed routine vaccination by age 16. Meningococcus conjugate vaccine is administered in 2 doses; each dose may be given at least 2 months apart for those with HIV, asplenia, or persistent complement-related disorders. A single dose may be recommended for travelers to areas where meningococcal disease is hyperendemic or epidemic, military recruits, or microbiologists. For those age ≥55 and older, meningococcal polysaccharide vaccine is recommended over meningococcal conjugate vaccine.
Influenza, MMR, diphtheria, pertussis, and pneumococcus also spread through droplet contact.
Case continued
If Ms. W had not previously received the meningococcus vaccine as part of adolescent immunizations, she could benefit from this vaccine because she plans to enter a residential substance use disorder treatment program.
Tobacco use
Patients with psychiatric illness are twice as likely to smoke compared with the general population.16 Adult smokers, especially those with chronic lung disease, are at higher risk for influenza and pneumococcal-related illness; they should be vaccinated against these illnesses regardless of age (as discussed in the “Age” section).
Case continued
Because she smokes, Ms. W should receive counseling on vaccinations, such as influenza and pneumonia, to lessen her risk of respiratory illnesses and downstream sepsis.
Conclusion
Ms. W’s case represents an unfortunately all-too-common scenario where her multifaceted biopsychosocial circumstances place her at high risk for vaccine-preventable conditions. Her weight is recorded and laboratory work ordered to evaluate her pregnancy status, blood counts, lipids, complete metabolic panel, lithium level, and HIV status. Fortunately, she had received her series of MMR, meningococcal, and Tdap vaccinations when she was younger. Influenza, HPV, HAV, HBV, and pneumococcal vaccinations were all recommended to her, all of which can be given on the same day (HAV and HBV often are available as a combined vaccine). Ms. W receives a renewal of her psychiatric medications and counseling on healthy living habits (eg, diet and exercise, quitting tobacco and alcohol use, and safe sex practices) and the importance of immunizations.
Vaccination is 1 of the 10 great public health achievements of the 20th century when one considers how immunization of vaccine-preventable diseases has reduced morbidity, mortality, and health-associated costs.17 As mental health professionals, we can help pass on the direct and indirect benefits of immunizations to an often underserved and undertreated population to help improve their health outcomes and quality of life.
Patients with chronic, severe mental illness live much shorter lives than the general population. The 25-year loss in life expectancy for people with chronic mental illness has been attributed to higher rates of cardiovascular disease driven by increased smoking, obesity, poverty, and poor nutrition.1 These individuals also face the added burden of struggling with a psychiatric condition that often interferes with their ability to make optimal preventative health decisions, including staying up to date on vaccinations.2 A recent review from Toronto, Canada, found that the influenza vaccination rates among homeless adults with mental illness—a population at high risk of respiratory illness—was only 6.7% compared with 31.1% for the general population of Ontario.3
Mental health professionals may serve as the only contacts to offer medical care to this vulnerable population, leading some psychiatric leaders to advocate that psychiatrists be considered primary care providers within accountable care organizations. Because most vaccines are easily available, mental health professionals should know about key immunizations to guide their patients accordingly.
In the United States, approximately 45,000 adults die annually from vaccine-preventable diseases, the majority from influenza.4 When combined with the most recent Adult Immunization Schedule and general recommendations adapted from the CDC,5,6 the mnemonic ARM SHOT allows for a quick assessment of risk factors to guide administration and education about most vaccinations (Table 1). ARM SHOT involves assessing the following components of an individual’s health status and living arrangements to determine one’s risk of contracting communicable diseases:
- Age
- Risk of exposure
- Medical conditions (comorbidities)
- Substance use history
- HIV status or other immunocompromised states
- Occupancy, or living arrangements
- Tobacco use.
We recommend keeping a copy of the Adult Immunization Schedule (age ≥19) and/or the immunization schedule for children and adolescents (age ≤18) close for quick reference. Here, we provide a case and then explore how each component of the ARM SHOT mnemonic applies in decision-making.
