User login
Longer edoxaban may benefit cancer patients with distal DVT
Patients with active cancer and newly diagnosed isolated distal deep vein thrombosis (DVT) who received 12 months of edoxaban (Savaysa) had fewer thrombotic events at 1 year than those who received 3 months of treatment, without significantly increased bleeding, in the ONCO-DVT trial.
However, lead author Yugo Yamashita, MD, of Kyoto University noted that caution is needed when determining anticoagulation strategies in individual patients with distal DVT, especially those with high risk for bleeding.
Dr. Yamashita presented the results at the annual congress of the European Society of Cardiology, and the trial was simultaneously published in the journal Circulation.
“This is the first and only randomized trial to show the superiority of longer duration over shorter duration of anticoagulation therapy for reducing thrombotic events in cancer patients with isolated distal DVT,” he said in a press briefing.
The results provide support for 12 months of edoxaban in patients with active cancer and isolated distal DVD, he said in an email.
However, “considering the risk of bleeding associated with anticoagulation therapy, physicians should make the decision of anticoagulation strategies for these patients based on risk-benefit balance of anticoagulation therapy in individual patients,” he stressed.
The take-home message for clinicians is that, “if you find minor DVT in cancer patients, please be careful, because their thrombotic risk was not low” in this trial, Dr. Yamashita said.
The study was conducted in Japan, so whether or not the results are generalizable to other populations is not clear. “Subgroup analysis based on body weight did not show any signal of different effect,” he noted, which suggests that the main results could be applied to other populations, including the U.S. population. However, “generalizability of the current results should be carried out carefully.”
Caution needed when translating findings into clinical practice
The assigned discussant, Teresa Lopez-Fernandez, MD, from La Paz University Hospital, Madrid, who was co-chairperson of 2022 ESC guidelines on cardio-oncology, noted that the optimal anticoagulation therapy strategy is unclear in patients with cancer and isolated distal DVT.
“2022 ESC guidelines on cardio-oncology and [European Society for Medical Oncology] guidelines from this year,” she said, “are both in agreement that we need to prolong anticoagulation [therapy to prevent venous thromboembolism (VTE)] when active cancer exists, and particularly in patients with metastatic cancer. The problem is that none of this text refers specifically to distal DVT.”
The ONCO-DVT trial sheds light on this, but there are a few points to consider when interpreting the findings.
Major bleeding was slightly increased in the 12-month vs 3-month edoxaban groups, although this was not statistically significant, she noted. Moreover, 75% of the patients were treated with low-dose edoxaban, mainly due to their low weight. Also, bleeding risk probably differs in different cancer types.
“These are important things that we need to keep in mind when we try to transfer this data to [inform] our clinical practice,” Dr. Lopez-Fernandez said.
She drew attention to a recent study based on RIETE registry data that suggests that “isolated distal DVT is a big problem for patients with cancer in comparison with noncancer patients, where it seems it’s a low-risk problem.”
The main takeaways from ONCO-DVT, Dr. Lopez-Fernandez said, are that it confirms that cancer-associated isolated distal DVT is a marker of poor prognosis, and it supports the need for extended anticoagulation in patients with active, ongoing cancer and isolated distal DVT.
However, “we need to be cautious to try to really understand what the bleeding risks of these patients are,” she said, “particularly because it is not always easy to transfer the results from an Asian population to other populations.”
There is also a need for further studies with other doses, with other novel oral anticoagulants, and in patients at high risk for bleeding, in clinical practice.
Dr. Yamashita said that the study suggests that there is a potential benefit of prolonged duration of anticoagulant therapy for some patients with isolated distal DVT, but not all patients should receive this dosing strategy, because some patients may be at high risk for bleeding or VTE recurrence. A subanalysis of data from ONCO-DVT study should shed further light on this.
“We need to individualize our risk stratification,” Dr. Lopez-Fernandez said, adding that notably, “a lot of patients in the 12-month group did not continue with the 12-month treatment,” which may have affected bleeding results. Dr. Yamashita agreed.
Study design and findings
From April 2019 to June 2022, the researchers enrolled and randomly assigned 604 patients with active cancer who had newly diagnosed isolated distal DVT, confirmed by ultrasonography, and were scheduled for DVT treatment with anticoagulation therapy, at 60 centers.
Active cancer was defined as a cancer diagnosis or cancer treatment (surgery, chemotherapy, radiotherapy, etc.) within 6 months of randomization, or current recurrence, local invasion, distant metastases, or hematopoietic malignancy without complete remission.
The most common reasons for ultrasonography were elevated D-dimer levels (62%) and suspected DVT because of symptoms (20%).
The patients had a mean age of 70.8 years and 28% were men. The most common cancer sites were ovaries (14%), uterus (13%), lung (11%), colon (9%), and pancreas (8%), followed by stomach, blood, and breast (each 5%).
The patients were randomly assigned 1:1 to receive 12 months or 3 months of oral edoxaban at a dose of 60 mg once daily or 30 mg once daily in patients with body weight of 60 kg or less, creatinine clearance of 30-50 mL/minute, or concomitant treatment with a potent P-glycoprotein inhibitor.
After excluding 3 patients who withdrew consent, 601 patients were included in the intention-to-treat population: 296 patients in the 12-month edoxaban group and 305 patients in the 3-month edoxaban group.
About 70% of patients had a body weight of 60 kg or less and about 22% had a creatinine clearance less than 50 mL/min. About three quarters received the lower dose of edoxaban.
In the 12-month edoxaban group, 223 patients completed the 1-year follow-up (66 patients had died and 7 were lost to follow-up). In the 3-month edoxaban group, 224 patients completed the 1-year follow-up (77 had died and 4 were lost to follow-up).
In the 12-month edoxaban group, 41% of the patients had discontinued treatment by 12 months. In the 3-month edoxaban group, 41% of patients had discontinued treatment by 3 months.
The primary endpoint – a symptomatic recurrent VTE event or VTE-related death – occurred in 3 of the 222 patients (1.2%) in the 12-month edoxaban group and in 22 of the 210 (8.5%) in the 3-month edoxaban group (odds ratio,0.13; 95% confidence interval, 0.03-0.44, P < .001). There were no VTE-related deaths.
The major secondary endpoint – major bleeding, according to International Society on Thrombosis and Hemostasis criteria – occurred in 28 of the 210 patients (10.2%) in the 12-month edoxaban group and in 22 of the 217 (7.6%) in the 3-month edoxaban group (OR, 1.34; 95% CI, 0.75-2.41, P = NS).
The researchers acknowledged that study limitations include an open-label design, a lower-than-expected primary endpoint rate, and less than high adherence to edoxaban, as well as the need for caution when generalizing the results to other populations.
The study was funded by Daiichi Sankyo. Dr. Yamashita disclosed receiving lecture fees from Bayer Healthcare, Bristol-Myers Squibb, Pfizer, and Daiichi Sankyo, and grant support from Bayer Healthcare and Daiichi Sankyo. Dr. Lopez-Fernandez disclosed receiving speaker fees from Phillips, Janssen, Daiichi Sankyo, Myocardial Solutions, AstraZeneca, Pfizer, Beigene, and Bayer not related to this study.
A version of this article appeared on Medscape.com.
Patients with active cancer and newly diagnosed isolated distal deep vein thrombosis (DVT) who received 12 months of edoxaban (Savaysa) had fewer thrombotic events at 1 year than those who received 3 months of treatment, without significantly increased bleeding, in the ONCO-DVT trial.
However, lead author Yugo Yamashita, MD, of Kyoto University noted that caution is needed when determining anticoagulation strategies in individual patients with distal DVT, especially those with high risk for bleeding.
Dr. Yamashita presented the results at the annual congress of the European Society of Cardiology, and the trial was simultaneously published in the journal Circulation.
“This is the first and only randomized trial to show the superiority of longer duration over shorter duration of anticoagulation therapy for reducing thrombotic events in cancer patients with isolated distal DVT,” he said in a press briefing.
The results provide support for 12 months of edoxaban in patients with active cancer and isolated distal DVD, he said in an email.
However, “considering the risk of bleeding associated with anticoagulation therapy, physicians should make the decision of anticoagulation strategies for these patients based on risk-benefit balance of anticoagulation therapy in individual patients,” he stressed.
The take-home message for clinicians is that, “if you find minor DVT in cancer patients, please be careful, because their thrombotic risk was not low” in this trial, Dr. Yamashita said.
The study was conducted in Japan, so whether or not the results are generalizable to other populations is not clear. “Subgroup analysis based on body weight did not show any signal of different effect,” he noted, which suggests that the main results could be applied to other populations, including the U.S. population. However, “generalizability of the current results should be carried out carefully.”
Caution needed when translating findings into clinical practice
The assigned discussant, Teresa Lopez-Fernandez, MD, from La Paz University Hospital, Madrid, who was co-chairperson of 2022 ESC guidelines on cardio-oncology, noted that the optimal anticoagulation therapy strategy is unclear in patients with cancer and isolated distal DVT.
“2022 ESC guidelines on cardio-oncology and [European Society for Medical Oncology] guidelines from this year,” she said, “are both in agreement that we need to prolong anticoagulation [therapy to prevent venous thromboembolism (VTE)] when active cancer exists, and particularly in patients with metastatic cancer. The problem is that none of this text refers specifically to distal DVT.”
The ONCO-DVT trial sheds light on this, but there are a few points to consider when interpreting the findings.
Major bleeding was slightly increased in the 12-month vs 3-month edoxaban groups, although this was not statistically significant, she noted. Moreover, 75% of the patients were treated with low-dose edoxaban, mainly due to their low weight. Also, bleeding risk probably differs in different cancer types.
“These are important things that we need to keep in mind when we try to transfer this data to [inform] our clinical practice,” Dr. Lopez-Fernandez said.
She drew attention to a recent study based on RIETE registry data that suggests that “isolated distal DVT is a big problem for patients with cancer in comparison with noncancer patients, where it seems it’s a low-risk problem.”
The main takeaways from ONCO-DVT, Dr. Lopez-Fernandez said, are that it confirms that cancer-associated isolated distal DVT is a marker of poor prognosis, and it supports the need for extended anticoagulation in patients with active, ongoing cancer and isolated distal DVT.
However, “we need to be cautious to try to really understand what the bleeding risks of these patients are,” she said, “particularly because it is not always easy to transfer the results from an Asian population to other populations.”
There is also a need for further studies with other doses, with other novel oral anticoagulants, and in patients at high risk for bleeding, in clinical practice.
Dr. Yamashita said that the study suggests that there is a potential benefit of prolonged duration of anticoagulant therapy for some patients with isolated distal DVT, but not all patients should receive this dosing strategy, because some patients may be at high risk for bleeding or VTE recurrence. A subanalysis of data from ONCO-DVT study should shed further light on this.
“We need to individualize our risk stratification,” Dr. Lopez-Fernandez said, adding that notably, “a lot of patients in the 12-month group did not continue with the 12-month treatment,” which may have affected bleeding results. Dr. Yamashita agreed.
Study design and findings
From April 2019 to June 2022, the researchers enrolled and randomly assigned 604 patients with active cancer who had newly diagnosed isolated distal DVT, confirmed by ultrasonography, and were scheduled for DVT treatment with anticoagulation therapy, at 60 centers.
Active cancer was defined as a cancer diagnosis or cancer treatment (surgery, chemotherapy, radiotherapy, etc.) within 6 months of randomization, or current recurrence, local invasion, distant metastases, or hematopoietic malignancy without complete remission.
The most common reasons for ultrasonography were elevated D-dimer levels (62%) and suspected DVT because of symptoms (20%).
The patients had a mean age of 70.8 years and 28% were men. The most common cancer sites were ovaries (14%), uterus (13%), lung (11%), colon (9%), and pancreas (8%), followed by stomach, blood, and breast (each 5%).
The patients were randomly assigned 1:1 to receive 12 months or 3 months of oral edoxaban at a dose of 60 mg once daily or 30 mg once daily in patients with body weight of 60 kg or less, creatinine clearance of 30-50 mL/minute, or concomitant treatment with a potent P-glycoprotein inhibitor.
After excluding 3 patients who withdrew consent, 601 patients were included in the intention-to-treat population: 296 patients in the 12-month edoxaban group and 305 patients in the 3-month edoxaban group.
About 70% of patients had a body weight of 60 kg or less and about 22% had a creatinine clearance less than 50 mL/min. About three quarters received the lower dose of edoxaban.
In the 12-month edoxaban group, 223 patients completed the 1-year follow-up (66 patients had died and 7 were lost to follow-up). In the 3-month edoxaban group, 224 patients completed the 1-year follow-up (77 had died and 4 were lost to follow-up).
In the 12-month edoxaban group, 41% of the patients had discontinued treatment by 12 months. In the 3-month edoxaban group, 41% of patients had discontinued treatment by 3 months.
The primary endpoint – a symptomatic recurrent VTE event or VTE-related death – occurred in 3 of the 222 patients (1.2%) in the 12-month edoxaban group and in 22 of the 210 (8.5%) in the 3-month edoxaban group (odds ratio,0.13; 95% confidence interval, 0.03-0.44, P < .001). There were no VTE-related deaths.
The major secondary endpoint – major bleeding, according to International Society on Thrombosis and Hemostasis criteria – occurred in 28 of the 210 patients (10.2%) in the 12-month edoxaban group and in 22 of the 217 (7.6%) in the 3-month edoxaban group (OR, 1.34; 95% CI, 0.75-2.41, P = NS).
The researchers acknowledged that study limitations include an open-label design, a lower-than-expected primary endpoint rate, and less than high adherence to edoxaban, as well as the need for caution when generalizing the results to other populations.
The study was funded by Daiichi Sankyo. Dr. Yamashita disclosed receiving lecture fees from Bayer Healthcare, Bristol-Myers Squibb, Pfizer, and Daiichi Sankyo, and grant support from Bayer Healthcare and Daiichi Sankyo. Dr. Lopez-Fernandez disclosed receiving speaker fees from Phillips, Janssen, Daiichi Sankyo, Myocardial Solutions, AstraZeneca, Pfizer, Beigene, and Bayer not related to this study.
A version of this article appeared on Medscape.com.
Patients with active cancer and newly diagnosed isolated distal deep vein thrombosis (DVT) who received 12 months of edoxaban (Savaysa) had fewer thrombotic events at 1 year than those who received 3 months of treatment, without significantly increased bleeding, in the ONCO-DVT trial.
However, lead author Yugo Yamashita, MD, of Kyoto University noted that caution is needed when determining anticoagulation strategies in individual patients with distal DVT, especially those with high risk for bleeding.
Dr. Yamashita presented the results at the annual congress of the European Society of Cardiology, and the trial was simultaneously published in the journal Circulation.
“This is the first and only randomized trial to show the superiority of longer duration over shorter duration of anticoagulation therapy for reducing thrombotic events in cancer patients with isolated distal DVT,” he said in a press briefing.
The results provide support for 12 months of edoxaban in patients with active cancer and isolated distal DVD, he said in an email.
However, “considering the risk of bleeding associated with anticoagulation therapy, physicians should make the decision of anticoagulation strategies for these patients based on risk-benefit balance of anticoagulation therapy in individual patients,” he stressed.
The take-home message for clinicians is that, “if you find minor DVT in cancer patients, please be careful, because their thrombotic risk was not low” in this trial, Dr. Yamashita said.
The study was conducted in Japan, so whether or not the results are generalizable to other populations is not clear. “Subgroup analysis based on body weight did not show any signal of different effect,” he noted, which suggests that the main results could be applied to other populations, including the U.S. population. However, “generalizability of the current results should be carried out carefully.”
Caution needed when translating findings into clinical practice
The assigned discussant, Teresa Lopez-Fernandez, MD, from La Paz University Hospital, Madrid, who was co-chairperson of 2022 ESC guidelines on cardio-oncology, noted that the optimal anticoagulation therapy strategy is unclear in patients with cancer and isolated distal DVT.
“2022 ESC guidelines on cardio-oncology and [European Society for Medical Oncology] guidelines from this year,” she said, “are both in agreement that we need to prolong anticoagulation [therapy to prevent venous thromboembolism (VTE)] when active cancer exists, and particularly in patients with metastatic cancer. The problem is that none of this text refers specifically to distal DVT.”
The ONCO-DVT trial sheds light on this, but there are a few points to consider when interpreting the findings.
Major bleeding was slightly increased in the 12-month vs 3-month edoxaban groups, although this was not statistically significant, she noted. Moreover, 75% of the patients were treated with low-dose edoxaban, mainly due to their low weight. Also, bleeding risk probably differs in different cancer types.
“These are important things that we need to keep in mind when we try to transfer this data to [inform] our clinical practice,” Dr. Lopez-Fernandez said.
She drew attention to a recent study based on RIETE registry data that suggests that “isolated distal DVT is a big problem for patients with cancer in comparison with noncancer patients, where it seems it’s a low-risk problem.”
The main takeaways from ONCO-DVT, Dr. Lopez-Fernandez said, are that it confirms that cancer-associated isolated distal DVT is a marker of poor prognosis, and it supports the need for extended anticoagulation in patients with active, ongoing cancer and isolated distal DVT.
However, “we need to be cautious to try to really understand what the bleeding risks of these patients are,” she said, “particularly because it is not always easy to transfer the results from an Asian population to other populations.”
There is also a need for further studies with other doses, with other novel oral anticoagulants, and in patients at high risk for bleeding, in clinical practice.
Dr. Yamashita said that the study suggests that there is a potential benefit of prolonged duration of anticoagulant therapy for some patients with isolated distal DVT, but not all patients should receive this dosing strategy, because some patients may be at high risk for bleeding or VTE recurrence. A subanalysis of data from ONCO-DVT study should shed further light on this.
“We need to individualize our risk stratification,” Dr. Lopez-Fernandez said, adding that notably, “a lot of patients in the 12-month group did not continue with the 12-month treatment,” which may have affected bleeding results. Dr. Yamashita agreed.
Study design and findings
From April 2019 to June 2022, the researchers enrolled and randomly assigned 604 patients with active cancer who had newly diagnosed isolated distal DVT, confirmed by ultrasonography, and were scheduled for DVT treatment with anticoagulation therapy, at 60 centers.
Active cancer was defined as a cancer diagnosis or cancer treatment (surgery, chemotherapy, radiotherapy, etc.) within 6 months of randomization, or current recurrence, local invasion, distant metastases, or hematopoietic malignancy without complete remission.
The most common reasons for ultrasonography were elevated D-dimer levels (62%) and suspected DVT because of symptoms (20%).
The patients had a mean age of 70.8 years and 28% were men. The most common cancer sites were ovaries (14%), uterus (13%), lung (11%), colon (9%), and pancreas (8%), followed by stomach, blood, and breast (each 5%).
The patients were randomly assigned 1:1 to receive 12 months or 3 months of oral edoxaban at a dose of 60 mg once daily or 30 mg once daily in patients with body weight of 60 kg or less, creatinine clearance of 30-50 mL/minute, or concomitant treatment with a potent P-glycoprotein inhibitor.
After excluding 3 patients who withdrew consent, 601 patients were included in the intention-to-treat population: 296 patients in the 12-month edoxaban group and 305 patients in the 3-month edoxaban group.
About 70% of patients had a body weight of 60 kg or less and about 22% had a creatinine clearance less than 50 mL/min. About three quarters received the lower dose of edoxaban.
In the 12-month edoxaban group, 223 patients completed the 1-year follow-up (66 patients had died and 7 were lost to follow-up). In the 3-month edoxaban group, 224 patients completed the 1-year follow-up (77 had died and 4 were lost to follow-up).
In the 12-month edoxaban group, 41% of the patients had discontinued treatment by 12 months. In the 3-month edoxaban group, 41% of patients had discontinued treatment by 3 months.
The primary endpoint – a symptomatic recurrent VTE event or VTE-related death – occurred in 3 of the 222 patients (1.2%) in the 12-month edoxaban group and in 22 of the 210 (8.5%) in the 3-month edoxaban group (odds ratio,0.13; 95% confidence interval, 0.03-0.44, P < .001). There were no VTE-related deaths.
The major secondary endpoint – major bleeding, according to International Society on Thrombosis and Hemostasis criteria – occurred in 28 of the 210 patients (10.2%) in the 12-month edoxaban group and in 22 of the 217 (7.6%) in the 3-month edoxaban group (OR, 1.34; 95% CI, 0.75-2.41, P = NS).
The researchers acknowledged that study limitations include an open-label design, a lower-than-expected primary endpoint rate, and less than high adherence to edoxaban, as well as the need for caution when generalizing the results to other populations.
The study was funded by Daiichi Sankyo. Dr. Yamashita disclosed receiving lecture fees from Bayer Healthcare, Bristol-Myers Squibb, Pfizer, and Daiichi Sankyo, and grant support from Bayer Healthcare and Daiichi Sankyo. Dr. Lopez-Fernandez disclosed receiving speaker fees from Phillips, Janssen, Daiichi Sankyo, Myocardial Solutions, AstraZeneca, Pfizer, Beigene, and Bayer not related to this study.
A version of this article appeared on Medscape.com.
FROM THE ESC CONGRESS 2023
Factor XI inhibitors: The promise of a truly safe anticoagulant?
The quest to find an anticoagulant that can prevent strokes, cardiovascular events, and venous thrombosis without significantly increasing risk of bleeding is something of a holy grail in cardiovascular medicine. Could the latest focus of interest in this field – the factor XI inhibitors – be the long–sought-after answer?
Topline results from the largest study so far of a factor XI inhibitor – released on Sep. 18 – are indeed very encouraging. The phase 2 AZALEA-TIMI 71 study was stopped early because of an “overwhelming” reduction in major and clinically relevant nonmajor bleeding shown with the factor XI inhibitor abelacimab (Anthos), compared with apixaban for patients with atrial fibrillation (AFib).
Very few other data from this study have yet been released. Full results are due to be presented at the scientific sessions of the American Heart Association in November. Researchers in the field are optimistic that this new class of drugs may allow millions more patients who are at risk of thrombotic events but are concerned about bleeding risk to be treated, with a consequent reduction in strokes and possibly cardiovascular events as well.
Why factor XI?
