User login
Management of Trauma and Burn Scars: The Dermatologist’s Role in Expanding Patient Access to Care
Hypertrophic scarring secondary to trauma, burns, and surgical interventions is a major source of morbidity worldwide and often is mechanically, aesthetically, and symptomatically debilitating. Modern advances in acute trauma care protocols have resulted in survival rates greater than 90% in both civilian and military populations.1,2 Patients with wounds that have historically proven fatal are now surviving and are confronted with the long-term sequelae of their injuries. With more than 52,000 service members injured in military engagements from 2001 to 2015 and 8.5 million civilians presenting annually with injury patterns at risk for hypertrophic scarring, it is paramount that we ensure access to safe and effective long-term scar care.2,3
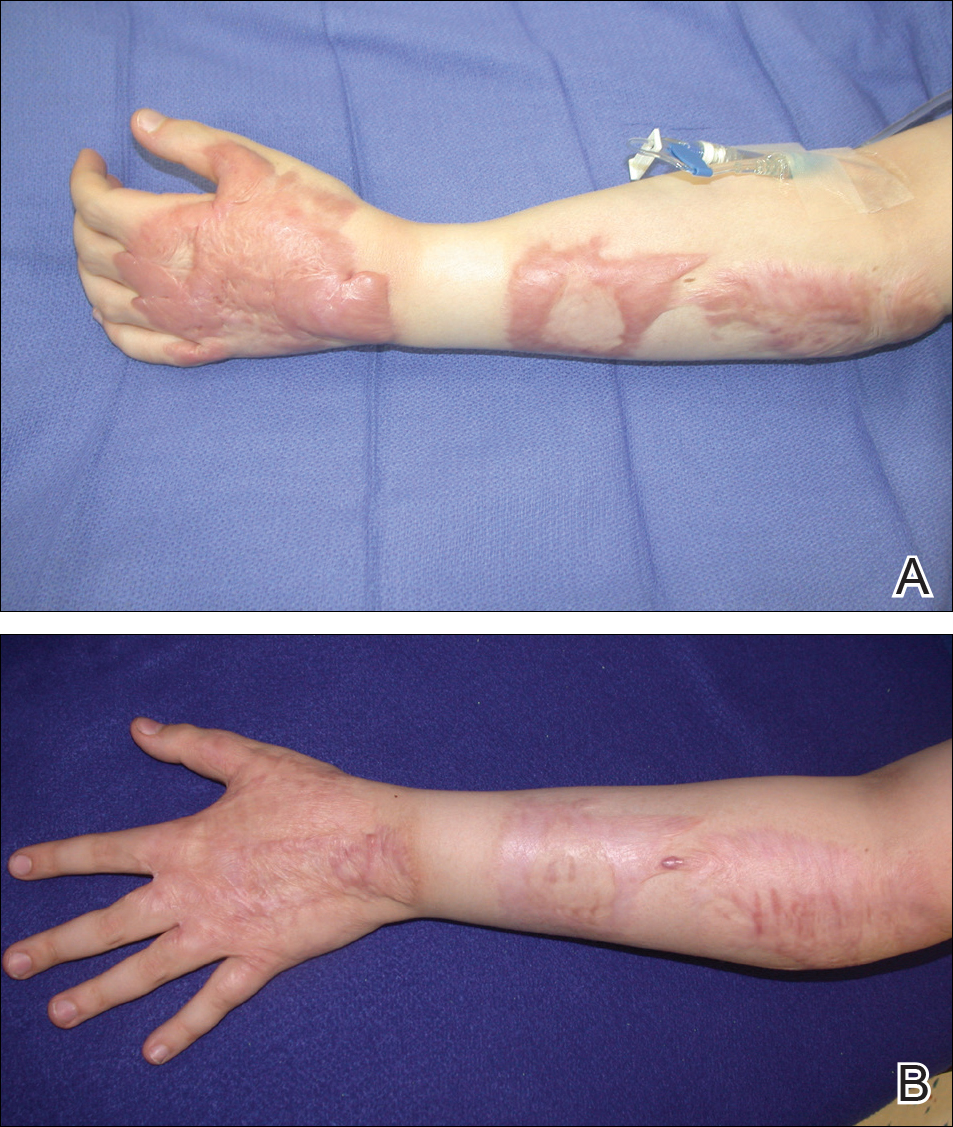
At its simplest level, hypertrophic scarring is believed to result from a disequilibrium between collagen production and degradation. This failure to properly transition through the stages of wound healing results in bothersome symptoms, a disfigured appearance, and mechanical dysfunction of the skin (Figure, A). Decreased elasticity and extensibility, increased dermal thickness, and scar contractures impair patient range of motion and functional mobility. Those affected commonly experience varying degrees of pruritus and dysesthesia along the scar. Combined with aesthetic variations in pigmentation, erythema, texture, and thickness, hypertrophic scarring often leads to long-term psychosocial impairment and decreased health-related quality of life.4
Treatment Approach
Treatment of hypertrophic scars requires a multimodal approach due to the spectrum of associated concerns and the natural recalcitrance of the scar to therapy. Protocols should be tailored to the individual but generally begin with tissue-conserving surgical interventions followed by selective photothermolysis of the scar vasculature. Subsequently, deep and superficial ablative fractional laser (AFL) treatment and local pharmacotherapy also are employed. Treatment can be accomplished in the outpatient setting under local anesthesia in a serial fashion. In the authors’ experience, these therapies behave in a synergistic fashion, achieving outcomes that far exceed the sum of their parts, often obviating the need for scar excision in the majority of cases (Figure, B).
Tissue-Conserving Surgical Intervention
Z-plasty is an indispensable surgical tool due to its ability to lengthen scars and reduce wound tension. Treatment is easily customizable to the patient and can be performed using the individual or multiple Z-plasty techniques. Undermining and step-off correction while suturing allow the physician to lower raised scars, elevate depressed scars, and obscure scar presence by minimizing the straight lines that draw the eye to the scar. Z-plasties rely on the creation and transposition of 2 triangular flaps and permit a 75% increase in length along the desired tension vector. As such, Z-plasties decrease wound tension and facilitate scar maturation.
Selective Photothermolysis of the Vasculature
Although there are several devices available to treat vascular and immature hypertrophic scars, the majority of studies have been conducted with the 595-nm pulsed dye laser. By preferentially heating oxyhemoglobin within the dermal microvasculature, the pulsed dye laser irreparably injures the vascular endothelium. The subsequent tissue hypoxia and collagen fiber heating results in collagen fiber realignment, normalization of collagen subtypes, and neocollagenesis.5 Pulsed dye laser therapy most effectively reduces erythema and pruritus; however, improvements in scar volume, pliability, and elasticity also have been reported.5 When targeting the fine vasculature of the scar, thermal confinement is critical to prevent injury to the surrounding dermis. As such, pulse widths of 0.45 to 1.5 milliseconds are routinely utilized with a fluence just sufficient to elicit transient purpura lasting 3 to 5 seconds. Employing a spot size of 7 to 10 mm, typical fluences range from 4.5 to 6.5 J/cm2. Engagement of the dynamic cooling device reduces the risk for complications, allowing the patient to proceed to the next step in their therapy regimen: the AFL.
Ablative Fractional Laser
The AFL creates a pixilated pattern of injury throughout the epidermis and dermis of the treatment area. Ablative fractional laser platforms include the 10,600-nm CO2 and 2940-nm erbium-doped YAG lasers, both targeting intracellular water. The AFL vaporizes columns of tissue, leaving minute vertical channels with narrow rims of protein coagulation referred to as microscopic treatment zones (MTZs).6 Scar collagen analysis after AFL treatment has shown a profile resembling unaffected skin.7 Consistently, patients report improvements in stiffness, range of motion, pain, pruritus, pigmentation, and erythema.Physician observers also have reported similar improvements in these end points.8,9 Recently, interim data from a prospective controlled trial were presented showing objective improvements in dermal thickness, elasticity, and extensibility after 3 treatments with the CO2 AFL.6 The UltraPulse CO2 laser (Lumenis) is the most well-studied and widely available AFL for scar therapy and as such we will outline common treatment parameters with this device. Of note, treatment end points may be generalized to any AFL.
The DeepFX UltraPulse configuration is utilized to achieve deep AFL therapy and has a fixed pulse width of 0.8 milliseconds, slightly less than the thermal relaxation time of the skin. The diameter of the MTZs is 120 µm, and MTZ density for scar treatment ranges from 1% to 10% with a goal depth of at least 80% of scar thickness. Maximal penetration of the AFL is 4 mm, which is directly proportional to fluence. The goal of deep AFL is the removal of scar tissue to facilitate remodeling and neocollagenesis. Superficial fractional ablation can then be achieved utilizing the ActiveFX UltraPulse configuration generating a 1.3-mm MTZ spot size. We commonly use a treatment level of 3 (82% density). Typical treatment energy ranges from 80 to 125 mJ, which correlates with depths of approximately 50 to 115 µm. With both configurations, the size and shape of the treatment area can be customized to the scar. In addition, frequency may be adjusted to control the speed of treatment while balancing the risk of bulk heating. The goal of superficial AFL is to minimize scar surface irregularities and ensure blending of deep AFL treatment. Once AFL treatment is complete, local pharmacotherapy can then be employed.
Pharmacotherapy
Intralesional corticosteroids have long represented the standard of care for hypertrophic scars, with concentrations between 2.5 and 40 mg/mL that are titrated to scar thickness and location to avoid unwanted atrophy. Visual blanching of the scar represents the clinical end point for treatment. Corticosteroids act by inhibiting fibroblast proliferation and enhancing collagen degradation.10 5-Fluorouracil (5-FU) also is used in scar management. In addition to inhibiting fibroblast proliferation and inducing fibroblast apoptosis, 5-FU inhibits myofibroblast proliferation, which is helpful in the prevention and treatment of scar contracture.11 As monotherapy, weekly injections with 1 to 3 mL of 50 mg/mL 5-FU has been safe and effective. Combination intralesional corticosteroid and 5-FU therapy has been reported and is associated with improved scar regression, reduced reoccurrence, and fewer side effects.11 In our experience, a 1:1 suspension is effective with appropriate titration of the corticosteroid component. Although less well defined, topical application of pharmacotherapy and massage to the newly created MTZs appears beneficial and offers another option for delivery of corticosteroids and 5-FU, in addition to a number of promising medications such as bimatoprost, poly-L-lactic acid, timolol, and rapamycin.12
Conclusion
Advances in laser surgery and our understanding of wound healing have created a paradigm shift in the treatment approach to trauma and burn scars. In lieu of extensive scar excisions, the summarized multimodal regimen emphasizing tissue conservation and autologous remodeling is gaining favor in the military, academic medical centers, and scar centers of excellence, but patients are finding local access to care difficult. Dermatologists are uniquely positioned to cost-effectively deliver this care in the outpatient setting utilizing devices and techniques they already possess. With the end goal of optimization of functional, symptomatic, and aesthetic state of the patient, it is critical that dermatologists seize this opportunity to truly make a difference for the military and civilian patients that need it most.
- American Burn Association, National Burn Repository. 2015 National burn repository report of data from 2005-2014. http://www.ameriburn.org/2015NBRAnnualReport.pdf. Accessed May 10, 2017.
- Centers for Disease Control and Prevention. 2013 National hospital ambulatory medical care survey emergency department summary tables. https://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf. Accessed May 10, 2017.
- Fischer H. A guide to U.S. Military casualty statistics: Operation Freedom’s Sentinel, Operation Inherent Resolve, Operation New Dawn, Operation Iraqi Freedom, and Operation Enduring Freedom. Congressional Research Service website. https://fas.org/sgp/crs/natsec/RS22452.pdf. Published August 7, 2015. Accessed May 10, 2017.
- Van Loey NE, Van Son MJ. Psychopathology and psychological problems in patients with burn scars: epidemiology and management. Am J Clin Dermatol. 2003;4:245-272.
- Vrijman C, van Drooge AM, Limpens J, et al. Laser and intense pulsed light therapy for the treatment of hypertrophic scars: a systematic review. Br J Dermatol. 2011;165:934-942.
- Miletta N, Lee K, Siwy K, et al. Objective improvement in burn scars after treatment with fractionated CO2 laser. Paper presented at: American Society for Laser Medicine and Surgery 36th Annual Conference; April 1-3, 2016; Boston, MA.
- Ozog DM, Liu A, Chaffins ML, et al. Evaluation of clinical results, histological architecture, and collagen expression following treatment of mature burn scars with a fraction carbon dioxide laser. JAMA Dermatol. 2013;149:50-57.
- Levi B, Ibrahim A, Mathews K, et al. The use of CO2 fractional photothermolysis for the treatment of burn scars. J Burn Care Res. 2016;37:106-114.
- van Drooge AM, Vrijman C, van der Veen W, et al. A randomized controlled pilot study on ablative fractional CO2 laser for consecutive patients presenting with various scar types. Dermatol Surg. 2015;41:371-377.
- Wang XQ, Lui YK, Wang ZY, et al. Antimitotic drug injections and radiotherapy: a review of the effectiveness of treatment for hypertrophic scars and keloids. Int J Low Extrem Wounds. 2008;7:151-159.
- Gupta S, Kalra A. Efficacy and safety of intralesional 5-fluorouracil in the treatment of keloids. Dermatology. 2002;204:130-132.
- Haedersdal M, Erlendsson AM, Paasch U, et al. Translational medicine in the field of AFL (AFXL)-assisted drug delivery: a critical review from basics to current clinical status. J Am Acad Dermatol. 2016;74:981-1004.
Hypertrophic scarring secondary to trauma, burns, and surgical interventions is a major source of morbidity worldwide and often is mechanically, aesthetically, and symptomatically debilitating. Modern advances in acute trauma care protocols have resulted in survival rates greater than 90% in both civilian and military populations.1,2 Patients with wounds that have historically proven fatal are now surviving and are confronted with the long-term sequelae of their injuries. With more than 52,000 service members injured in military engagements from 2001 to 2015 and 8.5 million civilians presenting annually with injury patterns at risk for hypertrophic scarring, it is paramount that we ensure access to safe and effective long-term scar care.2,3
At its simplest level, hypertrophic scarring is believed to result from a disequilibrium between collagen production and degradation. This failure to properly transition through the stages of wound healing results in bothersome symptoms, a disfigured appearance, and mechanical dysfunction of the skin (Figure, A). Decreased elasticity and extensibility, increased dermal thickness, and scar contractures impair patient range of motion and functional mobility. Those affected commonly experience varying degrees of pruritus and dysesthesia along the scar. Combined with aesthetic variations in pigmentation, erythema, texture, and thickness, hypertrophic scarring often leads to long-term psychosocial impairment and decreased health-related quality of life.4
Treatment Approach
Treatment of hypertrophic scars requires a multimodal approach due to the spectrum of associated concerns and the natural recalcitrance of the scar to therapy. Protocols should be tailored to the individual but generally begin with tissue-conserving surgical interventions followed by selective photothermolysis of the scar vasculature. Subsequently, deep and superficial ablative fractional laser (AFL) treatment and local pharmacotherapy also are employed. Treatment can be accomplished in the outpatient setting under local anesthesia in a serial fashion. In the authors’ experience, these therapies behave in a synergistic fashion, achieving outcomes that far exceed the sum of their parts, often obviating the need for scar excision in the majority of cases (Figure, B).
Tissue-Conserving Surgical Intervention
Z-plasty is an indispensable surgical tool due to its ability to lengthen scars and reduce wound tension. Treatment is easily customizable to the patient and can be performed using the individual or multiple Z-plasty techniques. Undermining and step-off correction while suturing allow the physician to lower raised scars, elevate depressed scars, and obscure scar presence by minimizing the straight lines that draw the eye to the scar. Z-plasties rely on the creation and transposition of 2 triangular flaps and permit a 75% increase in length along the desired tension vector. As such, Z-plasties decrease wound tension and facilitate scar maturation.
Selective Photothermolysis of the Vasculature
Although there are several devices available to treat vascular and immature hypertrophic scars, the majority of studies have been conducted with the 595-nm pulsed dye laser. By preferentially heating oxyhemoglobin within the dermal microvasculature, the pulsed dye laser irreparably injures the vascular endothelium. The subsequent tissue hypoxia and collagen fiber heating results in collagen fiber realignment, normalization of collagen subtypes, and neocollagenesis.5 Pulsed dye laser therapy most effectively reduces erythema and pruritus; however, improvements in scar volume, pliability, and elasticity also have been reported.5 When targeting the fine vasculature of the scar, thermal confinement is critical to prevent injury to the surrounding dermis. As such, pulse widths of 0.45 to 1.5 milliseconds are routinely utilized with a fluence just sufficient to elicit transient purpura lasting 3 to 5 seconds. Employing a spot size of 7 to 10 mm, typical fluences range from 4.5 to 6.5 J/cm2. Engagement of the dynamic cooling device reduces the risk for complications, allowing the patient to proceed to the next step in their therapy regimen: the AFL.