Case Evaluating risk, assess needs
Ms. W, age 24, has bipolar I disorder, most recently manic with psychotic features. She presents for follow-up in clinic after a 5-day hospitalization for mania and comorbid alcohol use disorder. Her medical comorbidities include asthma and active tobacco use. She is taking lurasidone, 20 mg/d, and lithium, 900 mg/d. Her case manager is working to place Ms. W in a residential substance use disorder treatment program. Ms. W is on a waiting list to establish care with a primary care physician and has a history of poor engagement with medical services in general; prior attempts to place her with a primary care physician failed.
In advance of Ms. W’s transfer to a residential treatment facility, you have been asked to place a Mantoux screening test for tuberculosis (purified protein derivative), which raises the important question about her susceptibility to infectious diseases in general. To protect Ms. W from preventable diseases for which vaccines are available, you review the ARM SHOT mnemonic to broadly assess her candidacy for vaccinations.
Age
Age may be the most important determinant of a patient’s need for vaccination (Table 2). The CDC immunization schedules account for age-specific risks for diseases, complications, and responses to vaccination (Figure 1).6
Influenza vaccination. Adults can have an intramuscular or intradermal inactivated influenza vaccination yearly in the fall or winter, unless they have an allergy to a vaccine component such as egg protein. Those with such an allergy can receive a recombinant influenza vaccine. Until the 2016 to 2017 flu season, an intranasal mist of live, attenuated influenza vaccine was available to healthy, non-pregnant women, ages 2 to 49, without high-risk medical conditions. However, the CDC dropped its recommendation for this vaccine because data showed it did not effectively prevent the flu.7 Individuals age ≥65 can receive either the standard- or high-dose inactivated influenza vaccination. The latter contains 4 times the amount of antigen with the intention of triggering a stronger immune response in older adults.
Pneumonia immunization. All patients age ≥65 should receive vaccinations for Streptococcus pneumoniae and its variants in the form of one 13-valent pneumococcal conjugate vaccine and, at least 1 year later, one 23-valent pneumococcal polysaccharide vaccine (PPSV23). Immunization reduces the morbidity and mortality from pneumococcal illness by decreasing the burden of a pneumonia, bacteremia, or meningitis infection. Adults, ages 19 to 64, with a chronic disease (referred to as “special populations” in CDC tables), such as diabetes, heart or lung disease, alcoholism, or cirrhosis, or those who smoke cigarettes, should receive PPSV23 with a second dose administered at least 5 years after the first. The CDC recommends a 1-time re-vaccination at age 65 for patients if >5 years have passed since the last PPSV23 and if the patient was younger than age 65 at the time of primary vaccine for S. pneumoniae. This can be a rather tricky clinical situation; the health care provider should verify a patient’s immunization history to ensure that she (he) is receiving only necessary vaccines. However, when the history cannot be verified, err on the side of inclusion, because risks are minimal.
Shingles vaccination. Adults age ≥60 who are not immunocompromised should receive a single dose of live attenuated vaccine from varicella-zoster virus (VZV) to limit the risk of shingles from a prior chickenpox infection. The vaccine is approximately 66.5% effective at preventing postherpetic neuralgia for up to 4.9 years. Individuals as young as age 50 may have the vaccine because the risk of herpes zoster radically increases from then on,8 although most insurers only cover VZV vaccination after age 60.
Tetanus, diphtheria, and acellular pertussis (Tdap) vaccine. All adults should complete the 3-dose primary vaccination series for tetanus, diphtheria, and pertussis (also known as whooping cough) and this should include 1 dose of Tdap. Administration of the primary series is staged so that the second dose is given 4 weeks after the initial dose and the final dose 6 to 12 months after the first dose. After receiving the primary series, adults should receive a tetanus-diphtheria booster dose every 10 years. For adults ages 19 to 64, the Advisory Committee on Immunization Practices (ACIP) recommends 1 dose of Tdap in place of a booster vaccination to decrease the transmission risk of pertussis to vulnerable persons, especially children.