In natural physiology, there are two ongoing processes: hemostasis – a set of actions that cause bleeding to stop after an injury – and thrombosis – a pathologic clotting process in which thrombus is formed and causes a stroke, MI, or deep venous thrombosis (DVT).
In patients prone to pathologic clotting, such as those with AFib, the balance of these two processes has shifted toward thrombosis, so anticoagulants are used to reduce the thrombotic risks. For many years, the only available oral anticoagulant was warfarin, a vitamin K antagonist that was very effective at preventing strokes but that comes with a high risk for bleeding, including intracranial hemorrhage (ICH) and fatal bleeding.
The introduction of the direct-acting anticoagulants (DOACs) a few years ago was a step forward in that these drugs have been shown to be as effective as warfarin but are associated with a lower risk of bleeding, particularly of ICH and fatal bleeding. But they still cause bleeding, and concerns over that risk of bleeding prevent millions of patients from taking these drugs and receiving protection against stroke.
John Alexander, MD, professor of medicine at Duke University Medical Center, Durham, N.C., a researcher active in this area, notes that “while the DOACs cause less bleeding than warfarin, they still cause two or three times more bleeding than placebo, and there is a huge, unmet need for safer anticoagulants that don’t cause as much bleeding. We are hopeful that factor XI inhibitors might be those anticoagulants.”
The lead investigator the AZALEA study, Christian Ruff, MD, professor of medicine at Brigham and Women’s Hospital, Boston, explained why it is thought that factor XI inhibitors may be different.
“There’s a lot of different clotting factors, and most of them converge in a central pathway. The problem, therefore, with anticoagulants used to date that block one of these factors is that they prevent clotting but also cause bleeding.
“It has been discovered that factor XI has a really unique position in the cascade of how our body forms clots in that it seems to be important in clot formation, but it doesn’t seem to play a major role in our ability to heal and repair blood vessels.”
Another doctor involved in the field, Manesh Patel, MD, chief of cardiology at Duke University Medical Center, added, “We think that factor XI inhibitors may prevent the pathologic formation of thrombosis while allowing formation of thrombus for natural hemostasis to prevent bleeding. That is why they are so promising.”
This correlates with epidemiologic data suggesting that patients with a genetic factor XI deficiency have low rates of stroke and MI but don’t appear to bleed spontaneously, Dr. Patel notes.
Candidates in development
The pharmaceutical industry is on the case with several factor XI inhibitors now in clinical development. At present, three main candidates lead the field. These are abelacimab (Anthos), a monoclonal antibody given by subcutaneous injection once a month; and two small molecules, milvexian (BMS/Janssen) and asundexian (Bayer), which are both given orally.
Phase 3 trials of these three factor XI inhibitors have recently started for a variety of thrombotic indications, including the prevention of stroke in patients with AFib, prevention of recurrent stroke in patients with ischemic stroke, and prevention of future cardiovascular events in patients with acute coronary syndrome (ACS).
Dr. Alexander, who has been involved in clinical trials of both milvexian and asundexian, commented: “We have pretty good data from a number of phase 2 trials now that these factor XI inhibitors at the doses used in these studies cause a lot less bleeding than therapeutic doses of DOACs and low-molecular-weight heparins.”
He pointed out that, in addition to the AZALEA trial with abelacimab, the phase 2 PACIFIC program of studies has shown less bleeding with asundexian than with apixaban in patients with AFib and a similar amount of bleeding as placebo in ACS/stroke patients on top of antiplatelet therapy. Milvexian has also shown similar results in the AXIOMATIC program of studies.
Dr. Ruff noted that the biggest need for new anticoagulants in general is in the AFib population. “Atrial fibrillation is one of the most common medical conditions in the world. Approximately one in every three people will develop AFib in their lifetime, and it is associated with more than a fivefold increased risk of stroke. But up to half of patients with AFib currently do not take anticoagulants because of concerns about bleeding risks, so these patients are being left unprotected from stroke risk.”
Dr. Ruff pointed out that the AZALEA study was the largest and longest study of a factor XI inhibitor to date; 1,287 patients were followed for a median of 2 years.
“This was the first trial of long-term administration of factor XI inhibitor against a full-dose DOAC, and it was stopped because of an overwhelming reduction in a major bleeding with abelacimab, compared with rivaroxaban,” he noted. “That is very encouraging. It looks like our quest to develop a safe anticoagulant with much lower rates of bleeding, compared with standard of care, seems to have been borne out. I think the field is very excited that we may finally have something that protects patients from thrombosis whilst being much safer than current agents.”
While all this sounds very promising, for these drugs to be successful, in addition to reducing bleeding risk, they will also have to be effective at preventing strokes and other thrombotic events.
“While we are pretty sure that factor XI inhibitors will cause less bleeding than current anticoagulants, what is unknown still is how effective they will be at preventing pathologic blood clots,” Dr. Alexander points out.
“We have some data from studies of these drugs in DVT prophylaxis after orthopedic surgery which suggest that they are effective in preventing blood clots in that scenario. But we don’t know yet about whether they can prevent pathologic blood clots that occur in AFib patients or in poststroke or post-ACS patients. Phase 3 studies are now underway with these three leading drug candidates which will answer some of these questions.”
Dr. Patel agrees that the efficacy data in the phase 3 trials will be key to the success of these drugs. “That is a very important part of the puzzle that is still missing,” he says.
Dr. Ruff notes that the AZALEA study will provide some data on efficacy. “But we already know that in the orthopedic surgery trials there was a 70%-80% reduction in VTE with abelacimab (at the 150-mg dose going forward) vs. prophylactic doses of low-molecular-weight heparin. And we know from the DOACs that the doses preventing clots on the venous side also translated into preventing strokes on the [AFib] side. So that is very encouraging,” Dr. Ruff adds.
Potential indications
The three leading factor XI inhibitors have slightly different phase 3 development programs.
Dr. Ruff notes that not every agent is being investigated in phase 3 trials for all the potential indications, but all three are going for the AFib indication. “This is by far the biggest population, the biggest market, and the biggest clinical need for these agents,” he says.
While the milvexian and asundexian trials are using an active comparator – pitting the factor XI inhibitors against apixaban in AFib patients – the Anthos LILAC trial is taking a slightly different approach and is comparing abelacimab with placebo in patients with AFib who are not currently taking an anticoagulant because of concerns about bleeding risk.
Janssen/BMS is conducting two other phase 3 trials of milvexian in their LIBREXIA phase 3 program. Those trials involve poststroke patients and ACS patients. Bayer is also involved in a poststroke trial of asundexian as part of its OCEANIC phase 3 program.
Dr. Ruff points out that anticoagulants currently do not have a large role in the poststroke or post-ACS population. “But the hope is that, if factor XI inhibitors are so safe, then there will be more enthusiasm about using an anticoagulant on top of antiplatelet therapy, which is the cornerstone of therapy in atherosclerotic cardiovascular disease.”
In addition to its phase 3 LILAC study in patients with AFib, Anthos is conducting two major phase 3 trials with abelacimab for the treatment of cancer-associated venous thromboembolism.
Dr. Ruff notes that the indication of postsurgery or general prevention of VTE is not being pursued at present.
“The orthopedic surgery studies were done mainly for dose finding and proof of principle reasons,” he explains. “In orthopedic surgery the window for anticoagulation is quite short – a few weeks or months. And for the prevention of recurrent VTE in general in the community, those people are at a relatively low risk of bleeding, so there may not be much advantage of the factor XI inhibitors, whereas AFib patients and those with stroke or ACS are usually older and have a much higher bleeding risk. I think this is where the advantages of an anticoagulant with a lower bleeding risk are most needed.”
Dr. Alexander points out that to date anticoagulants have shown more efficacy in venous clotting, which appears to be more dependent on coagulation factors and less dependent on platelets. “Atrial fibrillation is a mix between venous and arterial clotting, but it has more similarities to venous, so I think AFib is a place where new anticoagulants such as the factor XI inhibitors are more likely to have success,” he suggests.
“So far, anticoagulants have had a less clear long-term role in the poststroke and post-ACS populations, so these indications may be a more difficult goal,” he added.
The phase 3 studies are just starting and will take a few years before results are known.
Differences between the agents
The three factor XI inhibitors also have some differences. Dr. Ruff points out that most important will be the safety and efficacy of the drugs in phase 3 trials.
“Early data suggest that the various agents being developed may not have equal inhibition of factor XI. The monoclonal antibody abelacimab may produce a higher degree of inhibition than the small molecules. But we don’t know if that matters or not – whether we need to achieve a certain threshold to prevent stroke. The efficacy and safety data from the phase 3 trials are what will primarily guide use.”
There are also differences in formulations and dosage. Abelacimab is administered by subcutaneous injection once a month and has a long duration of activity, whereas the small molecules are taken orally and their duration of action is much shorter.
Dr. Ruff notes: “If these drugs cause bleeding, having a long-acting drug like abelacimab could be a disadvantage because we wouldn’t be able to stop it. But if they are very safe with regard to bleeding, then having the drug hang around for a long time is not necessarily a disadvantage, and it may improve compliance. These older patients often miss doses, and with a shorter-acting drug, that will mean they will be unprotected from stroke risk for a period of time, so there is a trade-off here.”
Dr. Ruff says that the AZALEA phase 2 study will provide some data on patients being managed around procedures. “The hope is that these drugs are so safe that they will not have to be stopped for procedures. And then the compliance issue of a once-a-month dosing would be an advantage.”
Dr. Patel says he believes there is a place for different formations. “Some patients may prefer a once-monthly injection; others will prefer a daily tablet. It may come down to patient preference, but a lot will depend on the study results with the different agents,” he commented.
What effect could these drugs have?
If these drugs do show efficacy in these phase 3 trials, what difference will they make to clinical practice? The potential appears to be very large.
“If these drugs are as effective at preventing strokes as DOACs, they will be a huge breakthrough, and there is good reason to think they would replace the DOACs,” Dr. Alexander says. “It would be a really big deal to have an anticoagulant that causes almost no bleeding and could prevent clots as well as the DOACs. This would enable a lot more patients to receive protection against stroke.”
Dr. Alexander believes the surgery studies are hopeful. “They show that the factor XI inhibitors are doing something to prevent blood clots. The big question is whether they are as effective as what we already have for the prevention of stroke and if not, what is the trade-off with bleeding?”
He points out that, even if the factor XI inhibitors are not as effective as DOACs but are found to be much safer, they might still have a potential clinical role, especially for those patients who currently do not take an anticoagulant because of concerns regarding bleeding.
But Dr. Patel points out that there is always the issue of costs with new drugs. “New drugs are always expensive. The DOACS are just about to become generic, and there will inevitably be concerns about access to an expensive new therapy.”
Dr. Alexander adds: “Yes, costs could be an issue, but a safer drug will definitely help to get more patients treated and in preventing more strokes, which would be a great thing.”
Dr. Patel has received grants from and acts as an adviser to Bayer (asundexian) and Janssen (milvexian). Dr. Alexander receives research funding from Bayer. Dr. Ruff receives research funding from Anthos for abelacimab trials, is on an AFib executive committee for BMS/Janssen, and has been on an advisory board for Bayer.
A version of this article first appeared on Medscape.com.
The quest to find an anticoagulant that can prevent strokes, cardiovascular events, and venous thrombosis without significantly increasing risk of bleeding is something of a holy grail in cardiovascular medicine. Could the latest focus of interest in this field – the factor XI inhibitors – be the long–sought-after answer?
Topline results from the largest study so far of a factor XI inhibitor – released on Sep. 18 – are indeed very encouraging. The phase 2 AZALEA-TIMI 71 study was stopped early because of an “overwhelming” reduction in major and clinically relevant nonmajor bleeding shown with the factor XI inhibitor abelacimab (Anthos), compared with apixaban for patients with atrial fibrillation (AFib).
Very few other data from this study have yet been released. Full results are due to be presented at the scientific sessions of the American Heart Association in November. Researchers in the field are optimistic that this new class of drugs may allow millions more patients who are at risk of thrombotic events but are concerned about bleeding risk to be treated, with a consequent reduction in strokes and possibly cardiovascular events as well.
Why factor XI?
In natural physiology, there are two ongoing processes: hemostasis – a set of actions that cause bleeding to stop after an injury – and thrombosis – a pathologic clotting process in which thrombus is formed and causes a stroke, MI, or deep venous thrombosis (DVT).
In patients prone to pathologic clotting, such as those with AFib, the balance of these two processes has shifted toward thrombosis, so anticoagulants are used to reduce the thrombotic risks. For many years, the only available oral anticoagulant was warfarin, a vitamin K antagonist that was very effective at preventing strokes but that comes with a high risk for bleeding, including intracranial hemorrhage (ICH) and fatal bleeding.
The introduction of the direct-acting anticoagulants (DOACs) a few years ago was a step forward in that these drugs have been shown to be as effective as warfarin but are associated with a lower risk of bleeding, particularly of ICH and fatal bleeding. But they still cause bleeding, and concerns over that risk of bleeding prevent millions of patients from taking these drugs and receiving protection against stroke.
John Alexander, MD, professor of medicine at Duke University Medical Center, Durham, N.C., a researcher active in this area, notes that “while the DOACs cause less bleeding than warfarin, they still cause two or three times more bleeding than placebo, and there is a huge, unmet need for safer anticoagulants that don’t cause as much bleeding. We are hopeful that factor XI inhibitors might be those anticoagulants.”
The lead investigator the AZALEA study, Christian Ruff, MD, professor of medicine at Brigham and Women’s Hospital, Boston, explained why it is thought that factor XI inhibitors may be different.
“There’s a lot of different clotting factors, and most of them converge in a central pathway. The problem, therefore, with anticoagulants used to date that block one of these factors is that they prevent clotting but also cause bleeding.
“It has been discovered that factor XI has a really unique position in the cascade of how our body forms clots in that it seems to be important in clot formation, but it doesn’t seem to play a major role in our ability to heal and repair blood vessels.”
Another doctor involved in the field, Manesh Patel, MD, chief of cardiology at Duke University Medical Center, added, “We think that factor XI inhibitors may prevent the pathologic formation of thrombosis while allowing formation of thrombus for natural hemostasis to prevent bleeding. That is why they are so promising.”
This correlates with epidemiologic data suggesting that patients with a genetic factor XI deficiency have low rates of stroke and MI but don’t appear to bleed spontaneously, Dr. Patel notes.
Candidates in development
The pharmaceutical industry is on the case with several factor XI inhibitors now in clinical development. At present, three main candidates lead the field. These are abelacimab (Anthos), a monoclonal antibody given by subcutaneous injection once a month; and two small molecules, milvexian (BMS/Janssen) and asundexian (Bayer), which are both given orally.
Phase 3 trials of these three factor XI inhibitors have recently started for a variety of thrombotic indications, including the prevention of stroke in patients with AFib, prevention of recurrent stroke in patients with ischemic stroke, and prevention of future cardiovascular events in patients with acute coronary syndrome (ACS).
Dr. Alexander, who has been involved in clinical trials of both milvexian and asundexian, commented: “We have pretty good data from a number of phase 2 trials now that these factor XI inhibitors at the doses used in these studies cause a lot less bleeding than therapeutic doses of DOACs and low-molecular-weight heparins.”
He pointed out that, in addition to the AZALEA trial with abelacimab, the phase 2 PACIFIC program of studies has shown less bleeding with asundexian than with apixaban in patients with AFib and a similar amount of bleeding as placebo in ACS/stroke patients on top of antiplatelet therapy. Milvexian has also shown similar results in the AXIOMATIC program of studies.
Dr. Ruff noted that the biggest need for new anticoagulants in general is in the AFib population. “Atrial fibrillation is one of the most common medical conditions in the world. Approximately one in every three people will develop AFib in their lifetime, and it is associated with more than a fivefold increased risk of stroke. But up to half of patients with AFib currently do not take anticoagulants because of concerns about bleeding risks, so these patients are being left unprotected from stroke risk.”
Dr. Ruff pointed out that the AZALEA study was the largest and longest study of a factor XI inhibitor to date; 1,287 patients were followed for a median of 2 years.
“This was the first trial of long-term administration of factor XI inhibitor against a full-dose DOAC, and it was stopped because of an overwhelming reduction in a major bleeding with abelacimab, compared with rivaroxaban,” he noted. “That is very encouraging. It looks like our quest to develop a safe anticoagulant with much lower rates of bleeding, compared with standard of care, seems to have been borne out. I think the field is very excited that we may finally have something that protects patients from thrombosis whilst being much safer than current agents.”
While all this sounds very promising, for these drugs to be successful, in addition to reducing bleeding risk, they will also have to be effective at preventing strokes and other thrombotic events.
“While we are pretty sure that factor XI inhibitors will cause less bleeding than current anticoagulants, what is unknown still is how effective they will be at preventing pathologic blood clots,” Dr. Alexander points out.
“We have some data from studies of these drugs in DVT prophylaxis after orthopedic surgery which suggest that they are effective in preventing blood clots in that scenario. But we don’t know yet about whether they can prevent pathologic blood clots that occur in AFib patients or in poststroke or post-ACS patients. Phase 3 studies are now underway with these three leading drug candidates which will answer some of these questions.”
Dr. Patel agrees that the efficacy data in the phase 3 trials will be key to the success of these drugs. “That is a very important part of the puzzle that is still missing,” he says.
Dr. Ruff notes that the AZALEA study will provide some data on efficacy. “But we already know that in the orthopedic surgery trials there was a 70%-80% reduction in VTE with abelacimab (at the 150-mg dose going forward) vs. prophylactic doses of low-molecular-weight heparin. And we know from the DOACs that the doses preventing clots on the venous side also translated into preventing strokes on the [AFib] side. So that is very encouraging,” Dr. Ruff adds.
Potential indications
The three leading factor XI inhibitors have slightly different phase 3 development programs.
Dr. Ruff notes that not every agent is being investigated in phase 3 trials for all the potential indications, but all three are going for the AFib indication. “This is by far the biggest population, the biggest market, and the biggest clinical need for these agents,” he says.
While the milvexian and asundexian trials are using an active comparator – pitting the factor XI inhibitors against apixaban in AFib patients – the Anthos LILAC trial is taking a slightly different approach and is comparing abelacimab with placebo in patients with AFib who are not currently taking an anticoagulant because of concerns about bleeding risk.
Janssen/BMS is conducting two other phase 3 trials of milvexian in their LIBREXIA phase 3 program. Those trials involve poststroke patients and ACS patients. Bayer is also involved in a poststroke trial of asundexian as part of its OCEANIC phase 3 program.
Dr. Ruff points out that anticoagulants currently do not have a large role in the poststroke or post-ACS population. “But the hope is that, if factor XI inhibitors are so safe, then there will be more enthusiasm about using an anticoagulant on top of antiplatelet therapy, which is the cornerstone of therapy in atherosclerotic cardiovascular disease.”
In addition to its phase 3 LILAC study in patients with AFib, Anthos is conducting two major phase 3 trials with abelacimab for the treatment of cancer-associated venous thromboembolism.
Dr. Ruff notes that the indication of postsurgery or general prevention of VTE is not being pursued at present.
“The orthopedic surgery studies were done mainly for dose finding and proof of principle reasons,” he explains. “In orthopedic surgery the window for anticoagulation is quite short – a few weeks or months. And for the prevention of recurrent VTE in general in the community, those people are at a relatively low risk of bleeding, so there may not be much advantage of the factor XI inhibitors, whereas AFib patients and those with stroke or ACS are usually older and have a much higher bleeding risk. I think this is where the advantages of an anticoagulant with a lower bleeding risk are most needed.”
Dr. Alexander points out that to date anticoagulants have shown more efficacy in venous clotting, which appears to be more dependent on coagulation factors and less dependent on platelets. “Atrial fibrillation is a mix between venous and arterial clotting, but it has more similarities to venous, so I think AFib is a place where new anticoagulants such as the factor XI inhibitors are more likely to have success,” he suggests.
“So far, anticoagulants have had a less clear long-term role in the poststroke and post-ACS populations, so these indications may be a more difficult goal,” he added.
The phase 3 studies are just starting and will take a few years before results are known.
Differences between the agents
The three factor XI inhibitors also have some differences. Dr. Ruff points out that most important will be the safety and efficacy of the drugs in phase 3 trials.
“Early data suggest that the various agents being developed may not have equal inhibition of factor XI. The monoclonal antibody abelacimab may produce a higher degree of inhibition than the small molecules. But we don’t know if that matters or not – whether we need to achieve a certain threshold to prevent stroke. The efficacy and safety data from the phase 3 trials are what will primarily guide use.”
There are also differences in formulations and dosage. Abelacimab is administered by subcutaneous injection once a month and has a long duration of activity, whereas the small molecules are taken orally and their duration of action is much shorter.
Dr. Ruff notes: “If these drugs cause bleeding, having a long-acting drug like abelacimab could be a disadvantage because we wouldn’t be able to stop it. But if they are very safe with regard to bleeding, then having the drug hang around for a long time is not necessarily a disadvantage, and it may improve compliance. These older patients often miss doses, and with a shorter-acting drug, that will mean they will be unprotected from stroke risk for a period of time, so there is a trade-off here.”
Dr. Ruff says that the AZALEA phase 2 study will provide some data on patients being managed around procedures. “The hope is that these drugs are so safe that they will not have to be stopped for procedures. And then the compliance issue of a once-a-month dosing would be an advantage.”
Dr. Patel says he believes there is a place for different formations. “Some patients may prefer a once-monthly injection; others will prefer a daily tablet. It may come down to patient preference, but a lot will depend on the study results with the different agents,” he commented.
What effect could these drugs have?
If these drugs do show efficacy in these phase 3 trials, what difference will they make to clinical practice? The potential appears to be very large.
“If these drugs are as effective at preventing strokes as DOACs, they will be a huge breakthrough, and there is good reason to think they would replace the DOACs,” Dr. Alexander says. “It would be a really big deal to have an anticoagulant that causes almost no bleeding and could prevent clots as well as the DOACs. This would enable a lot more patients to receive protection against stroke.”
Dr. Alexander believes the surgery studies are hopeful. “They show that the factor XI inhibitors are doing something to prevent blood clots. The big question is whether they are as effective as what we already have for the prevention of stroke and if not, what is the trade-off with bleeding?”