Ablative Fractional Laser
The AFL creates a pixilated pattern of injury throughout the epidermis and dermis of the treatment area. Ablative fractional laser platforms include the 10,600-nm CO2 and 2940-nm erbium-doped YAG lasers, both targeting intracellular water. The AFL vaporizes columns of tissue, leaving minute vertical channels with narrow rims of protein coagulation referred to as microscopic treatment zones (MTZs).6 Scar collagen analysis after AFL treatment has shown a profile resembling unaffected skin.7 Consistently, patients report improvements in stiffness, range of motion, pain, pruritus, pigmentation, and erythema.Physician observers also have reported similar improvements in these end points.8,9 Recently, interim data from a prospective controlled trial were presented showing objective improvements in dermal thickness, elasticity, and extensibility after 3 treatments with the CO2 AFL.6 The UltraPulse CO2 laser (Lumenis) is the most well-studied and widely available AFL for scar therapy and as such we will outline common treatment parameters with this device. Of note, treatment end points may be generalized to any AFL.
The DeepFX UltraPulse configuration is utilized to achieve deep AFL therapy and has a fixed pulse width of 0.8 milliseconds, slightly less than the thermal relaxation time of the skin. The diameter of the MTZs is 120 µm, and MTZ density for scar treatment ranges from 1% to 10% with a goal depth of at least 80% of scar thickness. Maximal penetration of the AFL is 4 mm, which is directly proportional to fluence. The goal of deep AFL is the removal of scar tissue to facilitate remodeling and neocollagenesis. Superficial fractional ablation can then be achieved utilizing the ActiveFX UltraPulse configuration generating a 1.3-mm MTZ spot size. We commonly use a treatment level of 3 (82% density). Typical treatment energy ranges from 80 to 125 mJ, which correlates with depths of approximately 50 to 115 µm. With both configurations, the size and shape of the treatment area can be customized to the scar. In addition, frequency may be adjusted to control the speed of treatment while balancing the risk of bulk heating. The goal of superficial AFL is to minimize scar surface irregularities and ensure blending of deep AFL treatment. Once AFL treatment is complete, local pharmacotherapy can then be employed.
Pharmacotherapy
Intralesional corticosteroids have long represented the standard of care for hypertrophic scars, with concentrations between 2.5 and 40 mg/mL that are titrated to scar thickness and location to avoid unwanted atrophy. Visual blanching of the scar represents the clinical end point for treatment. Corticosteroids act by inhibiting fibroblast proliferation and enhancing collagen degradation.10 5-Fluorouracil (5-FU) also is used in scar management. In addition to inhibiting fibroblast proliferation and inducing fibroblast apoptosis, 5-FU inhibits myofibroblast proliferation, which is helpful in the prevention and treatment of scar contracture.11 As monotherapy, weekly injections with 1 to 3 mL of 50 mg/mL 5-FU has been safe and effective. Combination intralesional corticosteroid and 5-FU therapy has been reported and is associated with improved scar regression, reduced reoccurrence, and fewer side effects.11 In our experience, a 1:1 suspension is effective with appropriate titration of the corticosteroid component. Although less well defined, topical application of pharmacotherapy and massage to the newly created MTZs appears beneficial and offers another option for delivery of corticosteroids and 5-FU, in addition to a number of promising medications such as bimatoprost, poly-L-lactic acid, timolol, and rapamycin.12
Conclusion
Advances in laser surgery and our understanding of wound healing have created a paradigm shift in the treatment approach to trauma and burn scars. In lieu of extensive scar excisions, the summarized multimodal regimen emphasizing tissue conservation and autologous remodeling is gaining favor in the military, academic medical centers, and scar centers of excellence, but patients are finding local access to care difficult. Dermatologists are uniquely positioned to cost-effectively deliver this care in the outpatient setting utilizing devices and techniques they already possess. With the end goal of optimization of functional, symptomatic, and aesthetic state of the patient, it is critical that dermatologists seize this opportunity to truly make a difference for the military and civilian patients that need it most.
Hypertrophic scarring secondary to trauma, burns, and surgical interventions is a major source of morbidity worldwide and often is mechanically, aesthetically, and symptomatically debilitating. Modern advances in acute trauma care protocols have resulted in survival rates greater than 90% in both civilian and military populations.1,2 Patients with wounds that have historically proven fatal are now surviving and are confronted with the long-term sequelae of their injuries. With more than 52,000 service members injured in military engagements from 2001 to 2015 and 8.5 million civilians presenting annually with injury patterns at risk for hypertrophic scarring, it is paramount that we ensure access to safe and effective long-term scar care.2,3
At its simplest level, hypertrophic scarring is believed to result from a disequilibrium between collagen production and degradation. This failure to properly transition through the stages of wound healing results in bothersome symptoms, a disfigured appearance, and mechanical dysfunction of the skin (Figure, A). Decreased elasticity and extensibility, increased dermal thickness, and scar contractures impair patient range of motion and functional mobility. Those affected commonly experience varying degrees of pruritus and dysesthesia along the scar. Combined with aesthetic variations in pigmentation, erythema, texture, and thickness, hypertrophic scarring often leads to long-term psychosocial impairment and decreased health-related quality of life.4
Treatment Approach
Treatment of hypertrophic scars requires a multimodal approach due to the spectrum of associated concerns and the natural recalcitrance of the scar to therapy. Protocols should be tailored to the individual but generally begin with tissue-conserving surgical interventions followed by selective photothermolysis of the scar vasculature. Subsequently, deep and superficial ablative fractional laser (AFL) treatment and local pharmacotherapy also are employed. Treatment can be accomplished in the outpatient setting under local anesthesia in a serial fashion. In the authors’ experience, these therapies behave in a synergistic fashion, achieving outcomes that far exceed the sum of their parts, often obviating the need for scar excision in the majority of cases (Figure, B).
Tissue-Conserving Surgical Intervention
Z-plasty is an indispensable surgical tool due to its ability to lengthen scars and reduce wound tension. Treatment is easily customizable to the patient and can be performed using the individual or multiple Z-plasty techniques. Undermining and step-off correction while suturing allow the physician to lower raised scars, elevate depressed scars, and obscure scar presence by minimizing the straight lines that draw the eye to the scar. Z-plasties rely on the creation and transposition of 2 triangular flaps and permit a 75% increase in length along the desired tension vector. As such, Z-plasties decrease wound tension and facilitate scar maturation.
Selective Photothermolysis of the Vasculature
Although there are several devices available to treat vascular and immature hypertrophic scars, the majority of studies have been conducted with the 595-nm pulsed dye laser. By preferentially heating oxyhemoglobin within the dermal microvasculature, the pulsed dye laser irreparably injures the vascular endothelium. The subsequent tissue hypoxia and collagen fiber heating results in collagen fiber realignment, normalization of collagen subtypes, and neocollagenesis.5 Pulsed dye laser therapy most effectively reduces erythema and pruritus; however, improvements in scar volume, pliability, and elasticity also have been reported.5 When targeting the fine vasculature of the scar, thermal confinement is critical to prevent injury to the surrounding dermis. As such, pulse widths of 0.45 to 1.5 milliseconds are routinely utilized with a fluence just sufficient to elicit transient purpura lasting 3 to 5 seconds. Employing a spot size of 7 to 10 mm, typical fluences range from 4.5 to 6.5 J/cm2. Engagement of the dynamic cooling device reduces the risk for complications, allowing the patient to proceed to the next step in their therapy regimen: the AFL.
Ablative Fractional Laser
The AFL creates a pixilated pattern of injury throughout the epidermis and dermis of the treatment area. Ablative fractional laser platforms include the 10,600-nm CO2 and 2940-nm erbium-doped YAG lasers, both targeting intracellular water. The AFL vaporizes columns of tissue, leaving minute vertical channels with narrow rims of protein coagulation referred to as microscopic treatment zones (MTZs).6 Scar collagen analysis after AFL treatment has shown a profile resembling unaffected skin.7 Consistently, patients report improvements in stiffness, range of motion, pain, pruritus, pigmentation, and erythema.Physician observers also have reported similar improvements in these end points.8,9 Recently, interim data from a prospective controlled trial were presented showing objective improvements in dermal thickness, elasticity, and extensibility after 3 treatments with the CO2 AFL.6 The UltraPulse CO2 laser (Lumenis) is the most well-studied and widely available AFL for scar therapy and as such we will outline common treatment parameters with this device. Of note, treatment end points may be generalized to any AFL.
The DeepFX UltraPulse configuration is utilized to achieve deep AFL therapy and has a fixed pulse width of 0.8 milliseconds, slightly less than the thermal relaxation time of the skin. The diameter of the MTZs is 120 µm, and MTZ density for scar treatment ranges from 1% to 10% with a goal depth of at least 80% of scar thickness. Maximal penetration of the AFL is 4 mm, which is directly proportional to fluence. The goal of deep AFL is the removal of scar tissue to facilitate remodeling and neocollagenesis. Superficial fractional ablation can then be achieved utilizing the ActiveFX UltraPulse configuration generating a 1.3-mm MTZ spot size. We commonly use a treatment level of 3 (82% density). Typical treatment energy ranges from 80 to 125 mJ, which correlates with depths of approximately 50 to 115 µm. With both configurations, the size and shape of the treatment area can be customized to the scar. In addition, frequency may be adjusted to control the speed of treatment while balancing the risk of bulk heating. The goal of superficial AFL is to minimize scar surface irregularities and ensure blending of deep AFL treatment. Once AFL treatment is complete, local pharmacotherapy can then be employed.
Pharmacotherapy
Intralesional corticosteroids have long represented the standard of care for hypertrophic scars, with concentrations between 2.5 and 40 mg/mL that are titrated to scar thickness and location to avoid unwanted atrophy. Visual blanching of the scar represents the clinical end point for treatment. Corticosteroids act by inhibiting fibroblast proliferation and enhancing collagen degradation.10 5-Fluorouracil (5-FU) also is used in scar management. In addition to inhibiting fibroblast proliferation and inducing fibroblast apoptosis, 5-FU inhibits myofibroblast proliferation, which is helpful in the prevention and treatment of scar contracture.11 As monotherapy, weekly injections with 1 to 3 mL of 50 mg/mL 5-FU has been safe and effective. Combination intralesional corticosteroid and 5-FU therapy has been reported and is associated with improved scar regression, reduced reoccurrence, and fewer side effects.11 In our experience, a 1:1 suspension is effective with appropriate titration of the corticosteroid component. Although less well defined, topical application of pharmacotherapy and massage to the newly created MTZs appears beneficial and offers another option for delivery of corticosteroids and 5-FU, in addition to a number of promising medications such as bimatoprost, poly-L-lactic acid, timolol, and rapamycin.12
Conclusion
Advances in laser surgery and our understanding of wound healing have created a paradigm shift in the treatment approach to trauma and burn scars. In lieu of extensive scar excisions, the summarized multimodal regimen emphasizing tissue conservation and autologous remodeling is gaining favor in the military, academic medical centers, and scar centers of excellence, but patients are finding local access to care difficult. Dermatologists are uniquely positioned to cost-effectively deliver this care in the outpatient setting utilizing devices and techniques they already possess. With the end goal of optimization of functional, symptomatic, and aesthetic state of the patient, it is critical that dermatologists seize this opportunity to truly make a difference for the military and civilian patients that need it most.
- American Burn Association, National Burn Repository. 2015 National burn repository report of data from 2005-2014. http://www.ameriburn.org/2015NBRAnnualReport.pdf. Accessed May 10, 2017.
- Centers for Disease Control and Prevention. 2013 National hospital ambulatory medical care survey emergency department summary tables. https://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf. Accessed May 10, 2017.
- Fischer H. A guide to U.S. Military casualty statistics: Operation Freedom’s Sentinel, Operation Inherent Resolve, Operation New Dawn, Operation Iraqi Freedom, and Operation Enduring Freedom. Congressional Research Service website. https://fas.org/sgp/crs/natsec/RS22452.pdf. Published August 7, 2015. Accessed May 10, 2017.
- Van Loey NE, Van Son MJ. Psychopathology and psychological problems in patients with burn scars: epidemiology and management. Am J Clin Dermatol. 2003;4:245-272.
- Vrijman C, van Drooge AM, Limpens J, et al. Laser and intense pulsed light therapy for the treatment of hypertrophic scars: a systematic review. Br J Dermatol. 2011;165:934-942.
- Miletta N, Lee K, Siwy K, et al. Objective improvement in burn scars after treatment with fractionated CO2 laser. Paper presented at: American Society for Laser Medicine and Surgery 36th Annual Conference; April 1-3, 2016; Boston, MA.
- Ozog DM, Liu A, Chaffins ML, et al. Evaluation of clinical results, histological architecture, and collagen expression following treatment of mature burn scars with a fraction carbon dioxide laser. JAMA Dermatol. 2013;149:50-57.
- Levi B, Ibrahim A, Mathews K, et al. The use of CO2 fractional photothermolysis for the treatment of burn scars. J Burn Care Res. 2016;37:106-114.
- van Drooge AM, Vrijman C, van der Veen W, et al. A randomized controlled pilot study on ablative fractional CO2 laser for consecutive patients presenting with various scar types. Dermatol Surg. 2015;41:371-377.
- Wang XQ, Lui YK, Wang ZY, et al. Antimitotic drug injections and radiotherapy: a review of the effectiveness of treatment for hypertrophic scars and keloids. Int J Low Extrem Wounds. 2008;7:151-159.
- Gupta S, Kalra A. Efficacy and safety of intralesional 5-fluorouracil in the treatment of keloids. Dermatology. 2002;204:130-132.
- Haedersdal M, Erlendsson AM, Paasch U, et al. Translational medicine in the field of AFL (AFXL)-assisted drug delivery: a critical review from basics to current clinical status. J Am Acad Dermatol. 2016;74:981-1004.
- American Burn Association, National Burn Repository. 2015 National burn repository report of data from 2005-2014. http://www.ameriburn.org/2015NBRAnnualReport.pdf. Accessed May 10, 2017.
- Centers for Disease Control and Prevention. 2013 National hospital ambulatory medical care survey emergency department summary tables. https://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf. Accessed May 10, 2017.
- Fischer H. A guide to U.S. Military casualty statistics: Operation Freedom’s Sentinel, Operation Inherent Resolve, Operation New Dawn, Operation Iraqi Freedom, and Operation Enduring Freedom. Congressional Research Service website. https://fas.org/sgp/crs/natsec/RS22452.pdf. Published August 7, 2015. Accessed May 10, 2017.
- Van Loey NE, Van Son MJ. Psychopathology and psychological problems in patients with burn scars: epidemiology and management. Am J Clin Dermatol. 2003;4:245-272.
- Vrijman C, van Drooge AM, Limpens J, et al. Laser and intense pulsed light therapy for the treatment of hypertrophic scars: a systematic review. Br J Dermatol. 2011;165:934-942.
- Miletta N, Lee K, Siwy K, et al. Objective improvement in burn scars after treatment with fractionated CO2 laser. Paper presented at: American Society for Laser Medicine and Surgery 36th Annual Conference; April 1-3, 2016; Boston, MA.
- Ozog DM, Liu A, Chaffins ML, et al. Evaluation of clinical results, histological architecture, and collagen expression following treatment of mature burn scars with a fraction carbon dioxide laser. JAMA Dermatol. 2013;149:50-57.
- Levi B, Ibrahim A, Mathews K, et al. The use of CO2 fractional photothermolysis for the treatment of burn scars. J Burn Care Res. 2016;37:106-114.
- van Drooge AM, Vrijman C, van der Veen W, et al. A randomized controlled pilot study on ablative fractional CO2 laser for consecutive patients presenting with various scar types. Dermatol Surg. 2015;41:371-377.
- Wang XQ, Lui YK, Wang ZY, et al. Antimitotic drug injections and radiotherapy: a review of the effectiveness of treatment for hypertrophic scars and keloids. Int J Low Extrem Wounds. 2008;7:151-159.
- Gupta S, Kalra A. Efficacy and safety of intralesional 5-fluorouracil in the treatment of keloids. Dermatology. 2002;204:130-132.
- Haedersdal M, Erlendsson AM, Paasch U, et al. Translational medicine in the field of AFL (AFXL)-assisted drug delivery: a critical review from basics to current clinical status. J Am Acad Dermatol. 2016;74:981-1004.
Practice Points
- Burn and trauma scarring represents a major source of morbidity in both the civilian and military populations worldwide and often is mechanically, aesthetically, and symptomatically debilitating.
- Advances in laser surgery and our understanding of wound healing have resulted in a scar therapy paradigm shift from large scar excisions and repair to a multimodal, tissue-conserving approach that relies on remodeling of the existing tissue.
- Dermatologists are uniquely positioned to increase patient access to cost-effective, outpatient-based burn and trauma scar care utilizing devices and techniques that they currently possess.