Human papillomavirus (HPV) immunization. The ACIP recommendation9 has been for children to receive routine vaccination for the 4 major strains of HPV—strains 6, 11, 16, and 18—starting at ages 11 to 12 to confer protection from HPV-associated diseases, such as genital warts, oropharyngeal cancer, and anal cancer; cancers of the cervix, vulva, and vagina in women; and penile cancer in men. Ideally, the vaccines are administered prior to HPV exposure from sexual contact. The quadrivalent HPV vaccine is safe and is administered as a 3-dose series, with the second and third doses given 2 and 6 months, respectively, after the initial dose. Adolescent girls also have the option of a bivalent HPV vaccine.
In 2016, the FDA approved a 9-valent HPV vaccine, a simpler 2-dose schedule for children ages 9 to 14 (2 doses at least 6 months apart). Leading cancer centers have endorsed this vaccine based on strong comparative data with the 3-dose regimen.10 For those not previously vaccinated, the HPV vaccine is available for women ages 13 to 26 and for men ages 13 to 21 (although men ages 22 to 26 can receive the vaccine, and it is recommended for men who have sex with men [MSM]). Women do not require Papanicolaou, serum pregnancy, HPV DNA, or HPV antibody tests prior to vaccination. If a woman becomes pregnant, remaining doses of the vaccine should be postponed until after delivery. Women still need to follow recommendations for cervical cancer screening because the HPV vaccine does not cover all genital strains of the virus. For sexually active individuals who might have HPV or genital warts, immunization has no clinical effect except to prevent other HPV strains.
Measles, mumps, and rubella (MMR) vaccine. All adults should receive, at minimum, 1 dose of MMR vaccination unless serological immunity can be verified or if contraindicated. Two doses of the vaccine are recommended for students attending post-high school institutions, health care personnel, and international travelers because they are at higher risk for exposure and transmission of measles and mumps. Individuals born before 1957 are considered immune to measles and mumps. A measles outbreak from December 2014 to February 201511 highlighted the importance of maintaining one’s immunity status for MMR.
Case continued
Based on Ms. W’s age, she should be offered vaccinations for influenza and opportunities to receive vaccinations for HPV, Tdap (the primary series, a Tdap or Td booster), and MMR, if appropriate and not completed previously.
Risk of exposure
Certain behaviors will increase the risk of exposure to and transmission of diseases communicable by blood and other bodily fluids (Table 3). These behaviors include needle injections (eg, during use of illicit drugs) and sexual activity with multiple partners, including MSM or promiscuity/impulsivity during a manic episode. A common consequence of risky behaviors is comorbid infection of HIV and viral hepatitis for those with substance use disorder or those who engage in high-risk sexual practices.12,13
Hepatitis B virus (HBV) immunization. Vaccination is one of the most effective ways to prevent HBV infection, which is why it is offered to all health care workers. HBV immunization is a 3-dose series in which the second and third doses are given 1 and 6 months after the initial doses, respectively. In addition to certain medical risk factors or conditions that indicate HBV vaccination, people should be offered the vaccine if they are in a higher risk occupation, travel, are of Asian or Pacific Islander ethnicity from an endemic area, or have any present or suspected sexually transmitted diseases.
Hepatitis A virus (HAV) vaccination. HAV is transmitted via fecal–oral routes, often from contaminated water or food, or through household or sexual contact with an infected person. Individuals should receive the HAV vaccine if they use illicit drugs by any route of administration, work with primates infected with HAV, travel to countries with unknown or high rates of HAV, or have chronic liver disease (ie, hepatitis, alcohol use disorder, or non-alcoholic fatty liver disease) or clotting deficiencies. The CDC Health Information for International Travel, commonly called the “Yellow Book,” publishes vaccination recommendations for those who plan travel to specific countries.14
Case continued
Ms. W’s history of mania (if such episodes included increased sexual activity) and substance use would make her a candidate for the HBV and HAV vaccinations and could also strengthen our previous recommendation that she receive the HPV vaccination.