He points out that, even if the factor XI inhibitors are not as effective as DOACs but are found to be much safer, they might still have a potential clinical role, especially for those patients who currently do not take an anticoagulant because of concerns regarding bleeding.
But Dr. Patel points out that there is always the issue of costs with new drugs. “New drugs are always expensive. The DOACS are just about to become generic, and there will inevitably be concerns about access to an expensive new therapy.”
Dr. Alexander adds: “Yes, costs could be an issue, but a safer drug will definitely help to get more patients treated and in preventing more strokes, which would be a great thing.”
Dr. Patel has received grants from and acts as an adviser to Bayer (asundexian) and Janssen (milvexian). Dr. Alexander receives research funding from Bayer. Dr. Ruff receives research funding from Anthos for abelacimab trials, is on an AFib executive committee for BMS/Janssen, and has been on an advisory board for Bayer.
A version of this article first appeared on Medscape.com.
The quest to find an anticoagulant that can prevent strokes, cardiovascular events, and venous thrombosis without significantly increasing risk of bleeding is something of a holy grail in cardiovascular medicine. Could the latest focus of interest in this field – the factor XI inhibitors – be the long–sought-after answer?
Topline results from the largest study so far of a factor XI inhibitor – released on Sep. 18 – are indeed very encouraging. The phase 2 AZALEA-TIMI 71 study was stopped early because of an “overwhelming” reduction in major and clinically relevant nonmajor bleeding shown with the factor XI inhibitor abelacimab (Anthos), compared with apixaban for patients with atrial fibrillation (AFib).
Very few other data from this study have yet been released. Full results are due to be presented at the scientific sessions of the American Heart Association in November. Researchers in the field are optimistic that this new class of drugs may allow millions more patients who are at risk of thrombotic events but are concerned about bleeding risk to be treated, with a consequent reduction in strokes and possibly cardiovascular events as well.
Why factor XI?
In natural physiology, there are two ongoing processes: hemostasis – a set of actions that cause bleeding to stop after an injury – and thrombosis – a pathologic clotting process in which thrombus is formed and causes a stroke, MI, or deep venous thrombosis (DVT).
In patients prone to pathologic clotting, such as those with AFib, the balance of these two processes has shifted toward thrombosis, so anticoagulants are used to reduce the thrombotic risks. For many years, the only available oral anticoagulant was warfarin, a vitamin K antagonist that was very effective at preventing strokes but that comes with a high risk for bleeding, including intracranial hemorrhage (ICH) and fatal bleeding.
The introduction of the direct-acting anticoagulants (DOACs) a few years ago was a step forward in that these drugs have been shown to be as effective as warfarin but are associated with a lower risk of bleeding, particularly of ICH and fatal bleeding. But they still cause bleeding, and concerns over that risk of bleeding prevent millions of patients from taking these drugs and receiving protection against stroke.
John Alexander, MD, professor of medicine at Duke University Medical Center, Durham, N.C., a researcher active in this area, notes that “while the DOACs cause less bleeding than warfarin, they still cause two or three times more bleeding than placebo, and there is a huge, unmet need for safer anticoagulants that don’t cause as much bleeding. We are hopeful that factor XI inhibitors might be those anticoagulants.”
The lead investigator the AZALEA study, Christian Ruff, MD, professor of medicine at Brigham and Women’s Hospital, Boston, explained why it is thought that factor XI inhibitors may be different.
“There’s a lot of different clotting factors, and most of them converge in a central pathway. The problem, therefore, with anticoagulants used to date that block one of these factors is that they prevent clotting but also cause bleeding.
“It has been discovered that factor XI has a really unique position in the cascade of how our body forms clots in that it seems to be important in clot formation, but it doesn’t seem to play a major role in our ability to heal and repair blood vessels.”
Another doctor involved in the field, Manesh Patel, MD, chief of cardiology at Duke University Medical Center, added, “We think that factor XI inhibitors may prevent the pathologic formation of thrombosis while allowing formation of thrombus for natural hemostasis to prevent bleeding. That is why they are so promising.”
This correlates with epidemiologic data suggesting that patients with a genetic factor XI deficiency have low rates of stroke and MI but don’t appear to bleed spontaneously, Dr. Patel notes.
Candidates in development
The pharmaceutical industry is on the case with several factor XI inhibitors now in clinical development. At present, three main candidates lead the field. These are abelacimab (Anthos), a monoclonal antibody given by subcutaneous injection once a month; and two small molecules, milvexian (BMS/Janssen) and asundexian (Bayer), which are both given orally.
Phase 3 trials of these three factor XI inhibitors have recently started for a variety of thrombotic indications, including the prevention of stroke in patients with AFib, prevention of recurrent stroke in patients with ischemic stroke, and prevention of future cardiovascular events in patients with acute coronary syndrome (ACS).
Dr. Alexander, who has been involved in clinical trials of both milvexian and asundexian, commented: “We have pretty good data from a number of phase 2 trials now that these factor XI inhibitors at the doses used in these studies cause a lot less bleeding than therapeutic doses of DOACs and low-molecular-weight heparins.”
He pointed out that, in addition to the AZALEA trial with abelacimab, the phase 2 PACIFIC program of studies has shown less bleeding with asundexian than with apixaban in patients with AFib and a similar amount of bleeding as placebo in ACS/stroke patients on top of antiplatelet therapy. Milvexian has also shown similar results in the AXIOMATIC program of studies.
Dr. Ruff noted that the biggest need for new anticoagulants in general is in the AFib population. “Atrial fibrillation is one of the most common medical conditions in the world. Approximately one in every three people will develop AFib in their lifetime, and it is associated with more than a fivefold increased risk of stroke. But up to half of patients with AFib currently do not take anticoagulants because of concerns about bleeding risks, so these patients are being left unprotected from stroke risk.”
Dr. Ruff pointed out that the AZALEA study was the largest and longest study of a factor XI inhibitor to date; 1,287 patients were followed for a median of 2 years.
“This was the first trial of long-term administration of factor XI inhibitor against a full-dose DOAC, and it was stopped because of an overwhelming reduction in a major bleeding with abelacimab, compared with rivaroxaban,” he noted. “That is very encouraging. It looks like our quest to develop a safe anticoagulant with much lower rates of bleeding, compared with standard of care, seems to have been borne out. I think the field is very excited that we may finally have something that protects patients from thrombosis whilst being much safer than current agents.”
While all this sounds very promising, for these drugs to be successful, in addition to reducing bleeding risk, they will also have to be effective at preventing strokes and other thrombotic events.
“While we are pretty sure that factor XI inhibitors will cause less bleeding than current anticoagulants, what is unknown still is how effective they will be at preventing pathologic blood clots,” Dr. Alexander points out.
“We have some data from studies of these drugs in DVT prophylaxis after orthopedic surgery which suggest that they are effective in preventing blood clots in that scenario. But we don’t know yet about whether they can prevent pathologic blood clots that occur in AFib patients or in poststroke or post-ACS patients. Phase 3 studies are now underway with these three leading drug candidates which will answer some of these questions.”
Dr. Patel agrees that the efficacy data in the phase 3 trials will be key to the success of these drugs. “That is a very important part of the puzzle that is still missing,” he says.
Dr. Ruff notes that the AZALEA study will provide some data on efficacy. “But we already know that in the orthopedic surgery trials there was a 70%-80% reduction in VTE with abelacimab (at the 150-mg dose going forward) vs. prophylactic doses of low-molecular-weight heparin. And we know from the DOACs that the doses preventing clots on the venous side also translated into preventing strokes on the [AFib] side. So that is very encouraging,” Dr. Ruff adds.
Potential indications
The three leading factor XI inhibitors have slightly different phase 3 development programs.
Dr. Ruff notes that not every agent is being investigated in phase 3 trials for all the potential indications, but all three are going for the AFib indication. “This is by far the biggest population, the biggest market, and the biggest clinical need for these agents,” he says.
While the milvexian and asundexian trials are using an active comparator – pitting the factor XI inhibitors against apixaban in AFib patients – the Anthos LILAC trial is taking a slightly different approach and is comparing abelacimab with placebo in patients with AFib who are not currently taking an anticoagulant because of concerns about bleeding risk.
Janssen/BMS is conducting two other phase 3 trials of milvexian in their LIBREXIA phase 3 program. Those trials involve poststroke patients and ACS patients. Bayer is also involved in a poststroke trial of asundexian as part of its OCEANIC phase 3 program.
Dr. Ruff points out that anticoagulants currently do not have a large role in the poststroke or post-ACS population. “But the hope is that, if factor XI inhibitors are so safe, then there will be more enthusiasm about using an anticoagulant on top of antiplatelet therapy, which is the cornerstone of therapy in atherosclerotic cardiovascular disease.”
In addition to its phase 3 LILAC study in patients with AFib, Anthos is conducting two major phase 3 trials with abelacimab for the treatment of cancer-associated venous thromboembolism.
Dr. Ruff notes that the indication of postsurgery or general prevention of VTE is not being pursued at present.
“The orthopedic surgery studies were done mainly for dose finding and proof of principle reasons,” he explains. “In orthopedic surgery the window for anticoagulation is quite short – a few weeks or months. And for the prevention of recurrent VTE in general in the community, those people are at a relatively low risk of bleeding, so there may not be much advantage of the factor XI inhibitors, whereas AFib patients and those with stroke or ACS are usually older and have a much higher bleeding risk. I think this is where the advantages of an anticoagulant with a lower bleeding risk are most needed.”
Dr. Alexander points out that to date anticoagulants have shown more efficacy in venous clotting, which appears to be more dependent on coagulation factors and less dependent on platelets. “Atrial fibrillation is a mix between venous and arterial clotting, but it has more similarities to venous, so I think AFib is a place where new anticoagulants such as the factor XI inhibitors are more likely to have success,” he suggests.
“So far, anticoagulants have had a less clear long-term role in the poststroke and post-ACS populations, so these indications may be a more difficult goal,” he added.
The phase 3 studies are just starting and will take a few years before results are known.
Differences between the agents
The three factor XI inhibitors also have some differences. Dr. Ruff points out that most important will be the safety and efficacy of the drugs in phase 3 trials.
“Early data suggest that the various agents being developed may not have equal inhibition of factor XI. The monoclonal antibody abelacimab may produce a higher degree of inhibition than the small molecules. But we don’t know if that matters or not – whether we need to achieve a certain threshold to prevent stroke. The efficacy and safety data from the phase 3 trials are what will primarily guide use.”
There are also differences in formulations and dosage. Abelacimab is administered by subcutaneous injection once a month and has a long duration of activity, whereas the small molecules are taken orally and their duration of action is much shorter.
Dr. Ruff notes: “If these drugs cause bleeding, having a long-acting drug like abelacimab could be a disadvantage because we wouldn’t be able to stop it. But if they are very safe with regard to bleeding, then having the drug hang around for a long time is not necessarily a disadvantage, and it may improve compliance. These older patients often miss doses, and with a shorter-acting drug, that will mean they will be unprotected from stroke risk for a period of time, so there is a trade-off here.”
Dr. Ruff says that the AZALEA phase 2 study will provide some data on patients being managed around procedures. “The hope is that these drugs are so safe that they will not have to be stopped for procedures. And then the compliance issue of a once-a-month dosing would be an advantage.”
Dr. Patel says he believes there is a place for different formations. “Some patients may prefer a once-monthly injection; others will prefer a daily tablet. It may come down to patient preference, but a lot will depend on the study results with the different agents,” he commented.
What effect could these drugs have?
If these drugs do show efficacy in these phase 3 trials, what difference will they make to clinical practice? The potential appears to be very large.
“If these drugs are as effective at preventing strokes as DOACs, they will be a huge breakthrough, and there is good reason to think they would replace the DOACs,” Dr. Alexander says. “It would be a really big deal to have an anticoagulant that causes almost no bleeding and could prevent clots as well as the DOACs. This would enable a lot more patients to receive protection against stroke.”
Dr. Alexander believes the surgery studies are hopeful. “They show that the factor XI inhibitors are doing something to prevent blood clots. The big question is whether they are as effective as what we already have for the prevention of stroke and if not, what is the trade-off with bleeding?”
He points out that, even if the factor XI inhibitors are not as effective as DOACs but are found to be much safer, they might still have a potential clinical role, especially for those patients who currently do not take an anticoagulant because of concerns regarding bleeding.
But Dr. Patel points out that there is always the issue of costs with new drugs. “New drugs are always expensive. The DOACS are just about to become generic, and there will inevitably be concerns about access to an expensive new therapy.”
Dr. Alexander adds: “Yes, costs could be an issue, but a safer drug will definitely help to get more patients treated and in preventing more strokes, which would be a great thing.”
Dr. Patel has received grants from and acts as an adviser to Bayer (asundexian) and Janssen (milvexian). Dr. Alexander receives research funding from Bayer. Dr. Ruff receives research funding from Anthos for abelacimab trials, is on an AFib executive committee for BMS/Janssen, and has been on an advisory board for Bayer.
A version of this article first appeared on Medscape.com.
Nationwide hematologists shortage: What’s being done?
Over decades, the shrinking pool of CHs – who are compensated far less than hematologist-oncologists – has put patients at risk without access to adequate and timely care. To alleviate this crisis, individual doctors and national organizations are taking action and making more resources available to CHs and their patients.
`Vicious cycle’
The root cause of the CH shortage can be traced to a dramatic reduction in the number of physicians trained in this field, as Leonard Valentino, MD, President of the National Bleeding Disorders Foundation in New York, explained in an interview.
“There is a vicious cycle where there’s not enough classical hematologists to be program directors, and therefore trainees are often steered to fellowships in oncology,” said Dr. Valentino.
According to data published in JAMA, in 1995 there were 74 classical hematology programs in the United States; by 2018, there were only 2, During this same time period, the number of combined hematology/oncology training programs (HOPs) nearly doubled, from 75 to 146. However, it is estimated that less than 5% of graduates of adult HOPs pursued a career in classical hematology, as reported in Blood Advances. This low percentage can be attributed, at least in part, to the emphasis that most HOPs place on oncology.
Dr. Valentino noted that financial pressures are also diverting medical students from becoming CHs, adding that a hematologist-oncologist can make three times the annual salary of a CH.
Furthermore, when CHs treat bleeding and clotting disorders, they often need to meet with a patient for a 60- to 90-minute initial consultation, then they go on to provide a lifetime of labor-intensive care.
“This work is neither verticalized [that is, supported by radiologists, surgeons, and a cadre of nurses], nor is it billable per hour on a scale comparable to what oncologists can charge,” Dr. Valentino explained.
The survey published in Blood Advances illustrates the consequences of such a disparity in income potential: 34% of hematology/oncology fellows surveyed were likely to enter solid tumor oncology, while 20% and 4.6% would proceed to malignant hematology and CH, respectively.
Toll on patients
Primary care doctors treat some common blood disorders, but they almost always refer more difficult or complicated cases to a shrinking population of CHs.

“For many Americans, it is getting more difficult to find providers who subspecialize in hemostasis and thrombosis disorders. Patients can expect prolonged waiting times to get evaluated after a referral” said Mukul Singal, MD, of the Indiana Hemophilia and Thrombosis Center in Indianapolis.
Dr. Singal said the shortage is so acute that “at many institutions, malignant hematologists or oncologists are having to staff in-patient hematology consult services and see outpatient classical hematology patients. General hematologist/oncologists or medical oncologists are often not as comfortable or experienced with dealing with some of the complex CH conditions.”
A working care model, without enough doctors
In 1975, responding to patient advocacy groups, the federal government began funding hemophilia treatment centers (HTCs). Such centers offer a comprehensive care model that gives patients access to practitioners and administrative staff with the expertise to help them stay as healthy as possible. According to the Centers for Disease Control and Prevention, people with hemophilia who used an HTC were 40% less likely to die of a hemophilia-related complication and 40% less likely to be hospitalized for bleeding complications, compared to those who did not receive such specialized care.
“HTCs are effective at keeping patients out of the hospital and engaged in their lives. Between 80% and 95% of hemophilia patients get their care from an HTC and more patients want more services from them,” said Joe Pugliese, president of the Hemophilia Alliance in Lansdale, Pa.
Expanding care to meet patient demand is challenged by the restrictions on doctors’ salaries. All 140 U.S.-based HTCs share a $4.9 million federal grant but, by law, they can’t pay any provider more than $211,000 a year. “These restrictions push many people to industry, leaving too few doctors to meet patient demand,” Mr. Pugliese explained.
The fact that most HTCs are located in or near major cities also presents patients with the challenge of commuting, sometimes across state lines, to see a specialist. However, an uptick in telemedicine has provided one bright spot for many patients, allowing care to be brought to them.
The Hemophilia Alliance is also working on a multifaceted approach to change the rules, so that CHs are offered better compensation. “We have lobbyists in Washington, as well as an advocacy committee and a payer committee working to better support the HTC model,” Mr. Pugliese said.
Beyond the paycheck: Supporting CHs and patients
As market and regulatory restrictions make it difficult to boost the pay of CHs, doctors and nonprofit organizations are collaborating to support young CHs and bring more into the field. The American Society of Hematology has started and fully funded the Hematology Focused Fellowship Training Program (HFFTP). This program pairs comprehensive classical hematology training with education in transfusion medicine, sickle cell disease, hemostasis/thrombosis, systems-based hematology, health equity research, and global health. According to the program’s website, HFFTP’s goal is to add 50 new academic hematologists nationwide by 2030, in an effort to “improve the lives of patients with blood and bone marrow disorders.”
Additionally, classic hematologists are aiming to attract younger physicians and trainees to their field by introducing them to the various rewarding aspects of dealing with patients with inherited, chronic blood diseases. Programs like the Partners Physicians Academy (PPA), a 5-day training course that is specifically designed to encourage and retain young hematology students as classical hematologists, are essential to this effort.
“Along with preparing physicians to work in an HTC, programs like the Hematology Focused Fellowship Training Program and the Partners Physicians Academy are so important because they might convince young doctors to stick with non–oncology-based hematology careers, through the right mix of knowing about exciting research like gene therapy, financial and mentorship support, and a desire to meet unmet medical need,” explained Dr. Valentino.
The next PPA is taking place Sept. 18-22 in Indianapolis.
Dr. Singal, Dr. Valentino, and Mr. Pugliese had no financial disclosures to report.
Over decades, the shrinking pool of CHs – who are compensated far less than hematologist-oncologists – has put patients at risk without access to adequate and timely care. To alleviate this crisis, individual doctors and national organizations are taking action and making more resources available to CHs and their patients.
`Vicious cycle’
The root cause of the CH shortage can be traced to a dramatic reduction in the number of physicians trained in this field, as Leonard Valentino, MD, President of the National Bleeding Disorders Foundation in New York, explained in an interview.
“There is a vicious cycle where there’s not enough classical hematologists to be program directors, and therefore trainees are often steered to fellowships in oncology,” said Dr. Valentino.
According to data published in JAMA, in 1995 there were 74 classical hematology programs in the United States; by 2018, there were only 2, During this same time period, the number of combined hematology/oncology training programs (HOPs) nearly doubled, from 75 to 146. However, it is estimated that less than 5% of graduates of adult HOPs pursued a career in classical hematology, as reported in Blood Advances. This low percentage can be attributed, at least in part, to the emphasis that most HOPs place on oncology.
Dr. Valentino noted that financial pressures are also diverting medical students from becoming CHs, adding that a hematologist-oncologist can make three times the annual salary of a CH.
Furthermore, when CHs treat bleeding and clotting disorders, they often need to meet with a patient for a 60- to 90-minute initial consultation, then they go on to provide a lifetime of labor-intensive care.
“This work is neither verticalized [that is, supported by radiologists, surgeons, and a cadre of nurses], nor is it billable per hour on a scale comparable to what oncologists can charge,” Dr. Valentino explained.
The survey published in Blood Advances illustrates the consequences of such a disparity in income potential: 34% of hematology/oncology fellows surveyed were likely to enter solid tumor oncology, while 20% and 4.6% would proceed to malignant hematology and CH, respectively.
Toll on patients
Primary care doctors treat some common blood disorders, but they almost always refer more difficult or complicated cases to a shrinking population of CHs.
“For many Americans, it is getting more difficult to find providers who subspecialize in hemostasis and thrombosis disorders. Patients can expect prolonged waiting times to get evaluated after a referral” said Mukul Singal, MD, of the Indiana Hemophilia and Thrombosis Center in Indianapolis.
Dr. Singal said the shortage is so acute that “at many institutions, malignant hematologists or oncologists are having to staff in-patient hematology consult services and see outpatient classical hematology patients. General hematologist/oncologists or medical oncologists are often not as comfortable or experienced with dealing with some of the complex CH conditions.”
A working care model, without enough doctors
In 1975, responding to patient advocacy groups, the federal government began funding hemophilia treatment centers (HTCs). Such centers offer a comprehensive care model that gives patients access to practitioners and administrative staff with the expertise to help them stay as healthy as possible. According to the Centers for Disease Control and Prevention, people with hemophilia who used an HTC were 40% less likely to die of a hemophilia-related complication and 40% less likely to be hospitalized for bleeding complications, compared to those who did not receive such specialized care.
“HTCs are effective at keeping patients out of the hospital and engaged in their lives. Between 80% and 95% of hemophilia patients get their care from an HTC and more patients want more services from them,” said Joe Pugliese, president of the Hemophilia Alliance in Lansdale, Pa.
Expanding care to meet patient demand is challenged by the restrictions on doctors’ salaries. All 140 U.S.-based HTCs share a $4.9 million federal grant but, by law, they can’t pay any provider more than $211,000 a year. “These restrictions push many people to industry, leaving too few doctors to meet patient demand,” Mr. Pugliese explained.
The fact that most HTCs are located in or near major cities also presents patients with the challenge of commuting, sometimes across state lines, to see a specialist. However, an uptick in telemedicine has provided one bright spot for many patients, allowing care to be brought to them.
The Hemophilia Alliance is also working on a multifaceted approach to change the rules, so that CHs are offered better compensation. “We have lobbyists in Washington, as well as an advocacy committee and a payer committee working to better support the HTC model,” Mr. Pugliese said.