In Vivo Reflectance Confocal Microscopy
Reflectance confocal microscopy (RCM) imaging received Category I Current Procedural Terminology (CPT) codes by the Centers for Medicare & Medicaid Services in January 2016 and can now be submitted to insurance companies with reimbursement comparable to a skin biopsy or a global skin pathology service.1 This fairly new technology is a US Food and Drug Administration–cleared noninvasive imaging modality that provides high-resolution in vivo cellular images of the skin. It has been shown to be efficacious in differentiating benign and malignant skin lesions, increasing diagnostic accuracy, and reducing the number of unnecessary skin biopsies that are performed. In addition to skin cancer diagnosis, RCM imaging also can help guide management of malignant lesions by detecting lateral margins prior to surgery as well as monitoring the lesion over time for treatment efficacy or recurrence. The potential impact of RCM imaging is tremendous, and reimbursement may lead to increased use in clinical practice to the benefit of our patients. Herein, we present a brief review of RCM imaging and reimbursement as well as the benefits and limitations of this new technology for dermatologists.
Reflectance Confocal Microscopy
In vivo RCM allows us to visualize the epidermis in real time on a cellular level down to the papillary dermis at a high resolution (×30) comparable to histologic examination. With optical sections 3- to 5-µm thick and a lateral resolution of 0.5 to 1.0 µm, RCM produces a stack of 500×500-µm2 images up to a depth of approximately 200 µm.2,3 At any chosen depth, these smaller images are stitched together with sophisticated software into a block, or mosaic, increasing the field of view to up to 8×8 mm2. Imaging is performed in en face planes oriented parallel to the skin surface, similar to dermoscopy.
Current CPT Guidelines and Reimbursement
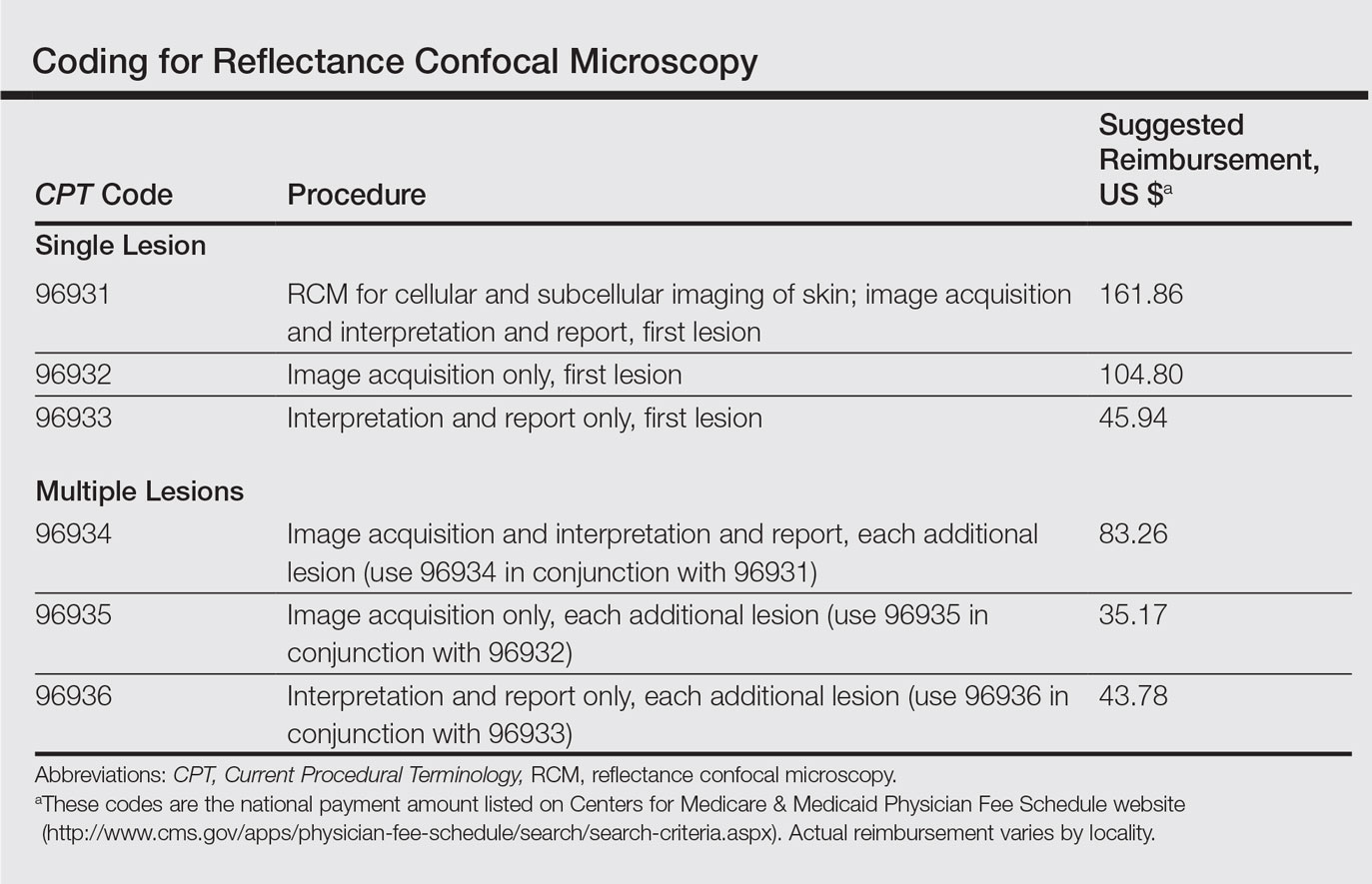
The CPT codes for RCM imaging provide reimbursement on a per-lesion basis and are similar to those used for skin biopsy and pathology (Table).1 Codes 96931 through 96933 are used for imaging of a single lesion on a patient. The first code—96931—is used when image acquisition, interpretation, and report creation are carried out by a single clinician. The next 2 codes are used when one clinician acquires the image—96932—comparable to the technical component of a pathology code, while another reads it and creates the report—96933—similar to a dermatopathologist billing for the professional component of a pathology report. For patients presenting with multiple lesions, the next 3 codes—96934, 96935, and 96936—are used in conjunction with the applicable first code for each additional lesion with similar global, technical, and professional components. Because these codes are not in the radiology or pathology sections of CPT, a single code cannot be used with modifier -TC (technical component) and modifier -26, as they are in those sections.
The wide-probe VivaScope 1500 (Caliber I.D., Inc) currently is the only confocal device that can be reported with a CPT code and routinely reimbursed. The handheld VivaScope 3000 (Caliber I.D., Inc) can only view a small stack and does not have the ability to acquire a full mosaic image; it is not covered by these codes.
Images can be viewed as a stack captured at the same horizontal position but at sequential depths or as a mosaic, which has a larger field of view but is limited to a single plane. To appropriately assess a lesion, clinicians must obtain a mosaic that needs to be assessed at multiple layers for a diagnosis to be made because it is a cross-section view.
Diagnosis
Studies have demonstrated the usefulness of RCM imaging in the diagnosis of a wide range of skin diseases, including melanoma and nonmelanoma skin cancers, infectious diseases, and inflammatory and autoimmune conditions, as well as wound healing and skin aging. Reflectance confocal microscopy imaging is not limited to the skin; it can be used to evaluate the hair, nails, oral mucosa, and other organs.
According to several studies, RCM imaging notably increases the diagnostic accuracy and detection rate of skin cancers over clinical and dermoscopic examination alone and therefore can act as an aid in differentiating lesions that are benign versus those that are suspicious and should be biopsied.
Reflectance confocal microscopy has been shown to have a mean sensitivity of 94% (range, 92%–96%) and specificity of 83% (range, 81%–84%) for all types of skin cancer when used with dermoscopy.4 In particular, for melanocytic lesions that are ambiguous on dermoscopy, RCM used in addition to dermoscopy increases the mean sensitivity and specificity for melanoma diagnosis to 93% (range, 89%–96%) and 76% (range, 68%–83%), respectively.5 Although these reported sensitivities are comparable to dermoscopy, the specificity is superior, especially for detecting hypomelanotic and amelanotic melanomas, which often lack specific features on dermoscopy.6-8
The combination of RCM with dermoscopy has reduced the number of unnecessary excisions of benign nevi by more than 50% when compared to dermoscopy alone.9 One study showed that the number needed to treat (ie, excise) a melanoma decreased from 14.6 with dermoscopy alone to 6.8 when guided by dermoscopy and RCM imaging.9 In a similar study, the number needed to treat dropped from 19.41 with dermoscopy alone to 6.25 with dermoscopy and RCM.10
These studies were not looking to evaluate RCM as a replacement test but rather as an add-on test to dermoscopy. Reflectance confocal microscopy imaging takes longer than dermoscopy for each lesion; therefore, RCM should only be used as an adjunctive tool to dermoscopy and not as an initial screening test. Consequentially, a dermatologist skilled in dermoscopy is essential in deciding which lesions would be appropriate for subsequent RCM imaging.
In Vivo Margin Mapping as an Adjunct to Surgery
Oftentimes, tumor margins are poorly defined and can be difficult to map clinically and dermoscopically. Studies have demonstrated the use of RCM in delineation of surgical margins prior to surgery or excisional biopsies.11,12 Alternatively, when complete removal at biopsy would be impractical (eg, for extremely large lesions or lesions located in cosmetically sensitive areas such as the face), RCM can be used to pick the best site for an appropriate biopsy, which decreases the chance of sampling error due to skip lesions and increases histologic accuracy.
Nonsurgical Treatment Monitoring
One advantage of RCM over conventional histology is that RCM imaging leaves the tissue intact, allowing dynamic changes to be studied over time, which is useful for monitoring nonmelanoma skin cancers and lentigo maligna being treated with noninvasive therapeutic modalities.13 If not as a definitive treatment, RCM can act as an adjunct for surgery by monitoring reduction in lesion size prior to Mohs micrographic surgery, thereby decreasing the resulting surgical defect.14
Limitations
Imaging Depth
Although RCM is a revolutionary device in the field of dermatology, it has several limitations. With a maximal imaging depth of 350 µm, the imaging resolution decreases substantially with depth, limiting accurate interpretation to 200 µm. Reflectance confocal microscopy can only image the superficial portion of a lesion; therefore, deep tumor margins cannot be assessed. Hypertrophic or hyperkeratotic lesions, including lesions on the palms and soles, also are unable to be imaged with RCM. This limitation in depth penetration makes treatment monitoring impossible for invasive lesions that extend into the dermal layer.
Difficult-to-Reach Areas
Another limitation is the difficulty imaging areas such as the ocular canthi, nasal alae, or helices of the ear due to the wide probe size on the VivaScope 1500. The advent of the smaller handheld VivaScope 3000 device allows for improved imaging of concave services and difficult lesions at the risk of less accurate imaging, low field of view, and no reimbursement at present.
False-Positive Results
Although RCM has been shown to be helpful in reducing unnecessary biopsies, there still is the issue of false-positives on imaging. False-positives most commonly occur in nevi with severe atypia or when Langerhans cells are present that cannot always be differentiated from melanocytic cells.3,15,16 One prospective study found 7 false-positive results from 63 sites using RCM for the diagnosis of lentigo malignas.16 False-negatives can occur in the presence of inflammatory infiltrates and scar tissue that can hide cellular morphology or in sampling errors due to skip lesions.3,16
Time Efficiency
The time required for acquisition of RCM mosaics and stacks followed by reading and interpretation can be substantial depending on the size and complexity of the lesion, which is a major limitation for use of RCM in busy dermatology practices; therefore, RCM should be reserved for lesions selected to undergo biopsy that are clinically equivocal for malignancy prior to RCM examination.17 It would not be cost-effective or time effective to evaluate lesions that either clinically or dermoscopically have a high probability of malignancy; however, patients and physicians may opt for increased specificity at the expense of time, particularly when a lesion is located on a cosmetically sensitive area, as patients can avoid initial histologic biopsy and gain the cosmetic benefit of going straight to surgery versus obtaining an initial diagnostic biopsy.
Cost
Lastly, the high cost involved in purchasing an RCM device and the training involved to use and interpret RCM images currently limits RCM to large academic centers. Reimbursement may make more widespread use feasible. In any event, RCM imaging should be part of the curriculum for both dermatology and pathology trainees.
Future Directions
In vivo RCM is a noninvasive imaging modality that allows for real-time evaluation of the skin. Used in conjunction with dermoscopy, RCM can substantially improve diagnostic accuracy and reduce the number of unnecessary biopsies. Now that RCM has finally gained foundational CPT codes and insurance reimbursement, there may be a growing demand for clinicians to incorporate this technology into their clinical practice.
- Current Procedural Terminology 2017, Professional Edition. Chicago IL: American Medical Association; 2016.
- Que SK, Fraga-Braghiroli N, Grant-Kels JM, et al. Through the looking glass: basics and principles of reflectance confocal microscopy [published online June 4, 2015]. J Am Acad Dermatol. 2015;73:276-284.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside [published online October 27, 2016]. Lasers Surg Med. 2017;49:7-19.
- Xiong YD, Ma S, Li X, et al. A meta-analysis of reflectance confocal microscopy for the diagnosis of malignant skin tumours. J Eur Acad Dermatol Venereol. 2016;30:1295-1302.
- Stevenson AD, Mickan S, Mallett S, et al. Systematic review of diagnostic accuracy of reflectance confocal microscopy for melanoma diagnosis in patients with clinically equivocal skin lesions. Dermatol Pract Concept. 2013;3:19-27.
- Busam KJ, Hester K, Charles C, et al. Detection of clinically amelanotic malignant melanoma and assessment of its margins by in vivo confocal scanning laser microscopy. Arch Dermatol. 2001;137:923-929.
- Losi A, Longo C, Cesinaro AM, et al. Hyporeflective pagetoid cells: a new clue for amelanotic melanoma diagnosis by reflectance confocal microscopy. Br J Dermatol. 2014;171:48-54.
- Guitera P, Menzies SQ, Argenziano G, et al. Dermoscopy and in vivo confocal microscopy are complementary techniques for the diagnosis of difficult amelanotic and light-coloured skin lesions [published online October 12, 2016]. Br J Dermatol. 2016;175:1311-1319.
- Pellacani G, Pepe P, Casari A, et al. Reflectance confocal microscopy as a second-level examination in skin oncology improves diagnostic accuracy and saves unnecessary excisions: a longitudinal prospective study. Br J Dermatol. 2014;171:1044-1051.
- Pellacani G, Witkowski A, Cesinaro AM, et al. Cost-benefit of reflectance confocal microscopy in the diagnostic performance of melanoma. J Eur Acad Dermatol Venereol. 2016;30:413-419.
- Champin J, Perrot JL, Cinotti E, et al. In vivo reflectance confocal microscopy to optimize the spaghetti technique for defining surgical margins of lentigo maligna. Dermatol Surg. 2014;40:247-256.
- Hibler BP, Cordova M, Wong RJ, et al. Intraoperative real-time reflectance confocal microscopy for guiding surgical margins of lentigo maligna melanoma. Dermatol Surg. 2015;41:980-983.
- Ulrich M, Lange-Asschenfeldt S, Gonzalez S. The use of reflectance confocal microscopy for monitoring response to therapy of skin malignancies. Dermatol Pract Concept. 2012;2:202a10.
- Torres A, Niemeyer A, Berkes B, et al. 5% imiquimod cream and reflectance-mode confocal microscopy as adjunct modalities to Mohs micrographic surgery for treatment of basal cell carcinoma. Dermatol Surg. 2004;30(12, pt 1):1462-1469.
- Hashemi P, Pulitzer MP, Scope A, et al. Langerhans cells and melanocytes share similar morphologic features under in vivo reflectance confocal microscopy: a challenge for melanoma diagnosis. J Am Acad Dermatol. 2012;66:452-462.
- Menge TD, Hibler BP, Cordova MA, et al. Concordance of handheld reflectance confocal microscopy (RCM) with histopathology in the diagnosis of lentigo maligna (LM): a prospective study. J Am Acad Dermatol. 2016;74:1114-1120.
- Borsari S, Pampena R, Lallas A, et al. Clinical indications for use of reflectance confocal microscopy for skin cancer diagnosis. JAMA Dermatol. 2016;152:1093-1098.
Reflectance confocal microscopy (RCM) imaging received Category I Current Procedural Terminology (CPT) codes by the Centers for Medicare & Medicaid Services in January 2016 and can now be submitted to insurance companies with reimbursement comparable to a skin biopsy or a global skin pathology service.1 This fairly new technology is a US Food and Drug Administration–cleared noninvasive imaging modality that provides high-resolution in vivo cellular images of the skin. It has been shown to be efficacious in differentiating benign and malignant skin lesions, increasing diagnostic accuracy, and reducing the number of unnecessary skin biopsies that are performed. In addition to skin cancer diagnosis, RCM imaging also can help guide management of malignant lesions by detecting lateral margins prior to surgery as well as monitoring the lesion over time for treatment efficacy or recurrence. The potential impact of RCM imaging is tremendous, and reimbursement may lead to increased use in clinical practice to the benefit of our patients. Herein, we present a brief review of RCM imaging and reimbursement as well as the benefits and limitations of this new technology for dermatologists.