Medical conditions
Patients with certain medical conditions may have difficulty fighting infections or become more susceptible to morbidity and mortality from coinfection with vaccine-preventable illnesses. Secondary effects of psychotropic medications that may carry implications for vaccine recommendations (eg, risk of agranulocytosis and impaired cell-medicated immunity with mirtazapine and clozapine or renal impairment from lithium use) are of particular concern in psychiatric patients.2
To help care for these patients, the CDC has developed a “medical conditions” schedule (Figure 2). This schedule makes vaccination recommendations for those with a weakened immune system, including patients with HIV, chronic obstructive pulmonary disease (COPD), diabetes, hepatitis, asplenia, end-stage renal disease, cardiac disease, and pregnancy.
Because patients with psychiatric illness face a greater risk of heart disease and diabetes, these conditions may warrant special reference on the schedule. The increased cardiometabolic risk factors in these patients may be due in part to genetics, socioeconomic status, lifestyle behaviors, and medications to treat their mental illness (eg, antipsychotics). Patients with bipolar disorder or schizophrenia in particular tend to have higher rates of COPD (mainly from chronic bronchitis) and asthma than the general population.12 Pay special attention to the indications schedule for those with chronic lung disease, especially patients who continue to smoke cigarettes.
Case continued
Because of Ms. W’s asthma, the CDC schedule recommends ensuring she is up to date on her influenza, pneumococcal, and Tdap vaccinations.
Substance use
Patients with combined psychiatric and substance use disorders (“dual diagnosis”) have lower rates of receiving preventive care than patients with either condition alone.15 Substance use can be behaviorally disinhibiting, leading to increased risk of exposures from sexual contact or other risky activities. The use of illicit substances can provide a nidus for infection depending on the route of administration and can result in negative effects on organ systems, compromising one’s ability to ward off infection.
Patients who use any illicit drugs, regardless of the method of delivery, should be recommended for HAV vaccination. For those with alcohol use disorder and/or chronic liver disease, and/or seeking treatment for substance use, hepatitis B screening and vaccination is recommended.
Case continued
From a substance use perspective, discussion of vaccination status for both hepatitis A and B would be important for Ms. W.
HIV or immunocompromised
Persons with severe mental illness have high rates of HIV, with almost 8 times the risk of exposure, compared with the general population due to myriad reasons, including greater rates of substance abuse, higher risk sexual behavior, and lack of awareness of HIV transmission.12,13 Patients with mental illness are also at risk of leukopenia and agranulocytosis from certain drugs used to treat their conditions, such as clozapine.
Pregnancy is a challenge for women with mental illness because of the pharmacologic risk and immune-system compromise to the mother and baby. A pregnant woman who has HIV with a CD4 count <200, or has a weakened immune system from an organ transplant or a similar condition, is a candidate for certain vaccines based on the Adult Immunization Schedule (Figure 2). However, these patients should avoid live vaccines, such as the intranasal mist of live influenza, MMR, VZV, and varicella, to avoid illness from these inoculations.
Case continued
Ms. W should undergo testing for pregnancy and HIV (and preferably other sexually transmitted infections per general preventive health guidelines) before receiving any live vaccinations.
Occupancy
Aside from direct transmission of bodily fluids, infectious diseases also can spread through droplets/secretions from the throat and respiratory tract. Close quarters or lengthy contact enhances communicability by droplets, and therefore people who reside in a communal living space (eg, individuals in substance use treatment facilities or those who reside in a nursing home) are most susceptible.
The bacterial disease Neisseria meningitidis (meningococcus) can spread through droplets and can cause pneumonia, bacteremia, and meningitis. Vaccination is indicated, and in some states is mandated, for college students who live in residence halls and missed routine vaccination by age 16. Meningococcus conjugate vaccine is administered in 2 doses; each dose may be given at least 2 months apart for those with HIV, asplenia, or persistent complement-related disorders. A single dose may be recommended for travelers to areas where meningococcal disease is hyperendemic or epidemic, military recruits, or microbiologists. For those age ≥55 and older, meningococcal polysaccharide vaccine is recommended over meningococcal conjugate vaccine.