Beyond the paycheck: Supporting CHs and patients
As market and regulatory restrictions make it difficult to boost the pay of CHs, doctors and nonprofit organizations are collaborating to support young CHs and bring more into the field. The American Society of Hematology has started and fully funded the Hematology Focused Fellowship Training Program (HFFTP). This program pairs comprehensive classical hematology training with education in transfusion medicine, sickle cell disease, hemostasis/thrombosis, systems-based hematology, health equity research, and global health. According to the program’s website, HFFTP’s goal is to add 50 new academic hematologists nationwide by 2030, in an effort to “improve the lives of patients with blood and bone marrow disorders.”
Additionally, classic hematologists are aiming to attract younger physicians and trainees to their field by introducing them to the various rewarding aspects of dealing with patients with inherited, chronic blood diseases. Programs like the Partners Physicians Academy (PPA), a 5-day training course that is specifically designed to encourage and retain young hematology students as classical hematologists, are essential to this effort.
“Along with preparing physicians to work in an HTC, programs like the Hematology Focused Fellowship Training Program and the Partners Physicians Academy are so important because they might convince young doctors to stick with non–oncology-based hematology careers, through the right mix of knowing about exciting research like gene therapy, financial and mentorship support, and a desire to meet unmet medical need,” explained Dr. Valentino.
The next PPA is taking place Sept. 18-22 in Indianapolis.
Dr. Singal, Dr. Valentino, and Mr. Pugliese had no financial disclosures to report.
Over decades, the shrinking pool of CHs – who are compensated far less than hematologist-oncologists – has put patients at risk without access to adequate and timely care. To alleviate this crisis, individual doctors and national organizations are taking action and making more resources available to CHs and their patients.
`Vicious cycle’
The root cause of the CH shortage can be traced to a dramatic reduction in the number of physicians trained in this field, as Leonard Valentino, MD, President of the National Bleeding Disorders Foundation in New York, explained in an interview.
“There is a vicious cycle where there’s not enough classical hematologists to be program directors, and therefore trainees are often steered to fellowships in oncology,” said Dr. Valentino.
According to data published in JAMA, in 1995 there were 74 classical hematology programs in the United States; by 2018, there were only 2, During this same time period, the number of combined hematology/oncology training programs (HOPs) nearly doubled, from 75 to 146. However, it is estimated that less than 5% of graduates of adult HOPs pursued a career in classical hematology, as reported in Blood Advances. This low percentage can be attributed, at least in part, to the emphasis that most HOPs place on oncology.
Dr. Valentino noted that financial pressures are also diverting medical students from becoming CHs, adding that a hematologist-oncologist can make three times the annual salary of a CH.
Furthermore, when CHs treat bleeding and clotting disorders, they often need to meet with a patient for a 60- to 90-minute initial consultation, then they go on to provide a lifetime of labor-intensive care.
“This work is neither verticalized [that is, supported by radiologists, surgeons, and a cadre of nurses], nor is it billable per hour on a scale comparable to what oncologists can charge,” Dr. Valentino explained.
The survey published in Blood Advances illustrates the consequences of such a disparity in income potential: 34% of hematology/oncology fellows surveyed were likely to enter solid tumor oncology, while 20% and 4.6% would proceed to malignant hematology and CH, respectively.
Toll on patients
Primary care doctors treat some common blood disorders, but they almost always refer more difficult or complicated cases to a shrinking population of CHs.
“For many Americans, it is getting more difficult to find providers who subspecialize in hemostasis and thrombosis disorders. Patients can expect prolonged waiting times to get evaluated after a referral” said Mukul Singal, MD, of the Indiana Hemophilia and Thrombosis Center in Indianapolis.
Dr. Singal said the shortage is so acute that “at many institutions, malignant hematologists or oncologists are having to staff in-patient hematology consult services and see outpatient classical hematology patients. General hematologist/oncologists or medical oncologists are often not as comfortable or experienced with dealing with some of the complex CH conditions.”
A working care model, without enough doctors
In 1975, responding to patient advocacy groups, the federal government began funding hemophilia treatment centers (HTCs). Such centers offer a comprehensive care model that gives patients access to practitioners and administrative staff with the expertise to help them stay as healthy as possible. According to the Centers for Disease Control and Prevention, people with hemophilia who used an HTC were 40% less likely to die of a hemophilia-related complication and 40% less likely to be hospitalized for bleeding complications, compared to those who did not receive such specialized care.
“HTCs are effective at keeping patients out of the hospital and engaged in their lives. Between 80% and 95% of hemophilia patients get their care from an HTC and more patients want more services from them,” said Joe Pugliese, president of the Hemophilia Alliance in Lansdale, Pa.
Expanding care to meet patient demand is challenged by the restrictions on doctors’ salaries. All 140 U.S.-based HTCs share a $4.9 million federal grant but, by law, they can’t pay any provider more than $211,000 a year. “These restrictions push many people to industry, leaving too few doctors to meet patient demand,” Mr. Pugliese explained.
The fact that most HTCs are located in or near major cities also presents patients with the challenge of commuting, sometimes across state lines, to see a specialist. However, an uptick in telemedicine has provided one bright spot for many patients, allowing care to be brought to them.
The Hemophilia Alliance is also working on a multifaceted approach to change the rules, so that CHs are offered better compensation. “We have lobbyists in Washington, as well as an advocacy committee and a payer committee working to better support the HTC model,” Mr. Pugliese said.
Beyond the paycheck: Supporting CHs and patients
As market and regulatory restrictions make it difficult to boost the pay of CHs, doctors and nonprofit organizations are collaborating to support young CHs and bring more into the field. The American Society of Hematology has started and fully funded the Hematology Focused Fellowship Training Program (HFFTP). This program pairs comprehensive classical hematology training with education in transfusion medicine, sickle cell disease, hemostasis/thrombosis, systems-based hematology, health equity research, and global health. According to the program’s website, HFFTP’s goal is to add 50 new academic hematologists nationwide by 2030, in an effort to “improve the lives of patients with blood and bone marrow disorders.”
Additionally, classic hematologists are aiming to attract younger physicians and trainees to their field by introducing them to the various rewarding aspects of dealing with patients with inherited, chronic blood diseases. Programs like the Partners Physicians Academy (PPA), a 5-day training course that is specifically designed to encourage and retain young hematology students as classical hematologists, are essential to this effort.
“Along with preparing physicians to work in an HTC, programs like the Hematology Focused Fellowship Training Program and the Partners Physicians Academy are so important because they might convince young doctors to stick with non–oncology-based hematology careers, through the right mix of knowing about exciting research like gene therapy, financial and mentorship support, and a desire to meet unmet medical need,” explained Dr. Valentino.
The next PPA is taking place Sept. 18-22 in Indianapolis.
Dr. Singal, Dr. Valentino, and Mr. Pugliese had no financial disclosures to report.
Steady VKA therapy beats switch to NOAC in frail AFib patients: FRAIL-AF
Switching frail patients with atrial fibrillation (AFib) from anticoagulation therapy with vitamin K antagonists (VKAs) to a novel oral anticoagulant (NOAC) resulted in more bleeding without any reduction in thromboembolic complications or all-cause mortality, randomized trial results show.
The study, FRAIL-AF, is the first randomized NOAC trial to exclusively include frail older patients, said lead author Linda P.T. Joosten, MD, Julius Center for Health Sciences and Primary Care in Utrecht, the Netherlands, and these unexpected findings provide evidence that goes beyond what is currently available.
“Data from the FRAIL-AF trial showed that switching from a VKA to a NOAC should not be considered without a clear indication in frail older patients with AF[ib], as switching to a NOAC leads to 69% more bleeding,” she concluded, without any benefit on secondary clinical endpoints, including thromboembolic events and all-cause mortality.
“The results turned out different than we expected,” Dr. Joosten said. “The hypothesis of this superiority trial was that switching from VKA therapy to a NOAC would result in less bleeding. However, we observed the opposite. After the interim analysis, the data and safety monitoring board advised to stop inclusion because switching from a VKA to a NOAC was clearly contraindicated with a hazard ratio of 1.69 and a highly significant P value of .001.”
Results of FRAIL-AF were presented at the annual congress of the European Society of Cardiology and published online in the journal Circulation.
Session moderator Renate B. Schnabel, MD, interventional cardiologist with University Heart & Vascular Center Hamburg (Germany), congratulated the researchers on these “astonishing” data.
“The thing I want to emphasize here is that, in the absence of randomized controlled trial data, we should be very cautious in extrapolating data from the landmark trials to populations not enrolled in those, and to rely on observational data only,” Dr. Schnabel told Dr. Joosten. “We need randomized controlled trials that sometimes give astonishing results.”
Frailty a clinical syndrome
Frailty is “a lot more than just aging, multiple comorbidities and polypharmacy,” Dr. Joosten explained. “It’s really a clinical syndrome, with people with a high biological vulnerability, dependency on significant others, and a reduced capacity to resist stressors, all leading to a reduced homeostatic reserve.”
Frailty is common in the community, with a prevalence of about 12%, she noted, “and even more important, AF[ib] in frail older people is very common, with a prevalence of 18%. And “without any doubt, we have to adequately anticoagulate frail AF[ib] patients, as they have a high stroke risk, with an incidence of 12.4% per year,” Dr. Joosten noted, compared with 3.9% per year among nonfrail AFib patients.
NOACs are preferred over VKAs in nonfrail AFib patients, after four major trials, RE-LY with dabigatran, ROCKET-AF with rivaroxaban, ARISTOTLE with apixaban, and ENGAGE-AF with edoxaban, showed that NOAC treatment resulted in less major bleeding while stroke risk was comparable with treatment with warfarin, she noted.
The 2023 European Heart Rhythm Association consensus document on management of arrhythmias in frailty syndrome concludes that the advantages of NOACs relative to VKAs are “likely consistent” in frail and nonfrail AFib patients, but the level of evidence is low.
So it’s unknown if NOACs are preferred over VKAs in frail AFib patients, “and it’s even more questionable whether patients on VKAs should switch to NOAC therapy,” Dr. Joosten said.
This new trial aimed to answer the question of whether switching frail AFib patients currently managed on a VKA to a NOAC would reduce bleeding. FRAIL-AF was a pragmatic, multicenter, open-label, randomized, controlled superiority trial.
Older AFib patients were deemed frail if they were aged 75 years or older and had a score of 3 or more on the validated Groningen Frailty Indicator (GFI). Patients with a glomerular filtration rate of less than 30 mL/min per 1.73 m2 or with valvular AFib were excluded.
Eligible patients were then assigned randomly to switch from their international normalized ratio (INR)–guided VKA treatment with either 1 mg acenocoumarol or 3 mg phenprocoumon, to a NOAC, or to continue VKA treatment. They were followed for 12 months for the primary outcome – major bleeding or clinically relevant nonmajor bleeding complication, whichever came first – accounting for death as a competing risk.
A total of 1,330 patients were randomly assigned between January 2018 and June 2022. Their mean age was 83 years, and they had a median GFI of 4. After randomization, 6 patients in the switch-to-NOAC arm, and 1 in the continue-VKA arm were found to have exclusion criteria, so in the end, 662 patients were switched from a VKA to NOAC, while 661 continued on VKA therapy. The choice of NOAC was made by the treating physician.
Major bleeding was defined as a fatal bleeding; bleeding in a critical area or organ; bleeding leading to transfusion; and/or bleeding leading to a fall in hemoglobin level of 2 g/dL (1.24 mmol/L) or more. Nonmajor bleeding was bleeding not considered major but requiring face-to-face consultation, hospitalization or increased level of care, or medical intervention.
After a prespecified futility analysis planned after 163 primary outcome events, the trial was halted when it was seen that there were 101 primary outcome events in the switch arm compared to 62 in the continue arm, Dr. Joosten said. The difference appeared to be driven by clinically relevant nonmajor bleeding.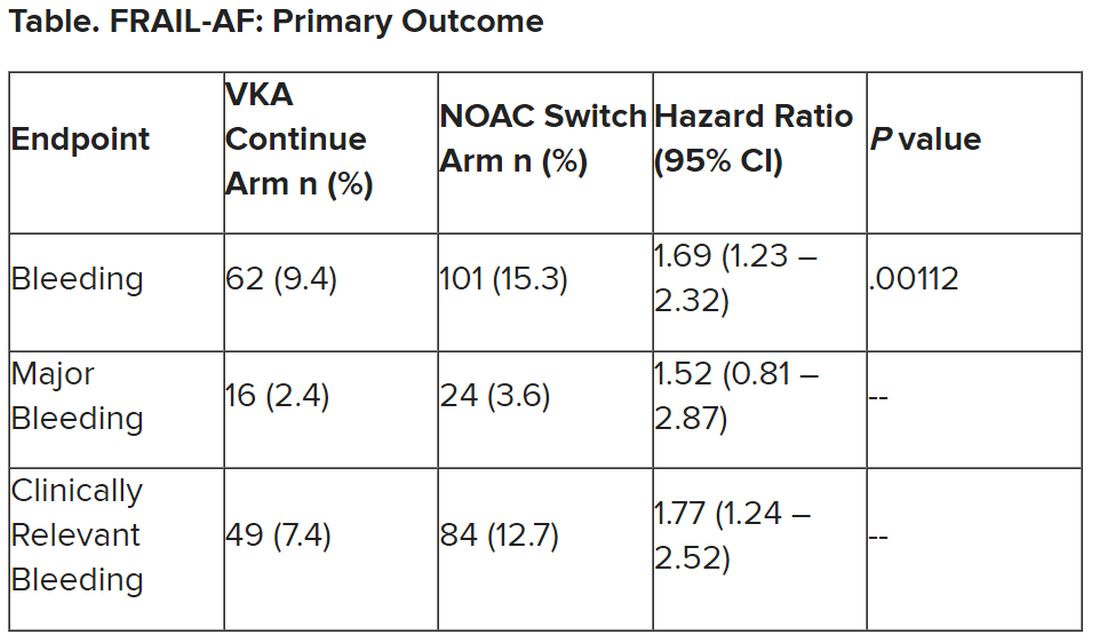
Secondary outcomes of thromboembolic events and all-cause mortality were similar between the groups.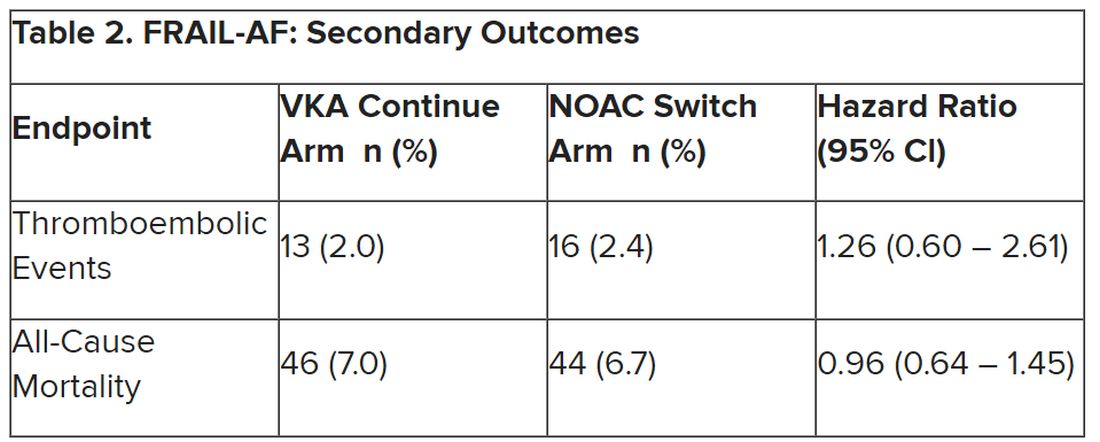
Completely different patients
Discussant at the meeting for the presentation was Isabelle C. Van Gelder, MD, University Medical Centre Groningen (the Netherlands). She said the results are important and relevant because it “provides data on an important gap of knowledge in our AF[ib] guidelines, and a note for all the cardiologists – this study was not done in the hospital. This trial was done in general practitioner practices, so that’s important to consider.”
Comparing FRAIL-AF patients with those of the four previous NOAC trials, “you see that enormous difference in age,” with an average age of 83 years versus 70-73 years in those trials. “These are completely different patients than have been included previously,” she said.
That GFI score of 4 or more includes patients on four or more different types of medication, as well as memory complaints, an inability to walk around the house, and problems with vision or hearing.
The finding of a 69% increase in bleeding with NOACs in FRAIL-AF was “completely unexpected, and I think that we as cardiologists and as NOAC believers did not expect it at all, but it is as clear as it is.” The curves don’t diverge immediately, but rather after 3 months or thereafter, “so it has nothing to do with the switching process. So why did it occur?”
The Netherlands has dedicated thrombosis services that might improve time in therapeutic range for VKA patients, but there is no real difference in TTRs in FRAIL-AF versus the other NOAC trials, Dr. Van Gelder noted.
The most likely suspect in her view is frailty itself, in particular the tendency for patients to be on a high number of medications. A previous study showed, for example, that polypharmacy could be used as a proxy for the effect of frailty on bleeding risk; patients on 10 or more medications had a higher risk for bleeding on treatment with rivaroxaban versus those on 4 or fewer medications.
“Therefore, in my view, why was there such a high risk of bleeding? It’s because these are other patients than we are normally used to treat, we as cardiologists,” although general practitioners see these patients all the time. “It’s all about frailty.”
NOACs are still relatively new drugs, with possible unknown interactions, she added. Because of their frailty and polypharmacy, these patients may benefit from INR control, Dr. Van Gelder speculated. “Therefore, I agree with them that we should be careful; if such old, frail patients survive on VKA, do not change medications and do not switch!”
The study was supported by the Dutch government with additional and unrestricted educational grants from Boehringer Ingelheim, BMS-Pfizer, Bayer, and Daiichi Sankyo. Dr. Joosten reported no relevant financial relationships. Dr. Van Gelder reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Switching frail patients with atrial fibrillation (AFib) from anticoagulation therapy with vitamin K antagonists (VKAs) to a novel oral anticoagulant (NOAC) resulted in more bleeding without any reduction in thromboembolic complications or all-cause mortality, randomized trial results show.
The study, FRAIL-AF, is the first randomized NOAC trial to exclusively include frail older patients, said lead author Linda P.T. Joosten, MD, Julius Center for Health Sciences and Primary Care in Utrecht, the Netherlands, and these unexpected findings provide evidence that goes beyond what is currently available.
“Data from the FRAIL-AF trial showed that switching from a VKA to a NOAC should not be considered without a clear indication in frail older patients with AF[ib], as switching to a NOAC leads to 69% more bleeding,” she concluded, without any benefit on secondary clinical endpoints, including thromboembolic events and all-cause mortality.
“The results turned out different than we expected,” Dr. Joosten said. “The hypothesis of this superiority trial was that switching from VKA therapy to a NOAC would result in less bleeding. However, we observed the opposite. After the interim analysis, the data and safety monitoring board advised to stop inclusion because switching from a VKA to a NOAC was clearly contraindicated with a hazard ratio of 1.69 and a highly significant P value of .001.”
Results of FRAIL-AF were presented at the annual congress of the European Society of Cardiology and published online in the journal Circulation.
Session moderator Renate B. Schnabel, MD, interventional cardiologist with University Heart & Vascular Center Hamburg (Germany), congratulated the researchers on these “astonishing” data.
“The thing I want to emphasize here is that, in the absence of randomized controlled trial data, we should be very cautious in extrapolating data from the landmark trials to populations not enrolled in those, and to rely on observational data only,” Dr. Schnabel told Dr. Joosten. “We need randomized controlled trials that sometimes give astonishing results.”
Frailty a clinical syndrome
Frailty is “a lot more than just aging, multiple comorbidities and polypharmacy,” Dr. Joosten explained. “It’s really a clinical syndrome, with people with a high biological vulnerability, dependency on significant others, and a reduced capacity to resist stressors, all leading to a reduced homeostatic reserve.”
Frailty is common in the community, with a prevalence of about 12%, she noted, “and even more important, AF[ib] in frail older people is very common, with a prevalence of 18%. And “without any doubt, we have to adequately anticoagulate frail AF[ib] patients, as they have a high stroke risk, with an incidence of 12.4% per year,” Dr. Joosten noted, compared with 3.9% per year among nonfrail AFib patients.
NOACs are preferred over VKAs in nonfrail AFib patients, after four major trials, RE-LY with dabigatran, ROCKET-AF with rivaroxaban, ARISTOTLE with apixaban, and ENGAGE-AF with edoxaban, showed that NOAC treatment resulted in less major bleeding while stroke risk was comparable with treatment with warfarin, she noted.
The 2023 European Heart Rhythm Association consensus document on management of arrhythmias in frailty syndrome concludes that the advantages of NOACs relative to VKAs are “likely consistent” in frail and nonfrail AFib patients, but the level of evidence is low.
So it’s unknown if NOACs are preferred over VKAs in frail AFib patients, “and it’s even more questionable whether patients on VKAs should switch to NOAC therapy,” Dr. Joosten said.
This new trial aimed to answer the question of whether switching frail AFib patients currently managed on a VKA to a NOAC would reduce bleeding. FRAIL-AF was a pragmatic, multicenter, open-label, randomized, controlled superiority trial.
Older AFib patients were deemed frail if they were aged 75 years or older and had a score of 3 or more on the validated Groningen Frailty Indicator (GFI). Patients with a glomerular filtration rate of less than 30 mL/min per 1.73 m2 or with valvular AFib were excluded.
Eligible patients were then assigned randomly to switch from their international normalized ratio (INR)–guided VKA treatment with either 1 mg acenocoumarol or 3 mg phenprocoumon, to a NOAC, or to continue VKA treatment. They were followed for 12 months for the primary outcome – major bleeding or clinically relevant nonmajor bleeding complication, whichever came first – accounting for death as a competing risk.
A total of 1,330 patients were randomly assigned between January 2018 and June 2022. Their mean age was 83 years, and they had a median GFI of 4. After randomization, 6 patients in the switch-to-NOAC arm, and 1 in the continue-VKA arm were found to have exclusion criteria, so in the end, 662 patients were switched from a VKA to NOAC, while 661 continued on VKA therapy. The choice of NOAC was made by the treating physician.