Reflectance Confocal Microscopy
In vivo RCM allows us to visualize the epidermis in real time on a cellular level down to the papillary dermis at a high resolution (×30) comparable to histologic examination. With optical sections 3- to 5-µm thick and a lateral resolution of 0.5 to 1.0 µm, RCM produces a stack of 500×500-µm2 images up to a depth of approximately 200 µm.2,3 At any chosen depth, these smaller images are stitched together with sophisticated software into a block, or mosaic, increasing the field of view to up to 8×8 mm2. Imaging is performed in en face planes oriented parallel to the skin surface, similar to dermoscopy.
Current CPT Guidelines and Reimbursement
The CPT codes for RCM imaging provide reimbursement on a per-lesion basis and are similar to those used for skin biopsy and pathology (Table).1 Codes 96931 through 96933 are used for imaging of a single lesion on a patient. The first code—96931—is used when image acquisition, interpretation, and report creation are carried out by a single clinician. The next 2 codes are used when one clinician acquires the image—96932—comparable to the technical component of a pathology code, while another reads it and creates the report—96933—similar to a dermatopathologist billing for the professional component of a pathology report. For patients presenting with multiple lesions, the next 3 codes—96934, 96935, and 96936—are used in conjunction with the applicable first code for each additional lesion with similar global, technical, and professional components. Because these codes are not in the radiology or pathology sections of CPT, a single code cannot be used with modifier -TC (technical component) and modifier -26, as they are in those sections.
The wide-probe VivaScope 1500 (Caliber I.D., Inc) currently is the only confocal device that can be reported with a CPT code and routinely reimbursed. The handheld VivaScope 3000 (Caliber I.D., Inc) can only view a small stack and does not have the ability to acquire a full mosaic image; it is not covered by these codes.
Images can be viewed as a stack captured at the same horizontal position but at sequential depths or as a mosaic, which has a larger field of view but is limited to a single plane. To appropriately assess a lesion, clinicians must obtain a mosaic that needs to be assessed at multiple layers for a diagnosis to be made because it is a cross-section view.
Diagnosis
Studies have demonstrated the usefulness of RCM imaging in the diagnosis of a wide range of skin diseases, including melanoma and nonmelanoma skin cancers, infectious diseases, and inflammatory and autoimmune conditions, as well as wound healing and skin aging. Reflectance confocal microscopy imaging is not limited to the skin; it can be used to evaluate the hair, nails, oral mucosa, and other organs.
According to several studies, RCM imaging notably increases the diagnostic accuracy and detection rate of skin cancers over clinical and dermoscopic examination alone and therefore can act as an aid in differentiating lesions that are benign versus those that are suspicious and should be biopsied.
Reflectance confocal microscopy has been shown to have a mean sensitivity of 94% (range, 92%–96%) and specificity of 83% (range, 81%–84%) for all types of skin cancer when used with dermoscopy.4 In particular, for melanocytic lesions that are ambiguous on dermoscopy, RCM used in addition to dermoscopy increases the mean sensitivity and specificity for melanoma diagnosis to 93% (range, 89%–96%) and 76% (range, 68%–83%), respectively.5 Although these reported sensitivities are comparable to dermoscopy, the specificity is superior, especially for detecting hypomelanotic and amelanotic melanomas, which often lack specific features on dermoscopy.6-8
The combination of RCM with dermoscopy has reduced the number of unnecessary excisions of benign nevi by more than 50% when compared to dermoscopy alone.9 One study showed that the number needed to treat (ie, excise) a melanoma decreased from 14.6 with dermoscopy alone to 6.8 when guided by dermoscopy and RCM imaging.9 In a similar study, the number needed to treat dropped from 19.41 with dermoscopy alone to 6.25 with dermoscopy and RCM.10
These studies were not looking to evaluate RCM as a replacement test but rather as an add-on test to dermoscopy. Reflectance confocal microscopy imaging takes longer than dermoscopy for each lesion; therefore, RCM should only be used as an adjunctive tool to dermoscopy and not as an initial screening test. Consequentially, a dermatologist skilled in dermoscopy is essential in deciding which lesions would be appropriate for subsequent RCM imaging.
In Vivo Margin Mapping as an Adjunct to Surgery
Oftentimes, tumor margins are poorly defined and can be difficult to map clinically and dermoscopically. Studies have demonstrated the use of RCM in delineation of surgical margins prior to surgery or excisional biopsies.11,12 Alternatively, when complete removal at biopsy would be impractical (eg, for extremely large lesions or lesions located in cosmetically sensitive areas such as the face), RCM can be used to pick the best site for an appropriate biopsy, which decreases the chance of sampling error due to skip lesions and increases histologic accuracy.
Nonsurgical Treatment Monitoring
One advantage of RCM over conventional histology is that RCM imaging leaves the tissue intact, allowing dynamic changes to be studied over time, which is useful for monitoring nonmelanoma skin cancers and lentigo maligna being treated with noninvasive therapeutic modalities.13 If not as a definitive treatment, RCM can act as an adjunct for surgery by monitoring reduction in lesion size prior to Mohs micrographic surgery, thereby decreasing the resulting surgical defect.14
Limitations
Imaging Depth
Although RCM is a revolutionary device in the field of dermatology, it has several limitations. With a maximal imaging depth of 350 µm, the imaging resolution decreases substantially with depth, limiting accurate interpretation to 200 µm. Reflectance confocal microscopy can only image the superficial portion of a lesion; therefore, deep tumor margins cannot be assessed. Hypertrophic or hyperkeratotic lesions, including lesions on the palms and soles, also are unable to be imaged with RCM. This limitation in depth penetration makes treatment monitoring impossible for invasive lesions that extend into the dermal layer.
Difficult-to-Reach Areas
Another limitation is the difficulty imaging areas such as the ocular canthi, nasal alae, or helices of the ear due to the wide probe size on the VivaScope 1500. The advent of the smaller handheld VivaScope 3000 device allows for improved imaging of concave services and difficult lesions at the risk of less accurate imaging, low field of view, and no reimbursement at present.
False-Positive Results
Although RCM has been shown to be helpful in reducing unnecessary biopsies, there still is the issue of false-positives on imaging. False-positives most commonly occur in nevi with severe atypia or when Langerhans cells are present that cannot always be differentiated from melanocytic cells.3,15,16 One prospective study found 7 false-positive results from 63 sites using RCM for the diagnosis of lentigo malignas.16 False-negatives can occur in the presence of inflammatory infiltrates and scar tissue that can hide cellular morphology or in sampling errors due to skip lesions.3,16
Time Efficiency
The time required for acquisition of RCM mosaics and stacks followed by reading and interpretation can be substantial depending on the size and complexity of the lesion, which is a major limitation for use of RCM in busy dermatology practices; therefore, RCM should be reserved for lesions selected to undergo biopsy that are clinically equivocal for malignancy prior to RCM examination.17 It would not be cost-effective or time effective to evaluate lesions that either clinically or dermoscopically have a high probability of malignancy; however, patients and physicians may opt for increased specificity at the expense of time, particularly when a lesion is located on a cosmetically sensitive area, as patients can avoid initial histologic biopsy and gain the cosmetic benefit of going straight to surgery versus obtaining an initial diagnostic biopsy.
Cost
Lastly, the high cost involved in purchasing an RCM device and the training involved to use and interpret RCM images currently limits RCM to large academic centers. Reimbursement may make more widespread use feasible. In any event, RCM imaging should be part of the curriculum for both dermatology and pathology trainees.
Future Directions
In vivo RCM is a noninvasive imaging modality that allows for real-time evaluation of the skin. Used in conjunction with dermoscopy, RCM can substantially improve diagnostic accuracy and reduce the number of unnecessary biopsies. Now that RCM has finally gained foundational CPT codes and insurance reimbursement, there may be a growing demand for clinicians to incorporate this technology into their clinical practice.
Reflectance confocal microscopy (RCM) imaging received Category I Current Procedural Terminology (CPT) codes by the Centers for Medicare & Medicaid Services in January 2016 and can now be submitted to insurance companies with reimbursement comparable to a skin biopsy or a global skin pathology service.1 This fairly new technology is a US Food and Drug Administration–cleared noninvasive imaging modality that provides high-resolution in vivo cellular images of the skin. It has been shown to be efficacious in differentiating benign and malignant skin lesions, increasing diagnostic accuracy, and reducing the number of unnecessary skin biopsies that are performed. In addition to skin cancer diagnosis, RCM imaging also can help guide management of malignant lesions by detecting lateral margins prior to surgery as well as monitoring the lesion over time for treatment efficacy or recurrence. The potential impact of RCM imaging is tremendous, and reimbursement may lead to increased use in clinical practice to the benefit of our patients. Herein, we present a brief review of RCM imaging and reimbursement as well as the benefits and limitations of this new technology for dermatologists.
Reflectance Confocal Microscopy
In vivo RCM allows us to visualize the epidermis in real time on a cellular level down to the papillary dermis at a high resolution (×30) comparable to histologic examination. With optical sections 3- to 5-µm thick and a lateral resolution of 0.5 to 1.0 µm, RCM produces a stack of 500×500-µm2 images up to a depth of approximately 200 µm.2,3 At any chosen depth, these smaller images are stitched together with sophisticated software into a block, or mosaic, increasing the field of view to up to 8×8 mm2. Imaging is performed in en face planes oriented parallel to the skin surface, similar to dermoscopy.
Current CPT Guidelines and Reimbursement
The CPT codes for RCM imaging provide reimbursement on a per-lesion basis and are similar to those used for skin biopsy and pathology (Table).1 Codes 96931 through 96933 are used for imaging of a single lesion on a patient. The first code—96931—is used when image acquisition, interpretation, and report creation are carried out by a single clinician. The next 2 codes are used when one clinician acquires the image—96932—comparable to the technical component of a pathology code, while another reads it and creates the report—96933—similar to a dermatopathologist billing for the professional component of a pathology report. For patients presenting with multiple lesions, the next 3 codes—96934, 96935, and 96936—are used in conjunction with the applicable first code for each additional lesion with similar global, technical, and professional components. Because these codes are not in the radiology or pathology sections of CPT, a single code cannot be used with modifier -TC (technical component) and modifier -26, as they are in those sections.
The wide-probe VivaScope 1500 (Caliber I.D., Inc) currently is the only confocal device that can be reported with a CPT code and routinely reimbursed. The handheld VivaScope 3000 (Caliber I.D., Inc) can only view a small stack and does not have the ability to acquire a full mosaic image; it is not covered by these codes.
Images can be viewed as a stack captured at the same horizontal position but at sequential depths or as a mosaic, which has a larger field of view but is limited to a single plane. To appropriately assess a lesion, clinicians must obtain a mosaic that needs to be assessed at multiple layers for a diagnosis to be made because it is a cross-section view.
Diagnosis
Studies have demonstrated the usefulness of RCM imaging in the diagnosis of a wide range of skin diseases, including melanoma and nonmelanoma skin cancers, infectious diseases, and inflammatory and autoimmune conditions, as well as wound healing and skin aging. Reflectance confocal microscopy imaging is not limited to the skin; it can be used to evaluate the hair, nails, oral mucosa, and other organs.
According to several studies, RCM imaging notably increases the diagnostic accuracy and detection rate of skin cancers over clinical and dermoscopic examination alone and therefore can act as an aid in differentiating lesions that are benign versus those that are suspicious and should be biopsied.
Reflectance confocal microscopy has been shown to have a mean sensitivity of 94% (range, 92%–96%) and specificity of 83% (range, 81%–84%) for all types of skin cancer when used with dermoscopy.4 In particular, for melanocytic lesions that are ambiguous on dermoscopy, RCM used in addition to dermoscopy increases the mean sensitivity and specificity for melanoma diagnosis to 93% (range, 89%–96%) and 76% (range, 68%–83%), respectively.5 Although these reported sensitivities are comparable to dermoscopy, the specificity is superior, especially for detecting hypomelanotic and amelanotic melanomas, which often lack specific features on dermoscopy.6-8
The combination of RCM with dermoscopy has reduced the number of unnecessary excisions of benign nevi by more than 50% when compared to dermoscopy alone.9 One study showed that the number needed to treat (ie, excise) a melanoma decreased from 14.6 with dermoscopy alone to 6.8 when guided by dermoscopy and RCM imaging.9 In a similar study, the number needed to treat dropped from 19.41 with dermoscopy alone to 6.25 with dermoscopy and RCM.10
These studies were not looking to evaluate RCM as a replacement test but rather as an add-on test to dermoscopy. Reflectance confocal microscopy imaging takes longer than dermoscopy for each lesion; therefore, RCM should only be used as an adjunctive tool to dermoscopy and not as an initial screening test. Consequentially, a dermatologist skilled in dermoscopy is essential in deciding which lesions would be appropriate for subsequent RCM imaging.
In Vivo Margin Mapping as an Adjunct to Surgery
Oftentimes, tumor margins are poorly defined and can be difficult to map clinically and dermoscopically. Studies have demonstrated the use of RCM in delineation of surgical margins prior to surgery or excisional biopsies.11,12 Alternatively, when complete removal at biopsy would be impractical (eg, for extremely large lesions or lesions located in cosmetically sensitive areas such as the face), RCM can be used to pick the best site for an appropriate biopsy, which decreases the chance of sampling error due to skip lesions and increases histologic accuracy.
Nonsurgical Treatment Monitoring
One advantage of RCM over conventional histology is that RCM imaging leaves the tissue intact, allowing dynamic changes to be studied over time, which is useful for monitoring nonmelanoma skin cancers and lentigo maligna being treated with noninvasive therapeutic modalities.13 If not as a definitive treatment, RCM can act as an adjunct for surgery by monitoring reduction in lesion size prior to Mohs micrographic surgery, thereby decreasing the resulting surgical defect.14
Limitations
Imaging Depth
Although RCM is a revolutionary device in the field of dermatology, it has several limitations. With a maximal imaging depth of 350 µm, the imaging resolution decreases substantially with depth, limiting accurate interpretation to 200 µm. Reflectance confocal microscopy can only image the superficial portion of a lesion; therefore, deep tumor margins cannot be assessed. Hypertrophic or hyperkeratotic lesions, including lesions on the palms and soles, also are unable to be imaged with RCM. This limitation in depth penetration makes treatment monitoring impossible for invasive lesions that extend into the dermal layer.
Difficult-to-Reach Areas
Another limitation is the difficulty imaging areas such as the ocular canthi, nasal alae, or helices of the ear due to the wide probe size on the VivaScope 1500. The advent of the smaller handheld VivaScope 3000 device allows for improved imaging of concave services and difficult lesions at the risk of less accurate imaging, low field of view, and no reimbursement at present.
False-Positive Results
Although RCM has been shown to be helpful in reducing unnecessary biopsies, there still is the issue of false-positives on imaging. False-positives most commonly occur in nevi with severe atypia or when Langerhans cells are present that cannot always be differentiated from melanocytic cells.3,15,16 One prospective study found 7 false-positive results from 63 sites using RCM for the diagnosis of lentigo malignas.16 False-negatives can occur in the presence of inflammatory infiltrates and scar tissue that can hide cellular morphology or in sampling errors due to skip lesions.3,16
Time Efficiency
The time required for acquisition of RCM mosaics and stacks followed by reading and interpretation can be substantial depending on the size and complexity of the lesion, which is a major limitation for use of RCM in busy dermatology practices; therefore, RCM should be reserved for lesions selected to undergo biopsy that are clinically equivocal for malignancy prior to RCM examination.17 It would not be cost-effective or time effective to evaluate lesions that either clinically or dermoscopically have a high probability of malignancy; however, patients and physicians may opt for increased specificity at the expense of time, particularly when a lesion is located on a cosmetically sensitive area, as patients can avoid initial histologic biopsy and gain the cosmetic benefit of going straight to surgery versus obtaining an initial diagnostic biopsy.
Cost
Lastly, the high cost involved in purchasing an RCM device and the training involved to use and interpret RCM images currently limits RCM to large academic centers. Reimbursement may make more widespread use feasible. In any event, RCM imaging should be part of the curriculum for both dermatology and pathology trainees.
Future Directions
In vivo RCM is a noninvasive imaging modality that allows for real-time evaluation of the skin. Used in conjunction with dermoscopy, RCM can substantially improve diagnostic accuracy and reduce the number of unnecessary biopsies. Now that RCM has finally gained foundational CPT codes and insurance reimbursement, there may be a growing demand for clinicians to incorporate this technology into their clinical practice.
- Current Procedural Terminology 2017, Professional Edition. Chicago IL: American Medical Association; 2016.
- Que SK, Fraga-Braghiroli N, Grant-Kels JM, et al. Through the looking glass: basics and principles of reflectance confocal microscopy [published online June 4, 2015]. J Am Acad Dermatol. 2015;73:276-284.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside [published online October 27, 2016]. Lasers Surg Med. 2017;49:7-19.