Influenza, MMR, diphtheria, pertussis, and pneumococcus also spread through droplet contact.
Case continued
If Ms. W had not previously received the meningococcus vaccine as part of adolescent immunizations, she could benefit from this vaccine because she plans to enter a residential substance use disorder treatment program.
Tobacco use
Patients with psychiatric illness are twice as likely to smoke compared with the general population.16 Adult smokers, especially those with chronic lung disease, are at higher risk for influenza and pneumococcal-related illness; they should be vaccinated against these illnesses regardless of age (as discussed in the “Age” section).
Case continued
Because she smokes, Ms. W should receive counseling on vaccinations, such as influenza and pneumonia, to lessen her risk of respiratory illnesses and downstream sepsis.
Conclusion
Ms. W’s case represents an unfortunately all-too-common scenario where her multifaceted biopsychosocial circumstances place her at high risk for vaccine-preventable conditions. Her weight is recorded and laboratory work ordered to evaluate her pregnancy status, blood counts, lipids, complete metabolic panel, lithium level, and HIV status. Fortunately, she had received her series of MMR, meningococcal, and Tdap vaccinations when she was younger. Influenza, HPV, HAV, HBV, and pneumococcal vaccinations were all recommended to her, all of which can be given on the same day (HAV and HBV often are available as a combined vaccine). Ms. W receives a renewal of her psychiatric medications and counseling on healthy living habits (eg, diet and exercise, quitting tobacco and alcohol use, and safe sex practices) and the importance of immunizations.
Vaccination is 1 of the 10 great public health achievements of the 20th century when one considers how immunization of vaccine-preventable diseases has reduced morbidity, mortality, and health-associated costs.17 As mental health professionals, we can help pass on the direct and indirect benefits of immunizations to an often underserved and undertreated population to help improve their health outcomes and quality of life.
1. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
2. Raj YP, Lloyd L. Adult immunizations. In: McCarron RM, Xiong GL, Keenan GR, et al, eds. Preventive medical care in psychiatry. Arlington, VA: American Psychiatric Publishing. 2015;215-227.
3. Young S, Dosani N, Whisler A, et al. Influenza vaccination rates among homeless adults with mental illness in Toronto. J Prim Care Community Health. 2015;6(3):211-214.
4. Kroger AT, Atkinson WL, Marcues EK, et al; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). General recommendations on immunization: recommendations on the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2006;55(RR-15):1-48.
5. Centers for Disease Control and Prevention. Recommended Adult Immunization by Vaccine and Age Group. http://www.cdc.gov/vaccines/schedules/hcp/adult.html. Updated February 27, 2017. Accessed February 1, 2017.
6. National Center for Immunization and Respiratory Diseases. General recommendations on immunization—recommendations on the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2011;60(2):1-64.
7. Centers for Disease Control and Prevention. ACIP votes down use of LAIV for 2016-2017 flu season. https://www.cdc.gov/media/releases/2016/s0622-laiv-flu.html. Updated June 22, 2016. Accessed February 1, 2017.
8. Hales CM, Harpaz, R, Ortega-Sanchez I, et al; Centers for Disease Control and Prevention. Update on recommendations for use of herpes zoster vaccine. MMWR Morb Mortal Wkly Rep. 2014;63(33):729-731.
9. Petrosky E, Bocchini Jr JA, Hariri S, et al; Centers for Disease Control and Prevention (CDC). Use of 9-valent human papillomavirus (HPV) vaccine: updated HPV vaccine recommendations of the advisory committee on immunization practices. MMWR Morb Mortal Wkly Rep. 2015;64(11)300-304.
10. Iversen OE, Miranda MJ, Ulied A, et al. Immunogenicity of the 9-valent HPV vaccine using 2-dose regimens in girls and boys vs a 3-dose regimen in women. JAMA. 2016;316(22):2411-2421.
11. Zipprich J, Winter K, Hacker J, et al; Centers for Disease Control and Prevention (CDC). Measles outbreak—California, December 2014-February 2015. MMWR Morb Mortal Wkly Rep. 2015;64(6):153-154.