Major bleeding was defined as a fatal bleeding; bleeding in a critical area or organ; bleeding leading to transfusion; and/or bleeding leading to a fall in hemoglobin level of 2 g/dL (1.24 mmol/L) or more. Nonmajor bleeding was bleeding not considered major but requiring face-to-face consultation, hospitalization or increased level of care, or medical intervention.
After a prespecified futility analysis planned after 163 primary outcome events, the trial was halted when it was seen that there were 101 primary outcome events in the switch arm compared to 62 in the continue arm, Dr. Joosten said. The difference appeared to be driven by clinically relevant nonmajor bleeding.
Secondary outcomes of thromboembolic events and all-cause mortality were similar between the groups.
Completely different patients
Discussant at the meeting for the presentation was Isabelle C. Van Gelder, MD, University Medical Centre Groningen (the Netherlands). She said the results are important and relevant because it “provides data on an important gap of knowledge in our AF[ib] guidelines, and a note for all the cardiologists – this study was not done in the hospital. This trial was done in general practitioner practices, so that’s important to consider.”
Comparing FRAIL-AF patients with those of the four previous NOAC trials, “you see that enormous difference in age,” with an average age of 83 years versus 70-73 years in those trials. “These are completely different patients than have been included previously,” she said.
That GFI score of 4 or more includes patients on four or more different types of medication, as well as memory complaints, an inability to walk around the house, and problems with vision or hearing.
The finding of a 69% increase in bleeding with NOACs in FRAIL-AF was “completely unexpected, and I think that we as cardiologists and as NOAC believers did not expect it at all, but it is as clear as it is.” The curves don’t diverge immediately, but rather after 3 months or thereafter, “so it has nothing to do with the switching process. So why did it occur?”
The Netherlands has dedicated thrombosis services that might improve time in therapeutic range for VKA patients, but there is no real difference in TTRs in FRAIL-AF versus the other NOAC trials, Dr. Van Gelder noted.
The most likely suspect in her view is frailty itself, in particular the tendency for patients to be on a high number of medications. A previous study showed, for example, that polypharmacy could be used as a proxy for the effect of frailty on bleeding risk; patients on 10 or more medications had a higher risk for bleeding on treatment with rivaroxaban versus those on 4 or fewer medications.
“Therefore, in my view, why was there such a high risk of bleeding? It’s because these are other patients than we are normally used to treat, we as cardiologists,” although general practitioners see these patients all the time. “It’s all about frailty.”
NOACs are still relatively new drugs, with possible unknown interactions, she added. Because of their frailty and polypharmacy, these patients may benefit from INR control, Dr. Van Gelder speculated. “Therefore, I agree with them that we should be careful; if such old, frail patients survive on VKA, do not change medications and do not switch!”
The study was supported by the Dutch government with additional and unrestricted educational grants from Boehringer Ingelheim, BMS-Pfizer, Bayer, and Daiichi Sankyo. Dr. Joosten reported no relevant financial relationships. Dr. Van Gelder reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Switching frail patients with atrial fibrillation (AFib) from anticoagulation therapy with vitamin K antagonists (VKAs) to a novel oral anticoagulant (NOAC) resulted in more bleeding without any reduction in thromboembolic complications or all-cause mortality, randomized trial results show.
The study, FRAIL-AF, is the first randomized NOAC trial to exclusively include frail older patients, said lead author Linda P.T. Joosten, MD, Julius Center for Health Sciences and Primary Care in Utrecht, the Netherlands, and these unexpected findings provide evidence that goes beyond what is currently available.
“Data from the FRAIL-AF trial showed that switching from a VKA to a NOAC should not be considered without a clear indication in frail older patients with AF[ib], as switching to a NOAC leads to 69% more bleeding,” she concluded, without any benefit on secondary clinical endpoints, including thromboembolic events and all-cause mortality.
“The results turned out different than we expected,” Dr. Joosten said. “The hypothesis of this superiority trial was that switching from VKA therapy to a NOAC would result in less bleeding. However, we observed the opposite. After the interim analysis, the data and safety monitoring board advised to stop inclusion because switching from a VKA to a NOAC was clearly contraindicated with a hazard ratio of 1.69 and a highly significant P value of .001.”
Results of FRAIL-AF were presented at the annual congress of the European Society of Cardiology and published online in the journal Circulation.
Session moderator Renate B. Schnabel, MD, interventional cardiologist with University Heart & Vascular Center Hamburg (Germany), congratulated the researchers on these “astonishing” data.
“The thing I want to emphasize here is that, in the absence of randomized controlled trial data, we should be very cautious in extrapolating data from the landmark trials to populations not enrolled in those, and to rely on observational data only,” Dr. Schnabel told Dr. Joosten. “We need randomized controlled trials that sometimes give astonishing results.”
Frailty a clinical syndrome
Frailty is “a lot more than just aging, multiple comorbidities and polypharmacy,” Dr. Joosten explained. “It’s really a clinical syndrome, with people with a high biological vulnerability, dependency on significant others, and a reduced capacity to resist stressors, all leading to a reduced homeostatic reserve.”
Frailty is common in the community, with a prevalence of about 12%, she noted, “and even more important, AF[ib] in frail older people is very common, with a prevalence of 18%. And “without any doubt, we have to adequately anticoagulate frail AF[ib] patients, as they have a high stroke risk, with an incidence of 12.4% per year,” Dr. Joosten noted, compared with 3.9% per year among nonfrail AFib patients.
NOACs are preferred over VKAs in nonfrail AFib patients, after four major trials, RE-LY with dabigatran, ROCKET-AF with rivaroxaban, ARISTOTLE with apixaban, and ENGAGE-AF with edoxaban, showed that NOAC treatment resulted in less major bleeding while stroke risk was comparable with treatment with warfarin, she noted.
The 2023 European Heart Rhythm Association consensus document on management of arrhythmias in frailty syndrome concludes that the advantages of NOACs relative to VKAs are “likely consistent” in frail and nonfrail AFib patients, but the level of evidence is low.
So it’s unknown if NOACs are preferred over VKAs in frail AFib patients, “and it’s even more questionable whether patients on VKAs should switch to NOAC therapy,” Dr. Joosten said.
This new trial aimed to answer the question of whether switching frail AFib patients currently managed on a VKA to a NOAC would reduce bleeding. FRAIL-AF was a pragmatic, multicenter, open-label, randomized, controlled superiority trial.
Older AFib patients were deemed frail if they were aged 75 years or older and had a score of 3 or more on the validated Groningen Frailty Indicator (GFI). Patients with a glomerular filtration rate of less than 30 mL/min per 1.73 m2 or with valvular AFib were excluded.
Eligible patients were then assigned randomly to switch from their international normalized ratio (INR)–guided VKA treatment with either 1 mg acenocoumarol or 3 mg phenprocoumon, to a NOAC, or to continue VKA treatment. They were followed for 12 months for the primary outcome – major bleeding or clinically relevant nonmajor bleeding complication, whichever came first – accounting for death as a competing risk.
A total of 1,330 patients were randomly assigned between January 2018 and June 2022. Their mean age was 83 years, and they had a median GFI of 4. After randomization, 6 patients in the switch-to-NOAC arm, and 1 in the continue-VKA arm were found to have exclusion criteria, so in the end, 662 patients were switched from a VKA to NOAC, while 661 continued on VKA therapy. The choice of NOAC was made by the treating physician.
Major bleeding was defined as a fatal bleeding; bleeding in a critical area or organ; bleeding leading to transfusion; and/or bleeding leading to a fall in hemoglobin level of 2 g/dL (1.24 mmol/L) or more. Nonmajor bleeding was bleeding not considered major but requiring face-to-face consultation, hospitalization or increased level of care, or medical intervention.
After a prespecified futility analysis planned after 163 primary outcome events, the trial was halted when it was seen that there were 101 primary outcome events in the switch arm compared to 62 in the continue arm, Dr. Joosten said. The difference appeared to be driven by clinically relevant nonmajor bleeding.
Secondary outcomes of thromboembolic events and all-cause mortality were similar between the groups.
Completely different patients
Discussant at the meeting for the presentation was Isabelle C. Van Gelder, MD, University Medical Centre Groningen (the Netherlands). She said the results are important and relevant because it “provides data on an important gap of knowledge in our AF[ib] guidelines, and a note for all the cardiologists – this study was not done in the hospital. This trial was done in general practitioner practices, so that’s important to consider.”
Comparing FRAIL-AF patients with those of the four previous NOAC trials, “you see that enormous difference in age,” with an average age of 83 years versus 70-73 years in those trials. “These are completely different patients than have been included previously,” she said.
That GFI score of 4 or more includes patients on four or more different types of medication, as well as memory complaints, an inability to walk around the house, and problems with vision or hearing.
The finding of a 69% increase in bleeding with NOACs in FRAIL-AF was “completely unexpected, and I think that we as cardiologists and as NOAC believers did not expect it at all, but it is as clear as it is.” The curves don’t diverge immediately, but rather after 3 months or thereafter, “so it has nothing to do with the switching process. So why did it occur?”
The Netherlands has dedicated thrombosis services that might improve time in therapeutic range for VKA patients, but there is no real difference in TTRs in FRAIL-AF versus the other NOAC trials, Dr. Van Gelder noted.
The most likely suspect in her view is frailty itself, in particular the tendency for patients to be on a high number of medications. A previous study showed, for example, that polypharmacy could be used as a proxy for the effect of frailty on bleeding risk; patients on 10 or more medications had a higher risk for bleeding on treatment with rivaroxaban versus those on 4 or fewer medications.
“Therefore, in my view, why was there such a high risk of bleeding? It’s because these are other patients than we are normally used to treat, we as cardiologists,” although general practitioners see these patients all the time. “It’s all about frailty.”
NOACs are still relatively new drugs, with possible unknown interactions, she added. Because of their frailty and polypharmacy, these patients may benefit from INR control, Dr. Van Gelder speculated. “Therefore, I agree with them that we should be careful; if such old, frail patients survive on VKA, do not change medications and do not switch!”
The study was supported by the Dutch government with additional and unrestricted educational grants from Boehringer Ingelheim, BMS-Pfizer, Bayer, and Daiichi Sankyo. Dr. Joosten reported no relevant financial relationships. Dr. Van Gelder reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE ESC CONGRESS 2023
Using JAK inhibitors for myelofibrosis
“We are thankfully starting to be blessed with more options than we’ve ever had,” he said, but “in the front-line proliferative setting, ruxolitinib has remained the standard of care.” It’s “well established in higher-risk patients and very much an option for very symptomatic lower-risk patients.”
Dr. Hunter helped his colleagues navigate the evolving field of JAK inhibition for myelofibrosis in a presentation titled “Choosing and Properly Using a JAK Inhibitor in Myelofibrosis,”at the Society of Hematologic Oncology annual meeting.
Ruxolitinib was the first JAK inhibitor for myelofibrosis on the U.S. market, approved in 2011. Two more have followed, fedratinib in 2019 and pacritinib in 2022.
A fourth JAK inhibitor for myelofibrosis, momelotinib, is under Food and Drug Administration review with a decision expected shortly.
JAK inhibitors disrupt a key pathogenic pathway in myelofibrosis and are a mainstay of treatment, but Dr. Hunter noted that they should not replace allogeneic transplants in patients who are candidates because transplants remain “the best way to achieve long term survival, especially in higher risk patients.”
He noted that not every patient needs a JAK inhibitor, especially “lower-risk, more asymptomatic patients who are predominantly manifesting with cytopenias. [They] are less likely to benefit.”
Dr. Hunter said that although ruxolitinib remains a treatment of choice, fedratinib “is certainly an option” with comparable rates of symptom control and splenomegaly reduction. Also, while ruxolitinib is dosed according to platelet levels, fedratinib allows for full dosing down to a platelet count of 50 x 109/L.
“But there’s more GI toxicity than with ruxolitinib, especially in the first couple of months,” he said, as well as a black box warning of Wernicke’s encephalopathy. “I generally put all my [fedratinib] patients on thiamine repletion as a precaution.”
One of the most challenging aspects of using JAK inhibitors for myelofibrosis is their tendency to cause cytopenia, particularly anemia and thrombocytopenia, which, ironically, are also hallmarks of myelofibrosis itself.
Although there’s an alternative low-dose ruxolitinib regimen that can be effective in anemic settings, the approval of pacritinib and most likely momelotinib is particularly helpful for cytopenic patients, “a population which historically has been very hard to treat with our prior agents,” Dr. Hunter said.
Pacritinib is approved specifically for patients with platelet counts below 50 x 109/L; momelotinib also included lower platelet counts in several studies. Both agents indirectly boost erythropoiesis with subsequent amelioration of anemia.
“Momelotinib is an important emerging agent for these more anemic patients,” with a spleen response comparable to ruxolitinib and significantly higher rates of transfusion independence, but with lower rates of symptom control, Dr. Hunter said.
Pacritinib “really helps extend the benefit of JAK inhibitors to a group of thrombocytopenic patients who have been hard to treat with ruxolitinib,” with the added potential of improving anemia, although, like fedratinib, it has more GI toxicity, he said.
There are multiple add-on options for JAK inhibitor patients with anemia, including luspatercept, an erythropoiesis-stimulating agent approved for anemia in patients with myelodysplastic syndromes; promising results were reported recently for myelofibrosis.
Fedratinib, pacritinib, and momelotinib all have activity in the second line after ruxolitinib failure, Dr. Hunter noted, but he cautioned that ruxolitinib must be tapered over a few weeks, not stopped abruptly, to avoid withdrawal symptoms. Some clinicians overlap JAK inhibitors a day or two to avoid issues.
“Clinical trials should still be considered in many of these settings,” he said, adding that emerging agents are under development, including multiple combination therapies, often with JAK inhibitors as the background.
No disclosure information was reported.
“We are thankfully starting to be blessed with more options than we’ve ever had,” he said, but “in the front-line proliferative setting, ruxolitinib has remained the standard of care.” It’s “well established in higher-risk patients and very much an option for very symptomatic lower-risk patients.”
Dr. Hunter helped his colleagues navigate the evolving field of JAK inhibition for myelofibrosis in a presentation titled “Choosing and Properly Using a JAK Inhibitor in Myelofibrosis,”at the Society of Hematologic Oncology annual meeting.
Ruxolitinib was the first JAK inhibitor for myelofibrosis on the U.S. market, approved in 2011. Two more have followed, fedratinib in 2019 and pacritinib in 2022.
A fourth JAK inhibitor for myelofibrosis, momelotinib, is under Food and Drug Administration review with a decision expected shortly.
JAK inhibitors disrupt a key pathogenic pathway in myelofibrosis and are a mainstay of treatment, but Dr. Hunter noted that they should not replace allogeneic transplants in patients who are candidates because transplants remain “the best way to achieve long term survival, especially in higher risk patients.”
He noted that not every patient needs a JAK inhibitor, especially “lower-risk, more asymptomatic patients who are predominantly manifesting with cytopenias. [They] are less likely to benefit.”
Dr. Hunter said that although ruxolitinib remains a treatment of choice, fedratinib “is certainly an option” with comparable rates of symptom control and splenomegaly reduction. Also, while ruxolitinib is dosed according to platelet levels, fedratinib allows for full dosing down to a platelet count of 50 x 109/L.
“But there’s more GI toxicity than with ruxolitinib, especially in the first couple of months,” he said, as well as a black box warning of Wernicke’s encephalopathy. “I generally put all my [fedratinib] patients on thiamine repletion as a precaution.”
One of the most challenging aspects of using JAK inhibitors for myelofibrosis is their tendency to cause cytopenia, particularly anemia and thrombocytopenia, which, ironically, are also hallmarks of myelofibrosis itself.
Although there’s an alternative low-dose ruxolitinib regimen that can be effective in anemic settings, the approval of pacritinib and most likely momelotinib is particularly helpful for cytopenic patients, “a population which historically has been very hard to treat with our prior agents,” Dr. Hunter said.
Pacritinib is approved specifically for patients with platelet counts below 50 x 109/L; momelotinib also included lower platelet counts in several studies. Both agents indirectly boost erythropoiesis with subsequent amelioration of anemia.
“Momelotinib is an important emerging agent for these more anemic patients,” with a spleen response comparable to ruxolitinib and significantly higher rates of transfusion independence, but with lower rates of symptom control, Dr. Hunter said.
Pacritinib “really helps extend the benefit of JAK inhibitors to a group of thrombocytopenic patients who have been hard to treat with ruxolitinib,” with the added potential of improving anemia, although, like fedratinib, it has more GI toxicity, he said.
There are multiple add-on options for JAK inhibitor patients with anemia, including luspatercept, an erythropoiesis-stimulating agent approved for anemia in patients with myelodysplastic syndromes; promising results were reported recently for myelofibrosis.
Fedratinib, pacritinib, and momelotinib all have activity in the second line after ruxolitinib failure, Dr. Hunter noted, but he cautioned that ruxolitinib must be tapered over a few weeks, not stopped abruptly, to avoid withdrawal symptoms. Some clinicians overlap JAK inhibitors a day or two to avoid issues.
“Clinical trials should still be considered in many of these settings,” he said, adding that emerging agents are under development, including multiple combination therapies, often with JAK inhibitors as the background.
No disclosure information was reported.
“We are thankfully starting to be blessed with more options than we’ve ever had,” he said, but “in the front-line proliferative setting, ruxolitinib has remained the standard of care.” It’s “well established in higher-risk patients and very much an option for very symptomatic lower-risk patients.”
Dr. Hunter helped his colleagues navigate the evolving field of JAK inhibition for myelofibrosis in a presentation titled “Choosing and Properly Using a JAK Inhibitor in Myelofibrosis,”at the Society of Hematologic Oncology annual meeting.
Ruxolitinib was the first JAK inhibitor for myelofibrosis on the U.S. market, approved in 2011. Two more have followed, fedratinib in 2019 and pacritinib in 2022.
A fourth JAK inhibitor for myelofibrosis, momelotinib, is under Food and Drug Administration review with a decision expected shortly.
JAK inhibitors disrupt a key pathogenic pathway in myelofibrosis and are a mainstay of treatment, but Dr. Hunter noted that they should not replace allogeneic transplants in patients who are candidates because transplants remain “the best way to achieve long term survival, especially in higher risk patients.”
He noted that not every patient needs a JAK inhibitor, especially “lower-risk, more asymptomatic patients who are predominantly manifesting with cytopenias. [They] are less likely to benefit.”
Dr. Hunter said that although ruxolitinib remains a treatment of choice, fedratinib “is certainly an option” with comparable rates of symptom control and splenomegaly reduction. Also, while ruxolitinib is dosed according to platelet levels, fedratinib allows for full dosing down to a platelet count of 50 x 109/L.
“But there’s more GI toxicity than with ruxolitinib, especially in the first couple of months,” he said, as well as a black box warning of Wernicke’s encephalopathy. “I generally put all my [fedratinib] patients on thiamine repletion as a precaution.”
One of the most challenging aspects of using JAK inhibitors for myelofibrosis is their tendency to cause cytopenia, particularly anemia and thrombocytopenia, which, ironically, are also hallmarks of myelofibrosis itself.
Although there’s an alternative low-dose ruxolitinib regimen that can be effective in anemic settings, the approval of pacritinib and most likely momelotinib is particularly helpful for cytopenic patients, “a population which historically has been very hard to treat with our prior agents,” Dr. Hunter said.
Pacritinib is approved specifically for patients with platelet counts below 50 x 109/L; momelotinib also included lower platelet counts in several studies. Both agents indirectly boost erythropoiesis with subsequent amelioration of anemia.
“Momelotinib is an important emerging agent for these more anemic patients,” with a spleen response comparable to ruxolitinib and significantly higher rates of transfusion independence, but with lower rates of symptom control, Dr. Hunter said.
Pacritinib “really helps extend the benefit of JAK inhibitors to a group of thrombocytopenic patients who have been hard to treat with ruxolitinib,” with the added potential of improving anemia, although, like fedratinib, it has more GI toxicity, he said.
There are multiple add-on options for JAK inhibitor patients with anemia, including luspatercept, an erythropoiesis-stimulating agent approved for anemia in patients with myelodysplastic syndromes; promising results were reported recently for myelofibrosis.
Fedratinib, pacritinib, and momelotinib all have activity in the second line after ruxolitinib failure, Dr. Hunter noted, but he cautioned that ruxolitinib must be tapered over a few weeks, not stopped abruptly, to avoid withdrawal symptoms. Some clinicians overlap JAK inhibitors a day or two to avoid issues.
“Clinical trials should still be considered in many of these settings,” he said, adding that emerging agents are under development, including multiple combination therapies, often with JAK inhibitors as the background.
No disclosure information was reported.
FROM SOHO 2023
CHP/CCUS: Low blood cancer risk for most patients
The reason is that patients will inevitably “go online and see that [the conditions are] associated with lots of bad things; it can really cause patients psychosocial harm if there is no one to explain what their risk is and also provide risk-specific management,” Dr. Weeks said at the annual meeting of the Society of Hematologic Oncology in Houston.
CHIP and CCUS are precursors of myeloid malignancies but for most patients, the risk of progression is less than 1%. CHIPS and CCUS are also associated with cardiovascular, rheumatologic, hepatic, and other diseases.
CHIP is defined by somatic mutations in myeloid malignancy driver genes with a variant allele fraction of 2% or more; CCUS is when those molecular features are accompanied by an unexplained and persistent anemia, thrombocytopenia, or neutropenia.
A small 2017 study suggested that about a third of patients with otherwise unexplained cytopenias have CCUS.
With the increasing use of next generation sequencing for tissue and liquid biopsies and other uses, the incidental diagnosis of both conditions is increasing.
Fortunately, Dr. Weeks’ group recently published a tool for predicting the risk of progression to myeloid malignancy.
Their “clonal hematopoiesis risk score” (CHRS) was developed and validated in over 400,000 healthy volunteers in the UK Biobank, with additional validation in cohorts from Dana Farber and the University of Pavia, Italy.
The CHRS incorporates eight high-risk genetic and clinical prognostic factors, including the type and number of genetic mutations in blood cells, factors related to red blood cell volume, and age over 65. It’s available online.