- Xiong YD, Ma S, Li X, et al. A meta-analysis of reflectance confocal microscopy for the diagnosis of malignant skin tumours. J Eur Acad Dermatol Venereol. 2016;30:1295-1302.
- Stevenson AD, Mickan S, Mallett S, et al. Systematic review of diagnostic accuracy of reflectance confocal microscopy for melanoma diagnosis in patients with clinically equivocal skin lesions. Dermatol Pract Concept. 2013;3:19-27.
- Busam KJ, Hester K, Charles C, et al. Detection of clinically amelanotic malignant melanoma and assessment of its margins by in vivo confocal scanning laser microscopy. Arch Dermatol. 2001;137:923-929.
- Losi A, Longo C, Cesinaro AM, et al. Hyporeflective pagetoid cells: a new clue for amelanotic melanoma diagnosis by reflectance confocal microscopy. Br J Dermatol. 2014;171:48-54.
- Guitera P, Menzies SQ, Argenziano G, et al. Dermoscopy and in vivo confocal microscopy are complementary techniques for the diagnosis of difficult amelanotic and light-coloured skin lesions [published online October 12, 2016]. Br J Dermatol. 2016;175:1311-1319.
- Pellacani G, Pepe P, Casari A, et al. Reflectance confocal microscopy as a second-level examination in skin oncology improves diagnostic accuracy and saves unnecessary excisions: a longitudinal prospective study. Br J Dermatol. 2014;171:1044-1051.
- Pellacani G, Witkowski A, Cesinaro AM, et al. Cost-benefit of reflectance confocal microscopy in the diagnostic performance of melanoma. J Eur Acad Dermatol Venereol. 2016;30:413-419.
- Champin J, Perrot JL, Cinotti E, et al. In vivo reflectance confocal microscopy to optimize the spaghetti technique for defining surgical margins of lentigo maligna. Dermatol Surg. 2014;40:247-256.
- Hibler BP, Cordova M, Wong RJ, et al. Intraoperative real-time reflectance confocal microscopy for guiding surgical margins of lentigo maligna melanoma. Dermatol Surg. 2015;41:980-983.
- Ulrich M, Lange-Asschenfeldt S, Gonzalez S. The use of reflectance confocal microscopy for monitoring response to therapy of skin malignancies. Dermatol Pract Concept. 2012;2:202a10.
- Torres A, Niemeyer A, Berkes B, et al. 5% imiquimod cream and reflectance-mode confocal microscopy as adjunct modalities to Mohs micrographic surgery for treatment of basal cell carcinoma. Dermatol Surg. 2004;30(12, pt 1):1462-1469.
- Hashemi P, Pulitzer MP, Scope A, et al. Langerhans cells and melanocytes share similar morphologic features under in vivo reflectance confocal microscopy: a challenge for melanoma diagnosis. J Am Acad Dermatol. 2012;66:452-462.
- Menge TD, Hibler BP, Cordova MA, et al. Concordance of handheld reflectance confocal microscopy (RCM) with histopathology in the diagnosis of lentigo maligna (LM): a prospective study. J Am Acad Dermatol. 2016;74:1114-1120.
- Borsari S, Pampena R, Lallas A, et al. Clinical indications for use of reflectance confocal microscopy for skin cancer diagnosis. JAMA Dermatol. 2016;152:1093-1098.
- Current Procedural Terminology 2017, Professional Edition. Chicago IL: American Medical Association; 2016.
- Que SK, Fraga-Braghiroli N, Grant-Kels JM, et al. Through the looking glass: basics and principles of reflectance confocal microscopy [published online June 4, 2015]. J Am Acad Dermatol. 2015;73:276-284.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside [published online October 27, 2016]. Lasers Surg Med. 2017;49:7-19.
- Xiong YD, Ma S, Li X, et al. A meta-analysis of reflectance confocal microscopy for the diagnosis of malignant skin tumours. J Eur Acad Dermatol Venereol. 2016;30:1295-1302.
- Stevenson AD, Mickan S, Mallett S, et al. Systematic review of diagnostic accuracy of reflectance confocal microscopy for melanoma diagnosis in patients with clinically equivocal skin lesions. Dermatol Pract Concept. 2013;3:19-27.
- Busam KJ, Hester K, Charles C, et al. Detection of clinically amelanotic malignant melanoma and assessment of its margins by in vivo confocal scanning laser microscopy. Arch Dermatol. 2001;137:923-929.
- Losi A, Longo C, Cesinaro AM, et al. Hyporeflective pagetoid cells: a new clue for amelanotic melanoma diagnosis by reflectance confocal microscopy. Br J Dermatol. 2014;171:48-54.
- Guitera P, Menzies SQ, Argenziano G, et al. Dermoscopy and in vivo confocal microscopy are complementary techniques for the diagnosis of difficult amelanotic and light-coloured skin lesions [published online October 12, 2016]. Br J Dermatol. 2016;175:1311-1319.
- Pellacani G, Pepe P, Casari A, et al. Reflectance confocal microscopy as a second-level examination in skin oncology improves diagnostic accuracy and saves unnecessary excisions: a longitudinal prospective study. Br J Dermatol. 2014;171:1044-1051.
- Pellacani G, Witkowski A, Cesinaro AM, et al. Cost-benefit of reflectance confocal microscopy in the diagnostic performance of melanoma. J Eur Acad Dermatol Venereol. 2016;30:413-419.
- Champin J, Perrot JL, Cinotti E, et al. In vivo reflectance confocal microscopy to optimize the spaghetti technique for defining surgical margins of lentigo maligna. Dermatol Surg. 2014;40:247-256.
- Hibler BP, Cordova M, Wong RJ, et al. Intraoperative real-time reflectance confocal microscopy for guiding surgical margins of lentigo maligna melanoma. Dermatol Surg. 2015;41:980-983.
- Ulrich M, Lange-Asschenfeldt S, Gonzalez S. The use of reflectance confocal microscopy for monitoring response to therapy of skin malignancies. Dermatol Pract Concept. 2012;2:202a10.
- Torres A, Niemeyer A, Berkes B, et al. 5% imiquimod cream and reflectance-mode confocal microscopy as adjunct modalities to Mohs micrographic surgery for treatment of basal cell carcinoma. Dermatol Surg. 2004;30(12, pt 1):1462-1469.
- Hashemi P, Pulitzer MP, Scope A, et al. Langerhans cells and melanocytes share similar morphologic features under in vivo reflectance confocal microscopy: a challenge for melanoma diagnosis. J Am Acad Dermatol. 2012;66:452-462.
- Menge TD, Hibler BP, Cordova MA, et al. Concordance of handheld reflectance confocal microscopy (RCM) with histopathology in the diagnosis of lentigo maligna (LM): a prospective study. J Am Acad Dermatol. 2016;74:1114-1120.
- Borsari S, Pampena R, Lallas A, et al. Clinical indications for use of reflectance confocal microscopy for skin cancer diagnosis. JAMA Dermatol. 2016;152:1093-1098.
Practice Points
- Reflectance confocal microscopy (RCM) recently received Category I Current Procedural Terminology codes for reimbursement comparable to a skin biopsy.
- When used in combination with dermoscopy, RCM has been shown to increase diagnostic accuracy of skin cancer.
- Reflectance confocal microscopy also is useful in surgical treatment planning and monitoring nonsurgical treatments over time.
- Limitations of RCM imaging include low imaging depth, difficulty in imaging certain areas of the skin, learning curve for interpreting these images, and the cost of equipment.

Stem cell therapy significantly improves ulcer healing
PORTLAND, ORE. – Treating chronic venous leg ulcers with mesenchymal stem cells and fibrin spray significantly improved wound healing, compared with vehicle control or saline plus conventional therapy, according to the results of a small randomized, controlled, double-blind pilot trial.
“Topical application of autologous, bone-marrow–derived mesenchymal stem cells may be an effective way to promote healing in patients with difficult-to-heal wounds,” said Ayman Grada, MD, of the department of dermatology at Boston University. “However, larger studies are needed to confirm this finding.”
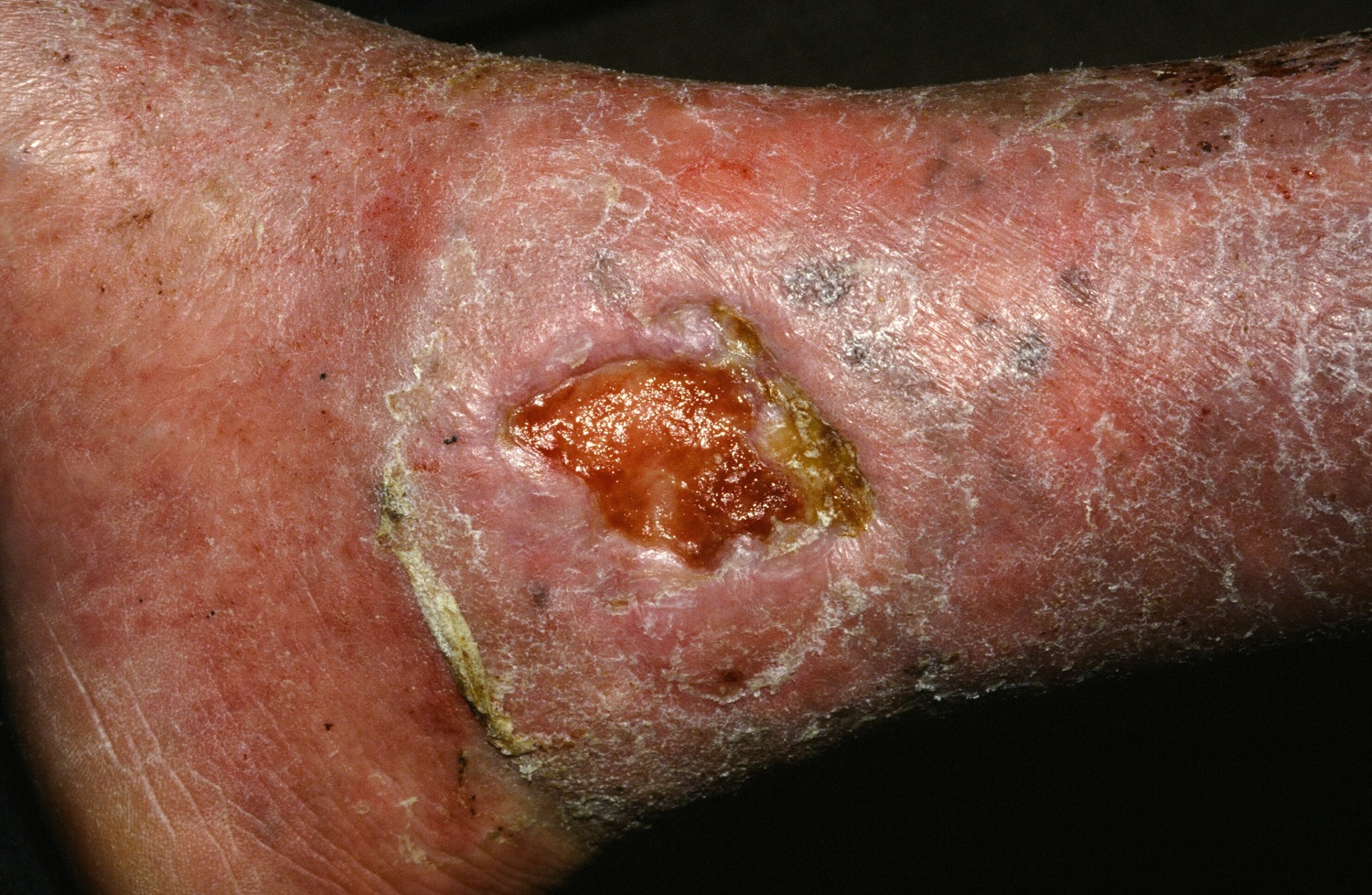
“Various treatment modalities have been used, but treatment outcomes are not always satisfactory,” said Dr. Grada. “In about 60% of cases, wounds fail to close, and there is also a high rate of recurrence.”
Preclinical work in several animal models indicated that applying mesenchymal stem cells to wounds accelerated healing through a variety of mechanisms, Dr. Grada noted. Based on that premise, he and his associates hypothesized that autologous cultured mesenchymal stem cells could accelerate wound healing in humans.
To test that idea, they randomly assigned the 11 trial participants to one of two control treatments or to the stem cell intervention. Four patients received normal saline with conventional standard care, three patients received fibrin spray plus conventional therapy, and four patients received conventional therapy plus autologous mesenchymal stem cells delivered in fibrin spray at a dose of 1 x 106 cells per square centimeter of wound surface. Patients were treated every 3 weeks, up to three times or until complete wound healing, and were followed for up to 24 weeks.
To acquire the stem cells, the researchers obtained 30- to 50-mL samples of bone marrow aspirate from the iliac crest, then separated and cultured the cells in-house. The controls underwent sham aspiration with needles that did not penetrate the bone, Dr. Grada said. At each 4-week follow-up visit, the investigators measured the perimeter and area of each wound and analyzed the results with public domain software called ImageJ. They calculated the linear advance of the wound margin by dividing change in area by average perimeter.
The healing rate of the intervention group outpaced that of either control group at each time point measured, Dr. Grada said. Average weekly healing rates by time point ranged between –0.002 cm and 0.006 cm for the saline group and between –0.05 cm and 0.01 cm for the fibrin spray group. Neither of these control groups achieved meaningful wound closure by week 24.
In contrast, stem cell recipients experienced consistent wound closure at rates of 0.11-0.13 cm per week. The study was too small for conventional statistical analysis, but a Bayesian time aggregated one-way analysis of variance yielded a statistically significant difference in healing rates among groups (P less than .0005).
Dr. Grada also discussed several case studies. An 82-year-old white woman with a decades-long history of venous ulcers experienced complete wound healing with mesenchymal stem cell therapy, which enabled her to become more independent within her long-term care facility. A 75-year-old African American woman achieved 80% wound healing with stem cell therapy after previously having failed to benefit from two applications of bioengineered skin.
Finally, a 39-year-old man with chronic, treatment-resistant venous ulcers achieved partial wound healing. “He has almost healed, with very thin epidermal coverage, but never to the point of no exudate and complete closure,” Dr. Grada said. “Therefore, we could not declare him healed, even though the ulcer was smaller at the end of the study.”
No patient in the study experienced adverse events from treatment. However, recruiting for the trial was difficult, because patients were reluctant to undergo bone marrow aspiration, Dr. Grada said.
Previous work indicates that the initial rate at which the wound heals dictates its final rate (J Am Acad Dermatol. 1993 Mar;28[3]:418-21), and that 4 weeks is enough to establish a healing trend, he noted. Dr. Grada concluded by quoting Hippocrates: “Natural forces within us are the true healers of disease.”
The National Institutes of Health supported the trial. Dr. Grada had no conflicts of interest.
PORTLAND, ORE. – Treating chronic venous leg ulcers with mesenchymal stem cells and fibrin spray significantly improved wound healing, compared with vehicle control or saline plus conventional therapy, according to the results of a small randomized, controlled, double-blind pilot trial.
“Topical application of autologous, bone-marrow–derived mesenchymal stem cells may be an effective way to promote healing in patients with difficult-to-heal wounds,” said Ayman Grada, MD, of the department of dermatology at Boston University. “However, larger studies are needed to confirm this finding.”
“Various treatment modalities have been used, but treatment outcomes are not always satisfactory,” said Dr. Grada. “In about 60% of cases, wounds fail to close, and there is also a high rate of recurrence.”
Preclinical work in several animal models indicated that applying mesenchymal stem cells to wounds accelerated healing through a variety of mechanisms, Dr. Grada noted. Based on that premise, he and his associates hypothesized that autologous cultured mesenchymal stem cells could accelerate wound healing in humans.
To test that idea, they randomly assigned the 11 trial participants to one of two control treatments or to the stem cell intervention. Four patients received normal saline with conventional standard care, three patients received fibrin spray plus conventional therapy, and four patients received conventional therapy plus autologous mesenchymal stem cells delivered in fibrin spray at a dose of 1 x 106 cells per square centimeter of wound surface. Patients were treated every 3 weeks, up to three times or until complete wound healing, and were followed for up to 24 weeks.
To acquire the stem cells, the researchers obtained 30- to 50-mL samples of bone marrow aspirate from the iliac crest, then separated and cultured the cells in-house. The controls underwent sham aspiration with needles that did not penetrate the bone, Dr. Grada said. At each 4-week follow-up visit, the investigators measured the perimeter and area of each wound and analyzed the results with public domain software called ImageJ. They calculated the linear advance of the wound margin by dividing change in area by average perimeter.
The healing rate of the intervention group outpaced that of either control group at each time point measured, Dr. Grada said. Average weekly healing rates by time point ranged between –0.002 cm and 0.006 cm for the saline group and between –0.05 cm and 0.01 cm for the fibrin spray group. Neither of these control groups achieved meaningful wound closure by week 24.