12. De Hert M, Correll CU, Bobes J, et al. Physical illness in patients with severe mental disorders. I. Prevalence, impact of medications and disparities in health care. World Psychiatry. 2011;10(1):52-77.
13. Rosenberg SD, Goodman LA, Osher FC, et al. Prevalence of HIV, hepatitis B, and hepatitis C in people with severe mental illness. Am J Public Health. 2001;91(1):31-37.
14. Centers for Disease for Control and Prevention. CDC yellow book 2018: health information for international travel. New York, NY: Oxford University Press; 2017.
15. Druss BG, Rosenheck RA, Desai MM, et al. Quality of preventive medical care for patients with mental disorders. Med Care. 2002;40(2):129-136.
16. Lasser K, Boyd J, Woolhandler S, et al. Smoking and mental illness: a population-based prevalence study. JAMA. 2000;284(20):2606-2610.
17. Centers for Disease Control and Prevention (CDC). Ten great public health achievements—United States, 2001-2010. MMWR Morb Mortal Wkly Rep. 2011;60(19);619-623.
1. Newcomer JW, Hennekens CH. Severe mental illness and risk of cardiovascular disease. JAMA. 2007;298(15):1794-1796.
2. Raj YP, Lloyd L. Adult immunizations. In: McCarron RM, Xiong GL, Keenan GR, et al, eds. Preventive medical care in psychiatry. Arlington, VA: American Psychiatric Publishing. 2015;215-227.
3. Young S, Dosani N, Whisler A, et al. Influenza vaccination rates among homeless adults with mental illness in Toronto. J Prim Care Community Health. 2015;6(3):211-214.
4. Kroger AT, Atkinson WL, Marcues EK, et al; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). General recommendations on immunization: recommendations on the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2006;55(RR-15):1-48.
5. Centers for Disease Control and Prevention. Recommended Adult Immunization by Vaccine and Age Group. http://www.cdc.gov/vaccines/schedules/hcp/adult.html. Updated February 27, 2017. Accessed February 1, 2017.
6. National Center for Immunization and Respiratory Diseases. General recommendations on immunization—recommendations on the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2011;60(2):1-64.
7. Centers for Disease Control and Prevention. ACIP votes down use of LAIV for 2016-2017 flu season. https://www.cdc.gov/media/releases/2016/s0622-laiv-flu.html. Updated June 22, 2016. Accessed February 1, 2017.
8. Hales CM, Harpaz, R, Ortega-Sanchez I, et al; Centers for Disease Control and Prevention. Update on recommendations for use of herpes zoster vaccine. MMWR Morb Mortal Wkly Rep. 2014;63(33):729-731.
9. Petrosky E, Bocchini Jr JA, Hariri S, et al; Centers for Disease Control and Prevention (CDC). Use of 9-valent human papillomavirus (HPV) vaccine: updated HPV vaccine recommendations of the advisory committee on immunization practices. MMWR Morb Mortal Wkly Rep. 2015;64(11)300-304.
10. Iversen OE, Miranda MJ, Ulied A, et al. Immunogenicity of the 9-valent HPV vaccine using 2-dose regimens in girls and boys vs a 3-dose regimen in women. JAMA. 2016;316(22):2411-2421.
11. Zipprich J, Winter K, Hacker J, et al; Centers for Disease Control and Prevention (CDC). Measles outbreak—California, December 2014-February 2015. MMWR Morb Mortal Wkly Rep. 2015;64(6):153-154.
12. De Hert M, Correll CU, Bobes J, et al. Physical illness in patients with severe mental disorders. I. Prevalence, impact of medications and disparities in health care. World Psychiatry. 2011;10(1):52-77.
13. Rosenberg SD, Goodman LA, Osher FC, et al. Prevalence of HIV, hepatitis B, and hepatitis C in people with severe mental illness. Am J Public Health. 2001;91(1):31-37.