“You just input the patient’s information and it spits out if the patient is low, intermediate, or high risk for progression to any myeloid malignancy,” Dr. Weeks told her audience.
High-risk patients have about a 50% 10-year cumulative incidence of myeloid malignancy. The large majority of patients are low risk, however, and have a 10-year cumulative incidence of less than 1%. Patients in the middle have a 10-year risk of about 8%.
The low-risk group “is the population of people who probably don’t need to see a specialist,” and can be followed with an annual CBC with their primary care doctors plus further workup with any clinical change. Patients should also be evaluated for cardiovascular and other comorbidity risks.
“It’s the high-risk group we worry most about,” Dr. Weeks said. “We see them more often and repeat the next-generation sequencing” annually with a CBC at least every 6 months and a bone marrow biopsy with any clinical change.
“This is the population we would shuttle towards a clinical trial, as this is the population most likely to benefit,” she said.
The overarching goal of the several ongoing studies in CHIP/CCUS is to find a way to prevent progression to blood cancer. They range from prospective cohorts and single arm pilot studies to randomized clinical trials. One trial is evaluating canakinumab to prevent progression. “Intervention in clonal hematopoiesis might have the dual benefit of both preventing hematologic malignancy as well as reducing [the] inflammatory comorbidities,” Dr. Weeks said.
The reason is that patients will inevitably “go online and see that [the conditions are] associated with lots of bad things; it can really cause patients psychosocial harm if there is no one to explain what their risk is and also provide risk-specific management,” Dr. Weeks said at the annual meeting of the Society of Hematologic Oncology in Houston.
CHIP and CCUS are precursors of myeloid malignancies but for most patients, the risk of progression is less than 1%. CHIPS and CCUS are also associated with cardiovascular, rheumatologic, hepatic, and other diseases.
CHIP is defined by somatic mutations in myeloid malignancy driver genes with a variant allele fraction of 2% or more; CCUS is when those molecular features are accompanied by an unexplained and persistent anemia, thrombocytopenia, or neutropenia.
A small 2017 study suggested that about a third of patients with otherwise unexplained cytopenias have CCUS.
With the increasing use of next generation sequencing for tissue and liquid biopsies and other uses, the incidental diagnosis of both conditions is increasing.
Fortunately, Dr. Weeks’ group recently published a tool for predicting the risk of progression to myeloid malignancy.
Their “clonal hematopoiesis risk score” (CHRS) was developed and validated in over 400,000 healthy volunteers in the UK Biobank, with additional validation in cohorts from Dana Farber and the University of Pavia, Italy.
The CHRS incorporates eight high-risk genetic and clinical prognostic factors, including the type and number of genetic mutations in blood cells, factors related to red blood cell volume, and age over 65. It’s available online.
“You just input the patient’s information and it spits out if the patient is low, intermediate, or high risk for progression to any myeloid malignancy,” Dr. Weeks told her audience.
High-risk patients have about a 50% 10-year cumulative incidence of myeloid malignancy. The large majority of patients are low risk, however, and have a 10-year cumulative incidence of less than 1%. Patients in the middle have a 10-year risk of about 8%.
The low-risk group “is the population of people who probably don’t need to see a specialist,” and can be followed with an annual CBC with their primary care doctors plus further workup with any clinical change. Patients should also be evaluated for cardiovascular and other comorbidity risks.
“It’s the high-risk group we worry most about,” Dr. Weeks said. “We see them more often and repeat the next-generation sequencing” annually with a CBC at least every 6 months and a bone marrow biopsy with any clinical change.
“This is the population we would shuttle towards a clinical trial, as this is the population most likely to benefit,” she said.
The overarching goal of the several ongoing studies in CHIP/CCUS is to find a way to prevent progression to blood cancer. They range from prospective cohorts and single arm pilot studies to randomized clinical trials. One trial is evaluating canakinumab to prevent progression. “Intervention in clonal hematopoiesis might have the dual benefit of both preventing hematologic malignancy as well as reducing [the] inflammatory comorbidities,” Dr. Weeks said.
The reason is that patients will inevitably “go online and see that [the conditions are] associated with lots of bad things; it can really cause patients psychosocial harm if there is no one to explain what their risk is and also provide risk-specific management,” Dr. Weeks said at the annual meeting of the Society of Hematologic Oncology in Houston.
CHIP and CCUS are precursors of myeloid malignancies but for most patients, the risk of progression is less than 1%. CHIPS and CCUS are also associated with cardiovascular, rheumatologic, hepatic, and other diseases.
CHIP is defined by somatic mutations in myeloid malignancy driver genes with a variant allele fraction of 2% or more; CCUS is when those molecular features are accompanied by an unexplained and persistent anemia, thrombocytopenia, or neutropenia.
A small 2017 study suggested that about a third of patients with otherwise unexplained cytopenias have CCUS.
With the increasing use of next generation sequencing for tissue and liquid biopsies and other uses, the incidental diagnosis of both conditions is increasing.
Fortunately, Dr. Weeks’ group recently published a tool for predicting the risk of progression to myeloid malignancy.
Their “clonal hematopoiesis risk score” (CHRS) was developed and validated in over 400,000 healthy volunteers in the UK Biobank, with additional validation in cohorts from Dana Farber and the University of Pavia, Italy.
The CHRS incorporates eight high-risk genetic and clinical prognostic factors, including the type and number of genetic mutations in blood cells, factors related to red blood cell volume, and age over 65. It’s available online.
“You just input the patient’s information and it spits out if the patient is low, intermediate, or high risk for progression to any myeloid malignancy,” Dr. Weeks told her audience.
High-risk patients have about a 50% 10-year cumulative incidence of myeloid malignancy. The large majority of patients are low risk, however, and have a 10-year cumulative incidence of less than 1%. Patients in the middle have a 10-year risk of about 8%.
The low-risk group “is the population of people who probably don’t need to see a specialist,” and can be followed with an annual CBC with their primary care doctors plus further workup with any clinical change. Patients should also be evaluated for cardiovascular and other comorbidity risks.
“It’s the high-risk group we worry most about,” Dr. Weeks said. “We see them more often and repeat the next-generation sequencing” annually with a CBC at least every 6 months and a bone marrow biopsy with any clinical change.
“This is the population we would shuttle towards a clinical trial, as this is the population most likely to benefit,” she said.
The overarching goal of the several ongoing studies in CHIP/CCUS is to find a way to prevent progression to blood cancer. They range from prospective cohorts and single arm pilot studies to randomized clinical trials. One trial is evaluating canakinumab to prevent progression. “Intervention in clonal hematopoiesis might have the dual benefit of both preventing hematologic malignancy as well as reducing [the] inflammatory comorbidities,” Dr. Weeks said.
FROM SOHO 2023
Medicare announces 10 drugs targeted for price cuts in 2026
People on Medicare may in 2026 see prices drop for 10 medicines, including pricey diabetes, cancer, blood clot, and arthritis treatments, if advocates for federal drug-price negotiations can implement their plans amid tough opposition.
It’s unclear at this time, though, how these negotiations will play out. The Chamber of Commerce has sided with pharmaceutical companies in bids to block direct Medicare negotiation of drug prices. Many influential Republicans in Congress oppose this plan, which has deep support from both Democrats and AARP.
While facing strong opposition to negotiations, the Centers for Medicare & Medicaid Services sought in its announcement to illustrate the high costs of the selected medicines.
CMS provided data on total Part D costs for selected medicines for the period from June 2022 to May 2023, along with tallies of the number of people taking these drugs. The 10 selected medicines are as follows:
- Eliquis (generic name: apixaban), used to prevent and treat serious blood clots. It is taken by about 3.7 million people through Part D plans. The estimated cost is $16.4 billion.
- Jardiance (generic name: empagliflozin), used for diabetes and heart failure. It is taken by almost 1.6 million people through Part D plans. The estimated cost is $7.06 billion.
- Xarelto (generic name: rivaroxaban), used for blood clots. It is taken by about 1.3 million people through Part D plans. The estimated cost is $6 billion.
- Januvia (generic name: sitagliptin), used for diabetes. It is taken by about 869,00 people through Part D plans. The estimated cost is $4.1 billion.
- Farxiga (generic name: dapagliflozin), used for diabetes, heart failure, and chronic kidney disease. It is taken by about 799,000 people through Part D plans. The estimated cost is almost $3.3 billion.
- Entresto (generic name: sacubitril/valsartan), used to treat heart failure. It is taken by 587,000 people through Part D plans. The estimated cost is $2.9 billion.
- Enbrel( generic name: etanercept), used for rheumatoid arthritis, psoriasis, and psoriatic arthritis. It is taken by 48,000 people through Part D plans. The estimated cost is $2.8 billion.
- Imbruvica (generic name: ibrutinib), used to treat some blood cancers. It is taken by about 20,000 people in Part D plans. The estimated cost is $2.7 billion.
- Stelara (generic name: ustekinumab), used to treat plaque psoriasis, psoriatic arthritis, or certain bowel conditions (Crohn’s disease, ulcerative colitis). It is used by about 22,000 people through Part D plans. The estimated cost is $2.6 billion.
- Fiasp; Fiasp FlexTouch; Fiasp PenFill; NovoLog; NovoLog FlexPen; NovoLog PenFill. These are forms of insulin used to treat diabetes. They are used by about 777,000 people through Part D plans. The estimated cost is $2.6 billion.
A vocal critic of Medicare drug negotiations, Joel White, president of the Council for Affordable Health Coverage, called the announcement of the 10 drugs selected for negotiation “a hollow victory lap.” A former Republican staffer on the House Ways and Means Committee, Mr. White aided with the development of the Medicare Part D plans and has kept tabs on the pharmacy programs since its launch in 2006.
“No one’s costs will go down now or for years because of this announcement” about Part D negotiations, Mr. White said in a statement.
According to its website, CAHC includes among its members the American Academy of Ophthalmology as well as some patient groups, drugmakers, such as Johnson & Johnson, and insurers and industry groups, such as the National Association of Manufacturers.
Separately, the influential Chamber of Commerce is making a strong push to at least delay the implementation of the Medicare Part D drug negotiations. On Aug. 28, the chamber released a letter sent to the Biden administration, raising concerns about a “rush” to implement the provisions of the Inflation Reduction Act.
The chamber also has filed suit to challenge the drug negotiation provisions of the Inflation Reduction Act, requesting that the court issue a preliminary injunction by Oct. 1, 2023.
Other pending legal challenges to direct Medicare drug negotiations include suits filed by Merck, Bristol-Myers Squibb, Johnson & Johnson, Boehringer Ingelheim, and AstraZeneca, according to an email from Pharmaceutical Research and Manufacturers of America. PhRMA also said it is a party to a case.
In addition, the three congressional Republicans with most direct influence over Medicare policy issued on Aug. 29 a joint statement outlining their objections to the planned negotiations on drug prices.
This drug-negotiation proposal is “an unworkable, legally dubious scheme that will lead to higher prices for new drugs coming to market, stifle the development of new cures, and destroy jobs,” said House Energy and Commerce Committee Chair Cathy McMorris Rodgers (R-Wash.), House Ways and Means Committee Chair Jason Smith (R-Mo.), and Senate Finance Committee Ranking Member Mike Crapo (R-Idaho).
Democrats were equally firm and vocal in their support of the negotiations. Senate Finance Chairman Ron Wyden (D-Ore.) issued a statement on Aug. 29 that said the release of the list of the 10 drugs selected for Medicare drug negotiations is part of a “seismic shift in the relationship between Big Pharma, the federal government, and seniors who are counting on lower prices.
“I will be following the negotiation process closely and will fight any attempt by Big Pharma to undo or undermine the progress that’s been made,” Mr. Wyden said.
In addition, AARP issued a statement of its continued support for Medicare drug negotiations.
“The No. 1 reason seniors skip or ration their prescriptions is because they can’t afford them. This must stop,” said AARP executive vice president and chief advocacy and engagement officer Nancy LeaMond in the statement. “The big drug companies and their allies continue suing to overturn the Medicare drug price negotiation program to keep up their price gouging. We can’t allow seniors to be Big Pharma’s cash machine anymore.”
A version of this article first appeared on Medscape.com.
People on Medicare may in 2026 see prices drop for 10 medicines, including pricey diabetes, cancer, blood clot, and arthritis treatments, if advocates for federal drug-price negotiations can implement their plans amid tough opposition.
It’s unclear at this time, though, how these negotiations will play out. The Chamber of Commerce has sided with pharmaceutical companies in bids to block direct Medicare negotiation of drug prices. Many influential Republicans in Congress oppose this plan, which has deep support from both Democrats and AARP.
While facing strong opposition to negotiations, the Centers for Medicare & Medicaid Services sought in its announcement to illustrate the high costs of the selected medicines.
CMS provided data on total Part D costs for selected medicines for the period from June 2022 to May 2023, along with tallies of the number of people taking these drugs. The 10 selected medicines are as follows:
- Eliquis (generic name: apixaban), used to prevent and treat serious blood clots. It is taken by about 3.7 million people through Part D plans. The estimated cost is $16.4 billion.
- Jardiance (generic name: empagliflozin), used for diabetes and heart failure. It is taken by almost 1.6 million people through Part D plans. The estimated cost is $7.06 billion.
- Xarelto (generic name: rivaroxaban), used for blood clots. It is taken by about 1.3 million people through Part D plans. The estimated cost is $6 billion.
- Januvia (generic name: sitagliptin), used for diabetes. It is taken by about 869,00 people through Part D plans. The estimated cost is $4.1 billion.
- Farxiga (generic name: dapagliflozin), used for diabetes, heart failure, and chronic kidney disease. It is taken by about 799,000 people through Part D plans. The estimated cost is almost $3.3 billion.
- Entresto (generic name: sacubitril/valsartan), used to treat heart failure. It is taken by 587,000 people through Part D plans. The estimated cost is $2.9 billion.
- Enbrel( generic name: etanercept), used for rheumatoid arthritis, psoriasis, and psoriatic arthritis. It is taken by 48,000 people through Part D plans. The estimated cost is $2.8 billion.
- Imbruvica (generic name: ibrutinib), used to treat some blood cancers. It is taken by about 20,000 people in Part D plans. The estimated cost is $2.7 billion.
- Stelara (generic name: ustekinumab), used to treat plaque psoriasis, psoriatic arthritis, or certain bowel conditions (Crohn’s disease, ulcerative colitis). It is used by about 22,000 people through Part D plans. The estimated cost is $2.6 billion.
- Fiasp; Fiasp FlexTouch; Fiasp PenFill; NovoLog; NovoLog FlexPen; NovoLog PenFill. These are forms of insulin used to treat diabetes. They are used by about 777,000 people through Part D plans. The estimated cost is $2.6 billion.
A vocal critic of Medicare drug negotiations, Joel White, president of the Council for Affordable Health Coverage, called the announcement of the 10 drugs selected for negotiation “a hollow victory lap.” A former Republican staffer on the House Ways and Means Committee, Mr. White aided with the development of the Medicare Part D plans and has kept tabs on the pharmacy programs since its launch in 2006.
“No one’s costs will go down now or for years because of this announcement” about Part D negotiations, Mr. White said in a statement.
According to its website, CAHC includes among its members the American Academy of Ophthalmology as well as some patient groups, drugmakers, such as Johnson & Johnson, and insurers and industry groups, such as the National Association of Manufacturers.
Separately, the influential Chamber of Commerce is making a strong push to at least delay the implementation of the Medicare Part D drug negotiations. On Aug. 28, the chamber released a letter sent to the Biden administration, raising concerns about a “rush” to implement the provisions of the Inflation Reduction Act.
The chamber also has filed suit to challenge the drug negotiation provisions of the Inflation Reduction Act, requesting that the court issue a preliminary injunction by Oct. 1, 2023.
Other pending legal challenges to direct Medicare drug negotiations include suits filed by Merck, Bristol-Myers Squibb, Johnson & Johnson, Boehringer Ingelheim, and AstraZeneca, according to an email from Pharmaceutical Research and Manufacturers of America. PhRMA also said it is a party to a case.
In addition, the three congressional Republicans with most direct influence over Medicare policy issued on Aug. 29 a joint statement outlining their objections to the planned negotiations on drug prices.
This drug-negotiation proposal is “an unworkable, legally dubious scheme that will lead to higher prices for new drugs coming to market, stifle the development of new cures, and destroy jobs,” said House Energy and Commerce Committee Chair Cathy McMorris Rodgers (R-Wash.), House Ways and Means Committee Chair Jason Smith (R-Mo.), and Senate Finance Committee Ranking Member Mike Crapo (R-Idaho).
Democrats were equally firm and vocal in their support of the negotiations. Senate Finance Chairman Ron Wyden (D-Ore.) issued a statement on Aug. 29 that said the release of the list of the 10 drugs selected for Medicare drug negotiations is part of a “seismic shift in the relationship between Big Pharma, the federal government, and seniors who are counting on lower prices.
“I will be following the negotiation process closely and will fight any attempt by Big Pharma to undo or undermine the progress that’s been made,” Mr. Wyden said.
In addition, AARP issued a statement of its continued support for Medicare drug negotiations.
“The No. 1 reason seniors skip or ration their prescriptions is because they can’t afford them. This must stop,” said AARP executive vice president and chief advocacy and engagement officer Nancy LeaMond in the statement. “The big drug companies and their allies continue suing to overturn the Medicare drug price negotiation program to keep up their price gouging. We can’t allow seniors to be Big Pharma’s cash machine anymore.”
A version of this article first appeared on Medscape.com.
People on Medicare may in 2026 see prices drop for 10 medicines, including pricey diabetes, cancer, blood clot, and arthritis treatments, if advocates for federal drug-price negotiations can implement their plans amid tough opposition.
It’s unclear at this time, though, how these negotiations will play out. The Chamber of Commerce has sided with pharmaceutical companies in bids to block direct Medicare negotiation of drug prices. Many influential Republicans in Congress oppose this plan, which has deep support from both Democrats and AARP.
While facing strong opposition to negotiations, the Centers for Medicare & Medicaid Services sought in its announcement to illustrate the high costs of the selected medicines.
CMS provided data on total Part D costs for selected medicines for the period from June 2022 to May 2023, along with tallies of the number of people taking these drugs. The 10 selected medicines are as follows:
- Eliquis (generic name: apixaban), used to prevent and treat serious blood clots. It is taken by about 3.7 million people through Part D plans. The estimated cost is $16.4 billion.
- Jardiance (generic name: empagliflozin), used for diabetes and heart failure. It is taken by almost 1.6 million people through Part D plans. The estimated cost is $7.06 billion.
- Xarelto (generic name: rivaroxaban), used for blood clots. It is taken by about 1.3 million people through Part D plans. The estimated cost is $6 billion.
- Januvia (generic name: sitagliptin), used for diabetes. It is taken by about 869,00 people through Part D plans. The estimated cost is $4.1 billion.
- Farxiga (generic name: dapagliflozin), used for diabetes, heart failure, and chronic kidney disease. It is taken by about 799,000 people through Part D plans. The estimated cost is almost $3.3 billion.
- Entresto (generic name: sacubitril/valsartan), used to treat heart failure. It is taken by 587,000 people through Part D plans. The estimated cost is $2.9 billion.
- Enbrel( generic name: etanercept), used for rheumatoid arthritis, psoriasis, and psoriatic arthritis. It is taken by 48,000 people through Part D plans. The estimated cost is $2.8 billion.
- Imbruvica (generic name: ibrutinib), used to treat some blood cancers. It is taken by about 20,000 people in Part D plans. The estimated cost is $2.7 billion.
- Stelara (generic name: ustekinumab), used to treat plaque psoriasis, psoriatic arthritis, or certain bowel conditions (Crohn’s disease, ulcerative colitis). It is used by about 22,000 people through Part D plans. The estimated cost is $2.6 billion.
- Fiasp; Fiasp FlexTouch; Fiasp PenFill; NovoLog; NovoLog FlexPen; NovoLog PenFill. These are forms of insulin used to treat diabetes. They are used by about 777,000 people through Part D plans. The estimated cost is $2.6 billion.
A vocal critic of Medicare drug negotiations, Joel White, president of the Council for Affordable Health Coverage, called the announcement of the 10 drugs selected for negotiation “a hollow victory lap.” A former Republican staffer on the House Ways and Means Committee, Mr. White aided with the development of the Medicare Part D plans and has kept tabs on the pharmacy programs since its launch in 2006.
“No one’s costs will go down now or for years because of this announcement” about Part D negotiations, Mr. White said in a statement.
According to its website, CAHC includes among its members the American Academy of Ophthalmology as well as some patient groups, drugmakers, such as Johnson & Johnson, and insurers and industry groups, such as the National Association of Manufacturers.
Separately, the influential Chamber of Commerce is making a strong push to at least delay the implementation of the Medicare Part D drug negotiations. On Aug. 28, the chamber released a letter sent to the Biden administration, raising concerns about a “rush” to implement the provisions of the Inflation Reduction Act.
The chamber also has filed suit to challenge the drug negotiation provisions of the Inflation Reduction Act, requesting that the court issue a preliminary injunction by Oct. 1, 2023.
Other pending legal challenges to direct Medicare drug negotiations include suits filed by Merck, Bristol-Myers Squibb, Johnson & Johnson, Boehringer Ingelheim, and AstraZeneca, according to an email from Pharmaceutical Research and Manufacturers of America. PhRMA also said it is a party to a case.
In addition, the three congressional Republicans with most direct influence over Medicare policy issued on Aug. 29 a joint statement outlining their objections to the planned negotiations on drug prices.
This drug-negotiation proposal is “an unworkable, legally dubious scheme that will lead to higher prices for new drugs coming to market, stifle the development of new cures, and destroy jobs,” said House Energy and Commerce Committee Chair Cathy McMorris Rodgers (R-Wash.), House Ways and Means Committee Chair Jason Smith (R-Mo.), and Senate Finance Committee Ranking Member Mike Crapo (R-Idaho).
Democrats were equally firm and vocal in their support of the negotiations. Senate Finance Chairman Ron Wyden (D-Ore.) issued a statement on Aug. 29 that said the release of the list of the 10 drugs selected for Medicare drug negotiations is part of a “seismic shift in the relationship between Big Pharma, the federal government, and seniors who are counting on lower prices.