In contrast, stem cell recipients experienced consistent wound closure at rates of 0.11-0.13 cm per week. The study was too small for conventional statistical analysis, but a Bayesian time aggregated one-way analysis of variance yielded a statistically significant difference in healing rates among groups (P less than .0005).
Dr. Grada also discussed several case studies. An 82-year-old white woman with a decades-long history of venous ulcers experienced complete wound healing with mesenchymal stem cell therapy, which enabled her to become more independent within her long-term care facility. A 75-year-old African American woman achieved 80% wound healing with stem cell therapy after previously having failed to benefit from two applications of bioengineered skin.
Finally, a 39-year-old man with chronic, treatment-resistant venous ulcers achieved partial wound healing. “He has almost healed, with very thin epidermal coverage, but never to the point of no exudate and complete closure,” Dr. Grada said. “Therefore, we could not declare him healed, even though the ulcer was smaller at the end of the study.”
No patient in the study experienced adverse events from treatment. However, recruiting for the trial was difficult, because patients were reluctant to undergo bone marrow aspiration, Dr. Grada said.
Previous work indicates that the initial rate at which the wound heals dictates its final rate (J Am Acad Dermatol. 1993 Mar;28[3]:418-21), and that 4 weeks is enough to establish a healing trend, he noted. Dr. Grada concluded by quoting Hippocrates: “Natural forces within us are the true healers of disease.”
The National Institutes of Health supported the trial. Dr. Grada had no conflicts of interest.
PORTLAND, ORE. – Treating chronic venous leg ulcers with mesenchymal stem cells and fibrin spray significantly improved wound healing, compared with vehicle control or saline plus conventional therapy, according to the results of a small randomized, controlled, double-blind pilot trial.
“Topical application of autologous, bone-marrow–derived mesenchymal stem cells may be an effective way to promote healing in patients with difficult-to-heal wounds,” said Ayman Grada, MD, of the department of dermatology at Boston University. “However, larger studies are needed to confirm this finding.”
“Various treatment modalities have been used, but treatment outcomes are not always satisfactory,” said Dr. Grada. “In about 60% of cases, wounds fail to close, and there is also a high rate of recurrence.”
Preclinical work in several animal models indicated that applying mesenchymal stem cells to wounds accelerated healing through a variety of mechanisms, Dr. Grada noted. Based on that premise, he and his associates hypothesized that autologous cultured mesenchymal stem cells could accelerate wound healing in humans.
To test that idea, they randomly assigned the 11 trial participants to one of two control treatments or to the stem cell intervention. Four patients received normal saline with conventional standard care, three patients received fibrin spray plus conventional therapy, and four patients received conventional therapy plus autologous mesenchymal stem cells delivered in fibrin spray at a dose of 1 x 106 cells per square centimeter of wound surface. Patients were treated every 3 weeks, up to three times or until complete wound healing, and were followed for up to 24 weeks.
To acquire the stem cells, the researchers obtained 30- to 50-mL samples of bone marrow aspirate from the iliac crest, then separated and cultured the cells in-house. The controls underwent sham aspiration with needles that did not penetrate the bone, Dr. Grada said. At each 4-week follow-up visit, the investigators measured the perimeter and area of each wound and analyzed the results with public domain software called ImageJ. They calculated the linear advance of the wound margin by dividing change in area by average perimeter.
The healing rate of the intervention group outpaced that of either control group at each time point measured, Dr. Grada said. Average weekly healing rates by time point ranged between –0.002 cm and 0.006 cm for the saline group and between –0.05 cm and 0.01 cm for the fibrin spray group. Neither of these control groups achieved meaningful wound closure by week 24.
In contrast, stem cell recipients experienced consistent wound closure at rates of 0.11-0.13 cm per week. The study was too small for conventional statistical analysis, but a Bayesian time aggregated one-way analysis of variance yielded a statistically significant difference in healing rates among groups (P less than .0005).
Dr. Grada also discussed several case studies. An 82-year-old white woman with a decades-long history of venous ulcers experienced complete wound healing with mesenchymal stem cell therapy, which enabled her to become more independent within her long-term care facility. A 75-year-old African American woman achieved 80% wound healing with stem cell therapy after previously having failed to benefit from two applications of bioengineered skin.
Finally, a 39-year-old man with chronic, treatment-resistant venous ulcers achieved partial wound healing. “He has almost healed, with very thin epidermal coverage, but never to the point of no exudate and complete closure,” Dr. Grada said. “Therefore, we could not declare him healed, even though the ulcer was smaller at the end of the study.”
No patient in the study experienced adverse events from treatment. However, recruiting for the trial was difficult, because patients were reluctant to undergo bone marrow aspiration, Dr. Grada said.
Previous work indicates that the initial rate at which the wound heals dictates its final rate (J Am Acad Dermatol. 1993 Mar;28[3]:418-21), and that 4 weeks is enough to establish a healing trend, he noted. Dr. Grada concluded by quoting Hippocrates: “Natural forces within us are the true healers of disease.”
The National Institutes of Health supported the trial. Dr. Grada had no conflicts of interest.
At SID 2017
Key clinical point: Treating chronic venous leg ulcers with mesenchymal stem cells and fibrin spray significantly improved wound healing, compared with vehicle control or saline plus conventional therapy.
Major finding: Neither control group achieved meaningful wound closure by week 24, while stem cell recipients experienced consistent wound closure at rates of 0.11-0.13 cm per week (P less than .0005 for difference in healing rates among groups).
Data source: A randomized, controlled, double-blind pilot trial of 11 patients.
Disclosures: The National Institutes of Health supported the study. Dr. Grada had no conflicts of interest.
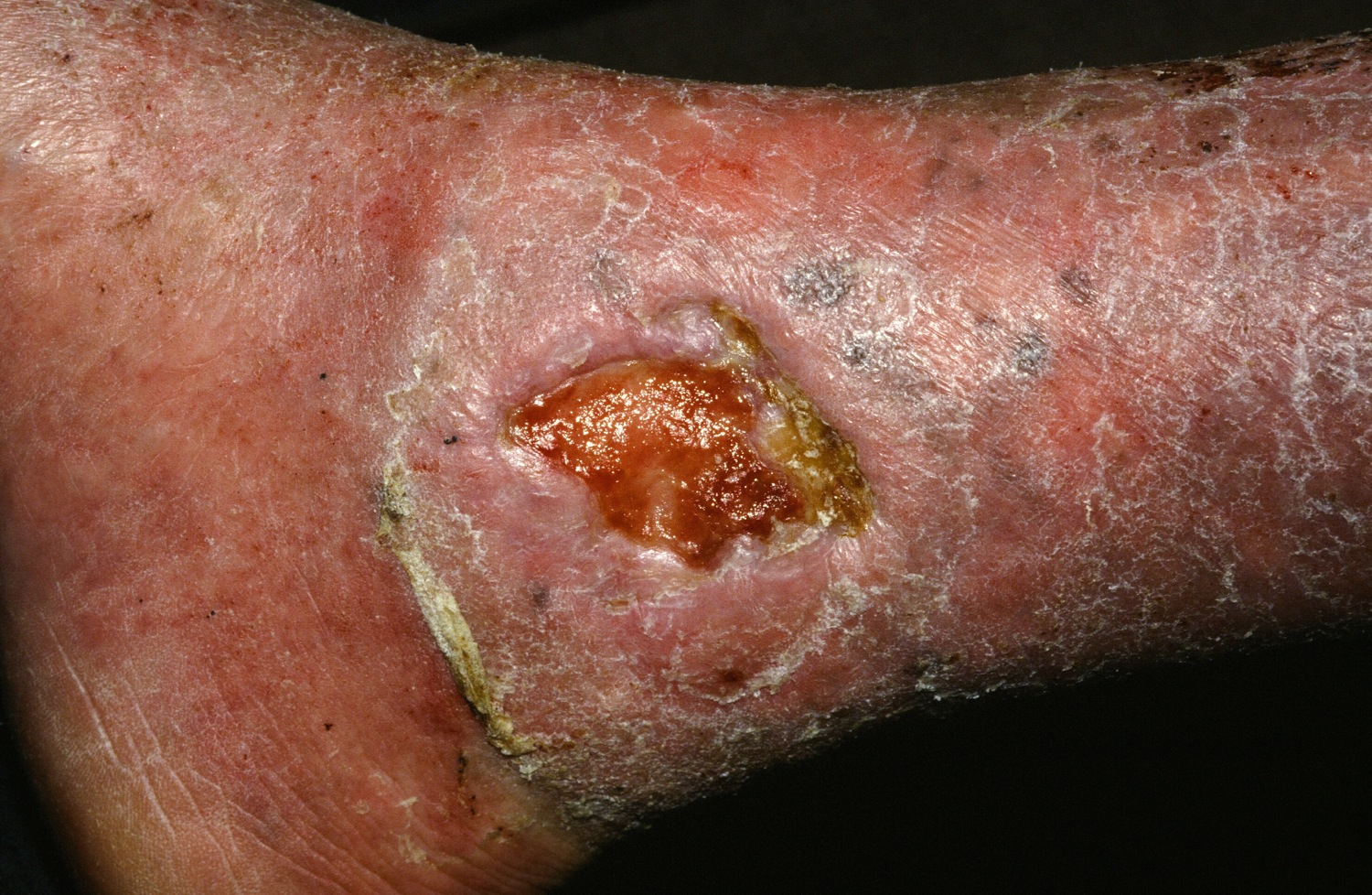
Canagliflozin gets boxed warning for amputation
The Food and Drug Administration has added a boxed warning to the label of diabetes drug canagliflozin for the risk of lower limb amputation.
The agency cited data from two clinical trials showing nearly double the risk of leg and foot amputations in patients treated with the canagliflozin, a sodium-glucose cotransporter 2 (SGLT2) inhibitor, compared with placebo, in a recent statement.![]()
The trials, which followed participants for an average of 5.7 and 2.1 years, respectively, showed that lower limb infections, gangrene, diabetic foot ulcers, and ischemia commonly occurred prior to the need for amputation.
The boxed warning advises physicians to consider a patient’s history of prior amputation, peripheral vascular disease, neuropathy, and diabetic foot ulcers before prescribing canagliflozin and to monitor patients for pain, tenderness, sores, ulcers, or infections on the feet or legs.
Consider discontinuing canagliflozin in these patients, as well as those with symptoms of hypotension, ketoacidosis, elevated serum potassium levels, severe urinary tract infections, hypoglycemia in combination with other prescription diabetes medicines, yeast infections, bone breaks, and increased cholesterol, according to the FDA.
The FDA first issued a safety communication on canagliflozin about a year ago but, at the time, did not advise assessing a patient’s risk for amputation.
Canagliflozin, marketed as Invokana, Invokamet, and Invokamet XR by Janssen Pharmaceuticals, was approved by the FDA in March 2013.
Adverse events involving canagliflozin – or any drug – should be reported to the FDA MedWatch program.
[email protected]
On Twitter @whitneymcknight
The Food and Drug Administration has added a boxed warning to the label of diabetes drug canagliflozin for the risk of lower limb amputation.
The agency cited data from two clinical trials showing nearly double the risk of leg and foot amputations in patients treated with the canagliflozin, a sodium-glucose cotransporter 2 (SGLT2) inhibitor, compared with placebo, in a recent statement.![]()
The trials, which followed participants for an average of 5.7 and 2.1 years, respectively, showed that lower limb infections, gangrene, diabetic foot ulcers, and ischemia commonly occurred prior to the need for amputation.
The boxed warning advises physicians to consider a patient’s history of prior amputation, peripheral vascular disease, neuropathy, and diabetic foot ulcers before prescribing canagliflozin and to monitor patients for pain, tenderness, sores, ulcers, or infections on the feet or legs.
Consider discontinuing canagliflozin in these patients, as well as those with symptoms of hypotension, ketoacidosis, elevated serum potassium levels, severe urinary tract infections, hypoglycemia in combination with other prescription diabetes medicines, yeast infections, bone breaks, and increased cholesterol, according to the FDA.
The FDA first issued a safety communication on canagliflozin about a year ago but, at the time, did not advise assessing a patient’s risk for amputation.
Canagliflozin, marketed as Invokana, Invokamet, and Invokamet XR by Janssen Pharmaceuticals, was approved by the FDA in March 2013.
Adverse events involving canagliflozin – or any drug – should be reported to the FDA MedWatch program.
[email protected]
On Twitter @whitneymcknight
The Food and Drug Administration has added a boxed warning to the label of diabetes drug canagliflozin for the risk of lower limb amputation.
The agency cited data from two clinical trials showing nearly double the risk of leg and foot amputations in patients treated with the canagliflozin, a sodium-glucose cotransporter 2 (SGLT2) inhibitor, compared with placebo, in a recent statement.![]()
The trials, which followed participants for an average of 5.7 and 2.1 years, respectively, showed that lower limb infections, gangrene, diabetic foot ulcers, and ischemia commonly occurred prior to the need for amputation.
The boxed warning advises physicians to consider a patient’s history of prior amputation, peripheral vascular disease, neuropathy, and diabetic foot ulcers before prescribing canagliflozin and to monitor patients for pain, tenderness, sores, ulcers, or infections on the feet or legs.
Consider discontinuing canagliflozin in these patients, as well as those with symptoms of hypotension, ketoacidosis, elevated serum potassium levels, severe urinary tract infections, hypoglycemia in combination with other prescription diabetes medicines, yeast infections, bone breaks, and increased cholesterol, according to the FDA.
The FDA first issued a safety communication on canagliflozin about a year ago but, at the time, did not advise assessing a patient’s risk for amputation.
Canagliflozin, marketed as Invokana, Invokamet, and Invokamet XR by Janssen Pharmaceuticals, was approved by the FDA in March 2013.
Adverse events involving canagliflozin – or any drug – should be reported to the FDA MedWatch program.
[email protected]
On Twitter @whitneymcknight
High allele level linked to lamotrigine-induced SCAR
, reported Byung-Keun Kim, MD, of Seoul National University and associates.
In a study of 18 Korean patients with lamotrigine-induced SCAR, a control group of Korean lamotrigine-tolerant patients, and a control group of the general Korean population, the frequency of the HLA-A*31:01 allele was significantly higher in the lamotrigine-induced SCAR patients than in the lamotrigine-tolerant patients (odds ratio, 11.43; P = .0037) or the other control group (OR, 7.27; P = .00034).
High levels of the HLA-A*31:01 allele also have been reported in Korean patients with carbamazepine-induced SCAR, suggesting an association with the HLA allele and drug-induced SCAR that is specific to ethnicity.
That idea is supported by reports that the HLA-B*15:02 allele is a well-known risk allele of carbamazepine-induced SCAR in Han Chinese and Southeast Asians and that other HLA alleles have been significantly associated with SCAR only with patients of European ancestry or only with patients of Mestizo Mexican ancestry, Dr. Kim and associates said.
The SCAR in this study were Stevens-Johnson syndrome, toxic epidermal necrolysis, and drug rash with eosinophilia and systemic syndrome, also known as DRESS.
Read more in the Annals of Allergy, Asthma & Immunology (2017 May;118[5]:629-30).
, reported Byung-Keun Kim, MD, of Seoul National University and associates.
In a study of 18 Korean patients with lamotrigine-induced SCAR, a control group of Korean lamotrigine-tolerant patients, and a control group of the general Korean population, the frequency of the HLA-A*31:01 allele was significantly higher in the lamotrigine-induced SCAR patients than in the lamotrigine-tolerant patients (odds ratio, 11.43; P = .0037) or the other control group (OR, 7.27; P = .00034).
High levels of the HLA-A*31:01 allele also have been reported in Korean patients with carbamazepine-induced SCAR, suggesting an association with the HLA allele and drug-induced SCAR that is specific to ethnicity.
That idea is supported by reports that the HLA-B*15:02 allele is a well-known risk allele of carbamazepine-induced SCAR in Han Chinese and Southeast Asians and that other HLA alleles have been significantly associated with SCAR only with patients of European ancestry or only with patients of Mestizo Mexican ancestry, Dr. Kim and associates said.
The SCAR in this study were Stevens-Johnson syndrome, toxic epidermal necrolysis, and drug rash with eosinophilia and systemic syndrome, also known as DRESS.
Read more in the Annals of Allergy, Asthma & Immunology (2017 May;118[5]:629-30).
, reported Byung-Keun Kim, MD, of Seoul National University and associates.
In a study of 18 Korean patients with lamotrigine-induced SCAR, a control group of Korean lamotrigine-tolerant patients, and a control group of the general Korean population, the frequency of the HLA-A*31:01 allele was significantly higher in the lamotrigine-induced SCAR patients than in the lamotrigine-tolerant patients (odds ratio, 11.43; P = .0037) or the other control group (OR, 7.27; P = .00034).
High levels of the HLA-A*31:01 allele also have been reported in Korean patients with carbamazepine-induced SCAR, suggesting an association with the HLA allele and drug-induced SCAR that is specific to ethnicity.