14. Centers for Disease for Control and Prevention. CDC yellow book 2018: health information for international travel. New York, NY: Oxford University Press; 2017.
15. Druss BG, Rosenheck RA, Desai MM, et al. Quality of preventive medical care for patients with mental disorders. Med Care. 2002;40(2):129-136.
16. Lasser K, Boyd J, Woolhandler S, et al. Smoking and mental illness: a population-based prevalence study. JAMA. 2000;284(20):2606-2610.
17. Centers for Disease Control and Prevention (CDC). Ten great public health achievements—United States, 2001-2010. MMWR Morb Mortal Wkly Rep. 2011;60(19);619-623.

A practical approach to interviewing a somatizing patient
Somatization is the experience of psychological distress in the form of bodil
Collecting a detailed history of physical symptoms can help the patient feel that you are listening to him (her) and that the chief concern is important. A detailed review of psychiatric symptoms (eg, hallucinations, paranoia, suicidality, etc.) should be deferred until later in the examination. Asking questions relating to psychiatric symptoms early could lead to further resistance by reinforcing negative preconceptions that the patient might have regarding mental illness.
Explicitly express empathy regarding physical symptoms throughout the interview to acknowledge any real suffering the patient is experiencing and to contradict any notion that psychiatric evaluation implies that the suffering could be imaginary.
Ask, “How has this illness affected your life?” This question helps make the connection between the patient’s physical state and social milieu. If somatization is confirmed, then the provider should assist the patient in reversing the arrow of causation. Although the ultimate goal is for the patient to understand how his (her) life has affected the symptoms, simply understanding that there are connections between the two is a start toward this goal.1
Explore the response to the previous question. Expand upon it to elicit a detailed social history, listening for any social stressors.
Obtain family and personal histories of allergies, substance abuse, and medical or psychiatric illness.
Review psychiatric symptoms. Make questions less jarring2 by adapting them to the patient’s situation, such as “Has your illness become so painful that at times you don’t even want to live?”
Perform cognitive and physical examinations. Conducting a physical examination could further reassure the patient that you are not ignoring physical complaints.
Educate the patient that the mind and body are connected and emotions affect how one feels physically. Use examples, such as “When I feel anxious, my heart beats faster” or “A headache might hurt more at work than at the beach.”
Elicit feedback and questions from the patient.
Discuss your treatment plan with the patient. Resistant patients with confirmed somatization disorders might accept psychiatric care as a means of dealing with the stress or pain of their physical symptoms.
Consider asking:
- What would you be doing if you weren’t in the hospital right now?
- Aside from your health, what’s the biggest challenge in your life?
- Everything has a good side and a bad side. Is there anything positive about dealing with your illness? Providing the patient with an example of negative aspects of a good thing (such as the calories in ice cream, the high cost of gold, etc.) can help make this point.
- What would your life look like if you didn’t have these symptoms?
1. Creed F, Guthrie E. Techniques for interviewing the somatising patient. Br J Psychiatry. 1993;162:467-471.
2. Carlat DJ. The psychiatric interview: a practical guide. 2nd ed. Philadelphia, PA: Lippincott, Williams, & Wilkins; 2005.
Somatization is the experience of psychological distress in the form of bodil
Collecting a detailed history of physical symptoms can help the patient feel that you are listening to him (her) and that the chief concern is important. A detailed review of psychiatric symptoms (eg, hallucinations, paranoia, suicidality, etc.) should be deferred until later in the examination. Asking questions relating to psychiatric symptoms early could lead to further resistance by reinforcing negative preconceptions that the patient might have regarding mental illness.
Explicitly express empathy regarding physical symptoms throughout the interview to acknowledge any real suffering the patient is experiencing and to contradict any notion that psychiatric evaluation implies that the suffering could be imaginary.
Ask, “How has this illness affected your life?” This question helps make the connection between the patient’s physical state and social milieu. If somatization is confirmed, then the provider should assist the patient in reversing the arrow of causation. Although the ultimate goal is for the patient to understand how his (her) life has affected the symptoms, simply understanding that there are connections between the two is a start toward this goal.1
Explore the response to the previous question. Expand upon it to elicit a detailed social history, listening for any social stressors.