“I will be following the negotiation process closely and will fight any attempt by Big Pharma to undo or undermine the progress that’s been made,” Mr. Wyden said.
In addition, AARP issued a statement of its continued support for Medicare drug negotiations.
“The No. 1 reason seniors skip or ration their prescriptions is because they can’t afford them. This must stop,” said AARP executive vice president and chief advocacy and engagement officer Nancy LeaMond in the statement. “The big drug companies and their allies continue suing to overturn the Medicare drug price negotiation program to keep up their price gouging. We can’t allow seniors to be Big Pharma’s cash machine anymore.”
A version of this article first appeared on Medscape.com.
PV: Novel rusfertide shows ‘impressive’ efficacy
“The results are surprisingly positive,” said senior author Ronald Hoffman, MD, of the Icahn School of Medicine at Mount Sinai, New York, in discussing the late-breaking research at a press briefing during the European Hematology Association Hybrid Congress 2023.
“Importantly, the study met all of its efficacy endpoints, including the proportion of responders, absence of phlebotomy eligibility, and hematocrit control,” Dr. Hoffman said.
PV, a relatively common clonal myeloproliferative neoplasm, is characterized by uncontrolled erythrocytosis, or excessive production of red blood cells, increasing the risk for serious complications such as thromboembolic and cardiovascular events – the most common causes of morbidity and mortality in this blood cancer.
To treat PV, the maintenance of hematocrit levels at below 45% is critical. However, the current standard of care, therapeutic phlebotomy, with or without cytoreductive agents, falls short in maintaining those lower levels in the majority of patients, Dr. Hoffman explained.
To improve responses, rusfertide was developed as a novel, synthetic form of hepcidin, a peptide hormone that is produced by the liver and functions to maintain iron homeostasis and control the formation of red blood cells.
“This is somewhat of a paradigm shift,” said Dr. Hoffman in the press briefing. “We’re trying to use a hormone made by the liver to control excessive red blood cell production from polycythemia vera.”
For the phase 2 REVIVE study evaluating rusfertide in PV, the authors enrolled 53 patients with PV who had a high phlebotomy burden while receiving the current standard of care. The study’s criteria called for patients to have received at least three therapeutic phlebotomies in the 28 weeks prior to enrollment, with or without concurrent cytoreductive agents.
During a first part of the study, patients received subcutaneous rusfertide once weekly over 28 weeks, during which period the dose was adjusted individually to achieve control of HCT levels below 45%.
The second part was a withdrawal phase extending from weeks 29 to 41, in which patients were randomized in a blinded fashion to either continue on rusfertide (n = 26) or receive a placebo (n = 27).
The patients had a median age of 58; they were 71.7% male, and 54.7% had previously been treated with therapeutic phlebotomy alone while 45.3% received therapeutic phlebotomy plus cytoreductive agents.
Patients were considered to be responders if they met three criteria, including having HCT control without phlebotomy eligibility, no therapeutic phlebotomy, and having completed 12 weeks of treatment.
At the end of the second phase, 69.2% of patients receiving rusfertide were responders versus just 18.5% in the placebo group (P = .0003).
Notably, the improvement with rusfertide was observed among those receiving therapeutic phlebotomy alone, as well as with cytoreductive agents (both P = .02).
Compared with placebo, rusfertide provided significant improvement in measures including the maintenance of response, the absence of the need for therapeutic phlebotomy, and persistent HCT control (P < .0001 for all).
Whereas the phlebotomy-free rate with rusfertide during the dose-finding weeks of 1-17 was 76.9% and in weeks 17-29, 87.3%, the rate increased in part 2 of the study to 92.3%.
Additional symptom benefits reported with rusfertide at week 29 versus baseline in part 1 of the study included significant improvements in concentration (P = .0018), itching (P = .0054), fatigue (P = .0074), and inactivity (P = .0005).
In terms of safety, rusfertide was generally well tolerated, with 83% of treatment-emergent adverse events (TEAEs) being grade 1-2, while 17% were grade 3, and none were grade 4 or 5.
The most common TEAEs consisted of injection-site reactions, which were localized, and grade 1-2 in severity. The incidence of reactions decreased with ongoing treatment. There were only two discontinuations resulting from TEAEs.
Among a total of 70 patients who were enrolled, 52 (74.3%) have continued to receive rusfertide for at least 1 year, 32 (45.7%) for at least 1.5 years, and 10 (14.3%) for at least 2 years, indicating the long-term tolerability of rusfertide.
Further commenting, first author Marina Kremyanskaya, MD, PhD, an assistant professor of medicine, hematology, and medical oncology at the Icahn School of Medicine at Mount Sinai, added that a key benefit is rusfertide’s tolerability with combination therapies, which is important in enabling the avoidance of phlebotomies.
“Many patients on cytoreductive therapies still require phlebotomies, and they can’t tolerate a dose increase, either due to cytopenias or other adverse reactions,” she said in an interview. “So adding rusfertide allows for better control of their hematocrits on a lower dose of their respective cytoreductive drug.”
“The combination treatment thus allows for elimination of phlebotomy requirements and potentially improves their symptoms,” Dr. Kremyanskaya said, adding that “using a lower dose of cytoreductive drug such as interferon or hydroxyurea could offer a symptomatic relief to patients as well.”
Overall, she agreed that the responses are remarkably positive.
“I think this is what is so impressive about this agent – basically everybody responds,” Dr. Kremyanskaya said. “When we first started treating patients, we were so impressed, as none of the other drugs we use to treat PV, or any other hematologic malignancy, come anywhere close to this response rate.”
In commenting on the study, Claire Harrison, MD, a professor of myeloproliferative neoplasms and deputy medical director of research at Guy’s and St Thomas’ NHS Foundation Trust in London, agreed that “these data show a strong signal for effectiveness of this therapy in controlling red cell proliferation in PV without inducing iron deficiency and adding to the symptom burden of patients.”
The alternative of phlebotomy “is painful and consumes patient time and hospital resources,” she said in an interview.
Dr. Harrison noted that an earlier signal suggested squamous cell cancer might be of potential concern, but the signal “has not re-emerged [suggesting] this does indeed seem to be a safe and extremely effective therapy.”
Further commenting on the study during the press briefing, Konstanze Döhner, MD, of the University of Ulm (Germany) added that “this is exciting data.”
“For a long time, we had no therapeutic options for PV, and now the field is rapidly developing,” she said.
In ongoing research, rusfertide is currently being studied in the phase 3, placebo-controlled VERIFY randomized trial.
The study was sponsored by Protagonist Therapeutics. Dr. Hoffman reports being on the advisory board for Protagonist Therapeutics, and Dr. Kremyanskaya is a consultant for Protagonist Therapeutics. Dr. Harrison had no disclosures to report.
“The results are surprisingly positive,” said senior author Ronald Hoffman, MD, of the Icahn School of Medicine at Mount Sinai, New York, in discussing the late-breaking research at a press briefing during the European Hematology Association Hybrid Congress 2023.
“Importantly, the study met all of its efficacy endpoints, including the proportion of responders, absence of phlebotomy eligibility, and hematocrit control,” Dr. Hoffman said.
PV, a relatively common clonal myeloproliferative neoplasm, is characterized by uncontrolled erythrocytosis, or excessive production of red blood cells, increasing the risk for serious complications such as thromboembolic and cardiovascular events – the most common causes of morbidity and mortality in this blood cancer.
To treat PV, the maintenance of hematocrit levels at below 45% is critical. However, the current standard of care, therapeutic phlebotomy, with or without cytoreductive agents, falls short in maintaining those lower levels in the majority of patients, Dr. Hoffman explained.
To improve responses, rusfertide was developed as a novel, synthetic form of hepcidin, a peptide hormone that is produced by the liver and functions to maintain iron homeostasis and control the formation of red blood cells.
“This is somewhat of a paradigm shift,” said Dr. Hoffman in the press briefing. “We’re trying to use a hormone made by the liver to control excessive red blood cell production from polycythemia vera.”
For the phase 2 REVIVE study evaluating rusfertide in PV, the authors enrolled 53 patients with PV who had a high phlebotomy burden while receiving the current standard of care. The study’s criteria called for patients to have received at least three therapeutic phlebotomies in the 28 weeks prior to enrollment, with or without concurrent cytoreductive agents.
During a first part of the study, patients received subcutaneous rusfertide once weekly over 28 weeks, during which period the dose was adjusted individually to achieve control of HCT levels below 45%.
The second part was a withdrawal phase extending from weeks 29 to 41, in which patients were randomized in a blinded fashion to either continue on rusfertide (n = 26) or receive a placebo (n = 27).
The patients had a median age of 58; they were 71.7% male, and 54.7% had previously been treated with therapeutic phlebotomy alone while 45.3% received therapeutic phlebotomy plus cytoreductive agents.
Patients were considered to be responders if they met three criteria, including having HCT control without phlebotomy eligibility, no therapeutic phlebotomy, and having completed 12 weeks of treatment.
At the end of the second phase, 69.2% of patients receiving rusfertide were responders versus just 18.5% in the placebo group (P = .0003).
Notably, the improvement with rusfertide was observed among those receiving therapeutic phlebotomy alone, as well as with cytoreductive agents (both P = .02).
Compared with placebo, rusfertide provided significant improvement in measures including the maintenance of response, the absence of the need for therapeutic phlebotomy, and persistent HCT control (P < .0001 for all).
Whereas the phlebotomy-free rate with rusfertide during the dose-finding weeks of 1-17 was 76.9% and in weeks 17-29, 87.3%, the rate increased in part 2 of the study to 92.3%.
Additional symptom benefits reported with rusfertide at week 29 versus baseline in part 1 of the study included significant improvements in concentration (P = .0018), itching (P = .0054), fatigue (P = .0074), and inactivity (P = .0005).
In terms of safety, rusfertide was generally well tolerated, with 83% of treatment-emergent adverse events (TEAEs) being grade 1-2, while 17% were grade 3, and none were grade 4 or 5.
The most common TEAEs consisted of injection-site reactions, which were localized, and grade 1-2 in severity. The incidence of reactions decreased with ongoing treatment. There were only two discontinuations resulting from TEAEs.
Among a total of 70 patients who were enrolled, 52 (74.3%) have continued to receive rusfertide for at least 1 year, 32 (45.7%) for at least 1.5 years, and 10 (14.3%) for at least 2 years, indicating the long-term tolerability of rusfertide.
Further commenting, first author Marina Kremyanskaya, MD, PhD, an assistant professor of medicine, hematology, and medical oncology at the Icahn School of Medicine at Mount Sinai, added that a key benefit is rusfertide’s tolerability with combination therapies, which is important in enabling the avoidance of phlebotomies.
“Many patients on cytoreductive therapies still require phlebotomies, and they can’t tolerate a dose increase, either due to cytopenias or other adverse reactions,” she said in an interview. “So adding rusfertide allows for better control of their hematocrits on a lower dose of their respective cytoreductive drug.”
“The combination treatment thus allows for elimination of phlebotomy requirements and potentially improves their symptoms,” Dr. Kremyanskaya said, adding that “using a lower dose of cytoreductive drug such as interferon or hydroxyurea could offer a symptomatic relief to patients as well.”
Overall, she agreed that the responses are remarkably positive.
“I think this is what is so impressive about this agent – basically everybody responds,” Dr. Kremyanskaya said. “When we first started treating patients, we were so impressed, as none of the other drugs we use to treat PV, or any other hematologic malignancy, come anywhere close to this response rate.”
In commenting on the study, Claire Harrison, MD, a professor of myeloproliferative neoplasms and deputy medical director of research at Guy’s and St Thomas’ NHS Foundation Trust in London, agreed that “these data show a strong signal for effectiveness of this therapy in controlling red cell proliferation in PV without inducing iron deficiency and adding to the symptom burden of patients.”
The alternative of phlebotomy “is painful and consumes patient time and hospital resources,” she said in an interview.
Dr. Harrison noted that an earlier signal suggested squamous cell cancer might be of potential concern, but the signal “has not re-emerged [suggesting] this does indeed seem to be a safe and extremely effective therapy.”
Further commenting on the study during the press briefing, Konstanze Döhner, MD, of the University of Ulm (Germany) added that “this is exciting data.”
“For a long time, we had no therapeutic options for PV, and now the field is rapidly developing,” she said.
In ongoing research, rusfertide is currently being studied in the phase 3, placebo-controlled VERIFY randomized trial.
The study was sponsored by Protagonist Therapeutics. Dr. Hoffman reports being on the advisory board for Protagonist Therapeutics, and Dr. Kremyanskaya is a consultant for Protagonist Therapeutics. Dr. Harrison had no disclosures to report.
“The results are surprisingly positive,” said senior author Ronald Hoffman, MD, of the Icahn School of Medicine at Mount Sinai, New York, in discussing the late-breaking research at a press briefing during the European Hematology Association Hybrid Congress 2023.
“Importantly, the study met all of its efficacy endpoints, including the proportion of responders, absence of phlebotomy eligibility, and hematocrit control,” Dr. Hoffman said.
PV, a relatively common clonal myeloproliferative neoplasm, is characterized by uncontrolled erythrocytosis, or excessive production of red blood cells, increasing the risk for serious complications such as thromboembolic and cardiovascular events – the most common causes of morbidity and mortality in this blood cancer.
To treat PV, the maintenance of hematocrit levels at below 45% is critical. However, the current standard of care, therapeutic phlebotomy, with or without cytoreductive agents, falls short in maintaining those lower levels in the majority of patients, Dr. Hoffman explained.
To improve responses, rusfertide was developed as a novel, synthetic form of hepcidin, a peptide hormone that is produced by the liver and functions to maintain iron homeostasis and control the formation of red blood cells.
“This is somewhat of a paradigm shift,” said Dr. Hoffman in the press briefing. “We’re trying to use a hormone made by the liver to control excessive red blood cell production from polycythemia vera.”
For the phase 2 REVIVE study evaluating rusfertide in PV, the authors enrolled 53 patients with PV who had a high phlebotomy burden while receiving the current standard of care. The study’s criteria called for patients to have received at least three therapeutic phlebotomies in the 28 weeks prior to enrollment, with or without concurrent cytoreductive agents.
During a first part of the study, patients received subcutaneous rusfertide once weekly over 28 weeks, during which period the dose was adjusted individually to achieve control of HCT levels below 45%.
The second part was a withdrawal phase extending from weeks 29 to 41, in which patients were randomized in a blinded fashion to either continue on rusfertide (n = 26) or receive a placebo (n = 27).
The patients had a median age of 58; they were 71.7% male, and 54.7% had previously been treated with therapeutic phlebotomy alone while 45.3% received therapeutic phlebotomy plus cytoreductive agents.
Patients were considered to be responders if they met three criteria, including having HCT control without phlebotomy eligibility, no therapeutic phlebotomy, and having completed 12 weeks of treatment.
At the end of the second phase, 69.2% of patients receiving rusfertide were responders versus just 18.5% in the placebo group (P = .0003).
Notably, the improvement with rusfertide was observed among those receiving therapeutic phlebotomy alone, as well as with cytoreductive agents (both P = .02).
Compared with placebo, rusfertide provided significant improvement in measures including the maintenance of response, the absence of the need for therapeutic phlebotomy, and persistent HCT control (P < .0001 for all).
Whereas the phlebotomy-free rate with rusfertide during the dose-finding weeks of 1-17 was 76.9% and in weeks 17-29, 87.3%, the rate increased in part 2 of the study to 92.3%.
Additional symptom benefits reported with rusfertide at week 29 versus baseline in part 1 of the study included significant improvements in concentration (P = .0018), itching (P = .0054), fatigue (P = .0074), and inactivity (P = .0005).
In terms of safety, rusfertide was generally well tolerated, with 83% of treatment-emergent adverse events (TEAEs) being grade 1-2, while 17% were grade 3, and none were grade 4 or 5.
The most common TEAEs consisted of injection-site reactions, which were localized, and grade 1-2 in severity. The incidence of reactions decreased with ongoing treatment. There were only two discontinuations resulting from TEAEs.
Among a total of 70 patients who were enrolled, 52 (74.3%) have continued to receive rusfertide for at least 1 year, 32 (45.7%) for at least 1.5 years, and 10 (14.3%) for at least 2 years, indicating the long-term tolerability of rusfertide.
Further commenting, first author Marina Kremyanskaya, MD, PhD, an assistant professor of medicine, hematology, and medical oncology at the Icahn School of Medicine at Mount Sinai, added that a key benefit is rusfertide’s tolerability with combination therapies, which is important in enabling the avoidance of phlebotomies.
“Many patients on cytoreductive therapies still require phlebotomies, and they can’t tolerate a dose increase, either due to cytopenias or other adverse reactions,” she said in an interview. “So adding rusfertide allows for better control of their hematocrits on a lower dose of their respective cytoreductive drug.”
“The combination treatment thus allows for elimination of phlebotomy requirements and potentially improves their symptoms,” Dr. Kremyanskaya said, adding that “using a lower dose of cytoreductive drug such as interferon or hydroxyurea could offer a symptomatic relief to patients as well.”
Overall, she agreed that the responses are remarkably positive.
“I think this is what is so impressive about this agent – basically everybody responds,” Dr. Kremyanskaya said. “When we first started treating patients, we were so impressed, as none of the other drugs we use to treat PV, or any other hematologic malignancy, come anywhere close to this response rate.”
In commenting on the study, Claire Harrison, MD, a professor of myeloproliferative neoplasms and deputy medical director of research at Guy’s and St Thomas’ NHS Foundation Trust in London, agreed that “these data show a strong signal for effectiveness of this therapy in controlling red cell proliferation in PV without inducing iron deficiency and adding to the symptom burden of patients.”
The alternative of phlebotomy “is painful and consumes patient time and hospital resources,” she said in an interview.
Dr. Harrison noted that an earlier signal suggested squamous cell cancer might be of potential concern, but the signal “has not re-emerged [suggesting] this does indeed seem to be a safe and extremely effective therapy.”
Further commenting on the study during the press briefing, Konstanze Döhner, MD, of the University of Ulm (Germany) added that “this is exciting data.”
“For a long time, we had no therapeutic options for PV, and now the field is rapidly developing,” she said.
In ongoing research, rusfertide is currently being studied in the phase 3, placebo-controlled VERIFY randomized trial.
The study was sponsored by Protagonist Therapeutics. Dr. Hoffman reports being on the advisory board for Protagonist Therapeutics, and Dr. Kremyanskaya is a consultant for Protagonist Therapeutics. Dr. Harrison had no disclosures to report.
FROM EHA 2023
Focus of new ASH VTE guidelines: Thrombophilia testing
according to new clinical practice guidelines released by the American Society of Hematology. Individuals with a family history of VTE and high-risk thrombophilia, and those with VTE at unusual body sites should also be tested, the guidelines panel agreed.
“These guidelines will potentially change practice – we know that providers and patients will make a shared treatment decision and we wanted to outline specific scenarios to guide that decision,” panel cochair and first author Saskia Middeldorp, MD, PhD, explained in a press release announcing the publication of the guidelines in Blood Advances.
Dr. Middeldorp is a professor of medicine and head of the department of internal medicine at Radboud University Medical Center, Nijmegen, the Netherlands.
The guidelines are the latest in an ASH series of VTE-related guidelines. ASH convened a multidisciplinary panel with clinical and methodological expertise to develop the guidelines, which were subject to public comment, and they “provide recommendations informed by case-based approaches and modeling to ensure the medical community can better diagnose and treat thrombophilia and people with the condition can make the best decisions for their care,” the press release explains.
Thrombophilia affects an estimated 10% of the population. Testing for the clotting disorder can be costly, and the use of testing to help guide treatment decisions is controversial.
“For decades there has been dispute about thrombophilia testing,” Dr. Middeldorp said. “We created a model about whether and when it would be useful to test for thrombophilia, and based on the model, we suggest it can be appropriate in [the specified] situations.
The panel agreed on 23 recommendations regarding thrombophilia testing and management. Most are based on “very low certainty” in the evidence because of modeling assumptions.
However, the panel agreed on a strong recommendation against testing the general population before starting combined oral contraceptives (COC), and a conditional recommendation for thrombophilia testing in:
- Patients with VTE associated with nonsurgical major transient or hormonal risk factors
- Patients with cerebral or splanchnic venous thrombosis in settings where anticoagulation would otherwise be discontinued
- Individuals with a family history of antithrombin, protein C, or protein S deficiency when considering thromboprophylaxis for minor provoking risk factors and for guidance related to the use of COC or hormone therapy
- Pregnant women with a family history of high-risk thrombophilia types
- Patients with cancer at low or intermediate risk of thrombosis and with a family history of VTE
“In all other instances, we suggest not testing for thrombophilia,” said Dr. Middeldorp.
The ASH guidelines largely mirror those of existing guidelines from a number of other organizations, but the recommendation in favor of testing for thrombophilia in patients with VTE provoked by a nonsurgical major transient risk factor or associated with COCs, hormone therapy, pregnancy or postpartum is new and “may cause considerable discussion, as many currently view these VTE episodes as provoked and are generally inclined to use short-term anticoagulation for such patients,” the guideline authors wrote.
“It is important to note, however, that most guidelines or guidance statements on thrombophilia testing did not distinguish between major and minor provoking risk factors, which current science suggests is appropriate,” they added.
Another novel recommendation is the suggestion to test for hereditary thrombophilia to guide the use of thromboprophylaxis during systemic treatment in ambulatory patients with cancer who are at low or intermediate risk for VTE and who have a family history of VTE.
“This new recommendation should be seen as a new application of an established risk stratification approach,” they said.
Additional research is urgently needed, particularly “large implementation studies comparing the impact, in terms of outcomes rates, among management strategies involving or not involving thrombophilia testing,” they noted.
The guideline was wholly funded by ASH. Dr. Middeldorp reported having no conflicts of interest.
according to new clinical practice guidelines released by the American Society of Hematology. Individuals with a family history of VTE and high-risk thrombophilia, and those with VTE at unusual body sites should also be tested, the guidelines panel agreed.
“These guidelines will potentially change practice – we know that providers and patients will make a shared treatment decision and we wanted to outline specific scenarios to guide that decision,” panel cochair and first author Saskia Middeldorp, MD, PhD, explained in a press release announcing the publication of the guidelines in Blood Advances.