That idea is supported by reports that the HLA-B*15:02 allele is a well-known risk allele of carbamazepine-induced SCAR in Han Chinese and Southeast Asians and that other HLA alleles have been significantly associated with SCAR only with patients of European ancestry or only with patients of Mestizo Mexican ancestry, Dr. Kim and associates said.
The SCAR in this study were Stevens-Johnson syndrome, toxic epidermal necrolysis, and drug rash with eosinophilia and systemic syndrome, also known as DRESS.
Read more in the Annals of Allergy, Asthma & Immunology (2017 May;118[5]:629-30).
FROM THE ANNALS OF ALLERGY, ASTHMA & IMMUNOLOGY
SJS, TEN occur less frequently in children than adults
Although incidences of Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are lower in children than in adults, the increased costs, lengths of stay, and mortality still pose a substantial health burden, said Derek Y. Hsu, of Northwestern University, Chicago, and his associates.
Using data from the 2009-2012 Nationwide Inpatient Sample of 1,687,172 pediatric admissions, “estimated frequencies per million children ranged from 4.3 to 5.8 for SJS, 0.6 to 1.4 for SJS/TEN, and 0 to 0.7 for TEN,” the study researchers reported. In adults, those numbers are 9.3, 1.9, and 1.6 per million adults per year, according to a 2016 study by the authors (J Invest Dermatol. 2016 Jul;136[7]:1387-97).
Pediatric SJS, SJS/TEN, and TEN mean hospital costs were $24,947, $63,787, and $102,243, respectively, compared with $10,496 for the control group.
The mean length of stay for patients with SJS, SJS/TEN, and TEN was 9.4 days, 15.7 days, and 20.4 days, compared with 4.6 days in children without these disorders, respectively, and they most often were discharged to their home or to other self-care.
“One in 10 children with SJS, SJS/TEN, and TEN underwent mechanical ventilation,” Mr. Hsu and his associates reported.
Mortality was 0% for SJS, 4% for SJS/TEN, and 16% for TEN.
Read more at the Journal of the American Academy of Dermatology (2017 May;76[5]:811-7).
Although incidences of Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are lower in children than in adults, the increased costs, lengths of stay, and mortality still pose a substantial health burden, said Derek Y. Hsu, of Northwestern University, Chicago, and his associates.
Using data from the 2009-2012 Nationwide Inpatient Sample of 1,687,172 pediatric admissions, “estimated frequencies per million children ranged from 4.3 to 5.8 for SJS, 0.6 to 1.4 for SJS/TEN, and 0 to 0.7 for TEN,” the study researchers reported. In adults, those numbers are 9.3, 1.9, and 1.6 per million adults per year, according to a 2016 study by the authors (J Invest Dermatol. 2016 Jul;136[7]:1387-97).
Pediatric SJS, SJS/TEN, and TEN mean hospital costs were $24,947, $63,787, and $102,243, respectively, compared with $10,496 for the control group.
The mean length of stay for patients with SJS, SJS/TEN, and TEN was 9.4 days, 15.7 days, and 20.4 days, compared with 4.6 days in children without these disorders, respectively, and they most often were discharged to their home or to other self-care.
“One in 10 children with SJS, SJS/TEN, and TEN underwent mechanical ventilation,” Mr. Hsu and his associates reported.
Mortality was 0% for SJS, 4% for SJS/TEN, and 16% for TEN.
Read more at the Journal of the American Academy of Dermatology (2017 May;76[5]:811-7).
Although incidences of Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are lower in children than in adults, the increased costs, lengths of stay, and mortality still pose a substantial health burden, said Derek Y. Hsu, of Northwestern University, Chicago, and his associates.
Using data from the 2009-2012 Nationwide Inpatient Sample of 1,687,172 pediatric admissions, “estimated frequencies per million children ranged from 4.3 to 5.8 for SJS, 0.6 to 1.4 for SJS/TEN, and 0 to 0.7 for TEN,” the study researchers reported. In adults, those numbers are 9.3, 1.9, and 1.6 per million adults per year, according to a 2016 study by the authors (J Invest Dermatol. 2016 Jul;136[7]:1387-97).
Pediatric SJS, SJS/TEN, and TEN mean hospital costs were $24,947, $63,787, and $102,243, respectively, compared with $10,496 for the control group.
The mean length of stay for patients with SJS, SJS/TEN, and TEN was 9.4 days, 15.7 days, and 20.4 days, compared with 4.6 days in children without these disorders, respectively, and they most often were discharged to their home or to other self-care.
“One in 10 children with SJS, SJS/TEN, and TEN underwent mechanical ventilation,” Mr. Hsu and his associates reported.
Mortality was 0% for SJS, 4% for SJS/TEN, and 16% for TEN.
Read more at the Journal of the American Academy of Dermatology (2017 May;76[5]:811-7).
Hidradenitis suppurativa diagnosis typically delayed in children
WASHINGTON – Children with hidradenitis suppurativa (HS) may suffer with symptoms for an average of 7 years before they are diagnosed, according to pediatric dermatologist Anna Yasmine Kirkorian, MD.
Data from a 2015 study showed that 73% of pediatric patients with HS were diagnosed more than 2 years after the onset of symptoms, said Dr. Kirkorian of the department of dermatology at Children’s National Health System and George Washington University, Washington. (Br J Dermatol. 2015 Dec;173[6]:1546-9).

Genetics can play a role in HS, likely via mutations in the gamma-secretase protein that leads to epidermal differentiation and immune regulation, Dr. Kirkorian said. Most of her patients with HS are black, and a recent study described a gamma-secretase mutation in a black family of a proband and four family members, she noted (JAMA Dermatol. 2015 Jun;151[6]:668-70). Gamma-secretase mutations also have been identified in Han Chinese populations, she said.
HS also is associated with precocious puberty. However, defining the age of onset of puberty can be difficulty because pubertal onset may vary between different ethnicities, noted Dr. Kirkorian. “Prepubertal children presenting with HS warrant an endocrinologic evaluation,” she said.
Dr. Kirkorian added that more research is needed to pinpoint the possible genetic component of HS and to identify genetic susceptibility that could lead to targeted treatment strategies.
The optimal treatment plan for pediatric HS is multimodal and addresses the comorbidities common with the condition, she said, and she predicted that specialized clinic or treatment centers that bring together areas, including psychiatry, wound care, pain management, surgery, endocrinology, and genetics, will evolve to serve these patients. To support these collaborative efforts, Dr. Kirkorian is a member of the Pediatric Dermatology Research Alliance (PeDRA), an organization formed to accelerate research on skin diseases in children.
The symposium was sponsored by AbbVie. Dr. Kirkorian had no financial conflicts to disclose. She is on the editorial board of Dermatology News.
WASHINGTON – Children with hidradenitis suppurativa (HS) may suffer with symptoms for an average of 7 years before they are diagnosed, according to pediatric dermatologist Anna Yasmine Kirkorian, MD.
Data from a 2015 study showed that 73% of pediatric patients with HS were diagnosed more than 2 years after the onset of symptoms, said Dr. Kirkorian of the department of dermatology at Children’s National Health System and George Washington University, Washington. (Br J Dermatol. 2015 Dec;173[6]:1546-9).
Genetics can play a role in HS, likely via mutations in the gamma-secretase protein that leads to epidermal differentiation and immune regulation, Dr. Kirkorian said. Most of her patients with HS are black, and a recent study described a gamma-secretase mutation in a black family of a proband and four family members, she noted (JAMA Dermatol. 2015 Jun;151[6]:668-70). Gamma-secretase mutations also have been identified in Han Chinese populations, she said.
HS also is associated with precocious puberty. However, defining the age of onset of puberty can be difficulty because pubertal onset may vary between different ethnicities, noted Dr. Kirkorian. “Prepubertal children presenting with HS warrant an endocrinologic evaluation,” she said.
Dr. Kirkorian added that more research is needed to pinpoint the possible genetic component of HS and to identify genetic susceptibility that could lead to targeted treatment strategies.
The optimal treatment plan for pediatric HS is multimodal and addresses the comorbidities common with the condition, she said, and she predicted that specialized clinic or treatment centers that bring together areas, including psychiatry, wound care, pain management, surgery, endocrinology, and genetics, will evolve to serve these patients. To support these collaborative efforts, Dr. Kirkorian is a member of the Pediatric Dermatology Research Alliance (PeDRA), an organization formed to accelerate research on skin diseases in children.
The symposium was sponsored by AbbVie. Dr. Kirkorian had no financial conflicts to disclose. She is on the editorial board of Dermatology News.
WASHINGTON – Children with hidradenitis suppurativa (HS) may suffer with symptoms for an average of 7 years before they are diagnosed, according to pediatric dermatologist Anna Yasmine Kirkorian, MD.
Data from a 2015 study showed that 73% of pediatric patients with HS were diagnosed more than 2 years after the onset of symptoms, said Dr. Kirkorian of the department of dermatology at Children’s National Health System and George Washington University, Washington. (Br J Dermatol. 2015 Dec;173[6]:1546-9).
Genetics can play a role in HS, likely via mutations in the gamma-secretase protein that leads to epidermal differentiation and immune regulation, Dr. Kirkorian said. Most of her patients with HS are black, and a recent study described a gamma-secretase mutation in a black family of a proband and four family members, she noted (JAMA Dermatol. 2015 Jun;151[6]:668-70). Gamma-secretase mutations also have been identified in Han Chinese populations, she said.
HS also is associated with precocious puberty. However, defining the age of onset of puberty can be difficulty because pubertal onset may vary between different ethnicities, noted Dr. Kirkorian. “Prepubertal children presenting with HS warrant an endocrinologic evaluation,” she said.
Dr. Kirkorian added that more research is needed to pinpoint the possible genetic component of HS and to identify genetic susceptibility that could lead to targeted treatment strategies.
The optimal treatment plan for pediatric HS is multimodal and addresses the comorbidities common with the condition, she said, and she predicted that specialized clinic or treatment centers that bring together areas, including psychiatry, wound care, pain management, surgery, endocrinology, and genetics, will evolve to serve these patients. To support these collaborative efforts, Dr. Kirkorian is a member of the Pediatric Dermatology Research Alliance (PeDRA), an organization formed to accelerate research on skin diseases in children.
The symposium was sponsored by AbbVie. Dr. Kirkorian had no financial conflicts to disclose. She is on the editorial board of Dermatology News.

Omalizumab effects rapid, often complete, clearance of refractory bullous pemphigoid
ORLANDO – Omalizumab, a monoclonal anti-IgE antibody, may be a good option for patients with treatment-refractory bullous pemphigoid.
Patients who received omalizumab (Xolair) experienced rapid improvements, with 30%-50% lesion clearance within a week and complete clearance by 3 weeks, Kenneth Yu, MD, said at the annual meeting of the American Academy of Dermatology. With regular injections, they were kept symptom free for months. Some patients did flare, but were then easily controlled on standard treatment. Omalizumab, approved by the Food and Drug Administration in 2003, is indicated for moderate to severe persistent asthma and chronic idiopathic urticaria.

“We have now treated six patients with omalizumab with very good results with five of them. These are not your garden-variety BP patients, but people with very treatment-resistant disease who have failed treatment with corticosteroids alone, and in combination with other immunosuppressants.”
The rapid clinical improvements, along with observations that eosinophilia decreased with treatment, “strengthen the evidence that BP is an IgE-mediated, organ-specific autoimmune disease,” said Dr. Yu, senior resident in dermatology at the University of Michigan, Ann Arbor.
“Would I use this as a first-line treatment for BP? Probably not. But if you are seeing someone who’s nonresponsive to therapy, you might want to check IgE and eosinophil levels and if those are elevated, you might consider omalizumab as an adjunct treatment – and you might observe a fairly dramatic response.”
Three of Dr. Yu’s patients received omalizumab as monotherapy, and three received it in conjunction with other immunosuppressants. He described their disease presentation, treatment, and progression.
In general, Dr. Yu reserves omalizumab for patients with refractory disease and two particular clinical characteristics: high eosinophil count and elevated serum IgE. The initial dosing is based on the asthma treatment nomogram for the drug; he titrates it according to clinical response. “We don’t alter the total dose given, but we do adjust the frequency with which we give it.”
His first patient was a 70-year-old woman with a 1-year history of poorly controlled BP; she had failed prednisone, azathioprine, and minocycline. She also had a history of steroid-related vertebral compression fractures. She presented with an eosinophil count of over 400 cells/microL.
“We treated her with subcutaneous injections of 300 mg every 2 weeks for 16 weeks,” Dr. Yu said. Within 1 week, she had a 44% reduction in blisters; within 4 weeks, she had gone from 50% body surface area involvement to 5%.
After eight injections, the patient was disease free and Dr. Yu discontinued treatment. She remained clear until week 32 after treatment initiation; she had a flare manifested by increased pruritus and recurrence of lesions. Dr. Yu restarted omalizumab and the lesions cleared within 2 weeks. From weeks 35-72, the patient received five more injections and remained disease free.
“After that, she did have another flare, so we used omalizumab again,” but without the same excellent results. “She had an initial decrease in pruritus, and symptom improvement, but her disease subsequently worsened. We restarted her on prednisone and azathioprine and she has done well.”
Dr. Yu said he made “a couple of interesting observations on this case.”
“We saw no real correlation between disease activity score, and the levels of serum IgG antibodies. But we did notice a parallel correlation with the level of eosinophil and disease severity and also treatment response,” he said. “It was quite clear that immediately after injection, she had a dramatic drop in eosinophils” from 1,600 to 60 cells/microL within 24 hours.
His next case was a 72-year-old woman with a history of somewhat controlled essential tremor, and 6 months of highly pruritic BP blistering. She had been treated with 60 mg/day prednisone, but didn’t tolerate it well, developing steroid-induced psychosis with agitation and violence, and a worsening of her tremor. The steroid was tapered to 40 mg/day and azathioprine was added, but she was did not respond to this change and continued to develop new blisters each day. She was admitted to the hospital for plasmapheresis, which was not helpful. Nor did she respond to six cycles of cyclophosphamide.
At that point, Dr. Yu drew IgE and eosinophil levels: Her absolute eosinophil count was 1,600 cells/microL and IgE was 287 units/mL. He then gave the patient 300 mg omalizumab subcutaneously.
“Ten days after a single injection, her blisters had almost completely resolved,” he said. “To briefly describe her disease course, the blisters went away, and she had resolution of her pruritus. She was discharged with 1 month of prednisone, but we tapered that and have been able to maintain her on omalizumab alone. She had one mild flare, which was readily controlled with prednisone. The last time we saw her, she was disease free.”
He also described four other steroid-refractory BP patients treated with omalizumab.“Their commonalities were that they all had steroid-refractory disease that was resistant to immunosuppressants, had a high level of IgE, and most of them also had eosinophilia.”
Dr. Yu’s descriptions:
• A 78-year-old woman with refractory BP of 1.5 years responded well to three initial injections spaced 6 and 4 weeks apart, and has been well maintained for 20 months with 300-mL injections administered once a month. One relapse was easily controlled.
• A 72-year-old woman with 3.5 years of refractory BP responded well to 375 mg injections every 4 weeks and has been symptom free for a year on that maintenance dose.
• A 55-year-old woman with a 7-month history of refractory BP experienced a 30% reduction in body surface area blistering within 1 week of her first 375-mg injection. By 3 weeks, she was clear. She had three injections, 2 weeks apart, and was disease free for 3 months.
• An 86-year-old woman with longstanding refractory BP experienced a 22% reduction in blister count within a week of her first 375-mL injection. After a series of injections every 2 weeks, however, she developed an exacerbation of her preexisting chronic obstructive pulmonary disease, which was due primarily to tapering her prednisone. However, she no longer uses omalizumab.
“It is difficult to make recommendations because of the limitations of our data,” Dr. Yu said. “But based on the small number of patients we have treated, I would consider using omalizumab in patients with resistant disease who have an elevated IgE and eosinophil count. The optimal dosing regimen is not yet determined. Our approach is to start out with the asthma dosing and titrate until we see improvement. We use the highest dose indicated for the patient’s weight and IgE levels, typically 300-375 mg subcutaneously every 2-8 weeks, and start tapering when the patient gets control.”
He had no financial disclosures.
[email protected]
On Twitter @Alz_Gal
ORLANDO – Omalizumab, a monoclonal anti-IgE antibody, may be a good option for patients with treatment-refractory bullous pemphigoid.
Patients who received omalizumab (Xolair) experienced rapid improvements, with 30%-50% lesion clearance within a week and complete clearance by 3 weeks, Kenneth Yu, MD, said at the annual meeting of the American Academy of Dermatology. With regular injections, they were kept symptom free for months. Some patients did flare, but were then easily controlled on standard treatment. Omalizumab, approved by the Food and Drug Administration in 2003, is indicated for moderate to severe persistent asthma and chronic idiopathic urticaria.
“We have now treated six patients with omalizumab with very good results with five of them. These are not your garden-variety BP patients, but people with very treatment-resistant disease who have failed treatment with corticosteroids alone, and in combination with other immunosuppressants.”