Obtain family and personal histories of allergies, substance abuse, and medical or psychiatric illness.
Review psychiatric symptoms. Make questions less jarring2 by adapting them to the patient’s situation, such as “Has your illness become so painful that at times you don’t even want to live?”
Perform cognitive and physical examinations. Conducting a physical examination could further reassure the patient that you are not ignoring physical complaints.
Educate the patient that the mind and body are connected and emotions affect how one feels physically. Use examples, such as “When I feel anxious, my heart beats faster” or “A headache might hurt more at work than at the beach.”
Elicit feedback and questions from the patient.
Discuss your treatment plan with the patient. Resistant patients with confirmed somatization disorders might accept psychiatric care as a means of dealing with the stress or pain of their physical symptoms.
Consider asking:
- What would you be doing if you weren’t in the hospital right now?
- Aside from your health, what’s the biggest challenge in your life?
- Everything has a good side and a bad side. Is there anything positive about dealing with your illness? Providing the patient with an example of negative aspects of a good thing (such as the calories in ice cream, the high cost of gold, etc.) can help make this point.
- What would your life look like if you didn’t have these symptoms?
Somatization is the experience of psychological distress in the form of bodil
Collecting a detailed history of physical symptoms can help the patient feel that you are listening to him (her) and that the chief concern is important. A detailed review of psychiatric symptoms (eg, hallucinations, paranoia, suicidality, etc.) should be deferred until later in the examination. Asking questions relating to psychiatric symptoms early could lead to further resistance by reinforcing negative preconceptions that the patient might have regarding mental illness.
Explicitly express empathy regarding physical symptoms throughout the interview to acknowledge any real suffering the patient is experiencing and to contradict any notion that psychiatric evaluation implies that the suffering could be imaginary.
Ask, “How has this illness affected your life?” This question helps make the connection between the patient’s physical state and social milieu. If somatization is confirmed, then the provider should assist the patient in reversing the arrow of causation. Although the ultimate goal is for the patient to understand how his (her) life has affected the symptoms, simply understanding that there are connections between the two is a start toward this goal.1
Explore the response to the previous question. Expand upon it to elicit a detailed social history, listening for any social stressors.
Obtain family and personal histories of allergies, substance abuse, and medical or psychiatric illness.
Review psychiatric symptoms. Make questions less jarring2 by adapting them to the patient’s situation, such as “Has your illness become so painful that at times you don’t even want to live?”
Perform cognitive and physical examinations. Conducting a physical examination could further reassure the patient that you are not ignoring physical complaints.
Educate the patient that the mind and body are connected and emotions affect how one feels physically. Use examples, such as “When I feel anxious, my heart beats faster” or “A headache might hurt more at work than at the beach.”
Elicit feedback and questions from the patient.
Discuss your treatment plan with the patient. Resistant patients with confirmed somatization disorders might accept psychiatric care as a means of dealing with the stress or pain of their physical symptoms.
Consider asking:
- What would you be doing if you weren’t in the hospital right now?
- Aside from your health, what’s the biggest challenge in your life?
- Everything has a good side and a bad side. Is there anything positive about dealing with your illness? Providing the patient with an example of negative aspects of a good thing (such as the calories in ice cream, the high cost of gold, etc.) can help make this point.
- What would your life look like if you didn’t have these symptoms?
1. Creed F, Guthrie E. Techniques for interviewing the somatising patient. Br J Psychiatry. 1993;162:467-471.
2. Carlat DJ. The psychiatric interview: a practical guide. 2nd ed. Philadelphia, PA: Lippincott, Williams, & Wilkins; 2005.
1. Creed F, Guthrie E. Techniques for interviewing the somatising patient. Br J Psychiatry. 1993;162:467-471.
2. Carlat DJ. The psychiatric interview: a practical guide. 2nd ed. Philadelphia, PA: Lippincott, Williams, & Wilkins; 2005.