Dr. Middeldorp is a professor of medicine and head of the department of internal medicine at Radboud University Medical Center, Nijmegen, the Netherlands.
The guidelines are the latest in an ASH series of VTE-related guidelines. ASH convened a multidisciplinary panel with clinical and methodological expertise to develop the guidelines, which were subject to public comment, and they “provide recommendations informed by case-based approaches and modeling to ensure the medical community can better diagnose and treat thrombophilia and people with the condition can make the best decisions for their care,” the press release explains.
Thrombophilia affects an estimated 10% of the population. Testing for the clotting disorder can be costly, and the use of testing to help guide treatment decisions is controversial.
“For decades there has been dispute about thrombophilia testing,” Dr. Middeldorp said. “We created a model about whether and when it would be useful to test for thrombophilia, and based on the model, we suggest it can be appropriate in [the specified] situations.
The panel agreed on 23 recommendations regarding thrombophilia testing and management. Most are based on “very low certainty” in the evidence because of modeling assumptions.
However, the panel agreed on a strong recommendation against testing the general population before starting combined oral contraceptives (COC), and a conditional recommendation for thrombophilia testing in:
- Patients with VTE associated with nonsurgical major transient or hormonal risk factors
- Patients with cerebral or splanchnic venous thrombosis in settings where anticoagulation would otherwise be discontinued
- Individuals with a family history of antithrombin, protein C, or protein S deficiency when considering thromboprophylaxis for minor provoking risk factors and for guidance related to the use of COC or hormone therapy
- Pregnant women with a family history of high-risk thrombophilia types
- Patients with cancer at low or intermediate risk of thrombosis and with a family history of VTE
“In all other instances, we suggest not testing for thrombophilia,” said Dr. Middeldorp.
The ASH guidelines largely mirror those of existing guidelines from a number of other organizations, but the recommendation in favor of testing for thrombophilia in patients with VTE provoked by a nonsurgical major transient risk factor or associated with COCs, hormone therapy, pregnancy or postpartum is new and “may cause considerable discussion, as many currently view these VTE episodes as provoked and are generally inclined to use short-term anticoagulation for such patients,” the guideline authors wrote.
“It is important to note, however, that most guidelines or guidance statements on thrombophilia testing did not distinguish between major and minor provoking risk factors, which current science suggests is appropriate,” they added.
Another novel recommendation is the suggestion to test for hereditary thrombophilia to guide the use of thromboprophylaxis during systemic treatment in ambulatory patients with cancer who are at low or intermediate risk for VTE and who have a family history of VTE.
“This new recommendation should be seen as a new application of an established risk stratification approach,” they said.
Additional research is urgently needed, particularly “large implementation studies comparing the impact, in terms of outcomes rates, among management strategies involving or not involving thrombophilia testing,” they noted.
The guideline was wholly funded by ASH. Dr. Middeldorp reported having no conflicts of interest.
according to new clinical practice guidelines released by the American Society of Hematology. Individuals with a family history of VTE and high-risk thrombophilia, and those with VTE at unusual body sites should also be tested, the guidelines panel agreed.
“These guidelines will potentially change practice – we know that providers and patients will make a shared treatment decision and we wanted to outline specific scenarios to guide that decision,” panel cochair and first author Saskia Middeldorp, MD, PhD, explained in a press release announcing the publication of the guidelines in Blood Advances.
Dr. Middeldorp is a professor of medicine and head of the department of internal medicine at Radboud University Medical Center, Nijmegen, the Netherlands.
The guidelines are the latest in an ASH series of VTE-related guidelines. ASH convened a multidisciplinary panel with clinical and methodological expertise to develop the guidelines, which were subject to public comment, and they “provide recommendations informed by case-based approaches and modeling to ensure the medical community can better diagnose and treat thrombophilia and people with the condition can make the best decisions for their care,” the press release explains.
Thrombophilia affects an estimated 10% of the population. Testing for the clotting disorder can be costly, and the use of testing to help guide treatment decisions is controversial.
“For decades there has been dispute about thrombophilia testing,” Dr. Middeldorp said. “We created a model about whether and when it would be useful to test for thrombophilia, and based on the model, we suggest it can be appropriate in [the specified] situations.
The panel agreed on 23 recommendations regarding thrombophilia testing and management. Most are based on “very low certainty” in the evidence because of modeling assumptions.
However, the panel agreed on a strong recommendation against testing the general population before starting combined oral contraceptives (COC), and a conditional recommendation for thrombophilia testing in:
- Patients with VTE associated with nonsurgical major transient or hormonal risk factors
- Patients with cerebral or splanchnic venous thrombosis in settings where anticoagulation would otherwise be discontinued
- Individuals with a family history of antithrombin, protein C, or protein S deficiency when considering thromboprophylaxis for minor provoking risk factors and for guidance related to the use of COC or hormone therapy
- Pregnant women with a family history of high-risk thrombophilia types
- Patients with cancer at low or intermediate risk of thrombosis and with a family history of VTE
“In all other instances, we suggest not testing for thrombophilia,” said Dr. Middeldorp.
The ASH guidelines largely mirror those of existing guidelines from a number of other organizations, but the recommendation in favor of testing for thrombophilia in patients with VTE provoked by a nonsurgical major transient risk factor or associated with COCs, hormone therapy, pregnancy or postpartum is new and “may cause considerable discussion, as many currently view these VTE episodes as provoked and are generally inclined to use short-term anticoagulation for such patients,” the guideline authors wrote.
“It is important to note, however, that most guidelines or guidance statements on thrombophilia testing did not distinguish between major and minor provoking risk factors, which current science suggests is appropriate,” they added.
Another novel recommendation is the suggestion to test for hereditary thrombophilia to guide the use of thromboprophylaxis during systemic treatment in ambulatory patients with cancer who are at low or intermediate risk for VTE and who have a family history of VTE.
“This new recommendation should be seen as a new application of an established risk stratification approach,” they said.
Additional research is urgently needed, particularly “large implementation studies comparing the impact, in terms of outcomes rates, among management strategies involving or not involving thrombophilia testing,” they noted.
The guideline was wholly funded by ASH. Dr. Middeldorp reported having no conflicts of interest.
FROM BLOOD ADVANCES
New update on left atrial appendage closure recommendations
An updated consensus statement on transcatheter left atrial appendage closure (LAAC) has put a newfound focus on patient selection for the procedure, specifically recommending that the procedure is appropriate for patients with nonvalvular atrial fibrillation who have risk for thromboembolism, aren’t well suited for direct oral anticoagulants (DOACs) and have a good chance of living for at least another year.
The statement, published online in the Journal of the Society for Cardiovascular Angiography & Interventions, also makes recommendations for how much experience operators should have, how many procedures they should perform to keep their skills up, and when and how to use imaging and prescribe DOACs, among other suggestions.
The statement represents the first updated guidance for LAAC since 2015. “Since then this field has really expanded and evolved,” writing group chair Jacqueline Saw, MD, said in an interview. “For instance, the indications are more matured and specific, and the procedural technical steps have matured. Imaging has also advanced, there’s more understanding about postprocedural care and there are also new devices that have been approved.”

Dr. Saw, an interventional cardiologist at Vancouver General Hospital and St. Paul’s Hospital, and a professor at the University of British Columbia in Vancouver, called the statement “a piece that puts everything together.”
“This document really summarizes the whole practice for doing transcatheter procedures,” she added, “so it’s all-in-one document in terms of recommendation of who we do the procedure for, how we should do it, how we should image and guide the procedure, and what complications to look out for and how to manage patients post procedure, be it with antithrombotic therapy and/or device surveillance.”
13 recommendations
In all, the statement carries 13 recommendations for LAAC. The Society for Cardiovascular Angiography & Interventions and the Heart Rhythm Society commissioned the writing group. The American College of Cardiology and Society of Cardiovascular Computed Tomography have endorsed the statement. The following are among the recommendations:
- Transcatheter LAAC is appropriate for patients with nonvalvular atrial fibrillation with high thromboembolic risk but for whom long-term oral anticoagulation may be contraindicated and who have at least 1 year’s life expectancy.
- Operators should have performed at least 50 prior left-sided ablations or structural procedures and at least 25 transseptal punctures (TSPs). Interventional-imaging physicians should have experience in guiding 25 or more TSPs before supporting LAAC procedures independently.
- To maintain skills, operators should do 25 or more TSPs and at least 12 LAACs over each 2-year period.
- On-site cardiovascular surgery backup should be available for new programs and for operators early in their learning curve.
- Baseline imaging with transesophageal echocardiography (TEE) or cardiac computed tomography should be performed before LAAC.
- Intraprocedural imaging guidance with TEE or intracardiac echocardiography.
- Follow labeling of each specific LAAC device for technical aspects of the procedure.
- Familiarity with avoiding, recognizing, and managing LAAC complications.
- Predischarge 2-dimensional TEE to rule out pericardial effusion and device embolization.
- Anticoagulation for device-related thrombus.
- Make all efforts to minimize peridevice leaks during implantation because their clinical impact and management isn’t well understood.
- Antithrombotic therapy with warfarin, DOAC, or dual-antiplatelet therapy after LAAC based on the studied regimen and instructions for each specific device, tailored to the bleeding risks for each patient.
- TEE or cardiac computed tomography at 45-90 days after LAAC for device surveillance to assess for peridevice leak and device-related thrombus.
The statement also includes precautionary recommendations. It advises against using routine closure of LAAC-associated iatrogenic atrial septal defects and states that combined procedures with LAAC, such as structural interventions and pulmonary vein isolation, should be avoided because randomized controlled trial data are pending.
“These recommendations are based upon data from updated publications and randomized trial data as well as large registries, including the National Cardiovascular Data Registry, so I think this is a very practical statement that puts all these pieces together for any budding interventionalist doing this procedure and even experienced operations,” Dr. Saw said.
Authors of an accompanying editorial agreed that the “rigorous standards” set out in the statement will help maintain “a high level of procedural safety in the setting of rapid expansion.”
The editorialists, Faisal M. Merchant, MD, of Emory University, Atlanta, and Mohamad Alkhouli, MD, professor of medicine at Mayo Clinic School of Medicine, Rochester, Minn., point out that the incidence of pericardial effusion has decreased from more than 5% in the pivotal Watchman trials to less than 1.5% in the most recent report from the National Cardiovascular Data Registry, which shows that more than 100,000 procedures have been performed in the United States.
But most important as the field moves forward, they stress, is patient selection. The recommendation of limiting patients to those with a life expectancy of 1 year “is a tacit recognition of the fact that the benefits of LAAC take time to accrue, and many older and frail patients are unlikely to derive meaningful benefit.”
Dr. Merchant and Dr. Alkhouli also note that there remains a conundrum in patient selection that remains from the original LAAC trials, which enrolled patients who were eligible for anticoagulation. “Somewhat paradoxically, after its approval, LAAC is mostly prescribed to patients who are not felt to be good anticoagulation candidates.” This leaves physicians “in the precarious position of extrapolating data to patients who were excluded from the original clinical trials.”
Therefore, the consensus statement “is right to put patient selection front and center in its recommendations, but as the field of LAAC comes of age, better evidence to support patient selection will be the real sign of maturity.”
Dr. Saw said she envisions another update over the next 2 years or so as ongoing clinical trials comparing DOAC and LAAC, namely the CHAMPION-AF and OPTION trials, report results.
Dr. Saw and Dr. Merchant, reported no conflicts of interest. Dr. Alkhouli has financial ties to Boston Scientific, Abbott, and Philips.
An updated consensus statement on transcatheter left atrial appendage closure (LAAC) has put a newfound focus on patient selection for the procedure, specifically recommending that the procedure is appropriate for patients with nonvalvular atrial fibrillation who have risk for thromboembolism, aren’t well suited for direct oral anticoagulants (DOACs) and have a good chance of living for at least another year.
The statement, published online in the Journal of the Society for Cardiovascular Angiography & Interventions, also makes recommendations for how much experience operators should have, how many procedures they should perform to keep their skills up, and when and how to use imaging and prescribe DOACs, among other suggestions.
The statement represents the first updated guidance for LAAC since 2015. “Since then this field has really expanded and evolved,” writing group chair Jacqueline Saw, MD, said in an interview. “For instance, the indications are more matured and specific, and the procedural technical steps have matured. Imaging has also advanced, there’s more understanding about postprocedural care and there are also new devices that have been approved.”
Dr. Saw, an interventional cardiologist at Vancouver General Hospital and St. Paul’s Hospital, and a professor at the University of British Columbia in Vancouver, called the statement “a piece that puts everything together.”
“This document really summarizes the whole practice for doing transcatheter procedures,” she added, “so it’s all-in-one document in terms of recommendation of who we do the procedure for, how we should do it, how we should image and guide the procedure, and what complications to look out for and how to manage patients post procedure, be it with antithrombotic therapy and/or device surveillance.”
13 recommendations
In all, the statement carries 13 recommendations for LAAC. The Society for Cardiovascular Angiography & Interventions and the Heart Rhythm Society commissioned the writing group. The American College of Cardiology and Society of Cardiovascular Computed Tomography have endorsed the statement. The following are among the recommendations:
- Transcatheter LAAC is appropriate for patients with nonvalvular atrial fibrillation with high thromboembolic risk but for whom long-term oral anticoagulation may be contraindicated and who have at least 1 year’s life expectancy.
- Operators should have performed at least 50 prior left-sided ablations or structural procedures and at least 25 transseptal punctures (TSPs). Interventional-imaging physicians should have experience in guiding 25 or more TSPs before supporting LAAC procedures independently.
- To maintain skills, operators should do 25 or more TSPs and at least 12 LAACs over each 2-year period.
- On-site cardiovascular surgery backup should be available for new programs and for operators early in their learning curve.
- Baseline imaging with transesophageal echocardiography (TEE) or cardiac computed tomography should be performed before LAAC.
- Intraprocedural imaging guidance with TEE or intracardiac echocardiography.
- Follow labeling of each specific LAAC device for technical aspects of the procedure.
- Familiarity with avoiding, recognizing, and managing LAAC complications.
- Predischarge 2-dimensional TEE to rule out pericardial effusion and device embolization.
- Anticoagulation for device-related thrombus.
- Make all efforts to minimize peridevice leaks during implantation because their clinical impact and management isn’t well understood.
- Antithrombotic therapy with warfarin, DOAC, or dual-antiplatelet therapy after LAAC based on the studied regimen and instructions for each specific device, tailored to the bleeding risks for each patient.
- TEE or cardiac computed tomography at 45-90 days after LAAC for device surveillance to assess for peridevice leak and device-related thrombus.
The statement also includes precautionary recommendations. It advises against using routine closure of LAAC-associated iatrogenic atrial septal defects and states that combined procedures with LAAC, such as structural interventions and pulmonary vein isolation, should be avoided because randomized controlled trial data are pending.
“These recommendations are based upon data from updated publications and randomized trial data as well as large registries, including the National Cardiovascular Data Registry, so I think this is a very practical statement that puts all these pieces together for any budding interventionalist doing this procedure and even experienced operations,” Dr. Saw said.
Authors of an accompanying editorial agreed that the “rigorous standards” set out in the statement will help maintain “a high level of procedural safety in the setting of rapid expansion.”
The editorialists, Faisal M. Merchant, MD, of Emory University, Atlanta, and Mohamad Alkhouli, MD, professor of medicine at Mayo Clinic School of Medicine, Rochester, Minn., point out that the incidence of pericardial effusion has decreased from more than 5% in the pivotal Watchman trials to less than 1.5% in the most recent report from the National Cardiovascular Data Registry, which shows that more than 100,000 procedures have been performed in the United States.
But most important as the field moves forward, they stress, is patient selection. The recommendation of limiting patients to those with a life expectancy of 1 year “is a tacit recognition of the fact that the benefits of LAAC take time to accrue, and many older and frail patients are unlikely to derive meaningful benefit.”
Dr. Merchant and Dr. Alkhouli also note that there remains a conundrum in patient selection that remains from the original LAAC trials, which enrolled patients who were eligible for anticoagulation. “Somewhat paradoxically, after its approval, LAAC is mostly prescribed to patients who are not felt to be good anticoagulation candidates.” This leaves physicians “in the precarious position of extrapolating data to patients who were excluded from the original clinical trials.”
Therefore, the consensus statement “is right to put patient selection front and center in its recommendations, but as the field of LAAC comes of age, better evidence to support patient selection will be the real sign of maturity.”
Dr. Saw said she envisions another update over the next 2 years or so as ongoing clinical trials comparing DOAC and LAAC, namely the CHAMPION-AF and OPTION trials, report results.
Dr. Saw and Dr. Merchant, reported no conflicts of interest. Dr. Alkhouli has financial ties to Boston Scientific, Abbott, and Philips.
An updated consensus statement on transcatheter left atrial appendage closure (LAAC) has put a newfound focus on patient selection for the procedure, specifically recommending that the procedure is appropriate for patients with nonvalvular atrial fibrillation who have risk for thromboembolism, aren’t well suited for direct oral anticoagulants (DOACs) and have a good chance of living for at least another year.
The statement, published online in the Journal of the Society for Cardiovascular Angiography & Interventions, also makes recommendations for how much experience operators should have, how many procedures they should perform to keep their skills up, and when and how to use imaging and prescribe DOACs, among other suggestions.
The statement represents the first updated guidance for LAAC since 2015. “Since then this field has really expanded and evolved,” writing group chair Jacqueline Saw, MD, said in an interview. “For instance, the indications are more matured and specific, and the procedural technical steps have matured. Imaging has also advanced, there’s more understanding about postprocedural care and there are also new devices that have been approved.”
Dr. Saw, an interventional cardiologist at Vancouver General Hospital and St. Paul’s Hospital, and a professor at the University of British Columbia in Vancouver, called the statement “a piece that puts everything together.”
“This document really summarizes the whole practice for doing transcatheter procedures,” she added, “so it’s all-in-one document in terms of recommendation of who we do the procedure for, how we should do it, how we should image and guide the procedure, and what complications to look out for and how to manage patients post procedure, be it with antithrombotic therapy and/or device surveillance.”
13 recommendations
In all, the statement carries 13 recommendations for LAAC. The Society for Cardiovascular Angiography & Interventions and the Heart Rhythm Society commissioned the writing group. The American College of Cardiology and Society of Cardiovascular Computed Tomography have endorsed the statement. The following are among the recommendations:
- Transcatheter LAAC is appropriate for patients with nonvalvular atrial fibrillation with high thromboembolic risk but for whom long-term oral anticoagulation may be contraindicated and who have at least 1 year’s life expectancy.
- Operators should have performed at least 50 prior left-sided ablations or structural procedures and at least 25 transseptal punctures (TSPs). Interventional-imaging physicians should have experience in guiding 25 or more TSPs before supporting LAAC procedures independently.
- To maintain skills, operators should do 25 or more TSPs and at least 12 LAACs over each 2-year period.
- On-site cardiovascular surgery backup should be available for new programs and for operators early in their learning curve.
- Baseline imaging with transesophageal echocardiography (TEE) or cardiac computed tomography should be performed before LAAC.
- Intraprocedural imaging guidance with TEE or intracardiac echocardiography.
- Follow labeling of each specific LAAC device for technical aspects of the procedure.
- Familiarity with avoiding, recognizing, and managing LAAC complications.
- Predischarge 2-dimensional TEE to rule out pericardial effusion and device embolization.
- Anticoagulation for device-related thrombus.
- Make all efforts to minimize peridevice leaks during implantation because their clinical impact and management isn’t well understood.
- Antithrombotic therapy with warfarin, DOAC, or dual-antiplatelet therapy after LAAC based on the studied regimen and instructions for each specific device, tailored to the bleeding risks for each patient.
- TEE or cardiac computed tomography at 45-90 days after LAAC for device surveillance to assess for peridevice leak and device-related thrombus.
The statement also includes precautionary recommendations. It advises against using routine closure of LAAC-associated iatrogenic atrial septal defects and states that combined procedures with LAAC, such as structural interventions and pulmonary vein isolation, should be avoided because randomized controlled trial data are pending.
“These recommendations are based upon data from updated publications and randomized trial data as well as large registries, including the National Cardiovascular Data Registry, so I think this is a very practical statement that puts all these pieces together for any budding interventionalist doing this procedure and even experienced operations,” Dr. Saw said.
Authors of an accompanying editorial agreed that the “rigorous standards” set out in the statement will help maintain “a high level of procedural safety in the setting of rapid expansion.”
The editorialists, Faisal M. Merchant, MD, of Emory University, Atlanta, and Mohamad Alkhouli, MD, professor of medicine at Mayo Clinic School of Medicine, Rochester, Minn., point out that the incidence of pericardial effusion has decreased from more than 5% in the pivotal Watchman trials to less than 1.5% in the most recent report from the National Cardiovascular Data Registry, which shows that more than 100,000 procedures have been performed in the United States.
But most important as the field moves forward, they stress, is patient selection. The recommendation of limiting patients to those with a life expectancy of 1 year “is a tacit recognition of the fact that the benefits of LAAC take time to accrue, and many older and frail patients are unlikely to derive meaningful benefit.”
Dr. Merchant and Dr. Alkhouli also note that there remains a conundrum in patient selection that remains from the original LAAC trials, which enrolled patients who were eligible for anticoagulation. “Somewhat paradoxically, after its approval, LAAC is mostly prescribed to patients who are not felt to be good anticoagulation candidates.” This leaves physicians “in the precarious position of extrapolating data to patients who were excluded from the original clinical trials.”
Therefore, the consensus statement “is right to put patient selection front and center in its recommendations, but as the field of LAAC comes of age, better evidence to support patient selection will be the real sign of maturity.”
Dr. Saw said she envisions another update over the next 2 years or so as ongoing clinical trials comparing DOAC and LAAC, namely the CHAMPION-AF and OPTION trials, report results.
Dr. Saw and Dr. Merchant, reported no conflicts of interest. Dr. Alkhouli has financial ties to Boston Scientific, Abbott, and Philips.
FROM THE JOURNAL OF THE SOCIETY FOR CARDIOVASCULAR ANGIOGRAPHY & INTERVENTIONS