The rapid clinical improvements, along with observations that eosinophilia decreased with treatment, “strengthen the evidence that BP is an IgE-mediated, organ-specific autoimmune disease,” said Dr. Yu, senior resident in dermatology at the University of Michigan, Ann Arbor.
“Would I use this as a first-line treatment for BP? Probably not. But if you are seeing someone who’s nonresponsive to therapy, you might want to check IgE and eosinophil levels and if those are elevated, you might consider omalizumab as an adjunct treatment – and you might observe a fairly dramatic response.”
Three of Dr. Yu’s patients received omalizumab as monotherapy, and three received it in conjunction with other immunosuppressants. He described their disease presentation, treatment, and progression.
In general, Dr. Yu reserves omalizumab for patients with refractory disease and two particular clinical characteristics: high eosinophil count and elevated serum IgE. The initial dosing is based on the asthma treatment nomogram for the drug; he titrates it according to clinical response. “We don’t alter the total dose given, but we do adjust the frequency with which we give it.”
His first patient was a 70-year-old woman with a 1-year history of poorly controlled BP; she had failed prednisone, azathioprine, and minocycline. She also had a history of steroid-related vertebral compression fractures. She presented with an eosinophil count of over 400 cells/microL.
“We treated her with subcutaneous injections of 300 mg every 2 weeks for 16 weeks,” Dr. Yu said. Within 1 week, she had a 44% reduction in blisters; within 4 weeks, she had gone from 50% body surface area involvement to 5%.
After eight injections, the patient was disease free and Dr. Yu discontinued treatment. She remained clear until week 32 after treatment initiation; she had a flare manifested by increased pruritus and recurrence of lesions. Dr. Yu restarted omalizumab and the lesions cleared within 2 weeks. From weeks 35-72, the patient received five more injections and remained disease free.
“After that, she did have another flare, so we used omalizumab again,” but without the same excellent results. “She had an initial decrease in pruritus, and symptom improvement, but her disease subsequently worsened. We restarted her on prednisone and azathioprine and she has done well.”
Dr. Yu said he made “a couple of interesting observations on this case.”
“We saw no real correlation between disease activity score, and the levels of serum IgG antibodies. But we did notice a parallel correlation with the level of eosinophil and disease severity and also treatment response,” he said. “It was quite clear that immediately after injection, she had a dramatic drop in eosinophils” from 1,600 to 60 cells/microL within 24 hours.
His next case was a 72-year-old woman with a history of somewhat controlled essential tremor, and 6 months of highly pruritic BP blistering. She had been treated with 60 mg/day prednisone, but didn’t tolerate it well, developing steroid-induced psychosis with agitation and violence, and a worsening of her tremor. The steroid was tapered to 40 mg/day and azathioprine was added, but she was did not respond to this change and continued to develop new blisters each day. She was admitted to the hospital for plasmapheresis, which was not helpful. Nor did she respond to six cycles of cyclophosphamide.
At that point, Dr. Yu drew IgE and eosinophil levels: Her absolute eosinophil count was 1,600 cells/microL and IgE was 287 units/mL. He then gave the patient 300 mg omalizumab subcutaneously.
“Ten days after a single injection, her blisters had almost completely resolved,” he said. “To briefly describe her disease course, the blisters went away, and she had resolution of her pruritus. She was discharged with 1 month of prednisone, but we tapered that and have been able to maintain her on omalizumab alone. She had one mild flare, which was readily controlled with prednisone. The last time we saw her, she was disease free.”
He also described four other steroid-refractory BP patients treated with omalizumab.“Their commonalities were that they all had steroid-refractory disease that was resistant to immunosuppressants, had a high level of IgE, and most of them also had eosinophilia.”
Dr. Yu’s descriptions:
• A 78-year-old woman with refractory BP of 1.5 years responded well to three initial injections spaced 6 and 4 weeks apart, and has been well maintained for 20 months with 300-mL injections administered once a month. One relapse was easily controlled.
• A 72-year-old woman with 3.5 years of refractory BP responded well to 375 mg injections every 4 weeks and has been symptom free for a year on that maintenance dose.
• A 55-year-old woman with a 7-month history of refractory BP experienced a 30% reduction in body surface area blistering within 1 week of her first 375-mg injection. By 3 weeks, she was clear. She had three injections, 2 weeks apart, and was disease free for 3 months.
• An 86-year-old woman with longstanding refractory BP experienced a 22% reduction in blister count within a week of her first 375-mL injection. After a series of injections every 2 weeks, however, she developed an exacerbation of her preexisting chronic obstructive pulmonary disease, which was due primarily to tapering her prednisone. However, she no longer uses omalizumab.
“It is difficult to make recommendations because of the limitations of our data,” Dr. Yu said. “But based on the small number of patients we have treated, I would consider using omalizumab in patients with resistant disease who have an elevated IgE and eosinophil count. The optimal dosing regimen is not yet determined. Our approach is to start out with the asthma dosing and titrate until we see improvement. We use the highest dose indicated for the patient’s weight and IgE levels, typically 300-375 mg subcutaneously every 2-8 weeks, and start tapering when the patient gets control.”
He had no financial disclosures.
[email protected]
On Twitter @Alz_Gal
ORLANDO – Omalizumab, a monoclonal anti-IgE antibody, may be a good option for patients with treatment-refractory bullous pemphigoid.
Patients who received omalizumab (Xolair) experienced rapid improvements, with 30%-50% lesion clearance within a week and complete clearance by 3 weeks, Kenneth Yu, MD, said at the annual meeting of the American Academy of Dermatology. With regular injections, they were kept symptom free for months. Some patients did flare, but were then easily controlled on standard treatment. Omalizumab, approved by the Food and Drug Administration in 2003, is indicated for moderate to severe persistent asthma and chronic idiopathic urticaria.
“We have now treated six patients with omalizumab with very good results with five of them. These are not your garden-variety BP patients, but people with very treatment-resistant disease who have failed treatment with corticosteroids alone, and in combination with other immunosuppressants.”
The rapid clinical improvements, along with observations that eosinophilia decreased with treatment, “strengthen the evidence that BP is an IgE-mediated, organ-specific autoimmune disease,” said Dr. Yu, senior resident in dermatology at the University of Michigan, Ann Arbor.
“Would I use this as a first-line treatment for BP? Probably not. But if you are seeing someone who’s nonresponsive to therapy, you might want to check IgE and eosinophil levels and if those are elevated, you might consider omalizumab as an adjunct treatment – and you might observe a fairly dramatic response.”
Three of Dr. Yu’s patients received omalizumab as monotherapy, and three received it in conjunction with other immunosuppressants. He described their disease presentation, treatment, and progression.
In general, Dr. Yu reserves omalizumab for patients with refractory disease and two particular clinical characteristics: high eosinophil count and elevated serum IgE. The initial dosing is based on the asthma treatment nomogram for the drug; he titrates it according to clinical response. “We don’t alter the total dose given, but we do adjust the frequency with which we give it.”
His first patient was a 70-year-old woman with a 1-year history of poorly controlled BP; she had failed prednisone, azathioprine, and minocycline. She also had a history of steroid-related vertebral compression fractures. She presented with an eosinophil count of over 400 cells/microL.
“We treated her with subcutaneous injections of 300 mg every 2 weeks for 16 weeks,” Dr. Yu said. Within 1 week, she had a 44% reduction in blisters; within 4 weeks, she had gone from 50% body surface area involvement to 5%.
After eight injections, the patient was disease free and Dr. Yu discontinued treatment. She remained clear until week 32 after treatment initiation; she had a flare manifested by increased pruritus and recurrence of lesions. Dr. Yu restarted omalizumab and the lesions cleared within 2 weeks. From weeks 35-72, the patient received five more injections and remained disease free.
“After that, she did have another flare, so we used omalizumab again,” but without the same excellent results. “She had an initial decrease in pruritus, and symptom improvement, but her disease subsequently worsened. We restarted her on prednisone and azathioprine and she has done well.”
Dr. Yu said he made “a couple of interesting observations on this case.”
“We saw no real correlation between disease activity score, and the levels of serum IgG antibodies. But we did notice a parallel correlation with the level of eosinophil and disease severity and also treatment response,” he said. “It was quite clear that immediately after injection, she had a dramatic drop in eosinophils” from 1,600 to 60 cells/microL within 24 hours.
His next case was a 72-year-old woman with a history of somewhat controlled essential tremor, and 6 months of highly pruritic BP blistering. She had been treated with 60 mg/day prednisone, but didn’t tolerate it well, developing steroid-induced psychosis with agitation and violence, and a worsening of her tremor. The steroid was tapered to 40 mg/day and azathioprine was added, but she was did not respond to this change and continued to develop new blisters each day. She was admitted to the hospital for plasmapheresis, which was not helpful. Nor did she respond to six cycles of cyclophosphamide.
At that point, Dr. Yu drew IgE and eosinophil levels: Her absolute eosinophil count was 1,600 cells/microL and IgE was 287 units/mL. He then gave the patient 300 mg omalizumab subcutaneously.
“Ten days after a single injection, her blisters had almost completely resolved,” he said. “To briefly describe her disease course, the blisters went away, and she had resolution of her pruritus. She was discharged with 1 month of prednisone, but we tapered that and have been able to maintain her on omalizumab alone. She had one mild flare, which was readily controlled with prednisone. The last time we saw her, she was disease free.”
He also described four other steroid-refractory BP patients treated with omalizumab.“Their commonalities were that they all had steroid-refractory disease that was resistant to immunosuppressants, had a high level of IgE, and most of them also had eosinophilia.”
Dr. Yu’s descriptions:
• A 78-year-old woman with refractory BP of 1.5 years responded well to three initial injections spaced 6 and 4 weeks apart, and has been well maintained for 20 months with 300-mL injections administered once a month. One relapse was easily controlled.
• A 72-year-old woman with 3.5 years of refractory BP responded well to 375 mg injections every 4 weeks and has been symptom free for a year on that maintenance dose.
• A 55-year-old woman with a 7-month history of refractory BP experienced a 30% reduction in body surface area blistering within 1 week of her first 375-mg injection. By 3 weeks, she was clear. She had three injections, 2 weeks apart, and was disease free for 3 months.
• An 86-year-old woman with longstanding refractory BP experienced a 22% reduction in blister count within a week of her first 375-mL injection. After a series of injections every 2 weeks, however, she developed an exacerbation of her preexisting chronic obstructive pulmonary disease, which was due primarily to tapering her prednisone. However, she no longer uses omalizumab.
“It is difficult to make recommendations because of the limitations of our data,” Dr. Yu said. “But based on the small number of patients we have treated, I would consider using omalizumab in patients with resistant disease who have an elevated IgE and eosinophil count. The optimal dosing regimen is not yet determined. Our approach is to start out with the asthma dosing and titrate until we see improvement. We use the highest dose indicated for the patient’s weight and IgE levels, typically 300-375 mg subcutaneously every 2-8 weeks, and start tapering when the patient gets control.”
He had no financial disclosures.
[email protected]
On Twitter @Alz_Gal
AT AAD 17
FDA grants breakthrough therapy status to rituximab for pemphigus vulgaris
The Food and Drug Administration has granted breakthrough therapy status to rituximab (Rituxan) for treating pemphigus vulgaris, according to the manufacturer.
Rituximab, a CD20-directed cytolytic antibody approved in 1997, is currently in a phase III study evaluating its efficacy for the pemphigus indication. It is approved in the United States for treating non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis (with methotrexate), granulomatosis with polyangiitis (Wegener’s granulomatosis), and microscopic polyangiitis (with glucocorticoids).![]()
The patients, who were experiencing their first episode of pemphigus vulgaris, were randomized to daily oral prednisone, tapered over a 12- to 18-month period, or rituximab administered intravenously (at days 0 and 14, and months 12 and 18), plus daily oral prednisone, tapered over 3 or 6 months. At 2 years, when they were no longer on therapy, 89% of those treated with rituximab and prednisone were in complete remission, compared with 34% of those treated with prednisone alone (P less than .0001).
The breakthrough therapy process is “designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s),” according to the FDA.
The study was supported by the French Ministry of Health, the French Society of Dermatology, and Roche, which owns Genentech. Genentech markets rituximab in the United States with Biogen and is conducting the phase III study.
The Food and Drug Administration has granted breakthrough therapy status to rituximab (Rituxan) for treating pemphigus vulgaris, according to the manufacturer.
Rituximab, a CD20-directed cytolytic antibody approved in 1997, is currently in a phase III study evaluating its efficacy for the pemphigus indication. It is approved in the United States for treating non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis (with methotrexate), granulomatosis with polyangiitis (Wegener’s granulomatosis), and microscopic polyangiitis (with glucocorticoids).![]()
The patients, who were experiencing their first episode of pemphigus vulgaris, were randomized to daily oral prednisone, tapered over a 12- to 18-month period, or rituximab administered intravenously (at days 0 and 14, and months 12 and 18), plus daily oral prednisone, tapered over 3 or 6 months. At 2 years, when they were no longer on therapy, 89% of those treated with rituximab and prednisone were in complete remission, compared with 34% of those treated with prednisone alone (P less than .0001).
The breakthrough therapy process is “designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s),” according to the FDA.
The study was supported by the French Ministry of Health, the French Society of Dermatology, and Roche, which owns Genentech. Genentech markets rituximab in the United States with Biogen and is conducting the phase III study.
The Food and Drug Administration has granted breakthrough therapy status to rituximab (Rituxan) for treating pemphigus vulgaris, according to the manufacturer.
Rituximab, a CD20-directed cytolytic antibody approved in 1997, is currently in a phase III study evaluating its efficacy for the pemphigus indication. It is approved in the United States for treating non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis (with methotrexate), granulomatosis with polyangiitis (Wegener’s granulomatosis), and microscopic polyangiitis (with glucocorticoids).![]()
The patients, who were experiencing their first episode of pemphigus vulgaris, were randomized to daily oral prednisone, tapered over a 12- to 18-month period, or rituximab administered intravenously (at days 0 and 14, and months 12 and 18), plus daily oral prednisone, tapered over 3 or 6 months. At 2 years, when they were no longer on therapy, 89% of those treated with rituximab and prednisone were in complete remission, compared with 34% of those treated with prednisone alone (P less than .0001).
The breakthrough therapy process is “designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s),” according to the FDA.
The study was supported by the French Ministry of Health, the French Society of Dermatology, and Roche, which owns Genentech. Genentech markets rituximab in the United States with Biogen and is conducting the phase III study.
VIDEO: Looking at keloids from a different perspective
ORLANDO – It may be time to start considering new options for treating keloids, according to Amy McMichael, MD, professor of dermatology at Wake Forest University, Winston-Salem, N.C.
At the annual meeting of the American Academy of Dermatology, Dr. McMichael discussed one of the highlights of the annual symposium of the Skin of Color Society, held right before the annual meeting, a presentation by Michael Tirgan, MD, who has treated patients with keloids for about 10 years.
Dr. Tirgan, an oncologist based in New York, has a large database and registry of patients and shared some interesting data at the symposium. “What he’s found is that those who come with the supermassive and massive keloids are those who have had the most surgery on their keloid,” Dr. McMichael said in a video interview at the meeting.
His findings shed light on what she described as “a new perspective in the way that we think about keloids” and a new approach to treatment – considering keloids as tumors – not just a scar.
Dr. McMichael had no relevant disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – It may be time to start considering new options for treating keloids, according to Amy McMichael, MD, professor of dermatology at Wake Forest University, Winston-Salem, N.C.
At the annual meeting of the American Academy of Dermatology, Dr. McMichael discussed one of the highlights of the annual symposium of the Skin of Color Society, held right before the annual meeting, a presentation by Michael Tirgan, MD, who has treated patients with keloids for about 10 years.
Dr. Tirgan, an oncologist based in New York, has a large database and registry of patients and shared some interesting data at the symposium. “What he’s found is that those who come with the supermassive and massive keloids are those who have had the most surgery on their keloid,” Dr. McMichael said in a video interview at the meeting.
His findings shed light on what she described as “a new perspective in the way that we think about keloids” and a new approach to treatment – considering keloids as tumors – not just a scar.
Dr. McMichael had no relevant disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – It may be time to start considering new options for treating keloids, according to Amy McMichael, MD, professor of dermatology at Wake Forest University, Winston-Salem, N.C.
At the annual meeting of the American Academy of Dermatology, Dr. McMichael discussed one of the highlights of the annual symposium of the Skin of Color Society, held right before the annual meeting, a presentation by Michael Tirgan, MD, who has treated patients with keloids for about 10 years.
Dr. Tirgan, an oncologist based in New York, has a large database and registry of patients and shared some interesting data at the symposium. “What he’s found is that those who come with the supermassive and massive keloids are those who have had the most surgery on their keloid,” Dr. McMichael said in a video interview at the meeting.
His findings shed light on what she described as “a new perspective in the way that we think about keloids” and a new approach to treatment – considering keloids as tumors – not just a scar.
Dr. McMichael had no relevant disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT AAD 17
